User login
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
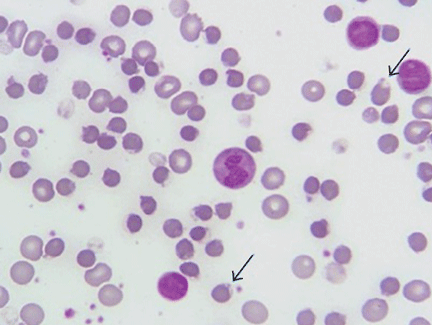
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
A 71-year-old woman came to the hospital because of generalized weakness, fatigue, and exertional dyspnea.
She had a history of anemia, recurrent urinary tract infections, and hyperactive bladder. She had been taking nitrofurantoin for a urinary tract infection and phenazopyridine for dysuria, and she noticed that her urine was dark-colored.
She was of northern European descent. She was unaware of any family history of blood-related disorders. She had been admitted to the hospital 6 weeks earlier for symptomatic anemia after taking nitrofurantoin for a urinary tract infection. At that time, she received 2 units of packed red blood cells and then was discharged. Follow-up blood work done 2 weeks later—including a glucose-6 phosphate dehydrogenase (G6PD) assay—was normal.
On physical examination, she was pale and weak. Her hemoglobin level was 5.5 g/dL (reference range 14.0–17.5), with normal white blood cell and platelet counts and an elevated reticulocyte count. A comprehensive metabolic panel showed elevated indirect bilirubin and lactate dehydrogenase levels. A direct Coombs test for autoimmune hemolytic anemia was negative, as was a haptoglobin assay to look for intravascular hemolytic anemia. G6PD levels were normal, yet a peripheral blood smear (Figure 1) showed features of G6PD deficiency.
What was the cause of her anemia?
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY
G6PD deficiency is an X-linked disorder1 that can present as hemolytic anemia. Symptoms of hemolysis can range from mild to severe on exposure to an inciting agent. Men are more commonly affected than women, and affected women are mostly heterozygous. The severity of hemolysis in heterozygous women depends on inactivation of the unaffected X chromosome in some cells.
When exposed to oxidizing agents, people with G6PD deficiency do not have enough nicotinamide adenine dinucleotide phosphate to protect red blood cells.2 This leads to oxidative denaturation of hemoglobin, formation of methemoglobin, and denaturation of globulin. These products are insoluble; they collect in red blood cells and are called Heinz bodies.3 When red blood cells containing Heinz bodies pass through the liver and spleen, the insoluble masses are taken up by macrophages, causing hemolysis and the formation of “bite cells”4 (so named because macrophages “bite” the Heinz bodies out of the red blood cells).
Patients with G6PD deficiency have all the clinical features of hemolytic anemia. On laboratory testing, the Coombs test is negative, the G6PD level is low, and the peripheral smear shows bite cells. The G6PD level is falsely normal or elevated during acute hemolysis because red blood cells deficient in G6PD are removed from circulation and replaced by young red blood cells. The G6PD level is also elevated after blood transfusion. Thus, the G6PD level should be tested 3 months after an acute event.
Hemolysis in G6PD is usually intermittent and self-limited. No treatment is needed except for avoidance of triggers and transfusion for symptomatic anemia. Of note, triggers include some of the drugs commonly used for urinary tract infections (sulfa drugs, nitrofurantoin, phenazopyridine) and antimalarials. Fava beans are also known to cause hemolytic crisis. A complete list of things to avoid can be found at www.g6pd.org/en/G6PDDeficiency/SafeUnsafe/DaEvitare_ISS-it.
There is no commercially available genetic testing kit for G6PD deficiency. Mutation analysis and G6PD gene sequencing are possible but are neither routinely done nor widely available.
BACK TO OUR PATIENT
Our patient’s hemolytic anemia was most likely drug-induced, secondary to a relative deficiency of G6PD. She had been taking nitrofurantoin and phenazopyridine; both of these are oxidizing agents and are known to cause acute hemolytic anemia in people with G6PD deficiency. The G6PD level can be normal after a recent blood transfusion and, as in our patient, during an acute episode of hemolysis.
Because of the strong suspicion of G6PD deficiency, both drugs were stopped when the patient was discharged from the hospital. She did not take either drug for 3 months. Her G6PD level was then retested and was found to be low, confirming the diagnosis. The patient was then advised not to take those drugs again. Since then, her hemoglobin level has remained stable and she has not needed any more blood transfusions.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.
- Mason PJ, Bautista JM, Gilsanz F. G6PD deficiency: the genotype-phenotype association. Blood Rev 2007; 21:267–283.
- Arese P, De Flora A. Pathophysiology of hemolysis in glucose-6-phosphate dehydrogenase deficiency. Semin Hematol 1990; 27:1–40.
- Jacob HS. Mechanisms of Heinz body formation and attachment to red cell membrane. Semin Hematol 1970; 7:341–354.
- Rifkind RA. Heinz body anemia: an ultrastructural study. II. Red cell sequestration and destruction. Blood 1965; 26:433–448.
