User login
THE CASE
A 37-year-old right-hand dominant woman came to our clinic seeking treatment for bilateral generalized hand cramping and weakness that she had been experiencing for approximately 2 to 3 years. She was dropping objects and had finger locking, yet had no numbness, tingling, or morning stiffness.
Ten months earlier, she had given birth to a healthy 3715 g girl. Our patient’s prenatal glucose tolerance test had been normal. Her pregnancy and delivery had been significant for oligohydramnios, failed post-term (41 weeks 4 days) induction, and emergent low transverse cesarean section due to fetal bradycardia. Since giving birth, our patient had 3 menstrual periods while breastfeeding. She had a copper intrauterine device inserted at her 6-week postpartum visit. She also had 2 truncal acrochordons removed 3 months postpartum. She had no history of neck trauma, overuse injury, or occupational exposures.
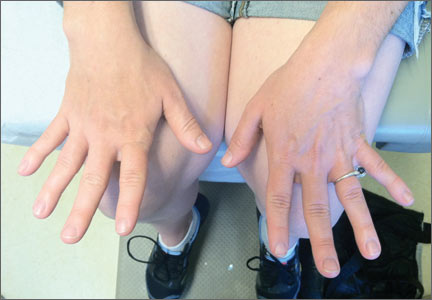
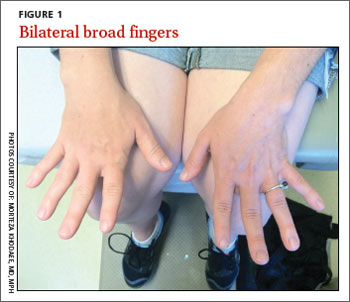
Her blood pressure and vital signs were within normal limits. Physical exam was notable for subtly coarse facial features and broad fingers (FIGURE 1).
She had normal wrist and hand joint range of motion; her wrist and hand strengths, including grip strength, were 5 out of 5. Tinel’s sign, Phalen’s maneuver, and Finkelstein’s test were negative.
Her upper extremity neurovascular exams were completely normal. Initial laboratory studies—including a comprehensive metabolic panel—were normal. The only exception was her creatine kinase, which was 265 U/L (normal, 24-195 U/L).
At a follow-up appointment 7 weeks later, we gathered a more detailed history and learned that over the past 2 to 3 years, the patient had noticed that her shoe and ring sizes had been increasing. She also mentioned some mild weight gain following her pregnancy.
Occasionally, she had generalized hand swelling, headaches, and saw floaters, but she denied losing peripheral vision. Additional lab work at this time revealed a fasting growth hormone (GH) level of 27.3 ng/mL (normal, 0.05-8 ng/mL) and an insulin-like growth factor 1 (IGF-1) level of 848 ng/mL (normal, 106-368 ng/mL). An anterior pituitary hormone panel and cortisol level were normal. A urine pregnancy test was negative.
THE DIAGNOSIS
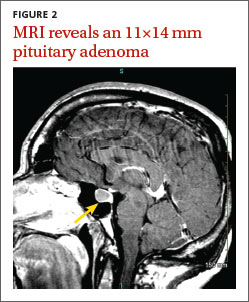
Magnetic resonance imaging (MRI) of our patient’s brain revealed a pituitary adenoma (FIGURE 2). Based on that and the patient’s elevated GH and IGF-1 levels, we diagnosed acromegaly due to a pituitary adenoma.
DISCUSSION
Acromegaly is a rare, progressively disfiguring disease with a prevalence of 40 cases per million people.1 It affects middle-aged adults, with no gender difference.2 In most cases, the cause is a benign pituitary adenoma.1-4
Physical changes include coarse facial features, generalized expansion of the skull, brow protrusion, ocular distension, prognathism, macroglossia, acral overgrowth, and dental malocclusion; these changes typically occur slowly over a long time period.1-5 For example, when we looked at the 3-year-old photo on our patient’s driver’s license, we noticed only subtle changes from her current appearance. Common clinical manifestations include headache, hyperpigmentation, hypertrichosis, hyperhidrosis, goiter, arthropathy, carpal tunnel syndrome, visual disturbances, and acrochordons.1,5
Acromegaly is associated with an increased risk of cardiovascular disease, metabolic disorders, infertility, sleep apnea, arthritis, thyroid tumors, colon adenomas, and carcinoma.1,2,4,5 Due to the insidious progression of acromegaly’s clinical manifestations, diagnosis is delayed for 4 to 10 years, on average.1 The diagnosis of acromegaly is typically based on an elevation of GH and IGF-1 levels.1,5 A brain MRI is essential in the diagnosis of a pituitary adenoma.1
Pregnancy among patients with acromegaly is uncommon. In fact, fewer than 150 cases have been reported in the literature.2,6 In most cases, it appears that pregnancy among patients with acromegaly is safe for mothers and newborns.6,7
The goals of treatment for acromegaly caused by a pituitary adenoma are to remove/ reduce the tumor and its mechanical effects, relieve symptoms, reduce serum GH and IGF-1, and restore pituitary function. Transsphenoidal surgical resection is the preferred treatment for pituitary adenomas.1,2,4 Radiation therapy and pharmacologic treatment may be necessary as adjuncts to surgery or for patients for whom surgery is contraindicated.1,4,5
Pharmacologic management of acromegaly includes dopamine agonists (cabergoline), somatostatin analogues (octreotide, lanreotide), and GH receptor antagonists (pegvisomant).1,3 Patients who receive effective early treatment of acromegaly have a life expectancy similar to that of the general population.1,5
Our patient
Our patient was referred to Neurosurgery and underwent transnasal transsphenoidal resection of the pituitary adenoma. Two weeks postop, her GH level had decreased to 0.66 ng/mL and her IGF-1 level was down to 386 ng/mL. Four months later, her GH (2.32 ng/mL) and IGF-1 levels (277 ng/mL) were within normal range and our patient reported improvement in all of her symptoms.
THE TAKEAWAY
Because it may take years for the classical clinical features of acromegaly such as coarse facial features, protruding jaw, and broad fingers to become apparent, diligent history taking is essential to diagnose the condition early. Patients may present with nonspecific and confusing symptoms such as muscle weakness.8 Early nonspecific symptoms and signs in the presence of normal basic laboratory tests should warrant an evaluation of fasting GH and IGF-1. Early treatment with surgery, radiation therapy, or pharmacotherapy may prevent or decrease the intensity of rheumatologic, cardiovascular, respiratory, and metabolic complications of acromegaly.1
1. Scacchi M, Cavagnini F. Acromegaly. Pituitary. 2006;9: 297-303.
2. Hossain B, Drake WM. Acromegaly. Medicine. 2009;37: 407-410.
3. Chan MR, Ziebert M, Maas DL, et al. “My rings won’t fit anymore”. Ectopic growth hormone-secreting tumor. Am Fam Physician. 2005;71:1766-1767.
4. Lake MG, Krook LS, Cruz SV. Pituitary adenomas: an overview. Am Fam Physician. 2013;88:319-327.
5. Vilar L, Valenzuela A, Ribeiro-Oliveira A Jr, et al. Multiple facets in the control of acromegaly. Pituitary. 2014;17 suppl 1:S11-S17.
6. Cheng V, Faiman C, Kennedy L, et al. Pregnancy and acromegaly: a review. Pituitary. 2012;15:59-63.
7. Caron P, Broussaud S, Bertherat J, et al. Acromegaly and pregnancy: a retrospective multicenter study of 59 pregnancies in 46 women. J Clin Endocrinol Metab. 2010;95:4680-4687.
8. Saguil A. Evaluation of the patient with muscle weakness. Am Fam Physician. 2005;71:1327-1336.
THE CASE
A 37-year-old right-hand dominant woman came to our clinic seeking treatment for bilateral generalized hand cramping and weakness that she had been experiencing for approximately 2 to 3 years. She was dropping objects and had finger locking, yet had no numbness, tingling, or morning stiffness.
Ten months earlier, she had given birth to a healthy 3715 g girl. Our patient’s prenatal glucose tolerance test had been normal. Her pregnancy and delivery had been significant for oligohydramnios, failed post-term (41 weeks 4 days) induction, and emergent low transverse cesarean section due to fetal bradycardia. Since giving birth, our patient had 3 menstrual periods while breastfeeding. She had a copper intrauterine device inserted at her 6-week postpartum visit. She also had 2 truncal acrochordons removed 3 months postpartum. She had no history of neck trauma, overuse injury, or occupational exposures.
Her blood pressure and vital signs were within normal limits. Physical exam was notable for subtly coarse facial features and broad fingers (FIGURE 1).
She had normal wrist and hand joint range of motion; her wrist and hand strengths, including grip strength, were 5 out of 5. Tinel’s sign, Phalen’s maneuver, and Finkelstein’s test were negative.
Her upper extremity neurovascular exams were completely normal. Initial laboratory studies—including a comprehensive metabolic panel—were normal. The only exception was her creatine kinase, which was 265 U/L (normal, 24-195 U/L).
At a follow-up appointment 7 weeks later, we gathered a more detailed history and learned that over the past 2 to 3 years, the patient had noticed that her shoe and ring sizes had been increasing. She also mentioned some mild weight gain following her pregnancy.
Occasionally, she had generalized hand swelling, headaches, and saw floaters, but she denied losing peripheral vision. Additional lab work at this time revealed a fasting growth hormone (GH) level of 27.3 ng/mL (normal, 0.05-8 ng/mL) and an insulin-like growth factor 1 (IGF-1) level of 848 ng/mL (normal, 106-368 ng/mL). An anterior pituitary hormone panel and cortisol level were normal. A urine pregnancy test was negative.
THE DIAGNOSIS
Magnetic resonance imaging (MRI) of our patient’s brain revealed a pituitary adenoma (FIGURE 2). Based on that and the patient’s elevated GH and IGF-1 levels, we diagnosed acromegaly due to a pituitary adenoma.
DISCUSSION
Acromegaly is a rare, progressively disfiguring disease with a prevalence of 40 cases per million people.1 It affects middle-aged adults, with no gender difference.2 In most cases, the cause is a benign pituitary adenoma.1-4
Physical changes include coarse facial features, generalized expansion of the skull, brow protrusion, ocular distension, prognathism, macroglossia, acral overgrowth, and dental malocclusion; these changes typically occur slowly over a long time period.1-5 For example, when we looked at the 3-year-old photo on our patient’s driver’s license, we noticed only subtle changes from her current appearance. Common clinical manifestations include headache, hyperpigmentation, hypertrichosis, hyperhidrosis, goiter, arthropathy, carpal tunnel syndrome, visual disturbances, and acrochordons.1,5
Acromegaly is associated with an increased risk of cardiovascular disease, metabolic disorders, infertility, sleep apnea, arthritis, thyroid tumors, colon adenomas, and carcinoma.1,2,4,5 Due to the insidious progression of acromegaly’s clinical manifestations, diagnosis is delayed for 4 to 10 years, on average.1 The diagnosis of acromegaly is typically based on an elevation of GH and IGF-1 levels.1,5 A brain MRI is essential in the diagnosis of a pituitary adenoma.1
Pregnancy among patients with acromegaly is uncommon. In fact, fewer than 150 cases have been reported in the literature.2,6 In most cases, it appears that pregnancy among patients with acromegaly is safe for mothers and newborns.6,7
The goals of treatment for acromegaly caused by a pituitary adenoma are to remove/ reduce the tumor and its mechanical effects, relieve symptoms, reduce serum GH and IGF-1, and restore pituitary function. Transsphenoidal surgical resection is the preferred treatment for pituitary adenomas.1,2,4 Radiation therapy and pharmacologic treatment may be necessary as adjuncts to surgery or for patients for whom surgery is contraindicated.1,4,5
Pharmacologic management of acromegaly includes dopamine agonists (cabergoline), somatostatin analogues (octreotide, lanreotide), and GH receptor antagonists (pegvisomant).1,3 Patients who receive effective early treatment of acromegaly have a life expectancy similar to that of the general population.1,5
Our patient
Our patient was referred to Neurosurgery and underwent transnasal transsphenoidal resection of the pituitary adenoma. Two weeks postop, her GH level had decreased to 0.66 ng/mL and her IGF-1 level was down to 386 ng/mL. Four months later, her GH (2.32 ng/mL) and IGF-1 levels (277 ng/mL) were within normal range and our patient reported improvement in all of her symptoms.
THE TAKEAWAY
Because it may take years for the classical clinical features of acromegaly such as coarse facial features, protruding jaw, and broad fingers to become apparent, diligent history taking is essential to diagnose the condition early. Patients may present with nonspecific and confusing symptoms such as muscle weakness.8 Early nonspecific symptoms and signs in the presence of normal basic laboratory tests should warrant an evaluation of fasting GH and IGF-1. Early treatment with surgery, radiation therapy, or pharmacotherapy may prevent or decrease the intensity of rheumatologic, cardiovascular, respiratory, and metabolic complications of acromegaly.1
THE CASE
A 37-year-old right-hand dominant woman came to our clinic seeking treatment for bilateral generalized hand cramping and weakness that she had been experiencing for approximately 2 to 3 years. She was dropping objects and had finger locking, yet had no numbness, tingling, or morning stiffness.
Ten months earlier, she had given birth to a healthy 3715 g girl. Our patient’s prenatal glucose tolerance test had been normal. Her pregnancy and delivery had been significant for oligohydramnios, failed post-term (41 weeks 4 days) induction, and emergent low transverse cesarean section due to fetal bradycardia. Since giving birth, our patient had 3 menstrual periods while breastfeeding. She had a copper intrauterine device inserted at her 6-week postpartum visit. She also had 2 truncal acrochordons removed 3 months postpartum. She had no history of neck trauma, overuse injury, or occupational exposures.
Her blood pressure and vital signs were within normal limits. Physical exam was notable for subtly coarse facial features and broad fingers (FIGURE 1).
She had normal wrist and hand joint range of motion; her wrist and hand strengths, including grip strength, were 5 out of 5. Tinel’s sign, Phalen’s maneuver, and Finkelstein’s test were negative.
Her upper extremity neurovascular exams were completely normal. Initial laboratory studies—including a comprehensive metabolic panel—were normal. The only exception was her creatine kinase, which was 265 U/L (normal, 24-195 U/L).
At a follow-up appointment 7 weeks later, we gathered a more detailed history and learned that over the past 2 to 3 years, the patient had noticed that her shoe and ring sizes had been increasing. She also mentioned some mild weight gain following her pregnancy.
Occasionally, she had generalized hand swelling, headaches, and saw floaters, but she denied losing peripheral vision. Additional lab work at this time revealed a fasting growth hormone (GH) level of 27.3 ng/mL (normal, 0.05-8 ng/mL) and an insulin-like growth factor 1 (IGF-1) level of 848 ng/mL (normal, 106-368 ng/mL). An anterior pituitary hormone panel and cortisol level were normal. A urine pregnancy test was negative.
THE DIAGNOSIS
Magnetic resonance imaging (MRI) of our patient’s brain revealed a pituitary adenoma (FIGURE 2). Based on that and the patient’s elevated GH and IGF-1 levels, we diagnosed acromegaly due to a pituitary adenoma.
DISCUSSION
Acromegaly is a rare, progressively disfiguring disease with a prevalence of 40 cases per million people.1 It affects middle-aged adults, with no gender difference.2 In most cases, the cause is a benign pituitary adenoma.1-4
Physical changes include coarse facial features, generalized expansion of the skull, brow protrusion, ocular distension, prognathism, macroglossia, acral overgrowth, and dental malocclusion; these changes typically occur slowly over a long time period.1-5 For example, when we looked at the 3-year-old photo on our patient’s driver’s license, we noticed only subtle changes from her current appearance. Common clinical manifestations include headache, hyperpigmentation, hypertrichosis, hyperhidrosis, goiter, arthropathy, carpal tunnel syndrome, visual disturbances, and acrochordons.1,5
Acromegaly is associated with an increased risk of cardiovascular disease, metabolic disorders, infertility, sleep apnea, arthritis, thyroid tumors, colon adenomas, and carcinoma.1,2,4,5 Due to the insidious progression of acromegaly’s clinical manifestations, diagnosis is delayed for 4 to 10 years, on average.1 The diagnosis of acromegaly is typically based on an elevation of GH and IGF-1 levels.1,5 A brain MRI is essential in the diagnosis of a pituitary adenoma.1
Pregnancy among patients with acromegaly is uncommon. In fact, fewer than 150 cases have been reported in the literature.2,6 In most cases, it appears that pregnancy among patients with acromegaly is safe for mothers and newborns.6,7
The goals of treatment for acromegaly caused by a pituitary adenoma are to remove/ reduce the tumor and its mechanical effects, relieve symptoms, reduce serum GH and IGF-1, and restore pituitary function. Transsphenoidal surgical resection is the preferred treatment for pituitary adenomas.1,2,4 Radiation therapy and pharmacologic treatment may be necessary as adjuncts to surgery or for patients for whom surgery is contraindicated.1,4,5
Pharmacologic management of acromegaly includes dopamine agonists (cabergoline), somatostatin analogues (octreotide, lanreotide), and GH receptor antagonists (pegvisomant).1,3 Patients who receive effective early treatment of acromegaly have a life expectancy similar to that of the general population.1,5
Our patient
Our patient was referred to Neurosurgery and underwent transnasal transsphenoidal resection of the pituitary adenoma. Two weeks postop, her GH level had decreased to 0.66 ng/mL and her IGF-1 level was down to 386 ng/mL. Four months later, her GH (2.32 ng/mL) and IGF-1 levels (277 ng/mL) were within normal range and our patient reported improvement in all of her symptoms.
THE TAKEAWAY
Because it may take years for the classical clinical features of acromegaly such as coarse facial features, protruding jaw, and broad fingers to become apparent, diligent history taking is essential to diagnose the condition early. Patients may present with nonspecific and confusing symptoms such as muscle weakness.8 Early nonspecific symptoms and signs in the presence of normal basic laboratory tests should warrant an evaluation of fasting GH and IGF-1. Early treatment with surgery, radiation therapy, or pharmacotherapy may prevent or decrease the intensity of rheumatologic, cardiovascular, respiratory, and metabolic complications of acromegaly.1
1. Scacchi M, Cavagnini F. Acromegaly. Pituitary. 2006;9: 297-303.
2. Hossain B, Drake WM. Acromegaly. Medicine. 2009;37: 407-410.
3. Chan MR, Ziebert M, Maas DL, et al. “My rings won’t fit anymore”. Ectopic growth hormone-secreting tumor. Am Fam Physician. 2005;71:1766-1767.
4. Lake MG, Krook LS, Cruz SV. Pituitary adenomas: an overview. Am Fam Physician. 2013;88:319-327.
5. Vilar L, Valenzuela A, Ribeiro-Oliveira A Jr, et al. Multiple facets in the control of acromegaly. Pituitary. 2014;17 suppl 1:S11-S17.
6. Cheng V, Faiman C, Kennedy L, et al. Pregnancy and acromegaly: a review. Pituitary. 2012;15:59-63.
7. Caron P, Broussaud S, Bertherat J, et al. Acromegaly and pregnancy: a retrospective multicenter study of 59 pregnancies in 46 women. J Clin Endocrinol Metab. 2010;95:4680-4687.
8. Saguil A. Evaluation of the patient with muscle weakness. Am Fam Physician. 2005;71:1327-1336.
1. Scacchi M, Cavagnini F. Acromegaly. Pituitary. 2006;9: 297-303.
2. Hossain B, Drake WM. Acromegaly. Medicine. 2009;37: 407-410.
3. Chan MR, Ziebert M, Maas DL, et al. “My rings won’t fit anymore”. Ectopic growth hormone-secreting tumor. Am Fam Physician. 2005;71:1766-1767.
4. Lake MG, Krook LS, Cruz SV. Pituitary adenomas: an overview. Am Fam Physician. 2013;88:319-327.
5. Vilar L, Valenzuela A, Ribeiro-Oliveira A Jr, et al. Multiple facets in the control of acromegaly. Pituitary. 2014;17 suppl 1:S11-S17.
6. Cheng V, Faiman C, Kennedy L, et al. Pregnancy and acromegaly: a review. Pituitary. 2012;15:59-63.
7. Caron P, Broussaud S, Bertherat J, et al. Acromegaly and pregnancy: a retrospective multicenter study of 59 pregnancies in 46 women. J Clin Endocrinol Metab. 2010;95:4680-4687.
8. Saguil A. Evaluation of the patient with muscle weakness. Am Fam Physician. 2005;71:1327-1336.