User login
IN THIS ARTICLE
- Presenting stages
- Diagnostic criteria
- Management of IP
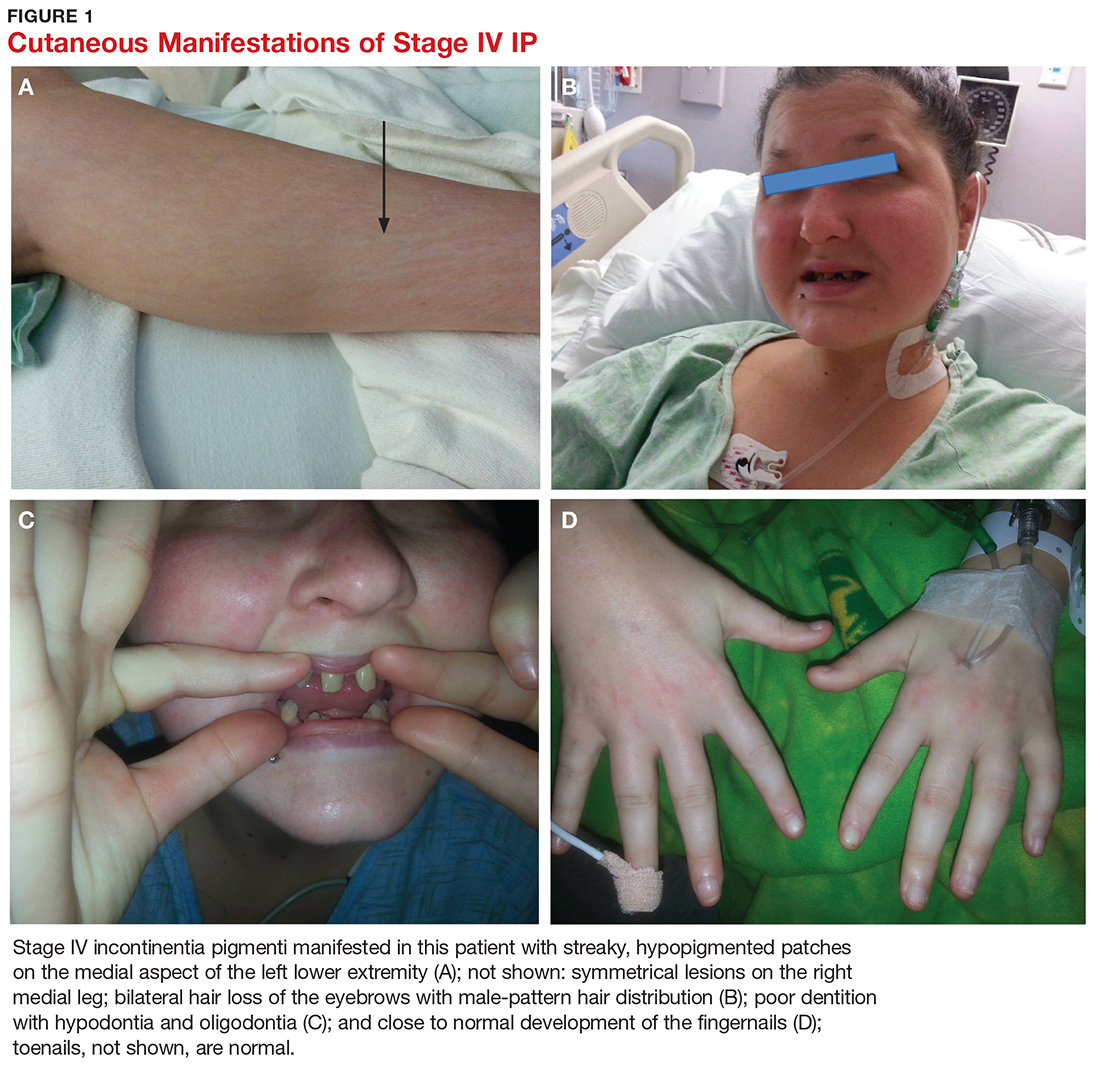
A 21-year-old woman with type 1 diabetes is admitted for recurrent diabetic ketoacidosis. Physical exam reveals hypopigmented, linear, streaky patches on the medial aspects of the bilateral lower legs (Figure 1A). The patient denies tenderness, pruritus, or paresthesia. There is obvious symmetrical hair loss on the lateral aspects of the eyebrows, as well as slightly wooly male-pattern hair distribution with patchy alopecia on the vertex of the head (Figure 1B). She has very poor dentition with hypodontia and malformed teeth (Figure 1C). Her fingernails and toenails appear normal, with no visible atrophy (Figure 1D). What explains her condition?
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare, X-linked dominant genodermatosis involving the cutaneous, ophthalmic, neurologic, and dental systems.1-3 It results from X-inactivation due to mutations in the NF-kappaB essential modulator (NEMO) gene with deletion of exons 4-10 in most cases. The NEMO gene encodes a regulatory component of the IkappaB kinase complex required to activate the NF-kappa B pathway, which is important for many immune, inflammatory, and apoptotic processes.4-6 This deletional mutation is typically lethal in normal 46,XY male karyotypes. Male fetuses with this mutation usually die in utero, making the reported cases predominantly female.4,7
The estimated incidence of IP is between 1/10,000 and 1/100,000.4 Due to the rarity of the condition, IP may be underrecognized and underdiagnosed.
CLINICAL PRESENTATION
Characteristic skin lesions of IP begin to develop at birth or in utero, in an evolving pattern that consists of four stages:
- The vesicular stage (stage I) is characterized by linear erythematous papules and blisters that manifest in newborns.
- The verrucous stage (stage II) begins as the blisters start to heal—usually after several weeks—and is distinguished by hyperkeratotic warty papules in linear or swirling distribution. This stage resolves on its own within months.
- The hyperpigmentation stage (stage III) is when swirling macules or patches develop. This hallmark stage of IP tends to remain static until adolescence.
- The hypopigmentation stage (stage IV) manifests with faded streaky patches, which may be subtly atrophic. This final stage usually develops in the second or third decade of life.2,3
All these cutaneous lesions follow Blaschko lines—invisible lines believed to result from embryonic cell migration that become visible with the manifestation of cutaneous or mucous lesions.6
Other associated cutaneous findings include patchy alopecia, nail dystrophy, and oral/dental anomalies such as hypodontia, oligodontia, and tooth deformities. In addition, ophthalmologic involvement can result in strabismus, cataracts, and retinal vascular changes that can lead to blindness. Central nervous system manifestations include seizures, cognitive impairment, and spastic paralysis.3
DIFFERENTIAL DIAGNOSIS
Because IP is uncommon, it may be easily overlooked or misdiagnosed as another, similar cutaneous manifestation. Cutaneous sarcoidosis, for example, is a skin lesion of noncaseating granuloma. It can present as patches, papules, ulcers, scars, ichthyosis, and alopecia. The development of cutaneous sarcoidosis can be idiopathic or iatrogenic, particularly in patients using anti-TNF therapy. The diagnosis is made clinically and can be confirmed pathologically.8
Stage I IP can also be confused with neonatal herpes simplex virus-1 (HSV-1) infection, given the similarities in vesicular morphology and linear distribution. The diagnosis of HSV-1 can be made based on history, physical exam, and pathology. Given the serious sequelae of neonatal HSV-1 infection, antiviral therapy should not be delayed until confirmation of the diagnosis in infants with vesicular eruptions.9
Erythema multiforme (EM) is another dermatologic condition frequently encountered in children and young adults. Its characteristic round target lesion usually has two rings surrounding the dusky-appearing central zone. Atypical lesions can be bullous or crusty, mimicking the appearance of stage I or II IP. EM is usually a self-limiting condition, but specific treatment may be required if the infectious agent is identified.10
Vitiligo, the development of white patches due to the loss of melanocytes, is another item in the differential. Although it most commonly involves the skin, the hair may also be affected. The diagnosis is made clinically and can be confirmed with skin biopsy if needed.11
DIAGNOSIS
Diagnostic criteria for IP have been proposed, with family history playing a role (see Table).2,3,12 Results of a case-study series indicate that 28% of patients with IP have a family history involving at least one first-degree female relative. IP was considered “sporadic” in 62% of cases studied.3

Without a family history of IP, at least one major criterion must be present to support the diagnosis. These include
- Neonatal rash (erythema, vesicles)
- Linear, atrophic, hairless lesions
- Hyperpigmentation (mainly on trunk, following Blaschko lines)
In a patient with a family history of IP, the presence of any major criterion strongly supports the diagnosis. These, as well as minor criteria, are outlined in the Table.2,3,12
In stages I and II of IP, pathologic features include spongiotic dermatitis with characteristic eosinophils and large dyskeratotic cells.3,13 In stage IV, skin biopsies may reveal slight atrophy and scattered apoptotic cells in the epidermis and epidermal hypopigmentation due to reduced melanocytes. The dermis typically appears thickened and is absent hair follicles and sweat glands.14 In a 2014 update, these pathologic features were proposed to be included in the major diagnostic criteria.12
TREATMENT/MANAGEMENT
Treatment of IP is centered on the involved organ systems. For cutaneous lesions, treatment is not usually necessary unless inflammation persists. In such cases, topical steroids or tacrolimus have been used with some success.15,16 In the vesicular stage, the patient should be monitored for bacterial infection, with appropriate prevention or treatment as necessary.
With other involved systems—such as dental, ophthalmologic, or neurologic (eg, seizures or other encephalopathy) anomalies—consultation and follow-up with the relevant specialist is warranted.
In this case, the patient denied family history of IP. She did have a history of infantile cataract and seizure. Her presenting signs were typical of stage IV IP: hypopigmented streaky patches on the skin of the lower legs, dental abnormalities, somewhat wooly hair, alopecia on the head, and loss of hair on the lateral aspects of the eyebrows. The uniqueness of this case is that the patient also had type 1 diabetes, a condition with a strong genetic predisposition. However, there is no evidence supporting an association between IP and either type of diabetes.
CONCLUSION
Although rare, when IP does occur, its manifestations are vast and severe enough to significantly reduce quality of life for patients; when it occurs in males, it is usually lethal. This genetic disorder can affect multiple body systems, making knowledge of its symptoms essential for proper diagnosis. Because its characteristic stages may be present at birth or in infancy, early identification and diagnosis of IP can help guide treatment intervention.
1. Roberts AP. Incontinentia pigmenti (Bloch-Sulzberger). Br Med. J. 1958;1(5079):1106-1107.
2. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30(1):53-59.
3. Hadj-Rabia S, Froidevaux N, Bodak D, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003; 139(9):1163-1170.
4. Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. Nature. 2000;405(6785):466-472.
5. Aradhya S, Courtois G, Rajkovic A, et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma). Am J Hum Genet. 2001;68(3):765-771.
6. Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89(1):26-36.
7. Kenwrick S, Woffendin H, Jakins T, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter Syndrome. Am J Hum Genet. 2001;69(6):1210-1217.
8. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician. 2002;65(8):1581-1584.
9. Faloyin M, Levitt J, Bercowitz E, et al. All that is vesicular is not herpes: incontinentia pigmenti masquerading as herpes simplex virus in a newborn. Pediatrics. 2004;114(2):e270-272.
10. Siedner-Weintraub Y, Gross I, David A, et al. Paediatric erythema multiforme: epidemiological, clinical and laboratory characteristics. Acta Derm Venereol. 2016 Nov 10. doi: 10.2340/00015555-2569.
11. Gawkrodger DJ, Ormerod AD, Shaw L, et al. Guideline for the diagnosis and management of vitiligo. Br J Dermatol. 2008;159(5):1051-1076.
12. Minic´ S, Trpinac D, Obradovic´ M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536-542.
13. Jean-Baptiste S, O’Toole EA, Chen M, et al. Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of incontinentia pigmenti. Clin Exp Immunol. 2002;127(3):470-478.
14. Hadj-Rabia S, Rimella A, Smahi A, et al. Clinical and histologic features of incontinentia pigmenti in adults with nuclear factor-κ B essential modulator gene mutations. J Am Acad Dermatol. 2011;64(3):508-515.
15. Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti. Clin Exp Dermatol. 2009;34(8):e611-613.
16. Jessup CJ, Morgan SC, Cohen LM, Viders DE. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8(10):944-946.
IN THIS ARTICLE
- Presenting stages
- Diagnostic criteria
- Management of IP
A 21-year-old woman with type 1 diabetes is admitted for recurrent diabetic ketoacidosis. Physical exam reveals hypopigmented, linear, streaky patches on the medial aspects of the bilateral lower legs (Figure 1A). The patient denies tenderness, pruritus, or paresthesia. There is obvious symmetrical hair loss on the lateral aspects of the eyebrows, as well as slightly wooly male-pattern hair distribution with patchy alopecia on the vertex of the head (Figure 1B). She has very poor dentition with hypodontia and malformed teeth (Figure 1C). Her fingernails and toenails appear normal, with no visible atrophy (Figure 1D). What explains her condition?
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare, X-linked dominant genodermatosis involving the cutaneous, ophthalmic, neurologic, and dental systems.1-3 It results from X-inactivation due to mutations in the NF-kappaB essential modulator (NEMO) gene with deletion of exons 4-10 in most cases. The NEMO gene encodes a regulatory component of the IkappaB kinase complex required to activate the NF-kappa B pathway, which is important for many immune, inflammatory, and apoptotic processes.4-6 This deletional mutation is typically lethal in normal 46,XY male karyotypes. Male fetuses with this mutation usually die in utero, making the reported cases predominantly female.4,7
The estimated incidence of IP is between 1/10,000 and 1/100,000.4 Due to the rarity of the condition, IP may be underrecognized and underdiagnosed.
CLINICAL PRESENTATION
Characteristic skin lesions of IP begin to develop at birth or in utero, in an evolving pattern that consists of four stages:
- The vesicular stage (stage I) is characterized by linear erythematous papules and blisters that manifest in newborns.
- The verrucous stage (stage II) begins as the blisters start to heal—usually after several weeks—and is distinguished by hyperkeratotic warty papules in linear or swirling distribution. This stage resolves on its own within months.
- The hyperpigmentation stage (stage III) is when swirling macules or patches develop. This hallmark stage of IP tends to remain static until adolescence.
- The hypopigmentation stage (stage IV) manifests with faded streaky patches, which may be subtly atrophic. This final stage usually develops in the second or third decade of life.2,3
All these cutaneous lesions follow Blaschko lines—invisible lines believed to result from embryonic cell migration that become visible with the manifestation of cutaneous or mucous lesions.6
Other associated cutaneous findings include patchy alopecia, nail dystrophy, and oral/dental anomalies such as hypodontia, oligodontia, and tooth deformities. In addition, ophthalmologic involvement can result in strabismus, cataracts, and retinal vascular changes that can lead to blindness. Central nervous system manifestations include seizures, cognitive impairment, and spastic paralysis.3
DIFFERENTIAL DIAGNOSIS
Because IP is uncommon, it may be easily overlooked or misdiagnosed as another, similar cutaneous manifestation. Cutaneous sarcoidosis, for example, is a skin lesion of noncaseating granuloma. It can present as patches, papules, ulcers, scars, ichthyosis, and alopecia. The development of cutaneous sarcoidosis can be idiopathic or iatrogenic, particularly in patients using anti-TNF therapy. The diagnosis is made clinically and can be confirmed pathologically.8
Stage I IP can also be confused with neonatal herpes simplex virus-1 (HSV-1) infection, given the similarities in vesicular morphology and linear distribution. The diagnosis of HSV-1 can be made based on history, physical exam, and pathology. Given the serious sequelae of neonatal HSV-1 infection, antiviral therapy should not be delayed until confirmation of the diagnosis in infants with vesicular eruptions.9
Erythema multiforme (EM) is another dermatologic condition frequently encountered in children and young adults. Its characteristic round target lesion usually has two rings surrounding the dusky-appearing central zone. Atypical lesions can be bullous or crusty, mimicking the appearance of stage I or II IP. EM is usually a self-limiting condition, but specific treatment may be required if the infectious agent is identified.10
Vitiligo, the development of white patches due to the loss of melanocytes, is another item in the differential. Although it most commonly involves the skin, the hair may also be affected. The diagnosis is made clinically and can be confirmed with skin biopsy if needed.11
DIAGNOSIS
Diagnostic criteria for IP have been proposed, with family history playing a role (see Table).2,3,12 Results of a case-study series indicate that 28% of patients with IP have a family history involving at least one first-degree female relative. IP was considered “sporadic” in 62% of cases studied.3
Without a family history of IP, at least one major criterion must be present to support the diagnosis. These include
- Neonatal rash (erythema, vesicles)
- Linear, atrophic, hairless lesions
- Hyperpigmentation (mainly on trunk, following Blaschko lines)
In a patient with a family history of IP, the presence of any major criterion strongly supports the diagnosis. These, as well as minor criteria, are outlined in the Table.2,3,12
In stages I and II of IP, pathologic features include spongiotic dermatitis with characteristic eosinophils and large dyskeratotic cells.3,13 In stage IV, skin biopsies may reveal slight atrophy and scattered apoptotic cells in the epidermis and epidermal hypopigmentation due to reduced melanocytes. The dermis typically appears thickened and is absent hair follicles and sweat glands.14 In a 2014 update, these pathologic features were proposed to be included in the major diagnostic criteria.12
TREATMENT/MANAGEMENT
Treatment of IP is centered on the involved organ systems. For cutaneous lesions, treatment is not usually necessary unless inflammation persists. In such cases, topical steroids or tacrolimus have been used with some success.15,16 In the vesicular stage, the patient should be monitored for bacterial infection, with appropriate prevention or treatment as necessary.
With other involved systems—such as dental, ophthalmologic, or neurologic (eg, seizures or other encephalopathy) anomalies—consultation and follow-up with the relevant specialist is warranted.
In this case, the patient denied family history of IP. She did have a history of infantile cataract and seizure. Her presenting signs were typical of stage IV IP: hypopigmented streaky patches on the skin of the lower legs, dental abnormalities, somewhat wooly hair, alopecia on the head, and loss of hair on the lateral aspects of the eyebrows. The uniqueness of this case is that the patient also had type 1 diabetes, a condition with a strong genetic predisposition. However, there is no evidence supporting an association between IP and either type of diabetes.
CONCLUSION
Although rare, when IP does occur, its manifestations are vast and severe enough to significantly reduce quality of life for patients; when it occurs in males, it is usually lethal. This genetic disorder can affect multiple body systems, making knowledge of its symptoms essential for proper diagnosis. Because its characteristic stages may be present at birth or in infancy, early identification and diagnosis of IP can help guide treatment intervention.
IN THIS ARTICLE
- Presenting stages
- Diagnostic criteria
- Management of IP
A 21-year-old woman with type 1 diabetes is admitted for recurrent diabetic ketoacidosis. Physical exam reveals hypopigmented, linear, streaky patches on the medial aspects of the bilateral lower legs (Figure 1A). The patient denies tenderness, pruritus, or paresthesia. There is obvious symmetrical hair loss on the lateral aspects of the eyebrows, as well as slightly wooly male-pattern hair distribution with patchy alopecia on the vertex of the head (Figure 1B). She has very poor dentition with hypodontia and malformed teeth (Figure 1C). Her fingernails and toenails appear normal, with no visible atrophy (Figure 1D). What explains her condition?
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare, X-linked dominant genodermatosis involving the cutaneous, ophthalmic, neurologic, and dental systems.1-3 It results from X-inactivation due to mutations in the NF-kappaB essential modulator (NEMO) gene with deletion of exons 4-10 in most cases. The NEMO gene encodes a regulatory component of the IkappaB kinase complex required to activate the NF-kappa B pathway, which is important for many immune, inflammatory, and apoptotic processes.4-6 This deletional mutation is typically lethal in normal 46,XY male karyotypes. Male fetuses with this mutation usually die in utero, making the reported cases predominantly female.4,7
The estimated incidence of IP is between 1/10,000 and 1/100,000.4 Due to the rarity of the condition, IP may be underrecognized and underdiagnosed.
CLINICAL PRESENTATION
Characteristic skin lesions of IP begin to develop at birth or in utero, in an evolving pattern that consists of four stages:
- The vesicular stage (stage I) is characterized by linear erythematous papules and blisters that manifest in newborns.
- The verrucous stage (stage II) begins as the blisters start to heal—usually after several weeks—and is distinguished by hyperkeratotic warty papules in linear or swirling distribution. This stage resolves on its own within months.
- The hyperpigmentation stage (stage III) is when swirling macules or patches develop. This hallmark stage of IP tends to remain static until adolescence.
- The hypopigmentation stage (stage IV) manifests with faded streaky patches, which may be subtly atrophic. This final stage usually develops in the second or third decade of life.2,3
All these cutaneous lesions follow Blaschko lines—invisible lines believed to result from embryonic cell migration that become visible with the manifestation of cutaneous or mucous lesions.6
Other associated cutaneous findings include patchy alopecia, nail dystrophy, and oral/dental anomalies such as hypodontia, oligodontia, and tooth deformities. In addition, ophthalmologic involvement can result in strabismus, cataracts, and retinal vascular changes that can lead to blindness. Central nervous system manifestations include seizures, cognitive impairment, and spastic paralysis.3
DIFFERENTIAL DIAGNOSIS
Because IP is uncommon, it may be easily overlooked or misdiagnosed as another, similar cutaneous manifestation. Cutaneous sarcoidosis, for example, is a skin lesion of noncaseating granuloma. It can present as patches, papules, ulcers, scars, ichthyosis, and alopecia. The development of cutaneous sarcoidosis can be idiopathic or iatrogenic, particularly in patients using anti-TNF therapy. The diagnosis is made clinically and can be confirmed pathologically.8
Stage I IP can also be confused with neonatal herpes simplex virus-1 (HSV-1) infection, given the similarities in vesicular morphology and linear distribution. The diagnosis of HSV-1 can be made based on history, physical exam, and pathology. Given the serious sequelae of neonatal HSV-1 infection, antiviral therapy should not be delayed until confirmation of the diagnosis in infants with vesicular eruptions.9
Erythema multiforme (EM) is another dermatologic condition frequently encountered in children and young adults. Its characteristic round target lesion usually has two rings surrounding the dusky-appearing central zone. Atypical lesions can be bullous or crusty, mimicking the appearance of stage I or II IP. EM is usually a self-limiting condition, but specific treatment may be required if the infectious agent is identified.10
Vitiligo, the development of white patches due to the loss of melanocytes, is another item in the differential. Although it most commonly involves the skin, the hair may also be affected. The diagnosis is made clinically and can be confirmed with skin biopsy if needed.11
DIAGNOSIS
Diagnostic criteria for IP have been proposed, with family history playing a role (see Table).2,3,12 Results of a case-study series indicate that 28% of patients with IP have a family history involving at least one first-degree female relative. IP was considered “sporadic” in 62% of cases studied.3
Without a family history of IP, at least one major criterion must be present to support the diagnosis. These include
- Neonatal rash (erythema, vesicles)
- Linear, atrophic, hairless lesions
- Hyperpigmentation (mainly on trunk, following Blaschko lines)
In a patient with a family history of IP, the presence of any major criterion strongly supports the diagnosis. These, as well as minor criteria, are outlined in the Table.2,3,12
In stages I and II of IP, pathologic features include spongiotic dermatitis with characteristic eosinophils and large dyskeratotic cells.3,13 In stage IV, skin biopsies may reveal slight atrophy and scattered apoptotic cells in the epidermis and epidermal hypopigmentation due to reduced melanocytes. The dermis typically appears thickened and is absent hair follicles and sweat glands.14 In a 2014 update, these pathologic features were proposed to be included in the major diagnostic criteria.12
TREATMENT/MANAGEMENT
Treatment of IP is centered on the involved organ systems. For cutaneous lesions, treatment is not usually necessary unless inflammation persists. In such cases, topical steroids or tacrolimus have been used with some success.15,16 In the vesicular stage, the patient should be monitored for bacterial infection, with appropriate prevention or treatment as necessary.
With other involved systems—such as dental, ophthalmologic, or neurologic (eg, seizures or other encephalopathy) anomalies—consultation and follow-up with the relevant specialist is warranted.
In this case, the patient denied family history of IP. She did have a history of infantile cataract and seizure. Her presenting signs were typical of stage IV IP: hypopigmented streaky patches on the skin of the lower legs, dental abnormalities, somewhat wooly hair, alopecia on the head, and loss of hair on the lateral aspects of the eyebrows. The uniqueness of this case is that the patient also had type 1 diabetes, a condition with a strong genetic predisposition. However, there is no evidence supporting an association between IP and either type of diabetes.
CONCLUSION
Although rare, when IP does occur, its manifestations are vast and severe enough to significantly reduce quality of life for patients; when it occurs in males, it is usually lethal. This genetic disorder can affect multiple body systems, making knowledge of its symptoms essential for proper diagnosis. Because its characteristic stages may be present at birth or in infancy, early identification and diagnosis of IP can help guide treatment intervention.
1. Roberts AP. Incontinentia pigmenti (Bloch-Sulzberger). Br Med. J. 1958;1(5079):1106-1107.
2. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30(1):53-59.
3. Hadj-Rabia S, Froidevaux N, Bodak D, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003; 139(9):1163-1170.
4. Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. Nature. 2000;405(6785):466-472.
5. Aradhya S, Courtois G, Rajkovic A, et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma). Am J Hum Genet. 2001;68(3):765-771.
6. Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89(1):26-36.
7. Kenwrick S, Woffendin H, Jakins T, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter Syndrome. Am J Hum Genet. 2001;69(6):1210-1217.
8. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician. 2002;65(8):1581-1584.
9. Faloyin M, Levitt J, Bercowitz E, et al. All that is vesicular is not herpes: incontinentia pigmenti masquerading as herpes simplex virus in a newborn. Pediatrics. 2004;114(2):e270-272.
10. Siedner-Weintraub Y, Gross I, David A, et al. Paediatric erythema multiforme: epidemiological, clinical and laboratory characteristics. Acta Derm Venereol. 2016 Nov 10. doi: 10.2340/00015555-2569.
11. Gawkrodger DJ, Ormerod AD, Shaw L, et al. Guideline for the diagnosis and management of vitiligo. Br J Dermatol. 2008;159(5):1051-1076.
12. Minic´ S, Trpinac D, Obradovic´ M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536-542.
13. Jean-Baptiste S, O’Toole EA, Chen M, et al. Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of incontinentia pigmenti. Clin Exp Immunol. 2002;127(3):470-478.
14. Hadj-Rabia S, Rimella A, Smahi A, et al. Clinical and histologic features of incontinentia pigmenti in adults with nuclear factor-κ B essential modulator gene mutations. J Am Acad Dermatol. 2011;64(3):508-515.
15. Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti. Clin Exp Dermatol. 2009;34(8):e611-613.
16. Jessup CJ, Morgan SC, Cohen LM, Viders DE. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8(10):944-946.
1. Roberts AP. Incontinentia pigmenti (Bloch-Sulzberger). Br Med. J. 1958;1(5079):1106-1107.
2. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30(1):53-59.
3. Hadj-Rabia S, Froidevaux N, Bodak D, et al. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003; 139(9):1163-1170.
4. Smahi A, Courtois G, Vabres P, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. Nature. 2000;405(6785):466-472.
5. Aradhya S, Courtois G, Rajkovic A, et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKK-gamma). Am J Hum Genet. 2001;68(3):765-771.
6. Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89(1):26-36.
7. Kenwrick S, Woffendin H, Jakins T, et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter Syndrome. Am J Hum Genet. 2001;69(6):1210-1217.
8. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician. 2002;65(8):1581-1584.
9. Faloyin M, Levitt J, Bercowitz E, et al. All that is vesicular is not herpes: incontinentia pigmenti masquerading as herpes simplex virus in a newborn. Pediatrics. 2004;114(2):e270-272.
10. Siedner-Weintraub Y, Gross I, David A, et al. Paediatric erythema multiforme: epidemiological, clinical and laboratory characteristics. Acta Derm Venereol. 2016 Nov 10. doi: 10.2340/00015555-2569.
11. Gawkrodger DJ, Ormerod AD, Shaw L, et al. Guideline for the diagnosis and management of vitiligo. Br J Dermatol. 2008;159(5):1051-1076.
12. Minic´ S, Trpinac D, Obradovic´ M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536-542.
13. Jean-Baptiste S, O’Toole EA, Chen M, et al. Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of incontinentia pigmenti. Clin Exp Immunol. 2002;127(3):470-478.
14. Hadj-Rabia S, Rimella A, Smahi A, et al. Clinical and histologic features of incontinentia pigmenti in adults with nuclear factor-κ B essential modulator gene mutations. J Am Acad Dermatol. 2011;64(3):508-515.
15. Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti. Clin Exp Dermatol. 2009;34(8):e611-613.
16. Jessup CJ, Morgan SC, Cohen LM, Viders DE. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8(10):944-946.