User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
How to treat depression, stress associated with infertility treatment
“I think it’s my fault we can’t get pregnant,” says Mrs. S, who has been referred by her gynecologist for evaluation of anxiety and depression. Mrs. S, age 33, and her husband have been trying to conceive their first child for 2 years.
The couple has undergone infertility workups, including a semen analysis and hysterosalpingography, and results have been within normal limits. The gynecologist recommended intercourse every other day, but Mr. S developed stress-related erectile dysfunction (which was treated with sildenafil).
Mrs. S has no personal or family history of depression. Her depression has worsened as she contemplates more invasive and expensive procedures, such as intrauterine insemination (IUI) and in vitro fertilization (IVF).
Her Beck Depression Inventory score of 22 indicates mild depression. She is not actively suicidal, but she sometimes doubts that life is worth living. She feels like a failure and wants to know if you think stress is contributing to her infertility.
Women with a 2- to 3-year history of infertility despite repeated treatments are at risk of stress, anxiety, and depression.1 Even if treatment eventually succeeds, anxiety often persists during pregnancy.2 Your knowledge of medical infertility treatments’ emotional toll will help you understand, educate, and support infertile women and their partners.
Infertility affects approximately 6 million U.S. women and their partners.3 As recently as the 1960s infertility was thought to be caused primarily by female psychological problems,4 such as neurotic, conflicted feelings about the transition to adulthood or about sex, pregnancy, labor, or motherhood.5,6
This belief changed as researchers identified organic causes of infertility, such as blocked fallopian tubes, sperm abnormalities, and anovulation. A definitive diagnosis can now be made in 85% to 90% of infertility cases, and two-thirds of couples can conceive after medical intervention.7
Age and fertility. Most experts recommend that women age >35 who wish to conceive seek gynecologic evaluation after 6 months of unsuccessful intercourse. Chances of becoming pregnant begin to decline at age 35 and drop sharply after age 40.8 Beyond age 43, the only infertility treatment likely to be successful is implanting an embryo created with an egg donated by a younger woman.
Stress and fertility
Infertility—failure to conceive after 1 year of regular unprotected intercourse—affects approximately 10% of the reproductive-age U.S. population (Box).3-8 Does stress affect a woman’s chance of becoming pregnant? Research into this question—voiced by Mrs. S—has produced conflicting results.5,9,10
Stress does not universally prevent pregnancy; women have conceived as a result of rape. However, chronic extreme stress—such as that imposed by war, imprisonment, or starvation—can change the menstrual cycle. Effects range from subtle luteal-phase deficiency to menses cessation.9 It may be that evolution favored females of species who could “turn off ” fertility during stressful times to conserve physical resources and “turn it back on” and bear offspring after the threat passed.
Neuroendocrine markers. Researchers examining the role of stress in infertility and its treatments have focused on the neuroendocrine system—particularly neurotransmitters such as prolactin, endorphin, norepinephrine, dopamine, and cortisol. Although chronic anxiety and depression have been linked in animal models to neuroendocrine mechanisms of infertility,4 findings in humans have been mixed (Table 1).11-15
Table 1
Does stress reduce fertility? Research results are mixed
| Study design (year of publication) | Results |
|---|---|
| Controlled prospective trial, 40 women undergoing IVF (1992)11 | IVF success rates were comparatively lower among women with high cortisol concentrations |
| Women with high prolactin concentrations had greater numbers of oocytes but lower fertilization rates | |
| Failure to conceive was associated with high depression symptom scores, maladaptive coping strategies, and avoidance behavior | |
| Controlled prospective trial, 330 infertile women (1993)12 | Depressed women had a lower pregnancy rate after a first IVF-ET, compared with nondepressed women |
| Uncontrolled prospective trial, 13 women without a history of infertility (1997)13 | Mean adrenaline, norepinephrine, and cortisol levels excreted in urine were not significantly different in menstrual cycles when women conceived, compared with nonconception cycles |
| Little relationship seen between psychological measures of mood state and excretion of adrenaline and cortisol | |
| Controlled, prospective trial, 49 infertile women (1997)14 | Patients who conceived with IVF-ET had lower systolic blood pressures and slower heart rates under stress-test conditions than did those who did not conceive |
| Controlled prospective trial, 40 women after successful IVF-ET (1998)15 | No difference in hormonal stress markers during first 27 days of pregnancy between women who later gave birth and those who experienced miscarriages |
| Physiologic stress hormone concentrations showed little association with psychological scores, and high anxiety and stress levels did not appear to prevent pregnancy | |
| IVF: In vitro fertilization | |
| IVF-ET: In vitro fertilization with embryo transplant | |
In one prospective, controlled, single-blind study, 184 women who had been trying to conceive for 1 to 2 years were randomly assigned to 10 sessions of group cognitive-behavioral therapy (CBT), a standard support group, or usual care. Sixty-four women withdrew before the study ended. After 1 year, women who received psychological interventions—47 in the CBT group and 48 in the standard support group—had statistically significant higher pregnancy rates, compared with 25 women who received usual care.16 Conversely, a literature review and evaluation of 25 studies found psychosocial interventions unlikely to improve pregnancy rates in infertile women.17
Methodologic problems. Most studies of stress’ influence on fertility are small, and many have methodologic problems.4 In some, researchers lumped together women whose infertility was caused by disparate diagnoses such as male-factor infertility, blocked fallopian tubes, and advanced age. Retrospective studies also must be interpreted with caution because:
- patients who did not become pregnant may have exaggerated the degree of their depression and its effects
- those with pre-existing medical problems would know they were unlikely to conceive and might have been more depressed before and during infertility treatments.18
Recommendation. When counseling patients about the role of stress in infertility and its treatment, we recommend emphasizing that:
- infertility can cause stress in many areas of life
- the effect of stress on fertility, if any, is likely to be minimal for most women.
Case continued: Strain and anger
You begin to see Mrs. S weekly for supportive therapy, using cognitive restructuring and relaxation techniques to alleviate her anxiety and depression. She decides not to start an antidepressant because she does not want to be on medication if she becomes pregnant.
During the next 2 months she finishes an unsuccessful IUI cycle and reports that her relationship with her husband has become strained. She avoids friends who have children and feels angry when she sees a pregnant woman. She dislikes going to family events because relatives sometimes ask, “When are you going to get pregnant?”
Her work as a manager is suffering because of her many visits to fertility specialists. Her Beck Depression Inventory score has increased to 33, indicating worsening depression.
Infertility’s psychological toll
Patients rarely accept infertility with equanimity, and their responses include shock, denial, anger, isolation, guilt, and grief.6 Some women say the experience of being infertile feels comparable to having cancer.20
The incidence of clinical major depression, poor self-esteem, and sexual dysfunction in women who undergo infertility evaluation does not differ significantly from that of their fertile peers.9 Even so, infertile women report a roller-coaster ride of emotions: hope as treatments are tried, despair when treatments fail.
Health care providers can add to the angst by telling women they have an “incompetent” cervix, “poor-quality” or “old” eggs, or “inadequate” mucus; these insensitive descriptions can lead women to blame themselves and feel ashamed, guilty, and depressed.4,5,18
Psychotherapy. Providing education and teaching skills such as relaxation training has been shown to reduce depressive symptoms more effectively than having patients discuss their thoughts and feelings about infertility.17 Helpful psychotherapies emphasize CBT and improved coping skills.
Negative coping strategies include escape/avoidance conduct or self-blame (such as, “I’m not getting pregnant because I work too hard”). Encourage patients to replace these with protective coping strategies, such as seeking social support and engaging in active problem-solving (“I reach out to friends who help comfort me, and I set limits with friends who make me feel bad about myself ”).21-23
Medication. Even though sadness and anxiety are normal responses to infertility, psychotropic medications might be appropriate after a thorough evaluation. Keep in mind, however, that selective serotonin reuptake inhibitors (SSRIs) can cause prolactinemia, which could interfere with ovulation.9 Miscarriage and stillbirth rates among women taking SSRIs are similar to those of the general population.24
Case continued: It takes two
Despite three IUI cycles over 12 months Mrs. S has not become pregnant. She considers IVF but is concerned about the cost and the less than 50% chance of success.
You encourage her to continue individual supportive and cognitive therapy and to consider couple’s therapy. She and her husband decide to attend a group for couples with infertility. She accepts your referral to RESOLVE, a national support program for infertile patients (see Related resources).
Problems facing infertile couples
Gender differences in coping style. Men and women experience infertility differently.
The women in infertile couples often are distressed, whereas the men tend to remain more confident that some kind of treatment will work. This imbalance can leave the woman feeling unsupported and the man feeling confused about why she is so upset about what he sees as just a medical problem to be solved.
When a couple’s infertility has been attributed to sperm abnormalities, however, the man’s stress level can equal the woman’s. Women tend to feel stress regardless of which partner is “at fault.”25
Grief reactions. The “loss” of a child never conceived generally goes unrecognized but has psychological consequences. Both partners can feel:
- low self-esteem
- sadness about being unable to experience parenting
- doubts about their femininity or masculinity
- regret over unfulfilled dreams.
Table 2
Fictions and facts about infertility
| Fiction | Fact |
|---|---|
| Infertility is a psychosomatic disorder | An organic cause is found in 85% to 90% of infertile couples7 |
| Infertility is a female problem | One-third of infertility cases are caused by female factors, one-third by male factors, and one-third by male and female factors or unknown causes26 |
| Infertility is epidemic | The number of patients seeking infertility treatment has increased dramatically in 20 years, but the infertility rate is stable3,5,18 |
| Infertility is rare | Approximately 10% of U.S. couples of childbearing age are infertile3 |
| If you adopt, you'll get pregnant | Conception rates are no higher following adoption than among childless couples7 |
Employment. Infertility treatments are time- and resource-intensive, and patients often miss work. Even while on the job, a woman distracted by infertility or treatment side effects might not perform as well as she could. Worries about job security add to her anxiety.
Finances. Infertility treatment is expensive and is not always covered by insurance. The American Society for Reproductive Medicine reports that the cost of an IVF cycle averages $12,400, and success rates are 26 (see Related resources).
To continue treatment, couples may take second jobs, acquire loans, deplete savings, or accumulate debt. Many couples—even with extraordinary effort—cannot afford to start or continue advanced infertility treatments.
Spirituality. Patients who believe that infertility is God’s punishment for past sins may experience a religious crisis. Those affiliated with religions that restrict assisted-reproductive technology may feel forced to choose between doctrinal dictates and their dreams of becoming parents.
Case continued: A new ‘RESOLVE’
Mrs. S enjoys her association with the online support of RESOLVE. Through message boards, she shares her concerns with other women undergoing infertility treatment. She also finds support from friends, although she continues to set limits such as declining invitations to baby showers. She practices relaxation techniques at home.
Since she and her husband have joined the group for infertile couples, their relationship has improved. Mrs. S feels that he better understands her fears after hearing other women in the group being “just as emotional.” He no longer tells her, “It’s just a medical problem.”
- National Infertility Association (RESOLVE). www.resolve.org.
- American Society for Reproductive Medicine. www.asrm.org.
- Sildenafil • Viagra
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Khademi A, Alleyassin A, Aghahosseini M, et al. Pretreatment Beck Depression Inventory score is an important predictor for post-treatment score in infertile patients: a before-after study. BMC Psychiatry 2005;5(1):25.-
2. Hjelmstedt A, Widstrom AM, Wramsby H, et al. Personality factors and emotional responses to pregnancy among IVF couples in early pregnancy: a comparative study. Acta Obstet Gynecol Scand 2003;82(2):152-61.
3. Abma J, Chandra A, Mosher W, et al. Fertility, family planning, and women’s health: new data from the 1995 National Survey of Family Growth. Vital and Health Statistics, Series 23, No. 19. National Center for Health Statistics, May 1997.
4. Wischmann TH. Psychogenic infertility-myths and facts. J Assist Reprod Genet 2003;20(12):485-94.
5. Burns LH, Covington SN, eds Infertility counseling: a comprehensive handbook for clinicians.. Pearl River, NY: Parthenon; 1999:122-35.
6. Stanton AL, Lobel M, Sears S, DeLuca RS. Psychosocial aspects of selected issues in women’s reproductive health: current status and future directions. J Consult Clin Psychol 2002;70(3):751-70.
7. National Infertility Association (RESOLVE) www.resolve.org. Accessed August 30, 2006.
8. Kasper DL, Braunwald E, Fauci A, et al. Harrison’s principles of internal medicine, 16th ed. New York: McGraw-Hill Professional; 2004.
9. Cedars M (ed) Infertility: practical pathways in obstetrics and gynecology. New York: McGraw-Hill; 2005:88-133.
10. Greil AL. Infertility and psychological distress: a critical review of the literature. Soc Sci Med 1997;45(11):1679-704.
11. Demyttenaere K, Nijs P, Evers-Kiebooms G, Koninckx PR. Coping and the ineffectiveness of coping influence the outcome of in vitro fertilization through stress responses. Psychoneuroendocrinology 1992;17(6):655-65.
12. Thiering P, Beaurepaire J, Jones M, et al. Mood state as a predictor of treatment outcome after in vitro fertilization/embryo transfer technology (IVF/ET). J Psychosom Res 1993;37(5):481-91.
13. Sanders KA, Bruce NW. A prospective study of psychological stress and fertility in women. Hum Reprod 1997;12(10):2324-9.
14. Facchinetti F, Matteo ML, Artini GP, et al. An increased vulnerability to stress is associated with a poor outcome of in vitro fertilization-embryo transfer treatment. Fertil Steril 1997;67(2):309-14.
15. Milad MP, Klock SC, Moses S, Chatterton R. Stress and anxiety do not result in pregnancy wastage. Hum Reprod 1998;13(8):2296-300.
16. Domar AD, Clapp D, Slawsby EA, et al. Impact of group psychological interventions on pregnancy rates in infertile women. Fertil Steril 2000;73(4):805-11.
17. Boivin J. A review to psychosocial interventions in infertility. Soc Sci Med 2003;57(12):2325-41.
18. Pasch LA. Confronting fertility problems: current research and future challenges. In: Baum A, Revenson TA, Singer JE (eds). Handbook of health psychology. Mahwah, NJ: Lawrence Erlbaum Associates; 2001:559-70.
19. Stewart DE, Boydell KM, McCarthy K, et al. A prospective study of the effectiveness of brief professionally-led support groups for infertility patients. Int J Psychiatry Med 1992;22(2):173-82.
20. Domar AD, Zuttermeister PC, Friedman R. The psychological impact of infertility: a comparison with patients with other medical conditions. J Psychosom Obstet Gynaecol 1993;14(suppl):45-52.
21. Litt MD, Tennen H, Affleck G, Klock S. Coping and cognitive factors in adaptation to in vitro fertilization failure. J Behav Med 1992;15(2):171-87.
22. Peterson BD, Newton CR, Rosen KH, Skaggs GE. The relationship between coping and depression in men and women referred for in vitro fertilization. Fertil Steril 2006;85(3):802-4.
23. Morrow KA, Thoreson RW, Penney LL. Predictors of psychological distress among infertility clinic patients. J Consult Clin Psychol 1995;63(1):163-7.
24. Hasser C, Brizendine L, Spielvogel A. SSRI use during pregnancy: do antidepressants’ benefits outweigh the risks? Current Psychiatry 2006;5(4):31-40.
25. Nachtigall RD, Becker G, Wozny M. The effects of gender-specific diagnosis on men’s and women’s response to infertility. Fertil Steril 1992;57(1):113-21.
26. American Society for Reproductive Medicine Information for patients. Is infertility treatment expensive? Available at: http://www.asrm.org/Patients/faqs.html#Q6. Accessed September 12, 2006
“I think it’s my fault we can’t get pregnant,” says Mrs. S, who has been referred by her gynecologist for evaluation of anxiety and depression. Mrs. S, age 33, and her husband have been trying to conceive their first child for 2 years.
The couple has undergone infertility workups, including a semen analysis and hysterosalpingography, and results have been within normal limits. The gynecologist recommended intercourse every other day, but Mr. S developed stress-related erectile dysfunction (which was treated with sildenafil).
Mrs. S has no personal or family history of depression. Her depression has worsened as she contemplates more invasive and expensive procedures, such as intrauterine insemination (IUI) and in vitro fertilization (IVF).
Her Beck Depression Inventory score of 22 indicates mild depression. She is not actively suicidal, but she sometimes doubts that life is worth living. She feels like a failure and wants to know if you think stress is contributing to her infertility.
Women with a 2- to 3-year history of infertility despite repeated treatments are at risk of stress, anxiety, and depression.1 Even if treatment eventually succeeds, anxiety often persists during pregnancy.2 Your knowledge of medical infertility treatments’ emotional toll will help you understand, educate, and support infertile women and their partners.
Infertility affects approximately 6 million U.S. women and their partners.3 As recently as the 1960s infertility was thought to be caused primarily by female psychological problems,4 such as neurotic, conflicted feelings about the transition to adulthood or about sex, pregnancy, labor, or motherhood.5,6
This belief changed as researchers identified organic causes of infertility, such as blocked fallopian tubes, sperm abnormalities, and anovulation. A definitive diagnosis can now be made in 85% to 90% of infertility cases, and two-thirds of couples can conceive after medical intervention.7
Age and fertility. Most experts recommend that women age >35 who wish to conceive seek gynecologic evaluation after 6 months of unsuccessful intercourse. Chances of becoming pregnant begin to decline at age 35 and drop sharply after age 40.8 Beyond age 43, the only infertility treatment likely to be successful is implanting an embryo created with an egg donated by a younger woman.
Stress and fertility
Infertility—failure to conceive after 1 year of regular unprotected intercourse—affects approximately 10% of the reproductive-age U.S. population (Box).3-8 Does stress affect a woman’s chance of becoming pregnant? Research into this question—voiced by Mrs. S—has produced conflicting results.5,9,10
Stress does not universally prevent pregnancy; women have conceived as a result of rape. However, chronic extreme stress—such as that imposed by war, imprisonment, or starvation—can change the menstrual cycle. Effects range from subtle luteal-phase deficiency to menses cessation.9 It may be that evolution favored females of species who could “turn off ” fertility during stressful times to conserve physical resources and “turn it back on” and bear offspring after the threat passed.
Neuroendocrine markers. Researchers examining the role of stress in infertility and its treatments have focused on the neuroendocrine system—particularly neurotransmitters such as prolactin, endorphin, norepinephrine, dopamine, and cortisol. Although chronic anxiety and depression have been linked in animal models to neuroendocrine mechanisms of infertility,4 findings in humans have been mixed (Table 1).11-15
Table 1
Does stress reduce fertility? Research results are mixed
| Study design (year of publication) | Results |
|---|---|
| Controlled prospective trial, 40 women undergoing IVF (1992)11 | IVF success rates were comparatively lower among women with high cortisol concentrations |
| Women with high prolactin concentrations had greater numbers of oocytes but lower fertilization rates | |
| Failure to conceive was associated with high depression symptom scores, maladaptive coping strategies, and avoidance behavior | |
| Controlled prospective trial, 330 infertile women (1993)12 | Depressed women had a lower pregnancy rate after a first IVF-ET, compared with nondepressed women |
| Uncontrolled prospective trial, 13 women without a history of infertility (1997)13 | Mean adrenaline, norepinephrine, and cortisol levels excreted in urine were not significantly different in menstrual cycles when women conceived, compared with nonconception cycles |
| Little relationship seen between psychological measures of mood state and excretion of adrenaline and cortisol | |
| Controlled, prospective trial, 49 infertile women (1997)14 | Patients who conceived with IVF-ET had lower systolic blood pressures and slower heart rates under stress-test conditions than did those who did not conceive |
| Controlled prospective trial, 40 women after successful IVF-ET (1998)15 | No difference in hormonal stress markers during first 27 days of pregnancy between women who later gave birth and those who experienced miscarriages |
| Physiologic stress hormone concentrations showed little association with psychological scores, and high anxiety and stress levels did not appear to prevent pregnancy | |
| IVF: In vitro fertilization | |
| IVF-ET: In vitro fertilization with embryo transplant | |
In one prospective, controlled, single-blind study, 184 women who had been trying to conceive for 1 to 2 years were randomly assigned to 10 sessions of group cognitive-behavioral therapy (CBT), a standard support group, or usual care. Sixty-four women withdrew before the study ended. After 1 year, women who received psychological interventions—47 in the CBT group and 48 in the standard support group—had statistically significant higher pregnancy rates, compared with 25 women who received usual care.16 Conversely, a literature review and evaluation of 25 studies found psychosocial interventions unlikely to improve pregnancy rates in infertile women.17
Methodologic problems. Most studies of stress’ influence on fertility are small, and many have methodologic problems.4 In some, researchers lumped together women whose infertility was caused by disparate diagnoses such as male-factor infertility, blocked fallopian tubes, and advanced age. Retrospective studies also must be interpreted with caution because:
- patients who did not become pregnant may have exaggerated the degree of their depression and its effects
- those with pre-existing medical problems would know they were unlikely to conceive and might have been more depressed before and during infertility treatments.18
Recommendation. When counseling patients about the role of stress in infertility and its treatment, we recommend emphasizing that:
- infertility can cause stress in many areas of life
- the effect of stress on fertility, if any, is likely to be minimal for most women.
Case continued: Strain and anger
You begin to see Mrs. S weekly for supportive therapy, using cognitive restructuring and relaxation techniques to alleviate her anxiety and depression. She decides not to start an antidepressant because she does not want to be on medication if she becomes pregnant.
During the next 2 months she finishes an unsuccessful IUI cycle and reports that her relationship with her husband has become strained. She avoids friends who have children and feels angry when she sees a pregnant woman. She dislikes going to family events because relatives sometimes ask, “When are you going to get pregnant?”
Her work as a manager is suffering because of her many visits to fertility specialists. Her Beck Depression Inventory score has increased to 33, indicating worsening depression.
Infertility’s psychological toll
Patients rarely accept infertility with equanimity, and their responses include shock, denial, anger, isolation, guilt, and grief.6 Some women say the experience of being infertile feels comparable to having cancer.20
The incidence of clinical major depression, poor self-esteem, and sexual dysfunction in women who undergo infertility evaluation does not differ significantly from that of their fertile peers.9 Even so, infertile women report a roller-coaster ride of emotions: hope as treatments are tried, despair when treatments fail.
Health care providers can add to the angst by telling women they have an “incompetent” cervix, “poor-quality” or “old” eggs, or “inadequate” mucus; these insensitive descriptions can lead women to blame themselves and feel ashamed, guilty, and depressed.4,5,18
Psychotherapy. Providing education and teaching skills such as relaxation training has been shown to reduce depressive symptoms more effectively than having patients discuss their thoughts and feelings about infertility.17 Helpful psychotherapies emphasize CBT and improved coping skills.
Negative coping strategies include escape/avoidance conduct or self-blame (such as, “I’m not getting pregnant because I work too hard”). Encourage patients to replace these with protective coping strategies, such as seeking social support and engaging in active problem-solving (“I reach out to friends who help comfort me, and I set limits with friends who make me feel bad about myself ”).21-23
Medication. Even though sadness and anxiety are normal responses to infertility, psychotropic medications might be appropriate after a thorough evaluation. Keep in mind, however, that selective serotonin reuptake inhibitors (SSRIs) can cause prolactinemia, which could interfere with ovulation.9 Miscarriage and stillbirth rates among women taking SSRIs are similar to those of the general population.24
Case continued: It takes two
Despite three IUI cycles over 12 months Mrs. S has not become pregnant. She considers IVF but is concerned about the cost and the less than 50% chance of success.
You encourage her to continue individual supportive and cognitive therapy and to consider couple’s therapy. She and her husband decide to attend a group for couples with infertility. She accepts your referral to RESOLVE, a national support program for infertile patients (see Related resources).
Problems facing infertile couples
Gender differences in coping style. Men and women experience infertility differently.
The women in infertile couples often are distressed, whereas the men tend to remain more confident that some kind of treatment will work. This imbalance can leave the woman feeling unsupported and the man feeling confused about why she is so upset about what he sees as just a medical problem to be solved.
When a couple’s infertility has been attributed to sperm abnormalities, however, the man’s stress level can equal the woman’s. Women tend to feel stress regardless of which partner is “at fault.”25
Grief reactions. The “loss” of a child never conceived generally goes unrecognized but has psychological consequences. Both partners can feel:
- low self-esteem
- sadness about being unable to experience parenting
- doubts about their femininity or masculinity
- regret over unfulfilled dreams.
Table 2
Fictions and facts about infertility
| Fiction | Fact |
|---|---|
| Infertility is a psychosomatic disorder | An organic cause is found in 85% to 90% of infertile couples7 |
| Infertility is a female problem | One-third of infertility cases are caused by female factors, one-third by male factors, and one-third by male and female factors or unknown causes26 |
| Infertility is epidemic | The number of patients seeking infertility treatment has increased dramatically in 20 years, but the infertility rate is stable3,5,18 |
| Infertility is rare | Approximately 10% of U.S. couples of childbearing age are infertile3 |
| If you adopt, you'll get pregnant | Conception rates are no higher following adoption than among childless couples7 |
Employment. Infertility treatments are time- and resource-intensive, and patients often miss work. Even while on the job, a woman distracted by infertility or treatment side effects might not perform as well as she could. Worries about job security add to her anxiety.
Finances. Infertility treatment is expensive and is not always covered by insurance. The American Society for Reproductive Medicine reports that the cost of an IVF cycle averages $12,400, and success rates are 26 (see Related resources).
To continue treatment, couples may take second jobs, acquire loans, deplete savings, or accumulate debt. Many couples—even with extraordinary effort—cannot afford to start or continue advanced infertility treatments.
Spirituality. Patients who believe that infertility is God’s punishment for past sins may experience a religious crisis. Those affiliated with religions that restrict assisted-reproductive technology may feel forced to choose between doctrinal dictates and their dreams of becoming parents.
Case continued: A new ‘RESOLVE’
Mrs. S enjoys her association with the online support of RESOLVE. Through message boards, she shares her concerns with other women undergoing infertility treatment. She also finds support from friends, although she continues to set limits such as declining invitations to baby showers. She practices relaxation techniques at home.
Since she and her husband have joined the group for infertile couples, their relationship has improved. Mrs. S feels that he better understands her fears after hearing other women in the group being “just as emotional.” He no longer tells her, “It’s just a medical problem.”
- National Infertility Association (RESOLVE). www.resolve.org.
- American Society for Reproductive Medicine. www.asrm.org.
- Sildenafil • Viagra
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
“I think it’s my fault we can’t get pregnant,” says Mrs. S, who has been referred by her gynecologist for evaluation of anxiety and depression. Mrs. S, age 33, and her husband have been trying to conceive their first child for 2 years.
The couple has undergone infertility workups, including a semen analysis and hysterosalpingography, and results have been within normal limits. The gynecologist recommended intercourse every other day, but Mr. S developed stress-related erectile dysfunction (which was treated with sildenafil).
Mrs. S has no personal or family history of depression. Her depression has worsened as she contemplates more invasive and expensive procedures, such as intrauterine insemination (IUI) and in vitro fertilization (IVF).
Her Beck Depression Inventory score of 22 indicates mild depression. She is not actively suicidal, but she sometimes doubts that life is worth living. She feels like a failure and wants to know if you think stress is contributing to her infertility.
Women with a 2- to 3-year history of infertility despite repeated treatments are at risk of stress, anxiety, and depression.1 Even if treatment eventually succeeds, anxiety often persists during pregnancy.2 Your knowledge of medical infertility treatments’ emotional toll will help you understand, educate, and support infertile women and their partners.
Infertility affects approximately 6 million U.S. women and their partners.3 As recently as the 1960s infertility was thought to be caused primarily by female psychological problems,4 such as neurotic, conflicted feelings about the transition to adulthood or about sex, pregnancy, labor, or motherhood.5,6
This belief changed as researchers identified organic causes of infertility, such as blocked fallopian tubes, sperm abnormalities, and anovulation. A definitive diagnosis can now be made in 85% to 90% of infertility cases, and two-thirds of couples can conceive after medical intervention.7
Age and fertility. Most experts recommend that women age >35 who wish to conceive seek gynecologic evaluation after 6 months of unsuccessful intercourse. Chances of becoming pregnant begin to decline at age 35 and drop sharply after age 40.8 Beyond age 43, the only infertility treatment likely to be successful is implanting an embryo created with an egg donated by a younger woman.
Stress and fertility
Infertility—failure to conceive after 1 year of regular unprotected intercourse—affects approximately 10% of the reproductive-age U.S. population (Box).3-8 Does stress affect a woman’s chance of becoming pregnant? Research into this question—voiced by Mrs. S—has produced conflicting results.5,9,10
Stress does not universally prevent pregnancy; women have conceived as a result of rape. However, chronic extreme stress—such as that imposed by war, imprisonment, or starvation—can change the menstrual cycle. Effects range from subtle luteal-phase deficiency to menses cessation.9 It may be that evolution favored females of species who could “turn off ” fertility during stressful times to conserve physical resources and “turn it back on” and bear offspring after the threat passed.
Neuroendocrine markers. Researchers examining the role of stress in infertility and its treatments have focused on the neuroendocrine system—particularly neurotransmitters such as prolactin, endorphin, norepinephrine, dopamine, and cortisol. Although chronic anxiety and depression have been linked in animal models to neuroendocrine mechanisms of infertility,4 findings in humans have been mixed (Table 1).11-15
Table 1
Does stress reduce fertility? Research results are mixed
| Study design (year of publication) | Results |
|---|---|
| Controlled prospective trial, 40 women undergoing IVF (1992)11 | IVF success rates were comparatively lower among women with high cortisol concentrations |
| Women with high prolactin concentrations had greater numbers of oocytes but lower fertilization rates | |
| Failure to conceive was associated with high depression symptom scores, maladaptive coping strategies, and avoidance behavior | |
| Controlled prospective trial, 330 infertile women (1993)12 | Depressed women had a lower pregnancy rate after a first IVF-ET, compared with nondepressed women |
| Uncontrolled prospective trial, 13 women without a history of infertility (1997)13 | Mean adrenaline, norepinephrine, and cortisol levels excreted in urine were not significantly different in menstrual cycles when women conceived, compared with nonconception cycles |
| Little relationship seen between psychological measures of mood state and excretion of adrenaline and cortisol | |
| Controlled, prospective trial, 49 infertile women (1997)14 | Patients who conceived with IVF-ET had lower systolic blood pressures and slower heart rates under stress-test conditions than did those who did not conceive |
| Controlled prospective trial, 40 women after successful IVF-ET (1998)15 | No difference in hormonal stress markers during first 27 days of pregnancy between women who later gave birth and those who experienced miscarriages |
| Physiologic stress hormone concentrations showed little association with psychological scores, and high anxiety and stress levels did not appear to prevent pregnancy | |
| IVF: In vitro fertilization | |
| IVF-ET: In vitro fertilization with embryo transplant | |
In one prospective, controlled, single-blind study, 184 women who had been trying to conceive for 1 to 2 years were randomly assigned to 10 sessions of group cognitive-behavioral therapy (CBT), a standard support group, or usual care. Sixty-four women withdrew before the study ended. After 1 year, women who received psychological interventions—47 in the CBT group and 48 in the standard support group—had statistically significant higher pregnancy rates, compared with 25 women who received usual care.16 Conversely, a literature review and evaluation of 25 studies found psychosocial interventions unlikely to improve pregnancy rates in infertile women.17
Methodologic problems. Most studies of stress’ influence on fertility are small, and many have methodologic problems.4 In some, researchers lumped together women whose infertility was caused by disparate diagnoses such as male-factor infertility, blocked fallopian tubes, and advanced age. Retrospective studies also must be interpreted with caution because:
- patients who did not become pregnant may have exaggerated the degree of their depression and its effects
- those with pre-existing medical problems would know they were unlikely to conceive and might have been more depressed before and during infertility treatments.18
Recommendation. When counseling patients about the role of stress in infertility and its treatment, we recommend emphasizing that:
- infertility can cause stress in many areas of life
- the effect of stress on fertility, if any, is likely to be minimal for most women.
Case continued: Strain and anger
You begin to see Mrs. S weekly for supportive therapy, using cognitive restructuring and relaxation techniques to alleviate her anxiety and depression. She decides not to start an antidepressant because she does not want to be on medication if she becomes pregnant.
During the next 2 months she finishes an unsuccessful IUI cycle and reports that her relationship with her husband has become strained. She avoids friends who have children and feels angry when she sees a pregnant woman. She dislikes going to family events because relatives sometimes ask, “When are you going to get pregnant?”
Her work as a manager is suffering because of her many visits to fertility specialists. Her Beck Depression Inventory score has increased to 33, indicating worsening depression.
Infertility’s psychological toll
Patients rarely accept infertility with equanimity, and their responses include shock, denial, anger, isolation, guilt, and grief.6 Some women say the experience of being infertile feels comparable to having cancer.20
The incidence of clinical major depression, poor self-esteem, and sexual dysfunction in women who undergo infertility evaluation does not differ significantly from that of their fertile peers.9 Even so, infertile women report a roller-coaster ride of emotions: hope as treatments are tried, despair when treatments fail.
Health care providers can add to the angst by telling women they have an “incompetent” cervix, “poor-quality” or “old” eggs, or “inadequate” mucus; these insensitive descriptions can lead women to blame themselves and feel ashamed, guilty, and depressed.4,5,18
Psychotherapy. Providing education and teaching skills such as relaxation training has been shown to reduce depressive symptoms more effectively than having patients discuss their thoughts and feelings about infertility.17 Helpful psychotherapies emphasize CBT and improved coping skills.
Negative coping strategies include escape/avoidance conduct or self-blame (such as, “I’m not getting pregnant because I work too hard”). Encourage patients to replace these with protective coping strategies, such as seeking social support and engaging in active problem-solving (“I reach out to friends who help comfort me, and I set limits with friends who make me feel bad about myself ”).21-23
Medication. Even though sadness and anxiety are normal responses to infertility, psychotropic medications might be appropriate after a thorough evaluation. Keep in mind, however, that selective serotonin reuptake inhibitors (SSRIs) can cause prolactinemia, which could interfere with ovulation.9 Miscarriage and stillbirth rates among women taking SSRIs are similar to those of the general population.24
Case continued: It takes two
Despite three IUI cycles over 12 months Mrs. S has not become pregnant. She considers IVF but is concerned about the cost and the less than 50% chance of success.
You encourage her to continue individual supportive and cognitive therapy and to consider couple’s therapy. She and her husband decide to attend a group for couples with infertility. She accepts your referral to RESOLVE, a national support program for infertile patients (see Related resources).
Problems facing infertile couples
Gender differences in coping style. Men and women experience infertility differently.
The women in infertile couples often are distressed, whereas the men tend to remain more confident that some kind of treatment will work. This imbalance can leave the woman feeling unsupported and the man feeling confused about why she is so upset about what he sees as just a medical problem to be solved.
When a couple’s infertility has been attributed to sperm abnormalities, however, the man’s stress level can equal the woman’s. Women tend to feel stress regardless of which partner is “at fault.”25
Grief reactions. The “loss” of a child never conceived generally goes unrecognized but has psychological consequences. Both partners can feel:
- low self-esteem
- sadness about being unable to experience parenting
- doubts about their femininity or masculinity
- regret over unfulfilled dreams.
Table 2
Fictions and facts about infertility
| Fiction | Fact |
|---|---|
| Infertility is a psychosomatic disorder | An organic cause is found in 85% to 90% of infertile couples7 |
| Infertility is a female problem | One-third of infertility cases are caused by female factors, one-third by male factors, and one-third by male and female factors or unknown causes26 |
| Infertility is epidemic | The number of patients seeking infertility treatment has increased dramatically in 20 years, but the infertility rate is stable3,5,18 |
| Infertility is rare | Approximately 10% of U.S. couples of childbearing age are infertile3 |
| If you adopt, you'll get pregnant | Conception rates are no higher following adoption than among childless couples7 |
Employment. Infertility treatments are time- and resource-intensive, and patients often miss work. Even while on the job, a woman distracted by infertility or treatment side effects might not perform as well as she could. Worries about job security add to her anxiety.
Finances. Infertility treatment is expensive and is not always covered by insurance. The American Society for Reproductive Medicine reports that the cost of an IVF cycle averages $12,400, and success rates are 26 (see Related resources).
To continue treatment, couples may take second jobs, acquire loans, deplete savings, or accumulate debt. Many couples—even with extraordinary effort—cannot afford to start or continue advanced infertility treatments.
Spirituality. Patients who believe that infertility is God’s punishment for past sins may experience a religious crisis. Those affiliated with religions that restrict assisted-reproductive technology may feel forced to choose between doctrinal dictates and their dreams of becoming parents.
Case continued: A new ‘RESOLVE’
Mrs. S enjoys her association with the online support of RESOLVE. Through message boards, she shares her concerns with other women undergoing infertility treatment. She also finds support from friends, although she continues to set limits such as declining invitations to baby showers. She practices relaxation techniques at home.
Since she and her husband have joined the group for infertile couples, their relationship has improved. Mrs. S feels that he better understands her fears after hearing other women in the group being “just as emotional.” He no longer tells her, “It’s just a medical problem.”
- National Infertility Association (RESOLVE). www.resolve.org.
- American Society for Reproductive Medicine. www.asrm.org.
- Sildenafil • Viagra
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Khademi A, Alleyassin A, Aghahosseini M, et al. Pretreatment Beck Depression Inventory score is an important predictor for post-treatment score in infertile patients: a before-after study. BMC Psychiatry 2005;5(1):25.-
2. Hjelmstedt A, Widstrom AM, Wramsby H, et al. Personality factors and emotional responses to pregnancy among IVF couples in early pregnancy: a comparative study. Acta Obstet Gynecol Scand 2003;82(2):152-61.
3. Abma J, Chandra A, Mosher W, et al. Fertility, family planning, and women’s health: new data from the 1995 National Survey of Family Growth. Vital and Health Statistics, Series 23, No. 19. National Center for Health Statistics, May 1997.
4. Wischmann TH. Psychogenic infertility-myths and facts. J Assist Reprod Genet 2003;20(12):485-94.
5. Burns LH, Covington SN, eds Infertility counseling: a comprehensive handbook for clinicians.. Pearl River, NY: Parthenon; 1999:122-35.
6. Stanton AL, Lobel M, Sears S, DeLuca RS. Psychosocial aspects of selected issues in women’s reproductive health: current status and future directions. J Consult Clin Psychol 2002;70(3):751-70.
7. National Infertility Association (RESOLVE) www.resolve.org. Accessed August 30, 2006.
8. Kasper DL, Braunwald E, Fauci A, et al. Harrison’s principles of internal medicine, 16th ed. New York: McGraw-Hill Professional; 2004.
9. Cedars M (ed) Infertility: practical pathways in obstetrics and gynecology. New York: McGraw-Hill; 2005:88-133.
10. Greil AL. Infertility and psychological distress: a critical review of the literature. Soc Sci Med 1997;45(11):1679-704.
11. Demyttenaere K, Nijs P, Evers-Kiebooms G, Koninckx PR. Coping and the ineffectiveness of coping influence the outcome of in vitro fertilization through stress responses. Psychoneuroendocrinology 1992;17(6):655-65.
12. Thiering P, Beaurepaire J, Jones M, et al. Mood state as a predictor of treatment outcome after in vitro fertilization/embryo transfer technology (IVF/ET). J Psychosom Res 1993;37(5):481-91.
13. Sanders KA, Bruce NW. A prospective study of psychological stress and fertility in women. Hum Reprod 1997;12(10):2324-9.
14. Facchinetti F, Matteo ML, Artini GP, et al. An increased vulnerability to stress is associated with a poor outcome of in vitro fertilization-embryo transfer treatment. Fertil Steril 1997;67(2):309-14.
15. Milad MP, Klock SC, Moses S, Chatterton R. Stress and anxiety do not result in pregnancy wastage. Hum Reprod 1998;13(8):2296-300.
16. Domar AD, Clapp D, Slawsby EA, et al. Impact of group psychological interventions on pregnancy rates in infertile women. Fertil Steril 2000;73(4):805-11.
17. Boivin J. A review to psychosocial interventions in infertility. Soc Sci Med 2003;57(12):2325-41.
18. Pasch LA. Confronting fertility problems: current research and future challenges. In: Baum A, Revenson TA, Singer JE (eds). Handbook of health psychology. Mahwah, NJ: Lawrence Erlbaum Associates; 2001:559-70.
19. Stewart DE, Boydell KM, McCarthy K, et al. A prospective study of the effectiveness of brief professionally-led support groups for infertility patients. Int J Psychiatry Med 1992;22(2):173-82.
20. Domar AD, Zuttermeister PC, Friedman R. The psychological impact of infertility: a comparison with patients with other medical conditions. J Psychosom Obstet Gynaecol 1993;14(suppl):45-52.
21. Litt MD, Tennen H, Affleck G, Klock S. Coping and cognitive factors in adaptation to in vitro fertilization failure. J Behav Med 1992;15(2):171-87.
22. Peterson BD, Newton CR, Rosen KH, Skaggs GE. The relationship between coping and depression in men and women referred for in vitro fertilization. Fertil Steril 2006;85(3):802-4.
23. Morrow KA, Thoreson RW, Penney LL. Predictors of psychological distress among infertility clinic patients. J Consult Clin Psychol 1995;63(1):163-7.
24. Hasser C, Brizendine L, Spielvogel A. SSRI use during pregnancy: do antidepressants’ benefits outweigh the risks? Current Psychiatry 2006;5(4):31-40.
25. Nachtigall RD, Becker G, Wozny M. The effects of gender-specific diagnosis on men’s and women’s response to infertility. Fertil Steril 1992;57(1):113-21.
26. American Society for Reproductive Medicine Information for patients. Is infertility treatment expensive? Available at: http://www.asrm.org/Patients/faqs.html#Q6. Accessed September 12, 2006
1. Khademi A, Alleyassin A, Aghahosseini M, et al. Pretreatment Beck Depression Inventory score is an important predictor for post-treatment score in infertile patients: a before-after study. BMC Psychiatry 2005;5(1):25.-
2. Hjelmstedt A, Widstrom AM, Wramsby H, et al. Personality factors and emotional responses to pregnancy among IVF couples in early pregnancy: a comparative study. Acta Obstet Gynecol Scand 2003;82(2):152-61.
3. Abma J, Chandra A, Mosher W, et al. Fertility, family planning, and women’s health: new data from the 1995 National Survey of Family Growth. Vital and Health Statistics, Series 23, No. 19. National Center for Health Statistics, May 1997.
4. Wischmann TH. Psychogenic infertility-myths and facts. J Assist Reprod Genet 2003;20(12):485-94.
5. Burns LH, Covington SN, eds Infertility counseling: a comprehensive handbook for clinicians.. Pearl River, NY: Parthenon; 1999:122-35.
6. Stanton AL, Lobel M, Sears S, DeLuca RS. Psychosocial aspects of selected issues in women’s reproductive health: current status and future directions. J Consult Clin Psychol 2002;70(3):751-70.
7. National Infertility Association (RESOLVE) www.resolve.org. Accessed August 30, 2006.
8. Kasper DL, Braunwald E, Fauci A, et al. Harrison’s principles of internal medicine, 16th ed. New York: McGraw-Hill Professional; 2004.
9. Cedars M (ed) Infertility: practical pathways in obstetrics and gynecology. New York: McGraw-Hill; 2005:88-133.
10. Greil AL. Infertility and psychological distress: a critical review of the literature. Soc Sci Med 1997;45(11):1679-704.
11. Demyttenaere K, Nijs P, Evers-Kiebooms G, Koninckx PR. Coping and the ineffectiveness of coping influence the outcome of in vitro fertilization through stress responses. Psychoneuroendocrinology 1992;17(6):655-65.
12. Thiering P, Beaurepaire J, Jones M, et al. Mood state as a predictor of treatment outcome after in vitro fertilization/embryo transfer technology (IVF/ET). J Psychosom Res 1993;37(5):481-91.
13. Sanders KA, Bruce NW. A prospective study of psychological stress and fertility in women. Hum Reprod 1997;12(10):2324-9.
14. Facchinetti F, Matteo ML, Artini GP, et al. An increased vulnerability to stress is associated with a poor outcome of in vitro fertilization-embryo transfer treatment. Fertil Steril 1997;67(2):309-14.
15. Milad MP, Klock SC, Moses S, Chatterton R. Stress and anxiety do not result in pregnancy wastage. Hum Reprod 1998;13(8):2296-300.
16. Domar AD, Clapp D, Slawsby EA, et al. Impact of group psychological interventions on pregnancy rates in infertile women. Fertil Steril 2000;73(4):805-11.
17. Boivin J. A review to psychosocial interventions in infertility. Soc Sci Med 2003;57(12):2325-41.
18. Pasch LA. Confronting fertility problems: current research and future challenges. In: Baum A, Revenson TA, Singer JE (eds). Handbook of health psychology. Mahwah, NJ: Lawrence Erlbaum Associates; 2001:559-70.
19. Stewart DE, Boydell KM, McCarthy K, et al. A prospective study of the effectiveness of brief professionally-led support groups for infertility patients. Int J Psychiatry Med 1992;22(2):173-82.
20. Domar AD, Zuttermeister PC, Friedman R. The psychological impact of infertility: a comparison with patients with other medical conditions. J Psychosom Obstet Gynaecol 1993;14(suppl):45-52.
21. Litt MD, Tennen H, Affleck G, Klock S. Coping and cognitive factors in adaptation to in vitro fertilization failure. J Behav Med 1992;15(2):171-87.
22. Peterson BD, Newton CR, Rosen KH, Skaggs GE. The relationship between coping and depression in men and women referred for in vitro fertilization. Fertil Steril 2006;85(3):802-4.
23. Morrow KA, Thoreson RW, Penney LL. Predictors of psychological distress among infertility clinic patients. J Consult Clin Psychol 1995;63(1):163-7.
24. Hasser C, Brizendine L, Spielvogel A. SSRI use during pregnancy: do antidepressants’ benefits outweigh the risks? Current Psychiatry 2006;5(4):31-40.
25. Nachtigall RD, Becker G, Wozny M. The effects of gender-specific diagnosis on men’s and women’s response to infertility. Fertil Steril 1992;57(1):113-21.
26. American Society for Reproductive Medicine Information for patients. Is infertility treatment expensive? Available at: http://www.asrm.org/Patients/faqs.html#Q6. Accessed September 12, 2006
New warnings on stimulants for ADHD: Cause for alarm?
In August the FDA called for new warnings on stimulants used for attention-deficit/hyperactivity disorder (ADHD). Amphetamines now carry black box warnings that say, “Misuse of amphetamines may cause sudden death and serious cardiovascular adverse events.” Amphetamines and methylphenidates used for ADHD include expanded information about cardiovascular risks at usual dosages for patients with heart conditions.
To examine the clinical implications of these warnings, Current Psychiatry hosted a conversation between ADHD experts Anthony Rostain, MD, MA, and Lenard Adler, MD.
Dr. Rostain: Changes to warnings on ADHD medications have many psychiatrists looking for guidance on using stimulants. Can you give us some background and discuss the labeling changes?
Dr. Adler: Stimulants have been used for more than 40 years as ADHD treatments, and they’ve been shown to be highly effective. The FDA, which monitors issues of cardiovascular safety and stimulants in an ongoing way, examined specific isolated cases and changed some of the warnings as a result.
Dr. Rostain: What should practicing psychiatrists be concerned about if they’re thinking of prescribing stimulants for an ADHD patient?
Dr. Adler: The take-home point is that stimulants—because of the way they work—have been known to have minor effects of increasing blood pressure and pulse (Box 1).1-3 Clinicians have known about issues regarding stimulant use by patients with pre-existing cardiovascular conditions, but now the warnings are more formal for the methylphenidate and amphetamine products.
Dr. Rostain: An FDA committee recommended black box warnings on all stimulants used for ADHD, but the FDA decided instead to clarify warnings in prescription information for some medications. What was the FDA process?
Dr. Adler: The discussion was internal at the FDA, so I can’t say what their thinking was. The black box warning on amphetamines notes two issues. One is the potential for abuse and diversion, and the other warns of potential for sudden death and serious cardiovascular effects if the drug is misused. A warning has also been placed on all methylphenidate products regarding cardiovascular risk for patients with pre-existing cardiovascular conditions, but it is not a black box warning.
in healthy children and adults
Researchers at Massachusetts General Hospital have examined the effects of ADHD medications on blood pressure and heart rate in children and adults.
Children and adolescents. The first study1 was a 1-year extension of an open-label trial of once-daily, osmotic-release methylphenidate (MPH) in 432 children (age 6 to 13) with ADHD. Their blood pressure and heart rate were recorded at baseline and monthly.
At 12 months, MPH use at 18 to 54 mg/d was associated with minor but statistically significant mean increases in:
- systolic blood pressure (3.3 mm Hg [P<0.001])
- diastolic blood pressure (1.5 mm Hg [P<0.001])
- heart rate (3.9 bpm [P<0.0001]).
Adults. In a 24-month study,2 223 healthy adults with ADHD (age≥18) received mixed amphetamine salts extended-release (MAS XR) in an open-label extension of a 4-week, double-blind, placebo-controlled trial. MAS XR was started at 20 mg/d for 1 week, then increased up to 60 mg/d based on therapeutic effect, as measured by the ADHD Rating Scale IV.
Blood pressure and pulse were measured at baseline, weekly, then monthly, and 12-lead ECGs were obtained at baseline, weekly, then at 3- and 6-month intervals. Changes after 2 years were small and not statistically significant:
- systolic blood pressure (2.3±12.5 mm Hg)
- diastolic blood pressure (1.3±9.2 mm Hg)
- pulse (2.1±13.4 bpm).
A clinically insignificant increase was observed in the mean QTcB interval (7.2 msec; P<0.001), although no patient’s QTcB interval exceeded 480 msec. Seven patients dropped out because of cardiovascular side effects (5 with hypertension, and 2 with palpitation/tachycardia), which were not reported as being serious.
Stimulants and nonstimulants. In another study,3 the same researchers analyzed the cardiovascular effects of three stimulants (methylphenidate, amphetamine compounds, and pemoline) and two nonstimulants (bupropion and desipramine) used to treat ADHD in adults. Data on a total of 125 patients (mean age 39±9 years) from three previous placebo-controlled studies were re-examined for the medications’ effects on blood pressure and heart rate.
Minor but statistically significant changes in blood pressure and heart rate were found to be associated with both stimulant and nonstimulant medications:
- systolic blood pressure (bupropion, +5.9 mm Hg [P<0.05]; amphetamine, +5.4 mm Hg [P<0.05])
- diastolic blood pressure (desipramine, +7.1 mm Hg [P<0.05])
- heart rate (bupropion, +6.9 bpm [P<0.05]; amphetamine, +7.3 bpm [P<0.05]; methylphenidate, +4.5 bpm [P<0.05]).
In the last two studies, the authors concluded that although the cardiovascular effects of ADHD medications in healthy adults were minimal, clinicians should monitor vital signs at baseline and periodically during treatment.
Dr. Rostain: How were the warnings clarified?
Dr. Adler: The FDA has changed the language. Now physicians are warned that sudden death can occur at usual doses in patients with a pre-existing structural cardiac abnormality or other serious heart problem. So, stimulants generally should not be used in children or adolescents with known serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, or other serious cardiac problems that may place them at increased vulnerability to the sympathomimetic effects of a stimulant drug.
Dr. Rostain: What about adults?
Dr. Adler: The language is the same for adults. Adults have a greater likelihood than children of having a history of serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, coronary artery disease, or other cardiac problems. Adults with such abnormalities generally should not be treated with stimulant drugs.
Dr. Rostain: What’s the impact for clinicians?
Dr. Adler: Clinicians have known that stimulants should not be used in patients with significant pre-existing cardiovascular conditions. That generally includes structural abnormalities such as serious heart murmurs and abnormalities of the electro-conduction of the impulse through the heart. When patients present with a history of cardiac abnormalities, clinicians should speak to the pediatrician, primary care physician (PCP), or cardiologist, go over the risk factors, and decide whether these medications can be prescribed for the patient.
Dr. Rostain: Should psychiatrists perform screening tests before prescribing stimulants? When should they consult with a specialist?
Dr. Adler: There is no recommendation in the prescribing information. But clearly a clinician should determine whether a patient has structural cardiac abnormalities or serious heart problems. That means taking a history about heart murmur, syncope, or other serious heart problems. Also, you want to know if the patient is hypertensive. The burden is on the prescribing clinician.
Dr. Rostain: Suppose you have a patient with hypertension or a history of a heart condition, should that patient first be evaluated by a cardiologist? What about a screening ECG?
Dr. Adler: There are no specific recommendations. If clinicians have questions about prescribing the medication, they should consult with the patient’s PCP or cardiologist.
Dr. Rostain: Let’s say the patient has some heart issues, but the PCP or pediatrician gives the goahead to prescribe stimulants. What sort of monitoring do you recommend?
Dr. Adler: I can’t answer that directly. Clearly, you’re going to want to partner with the PCP to establish a plan of how to carefully monitor this patient. FDA guidelines recommend ongoing blood pressure monitoring, especially if the patient is hypertensive, but do not specify how often.
Dr. Rostain: What alternatives do psychiatrists have when treating ADHD in patients in whom stimulants may pose some risk?
Dr. Adler: The only approved nonstimulant ADHD medication is atomoxetine, the labeling of which carries language about possible effects on blood pressure. The FDA warning about structural cardiac abnormalities has not been extended to atomoxetine, but blood pressure needs to be monitored. Whether our medical colleagues feel comfortable using a nonstimulant in patients with structural cardiac abnormalities has not been determined.
Dr. Rostain: In the absence of guidelines in the new warnings on stimulants, are there any studies to help clinicians with treatment and monitoring?
Dr. Adler: There’s very little data. A group at Massachusetts General Hospital has been studying the effects of ADHD medication on adults with hypertension (Box 2).4 That’s a different issue than a structural cardiac abnormality, but at least we have some data. This group found that you can safely give stimulants to hypertensive patients by partnering with medical colleagues and monitoring the patient carefully. Antihypertensive dosages may need to be adjusted during psychostimulant treatment.
Dr. Rostain: How do you choose a medication if your patient has a structural heart abnormality?
Dr. Adler: Again, we don’t have a lot of data. The decision would depend on the cardiac abnormality and the consulting physician’s comfort level. Keep in mind that psychostimulants have a short duration of effect, so the effects of the medication can dissipate fairly quickly. Again, the decision to medicate a patient with pre-existing cardiac abnormalities must be done with medical guidance.
In a short-term, open-label trial by Wilens et al,4 13 adults with ADHD and hypertension received mixed amphetamine salts extended-release (MAS-XR), up to 60 mg/d, for 6 weeks (phase 1), then discontinued MAS-XR for 2 weeks (phase 2). All patients had normal blood pressure (<135/85 mm Hg) for at least 4 weeks before entering the study and received a comprehensive clinical assessment, including ECG. Blood pressure was measured manually at each clinic visit.
Single episodes of hypertension (>140/90 mm Hg) occurred at similar rates in each phase, but these episodes were not sustained at any two consecutive visits. Group mean systolic and diastolic blood pressures and pulse did not increase during stimulant treatment. No clinically significant ECG changes were observed, and no serious adverse events occurred.
The authors concluded that this preliminary trial suggests that adults with ADHD and controlled hypertension can be safely treated with stimulant medications.
Dr. Rostain: So are you saying clinicians should make decisions about prescribing stimulants for patients with ADHD on a case-by-case basis?
Dr. Adler: Exactly.
Dr. Rostain: What about children and adolescents who have unknown structural heart defects? A lot of parents are concerned about reports of sudden cardiac death in young athletes, such as when playing soccer or basketball. Is there any way for practitioners to protect children with ADHD from an unexpected event?
Dr. Adler: In general, stimulants are safe medications, but we don’t have guidelines to help us determine who will need an ECG and who will not. Children are less likely to have had an ECG in the past than an adult, so it’s important to do a history, obtain input from the pediatrician or PCP, and clearly review the risks and benefits of medication therapy with the patient’s family.
Dr. Rostain: What would you advise clinicians to tell parents of children with ADHD or adult patients who have concerns about the new labeling on stimulants?
Dr. Adler: It would be a shame if patients were not receiving treatment for ADHD because of unfounded medical concerns. When these medications are used appropriately, they have dramatic and positive affects on ADHD.
ADHD is common and highly impairing. Deciding not to treat it has serious consequences in terms of divorce, separation, underperformance in school and on the job, unemployment, smoking, substance use, and issues with motor vehicle accidents and driving.
The goal of treatment is for our patients to get better, and ADHD is highly treatable with medication. But we must be cognizant of the warnings and prescribe medications appropriately. The message is that we’ve got to work collaboratively with our partners in medicine and, in the absence of guidelines, use good common sense.
Related resources
- Wilens TE, Hammerness PG, Biederman J, et al. Blood pressure changes associated with medication treatment of adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry 2005;66:253-9.
Drug brand names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Methylphenidate • Concerta, Ritalin
- Mixed amphetamine salts • Adderall
- Pemoline • Cylert
Disclosures
Dr. Adler is a consultant to and receives grant/research support from Abbott Laboratories, Cephalon, Cortex Pharmaceuticals, Eli Lilly and Company, New River Pharmaceuticals, Novartis Pharmaceuticals Corp., Ortho-McNeil, Pfizer, and Shire. He also receives grant/research support from Bristol-Myers Squibb and Merck and Co., and is a speaker for Eli Lilly and Company.
Dr. Rostain is a consultant to Shire and a speaker for Eli Lilly and Company and Ortho-McNeil.
1. Wilens TE, Biederman J, Lerner M. Concerta Study Group. Effects of once-daily osmotic-release methylphenidate on blood pressure and heart rate in children with attention-deficit/hyperactivity disorder: results from a one-year follow-up study. J Clin Psychopharmacol 2004;24(1):36-41.
2. Biederman J, Spencer TJ, Wilens TE, et al. Long-term safety and effectiveness of mixed amphetamine salts extended release in adults with ADHD. CNS Spectr 2005;10(12 suppl 20):16-25.
3. Wilens TE, Hammerness PG, Biederman J, et al. Blood pressure changes associated with medication treatment of adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry 2005;66(2):253-9.
4. Wilens TE, Zusman RM, Hammerness PG, et al. An open-label study of the tolerability of mixed amphetamine salts in adults with attention-deficit/ hyperactivity disorder and treated primary essential hypertension. J Clin Psychiatry 2006;67(5):696-702.
Dr. Adler is associate professor of psychiatry and director of the adult ADHD program at New York University Medical Center. He recently published a book for patients, Scattered Minds: Hope and Help for Adults with ADHD.
Dr. Rostain is associate professor of psychiatry and pediatrics and director of education, department of psychiatry, University of Pennsylvania School of Medicine, Philadelphia.
In August the FDA called for new warnings on stimulants used for attention-deficit/hyperactivity disorder (ADHD). Amphetamines now carry black box warnings that say, “Misuse of amphetamines may cause sudden death and serious cardiovascular adverse events.” Amphetamines and methylphenidates used for ADHD include expanded information about cardiovascular risks at usual dosages for patients with heart conditions.
To examine the clinical implications of these warnings, Current Psychiatry hosted a conversation between ADHD experts Anthony Rostain, MD, MA, and Lenard Adler, MD.
Dr. Rostain: Changes to warnings on ADHD medications have many psychiatrists looking for guidance on using stimulants. Can you give us some background and discuss the labeling changes?
Dr. Adler: Stimulants have been used for more than 40 years as ADHD treatments, and they’ve been shown to be highly effective. The FDA, which monitors issues of cardiovascular safety and stimulants in an ongoing way, examined specific isolated cases and changed some of the warnings as a result.
Dr. Rostain: What should practicing psychiatrists be concerned about if they’re thinking of prescribing stimulants for an ADHD patient?
Dr. Adler: The take-home point is that stimulants—because of the way they work—have been known to have minor effects of increasing blood pressure and pulse (Box 1).1-3 Clinicians have known about issues regarding stimulant use by patients with pre-existing cardiovascular conditions, but now the warnings are more formal for the methylphenidate and amphetamine products.
Dr. Rostain: An FDA committee recommended black box warnings on all stimulants used for ADHD, but the FDA decided instead to clarify warnings in prescription information for some medications. What was the FDA process?
Dr. Adler: The discussion was internal at the FDA, so I can’t say what their thinking was. The black box warning on amphetamines notes two issues. One is the potential for abuse and diversion, and the other warns of potential for sudden death and serious cardiovascular effects if the drug is misused. A warning has also been placed on all methylphenidate products regarding cardiovascular risk for patients with pre-existing cardiovascular conditions, but it is not a black box warning.
in healthy children and adults
Researchers at Massachusetts General Hospital have examined the effects of ADHD medications on blood pressure and heart rate in children and adults.
Children and adolescents. The first study1 was a 1-year extension of an open-label trial of once-daily, osmotic-release methylphenidate (MPH) in 432 children (age 6 to 13) with ADHD. Their blood pressure and heart rate were recorded at baseline and monthly.
At 12 months, MPH use at 18 to 54 mg/d was associated with minor but statistically significant mean increases in:
- systolic blood pressure (3.3 mm Hg [P<0.001])
- diastolic blood pressure (1.5 mm Hg [P<0.001])
- heart rate (3.9 bpm [P<0.0001]).
Adults. In a 24-month study,2 223 healthy adults with ADHD (age≥18) received mixed amphetamine salts extended-release (MAS XR) in an open-label extension of a 4-week, double-blind, placebo-controlled trial. MAS XR was started at 20 mg/d for 1 week, then increased up to 60 mg/d based on therapeutic effect, as measured by the ADHD Rating Scale IV.
Blood pressure and pulse were measured at baseline, weekly, then monthly, and 12-lead ECGs were obtained at baseline, weekly, then at 3- and 6-month intervals. Changes after 2 years were small and not statistically significant:
- systolic blood pressure (2.3±12.5 mm Hg)
- diastolic blood pressure (1.3±9.2 mm Hg)
- pulse (2.1±13.4 bpm).
A clinically insignificant increase was observed in the mean QTcB interval (7.2 msec; P<0.001), although no patient’s QTcB interval exceeded 480 msec. Seven patients dropped out because of cardiovascular side effects (5 with hypertension, and 2 with palpitation/tachycardia), which were not reported as being serious.
Stimulants and nonstimulants. In another study,3 the same researchers analyzed the cardiovascular effects of three stimulants (methylphenidate, amphetamine compounds, and pemoline) and two nonstimulants (bupropion and desipramine) used to treat ADHD in adults. Data on a total of 125 patients (mean age 39±9 years) from three previous placebo-controlled studies were re-examined for the medications’ effects on blood pressure and heart rate.
Minor but statistically significant changes in blood pressure and heart rate were found to be associated with both stimulant and nonstimulant medications:
- systolic blood pressure (bupropion, +5.9 mm Hg [P<0.05]; amphetamine, +5.4 mm Hg [P<0.05])
- diastolic blood pressure (desipramine, +7.1 mm Hg [P<0.05])
- heart rate (bupropion, +6.9 bpm [P<0.05]; amphetamine, +7.3 bpm [P<0.05]; methylphenidate, +4.5 bpm [P<0.05]).
In the last two studies, the authors concluded that although the cardiovascular effects of ADHD medications in healthy adults were minimal, clinicians should monitor vital signs at baseline and periodically during treatment.
Dr. Rostain: How were the warnings clarified?
Dr. Adler: The FDA has changed the language. Now physicians are warned that sudden death can occur at usual doses in patients with a pre-existing structural cardiac abnormality or other serious heart problem. So, stimulants generally should not be used in children or adolescents with known serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, or other serious cardiac problems that may place them at increased vulnerability to the sympathomimetic effects of a stimulant drug.
Dr. Rostain: What about adults?
Dr. Adler: The language is the same for adults. Adults have a greater likelihood than children of having a history of serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, coronary artery disease, or other cardiac problems. Adults with such abnormalities generally should not be treated with stimulant drugs.
Dr. Rostain: What’s the impact for clinicians?
Dr. Adler: Clinicians have known that stimulants should not be used in patients with significant pre-existing cardiovascular conditions. That generally includes structural abnormalities such as serious heart murmurs and abnormalities of the electro-conduction of the impulse through the heart. When patients present with a history of cardiac abnormalities, clinicians should speak to the pediatrician, primary care physician (PCP), or cardiologist, go over the risk factors, and decide whether these medications can be prescribed for the patient.
Dr. Rostain: Should psychiatrists perform screening tests before prescribing stimulants? When should they consult with a specialist?
Dr. Adler: There is no recommendation in the prescribing information. But clearly a clinician should determine whether a patient has structural cardiac abnormalities or serious heart problems. That means taking a history about heart murmur, syncope, or other serious heart problems. Also, you want to know if the patient is hypertensive. The burden is on the prescribing clinician.
Dr. Rostain: Suppose you have a patient with hypertension or a history of a heart condition, should that patient first be evaluated by a cardiologist? What about a screening ECG?
Dr. Adler: There are no specific recommendations. If clinicians have questions about prescribing the medication, they should consult with the patient’s PCP or cardiologist.
Dr. Rostain: Let’s say the patient has some heart issues, but the PCP or pediatrician gives the goahead to prescribe stimulants. What sort of monitoring do you recommend?
Dr. Adler: I can’t answer that directly. Clearly, you’re going to want to partner with the PCP to establish a plan of how to carefully monitor this patient. FDA guidelines recommend ongoing blood pressure monitoring, especially if the patient is hypertensive, but do not specify how often.
Dr. Rostain: What alternatives do psychiatrists have when treating ADHD in patients in whom stimulants may pose some risk?
Dr. Adler: The only approved nonstimulant ADHD medication is atomoxetine, the labeling of which carries language about possible effects on blood pressure. The FDA warning about structural cardiac abnormalities has not been extended to atomoxetine, but blood pressure needs to be monitored. Whether our medical colleagues feel comfortable using a nonstimulant in patients with structural cardiac abnormalities has not been determined.
Dr. Rostain: In the absence of guidelines in the new warnings on stimulants, are there any studies to help clinicians with treatment and monitoring?
Dr. Adler: There’s very little data. A group at Massachusetts General Hospital has been studying the effects of ADHD medication on adults with hypertension (Box 2).4 That’s a different issue than a structural cardiac abnormality, but at least we have some data. This group found that you can safely give stimulants to hypertensive patients by partnering with medical colleagues and monitoring the patient carefully. Antihypertensive dosages may need to be adjusted during psychostimulant treatment.
Dr. Rostain: How do you choose a medication if your patient has a structural heart abnormality?
Dr. Adler: Again, we don’t have a lot of data. The decision would depend on the cardiac abnormality and the consulting physician’s comfort level. Keep in mind that psychostimulants have a short duration of effect, so the effects of the medication can dissipate fairly quickly. Again, the decision to medicate a patient with pre-existing cardiac abnormalities must be done with medical guidance.
In a short-term, open-label trial by Wilens et al,4 13 adults with ADHD and hypertension received mixed amphetamine salts extended-release (MAS-XR), up to 60 mg/d, for 6 weeks (phase 1), then discontinued MAS-XR for 2 weeks (phase 2). All patients had normal blood pressure (<135/85 mm Hg) for at least 4 weeks before entering the study and received a comprehensive clinical assessment, including ECG. Blood pressure was measured manually at each clinic visit.
Single episodes of hypertension (>140/90 mm Hg) occurred at similar rates in each phase, but these episodes were not sustained at any two consecutive visits. Group mean systolic and diastolic blood pressures and pulse did not increase during stimulant treatment. No clinically significant ECG changes were observed, and no serious adverse events occurred.
The authors concluded that this preliminary trial suggests that adults with ADHD and controlled hypertension can be safely treated with stimulant medications.
Dr. Rostain: So are you saying clinicians should make decisions about prescribing stimulants for patients with ADHD on a case-by-case basis?
Dr. Adler: Exactly.
Dr. Rostain: What about children and adolescents who have unknown structural heart defects? A lot of parents are concerned about reports of sudden cardiac death in young athletes, such as when playing soccer or basketball. Is there any way for practitioners to protect children with ADHD from an unexpected event?
Dr. Adler: In general, stimulants are safe medications, but we don’t have guidelines to help us determine who will need an ECG and who will not. Children are less likely to have had an ECG in the past than an adult, so it’s important to do a history, obtain input from the pediatrician or PCP, and clearly review the risks and benefits of medication therapy with the patient’s family.
Dr. Rostain: What would you advise clinicians to tell parents of children with ADHD or adult patients who have concerns about the new labeling on stimulants?
Dr. Adler: It would be a shame if patients were not receiving treatment for ADHD because of unfounded medical concerns. When these medications are used appropriately, they have dramatic and positive affects on ADHD.
ADHD is common and highly impairing. Deciding not to treat it has serious consequences in terms of divorce, separation, underperformance in school and on the job, unemployment, smoking, substance use, and issues with motor vehicle accidents and driving.
The goal of treatment is for our patients to get better, and ADHD is highly treatable with medication. But we must be cognizant of the warnings and prescribe medications appropriately. The message is that we’ve got to work collaboratively with our partners in medicine and, in the absence of guidelines, use good common sense.
Related resources
- Wilens TE, Hammerness PG, Biederman J, et al. Blood pressure changes associated with medication treatment of adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry 2005;66:253-9.
Drug brand names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Methylphenidate • Concerta, Ritalin
- Mixed amphetamine salts • Adderall
- Pemoline • Cylert
Disclosures
Dr. Adler is a consultant to and receives grant/research support from Abbott Laboratories, Cephalon, Cortex Pharmaceuticals, Eli Lilly and Company, New River Pharmaceuticals, Novartis Pharmaceuticals Corp., Ortho-McNeil, Pfizer, and Shire. He also receives grant/research support from Bristol-Myers Squibb and Merck and Co., and is a speaker for Eli Lilly and Company.
Dr. Rostain is a consultant to Shire and a speaker for Eli Lilly and Company and Ortho-McNeil.
In August the FDA called for new warnings on stimulants used for attention-deficit/hyperactivity disorder (ADHD). Amphetamines now carry black box warnings that say, “Misuse of amphetamines may cause sudden death and serious cardiovascular adverse events.” Amphetamines and methylphenidates used for ADHD include expanded information about cardiovascular risks at usual dosages for patients with heart conditions.
To examine the clinical implications of these warnings, Current Psychiatry hosted a conversation between ADHD experts Anthony Rostain, MD, MA, and Lenard Adler, MD.
Dr. Rostain: Changes to warnings on ADHD medications have many psychiatrists looking for guidance on using stimulants. Can you give us some background and discuss the labeling changes?
Dr. Adler: Stimulants have been used for more than 40 years as ADHD treatments, and they’ve been shown to be highly effective. The FDA, which monitors issues of cardiovascular safety and stimulants in an ongoing way, examined specific isolated cases and changed some of the warnings as a result.
Dr. Rostain: What should practicing psychiatrists be concerned about if they’re thinking of prescribing stimulants for an ADHD patient?
Dr. Adler: The take-home point is that stimulants—because of the way they work—have been known to have minor effects of increasing blood pressure and pulse (Box 1).1-3 Clinicians have known about issues regarding stimulant use by patients with pre-existing cardiovascular conditions, but now the warnings are more formal for the methylphenidate and amphetamine products.
Dr. Rostain: An FDA committee recommended black box warnings on all stimulants used for ADHD, but the FDA decided instead to clarify warnings in prescription information for some medications. What was the FDA process?
Dr. Adler: The discussion was internal at the FDA, so I can’t say what their thinking was. The black box warning on amphetamines notes two issues. One is the potential for abuse and diversion, and the other warns of potential for sudden death and serious cardiovascular effects if the drug is misused. A warning has also been placed on all methylphenidate products regarding cardiovascular risk for patients with pre-existing cardiovascular conditions, but it is not a black box warning.
in healthy children and adults
Researchers at Massachusetts General Hospital have examined the effects of ADHD medications on blood pressure and heart rate in children and adults.
Children and adolescents. The first study1 was a 1-year extension of an open-label trial of once-daily, osmotic-release methylphenidate (MPH) in 432 children (age 6 to 13) with ADHD. Their blood pressure and heart rate were recorded at baseline and monthly.
At 12 months, MPH use at 18 to 54 mg/d was associated with minor but statistically significant mean increases in:
- systolic blood pressure (3.3 mm Hg [P<0.001])
- diastolic blood pressure (1.5 mm Hg [P<0.001])
- heart rate (3.9 bpm [P<0.0001]).
Adults. In a 24-month study,2 223 healthy adults with ADHD (age≥18) received mixed amphetamine salts extended-release (MAS XR) in an open-label extension of a 4-week, double-blind, placebo-controlled trial. MAS XR was started at 20 mg/d for 1 week, then increased up to 60 mg/d based on therapeutic effect, as measured by the ADHD Rating Scale IV.
Blood pressure and pulse were measured at baseline, weekly, then monthly, and 12-lead ECGs were obtained at baseline, weekly, then at 3- and 6-month intervals. Changes after 2 years were small and not statistically significant:
- systolic blood pressure (2.3±12.5 mm Hg)
- diastolic blood pressure (1.3±9.2 mm Hg)
- pulse (2.1±13.4 bpm).
A clinically insignificant increase was observed in the mean QTcB interval (7.2 msec; P<0.001), although no patient’s QTcB interval exceeded 480 msec. Seven patients dropped out because of cardiovascular side effects (5 with hypertension, and 2 with palpitation/tachycardia), which were not reported as being serious.
Stimulants and nonstimulants. In another study,3 the same researchers analyzed the cardiovascular effects of three stimulants (methylphenidate, amphetamine compounds, and pemoline) and two nonstimulants (bupropion and desipramine) used to treat ADHD in adults. Data on a total of 125 patients (mean age 39±9 years) from three previous placebo-controlled studies were re-examined for the medications’ effects on blood pressure and heart rate.
Minor but statistically significant changes in blood pressure and heart rate were found to be associated with both stimulant and nonstimulant medications:
- systolic blood pressure (bupropion, +5.9 mm Hg [P<0.05]; amphetamine, +5.4 mm Hg [P<0.05])
- diastolic blood pressure (desipramine, +7.1 mm Hg [P<0.05])
- heart rate (bupropion, +6.9 bpm [P<0.05]; amphetamine, +7.3 bpm [P<0.05]; methylphenidate, +4.5 bpm [P<0.05]).
In the last two studies, the authors concluded that although the cardiovascular effects of ADHD medications in healthy adults were minimal, clinicians should monitor vital signs at baseline and periodically during treatment.
Dr. Rostain: How were the warnings clarified?
Dr. Adler: The FDA has changed the language. Now physicians are warned that sudden death can occur at usual doses in patients with a pre-existing structural cardiac abnormality or other serious heart problem. So, stimulants generally should not be used in children or adolescents with known serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, or other serious cardiac problems that may place them at increased vulnerability to the sympathomimetic effects of a stimulant drug.
Dr. Rostain: What about adults?
Dr. Adler: The language is the same for adults. Adults have a greater likelihood than children of having a history of serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, coronary artery disease, or other cardiac problems. Adults with such abnormalities generally should not be treated with stimulant drugs.
Dr. Rostain: What’s the impact for clinicians?
Dr. Adler: Clinicians have known that stimulants should not be used in patients with significant pre-existing cardiovascular conditions. That generally includes structural abnormalities such as serious heart murmurs and abnormalities of the electro-conduction of the impulse through the heart. When patients present with a history of cardiac abnormalities, clinicians should speak to the pediatrician, primary care physician (PCP), or cardiologist, go over the risk factors, and decide whether these medications can be prescribed for the patient.
Dr. Rostain: Should psychiatrists perform screening tests before prescribing stimulants? When should they consult with a specialist?
Dr. Adler: There is no recommendation in the prescribing information. But clearly a clinician should determine whether a patient has structural cardiac abnormalities or serious heart problems. That means taking a history about heart murmur, syncope, or other serious heart problems. Also, you want to know if the patient is hypertensive. The burden is on the prescribing clinician.
Dr. Rostain: Suppose you have a patient with hypertension or a history of a heart condition, should that patient first be evaluated by a cardiologist? What about a screening ECG?
Dr. Adler: There are no specific recommendations. If clinicians have questions about prescribing the medication, they should consult with the patient’s PCP or cardiologist.
Dr. Rostain: Let’s say the patient has some heart issues, but the PCP or pediatrician gives the goahead to prescribe stimulants. What sort of monitoring do you recommend?
Dr. Adler: I can’t answer that directly. Clearly, you’re going to want to partner with the PCP to establish a plan of how to carefully monitor this patient. FDA guidelines recommend ongoing blood pressure monitoring, especially if the patient is hypertensive, but do not specify how often.
Dr. Rostain: What alternatives do psychiatrists have when treating ADHD in patients in whom stimulants may pose some risk?
Dr. Adler: The only approved nonstimulant ADHD medication is atomoxetine, the labeling of which carries language about possible effects on blood pressure. The FDA warning about structural cardiac abnormalities has not been extended to atomoxetine, but blood pressure needs to be monitored. Whether our medical colleagues feel comfortable using a nonstimulant in patients with structural cardiac abnormalities has not been determined.
Dr. Rostain: In the absence of guidelines in the new warnings on stimulants, are there any studies to help clinicians with treatment and monitoring?
Dr. Adler: There’s very little data. A group at Massachusetts General Hospital has been studying the effects of ADHD medication on adults with hypertension (Box 2).4 That’s a different issue than a structural cardiac abnormality, but at least we have some data. This group found that you can safely give stimulants to hypertensive patients by partnering with medical colleagues and monitoring the patient carefully. Antihypertensive dosages may need to be adjusted during psychostimulant treatment.
Dr. Rostain: How do you choose a medication if your patient has a structural heart abnormality?
Dr. Adler: Again, we don’t have a lot of data. The decision would depend on the cardiac abnormality and the consulting physician’s comfort level. Keep in mind that psychostimulants have a short duration of effect, so the effects of the medication can dissipate fairly quickly. Again, the decision to medicate a patient with pre-existing cardiac abnormalities must be done with medical guidance.
In a short-term, open-label trial by Wilens et al,4 13 adults with ADHD and hypertension received mixed amphetamine salts extended-release (MAS-XR), up to 60 mg/d, for 6 weeks (phase 1), then discontinued MAS-XR for 2 weeks (phase 2). All patients had normal blood pressure (<135/85 mm Hg) for at least 4 weeks before entering the study and received a comprehensive clinical assessment, including ECG. Blood pressure was measured manually at each clinic visit.
Single episodes of hypertension (>140/90 mm Hg) occurred at similar rates in each phase, but these episodes were not sustained at any two consecutive visits. Group mean systolic and diastolic blood pressures and pulse did not increase during stimulant treatment. No clinically significant ECG changes were observed, and no serious adverse events occurred.
The authors concluded that this preliminary trial suggests that adults with ADHD and controlled hypertension can be safely treated with stimulant medications.
Dr. Rostain: So are you saying clinicians should make decisions about prescribing stimulants for patients with ADHD on a case-by-case basis?
Dr. Adler: Exactly.
Dr. Rostain: What about children and adolescents who have unknown structural heart defects? A lot of parents are concerned about reports of sudden cardiac death in young athletes, such as when playing soccer or basketball. Is there any way for practitioners to protect children with ADHD from an unexpected event?
Dr. Adler: In general, stimulants are safe medications, but we don’t have guidelines to help us determine who will need an ECG and who will not. Children are less likely to have had an ECG in the past than an adult, so it’s important to do a history, obtain input from the pediatrician or PCP, and clearly review the risks and benefits of medication therapy with the patient’s family.
Dr. Rostain: What would you advise clinicians to tell parents of children with ADHD or adult patients who have concerns about the new labeling on stimulants?
Dr. Adler: It would be a shame if patients were not receiving treatment for ADHD because of unfounded medical concerns. When these medications are used appropriately, they have dramatic and positive affects on ADHD.
ADHD is common and highly impairing. Deciding not to treat it has serious consequences in terms of divorce, separation, underperformance in school and on the job, unemployment, smoking, substance use, and issues with motor vehicle accidents and driving.
The goal of treatment is for our patients to get better, and ADHD is highly treatable with medication. But we must be cognizant of the warnings and prescribe medications appropriately. The message is that we’ve got to work collaboratively with our partners in medicine and, in the absence of guidelines, use good common sense.
Related resources
- Wilens TE, Hammerness PG, Biederman J, et al. Blood pressure changes associated with medication treatment of adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry 2005;66:253-9.
Drug brand names
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Methylphenidate • Concerta, Ritalin
- Mixed amphetamine salts • Adderall
- Pemoline • Cylert
Disclosures
Dr. Adler is a consultant to and receives grant/research support from Abbott Laboratories, Cephalon, Cortex Pharmaceuticals, Eli Lilly and Company, New River Pharmaceuticals, Novartis Pharmaceuticals Corp., Ortho-McNeil, Pfizer, and Shire. He also receives grant/research support from Bristol-Myers Squibb and Merck and Co., and is a speaker for Eli Lilly and Company.
Dr. Rostain is a consultant to Shire and a speaker for Eli Lilly and Company and Ortho-McNeil.
1. Wilens TE, Biederman J, Lerner M. Concerta Study Group. Effects of once-daily osmotic-release methylphenidate on blood pressure and heart rate in children with attention-deficit/hyperactivity disorder: results from a one-year follow-up study. J Clin Psychopharmacol 2004;24(1):36-41.
2. Biederman J, Spencer TJ, Wilens TE, et al. Long-term safety and effectiveness of mixed amphetamine salts extended release in adults with ADHD. CNS Spectr 2005;10(12 suppl 20):16-25.
3. Wilens TE, Hammerness PG, Biederman J, et al. Blood pressure changes associated with medication treatment of adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry 2005;66(2):253-9.
4. Wilens TE, Zusman RM, Hammerness PG, et al. An open-label study of the tolerability of mixed amphetamine salts in adults with attention-deficit/ hyperactivity disorder and treated primary essential hypertension. J Clin Psychiatry 2006;67(5):696-702.
Dr. Adler is associate professor of psychiatry and director of the adult ADHD program at New York University Medical Center. He recently published a book for patients, Scattered Minds: Hope and Help for Adults with ADHD.
Dr. Rostain is associate professor of psychiatry and pediatrics and director of education, department of psychiatry, University of Pennsylvania School of Medicine, Philadelphia.
1. Wilens TE, Biederman J, Lerner M. Concerta Study Group. Effects of once-daily osmotic-release methylphenidate on blood pressure and heart rate in children with attention-deficit/hyperactivity disorder: results from a one-year follow-up study. J Clin Psychopharmacol 2004;24(1):36-41.
2. Biederman J, Spencer TJ, Wilens TE, et al. Long-term safety and effectiveness of mixed amphetamine salts extended release in adults with ADHD. CNS Spectr 2005;10(12 suppl 20):16-25.
3. Wilens TE, Hammerness PG, Biederman J, et al. Blood pressure changes associated with medication treatment of adults with attention-deficit/hyperactivity disorder. J Clin Psychiatry 2005;66(2):253-9.
4. Wilens TE, Zusman RM, Hammerness PG, et al. An open-label study of the tolerability of mixed amphetamine salts in adults with attention-deficit/ hyperactivity disorder and treated primary essential hypertension. J Clin Psychiatry 2006;67(5):696-702.
Dr. Adler is associate professor of psychiatry and director of the adult ADHD program at New York University Medical Center. He recently published a book for patients, Scattered Minds: Hope and Help for Adults with ADHD.
Dr. Rostain is associate professor of psychiatry and pediatrics and director of education, department of psychiatry, University of Pennsylvania School of Medicine, Philadelphia.
Treatment-resistant depression: Are atypical antipsychotics effective and safe enough?
Adding second-generation antipsychotics (SGAs) may boost the effectiveness of antidepressants in treatment-resistant unipolar major depression. Exactly when to try SGAs remains unclear, however, given their potential for adverse effects.
Major depression often is severe and chronic, and many patients remain ill even after multiple rounds of treatment. For patients without psychosis, where do SGAs fit into an algorithm for treatment-resistant depression?
This article examines the evidence on antipsychotic augmentation and discusses issues to consider—effectiveness, adverse effects, therapeutic dosages, and the patient’s quality of life—in making your clinical decisions.
Antidepressants alone
An optimal trial. Most depressed patients do not experience full response after initial antidepressant treatment, even with optimal therapeutic trials. An optimal trial means maintaining the maximum tolerated dosage within the antidepressant’s typical therapeutic range for at least 3 weeks.1 Reported remission rates from initial and second-line treatments include:
- one-third of patients after a vigorous initial trial of citalopram in a National Institute of Mental Health study2
- 20% to 30% of patients given citalopram plus bupropion or buspirone3 or switched to bupropion, sertraline, or venlafaxine4
- 50% of patients treated for depression in a primary care practice during the first 2 years after an initial antidepressant prescription.5
Subsequent options. In addition to various monotherapies and combinations, many options have been proposed for managing nonresponse to initial antidepressant therapy (Table 1). These include:
- augmenting with lithium, thyroid hormone, pindolol, or estrogen
- switching to a drug in another therapeutic class, such as a tricyclic antidepressant or monoamine oxidase inhibitor
- adding cognitive-behavioral therapy.7
Depression is often chronic and disabling. Selective serotonin reuptake inhibitors (SSRIs) are the mainstay of treatment, but recent data suggest that:
- few patients achieve therapeutic remission with initial SSRIs
- relapse or recurrence after remission is common.6
Clinically, this means psychiatrists contend with treatment resistance in nearly all patients with major depression.
Chronic, inadequately treated depression has a pervasive, adverse effect on patients’ quality of life, impairing the ability to work and perform social roles such as parenting. Even when an antidepressant produces partial response, considerable impairment remains. Depressed patients who do not achieve full therapeutic remission remain in this partially remitted, disabled state throughout treatment.8
Aggressive and persistent management is the key to effectively treating major depression.
Therapeutic suggestions when an SSRI does not lead to remission*
| Pharmacotherapy | Example | Recommended dosing |
|---|---|---|
| Monotherapy | An SNRI such as: | |
| Duloxetine | 30 to 120 mg/d | |
| Venlafaxine XR | 150 to 375 mg/d | |
| Combination therapies with SSRIs | Bupropion | 200 to 400 mg/d |
| Buspirone | 30 to 60 mg/d | |
| Augmentation | Lithium | 900 to 1,200 mg/d |
| Thyroid hormone | 25 mcg/d | |
| Pindolol | 5 to 30 mg bid | |
| Estrogen (such as 17a-estradiol) | 100 mcg/d | |
| Switch to another Tricyclic antidepressant class | Tricyclic | |
| Imipramine | 150 to 250 mg/d* | |
| Nortriptyline | 75 to 200 mg/d* | |
| Desipramine | 150 to 250 mg/d* | |
| MAOI | ||
| Phenelzine | 30 to 60 mg/d | |
| Tranylcypromine | 20 to 60 mg/d | |
| Selegiline (patch) | 9 to 12 mg/patch/day | |
| * Suggestions are not listed in stepwise order | ||
| MAOI: monoamine oxidase inhibitor | ||
| SNRI: serotonin-norepinephrine reuptake inhibitor | ||
| SSRI: selective serotonin reuptake inhibitor | ||
Atypicals for unipolar depression
Why atypicals? Researchers are investigating the use of SGAs in treatment-resistant mood disorders because of these drugs’ unique psychopharmacologic properties (Box 2).9-11
Except for clozapine, all available SGAs—aripiprazole, olanzapine, quetiapine, risperidone, and ziprasidone—are FDA-approved for acute bipolar mania. Evidence also strongly supports the benefits of quetiapine12 and the fixed-dose olanzapine/ fluoxetine combination13 for bipolar depression. Olanzapine/fluoxetine—originally studied for use in treatment-resistant unipolar depression—is approved for bipolar depression.14
Robust response. An uncontrolled case series first suggested that an SGA might help treat unipolar depression after initial selective serotonin reuptake inhibitors (SSRIs) fail to achieve remission. Ostroff and Nelson15 enrolled 8 outpatients (5 men, 3 women, ages 36 to 75) with nonpsychotic unipolar major depression that did not respond to initial fluoxetine or paroxetine. Patients had been taking fluoxetine, 20 to 40 mg/d, for 6 weeks to 4 months or paroxetine, 10 to 30 mg/d, for 2 to 8 weeks.
Patients reported a robust clinical effect within days after risperidone, 0.5 to 1.0 mg/d, was added to the SSRIs. Depression symptoms dropped to remission levels within 1 week, as measured by baseline and follow-up Hamilton Rating Scale for Depression (HAM-D) scores.
Olanzapine/fluoxetine. Our group subsequently enrolled 28 nonpsychotic patients with unipolar depression in a double-blind, placebo-controlled trial.14 We first treated these patients—who had not responded adequately to an SSRI or an antidepressant from another class—with open-label fluoxetine, up to 60 mg/d. Those whose scores on depression rating scales improved by ≥30% were excluded from the double-blind phase, when we randomly assigned the remaining patients to:
- olanzapine, mean 12.5 mg/d, plus placebo (n=8)
- a continuation of fluoxetine, mean 52 mg/d, plus placebo (n=10)
- or olanzapine/fluoxetine, mean 13.5/52 mg/d (n=10).
Final depression remission rates (HAM-D score ≤8 for 2 weeks) were:
- 60% with olanzapine/fluoxetine
- 25% with olanzapine alone
- 20% with continuation fluoxetine.
Until recently, researchers had been unable to replicate these results or extend this study in larger populations because of high response rates in the monotherapy treatment groups.16,17 In May 2006, however, Thase et al18 presented data from a large-scale replication trial that confirmed the finding of a more robust effect with fixed-dose olanzapine/fluoxetine in unipolar major depression, compared with olanzapine or fluoxetine monotherapy.
Second-generation antipsychotics (SGAs) differ from first-generation antipsychotics (FGAs) in their putative mechanisms of action.
FGAs’ antipsychotic effects depend largely on central dopamine type 2 (D2) receptor blockade. Their additional receptor-binding characteristics—blocking cholinergic, histamine, and alpha adrenergic receptors—appear to confer side effects but no added therapeutic benefit.9
SGAs bind weakly to D2 receptors and in varying degrees to serotonin (5-HT) receptors, including 5-HT subtypes 1A, 2A, 2C, 5, 7, and others. The SGAs also have other transmitter effects.10 On balance, the SGAs’ effects are more complex than those of the FGAs.
SGAs are called “atypical” because their beneficial and adverse clinical actions do not follow the FGAs’ usual pattern. FGAs’ relative potency in reducing psychosis is proportional to the propensity to cause extrapyramidal symptoms (EPS). Both the clinical effect and EPS are functions of D2 receptor blockade.11 In contrast, clozapine—the prototypical SGA—is a potent antipsychotic that exerts essentially no EPS.
Compared with FGAs, clozapine’s more complicated psychopharmacology has been shown to produce an enhanced effect on negative, cognitive, and mood symptoms in some patients with schizophrenia.10
- sertraline, 100 to 200 mg/d
- sertraline plus ziprasidone, 80 mg/d
- or sertraline plus ziprasidone, 160 mg/d.
Risperidone. One three-phase study21 evaluated the long-term efficacy of adding risperidone to citalopram in 489 patients with treatment-resistant depression. The design was:
- phase 1: 4 to 6 weeks of open-label citalopram, 20 to 60 mg/d (N=489)
- phase 2: 4 to 6 weeks of citalopram plus open-label risperidone, 0.25 to 2 mg/d (N=386)
- phase 3: 24 weeks of citalopram plus double-blind risperidone or placebo (N=241).
Median time to relapse in phase 3 was 102 days with risperidone augmentation and 85 days with placebo—not a statistically significant difference. Relapse rates were 53.3% with risperidone and 54.6% in the control group. These results suggest that risperidone had an initial acute effect that was not sustained.
In another study,22 463 depressed patients received an optimized antidepressant trial. The 274 who did not respond sufficiently were randomly assigned to risperidone, 1 to 2 mg/d, or placebo for 6 weeks. Mean HAM-D scores fell from 24.2 to 15.2 in the risperidone group and from 24.6 to 17.5 in the control group—a modest but statistically significant difference in favor of risperidone. The baseline-toendpoint change in this study is similar to that reported in a trial of risperidone, 1 to 4 mg/d, plus paroxetine, 20 to 40 mg/d, in bipolar depression.23
Shelton24 compared the effectiveness of adding risperidone or bupropion to SSRIs and serotonin norepinephrine reuptake inhibitors (SNRIs) for 6 weeks. Risperidone and bupropion were similarly effective as augmentation, but risperidone had a more rapid effect—producing statistically significant greater benefits within the first week of treatment.
Aripiprazole. Two open-label trials showed that aripiprazole combined with SSRIs exerts generally beneficial effects in treatment-resistant depression.25,26 Simon and Nemeroff25 began by adding aripiprazole at 10 mg/d, but emerging akathisia prompted them to reduce the starting dosage to 2.5 mg/d.
Barbee et al27 reported the results of a retrospective case series of aripiprazole augmentation in depressed patients who had not responded adequately to multiple medication trials, including SGAs. Fourteen of 30 patients (46.7%) were rated “much improved” or “very improved” with added aripiprazole, based on Prospective Global Assessment of Functioning and Clinical Global Impressions- Improvement scores. But 9 patients (30%) did not complete the full course of therapy, and 6 of the 14 responders (42.9%) relapsed while taking aripiprazole. The net response rate across 6 weeks was 27%.
Although this study involved only aripiprazole, the results suggest that trying a second SGA may not be more effective after a first SGA fails to improve treatment-resistant depression.
Quetiapine. A 9-week, open-label, variable-dose study of 11 patients28 first suggested that augmenting SSRIs with quetiapine could improve residual anxiety in resistant depression. Subsequently, 112 patients with major depression and anxiety were randomly assigned to single-blind treatment with paroxetine, ≥60 mg/d, with or without quetiapine, ≥200 mg/d. After 8 weeks, the 58 patients receiving quetiapine augmentation showed greater improvement than the 54 receiving SSRI monotherapy, based on Hamilton Anxiety Scale (HAM-A) and HAM-D scores.29
Adding quetiapine to antidepressant therapy was then examined in a randomized, placebo-controlled trial by McIntyre et al.30 Fifty-eight patients with unipolar depression who had not responded adequately after 6 weeks of SSRI or SNRI therapy were randomly assigned to quetiapine, 50 to 600 mg/d (mean dose 202±93 mg/d) or placebo for 8 weeks. Adjunctive quetiapine was significantly more effective than placebo, as measured by HAM-D scores. Patients receiving quetiapine also showed significantly better HAM-A scores at all points except week 8.
The dropout rate was relatively high for both groups—11 of 29 (38%) receiving quetiapine and 13 of 29 (45%) receiving placebo. The main reasons for discontinuation were side effects with quetiapine (sedation, dry mouth, and weight gain) and lack of effect with placebo.
These results are similar to those of another double-blind, placebo-controlled trial,31 in which 32 patients with SSRI/SNRI-resistant depression received adjunctive quetiapine, 200 to 400 mg/d (mean 268 mg/d) or placebo for 8 weeks.
Though small, these studies indicate that quetiapine may be effective as augmentation for treatment-resistant unipolar depression. Controlled data from a larger study are needed.
Discussion. Because of insufficient data, we do not know if SGAs are equivalent when used to augment antidepressant therapy in unipolar major depression. Olanzapine has been studied more than other SGAs in treatmentresistant depression and has shown efficacy in several—but not all—short- and long-term augmentation trials. Evidence on other SGAs is limited, and no head-to-head comparisons have been reported.
Adverse effects
Some SGAs may be effective in treatment-resistant depression, but any discussion of using them must also include their potential for adverse effects.
Weight gain and subsequent metabolic syndrome have been associated with olanzapine and—to a lesser degree—with quetiapine and risperidone. Ziprasidone and aripiprazole have relatively little effect on patients’ weight.
Extrapyramidal symptoms. All SGAs carry a risk of tardive dyskinesia. The risk is lower with SGAs than with first-generation antipsychotics (FGAs) but is an important clinical consideration.32
Hyperprolactinemia. Risperidone has been associated with an elevated risk of hyperprolactinemia, although less than that associated with FGAs.33 This risk does not appear to be a problem with quetiapine34 and aripiprazole;35 it is low with olanzapine (except at higher dosages);36 and the prolactin increase associated with ziprasidone may resolve within the first month of treatment.37
Prescribing rationale
‘Overcautious’ treatment. Even with careful management of side effects, SGAs are not preferred to strategies such as switching antidepressants or adding bupropion for treatment-resistant unipolar depression. But do not exclude SGAs solely because of their potential for adverse effects.
I am concerned about anecdotal reports of overcautious clinicians basing medication choices largely on safety—and, by extension, legal—considerations rather than on effectiveness. Certainly, safety concerns should prevail when two options are equally effective. But we do our patients no service by selecting ineffective drugs just because they have a low potential for adverse effects or by dosing effective drugs below the therapeutic range (Table 2).
When a drug is effective and may be the best choice for the patient, the question becomes, “Can I manage the potential for adverse effects?” When prescribing SGAs, it is important to monitor patients’ weight and serum lipid and glucose levels and regularly to look for abnormal involuntary movements.
An important question remains: Where do SGAs belong in the hierarchy of treatment options? Unfortunately, treatment guidelines for depression do not typically mention antipsychotics. Because of relative safety issues, two trials of monotherapies of different classes and, perhaps, combination therapy with bupropion would come before SGAs. However, it remains unclear exactly where.
SGAs probably belong ahead of electroconvulsive therapy or vagal nerve stimulation. But should they come before augmentation with lithium or thyroid hormone? Or, for that matter, trials of tricyclics or monoamine oxidase inhibitors?
Unfortunately, the available evidence provides little guidance. For a list of therapeutic algorithms developed for treatment-resistant depression, see Related resources.
Table 2
Recommended dosing of SGAs to augment antidepressant therapy
| Medication | Therapeutic range (mg/d) |
|---|---|
| Aripiprazole | 5 to 30 |
| Olanzapine | 5 to 20 |
| Quetiapine | 100 to 400 |
| Risperidone | 2 to 4 |
| Ziprasidone | 80 to 160 |
Algorithms for treatment-resistant depression
- Trivedi MH, Kern JK, Grannemann BD, et al. A computerized clinical decision support system as a means of implementing depression guidelines. Psychiatr Serv 2004;55(8);879-85.
- Rush AJ, Crismon ML, Kashner TM, et al. Texas Medication Algorithm Project, phase 3 (TMAP-3): rationale and study design. J Clin Psychiatry 2003;64(4);357-69.
- Trivedi M. Algorithms in clinical psychiatry: a stepped approach toward the path to recovery. Psychopharmacol Bull 2002;36(suppl 2);142-9.
- Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry 2001;62(suppl 6);22-9.
- Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on Medication Treatment of Major Depressive Disorder. J Clin Psychiatry 1999;60(3);142-56.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Buspirone • BuSpar
- Citalopram • Celexa
- Clozapine • Clozaril
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Imipramine • Tofranil
- Lithium • various
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Selegiline (patch) • EMSAM
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Shelton receives grant/research support from Eli Lilly and Co., GlaxoSmithKline, Pfizer, Janssen Pharmaceutica, sanofi-aventis, Wyeth Pharmaceuticals, AstraZeneca Pharmaceuticals, and Abbott Laboratories. He is a consultant to Pfizer and Janssen Pharmaceutica and a speaker for Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Pfizer, GlaxoSmithKline, Solvay Pharmaceuticals, Wyeth Pharmaceuticals, and Abbott Laboratories.
1. Shelton RC. The use of antidepressants in novel combination therapies. J Clin Psychiatry 2003;64(suppl 2):14-8.
2. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
3. Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006;354(12):1243-52.
4. Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006;354(12):1231-42.
5. Simon GE, Heiligenstein J, Revicki D, et al. Long-term outcomes of initial antidepressant drug choice in a “real world” randomized trial. Arch Fam Med 1999;8(4):319-25.
6. Nierenberg AA, Petersen TJ, Alpert JE. Prevention of relapse and recurrence in depression: the role of long-term pharmacotherapy and psychotherapy. J Clin Psychiatry 2003;64(suppl 15):13-17.
7. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342(20):1462-70.
8. Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990-2020: Global Burden of Disease Study. Lancet 1997;349(9064):1498-1504.
9. Bennett MR. Monoaminergic synapses and schizophrenia: 45 years of neuroleptics. J Psychopharmacol 1998;12(3):289-304.
10. Meltzer HY. The role of serotonin in antipsychotic drug action. Neuropsychopharmacology 1999;21(2 suppl):106S-115S.
11. Meltzer HY, Bastani B, Ramirez L, Matsubara S. Clozapine: new research on efficacy and mechanism of action. Eur Arch Psychiatry Neurol Sci 1989;238(5-6):332-9.
12. Hirschfeld RM, Weisler RH, Raines SR, Macfadden W. for the BOLDER Study Group. Quetiapine in the treatment of anxiety in patients with bipolar I or II depression: a secondary analysis from a randomized, double-blind, placebo-controlled study. J Clin Psychiatry 2006;67(3):355-62.
13. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60(11):1079-88.
14. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158(1):131-4.
15. Ostroff RB, Nelson JC. Risperidone augmentation of selective serotonin reuptake inhibitors in major depression. J Clin Psychiatry 1999;60(4):256-9.
16. Shelton RC. The combination of olanzapine and fluoxetine in mood disorders. Expert Opin Pharmacother 2003;4(7):1175-83.
17. Shelton RC, Williamson DJ, Corya SA, et al. Olanzapine/fluoxetine combination for treatment-resistant depression: a controlled study of SSRI and nortriptyline resistance. J Clin Psychiatry 2005;66(10):1289-97.
18. Thase ME, Corya SA, Olawale O, et al. Olanzapine/fluoxetine combination, olanzapine, and fluoxetine in treatment-resistant major depressive disorder. Presented at: Society of Biological Psychiatry annual meeting; May 18-20, 2006; Toronto, Ontario.
19. Nemeroff CB. Use of atypical antipsychotics in refractory depression and anxiety. J Clin Psychiatry 2005;66(suppl 8):13-21.
20. Dunner DL, Amsterdam JD, Shelton RC, et al. Adjunctive ziprasidone in treatment-resistant depression: a randomized, double-blind, 8-week pilot study. Presented at: American College of Neuropsychopharmacology annual meeting; December 12-16, 2004; San Juan, PR.
21. Rapaport MH, Gharabawi GM, Canuso CM, et al. Effects of risperidone augmentation in patients with treatment-resistant depression: results of open-label treatment followed by double-blind continuation. Neuropsychopharmacology 2006 (in press; advance online publication June 7, 2006; doi:10.1038/sj.npp.1301113).
22. Gharabawi G, Canuso C, Pandina G, et al. A double-blind placebocontrolled study of adjunctive risperidone for treatment-resistant major depressive disorder. Int J Neuropsychopharmacol 2006;9(suppl 1):S236.-
23. Shelton RC, Stahl SM. Risperidone and paroxetine given singly and in combination for bipolar depression. J Clin Psychiatry 2004;65(12):1715-9.
24. Shelton RC. A comparison of risperidone and bupropion augmentation of serotonin reuptake inhibitors in treatment-resistant unipolar major depression. Presented at: Society of Biological Psychiatry annual meeting; May 18-20, 2006; Toronto, Ontario.
25. Simon JS, Nemeroff CB. Aripiprazole augmentation of antidepressants for the treatment of partially responding and nonresponding patients with major depressive disorder. J Clin Psychiatry 2005;66(10):1216-20.
26. Papakostas GI, Petersen TJ, Kinrys G, et al. Aripiprazole augmentation of selective serotonin reuptake inhibitors for treatment-resistant major depressive disorder. J Clin Psychiatry 2005;66(10):1326-30.
27. Barbee JG, Conrad EJ, Jamhour NJ. Aripiprazole augmentation in treatment-resistant depression. Ann Clin Psychiatry 2004;16:189-94.
28. Adson DE, Kushner MG, Eiben KM, Schulz SC. Preliminary experience with adjunctive quetiapine in patients receiving selective serotonin reuptake inhibitors. Depress Anxiety 2004;19:121-6.
29. Yargic LI, Corapcioglu A, Kocabasoglu N, et al. A prospective randomized single-blind, multicenter trial comparing the efficacy and safety of paroxetine with and without quetiapine therapy in depression associated with anxiety. Int J Psychiatry Clin Pract 2004;8(4):205-11.
30. McIntyre A, Gendron A, McIntyre A. Quetiapine augmentation of SSRIs/SNRIs in major depression with anxiety. Poster presented at: American Psychiatric Association annual meeting; May 2006; Toronto, Ontario.
31. Mattingly G, Ilivicky H, Canale J, Anderson R. Quetiapine combination for treatment-resistant depression. Poster presented at: American Psychiatric Association annual meeting; May 2006; Toronto, Ontario.
32. Keck PE, Jr, McElroy SL, Strakowski SM, Soutullo CA. Antipsychotics in the treatment of mood disorders and risk of tardive dyskinesia. J Clin Psychiatry 2000;61(suppl 4):33-8.
33. Haddad PM, Wieck A. Antipsychotic-induced hyperprolactinaemia: mechanisms, clinical features and management. Drugs 2004;64(20):2291-314.
34. Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry 1997;42(4):233-46.
35. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63(9):763-71.
36. Tollefson GD, Kuntz AJ. Review of recent clinical studies with olanzapine. Br J Psychiatry Suppl 1990;(37):30-5.
37. Goff DC, Posever T, Herz L, et al. An exploratory haloperidolcontrolled dose-finding study of ziprasidone in hospitalized patients with schizophrenia or schizoaffective disorder. J Clin Psychopharmacol 1998;18(4):296-304.
Adding second-generation antipsychotics (SGAs) may boost the effectiveness of antidepressants in treatment-resistant unipolar major depression. Exactly when to try SGAs remains unclear, however, given their potential for adverse effects.
Major depression often is severe and chronic, and many patients remain ill even after multiple rounds of treatment. For patients without psychosis, where do SGAs fit into an algorithm for treatment-resistant depression?
This article examines the evidence on antipsychotic augmentation and discusses issues to consider—effectiveness, adverse effects, therapeutic dosages, and the patient’s quality of life—in making your clinical decisions.
Antidepressants alone
An optimal trial. Most depressed patients do not experience full response after initial antidepressant treatment, even with optimal therapeutic trials. An optimal trial means maintaining the maximum tolerated dosage within the antidepressant’s typical therapeutic range for at least 3 weeks.1 Reported remission rates from initial and second-line treatments include:
- one-third of patients after a vigorous initial trial of citalopram in a National Institute of Mental Health study2
- 20% to 30% of patients given citalopram plus bupropion or buspirone3 or switched to bupropion, sertraline, or venlafaxine4
- 50% of patients treated for depression in a primary care practice during the first 2 years after an initial antidepressant prescription.5
Subsequent options. In addition to various monotherapies and combinations, many options have been proposed for managing nonresponse to initial antidepressant therapy (Table 1). These include:
- augmenting with lithium, thyroid hormone, pindolol, or estrogen
- switching to a drug in another therapeutic class, such as a tricyclic antidepressant or monoamine oxidase inhibitor
- adding cognitive-behavioral therapy.7
Depression is often chronic and disabling. Selective serotonin reuptake inhibitors (SSRIs) are the mainstay of treatment, but recent data suggest that:
- few patients achieve therapeutic remission with initial SSRIs
- relapse or recurrence after remission is common.6
Clinically, this means psychiatrists contend with treatment resistance in nearly all patients with major depression.
Chronic, inadequately treated depression has a pervasive, adverse effect on patients’ quality of life, impairing the ability to work and perform social roles such as parenting. Even when an antidepressant produces partial response, considerable impairment remains. Depressed patients who do not achieve full therapeutic remission remain in this partially remitted, disabled state throughout treatment.8
Aggressive and persistent management is the key to effectively treating major depression.
Therapeutic suggestions when an SSRI does not lead to remission*
| Pharmacotherapy | Example | Recommended dosing |
|---|---|---|
| Monotherapy | An SNRI such as: | |
| Duloxetine | 30 to 120 mg/d | |
| Venlafaxine XR | 150 to 375 mg/d | |
| Combination therapies with SSRIs | Bupropion | 200 to 400 mg/d |
| Buspirone | 30 to 60 mg/d | |
| Augmentation | Lithium | 900 to 1,200 mg/d |
| Thyroid hormone | 25 mcg/d | |
| Pindolol | 5 to 30 mg bid | |
| Estrogen (such as 17a-estradiol) | 100 mcg/d | |
| Switch to another Tricyclic antidepressant class | Tricyclic | |
| Imipramine | 150 to 250 mg/d* | |
| Nortriptyline | 75 to 200 mg/d* | |
| Desipramine | 150 to 250 mg/d* | |
| MAOI | ||
| Phenelzine | 30 to 60 mg/d | |
| Tranylcypromine | 20 to 60 mg/d | |
| Selegiline (patch) | 9 to 12 mg/patch/day | |
| * Suggestions are not listed in stepwise order | ||
| MAOI: monoamine oxidase inhibitor | ||
| SNRI: serotonin-norepinephrine reuptake inhibitor | ||
| SSRI: selective serotonin reuptake inhibitor | ||
Atypicals for unipolar depression
Why atypicals? Researchers are investigating the use of SGAs in treatment-resistant mood disorders because of these drugs’ unique psychopharmacologic properties (Box 2).9-11
Except for clozapine, all available SGAs—aripiprazole, olanzapine, quetiapine, risperidone, and ziprasidone—are FDA-approved for acute bipolar mania. Evidence also strongly supports the benefits of quetiapine12 and the fixed-dose olanzapine/ fluoxetine combination13 for bipolar depression. Olanzapine/fluoxetine—originally studied for use in treatment-resistant unipolar depression—is approved for bipolar depression.14
Robust response. An uncontrolled case series first suggested that an SGA might help treat unipolar depression after initial selective serotonin reuptake inhibitors (SSRIs) fail to achieve remission. Ostroff and Nelson15 enrolled 8 outpatients (5 men, 3 women, ages 36 to 75) with nonpsychotic unipolar major depression that did not respond to initial fluoxetine or paroxetine. Patients had been taking fluoxetine, 20 to 40 mg/d, for 6 weeks to 4 months or paroxetine, 10 to 30 mg/d, for 2 to 8 weeks.
Patients reported a robust clinical effect within days after risperidone, 0.5 to 1.0 mg/d, was added to the SSRIs. Depression symptoms dropped to remission levels within 1 week, as measured by baseline and follow-up Hamilton Rating Scale for Depression (HAM-D) scores.
Olanzapine/fluoxetine. Our group subsequently enrolled 28 nonpsychotic patients with unipolar depression in a double-blind, placebo-controlled trial.14 We first treated these patients—who had not responded adequately to an SSRI or an antidepressant from another class—with open-label fluoxetine, up to 60 mg/d. Those whose scores on depression rating scales improved by ≥30% were excluded from the double-blind phase, when we randomly assigned the remaining patients to:
- olanzapine, mean 12.5 mg/d, plus placebo (n=8)
- a continuation of fluoxetine, mean 52 mg/d, plus placebo (n=10)
- or olanzapine/fluoxetine, mean 13.5/52 mg/d (n=10).
Final depression remission rates (HAM-D score ≤8 for 2 weeks) were:
- 60% with olanzapine/fluoxetine
- 25% with olanzapine alone
- 20% with continuation fluoxetine.
Until recently, researchers had been unable to replicate these results or extend this study in larger populations because of high response rates in the monotherapy treatment groups.16,17 In May 2006, however, Thase et al18 presented data from a large-scale replication trial that confirmed the finding of a more robust effect with fixed-dose olanzapine/fluoxetine in unipolar major depression, compared with olanzapine or fluoxetine monotherapy.
Second-generation antipsychotics (SGAs) differ from first-generation antipsychotics (FGAs) in their putative mechanisms of action.
FGAs’ antipsychotic effects depend largely on central dopamine type 2 (D2) receptor blockade. Their additional receptor-binding characteristics—blocking cholinergic, histamine, and alpha adrenergic receptors—appear to confer side effects but no added therapeutic benefit.9
SGAs bind weakly to D2 receptors and in varying degrees to serotonin (5-HT) receptors, including 5-HT subtypes 1A, 2A, 2C, 5, 7, and others. The SGAs also have other transmitter effects.10 On balance, the SGAs’ effects are more complex than those of the FGAs.
SGAs are called “atypical” because their beneficial and adverse clinical actions do not follow the FGAs’ usual pattern. FGAs’ relative potency in reducing psychosis is proportional to the propensity to cause extrapyramidal symptoms (EPS). Both the clinical effect and EPS are functions of D2 receptor blockade.11 In contrast, clozapine—the prototypical SGA—is a potent antipsychotic that exerts essentially no EPS.
Compared with FGAs, clozapine’s more complicated psychopharmacology has been shown to produce an enhanced effect on negative, cognitive, and mood symptoms in some patients with schizophrenia.10
- sertraline, 100 to 200 mg/d
- sertraline plus ziprasidone, 80 mg/d
- or sertraline plus ziprasidone, 160 mg/d.
Risperidone. One three-phase study21 evaluated the long-term efficacy of adding risperidone to citalopram in 489 patients with treatment-resistant depression. The design was:
- phase 1: 4 to 6 weeks of open-label citalopram, 20 to 60 mg/d (N=489)
- phase 2: 4 to 6 weeks of citalopram plus open-label risperidone, 0.25 to 2 mg/d (N=386)
- phase 3: 24 weeks of citalopram plus double-blind risperidone or placebo (N=241).
Median time to relapse in phase 3 was 102 days with risperidone augmentation and 85 days with placebo—not a statistically significant difference. Relapse rates were 53.3% with risperidone and 54.6% in the control group. These results suggest that risperidone had an initial acute effect that was not sustained.
In another study,22 463 depressed patients received an optimized antidepressant trial. The 274 who did not respond sufficiently were randomly assigned to risperidone, 1 to 2 mg/d, or placebo for 6 weeks. Mean HAM-D scores fell from 24.2 to 15.2 in the risperidone group and from 24.6 to 17.5 in the control group—a modest but statistically significant difference in favor of risperidone. The baseline-toendpoint change in this study is similar to that reported in a trial of risperidone, 1 to 4 mg/d, plus paroxetine, 20 to 40 mg/d, in bipolar depression.23
Shelton24 compared the effectiveness of adding risperidone or bupropion to SSRIs and serotonin norepinephrine reuptake inhibitors (SNRIs) for 6 weeks. Risperidone and bupropion were similarly effective as augmentation, but risperidone had a more rapid effect—producing statistically significant greater benefits within the first week of treatment.
Aripiprazole. Two open-label trials showed that aripiprazole combined with SSRIs exerts generally beneficial effects in treatment-resistant depression.25,26 Simon and Nemeroff25 began by adding aripiprazole at 10 mg/d, but emerging akathisia prompted them to reduce the starting dosage to 2.5 mg/d.
Barbee et al27 reported the results of a retrospective case series of aripiprazole augmentation in depressed patients who had not responded adequately to multiple medication trials, including SGAs. Fourteen of 30 patients (46.7%) were rated “much improved” or “very improved” with added aripiprazole, based on Prospective Global Assessment of Functioning and Clinical Global Impressions- Improvement scores. But 9 patients (30%) did not complete the full course of therapy, and 6 of the 14 responders (42.9%) relapsed while taking aripiprazole. The net response rate across 6 weeks was 27%.
Although this study involved only aripiprazole, the results suggest that trying a second SGA may not be more effective after a first SGA fails to improve treatment-resistant depression.
Quetiapine. A 9-week, open-label, variable-dose study of 11 patients28 first suggested that augmenting SSRIs with quetiapine could improve residual anxiety in resistant depression. Subsequently, 112 patients with major depression and anxiety were randomly assigned to single-blind treatment with paroxetine, ≥60 mg/d, with or without quetiapine, ≥200 mg/d. After 8 weeks, the 58 patients receiving quetiapine augmentation showed greater improvement than the 54 receiving SSRI monotherapy, based on Hamilton Anxiety Scale (HAM-A) and HAM-D scores.29
Adding quetiapine to antidepressant therapy was then examined in a randomized, placebo-controlled trial by McIntyre et al.30 Fifty-eight patients with unipolar depression who had not responded adequately after 6 weeks of SSRI or SNRI therapy were randomly assigned to quetiapine, 50 to 600 mg/d (mean dose 202±93 mg/d) or placebo for 8 weeks. Adjunctive quetiapine was significantly more effective than placebo, as measured by HAM-D scores. Patients receiving quetiapine also showed significantly better HAM-A scores at all points except week 8.
The dropout rate was relatively high for both groups—11 of 29 (38%) receiving quetiapine and 13 of 29 (45%) receiving placebo. The main reasons for discontinuation were side effects with quetiapine (sedation, dry mouth, and weight gain) and lack of effect with placebo.
These results are similar to those of another double-blind, placebo-controlled trial,31 in which 32 patients with SSRI/SNRI-resistant depression received adjunctive quetiapine, 200 to 400 mg/d (mean 268 mg/d) or placebo for 8 weeks.
Though small, these studies indicate that quetiapine may be effective as augmentation for treatment-resistant unipolar depression. Controlled data from a larger study are needed.
Discussion. Because of insufficient data, we do not know if SGAs are equivalent when used to augment antidepressant therapy in unipolar major depression. Olanzapine has been studied more than other SGAs in treatmentresistant depression and has shown efficacy in several—but not all—short- and long-term augmentation trials. Evidence on other SGAs is limited, and no head-to-head comparisons have been reported.
Adverse effects
Some SGAs may be effective in treatment-resistant depression, but any discussion of using them must also include their potential for adverse effects.
Weight gain and subsequent metabolic syndrome have been associated with olanzapine and—to a lesser degree—with quetiapine and risperidone. Ziprasidone and aripiprazole have relatively little effect on patients’ weight.
Extrapyramidal symptoms. All SGAs carry a risk of tardive dyskinesia. The risk is lower with SGAs than with first-generation antipsychotics (FGAs) but is an important clinical consideration.32
Hyperprolactinemia. Risperidone has been associated with an elevated risk of hyperprolactinemia, although less than that associated with FGAs.33 This risk does not appear to be a problem with quetiapine34 and aripiprazole;35 it is low with olanzapine (except at higher dosages);36 and the prolactin increase associated with ziprasidone may resolve within the first month of treatment.37
Prescribing rationale
‘Overcautious’ treatment. Even with careful management of side effects, SGAs are not preferred to strategies such as switching antidepressants or adding bupropion for treatment-resistant unipolar depression. But do not exclude SGAs solely because of their potential for adverse effects.
I am concerned about anecdotal reports of overcautious clinicians basing medication choices largely on safety—and, by extension, legal—considerations rather than on effectiveness. Certainly, safety concerns should prevail when two options are equally effective. But we do our patients no service by selecting ineffective drugs just because they have a low potential for adverse effects or by dosing effective drugs below the therapeutic range (Table 2).
When a drug is effective and may be the best choice for the patient, the question becomes, “Can I manage the potential for adverse effects?” When prescribing SGAs, it is important to monitor patients’ weight and serum lipid and glucose levels and regularly to look for abnormal involuntary movements.
An important question remains: Where do SGAs belong in the hierarchy of treatment options? Unfortunately, treatment guidelines for depression do not typically mention antipsychotics. Because of relative safety issues, two trials of monotherapies of different classes and, perhaps, combination therapy with bupropion would come before SGAs. However, it remains unclear exactly where.
SGAs probably belong ahead of electroconvulsive therapy or vagal nerve stimulation. But should they come before augmentation with lithium or thyroid hormone? Or, for that matter, trials of tricyclics or monoamine oxidase inhibitors?
Unfortunately, the available evidence provides little guidance. For a list of therapeutic algorithms developed for treatment-resistant depression, see Related resources.
Table 2
Recommended dosing of SGAs to augment antidepressant therapy
| Medication | Therapeutic range (mg/d) |
|---|---|
| Aripiprazole | 5 to 30 |
| Olanzapine | 5 to 20 |
| Quetiapine | 100 to 400 |
| Risperidone | 2 to 4 |
| Ziprasidone | 80 to 160 |
Algorithms for treatment-resistant depression
- Trivedi MH, Kern JK, Grannemann BD, et al. A computerized clinical decision support system as a means of implementing depression guidelines. Psychiatr Serv 2004;55(8);879-85.
- Rush AJ, Crismon ML, Kashner TM, et al. Texas Medication Algorithm Project, phase 3 (TMAP-3): rationale and study design. J Clin Psychiatry 2003;64(4);357-69.
- Trivedi M. Algorithms in clinical psychiatry: a stepped approach toward the path to recovery. Psychopharmacol Bull 2002;36(suppl 2);142-9.
- Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry 2001;62(suppl 6);22-9.
- Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on Medication Treatment of Major Depressive Disorder. J Clin Psychiatry 1999;60(3);142-56.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Buspirone • BuSpar
- Citalopram • Celexa
- Clozapine • Clozaril
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Imipramine • Tofranil
- Lithium • various
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Selegiline (patch) • EMSAM
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Shelton receives grant/research support from Eli Lilly and Co., GlaxoSmithKline, Pfizer, Janssen Pharmaceutica, sanofi-aventis, Wyeth Pharmaceuticals, AstraZeneca Pharmaceuticals, and Abbott Laboratories. He is a consultant to Pfizer and Janssen Pharmaceutica and a speaker for Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Pfizer, GlaxoSmithKline, Solvay Pharmaceuticals, Wyeth Pharmaceuticals, and Abbott Laboratories.
Adding second-generation antipsychotics (SGAs) may boost the effectiveness of antidepressants in treatment-resistant unipolar major depression. Exactly when to try SGAs remains unclear, however, given their potential for adverse effects.
Major depression often is severe and chronic, and many patients remain ill even after multiple rounds of treatment. For patients without psychosis, where do SGAs fit into an algorithm for treatment-resistant depression?
This article examines the evidence on antipsychotic augmentation and discusses issues to consider—effectiveness, adverse effects, therapeutic dosages, and the patient’s quality of life—in making your clinical decisions.
Antidepressants alone
An optimal trial. Most depressed patients do not experience full response after initial antidepressant treatment, even with optimal therapeutic trials. An optimal trial means maintaining the maximum tolerated dosage within the antidepressant’s typical therapeutic range for at least 3 weeks.1 Reported remission rates from initial and second-line treatments include:
- one-third of patients after a vigorous initial trial of citalopram in a National Institute of Mental Health study2
- 20% to 30% of patients given citalopram plus bupropion or buspirone3 or switched to bupropion, sertraline, or venlafaxine4
- 50% of patients treated for depression in a primary care practice during the first 2 years after an initial antidepressant prescription.5
Subsequent options. In addition to various monotherapies and combinations, many options have been proposed for managing nonresponse to initial antidepressant therapy (Table 1). These include:
- augmenting with lithium, thyroid hormone, pindolol, or estrogen
- switching to a drug in another therapeutic class, such as a tricyclic antidepressant or monoamine oxidase inhibitor
- adding cognitive-behavioral therapy.7
Depression is often chronic and disabling. Selective serotonin reuptake inhibitors (SSRIs) are the mainstay of treatment, but recent data suggest that:
- few patients achieve therapeutic remission with initial SSRIs
- relapse or recurrence after remission is common.6
Clinically, this means psychiatrists contend with treatment resistance in nearly all patients with major depression.
Chronic, inadequately treated depression has a pervasive, adverse effect on patients’ quality of life, impairing the ability to work and perform social roles such as parenting. Even when an antidepressant produces partial response, considerable impairment remains. Depressed patients who do not achieve full therapeutic remission remain in this partially remitted, disabled state throughout treatment.8
Aggressive and persistent management is the key to effectively treating major depression.
Therapeutic suggestions when an SSRI does not lead to remission*
| Pharmacotherapy | Example | Recommended dosing |
|---|---|---|
| Monotherapy | An SNRI such as: | |
| Duloxetine | 30 to 120 mg/d | |
| Venlafaxine XR | 150 to 375 mg/d | |
| Combination therapies with SSRIs | Bupropion | 200 to 400 mg/d |
| Buspirone | 30 to 60 mg/d | |
| Augmentation | Lithium | 900 to 1,200 mg/d |
| Thyroid hormone | 25 mcg/d | |
| Pindolol | 5 to 30 mg bid | |
| Estrogen (such as 17a-estradiol) | 100 mcg/d | |
| Switch to another Tricyclic antidepressant class | Tricyclic | |
| Imipramine | 150 to 250 mg/d* | |
| Nortriptyline | 75 to 200 mg/d* | |
| Desipramine | 150 to 250 mg/d* | |
| MAOI | ||
| Phenelzine | 30 to 60 mg/d | |
| Tranylcypromine | 20 to 60 mg/d | |
| Selegiline (patch) | 9 to 12 mg/patch/day | |
| * Suggestions are not listed in stepwise order | ||
| MAOI: monoamine oxidase inhibitor | ||
| SNRI: serotonin-norepinephrine reuptake inhibitor | ||
| SSRI: selective serotonin reuptake inhibitor | ||
Atypicals for unipolar depression
Why atypicals? Researchers are investigating the use of SGAs in treatment-resistant mood disorders because of these drugs’ unique psychopharmacologic properties (Box 2).9-11
Except for clozapine, all available SGAs—aripiprazole, olanzapine, quetiapine, risperidone, and ziprasidone—are FDA-approved for acute bipolar mania. Evidence also strongly supports the benefits of quetiapine12 and the fixed-dose olanzapine/ fluoxetine combination13 for bipolar depression. Olanzapine/fluoxetine—originally studied for use in treatment-resistant unipolar depression—is approved for bipolar depression.14
Robust response. An uncontrolled case series first suggested that an SGA might help treat unipolar depression after initial selective serotonin reuptake inhibitors (SSRIs) fail to achieve remission. Ostroff and Nelson15 enrolled 8 outpatients (5 men, 3 women, ages 36 to 75) with nonpsychotic unipolar major depression that did not respond to initial fluoxetine or paroxetine. Patients had been taking fluoxetine, 20 to 40 mg/d, for 6 weeks to 4 months or paroxetine, 10 to 30 mg/d, for 2 to 8 weeks.
Patients reported a robust clinical effect within days after risperidone, 0.5 to 1.0 mg/d, was added to the SSRIs. Depression symptoms dropped to remission levels within 1 week, as measured by baseline and follow-up Hamilton Rating Scale for Depression (HAM-D) scores.
Olanzapine/fluoxetine. Our group subsequently enrolled 28 nonpsychotic patients with unipolar depression in a double-blind, placebo-controlled trial.14 We first treated these patients—who had not responded adequately to an SSRI or an antidepressant from another class—with open-label fluoxetine, up to 60 mg/d. Those whose scores on depression rating scales improved by ≥30% were excluded from the double-blind phase, when we randomly assigned the remaining patients to:
- olanzapine, mean 12.5 mg/d, plus placebo (n=8)
- a continuation of fluoxetine, mean 52 mg/d, plus placebo (n=10)
- or olanzapine/fluoxetine, mean 13.5/52 mg/d (n=10).
Final depression remission rates (HAM-D score ≤8 for 2 weeks) were:
- 60% with olanzapine/fluoxetine
- 25% with olanzapine alone
- 20% with continuation fluoxetine.
Until recently, researchers had been unable to replicate these results or extend this study in larger populations because of high response rates in the monotherapy treatment groups.16,17 In May 2006, however, Thase et al18 presented data from a large-scale replication trial that confirmed the finding of a more robust effect with fixed-dose olanzapine/fluoxetine in unipolar major depression, compared with olanzapine or fluoxetine monotherapy.
Second-generation antipsychotics (SGAs) differ from first-generation antipsychotics (FGAs) in their putative mechanisms of action.
FGAs’ antipsychotic effects depend largely on central dopamine type 2 (D2) receptor blockade. Their additional receptor-binding characteristics—blocking cholinergic, histamine, and alpha adrenergic receptors—appear to confer side effects but no added therapeutic benefit.9
SGAs bind weakly to D2 receptors and in varying degrees to serotonin (5-HT) receptors, including 5-HT subtypes 1A, 2A, 2C, 5, 7, and others. The SGAs also have other transmitter effects.10 On balance, the SGAs’ effects are more complex than those of the FGAs.
SGAs are called “atypical” because their beneficial and adverse clinical actions do not follow the FGAs’ usual pattern. FGAs’ relative potency in reducing psychosis is proportional to the propensity to cause extrapyramidal symptoms (EPS). Both the clinical effect and EPS are functions of D2 receptor blockade.11 In contrast, clozapine—the prototypical SGA—is a potent antipsychotic that exerts essentially no EPS.
Compared with FGAs, clozapine’s more complicated psychopharmacology has been shown to produce an enhanced effect on negative, cognitive, and mood symptoms in some patients with schizophrenia.10
- sertraline, 100 to 200 mg/d
- sertraline plus ziprasidone, 80 mg/d
- or sertraline plus ziprasidone, 160 mg/d.
Risperidone. One three-phase study21 evaluated the long-term efficacy of adding risperidone to citalopram in 489 patients with treatment-resistant depression. The design was:
- phase 1: 4 to 6 weeks of open-label citalopram, 20 to 60 mg/d (N=489)
- phase 2: 4 to 6 weeks of citalopram plus open-label risperidone, 0.25 to 2 mg/d (N=386)
- phase 3: 24 weeks of citalopram plus double-blind risperidone or placebo (N=241).
Median time to relapse in phase 3 was 102 days with risperidone augmentation and 85 days with placebo—not a statistically significant difference. Relapse rates were 53.3% with risperidone and 54.6% in the control group. These results suggest that risperidone had an initial acute effect that was not sustained.
In another study,22 463 depressed patients received an optimized antidepressant trial. The 274 who did not respond sufficiently were randomly assigned to risperidone, 1 to 2 mg/d, or placebo for 6 weeks. Mean HAM-D scores fell from 24.2 to 15.2 in the risperidone group and from 24.6 to 17.5 in the control group—a modest but statistically significant difference in favor of risperidone. The baseline-toendpoint change in this study is similar to that reported in a trial of risperidone, 1 to 4 mg/d, plus paroxetine, 20 to 40 mg/d, in bipolar depression.23
Shelton24 compared the effectiveness of adding risperidone or bupropion to SSRIs and serotonin norepinephrine reuptake inhibitors (SNRIs) for 6 weeks. Risperidone and bupropion were similarly effective as augmentation, but risperidone had a more rapid effect—producing statistically significant greater benefits within the first week of treatment.
Aripiprazole. Two open-label trials showed that aripiprazole combined with SSRIs exerts generally beneficial effects in treatment-resistant depression.25,26 Simon and Nemeroff25 began by adding aripiprazole at 10 mg/d, but emerging akathisia prompted them to reduce the starting dosage to 2.5 mg/d.
Barbee et al27 reported the results of a retrospective case series of aripiprazole augmentation in depressed patients who had not responded adequately to multiple medication trials, including SGAs. Fourteen of 30 patients (46.7%) were rated “much improved” or “very improved” with added aripiprazole, based on Prospective Global Assessment of Functioning and Clinical Global Impressions- Improvement scores. But 9 patients (30%) did not complete the full course of therapy, and 6 of the 14 responders (42.9%) relapsed while taking aripiprazole. The net response rate across 6 weeks was 27%.
Although this study involved only aripiprazole, the results suggest that trying a second SGA may not be more effective after a first SGA fails to improve treatment-resistant depression.
Quetiapine. A 9-week, open-label, variable-dose study of 11 patients28 first suggested that augmenting SSRIs with quetiapine could improve residual anxiety in resistant depression. Subsequently, 112 patients with major depression and anxiety were randomly assigned to single-blind treatment with paroxetine, ≥60 mg/d, with or without quetiapine, ≥200 mg/d. After 8 weeks, the 58 patients receiving quetiapine augmentation showed greater improvement than the 54 receiving SSRI monotherapy, based on Hamilton Anxiety Scale (HAM-A) and HAM-D scores.29
Adding quetiapine to antidepressant therapy was then examined in a randomized, placebo-controlled trial by McIntyre et al.30 Fifty-eight patients with unipolar depression who had not responded adequately after 6 weeks of SSRI or SNRI therapy were randomly assigned to quetiapine, 50 to 600 mg/d (mean dose 202±93 mg/d) or placebo for 8 weeks. Adjunctive quetiapine was significantly more effective than placebo, as measured by HAM-D scores. Patients receiving quetiapine also showed significantly better HAM-A scores at all points except week 8.
The dropout rate was relatively high for both groups—11 of 29 (38%) receiving quetiapine and 13 of 29 (45%) receiving placebo. The main reasons for discontinuation were side effects with quetiapine (sedation, dry mouth, and weight gain) and lack of effect with placebo.
These results are similar to those of another double-blind, placebo-controlled trial,31 in which 32 patients with SSRI/SNRI-resistant depression received adjunctive quetiapine, 200 to 400 mg/d (mean 268 mg/d) or placebo for 8 weeks.
Though small, these studies indicate that quetiapine may be effective as augmentation for treatment-resistant unipolar depression. Controlled data from a larger study are needed.
Discussion. Because of insufficient data, we do not know if SGAs are equivalent when used to augment antidepressant therapy in unipolar major depression. Olanzapine has been studied more than other SGAs in treatmentresistant depression and has shown efficacy in several—but not all—short- and long-term augmentation trials. Evidence on other SGAs is limited, and no head-to-head comparisons have been reported.
Adverse effects
Some SGAs may be effective in treatment-resistant depression, but any discussion of using them must also include their potential for adverse effects.
Weight gain and subsequent metabolic syndrome have been associated with olanzapine and—to a lesser degree—with quetiapine and risperidone. Ziprasidone and aripiprazole have relatively little effect on patients’ weight.
Extrapyramidal symptoms. All SGAs carry a risk of tardive dyskinesia. The risk is lower with SGAs than with first-generation antipsychotics (FGAs) but is an important clinical consideration.32
Hyperprolactinemia. Risperidone has been associated with an elevated risk of hyperprolactinemia, although less than that associated with FGAs.33 This risk does not appear to be a problem with quetiapine34 and aripiprazole;35 it is low with olanzapine (except at higher dosages);36 and the prolactin increase associated with ziprasidone may resolve within the first month of treatment.37
Prescribing rationale
‘Overcautious’ treatment. Even with careful management of side effects, SGAs are not preferred to strategies such as switching antidepressants or adding bupropion for treatment-resistant unipolar depression. But do not exclude SGAs solely because of their potential for adverse effects.
I am concerned about anecdotal reports of overcautious clinicians basing medication choices largely on safety—and, by extension, legal—considerations rather than on effectiveness. Certainly, safety concerns should prevail when two options are equally effective. But we do our patients no service by selecting ineffective drugs just because they have a low potential for adverse effects or by dosing effective drugs below the therapeutic range (Table 2).
When a drug is effective and may be the best choice for the patient, the question becomes, “Can I manage the potential for adverse effects?” When prescribing SGAs, it is important to monitor patients’ weight and serum lipid and glucose levels and regularly to look for abnormal involuntary movements.
An important question remains: Where do SGAs belong in the hierarchy of treatment options? Unfortunately, treatment guidelines for depression do not typically mention antipsychotics. Because of relative safety issues, two trials of monotherapies of different classes and, perhaps, combination therapy with bupropion would come before SGAs. However, it remains unclear exactly where.
SGAs probably belong ahead of electroconvulsive therapy or vagal nerve stimulation. But should they come before augmentation with lithium or thyroid hormone? Or, for that matter, trials of tricyclics or monoamine oxidase inhibitors?
Unfortunately, the available evidence provides little guidance. For a list of therapeutic algorithms developed for treatment-resistant depression, see Related resources.
Table 2
Recommended dosing of SGAs to augment antidepressant therapy
| Medication | Therapeutic range (mg/d) |
|---|---|
| Aripiprazole | 5 to 30 |
| Olanzapine | 5 to 20 |
| Quetiapine | 100 to 400 |
| Risperidone | 2 to 4 |
| Ziprasidone | 80 to 160 |
Algorithms for treatment-resistant depression
- Trivedi MH, Kern JK, Grannemann BD, et al. A computerized clinical decision support system as a means of implementing depression guidelines. Psychiatr Serv 2004;55(8);879-85.
- Rush AJ, Crismon ML, Kashner TM, et al. Texas Medication Algorithm Project, phase 3 (TMAP-3): rationale and study design. J Clin Psychiatry 2003;64(4);357-69.
- Trivedi M. Algorithms in clinical psychiatry: a stepped approach toward the path to recovery. Psychopharmacol Bull 2002;36(suppl 2);142-9.
- Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry 2001;62(suppl 6);22-9.
- Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: report of the Texas Consensus Conference Panel on Medication Treatment of Major Depressive Disorder. J Clin Psychiatry 1999;60(3);142-56.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Buspirone • BuSpar
- Citalopram • Celexa
- Clozapine • Clozaril
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Imipramine • Tofranil
- Lithium • various
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Olanzapine/fluoxetine • Symbyax
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Selegiline (patch) • EMSAM
- Sertraline • Zoloft
- Tranylcypromine • Parnate
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Shelton receives grant/research support from Eli Lilly and Co., GlaxoSmithKline, Pfizer, Janssen Pharmaceutica, sanofi-aventis, Wyeth Pharmaceuticals, AstraZeneca Pharmaceuticals, and Abbott Laboratories. He is a consultant to Pfizer and Janssen Pharmaceutica and a speaker for Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Pfizer, GlaxoSmithKline, Solvay Pharmaceuticals, Wyeth Pharmaceuticals, and Abbott Laboratories.
1. Shelton RC. The use of antidepressants in novel combination therapies. J Clin Psychiatry 2003;64(suppl 2):14-8.
2. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
3. Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006;354(12):1243-52.
4. Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006;354(12):1231-42.
5. Simon GE, Heiligenstein J, Revicki D, et al. Long-term outcomes of initial antidepressant drug choice in a “real world” randomized trial. Arch Fam Med 1999;8(4):319-25.
6. Nierenberg AA, Petersen TJ, Alpert JE. Prevention of relapse and recurrence in depression: the role of long-term pharmacotherapy and psychotherapy. J Clin Psychiatry 2003;64(suppl 15):13-17.
7. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342(20):1462-70.
8. Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990-2020: Global Burden of Disease Study. Lancet 1997;349(9064):1498-1504.
9. Bennett MR. Monoaminergic synapses and schizophrenia: 45 years of neuroleptics. J Psychopharmacol 1998;12(3):289-304.
10. Meltzer HY. The role of serotonin in antipsychotic drug action. Neuropsychopharmacology 1999;21(2 suppl):106S-115S.
11. Meltzer HY, Bastani B, Ramirez L, Matsubara S. Clozapine: new research on efficacy and mechanism of action. Eur Arch Psychiatry Neurol Sci 1989;238(5-6):332-9.
12. Hirschfeld RM, Weisler RH, Raines SR, Macfadden W. for the BOLDER Study Group. Quetiapine in the treatment of anxiety in patients with bipolar I or II depression: a secondary analysis from a randomized, double-blind, placebo-controlled study. J Clin Psychiatry 2006;67(3):355-62.
13. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60(11):1079-88.
14. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158(1):131-4.
15. Ostroff RB, Nelson JC. Risperidone augmentation of selective serotonin reuptake inhibitors in major depression. J Clin Psychiatry 1999;60(4):256-9.
16. Shelton RC. The combination of olanzapine and fluoxetine in mood disorders. Expert Opin Pharmacother 2003;4(7):1175-83.
17. Shelton RC, Williamson DJ, Corya SA, et al. Olanzapine/fluoxetine combination for treatment-resistant depression: a controlled study of SSRI and nortriptyline resistance. J Clin Psychiatry 2005;66(10):1289-97.
18. Thase ME, Corya SA, Olawale O, et al. Olanzapine/fluoxetine combination, olanzapine, and fluoxetine in treatment-resistant major depressive disorder. Presented at: Society of Biological Psychiatry annual meeting; May 18-20, 2006; Toronto, Ontario.
19. Nemeroff CB. Use of atypical antipsychotics in refractory depression and anxiety. J Clin Psychiatry 2005;66(suppl 8):13-21.
20. Dunner DL, Amsterdam JD, Shelton RC, et al. Adjunctive ziprasidone in treatment-resistant depression: a randomized, double-blind, 8-week pilot study. Presented at: American College of Neuropsychopharmacology annual meeting; December 12-16, 2004; San Juan, PR.
21. Rapaport MH, Gharabawi GM, Canuso CM, et al. Effects of risperidone augmentation in patients with treatment-resistant depression: results of open-label treatment followed by double-blind continuation. Neuropsychopharmacology 2006 (in press; advance online publication June 7, 2006; doi:10.1038/sj.npp.1301113).
22. Gharabawi G, Canuso C, Pandina G, et al. A double-blind placebocontrolled study of adjunctive risperidone for treatment-resistant major depressive disorder. Int J Neuropsychopharmacol 2006;9(suppl 1):S236.-
23. Shelton RC, Stahl SM. Risperidone and paroxetine given singly and in combination for bipolar depression. J Clin Psychiatry 2004;65(12):1715-9.
24. Shelton RC. A comparison of risperidone and bupropion augmentation of serotonin reuptake inhibitors in treatment-resistant unipolar major depression. Presented at: Society of Biological Psychiatry annual meeting; May 18-20, 2006; Toronto, Ontario.
25. Simon JS, Nemeroff CB. Aripiprazole augmentation of antidepressants for the treatment of partially responding and nonresponding patients with major depressive disorder. J Clin Psychiatry 2005;66(10):1216-20.
26. Papakostas GI, Petersen TJ, Kinrys G, et al. Aripiprazole augmentation of selective serotonin reuptake inhibitors for treatment-resistant major depressive disorder. J Clin Psychiatry 2005;66(10):1326-30.
27. Barbee JG, Conrad EJ, Jamhour NJ. Aripiprazole augmentation in treatment-resistant depression. Ann Clin Psychiatry 2004;16:189-94.
28. Adson DE, Kushner MG, Eiben KM, Schulz SC. Preliminary experience with adjunctive quetiapine in patients receiving selective serotonin reuptake inhibitors. Depress Anxiety 2004;19:121-6.
29. Yargic LI, Corapcioglu A, Kocabasoglu N, et al. A prospective randomized single-blind, multicenter trial comparing the efficacy and safety of paroxetine with and without quetiapine therapy in depression associated with anxiety. Int J Psychiatry Clin Pract 2004;8(4):205-11.
30. McIntyre A, Gendron A, McIntyre A. Quetiapine augmentation of SSRIs/SNRIs in major depression with anxiety. Poster presented at: American Psychiatric Association annual meeting; May 2006; Toronto, Ontario.
31. Mattingly G, Ilivicky H, Canale J, Anderson R. Quetiapine combination for treatment-resistant depression. Poster presented at: American Psychiatric Association annual meeting; May 2006; Toronto, Ontario.
32. Keck PE, Jr, McElroy SL, Strakowski SM, Soutullo CA. Antipsychotics in the treatment of mood disorders and risk of tardive dyskinesia. J Clin Psychiatry 2000;61(suppl 4):33-8.
33. Haddad PM, Wieck A. Antipsychotic-induced hyperprolactinaemia: mechanisms, clinical features and management. Drugs 2004;64(20):2291-314.
34. Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry 1997;42(4):233-46.
35. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63(9):763-71.
36. Tollefson GD, Kuntz AJ. Review of recent clinical studies with olanzapine. Br J Psychiatry Suppl 1990;(37):30-5.
37. Goff DC, Posever T, Herz L, et al. An exploratory haloperidolcontrolled dose-finding study of ziprasidone in hospitalized patients with schizophrenia or schizoaffective disorder. J Clin Psychopharmacol 1998;18(4):296-304.
1. Shelton RC. The use of antidepressants in novel combination therapies. J Clin Psychiatry 2003;64(suppl 2):14-8.
2. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
3. Trivedi MH, Fava M, Wisniewski SR, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med 2006;354(12):1243-52.
4. Rush AJ, Trivedi MH, Wisniewski SR, et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 2006;354(12):1231-42.
5. Simon GE, Heiligenstein J, Revicki D, et al. Long-term outcomes of initial antidepressant drug choice in a “real world” randomized trial. Arch Fam Med 1999;8(4):319-25.
6. Nierenberg AA, Petersen TJ, Alpert JE. Prevention of relapse and recurrence in depression: the role of long-term pharmacotherapy and psychotherapy. J Clin Psychiatry 2003;64(suppl 15):13-17.
7. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342(20):1462-70.
8. Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990-2020: Global Burden of Disease Study. Lancet 1997;349(9064):1498-1504.
9. Bennett MR. Monoaminergic synapses and schizophrenia: 45 years of neuroleptics. J Psychopharmacol 1998;12(3):289-304.
10. Meltzer HY. The role of serotonin in antipsychotic drug action. Neuropsychopharmacology 1999;21(2 suppl):106S-115S.
11. Meltzer HY, Bastani B, Ramirez L, Matsubara S. Clozapine: new research on efficacy and mechanism of action. Eur Arch Psychiatry Neurol Sci 1989;238(5-6):332-9.
12. Hirschfeld RM, Weisler RH, Raines SR, Macfadden W. for the BOLDER Study Group. Quetiapine in the treatment of anxiety in patients with bipolar I or II depression: a secondary analysis from a randomized, double-blind, placebo-controlled study. J Clin Psychiatry 2006;67(3):355-62.
13. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60(11):1079-88.
14. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158(1):131-4.
15. Ostroff RB, Nelson JC. Risperidone augmentation of selective serotonin reuptake inhibitors in major depression. J Clin Psychiatry 1999;60(4):256-9.
16. Shelton RC. The combination of olanzapine and fluoxetine in mood disorders. Expert Opin Pharmacother 2003;4(7):1175-83.
17. Shelton RC, Williamson DJ, Corya SA, et al. Olanzapine/fluoxetine combination for treatment-resistant depression: a controlled study of SSRI and nortriptyline resistance. J Clin Psychiatry 2005;66(10):1289-97.
18. Thase ME, Corya SA, Olawale O, et al. Olanzapine/fluoxetine combination, olanzapine, and fluoxetine in treatment-resistant major depressive disorder. Presented at: Society of Biological Psychiatry annual meeting; May 18-20, 2006; Toronto, Ontario.
19. Nemeroff CB. Use of atypical antipsychotics in refractory depression and anxiety. J Clin Psychiatry 2005;66(suppl 8):13-21.
20. Dunner DL, Amsterdam JD, Shelton RC, et al. Adjunctive ziprasidone in treatment-resistant depression: a randomized, double-blind, 8-week pilot study. Presented at: American College of Neuropsychopharmacology annual meeting; December 12-16, 2004; San Juan, PR.
21. Rapaport MH, Gharabawi GM, Canuso CM, et al. Effects of risperidone augmentation in patients with treatment-resistant depression: results of open-label treatment followed by double-blind continuation. Neuropsychopharmacology 2006 (in press; advance online publication June 7, 2006; doi:10.1038/sj.npp.1301113).
22. Gharabawi G, Canuso C, Pandina G, et al. A double-blind placebocontrolled study of adjunctive risperidone for treatment-resistant major depressive disorder. Int J Neuropsychopharmacol 2006;9(suppl 1):S236.-
23. Shelton RC, Stahl SM. Risperidone and paroxetine given singly and in combination for bipolar depression. J Clin Psychiatry 2004;65(12):1715-9.
24. Shelton RC. A comparison of risperidone and bupropion augmentation of serotonin reuptake inhibitors in treatment-resistant unipolar major depression. Presented at: Society of Biological Psychiatry annual meeting; May 18-20, 2006; Toronto, Ontario.
25. Simon JS, Nemeroff CB. Aripiprazole augmentation of antidepressants for the treatment of partially responding and nonresponding patients with major depressive disorder. J Clin Psychiatry 2005;66(10):1216-20.
26. Papakostas GI, Petersen TJ, Kinrys G, et al. Aripiprazole augmentation of selective serotonin reuptake inhibitors for treatment-resistant major depressive disorder. J Clin Psychiatry 2005;66(10):1326-30.
27. Barbee JG, Conrad EJ, Jamhour NJ. Aripiprazole augmentation in treatment-resistant depression. Ann Clin Psychiatry 2004;16:189-94.
28. Adson DE, Kushner MG, Eiben KM, Schulz SC. Preliminary experience with adjunctive quetiapine in patients receiving selective serotonin reuptake inhibitors. Depress Anxiety 2004;19:121-6.
29. Yargic LI, Corapcioglu A, Kocabasoglu N, et al. A prospective randomized single-blind, multicenter trial comparing the efficacy and safety of paroxetine with and without quetiapine therapy in depression associated with anxiety. Int J Psychiatry Clin Pract 2004;8(4):205-11.
30. McIntyre A, Gendron A, McIntyre A. Quetiapine augmentation of SSRIs/SNRIs in major depression with anxiety. Poster presented at: American Psychiatric Association annual meeting; May 2006; Toronto, Ontario.
31. Mattingly G, Ilivicky H, Canale J, Anderson R. Quetiapine combination for treatment-resistant depression. Poster presented at: American Psychiatric Association annual meeting; May 2006; Toronto, Ontario.
32. Keck PE, Jr, McElroy SL, Strakowski SM, Soutullo CA. Antipsychotics in the treatment of mood disorders and risk of tardive dyskinesia. J Clin Psychiatry 2000;61(suppl 4):33-8.
33. Haddad PM, Wieck A. Antipsychotic-induced hyperprolactinaemia: mechanisms, clinical features and management. Drugs 2004;64(20):2291-314.
34. Arvanitis LA, Miller BG. Multiple fixed doses of “Seroquel” (quetiapine) in patients with acute exacerbation of schizophrenia: a comparison with haloperidol and placebo. The Seroquel Trial 13 Study Group. Biol Psychiatry 1997;42(4):233-46.
35. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63(9):763-71.
36. Tollefson GD, Kuntz AJ. Review of recent clinical studies with olanzapine. Br J Psychiatry Suppl 1990;(37):30-5.
37. Goff DC, Posever T, Herz L, et al. An exploratory haloperidolcontrolled dose-finding study of ziprasidone in hospitalized patients with schizophrenia or schizoaffective disorder. J Clin Psychopharmacol 1998;18(4):296-304.
Crisis debriefing: What helps, and what might not
Debriefing interventions have sprung from the understandable desire to reduce—if not eliminate—victims’ suffering after traumatic loss. Unfortunately, no compelling evidence has shown that an intervention given within a few days of a traumatic event can prevent significant psychological distress.
Evidence does suggest, however, that components of psychological debriefing discussed here may help you provide effective “first aid” to trauma victims and identify persons at risk for chronic psychological problems.
Complicated grief reactions
Death of a family member or close friend is among life’s most painful loses. When death occurs unexpectedly—as from violence, accident, natural disaster, or suicide—survivors’ emotional and psychological response can be pronounced.
Most survivors report great distress immediately after trauma or traumatic loss, but only an estimated 9% develop chronic psychopathology,1 such as complicated grief (Table 1).2,3 If the death was violent, surviving loved ones may experience complicated grief and posttraumatic stress disorder (PTSD)4 (Table 2).
Complicated grief is associated with considerable morbidity and risk of physical illness.5 PTSD develops in approximately one-third of cases involving sudden, unexpected death of a close friend or relative1 and can result in comorbid—but distinguishable—reactions to the loss (Box).6
Evidence-based secondary and tertiary intervention protocols have been developed for PTSD,7 but no practice guideline exists for treating or preventing complicated grief. Few controlled trials have been done.8
Table 1
Clinical features of complicated grief
|
| Source: Reference 3 |
Clinical features of posttraumatic stress disorder
|
| Source: DSM-IV-TR |
- educating individuals about stress reactions and how to cope with them
- instilling messages that stress reactions are normal
- helping affected persons process and share their emotions
- providing information about and opportunity for further intervention, if needed.
Traumatic loss. Although complicated grief (CG) and posttraumatic stress disorder (PTSD) can both develop following the loss of a loved one from a traumatic event, CG also can develop after expected deaths from natural causes. PTSD is exceedingly uncommon if a loved one’s death did not result from homicide, suicide, or accident, whereas CG can occur when the loss was not particularly violent or sudden.
Avoidance vs preoccupation. The fundamental difference between CG (Table 1) and PTSD (Table 2) symptoms is the degree that survivors avoid cues and contexts that remind them of their loss.
Those with PTSD go to great lengths to avoid thinking about the traumatic event and actively avoid situations that may remind them of it. This avoidance, paradoxically, exacerbates intrusive memories, as trying not to think about something increases the frequency of those thoughts.
Individuals with CG do not avoid reminders of the deceased. Quite the opposite, they seek out reminders (such as photos or recordings) and find solace in them. Reminders may contribute to their ongoing rumination or preoccupation, in which they retreat into memories of the deceased rather than engage in present life.
Hyperarousal symptoms that are required for PTSD diagnosis are largely absent in CG. Even when persons with CG experience arousal, it is not akin to scanning the environment for danger or threat, as is typical with PTSD. Persons with CG have a pronounced negative affect and bereavement-related depression, rather than an exaggerated startle response or heightened physiologic reactivity.
Does debriefing work?
Debriefing is designed not to address the intense but transient emotional reactions that can be expected immediately following traumatic loss but to prevent protracted, incapacitating distress. For an early intervention to be considered effective, it must be associated with greater or more expedient symptom recovery compared with natural remission. Controlled clinical trials are necessary to determine if this is the case.9
Control groups are essential when studying treatment outcomes of early crisis interventions. Simply documenting improvement among treated individuals is insufficient because substantial symptom remission is the norm and chronic psychopathology is comparatively rare. Thus, early interventions studies should at least:
- include a treatment group and a no-treatment or wait-list control condition
- randomly assign participants to avoid selfselection biases.
Debriefing for traumatic loss. Debriefing-based interventions have been used after mass violence and other large-scale traumatic events that may trigger complicated grief reactions.10 Most studies have not evaluated the impact of debriefing on complicated grief specifically but have focused on PTSD, anxiety, and depression. Typical published accounts of debriefing-based interventions for grief responses11 have been anecdotal, qualitative, and uncontrolled.
One rare controlled study of debriefing12 was designed to target emotional difficulties in women following early miscarriage. The one-half of participants who were debriefed 2 weeks after miscarriage perceived debriefing to be helpful. Despite significant improvement in early intrusion and avoidance scores, however, the women who were debriefed showed no greater improvement after 4 months than did a nondebriefed control group. The investigators concluded that debriefing did not influence post-loss adaptation.
A wider search. In the absence of randomized, controlled trials (RCTs) of debriefing-based interventions for traumatic loss, we turn to the larger debriefing literature. Nearly all debriefing studies have focused on PTSD symptoms rather than grief responses.
A number of peer-reviewed studies suggest that psychological debriefing is an effective intervention. These studies13,14 are characterized by dramatic symptom reductions following the intervention. Unfortunately, nearly all lack a control group, and the few that were controlled14 were not randomized. Studies reviewed by Everly et al15 also contain fundamental flaws, such as lack of random assignment, failure to assess individuals prior to the intervention, and lack of control groups.
None of the few RCTs of psychological debriefing conducted in traumatized populations show that it accelerates recovery in treated persons compared with nontreated controls.16 All of the studies17-22 included untreated control conditions, and participants were randomly assigned. Without exception, debriefed participants did not show superior improvement, and in two studies they showed worse outcomes than did untreated controls.17,21
Focused interventions
To provide optimal care to our patients, we must base our decisions on rigorous empirical study. In the case of debriefing, available well-controlled trials lead us to conclude that debriefings are inert.
To be clear, we are not philosophically opposed to early intervention for traumatic loss. We believe researchers must continue to develop and study interventions that can stave off chronic pathology among those at risk after traumatic loss.
Thus, clinicians and researchers face the same imperative: to accurately and efficiently identify persons at risk. Indiscriminately debriefing all persons who experience traumatic loss—without regard to risk—is not the most judicial use of clinical resources. Nor is it likely to advance our understanding of risk factors and resiliency in loss or of treatment efficacy.
Grief literature indicates that broadly applying interventions to anyone who has experienced loss does not help and may in fact exacerbate grief symptoms. Focused interventions for persons most at risk for complicated grief are more effective.23
Practice recommendations
Given the limited evidence, the recommendations that follow are preliminary and based on the few early interventions for trauma that have produced superior outcomes compared with untreated controls.24,25 In general, these interventions used:
- cognitive-behavioral techniques (education, promotion of adaptive coping strategies)
- exposure exercises for survivors who were using maladaptive avoidant coping strategies
- homework to reinforce therapeutic activities initiated in session.
‘Psychological first aid.’ Although immediate formal treatment is not recommended, a National Institute of Mental Health consensus conference26 recommended offering trauma victims “psychological first aid” (Table 3) when feasible. Psychological first aid is not intended to prevent chronic psychopathology but to provide:
- immediate emotional and informational support
- psychoeducational materials that describe common sequelae of trauma
- information about how and where to get help, if desired.
Recommended components of ‘psychological first aid’
|
| Source: National Institute of Mental Health, reference 15 |
Screening for risk factors. When victims seek your professional support or services immediately after a traumatic event, screen for risk factors for complicated grief, PTSD, or other chronic difficulties. Complicated grief is a relatively new diagnosis, and research on its risk factors is preliminary. The literature suggests, however, that risk factors may include:
- childhood abuse and neglect
- childhood separation anxiety
- loss of a child
- excessive interpersonal dependency or insecure attachment styles.6
In the weeks and months after the traumatic event, we recommend screening the most distressed victims for risk of developing chronic psychopathology. The National Center for PTSD offers self-report measures appropriate for various populations (such as children or adults) and trauma contexts (such as combat) (see Related resources). The Inventory of Complicated Grief28 is useful for screening for CG.
Empirically informed CBT. Provide brief cognitive behavioral interventions only for persons at risk and only after sufficient time has passed to allow you to differentiate between normal grief and abnormal responses. Early interventions that have shown promising outcomes typically have been delivered approximately 2 weeks after the traumatic exposure.24,25
Brief, multi-session CBT given several days to a few weeks after the trauma has been associated with improved posttraumatic adjustment.24,25 Interventions that appear to be most promising for patients who meet criteria for CG combine:
- psychoeducation
- exposure therapy for those having difficulty grasping the reality of their loss
- and behavioral activation techniques.29
Focus psychoeducation on how maladaptive strategies (such as avoiding trauma cues) can prolong trauma-related distress. Structure early interventions to encourage home-based exercises (such as exposure). These may reduce victims’ reliance on maladaptive strategies, accelerate therapeutic effects, and promote the generalization of treatment gains.24-25
- Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004.
- National Center for PTSD:
- Shear K, Frank E, Houck PR, Reynolds CF 3rd. Treatment of complicated grief: a randomized controlled trial. JAMA 2005;293(21):2601-8.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Breslau N, Kessler RC, Chilcoat HD, et al. Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Arch Gen Psychiatry 1998;55(7):626-32.
2. Latham AE, Prigerson HG. Suicidality and bereavement: complicated grief as psychiatric disorder presenting greatest risk for suicidality. Suicide Life Threat Behav 2004;34(4):350-62.
3. Gray MJ, Prigerson HG, Litz BT. Conceptual and definitional issues in complicated grief. In: Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004:65-84.
4. Neria Y, Litz BT. Bereavement by traumatic means: the complex synergy of trauma and grief. Journal of Loss and Trauma 2004;9(1):73-87.
5. Silverman GK, Jacobs SC, Kasl SV, et al. Quality of life impairments associated with diagnostic criteria for traumatic grief. Psychol Med 2000;30(4):857-62.
6. Lichtenthal WG, Cruess DG, Prigerson HG. A case for establishing complicated grief as a distinct mental disorder in DSM-V. Clin Psychol Rev 2004;24(6):637-62.
7. American Psychiatric Association. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry 2004;161(11 suppl):3-31.
8. Schut H, Stroebe MS, van den Bout J, Terheggen M. The efficacy of bereavement interventions: Determining who benefits. In: Stroebe MS, Hansson RO, Stroebe W, Schut H (eds). Handbook of bereavement research: consequences, coping, and care. Washington, DC: American Psychological Association; 2001:705-37.
9. Litz BT, Gray MJ. Early intervention for trauma in adults: a framework for first aid and secondary prevention. In: Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004:87-111.
10. Litz BT, Gray MJ. Early intervention for mass violence: What is the evidence? What should be done? Cognit Behav Pract 2002;9(4):266-72.
11. Webb NB. Groups for children traumatically bereaved by the attacks of September 11, 2001. Int J Group Psychother 2005;55(3):355-74.
12. Lee C, Slade P, Lygo V. The influence of psychological debriefing on emotional adaptation in women following early miscarriage: a preliminary study. Br J Med Psychol 1996;69(1):47-58.
13. Robinson R, Mitchell J. Getting some balance back into the debriefing debate. Bull Aust Psychol Soc 1995;17:5-10.
14. Jenkins S. Social support and debriefing efficacy among emergency medical workers after a mass shooting incident. J Soc Behav Personality 1996;11:477-92.
15. Everly GS, Flannery RB, Mitchell JT. Critical incident stress management (CISM): A review of the literature. Aggression Violent Behav 2000;5(1):23-40.
16. Litz B, Gray M, Bryant R, Adler A. Early intervention for trauma: Current status and future directions. Clin Psychol Sci Pract 2002;9(2):112-34.
17. Bisson J, Jenkins P, Alexander J, Bannister C. Randomized controlled trial of psychological debriefing for victims of acute burn trauma. Br J Psychiatry 1997;171:78-81.
18. Conlon L, Fahy T, Conroy R. PTSD in ambulant RTA victims: A randomized controlled trial of debriefing. J Psychosom Res 1999;46:37-44.
19. Deahl M, Srinivasan M, Jones N, et al. Preventing psychological trauma in soldiers: the role of operational stress training and psychological debriefing. Br J Med Psychol 2000;73:77-85.
20. Hobbs M, Mayou R, Harrison B, Warlock P. A randomized trial of psychological debriefing for victims of road traffic accidents. BMJ 1996;313:1438-9.
21. Mayou R, Ehlers A, Hobbs M. Psychological debriefing for road traffic accident victims: three-year follow-up of a randomized controlled trial. Br J Psychiatry 2000;176:589-93.
22. Rose S, Brewin C, Andrews B, Kirk M. A randomized controlled trial of individual psychological debriefing for victims of violent crime. Psychol Med 1999;29:793-9.
23. Schut H, Stroebe MS, van den Bout J, Terheggen M. The efficacy of bereavement interventions: determining who benefits. In: Stroebe MS, Hansson RO, Stroebe W, Schut H (eds). Handbook of bereavement research: Consequences, coping, and care. Washington, DC: American Psychological Association; 2001:705-37.
24. Bryant RA, Harvey AG, Dang S, et al. Treatment of acute stress disorder: a comparison of cognitive-behavioral therapy and supportive counseling. J Consult Clin Psychol 1998;66(5):862-6.
25. Foa EB, Hearst-Ikeda D, Perry KJ. Evaluation of a brief cognitive-behavioral program for the prevention of chronic PTSD in recent assault victims. J Consult Clin Psychol 1995;63(6):948.-
26. National Institute of Mental Health. Mental health and mass violence: evidence-based early psychological intervention for victims/survivors of mass violence. A workshop to reach consensus on best practices. NIH publication No. 02-5138. Washington, DC: U.S. Government Printing Office; 2002:13. Available at: www.nimh.nih.gov/healthinformation/massviolence_intervention.cfm. Accessed August 24, 2006.
27. Ozer EJ, Best SR, Lipsey TL, Weiss DS. Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull 2003;129(1):52-73.
28. Prigerson HG, Maciejewski PK, Reynolds CF, III. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res 1995;59(1-2):65-79.
29. Shear K, Frank E, Houck PR, Reynolds CF, 3rd. Treatment of complicated grief: a randomized controlled trial. JAMA 2005;293(21):2601-8.
Debriefing interventions have sprung from the understandable desire to reduce—if not eliminate—victims’ suffering after traumatic loss. Unfortunately, no compelling evidence has shown that an intervention given within a few days of a traumatic event can prevent significant psychological distress.
Evidence does suggest, however, that components of psychological debriefing discussed here may help you provide effective “first aid” to trauma victims and identify persons at risk for chronic psychological problems.
Complicated grief reactions
Death of a family member or close friend is among life’s most painful loses. When death occurs unexpectedly—as from violence, accident, natural disaster, or suicide—survivors’ emotional and psychological response can be pronounced.
Most survivors report great distress immediately after trauma or traumatic loss, but only an estimated 9% develop chronic psychopathology,1 such as complicated grief (Table 1).2,3 If the death was violent, surviving loved ones may experience complicated grief and posttraumatic stress disorder (PTSD)4 (Table 2).
Complicated grief is associated with considerable morbidity and risk of physical illness.5 PTSD develops in approximately one-third of cases involving sudden, unexpected death of a close friend or relative1 and can result in comorbid—but distinguishable—reactions to the loss (Box).6
Evidence-based secondary and tertiary intervention protocols have been developed for PTSD,7 but no practice guideline exists for treating or preventing complicated grief. Few controlled trials have been done.8
Table 1
Clinical features of complicated grief
|
| Source: Reference 3 |
Clinical features of posttraumatic stress disorder
|
| Source: DSM-IV-TR |
- educating individuals about stress reactions and how to cope with them
- instilling messages that stress reactions are normal
- helping affected persons process and share their emotions
- providing information about and opportunity for further intervention, if needed.
Traumatic loss. Although complicated grief (CG) and posttraumatic stress disorder (PTSD) can both develop following the loss of a loved one from a traumatic event, CG also can develop after expected deaths from natural causes. PTSD is exceedingly uncommon if a loved one’s death did not result from homicide, suicide, or accident, whereas CG can occur when the loss was not particularly violent or sudden.
Avoidance vs preoccupation. The fundamental difference between CG (Table 1) and PTSD (Table 2) symptoms is the degree that survivors avoid cues and contexts that remind them of their loss.
Those with PTSD go to great lengths to avoid thinking about the traumatic event and actively avoid situations that may remind them of it. This avoidance, paradoxically, exacerbates intrusive memories, as trying not to think about something increases the frequency of those thoughts.
Individuals with CG do not avoid reminders of the deceased. Quite the opposite, they seek out reminders (such as photos or recordings) and find solace in them. Reminders may contribute to their ongoing rumination or preoccupation, in which they retreat into memories of the deceased rather than engage in present life.
Hyperarousal symptoms that are required for PTSD diagnosis are largely absent in CG. Even when persons with CG experience arousal, it is not akin to scanning the environment for danger or threat, as is typical with PTSD. Persons with CG have a pronounced negative affect and bereavement-related depression, rather than an exaggerated startle response or heightened physiologic reactivity.
Does debriefing work?
Debriefing is designed not to address the intense but transient emotional reactions that can be expected immediately following traumatic loss but to prevent protracted, incapacitating distress. For an early intervention to be considered effective, it must be associated with greater or more expedient symptom recovery compared with natural remission. Controlled clinical trials are necessary to determine if this is the case.9
Control groups are essential when studying treatment outcomes of early crisis interventions. Simply documenting improvement among treated individuals is insufficient because substantial symptom remission is the norm and chronic psychopathology is comparatively rare. Thus, early interventions studies should at least:
- include a treatment group and a no-treatment or wait-list control condition
- randomly assign participants to avoid selfselection biases.
Debriefing for traumatic loss. Debriefing-based interventions have been used after mass violence and other large-scale traumatic events that may trigger complicated grief reactions.10 Most studies have not evaluated the impact of debriefing on complicated grief specifically but have focused on PTSD, anxiety, and depression. Typical published accounts of debriefing-based interventions for grief responses11 have been anecdotal, qualitative, and uncontrolled.
One rare controlled study of debriefing12 was designed to target emotional difficulties in women following early miscarriage. The one-half of participants who were debriefed 2 weeks after miscarriage perceived debriefing to be helpful. Despite significant improvement in early intrusion and avoidance scores, however, the women who were debriefed showed no greater improvement after 4 months than did a nondebriefed control group. The investigators concluded that debriefing did not influence post-loss adaptation.
A wider search. In the absence of randomized, controlled trials (RCTs) of debriefing-based interventions for traumatic loss, we turn to the larger debriefing literature. Nearly all debriefing studies have focused on PTSD symptoms rather than grief responses.
A number of peer-reviewed studies suggest that psychological debriefing is an effective intervention. These studies13,14 are characterized by dramatic symptom reductions following the intervention. Unfortunately, nearly all lack a control group, and the few that were controlled14 were not randomized. Studies reviewed by Everly et al15 also contain fundamental flaws, such as lack of random assignment, failure to assess individuals prior to the intervention, and lack of control groups.
None of the few RCTs of psychological debriefing conducted in traumatized populations show that it accelerates recovery in treated persons compared with nontreated controls.16 All of the studies17-22 included untreated control conditions, and participants were randomly assigned. Without exception, debriefed participants did not show superior improvement, and in two studies they showed worse outcomes than did untreated controls.17,21
Focused interventions
To provide optimal care to our patients, we must base our decisions on rigorous empirical study. In the case of debriefing, available well-controlled trials lead us to conclude that debriefings are inert.
To be clear, we are not philosophically opposed to early intervention for traumatic loss. We believe researchers must continue to develop and study interventions that can stave off chronic pathology among those at risk after traumatic loss.
Thus, clinicians and researchers face the same imperative: to accurately and efficiently identify persons at risk. Indiscriminately debriefing all persons who experience traumatic loss—without regard to risk—is not the most judicial use of clinical resources. Nor is it likely to advance our understanding of risk factors and resiliency in loss or of treatment efficacy.
Grief literature indicates that broadly applying interventions to anyone who has experienced loss does not help and may in fact exacerbate grief symptoms. Focused interventions for persons most at risk for complicated grief are more effective.23
Practice recommendations
Given the limited evidence, the recommendations that follow are preliminary and based on the few early interventions for trauma that have produced superior outcomes compared with untreated controls.24,25 In general, these interventions used:
- cognitive-behavioral techniques (education, promotion of adaptive coping strategies)
- exposure exercises for survivors who were using maladaptive avoidant coping strategies
- homework to reinforce therapeutic activities initiated in session.
‘Psychological first aid.’ Although immediate formal treatment is not recommended, a National Institute of Mental Health consensus conference26 recommended offering trauma victims “psychological first aid” (Table 3) when feasible. Psychological first aid is not intended to prevent chronic psychopathology but to provide:
- immediate emotional and informational support
- psychoeducational materials that describe common sequelae of trauma
- information about how and where to get help, if desired.
Recommended components of ‘psychological first aid’
|
| Source: National Institute of Mental Health, reference 15 |
Screening for risk factors. When victims seek your professional support or services immediately after a traumatic event, screen for risk factors for complicated grief, PTSD, or other chronic difficulties. Complicated grief is a relatively new diagnosis, and research on its risk factors is preliminary. The literature suggests, however, that risk factors may include:
- childhood abuse and neglect
- childhood separation anxiety
- loss of a child
- excessive interpersonal dependency or insecure attachment styles.6
In the weeks and months after the traumatic event, we recommend screening the most distressed victims for risk of developing chronic psychopathology. The National Center for PTSD offers self-report measures appropriate for various populations (such as children or adults) and trauma contexts (such as combat) (see Related resources). The Inventory of Complicated Grief28 is useful for screening for CG.
Empirically informed CBT. Provide brief cognitive behavioral interventions only for persons at risk and only after sufficient time has passed to allow you to differentiate between normal grief and abnormal responses. Early interventions that have shown promising outcomes typically have been delivered approximately 2 weeks after the traumatic exposure.24,25
Brief, multi-session CBT given several days to a few weeks after the trauma has been associated with improved posttraumatic adjustment.24,25 Interventions that appear to be most promising for patients who meet criteria for CG combine:
- psychoeducation
- exposure therapy for those having difficulty grasping the reality of their loss
- and behavioral activation techniques.29
Focus psychoeducation on how maladaptive strategies (such as avoiding trauma cues) can prolong trauma-related distress. Structure early interventions to encourage home-based exercises (such as exposure). These may reduce victims’ reliance on maladaptive strategies, accelerate therapeutic effects, and promote the generalization of treatment gains.24-25
- Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004.
- National Center for PTSD:
- Shear K, Frank E, Houck PR, Reynolds CF 3rd. Treatment of complicated grief: a randomized controlled trial. JAMA 2005;293(21):2601-8.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Debriefing interventions have sprung from the understandable desire to reduce—if not eliminate—victims’ suffering after traumatic loss. Unfortunately, no compelling evidence has shown that an intervention given within a few days of a traumatic event can prevent significant psychological distress.
Evidence does suggest, however, that components of psychological debriefing discussed here may help you provide effective “first aid” to trauma victims and identify persons at risk for chronic psychological problems.
Complicated grief reactions
Death of a family member or close friend is among life’s most painful loses. When death occurs unexpectedly—as from violence, accident, natural disaster, or suicide—survivors’ emotional and psychological response can be pronounced.
Most survivors report great distress immediately after trauma or traumatic loss, but only an estimated 9% develop chronic psychopathology,1 such as complicated grief (Table 1).2,3 If the death was violent, surviving loved ones may experience complicated grief and posttraumatic stress disorder (PTSD)4 (Table 2).
Complicated grief is associated with considerable morbidity and risk of physical illness.5 PTSD develops in approximately one-third of cases involving sudden, unexpected death of a close friend or relative1 and can result in comorbid—but distinguishable—reactions to the loss (Box).6
Evidence-based secondary and tertiary intervention protocols have been developed for PTSD,7 but no practice guideline exists for treating or preventing complicated grief. Few controlled trials have been done.8
Table 1
Clinical features of complicated grief
|
| Source: Reference 3 |
Clinical features of posttraumatic stress disorder
|
| Source: DSM-IV-TR |
- educating individuals about stress reactions and how to cope with them
- instilling messages that stress reactions are normal
- helping affected persons process and share their emotions
- providing information about and opportunity for further intervention, if needed.
Traumatic loss. Although complicated grief (CG) and posttraumatic stress disorder (PTSD) can both develop following the loss of a loved one from a traumatic event, CG also can develop after expected deaths from natural causes. PTSD is exceedingly uncommon if a loved one’s death did not result from homicide, suicide, or accident, whereas CG can occur when the loss was not particularly violent or sudden.
Avoidance vs preoccupation. The fundamental difference between CG (Table 1) and PTSD (Table 2) symptoms is the degree that survivors avoid cues and contexts that remind them of their loss.
Those with PTSD go to great lengths to avoid thinking about the traumatic event and actively avoid situations that may remind them of it. This avoidance, paradoxically, exacerbates intrusive memories, as trying not to think about something increases the frequency of those thoughts.
Individuals with CG do not avoid reminders of the deceased. Quite the opposite, they seek out reminders (such as photos or recordings) and find solace in them. Reminders may contribute to their ongoing rumination or preoccupation, in which they retreat into memories of the deceased rather than engage in present life.
Hyperarousal symptoms that are required for PTSD diagnosis are largely absent in CG. Even when persons with CG experience arousal, it is not akin to scanning the environment for danger or threat, as is typical with PTSD. Persons with CG have a pronounced negative affect and bereavement-related depression, rather than an exaggerated startle response or heightened physiologic reactivity.
Does debriefing work?
Debriefing is designed not to address the intense but transient emotional reactions that can be expected immediately following traumatic loss but to prevent protracted, incapacitating distress. For an early intervention to be considered effective, it must be associated with greater or more expedient symptom recovery compared with natural remission. Controlled clinical trials are necessary to determine if this is the case.9
Control groups are essential when studying treatment outcomes of early crisis interventions. Simply documenting improvement among treated individuals is insufficient because substantial symptom remission is the norm and chronic psychopathology is comparatively rare. Thus, early interventions studies should at least:
- include a treatment group and a no-treatment or wait-list control condition
- randomly assign participants to avoid selfselection biases.
Debriefing for traumatic loss. Debriefing-based interventions have been used after mass violence and other large-scale traumatic events that may trigger complicated grief reactions.10 Most studies have not evaluated the impact of debriefing on complicated grief specifically but have focused on PTSD, anxiety, and depression. Typical published accounts of debriefing-based interventions for grief responses11 have been anecdotal, qualitative, and uncontrolled.
One rare controlled study of debriefing12 was designed to target emotional difficulties in women following early miscarriage. The one-half of participants who were debriefed 2 weeks after miscarriage perceived debriefing to be helpful. Despite significant improvement in early intrusion and avoidance scores, however, the women who were debriefed showed no greater improvement after 4 months than did a nondebriefed control group. The investigators concluded that debriefing did not influence post-loss adaptation.
A wider search. In the absence of randomized, controlled trials (RCTs) of debriefing-based interventions for traumatic loss, we turn to the larger debriefing literature. Nearly all debriefing studies have focused on PTSD symptoms rather than grief responses.
A number of peer-reviewed studies suggest that psychological debriefing is an effective intervention. These studies13,14 are characterized by dramatic symptom reductions following the intervention. Unfortunately, nearly all lack a control group, and the few that were controlled14 were not randomized. Studies reviewed by Everly et al15 also contain fundamental flaws, such as lack of random assignment, failure to assess individuals prior to the intervention, and lack of control groups.
None of the few RCTs of psychological debriefing conducted in traumatized populations show that it accelerates recovery in treated persons compared with nontreated controls.16 All of the studies17-22 included untreated control conditions, and participants were randomly assigned. Without exception, debriefed participants did not show superior improvement, and in two studies they showed worse outcomes than did untreated controls.17,21
Focused interventions
To provide optimal care to our patients, we must base our decisions on rigorous empirical study. In the case of debriefing, available well-controlled trials lead us to conclude that debriefings are inert.
To be clear, we are not philosophically opposed to early intervention for traumatic loss. We believe researchers must continue to develop and study interventions that can stave off chronic pathology among those at risk after traumatic loss.
Thus, clinicians and researchers face the same imperative: to accurately and efficiently identify persons at risk. Indiscriminately debriefing all persons who experience traumatic loss—without regard to risk—is not the most judicial use of clinical resources. Nor is it likely to advance our understanding of risk factors and resiliency in loss or of treatment efficacy.
Grief literature indicates that broadly applying interventions to anyone who has experienced loss does not help and may in fact exacerbate grief symptoms. Focused interventions for persons most at risk for complicated grief are more effective.23
Practice recommendations
Given the limited evidence, the recommendations that follow are preliminary and based on the few early interventions for trauma that have produced superior outcomes compared with untreated controls.24,25 In general, these interventions used:
- cognitive-behavioral techniques (education, promotion of adaptive coping strategies)
- exposure exercises for survivors who were using maladaptive avoidant coping strategies
- homework to reinforce therapeutic activities initiated in session.
‘Psychological first aid.’ Although immediate formal treatment is not recommended, a National Institute of Mental Health consensus conference26 recommended offering trauma victims “psychological first aid” (Table 3) when feasible. Psychological first aid is not intended to prevent chronic psychopathology but to provide:
- immediate emotional and informational support
- psychoeducational materials that describe common sequelae of trauma
- information about how and where to get help, if desired.
Recommended components of ‘psychological first aid’
|
| Source: National Institute of Mental Health, reference 15 |
Screening for risk factors. When victims seek your professional support or services immediately after a traumatic event, screen for risk factors for complicated grief, PTSD, or other chronic difficulties. Complicated grief is a relatively new diagnosis, and research on its risk factors is preliminary. The literature suggests, however, that risk factors may include:
- childhood abuse and neglect
- childhood separation anxiety
- loss of a child
- excessive interpersonal dependency or insecure attachment styles.6
In the weeks and months after the traumatic event, we recommend screening the most distressed victims for risk of developing chronic psychopathology. The National Center for PTSD offers self-report measures appropriate for various populations (such as children or adults) and trauma contexts (such as combat) (see Related resources). The Inventory of Complicated Grief28 is useful for screening for CG.
Empirically informed CBT. Provide brief cognitive behavioral interventions only for persons at risk and only after sufficient time has passed to allow you to differentiate between normal grief and abnormal responses. Early interventions that have shown promising outcomes typically have been delivered approximately 2 weeks after the traumatic exposure.24,25
Brief, multi-session CBT given several days to a few weeks after the trauma has been associated with improved posttraumatic adjustment.24,25 Interventions that appear to be most promising for patients who meet criteria for CG combine:
- psychoeducation
- exposure therapy for those having difficulty grasping the reality of their loss
- and behavioral activation techniques.29
Focus psychoeducation on how maladaptive strategies (such as avoiding trauma cues) can prolong trauma-related distress. Structure early interventions to encourage home-based exercises (such as exposure). These may reduce victims’ reliance on maladaptive strategies, accelerate therapeutic effects, and promote the generalization of treatment gains.24-25
- Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004.
- National Center for PTSD:
- Shear K, Frank E, Houck PR, Reynolds CF 3rd. Treatment of complicated grief: a randomized controlled trial. JAMA 2005;293(21):2601-8.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Breslau N, Kessler RC, Chilcoat HD, et al. Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Arch Gen Psychiatry 1998;55(7):626-32.
2. Latham AE, Prigerson HG. Suicidality and bereavement: complicated grief as psychiatric disorder presenting greatest risk for suicidality. Suicide Life Threat Behav 2004;34(4):350-62.
3. Gray MJ, Prigerson HG, Litz BT. Conceptual and definitional issues in complicated grief. In: Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004:65-84.
4. Neria Y, Litz BT. Bereavement by traumatic means: the complex synergy of trauma and grief. Journal of Loss and Trauma 2004;9(1):73-87.
5. Silverman GK, Jacobs SC, Kasl SV, et al. Quality of life impairments associated with diagnostic criteria for traumatic grief. Psychol Med 2000;30(4):857-62.
6. Lichtenthal WG, Cruess DG, Prigerson HG. A case for establishing complicated grief as a distinct mental disorder in DSM-V. Clin Psychol Rev 2004;24(6):637-62.
7. American Psychiatric Association. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry 2004;161(11 suppl):3-31.
8. Schut H, Stroebe MS, van den Bout J, Terheggen M. The efficacy of bereavement interventions: Determining who benefits. In: Stroebe MS, Hansson RO, Stroebe W, Schut H (eds). Handbook of bereavement research: consequences, coping, and care. Washington, DC: American Psychological Association; 2001:705-37.
9. Litz BT, Gray MJ. Early intervention for trauma in adults: a framework for first aid and secondary prevention. In: Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004:87-111.
10. Litz BT, Gray MJ. Early intervention for mass violence: What is the evidence? What should be done? Cognit Behav Pract 2002;9(4):266-72.
11. Webb NB. Groups for children traumatically bereaved by the attacks of September 11, 2001. Int J Group Psychother 2005;55(3):355-74.
12. Lee C, Slade P, Lygo V. The influence of psychological debriefing on emotional adaptation in women following early miscarriage: a preliminary study. Br J Med Psychol 1996;69(1):47-58.
13. Robinson R, Mitchell J. Getting some balance back into the debriefing debate. Bull Aust Psychol Soc 1995;17:5-10.
14. Jenkins S. Social support and debriefing efficacy among emergency medical workers after a mass shooting incident. J Soc Behav Personality 1996;11:477-92.
15. Everly GS, Flannery RB, Mitchell JT. Critical incident stress management (CISM): A review of the literature. Aggression Violent Behav 2000;5(1):23-40.
16. Litz B, Gray M, Bryant R, Adler A. Early intervention for trauma: Current status and future directions. Clin Psychol Sci Pract 2002;9(2):112-34.
17. Bisson J, Jenkins P, Alexander J, Bannister C. Randomized controlled trial of psychological debriefing for victims of acute burn trauma. Br J Psychiatry 1997;171:78-81.
18. Conlon L, Fahy T, Conroy R. PTSD in ambulant RTA victims: A randomized controlled trial of debriefing. J Psychosom Res 1999;46:37-44.
19. Deahl M, Srinivasan M, Jones N, et al. Preventing psychological trauma in soldiers: the role of operational stress training and psychological debriefing. Br J Med Psychol 2000;73:77-85.
20. Hobbs M, Mayou R, Harrison B, Warlock P. A randomized trial of psychological debriefing for victims of road traffic accidents. BMJ 1996;313:1438-9.
21. Mayou R, Ehlers A, Hobbs M. Psychological debriefing for road traffic accident victims: three-year follow-up of a randomized controlled trial. Br J Psychiatry 2000;176:589-93.
22. Rose S, Brewin C, Andrews B, Kirk M. A randomized controlled trial of individual psychological debriefing for victims of violent crime. Psychol Med 1999;29:793-9.
23. Schut H, Stroebe MS, van den Bout J, Terheggen M. The efficacy of bereavement interventions: determining who benefits. In: Stroebe MS, Hansson RO, Stroebe W, Schut H (eds). Handbook of bereavement research: Consequences, coping, and care. Washington, DC: American Psychological Association; 2001:705-37.
24. Bryant RA, Harvey AG, Dang S, et al. Treatment of acute stress disorder: a comparison of cognitive-behavioral therapy and supportive counseling. J Consult Clin Psychol 1998;66(5):862-6.
25. Foa EB, Hearst-Ikeda D, Perry KJ. Evaluation of a brief cognitive-behavioral program for the prevention of chronic PTSD in recent assault victims. J Consult Clin Psychol 1995;63(6):948.-
26. National Institute of Mental Health. Mental health and mass violence: evidence-based early psychological intervention for victims/survivors of mass violence. A workshop to reach consensus on best practices. NIH publication No. 02-5138. Washington, DC: U.S. Government Printing Office; 2002:13. Available at: www.nimh.nih.gov/healthinformation/massviolence_intervention.cfm. Accessed August 24, 2006.
27. Ozer EJ, Best SR, Lipsey TL, Weiss DS. Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull 2003;129(1):52-73.
28. Prigerson HG, Maciejewski PK, Reynolds CF, III. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res 1995;59(1-2):65-79.
29. Shear K, Frank E, Houck PR, Reynolds CF, 3rd. Treatment of complicated grief: a randomized controlled trial. JAMA 2005;293(21):2601-8.
1. Breslau N, Kessler RC, Chilcoat HD, et al. Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Arch Gen Psychiatry 1998;55(7):626-32.
2. Latham AE, Prigerson HG. Suicidality and bereavement: complicated grief as psychiatric disorder presenting greatest risk for suicidality. Suicide Life Threat Behav 2004;34(4):350-62.
3. Gray MJ, Prigerson HG, Litz BT. Conceptual and definitional issues in complicated grief. In: Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004:65-84.
4. Neria Y, Litz BT. Bereavement by traumatic means: the complex synergy of trauma and grief. Journal of Loss and Trauma 2004;9(1):73-87.
5. Silverman GK, Jacobs SC, Kasl SV, et al. Quality of life impairments associated with diagnostic criteria for traumatic grief. Psychol Med 2000;30(4):857-62.
6. Lichtenthal WG, Cruess DG, Prigerson HG. A case for establishing complicated grief as a distinct mental disorder in DSM-V. Clin Psychol Rev 2004;24(6):637-62.
7. American Psychiatric Association. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. Am J Psychiatry 2004;161(11 suppl):3-31.
8. Schut H, Stroebe MS, van den Bout J, Terheggen M. The efficacy of bereavement interventions: Determining who benefits. In: Stroebe MS, Hansson RO, Stroebe W, Schut H (eds). Handbook of bereavement research: consequences, coping, and care. Washington, DC: American Psychological Association; 2001:705-37.
9. Litz BT, Gray MJ. Early intervention for trauma in adults: a framework for first aid and secondary prevention. In: Litz BT (ed). Early intervention for trauma and traumatic loss. New York: Guilford Press; 2004:87-111.
10. Litz BT, Gray MJ. Early intervention for mass violence: What is the evidence? What should be done? Cognit Behav Pract 2002;9(4):266-72.
11. Webb NB. Groups for children traumatically bereaved by the attacks of September 11, 2001. Int J Group Psychother 2005;55(3):355-74.
12. Lee C, Slade P, Lygo V. The influence of psychological debriefing on emotional adaptation in women following early miscarriage: a preliminary study. Br J Med Psychol 1996;69(1):47-58.
13. Robinson R, Mitchell J. Getting some balance back into the debriefing debate. Bull Aust Psychol Soc 1995;17:5-10.
14. Jenkins S. Social support and debriefing efficacy among emergency medical workers after a mass shooting incident. J Soc Behav Personality 1996;11:477-92.
15. Everly GS, Flannery RB, Mitchell JT. Critical incident stress management (CISM): A review of the literature. Aggression Violent Behav 2000;5(1):23-40.
16. Litz B, Gray M, Bryant R, Adler A. Early intervention for trauma: Current status and future directions. Clin Psychol Sci Pract 2002;9(2):112-34.
17. Bisson J, Jenkins P, Alexander J, Bannister C. Randomized controlled trial of psychological debriefing for victims of acute burn trauma. Br J Psychiatry 1997;171:78-81.
18. Conlon L, Fahy T, Conroy R. PTSD in ambulant RTA victims: A randomized controlled trial of debriefing. J Psychosom Res 1999;46:37-44.
19. Deahl M, Srinivasan M, Jones N, et al. Preventing psychological trauma in soldiers: the role of operational stress training and psychological debriefing. Br J Med Psychol 2000;73:77-85.
20. Hobbs M, Mayou R, Harrison B, Warlock P. A randomized trial of psychological debriefing for victims of road traffic accidents. BMJ 1996;313:1438-9.
21. Mayou R, Ehlers A, Hobbs M. Psychological debriefing for road traffic accident victims: three-year follow-up of a randomized controlled trial. Br J Psychiatry 2000;176:589-93.
22. Rose S, Brewin C, Andrews B, Kirk M. A randomized controlled trial of individual psychological debriefing for victims of violent crime. Psychol Med 1999;29:793-9.
23. Schut H, Stroebe MS, van den Bout J, Terheggen M. The efficacy of bereavement interventions: determining who benefits. In: Stroebe MS, Hansson RO, Stroebe W, Schut H (eds). Handbook of bereavement research: Consequences, coping, and care. Washington, DC: American Psychological Association; 2001:705-37.
24. Bryant RA, Harvey AG, Dang S, et al. Treatment of acute stress disorder: a comparison of cognitive-behavioral therapy and supportive counseling. J Consult Clin Psychol 1998;66(5):862-6.
25. Foa EB, Hearst-Ikeda D, Perry KJ. Evaluation of a brief cognitive-behavioral program for the prevention of chronic PTSD in recent assault victims. J Consult Clin Psychol 1995;63(6):948.-
26. National Institute of Mental Health. Mental health and mass violence: evidence-based early psychological intervention for victims/survivors of mass violence. A workshop to reach consensus on best practices. NIH publication No. 02-5138. Washington, DC: U.S. Government Printing Office; 2002:13. Available at: www.nimh.nih.gov/healthinformation/massviolence_intervention.cfm. Accessed August 24, 2006.
27. Ozer EJ, Best SR, Lipsey TL, Weiss DS. Predictors of posttraumatic stress disorder and symptoms in adults: a meta-analysis. Psychol Bull 2003;129(1):52-73.
28. Prigerson HG, Maciejewski PK, Reynolds CF, III. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res 1995;59(1-2):65-79.
29. Shear K, Frank E, Houck PR, Reynolds CF, 3rd. Treatment of complicated grief: a randomized controlled trial. JAMA 2005;293(21):2601-8.
Which antipsychotic do I choose next?
CATIE phase 2 offers insights on efficacy an tolerability
After nearly 3 out of 4 phase 1 patients stopped taking their assigned antipsychotics within 18 months, researchers in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) braced themselves for phase 2.February 2006)
CATIE’s eligibility criteria are broad and include schizophrenia patients with comorbid conditions such as substance abuse and mood disorders. The primary outcome measure is all-cause treatment discontinuation, which incorporates efficacy, safety, tolerability, patient choice, and clinician choice (Table 1).
Phase 1 compared the efficacy and safety of four second-generation antipsychotics (SGA) and one first-generation antipsychotic (FGA).3 Nasrallah concluded that—despite the high discontinuation rate in that phase—there were “no winners or losers” among the five antipsychotics. The results, Nasrallah concluded:
- provide a compelling rationale for clinicians to match medication profiles to individual patients
- support the need for clinicians to have choices among medications when treating patients with schizophrenia.4
Table 1
Drug discontinuation patterns in CATIE phase 1
| Measures | Findings after 18 months | |
|---|---|---|
| % of patients who discontinued medication for any reason | Olanzapine (64%) | Ziprasidone (79%) |
| Risperidone (74%) | Quetiapine (82%) | |
| Perphenazine (75%) | ||
| Time to discontinuation for any reason | Longest (most favorable) with olanzapine, but not statistically longer with olanzapine than with ziprasidone or perphenazine | |
| No statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | ||
| Time to discontinuation for lack of efficacy* | Longer with olanzapine; no statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | |
| Time to discontinuation for intolerable side effects | No statistical difference among agents | |
| Rate of discontinuation for intolerable side effects | Highest (19%) with olanzapine (primarily because of weight gain or metabolic effects with this medication) | |
| Rate of discontinuation for extrapyramidal effects | Highest (8%) with perphenazine | |
| Rate of discontinuation for intolerability (overall) | Lowest with risperidone (10%) | |
| * Nonequivalent dosing in CATIE phase 1 is an ongoing debate. | ||
What to do next?
When an initial antipsychotic proves inadequate or causes intolerable side effects, how do you choose a more efficacious or tolerable medication? Phase 2 offered CATIE patients and their clinicians two choices—an efficacy and a tolerability pathway (Figure).1,2
CATIE phase 2: Distribution of patients in efficacy and tolerability pathways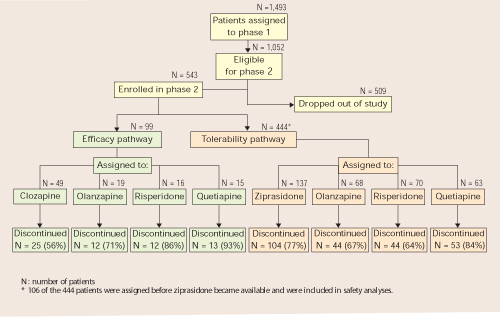
Efficacy pathway. Patients who chose the efficacy pathway were randomly assigned to clozapine (50%) or olanzapine, risperidone, or quetiapine.1 Researchers selected clozapine as the major efficacy comparator because of its robust effects in treatment-refractory schizophrenia. Clozapine was given open-label because of its safety monitoring requirements; other treatments were double-blind.
As in phase 1, the primary outcome measure was time until discontinuation for any reason. Secondary outcome measures included time to discontinuation because of side effects, patient choice, or lack of efficacy.
Tolerability pathway. Patients who chose the tolerability pathway were randomly assigned to double-blind treatment with ziprasidone, olanzapine, risperidone, or quetiapine.2 Ziprasidone was the major comparator because of clinical data showing a favorable tolerability profile.
The primary outcome measure was time to discontinuation for any reason. Secondary outcomes included reason for discontinuation (as determined by the study clinician), symptomatic ratings, and evaluations of adverse effects.
Trial duration. No patients in either pathway received the same antipsychotics they had taken in phase 1. All patients could continue treatment through the 18 months of the CATIE trial or until they completed 6 months in phase 2.
Efficacy pathway results
Discontinuation. Consistent with literature about its efficacy in treatment-refractory schizophrenia, clozapine showed a robust clinical effect. Overall, more patients receiving clozapine stayed on treatment and for longer periods, compared with patients receiving olanzapine, risperidone, or quetiapine (Table 2).
On secondary measures, discontinuation for lack of efficacy was significantly lower with clozapine (11%) than with:
- olanzapine (35%)
- risperidone or quetiapine (each at 43%).
Discontinuation rates because of adverse effects or by patient choice were the same across all medications (Table 3). Patients on clozapine achieved better ratings in overall psychotic symptoms, positive symptoms, and general function, but not in negative symptoms.
Weight gain. On average, patients gained more weight while taking olanzapine (+1.1 lb/mo) than with:
- risperidone (+0.5 lb/mo)
- clozapine (+0.5 lb/mo)
- quetiapine (+0.5 lb/mo)
Differences in weight gain—or in metabolic parameters or other adverse effects—were not statistically significant, however.
Table 2
Phase 2 efficacy pathway: Discontinuation for any reason
| Measure | Clozapine | Olanzapine | Risperidone | Quetiapine |
|---|---|---|---|---|
| How many patients discontinued | 25 of 49 (56%) | 12 of 19 (71%) | 12 of 16 (86%) | 13 of 15 (93%) |
| Median time to discontinuation | 10.5 months | 2.7 months | 2.8 months | 3.3 months |
Table 3
Reasons patients stopped taking their medications in CATIE phase 2
| Reason | Efficacy pathway | Tolerability pathway |
|---|---|---|
| All cause | 69% | 74% |
| Lack of efficacy | 26% | 29% |
| Lack of tolerability | 10% | 15% |
| Patient choice | 26% | 24% |
Tolerability pathway results
Discontinuation. Patients in the tolerability pathway took olanzapine or risperidone significantly longer—median 6.3 and 7 months, respectively— compared with ziprasidone (4 months) or quetiapine (2.8 months).
- Time to discontinuation during phase 2 was the same across all drugs among patients who entered phase 2 because of intolerable side effects in phase 1.
- Time to discontinuation because of side effects also was similar whether patients discontinued phase 1 for lack of efficacy or intolerable side effects. Patients stopped treatment in the efficacy and tolerability pathways for similar reasons (Table 3).
Weight gain. Patients taking olanzapine gained more weight (average +1.3 lb/mo) than did those taking the other drugs. Patients taking ziprasidone lost weight (average –1.7 lb/mo). Among 61 patients who gained weight during phase 1, 42% of those switched to ziprasidone lost weight in phase 2, as did:
- 20% of those switched to risperidone
- 7% of those switched to quetiapine.
Among those switched to olanzapine in phase 2, no one lost weight and 2% gained weight.
Metabolic effects. Some parameters changed, depending on drug assignment:
- prolactin increased in patients switched to risperidone
- cholesterol and triglycerides increased in patients switched to olanzapine or quetiapine but decreased in those switched to risperidone or ziprasidone
- QTc interval measurements showed no difference across all drugs.
Methodologic caveats
When considering how CATIE’s phase 2 findings might apply to clinical practice, keep in mind four caveats about the study’s design.
Clozapine was given open-label, yet quetiapine, olanzapine, and risperidone were given double-blind in the efficacy pathway. This pathway’s findings are consistent with what we know about clozapine and other SGAs in treatment-refractory schizophrenia, but how the open-label design affected clozapine therapy outcomes is unclear.
Were patients who knew they were taking clozapine more willing to “stay the course” than were patients in the pathway’s double-blind arm?
Discontinuation rates remained high. The 74% “overall discontinuation rate” in phase 1 surprised many psychiatrists because of the perceived high rate at which patients did not adhere to the first medications they received. To some extent, the word “discontinuation” is imprecise, however, because this group includes patients who did not drop out of treatment altogether but chose to move on to phase 2.
It is important to note, however, that nearly one-half of phase 1 patients who were eligible to enter phase 2 (509 of 1,052) did not. This group represents the true drop-out rate, which is substantial. The high rates of discontinuation seen in phase 1 also occurred in both phase 2 pathways (Table 3).
Few patients entered the efficacy pathway. In an approach designed to reflect routine clinical practice, the researchers recommended the efficacy pathway to patients who discontinued phase 1 because of lack of efficacy and the tolerability pathway to those who discontinued phase 1 because of intolerability. Many patients did not follow the recommendations, however, and seemed to choose their pathways based on whether they wanted a chance to receive clozapine or ziprasidone in phase 2.
Thus, among the 543 phase 1 patients who enrolled in phase 2, 99 (18%) entered the efficacy pathway, and 444 (82%) entered the tolerability pathway. The efficacy pathway included 85 patients who discontinued phase 1 for lack of efficacy and 5 for lack of tolerability. The tolerability pathway included 184 patients who discontinued phase 1 for lack of efficacy and 168 for lack of tolerability.
Dosages may not have been equivalent. SGAs’ dosing equivalency is unknown,5,6 which impedes our ability to interpret comparative studies such as CATIE. The study’s designers developed the its dosing ranges by careful consideration, including recommendations from each SGA’s manufacturer. As Nasrallah described,4 the trial’s dosages were not universally consistent with FDA-approved ranges or usual clinical practice (Table 4). In phase 2, for example, ziprasidone dosages were less than psychiatrists usually use, and quetiapine dosages were greater than usual.
Fortunately, studies are underway to determine each SGA’s optimum dosing. This work will help us understand what we can expect when we increase an antipsychotic’s dosage—a key step towards understanding dosing equivalency.
Table 4
Mean modal antipsychotic dosages (mg/d) in CATIE phase 2 pathways*
| Clozapine | Ziprasidone | Olanzapine | Risperidone | Quetiapine | |
|---|---|---|---|---|---|
| Efficacy pathway | 332 | — | 23.4 | 4.8 | 642.9 |
| Tolerability pathway | — | 115.9 | 20.5 | 4.15 | 65.2 |
| * 800 mg/d of quetiapine and 160 mg/d of ziprasidone are generally regarded as therapeutically equivalent to 20 mg/d of olanzapine. | |||||
What clinicians can expect
A recent analysis helps put CATIE’s findings in perspective. Citrome and Stroup7 quantified the results of phase 1 and 2 with respect to:
- number needed to treat (NNT)—how many patients a clinician needs to treat with drug A to see one additional benefit, compared with drug B
- number needed to harm (NNH)—how many patients a clinician needs to treat with drug A to see a given adverse effect, compared with drug B.
In this analysis, the NNT for olanzapine (5.5 to 10) was lowest among the drugs compared in phase 1, and the NNT for clozapine (3) was lowest among those compared in phase 2. A lower number means that, overall, clinicians can expect a more robust treatment response.
On the other hand, the NNH for olanzapine in weight gain and metabolic disturbances (12.4 to 17.7) was the lowest in phase 1, indicating that clinicians can expect more weight gain and metabolic effects with olanzapine than with other SGAs. Ziprasidone had the highest NNH (106 to 208) among the agents in phase 2 for avoiding discontinuation because of weight gain or metabolic disturbances. In other words, ziprasidone appears less likely than other SGAs to cause metabolic problems.
These risk-attribution measures demonstrate the dilemma clinicians face when trying to match schizophrenia patients with antipsychotics. CATIE was “an N of 1,493” subjects, whereas each patient we see in clinical practice is “an N of 1.” One patient may need a more-robust response; another may need improved tolerability.
We strive for balance, seeking to optimize efficacy—often by raising the dosage—while minimizing adverse effects.
What to tell patients
CATIE phases 1 and 2 provide a compelling rationale for individualized treatment, which should be standard clinical practice for schizophrenia:
- All drugs used in phases 1 and 2 worked.
- All showed noteworthy adverse effects that were different for each drug.
- Different patients responded differently to each drug.
Using our clinical judgment and available information, we must match—as best we can—the individual patient’s characteristics with the antipsychotics’ risk: benefit profiles. CATIE phases 1 and 2 provide independent information on the comparative efficacy and tolerability of each medication.
The CATIE investigators and NIMH have done a great service to our field in providing a rich repository of timely information to inform clinical practice. But the CATIE study was not designed to answer all our questions about treating schizophrenia.8,9 Clinicians and patients need to look elsewhere for guidance on the roles of:
- psychosocial treatments
- recovery and the therapeutic alliance in maximizing outcomes
- long-acting SGA formulations
- aripiprazole (addressed in CATIE phase 3)
- SGAs in first-episode schizophrenia
- FGAs when a patient does not adequately respond to an initial SGA.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). www.CATIE.unc.edu.
- Lieberman JA. What the CATIE study means in clinical practice. Psychiatr Serv 2006;57(8):1075.
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Buckley receives research/grant support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Pfizer, and Solvay Pharmaceuticals, and is a consultant to Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Merck & Co., and Pfizer.
Acknowledgement
The author thanks Del Miller, MD, for comments given on a draft of this paper.
1. Stroup TS, Lieberman JA, McEvoy JP, et al for the CATIE investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry 2006;163:611-22.
2. McEvoy JP, Lieberman JA, Stroup TS, et al for the CATIE investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 2006;163:600-10.
3. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
4. Nasrallah HA. CATIE’s surprises: In antipsychotics’ square-off, were there winners or losers? Current Psychiatry 2006;5(2):52-65.
5. Buckley PF. Dosing equivalency of second-generation antipsychotics. J Clin Psychopharmacol 2005;25(5):501-2.
6. Davis JM. The choice of drugs for schizophrenia. N Engl J Med 2006;354(5):518-20.
7. Citrome L, Stroup TS. Schizophrenia clinical antipsychotic trials intervention effectiveness and number needed to treat: How can CATIE inform clinicians? Int J Clin Pract 2006 (in press).
8. Ragins M. Should the CATIE study be a wake-up call? Psychiatr Serv 2005;56:1489.-
9. Lieberman JA, Hsiao J. Interpreting the results of the CATIE study. Psychiatr Serv 2006;57:139.-
CATIE phase 2 offers insights on efficacy an tolerability
After nearly 3 out of 4 phase 1 patients stopped taking their assigned antipsychotics within 18 months, researchers in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) braced themselves for phase 2.February 2006)
CATIE’s eligibility criteria are broad and include schizophrenia patients with comorbid conditions such as substance abuse and mood disorders. The primary outcome measure is all-cause treatment discontinuation, which incorporates efficacy, safety, tolerability, patient choice, and clinician choice (Table 1).
Phase 1 compared the efficacy and safety of four second-generation antipsychotics (SGA) and one first-generation antipsychotic (FGA).3 Nasrallah concluded that—despite the high discontinuation rate in that phase—there were “no winners or losers” among the five antipsychotics. The results, Nasrallah concluded:
- provide a compelling rationale for clinicians to match medication profiles to individual patients
- support the need for clinicians to have choices among medications when treating patients with schizophrenia.4
Table 1
Drug discontinuation patterns in CATIE phase 1
| Measures | Findings after 18 months | |
|---|---|---|
| % of patients who discontinued medication for any reason | Olanzapine (64%) | Ziprasidone (79%) |
| Risperidone (74%) | Quetiapine (82%) | |
| Perphenazine (75%) | ||
| Time to discontinuation for any reason | Longest (most favorable) with olanzapine, but not statistically longer with olanzapine than with ziprasidone or perphenazine | |
| No statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | ||
| Time to discontinuation for lack of efficacy* | Longer with olanzapine; no statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | |
| Time to discontinuation for intolerable side effects | No statistical difference among agents | |
| Rate of discontinuation for intolerable side effects | Highest (19%) with olanzapine (primarily because of weight gain or metabolic effects with this medication) | |
| Rate of discontinuation for extrapyramidal effects | Highest (8%) with perphenazine | |
| Rate of discontinuation for intolerability (overall) | Lowest with risperidone (10%) | |
| * Nonequivalent dosing in CATIE phase 1 is an ongoing debate. | ||
What to do next?
When an initial antipsychotic proves inadequate or causes intolerable side effects, how do you choose a more efficacious or tolerable medication? Phase 2 offered CATIE patients and their clinicians two choices—an efficacy and a tolerability pathway (Figure).1,2
CATIE phase 2: Distribution of patients in efficacy and tolerability pathways
Efficacy pathway. Patients who chose the efficacy pathway were randomly assigned to clozapine (50%) or olanzapine, risperidone, or quetiapine.1 Researchers selected clozapine as the major efficacy comparator because of its robust effects in treatment-refractory schizophrenia. Clozapine was given open-label because of its safety monitoring requirements; other treatments were double-blind.
As in phase 1, the primary outcome measure was time until discontinuation for any reason. Secondary outcome measures included time to discontinuation because of side effects, patient choice, or lack of efficacy.
Tolerability pathway. Patients who chose the tolerability pathway were randomly assigned to double-blind treatment with ziprasidone, olanzapine, risperidone, or quetiapine.2 Ziprasidone was the major comparator because of clinical data showing a favorable tolerability profile.
The primary outcome measure was time to discontinuation for any reason. Secondary outcomes included reason for discontinuation (as determined by the study clinician), symptomatic ratings, and evaluations of adverse effects.
Trial duration. No patients in either pathway received the same antipsychotics they had taken in phase 1. All patients could continue treatment through the 18 months of the CATIE trial or until they completed 6 months in phase 2.
Efficacy pathway results
Discontinuation. Consistent with literature about its efficacy in treatment-refractory schizophrenia, clozapine showed a robust clinical effect. Overall, more patients receiving clozapine stayed on treatment and for longer periods, compared with patients receiving olanzapine, risperidone, or quetiapine (Table 2).
On secondary measures, discontinuation for lack of efficacy was significantly lower with clozapine (11%) than with:
- olanzapine (35%)
- risperidone or quetiapine (each at 43%).
Discontinuation rates because of adverse effects or by patient choice were the same across all medications (Table 3). Patients on clozapine achieved better ratings in overall psychotic symptoms, positive symptoms, and general function, but not in negative symptoms.
Weight gain. On average, patients gained more weight while taking olanzapine (+1.1 lb/mo) than with:
- risperidone (+0.5 lb/mo)
- clozapine (+0.5 lb/mo)
- quetiapine (+0.5 lb/mo)
Differences in weight gain—or in metabolic parameters or other adverse effects—were not statistically significant, however.
Table 2
Phase 2 efficacy pathway: Discontinuation for any reason
| Measure | Clozapine | Olanzapine | Risperidone | Quetiapine |
|---|---|---|---|---|
| How many patients discontinued | 25 of 49 (56%) | 12 of 19 (71%) | 12 of 16 (86%) | 13 of 15 (93%) |
| Median time to discontinuation | 10.5 months | 2.7 months | 2.8 months | 3.3 months |
Table 3
Reasons patients stopped taking their medications in CATIE phase 2
| Reason | Efficacy pathway | Tolerability pathway |
|---|---|---|
| All cause | 69% | 74% |
| Lack of efficacy | 26% | 29% |
| Lack of tolerability | 10% | 15% |
| Patient choice | 26% | 24% |
Tolerability pathway results
Discontinuation. Patients in the tolerability pathway took olanzapine or risperidone significantly longer—median 6.3 and 7 months, respectively— compared with ziprasidone (4 months) or quetiapine (2.8 months).
- Time to discontinuation during phase 2 was the same across all drugs among patients who entered phase 2 because of intolerable side effects in phase 1.
- Time to discontinuation because of side effects also was similar whether patients discontinued phase 1 for lack of efficacy or intolerable side effects. Patients stopped treatment in the efficacy and tolerability pathways for similar reasons (Table 3).
Weight gain. Patients taking olanzapine gained more weight (average +1.3 lb/mo) than did those taking the other drugs. Patients taking ziprasidone lost weight (average –1.7 lb/mo). Among 61 patients who gained weight during phase 1, 42% of those switched to ziprasidone lost weight in phase 2, as did:
- 20% of those switched to risperidone
- 7% of those switched to quetiapine.
Among those switched to olanzapine in phase 2, no one lost weight and 2% gained weight.
Metabolic effects. Some parameters changed, depending on drug assignment:
- prolactin increased in patients switched to risperidone
- cholesterol and triglycerides increased in patients switched to olanzapine or quetiapine but decreased in those switched to risperidone or ziprasidone
- QTc interval measurements showed no difference across all drugs.
Methodologic caveats
When considering how CATIE’s phase 2 findings might apply to clinical practice, keep in mind four caveats about the study’s design.
Clozapine was given open-label, yet quetiapine, olanzapine, and risperidone were given double-blind in the efficacy pathway. This pathway’s findings are consistent with what we know about clozapine and other SGAs in treatment-refractory schizophrenia, but how the open-label design affected clozapine therapy outcomes is unclear.
Were patients who knew they were taking clozapine more willing to “stay the course” than were patients in the pathway’s double-blind arm?
Discontinuation rates remained high. The 74% “overall discontinuation rate” in phase 1 surprised many psychiatrists because of the perceived high rate at which patients did not adhere to the first medications they received. To some extent, the word “discontinuation” is imprecise, however, because this group includes patients who did not drop out of treatment altogether but chose to move on to phase 2.
It is important to note, however, that nearly one-half of phase 1 patients who were eligible to enter phase 2 (509 of 1,052) did not. This group represents the true drop-out rate, which is substantial. The high rates of discontinuation seen in phase 1 also occurred in both phase 2 pathways (Table 3).
Few patients entered the efficacy pathway. In an approach designed to reflect routine clinical practice, the researchers recommended the efficacy pathway to patients who discontinued phase 1 because of lack of efficacy and the tolerability pathway to those who discontinued phase 1 because of intolerability. Many patients did not follow the recommendations, however, and seemed to choose their pathways based on whether they wanted a chance to receive clozapine or ziprasidone in phase 2.
Thus, among the 543 phase 1 patients who enrolled in phase 2, 99 (18%) entered the efficacy pathway, and 444 (82%) entered the tolerability pathway. The efficacy pathway included 85 patients who discontinued phase 1 for lack of efficacy and 5 for lack of tolerability. The tolerability pathway included 184 patients who discontinued phase 1 for lack of efficacy and 168 for lack of tolerability.
Dosages may not have been equivalent. SGAs’ dosing equivalency is unknown,5,6 which impedes our ability to interpret comparative studies such as CATIE. The study’s designers developed the its dosing ranges by careful consideration, including recommendations from each SGA’s manufacturer. As Nasrallah described,4 the trial’s dosages were not universally consistent with FDA-approved ranges or usual clinical practice (Table 4). In phase 2, for example, ziprasidone dosages were less than psychiatrists usually use, and quetiapine dosages were greater than usual.
Fortunately, studies are underway to determine each SGA’s optimum dosing. This work will help us understand what we can expect when we increase an antipsychotic’s dosage—a key step towards understanding dosing equivalency.
Table 4
Mean modal antipsychotic dosages (mg/d) in CATIE phase 2 pathways*
| Clozapine | Ziprasidone | Olanzapine | Risperidone | Quetiapine | |
|---|---|---|---|---|---|
| Efficacy pathway | 332 | — | 23.4 | 4.8 | 642.9 |
| Tolerability pathway | — | 115.9 | 20.5 | 4.15 | 65.2 |
| * 800 mg/d of quetiapine and 160 mg/d of ziprasidone are generally regarded as therapeutically equivalent to 20 mg/d of olanzapine. | |||||
What clinicians can expect
A recent analysis helps put CATIE’s findings in perspective. Citrome and Stroup7 quantified the results of phase 1 and 2 with respect to:
- number needed to treat (NNT)—how many patients a clinician needs to treat with drug A to see one additional benefit, compared with drug B
- number needed to harm (NNH)—how many patients a clinician needs to treat with drug A to see a given adverse effect, compared with drug B.
In this analysis, the NNT for olanzapine (5.5 to 10) was lowest among the drugs compared in phase 1, and the NNT for clozapine (3) was lowest among those compared in phase 2. A lower number means that, overall, clinicians can expect a more robust treatment response.
On the other hand, the NNH for olanzapine in weight gain and metabolic disturbances (12.4 to 17.7) was the lowest in phase 1, indicating that clinicians can expect more weight gain and metabolic effects with olanzapine than with other SGAs. Ziprasidone had the highest NNH (106 to 208) among the agents in phase 2 for avoiding discontinuation because of weight gain or metabolic disturbances. In other words, ziprasidone appears less likely than other SGAs to cause metabolic problems.
These risk-attribution measures demonstrate the dilemma clinicians face when trying to match schizophrenia patients with antipsychotics. CATIE was “an N of 1,493” subjects, whereas each patient we see in clinical practice is “an N of 1.” One patient may need a more-robust response; another may need improved tolerability.
We strive for balance, seeking to optimize efficacy—often by raising the dosage—while minimizing adverse effects.
What to tell patients
CATIE phases 1 and 2 provide a compelling rationale for individualized treatment, which should be standard clinical practice for schizophrenia:
- All drugs used in phases 1 and 2 worked.
- All showed noteworthy adverse effects that were different for each drug.
- Different patients responded differently to each drug.
Using our clinical judgment and available information, we must match—as best we can—the individual patient’s characteristics with the antipsychotics’ risk: benefit profiles. CATIE phases 1 and 2 provide independent information on the comparative efficacy and tolerability of each medication.
The CATIE investigators and NIMH have done a great service to our field in providing a rich repository of timely information to inform clinical practice. But the CATIE study was not designed to answer all our questions about treating schizophrenia.8,9 Clinicians and patients need to look elsewhere for guidance on the roles of:
- psychosocial treatments
- recovery and the therapeutic alliance in maximizing outcomes
- long-acting SGA formulations
- aripiprazole (addressed in CATIE phase 3)
- SGAs in first-episode schizophrenia
- FGAs when a patient does not adequately respond to an initial SGA.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). www.CATIE.unc.edu.
- Lieberman JA. What the CATIE study means in clinical practice. Psychiatr Serv 2006;57(8):1075.
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Buckley receives research/grant support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Pfizer, and Solvay Pharmaceuticals, and is a consultant to Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Merck & Co., and Pfizer.
Acknowledgement
The author thanks Del Miller, MD, for comments given on a draft of this paper.
CATIE phase 2 offers insights on efficacy an tolerability
After nearly 3 out of 4 phase 1 patients stopped taking their assigned antipsychotics within 18 months, researchers in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) braced themselves for phase 2.February 2006)
CATIE’s eligibility criteria are broad and include schizophrenia patients with comorbid conditions such as substance abuse and mood disorders. The primary outcome measure is all-cause treatment discontinuation, which incorporates efficacy, safety, tolerability, patient choice, and clinician choice (Table 1).
Phase 1 compared the efficacy and safety of four second-generation antipsychotics (SGA) and one first-generation antipsychotic (FGA).3 Nasrallah concluded that—despite the high discontinuation rate in that phase—there were “no winners or losers” among the five antipsychotics. The results, Nasrallah concluded:
- provide a compelling rationale for clinicians to match medication profiles to individual patients
- support the need for clinicians to have choices among medications when treating patients with schizophrenia.4
Table 1
Drug discontinuation patterns in CATIE phase 1
| Measures | Findings after 18 months | |
|---|---|---|
| % of patients who discontinued medication for any reason | Olanzapine (64%) | Ziprasidone (79%) |
| Risperidone (74%) | Quetiapine (82%) | |
| Perphenazine (75%) | ||
| Time to discontinuation for any reason | Longest (most favorable) with olanzapine, but not statistically longer with olanzapine than with ziprasidone or perphenazine | |
| No statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | ||
| Time to discontinuation for lack of efficacy* | Longer with olanzapine; no statistical difference among risperidone, quetiapine, ziprasidone, and perphenazine | |
| Time to discontinuation for intolerable side effects | No statistical difference among agents | |
| Rate of discontinuation for intolerable side effects | Highest (19%) with olanzapine (primarily because of weight gain or metabolic effects with this medication) | |
| Rate of discontinuation for extrapyramidal effects | Highest (8%) with perphenazine | |
| Rate of discontinuation for intolerability (overall) | Lowest with risperidone (10%) | |
| * Nonequivalent dosing in CATIE phase 1 is an ongoing debate. | ||
What to do next?
When an initial antipsychotic proves inadequate or causes intolerable side effects, how do you choose a more efficacious or tolerable medication? Phase 2 offered CATIE patients and their clinicians two choices—an efficacy and a tolerability pathway (Figure).1,2
CATIE phase 2: Distribution of patients in efficacy and tolerability pathways
Efficacy pathway. Patients who chose the efficacy pathway were randomly assigned to clozapine (50%) or olanzapine, risperidone, or quetiapine.1 Researchers selected clozapine as the major efficacy comparator because of its robust effects in treatment-refractory schizophrenia. Clozapine was given open-label because of its safety monitoring requirements; other treatments were double-blind.
As in phase 1, the primary outcome measure was time until discontinuation for any reason. Secondary outcome measures included time to discontinuation because of side effects, patient choice, or lack of efficacy.
Tolerability pathway. Patients who chose the tolerability pathway were randomly assigned to double-blind treatment with ziprasidone, olanzapine, risperidone, or quetiapine.2 Ziprasidone was the major comparator because of clinical data showing a favorable tolerability profile.
The primary outcome measure was time to discontinuation for any reason. Secondary outcomes included reason for discontinuation (as determined by the study clinician), symptomatic ratings, and evaluations of adverse effects.
Trial duration. No patients in either pathway received the same antipsychotics they had taken in phase 1. All patients could continue treatment through the 18 months of the CATIE trial or until they completed 6 months in phase 2.
Efficacy pathway results
Discontinuation. Consistent with literature about its efficacy in treatment-refractory schizophrenia, clozapine showed a robust clinical effect. Overall, more patients receiving clozapine stayed on treatment and for longer periods, compared with patients receiving olanzapine, risperidone, or quetiapine (Table 2).
On secondary measures, discontinuation for lack of efficacy was significantly lower with clozapine (11%) than with:
- olanzapine (35%)
- risperidone or quetiapine (each at 43%).
Discontinuation rates because of adverse effects or by patient choice were the same across all medications (Table 3). Patients on clozapine achieved better ratings in overall psychotic symptoms, positive symptoms, and general function, but not in negative symptoms.
Weight gain. On average, patients gained more weight while taking olanzapine (+1.1 lb/mo) than with:
- risperidone (+0.5 lb/mo)
- clozapine (+0.5 lb/mo)
- quetiapine (+0.5 lb/mo)
Differences in weight gain—or in metabolic parameters or other adverse effects—were not statistically significant, however.
Table 2
Phase 2 efficacy pathway: Discontinuation for any reason
| Measure | Clozapine | Olanzapine | Risperidone | Quetiapine |
|---|---|---|---|---|
| How many patients discontinued | 25 of 49 (56%) | 12 of 19 (71%) | 12 of 16 (86%) | 13 of 15 (93%) |
| Median time to discontinuation | 10.5 months | 2.7 months | 2.8 months | 3.3 months |
Table 3
Reasons patients stopped taking their medications in CATIE phase 2
| Reason | Efficacy pathway | Tolerability pathway |
|---|---|---|
| All cause | 69% | 74% |
| Lack of efficacy | 26% | 29% |
| Lack of tolerability | 10% | 15% |
| Patient choice | 26% | 24% |
Tolerability pathway results
Discontinuation. Patients in the tolerability pathway took olanzapine or risperidone significantly longer—median 6.3 and 7 months, respectively— compared with ziprasidone (4 months) or quetiapine (2.8 months).
- Time to discontinuation during phase 2 was the same across all drugs among patients who entered phase 2 because of intolerable side effects in phase 1.
- Time to discontinuation because of side effects also was similar whether patients discontinued phase 1 for lack of efficacy or intolerable side effects. Patients stopped treatment in the efficacy and tolerability pathways for similar reasons (Table 3).
Weight gain. Patients taking olanzapine gained more weight (average +1.3 lb/mo) than did those taking the other drugs. Patients taking ziprasidone lost weight (average –1.7 lb/mo). Among 61 patients who gained weight during phase 1, 42% of those switched to ziprasidone lost weight in phase 2, as did:
- 20% of those switched to risperidone
- 7% of those switched to quetiapine.
Among those switched to olanzapine in phase 2, no one lost weight and 2% gained weight.
Metabolic effects. Some parameters changed, depending on drug assignment:
- prolactin increased in patients switched to risperidone
- cholesterol and triglycerides increased in patients switched to olanzapine or quetiapine but decreased in those switched to risperidone or ziprasidone
- QTc interval measurements showed no difference across all drugs.
Methodologic caveats
When considering how CATIE’s phase 2 findings might apply to clinical practice, keep in mind four caveats about the study’s design.
Clozapine was given open-label, yet quetiapine, olanzapine, and risperidone were given double-blind in the efficacy pathway. This pathway’s findings are consistent with what we know about clozapine and other SGAs in treatment-refractory schizophrenia, but how the open-label design affected clozapine therapy outcomes is unclear.
Were patients who knew they were taking clozapine more willing to “stay the course” than were patients in the pathway’s double-blind arm?
Discontinuation rates remained high. The 74% “overall discontinuation rate” in phase 1 surprised many psychiatrists because of the perceived high rate at which patients did not adhere to the first medications they received. To some extent, the word “discontinuation” is imprecise, however, because this group includes patients who did not drop out of treatment altogether but chose to move on to phase 2.
It is important to note, however, that nearly one-half of phase 1 patients who were eligible to enter phase 2 (509 of 1,052) did not. This group represents the true drop-out rate, which is substantial. The high rates of discontinuation seen in phase 1 also occurred in both phase 2 pathways (Table 3).
Few patients entered the efficacy pathway. In an approach designed to reflect routine clinical practice, the researchers recommended the efficacy pathway to patients who discontinued phase 1 because of lack of efficacy and the tolerability pathway to those who discontinued phase 1 because of intolerability. Many patients did not follow the recommendations, however, and seemed to choose their pathways based on whether they wanted a chance to receive clozapine or ziprasidone in phase 2.
Thus, among the 543 phase 1 patients who enrolled in phase 2, 99 (18%) entered the efficacy pathway, and 444 (82%) entered the tolerability pathway. The efficacy pathway included 85 patients who discontinued phase 1 for lack of efficacy and 5 for lack of tolerability. The tolerability pathway included 184 patients who discontinued phase 1 for lack of efficacy and 168 for lack of tolerability.
Dosages may not have been equivalent. SGAs’ dosing equivalency is unknown,5,6 which impedes our ability to interpret comparative studies such as CATIE. The study’s designers developed the its dosing ranges by careful consideration, including recommendations from each SGA’s manufacturer. As Nasrallah described,4 the trial’s dosages were not universally consistent with FDA-approved ranges or usual clinical practice (Table 4). In phase 2, for example, ziprasidone dosages were less than psychiatrists usually use, and quetiapine dosages were greater than usual.
Fortunately, studies are underway to determine each SGA’s optimum dosing. This work will help us understand what we can expect when we increase an antipsychotic’s dosage—a key step towards understanding dosing equivalency.
Table 4
Mean modal antipsychotic dosages (mg/d) in CATIE phase 2 pathways*
| Clozapine | Ziprasidone | Olanzapine | Risperidone | Quetiapine | |
|---|---|---|---|---|---|
| Efficacy pathway | 332 | — | 23.4 | 4.8 | 642.9 |
| Tolerability pathway | — | 115.9 | 20.5 | 4.15 | 65.2 |
| * 800 mg/d of quetiapine and 160 mg/d of ziprasidone are generally regarded as therapeutically equivalent to 20 mg/d of olanzapine. | |||||
What clinicians can expect
A recent analysis helps put CATIE’s findings in perspective. Citrome and Stroup7 quantified the results of phase 1 and 2 with respect to:
- number needed to treat (NNT)—how many patients a clinician needs to treat with drug A to see one additional benefit, compared with drug B
- number needed to harm (NNH)—how many patients a clinician needs to treat with drug A to see a given adverse effect, compared with drug B.
In this analysis, the NNT for olanzapine (5.5 to 10) was lowest among the drugs compared in phase 1, and the NNT for clozapine (3) was lowest among those compared in phase 2. A lower number means that, overall, clinicians can expect a more robust treatment response.
On the other hand, the NNH for olanzapine in weight gain and metabolic disturbances (12.4 to 17.7) was the lowest in phase 1, indicating that clinicians can expect more weight gain and metabolic effects with olanzapine than with other SGAs. Ziprasidone had the highest NNH (106 to 208) among the agents in phase 2 for avoiding discontinuation because of weight gain or metabolic disturbances. In other words, ziprasidone appears less likely than other SGAs to cause metabolic problems.
These risk-attribution measures demonstrate the dilemma clinicians face when trying to match schizophrenia patients with antipsychotics. CATIE was “an N of 1,493” subjects, whereas each patient we see in clinical practice is “an N of 1.” One patient may need a more-robust response; another may need improved tolerability.
We strive for balance, seeking to optimize efficacy—often by raising the dosage—while minimizing adverse effects.
What to tell patients
CATIE phases 1 and 2 provide a compelling rationale for individualized treatment, which should be standard clinical practice for schizophrenia:
- All drugs used in phases 1 and 2 worked.
- All showed noteworthy adverse effects that were different for each drug.
- Different patients responded differently to each drug.
Using our clinical judgment and available information, we must match—as best we can—the individual patient’s characteristics with the antipsychotics’ risk: benefit profiles. CATIE phases 1 and 2 provide independent information on the comparative efficacy and tolerability of each medication.
The CATIE investigators and NIMH have done a great service to our field in providing a rich repository of timely information to inform clinical practice. But the CATIE study was not designed to answer all our questions about treating schizophrenia.8,9 Clinicians and patients need to look elsewhere for guidance on the roles of:
- psychosocial treatments
- recovery and the therapeutic alliance in maximizing outcomes
- long-acting SGA formulations
- aripiprazole (addressed in CATIE phase 3)
- SGAs in first-episode schizophrenia
- FGAs when a patient does not adequately respond to an initial SGA.
Related resources
- Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE). www.CATIE.unc.edu.
- Lieberman JA. What the CATIE study means in clinical practice. Psychiatr Serv 2006;57(8):1075.
Drug brand names
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Buckley receives research/grant support from AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Pfizer, and Solvay Pharmaceuticals, and is a consultant to Abbott Laboratories, Alamo Pharmaceuticals, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly & Co., Janssen Pharmaceutica, Merck & Co., and Pfizer.
Acknowledgement
The author thanks Del Miller, MD, for comments given on a draft of this paper.
1. Stroup TS, Lieberman JA, McEvoy JP, et al for the CATIE investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry 2006;163:611-22.
2. McEvoy JP, Lieberman JA, Stroup TS, et al for the CATIE investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 2006;163:600-10.
3. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
4. Nasrallah HA. CATIE’s surprises: In antipsychotics’ square-off, were there winners or losers? Current Psychiatry 2006;5(2):52-65.
5. Buckley PF. Dosing equivalency of second-generation antipsychotics. J Clin Psychopharmacol 2005;25(5):501-2.
6. Davis JM. The choice of drugs for schizophrenia. N Engl J Med 2006;354(5):518-20.
7. Citrome L, Stroup TS. Schizophrenia clinical antipsychotic trials intervention effectiveness and number needed to treat: How can CATIE inform clinicians? Int J Clin Pract 2006 (in press).
8. Ragins M. Should the CATIE study be a wake-up call? Psychiatr Serv 2005;56:1489.-
9. Lieberman JA, Hsiao J. Interpreting the results of the CATIE study. Psychiatr Serv 2006;57:139.-
1. Stroup TS, Lieberman JA, McEvoy JP, et al for the CATIE investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry 2006;163:611-22.
2. McEvoy JP, Lieberman JA, Stroup TS, et al for the CATIE investigators. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 2006;163:600-10.
3. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353:1209-23.
4. Nasrallah HA. CATIE’s surprises: In antipsychotics’ square-off, were there winners or losers? Current Psychiatry 2006;5(2):52-65.
5. Buckley PF. Dosing equivalency of second-generation antipsychotics. J Clin Psychopharmacol 2005;25(5):501-2.
6. Davis JM. The choice of drugs for schizophrenia. N Engl J Med 2006;354(5):518-20.
7. Citrome L, Stroup TS. Schizophrenia clinical antipsychotic trials intervention effectiveness and number needed to treat: How can CATIE inform clinicians? Int J Clin Pract 2006 (in press).
8. Ragins M. Should the CATIE study be a wake-up call? Psychiatr Serv 2005;56:1489.-
9. Lieberman JA, Hsiao J. Interpreting the results of the CATIE study. Psychiatr Serv 2006;57:139.-
Is your patient too sick to work?
Ms. S, age 34, is a hard-working, single mother of two who works full-time at a local factory. She has recurrent major depression and has been struggling for 2 months. As you write the script to increase her fluoxetine dosage, she asks, “Hey, Doc, can you sign this form so I can have some time off work?”
If you feel uncomfortable signing work forms, approach your patients’ employment issues as you would any medical problem. Your job is to assess capacity to work, and the employer—based on restrictions you write in the medical report—decides if an accommodation can be made.
By answering five questions (Table 1), you can make informed decisions about your patients’ ability to work while:
- minimizing recovery time
- maintaining their daily structure and functioning
- reducing risk of chronic disability.
Table 1
Can a mentally ill patient work? 5 questions to consider
| What is the employee’s diagnosis? |
| What work can the employee do today? |
| To reduce risk of medical harm, what work should the employee not do? |
| Does an impairment prevent the employee from performing essential job functions? |
| What must occur to get the patient back to work? |
QUESTION 1: What is the employee’s diagnosis?
Diagnosis gives you a framework to understand what an employee can and cannot do on the job. Because Ms. S’ diagnosis is major depressive disorder, she may have low mood, low energy, decreased concentration, slowed movement, and disturbed sleep.
But diagnosis alone does not mean she is unable to work. Being separated from work can destabilize a person’s life (Box),1-4 and debilitating depressive symptoms can change from day to day.
QUESTION 2: What work can the employee do today?
Four capacities are needed to function in the workplace:
- understanding and memory
- sustained concentration and persistence
- social interaction
- adaptation.5
A mildly depressed custodian could follow instructions, perform repetitive tasks, and keep pace on a daily shift. A depressed school principal, on the other hand, might lack the decision-making and social skills to relate to teachers, students, and parents. If she can follow instructions and perform simple tasks, however, she might be able to return to work and catch up on paperwork. She could postpone meetings and appointments until she feels well enough resume her full duties.
For almost any job, an employee must have understanding and memory to follow instructions and perform simple tasks.
Table 2
Can your patient work? Assess work functions by required capacities
| Work function* | Capacity required to perform work function† |
|---|---|
| Comprehend and follow instructions | Understanding and memory |
| Perform simple and repetitive tasks | Understanding and memory |
| Maintain a work pace appropriate to a given workload | Sustained concentration and persistence |
| Perform complex or varied tasks | Understanding and memory; adaptation |
| Relate to other people beyond giving and receiving instructions | Social interaction; adaptation |
| Influence people | Social interaction; adaptation |
| Make generalizations, evaluations, or decisions without immediate supervision | Understanding and memory; adaptation |
| Accept and carry out responsibility for direction, control, and planning | Understanding and memory; adaptation |
| *Defined by the California Division of Industrial Accidents. | |
| †Analogous capacities developed by the American Medical Association and Social Security Administration | |
| Source: References 5 and 6 | |
Case continued: assessing capacity
To assess ability to comprehend and follow instructions, you could ask Ms. S to:
- take the yellow book off the bookshelf with her right hand
- turn to page 23
- set the book down on your desk.
To assess ability to perform simple tasks, you might ask Ms. S to describe what she does on a typical day. If she brushes her teeth, makes meals, does laundry, and buys groceries, she can probably do similar tasks at work. If she’s lying on the couch, staring at the wall, and neglecting self care, she might not have the motivation or concentration to complete simple tasks.
It might help to know how Ms. S arrived to see you. Driving is a more complicated task than having a friend or family member bring her. Ms. S’ story may include inconsistencies, and ideally you would have her sign releases to obtain collateral history from family, friends, or perhaps her supervisor.
Watching Ms. S leave can offer information about her functioning. Does she talk to anyone? Does she look the same as when she was in the office? Did she appear slowed when you saw her, yet could easily walk to the car and drive off?
QUESTION 3: To reduce risk of medical harm, what work should the patient not do?
A suicidal or homicidal patient may need to be hospitalized and should not go to work. Someone who is neglecting self care—such as eating—probably does not belong at work and could be at risk for harm.
Symptoms such as decreased concentration, psychomotor slowing, and decreased alertness are absolute contraindications for hazardous jobs that require sustained concentration and quick decisions—such as driving fork lifts or operating heavy machinery.
If potential exists for harm, recommend that the employee be treated before you re-evaluate return to work. Ideally, a case manager from the employer’s occupational health provider would check with the employee during treatment to reassess safety factors and facilitate a smooth return to work.
If medical harm is unlikely, recommending time off work becomes “medical discretion” and is not necessarily “medically required.”7
QUESTION 4: Does an impairment prevent the patient from performing essential job functions?
Impairment—a medical term—is often confused with disability, an administrative term:
- Impairment is “a loss, loss of use, or derangement of a body part, organ system, or organ function.”
- Disability is “an alteration of an individual’s capacity to meet personal, social, or occupational demands because of an impairment.”5
The longer a person is away from work with an injury or illness, the less likely he or she will return:
- 50% of persons off work for 8 weeks will not return.
- >85% of persons off work for 6 months will not return to long-term employment and are at risk for long-term disability.1
Unemployment increases mortality rates, physical and mental illness, and use of medical services.2 The unemployed may be more likely than the employed to visit physicians, take medications, or be admitted to hospitals.3
A disability mindset can develop after only 2 to 4 weeks off work, even in capable workers. An estimated 60% to 80% of time away from work is medically unnecessary.4 Returning to work as soon as possible after an illness or injury maximizes health outcomes and ability to function.
An employee who can perform essential job functions despite an impairment should stay at work. If he or she cannot do those functions, clearly state the impairment—such as decreased concentration, problems with persistence and pace—on the medical form so that the employer can decide if an accommodation can be made.
If essential job functions are high-order—such as air traffic control—even slight impairment could prevent the employee from safely doing the job. An impaired air traffic controller probably could do less-complex activities, however, such as clerical work.
Medical discretion. Most depressed patients can follow instructions and complete simple tasks, but many cannot keep up with the usual work pace because of low energy and slowed thinking and movements. Using medical discretion to recommend a short time off might help a depressed person return to full productivity more quickly if intensive treatment is available.
Limit discretionary time off to short periods when a treatment program is available. When blanket restrictions are written, the patient too often sits idle at home, getting worse and not better. At least excuse your patient from work to attend medical appointments and engage in depression treatment.
QUESTION 5: What must occur to get the patient back to work?
Returning to work as soon as possible can be therapeutic. Having a regular routine and daily structure gives the depressed person a sense of normalcy not found while sitting at home. Beyond stating the impairment on the medical form, suggest possible accommodations the employer could make to expedite return to work.
For your depressed patient, you could suggest reduced work hours (to accommodate low energy) or allowing more time to complete tasks. Whatever the diagnosis, addressing conflict in the workplace often helps. Strategies include:
- working individually with your patient
- alerting the patient to the employer’s conflict resolution policies and employee assistance programs.
CASE RESOLUTION: WORK AS THERAPY
Ms. S shows good eye contact, is appropriately dressed, shows no psychomotor abnormalities, and is not suicidal or homicidal. Mental status exam is normal. You determine that she can follow instructions, perform tasks, and keep up with her workload. She can probably do her job but initially might have difficulties because of depression’s effects on socialization and executive functioning.
You encourage Ms. S to return to work for the therapeutic benefits of a daily routine, but you suggest she postpone big projects or major decisions until she feels better.
Reassure your patient when you find no compelling reason why she cannot return to work. Explain that feeling ambivalent about maintaining function at work is normal, and staying at work is crucial to confidence and self-esteem.
Recognize the potential attraction of secondary gain—such as attention from family and medical providers and financial incentives to stay off work—but also normalize the experience for your patient. Help her regain her independence and start thinking about how she can improve her work situation.
Related resources
- Academy of Organizational and Occupational Psychiatry. www.aoop.org.
- National Partnership for Workplace Mental Health. www.workplacementalhealth.org
- Talmage JB, Melhorn JM (eds). A physician’s guide to return to work. Chicago: AMA Press; 2005.
- Fluoxetine • Prozac
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pilley M. How the primary care physician can help patients negotiate the return-to-work disability dilemma. In: Talmage JB, Melhorn JM, eds. A physician’s guide to return to work. Chicago: AMA Press; 2005:151.
2. Mathers CD, Schofield DJ. The health consequences of unemployment: the evidence. Med J Aust 1998;168:178-82.
3. Jin RL, Shah CP, Svoboda TJ. The impact of unemployment on health: a review of the evidence. Can Med Assoc J 1995;153:529-40.
4. Christian J. Reducing disability days: healing more than the injury. Journal of Workers Compensation 2000;9:30-55.
5. Cocchiarella L, Andersson BJ. Mental and behavioral disorders. In: Cocchiarella L, Andersson BJ, eds. Guides to the evaluation of permanent impairment, 5th ed. Chicago: American Medical Association; 2001:361–4.
6. Enelow AJ. Psychiatric disorders and work function. Psychiatr Ann 1991;21(1):27-35.
7. American College of Occupational and Environmental Medicine. Preventing needless work disability by helping people stay employed. Available at: http://www.acoem.org/position/statements.asp?CATA_ID=100. Accessed Aug. 8, 2006.
8. United States Department of Labor. Office of Disability Employment Policy. Glossary of commonly used terms. Available at: http://www.dol.gov/odep/pubs/ek00/glossary.htm. Accessed Aug. 8, 2006.
Ms. S, age 34, is a hard-working, single mother of two who works full-time at a local factory. She has recurrent major depression and has been struggling for 2 months. As you write the script to increase her fluoxetine dosage, she asks, “Hey, Doc, can you sign this form so I can have some time off work?”
If you feel uncomfortable signing work forms, approach your patients’ employment issues as you would any medical problem. Your job is to assess capacity to work, and the employer—based on restrictions you write in the medical report—decides if an accommodation can be made.
By answering five questions (Table 1), you can make informed decisions about your patients’ ability to work while:
- minimizing recovery time
- maintaining their daily structure and functioning
- reducing risk of chronic disability.
Table 1
Can a mentally ill patient work? 5 questions to consider
| What is the employee’s diagnosis? |
| What work can the employee do today? |
| To reduce risk of medical harm, what work should the employee not do? |
| Does an impairment prevent the employee from performing essential job functions? |
| What must occur to get the patient back to work? |
QUESTION 1: What is the employee’s diagnosis?
Diagnosis gives you a framework to understand what an employee can and cannot do on the job. Because Ms. S’ diagnosis is major depressive disorder, she may have low mood, low energy, decreased concentration, slowed movement, and disturbed sleep.
But diagnosis alone does not mean she is unable to work. Being separated from work can destabilize a person’s life (Box),1-4 and debilitating depressive symptoms can change from day to day.
QUESTION 2: What work can the employee do today?
Four capacities are needed to function in the workplace:
- understanding and memory
- sustained concentration and persistence
- social interaction
- adaptation.5
A mildly depressed custodian could follow instructions, perform repetitive tasks, and keep pace on a daily shift. A depressed school principal, on the other hand, might lack the decision-making and social skills to relate to teachers, students, and parents. If she can follow instructions and perform simple tasks, however, she might be able to return to work and catch up on paperwork. She could postpone meetings and appointments until she feels well enough resume her full duties.
For almost any job, an employee must have understanding and memory to follow instructions and perform simple tasks.
Table 2
Can your patient work? Assess work functions by required capacities
| Work function* | Capacity required to perform work function† |
|---|---|
| Comprehend and follow instructions | Understanding and memory |
| Perform simple and repetitive tasks | Understanding and memory |
| Maintain a work pace appropriate to a given workload | Sustained concentration and persistence |
| Perform complex or varied tasks | Understanding and memory; adaptation |
| Relate to other people beyond giving and receiving instructions | Social interaction; adaptation |
| Influence people | Social interaction; adaptation |
| Make generalizations, evaluations, or decisions without immediate supervision | Understanding and memory; adaptation |
| Accept and carry out responsibility for direction, control, and planning | Understanding and memory; adaptation |
| *Defined by the California Division of Industrial Accidents. | |
| †Analogous capacities developed by the American Medical Association and Social Security Administration | |
| Source: References 5 and 6 | |
Case continued: assessing capacity
To assess ability to comprehend and follow instructions, you could ask Ms. S to:
- take the yellow book off the bookshelf with her right hand
- turn to page 23
- set the book down on your desk.
To assess ability to perform simple tasks, you might ask Ms. S to describe what she does on a typical day. If she brushes her teeth, makes meals, does laundry, and buys groceries, she can probably do similar tasks at work. If she’s lying on the couch, staring at the wall, and neglecting self care, she might not have the motivation or concentration to complete simple tasks.
It might help to know how Ms. S arrived to see you. Driving is a more complicated task than having a friend or family member bring her. Ms. S’ story may include inconsistencies, and ideally you would have her sign releases to obtain collateral history from family, friends, or perhaps her supervisor.
Watching Ms. S leave can offer information about her functioning. Does she talk to anyone? Does she look the same as when she was in the office? Did she appear slowed when you saw her, yet could easily walk to the car and drive off?
QUESTION 3: To reduce risk of medical harm, what work should the patient not do?
A suicidal or homicidal patient may need to be hospitalized and should not go to work. Someone who is neglecting self care—such as eating—probably does not belong at work and could be at risk for harm.
Symptoms such as decreased concentration, psychomotor slowing, and decreased alertness are absolute contraindications for hazardous jobs that require sustained concentration and quick decisions—such as driving fork lifts or operating heavy machinery.
If potential exists for harm, recommend that the employee be treated before you re-evaluate return to work. Ideally, a case manager from the employer’s occupational health provider would check with the employee during treatment to reassess safety factors and facilitate a smooth return to work.
If medical harm is unlikely, recommending time off work becomes “medical discretion” and is not necessarily “medically required.”7
QUESTION 4: Does an impairment prevent the patient from performing essential job functions?
Impairment—a medical term—is often confused with disability, an administrative term:
- Impairment is “a loss, loss of use, or derangement of a body part, organ system, or organ function.”
- Disability is “an alteration of an individual’s capacity to meet personal, social, or occupational demands because of an impairment.”5
The longer a person is away from work with an injury or illness, the less likely he or she will return:
- 50% of persons off work for 8 weeks will not return.
- >85% of persons off work for 6 months will not return to long-term employment and are at risk for long-term disability.1
Unemployment increases mortality rates, physical and mental illness, and use of medical services.2 The unemployed may be more likely than the employed to visit physicians, take medications, or be admitted to hospitals.3
A disability mindset can develop after only 2 to 4 weeks off work, even in capable workers. An estimated 60% to 80% of time away from work is medically unnecessary.4 Returning to work as soon as possible after an illness or injury maximizes health outcomes and ability to function.
An employee who can perform essential job functions despite an impairment should stay at work. If he or she cannot do those functions, clearly state the impairment—such as decreased concentration, problems with persistence and pace—on the medical form so that the employer can decide if an accommodation can be made.
If essential job functions are high-order—such as air traffic control—even slight impairment could prevent the employee from safely doing the job. An impaired air traffic controller probably could do less-complex activities, however, such as clerical work.
Medical discretion. Most depressed patients can follow instructions and complete simple tasks, but many cannot keep up with the usual work pace because of low energy and slowed thinking and movements. Using medical discretion to recommend a short time off might help a depressed person return to full productivity more quickly if intensive treatment is available.
Limit discretionary time off to short periods when a treatment program is available. When blanket restrictions are written, the patient too often sits idle at home, getting worse and not better. At least excuse your patient from work to attend medical appointments and engage in depression treatment.
QUESTION 5: What must occur to get the patient back to work?
Returning to work as soon as possible can be therapeutic. Having a regular routine and daily structure gives the depressed person a sense of normalcy not found while sitting at home. Beyond stating the impairment on the medical form, suggest possible accommodations the employer could make to expedite return to work.
For your depressed patient, you could suggest reduced work hours (to accommodate low energy) or allowing more time to complete tasks. Whatever the diagnosis, addressing conflict in the workplace often helps. Strategies include:
- working individually with your patient
- alerting the patient to the employer’s conflict resolution policies and employee assistance programs.
CASE RESOLUTION: WORK AS THERAPY
Ms. S shows good eye contact, is appropriately dressed, shows no psychomotor abnormalities, and is not suicidal or homicidal. Mental status exam is normal. You determine that she can follow instructions, perform tasks, and keep up with her workload. She can probably do her job but initially might have difficulties because of depression’s effects on socialization and executive functioning.
You encourage Ms. S to return to work for the therapeutic benefits of a daily routine, but you suggest she postpone big projects or major decisions until she feels better.
Reassure your patient when you find no compelling reason why she cannot return to work. Explain that feeling ambivalent about maintaining function at work is normal, and staying at work is crucial to confidence and self-esteem.
Recognize the potential attraction of secondary gain—such as attention from family and medical providers and financial incentives to stay off work—but also normalize the experience for your patient. Help her regain her independence and start thinking about how she can improve her work situation.
Related resources
- Academy of Organizational and Occupational Psychiatry. www.aoop.org.
- National Partnership for Workplace Mental Health. www.workplacementalhealth.org
- Talmage JB, Melhorn JM (eds). A physician’s guide to return to work. Chicago: AMA Press; 2005.
- Fluoxetine • Prozac
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. S, age 34, is a hard-working, single mother of two who works full-time at a local factory. She has recurrent major depression and has been struggling for 2 months. As you write the script to increase her fluoxetine dosage, she asks, “Hey, Doc, can you sign this form so I can have some time off work?”
If you feel uncomfortable signing work forms, approach your patients’ employment issues as you would any medical problem. Your job is to assess capacity to work, and the employer—based on restrictions you write in the medical report—decides if an accommodation can be made.
By answering five questions (Table 1), you can make informed decisions about your patients’ ability to work while:
- minimizing recovery time
- maintaining their daily structure and functioning
- reducing risk of chronic disability.
Table 1
Can a mentally ill patient work? 5 questions to consider
| What is the employee’s diagnosis? |
| What work can the employee do today? |
| To reduce risk of medical harm, what work should the employee not do? |
| Does an impairment prevent the employee from performing essential job functions? |
| What must occur to get the patient back to work? |
QUESTION 1: What is the employee’s diagnosis?
Diagnosis gives you a framework to understand what an employee can and cannot do on the job. Because Ms. S’ diagnosis is major depressive disorder, she may have low mood, low energy, decreased concentration, slowed movement, and disturbed sleep.
But diagnosis alone does not mean she is unable to work. Being separated from work can destabilize a person’s life (Box),1-4 and debilitating depressive symptoms can change from day to day.
QUESTION 2: What work can the employee do today?
Four capacities are needed to function in the workplace:
- understanding and memory
- sustained concentration and persistence
- social interaction
- adaptation.5
A mildly depressed custodian could follow instructions, perform repetitive tasks, and keep pace on a daily shift. A depressed school principal, on the other hand, might lack the decision-making and social skills to relate to teachers, students, and parents. If she can follow instructions and perform simple tasks, however, she might be able to return to work and catch up on paperwork. She could postpone meetings and appointments until she feels well enough resume her full duties.
For almost any job, an employee must have understanding and memory to follow instructions and perform simple tasks.
Table 2
Can your patient work? Assess work functions by required capacities
| Work function* | Capacity required to perform work function† |
|---|---|
| Comprehend and follow instructions | Understanding and memory |
| Perform simple and repetitive tasks | Understanding and memory |
| Maintain a work pace appropriate to a given workload | Sustained concentration and persistence |
| Perform complex or varied tasks | Understanding and memory; adaptation |
| Relate to other people beyond giving and receiving instructions | Social interaction; adaptation |
| Influence people | Social interaction; adaptation |
| Make generalizations, evaluations, or decisions without immediate supervision | Understanding and memory; adaptation |
| Accept and carry out responsibility for direction, control, and planning | Understanding and memory; adaptation |
| *Defined by the California Division of Industrial Accidents. | |
| †Analogous capacities developed by the American Medical Association and Social Security Administration | |
| Source: References 5 and 6 | |
Case continued: assessing capacity
To assess ability to comprehend and follow instructions, you could ask Ms. S to:
- take the yellow book off the bookshelf with her right hand
- turn to page 23
- set the book down on your desk.
To assess ability to perform simple tasks, you might ask Ms. S to describe what she does on a typical day. If she brushes her teeth, makes meals, does laundry, and buys groceries, she can probably do similar tasks at work. If she’s lying on the couch, staring at the wall, and neglecting self care, she might not have the motivation or concentration to complete simple tasks.
It might help to know how Ms. S arrived to see you. Driving is a more complicated task than having a friend or family member bring her. Ms. S’ story may include inconsistencies, and ideally you would have her sign releases to obtain collateral history from family, friends, or perhaps her supervisor.
Watching Ms. S leave can offer information about her functioning. Does she talk to anyone? Does she look the same as when she was in the office? Did she appear slowed when you saw her, yet could easily walk to the car and drive off?
QUESTION 3: To reduce risk of medical harm, what work should the patient not do?
A suicidal or homicidal patient may need to be hospitalized and should not go to work. Someone who is neglecting self care—such as eating—probably does not belong at work and could be at risk for harm.
Symptoms such as decreased concentration, psychomotor slowing, and decreased alertness are absolute contraindications for hazardous jobs that require sustained concentration and quick decisions—such as driving fork lifts or operating heavy machinery.
If potential exists for harm, recommend that the employee be treated before you re-evaluate return to work. Ideally, a case manager from the employer’s occupational health provider would check with the employee during treatment to reassess safety factors and facilitate a smooth return to work.
If medical harm is unlikely, recommending time off work becomes “medical discretion” and is not necessarily “medically required.”7
QUESTION 4: Does an impairment prevent the patient from performing essential job functions?
Impairment—a medical term—is often confused with disability, an administrative term:
- Impairment is “a loss, loss of use, or derangement of a body part, organ system, or organ function.”
- Disability is “an alteration of an individual’s capacity to meet personal, social, or occupational demands because of an impairment.”5
The longer a person is away from work with an injury or illness, the less likely he or she will return:
- 50% of persons off work for 8 weeks will not return.
- >85% of persons off work for 6 months will not return to long-term employment and are at risk for long-term disability.1
Unemployment increases mortality rates, physical and mental illness, and use of medical services.2 The unemployed may be more likely than the employed to visit physicians, take medications, or be admitted to hospitals.3
A disability mindset can develop after only 2 to 4 weeks off work, even in capable workers. An estimated 60% to 80% of time away from work is medically unnecessary.4 Returning to work as soon as possible after an illness or injury maximizes health outcomes and ability to function.
An employee who can perform essential job functions despite an impairment should stay at work. If he or she cannot do those functions, clearly state the impairment—such as decreased concentration, problems with persistence and pace—on the medical form so that the employer can decide if an accommodation can be made.
If essential job functions are high-order—such as air traffic control—even slight impairment could prevent the employee from safely doing the job. An impaired air traffic controller probably could do less-complex activities, however, such as clerical work.
Medical discretion. Most depressed patients can follow instructions and complete simple tasks, but many cannot keep up with the usual work pace because of low energy and slowed thinking and movements. Using medical discretion to recommend a short time off might help a depressed person return to full productivity more quickly if intensive treatment is available.
Limit discretionary time off to short periods when a treatment program is available. When blanket restrictions are written, the patient too often sits idle at home, getting worse and not better. At least excuse your patient from work to attend medical appointments and engage in depression treatment.
QUESTION 5: What must occur to get the patient back to work?
Returning to work as soon as possible can be therapeutic. Having a regular routine and daily structure gives the depressed person a sense of normalcy not found while sitting at home. Beyond stating the impairment on the medical form, suggest possible accommodations the employer could make to expedite return to work.
For your depressed patient, you could suggest reduced work hours (to accommodate low energy) or allowing more time to complete tasks. Whatever the diagnosis, addressing conflict in the workplace often helps. Strategies include:
- working individually with your patient
- alerting the patient to the employer’s conflict resolution policies and employee assistance programs.
CASE RESOLUTION: WORK AS THERAPY
Ms. S shows good eye contact, is appropriately dressed, shows no psychomotor abnormalities, and is not suicidal or homicidal. Mental status exam is normal. You determine that she can follow instructions, perform tasks, and keep up with her workload. She can probably do her job but initially might have difficulties because of depression’s effects on socialization and executive functioning.
You encourage Ms. S to return to work for the therapeutic benefits of a daily routine, but you suggest she postpone big projects or major decisions until she feels better.
Reassure your patient when you find no compelling reason why she cannot return to work. Explain that feeling ambivalent about maintaining function at work is normal, and staying at work is crucial to confidence and self-esteem.
Recognize the potential attraction of secondary gain—such as attention from family and medical providers and financial incentives to stay off work—but also normalize the experience for your patient. Help her regain her independence and start thinking about how she can improve her work situation.
Related resources
- Academy of Organizational and Occupational Psychiatry. www.aoop.org.
- National Partnership for Workplace Mental Health. www.workplacementalhealth.org
- Talmage JB, Melhorn JM (eds). A physician’s guide to return to work. Chicago: AMA Press; 2005.
- Fluoxetine • Prozac
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pilley M. How the primary care physician can help patients negotiate the return-to-work disability dilemma. In: Talmage JB, Melhorn JM, eds. A physician’s guide to return to work. Chicago: AMA Press; 2005:151.
2. Mathers CD, Schofield DJ. The health consequences of unemployment: the evidence. Med J Aust 1998;168:178-82.
3. Jin RL, Shah CP, Svoboda TJ. The impact of unemployment on health: a review of the evidence. Can Med Assoc J 1995;153:529-40.
4. Christian J. Reducing disability days: healing more than the injury. Journal of Workers Compensation 2000;9:30-55.
5. Cocchiarella L, Andersson BJ. Mental and behavioral disorders. In: Cocchiarella L, Andersson BJ, eds. Guides to the evaluation of permanent impairment, 5th ed. Chicago: American Medical Association; 2001:361–4.
6. Enelow AJ. Psychiatric disorders and work function. Psychiatr Ann 1991;21(1):27-35.
7. American College of Occupational and Environmental Medicine. Preventing needless work disability by helping people stay employed. Available at: http://www.acoem.org/position/statements.asp?CATA_ID=100. Accessed Aug. 8, 2006.
8. United States Department of Labor. Office of Disability Employment Policy. Glossary of commonly used terms. Available at: http://www.dol.gov/odep/pubs/ek00/glossary.htm. Accessed Aug. 8, 2006.
1. Pilley M. How the primary care physician can help patients negotiate the return-to-work disability dilemma. In: Talmage JB, Melhorn JM, eds. A physician’s guide to return to work. Chicago: AMA Press; 2005:151.
2. Mathers CD, Schofield DJ. The health consequences of unemployment: the evidence. Med J Aust 1998;168:178-82.
3. Jin RL, Shah CP, Svoboda TJ. The impact of unemployment on health: a review of the evidence. Can Med Assoc J 1995;153:529-40.
4. Christian J. Reducing disability days: healing more than the injury. Journal of Workers Compensation 2000;9:30-55.
5. Cocchiarella L, Andersson BJ. Mental and behavioral disorders. In: Cocchiarella L, Andersson BJ, eds. Guides to the evaluation of permanent impairment, 5th ed. Chicago: American Medical Association; 2001:361–4.
6. Enelow AJ. Psychiatric disorders and work function. Psychiatr Ann 1991;21(1):27-35.
7. American College of Occupational and Environmental Medicine. Preventing needless work disability by helping people stay employed. Available at: http://www.acoem.org/position/statements.asp?CATA_ID=100. Accessed Aug. 8, 2006.
8. United States Department of Labor. Office of Disability Employment Policy. Glossary of commonly used terms. Available at: http://www.dol.gov/odep/pubs/ek00/glossary.htm. Accessed Aug. 8, 2006.
Make the most of the ‘15-minute med-check’
With today’s practice environment, most patient visits are limited to 15 minutes. Make the most of that time with the patient by following these guidelines organized by the mnemonic MEDCHECK. Try to cover all eight guidelines during each appointment, even if briefly.
Medication. Begin with an open-ended question to elicit the patient’s thoughts on his or her treatment, such as, “How’s the medication working for you?” Also ask what he or she expects to accomplish during the session. With the patient’s permission, get the family’s perspective on how the patient is doing.
Environmental changes. Learn about events in your patient’s life and how he or she is coping with them. Try to uncover information about stressors—such as a new job—or positive changes such as an old friend returning to the area. Finding a topic that the patient likes to talk about—a favorite activity or television show, for example—can help monitor improvement over time.
Diagnoses. Continually reassess the primary diagnosis and look for evidence of a medical illness, medication side effects, or secondary psychiatric conditions—especially alcohol or drug abuse.
Coordination of care. Update the patient’s file on dealings with therapists, case managers, and other physicians.
Handouts. Provide handouts and/or Web sites describing a medication’s therapeutic and side effects. Get handouts from numerous sources or develop information sheets and adapt them to your patient population. Include generic and brand names of medications to avoid confusion.
Empathy. Conveying empathy for the patient’s problems or pleasures is crucial to a strong therapeutic alliance and effective treatment.
Costs. Don’t ignore medication costs. Being up-to-date on formulary options helps patients get needed prescriptions.
Knowledge. End the session by asking the patient to summarize the medication plan to ensure that he or she knows what to do.
The MEDCHECK guidelines do not take into account necessary tasks outside of the session:
- Schedule time before your appointments to review charts and recall information from a patient’s last visit. If you cannot update the chart during the visit, reserve a few minutes later for documentation.
- Take periodic breaks to return phone calls or e-mails or take a short walk.
- Read up on relevant treatment guidelines to ensure you are providing evidence-based care.
- Finally, reserve time to be an advocate for your patient by addressing any administrative short-comings or removing obstacles to therapeutic recommendations.
Of course, short visits are not appropriate for all patients. Give more time to patients in crisis or to complicated cases such as children, pregnant women, or those needing interpreters.
Dr. Moffic is professor of psychiatry and behavioral medicine, Medical College of Wisconsin, Milwaukee.
With today’s practice environment, most patient visits are limited to 15 minutes. Make the most of that time with the patient by following these guidelines organized by the mnemonic MEDCHECK. Try to cover all eight guidelines during each appointment, even if briefly.
Medication. Begin with an open-ended question to elicit the patient’s thoughts on his or her treatment, such as, “How’s the medication working for you?” Also ask what he or she expects to accomplish during the session. With the patient’s permission, get the family’s perspective on how the patient is doing.
Environmental changes. Learn about events in your patient’s life and how he or she is coping with them. Try to uncover information about stressors—such as a new job—or positive changes such as an old friend returning to the area. Finding a topic that the patient likes to talk about—a favorite activity or television show, for example—can help monitor improvement over time.
Diagnoses. Continually reassess the primary diagnosis and look for evidence of a medical illness, medication side effects, or secondary psychiatric conditions—especially alcohol or drug abuse.
Coordination of care. Update the patient’s file on dealings with therapists, case managers, and other physicians.
Handouts. Provide handouts and/or Web sites describing a medication’s therapeutic and side effects. Get handouts from numerous sources or develop information sheets and adapt them to your patient population. Include generic and brand names of medications to avoid confusion.
Empathy. Conveying empathy for the patient’s problems or pleasures is crucial to a strong therapeutic alliance and effective treatment.
Costs. Don’t ignore medication costs. Being up-to-date on formulary options helps patients get needed prescriptions.
Knowledge. End the session by asking the patient to summarize the medication plan to ensure that he or she knows what to do.
The MEDCHECK guidelines do not take into account necessary tasks outside of the session:
- Schedule time before your appointments to review charts and recall information from a patient’s last visit. If you cannot update the chart during the visit, reserve a few minutes later for documentation.
- Take periodic breaks to return phone calls or e-mails or take a short walk.
- Read up on relevant treatment guidelines to ensure you are providing evidence-based care.
- Finally, reserve time to be an advocate for your patient by addressing any administrative short-comings or removing obstacles to therapeutic recommendations.
Of course, short visits are not appropriate for all patients. Give more time to patients in crisis or to complicated cases such as children, pregnant women, or those needing interpreters.
With today’s practice environment, most patient visits are limited to 15 minutes. Make the most of that time with the patient by following these guidelines organized by the mnemonic MEDCHECK. Try to cover all eight guidelines during each appointment, even if briefly.
Medication. Begin with an open-ended question to elicit the patient’s thoughts on his or her treatment, such as, “How’s the medication working for you?” Also ask what he or she expects to accomplish during the session. With the patient’s permission, get the family’s perspective on how the patient is doing.
Environmental changes. Learn about events in your patient’s life and how he or she is coping with them. Try to uncover information about stressors—such as a new job—or positive changes such as an old friend returning to the area. Finding a topic that the patient likes to talk about—a favorite activity or television show, for example—can help monitor improvement over time.
Diagnoses. Continually reassess the primary diagnosis and look for evidence of a medical illness, medication side effects, or secondary psychiatric conditions—especially alcohol or drug abuse.
Coordination of care. Update the patient’s file on dealings with therapists, case managers, and other physicians.
Handouts. Provide handouts and/or Web sites describing a medication’s therapeutic and side effects. Get handouts from numerous sources or develop information sheets and adapt them to your patient population. Include generic and brand names of medications to avoid confusion.
Empathy. Conveying empathy for the patient’s problems or pleasures is crucial to a strong therapeutic alliance and effective treatment.
Costs. Don’t ignore medication costs. Being up-to-date on formulary options helps patients get needed prescriptions.
Knowledge. End the session by asking the patient to summarize the medication plan to ensure that he or she knows what to do.
The MEDCHECK guidelines do not take into account necessary tasks outside of the session:
- Schedule time before your appointments to review charts and recall information from a patient’s last visit. If you cannot update the chart during the visit, reserve a few minutes later for documentation.
- Take periodic breaks to return phone calls or e-mails or take a short walk.
- Read up on relevant treatment guidelines to ensure you are providing evidence-based care.
- Finally, reserve time to be an advocate for your patient by addressing any administrative short-comings or removing obstacles to therapeutic recommendations.
Of course, short visits are not appropriate for all patients. Give more time to patients in crisis or to complicated cases such as children, pregnant women, or those needing interpreters.
Dr. Moffic is professor of psychiatry and behavioral medicine, Medical College of Wisconsin, Milwaukee.
Dr. Moffic is professor of psychiatry and behavioral medicine, Medical College of Wisconsin, Milwaukee.
Brain workouts boost attention
Research over the past decade suggests that the brain, like muscles, might get stronger after a good workout. Some evidence suggests that patients with attention-deficit/hyperactivity disorder (ADHD) can improve focus and concentration with mental exercises.
Research has unveiled the brain’s remarkable capacity for structural change as the result of experience. Repeating a specific brain function enhances neural mechanisms by increasing synapse formation or generating new neurons.1 Increased use of neurons likely activates growth factor proteins that stimulate neural growth.
Healing the brain by exercising an impaired region is of great interest to physicians. For example, a group at Yale University showed that children with dyslexia who receive proper reading instruction read more fluently and show increased activity in the left hemisphere regions that decode words.2 Other research has shown that constraint-induced movement therapy—when a stroke patient is forced to use his or her impaired arm or leg by restraining the good one—can “reawaken” parts of the brain damaged by a stroke.3 Researchers also are studying whether intellectual exercises such as reading or learning new skills can forestall Alzheimer’s disease.
Neurofeedback basics
Can the brain’s malleability help children with ADHD? Imaging studies show that children with ADHD have structural and functional deficits in the prefrontal cortex, the area associated with attention. Stimulant medications prescribed for ADHD compensate for this defect by increasing dopaminergic neuron activity in the prefrontal cortex.
Neurofeedback—also called EEG biofeedback—also could alleviate ADHD symptoms. For 30 years, neurofeedback has been studied as an ADHD treatment with promising results.5 Improvement in attention, concentration, and working memory has been reported in up to 75% of cases in the literature, although randomized controlled trials have not been conducted.
Neurofeedback teaches an individual to regulate the electrical activity of his or her brain with mental exercises.4 EEG frequencies generally can be divided into four basic rhythms:
- beta rhythm is alert and focused
- alpha is relaxed
- theta is between awake and asleep
- delta is deep sleep (Figure 1).
The patient aims to spend more time in the alert beta rhythm and less time in the slower, more relaxed rhythms by thinking thoughts that generate the appropriate rhythm. More time spent in beta rhythm means better attention and concentration. A computer monitoring EEG frequencies helps the patient learn to regulate his or her brain rhythms.
Figure 1 EEG frequencies characterizing 4 basic brain rhythms
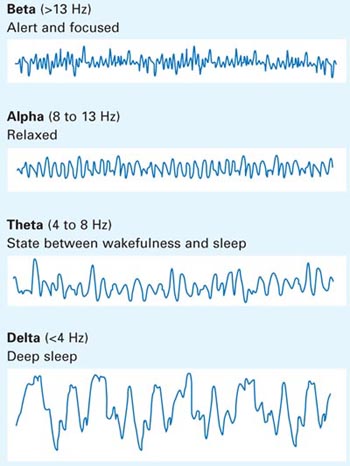
Adhd help
A University of Montreal research group recently completed an open, randomized, neurofeedback trial of 20 children ages 8 to 12 with ADHD who do not take psychostimulants or other medications for ADHD.6 Fifteen received neurofeedback therapy consisting of 40 one-hour sessions over 15 weeks. Five children who did not receive treatment served as the control group. All subjects took several neuropsychological tests measuring attention and hyperactivity before and after the study (Figure 2a). On average the neurofeedback group scored significantly higher on all measures of attention without side effects compared with the control group.
A comparison of functional brain imaging scans taken before and after the study showed increased anterior cingulate gyrus activity in the frontal cortex in the neurofeedback group but not the controls (Figure 2b). Subjects took the Counting Stroop Test—a mental exercise that involves the anterior cingulate gyrus—while in the scanner. The imaging studies were averaged within the groups, and the before and after scans were subtracted from each other. The increased activity translates into improved attention and concentration and decreased impulsivity, allowing children to perform better in school, get into less trouble, and have better relationships with parents.
Although we need more studies, this research suggests that neurofeedback might be a treatment option for patients with ADHD who cannot tolerate or do not wish to take medications.
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
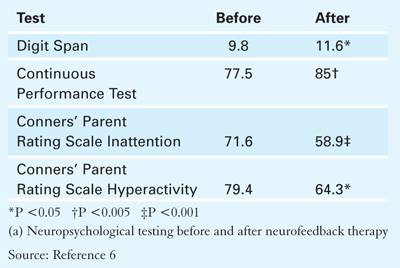
A. Neuropsychological testing
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
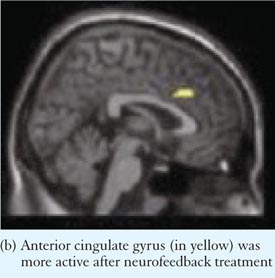
B. Imaging study
1. Gage FH. Brain, repair yourself. Sci Am 2003;289(3):46-53.
2. Shaywitz SE, Shaywitz BA. Dyslexia (specific reading disability). Biol Psychiatry 2005;57(11):1301-9.
3. Taub E, Uswatte G. Constraint-induced movement therapy: bridging from the primate laboratory to the stroke rehabilitation laboratory. J Rehabil Med 2003(41 suppl);34-40.
4. Kraft U. Train your brain. Sci Am Mind 2006;17(1):58-63.
5. Monastra VJ. Electroencephalographic biofeedback (neurotherapy) as a treatment for attention deficit hyperactivity disorder: rationale and empirical foundation. Child Adolesc Psychiatr Clin N Am 2005;14(1):55-82.
6. Levesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett 2006;394(3):216-21.
Research over the past decade suggests that the brain, like muscles, might get stronger after a good workout. Some evidence suggests that patients with attention-deficit/hyperactivity disorder (ADHD) can improve focus and concentration with mental exercises.
Research has unveiled the brain’s remarkable capacity for structural change as the result of experience. Repeating a specific brain function enhances neural mechanisms by increasing synapse formation or generating new neurons.1 Increased use of neurons likely activates growth factor proteins that stimulate neural growth.
Healing the brain by exercising an impaired region is of great interest to physicians. For example, a group at Yale University showed that children with dyslexia who receive proper reading instruction read more fluently and show increased activity in the left hemisphere regions that decode words.2 Other research has shown that constraint-induced movement therapy—when a stroke patient is forced to use his or her impaired arm or leg by restraining the good one—can “reawaken” parts of the brain damaged by a stroke.3 Researchers also are studying whether intellectual exercises such as reading or learning new skills can forestall Alzheimer’s disease.
Neurofeedback basics
Can the brain’s malleability help children with ADHD? Imaging studies show that children with ADHD have structural and functional deficits in the prefrontal cortex, the area associated with attention. Stimulant medications prescribed for ADHD compensate for this defect by increasing dopaminergic neuron activity in the prefrontal cortex.
Neurofeedback—also called EEG biofeedback—also could alleviate ADHD symptoms. For 30 years, neurofeedback has been studied as an ADHD treatment with promising results.5 Improvement in attention, concentration, and working memory has been reported in up to 75% of cases in the literature, although randomized controlled trials have not been conducted.
Neurofeedback teaches an individual to regulate the electrical activity of his or her brain with mental exercises.4 EEG frequencies generally can be divided into four basic rhythms:
- beta rhythm is alert and focused
- alpha is relaxed
- theta is between awake and asleep
- delta is deep sleep (Figure 1).
The patient aims to spend more time in the alert beta rhythm and less time in the slower, more relaxed rhythms by thinking thoughts that generate the appropriate rhythm. More time spent in beta rhythm means better attention and concentration. A computer monitoring EEG frequencies helps the patient learn to regulate his or her brain rhythms.
Figure 1 EEG frequencies characterizing 4 basic brain rhythms
Adhd help
A University of Montreal research group recently completed an open, randomized, neurofeedback trial of 20 children ages 8 to 12 with ADHD who do not take psychostimulants or other medications for ADHD.6 Fifteen received neurofeedback therapy consisting of 40 one-hour sessions over 15 weeks. Five children who did not receive treatment served as the control group. All subjects took several neuropsychological tests measuring attention and hyperactivity before and after the study (Figure 2a). On average the neurofeedback group scored significantly higher on all measures of attention without side effects compared with the control group.
A comparison of functional brain imaging scans taken before and after the study showed increased anterior cingulate gyrus activity in the frontal cortex in the neurofeedback group but not the controls (Figure 2b). Subjects took the Counting Stroop Test—a mental exercise that involves the anterior cingulate gyrus—while in the scanner. The imaging studies were averaged within the groups, and the before and after scans were subtracted from each other. The increased activity translates into improved attention and concentration and decreased impulsivity, allowing children to perform better in school, get into less trouble, and have better relationships with parents.
Although we need more studies, this research suggests that neurofeedback might be a treatment option for patients with ADHD who cannot tolerate or do not wish to take medications.
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
A. Neuropsychological testing
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
B. Imaging study
Research over the past decade suggests that the brain, like muscles, might get stronger after a good workout. Some evidence suggests that patients with attention-deficit/hyperactivity disorder (ADHD) can improve focus and concentration with mental exercises.
Research has unveiled the brain’s remarkable capacity for structural change as the result of experience. Repeating a specific brain function enhances neural mechanisms by increasing synapse formation or generating new neurons.1 Increased use of neurons likely activates growth factor proteins that stimulate neural growth.
Healing the brain by exercising an impaired region is of great interest to physicians. For example, a group at Yale University showed that children with dyslexia who receive proper reading instruction read more fluently and show increased activity in the left hemisphere regions that decode words.2 Other research has shown that constraint-induced movement therapy—when a stroke patient is forced to use his or her impaired arm or leg by restraining the good one—can “reawaken” parts of the brain damaged by a stroke.3 Researchers also are studying whether intellectual exercises such as reading or learning new skills can forestall Alzheimer’s disease.
Neurofeedback basics
Can the brain’s malleability help children with ADHD? Imaging studies show that children with ADHD have structural and functional deficits in the prefrontal cortex, the area associated with attention. Stimulant medications prescribed for ADHD compensate for this defect by increasing dopaminergic neuron activity in the prefrontal cortex.
Neurofeedback—also called EEG biofeedback—also could alleviate ADHD symptoms. For 30 years, neurofeedback has been studied as an ADHD treatment with promising results.5 Improvement in attention, concentration, and working memory has been reported in up to 75% of cases in the literature, although randomized controlled trials have not been conducted.
Neurofeedback teaches an individual to regulate the electrical activity of his or her brain with mental exercises.4 EEG frequencies generally can be divided into four basic rhythms:
- beta rhythm is alert and focused
- alpha is relaxed
- theta is between awake and asleep
- delta is deep sleep (Figure 1).
The patient aims to spend more time in the alert beta rhythm and less time in the slower, more relaxed rhythms by thinking thoughts that generate the appropriate rhythm. More time spent in beta rhythm means better attention and concentration. A computer monitoring EEG frequencies helps the patient learn to regulate his or her brain rhythms.
Figure 1 EEG frequencies characterizing 4 basic brain rhythms
Adhd help
A University of Montreal research group recently completed an open, randomized, neurofeedback trial of 20 children ages 8 to 12 with ADHD who do not take psychostimulants or other medications for ADHD.6 Fifteen received neurofeedback therapy consisting of 40 one-hour sessions over 15 weeks. Five children who did not receive treatment served as the control group. All subjects took several neuropsychological tests measuring attention and hyperactivity before and after the study (Figure 2a). On average the neurofeedback group scored significantly higher on all measures of attention without side effects compared with the control group.
A comparison of functional brain imaging scans taken before and after the study showed increased anterior cingulate gyrus activity in the frontal cortex in the neurofeedback group but not the controls (Figure 2b). Subjects took the Counting Stroop Test—a mental exercise that involves the anterior cingulate gyrus—while in the scanner. The imaging studies were averaged within the groups, and the before and after scans were subtracted from each other. The increased activity translates into improved attention and concentration and decreased impulsivity, allowing children to perform better in school, get into less trouble, and have better relationships with parents.
Although we need more studies, this research suggests that neurofeedback might be a treatment option for patients with ADHD who cannot tolerate or do not wish to take medications.
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
A. Neuropsychological testing
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
B. Imaging study
1. Gage FH. Brain, repair yourself. Sci Am 2003;289(3):46-53.
2. Shaywitz SE, Shaywitz BA. Dyslexia (specific reading disability). Biol Psychiatry 2005;57(11):1301-9.
3. Taub E, Uswatte G. Constraint-induced movement therapy: bridging from the primate laboratory to the stroke rehabilitation laboratory. J Rehabil Med 2003(41 suppl);34-40.
4. Kraft U. Train your brain. Sci Am Mind 2006;17(1):58-63.
5. Monastra VJ. Electroencephalographic biofeedback (neurotherapy) as a treatment for attention deficit hyperactivity disorder: rationale and empirical foundation. Child Adolesc Psychiatr Clin N Am 2005;14(1):55-82.
6. Levesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett 2006;394(3):216-21.
1. Gage FH. Brain, repair yourself. Sci Am 2003;289(3):46-53.
2. Shaywitz SE, Shaywitz BA. Dyslexia (specific reading disability). Biol Psychiatry 2005;57(11):1301-9.
3. Taub E, Uswatte G. Constraint-induced movement therapy: bridging from the primate laboratory to the stroke rehabilitation laboratory. J Rehabil Med 2003(41 suppl);34-40.
4. Kraft U. Train your brain. Sci Am Mind 2006;17(1):58-63.
5. Monastra VJ. Electroencephalographic biofeedback (neurotherapy) as a treatment for attention deficit hyperactivity disorder: rationale and empirical foundation. Child Adolesc Psychiatr Clin N Am 2005;14(1):55-82.
6. Levesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett 2006;394(3):216-21.
Postpartum depression or medical problem?
Many medical conditions common among new mothers can cause depressed mood, fatigue, and other symptoms that suggest postpartum depression. To help you quickly pinpoint the source of a new mother’s depressive symptoms and plan treatment, this article reviews:
- new-onset or pre-existing neurologic, cardiovascular, thyroid, and other conditions that mimic postpartum depression
- risk factors and clinical features that distinguish postpartum depression from other psychiatric disorders
- laboratory tests that confirm or rule out medical problems.
Case: ‘I can’t sleep’
Mrs. A, age 40, sleeps 2 hours nightly at most. Awakened by her 3-month-old daughter’s overnight crying, she lies awake and ruminates over the day’s events. Throughout the day, she fears she cannot care for her baby and 2-year-old son, and she depends on a family member to stay home with her. Financial concerns force her back to work 3 months after giving birth, but she is so despondent that she can barely function.
Mrs. A’s primary care physician diagnoses primary insomnia and prescribes mirtazapine and zolpidem, 15 and 10 mg each night, respectively, but her sleep disturbance persists after 6 weeks. The physician adds the hypnotic temazepam, 15 mg at night, and the sedating anticonvulsant gabapentin, 300 mg at bedtime. Both are titrated over 6 months to 45 mg and 1,800 mg at bedtime, respectively, but Mrs. A continues to lose sleep.
After 6 months, the doctor stops mirtazapine because Mrs. A has gained 20 lb. A switch to sertraline, 25 mg/d, has no effect.
Eighteen months after symptom onset, Mrs. A still sleeps poorly, even though her daughter—now age 2—sleeps through the night. Her depressed mood—undiagnosed by the physician—continues to worsen. She sees a psychiatrist after routine blood tests and a sleep study reveal no medical cause for her insomnia.
Is it postpartum depression?
Mrs. A’s despondent mood, sleep disturbances, feelings of inadequacy as a parent, and impaired concentration suggest postpartum depression. Ego-dystonic obsessive thoughts of harming the infant might emerge, although nonpsychotic patients rarely act upon them.1
Finding risk factors for postpartum depression can clarify the diagnosis. Ask the patient:
- When did you first notice symptoms? DSMIV-TR says postpartum depression usually begins within 4 weeks of giving birth,2 but most researchers define the postpartum period as ≤6 months after delivery.1,3 Mrs. A’s depression and insomnia started 3 months after childbirth.
- Have you been depressed before? Women with past postpartum or other depressive episodes face a high risk of recurrence after subsequent pregnancies.1,3 Active eating disorder during pregnancy4 and past premenstrual dysphoric disorder also are risk factors.1,3
- Has anyone in your family had depression? This increases postpartum depression risk.5
- Who is helping you? Psychosocial stress and lack of social support can fuel postpartum depression.1,3 Mrs. A gets practical help from family members, but life’s pressures are taking their toll.
Is it another mental illness?
Screen women with postpartum depressive symptoms for anxiety, which is highly comorbid with depression.6
Include bipolar disorder in the differential diagnosis. Ask new mothers with depressive symptoms if they feel inexplicably happy, irritable, or unusually energetic at times. Also screen for postpartum psychosis, which can progress to bipolar disorder7 and—worse—greatly increase the risk of infanticide.
The Edinburgh Postnatal Depression Scale,8 a 10-item self-report screening tool that takes about 5 minutes to complete, can help identify postpartum depression (see Related resources).
Case continued: A postpartum headache
During our initial interview, Mrs. A denies thoughts of harming herself or her children, and psychotic symptoms are not apparent. She reports no past depressive or anxiety episodes and does not use alcohol or illicit drugs. Her sister has a history of depression (not postpartum).
During review of systems, Mrs. A complains of persistent headaches. Brain MRI reveals a 4.5×5 mm microadenoma in the pituitary gland. We refer her to an endocrinologist, who obtains prolactin readings of 92 and 122.4 ng/mL (normal range, 2.8 to 29.2 ng/mL).
Discussion. Mrs. A had few predictive factors for postpartum depression, an atypical presentation with insomnia as the main symptom, and incomplete response after 18 months of treatment. These findings—plus her elevated prolactin and brain MRI results—suggest a medical cause.
Is it a medical problem?
Pre-existing or new-onset postpartum medical conditions can confound the diagnosis.
- Fatigue can mimic depression’s neurovegetative signs (poor energy, decreased appetite, sleep). Common causes include sleep deprivation, thyroid disorders, anemia, cardiomyopathy, and infections (Table 1).9
- Weight change could signal a medical condition whose symptoms resemble postpartum depression—such as diabetes or human immunodeficiency virus (HIV) (Table 2).
- Other disorders—including neurologic diseases, prolactinomas, systemic lupus erythematosus, diabetes, and rheumatoid arthritis—can cause depressive and other psychiatric symptoms (Table 3).
Recognizing the following disorders’ physical signs is key to uncovering a medical cause for postpartum depressive symptoms.
Thyroid disease. Postpartum thyroiditis (PPT) can occur 1 to 3 months after delivery,10 often recurs after subsequent pregnancies,11 and can progress to permanent hypothyroidism within 5 years.10 Hypothyroidism can cause cognitive slowing, depression, and psychosis, and acute mania has been reported with severe hypothyroidism secondary to PPT.12
Find out if the patient tested positive early in gestation for thyroid antibodies, as this may predict postpartum depression.
Multiple sclerosis (MS) can cause anxiety, mania, depression, and cognitive impairment.13 Drugs used to treat MS—such as steroids or interferon—can induce depression.
Relapses are infrequent during pregnancy but increase significantly within 3 months after giving birth14 in about one-third of women with active MS before pregnancy.15 Gait ataxia, sensory loss, numbness, hyperactive reflexes or spasticity, bladder dysfunction, visual impairment, disordered ocular motility, and fatigue are prominent clinical signs of MS.16
Myasthenia gravis (MG). Women who become pregnant within 1 year after diagnosis run a high risk of MG exacerbation.17
Fatigue and muscular weakness caused by MG can mimic depression, and adjusting to this debilitating illness can cause depression. Double vision, droopy eyelids, and muscle weakness alleviated by rest but worsened by activity are pathognomonic signs.16
Other neurologic diseases. Pre-existing seizure disorders can worsen after giving birth and cause depression.14
Subtle presentations of brain tumors include cognitive deficits, mood disturbance, and personality change. A left frontal lobe tumor can cause depression.
Ask the patient if she has had headaches, visual symptoms, vomiting, seizures, or focal neurologic deficits—any of these could signal a primary brain tumor or intracranial hemorrhage.
Prolactinomas, the most common pituitary tumor in pregnant and postpartum women, enlarge during pregnancy and regress after delivery.14 Depression, anxiety, apathy, and personality changes may stem from the pituitary tumor, its treatment, or changes in the hypothalamic-pituitary-end organ axis.18 Typical amenorrhea-galactorrhea syndrome resembles postpartum physiologic changes.
Headaches are common, and compression of the optic chiasm with macrodenomas causes visual field changes.
Systemic lupus erythematosus (SLE), most prevalent in young women, might flare during pregnancy and within 6 weeks after giving birth.11 Headaches, seizures, or cerebrovascular events with comorbid mood disorders, delirium, dementia, psychosis, or anxiety can signal SLE.13
Suspect SLE if the patient presents with fatigue, “butterfly” face rash, or joint pain. Test for renal or cardiopulmonary involvement.
Rheumatoid arthritis (RA). Because inflammatory activity is heightened after childbirth, postpartum women—particularly after bearing a first child—face a five-fold risk of RA compared with other women.11 Breast-feeding might worsen RA, presumably by increasing prolactin production.
Physical limitations caused by RA can cause depression. Symmetric joint pain associated with morning stiffness—especially in the fingers, hands, or knees—might signal RA.
Anemia. Increased need for iron and folic acid during pregnancy can lead to anemia. Neuropsychiatric manifestations of folate deficiency range from mild irritability to severe depression, dementia, psychosis, and confusion.19 Vitamin B12 deficiency can lead to megaloblastic anemia or neurologic problems such as peripheral neuropathy, as well as depression, delirium, or dementia.19
Ask the patient about:
- alcohol dependence, malnourishment, chronic illness, inflammatory bowel disease, gastric bypass or other gastric surgery, which can impair vitamin B12 absorption
- use of anticonvulsants such as carbamazepine or valproic acid, which can decrease folate.
When not in hypotensive circulatory shock, patients with adrenal insufficiency might present with depression, delirium, or psychosis.13 Ask the patient if she is having lactation problems and irregular periods, which could signal a pituitary problem.
Peripartum cardiomyopathy—an acute dilated cardiomyopathy— appears ≤6 months after delivery and may cause fatigue.10,20 Check for shortness of breath at night and with exertion, palpitations, and extremity swelling.
Gestational diabetes. Pregnancy-induced insulin resistance leads to gestational diabetes mellitus. Women with gestational diabetes can develop type 2 diabetes after giving birth.10
Blood sugar fluctuations can cause depression, irritability, or memory problems. Depression can sabotage adherence to diet and treatment, leading to poor glycemic control.
Ask the patient if she was diagnosed with gestational diabetes and if she is experiencing fatigue, excessive thirst, frequent urination, blurred vision, headaches, excessive hunger, or unexplainable weight loss.
Primary biliary cirrhosis is most prevalent in women ages 35 to 60 and may cause depression.20 Pruritus, fatigue, jaundice, and liver abnormalities point to this autoimmune disease, and postpartum exacerbations have been reported.21
HIV infection often leads to cognitive loss and depression with suicidal thoughts.13 Highly active antiretroviral medications commonly cause agitation, pain, mood changes, and insomnia.
Ask the patient is she is HIV positive. Watch for weight loss, fever, anorexia, and recurrent infections.
Substance abuse. Intoxication, withdrawal, or long-term alcohol or drug use can contribute to depression. Women at high risk for substance abuse disorder might not adhere to psychiatric treatment and may be prone to sexually transmitted diseases. If possible, see the patient every 3 to 4 weeks during the postpartum period.
Pain—if not adequately controlled—can fuel depression. Ask the patient if she has chronic pain or suffered a severe injury.
Table 1
Possible tests if postpartum patient is constantly fatigued
| Laboratory test | Confirms or rules out | Order if patient also presents with: |
|---|---|---|
| Acetylcholine receptor antibodies | Myasthenia gravis | Double vision, droopy eyelids, muscle weakness |
| Alkaline phosphatase | Primary biliary cirrhosis | Jaundice, pruritus |
| Antimitochondrial antibody | Primary biliary cirrhosis | Jaundice, pruritus |
| Antinuclear antibody | Systemic lupus erythematosus | ‘Butterfly’ facial rash, joint pain, morning stiffness |
| CBC | Microcytic anemia, megaloblastic anemia | Pallor, low energy, peripheral neuropathy, shortness of breath |
| Electrolytes | Adrenal insufficiency, renal disease | Low blood pressure, seizures, skin pigmentation |
| Glucose (fasting or glucose tolerance) | Type 1 or 2 diabetes mellitus | Blurred vision, excessive thirst/hunger, headaches, frequent urination, unexplainable weight loss |
| HIV | HIV infection/AIDS | Anorexia, recurrent infections, weight loss |
| Liver function tests | Alcohol abuse, hepatitis, primary biliary cirrhosis | Asterixis (flapping tremor), easy bruising, jaundice, pruritus, spider telangiectasias |
| Lumbar puncture | Multiple sclerosis | Bladder dysfunction, gait ataxia, ocular signs, sensory loss, spasticity |
Possible tests if postpartum patient has lost or gained weight
| Laboratory test | Confirms or rules out | Order if patient also presents with: |
|---|---|---|
| Antithyroid antibody | Postpartum thyroiditis | Constipation, dry skin, hair loss, lethargy, memory loss |
| Glucose (fasting or glucose tolerance) | Type 1 or 2 diabetes mellitus | Blurred vision, excessive thirst/hunger, fatigue, frequent urination, headaches |
| HIV | HIV infection/AIDS | Anorexia, fatigue, recurrent infections |
| TSH±thyroid panel | Hypothyroidism | Constipation, dry skin, hair loss, lethargy |
| TSH±thyroid panel | Hyperthyroidism | Agitation, anxiety, heat intolerance, palpitations |
Possible tests if postpartum patient has other physical symptoms
| Laboratory test | Confirms or rules out | Order if patient presents with: |
|---|---|---|
| Blood urea nitrogen/creatinine | Renal disease, dehydration | Back pain, frequent urination or oliguria, low blood pressure |
| Brain MRI | Brain tumors, white matter disease | Focal deficits, headaches, seizures, vision problems, vomiting |
| C-reactive protein | Rheumatoid arthritis | Joint pain, morning stiffness |
| ECG | Cardiomyopathy | Extremity swelling, palpitations, shortness of breath at night and with exertion |
| Erythrocyte sedimentation rate | Rheumatoid arthritis, SLE | ‘Butterfly’ facial rash, joint pain |
| Folate | Folate deficiency | Ataxia, loss of vibration and position sense, peripheral neuropathy, weakness |
| Prolactin | Prolactinoma, hypopituitarism | Amenorrhea/galactorrhea, headache, visual field loss |
| Rapid plasma reagin | Syphilis | Ataxic wide-based gait, loss of position, deep pain and temperature sensation, palmar/plantar rash |
| Rheumatoid factor | Rheumatoid arthritis | Morning stiffness, symmetric joint pain |
| Urinalysis | Urinary infection, diabetes, renal disease | Burning or difficulty with voiding, dark-colored urine, frequent urination |
| Urine drug screen | Substance abuse disorder | Erratic behavior, irritability or aggression; violence, mental status changes |
| Vitamin B12 | Anemia, malnutrition, inflammatory bowel disease | Loss of position or vibratory sensation, mood and cognitive changes, tingling and numbness in hands and feet |
| SLE: Systemic lupus erythematosus | ||
Determining a medical cause
Laboratory and neuroimaging findings—obtained in concert with the patient’s primary care physician—will help confirm or rule out a medical problem (Table 4). Consult with a neurologist, endocrinologist or rheumatologist if indicated.
Table 4
Findings that signal a possible postpartum medical problem
| Laboratory finding | Could signal … |
|---|---|
| Low hemoglobin, hematocrit and mean cell volume (MCV) values | Microcytic anemia |
| MCV >100 mm3 | Megaloblastic anemia |
| Positive anticardiolipin or antinuclear antibody | Systemic lupus erythematosus |
| Blood urea nitrogen >20 mg/dL, creatinine >1.5 mg/dL | Acute or chronic renal failure |
| Low specific gravity on urinalysis | Diabetes insipidus or renal tubular abnormalities |
| Proteinuria with glycosuria | Diabetes mellitus |
| Proteinuria with protein or cellular casts | Systemic lupus erythematosus |
| Hyponatremia and hyperkalemia | Adrenocortical insufficiency |
| Hypo/hypernatremia | Seizures |
| Albumin | Malnutrition |
| SGOT/SGPT >35 u/L (each) | Alcohol abuse disorder, hepatitis, hepatic encephalopathy |
| Alkaline phosphatase >120 u/L, positive antimitochondrial antibody | Primary biliary cirrhosis |
| Erythrocyte sedimentation rate >20 mm/hr | Systemic lupus erythematosus, rheumatoid arthritis |
| Positive rheumatoid factor | Rheumatoid arthritis |
| Prolactin >24 ng/mL | Prolactinoma |
| TSH >5 µu/mL | Hypothyroidism |
| TSH | Hyperthyroidism |
| IgG >1.4 mg/dL, oligoclonal bands, myelin basic protein in CSF | Multiple sclerosis |
| White matter hyperintensities in brain MRI | Multiple sclerosis, CNS vasculitis, tumors |
| Source: Reference 5 | |
Case: will the tumor resolve?
Mrs. A’s endocrinologist prescribes bromocriptine to manage her hyperprolactinemia, but she refuses to start the dopamine agonist after the doctor explains that it might cause psychosis.
Working closely, the psychiatrist and endocrinologist postpone bromocriptine therapy to see if the prolactinoma will resolve without treatment. They order brain MRIs every 6 months to track the tumor.
Mrs. A starts weekly psychodynamic therapy, during which she explores her fear of failure as a mother. Within 2 months, she recognizes that she has set unrealistically high expectations for herself. Adopting a supportive approach, the therapist encourages her to go on dates with her husband and run errands or relax alone for 2 hours each weekend.
The psychiatrist discusses sleep hygiene and adds quetiapine, 25 mg at bedtime; reduces gabapentin over 3 months to 300 mg nightly; and titrates sertraline to 100 mg/d. The psychiatrist also weans Mrs. A off temazepam over 3 months, watching closely for withdrawal symptoms.
At the psychiatrist’s suggestion, Mrs. A. resumes exercising at a gym four to five times a week. Mrs. A reduces zolpidem use—taking it only as needed for insomnia—then tapers off gabapentin. Quetiapine is discontinued.
After 4 months, psychotherapy sessions are decreased to biweekly. Prolactin is 66.6 ng/mL at 3 months, then normalizes to 23.4 ng/mL at 6 months. Six months later, brain MRI shows no change in baseline tumor size. The endocrinologist continues semiannual brain MRI and prolactin testing to see if the tumor will shrink without surgery.
Nearly 1 year after presentation, Mrs. A’s depression is in remission.
- Edinburgh Postnatal Depression Scale. http://www.drgrelling.com/Downloads.htm (click on “Edinburgh Postnatal Depression Scale” under “Resources for professionals”).
- Postpartum Support International. www.postpartum.net.
- Bromocriptine • Parlodel
- Carbamazepine • Tegretol, others
- Gabapentin • Neurontin
- Mirtazapine • Remeron
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Temazepam • Restoril
- Valproic acid • Depakene
- Zolpidem • Ambien
Dr. Seritan reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Miller LJ. Postpartum depression. JAMA 2002;287:762-5.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000:204.
3. Burt V, Hendrick V. Clinical manual of women’s mental health. Arlington, VA: American Psychiatric Publishing; 2005:79-100.
4. Franko DL, Blais MA, Becker AE, et al. Pregnancy complications and neonatal outcomes in women with eating disorders. Am J Psychiatry 2001;158:1461-6.
5. Berga SL, Parry BL, Cyranowski JL. Psychiatry and reproductive medicine. In: Sadock BJ, Sadock VA, eds. Comprehensive textbook of psychiatry, 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
6. Altshuler LL, Hendrick V, Cohen L. An update on mood and anxiety disorders during pregnancy and the postpartum period. Prim Care Companion J Clin Psychiatry 2000;2:217-22.
7. Chaudron LH. Pies RW: The relationship between postpartum psychosis and bipolar disorder: A review. J Clin Psychiatry 2003;64:1284-92.
8. Cox JL, Holden JM, Sagvosky R. Detection of postnatal depression: Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry 1987;150:782-6.
9. Atkinson LS, Baxley EG. Postpartum fatigue. Am Fam Physician 1994;50:113-18.
10. Kaaja RJ, Greer IA. Manifestations of chronic disease during pregnancy. JAMA 2005;294:2751-7.
11. Stagnaro-Green A. Postpartum thyroiditis. Best Pract Res Clin Endocrinol Metab 2004;18:303-16.
12. Stowell CP, Barnhill JW. Acute mania in the setting of severe hypothyroidism. Psychosomatics 2005;46:259-61.
13. Sadock BJ, Sadock VA. Consultation-liaison psychiatry (Chapter 284). In: Synopsis of psychiatry, 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2003:844-7.
14. Karnad DR, Guntupalli KK. Neurologic disorders in pregnancy. Crit Care Med 2005;33:S362-S371.
15. Vukusic S, Confavreux C. Multiple sclerosis and pregnancy. Rev Neurol 2006;162:299-309.
16. Kaufman DM. Clinical Neurology for Psychiatrists. Philadelphia: WB Saunders; 2001.
17. Ramirez C, de Seze J, Delrieu O, et al. [Myasthenia gravis and pregnancy: clinical course and management of delivery and the postpartum phase.] Rev Neurol (Paris) 2006;162:330-8 (French).
18. Weitzner MA, Kanfer S, Booth-Jones M. Apathy and pituitary disease: it has nothing to do with depression. J Neuropsychiatry Clin Neurosci 2005;17:159-66.
19. Peselow E. Other pharmacological and biological therapies. In: Sadock BJ, Sadock VA, eds. Comprehensive textbook of psychiatry, 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
20. Kasper DL, Braunwald E, Fauci A, et al. Harrison’s principles of internal medicine, 16th ed. New York: McGraw-Hill; 2004.
21. Ohba K, Omagari K, Kusakari C, et al. Flare-up of autoimmune hepatitis after delivery in a patient with primary biliary irrhosis: postpartum overlap syndrome of primary biliary cirrhosis and autoimmune hepatitis. Dig Dis Sci 2005;50:201-6.
Many medical conditions common among new mothers can cause depressed mood, fatigue, and other symptoms that suggest postpartum depression. To help you quickly pinpoint the source of a new mother’s depressive symptoms and plan treatment, this article reviews:
- new-onset or pre-existing neurologic, cardiovascular, thyroid, and other conditions that mimic postpartum depression
- risk factors and clinical features that distinguish postpartum depression from other psychiatric disorders
- laboratory tests that confirm or rule out medical problems.
Case: ‘I can’t sleep’
Mrs. A, age 40, sleeps 2 hours nightly at most. Awakened by her 3-month-old daughter’s overnight crying, she lies awake and ruminates over the day’s events. Throughout the day, she fears she cannot care for her baby and 2-year-old son, and she depends on a family member to stay home with her. Financial concerns force her back to work 3 months after giving birth, but she is so despondent that she can barely function.
Mrs. A’s primary care physician diagnoses primary insomnia and prescribes mirtazapine and zolpidem, 15 and 10 mg each night, respectively, but her sleep disturbance persists after 6 weeks. The physician adds the hypnotic temazepam, 15 mg at night, and the sedating anticonvulsant gabapentin, 300 mg at bedtime. Both are titrated over 6 months to 45 mg and 1,800 mg at bedtime, respectively, but Mrs. A continues to lose sleep.
After 6 months, the doctor stops mirtazapine because Mrs. A has gained 20 lb. A switch to sertraline, 25 mg/d, has no effect.
Eighteen months after symptom onset, Mrs. A still sleeps poorly, even though her daughter—now age 2—sleeps through the night. Her depressed mood—undiagnosed by the physician—continues to worsen. She sees a psychiatrist after routine blood tests and a sleep study reveal no medical cause for her insomnia.
Is it postpartum depression?
Mrs. A’s despondent mood, sleep disturbances, feelings of inadequacy as a parent, and impaired concentration suggest postpartum depression. Ego-dystonic obsessive thoughts of harming the infant might emerge, although nonpsychotic patients rarely act upon them.1
Finding risk factors for postpartum depression can clarify the diagnosis. Ask the patient:
- When did you first notice symptoms? DSMIV-TR says postpartum depression usually begins within 4 weeks of giving birth,2 but most researchers define the postpartum period as ≤6 months after delivery.1,3 Mrs. A’s depression and insomnia started 3 months after childbirth.
- Have you been depressed before? Women with past postpartum or other depressive episodes face a high risk of recurrence after subsequent pregnancies.1,3 Active eating disorder during pregnancy4 and past premenstrual dysphoric disorder also are risk factors.1,3
- Has anyone in your family had depression? This increases postpartum depression risk.5
- Who is helping you? Psychosocial stress and lack of social support can fuel postpartum depression.1,3 Mrs. A gets practical help from family members, but life’s pressures are taking their toll.
Is it another mental illness?
Screen women with postpartum depressive symptoms for anxiety, which is highly comorbid with depression.6
Include bipolar disorder in the differential diagnosis. Ask new mothers with depressive symptoms if they feel inexplicably happy, irritable, or unusually energetic at times. Also screen for postpartum psychosis, which can progress to bipolar disorder7 and—worse—greatly increase the risk of infanticide.
The Edinburgh Postnatal Depression Scale,8 a 10-item self-report screening tool that takes about 5 minutes to complete, can help identify postpartum depression (see Related resources).
Case continued: A postpartum headache
During our initial interview, Mrs. A denies thoughts of harming herself or her children, and psychotic symptoms are not apparent. She reports no past depressive or anxiety episodes and does not use alcohol or illicit drugs. Her sister has a history of depression (not postpartum).
During review of systems, Mrs. A complains of persistent headaches. Brain MRI reveals a 4.5×5 mm microadenoma in the pituitary gland. We refer her to an endocrinologist, who obtains prolactin readings of 92 and 122.4 ng/mL (normal range, 2.8 to 29.2 ng/mL).
Discussion. Mrs. A had few predictive factors for postpartum depression, an atypical presentation with insomnia as the main symptom, and incomplete response after 18 months of treatment. These findings—plus her elevated prolactin and brain MRI results—suggest a medical cause.
Is it a medical problem?
Pre-existing or new-onset postpartum medical conditions can confound the diagnosis.
- Fatigue can mimic depression’s neurovegetative signs (poor energy, decreased appetite, sleep). Common causes include sleep deprivation, thyroid disorders, anemia, cardiomyopathy, and infections (Table 1).9
- Weight change could signal a medical condition whose symptoms resemble postpartum depression—such as diabetes or human immunodeficiency virus (HIV) (Table 2).
- Other disorders—including neurologic diseases, prolactinomas, systemic lupus erythematosus, diabetes, and rheumatoid arthritis—can cause depressive and other psychiatric symptoms (Table 3).
Recognizing the following disorders’ physical signs is key to uncovering a medical cause for postpartum depressive symptoms.
Thyroid disease. Postpartum thyroiditis (PPT) can occur 1 to 3 months after delivery,10 often recurs after subsequent pregnancies,11 and can progress to permanent hypothyroidism within 5 years.10 Hypothyroidism can cause cognitive slowing, depression, and psychosis, and acute mania has been reported with severe hypothyroidism secondary to PPT.12
Find out if the patient tested positive early in gestation for thyroid antibodies, as this may predict postpartum depression.
Multiple sclerosis (MS) can cause anxiety, mania, depression, and cognitive impairment.13 Drugs used to treat MS—such as steroids or interferon—can induce depression.
Relapses are infrequent during pregnancy but increase significantly within 3 months after giving birth14 in about one-third of women with active MS before pregnancy.15 Gait ataxia, sensory loss, numbness, hyperactive reflexes or spasticity, bladder dysfunction, visual impairment, disordered ocular motility, and fatigue are prominent clinical signs of MS.16
Myasthenia gravis (MG). Women who become pregnant within 1 year after diagnosis run a high risk of MG exacerbation.17
Fatigue and muscular weakness caused by MG can mimic depression, and adjusting to this debilitating illness can cause depression. Double vision, droopy eyelids, and muscle weakness alleviated by rest but worsened by activity are pathognomonic signs.16
Other neurologic diseases. Pre-existing seizure disorders can worsen after giving birth and cause depression.14
Subtle presentations of brain tumors include cognitive deficits, mood disturbance, and personality change. A left frontal lobe tumor can cause depression.
Ask the patient if she has had headaches, visual symptoms, vomiting, seizures, or focal neurologic deficits—any of these could signal a primary brain tumor or intracranial hemorrhage.
Prolactinomas, the most common pituitary tumor in pregnant and postpartum women, enlarge during pregnancy and regress after delivery.14 Depression, anxiety, apathy, and personality changes may stem from the pituitary tumor, its treatment, or changes in the hypothalamic-pituitary-end organ axis.18 Typical amenorrhea-galactorrhea syndrome resembles postpartum physiologic changes.
Headaches are common, and compression of the optic chiasm with macrodenomas causes visual field changes.
Systemic lupus erythematosus (SLE), most prevalent in young women, might flare during pregnancy and within 6 weeks after giving birth.11 Headaches, seizures, or cerebrovascular events with comorbid mood disorders, delirium, dementia, psychosis, or anxiety can signal SLE.13
Suspect SLE if the patient presents with fatigue, “butterfly” face rash, or joint pain. Test for renal or cardiopulmonary involvement.
Rheumatoid arthritis (RA). Because inflammatory activity is heightened after childbirth, postpartum women—particularly after bearing a first child—face a five-fold risk of RA compared with other women.11 Breast-feeding might worsen RA, presumably by increasing prolactin production.
Physical limitations caused by RA can cause depression. Symmetric joint pain associated with morning stiffness—especially in the fingers, hands, or knees—might signal RA.
Anemia. Increased need for iron and folic acid during pregnancy can lead to anemia. Neuropsychiatric manifestations of folate deficiency range from mild irritability to severe depression, dementia, psychosis, and confusion.19 Vitamin B12 deficiency can lead to megaloblastic anemia or neurologic problems such as peripheral neuropathy, as well as depression, delirium, or dementia.19
Ask the patient about:
- alcohol dependence, malnourishment, chronic illness, inflammatory bowel disease, gastric bypass or other gastric surgery, which can impair vitamin B12 absorption
- use of anticonvulsants such as carbamazepine or valproic acid, which can decrease folate.
When not in hypotensive circulatory shock, patients with adrenal insufficiency might present with depression, delirium, or psychosis.13 Ask the patient if she is having lactation problems and irregular periods, which could signal a pituitary problem.
Peripartum cardiomyopathy—an acute dilated cardiomyopathy— appears ≤6 months after delivery and may cause fatigue.10,20 Check for shortness of breath at night and with exertion, palpitations, and extremity swelling.
Gestational diabetes. Pregnancy-induced insulin resistance leads to gestational diabetes mellitus. Women with gestational diabetes can develop type 2 diabetes after giving birth.10
Blood sugar fluctuations can cause depression, irritability, or memory problems. Depression can sabotage adherence to diet and treatment, leading to poor glycemic control.
Ask the patient if she was diagnosed with gestational diabetes and if she is experiencing fatigue, excessive thirst, frequent urination, blurred vision, headaches, excessive hunger, or unexplainable weight loss.
Primary biliary cirrhosis is most prevalent in women ages 35 to 60 and may cause depression.20 Pruritus, fatigue, jaundice, and liver abnormalities point to this autoimmune disease, and postpartum exacerbations have been reported.21
HIV infection often leads to cognitive loss and depression with suicidal thoughts.13 Highly active antiretroviral medications commonly cause agitation, pain, mood changes, and insomnia.
Ask the patient is she is HIV positive. Watch for weight loss, fever, anorexia, and recurrent infections.
Substance abuse. Intoxication, withdrawal, or long-term alcohol or drug use can contribute to depression. Women at high risk for substance abuse disorder might not adhere to psychiatric treatment and may be prone to sexually transmitted diseases. If possible, see the patient every 3 to 4 weeks during the postpartum period.
Pain—if not adequately controlled—can fuel depression. Ask the patient if she has chronic pain or suffered a severe injury.
Table 1
Possible tests if postpartum patient is constantly fatigued
| Laboratory test | Confirms or rules out | Order if patient also presents with: |
|---|---|---|
| Acetylcholine receptor antibodies | Myasthenia gravis | Double vision, droopy eyelids, muscle weakness |
| Alkaline phosphatase | Primary biliary cirrhosis | Jaundice, pruritus |
| Antimitochondrial antibody | Primary biliary cirrhosis | Jaundice, pruritus |
| Antinuclear antibody | Systemic lupus erythematosus | ‘Butterfly’ facial rash, joint pain, morning stiffness |
| CBC | Microcytic anemia, megaloblastic anemia | Pallor, low energy, peripheral neuropathy, shortness of breath |
| Electrolytes | Adrenal insufficiency, renal disease | Low blood pressure, seizures, skin pigmentation |
| Glucose (fasting or glucose tolerance) | Type 1 or 2 diabetes mellitus | Blurred vision, excessive thirst/hunger, headaches, frequent urination, unexplainable weight loss |
| HIV | HIV infection/AIDS | Anorexia, recurrent infections, weight loss |
| Liver function tests | Alcohol abuse, hepatitis, primary biliary cirrhosis | Asterixis (flapping tremor), easy bruising, jaundice, pruritus, spider telangiectasias |
| Lumbar puncture | Multiple sclerosis | Bladder dysfunction, gait ataxia, ocular signs, sensory loss, spasticity |
Possible tests if postpartum patient has lost or gained weight
| Laboratory test | Confirms or rules out | Order if patient also presents with: |
|---|---|---|
| Antithyroid antibody | Postpartum thyroiditis | Constipation, dry skin, hair loss, lethargy, memory loss |
| Glucose (fasting or glucose tolerance) | Type 1 or 2 diabetes mellitus | Blurred vision, excessive thirst/hunger, fatigue, frequent urination, headaches |
| HIV | HIV infection/AIDS | Anorexia, fatigue, recurrent infections |
| TSH±thyroid panel | Hypothyroidism | Constipation, dry skin, hair loss, lethargy |
| TSH±thyroid panel | Hyperthyroidism | Agitation, anxiety, heat intolerance, palpitations |
Possible tests if postpartum patient has other physical symptoms
| Laboratory test | Confirms or rules out | Order if patient presents with: |
|---|---|---|
| Blood urea nitrogen/creatinine | Renal disease, dehydration | Back pain, frequent urination or oliguria, low blood pressure |
| Brain MRI | Brain tumors, white matter disease | Focal deficits, headaches, seizures, vision problems, vomiting |
| C-reactive protein | Rheumatoid arthritis | Joint pain, morning stiffness |
| ECG | Cardiomyopathy | Extremity swelling, palpitations, shortness of breath at night and with exertion |
| Erythrocyte sedimentation rate | Rheumatoid arthritis, SLE | ‘Butterfly’ facial rash, joint pain |
| Folate | Folate deficiency | Ataxia, loss of vibration and position sense, peripheral neuropathy, weakness |
| Prolactin | Prolactinoma, hypopituitarism | Amenorrhea/galactorrhea, headache, visual field loss |
| Rapid plasma reagin | Syphilis | Ataxic wide-based gait, loss of position, deep pain and temperature sensation, palmar/plantar rash |
| Rheumatoid factor | Rheumatoid arthritis | Morning stiffness, symmetric joint pain |
| Urinalysis | Urinary infection, diabetes, renal disease | Burning or difficulty with voiding, dark-colored urine, frequent urination |
| Urine drug screen | Substance abuse disorder | Erratic behavior, irritability or aggression; violence, mental status changes |
| Vitamin B12 | Anemia, malnutrition, inflammatory bowel disease | Loss of position or vibratory sensation, mood and cognitive changes, tingling and numbness in hands and feet |
| SLE: Systemic lupus erythematosus | ||
Determining a medical cause
Laboratory and neuroimaging findings—obtained in concert with the patient’s primary care physician—will help confirm or rule out a medical problem (Table 4). Consult with a neurologist, endocrinologist or rheumatologist if indicated.
Table 4
Findings that signal a possible postpartum medical problem
| Laboratory finding | Could signal … |
|---|---|
| Low hemoglobin, hematocrit and mean cell volume (MCV) values | Microcytic anemia |
| MCV >100 mm3 | Megaloblastic anemia |
| Positive anticardiolipin or antinuclear antibody | Systemic lupus erythematosus |
| Blood urea nitrogen >20 mg/dL, creatinine >1.5 mg/dL | Acute or chronic renal failure |
| Low specific gravity on urinalysis | Diabetes insipidus or renal tubular abnormalities |
| Proteinuria with glycosuria | Diabetes mellitus |
| Proteinuria with protein or cellular casts | Systemic lupus erythematosus |
| Hyponatremia and hyperkalemia | Adrenocortical insufficiency |
| Hypo/hypernatremia | Seizures |
| Albumin | Malnutrition |
| SGOT/SGPT >35 u/L (each) | Alcohol abuse disorder, hepatitis, hepatic encephalopathy |
| Alkaline phosphatase >120 u/L, positive antimitochondrial antibody | Primary biliary cirrhosis |
| Erythrocyte sedimentation rate >20 mm/hr | Systemic lupus erythematosus, rheumatoid arthritis |
| Positive rheumatoid factor | Rheumatoid arthritis |
| Prolactin >24 ng/mL | Prolactinoma |
| TSH >5 µu/mL | Hypothyroidism |
| TSH | Hyperthyroidism |
| IgG >1.4 mg/dL, oligoclonal bands, myelin basic protein in CSF | Multiple sclerosis |
| White matter hyperintensities in brain MRI | Multiple sclerosis, CNS vasculitis, tumors |
| Source: Reference 5 | |
Case: will the tumor resolve?
Mrs. A’s endocrinologist prescribes bromocriptine to manage her hyperprolactinemia, but she refuses to start the dopamine agonist after the doctor explains that it might cause psychosis.
Working closely, the psychiatrist and endocrinologist postpone bromocriptine therapy to see if the prolactinoma will resolve without treatment. They order brain MRIs every 6 months to track the tumor.
Mrs. A starts weekly psychodynamic therapy, during which she explores her fear of failure as a mother. Within 2 months, she recognizes that she has set unrealistically high expectations for herself. Adopting a supportive approach, the therapist encourages her to go on dates with her husband and run errands or relax alone for 2 hours each weekend.
The psychiatrist discusses sleep hygiene and adds quetiapine, 25 mg at bedtime; reduces gabapentin over 3 months to 300 mg nightly; and titrates sertraline to 100 mg/d. The psychiatrist also weans Mrs. A off temazepam over 3 months, watching closely for withdrawal symptoms.
At the psychiatrist’s suggestion, Mrs. A. resumes exercising at a gym four to five times a week. Mrs. A reduces zolpidem use—taking it only as needed for insomnia—then tapers off gabapentin. Quetiapine is discontinued.
After 4 months, psychotherapy sessions are decreased to biweekly. Prolactin is 66.6 ng/mL at 3 months, then normalizes to 23.4 ng/mL at 6 months. Six months later, brain MRI shows no change in baseline tumor size. The endocrinologist continues semiannual brain MRI and prolactin testing to see if the tumor will shrink without surgery.
Nearly 1 year after presentation, Mrs. A’s depression is in remission.
- Edinburgh Postnatal Depression Scale. http://www.drgrelling.com/Downloads.htm (click on “Edinburgh Postnatal Depression Scale” under “Resources for professionals”).
- Postpartum Support International. www.postpartum.net.
- Bromocriptine • Parlodel
- Carbamazepine • Tegretol, others
- Gabapentin • Neurontin
- Mirtazapine • Remeron
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Temazepam • Restoril
- Valproic acid • Depakene
- Zolpidem • Ambien
Dr. Seritan reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
Many medical conditions common among new mothers can cause depressed mood, fatigue, and other symptoms that suggest postpartum depression. To help you quickly pinpoint the source of a new mother’s depressive symptoms and plan treatment, this article reviews:
- new-onset or pre-existing neurologic, cardiovascular, thyroid, and other conditions that mimic postpartum depression
- risk factors and clinical features that distinguish postpartum depression from other psychiatric disorders
- laboratory tests that confirm or rule out medical problems.
Case: ‘I can’t sleep’
Mrs. A, age 40, sleeps 2 hours nightly at most. Awakened by her 3-month-old daughter’s overnight crying, she lies awake and ruminates over the day’s events. Throughout the day, she fears she cannot care for her baby and 2-year-old son, and she depends on a family member to stay home with her. Financial concerns force her back to work 3 months after giving birth, but she is so despondent that she can barely function.
Mrs. A’s primary care physician diagnoses primary insomnia and prescribes mirtazapine and zolpidem, 15 and 10 mg each night, respectively, but her sleep disturbance persists after 6 weeks. The physician adds the hypnotic temazepam, 15 mg at night, and the sedating anticonvulsant gabapentin, 300 mg at bedtime. Both are titrated over 6 months to 45 mg and 1,800 mg at bedtime, respectively, but Mrs. A continues to lose sleep.
After 6 months, the doctor stops mirtazapine because Mrs. A has gained 20 lb. A switch to sertraline, 25 mg/d, has no effect.
Eighteen months after symptom onset, Mrs. A still sleeps poorly, even though her daughter—now age 2—sleeps through the night. Her depressed mood—undiagnosed by the physician—continues to worsen. She sees a psychiatrist after routine blood tests and a sleep study reveal no medical cause for her insomnia.
Is it postpartum depression?
Mrs. A’s despondent mood, sleep disturbances, feelings of inadequacy as a parent, and impaired concentration suggest postpartum depression. Ego-dystonic obsessive thoughts of harming the infant might emerge, although nonpsychotic patients rarely act upon them.1
Finding risk factors for postpartum depression can clarify the diagnosis. Ask the patient:
- When did you first notice symptoms? DSMIV-TR says postpartum depression usually begins within 4 weeks of giving birth,2 but most researchers define the postpartum period as ≤6 months after delivery.1,3 Mrs. A’s depression and insomnia started 3 months after childbirth.
- Have you been depressed before? Women with past postpartum or other depressive episodes face a high risk of recurrence after subsequent pregnancies.1,3 Active eating disorder during pregnancy4 and past premenstrual dysphoric disorder also are risk factors.1,3
- Has anyone in your family had depression? This increases postpartum depression risk.5
- Who is helping you? Psychosocial stress and lack of social support can fuel postpartum depression.1,3 Mrs. A gets practical help from family members, but life’s pressures are taking their toll.
Is it another mental illness?
Screen women with postpartum depressive symptoms for anxiety, which is highly comorbid with depression.6
Include bipolar disorder in the differential diagnosis. Ask new mothers with depressive symptoms if they feel inexplicably happy, irritable, or unusually energetic at times. Also screen for postpartum psychosis, which can progress to bipolar disorder7 and—worse—greatly increase the risk of infanticide.
The Edinburgh Postnatal Depression Scale,8 a 10-item self-report screening tool that takes about 5 minutes to complete, can help identify postpartum depression (see Related resources).
Case continued: A postpartum headache
During our initial interview, Mrs. A denies thoughts of harming herself or her children, and psychotic symptoms are not apparent. She reports no past depressive or anxiety episodes and does not use alcohol or illicit drugs. Her sister has a history of depression (not postpartum).
During review of systems, Mrs. A complains of persistent headaches. Brain MRI reveals a 4.5×5 mm microadenoma in the pituitary gland. We refer her to an endocrinologist, who obtains prolactin readings of 92 and 122.4 ng/mL (normal range, 2.8 to 29.2 ng/mL).
Discussion. Mrs. A had few predictive factors for postpartum depression, an atypical presentation with insomnia as the main symptom, and incomplete response after 18 months of treatment. These findings—plus her elevated prolactin and brain MRI results—suggest a medical cause.
Is it a medical problem?
Pre-existing or new-onset postpartum medical conditions can confound the diagnosis.
- Fatigue can mimic depression’s neurovegetative signs (poor energy, decreased appetite, sleep). Common causes include sleep deprivation, thyroid disorders, anemia, cardiomyopathy, and infections (Table 1).9
- Weight change could signal a medical condition whose symptoms resemble postpartum depression—such as diabetes or human immunodeficiency virus (HIV) (Table 2).
- Other disorders—including neurologic diseases, prolactinomas, systemic lupus erythematosus, diabetes, and rheumatoid arthritis—can cause depressive and other psychiatric symptoms (Table 3).
Recognizing the following disorders’ physical signs is key to uncovering a medical cause for postpartum depressive symptoms.
Thyroid disease. Postpartum thyroiditis (PPT) can occur 1 to 3 months after delivery,10 often recurs after subsequent pregnancies,11 and can progress to permanent hypothyroidism within 5 years.10 Hypothyroidism can cause cognitive slowing, depression, and psychosis, and acute mania has been reported with severe hypothyroidism secondary to PPT.12
Find out if the patient tested positive early in gestation for thyroid antibodies, as this may predict postpartum depression.
Multiple sclerosis (MS) can cause anxiety, mania, depression, and cognitive impairment.13 Drugs used to treat MS—such as steroids or interferon—can induce depression.
Relapses are infrequent during pregnancy but increase significantly within 3 months after giving birth14 in about one-third of women with active MS before pregnancy.15 Gait ataxia, sensory loss, numbness, hyperactive reflexes or spasticity, bladder dysfunction, visual impairment, disordered ocular motility, and fatigue are prominent clinical signs of MS.16
Myasthenia gravis (MG). Women who become pregnant within 1 year after diagnosis run a high risk of MG exacerbation.17
Fatigue and muscular weakness caused by MG can mimic depression, and adjusting to this debilitating illness can cause depression. Double vision, droopy eyelids, and muscle weakness alleviated by rest but worsened by activity are pathognomonic signs.16
Other neurologic diseases. Pre-existing seizure disorders can worsen after giving birth and cause depression.14
Subtle presentations of brain tumors include cognitive deficits, mood disturbance, and personality change. A left frontal lobe tumor can cause depression.
Ask the patient if she has had headaches, visual symptoms, vomiting, seizures, or focal neurologic deficits—any of these could signal a primary brain tumor or intracranial hemorrhage.
Prolactinomas, the most common pituitary tumor in pregnant and postpartum women, enlarge during pregnancy and regress after delivery.14 Depression, anxiety, apathy, and personality changes may stem from the pituitary tumor, its treatment, or changes in the hypothalamic-pituitary-end organ axis.18 Typical amenorrhea-galactorrhea syndrome resembles postpartum physiologic changes.
Headaches are common, and compression of the optic chiasm with macrodenomas causes visual field changes.
Systemic lupus erythematosus (SLE), most prevalent in young women, might flare during pregnancy and within 6 weeks after giving birth.11 Headaches, seizures, or cerebrovascular events with comorbid mood disorders, delirium, dementia, psychosis, or anxiety can signal SLE.13
Suspect SLE if the patient presents with fatigue, “butterfly” face rash, or joint pain. Test for renal or cardiopulmonary involvement.
Rheumatoid arthritis (RA). Because inflammatory activity is heightened after childbirth, postpartum women—particularly after bearing a first child—face a five-fold risk of RA compared with other women.11 Breast-feeding might worsen RA, presumably by increasing prolactin production.
Physical limitations caused by RA can cause depression. Symmetric joint pain associated with morning stiffness—especially in the fingers, hands, or knees—might signal RA.
Anemia. Increased need for iron and folic acid during pregnancy can lead to anemia. Neuropsychiatric manifestations of folate deficiency range from mild irritability to severe depression, dementia, psychosis, and confusion.19 Vitamin B12 deficiency can lead to megaloblastic anemia or neurologic problems such as peripheral neuropathy, as well as depression, delirium, or dementia.19
Ask the patient about:
- alcohol dependence, malnourishment, chronic illness, inflammatory bowel disease, gastric bypass or other gastric surgery, which can impair vitamin B12 absorption
- use of anticonvulsants such as carbamazepine or valproic acid, which can decrease folate.
When not in hypotensive circulatory shock, patients with adrenal insufficiency might present with depression, delirium, or psychosis.13 Ask the patient if she is having lactation problems and irregular periods, which could signal a pituitary problem.
Peripartum cardiomyopathy—an acute dilated cardiomyopathy— appears ≤6 months after delivery and may cause fatigue.10,20 Check for shortness of breath at night and with exertion, palpitations, and extremity swelling.
Gestational diabetes. Pregnancy-induced insulin resistance leads to gestational diabetes mellitus. Women with gestational diabetes can develop type 2 diabetes after giving birth.10
Blood sugar fluctuations can cause depression, irritability, or memory problems. Depression can sabotage adherence to diet and treatment, leading to poor glycemic control.
Ask the patient if she was diagnosed with gestational diabetes and if she is experiencing fatigue, excessive thirst, frequent urination, blurred vision, headaches, excessive hunger, or unexplainable weight loss.
Primary biliary cirrhosis is most prevalent in women ages 35 to 60 and may cause depression.20 Pruritus, fatigue, jaundice, and liver abnormalities point to this autoimmune disease, and postpartum exacerbations have been reported.21
HIV infection often leads to cognitive loss and depression with suicidal thoughts.13 Highly active antiretroviral medications commonly cause agitation, pain, mood changes, and insomnia.
Ask the patient is she is HIV positive. Watch for weight loss, fever, anorexia, and recurrent infections.
Substance abuse. Intoxication, withdrawal, or long-term alcohol or drug use can contribute to depression. Women at high risk for substance abuse disorder might not adhere to psychiatric treatment and may be prone to sexually transmitted diseases. If possible, see the patient every 3 to 4 weeks during the postpartum period.
Pain—if not adequately controlled—can fuel depression. Ask the patient if she has chronic pain or suffered a severe injury.
Table 1
Possible tests if postpartum patient is constantly fatigued
| Laboratory test | Confirms or rules out | Order if patient also presents with: |
|---|---|---|
| Acetylcholine receptor antibodies | Myasthenia gravis | Double vision, droopy eyelids, muscle weakness |
| Alkaline phosphatase | Primary biliary cirrhosis | Jaundice, pruritus |
| Antimitochondrial antibody | Primary biliary cirrhosis | Jaundice, pruritus |
| Antinuclear antibody | Systemic lupus erythematosus | ‘Butterfly’ facial rash, joint pain, morning stiffness |
| CBC | Microcytic anemia, megaloblastic anemia | Pallor, low energy, peripheral neuropathy, shortness of breath |
| Electrolytes | Adrenal insufficiency, renal disease | Low blood pressure, seizures, skin pigmentation |
| Glucose (fasting or glucose tolerance) | Type 1 or 2 diabetes mellitus | Blurred vision, excessive thirst/hunger, headaches, frequent urination, unexplainable weight loss |
| HIV | HIV infection/AIDS | Anorexia, recurrent infections, weight loss |
| Liver function tests | Alcohol abuse, hepatitis, primary biliary cirrhosis | Asterixis (flapping tremor), easy bruising, jaundice, pruritus, spider telangiectasias |
| Lumbar puncture | Multiple sclerosis | Bladder dysfunction, gait ataxia, ocular signs, sensory loss, spasticity |
Possible tests if postpartum patient has lost or gained weight
| Laboratory test | Confirms or rules out | Order if patient also presents with: |
|---|---|---|
| Antithyroid antibody | Postpartum thyroiditis | Constipation, dry skin, hair loss, lethargy, memory loss |
| Glucose (fasting or glucose tolerance) | Type 1 or 2 diabetes mellitus | Blurred vision, excessive thirst/hunger, fatigue, frequent urination, headaches |
| HIV | HIV infection/AIDS | Anorexia, fatigue, recurrent infections |
| TSH±thyroid panel | Hypothyroidism | Constipation, dry skin, hair loss, lethargy |
| TSH±thyroid panel | Hyperthyroidism | Agitation, anxiety, heat intolerance, palpitations |
Possible tests if postpartum patient has other physical symptoms
| Laboratory test | Confirms or rules out | Order if patient presents with: |
|---|---|---|
| Blood urea nitrogen/creatinine | Renal disease, dehydration | Back pain, frequent urination or oliguria, low blood pressure |
| Brain MRI | Brain tumors, white matter disease | Focal deficits, headaches, seizures, vision problems, vomiting |
| C-reactive protein | Rheumatoid arthritis | Joint pain, morning stiffness |
| ECG | Cardiomyopathy | Extremity swelling, palpitations, shortness of breath at night and with exertion |
| Erythrocyte sedimentation rate | Rheumatoid arthritis, SLE | ‘Butterfly’ facial rash, joint pain |
| Folate | Folate deficiency | Ataxia, loss of vibration and position sense, peripheral neuropathy, weakness |
| Prolactin | Prolactinoma, hypopituitarism | Amenorrhea/galactorrhea, headache, visual field loss |
| Rapid plasma reagin | Syphilis | Ataxic wide-based gait, loss of position, deep pain and temperature sensation, palmar/plantar rash |
| Rheumatoid factor | Rheumatoid arthritis | Morning stiffness, symmetric joint pain |
| Urinalysis | Urinary infection, diabetes, renal disease | Burning or difficulty with voiding, dark-colored urine, frequent urination |
| Urine drug screen | Substance abuse disorder | Erratic behavior, irritability or aggression; violence, mental status changes |
| Vitamin B12 | Anemia, malnutrition, inflammatory bowel disease | Loss of position or vibratory sensation, mood and cognitive changes, tingling and numbness in hands and feet |
| SLE: Systemic lupus erythematosus | ||
Determining a medical cause
Laboratory and neuroimaging findings—obtained in concert with the patient’s primary care physician—will help confirm or rule out a medical problem (Table 4). Consult with a neurologist, endocrinologist or rheumatologist if indicated.
Table 4
Findings that signal a possible postpartum medical problem
| Laboratory finding | Could signal … |
|---|---|
| Low hemoglobin, hematocrit and mean cell volume (MCV) values | Microcytic anemia |
| MCV >100 mm3 | Megaloblastic anemia |
| Positive anticardiolipin or antinuclear antibody | Systemic lupus erythematosus |
| Blood urea nitrogen >20 mg/dL, creatinine >1.5 mg/dL | Acute or chronic renal failure |
| Low specific gravity on urinalysis | Diabetes insipidus or renal tubular abnormalities |
| Proteinuria with glycosuria | Diabetes mellitus |
| Proteinuria with protein or cellular casts | Systemic lupus erythematosus |
| Hyponatremia and hyperkalemia | Adrenocortical insufficiency |
| Hypo/hypernatremia | Seizures |
| Albumin | Malnutrition |
| SGOT/SGPT >35 u/L (each) | Alcohol abuse disorder, hepatitis, hepatic encephalopathy |
| Alkaline phosphatase >120 u/L, positive antimitochondrial antibody | Primary biliary cirrhosis |
| Erythrocyte sedimentation rate >20 mm/hr | Systemic lupus erythematosus, rheumatoid arthritis |
| Positive rheumatoid factor | Rheumatoid arthritis |
| Prolactin >24 ng/mL | Prolactinoma |
| TSH >5 µu/mL | Hypothyroidism |
| TSH | Hyperthyroidism |
| IgG >1.4 mg/dL, oligoclonal bands, myelin basic protein in CSF | Multiple sclerosis |
| White matter hyperintensities in brain MRI | Multiple sclerosis, CNS vasculitis, tumors |
| Source: Reference 5 | |
Case: will the tumor resolve?
Mrs. A’s endocrinologist prescribes bromocriptine to manage her hyperprolactinemia, but she refuses to start the dopamine agonist after the doctor explains that it might cause psychosis.
Working closely, the psychiatrist and endocrinologist postpone bromocriptine therapy to see if the prolactinoma will resolve without treatment. They order brain MRIs every 6 months to track the tumor.
Mrs. A starts weekly psychodynamic therapy, during which she explores her fear of failure as a mother. Within 2 months, she recognizes that she has set unrealistically high expectations for herself. Adopting a supportive approach, the therapist encourages her to go on dates with her husband and run errands or relax alone for 2 hours each weekend.
The psychiatrist discusses sleep hygiene and adds quetiapine, 25 mg at bedtime; reduces gabapentin over 3 months to 300 mg nightly; and titrates sertraline to 100 mg/d. The psychiatrist also weans Mrs. A off temazepam over 3 months, watching closely for withdrawal symptoms.
At the psychiatrist’s suggestion, Mrs. A. resumes exercising at a gym four to five times a week. Mrs. A reduces zolpidem use—taking it only as needed for insomnia—then tapers off gabapentin. Quetiapine is discontinued.
After 4 months, psychotherapy sessions are decreased to biweekly. Prolactin is 66.6 ng/mL at 3 months, then normalizes to 23.4 ng/mL at 6 months. Six months later, brain MRI shows no change in baseline tumor size. The endocrinologist continues semiannual brain MRI and prolactin testing to see if the tumor will shrink without surgery.
Nearly 1 year after presentation, Mrs. A’s depression is in remission.
- Edinburgh Postnatal Depression Scale. http://www.drgrelling.com/Downloads.htm (click on “Edinburgh Postnatal Depression Scale” under “Resources for professionals”).
- Postpartum Support International. www.postpartum.net.
- Bromocriptine • Parlodel
- Carbamazepine • Tegretol, others
- Gabapentin • Neurontin
- Mirtazapine • Remeron
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Temazepam • Restoril
- Valproic acid • Depakene
- Zolpidem • Ambien
Dr. Seritan reports no financial relationship with any company whose products are mentioned in this article, or with manufacturers of competing products.
1. Miller LJ. Postpartum depression. JAMA 2002;287:762-5.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000:204.
3. Burt V, Hendrick V. Clinical manual of women’s mental health. Arlington, VA: American Psychiatric Publishing; 2005:79-100.
4. Franko DL, Blais MA, Becker AE, et al. Pregnancy complications and neonatal outcomes in women with eating disorders. Am J Psychiatry 2001;158:1461-6.
5. Berga SL, Parry BL, Cyranowski JL. Psychiatry and reproductive medicine. In: Sadock BJ, Sadock VA, eds. Comprehensive textbook of psychiatry, 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
6. Altshuler LL, Hendrick V, Cohen L. An update on mood and anxiety disorders during pregnancy and the postpartum period. Prim Care Companion J Clin Psychiatry 2000;2:217-22.
7. Chaudron LH. Pies RW: The relationship between postpartum psychosis and bipolar disorder: A review. J Clin Psychiatry 2003;64:1284-92.
8. Cox JL, Holden JM, Sagvosky R. Detection of postnatal depression: Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry 1987;150:782-6.
9. Atkinson LS, Baxley EG. Postpartum fatigue. Am Fam Physician 1994;50:113-18.
10. Kaaja RJ, Greer IA. Manifestations of chronic disease during pregnancy. JAMA 2005;294:2751-7.
11. Stagnaro-Green A. Postpartum thyroiditis. Best Pract Res Clin Endocrinol Metab 2004;18:303-16.
12. Stowell CP, Barnhill JW. Acute mania in the setting of severe hypothyroidism. Psychosomatics 2005;46:259-61.
13. Sadock BJ, Sadock VA. Consultation-liaison psychiatry (Chapter 284). In: Synopsis of psychiatry, 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2003:844-7.
14. Karnad DR, Guntupalli KK. Neurologic disorders in pregnancy. Crit Care Med 2005;33:S362-S371.
15. Vukusic S, Confavreux C. Multiple sclerosis and pregnancy. Rev Neurol 2006;162:299-309.
16. Kaufman DM. Clinical Neurology for Psychiatrists. Philadelphia: WB Saunders; 2001.
17. Ramirez C, de Seze J, Delrieu O, et al. [Myasthenia gravis and pregnancy: clinical course and management of delivery and the postpartum phase.] Rev Neurol (Paris) 2006;162:330-8 (French).
18. Weitzner MA, Kanfer S, Booth-Jones M. Apathy and pituitary disease: it has nothing to do with depression. J Neuropsychiatry Clin Neurosci 2005;17:159-66.
19. Peselow E. Other pharmacological and biological therapies. In: Sadock BJ, Sadock VA, eds. Comprehensive textbook of psychiatry, 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
20. Kasper DL, Braunwald E, Fauci A, et al. Harrison’s principles of internal medicine, 16th ed. New York: McGraw-Hill; 2004.
21. Ohba K, Omagari K, Kusakari C, et al. Flare-up of autoimmune hepatitis after delivery in a patient with primary biliary irrhosis: postpartum overlap syndrome of primary biliary cirrhosis and autoimmune hepatitis. Dig Dis Sci 2005;50:201-6.
1. Miller LJ. Postpartum depression. JAMA 2002;287:762-5.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000:204.
3. Burt V, Hendrick V. Clinical manual of women’s mental health. Arlington, VA: American Psychiatric Publishing; 2005:79-100.
4. Franko DL, Blais MA, Becker AE, et al. Pregnancy complications and neonatal outcomes in women with eating disorders. Am J Psychiatry 2001;158:1461-6.
5. Berga SL, Parry BL, Cyranowski JL. Psychiatry and reproductive medicine. In: Sadock BJ, Sadock VA, eds. Comprehensive textbook of psychiatry, 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
6. Altshuler LL, Hendrick V, Cohen L. An update on mood and anxiety disorders during pregnancy and the postpartum period. Prim Care Companion J Clin Psychiatry 2000;2:217-22.
7. Chaudron LH. Pies RW: The relationship between postpartum psychosis and bipolar disorder: A review. J Clin Psychiatry 2003;64:1284-92.
8. Cox JL, Holden JM, Sagvosky R. Detection of postnatal depression: Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry 1987;150:782-6.
9. Atkinson LS, Baxley EG. Postpartum fatigue. Am Fam Physician 1994;50:113-18.
10. Kaaja RJ, Greer IA. Manifestations of chronic disease during pregnancy. JAMA 2005;294:2751-7.
11. Stagnaro-Green A. Postpartum thyroiditis. Best Pract Res Clin Endocrinol Metab 2004;18:303-16.
12. Stowell CP, Barnhill JW. Acute mania in the setting of severe hypothyroidism. Psychosomatics 2005;46:259-61.
13. Sadock BJ, Sadock VA. Consultation-liaison psychiatry (Chapter 284). In: Synopsis of psychiatry, 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2003:844-7.
14. Karnad DR, Guntupalli KK. Neurologic disorders in pregnancy. Crit Care Med 2005;33:S362-S371.
15. Vukusic S, Confavreux C. Multiple sclerosis and pregnancy. Rev Neurol 2006;162:299-309.
16. Kaufman DM. Clinical Neurology for Psychiatrists. Philadelphia: WB Saunders; 2001.
17. Ramirez C, de Seze J, Delrieu O, et al. [Myasthenia gravis and pregnancy: clinical course and management of delivery and the postpartum phase.] Rev Neurol (Paris) 2006;162:330-8 (French).
18. Weitzner MA, Kanfer S, Booth-Jones M. Apathy and pituitary disease: it has nothing to do with depression. J Neuropsychiatry Clin Neurosci 2005;17:159-66.
19. Peselow E. Other pharmacological and biological therapies. In: Sadock BJ, Sadock VA, eds. Comprehensive textbook of psychiatry, 8th ed. Philadelphia: Lippincott Williams & Wilkins; 2005.
20. Kasper DL, Braunwald E, Fauci A, et al. Harrison’s principles of internal medicine, 16th ed. New York: McGraw-Hill; 2004.
21. Ohba K, Omagari K, Kusakari C, et al. Flare-up of autoimmune hepatitis after delivery in a patient with primary biliary irrhosis: postpartum overlap syndrome of primary biliary cirrhosis and autoimmune hepatitis. Dig Dis Sci 2005;50:201-6.
Our mission: To meet your needs
Many of you know me from my published research and books, national presentations, or CME broadcasts. What you might not know is how much I enjoy the clinical practice of psychiatry and helping seriously ill patients regain their wellness and return to social and vocational functioning.
Over the years, my patients have been my best teachers about disease burden, effective treatment strategies, coping with illness, and recovery. I never cease to be amazed at how vital a strong physician-patient alliance is to achieving successful clinical outcomes. My patients also have been my best source of research questions about the causes and treatment of psychiatric brain disorders. The more I learn, the more I am humbled by how much remains to be discovered in our field.
The mission of clinically focused journals such as Current Psychiatry is to inform readers about practical applications of the latest medical advances. That’s why many of you read Current Psychiatry from cover to cover. Psychiatry is a vibrant, rapidly growing medical specialty with a solid neuroscience foundation and a promising future, but we face many unmet needs in daily practice. Most relate to the diagnosis and treatment of common psychiatric disorders.
Needed: Diagnostic clarity
The schema of psychiatric diagnosis needs to become anchored in scientific evidence, for example. DSMIV-TR’s clusters of inclusion and exclusion symptom-focused criteria are unsatisfactory and not validated by empiric biomedical findings. Many patients have multiple comorbidities, which raises questions about:
- which illness is primary or secondary
- what is the shared neurobiology
- why does a class of drugs approved for one diagnosis—such as selective serotonin reuptake inhibitors, atypical antipsychotics, or anticonvulsants—help many other symptoms or diagnoses?
When it comes to personality disorders, why do axis II symptoms emerge during an acute axis I onset and disappear when the axis I episode remits? Why, on the other hand, do the features of “real” axis II disorders endure for a lifetime? Why the distinction between axis I and II anyway?
Why do addictive disorders such as substance abuse plague so many of our patients? Why do symptoms such as insomnia, anxiety, dysphoria, or aggression occur in disparate psychiatric disorders such as anxiety, mood, psychotic, or personality disorders?
Every day these nosologic questions challenge us and influence our management decisions and prognostic formulations. Much research is needed because specific, effective treatment requires diagnostic clarity and validity.
Needed: More effective treatments
This brings us to a perennial unmet need: more effective treatments. Psychiatry is experiencing a relative drought of innovative biological and psychological treatments. Pharmacologic agents are approved for a limited number of DSM-IV-TR diagnoses, which has created an extensive “black market psychopharmacology.” Approved psychotropics are used for unapproved indications, in unapproved doses, and in unapproved combinations—particularly in child and adolescent psychiatry with its serious dearth of controlled studies. We urgently need:
- nondopaminergic approaches to schizophrenia (where so many clues point to glutamate pathways) that can effectively treat cognitive deficits and negative symptoms
- antidepressants with rapid onset of action, especially for patients with suicidal intent
- an unambiguously effective mood stabilizer which, as monotherapy, restores balance to all phases of bipolar disorder
- effective treatment for alcoholism and drug dependencies.
We also await breakthrough medications for brain disorders with no known treatments, such as antisocial behavior, hypochondriasis, autism, pedophilia, and dissociative disorders, to name a few. Finally, we need drugs designed to avoid metabolic complications and other serious illnesses that can disrupt quality of life and lead to premature death.
Needed: Medical care for the mentally ill
Speaking of early mortality—and its association with chronic psychosis, mood, and anxiety disorders—another unmet need is the integration of primary care and psychiatric care for public-sector patients whose nonpsychiatric medical needs are woefully neglected. The mental health system is widely described as “broken,” and to fix it we need to advocate tirelessly for a more rational system of comprehensive medical care for our seriously mentally ill patients.
We also need to advocate for parity in reimbursement for treating psychiatric brain disorders, to eliminate the stigma of having an emotional or behavioral ailment, and to end the shameful incarceration of the criminally mentally ill in lieu of proper and humane treatment in a medical setting.
What do you need?
Current Psychiatry will continue to bring you the latest advances regarding the causes and treatments of psychiatric illness. Please contact me by:
- e-mail ([email protected])
- or fax (201-391-2778) about your major unmet clinical needs. Besides publishing your letters, I pledge that we will listen to you and meet your needs. That, ultimately, is Current Psychiatry’s mission.
I look forward to hearing from you.

Many of you know me from my published research and books, national presentations, or CME broadcasts. What you might not know is how much I enjoy the clinical practice of psychiatry and helping seriously ill patients regain their wellness and return to social and vocational functioning.
Over the years, my patients have been my best teachers about disease burden, effective treatment strategies, coping with illness, and recovery. I never cease to be amazed at how vital a strong physician-patient alliance is to achieving successful clinical outcomes. My patients also have been my best source of research questions about the causes and treatment of psychiatric brain disorders. The more I learn, the more I am humbled by how much remains to be discovered in our field.
The mission of clinically focused journals such as Current Psychiatry is to inform readers about practical applications of the latest medical advances. That’s why many of you read Current Psychiatry from cover to cover. Psychiatry is a vibrant, rapidly growing medical specialty with a solid neuroscience foundation and a promising future, but we face many unmet needs in daily practice. Most relate to the diagnosis and treatment of common psychiatric disorders.
Needed: Diagnostic clarity
The schema of psychiatric diagnosis needs to become anchored in scientific evidence, for example. DSMIV-TR’s clusters of inclusion and exclusion symptom-focused criteria are unsatisfactory and not validated by empiric biomedical findings. Many patients have multiple comorbidities, which raises questions about:
- which illness is primary or secondary
- what is the shared neurobiology
- why does a class of drugs approved for one diagnosis—such as selective serotonin reuptake inhibitors, atypical antipsychotics, or anticonvulsants—help many other symptoms or diagnoses?
When it comes to personality disorders, why do axis II symptoms emerge during an acute axis I onset and disappear when the axis I episode remits? Why, on the other hand, do the features of “real” axis II disorders endure for a lifetime? Why the distinction between axis I and II anyway?
Why do addictive disorders such as substance abuse plague so many of our patients? Why do symptoms such as insomnia, anxiety, dysphoria, or aggression occur in disparate psychiatric disorders such as anxiety, mood, psychotic, or personality disorders?
Every day these nosologic questions challenge us and influence our management decisions and prognostic formulations. Much research is needed because specific, effective treatment requires diagnostic clarity and validity.
Needed: More effective treatments
This brings us to a perennial unmet need: more effective treatments. Psychiatry is experiencing a relative drought of innovative biological and psychological treatments. Pharmacologic agents are approved for a limited number of DSM-IV-TR diagnoses, which has created an extensive “black market psychopharmacology.” Approved psychotropics are used for unapproved indications, in unapproved doses, and in unapproved combinations—particularly in child and adolescent psychiatry with its serious dearth of controlled studies. We urgently need:
- nondopaminergic approaches to schizophrenia (where so many clues point to glutamate pathways) that can effectively treat cognitive deficits and negative symptoms
- antidepressants with rapid onset of action, especially for patients with suicidal intent
- an unambiguously effective mood stabilizer which, as monotherapy, restores balance to all phases of bipolar disorder
- effective treatment for alcoholism and drug dependencies.
We also await breakthrough medications for brain disorders with no known treatments, such as antisocial behavior, hypochondriasis, autism, pedophilia, and dissociative disorders, to name a few. Finally, we need drugs designed to avoid metabolic complications and other serious illnesses that can disrupt quality of life and lead to premature death.
Needed: Medical care for the mentally ill
Speaking of early mortality—and its association with chronic psychosis, mood, and anxiety disorders—another unmet need is the integration of primary care and psychiatric care for public-sector patients whose nonpsychiatric medical needs are woefully neglected. The mental health system is widely described as “broken,” and to fix it we need to advocate tirelessly for a more rational system of comprehensive medical care for our seriously mentally ill patients.
We also need to advocate for parity in reimbursement for treating psychiatric brain disorders, to eliminate the stigma of having an emotional or behavioral ailment, and to end the shameful incarceration of the criminally mentally ill in lieu of proper and humane treatment in a medical setting.
What do you need?
Current Psychiatry will continue to bring you the latest advances regarding the causes and treatments of psychiatric illness. Please contact me by:
- e-mail ([email protected])
- or fax (201-391-2778) about your major unmet clinical needs. Besides publishing your letters, I pledge that we will listen to you and meet your needs. That, ultimately, is Current Psychiatry’s mission.
I look forward to hearing from you.
Many of you know me from my published research and books, national presentations, or CME broadcasts. What you might not know is how much I enjoy the clinical practice of psychiatry and helping seriously ill patients regain their wellness and return to social and vocational functioning.
Over the years, my patients have been my best teachers about disease burden, effective treatment strategies, coping with illness, and recovery. I never cease to be amazed at how vital a strong physician-patient alliance is to achieving successful clinical outcomes. My patients also have been my best source of research questions about the causes and treatment of psychiatric brain disorders. The more I learn, the more I am humbled by how much remains to be discovered in our field.
The mission of clinically focused journals such as Current Psychiatry is to inform readers about practical applications of the latest medical advances. That’s why many of you read Current Psychiatry from cover to cover. Psychiatry is a vibrant, rapidly growing medical specialty with a solid neuroscience foundation and a promising future, but we face many unmet needs in daily practice. Most relate to the diagnosis and treatment of common psychiatric disorders.
Needed: Diagnostic clarity
The schema of psychiatric diagnosis needs to become anchored in scientific evidence, for example. DSMIV-TR’s clusters of inclusion and exclusion symptom-focused criteria are unsatisfactory and not validated by empiric biomedical findings. Many patients have multiple comorbidities, which raises questions about:
- which illness is primary or secondary
- what is the shared neurobiology
- why does a class of drugs approved for one diagnosis—such as selective serotonin reuptake inhibitors, atypical antipsychotics, or anticonvulsants—help many other symptoms or diagnoses?
When it comes to personality disorders, why do axis II symptoms emerge during an acute axis I onset and disappear when the axis I episode remits? Why, on the other hand, do the features of “real” axis II disorders endure for a lifetime? Why the distinction between axis I and II anyway?
Why do addictive disorders such as substance abuse plague so many of our patients? Why do symptoms such as insomnia, anxiety, dysphoria, or aggression occur in disparate psychiatric disorders such as anxiety, mood, psychotic, or personality disorders?
Every day these nosologic questions challenge us and influence our management decisions and prognostic formulations. Much research is needed because specific, effective treatment requires diagnostic clarity and validity.
Needed: More effective treatments
This brings us to a perennial unmet need: more effective treatments. Psychiatry is experiencing a relative drought of innovative biological and psychological treatments. Pharmacologic agents are approved for a limited number of DSM-IV-TR diagnoses, which has created an extensive “black market psychopharmacology.” Approved psychotropics are used for unapproved indications, in unapproved doses, and in unapproved combinations—particularly in child and adolescent psychiatry with its serious dearth of controlled studies. We urgently need:
- nondopaminergic approaches to schizophrenia (where so many clues point to glutamate pathways) that can effectively treat cognitive deficits and negative symptoms
- antidepressants with rapid onset of action, especially for patients with suicidal intent
- an unambiguously effective mood stabilizer which, as monotherapy, restores balance to all phases of bipolar disorder
- effective treatment for alcoholism and drug dependencies.
We also await breakthrough medications for brain disorders with no known treatments, such as antisocial behavior, hypochondriasis, autism, pedophilia, and dissociative disorders, to name a few. Finally, we need drugs designed to avoid metabolic complications and other serious illnesses that can disrupt quality of life and lead to premature death.
Needed: Medical care for the mentally ill
Speaking of early mortality—and its association with chronic psychosis, mood, and anxiety disorders—another unmet need is the integration of primary care and psychiatric care for public-sector patients whose nonpsychiatric medical needs are woefully neglected. The mental health system is widely described as “broken,” and to fix it we need to advocate tirelessly for a more rational system of comprehensive medical care for our seriously mentally ill patients.
We also need to advocate for parity in reimbursement for treating psychiatric brain disorders, to eliminate the stigma of having an emotional or behavioral ailment, and to end the shameful incarceration of the criminally mentally ill in lieu of proper and humane treatment in a medical setting.
What do you need?
Current Psychiatry will continue to bring you the latest advances regarding the causes and treatments of psychiatric illness. Please contact me by:
- e-mail ([email protected])
- or fax (201-391-2778) about your major unmet clinical needs. Besides publishing your letters, I pledge that we will listen to you and meet your needs. That, ultimately, is Current Psychiatry’s mission.
I look forward to hearing from you.