User login
Vaccines are marvelous, and there are many well documented success stories, including rotavirus (RV) vaccines, where a live vaccine is administered to the gastrointestinal mucosa via oral drops. Antigens presented at the mucosal/epithelial surface not only induce systemic serum IgG – as do injectable vaccines – but also induce secretory IgA (sIgA), which is most helpful in diseases that directly affect the mucosa.

Mucosal vs. systemic immunity
Antibody being present on mucosal surfaces (point of initial pathogen contact) has a chance to neutralize the pathogen before it gains a foothold. Pathogen-specific mucosal lymphoid elements (e.g. in Peyer’s patches in the gut) also appear critical for optimal protection.1 The presence of both mucosal immune elements means that infection is severely limited or at times entirely prevented. So virus entering the GI tract causes minimal to no gut lining injury. Hence, there is no or mostly reduced vomiting/diarrhea. A downside of mucosally-administered live vaccines is that preexisting antibody to the vaccine antigens can reduce or block vaccine virus replication in the vaccinee, blunting or preventing protection. Note: Preexisting antibody also affects injectable live vaccines, such as the measles vaccine, similarly.
Classic injectable live or nonlive vaccines provide their most potent protection via systemic cellular responses antibody and/or antibodies in serum and extracellular fluid (ECF) where IgG and IgM are in highest concentrations. So even successful injectable vaccines still allow mucosal infection to start but then intercept further spread and prevent most of the downstream damage (think pertussis) or neutralize an infection-generated toxin (pertussis or tetanus). It usually is only after infection-induced damage occurs that systemic IgG and IgM gain better access to respiratory epithelial surfaces, but still only at a fraction of circulating concentrations. Indeed, pertussis vaccine–induced systemic immunity allows the pathogen to attack and replicate in/on host surface cells, causing toxin release and variable amounts of local mucosal injury/inflammation before vaccine-induced systemic immunity gains adequate access to the pathogen and/or to its toxin which may enter systemic circulation.
Live attenuated influenza vaccine (LAIV) induces mucosal immunity
Another “standard” vaccine that induces mucosal immunity – LAIV – was developed to improve on protection afforded by injectable influenza vaccines (IIVs), but LAIV has had hiccups in the United States. One example is several years of negligible protection against H1N1 disease. As long as LAIV’s vaccine strain had reasonably matched the circulating strains, LAIV worked at least as well as injectable influenza vaccine, and even offered some cross-protection against mildly mismatched strains. But after a number of years of LAIV use, vaccine effectiveness in the United States vs. H1N1 strains appeared to fade due to previously undetected but significant changes in the circulating H1N1 strain. The lesson is that mucosal immunity’s advantages are lost if too much change occurs in the pathogen target for sIgA and mucosally-associated lymphoid tissue cells (MALT)).
Other vaccines likely need to induce mucosal immunity
Protection at the mucosal level will likely be needed for success against norovirus, parainfluenza, respiratory syncytial virus (RSV), Neisseria gonorrhea, and chlamydia. Another helpful aspect of mucosal immunity is that immune cells and sIgA not only reside on the mucosa where the antigen was originally presented, but there is also a reasonable chance that these components will traffic to other mucosal surfaces.2 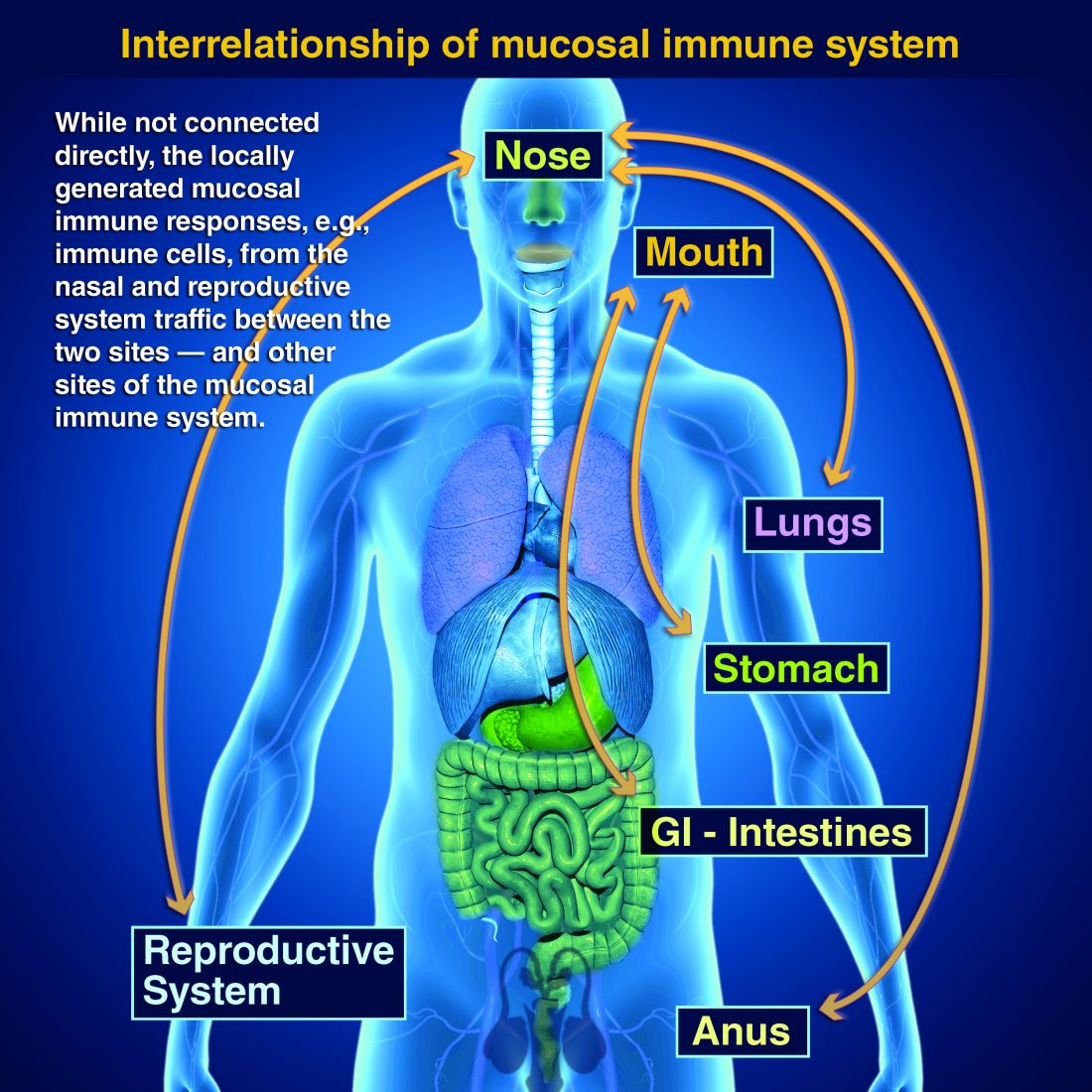
So intranasal vaccine could be expected to protect distant mucosal surfaces (urogenital, GI, and respiratory), leading to vaccine-induced systemic antibody plus mucosal immunity (sIGA and MALT responses) at each site.
Let’s look at a novel “two-site” chlamydia vaccine
Recently a phase 1 chlamydia vaccine that used a novel two-pronged administration site/schedule was successful at inducing both mucosal and systemic immunity in a proof-of-concept study – achieving the best of both worlds.3 This may be a template for vaccines in years to come. British investigators studied 50 healthy women aged 19-45 years in a double-blind, parallel, randomized, placebo-controlled trial that used a recombinant chlamydia protein subunit antigen (CTH522). The vaccine schedule involved three injectable priming doses followed soon thereafter by two intranasal boosting doses. There were three groups:
1. CTH522 adjuvanted with CAF01 liposomes (CTH522:CAF01).
2. CTH522 adjuvanted with aluminum hydroxide (CTH522:AH).
3. Placebo (saline).
The intramuscular (IM) priming schedule was 0, 1, and 4 months. The intranasal vaccine booster doses or placebo were given at 4.5 and 5 months. No related serious adverse reactions occurred. For injectable dosing, the most frequent adverse event was mild local injection-site reactions in all subjects in both vaccine groups vs. in 60% of placebo recipients (P = .053). The adjuvants were the likely cause for local reactions. Intranasal doses had local reactions in 47% of both vaccine groups and 60% of placebo recipients; P = 1.000).
Both vaccines produced systemic IgG seroconversion (including neutralizing antibody) plus small amounts of IgG in the nasal cavity and genital tract in all vaccine recipients; no placebo recipient seroconverted. Interestingly, liposomally-adjuvanted vaccine produced a more rapid systemic IgG response and higher serum titers than the alum-adjuvanted vaccine. Likewise, the IM liposomal vaccine also induced higher but still small mucosal IgG antibody responses (P = .0091). Intranasal IM-induced IgG titers were not boosted by later intranasal vaccine dosing.
Subjects getting liposomal vaccine (but not alum vaccine or placebo) boosters had detectable sIgA titers in both nasal and genital tract secretions. Liposomal vaccine recipients also had fivefold to sixfold higher median titers than alum vaccine recipients after the priming dose, and these higher titers persisted to the end of the study. All liposomal vaccine recipients developed antichlamydial cell-mediated responses vs. 57% alum-adjuvanted vaccine recipients. (P = .01). So both use of two-site dosing and the liposomal adjuvant appeared critical to better responses.

In summary
While this candidate vaccine has hurdles to overcome before coming into routine use, the proof-of-principle that a combination injectable-intranasal vaccine schedule can induce robust systemic and mucosal immunity when given with an appropriate adjuvant is very promising. Adding more vaccines to the schedule then becomes an issue, but that is one of those “good” problems we can deal with later.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines, receives funding from GlaxoSmithKline for studies on pneumococcal and rotavirus vaccines, and from Pfizer for a study on pneumococcal vaccine on which Dr. Harrison is a sub-investigator. The hospital also receives Centers for Disease Control and Prevention funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus, and also for rotavirus. Email Dr. Harrison at [email protected].
References
1. PLOS Biology. 2012 Sep 1. doi: 10.1371/journal.pbio.1001397.
2. Mucosal Immunity in the Human Female Reproductive Tract in “Mucosal Immunology,” 4th ed., Volume 2 (Cambridge, MA: Academic Press, 2015, pp. 2097-124).
3. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30279-8.
Vaccines are marvelous, and there are many well documented success stories, including rotavirus (RV) vaccines, where a live vaccine is administered to the gastrointestinal mucosa via oral drops. Antigens presented at the mucosal/epithelial surface not only induce systemic serum IgG – as do injectable vaccines – but also induce secretory IgA (sIgA), which is most helpful in diseases that directly affect the mucosa.
Mucosal vs. systemic immunity
Antibody being present on mucosal surfaces (point of initial pathogen contact) has a chance to neutralize the pathogen before it gains a foothold. Pathogen-specific mucosal lymphoid elements (e.g. in Peyer’s patches in the gut) also appear critical for optimal protection.1 The presence of both mucosal immune elements means that infection is severely limited or at times entirely prevented. So virus entering the GI tract causes minimal to no gut lining injury. Hence, there is no or mostly reduced vomiting/diarrhea. A downside of mucosally-administered live vaccines is that preexisting antibody to the vaccine antigens can reduce or block vaccine virus replication in the vaccinee, blunting or preventing protection. Note: Preexisting antibody also affects injectable live vaccines, such as the measles vaccine, similarly.
Classic injectable live or nonlive vaccines provide their most potent protection via systemic cellular responses antibody and/or antibodies in serum and extracellular fluid (ECF) where IgG and IgM are in highest concentrations. So even successful injectable vaccines still allow mucosal infection to start but then intercept further spread and prevent most of the downstream damage (think pertussis) or neutralize an infection-generated toxin (pertussis or tetanus). It usually is only after infection-induced damage occurs that systemic IgG and IgM gain better access to respiratory epithelial surfaces, but still only at a fraction of circulating concentrations. Indeed, pertussis vaccine–induced systemic immunity allows the pathogen to attack and replicate in/on host surface cells, causing toxin release and variable amounts of local mucosal injury/inflammation before vaccine-induced systemic immunity gains adequate access to the pathogen and/or to its toxin which may enter systemic circulation.
Live attenuated influenza vaccine (LAIV) induces mucosal immunity
Another “standard” vaccine that induces mucosal immunity – LAIV – was developed to improve on protection afforded by injectable influenza vaccines (IIVs), but LAIV has had hiccups in the United States. One example is several years of negligible protection against H1N1 disease. As long as LAIV’s vaccine strain had reasonably matched the circulating strains, LAIV worked at least as well as injectable influenza vaccine, and even offered some cross-protection against mildly mismatched strains. But after a number of years of LAIV use, vaccine effectiveness in the United States vs. H1N1 strains appeared to fade due to previously undetected but significant changes in the circulating H1N1 strain. The lesson is that mucosal immunity’s advantages are lost if too much change occurs in the pathogen target for sIgA and mucosally-associated lymphoid tissue cells (MALT)).
Other vaccines likely need to induce mucosal immunity
Protection at the mucosal level will likely be needed for success against norovirus, parainfluenza, respiratory syncytial virus (RSV), Neisseria gonorrhea, and chlamydia. Another helpful aspect of mucosal immunity is that immune cells and sIgA not only reside on the mucosa where the antigen was originally presented, but there is also a reasonable chance that these components will traffic to other mucosal surfaces.2
So intranasal vaccine could be expected to protect distant mucosal surfaces (urogenital, GI, and respiratory), leading to vaccine-induced systemic antibody plus mucosal immunity (sIGA and MALT responses) at each site.
Let’s look at a novel “two-site” chlamydia vaccine
Recently a phase 1 chlamydia vaccine that used a novel two-pronged administration site/schedule was successful at inducing both mucosal and systemic immunity in a proof-of-concept study – achieving the best of both worlds.3 This may be a template for vaccines in years to come. British investigators studied 50 healthy women aged 19-45 years in a double-blind, parallel, randomized, placebo-controlled trial that used a recombinant chlamydia protein subunit antigen (CTH522). The vaccine schedule involved three injectable priming doses followed soon thereafter by two intranasal boosting doses. There were three groups:
1. CTH522 adjuvanted with CAF01 liposomes (CTH522:CAF01).
2. CTH522 adjuvanted with aluminum hydroxide (CTH522:AH).
3. Placebo (saline).
The intramuscular (IM) priming schedule was 0, 1, and 4 months. The intranasal vaccine booster doses or placebo were given at 4.5 and 5 months. No related serious adverse reactions occurred. For injectable dosing, the most frequent adverse event was mild local injection-site reactions in all subjects in both vaccine groups vs. in 60% of placebo recipients (P = .053). The adjuvants were the likely cause for local reactions. Intranasal doses had local reactions in 47% of both vaccine groups and 60% of placebo recipients; P = 1.000).
Both vaccines produced systemic IgG seroconversion (including neutralizing antibody) plus small amounts of IgG in the nasal cavity and genital tract in all vaccine recipients; no placebo recipient seroconverted. Interestingly, liposomally-adjuvanted vaccine produced a more rapid systemic IgG response and higher serum titers than the alum-adjuvanted vaccine. Likewise, the IM liposomal vaccine also induced higher but still small mucosal IgG antibody responses (P = .0091). Intranasal IM-induced IgG titers were not boosted by later intranasal vaccine dosing.
Subjects getting liposomal vaccine (but not alum vaccine or placebo) boosters had detectable sIgA titers in both nasal and genital tract secretions. Liposomal vaccine recipients also had fivefold to sixfold higher median titers than alum vaccine recipients after the priming dose, and these higher titers persisted to the end of the study. All liposomal vaccine recipients developed antichlamydial cell-mediated responses vs. 57% alum-adjuvanted vaccine recipients. (P = .01). So both use of two-site dosing and the liposomal adjuvant appeared critical to better responses.
In summary
While this candidate vaccine has hurdles to overcome before coming into routine use, the proof-of-principle that a combination injectable-intranasal vaccine schedule can induce robust systemic and mucosal immunity when given with an appropriate adjuvant is very promising. Adding more vaccines to the schedule then becomes an issue, but that is one of those “good” problems we can deal with later.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines, receives funding from GlaxoSmithKline for studies on pneumococcal and rotavirus vaccines, and from Pfizer for a study on pneumococcal vaccine on which Dr. Harrison is a sub-investigator. The hospital also receives Centers for Disease Control and Prevention funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus, and also for rotavirus. Email Dr. Harrison at [email protected].
References
1. PLOS Biology. 2012 Sep 1. doi: 10.1371/journal.pbio.1001397.
2. Mucosal Immunity in the Human Female Reproductive Tract in “Mucosal Immunology,” 4th ed., Volume 2 (Cambridge, MA: Academic Press, 2015, pp. 2097-124).
3. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30279-8.
Vaccines are marvelous, and there are many well documented success stories, including rotavirus (RV) vaccines, where a live vaccine is administered to the gastrointestinal mucosa via oral drops. Antigens presented at the mucosal/epithelial surface not only induce systemic serum IgG – as do injectable vaccines – but also induce secretory IgA (sIgA), which is most helpful in diseases that directly affect the mucosa.
Mucosal vs. systemic immunity
Antibody being present on mucosal surfaces (point of initial pathogen contact) has a chance to neutralize the pathogen before it gains a foothold. Pathogen-specific mucosal lymphoid elements (e.g. in Peyer’s patches in the gut) also appear critical for optimal protection.1 The presence of both mucosal immune elements means that infection is severely limited or at times entirely prevented. So virus entering the GI tract causes minimal to no gut lining injury. Hence, there is no or mostly reduced vomiting/diarrhea. A downside of mucosally-administered live vaccines is that preexisting antibody to the vaccine antigens can reduce or block vaccine virus replication in the vaccinee, blunting or preventing protection. Note: Preexisting antibody also affects injectable live vaccines, such as the measles vaccine, similarly.
Classic injectable live or nonlive vaccines provide their most potent protection via systemic cellular responses antibody and/or antibodies in serum and extracellular fluid (ECF) where IgG and IgM are in highest concentrations. So even successful injectable vaccines still allow mucosal infection to start but then intercept further spread and prevent most of the downstream damage (think pertussis) or neutralize an infection-generated toxin (pertussis or tetanus). It usually is only after infection-induced damage occurs that systemic IgG and IgM gain better access to respiratory epithelial surfaces, but still only at a fraction of circulating concentrations. Indeed, pertussis vaccine–induced systemic immunity allows the pathogen to attack and replicate in/on host surface cells, causing toxin release and variable amounts of local mucosal injury/inflammation before vaccine-induced systemic immunity gains adequate access to the pathogen and/or to its toxin which may enter systemic circulation.
Live attenuated influenza vaccine (LAIV) induces mucosal immunity
Another “standard” vaccine that induces mucosal immunity – LAIV – was developed to improve on protection afforded by injectable influenza vaccines (IIVs), but LAIV has had hiccups in the United States. One example is several years of negligible protection against H1N1 disease. As long as LAIV’s vaccine strain had reasonably matched the circulating strains, LAIV worked at least as well as injectable influenza vaccine, and even offered some cross-protection against mildly mismatched strains. But after a number of years of LAIV use, vaccine effectiveness in the United States vs. H1N1 strains appeared to fade due to previously undetected but significant changes in the circulating H1N1 strain. The lesson is that mucosal immunity’s advantages are lost if too much change occurs in the pathogen target for sIgA and mucosally-associated lymphoid tissue cells (MALT)).
Other vaccines likely need to induce mucosal immunity
Protection at the mucosal level will likely be needed for success against norovirus, parainfluenza, respiratory syncytial virus (RSV), Neisseria gonorrhea, and chlamydia. Another helpful aspect of mucosal immunity is that immune cells and sIgA not only reside on the mucosa where the antigen was originally presented, but there is also a reasonable chance that these components will traffic to other mucosal surfaces.2
So intranasal vaccine could be expected to protect distant mucosal surfaces (urogenital, GI, and respiratory), leading to vaccine-induced systemic antibody plus mucosal immunity (sIGA and MALT responses) at each site.
Let’s look at a novel “two-site” chlamydia vaccine
Recently a phase 1 chlamydia vaccine that used a novel two-pronged administration site/schedule was successful at inducing both mucosal and systemic immunity in a proof-of-concept study – achieving the best of both worlds.3 This may be a template for vaccines in years to come. British investigators studied 50 healthy women aged 19-45 years in a double-blind, parallel, randomized, placebo-controlled trial that used a recombinant chlamydia protein subunit antigen (CTH522). The vaccine schedule involved three injectable priming doses followed soon thereafter by two intranasal boosting doses. There were three groups:
1. CTH522 adjuvanted with CAF01 liposomes (CTH522:CAF01).
2. CTH522 adjuvanted with aluminum hydroxide (CTH522:AH).
3. Placebo (saline).
The intramuscular (IM) priming schedule was 0, 1, and 4 months. The intranasal vaccine booster doses or placebo were given at 4.5 and 5 months. No related serious adverse reactions occurred. For injectable dosing, the most frequent adverse event was mild local injection-site reactions in all subjects in both vaccine groups vs. in 60% of placebo recipients (P = .053). The adjuvants were the likely cause for local reactions. Intranasal doses had local reactions in 47% of both vaccine groups and 60% of placebo recipients; P = 1.000).
Both vaccines produced systemic IgG seroconversion (including neutralizing antibody) plus small amounts of IgG in the nasal cavity and genital tract in all vaccine recipients; no placebo recipient seroconverted. Interestingly, liposomally-adjuvanted vaccine produced a more rapid systemic IgG response and higher serum titers than the alum-adjuvanted vaccine. Likewise, the IM liposomal vaccine also induced higher but still small mucosal IgG antibody responses (P = .0091). Intranasal IM-induced IgG titers were not boosted by later intranasal vaccine dosing.
Subjects getting liposomal vaccine (but not alum vaccine or placebo) boosters had detectable sIgA titers in both nasal and genital tract secretions. Liposomal vaccine recipients also had fivefold to sixfold higher median titers than alum vaccine recipients after the priming dose, and these higher titers persisted to the end of the study. All liposomal vaccine recipients developed antichlamydial cell-mediated responses vs. 57% alum-adjuvanted vaccine recipients. (P = .01). So both use of two-site dosing and the liposomal adjuvant appeared critical to better responses.
In summary
While this candidate vaccine has hurdles to overcome before coming into routine use, the proof-of-principle that a combination injectable-intranasal vaccine schedule can induce robust systemic and mucosal immunity when given with an appropriate adjuvant is very promising. Adding more vaccines to the schedule then becomes an issue, but that is one of those “good” problems we can deal with later.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospital-Kansas City, Mo. Children’s Mercy Hospital receives grant funding to study two candidate RSV vaccines, receives funding from GlaxoSmithKline for studies on pneumococcal and rotavirus vaccines, and from Pfizer for a study on pneumococcal vaccine on which Dr. Harrison is a sub-investigator. The hospital also receives Centers for Disease Control and Prevention funding under the New Vaccine Surveillance Network for multicenter surveillance of acute respiratory infections, including influenza, RSV, and parainfluenza virus, and also for rotavirus. Email Dr. Harrison at [email protected].
References
1. PLOS Biology. 2012 Sep 1. doi: 10.1371/journal.pbio.1001397.
2. Mucosal Immunity in the Human Female Reproductive Tract in “Mucosal Immunology,” 4th ed., Volume 2 (Cambridge, MA: Academic Press, 2015, pp. 2097-124).
3. Lancet Infect Dis. 2019. doi: 10.1016/S1473-3099(19)30279-8.