User login
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE

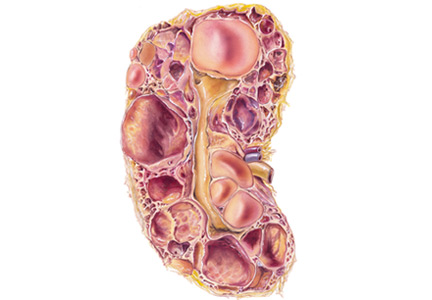
ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25

Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).

A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
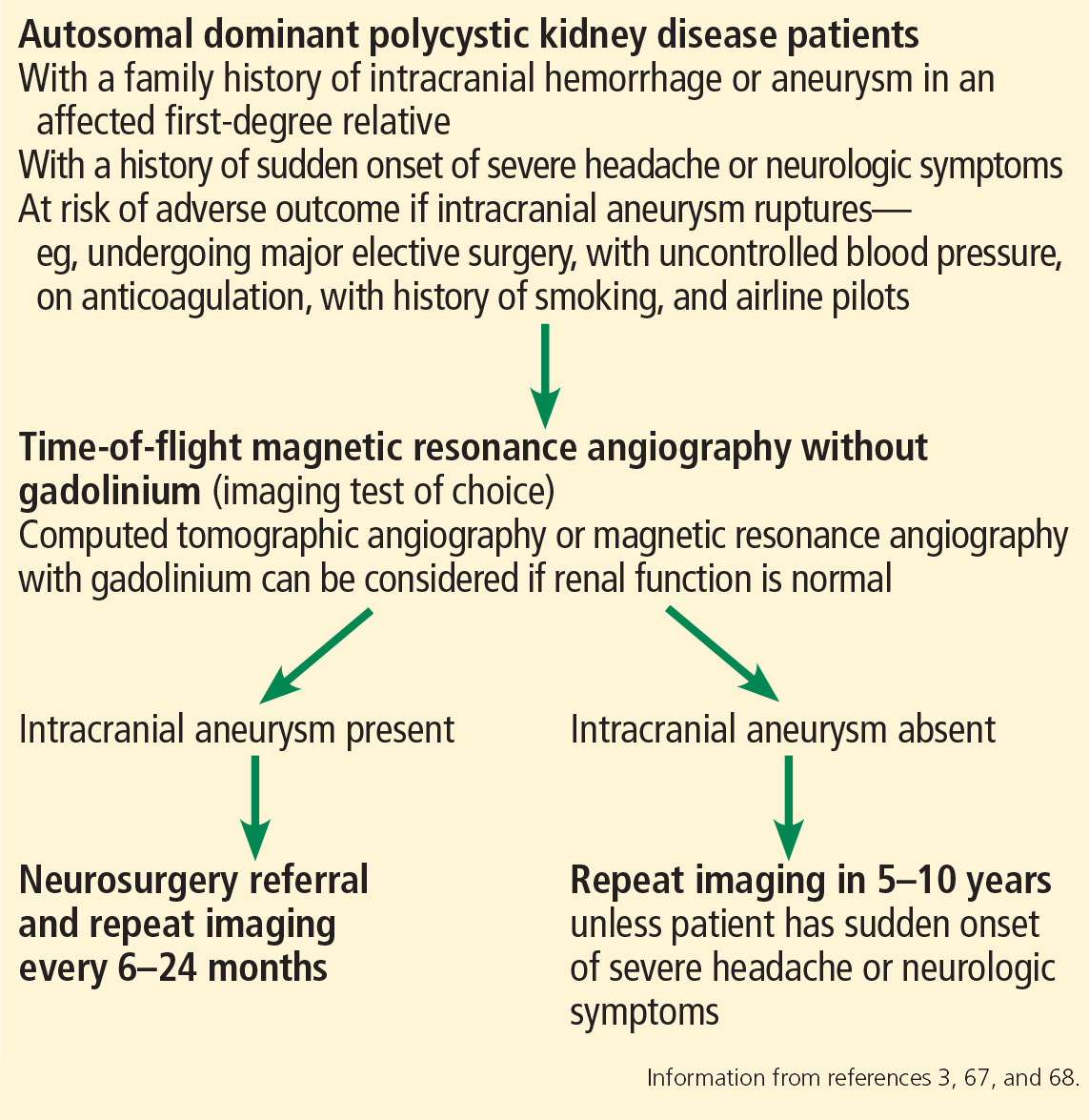
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE
ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25
Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).
A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
Autosomal dominant polycystic kidney disease (ADPKD) has significant extrarenal manifestations. Hypertension is a common complication, arises early in the course of the disease, and is implicated in the development of left ventricular hypertrophy. Patients with ADPKD are also at risk of other cardiovascular complications (Table 1).
This article reviews the timely diagnosis of these common ADPKD complications and how to manage them.
ADPKD ACCOUNTS FOR 10% OF END-STAGE RENAL DISEASE
ADPKD is a genetic condition characterized by multiple renal cysts.1 Progressive enlargement of these cysts leads to a gradual decline in kidney function and eventually end-stage renal disease by the fifth or sixth decade of life.2 Worldwide, about 12.5 million people have ADPKD, and it accounts for about 10% of cases of end-stage renal disease.1,3,4
ADPKD has a variety of clinical presentations, including (in decreasing order of frequency) hypertension, flank pain, abdominal masses, urinary tract infection, renal failure, renal stones, and cerebrovascular accidents.2
Extrarenal complications are common and include hepatic cysts, hypertension, left ventricular hypertrophy, valvular heart disease, intracranial and extracranial aneurysms, pancreatic cysts, and diverticulosis.1–5
Less-common complications are dissection of the aorta and the internal carotid, vertebral, and iliac arteries6–10; aneurysm of the coronary, popliteal, and splenic arteries11–14; atrial myxoma15; cardiomyopathy16; pericardial effusion17; intracranial arterial dolichoectasia18; arachnoid cysts2; and intraoperative inferior vena cava syndrome (normally in ADPKD patients, pressure on the inferior vena cava results in compensatory sympathetic overactivity to maintain blood pressure), which occurs due to reduced sympathetic output under the influence of epidural or general anesthesia.19
Cardiovascular complications, especially cardiac hypertrophy and coronary artery disease, are now the leading cause of death in patients with ADPKD, as renal replacement therapy has improved and made death from end-stage renal disease less common.20,21
HYPERTENSION IN ADPKD
Hypertension is the most frequent initial presentation of ADPKD, occurring in 50% to 75% of cases and usually preceding the onset of renal failure.2,22 Hypertension is more common in male ADPKD patients, begins early in the course of the disease, and is diagnosed around the fourth decade of life.21
In a study in 2007, de Almeida et al23 used 24-hour ambulatory blood pressure monitoring early in the course of ADPKD and found significantly higher systolic, diastolic, and mean 24-hour blood pressures in ADPKD patients who had normal in-office blood pressure than in normotensive controls. In addition, nighttime systolic, nighttime diastolic, and nighttime mean blood pressures were significantly higher in the ADPKD group.
Hypertension is strongly associated with an accelerated decline in renal function to end-stage renal disease, development of left ventricular hypertrophy, and cardiovascular death.20,24
Although a prospective study25 showed a strong association between renal stones and hypertension in ADPKD, the relation between them is not clear. The incidence of renal stones is higher in hypertensive than in normotensive ADPKD patients, although evidence has to be established whether nephrolithiasis is a risk factor for hypertension or the other way around.25
Hypertension in ADPKD is multifactorial (Figure 1). The major factors associated with its development are increased activation of the renin-angiotensin-aldosterone system (RAAS); overexpression of endothelin receptor subtype A (ET-A) in cystic kidneys; increased production of endothelin 1 (ET-1); and sodium retention.26–31
The renin-angiotensin-aldosterone system
Activation of the RAAS plays a major role in the development and maintenance of hypertension in ADPKD. This is thought to be mainly due to progressive enlargement of renal cysts, which causes renal arteriolar attenuation and ischemia secondary to pressure effects, which in turn activates the RAAS.26,30,32–34 Two studies in patients with normal renal function found that cyst growth and increasing kidney volume have a strong relationship with the development of hypertension and declining kidney function.35,36
Ectopic secretion of RAAS components in polycystic kidneys has also been implicated in the development of hypertension, whereby renin, angiotensinogen, angiotensin-converting enzyme (ACE), angiotensin II, and angiotensin II receptors are produced in the epithelium of cysts and dilated tubules in polycystic kidneys.37–39 Proximal renal cysts and tubules produce ectopic angiotensinogen, which is converted to angiotensin I by renin in distal renal cysts. Angiotensin I is converted to angiotensin II by ACE in distal tubules, which in turn stimulates angiotensin II receptors, causing sodium and water retention in distal tubules.37 This may be responsible for hypertension in the initial stages; however, RAAS hyperactivity due to renal injury may predominate during later stages.37
Increased RAAS activity also increases sympathetic output, which in turn raises catecholamine levels and blood pressure.34 A study showed higher levels of plasma catecholamines in ADPKD hypertensive patients irrespective of renal function than in patients with essential hypertension.40
ET-A receptor and ET-1
A few studies have shown that in ADPKD patients, increased density of ET-A receptors and overproduction of ET-1, a potent vasoconstrictor, play a significant role in the development of hypertension and gradual loss of kidney function due to cyst enlargement and interstitial scarring.28,29 Ong et al29 found that expression of ET-A receptors is increased in smooth muscle cells of renal arteries, glomerular mesangial cells, and cyst epithelia in ADPKD.
Sodium retention
Studies in ADPKD patients with preserved renal function have linked high blood pressure to sodium retention and volume expansion.30,31,41 However, this phenomenon reverses when there is significant renal impairment in ADPKD.
As evidence of this, a study demonstrated significantly more natriuresis in patients with renal failure due to ADPKD than in patients with a similar degree of renal failure due to chronic glomerulonephritis.31 Moreover, another study found that the prevalence of hypertension is higher in ADPKD patients than in those with other nephropathies with preserved renal function, but this association reverses with significant decline in kidney function.22
MANAGING HYPERTENSION IN ADPKD
Early diagnosis of hypertension and effective control of it, even before ADPKD is diagnosed, is crucial to reduce cardiovascular mortality. Aggressive blood pressure control in the prehypertensive phase of ADPKD will also help reduce the incidence of left ventricular hypertrophy and mitral regurgitation and slow the progression of renal failure (Figure 2).
A meta-analysis42 revealed hypertension to be present in 20% of ADPKD patients younger than 21, and many of them were undiagnosed. This study also suggests that patients at risk of hypertension (ie, all patients with ADPKD) should be routinely screened for it.
Ambulatory blood pressure monitoring may play an important role in diagnosing hypertension early in the prehypertensive stage of ADPKD.23
Target blood pressures: No consensus
Two well-powered double-blind, placebo-controlled trials, known as HALT-PKD Study A and HALT-PKD Study B, tested the effects of 2 different blood pressure targets and of monotherapy with an ACE inhibitor vs combination therapy with an ACE inhibitor plus an angiotensin II receptor blocker (ARB) on renal function, total kidney volume, left ventricular mass index, and urinary albumin excretion in the early (estimated glomerular filtration rate [eGFR] > 60 mL/min) and late (eGFR 25–60 mL/min) stages of ADPKD, respectively.43,44
HALT-PKD Study A43 found that, in the early stages of ADPKD with preserved renal function, meticulous control of blood pressure (95–110/60–75 mm Hg) was strongly correlated with significant reductions in left ventricular mass index, albuminuria, and rate of total kidney volume growth without remarkable alteration in renal function compared with standard blood pressure control (120–130/70–80 mm Hg). However, no notable differences were observed between the ACE inhibitor and ACE inhibitor-plus-ARB groups.43
Despite the evidence, universal consensus guidelines are lacking, and the available guidelines on hypertension management have different blood pressure goals in patients with chronic kidney disease.
The eighth Joint National Committee guideline of 2014 recommends a blood pressure goal of less than 140/90 mm Hg in patients with diabetic and nondiabetic chronic kidney disease.45
The National Institute for Health and Care Excellence 2011 guideline recommends a blood pressure goal of less than 130/80 mm Hg in chronic kidney disease patients.46
The European Society of Hypertension and European Society of Cardiology joint 2013 guideline recommends a systolic blood pressure goal of less than 140 mm Hg in diabetic and nondiabetic patients with chronic kidney disease.47
The 2016 Kidney Health Australia-Caring for Australians With Renal Impairment guideline for diagnosis and management of ADPKD48 recommends a lower blood pressure goal of 96–110/60–75 mm Hg in patients with an eGFR greater than 60 mL/min/1.73 m2 who can tolerate it without side effects, which is based on the findings of HALT-PKD Study A.43
Helal et al recommend that blood pressure be controlled to less than 130/80 mm Hg, until there is more evidence for a safe and effective target blood pressure goal in ADPKD patients.49
We recommend a target blood pressure less than 110/75 mm Hg in hypertensive ADPKD patients with preserved renal function who can tolerate this level, and less than 130/80 mm Hg in ADPKD patients with stage 3 chronic kidney disease. These targets can be achieved with ACE inhibitor or ARB monotherapy.43,44 However, no studies have established the safest lower limit of target blood pressure in ADPKD.
ACE inhibitors, ARBs are mainstays
Mainstays of antihypertensive drug therapy in ADPKD are ACE inhibitors and ARBs.
HALT-PKD Study B44 demonstrated that, in the late stages of ADPKD, target blood pressure control (110–130/70–80 mm Hg) can be attained with ACE inhibitor monotherapy or with an ACE inhibitor plus an ARB, but the latter produced no additive benefit.
Patch et al,50 in a retrospective cohort study, showed that broadening the spectrum of antihypertensive therapy decreases mortality in ADPKD patients. Evaluating ADPKD patients from the UK General Practice Research Database between 1991 and 2008, they found a trend toward lower mortality rates as the number of antihypertensive drugs prescribed within 1 year increased. They also observed that the prescription of RAAS-blocking agents increased from 7% in 1991 to 46% in 2008.50
However, a 3-year prospective randomized double-blind study compared the effects of the ACE inhibitor ramipril and the beta-blocker metoprolol in hypertensive ADPKD patients.51 The results showed that effective blood pressure control could be achieved in both groups with no significant differences in left ventricular mass index, albuminuria, or kidney function.51
Treatment strategies
Lifestyle modification is the initial approach to the management of hypertension before starting drug therapy. Lifestyle changes include dietary salt restriction to less than 6 g/day, weight reduction, regular exercise, increased fluid intake (up to 3 L/day or to satisfy thirst), smoking cessation, and avoidance of caffeine.47–49
ACE inhibitors are first-line drugs in hypertensive ADPKD patients.
ARBs can also be considered, but there is no role for dual ACE inhibitor and ARB therapy.43,48 A study found ACE inhibitors to be more cost-effective and to decrease mortality rates to a greater extent than ARBs.52
Beta-blockers or calcium channel blockers should be considered instead if ACE inhibitors and ARBs are contraindicated, or as add-on drugs if ACE inhibitors and ARBs fail to reduce blood pressure adequately.48,49
Diuretics are third-line agents. Thiazides are preferred in ADPKD patients with normal renal function and loop diuretics in those with impaired renal function.49
LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Increased left ventricular mass is an indirect indicator of untreated hypertension, and it often goes unnoticed in patients with undiagnosed ADPKD. Left ventricular hypertrophy is associated with arrhythmias and heart failure, which contribute significantly to cardiovascular mortality and adverse renal outcomes.20,24
A 5-year randomized clinical trial by Cadnapaphornchai et al36 in ADPKD patients between 4 and 21 years of age showed strong correlations between hypertension, left ventricular mass index, and kidney volume and a negative correlation between left ventricular mass index and renal function.
Several factors are thought to contribute to left ventricular hypertrophy in ADPKD (Figure 1).
Hypertension. Two studies of 24-hour ambulatory blood pressure monitoring showed that nocturnal blood pressures decreased less in normotensive and hypertensive ADPKD patients than in normotensive and hypertensive controls.23,53 This persistent elevation of nocturnal blood pressure may contribute to the development and progression of left ventricular hypertrophy.
On the other hand, Valero et al54 reported that the left ventricular mass index was strongly associated with ambulatory systolic blood pressure rather than elevated nocturnal blood pressure in ADPKD patients compared with healthy controls.
FGF23. High levels of fibroblast growth factor 23 (FGF23) have been shown to be strongly associated with left ventricular hypertrophy in ADPKD. Experimental studies have shown that FGF23 is directly involved in the pathogenesis of left ventricular hypertrophy through stimulation of the calcineurin-nuclear factor of activated T cells pathway.
Faul et al55 induced cardiac hypertrophy in mice that were deficient in klotho (a transmembrane protein that increases FGF23 affinity for FGF receptors) by injecting FGF23 intravenously.
Yildiz et al56 observed higher levels of FGF23 in hypertensive and normotensive ADPKD patients with normal renal function than in healthy controls. They also found a lower elasticity index in the large and small arteries in normotensive and hypertensive ADPKD patients, which accounts for vascular dysfunction. High FGF23 levels may be responsible for the left ventricular hypertrophy seen in normotensive ADPKD patients with preserved renal function.
Polymorphisms in the ACE gene have been implicated in the development of cardiac hypertrophy in ADPKD.
Wanic-Kossowska et al57 studied the association between ACE gene polymorphisms and cardiovascular complications in ADPKD patients. They found a higher prevalence of the homozygous DD genotype among ADPKD patients with end-stage renal disease than in those in the early stages of chronic kidney disease in ADPKD. Also, the DD genotype has been shown to be more strongly associated with left ventricular hypertrophy and left ventricular dysfunction than other (II or ID) genotypes. These findings suggest that the DD genotype carries higher risk for the development of end-stage renal disease, left ventricular hypertrophy, and other cardiovascular complications.
MANAGING LEFT VENTRICULAR HYPERTROPHY IN ADPKD
Preventing and halting progression of left ventricular hypertrophy primarily involves effective blood pressure control, especially in the early stages of ADPKD (Figure 2).
A 7-year prospective randomized trial in ADPKD patients with established hypertension and left ventricular hypertrophy proved that aggressive (< 120/80 mm Hg) compared with standard blood pressure control (135–140/85–90 mm Hg) significantly reduces left ventricular mass index. ACE inhibitors were preferred over calcium channel blockers.58
HALT-PKD Study A showed that a significant decrease in left ventricular mass index can be achieved by aggressive blood pressure control (95–110/60–75 mm Hg) with an ACE inhibitor alone or in combination with an ARB in the early stages of ADPKD with preserved renal function.43
A 5-year randomized clinical trial in children with borderline hypertension treated with an ACE inhibitor for effective control of blood pressure showed no change in left ventricular mass index or renal function.36,59
These results support starting ACE inhibitor therapy early in the disease process when blood pressure is still normal or borderline to prevent the progression of left ventricular hypertrophy or worsening kidney function.
Since FGF23 is directly involved in the causation of left ventricular hypertrophy, FGF receptors may be potential therapeutic targets to prevent left ventricular hypertrophy in ADPKD. An FGF receptor blocker was shown to decrease left ventricular hypertrophy in rats with chronic kidney disease without affecting blood pressure.55
INTRACRANIAL ANEURYSM IN ADPKD
Intracranial aneurysm is the most dangerous complication of ADPKD. When an aneurysm ruptures, the mortality rate is 4% to 7%, and 50% of survivors are left with residual neurologic deficits.5,60,61
In various studies, the prevalence of intracranial aneurysm in ADPKD ranged from 4% to 41.2%, compared with 1% in the general population.5,62,63 On follow-up ranging from 18 months to about 10 years, the incidence of new intracranial aneurysm was 2.6% to 13.3% in patients with previously normal findings on magnetic resonance angiography and 25% in patients with a history of intracranial aneurysm.62,64,65
The most common sites are the middle cerebral artery (45%), internal carotid artery (40.5%), and anterior communicating artery (35.1%).66 (The numbers add up to more than 100% because some patients have aneurysms in more than 1 site.) The mean size of a ruptured aneurysm was 6 mm per a recent systematic review.66 Intracranial aneurysms 6 mm or larger are at highest risk of rupture.66
SCREENING FOR INTRACRANIAL ANEURYSM
Timely screening and intervention for intracranial aneurysm is crucial to prevent death from intracranial hemorrhage.
Currently, there are no standard guidelines for screening and follow-up of intracranial aneurysm in ADPKD patients. However, some recommendations are available from the ADPKD Kidney Disease Improving Global Outcomes Controversies Conference3 and Kidney Health Australia—Caring for Australasians With Renal Impairment ADPKD guidelines67 (Figure 3).
Imaging tests
Magnetic resonance angiography (MRA) with gadolinium enhancement and computed tomographic angiography (CTA) are recommended for screening in ADPKD patients with normal renal function,67 but time-of-flight MRA without gadolinium is the imaging test of choice because it is noninvasive and poses no risk of nephrotoxicity or contrast allergy.3,68 Further, gadolinium should be avoided in patients whose eGFR is 30 mL/min/1.73 m2 or less because of risk of nephrogenic systemic sclerosis and fibrosis.67,68
The sensitivity of time-of-flight MRA screening for intracranial aneurysm varies depending on the size of aneurysm; 67% for those less than 3 mm, 79% for those 3 to 5 mm, and 95% for those larger than 5 mm.69 The sensitivity of CTA screening is 95% for aneurysms larger than 7 mm and 53% for those measuring 2 mm.70,71 The specificity of CTA screening was reported to be 98.9% overall.71
When to screen
Screening for intracranial aneurysm is recommended at the time of ADPKD diagnosis for all high-risk patients, ie, those who have a family history of intracranial hemorrhage or aneurysm in an affected first-degree relative.67 It is also recommended for ADPKD patients with a history of sudden-onset severe headache or neurologic symptoms.67 A third group for whom screening is recommended is ADPKD patients who have no family history of intracranial aneurysm or hemorrhage but who are at risk of poor outcome if an intracranial aneurysm ruptures (eg, those undergoing major elective surgery, with uncontrolled blood pressure, on anticoagulation, with a history of or current smoking, and airline pilots).67
Patients found to have an intracranial aneurysm on screening should be referred to a neurosurgeon and should undergo repeat MRA or CTA imaging every 6 to 24 months.3 High-risk ADPKD patients with normal findings on initial screening should have repeat MRA or CTA screening in 5 to 10 years unless they suffer from sudden-onset severe headache or neurologic symptoms.65,67
Both smoking and high blood pressure increase the risk of formation and growth of intracranial aneurysm. Hence, meticulous control of blood pressure and smoking cessation are recommended in ADPKD patients.3,67
CARDIAC VALVULAR ABNORMALITIES IN ADPKD
Of the valvular abnormalities that complicate ADPKD, the more common ones are mitral valve prolapse and mitral and aortic regurgitation. The less common ones are tricuspid valve prolapse and tricuspid regurgitation.72–74
The pathophysiology underlying these valvular abnormalities is unclear. However, defective collagen synthesis and myxomatous degeneration have been demonstrated in histopathologic examination of affected valvular tissue.75 Also, ACE gene polymorphism, especially the DD genotype, has been shown to be associated with cardiac valvular calcifications and valvular insufficiency.57
Lumiaho et al72 found a higher prevalence of mitral valve prolapse, mitral regurgitation, and left ventricular hypertrophy in patients with ADPKD type 1 (due to abnormalities in PDK1) than in unaffected family members and healthy controls. The investigators speculated that mitral regurgitation is caused by the high blood pressure observed in ADPKD type 1 patients, since hypertension causes left ventricular hypertrophy and left ventricular dilatation. The severity of renal failure was related to mitral regurgitation but not mitral valve prolapse.
Similarly, Gabow et al24 showed that there is no significant relationship between mitral valve prolapse and progression of renal disease in ADPKD.
Interestingly, Fick et al20 found that mitral valve prolapse has no significant effect on cardiovascular mortality.
CORONARY ARTERY DISEASE AND ANEURYSM IN ADPKD
Atherosclerosis sets in early in ADPKD, resulting in coronary artery disease and adverse cardiovascular outcomes.
Coronary flow velocity reserve is the ability of coronary arteries to dilate in response to myocardial oxygen demand. Atherosclerosis decreases this reserve in ADPKD patients, as shown in several studies.
Turkmen et al, in a series of studies,76–78 found that ADPKD patients had significantly less coronary flow velocity reserve, thicker carotid intima media (a surrogate marker of atherosclerosis), and greater insulin resistance than healthy controls. These findings imply that atherosclerosis begins very early in the course of ADPKD and has remarkable effects on cardiovascular morbidity and mortality.76
Aneurysm. Although the risk of extracranial aneurysm is higher with ADPKD, coronary artery aneurysm is uncommon. The pathogenesis of coronary aneurysm has been linked to abnormal expression of the proteins polycystin 1 and polycystin 2 in vascular smooth muscle.11,79 The PKD1 and PKD2 genes encode polycystin 1 and 2, respectively, in ADPKD. These polycystins are also expressed in the liver, kidneys, and myocardium and are involved in the regulation of intracellular calcium, stretch-activated ion channels, and vascular smooth muscle cell proliferation and apoptosis.11,16 Abnormally expressed polycystin in ADPKD therefore has an impact on arterial wall integrity, resulting in focal medial defects in the vasculature that eventually develop into micro- and macroaneurysms.11
Hadimeri et al79 found a higher prevalence of coronary aneurysm and ectasia in ADPKD patients than in controls. Most coronary aneurysms are smaller than 1 cm; however, a coronary aneurysm measuring 4 cm in diameter was found at autopsy of an ADPKD patient.11
Spontaneous coronary artery dissection is very rare in the general population, but Bobrie et al reported a case of it in an ADPKD patient.9
ATRIAL MYXOMA, CARDIOMYOPATHY, AND PERICARDIAL EFFUSION IN ADPKD
Atrial myxoma in ADPKD patients has been described in 2 case reports.15,80 However, the association of atrial myxoma with ADPKD is poorly understood and may be a coincidental finding.
Cardiomyopathy in ADPKD has been linked to abnormalities in the intracellular calcium pathway, although a clear picture of its involvement has yet to be established.
Paavola et al16 described the pathophysiology of ADPKD-associated cardiomyopathy in PKD2 mutant zebrafish lacking polycystin 2. These mutants showed decreased cardiac output and had atrioventricular blocks. The findings were attributed to abnormal intracellular calcium cycling. These findings correlated well with the frequent finding of idiopathic dilated cardiomyopathy in ADPKD patients, especially with PKD2 mutations.16 Also, 2 cases of dilated cardiomyopathy in ADPKD have been reported and thought to be related to PKD2 mutations.81,82
Pericardial effusion. Even though the exact pathophysiology of pericardial effusion in ADPKD is unknown, it has been theorized to be related to defects in connective tissue and extracellular matrix due to PKD1 and PKD2 mutations. These abnormalities increase the compliance and impair the recoil capacity of connective tissue, which results in unusual distention of the parietal pericardium. This abnormal distention of the parietal pericardium together with increased extracellular volume may lead to pericardial effusion.17
Qian et al17 found a higher prevalence of pericardial effusion in ADPKD patients. It was generally asymptomatic, and the cause was attributed to these connective tissue and extracellular matrix abnormalities.
EMERGING THERAPIES AND TESTS
Recent trials have investigated the effects of vasopressin receptor antagonists, specifically V2 receptor blockers in ADPKD and its complications.
Tolvaptan has been shown to slow the rate of increase in cyst size and total kidney volume.83,84 Also, a correlation between kidney size and diastolic blood pressure has been observed in ADPKD patients.85 Reducing cyst volume may reduce pressure effects and decrease renal ischemia, which in turn may reduce RAAS activation; however, the evidence to support this hypothesis is poor. A major clinical trial of tolvaptan in ADPKD patients showed no effect on blood pressure control, but the drug slowed the rate of increase in total kidney volume and fall in eGFR.83
Endothelin receptor antagonists are also in the preliminary stage of development for use in ADPKD. The effects of acute blockade of the renal endothelial system with bosentan were investigated in animal models by Hocher et al.28 This study showed a greater reduction in mean arterial pressure after bosentan administration, resulting in significantly decreased GFR and renal blood flow. Nonetheless, the mean arterial pressure-lowering effect of bosentan was more marked than the reductions in GFR and renal blood flow.
Raina et al,86 in a pilot cross-sectional analysis, showed that urinary excretion of ET-1 is increased in ADPKD patients, and may serve as a surrogate marker for ET-1 in renal tissue and a noninvasive marker of early kidney injury.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
- Ha SK, Park CH, Kna JS, et al. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Yonsei Med J 1997; 38:111–116.
- Romao EA, Moyses Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Brazilian J Med Biol Res 2006; 39:533–538.
- Chapman AB, Devuyst O, Eckardt KU, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2015; 88:17–27.
- Perrone RD, Ruthazer R, Terrin NC. Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am J Kidney Dis 2001; 38:777–784.
- Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev 2013; 9:2–11.
- Silverio A, Prota C, Di Maio M, et al. Aortic dissection in patients with autosomal dominant polycystic kidney disease: a series of two cases and a review of the literature. Nephrology 2015; 20:229–235.
- Ramineni R, Daniel GK. Use of endovascular stent-graft repair for type B aortic dissection in polycystic kidney disease. J Invas Cardiol 2010; 22:E171–E174.
- Courtois A, Nusgens BV, Delvenne P, et al. Dissection of iliac artery in a patient with autosomal dominant polycystic kidney disease: a case report. Aorta 2013; 1:123–125.
- Bobrie G, Brunet-Bourgin F, Alamowitch S, et al. Spontaneous artery dissection: is it part of the spectrum of autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 1998; 13:2138–2141.
- Minami T, Karube N, Sakamoto A. [Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease; report of a case]. Kyobu Geka 2009; 62:924–927.
- Ohara K, Kimura T, Karasawa T, et al. A large coronary aneurysm and its probable precursor lesions in a patient with autosomal dominant polycystic kidney disease: an implication for the process of aneurysmogenesis. Pathol Int 2012; 62:758–762.
- Al-Hakim W, Goldsmith DJ. Bilateral popliteal aneurysms complicating adult polycystic kidney disease in a patient with a marfanoid habitus. Postgrad Med J 2003; 79:474–475.
- Kanagasundaram NS, Perry EP, Turney JH. Aneurysm of the splenic artery in a patient with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1999; 14:183–184.
- Kang YR, Ahn JH, Kim KH, Choi YM, Choi J, Park JR. Multiple cardiovascular manifestations in a patient with autosomal dominant polycystic kidney disease. J Cardiovasc Ultrasound 2014; 22:144–147.
- Iglesias D, Fraga AR, Arrizurieta E, et al. Atrial myxoma in a woman with autosomal dominant polycystic kidney disease type 2. Am J Kidney Dis 1997; 29:164–165.
- Paavola J, Schliffke S, Rossetti S, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 2013; 58:199–208.
- Qian Q, Hartman RP, King BF, Torres VE. Increased occurrence of pericardial effusion in patients with autosomal dominant polycystic kidney disease.
- Schievink WI, Torres VE, Wiebers DO, Huston J 3rd. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1997; 8:1298–1303.
- Pierre SA, Jaeger MT, Siemens DR. Intra-operative inferior vena cava syndrome in a patient with autosomal dominant polycystic kidney disease. World J Urol 2006; 24:110–112.
- Fick GM, Johnson AM, Hammond WS, Gabow PA. Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1995; 5:2048–2056.
- Helal I, Reed B, Mettler P, et al. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am J Nephrol 2012; 36:362–370.
- Calabrese G, Vagelli G, Cristofano C, Barsotti G. Behaviour of arterial pressure in different stages of polycystic kidney disease. Nephron 1982; 32:207–208.
- de Almeida EA, de Oliveira EI, Lopes JA, Almeida AG, Lopes MG, Prata MM. Ambulatory blood pressure measurement in young normotensive patients with autosomal dominant polycystic kidney disease. Rev Port Cardiol 2007; 26:235–243.
- Gabow PA, Johnson AM, Kaehny WD, et al. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 41:1311–1319.
- Bajrami V, Idrizi A, Roshi E, Barbullushi M. Association between nephrolithiasis, hypertension and obesity in polycystic kidney disease. Open Access Maced J Med Sci 2016; 4:43–46.
- Chapman AB, Johnson A, Gabow PA, Schrier RW. The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 1990; 323:1091–1096.
- Nakamura T, Ebihara I, Fukui M, et al. Increased endothelin and endothelin receptor mRNA expression in polycystic kidneys of cpk mice. J Am Soc Nephrol 1993; 4:1064–1072.
- Hocher B, Zart R, Schwarz A, et al. Renal endothelin system in polycystic kidney disease. J Am Soc Nephrol 1998; 9:1169–1177.
- Ong AC, Newby LJ, Dashwood MR. Expression and cellular localisation of renal endothelin-1 and endothelin receptor subtypes in autosomal-dominant polycystic kidney disease. Nephron Exper Nephrol 2003; 93:e80.
- Nash DA Jr. Hypertension in polycystic kidney disease without renal failure. Arch Intern Med 1977; 137:1571–1575.
- D’Angelo A, Mioni G, Ossi E, Lupo A, Valvo E, Maschio G. Alterations in renal tubular sodium and water transport in polycystic kidney disease. Clin Nephrol 1975; 3:99–105.
- Ettinger A, Kahn PC, Wise HM Jr. The importance of selective renal angiography in the diagnosis of polycystic disease. J Urol 1969; 102:156–161.
- Cornell SH. Angiography in polycystic disease of the kidneys. J Urol 1970; 103:24–26.
- Schrier RW. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2009; 20:1888–1893.
- Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int 2003; 64:1035–1045.
- Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Prospective change in renal volume and function in children with ADPKD. Clin J Am Soc Nephrol 2009; 4:820–829.
- Loghman-Adham M, Soto CE, Inagami T, Cassis L. The intrarenal renin-angiotensin system in autosomal dominant polycystic kidney disease. Am J Physiol Renal Physiol 2004; 287:F775–F788.
- Torres VE, Donovan KA, Scicli G, et al. Synthesis of renin by tubulocystic epithelium in autosomal-dominant polycystic kidney disease. Kidney Int 1992; 42:364–373.
- Graham PC, Lindop GB. The anatomy of the renin-secreting cell in adult polycystic kidney disease. Kidney Int 1988; 33:1084–1090.
- Cerasola G, Vecchi M, Mule G, et al. Sympathetic activity and blood pressure pattern in autosomal dominant polycystic kidney disease hypertensives. Am J Nephrol 1998; 18:391–398.
- Valvo E, Gammaro L, Tessitore N, et al. Hypertension of polycystic kidney disease: mechanisms and hemodynamic alterations. Am J Nephrol 1985; 5:176–181.
- Marlais M, Cuthell O, Langan D, Dudley J, Sinha MD, Winyard PJ. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch Dis Child 2016; 101:1142–1147.
- Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2255–2266.
- Torres VE, Abebe KZ, Chapman AB, et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med 2014; 371:2267–2276.
- James PA, Oparil S, Carter BL, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA 2014; 311:507–520.
- Ritchie LD, Campbell NC, Murchie P. New NICE guidelines for hypertension. BMJ 2011; 343:d5644.
- Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013; 34:2159–2219.
- Rangan GK, Alexander SI, Campbell KL, et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrology 2016; 21:705–716.
- Helal I, Al-Rowaie F, Abderrahim E, Kheder A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J Kidney Dis Transpl 2017; 28:253–260.
- Patch C, Charlton J, Roderick PJ, Gulliford MC. Use of antihypertensive medications and mortality of patients with autosomal dominant polycystic kidney disease: a population-based study. Am J Kidney Dis 2011; 57:856–862.
- Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2008; 23:573–579.
- Clark LA, Whitmire S, Patton S, Clark C, Blanchette CM, Howden R. Cost-effectiveness of angiotensin-converting enzyme inhibitors versus angiotensin II receptor blockers as first-line treatment in autosomal dominant polycystic kidney disease. J Med Econ 2017:1–17.
- Li Kam Wa TC, Macnicol AM, Watson ML. Ambulatory blood pressure in hypertensive patients with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12:2075–2080.
- Valero FA, Martinez-Vea A, Bardaji A, et al. Ambulatory blood pressure and left ventricular mass in normotensive patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1999; 10:1020–1026.
- Faul C, Amaral AP, Oskouei B, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest 2011; 121:4393–4408.
- Yildiz A, Gul CB, Ersoy A, Asiltas B, Ermurat S, Dogan S, et al. Arterial dysfunction in early autosomal dominant polycystic kidney disease independent of fibroblast growth factor 23. Iranian J Kidney Dis 2014; 8:443–449.
- Wanic-Kossowska M, Posnik B, Kobelski M, et al. The polymorphism of the ACE gene affects left ventricular hypertrophy and causes disturbances in left ventricular systolic/diastolic function in patients with autosomal dominant polycystic kidney disease. ScientificWorldJournal 2014; 2014:707658.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Cadnapaphornchai MA. Hypertension in children with autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9:21–26.
- Schievink WI, Prendergast V, Zabramski JM. Rupture of a previously documented small asymptomatic intracranial aneurysm in a patient with autosomal dominant polycystic kidney disease. Case report. J Neurosurg 1998; 89:479–482.
- Graf S, Schischma A, Eberhardt KE, Istel R, Stiasny B, Schulze BD. Intracranial aneurysms and dolichoectasia in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2002; 17:819–823.
- Nakajima F, Shibahara N, Arai M, Gohji K, Ueda H, Katsuoka Y. Intracranial aneurysms and autosomal dominant polycystic kidney disease: followup study by magnetic resonance angiography. J Urol 2000; 164:311–313.
- Wakabayashi T, Fujita S, Ohbora Y, Suyama T, Tamaki N, Matsumoto S. Polycystic kidney disease and intracranial aneurysms. Early angiographic diagnosis and early operation for the unruptured aneurysm. J Neurosurg 1983; 58:488–491.
- Belz MM, Fick-Brosnahan GM, Hughes RL, et al. Recurrence of intracranial aneurysms in autosomal-dominant polycystic kidney disease. Kidney Int 2003; 63:1824–1830.
- Schrier RW, Belz MM, Johnson AM, et al. Repeat imaging for intracranial aneurysms in patients with autosomal dominant polycystic kidney disease with initially negative studies: a prospective ten-year follow-up. J Am Soc Nephrol 2004; 15:1023–1028.
- Cagnazzo F, Gambacciani C, Morganti R, Perrini P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: prevalence, risk of rupture, and management. A systematic review. Acta Neurochirurgica 2017; 5:811–821.
- Lee VW, Dexter MA, Mai J, Vladica P, Lopez-Vargas P, Rangan GK. KHA-CARI autosomal dominant polycystic kidney disease guideline: management of intracranial aneurysms. Semin Nephrol 2015; 35:612–617.
- Rozenfeld MN, Ansari SA, Shaibani A, Russell EJ, Mohan P, Hurley MC. Should patients with autosomal dominant polycystic kidney disease be screened for cerebral aneurysms? AJNR Am J Neuroradiol 2014; 35:3–9.
- Hiratsuka Y, Miki H, Kiriyama I, et al. Diagnosis of unruptured intracranial aneurysms: 3T MR angiography versus 64-channel multi-detector row CT angiography. Magn Reson Med Sci 2008; 7:169–178.
- van Gelder JM. Computed tomographic angiography for detecting cerebral aneurysms: implications of aneurysm size distribution for the sensitivity, specificity, and likelihood ratios. Neurosurgery 2003; 53:597–605.
- Villablanca JP, Jahan R, Hooshi P, et al. Detection and characterization of very small cerebral aneurysms by using 2D and 3D helical CT angiography. AJNR Am J Neuroradiol 2002; 23:1187–1198.
- Lumiaho A, Ikäheimo R, Miettinen R, et al. Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1. Am J Kidney Dis 2001; 38:1208–1216.
- Timio M, Monarca C, Pede S, Gentili S, Verdura C, Lolli S. The spectrum of cardiovascular abnormalities in autosomal dominant polycystic kidney disease: a 10-year follow-up in a five-generation kindred. Clin Nephrol 1992; 37:245–251.
- Hossack KF, Leddy CL, Johnson AM, Schrier RW, Gabow PA. Echocardiographic findings in autosomal dominant polycystic kidney disease. N Engl J Med 1988; 319:907–912.
- Leier CV, Baker PB, Kilman JW, Wooley CF. Cardiovascular abnormalities associated with adult polycystic kidney disease. Ann Intern Med 1984; 100:683–688.
- Turkmen K, Oflaz H, Uslu B, et al. Coronary flow velocity reserve and carotid intima media thickness in patients with autosomal dominant polycystic kidney disease: from impaired tubules to impaired carotid and coronary arteries. Clin J Am Soc Nephrol 2008; 3:986–991.
- Turkmen K, Tufan F, Alpay N, et al. Insulin resistance and coronary flow velocity reserve in patients with autosomal dominant polycystic kidney disease. Intern Med J 2012; 42:146–153.
- Turkmen K, Tufan F, Selcuk E, Akpinar T, Oflaz H, Ecder T. Neutrophil-to-lymphocyte ratio, insulin resistance, and endothelial dysfunction in patients with autosomal dominant polycystic kidney disease. Indian J Nephrol 2013; 23:34–40.
- Hadimeri H, Lamm C, Nyberg G. Coronary aneurysms in patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1998; 9:837–841.
- Earle K, Hoffbrand BI. Adult dominant polycystic kidney disease and atrial myxoma. Nephron 1989; 52:197.
- Chung BM, Chong S, Lee W-S, Hwang S-N. Autosomal dominant polycystic kidney disease combined with intracranial aneurysm and dilated cardiomyopathy: a case report. J Korean Soc Radiol 2014; 71:84–88.
- Mariathasan DAL, Kumanan T. Adult polycystic kidney disease and idiopathic dilated cardiomyopathy: a rare genetic association. J Ceylon Coll Physicians 2016: 46:42–44.
- Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 2012; 367:2407–2018.
- Higashihara E, Torres VE, Chapman AB, et al. Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol 2011; 6:2499–2507.
- Sans Atxer L, Roca-Cusachs A, Torra R, et al. Relationship between renal size and blood pressure profile in patients with autosomal dominant polycystic kidney disease without renal failure. Nefrologia 2010; 30:567–572. In Spanish.
- Raina R, Lou L, Berger B, et al. Relationship of urinary endothelin-1 with estimated glomerular filtration rate in autosomal dominant polycystic kidney disease: a pilot cross-sectional analysis. BMC Nephrol 2016; 17:22.
KEY POINTS
- Hypertension and left ventricular hypertrophy are common complications of ADPKD.
- Cardiovascular disease is a major cause of morbidity and death in ADPKD.
- Early diagnosis and aggressive management of high blood pressure, specifically with agents that block the renin-angiotensin-aldosterone system, are necessary to prevent left ventricular hypertrophy and progression of renal failure in ADPKD.
- Timely screening and intervention for intracranial aneurysm would lessen the rates of morbidity and death from intracranial hemorrhage.