User login
Reassuring findings on neurodevelopmental outcomes in HIV-exposed children
DURBAN, SOUTH AFRICA – Children exposed to HIV in utero but uninfected at birth have neurodevelopmental test scores at age 24 months that are comparable with those of unexposed children, based on a study conducted in Botswana and presented by Jean Leidner at the 21st International AIDS Conference.
“These results provide reassurance regarding the potential effects of in-utero HIV and antiretroviral exposure,” declared Ms. Leidner, CEO of Goodtables Data Consulting in Norman, Okla., and the Botswana Harvard AIDS Institute Partnership.

The two groups of children had virtually identical scores on the cognitive, gross motor, fine motor, expressive language, and receptive language domains measured in the Bayley-III. The same was true for scores on the fine motor, locomotor, language, and personal-social elements of the Developmental Milestone Checklist.
The two groups of children differed in other ways; 17% of the uninfected children exposed to HIV in utero and 8% of the controls were low birth weight. The HIV-exposed children are being raised in a more challenging environment: just 49% have electricity in the home, compared with 64% of control families. Moreover, 53% of the HIV-exposed children and 33% of the controls live under conditions of moderate-to-severe food uncertainty.
Only 8% of the HIV-infected mothers breastfed, whereas breastfeeding was universal among the control group.
More than 99% of the HIV-infected mothers took antiretroviral medication antenatally. Roughly two-thirds were on zidovudine (Retrovir) monotherapy, the rest on a three-drug regimen of nevirapine (Viramune) plus lamivudine/zidovudine (Combivir). These are older antiretrovirals. Additional neurodevelopmental studies are warranted in children with in-utero exposure to newer agents, as well as in older children, Ms. Leidner said.
She reported having no financial conflicts regarding this study, which was funded by the National Institute of Mental Health.
DURBAN, SOUTH AFRICA – Children exposed to HIV in utero but uninfected at birth have neurodevelopmental test scores at age 24 months that are comparable with those of unexposed children, based on a study conducted in Botswana and presented by Jean Leidner at the 21st International AIDS Conference.
“These results provide reassurance regarding the potential effects of in-utero HIV and antiretroviral exposure,” declared Ms. Leidner, CEO of Goodtables Data Consulting in Norman, Okla., and the Botswana Harvard AIDS Institute Partnership.
The two groups of children had virtually identical scores on the cognitive, gross motor, fine motor, expressive language, and receptive language domains measured in the Bayley-III. The same was true for scores on the fine motor, locomotor, language, and personal-social elements of the Developmental Milestone Checklist.
The two groups of children differed in other ways; 17% of the uninfected children exposed to HIV in utero and 8% of the controls were low birth weight. The HIV-exposed children are being raised in a more challenging environment: just 49% have electricity in the home, compared with 64% of control families. Moreover, 53% of the HIV-exposed children and 33% of the controls live under conditions of moderate-to-severe food uncertainty.
Only 8% of the HIV-infected mothers breastfed, whereas breastfeeding was universal among the control group.
More than 99% of the HIV-infected mothers took antiretroviral medication antenatally. Roughly two-thirds were on zidovudine (Retrovir) monotherapy, the rest on a three-drug regimen of nevirapine (Viramune) plus lamivudine/zidovudine (Combivir). These are older antiretrovirals. Additional neurodevelopmental studies are warranted in children with in-utero exposure to newer agents, as well as in older children, Ms. Leidner said.
She reported having no financial conflicts regarding this study, which was funded by the National Institute of Mental Health.
DURBAN, SOUTH AFRICA – Children exposed to HIV in utero but uninfected at birth have neurodevelopmental test scores at age 24 months that are comparable with those of unexposed children, based on a study conducted in Botswana and presented by Jean Leidner at the 21st International AIDS Conference.
“These results provide reassurance regarding the potential effects of in-utero HIV and antiretroviral exposure,” declared Ms. Leidner, CEO of Goodtables Data Consulting in Norman, Okla., and the Botswana Harvard AIDS Institute Partnership.
The two groups of children had virtually identical scores on the cognitive, gross motor, fine motor, expressive language, and receptive language domains measured in the Bayley-III. The same was true for scores on the fine motor, locomotor, language, and personal-social elements of the Developmental Milestone Checklist.
The two groups of children differed in other ways; 17% of the uninfected children exposed to HIV in utero and 8% of the controls were low birth weight. The HIV-exposed children are being raised in a more challenging environment: just 49% have electricity in the home, compared with 64% of control families. Moreover, 53% of the HIV-exposed children and 33% of the controls live under conditions of moderate-to-severe food uncertainty.
Only 8% of the HIV-infected mothers breastfed, whereas breastfeeding was universal among the control group.
More than 99% of the HIV-infected mothers took antiretroviral medication antenatally. Roughly two-thirds were on zidovudine (Retrovir) monotherapy, the rest on a three-drug regimen of nevirapine (Viramune) plus lamivudine/zidovudine (Combivir). These are older antiretrovirals. Additional neurodevelopmental studies are warranted in children with in-utero exposure to newer agents, as well as in older children, Ms. Leidner said.
She reported having no financial conflicts regarding this study, which was funded by the National Institute of Mental Health.
Key clinical point:
Major finding: In-utero exposure to maternal HIV and antiretroviral drugs had no measurable adverse neurodevelopmental effects at age 24 months in uninfected children.
Data source: 337 uninfected children exposed to HIV in-utero and 387 children unexposed to HIV in utero.
Disclosures: The National Institute of Mental Health funded the study. The presenter reported having no financial conflicts of interest.
Prospects brighten for an HIV vaccine
DURBAN, SOUTH AFRICA – A new optimism regarding the possibility of creating a safe and effective HIV preventive vaccine was very much in evidence at the 21st International AIDS Conference.
“The HIV vaccine field is open for business,” an exuberant Larry Corey, MD, declared in a plenary address highlighting recent major progress in HIV vaccine development.
Three extremely important HIV vaccine efficacy clinical trials testing diverse promising strategies are now either in progress or soon to start, noted Dr. Corey, professor of laboratory medicine and medicine at the University of Washington and emeritus director of the Fred Hutchinson Cancer Research Center in Seattle.

“We are finally moving the needle forward with human efficacy trials that are commensurate with the need for developing an HIV vaccine,” Dr. Corey said.
He emphasized that HIV is “still the world’s most pressing global health issue,” with more than 45,000 new infections occurring annually in the United States and more than 2 million annually worldwide. And while numerous nonvaccine prevention methods have been developed, they share a major limitation: Their extended effectiveness requires continuous adherence.
“With asymptomatic acquisition, prolonged subclinical infection, and sexual transmission, getting to an AIDS-free generation will require a vaccine,” Dr. Corey predicted.
After years of discouragingly negative HIV vaccine studies, researchers finally turned a corner in 2009 with the reported results of the U.S. Military HIV Research Program–led RV144 trial, commonly known as the Thai Trial, Anthony S. Fauci, MD, recalled in an interview.
The trial, which randomized more than 16,000 young adult Thais, showed a modest 31% efficacy at 3.5 years, but a more substantial and encouraging 60% efficacy at 12 months (N Engl J Med. 2009 Dec 3;361[23]:2209-20). More importantly, the Thai trial opened up a whole new avenue to HIV vaccine development.

“We thought the Thai vaccine would induce neutralizing antibodies, but it didn’t. Instead, it induced nonneutralizing antibodies against a component of the V1V2 loop region of the HIV envelope, which was associated with protection. So the good news about that study was that even though that vaccine wasn’t efficacious enough to make it a usable vaccine, it gave us something we could improve upon by using the same platform and enhancing the response to provide greater depth, breadth, and durability,” explained Dr. Fauci, director of the National Institute of Allergy and Infectious Diseases (NIAID).
The improved vaccine consists of a canarypox-based vaccine called ALVAC-HIV and a bivalent gp120 protein subunit vaccine with MF59, a different adjuvant from that used in the Thai trial, in an effort to achieve a more robust immune response. Also, the four-injection series studied in the Thai trial is now bolstered by a booster injection at the 12-month mark. This vaccine has been altered to be specific to HIV clade C, the predominant HIV subtype in southern Africa, where the bulk of new HIV infections occur.
At the AIDS 2016 conference, Linda-Gail Bekker, MD, presented the primary immunogenicity results from the HIV Vaccine Trials Network (HVTN) 100 trial, a phase 1/2, double-blind, placebo-controlled study of the improved version of the Thai trial vaccine, known as the Clade C ALVAC-(vCP2438) and bivalent subtype C gp120/MF59 vaccine, in 252 HIV-uninfected South African adults.

The ALVAC/protein vaccine achieved cellular and humoral immune responses that exceeded all four predetermined criteria as correlates of protection. As a result, a pivotal phase III, randomized, double-blind, placebo-controlled vaccine efficacy trial known as HVTN 702 got the green light to begin in November 2016 in 5,400 HIV-negative adults in South Africa. Participants will be assessed at 24 and 36 months of follow-up, announced Dr. Bekker, cochair of HVTN 702, International AIDS Society president-elect, and deputy director of the Desmond Tutu HIV Center in Cape Town, South Africa.
As a principal investigator in the NIAID-supported HIV Vaccine Trials Network, Dr. Corey has been deeply involved in the development of this vaccine. He also is chair of the ongoing HVTN 703 and 704 phase IIb trials, testing an entirely different vaccine approach. The hypothesis being tested in HVTN 703 and 704 is that a passively infused monoclonal antibody can protect against HIV infection in 2,400 men who have sex with men and transgender men in North and South America, as well as in 1,500 women in sub-Saharan Africa. Both studies began in spring 2016.
“Every card-carrying virologist feels this should work,” according to Dr. Corey.
The rationale for having two study populations is that investigators suspect the effects of the antibody may vary depending upon whether the route of HIV acquisition is rectal or vaginal, he explained.
The monoclonal antibody contains VRC01, which effectively blocks viral binding to CD4 cells. Study participants will receive an intravenous infusion of VRC01 at 10 or 30 mg/kg or placebo every 2 months. If the results are positive and a second-generation product and delivery system can be developed, antibody-mediated prevention could also have a major potential role in interrupting maternal to child transmission of HIV resulting from intrapartum exposure or breastfeeding.
Dr. Corey also highlighted a third strategy of HIV vaccine development, one at an earlier stage. Investigators at Johnson & Johnson, in collaboration with the NIAID, HVTN, and other partners, are pursuing a multi-clade approach, one designed to protect against all clades of HIV found around the world. This strategy entails first giving an adenovirus serotype 26–vectored vaccine to prime the immune system, following up with administration of several boosters containing mosaic inserts to increase the response. This vaccine is in phase I studies with no results yet.
Dr. Fauci is not sure which if any of these three approaches will yield a safe and effective vaccine for HIV prevention.
“It’s important to realize that this is a very difficult scientific challenge,” he said. “The body does not readily make an adequate immune response against HIV, unlike virtually any other viral infection. Even the serious ones that cause a degree of morbidity and mortality – smallpox, measles, rubella, polio – ultimately the body does make a good immune response and allows us to clear the virus and leaves us with protection against subsequent exposure to the same virus. We don’t have that advantage with HIV. So it’s going to be difficult to get a safe and effective HIV vaccine, but I think the scientific challenge is worth going after and there’s a reasonable chance we might get there.”
Dr. Corey, Dr. Fauci, and Dr. Bekker reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – A new optimism regarding the possibility of creating a safe and effective HIV preventive vaccine was very much in evidence at the 21st International AIDS Conference.
“The HIV vaccine field is open for business,” an exuberant Larry Corey, MD, declared in a plenary address highlighting recent major progress in HIV vaccine development.
Three extremely important HIV vaccine efficacy clinical trials testing diverse promising strategies are now either in progress or soon to start, noted Dr. Corey, professor of laboratory medicine and medicine at the University of Washington and emeritus director of the Fred Hutchinson Cancer Research Center in Seattle.
“We are finally moving the needle forward with human efficacy trials that are commensurate with the need for developing an HIV vaccine,” Dr. Corey said.
He emphasized that HIV is “still the world’s most pressing global health issue,” with more than 45,000 new infections occurring annually in the United States and more than 2 million annually worldwide. And while numerous nonvaccine prevention methods have been developed, they share a major limitation: Their extended effectiveness requires continuous adherence.
“With asymptomatic acquisition, prolonged subclinical infection, and sexual transmission, getting to an AIDS-free generation will require a vaccine,” Dr. Corey predicted.
After years of discouragingly negative HIV vaccine studies, researchers finally turned a corner in 2009 with the reported results of the U.S. Military HIV Research Program–led RV144 trial, commonly known as the Thai Trial, Anthony S. Fauci, MD, recalled in an interview.
The trial, which randomized more than 16,000 young adult Thais, showed a modest 31% efficacy at 3.5 years, but a more substantial and encouraging 60% efficacy at 12 months (N Engl J Med. 2009 Dec 3;361[23]:2209-20). More importantly, the Thai trial opened up a whole new avenue to HIV vaccine development.
“We thought the Thai vaccine would induce neutralizing antibodies, but it didn’t. Instead, it induced nonneutralizing antibodies against a component of the V1V2 loop region of the HIV envelope, which was associated with protection. So the good news about that study was that even though that vaccine wasn’t efficacious enough to make it a usable vaccine, it gave us something we could improve upon by using the same platform and enhancing the response to provide greater depth, breadth, and durability,” explained Dr. Fauci, director of the National Institute of Allergy and Infectious Diseases (NIAID).
The improved vaccine consists of a canarypox-based vaccine called ALVAC-HIV and a bivalent gp120 protein subunit vaccine with MF59, a different adjuvant from that used in the Thai trial, in an effort to achieve a more robust immune response. Also, the four-injection series studied in the Thai trial is now bolstered by a booster injection at the 12-month mark. This vaccine has been altered to be specific to HIV clade C, the predominant HIV subtype in southern Africa, where the bulk of new HIV infections occur.
At the AIDS 2016 conference, Linda-Gail Bekker, MD, presented the primary immunogenicity results from the HIV Vaccine Trials Network (HVTN) 100 trial, a phase 1/2, double-blind, placebo-controlled study of the improved version of the Thai trial vaccine, known as the Clade C ALVAC-(vCP2438) and bivalent subtype C gp120/MF59 vaccine, in 252 HIV-uninfected South African adults.
The ALVAC/protein vaccine achieved cellular and humoral immune responses that exceeded all four predetermined criteria as correlates of protection. As a result, a pivotal phase III, randomized, double-blind, placebo-controlled vaccine efficacy trial known as HVTN 702 got the green light to begin in November 2016 in 5,400 HIV-negative adults in South Africa. Participants will be assessed at 24 and 36 months of follow-up, announced Dr. Bekker, cochair of HVTN 702, International AIDS Society president-elect, and deputy director of the Desmond Tutu HIV Center in Cape Town, South Africa.
As a principal investigator in the NIAID-supported HIV Vaccine Trials Network, Dr. Corey has been deeply involved in the development of this vaccine. He also is chair of the ongoing HVTN 703 and 704 phase IIb trials, testing an entirely different vaccine approach. The hypothesis being tested in HVTN 703 and 704 is that a passively infused monoclonal antibody can protect against HIV infection in 2,400 men who have sex with men and transgender men in North and South America, as well as in 1,500 women in sub-Saharan Africa. Both studies began in spring 2016.
“Every card-carrying virologist feels this should work,” according to Dr. Corey.
The rationale for having two study populations is that investigators suspect the effects of the antibody may vary depending upon whether the route of HIV acquisition is rectal or vaginal, he explained.
The monoclonal antibody contains VRC01, which effectively blocks viral binding to CD4 cells. Study participants will receive an intravenous infusion of VRC01 at 10 or 30 mg/kg or placebo every 2 months. If the results are positive and a second-generation product and delivery system can be developed, antibody-mediated prevention could also have a major potential role in interrupting maternal to child transmission of HIV resulting from intrapartum exposure or breastfeeding.
Dr. Corey also highlighted a third strategy of HIV vaccine development, one at an earlier stage. Investigators at Johnson & Johnson, in collaboration with the NIAID, HVTN, and other partners, are pursuing a multi-clade approach, one designed to protect against all clades of HIV found around the world. This strategy entails first giving an adenovirus serotype 26–vectored vaccine to prime the immune system, following up with administration of several boosters containing mosaic inserts to increase the response. This vaccine is in phase I studies with no results yet.
Dr. Fauci is not sure which if any of these three approaches will yield a safe and effective vaccine for HIV prevention.
“It’s important to realize that this is a very difficult scientific challenge,” he said. “The body does not readily make an adequate immune response against HIV, unlike virtually any other viral infection. Even the serious ones that cause a degree of morbidity and mortality – smallpox, measles, rubella, polio – ultimately the body does make a good immune response and allows us to clear the virus and leaves us with protection against subsequent exposure to the same virus. We don’t have that advantage with HIV. So it’s going to be difficult to get a safe and effective HIV vaccine, but I think the scientific challenge is worth going after and there’s a reasonable chance we might get there.”
Dr. Corey, Dr. Fauci, and Dr. Bekker reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – A new optimism regarding the possibility of creating a safe and effective HIV preventive vaccine was very much in evidence at the 21st International AIDS Conference.
“The HIV vaccine field is open for business,” an exuberant Larry Corey, MD, declared in a plenary address highlighting recent major progress in HIV vaccine development.
Three extremely important HIV vaccine efficacy clinical trials testing diverse promising strategies are now either in progress or soon to start, noted Dr. Corey, professor of laboratory medicine and medicine at the University of Washington and emeritus director of the Fred Hutchinson Cancer Research Center in Seattle.
“We are finally moving the needle forward with human efficacy trials that are commensurate with the need for developing an HIV vaccine,” Dr. Corey said.
He emphasized that HIV is “still the world’s most pressing global health issue,” with more than 45,000 new infections occurring annually in the United States and more than 2 million annually worldwide. And while numerous nonvaccine prevention methods have been developed, they share a major limitation: Their extended effectiveness requires continuous adherence.
“With asymptomatic acquisition, prolonged subclinical infection, and sexual transmission, getting to an AIDS-free generation will require a vaccine,” Dr. Corey predicted.
After years of discouragingly negative HIV vaccine studies, researchers finally turned a corner in 2009 with the reported results of the U.S. Military HIV Research Program–led RV144 trial, commonly known as the Thai Trial, Anthony S. Fauci, MD, recalled in an interview.
The trial, which randomized more than 16,000 young adult Thais, showed a modest 31% efficacy at 3.5 years, but a more substantial and encouraging 60% efficacy at 12 months (N Engl J Med. 2009 Dec 3;361[23]:2209-20). More importantly, the Thai trial opened up a whole new avenue to HIV vaccine development.
“We thought the Thai vaccine would induce neutralizing antibodies, but it didn’t. Instead, it induced nonneutralizing antibodies against a component of the V1V2 loop region of the HIV envelope, which was associated with protection. So the good news about that study was that even though that vaccine wasn’t efficacious enough to make it a usable vaccine, it gave us something we could improve upon by using the same platform and enhancing the response to provide greater depth, breadth, and durability,” explained Dr. Fauci, director of the National Institute of Allergy and Infectious Diseases (NIAID).
The improved vaccine consists of a canarypox-based vaccine called ALVAC-HIV and a bivalent gp120 protein subunit vaccine with MF59, a different adjuvant from that used in the Thai trial, in an effort to achieve a more robust immune response. Also, the four-injection series studied in the Thai trial is now bolstered by a booster injection at the 12-month mark. This vaccine has been altered to be specific to HIV clade C, the predominant HIV subtype in southern Africa, where the bulk of new HIV infections occur.
At the AIDS 2016 conference, Linda-Gail Bekker, MD, presented the primary immunogenicity results from the HIV Vaccine Trials Network (HVTN) 100 trial, a phase 1/2, double-blind, placebo-controlled study of the improved version of the Thai trial vaccine, known as the Clade C ALVAC-(vCP2438) and bivalent subtype C gp120/MF59 vaccine, in 252 HIV-uninfected South African adults.
The ALVAC/protein vaccine achieved cellular and humoral immune responses that exceeded all four predetermined criteria as correlates of protection. As a result, a pivotal phase III, randomized, double-blind, placebo-controlled vaccine efficacy trial known as HVTN 702 got the green light to begin in November 2016 in 5,400 HIV-negative adults in South Africa. Participants will be assessed at 24 and 36 months of follow-up, announced Dr. Bekker, cochair of HVTN 702, International AIDS Society president-elect, and deputy director of the Desmond Tutu HIV Center in Cape Town, South Africa.
As a principal investigator in the NIAID-supported HIV Vaccine Trials Network, Dr. Corey has been deeply involved in the development of this vaccine. He also is chair of the ongoing HVTN 703 and 704 phase IIb trials, testing an entirely different vaccine approach. The hypothesis being tested in HVTN 703 and 704 is that a passively infused monoclonal antibody can protect against HIV infection in 2,400 men who have sex with men and transgender men in North and South America, as well as in 1,500 women in sub-Saharan Africa. Both studies began in spring 2016.
“Every card-carrying virologist feels this should work,” according to Dr. Corey.
The rationale for having two study populations is that investigators suspect the effects of the antibody may vary depending upon whether the route of HIV acquisition is rectal or vaginal, he explained.
The monoclonal antibody contains VRC01, which effectively blocks viral binding to CD4 cells. Study participants will receive an intravenous infusion of VRC01 at 10 or 30 mg/kg or placebo every 2 months. If the results are positive and a second-generation product and delivery system can be developed, antibody-mediated prevention could also have a major potential role in interrupting maternal to child transmission of HIV resulting from intrapartum exposure or breastfeeding.
Dr. Corey also highlighted a third strategy of HIV vaccine development, one at an earlier stage. Investigators at Johnson & Johnson, in collaboration with the NIAID, HVTN, and other partners, are pursuing a multi-clade approach, one designed to protect against all clades of HIV found around the world. This strategy entails first giving an adenovirus serotype 26–vectored vaccine to prime the immune system, following up with administration of several boosters containing mosaic inserts to increase the response. This vaccine is in phase I studies with no results yet.
Dr. Fauci is not sure which if any of these three approaches will yield a safe and effective vaccine for HIV prevention.
“It’s important to realize that this is a very difficult scientific challenge,” he said. “The body does not readily make an adequate immune response against HIV, unlike virtually any other viral infection. Even the serious ones that cause a degree of morbidity and mortality – smallpox, measles, rubella, polio – ultimately the body does make a good immune response and allows us to clear the virus and leaves us with protection against subsequent exposure to the same virus. We don’t have that advantage with HIV. So it’s going to be difficult to get a safe and effective HIV vaccine, but I think the scientific challenge is worth going after and there’s a reasonable chance we might get there.”
Dr. Corey, Dr. Fauci, and Dr. Bekker reported having no financial conflicts of interest.
EXPERT ANALYSIS FROM AIDS 2016

ARIA trial: Triumeq proves a winner in HIV-infected women
DURBAN, SOUTH AFRICA – A novel single-tablet, triple-antiretroviral combination regimen outperformed the well-established combination of ritonavir-boosted atazanavir plus tenofovir/emtricitabine in the ARIA trial, a phase IIIb study conducted in treatment-naive women with HIV infection.
The fixed-dose once-daily combination of dolutegravir 50 mg/abacavir 600 mg/lamivudine 300 mg, marketed in the United States as Triumeq since its approval in 2014, showed superior efficacy and a more favorable safety profile than did ritonavir-boosted atazanavir (Reyataz) plus tenofovir/emtricitabine (Truvada), Catherine Orrell, MD, reported at the 21st International AIDS Conference.

ARIA was a 48-week open-label randomized trial involving 495 treatment-naive HIV-infected women in the United States and 11 other countries. The primary endpoint – a plasma HIV RNA viral load below 50 copies/mL after 48 weeks of treatment – was achieved in 82% of patients on dolutegravir/abacavir/lamivudine (DTG/ABC/3TC), compared with 71% on ritonavir-boosted atazanavir plus tenofovir/emtricitabine (ATV+RTV+FTC/TDF).
Of note, the difference in efficacy was even more pronounced among women with a baseline viral load in excess of 100,000 copies/mL: an 80% success rate in attaining a viral load of less than 50 copies/mL at 48 weeks in the DTG/ABC/3TC group, compared with 64% in the comparator arm. Women with this baseline massive viral load comprised 28% of study participants, added Dr. Orrell of the University of Cape Town, South Africa.
To be eligible for the study, women had to be negative for an HLA-B*5701 genetic screen for allergic hypersensitivity to abacavir.
A particularly attractive feature of DTG/ABC/3TC is that dolutegravir, an unboosted integrin strand transfer inhibitor, provides a high barrier to development of drug resistance. Indeed, no subjects in the dolutegravir arm developed treatment-emergent primary integrin strand transfer inhibitor or abacavir/lamivudine resistance mutations, according to Dr. Orrell.
The superior efficacy of the single-tablet regimen was driven in part by fewer discontinuations due to adverse events: a 4% rate versus 7% in the comparator arm. Another key factor was the substantially lower virologic failure rate in DTG/ABC/3TC-treated women: 6% versus 14% in the comparator arm.
The single-tablet regimen also had a better safety profile. The combined rate of the most common drug-related adverse events – nausea, diarrhea, headache, and jaundice – was 22% in the DTG/ABC/3TC group compared with 38% with ATV+RTV+FTC/TDF.

The incidence of treatment-emergent psychiatric events – insomnia, anxiety, depression, or suicidal ideation – was roughly 14% in each treatment arm. That’s an important finding because some other studies have found an increase in psychiatric events in patients receiving integrin strand transfer inhibitors.
“The overall results, I think, are important for the field,” Kimberly Smith, MD, said at a press conference highlighting the ARIA trial.
“Women are often underrepresented in HIV clinical trials even though they bear much of the burden of the HIV epidemic,” added Dr. Smith, vice president for global medical strategy and head of research and development at ViiV Healthcare in Research Triangle Park, N.C.
“This fixed-dose combination is a winner,” said Salim Abdool Karim, MD, director of the Center for the AIDS Program of Research in South Africa, Durban, who chaired the press conference.
Dr. Orrell received a research grant from ViiV Healthcare, which sponsored the ARIA study.
DURBAN, SOUTH AFRICA – A novel single-tablet, triple-antiretroviral combination regimen outperformed the well-established combination of ritonavir-boosted atazanavir plus tenofovir/emtricitabine in the ARIA trial, a phase IIIb study conducted in treatment-naive women with HIV infection.
The fixed-dose once-daily combination of dolutegravir 50 mg/abacavir 600 mg/lamivudine 300 mg, marketed in the United States as Triumeq since its approval in 2014, showed superior efficacy and a more favorable safety profile than did ritonavir-boosted atazanavir (Reyataz) plus tenofovir/emtricitabine (Truvada), Catherine Orrell, MD, reported at the 21st International AIDS Conference.
ARIA was a 48-week open-label randomized trial involving 495 treatment-naive HIV-infected women in the United States and 11 other countries. The primary endpoint – a plasma HIV RNA viral load below 50 copies/mL after 48 weeks of treatment – was achieved in 82% of patients on dolutegravir/abacavir/lamivudine (DTG/ABC/3TC), compared with 71% on ritonavir-boosted atazanavir plus tenofovir/emtricitabine (ATV+RTV+FTC/TDF).
Of note, the difference in efficacy was even more pronounced among women with a baseline viral load in excess of 100,000 copies/mL: an 80% success rate in attaining a viral load of less than 50 copies/mL at 48 weeks in the DTG/ABC/3TC group, compared with 64% in the comparator arm. Women with this baseline massive viral load comprised 28% of study participants, added Dr. Orrell of the University of Cape Town, South Africa.
To be eligible for the study, women had to be negative for an HLA-B*5701 genetic screen for allergic hypersensitivity to abacavir.
A particularly attractive feature of DTG/ABC/3TC is that dolutegravir, an unboosted integrin strand transfer inhibitor, provides a high barrier to development of drug resistance. Indeed, no subjects in the dolutegravir arm developed treatment-emergent primary integrin strand transfer inhibitor or abacavir/lamivudine resistance mutations, according to Dr. Orrell.
The superior efficacy of the single-tablet regimen was driven in part by fewer discontinuations due to adverse events: a 4% rate versus 7% in the comparator arm. Another key factor was the substantially lower virologic failure rate in DTG/ABC/3TC-treated women: 6% versus 14% in the comparator arm.
The single-tablet regimen also had a better safety profile. The combined rate of the most common drug-related adverse events – nausea, diarrhea, headache, and jaundice – was 22% in the DTG/ABC/3TC group compared with 38% with ATV+RTV+FTC/TDF.
The incidence of treatment-emergent psychiatric events – insomnia, anxiety, depression, or suicidal ideation – was roughly 14% in each treatment arm. That’s an important finding because some other studies have found an increase in psychiatric events in patients receiving integrin strand transfer inhibitors.
“The overall results, I think, are important for the field,” Kimberly Smith, MD, said at a press conference highlighting the ARIA trial.
“Women are often underrepresented in HIV clinical trials even though they bear much of the burden of the HIV epidemic,” added Dr. Smith, vice president for global medical strategy and head of research and development at ViiV Healthcare in Research Triangle Park, N.C.
“This fixed-dose combination is a winner,” said Salim Abdool Karim, MD, director of the Center for the AIDS Program of Research in South Africa, Durban, who chaired the press conference.
Dr. Orrell received a research grant from ViiV Healthcare, which sponsored the ARIA study.
DURBAN, SOUTH AFRICA – A novel single-tablet, triple-antiretroviral combination regimen outperformed the well-established combination of ritonavir-boosted atazanavir plus tenofovir/emtricitabine in the ARIA trial, a phase IIIb study conducted in treatment-naive women with HIV infection.
The fixed-dose once-daily combination of dolutegravir 50 mg/abacavir 600 mg/lamivudine 300 mg, marketed in the United States as Triumeq since its approval in 2014, showed superior efficacy and a more favorable safety profile than did ritonavir-boosted atazanavir (Reyataz) plus tenofovir/emtricitabine (Truvada), Catherine Orrell, MD, reported at the 21st International AIDS Conference.
ARIA was a 48-week open-label randomized trial involving 495 treatment-naive HIV-infected women in the United States and 11 other countries. The primary endpoint – a plasma HIV RNA viral load below 50 copies/mL after 48 weeks of treatment – was achieved in 82% of patients on dolutegravir/abacavir/lamivudine (DTG/ABC/3TC), compared with 71% on ritonavir-boosted atazanavir plus tenofovir/emtricitabine (ATV+RTV+FTC/TDF).
Of note, the difference in efficacy was even more pronounced among women with a baseline viral load in excess of 100,000 copies/mL: an 80% success rate in attaining a viral load of less than 50 copies/mL at 48 weeks in the DTG/ABC/3TC group, compared with 64% in the comparator arm. Women with this baseline massive viral load comprised 28% of study participants, added Dr. Orrell of the University of Cape Town, South Africa.
To be eligible for the study, women had to be negative for an HLA-B*5701 genetic screen for allergic hypersensitivity to abacavir.
A particularly attractive feature of DTG/ABC/3TC is that dolutegravir, an unboosted integrin strand transfer inhibitor, provides a high barrier to development of drug resistance. Indeed, no subjects in the dolutegravir arm developed treatment-emergent primary integrin strand transfer inhibitor or abacavir/lamivudine resistance mutations, according to Dr. Orrell.
The superior efficacy of the single-tablet regimen was driven in part by fewer discontinuations due to adverse events: a 4% rate versus 7% in the comparator arm. Another key factor was the substantially lower virologic failure rate in DTG/ABC/3TC-treated women: 6% versus 14% in the comparator arm.
The single-tablet regimen also had a better safety profile. The combined rate of the most common drug-related adverse events – nausea, diarrhea, headache, and jaundice – was 22% in the DTG/ABC/3TC group compared with 38% with ATV+RTV+FTC/TDF.
The incidence of treatment-emergent psychiatric events – insomnia, anxiety, depression, or suicidal ideation – was roughly 14% in each treatment arm. That’s an important finding because some other studies have found an increase in psychiatric events in patients receiving integrin strand transfer inhibitors.
“The overall results, I think, are important for the field,” Kimberly Smith, MD, said at a press conference highlighting the ARIA trial.
“Women are often underrepresented in HIV clinical trials even though they bear much of the burden of the HIV epidemic,” added Dr. Smith, vice president for global medical strategy and head of research and development at ViiV Healthcare in Research Triangle Park, N.C.
“This fixed-dose combination is a winner,” said Salim Abdool Karim, MD, director of the Center for the AIDS Program of Research in South Africa, Durban, who chaired the press conference.
Dr. Orrell received a research grant from ViiV Healthcare, which sponsored the ARIA study.
AT AIDS 2016
Key clinical point: The once-daily fixed-dose combination of dolutegravir/abacavir/lamivudine is significantly more effective and has a better safety profile than does ritonavir-boosted atazanavir plus tenofovir/emtricitabine in treatment-naive women with HIV infection.
Major finding: Eighty-two percent of women assigned to dolutegravir/abacavir/lamivudine had a plasma HIV RNA viral load below 50 copies/mL after 48 weeks of treatment, compared with 71% on ritonavir-boosted atazanavir plus tenofovir/emtricitabine.
Data source: A phase IIIb open-label randomized trial of 495 HIV-infected women.
Disclosures: The presenter received a research grant from ViiV Healthcare, which sponsored the study.

Oral HIV PrEP also protects against herpes
DURBAN, SOUTH AFRICA – Oral antiretroviral pre-exposure prophylaxis (PrEP) against HIV also reduces the risk of acquiring herpes simplex virus type 2, according to research presented at the 21st International AIDS Conference.
“Given the limited interventions for primary prevention of HSV-2, efficacy against HSV-2 provides additional benefit to oral PrEP,” observed Connie Celum, MD, professor of global health and medicine at the University of Washington, Seattle.

HSV-2 is, however, the only non-HIV sexually transmitted infection whose incidence is reduced by PrEP with emtricitabine/tenofovir (Truvada), the sole approved agent for oral HIV PrEP, she added.
Dr. Celum was lead author in a report from the landmark Partners PrEP study of HIV serodiscordant heterosexual Kenyan and Ugandan couples, which demonstrated that oral PrEP provided a 33% reduction in the risk of HSV-2 infection in participants with a known HSV-2-positive partner (Ann Intern Med. 2014 Jul 1;161[1]:11-9. doi: 10.7326/M13-2471).
Tenofovir has been shown to have in vitro activity against HSV-2, providing biologic plausibility to the Partners PrEP study finding, but the 90% effective concentration of the drug required to achieve strong anti-HSV-2 activity in the laboratory makes it likely that good adherence to daily oral PrEP is necessary to see the clinical benefit in terms of reduced HSV-2 acquisition, Dr. Celum said.
She also addressed other questions about the interaction between PrEP and non-HIV STIs she often receives from physicians who provide care for HIV-infected or at-risk patients.
Do STIs reduce the efficacy of oral PrEP? “My answer would be no,” Dr. Celum said. She noted that no difference in PrEP efficacy was seen between patients with and without STIs in Partners PrEP or the French IPERGAY study.
Does PrEP increase the rate of other STIs? The concern here has been that PrEP’s beneficial effect in reducing the risk of HIV infection could be counteracted by a compensatory increase in unsafe sexual practices among PrEP users, with a resultant increase in other STIs. That didn’t occur, however, in the recently reported UK PROUD randomized study of 544 men who have sex with men (MSM), which showed no increase in other STIs with the addition of daily oral PrEP (Lancet. 2016 Jan 2;387[10013]:53-60. doi: 10.1016/S0140-6736[15]00056-2).
Are STIs useful in selecting patients for PrEP by serving as a marker of increased risk for HIV? The answer is a strong yes for MSM. The iPrEX study documented that the number of MSM and transgender women who needed to be treated with PrEP for 1 year to prevent one additional case of HIV infection dropped from 62 for the group as a whole to 36 for those self-reporting condomless receptive anal intercourse and 41 for those with another STI (Lancet Infect Dis. 2014 Jun;14[6]:468-75. doi: 10.1016/S1473-3099[14]70025-8). To cast a wide net for potential beneficiaries, most PrEP programs targeting MSM offer PrEP to those with other STIs or who report engaging in condomless anal sex, Dr. Celum said.
Can PrEP programs reduce STIs through engagement in care? “I think there’s a great opportunity here,” she said. “Most programs are rolling out PrEP with quarterly visits for refills. And I think particularly in MSM and probably in young women, STI testing at those visits provides an opportunity for earlier diagnosis and treatment of STIs as well as for partner notification.”
Dr. Celum reported having no financial conflicts regarding her presentation.
DURBAN, SOUTH AFRICA – Oral antiretroviral pre-exposure prophylaxis (PrEP) against HIV also reduces the risk of acquiring herpes simplex virus type 2, according to research presented at the 21st International AIDS Conference.
“Given the limited interventions for primary prevention of HSV-2, efficacy against HSV-2 provides additional benefit to oral PrEP,” observed Connie Celum, MD, professor of global health and medicine at the University of Washington, Seattle.
HSV-2 is, however, the only non-HIV sexually transmitted infection whose incidence is reduced by PrEP with emtricitabine/tenofovir (Truvada), the sole approved agent for oral HIV PrEP, she added.
Dr. Celum was lead author in a report from the landmark Partners PrEP study of HIV serodiscordant heterosexual Kenyan and Ugandan couples, which demonstrated that oral PrEP provided a 33% reduction in the risk of HSV-2 infection in participants with a known HSV-2-positive partner (Ann Intern Med. 2014 Jul 1;161[1]:11-9. doi: 10.7326/M13-2471).
Tenofovir has been shown to have in vitro activity against HSV-2, providing biologic plausibility to the Partners PrEP study finding, but the 90% effective concentration of the drug required to achieve strong anti-HSV-2 activity in the laboratory makes it likely that good adherence to daily oral PrEP is necessary to see the clinical benefit in terms of reduced HSV-2 acquisition, Dr. Celum said.
She also addressed other questions about the interaction between PrEP and non-HIV STIs she often receives from physicians who provide care for HIV-infected or at-risk patients.
Do STIs reduce the efficacy of oral PrEP? “My answer would be no,” Dr. Celum said. She noted that no difference in PrEP efficacy was seen between patients with and without STIs in Partners PrEP or the French IPERGAY study.
Does PrEP increase the rate of other STIs? The concern here has been that PrEP’s beneficial effect in reducing the risk of HIV infection could be counteracted by a compensatory increase in unsafe sexual practices among PrEP users, with a resultant increase in other STIs. That didn’t occur, however, in the recently reported UK PROUD randomized study of 544 men who have sex with men (MSM), which showed no increase in other STIs with the addition of daily oral PrEP (Lancet. 2016 Jan 2;387[10013]:53-60. doi: 10.1016/S0140-6736[15]00056-2).
Are STIs useful in selecting patients for PrEP by serving as a marker of increased risk for HIV? The answer is a strong yes for MSM. The iPrEX study documented that the number of MSM and transgender women who needed to be treated with PrEP for 1 year to prevent one additional case of HIV infection dropped from 62 for the group as a whole to 36 for those self-reporting condomless receptive anal intercourse and 41 for those with another STI (Lancet Infect Dis. 2014 Jun;14[6]:468-75. doi: 10.1016/S1473-3099[14]70025-8). To cast a wide net for potential beneficiaries, most PrEP programs targeting MSM offer PrEP to those with other STIs or who report engaging in condomless anal sex, Dr. Celum said.
Can PrEP programs reduce STIs through engagement in care? “I think there’s a great opportunity here,” she said. “Most programs are rolling out PrEP with quarterly visits for refills. And I think particularly in MSM and probably in young women, STI testing at those visits provides an opportunity for earlier diagnosis and treatment of STIs as well as for partner notification.”
Dr. Celum reported having no financial conflicts regarding her presentation.
DURBAN, SOUTH AFRICA – Oral antiretroviral pre-exposure prophylaxis (PrEP) against HIV also reduces the risk of acquiring herpes simplex virus type 2, according to research presented at the 21st International AIDS Conference.
“Given the limited interventions for primary prevention of HSV-2, efficacy against HSV-2 provides additional benefit to oral PrEP,” observed Connie Celum, MD, professor of global health and medicine at the University of Washington, Seattle.
HSV-2 is, however, the only non-HIV sexually transmitted infection whose incidence is reduced by PrEP with emtricitabine/tenofovir (Truvada), the sole approved agent for oral HIV PrEP, she added.
Dr. Celum was lead author in a report from the landmark Partners PrEP study of HIV serodiscordant heterosexual Kenyan and Ugandan couples, which demonstrated that oral PrEP provided a 33% reduction in the risk of HSV-2 infection in participants with a known HSV-2-positive partner (Ann Intern Med. 2014 Jul 1;161[1]:11-9. doi: 10.7326/M13-2471).
Tenofovir has been shown to have in vitro activity against HSV-2, providing biologic plausibility to the Partners PrEP study finding, but the 90% effective concentration of the drug required to achieve strong anti-HSV-2 activity in the laboratory makes it likely that good adherence to daily oral PrEP is necessary to see the clinical benefit in terms of reduced HSV-2 acquisition, Dr. Celum said.
She also addressed other questions about the interaction between PrEP and non-HIV STIs she often receives from physicians who provide care for HIV-infected or at-risk patients.
Do STIs reduce the efficacy of oral PrEP? “My answer would be no,” Dr. Celum said. She noted that no difference in PrEP efficacy was seen between patients with and without STIs in Partners PrEP or the French IPERGAY study.
Does PrEP increase the rate of other STIs? The concern here has been that PrEP’s beneficial effect in reducing the risk of HIV infection could be counteracted by a compensatory increase in unsafe sexual practices among PrEP users, with a resultant increase in other STIs. That didn’t occur, however, in the recently reported UK PROUD randomized study of 544 men who have sex with men (MSM), which showed no increase in other STIs with the addition of daily oral PrEP (Lancet. 2016 Jan 2;387[10013]:53-60. doi: 10.1016/S0140-6736[15]00056-2).
Are STIs useful in selecting patients for PrEP by serving as a marker of increased risk for HIV? The answer is a strong yes for MSM. The iPrEX study documented that the number of MSM and transgender women who needed to be treated with PrEP for 1 year to prevent one additional case of HIV infection dropped from 62 for the group as a whole to 36 for those self-reporting condomless receptive anal intercourse and 41 for those with another STI (Lancet Infect Dis. 2014 Jun;14[6]:468-75. doi: 10.1016/S1473-3099[14]70025-8). To cast a wide net for potential beneficiaries, most PrEP programs targeting MSM offer PrEP to those with other STIs or who report engaging in condomless anal sex, Dr. Celum said.
Can PrEP programs reduce STIs through engagement in care? “I think there’s a great opportunity here,” she said. “Most programs are rolling out PrEP with quarterly visits for refills. And I think particularly in MSM and probably in young women, STI testing at those visits provides an opportunity for earlier diagnosis and treatment of STIs as well as for partner notification.”
Dr. Celum reported having no financial conflicts regarding her presentation.
EXPERT ANALYSIS FROM AIDS 2016

Postpartum HIV treatment reduces key maternal illnesses
DURBAN, SOUTH AFRICA – HIV-infected women who remained on antiretroviral therapy throughout the postpartum period reduced their risk of clinical stage 2 or 3 HIV disease events by 53%, compared with those who stopped treatment postpartum in the PROMISE 1077HS trial, Judith Currier, MD, reported at the 21st International AIDS Conference.
PROMISE (Promoting Maternal and Infant Survival Everywhere) is an ongoing multinational, multicomponent series of clinical trials. PROMISE 1077HS was designed to assess the risks and benefits of continued antiretroviral therapy (ART), compared with stopping therapy among nonbreastfeeding women postpartum, explained Dr. Currier, professor of medicine and chief of infectious diseases at the University of California, Los Angeles.

PROMISE 1077HS included 1,653 HIV-positive women in the United States and seven low- or middle-income countries. All participants were relatively healthy as evidenced by their median CD4+ count of 550 cells/mm3 prior to starting ART in pregnancy. None were planning to breastfeed. Upon delivery, the women were randomized to continue ART – the chief regimen was ritonavir-boosted lopinavir (Kaletra) plus tenofovir/emtricitabine (Truvada) – or stop therapy, restarting only when their CD4+ count fell below 350 cells/mm3.
Participants were prospectively followed for a median of 2.3 years post delivery. At that point, in summer 2015, the results of the landmark START trial were released (N Engl J Med. 2015 Aug 27;373[9]:795-807), paving the way for the current global strategy of ART for life in HIV-infected patients regardless of their CD4+ cell count.
The primary efficacy endpoint in PROMISE 1077HS was a composite of an AIDS event, death, or a serious cardiovascular, renal, or hepatic event. In this relatively healthy population, too few of these events occurred to allow the researchers to draw conclusions (four in the continued-ART group and six in the controls who stopped ART post partum).
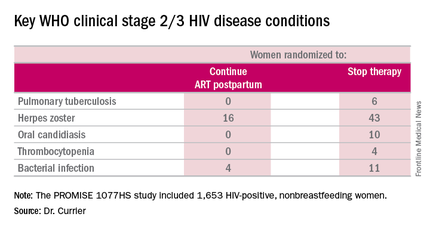
But the secondary endpoint of time to World Health Organization (WHO) clinical stage 2 or 3 HIV disease events was a different story. A total of 39 of these events occurred in the continued-ART group, for a rate of 2.02% per year, compared with 80 events and a rate of 4.36% per year in controls. That difference translated into a 53% relative risk reduction. Some key WHO clinical stage 2 and 3 HIV disease events include pulmonary tuberculosis, herpes zoster, oral candidiasis, thrombocytopenia, and bacterial infection.
Fully 23% of patients in the continued-ART group had laboratory-confirmed virologic failure as defined by a viral load of HIV RNA in excess of 1,000 copies/mL after at least 24 weeks of postpartum ART. Additionally, resistance testing indicated two-thirds of affected patients showed no evidence of resistance at the time of virologic failure, meaning their viremia was due to nonadherence to ART.
“This virologic failure rate highlights the importance of the challenge of adherence in this population,” Dr. Currier said. “Interventions to improve adherence as well as studies to examine newer regimens with a high genetic barrier to resistance are needed to ensure maximal long-term benefit from this strategy of continued ART postpartum.”
The PROMISE studies are funded by the National Institutes of Health. Dr. Currier reported having no financial disclosures.
DURBAN, SOUTH AFRICA – HIV-infected women who remained on antiretroviral therapy throughout the postpartum period reduced their risk of clinical stage 2 or 3 HIV disease events by 53%, compared with those who stopped treatment postpartum in the PROMISE 1077HS trial, Judith Currier, MD, reported at the 21st International AIDS Conference.
PROMISE (Promoting Maternal and Infant Survival Everywhere) is an ongoing multinational, multicomponent series of clinical trials. PROMISE 1077HS was designed to assess the risks and benefits of continued antiretroviral therapy (ART), compared with stopping therapy among nonbreastfeeding women postpartum, explained Dr. Currier, professor of medicine and chief of infectious diseases at the University of California, Los Angeles.
PROMISE 1077HS included 1,653 HIV-positive women in the United States and seven low- or middle-income countries. All participants were relatively healthy as evidenced by their median CD4+ count of 550 cells/mm3 prior to starting ART in pregnancy. None were planning to breastfeed. Upon delivery, the women were randomized to continue ART – the chief regimen was ritonavir-boosted lopinavir (Kaletra) plus tenofovir/emtricitabine (Truvada) – or stop therapy, restarting only when their CD4+ count fell below 350 cells/mm3.
Participants were prospectively followed for a median of 2.3 years post delivery. At that point, in summer 2015, the results of the landmark START trial were released (N Engl J Med. 2015 Aug 27;373[9]:795-807), paving the way for the current global strategy of ART for life in HIV-infected patients regardless of their CD4+ cell count.
The primary efficacy endpoint in PROMISE 1077HS was a composite of an AIDS event, death, or a serious cardiovascular, renal, or hepatic event. In this relatively healthy population, too few of these events occurred to allow the researchers to draw conclusions (four in the continued-ART group and six in the controls who stopped ART post partum).
But the secondary endpoint of time to World Health Organization (WHO) clinical stage 2 or 3 HIV disease events was a different story. A total of 39 of these events occurred in the continued-ART group, for a rate of 2.02% per year, compared with 80 events and a rate of 4.36% per year in controls. That difference translated into a 53% relative risk reduction. Some key WHO clinical stage 2 and 3 HIV disease events include pulmonary tuberculosis, herpes zoster, oral candidiasis, thrombocytopenia, and bacterial infection.
Fully 23% of patients in the continued-ART group had laboratory-confirmed virologic failure as defined by a viral load of HIV RNA in excess of 1,000 copies/mL after at least 24 weeks of postpartum ART. Additionally, resistance testing indicated two-thirds of affected patients showed no evidence of resistance at the time of virologic failure, meaning their viremia was due to nonadherence to ART.
“This virologic failure rate highlights the importance of the challenge of adherence in this population,” Dr. Currier said. “Interventions to improve adherence as well as studies to examine newer regimens with a high genetic barrier to resistance are needed to ensure maximal long-term benefit from this strategy of continued ART postpartum.”
The PROMISE studies are funded by the National Institutes of Health. Dr. Currier reported having no financial disclosures.
DURBAN, SOUTH AFRICA – HIV-infected women who remained on antiretroviral therapy throughout the postpartum period reduced their risk of clinical stage 2 or 3 HIV disease events by 53%, compared with those who stopped treatment postpartum in the PROMISE 1077HS trial, Judith Currier, MD, reported at the 21st International AIDS Conference.
PROMISE (Promoting Maternal and Infant Survival Everywhere) is an ongoing multinational, multicomponent series of clinical trials. PROMISE 1077HS was designed to assess the risks and benefits of continued antiretroviral therapy (ART), compared with stopping therapy among nonbreastfeeding women postpartum, explained Dr. Currier, professor of medicine and chief of infectious diseases at the University of California, Los Angeles.
PROMISE 1077HS included 1,653 HIV-positive women in the United States and seven low- or middle-income countries. All participants were relatively healthy as evidenced by their median CD4+ count of 550 cells/mm3 prior to starting ART in pregnancy. None were planning to breastfeed. Upon delivery, the women were randomized to continue ART – the chief regimen was ritonavir-boosted lopinavir (Kaletra) plus tenofovir/emtricitabine (Truvada) – or stop therapy, restarting only when their CD4+ count fell below 350 cells/mm3.
Participants were prospectively followed for a median of 2.3 years post delivery. At that point, in summer 2015, the results of the landmark START trial were released (N Engl J Med. 2015 Aug 27;373[9]:795-807), paving the way for the current global strategy of ART for life in HIV-infected patients regardless of their CD4+ cell count.
The primary efficacy endpoint in PROMISE 1077HS was a composite of an AIDS event, death, or a serious cardiovascular, renal, or hepatic event. In this relatively healthy population, too few of these events occurred to allow the researchers to draw conclusions (four in the continued-ART group and six in the controls who stopped ART post partum).
But the secondary endpoint of time to World Health Organization (WHO) clinical stage 2 or 3 HIV disease events was a different story. A total of 39 of these events occurred in the continued-ART group, for a rate of 2.02% per year, compared with 80 events and a rate of 4.36% per year in controls. That difference translated into a 53% relative risk reduction. Some key WHO clinical stage 2 and 3 HIV disease events include pulmonary tuberculosis, herpes zoster, oral candidiasis, thrombocytopenia, and bacterial infection.
Fully 23% of patients in the continued-ART group had laboratory-confirmed virologic failure as defined by a viral load of HIV RNA in excess of 1,000 copies/mL after at least 24 weeks of postpartum ART. Additionally, resistance testing indicated two-thirds of affected patients showed no evidence of resistance at the time of virologic failure, meaning their viremia was due to nonadherence to ART.
“This virologic failure rate highlights the importance of the challenge of adherence in this population,” Dr. Currier said. “Interventions to improve adherence as well as studies to examine newer regimens with a high genetic barrier to resistance are needed to ensure maximal long-term benefit from this strategy of continued ART postpartum.”
The PROMISE studies are funded by the National Institutes of Health. Dr. Currier reported having no financial disclosures.
AT AIDS 2016
Key clinical point: Women with HIV should continue antiretroviral therapy post partum.
Major finding: HIV-infected women who continued antiretroviral therapy post partum experienced 53% fewer WHO clinical stage 2 or 3 HIV disease events than women assigned to stop therapy after delivery.
Data source: The PROMISE 1077HS study included 1,653 HIV-positive, nonbreastfeeding women randomized to either continue or stop antiretroviral therapy post partum.
Disclosures: The PROMISE studies are funded by the National Institutes of Health. Dr. Currier reported having no financial disclosures.

Is stem-cell transplant curative for HIV infection?
DURBAN, SOUTH AFRICA – The 15 HIV-infected patients who have undergone allogeneic stem-cell transplant for life-threatening hematologic cancers under the auspices of the European EpiStem Consortium have uniformly demonstrated a profound and durable reduction in viral reservoir to a degree that hasn’t been approached by any other investigational cure strategy, Annemarie Wensing, MD, said at the 21st International AIDS Conference.
“We see an enormous reduction in the viral reservoir, and in two patients we cannot find any viable HIV in the blood using ultrasensitive tests. But we don’t know whether these patients are cured because they are still on antiretroviral therapy,” said Dr. Wensing of Utrecht (The Netherlands) University.

Non-Hodgkin’s lymphoma and Hodgkin’s lymphoma are 7-9 times more frequent in HIV-positive patients than in the general population. But allogeneic stem cell transplantation is an even higher-risk treatment in HIV-positive patients with life-threatening leukemia or lymphoma than in the HIV-negative population. Only 6 of the 15 EuroStem patients remain alive. Eight died within 4 months of the procedure and another died 2.5 years post-transplant, all from progression of their cancer or as a result of opportunistic infections arising during the immunosuppressive chemoablation that’s central to stem-cell transplantation. However, 3 of the 15 patients have survived longer than 3 years. In two of them, no HIV can be detected in blood or intestinal tissue using ultrasensitive tests, while in the third there is “only a slight trace,” according to Dr. Wensing, a clinical virologist.
EpiStem (the European Project to Guide and Investigate the Potential for HIV Cure by Stem-Cell Transplantation) is a multinational collaboration of European oncologists, infectious disease physicians, and other specialists. It was formed in response to the successful outcome of allogeneic stem cell transplantation for acute myeloid leukemia in HIV-positive Timothy Brown, more famously known as “the Berlin patient” (N Engl J Med. 2009 Feb 12;360(7):692-8). He has thus far survived 7 years off antiretroviral therapy.
Much has been made of the fact that Mr. Brown’s donor cells were homozygous for the CCR5 delta32 mutation, which confers natural resistance to HIV infection because it prevents the virus from infecting T cells. Only 1% or less of the population is homozygous for this mutation. But Dr. Wensing isn’t convinced that using donor cells with the mutation is a prerequisite for success. Indeed, while 4 of the 15 EpiStem patients received stem cells from donors homozygous for the mutation and another got donor cells heterozygous for the CCR5 delta32 mutation, the other 10 received stem cells capable of being infected by HIV – yet all 15 experienced an enormous reduction in their viral reservoir. And two of the three patients who have survived longer than 3 years got stem cells without the CCR5 delta32 mutation.
Dr. Wensing observed that a common denominator shared by Timothy Brown and the two EpiStem patients who have trace or undetectable HIV in blood or tissue samples more than 3 years post-transplant is that all three developed severe graft-versus-host disease in conjunction with their stem cell transplantation. She suspects this may have helped them to clear the infection, a hypothesis she intends to pursue further as EpiStem gathers more patients.
Eventually, if patients continue to test negative for HIV using ultrasensitive tests, it will be time to have a discussion with patients and their treating physicians as to whether they should continue on antiretroviral therapy.
“In the end it’s the patients’ decision, but they should be very well counseled because it can have medical and also psychological consequences if HIV returns,” she said.
EpiStem is funded by the American Foundation for AIDS Research Conssortium on HIV Eradication. Dr. Wensing reported having no financial conflicts regarding her presentation.
DURBAN, SOUTH AFRICA – The 15 HIV-infected patients who have undergone allogeneic stem-cell transplant for life-threatening hematologic cancers under the auspices of the European EpiStem Consortium have uniformly demonstrated a profound and durable reduction in viral reservoir to a degree that hasn’t been approached by any other investigational cure strategy, Annemarie Wensing, MD, said at the 21st International AIDS Conference.
“We see an enormous reduction in the viral reservoir, and in two patients we cannot find any viable HIV in the blood using ultrasensitive tests. But we don’t know whether these patients are cured because they are still on antiretroviral therapy,” said Dr. Wensing of Utrecht (The Netherlands) University.
Non-Hodgkin’s lymphoma and Hodgkin’s lymphoma are 7-9 times more frequent in HIV-positive patients than in the general population. But allogeneic stem cell transplantation is an even higher-risk treatment in HIV-positive patients with life-threatening leukemia or lymphoma than in the HIV-negative population. Only 6 of the 15 EuroStem patients remain alive. Eight died within 4 months of the procedure and another died 2.5 years post-transplant, all from progression of their cancer or as a result of opportunistic infections arising during the immunosuppressive chemoablation that’s central to stem-cell transplantation. However, 3 of the 15 patients have survived longer than 3 years. In two of them, no HIV can be detected in blood or intestinal tissue using ultrasensitive tests, while in the third there is “only a slight trace,” according to Dr. Wensing, a clinical virologist.
EpiStem (the European Project to Guide and Investigate the Potential for HIV Cure by Stem-Cell Transplantation) is a multinational collaboration of European oncologists, infectious disease physicians, and other specialists. It was formed in response to the successful outcome of allogeneic stem cell transplantation for acute myeloid leukemia in HIV-positive Timothy Brown, more famously known as “the Berlin patient” (N Engl J Med. 2009 Feb 12;360(7):692-8). He has thus far survived 7 years off antiretroviral therapy.
Much has been made of the fact that Mr. Brown’s donor cells were homozygous for the CCR5 delta32 mutation, which confers natural resistance to HIV infection because it prevents the virus from infecting T cells. Only 1% or less of the population is homozygous for this mutation. But Dr. Wensing isn’t convinced that using donor cells with the mutation is a prerequisite for success. Indeed, while 4 of the 15 EpiStem patients received stem cells from donors homozygous for the mutation and another got donor cells heterozygous for the CCR5 delta32 mutation, the other 10 received stem cells capable of being infected by HIV – yet all 15 experienced an enormous reduction in their viral reservoir. And two of the three patients who have survived longer than 3 years got stem cells without the CCR5 delta32 mutation.
Dr. Wensing observed that a common denominator shared by Timothy Brown and the two EpiStem patients who have trace or undetectable HIV in blood or tissue samples more than 3 years post-transplant is that all three developed severe graft-versus-host disease in conjunction with their stem cell transplantation. She suspects this may have helped them to clear the infection, a hypothesis she intends to pursue further as EpiStem gathers more patients.
Eventually, if patients continue to test negative for HIV using ultrasensitive tests, it will be time to have a discussion with patients and their treating physicians as to whether they should continue on antiretroviral therapy.
“In the end it’s the patients’ decision, but they should be very well counseled because it can have medical and also psychological consequences if HIV returns,” she said.
EpiStem is funded by the American Foundation for AIDS Research Conssortium on HIV Eradication. Dr. Wensing reported having no financial conflicts regarding her presentation.
DURBAN, SOUTH AFRICA – The 15 HIV-infected patients who have undergone allogeneic stem-cell transplant for life-threatening hematologic cancers under the auspices of the European EpiStem Consortium have uniformly demonstrated a profound and durable reduction in viral reservoir to a degree that hasn’t been approached by any other investigational cure strategy, Annemarie Wensing, MD, said at the 21st International AIDS Conference.
“We see an enormous reduction in the viral reservoir, and in two patients we cannot find any viable HIV in the blood using ultrasensitive tests. But we don’t know whether these patients are cured because they are still on antiretroviral therapy,” said Dr. Wensing of Utrecht (The Netherlands) University.
Non-Hodgkin’s lymphoma and Hodgkin’s lymphoma are 7-9 times more frequent in HIV-positive patients than in the general population. But allogeneic stem cell transplantation is an even higher-risk treatment in HIV-positive patients with life-threatening leukemia or lymphoma than in the HIV-negative population. Only 6 of the 15 EuroStem patients remain alive. Eight died within 4 months of the procedure and another died 2.5 years post-transplant, all from progression of their cancer or as a result of opportunistic infections arising during the immunosuppressive chemoablation that’s central to stem-cell transplantation. However, 3 of the 15 patients have survived longer than 3 years. In two of them, no HIV can be detected in blood or intestinal tissue using ultrasensitive tests, while in the third there is “only a slight trace,” according to Dr. Wensing, a clinical virologist.
EpiStem (the European Project to Guide and Investigate the Potential for HIV Cure by Stem-Cell Transplantation) is a multinational collaboration of European oncologists, infectious disease physicians, and other specialists. It was formed in response to the successful outcome of allogeneic stem cell transplantation for acute myeloid leukemia in HIV-positive Timothy Brown, more famously known as “the Berlin patient” (N Engl J Med. 2009 Feb 12;360(7):692-8). He has thus far survived 7 years off antiretroviral therapy.
Much has been made of the fact that Mr. Brown’s donor cells were homozygous for the CCR5 delta32 mutation, which confers natural resistance to HIV infection because it prevents the virus from infecting T cells. Only 1% or less of the population is homozygous for this mutation. But Dr. Wensing isn’t convinced that using donor cells with the mutation is a prerequisite for success. Indeed, while 4 of the 15 EpiStem patients received stem cells from donors homozygous for the mutation and another got donor cells heterozygous for the CCR5 delta32 mutation, the other 10 received stem cells capable of being infected by HIV – yet all 15 experienced an enormous reduction in their viral reservoir. And two of the three patients who have survived longer than 3 years got stem cells without the CCR5 delta32 mutation.
Dr. Wensing observed that a common denominator shared by Timothy Brown and the two EpiStem patients who have trace or undetectable HIV in blood or tissue samples more than 3 years post-transplant is that all three developed severe graft-versus-host disease in conjunction with their stem cell transplantation. She suspects this may have helped them to clear the infection, a hypothesis she intends to pursue further as EpiStem gathers more patients.
Eventually, if patients continue to test negative for HIV using ultrasensitive tests, it will be time to have a discussion with patients and their treating physicians as to whether they should continue on antiretroviral therapy.
“In the end it’s the patients’ decision, but they should be very well counseled because it can have medical and also psychological consequences if HIV returns,” she said.
EpiStem is funded by the American Foundation for AIDS Research Conssortium on HIV Eradication. Dr. Wensing reported having no financial conflicts regarding her presentation.
AT AIDS 2016
Key clinical point: It doesn’t appear to be necessary to use donor stem cells that are homozygous for the CCR5 delta32 mutation to achieve enormous sustained reductions in the viral reservoir in HIV-infected patients undergoing allogeneic stem cell transplantation for hematologic cancers.
Major finding: Two of three patients in a European series who have survived for longer than 3 years after stem-cell transplantation with undetectable or only trace HIV in their blood received donor cells lacking the rare CCR5 delta32 mutation.
Data source: EpiStem is an ongoing observational study of HIV-infected patients who undergo allogeneic stem cell transplantation for life-threatening hematologic cancers.
Disclosures: The EpiStem project is funded by the American Foundation for AIDS Research Conssortium on HIV Eradication. The presenter reported having no financial conflicts regarding her presentation.

Maraviroc shows potential for HIV PrEP in women
DURBAN, SOUTH AFRICA – Maraviroc-containing regimens for daily oral pre-exposure prophylaxis in women at risk for HIV infection showed good safety and tolerability in a phase 2 study, the first randomized trial of PrEP ever conducted in U.S. women, Roy M. Gulick, MD, reported at the 21st International AIDS Conference.
No new HIV infections occurred in the 188 women who participated in the 48-week, randomized, double-blind, placebo-controlled study known as the HPTN 069/ACTG A5305 trial.

However, the results shouldn’t be taken as evidence of efficacy. The relatively low 2% incidence of new STIs diagnosed during 48 weeks of close followup – three cases of chlamydia, one of gonorrhea – suggests that the study population probably wasn’t at high risk for acquiring HIV. Further, the study wasn’t powered to determine efficacy. That determination will have to await a larger phase 3 trial, observed Dr. Gulick, professor of medicine at Cornell University in New York.
Maraviroc (Selzentry) is categorized as an HIV entry inhibitor. It’s an antagonist of the CCR5 receptor found on the surface of T cells, which is the route of HIV infection. The rationale for exploring the drug for HIV PrEP, according to Dr. Gulick, is that it concentrates in both the genital tract and rectum, doesn’t select for drug-resistant viral strains, is well tolerated, and it isn’t commonly used for treatment of HIV infection.
Additional options for oral daily HIV PrEP are clearly desirable, he added. The only approved agent is Truvada (tenofovir/emtricitabine), which is often used in HIV therapy as well, and there is concern it may select for drug resistance. Plus, it has renal, GI, and bone side effects.
Study participants were HIV-negative adult women who were born female and considered at risk for HIV acquisition because of a history of condomless vaginal or anal intercourse with at least one HIV-positive or unknown status man within the previous 90 days. The women were randomized to one of four study arms: maraviroc at the standard dose of 300 mg/day plus two placebo pills; maraviroc plus emtricitabine at the standard dose of 200 mg/day plus one placebo pill; maraviroc plus tenofovir at 300 mg/day plus a placebo pill, or a control regimen of fixed-dose Truvada (tenofovir 300 mg/emtricitabine 200 mg) plus two placebo pills. Thus, everyone took three pills once daily.
The three-pill regimen might help explain the less than stellar patient adherence. Study drugs were detectable – and not necessarily at therapeutic levels – in the plasma of 65% of subjects at 24 weeks and 60% at 48 weeks, with no differences between the study arms.
Maraviroc alone was associated with fewer grade 2-4 adverse events than the other regimens.
There were 11 grade 3 or 4 adverse events deemed by investigators to be related to study drugs. They included abnormal weight loss, depression, hypophosphatemia, a rise in LDL cholesterol, headache, vitamin D deficiency, back pain, two spontaneous abortions, and two dissimilar cases of congenital anomaly, with no obvious differences between the study groups in rate or pattern. Rates of specific renal and GI toxicities were comparable across the four study arms.
Earlier in 2016, Dr. Gulick presented the results of the men’s arm of HPTN 069/ACTG A5305, a parallel 48-week randomized trial in 406 men who have sex with men. Five men in the maraviroc monotherapy arm seroconverted during the 48-week study, for an incidence of 1.4%. All had no or low plasma drug concentrations, and all five were infected with HIV lacking antiretroviral drug resistance.
Dr. Gulick and coinvestigators plan to present the findings of an analysis of rectal and vaginal biopsies from 42 women in the trial, along with a bone mineral density substudy in 200 men and 200 women men in the trial, plus detailed quality-of-life, behavioral, and adherence data in the full men’s and women’s cohorts.
The trial was sponsored by the HIV Prevention Trials Network and the AIDS Clinical Trials Group with funding from the National Institute of Allergic and Infectious Diseases. Dr. Gulick reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – Maraviroc-containing regimens for daily oral pre-exposure prophylaxis in women at risk for HIV infection showed good safety and tolerability in a phase 2 study, the first randomized trial of PrEP ever conducted in U.S. women, Roy M. Gulick, MD, reported at the 21st International AIDS Conference.
No new HIV infections occurred in the 188 women who participated in the 48-week, randomized, double-blind, placebo-controlled study known as the HPTN 069/ACTG A5305 trial.
However, the results shouldn’t be taken as evidence of efficacy. The relatively low 2% incidence of new STIs diagnosed during 48 weeks of close followup – three cases of chlamydia, one of gonorrhea – suggests that the study population probably wasn’t at high risk for acquiring HIV. Further, the study wasn’t powered to determine efficacy. That determination will have to await a larger phase 3 trial, observed Dr. Gulick, professor of medicine at Cornell University in New York.
Maraviroc (Selzentry) is categorized as an HIV entry inhibitor. It’s an antagonist of the CCR5 receptor found on the surface of T cells, which is the route of HIV infection. The rationale for exploring the drug for HIV PrEP, according to Dr. Gulick, is that it concentrates in both the genital tract and rectum, doesn’t select for drug-resistant viral strains, is well tolerated, and it isn’t commonly used for treatment of HIV infection.
Additional options for oral daily HIV PrEP are clearly desirable, he added. The only approved agent is Truvada (tenofovir/emtricitabine), which is often used in HIV therapy as well, and there is concern it may select for drug resistance. Plus, it has renal, GI, and bone side effects.
Study participants were HIV-negative adult women who were born female and considered at risk for HIV acquisition because of a history of condomless vaginal or anal intercourse with at least one HIV-positive or unknown status man within the previous 90 days. The women were randomized to one of four study arms: maraviroc at the standard dose of 300 mg/day plus two placebo pills; maraviroc plus emtricitabine at the standard dose of 200 mg/day plus one placebo pill; maraviroc plus tenofovir at 300 mg/day plus a placebo pill, or a control regimen of fixed-dose Truvada (tenofovir 300 mg/emtricitabine 200 mg) plus two placebo pills. Thus, everyone took three pills once daily.
The three-pill regimen might help explain the less than stellar patient adherence. Study drugs were detectable – and not necessarily at therapeutic levels – in the plasma of 65% of subjects at 24 weeks and 60% at 48 weeks, with no differences between the study arms.
Maraviroc alone was associated with fewer grade 2-4 adverse events than the other regimens.
There were 11 grade 3 or 4 adverse events deemed by investigators to be related to study drugs. They included abnormal weight loss, depression, hypophosphatemia, a rise in LDL cholesterol, headache, vitamin D deficiency, back pain, two spontaneous abortions, and two dissimilar cases of congenital anomaly, with no obvious differences between the study groups in rate or pattern. Rates of specific renal and GI toxicities were comparable across the four study arms.
Earlier in 2016, Dr. Gulick presented the results of the men’s arm of HPTN 069/ACTG A5305, a parallel 48-week randomized trial in 406 men who have sex with men. Five men in the maraviroc monotherapy arm seroconverted during the 48-week study, for an incidence of 1.4%. All had no or low plasma drug concentrations, and all five were infected with HIV lacking antiretroviral drug resistance.
Dr. Gulick and coinvestigators plan to present the findings of an analysis of rectal and vaginal biopsies from 42 women in the trial, along with a bone mineral density substudy in 200 men and 200 women men in the trial, plus detailed quality-of-life, behavioral, and adherence data in the full men’s and women’s cohorts.
The trial was sponsored by the HIV Prevention Trials Network and the AIDS Clinical Trials Group with funding from the National Institute of Allergic and Infectious Diseases. Dr. Gulick reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – Maraviroc-containing regimens for daily oral pre-exposure prophylaxis in women at risk for HIV infection showed good safety and tolerability in a phase 2 study, the first randomized trial of PrEP ever conducted in U.S. women, Roy M. Gulick, MD, reported at the 21st International AIDS Conference.
No new HIV infections occurred in the 188 women who participated in the 48-week, randomized, double-blind, placebo-controlled study known as the HPTN 069/ACTG A5305 trial.
However, the results shouldn’t be taken as evidence of efficacy. The relatively low 2% incidence of new STIs diagnosed during 48 weeks of close followup – three cases of chlamydia, one of gonorrhea – suggests that the study population probably wasn’t at high risk for acquiring HIV. Further, the study wasn’t powered to determine efficacy. That determination will have to await a larger phase 3 trial, observed Dr. Gulick, professor of medicine at Cornell University in New York.
Maraviroc (Selzentry) is categorized as an HIV entry inhibitor. It’s an antagonist of the CCR5 receptor found on the surface of T cells, which is the route of HIV infection. The rationale for exploring the drug for HIV PrEP, according to Dr. Gulick, is that it concentrates in both the genital tract and rectum, doesn’t select for drug-resistant viral strains, is well tolerated, and it isn’t commonly used for treatment of HIV infection.
Additional options for oral daily HIV PrEP are clearly desirable, he added. The only approved agent is Truvada (tenofovir/emtricitabine), which is often used in HIV therapy as well, and there is concern it may select for drug resistance. Plus, it has renal, GI, and bone side effects.
Study participants were HIV-negative adult women who were born female and considered at risk for HIV acquisition because of a history of condomless vaginal or anal intercourse with at least one HIV-positive or unknown status man within the previous 90 days. The women were randomized to one of four study arms: maraviroc at the standard dose of 300 mg/day plus two placebo pills; maraviroc plus emtricitabine at the standard dose of 200 mg/day plus one placebo pill; maraviroc plus tenofovir at 300 mg/day plus a placebo pill, or a control regimen of fixed-dose Truvada (tenofovir 300 mg/emtricitabine 200 mg) plus two placebo pills. Thus, everyone took three pills once daily.
The three-pill regimen might help explain the less than stellar patient adherence. Study drugs were detectable – and not necessarily at therapeutic levels – in the plasma of 65% of subjects at 24 weeks and 60% at 48 weeks, with no differences between the study arms.
Maraviroc alone was associated with fewer grade 2-4 adverse events than the other regimens.
There were 11 grade 3 or 4 adverse events deemed by investigators to be related to study drugs. They included abnormal weight loss, depression, hypophosphatemia, a rise in LDL cholesterol, headache, vitamin D deficiency, back pain, two spontaneous abortions, and two dissimilar cases of congenital anomaly, with no obvious differences between the study groups in rate or pattern. Rates of specific renal and GI toxicities were comparable across the four study arms.
Earlier in 2016, Dr. Gulick presented the results of the men’s arm of HPTN 069/ACTG A5305, a parallel 48-week randomized trial in 406 men who have sex with men. Five men in the maraviroc monotherapy arm seroconverted during the 48-week study, for an incidence of 1.4%. All had no or low plasma drug concentrations, and all five were infected with HIV lacking antiretroviral drug resistance.
Dr. Gulick and coinvestigators plan to present the findings of an analysis of rectal and vaginal biopsies from 42 women in the trial, along with a bone mineral density substudy in 200 men and 200 women men in the trial, plus detailed quality-of-life, behavioral, and adherence data in the full men’s and women’s cohorts.
The trial was sponsored by the HIV Prevention Trials Network and the AIDS Clinical Trials Group with funding from the National Institute of Allergic and Infectious Diseases. Dr. Gulick reported having no financial conflicts of interest.
AT AIDS 2016
Key clinical point: Maraviroc shows promise as an alternative to Truvada for HIV pre-exposure prophylaxis.
Major finding: No new HIV infections occurred in at-risk women in a 48-week study of daily oral HIV pre-exposure prophylaxis comparing three maraviroc-containing regimens and tenofovir/emtricitabine.
Data source: This was a randomized, double-blind, placebo-controlled, multicenter, 48-week clinical trial involving 188 women at risk for HIV infection.
Disclosures: The trial was sponsored by the HIV Prevention Trials Network and the AIDS Clinical Trials Group with funding from the National Institute of Allergy and Infectious Diseases. The presenter reported having no financial conflicts of interest.

Long-acting all-injectable HIV therapy successful in phase 2b
DURBAN, SOUTH AFRICA – Long-acting injectable antiretroviral therapy with cabotegravir and rilpivirine in nanosuspension successfully suppressed HIV-infected patients’ plasma viral load to fewer than 50 copies/mL for 48 weeks as maintenance therapy in the LATTE-2 trial, David A. Margolis, MD, reported at the 21st International AIDS Conference.
The intramuscular gluteal injections given every 4 or 8 weeks also proved safe and extremely well tolerated. At week 48, more than 97% of patients on injectable maintenance antiretroviral therapy (ART) reported a high degree of satisfaction with their treatment regimen on a structured questionnaire and expressed a willingness to continue on injectable therapy in the future, according to Dr. Margolis of Viiv Healthcare in Research Triangle Park, N.C.

Cabotegravir is an HIV integrase inhibitor, rilpivirine a nonnucleoside reverse transcriptase inhibitor. Rilpivirine is already approved as Edurant, a once-daily 25-mg tablet for oral therapy. Cabotegravir is under development as a 30-mg once-daily tablet. As oral agents, each has a half-life of roughly 40 hours. The two-drug oral combination showed a high degree of safety and efficacy through 96 weeks of follow-up in the LATTE-1 study, previously reported by Dr. Margolis (Lancet Infect Dis. 2015 Oct;15[10]):1145-55) .
In addition, the two antiretroviral agents are being developed as long-acting injectables with half-lives of 20-90 days. LATTE-2 was a phase IIb, open-label, multicenter study that began with 309 HIV-positive, ART-naive subjects who participated in a 20-week induction phase during which they received 30 mg/day of oral cabotegravir plus abacavir/lamivudine to induce HIV viral suppression. During the last 4 weeks of the induction phase, participants also received oral rilpivirine at 25 mg once daily to ensure they could tolerate the drug.
At the end of the 20-week induction phase, 228 patients with an HIV viral load below 50 copies/mL were randomized 2:2:1 to maintenance therapy with intramuscular injections of long-acting cabotegravir at 200 mg/mL and long-acting rilpivirine at 300 mg/mL in nanosuspension every 4 or 8 weeks or to the oral once-daily versions of the two drugs.
After 48 weeks of maintenance therapy, an HIV RNA load of less than 50 copies/mL was present in 92% of subjects on injections every 8 weeks, 91% with injections every 4 weeks, and 89% of those on oral daily therapy. Moreover, of the eight virologic nonresponders at 48 weeks in the 8-week-injection schedule, six remained in the study and five of them have consistently achieved a viral load below 50 copies/mL in subsequent visits through week 72.
Viral failure defined as an HIV RNA viral load greater than 200 copies/mL on two occasions occurred in 1% of patients on the 8-week interval schedule, 1% of those on oral therapy, and no one on IM injections every 4 weeks. One patient on every-8-week schedule developed treatment-emergent viral resistance.
More than 80% of patients in both IM injection study arms experienced painful injection site reactions after their first treatment session, 82% of which were mild and 17% moderate. Ninety percent of these reactions resolved within 7 days; median duration was 3 days. Two patients withdrew from the study as a result of these reactions. After the first round of injections, the incidence of injection site reactions fell to 25%-30%.
Other adverse events consisted of fever, fatigue, headache, rash, and flu-like symptoms, each with an incidence of 2%-4%.
Based upon the LATTE-2 outcomes, the once-per-month injection regimen has been selected for the forthcoming pivotal phase III randomized clinical trials of long-acting cabotegravir/rilpivirine for ART, Dr. Margolis said.
The pharmaceutical industry is pursuing long-acting injectable ART both for pre-exposure prophylaxis (PrEP) and for treatment of HIV-infected patients. Clinicians and patients alike are eager for this option, especially for PrEP, where adherence to daily oral therapy has been suboptimal in many studies.
But there is a fly in the ointment. One of the most talked about studies presented at AIDS 2016 was Dr. Ian McGowan’s report that low levels of rilpivirine were found in plasma and female genital tract fluids more than 18 months after a single IM 1,200-mg dose of rilpivirine in seven participants in a long-acting PrEP study.

“There is certainly the possibility that extended periods of drug availability at perhaps subtherapeutic concentrations might increase the risk of ART resistance in individuals who seroconvert after exposure to long-acting PrEP,” cautioned Dr. McGowan, professor of medicine at the University of Pittsburgh.
No similar studies have as yet been done with other candidate long-acting injectables, he said.
“It’s clear, I think, that as we move forward with this exciting field of long-acting injectables we really need to better characterize the terminal half-life of these products so that we can better inform management of the pharmacokinetic tail and hopefully avoid the potential for ART resistance,” Dr. McGowan added.
His study was funded by the Bill and Melinda Gates Foundation.
Dr. Margolis is an employee of Viiv Healthcare, the LATTE-2 study sponsor.
DURBAN, SOUTH AFRICA – Long-acting injectable antiretroviral therapy with cabotegravir and rilpivirine in nanosuspension successfully suppressed HIV-infected patients’ plasma viral load to fewer than 50 copies/mL for 48 weeks as maintenance therapy in the LATTE-2 trial, David A. Margolis, MD, reported at the 21st International AIDS Conference.
The intramuscular gluteal injections given every 4 or 8 weeks also proved safe and extremely well tolerated. At week 48, more than 97% of patients on injectable maintenance antiretroviral therapy (ART) reported a high degree of satisfaction with their treatment regimen on a structured questionnaire and expressed a willingness to continue on injectable therapy in the future, according to Dr. Margolis of Viiv Healthcare in Research Triangle Park, N.C.
Cabotegravir is an HIV integrase inhibitor, rilpivirine a nonnucleoside reverse transcriptase inhibitor. Rilpivirine is already approved as Edurant, a once-daily 25-mg tablet for oral therapy. Cabotegravir is under development as a 30-mg once-daily tablet. As oral agents, each has a half-life of roughly 40 hours. The two-drug oral combination showed a high degree of safety and efficacy through 96 weeks of follow-up in the LATTE-1 study, previously reported by Dr. Margolis (Lancet Infect Dis. 2015 Oct;15[10]):1145-55) .
In addition, the two antiretroviral agents are being developed as long-acting injectables with half-lives of 20-90 days. LATTE-2 was a phase IIb, open-label, multicenter study that began with 309 HIV-positive, ART-naive subjects who participated in a 20-week induction phase during which they received 30 mg/day of oral cabotegravir plus abacavir/lamivudine to induce HIV viral suppression. During the last 4 weeks of the induction phase, participants also received oral rilpivirine at 25 mg once daily to ensure they could tolerate the drug.
At the end of the 20-week induction phase, 228 patients with an HIV viral load below 50 copies/mL were randomized 2:2:1 to maintenance therapy with intramuscular injections of long-acting cabotegravir at 200 mg/mL and long-acting rilpivirine at 300 mg/mL in nanosuspension every 4 or 8 weeks or to the oral once-daily versions of the two drugs.
After 48 weeks of maintenance therapy, an HIV RNA load of less than 50 copies/mL was present in 92% of subjects on injections every 8 weeks, 91% with injections every 4 weeks, and 89% of those on oral daily therapy. Moreover, of the eight virologic nonresponders at 48 weeks in the 8-week-injection schedule, six remained in the study and five of them have consistently achieved a viral load below 50 copies/mL in subsequent visits through week 72.
Viral failure defined as an HIV RNA viral load greater than 200 copies/mL on two occasions occurred in 1% of patients on the 8-week interval schedule, 1% of those on oral therapy, and no one on IM injections every 4 weeks. One patient on every-8-week schedule developed treatment-emergent viral resistance.
More than 80% of patients in both IM injection study arms experienced painful injection site reactions after their first treatment session, 82% of which were mild and 17% moderate. Ninety percent of these reactions resolved within 7 days; median duration was 3 days. Two patients withdrew from the study as a result of these reactions. After the first round of injections, the incidence of injection site reactions fell to 25%-30%.
Other adverse events consisted of fever, fatigue, headache, rash, and flu-like symptoms, each with an incidence of 2%-4%.
Based upon the LATTE-2 outcomes, the once-per-month injection regimen has been selected for the forthcoming pivotal phase III randomized clinical trials of long-acting cabotegravir/rilpivirine for ART, Dr. Margolis said.
The pharmaceutical industry is pursuing long-acting injectable ART both for pre-exposure prophylaxis (PrEP) and for treatment of HIV-infected patients. Clinicians and patients alike are eager for this option, especially for PrEP, where adherence to daily oral therapy has been suboptimal in many studies.
But there is a fly in the ointment. One of the most talked about studies presented at AIDS 2016 was Dr. Ian McGowan’s report that low levels of rilpivirine were found in plasma and female genital tract fluids more than 18 months after a single IM 1,200-mg dose of rilpivirine in seven participants in a long-acting PrEP study.
“There is certainly the possibility that extended periods of drug availability at perhaps subtherapeutic concentrations might increase the risk of ART resistance in individuals who seroconvert after exposure to long-acting PrEP,” cautioned Dr. McGowan, professor of medicine at the University of Pittsburgh.
No similar studies have as yet been done with other candidate long-acting injectables, he said.
“It’s clear, I think, that as we move forward with this exciting field of long-acting injectables we really need to better characterize the terminal half-life of these products so that we can better inform management of the pharmacokinetic tail and hopefully avoid the potential for ART resistance,” Dr. McGowan added.
His study was funded by the Bill and Melinda Gates Foundation.
Dr. Margolis is an employee of Viiv Healthcare, the LATTE-2 study sponsor.
DURBAN, SOUTH AFRICA – Long-acting injectable antiretroviral therapy with cabotegravir and rilpivirine in nanosuspension successfully suppressed HIV-infected patients’ plasma viral load to fewer than 50 copies/mL for 48 weeks as maintenance therapy in the LATTE-2 trial, David A. Margolis, MD, reported at the 21st International AIDS Conference.
The intramuscular gluteal injections given every 4 or 8 weeks also proved safe and extremely well tolerated. At week 48, more than 97% of patients on injectable maintenance antiretroviral therapy (ART) reported a high degree of satisfaction with their treatment regimen on a structured questionnaire and expressed a willingness to continue on injectable therapy in the future, according to Dr. Margolis of Viiv Healthcare in Research Triangle Park, N.C.
Cabotegravir is an HIV integrase inhibitor, rilpivirine a nonnucleoside reverse transcriptase inhibitor. Rilpivirine is already approved as Edurant, a once-daily 25-mg tablet for oral therapy. Cabotegravir is under development as a 30-mg once-daily tablet. As oral agents, each has a half-life of roughly 40 hours. The two-drug oral combination showed a high degree of safety and efficacy through 96 weeks of follow-up in the LATTE-1 study, previously reported by Dr. Margolis (Lancet Infect Dis. 2015 Oct;15[10]):1145-55) .
In addition, the two antiretroviral agents are being developed as long-acting injectables with half-lives of 20-90 days. LATTE-2 was a phase IIb, open-label, multicenter study that began with 309 HIV-positive, ART-naive subjects who participated in a 20-week induction phase during which they received 30 mg/day of oral cabotegravir plus abacavir/lamivudine to induce HIV viral suppression. During the last 4 weeks of the induction phase, participants also received oral rilpivirine at 25 mg once daily to ensure they could tolerate the drug.
At the end of the 20-week induction phase, 228 patients with an HIV viral load below 50 copies/mL were randomized 2:2:1 to maintenance therapy with intramuscular injections of long-acting cabotegravir at 200 mg/mL and long-acting rilpivirine at 300 mg/mL in nanosuspension every 4 or 8 weeks or to the oral once-daily versions of the two drugs.
After 48 weeks of maintenance therapy, an HIV RNA load of less than 50 copies/mL was present in 92% of subjects on injections every 8 weeks, 91% with injections every 4 weeks, and 89% of those on oral daily therapy. Moreover, of the eight virologic nonresponders at 48 weeks in the 8-week-injection schedule, six remained in the study and five of them have consistently achieved a viral load below 50 copies/mL in subsequent visits through week 72.
Viral failure defined as an HIV RNA viral load greater than 200 copies/mL on two occasions occurred in 1% of patients on the 8-week interval schedule, 1% of those on oral therapy, and no one on IM injections every 4 weeks. One patient on every-8-week schedule developed treatment-emergent viral resistance.
More than 80% of patients in both IM injection study arms experienced painful injection site reactions after their first treatment session, 82% of which were mild and 17% moderate. Ninety percent of these reactions resolved within 7 days; median duration was 3 days. Two patients withdrew from the study as a result of these reactions. After the first round of injections, the incidence of injection site reactions fell to 25%-30%.
Other adverse events consisted of fever, fatigue, headache, rash, and flu-like symptoms, each with an incidence of 2%-4%.
Based upon the LATTE-2 outcomes, the once-per-month injection regimen has been selected for the forthcoming pivotal phase III randomized clinical trials of long-acting cabotegravir/rilpivirine for ART, Dr. Margolis said.
The pharmaceutical industry is pursuing long-acting injectable ART both for pre-exposure prophylaxis (PrEP) and for treatment of HIV-infected patients. Clinicians and patients alike are eager for this option, especially for PrEP, where adherence to daily oral therapy has been suboptimal in many studies.
But there is a fly in the ointment. One of the most talked about studies presented at AIDS 2016 was Dr. Ian McGowan’s report that low levels of rilpivirine were found in plasma and female genital tract fluids more than 18 months after a single IM 1,200-mg dose of rilpivirine in seven participants in a long-acting PrEP study.
“There is certainly the possibility that extended periods of drug availability at perhaps subtherapeutic concentrations might increase the risk of ART resistance in individuals who seroconvert after exposure to long-acting PrEP,” cautioned Dr. McGowan, professor of medicine at the University of Pittsburgh.
No similar studies have as yet been done with other candidate long-acting injectables, he said.
“It’s clear, I think, that as we move forward with this exciting field of long-acting injectables we really need to better characterize the terminal half-life of these products so that we can better inform management of the pharmacokinetic tail and hopefully avoid the potential for ART resistance,” Dr. McGowan added.
His study was funded by the Bill and Melinda Gates Foundation.
Dr. Margolis is an employee of Viiv Healthcare, the LATTE-2 study sponsor.
AT AIDS 2016
Key clinical point: The era of all-injectable, long-acting antiretroviral therapy for HIV may be drawing closer.
Major finding: More than 90% of HIV-infected patients maintained a viral load below 50 copies/mL throughout 48 weeks of maintenance therapy with intramuscular injections of cabotegravir and rilpivirine regardless of whether given every 4 or 8 weeks.
Data source: LATTE-2 was a phase IIb, open-label, multicenter study in which 228 HIV-infected patients with an HIV RNA viral load below 50 copies/mL on oral therapy at baseline were randomized to long-acting injectable antiretroviral therapy either every 4 or 8 weeks or to daily oral therapy.
Disclosures: The presenter is an employee of ViiV Healthcare, the study sponsor.

With HIV PrEP, benefits outweigh resistance risk
DURBAN, SOUTH AFRICA – For physicians who are leery of prescribing oral daily pre-exposure prophylaxis (PrEP) against HIV infection because of concern that it will promote drug-resistant viral strains, Robert Grant, MD, has a reassuring message.
It’s a message based upon his systematic review of published resistance testing results in all the randomized, placebo-controlled PrEP trials.
“For clinicians who are anxious, I can say based on this excellent work that if your primary concern is drug resistance – and I’m not suggesting that it should be – but if you are very concerned about drug resistance, then you should be a PrEP advocate, because in the end, your population will have less drug resistance,” Dr. Grant said at the 21st International AIDS Conference.
The overall rate of resistance to emtricitabine or tenofovir – the antiretrovirals contained in fixed-dose combination in Truvada, the only FDA-approved agent for HIV PrEP – was five cases in 9,222 trial participants, for a risk of 0.05%. The number needed to treat in order to prevent one HIV infection was 13-60, depending upon the adherence rate in a given trial.
In contrast, the number needed to harm by causing emergence of a drug-resistant strain of HIV was 1,844, reported Dr. Grant, professor of medicine at the University of California, San Francisco.
Drug resistance during PrEP is rare. It occurs mainly when PrEP is started or restarted during an acute HIV infection. Almost all of the resistance is to emtricitabine. Those emtricitabine-resistant HIV infections are treatable.
The randomized trials of PrEP used several methods of screening for acute HIV infection: rapid second- and third-generation antibody assays, highly sensitive HIV nucleic acid genotypic assays capable of detecting resistant viral variants present in very low abundance, and a clinical screen.
The use of one or the other of the laboratory approaches is common practice in the United States and Europe. But these tools are much less frequently available in sub-Saharan Africa and elsewhere the HIV epidemic is hitting hardest. That is a setting where a clinical screening program could be of great value.
“The majority of people with acute HIV infection will have some sort of symptoms of an acute viral syndrome. They’re nonspecific symptoms: a flu-like illness, fever, sore throat, headache, a rash,” he observed.
Dr. Grant was first author of the iPrEx OLE study, the only published trial to examine a strategy of clinical screening for acute viral syndromes and acute HIV infection in PrEP candidates (Lancet Infect Dis. 2014 Sep;14[9]:820-9).
Thirty of 1,603 PrEP candidates (1.9%) who underwent clinical screening had PrEP deferred because of an acute viral syndrome. Two of those 30 patients subsequently proved to have acute HIV infection on laboratory testing, 25 of the HIV-negative patients had a delayed start of PrEP, and 3 of the 30 never started PrEP. Thus, clinical screening had 100% sensitivity, 98% specificity, 100% negative predictive value, and a 6.7% positive predictive value, according to Dr. Grant.
The World Health Organization sponsored his systematic review. Dr. Grant reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – For physicians who are leery of prescribing oral daily pre-exposure prophylaxis (PrEP) against HIV infection because of concern that it will promote drug-resistant viral strains, Robert Grant, MD, has a reassuring message.
It’s a message based upon his systematic review of published resistance testing results in all the randomized, placebo-controlled PrEP trials.
“For clinicians who are anxious, I can say based on this excellent work that if your primary concern is drug resistance – and I’m not suggesting that it should be – but if you are very concerned about drug resistance, then you should be a PrEP advocate, because in the end, your population will have less drug resistance,” Dr. Grant said at the 21st International AIDS Conference.
The overall rate of resistance to emtricitabine or tenofovir – the antiretrovirals contained in fixed-dose combination in Truvada, the only FDA-approved agent for HIV PrEP – was five cases in 9,222 trial participants, for a risk of 0.05%. The number needed to treat in order to prevent one HIV infection was 13-60, depending upon the adherence rate in a given trial.
In contrast, the number needed to harm by causing emergence of a drug-resistant strain of HIV was 1,844, reported Dr. Grant, professor of medicine at the University of California, San Francisco.
Drug resistance during PrEP is rare. It occurs mainly when PrEP is started or restarted during an acute HIV infection. Almost all of the resistance is to emtricitabine. Those emtricitabine-resistant HIV infections are treatable.
The randomized trials of PrEP used several methods of screening for acute HIV infection: rapid second- and third-generation antibody assays, highly sensitive HIV nucleic acid genotypic assays capable of detecting resistant viral variants present in very low abundance, and a clinical screen.
The use of one or the other of the laboratory approaches is common practice in the United States and Europe. But these tools are much less frequently available in sub-Saharan Africa and elsewhere the HIV epidemic is hitting hardest. That is a setting where a clinical screening program could be of great value.
“The majority of people with acute HIV infection will have some sort of symptoms of an acute viral syndrome. They’re nonspecific symptoms: a flu-like illness, fever, sore throat, headache, a rash,” he observed.
Dr. Grant was first author of the iPrEx OLE study, the only published trial to examine a strategy of clinical screening for acute viral syndromes and acute HIV infection in PrEP candidates (Lancet Infect Dis. 2014 Sep;14[9]:820-9).
Thirty of 1,603 PrEP candidates (1.9%) who underwent clinical screening had PrEP deferred because of an acute viral syndrome. Two of those 30 patients subsequently proved to have acute HIV infection on laboratory testing, 25 of the HIV-negative patients had a delayed start of PrEP, and 3 of the 30 never started PrEP. Thus, clinical screening had 100% sensitivity, 98% specificity, 100% negative predictive value, and a 6.7% positive predictive value, according to Dr. Grant.
The World Health Organization sponsored his systematic review. Dr. Grant reported having no financial conflicts of interest.
DURBAN, SOUTH AFRICA – For physicians who are leery of prescribing oral daily pre-exposure prophylaxis (PrEP) against HIV infection because of concern that it will promote drug-resistant viral strains, Robert Grant, MD, has a reassuring message.
It’s a message based upon his systematic review of published resistance testing results in all the randomized, placebo-controlled PrEP trials.
“For clinicians who are anxious, I can say based on this excellent work that if your primary concern is drug resistance – and I’m not suggesting that it should be – but if you are very concerned about drug resistance, then you should be a PrEP advocate, because in the end, your population will have less drug resistance,” Dr. Grant said at the 21st International AIDS Conference.
The overall rate of resistance to emtricitabine or tenofovir – the antiretrovirals contained in fixed-dose combination in Truvada, the only FDA-approved agent for HIV PrEP – was five cases in 9,222 trial participants, for a risk of 0.05%. The number needed to treat in order to prevent one HIV infection was 13-60, depending upon the adherence rate in a given trial.
In contrast, the number needed to harm by causing emergence of a drug-resistant strain of HIV was 1,844, reported Dr. Grant, professor of medicine at the University of California, San Francisco.
Drug resistance during PrEP is rare. It occurs mainly when PrEP is started or restarted during an acute HIV infection. Almost all of the resistance is to emtricitabine. Those emtricitabine-resistant HIV infections are treatable.
The randomized trials of PrEP used several methods of screening for acute HIV infection: rapid second- and third-generation antibody assays, highly sensitive HIV nucleic acid genotypic assays capable of detecting resistant viral variants present in very low abundance, and a clinical screen.
The use of one or the other of the laboratory approaches is common practice in the United States and Europe. But these tools are much less frequently available in sub-Saharan Africa and elsewhere the HIV epidemic is hitting hardest. That is a setting where a clinical screening program could be of great value.
“The majority of people with acute HIV infection will have some sort of symptoms of an acute viral syndrome. They’re nonspecific symptoms: a flu-like illness, fever, sore throat, headache, a rash,” he observed.
Dr. Grant was first author of the iPrEx OLE study, the only published trial to examine a strategy of clinical screening for acute viral syndromes and acute HIV infection in PrEP candidates (Lancet Infect Dis. 2014 Sep;14[9]:820-9).
Thirty of 1,603 PrEP candidates (1.9%) who underwent clinical screening had PrEP deferred because of an acute viral syndrome. Two of those 30 patients subsequently proved to have acute HIV infection on laboratory testing, 25 of the HIV-negative patients had a delayed start of PrEP, and 3 of the 30 never started PrEP. Thus, clinical screening had 100% sensitivity, 98% specificity, 100% negative predictive value, and a 6.7% positive predictive value, according to Dr. Grant.
The World Health Organization sponsored his systematic review. Dr. Grant reported having no financial conflicts of interest.
AT AIDS 2016
Key clinical point: The benefit of oral daily chemoprophylaxis against HIV infection in at-risk individuals far outweighs the risk.
Major finding: The number needed to treat with oral daily HIV pre-exposure prophylaxis in order to prevent one HIV infection is 13-60; the number needed to harm by causing a case of drug-resistant HIV is 1,844.
Data source: This was a systematic review of six published randomized, placebo-controlled trials of HIV pre-exposure prophylaxis totaling 15,894 participants.
Disclosures: The World Health Organization sponsored the review. Dr. Grant reported having no financial conflicts of interest.
Ombitasvir/paritaprevir-based HCV regimens perform well in HIV coinfection
DURBAN, SOUTH AFRICA – The all-oral, interferon-free, triple- or double-direct-acting antiviral regimens informally known as 3D and 2D proved effective and safe for treatment of chronic hepatitis C due to HCV genotypes 1 and 4 in HIV-coinfected patients taking various antiretroviral combinations in the phase III, multicenter TURQUOISE-1, Part 2 trial, Jürgen K. Rockstroh, MD, reported at the 21st International AIDS Conference.
TURQUOISE-1, Part 2 included 228 patients coinfected with HCV/HIV. Of the 154 with HCV genotype 1a and 44 with genotype 1b, 21 of whom had compensated cirrhosis, 98% of subjects achieved a sustained virologic response (SVR) 12 weeks after conclusion of the 3D regimen, or SVR12, in a modified intent-to-treat analysis. A handful of patients with missing data, or those who stopped treatment for reasons other than virologic failure, were excluded from the analysis.

In the 28 patients coinfected with HCV genotype 4, none of whom had cirrhosis, the modified SVR12 rate was 100%.
The so-called 3D regimen consists of the NS5A inhibitor ombitasvir coformulated in once-daily, fixed-dose fashion with the NS3/4A protease inhibitor paritaprevir boosted with ritonavir, along with twice-daily dasabuvir, a nonnucleoside NS5B RNA polymerase inhibitor. This combination has received Food and Drug Administration approval and is marketed as Viekira Pak for treatment of HCV genotype 1 in HIV-coinfected patients on the basis of the TURQUOISE-1, Part 1a trial (JAMA 2015;313[12]:1223-32). But this study was confined to 63 U.S. patients, and European investigators wanted to see how 3D worked in a larger, more diverse population, explained Dr. Rockstroh, professor of medicine and head of the outpatient HIV clinic at the University of Bonn (Germany).
In TURQUOISE-1, Part 2, patients with HCV genotype 1a without cirrhosis received 12 weeks of the 3D regimen plus ribavirin, while those with compensated cirrhosis got 24 weeks. Patients with genotype 1b received 12 weeks of 3D without ribavirin, regardless of whether or not they had compensated cirrhosis.
Because dasabuvir is ineffective against HCV genotype 4, those patients got 12 weeks of the 2D regimen – the coformulated ombitasvir/paritaprevir/ritonavir – along with ribavirin.
All subjects were on antiretroviral regimens based upon either darunavir (Prezista), atazanavir (Reyataz), raltegravir (Isentress), or dolutegravir (Tivicay). Nearly all (97%) of the patients had an HIV RNA viral load of less than 40 copies/mL at baseline. Those on ritonavir-boosted antiretroviral therapy discontinued the ritonavir and received it instead via their 3D or 2D hepatitis C therapy.
HIV suppression was well maintained throughout the study. Intermittent episodes of HIV viremia affected 4% of participants, but in all cases the peak was less than 200 copies/mL. None of the episodes met standard criteria for HIV genotypic resistance testing, according to Dr. Rockstroh.
In contrast to the former standard therapy consisting of pegylated interferon and ribavirin, which had an SVR of about 30% in HCV/HIV coinfected patients and was fraught with serious side effects, no one discontinued treatment due to adverse events in TURQUOISE-1, Part 2. No cases of hepatic decompensation occurred. The only serious adverse event deemed possibly treatment related was a single case of depression. Mild fatigue, nausea, diarrhea, insomnia, headache, or itching each occurred in 10%-20% of subjects.
As effective antiretroviral therapy enables HIV-infected patients to have a lifespan approaching normal, HCV coinfection has emerged as a major issue. Roughly one-third of HIV-positive individuals are coinfected with HCV, and the rate of death due to HCV-related liver disease in coinfected patients exceeds the rate of death due to HIV. HCV genotype 1 accounts for three-quarters of chronic hepatitis C in the United States, while genotype 4 predominates in Africa, where the bulk of HIV infections occur.
During the AIDS 2016 conference, the Food and Drug Administration approved Viekira XR, an extended-release coformulation of the 3D regimen. Each tablet contains 200 mg of dasabuvir, 8.33 mg of ombitasvir, 50 mg of paritaprevir, and 33.33 mg of ritonavir. The standard dosing is three oral tablets taken once daily.
The TURQUOISE-1, Part 2 trial was sponsored by AbbVie. Dr. Rockstroh is a consultant to AbbVie and seven other pharmaceutical companies.
DURBAN, SOUTH AFRICA – The all-oral, interferon-free, triple- or double-direct-acting antiviral regimens informally known as 3D and 2D proved effective and safe for treatment of chronic hepatitis C due to HCV genotypes 1 and 4 in HIV-coinfected patients taking various antiretroviral combinations in the phase III, multicenter TURQUOISE-1, Part 2 trial, Jürgen K. Rockstroh, MD, reported at the 21st International AIDS Conference.
TURQUOISE-1, Part 2 included 228 patients coinfected with HCV/HIV. Of the 154 with HCV genotype 1a and 44 with genotype 1b, 21 of whom had compensated cirrhosis, 98% of subjects achieved a sustained virologic response (SVR) 12 weeks after conclusion of the 3D regimen, or SVR12, in a modified intent-to-treat analysis. A handful of patients with missing data, or those who stopped treatment for reasons other than virologic failure, were excluded from the analysis.
In the 28 patients coinfected with HCV genotype 4, none of whom had cirrhosis, the modified SVR12 rate was 100%.
The so-called 3D regimen consists of the NS5A inhibitor ombitasvir coformulated in once-daily, fixed-dose fashion with the NS3/4A protease inhibitor paritaprevir boosted with ritonavir, along with twice-daily dasabuvir, a nonnucleoside NS5B RNA polymerase inhibitor. This combination has received Food and Drug Administration approval and is marketed as Viekira Pak for treatment of HCV genotype 1 in HIV-coinfected patients on the basis of the TURQUOISE-1, Part 1a trial (JAMA 2015;313[12]:1223-32). But this study was confined to 63 U.S. patients, and European investigators wanted to see how 3D worked in a larger, more diverse population, explained Dr. Rockstroh, professor of medicine and head of the outpatient HIV clinic at the University of Bonn (Germany).
In TURQUOISE-1, Part 2, patients with HCV genotype 1a without cirrhosis received 12 weeks of the 3D regimen plus ribavirin, while those with compensated cirrhosis got 24 weeks. Patients with genotype 1b received 12 weeks of 3D without ribavirin, regardless of whether or not they had compensated cirrhosis.
Because dasabuvir is ineffective against HCV genotype 4, those patients got 12 weeks of the 2D regimen – the coformulated ombitasvir/paritaprevir/ritonavir – along with ribavirin.
All subjects were on antiretroviral regimens based upon either darunavir (Prezista), atazanavir (Reyataz), raltegravir (Isentress), or dolutegravir (Tivicay). Nearly all (97%) of the patients had an HIV RNA viral load of less than 40 copies/mL at baseline. Those on ritonavir-boosted antiretroviral therapy discontinued the ritonavir and received it instead via their 3D or 2D hepatitis C therapy.
HIV suppression was well maintained throughout the study. Intermittent episodes of HIV viremia affected 4% of participants, but in all cases the peak was less than 200 copies/mL. None of the episodes met standard criteria for HIV genotypic resistance testing, according to Dr. Rockstroh.
In contrast to the former standard therapy consisting of pegylated interferon and ribavirin, which had an SVR of about 30% in HCV/HIV coinfected patients and was fraught with serious side effects, no one discontinued treatment due to adverse events in TURQUOISE-1, Part 2. No cases of hepatic decompensation occurred. The only serious adverse event deemed possibly treatment related was a single case of depression. Mild fatigue, nausea, diarrhea, insomnia, headache, or itching each occurred in 10%-20% of subjects.
As effective antiretroviral therapy enables HIV-infected patients to have a lifespan approaching normal, HCV coinfection has emerged as a major issue. Roughly one-third of HIV-positive individuals are coinfected with HCV, and the rate of death due to HCV-related liver disease in coinfected patients exceeds the rate of death due to HIV. HCV genotype 1 accounts for three-quarters of chronic hepatitis C in the United States, while genotype 4 predominates in Africa, where the bulk of HIV infections occur.
During the AIDS 2016 conference, the Food and Drug Administration approved Viekira XR, an extended-release coformulation of the 3D regimen. Each tablet contains 200 mg of dasabuvir, 8.33 mg of ombitasvir, 50 mg of paritaprevir, and 33.33 mg of ritonavir. The standard dosing is three oral tablets taken once daily.
The TURQUOISE-1, Part 2 trial was sponsored by AbbVie. Dr. Rockstroh is a consultant to AbbVie and seven other pharmaceutical companies.
DURBAN, SOUTH AFRICA – The all-oral, interferon-free, triple- or double-direct-acting antiviral regimens informally known as 3D and 2D proved effective and safe for treatment of chronic hepatitis C due to HCV genotypes 1 and 4 in HIV-coinfected patients taking various antiretroviral combinations in the phase III, multicenter TURQUOISE-1, Part 2 trial, Jürgen K. Rockstroh, MD, reported at the 21st International AIDS Conference.
TURQUOISE-1, Part 2 included 228 patients coinfected with HCV/HIV. Of the 154 with HCV genotype 1a and 44 with genotype 1b, 21 of whom had compensated cirrhosis, 98% of subjects achieved a sustained virologic response (SVR) 12 weeks after conclusion of the 3D regimen, or SVR12, in a modified intent-to-treat analysis. A handful of patients with missing data, or those who stopped treatment for reasons other than virologic failure, were excluded from the analysis.
In the 28 patients coinfected with HCV genotype 4, none of whom had cirrhosis, the modified SVR12 rate was 100%.
The so-called 3D regimen consists of the NS5A inhibitor ombitasvir coformulated in once-daily, fixed-dose fashion with the NS3/4A protease inhibitor paritaprevir boosted with ritonavir, along with twice-daily dasabuvir, a nonnucleoside NS5B RNA polymerase inhibitor. This combination has received Food and Drug Administration approval and is marketed as Viekira Pak for treatment of HCV genotype 1 in HIV-coinfected patients on the basis of the TURQUOISE-1, Part 1a trial (JAMA 2015;313[12]:1223-32). But this study was confined to 63 U.S. patients, and European investigators wanted to see how 3D worked in a larger, more diverse population, explained Dr. Rockstroh, professor of medicine and head of the outpatient HIV clinic at the University of Bonn (Germany).
In TURQUOISE-1, Part 2, patients with HCV genotype 1a without cirrhosis received 12 weeks of the 3D regimen plus ribavirin, while those with compensated cirrhosis got 24 weeks. Patients with genotype 1b received 12 weeks of 3D without ribavirin, regardless of whether or not they had compensated cirrhosis.
Because dasabuvir is ineffective against HCV genotype 4, those patients got 12 weeks of the 2D regimen – the coformulated ombitasvir/paritaprevir/ritonavir – along with ribavirin.
All subjects were on antiretroviral regimens based upon either darunavir (Prezista), atazanavir (Reyataz), raltegravir (Isentress), or dolutegravir (Tivicay). Nearly all (97%) of the patients had an HIV RNA viral load of less than 40 copies/mL at baseline. Those on ritonavir-boosted antiretroviral therapy discontinued the ritonavir and received it instead via their 3D or 2D hepatitis C therapy.
HIV suppression was well maintained throughout the study. Intermittent episodes of HIV viremia affected 4% of participants, but in all cases the peak was less than 200 copies/mL. None of the episodes met standard criteria for HIV genotypic resistance testing, according to Dr. Rockstroh.
In contrast to the former standard therapy consisting of pegylated interferon and ribavirin, which had an SVR of about 30% in HCV/HIV coinfected patients and was fraught with serious side effects, no one discontinued treatment due to adverse events in TURQUOISE-1, Part 2. No cases of hepatic decompensation occurred. The only serious adverse event deemed possibly treatment related was a single case of depression. Mild fatigue, nausea, diarrhea, insomnia, headache, or itching each occurred in 10%-20% of subjects.
As effective antiretroviral therapy enables HIV-infected patients to have a lifespan approaching normal, HCV coinfection has emerged as a major issue. Roughly one-third of HIV-positive individuals are coinfected with HCV, and the rate of death due to HCV-related liver disease in coinfected patients exceeds the rate of death due to HIV. HCV genotype 1 accounts for three-quarters of chronic hepatitis C in the United States, while genotype 4 predominates in Africa, where the bulk of HIV infections occur.
During the AIDS 2016 conference, the Food and Drug Administration approved Viekira XR, an extended-release coformulation of the 3D regimen. Each tablet contains 200 mg of dasabuvir, 8.33 mg of ombitasvir, 50 mg of paritaprevir, and 33.33 mg of ritonavir. The standard dosing is three oral tablets taken once daily.
The TURQUOISE-1, Part 2 trial was sponsored by AbbVie. Dr. Rockstroh is a consultant to AbbVie and seven other pharmaceutical companies.
AT AIDS 2016
Key clinical point: All-oral regimens featuring two or three direct-acting-antiviral agents proved safe, effective, and well tolerated for treatment of chronic hepatitis C due to genotypes 1 or 4 in patients coinfected with HIV.
Major finding: The sustained virologic response rate 12 weeks after completion of an all-oral, triple-direct-acting antiviral regimen for chronic hepatitis C due to genotype 1 in HIV-coinfected patients was 98%, while the corresponding rate was 100% in coinfected patients with genotype 4 disease.
Data source: The TURQUOISE-1, Part 2 trial, a phase III multicenter study of all-oral, triple- or double-direct-acting antiviral regimens in 228 patients coinfected with HIV and hepatitis C genotypes 1 or 4.
Disclosures: The TURQUOISE-1, Part 2 trial was sponsored by AbbVie. The presenter is a consultant to the company.
