User login
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
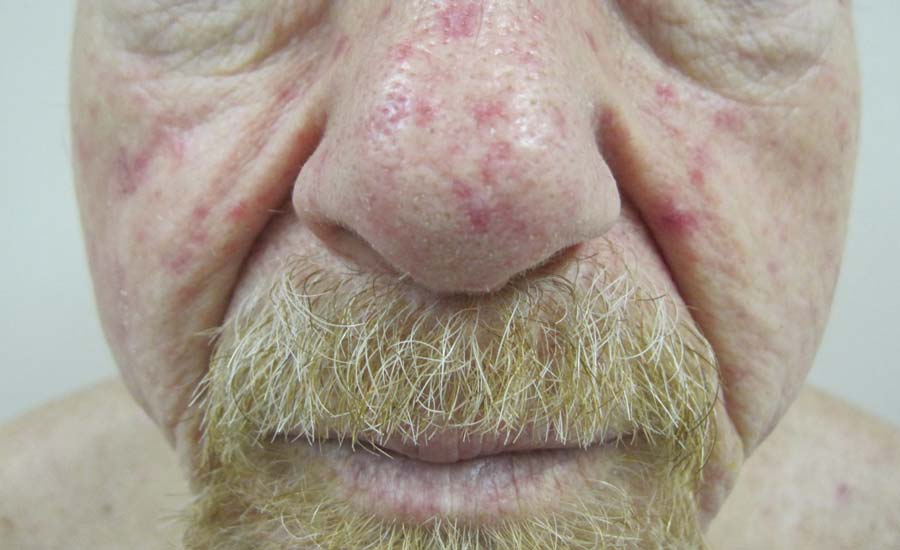
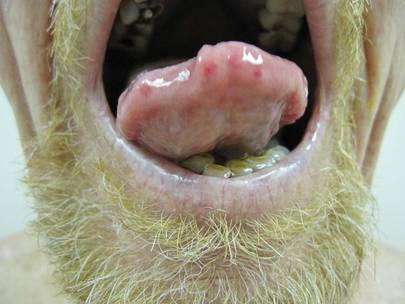
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
The Diagnosis: Hereditary Hemorrhagic Telangiectasia
Physical examination of our patient revealed multiple fine punctate telangiectases on the bilateral cheeks and the dorsum of the nose. Further examination revealed matlike telangiectases on the distal aspect of the tongue (Figure), buccal mucosa, palms, and fingers. Since diagnosis he has experienced several episodes of severe gastrointestinal tract bleeding. He underwent an esophagogastroduodenoscopy and was found to have bleeding gastric antral vascular ectasia that was treated with argon plasma coagulation. One year later he had a second esophagogastroduodenoscopy performed because of heme-positive stools and was found to have additional bleeding vascular ectasia that was treated again with argon plasma coagulation.
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is an autosomal-dominant disorder that is characterized by mucocutaneous and visceral telangiectases, recurrent hemorrhages, and a familial occurrence.1 In North America, the overall incidence is approximately 1 per 5000 to 10,000 individuals per year.2 Although this disorder generally has an autosomal-dominant transmission, approximately 20% of cases do not have a familial component.2
Clinically these patients most commonly present with a recurrent episode of epistaxis that occurs within the first 2 decades of life. Shortly after the onset of these recurrent episodes, patients begin to develop punctate or splinterlike telangiectases on the lips, oral mucosa, upper extremities, nail beds, and trunk. These cutaneous telangiectases are a cosmetic problem and can sometimes cause hemorrhage, especially from the tongue and fingers.1 The serious complications of HHT arise from internal organ involvement. Gastrointestinal telangiectases and arteriovenous malformations (AVMs) can result in acute gastrointestinal hemorrhages or iron deficiency anemia in approximately 16% of patients. Central nervous system AVMs result in migraines, brain abscesses, paraparesis, ischemia, strokes, transient ischemic attacks, seizures, and both intracerebral and subarachnoid hemorrhage. Pulmonary AVMs can cause a right-to-left shunt, leading to hypoxemia and embolic events due to the bypass of the lungs, which act as a filtering capillary bed.3
Genetic linkage analysis has revealed 2 genes that are responsible for HHT. The first gene, ENG, encodes endoglin and is found on band 9q33-34.3 The second gene, ALK1, encodes activin receptorlike kinase 1 and is found on band 12q11-12. Both of these gene products are involved with vascular remodeling. They are integral membrane glycoproteins mainly expressed on vascular endothelial cells and act as surface receptors for transforming growth factor b. Mutations in ALK1 are associated with a generally more benign course, whereas mutations in ENG more commonly have pulmonary AVMs.3
The diagnosis of HHT is established if 3 of the following features are present: (1) epistaxis (ie, spontaneous recurrent nosebleeds); (2) telangiectases in characteristic sites (ie, oral cavity, lips, nose, fingers); (3) visceral lesions, such as gastrointestinal telangiectases (with or without bleeding), pulmonary AVM, hepatic AVM, cerebral AVM, or spinal AVM; (4) family history (ie, a first-degree relative with HHT).3 Similar diseases with telangiectases should be considered in the differential diagnosis including CREST syndrome (characterized by calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, and telangiectasia), generalized essential telangiectasia, and ataxia telangiectasia.2
Patients diagnosed with HHT who have a family history of the disease should be evaluated for pulmonary AVM through chest computed tomography and pulmonary angiography because they are at the highest risk. Magnetic resonance imaging is useful to rule out central nervous system involvement. Treatment of cutaneous telangiectases consists of electrocauterization; sclerotherapy; and a variety of lasers and light sources including the Nd:YAG laser, intense pulsed light, argon laser, or pulsed dye laser.1,2,4 An extremely useful resource for patients with HHT and family members is the HHT Foundation (http://curehht.org).
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
1. Fernández-Jorge B, Del Pozo Losada J, Paradela S, et al. Treatment of cutaneous and mucosal telangiectases in hereditary hemorrhagic telangiectasia: report of three cases. J Cosmet Laser Ther. 2007;9:29-33.
2. Lee HE, Sagong C, Yeo KY, et al. A case of hereditary hemorrhagic telangiectasia. Ann Dermatol. 2009;21:206-208.
3. Garzon MC, Huang JT, Enjolras O, et al. Vascular malformations. part II: associated syndromes. J Am Acad Dermatol. 2007;56:541-564.
4. Sato Y, Takayama T, Takahari D, et al. Successful treatment for gastro-intestinal bleeding of Osler-Weber-Rendu disease by argon plasma coagulation using double-balloon enteroscopy. Endoscopy. 2008;40(suppl 2):E228-E229.
A 68-year-old man presented for evaluation of numerous telangiectases that developed on the cheeks, nose, lips, tongue, and fingers. He denied any history of gastrointestinal tract bleeding but admitted to numerous nosebleeds in the recent past. Family history indicated that his maternal grandmother died of an aneurysm, his mother died of a cerebral hemorrhage, 2 paternal cousins had hemochromatosis, and 2 brothers were healthy.