Two days ago, a 27-year-old woman noticed that her vision was blurry in her right eye. She has come to see her primary care physician for advice. This is the first time this has happened to her. She describes seeing a grayish blur over the center of her vision, but she has not noted any other symptoms except for some soreness around the right eye, which is worse with eye movements.
How should she be assessed and treated?
IMPORTANT TO RECOGNIZE
Sudden vision loss is one of the more common problems encountered in ophthalmology and neurology.
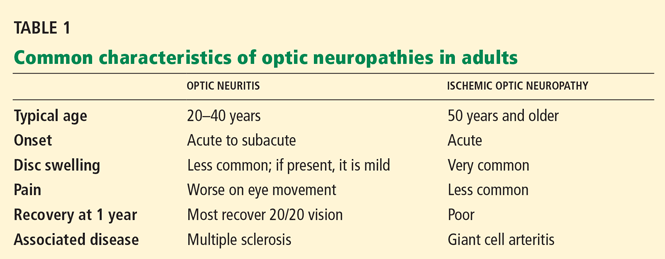
Optic neuritis, a demyelinating inflammatory condition that causes acute vision loss, is associated with multiple sclerosis (MS), and recognizing its classic clinical manifestations early is important so that appropriate diagnostic testing (magnetic resonance imaging [MRI]) and treatment (corticosteroids and immunomodulators) can be started.
Although a comprehensive review of all the optic neuropathies is beyond the scope of this paper, in the pages that follow we review some of the most common causes, which may be first seen by the general internist.
FOUR SUBTYPES OF OPTIC NEURITIS
There are four subtypes of optic neuritis:
Figure 1. In retrobulbar optic neuritis, the inflammation and demyelination occur behind the globe of the eye. The optic disc appears normal with no signs of swelling or pallor.Retrobulbar neuritis (Figure 1), or inflammation of the optic nerve behind the eye, is the form most commonly associated with MS.
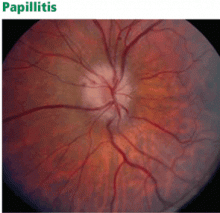
Papillitis (Figure 2), or inflammation of the optic disc, can also be associated with MS.
Perineuritis is inflammation of the optic nerve sheath, sparing the optic nerve itself. Usually, patients are older, and vision loss is mild to moderate. Perineuritis is commonly due to infectious or inflammatory conditions, eg, syphilis or sarcoidosis.
Figure 2. In papillitis, mild swelling and elevation of the optic disc can be seen. The small splinter hemorrhage seen at 10 o’clock is not typical of optic neuritis associated with multiple sclerosis.Neuroretinitis may occur at any age. There is concomitant swelling of the optic nerve and macula. Exudates that form around the macula give the appearance of a star.
Perineuritis and neuroretinitis are not associated with MS, and if they are found they suggest another etiology. In the rest of this review, “optic neuritis” means retrobulbar optic neuritis, the form most commonly seen in clinical practice.
MOST COMMON IN YOUNG WOMEN
Acute demyelinating optic neuritis most often affects women in their 20s and 30s.1–3 Studies in the United States have estimated its annual incidence to be 5.1 to 6.4 per 100,000.4,5 The incidence is higher in populations at higher latitudes and lower near the equator. It is less common in blacks than in whites.6
In children, optic neuritis is not as strongly associated with MS, especially when there is optic disc swelling or bilateral involvement. Most children have a good visual outcome, although approximately 20% may be visually disabled.7–9
FEATURES: VISION LOSS AND EYE PAIN
Most of our current knowledge of the clinical features of optic neuritis comes from the Optic Neuritis Treatment Trial (ONTT),10 conducted in the 1990s. This trial enrolled 457 patients 18 to 46 years old who had acute unilateral optic neuritis. The patients had to have symptoms consistent with acute unilateral optic neuritis for 8 days or less. They could not have evidence of any systemic disease (except for MS) or have received prior treatment for MS. Therefore, this study was quite representative of the patients with optic neuritis that one might encounter in the clinic and is highly important in both characterizing optic neuritis and defining its treatment.
The study found that the two most common symptoms are vision loss and eye pain.
Vision loss in optic neuritis typically occurs over several hours to days, and vision reaches a nadir within 1 to 2 weeks. Typically, patients begin to recover 2 to 4 weeks after the onset of the vision loss. The optic nerve may take up to 6 to 12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.11 More than two-thirds of patients have at least 20/20 vision once they have fully recovered from the optic neuritis. Only 3% of patients become completely blind.
Eye pain is very common in optic neuritis (it affected 87% of patients in the ONTT) and typically worsens with eye movement. The eye is also sore to touch. The pain generally begins at the same time as the visual loss and improves along with it. Eye movements also may bring about photopsia (flickering or flashes of light), a symptom reported by 30% of the ONTT participants.
Loss of color vision out of proportion to the loss of visual acuity is characteristic of optic neuropathies. In the ONTT, 88% of the involved eyes had abnormal color vision as assessed by the Ishihara test (using pseudoisochromatic plates), and 94% as assessed by the Farnsworth-Munsell 100 hue test, which is more sensitive but cumbersome. The most common patterns of color vision loss in optic nerve disease are loss of red (protanopia) and green (deutranopia).
A good way to screen for loss of color vision is to test for color desaturation. First, ask the patient to fixate with the normal eye on a bright red object (for example, the cap from a bottle of ophthalmic mydriatic drops or a pen cap). Then ask the patient to compare the intensity of the redness between the good eye and the affected eye. The patient can quantify the color desaturation by rating what percentage of red is lost in the affected eye compared with the normal eye.
Temporary exacerbations of visual problems during fever (the Uhthoff phenomenon) can occur in patients who have had optic neuritis. These transient pseudoexacerbations are not new episodes of optic neuritis and should resolve after the body temperature returns to normal.
A relative afferent pupillary defect should be seen in the involved eye in all patients with optic neuritis if the other eye is uninvolved and healthy.12 The best way to elicit this sign is to perform the swinging light test in a dark room with the patient fixating at a distant target, which prevents miosis due to accommodation. When the light is swung to the involved eye, the pupil dilates because less light stimulus reaches the midbrain through the affected optic nerve. As the optic nerve heals and recovers, this sign may become subtle, but it persists in more than 90% of cases.12
Findings on funduscopy
Examination of the fundus is helpful in the clinical diagnosis of optic neuritis.
Two-thirds of the ONTT patients had retrobulbar neuritis, and one-third had papillitis. If optic nerve swelling is present, it is typically mild.
Peripapillary hemorrhages were exceedingly rare in the cases of papillitis (only 6%) and were associated with a very low to zero risk of developing MS. If peripapillary hemorrhages are found on examination, one should consider another diagnosis, such as anterior ischemic optic neuropathy.11
CASE CONTINUED
Our patient undergoes a neurologic examination, which reveals an afferent pupillary defect in the right eye and visual acuity of 20/100 in the right eye and 20/20 in the left. Visual fields are normal in the left eye as assessed by confrontation, but there is a central scotoma in the right.
OTHER TYPES OF NEUROPATHY
Optic neuritis is a clinical diagnosis based on the history and findings on examination. If the patient does not have its typical features (Table 1), then other diagnoses should be pursued with serologic and cerebrospinal fluid studies.
The following should be included in the differential diagnosis of optic neuritis:
Ischemic optic neuropathy
Ischemic optic neuropathy is more common in patients age 50 and older, whereas optic neuritis is more common in younger patients. Most patients with ischemic optic neuropathy have hypertension, hypercholesterolemia, diabetes mellitus, obstructive sleep apnea, or other vascular risk factors. The disease has several important subtypes, as discussed below.
Figure 3. Left, fundus photo several weeks after an attack of nonarteritic anterior ischemic optic neuropathy demonstrating pallor of the superior half of the disc. Middle, an associated inferior altitudinal defect. Right, sectoral swelling of the disc with flame or splinter hemorrhages is commonly seen. This is not typical of demyelinating optic neuritis.Nonarteritic anterior ischemic optic neuropathy is the most common form. Typically, there is acute onset of painless vision loss in one eye and an associated altitudinal field defect (Figure 3). For example, if the superior rim of the optic nerve acutely develops swelling and then becomes pale, a corresponding altitudinal cut would develop in the inferior visual field, respecting the horizontal meridian. Many patients first notice the vision loss upon waking up in the morning.13
Although patients with nonarteritic anterior ischemic optic neuropathy typically have vasculopathic risk factors such as hypertension, diabetes mellitus, peripheral vascular disease, or hypercholesterolemia, there is no proven causation between the two. The age of these patients ranges from 50 to 70, with an average age of 66.
The disc appears swollen and may have flame or splinter hemorrhages (Figure 3). The cup of the involved disc is typically small. The visual loss is believed to be the result of poor perfusion in the circulation of the posterior ciliary artery, which supplies the optic nerve head.1 If the other eye also has a small cup, it is considered to be at risk of future ischemic events. In one study,14 the opposite eye became involved within the next 5 years in 14.7% of all cases. The risk of recurrent disease in the same eye is low (6.4% in another study15).
Arteritic anterior ischemic optic neuropathy is more common in patients over age 70 and is usually due to giant cell arteritis, which has a significant association with polymyalgia rheumatica. Patients may have jaw claudication, proximal myalgia and arthralgia, scalp tenderness, headache, fatigue, and a significantly elevated erythrocyte sedimentation rate and C-reactive protein level. These features should be looked for in the review of systems, although patients may not have all of them.
The funduscopic examination may reveal a pale, swollen disc, peripapillary hemorrhages, branch or central retinal artery occlusions, or cotton-wool spots.
Temporal artery biopsy is the gold standard for diagnosis, but treatment with corticosteroids should not be delayed pending biopsy or other test results.1
Thrombocytosis has been associated with a higher risk of permanent vision loss in patients with giant cell arteritis.16
Posterior ischemic optic neuropathy is the least common form of ischemic optic neuropathy. This diagnosis should be entertained in older patients who report acute, painless vision loss but have a normal funduscopic examination. Giant cell arteritis must be considered first in this setting.
Bilateral posterior ischemic optic neuropathy can occur (although rarely) in patients undergoing cardiac or spinal surgery.17 Risk factors thought to be associated with perioperative disease include anemia, hypotension, substantial blood loss during the surgery, surgeries longer than 6.5 hours, carotid atherosclerosis, and diabetes.18
There are no effective treatments for most ischemic optic neuropathies with the crucial exception of giant cell arteritis.
Neuromyelitis optica (Devic disease)
Neuromyelitis optica (Devic disease) is a combination of optic neuritis and transverse myelitis (Table 2). Clinically, the disease spares the nervous system except for the optic nerves and spinal cord. The onset of the optic neuritis may precede or follow the onset of the transverse myelitis by up to 2 to 4 years.19 Usually, the optic neuritis is bilateral and retrobulbar and results in severe vision loss, worse than that seen in patients with MS.19,20
The transverse myelitis may cause paraplegia or quadriplegia, depending on the location of the lesion in the spinal cord (cervical vs thoracic). The transverse myelitis in neuromyelitis optica is distinct from that seen in MS. In neuromyelitis optica, the transverse myelitis is longitudinally extensive, spanning more than three vertebral bodies in length. In MS, spinal cord lesions usually are more discrete and involve one or two spinal cord segments.21
Recently, serum neuromyelitis optica immunoglobulin G (IgG) antibody has been shown to be a significant biomarker of this disease. Its sensitivity ranges from approximately 60% to 70% and its specificity is greater than 90%.22 This antibody binds to aquaporin-4, an important water-channel protein in the blood-brain barrier of the central nervous system, and evidence suggests that it is involved in the pathogenesis of the disease.23
Initially, it was proposed that MRI of the brain had to be normal for neuromyelitis optica to be diagnosed.21 However, the proposed 2006 criteria allow for some abnormal T2 and fluid-attenuated inversion recovery (FLAIR) hyperintensities in the periaqueductal gray matter and diencephalon.22
The spinal fluid in neuromyelitis optica may show a pleocytosis larger than that seen in MS (> 50 white blood cells per mm3) and may have a significant neutrophilic component.21 Oligoclonal bands are not typically present.
It is still debated whether neuromyelitis optica is a separate disease from MS or a subset of it. The implications of this debate may affect its management, as discussed below.
Inflammatory optic neuropathies
Inflammatory optic neuropathies can be caused by infections (eg, syphilis, cat scratch disease) or by noninfectious conditions (eg, sarcoidosis). Lyme disease is rarely a cause of retrobulbar optic neuritis but may cause papillitis.24 West Nile virus has also been reported to cause optic neuritis.25 Lupus may cause an optic neuropathy by inflammatory or ischemic mechanisms.26
Compressive optic neuropathies
Compressive optic neuropathies may be due to mass lesions, tumors, thyroid eye disease, or other orbital processes. MRI of the brain and orbits will confirm or rule out diagnoses associated with compressive optic neuropathy.
Genetic causes
Genetic causes of optic neuropathy include the Leber and Kjer hereditary optic neuropathies.
Leber optic neuropathy involves subacute and painless vision loss, with sequential involvement of both eyes over a period of weeks to months. This disorder predominantly affects men (80%–90% of patients) and is inherited from maternal mitochondrial DNA. The three most common mutations implicated in Leber optic neuropathy (located at base pairs 11,778, 3,460, and 14,484 in the mitochondrial DNA) are involved in more than 90% of cases. The prognosis for recovery varies depending on the genotype.27 These genes encode proteins that are part of complex I of the mitochondrial respiratory chain.28 Funduscopic examination most commonly shows circumpapillary telangiectasia, although up to one-third of patients can have a normal-appearing disc initially. Central vision is affected more severely than peripheral vision.29
Kjer autosomal-dominant optic atrophy is the most common hereditary optic neuropathy. This disease primarily affects children in the first decade of life with slowly progressive loss of vision. As with other optic neuropathies, there will eventually be pallor of the optic disc, a cecocentral scotoma, and loss of color perception. The OPA1 gene located on chromosome 3q28 has been implicated in most patients with dominant optic atrophy; a test is commercially available for diagnosis.30,31
Toxic and metabolic causes
Many agents can cause optic neuropathy. Toxins strongly associated with optic neuropathy include carbon monoxide, methanol, ethylene glycol, perchloroethylene, and tobacco. Drugs linked to optic neuropathy are ethambutol (Myambutol), clioquinol (Vioform), isoniazid (Nydrazid), amiodarone (Cordarone), linezolid (Zyvox), methotrexate, sildenafil (Viagra), oxymetazoline (contained in Afrin and other nasal sprays), and infliximab (Remicade).32–37 Additionally, several chemotherapeutic agents are known to cause optic atrophy, including vincristine (Oncovin), cisplatin (Platinol), carboplatin (Paraplatin), and paclitaxel (Abraxane, Onxol).
Nutritional deficiencies are presumed to have played a significant role in the endemics of optic neuropathy that have occurred in poor countries, such as in Cuba during the 1990s.38 Most nutritional optic neuropathies appear to be exacerbated by tobacco.39
MRI ASSESSES RISK OF MS
The diagnosis of optic neuritis is clinical, based on the history and physical findings.
However, MRI of the brain and orbits with gadolinium contrast has become the cornerstone of the evaluation in patients with optic neuritis. And MRI not only helps confirm the clinical diagnosis, but it also more importantly offers very strong prognostic information about the risk of future demyelinating events and MS.
Gadolinium-enhanced fat-saturated T1-weighted MRI of the orbits is the best sequence to show the inflammation of the optic nerve in optic neuritis (fat saturation is necessary to hide the bright signal of the orbital fat tissue).
Contrast-enhanced MRI can also help differentiate optic neuritis from nonarteritic anterior ischemic optic neuropathy. MRI of the orbits with gadolinium contrast shows enhancement of the affected optic nerve in approximately 95% of cases of optic neuritis, whereas optic nerve enhancement rarely occurs in nonarteritic anterior ischemic optic neuropathy.40
Brain MRI may show other white matter lesions (either hyperintensities on T2-weighted images or enhancement of T1-weighted images postcontrast), which denote a higher risk of developing MS. In 15-year follow-up data from the ONTT, monosymptomatic patients with no white matter lesions had a 25% risk of MS (defined at the time the ONTT was conducted as a second demyelinating event), while those with one lesion or more had a 72% risk.41
An earlier, prospective study in 102 Italian patients with optic neuritis found the risk of developing MS to be about 36% at 6 years and 42% at 8 years (using the Posner diagnostic criteria). When brain MRI data were analyzed, those with one or more lesions had a 52% risk of developing MS at 8 years, whereas those with no MRI lesions did not develop MS.42
Other studies have stratified the risk of MS in patients with clinically isolated syndromes (including not only optic neuritis, but also other neurologic symptoms such as brainstem, motor, or sensory deficits). At mean follow-ups ranging from 5 to 14 years, the risk of developing MS was 8% to 24% in patients with normal findings on brain MRI compared with 56% to 88% in those with abnormal MRI findings.43,44
Optic neuritis patients with atypical white matter lesions on brain MRI may benefit from lumbar puncture to look for oligoclonal bands, to measure the IgG index and the IgG synthesis rate, and to test for myelin basic protein in the cerebrospinal fluid. Of patients with acute optic neuritis, 79% have abnormalities in their cerebrospinal fluid. Oligoclonal bands are present in 69%, and for patients with oligoclonal bands, the 5-year probability of developing MS is estimated to be 65%, compared with 10% in those without bands. If the patient has no oligoclonal bands and has normal findings on brain MRI, he or she will not have MS 5 years later.45–47
Patients with optic neuritis who have no white matter lesions on brain MRI may be further risk-stratified on the basis of their clinical findings. In the ONTT 15-year follow-up, MS did not develop in any patient who had no brain lesions on baseline MRI, no prior optic neuritis in the contralateral eye, and no prior neurologic symptoms or signs, even if the patient had severe disc swelling (eg, peripapillary hemorrhage or retinal exudates) or if vision was reduced to no light perception.41
CASE CONTINUED: FINDINGS ON MRI
Figure 4. The patient’s magnetic resonance image. Top, an axial T2 image with contrast; bottom, sagittal T1 image with contrast. The white matter lesions indicate she is at risk of developing multiple sclerosis.
Our patient undergoes MRI, which shows lesions on axial T2 and sagittal T1 imaging with contrast (Figure 4). Of note, there are significant lesions perpendicular to the corpus callosum (Dawson fingers), some of which enhance with contrast. The enhancement indicates breakdown of the blood-brain barrier and suggests that there is active inflammation in the white matter.
Patients in the ONTT were randomized to receive one of three treatments:
Oral prednisone 1 mg/kg/day for 14 days and then tapered over 4 days
Intravenous methylprednisolone (Solu-Medrol) 250 mg four times per day for 3 days followed by oral prednisone 1 mg/kg/day for 11 days and then tapered for 4 days
Oral placebo for 14 days.
The primary visual outcomes measured were visual acuity and contrast sensitivity.48
Patients who received intravenous methylprednisolone recovered their visual function faster, although the visual outcomes after 6 months were no better with methylprednisolone than with placebo or oral prednisone. Intravenous methylprednisolone also reduced the risk of MS within the first 2 years in patients with high-risk brain MRIs.
Surprisingly, patients in the oral prednisone group had a higher risk of recurrent optic neuritis in both eyes than did patients given intravenous methylprednisolone or placebo (30% at 2 years with oral prednisone vs 16% with placebo and 13% with intravenous methylprednisolone).48 At 10 years, the risk of recurrent optic neuritis was still higher in the oral prednisone group (44%) than in the intravenous methylprednisolone group (29%) (P = .03). However, the difference between the oral prednisone and placebo groups was no longer statistically significant (P = .07).49 Oral prednisone alone is therefore contraindicated in the treatment of typical unilateral demyelinating optic neuritis.
Many patients can now be treated with intravenous infusions of methylprednisolone at home for episodes of optic neuritis.
Risks vs benefits of corticosteroid therapy
When deciding whether to treat an individual patient who has optic neuritis with intravenous corticosteroids, one should consider all the benefits and risks.
Corticosteroids do not affect long-term visual outcome, although they do hasten recovery. Patients with mild vision loss (visual acuity better than 20/40), no significant visual field loss, and no enhancing lesions on brain MRI can forgo therapy with intravenous corticosteroids.
On the other hand, we strongly favor intravenous corticosteroid treatment in patients who have both acute optic neuritis and high signal abnormalities on brain MRI, since it may delay the onset of MS. In addition, patients with severe vision loss should receive intravenous corticosteroids to hasten their recovery. In the rare circumstance in which intravenous corticosteroids are not available, high-dose oral methyl-prednisolone (500 mg daily for 5 days and then tapered over 10 days) may be acceptable.50
The side effects of corticosteroids are minimal when they are given for a brief time in otherwise healthy patients. The most common side effects are mood changes, facial flushing, sleep perturbations, weight gain, and dyspepsia.48
IMMUNOGLOBUL IN: LITTLE BENEFIT
In a double-blind, randomized trial, patients were treated with intravenous immunoglobulin 0.4 g/kg or placebo on days 0, 1, 2, 30, and 60. No difference was found in the primary outcomes of contrast sensitivity, visual acuity, or color vision from 1 week up to 6 months. There was also no significant difference in MRI outcomes between the two groups. The number of relapses was similar between both groups after 6 months.51,52
PLASMA EXCHANGE: FEW DATA
Data on plasma exchange are too scarce for us to make any recommendations. In one trial in 10 patients with severe optic neuritis, 3 patients appeared to benefit from plasma exchange. All patients had already received two doses of intravenous steroids.53
IMMUNOMODULATORY THERAPY MAY PREVENT MULTIPLE SCLEROSIS IN SOME
The most important clinical decision to make in patients with optic neuritis is whether to begin immunomodulatory therapy. Patients who may benefit the most from immunomodulatory therapy are those with abnormal white matter lesions on brain MRI, as they are at higher risk of developing MS.
Collectively, data from three studies indicate that early treatment in patients with clinically isolated syndromes, such as optic neuritis, reduces the rate of MS to 35% (from 50% without treatment) and reduces the number of new active lesions on MRI by approximately 50%.54–56
In addition, the Betaferon/Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT) trial57 found that at 3 years the rate of disability was 40% lower in patients who started immunomodulatory therapy (interferon beta-1b; Betaseron) early vs later. (Early treatment meant starting within 60 days of the clinically isolated syndrome; late treatment began 2 years later.) This study suggests that early treatment may reduce future disability, although these results need to be confirmed in prospective trials.
Therefore, once the diagnosis is secure, patients with optic neuritis should be referred to a neurologist with experience in treating MS to begin treatment with immunomodulatory therapy, such as glatiramer acetate (Copaxone), interferon beta-1a (Avonex, Refib), or interferon beta-1b (Betaseron).
Patients who have a normal MRI of the brain may consider deferring therapy, since they are at low risk of developing MS. These patients should undergo surveillance MRI (at least annually at first) to look for the development of white matter lesions, as the ONTT showed even this cohort has a 22% risk of developing MS.
If neuromyelitis optica is suspected (ie, in patients with severe unilateral or bilateral vision loss, recurrent optic neuritis, paraplegia, or quadriplegia), the serum neuromyelitis optic antibody can be tested, keeping in mind that 30% to 40% of patients with neuromyelitis optica will be seronegative for this antibody. Other tests supporting the diagnosis of neuromyelitis optica may include an MRI of the spine showing longitudinally extensive transverse myelitis, a polymorphonuclear pleocytosis in the cerebrospinal fluid, absent oligoclonal bands in the cerebrospinal fluid, and normal MRI of the brain (with some possible signal abnormalities in the periaqueductal gray matter and around the diencephalon).
Because neuromyelitis optica is believed to be mediated primarily by the humoral immune system, immunomodulatory therapy is not a first-line treatment. Patients with neuromyelitis optica can be treated initially with corticosteroids, intravenous immunoglobulin therapy, plasma exchange, or immunosuppressive agents such as azathioprine (Imuran), rituximab (Rituxan), or cyclophosphamide (Cytoxan). The choice of medication should be deferred to a neurologist familiar with treatment of this disorder.
The risk of MS may be lower in children than in adults. One large, retrospective study found the cumulative risk of developing MS (the study predated the McDonald criteria) was 13% at 10 years and 19% by 20 years.58 More recently, a large series from Toronto reported a higher rate of MS development in children with optic neuritis (36% at two years).59 By comparison, studies of adults with unilateral optic neuritis found a 38% to 39% risk of converting to MS at 10 years.5,41 The use of immunomodulatory therapies to reduce the risk of MS has not been well studied in children, since the prevalence is low in this age group.
The most common side effects of the beta-interferons are flulike symptoms (fatigue, myalgia), injection site reactions, and elevations of aminotransferase levels. Most patients are able to tolerate the side effects if the beta-interferon is taken with acetaminophen (Tylenol) or with over-the-counter nonsteroidal anti-inflammatory drugs.
Glatiramer acetate does not cause flulike symptoms or elevate aminotransferases, but it does require more frequent injections. Rarely, it may cause an idiosyncratic panic-like attack.
CASE CONTINUED
The best therapeutic regimen for this patient would be intravenous methylprednisolone, and subsequently a disease-modifying, immunomodulatory agent (interferon beta or glatiramer acetate). Our patient chose to start therapy with interferon beta-1a 30 μg intramuscularly once a week. She has been tolerating the medication well and has had no new neurologic or visual symptoms for the past 2 years.
References
Liu GT. Visual loss: optic neuropathies. In: Liu GT, Volpe NJ, Galetta SL, editors. Neuro–Ophthalmology: Diagnosis and Management. Philadelphia, PA: WB Saunders, 2001:103–187.
Wray SH. Optic neuritis. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Philadelphia, PA: WB Saunders, 1994:2539–2568.
Optic Neuritis Study Group. The clinical profile of optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol1991; 109:1673–1678.
Percy AK, Nobrega FT, Kurland LT. Optic neuritis and multiple sclerosis: an epidemiologic study. Arch Ophthalmol1972; 87:135–139.
Rodriguez M, Siva A, Cross SA, O’Brien PC, Kurland LT. Optic neuritis: a population–based study in Olmsted County, Minnesota. Neurology1995; 45:244–250.
Phillips PH, Newman NJ, Lynn MJ. Optic neuritis in African Americans. Arch Neurol1998; 55:186–192.
Brady KM, Brar AS, Lee AG, Coats DK, Paysse EA, Steinkuller PG. Optic neuritis in children: clinical features and visual outcome. J AAPOS1999; 3:98–103.
Kriss A, Francis DA, Cuendet B, et al. Recovery after optic neuritis in childhood. J Neurol Neurosurg Psychiatry1988; 51:1253–1258.
Beck RW. The Optic Neuritis Treatment Trial. Arch Ophthalmol1988; 106:1051–1053.
Optic Neuritis Study Group. Visual function 15 years after optic neuritis. Ophthalmology2008; 115:1079–1082.
Cox TA, Thompson HS, Corbett JJ. Relative afferent pupillary defects in optic neuritis. Am J Ophthalmol1981; 92:685–690.
Arnold AC. Ischemic optic neuropathies. Ophthalmol Clin North Am2001; 14:83–98.
Newman NJ, Scherer R, Langenberg P, et al. The fellow eye in NAION: report from the ischemic optic neuropathy decompression trial follow–up study. Am J Ophthalmol2002; 134:317–328.
Hayreh SS, Podhajsky PA, Zimmerman B. Ipsilateral recurrence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol2001; 132:734–742.
Liozon E, Herrmann F, Ly K, et al. Risk factors for visual loss in giant cell (temporal) arteritis: a prospective study of 174 patients. Am J Med2001; 111:211–217.
Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol2005; 50:15–26.
American Society of Anesthesiologists Task Force on Perioperative Blindness. Practice advisory for perioperative visual loss associated with spine surgery: a report by the American Society of Anesthesiologists Task Force on Perioperative Blindness. Anesthesiology2006; 104:1319–1328.
Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology2007; 114:810–815.
Papais-Alvarenga RM, Carellos SC, Alvarenga MP, Holander C, Bichara RP, Thuler LC. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol2008; 126:12–16.
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology1999; 53:1107–1114.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology2006; 66:1485–1489.
Takahashi T, Fujihara K, Nakashima I, et al. Anti–aquaporin–4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain2007; 130:1235–1243.
Sibony P, Halperin J, Coyle PK, Patel K. Reactive Lyme serology in optic neuritis. J Neuroophthalmol2005; 25:71–82.
Anninger WV, Lomeo MD, Dingle J, Epstein AD, Lubow M. West Nile virus–associated optic neuritis and chorioretinitis. Am J Ophthalmol2003; 136:1183–1185.
Jabs DA, Miller NR, Newman SA, Johnson MA, Stevens MB. Optic neuropathy in systemic lupus erythematosus. Arch Ophthalmol1986; 104:564–568.
Howell N. LHON and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol2003; 37:94–108.
Newman NJ. Hereditary optic neuropathies. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. Philadelphia, PA: Lippincott Williams & Wilkins, 2005;465–501.
Smith JL, Hoyt WF, Susac JO. Ocular fundus in acute Leber optic neuropathy. Arch Ophthalmol1973; 90:349–354.
Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol2003; 87:48–53.
Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin– related GTPase, is mutated in autosomal dominant atrophy linked to chromosome 3q28. Nat Genet2000; 26:211–215.
McKinley SH, Foroozan R. Optic neuropathy associated with linezolid treatment. J Neuroophthalmol2005; 25:18–21.
Melamud A, Kosmorsky GS, Lee MS. Ocular ethambutol toxicity. Mayo Clin Proc2003; 78:1409–1411.
Kerrison JB. Optic neuropathies caused by toxins and adverse drug reactions. Ophthalmol Clin North Am2004; 17:481–488.
Pomeranz HD, Bhavsar AR. Nonarteritic ischemic optic neuropathy developing soon after use of sildenafil (Viagra): a report of seven new cases. J Neuroophthalmol2005; 25:9–13.
Fivgas GD, Newman NJ. Anterior ischemic optic neuropathy following the use of a nasal decongestant. Am J Ophthalmol1999; 127:104–106.
The Cuba Neuropathy Field Investigation Team. Epidemic optic neuropathy in Cuba—clinical characterization and risk factors. N Engl J Med1995; 333:1176–1182.
Solberg Y, Rosner M, Belkin M. The association between cigarette smoking and ocular diseases. Surv Ophthalmol1998; 42:535–547.
Rizzo JF, Andreoli CM, Rabinov JD. Use of magnetic resonance imaging to differentiate optic neuritis and nonarteritic anterior ischemic optic neuropathy. Ophthalmology2002; 109:1679–1684.
The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final Optic Neuritis Treatment Trial follow-up. Arch Neurol2008; 65:727–732.
Ghezzi A, Martinelli V, Torri V, et al. Long–term follow–up of isolated optic neuritis: the risk of developing multiple sclerosis, its outcome, and the prognostic role of paraclinical tests. J Neurol1999; 246:770– 775.
Brex PA, Ciccarelli O, O'Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med2002; 346:158–164.
Tintore M, Rovira A, Rio J, et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology2006; 67:968–972.
Söderström M, Ya–Ping J, Hillert J. Optic neuritis: prognosis for multiple sclerosis from MRI, CSF, and HLA findings. Neurology1998; 50:708–714.
Frederiksen JL, Madsen HO, Ryder LP, Larsson HB, Morling N, Svejgaard A. HLA typing in acute optic neuritis: relation to multiple sclerosis and magnetic resonance imaging findings. Arch Neurol1997; 54:76–80.
Frederiksen JL, Larsson HB, Oleson J. Correlation of magnetic resonance imaging and CSF findings in patients with acute monosymptomatic optic neuritis. Acta Neurol Scand1992; 86:317–322.
Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med1992; 326:581–588.
Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol2003; 121:944–949.
Sellebjerg F, Nielsen HS, Frederiksen JL, Olesen J. A randomized, controlled trial of oral high-dose methylprednisolone in acute optic neuritis. Neurology1999; 52:1479–1484.
Noseworthy JH, O’Brien PC, Peterson TM, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology2001; 56:1514–1522.
Roed HG, Langkilde A, Sellebjerg F, et al. A double–blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology2005; 64:804–810.
Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology2004; 63:1081–1083.
CHAMPS Study Group. Interferon beta-1a for optic neuritis patients at high risk for multiple sclerosis. Am J Ophthalmol2001; 132:463– 471.
Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet2001; 357:1576–1582.
Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology2006; 67:1242–1249.
Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet2007; 370:389–397.
Lucchinetti CF, Kiers L, O’Duffy A, et al. Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology1997; 49:1413–1418.
Wilejto M, Shroff M, Buncic JR, Kennedy J, Goia C, Banwell B. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology2006; 67:258–262.
Benjamin J. Osborne, MD Assistant Professor of Neurology and Ophthalmology, Departments of Neurology and Ophthalmology, Georgetown University Medical Center, Washington, DC
Nicholas J. Volpe, MD Niessen Professor of Ophthalmology and Neurology, Vice Chair for Clinical Practice and Residency Program Director, Division of Neuro-Ophthalmology, University of Pennsylvania School of Medicine, Scheie Eye Institute, Philadelphia, PA
Address: Nicholas J. Volpe, MD, Scheie Eye Institute; 39th and Market Streets, Philadelphia, PA 19104; e-mail [email protected]
Benjamin J. Osborne, MD Assistant Professor of Neurology and Ophthalmology, Departments of Neurology and Ophthalmology, Georgetown University Medical Center, Washington, DC
Nicholas J. Volpe, MD Niessen Professor of Ophthalmology and Neurology, Vice Chair for Clinical Practice and Residency Program Director, Division of Neuro-Ophthalmology, University of Pennsylvania School of Medicine, Scheie Eye Institute, Philadelphia, PA
Address: Nicholas J. Volpe, MD, Scheie Eye Institute; 39th and Market Streets, Philadelphia, PA 19104; e-mail [email protected]
Author and Disclosure Information
Benjamin J. Osborne, MD Assistant Professor of Neurology and Ophthalmology, Departments of Neurology and Ophthalmology, Georgetown University Medical Center, Washington, DC
Nicholas J. Volpe, MD Niessen Professor of Ophthalmology and Neurology, Vice Chair for Clinical Practice and Residency Program Director, Division of Neuro-Ophthalmology, University of Pennsylvania School of Medicine, Scheie Eye Institute, Philadelphia, PA
Address: Nicholas J. Volpe, MD, Scheie Eye Institute; 39th and Market Streets, Philadelphia, PA 19104; e-mail [email protected]
Two days ago, a 27-year-old woman noticed that her vision was blurry in her right eye. She has come to see her primary care physician for advice. This is the first time this has happened to her. She describes seeing a grayish blur over the center of her vision, but she has not noted any other symptoms except for some soreness around the right eye, which is worse with eye movements.
How should she be assessed and treated?
IMPORTANT TO RECOGNIZE
Sudden vision loss is one of the more common problems encountered in ophthalmology and neurology.
Optic neuritis, a demyelinating inflammatory condition that causes acute vision loss, is associated with multiple sclerosis (MS), and recognizing its classic clinical manifestations early is important so that appropriate diagnostic testing (magnetic resonance imaging [MRI]) and treatment (corticosteroids and immunomodulators) can be started.
Although a comprehensive review of all the optic neuropathies is beyond the scope of this paper, in the pages that follow we review some of the most common causes, which may be first seen by the general internist.
FOUR SUBTYPES OF OPTIC NEURITIS
There are four subtypes of optic neuritis:
Figure 1. In retrobulbar optic neuritis, the inflammation and demyelination occur behind the globe of the eye. The optic disc appears normal with no signs of swelling or pallor.Retrobulbar neuritis (Figure 1), or inflammation of the optic nerve behind the eye, is the form most commonly associated with MS.
Papillitis (Figure 2), or inflammation of the optic disc, can also be associated with MS.
Perineuritis is inflammation of the optic nerve sheath, sparing the optic nerve itself. Usually, patients are older, and vision loss is mild to moderate. Perineuritis is commonly due to infectious or inflammatory conditions, eg, syphilis or sarcoidosis.
Figure 2. In papillitis, mild swelling and elevation of the optic disc can be seen. The small splinter hemorrhage seen at 10 o’clock is not typical of optic neuritis associated with multiple sclerosis.Neuroretinitis may occur at any age. There is concomitant swelling of the optic nerve and macula. Exudates that form around the macula give the appearance of a star.
Perineuritis and neuroretinitis are not associated with MS, and if they are found they suggest another etiology. In the rest of this review, “optic neuritis” means retrobulbar optic neuritis, the form most commonly seen in clinical practice.
MOST COMMON IN YOUNG WOMEN
Acute demyelinating optic neuritis most often affects women in their 20s and 30s.1–3 Studies in the United States have estimated its annual incidence to be 5.1 to 6.4 per 100,000.4,5 The incidence is higher in populations at higher latitudes and lower near the equator. It is less common in blacks than in whites.6
In children, optic neuritis is not as strongly associated with MS, especially when there is optic disc swelling or bilateral involvement. Most children have a good visual outcome, although approximately 20% may be visually disabled.7–9
FEATURES: VISION LOSS AND EYE PAIN
Most of our current knowledge of the clinical features of optic neuritis comes from the Optic Neuritis Treatment Trial (ONTT),10 conducted in the 1990s. This trial enrolled 457 patients 18 to 46 years old who had acute unilateral optic neuritis. The patients had to have symptoms consistent with acute unilateral optic neuritis for 8 days or less. They could not have evidence of any systemic disease (except for MS) or have received prior treatment for MS. Therefore, this study was quite representative of the patients with optic neuritis that one might encounter in the clinic and is highly important in both characterizing optic neuritis and defining its treatment.
The study found that the two most common symptoms are vision loss and eye pain.
Vision loss in optic neuritis typically occurs over several hours to days, and vision reaches a nadir within 1 to 2 weeks. Typically, patients begin to recover 2 to 4 weeks after the onset of the vision loss. The optic nerve may take up to 6 to 12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.11 More than two-thirds of patients have at least 20/20 vision once they have fully recovered from the optic neuritis. Only 3% of patients become completely blind.
Eye pain is very common in optic neuritis (it affected 87% of patients in the ONTT) and typically worsens with eye movement. The eye is also sore to touch. The pain generally begins at the same time as the visual loss and improves along with it. Eye movements also may bring about photopsia (flickering or flashes of light), a symptom reported by 30% of the ONTT participants.
Loss of color vision out of proportion to the loss of visual acuity is characteristic of optic neuropathies. In the ONTT, 88% of the involved eyes had abnormal color vision as assessed by the Ishihara test (using pseudoisochromatic plates), and 94% as assessed by the Farnsworth-Munsell 100 hue test, which is more sensitive but cumbersome. The most common patterns of color vision loss in optic nerve disease are loss of red (protanopia) and green (deutranopia).
A good way to screen for loss of color vision is to test for color desaturation. First, ask the patient to fixate with the normal eye on a bright red object (for example, the cap from a bottle of ophthalmic mydriatic drops or a pen cap). Then ask the patient to compare the intensity of the redness between the good eye and the affected eye. The patient can quantify the color desaturation by rating what percentage of red is lost in the affected eye compared with the normal eye.
Temporary exacerbations of visual problems during fever (the Uhthoff phenomenon) can occur in patients who have had optic neuritis. These transient pseudoexacerbations are not new episodes of optic neuritis and should resolve after the body temperature returns to normal.
A relative afferent pupillary defect should be seen in the involved eye in all patients with optic neuritis if the other eye is uninvolved and healthy.12 The best way to elicit this sign is to perform the swinging light test in a dark room with the patient fixating at a distant target, which prevents miosis due to accommodation. When the light is swung to the involved eye, the pupil dilates because less light stimulus reaches the midbrain through the affected optic nerve. As the optic nerve heals and recovers, this sign may become subtle, but it persists in more than 90% of cases.12
Findings on funduscopy
Examination of the fundus is helpful in the clinical diagnosis of optic neuritis.
Two-thirds of the ONTT patients had retrobulbar neuritis, and one-third had papillitis. If optic nerve swelling is present, it is typically mild.
Peripapillary hemorrhages were exceedingly rare in the cases of papillitis (only 6%) and were associated with a very low to zero risk of developing MS. If peripapillary hemorrhages are found on examination, one should consider another diagnosis, such as anterior ischemic optic neuropathy.11
CASE CONTINUED
Our patient undergoes a neurologic examination, which reveals an afferent pupillary defect in the right eye and visual acuity of 20/100 in the right eye and 20/20 in the left. Visual fields are normal in the left eye as assessed by confrontation, but there is a central scotoma in the right.
OTHER TYPES OF NEUROPATHY
Optic neuritis is a clinical diagnosis based on the history and findings on examination. If the patient does not have its typical features (Table 1), then other diagnoses should be pursued with serologic and cerebrospinal fluid studies.
The following should be included in the differential diagnosis of optic neuritis:
Ischemic optic neuropathy
Ischemic optic neuropathy is more common in patients age 50 and older, whereas optic neuritis is more common in younger patients. Most patients with ischemic optic neuropathy have hypertension, hypercholesterolemia, diabetes mellitus, obstructive sleep apnea, or other vascular risk factors. The disease has several important subtypes, as discussed below.
Figure 3. Left, fundus photo several weeks after an attack of nonarteritic anterior ischemic optic neuropathy demonstrating pallor of the superior half of the disc. Middle, an associated inferior altitudinal defect. Right, sectoral swelling of the disc with flame or splinter hemorrhages is commonly seen. This is not typical of demyelinating optic neuritis.Nonarteritic anterior ischemic optic neuropathy is the most common form. Typically, there is acute onset of painless vision loss in one eye and an associated altitudinal field defect (Figure 3). For example, if the superior rim of the optic nerve acutely develops swelling and then becomes pale, a corresponding altitudinal cut would develop in the inferior visual field, respecting the horizontal meridian. Many patients first notice the vision loss upon waking up in the morning.13
Although patients with nonarteritic anterior ischemic optic neuropathy typically have vasculopathic risk factors such as hypertension, diabetes mellitus, peripheral vascular disease, or hypercholesterolemia, there is no proven causation between the two. The age of these patients ranges from 50 to 70, with an average age of 66.
The disc appears swollen and may have flame or splinter hemorrhages (Figure 3). The cup of the involved disc is typically small. The visual loss is believed to be the result of poor perfusion in the circulation of the posterior ciliary artery, which supplies the optic nerve head.1 If the other eye also has a small cup, it is considered to be at risk of future ischemic events. In one study,14 the opposite eye became involved within the next 5 years in 14.7% of all cases. The risk of recurrent disease in the same eye is low (6.4% in another study15).
Arteritic anterior ischemic optic neuropathy is more common in patients over age 70 and is usually due to giant cell arteritis, which has a significant association with polymyalgia rheumatica. Patients may have jaw claudication, proximal myalgia and arthralgia, scalp tenderness, headache, fatigue, and a significantly elevated erythrocyte sedimentation rate and C-reactive protein level. These features should be looked for in the review of systems, although patients may not have all of them.
The funduscopic examination may reveal a pale, swollen disc, peripapillary hemorrhages, branch or central retinal artery occlusions, or cotton-wool spots.
Temporal artery biopsy is the gold standard for diagnosis, but treatment with corticosteroids should not be delayed pending biopsy or other test results.1
Thrombocytosis has been associated with a higher risk of permanent vision loss in patients with giant cell arteritis.16
Posterior ischemic optic neuropathy is the least common form of ischemic optic neuropathy. This diagnosis should be entertained in older patients who report acute, painless vision loss but have a normal funduscopic examination. Giant cell arteritis must be considered first in this setting.
Bilateral posterior ischemic optic neuropathy can occur (although rarely) in patients undergoing cardiac or spinal surgery.17 Risk factors thought to be associated with perioperative disease include anemia, hypotension, substantial blood loss during the surgery, surgeries longer than 6.5 hours, carotid atherosclerosis, and diabetes.18
There are no effective treatments for most ischemic optic neuropathies with the crucial exception of giant cell arteritis.
Neuromyelitis optica (Devic disease)
Neuromyelitis optica (Devic disease) is a combination of optic neuritis and transverse myelitis (Table 2). Clinically, the disease spares the nervous system except for the optic nerves and spinal cord. The onset of the optic neuritis may precede or follow the onset of the transverse myelitis by up to 2 to 4 years.19 Usually, the optic neuritis is bilateral and retrobulbar and results in severe vision loss, worse than that seen in patients with MS.19,20
The transverse myelitis may cause paraplegia or quadriplegia, depending on the location of the lesion in the spinal cord (cervical vs thoracic). The transverse myelitis in neuromyelitis optica is distinct from that seen in MS. In neuromyelitis optica, the transverse myelitis is longitudinally extensive, spanning more than three vertebral bodies in length. In MS, spinal cord lesions usually are more discrete and involve one or two spinal cord segments.21
Recently, serum neuromyelitis optica immunoglobulin G (IgG) antibody has been shown to be a significant biomarker of this disease. Its sensitivity ranges from approximately 60% to 70% and its specificity is greater than 90%.22 This antibody binds to aquaporin-4, an important water-channel protein in the blood-brain barrier of the central nervous system, and evidence suggests that it is involved in the pathogenesis of the disease.23
Initially, it was proposed that MRI of the brain had to be normal for neuromyelitis optica to be diagnosed.21 However, the proposed 2006 criteria allow for some abnormal T2 and fluid-attenuated inversion recovery (FLAIR) hyperintensities in the periaqueductal gray matter and diencephalon.22
The spinal fluid in neuromyelitis optica may show a pleocytosis larger than that seen in MS (> 50 white blood cells per mm3) and may have a significant neutrophilic component.21 Oligoclonal bands are not typically present.
It is still debated whether neuromyelitis optica is a separate disease from MS or a subset of it. The implications of this debate may affect its management, as discussed below.
Inflammatory optic neuropathies
Inflammatory optic neuropathies can be caused by infections (eg, syphilis, cat scratch disease) or by noninfectious conditions (eg, sarcoidosis). Lyme disease is rarely a cause of retrobulbar optic neuritis but may cause papillitis.24 West Nile virus has also been reported to cause optic neuritis.25 Lupus may cause an optic neuropathy by inflammatory or ischemic mechanisms.26
Compressive optic neuropathies
Compressive optic neuropathies may be due to mass lesions, tumors, thyroid eye disease, or other orbital processes. MRI of the brain and orbits will confirm or rule out diagnoses associated with compressive optic neuropathy.
Genetic causes
Genetic causes of optic neuropathy include the Leber and Kjer hereditary optic neuropathies.
Leber optic neuropathy involves subacute and painless vision loss, with sequential involvement of both eyes over a period of weeks to months. This disorder predominantly affects men (80%–90% of patients) and is inherited from maternal mitochondrial DNA. The three most common mutations implicated in Leber optic neuropathy (located at base pairs 11,778, 3,460, and 14,484 in the mitochondrial DNA) are involved in more than 90% of cases. The prognosis for recovery varies depending on the genotype.27 These genes encode proteins that are part of complex I of the mitochondrial respiratory chain.28 Funduscopic examination most commonly shows circumpapillary telangiectasia, although up to one-third of patients can have a normal-appearing disc initially. Central vision is affected more severely than peripheral vision.29
Kjer autosomal-dominant optic atrophy is the most common hereditary optic neuropathy. This disease primarily affects children in the first decade of life with slowly progressive loss of vision. As with other optic neuropathies, there will eventually be pallor of the optic disc, a cecocentral scotoma, and loss of color perception. The OPA1 gene located on chromosome 3q28 has been implicated in most patients with dominant optic atrophy; a test is commercially available for diagnosis.30,31
Toxic and metabolic causes
Many agents can cause optic neuropathy. Toxins strongly associated with optic neuropathy include carbon monoxide, methanol, ethylene glycol, perchloroethylene, and tobacco. Drugs linked to optic neuropathy are ethambutol (Myambutol), clioquinol (Vioform), isoniazid (Nydrazid), amiodarone (Cordarone), linezolid (Zyvox), methotrexate, sildenafil (Viagra), oxymetazoline (contained in Afrin and other nasal sprays), and infliximab (Remicade).32–37 Additionally, several chemotherapeutic agents are known to cause optic atrophy, including vincristine (Oncovin), cisplatin (Platinol), carboplatin (Paraplatin), and paclitaxel (Abraxane, Onxol).
Nutritional deficiencies are presumed to have played a significant role in the endemics of optic neuropathy that have occurred in poor countries, such as in Cuba during the 1990s.38 Most nutritional optic neuropathies appear to be exacerbated by tobacco.39
MRI ASSESSES RISK OF MS
The diagnosis of optic neuritis is clinical, based on the history and physical findings.
However, MRI of the brain and orbits with gadolinium contrast has become the cornerstone of the evaluation in patients with optic neuritis. And MRI not only helps confirm the clinical diagnosis, but it also more importantly offers very strong prognostic information about the risk of future demyelinating events and MS.
Gadolinium-enhanced fat-saturated T1-weighted MRI of the orbits is the best sequence to show the inflammation of the optic nerve in optic neuritis (fat saturation is necessary to hide the bright signal of the orbital fat tissue).
Contrast-enhanced MRI can also help differentiate optic neuritis from nonarteritic anterior ischemic optic neuropathy. MRI of the orbits with gadolinium contrast shows enhancement of the affected optic nerve in approximately 95% of cases of optic neuritis, whereas optic nerve enhancement rarely occurs in nonarteritic anterior ischemic optic neuropathy.40
Brain MRI may show other white matter lesions (either hyperintensities on T2-weighted images or enhancement of T1-weighted images postcontrast), which denote a higher risk of developing MS. In 15-year follow-up data from the ONTT, monosymptomatic patients with no white matter lesions had a 25% risk of MS (defined at the time the ONTT was conducted as a second demyelinating event), while those with one lesion or more had a 72% risk.41
An earlier, prospective study in 102 Italian patients with optic neuritis found the risk of developing MS to be about 36% at 6 years and 42% at 8 years (using the Posner diagnostic criteria). When brain MRI data were analyzed, those with one or more lesions had a 52% risk of developing MS at 8 years, whereas those with no MRI lesions did not develop MS.42
Other studies have stratified the risk of MS in patients with clinically isolated syndromes (including not only optic neuritis, but also other neurologic symptoms such as brainstem, motor, or sensory deficits). At mean follow-ups ranging from 5 to 14 years, the risk of developing MS was 8% to 24% in patients with normal findings on brain MRI compared with 56% to 88% in those with abnormal MRI findings.43,44
Optic neuritis patients with atypical white matter lesions on brain MRI may benefit from lumbar puncture to look for oligoclonal bands, to measure the IgG index and the IgG synthesis rate, and to test for myelin basic protein in the cerebrospinal fluid. Of patients with acute optic neuritis, 79% have abnormalities in their cerebrospinal fluid. Oligoclonal bands are present in 69%, and for patients with oligoclonal bands, the 5-year probability of developing MS is estimated to be 65%, compared with 10% in those without bands. If the patient has no oligoclonal bands and has normal findings on brain MRI, he or she will not have MS 5 years later.45–47
Patients with optic neuritis who have no white matter lesions on brain MRI may be further risk-stratified on the basis of their clinical findings. In the ONTT 15-year follow-up, MS did not develop in any patient who had no brain lesions on baseline MRI, no prior optic neuritis in the contralateral eye, and no prior neurologic symptoms or signs, even if the patient had severe disc swelling (eg, peripapillary hemorrhage or retinal exudates) or if vision was reduced to no light perception.41
CASE CONTINUED: FINDINGS ON MRI
Figure 4. The patient’s magnetic resonance image. Top, an axial T2 image with contrast; bottom, sagittal T1 image with contrast. The white matter lesions indicate she is at risk of developing multiple sclerosis.
Our patient undergoes MRI, which shows lesions on axial T2 and sagittal T1 imaging with contrast (Figure 4). Of note, there are significant lesions perpendicular to the corpus callosum (Dawson fingers), some of which enhance with contrast. The enhancement indicates breakdown of the blood-brain barrier and suggests that there is active inflammation in the white matter.
Patients in the ONTT were randomized to receive one of three treatments:
Oral prednisone 1 mg/kg/day for 14 days and then tapered over 4 days
Intravenous methylprednisolone (Solu-Medrol) 250 mg four times per day for 3 days followed by oral prednisone 1 mg/kg/day for 11 days and then tapered for 4 days
Oral placebo for 14 days.
The primary visual outcomes measured were visual acuity and contrast sensitivity.48
Patients who received intravenous methylprednisolone recovered their visual function faster, although the visual outcomes after 6 months were no better with methylprednisolone than with placebo or oral prednisone. Intravenous methylprednisolone also reduced the risk of MS within the first 2 years in patients with high-risk brain MRIs.
Surprisingly, patients in the oral prednisone group had a higher risk of recurrent optic neuritis in both eyes than did patients given intravenous methylprednisolone or placebo (30% at 2 years with oral prednisone vs 16% with placebo and 13% with intravenous methylprednisolone).48 At 10 years, the risk of recurrent optic neuritis was still higher in the oral prednisone group (44%) than in the intravenous methylprednisolone group (29%) (P = .03). However, the difference between the oral prednisone and placebo groups was no longer statistically significant (P = .07).49 Oral prednisone alone is therefore contraindicated in the treatment of typical unilateral demyelinating optic neuritis.
Many patients can now be treated with intravenous infusions of methylprednisolone at home for episodes of optic neuritis.
Risks vs benefits of corticosteroid therapy
When deciding whether to treat an individual patient who has optic neuritis with intravenous corticosteroids, one should consider all the benefits and risks.
Corticosteroids do not affect long-term visual outcome, although they do hasten recovery. Patients with mild vision loss (visual acuity better than 20/40), no significant visual field loss, and no enhancing lesions on brain MRI can forgo therapy with intravenous corticosteroids.
On the other hand, we strongly favor intravenous corticosteroid treatment in patients who have both acute optic neuritis and high signal abnormalities on brain MRI, since it may delay the onset of MS. In addition, patients with severe vision loss should receive intravenous corticosteroids to hasten their recovery. In the rare circumstance in which intravenous corticosteroids are not available, high-dose oral methyl-prednisolone (500 mg daily for 5 days and then tapered over 10 days) may be acceptable.50
The side effects of corticosteroids are minimal when they are given for a brief time in otherwise healthy patients. The most common side effects are mood changes, facial flushing, sleep perturbations, weight gain, and dyspepsia.48
IMMUNOGLOBUL IN: LITTLE BENEFIT
In a double-blind, randomized trial, patients were treated with intravenous immunoglobulin 0.4 g/kg or placebo on days 0, 1, 2, 30, and 60. No difference was found in the primary outcomes of contrast sensitivity, visual acuity, or color vision from 1 week up to 6 months. There was also no significant difference in MRI outcomes between the two groups. The number of relapses was similar between both groups after 6 months.51,52
PLASMA EXCHANGE: FEW DATA
Data on plasma exchange are too scarce for us to make any recommendations. In one trial in 10 patients with severe optic neuritis, 3 patients appeared to benefit from plasma exchange. All patients had already received two doses of intravenous steroids.53
IMMUNOMODULATORY THERAPY MAY PREVENT MULTIPLE SCLEROSIS IN SOME
The most important clinical decision to make in patients with optic neuritis is whether to begin immunomodulatory therapy. Patients who may benefit the most from immunomodulatory therapy are those with abnormal white matter lesions on brain MRI, as they are at higher risk of developing MS.
Collectively, data from three studies indicate that early treatment in patients with clinically isolated syndromes, such as optic neuritis, reduces the rate of MS to 35% (from 50% without treatment) and reduces the number of new active lesions on MRI by approximately 50%.54–56
In addition, the Betaferon/Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT) trial57 found that at 3 years the rate of disability was 40% lower in patients who started immunomodulatory therapy (interferon beta-1b; Betaseron) early vs later. (Early treatment meant starting within 60 days of the clinically isolated syndrome; late treatment began 2 years later.) This study suggests that early treatment may reduce future disability, although these results need to be confirmed in prospective trials.
Therefore, once the diagnosis is secure, patients with optic neuritis should be referred to a neurologist with experience in treating MS to begin treatment with immunomodulatory therapy, such as glatiramer acetate (Copaxone), interferon beta-1a (Avonex, Refib), or interferon beta-1b (Betaseron).
Patients who have a normal MRI of the brain may consider deferring therapy, since they are at low risk of developing MS. These patients should undergo surveillance MRI (at least annually at first) to look for the development of white matter lesions, as the ONTT showed even this cohort has a 22% risk of developing MS.
If neuromyelitis optica is suspected (ie, in patients with severe unilateral or bilateral vision loss, recurrent optic neuritis, paraplegia, or quadriplegia), the serum neuromyelitis optic antibody can be tested, keeping in mind that 30% to 40% of patients with neuromyelitis optica will be seronegative for this antibody. Other tests supporting the diagnosis of neuromyelitis optica may include an MRI of the spine showing longitudinally extensive transverse myelitis, a polymorphonuclear pleocytosis in the cerebrospinal fluid, absent oligoclonal bands in the cerebrospinal fluid, and normal MRI of the brain (with some possible signal abnormalities in the periaqueductal gray matter and around the diencephalon).
Because neuromyelitis optica is believed to be mediated primarily by the humoral immune system, immunomodulatory therapy is not a first-line treatment. Patients with neuromyelitis optica can be treated initially with corticosteroids, intravenous immunoglobulin therapy, plasma exchange, or immunosuppressive agents such as azathioprine (Imuran), rituximab (Rituxan), or cyclophosphamide (Cytoxan). The choice of medication should be deferred to a neurologist familiar with treatment of this disorder.
The risk of MS may be lower in children than in adults. One large, retrospective study found the cumulative risk of developing MS (the study predated the McDonald criteria) was 13% at 10 years and 19% by 20 years.58 More recently, a large series from Toronto reported a higher rate of MS development in children with optic neuritis (36% at two years).59 By comparison, studies of adults with unilateral optic neuritis found a 38% to 39% risk of converting to MS at 10 years.5,41 The use of immunomodulatory therapies to reduce the risk of MS has not been well studied in children, since the prevalence is low in this age group.
The most common side effects of the beta-interferons are flulike symptoms (fatigue, myalgia), injection site reactions, and elevations of aminotransferase levels. Most patients are able to tolerate the side effects if the beta-interferon is taken with acetaminophen (Tylenol) or with over-the-counter nonsteroidal anti-inflammatory drugs.
Glatiramer acetate does not cause flulike symptoms or elevate aminotransferases, but it does require more frequent injections. Rarely, it may cause an idiosyncratic panic-like attack.
CASE CONTINUED
The best therapeutic regimen for this patient would be intravenous methylprednisolone, and subsequently a disease-modifying, immunomodulatory agent (interferon beta or glatiramer acetate). Our patient chose to start therapy with interferon beta-1a 30 μg intramuscularly once a week. She has been tolerating the medication well and has had no new neurologic or visual symptoms for the past 2 years.
Two days ago, a 27-year-old woman noticed that her vision was blurry in her right eye. She has come to see her primary care physician for advice. This is the first time this has happened to her. She describes seeing a grayish blur over the center of her vision, but she has not noted any other symptoms except for some soreness around the right eye, which is worse with eye movements.
How should she be assessed and treated?
IMPORTANT TO RECOGNIZE
Sudden vision loss is one of the more common problems encountered in ophthalmology and neurology.
Optic neuritis, a demyelinating inflammatory condition that causes acute vision loss, is associated with multiple sclerosis (MS), and recognizing its classic clinical manifestations early is important so that appropriate diagnostic testing (magnetic resonance imaging [MRI]) and treatment (corticosteroids and immunomodulators) can be started.
Although a comprehensive review of all the optic neuropathies is beyond the scope of this paper, in the pages that follow we review some of the most common causes, which may be first seen by the general internist.
FOUR SUBTYPES OF OPTIC NEURITIS
There are four subtypes of optic neuritis:
Figure 1. In retrobulbar optic neuritis, the inflammation and demyelination occur behind the globe of the eye. The optic disc appears normal with no signs of swelling or pallor.Retrobulbar neuritis (Figure 1), or inflammation of the optic nerve behind the eye, is the form most commonly associated with MS.
Papillitis (Figure 2), or inflammation of the optic disc, can also be associated with MS.
Perineuritis is inflammation of the optic nerve sheath, sparing the optic nerve itself. Usually, patients are older, and vision loss is mild to moderate. Perineuritis is commonly due to infectious or inflammatory conditions, eg, syphilis or sarcoidosis.
Figure 2. In papillitis, mild swelling and elevation of the optic disc can be seen. The small splinter hemorrhage seen at 10 o’clock is not typical of optic neuritis associated with multiple sclerosis.Neuroretinitis may occur at any age. There is concomitant swelling of the optic nerve and macula. Exudates that form around the macula give the appearance of a star.
Perineuritis and neuroretinitis are not associated with MS, and if they are found they suggest another etiology. In the rest of this review, “optic neuritis” means retrobulbar optic neuritis, the form most commonly seen in clinical practice.
MOST COMMON IN YOUNG WOMEN
Acute demyelinating optic neuritis most often affects women in their 20s and 30s.1–3 Studies in the United States have estimated its annual incidence to be 5.1 to 6.4 per 100,000.4,5 The incidence is higher in populations at higher latitudes and lower near the equator. It is less common in blacks than in whites.6
In children, optic neuritis is not as strongly associated with MS, especially when there is optic disc swelling or bilateral involvement. Most children have a good visual outcome, although approximately 20% may be visually disabled.7–9
FEATURES: VISION LOSS AND EYE PAIN
Most of our current knowledge of the clinical features of optic neuritis comes from the Optic Neuritis Treatment Trial (ONTT),10 conducted in the 1990s. This trial enrolled 457 patients 18 to 46 years old who had acute unilateral optic neuritis. The patients had to have symptoms consistent with acute unilateral optic neuritis for 8 days or less. They could not have evidence of any systemic disease (except for MS) or have received prior treatment for MS. Therefore, this study was quite representative of the patients with optic neuritis that one might encounter in the clinic and is highly important in both characterizing optic neuritis and defining its treatment.
The study found that the two most common symptoms are vision loss and eye pain.
Vision loss in optic neuritis typically occurs over several hours to days, and vision reaches a nadir within 1 to 2 weeks. Typically, patients begin to recover 2 to 4 weeks after the onset of the vision loss. The optic nerve may take up to 6 to 12 months to heal completely, but most patients recover as much vision as they are going to within the first few months.11 More than two-thirds of patients have at least 20/20 vision once they have fully recovered from the optic neuritis. Only 3% of patients become completely blind.
Eye pain is very common in optic neuritis (it affected 87% of patients in the ONTT) and typically worsens with eye movement. The eye is also sore to touch. The pain generally begins at the same time as the visual loss and improves along with it. Eye movements also may bring about photopsia (flickering or flashes of light), a symptom reported by 30% of the ONTT participants.
Loss of color vision out of proportion to the loss of visual acuity is characteristic of optic neuropathies. In the ONTT, 88% of the involved eyes had abnormal color vision as assessed by the Ishihara test (using pseudoisochromatic plates), and 94% as assessed by the Farnsworth-Munsell 100 hue test, which is more sensitive but cumbersome. The most common patterns of color vision loss in optic nerve disease are loss of red (protanopia) and green (deutranopia).
A good way to screen for loss of color vision is to test for color desaturation. First, ask the patient to fixate with the normal eye on a bright red object (for example, the cap from a bottle of ophthalmic mydriatic drops or a pen cap). Then ask the patient to compare the intensity of the redness between the good eye and the affected eye. The patient can quantify the color desaturation by rating what percentage of red is lost in the affected eye compared with the normal eye.
Temporary exacerbations of visual problems during fever (the Uhthoff phenomenon) can occur in patients who have had optic neuritis. These transient pseudoexacerbations are not new episodes of optic neuritis and should resolve after the body temperature returns to normal.
A relative afferent pupillary defect should be seen in the involved eye in all patients with optic neuritis if the other eye is uninvolved and healthy.12 The best way to elicit this sign is to perform the swinging light test in a dark room with the patient fixating at a distant target, which prevents miosis due to accommodation. When the light is swung to the involved eye, the pupil dilates because less light stimulus reaches the midbrain through the affected optic nerve. As the optic nerve heals and recovers, this sign may become subtle, but it persists in more than 90% of cases.12
Findings on funduscopy
Examination of the fundus is helpful in the clinical diagnosis of optic neuritis.
Two-thirds of the ONTT patients had retrobulbar neuritis, and one-third had papillitis. If optic nerve swelling is present, it is typically mild.
Peripapillary hemorrhages were exceedingly rare in the cases of papillitis (only 6%) and were associated with a very low to zero risk of developing MS. If peripapillary hemorrhages are found on examination, one should consider another diagnosis, such as anterior ischemic optic neuropathy.11
CASE CONTINUED
Our patient undergoes a neurologic examination, which reveals an afferent pupillary defect in the right eye and visual acuity of 20/100 in the right eye and 20/20 in the left. Visual fields are normal in the left eye as assessed by confrontation, but there is a central scotoma in the right.
OTHER TYPES OF NEUROPATHY
Optic neuritis is a clinical diagnosis based on the history and findings on examination. If the patient does not have its typical features (Table 1), then other diagnoses should be pursued with serologic and cerebrospinal fluid studies.
The following should be included in the differential diagnosis of optic neuritis:
Ischemic optic neuropathy
Ischemic optic neuropathy is more common in patients age 50 and older, whereas optic neuritis is more common in younger patients. Most patients with ischemic optic neuropathy have hypertension, hypercholesterolemia, diabetes mellitus, obstructive sleep apnea, or other vascular risk factors. The disease has several important subtypes, as discussed below.
Figure 3. Left, fundus photo several weeks after an attack of nonarteritic anterior ischemic optic neuropathy demonstrating pallor of the superior half of the disc. Middle, an associated inferior altitudinal defect. Right, sectoral swelling of the disc with flame or splinter hemorrhages is commonly seen. This is not typical of demyelinating optic neuritis.Nonarteritic anterior ischemic optic neuropathy is the most common form. Typically, there is acute onset of painless vision loss in one eye and an associated altitudinal field defect (Figure 3). For example, if the superior rim of the optic nerve acutely develops swelling and then becomes pale, a corresponding altitudinal cut would develop in the inferior visual field, respecting the horizontal meridian. Many patients first notice the vision loss upon waking up in the morning.13
Although patients with nonarteritic anterior ischemic optic neuropathy typically have vasculopathic risk factors such as hypertension, diabetes mellitus, peripheral vascular disease, or hypercholesterolemia, there is no proven causation between the two. The age of these patients ranges from 50 to 70, with an average age of 66.
The disc appears swollen and may have flame or splinter hemorrhages (Figure 3). The cup of the involved disc is typically small. The visual loss is believed to be the result of poor perfusion in the circulation of the posterior ciliary artery, which supplies the optic nerve head.1 If the other eye also has a small cup, it is considered to be at risk of future ischemic events. In one study,14 the opposite eye became involved within the next 5 years in 14.7% of all cases. The risk of recurrent disease in the same eye is low (6.4% in another study15).
Arteritic anterior ischemic optic neuropathy is more common in patients over age 70 and is usually due to giant cell arteritis, which has a significant association with polymyalgia rheumatica. Patients may have jaw claudication, proximal myalgia and arthralgia, scalp tenderness, headache, fatigue, and a significantly elevated erythrocyte sedimentation rate and C-reactive protein level. These features should be looked for in the review of systems, although patients may not have all of them.
The funduscopic examination may reveal a pale, swollen disc, peripapillary hemorrhages, branch or central retinal artery occlusions, or cotton-wool spots.
Temporal artery biopsy is the gold standard for diagnosis, but treatment with corticosteroids should not be delayed pending biopsy or other test results.1
Thrombocytosis has been associated with a higher risk of permanent vision loss in patients with giant cell arteritis.16
Posterior ischemic optic neuropathy is the least common form of ischemic optic neuropathy. This diagnosis should be entertained in older patients who report acute, painless vision loss but have a normal funduscopic examination. Giant cell arteritis must be considered first in this setting.
Bilateral posterior ischemic optic neuropathy can occur (although rarely) in patients undergoing cardiac or spinal surgery.17 Risk factors thought to be associated with perioperative disease include anemia, hypotension, substantial blood loss during the surgery, surgeries longer than 6.5 hours, carotid atherosclerosis, and diabetes.18
There are no effective treatments for most ischemic optic neuropathies with the crucial exception of giant cell arteritis.
Neuromyelitis optica (Devic disease)
Neuromyelitis optica (Devic disease) is a combination of optic neuritis and transverse myelitis (Table 2). Clinically, the disease spares the nervous system except for the optic nerves and spinal cord. The onset of the optic neuritis may precede or follow the onset of the transverse myelitis by up to 2 to 4 years.19 Usually, the optic neuritis is bilateral and retrobulbar and results in severe vision loss, worse than that seen in patients with MS.19,20
The transverse myelitis may cause paraplegia or quadriplegia, depending on the location of the lesion in the spinal cord (cervical vs thoracic). The transverse myelitis in neuromyelitis optica is distinct from that seen in MS. In neuromyelitis optica, the transverse myelitis is longitudinally extensive, spanning more than three vertebral bodies in length. In MS, spinal cord lesions usually are more discrete and involve one or two spinal cord segments.21
Recently, serum neuromyelitis optica immunoglobulin G (IgG) antibody has been shown to be a significant biomarker of this disease. Its sensitivity ranges from approximately 60% to 70% and its specificity is greater than 90%.22 This antibody binds to aquaporin-4, an important water-channel protein in the blood-brain barrier of the central nervous system, and evidence suggests that it is involved in the pathogenesis of the disease.23
Initially, it was proposed that MRI of the brain had to be normal for neuromyelitis optica to be diagnosed.21 However, the proposed 2006 criteria allow for some abnormal T2 and fluid-attenuated inversion recovery (FLAIR) hyperintensities in the periaqueductal gray matter and diencephalon.22
The spinal fluid in neuromyelitis optica may show a pleocytosis larger than that seen in MS (> 50 white blood cells per mm3) and may have a significant neutrophilic component.21 Oligoclonal bands are not typically present.
It is still debated whether neuromyelitis optica is a separate disease from MS or a subset of it. The implications of this debate may affect its management, as discussed below.
Inflammatory optic neuropathies
Inflammatory optic neuropathies can be caused by infections (eg, syphilis, cat scratch disease) or by noninfectious conditions (eg, sarcoidosis). Lyme disease is rarely a cause of retrobulbar optic neuritis but may cause papillitis.24 West Nile virus has also been reported to cause optic neuritis.25 Lupus may cause an optic neuropathy by inflammatory or ischemic mechanisms.26
Compressive optic neuropathies
Compressive optic neuropathies may be due to mass lesions, tumors, thyroid eye disease, or other orbital processes. MRI of the brain and orbits will confirm or rule out diagnoses associated with compressive optic neuropathy.
Genetic causes
Genetic causes of optic neuropathy include the Leber and Kjer hereditary optic neuropathies.
Leber optic neuropathy involves subacute and painless vision loss, with sequential involvement of both eyes over a period of weeks to months. This disorder predominantly affects men (80%–90% of patients) and is inherited from maternal mitochondrial DNA. The three most common mutations implicated in Leber optic neuropathy (located at base pairs 11,778, 3,460, and 14,484 in the mitochondrial DNA) are involved in more than 90% of cases. The prognosis for recovery varies depending on the genotype.27 These genes encode proteins that are part of complex I of the mitochondrial respiratory chain.28 Funduscopic examination most commonly shows circumpapillary telangiectasia, although up to one-third of patients can have a normal-appearing disc initially. Central vision is affected more severely than peripheral vision.29
Kjer autosomal-dominant optic atrophy is the most common hereditary optic neuropathy. This disease primarily affects children in the first decade of life with slowly progressive loss of vision. As with other optic neuropathies, there will eventually be pallor of the optic disc, a cecocentral scotoma, and loss of color perception. The OPA1 gene located on chromosome 3q28 has been implicated in most patients with dominant optic atrophy; a test is commercially available for diagnosis.30,31
Toxic and metabolic causes
Many agents can cause optic neuropathy. Toxins strongly associated with optic neuropathy include carbon monoxide, methanol, ethylene glycol, perchloroethylene, and tobacco. Drugs linked to optic neuropathy are ethambutol (Myambutol), clioquinol (Vioform), isoniazid (Nydrazid), amiodarone (Cordarone), linezolid (Zyvox), methotrexate, sildenafil (Viagra), oxymetazoline (contained in Afrin and other nasal sprays), and infliximab (Remicade).32–37 Additionally, several chemotherapeutic agents are known to cause optic atrophy, including vincristine (Oncovin), cisplatin (Platinol), carboplatin (Paraplatin), and paclitaxel (Abraxane, Onxol).
Nutritional deficiencies are presumed to have played a significant role in the endemics of optic neuropathy that have occurred in poor countries, such as in Cuba during the 1990s.38 Most nutritional optic neuropathies appear to be exacerbated by tobacco.39
MRI ASSESSES RISK OF MS
The diagnosis of optic neuritis is clinical, based on the history and physical findings.
However, MRI of the brain and orbits with gadolinium contrast has become the cornerstone of the evaluation in patients with optic neuritis. And MRI not only helps confirm the clinical diagnosis, but it also more importantly offers very strong prognostic information about the risk of future demyelinating events and MS.
Gadolinium-enhanced fat-saturated T1-weighted MRI of the orbits is the best sequence to show the inflammation of the optic nerve in optic neuritis (fat saturation is necessary to hide the bright signal of the orbital fat tissue).
Contrast-enhanced MRI can also help differentiate optic neuritis from nonarteritic anterior ischemic optic neuropathy. MRI of the orbits with gadolinium contrast shows enhancement of the affected optic nerve in approximately 95% of cases of optic neuritis, whereas optic nerve enhancement rarely occurs in nonarteritic anterior ischemic optic neuropathy.40
Brain MRI may show other white matter lesions (either hyperintensities on T2-weighted images or enhancement of T1-weighted images postcontrast), which denote a higher risk of developing MS. In 15-year follow-up data from the ONTT, monosymptomatic patients with no white matter lesions had a 25% risk of MS (defined at the time the ONTT was conducted as a second demyelinating event), while those with one lesion or more had a 72% risk.41
An earlier, prospective study in 102 Italian patients with optic neuritis found the risk of developing MS to be about 36% at 6 years and 42% at 8 years (using the Posner diagnostic criteria). When brain MRI data were analyzed, those with one or more lesions had a 52% risk of developing MS at 8 years, whereas those with no MRI lesions did not develop MS.42
Other studies have stratified the risk of MS in patients with clinically isolated syndromes (including not only optic neuritis, but also other neurologic symptoms such as brainstem, motor, or sensory deficits). At mean follow-ups ranging from 5 to 14 years, the risk of developing MS was 8% to 24% in patients with normal findings on brain MRI compared with 56% to 88% in those with abnormal MRI findings.43,44
Optic neuritis patients with atypical white matter lesions on brain MRI may benefit from lumbar puncture to look for oligoclonal bands, to measure the IgG index and the IgG synthesis rate, and to test for myelin basic protein in the cerebrospinal fluid. Of patients with acute optic neuritis, 79% have abnormalities in their cerebrospinal fluid. Oligoclonal bands are present in 69%, and for patients with oligoclonal bands, the 5-year probability of developing MS is estimated to be 65%, compared with 10% in those without bands. If the patient has no oligoclonal bands and has normal findings on brain MRI, he or she will not have MS 5 years later.45–47
Patients with optic neuritis who have no white matter lesions on brain MRI may be further risk-stratified on the basis of their clinical findings. In the ONTT 15-year follow-up, MS did not develop in any patient who had no brain lesions on baseline MRI, no prior optic neuritis in the contralateral eye, and no prior neurologic symptoms or signs, even if the patient had severe disc swelling (eg, peripapillary hemorrhage or retinal exudates) or if vision was reduced to no light perception.41
CASE CONTINUED: FINDINGS ON MRI
Figure 4. The patient’s magnetic resonance image. Top, an axial T2 image with contrast; bottom, sagittal T1 image with contrast. The white matter lesions indicate she is at risk of developing multiple sclerosis.
Our patient undergoes MRI, which shows lesions on axial T2 and sagittal T1 imaging with contrast (Figure 4). Of note, there are significant lesions perpendicular to the corpus callosum (Dawson fingers), some of which enhance with contrast. The enhancement indicates breakdown of the blood-brain barrier and suggests that there is active inflammation in the white matter.
Patients in the ONTT were randomized to receive one of three treatments:
Oral prednisone 1 mg/kg/day for 14 days and then tapered over 4 days
Intravenous methylprednisolone (Solu-Medrol) 250 mg four times per day for 3 days followed by oral prednisone 1 mg/kg/day for 11 days and then tapered for 4 days
Oral placebo for 14 days.
The primary visual outcomes measured were visual acuity and contrast sensitivity.48
Patients who received intravenous methylprednisolone recovered their visual function faster, although the visual outcomes after 6 months were no better with methylprednisolone than with placebo or oral prednisone. Intravenous methylprednisolone also reduced the risk of MS within the first 2 years in patients with high-risk brain MRIs.
Surprisingly, patients in the oral prednisone group had a higher risk of recurrent optic neuritis in both eyes than did patients given intravenous methylprednisolone or placebo (30% at 2 years with oral prednisone vs 16% with placebo and 13% with intravenous methylprednisolone).48 At 10 years, the risk of recurrent optic neuritis was still higher in the oral prednisone group (44%) than in the intravenous methylprednisolone group (29%) (P = .03). However, the difference between the oral prednisone and placebo groups was no longer statistically significant (P = .07).49 Oral prednisone alone is therefore contraindicated in the treatment of typical unilateral demyelinating optic neuritis.
Many patients can now be treated with intravenous infusions of methylprednisolone at home for episodes of optic neuritis.
Risks vs benefits of corticosteroid therapy
When deciding whether to treat an individual patient who has optic neuritis with intravenous corticosteroids, one should consider all the benefits and risks.
Corticosteroids do not affect long-term visual outcome, although they do hasten recovery. Patients with mild vision loss (visual acuity better than 20/40), no significant visual field loss, and no enhancing lesions on brain MRI can forgo therapy with intravenous corticosteroids.
On the other hand, we strongly favor intravenous corticosteroid treatment in patients who have both acute optic neuritis and high signal abnormalities on brain MRI, since it may delay the onset of MS. In addition, patients with severe vision loss should receive intravenous corticosteroids to hasten their recovery. In the rare circumstance in which intravenous corticosteroids are not available, high-dose oral methyl-prednisolone (500 mg daily for 5 days and then tapered over 10 days) may be acceptable.50
The side effects of corticosteroids are minimal when they are given for a brief time in otherwise healthy patients. The most common side effects are mood changes, facial flushing, sleep perturbations, weight gain, and dyspepsia.48
IMMUNOGLOBUL IN: LITTLE BENEFIT
In a double-blind, randomized trial, patients were treated with intravenous immunoglobulin 0.4 g/kg or placebo on days 0, 1, 2, 30, and 60. No difference was found in the primary outcomes of contrast sensitivity, visual acuity, or color vision from 1 week up to 6 months. There was also no significant difference in MRI outcomes between the two groups. The number of relapses was similar between both groups after 6 months.51,52
PLASMA EXCHANGE: FEW DATA
Data on plasma exchange are too scarce for us to make any recommendations. In one trial in 10 patients with severe optic neuritis, 3 patients appeared to benefit from plasma exchange. All patients had already received two doses of intravenous steroids.53
IMMUNOMODULATORY THERAPY MAY PREVENT MULTIPLE SCLEROSIS IN SOME
The most important clinical decision to make in patients with optic neuritis is whether to begin immunomodulatory therapy. Patients who may benefit the most from immunomodulatory therapy are those with abnormal white matter lesions on brain MRI, as they are at higher risk of developing MS.
Collectively, data from three studies indicate that early treatment in patients with clinically isolated syndromes, such as optic neuritis, reduces the rate of MS to 35% (from 50% without treatment) and reduces the number of new active lesions on MRI by approximately 50%.54–56
In addition, the Betaferon/Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment (BENEFIT) trial57 found that at 3 years the rate of disability was 40% lower in patients who started immunomodulatory therapy (interferon beta-1b; Betaseron) early vs later. (Early treatment meant starting within 60 days of the clinically isolated syndrome; late treatment began 2 years later.) This study suggests that early treatment may reduce future disability, although these results need to be confirmed in prospective trials.
Therefore, once the diagnosis is secure, patients with optic neuritis should be referred to a neurologist with experience in treating MS to begin treatment with immunomodulatory therapy, such as glatiramer acetate (Copaxone), interferon beta-1a (Avonex, Refib), or interferon beta-1b (Betaseron).
Patients who have a normal MRI of the brain may consider deferring therapy, since they are at low risk of developing MS. These patients should undergo surveillance MRI (at least annually at first) to look for the development of white matter lesions, as the ONTT showed even this cohort has a 22% risk of developing MS.
If neuromyelitis optica is suspected (ie, in patients with severe unilateral or bilateral vision loss, recurrent optic neuritis, paraplegia, or quadriplegia), the serum neuromyelitis optic antibody can be tested, keeping in mind that 30% to 40% of patients with neuromyelitis optica will be seronegative for this antibody. Other tests supporting the diagnosis of neuromyelitis optica may include an MRI of the spine showing longitudinally extensive transverse myelitis, a polymorphonuclear pleocytosis in the cerebrospinal fluid, absent oligoclonal bands in the cerebrospinal fluid, and normal MRI of the brain (with some possible signal abnormalities in the periaqueductal gray matter and around the diencephalon).
Because neuromyelitis optica is believed to be mediated primarily by the humoral immune system, immunomodulatory therapy is not a first-line treatment. Patients with neuromyelitis optica can be treated initially with corticosteroids, intravenous immunoglobulin therapy, plasma exchange, or immunosuppressive agents such as azathioprine (Imuran), rituximab (Rituxan), or cyclophosphamide (Cytoxan). The choice of medication should be deferred to a neurologist familiar with treatment of this disorder.
The risk of MS may be lower in children than in adults. One large, retrospective study found the cumulative risk of developing MS (the study predated the McDonald criteria) was 13% at 10 years and 19% by 20 years.58 More recently, a large series from Toronto reported a higher rate of MS development in children with optic neuritis (36% at two years).59 By comparison, studies of adults with unilateral optic neuritis found a 38% to 39% risk of converting to MS at 10 years.5,41 The use of immunomodulatory therapies to reduce the risk of MS has not been well studied in children, since the prevalence is low in this age group.
The most common side effects of the beta-interferons are flulike symptoms (fatigue, myalgia), injection site reactions, and elevations of aminotransferase levels. Most patients are able to tolerate the side effects if the beta-interferon is taken with acetaminophen (Tylenol) or with over-the-counter nonsteroidal anti-inflammatory drugs.
Glatiramer acetate does not cause flulike symptoms or elevate aminotransferases, but it does require more frequent injections. Rarely, it may cause an idiosyncratic panic-like attack.
CASE CONTINUED
The best therapeutic regimen for this patient would be intravenous methylprednisolone, and subsequently a disease-modifying, immunomodulatory agent (interferon beta or glatiramer acetate). Our patient chose to start therapy with interferon beta-1a 30 μg intramuscularly once a week. She has been tolerating the medication well and has had no new neurologic or visual symptoms for the past 2 years.
References
Liu GT. Visual loss: optic neuropathies. In: Liu GT, Volpe NJ, Galetta SL, editors. Neuro–Ophthalmology: Diagnosis and Management. Philadelphia, PA: WB Saunders, 2001:103–187.
Wray SH. Optic neuritis. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Philadelphia, PA: WB Saunders, 1994:2539–2568.
Optic Neuritis Study Group. The clinical profile of optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol1991; 109:1673–1678.
Percy AK, Nobrega FT, Kurland LT. Optic neuritis and multiple sclerosis: an epidemiologic study. Arch Ophthalmol1972; 87:135–139.
Rodriguez M, Siva A, Cross SA, O’Brien PC, Kurland LT. Optic neuritis: a population–based study in Olmsted County, Minnesota. Neurology1995; 45:244–250.
Phillips PH, Newman NJ, Lynn MJ. Optic neuritis in African Americans. Arch Neurol1998; 55:186–192.
Brady KM, Brar AS, Lee AG, Coats DK, Paysse EA, Steinkuller PG. Optic neuritis in children: clinical features and visual outcome. J AAPOS1999; 3:98–103.
Kriss A, Francis DA, Cuendet B, et al. Recovery after optic neuritis in childhood. J Neurol Neurosurg Psychiatry1988; 51:1253–1258.
Beck RW. The Optic Neuritis Treatment Trial. Arch Ophthalmol1988; 106:1051–1053.
Optic Neuritis Study Group. Visual function 15 years after optic neuritis. Ophthalmology2008; 115:1079–1082.
Cox TA, Thompson HS, Corbett JJ. Relative afferent pupillary defects in optic neuritis. Am J Ophthalmol1981; 92:685–690.
Arnold AC. Ischemic optic neuropathies. Ophthalmol Clin North Am2001; 14:83–98.
Newman NJ, Scherer R, Langenberg P, et al. The fellow eye in NAION: report from the ischemic optic neuropathy decompression trial follow–up study. Am J Ophthalmol2002; 134:317–328.
Hayreh SS, Podhajsky PA, Zimmerman B. Ipsilateral recurrence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol2001; 132:734–742.
Liozon E, Herrmann F, Ly K, et al. Risk factors for visual loss in giant cell (temporal) arteritis: a prospective study of 174 patients. Am J Med2001; 111:211–217.
Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol2005; 50:15–26.
American Society of Anesthesiologists Task Force on Perioperative Blindness. Practice advisory for perioperative visual loss associated with spine surgery: a report by the American Society of Anesthesiologists Task Force on Perioperative Blindness. Anesthesiology2006; 104:1319–1328.
Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology2007; 114:810–815.
Papais-Alvarenga RM, Carellos SC, Alvarenga MP, Holander C, Bichara RP, Thuler LC. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol2008; 126:12–16.
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology1999; 53:1107–1114.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology2006; 66:1485–1489.
Takahashi T, Fujihara K, Nakashima I, et al. Anti–aquaporin–4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain2007; 130:1235–1243.
Sibony P, Halperin J, Coyle PK, Patel K. Reactive Lyme serology in optic neuritis. J Neuroophthalmol2005; 25:71–82.
Anninger WV, Lomeo MD, Dingle J, Epstein AD, Lubow M. West Nile virus–associated optic neuritis and chorioretinitis. Am J Ophthalmol2003; 136:1183–1185.
Jabs DA, Miller NR, Newman SA, Johnson MA, Stevens MB. Optic neuropathy in systemic lupus erythematosus. Arch Ophthalmol1986; 104:564–568.
Howell N. LHON and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol2003; 37:94–108.
Newman NJ. Hereditary optic neuropathies. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. Philadelphia, PA: Lippincott Williams & Wilkins, 2005;465–501.
Smith JL, Hoyt WF, Susac JO. Ocular fundus in acute Leber optic neuropathy. Arch Ophthalmol1973; 90:349–354.
Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol2003; 87:48–53.
Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin– related GTPase, is mutated in autosomal dominant atrophy linked to chromosome 3q28. Nat Genet2000; 26:211–215.
McKinley SH, Foroozan R. Optic neuropathy associated with linezolid treatment. J Neuroophthalmol2005; 25:18–21.
Melamud A, Kosmorsky GS, Lee MS. Ocular ethambutol toxicity. Mayo Clin Proc2003; 78:1409–1411.
Kerrison JB. Optic neuropathies caused by toxins and adverse drug reactions. Ophthalmol Clin North Am2004; 17:481–488.
Pomeranz HD, Bhavsar AR. Nonarteritic ischemic optic neuropathy developing soon after use of sildenafil (Viagra): a report of seven new cases. J Neuroophthalmol2005; 25:9–13.
Fivgas GD, Newman NJ. Anterior ischemic optic neuropathy following the use of a nasal decongestant. Am J Ophthalmol1999; 127:104–106.
The Cuba Neuropathy Field Investigation Team. Epidemic optic neuropathy in Cuba—clinical characterization and risk factors. N Engl J Med1995; 333:1176–1182.
Solberg Y, Rosner M, Belkin M. The association between cigarette smoking and ocular diseases. Surv Ophthalmol1998; 42:535–547.
Rizzo JF, Andreoli CM, Rabinov JD. Use of magnetic resonance imaging to differentiate optic neuritis and nonarteritic anterior ischemic optic neuropathy. Ophthalmology2002; 109:1679–1684.
The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final Optic Neuritis Treatment Trial follow-up. Arch Neurol2008; 65:727–732.
Ghezzi A, Martinelli V, Torri V, et al. Long–term follow–up of isolated optic neuritis: the risk of developing multiple sclerosis, its outcome, and the prognostic role of paraclinical tests. J Neurol1999; 246:770– 775.
Brex PA, Ciccarelli O, O'Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med2002; 346:158–164.
Tintore M, Rovira A, Rio J, et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology2006; 67:968–972.
Söderström M, Ya–Ping J, Hillert J. Optic neuritis: prognosis for multiple sclerosis from MRI, CSF, and HLA findings. Neurology1998; 50:708–714.
Frederiksen JL, Madsen HO, Ryder LP, Larsson HB, Morling N, Svejgaard A. HLA typing in acute optic neuritis: relation to multiple sclerosis and magnetic resonance imaging findings. Arch Neurol1997; 54:76–80.
Frederiksen JL, Larsson HB, Oleson J. Correlation of magnetic resonance imaging and CSF findings in patients with acute monosymptomatic optic neuritis. Acta Neurol Scand1992; 86:317–322.
Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med1992; 326:581–588.
Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol2003; 121:944–949.
Sellebjerg F, Nielsen HS, Frederiksen JL, Olesen J. A randomized, controlled trial of oral high-dose methylprednisolone in acute optic neuritis. Neurology1999; 52:1479–1484.
Noseworthy JH, O’Brien PC, Peterson TM, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology2001; 56:1514–1522.
Roed HG, Langkilde A, Sellebjerg F, et al. A double–blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology2005; 64:804–810.
Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology2004; 63:1081–1083.
CHAMPS Study Group. Interferon beta-1a for optic neuritis patients at high risk for multiple sclerosis. Am J Ophthalmol2001; 132:463– 471.
Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet2001; 357:1576–1582.
Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology2006; 67:1242–1249.
Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet2007; 370:389–397.
Lucchinetti CF, Kiers L, O’Duffy A, et al. Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology1997; 49:1413–1418.
Wilejto M, Shroff M, Buncic JR, Kennedy J, Goia C, Banwell B. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology2006; 67:258–262.
References
Liu GT. Visual loss: optic neuropathies. In: Liu GT, Volpe NJ, Galetta SL, editors. Neuro–Ophthalmology: Diagnosis and Management. Philadelphia, PA: WB Saunders, 2001:103–187.
Wray SH. Optic neuritis. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Philadelphia, PA: WB Saunders, 1994:2539–2568.
Optic Neuritis Study Group. The clinical profile of optic neuritis: experience of the Optic Neuritis Treatment Trial. Arch Ophthalmol1991; 109:1673–1678.
Percy AK, Nobrega FT, Kurland LT. Optic neuritis and multiple sclerosis: an epidemiologic study. Arch Ophthalmol1972; 87:135–139.
Rodriguez M, Siva A, Cross SA, O’Brien PC, Kurland LT. Optic neuritis: a population–based study in Olmsted County, Minnesota. Neurology1995; 45:244–250.
Phillips PH, Newman NJ, Lynn MJ. Optic neuritis in African Americans. Arch Neurol1998; 55:186–192.
Brady KM, Brar AS, Lee AG, Coats DK, Paysse EA, Steinkuller PG. Optic neuritis in children: clinical features and visual outcome. J AAPOS1999; 3:98–103.
Kriss A, Francis DA, Cuendet B, et al. Recovery after optic neuritis in childhood. J Neurol Neurosurg Psychiatry1988; 51:1253–1258.
Beck RW. The Optic Neuritis Treatment Trial. Arch Ophthalmol1988; 106:1051–1053.
Optic Neuritis Study Group. Visual function 15 years after optic neuritis. Ophthalmology2008; 115:1079–1082.
Cox TA, Thompson HS, Corbett JJ. Relative afferent pupillary defects in optic neuritis. Am J Ophthalmol1981; 92:685–690.
Arnold AC. Ischemic optic neuropathies. Ophthalmol Clin North Am2001; 14:83–98.
Newman NJ, Scherer R, Langenberg P, et al. The fellow eye in NAION: report from the ischemic optic neuropathy decompression trial follow–up study. Am J Ophthalmol2002; 134:317–328.
Hayreh SS, Podhajsky PA, Zimmerman B. Ipsilateral recurrence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol2001; 132:734–742.
Liozon E, Herrmann F, Ly K, et al. Risk factors for visual loss in giant cell (temporal) arteritis: a prospective study of 174 patients. Am J Med2001; 111:211–217.
Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol2005; 50:15–26.
American Society of Anesthesiologists Task Force on Perioperative Blindness. Practice advisory for perioperative visual loss associated with spine surgery: a report by the American Society of Anesthesiologists Task Force on Perioperative Blindness. Anesthesiology2006; 104:1319–1328.
Merle H, Olindo S, Bonnan M, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology2007; 114:810–815.
Papais-Alvarenga RM, Carellos SC, Alvarenga MP, Holander C, Bichara RP, Thuler LC. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol2008; 126:12–16.
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology1999; 53:1107–1114.
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology2006; 66:1485–1489.
Takahashi T, Fujihara K, Nakashima I, et al. Anti–aquaporin–4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain2007; 130:1235–1243.
Sibony P, Halperin J, Coyle PK, Patel K. Reactive Lyme serology in optic neuritis. J Neuroophthalmol2005; 25:71–82.
Anninger WV, Lomeo MD, Dingle J, Epstein AD, Lubow M. West Nile virus–associated optic neuritis and chorioretinitis. Am J Ophthalmol2003; 136:1183–1185.
Jabs DA, Miller NR, Newman SA, Johnson MA, Stevens MB. Optic neuropathy in systemic lupus erythematosus. Arch Ophthalmol1986; 104:564–568.
Howell N. LHON and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol2003; 37:94–108.
Newman NJ. Hereditary optic neuropathies. In: Miller NR, Newman NJ, editors. Walsh and Hoyt’s Clinical Neuro-ophthalmology. Philadelphia, PA: Lippincott Williams & Wilkins, 2005;465–501.
Smith JL, Hoyt WF, Susac JO. Ocular fundus in acute Leber optic neuropathy. Arch Ophthalmol1973; 90:349–354.
Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol2003; 87:48–53.
Alexander C, Votruba M, Pesch UE, et al. OPA1, encoding a dynamin– related GTPase, is mutated in autosomal dominant atrophy linked to chromosome 3q28. Nat Genet2000; 26:211–215.
McKinley SH, Foroozan R. Optic neuropathy associated with linezolid treatment. J Neuroophthalmol2005; 25:18–21.
Melamud A, Kosmorsky GS, Lee MS. Ocular ethambutol toxicity. Mayo Clin Proc2003; 78:1409–1411.
Kerrison JB. Optic neuropathies caused by toxins and adverse drug reactions. Ophthalmol Clin North Am2004; 17:481–488.
Pomeranz HD, Bhavsar AR. Nonarteritic ischemic optic neuropathy developing soon after use of sildenafil (Viagra): a report of seven new cases. J Neuroophthalmol2005; 25:9–13.
Fivgas GD, Newman NJ. Anterior ischemic optic neuropathy following the use of a nasal decongestant. Am J Ophthalmol1999; 127:104–106.
The Cuba Neuropathy Field Investigation Team. Epidemic optic neuropathy in Cuba—clinical characterization and risk factors. N Engl J Med1995; 333:1176–1182.
Solberg Y, Rosner M, Belkin M. The association between cigarette smoking and ocular diseases. Surv Ophthalmol1998; 42:535–547.
Rizzo JF, Andreoli CM, Rabinov JD. Use of magnetic resonance imaging to differentiate optic neuritis and nonarteritic anterior ischemic optic neuropathy. Ophthalmology2002; 109:1679–1684.
The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final Optic Neuritis Treatment Trial follow-up. Arch Neurol2008; 65:727–732.
Ghezzi A, Martinelli V, Torri V, et al. Long–term follow–up of isolated optic neuritis: the risk of developing multiple sclerosis, its outcome, and the prognostic role of paraclinical tests. J Neurol1999; 246:770– 775.
Brex PA, Ciccarelli O, O'Riordan JI, Sailer M, Thompson AJ, Miller DH. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med2002; 346:158–164.
Tintore M, Rovira A, Rio J, et al. Baseline MRI predicts future attacks and disability in clinically isolated syndromes. Neurology2006; 67:968–972.
Söderström M, Ya–Ping J, Hillert J. Optic neuritis: prognosis for multiple sclerosis from MRI, CSF, and HLA findings. Neurology1998; 50:708–714.
Frederiksen JL, Madsen HO, Ryder LP, Larsson HB, Morling N, Svejgaard A. HLA typing in acute optic neuritis: relation to multiple sclerosis and magnetic resonance imaging findings. Arch Neurol1997; 54:76–80.
Frederiksen JL, Larsson HB, Oleson J. Correlation of magnetic resonance imaging and CSF findings in patients with acute monosymptomatic optic neuritis. Acta Neurol Scand1992; 86:317–322.
Beck RW, Cleary PA, Anderson MM, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med1992; 326:581–588.
Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol2003; 121:944–949.
Sellebjerg F, Nielsen HS, Frederiksen JL, Olesen J. A randomized, controlled trial of oral high-dose methylprednisolone in acute optic neuritis. Neurology1999; 52:1479–1484.
Noseworthy JH, O’Brien PC, Peterson TM, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology2001; 56:1514–1522.
Roed HG, Langkilde A, Sellebjerg F, et al. A double–blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology2005; 64:804–810.
Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology2004; 63:1081–1083.
CHAMPS Study Group. Interferon beta-1a for optic neuritis patients at high risk for multiple sclerosis. Am J Ophthalmol2001; 132:463– 471.
Comi G, Filippi M, Barkhof F, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet2001; 357:1576–1582.
Kappos L, Polman CH, Freedman MS, et al. Treatment with interferon beta-1b delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology2006; 67:1242–1249.
Kappos L, Freedman MS, Polman CH, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet2007; 370:389–397.
Lucchinetti CF, Kiers L, O’Duffy A, et al. Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology1997; 49:1413–1418.
Wilejto M, Shroff M, Buncic JR, Kennedy J, Goia C, Banwell B. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology2006; 67:258–262.
Optic neuritis is most common in women in their 20s and 30s, whereas ischemic optic neuropathy, which is more common, primarily affects older people.
The diagnosis of optic neuritis is primarily clinical, but magnetic resonance imaging confirms the diagnosis and, more importantly, assesses the risk of MS.
Intravenous methylprednisolone (Solu-Medrol) does not affect the long-term visual outcome, but it speeds visual recovery and reduces the risk of MS. Surprisingly, oral prednisone seems to increase the risk of recurrent optic neuritis and is therefore contraindicated.
Early treatment with interferon beta reduces the risk of MS and should be considered in patients at high risk.