User login
Clinical Conundrum
A 62‐year‐old man with psoriasis for more than 30 years presented to the emergency department with a scaly, pruritic rash involving his face, trunk, and extremities that he had had for the past 10 days. The rash was spreading and not responding to application of clobetasol ointment, which had helped his psoriasis in the past. He also reported mild pharyngitis, headache, and myalgias.
A patient with a chronic skin condition presenting with a new rash means the clinician must consider whether it is an alternative manifestation of the chronic disorder or a new illness. Psoriasis takes many forms including guttate psoriasis, which presents with small, droplike plaques and frequently follows respiratory infections (particularly those caused by Streptococcus). Well‐controlled psoriasis rarely transforms after 3 decades, so I would consider other conditions. The tempo of illness makes certain life‐threatening syndromes, including Stevens‐Johnson, toxic shock, and purpura fulminans, unlikely. An allergic reaction, atopic dermatitis, or medication reaction is possible. Infections, either systemic (eg, syphilis) or dermatologic (eg, scabies), should be considered. Photosensitivity could involve the sun‐exposed areas, such as the extremities and face. Seborrheic dermatitis can cause scaling lesions of the face and trunk but not the extremities. Vasculitis merits consideration, but dependent regions are typically affected more than the head. Mycosis fungoides or a paraneoplastic phenomenon could cause a diffuse rash in this age group.
The patient had diabetes mellitus, hypertension, diverticulosis, and depression. Three months earlier he had undergone surgical drainage of a perirectal abscess. His usual medications were lovastatin, paroxetine, insulin, hydrochlorothiazide, and lisinopril. Three weeks previously he had completed a 10‐day course of trimethoprim/sulfamethoxazole for an upper respiratory infection. Otherwise, he was taking no new medications. He was allergic to penicillin. He denied substance abuse, recent travel, or risk factors for human immunodeficiency virus (HIV) infection. He worked as an automobile painter, lived with his wife, and had a pet dog.
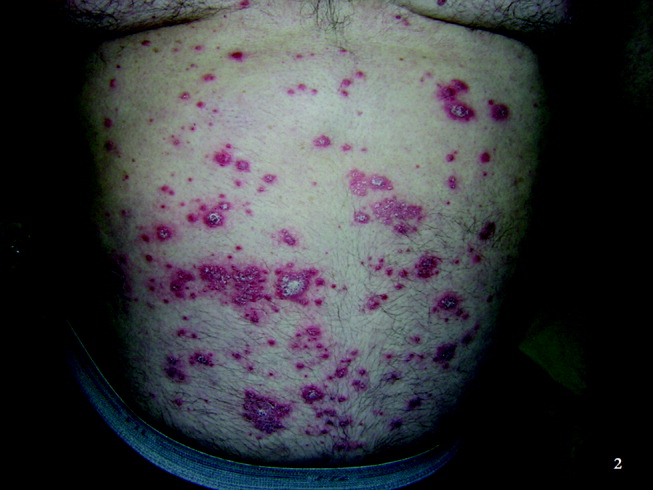
Physical examination revealed a well‐appearing man with normal vital signs. His skin had well‐defined circumscribed pink plaques, mostly 1‐2 cm in size, with thick, silvery scales in the ears and on the dorsal and ventral arms and legs, chest, back, face, and scalp. There were no pustules or other signs of infection (Figs. 1and 2). The nails exhibited distal onycholysis, oil spots, and rare pits. His posterior pharynx was mildly erythematous. The results of cardiovascular, pulmonary, and abdominal examinations were normal.

Although other scaling skin conditions such as eczema, irritant dermatitis, or malignancy remain possible, his rash is most consistent with widespread psoriasis. I would consider immunological changes that may have caused a remarkably altered and more severe expression of his chronic disease, for example, recent steroid therapy or HIV infection. The company a rash keeps helps frame the differential diagnosis. Based on the patient's well appearance, the time course, his minimal systemic symptoms, and the appearance of the rash, my leading considerations are psoriasis or an allergic dermatitis. Cutaneous T‐cell malignancy, with its indolent and sometimes protean manifestations, remains possible in a patient of his age. I would now consult a dermatologist for 3 reasons: this patient has a chronic disease that I do not manage beyond basic treatments (eg, topical steroids), he has an undiagnosed illness with substantial dermatologic manifestations, and he may need a skin biopsy for definitive diagnosis.
The dermatology team diagnosed a guttate psoriasis flare, possibly associated with streptococcal pharyngitis. The differential diagnosis included secondary syphilis, although the team believed this was less likely. The dermatology team recommended obtaining a throat culture, streptozyme assay, and rapid plasma reagin and prescribed oral erythromycin and topical steroid ointment under a sauna suit.
I would follow his response to the prescribed steroid treatments. If the patient's course deviates from the dermatologists' expectations, I would request a skin biopsy and undertake further evaluations in search of an underlying systemic disease.
The patient followed up in the dermatology clinic 3 weeks later. His rash had worsened, and he had developed patchy alopecia and progressive edema of the face, ears, and eyes. He denied mouth or tongue swelling, difficulty breathing, or hives. The streptozyme assay was positive, but the other laboratory test results were negative.
The dermatology team diagnosed a severely inflammatory psoriasis flare and prescribed an oral retinoid, acitretin, and referred him for ultraviolet light therapy. He was unable to travel for phototherapy, and acitretin was discontinued after 1 week because of elevated serum transaminase levels. The dermatologists then prescribed oral cyclosporine.
The progression of disease despite standard treatment suggests a nonpsoriatic condition. Although medications could cause the abnormal liver tests, so could another underlying illness that involves the liver. An infiltrative disorder of the skin with hair follicle destruction and local lymphedema could explain both alopecia and facial edema.
I am unable account for his clinical features with a single disease, so the differential remains broad, including severe psoriasis, an infiltrating cutaneous malignancy, or a toxic exposure. Arsenic poisoning causes hyperkeratotic skin lesions, although he lacks the associated gastrointestinal and neurological symptoms. I would not have added the potentially toxic cyclosporine.
When he returned to dermatology clinic 1 week later, his rash and facial swelling had worsened. He also reported muscle and joint aches, fatigue, lightheadedness, anorexia, nausea, abdominal pain, diarrhea, and dyspnea on exertion. He denied fever, chills, and night sweats.
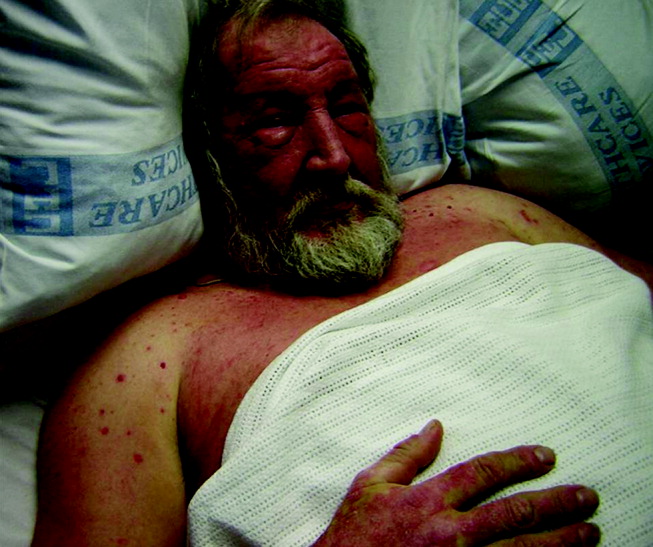
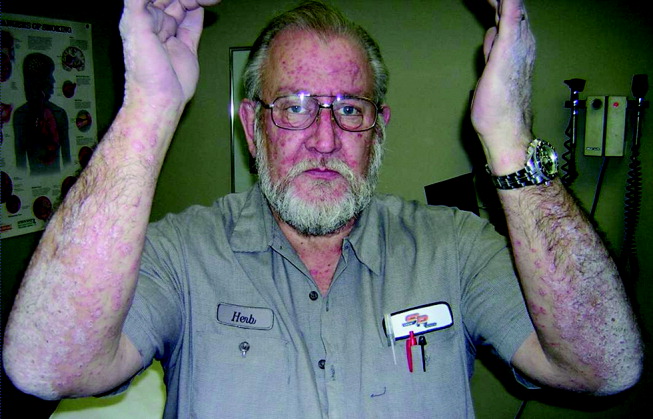
He appeared ill and used a cane to arise and walk. His vital signs and oxygen saturation were normal. He had marked swelling of his face, diffuse erythema and swelling on the chest, and widespread scaly, erythematous plaques (Fig. 3). The proximal nail folds of his fingers were erythematous, with ragged cuticles. His abdomen was mildly distended, but the rest of the physical examination was normal.
He has become too systemically ill to attribute his condition to psoriasis. The nail findings suggest dermatomyositis, which could explain many of his findings. The diffuse erythema and his difficulty walking are consistent with its skin and muscle involvement. Dyspnea could be explained by dermatomyositis‐associated interstitial lung disease. A dermatomyositis‐associated hematological or solid malignancy could account for his multisystem ailments and functional decline. A point against dermatomyositis is the relatively explosive onset of his disease. He should be carefully examined for any motor weakness. With his progressive erythroderma, I am also concerned about an advancing cutaneous T‐cell lymphoma (with leukemic transformation).
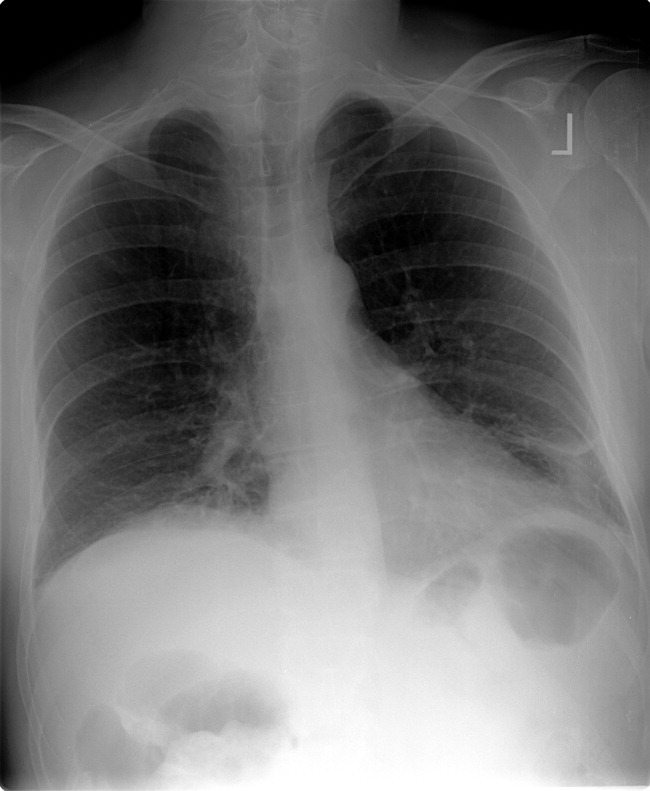
Blood tests revealed the following values: white‐blood‐cell count, 8700/L; hematocrit, 46%; platelet count, 172,000/L; blood urea nitrogen, 26 mg/dL; creatinine, 1.0 mg/dL; glucose, 199 mg/dL; albumin, 3.1 g/dL; alkaline phosphatase, 172 U/L (normal range 45‐129); alanine aminotransferase, 75 U/L (normal range 0‐39 U/L); aspartate aminotransferase, 263 U/L (normal range 0‐37 U/L); total bilirubin, 1.1 mg/dL; prothrombin time, 16 seconds (normal range 11.7‐14.3 seconds), and serum creatinine, kinase, 4253 U/L (normal range 0‐194 U/L). HIV serology was negative. Urinalysis revealed trace protein. The results of chest radiographs and an electrocardiogram were normal.
The liver function tests results are consistent with medication effects or liver involvement in a systemic disease. The creatinine kinase elevation is consistent with a myopathy such as dermatomyositis. A skin biopsy would still be useful. Depending on those results, he may need a muscle biopsy, urine heavy metal testing, and computed tomography body imaging. Considering his transaminase and creatinine kinase elevations, I would discontinue lovastatin.
The patient was hospitalized. Further questioning revealed that he had typical Raynaud's phenomenon and odynophagia. A detailed neurological examination showed weakness (3/5) of the triceps and iliopsoas muscles and difficulty rising from a chair without using his arms. Dermatoscopic examination of the proximal nail folds showed dilated capillary loops and foci of hemorrhage.
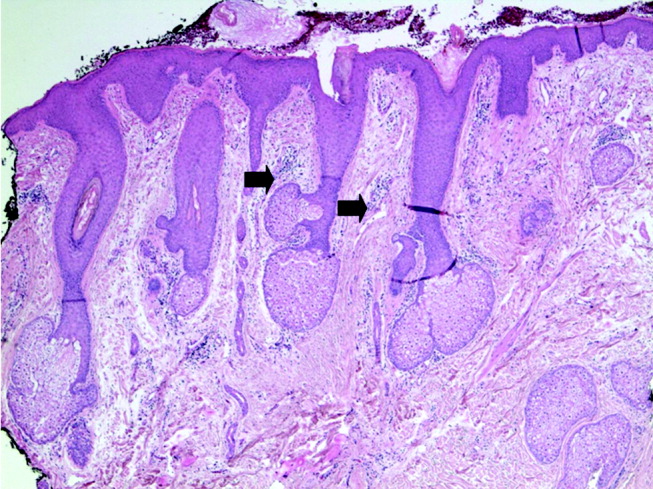
Blood tests showed a lactate dehydrogenase level of 456 U/L (normal range 0‐249 U/L) and an aldolase of 38 U/L (normal range 1.2‐7.6 U/L). Tests for antinuclear antibodies, anti‐Jo antibody, and antimyeloperoxidase antibodies were negative. Two skin biopsies were interpreted by general pathology as consistent with partially treated psoriasis, whereas another showed nonspecific changes with minimal superficial perivascular lymphohistiocytic inflammation (Fig. 4). Lisinopril was discontinued because of its possible contribution to the facial edema.
Dermatomyositis is now the leading diagnosis. Characteristic features include his proximal muscle weakness, Raynaud's phenomenon, and dilated nailfold capillary loops. I am not overly dissuaded by the negative antinuclear antibodies, but because of additional atypical features (ie, extensive cutaneous edema, rapid onset, illness severity, prominent gastrointestinal symptoms), a confirmatory muscle biopsy is needed. Endoscopy of the proximal aerodigestive tract would help evaluate the odynophagia. There is little to suggest infection, malignancy, or metabolic derangement.
The inpatient medical team considered myositis related to retinoid or cyclosporine therapy. They discontinued cyclosporine and began systemic corticosteroid therapy. Within a few days, the patient's rash, muscle pain, and weakness improved, and the elevated transaminase and creatinine kinase levels decreased.
Dermatology recommended an evaluation for dermatomyositis‐associated malignancy, but the medicine team and rheumatology consultants, noting the lack of classic skin findings (heliotrope rash and Gottron's papules) and the uncharacteristically rapid onset and improvement of myositis, suggested delaying the evaluation until dermatomyositis was proven.
An immediate improvement in symptoms with steroids is nonspecific, often occurring in autoimmune, infectious, and neoplastic diseases. This juncture in the case is common in complex multisystem illnesses, where various consultants may arrive at differing conclusions. With both typical and atypical features of dermatomyositis, where should one set the therapeutic threshold, that is, the point where one ends testing, accepts a diagnosis, and initiates treatment? Several factors raise the level of certainty I would require. First, dermatomyositis is quite rare. Adding atypical features further increases the burden of proof for that illness. Second, the existence of alternative possibilities (admittedly of equal uncertainty) gives me some pause. Finally, the toxicity of the proposed treatments raises the therapeutic threshold. Acknowledging that empiric treatment may be indicated for a severely ill patient at a lower level of certainty, I would hesitate to commit a patient to long‐term steroids without being confident of the diagnosis. I would therefore require a muscle biopsy, or at least electromyography to support or exclude dermatomyositis.
The patient was discharged from the hospital on high‐dose prednisone. He underwent electromyography, which revealed inflammatory myopathic changes more apparent in the proximal than distal muscles. These findings were thought to be compatible with dermatomyositis, although the fibrillations and positive sharp waves characteristic of acute inflammation were absent, perhaps because of corticosteroid therapy.
The patient mistakenly stopped taking his prednisone. Within days, his weakness and skin rash worsened, and he developed nausea with vomiting. He returned to clinic, where his creatinine kinase level was again found to be elevated, and he was rehospitalized. Oral corticosteroid therapy was restarted with prompt improvement. On review of the original skin biopsies, a dermatopathologist observed areas of thickened dermal collagen and a superficial and deep perivascular lymphocytic infiltrate, both consistent with connective tissue disease.
These 3 additional findings (ie, electromyography results, temporally established steroid responsiveness, and the new skin biopsy interpretation) in aggregate support the diagnosis of dermatomyositis, but the nausea and vomiting are unusual. I would discuss these results with a rheumatologist and still request a confirmatory muscle biopsy. Because diagnosing dermatomyositis should prompt consideration of seeking an underlying malignancy in a patient of this age group, I would repeat a targeted history and physical examination along with age‐ and risk‐factor‐appropriate screening. If muscle biopsy results are not definitive, finding an underlying malignancy would lend support to dermatomyositis.
While hospitalized, the patient complained of continued odynophagia and was noted to have oral candidiasis. Upper endoscopy, undertaken to evaluate for esophageal candidiasis, revealed a mass at the gastroesophageal junction. Biopsy revealed gastric‐type adenocarcinoma. An abdominal computed tomography scan demonstrated 3 hypodense hepatic lesions, evidence of cirrhosis, and ascites. Cytology of paracentesis fluid revealed cells compatible with adenocarcinoma. The patient died in hospice care 2 weeks later.
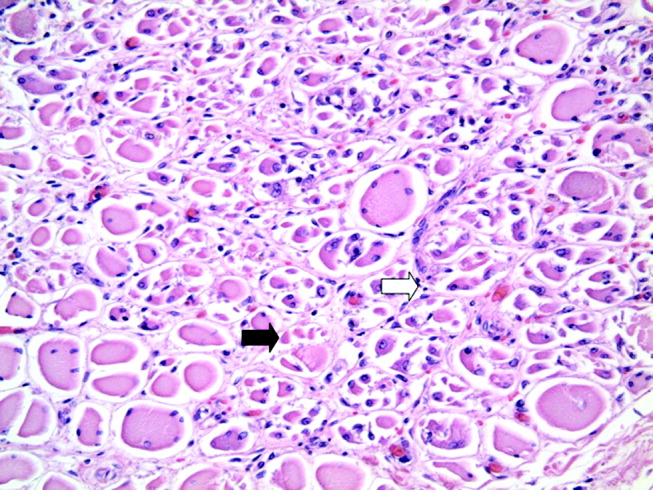
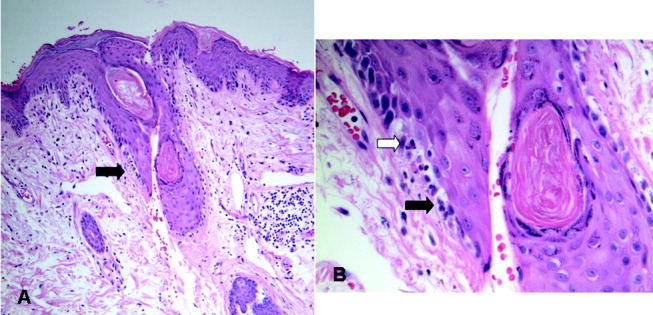
At autopsy, he had metastatic gastric‐type adenocarcinoma. A muscle biopsy (Fig. 5) revealed muscle atrophy with small foci of lymphocytic infiltrates, most compatible with dermatomyositis. Another dermatopathologist reviewed the skin biopsies and noted interface dermatitis, which is typical of connective tissue diseases like dermatomyositis (Fig. 6A,B).
COMMENTARY
Dermatomyositis is an idiopathic inflammatory myopathy characterized by endomysial inflammation and muscle weakness and differentiated from other myopathies by the presence of a rash.1 Muscle disease may manifest with or precede the rash, but up to 40% of patients present with skin manifestations alone, an entity called amyopathic dermatomyositis.2 When present, the myositis generally develops over months, but the onset can be acute.1 The weakness is typically symmetrical and proximal,1 and many patients have oropharyngeal dysphagia.3
The characteristic rash is erythematous, symmetrical, and photodistributed.4 Classic cutaneous findings are the heliotrope rash (violaceous eyelid erythema), which is pathognomonic but uncommon, and the more common Gottron's papules (violaceous, slightly elevated papules and plaques on bony prominences and extensor surfaces, especially the knuckles).4 Other findings include periorbital edema, scalp dermatitis, poikiloderma (ie, hyperpigmentation, hypopigmentation, atrophy, and telangiectasia), periungual erythema, and dystrophic cuticles.2 The cutaneous manifestations of dermatomyositis may be similar to those of psoriasis, systemic lupus erythematosus, lichen planus, rosacea, polymorphous light eruption, drug eruption, atopic dermatitis, seborrheic dermatitis, or allergic contact dermatitis.4
Diagnosing dermatomyositis requires considering clinical, laboratory, electromyographical, and histological evidence, as there are no widely accepted, validated diagnostic criteria.1, 5 The diagnosis is usually suspected if there is a characteristic rash and symptoms of myositis (eg, proximal muscle weakness, myalgias, fatigue, or an inability to swallow). When the patient has an atypical rash, skin biopsy can differentiate dermatomyositis from other conditions, except lupus, which shares the key finding of interface dermatitis.2 The histological findings can be variable and subtle,6 so consultation with a dermatopathologist may be helpful.
Myositis may be confirmed by various studies. Most patients have elevated muscle enzymes (ie, creatinine kinase, aldolase, lactate dehydrogenase, or transaminases)1; for those who do not, magnetic resonance imaging can be helpful in detecting muscle involvement and locating the best site for muscle biopsy.7 Electromyography reveals nonspecific muscle membrane instability.8 Muscle biopsy shows muscle fiber necrosis, perifascicular atrophy, and perivascular and perifascicular lymphocytic infiltrates. These can be patchy, diminished by steroid use, and occasionally seen in noninflammatory muscular dystrophies.8 For a patient with typical myositis and a characteristic rash, muscle biopsy may be unnecessary.1
The clinical utility of serologic testing for diagnosing dermatomyositis is controversial.2 Myositis‐specific antibody testing is insensitive but specific; these antibodies include Jo‐1, an antisynthetase antibody that predicts incomplete response to therapy and lung involvement, and Mi‐2, which is associated with better response to therapy.2, 9, 10 The sensitivity and specificity of antinuclear antibodies are both approximately 60%.10
Patients with dermatomyositis have higher rates of cancers than age‐matched controls, and nearly 25% of patients are diagnosed with a malignancy at some point during the course of the disease.11 Malignancies are typically solid tumors that manifest within 3 years of the diagnosis,1214 although the increased risk may exist for at least 5 years.14 There is a 10‐fold higher risk of ovarian cancer in women with dermatomyositis.12, 15 Other associated malignancies include lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma.14
Recommendations for screening affected patients for cancer have changed over the years, with increasing evidence of an association between dermatomyositis and malignancy and evolving improvements in diagnostic techniques.16 Many authorities recommend that all adult patients with dermatomyositis be evaluated for cancer, including a complete physical examination, basic hematological tests, age‐ and sex‐appropriate screening (eg, mammography, pap smear, and colonoscopy), and chest x‐ray.16 Some would add upper endoscopy; imaging of the chest, abdomen, and pelvis; gynecological examination; and serum CA‐125 level to better evaluate for the most common malignancies (ie, ovarian, gastric, lung, and pancreatic carcinomas and non‐Hodgkins lymphoma).12, 1720
In 19% of adults, dermatomyositis overlaps with other autoimmune disorders, usually systemic lupus erythematosus and systemic sclerosis.21 These manifest as Raynaud's phenomenon, arthritis, esophageal dysmotility, renal disease, or neuropathy.21 Other potentially serious systemic manifestations of dermatomyositis include proximal dysphagia from pharyngeal myopathy; distal dysphagia from esophageal dysmotility in systemic sclerosis overlap; pulmonary disease from autoimmune interstitial lung disease or aspiration; cardiac disease from conduction abnormalities, myocarditis, pericarditis, and valvular disease; and rhabdomyolysis.2
Treatment of dermatomyositis requires systemic immunosuppression with 1 or more agents. The prognosis of dermatomyositis is variable. Mortality at 5 years ranges from 23% to 73%. At least a third of patients are left with mild to severe disability.1 In addition to older age, predictors of poor outcome include male sex, dysphagia, longstanding symptoms before treatment, pulmonary or cardiac involvement, and presence of antisynthetase antibodies.22
Dermatomyositis is often treated in the outpatient setting, but there are many reasons for hospitalization. Complications of treatment, like infection or adverse effects of medications, could result in hospitalization. Treatment with intravenous pulse corticosteroids or IVIG may require inpatient administration if no infusion center is available. Other indications for inpatient evaluation include the consequences of various malignancies and the more severe expression of systemic complications of dermatomyositis (eg, dysphagia and pulmonary, cardiac, or renal disease).
Every parent knows the plaintive backseat whine, Are we there, yet? Clinicians may also experience this feeling when attempting to diagnose a perplexing illness, especially one that lacks a definitive diagnostic test. It was easy for this patient's doctors to assume initially that his new rash was a manifestation of his long‐standing psoriasis. Having done so, they could understandably attribute the subsequent findings to either evolution of this disease or to consequences of the prescribed treatments, rather than considering a novel diagnosis. Only when faced with new (or newly appreciated) findings suggesting myopathy did the clinicians (and our discussant) consider the diagnosis of dermatomyositis. Even then, the primary inpatient medical team and their consultants were unsure when they had sufficient evidence to be certain.
Several factors compounded the difficulty of making a diagnosis in this case: the clinicians were dealing with a rare disease; they were considering alternative diagnoses (ie, psoriasis or a toxic effect of medication); and the disease presented somewhat atypically. The clinicians initially failed to consider and then accept the correct diagnosis because the patient's rash was not classic, his biopsy was interpreted as nonspecific, and he lacked myositis at presentation. Furthermore, when the generalists sought expert assistance, they encountered a difference of opinion among the consultants. These complex situations should goad the clinician into carefully considering the therapeutic threshold, that is, the transition point from diagnostic testing to therapeutic intervention.23 With complex cases like this, it may be difficult to know when one has reached a strongly supported diagnosis, and frequently asking whether we are there yet may be appropriate.
Take‐Home Points for the Hospitalist
-
A skin rash, which may have typical or atypical features, distinguishes dermatomyositis from other acquired myopathies.
-
Consider consultation with pathology specialists for skin and muscle biopsies.
-
Ovarian, lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma are the most common cancers associated with dermatomyositis.
-
In addition to age‐appropriate cancer screening, consider obtaining upper endoscopy, imaging of the chest/abdomen/pelvis, and CA‐125.
-
Patients with dermatomyositis and no obvious concurrent malignancy need long‐term outpatient follow‐up for repeated malignancy screening.
- ,.Polymyositis and dermatomyositis.Lancet.2003;362:971–982.
- .Dermatomyositis.Lancet.2000;355:53–47.
- ,,,.Oropharyngeal dysphagia in polymyositis/dermatomyositis.Clin Neurol Neurosurg.2004;107(1):32–37.
- ,.Skin involvement in dermatomyositis.Curr Opin Rheumatol.2003;15:714–22.
- ,,,,,.Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients.Medicine (Baltimore).2005;84:231–249.
- .Skin Pathology.2nd ed.New York:Churchill Livingstone;2002.
- ,.Utility of magnetic resonance imaging in the evaluation of patients with inflammatory myopathies.Curr Rheumatol Rep.2001;3:334–245.
- ,,.Is it really myositis? A consideration of the differential diagnosis.Curr Opin Rheumatol2004;16:684–691.
- .Idiopathic inflammatory myopathy: autoantibody update.Curr Rheumatol Rep.2002;4:434–441.
- ,,.Laboratory assessment in musculoskeletal disorders.Best Pract Res Clin Rheumatol.2003;17:475–494.
- ,.Dermatomyositis.Clin Dermatol.2006;24:363–373.
- ,,, et al.Frequency of specific cancer types in dermatomyositis and polymyositis: a population‐based study.Lancet.2001;357:96–100.
- ,,, et al.Cancer‐associated myositis: clinical features and prognostic signs.Ann N Y Acad Sci.2005;1051:64–71.
- ,,,,.Incidence of malignant disease in biopsy‐proven inflammatory myopathy. A population‐based cohort study.Ann Intern Med.2001;134:1087–1095.
- ,,.Risk of cancer in patients with dermatomyositis or polymyositis, and follow‐up implications: a Scottish population‐based cohort study.Br J Cancer.2001;85 (1):41–45.
- .When and how should the patient with dermatomyositis or amyopathic dermatomyositis be assessed for possible cancer?Arch Dermatol.2002;138:969–971.
- ,,.Ovarian cancer in patients with dermatomyositis.Medicine (Baltimore).1994;73(3):153–160.
- ,,,.Dermatomyositis sine myositis: association with malignancy.J Rheumatol.1996;23 (1):101–105.
- ,,, et al.Tumor antigen markers for the detection of solid cancers in inflammatory myopathies.Cancer Epidemiol Biomarkers Prev.2005;14:1279–1282.
- ,,, et al.Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients.Arch Dermatol.2002;138:885–890.
- ,,,,,.Dermatomyositis: a dermatology‐based case series.J Am Acad Dermatol.1998;38:397–404.
- ,,, et al.Long‐term outcome in polymyositis and dermatomyositis.Ann Rheum Dis.2006;65:1456–1461.
- .Our stubborn quest for diagnostic certainty. A cause of excessive testing.N Engl J Med.1989;320:1489–1491.
A 62‐year‐old man with psoriasis for more than 30 years presented to the emergency department with a scaly, pruritic rash involving his face, trunk, and extremities that he had had for the past 10 days. The rash was spreading and not responding to application of clobetasol ointment, which had helped his psoriasis in the past. He also reported mild pharyngitis, headache, and myalgias.
A patient with a chronic skin condition presenting with a new rash means the clinician must consider whether it is an alternative manifestation of the chronic disorder or a new illness. Psoriasis takes many forms including guttate psoriasis, which presents with small, droplike plaques and frequently follows respiratory infections (particularly those caused by Streptococcus). Well‐controlled psoriasis rarely transforms after 3 decades, so I would consider other conditions. The tempo of illness makes certain life‐threatening syndromes, including Stevens‐Johnson, toxic shock, and purpura fulminans, unlikely. An allergic reaction, atopic dermatitis, or medication reaction is possible. Infections, either systemic (eg, syphilis) or dermatologic (eg, scabies), should be considered. Photosensitivity could involve the sun‐exposed areas, such as the extremities and face. Seborrheic dermatitis can cause scaling lesions of the face and trunk but not the extremities. Vasculitis merits consideration, but dependent regions are typically affected more than the head. Mycosis fungoides or a paraneoplastic phenomenon could cause a diffuse rash in this age group.
The patient had diabetes mellitus, hypertension, diverticulosis, and depression. Three months earlier he had undergone surgical drainage of a perirectal abscess. His usual medications were lovastatin, paroxetine, insulin, hydrochlorothiazide, and lisinopril. Three weeks previously he had completed a 10‐day course of trimethoprim/sulfamethoxazole for an upper respiratory infection. Otherwise, he was taking no new medications. He was allergic to penicillin. He denied substance abuse, recent travel, or risk factors for human immunodeficiency virus (HIV) infection. He worked as an automobile painter, lived with his wife, and had a pet dog.
Physical examination revealed a well‐appearing man with normal vital signs. His skin had well‐defined circumscribed pink plaques, mostly 1‐2 cm in size, with thick, silvery scales in the ears and on the dorsal and ventral arms and legs, chest, back, face, and scalp. There were no pustules or other signs of infection (Figs. 1and 2). The nails exhibited distal onycholysis, oil spots, and rare pits. His posterior pharynx was mildly erythematous. The results of cardiovascular, pulmonary, and abdominal examinations were normal.
Although other scaling skin conditions such as eczema, irritant dermatitis, or malignancy remain possible, his rash is most consistent with widespread psoriasis. I would consider immunological changes that may have caused a remarkably altered and more severe expression of his chronic disease, for example, recent steroid therapy or HIV infection. The company a rash keeps helps frame the differential diagnosis. Based on the patient's well appearance, the time course, his minimal systemic symptoms, and the appearance of the rash, my leading considerations are psoriasis or an allergic dermatitis. Cutaneous T‐cell malignancy, with its indolent and sometimes protean manifestations, remains possible in a patient of his age. I would now consult a dermatologist for 3 reasons: this patient has a chronic disease that I do not manage beyond basic treatments (eg, topical steroids), he has an undiagnosed illness with substantial dermatologic manifestations, and he may need a skin biopsy for definitive diagnosis.
The dermatology team diagnosed a guttate psoriasis flare, possibly associated with streptococcal pharyngitis. The differential diagnosis included secondary syphilis, although the team believed this was less likely. The dermatology team recommended obtaining a throat culture, streptozyme assay, and rapid plasma reagin and prescribed oral erythromycin and topical steroid ointment under a sauna suit.
I would follow his response to the prescribed steroid treatments. If the patient's course deviates from the dermatologists' expectations, I would request a skin biopsy and undertake further evaluations in search of an underlying systemic disease.
The patient followed up in the dermatology clinic 3 weeks later. His rash had worsened, and he had developed patchy alopecia and progressive edema of the face, ears, and eyes. He denied mouth or tongue swelling, difficulty breathing, or hives. The streptozyme assay was positive, but the other laboratory test results were negative.
The dermatology team diagnosed a severely inflammatory psoriasis flare and prescribed an oral retinoid, acitretin, and referred him for ultraviolet light therapy. He was unable to travel for phototherapy, and acitretin was discontinued after 1 week because of elevated serum transaminase levels. The dermatologists then prescribed oral cyclosporine.
The progression of disease despite standard treatment suggests a nonpsoriatic condition. Although medications could cause the abnormal liver tests, so could another underlying illness that involves the liver. An infiltrative disorder of the skin with hair follicle destruction and local lymphedema could explain both alopecia and facial edema.
I am unable account for his clinical features with a single disease, so the differential remains broad, including severe psoriasis, an infiltrating cutaneous malignancy, or a toxic exposure. Arsenic poisoning causes hyperkeratotic skin lesions, although he lacks the associated gastrointestinal and neurological symptoms. I would not have added the potentially toxic cyclosporine.
When he returned to dermatology clinic 1 week later, his rash and facial swelling had worsened. He also reported muscle and joint aches, fatigue, lightheadedness, anorexia, nausea, abdominal pain, diarrhea, and dyspnea on exertion. He denied fever, chills, and night sweats.
He appeared ill and used a cane to arise and walk. His vital signs and oxygen saturation were normal. He had marked swelling of his face, diffuse erythema and swelling on the chest, and widespread scaly, erythematous plaques (Fig. 3). The proximal nail folds of his fingers were erythematous, with ragged cuticles. His abdomen was mildly distended, but the rest of the physical examination was normal.
He has become too systemically ill to attribute his condition to psoriasis. The nail findings suggest dermatomyositis, which could explain many of his findings. The diffuse erythema and his difficulty walking are consistent with its skin and muscle involvement. Dyspnea could be explained by dermatomyositis‐associated interstitial lung disease. A dermatomyositis‐associated hematological or solid malignancy could account for his multisystem ailments and functional decline. A point against dermatomyositis is the relatively explosive onset of his disease. He should be carefully examined for any motor weakness. With his progressive erythroderma, I am also concerned about an advancing cutaneous T‐cell lymphoma (with leukemic transformation).
Blood tests revealed the following values: white‐blood‐cell count, 8700/L; hematocrit, 46%; platelet count, 172,000/L; blood urea nitrogen, 26 mg/dL; creatinine, 1.0 mg/dL; glucose, 199 mg/dL; albumin, 3.1 g/dL; alkaline phosphatase, 172 U/L (normal range 45‐129); alanine aminotransferase, 75 U/L (normal range 0‐39 U/L); aspartate aminotransferase, 263 U/L (normal range 0‐37 U/L); total bilirubin, 1.1 mg/dL; prothrombin time, 16 seconds (normal range 11.7‐14.3 seconds), and serum creatinine, kinase, 4253 U/L (normal range 0‐194 U/L). HIV serology was negative. Urinalysis revealed trace protein. The results of chest radiographs and an electrocardiogram were normal.
The liver function tests results are consistent with medication effects or liver involvement in a systemic disease. The creatinine kinase elevation is consistent with a myopathy such as dermatomyositis. A skin biopsy would still be useful. Depending on those results, he may need a muscle biopsy, urine heavy metal testing, and computed tomography body imaging. Considering his transaminase and creatinine kinase elevations, I would discontinue lovastatin.
The patient was hospitalized. Further questioning revealed that he had typical Raynaud's phenomenon and odynophagia. A detailed neurological examination showed weakness (3/5) of the triceps and iliopsoas muscles and difficulty rising from a chair without using his arms. Dermatoscopic examination of the proximal nail folds showed dilated capillary loops and foci of hemorrhage.
Blood tests showed a lactate dehydrogenase level of 456 U/L (normal range 0‐249 U/L) and an aldolase of 38 U/L (normal range 1.2‐7.6 U/L). Tests for antinuclear antibodies, anti‐Jo antibody, and antimyeloperoxidase antibodies were negative. Two skin biopsies were interpreted by general pathology as consistent with partially treated psoriasis, whereas another showed nonspecific changes with minimal superficial perivascular lymphohistiocytic inflammation (Fig. 4). Lisinopril was discontinued because of its possible contribution to the facial edema.
Dermatomyositis is now the leading diagnosis. Characteristic features include his proximal muscle weakness, Raynaud's phenomenon, and dilated nailfold capillary loops. I am not overly dissuaded by the negative antinuclear antibodies, but because of additional atypical features (ie, extensive cutaneous edema, rapid onset, illness severity, prominent gastrointestinal symptoms), a confirmatory muscle biopsy is needed. Endoscopy of the proximal aerodigestive tract would help evaluate the odynophagia. There is little to suggest infection, malignancy, or metabolic derangement.
The inpatient medical team considered myositis related to retinoid or cyclosporine therapy. They discontinued cyclosporine and began systemic corticosteroid therapy. Within a few days, the patient's rash, muscle pain, and weakness improved, and the elevated transaminase and creatinine kinase levels decreased.
Dermatology recommended an evaluation for dermatomyositis‐associated malignancy, but the medicine team and rheumatology consultants, noting the lack of classic skin findings (heliotrope rash and Gottron's papules) and the uncharacteristically rapid onset and improvement of myositis, suggested delaying the evaluation until dermatomyositis was proven.
An immediate improvement in symptoms with steroids is nonspecific, often occurring in autoimmune, infectious, and neoplastic diseases. This juncture in the case is common in complex multisystem illnesses, where various consultants may arrive at differing conclusions. With both typical and atypical features of dermatomyositis, where should one set the therapeutic threshold, that is, the point where one ends testing, accepts a diagnosis, and initiates treatment? Several factors raise the level of certainty I would require. First, dermatomyositis is quite rare. Adding atypical features further increases the burden of proof for that illness. Second, the existence of alternative possibilities (admittedly of equal uncertainty) gives me some pause. Finally, the toxicity of the proposed treatments raises the therapeutic threshold. Acknowledging that empiric treatment may be indicated for a severely ill patient at a lower level of certainty, I would hesitate to commit a patient to long‐term steroids without being confident of the diagnosis. I would therefore require a muscle biopsy, or at least electromyography to support or exclude dermatomyositis.
The patient was discharged from the hospital on high‐dose prednisone. He underwent electromyography, which revealed inflammatory myopathic changes more apparent in the proximal than distal muscles. These findings were thought to be compatible with dermatomyositis, although the fibrillations and positive sharp waves characteristic of acute inflammation were absent, perhaps because of corticosteroid therapy.
The patient mistakenly stopped taking his prednisone. Within days, his weakness and skin rash worsened, and he developed nausea with vomiting. He returned to clinic, where his creatinine kinase level was again found to be elevated, and he was rehospitalized. Oral corticosteroid therapy was restarted with prompt improvement. On review of the original skin biopsies, a dermatopathologist observed areas of thickened dermal collagen and a superficial and deep perivascular lymphocytic infiltrate, both consistent with connective tissue disease.
These 3 additional findings (ie, electromyography results, temporally established steroid responsiveness, and the new skin biopsy interpretation) in aggregate support the diagnosis of dermatomyositis, but the nausea and vomiting are unusual. I would discuss these results with a rheumatologist and still request a confirmatory muscle biopsy. Because diagnosing dermatomyositis should prompt consideration of seeking an underlying malignancy in a patient of this age group, I would repeat a targeted history and physical examination along with age‐ and risk‐factor‐appropriate screening. If muscle biopsy results are not definitive, finding an underlying malignancy would lend support to dermatomyositis.
While hospitalized, the patient complained of continued odynophagia and was noted to have oral candidiasis. Upper endoscopy, undertaken to evaluate for esophageal candidiasis, revealed a mass at the gastroesophageal junction. Biopsy revealed gastric‐type adenocarcinoma. An abdominal computed tomography scan demonstrated 3 hypodense hepatic lesions, evidence of cirrhosis, and ascites. Cytology of paracentesis fluid revealed cells compatible with adenocarcinoma. The patient died in hospice care 2 weeks later.
At autopsy, he had metastatic gastric‐type adenocarcinoma. A muscle biopsy (Fig. 5) revealed muscle atrophy with small foci of lymphocytic infiltrates, most compatible with dermatomyositis. Another dermatopathologist reviewed the skin biopsies and noted interface dermatitis, which is typical of connective tissue diseases like dermatomyositis (Fig. 6A,B).
COMMENTARY
Dermatomyositis is an idiopathic inflammatory myopathy characterized by endomysial inflammation and muscle weakness and differentiated from other myopathies by the presence of a rash.1 Muscle disease may manifest with or precede the rash, but up to 40% of patients present with skin manifestations alone, an entity called amyopathic dermatomyositis.2 When present, the myositis generally develops over months, but the onset can be acute.1 The weakness is typically symmetrical and proximal,1 and many patients have oropharyngeal dysphagia.3
The characteristic rash is erythematous, symmetrical, and photodistributed.4 Classic cutaneous findings are the heliotrope rash (violaceous eyelid erythema), which is pathognomonic but uncommon, and the more common Gottron's papules (violaceous, slightly elevated papules and plaques on bony prominences and extensor surfaces, especially the knuckles).4 Other findings include periorbital edema, scalp dermatitis, poikiloderma (ie, hyperpigmentation, hypopigmentation, atrophy, and telangiectasia), periungual erythema, and dystrophic cuticles.2 The cutaneous manifestations of dermatomyositis may be similar to those of psoriasis, systemic lupus erythematosus, lichen planus, rosacea, polymorphous light eruption, drug eruption, atopic dermatitis, seborrheic dermatitis, or allergic contact dermatitis.4
Diagnosing dermatomyositis requires considering clinical, laboratory, electromyographical, and histological evidence, as there are no widely accepted, validated diagnostic criteria.1, 5 The diagnosis is usually suspected if there is a characteristic rash and symptoms of myositis (eg, proximal muscle weakness, myalgias, fatigue, or an inability to swallow). When the patient has an atypical rash, skin biopsy can differentiate dermatomyositis from other conditions, except lupus, which shares the key finding of interface dermatitis.2 The histological findings can be variable and subtle,6 so consultation with a dermatopathologist may be helpful.
Myositis may be confirmed by various studies. Most patients have elevated muscle enzymes (ie, creatinine kinase, aldolase, lactate dehydrogenase, or transaminases)1; for those who do not, magnetic resonance imaging can be helpful in detecting muscle involvement and locating the best site for muscle biopsy.7 Electromyography reveals nonspecific muscle membrane instability.8 Muscle biopsy shows muscle fiber necrosis, perifascicular atrophy, and perivascular and perifascicular lymphocytic infiltrates. These can be patchy, diminished by steroid use, and occasionally seen in noninflammatory muscular dystrophies.8 For a patient with typical myositis and a characteristic rash, muscle biopsy may be unnecessary.1
The clinical utility of serologic testing for diagnosing dermatomyositis is controversial.2 Myositis‐specific antibody testing is insensitive but specific; these antibodies include Jo‐1, an antisynthetase antibody that predicts incomplete response to therapy and lung involvement, and Mi‐2, which is associated with better response to therapy.2, 9, 10 The sensitivity and specificity of antinuclear antibodies are both approximately 60%.10
Patients with dermatomyositis have higher rates of cancers than age‐matched controls, and nearly 25% of patients are diagnosed with a malignancy at some point during the course of the disease.11 Malignancies are typically solid tumors that manifest within 3 years of the diagnosis,1214 although the increased risk may exist for at least 5 years.14 There is a 10‐fold higher risk of ovarian cancer in women with dermatomyositis.12, 15 Other associated malignancies include lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma.14
Recommendations for screening affected patients for cancer have changed over the years, with increasing evidence of an association between dermatomyositis and malignancy and evolving improvements in diagnostic techniques.16 Many authorities recommend that all adult patients with dermatomyositis be evaluated for cancer, including a complete physical examination, basic hematological tests, age‐ and sex‐appropriate screening (eg, mammography, pap smear, and colonoscopy), and chest x‐ray.16 Some would add upper endoscopy; imaging of the chest, abdomen, and pelvis; gynecological examination; and serum CA‐125 level to better evaluate for the most common malignancies (ie, ovarian, gastric, lung, and pancreatic carcinomas and non‐Hodgkins lymphoma).12, 1720
In 19% of adults, dermatomyositis overlaps with other autoimmune disorders, usually systemic lupus erythematosus and systemic sclerosis.21 These manifest as Raynaud's phenomenon, arthritis, esophageal dysmotility, renal disease, or neuropathy.21 Other potentially serious systemic manifestations of dermatomyositis include proximal dysphagia from pharyngeal myopathy; distal dysphagia from esophageal dysmotility in systemic sclerosis overlap; pulmonary disease from autoimmune interstitial lung disease or aspiration; cardiac disease from conduction abnormalities, myocarditis, pericarditis, and valvular disease; and rhabdomyolysis.2
Treatment of dermatomyositis requires systemic immunosuppression with 1 or more agents. The prognosis of dermatomyositis is variable. Mortality at 5 years ranges from 23% to 73%. At least a third of patients are left with mild to severe disability.1 In addition to older age, predictors of poor outcome include male sex, dysphagia, longstanding symptoms before treatment, pulmonary or cardiac involvement, and presence of antisynthetase antibodies.22
Dermatomyositis is often treated in the outpatient setting, but there are many reasons for hospitalization. Complications of treatment, like infection or adverse effects of medications, could result in hospitalization. Treatment with intravenous pulse corticosteroids or IVIG may require inpatient administration if no infusion center is available. Other indications for inpatient evaluation include the consequences of various malignancies and the more severe expression of systemic complications of dermatomyositis (eg, dysphagia and pulmonary, cardiac, or renal disease).
Every parent knows the plaintive backseat whine, Are we there, yet? Clinicians may also experience this feeling when attempting to diagnose a perplexing illness, especially one that lacks a definitive diagnostic test. It was easy for this patient's doctors to assume initially that his new rash was a manifestation of his long‐standing psoriasis. Having done so, they could understandably attribute the subsequent findings to either evolution of this disease or to consequences of the prescribed treatments, rather than considering a novel diagnosis. Only when faced with new (or newly appreciated) findings suggesting myopathy did the clinicians (and our discussant) consider the diagnosis of dermatomyositis. Even then, the primary inpatient medical team and their consultants were unsure when they had sufficient evidence to be certain.
Several factors compounded the difficulty of making a diagnosis in this case: the clinicians were dealing with a rare disease; they were considering alternative diagnoses (ie, psoriasis or a toxic effect of medication); and the disease presented somewhat atypically. The clinicians initially failed to consider and then accept the correct diagnosis because the patient's rash was not classic, his biopsy was interpreted as nonspecific, and he lacked myositis at presentation. Furthermore, when the generalists sought expert assistance, they encountered a difference of opinion among the consultants. These complex situations should goad the clinician into carefully considering the therapeutic threshold, that is, the transition point from diagnostic testing to therapeutic intervention.23 With complex cases like this, it may be difficult to know when one has reached a strongly supported diagnosis, and frequently asking whether we are there yet may be appropriate.
Take‐Home Points for the Hospitalist
-
A skin rash, which may have typical or atypical features, distinguishes dermatomyositis from other acquired myopathies.
-
Consider consultation with pathology specialists for skin and muscle biopsies.
-
Ovarian, lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma are the most common cancers associated with dermatomyositis.
-
In addition to age‐appropriate cancer screening, consider obtaining upper endoscopy, imaging of the chest/abdomen/pelvis, and CA‐125.
-
Patients with dermatomyositis and no obvious concurrent malignancy need long‐term outpatient follow‐up for repeated malignancy screening.
A 62‐year‐old man with psoriasis for more than 30 years presented to the emergency department with a scaly, pruritic rash involving his face, trunk, and extremities that he had had for the past 10 days. The rash was spreading and not responding to application of clobetasol ointment, which had helped his psoriasis in the past. He also reported mild pharyngitis, headache, and myalgias.
A patient with a chronic skin condition presenting with a new rash means the clinician must consider whether it is an alternative manifestation of the chronic disorder or a new illness. Psoriasis takes many forms including guttate psoriasis, which presents with small, droplike plaques and frequently follows respiratory infections (particularly those caused by Streptococcus). Well‐controlled psoriasis rarely transforms after 3 decades, so I would consider other conditions. The tempo of illness makes certain life‐threatening syndromes, including Stevens‐Johnson, toxic shock, and purpura fulminans, unlikely. An allergic reaction, atopic dermatitis, or medication reaction is possible. Infections, either systemic (eg, syphilis) or dermatologic (eg, scabies), should be considered. Photosensitivity could involve the sun‐exposed areas, such as the extremities and face. Seborrheic dermatitis can cause scaling lesions of the face and trunk but not the extremities. Vasculitis merits consideration, but dependent regions are typically affected more than the head. Mycosis fungoides or a paraneoplastic phenomenon could cause a diffuse rash in this age group.
The patient had diabetes mellitus, hypertension, diverticulosis, and depression. Three months earlier he had undergone surgical drainage of a perirectal abscess. His usual medications were lovastatin, paroxetine, insulin, hydrochlorothiazide, and lisinopril. Three weeks previously he had completed a 10‐day course of trimethoprim/sulfamethoxazole for an upper respiratory infection. Otherwise, he was taking no new medications. He was allergic to penicillin. He denied substance abuse, recent travel, or risk factors for human immunodeficiency virus (HIV) infection. He worked as an automobile painter, lived with his wife, and had a pet dog.
Physical examination revealed a well‐appearing man with normal vital signs. His skin had well‐defined circumscribed pink plaques, mostly 1‐2 cm in size, with thick, silvery scales in the ears and on the dorsal and ventral arms and legs, chest, back, face, and scalp. There were no pustules or other signs of infection (Figs. 1and 2). The nails exhibited distal onycholysis, oil spots, and rare pits. His posterior pharynx was mildly erythematous. The results of cardiovascular, pulmonary, and abdominal examinations were normal.
Although other scaling skin conditions such as eczema, irritant dermatitis, or malignancy remain possible, his rash is most consistent with widespread psoriasis. I would consider immunological changes that may have caused a remarkably altered and more severe expression of his chronic disease, for example, recent steroid therapy or HIV infection. The company a rash keeps helps frame the differential diagnosis. Based on the patient's well appearance, the time course, his minimal systemic symptoms, and the appearance of the rash, my leading considerations are psoriasis or an allergic dermatitis. Cutaneous T‐cell malignancy, with its indolent and sometimes protean manifestations, remains possible in a patient of his age. I would now consult a dermatologist for 3 reasons: this patient has a chronic disease that I do not manage beyond basic treatments (eg, topical steroids), he has an undiagnosed illness with substantial dermatologic manifestations, and he may need a skin biopsy for definitive diagnosis.
The dermatology team diagnosed a guttate psoriasis flare, possibly associated with streptococcal pharyngitis. The differential diagnosis included secondary syphilis, although the team believed this was less likely. The dermatology team recommended obtaining a throat culture, streptozyme assay, and rapid plasma reagin and prescribed oral erythromycin and topical steroid ointment under a sauna suit.
I would follow his response to the prescribed steroid treatments. If the patient's course deviates from the dermatologists' expectations, I would request a skin biopsy and undertake further evaluations in search of an underlying systemic disease.
The patient followed up in the dermatology clinic 3 weeks later. His rash had worsened, and he had developed patchy alopecia and progressive edema of the face, ears, and eyes. He denied mouth or tongue swelling, difficulty breathing, or hives. The streptozyme assay was positive, but the other laboratory test results were negative.
The dermatology team diagnosed a severely inflammatory psoriasis flare and prescribed an oral retinoid, acitretin, and referred him for ultraviolet light therapy. He was unable to travel for phototherapy, and acitretin was discontinued after 1 week because of elevated serum transaminase levels. The dermatologists then prescribed oral cyclosporine.
The progression of disease despite standard treatment suggests a nonpsoriatic condition. Although medications could cause the abnormal liver tests, so could another underlying illness that involves the liver. An infiltrative disorder of the skin with hair follicle destruction and local lymphedema could explain both alopecia and facial edema.
I am unable account for his clinical features with a single disease, so the differential remains broad, including severe psoriasis, an infiltrating cutaneous malignancy, or a toxic exposure. Arsenic poisoning causes hyperkeratotic skin lesions, although he lacks the associated gastrointestinal and neurological symptoms. I would not have added the potentially toxic cyclosporine.
When he returned to dermatology clinic 1 week later, his rash and facial swelling had worsened. He also reported muscle and joint aches, fatigue, lightheadedness, anorexia, nausea, abdominal pain, diarrhea, and dyspnea on exertion. He denied fever, chills, and night sweats.
He appeared ill and used a cane to arise and walk. His vital signs and oxygen saturation were normal. He had marked swelling of his face, diffuse erythema and swelling on the chest, and widespread scaly, erythematous plaques (Fig. 3). The proximal nail folds of his fingers were erythematous, with ragged cuticles. His abdomen was mildly distended, but the rest of the physical examination was normal.
He has become too systemically ill to attribute his condition to psoriasis. The nail findings suggest dermatomyositis, which could explain many of his findings. The diffuse erythema and his difficulty walking are consistent with its skin and muscle involvement. Dyspnea could be explained by dermatomyositis‐associated interstitial lung disease. A dermatomyositis‐associated hematological or solid malignancy could account for his multisystem ailments and functional decline. A point against dermatomyositis is the relatively explosive onset of his disease. He should be carefully examined for any motor weakness. With his progressive erythroderma, I am also concerned about an advancing cutaneous T‐cell lymphoma (with leukemic transformation).
Blood tests revealed the following values: white‐blood‐cell count, 8700/L; hematocrit, 46%; platelet count, 172,000/L; blood urea nitrogen, 26 mg/dL; creatinine, 1.0 mg/dL; glucose, 199 mg/dL; albumin, 3.1 g/dL; alkaline phosphatase, 172 U/L (normal range 45‐129); alanine aminotransferase, 75 U/L (normal range 0‐39 U/L); aspartate aminotransferase, 263 U/L (normal range 0‐37 U/L); total bilirubin, 1.1 mg/dL; prothrombin time, 16 seconds (normal range 11.7‐14.3 seconds), and serum creatinine, kinase, 4253 U/L (normal range 0‐194 U/L). HIV serology was negative. Urinalysis revealed trace protein. The results of chest radiographs and an electrocardiogram were normal.
The liver function tests results are consistent with medication effects or liver involvement in a systemic disease. The creatinine kinase elevation is consistent with a myopathy such as dermatomyositis. A skin biopsy would still be useful. Depending on those results, he may need a muscle biopsy, urine heavy metal testing, and computed tomography body imaging. Considering his transaminase and creatinine kinase elevations, I would discontinue lovastatin.
The patient was hospitalized. Further questioning revealed that he had typical Raynaud's phenomenon and odynophagia. A detailed neurological examination showed weakness (3/5) of the triceps and iliopsoas muscles and difficulty rising from a chair without using his arms. Dermatoscopic examination of the proximal nail folds showed dilated capillary loops and foci of hemorrhage.
Blood tests showed a lactate dehydrogenase level of 456 U/L (normal range 0‐249 U/L) and an aldolase of 38 U/L (normal range 1.2‐7.6 U/L). Tests for antinuclear antibodies, anti‐Jo antibody, and antimyeloperoxidase antibodies were negative. Two skin biopsies were interpreted by general pathology as consistent with partially treated psoriasis, whereas another showed nonspecific changes with minimal superficial perivascular lymphohistiocytic inflammation (Fig. 4). Lisinopril was discontinued because of its possible contribution to the facial edema.
Dermatomyositis is now the leading diagnosis. Characteristic features include his proximal muscle weakness, Raynaud's phenomenon, and dilated nailfold capillary loops. I am not overly dissuaded by the negative antinuclear antibodies, but because of additional atypical features (ie, extensive cutaneous edema, rapid onset, illness severity, prominent gastrointestinal symptoms), a confirmatory muscle biopsy is needed. Endoscopy of the proximal aerodigestive tract would help evaluate the odynophagia. There is little to suggest infection, malignancy, or metabolic derangement.
The inpatient medical team considered myositis related to retinoid or cyclosporine therapy. They discontinued cyclosporine and began systemic corticosteroid therapy. Within a few days, the patient's rash, muscle pain, and weakness improved, and the elevated transaminase and creatinine kinase levels decreased.
Dermatology recommended an evaluation for dermatomyositis‐associated malignancy, but the medicine team and rheumatology consultants, noting the lack of classic skin findings (heliotrope rash and Gottron's papules) and the uncharacteristically rapid onset and improvement of myositis, suggested delaying the evaluation until dermatomyositis was proven.
An immediate improvement in symptoms with steroids is nonspecific, often occurring in autoimmune, infectious, and neoplastic diseases. This juncture in the case is common in complex multisystem illnesses, where various consultants may arrive at differing conclusions. With both typical and atypical features of dermatomyositis, where should one set the therapeutic threshold, that is, the point where one ends testing, accepts a diagnosis, and initiates treatment? Several factors raise the level of certainty I would require. First, dermatomyositis is quite rare. Adding atypical features further increases the burden of proof for that illness. Second, the existence of alternative possibilities (admittedly of equal uncertainty) gives me some pause. Finally, the toxicity of the proposed treatments raises the therapeutic threshold. Acknowledging that empiric treatment may be indicated for a severely ill patient at a lower level of certainty, I would hesitate to commit a patient to long‐term steroids without being confident of the diagnosis. I would therefore require a muscle biopsy, or at least electromyography to support or exclude dermatomyositis.
The patient was discharged from the hospital on high‐dose prednisone. He underwent electromyography, which revealed inflammatory myopathic changes more apparent in the proximal than distal muscles. These findings were thought to be compatible with dermatomyositis, although the fibrillations and positive sharp waves characteristic of acute inflammation were absent, perhaps because of corticosteroid therapy.
The patient mistakenly stopped taking his prednisone. Within days, his weakness and skin rash worsened, and he developed nausea with vomiting. He returned to clinic, where his creatinine kinase level was again found to be elevated, and he was rehospitalized. Oral corticosteroid therapy was restarted with prompt improvement. On review of the original skin biopsies, a dermatopathologist observed areas of thickened dermal collagen and a superficial and deep perivascular lymphocytic infiltrate, both consistent with connective tissue disease.
These 3 additional findings (ie, electromyography results, temporally established steroid responsiveness, and the new skin biopsy interpretation) in aggregate support the diagnosis of dermatomyositis, but the nausea and vomiting are unusual. I would discuss these results with a rheumatologist and still request a confirmatory muscle biopsy. Because diagnosing dermatomyositis should prompt consideration of seeking an underlying malignancy in a patient of this age group, I would repeat a targeted history and physical examination along with age‐ and risk‐factor‐appropriate screening. If muscle biopsy results are not definitive, finding an underlying malignancy would lend support to dermatomyositis.
While hospitalized, the patient complained of continued odynophagia and was noted to have oral candidiasis. Upper endoscopy, undertaken to evaluate for esophageal candidiasis, revealed a mass at the gastroesophageal junction. Biopsy revealed gastric‐type adenocarcinoma. An abdominal computed tomography scan demonstrated 3 hypodense hepatic lesions, evidence of cirrhosis, and ascites. Cytology of paracentesis fluid revealed cells compatible with adenocarcinoma. The patient died in hospice care 2 weeks later.
At autopsy, he had metastatic gastric‐type adenocarcinoma. A muscle biopsy (Fig. 5) revealed muscle atrophy with small foci of lymphocytic infiltrates, most compatible with dermatomyositis. Another dermatopathologist reviewed the skin biopsies and noted interface dermatitis, which is typical of connective tissue diseases like dermatomyositis (Fig. 6A,B).
COMMENTARY
Dermatomyositis is an idiopathic inflammatory myopathy characterized by endomysial inflammation and muscle weakness and differentiated from other myopathies by the presence of a rash.1 Muscle disease may manifest with or precede the rash, but up to 40% of patients present with skin manifestations alone, an entity called amyopathic dermatomyositis.2 When present, the myositis generally develops over months, but the onset can be acute.1 The weakness is typically symmetrical and proximal,1 and many patients have oropharyngeal dysphagia.3
The characteristic rash is erythematous, symmetrical, and photodistributed.4 Classic cutaneous findings are the heliotrope rash (violaceous eyelid erythema), which is pathognomonic but uncommon, and the more common Gottron's papules (violaceous, slightly elevated papules and plaques on bony prominences and extensor surfaces, especially the knuckles).4 Other findings include periorbital edema, scalp dermatitis, poikiloderma (ie, hyperpigmentation, hypopigmentation, atrophy, and telangiectasia), periungual erythema, and dystrophic cuticles.2 The cutaneous manifestations of dermatomyositis may be similar to those of psoriasis, systemic lupus erythematosus, lichen planus, rosacea, polymorphous light eruption, drug eruption, atopic dermatitis, seborrheic dermatitis, or allergic contact dermatitis.4
Diagnosing dermatomyositis requires considering clinical, laboratory, electromyographical, and histological evidence, as there are no widely accepted, validated diagnostic criteria.1, 5 The diagnosis is usually suspected if there is a characteristic rash and symptoms of myositis (eg, proximal muscle weakness, myalgias, fatigue, or an inability to swallow). When the patient has an atypical rash, skin biopsy can differentiate dermatomyositis from other conditions, except lupus, which shares the key finding of interface dermatitis.2 The histological findings can be variable and subtle,6 so consultation with a dermatopathologist may be helpful.
Myositis may be confirmed by various studies. Most patients have elevated muscle enzymes (ie, creatinine kinase, aldolase, lactate dehydrogenase, or transaminases)1; for those who do not, magnetic resonance imaging can be helpful in detecting muscle involvement and locating the best site for muscle biopsy.7 Electromyography reveals nonspecific muscle membrane instability.8 Muscle biopsy shows muscle fiber necrosis, perifascicular atrophy, and perivascular and perifascicular lymphocytic infiltrates. These can be patchy, diminished by steroid use, and occasionally seen in noninflammatory muscular dystrophies.8 For a patient with typical myositis and a characteristic rash, muscle biopsy may be unnecessary.1
The clinical utility of serologic testing for diagnosing dermatomyositis is controversial.2 Myositis‐specific antibody testing is insensitive but specific; these antibodies include Jo‐1, an antisynthetase antibody that predicts incomplete response to therapy and lung involvement, and Mi‐2, which is associated with better response to therapy.2, 9, 10 The sensitivity and specificity of antinuclear antibodies are both approximately 60%.10
Patients with dermatomyositis have higher rates of cancers than age‐matched controls, and nearly 25% of patients are diagnosed with a malignancy at some point during the course of the disease.11 Malignancies are typically solid tumors that manifest within 3 years of the diagnosis,1214 although the increased risk may exist for at least 5 years.14 There is a 10‐fold higher risk of ovarian cancer in women with dermatomyositis.12, 15 Other associated malignancies include lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma.14
Recommendations for screening affected patients for cancer have changed over the years, with increasing evidence of an association between dermatomyositis and malignancy and evolving improvements in diagnostic techniques.16 Many authorities recommend that all adult patients with dermatomyositis be evaluated for cancer, including a complete physical examination, basic hematological tests, age‐ and sex‐appropriate screening (eg, mammography, pap smear, and colonoscopy), and chest x‐ray.16 Some would add upper endoscopy; imaging of the chest, abdomen, and pelvis; gynecological examination; and serum CA‐125 level to better evaluate for the most common malignancies (ie, ovarian, gastric, lung, and pancreatic carcinomas and non‐Hodgkins lymphoma).12, 1720
In 19% of adults, dermatomyositis overlaps with other autoimmune disorders, usually systemic lupus erythematosus and systemic sclerosis.21 These manifest as Raynaud's phenomenon, arthritis, esophageal dysmotility, renal disease, or neuropathy.21 Other potentially serious systemic manifestations of dermatomyositis include proximal dysphagia from pharyngeal myopathy; distal dysphagia from esophageal dysmotility in systemic sclerosis overlap; pulmonary disease from autoimmune interstitial lung disease or aspiration; cardiac disease from conduction abnormalities, myocarditis, pericarditis, and valvular disease; and rhabdomyolysis.2
Treatment of dermatomyositis requires systemic immunosuppression with 1 or more agents. The prognosis of dermatomyositis is variable. Mortality at 5 years ranges from 23% to 73%. At least a third of patients are left with mild to severe disability.1 In addition to older age, predictors of poor outcome include male sex, dysphagia, longstanding symptoms before treatment, pulmonary or cardiac involvement, and presence of antisynthetase antibodies.22
Dermatomyositis is often treated in the outpatient setting, but there are many reasons for hospitalization. Complications of treatment, like infection or adverse effects of medications, could result in hospitalization. Treatment with intravenous pulse corticosteroids or IVIG may require inpatient administration if no infusion center is available. Other indications for inpatient evaluation include the consequences of various malignancies and the more severe expression of systemic complications of dermatomyositis (eg, dysphagia and pulmonary, cardiac, or renal disease).
Every parent knows the plaintive backseat whine, Are we there, yet? Clinicians may also experience this feeling when attempting to diagnose a perplexing illness, especially one that lacks a definitive diagnostic test. It was easy for this patient's doctors to assume initially that his new rash was a manifestation of his long‐standing psoriasis. Having done so, they could understandably attribute the subsequent findings to either evolution of this disease or to consequences of the prescribed treatments, rather than considering a novel diagnosis. Only when faced with new (or newly appreciated) findings suggesting myopathy did the clinicians (and our discussant) consider the diagnosis of dermatomyositis. Even then, the primary inpatient medical team and their consultants were unsure when they had sufficient evidence to be certain.
Several factors compounded the difficulty of making a diagnosis in this case: the clinicians were dealing with a rare disease; they were considering alternative diagnoses (ie, psoriasis or a toxic effect of medication); and the disease presented somewhat atypically. The clinicians initially failed to consider and then accept the correct diagnosis because the patient's rash was not classic, his biopsy was interpreted as nonspecific, and he lacked myositis at presentation. Furthermore, when the generalists sought expert assistance, they encountered a difference of opinion among the consultants. These complex situations should goad the clinician into carefully considering the therapeutic threshold, that is, the transition point from diagnostic testing to therapeutic intervention.23 With complex cases like this, it may be difficult to know when one has reached a strongly supported diagnosis, and frequently asking whether we are there yet may be appropriate.
Take‐Home Points for the Hospitalist
-
A skin rash, which may have typical or atypical features, distinguishes dermatomyositis from other acquired myopathies.
-
Consider consultation with pathology specialists for skin and muscle biopsies.
-
Ovarian, lung, gastric, colorectal, pancreatic, and breast carcinomas and non‐Hodgkin's lymphoma are the most common cancers associated with dermatomyositis.
-
In addition to age‐appropriate cancer screening, consider obtaining upper endoscopy, imaging of the chest/abdomen/pelvis, and CA‐125.
-
Patients with dermatomyositis and no obvious concurrent malignancy need long‐term outpatient follow‐up for repeated malignancy screening.
- ,.Polymyositis and dermatomyositis.Lancet.2003;362:971–982.
- .Dermatomyositis.Lancet.2000;355:53–47.
- ,,,.Oropharyngeal dysphagia in polymyositis/dermatomyositis.Clin Neurol Neurosurg.2004;107(1):32–37.
- ,.Skin involvement in dermatomyositis.Curr Opin Rheumatol.2003;15:714–22.
- ,,,,,.Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients.Medicine (Baltimore).2005;84:231–249.
- .Skin Pathology.2nd ed.New York:Churchill Livingstone;2002.
- ,.Utility of magnetic resonance imaging in the evaluation of patients with inflammatory myopathies.Curr Rheumatol Rep.2001;3:334–245.
- ,,.Is it really myositis? A consideration of the differential diagnosis.Curr Opin Rheumatol2004;16:684–691.
- .Idiopathic inflammatory myopathy: autoantibody update.Curr Rheumatol Rep.2002;4:434–441.
- ,,.Laboratory assessment in musculoskeletal disorders.Best Pract Res Clin Rheumatol.2003;17:475–494.
- ,.Dermatomyositis.Clin Dermatol.2006;24:363–373.
- ,,, et al.Frequency of specific cancer types in dermatomyositis and polymyositis: a population‐based study.Lancet.2001;357:96–100.
- ,,, et al.Cancer‐associated myositis: clinical features and prognostic signs.Ann N Y Acad Sci.2005;1051:64–71.
- ,,,,.Incidence of malignant disease in biopsy‐proven inflammatory myopathy. A population‐based cohort study.Ann Intern Med.2001;134:1087–1095.
- ,,.Risk of cancer in patients with dermatomyositis or polymyositis, and follow‐up implications: a Scottish population‐based cohort study.Br J Cancer.2001;85 (1):41–45.
- .When and how should the patient with dermatomyositis or amyopathic dermatomyositis be assessed for possible cancer?Arch Dermatol.2002;138:969–971.
- ,,.Ovarian cancer in patients with dermatomyositis.Medicine (Baltimore).1994;73(3):153–160.
- ,,,.Dermatomyositis sine myositis: association with malignancy.J Rheumatol.1996;23 (1):101–105.
- ,,, et al.Tumor antigen markers for the detection of solid cancers in inflammatory myopathies.Cancer Epidemiol Biomarkers Prev.2005;14:1279–1282.
- ,,, et al.Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients.Arch Dermatol.2002;138:885–890.
- ,,,,,.Dermatomyositis: a dermatology‐based case series.J Am Acad Dermatol.1998;38:397–404.
- ,,, et al.Long‐term outcome in polymyositis and dermatomyositis.Ann Rheum Dis.2006;65:1456–1461.
- .Our stubborn quest for diagnostic certainty. A cause of excessive testing.N Engl J Med.1989;320:1489–1491.
- ,.Polymyositis and dermatomyositis.Lancet.2003;362:971–982.
- .Dermatomyositis.Lancet.2000;355:53–47.
- ,,,.Oropharyngeal dysphagia in polymyositis/dermatomyositis.Clin Neurol Neurosurg.2004;107(1):32–37.
- ,.Skin involvement in dermatomyositis.Curr Opin Rheumatol.2003;15:714–22.
- ,,,,,.Novel classification of idiopathic inflammatory myopathies based on overlap syndrome features and autoantibodies: analysis of 100 French Canadian patients.Medicine (Baltimore).2005;84:231–249.
- .Skin Pathology.2nd ed.New York:Churchill Livingstone;2002.
- ,.Utility of magnetic resonance imaging in the evaluation of patients with inflammatory myopathies.Curr Rheumatol Rep.2001;3:334–245.
- ,,.Is it really myositis? A consideration of the differential diagnosis.Curr Opin Rheumatol2004;16:684–691.
- .Idiopathic inflammatory myopathy: autoantibody update.Curr Rheumatol Rep.2002;4:434–441.
- ,,.Laboratory assessment in musculoskeletal disorders.Best Pract Res Clin Rheumatol.2003;17:475–494.
- ,.Dermatomyositis.Clin Dermatol.2006;24:363–373.
- ,,, et al.Frequency of specific cancer types in dermatomyositis and polymyositis: a population‐based study.Lancet.2001;357:96–100.
- ,,, et al.Cancer‐associated myositis: clinical features and prognostic signs.Ann N Y Acad Sci.2005;1051:64–71.
- ,,,,.Incidence of malignant disease in biopsy‐proven inflammatory myopathy. A population‐based cohort study.Ann Intern Med.2001;134:1087–1095.
- ,,.Risk of cancer in patients with dermatomyositis or polymyositis, and follow‐up implications: a Scottish population‐based cohort study.Br J Cancer.2001;85 (1):41–45.
- .When and how should the patient with dermatomyositis or amyopathic dermatomyositis be assessed for possible cancer?Arch Dermatol.2002;138:969–971.
- ,,.Ovarian cancer in patients with dermatomyositis.Medicine (Baltimore).1994;73(3):153–160.
- ,,,.Dermatomyositis sine myositis: association with malignancy.J Rheumatol.1996;23 (1):101–105.
- ,,, et al.Tumor antigen markers for the detection of solid cancers in inflammatory myopathies.Cancer Epidemiol Biomarkers Prev.2005;14:1279–1282.
- ,,, et al.Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients.Arch Dermatol.2002;138:885–890.
- ,,,,,.Dermatomyositis: a dermatology‐based case series.J Am Acad Dermatol.1998;38:397–404.
- ,,, et al.Long‐term outcome in polymyositis and dermatomyositis.Ann Rheum Dis.2006;65:1456–1461.
- .Our stubborn quest for diagnostic certainty. A cause of excessive testing.N Engl J Med.1989;320:1489–1491.
Clinical Conundrum
A 56‐year‐old woman from Colombia presented to the emergency department after 24 hours of abdominal pain. One week before, she had experienced similar pain that lasted for 4 hours and spontaneously resolved. She was nauseated but had no vomiting. She reported an unintentional 14‐pound weight loss over the preceding 3 weeks. She denied fever, chills, night sweats, diarrhea, constipation, dysuria, or jaundice.
In a middle‐aged woman with abdominal pain and nausea, diagnostic considerations include gallbladder disease, diseases of the bowel (such as a partial small‐bowel obstruction or inflammatory conditions), hepatic or pancreatic conditions, and nongastrointestinal ailments such as cardiac ischemia. Knowing the specific location of pain, its quality, precipitating factors, and accompanying systemic symptoms may help to narrow the diagnosis. The unintentional weight loss preceding the onset of pain may be an important clue because it suggests a systemic condition, and in a South American immigrantparticularly if she has traveled recentlyit is important to consider parasitic illnesses. The absence of fever makes some infections such as tuberculosis and malaria less likely. At this point, in addition to a thorough history and physical, laboratory tests should include a complete blood count (with quantification of eosinophils) and a metabolic panel with liver enzymes and albumin.
The patient described pain in the midline, just inferior to the umbilicus. The pain was constant, developed without any particular provocation, and was not related to meals or exertion. There were no constitutional symptoms aside from weight loss. She had a history of bipolar disorder, hypothyroidism, osteoarthritis, and chronic sinusitis and had previously undergone cholecystectomy and abdominal hysterectomy. She was taking levothyroxine, montelukast, bupropion, oxcarbazepine, fexofenadine, meloxicam, zolpidem, and, as needed, acetaminophen. She had recently completed a 10‐day course of levofloxacin for acute sinusitis. She had immigrated to the United States 10 years earlier and lived with her husband and daughter. She denied the use of tobacco, alcohol or illicit drugs. She had visited Colombia 6 months earlier but had no other recent travel history.
The history of cholecystectomy makes a biliary tract process unlikely. Its location reduces the likelihood of a hepatic or pancreatic process, but I would like to see the liver enzymes, especially given her recent acetaminophen use. The comorbid illnessesparticularly her bipolar disordermay be relevant because psychiatric illness might be associated with medication overuse or undisclosed toxic ingestions. For example, excess thyroxine might lead to weight loss while overuse of nonsteroidal anti‐inflammatory drugs such as meloxicam can cause intestinal ulceration, not only in the upper tract, but also in the colon. Undisclosed ingestions may also be associated with abdominal symptoms. Her surgical history makes adhesions with a secondary partial bowel obstruction possible. With no travel outside this country in the last 6 months, exotic infections are less likely. Finally, the recent course of levofloxacin may be relevant because many antibiotics are associated with nonspecific abdominal symptoms, and Clostridium difficile colitis occasionally presents without diarrhea.
The patient reported taking her medications as prescribed and denied ingesting other medications. On physical examination, she had a temperature of 98.9F, a pulse of 81 beats per minute, a blood pressure of 110/80 mm Hg, and a respiratory rate of 16 respirations per minute. She had a normal oxygen saturation while breathing ambient air. Her weight was 58 kg. There was no scleral icterus or jugular venous distension. She had a small painless ulcer involving the hard palate. Her lungs were clear to auscultation, and cardiac examination was normal. The abdomen was soft, bowel sounds were present, and there was moderate tenderness to palpation inferior to the umbilicus. There was no rebound or guarding, hepatosplenomegaly or other masses. There was no peripheral edema and no lymphadenopathy. Neurological examination was normal.
The oral ulcer may or may not be related to the clinical presentation because oral ulcers, whether painful or painless, are ubiquitous and may be isolated or may be associated with a wide range of infectious and noninfectious systemic diseases. Although some systemic causes of mucocutaneous ulcers are associated with weight loss (including Crohn's disease, Behcet's disease, celiac sprue, human immunodeficiency virus [HIV], herpesviruses, syphilis, and systemic lupus erythematosus [SLE], among others), the lack of specificity of this finding limits its diagnostic utility. However, it is reasonable to ask whether the patient has noted frequent ulceration in the mouth or genitalia, as recurrent or severe ulcerations may narrow the diagnostic considerations. On the other hand, the focal nature of the pain inferior to the umbilicus suggests a discrete process in the abdomen or pelvis, such as an abscess, mass, or localized area of bowel inflammation. A plain abdominal film is likely to be low yield in this situation, so pursuing computed tomography is appropriate. Not all patients with focal abdominal pain require abdominal imaging, but in the context of weight loss and persistent symptoms for more than a week, imaging is prudent in this case.
The patient denied genital ulceration but did report painless oral ulcers over the preceding months. Laboratory evaluation revealed a white‐cell count of 1000/mm3, of which 6% were neutrophils, 5% were band forms, 36% were lymphocytes, and 47% were monocytes. The absolute neutrophil count was 110/mm3. Hemoglobin level was 10.2 g/dL with a mean corpuscular volume of 90 m3, and the platelet count was 151,000/mm3. Other results of laboratory studies were: sodium, 140 mmol/L; potassium, 3.8 mmol/L; chloride, 96 mmol/L; bicarbonate, 23 mmol/L; blood urea nitrogen, 13 mg/dL; creatinine, 0.4 mg/dL; lipase, 32 U/L (normal range, 13‐60); amylase, 73 U/L (normal range, 30‐110); albumin, 4.0 g/dL; aspartate aminotransferase, 779 U/L (normal range, 13‐35); alanine aminotransferase, 330 U/L (normal range, 7‐35); alkaline phosphatase, 510 U/L (normal range, 35‐104); and total bilirubin, 0.9 mg/dL (normal range, 0.1‐1.2). The lactate dehydrogenase level was 200 U/L (normal range, 135‐214). The corrected reticulocyte count was 1.6% (normal range, 0.3‐2.3), and haptoglobin was 190 mg/dL (normal range, 43‐212). A direct Coomb's test was positive. The erythrocyte sedimentation rate was 113 mm/hour (normal range, 1‐25). Urinalysis was normal without evidence of protein or blood.
Laboratory abnormalities include elevated transaminases and alkaline phosphatase, a markedly elevated erythrocyte sedimentation rate, and profound leukopenia with neutropenia. The patient is anemic, which may elevate the sedimentation rate but not typically to this degree. The patient is not febrile, but if she were to develop a fever, empiric antibiotics would be prudent. The normal albumin and bilirubin suggest that hepatic synthetic and excretory functions remain intact. Although the direct Coombs test is positive, the reticulocyte and lactate dehydrogenase levels argue against brisk hemolysis; this abnormality may simply be a marker of nonspecific immune activation. A variety of infections can cause neutropenia and liver enzyme abnormalities including parasites (malaria or leishmaniasis), viruses (cytomegalovirus or Epstein‐Barr virus [EBV]), tick‐borne bacterial infections (ehrlichiosis or rickettsial infection), and granulomatous infections (tuberculosis). Malignant infiltration of the reticuloendothelial system can also lead to cytopenias and liver enzyme abnormalities. Autoimmunity remains a consideration, as SLE may lead to cytopenias, oral ulcers, and nonspecific immune phenomena. Rather than ordering a large number of blood tests, I favor a targeted approach with abdominal computed tomography followed by biopsy of either the liver or bone marrow.
Chest radiography revealed no abnormalities. Computed tomography of the chest, abdomen, and pelvis with intravenous and oral contrast demonstrated concentric wall thickening of the transverse colon, but no evidence of obstruction or free air. The patient was treated with intravenous fluids, morphine, and cefepime. Bone marrow biopsy was performed, which demonstrated a hypercellular marrow with increased myeloid precursors and a left shift and megakaryocytic hyperplasia. Flow cytometry revealed no abnormally restricted clonal populations. A concerted search for an infectious etiology of the patient's neutropenia was unrevealing, including tests for HIV, cytomegalovirus, hepatitis A, hepatitis B, hepatitis C, Mycoplasma pneumoniae, EBV, and parvovirus B19.
I hope blood cultures were drawn prior to the initiation of antibiotics. Hypercellularity of the bone marrow in the context of leukopenia raises concern that white blood cells are being destroyed peripherally. Autoimmunity against neutrophils can be transiently induced by viruses such as HIV, hepatitis B, and EBV, but these infections have been excluded. Testing for antinuclear antibodies is reasonable. A normal‐sized spleen on the abdominal CT excludes hypersplenism. Colonic thickening can be associated with infection, ischemia, inflammatory bowel disease, and malignancy. The question is whether the colonic thickening is part of the same disease process causing the leukopenia and liver enzyme elevation or whether it represents a secondary infectious process in the setting of neutropenia (such as Clostridium difficile infection or typhlitis). Testing for stool pathogens (including ova and parasites) is certainly appropriate, and consideration of a colonoscopy with biopsy is reasonable, provided that appropriate antimicrobial coverage remains in place.
Blood cultures obtained prior to starting antibiotics were negative. The patient's abdominal pain improved, and she was discharged home to have close follow‐up with a hematologist. The results of her liver function tests improved, and her absolute neutrophil count was 230/mm3 at the time of discharge. Her neutropenia was believed to be secondary to peripheral destruction from a viral, drug‐mediated, or autoimmune process. Oxcarbazepine (Trileptal) was discontinued, as it was believed to be the medication most likely to be responsible. She returned to the hospital 3 days later with recurrence of her abdominal pain and diarrhea. She remained afebrile. Additional history revealed arthralgias over the previous 2 months, mild alopecia, and prior symptoms suggestive of Raynaud's phenomenon. Stool studies failed to establish an infectious etiology for the diarrhea, and her continued neutropenia responded appropriately to treatment with subcutaneous filgrastim. Colonoscopy could be performed only to the hepatic flexure and revealed no abnormalities. A serologic test for antinuclear antibodies was positive at a titer of 1:640 in a homogenous pattern, and a test for antineutrophil cytoplasmic antibodies was negative. Complement levels were normal, and tests for cryoglobulins, rapid plasma reagin, anticardiolipin antibody, lupus anticoagulant, rheumatoid factor, and antibodies to extractable nuclear antigens were all negative.
Raynaud's phenomenon is consistent with lupus. Double‐stranded DNA antibodies should be sent, although the urine did not demonstrate protein or an active sediment. Systemic sclerosis and the CREST syndrome is strongly associated with Raynaud's phenomenon and high‐titer ANA, but the patient does not have sclerodactyly, which is generally the earliest skin involvement. Autoimmune hepatitis is often associated with high‐titer ANA but does not fit this clinical picture. Given that the patient's presentation included segmental bowel wall thickening and a transient but marked liver enzyme elevation with AST predominance, I am concerned about vasculitis of the abdominal vasculature and would strongly consider a mesenteric angiogram.
To exclude mesenteric vasculitis, the patient underwent magnetic resonance angiography of the abdomen, the results of which were normal. A repeat test for antinuclear antibodies was positive at a titer of 1:2560 in a uniform pattern. A test for anti‐double‐stranded DNA was positive at 1370 U/mL. The patient was diagnosed with systemic lupus and probable lupus enteritis, and therapy with oral prednisone (10 mg daily) and hydroxychloroquine was initiated. She had prompt improvement in her abdominal pain, and was discharged home. Five months later she developed proteinuria and underwent a renal biopsy, which showed minor, nonspecific glomerular abnormalities, suggesting possible mild lupus nephritis. Eight months after her initial presentation, she remains free of abdominal pain and has regained the weight she had initially lost. Her oral ulcers have resolved, and her blood counts have normalized. Her serum creatinine has remained normal. She is now maintained on prednisone (15 mg daily), hydroxychloroquine, and mycophenolate mofetil.
COMMENTARY
A diagnosis of systemic lupus erythematosus (SLE) provided a unifying explanation for the patient's findings. Indeed, she manifested 4 of the 11 American College of Rheumatology criteria for systemic lupus (oral ulcers, leukopenia, positive anti‐DNA, and positive ANA), meeting criteria for a definite diagnosis of SLE. She additionally had multiple other features suggestive of lupus including Raynaud's phenomenon, arthralgias, alopecia, mild thrombocytopenia, and a positive Coombs' test (although the normal reticulocyte count, lactate dehydrogenase, and haptoglobin were most consistent with anemia of a chronic disease).
The protean manifestations of SLE can present significant diagnostic challenges. In this case, physicians were immediately drawn to the patient's acute abdominal pain and severe neutropenia and failed to recognize more subtle disease manifestations that may have aided in establishing a unifying diagnosis sooner. The initial history and review of systems did not disclose arthralgias, alopecia, or Raynaud's phenomenon. In an era of increasing use of hospitalists, which creates potential discontinuity between inpatient and outpatient physicians, a thorough history and review of systems may be particularly important in diagnosing acute manifestations of chronic systemic disease. Inpatient physicians may be overly focused on the small subset of acute complaints leading to hospitalization, without considering the larger constellation of symptoms that may facilitate accurate diagnosis. Our discussant quickly recognized the multisystem nature of the patient's illness and appropriately focused on infectious, neoplastic, and autoimmune categories of disease as being most likely. When infectious and neoplastic conditions were excluded with reasonable certainty, a directed serologic investigation for autoimmune disease was requested, culminating in a diagnosis of SLE.
Involvement of the skin as well as hematologic, renal, and musculoskeletal systems in SLE is commonly recognized, whereas gastrointestinal involvement is perceived to occur much less frequently. However, abdominal pain occurs in up to 40% of patients with lupus.14 Abdominal pain in lupus patients can arise from non‐lupus‐related conditions as well as lupus‐related entities, including serositis, mesenteric vasculitis with or without infarction, mesenteric thrombosis, pancreatitis, inflammatory bowel disease, and adverse medication effects including peptic ulcer disease. Abnormal liver chemistries, as seen in our patient, occur in 20%‐50% of patients with lupus and may be due to lupus hepatitis, concomitant autoimmune hepatitis, or medications including NSAIDs.5, 6 Oral ulcers and leukopenia are likewise common in SLE, with each seen in up to half of patients.4, 7, 8 Leukopenia in SLE may a result of neutropenia, lymphocytopenia, or both. However, severe neutropenia (ie, absolute count less than 500/L), as seen in our case, is more often a result of myelotoxicity from immunosuppressive therapy, rather than SLE itself.9
Lupus enteritis represents bowel microischemia from small‐vessel arteritis or venulitis that often is not evident on conventional mesenteric angiography.4, 10, 11 The reported prevalence of intestinal vasculitis in patients with SLE varies widely, depending on the characteristics of lupus patients sampled in individual studies. Intestinal vasculitis affects 0.2%‐0.5% of SLE patients in general,4, 12 whereas among SLE patients with active disease and an acute abdomen, vasculitis has been reported in up to 53% of patients.10 Antiphospholipid antibodies, antibodies to extractable nuclear antigens, the SLE Disease Activity Index, complement levels, erythrocyte sedimentation rate, C‐reactive protein, and anti‐double‐stranded DNA do not reliably differentiate lupus enteritis from acute abdominal pain due to other etiologies in patients with SLE.11 However, a concomitant drop in the white blood cell count at the onset of symptoms may be useful in distinguishing lupus enteritis from other causes of acute abdominal pain among lupus patients.11 Computed tomography findings consistent with lupus enteritis are nonspecific and include bowel‐wall thickening, submucosal edema (eg, target sign), dilatation of intestinal segments, engorgement of mesenteric vessels, and increased attenuation of mesenteric fat.13 Colonoscopy may reveal areas of ischemia and ulceration, and biopsy can confirm intestinal vasculitis. However, intestinal involvement may be segmental, and pathologic confirmation may be difficult. Contrast enema, gallium scanning, and indium‐labeled white cell scanning may be useful, but lack specificity. No controlled trials to date have evaluated the optimal therapy for lupus enteritis, but pulsed methylprednisolone is often recommended.4 Cyclophosphamide, azathioprine, methotrexate, and cyclosporine have also been used as adjunctive agents. Patients may progress to intestinal infarction and perforation, which augurs a poor prognosis, and early surgical exploration should be considered in severely ill patients.10 Death may occur in more than two‐thirds of patients whose disease progresses to intestinal perforation.1
In summary, a multisystem disease such as SLE requires a comprehensive history, physical exam, and review of systems to establish a correct diagnosis. In our case, an extensive evaluation was necessary to exclude other etiologies of abdominal pain and systemic illness, particularly as infectious and neoplastic conditions occur far more often than lupus enteritis in the general population. However, profound laboratory abnormalities may have preoccupied the attention of treating physicians, leading them to overlook less obvious but important historical and physical findings suggestive of SLE. The cohesively abnormal forest may thus have been obscured by erratically abnormal individual trees. Gastrointestinal symptoms may be underrecognized in SLE. When these result for lupus enteritis, timely recognition may be lifesaving.
- ,.The gastrointestinal manifestations of systemic lupus erythematosus: a review of the literature.Semin Arthritis Rheum.1980;9:237.
- ,,.Acute abdominal complications of systemic lupus erythematosus and polyarteritis nodosa.Am J Med.1982;73:525–531.
- ,.Acute gastrointestinal manifestations of systemic lupus erythematosus.Can J Surg.1987;30:185–188.
- ,,.A review of gastrointestinal manifestations of systemic lupus erythematosus.Rheumatology.1999;38:917–932.
- ,.Connective tissue disease and the liver.J Clin Gastroenterol.2002;35:345–349.
- ,,.The spectrum of liver disease in systemic lupus erythematosus: report of 33 histologically‐proved cases and review of the literature.Am J Med.1980;69:187–194.
- ,.Hematologic aspects of systemic lupus erythematosus: current concepts.Ann Intern Med.1977;86:220–229.
- ,.Prevalence and significance of hematological abnormalities in patients with systemic lupus erythematosus.Q J Med1991;80:605–12.
- ,,, et al.Moderate and severe neutropenia in patients with systemic lupus erythematosus.Rheumatology.2006;45:994–998.
- ,,, et al.Acute abdomen in systemic lupus erythematosus: the importance of early laparotomy.Am J Med.1997;103:100–105.
- ,,, et al.Acute abdominal pain in systemic lupus erythematosus: focus on lupus enteritis (gastrointestinal vasculitis).Ann Rheum Dis,2002;61:547–550.
- ,,, et al.Vasculitis in systemic lupus erythematosus.Lupus.1997;6:235–242.
- ,,, et al.CT features of systemic lupus erythematosus in patients with acute abdominal pain: emphasis on ischemic bowel disease.Radiology.1999;211:203–209.
A 56‐year‐old woman from Colombia presented to the emergency department after 24 hours of abdominal pain. One week before, she had experienced similar pain that lasted for 4 hours and spontaneously resolved. She was nauseated but had no vomiting. She reported an unintentional 14‐pound weight loss over the preceding 3 weeks. She denied fever, chills, night sweats, diarrhea, constipation, dysuria, or jaundice.
In a middle‐aged woman with abdominal pain and nausea, diagnostic considerations include gallbladder disease, diseases of the bowel (such as a partial small‐bowel obstruction or inflammatory conditions), hepatic or pancreatic conditions, and nongastrointestinal ailments such as cardiac ischemia. Knowing the specific location of pain, its quality, precipitating factors, and accompanying systemic symptoms may help to narrow the diagnosis. The unintentional weight loss preceding the onset of pain may be an important clue because it suggests a systemic condition, and in a South American immigrantparticularly if she has traveled recentlyit is important to consider parasitic illnesses. The absence of fever makes some infections such as tuberculosis and malaria less likely. At this point, in addition to a thorough history and physical, laboratory tests should include a complete blood count (with quantification of eosinophils) and a metabolic panel with liver enzymes and albumin.
The patient described pain in the midline, just inferior to the umbilicus. The pain was constant, developed without any particular provocation, and was not related to meals or exertion. There were no constitutional symptoms aside from weight loss. She had a history of bipolar disorder, hypothyroidism, osteoarthritis, and chronic sinusitis and had previously undergone cholecystectomy and abdominal hysterectomy. She was taking levothyroxine, montelukast, bupropion, oxcarbazepine, fexofenadine, meloxicam, zolpidem, and, as needed, acetaminophen. She had recently completed a 10‐day course of levofloxacin for acute sinusitis. She had immigrated to the United States 10 years earlier and lived with her husband and daughter. She denied the use of tobacco, alcohol or illicit drugs. She had visited Colombia 6 months earlier but had no other recent travel history.
The history of cholecystectomy makes a biliary tract process unlikely. Its location reduces the likelihood of a hepatic or pancreatic process, but I would like to see the liver enzymes, especially given her recent acetaminophen use. The comorbid illnessesparticularly her bipolar disordermay be relevant because psychiatric illness might be associated with medication overuse or undisclosed toxic ingestions. For example, excess thyroxine might lead to weight loss while overuse of nonsteroidal anti‐inflammatory drugs such as meloxicam can cause intestinal ulceration, not only in the upper tract, but also in the colon. Undisclosed ingestions may also be associated with abdominal symptoms. Her surgical history makes adhesions with a secondary partial bowel obstruction possible. With no travel outside this country in the last 6 months, exotic infections are less likely. Finally, the recent course of levofloxacin may be relevant because many antibiotics are associated with nonspecific abdominal symptoms, and Clostridium difficile colitis occasionally presents without diarrhea.
The patient reported taking her medications as prescribed and denied ingesting other medications. On physical examination, she had a temperature of 98.9F, a pulse of 81 beats per minute, a blood pressure of 110/80 mm Hg, and a respiratory rate of 16 respirations per minute. She had a normal oxygen saturation while breathing ambient air. Her weight was 58 kg. There was no scleral icterus or jugular venous distension. She had a small painless ulcer involving the hard palate. Her lungs were clear to auscultation, and cardiac examination was normal. The abdomen was soft, bowel sounds were present, and there was moderate tenderness to palpation inferior to the umbilicus. There was no rebound or guarding, hepatosplenomegaly or other masses. There was no peripheral edema and no lymphadenopathy. Neurological examination was normal.
The oral ulcer may or may not be related to the clinical presentation because oral ulcers, whether painful or painless, are ubiquitous and may be isolated or may be associated with a wide range of infectious and noninfectious systemic diseases. Although some systemic causes of mucocutaneous ulcers are associated with weight loss (including Crohn's disease, Behcet's disease, celiac sprue, human immunodeficiency virus [HIV], herpesviruses, syphilis, and systemic lupus erythematosus [SLE], among others), the lack of specificity of this finding limits its diagnostic utility. However, it is reasonable to ask whether the patient has noted frequent ulceration in the mouth or genitalia, as recurrent or severe ulcerations may narrow the diagnostic considerations. On the other hand, the focal nature of the pain inferior to the umbilicus suggests a discrete process in the abdomen or pelvis, such as an abscess, mass, or localized area of bowel inflammation. A plain abdominal film is likely to be low yield in this situation, so pursuing computed tomography is appropriate. Not all patients with focal abdominal pain require abdominal imaging, but in the context of weight loss and persistent symptoms for more than a week, imaging is prudent in this case.
The patient denied genital ulceration but did report painless oral ulcers over the preceding months. Laboratory evaluation revealed a white‐cell count of 1000/mm3, of which 6% were neutrophils, 5% were band forms, 36% were lymphocytes, and 47% were monocytes. The absolute neutrophil count was 110/mm3. Hemoglobin level was 10.2 g/dL with a mean corpuscular volume of 90 m3, and the platelet count was 151,000/mm3. Other results of laboratory studies were: sodium, 140 mmol/L; potassium, 3.8 mmol/L; chloride, 96 mmol/L; bicarbonate, 23 mmol/L; blood urea nitrogen, 13 mg/dL; creatinine, 0.4 mg/dL; lipase, 32 U/L (normal range, 13‐60); amylase, 73 U/L (normal range, 30‐110); albumin, 4.0 g/dL; aspartate aminotransferase, 779 U/L (normal range, 13‐35); alanine aminotransferase, 330 U/L (normal range, 7‐35); alkaline phosphatase, 510 U/L (normal range, 35‐104); and total bilirubin, 0.9 mg/dL (normal range, 0.1‐1.2). The lactate dehydrogenase level was 200 U/L (normal range, 135‐214). The corrected reticulocyte count was 1.6% (normal range, 0.3‐2.3), and haptoglobin was 190 mg/dL (normal range, 43‐212). A direct Coomb's test was positive. The erythrocyte sedimentation rate was 113 mm/hour (normal range, 1‐25). Urinalysis was normal without evidence of protein or blood.
Laboratory abnormalities include elevated transaminases and alkaline phosphatase, a markedly elevated erythrocyte sedimentation rate, and profound leukopenia with neutropenia. The patient is anemic, which may elevate the sedimentation rate but not typically to this degree. The patient is not febrile, but if she were to develop a fever, empiric antibiotics would be prudent. The normal albumin and bilirubin suggest that hepatic synthetic and excretory functions remain intact. Although the direct Coombs test is positive, the reticulocyte and lactate dehydrogenase levels argue against brisk hemolysis; this abnormality may simply be a marker of nonspecific immune activation. A variety of infections can cause neutropenia and liver enzyme abnormalities including parasites (malaria or leishmaniasis), viruses (cytomegalovirus or Epstein‐Barr virus [EBV]), tick‐borne bacterial infections (ehrlichiosis or rickettsial infection), and granulomatous infections (tuberculosis). Malignant infiltration of the reticuloendothelial system can also lead to cytopenias and liver enzyme abnormalities. Autoimmunity remains a consideration, as SLE may lead to cytopenias, oral ulcers, and nonspecific immune phenomena. Rather than ordering a large number of blood tests, I favor a targeted approach with abdominal computed tomography followed by biopsy of either the liver or bone marrow.
Chest radiography revealed no abnormalities. Computed tomography of the chest, abdomen, and pelvis with intravenous and oral contrast demonstrated concentric wall thickening of the transverse colon, but no evidence of obstruction or free air. The patient was treated with intravenous fluids, morphine, and cefepime. Bone marrow biopsy was performed, which demonstrated a hypercellular marrow with increased myeloid precursors and a left shift and megakaryocytic hyperplasia. Flow cytometry revealed no abnormally restricted clonal populations. A concerted search for an infectious etiology of the patient's neutropenia was unrevealing, including tests for HIV, cytomegalovirus, hepatitis A, hepatitis B, hepatitis C, Mycoplasma pneumoniae, EBV, and parvovirus B19.
I hope blood cultures were drawn prior to the initiation of antibiotics. Hypercellularity of the bone marrow in the context of leukopenia raises concern that white blood cells are being destroyed peripherally. Autoimmunity against neutrophils can be transiently induced by viruses such as HIV, hepatitis B, and EBV, but these infections have been excluded. Testing for antinuclear antibodies is reasonable. A normal‐sized spleen on the abdominal CT excludes hypersplenism. Colonic thickening can be associated with infection, ischemia, inflammatory bowel disease, and malignancy. The question is whether the colonic thickening is part of the same disease process causing the leukopenia and liver enzyme elevation or whether it represents a secondary infectious process in the setting of neutropenia (such as Clostridium difficile infection or typhlitis). Testing for stool pathogens (including ova and parasites) is certainly appropriate, and consideration of a colonoscopy with biopsy is reasonable, provided that appropriate antimicrobial coverage remains in place.
Blood cultures obtained prior to starting antibiotics were negative. The patient's abdominal pain improved, and she was discharged home to have close follow‐up with a hematologist. The results of her liver function tests improved, and her absolute neutrophil count was 230/mm3 at the time of discharge. Her neutropenia was believed to be secondary to peripheral destruction from a viral, drug‐mediated, or autoimmune process. Oxcarbazepine (Trileptal) was discontinued, as it was believed to be the medication most likely to be responsible. She returned to the hospital 3 days later with recurrence of her abdominal pain and diarrhea. She remained afebrile. Additional history revealed arthralgias over the previous 2 months, mild alopecia, and prior symptoms suggestive of Raynaud's phenomenon. Stool studies failed to establish an infectious etiology for the diarrhea, and her continued neutropenia responded appropriately to treatment with subcutaneous filgrastim. Colonoscopy could be performed only to the hepatic flexure and revealed no abnormalities. A serologic test for antinuclear antibodies was positive at a titer of 1:640 in a homogenous pattern, and a test for antineutrophil cytoplasmic antibodies was negative. Complement levels were normal, and tests for cryoglobulins, rapid plasma reagin, anticardiolipin antibody, lupus anticoagulant, rheumatoid factor, and antibodies to extractable nuclear antigens were all negative.
Raynaud's phenomenon is consistent with lupus. Double‐stranded DNA antibodies should be sent, although the urine did not demonstrate protein or an active sediment. Systemic sclerosis and the CREST syndrome is strongly associated with Raynaud's phenomenon and high‐titer ANA, but the patient does not have sclerodactyly, which is generally the earliest skin involvement. Autoimmune hepatitis is often associated with high‐titer ANA but does not fit this clinical picture. Given that the patient's presentation included segmental bowel wall thickening and a transient but marked liver enzyme elevation with AST predominance, I am concerned about vasculitis of the abdominal vasculature and would strongly consider a mesenteric angiogram.
To exclude mesenteric vasculitis, the patient underwent magnetic resonance angiography of the abdomen, the results of which were normal. A repeat test for antinuclear antibodies was positive at a titer of 1:2560 in a uniform pattern. A test for anti‐double‐stranded DNA was positive at 1370 U/mL. The patient was diagnosed with systemic lupus and probable lupus enteritis, and therapy with oral prednisone (10 mg daily) and hydroxychloroquine was initiated. She had prompt improvement in her abdominal pain, and was discharged home. Five months later she developed proteinuria and underwent a renal biopsy, which showed minor, nonspecific glomerular abnormalities, suggesting possible mild lupus nephritis. Eight months after her initial presentation, she remains free of abdominal pain and has regained the weight she had initially lost. Her oral ulcers have resolved, and her blood counts have normalized. Her serum creatinine has remained normal. She is now maintained on prednisone (15 mg daily), hydroxychloroquine, and mycophenolate mofetil.
COMMENTARY
A diagnosis of systemic lupus erythematosus (SLE) provided a unifying explanation for the patient's findings. Indeed, she manifested 4 of the 11 American College of Rheumatology criteria for systemic lupus (oral ulcers, leukopenia, positive anti‐DNA, and positive ANA), meeting criteria for a definite diagnosis of SLE. She additionally had multiple other features suggestive of lupus including Raynaud's phenomenon, arthralgias, alopecia, mild thrombocytopenia, and a positive Coombs' test (although the normal reticulocyte count, lactate dehydrogenase, and haptoglobin were most consistent with anemia of a chronic disease).
The protean manifestations of SLE can present significant diagnostic challenges. In this case, physicians were immediately drawn to the patient's acute abdominal pain and severe neutropenia and failed to recognize more subtle disease manifestations that may have aided in establishing a unifying diagnosis sooner. The initial history and review of systems did not disclose arthralgias, alopecia, or Raynaud's phenomenon. In an era of increasing use of hospitalists, which creates potential discontinuity between inpatient and outpatient physicians, a thorough history and review of systems may be particularly important in diagnosing acute manifestations of chronic systemic disease. Inpatient physicians may be overly focused on the small subset of acute complaints leading to hospitalization, without considering the larger constellation of symptoms that may facilitate accurate diagnosis. Our discussant quickly recognized the multisystem nature of the patient's illness and appropriately focused on infectious, neoplastic, and autoimmune categories of disease as being most likely. When infectious and neoplastic conditions were excluded with reasonable certainty, a directed serologic investigation for autoimmune disease was requested, culminating in a diagnosis of SLE.
Involvement of the skin as well as hematologic, renal, and musculoskeletal systems in SLE is commonly recognized, whereas gastrointestinal involvement is perceived to occur much less frequently. However, abdominal pain occurs in up to 40% of patients with lupus.14 Abdominal pain in lupus patients can arise from non‐lupus‐related conditions as well as lupus‐related entities, including serositis, mesenteric vasculitis with or without infarction, mesenteric thrombosis, pancreatitis, inflammatory bowel disease, and adverse medication effects including peptic ulcer disease. Abnormal liver chemistries, as seen in our patient, occur in 20%‐50% of patients with lupus and may be due to lupus hepatitis, concomitant autoimmune hepatitis, or medications including NSAIDs.5, 6 Oral ulcers and leukopenia are likewise common in SLE, with each seen in up to half of patients.4, 7, 8 Leukopenia in SLE may a result of neutropenia, lymphocytopenia, or both. However, severe neutropenia (ie, absolute count less than 500/L), as seen in our case, is more often a result of myelotoxicity from immunosuppressive therapy, rather than SLE itself.9
Lupus enteritis represents bowel microischemia from small‐vessel arteritis or venulitis that often is not evident on conventional mesenteric angiography.4, 10, 11 The reported prevalence of intestinal vasculitis in patients with SLE varies widely, depending on the characteristics of lupus patients sampled in individual studies. Intestinal vasculitis affects 0.2%‐0.5% of SLE patients in general,4, 12 whereas among SLE patients with active disease and an acute abdomen, vasculitis has been reported in up to 53% of patients.10 Antiphospholipid antibodies, antibodies to extractable nuclear antigens, the SLE Disease Activity Index, complement levels, erythrocyte sedimentation rate, C‐reactive protein, and anti‐double‐stranded DNA do not reliably differentiate lupus enteritis from acute abdominal pain due to other etiologies in patients with SLE.11 However, a concomitant drop in the white blood cell count at the onset of symptoms may be useful in distinguishing lupus enteritis from other causes of acute abdominal pain among lupus patients.11 Computed tomography findings consistent with lupus enteritis are nonspecific and include bowel‐wall thickening, submucosal edema (eg, target sign), dilatation of intestinal segments, engorgement of mesenteric vessels, and increased attenuation of mesenteric fat.13 Colonoscopy may reveal areas of ischemia and ulceration, and biopsy can confirm intestinal vasculitis. However, intestinal involvement may be segmental, and pathologic confirmation may be difficult. Contrast enema, gallium scanning, and indium‐labeled white cell scanning may be useful, but lack specificity. No controlled trials to date have evaluated the optimal therapy for lupus enteritis, but pulsed methylprednisolone is often recommended.4 Cyclophosphamide, azathioprine, methotrexate, and cyclosporine have also been used as adjunctive agents. Patients may progress to intestinal infarction and perforation, which augurs a poor prognosis, and early surgical exploration should be considered in severely ill patients.10 Death may occur in more than two‐thirds of patients whose disease progresses to intestinal perforation.1
In summary, a multisystem disease such as SLE requires a comprehensive history, physical exam, and review of systems to establish a correct diagnosis. In our case, an extensive evaluation was necessary to exclude other etiologies of abdominal pain and systemic illness, particularly as infectious and neoplastic conditions occur far more often than lupus enteritis in the general population. However, profound laboratory abnormalities may have preoccupied the attention of treating physicians, leading them to overlook less obvious but important historical and physical findings suggestive of SLE. The cohesively abnormal forest may thus have been obscured by erratically abnormal individual trees. Gastrointestinal symptoms may be underrecognized in SLE. When these result for lupus enteritis, timely recognition may be lifesaving.
A 56‐year‐old woman from Colombia presented to the emergency department after 24 hours of abdominal pain. One week before, she had experienced similar pain that lasted for 4 hours and spontaneously resolved. She was nauseated but had no vomiting. She reported an unintentional 14‐pound weight loss over the preceding 3 weeks. She denied fever, chills, night sweats, diarrhea, constipation, dysuria, or jaundice.
In a middle‐aged woman with abdominal pain and nausea, diagnostic considerations include gallbladder disease, diseases of the bowel (such as a partial small‐bowel obstruction or inflammatory conditions), hepatic or pancreatic conditions, and nongastrointestinal ailments such as cardiac ischemia. Knowing the specific location of pain, its quality, precipitating factors, and accompanying systemic symptoms may help to narrow the diagnosis. The unintentional weight loss preceding the onset of pain may be an important clue because it suggests a systemic condition, and in a South American immigrantparticularly if she has traveled recentlyit is important to consider parasitic illnesses. The absence of fever makes some infections such as tuberculosis and malaria less likely. At this point, in addition to a thorough history and physical, laboratory tests should include a complete blood count (with quantification of eosinophils) and a metabolic panel with liver enzymes and albumin.
The patient described pain in the midline, just inferior to the umbilicus. The pain was constant, developed without any particular provocation, and was not related to meals or exertion. There were no constitutional symptoms aside from weight loss. She had a history of bipolar disorder, hypothyroidism, osteoarthritis, and chronic sinusitis and had previously undergone cholecystectomy and abdominal hysterectomy. She was taking levothyroxine, montelukast, bupropion, oxcarbazepine, fexofenadine, meloxicam, zolpidem, and, as needed, acetaminophen. She had recently completed a 10‐day course of levofloxacin for acute sinusitis. She had immigrated to the United States 10 years earlier and lived with her husband and daughter. She denied the use of tobacco, alcohol or illicit drugs. She had visited Colombia 6 months earlier but had no other recent travel history.
The history of cholecystectomy makes a biliary tract process unlikely. Its location reduces the likelihood of a hepatic or pancreatic process, but I would like to see the liver enzymes, especially given her recent acetaminophen use. The comorbid illnessesparticularly her bipolar disordermay be relevant because psychiatric illness might be associated with medication overuse or undisclosed toxic ingestions. For example, excess thyroxine might lead to weight loss while overuse of nonsteroidal anti‐inflammatory drugs such as meloxicam can cause intestinal ulceration, not only in the upper tract, but also in the colon. Undisclosed ingestions may also be associated with abdominal symptoms. Her surgical history makes adhesions with a secondary partial bowel obstruction possible. With no travel outside this country in the last 6 months, exotic infections are less likely. Finally, the recent course of levofloxacin may be relevant because many antibiotics are associated with nonspecific abdominal symptoms, and Clostridium difficile colitis occasionally presents without diarrhea.
The patient reported taking her medications as prescribed and denied ingesting other medications. On physical examination, she had a temperature of 98.9F, a pulse of 81 beats per minute, a blood pressure of 110/80 mm Hg, and a respiratory rate of 16 respirations per minute. She had a normal oxygen saturation while breathing ambient air. Her weight was 58 kg. There was no scleral icterus or jugular venous distension. She had a small painless ulcer involving the hard palate. Her lungs were clear to auscultation, and cardiac examination was normal. The abdomen was soft, bowel sounds were present, and there was moderate tenderness to palpation inferior to the umbilicus. There was no rebound or guarding, hepatosplenomegaly or other masses. There was no peripheral edema and no lymphadenopathy. Neurological examination was normal.
The oral ulcer may or may not be related to the clinical presentation because oral ulcers, whether painful or painless, are ubiquitous and may be isolated or may be associated with a wide range of infectious and noninfectious systemic diseases. Although some systemic causes of mucocutaneous ulcers are associated with weight loss (including Crohn's disease, Behcet's disease, celiac sprue, human immunodeficiency virus [HIV], herpesviruses, syphilis, and systemic lupus erythematosus [SLE], among others), the lack of specificity of this finding limits its diagnostic utility. However, it is reasonable to ask whether the patient has noted frequent ulceration in the mouth or genitalia, as recurrent or severe ulcerations may narrow the diagnostic considerations. On the other hand, the focal nature of the pain inferior to the umbilicus suggests a discrete process in the abdomen or pelvis, such as an abscess, mass, or localized area of bowel inflammation. A plain abdominal film is likely to be low yield in this situation, so pursuing computed tomography is appropriate. Not all patients with focal abdominal pain require abdominal imaging, but in the context of weight loss and persistent symptoms for more than a week, imaging is prudent in this case.
The patient denied genital ulceration but did report painless oral ulcers over the preceding months. Laboratory evaluation revealed a white‐cell count of 1000/mm3, of which 6% were neutrophils, 5% were band forms, 36% were lymphocytes, and 47% were monocytes. The absolute neutrophil count was 110/mm3. Hemoglobin level was 10.2 g/dL with a mean corpuscular volume of 90 m3, and the platelet count was 151,000/mm3. Other results of laboratory studies were: sodium, 140 mmol/L; potassium, 3.8 mmol/L; chloride, 96 mmol/L; bicarbonate, 23 mmol/L; blood urea nitrogen, 13 mg/dL; creatinine, 0.4 mg/dL; lipase, 32 U/L (normal range, 13‐60); amylase, 73 U/L (normal range, 30‐110); albumin, 4.0 g/dL; aspartate aminotransferase, 779 U/L (normal range, 13‐35); alanine aminotransferase, 330 U/L (normal range, 7‐35); alkaline phosphatase, 510 U/L (normal range, 35‐104); and total bilirubin, 0.9 mg/dL (normal range, 0.1‐1.2). The lactate dehydrogenase level was 200 U/L (normal range, 135‐214). The corrected reticulocyte count was 1.6% (normal range, 0.3‐2.3), and haptoglobin was 190 mg/dL (normal range, 43‐212). A direct Coomb's test was positive. The erythrocyte sedimentation rate was 113 mm/hour (normal range, 1‐25). Urinalysis was normal without evidence of protein or blood.
Laboratory abnormalities include elevated transaminases and alkaline phosphatase, a markedly elevated erythrocyte sedimentation rate, and profound leukopenia with neutropenia. The patient is anemic, which may elevate the sedimentation rate but not typically to this degree. The patient is not febrile, but if she were to develop a fever, empiric antibiotics would be prudent. The normal albumin and bilirubin suggest that hepatic synthetic and excretory functions remain intact. Although the direct Coombs test is positive, the reticulocyte and lactate dehydrogenase levels argue against brisk hemolysis; this abnormality may simply be a marker of nonspecific immune activation. A variety of infections can cause neutropenia and liver enzyme abnormalities including parasites (malaria or leishmaniasis), viruses (cytomegalovirus or Epstein‐Barr virus [EBV]), tick‐borne bacterial infections (ehrlichiosis or rickettsial infection), and granulomatous infections (tuberculosis). Malignant infiltration of the reticuloendothelial system can also lead to cytopenias and liver enzyme abnormalities. Autoimmunity remains a consideration, as SLE may lead to cytopenias, oral ulcers, and nonspecific immune phenomena. Rather than ordering a large number of blood tests, I favor a targeted approach with abdominal computed tomography followed by biopsy of either the liver or bone marrow.
Chest radiography revealed no abnormalities. Computed tomography of the chest, abdomen, and pelvis with intravenous and oral contrast demonstrated concentric wall thickening of the transverse colon, but no evidence of obstruction or free air. The patient was treated with intravenous fluids, morphine, and cefepime. Bone marrow biopsy was performed, which demonstrated a hypercellular marrow with increased myeloid precursors and a left shift and megakaryocytic hyperplasia. Flow cytometry revealed no abnormally restricted clonal populations. A concerted search for an infectious etiology of the patient's neutropenia was unrevealing, including tests for HIV, cytomegalovirus, hepatitis A, hepatitis B, hepatitis C, Mycoplasma pneumoniae, EBV, and parvovirus B19.
I hope blood cultures were drawn prior to the initiation of antibiotics. Hypercellularity of the bone marrow in the context of leukopenia raises concern that white blood cells are being destroyed peripherally. Autoimmunity against neutrophils can be transiently induced by viruses such as HIV, hepatitis B, and EBV, but these infections have been excluded. Testing for antinuclear antibodies is reasonable. A normal‐sized spleen on the abdominal CT excludes hypersplenism. Colonic thickening can be associated with infection, ischemia, inflammatory bowel disease, and malignancy. The question is whether the colonic thickening is part of the same disease process causing the leukopenia and liver enzyme elevation or whether it represents a secondary infectious process in the setting of neutropenia (such as Clostridium difficile infection or typhlitis). Testing for stool pathogens (including ova and parasites) is certainly appropriate, and consideration of a colonoscopy with biopsy is reasonable, provided that appropriate antimicrobial coverage remains in place.
Blood cultures obtained prior to starting antibiotics were negative. The patient's abdominal pain improved, and she was discharged home to have close follow‐up with a hematologist. The results of her liver function tests improved, and her absolute neutrophil count was 230/mm3 at the time of discharge. Her neutropenia was believed to be secondary to peripheral destruction from a viral, drug‐mediated, or autoimmune process. Oxcarbazepine (Trileptal) was discontinued, as it was believed to be the medication most likely to be responsible. She returned to the hospital 3 days later with recurrence of her abdominal pain and diarrhea. She remained afebrile. Additional history revealed arthralgias over the previous 2 months, mild alopecia, and prior symptoms suggestive of Raynaud's phenomenon. Stool studies failed to establish an infectious etiology for the diarrhea, and her continued neutropenia responded appropriately to treatment with subcutaneous filgrastim. Colonoscopy could be performed only to the hepatic flexure and revealed no abnormalities. A serologic test for antinuclear antibodies was positive at a titer of 1:640 in a homogenous pattern, and a test for antineutrophil cytoplasmic antibodies was negative. Complement levels were normal, and tests for cryoglobulins, rapid plasma reagin, anticardiolipin antibody, lupus anticoagulant, rheumatoid factor, and antibodies to extractable nuclear antigens were all negative.
Raynaud's phenomenon is consistent with lupus. Double‐stranded DNA antibodies should be sent, although the urine did not demonstrate protein or an active sediment. Systemic sclerosis and the CREST syndrome is strongly associated with Raynaud's phenomenon and high‐titer ANA, but the patient does not have sclerodactyly, which is generally the earliest skin involvement. Autoimmune hepatitis is often associated with high‐titer ANA but does not fit this clinical picture. Given that the patient's presentation included segmental bowel wall thickening and a transient but marked liver enzyme elevation with AST predominance, I am concerned about vasculitis of the abdominal vasculature and would strongly consider a mesenteric angiogram.
To exclude mesenteric vasculitis, the patient underwent magnetic resonance angiography of the abdomen, the results of which were normal. A repeat test for antinuclear antibodies was positive at a titer of 1:2560 in a uniform pattern. A test for anti‐double‐stranded DNA was positive at 1370 U/mL. The patient was diagnosed with systemic lupus and probable lupus enteritis, and therapy with oral prednisone (10 mg daily) and hydroxychloroquine was initiated. She had prompt improvement in her abdominal pain, and was discharged home. Five months later she developed proteinuria and underwent a renal biopsy, which showed minor, nonspecific glomerular abnormalities, suggesting possible mild lupus nephritis. Eight months after her initial presentation, she remains free of abdominal pain and has regained the weight she had initially lost. Her oral ulcers have resolved, and her blood counts have normalized. Her serum creatinine has remained normal. She is now maintained on prednisone (15 mg daily), hydroxychloroquine, and mycophenolate mofetil.
COMMENTARY
A diagnosis of systemic lupus erythematosus (SLE) provided a unifying explanation for the patient's findings. Indeed, she manifested 4 of the 11 American College of Rheumatology criteria for systemic lupus (oral ulcers, leukopenia, positive anti‐DNA, and positive ANA), meeting criteria for a definite diagnosis of SLE. She additionally had multiple other features suggestive of lupus including Raynaud's phenomenon, arthralgias, alopecia, mild thrombocytopenia, and a positive Coombs' test (although the normal reticulocyte count, lactate dehydrogenase, and haptoglobin were most consistent with anemia of a chronic disease).
The protean manifestations of SLE can present significant diagnostic challenges. In this case, physicians were immediately drawn to the patient's acute abdominal pain and severe neutropenia and failed to recognize more subtle disease manifestations that may have aided in establishing a unifying diagnosis sooner. The initial history and review of systems did not disclose arthralgias, alopecia, or Raynaud's phenomenon. In an era of increasing use of hospitalists, which creates potential discontinuity between inpatient and outpatient physicians, a thorough history and review of systems may be particularly important in diagnosing acute manifestations of chronic systemic disease. Inpatient physicians may be overly focused on the small subset of acute complaints leading to hospitalization, without considering the larger constellation of symptoms that may facilitate accurate diagnosis. Our discussant quickly recognized the multisystem nature of the patient's illness and appropriately focused on infectious, neoplastic, and autoimmune categories of disease as being most likely. When infectious and neoplastic conditions were excluded with reasonable certainty, a directed serologic investigation for autoimmune disease was requested, culminating in a diagnosis of SLE.
Involvement of the skin as well as hematologic, renal, and musculoskeletal systems in SLE is commonly recognized, whereas gastrointestinal involvement is perceived to occur much less frequently. However, abdominal pain occurs in up to 40% of patients with lupus.14 Abdominal pain in lupus patients can arise from non‐lupus‐related conditions as well as lupus‐related entities, including serositis, mesenteric vasculitis with or without infarction, mesenteric thrombosis, pancreatitis, inflammatory bowel disease, and adverse medication effects including peptic ulcer disease. Abnormal liver chemistries, as seen in our patient, occur in 20%‐50% of patients with lupus and may be due to lupus hepatitis, concomitant autoimmune hepatitis, or medications including NSAIDs.5, 6 Oral ulcers and leukopenia are likewise common in SLE, with each seen in up to half of patients.4, 7, 8 Leukopenia in SLE may a result of neutropenia, lymphocytopenia, or both. However, severe neutropenia (ie, absolute count less than 500/L), as seen in our case, is more often a result of myelotoxicity from immunosuppressive therapy, rather than SLE itself.9
Lupus enteritis represents bowel microischemia from small‐vessel arteritis or venulitis that often is not evident on conventional mesenteric angiography.4, 10, 11 The reported prevalence of intestinal vasculitis in patients with SLE varies widely, depending on the characteristics of lupus patients sampled in individual studies. Intestinal vasculitis affects 0.2%‐0.5% of SLE patients in general,4, 12 whereas among SLE patients with active disease and an acute abdomen, vasculitis has been reported in up to 53% of patients.10 Antiphospholipid antibodies, antibodies to extractable nuclear antigens, the SLE Disease Activity Index, complement levels, erythrocyte sedimentation rate, C‐reactive protein, and anti‐double‐stranded DNA do not reliably differentiate lupus enteritis from acute abdominal pain due to other etiologies in patients with SLE.11 However, a concomitant drop in the white blood cell count at the onset of symptoms may be useful in distinguishing lupus enteritis from other causes of acute abdominal pain among lupus patients.11 Computed tomography findings consistent with lupus enteritis are nonspecific and include bowel‐wall thickening, submucosal edema (eg, target sign), dilatation of intestinal segments, engorgement of mesenteric vessels, and increased attenuation of mesenteric fat.13 Colonoscopy may reveal areas of ischemia and ulceration, and biopsy can confirm intestinal vasculitis. However, intestinal involvement may be segmental, and pathologic confirmation may be difficult. Contrast enema, gallium scanning, and indium‐labeled white cell scanning may be useful, but lack specificity. No controlled trials to date have evaluated the optimal therapy for lupus enteritis, but pulsed methylprednisolone is often recommended.4 Cyclophosphamide, azathioprine, methotrexate, and cyclosporine have also been used as adjunctive agents. Patients may progress to intestinal infarction and perforation, which augurs a poor prognosis, and early surgical exploration should be considered in severely ill patients.10 Death may occur in more than two‐thirds of patients whose disease progresses to intestinal perforation.1
In summary, a multisystem disease such as SLE requires a comprehensive history, physical exam, and review of systems to establish a correct diagnosis. In our case, an extensive evaluation was necessary to exclude other etiologies of abdominal pain and systemic illness, particularly as infectious and neoplastic conditions occur far more often than lupus enteritis in the general population. However, profound laboratory abnormalities may have preoccupied the attention of treating physicians, leading them to overlook less obvious but important historical and physical findings suggestive of SLE. The cohesively abnormal forest may thus have been obscured by erratically abnormal individual trees. Gastrointestinal symptoms may be underrecognized in SLE. When these result for lupus enteritis, timely recognition may be lifesaving.
- ,.The gastrointestinal manifestations of systemic lupus erythematosus: a review of the literature.Semin Arthritis Rheum.1980;9:237.
- ,,.Acute abdominal complications of systemic lupus erythematosus and polyarteritis nodosa.Am J Med.1982;73:525–531.
- ,.Acute gastrointestinal manifestations of systemic lupus erythematosus.Can J Surg.1987;30:185–188.
- ,,.A review of gastrointestinal manifestations of systemic lupus erythematosus.Rheumatology.1999;38:917–932.
- ,.Connective tissue disease and the liver.J Clin Gastroenterol.2002;35:345–349.
- ,,.The spectrum of liver disease in systemic lupus erythematosus: report of 33 histologically‐proved cases and review of the literature.Am J Med.1980;69:187–194.
- ,.Hematologic aspects of systemic lupus erythematosus: current concepts.Ann Intern Med.1977;86:220–229.
- ,.Prevalence and significance of hematological abnormalities in patients with systemic lupus erythematosus.Q J Med1991;80:605–12.
- ,,, et al.Moderate and severe neutropenia in patients with systemic lupus erythematosus.Rheumatology.2006;45:994–998.
- ,,, et al.Acute abdomen in systemic lupus erythematosus: the importance of early laparotomy.Am J Med.1997;103:100–105.
- ,,, et al.Acute abdominal pain in systemic lupus erythematosus: focus on lupus enteritis (gastrointestinal vasculitis).Ann Rheum Dis,2002;61:547–550.
- ,,, et al.Vasculitis in systemic lupus erythematosus.Lupus.1997;6:235–242.
- ,,, et al.CT features of systemic lupus erythematosus in patients with acute abdominal pain: emphasis on ischemic bowel disease.Radiology.1999;211:203–209.
- ,.The gastrointestinal manifestations of systemic lupus erythematosus: a review of the literature.Semin Arthritis Rheum.1980;9:237.
- ,,.Acute abdominal complications of systemic lupus erythematosus and polyarteritis nodosa.Am J Med.1982;73:525–531.
- ,.Acute gastrointestinal manifestations of systemic lupus erythematosus.Can J Surg.1987;30:185–188.
- ,,.A review of gastrointestinal manifestations of systemic lupus erythematosus.Rheumatology.1999;38:917–932.
- ,.Connective tissue disease and the liver.J Clin Gastroenterol.2002;35:345–349.
- ,,.The spectrum of liver disease in systemic lupus erythematosus: report of 33 histologically‐proved cases and review of the literature.Am J Med.1980;69:187–194.
- ,.Hematologic aspects of systemic lupus erythematosus: current concepts.Ann Intern Med.1977;86:220–229.
- ,.Prevalence and significance of hematological abnormalities in patients with systemic lupus erythematosus.Q J Med1991;80:605–12.
- ,,, et al.Moderate and severe neutropenia in patients with systemic lupus erythematosus.Rheumatology.2006;45:994–998.
- ,,, et al.Acute abdomen in systemic lupus erythematosus: the importance of early laparotomy.Am J Med.1997;103:100–105.
- ,,, et al.Acute abdominal pain in systemic lupus erythematosus: focus on lupus enteritis (gastrointestinal vasculitis).Ann Rheum Dis,2002;61:547–550.
- ,,, et al.Vasculitis in systemic lupus erythematosus.Lupus.1997;6:235–242.
- ,,, et al.CT features of systemic lupus erythematosus in patients with acute abdominal pain: emphasis on ischemic bowel disease.Radiology.1999;211:203–209.
Clinical Conundrum
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
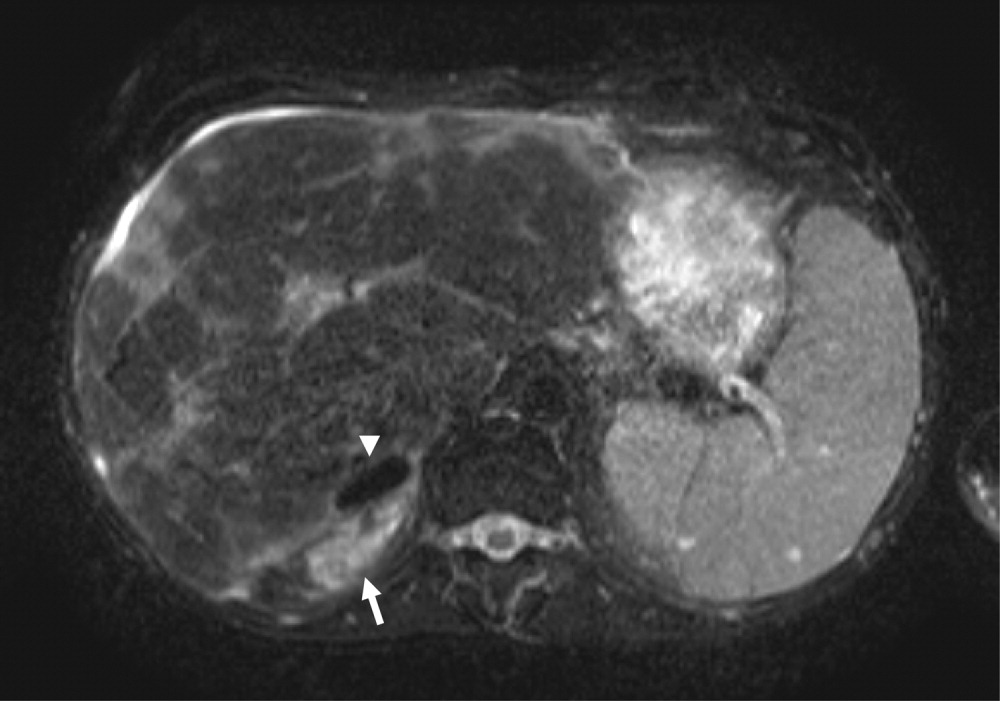
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
56‐year‐old man with a history of chronic liver disease of unknown etiology was referred for evaluation of intermittent low‐grade fevers, constipation, and an unintentional weight loss of 20‐kg during the previous 9 months. Three weeks prior to presentation, he was admitted to his local hospital for these symptoms and was treated empirically with cefotaxime for 6 days, but his symptoms persisted.
The patient's age and sex make him statistically at risk for vascular disease as well as malignancy. The history of chronic liver disease of unknown etiology is intriguing. In evaluating a patient with chronic liver disease, I want to know about alcohol consumption, intravenous drug use, family history, viral hepatitis serology, and antinuclear antibody testing. Chronic liver disease places this patient at increased risk for infection because portal hypertension causes blood to bypass a large part of the reticuloendothelial system (liver and spleen), therefore increasing the risk of sustained bacteremia.
Regarding his chronic low‐grade fever, I would like to know about his country of origin, travel history, occupational history, risk factors for human immunodeficiency virus (HIV) and tuberculosis, and any symptoms or signs of rheumatologic disease. Constipation and weight loss can be a result of malignancy (eg, hepatocellular carcinoma, colorectal cancer), vascular disease (eg, mesenteric thrombosis), or metabolic derangement (eg, hypercalcemia).
The patient had a history of recurrent episodes of ascites and low‐grade fevers. He first developed ascites, abdominal pain, low‐grade fevers, and pedal edema 20 years ago. These signs and symptoms resolved spontaneously, but similar episodes have recurred every 46 years since. Each time, diagnostic evaluation failed to reveal a specific etiology.
Twelve years prior to presentation, the patient was evaluated for chronic liver disease. Diagnostic tests at that time included viral hepatitis serology, ceruloplasmin, ferritin, alpha‐1‐antitrypsin, antimitochondrial antibody, and antinuclear antibody testing, all the results of which were within the normal range. The patient denied consumption of alcohol, medications, or toxic substances. Percutaneous liver biopsy demonstrated focal parenchymal scarring interspersed with areas of normal parenchyma, consistent with focal ischemic injury (Fig. 1).
The duration of the patient's symptoms is striking. A unifying diagnosis for this patient must explain his chronic liver disease, periodic fevers, ascites, and abdominal pain that started at a relatively young age. Conditions to consider include hepatitis B or C, hemochromatosis, Wilson's disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, alpha‐1‐antitrypsin deficiency, and drug or toxin exposure. Venoocclusive disease of the liver and chronic congestive hepatopathy (from heart failure or constrictive pericarditis) are especially attractive possibilities, given the findings of focal ischemic injury on liver biopsy.
Recurrent fever and abdominal pain can occur because of familial Mediterranean fever, which results from a genetic abnormality and causes recurrent peritoneal inflammation associated with fever and ascites. Although unlikely in this case, familial Mediterranean fever can cause secondary amyloidosis with liver involvement.
The patient reported episodic, vague abdominal pain, nausea, anorexia, night sweats, hair thinning, extreme fatigue, and lightheadedness. He had no known allergies, and his medications included propranolol, lactulose, docusate, and omeprazole. He was white, born in the United States, and a lawyer, but he had not worked during the previous 4 months. He was married and monogamous, and an HIV antibody test 4 months prior was negative. He had a remote history of tobacco and alcohol use between the 1960s and the 1980s. He denied intravenous drug use. His family history was only remarkable for a father with coronary artery disease.
With fever, the hypothalamic set point for temperature increases. Night sweats usually indicate an exaggeration of the normal diurnal drop in the hypothalamic set point for temperature, with dissipation of increased heat (caused by fever) through evaporation of perspiration. Unfortunately, night sweats are not specific to any particular cause of fever. Fatigue is equally nonspecific but could result from anemia, hypothyroidism, or adrenal insufficiency or could be a side effect of the propranolol. The lack of a family history makes hereditary periodic fevers unlikely.
The patient appeared chronically ill. His temperature was 35.2C, blood pressure 71/53 mm Hg, heart rate 84 beats per minute, respiratory rate 14 breaths per minute, and oxygen saturation 99% while breathing room air. His weight was 47 kg. Examination of the patient's head and neck revealed bitemporal wasting but no scleral icterus, and the oropharynx was clear. There was no thyromegaly or lymphadenopathy. The findings of the cardiopulmonary examination was normal. The abdomen was soft with mild diffuse tenderness. There was no organomegaly or obvious ascites. His extremities were warm and without edema or cyanosis. He was dark‐skinned and had rare spider angiomas. The results of his neurological examination were normal.
Sepsis, drug ingestion (particularly vasodilators), environmental exposure, and endocrine abnormalities such as adrenal insufficiency and hypothyroidism can all cause both hypothermia and hypotension. Adrenal insufficiency is especially intriguing becauase it is also associated with malaise, abdominal pain, and hyperpigmentation. Explaining both adrenal insufficiency and chronic liver disease is more difficult. Hemochromatosis can cause cirrhotic liver disease, adrenal and thyroid insufficiency, and dark skin, but the patient's normal ferritin and liver biopsy findings make this disease unlikely.
The results of the laboratory studies were: white‐cell count, 4900/mm3, with a normal differential count; hemoglobin, 11.0 g/dL; platelet count, 52,000/mm3; mean corpuscular volume, 89 m3; sodium, 131 mmol/L; potassium, 5.0 mmol/L; chloride, 101 mmol/L; bicarbonate, 21 mmol/L; blood urea nitrogen, 31 mg/dL; creatinine, 1.8 mg/dL; aspartate aminotransferase, 45 U/L (normal range 1641 U/L); alanine aminotransferase, 30 U/L (normal range 1259 U/L); alkaline phosphatase, 587 U/L (normal range 29111 U/L); total bilirubin, 1.1 mg/dL (normal range 0.31.3 mg/dL); gamma‐glutamyl transferase, 169 U/L (normal range 771 U/L); lactate dehydrogenase, 127 IU/L (normal range 91185 IU/L); thyroid‐stimulating hormone, 3.1 mIU/L (normal range 0.54.7 mIU/L). Coagulation studies revealed a prothrombin time of 12 seconds (international normalized ratio [INR] 1.1) and an activated partial thromboplastin time (aPTT) of greater than 100 seconds. Urinalysis and chest radiography were unremarkable.
The low sodium, high potassium, and relatively low bicarbonate levels are all compatible with adrenal insufficiency. When present, the combination of hyponatremia (primarily from glucocorticoid deficiency) and hyperkalemia (from mineralocorticoid deficiency) suggests the adrenal insufficiency is primary, rather than from the pituitary. The differential diagnosis of primary adrenal insufficiency includes autoimmune disease, granulomatous disease, and tumor.
Most interesting is the isolated prolongation of the aPTT, making adrenal hemorrhage another possibility as a cause of the adrenal insufficiency. Isolated elevation of the aPTT suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which would interfere with the test. Heparin administration (which may not be immediately obvious, as in the case of a heparin lock of an intravenous line) and von Willebrand disease (from loss of the normal von Willebrand factorassociated prevention of factor VIII proteolysis) can also cause isolated prolongation of the aPTT.
Tumor, perhaps hepatocellular cancer, remains a possible explanation for the elevated alkaline phosphatase, with possible adrenal involvement. Amyloidosis and diffuse granulomatous disease (either infectious or noninfectious, such as sarcoidosis) can cause elevation in alkaline phosphatase. At this time, I would rule out adrenal insufficiency, further evaluate the elevated aPTT, and image the liver and adrenal glands.
The patient was hospitalized and given intravenous fluids. His blood pressure increased to 90/54 mm Hg. Further testing revealed an alpha‐fetoprotein of 1.5 g/dL (normal range 6.4 g/dL), an erythrocyte sedimentation rate of greater than 100 mm/s, and normal results of an antinuclear antibody test. Serum cortisol, drawn at 6 a.m., was 3 ng/dL; 60 minutes after cosyntropin stimulation, serum cortisol was 1 ng/dL. An ultrasound of the liver revealed chronic hepatic vein thrombosis.
The low absolute values and the failure of serum cortisol to respond to cosyntropin confirm the diagnosis of adrenal glucocorticoid deficiency. Hepatic vein thrombosis (Budd‐Chiari syndrome) is an unusual occurrence, often associated with a hypercoagulable state or tumor. How can we put these new findings together with the rest of the patient's abnormalities?
Primary antiphospholipid antibody syndrome is the most attractive unifying diagnosis because it appears to explain the most abnormalities with the fewest diagnoses. This syndrome includes arterial and venous thrombosis, thrombocytopenia, and isolated elevation of the aPTT and has been associated with hepatic vein thrombosis (acute and chronic) and adrenal insufficiency (from adrenal hemorrhage as a result of adrenal vein thrombosis). The histological findings of focal ischemic injury, seen on the patient's liver biopsy, are likely explained by hepatic venoocclusive disease.
Magnetic resonance imaging (MRI) of the abdomen (Fig. 2) demonstrated adrenal hemorrhage in the right adrenal gland. The patient's aPTT remained elevated even after his serum was mixed with normal serum, thereby excluding a factor deficiency. The results of a dilute Russell's viper venom time test (which tests the phospholipid‐dependent portion of the coagulation cascade) also showed elevation. The addition of phospholipids to the patient's serum corrected the aPTT, and a screen for factor inhibitors was negative. An anticardiolipin antibody (IgG) test was positive at 59.0 U (normal 0.42.3 U). These findings confirmed the presence of antiphospholipid antibodies.
The findings of a bone marrow biopsy, performed to exclude infiltrative diseases, were normal. The patient was diagnosed with primary antiphospholipid syndrome. Hydrocortisone and fludrocortisone were initiated, with the intention to continue them indefinitely. The patient was also started on intravenous heparin, which continued until he achieved a goal INR of 2.03.0 on warfarin. The patient was counseled on the importance of lifelong warfarin therapy given his diagnosis of antiphospholipid syndrome with hepatic vein and adrenal vein thromboses. On follow‐up 6 months after discharge, the patient's hypotension and fatigue had resolved, his alkaline phosphatase level had decreased substantially, and he had returned to work as a lawyer.
COMMENTARY
The diagnosis of a complex case with numerous clinical and laboratory abnormalities can be very difficult. The discussant successfully came to the correct diagnosis because he carefully evaluated each piece of evidence and did not fall prey to faulty triggering, the generation of diagnostic hypotheses based on selected pieces of clinical data.1 In the diagnostic process, physicians trigger new diagnostic possibilities and discard initial hypotheses as new findings emerge. Often, because of heuristic (analytic) biases, physicians fall victim to faulty triggering when evaluating patients.2 When confronted with a trigger feature such as night sweats, many physicians increase their consideration of tuberculosis or lymphoma at the expense of more common diagnoses, even though, as the discussant pointed out, any patient with fever can have this symptom.3 Whereas faulty use of trigger features may make physicians inappropriately consider uncommon diseases, a distinguishing feature limits the number of diagnostic possibilities and significantly changesincreases or decreasesthe likelihood of there being a rare disease.4 By correctly using the distinguishing feature of an elevated aPTT in the context of the patient's diverse clinical features, the discussant was able to arrive at a single, unifying diagnosis of antiphospholipid syndrome.
Antiphospholipid syndrome is arterial or venous thrombosis associated with significantly elevated antiphospholipid antibodies. Isolated prolongation of the aPTT is often the first clue to the presence of antiphospholipid antibodies, which interfere with phospholipid‐dependent coagulation assays.5 Antiphospholipid syndrome is considered primary if it is not associated with a known underlying disease or medication. Antiphospholipid syndrome is secondary if it is associated with certain diseases such as systemic lupus erythematosus and malignancy or with an adverse effect of medication. Although the prevalence of antiphospholipid antibodies is 1%5% in young, apparently healthy control subjects, it is higher in elderly patients with chronic diseases.6 It remains unclear why only certain patients with antiphospholipid antibodies manifest the syndrome, though having vascular risk factors may increase the risk of developing thrombosis in the presence of antiphospholipid antibodies.7
Three types of antiphospholipid antibody tests are currently in clinical use: lupus anticoagulants (measured by prolonged clotting time in a phospholipid‐dependent clotting test, such as the aPTT), anticardiolipin antibodies, and anti‐2‐glycoprotein I antibodies. All 3 tests are plagued by not being standardized between hospitals and laboratories and have limited sensitivity and specificity.8, 9 Lupus anticoagulants are most closely associated with thrombosis. Although a prolonged aPTT in the presence of thrombosis is often the first clue to the presence of lupus anticoagulants, only 30%40% of patients with the syndrome have this laboratory abnormality.10 Therefore, a normal aPTT result does not rule out the presence of antiphospholipid antibodies, and other tests of lupus anticoagulants, such as the dilute Russell viper venom time, should be performed.8, 9 There are many types of anticardiolipin antibodies of varying immunoglobulin isotypes, which all share the ability to bind cardiolipin in vitro. The IgG isotypes (as in our patient) are thought to be most closely associated with thrombosis, and it is known that high titers of anticardiolipin antibodies have much better discriminatory value than low titers.810 There is little data on the anti‐2‐glycoprotein I antibodies, but preliminary data suggest these antibodies may be more specific for the antiphospholipid syndrome.11
The antiphospholipid syndrome has classically been associated with lower‐extremity deep venous thrombosis, recurrent fetal loss, thrombocytopenia, and livedo reticularis.10 However, depending on the size and distribution of the vasculature involved and the extent and chronicity of involvement, antiphospholipid syndrome can result in manifestation of a wide range of diseases. Acute presentations such as thrombotic disease of the gastrointestinal, cardiac, and central nervous systems can be rapid and catastrophic. A more chronic and indolent course can lead to progressive organ dysfunction, as in this patient, with chronic liver disease resulting from recurrent episodes of hepatic venoocclusive disease and chronic hepatic vein thrombosis, a rare but well‐described complication of antiphospholipid syndrome.12, 13 It is unclear why the course of our patient's hepatic vein thrombosis waxed and waned so much. We hypothesized that he had episodes of microvascular hepatic venous thrombosis that led to transient hepatic dysfunction, with subsequent recovery upon spontaneous recanalization of hepatic veins or with healing and regeneration of liver tissue.
Treatment of antiphospholipid syndrome is controversial. Although prior reports suggested that patients with this syndrome were at higher risk for recurrent thrombosis when treated with the usual dose of warfarin (target INR 2.03.0), 2 randomized trial showed there was no difference in the recurrence of thrombosis between moderate‐intensity treatment with warfarin and high‐intensity treatment with warfarin.14, 15 Our patient was treated with warfarin to a moderate‐intensity target INR of 2.03.0 because he had liver disease and adrenal hemorrhage. Although he has done well, it is important that he be continuously reassessed, as should all patients with similar conditions, for the risk and recurrence of thrombosis weighed against the risk of bleeding.
Adrenal insufficiency is another rare complication of antiphospholipid syndrome. It was first described as such in 198016 and has since been reported in both children and adults.1719 Abdominal pain and hypotension were the most common findings (55% and 54%, respectively) in one case series of 86 patients with adrenal insufficiency from antiphospholipid syndrome.20 Fever, nausea, vomiting, weakness, fatigue, lethargy, and altered mental status were also variably present. Loss of adrenal function is most often a result of adrenal hemorrhage, which is best detected by MRI of the adrenal glands.21
The vascular anatomy of the adrenal gland is unusual. Multiple arteries supply the gland, but only one central vein provides drainage, making the gland relatively vulnerable to hemorrhagic infarction.22 Most cases of adrenal insufficiency from antiphospholipid syndrome are thought to be a result of adrenal vein thrombosis. The MRI showed that only the right adrenal gland of our patient had evidence of hemorrhage. Because both adrenal glands must be damaged before adrenal insufficiency results, it is probable that the left adrenal gland was damaged because of prior episodes of infarction and/or hemorrhage, but remote damage could not be detected by MRI. Of note, antiphospholipid antibodies directed against cholesterol‐rich proteins in the adrenal gland can also cause a locally active procoagulant state with microvascular venous thrombosis and subsequent postinfarction hemorrhage, which is another way in which the left adrenal gland could have been damaged without showing up radiographically.23
As for other types of adrenal insufficiency, the primary treatment for adrenal insufficiency from antiphospholipid syndrome is rapid corticosteroid replacement, with the addition of anticoagulants to treat the hypercoagulable state of the antiphospholipid syndrome. Adrenal insufficiency is temporary in some cases.24 Mortality from adrenal insufficiency due to antiphospholipid syndrome may be higher than that from other forms of adrenal insufficiency.22 Therefore, screening for adrenal insufficiency is critical for any patient with suspected or documented antiphospholipid syndrome who presents with abdominal pain, weakness, electrolyte abnormalities, or unexplained hypotension.
This case illustrates the importance, as the key to diagnosis, of determining a distinguishing feature such as a prolonged aPTT from among the multitude of abnormalities that could have led the diagnostic process astray. Occasionally, a single clinical or laboratory abnormality, such as the elevated aPTT in our patient, is so valuable in the assessment of a difficult case that it significantly increases the likelihood of an uncommon condition and leads to the correct final diagnosis, thereby becoming the pivotal distinguishing feature.
Key Points
-
Hypercoagulability can lead to adrenal insufficiency by causing adrenal vein thrombosis and adrenal infarction. Therefore, hypercoagulable states, such as antiphospholipid syndrome, should be considered for patients who present with symptoms or signs of unexplained adrenal insufficiency.
-
Isolated elevation of activated partial thromboplastin time (aPTT) suggests deficiency or inhibition of the factors involved in the intrinsic pathway (factors VIII, IX, XI, and XII) or the presence of an antiphospholipid antibody, which interferes with this test. Heparin administration and von Willebrand disease can also cause isolated prolongation of the aPTT.
-
Treatment of the antiphospholipid syndrome is controversial, but according to the results of 2 recent randomized, controlled trials, patients with this syndrome who have had their first episode of thrombosis should be treated with warfarin, with a goal INR of 2.03.0.
-
When interpreted incorrectly, trigger features such as night sweats cause clinicians to inappropriately consider a rare diagnosis, even though common diagnoses may be more likely. On the other hand, distinguishing features, such as the prolonged aPTT in this patient, truly do increase or decrease the probability of a rare diagnosis.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.
- .Diagnostic reasoning.Ann Intern Med.1989;110:893–900.
- ,.Cognitive errors in diagnosis: instantiation, classification, and consequences.Am J Med.1989;86:433–441.
- ,,.Diagnosing night sweats.Am Fam Physician.2003;67:1019–1024.
- ,.When you hear hoof beats: four principles for separating zebras from horses.J Am Board Fam Pract.2000;13:424–429.
- ,,.The antiphospholipid syndrome.N Engl J Med.2002;346:752–763.
- .Epidemiology of the antiphospholipid antibody syndrome.J Autoimmun.2000;15:145–151.
- ,,,,.Antiphospholipid syndrome and asymptomatic carriers of antiphospholipid antibody: prospective analysis of 404 individuals.J Rheumatol.2004;31:1560–1567.
- ,,.Management of antiphospholipid antibody syndrome: a systematic review.JAMA2006;295:1050–7.
- ,,, et al.International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS).J Thromb Haemost.2006;4:295–306.
- .Antiphospholipid syndrome.Dis Mon.2003;49:696–741.
- ,,, et al.Value of autoantibodies to beta(2)‐glycoprotein 1 in the diagnosis of antiphospholipid syndrome.Rheumatology (Oxford).2002;41:550–553.
- ,,, et al.Budd‐Chiari syndrome secondary to antiphospholipid syndrome: clinical and immunologic characteristics of 43 patients.Medicine (Baltimore).2001;80:345–354.
- ,,.The Budd‐Chiari syndrome.N Engl J Med.2004;350:578–585.
- ,,, et al.A randomized clinical trial of high‐intensity warfarin vs. conventional antithrombotic therapy for the prevention of recurrent thrombosis in patients with the antiphospholipid syndrome (WAPS).J Thromb Haemost.2005;3:848–853.
- ,,, et al.A comparison of two intensities of warfarin for the prevention of recurrent thrombosis in patients with the antiphospholipid antibody syndrome.N Engl J Med.2003;349:1133–1138.
- ,,.Thrombosis in patients with the lupus anticoagulant.Ann Intern Med.1980;92:156–159.
- ,,,,,.Spontaneous adrenal hemorrhage associated with transient antiphospholipid antibody in a child.Clin Pediatr (Phila).2001;40:347–350.
- ,,,,,.Association of primary antiphospholipid syndrome with primary adrenal insufficiency.J Rheumatol.1996;23:1286–1287.
- ,.Adrenal insufficiency in the antiphospholipid antibody syndrome.Semin Arthritis Rheum.1995;25:109–116.
- ,,, et al.Adrenal involvement in the antiphospholipid syndrome: clinical and immunologic characteristics of 86 patients.Medicine (Baltimore).2003;82:106–118.
- ,,.Adrenal hemorrhage in patients with primary antiphospholipid syndrome: imaging findings.AJR Am J Roentgenol.1995;165:361–364.
- ,,.Adrenal hemorrhage: a 25‐year experience at the Mayo Clinic.Mayo Clin Proc.2001;76:161–168.
- ,,,,.Antiphospholipid syndrome and endocrine damage: why bilateral adrenal thrombosis?Eur J Haematol.2003;71:299–302.
- ,.Reversible adrenal insufficiency after adrenal hemorrhage.Ann Intern Med.1993;119:439–440.
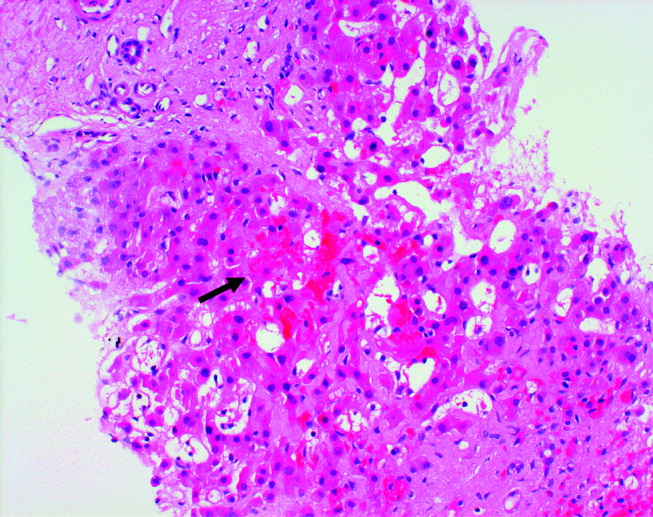
Clinical Conundrum
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
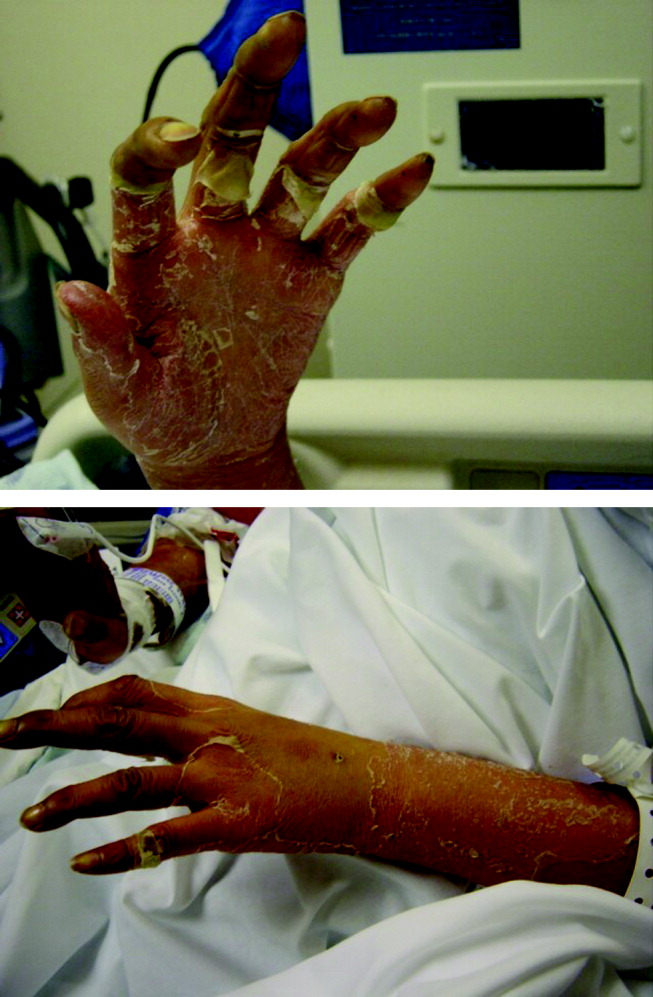
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.

COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.
COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
A 20‐year‐old woman presented to the emergency department after 2 days of epistaxis and vaginal bleeding.
A young woman is more likely to present with infection, toxic exposure, or rheumatologic disease than with a degenerative disease or malignancy. Her bleeding may relate to a platelet abnormality, either quantitative or qualitative. I would pursue her bleeding and menstrual history further.
The patient was healthy until 2 months previously, when she noted arthralgia of her shoulders, wrists, elbows, knees, and ankles. She was examined by a rheumatologist who detected mild arthritis in her left wrist and proximal interphalangeal joints. The rest of her joints were normal. Rheumatoid factor and ANA were positive, and the erythrocyte sedimentation rate was 122 mm/hour. She was diagnosed with possible systemic lupus erythematosus and was placed on a nonsteroidal anti‐inflammatory agent. At a follow‐up visit 1 month prior to admission, her arthralgia had markedly improved. Two weeks prior to admission, the patient began to feel fatigued. Two days prior to admission, she developed epistaxis and what she thought was her menses, though bleeding was heavier than usual and associated with the passage of red clots. On the day of admission the vaginal bleeding worsened, and emergency personnel transported the patient to the hospital.
The diagnosis of systemic lupus erythematosus (SLE) is not engraved in stone. One must be vigilant for other diseases masquerading as SLE while continuing to build a case for it. As more criteria are fulfilled, the probability of lupus increases, yet no findings, alone or in combination, are pathognomonic of this protean disease. This patient's age, sex, and serology are compatible with SLE; otherwise, her presentation is nonspecific. I would request a complete blood count, coagulation tests, and additional serological tests.
The quantity of the bleeding is described, but this does not help decipher its etiology. Excess bleeding may be a result of one or more of 3 broad etiologies: problems with platelets (quantitative or qualitative), with clotting factors (quantitative or qualitative), or with blood vessels (trauma, vasculitis, or diseases affecting collagen). Because quantitative and qualitative factor disorders generally do not present with mucosal bleeding, I am thinking more about platelet problems and about processes that damage the microvasculature. If this woman has lupus, immunologic thrombocytopenia may be the cause of mucosal bleeding.
The patient had no previous medical problems and had never been pregnant. Her only medication was sulindac twice daily for the past month. She was born in Hong Kong, graduated from high school in San Francisco, and attended junior college. She lived with her parents and brother and denied alcohol, tobacco, or recreational drug use but had recently obtained a tattoo on her lower back. There was no family history of autoimmune or bleeding disorders, and a review of systems was notable for dyspnea with minimal exertion and fatigue which worsened in the past 2 days. She had no prior episodes of abnormal bleeding or clotting.
Tattoos may be surrogates for other high‐risk behaviors and suggest an increased risk of hepatitis and sexually transmitted diseases. I want to know her sexual history and other risk factors for human immunodeficiency virus infection. The dyspnea and fatigue are likely the result of anemia, but I am also considering cardiac disease. Though SLE remains a possibility, I cannot assume the presence of a lupus anticoagulant with antiphospholipid syndrome without a history of infertility or recurrent miscarriages.
On arrival at the emergency department, the patient had a blood pressure of 78/46 mm Hg, a pulse of 120 beats/min, a temperature of 34C, 14 respirations per minute, and oxygen saturation of 99% while breathing supplemental oxygen through a nonrebreather mask. Systolic blood pressure improved to 90 mmHg after 4 L of normal saline was administered. The patient was pale but alert. There was crusted blood in her mouth and nostrils without active bleeding or petechiae. Her tongue was pierced with a ring, and sclerae were anicteric. Bleeding was noted from both nipples. There was no heart murmur or gallop, and jugular venous pressure was not elevated. Pulmonary exam revealed bibasilar crackles. Abdomen was soft, not tender, and without hepatosplenomegaly, and her umbilicus was pierced by a ring. Genitourinary exam revealed scant vaginal discharge and clotted blood in the vagina. Skin demonstrated no petechiae, ecchymoses, or stigmata of liver disease. Neurological and joint exams were normal.
It is hard to conceive of vaginal bleeding producing this profound a degree of hypotension. The patient may have additional occult sites of bleeding, or she may have a distributive cause of hypotension such as sepsis or adrenal hemorrhage with resultant adrenal insufficiency. Breast bleeding is unusual, even with profound thrombocytopenia, and I wonder about a concomitant factor deficiency. Furthermore, if thrombocytopenia was the sole reason for the bleeding, I would have expected petechiae. Diffuse vascular injury, such as from lupus or vasculitis, would be an unusual cause of profound bleeding unless there was also disseminated intravascular coagulation.
Laboratory studies revealed a white count of 2000/mm3, of which 42% were neutrophils, 40% bands, 8% lymphocytes, and 10% monocytes. Hematocrit was 17.6%, platelets 35,000/mm3. Sodium was 124 mmol/L, potassium 6 mmol/L, chloride 92 mmol/L, bicarbonate 10 mmol/L, blood urea nitrogen 122 mg/dL (43.5 mmol/L), and creatinine 3.4 mg/dL (300 mol/L). Blood glucose was 44 mg/dL (2.44 mmol/L). Total bilirubin was 3.0 mg/dL (51.3 mol/L; normal range, 0.1‐1.5), alkaline phosphatase 105 U/L (normal range, 39‐117), aspartate aminotransferase 849 U/L (normal range, 8‐31), alanine aminotransferase 261 U/L (normal range, 7‐31), international normalized unit (INR) 2.9, and partial thromboplastin time (PTT) 34.2 seconds.
The combination of profound hypotension, electrolyte abnormalities, hypoglycemia, and hypothermia makes adrenal insufficiency a consideration. I would perform a cortrosyn stimulation test and start glucocorticoid and perhaps mineralocorticoid replacement. In addition, there is renal failure and metabolic acidosis, with a calculated anion gap of 22. The anion gap may be from lactic acidosis secondary to hypotension and hypoperfusion. The abnormal transaminases and bilirubin could relate to infectious hepatitis or systemic infection. Although ischemia could explain these findings, it is rare for a 20‐year‐old to develop ischemic hepatopathy. Thrombocytopenia this moderate may augment the volume of blood loss, but spontaneous bleeding because of thrombocytopenia is unusual until the platelet count falls below 20,000/mm3. Furthermore, the elevated INR points to a mixed coagulopathy. Interpretation of the INR is complicated by the fact she has liver disease, and I am most concerned about acute disseminated intravascular coagulation (DIC) or impending fulminant hepatic failure. This is not the pattern seen with antiphospholipid antibody syndrome, in which the INR tends to be preserved and the PTT prolonged.
Urine dipstick testing demonstrated a specific gravity of 1.015, trace leukocyte esterase, 2+ protein, and 3+ blood, and microscopy revealed 2 white blood cells and 38 red blood cells per high‐power field, many bacteria, and no casts. Creatine kinase was 20,599 U/L, with a myocardial fraction of 1.4%. Lipase was normal, lactate was 7.3 mmol/L, and serum pregnancy test was negative.
Although there is proteinuria and hematuria, we do not have solid evidence of glomerulonephritis. Although the red cells could be a contaminant from her vaginal bleeding, I would examine her sediment carefully for dysmorphic red cells, recognizing that only a quarter of people with glomerulonephritis have red‐cell casts. A urine protein‐to‐creatinine ratio would be useful for estimating the degree of proteinuria. The elevated creatine kinase indicates rhabdomyolysis. In a previously healthy young woman without evidence of cardiogenic shock, it would be unusual for hypotension to result in rhabdomyolysis. Infection and metabolic derangements are possible etiologies of rhabdomyolysis. Alternatively, coagulopathy might have produced intramuscular bleeding. The constellation of thrombocytopenia, anemia, and renal failure raises my suspicion that there is a thrombotic microangiopathy, such as thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS). I would inspect a peripheral‐blood smear for schistocytes and evidence of microangiopathy.
The chest radiograph demonstrated low lung volumes, patchy areas of consolidation, and pulmonary edema. Heart size was normal, and there were no pleural effusions. On the first hospital day the patient required mechanical ventilation because of respiratory failure. She received 5 units of packed red blood cells, 2 units of fresh frozen plasma, and 1 unit of platelets. Vasopressor infusion was started, and a vascular catheter was placed for hemodialysis. Blood, respiratory, and urine cultures were sent, and methylprednisolone, piperacillin/tazobactam, and vancomycin were administered. D‐dimer was greater than 10,000 ng/mL, fibrinogen was 178 mg/dL, and lactate dehydrogenase was 1671 U/L (27 kat/L). The peripheral‐blood smear demonstrated 1+ schistocytes and no spherocytes. There were fewer white blood cells with bands and myelocytes, but no blasts.
The presence of schistocytes and the elevated lactate dehydrogenase point to a microangiopathic hemolytic process. Causes of microangiopathic hemolytic anemia include TTP, HUS, DIC, paraneoplastic conditions, and endothelial damage from malignant hypertension or scleroderma renal crisis. The INR and PTT will usually be normal in TTP and HUS. The depressed fibrinogen and elevated D‐dimer suggest that in response to severe bleeding, she is also clotting. DIC, possibly from a severe infection, would explain these findings. Alternatively, the multisystem organ failure may represent progression of SLE.
Additional serology studies detected antinuclear antibodies at 1:320 with a speckled pattern. Rheumatoid factor was not present, but antidouble‐stranded DNA and antiSmith antibodies were elevated. C3 was 30 mg/dL (normal range, 90‐180), C4 was 24 mg/dL (normal range, 16‐47), and the erythrocyte sedimentation rate was 53 mm/h.
The results of the additional lab tests support a diagnosis of lupus and thus a lupus flare, but I agree that antibiotics should be empirically administered while searching for an underlying infection that might mimic lupus. Apart from infection, severe lupus may be complicated by widespread vasculitis or catastrophic antiphospholipid antibody syndrome, which would necessitate high‐dose immunosuppressive therapy and anticoagulation, respectively.
Tests for antiphospholipid antibodies including lupus anticoagulant and for anticardiolipin antibodies were negative. The patient continued to require vasopressors, hemodialysis, and mechanical ventilation. On the fourth hospital day she developed a morbilliform rash over her trunk, face, and extremities. Skin over her right buttock became indurated and tender. On the sixth day of hospitalization the skin on her face, extremities, and palms began to desquamate (Fig. 1).
Regarding the rash, it is hard to differentiate the chicken from the egg. The rash may be a reaction to medication, or it may be a clue to a multiorgan disease. I am considering severe skin reactions like Stevens‐Johnson as well as bacterial toxin‐mediated diseases such as toxic shock syndrome. The criteria for toxic shock syndrome with multisystem involvement are very similar to those for lupus. In this case, a desquamating rash occurring on the heel of a multiorgan illness definitely points to toxic shock syndrome. In staphylococcal toxic shock cases, blood cultures are frequently negative, and the origin may elude detection, but of the sources identified, most have been wounds and soft‐tissue infections.
On hospital day 4, blood cultures from admission grew oxacillin‐sensitive Staphylococcus aureus in 4 of the 4 bottles. Magnetic resonance imaging of the thigh demonstrated extensive necrosis of multiple muscles (Fig. 2). The patient underwent muscle debridement in the operating room, and Gram's stain of the debrided muscle revealed Gram‐positive cocci. Following surgery, she rapidly improved. She no longer required dialysis and was eventually discharged home after completing a prolonged course of intravenous anti‐Staphylococcal antibiotics at a rehabilitation facility. Follow‐up urine testing on 2 occasions revealed 1.6 and 1.4 g of protein in 24‐hour collections, but serum creatinine remained normal, and microscopy demonstrated no dysmorphic red cells or red‐cell casts. Performance of a kidney biopsy was deferred. Other than transient arthralgia and malar rash, her lupus has been quiescent, and her prednisone dose was tapered to 5 mg daily. Six months after discharge she returned to school.
COMMENTARY
Using the American College of Rheumatology (ACR) definition, systemic lupus erythematosus (SLE) is diagnosed when at least 4 criteria are met with a sensitivity and specificity above 95%. These criteria were developed for study purposes to differentiate SLE from other rheumatic diseases. At disease onset a patient may not meet the ACR threshold, but delaying treatment may be harmful. Data conflict on the probability of such patients eventually being classified as having SLE, with estimates ranging from less than 10% to more than 60%.1, 2 With SLE prominent in the differential diagnosis of a critically ill patient, hospitalists must consider the 3 most common causes of death in lupus patients: lupus crisis, severe infection, and thrombosis.3
Most exacerbations of SLE occur in one system, most commonly the musculoskeletal system, and are mild. However, 10% of patients a year will require high‐dose corticosteroids or cytotoxic agents for severe flares that can occur in any system affected by lupus and in 15% of cases may involve multiple sites simultaneously.4, 5 Diagnosing lupus flares remains challenging. Although pulmonary hemorrhage and red blood cell casts may strongly implicate active lupus in the lungs or kidneys, specific clinical and laboratory markers of lupus crisis are lacking. Several global indices reliably measure current disease status but are cumbersome, cannot be relied on solely for treatment decisions and have not been well studied in hospitalized patients.68 Fever, once a dependable harbinger of active lupus,9 cannot reliably discriminate lupus flares from infection. In 2 studies, Rovin et al. found that infection accounted for fever in all but one SLE outpatient taking prednisone and that in hospitalized SLE patients, failure of fevers to resolve within 48 hours of administering 20‐40 mg of prednisone daily strongly suggested infection.10 The laboratory findings provided general support for there being an SLE flare or an infection, but, as the discussant pointed out, these cannot be relied on exclusively to discriminate between the two. Results that suggest infection in an SLE patient include leukocytosis, increased band forms or metamyelocytes, and possibly elevated C‐reactive protein. Findings favoring SLE flare include leukopenia, low C3 or C4 (particularly for nephritis or hematologic flares) and elevated anti‐double‐stranded DNA antibodies for nephritis.1113 Without a clear gold standard for definitively determining a lupus crisis, it is diagnosed when clinical manifestations fit a pattern seen in SLE (nephritis, cerebritis, serositis, vasculitis, pneumonitis), the results of serology studies support this conclusion, and other plausible diagnoses are excluded.
Infection and active disease account for most ICU admissions of lupus patients. SLE and infection intertwine in 3 ways. First, SLE patients are predisposed to infection, possibly because of a variety of identified genetic abnormalities of immune function.14 Although community‐acquired bacteria and viruses account for most infections, lupus patients are vulnerable to a wide array of atypical and opportunistic pathogens. Clinical factors that augment this intrinsic risk include severity of the underlying SLE, flares of the central nervous system or kidneys, and use of immunosuppressive agents.14 The latter deserves particular attention, as a recent study found more than 90% of SLE patients admitted to an ICU with severe infection were taking corticosteroids prior to hospitalization.15 Second, infection may trigger a lupus flare. Third, features of severe lupus flares and infection may overlap. Differentiating between the 2 may be difficult, and the stakes are high, as SLE patients admitted to ICUs have a risk of death that is substantially higher (47%) than that of those without SLE (29%) and much greater than the overall risk of death for those with SLE, for whom 10‐year survival exceeds 90%.15
In addition to lupus crisis and infection, the differential diagnosis of acute multisystem disease in a patient with SLE includes catastrophic antiphospholipid syndrome (APS) and thrombotic thrombocytopenic purpura, 2 thrombotic microangiopathies to which SLE patients are predisposed. Thrombocytopenia and hemolytic anemia with schistocytes should raise suspicion of these diagnoses. Additional findings for TTP include fevers, altered mental status, acute renal failure, and elevated serum lactate dehydrogenase; however, prothrombin time should not be prolonged. Lupus anticoagulant or anticardiolipin antibodies are found in up to 30% of lupus patients, of whom 50%‐70% develop APS within 20 years, characterized by thrombosis or spontaneous abortions in the presence of antiphospholipid antibodies.16 Catastrophic APS is a rare subset of APS involving thromboses of multiple organs simultaneously and has a mortality rate of 50%.
In the present patient, an elevated INR, bleeding, hypotension, and the absence of antiphospholipid antibodies argued against TTP and APS, leading the discussant to focus on SLE and sepsis. Arthralgia, cytopenia, and the results of serology studies suggested a lupus crisis, but hypothermia, hypotension, and DIC pointed to severe infection. Empiric treatment of both conditions with corticosteroids and broad‐spectrum antibiotics was indicated, and ultimately the patient's condition was found to meet criteria for toxic shock syndrome (TSS) and SLE. TSS has rarely been reported in SLE1718 and poses a particularly difficult diagnostic challenge because a severe lupus flare can meet the diagnostic criteria for TSS (Table 1), especially early on, before the characteristic desquamating rash appears. Acuity of the illness increased the ante in this challenging case. Afraid not to treat a potentially life‐threatening condition, empiric treatment of severe lupus and sepsis was initiated. Attention then shifted to fraying, or unraveling, the knot linking infection and lupus. Ultimately, diagnoses of both TSS and SLE were established.
|
| 1. Fever > 38.9C |
| 2. Hypotension (SBP 90 mm Hg) |
| 3. Diffuse erythroderma |
| 4. Desquamation, particularly of palms and soles (occurring 1‐2 weeks after onset of illness) |
| 5. Involvement of 3 or more systems: |
| GI (vomiting or diarrhea at onset) |
| Muscular (CK > twice the upper limit of normal or severe myalgia) |
| Mucus membranes (vaginal, oropharyngeal, or conjunctival hyperemia) |
| Renal (pyuria; BUN or creatinine > twice the upper limit of normal) |
| Hepatic (bilirubin or transaminases > twice the upper limit of normal) |
| Hematologic (platelets 100,000/mm3) |
| Central nervous system (altered mental status without localizing deficits unexplained by hypotension or fever) |
| In addition, negative cultures of blood, throat, and cerebrospinal fluid are expected (except for blood cultures in S. aureus TSS, which may be positive). |
Acknowledgements
The authors thank Michael Chan, MD, and Shelley Gordon, MD, for their input on this manuscript.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.
- ,.Incomplete lupus erythematosus.Arch Intern Med.1989;149:2473–2476.
- ,,.Systemic lupus erythematosus. Differences between patients who do, and who do not, fulfill classification criteria at the time of diagnosis.J Rheumatol.1980;7:831–837.
- ,,, et al.Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients.Medicine (Baltimore).2003;82:299–308.
- ,,,,.Definition and treatment of lupus flares measured by the BILAG index.Rheumatology.2003;42:1372–1379.
- ,,,,.The occurrence, nature and distributions of flares in a cohort of patients with systemic lupus erythematosus: a rheumatologic view.Br J Rheumatol.1995;34:257–260.
- ,,.Comparison of the validity and sensitivity to change of 5 activity indices in systemic lupus erythematosus.J Rheumatol.2000;27:664–670.
- ,,.Serologically active clinically quiescent systemic lupus erythematosus—predictors of clinical flares.J Rheumatol.1994;21:2239–2241.
- ,,,,,.Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail.Arthritis Rheum.1996;39:370–378.
- ,,.Fever in systemic lupus erythematosus.Am J Med.1979;67:935–940.
- ,,, et al.Clinical significance of fever in the systemic lupus erythematosus patient receiving steroid therapy.Kidney Int.2005;68:747–759.
- ,,.Lupus nephritis flares.Lupus.2005;14:49–52.
- ,,,.A decrease in complement is associated with increased renal and hematologic activity in patients with systemic lupus erythematosus.Arthritis Rheum.2001;44:2350–2357.
- ,,,.Definition, incidence, and clinical description of flare in systemic lupus erythematosus. A prospective cohort study.Arthritis Rheum.1991;34:937–944.
- ,.Infections and SLE.Autoimmunity.2005;38:473–485.
- ,,, et al.Outcome and prognostic factors in critically ill patients with systemic lupus erythematosus: a retrospective study.Critical Care.2005;9:R177–R183.
- ,,.The Antiphospholipid Syndrome.N Engl J Med.2002;346:752–763.
- ,,.Toxic shock syndrome in a patient with systemic lupus erythematosus.Can Med Assoc J.1983;129:1201–1202.
- ,,.Toxic shock syndrome in a patient with breast cancer and systemic lupus erythematosus.Eur J Surg Oncol.2001;27:330–331.
- Case definitions for infectious conditions under public health surveillance.MMWR Recomm Rep.1997;46(RR‐10):39.
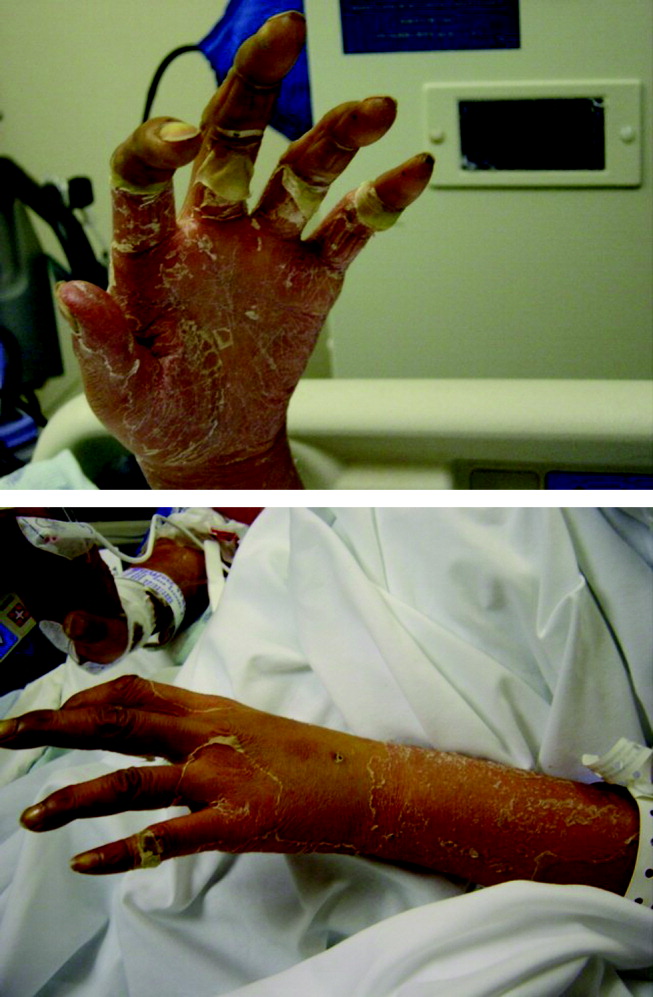
Clinical Conundrum
A 35‐year‐old man presented to the emergency department of a community hospital with 3 days of nausea, vomiting, abdominal pain, and diarrhea. He had developed a sore throat, nasal congestion, and green sputum on a business trip to Las Vegas 10 days prior. He then traveled to Shanghai, China, where he developed frequent diarrhea with mucous and urgency. The stool was mustard colored without blood or melena. He became nauseated and unable to keep down food or fluids. He noted a 4‐kg weight loss since the beginning of his symptoms. He had not traveled outside Shanghai or eaten exotic foods. His travel companions remained unaffected. The patient had had type I diabetes mellitus for 23 years with known retinopathy and microalbuminuria. He had hypertension and hyperlipidemia but was otherwise healthy. He was a married traveling salesman with 3 healthy children. He did not smoke or drink and reported no drug use or extramarital relations. He had no known allergies. His medications included insulin, lisinopril, atorvastatin, and valsartan. He was afebrile, his vital signs were stable and the physical examination was unremarkable.
The patient presents with gastrointestinal symptoms following a trip to China. He may have an infection that began in the respiratory system and now is causing some gastrointestinal symptoms, like Legionnella. If he had received antibiotics, the diarrheal illness could be a complication. With his recent travel to China, typical enteric pathogens would have to be considered: enterotoxigenic E. coli, Shigella, Salmonella, Campylobacter, or perhaps Giardia.
He was admitted and treated with intravenous fluids and ciprofloxacin for presumed gastroenteritis. He was discharged the next day but returned 2 days later because of continued nausea and vomiting and limited oral intake. He was febrile, to 38.9C, with chills. The results of an abdominal exam were normal. Stool studies for Salmonella, Shigella, Campylobacter, Yersinia, enterotoxigenic E. coli, Giardia, and Cryptosporidium were negative. No blood parasites were seen. The white‐cell count was 3900/mm3 with a normal differential count. Hemoglobin level, platelet count, serum electrolytes and creatinine were normal. He was readmitted for intravenous fluids.
The patient was treated for traveler's diarrhea, although this is not a typical case and is now becoming a protracted illness. Amebiasis would be a consideration, as well as typhoid or perhaps an abdominal abscess. The normal platelet count reduces the likelihood of hemolytic uremic syndrome, as does the absence of bloody diarrhea. I would evaluate for a systemic illness: check for adenopathy, do a thorough abdominal exam, and get a chest radiogram, blood cultures, and liver function tests. I am also concerned about metabolic abnormalities that could occur as a consequence of the diarrhea.
Over 3 days, oliguria developed along with urinary hesitancy, a 9‐kg weight gain, and the development of marked edema. Blood pressure and heart rate remained normal, and a chest radiograph was clear. Liver function tests were normal. A urinary catheter was inserted. Urinalysis revealed a specific gravity of 1.031, protein of 100 mg/dL, and trace glucose but was otherwise negative; no casts or cells were seen in the sediment. Chemistries included sodium of 133 mmol/L, potassium of 3.9 mmol/L, and serum bicarbonate of 18.4 mmol/L. Blood and urine cultures were sterile. The creatinine increased from 1.0 mg/dL (88.4 mol/L) to 1.3 mg/dL (115 mol/L). He was transferred to a tertiary care hospital for renal consultation because of concerns of impending renal failure and for consideration of a kidney biopsy.
In a typical case of a malabsorptive diarrhea, the patient could be volume depleted, but in this case he has gained 9‐kg and is grossly edematous. The chest radiograph and liver tests point to renal rather than cardiac or hepatologic causes for the edema. A glomerulonephritis may be driving the salt and water retention.
The proteinuria could be related to hemodynamics, or it could be from a glomerular lesion secondary to immune complexes. The specific gravity of 1.031 indicates the kidney is able to concentrate, and we are not seeing acute tubular necrosis. There is only minimal elevation in creatinine at this point. Quantitation or estimation of the degree of proteinuria by a protein‐to‐creatinine ratio would be helpful.
A further workup should include additional blood cultures and a CT scan of the lungs and abdomen to look for occult infection. Is he unfortunate enough to have developed a malignancy? Is this a connective tissue disease? Reexamination of the urine sediment is important to evaluate for glomerulonephritis.
The patient reported ongoing nausea and vomiting, but his diarrhea resolved. He was tachypneic, with a respiratory rate of 26 breaths/minute and an oxygen saturation of 98% breathing ambient air. His temperature was 37.1C, heart rate 84 beats/minute, and blood pressure 120/65 mm Hg. His mucous membranes were moist, and his jugular venous pressure was 6 cm. No lymphadenopathy was present. The heart and lungs were normal. The abdomen was soft, nontender, and without organomegaly, masses, or shifting dullness; bowel sounds were hypoactive. Severe edema of his legs, sacrum, hands, arms, and orbits was noted. His right hand and wrist were painful with limited mobility and small joint effusions of the wrist and metacarpophalangeal joints, but without erythema or warmth. Small petechiae were noted on his eyelids; skin examination was otherwise unremarkable. He had been given ciprofloxacin, phenergan, calcium carbonate, and pantoprazole prior to transfer. Stool was negative for occult blood.
His lungs are clear, but it is possible to have early pulmonary congestion with normal breath sounds. As he is normotensive and has a normal JVP, I would not give further intravenous fluid. Unless he has evidence of symptomatic pulmonary edema, I would not give diuretics but would simply observe his course. At this point I would ultrasound his kidneys to make sure there is no obstruction. The proteinuria could be a result of underlying diabetic glomerulopathy that may predispose to fluid retention.
He has significant oliguria but only a mild rise in creatinine. As the referring physicians requested a biopsy, we should consider it. A decision to biopsy the kidney would rest on the degree of proteinuria and the activity of the sediment. For example, if red blood cell casts or dysmorphic red cells were present, postinfectious glomerulonephritis or IgA nephropathy would be more likely. However, if the proteinuria is in the non‐nephrotic range and the sediment is nonreactive, the yield of a biopsy would be low.
His right hand and wrist are painful with limited mobility. This could be a sequela of endocarditis, although the absence of a murmur and negative blood cultures make it unlikely. He could have an infectious arthritis, although this usually presents more dramatically. He could have gout or pseudogout, which could be determined by joint aspiration. Finally, could this be iatrogenic? A drug reaction could explain some of the features, including rash, fever, joint symptoms, and renal abnormalities.
Repeat urinalysis revealed protein of more than 300 mg/dL, hematuria (1+), mucous, renal tubular epithelial cells, renal tubular epithelial cell casts, and granular casts. Eosinophiluria was absent. Laboratory evaluation revealed a hemoglobin level of 10.7 g/dL (decreased from 13 g/dL on initial presentation), white‐cell count of 7300/mm3, platelet count of 337,000/mm3, serum creatinine of 1.2 mg/dL (106 mol/L), blood urea nitrogen of 23 mg/dL (8.2 mol.L), total serum protein of 6.3 g/dL (normal range, 6.3‐8.7), and albumin of 2.7 g/dL (normal range, 3.2‐5.2). Other liver tests and serum electrolytes were normal.
This degree of proteinuria is significant, but it is unclear whether this is related to the underlying disease process or his advancing diabetes. He has some hematuria, but that could be from the urinary catheter. It would be helpful to know if the red blood cells are dysmorphic, which would point to a glomerulonephritis. He has renal cells, renal cell casts, and granular casts, which are nonspecific. He has a mild anemia, which is unexplained, but could relate to phlebotomy or overhydration. The hypoalbuminemia may be a result of renal losses or a catabolic state.
A renal ultrasonogram was normal, apart from evidence of bilateral pleural effusions. Antinuclear antibody and rheumatoid factor test results were negative, as were those for hepatitis A, hepatitis B surface antigen, and hepatitis C antibodies. Antistreptolysin O and antideoxyribonuclease B titers were normal. Total complement Ch50 was low at 29 U/mL (normal range, 30‐75) as was complement factor C3 at 67 mg/dL (normal range, 90‐180). Complement factor C4 was normal. Serum and urine electrophoresis revealed no monoclonal protein spike. Vitamin B12 and serum folate were normal, serum ferritin was 584 ng/mL (normal range, 30‐400), iron serum was 29 g/dL (normal range, 45‐160), transferrin saturation was 16%, and total iron‐binding capacity was 164 g/dL (normal range, 250‐450). The reticulocyte count was 2.9% with an absolute reticulocyte count of 102/cm3 and a reticulocyte production index of 0.96 (normal range, 1.0‐2.0).
It is reassuring that his urinary tract ultrasound is normal. In addition to edema, he has bilateral effusions, which are probably transudative, related to fluid overload. The urinalysis does not suggest a rapidly progressive glomerulonephritis, but autoimmune disease is still in the differential.
He has a mild complement C3 deficiency. In nephrology we think of lupus, infective endocarditis, cryoglobulinemia, and specific glomerular lesions such as membranoproliferative glomerulonephritis and postinfectious glomerulonephritis as being associated with the development of circulating immune complexes that may lead to low complement levels. There is no evidence of a paraprotein, but testing for cryoglobulins should be considered. Cryoglobulins are associated with hepatitis C but may be induced by a variety of infections. Acting like immune complexes, they can lead to low complement levels and could cause some of this patient's symptoms. However, this whole illness seems most likely to be secondary to infection. The normal antistreptolysin O and antideoxyribonuclease B titers make streptococcal disease unlikely, but another bacterial infection could cause postinfectious glomerulonephritis.
Over the course of his 5‐day hospital stay, the patient received furosemide with increased urine output and normalization of his serum creatinine to its baseline level of 1.0 mg.dL (88.4 mol/L). Proteinuria resolved to 44 mg/dL. A kidney biopsy was not performed. The parvovirus IgG index, checked because of anemia and oligoarthralgias, was 3.67 (normal 0‐1.10), and the IgM index was 8.13 (normal 0‐1.10), suggesting recent infection. The patient was discharged after 5 days. His edema had resolved on discharge; he continued to be nauseated but was able to eat and drink normally. Six months after his hospitalization, his symptoms had completely resolved.
Parvovirus! It could cause the pulmonary infection and the gastroenteric symptoms. Parvovirus usually causes more anemia than nausea and vomiting. We see it occasionally in our transplant patients. The underlying diabetic nephropathy may have made him more symptomatic with a superimposed glomerulonephritis. The most important pedagogic point is that he did well with a very conservative approach, and the possible iatrogenic consequences of a kidney biopsy, had it been performed, were avoided.
COMMENTARY
Parvovirus B19 is endemic, with as many as 80% of adults showing serologic evidence of past infection. Although most adults with detectable B19‐specific IgG do not recall having had specific symptoms, a number of syndromes have been associated with acute infection.1, 2 Parvovirus B19 should be included in the differential for postinfectious glomerulonephritis, especially if a patient presents with marked edema with preserved renal function.
Human parvovirus B19, a member of the erythrovirus genus, is a nonenveloped single‐stranded DNA virus that propagates in erythroid progenitor cells, arresting erythropoiesis.3 The cellular receptor for the virus is globoside (erythrocyte P antigen), a neutral glycosphingolipid densely present on erythroid cells and also found on hepatocytes, nephrons, and bowel mucosa.3, 4
The most common clinical presentation of parvovirus B19 in children is erythema infectiosum, or fifth disease.3 In adults, the infection is known to cause symmetric polyarthropathy, rash, malaise, coryza, headache, and gastrointestinal symptoms (nausea, abdominal pain) and may mimic systemic lupus erythematosus.1, 3 In patients with sickle cell anemia or other chronic hemolytic disorders, parvovirus B19 can cause a transient aplastic crisis.3 Immunosuppressed patients (eg, organ transplant recipients, patients with certain cancers or advanced AIDS) may develop chronic infection and anemia because of an inability to mount an immune response to clear viremia. Mild anemia or pancytopenia is frequently observed in normal infected hosts.
The syndrome of renal involvement in parvovirus B19 includes the typical features of fever, a maculopapular or reticular erythematous rash on the face or extremities, and polyarthritis, accompanied by oliguria that leads to systemic edema. Mild pancytopenia, proteinuria, hematuria, and hypocomplementemia are often present. Creatinine is usually normal or near normal. These symptoms typically appear 1‐2 weeks after the initial viral syndrome.5, 6 With supportive care, most recover spontaneously, although chronic kidney disease has been reported.7, 8
Published kidney biopsy findings of parvovirus B19 show endocapillary or mesangial proliferative glomerulonephritis with subendothelial electron‐dense deposits and granular deposition of C3, IgG, or IgM along the capillary walls and mesangium. These lesions suggest immune complex deposition and are consistent with postinfectious glomerulonephritis.5, 9, 10 Indeed, increased levels of circulating immune complexes have been seen during acute parvovirus B19 infection.6, 9 It is likely that the protracted symptoms our patient experienced resulted from the formation, circulation, and deposition of immune complexes. The presence of globoside in the kidneys and bowel also raises the possibility of direct infection of these organs.
Postinfectious glomerulonephritis is often thought to be synonymous with poststreptococcal glomerulonephritis. However, viruses, including hepatitis B and C viruses, human immunodeficiency virus, cytomegalovirus, hantavirus, and parvovirus B19 may cause postinfectious glomerulonephritis. As with poststreptococcal glomerulonephritis, glomerular disease associated with viral infection appears to be mediated by the immune complexes. The pathogenic series of events leading to glomerular injury includes formation of circulating immune complexes with subsequent deposition in the glomerulus, or formation of in situ antigen‐antibody reactions.11 Immune complexes in the glomerulus lead to activation of the complement cascade, which in turn leads to hypocomplementemia, as the complement cascade is activated faster than the synthesis of new complement proteins.12 Histologically, a number of different renal lesions may be seen in postviral glomerulonephritis, including membranous, membranoproliferative, and mesangial glomerulonephritis, as well as focal segmental glomerulosclerosis.
Our patient presented with symptoms compatible with but not specific for parvovirus B19. Using a pattern recognition approach to diagnosis, our discussant correctly identified the disease pattern as a postinfectious glomerulonephritis but was unable to identify the correct pathogen, as bacterial infections were the main focus of concern, and viruses, parvovirus B19 in particular, were not considered. The clinical pattern of arthralgia, gastrointestinal symptoms, fever combined with anemia or pancytopenia, and hypocomplementemia is typical of the clues for parvovirus B19. Although renal involvement is unusual, the presence of oliguria, hematuria, and edema with minimal creatinine elevation is typical of parvovirus renal disease.
An essential part of clinical judgment is carefully determining which of a patient's often myriad complaints must be considered part of the disease process. Common and nonspecific signs and symptoms often fall off the clinician's radar screen. In this instance, several of the hallmark features of parvovirus B19 disease were dismissed by our discussant as due to the patient's previous medical conditions or hospital‐related interventions. Anemia (due to interruption of erythropoiesis by parvovirus B19 replication) was attributed to hydration or phlebotomy, fluid retention was attributed to advancing diabetes, and hematuria was attributed to a urinary catheter. It is important to evaluate the entire clinical picture prior to excluding potential clues to the diagnosis. Another reasonable approach would have been to choose a less general sign or symptom to narrow the possible diagnoses. For example, had the wrist arthralgia been more central in the discussant's thoughts, parvovirus B19 might have appeared on the differential.
Finally, the discussant wrestled with the decision to perform a renal biopsy for a definitive diagnosis versus the potential complications of the procedure. In this case, it was possible to achieve a clinical diagnosis, support it with serologic evidence, and thus avoid the need for biopsy. The current medical climate emphasizes the importance of reaching a definitive diagnosis as rapidly as possible. There are pressures to act quickly and utilize technology that may add both cost and risk. This case emphasizes the value of clinical reasoning and patience, which led to a correct diagnosis and a favorable outcome without the need for invasive procedures. Clinical acumen must occasionally include avoiding the temptation to perform the next test and merely standing at the patient's bedside instead.
- ,,, et al.Clinical manifestations of human parvovirus B19 in adults.Arch Intern Med.1989;149:1153‐1156.
- ,.The prevalence of antibody to human parvovirus B19 in England and Wales.J Med Microbiol.1999;25:25‐28.
- ,.Parvovirus B19.N Engl J Med.2004;350:586‐597.
- ,,.Multiple glycosphingolipids determine the tissue tropism of parvovirus B19.J Infect Dis.1995;172:1198‐1205.
- ,,,.Renal involvement induced by human parvovirus B19 infection.Nephron.2001;89:280‐285.
- ,,, et al.Association of parvovirus B19 infection with acute glomerulonephritis in the healthy adults: case report and review of the literature.Clin Nephrol.2002;57:69‐73.
- .Renal involvement in human parvovirus B19 infection.Pediatr Nephrol.2003;18:966‐967.
- ,,, et al.Acute glomerulonephritis after human parvovirus B19 infection.Am J Kidney Dis.2000;35:1‐8.
- ,,,,.Human parvovirus B19 and renal disease?Neth J Med.2000;56:163‐165.
- ,,, et al.Nephrotic syndrome associated with human parvovirus B19 infection.Pediatr Nephrol.2003;18:280‐282.
- ,.Glomerulonephritis.Lancet.2005;365:1797‐1806.
- .Complement and the kidney.J Immunol.2003;171:3319‐3324.
A 35‐year‐old man presented to the emergency department of a community hospital with 3 days of nausea, vomiting, abdominal pain, and diarrhea. He had developed a sore throat, nasal congestion, and green sputum on a business trip to Las Vegas 10 days prior. He then traveled to Shanghai, China, where he developed frequent diarrhea with mucous and urgency. The stool was mustard colored without blood or melena. He became nauseated and unable to keep down food or fluids. He noted a 4‐kg weight loss since the beginning of his symptoms. He had not traveled outside Shanghai or eaten exotic foods. His travel companions remained unaffected. The patient had had type I diabetes mellitus for 23 years with known retinopathy and microalbuminuria. He had hypertension and hyperlipidemia but was otherwise healthy. He was a married traveling salesman with 3 healthy children. He did not smoke or drink and reported no drug use or extramarital relations. He had no known allergies. His medications included insulin, lisinopril, atorvastatin, and valsartan. He was afebrile, his vital signs were stable and the physical examination was unremarkable.
The patient presents with gastrointestinal symptoms following a trip to China. He may have an infection that began in the respiratory system and now is causing some gastrointestinal symptoms, like Legionnella. If he had received antibiotics, the diarrheal illness could be a complication. With his recent travel to China, typical enteric pathogens would have to be considered: enterotoxigenic E. coli, Shigella, Salmonella, Campylobacter, or perhaps Giardia.
He was admitted and treated with intravenous fluids and ciprofloxacin for presumed gastroenteritis. He was discharged the next day but returned 2 days later because of continued nausea and vomiting and limited oral intake. He was febrile, to 38.9C, with chills. The results of an abdominal exam were normal. Stool studies for Salmonella, Shigella, Campylobacter, Yersinia, enterotoxigenic E. coli, Giardia, and Cryptosporidium were negative. No blood parasites were seen. The white‐cell count was 3900/mm3 with a normal differential count. Hemoglobin level, platelet count, serum electrolytes and creatinine were normal. He was readmitted for intravenous fluids.
The patient was treated for traveler's diarrhea, although this is not a typical case and is now becoming a protracted illness. Amebiasis would be a consideration, as well as typhoid or perhaps an abdominal abscess. The normal platelet count reduces the likelihood of hemolytic uremic syndrome, as does the absence of bloody diarrhea. I would evaluate for a systemic illness: check for adenopathy, do a thorough abdominal exam, and get a chest radiogram, blood cultures, and liver function tests. I am also concerned about metabolic abnormalities that could occur as a consequence of the diarrhea.
Over 3 days, oliguria developed along with urinary hesitancy, a 9‐kg weight gain, and the development of marked edema. Blood pressure and heart rate remained normal, and a chest radiograph was clear. Liver function tests were normal. A urinary catheter was inserted. Urinalysis revealed a specific gravity of 1.031, protein of 100 mg/dL, and trace glucose but was otherwise negative; no casts or cells were seen in the sediment. Chemistries included sodium of 133 mmol/L, potassium of 3.9 mmol/L, and serum bicarbonate of 18.4 mmol/L. Blood and urine cultures were sterile. The creatinine increased from 1.0 mg/dL (88.4 mol/L) to 1.3 mg/dL (115 mol/L). He was transferred to a tertiary care hospital for renal consultation because of concerns of impending renal failure and for consideration of a kidney biopsy.
In a typical case of a malabsorptive diarrhea, the patient could be volume depleted, but in this case he has gained 9‐kg and is grossly edematous. The chest radiograph and liver tests point to renal rather than cardiac or hepatologic causes for the edema. A glomerulonephritis may be driving the salt and water retention.
The proteinuria could be related to hemodynamics, or it could be from a glomerular lesion secondary to immune complexes. The specific gravity of 1.031 indicates the kidney is able to concentrate, and we are not seeing acute tubular necrosis. There is only minimal elevation in creatinine at this point. Quantitation or estimation of the degree of proteinuria by a protein‐to‐creatinine ratio would be helpful.
A further workup should include additional blood cultures and a CT scan of the lungs and abdomen to look for occult infection. Is he unfortunate enough to have developed a malignancy? Is this a connective tissue disease? Reexamination of the urine sediment is important to evaluate for glomerulonephritis.
The patient reported ongoing nausea and vomiting, but his diarrhea resolved. He was tachypneic, with a respiratory rate of 26 breaths/minute and an oxygen saturation of 98% breathing ambient air. His temperature was 37.1C, heart rate 84 beats/minute, and blood pressure 120/65 mm Hg. His mucous membranes were moist, and his jugular venous pressure was 6 cm. No lymphadenopathy was present. The heart and lungs were normal. The abdomen was soft, nontender, and without organomegaly, masses, or shifting dullness; bowel sounds were hypoactive. Severe edema of his legs, sacrum, hands, arms, and orbits was noted. His right hand and wrist were painful with limited mobility and small joint effusions of the wrist and metacarpophalangeal joints, but without erythema or warmth. Small petechiae were noted on his eyelids; skin examination was otherwise unremarkable. He had been given ciprofloxacin, phenergan, calcium carbonate, and pantoprazole prior to transfer. Stool was negative for occult blood.
His lungs are clear, but it is possible to have early pulmonary congestion with normal breath sounds. As he is normotensive and has a normal JVP, I would not give further intravenous fluid. Unless he has evidence of symptomatic pulmonary edema, I would not give diuretics but would simply observe his course. At this point I would ultrasound his kidneys to make sure there is no obstruction. The proteinuria could be a result of underlying diabetic glomerulopathy that may predispose to fluid retention.
He has significant oliguria but only a mild rise in creatinine. As the referring physicians requested a biopsy, we should consider it. A decision to biopsy the kidney would rest on the degree of proteinuria and the activity of the sediment. For example, if red blood cell casts or dysmorphic red cells were present, postinfectious glomerulonephritis or IgA nephropathy would be more likely. However, if the proteinuria is in the non‐nephrotic range and the sediment is nonreactive, the yield of a biopsy would be low.
His right hand and wrist are painful with limited mobility. This could be a sequela of endocarditis, although the absence of a murmur and negative blood cultures make it unlikely. He could have an infectious arthritis, although this usually presents more dramatically. He could have gout or pseudogout, which could be determined by joint aspiration. Finally, could this be iatrogenic? A drug reaction could explain some of the features, including rash, fever, joint symptoms, and renal abnormalities.
Repeat urinalysis revealed protein of more than 300 mg/dL, hematuria (1+), mucous, renal tubular epithelial cells, renal tubular epithelial cell casts, and granular casts. Eosinophiluria was absent. Laboratory evaluation revealed a hemoglobin level of 10.7 g/dL (decreased from 13 g/dL on initial presentation), white‐cell count of 7300/mm3, platelet count of 337,000/mm3, serum creatinine of 1.2 mg/dL (106 mol/L), blood urea nitrogen of 23 mg/dL (8.2 mol.L), total serum protein of 6.3 g/dL (normal range, 6.3‐8.7), and albumin of 2.7 g/dL (normal range, 3.2‐5.2). Other liver tests and serum electrolytes were normal.
This degree of proteinuria is significant, but it is unclear whether this is related to the underlying disease process or his advancing diabetes. He has some hematuria, but that could be from the urinary catheter. It would be helpful to know if the red blood cells are dysmorphic, which would point to a glomerulonephritis. He has renal cells, renal cell casts, and granular casts, which are nonspecific. He has a mild anemia, which is unexplained, but could relate to phlebotomy or overhydration. The hypoalbuminemia may be a result of renal losses or a catabolic state.
A renal ultrasonogram was normal, apart from evidence of bilateral pleural effusions. Antinuclear antibody and rheumatoid factor test results were negative, as were those for hepatitis A, hepatitis B surface antigen, and hepatitis C antibodies. Antistreptolysin O and antideoxyribonuclease B titers were normal. Total complement Ch50 was low at 29 U/mL (normal range, 30‐75) as was complement factor C3 at 67 mg/dL (normal range, 90‐180). Complement factor C4 was normal. Serum and urine electrophoresis revealed no monoclonal protein spike. Vitamin B12 and serum folate were normal, serum ferritin was 584 ng/mL (normal range, 30‐400), iron serum was 29 g/dL (normal range, 45‐160), transferrin saturation was 16%, and total iron‐binding capacity was 164 g/dL (normal range, 250‐450). The reticulocyte count was 2.9% with an absolute reticulocyte count of 102/cm3 and a reticulocyte production index of 0.96 (normal range, 1.0‐2.0).
It is reassuring that his urinary tract ultrasound is normal. In addition to edema, he has bilateral effusions, which are probably transudative, related to fluid overload. The urinalysis does not suggest a rapidly progressive glomerulonephritis, but autoimmune disease is still in the differential.
He has a mild complement C3 deficiency. In nephrology we think of lupus, infective endocarditis, cryoglobulinemia, and specific glomerular lesions such as membranoproliferative glomerulonephritis and postinfectious glomerulonephritis as being associated with the development of circulating immune complexes that may lead to low complement levels. There is no evidence of a paraprotein, but testing for cryoglobulins should be considered. Cryoglobulins are associated with hepatitis C but may be induced by a variety of infections. Acting like immune complexes, they can lead to low complement levels and could cause some of this patient's symptoms. However, this whole illness seems most likely to be secondary to infection. The normal antistreptolysin O and antideoxyribonuclease B titers make streptococcal disease unlikely, but another bacterial infection could cause postinfectious glomerulonephritis.
Over the course of his 5‐day hospital stay, the patient received furosemide with increased urine output and normalization of his serum creatinine to its baseline level of 1.0 mg.dL (88.4 mol/L). Proteinuria resolved to 44 mg/dL. A kidney biopsy was not performed. The parvovirus IgG index, checked because of anemia and oligoarthralgias, was 3.67 (normal 0‐1.10), and the IgM index was 8.13 (normal 0‐1.10), suggesting recent infection. The patient was discharged after 5 days. His edema had resolved on discharge; he continued to be nauseated but was able to eat and drink normally. Six months after his hospitalization, his symptoms had completely resolved.
Parvovirus! It could cause the pulmonary infection and the gastroenteric symptoms. Parvovirus usually causes more anemia than nausea and vomiting. We see it occasionally in our transplant patients. The underlying diabetic nephropathy may have made him more symptomatic with a superimposed glomerulonephritis. The most important pedagogic point is that he did well with a very conservative approach, and the possible iatrogenic consequences of a kidney biopsy, had it been performed, were avoided.
COMMENTARY
Parvovirus B19 is endemic, with as many as 80% of adults showing serologic evidence of past infection. Although most adults with detectable B19‐specific IgG do not recall having had specific symptoms, a number of syndromes have been associated with acute infection.1, 2 Parvovirus B19 should be included in the differential for postinfectious glomerulonephritis, especially if a patient presents with marked edema with preserved renal function.
Human parvovirus B19, a member of the erythrovirus genus, is a nonenveloped single‐stranded DNA virus that propagates in erythroid progenitor cells, arresting erythropoiesis.3 The cellular receptor for the virus is globoside (erythrocyte P antigen), a neutral glycosphingolipid densely present on erythroid cells and also found on hepatocytes, nephrons, and bowel mucosa.3, 4
The most common clinical presentation of parvovirus B19 in children is erythema infectiosum, or fifth disease.3 In adults, the infection is known to cause symmetric polyarthropathy, rash, malaise, coryza, headache, and gastrointestinal symptoms (nausea, abdominal pain) and may mimic systemic lupus erythematosus.1, 3 In patients with sickle cell anemia or other chronic hemolytic disorders, parvovirus B19 can cause a transient aplastic crisis.3 Immunosuppressed patients (eg, organ transplant recipients, patients with certain cancers or advanced AIDS) may develop chronic infection and anemia because of an inability to mount an immune response to clear viremia. Mild anemia or pancytopenia is frequently observed in normal infected hosts.
The syndrome of renal involvement in parvovirus B19 includes the typical features of fever, a maculopapular or reticular erythematous rash on the face or extremities, and polyarthritis, accompanied by oliguria that leads to systemic edema. Mild pancytopenia, proteinuria, hematuria, and hypocomplementemia are often present. Creatinine is usually normal or near normal. These symptoms typically appear 1‐2 weeks after the initial viral syndrome.5, 6 With supportive care, most recover spontaneously, although chronic kidney disease has been reported.7, 8
Published kidney biopsy findings of parvovirus B19 show endocapillary or mesangial proliferative glomerulonephritis with subendothelial electron‐dense deposits and granular deposition of C3, IgG, or IgM along the capillary walls and mesangium. These lesions suggest immune complex deposition and are consistent with postinfectious glomerulonephritis.5, 9, 10 Indeed, increased levels of circulating immune complexes have been seen during acute parvovirus B19 infection.6, 9 It is likely that the protracted symptoms our patient experienced resulted from the formation, circulation, and deposition of immune complexes. The presence of globoside in the kidneys and bowel also raises the possibility of direct infection of these organs.
Postinfectious glomerulonephritis is often thought to be synonymous with poststreptococcal glomerulonephritis. However, viruses, including hepatitis B and C viruses, human immunodeficiency virus, cytomegalovirus, hantavirus, and parvovirus B19 may cause postinfectious glomerulonephritis. As with poststreptococcal glomerulonephritis, glomerular disease associated with viral infection appears to be mediated by the immune complexes. The pathogenic series of events leading to glomerular injury includes formation of circulating immune complexes with subsequent deposition in the glomerulus, or formation of in situ antigen‐antibody reactions.11 Immune complexes in the glomerulus lead to activation of the complement cascade, which in turn leads to hypocomplementemia, as the complement cascade is activated faster than the synthesis of new complement proteins.12 Histologically, a number of different renal lesions may be seen in postviral glomerulonephritis, including membranous, membranoproliferative, and mesangial glomerulonephritis, as well as focal segmental glomerulosclerosis.
Our patient presented with symptoms compatible with but not specific for parvovirus B19. Using a pattern recognition approach to diagnosis, our discussant correctly identified the disease pattern as a postinfectious glomerulonephritis but was unable to identify the correct pathogen, as bacterial infections were the main focus of concern, and viruses, parvovirus B19 in particular, were not considered. The clinical pattern of arthralgia, gastrointestinal symptoms, fever combined with anemia or pancytopenia, and hypocomplementemia is typical of the clues for parvovirus B19. Although renal involvement is unusual, the presence of oliguria, hematuria, and edema with minimal creatinine elevation is typical of parvovirus renal disease.
An essential part of clinical judgment is carefully determining which of a patient's often myriad complaints must be considered part of the disease process. Common and nonspecific signs and symptoms often fall off the clinician's radar screen. In this instance, several of the hallmark features of parvovirus B19 disease were dismissed by our discussant as due to the patient's previous medical conditions or hospital‐related interventions. Anemia (due to interruption of erythropoiesis by parvovirus B19 replication) was attributed to hydration or phlebotomy, fluid retention was attributed to advancing diabetes, and hematuria was attributed to a urinary catheter. It is important to evaluate the entire clinical picture prior to excluding potential clues to the diagnosis. Another reasonable approach would have been to choose a less general sign or symptom to narrow the possible diagnoses. For example, had the wrist arthralgia been more central in the discussant's thoughts, parvovirus B19 might have appeared on the differential.
Finally, the discussant wrestled with the decision to perform a renal biopsy for a definitive diagnosis versus the potential complications of the procedure. In this case, it was possible to achieve a clinical diagnosis, support it with serologic evidence, and thus avoid the need for biopsy. The current medical climate emphasizes the importance of reaching a definitive diagnosis as rapidly as possible. There are pressures to act quickly and utilize technology that may add both cost and risk. This case emphasizes the value of clinical reasoning and patience, which led to a correct diagnosis and a favorable outcome without the need for invasive procedures. Clinical acumen must occasionally include avoiding the temptation to perform the next test and merely standing at the patient's bedside instead.
A 35‐year‐old man presented to the emergency department of a community hospital with 3 days of nausea, vomiting, abdominal pain, and diarrhea. He had developed a sore throat, nasal congestion, and green sputum on a business trip to Las Vegas 10 days prior. He then traveled to Shanghai, China, where he developed frequent diarrhea with mucous and urgency. The stool was mustard colored without blood or melena. He became nauseated and unable to keep down food or fluids. He noted a 4‐kg weight loss since the beginning of his symptoms. He had not traveled outside Shanghai or eaten exotic foods. His travel companions remained unaffected. The patient had had type I diabetes mellitus for 23 years with known retinopathy and microalbuminuria. He had hypertension and hyperlipidemia but was otherwise healthy. He was a married traveling salesman with 3 healthy children. He did not smoke or drink and reported no drug use or extramarital relations. He had no known allergies. His medications included insulin, lisinopril, atorvastatin, and valsartan. He was afebrile, his vital signs were stable and the physical examination was unremarkable.
The patient presents with gastrointestinal symptoms following a trip to China. He may have an infection that began in the respiratory system and now is causing some gastrointestinal symptoms, like Legionnella. If he had received antibiotics, the diarrheal illness could be a complication. With his recent travel to China, typical enteric pathogens would have to be considered: enterotoxigenic E. coli, Shigella, Salmonella, Campylobacter, or perhaps Giardia.
He was admitted and treated with intravenous fluids and ciprofloxacin for presumed gastroenteritis. He was discharged the next day but returned 2 days later because of continued nausea and vomiting and limited oral intake. He was febrile, to 38.9C, with chills. The results of an abdominal exam were normal. Stool studies for Salmonella, Shigella, Campylobacter, Yersinia, enterotoxigenic E. coli, Giardia, and Cryptosporidium were negative. No blood parasites were seen. The white‐cell count was 3900/mm3 with a normal differential count. Hemoglobin level, platelet count, serum electrolytes and creatinine were normal. He was readmitted for intravenous fluids.
The patient was treated for traveler's diarrhea, although this is not a typical case and is now becoming a protracted illness. Amebiasis would be a consideration, as well as typhoid or perhaps an abdominal abscess. The normal platelet count reduces the likelihood of hemolytic uremic syndrome, as does the absence of bloody diarrhea. I would evaluate for a systemic illness: check for adenopathy, do a thorough abdominal exam, and get a chest radiogram, blood cultures, and liver function tests. I am also concerned about metabolic abnormalities that could occur as a consequence of the diarrhea.
Over 3 days, oliguria developed along with urinary hesitancy, a 9‐kg weight gain, and the development of marked edema. Blood pressure and heart rate remained normal, and a chest radiograph was clear. Liver function tests were normal. A urinary catheter was inserted. Urinalysis revealed a specific gravity of 1.031, protein of 100 mg/dL, and trace glucose but was otherwise negative; no casts or cells were seen in the sediment. Chemistries included sodium of 133 mmol/L, potassium of 3.9 mmol/L, and serum bicarbonate of 18.4 mmol/L. Blood and urine cultures were sterile. The creatinine increased from 1.0 mg/dL (88.4 mol/L) to 1.3 mg/dL (115 mol/L). He was transferred to a tertiary care hospital for renal consultation because of concerns of impending renal failure and for consideration of a kidney biopsy.
In a typical case of a malabsorptive diarrhea, the patient could be volume depleted, but in this case he has gained 9‐kg and is grossly edematous. The chest radiograph and liver tests point to renal rather than cardiac or hepatologic causes for the edema. A glomerulonephritis may be driving the salt and water retention.
The proteinuria could be related to hemodynamics, or it could be from a glomerular lesion secondary to immune complexes. The specific gravity of 1.031 indicates the kidney is able to concentrate, and we are not seeing acute tubular necrosis. There is only minimal elevation in creatinine at this point. Quantitation or estimation of the degree of proteinuria by a protein‐to‐creatinine ratio would be helpful.
A further workup should include additional blood cultures and a CT scan of the lungs and abdomen to look for occult infection. Is he unfortunate enough to have developed a malignancy? Is this a connective tissue disease? Reexamination of the urine sediment is important to evaluate for glomerulonephritis.
The patient reported ongoing nausea and vomiting, but his diarrhea resolved. He was tachypneic, with a respiratory rate of 26 breaths/minute and an oxygen saturation of 98% breathing ambient air. His temperature was 37.1C, heart rate 84 beats/minute, and blood pressure 120/65 mm Hg. His mucous membranes were moist, and his jugular venous pressure was 6 cm. No lymphadenopathy was present. The heart and lungs were normal. The abdomen was soft, nontender, and without organomegaly, masses, or shifting dullness; bowel sounds were hypoactive. Severe edema of his legs, sacrum, hands, arms, and orbits was noted. His right hand and wrist were painful with limited mobility and small joint effusions of the wrist and metacarpophalangeal joints, but without erythema or warmth. Small petechiae were noted on his eyelids; skin examination was otherwise unremarkable. He had been given ciprofloxacin, phenergan, calcium carbonate, and pantoprazole prior to transfer. Stool was negative for occult blood.
His lungs are clear, but it is possible to have early pulmonary congestion with normal breath sounds. As he is normotensive and has a normal JVP, I would not give further intravenous fluid. Unless he has evidence of symptomatic pulmonary edema, I would not give diuretics but would simply observe his course. At this point I would ultrasound his kidneys to make sure there is no obstruction. The proteinuria could be a result of underlying diabetic glomerulopathy that may predispose to fluid retention.
He has significant oliguria but only a mild rise in creatinine. As the referring physicians requested a biopsy, we should consider it. A decision to biopsy the kidney would rest on the degree of proteinuria and the activity of the sediment. For example, if red blood cell casts or dysmorphic red cells were present, postinfectious glomerulonephritis or IgA nephropathy would be more likely. However, if the proteinuria is in the non‐nephrotic range and the sediment is nonreactive, the yield of a biopsy would be low.
His right hand and wrist are painful with limited mobility. This could be a sequela of endocarditis, although the absence of a murmur and negative blood cultures make it unlikely. He could have an infectious arthritis, although this usually presents more dramatically. He could have gout or pseudogout, which could be determined by joint aspiration. Finally, could this be iatrogenic? A drug reaction could explain some of the features, including rash, fever, joint symptoms, and renal abnormalities.
Repeat urinalysis revealed protein of more than 300 mg/dL, hematuria (1+), mucous, renal tubular epithelial cells, renal tubular epithelial cell casts, and granular casts. Eosinophiluria was absent. Laboratory evaluation revealed a hemoglobin level of 10.7 g/dL (decreased from 13 g/dL on initial presentation), white‐cell count of 7300/mm3, platelet count of 337,000/mm3, serum creatinine of 1.2 mg/dL (106 mol/L), blood urea nitrogen of 23 mg/dL (8.2 mol.L), total serum protein of 6.3 g/dL (normal range, 6.3‐8.7), and albumin of 2.7 g/dL (normal range, 3.2‐5.2). Other liver tests and serum electrolytes were normal.
This degree of proteinuria is significant, but it is unclear whether this is related to the underlying disease process or his advancing diabetes. He has some hematuria, but that could be from the urinary catheter. It would be helpful to know if the red blood cells are dysmorphic, which would point to a glomerulonephritis. He has renal cells, renal cell casts, and granular casts, which are nonspecific. He has a mild anemia, which is unexplained, but could relate to phlebotomy or overhydration. The hypoalbuminemia may be a result of renal losses or a catabolic state.
A renal ultrasonogram was normal, apart from evidence of bilateral pleural effusions. Antinuclear antibody and rheumatoid factor test results were negative, as were those for hepatitis A, hepatitis B surface antigen, and hepatitis C antibodies. Antistreptolysin O and antideoxyribonuclease B titers were normal. Total complement Ch50 was low at 29 U/mL (normal range, 30‐75) as was complement factor C3 at 67 mg/dL (normal range, 90‐180). Complement factor C4 was normal. Serum and urine electrophoresis revealed no monoclonal protein spike. Vitamin B12 and serum folate were normal, serum ferritin was 584 ng/mL (normal range, 30‐400), iron serum was 29 g/dL (normal range, 45‐160), transferrin saturation was 16%, and total iron‐binding capacity was 164 g/dL (normal range, 250‐450). The reticulocyte count was 2.9% with an absolute reticulocyte count of 102/cm3 and a reticulocyte production index of 0.96 (normal range, 1.0‐2.0).
It is reassuring that his urinary tract ultrasound is normal. In addition to edema, he has bilateral effusions, which are probably transudative, related to fluid overload. The urinalysis does not suggest a rapidly progressive glomerulonephritis, but autoimmune disease is still in the differential.
He has a mild complement C3 deficiency. In nephrology we think of lupus, infective endocarditis, cryoglobulinemia, and specific glomerular lesions such as membranoproliferative glomerulonephritis and postinfectious glomerulonephritis as being associated with the development of circulating immune complexes that may lead to low complement levels. There is no evidence of a paraprotein, but testing for cryoglobulins should be considered. Cryoglobulins are associated with hepatitis C but may be induced by a variety of infections. Acting like immune complexes, they can lead to low complement levels and could cause some of this patient's symptoms. However, this whole illness seems most likely to be secondary to infection. The normal antistreptolysin O and antideoxyribonuclease B titers make streptococcal disease unlikely, but another bacterial infection could cause postinfectious glomerulonephritis.
Over the course of his 5‐day hospital stay, the patient received furosemide with increased urine output and normalization of his serum creatinine to its baseline level of 1.0 mg.dL (88.4 mol/L). Proteinuria resolved to 44 mg/dL. A kidney biopsy was not performed. The parvovirus IgG index, checked because of anemia and oligoarthralgias, was 3.67 (normal 0‐1.10), and the IgM index was 8.13 (normal 0‐1.10), suggesting recent infection. The patient was discharged after 5 days. His edema had resolved on discharge; he continued to be nauseated but was able to eat and drink normally. Six months after his hospitalization, his symptoms had completely resolved.
Parvovirus! It could cause the pulmonary infection and the gastroenteric symptoms. Parvovirus usually causes more anemia than nausea and vomiting. We see it occasionally in our transplant patients. The underlying diabetic nephropathy may have made him more symptomatic with a superimposed glomerulonephritis. The most important pedagogic point is that he did well with a very conservative approach, and the possible iatrogenic consequences of a kidney biopsy, had it been performed, were avoided.
COMMENTARY
Parvovirus B19 is endemic, with as many as 80% of adults showing serologic evidence of past infection. Although most adults with detectable B19‐specific IgG do not recall having had specific symptoms, a number of syndromes have been associated with acute infection.1, 2 Parvovirus B19 should be included in the differential for postinfectious glomerulonephritis, especially if a patient presents with marked edema with preserved renal function.
Human parvovirus B19, a member of the erythrovirus genus, is a nonenveloped single‐stranded DNA virus that propagates in erythroid progenitor cells, arresting erythropoiesis.3 The cellular receptor for the virus is globoside (erythrocyte P antigen), a neutral glycosphingolipid densely present on erythroid cells and also found on hepatocytes, nephrons, and bowel mucosa.3, 4
The most common clinical presentation of parvovirus B19 in children is erythema infectiosum, or fifth disease.3 In adults, the infection is known to cause symmetric polyarthropathy, rash, malaise, coryza, headache, and gastrointestinal symptoms (nausea, abdominal pain) and may mimic systemic lupus erythematosus.1, 3 In patients with sickle cell anemia or other chronic hemolytic disorders, parvovirus B19 can cause a transient aplastic crisis.3 Immunosuppressed patients (eg, organ transplant recipients, patients with certain cancers or advanced AIDS) may develop chronic infection and anemia because of an inability to mount an immune response to clear viremia. Mild anemia or pancytopenia is frequently observed in normal infected hosts.
The syndrome of renal involvement in parvovirus B19 includes the typical features of fever, a maculopapular or reticular erythematous rash on the face or extremities, and polyarthritis, accompanied by oliguria that leads to systemic edema. Mild pancytopenia, proteinuria, hematuria, and hypocomplementemia are often present. Creatinine is usually normal or near normal. These symptoms typically appear 1‐2 weeks after the initial viral syndrome.5, 6 With supportive care, most recover spontaneously, although chronic kidney disease has been reported.7, 8
Published kidney biopsy findings of parvovirus B19 show endocapillary or mesangial proliferative glomerulonephritis with subendothelial electron‐dense deposits and granular deposition of C3, IgG, or IgM along the capillary walls and mesangium. These lesions suggest immune complex deposition and are consistent with postinfectious glomerulonephritis.5, 9, 10 Indeed, increased levels of circulating immune complexes have been seen during acute parvovirus B19 infection.6, 9 It is likely that the protracted symptoms our patient experienced resulted from the formation, circulation, and deposition of immune complexes. The presence of globoside in the kidneys and bowel also raises the possibility of direct infection of these organs.
Postinfectious glomerulonephritis is often thought to be synonymous with poststreptococcal glomerulonephritis. However, viruses, including hepatitis B and C viruses, human immunodeficiency virus, cytomegalovirus, hantavirus, and parvovirus B19 may cause postinfectious glomerulonephritis. As with poststreptococcal glomerulonephritis, glomerular disease associated with viral infection appears to be mediated by the immune complexes. The pathogenic series of events leading to glomerular injury includes formation of circulating immune complexes with subsequent deposition in the glomerulus, or formation of in situ antigen‐antibody reactions.11 Immune complexes in the glomerulus lead to activation of the complement cascade, which in turn leads to hypocomplementemia, as the complement cascade is activated faster than the synthesis of new complement proteins.12 Histologically, a number of different renal lesions may be seen in postviral glomerulonephritis, including membranous, membranoproliferative, and mesangial glomerulonephritis, as well as focal segmental glomerulosclerosis.
Our patient presented with symptoms compatible with but not specific for parvovirus B19. Using a pattern recognition approach to diagnosis, our discussant correctly identified the disease pattern as a postinfectious glomerulonephritis but was unable to identify the correct pathogen, as bacterial infections were the main focus of concern, and viruses, parvovirus B19 in particular, were not considered. The clinical pattern of arthralgia, gastrointestinal symptoms, fever combined with anemia or pancytopenia, and hypocomplementemia is typical of the clues for parvovirus B19. Although renal involvement is unusual, the presence of oliguria, hematuria, and edema with minimal creatinine elevation is typical of parvovirus renal disease.
An essential part of clinical judgment is carefully determining which of a patient's often myriad complaints must be considered part of the disease process. Common and nonspecific signs and symptoms often fall off the clinician's radar screen. In this instance, several of the hallmark features of parvovirus B19 disease were dismissed by our discussant as due to the patient's previous medical conditions or hospital‐related interventions. Anemia (due to interruption of erythropoiesis by parvovirus B19 replication) was attributed to hydration or phlebotomy, fluid retention was attributed to advancing diabetes, and hematuria was attributed to a urinary catheter. It is important to evaluate the entire clinical picture prior to excluding potential clues to the diagnosis. Another reasonable approach would have been to choose a less general sign or symptom to narrow the possible diagnoses. For example, had the wrist arthralgia been more central in the discussant's thoughts, parvovirus B19 might have appeared on the differential.
Finally, the discussant wrestled with the decision to perform a renal biopsy for a definitive diagnosis versus the potential complications of the procedure. In this case, it was possible to achieve a clinical diagnosis, support it with serologic evidence, and thus avoid the need for biopsy. The current medical climate emphasizes the importance of reaching a definitive diagnosis as rapidly as possible. There are pressures to act quickly and utilize technology that may add both cost and risk. This case emphasizes the value of clinical reasoning and patience, which led to a correct diagnosis and a favorable outcome without the need for invasive procedures. Clinical acumen must occasionally include avoiding the temptation to perform the next test and merely standing at the patient's bedside instead.
- ,,, et al.Clinical manifestations of human parvovirus B19 in adults.Arch Intern Med.1989;149:1153‐1156.
- ,.The prevalence of antibody to human parvovirus B19 in England and Wales.J Med Microbiol.1999;25:25‐28.
- ,.Parvovirus B19.N Engl J Med.2004;350:586‐597.
- ,,.Multiple glycosphingolipids determine the tissue tropism of parvovirus B19.J Infect Dis.1995;172:1198‐1205.
- ,,,.Renal involvement induced by human parvovirus B19 infection.Nephron.2001;89:280‐285.
- ,,, et al.Association of parvovirus B19 infection with acute glomerulonephritis in the healthy adults: case report and review of the literature.Clin Nephrol.2002;57:69‐73.
- .Renal involvement in human parvovirus B19 infection.Pediatr Nephrol.2003;18:966‐967.
- ,,, et al.Acute glomerulonephritis after human parvovirus B19 infection.Am J Kidney Dis.2000;35:1‐8.
- ,,,,.Human parvovirus B19 and renal disease?Neth J Med.2000;56:163‐165.
- ,,, et al.Nephrotic syndrome associated with human parvovirus B19 infection.Pediatr Nephrol.2003;18:280‐282.
- ,.Glomerulonephritis.Lancet.2005;365:1797‐1806.
- .Complement and the kidney.J Immunol.2003;171:3319‐3324.
- ,,, et al.Clinical manifestations of human parvovirus B19 in adults.Arch Intern Med.1989;149:1153‐1156.
- ,.The prevalence of antibody to human parvovirus B19 in England and Wales.J Med Microbiol.1999;25:25‐28.
- ,.Parvovirus B19.N Engl J Med.2004;350:586‐597.
- ,,.Multiple glycosphingolipids determine the tissue tropism of parvovirus B19.J Infect Dis.1995;172:1198‐1205.
- ,,,.Renal involvement induced by human parvovirus B19 infection.Nephron.2001;89:280‐285.
- ,,, et al.Association of parvovirus B19 infection with acute glomerulonephritis in the healthy adults: case report and review of the literature.Clin Nephrol.2002;57:69‐73.
- .Renal involvement in human parvovirus B19 infection.Pediatr Nephrol.2003;18:966‐967.
- ,,, et al.Acute glomerulonephritis after human parvovirus B19 infection.Am J Kidney Dis.2000;35:1‐8.
- ,,,,.Human parvovirus B19 and renal disease?Neth J Med.2000;56:163‐165.
- ,,, et al.Nephrotic syndrome associated with human parvovirus B19 infection.Pediatr Nephrol.2003;18:280‐282.
- ,.Glomerulonephritis.Lancet.2005;365:1797‐1806.
- .Complement and the kidney.J Immunol.2003;171:3319‐3324.
Clinical Conundrum
A 45‐year‐old man who immigrated to Canada from Ghana at the age of 33 presented with a 2‐year history of progressive cognitive changes. He had bifrontal headache, right‐sided scalp paresthesias, and a 40‐pound weight loss. He was unable to perform his job as an auto parts worker. His wife noticed short‐ and long‐term memory problems and poor concentration. On physical exam he had no focal neurological findings but his score on the Mini‐Mental Status Exam (MMSE) was 23/30, with deficits in attention and recall.
The first important element of this illness is its chronicity. His symptoms progressed slowly over 2 years. Second, aside from his neurological problems, he is an otherwise healthy young, African‐born male. This clinical picture could be the early presentation of a demyelinating, infiltrative, or vascular illness. If vascular, it is more likely a vasculitis than atherosclerotic disease. Malignancy and infection are definitely in the differential, but at this point, I think they are less likely to be the cause, given the tempo of presentation. I would begin my investigations with basic blood work and a computerized tomography (CT) scan of his brain.
A CT scan of the head with contrast demonstrated an enlarged left lateral ventricle with no evidence of obstruction in the foramen of Munro.
The radiological findings of communicating hydrocephalus with normal parenchyma imply a disease that affects the leptomeningeal space. Given that we are looking at an illness that can change cerebral spinal fluid (CSF) flow rather than primary parenchymal disease, demyelinating and vascular illnesses are less likely etiologies, and infiltrative diseases move up on my list. Malignancy and infectious diseases remain in the differential.
He disappeared to follow up for 1 year, during which he returned to Ghana and experienced progressive neurological deterioration, with incontinence, gait instability, and inability to converse clearly and perform activities of daily living. On his return to Canada, an urgent CT scan and magnetic resonance imaging (MRI) of the brain demonstrated ongoing and unchanged hydrocephalus with aqueductal stenosis. A referral was made to a neurosurgeon for insertion of a ventriculoperitoneal shunt. A routine preoperative chest radiograph demonstrated new bilateral upper‐zone reticulonodular changes.
He had no respiratory symptoms, fevers, or lymphadenopathy. His occupational history revealed no exposure to asbestos, silica, farms, or mines. He had no history of either respiratory or neurological illness in the past and no travel other than to Ghana and Toronto. When he immigrated to Toronto, Canada, 12 years before, he had a normal chest radiograph and negative PPD tuberculin skin test.
Many illnesses produce asymptomatic changes on chest x‐ray. Oslerian principles would suggest that we should think of a single diagnosis to explain both nodular lung disease and more than 3 years of a progressive disease affecting the leptomeninges. It is unlikely that tuberculosis, other fungal diseases, or malignancy would result in the chest and brain pathology over a 3‐year period without other sequelae. Sarcoidosis could cause both chronic leptomeningeal changes and the radiographic lung findings. The next steps in investigating this patient should include measurement of angiotensin‐converting enzyme (ACE) and serum calcium and pulmonary function tests. I would ultimately send him for a pathological biopsy of his lung tissue to confirm noncaseating granuloma and exclude infection and malignancy.
Complete blood count, renal and liver biochemistry, and calcium were normal. An ACE level was elevated at 69 g/L (normal 40 g/L). A human immunodeficiency virus (HIV‐1 and HIV‐2) test, tuberculin skin test, and syphilis serology were negative. A CT scan of the chest demonstrated bilateral upper‐zone reticulonodular changes and diffuse lymphadenopathy (Fig. 1). Pulmonary function tests (PFTs) demonstrated a forced expiratory volume 1 (FEV1) of 3.4 L (94%), forced vital capacity (FVC) of 4.0 L (83%), an FEV1/FVC of 87%, total lung capacity (TLC) of 92% predicted, and diffusion capacity (DLCO) of 67% predicted. An MRI with gadolinium (Fig. 2) demonstrated hydrocephalus, mild basal leptomeningeal enhancement around the perivascular spaces into the subinsular region, and an increased T2 signal in periventricular white matter.
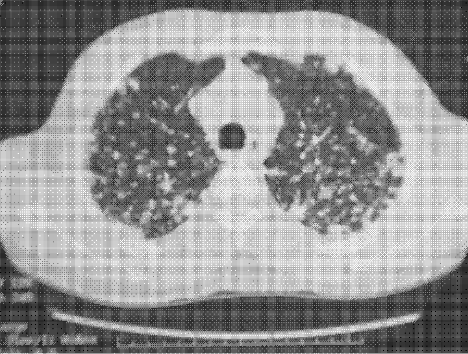
A bronchoscopy with bronchoalveolar lavage and transbronchial biopsies were performed. Pathology (Fig. 3) demonstrated non‐caseating epitheliod granulomas, with negative special stains for acid‐fast bacilli (AFB) and fungus, and negative fungal and AFB cultures of the bronchial alveolar lavage.
With negative tests for infectious causes such as tuberculosis, I think there is now enough evidence that this patient has sarcoidosis involving the lung and leptomeninges. At this point I would start therapy with steroids.
The patient was started on prednisone 40 mg po qd, and his neurological symptoms improved markedly over the course of 1‐2 months, with normalization of his MMSE and a return to cognitive baseline. As his symptoms stabilized with no change in CT imaging, he returned to work, and over the course of 2 years his prednisone dosage was tapered to 10 mg po od. While on prednisone he developed hypertension and hyperglycemia. He continued to have no respiratory symptoms.
He was cognitively at baseline until 20 months later, when he was readmitted to the hospital with a 2‐week history of worsening headache, increased confusion, poor memory, and wandering. His MMSE had deteriorated to 19/30, with deficits again in memory and attention.
First, we can say with reasonable confidence that the diagnosis of sarcoid was correct. His long and sustained response to steroids, plus the absence of the unmasking of an infectious or malignant disease, supports this conclusion. However, he is now exhibiting an apparent relapse that mimics his presentation 3 years earlier. The question is whether he is suffering from a flare of his disease or whether a second illness has occurred. The most obvious second illness is an opportunistic infection after years of steroid use. I would certainly repeat the angiotensin‐converting enzyme and serum calcium tests and repeat the imaging of his lungs and central nervous system. He also warrants a lumbar puncture with CSF culture, stain, and PCR for opportunistic infections. If these studies are inconclusive and do not specifically suggest relapsing sarcoid, I would once again consider biopsy of tissue from either a lung or leptomeninges.
An MRI with gadolinium looked unchanged from the previous one. A lumbar puncture was performed, and his CSF demonstrated 3 WBCs, no RBCs, normal glucose, and elevated protein at 1.17 g/L, and tests for bacteria, TB, fungi, and viruses were all negative. Repeat blood work was unremarkable, and the ACE level was 2 g/L.
A chest radiograph (Fig. 4a) and CT chest (Fig. 4b) showed marked deterioration, with increased diffuse airspace opacities, interstitial nodularity, and small apical bullae. His PFTs showed some deterioration, with FEV1 2.52 L (73%), FVC 3.29 L (73%), FEV1/FVC 76%, TLC 70% predicted, DLCO 72% predicted. However, he still had no respiratory symptoms.
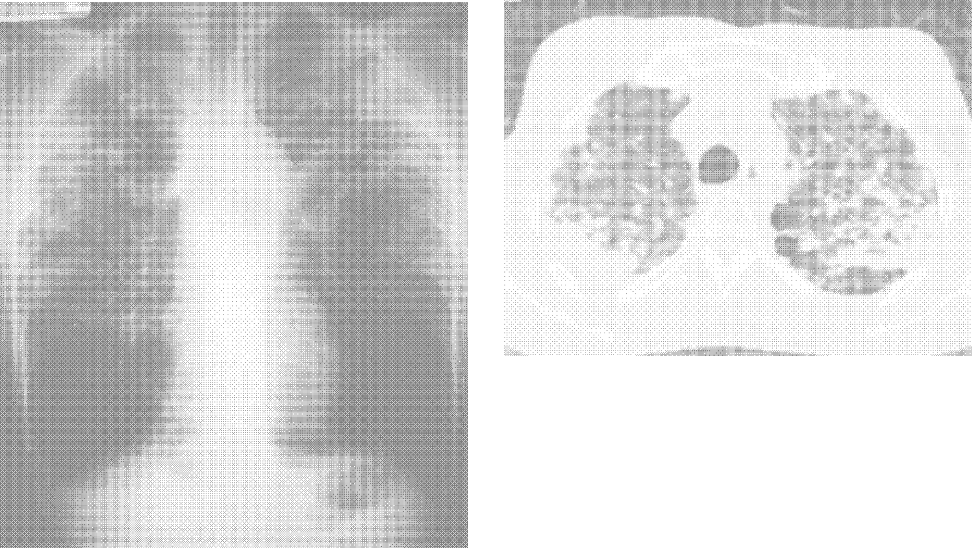
The changes on lumbar puncture are nonspecific. The ACE level is now very low, making sarcoidosis unlikely but not impossible. The chest imaging shows features, specifically interstitial nodularity, consistent with ongoing or relapsing sarcoidosis, but the extensive apical bullae are not characteristic. My best guess is that this patient's illness is not simply relapsing sarcoid but represents superimposed opportunistic infectious disease. I would not reintroduce steroids without pursuing a definitive diagnosis with tissue pathology.
He was placed on prednisone 60 mg po qd and started on trimethoprim‐sulfamethoxazole for Pneumocystis pneumonia (PCP) prophylaxis. He showed modest improvement in his neurological status. A repeat bronchoscopy was not performed. Four months later he was seen by his pulmonologist. He remained without respiratory symptoms and was neurologically unchanged, and a chest radiograph showed no change. He was continued on prednisone 60 mg po qd.
Three weeks later, he was admitted to the hospital with a 2‐week history of anorexia, fatigue, night sweats, right‐sided pleuritic chest pain with productive cough, increasing dyspnea, and no hemoptysis. On admission he was hypoxic with evidence of respiratory distress, and his chest radiograph showed evidence of new right‐sided airspace disease with an associated large right pleural effusion. Initial labs demonstrated a leukocytosis.
I am now very suspicious that this illness is not relapsed sarcoidosis based on his prior clinical response to high‐dose prednisone and that he currently is showing no neurological improvement. His recent clinical deterioration on this very high dose of prednisone makes me think that opportunistic lung infection or disseminated disease is definitely the cause, although the differential is broad. In addition to the typical viral and bacterial causes of community‐acquired pneumonia, this could be caused by unusual bacterial pathogens, tuberculosis, nontuberculous mycobacteria, or fungal diseases including Candida, Aspergillus and dimorphic fungi. I would begin empiric therapy with antibiotics, obtain pleural fluid for examination and culture, and blood cultures.
The patient was treated with a respiratory fluoroquinolone, and blood and sputum cultures were performed. A right thoracentesis removed 300 cc of yellow exudate, with negative gram stain and initial culture. Over the next 24 hours, the patient deteriorated rapidly, with progressive hypoxia and clinical and radiological (Fig. 5) evidence of acute respiratory distress syndrome (ARDS). He required endotracheal intubation with mechanical ventilation.
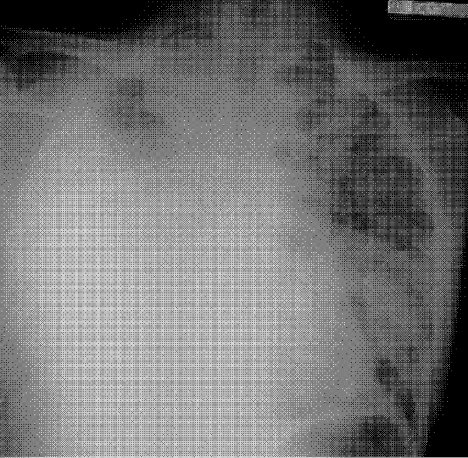
He has a progressive illness not responsive to broad‐spectrum antibiotics, and he has deteriorated. At this point it is imperative that he undergo bronchoscopy and transbronchial biopsy.
Bronchoscopy demonstrated secretions from the right lower lobe. Gram stain from a bronchoalveolar lavage from the right lower lobe was negative, and cultures showed no growth after 24 hours. Immediately after bronchoscopy a third‐generation cephalosporin was empirically added. The next day the patient developed hypotension and was started on norepinephrine. Over the subsequent 48 hours, he developed progressive multiorgan failure. Despite multiple vasopressors, high‐frequency oscillator ventilation, broad‐spectrum antimicrobials, and activated protein C, he died in the intensive care unit. At the time of death, all blood cultures were negative, abdominal CT scans showed no intraabdominal infections, and the BAL performed on admission demonstrated negative gram stain, fungal stain, AFB stain, and PCP and no growth from fungal or bacterial cultures.
I think it is an unavoidable conclusion that this man's progressive systemic inflammatory response syndrome and ultimate multiorgan failure was caused by an opportunistic pathogen that was not antibiotic responsive and not identified from the extensive range of infectious disease studies performed. Despite all the negative studies, there still might be either mycobacterial illness or fungal illness. With negative cultures, Candida or Aspergillus infection is unlikely. Other opportunistic fungi like Blastomyces, Histoplasma, and Cryptococcus are certainly in the differential because these organisms can be notoriously difficult to detect on routine surveying such as bronchoalveolar lavage or lumbar puncture. Blastomyces and Histoplasma are both endemic in the area of Canada where the patient resided. I would also keep the zygomycoses in the differential.
Five days after death, fungal culture was reported demonstrating Blastomyces dermatitidis. Postmortem demonstrated disseminated blastomycosis causing severe bilateral pneumonia (Fig. 6a), empyema of right lung, and involvement of the thyroid, heart, liver, spleen, and kidneys. There was also evidence of active CNS blastomycosis involving the meninges and cerebral cortex and diencephalon (Fig. 6b). As well as active blastomycosis, the leptomeninges demonstrated fibrosis and old granulomas that did not contain an organism.

COMMENTARY
This case describes a 45‐year‐old man who presented with chronic cognitive symptoms associated with hydrocephalus. The first step in establishing the diagnosis was made by realizing that a communicating hydrocephalus with no parenchymal CNS disease was highly suggestive of a leptomeningeal process. This narrowed the differential diagnosis to an infiltrative disease affecting the leptomeninges. The next step involved the discovery of an upper‐lobe interstitial lung process, establishing sarcoidosis as the most likely unifying diagnosis. This was confirmed with transbronchial biopsies showing noncaseating granulomas and by the sustained response to treatment with corticosteroids. Unfortunately, after a 2‐year remission, he developed a recurrence of both the neurological and respiratory findings. When his symptoms progressed despite higher doses of corticosteroids, it became apparent that the etiology of his clinical deterioration was not recurrent disease. Instead, the deterioration was caused by disseminated blastomycosis, an opportunistic infection that developed as a result of the immunosuppressants used to treat the sarcoidosis.
With the final diagnosis of blastomycosis, one question about this case becomes: Could it have been blastomycosis and not sarcoid that was responsible for his original neurological presentation? Blastomyces dermatitidis is a thermally dimorphic fungus that causes disease from inhalation of airborne spores found in soil. Areas of North America in which it is endemic include regions bordering the Mississipi and Ohio rivers, as well as the areas bordering the Great Lakes.1 The patient in this case lived in metropolitan Toronto, on Lake Ontario, where blastomycosis is an important yet underreported disease.24 He likely was exposed to blastomyces in Toronto, which in immunocompromised patients may be followed after weeks to months by dissemination to other body sites including the dermis, bones, joints, urogenital system, and, rarely, the central nervous system (CNS) and liver.5 Like sarcoidosis, infection with blastomycosis can produce pathologic evidence of noncaseating granulomatous inflammation. However, as the discussant astutely pointed out, it would be unusual for this patient to have clinically inapparent blastomycosis for almost 2 years while on high‐dose prednisone. The initial diagnosis of sarcoid likely was correct.
CNS disease caused by Blastomyces dermatitidis is quite rare, with only 22 reported cases of meningoencephalitis in the literature.6 As this case demonstrates, CNS blastomycosis is very difficult to diagnose because of the absence of sensitive serologic markers and the difficulty of isolating the organism from blood and cerebrospinal fluid. CSF sampling from lumbar puncture led to its diagnosis in only 2 of the 22 reported cases.7 Furthermore, reliable CSF cultures are usually only obtained via ventricular sampling or tissue biopsy, which itself is limited by the organism's predilection for deep structures of the cerebrum, midbrain, and basal meninges.6 Blastomyces involving the CNS rarely occurs in isolation. In the patient's case, during his neurological deterioration, there was clear radiological evidence of progressive pulmonary pathology despite his being asymptomatic, and as the discussant suggests, pulmonary investigations were warranted.
Pulmonary manifestations of blastomycosis are variable. Acute infections most commonly resemble pneumonia, whereas chronic disease may show reticulonodular changes indistinguishable from sarcoidosis. Severe cases have been shown to progress to respiratory failure with acute respiratory distress syndrome (ARDS).1 The diagnosis is usually established through culture of noninvasive (sensitivity 86%) or bronchoalveolar lavage (sensitivity 92%) specimens.8 However, blastomyces will take between 5 and 30 days to grow in culture.1 In cases where the diagnosis needs to be established quickly, a KOH smear can be done looking for broad‐based budding yeast. Although the yield of this test is lower (0%‐50%), the results can be available within 24 hours.9 As these tests are not always routinely performed, direct communication with the pathologist is recommended if a rapid diagnosis is needed.
The major challenge of this case lay in distinguishing between the recurrence of an old disease and the complications of its treatment. In this case the discussant addresses strategies that might be useful in differentiating recurrent sarcoidosis from an opportunistic infection like blastomycosis. The first issue is the steroid therapy. The exact dose of steroids required to compromise the immune system enough to yield infections is not known. However, in a meta‐analysis of 71 controlled clinical trials performed with steroids, Stuck et. al. were able to show that the occurrence of opportunistic infections depended on both the amount of daily steroid and the cumulative dose.10 Opportunistic infections were unlikely to occur in patients given a mean daily dose of less than 10 mg/day or a cumulative dose of less than 700 mg of prednisone. Although the patient in the present case was only on 10 mg/day of prednisone, his mean daily dose was more than 10 mg/day, and his cumulative dose far exceeded 700 mg. Therefore, an opportunistic infection should have been strongly considered.
The other item used to help distinguish between the 2 diseases was serum angiotensin‐converting enzyme (ACE) level. ACE is an enzyme produced by the epithelial cells of the granulomas in sarcoidosis. ACE alone is inadequate for diagnosis, with a reported sensitivity of 40%‐90%, depending on the population studied and on the definition of normal.1114 Even an ACE level more than twice the normal is not diagnostic for sarcoidosis, with elevated levels found in histoplasmosis, silicosis, tuberculosis, Gaucher's disease, and other disorders.15 Rather than as a diagnostic test, ACE level instead is used to follow disease activity in sarcoidosis, as ACE level often reflects the granuloma burden.1618 The low levels at the initial recurrence suggests the symptoms were not a result of active sarcoid, especially considering that if ACE levels are originally elevated with sarcoidosis, they are almost always elevated again when the disease recurs.14 Normal levels of ACE in sarcoid patients with previously elevated ACE levels should therefore prompt a search for an alternate diagnosis.
This case is an example of the therapy causing a complication that mimics the disease it was intended to cure. When any patient deteriorates while on steroids, the clinician must ask the age‐old question: should more steroids be prescribed or less? As in this case, the answer is not always apparent. Safe decisions in these situations demand awareness of the opportunistic infections endemic to the area and a willingness to perform early invasive procedures (in this case bronchoscopy) to obtain samples to make a definitive diagnosis. By doing so, the devastating chain of events that occurred here hopefully can be avoided.
Acknowledgements
The authors would like to acknowledge Dr. Eleanor Latta and Dr. Serge Jothy, Department of Pathology, St. Michael's Hospital, University of Toronto, for contributing the pathological images.
- ,,.Blastomycosis.Infect Dis Clin North Am.2003;17(1):21,40, vii.
- ,,,,,.Novel cases of blastomycosis acquired in Toronto, Ontario.CMAJ.2000;163:1309–1312.
- ,,,,,.Blastomycosis acquired by three children in Toronto.Can J Infect Dis Med Micro.2002;13(4):259–263.
- ,,, et al.Blastomycosis in Ontario, 1994‐2003.Emerg Infect Dis.2006;12(2):274–279.
- ,,, et al.Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals.Clin Infect Dis.2002;34:1310–1316.
- ,,,.Meningoencephalitis due to Blastomyces dermatitidis: case report and literature review.Mayo Clin Proc.2000;75:403–408.
- ,,,.Chronic blastomycotic meningitis.Am J Med.1981;71:501–505.
- ,.Pulmonary blastomycosis: an appraisal of diagnostic techniques.Chest.2002;121:768–773.
- ,,.Blastomycosis as an etiology of acute lung injury.South Med J.1998;91:861–863.
- ,,.Risk of infectious complications in patients taking glucocorticosteroids.Rev Infect Dis.1989;11:954–963.
- .Elevation of serum angiotensin‐converting‐enzyme (ACE) level in sarcoidosis.Am J Med.1975;59:365–372.
- ,,,.Elevated serum angiotensin I converting enzyme in sarcoidosis.Am Rev Respir Dis.1976;114:525–528.
- ,,.Serum angiotensin‐converting enzyme (SACE) in sarcoidosis and other granulomatous disorders.Lancet.1978;2:1331–1334.
- ,.Serum angiotensin converting enzyme in sarcoidosis: sensitivity and specificity in diagnosis: correlations with disease activity, duration, extra‐thoracic involvement, radiographic type and therapy.Q J Med.1985;55(218):253–270.
- Statement on sarcoidosis.Joint Statement of the American Thoracic Society (ATS), theEuropean Respiratory Society (ERS) and theWorld Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999.Am J Respir Crit Care Med.1999;160:736–755.
- ,,.Value of serial measurement of serum angiotensin converting enzyme in the management of sarcoidosis.Am J Med.1981;70(1):44–50.
- ,,,.Serum angiotensin‐converting enzyme (SACE) activity as an indicator of total body granuloma load and prognosis in sarcoidosis.Sarcoidosis.1987;4(2):142–148.
- ,,.Serum angiotensin converting enzyme in sarcoidosis: clinical significance.Isr J Med Sci.1977;13:1001–1006.
A 45‐year‐old man who immigrated to Canada from Ghana at the age of 33 presented with a 2‐year history of progressive cognitive changes. He had bifrontal headache, right‐sided scalp paresthesias, and a 40‐pound weight loss. He was unable to perform his job as an auto parts worker. His wife noticed short‐ and long‐term memory problems and poor concentration. On physical exam he had no focal neurological findings but his score on the Mini‐Mental Status Exam (MMSE) was 23/30, with deficits in attention and recall.
The first important element of this illness is its chronicity. His symptoms progressed slowly over 2 years. Second, aside from his neurological problems, he is an otherwise healthy young, African‐born male. This clinical picture could be the early presentation of a demyelinating, infiltrative, or vascular illness. If vascular, it is more likely a vasculitis than atherosclerotic disease. Malignancy and infection are definitely in the differential, but at this point, I think they are less likely to be the cause, given the tempo of presentation. I would begin my investigations with basic blood work and a computerized tomography (CT) scan of his brain.
A CT scan of the head with contrast demonstrated an enlarged left lateral ventricle with no evidence of obstruction in the foramen of Munro.
The radiological findings of communicating hydrocephalus with normal parenchyma imply a disease that affects the leptomeningeal space. Given that we are looking at an illness that can change cerebral spinal fluid (CSF) flow rather than primary parenchymal disease, demyelinating and vascular illnesses are less likely etiologies, and infiltrative diseases move up on my list. Malignancy and infectious diseases remain in the differential.
He disappeared to follow up for 1 year, during which he returned to Ghana and experienced progressive neurological deterioration, with incontinence, gait instability, and inability to converse clearly and perform activities of daily living. On his return to Canada, an urgent CT scan and magnetic resonance imaging (MRI) of the brain demonstrated ongoing and unchanged hydrocephalus with aqueductal stenosis. A referral was made to a neurosurgeon for insertion of a ventriculoperitoneal shunt. A routine preoperative chest radiograph demonstrated new bilateral upper‐zone reticulonodular changes.
He had no respiratory symptoms, fevers, or lymphadenopathy. His occupational history revealed no exposure to asbestos, silica, farms, or mines. He had no history of either respiratory or neurological illness in the past and no travel other than to Ghana and Toronto. When he immigrated to Toronto, Canada, 12 years before, he had a normal chest radiograph and negative PPD tuberculin skin test.
Many illnesses produce asymptomatic changes on chest x‐ray. Oslerian principles would suggest that we should think of a single diagnosis to explain both nodular lung disease and more than 3 years of a progressive disease affecting the leptomeninges. It is unlikely that tuberculosis, other fungal diseases, or malignancy would result in the chest and brain pathology over a 3‐year period without other sequelae. Sarcoidosis could cause both chronic leptomeningeal changes and the radiographic lung findings. The next steps in investigating this patient should include measurement of angiotensin‐converting enzyme (ACE) and serum calcium and pulmonary function tests. I would ultimately send him for a pathological biopsy of his lung tissue to confirm noncaseating granuloma and exclude infection and malignancy.
Complete blood count, renal and liver biochemistry, and calcium were normal. An ACE level was elevated at 69 g/L (normal 40 g/L). A human immunodeficiency virus (HIV‐1 and HIV‐2) test, tuberculin skin test, and syphilis serology were negative. A CT scan of the chest demonstrated bilateral upper‐zone reticulonodular changes and diffuse lymphadenopathy (Fig. 1). Pulmonary function tests (PFTs) demonstrated a forced expiratory volume 1 (FEV1) of 3.4 L (94%), forced vital capacity (FVC) of 4.0 L (83%), an FEV1/FVC of 87%, total lung capacity (TLC) of 92% predicted, and diffusion capacity (DLCO) of 67% predicted. An MRI with gadolinium (Fig. 2) demonstrated hydrocephalus, mild basal leptomeningeal enhancement around the perivascular spaces into the subinsular region, and an increased T2 signal in periventricular white matter.
A bronchoscopy with bronchoalveolar lavage and transbronchial biopsies were performed. Pathology (Fig. 3) demonstrated non‐caseating epitheliod granulomas, with negative special stains for acid‐fast bacilli (AFB) and fungus, and negative fungal and AFB cultures of the bronchial alveolar lavage.
With negative tests for infectious causes such as tuberculosis, I think there is now enough evidence that this patient has sarcoidosis involving the lung and leptomeninges. At this point I would start therapy with steroids.
The patient was started on prednisone 40 mg po qd, and his neurological symptoms improved markedly over the course of 1‐2 months, with normalization of his MMSE and a return to cognitive baseline. As his symptoms stabilized with no change in CT imaging, he returned to work, and over the course of 2 years his prednisone dosage was tapered to 10 mg po od. While on prednisone he developed hypertension and hyperglycemia. He continued to have no respiratory symptoms.
He was cognitively at baseline until 20 months later, when he was readmitted to the hospital with a 2‐week history of worsening headache, increased confusion, poor memory, and wandering. His MMSE had deteriorated to 19/30, with deficits again in memory and attention.
First, we can say with reasonable confidence that the diagnosis of sarcoid was correct. His long and sustained response to steroids, plus the absence of the unmasking of an infectious or malignant disease, supports this conclusion. However, he is now exhibiting an apparent relapse that mimics his presentation 3 years earlier. The question is whether he is suffering from a flare of his disease or whether a second illness has occurred. The most obvious second illness is an opportunistic infection after years of steroid use. I would certainly repeat the angiotensin‐converting enzyme and serum calcium tests and repeat the imaging of his lungs and central nervous system. He also warrants a lumbar puncture with CSF culture, stain, and PCR for opportunistic infections. If these studies are inconclusive and do not specifically suggest relapsing sarcoid, I would once again consider biopsy of tissue from either a lung or leptomeninges.
An MRI with gadolinium looked unchanged from the previous one. A lumbar puncture was performed, and his CSF demonstrated 3 WBCs, no RBCs, normal glucose, and elevated protein at 1.17 g/L, and tests for bacteria, TB, fungi, and viruses were all negative. Repeat blood work was unremarkable, and the ACE level was 2 g/L.
A chest radiograph (Fig. 4a) and CT chest (Fig. 4b) showed marked deterioration, with increased diffuse airspace opacities, interstitial nodularity, and small apical bullae. His PFTs showed some deterioration, with FEV1 2.52 L (73%), FVC 3.29 L (73%), FEV1/FVC 76%, TLC 70% predicted, DLCO 72% predicted. However, he still had no respiratory symptoms.
The changes on lumbar puncture are nonspecific. The ACE level is now very low, making sarcoidosis unlikely but not impossible. The chest imaging shows features, specifically interstitial nodularity, consistent with ongoing or relapsing sarcoidosis, but the extensive apical bullae are not characteristic. My best guess is that this patient's illness is not simply relapsing sarcoid but represents superimposed opportunistic infectious disease. I would not reintroduce steroids without pursuing a definitive diagnosis with tissue pathology.
He was placed on prednisone 60 mg po qd and started on trimethoprim‐sulfamethoxazole for Pneumocystis pneumonia (PCP) prophylaxis. He showed modest improvement in his neurological status. A repeat bronchoscopy was not performed. Four months later he was seen by his pulmonologist. He remained without respiratory symptoms and was neurologically unchanged, and a chest radiograph showed no change. He was continued on prednisone 60 mg po qd.
Three weeks later, he was admitted to the hospital with a 2‐week history of anorexia, fatigue, night sweats, right‐sided pleuritic chest pain with productive cough, increasing dyspnea, and no hemoptysis. On admission he was hypoxic with evidence of respiratory distress, and his chest radiograph showed evidence of new right‐sided airspace disease with an associated large right pleural effusion. Initial labs demonstrated a leukocytosis.
I am now very suspicious that this illness is not relapsed sarcoidosis based on his prior clinical response to high‐dose prednisone and that he currently is showing no neurological improvement. His recent clinical deterioration on this very high dose of prednisone makes me think that opportunistic lung infection or disseminated disease is definitely the cause, although the differential is broad. In addition to the typical viral and bacterial causes of community‐acquired pneumonia, this could be caused by unusual bacterial pathogens, tuberculosis, nontuberculous mycobacteria, or fungal diseases including Candida, Aspergillus and dimorphic fungi. I would begin empiric therapy with antibiotics, obtain pleural fluid for examination and culture, and blood cultures.
The patient was treated with a respiratory fluoroquinolone, and blood and sputum cultures were performed. A right thoracentesis removed 300 cc of yellow exudate, with negative gram stain and initial culture. Over the next 24 hours, the patient deteriorated rapidly, with progressive hypoxia and clinical and radiological (Fig. 5) evidence of acute respiratory distress syndrome (ARDS). He required endotracheal intubation with mechanical ventilation.
He has a progressive illness not responsive to broad‐spectrum antibiotics, and he has deteriorated. At this point it is imperative that he undergo bronchoscopy and transbronchial biopsy.
Bronchoscopy demonstrated secretions from the right lower lobe. Gram stain from a bronchoalveolar lavage from the right lower lobe was negative, and cultures showed no growth after 24 hours. Immediately after bronchoscopy a third‐generation cephalosporin was empirically added. The next day the patient developed hypotension and was started on norepinephrine. Over the subsequent 48 hours, he developed progressive multiorgan failure. Despite multiple vasopressors, high‐frequency oscillator ventilation, broad‐spectrum antimicrobials, and activated protein C, he died in the intensive care unit. At the time of death, all blood cultures were negative, abdominal CT scans showed no intraabdominal infections, and the BAL performed on admission demonstrated negative gram stain, fungal stain, AFB stain, and PCP and no growth from fungal or bacterial cultures.
I think it is an unavoidable conclusion that this man's progressive systemic inflammatory response syndrome and ultimate multiorgan failure was caused by an opportunistic pathogen that was not antibiotic responsive and not identified from the extensive range of infectious disease studies performed. Despite all the negative studies, there still might be either mycobacterial illness or fungal illness. With negative cultures, Candida or Aspergillus infection is unlikely. Other opportunistic fungi like Blastomyces, Histoplasma, and Cryptococcus are certainly in the differential because these organisms can be notoriously difficult to detect on routine surveying such as bronchoalveolar lavage or lumbar puncture. Blastomyces and Histoplasma are both endemic in the area of Canada where the patient resided. I would also keep the zygomycoses in the differential.
Five days after death, fungal culture was reported demonstrating Blastomyces dermatitidis. Postmortem demonstrated disseminated blastomycosis causing severe bilateral pneumonia (Fig. 6a), empyema of right lung, and involvement of the thyroid, heart, liver, spleen, and kidneys. There was also evidence of active CNS blastomycosis involving the meninges and cerebral cortex and diencephalon (Fig. 6b). As well as active blastomycosis, the leptomeninges demonstrated fibrosis and old granulomas that did not contain an organism.
COMMENTARY
This case describes a 45‐year‐old man who presented with chronic cognitive symptoms associated with hydrocephalus. The first step in establishing the diagnosis was made by realizing that a communicating hydrocephalus with no parenchymal CNS disease was highly suggestive of a leptomeningeal process. This narrowed the differential diagnosis to an infiltrative disease affecting the leptomeninges. The next step involved the discovery of an upper‐lobe interstitial lung process, establishing sarcoidosis as the most likely unifying diagnosis. This was confirmed with transbronchial biopsies showing noncaseating granulomas and by the sustained response to treatment with corticosteroids. Unfortunately, after a 2‐year remission, he developed a recurrence of both the neurological and respiratory findings. When his symptoms progressed despite higher doses of corticosteroids, it became apparent that the etiology of his clinical deterioration was not recurrent disease. Instead, the deterioration was caused by disseminated blastomycosis, an opportunistic infection that developed as a result of the immunosuppressants used to treat the sarcoidosis.
With the final diagnosis of blastomycosis, one question about this case becomes: Could it have been blastomycosis and not sarcoid that was responsible for his original neurological presentation? Blastomyces dermatitidis is a thermally dimorphic fungus that causes disease from inhalation of airborne spores found in soil. Areas of North America in which it is endemic include regions bordering the Mississipi and Ohio rivers, as well as the areas bordering the Great Lakes.1 The patient in this case lived in metropolitan Toronto, on Lake Ontario, where blastomycosis is an important yet underreported disease.24 He likely was exposed to blastomyces in Toronto, which in immunocompromised patients may be followed after weeks to months by dissemination to other body sites including the dermis, bones, joints, urogenital system, and, rarely, the central nervous system (CNS) and liver.5 Like sarcoidosis, infection with blastomycosis can produce pathologic evidence of noncaseating granulomatous inflammation. However, as the discussant astutely pointed out, it would be unusual for this patient to have clinically inapparent blastomycosis for almost 2 years while on high‐dose prednisone. The initial diagnosis of sarcoid likely was correct.
CNS disease caused by Blastomyces dermatitidis is quite rare, with only 22 reported cases of meningoencephalitis in the literature.6 As this case demonstrates, CNS blastomycosis is very difficult to diagnose because of the absence of sensitive serologic markers and the difficulty of isolating the organism from blood and cerebrospinal fluid. CSF sampling from lumbar puncture led to its diagnosis in only 2 of the 22 reported cases.7 Furthermore, reliable CSF cultures are usually only obtained via ventricular sampling or tissue biopsy, which itself is limited by the organism's predilection for deep structures of the cerebrum, midbrain, and basal meninges.6 Blastomyces involving the CNS rarely occurs in isolation. In the patient's case, during his neurological deterioration, there was clear radiological evidence of progressive pulmonary pathology despite his being asymptomatic, and as the discussant suggests, pulmonary investigations were warranted.
Pulmonary manifestations of blastomycosis are variable. Acute infections most commonly resemble pneumonia, whereas chronic disease may show reticulonodular changes indistinguishable from sarcoidosis. Severe cases have been shown to progress to respiratory failure with acute respiratory distress syndrome (ARDS).1 The diagnosis is usually established through culture of noninvasive (sensitivity 86%) or bronchoalveolar lavage (sensitivity 92%) specimens.8 However, blastomyces will take between 5 and 30 days to grow in culture.1 In cases where the diagnosis needs to be established quickly, a KOH smear can be done looking for broad‐based budding yeast. Although the yield of this test is lower (0%‐50%), the results can be available within 24 hours.9 As these tests are not always routinely performed, direct communication with the pathologist is recommended if a rapid diagnosis is needed.
The major challenge of this case lay in distinguishing between the recurrence of an old disease and the complications of its treatment. In this case the discussant addresses strategies that might be useful in differentiating recurrent sarcoidosis from an opportunistic infection like blastomycosis. The first issue is the steroid therapy. The exact dose of steroids required to compromise the immune system enough to yield infections is not known. However, in a meta‐analysis of 71 controlled clinical trials performed with steroids, Stuck et. al. were able to show that the occurrence of opportunistic infections depended on both the amount of daily steroid and the cumulative dose.10 Opportunistic infections were unlikely to occur in patients given a mean daily dose of less than 10 mg/day or a cumulative dose of less than 700 mg of prednisone. Although the patient in the present case was only on 10 mg/day of prednisone, his mean daily dose was more than 10 mg/day, and his cumulative dose far exceeded 700 mg. Therefore, an opportunistic infection should have been strongly considered.
The other item used to help distinguish between the 2 diseases was serum angiotensin‐converting enzyme (ACE) level. ACE is an enzyme produced by the epithelial cells of the granulomas in sarcoidosis. ACE alone is inadequate for diagnosis, with a reported sensitivity of 40%‐90%, depending on the population studied and on the definition of normal.1114 Even an ACE level more than twice the normal is not diagnostic for sarcoidosis, with elevated levels found in histoplasmosis, silicosis, tuberculosis, Gaucher's disease, and other disorders.15 Rather than as a diagnostic test, ACE level instead is used to follow disease activity in sarcoidosis, as ACE level often reflects the granuloma burden.1618 The low levels at the initial recurrence suggests the symptoms were not a result of active sarcoid, especially considering that if ACE levels are originally elevated with sarcoidosis, they are almost always elevated again when the disease recurs.14 Normal levels of ACE in sarcoid patients with previously elevated ACE levels should therefore prompt a search for an alternate diagnosis.
This case is an example of the therapy causing a complication that mimics the disease it was intended to cure. When any patient deteriorates while on steroids, the clinician must ask the age‐old question: should more steroids be prescribed or less? As in this case, the answer is not always apparent. Safe decisions in these situations demand awareness of the opportunistic infections endemic to the area and a willingness to perform early invasive procedures (in this case bronchoscopy) to obtain samples to make a definitive diagnosis. By doing so, the devastating chain of events that occurred here hopefully can be avoided.
Acknowledgements
The authors would like to acknowledge Dr. Eleanor Latta and Dr. Serge Jothy, Department of Pathology, St. Michael's Hospital, University of Toronto, for contributing the pathological images.
A 45‐year‐old man who immigrated to Canada from Ghana at the age of 33 presented with a 2‐year history of progressive cognitive changes. He had bifrontal headache, right‐sided scalp paresthesias, and a 40‐pound weight loss. He was unable to perform his job as an auto parts worker. His wife noticed short‐ and long‐term memory problems and poor concentration. On physical exam he had no focal neurological findings but his score on the Mini‐Mental Status Exam (MMSE) was 23/30, with deficits in attention and recall.
The first important element of this illness is its chronicity. His symptoms progressed slowly over 2 years. Second, aside from his neurological problems, he is an otherwise healthy young, African‐born male. This clinical picture could be the early presentation of a demyelinating, infiltrative, or vascular illness. If vascular, it is more likely a vasculitis than atherosclerotic disease. Malignancy and infection are definitely in the differential, but at this point, I think they are less likely to be the cause, given the tempo of presentation. I would begin my investigations with basic blood work and a computerized tomography (CT) scan of his brain.
A CT scan of the head with contrast demonstrated an enlarged left lateral ventricle with no evidence of obstruction in the foramen of Munro.
The radiological findings of communicating hydrocephalus with normal parenchyma imply a disease that affects the leptomeningeal space. Given that we are looking at an illness that can change cerebral spinal fluid (CSF) flow rather than primary parenchymal disease, demyelinating and vascular illnesses are less likely etiologies, and infiltrative diseases move up on my list. Malignancy and infectious diseases remain in the differential.
He disappeared to follow up for 1 year, during which he returned to Ghana and experienced progressive neurological deterioration, with incontinence, gait instability, and inability to converse clearly and perform activities of daily living. On his return to Canada, an urgent CT scan and magnetic resonance imaging (MRI) of the brain demonstrated ongoing and unchanged hydrocephalus with aqueductal stenosis. A referral was made to a neurosurgeon for insertion of a ventriculoperitoneal shunt. A routine preoperative chest radiograph demonstrated new bilateral upper‐zone reticulonodular changes.
He had no respiratory symptoms, fevers, or lymphadenopathy. His occupational history revealed no exposure to asbestos, silica, farms, or mines. He had no history of either respiratory or neurological illness in the past and no travel other than to Ghana and Toronto. When he immigrated to Toronto, Canada, 12 years before, he had a normal chest radiograph and negative PPD tuberculin skin test.
Many illnesses produce asymptomatic changes on chest x‐ray. Oslerian principles would suggest that we should think of a single diagnosis to explain both nodular lung disease and more than 3 years of a progressive disease affecting the leptomeninges. It is unlikely that tuberculosis, other fungal diseases, or malignancy would result in the chest and brain pathology over a 3‐year period without other sequelae. Sarcoidosis could cause both chronic leptomeningeal changes and the radiographic lung findings. The next steps in investigating this patient should include measurement of angiotensin‐converting enzyme (ACE) and serum calcium and pulmonary function tests. I would ultimately send him for a pathological biopsy of his lung tissue to confirm noncaseating granuloma and exclude infection and malignancy.
Complete blood count, renal and liver biochemistry, and calcium were normal. An ACE level was elevated at 69 g/L (normal 40 g/L). A human immunodeficiency virus (HIV‐1 and HIV‐2) test, tuberculin skin test, and syphilis serology were negative. A CT scan of the chest demonstrated bilateral upper‐zone reticulonodular changes and diffuse lymphadenopathy (Fig. 1). Pulmonary function tests (PFTs) demonstrated a forced expiratory volume 1 (FEV1) of 3.4 L (94%), forced vital capacity (FVC) of 4.0 L (83%), an FEV1/FVC of 87%, total lung capacity (TLC) of 92% predicted, and diffusion capacity (DLCO) of 67% predicted. An MRI with gadolinium (Fig. 2) demonstrated hydrocephalus, mild basal leptomeningeal enhancement around the perivascular spaces into the subinsular region, and an increased T2 signal in periventricular white matter.
A bronchoscopy with bronchoalveolar lavage and transbronchial biopsies were performed. Pathology (Fig. 3) demonstrated non‐caseating epitheliod granulomas, with negative special stains for acid‐fast bacilli (AFB) and fungus, and negative fungal and AFB cultures of the bronchial alveolar lavage.
With negative tests for infectious causes such as tuberculosis, I think there is now enough evidence that this patient has sarcoidosis involving the lung and leptomeninges. At this point I would start therapy with steroids.
The patient was started on prednisone 40 mg po qd, and his neurological symptoms improved markedly over the course of 1‐2 months, with normalization of his MMSE and a return to cognitive baseline. As his symptoms stabilized with no change in CT imaging, he returned to work, and over the course of 2 years his prednisone dosage was tapered to 10 mg po od. While on prednisone he developed hypertension and hyperglycemia. He continued to have no respiratory symptoms.
He was cognitively at baseline until 20 months later, when he was readmitted to the hospital with a 2‐week history of worsening headache, increased confusion, poor memory, and wandering. His MMSE had deteriorated to 19/30, with deficits again in memory and attention.
First, we can say with reasonable confidence that the diagnosis of sarcoid was correct. His long and sustained response to steroids, plus the absence of the unmasking of an infectious or malignant disease, supports this conclusion. However, he is now exhibiting an apparent relapse that mimics his presentation 3 years earlier. The question is whether he is suffering from a flare of his disease or whether a second illness has occurred. The most obvious second illness is an opportunistic infection after years of steroid use. I would certainly repeat the angiotensin‐converting enzyme and serum calcium tests and repeat the imaging of his lungs and central nervous system. He also warrants a lumbar puncture with CSF culture, stain, and PCR for opportunistic infections. If these studies are inconclusive and do not specifically suggest relapsing sarcoid, I would once again consider biopsy of tissue from either a lung or leptomeninges.
An MRI with gadolinium looked unchanged from the previous one. A lumbar puncture was performed, and his CSF demonstrated 3 WBCs, no RBCs, normal glucose, and elevated protein at 1.17 g/L, and tests for bacteria, TB, fungi, and viruses were all negative. Repeat blood work was unremarkable, and the ACE level was 2 g/L.
A chest radiograph (Fig. 4a) and CT chest (Fig. 4b) showed marked deterioration, with increased diffuse airspace opacities, interstitial nodularity, and small apical bullae. His PFTs showed some deterioration, with FEV1 2.52 L (73%), FVC 3.29 L (73%), FEV1/FVC 76%, TLC 70% predicted, DLCO 72% predicted. However, he still had no respiratory symptoms.
The changes on lumbar puncture are nonspecific. The ACE level is now very low, making sarcoidosis unlikely but not impossible. The chest imaging shows features, specifically interstitial nodularity, consistent with ongoing or relapsing sarcoidosis, but the extensive apical bullae are not characteristic. My best guess is that this patient's illness is not simply relapsing sarcoid but represents superimposed opportunistic infectious disease. I would not reintroduce steroids without pursuing a definitive diagnosis with tissue pathology.
He was placed on prednisone 60 mg po qd and started on trimethoprim‐sulfamethoxazole for Pneumocystis pneumonia (PCP) prophylaxis. He showed modest improvement in his neurological status. A repeat bronchoscopy was not performed. Four months later he was seen by his pulmonologist. He remained without respiratory symptoms and was neurologically unchanged, and a chest radiograph showed no change. He was continued on prednisone 60 mg po qd.
Three weeks later, he was admitted to the hospital with a 2‐week history of anorexia, fatigue, night sweats, right‐sided pleuritic chest pain with productive cough, increasing dyspnea, and no hemoptysis. On admission he was hypoxic with evidence of respiratory distress, and his chest radiograph showed evidence of new right‐sided airspace disease with an associated large right pleural effusion. Initial labs demonstrated a leukocytosis.
I am now very suspicious that this illness is not relapsed sarcoidosis based on his prior clinical response to high‐dose prednisone and that he currently is showing no neurological improvement. His recent clinical deterioration on this very high dose of prednisone makes me think that opportunistic lung infection or disseminated disease is definitely the cause, although the differential is broad. In addition to the typical viral and bacterial causes of community‐acquired pneumonia, this could be caused by unusual bacterial pathogens, tuberculosis, nontuberculous mycobacteria, or fungal diseases including Candida, Aspergillus and dimorphic fungi. I would begin empiric therapy with antibiotics, obtain pleural fluid for examination and culture, and blood cultures.
The patient was treated with a respiratory fluoroquinolone, and blood and sputum cultures were performed. A right thoracentesis removed 300 cc of yellow exudate, with negative gram stain and initial culture. Over the next 24 hours, the patient deteriorated rapidly, with progressive hypoxia and clinical and radiological (Fig. 5) evidence of acute respiratory distress syndrome (ARDS). He required endotracheal intubation with mechanical ventilation.
He has a progressive illness not responsive to broad‐spectrum antibiotics, and he has deteriorated. At this point it is imperative that he undergo bronchoscopy and transbronchial biopsy.
Bronchoscopy demonstrated secretions from the right lower lobe. Gram stain from a bronchoalveolar lavage from the right lower lobe was negative, and cultures showed no growth after 24 hours. Immediately after bronchoscopy a third‐generation cephalosporin was empirically added. The next day the patient developed hypotension and was started on norepinephrine. Over the subsequent 48 hours, he developed progressive multiorgan failure. Despite multiple vasopressors, high‐frequency oscillator ventilation, broad‐spectrum antimicrobials, and activated protein C, he died in the intensive care unit. At the time of death, all blood cultures were negative, abdominal CT scans showed no intraabdominal infections, and the BAL performed on admission demonstrated negative gram stain, fungal stain, AFB stain, and PCP and no growth from fungal or bacterial cultures.
I think it is an unavoidable conclusion that this man's progressive systemic inflammatory response syndrome and ultimate multiorgan failure was caused by an opportunistic pathogen that was not antibiotic responsive and not identified from the extensive range of infectious disease studies performed. Despite all the negative studies, there still might be either mycobacterial illness or fungal illness. With negative cultures, Candida or Aspergillus infection is unlikely. Other opportunistic fungi like Blastomyces, Histoplasma, and Cryptococcus are certainly in the differential because these organisms can be notoriously difficult to detect on routine surveying such as bronchoalveolar lavage or lumbar puncture. Blastomyces and Histoplasma are both endemic in the area of Canada where the patient resided. I would also keep the zygomycoses in the differential.
Five days after death, fungal culture was reported demonstrating Blastomyces dermatitidis. Postmortem demonstrated disseminated blastomycosis causing severe bilateral pneumonia (Fig. 6a), empyema of right lung, and involvement of the thyroid, heart, liver, spleen, and kidneys. There was also evidence of active CNS blastomycosis involving the meninges and cerebral cortex and diencephalon (Fig. 6b). As well as active blastomycosis, the leptomeninges demonstrated fibrosis and old granulomas that did not contain an organism.
COMMENTARY
This case describes a 45‐year‐old man who presented with chronic cognitive symptoms associated with hydrocephalus. The first step in establishing the diagnosis was made by realizing that a communicating hydrocephalus with no parenchymal CNS disease was highly suggestive of a leptomeningeal process. This narrowed the differential diagnosis to an infiltrative disease affecting the leptomeninges. The next step involved the discovery of an upper‐lobe interstitial lung process, establishing sarcoidosis as the most likely unifying diagnosis. This was confirmed with transbronchial biopsies showing noncaseating granulomas and by the sustained response to treatment with corticosteroids. Unfortunately, after a 2‐year remission, he developed a recurrence of both the neurological and respiratory findings. When his symptoms progressed despite higher doses of corticosteroids, it became apparent that the etiology of his clinical deterioration was not recurrent disease. Instead, the deterioration was caused by disseminated blastomycosis, an opportunistic infection that developed as a result of the immunosuppressants used to treat the sarcoidosis.
With the final diagnosis of blastomycosis, one question about this case becomes: Could it have been blastomycosis and not sarcoid that was responsible for his original neurological presentation? Blastomyces dermatitidis is a thermally dimorphic fungus that causes disease from inhalation of airborne spores found in soil. Areas of North America in which it is endemic include regions bordering the Mississipi and Ohio rivers, as well as the areas bordering the Great Lakes.1 The patient in this case lived in metropolitan Toronto, on Lake Ontario, where blastomycosis is an important yet underreported disease.24 He likely was exposed to blastomyces in Toronto, which in immunocompromised patients may be followed after weeks to months by dissemination to other body sites including the dermis, bones, joints, urogenital system, and, rarely, the central nervous system (CNS) and liver.5 Like sarcoidosis, infection with blastomycosis can produce pathologic evidence of noncaseating granulomatous inflammation. However, as the discussant astutely pointed out, it would be unusual for this patient to have clinically inapparent blastomycosis for almost 2 years while on high‐dose prednisone. The initial diagnosis of sarcoid likely was correct.
CNS disease caused by Blastomyces dermatitidis is quite rare, with only 22 reported cases of meningoencephalitis in the literature.6 As this case demonstrates, CNS blastomycosis is very difficult to diagnose because of the absence of sensitive serologic markers and the difficulty of isolating the organism from blood and cerebrospinal fluid. CSF sampling from lumbar puncture led to its diagnosis in only 2 of the 22 reported cases.7 Furthermore, reliable CSF cultures are usually only obtained via ventricular sampling or tissue biopsy, which itself is limited by the organism's predilection for deep structures of the cerebrum, midbrain, and basal meninges.6 Blastomyces involving the CNS rarely occurs in isolation. In the patient's case, during his neurological deterioration, there was clear radiological evidence of progressive pulmonary pathology despite his being asymptomatic, and as the discussant suggests, pulmonary investigations were warranted.
Pulmonary manifestations of blastomycosis are variable. Acute infections most commonly resemble pneumonia, whereas chronic disease may show reticulonodular changes indistinguishable from sarcoidosis. Severe cases have been shown to progress to respiratory failure with acute respiratory distress syndrome (ARDS).1 The diagnosis is usually established through culture of noninvasive (sensitivity 86%) or bronchoalveolar lavage (sensitivity 92%) specimens.8 However, blastomyces will take between 5 and 30 days to grow in culture.1 In cases where the diagnosis needs to be established quickly, a KOH smear can be done looking for broad‐based budding yeast. Although the yield of this test is lower (0%‐50%), the results can be available within 24 hours.9 As these tests are not always routinely performed, direct communication with the pathologist is recommended if a rapid diagnosis is needed.
The major challenge of this case lay in distinguishing between the recurrence of an old disease and the complications of its treatment. In this case the discussant addresses strategies that might be useful in differentiating recurrent sarcoidosis from an opportunistic infection like blastomycosis. The first issue is the steroid therapy. The exact dose of steroids required to compromise the immune system enough to yield infections is not known. However, in a meta‐analysis of 71 controlled clinical trials performed with steroids, Stuck et. al. were able to show that the occurrence of opportunistic infections depended on both the amount of daily steroid and the cumulative dose.10 Opportunistic infections were unlikely to occur in patients given a mean daily dose of less than 10 mg/day or a cumulative dose of less than 700 mg of prednisone. Although the patient in the present case was only on 10 mg/day of prednisone, his mean daily dose was more than 10 mg/day, and his cumulative dose far exceeded 700 mg. Therefore, an opportunistic infection should have been strongly considered.
The other item used to help distinguish between the 2 diseases was serum angiotensin‐converting enzyme (ACE) level. ACE is an enzyme produced by the epithelial cells of the granulomas in sarcoidosis. ACE alone is inadequate for diagnosis, with a reported sensitivity of 40%‐90%, depending on the population studied and on the definition of normal.1114 Even an ACE level more than twice the normal is not diagnostic for sarcoidosis, with elevated levels found in histoplasmosis, silicosis, tuberculosis, Gaucher's disease, and other disorders.15 Rather than as a diagnostic test, ACE level instead is used to follow disease activity in sarcoidosis, as ACE level often reflects the granuloma burden.1618 The low levels at the initial recurrence suggests the symptoms were not a result of active sarcoid, especially considering that if ACE levels are originally elevated with sarcoidosis, they are almost always elevated again when the disease recurs.14 Normal levels of ACE in sarcoid patients with previously elevated ACE levels should therefore prompt a search for an alternate diagnosis.
This case is an example of the therapy causing a complication that mimics the disease it was intended to cure. When any patient deteriorates while on steroids, the clinician must ask the age‐old question: should more steroids be prescribed or less? As in this case, the answer is not always apparent. Safe decisions in these situations demand awareness of the opportunistic infections endemic to the area and a willingness to perform early invasive procedures (in this case bronchoscopy) to obtain samples to make a definitive diagnosis. By doing so, the devastating chain of events that occurred here hopefully can be avoided.
Acknowledgements
The authors would like to acknowledge Dr. Eleanor Latta and Dr. Serge Jothy, Department of Pathology, St. Michael's Hospital, University of Toronto, for contributing the pathological images.
- ,,.Blastomycosis.Infect Dis Clin North Am.2003;17(1):21,40, vii.
- ,,,,,.Novel cases of blastomycosis acquired in Toronto, Ontario.CMAJ.2000;163:1309–1312.
- ,,,,,.Blastomycosis acquired by three children in Toronto.Can J Infect Dis Med Micro.2002;13(4):259–263.
- ,,, et al.Blastomycosis in Ontario, 1994‐2003.Emerg Infect Dis.2006;12(2):274–279.
- ,,, et al.Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals.Clin Infect Dis.2002;34:1310–1316.
- ,,,.Meningoencephalitis due to Blastomyces dermatitidis: case report and literature review.Mayo Clin Proc.2000;75:403–408.
- ,,,.Chronic blastomycotic meningitis.Am J Med.1981;71:501–505.
- ,.Pulmonary blastomycosis: an appraisal of diagnostic techniques.Chest.2002;121:768–773.
- ,,.Blastomycosis as an etiology of acute lung injury.South Med J.1998;91:861–863.
- ,,.Risk of infectious complications in patients taking glucocorticosteroids.Rev Infect Dis.1989;11:954–963.
- .Elevation of serum angiotensin‐converting‐enzyme (ACE) level in sarcoidosis.Am J Med.1975;59:365–372.
- ,,,.Elevated serum angiotensin I converting enzyme in sarcoidosis.Am Rev Respir Dis.1976;114:525–528.
- ,,.Serum angiotensin‐converting enzyme (SACE) in sarcoidosis and other granulomatous disorders.Lancet.1978;2:1331–1334.
- ,.Serum angiotensin converting enzyme in sarcoidosis: sensitivity and specificity in diagnosis: correlations with disease activity, duration, extra‐thoracic involvement, radiographic type and therapy.Q J Med.1985;55(218):253–270.
- Statement on sarcoidosis.Joint Statement of the American Thoracic Society (ATS), theEuropean Respiratory Society (ERS) and theWorld Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999.Am J Respir Crit Care Med.1999;160:736–755.
- ,,.Value of serial measurement of serum angiotensin converting enzyme in the management of sarcoidosis.Am J Med.1981;70(1):44–50.
- ,,,.Serum angiotensin‐converting enzyme (SACE) activity as an indicator of total body granuloma load and prognosis in sarcoidosis.Sarcoidosis.1987;4(2):142–148.
- ,,.Serum angiotensin converting enzyme in sarcoidosis: clinical significance.Isr J Med Sci.1977;13:1001–1006.
- ,,.Blastomycosis.Infect Dis Clin North Am.2003;17(1):21,40, vii.
- ,,,,,.Novel cases of blastomycosis acquired in Toronto, Ontario.CMAJ.2000;163:1309–1312.
- ,,,,,.Blastomycosis acquired by three children in Toronto.Can J Infect Dis Med Micro.2002;13(4):259–263.
- ,,, et al.Blastomycosis in Ontario, 1994‐2003.Emerg Infect Dis.2006;12(2):274–279.
- ,,, et al.Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals.Clin Infect Dis.2002;34:1310–1316.
- ,,,.Meningoencephalitis due to Blastomyces dermatitidis: case report and literature review.Mayo Clin Proc.2000;75:403–408.
- ,,,.Chronic blastomycotic meningitis.Am J Med.1981;71:501–505.
- ,.Pulmonary blastomycosis: an appraisal of diagnostic techniques.Chest.2002;121:768–773.
- ,,.Blastomycosis as an etiology of acute lung injury.South Med J.1998;91:861–863.
- ,,.Risk of infectious complications in patients taking glucocorticosteroids.Rev Infect Dis.1989;11:954–963.
- .Elevation of serum angiotensin‐converting‐enzyme (ACE) level in sarcoidosis.Am J Med.1975;59:365–372.
- ,,,.Elevated serum angiotensin I converting enzyme in sarcoidosis.Am Rev Respir Dis.1976;114:525–528.
- ,,.Serum angiotensin‐converting enzyme (SACE) in sarcoidosis and other granulomatous disorders.Lancet.1978;2:1331–1334.
- ,.Serum angiotensin converting enzyme in sarcoidosis: sensitivity and specificity in diagnosis: correlations with disease activity, duration, extra‐thoracic involvement, radiographic type and therapy.Q J Med.1985;55(218):253–270.
- Statement on sarcoidosis.Joint Statement of the American Thoracic Society (ATS), theEuropean Respiratory Society (ERS) and theWorld Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999.Am J Respir Crit Care Med.1999;160:736–755.
- ,,.Value of serial measurement of serum angiotensin converting enzyme in the management of sarcoidosis.Am J Med.1981;70(1):44–50.
- ,,,.Serum angiotensin‐converting enzyme (SACE) activity as an indicator of total body granuloma load and prognosis in sarcoidosis.Sarcoidosis.1987;4(2):142–148.
- ,,.Serum angiotensin converting enzyme in sarcoidosis: clinical significance.Isr J Med Sci.1977;13:1001–1006.
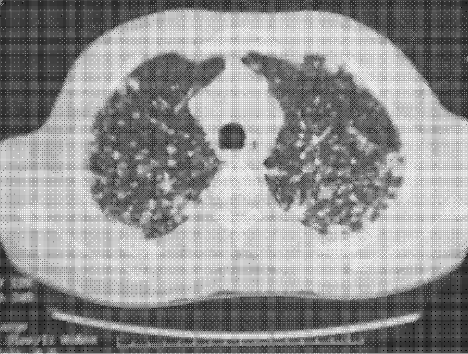
Clinical Conundrum
A 47‐year‐old woman was brought to the emergency department by her family because of 1 week of abdominal pain. The pain had begun in the epigastrium but had spread across the abdomen. She described it as constant and 10 of 10 in intensity but could not identify aggravating or alleviating factors. She also complained of nausea and vomiting, beginning 4 days prior to presentation, occurring 25 times per day. She noted poor oral intake and mild diarrhea. She denied melena or hematochezia. She reported no recent fever, dysuria, chills, or night sweats; however, she reported upper respiratory symptoms 2 weeks prior to presentation. On the day of presentation, her family felt she was becoming increasingly lethargic.
Epigastric pain in a middle‐aged woman suggests several possible diagnoses. Conditions such as acute cholecystitis begin abruptly, whereas small bowel obstruction, appendicitis, and diverticulitis start gradually. Nausea and vomiting are common concomitants of abdominal pain and are nonspecific. The absence of fever and chills is reassuring. Of greatest concern is the mental status. Initially, I think of enterohemorrhagic E. coli syndromes with associated glomerulonephritis. With a more systemic metabolic abnormality such as this, the rapid development of the disease tends to exaggerate symptoms.
The patient had a history of nephrolithiasis and underwent total abdominal hysterectomy and bilateral salpingo‐oopherectomy secondary to uterine fibroids in the past. She took occasional acetaminophen, smoked two cigarettes per day, and rarely consumed alcohol. Temperature was 38.5C, heart rate was 160 beats/minute, respiratory rate was 28/minute, and blood pressure was 92/52 mm Hg; oxygen saturation was 100% breathing 2 L of oxygen by nasal cannula. She was a moderately obese African American woman in moderate distress, lying in bed moaning. Mucous membranes were dry. There was no lymphadenopathy or thyromegaly. Heart rate was regular without appreciable murmur, rub, or gallop. Lungs were clear. Abdomen was soft and nondistended, with diffuse tenderness to palpation; bowel sounds were present; there was no rebound or guarding. She had normal rectal tone with brown, guaiac‐negative stool. There was no costovertebral angle tenderness. She was oriented to person, place, and time but lethargic; deep tendon reflexes were 3+ bilaterally, and no focal signs were elicited.
Renal stones certainly produce abdominal pain, and the rare patient undergoes laparotomy for this reason. The hysterectomy tells us that small bowel obstruction could be a reason for her symptoms, although abnormal mental status would not be expected without additional problems such as infection. The tachycardia seems out of proportion to her temperature. Hyperpnea and absent respiratory symptoms, along with hypotension and tachycardia, suggest a sepsis syndrome. Her physical exam confirms dehydration. Examination of the abdomen makes me speculate about whether she has a nonsurgical cause of acute abdomen. The lethargy remains unexplained. Sepsis syndrome, possibly from a perinephric abscess, is my leading diagnosis.
White blood cell count was 15.9/mm3 with 78% neutrophils, a hemoglobin of 14.3 g/dL with a MCV of 76 and a platelet count of 320/mm3. Sodium was 159 mmol/L, chloride 128 mmol/L, bicarbonate 19 mmol/L, blood urea nitrogen 120 mmol/L, creatinine 3.1 mg/dL, calcium 11.7 mg/dL, albumin 3.3 g/dL, serum aspartate aminotransferase 65 U/L, serum alanine aminotransferase 72 U/L, total bilirubin 0.7 mg/dL, amylase 137 U/L (normal 30100), and lipase 92 IU/dL (normal 424). Urine obtained from a Foley catheter revealed negative nitrite and leukocyte esterase, 5075 red blood cells, and 1025 white blood cells per high‐powered field.
The elevated serum sodium is likely contributing to her abnormal mental status. It is unusual for a previously healthy and conscious woman to become this hypernatremic because persons with a normal mental status will defend their sodium balance strenuously, assuming regulatory mechanisms are intact. Generally, this level of hypernatremia indicates 2 things. One, a patient was not allowed, or did not seek access to, free water. The other is the presence of diabetes insipidus. It is unlikely she became this dehydrated from the initial gastrointestinal episode as described. The low MCV suggests she may be a thalassemia carrier, as microcytosis with iron deficiency typically does not occur until the patient is anemic, although she may be when rehydrated. Serum calcium, while elevated, also will likely return to the normal range with hydration. The metabolic abnormalities strongly suggest a problem in the central nervous system. The hematuria in the urinalysis continues to raise the possibility of nephrolithiasis as a cause of abdominal pain, though it does not fit well with the rest of the patient's clinical picture. The hematuria and pyuria both could still indicate a urinary tract infection such as pyelonephritis or perinephric abscess causing a sepsis syndrome.
An acute abdominal series and chest radiograph revealed a paucity of gas in the abdomen but no free air under the diaphragm or active cardiopulmonary disease. Abdominal ultrasound showed cholelithiasis without biliary dilation. There was no evidence of hydronephrosis, hydroureter, or perinephric abscess. A noncontrast abdominal‐pelvic computed tomography (CT) scan demonstrated no peripancreatic stranding or fluid collection and no nephrolithiasis or fluid collection suggestive of abscess. The admission electrocardiogram, read as sinus tachycardia with a rate of 160, is displayed in Figure 1.
I have long believed that unexplained sinus tachycardia is one of the most ominous rhythms in clinical medicine; it is expected after vigorous exercise, among other situations, but not in the condition in which this woman finds herself. The nature of the tracing does not indicate the likelihood of a supraventricular arrhythmia, particularly atrial flutter, which should be considered given the rate. The absence of free air under the diaphragm on chest radiography is reassuring. Though the pancreatic enzymes are mildly elevated, they are usually far more striking in gallstone pancreatitis. Hypercalcemia may result in abdominal pain by several mechanisms. I remain concerned about her central nervous system.
The patient was admitted to the intensive care unit (ICU), where she received intravenous antibiotics and aggressive rehydration. The following morning, she continued to complain of abdominal pain. Her systolic blood pressure was 115 mmHg, and her heart rate ranged between 140 and 150 beats/minute. The remainder of her physical exam was unchanged. Repeat laboratory tests revealed a white blood cell count of 14.7/mm3, a blood urea nitrogen of 66 mg/dL, a creatinine of 1.3 mg.dL, amylase of 67 IU/L, and lipase of 70 IU/dL. A contrast‐enhanced abdominal‐pelvic CT scan did not reveal intra‐abdominal pathology. Blood and urine cultures obtained at admission were negative for any growth.
The patient was appropriately admitted to the ICU. When caring for a critically ill patient, establishing a diagnosis is less important initially than addressing treatable conditions with dispatch. The negative CT scans rule out previously entertained diagnoses like nephrolithiasis and perinephric abscess. It is possible that the initially positive urinalysis was a result of urinary catheter placement trauma. Given the course to date, I believe this patient likely has a nonsurgical cause of abdominal pain. I am considering entities such as lead intoxication, hypercalcemia, a tear of the rectus abdominus caused by vomiting, systemic vasculitis, or a hypercoagulable state leading to intra‐abdominal venous thrombosis.
By hospital day 3 her sodium decreased to 149 mmol/L and her creatinine was 1.0 mg/dL. Abdominal pain persisted, unchanged from admission. Her systolic blood pressure had stabilized at 120 mmHg, but the heart rate remained near 150 beats/minute. Her abdomen remained soft and nondistended on exam but diffusely tender to palpation. Her amylase and lipase continued to decrease, and her repeat electrocardiogram demonstrated tachycardia with a rate of 144.
We are gratified to see that her serum sodium has waned but not with the persistence of the tachycardia. It must be assumed that this patient has an infectious disease that we are not clever enough to diagnose at this time. I am also considering an autoimmune process, such as systemic lupus erythematosus. It is difficult to envision a neoplastic disorder causing these problems. The differential remains broad, however, because we have not ruled out metabolic or endocrine causes. It is difficult to imagine she could have Addison's diseasea common cause of severe abdominal pain, tachycardia, and hypotensiongiven her serum sodium level. Hyperthyroidism has been known to produce mild hypercalcemia and abdominal complaints and is an intriguing possibility. The striking elevation of her serum sodium makes me consider the possibility of a problem in the posterior pituitary gland such as sarcoidosis. I cannot explain how sarcoidosis would cause her abdominal pain, unless the hypercalcemia were related. The tachycardia remains of concern, especially if she is otherwise improving. Thus, I would likely administer a small dose of adenosine to ascertain that this is not a different supraventricular tachycardia. In sinus tachycardia, the rate is usually attendant to the clinical picture and thus begs explanation given her clinical improvement.
After receiving 6 mg of intravenous adenosine, the patient's heart rate declined; atrial flutter waves were observed.
This case nicely demonstrates a key teaching point: a fast regular heart rate of about 150, irrespective of the electrocardiogram, suggests atrial flutter. Who gets atrial flutter? Patients with chronic lung disease, myocardial ischemia (albeit rarely), alcohol‐induced cardiomyopathy, and infiltrative cardiac disorders do. Additionally, we also have to consider thyroid dysfunction.
If forced to come up with a single unifying diagnosis at this point, I would have to say this patient most likely has sarcoidosis because this entity would account for modest hypercalcemia, the myocardial conduction disturbance, and hypernatremia because of diabetes insipidus; furthermore, it would fit the patient's demographic profile. However, I am also concerned about hyperthyroidism and would not proceed until thyroid function studies were obtained.
Thyroid studies revealed thyroid stimulating hormone of less than 0.01 mU/L (normal range, 0.305.50), free thyroxine (T4) of 5.81 ng.dL (normal range, 0.731.79), free triiodothyronine (T3) of 15.7 pg/mL (normal range, 2.85.3), and total triiodothyronine (T3) of 218 ng/dL (normal range, 95170). The patient was diagnosed with thyroid crisis and was started on propranolol, propylthiouracil, hydrocortisone, and a saturated solution of potassium iodine. Thyroid stimulating immunoglobulins were obtained and found to be markedly elevated (3.4 TSI index; normal 1.3), suggestive of Grave's disease. Over the next several days, the patient's abdominal pain and tachycardia resolved. Her mental status returned to normal. A workup for her microcytic anemia revealed beta thalassemia trait. The patient was discharged home on hospital day 9 and has done well as an outpatient.
COMMENTARY
As Sir Zachary Cope stated in his classic text Cope's Early Diagnosis of the Acute Abdomen, [I]t is only by thorough history taking and physical examination that one can propound a diagnosis.1 When first presented with a patient whose chief complaint is abdominal pain, physicians tend to focus on the disorders of both the hollow and solid organs of the abdomen as potential sources of the pain. The differential diagnosis traditionally includes disorders such as cholecystitis, peptic ulcer disease, pancreatitis, small bowel obstruction, bowel ischemia or perforation, splenic abscess and infarct, nephrolithiasis, diverticulitis, and appendicitis, all of which were initially considered by the clinicians involved in this case. But as our discussant pointed out, in this case the differential needed to be broadened to include less common disorders, particularly given the patient's altered mental status, numerous electrolyte abnormalities, and lethargy and the lack of explanation provided by the physical examination and sophisticated imaging studies.
Specifically, a myriad of systemic diseases and metabolic derangements can cause abdominal complaints and mimic surgical abdominal disease, including hypercalcemia, acute intermittent porphyria, diabetic ketoacidosis, lead intoxication, familial Mediterranean fever, vasculopathies, adrenal insufficiency, and hyperthyroidism. Unfortunately, the frequency with which abdominal pain occurs in many of these less common disease processes and the pathophysiology that underlies its occurrence are not well defined. For example, abdominal pain is well described as a typical manifestation of both diabetic ketoacidosis and lead poisoning, but the pathophysiology behind its occurrence is poorly understood in both. Further, as a manifestation of thyrotoxicosis and as one of the diagnostic criteria for thyroid storm, the reported prevalence of abdominal pain in this condition is variable, ranging from rare to 20%47%.24 Also, although other gastrointestinal manifestations of hyperthyroidism (such as nausea, vomiting, and hyperdefecation) are thought to be the result of the effect of excess thyroid hormone on gastrointestinal motility, it is unclear whether this similar mechanism is responsible for the perception of abdominal pain.4
An important clue to the underlying diagnosis in this case was the patient's marked tachycardia. Classically, a persistent heart rate of 150 should raise suspicion of atrial flutter with a 2:1 conduction block, as was eventually discovered in this case. Adenosine, in addition to other vagal maneuvers such as carotid massage or Valsalva that also block atrioventricular (AV) node conduction, has been recognized as a safe and effective means of establishing a diagnosis in tachyarrhythmias.5 In AV nodal‐dependent tachycardias, such as AV node reentrant tachycardia or AV reentrant tachycardia, adenosine will often terminate the tachyarrhythmia by blocking the anterograde limb of the reentrant circuit. In AV nodeindependent tachyarrhythmias, such as atrial flutter or atrial fibrillation, adenosine will not terminate the rhythm. However, in the case of flutter, blocking the AV node will usually transiently unmask the underlying P waves, thereby facilitating the diagnosis.5, 6
In this patient, the discovery of atrial flutter was the main clue that thyrotoxicosis may provide the unifying diagnosis. Thyroid hormone has a direct positive cardiac chronotropic effect, resulting in the increased resting heart characteristic of thyrotoxicosis. Specifically, this hormone increases sinoatrial‐node firing, shortens the refractory period of conduction tissue within the heart, and decreases the electrical threshold for atrial excitation. In addition to predisposing to sinus tachycardia (the most common rhythm associated with this disorder), thyrotoxicosis is also associated with atrial tachycardias such as atrial flutter and, more classically, atrial fibrillation.7, 8 Though no studies have specifically evaluated the incidence of atrial flutter in thyrotoxicosis, atrial fibrillation has been found in 9%22% of these patients.7
Finally, several of the patient's electrolyte derangements could explain some of her clinical findings and are clues to the underlying diagnosis. She initially presented with a mild hypercalcemia that persisted even after hydration. Potential explanations include her severe dehydration or her underlying thyrotoxicosis because hypercalcemia is present in up to 20% of patients with hyperthyroidism.9, 10 However, the presence of significant hypercalcemia in the setting of thyrotoxicosis may actually make the diagnosis of thyrotoxicosis more difficult, masking the hypermetabolic signs and symptoms of the hyperthyroid state.11 Interestingly, coexistent primary hyperparathyroidism does occur in a few of these patients, but it likely was not an underlying cause in our patient given that her calcium normalized after receipt of propylthiouracil therapy.12
The patient's marked hypernatremia is more difficult to explain. She may have developed nephrogenic diabetes insipidus secondary to hypercalcemia, explained by a renal concentrating defect that can become evident once the calcium is persistently above 11 mg/dL.13 Combined with her altered mental status, which likely limited her ability to access free water, this may be enough to explain her marked hypernatremia. Her rapid improvement with rehydration is also consistent with this explanation, mediated through the improvement of her serum free calcium.
This case highlights the importance of using all the clinical clues provided by the history, physical exam, and laboratory and imaging studies when generating an initial differential diagnosis, as well as the importance of being willing to appropriately broaden and narrow the list of possibilities as a case evolves. When this patient was initially evaluated by physicians in the emergency department, they believed her symptoms were most consistent with generalized peritonitis that was likely secondary to an infectious or inflammatory intra‐abdominal process such as pancreatitis (especially in light of her mildly elevated lipase and amylase), appendicitis, or diverticulitis. When the medical team in the intensive care unit assumed care of this patient, members of the team failed to recognize several of the early clues, including the patient's markedly abnormal mental status, electrolyte derangements, and persistent tachycardia despite aggressive rehydration, which suggested the possibility of alternative, and less common, etiologies of her abdominal pain. Instead, they continued to aggressively pursue the possibility of the initial differential diagnosis, even repeating some of the previously negative studies from the emergency department. This case illustrates the importance of constantly reevaluating the available information from physical examination and laboratory and imaging studies and not falling victim to intellectual blind spots created by suggested diagnoses by other care providers. Fortunately for this patient, her thyroid crisis was diagnosed, albeit with some delay, before any long‐term complications occurred.
- Silen W, ed.Cope's Early Diagnosis of the Acute Abdomen.19th ed.New York:Oxford University Press;1995:4.
- ,.An unusual cause of abdominal pain in young woman.Ann Emerg Med.1991;20:574–582.
- .Vomiting, nausea and abdominal pain: unrecognized symptoms of thyrotoxicosis.J Fam Prac.1989;24:382–386.
- Powell DW,Alpers DH,Yamada,Owyang C,Laine L, eds.Textbook of Gastroenterology.3rd ed.Philadelphia, Pa:Lippincott Williams 783,2516.
- ,,.Usefulness of adenosine in diagnosis of tachyarrhythmias.Am J Cardiol.1995;75:952–955.
- ,,,,.Supraventricular tachycardia.Med Clin North Am.2001;85:193–223.
- .Thyrotoxicosis and the heart.N Engl J Med.1992;327:94–8.
- ,.Thyrotoxicosis and the heart.Endocrinol Metab Clin North Am.1998;27:51–62.
- ,,,.Treatment of thyrotoxic hypercalcemia with propranolol.N Engl J Med.1976;294:431.
- ,,,.Ionized and total plasma calcium and parathyroid hormone in hyperthyroidism.Ann Intern Med.1976;84:668.
- ,.Hypercalcemic crisis.Med Clin North Am.1995;79:79–92.
- ,,.Thyrotoxicosis, hypercalcemia, and secondary hyperparathyroidism.Arch Intern Med.1979;139:661–663.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
A 47‐year‐old woman was brought to the emergency department by her family because of 1 week of abdominal pain. The pain had begun in the epigastrium but had spread across the abdomen. She described it as constant and 10 of 10 in intensity but could not identify aggravating or alleviating factors. She also complained of nausea and vomiting, beginning 4 days prior to presentation, occurring 25 times per day. She noted poor oral intake and mild diarrhea. She denied melena or hematochezia. She reported no recent fever, dysuria, chills, or night sweats; however, she reported upper respiratory symptoms 2 weeks prior to presentation. On the day of presentation, her family felt she was becoming increasingly lethargic.
Epigastric pain in a middle‐aged woman suggests several possible diagnoses. Conditions such as acute cholecystitis begin abruptly, whereas small bowel obstruction, appendicitis, and diverticulitis start gradually. Nausea and vomiting are common concomitants of abdominal pain and are nonspecific. The absence of fever and chills is reassuring. Of greatest concern is the mental status. Initially, I think of enterohemorrhagic E. coli syndromes with associated glomerulonephritis. With a more systemic metabolic abnormality such as this, the rapid development of the disease tends to exaggerate symptoms.
The patient had a history of nephrolithiasis and underwent total abdominal hysterectomy and bilateral salpingo‐oopherectomy secondary to uterine fibroids in the past. She took occasional acetaminophen, smoked two cigarettes per day, and rarely consumed alcohol. Temperature was 38.5C, heart rate was 160 beats/minute, respiratory rate was 28/minute, and blood pressure was 92/52 mm Hg; oxygen saturation was 100% breathing 2 L of oxygen by nasal cannula. She was a moderately obese African American woman in moderate distress, lying in bed moaning. Mucous membranes were dry. There was no lymphadenopathy or thyromegaly. Heart rate was regular without appreciable murmur, rub, or gallop. Lungs were clear. Abdomen was soft and nondistended, with diffuse tenderness to palpation; bowel sounds were present; there was no rebound or guarding. She had normal rectal tone with brown, guaiac‐negative stool. There was no costovertebral angle tenderness. She was oriented to person, place, and time but lethargic; deep tendon reflexes were 3+ bilaterally, and no focal signs were elicited.
Renal stones certainly produce abdominal pain, and the rare patient undergoes laparotomy for this reason. The hysterectomy tells us that small bowel obstruction could be a reason for her symptoms, although abnormal mental status would not be expected without additional problems such as infection. The tachycardia seems out of proportion to her temperature. Hyperpnea and absent respiratory symptoms, along with hypotension and tachycardia, suggest a sepsis syndrome. Her physical exam confirms dehydration. Examination of the abdomen makes me speculate about whether she has a nonsurgical cause of acute abdomen. The lethargy remains unexplained. Sepsis syndrome, possibly from a perinephric abscess, is my leading diagnosis.
White blood cell count was 15.9/mm3 with 78% neutrophils, a hemoglobin of 14.3 g/dL with a MCV of 76 and a platelet count of 320/mm3. Sodium was 159 mmol/L, chloride 128 mmol/L, bicarbonate 19 mmol/L, blood urea nitrogen 120 mmol/L, creatinine 3.1 mg/dL, calcium 11.7 mg/dL, albumin 3.3 g/dL, serum aspartate aminotransferase 65 U/L, serum alanine aminotransferase 72 U/L, total bilirubin 0.7 mg/dL, amylase 137 U/L (normal 30100), and lipase 92 IU/dL (normal 424). Urine obtained from a Foley catheter revealed negative nitrite and leukocyte esterase, 5075 red blood cells, and 1025 white blood cells per high‐powered field.
The elevated serum sodium is likely contributing to her abnormal mental status. It is unusual for a previously healthy and conscious woman to become this hypernatremic because persons with a normal mental status will defend their sodium balance strenuously, assuming regulatory mechanisms are intact. Generally, this level of hypernatremia indicates 2 things. One, a patient was not allowed, or did not seek access to, free water. The other is the presence of diabetes insipidus. It is unlikely she became this dehydrated from the initial gastrointestinal episode as described. The low MCV suggests she may be a thalassemia carrier, as microcytosis with iron deficiency typically does not occur until the patient is anemic, although she may be when rehydrated. Serum calcium, while elevated, also will likely return to the normal range with hydration. The metabolic abnormalities strongly suggest a problem in the central nervous system. The hematuria in the urinalysis continues to raise the possibility of nephrolithiasis as a cause of abdominal pain, though it does not fit well with the rest of the patient's clinical picture. The hematuria and pyuria both could still indicate a urinary tract infection such as pyelonephritis or perinephric abscess causing a sepsis syndrome.
An acute abdominal series and chest radiograph revealed a paucity of gas in the abdomen but no free air under the diaphragm or active cardiopulmonary disease. Abdominal ultrasound showed cholelithiasis without biliary dilation. There was no evidence of hydronephrosis, hydroureter, or perinephric abscess. A noncontrast abdominal‐pelvic computed tomography (CT) scan demonstrated no peripancreatic stranding or fluid collection and no nephrolithiasis or fluid collection suggestive of abscess. The admission electrocardiogram, read as sinus tachycardia with a rate of 160, is displayed in Figure 1.
I have long believed that unexplained sinus tachycardia is one of the most ominous rhythms in clinical medicine; it is expected after vigorous exercise, among other situations, but not in the condition in which this woman finds herself. The nature of the tracing does not indicate the likelihood of a supraventricular arrhythmia, particularly atrial flutter, which should be considered given the rate. The absence of free air under the diaphragm on chest radiography is reassuring. Though the pancreatic enzymes are mildly elevated, they are usually far more striking in gallstone pancreatitis. Hypercalcemia may result in abdominal pain by several mechanisms. I remain concerned about her central nervous system.
The patient was admitted to the intensive care unit (ICU), where she received intravenous antibiotics and aggressive rehydration. The following morning, she continued to complain of abdominal pain. Her systolic blood pressure was 115 mmHg, and her heart rate ranged between 140 and 150 beats/minute. The remainder of her physical exam was unchanged. Repeat laboratory tests revealed a white blood cell count of 14.7/mm3, a blood urea nitrogen of 66 mg/dL, a creatinine of 1.3 mg.dL, amylase of 67 IU/L, and lipase of 70 IU/dL. A contrast‐enhanced abdominal‐pelvic CT scan did not reveal intra‐abdominal pathology. Blood and urine cultures obtained at admission were negative for any growth.
The patient was appropriately admitted to the ICU. When caring for a critically ill patient, establishing a diagnosis is less important initially than addressing treatable conditions with dispatch. The negative CT scans rule out previously entertained diagnoses like nephrolithiasis and perinephric abscess. It is possible that the initially positive urinalysis was a result of urinary catheter placement trauma. Given the course to date, I believe this patient likely has a nonsurgical cause of abdominal pain. I am considering entities such as lead intoxication, hypercalcemia, a tear of the rectus abdominus caused by vomiting, systemic vasculitis, or a hypercoagulable state leading to intra‐abdominal venous thrombosis.
By hospital day 3 her sodium decreased to 149 mmol/L and her creatinine was 1.0 mg/dL. Abdominal pain persisted, unchanged from admission. Her systolic blood pressure had stabilized at 120 mmHg, but the heart rate remained near 150 beats/minute. Her abdomen remained soft and nondistended on exam but diffusely tender to palpation. Her amylase and lipase continued to decrease, and her repeat electrocardiogram demonstrated tachycardia with a rate of 144.
We are gratified to see that her serum sodium has waned but not with the persistence of the tachycardia. It must be assumed that this patient has an infectious disease that we are not clever enough to diagnose at this time. I am also considering an autoimmune process, such as systemic lupus erythematosus. It is difficult to envision a neoplastic disorder causing these problems. The differential remains broad, however, because we have not ruled out metabolic or endocrine causes. It is difficult to imagine she could have Addison's diseasea common cause of severe abdominal pain, tachycardia, and hypotensiongiven her serum sodium level. Hyperthyroidism has been known to produce mild hypercalcemia and abdominal complaints and is an intriguing possibility. The striking elevation of her serum sodium makes me consider the possibility of a problem in the posterior pituitary gland such as sarcoidosis. I cannot explain how sarcoidosis would cause her abdominal pain, unless the hypercalcemia were related. The tachycardia remains of concern, especially if she is otherwise improving. Thus, I would likely administer a small dose of adenosine to ascertain that this is not a different supraventricular tachycardia. In sinus tachycardia, the rate is usually attendant to the clinical picture and thus begs explanation given her clinical improvement.
After receiving 6 mg of intravenous adenosine, the patient's heart rate declined; atrial flutter waves were observed.
This case nicely demonstrates a key teaching point: a fast regular heart rate of about 150, irrespective of the electrocardiogram, suggests atrial flutter. Who gets atrial flutter? Patients with chronic lung disease, myocardial ischemia (albeit rarely), alcohol‐induced cardiomyopathy, and infiltrative cardiac disorders do. Additionally, we also have to consider thyroid dysfunction.
If forced to come up with a single unifying diagnosis at this point, I would have to say this patient most likely has sarcoidosis because this entity would account for modest hypercalcemia, the myocardial conduction disturbance, and hypernatremia because of diabetes insipidus; furthermore, it would fit the patient's demographic profile. However, I am also concerned about hyperthyroidism and would not proceed until thyroid function studies were obtained.
Thyroid studies revealed thyroid stimulating hormone of less than 0.01 mU/L (normal range, 0.305.50), free thyroxine (T4) of 5.81 ng.dL (normal range, 0.731.79), free triiodothyronine (T3) of 15.7 pg/mL (normal range, 2.85.3), and total triiodothyronine (T3) of 218 ng/dL (normal range, 95170). The patient was diagnosed with thyroid crisis and was started on propranolol, propylthiouracil, hydrocortisone, and a saturated solution of potassium iodine. Thyroid stimulating immunoglobulins were obtained and found to be markedly elevated (3.4 TSI index; normal 1.3), suggestive of Grave's disease. Over the next several days, the patient's abdominal pain and tachycardia resolved. Her mental status returned to normal. A workup for her microcytic anemia revealed beta thalassemia trait. The patient was discharged home on hospital day 9 and has done well as an outpatient.
COMMENTARY
As Sir Zachary Cope stated in his classic text Cope's Early Diagnosis of the Acute Abdomen, [I]t is only by thorough history taking and physical examination that one can propound a diagnosis.1 When first presented with a patient whose chief complaint is abdominal pain, physicians tend to focus on the disorders of both the hollow and solid organs of the abdomen as potential sources of the pain. The differential diagnosis traditionally includes disorders such as cholecystitis, peptic ulcer disease, pancreatitis, small bowel obstruction, bowel ischemia or perforation, splenic abscess and infarct, nephrolithiasis, diverticulitis, and appendicitis, all of which were initially considered by the clinicians involved in this case. But as our discussant pointed out, in this case the differential needed to be broadened to include less common disorders, particularly given the patient's altered mental status, numerous electrolyte abnormalities, and lethargy and the lack of explanation provided by the physical examination and sophisticated imaging studies.
Specifically, a myriad of systemic diseases and metabolic derangements can cause abdominal complaints and mimic surgical abdominal disease, including hypercalcemia, acute intermittent porphyria, diabetic ketoacidosis, lead intoxication, familial Mediterranean fever, vasculopathies, adrenal insufficiency, and hyperthyroidism. Unfortunately, the frequency with which abdominal pain occurs in many of these less common disease processes and the pathophysiology that underlies its occurrence are not well defined. For example, abdominal pain is well described as a typical manifestation of both diabetic ketoacidosis and lead poisoning, but the pathophysiology behind its occurrence is poorly understood in both. Further, as a manifestation of thyrotoxicosis and as one of the diagnostic criteria for thyroid storm, the reported prevalence of abdominal pain in this condition is variable, ranging from rare to 20%47%.24 Also, although other gastrointestinal manifestations of hyperthyroidism (such as nausea, vomiting, and hyperdefecation) are thought to be the result of the effect of excess thyroid hormone on gastrointestinal motility, it is unclear whether this similar mechanism is responsible for the perception of abdominal pain.4
An important clue to the underlying diagnosis in this case was the patient's marked tachycardia. Classically, a persistent heart rate of 150 should raise suspicion of atrial flutter with a 2:1 conduction block, as was eventually discovered in this case. Adenosine, in addition to other vagal maneuvers such as carotid massage or Valsalva that also block atrioventricular (AV) node conduction, has been recognized as a safe and effective means of establishing a diagnosis in tachyarrhythmias.5 In AV nodal‐dependent tachycardias, such as AV node reentrant tachycardia or AV reentrant tachycardia, adenosine will often terminate the tachyarrhythmia by blocking the anterograde limb of the reentrant circuit. In AV nodeindependent tachyarrhythmias, such as atrial flutter or atrial fibrillation, adenosine will not terminate the rhythm. However, in the case of flutter, blocking the AV node will usually transiently unmask the underlying P waves, thereby facilitating the diagnosis.5, 6
In this patient, the discovery of atrial flutter was the main clue that thyrotoxicosis may provide the unifying diagnosis. Thyroid hormone has a direct positive cardiac chronotropic effect, resulting in the increased resting heart characteristic of thyrotoxicosis. Specifically, this hormone increases sinoatrial‐node firing, shortens the refractory period of conduction tissue within the heart, and decreases the electrical threshold for atrial excitation. In addition to predisposing to sinus tachycardia (the most common rhythm associated with this disorder), thyrotoxicosis is also associated with atrial tachycardias such as atrial flutter and, more classically, atrial fibrillation.7, 8 Though no studies have specifically evaluated the incidence of atrial flutter in thyrotoxicosis, atrial fibrillation has been found in 9%22% of these patients.7
Finally, several of the patient's electrolyte derangements could explain some of her clinical findings and are clues to the underlying diagnosis. She initially presented with a mild hypercalcemia that persisted even after hydration. Potential explanations include her severe dehydration or her underlying thyrotoxicosis because hypercalcemia is present in up to 20% of patients with hyperthyroidism.9, 10 However, the presence of significant hypercalcemia in the setting of thyrotoxicosis may actually make the diagnosis of thyrotoxicosis more difficult, masking the hypermetabolic signs and symptoms of the hyperthyroid state.11 Interestingly, coexistent primary hyperparathyroidism does occur in a few of these patients, but it likely was not an underlying cause in our patient given that her calcium normalized after receipt of propylthiouracil therapy.12
The patient's marked hypernatremia is more difficult to explain. She may have developed nephrogenic diabetes insipidus secondary to hypercalcemia, explained by a renal concentrating defect that can become evident once the calcium is persistently above 11 mg/dL.13 Combined with her altered mental status, which likely limited her ability to access free water, this may be enough to explain her marked hypernatremia. Her rapid improvement with rehydration is also consistent with this explanation, mediated through the improvement of her serum free calcium.
This case highlights the importance of using all the clinical clues provided by the history, physical exam, and laboratory and imaging studies when generating an initial differential diagnosis, as well as the importance of being willing to appropriately broaden and narrow the list of possibilities as a case evolves. When this patient was initially evaluated by physicians in the emergency department, they believed her symptoms were most consistent with generalized peritonitis that was likely secondary to an infectious or inflammatory intra‐abdominal process such as pancreatitis (especially in light of her mildly elevated lipase and amylase), appendicitis, or diverticulitis. When the medical team in the intensive care unit assumed care of this patient, members of the team failed to recognize several of the early clues, including the patient's markedly abnormal mental status, electrolyte derangements, and persistent tachycardia despite aggressive rehydration, which suggested the possibility of alternative, and less common, etiologies of her abdominal pain. Instead, they continued to aggressively pursue the possibility of the initial differential diagnosis, even repeating some of the previously negative studies from the emergency department. This case illustrates the importance of constantly reevaluating the available information from physical examination and laboratory and imaging studies and not falling victim to intellectual blind spots created by suggested diagnoses by other care providers. Fortunately for this patient, her thyroid crisis was diagnosed, albeit with some delay, before any long‐term complications occurred.
A 47‐year‐old woman was brought to the emergency department by her family because of 1 week of abdominal pain. The pain had begun in the epigastrium but had spread across the abdomen. She described it as constant and 10 of 10 in intensity but could not identify aggravating or alleviating factors. She also complained of nausea and vomiting, beginning 4 days prior to presentation, occurring 25 times per day. She noted poor oral intake and mild diarrhea. She denied melena or hematochezia. She reported no recent fever, dysuria, chills, or night sweats; however, she reported upper respiratory symptoms 2 weeks prior to presentation. On the day of presentation, her family felt she was becoming increasingly lethargic.
Epigastric pain in a middle‐aged woman suggests several possible diagnoses. Conditions such as acute cholecystitis begin abruptly, whereas small bowel obstruction, appendicitis, and diverticulitis start gradually. Nausea and vomiting are common concomitants of abdominal pain and are nonspecific. The absence of fever and chills is reassuring. Of greatest concern is the mental status. Initially, I think of enterohemorrhagic E. coli syndromes with associated glomerulonephritis. With a more systemic metabolic abnormality such as this, the rapid development of the disease tends to exaggerate symptoms.
The patient had a history of nephrolithiasis and underwent total abdominal hysterectomy and bilateral salpingo‐oopherectomy secondary to uterine fibroids in the past. She took occasional acetaminophen, smoked two cigarettes per day, and rarely consumed alcohol. Temperature was 38.5C, heart rate was 160 beats/minute, respiratory rate was 28/minute, and blood pressure was 92/52 mm Hg; oxygen saturation was 100% breathing 2 L of oxygen by nasal cannula. She was a moderately obese African American woman in moderate distress, lying in bed moaning. Mucous membranes were dry. There was no lymphadenopathy or thyromegaly. Heart rate was regular without appreciable murmur, rub, or gallop. Lungs were clear. Abdomen was soft and nondistended, with diffuse tenderness to palpation; bowel sounds were present; there was no rebound or guarding. She had normal rectal tone with brown, guaiac‐negative stool. There was no costovertebral angle tenderness. She was oriented to person, place, and time but lethargic; deep tendon reflexes were 3+ bilaterally, and no focal signs were elicited.
Renal stones certainly produce abdominal pain, and the rare patient undergoes laparotomy for this reason. The hysterectomy tells us that small bowel obstruction could be a reason for her symptoms, although abnormal mental status would not be expected without additional problems such as infection. The tachycardia seems out of proportion to her temperature. Hyperpnea and absent respiratory symptoms, along with hypotension and tachycardia, suggest a sepsis syndrome. Her physical exam confirms dehydration. Examination of the abdomen makes me speculate about whether she has a nonsurgical cause of acute abdomen. The lethargy remains unexplained. Sepsis syndrome, possibly from a perinephric abscess, is my leading diagnosis.
White blood cell count was 15.9/mm3 with 78% neutrophils, a hemoglobin of 14.3 g/dL with a MCV of 76 and a platelet count of 320/mm3. Sodium was 159 mmol/L, chloride 128 mmol/L, bicarbonate 19 mmol/L, blood urea nitrogen 120 mmol/L, creatinine 3.1 mg/dL, calcium 11.7 mg/dL, albumin 3.3 g/dL, serum aspartate aminotransferase 65 U/L, serum alanine aminotransferase 72 U/L, total bilirubin 0.7 mg/dL, amylase 137 U/L (normal 30100), and lipase 92 IU/dL (normal 424). Urine obtained from a Foley catheter revealed negative nitrite and leukocyte esterase, 5075 red blood cells, and 1025 white blood cells per high‐powered field.
The elevated serum sodium is likely contributing to her abnormal mental status. It is unusual for a previously healthy and conscious woman to become this hypernatremic because persons with a normal mental status will defend their sodium balance strenuously, assuming regulatory mechanisms are intact. Generally, this level of hypernatremia indicates 2 things. One, a patient was not allowed, or did not seek access to, free water. The other is the presence of diabetes insipidus. It is unlikely she became this dehydrated from the initial gastrointestinal episode as described. The low MCV suggests she may be a thalassemia carrier, as microcytosis with iron deficiency typically does not occur until the patient is anemic, although she may be when rehydrated. Serum calcium, while elevated, also will likely return to the normal range with hydration. The metabolic abnormalities strongly suggest a problem in the central nervous system. The hematuria in the urinalysis continues to raise the possibility of nephrolithiasis as a cause of abdominal pain, though it does not fit well with the rest of the patient's clinical picture. The hematuria and pyuria both could still indicate a urinary tract infection such as pyelonephritis or perinephric abscess causing a sepsis syndrome.
An acute abdominal series and chest radiograph revealed a paucity of gas in the abdomen but no free air under the diaphragm or active cardiopulmonary disease. Abdominal ultrasound showed cholelithiasis without biliary dilation. There was no evidence of hydronephrosis, hydroureter, or perinephric abscess. A noncontrast abdominal‐pelvic computed tomography (CT) scan demonstrated no peripancreatic stranding or fluid collection and no nephrolithiasis or fluid collection suggestive of abscess. The admission electrocardiogram, read as sinus tachycardia with a rate of 160, is displayed in Figure 1.
I have long believed that unexplained sinus tachycardia is one of the most ominous rhythms in clinical medicine; it is expected after vigorous exercise, among other situations, but not in the condition in which this woman finds herself. The nature of the tracing does not indicate the likelihood of a supraventricular arrhythmia, particularly atrial flutter, which should be considered given the rate. The absence of free air under the diaphragm on chest radiography is reassuring. Though the pancreatic enzymes are mildly elevated, they are usually far more striking in gallstone pancreatitis. Hypercalcemia may result in abdominal pain by several mechanisms. I remain concerned about her central nervous system.
The patient was admitted to the intensive care unit (ICU), where she received intravenous antibiotics and aggressive rehydration. The following morning, she continued to complain of abdominal pain. Her systolic blood pressure was 115 mmHg, and her heart rate ranged between 140 and 150 beats/minute. The remainder of her physical exam was unchanged. Repeat laboratory tests revealed a white blood cell count of 14.7/mm3, a blood urea nitrogen of 66 mg/dL, a creatinine of 1.3 mg.dL, amylase of 67 IU/L, and lipase of 70 IU/dL. A contrast‐enhanced abdominal‐pelvic CT scan did not reveal intra‐abdominal pathology. Blood and urine cultures obtained at admission were negative for any growth.
The patient was appropriately admitted to the ICU. When caring for a critically ill patient, establishing a diagnosis is less important initially than addressing treatable conditions with dispatch. The negative CT scans rule out previously entertained diagnoses like nephrolithiasis and perinephric abscess. It is possible that the initially positive urinalysis was a result of urinary catheter placement trauma. Given the course to date, I believe this patient likely has a nonsurgical cause of abdominal pain. I am considering entities such as lead intoxication, hypercalcemia, a tear of the rectus abdominus caused by vomiting, systemic vasculitis, or a hypercoagulable state leading to intra‐abdominal venous thrombosis.
By hospital day 3 her sodium decreased to 149 mmol/L and her creatinine was 1.0 mg/dL. Abdominal pain persisted, unchanged from admission. Her systolic blood pressure had stabilized at 120 mmHg, but the heart rate remained near 150 beats/minute. Her abdomen remained soft and nondistended on exam but diffusely tender to palpation. Her amylase and lipase continued to decrease, and her repeat electrocardiogram demonstrated tachycardia with a rate of 144.
We are gratified to see that her serum sodium has waned but not with the persistence of the tachycardia. It must be assumed that this patient has an infectious disease that we are not clever enough to diagnose at this time. I am also considering an autoimmune process, such as systemic lupus erythematosus. It is difficult to envision a neoplastic disorder causing these problems. The differential remains broad, however, because we have not ruled out metabolic or endocrine causes. It is difficult to imagine she could have Addison's diseasea common cause of severe abdominal pain, tachycardia, and hypotensiongiven her serum sodium level. Hyperthyroidism has been known to produce mild hypercalcemia and abdominal complaints and is an intriguing possibility. The striking elevation of her serum sodium makes me consider the possibility of a problem in the posterior pituitary gland such as sarcoidosis. I cannot explain how sarcoidosis would cause her abdominal pain, unless the hypercalcemia were related. The tachycardia remains of concern, especially if she is otherwise improving. Thus, I would likely administer a small dose of adenosine to ascertain that this is not a different supraventricular tachycardia. In sinus tachycardia, the rate is usually attendant to the clinical picture and thus begs explanation given her clinical improvement.
After receiving 6 mg of intravenous adenosine, the patient's heart rate declined; atrial flutter waves were observed.
This case nicely demonstrates a key teaching point: a fast regular heart rate of about 150, irrespective of the electrocardiogram, suggests atrial flutter. Who gets atrial flutter? Patients with chronic lung disease, myocardial ischemia (albeit rarely), alcohol‐induced cardiomyopathy, and infiltrative cardiac disorders do. Additionally, we also have to consider thyroid dysfunction.
If forced to come up with a single unifying diagnosis at this point, I would have to say this patient most likely has sarcoidosis because this entity would account for modest hypercalcemia, the myocardial conduction disturbance, and hypernatremia because of diabetes insipidus; furthermore, it would fit the patient's demographic profile. However, I am also concerned about hyperthyroidism and would not proceed until thyroid function studies were obtained.
Thyroid studies revealed thyroid stimulating hormone of less than 0.01 mU/L (normal range, 0.305.50), free thyroxine (T4) of 5.81 ng.dL (normal range, 0.731.79), free triiodothyronine (T3) of 15.7 pg/mL (normal range, 2.85.3), and total triiodothyronine (T3) of 218 ng/dL (normal range, 95170). The patient was diagnosed with thyroid crisis and was started on propranolol, propylthiouracil, hydrocortisone, and a saturated solution of potassium iodine. Thyroid stimulating immunoglobulins were obtained and found to be markedly elevated (3.4 TSI index; normal 1.3), suggestive of Grave's disease. Over the next several days, the patient's abdominal pain and tachycardia resolved. Her mental status returned to normal. A workup for her microcytic anemia revealed beta thalassemia trait. The patient was discharged home on hospital day 9 and has done well as an outpatient.
COMMENTARY
As Sir Zachary Cope stated in his classic text Cope's Early Diagnosis of the Acute Abdomen, [I]t is only by thorough history taking and physical examination that one can propound a diagnosis.1 When first presented with a patient whose chief complaint is abdominal pain, physicians tend to focus on the disorders of both the hollow and solid organs of the abdomen as potential sources of the pain. The differential diagnosis traditionally includes disorders such as cholecystitis, peptic ulcer disease, pancreatitis, small bowel obstruction, bowel ischemia or perforation, splenic abscess and infarct, nephrolithiasis, diverticulitis, and appendicitis, all of which were initially considered by the clinicians involved in this case. But as our discussant pointed out, in this case the differential needed to be broadened to include less common disorders, particularly given the patient's altered mental status, numerous electrolyte abnormalities, and lethargy and the lack of explanation provided by the physical examination and sophisticated imaging studies.
Specifically, a myriad of systemic diseases and metabolic derangements can cause abdominal complaints and mimic surgical abdominal disease, including hypercalcemia, acute intermittent porphyria, diabetic ketoacidosis, lead intoxication, familial Mediterranean fever, vasculopathies, adrenal insufficiency, and hyperthyroidism. Unfortunately, the frequency with which abdominal pain occurs in many of these less common disease processes and the pathophysiology that underlies its occurrence are not well defined. For example, abdominal pain is well described as a typical manifestation of both diabetic ketoacidosis and lead poisoning, but the pathophysiology behind its occurrence is poorly understood in both. Further, as a manifestation of thyrotoxicosis and as one of the diagnostic criteria for thyroid storm, the reported prevalence of abdominal pain in this condition is variable, ranging from rare to 20%47%.24 Also, although other gastrointestinal manifestations of hyperthyroidism (such as nausea, vomiting, and hyperdefecation) are thought to be the result of the effect of excess thyroid hormone on gastrointestinal motility, it is unclear whether this similar mechanism is responsible for the perception of abdominal pain.4
An important clue to the underlying diagnosis in this case was the patient's marked tachycardia. Classically, a persistent heart rate of 150 should raise suspicion of atrial flutter with a 2:1 conduction block, as was eventually discovered in this case. Adenosine, in addition to other vagal maneuvers such as carotid massage or Valsalva that also block atrioventricular (AV) node conduction, has been recognized as a safe and effective means of establishing a diagnosis in tachyarrhythmias.5 In AV nodal‐dependent tachycardias, such as AV node reentrant tachycardia or AV reentrant tachycardia, adenosine will often terminate the tachyarrhythmia by blocking the anterograde limb of the reentrant circuit. In AV nodeindependent tachyarrhythmias, such as atrial flutter or atrial fibrillation, adenosine will not terminate the rhythm. However, in the case of flutter, blocking the AV node will usually transiently unmask the underlying P waves, thereby facilitating the diagnosis.5, 6
In this patient, the discovery of atrial flutter was the main clue that thyrotoxicosis may provide the unifying diagnosis. Thyroid hormone has a direct positive cardiac chronotropic effect, resulting in the increased resting heart characteristic of thyrotoxicosis. Specifically, this hormone increases sinoatrial‐node firing, shortens the refractory period of conduction tissue within the heart, and decreases the electrical threshold for atrial excitation. In addition to predisposing to sinus tachycardia (the most common rhythm associated with this disorder), thyrotoxicosis is also associated with atrial tachycardias such as atrial flutter and, more classically, atrial fibrillation.7, 8 Though no studies have specifically evaluated the incidence of atrial flutter in thyrotoxicosis, atrial fibrillation has been found in 9%22% of these patients.7
Finally, several of the patient's electrolyte derangements could explain some of her clinical findings and are clues to the underlying diagnosis. She initially presented with a mild hypercalcemia that persisted even after hydration. Potential explanations include her severe dehydration or her underlying thyrotoxicosis because hypercalcemia is present in up to 20% of patients with hyperthyroidism.9, 10 However, the presence of significant hypercalcemia in the setting of thyrotoxicosis may actually make the diagnosis of thyrotoxicosis more difficult, masking the hypermetabolic signs and symptoms of the hyperthyroid state.11 Interestingly, coexistent primary hyperparathyroidism does occur in a few of these patients, but it likely was not an underlying cause in our patient given that her calcium normalized after receipt of propylthiouracil therapy.12
The patient's marked hypernatremia is more difficult to explain. She may have developed nephrogenic diabetes insipidus secondary to hypercalcemia, explained by a renal concentrating defect that can become evident once the calcium is persistently above 11 mg/dL.13 Combined with her altered mental status, which likely limited her ability to access free water, this may be enough to explain her marked hypernatremia. Her rapid improvement with rehydration is also consistent with this explanation, mediated through the improvement of her serum free calcium.
This case highlights the importance of using all the clinical clues provided by the history, physical exam, and laboratory and imaging studies when generating an initial differential diagnosis, as well as the importance of being willing to appropriately broaden and narrow the list of possibilities as a case evolves. When this patient was initially evaluated by physicians in the emergency department, they believed her symptoms were most consistent with generalized peritonitis that was likely secondary to an infectious or inflammatory intra‐abdominal process such as pancreatitis (especially in light of her mildly elevated lipase and amylase), appendicitis, or diverticulitis. When the medical team in the intensive care unit assumed care of this patient, members of the team failed to recognize several of the early clues, including the patient's markedly abnormal mental status, electrolyte derangements, and persistent tachycardia despite aggressive rehydration, which suggested the possibility of alternative, and less common, etiologies of her abdominal pain. Instead, they continued to aggressively pursue the possibility of the initial differential diagnosis, even repeating some of the previously negative studies from the emergency department. This case illustrates the importance of constantly reevaluating the available information from physical examination and laboratory and imaging studies and not falling victim to intellectual blind spots created by suggested diagnoses by other care providers. Fortunately for this patient, her thyroid crisis was diagnosed, albeit with some delay, before any long‐term complications occurred.
- Silen W, ed.Cope's Early Diagnosis of the Acute Abdomen.19th ed.New York:Oxford University Press;1995:4.
- ,.An unusual cause of abdominal pain in young woman.Ann Emerg Med.1991;20:574–582.
- .Vomiting, nausea and abdominal pain: unrecognized symptoms of thyrotoxicosis.J Fam Prac.1989;24:382–386.
- Powell DW,Alpers DH,Yamada,Owyang C,Laine L, eds.Textbook of Gastroenterology.3rd ed.Philadelphia, Pa:Lippincott Williams 783,2516.
- ,,.Usefulness of adenosine in diagnosis of tachyarrhythmias.Am J Cardiol.1995;75:952–955.
- ,,,,.Supraventricular tachycardia.Med Clin North Am.2001;85:193–223.
- .Thyrotoxicosis and the heart.N Engl J Med.1992;327:94–8.
- ,.Thyrotoxicosis and the heart.Endocrinol Metab Clin North Am.1998;27:51–62.
- ,,,.Treatment of thyrotoxic hypercalcemia with propranolol.N Engl J Med.1976;294:431.
- ,,,.Ionized and total plasma calcium and parathyroid hormone in hyperthyroidism.Ann Intern Med.1976;84:668.
- ,.Hypercalcemic crisis.Med Clin North Am.1995;79:79–92.
- ,,.Thyrotoxicosis, hypercalcemia, and secondary hyperparathyroidism.Arch Intern Med.1979;139:661–663.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
- Silen W, ed.Cope's Early Diagnosis of the Acute Abdomen.19th ed.New York:Oxford University Press;1995:4.
- ,.An unusual cause of abdominal pain in young woman.Ann Emerg Med.1991;20:574–582.
- .Vomiting, nausea and abdominal pain: unrecognized symptoms of thyrotoxicosis.J Fam Prac.1989;24:382–386.
- Powell DW,Alpers DH,Yamada,Owyang C,Laine L, eds.Textbook of Gastroenterology.3rd ed.Philadelphia, Pa:Lippincott Williams 783,2516.
- ,,.Usefulness of adenosine in diagnosis of tachyarrhythmias.Am J Cardiol.1995;75:952–955.
- ,,,,.Supraventricular tachycardia.Med Clin North Am.2001;85:193–223.
- .Thyrotoxicosis and the heart.N Engl J Med.1992;327:94–8.
- ,.Thyrotoxicosis and the heart.Endocrinol Metab Clin North Am.1998;27:51–62.
- ,,,.Treatment of thyrotoxic hypercalcemia with propranolol.N Engl J Med.1976;294:431.
- ,,,.Ionized and total plasma calcium and parathyroid hormone in hyperthyroidism.Ann Intern Med.1976;84:668.
- ,.Hypercalcemic crisis.Med Clin North Am.1995;79:79–92.
- ,,.Thyrotoxicosis, hypercalcemia, and secondary hyperparathyroidism.Arch Intern Med.1979;139:661–663.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
Clinical Conundrum
A 46‐year‐old African American man presented to the emergency department with severe chest pain that awakened him from sleep. The pain was substernal, radiated to the neck and back, and was continuous, lasting approximately 1 hour. It was associated with nausea, diaphoresis, and dizziness. The patient denied dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysphagia, odynophagia, vomiting, fever, chills, or headache. He denied recent recreational drug use. He works as a landscaper.
Substernal chest pain radiating to the neck and back implicates structures in the middle mediastinum, chiefly the heart, aorta, esophagus, pulmonary arteries, and mediastinal pleura. The presence of nausea and diaphoresis suggests a vagal response to pain.
The sudden onset of symptoms and the lack of fevers and chills make infectious causes in the mediastinum such as mediastinitis and pneumonia less likely. Acute pulmonary embolism often presents with pleuritic pain and dyspnea, features not present in this patient.
The absence of odynophagia and dysphagia makes esophageal rupture or perforation of esophageal diverticula unlikely. Likewise, the absence of dyspnea, orthopnea, and paroxysmal nocturnal dyspnea makes it unlikely to be acute left ventricular failure from a sudden rise in left atrial pressure. Such a scenario may occur in the setting of a myocardial infarction or rupture of a papillary muscle, chordae tendineae, or sinus of Valsalva.
Dissection of the aorta with or without involvement of the aortic root merits strong consideration. Dissection involving the carotid or vertebral arteries could explain the patient's dizziness. Physical stresses in a landscaper may contribute to elevation in blood pressure and set the stage for an aortic dissection, especially with other risk factors.
The patient has a history of a positive PPD and was treated with isoniazid for 6 months. His mother had a history of hypertension and died from a myocardial infarction at age 64. His father's medical history is unknown. He has a history of alcohol abuse but has been abstinent for more than a year. He smoked marijuana and tobacco occasionally, with a 15 pack‐year cigarette history. He also stated that the last time he used cocaine was 1 year prior to admission.
That his mother succumbed to a myocardial infarction as well as having been hypertensive could be important family history risk factors given the patient's symptoms. Furthermore, the concurrent use of alcohol and tobacco by the patient increases his risk of developing severe hypertension. The use of cocaine is associated with sudden elevations in systemic blood pressure, which predispose to intimal damage in the aorta, especially if other risk factors are present. Marijuana smoking has been implicated in pulmonary aspergillosis, but this sudden presentation in the absence of pulmonary symptoms makes it most unlikely. Optimal therapy of a positive PPD should not predispose the patient to acute exudative pericarditis. Thus far the features suggest an acute vascular episode without significant compromise of cardiac output.
The patient was alert and in mild distress from chest discomfort. He was afebrile, with a blood pressure of 136/64 in the right arm and 139/63 in the left arm. Heart rate was 60 beats per minute, and the respiratory rate was 12 beats per minute, with an oxygen saturation of 99% while breathing room air. Examination of the head and neck revealed no signs of trauma. Jugular venous waveforms were normal, and carotid artery pulsations had normal strength and upstroke without audible bruits. The lungs were clear to auscultation. Heart sounds were notable for a 3/6 diastolic murmur heard best at the right sternal border. Rate and rhythm were regular, and the apical impulse was sustained but not displaced. The peripheral pulses were present and equal in quality throughout. The findings of the abdominal exam were normal, and the digital rectal examination was negative for occult blood. The findings of the neurologic, musculoskeletal, and dermatologic exams also were normal.
A slightly elevated pulse pressure without a significant difference in upper extremity blood pressure could be a result of aortic regurgitation, sinus of Valsalva rupture, or a high‐cardiac‐output state, as seen in thyrotoxicosis, anemia, or arteriovenous fistula.
The presence of a 3/6 diastolic murmur at the sternal border, however, favors conditions that cause aortic valve regurgitation and, less commonly, pulmonary valve regurgitation, or turbulent flow across the tricuspid valve. The murmur of pulmonary and aortic valve regurgitation can be difficult to distinguish by auscultation; however, the absence of other findings such as a right ventricular heave, elevated jugular venous pressure, or primary lung disease do not support a pulmonary valve etiology. Turbulent flow across the tricuspid valve can be seen in high‐output states, large atrial septal defects, and tricuspid stenosis. This type of murmur is heard best at the lower sternal border and tends to increase with inspiration. An early diastolic murmur would suggest aortic regurgitation, either from aortic valve or aortic root pathology. Aortic regurgitation would contribute to a sustained apical impulse.
Clear lungs and the absence of tachycardia suggest that left ventricle function is not severely compromised. These findings argue against acute rupture of the sinus of Valsalva, a condition that normally causes biventricular failure. The presence of equal peripheral pulses does not exclude the diagnosis of aortic dissection, as the dissection may have occurred proximal to the origins of the right innominate and the left subclavian arteries.
Initial laboratory studies revealed a hematocrit of 34.9, a leukocyte count of 6800/mm3, and a platelet count of 195 000/mm3. The levels of serum electrolytes, serum creatinine, blood urea nitrogen, and initial cardiac enzymes were normal. The urine drug screen was negative. The electrocardiogram showed evidence of left ventricular hypertrophy (Figure 1), and portable chest radiography revealed an enlarged cardiac silhouette and a widened mediastinum (Figure 2).
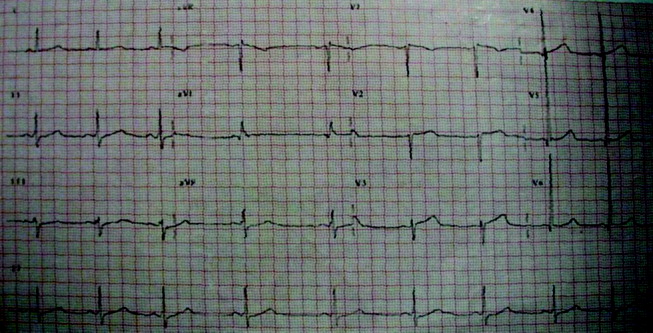
The presence of a widened mediastinum is consistent with aortic dissection but may also suggest a mass, aortic aneurysm, infiltrative disease, or a collection of fluid (eg, blood). A normal level of cardiac enzymes and the absence of ischemic findings on electrocardiography make myocardial infarction less likely. Given his hemodynamic stability, he is unlikely to have suffered cardiac rupture; however, he may still have a dissection of the aorta or a rupture of the sinus of Valsalva. The slight decline in hematocrit is compatible with either a mediastinal or pericardial collection of blood.
The patient was emergently sent for computerized axial tomography of the chest, which revealed a dilated aortic root at 6 cm but no evidence of aortic dissection (Figure 3). The lung fields were normal.
Despite a negative test, the presence of severe acute chest pain in the presence of a widened mediastinum is still concerning for aortic dissection. If the dissection involved the aortic valve annulus, the resulting acute regurgitation can not be severe, given the absence of left ventricular heart failure. Likewise, the presence of normal lung fields suggests the patient has no acute elevation of left ventricular end diastolic pressures such as is seen in ventricle septal defect, papillary muscle dysfunction, or acute mitral valvular lesion.
Cardiology consultants were emergently consulted and performed a transesophageal echocardiogram that confirmed a dilated aortic root (6.3 cm) without evidence of dissection. The patient was noted to have moderate to severe aortic regurgitation and a dilated left ventricle with moderate hypertrophy. A trace effusion was noted, and the left ventricular ejection fraction was estimated to be 50%.
The presence of moderate to severe aortic regurgitation with a dilated aortic root suggests 3 possibilities: undiagnosed dissection of the aortic root with preexisting aortic insufficiency (eg, bicuspid aortic valve, rheumatic valve disease, previous endocarditis); infectious (eg, syphilitic) or noninfectious (eg, ankylosing spondylitis, Takayasu's arteritis) meso‐aortitis causing aortic dilatation and subsequent regurgitation; or, finally, connective tissue diseases (eg, Marfan's syndrome, Ehlers‐Danlos), which can cause premature degeneration of the aortic media. The acuity of the patient's symptoms and the lack of systemic findings make a connective tissue or inflammatory disease unlikely. The clinical index of suspicion for aortic dissection needs to remain very high, as failure to make an expedient diagnosis may lead to complications and a deleterious outcome. Aortography may help to define this lesion.
The cardiothoracic surgical team was consulted and recommended aortic root and valve replacement. The patient was observed overnight and scheduled for preoperative cardiac catheterization the following morning. Aortogram revealed moderate to severe aortic insufficiency and a small dissection flap on the lesser curvature of the aorta, above the left coronary ostia (Figure 4). Coronary angiography revealed a 50% stenosis of the ostia of the first and second diagonal arteries, with no other flow‐limiting lesions.
The study has clearly demonstrated that the patient suffered an acute aortic dissection without involvement of the aortic annulus. Given the absence of left ventricular failure, it appears the aortic regurgitation was chronic and secondary to a previously existing aortic aneurysm. Asymptomatic congenital defects of the meso‐aorta such as cystic medical necrosis can predispose to aortic dissection at a relatively young age. However, this patient's condition may have been aggravated by drug abuse, paroxysmal elevation of blood pressure during landscaping, or other risk factors. Surgical correction of the dissection and aortic regurgitation is necessary.
The patient underwent aortic valve replacement with a 25‐mm St. Jude mechanical valve and an ascending aortic transection repair with a 32‐mm Dacron tube graft. On postoperative day 9, the patient was discharged home in stable condition.
COMMENTARY
Ensuring both accuracy and efficiency when making a diagnosis can be difficult, particularly when patients present in an atypical fashion or when diagnostic testing yields inconclusive results. Thus, a physician must sift through each clinical clue, remembering that although certain findings are pathognomonic for a disease process, a constellation of signs and symptoms when present can be equally diagnostic.
The initial likelihood of disease (ie, pretest probability) is generated by the history and physical and laboratory examinations. If the pretest probability is high and the subsequent diagnostic test is positive, the modified likelihood of disease (post‐test probability) is nearly 100%. If, however, the pretest probability is high and the diagnostic test is negative, the likelihood of disease is less clear. In these situations, the physician can either review the initial findings that generated the pretest probability or perform an additional diagnostic test of higher sensitivity.
In this exercise, a 46‐year‐old man presented to the emergency department with severe chest pain and findings characteristic of aortic dissection. The physicians appropriately sent him for chest computerized tomography (CT) because of a high pretest likelihood of aortic dissection. Because this test did not confirm the presence of aortic dissection, the patient underwent transesophageal echocardiography (TEE), a test of equal or greater sensitivity.1, 2 This test was also negative; however, a small dissection flap was subsequently found during cardiac catheterization. In this case, a test of lower sensitivity and specificity confirmed the diagnosis of dissection,3 demonstrating the possibility that either CT or TEE can be misinterpreted. Indeed, a final review of the TEE, showed the dissection flap, albeit small, had been missed. In this case, a diagnostic error was made that delayed the diagnosis when sufficient information was available earlier. Diagnostic errors are prevalent in medical practice and are commonly the result of numerous factors, though cognitive problems appear to be the largest contributor to this process.4 In particular, faulty synthesis of information, rather than inadequate medical knowledge, is the most common cause of cognitive medical errors. The error made in this case likely falls under the subcategory of faulty test detection or perception and subsequent premature closure (failure to consider other possibilities once an initial diagnosis of uncomplicated aortic aneurysm had been reached).4
Though medical errors cannot be completely eliminated, cases such as this should be reviewed to understand the cognitive processes that may lead to an erroneous diagnosis. In addition to the false‐negative TEE finding, this patient also had a coexisting condition that may have preoccupied the medical team. The thoracic aortic aneurysm seen by all imaging modalities was large and required intervention regardless of the presence of a dissection. However, this chronic condition became the focus of treatment, and the acute event that precipitated admission was missed. Perhaps if the primary team maintained a very high index of suspicion for dissection and conveyed this to the cardiology consultants, a meticulous review of the TEE would have then followed that may have uncovered the subtle findings of the dissection flap.
Fortunately, definitive treatment with surgical aortic root and valve replacement was performed in a timely manner, as the consequences of a delayed diagnosis in this situation could have been catastrophic. The mortality rate of a type A dissection is extremely high initially (1%‐2% per hour),5, 6 and thus surgical intervention is typically performed immediately after the diagnosis and the extent of this disease is established, rather than the following morning.7
This case highlights not only the problems resulting in diagnostic errors but also exemplifies the thought process required to make a challenging diagnosis. Our case discussant was able to avoid cognitive pitfalls by presenting a broad differential diagnosis and reevaluating the diagnosis with each additional piece of information provided.8 An experienced clinician should realize that patients with an extremely high pretest probability of disease and a negative diagnostic test should be further investigated, regardless of the test sensitivity. Furthermore, time‐honored methods such as history taking, physical examination. and thoughtful analyses should remain critical tools in the process of reaching an accurate diagnosis despite technological advances in diagnostic testing.
- ,,, et al.The diagnosis of thoracic aortic dissection by noninvasive imaging procedures.N Engl J Med.1993;328(1):1–9.
- ,,, et al.Accuracy of biplane and multiplane transesophageal echocardiography in diagnosis of typical acute aortic dissection and intramural hematoma.J Am Coll Cardiol.1996;28:627–636.
- ,.Aortic dissection: new frontiers in diagnosis and management: Part I: from etiology to diagnostic strategies.Circulation.2003;108:628–635.
- ,,.Diagnostic error in internal medicine.Arch Intern Med.2005;165:1493–1499.
- ,,.Dissecting aneurysm of the aorta: a review of 505 cases.Medicine (Baltimore).1958;37(3):217–279.
- ,,, et al.The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease.JAMA.2000;283:897–903.
- ,.Surgery of the thoracic aorta.N Engl J Med.1997;336:1876–1888.
- .Clinical reasoning in medicine. In:Higgs J,Jones MA, eds.Clinical Reasoning in the Health Professions.Woburn, Mass:Butterworth‐Heinemann;1995:49–59.
A 46‐year‐old African American man presented to the emergency department with severe chest pain that awakened him from sleep. The pain was substernal, radiated to the neck and back, and was continuous, lasting approximately 1 hour. It was associated with nausea, diaphoresis, and dizziness. The patient denied dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysphagia, odynophagia, vomiting, fever, chills, or headache. He denied recent recreational drug use. He works as a landscaper.
Substernal chest pain radiating to the neck and back implicates structures in the middle mediastinum, chiefly the heart, aorta, esophagus, pulmonary arteries, and mediastinal pleura. The presence of nausea and diaphoresis suggests a vagal response to pain.
The sudden onset of symptoms and the lack of fevers and chills make infectious causes in the mediastinum such as mediastinitis and pneumonia less likely. Acute pulmonary embolism often presents with pleuritic pain and dyspnea, features not present in this patient.
The absence of odynophagia and dysphagia makes esophageal rupture or perforation of esophageal diverticula unlikely. Likewise, the absence of dyspnea, orthopnea, and paroxysmal nocturnal dyspnea makes it unlikely to be acute left ventricular failure from a sudden rise in left atrial pressure. Such a scenario may occur in the setting of a myocardial infarction or rupture of a papillary muscle, chordae tendineae, or sinus of Valsalva.
Dissection of the aorta with or without involvement of the aortic root merits strong consideration. Dissection involving the carotid or vertebral arteries could explain the patient's dizziness. Physical stresses in a landscaper may contribute to elevation in blood pressure and set the stage for an aortic dissection, especially with other risk factors.
The patient has a history of a positive PPD and was treated with isoniazid for 6 months. His mother had a history of hypertension and died from a myocardial infarction at age 64. His father's medical history is unknown. He has a history of alcohol abuse but has been abstinent for more than a year. He smoked marijuana and tobacco occasionally, with a 15 pack‐year cigarette history. He also stated that the last time he used cocaine was 1 year prior to admission.
That his mother succumbed to a myocardial infarction as well as having been hypertensive could be important family history risk factors given the patient's symptoms. Furthermore, the concurrent use of alcohol and tobacco by the patient increases his risk of developing severe hypertension. The use of cocaine is associated with sudden elevations in systemic blood pressure, which predispose to intimal damage in the aorta, especially if other risk factors are present. Marijuana smoking has been implicated in pulmonary aspergillosis, but this sudden presentation in the absence of pulmonary symptoms makes it most unlikely. Optimal therapy of a positive PPD should not predispose the patient to acute exudative pericarditis. Thus far the features suggest an acute vascular episode without significant compromise of cardiac output.
The patient was alert and in mild distress from chest discomfort. He was afebrile, with a blood pressure of 136/64 in the right arm and 139/63 in the left arm. Heart rate was 60 beats per minute, and the respiratory rate was 12 beats per minute, with an oxygen saturation of 99% while breathing room air. Examination of the head and neck revealed no signs of trauma. Jugular venous waveforms were normal, and carotid artery pulsations had normal strength and upstroke without audible bruits. The lungs were clear to auscultation. Heart sounds were notable for a 3/6 diastolic murmur heard best at the right sternal border. Rate and rhythm were regular, and the apical impulse was sustained but not displaced. The peripheral pulses were present and equal in quality throughout. The findings of the abdominal exam were normal, and the digital rectal examination was negative for occult blood. The findings of the neurologic, musculoskeletal, and dermatologic exams also were normal.
A slightly elevated pulse pressure without a significant difference in upper extremity blood pressure could be a result of aortic regurgitation, sinus of Valsalva rupture, or a high‐cardiac‐output state, as seen in thyrotoxicosis, anemia, or arteriovenous fistula.
The presence of a 3/6 diastolic murmur at the sternal border, however, favors conditions that cause aortic valve regurgitation and, less commonly, pulmonary valve regurgitation, or turbulent flow across the tricuspid valve. The murmur of pulmonary and aortic valve regurgitation can be difficult to distinguish by auscultation; however, the absence of other findings such as a right ventricular heave, elevated jugular venous pressure, or primary lung disease do not support a pulmonary valve etiology. Turbulent flow across the tricuspid valve can be seen in high‐output states, large atrial septal defects, and tricuspid stenosis. This type of murmur is heard best at the lower sternal border and tends to increase with inspiration. An early diastolic murmur would suggest aortic regurgitation, either from aortic valve or aortic root pathology. Aortic regurgitation would contribute to a sustained apical impulse.
Clear lungs and the absence of tachycardia suggest that left ventricle function is not severely compromised. These findings argue against acute rupture of the sinus of Valsalva, a condition that normally causes biventricular failure. The presence of equal peripheral pulses does not exclude the diagnosis of aortic dissection, as the dissection may have occurred proximal to the origins of the right innominate and the left subclavian arteries.
Initial laboratory studies revealed a hematocrit of 34.9, a leukocyte count of 6800/mm3, and a platelet count of 195 000/mm3. The levels of serum electrolytes, serum creatinine, blood urea nitrogen, and initial cardiac enzymes were normal. The urine drug screen was negative. The electrocardiogram showed evidence of left ventricular hypertrophy (Figure 1), and portable chest radiography revealed an enlarged cardiac silhouette and a widened mediastinum (Figure 2).
The presence of a widened mediastinum is consistent with aortic dissection but may also suggest a mass, aortic aneurysm, infiltrative disease, or a collection of fluid (eg, blood). A normal level of cardiac enzymes and the absence of ischemic findings on electrocardiography make myocardial infarction less likely. Given his hemodynamic stability, he is unlikely to have suffered cardiac rupture; however, he may still have a dissection of the aorta or a rupture of the sinus of Valsalva. The slight decline in hematocrit is compatible with either a mediastinal or pericardial collection of blood.
The patient was emergently sent for computerized axial tomography of the chest, which revealed a dilated aortic root at 6 cm but no evidence of aortic dissection (Figure 3). The lung fields were normal.
Despite a negative test, the presence of severe acute chest pain in the presence of a widened mediastinum is still concerning for aortic dissection. If the dissection involved the aortic valve annulus, the resulting acute regurgitation can not be severe, given the absence of left ventricular heart failure. Likewise, the presence of normal lung fields suggests the patient has no acute elevation of left ventricular end diastolic pressures such as is seen in ventricle septal defect, papillary muscle dysfunction, or acute mitral valvular lesion.
Cardiology consultants were emergently consulted and performed a transesophageal echocardiogram that confirmed a dilated aortic root (6.3 cm) without evidence of dissection. The patient was noted to have moderate to severe aortic regurgitation and a dilated left ventricle with moderate hypertrophy. A trace effusion was noted, and the left ventricular ejection fraction was estimated to be 50%.
The presence of moderate to severe aortic regurgitation with a dilated aortic root suggests 3 possibilities: undiagnosed dissection of the aortic root with preexisting aortic insufficiency (eg, bicuspid aortic valve, rheumatic valve disease, previous endocarditis); infectious (eg, syphilitic) or noninfectious (eg, ankylosing spondylitis, Takayasu's arteritis) meso‐aortitis causing aortic dilatation and subsequent regurgitation; or, finally, connective tissue diseases (eg, Marfan's syndrome, Ehlers‐Danlos), which can cause premature degeneration of the aortic media. The acuity of the patient's symptoms and the lack of systemic findings make a connective tissue or inflammatory disease unlikely. The clinical index of suspicion for aortic dissection needs to remain very high, as failure to make an expedient diagnosis may lead to complications and a deleterious outcome. Aortography may help to define this lesion.
The cardiothoracic surgical team was consulted and recommended aortic root and valve replacement. The patient was observed overnight and scheduled for preoperative cardiac catheterization the following morning. Aortogram revealed moderate to severe aortic insufficiency and a small dissection flap on the lesser curvature of the aorta, above the left coronary ostia (Figure 4). Coronary angiography revealed a 50% stenosis of the ostia of the first and second diagonal arteries, with no other flow‐limiting lesions.
The study has clearly demonstrated that the patient suffered an acute aortic dissection without involvement of the aortic annulus. Given the absence of left ventricular failure, it appears the aortic regurgitation was chronic and secondary to a previously existing aortic aneurysm. Asymptomatic congenital defects of the meso‐aorta such as cystic medical necrosis can predispose to aortic dissection at a relatively young age. However, this patient's condition may have been aggravated by drug abuse, paroxysmal elevation of blood pressure during landscaping, or other risk factors. Surgical correction of the dissection and aortic regurgitation is necessary.
The patient underwent aortic valve replacement with a 25‐mm St. Jude mechanical valve and an ascending aortic transection repair with a 32‐mm Dacron tube graft. On postoperative day 9, the patient was discharged home in stable condition.
COMMENTARY
Ensuring both accuracy and efficiency when making a diagnosis can be difficult, particularly when patients present in an atypical fashion or when diagnostic testing yields inconclusive results. Thus, a physician must sift through each clinical clue, remembering that although certain findings are pathognomonic for a disease process, a constellation of signs and symptoms when present can be equally diagnostic.
The initial likelihood of disease (ie, pretest probability) is generated by the history and physical and laboratory examinations. If the pretest probability is high and the subsequent diagnostic test is positive, the modified likelihood of disease (post‐test probability) is nearly 100%. If, however, the pretest probability is high and the diagnostic test is negative, the likelihood of disease is less clear. In these situations, the physician can either review the initial findings that generated the pretest probability or perform an additional diagnostic test of higher sensitivity.
In this exercise, a 46‐year‐old man presented to the emergency department with severe chest pain and findings characteristic of aortic dissection. The physicians appropriately sent him for chest computerized tomography (CT) because of a high pretest likelihood of aortic dissection. Because this test did not confirm the presence of aortic dissection, the patient underwent transesophageal echocardiography (TEE), a test of equal or greater sensitivity.1, 2 This test was also negative; however, a small dissection flap was subsequently found during cardiac catheterization. In this case, a test of lower sensitivity and specificity confirmed the diagnosis of dissection,3 demonstrating the possibility that either CT or TEE can be misinterpreted. Indeed, a final review of the TEE, showed the dissection flap, albeit small, had been missed. In this case, a diagnostic error was made that delayed the diagnosis when sufficient information was available earlier. Diagnostic errors are prevalent in medical practice and are commonly the result of numerous factors, though cognitive problems appear to be the largest contributor to this process.4 In particular, faulty synthesis of information, rather than inadequate medical knowledge, is the most common cause of cognitive medical errors. The error made in this case likely falls under the subcategory of faulty test detection or perception and subsequent premature closure (failure to consider other possibilities once an initial diagnosis of uncomplicated aortic aneurysm had been reached).4
Though medical errors cannot be completely eliminated, cases such as this should be reviewed to understand the cognitive processes that may lead to an erroneous diagnosis. In addition to the false‐negative TEE finding, this patient also had a coexisting condition that may have preoccupied the medical team. The thoracic aortic aneurysm seen by all imaging modalities was large and required intervention regardless of the presence of a dissection. However, this chronic condition became the focus of treatment, and the acute event that precipitated admission was missed. Perhaps if the primary team maintained a very high index of suspicion for dissection and conveyed this to the cardiology consultants, a meticulous review of the TEE would have then followed that may have uncovered the subtle findings of the dissection flap.
Fortunately, definitive treatment with surgical aortic root and valve replacement was performed in a timely manner, as the consequences of a delayed diagnosis in this situation could have been catastrophic. The mortality rate of a type A dissection is extremely high initially (1%‐2% per hour),5, 6 and thus surgical intervention is typically performed immediately after the diagnosis and the extent of this disease is established, rather than the following morning.7
This case highlights not only the problems resulting in diagnostic errors but also exemplifies the thought process required to make a challenging diagnosis. Our case discussant was able to avoid cognitive pitfalls by presenting a broad differential diagnosis and reevaluating the diagnosis with each additional piece of information provided.8 An experienced clinician should realize that patients with an extremely high pretest probability of disease and a negative diagnostic test should be further investigated, regardless of the test sensitivity. Furthermore, time‐honored methods such as history taking, physical examination. and thoughtful analyses should remain critical tools in the process of reaching an accurate diagnosis despite technological advances in diagnostic testing.
A 46‐year‐old African American man presented to the emergency department with severe chest pain that awakened him from sleep. The pain was substernal, radiated to the neck and back, and was continuous, lasting approximately 1 hour. It was associated with nausea, diaphoresis, and dizziness. The patient denied dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysphagia, odynophagia, vomiting, fever, chills, or headache. He denied recent recreational drug use. He works as a landscaper.
Substernal chest pain radiating to the neck and back implicates structures in the middle mediastinum, chiefly the heart, aorta, esophagus, pulmonary arteries, and mediastinal pleura. The presence of nausea and diaphoresis suggests a vagal response to pain.
The sudden onset of symptoms and the lack of fevers and chills make infectious causes in the mediastinum such as mediastinitis and pneumonia less likely. Acute pulmonary embolism often presents with pleuritic pain and dyspnea, features not present in this patient.
The absence of odynophagia and dysphagia makes esophageal rupture or perforation of esophageal diverticula unlikely. Likewise, the absence of dyspnea, orthopnea, and paroxysmal nocturnal dyspnea makes it unlikely to be acute left ventricular failure from a sudden rise in left atrial pressure. Such a scenario may occur in the setting of a myocardial infarction or rupture of a papillary muscle, chordae tendineae, or sinus of Valsalva.
Dissection of the aorta with or without involvement of the aortic root merits strong consideration. Dissection involving the carotid or vertebral arteries could explain the patient's dizziness. Physical stresses in a landscaper may contribute to elevation in blood pressure and set the stage for an aortic dissection, especially with other risk factors.
The patient has a history of a positive PPD and was treated with isoniazid for 6 months. His mother had a history of hypertension and died from a myocardial infarction at age 64. His father's medical history is unknown. He has a history of alcohol abuse but has been abstinent for more than a year. He smoked marijuana and tobacco occasionally, with a 15 pack‐year cigarette history. He also stated that the last time he used cocaine was 1 year prior to admission.
That his mother succumbed to a myocardial infarction as well as having been hypertensive could be important family history risk factors given the patient's symptoms. Furthermore, the concurrent use of alcohol and tobacco by the patient increases his risk of developing severe hypertension. The use of cocaine is associated with sudden elevations in systemic blood pressure, which predispose to intimal damage in the aorta, especially if other risk factors are present. Marijuana smoking has been implicated in pulmonary aspergillosis, but this sudden presentation in the absence of pulmonary symptoms makes it most unlikely. Optimal therapy of a positive PPD should not predispose the patient to acute exudative pericarditis. Thus far the features suggest an acute vascular episode without significant compromise of cardiac output.
The patient was alert and in mild distress from chest discomfort. He was afebrile, with a blood pressure of 136/64 in the right arm and 139/63 in the left arm. Heart rate was 60 beats per minute, and the respiratory rate was 12 beats per minute, with an oxygen saturation of 99% while breathing room air. Examination of the head and neck revealed no signs of trauma. Jugular venous waveforms were normal, and carotid artery pulsations had normal strength and upstroke without audible bruits. The lungs were clear to auscultation. Heart sounds were notable for a 3/6 diastolic murmur heard best at the right sternal border. Rate and rhythm were regular, and the apical impulse was sustained but not displaced. The peripheral pulses were present and equal in quality throughout. The findings of the abdominal exam were normal, and the digital rectal examination was negative for occult blood. The findings of the neurologic, musculoskeletal, and dermatologic exams also were normal.
A slightly elevated pulse pressure without a significant difference in upper extremity blood pressure could be a result of aortic regurgitation, sinus of Valsalva rupture, or a high‐cardiac‐output state, as seen in thyrotoxicosis, anemia, or arteriovenous fistula.
The presence of a 3/6 diastolic murmur at the sternal border, however, favors conditions that cause aortic valve regurgitation and, less commonly, pulmonary valve regurgitation, or turbulent flow across the tricuspid valve. The murmur of pulmonary and aortic valve regurgitation can be difficult to distinguish by auscultation; however, the absence of other findings such as a right ventricular heave, elevated jugular venous pressure, or primary lung disease do not support a pulmonary valve etiology. Turbulent flow across the tricuspid valve can be seen in high‐output states, large atrial septal defects, and tricuspid stenosis. This type of murmur is heard best at the lower sternal border and tends to increase with inspiration. An early diastolic murmur would suggest aortic regurgitation, either from aortic valve or aortic root pathology. Aortic regurgitation would contribute to a sustained apical impulse.
Clear lungs and the absence of tachycardia suggest that left ventricle function is not severely compromised. These findings argue against acute rupture of the sinus of Valsalva, a condition that normally causes biventricular failure. The presence of equal peripheral pulses does not exclude the diagnosis of aortic dissection, as the dissection may have occurred proximal to the origins of the right innominate and the left subclavian arteries.
Initial laboratory studies revealed a hematocrit of 34.9, a leukocyte count of 6800/mm3, and a platelet count of 195 000/mm3. The levels of serum electrolytes, serum creatinine, blood urea nitrogen, and initial cardiac enzymes were normal. The urine drug screen was negative. The electrocardiogram showed evidence of left ventricular hypertrophy (Figure 1), and portable chest radiography revealed an enlarged cardiac silhouette and a widened mediastinum (Figure 2).
The presence of a widened mediastinum is consistent with aortic dissection but may also suggest a mass, aortic aneurysm, infiltrative disease, or a collection of fluid (eg, blood). A normal level of cardiac enzymes and the absence of ischemic findings on electrocardiography make myocardial infarction less likely. Given his hemodynamic stability, he is unlikely to have suffered cardiac rupture; however, he may still have a dissection of the aorta or a rupture of the sinus of Valsalva. The slight decline in hematocrit is compatible with either a mediastinal or pericardial collection of blood.
The patient was emergently sent for computerized axial tomography of the chest, which revealed a dilated aortic root at 6 cm but no evidence of aortic dissection (Figure 3). The lung fields were normal.
Despite a negative test, the presence of severe acute chest pain in the presence of a widened mediastinum is still concerning for aortic dissection. If the dissection involved the aortic valve annulus, the resulting acute regurgitation can not be severe, given the absence of left ventricular heart failure. Likewise, the presence of normal lung fields suggests the patient has no acute elevation of left ventricular end diastolic pressures such as is seen in ventricle septal defect, papillary muscle dysfunction, or acute mitral valvular lesion.
Cardiology consultants were emergently consulted and performed a transesophageal echocardiogram that confirmed a dilated aortic root (6.3 cm) without evidence of dissection. The patient was noted to have moderate to severe aortic regurgitation and a dilated left ventricle with moderate hypertrophy. A trace effusion was noted, and the left ventricular ejection fraction was estimated to be 50%.
The presence of moderate to severe aortic regurgitation with a dilated aortic root suggests 3 possibilities: undiagnosed dissection of the aortic root with preexisting aortic insufficiency (eg, bicuspid aortic valve, rheumatic valve disease, previous endocarditis); infectious (eg, syphilitic) or noninfectious (eg, ankylosing spondylitis, Takayasu's arteritis) meso‐aortitis causing aortic dilatation and subsequent regurgitation; or, finally, connective tissue diseases (eg, Marfan's syndrome, Ehlers‐Danlos), which can cause premature degeneration of the aortic media. The acuity of the patient's symptoms and the lack of systemic findings make a connective tissue or inflammatory disease unlikely. The clinical index of suspicion for aortic dissection needs to remain very high, as failure to make an expedient diagnosis may lead to complications and a deleterious outcome. Aortography may help to define this lesion.
The cardiothoracic surgical team was consulted and recommended aortic root and valve replacement. The patient was observed overnight and scheduled for preoperative cardiac catheterization the following morning. Aortogram revealed moderate to severe aortic insufficiency and a small dissection flap on the lesser curvature of the aorta, above the left coronary ostia (Figure 4). Coronary angiography revealed a 50% stenosis of the ostia of the first and second diagonal arteries, with no other flow‐limiting lesions.
The study has clearly demonstrated that the patient suffered an acute aortic dissection without involvement of the aortic annulus. Given the absence of left ventricular failure, it appears the aortic regurgitation was chronic and secondary to a previously existing aortic aneurysm. Asymptomatic congenital defects of the meso‐aorta such as cystic medical necrosis can predispose to aortic dissection at a relatively young age. However, this patient's condition may have been aggravated by drug abuse, paroxysmal elevation of blood pressure during landscaping, or other risk factors. Surgical correction of the dissection and aortic regurgitation is necessary.
The patient underwent aortic valve replacement with a 25‐mm St. Jude mechanical valve and an ascending aortic transection repair with a 32‐mm Dacron tube graft. On postoperative day 9, the patient was discharged home in stable condition.
COMMENTARY
Ensuring both accuracy and efficiency when making a diagnosis can be difficult, particularly when patients present in an atypical fashion or when diagnostic testing yields inconclusive results. Thus, a physician must sift through each clinical clue, remembering that although certain findings are pathognomonic for a disease process, a constellation of signs and symptoms when present can be equally diagnostic.
The initial likelihood of disease (ie, pretest probability) is generated by the history and physical and laboratory examinations. If the pretest probability is high and the subsequent diagnostic test is positive, the modified likelihood of disease (post‐test probability) is nearly 100%. If, however, the pretest probability is high and the diagnostic test is negative, the likelihood of disease is less clear. In these situations, the physician can either review the initial findings that generated the pretest probability or perform an additional diagnostic test of higher sensitivity.
In this exercise, a 46‐year‐old man presented to the emergency department with severe chest pain and findings characteristic of aortic dissection. The physicians appropriately sent him for chest computerized tomography (CT) because of a high pretest likelihood of aortic dissection. Because this test did not confirm the presence of aortic dissection, the patient underwent transesophageal echocardiography (TEE), a test of equal or greater sensitivity.1, 2 This test was also negative; however, a small dissection flap was subsequently found during cardiac catheterization. In this case, a test of lower sensitivity and specificity confirmed the diagnosis of dissection,3 demonstrating the possibility that either CT or TEE can be misinterpreted. Indeed, a final review of the TEE, showed the dissection flap, albeit small, had been missed. In this case, a diagnostic error was made that delayed the diagnosis when sufficient information was available earlier. Diagnostic errors are prevalent in medical practice and are commonly the result of numerous factors, though cognitive problems appear to be the largest contributor to this process.4 In particular, faulty synthesis of information, rather than inadequate medical knowledge, is the most common cause of cognitive medical errors. The error made in this case likely falls under the subcategory of faulty test detection or perception and subsequent premature closure (failure to consider other possibilities once an initial diagnosis of uncomplicated aortic aneurysm had been reached).4
Though medical errors cannot be completely eliminated, cases such as this should be reviewed to understand the cognitive processes that may lead to an erroneous diagnosis. In addition to the false‐negative TEE finding, this patient also had a coexisting condition that may have preoccupied the medical team. The thoracic aortic aneurysm seen by all imaging modalities was large and required intervention regardless of the presence of a dissection. However, this chronic condition became the focus of treatment, and the acute event that precipitated admission was missed. Perhaps if the primary team maintained a very high index of suspicion for dissection and conveyed this to the cardiology consultants, a meticulous review of the TEE would have then followed that may have uncovered the subtle findings of the dissection flap.
Fortunately, definitive treatment with surgical aortic root and valve replacement was performed in a timely manner, as the consequences of a delayed diagnosis in this situation could have been catastrophic. The mortality rate of a type A dissection is extremely high initially (1%‐2% per hour),5, 6 and thus surgical intervention is typically performed immediately after the diagnosis and the extent of this disease is established, rather than the following morning.7
This case highlights not only the problems resulting in diagnostic errors but also exemplifies the thought process required to make a challenging diagnosis. Our case discussant was able to avoid cognitive pitfalls by presenting a broad differential diagnosis and reevaluating the diagnosis with each additional piece of information provided.8 An experienced clinician should realize that patients with an extremely high pretest probability of disease and a negative diagnostic test should be further investigated, regardless of the test sensitivity. Furthermore, time‐honored methods such as history taking, physical examination. and thoughtful analyses should remain critical tools in the process of reaching an accurate diagnosis despite technological advances in diagnostic testing.
- ,,, et al.The diagnosis of thoracic aortic dissection by noninvasive imaging procedures.N Engl J Med.1993;328(1):1–9.
- ,,, et al.Accuracy of biplane and multiplane transesophageal echocardiography in diagnosis of typical acute aortic dissection and intramural hematoma.J Am Coll Cardiol.1996;28:627–636.
- ,.Aortic dissection: new frontiers in diagnosis and management: Part I: from etiology to diagnostic strategies.Circulation.2003;108:628–635.
- ,,.Diagnostic error in internal medicine.Arch Intern Med.2005;165:1493–1499.
- ,,.Dissecting aneurysm of the aorta: a review of 505 cases.Medicine (Baltimore).1958;37(3):217–279.
- ,,, et al.The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease.JAMA.2000;283:897–903.
- ,.Surgery of the thoracic aorta.N Engl J Med.1997;336:1876–1888.
- .Clinical reasoning in medicine. In:Higgs J,Jones MA, eds.Clinical Reasoning in the Health Professions.Woburn, Mass:Butterworth‐Heinemann;1995:49–59.
- ,,, et al.The diagnosis of thoracic aortic dissection by noninvasive imaging procedures.N Engl J Med.1993;328(1):1–9.
- ,,, et al.Accuracy of biplane and multiplane transesophageal echocardiography in diagnosis of typical acute aortic dissection and intramural hematoma.J Am Coll Cardiol.1996;28:627–636.
- ,.Aortic dissection: new frontiers in diagnosis and management: Part I: from etiology to diagnostic strategies.Circulation.2003;108:628–635.
- ,,.Diagnostic error in internal medicine.Arch Intern Med.2005;165:1493–1499.
- ,,.Dissecting aneurysm of the aorta: a review of 505 cases.Medicine (Baltimore).1958;37(3):217–279.
- ,,, et al.The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease.JAMA.2000;283:897–903.
- ,.Surgery of the thoracic aorta.N Engl J Med.1997;336:1876–1888.
- .Clinical reasoning in medicine. In:Higgs J,Jones MA, eds.Clinical Reasoning in the Health Professions.Woburn, Mass:Butterworth‐Heinemann;1995:49–59.
Clinical Conundrum
A 49‐year‐old man presented with 2 days of chills, fever, anorexia, and increased cough and dyspnea. The patient had a history of chronic obstructive pulmonary disease (COPD) and noted that his cough and dyspnea had increased above normal for several days. He was now dyspneic with minimal activity and had slept at a 45‐degree incline the night prior to evaluation due to dyspnea. He noted less improvement than usual with the use of his metered dose inhaler. His cough was occasionally productive of small amounts of white phlegm. He had vomited once. During a coughing episode the patient experienced a sudden onset of sharp right upper quadrant abdominal pain that worsened with coughing and sudden position changes. The patient denied a prior history of abdominal pain or surgery. The patient's last bowel movement was 2 days prior to admission. He denied melena or bright red blood per rectum.
My initial differential diagnosis for this patient's dyspnea and cough is pneumonia, acute exacerbation of COPD, or congestive heart failure. The presence of fever and anorexia increases the likelihood of infectious etiologies, whereas the presence of orthopnea points toward congestive heart failure. Noncardiac processessuch as a large pleural effusion or apical lung diseasecould also cause orthopnea. His abdominal pain could be a result of pneumonia alone (perhaps in the right lower lobe with diaphragmatic irritation), but I am also considering complications of pneumonia such as empyema. Although his abdominal pain, dyspnea, and cough could also be a result of hepatobiliary disease, a perforated viscus, or pancreatitis, we currently have little reason to suspect a direct abdominal etiology. My top diagnosis is community‐acquired pneumonia, perhaps accompanied by pleural effusion.
His medical history was significant for dilated cardiomyopathy and heavy alcohol use. His medications included various meter‐dosed inhalers, bupropion, digoxin, spironolactone, lisinopril, and metoprolol. He had never received corticosteroid therapy and had not previously been hospitalized for COPD‐related problems. He had smoked one pack of cigarettes daily for 40 years.
Heavy alcohol use is associated with an increased risk of several pulmonary infections such as gram‐negative necrotizing pneumonia (classically, Klebsiella pneumoniae), pneumococcal pneumonia, aspiration pneumonia, anaerobic lung abscesses, and tuberculosis. Given his right upper quadrant pain, acute alcoholic hepatitis and alcohol‐related pancreatitis enter the differential. His history of cardiomyopathy makes me consider congestive heart failure as more likely than before, and perhaps his abdominal pain is a result of hepatic congestion from right heart failure. His fever, however, cannot be attributed to cardiac failure. Less likely diagnoses include ischemic conditions related to his cardiomyopathy such as mesenteric ischemia from low perfusion or embolism from a cardiac thrombus. A pulmonary infection remains the most likely diagnosis.
He was an ill‐appearing man in moderate respiratory distress, looking older than his stated age. His temperature was 38.4C, heart rate 129 beats/minute, blood pressure 85/56 mm Hg, respiratory rate 24 breaths/minute, and oxygen saturation 92% on room air. A cardiovascular exam revealed no murmur, gallop, or rub. The jugular venous pulse was not elevated. His lungs were clear to auscultation. Abdominal exam revealed right‐sided abdominal tenderness that appeared to localize to the rectus sheath. Otherwise, the abdomen was soft, with normal bowel sounds and no organomegaly. Rectal examination revealed guaiac negative stool and no focal tenderness. His extremities were normal.
His vital signs are worrisome for impending cardiovascular collapse and shock, possibly due to sepsis. The relatively nonfocal cardiopulmonary exam is surprising given his initial symptoms and makes me wonder if his dyspnea is primarily related to an abdominal process leading to diaphragmatic irritation rather than to a thoracic process. Congestive heart failure seems unlikely given the lack of supportive physical examination findings. His abdominal exam findings are puzzling. Although his abdominal wall tenderness could be benignperhaps from muscular strain or a tear from coughingit could represent a more worrisome process such as infection or a hematoma in the abdominal wall muscles. Mesenteric ischemia is still possible, as the exam is often unimpressive. A hepatic abscess or subphrenic abscess should be considered, as physical exam findings in these conditions can be subtle.
My differential remains relatively unchanged, but I have now put consideration of a hepatic or subphrenic abscess higher on my list. Early empiric broad‐spectrum antibiotics seem necessary.
He had a white blood cell count of 26,700/mL with 92% neutrophils, a hemoglobin of 14.6 g/dL, and a platelet count of 312,000/mL. Sodium was 134 mmol/L, potassium was 4.3 mmol/L, chloride was 94 mmol/L, bicarbonate was 23 mmol/L, blood urea nitrogen was 23 mg/dL, and creatinine was 2.1 mg/dL. The results of the calcium, protein, albumin, and liver function tests were normal. Urinalysis was negative for protein and red blood cells. An electrocardiogram revealed sinus tachycardia. A chest radiograph at admission revealed mild opacities in both lower lobes and the right middle lobe consistent with either atelectasis or pneumonia (Fig. 1). A very small left effusion was also identified.
The additional data reinforce my clinical impression that this process is likely to be infectious. The chest radiograph is consistent with community‐acquired pneumonia, possibly from an atypical pathogen. Given his elevated creatinine, I am also considering a pulmonary‐renal syndrome such as vasculitis, though hematuria was not present. A subphrenic abscess, mesenteric ischemia, or an abdominal wall process (because his abdominal tenderness on exam still needs an explanation) remain possibilities; my suspicion would increase if he does not respond appropriately to therapy for community‐acquired pneumonia.
The clinical team's working diagnosis also was community‐acquired pneumonia. Blood and sputum cultures were obtained, and the patient was treated with intravenous ceftriaxone, azithromycin, and intravenous fluid. By the second day, his creatinine had normalized; however, his hypoxemia had worsened, and he now required supplemental oxygen. His temperature was 39.3C, and his heart rate was 150 beats/minute. The findings of an abdominal ultrasound of the kidneys, spleen, and right upper quadrant were normal.
It is too early to say the patient has failed therapy because a patient can get worse before getting better during the course of antibiotic therapy for community‐acquired pneumonia. Fever, for example, may take up to 7 days to resolve, depending on host factors and the pathogen. Though I typically wait about 72 hours before assuming a patient is not appropriately responding to therapy, the additional information has made me concerned. The degree of tachycardia is significant and warrants an EKG to exclude an arrthymia. I would also repeat the chest radiograph to evaluate for worsening infiltrates or increased pleural effusion.
On the third hospital day, the patient's abdominal pain had decreased with analgesia, but his fever, cough, and dyspnea remained largely unchanged. Antibiotics were changed to intravenous levofloxacin. A repeat chest radiograph revealed elevation of the right hemidiaphragm and bilateral effusions (Fig. 2). An electrocardiogram revealed sinus tachycardia. Blood cultures revealed no growth, and sputum cultures grew oral flora.
A significantly elevated right hemidiaphragm makes me reconsider the diagnosis of simple community‐acquired pneumonia. The differential diagnosis for an elevated hemidiaphragm is best considered by location in relation to the diaphragm. Causes above the diaphragm include rib fracture, atelectasis, pleural thickening, and volume loss of the lung for another reason (e.g., surgery, bronchial obstruction due to tumor or mucus plugging), as well as mimics such as a densely consolidated pneumonia, pulmonary infarction, or a subpulmonary effusion. Diaphragmatic causes include eventration, rupture, phrenic nerve weakness, and intrinsic weakness because of neuromuscular disease (usually bilateral). Causes below the diaphragm that must be considered are subphrenic or liver abscess, liver (and other abdominal) malignancy, pancreatic pseudocyst, and distended bowel. Given the clinical picture, I am focusing below the diaphragmespecially on a possible hepatic or subphrenic abscess (which could be missed on ultrasound) and mimics of it such as dense consolidation or a subpulmonary effusion. Given the lack of response to antibiotics, I need to consider an infection that is not being treated, either because of location (abscess, effusion) or microbiology (tuberculosis, a parasite, a fungus, resistant bacteria). After confirming that the patient has a substantive pleural effusion, he needs a thoracentesis.
On the fourth hospital day, his temperature was 38.8C, and his white blood cell count was 21,000/mL. A right‐sided thoracentesis was performed; approximately 250 cc of fluid was obtained. Pleural fluid analysis revealed bloody fluid, with a white blood cell count of 16,750/mL with 94% neutrophils, 40,000 red blood cells/mL, lactate dehydrogenase of 278 U/L (normal serum value 80200 U/L), protein of 3.7 g/dL, and glucose of 81 mg/dL. A pleural fluid pH was not obtained. A gram stain revealed many white blood cells with no organisms noted. Serum protein was 7.4 g/dL. These results were thought to represent an exudative parapneumonic effusion; levofloxacin and supplemental oxygen were continued.
The pleural fluid appears exudative, but I am not sure this man has a parapneumonic effusion because, despite clinical deterioration, an obvious infiltrate is not seen on interval chest radiography. We must look closely at the fluid because this is a bloody effusion and somewhat atypical for a parapneumonic effusion. Also, the effusion does not appear large enough to explain why he has not improved on the current antibiotics. We should thus reconsider our diagnosis and management. I would obtain additional imaging (such as an abdominal and chest computed tomography [CT]) and perhaps obtain a consultation from the pulmonary team regarding the postulated initial diagnosis of pneumonia with effusion.
On the fifth day of hospitalization, the patient's dyspnea and cough persisted but were improved. His abdominal pain was minimal and felt improved with flatus. Fever continued to 38.8C, and the white blood cell count was 20,000/mL. On examination the patient had decreased breath sounds at the right base and bibasilar crackles. His abdomen was soft, with tenderness in his right upper quadrant only with deep palpation; bowel sounds remained. An ultrasound of the chest was performed to look for a loculated effusion; however, no fluid was identified. The pulmonary consultant thought it likely that the patient had a subpulmonic effusion and recommended CT of the abdomen and chest.
His right upper quadrant tenderness is still unexplained. I would agree with the CT, primarily to evaluate other causes of his elevated diaphragm such as subphrenic or hepatic abscess. For now, I would make no change in antibiotic therapy.
On the sixth hospital day, the patient had an episode of bilious emesis. Chest and abdominal CT revealed collapse of the right middle and lower lobes with a small adjacent effusion, and a 6 6 16 cm abscess intimately opposed to the right lobe of the liver. Extending from the inferior extent of the abscess was a tubular thick‐walled structure connecting to the cecum that was suspicious of a thickened inflamed appendix. There was periappendiceal stranding suggesting inflammation. The small bowel was diffusely dilated up to 4.5 cm, suggesting a small bowel obstruction.
I suspect that his abscess is related to a perforated appendix and that the dilated small bowel is most likely a result of localized irritation of the bowel by the abscess and appendicitis. The collapsed lung is most likely due to local inflammation from the subdiaphragmatic abscess. Treatment should now be changed substantially. I would ask a surgeon to evaluate the patient because the most likely diagnosis is perforated appendicitis with abscess formation.
When the periappendiceal abscess was drained percutaneously, 190 mL of purulent fluid was removed. The cultures were positive for Klebsiella pneumonia, Enterococcus faecalis,and Streptococcus milleri. The patient was given 6 weeks of intravenous antibiotics with improvement in his clinical symptoms. During the interval the findings on his chest radiograph resolved completely. A laproscopic appendectomy 3 months later revealed significant right lower quadrant adhesions. The pathology specimen identified a distorted appendix with regeneration consistent with prior appendicitis. The patient was contacted 4 months after his surgery, and he reported that he was doing well, with no cardiopulmonary or gastrointestinal symptoms.
COMMENTARY
Community‐acquired pneumonia (CAP) is a common cause of acute illness and accounts for nearly 1 million admissions per year in the United States.1 The diagnosis of CAP is made when symptoms including dyspnea, fever, cough, or leukocytosis are present, with confirmation provided by a chest radiograph. Often the diagnosis is clear; however, there is no pathognomonic constellation of signs or symptoms that establish the diagnosis with certainty.2 Many physicians learn that pneumoniaespecially lower‐lobe pneumoniacan lead to abdominal findings such as upper quadrant pain, vomiting, and tenderness to palpation. Conversely, the patient discussed above illustrates that a primary abdominal process can also result in a symptom complex that mimics pneumonia.
The prevalence of CAP coupled with the inherent uncertainty of a clinical diagnosis of CAP leads to an important question: How long is too long before questioning the diagnosis? An analysis of the pneumonia Patient Outcomes Research Trial (PORT) limited to inpatients with CAP examined time to clinical stability. For the majority of patients, abnormal vital signs resolved within 23 days.3 In this study, 29% of patients had severe disease, and not surprisingly, these patients took longer to improve. Using the pneumonia severity index score, which accounts for age, comorbidity, abnormal vital signs, and laboratory data, the patient described in this article would be considered at high risk for death and complication with an estimated mortality of 9%.4 Using a combination of defervescence, resolution of tachycardia, tachypnea, and hypoxemia as markers of clinical stability, a patient like ours should respond within 4 days (with a range of 27 days). On the basis of these dataand the discrepancy between the patient's severe illness and relatively minor pulmonary infiltratesit seems reasonable to have considered this patient as failing CAP therapy as early as the fourth day of hospitalization.
In approximately 10% of hospitalized patients with CAP, the clinical course is protracted.5 When patients do not improve as quickly as expected, the reasons that could explain this should be investigated. In a cohort of 49 patients with CAP who failed therapy the most common reasons for failure to improve were severity of the pneumonia and drug resistance.6 A multicenter study found that the incidence of resistance to penicillin by Streptococcus pneumoniae, the most common bacterial pathogen in CAP, was 30%, with a 4% in vitro resistance rate to ceftriaxone.7 How well in vitro resistance predicts clinical response, however, is unclear. Risk factors for antibiotic resistance include close exposure to children, recent antibiotic use, and recent hospitalization. Immunosuppressive conditions should also be considered in patients who fail to improve. Suppurative complications of pneumoniasuch as empyema, parapneumonic effusion, and lung abscessalso delay recovery.
Another consideration in a patient with what appears to be a nonresolving pneumonia with pleural effusion is that the initial diagnosis is incorrect and the cause is extrathoracic. Pulmonary and cardiac diseases account for more than 90% of effusions, whereas less than 5% of pleural effusions result from intraabdominal causes.8 When should intraabdominal diseases be sought in patients with an effusion, fever, dyspnea, and cough? Light suggests that intraabdominal pathology should be investigated in patients who have pleural effusions without significant parenchymal disease.8 This point is underscored by the experience of our patient, whose chest radiographs showed, despite clinical decline, minimal airspace disease.
Several abdominal entities cause pleural effusion. Pancreatitis, either acute or chronic, with pseudocyst formation is the most common abdominal cause of exudative pleural effusions. Approximately 10% of patients with pancreatic disease will develop effusions, usually left‐sided.9 These left‐sided effusions are also seen in splenic abscesses, usually as a result of endocarditis. Intrahepatic abscess is associated with effusions in 20% of patients.10 A subphrenic abscess, as seen in our patient, is an uncommon cause of exudative pleural effusions. Historically, subphrenic abscesses resulted from a perforated viscus, with ruptured appendicitis the most common cause,11 followed by perforated peptic ulcers and biliary tract disease. With the advent of antibiotics, the causes of subphrenic abscess changed considerably, with the majority of current cases resulting from postsurgical complications.12 The findings of a chest radiograph are abnormal in 80% of patients with subphrenic abscess;1214 an elevated hemidiaphragm and pleural effusion are found in the majority of cases. The symptoms of a subphrenic abscess are nonspecific, and patient's complaints are equally split between predominantly thoracic and predomninantly abdominal complaints.15
Appendicitis, a common disease predominantly of the young, may lead to atypical presentations in older individuals. In a retrospective analysis of 113 patients older than 60 years with appendicitis, 70% presented in an atypical fashion.16 Typical symptoms include right lower quadrant pain, fever, anorexia and a white blood cell count greater than 10,000/mL. Fever was the most frequently absent symptom, seen in only 37% of older patients. In this cohort, approximately one third of older patients waited more than 48 hours prior to presentation. The time between symptom onset and clinical presentation is a strong predictor of perforation risk.17 As in this case, roughly 2% of patients with acute appendicitis will present with perforation and abscess formation.18 In such patients the management is initially conservative. Percutaneous drainage and broad spectrum antibiotics are the treatment of choice, followed by an interval appendectomy in 612 weeks.19 The rationale for delayed surgery is that earlier surgery may disseminate a localized inflammatory process.20
Community‐acquired pneumonia is a more frequent cause of hospital admission than is intraabdominal abscess. Physicians often face the dilemma of when to pursue alternative diagnoses after a patient who is thought to have an atypical presentation of a common disease (ie, CAP) fails to respond to conventional therapy. Although clinicians learn that right upper quadrant pain may be a symptom of pneumonia, our patient revealed that abdominal causes may mimic pneumonia and produce a pleural effusion. Determining whether the primary disease originates above or below the diaphragm is critical to guiding therapy. When patients fail to respond adequately to therapy, clinicians should set a low threshold for deciding to image the abdomen in a patient with modest pulmonary infiltrates, pleural effusion, and abdominal pain.
- ,,, et al.The cost of treating community‐acquired pneumonia.Clin Ther.1998;20:820–827.
- ,,.Does this patient have community‐acquired pneumonia? Diagnosing pneumonia by history and physical examination.JAMA.1997;278:1440–1445.
- ,,, et al.Time to clinical stability in patients hospitalized with community acquired pneumonia. Implications for practice guidelines.JAMA.1998;279:1452–1457.
- ,,, et al.A prediction rule to identify low‐risk patients with community‐acquired pneumonia.N Engl J Med.1997;336:243–250.
- ,,, et al.Utility of fiberoptic bronchoscopy in non resolving pneumonia.Chest.1990;98:1322–1326.
- ,,, et al.Antimicrobial treatment failures in patients with community acquired pneumonia. Causes and prognostic implications.Am J Respir Crit Care Med.2000;162:154–160.
- ,,, et al.Antimicrobial resistance with Streptococcus pneumoniae in the United States, 1997–98.Emerg Infect Dis.1999;5:757–765.
- ,.Pleural effusion. In:Murray JF,Nadel JA, eds.Textbook of respiratory medicine. 3rd ed.Philadelphia:WB Saunders,2000:2013–2041.
- ,,.Significance of pleural effusion in patients with acute pancreatitis.Am J Gastroenterol.1992;87:871–874.
- .Exudative pleural effusions secondary to gastrointestinal diseases.Clin Chest Med.1985;6(1):103–111.
- .Subphrenic abscess.Ann Surg.1963;158:240–248.
- ,,,.Upper abdominal abscess: a continuing and deadly problem.Am J Roentgenol.1980;134:759–765.
- .Subphrenic abscess. A clinical study of 101 cases.Acta Chir Scand.1959;117:388–408.
- ,,.Subphrenic abscess a continuing hazard.Am J Surg.1969:117–122.
- ,.Subphrenic abscess: a thoracoabdominal clinical complex. The changing picture with antibiotics.Am J Surg.1964;108:165–172.
- ,.What have we learned over the past 20 years about appendicitis in the elderly.Am J Surg.2003;185:198–201.
- ,,, et al.Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies.Am Surg.2000;66:548–554.
- ,,.Appendicitis with a palpable mass.Ann Surg.1981;193:227–229.
- ,,, et al.Nonoperative management of perforated appendicitis without periappendiceal mass.Am J Surg.2000;179:177–181.
- ,,.Appendix. In:Townsend CM, ed.Sabiston textbook of surgery. The biologic basis of modern surgical practice. 16th ed.Philadelphia:W. B. Saunders,2001:917–928.
A 49‐year‐old man presented with 2 days of chills, fever, anorexia, and increased cough and dyspnea. The patient had a history of chronic obstructive pulmonary disease (COPD) and noted that his cough and dyspnea had increased above normal for several days. He was now dyspneic with minimal activity and had slept at a 45‐degree incline the night prior to evaluation due to dyspnea. He noted less improvement than usual with the use of his metered dose inhaler. His cough was occasionally productive of small amounts of white phlegm. He had vomited once. During a coughing episode the patient experienced a sudden onset of sharp right upper quadrant abdominal pain that worsened with coughing and sudden position changes. The patient denied a prior history of abdominal pain or surgery. The patient's last bowel movement was 2 days prior to admission. He denied melena or bright red blood per rectum.
My initial differential diagnosis for this patient's dyspnea and cough is pneumonia, acute exacerbation of COPD, or congestive heart failure. The presence of fever and anorexia increases the likelihood of infectious etiologies, whereas the presence of orthopnea points toward congestive heart failure. Noncardiac processessuch as a large pleural effusion or apical lung diseasecould also cause orthopnea. His abdominal pain could be a result of pneumonia alone (perhaps in the right lower lobe with diaphragmatic irritation), but I am also considering complications of pneumonia such as empyema. Although his abdominal pain, dyspnea, and cough could also be a result of hepatobiliary disease, a perforated viscus, or pancreatitis, we currently have little reason to suspect a direct abdominal etiology. My top diagnosis is community‐acquired pneumonia, perhaps accompanied by pleural effusion.
His medical history was significant for dilated cardiomyopathy and heavy alcohol use. His medications included various meter‐dosed inhalers, bupropion, digoxin, spironolactone, lisinopril, and metoprolol. He had never received corticosteroid therapy and had not previously been hospitalized for COPD‐related problems. He had smoked one pack of cigarettes daily for 40 years.
Heavy alcohol use is associated with an increased risk of several pulmonary infections such as gram‐negative necrotizing pneumonia (classically, Klebsiella pneumoniae), pneumococcal pneumonia, aspiration pneumonia, anaerobic lung abscesses, and tuberculosis. Given his right upper quadrant pain, acute alcoholic hepatitis and alcohol‐related pancreatitis enter the differential. His history of cardiomyopathy makes me consider congestive heart failure as more likely than before, and perhaps his abdominal pain is a result of hepatic congestion from right heart failure. His fever, however, cannot be attributed to cardiac failure. Less likely diagnoses include ischemic conditions related to his cardiomyopathy such as mesenteric ischemia from low perfusion or embolism from a cardiac thrombus. A pulmonary infection remains the most likely diagnosis.
He was an ill‐appearing man in moderate respiratory distress, looking older than his stated age. His temperature was 38.4C, heart rate 129 beats/minute, blood pressure 85/56 mm Hg, respiratory rate 24 breaths/minute, and oxygen saturation 92% on room air. A cardiovascular exam revealed no murmur, gallop, or rub. The jugular venous pulse was not elevated. His lungs were clear to auscultation. Abdominal exam revealed right‐sided abdominal tenderness that appeared to localize to the rectus sheath. Otherwise, the abdomen was soft, with normal bowel sounds and no organomegaly. Rectal examination revealed guaiac negative stool and no focal tenderness. His extremities were normal.
His vital signs are worrisome for impending cardiovascular collapse and shock, possibly due to sepsis. The relatively nonfocal cardiopulmonary exam is surprising given his initial symptoms and makes me wonder if his dyspnea is primarily related to an abdominal process leading to diaphragmatic irritation rather than to a thoracic process. Congestive heart failure seems unlikely given the lack of supportive physical examination findings. His abdominal exam findings are puzzling. Although his abdominal wall tenderness could be benignperhaps from muscular strain or a tear from coughingit could represent a more worrisome process such as infection or a hematoma in the abdominal wall muscles. Mesenteric ischemia is still possible, as the exam is often unimpressive. A hepatic abscess or subphrenic abscess should be considered, as physical exam findings in these conditions can be subtle.
My differential remains relatively unchanged, but I have now put consideration of a hepatic or subphrenic abscess higher on my list. Early empiric broad‐spectrum antibiotics seem necessary.
He had a white blood cell count of 26,700/mL with 92% neutrophils, a hemoglobin of 14.6 g/dL, and a platelet count of 312,000/mL. Sodium was 134 mmol/L, potassium was 4.3 mmol/L, chloride was 94 mmol/L, bicarbonate was 23 mmol/L, blood urea nitrogen was 23 mg/dL, and creatinine was 2.1 mg/dL. The results of the calcium, protein, albumin, and liver function tests were normal. Urinalysis was negative for protein and red blood cells. An electrocardiogram revealed sinus tachycardia. A chest radiograph at admission revealed mild opacities in both lower lobes and the right middle lobe consistent with either atelectasis or pneumonia (Fig. 1). A very small left effusion was also identified.
The additional data reinforce my clinical impression that this process is likely to be infectious. The chest radiograph is consistent with community‐acquired pneumonia, possibly from an atypical pathogen. Given his elevated creatinine, I am also considering a pulmonary‐renal syndrome such as vasculitis, though hematuria was not present. A subphrenic abscess, mesenteric ischemia, or an abdominal wall process (because his abdominal tenderness on exam still needs an explanation) remain possibilities; my suspicion would increase if he does not respond appropriately to therapy for community‐acquired pneumonia.
The clinical team's working diagnosis also was community‐acquired pneumonia. Blood and sputum cultures were obtained, and the patient was treated with intravenous ceftriaxone, azithromycin, and intravenous fluid. By the second day, his creatinine had normalized; however, his hypoxemia had worsened, and he now required supplemental oxygen. His temperature was 39.3C, and his heart rate was 150 beats/minute. The findings of an abdominal ultrasound of the kidneys, spleen, and right upper quadrant were normal.
It is too early to say the patient has failed therapy because a patient can get worse before getting better during the course of antibiotic therapy for community‐acquired pneumonia. Fever, for example, may take up to 7 days to resolve, depending on host factors and the pathogen. Though I typically wait about 72 hours before assuming a patient is not appropriately responding to therapy, the additional information has made me concerned. The degree of tachycardia is significant and warrants an EKG to exclude an arrthymia. I would also repeat the chest radiograph to evaluate for worsening infiltrates or increased pleural effusion.
On the third hospital day, the patient's abdominal pain had decreased with analgesia, but his fever, cough, and dyspnea remained largely unchanged. Antibiotics were changed to intravenous levofloxacin. A repeat chest radiograph revealed elevation of the right hemidiaphragm and bilateral effusions (Fig. 2). An electrocardiogram revealed sinus tachycardia. Blood cultures revealed no growth, and sputum cultures grew oral flora.
A significantly elevated right hemidiaphragm makes me reconsider the diagnosis of simple community‐acquired pneumonia. The differential diagnosis for an elevated hemidiaphragm is best considered by location in relation to the diaphragm. Causes above the diaphragm include rib fracture, atelectasis, pleural thickening, and volume loss of the lung for another reason (e.g., surgery, bronchial obstruction due to tumor or mucus plugging), as well as mimics such as a densely consolidated pneumonia, pulmonary infarction, or a subpulmonary effusion. Diaphragmatic causes include eventration, rupture, phrenic nerve weakness, and intrinsic weakness because of neuromuscular disease (usually bilateral). Causes below the diaphragm that must be considered are subphrenic or liver abscess, liver (and other abdominal) malignancy, pancreatic pseudocyst, and distended bowel. Given the clinical picture, I am focusing below the diaphragmespecially on a possible hepatic or subphrenic abscess (which could be missed on ultrasound) and mimics of it such as dense consolidation or a subpulmonary effusion. Given the lack of response to antibiotics, I need to consider an infection that is not being treated, either because of location (abscess, effusion) or microbiology (tuberculosis, a parasite, a fungus, resistant bacteria). After confirming that the patient has a substantive pleural effusion, he needs a thoracentesis.
On the fourth hospital day, his temperature was 38.8C, and his white blood cell count was 21,000/mL. A right‐sided thoracentesis was performed; approximately 250 cc of fluid was obtained. Pleural fluid analysis revealed bloody fluid, with a white blood cell count of 16,750/mL with 94% neutrophils, 40,000 red blood cells/mL, lactate dehydrogenase of 278 U/L (normal serum value 80200 U/L), protein of 3.7 g/dL, and glucose of 81 mg/dL. A pleural fluid pH was not obtained. A gram stain revealed many white blood cells with no organisms noted. Serum protein was 7.4 g/dL. These results were thought to represent an exudative parapneumonic effusion; levofloxacin and supplemental oxygen were continued.
The pleural fluid appears exudative, but I am not sure this man has a parapneumonic effusion because, despite clinical deterioration, an obvious infiltrate is not seen on interval chest radiography. We must look closely at the fluid because this is a bloody effusion and somewhat atypical for a parapneumonic effusion. Also, the effusion does not appear large enough to explain why he has not improved on the current antibiotics. We should thus reconsider our diagnosis and management. I would obtain additional imaging (such as an abdominal and chest computed tomography [CT]) and perhaps obtain a consultation from the pulmonary team regarding the postulated initial diagnosis of pneumonia with effusion.
On the fifth day of hospitalization, the patient's dyspnea and cough persisted but were improved. His abdominal pain was minimal and felt improved with flatus. Fever continued to 38.8C, and the white blood cell count was 20,000/mL. On examination the patient had decreased breath sounds at the right base and bibasilar crackles. His abdomen was soft, with tenderness in his right upper quadrant only with deep palpation; bowel sounds remained. An ultrasound of the chest was performed to look for a loculated effusion; however, no fluid was identified. The pulmonary consultant thought it likely that the patient had a subpulmonic effusion and recommended CT of the abdomen and chest.
His right upper quadrant tenderness is still unexplained. I would agree with the CT, primarily to evaluate other causes of his elevated diaphragm such as subphrenic or hepatic abscess. For now, I would make no change in antibiotic therapy.
On the sixth hospital day, the patient had an episode of bilious emesis. Chest and abdominal CT revealed collapse of the right middle and lower lobes with a small adjacent effusion, and a 6 6 16 cm abscess intimately opposed to the right lobe of the liver. Extending from the inferior extent of the abscess was a tubular thick‐walled structure connecting to the cecum that was suspicious of a thickened inflamed appendix. There was periappendiceal stranding suggesting inflammation. The small bowel was diffusely dilated up to 4.5 cm, suggesting a small bowel obstruction.
I suspect that his abscess is related to a perforated appendix and that the dilated small bowel is most likely a result of localized irritation of the bowel by the abscess and appendicitis. The collapsed lung is most likely due to local inflammation from the subdiaphragmatic abscess. Treatment should now be changed substantially. I would ask a surgeon to evaluate the patient because the most likely diagnosis is perforated appendicitis with abscess formation.
When the periappendiceal abscess was drained percutaneously, 190 mL of purulent fluid was removed. The cultures were positive for Klebsiella pneumonia, Enterococcus faecalis,and Streptococcus milleri. The patient was given 6 weeks of intravenous antibiotics with improvement in his clinical symptoms. During the interval the findings on his chest radiograph resolved completely. A laproscopic appendectomy 3 months later revealed significant right lower quadrant adhesions. The pathology specimen identified a distorted appendix with regeneration consistent with prior appendicitis. The patient was contacted 4 months after his surgery, and he reported that he was doing well, with no cardiopulmonary or gastrointestinal symptoms.
COMMENTARY
Community‐acquired pneumonia (CAP) is a common cause of acute illness and accounts for nearly 1 million admissions per year in the United States.1 The diagnosis of CAP is made when symptoms including dyspnea, fever, cough, or leukocytosis are present, with confirmation provided by a chest radiograph. Often the diagnosis is clear; however, there is no pathognomonic constellation of signs or symptoms that establish the diagnosis with certainty.2 Many physicians learn that pneumoniaespecially lower‐lobe pneumoniacan lead to abdominal findings such as upper quadrant pain, vomiting, and tenderness to palpation. Conversely, the patient discussed above illustrates that a primary abdominal process can also result in a symptom complex that mimics pneumonia.
The prevalence of CAP coupled with the inherent uncertainty of a clinical diagnosis of CAP leads to an important question: How long is too long before questioning the diagnosis? An analysis of the pneumonia Patient Outcomes Research Trial (PORT) limited to inpatients with CAP examined time to clinical stability. For the majority of patients, abnormal vital signs resolved within 23 days.3 In this study, 29% of patients had severe disease, and not surprisingly, these patients took longer to improve. Using the pneumonia severity index score, which accounts for age, comorbidity, abnormal vital signs, and laboratory data, the patient described in this article would be considered at high risk for death and complication with an estimated mortality of 9%.4 Using a combination of defervescence, resolution of tachycardia, tachypnea, and hypoxemia as markers of clinical stability, a patient like ours should respond within 4 days (with a range of 27 days). On the basis of these dataand the discrepancy between the patient's severe illness and relatively minor pulmonary infiltratesit seems reasonable to have considered this patient as failing CAP therapy as early as the fourth day of hospitalization.
In approximately 10% of hospitalized patients with CAP, the clinical course is protracted.5 When patients do not improve as quickly as expected, the reasons that could explain this should be investigated. In a cohort of 49 patients with CAP who failed therapy the most common reasons for failure to improve were severity of the pneumonia and drug resistance.6 A multicenter study found that the incidence of resistance to penicillin by Streptococcus pneumoniae, the most common bacterial pathogen in CAP, was 30%, with a 4% in vitro resistance rate to ceftriaxone.7 How well in vitro resistance predicts clinical response, however, is unclear. Risk factors for antibiotic resistance include close exposure to children, recent antibiotic use, and recent hospitalization. Immunosuppressive conditions should also be considered in patients who fail to improve. Suppurative complications of pneumoniasuch as empyema, parapneumonic effusion, and lung abscessalso delay recovery.
Another consideration in a patient with what appears to be a nonresolving pneumonia with pleural effusion is that the initial diagnosis is incorrect and the cause is extrathoracic. Pulmonary and cardiac diseases account for more than 90% of effusions, whereas less than 5% of pleural effusions result from intraabdominal causes.8 When should intraabdominal diseases be sought in patients with an effusion, fever, dyspnea, and cough? Light suggests that intraabdominal pathology should be investigated in patients who have pleural effusions without significant parenchymal disease.8 This point is underscored by the experience of our patient, whose chest radiographs showed, despite clinical decline, minimal airspace disease.
Several abdominal entities cause pleural effusion. Pancreatitis, either acute or chronic, with pseudocyst formation is the most common abdominal cause of exudative pleural effusions. Approximately 10% of patients with pancreatic disease will develop effusions, usually left‐sided.9 These left‐sided effusions are also seen in splenic abscesses, usually as a result of endocarditis. Intrahepatic abscess is associated with effusions in 20% of patients.10 A subphrenic abscess, as seen in our patient, is an uncommon cause of exudative pleural effusions. Historically, subphrenic abscesses resulted from a perforated viscus, with ruptured appendicitis the most common cause,11 followed by perforated peptic ulcers and biliary tract disease. With the advent of antibiotics, the causes of subphrenic abscess changed considerably, with the majority of current cases resulting from postsurgical complications.12 The findings of a chest radiograph are abnormal in 80% of patients with subphrenic abscess;1214 an elevated hemidiaphragm and pleural effusion are found in the majority of cases. The symptoms of a subphrenic abscess are nonspecific, and patient's complaints are equally split between predominantly thoracic and predomninantly abdominal complaints.15
Appendicitis, a common disease predominantly of the young, may lead to atypical presentations in older individuals. In a retrospective analysis of 113 patients older than 60 years with appendicitis, 70% presented in an atypical fashion.16 Typical symptoms include right lower quadrant pain, fever, anorexia and a white blood cell count greater than 10,000/mL. Fever was the most frequently absent symptom, seen in only 37% of older patients. In this cohort, approximately one third of older patients waited more than 48 hours prior to presentation. The time between symptom onset and clinical presentation is a strong predictor of perforation risk.17 As in this case, roughly 2% of patients with acute appendicitis will present with perforation and abscess formation.18 In such patients the management is initially conservative. Percutaneous drainage and broad spectrum antibiotics are the treatment of choice, followed by an interval appendectomy in 612 weeks.19 The rationale for delayed surgery is that earlier surgery may disseminate a localized inflammatory process.20
Community‐acquired pneumonia is a more frequent cause of hospital admission than is intraabdominal abscess. Physicians often face the dilemma of when to pursue alternative diagnoses after a patient who is thought to have an atypical presentation of a common disease (ie, CAP) fails to respond to conventional therapy. Although clinicians learn that right upper quadrant pain may be a symptom of pneumonia, our patient revealed that abdominal causes may mimic pneumonia and produce a pleural effusion. Determining whether the primary disease originates above or below the diaphragm is critical to guiding therapy. When patients fail to respond adequately to therapy, clinicians should set a low threshold for deciding to image the abdomen in a patient with modest pulmonary infiltrates, pleural effusion, and abdominal pain.
A 49‐year‐old man presented with 2 days of chills, fever, anorexia, and increased cough and dyspnea. The patient had a history of chronic obstructive pulmonary disease (COPD) and noted that his cough and dyspnea had increased above normal for several days. He was now dyspneic with minimal activity and had slept at a 45‐degree incline the night prior to evaluation due to dyspnea. He noted less improvement than usual with the use of his metered dose inhaler. His cough was occasionally productive of small amounts of white phlegm. He had vomited once. During a coughing episode the patient experienced a sudden onset of sharp right upper quadrant abdominal pain that worsened with coughing and sudden position changes. The patient denied a prior history of abdominal pain or surgery. The patient's last bowel movement was 2 days prior to admission. He denied melena or bright red blood per rectum.
My initial differential diagnosis for this patient's dyspnea and cough is pneumonia, acute exacerbation of COPD, or congestive heart failure. The presence of fever and anorexia increases the likelihood of infectious etiologies, whereas the presence of orthopnea points toward congestive heart failure. Noncardiac processessuch as a large pleural effusion or apical lung diseasecould also cause orthopnea. His abdominal pain could be a result of pneumonia alone (perhaps in the right lower lobe with diaphragmatic irritation), but I am also considering complications of pneumonia such as empyema. Although his abdominal pain, dyspnea, and cough could also be a result of hepatobiliary disease, a perforated viscus, or pancreatitis, we currently have little reason to suspect a direct abdominal etiology. My top diagnosis is community‐acquired pneumonia, perhaps accompanied by pleural effusion.
His medical history was significant for dilated cardiomyopathy and heavy alcohol use. His medications included various meter‐dosed inhalers, bupropion, digoxin, spironolactone, lisinopril, and metoprolol. He had never received corticosteroid therapy and had not previously been hospitalized for COPD‐related problems. He had smoked one pack of cigarettes daily for 40 years.
Heavy alcohol use is associated with an increased risk of several pulmonary infections such as gram‐negative necrotizing pneumonia (classically, Klebsiella pneumoniae), pneumococcal pneumonia, aspiration pneumonia, anaerobic lung abscesses, and tuberculosis. Given his right upper quadrant pain, acute alcoholic hepatitis and alcohol‐related pancreatitis enter the differential. His history of cardiomyopathy makes me consider congestive heart failure as more likely than before, and perhaps his abdominal pain is a result of hepatic congestion from right heart failure. His fever, however, cannot be attributed to cardiac failure. Less likely diagnoses include ischemic conditions related to his cardiomyopathy such as mesenteric ischemia from low perfusion or embolism from a cardiac thrombus. A pulmonary infection remains the most likely diagnosis.
He was an ill‐appearing man in moderate respiratory distress, looking older than his stated age. His temperature was 38.4C, heart rate 129 beats/minute, blood pressure 85/56 mm Hg, respiratory rate 24 breaths/minute, and oxygen saturation 92% on room air. A cardiovascular exam revealed no murmur, gallop, or rub. The jugular venous pulse was not elevated. His lungs were clear to auscultation. Abdominal exam revealed right‐sided abdominal tenderness that appeared to localize to the rectus sheath. Otherwise, the abdomen was soft, with normal bowel sounds and no organomegaly. Rectal examination revealed guaiac negative stool and no focal tenderness. His extremities were normal.
His vital signs are worrisome for impending cardiovascular collapse and shock, possibly due to sepsis. The relatively nonfocal cardiopulmonary exam is surprising given his initial symptoms and makes me wonder if his dyspnea is primarily related to an abdominal process leading to diaphragmatic irritation rather than to a thoracic process. Congestive heart failure seems unlikely given the lack of supportive physical examination findings. His abdominal exam findings are puzzling. Although his abdominal wall tenderness could be benignperhaps from muscular strain or a tear from coughingit could represent a more worrisome process such as infection or a hematoma in the abdominal wall muscles. Mesenteric ischemia is still possible, as the exam is often unimpressive. A hepatic abscess or subphrenic abscess should be considered, as physical exam findings in these conditions can be subtle.
My differential remains relatively unchanged, but I have now put consideration of a hepatic or subphrenic abscess higher on my list. Early empiric broad‐spectrum antibiotics seem necessary.
He had a white blood cell count of 26,700/mL with 92% neutrophils, a hemoglobin of 14.6 g/dL, and a platelet count of 312,000/mL. Sodium was 134 mmol/L, potassium was 4.3 mmol/L, chloride was 94 mmol/L, bicarbonate was 23 mmol/L, blood urea nitrogen was 23 mg/dL, and creatinine was 2.1 mg/dL. The results of the calcium, protein, albumin, and liver function tests were normal. Urinalysis was negative for protein and red blood cells. An electrocardiogram revealed sinus tachycardia. A chest radiograph at admission revealed mild opacities in both lower lobes and the right middle lobe consistent with either atelectasis or pneumonia (Fig. 1). A very small left effusion was also identified.
The additional data reinforce my clinical impression that this process is likely to be infectious. The chest radiograph is consistent with community‐acquired pneumonia, possibly from an atypical pathogen. Given his elevated creatinine, I am also considering a pulmonary‐renal syndrome such as vasculitis, though hematuria was not present. A subphrenic abscess, mesenteric ischemia, or an abdominal wall process (because his abdominal tenderness on exam still needs an explanation) remain possibilities; my suspicion would increase if he does not respond appropriately to therapy for community‐acquired pneumonia.
The clinical team's working diagnosis also was community‐acquired pneumonia. Blood and sputum cultures were obtained, and the patient was treated with intravenous ceftriaxone, azithromycin, and intravenous fluid. By the second day, his creatinine had normalized; however, his hypoxemia had worsened, and he now required supplemental oxygen. His temperature was 39.3C, and his heart rate was 150 beats/minute. The findings of an abdominal ultrasound of the kidneys, spleen, and right upper quadrant were normal.
It is too early to say the patient has failed therapy because a patient can get worse before getting better during the course of antibiotic therapy for community‐acquired pneumonia. Fever, for example, may take up to 7 days to resolve, depending on host factors and the pathogen. Though I typically wait about 72 hours before assuming a patient is not appropriately responding to therapy, the additional information has made me concerned. The degree of tachycardia is significant and warrants an EKG to exclude an arrthymia. I would also repeat the chest radiograph to evaluate for worsening infiltrates or increased pleural effusion.
On the third hospital day, the patient's abdominal pain had decreased with analgesia, but his fever, cough, and dyspnea remained largely unchanged. Antibiotics were changed to intravenous levofloxacin. A repeat chest radiograph revealed elevation of the right hemidiaphragm and bilateral effusions (Fig. 2). An electrocardiogram revealed sinus tachycardia. Blood cultures revealed no growth, and sputum cultures grew oral flora.
A significantly elevated right hemidiaphragm makes me reconsider the diagnosis of simple community‐acquired pneumonia. The differential diagnosis for an elevated hemidiaphragm is best considered by location in relation to the diaphragm. Causes above the diaphragm include rib fracture, atelectasis, pleural thickening, and volume loss of the lung for another reason (e.g., surgery, bronchial obstruction due to tumor or mucus plugging), as well as mimics such as a densely consolidated pneumonia, pulmonary infarction, or a subpulmonary effusion. Diaphragmatic causes include eventration, rupture, phrenic nerve weakness, and intrinsic weakness because of neuromuscular disease (usually bilateral). Causes below the diaphragm that must be considered are subphrenic or liver abscess, liver (and other abdominal) malignancy, pancreatic pseudocyst, and distended bowel. Given the clinical picture, I am focusing below the diaphragmespecially on a possible hepatic or subphrenic abscess (which could be missed on ultrasound) and mimics of it such as dense consolidation or a subpulmonary effusion. Given the lack of response to antibiotics, I need to consider an infection that is not being treated, either because of location (abscess, effusion) or microbiology (tuberculosis, a parasite, a fungus, resistant bacteria). After confirming that the patient has a substantive pleural effusion, he needs a thoracentesis.
On the fourth hospital day, his temperature was 38.8C, and his white blood cell count was 21,000/mL. A right‐sided thoracentesis was performed; approximately 250 cc of fluid was obtained. Pleural fluid analysis revealed bloody fluid, with a white blood cell count of 16,750/mL with 94% neutrophils, 40,000 red blood cells/mL, lactate dehydrogenase of 278 U/L (normal serum value 80200 U/L), protein of 3.7 g/dL, and glucose of 81 mg/dL. A pleural fluid pH was not obtained. A gram stain revealed many white blood cells with no organisms noted. Serum protein was 7.4 g/dL. These results were thought to represent an exudative parapneumonic effusion; levofloxacin and supplemental oxygen were continued.
The pleural fluid appears exudative, but I am not sure this man has a parapneumonic effusion because, despite clinical deterioration, an obvious infiltrate is not seen on interval chest radiography. We must look closely at the fluid because this is a bloody effusion and somewhat atypical for a parapneumonic effusion. Also, the effusion does not appear large enough to explain why he has not improved on the current antibiotics. We should thus reconsider our diagnosis and management. I would obtain additional imaging (such as an abdominal and chest computed tomography [CT]) and perhaps obtain a consultation from the pulmonary team regarding the postulated initial diagnosis of pneumonia with effusion.
On the fifth day of hospitalization, the patient's dyspnea and cough persisted but were improved. His abdominal pain was minimal and felt improved with flatus. Fever continued to 38.8C, and the white blood cell count was 20,000/mL. On examination the patient had decreased breath sounds at the right base and bibasilar crackles. His abdomen was soft, with tenderness in his right upper quadrant only with deep palpation; bowel sounds remained. An ultrasound of the chest was performed to look for a loculated effusion; however, no fluid was identified. The pulmonary consultant thought it likely that the patient had a subpulmonic effusion and recommended CT of the abdomen and chest.
His right upper quadrant tenderness is still unexplained. I would agree with the CT, primarily to evaluate other causes of his elevated diaphragm such as subphrenic or hepatic abscess. For now, I would make no change in antibiotic therapy.
On the sixth hospital day, the patient had an episode of bilious emesis. Chest and abdominal CT revealed collapse of the right middle and lower lobes with a small adjacent effusion, and a 6 6 16 cm abscess intimately opposed to the right lobe of the liver. Extending from the inferior extent of the abscess was a tubular thick‐walled structure connecting to the cecum that was suspicious of a thickened inflamed appendix. There was periappendiceal stranding suggesting inflammation. The small bowel was diffusely dilated up to 4.5 cm, suggesting a small bowel obstruction.
I suspect that his abscess is related to a perforated appendix and that the dilated small bowel is most likely a result of localized irritation of the bowel by the abscess and appendicitis. The collapsed lung is most likely due to local inflammation from the subdiaphragmatic abscess. Treatment should now be changed substantially. I would ask a surgeon to evaluate the patient because the most likely diagnosis is perforated appendicitis with abscess formation.
When the periappendiceal abscess was drained percutaneously, 190 mL of purulent fluid was removed. The cultures were positive for Klebsiella pneumonia, Enterococcus faecalis,and Streptococcus milleri. The patient was given 6 weeks of intravenous antibiotics with improvement in his clinical symptoms. During the interval the findings on his chest radiograph resolved completely. A laproscopic appendectomy 3 months later revealed significant right lower quadrant adhesions. The pathology specimen identified a distorted appendix with regeneration consistent with prior appendicitis. The patient was contacted 4 months after his surgery, and he reported that he was doing well, with no cardiopulmonary or gastrointestinal symptoms.
COMMENTARY
Community‐acquired pneumonia (CAP) is a common cause of acute illness and accounts for nearly 1 million admissions per year in the United States.1 The diagnosis of CAP is made when symptoms including dyspnea, fever, cough, or leukocytosis are present, with confirmation provided by a chest radiograph. Often the diagnosis is clear; however, there is no pathognomonic constellation of signs or symptoms that establish the diagnosis with certainty.2 Many physicians learn that pneumoniaespecially lower‐lobe pneumoniacan lead to abdominal findings such as upper quadrant pain, vomiting, and tenderness to palpation. Conversely, the patient discussed above illustrates that a primary abdominal process can also result in a symptom complex that mimics pneumonia.
The prevalence of CAP coupled with the inherent uncertainty of a clinical diagnosis of CAP leads to an important question: How long is too long before questioning the diagnosis? An analysis of the pneumonia Patient Outcomes Research Trial (PORT) limited to inpatients with CAP examined time to clinical stability. For the majority of patients, abnormal vital signs resolved within 23 days.3 In this study, 29% of patients had severe disease, and not surprisingly, these patients took longer to improve. Using the pneumonia severity index score, which accounts for age, comorbidity, abnormal vital signs, and laboratory data, the patient described in this article would be considered at high risk for death and complication with an estimated mortality of 9%.4 Using a combination of defervescence, resolution of tachycardia, tachypnea, and hypoxemia as markers of clinical stability, a patient like ours should respond within 4 days (with a range of 27 days). On the basis of these dataand the discrepancy between the patient's severe illness and relatively minor pulmonary infiltratesit seems reasonable to have considered this patient as failing CAP therapy as early as the fourth day of hospitalization.
In approximately 10% of hospitalized patients with CAP, the clinical course is protracted.5 When patients do not improve as quickly as expected, the reasons that could explain this should be investigated. In a cohort of 49 patients with CAP who failed therapy the most common reasons for failure to improve were severity of the pneumonia and drug resistance.6 A multicenter study found that the incidence of resistance to penicillin by Streptococcus pneumoniae, the most common bacterial pathogen in CAP, was 30%, with a 4% in vitro resistance rate to ceftriaxone.7 How well in vitro resistance predicts clinical response, however, is unclear. Risk factors for antibiotic resistance include close exposure to children, recent antibiotic use, and recent hospitalization. Immunosuppressive conditions should also be considered in patients who fail to improve. Suppurative complications of pneumoniasuch as empyema, parapneumonic effusion, and lung abscessalso delay recovery.
Another consideration in a patient with what appears to be a nonresolving pneumonia with pleural effusion is that the initial diagnosis is incorrect and the cause is extrathoracic. Pulmonary and cardiac diseases account for more than 90% of effusions, whereas less than 5% of pleural effusions result from intraabdominal causes.8 When should intraabdominal diseases be sought in patients with an effusion, fever, dyspnea, and cough? Light suggests that intraabdominal pathology should be investigated in patients who have pleural effusions without significant parenchymal disease.8 This point is underscored by the experience of our patient, whose chest radiographs showed, despite clinical decline, minimal airspace disease.
Several abdominal entities cause pleural effusion. Pancreatitis, either acute or chronic, with pseudocyst formation is the most common abdominal cause of exudative pleural effusions. Approximately 10% of patients with pancreatic disease will develop effusions, usually left‐sided.9 These left‐sided effusions are also seen in splenic abscesses, usually as a result of endocarditis. Intrahepatic abscess is associated with effusions in 20% of patients.10 A subphrenic abscess, as seen in our patient, is an uncommon cause of exudative pleural effusions. Historically, subphrenic abscesses resulted from a perforated viscus, with ruptured appendicitis the most common cause,11 followed by perforated peptic ulcers and biliary tract disease. With the advent of antibiotics, the causes of subphrenic abscess changed considerably, with the majority of current cases resulting from postsurgical complications.12 The findings of a chest radiograph are abnormal in 80% of patients with subphrenic abscess;1214 an elevated hemidiaphragm and pleural effusion are found in the majority of cases. The symptoms of a subphrenic abscess are nonspecific, and patient's complaints are equally split between predominantly thoracic and predomninantly abdominal complaints.15
Appendicitis, a common disease predominantly of the young, may lead to atypical presentations in older individuals. In a retrospective analysis of 113 patients older than 60 years with appendicitis, 70% presented in an atypical fashion.16 Typical symptoms include right lower quadrant pain, fever, anorexia and a white blood cell count greater than 10,000/mL. Fever was the most frequently absent symptom, seen in only 37% of older patients. In this cohort, approximately one third of older patients waited more than 48 hours prior to presentation. The time between symptom onset and clinical presentation is a strong predictor of perforation risk.17 As in this case, roughly 2% of patients with acute appendicitis will present with perforation and abscess formation.18 In such patients the management is initially conservative. Percutaneous drainage and broad spectrum antibiotics are the treatment of choice, followed by an interval appendectomy in 612 weeks.19 The rationale for delayed surgery is that earlier surgery may disseminate a localized inflammatory process.20
Community‐acquired pneumonia is a more frequent cause of hospital admission than is intraabdominal abscess. Physicians often face the dilemma of when to pursue alternative diagnoses after a patient who is thought to have an atypical presentation of a common disease (ie, CAP) fails to respond to conventional therapy. Although clinicians learn that right upper quadrant pain may be a symptom of pneumonia, our patient revealed that abdominal causes may mimic pneumonia and produce a pleural effusion. Determining whether the primary disease originates above or below the diaphragm is critical to guiding therapy. When patients fail to respond adequately to therapy, clinicians should set a low threshold for deciding to image the abdomen in a patient with modest pulmonary infiltrates, pleural effusion, and abdominal pain.
- ,,, et al.The cost of treating community‐acquired pneumonia.Clin Ther.1998;20:820–827.
- ,,.Does this patient have community‐acquired pneumonia? Diagnosing pneumonia by history and physical examination.JAMA.1997;278:1440–1445.
- ,,, et al.Time to clinical stability in patients hospitalized with community acquired pneumonia. Implications for practice guidelines.JAMA.1998;279:1452–1457.
- ,,, et al.A prediction rule to identify low‐risk patients with community‐acquired pneumonia.N Engl J Med.1997;336:243–250.
- ,,, et al.Utility of fiberoptic bronchoscopy in non resolving pneumonia.Chest.1990;98:1322–1326.
- ,,, et al.Antimicrobial treatment failures in patients with community acquired pneumonia. Causes and prognostic implications.Am J Respir Crit Care Med.2000;162:154–160.
- ,,, et al.Antimicrobial resistance with Streptococcus pneumoniae in the United States, 1997–98.Emerg Infect Dis.1999;5:757–765.
- ,.Pleural effusion. In:Murray JF,Nadel JA, eds.Textbook of respiratory medicine. 3rd ed.Philadelphia:WB Saunders,2000:2013–2041.
- ,,.Significance of pleural effusion in patients with acute pancreatitis.Am J Gastroenterol.1992;87:871–874.
- .Exudative pleural effusions secondary to gastrointestinal diseases.Clin Chest Med.1985;6(1):103–111.
- .Subphrenic abscess.Ann Surg.1963;158:240–248.
- ,,,.Upper abdominal abscess: a continuing and deadly problem.Am J Roentgenol.1980;134:759–765.
- .Subphrenic abscess. A clinical study of 101 cases.Acta Chir Scand.1959;117:388–408.
- ,,.Subphrenic abscess a continuing hazard.Am J Surg.1969:117–122.
- ,.Subphrenic abscess: a thoracoabdominal clinical complex. The changing picture with antibiotics.Am J Surg.1964;108:165–172.
- ,.What have we learned over the past 20 years about appendicitis in the elderly.Am J Surg.2003;185:198–201.
- ,,, et al.Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies.Am Surg.2000;66:548–554.
- ,,.Appendicitis with a palpable mass.Ann Surg.1981;193:227–229.
- ,,, et al.Nonoperative management of perforated appendicitis without periappendiceal mass.Am J Surg.2000;179:177–181.
- ,,.Appendix. In:Townsend CM, ed.Sabiston textbook of surgery. The biologic basis of modern surgical practice. 16th ed.Philadelphia:W. B. Saunders,2001:917–928.
- ,,, et al.The cost of treating community‐acquired pneumonia.Clin Ther.1998;20:820–827.
- ,,.Does this patient have community‐acquired pneumonia? Diagnosing pneumonia by history and physical examination.JAMA.1997;278:1440–1445.
- ,,, et al.Time to clinical stability in patients hospitalized with community acquired pneumonia. Implications for practice guidelines.JAMA.1998;279:1452–1457.
- ,,, et al.A prediction rule to identify low‐risk patients with community‐acquired pneumonia.N Engl J Med.1997;336:243–250.
- ,,, et al.Utility of fiberoptic bronchoscopy in non resolving pneumonia.Chest.1990;98:1322–1326.
- ,,, et al.Antimicrobial treatment failures in patients with community acquired pneumonia. Causes and prognostic implications.Am J Respir Crit Care Med.2000;162:154–160.
- ,,, et al.Antimicrobial resistance with Streptococcus pneumoniae in the United States, 1997–98.Emerg Infect Dis.1999;5:757–765.
- ,.Pleural effusion. In:Murray JF,Nadel JA, eds.Textbook of respiratory medicine. 3rd ed.Philadelphia:WB Saunders,2000:2013–2041.
- ,,.Significance of pleural effusion in patients with acute pancreatitis.Am J Gastroenterol.1992;87:871–874.
- .Exudative pleural effusions secondary to gastrointestinal diseases.Clin Chest Med.1985;6(1):103–111.
- .Subphrenic abscess.Ann Surg.1963;158:240–248.
- ,,,.Upper abdominal abscess: a continuing and deadly problem.Am J Roentgenol.1980;134:759–765.
- .Subphrenic abscess. A clinical study of 101 cases.Acta Chir Scand.1959;117:388–408.
- ,,.Subphrenic abscess a continuing hazard.Am J Surg.1969:117–122.
- ,.Subphrenic abscess: a thoracoabdominal clinical complex. The changing picture with antibiotics.Am J Surg.1964;108:165–172.
- ,.What have we learned over the past 20 years about appendicitis in the elderly.Am J Surg.2003;185:198–201.
- ,,, et al.Appendicitis: why so complicated? Analysis of 5755 consecutive appendectomies.Am Surg.2000;66:548–554.
- ,,.Appendicitis with a palpable mass.Ann Surg.1981;193:227–229.
- ,,, et al.Nonoperative management of perforated appendicitis without periappendiceal mass.Am J Surg.2000;179:177–181.
- ,,.Appendix. In:Townsend CM, ed.Sabiston textbook of surgery. The biologic basis of modern surgical practice. 16th ed.Philadelphia:W. B. Saunders,2001:917–928.