User login
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
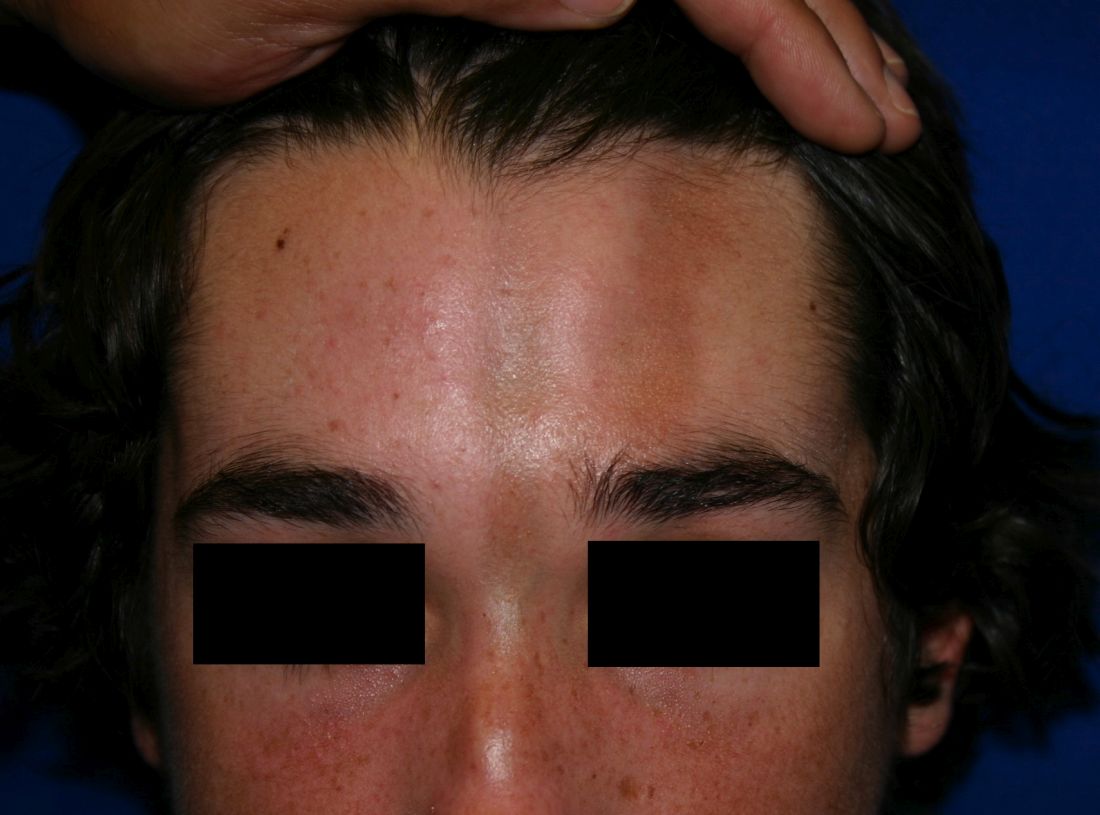
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9

Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.

Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9
Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
Morphea, also known as localized scleroderma, is a rare fibrosing disorder of the skin and the underlying tissue that encompasses a variety of distinct subtypes classified by pattern and depth of lesion involvement. It may involve fat, fascia, muscle, and bone, and rarely, the central nervous system. Morphea is easily differentiated from systemic sclerosis by its primarily cutaneous involvement, although a minority of patients may have associated extracutaneous findings. Systemic sclerosis describes a well-defined disorder of skin sclerosis with a specific pattern of internal organ involvement.
Classification of the different subtypes of morphea are based on clinical presentation of the lesions. The most widely used system characterizes morphea into linear, circumscribed, generalized, pansclerotic, and mixed morphea subtypes.1 Mixed morphea describes the presence of two or more patterns of disease and affects 15% of patients. Morphea lesions initially present as erythematous to violaceous patches and plaques that eventually become white and sclerotic, with resulting destruction of the surrounding structures.
Linear scleroderma is the most common subtype of morphea in children and adolescents, affecting 42%-67% of children with morphea.1 It is characterized by linear plaques, often on the extremities, face, or scalp, that tend to follow Blaschko lines.4 These lesions may extend past the dermis to the subcutaneous tissue, muscle, and even bone, resulting in significant deformities. When on the scalp or face, particularly the forehead, the linear lesion may be referred to as the en coup de sabre variant. Ocular and CNS involvement should be of concern in these patients. When subcutaneous atrophy on the unilateral face is present with unaffected overlaying skin, this is known as the Parry-Romberg syndrome or progressive hemifacial atrophy. Involvement of the extremities is common, and unfortunately, may lead to muscle atrophy of the affected limb, contractures in areas overlying joint spaces, and occasionally limb length discrepancies.
Circumscribed morphea describes three or fewer discrete, oval, or round indurated plaques, with central whitening and a violaceous periphery. They generally are found on the trunk. When lesions have deeper involvement, delving past the dermis to involve the underlying fascia and muscle, the patient may experience a “bound down” sensation. Most lesions soften over 3-5 years.
Generalized morphea is used to describe the presence of at least four plaques, larger than 3 cm, that become confluent in at least two different locations on the body. Patients with generalized morphea have higher rates of systemic symptoms such as arthritis and fatigue.
Pansclerotic morphea, the most severe subtype, is characterized by significant body surface area involvement coupled with deep depth of involvement, often to the bone. The widespread blistering associated with pansclerotic morphea may lead to chronic ulceration and, later on, a higher risk of squamous cell carcinoma development. Despite its extensive distribution, pansclerotic morphea does not cause the severe organ and vascular fibrosis that is characteristically seen in systemic sclerosis. Raynaud’s phenomenon, abnormal nailfold capillaries, and sclerodactyly also will be absent in pansclerotic morphea.
Extracutaneous findings are present in up to 22% of patients with morphea.5 Arthritis is the most common associated finding, and often is correlated with a positive rheumatoid factor. Neurologic involvement most frequently is seen in patients with facial morphea and may present as seizures, as in this patient. MRI abnormalities such as calcifications and white matter changes may be seen. Other common extracutaneous features include fatigue, vascular abnormalities, and ocular findings, such as uveitis.
Morphea and systemic sclerosis appear similar on histology. In early morphea, lymphocytic perivascular infiltrates may be seen in the reticular dermis. In late morphea, the inflammatory cells are replaced by an abundance of collagen bundles infiltrating the dermis.
although the instigating factor activating this pathway is unknown. Multiple factors have been associated with the development of morphea, including autoimmunity, trauma, Borrelia and cytomegalovirus infections, radiation, and certain medications in case reports. Patients with morphea have higher rates of concomitant autoimmune diseases than that found in the general population6 and also have higher rates of autoantibody positivity. In a 750-patient, multicenter study of children with morphea, 42% of patients had positive antinuclear antibodies.7
Diagnosis
Morphea is diagnosed clinically, based on the characteristic appearance of the lesions. A biopsy may be helpful if the presentation is atypical. Although patients with morphea have higher rates of autoantibody positivity, there are no specific laboratory tests that consistently or reliably offer diagnostic value.8 Imaging modalities such as MRI may be utilized to view depth of involvement. Other noninvasive measures, such as thermography and ultrasonography, may be used to determine disease activity.9
Treatment
Treatment for morphea often is multidisciplinary and depends on the severity of involvement and extent to which it impedes functionality and quality of life. Localized plaques that do not restrict movement may be treated with topical corticosteroids, calcipotriene, and tacrolimus. However, topical corticosteroids should be discontinued if there are no signs of improvement in 2-3 months.
For patients with deforming or functionally significant disease, systemic treatment is advised. Methotrexate with or without systemic corticosteroids has been frequently studied, and is the most commonly recommended systemic therapy.11 Some experts have recommended treatment for at least 2-3 years, with at least 1 year of disease inactivity, before discontinuing treatment. Despite this duration of treatment, up to one-quarter of patients, especially those with linear morphea, will still experience recurrence of disease. Management of morphea may be aided by rheumatology and/or dermatology consultation.
Ms. Han is a medical student at the University of California, San Diego. Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children’s Hospital–San Diego. He is vice chair of the department of dermatology and a professor of dermatology and pediatrics at the University of California, San Diego. Ms. Han and Dr. Eichenfield had no conflicts of interest or financial disclosures.
References
1. Fett N et al. J Am Acad Dermatol. 2011 Feb;64(2):217-28.
2. Condie D et al. Arthritis Rheumatol. 2014 Dec;66(12):3496-504.
3. Zulian F et al. J Pediatr. 2006 Aug;149(2):248-51.
4. Weibel L et al. Br J Dermatol. 2008 Jul;159(1):175-81.
5. Zulian F et al. Arthritis Rheum. 2005 Sep;52(9):2873-81.
6. Leitenberger JJ et al. Arch Dermatol. 2009 May;145(5):545-50.
7. Zulian F et al. Rheumatology (Oxford). 2006 May;45(5):614-20.
8. Dharamsi JW et al. JAMA Dermatol. 2013 Oct;149(10):1159-65.
9. Zulian F et al. Curr Opin Rheumatol. 2013 Sep;25(5):643-50.
10. Pope E et al. Pediatr Clin North Am. 2014 Apr;61(2):309-19.
11. Strickland N et al. Am Acad Dermatol. 2015 Apr; 72(4): 727-8.
12. Schoch JJ et al. Pediatr Dermatol. 2018. 35(1): 43-6.
A 14-year-old patient presents to a dermatology clinic for a depression on his forehead, which has been there for about 2 years. A few years ago, he used to have a pruritic pink lesion on the forehead where the depression is now. He denies any symptoms.