User login
Case Report
A 5 year‐old girl presented to an emergency department (ED) with a 2‐day history of more than 12 episodes of nonbilious emesis. She received intravenous (IV) promethazine and 400 cc of normal saline and was discharged home. When emesis recurred the next morning, her pediatrician referred her for admission to our hospital for further hydration. The patient had neither fever nor rash but did report periumbilical pain and a few loose stools. She did not receive any medications other than the single dose of promethazine. Parents denied other toxic ingestion. The patient's family members had had a diarrheal illness over the past days.
The patient was born at term, small for gestational age, with uncomplicated pregnancy and delivery. She had normal growth and development and previously had been healthy, although her mother reported several prior ED visits for vomiting, including emesis with upper respiratory illnesses (URIs). There was no family history of gastrointestinal, metabolic, or renal disease. The patient lived with her parents and brother and attended kindergarten.
On examination, her temperature was 98.6F, blood pressure was 103/66, heart rate was 88, and respiratory rate was 24. She weighed 17.8 kg and was 115 cm tall (35th and 75th percentiles, respectively). She was alert and cooperative. Her breath had a ketotic odor. She had somewhat sunken eyes and dry mucous membranes. Her neck was supple, without lymphadenopathy. Capillary refill time was 3 seconds. Her lungs were clear to auscultation. Her abdomen was soft, nontender to palpation, and nondistended, and bowel sounds were hyperactive. Liver and spleen were of normal size. There was no edema, clubbing, nor cyanosis in the extremities. The patient was very fair‐skinned and did not have rashes, bruises, or other skin lesions. The results of her neurologic exam were entirely normal. Initial serum chemistry results were: sodium, 140 mEq/L; potassium, 5.9 mEq/L (hemolyzed); chloride, 107 mEq/L; bicarbonate, 13 mEq/L; glucose, 52 mg/dL; BUN, 19 mg/dL; creatinine, 0.5 mg/dL; and calcium, 10.5 mg/dL. Other laboratory analyses were ordered including urinalysis (UA), serum lactate, stool culture, rotaviral test, and fecal white blood cell count.
RESULTS
Further laboratory analysis revealed a urine pH of 5.5, specific gravity of 1.023, and 3+ ketones, with an otherwise normal UA. Blood lactic acid and pyruvic acid levels were normal at 0.8 and 0.11 mmol/L, respectively. All stool studies were negative. Tests to determine plasma quantitative amino acid and urine quantitative organic acid levels were ordered. The clinical course was benign, with recovery and normalization of blood chemistry values within 24 hours of IV hydration. The patient was discharged home the next day. Results of biochemical genetics laboratory testing were available 5 days later (Table 1). Plasma amino acids showed a decreased level of alanine, with elevated levels of leucine, isoleucine, and valine. L‐alloisoleucine, which is not normally in plasma but is pathognomic for maple syrup urine disease (MSUD), was detected. Urine organic acid test results were notable for ketonuria, with elevated branched‐chain 2‐hydroxy and 2‐oxo acids consistent with a diagnosis of MSUD (Table 1).
| Measured (mol/L) | Normal Range | |
|---|---|---|
| Plasma amino acids | ||
| Alanine | 103 | 246‐486 |
| Leucine | 434 | 61‐168 |
| Valine | 576 | 110‐279 |
| Isoleucine | 280 | 39‐88 |
| L‐Alloisoleucine | 28 | 0 |
| Measured (mmol/mol creatinine) | Normal Range | |
| Urine organic acids | ||
| 3‐Hydroxybutyric | 13,000 | 0‐10 |
| 2‐Hydroxyisovaleric | 16 | 0‐6 |
| 2‐Hydroxyisocaproic | 0 | 0‐2 |
| 2‐Hydroxy‐3‐methylvaleric | 0 | 0‐2 |
| 2‐Oxoisovaleric | 0 | 0‐2 |
| 2‐Oxoisocaproic | 0 | 0‐2 |
| 2‐Oxo‐3‐methylvaleric | 41 | 0‐2 |
The patient has done very well on follow‐up. Modest restriction of protein intake was instituted (2 g/kg daily), and approximately 1 month after hospitalization a trial of thiamine was started. Plasma amino acid and urine organic acids have been normal on subsequent testing while the patient has been clinically well. Whole‐body leucine oxidation was estimated by quantitation of 13CO2 after administration of an oral bolus of 1‐13C‐leucine, following the protocol of Elsas et al.1 The enrichment of 13CO2 indicated oxidation of approximately 11% of the administered leucine over 3 hours, which was at the lower end of the normal range, with no increase after thiamine supplementation for 1 month. The patient did have 2 episodes of vomiting associated with mild intercurrent illness 1 month and 1 year after hospitalization. Care was sought promptly at urgent care centers. Ketonuria was documented; however, no blood tests were ordered. There were no changes in mental status, and oral rehydration was successful. Molecular analysis of the branched chain ketoacid dehydrogenase complex E1, E1, and E2 subunit genes did not reveal any mutations in the coding regions, although 3 sequence variations were observed in E1 (2 silent changes, c.972C>T [Phe324Phe] and c.1221A>G [Leu407Leu], and c.376G>T [Gly126Cys], at this time of unclear significance).
DISCUSSION
We report the case of a young girl who presented with what was initially labeled simple gastroenteritis. It is important to note, however, that she had several days of repeated emesis, no fever, and minimal diarrhea, along with multiple previous episodes of vomiting illnesses requiring ED visits. Combined, these prompted further evaluation of her acidosis. Maple syrup urine disease, a congenital condition that can be lethal, went undiagnosed in this patient until this admission when she was 5 years old.
Metabolic Acidosis
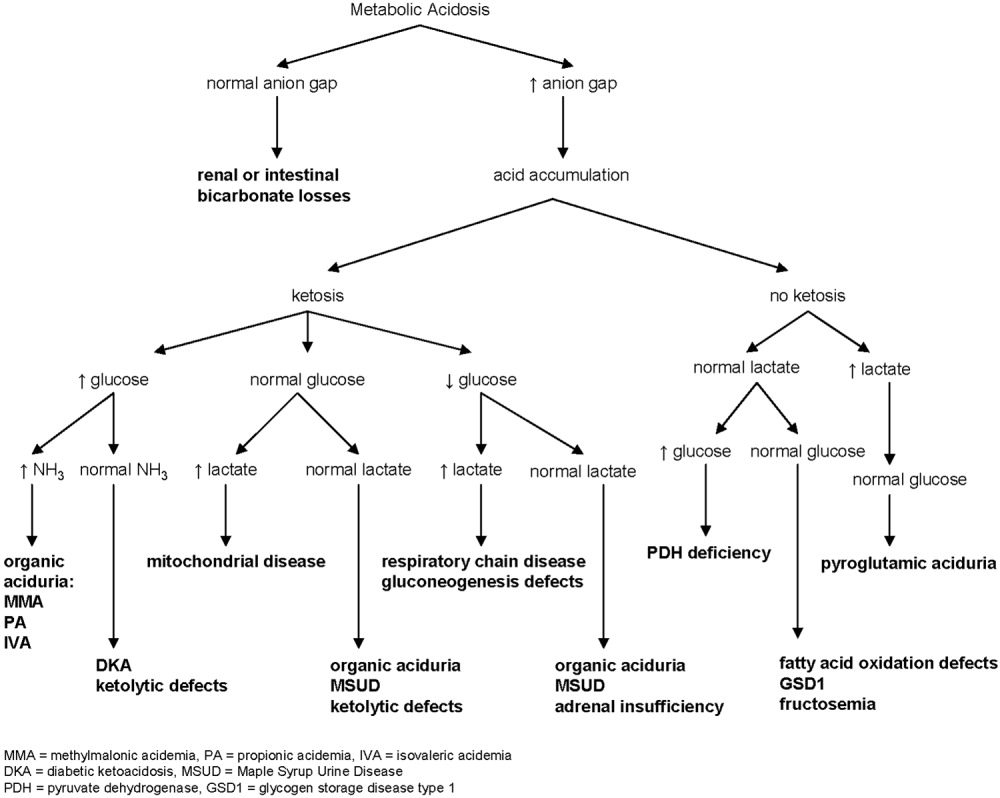
Metabolic acidosis is a very common laboratory abnormality caused by 1 of 3 basic mechanisms: loss of bicarbonate, impaired renal acid excretion, or the addition of either endogenous or exogenous acids to the body. Common causes of nonanion gap metabolic acidosis in children include diarrhea and renal tubular acidosis (RTA). Increased anion gap is associated with lactic acidosis, ketoacidosis, and ingestion of such substances as methanol, ethylene glycol, acetylsalicylic acid, and bismuth subsalicylate. Inborn errors of metabolism cause production of ketoacids, lactic acid, and other organic anions. This can occur chronically or during acute decompensation with illness, stress, or therapy noncompliance.2 Serum anion gap, glucose, ketones, lactate, and ammonia can help to elucidate the specific etiology of metabolic acidosis (Fig. 1).
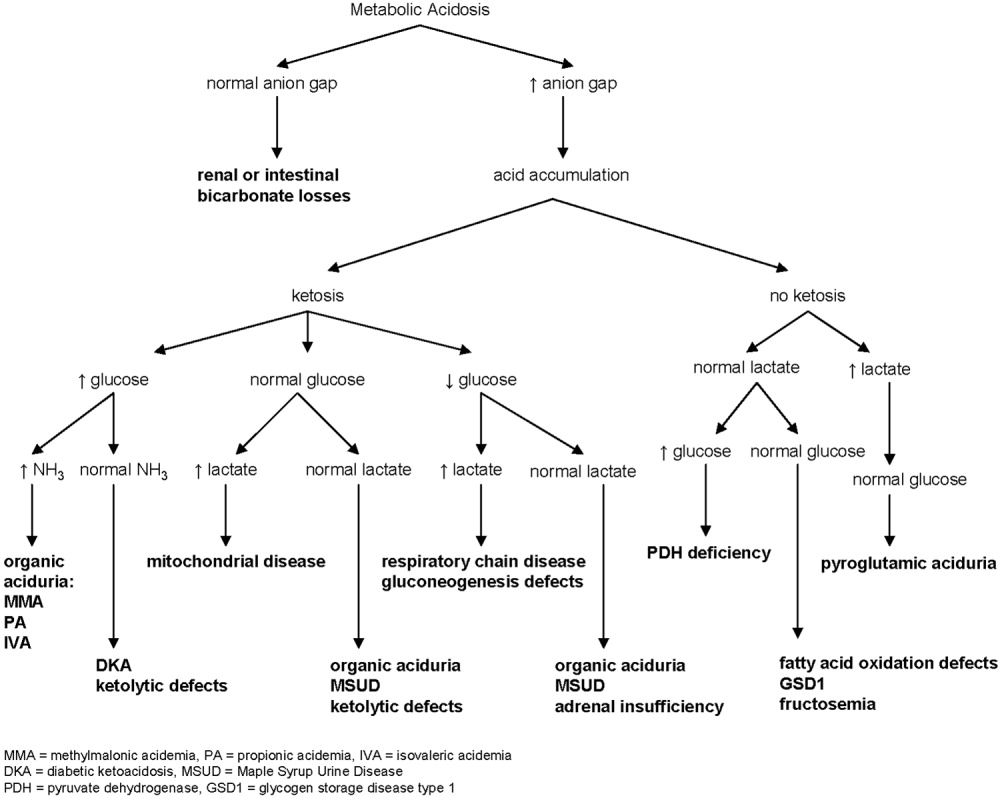
In our patient whose anion gap was at the upper limit of normal, both increased gap and non‐anion gap were considered as causes of the metabolic acidosis. Diarrhea was minimal, and there was no history of toxin or medication ingestion other than promethazine. RTA was unlikely given the borderline high serum anion gap and normal UA. Lactate level was normal, and examination did not find evidence of profound dehydration, both refuting that she had lactic acidosis severe enough to account for a serum bicarbonate of 13 mEq/L. Inborn errors of metabolism were therefore strongly considered.
Maple Syrup Urine Disease
Background
MSUD, or branched‐chain ketoaciduria, is a disease resulting from defects in the catabolic pathway of the branched‐chain amino acids (BCAAs) isoleucine, leucine, and valine. The deficient enzyme is the branched‐chain alpha‐ketoacid dehydrogenase complex (BCKDC), an enzyme system responsible for oxidative decarboxylation of the 2‐oxoacid transamination products of isoleucine, leucine, and valine. BCKDC is made up of 4 subunits (E1, E1, E2, and E3); its coenzymes include thiamine (vitamin B1) and lipoic acid. Deficiency of BCKDC leads to accumulation of BCAAs and the related branched chain oxoacids and organic acid intermediates, including one (sotalone) that lends a sweet odor reminiscent of maple syrup to sweat, cerumen, and urine. MSUD is autosomal recessive, with more than 100 specific mutations identified in the 4 genes encoding BCKDC.3 MSUD is a rare disease, occurring in 1 of every 180,000 newborns in the United States.2
Clinical Phenotypes
MSUD has been divided into at least 5 clinical phenotypes,4 although in several cases distinctions are not clear. Differences result from variation in the severity of enzyme deficiency. Classic MSUD is the most severe form, with less than 2% of normal BCKDC function; it presents in the first week of life with poor feeding and neurologic signs such as hypo/hypertonia, seizures, lethargy, and coma.5 General laboratory findings are nonspecific except for ketoacidosis. This form is rapidly fatal in the first months of life if not treated. Intermediate MSUD is milder, having 3%‐30% of BCKDC activity. Patients manifest variable degrees of retardation, developmental delay, and failure to thrive, often without signs of ketoacidosis. Thiamine‐responsive MSUD is distinguished by the favorable response to high‐dose thiamine supplementation with significant reduction in BCAA level.6 Although it is reasonable to try treatment with thiamine in most cases of MSUD, responsive patients are very rare. MSUD due to a deficiency of the E3 subunit is the rarest form, described in fewer than 10 patients.7 Intermittent MSUD is the least severe form, with 5%‐20% BCKDC activity. Children develop with normal growth and intelligence but are at risk of acute metabolic decompensation during catabolic states such as stress, infection, or surgery. Recurrent episodes of ketoacidosis, ataxia, and lethargy can lead to coma and death if untreated.8 Initial symptoms usually occur by 2 years of age but have appeared as late as the fifth decade of life. Between episodes, a normal diet is tolerated without elevation of BCAA level.
Long‐term morbidity and mortality in MSUD is neurologic. Death is often a result of brain edema that is generally attributed to the osmotic effects of leucine and amino acid imbalance; however, pathophysiologic mechanisms remain unclear.9 Progressive white matter changes are thought to result from chronic exposure to leucine. Levels of some amino acids and neurotransmitters are reduced in MSUD, which may play a role in causing encephalopathies and coma.10
Diagnosis
Elevation of plasma BCAA level can be directly assessed by standard plasma amino acid analysis; reduction of alanine is also characteristic. L‐alloisoleucine is the most sensitive and specific marker of MSUD and is pathognomic for MSUD.1112 In most cases of intermittent MSUD it is detectable at all times, including when BCAA level is normal, but in some cases it may be absent between episodes. Urine organic acids show elevation of the 2‐oxoacids corresponding to leucine, valine, and isoleucine and the corresponding 2‐hydroxy‐acids. Modern newborn screening programs generally ascertain MSUD by liquid chromatographytandem mass spectrometry, which detects elevated levels of leucine, isoleucine, and L‐alloisoleucine or the ratio of these to alanine. BCAA concentration is elevated in plasma within hours of birth; however, it is unclear how often intermittent MSUD might be missed in newborn screening.
Treatment
Therapy consists of dietary control (limited protein intake) and, in some cases, thiamine supplementation. The goal is to limit BCAA so as not to overwhelm the capacity of the BCKDC. However, BCAAs are essential, and the challenge is therefore to provide appropriate amounts of protein to sustain growth without exceeding the individual's metabolic capacity. Generally, the amount of dietary protein tolerated is insufficient to provide enough of the other essential amino acids, so supplemental amino acids are needed. During acute episodes a BCAA‐free diet, sometimes with insulin and glucose, is used to encourage BCAA removal. If this anabolic approach is unsuccessful, dialysis may be used. Orthotopic liver transplantation has also been performed.1315 In all cases so far, BCAA level normalized after transplantation, and metabolic control was sustainable on a regular diet without protein restriction.13, 1618
Lessons for the Physician
Vomiting without true diarrhea deserves careful evaluation. Inborn errors of metabolism are individually rare but as a group are fairly common. One study noted an incidence of 15.7 per 100,000 births detected on tandem mass spectrometry screening, whereas rates of clinical detection are lower.19 This case illustrates how symptoms may be nonspecific and can easily mimic simple gastroenteritis. The following may be helpful when evaluating a patient with isolated emesis:
-
History: Does the patient have a history of emesis with illnesses that usually do not cause vomiting, such as an asthma flare or URI? Has the patient required emergency treatment or IV fluids for bouts of emesis in the past? Could toxins or medications that cause metabolic acidosis have been ingested?
-
Physical exam: Skin lesions, odors, and hepatomegaly are sometimes found in metabolic disorders. Commonly recognized odors include the musty smell of phenylketonuria and the sweet maple syrup smell of MSUD.
-
Laboratory studies: UA and calculation of anion gap are first steps to take to eliminate more common causes of metabolic acidosis in children.
-
Reliance on newborn screening: Patients with intermittent MSUD may have normal BCAA levels between acute episodes. Newborn screening tests may not identify these patients. The physician must maintain a degree of suspicion in approaching an acute illness that might indicate a metabolic disease, even in a child who has had negative expanded newborn screening.
Unusual disorders may masquerade as a simple problem. Common laboratory tests and a thorough history and exam can help to differentiate between simple gastroenteritis and inborn metabolic error and guide further diagnostic evaluation.
- ,,.Practical methods to estimate whole body leucine oxidation in maple syrup urine disease.Pediatr Res.1993;33:445–451.
- Behrman RE,Kliegman RM,Jenson HB, editors.Nelson Textbook of Pediatrics.Philadelphia:Saunders;2004:224–235,409–418.
- ,,.Lessons from genetic disorders of branched‐chain amino acid metabolism.J Nutr.2006;136(suppl 1):243S–249S.
- ,.Maple syrup urine disease (branched‐chain ketoaciduria). In:Scriver CR,Beaudet AL,Sly WS,Valle D,Vogelstein B,Childs B, editors.The Metabolic and Molecular Basis of Inherited Disease.New York:McGraw‐Hill;2001:1971–2006.
- ,,.A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance.Pediatrics.1954;14:462–467.
- ,,,.Thiamine‐responsive maple‐syrup‐urine disease.Lancet.1971;1:310–312.
- ,,.Deficiency of dihydrolipoyl dehydrogenase (a component of the pyruvate and ‐ketoglutarate dehydrogenase complexes): a cause of congenital chronic lactic acidosis in infancy.Pediatr Res.1977;11:1198–202.
- ,,.Late‐onset branched‐chain ketoaciduria: (maple syrup urine disease).J Lancet.1966;86(3):149–152.
- ,,,.Cerebral edema causing death in children with maple syrup urine disease.J Pediatr.1991;119:42–45.
- ,,,,,,.Glutamate and γ‐aminobutyric acid neurotransmitter systems in the acute phase of maple syrup urine disease and citrullinemia encephalopathies in newborn calves.J Neurochem.1992;59:582–590.
- ,,,Maple syrup urine disease, with particular reference to dietotherapy.Pediatrics.1964;34:454–472.
- ,,,.Significance of L‐alloisoleucine in plasma for diagnosis of maple syrup urine disease.Clin Chem.1999;45:1734–40.
- ,,, et al.Mid‐term outcome of 2 cases with maple syrup urine disease: role of liver transplantation in the treatment.Arch Pediatr.1994;1:730–734.
- , et al.Transplantation for maple syrup urine disease (MSUD) and methylmalonic acidopathy (MMA).J Inherit Metab Dis.1997;20(suppl 1):37.
- ,,,.Liver transplantation in maple syrup urine disease.Eur J Pediatrics.1999;158(suppl 2):S60–S64.
- ,,, et al.Elective liver transplantation for the treatment of classical maple syrup urine disease.Am J Transplant.2006;6:557–564.
- ,,,,,.Domino liver transplantation in maple syrup urine disease.Liver Transpl.2006;12:876–882.
- ,,,.Branched‐chain L‐amino acid metabolism in classical maple syrup urine disease after orthotopic liver transplantation.J Inherit Metab Dis.2000;23:805–818.
- ,,,.Screening newborns for inborn errors of metabolism by tandem mass spectrometry.N Engl J Med.2003;348:2304–2312.
A 5 year‐old girl presented to an emergency department (ED) with a 2‐day history of more than 12 episodes of nonbilious emesis. She received intravenous (IV) promethazine and 400 cc of normal saline and was discharged home. When emesis recurred the next morning, her pediatrician referred her for admission to our hospital for further hydration. The patient had neither fever nor rash but did report periumbilical pain and a few loose stools. She did not receive any medications other than the single dose of promethazine. Parents denied other toxic ingestion. The patient's family members had had a diarrheal illness over the past days.
The patient was born at term, small for gestational age, with uncomplicated pregnancy and delivery. She had normal growth and development and previously had been healthy, although her mother reported several prior ED visits for vomiting, including emesis with upper respiratory illnesses (URIs). There was no family history of gastrointestinal, metabolic, or renal disease. The patient lived with her parents and brother and attended kindergarten.
On examination, her temperature was 98.6F, blood pressure was 103/66, heart rate was 88, and respiratory rate was 24. She weighed 17.8 kg and was 115 cm tall (35th and 75th percentiles, respectively). She was alert and cooperative. Her breath had a ketotic odor. She had somewhat sunken eyes and dry mucous membranes. Her neck was supple, without lymphadenopathy. Capillary refill time was 3 seconds. Her lungs were clear to auscultation. Her abdomen was soft, nontender to palpation, and nondistended, and bowel sounds were hyperactive. Liver and spleen were of normal size. There was no edema, clubbing, nor cyanosis in the extremities. The patient was very fair‐skinned and did not have rashes, bruises, or other skin lesions. The results of her neurologic exam were entirely normal. Initial serum chemistry results were: sodium, 140 mEq/L; potassium, 5.9 mEq/L (hemolyzed); chloride, 107 mEq/L; bicarbonate, 13 mEq/L; glucose, 52 mg/dL; BUN, 19 mg/dL; creatinine, 0.5 mg/dL; and calcium, 10.5 mg/dL. Other laboratory analyses were ordered including urinalysis (UA), serum lactate, stool culture, rotaviral test, and fecal white blood cell count.
RESULTS
Further laboratory analysis revealed a urine pH of 5.5, specific gravity of 1.023, and 3+ ketones, with an otherwise normal UA. Blood lactic acid and pyruvic acid levels were normal at 0.8 and 0.11 mmol/L, respectively. All stool studies were negative. Tests to determine plasma quantitative amino acid and urine quantitative organic acid levels were ordered. The clinical course was benign, with recovery and normalization of blood chemistry values within 24 hours of IV hydration. The patient was discharged home the next day. Results of biochemical genetics laboratory testing were available 5 days later (Table 1). Plasma amino acids showed a decreased level of alanine, with elevated levels of leucine, isoleucine, and valine. L‐alloisoleucine, which is not normally in plasma but is pathognomic for maple syrup urine disease (MSUD), was detected. Urine organic acid test results were notable for ketonuria, with elevated branched‐chain 2‐hydroxy and 2‐oxo acids consistent with a diagnosis of MSUD (Table 1).
| Measured (mol/L) | Normal Range | |
|---|---|---|
| Plasma amino acids | ||
| Alanine | 103 | 246‐486 |
| Leucine | 434 | 61‐168 |
| Valine | 576 | 110‐279 |
| Isoleucine | 280 | 39‐88 |
| L‐Alloisoleucine | 28 | 0 |
| Measured (mmol/mol creatinine) | Normal Range | |
| Urine organic acids | ||
| 3‐Hydroxybutyric | 13,000 | 0‐10 |
| 2‐Hydroxyisovaleric | 16 | 0‐6 |
| 2‐Hydroxyisocaproic | 0 | 0‐2 |
| 2‐Hydroxy‐3‐methylvaleric | 0 | 0‐2 |
| 2‐Oxoisovaleric | 0 | 0‐2 |
| 2‐Oxoisocaproic | 0 | 0‐2 |
| 2‐Oxo‐3‐methylvaleric | 41 | 0‐2 |
The patient has done very well on follow‐up. Modest restriction of protein intake was instituted (2 g/kg daily), and approximately 1 month after hospitalization a trial of thiamine was started. Plasma amino acid and urine organic acids have been normal on subsequent testing while the patient has been clinically well. Whole‐body leucine oxidation was estimated by quantitation of 13CO2 after administration of an oral bolus of 1‐13C‐leucine, following the protocol of Elsas et al.1 The enrichment of 13CO2 indicated oxidation of approximately 11% of the administered leucine over 3 hours, which was at the lower end of the normal range, with no increase after thiamine supplementation for 1 month. The patient did have 2 episodes of vomiting associated with mild intercurrent illness 1 month and 1 year after hospitalization. Care was sought promptly at urgent care centers. Ketonuria was documented; however, no blood tests were ordered. There were no changes in mental status, and oral rehydration was successful. Molecular analysis of the branched chain ketoacid dehydrogenase complex E1, E1, and E2 subunit genes did not reveal any mutations in the coding regions, although 3 sequence variations were observed in E1 (2 silent changes, c.972C>T [Phe324Phe] and c.1221A>G [Leu407Leu], and c.376G>T [Gly126Cys], at this time of unclear significance).
DISCUSSION
We report the case of a young girl who presented with what was initially labeled simple gastroenteritis. It is important to note, however, that she had several days of repeated emesis, no fever, and minimal diarrhea, along with multiple previous episodes of vomiting illnesses requiring ED visits. Combined, these prompted further evaluation of her acidosis. Maple syrup urine disease, a congenital condition that can be lethal, went undiagnosed in this patient until this admission when she was 5 years old.
Metabolic Acidosis
Metabolic acidosis is a very common laboratory abnormality caused by 1 of 3 basic mechanisms: loss of bicarbonate, impaired renal acid excretion, or the addition of either endogenous or exogenous acids to the body. Common causes of nonanion gap metabolic acidosis in children include diarrhea and renal tubular acidosis (RTA). Increased anion gap is associated with lactic acidosis, ketoacidosis, and ingestion of such substances as methanol, ethylene glycol, acetylsalicylic acid, and bismuth subsalicylate. Inborn errors of metabolism cause production of ketoacids, lactic acid, and other organic anions. This can occur chronically or during acute decompensation with illness, stress, or therapy noncompliance.2 Serum anion gap, glucose, ketones, lactate, and ammonia can help to elucidate the specific etiology of metabolic acidosis (Fig. 1).
In our patient whose anion gap was at the upper limit of normal, both increased gap and non‐anion gap were considered as causes of the metabolic acidosis. Diarrhea was minimal, and there was no history of toxin or medication ingestion other than promethazine. RTA was unlikely given the borderline high serum anion gap and normal UA. Lactate level was normal, and examination did not find evidence of profound dehydration, both refuting that she had lactic acidosis severe enough to account for a serum bicarbonate of 13 mEq/L. Inborn errors of metabolism were therefore strongly considered.
Maple Syrup Urine Disease
Background
MSUD, or branched‐chain ketoaciduria, is a disease resulting from defects in the catabolic pathway of the branched‐chain amino acids (BCAAs) isoleucine, leucine, and valine. The deficient enzyme is the branched‐chain alpha‐ketoacid dehydrogenase complex (BCKDC), an enzyme system responsible for oxidative decarboxylation of the 2‐oxoacid transamination products of isoleucine, leucine, and valine. BCKDC is made up of 4 subunits (E1, E1, E2, and E3); its coenzymes include thiamine (vitamin B1) and lipoic acid. Deficiency of BCKDC leads to accumulation of BCAAs and the related branched chain oxoacids and organic acid intermediates, including one (sotalone) that lends a sweet odor reminiscent of maple syrup to sweat, cerumen, and urine. MSUD is autosomal recessive, with more than 100 specific mutations identified in the 4 genes encoding BCKDC.3 MSUD is a rare disease, occurring in 1 of every 180,000 newborns in the United States.2
Clinical Phenotypes
MSUD has been divided into at least 5 clinical phenotypes,4 although in several cases distinctions are not clear. Differences result from variation in the severity of enzyme deficiency. Classic MSUD is the most severe form, with less than 2% of normal BCKDC function; it presents in the first week of life with poor feeding and neurologic signs such as hypo/hypertonia, seizures, lethargy, and coma.5 General laboratory findings are nonspecific except for ketoacidosis. This form is rapidly fatal in the first months of life if not treated. Intermediate MSUD is milder, having 3%‐30% of BCKDC activity. Patients manifest variable degrees of retardation, developmental delay, and failure to thrive, often without signs of ketoacidosis. Thiamine‐responsive MSUD is distinguished by the favorable response to high‐dose thiamine supplementation with significant reduction in BCAA level.6 Although it is reasonable to try treatment with thiamine in most cases of MSUD, responsive patients are very rare. MSUD due to a deficiency of the E3 subunit is the rarest form, described in fewer than 10 patients.7 Intermittent MSUD is the least severe form, with 5%‐20% BCKDC activity. Children develop with normal growth and intelligence but are at risk of acute metabolic decompensation during catabolic states such as stress, infection, or surgery. Recurrent episodes of ketoacidosis, ataxia, and lethargy can lead to coma and death if untreated.8 Initial symptoms usually occur by 2 years of age but have appeared as late as the fifth decade of life. Between episodes, a normal diet is tolerated without elevation of BCAA level.
Long‐term morbidity and mortality in MSUD is neurologic. Death is often a result of brain edema that is generally attributed to the osmotic effects of leucine and amino acid imbalance; however, pathophysiologic mechanisms remain unclear.9 Progressive white matter changes are thought to result from chronic exposure to leucine. Levels of some amino acids and neurotransmitters are reduced in MSUD, which may play a role in causing encephalopathies and coma.10
Diagnosis
Elevation of plasma BCAA level can be directly assessed by standard plasma amino acid analysis; reduction of alanine is also characteristic. L‐alloisoleucine is the most sensitive and specific marker of MSUD and is pathognomic for MSUD.1112 In most cases of intermittent MSUD it is detectable at all times, including when BCAA level is normal, but in some cases it may be absent between episodes. Urine organic acids show elevation of the 2‐oxoacids corresponding to leucine, valine, and isoleucine and the corresponding 2‐hydroxy‐acids. Modern newborn screening programs generally ascertain MSUD by liquid chromatographytandem mass spectrometry, which detects elevated levels of leucine, isoleucine, and L‐alloisoleucine or the ratio of these to alanine. BCAA concentration is elevated in plasma within hours of birth; however, it is unclear how often intermittent MSUD might be missed in newborn screening.
Treatment
Therapy consists of dietary control (limited protein intake) and, in some cases, thiamine supplementation. The goal is to limit BCAA so as not to overwhelm the capacity of the BCKDC. However, BCAAs are essential, and the challenge is therefore to provide appropriate amounts of protein to sustain growth without exceeding the individual's metabolic capacity. Generally, the amount of dietary protein tolerated is insufficient to provide enough of the other essential amino acids, so supplemental amino acids are needed. During acute episodes a BCAA‐free diet, sometimes with insulin and glucose, is used to encourage BCAA removal. If this anabolic approach is unsuccessful, dialysis may be used. Orthotopic liver transplantation has also been performed.1315 In all cases so far, BCAA level normalized after transplantation, and metabolic control was sustainable on a regular diet without protein restriction.13, 1618
Lessons for the Physician
Vomiting without true diarrhea deserves careful evaluation. Inborn errors of metabolism are individually rare but as a group are fairly common. One study noted an incidence of 15.7 per 100,000 births detected on tandem mass spectrometry screening, whereas rates of clinical detection are lower.19 This case illustrates how symptoms may be nonspecific and can easily mimic simple gastroenteritis. The following may be helpful when evaluating a patient with isolated emesis:
-
History: Does the patient have a history of emesis with illnesses that usually do not cause vomiting, such as an asthma flare or URI? Has the patient required emergency treatment or IV fluids for bouts of emesis in the past? Could toxins or medications that cause metabolic acidosis have been ingested?
-
Physical exam: Skin lesions, odors, and hepatomegaly are sometimes found in metabolic disorders. Commonly recognized odors include the musty smell of phenylketonuria and the sweet maple syrup smell of MSUD.
-
Laboratory studies: UA and calculation of anion gap are first steps to take to eliminate more common causes of metabolic acidosis in children.
-
Reliance on newborn screening: Patients with intermittent MSUD may have normal BCAA levels between acute episodes. Newborn screening tests may not identify these patients. The physician must maintain a degree of suspicion in approaching an acute illness that might indicate a metabolic disease, even in a child who has had negative expanded newborn screening.
Unusual disorders may masquerade as a simple problem. Common laboratory tests and a thorough history and exam can help to differentiate between simple gastroenteritis and inborn metabolic error and guide further diagnostic evaluation.
A 5 year‐old girl presented to an emergency department (ED) with a 2‐day history of more than 12 episodes of nonbilious emesis. She received intravenous (IV) promethazine and 400 cc of normal saline and was discharged home. When emesis recurred the next morning, her pediatrician referred her for admission to our hospital for further hydration. The patient had neither fever nor rash but did report periumbilical pain and a few loose stools. She did not receive any medications other than the single dose of promethazine. Parents denied other toxic ingestion. The patient's family members had had a diarrheal illness over the past days.
The patient was born at term, small for gestational age, with uncomplicated pregnancy and delivery. She had normal growth and development and previously had been healthy, although her mother reported several prior ED visits for vomiting, including emesis with upper respiratory illnesses (URIs). There was no family history of gastrointestinal, metabolic, or renal disease. The patient lived with her parents and brother and attended kindergarten.
On examination, her temperature was 98.6F, blood pressure was 103/66, heart rate was 88, and respiratory rate was 24. She weighed 17.8 kg and was 115 cm tall (35th and 75th percentiles, respectively). She was alert and cooperative. Her breath had a ketotic odor. She had somewhat sunken eyes and dry mucous membranes. Her neck was supple, without lymphadenopathy. Capillary refill time was 3 seconds. Her lungs were clear to auscultation. Her abdomen was soft, nontender to palpation, and nondistended, and bowel sounds were hyperactive. Liver and spleen were of normal size. There was no edema, clubbing, nor cyanosis in the extremities. The patient was very fair‐skinned and did not have rashes, bruises, or other skin lesions. The results of her neurologic exam were entirely normal. Initial serum chemistry results were: sodium, 140 mEq/L; potassium, 5.9 mEq/L (hemolyzed); chloride, 107 mEq/L; bicarbonate, 13 mEq/L; glucose, 52 mg/dL; BUN, 19 mg/dL; creatinine, 0.5 mg/dL; and calcium, 10.5 mg/dL. Other laboratory analyses were ordered including urinalysis (UA), serum lactate, stool culture, rotaviral test, and fecal white blood cell count.
RESULTS
Further laboratory analysis revealed a urine pH of 5.5, specific gravity of 1.023, and 3+ ketones, with an otherwise normal UA. Blood lactic acid and pyruvic acid levels were normal at 0.8 and 0.11 mmol/L, respectively. All stool studies were negative. Tests to determine plasma quantitative amino acid and urine quantitative organic acid levels were ordered. The clinical course was benign, with recovery and normalization of blood chemistry values within 24 hours of IV hydration. The patient was discharged home the next day. Results of biochemical genetics laboratory testing were available 5 days later (Table 1). Plasma amino acids showed a decreased level of alanine, with elevated levels of leucine, isoleucine, and valine. L‐alloisoleucine, which is not normally in plasma but is pathognomic for maple syrup urine disease (MSUD), was detected. Urine organic acid test results were notable for ketonuria, with elevated branched‐chain 2‐hydroxy and 2‐oxo acids consistent with a diagnosis of MSUD (Table 1).
| Measured (mol/L) | Normal Range | |
|---|---|---|
| Plasma amino acids | ||
| Alanine | 103 | 246‐486 |
| Leucine | 434 | 61‐168 |
| Valine | 576 | 110‐279 |
| Isoleucine | 280 | 39‐88 |
| L‐Alloisoleucine | 28 | 0 |
| Measured (mmol/mol creatinine) | Normal Range | |
| Urine organic acids | ||
| 3‐Hydroxybutyric | 13,000 | 0‐10 |
| 2‐Hydroxyisovaleric | 16 | 0‐6 |
| 2‐Hydroxyisocaproic | 0 | 0‐2 |
| 2‐Hydroxy‐3‐methylvaleric | 0 | 0‐2 |
| 2‐Oxoisovaleric | 0 | 0‐2 |
| 2‐Oxoisocaproic | 0 | 0‐2 |
| 2‐Oxo‐3‐methylvaleric | 41 | 0‐2 |
The patient has done very well on follow‐up. Modest restriction of protein intake was instituted (2 g/kg daily), and approximately 1 month after hospitalization a trial of thiamine was started. Plasma amino acid and urine organic acids have been normal on subsequent testing while the patient has been clinically well. Whole‐body leucine oxidation was estimated by quantitation of 13CO2 after administration of an oral bolus of 1‐13C‐leucine, following the protocol of Elsas et al.1 The enrichment of 13CO2 indicated oxidation of approximately 11% of the administered leucine over 3 hours, which was at the lower end of the normal range, with no increase after thiamine supplementation for 1 month. The patient did have 2 episodes of vomiting associated with mild intercurrent illness 1 month and 1 year after hospitalization. Care was sought promptly at urgent care centers. Ketonuria was documented; however, no blood tests were ordered. There were no changes in mental status, and oral rehydration was successful. Molecular analysis of the branched chain ketoacid dehydrogenase complex E1, E1, and E2 subunit genes did not reveal any mutations in the coding regions, although 3 sequence variations were observed in E1 (2 silent changes, c.972C>T [Phe324Phe] and c.1221A>G [Leu407Leu], and c.376G>T [Gly126Cys], at this time of unclear significance).
DISCUSSION
We report the case of a young girl who presented with what was initially labeled simple gastroenteritis. It is important to note, however, that she had several days of repeated emesis, no fever, and minimal diarrhea, along with multiple previous episodes of vomiting illnesses requiring ED visits. Combined, these prompted further evaluation of her acidosis. Maple syrup urine disease, a congenital condition that can be lethal, went undiagnosed in this patient until this admission when she was 5 years old.
Metabolic Acidosis
Metabolic acidosis is a very common laboratory abnormality caused by 1 of 3 basic mechanisms: loss of bicarbonate, impaired renal acid excretion, or the addition of either endogenous or exogenous acids to the body. Common causes of nonanion gap metabolic acidosis in children include diarrhea and renal tubular acidosis (RTA). Increased anion gap is associated with lactic acidosis, ketoacidosis, and ingestion of such substances as methanol, ethylene glycol, acetylsalicylic acid, and bismuth subsalicylate. Inborn errors of metabolism cause production of ketoacids, lactic acid, and other organic anions. This can occur chronically or during acute decompensation with illness, stress, or therapy noncompliance.2 Serum anion gap, glucose, ketones, lactate, and ammonia can help to elucidate the specific etiology of metabolic acidosis (Fig. 1).
In our patient whose anion gap was at the upper limit of normal, both increased gap and non‐anion gap were considered as causes of the metabolic acidosis. Diarrhea was minimal, and there was no history of toxin or medication ingestion other than promethazine. RTA was unlikely given the borderline high serum anion gap and normal UA. Lactate level was normal, and examination did not find evidence of profound dehydration, both refuting that she had lactic acidosis severe enough to account for a serum bicarbonate of 13 mEq/L. Inborn errors of metabolism were therefore strongly considered.
Maple Syrup Urine Disease
Background
MSUD, or branched‐chain ketoaciduria, is a disease resulting from defects in the catabolic pathway of the branched‐chain amino acids (BCAAs) isoleucine, leucine, and valine. The deficient enzyme is the branched‐chain alpha‐ketoacid dehydrogenase complex (BCKDC), an enzyme system responsible for oxidative decarboxylation of the 2‐oxoacid transamination products of isoleucine, leucine, and valine. BCKDC is made up of 4 subunits (E1, E1, E2, and E3); its coenzymes include thiamine (vitamin B1) and lipoic acid. Deficiency of BCKDC leads to accumulation of BCAAs and the related branched chain oxoacids and organic acid intermediates, including one (sotalone) that lends a sweet odor reminiscent of maple syrup to sweat, cerumen, and urine. MSUD is autosomal recessive, with more than 100 specific mutations identified in the 4 genes encoding BCKDC.3 MSUD is a rare disease, occurring in 1 of every 180,000 newborns in the United States.2
Clinical Phenotypes
MSUD has been divided into at least 5 clinical phenotypes,4 although in several cases distinctions are not clear. Differences result from variation in the severity of enzyme deficiency. Classic MSUD is the most severe form, with less than 2% of normal BCKDC function; it presents in the first week of life with poor feeding and neurologic signs such as hypo/hypertonia, seizures, lethargy, and coma.5 General laboratory findings are nonspecific except for ketoacidosis. This form is rapidly fatal in the first months of life if not treated. Intermediate MSUD is milder, having 3%‐30% of BCKDC activity. Patients manifest variable degrees of retardation, developmental delay, and failure to thrive, often without signs of ketoacidosis. Thiamine‐responsive MSUD is distinguished by the favorable response to high‐dose thiamine supplementation with significant reduction in BCAA level.6 Although it is reasonable to try treatment with thiamine in most cases of MSUD, responsive patients are very rare. MSUD due to a deficiency of the E3 subunit is the rarest form, described in fewer than 10 patients.7 Intermittent MSUD is the least severe form, with 5%‐20% BCKDC activity. Children develop with normal growth and intelligence but are at risk of acute metabolic decompensation during catabolic states such as stress, infection, or surgery. Recurrent episodes of ketoacidosis, ataxia, and lethargy can lead to coma and death if untreated.8 Initial symptoms usually occur by 2 years of age but have appeared as late as the fifth decade of life. Between episodes, a normal diet is tolerated without elevation of BCAA level.
Long‐term morbidity and mortality in MSUD is neurologic. Death is often a result of brain edema that is generally attributed to the osmotic effects of leucine and amino acid imbalance; however, pathophysiologic mechanisms remain unclear.9 Progressive white matter changes are thought to result from chronic exposure to leucine. Levels of some amino acids and neurotransmitters are reduced in MSUD, which may play a role in causing encephalopathies and coma.10
Diagnosis
Elevation of plasma BCAA level can be directly assessed by standard plasma amino acid analysis; reduction of alanine is also characteristic. L‐alloisoleucine is the most sensitive and specific marker of MSUD and is pathognomic for MSUD.1112 In most cases of intermittent MSUD it is detectable at all times, including when BCAA level is normal, but in some cases it may be absent between episodes. Urine organic acids show elevation of the 2‐oxoacids corresponding to leucine, valine, and isoleucine and the corresponding 2‐hydroxy‐acids. Modern newborn screening programs generally ascertain MSUD by liquid chromatographytandem mass spectrometry, which detects elevated levels of leucine, isoleucine, and L‐alloisoleucine or the ratio of these to alanine. BCAA concentration is elevated in plasma within hours of birth; however, it is unclear how often intermittent MSUD might be missed in newborn screening.
Treatment
Therapy consists of dietary control (limited protein intake) and, in some cases, thiamine supplementation. The goal is to limit BCAA so as not to overwhelm the capacity of the BCKDC. However, BCAAs are essential, and the challenge is therefore to provide appropriate amounts of protein to sustain growth without exceeding the individual's metabolic capacity. Generally, the amount of dietary protein tolerated is insufficient to provide enough of the other essential amino acids, so supplemental amino acids are needed. During acute episodes a BCAA‐free diet, sometimes with insulin and glucose, is used to encourage BCAA removal. If this anabolic approach is unsuccessful, dialysis may be used. Orthotopic liver transplantation has also been performed.1315 In all cases so far, BCAA level normalized after transplantation, and metabolic control was sustainable on a regular diet without protein restriction.13, 1618
Lessons for the Physician
Vomiting without true diarrhea deserves careful evaluation. Inborn errors of metabolism are individually rare but as a group are fairly common. One study noted an incidence of 15.7 per 100,000 births detected on tandem mass spectrometry screening, whereas rates of clinical detection are lower.19 This case illustrates how symptoms may be nonspecific and can easily mimic simple gastroenteritis. The following may be helpful when evaluating a patient with isolated emesis:
-
History: Does the patient have a history of emesis with illnesses that usually do not cause vomiting, such as an asthma flare or URI? Has the patient required emergency treatment or IV fluids for bouts of emesis in the past? Could toxins or medications that cause metabolic acidosis have been ingested?
-
Physical exam: Skin lesions, odors, and hepatomegaly are sometimes found in metabolic disorders. Commonly recognized odors include the musty smell of phenylketonuria and the sweet maple syrup smell of MSUD.
-
Laboratory studies: UA and calculation of anion gap are first steps to take to eliminate more common causes of metabolic acidosis in children.
-
Reliance on newborn screening: Patients with intermittent MSUD may have normal BCAA levels between acute episodes. Newborn screening tests may not identify these patients. The physician must maintain a degree of suspicion in approaching an acute illness that might indicate a metabolic disease, even in a child who has had negative expanded newborn screening.
Unusual disorders may masquerade as a simple problem. Common laboratory tests and a thorough history and exam can help to differentiate between simple gastroenteritis and inborn metabolic error and guide further diagnostic evaluation.
- ,,.Practical methods to estimate whole body leucine oxidation in maple syrup urine disease.Pediatr Res.1993;33:445–451.
- Behrman RE,Kliegman RM,Jenson HB, editors.Nelson Textbook of Pediatrics.Philadelphia:Saunders;2004:224–235,409–418.
- ,,.Lessons from genetic disorders of branched‐chain amino acid metabolism.J Nutr.2006;136(suppl 1):243S–249S.
- ,.Maple syrup urine disease (branched‐chain ketoaciduria). In:Scriver CR,Beaudet AL,Sly WS,Valle D,Vogelstein B,Childs B, editors.The Metabolic and Molecular Basis of Inherited Disease.New York:McGraw‐Hill;2001:1971–2006.
- ,,.A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance.Pediatrics.1954;14:462–467.
- ,,,.Thiamine‐responsive maple‐syrup‐urine disease.Lancet.1971;1:310–312.
- ,,.Deficiency of dihydrolipoyl dehydrogenase (a component of the pyruvate and ‐ketoglutarate dehydrogenase complexes): a cause of congenital chronic lactic acidosis in infancy.Pediatr Res.1977;11:1198–202.
- ,,.Late‐onset branched‐chain ketoaciduria: (maple syrup urine disease).J Lancet.1966;86(3):149–152.
- ,,,.Cerebral edema causing death in children with maple syrup urine disease.J Pediatr.1991;119:42–45.
- ,,,,,,.Glutamate and γ‐aminobutyric acid neurotransmitter systems in the acute phase of maple syrup urine disease and citrullinemia encephalopathies in newborn calves.J Neurochem.1992;59:582–590.
- ,,,Maple syrup urine disease, with particular reference to dietotherapy.Pediatrics.1964;34:454–472.
- ,,,.Significance of L‐alloisoleucine in plasma for diagnosis of maple syrup urine disease.Clin Chem.1999;45:1734–40.
- ,,, et al.Mid‐term outcome of 2 cases with maple syrup urine disease: role of liver transplantation in the treatment.Arch Pediatr.1994;1:730–734.
- , et al.Transplantation for maple syrup urine disease (MSUD) and methylmalonic acidopathy (MMA).J Inherit Metab Dis.1997;20(suppl 1):37.
- ,,,.Liver transplantation in maple syrup urine disease.Eur J Pediatrics.1999;158(suppl 2):S60–S64.
- ,,, et al.Elective liver transplantation for the treatment of classical maple syrup urine disease.Am J Transplant.2006;6:557–564.
- ,,,,,.Domino liver transplantation in maple syrup urine disease.Liver Transpl.2006;12:876–882.
- ,,,.Branched‐chain L‐amino acid metabolism in classical maple syrup urine disease after orthotopic liver transplantation.J Inherit Metab Dis.2000;23:805–818.
- ,,,.Screening newborns for inborn errors of metabolism by tandem mass spectrometry.N Engl J Med.2003;348:2304–2312.
- ,,.Practical methods to estimate whole body leucine oxidation in maple syrup urine disease.Pediatr Res.1993;33:445–451.
- Behrman RE,Kliegman RM,Jenson HB, editors.Nelson Textbook of Pediatrics.Philadelphia:Saunders;2004:224–235,409–418.
- ,,.Lessons from genetic disorders of branched‐chain amino acid metabolism.J Nutr.2006;136(suppl 1):243S–249S.
- ,.Maple syrup urine disease (branched‐chain ketoaciduria). In:Scriver CR,Beaudet AL,Sly WS,Valle D,Vogelstein B,Childs B, editors.The Metabolic and Molecular Basis of Inherited Disease.New York:McGraw‐Hill;2001:1971–2006.
- ,,.A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance.Pediatrics.1954;14:462–467.
- ,,,.Thiamine‐responsive maple‐syrup‐urine disease.Lancet.1971;1:310–312.
- ,,.Deficiency of dihydrolipoyl dehydrogenase (a component of the pyruvate and ‐ketoglutarate dehydrogenase complexes): a cause of congenital chronic lactic acidosis in infancy.Pediatr Res.1977;11:1198–202.
- ,,.Late‐onset branched‐chain ketoaciduria: (maple syrup urine disease).J Lancet.1966;86(3):149–152.
- ,,,.Cerebral edema causing death in children with maple syrup urine disease.J Pediatr.1991;119:42–45.
- ,,,,,,.Glutamate and γ‐aminobutyric acid neurotransmitter systems in the acute phase of maple syrup urine disease and citrullinemia encephalopathies in newborn calves.J Neurochem.1992;59:582–590.
- ,,,Maple syrup urine disease, with particular reference to dietotherapy.Pediatrics.1964;34:454–472.
- ,,,.Significance of L‐alloisoleucine in plasma for diagnosis of maple syrup urine disease.Clin Chem.1999;45:1734–40.
- ,,, et al.Mid‐term outcome of 2 cases with maple syrup urine disease: role of liver transplantation in the treatment.Arch Pediatr.1994;1:730–734.
- , et al.Transplantation for maple syrup urine disease (MSUD) and methylmalonic acidopathy (MMA).J Inherit Metab Dis.1997;20(suppl 1):37.
- ,,,.Liver transplantation in maple syrup urine disease.Eur J Pediatrics.1999;158(suppl 2):S60–S64.
- ,,, et al.Elective liver transplantation for the treatment of classical maple syrup urine disease.Am J Transplant.2006;6:557–564.
- ,,,,,.Domino liver transplantation in maple syrup urine disease.Liver Transpl.2006;12:876–882.
- ,,,.Branched‐chain L‐amino acid metabolism in classical maple syrup urine disease after orthotopic liver transplantation.J Inherit Metab Dis.2000;23:805–818.
- ,,,.Screening newborns for inborn errors of metabolism by tandem mass spectrometry.N Engl J Med.2003;348:2304–2312.