User login
Leadership & Professional Development: Cultivating Habits for the Hospitalist
“We are what we repeatedly do. Excellence, then, is not an act, but a habit.”
—Will Durant
We are a collection of our habits—the routine, repetitive, subconscious behaviors we perform on a daily basis. Some of these behaviors are positive, others less so. Habits allow us to perform tasks automatically, without the need for active decision making. Amidst a constantly changing clinical environment, cultivating consistent habits can improve our adherence to best practices and free cognitive effort toward more challenging diagnostic or therapeutic tasks.
Establishing habits requires practice and intentionality. First, identify those habits that are desirable in your personal and professional life. Next, find a method to develop the habit. Then, hold yourself accountable as you work to embed the habit. Simple? Not quite.
In “The Power of Habit,” author Charles Duhigg introduces habit loops as a way to successfully develop this practice.1 Habit loops—sequences comprising a cue, routine, and reward—are integral to developing routines that support professional and personal aspects of hospitalist life. Consider a hospitalist seeking to develop a prerounds routine to increase efficiency and limit missed patient information. First, the clinician should identify a cue to start the routine, such as sitting down to log in at a specific workstation. Second, a sequence of actions is “chunked” into a consistent order, such as a review of vital signs, clinical notes, and patient labs. After the routine is completed, the clinician should finish with a reward, such as a cup of coffee after rounds. Want to set up a habit for ensuring learning goals are set with trainees at the beginning of every block? Set a calendar reminder for this on the first day, standardize how you communicate goals, and reward yourself with a team lunch at the end of the rotation. What if it’s a busy first day on service? Doesn’t matter. As Clay Christensen notes in “How Will You Measure Your Life?,” making one commitment to a habit is easier than deciding whether or not to engage in the routine every time new circumstances arise.2 The intentionality that comes with this act ensures consistency in the practice.
As a busy hospitalist, establishing habits for personal and professional development requires cues and rewards. For example, do you want to cement a habit of reading the latest journal articles or carving out time each day to reflect on your work? Then cultivate the routine by creating a cue, such as a dashboard on a wall to visualize how many articles you’ve read this week or whether you’ve paused to reflect on your rotation. Reinforce the routine by creating a reward: a walk outside, time with family, or another activity you enjoy. Pair the same reward with the same routine to strengthen the habit loop.
A few additional tips for cultivating habits: it is useful to pair an existing reliable habit, or “anchor habit,” with a new one, such as a short meditation after brushing your teeth.3 Doing so reinforces behaviors in a positive way. You may use the same principles to lose unwanted habits (eg, checking e-mail excessively) by removing cues, such as turning off notifications or using airplane mode and rewarding yourself when you see the behavior through.
Habits are larger than behaviors; they can impact your personal and professional life in important ways. By actively creating habits that align with your long-term priorities, you can create a safety net if and when change arrives. Understanding the psychology of habits and employing cues and rewards effectively can lead hospitalists to create positive routines that improve their clinical practice and personal lives.
1. Duhigg C. The Power of Habit: Why We Do What We Do in Life and Business. Random House; 2012.
2. Christensen CM. How Will You Measure Your Life? (Harvard Business Review Classics). Harvard Business Review Press; 2017.
3. Fogg B. Tiny Habits w/Dr. BJ Fogg-Behavior Change: Tiny Habits; 2011.
“We are what we repeatedly do. Excellence, then, is not an act, but a habit.”
—Will Durant
We are a collection of our habits—the routine, repetitive, subconscious behaviors we perform on a daily basis. Some of these behaviors are positive, others less so. Habits allow us to perform tasks automatically, without the need for active decision making. Amidst a constantly changing clinical environment, cultivating consistent habits can improve our adherence to best practices and free cognitive effort toward more challenging diagnostic or therapeutic tasks.
Establishing habits requires practice and intentionality. First, identify those habits that are desirable in your personal and professional life. Next, find a method to develop the habit. Then, hold yourself accountable as you work to embed the habit. Simple? Not quite.
In “The Power of Habit,” author Charles Duhigg introduces habit loops as a way to successfully develop this practice.1 Habit loops—sequences comprising a cue, routine, and reward—are integral to developing routines that support professional and personal aspects of hospitalist life. Consider a hospitalist seeking to develop a prerounds routine to increase efficiency and limit missed patient information. First, the clinician should identify a cue to start the routine, such as sitting down to log in at a specific workstation. Second, a sequence of actions is “chunked” into a consistent order, such as a review of vital signs, clinical notes, and patient labs. After the routine is completed, the clinician should finish with a reward, such as a cup of coffee after rounds. Want to set up a habit for ensuring learning goals are set with trainees at the beginning of every block? Set a calendar reminder for this on the first day, standardize how you communicate goals, and reward yourself with a team lunch at the end of the rotation. What if it’s a busy first day on service? Doesn’t matter. As Clay Christensen notes in “How Will You Measure Your Life?,” making one commitment to a habit is easier than deciding whether or not to engage in the routine every time new circumstances arise.2 The intentionality that comes with this act ensures consistency in the practice.
As a busy hospitalist, establishing habits for personal and professional development requires cues and rewards. For example, do you want to cement a habit of reading the latest journal articles or carving out time each day to reflect on your work? Then cultivate the routine by creating a cue, such as a dashboard on a wall to visualize how many articles you’ve read this week or whether you’ve paused to reflect on your rotation. Reinforce the routine by creating a reward: a walk outside, time with family, or another activity you enjoy. Pair the same reward with the same routine to strengthen the habit loop.
A few additional tips for cultivating habits: it is useful to pair an existing reliable habit, or “anchor habit,” with a new one, such as a short meditation after brushing your teeth.3 Doing so reinforces behaviors in a positive way. You may use the same principles to lose unwanted habits (eg, checking e-mail excessively) by removing cues, such as turning off notifications or using airplane mode and rewarding yourself when you see the behavior through.
Habits are larger than behaviors; they can impact your personal and professional life in important ways. By actively creating habits that align with your long-term priorities, you can create a safety net if and when change arrives. Understanding the psychology of habits and employing cues and rewards effectively can lead hospitalists to create positive routines that improve their clinical practice and personal lives.
“We are what we repeatedly do. Excellence, then, is not an act, but a habit.”
—Will Durant
We are a collection of our habits—the routine, repetitive, subconscious behaviors we perform on a daily basis. Some of these behaviors are positive, others less so. Habits allow us to perform tasks automatically, without the need for active decision making. Amidst a constantly changing clinical environment, cultivating consistent habits can improve our adherence to best practices and free cognitive effort toward more challenging diagnostic or therapeutic tasks.
Establishing habits requires practice and intentionality. First, identify those habits that are desirable in your personal and professional life. Next, find a method to develop the habit. Then, hold yourself accountable as you work to embed the habit. Simple? Not quite.
In “The Power of Habit,” author Charles Duhigg introduces habit loops as a way to successfully develop this practice.1 Habit loops—sequences comprising a cue, routine, and reward—are integral to developing routines that support professional and personal aspects of hospitalist life. Consider a hospitalist seeking to develop a prerounds routine to increase efficiency and limit missed patient information. First, the clinician should identify a cue to start the routine, such as sitting down to log in at a specific workstation. Second, a sequence of actions is “chunked” into a consistent order, such as a review of vital signs, clinical notes, and patient labs. After the routine is completed, the clinician should finish with a reward, such as a cup of coffee after rounds. Want to set up a habit for ensuring learning goals are set with trainees at the beginning of every block? Set a calendar reminder for this on the first day, standardize how you communicate goals, and reward yourself with a team lunch at the end of the rotation. What if it’s a busy first day on service? Doesn’t matter. As Clay Christensen notes in “How Will You Measure Your Life?,” making one commitment to a habit is easier than deciding whether or not to engage in the routine every time new circumstances arise.2 The intentionality that comes with this act ensures consistency in the practice.
As a busy hospitalist, establishing habits for personal and professional development requires cues and rewards. For example, do you want to cement a habit of reading the latest journal articles or carving out time each day to reflect on your work? Then cultivate the routine by creating a cue, such as a dashboard on a wall to visualize how many articles you’ve read this week or whether you’ve paused to reflect on your rotation. Reinforce the routine by creating a reward: a walk outside, time with family, or another activity you enjoy. Pair the same reward with the same routine to strengthen the habit loop.
A few additional tips for cultivating habits: it is useful to pair an existing reliable habit, or “anchor habit,” with a new one, such as a short meditation after brushing your teeth.3 Doing so reinforces behaviors in a positive way. You may use the same principles to lose unwanted habits (eg, checking e-mail excessively) by removing cues, such as turning off notifications or using airplane mode and rewarding yourself when you see the behavior through.
Habits are larger than behaviors; they can impact your personal and professional life in important ways. By actively creating habits that align with your long-term priorities, you can create a safety net if and when change arrives. Understanding the psychology of habits and employing cues and rewards effectively can lead hospitalists to create positive routines that improve their clinical practice and personal lives.
1. Duhigg C. The Power of Habit: Why We Do What We Do in Life and Business. Random House; 2012.
2. Christensen CM. How Will You Measure Your Life? (Harvard Business Review Classics). Harvard Business Review Press; 2017.
3. Fogg B. Tiny Habits w/Dr. BJ Fogg-Behavior Change: Tiny Habits; 2011.
1. Duhigg C. The Power of Habit: Why We Do What We Do in Life and Business. Random House; 2012.
2. Christensen CM. How Will You Measure Your Life? (Harvard Business Review Classics). Harvard Business Review Press; 2017.
3. Fogg B. Tiny Habits w/Dr. BJ Fogg-Behavior Change: Tiny Habits; 2011.
© 2020 Society of Hospital Medicine
How should urine electrolytes be ordered and interpreted in acute kidney injury and electrolyte abnormalities?
The case
A 50-year old woman naive to the health care system presents to the ED with nausea, malaise, and decreased exercise tolerance for several weeks. Physical exam reveals mild bilateral lower extremity edema. Her labs are notable for an elevated creatinine of 7.0. She is admitted for work-up of her renal disease.
Nephrology was consulted and recommended obtaining urine electrolytes. The admitting hospitalist is unsure which urine electrolytes are appropriate to order, and in turn orders all of the urine electrolytes in the order set.
Which urine electrolytes should be ordered in various clinical contexts?

Introduction
Hospitalists have been on the forefront of efforts to tailor testing and resource utilization to eliminate wasteful practices in health care. To order and interpret diagnostic tests appropriately, a hospitalist needs to have a thorough understanding of the diagnostic utility of laboratory tests. There is a lack of clear diagnostic guidelines, so ordering all the urine electrolytes in a “blanket” strategy is a common practice. We will discuss the diagnostic utility of each of the urine electrolytes in a variety of clinical scenarios.
Acute kidney injury
Both the fractional excretion of sodium (FENa) and the fractional excretion of urea (FEUrea) have long been used as part of the standard work-up for determining if acute kidney injury (AKI) is due to prerenal causes. Although these markers prove to be beneficial in the work-up of AKI, both the FENa and FEUrea have several limitations.
FENa measures the ratio of sodium excreted in the urine compared to how much is filtered through the kidney. A FENa of less than 1% in oliguric patients may indicate prerenal azotemia, as an increased reabsorption of sodium is the appropriate response of functioning nephrons to decreased renal perfusion. Values greater than 3% may be consistent with acute tubular necrosis (ATN) due to inappropriate sodium excretion in the setting of tubular damage.
Importantly, a FENa value of less than 1% occurs in a number of conditions other than prerenal azotemia due to dehydration, including hypervolemic prerenal states such as cirrhosis or heart failure; AKI due to radiocontrast or heme pigments; acute glomerulonephritis; transition from prerenal to postischemic ATN or sepsis, and in acute interstitial nephritis (AIN).1,2 Approximately 10% of patients with nonoliguric ATN have a FENa less than 1.0%. Moreover, use of diuretics can falsely elevate the FENa due to inhibition of sodium reabsorption. FENa values above 3% can occur in volume contraction in patients with chronic kidney disease (CKD) or in elderly patients as their sodium reabsorption is impaired.3 Acute volume loss (e.g. blood loss), or more commonly, administration of diuretics or intravenous fluids, can also alter the interpretation of the FENa.2
Many of the limitations of the FENa also apply to the FEUrea, including interpretation in the elderly and use in acute volume changes. However, the FEUrea has unique limitations, particularly in patients with sepsis, as cytokines released in sepsis may interfere with urea transporters in the kidney and colon.2 Its interpretation also relies on intact functioning of the proximal tubule, which can be altered in many conditions including uncontrolled diabetes. Overall, the FENa and FEUrea can be helpful to determine the etiology of AKI, but only in certain clinical scenarios.
Hyponatremia
Hyponatremia is the most common electrolyte abnormality in hospitalized patients, with a prevalence of up to 30% in critically ill patients.4 It often is acquired during the hospitalization itself. A detailed history and physical exam, including careful assessment of volume status, is as important as laboratory values in establishing the cause of hyponatremia.
Urine sodium and urine osmolality are measured to understand whether the renin-aldosterone-angiotensin system (RAAS) and antidiuretic hormone (ADH) are activated. If renal blood flow or renal delivery of sodium is decreased, renin secretion from the juxtaglomerular apparatus will be activated, ultimately leading to increased reabsorption of sodium in the distal tubules and collecting ducts. Thus, low urine sodium signals that the RAAS is activated due to decreased serum sodium concentration or decreased renal blood flow from hypovolemia or low effective arterial circulation from cirrhosis or heart failure.
Most causes of hyponatremia will have low urine sodium values, including hypovolemia, cirrhosis, heart failure, “tea-and-toast” diet, beer potomania, and primary polydipsia. However, the urine sodium may be unreliable in patients who are not oliguric or who have CKD.
Diuretic-induced hyponatremia from thiazide or loop diuretics will likely have elevated urine sodium levels. Similarly, the syndrome of inappropriate antidiuretic hormone secretion (SIADH) will have an elevated urine sodium above 20-40 mEq/L.
Urine osmolality becomes elevated when ADH is secreted in response to reduced plasma volume or increased plasma osmolality. Urine osmolality is low in cases such as primary polydipsia, which creates a maximally dilute urine of 40-100 mEq/L, and in tea-and-toast diets or beer potomania due to low solute intake. Urine osmolality can be elevated in hypovolemic states as well as SIADH, and is variable in hypothyroidism and selective serotonin reuptake inhibitor administration. Thus, urine sodium, and not urine osmolality, is the most useful differentiator between SIADH and hypovolemic states.
In a study of 555 patients with hyponatremia secondary to SIADH, mean urine sodium was found to be 72 (range 30-251) and the median urine osmolality was 379 (range 123-1019).5
In cases of marked hyperglycemia, serum osmolality should be measured to evaluate hyperglycemia as a cause of hyperosmolar hyponatremia. Pseudohyponatremia in the setting of hyperlipidemia, hypertriglyceridemia, or hyperparaproteinemia represents a laboratory artifact due to lower plasma water concentration in the specimen sample and should be excluded.
Hypokalemia
About 20% of patients are hypokalemic during an inpatient hospitalization. There is a broad differential for hypokalemia, including medical, nutritional, and medication-related causes. Exogenous insulin administration or endogenous production in cases of refeeding syndrome drives potassium intracellularly via the N+/K+ ATPase. Increased sympathetic activity from alcohol withdrawal, acute myocardial infarction, head injury, or thyroid imbalance, as well as iatrogenic causes such as albuterol administration, also drive potassium intracellularly. Diarrhea and nasogastric tube suction lead to gastrointestinal (GI) potassium losses, while antibiotics, chemotherapeutic agents, and diuretics can cause hypokalemia through renal potassium wasting. Hyperaldosteronism and renal tubular acidosis are less common causes.6
The history, review of medications, physical exam, and initial basic laboratory testing (electrolytes, BUN, creatinine, magnesium) should assess for pseudohypokalemia, poor oral intake, diuretic use, acid-base disturbances, or GI losses.
Measuring urine potassium is useful in the work-up of the hypokalemic patient when these conditions are not evident. Urine potassium – either 24-hour or spot urine potassium-to-creatinine ratio – can help determine if urinary potassium wasting is a factor. Potassium is excreted at a near constant rate throughout the day. A urine potassium-to-creatinine ratio corrects for variations in urine volume. When this ratio is greater than 13 mEq/g, renal potassium losses should be suspected. If the ratio is less than 13 mEq/g, hypokalemia is likely due to transcellular potassium shifts, GI losses, diuretics, or poor intake.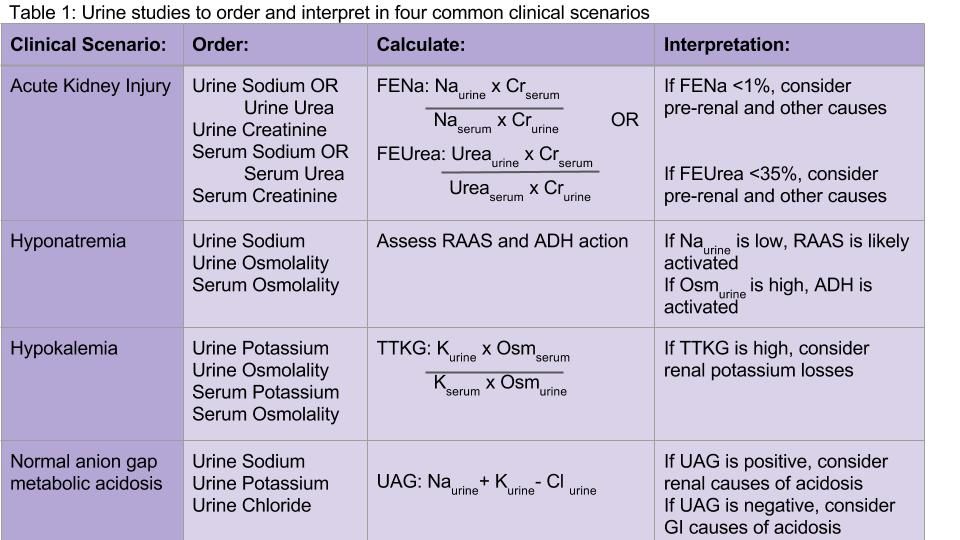
Hyperkalemia
Several concepts in hypokalemia are relevant to hyperkalemia. Redistribution of potassium into the extracellular fluid can cause hyperkalemia when the body tries to counterbalance low extracellular pH by potassium-hydrogen exchange. Medications may cause an extracellular shift of potassium (e.g. digoxin) or induce diminished potassium excretion (e.g. NSAIDs, spironolactone, ACE/ARBs).
CKD and end-stage kidney disease are common causes of hyperkalemia in the hospitalized patient – as functioning nephrons decrease, poor Na-K exchange ensues. Hypoaldosteronism and type 4 renal tubular acidosis are also on the differential diagnosis. Pseudohyperkalemia secondary to thrombocytosis, erythrocytosis, or activated platelets should be considered and evaluated.
Appropriate renal excretion of potassium is mediated by the connecting segment between the distal tubule and the collecting duct, and the cortical collecting duct itself. There are four major causes of hyperkalemia due to reduced urinary potassium secretion: reduced aldosterone secretion, reduced response to aldosterone, reduced distal sodium and water delivery (often related to low effective arterial blood volume), and kidney injury.6
Measurement of 24-hour urinary potassium excretion is of limited utility in patients with persistent stable hyperkalemia because urinary potassium excretion is related to potassium intake. The TTKG was previously used to assess the degree of aldosterone activity by estimating the potassium concentration in the cortical collecting tubule. However, some assumptions upon which this calculation was based have been considered invalid by the original studies’ authors, and the TTKG to evaluate potassium abnormalities is no longer uniformly recommended.7,8 Ultimately, if patients have persistent hyperkalemia, work-up for hypoaldosteronism should be considered.
Normal anion gap metabolic acidosis
The urine anion gap (UAG) is used to determine the cause of normal anion gap hyperchloremic metabolic acidosis by indirectly measuring urinary excretion of ammonium. To maintain a normal acid/base balance, hydrogen ions are excreted in the urine with simultaneous reabsorption of bicarbonate. Hydrogen ions are bound to ammonia (NH3) to form ammonium (NH4+), which is excreted as NH4Cl in the urine.
The UAG is calculated by adding urine sodium and urine potassium and subtracting urine chloride (see Table 1). In a patient without an acid/base disturbance, the UAG is positive because more Na and K is absorbed in the gastrointestinal system compared to Cl, and thus more Na and K is excreted in the urine. In a normal anion gap metabolic acidosis through an acid load or bicarbonate loss, the normal response of the kidney is to excrete more hydrogen ions, resulting in more chloride excretion as NH4Cl. This leads to a negative urine anion gap, as Cl excretion outweighs Na and K excretion. When NH4+ excretion is impaired, such as in distal renal tubular acidosis (RTA), the urine anion gap will remain positive despite the metabolic acidosis. Thus, a positive UAG points to renal causes of the normal anion gap metabolic acidosis, whereas a negative UAG points to extrarenal causes such as bicarbonate losses in the GI tract.9
Additional considerations
Urine studies can also be useful for assessment of proteinuria and albuminuria in a patient with CKD or diabetes, diagnosis of plasma cell dyscrasias, the diagnosis and prevention of nephrolithiasis, and a wide variety of other conditions.
Back to the case
Our patient was admitted with an elevated creatinine of unclear chronicity, and subacute symptoms of uremia. Because she was oliguric, urine and serum sodium and creatinine were measured before intravenous fluids were administered. Her FENa was 2%, which was not consistent with prerenal azotemia or ATN. She was found to have CKD secondary to previously undiagnosed diabetes. Upon further questioning, she had been taking high-dose NSAIDs for her chronic knee pain. Her renal function improved mildly by withholding NSAIDs, and she was discharged with appropriate nephrology follow-up.
Bottom line
Urine electrolytes have specific indications and utilities for different clinical scenarios, and should be ordered in a targeted manner that can aide in diagnosing AKI, hyponatremia, hypokalemia, and normal anion gap metabolic acidosis.
Dr. Tummalapalli, Dr. Krouss, and Dr. Goetz are hospitalists in the department of medicine at the Icahn School of Medicine at Mount Sinai in New York City.
References
1. Brosius FC, Lau K. Low fractional excretion of sodium in acute renal failure: role of timing of the test and ischemia. Am J Nephrol. 1986;6(6):450-7.
2. Gottfried J, Weisen J, Raina R, Nally J. Finding the cause of acute kidney injury: Which index of fractional excretion is better? Cleve Clin J Med. 2012;79(2):121-6.
3. Steiner, RW. Interpreting the fractional excretion of sodium. Am J Med. 1984;77(4):699-702.
4. DeVita MV, Gardenswartz MH, Konecky A, Zabetakis PM. Incidence and etiology of hyponatremia in an intensive care unit. Clin Nephrol. 1990;34(4):163-6.
5. Shepshelovich D, Leibovitch C, Klein A, et al. The syndrome of inappropriate antidiuretic hormone secretion: distribution and characterization according to etiologies. Eur J Int Med. 2015;26(10):819-24.
6. Mount DB. Fluid and Electrolyte Disturbances. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J. eds. Harrison’s Principles of Internal Medicine, 19e. New York, NY: McGraw-Hill; 2015.
7. Kamel KS. Intrarenal urea recycling leads to a higher rate of renal excretion of potassium: an hypothesis with clinical implications. Curr Opin Nephrol Hypertens. 2011 Sep;20(5):547-54.
8. Kamel KS, Davids MR, Lin S-H, Halperin ML. Interpretation of Electrolyte and Acid-Base Parameters in Blood and Urine. In: Brenner and Rector’s The Kidney, 27, 804-45.e2. Philadelphia, PA: Elsevier; 2016.
9. Goldstein MB, Bear R, Richardson RMA, Marsden PA, Halperin ML. The Urine Anion Gap: a clinically useful index of ammonium excretion. Am J Med Sci. 1986;198-202.
Key Points:
• In acute kidney injury, the FENa and FEUrea may be calculated to distinguish prerenal azotemia from ATN; however, FENa and FEUrea may be low in a wide variety of conditions other than prerenal azotemia.
• Urine sodium and osmolality values are helpful in diagnosing the cause of hyponatremia, but have a number of limitations in nonoliguric patients and those with CKD.
• An elevated transtubular potassium gradient (TTKG) may indicate renal loss of potassium in patients with hypokalemia.
• A positive urine anion gap (UAG) in the setting of a normal anion gap metabolic acidosis points to renal causes of the metabolic acidosis, whereas a negative UAG points to extrarenal causes such as bicarbonate losses in the GI tract.Ad
Additional Reading:
Goldstein MB, Bear R, Richardson RMA, Marsden PA, Halperin ML. The Urine Anion Gap: A Clinically Useful Index of Ammonium Excretion. Am J Med Sci. 1986;198-202.
Gotfried J, Wiesen J, Raina R, Nally Jr JV. Finding the cause of acute kidney injury: which index of fractional excretion is better. Cleve Clin J Med. 2012;79(2):121-126.
Kamel KS, Davids MR, Lin S-H, Halperin ML. Interpretation of Electrolyte and Acid-Base Parameters in Blood and Urine. In: Brenner and Rector’s The Kidney, 27, 804-845.e2. Philadelphia, PA: Elsevier; 2016.
The case
A 50-year old woman naive to the health care system presents to the ED with nausea, malaise, and decreased exercise tolerance for several weeks. Physical exam reveals mild bilateral lower extremity edema. Her labs are notable for an elevated creatinine of 7.0. She is admitted for work-up of her renal disease.
Nephrology was consulted and recommended obtaining urine electrolytes. The admitting hospitalist is unsure which urine electrolytes are appropriate to order, and in turn orders all of the urine electrolytes in the order set.
Which urine electrolytes should be ordered in various clinical contexts?
Introduction
Hospitalists have been on the forefront of efforts to tailor testing and resource utilization to eliminate wasteful practices in health care. To order and interpret diagnostic tests appropriately, a hospitalist needs to have a thorough understanding of the diagnostic utility of laboratory tests. There is a lack of clear diagnostic guidelines, so ordering all the urine electrolytes in a “blanket” strategy is a common practice. We will discuss the diagnostic utility of each of the urine electrolytes in a variety of clinical scenarios.
Acute kidney injury
Both the fractional excretion of sodium (FENa) and the fractional excretion of urea (FEUrea) have long been used as part of the standard work-up for determining if acute kidney injury (AKI) is due to prerenal causes. Although these markers prove to be beneficial in the work-up of AKI, both the FENa and FEUrea have several limitations.
FENa measures the ratio of sodium excreted in the urine compared to how much is filtered through the kidney. A FENa of less than 1% in oliguric patients may indicate prerenal azotemia, as an increased reabsorption of sodium is the appropriate response of functioning nephrons to decreased renal perfusion. Values greater than 3% may be consistent with acute tubular necrosis (ATN) due to inappropriate sodium excretion in the setting of tubular damage.
Importantly, a FENa value of less than 1% occurs in a number of conditions other than prerenal azotemia due to dehydration, including hypervolemic prerenal states such as cirrhosis or heart failure; AKI due to radiocontrast or heme pigments; acute glomerulonephritis; transition from prerenal to postischemic ATN or sepsis, and in acute interstitial nephritis (AIN).1,2 Approximately 10% of patients with nonoliguric ATN have a FENa less than 1.0%. Moreover, use of diuretics can falsely elevate the FENa due to inhibition of sodium reabsorption. FENa values above 3% can occur in volume contraction in patients with chronic kidney disease (CKD) or in elderly patients as their sodium reabsorption is impaired.3 Acute volume loss (e.g. blood loss), or more commonly, administration of diuretics or intravenous fluids, can also alter the interpretation of the FENa.2
Many of the limitations of the FENa also apply to the FEUrea, including interpretation in the elderly and use in acute volume changes. However, the FEUrea has unique limitations, particularly in patients with sepsis, as cytokines released in sepsis may interfere with urea transporters in the kidney and colon.2 Its interpretation also relies on intact functioning of the proximal tubule, which can be altered in many conditions including uncontrolled diabetes. Overall, the FENa and FEUrea can be helpful to determine the etiology of AKI, but only in certain clinical scenarios.
Hyponatremia
Hyponatremia is the most common electrolyte abnormality in hospitalized patients, with a prevalence of up to 30% in critically ill patients.4 It often is acquired during the hospitalization itself. A detailed history and physical exam, including careful assessment of volume status, is as important as laboratory values in establishing the cause of hyponatremia.
Urine sodium and urine osmolality are measured to understand whether the renin-aldosterone-angiotensin system (RAAS) and antidiuretic hormone (ADH) are activated. If renal blood flow or renal delivery of sodium is decreased, renin secretion from the juxtaglomerular apparatus will be activated, ultimately leading to increased reabsorption of sodium in the distal tubules and collecting ducts. Thus, low urine sodium signals that the RAAS is activated due to decreased serum sodium concentration or decreased renal blood flow from hypovolemia or low effective arterial circulation from cirrhosis or heart failure.
Most causes of hyponatremia will have low urine sodium values, including hypovolemia, cirrhosis, heart failure, “tea-and-toast” diet, beer potomania, and primary polydipsia. However, the urine sodium may be unreliable in patients who are not oliguric or who have CKD.
Diuretic-induced hyponatremia from thiazide or loop diuretics will likely have elevated urine sodium levels. Similarly, the syndrome of inappropriate antidiuretic hormone secretion (SIADH) will have an elevated urine sodium above 20-40 mEq/L.
Urine osmolality becomes elevated when ADH is secreted in response to reduced plasma volume or increased plasma osmolality. Urine osmolality is low in cases such as primary polydipsia, which creates a maximally dilute urine of 40-100 mEq/L, and in tea-and-toast diets or beer potomania due to low solute intake. Urine osmolality can be elevated in hypovolemic states as well as SIADH, and is variable in hypothyroidism and selective serotonin reuptake inhibitor administration. Thus, urine sodium, and not urine osmolality, is the most useful differentiator between SIADH and hypovolemic states.
In a study of 555 patients with hyponatremia secondary to SIADH, mean urine sodium was found to be 72 (range 30-251) and the median urine osmolality was 379 (range 123-1019).5
In cases of marked hyperglycemia, serum osmolality should be measured to evaluate hyperglycemia as a cause of hyperosmolar hyponatremia. Pseudohyponatremia in the setting of hyperlipidemia, hypertriglyceridemia, or hyperparaproteinemia represents a laboratory artifact due to lower plasma water concentration in the specimen sample and should be excluded.
Hypokalemia
About 20% of patients are hypokalemic during an inpatient hospitalization. There is a broad differential for hypokalemia, including medical, nutritional, and medication-related causes. Exogenous insulin administration or endogenous production in cases of refeeding syndrome drives potassium intracellularly via the N+/K+ ATPase. Increased sympathetic activity from alcohol withdrawal, acute myocardial infarction, head injury, or thyroid imbalance, as well as iatrogenic causes such as albuterol administration, also drive potassium intracellularly. Diarrhea and nasogastric tube suction lead to gastrointestinal (GI) potassium losses, while antibiotics, chemotherapeutic agents, and diuretics can cause hypokalemia through renal potassium wasting. Hyperaldosteronism and renal tubular acidosis are less common causes.6
The history, review of medications, physical exam, and initial basic laboratory testing (electrolytes, BUN, creatinine, magnesium) should assess for pseudohypokalemia, poor oral intake, diuretic use, acid-base disturbances, or GI losses.
Measuring urine potassium is useful in the work-up of the hypokalemic patient when these conditions are not evident. Urine potassium – either 24-hour or spot urine potassium-to-creatinine ratio – can help determine if urinary potassium wasting is a factor. Potassium is excreted at a near constant rate throughout the day. A urine potassium-to-creatinine ratio corrects for variations in urine volume. When this ratio is greater than 13 mEq/g, renal potassium losses should be suspected. If the ratio is less than 13 mEq/g, hypokalemia is likely due to transcellular potassium shifts, GI losses, diuretics, or poor intake.
Hyperkalemia
Several concepts in hypokalemia are relevant to hyperkalemia. Redistribution of potassium into the extracellular fluid can cause hyperkalemia when the body tries to counterbalance low extracellular pH by potassium-hydrogen exchange. Medications may cause an extracellular shift of potassium (e.g. digoxin) or induce diminished potassium excretion (e.g. NSAIDs, spironolactone, ACE/ARBs).
CKD and end-stage kidney disease are common causes of hyperkalemia in the hospitalized patient – as functioning nephrons decrease, poor Na-K exchange ensues. Hypoaldosteronism and type 4 renal tubular acidosis are also on the differential diagnosis. Pseudohyperkalemia secondary to thrombocytosis, erythrocytosis, or activated platelets should be considered and evaluated.
Appropriate renal excretion of potassium is mediated by the connecting segment between the distal tubule and the collecting duct, and the cortical collecting duct itself. There are four major causes of hyperkalemia due to reduced urinary potassium secretion: reduced aldosterone secretion, reduced response to aldosterone, reduced distal sodium and water delivery (often related to low effective arterial blood volume), and kidney injury.6
Measurement of 24-hour urinary potassium excretion is of limited utility in patients with persistent stable hyperkalemia because urinary potassium excretion is related to potassium intake. The TTKG was previously used to assess the degree of aldosterone activity by estimating the potassium concentration in the cortical collecting tubule. However, some assumptions upon which this calculation was based have been considered invalid by the original studies’ authors, and the TTKG to evaluate potassium abnormalities is no longer uniformly recommended.7,8 Ultimately, if patients have persistent hyperkalemia, work-up for hypoaldosteronism should be considered.
Normal anion gap metabolic acidosis
The urine anion gap (UAG) is used to determine the cause of normal anion gap hyperchloremic metabolic acidosis by indirectly measuring urinary excretion of ammonium. To maintain a normal acid/base balance, hydrogen ions are excreted in the urine with simultaneous reabsorption of bicarbonate. Hydrogen ions are bound to ammonia (NH3) to form ammonium (NH4+), which is excreted as NH4Cl in the urine.
The UAG is calculated by adding urine sodium and urine potassium and subtracting urine chloride (see Table 1). In a patient without an acid/base disturbance, the UAG is positive because more Na and K is absorbed in the gastrointestinal system compared to Cl, and thus more Na and K is excreted in the urine. In a normal anion gap metabolic acidosis through an acid load or bicarbonate loss, the normal response of the kidney is to excrete more hydrogen ions, resulting in more chloride excretion as NH4Cl. This leads to a negative urine anion gap, as Cl excretion outweighs Na and K excretion. When NH4+ excretion is impaired, such as in distal renal tubular acidosis (RTA), the urine anion gap will remain positive despite the metabolic acidosis. Thus, a positive UAG points to renal causes of the normal anion gap metabolic acidosis, whereas a negative UAG points to extrarenal causes such as bicarbonate losses in the GI tract.9
Additional considerations
Urine studies can also be useful for assessment of proteinuria and albuminuria in a patient with CKD or diabetes, diagnosis of plasma cell dyscrasias, the diagnosis and prevention of nephrolithiasis, and a wide variety of other conditions.
Back to the case
Our patient was admitted with an elevated creatinine of unclear chronicity, and subacute symptoms of uremia. Because she was oliguric, urine and serum sodium and creatinine were measured before intravenous fluids were administered. Her FENa was 2%, which was not consistent with prerenal azotemia or ATN. She was found to have CKD secondary to previously undiagnosed diabetes. Upon further questioning, she had been taking high-dose NSAIDs for her chronic knee pain. Her renal function improved mildly by withholding NSAIDs, and she was discharged with appropriate nephrology follow-up.
Bottom line
Urine electrolytes have specific indications and utilities for different clinical scenarios, and should be ordered in a targeted manner that can aide in diagnosing AKI, hyponatremia, hypokalemia, and normal anion gap metabolic acidosis.
Dr. Tummalapalli, Dr. Krouss, and Dr. Goetz are hospitalists in the department of medicine at the Icahn School of Medicine at Mount Sinai in New York City.
References
1. Brosius FC, Lau K. Low fractional excretion of sodium in acute renal failure: role of timing of the test and ischemia. Am J Nephrol. 1986;6(6):450-7.
2. Gottfried J, Weisen J, Raina R, Nally J. Finding the cause of acute kidney injury: Which index of fractional excretion is better? Cleve Clin J Med. 2012;79(2):121-6.
3. Steiner, RW. Interpreting the fractional excretion of sodium. Am J Med. 1984;77(4):699-702.
4. DeVita MV, Gardenswartz MH, Konecky A, Zabetakis PM. Incidence and etiology of hyponatremia in an intensive care unit. Clin Nephrol. 1990;34(4):163-6.
5. Shepshelovich D, Leibovitch C, Klein A, et al. The syndrome of inappropriate antidiuretic hormone secretion: distribution and characterization according to etiologies. Eur J Int Med. 2015;26(10):819-24.
6. Mount DB. Fluid and Electrolyte Disturbances. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J. eds. Harrison’s Principles of Internal Medicine, 19e. New York, NY: McGraw-Hill; 2015.
7. Kamel KS. Intrarenal urea recycling leads to a higher rate of renal excretion of potassium: an hypothesis with clinical implications. Curr Opin Nephrol Hypertens. 2011 Sep;20(5):547-54.
8. Kamel KS, Davids MR, Lin S-H, Halperin ML. Interpretation of Electrolyte and Acid-Base Parameters in Blood and Urine. In: Brenner and Rector’s The Kidney, 27, 804-45.e2. Philadelphia, PA: Elsevier; 2016.
9. Goldstein MB, Bear R, Richardson RMA, Marsden PA, Halperin ML. The Urine Anion Gap: a clinically useful index of ammonium excretion. Am J Med Sci. 1986;198-202.
Key Points:
• In acute kidney injury, the FENa and FEUrea may be calculated to distinguish prerenal azotemia from ATN; however, FENa and FEUrea may be low in a wide variety of conditions other than prerenal azotemia.
• Urine sodium and osmolality values are helpful in diagnosing the cause of hyponatremia, but have a number of limitations in nonoliguric patients and those with CKD.
• An elevated transtubular potassium gradient (TTKG) may indicate renal loss of potassium in patients with hypokalemia.
• A positive urine anion gap (UAG) in the setting of a normal anion gap metabolic acidosis points to renal causes of the metabolic acidosis, whereas a negative UAG points to extrarenal causes such as bicarbonate losses in the GI tract.Ad
Additional Reading:
Goldstein MB, Bear R, Richardson RMA, Marsden PA, Halperin ML. The Urine Anion Gap: A Clinically Useful Index of Ammonium Excretion. Am J Med Sci. 1986;198-202.
Gotfried J, Wiesen J, Raina R, Nally Jr JV. Finding the cause of acute kidney injury: which index of fractional excretion is better. Cleve Clin J Med. 2012;79(2):121-126.
Kamel KS, Davids MR, Lin S-H, Halperin ML. Interpretation of Electrolyte and Acid-Base Parameters in Blood and Urine. In: Brenner and Rector’s The Kidney, 27, 804-845.e2. Philadelphia, PA: Elsevier; 2016.
The case
A 50-year old woman naive to the health care system presents to the ED with nausea, malaise, and decreased exercise tolerance for several weeks. Physical exam reveals mild bilateral lower extremity edema. Her labs are notable for an elevated creatinine of 7.0. She is admitted for work-up of her renal disease.
Nephrology was consulted and recommended obtaining urine electrolytes. The admitting hospitalist is unsure which urine electrolytes are appropriate to order, and in turn orders all of the urine electrolytes in the order set.
Which urine electrolytes should be ordered in various clinical contexts?
Introduction
Hospitalists have been on the forefront of efforts to tailor testing and resource utilization to eliminate wasteful practices in health care. To order and interpret diagnostic tests appropriately, a hospitalist needs to have a thorough understanding of the diagnostic utility of laboratory tests. There is a lack of clear diagnostic guidelines, so ordering all the urine electrolytes in a “blanket” strategy is a common practice. We will discuss the diagnostic utility of each of the urine electrolytes in a variety of clinical scenarios.
Acute kidney injury
Both the fractional excretion of sodium (FENa) and the fractional excretion of urea (FEUrea) have long been used as part of the standard work-up for determining if acute kidney injury (AKI) is due to prerenal causes. Although these markers prove to be beneficial in the work-up of AKI, both the FENa and FEUrea have several limitations.
FENa measures the ratio of sodium excreted in the urine compared to how much is filtered through the kidney. A FENa of less than 1% in oliguric patients may indicate prerenal azotemia, as an increased reabsorption of sodium is the appropriate response of functioning nephrons to decreased renal perfusion. Values greater than 3% may be consistent with acute tubular necrosis (ATN) due to inappropriate sodium excretion in the setting of tubular damage.
Importantly, a FENa value of less than 1% occurs in a number of conditions other than prerenal azotemia due to dehydration, including hypervolemic prerenal states such as cirrhosis or heart failure; AKI due to radiocontrast or heme pigments; acute glomerulonephritis; transition from prerenal to postischemic ATN or sepsis, and in acute interstitial nephritis (AIN).1,2 Approximately 10% of patients with nonoliguric ATN have a FENa less than 1.0%. Moreover, use of diuretics can falsely elevate the FENa due to inhibition of sodium reabsorption. FENa values above 3% can occur in volume contraction in patients with chronic kidney disease (CKD) or in elderly patients as their sodium reabsorption is impaired.3 Acute volume loss (e.g. blood loss), or more commonly, administration of diuretics or intravenous fluids, can also alter the interpretation of the FENa.2
Many of the limitations of the FENa also apply to the FEUrea, including interpretation in the elderly and use in acute volume changes. However, the FEUrea has unique limitations, particularly in patients with sepsis, as cytokines released in sepsis may interfere with urea transporters in the kidney and colon.2 Its interpretation also relies on intact functioning of the proximal tubule, which can be altered in many conditions including uncontrolled diabetes. Overall, the FENa and FEUrea can be helpful to determine the etiology of AKI, but only in certain clinical scenarios.
Hyponatremia
Hyponatremia is the most common electrolyte abnormality in hospitalized patients, with a prevalence of up to 30% in critically ill patients.4 It often is acquired during the hospitalization itself. A detailed history and physical exam, including careful assessment of volume status, is as important as laboratory values in establishing the cause of hyponatremia.
Urine sodium and urine osmolality are measured to understand whether the renin-aldosterone-angiotensin system (RAAS) and antidiuretic hormone (ADH) are activated. If renal blood flow or renal delivery of sodium is decreased, renin secretion from the juxtaglomerular apparatus will be activated, ultimately leading to increased reabsorption of sodium in the distal tubules and collecting ducts. Thus, low urine sodium signals that the RAAS is activated due to decreased serum sodium concentration or decreased renal blood flow from hypovolemia or low effective arterial circulation from cirrhosis or heart failure.
Most causes of hyponatremia will have low urine sodium values, including hypovolemia, cirrhosis, heart failure, “tea-and-toast” diet, beer potomania, and primary polydipsia. However, the urine sodium may be unreliable in patients who are not oliguric or who have CKD.
Diuretic-induced hyponatremia from thiazide or loop diuretics will likely have elevated urine sodium levels. Similarly, the syndrome of inappropriate antidiuretic hormone secretion (SIADH) will have an elevated urine sodium above 20-40 mEq/L.
Urine osmolality becomes elevated when ADH is secreted in response to reduced plasma volume or increased plasma osmolality. Urine osmolality is low in cases such as primary polydipsia, which creates a maximally dilute urine of 40-100 mEq/L, and in tea-and-toast diets or beer potomania due to low solute intake. Urine osmolality can be elevated in hypovolemic states as well as SIADH, and is variable in hypothyroidism and selective serotonin reuptake inhibitor administration. Thus, urine sodium, and not urine osmolality, is the most useful differentiator between SIADH and hypovolemic states.
In a study of 555 patients with hyponatremia secondary to SIADH, mean urine sodium was found to be 72 (range 30-251) and the median urine osmolality was 379 (range 123-1019).5
In cases of marked hyperglycemia, serum osmolality should be measured to evaluate hyperglycemia as a cause of hyperosmolar hyponatremia. Pseudohyponatremia in the setting of hyperlipidemia, hypertriglyceridemia, or hyperparaproteinemia represents a laboratory artifact due to lower plasma water concentration in the specimen sample and should be excluded.
Hypokalemia
About 20% of patients are hypokalemic during an inpatient hospitalization. There is a broad differential for hypokalemia, including medical, nutritional, and medication-related causes. Exogenous insulin administration or endogenous production in cases of refeeding syndrome drives potassium intracellularly via the N+/K+ ATPase. Increased sympathetic activity from alcohol withdrawal, acute myocardial infarction, head injury, or thyroid imbalance, as well as iatrogenic causes such as albuterol administration, also drive potassium intracellularly. Diarrhea and nasogastric tube suction lead to gastrointestinal (GI) potassium losses, while antibiotics, chemotherapeutic agents, and diuretics can cause hypokalemia through renal potassium wasting. Hyperaldosteronism and renal tubular acidosis are less common causes.6
The history, review of medications, physical exam, and initial basic laboratory testing (electrolytes, BUN, creatinine, magnesium) should assess for pseudohypokalemia, poor oral intake, diuretic use, acid-base disturbances, or GI losses.
Measuring urine potassium is useful in the work-up of the hypokalemic patient when these conditions are not evident. Urine potassium – either 24-hour or spot urine potassium-to-creatinine ratio – can help determine if urinary potassium wasting is a factor. Potassium is excreted at a near constant rate throughout the day. A urine potassium-to-creatinine ratio corrects for variations in urine volume. When this ratio is greater than 13 mEq/g, renal potassium losses should be suspected. If the ratio is less than 13 mEq/g, hypokalemia is likely due to transcellular potassium shifts, GI losses, diuretics, or poor intake.
Hyperkalemia
Several concepts in hypokalemia are relevant to hyperkalemia. Redistribution of potassium into the extracellular fluid can cause hyperkalemia when the body tries to counterbalance low extracellular pH by potassium-hydrogen exchange. Medications may cause an extracellular shift of potassium (e.g. digoxin) or induce diminished potassium excretion (e.g. NSAIDs, spironolactone, ACE/ARBs).
CKD and end-stage kidney disease are common causes of hyperkalemia in the hospitalized patient – as functioning nephrons decrease, poor Na-K exchange ensues. Hypoaldosteronism and type 4 renal tubular acidosis are also on the differential diagnosis. Pseudohyperkalemia secondary to thrombocytosis, erythrocytosis, or activated platelets should be considered and evaluated.
Appropriate renal excretion of potassium is mediated by the connecting segment between the distal tubule and the collecting duct, and the cortical collecting duct itself. There are four major causes of hyperkalemia due to reduced urinary potassium secretion: reduced aldosterone secretion, reduced response to aldosterone, reduced distal sodium and water delivery (often related to low effective arterial blood volume), and kidney injury.6
Measurement of 24-hour urinary potassium excretion is of limited utility in patients with persistent stable hyperkalemia because urinary potassium excretion is related to potassium intake. The TTKG was previously used to assess the degree of aldosterone activity by estimating the potassium concentration in the cortical collecting tubule. However, some assumptions upon which this calculation was based have been considered invalid by the original studies’ authors, and the TTKG to evaluate potassium abnormalities is no longer uniformly recommended.7,8 Ultimately, if patients have persistent hyperkalemia, work-up for hypoaldosteronism should be considered.
Normal anion gap metabolic acidosis
The urine anion gap (UAG) is used to determine the cause of normal anion gap hyperchloremic metabolic acidosis by indirectly measuring urinary excretion of ammonium. To maintain a normal acid/base balance, hydrogen ions are excreted in the urine with simultaneous reabsorption of bicarbonate. Hydrogen ions are bound to ammonia (NH3) to form ammonium (NH4+), which is excreted as NH4Cl in the urine.
The UAG is calculated by adding urine sodium and urine potassium and subtracting urine chloride (see Table 1). In a patient without an acid/base disturbance, the UAG is positive because more Na and K is absorbed in the gastrointestinal system compared to Cl, and thus more Na and K is excreted in the urine. In a normal anion gap metabolic acidosis through an acid load or bicarbonate loss, the normal response of the kidney is to excrete more hydrogen ions, resulting in more chloride excretion as NH4Cl. This leads to a negative urine anion gap, as Cl excretion outweighs Na and K excretion. When NH4+ excretion is impaired, such as in distal renal tubular acidosis (RTA), the urine anion gap will remain positive despite the metabolic acidosis. Thus, a positive UAG points to renal causes of the normal anion gap metabolic acidosis, whereas a negative UAG points to extrarenal causes such as bicarbonate losses in the GI tract.9
Additional considerations
Urine studies can also be useful for assessment of proteinuria and albuminuria in a patient with CKD or diabetes, diagnosis of plasma cell dyscrasias, the diagnosis and prevention of nephrolithiasis, and a wide variety of other conditions.
Back to the case
Our patient was admitted with an elevated creatinine of unclear chronicity, and subacute symptoms of uremia. Because she was oliguric, urine and serum sodium and creatinine were measured before intravenous fluids were administered. Her FENa was 2%, which was not consistent with prerenal azotemia or ATN. She was found to have CKD secondary to previously undiagnosed diabetes. Upon further questioning, she had been taking high-dose NSAIDs for her chronic knee pain. Her renal function improved mildly by withholding NSAIDs, and she was discharged with appropriate nephrology follow-up.
Bottom line
Urine electrolytes have specific indications and utilities for different clinical scenarios, and should be ordered in a targeted manner that can aide in diagnosing AKI, hyponatremia, hypokalemia, and normal anion gap metabolic acidosis.
Dr. Tummalapalli, Dr. Krouss, and Dr. Goetz are hospitalists in the department of medicine at the Icahn School of Medicine at Mount Sinai in New York City.
References
1. Brosius FC, Lau K. Low fractional excretion of sodium in acute renal failure: role of timing of the test and ischemia. Am J Nephrol. 1986;6(6):450-7.
2. Gottfried J, Weisen J, Raina R, Nally J. Finding the cause of acute kidney injury: Which index of fractional excretion is better? Cleve Clin J Med. 2012;79(2):121-6.
3. Steiner, RW. Interpreting the fractional excretion of sodium. Am J Med. 1984;77(4):699-702.
4. DeVita MV, Gardenswartz MH, Konecky A, Zabetakis PM. Incidence and etiology of hyponatremia in an intensive care unit. Clin Nephrol. 1990;34(4):163-6.
5. Shepshelovich D, Leibovitch C, Klein A, et al. The syndrome of inappropriate antidiuretic hormone secretion: distribution and characterization according to etiologies. Eur J Int Med. 2015;26(10):819-24.
6. Mount DB. Fluid and Electrolyte Disturbances. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J. eds. Harrison’s Principles of Internal Medicine, 19e. New York, NY: McGraw-Hill; 2015.
7. Kamel KS. Intrarenal urea recycling leads to a higher rate of renal excretion of potassium: an hypothesis with clinical implications. Curr Opin Nephrol Hypertens. 2011 Sep;20(5):547-54.
8. Kamel KS, Davids MR, Lin S-H, Halperin ML. Interpretation of Electrolyte and Acid-Base Parameters in Blood and Urine. In: Brenner and Rector’s The Kidney, 27, 804-45.e2. Philadelphia, PA: Elsevier; 2016.
9. Goldstein MB, Bear R, Richardson RMA, Marsden PA, Halperin ML. The Urine Anion Gap: a clinically useful index of ammonium excretion. Am J Med Sci. 1986;198-202.
Key Points:
• In acute kidney injury, the FENa and FEUrea may be calculated to distinguish prerenal azotemia from ATN; however, FENa and FEUrea may be low in a wide variety of conditions other than prerenal azotemia.
• Urine sodium and osmolality values are helpful in diagnosing the cause of hyponatremia, but have a number of limitations in nonoliguric patients and those with CKD.
• An elevated transtubular potassium gradient (TTKG) may indicate renal loss of potassium in patients with hypokalemia.
• A positive urine anion gap (UAG) in the setting of a normal anion gap metabolic acidosis points to renal causes of the metabolic acidosis, whereas a negative UAG points to extrarenal causes such as bicarbonate losses in the GI tract.Ad
Additional Reading:
Goldstein MB, Bear R, Richardson RMA, Marsden PA, Halperin ML. The Urine Anion Gap: A Clinically Useful Index of Ammonium Excretion. Am J Med Sci. 1986;198-202.
Gotfried J, Wiesen J, Raina R, Nally Jr JV. Finding the cause of acute kidney injury: which index of fractional excretion is better. Cleve Clin J Med. 2012;79(2):121-126.
Kamel KS, Davids MR, Lin S-H, Halperin ML. Interpretation of Electrolyte and Acid-Base Parameters in Blood and Urine. In: Brenner and Rector’s The Kidney, 27, 804-845.e2. Philadelphia, PA: Elsevier; 2016.