User login
A change of heart
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
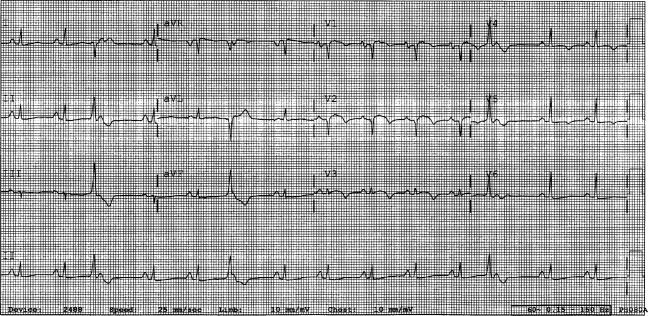
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
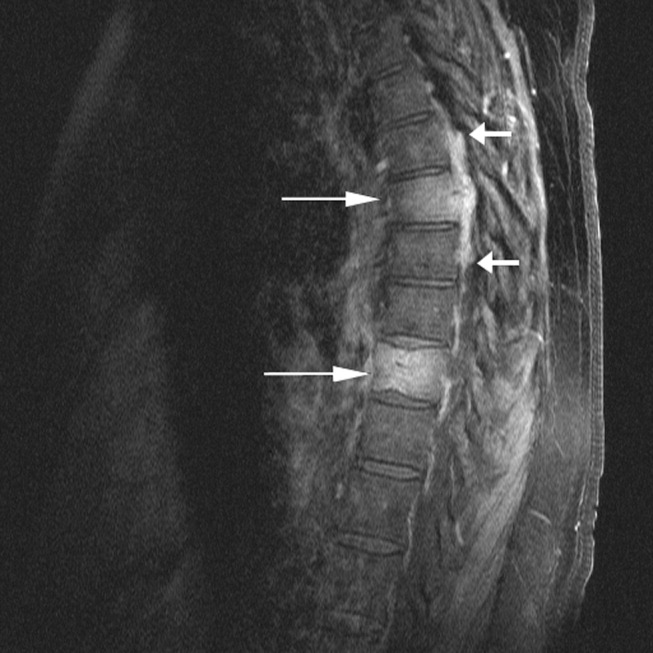
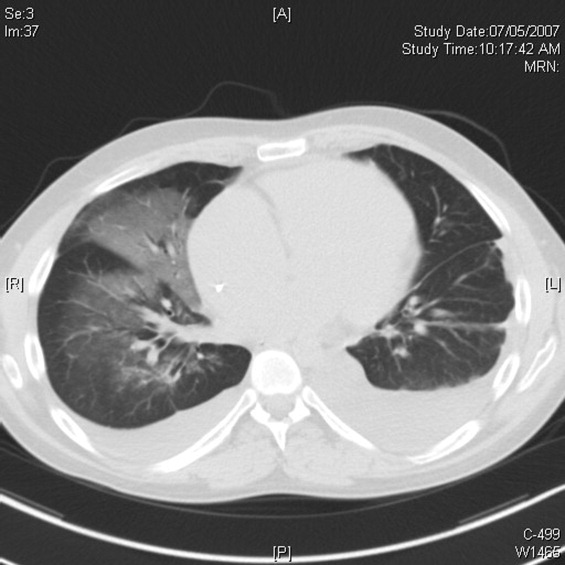
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
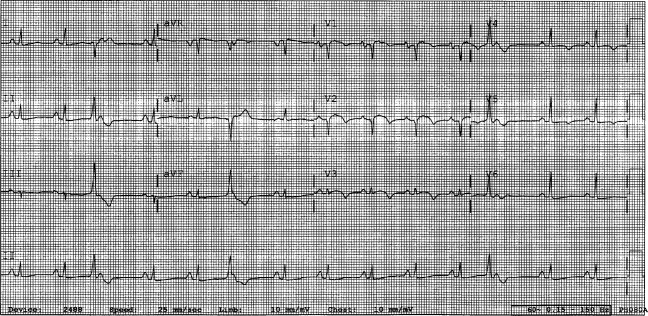
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
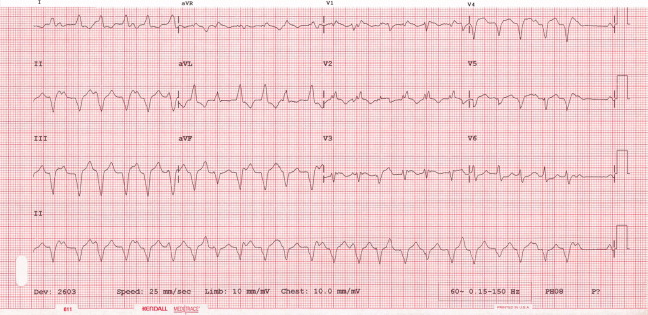
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
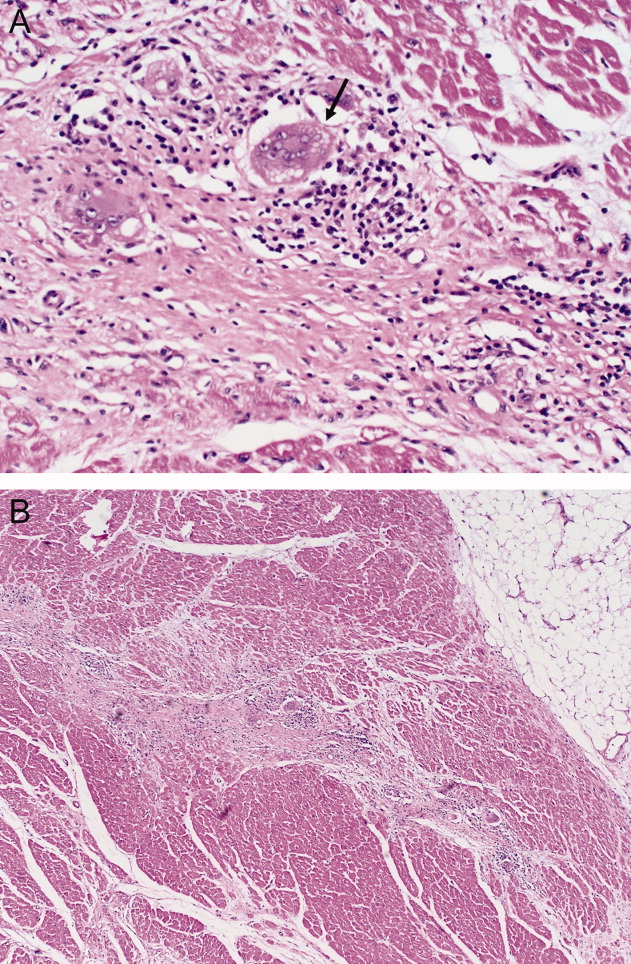
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 29‐year‐old man developed palpitations and dyspnea while loading boxes into a truck. In the emergency department, telemetry demonstrated a wide‐complex tachycardia at a rate of 204 beats per minute. The patient spontaneously cardioverted to sinus rhythm (Figure 1) before direct current cardioversion was performed.
Wide‐complex tachycardia is usually explained by a supraventricular tachycardia with aberrant ventricular conduction or a ventricular tachycardia. Although algorithms exist to guide the clinician in parsing out those etiologies, often the knowledge of underlying structural cardiac disease is most informative. In patients with a history of myocardial infarction, greater than 95% of wide‐complex tachycardia is ventricular tachycardia. The ventricular ectopy, T‐wave inversion or flattening, and poor R‐wave progression are suggestive of a cardiomyopathy, either acute or chronic. A pressing concern, especially with the Q waves and concave ST morphology in V1 and V2, would be coronary ischemia. His age makes this less likely, but an aberrant coronary circulation or drug use could account for it.
Over the past 2 years, the patient had several episodes of sustained palpitations, which terminated after several minutes. Previously, the patient exercised frequently including playing rugby in college. However, over the past year he experienced difficulty climbing stairs due to shortness of breath, which he attributed to deconditioning and smoking. He had no significant medical history, was not taking any medications, nor did he use recreational stimulants. He drank alcohol occasionally. He had no risk factors for the human immunodeficiency virus (HIV). Both of the patient's parents were alive and well. There was no family history of sudden cardiac death.
The duration of symptoms suggests that this is a chronic cardiomyopathy rather than acute myocarditis or acute ischemia, acknowledging that either one could be superimposed. The absence of family history lowers the likelihood of heritable causes of arrhythmia that may accompany a structurally normal (eg, long QT syndrome) or abnormal (eg, hypertrophic cardiomyopathy) heart, although penetrance can be variable. What might account for a cardiomyopathy in a young person? Most cases are probably idiopathic, but etiologies that diverge from the usual suspects of coronary artery disease, hypertension, and valvular disease, which affect an older population, include antecedent viral myocarditis, substance abuse, HIV, or infiltrative disorders such as sarcoidosis.
The patient's pulse was 92 beats per minute and regular and the blood pressure was 96/52 mm Hg. The jugular venous pressure was elevated with prominent v‐waves, the point of maximal impulse was diffuse, there were no extra heart sounds or murmurs, and an enlarged liver was detected. An echocardiogram demonstrated left ventricular dysfunction with an ejection fraction of 30%, severe enlargement of the right atrium and right ventricle, and moderate tricuspid regurgitation. Cardiac catheterization revealed normal coronary arteries without evidence of pulmonary hypertension or intracardiac shunt.
The physical examination and echocardiographic findings of right‐sided failure are unusual given the absence of pulmonary hypertension or intracardiac shunt, and could prompt repeat of the hemodynamic measurements and/or investigations for pulmonary disease that may account for right‐sided pressure overload (in addition to that caused by left ventricular failure). An alternative explanation would be a cardiomyopathic process that preferentially involves the right side of the heart, such as arrhythmogenic right ventricular dysplasia (ARVD), but that would not satisfactorily explain the significant decline in left ventricular function. An acute right ventricular infarction could cause his acute symptoms and his examination and echocardiographic findings, but not the underlying chronic illness. It is common to see patients with long‐standing biventricular failure who present with prominent signs of right‐sided failure (elevated neck veins, hepatomegaly, and edema) but limited or no signs of left‐sided failure (rales) to match their degree of volume overload or dyspnea.
Cardiac magnetic resonance imaging (MRI) revealed a dilated right ventricle with extensive hyperenhancement, a right ventricular ejection fraction of 9%, and moderate left ventricular dysfunction (Figure 2). Electrophysiology testing induced both nonsustained polymorphic and monomorphic ventricular tachycardia. Late potentials were detected on a signal‐averaged electrocardiogram. A single‐chamber cardioverter defibrillator was implanted and the patient was discharged on carvedilol, lisinopril, and spironolactone. An HIV‐1 antibody was negative and a thyroid‐stimulating hormone concentration was within normal limits.
Assuming that accurate evaluation of the pulmonary circulation has been undertaken to exclude pulmonary hypertension, the enlarged and hyperenhanced right ventricle on MRI suggests a process that preferentially infiltrates the right ventricular myocardium, and may secondarily affect the left ventricle either by further infiltration or as a consequence of altered mechanics from the highly dysfunctional right ventricle. ARVD affects the right ventricle, but it is possible that another infiltrative cardiomyopathy, such as sarcoid or an antecedent viral infection, could be restricted in its distribution. Late‐potentials identified on signal average electrocardiograms indicate areas of abnormal conduction that may serve as substrate for reentrant ventricular arrhythmias. They are, however, nonspecific, as they are seen in a variety of myocardial diseases.
The patient continued to have progressive dyspnea and was readmitted after receiving an appropriate implantable cardioverter defibrillator shock for ventricular tachycardia. Recurrent slow ventricular tachycardia (Figure 3) was treated with supplemental beta‐blockade and amiodarone (10 g total). Repeat echocardiography demonstrated severe left ventricular dysfunction with an ejection fraction of less than 15%. There were no recurrences of ventricular arrhythmias and the patient was discharged and referred for cardiac transplant evaluation for ARVD.
This degree of left ventricular dysfunction is unlikely to be accounted for by altered mechanics and interactions from a failing right ventricle alone and frames this as a biventricular cardiomyopathy, which has an extensive differential diagnosis and requires information from the general medical evaluation.
On routine laboratory testing 6 months later, a serum aspartate aminotransferase of 79 units/L and a serum alanine aminotransferase of 118 units/L were found. Bilirubin, albumin, and alkaline phosphatase were normal. The transaminase levels had been normal on initial evaluation. The patient reported that 2 paternal uncles had end‐stage nonalcoholic cirrhosis. Transjugular liver biopsy was consistent with mild lobular hepatitis with mild portal fibrosis with a few lobular collections of mononuclear cells. There was no evidence of iron overload. The hepatic venogram and transhepatic pressure gradient (2 mm Hg) were normal.
The elevated transaminase levels could be due to amiodarone‐associated hepatotoxicity, hepatic congestion, or a primary liver disease. It is important to consider combined cardiohepatic syndromes such as hemochromatosis, sarcoidosis, or amyloidosis. The relatively normal liver histology and normal hepatic hemodynamics do not suggest a significant primary intrinsic liver disease. The 2 uncles with cirrhosis could suggest a heritable liver disease, although cirrhosis in multiple family members is frequently accounted for by shared habits such as alcohol consumption or excessive caloric intake. Liver disorders with a genetic component, such as hemochromatosis, Wilson's disease, and alpha‐1‐antitrypsin deficiency are mostly autosomal recessive, which would make this pattern of transmission unusual. Furthermore, aside from hemochromatosis, these genetic hepatic disorders have few cardiac manifestations. Right‐sided congestion and amiodarone appear to be the most likely explanations of his liver abnormalities.
Pulmonary function testing revealed normal lung volumes without obstruction, but the diffusing capacity for carbon monoxide was substantially reduced. Computed tomography of the chest identified scattered ground‐glass opacities as well as small nodules with an upper lobe distribution (Figure 4). Although not reported on the initial interpretation, review of a chest x‐ray taken 6 months previously also demonstrated small nodules in the upper lobe distribution. Bronchoscopic examination was normal. Bronchioalveolar lavage fluid stains and cultures for bacteria, mycobacteria, Pneumocystis, and fungus were negative. Transbronchial biopsies of the right middle lobe had no evidence of infection, malignancy, or granulomatous inflammation. The patient continued to have progressive New York Heart Association Class IV heart failure symptoms. Repeat right heart catheterization was notable for a cardiac index of 1.4 L/minute/m2. The mean pulmonary artery pressure was 20 mm Hg. An intraaortic balloon pump was placed for refractory cardiogenic shock.
The reduced diffusion capacity and ground‐glass opacities suggest an interstitial process, which may have been missed on transbronchial biopsy because of sampling error. His pulmonary disease is likely another manifestation of his infiltrative cardiac disease. The constellation of cardiac, pulmonary, and hepatic involvement in the context of progressive dyspnea over 2 years is suggestive of sarcoidosis although the absence of hilar lymphadenopathy and 2 biopsy specimens without granulomas argue against the diagnosis, and the effects of amiodarone on the latter 2 organs cannot be ignored. On the limited menu of pharmacologic treatments that may treat this severe and progressive cardiomyopathy are steroids, which makes a diligent search for a steroid‐responsive syndrome important. Therefore, despite the negative studies, sarcoidosis must be investigated to the fullest extent with either an endomyocardial biopsy or surgical lung biopsy.
The patient underwent cardiac transplantation. The native heart was found to have right ventricular thinning, which was most notable at the right ventricular outflow tract. Microscopic examination revealed extensive fibrosis and granulomatous inflammation (Figure 5) with scarring typical of cardiac sarcoidosis. Six months after cardiac transplantation, the patient is doing well on prednisone, tacrolimus, and mycophenolate mofetil. Follow‐up chest x‐rays show resolution of the pulmonary nodules.
COMMENTARY
Cardiomyopathy in a young person is a relatively uncommon clinical event that prompts consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in older adults. This case highlights the challenges of arriving at a diagnosis in the absence of a gold standard, and the greater challenges of modifying initial diagnostic impressions as new clinical data become available.
After encountering ventricular tachycardia and right ventricular dysfunction in a young patient, the clinicians arrived at the diagnosis of ARVD. This rare and progressive disorder is associated with up to 20% of ventricular arrhythmias and sudden death in the young,1, 2 but can be challenging to diagnose. Despite common referrals for cardiac MRI to exclude ARVD, cardiac MRI is not the gold standard for diagnosis and is the most common method of misdiagnosis of ARVD.3 A diagnosis of ARVD requires the presence of 2 major, 1 major and 2 minor, or 4 minor International Task Force criteria (Table 1).4, 5 While the diagnostic criteria provide standardization across populations (eg, in clinical studies), additional considerations are needed in the management of individual patients. Scoring systems serve as a tool, but the final diagnosis requires balancing such criteria with competing hypotheses. This dilemma is familiar to clinicians considering other less common conditions such as amyotrophic lateral sclerosis (World Neurology Foundation), rheumatic fever (Jones criteria), or systemic lupus erythematosus (American College of Rheumatology). This patient's cardiac MRI findings, precordial T‐wave inversions, frequent ventricular ectopy, and late potentials on a signal‐averaged electrocardiogram fulfilled the International Task Force criteria for a diagnosis of ARVD. Discordant information included the right bundle branch pattern of the ventricular tachycardia, which suggested left ventricular origin, as opposed to the more common left bundle branch pattern observed in ARVD, and the absence of a family history. In addition, in U.S. populations only 25% of cases present with heart failure and fewer than 5% develop biventricular failure.6 Nonetheless, this patient's imaging evidence of right ventricular structural abnormalities and dysfunction and electrocardiographic abnormalities coupled with the absence of obvious systemic disease made ARVD the logical working diagnosis.
| Major | Minor | |
|---|---|---|
| ||
| I. Global and/or regional dysfunction and structural alterations | Severe dilation and reduction of right ventricular ejection fraction, localized right ventricular aneurysms | Mild right ventricular dilatation and/or reduced ejection fraction |
| II. Endomyocardial biopsy | Fibrofatty replacement of myocardium | |
| III. Repolarization abnormalities | T‐wave inversion in leads V1‐V3 or beyond | |
| IV. Depolarization/conduction abnormalities | Epsilon waves or localized QRS prolongation (>110 msec) in leads V1‐V3 | Late potentials on signal‐averaged electrocardiogram |
| V. Arrhythmias | Left bundle branch block‐type ventricular tachycardia (sustained and nonsustained) or frequent ventricular extra systoles (>1,000/24 hours) | |
| VI. Family history | Familial disease confirmed at necropsy or surgery | Familial history of premature sudden death (<35 years old) or clinical diagnosis based on present criteria |
When more widespread manifestations developed, namely hepatic and pulmonary abnormalities, each was investigated with imaging and biopsy. Once a multisystem illness became apparent, the discussant reframed the patient's illness to include other diagnostic possibilities. In practice it is difficult to reverse a working diagnosis despite contradictory evidence because of the common pitfall of anchoring bias. Tversky and Kahneman7 were the first to describe the cognitive processes behind probability assessment and decision making in time‐sensitive situations. Under these conditions, decision makers tend to focus on the first symptom, striking feature, or diagnosis and anchor subsequent probabilities to that initial presentation. Once a decision or diagnosis has been reached, clinicians tend to interpret subsequent findings in the context of the original diagnosis rather than reevaluating their initial impression. In the setting of a known diagnosis of ARVD, 3 separate diagnoses (ARVD, amiodarone‐associated lung injury, and amiodarone‐induced hepatic dysfunction) were considered by the treating physicians. The initial diagnosis of ARVD followed by the sequential rather than simultaneous manifestations of sarcoidosis made arriving at the revised diagnosis even more challenging.
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular dysplasia.8, 9 The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, myocarditis, idiopathic cardiomyopathy, and sarcoidosis. Cardiac sarcoidosis can present as ventricular ectopy, sustained ventricular arrhythmias, asymptomatic ventricular dysfunction, heart failure, or sudden death.10 Although 25% of patients with sarcoidosis have evidence of cardiac involvement at autopsy, only 5% have clinical manifestations.11 Those patients with clinical evidence of cardiac sarcoidosis have a wide range of clinical findings (Table 2). While the patient's cardiomyopathy was advanced, it is possible that earlier administration of corticosteroid therapy may have arrested his progressive biventricular failure. As clinicians, we should always remember to force ourselves to broaden our differential diagnosis when new findings become available, especially those that point to a systemicrather than an organ‐specificdisorder. In this case, while the original diagnostic findings were accurate and strongly suggested ARVD, a change of heart was needed to arrive at the ultimate diagnosis.
| Clinical Manifestation | Prevalence (%) |
|---|---|
| Atrioventricular block | 40 |
| Bundle branch block | 40 |
| Supraventricular tachycardia | 20 |
| Ventricular arrhythmias | 25 |
| Heart failure | 25 |
| Sudden cardiac death | 35 |
KEY POINTS FOR HOSPITALISTS
-
Cardiomyopathy in a young person requires consideration of a broad differential diagnosis that is notably different from the most common etiologies of cardiomyopathy in the elderly.
-
Anchoring bias is a common pitfall in clinical decision making. When new or contradictory findings are uncovered, clinicians should reevaluate their initial impression to ensure it remains the most likely diagnosis.
-
Cardiac sarcoidosis is a mimic of ARVD and should be considered when evaluating a patient for right ventricular cardiomyopathy. The differential diagnosis of ARVD includes idiopathic ventricular tachycardia, right ventricular outflow tract tachycardia, myocarditis, idiopathic dilated cardiomyopathy, and sarcoidosis.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.
- ,,, et al.Right ventricular dysplasia: a report of 24 adult cases.Circulation.1982;65:384–398.
- ,,,,.Right ventricular cardiomyopathy and sudden death in young people.N Engl J Med.1988;318:129–133.
- ,,, et al.Misdiagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy.J Cardiovasc Electrophysiol.2004;15:300–306.
- ,,, et al.Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.Br Heart J.1994;71:215–218.
- ,,, et al.Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia.Heart Rhythm.2005;2:1188–1194.
- ,,, et al.Arrhythmogenic right ventricular dysplasia: a United States experience.Circulation.2005;112:3823–3832.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,.Unusual presentation of cardiac sarcoidosis.Congest Heart Fail.2007;13:116–118.
- ,,, et al.Cardiac sarcoidosis mimicking right ventricular dysplasia.Circ J.2003;67:169–171.
- ,,,,.Refractory ventricular tachycardia secondary to cardiac sarcoid: electrophysiologic characteristics, mapping, and ablation.Heart Rhythm.2006;3:924–929.
- ,.Sarcoid heart disease: clinical course and treatment.Int J Cardiol.2004;97:173–182.
Short of breath, not short of diagnoses
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 71‐year‐old African‐American woman presented to the emergency department with chest pain, shortness of breath, and cough. She had initially presented to her primary care physician 2 weeks previously complaining of worsening cough and shortness of breath and was told to continue her inhaled albuterol and glucocorticoids and was prescribed a prednisone taper and an unknown course of antibiotics. She noted no improvement in her symptoms despite compliance with this treatment. Three days prior to admission she described the gradual onset of left‐sided pleuritic chest pain with continued cough, associated with yellow sputum and worsening dyspnea. Review of systems was remarkable for generalized weakness and malaise. She denied fever, chills, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, diarrhea, nausea, vomiting, or abdominal pain.
Her past medical history included a diagnosis of chronic obstructive pulmonary disease (COPD) but pulmonary function tests 7 years prior to admission showed an forced expiratory volume in the first second (FEV1)/forced vital capacity (FVC) ratio of 81%. She had a 30 pack‐year history of smoking, but quit 35 years ago. The patient also carried a diagnosis of heart failure, but an echocardiogram done 1 year ago demonstrated a left ventricular ejection fraction of 65% to 70% without diastolic dysfunction but mild right ventricular dilation and hypertrophy. Additionally, she had known nonobstructive coronary atherosclerotic heart disease, dyslipidemia, hypertension, morbid obesity, depression, and a documented chronic right hemidiaphragm elevation.
At this point the history suggests that the patient does not have a clear diagnosis of COPD. The lack of definitive spirometry evidence of chronic airway obstruction concerns me; I think that she may have been mistakenly treated with chronic inhaled steroids and doses of antibiotics for an acute exacerbation of chronic lung disease. Additional review of her history gives some indication of advanced lung disease, with her recent echocardiogram showing strain on the right ventricle with right ventricular hypertrophy and dilation, but there is no mention of the presence or severity of pulmonary hypertension. Nonetheless, I would be concerned that she probably has underlying significant cor pulmonale.
The patient now re‐presents with a worsening of her pulmonary symptoms. Her left‐sided pleuritic pain would make me concerned that she had a pulmonary embolus (PE). This morbidly obese patient with new pulmonary symptoms, right ventricular strain on her previous echocardiogram, and a persistent elevated right hemidiaphragm suggests a presentation of another PE.
At this time I cannot rule out other common possibilities such as infectious pneumonia. If she does have pneumonia, I would be concerned she could be harboring a multidrug‐resistant bacterial infection given her recent course of antibiotics in addition to her use of both chronic inhaled and intermittent oral glucocorticoids.
After gathering the rest of her full medical history, I would focus my physical exam on looking for evidence of parenchymal lung disease, signs of pulmonary hypertension, and pneumonia.
Her surgical history includes a previous hysterectomy, cholecystectomy, hernia repair, and left hepatic lobectomy for a benign mass. Her outpatient medications were ibuprofen, bupropion, fluvastatin, atenolol, potassium, aspirin, clopidogrel, albuterol inhaler, fluticasone/salmeterol inhaler, and omeprazole. She reports an allergy to penicillin and to sulfa drugs. Her mother died of an unknown cancer at age 77 years. She denied any international travel and she has always lived in Georgia.
The patient has been retired since 1992, having previously worked for the U.S. Postal Service. She admits to occasional alcohol intake (2 to 3 drinks a month). No recent travel, surgery, or prolonged immobilization was noted.
On initial examination she was alert and mentally appropriate, but appeared to be in mild respiratory distress with a respiratory rate of 28 breaths/minute. Her blood pressure (BP) was 99/70, heart rate 102, temperature of 38.2C, and oxygen saturation of 93% on room air and 97% on 2 L of oxygen via nasal cannula. Auscultation of her lungs revealed crackles over her left anterior lung field, bronchial breath sounds in the left posterior midlung, and bibasilar crackles. No wheezing was noted. Her cardiovascular exam and the remainder of her physical exam were unremarkable except for morbid obesity.
While my initial thoughts were leaning toward an exacerbation of chronic lung disease or possibly a new PE, at this moment, infection seems more likely. Indeed, her pulmonary findings suggest a left‐sided inflammatory process, and her vital signs meet criteria for systemic inflammatory response syndrome (SIRS). My primary concern is sepsis due to a drug‐resistant bacterial infection, including Staphylococcus aureus or gram‐negative bacteria or possibly more unusual organisms such as Nocardia or fungi, due to her recent use of antibiotics and chronic inhaled steroid use and recent course of oral glucocorticoids.
Conversely, the SIRS could be a manifestation of a noninfectious lung process such as acute interstitial pneumonia or an eosinophilic pneumonia. Given the diagnostic complexity, I would strongly consider consulting a pulmonologist if the patient did not improve quickly. At this point, I would like to review a posterior‐anterior (PA) and lateral chest radiograph, and room air arterial blood gas (ABG) in addition to basic laboratory test values.
Laboratory data obtained on admission was remarkable for a white blood cell (WBC) count of 26,500/L with 75% neutrophils and 6% eosinophils. Hemoglobin was 14.4 gm/dL. Platelet count was 454,000/L. Serum chemistries showed a sodium of 137 mEq/dL, potassium 4.3 mEq/dL, Cl 108 mEq/dL, bicarbonate 19 mEq/dL, blood urea nitrogen (BUN) 8 mg/dL, creatinine 1.0 mg/dL, and glucose 137 mg/dL. Cardiac enzymes were normal. Calcium was 9.8 mg/dL, albumin 2.7 gm/dL, total protein 6.9 gm/dL, AST 36 U/L, ALT 54 U/L and the bilirubin was normal. Chest radiograph (Figure 1) demonstrated a left perihilar infiltrate with air bronchograms and marked right hemidiaphragm elevation as seen on previous films. Unchanged increased interstitial markings were also present. Her electrocardiogram (ECG) showed normal sinus rhythm, normal axis, and QRS duration with nonspecific diffuse T‐wave abnormalities.
Given her presentation, I am worried about how well she is oxygenating and ventilating. An ABG should be done to assess her status more accurately. An albumin of 2.7 gm/dL indicates that she is fairly sick. I would not hesitate to consider testing the patient for human immunodeficiency virus (HIV) given how this information would dramatically change the differential diagnoses of her pulmonary process.
I am still most concerned about sepsis secondary to pneumonia in this patient with multiple chronic comorbidities, underlying chronic lung disease, receiving chronic inhaled glucocorticoids and a recent course of oral glucocorticoids and antibiotics. While I would initiate hydration I do not see a clear indication for early goal‐directed therapy for severe sepsis. In addition to obtaining an ABG and starting intravenous fluids, I would also draw blood cultures, send sputum for gram stain, culture, and sensitivity, and perform a urinalysis. I would also administer empiric antibiotics as quickly as possible based on a number of pneumonia clinical studies suggesting improved outcomes with early antibiotic administration. Because of her use of antibiotics and both inhaled and oral glucocorticoids, she is at higher risk for potentially multidrug‐resistant bacterial pathogens, including Staphyloccocus aureus and gram‐negative bacteria such as Pseudomonas and Klebsiella (Table 1). Therefore, I would initially cover her broadly for these organisms.
| Meets Any of the Following |
|---|
| Antimicrobial therapy in the preceding 90 days |
| Current hospitalization of 5 days or more |
| High frequency of antibiotic resistance in the community or in the specific hospital unit |
| Presence of risk factors for healthcare‐associated pneumonia (HCAP) |
| Hospitalization for >2 days in the preceding 90 days |
| Residence in nursing home or long‐term care facility (LTAC) for at least 5 days in last 90 days |
| Home infusion therapy including intravenous antibiotics within 30 days |
| Home wound care within 30 days |
| Chronic hemodialysis in hospital or clinic within 30 days |
| Family member with multidrug‐resistant pathogen |
| Immunosuppressive disease and/or therapy |
In addition to initial treatment choice, the inpatient triage decision is another important issue, especially at a community hospital where intensive care unit (ICU) resources are rare and often the admission decision is between sending a moderately sick patient to a regular floor bed or the medical ICU. Both the American Thoracic Society and Infectious Diseases Society of America support an ICU triage protocol in their guidelines for the management of community‐acquired pneumonia in adults that utilizes the following 9 minor criteria, of which the presence of at least 3 should support ICU admission: respiratory rate 30 breaths/minute; oxygenation index (pressure of oxygen [PaO2]/fraction of inspired oxygen [FiO2] ratio) 250; multilobar infiltrates; confusion/disorientation; uremia (BUN level 20 mg/dL); leukopenia (WBC count <4,000 cells/mm3); thrombocytopenia (platelet count <100,000 cells/mm3); hypothermia (core temperature <36C); and hypotension requiring aggressive fluid. Despite the absence of these criteria in this patient, it is important to note that no triage protocol has been adequately prospectively validated. Retrospective study of the minor criteria has found that the presence of at least 2 of the following 3 clinical criteria to have the highest specificity for predicting cardiopulmonary decompensation and subsequent need for ICU care: (1) initial hypotension (BP <90/60) on presentation with response to initial intravenous fluids to a BP >90/60; (2) oxygenation failure as indicated by PaO2/FiO2 ratio less than 250; or (3) the presence of multilobar or bilateral infiltrates on chest radiography.
I also want to comment on the relative elevation of her calcium, especially given the low albumin. This may simply be due to volume depletion, as many older patients have asymptomatic mild primary hyperparathyroidism. However, this elevated calcium may be a clue to the underlying lung process. Granulomatous lung disease due to tuberculosis or fungal infection could yield elevated calcium levels via increases in macrophage production of the active vitamin D metabolite calcitriol. This will need to be followed and a parathormone (PTH) level would be the best first test to request if the calcium level remains elevated. If the PTH level is suppressed, granulomatous disease or malignancy would be the more likely cause.
The patient was admitted with a presumptive diagnosis of community‐acquired pneumonia, was started on ceftriaxone and azithromycin, and given intravenous fluids, oxygen, and continued on inhaled salmeterol/fluticasone. Sputum was ordered for gram stain, culture, and sensitivity, and blood cultures were obtained. Urinalysis showed 1‐5 WBCs/high‐power field. Venous thromboembolism prophylaxis was initiated with subcutaneous heparin 5,000 units 8 hours. Her blood pressure normalized rapidly and during the next few days she stated she was feeling better. Despite continued significant wheezing her oxygen saturation remained at 98% on 2 L of oxygen via nasal cannula and she was less tachypneic. Attempts at obtaining an ABG were unsuccessful, and the patient subsequently refused additional attempts. Over the first few days her WBC count remained elevated above 20,000/L, with worsening bandemia (11%), and fever ranging from 38C to 39C. Sputum analysis was initially unsuccessful and blood cultures remained negative.
I am concerned about the persistent fever and elevated WBC count, and want to emphasize that I might have treated her with broader spectrum antibiotics to cover additional multidrug‐resistant bacterial organisms. I would have initially ordered vancomycin to cover methicillin resistant Staphylococcus aureus (MRSA) plus 2 additional antibiotics that cover multidrug‐resistant gram negative pathogens including Pseudomonas aeruginosa.
On the fifth hospital day, her WBC count dropped to 13,400/L and she defervesced. However, her respiratory status worsened during that same day with increased tachypnea. Of note, no results were reported from the initial sputum cultures and they were reordered and a noncontrast chest computed tomography (CT) was also ordered.
I think at this point, even though she has remained stable hemodynamically and oxygenating easily with supplemental oxygen, the question of whether or not her primary process is infectious or noninfectious lingers. I agree with obtaining a chest CT scan.
I am not surprised that sputum was not evaluated despite the orders. Among hospitalized patients with pneumonia, we frequently find that about a third of the time sputum cannot be obtained, about a third of the time it is obtained but the quality is unsatisfactory, and only a third of the time does the sputum sample meet criteria (less than 5 squamous epithelial cells per high‐power field) for adequate interpretation of the gram‐stain and culture result. Unfortunately, no one has developed a better way to improve this process. Nonetheless, I believe we do not try hard enough to obtain sputum in the first hours of evaluating our patients. I joke with our internal medicine residents that they should carry a sputum cup with them when they evaluate a patient with possible pneumonia. One recent prospective study of the value of sputum gram‐staining in community‐acquired pneumonia has found it to be highly specific for identifying Streptococcus pneumoniae or Haemophilus influenzae pneumonia.
The CT scan (Figure 2) performed on hospital day 6 demonstrated consolidation in the left upper lobe with areas of cavitation. There was also interstitial infiltrate extending into the lingula. Elevation of the right hemidiaphragm with atelectasis in both lung bases was also noted. A small effusion was present on the left and possibly a minimal effusion on the right as well. There was no pericardial effusion and only a few small pretracheal and periaortic lymph nodes were noted.

Given her failure to improve significantly after 6 days of antibiotic treatment, and her recent use of glucocorticoids, I would expand my diagnostic considerations to include other necrotizing bacterial infections, tuberculosis, fungus, and Nocardia.
Given the results of the CT scan she was placed in respiratory isolation to rule out active pulmonary tuberculosis. Though tachypneic, her blood pressure and pulse remained stable. However, her oxygen saturation deteriorated, declining to 92% on 2 L of oxygen via nasal cannula during hospital days 6 and 7. Subsequent successful attempts at collecting sputum yielded rapid growth of yeast (not Cryptococcus spp.). Pulmonary and infectious disease consultations were obtained and vancomycin was added to her regimen. The patient subsequently agreed to undergo diagnostic bronchoscopy.
I agree with obtaining input from expert consultants. I think we too often underutilize consultation in patients that are better but not completely better when we are not entirely sure what is going on. Evidence of noncryptococcus yeast in sputum may sometimes indicate colonization with Candida spp. without any significant clinical consequence. This finding may alternatively suggest the possibility of a true fungal pneumonia caused by 1 of the dimorphic fungi, including Histoplasma capsulatum, Paracoccidioides brasiliensis, Blastomyces dermatitides, or Coccidioides immitis. However, in this case there is not a strong epidemiologic patient history of exposure to any of these types of fungi.
Three sputum smears were negative for acid fast bacilli (AFB). Bronchoscopy revealed grossly abnormal mucosa in the left upper lobe and bronchomalacia, but no obstructive lesions. A transthoracic echocardiogram was ordered to evaluate her degree of pulmonary hypertension.
The 3 sputum specimens that were negative for AFB despite cavitary lung disease have high sensitivity for ruling out pulmonary tuberculosis. In addition, given the absence of any bacterial pathogen isolated from these specimens, I would pursue the possibility of other potential fungal pathogens given the patient's subacute course, history of using inhaled and oral corticosteroids, sputum results, and the presence of a cavitary lesion on her CT scan images.
Cytologic examination of the bronchoalveolar lavage (BAL) sample showed a cell differential of 1% bands, 58% neutrophils, 9% lymphs, and 27% eosinophils. The routine postbronchoscopy chest radiograph showed complete opacification of the left lung. The patient's WBC count rose to 26,000/L but she remained afebrile. Echocardiogram was reported to be of very poor quality due to her obesity. The cardiologist reviewing the echocardiogram called the attending physicians and stated there was possibly something in the left pulmonary artery and aortic dissection could not be ruled out.
The presence of eosinophilia on BAL may be a very important clue as to what lung pathology she has. In fact, eosinophilia in this setting may indicate the possibility of parasitic or fungal infection of the lung, or inflammation of the airway associated to drug toxicity, asthma, or environmental toxin exposure. With this additional information, I am concerned that she may be harboring an atypical infection such as an invasive fungus. The echocardiogram results are unclear to me but will need to be clarified with additional testing.
The interpretation of the transbronchial biopsy specimen was limited but suggested invasive pseudomembranous tracheal bronchitis due to aspergillosis. The routine hematoxylin and eosin stain showed portions of alveolar lung tissue and some collapsed submucosal bronchial glands with relatively normal‐looking lung tissue but along the edge of the spaces were obvious fungal organisms. The Gomori's methenamine silver (GMS) stain suggested the presence of Aspergillus organisms (Figure 3). Fungal cultures were also negative for any of the other dimorphic fungi or for molds.

Despite the negative culture results, the overall clinical picture suggests a necrotizing pneumonia caused by an invasive Aspergillus affecting both the bronchial tree and the lower respiratory tract. Generally, necrotizing pneumonias usually have a slow response to antimicrobial therapy. Given the inherent difficulty in differentiating clearly between invasive and noninvasive disease based on a transbronchial biopsy specimen, initiating antifungal therapy for invasive aspergillosis is appropriate in this patient. This patient's recent use of oral glucocorticoids and chronic use of inhaled glucocorticoids are both potential risk factors that predisposed this patient to develop invasive aspergillosis.
Many times we simply follow treatment guidelines for different categories of pneumonia, and have limited or inadequate clinical information to make more definitive diagnoses. While we need these treatment protocols, physicians must avoid falling into the trap that antibiotics treat all infectious etiologies in the lung and we should make reasonable efforts to pin down the etiology. All of us have been fooled by atypical presentations of tuberculosis, fungus, and noninfectious diseases of the lung. I think it behooves us to be vigilant about alternative diagnoses and consider pursuing additional studies whenever the clinical response to initial treatment does not meet our expectations.
Subsequently, the patient's additional cultures remained negative. The official echocardiogram report was read as questionable PE in the pulmonary artery. A spiral CT angiogram revealed a pulmonary artery embolus in the left upper lobe and she was treated with anticoagulation. Her shortness of breath improved steadily and she was successfully discharged after receiving 9 days of oral voriconazole. Outpatient pulmonary function testing documented the presence of chronic obstructive lung disease. She completed a 5‐month course of voriconazole therapy with significant clinical and radiologic improvement of her pulmonary infiltrate. She also completed a 12‐month treatment with warfarin for the concomitant pulmonary embolism. On follow‐up at 12 months she was doing well.
COMMENTARY
Aspergillosis caused particularly by Aspergillus fumigatus is considered an emerging infectious disease that frequently produces significant morbidity and mortality among immunocompromised patients.1, 2 The most frequently‐affected organs by this fungal pathogen include the lung and the central nervous system. There are 3 pathogenic mechanisms of Aspergillus infection of the lung: colonization, hypersensitivity reaction, and invasive aspergillosis.1
Invasive pulmonary aspergillosis is predominantly seen among individuals with severe degrees of immunosuppression as a result of solid‐organ transplantation, immunosuppressive therapies for autoimmune diseases, systemic glucocorticoids, and chemotherapy for hematologic malignancies. Mortality due to invasive aspergillosis continues to be very high (>58%) despite our improved ability to diagnose this condition and newer therapies to treat immunocompromised individuals.1 Invasive aspergillosis can manifest clinically in multiple ways. These include: (1) an invasive vascular process in which fungal organisms invade blood vessels, causing a rapidly progressive and often fatal illness; (2) necrotizing pseudomembranous tracheal bronchitis; (3) chronic necrotizing aspergillosis; (4) bronchopleural fistula; or (5) empyema.35 In our case, while the pathologic findings were most suggestive of an invasive pseudomembranous tracheal bronchitis, the overall clinical picture was most compatible with a necrotizing pneumonia due to invasive aspergillosis.
In addition to the traditional identified risk factors for invasive pulmonary aspergillosis, a number of reports during the last decade have demonstrated the occurrence of invasive aspergillosis in patients with COPD.14 A systematic review of the literature demonstrated that among 1,941 patients with invasive aspergillosis, 26 (1.3%) had evidence of COPD as the main risk factor for developing invasive aspergillosis.1 A single report has associated the potential use of inhaled steroids with the occurrence of invasive aspergillosis in this patient population.2 However, other factors that may promote increased susceptibility to invasive fungal infection among patients with COPD include the use of long‐term or repeated short‐term glucocorticoid treatments, and the presence of multiple additional comorbidities, which may be found in this same population such as diabetes, malnutrition, or end‐stage renal disease.3, 4 Most reported series have demonstrated a high mortality rate of invasive pulmonary aspergillosis in patients with COPD.14
The diagnosis of invasive pulmonary aspergillosis represents a significant clinical challenge. Diagnostic algorithms incorporating CT, antigen detection testing (for serum galactomannan and ‐glucan) as well as polymerase chain reaction diagnostic testing appear to be beneficial in the early diagnosis of invasive aspergillosis in particular settings such as in allogeneic hematopoietic stem cell transplantation.5 The role of antigen testing to identify early invasive aspergillosis in patients with COPD remains uncertain since it has been evaluated in a limited number of patients and therefore clinical suspicion is critical to push clinicians to pursue invasive tissue biopsy and cultures to confirm the diagnosis.3, 4
Based on the available clinical case series and in our case, invasive pulmonary aspergillosis should be suspected in COPD patients with rapidly progressive pneumonia not responding to antibacterial therapy and who have received oral or inhaled glucocorticoids in the recent past. In addition, this case also illustrates that occasionally, patients present with more than 1 life‐threatening diagnosis. This patient was also diagnosed with PE despite adequate prophylaxis. In addition to the well‐known clinical risk factors of obesity and lung disease, the underlying infection may have contributed to a systemic or local hypercoagulable condition that further increased her risk for venous thromboembolism.
KEY TEACHING POINTS
-
Clinicians should remember to consider a broad differential in patients presenting with pneumonia, including the possibility of fungal pathogens in patients with known risk factors and in patients with multiple, potentially immunosuppressive comorbidities, or in patients who do not improve on standard antibiotic therapy.
-
There is some evidence of an association between COPD and invasive aspergillosis, likely due to the frequent use of oral corticosteroids and/or chronic inhaled steroids in this population.
- ,,.Aspergillosis case‐fatality rate: systematic review of the literature.Clin Infect Dis.2001;32:358–366.
- ,,,,.Invasive pulmonary filamentous fungal infection in a patient receiving inhaled corticosteroid therapy.Clin Infect Dis.2002;35:e54–e56.
- ,,, et al.Invasive pulmonary aspergillosis in chronic obstructive pulmonary disease: an emerging fungal pathogen.Clin Microbiol Infect.2005;11:427–429.
- ,,,,,.Invasive pulmonary aspergillosis in patients with chronic obstructive pulmonary disease: report of eight cases and review.Clin Infect Dis.1998;26:1473–1475.
- ,.Current approaches to diagnosis and treatment to invasive aspergillosis.Am J Respir Crit Care Med.2006;173:707–717.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 71‐year‐old African‐American woman presented to the emergency department with chest pain, shortness of breath, and cough. She had initially presented to her primary care physician 2 weeks previously complaining of worsening cough and shortness of breath and was told to continue her inhaled albuterol and glucocorticoids and was prescribed a prednisone taper and an unknown course of antibiotics. She noted no improvement in her symptoms despite compliance with this treatment. Three days prior to admission she described the gradual onset of left‐sided pleuritic chest pain with continued cough, associated with yellow sputum and worsening dyspnea. Review of systems was remarkable for generalized weakness and malaise. She denied fever, chills, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, diarrhea, nausea, vomiting, or abdominal pain.
Her past medical history included a diagnosis of chronic obstructive pulmonary disease (COPD) but pulmonary function tests 7 years prior to admission showed an forced expiratory volume in the first second (FEV1)/forced vital capacity (FVC) ratio of 81%. She had a 30 pack‐year history of smoking, but quit 35 years ago. The patient also carried a diagnosis of heart failure, but an echocardiogram done 1 year ago demonstrated a left ventricular ejection fraction of 65% to 70% without diastolic dysfunction but mild right ventricular dilation and hypertrophy. Additionally, she had known nonobstructive coronary atherosclerotic heart disease, dyslipidemia, hypertension, morbid obesity, depression, and a documented chronic right hemidiaphragm elevation.
At this point the history suggests that the patient does not have a clear diagnosis of COPD. The lack of definitive spirometry evidence of chronic airway obstruction concerns me; I think that she may have been mistakenly treated with chronic inhaled steroids and doses of antibiotics for an acute exacerbation of chronic lung disease. Additional review of her history gives some indication of advanced lung disease, with her recent echocardiogram showing strain on the right ventricle with right ventricular hypertrophy and dilation, but there is no mention of the presence or severity of pulmonary hypertension. Nonetheless, I would be concerned that she probably has underlying significant cor pulmonale.
The patient now re‐presents with a worsening of her pulmonary symptoms. Her left‐sided pleuritic pain would make me concerned that she had a pulmonary embolus (PE). This morbidly obese patient with new pulmonary symptoms, right ventricular strain on her previous echocardiogram, and a persistent elevated right hemidiaphragm suggests a presentation of another PE.
At this time I cannot rule out other common possibilities such as infectious pneumonia. If she does have pneumonia, I would be concerned she could be harboring a multidrug‐resistant bacterial infection given her recent course of antibiotics in addition to her use of both chronic inhaled and intermittent oral glucocorticoids.
After gathering the rest of her full medical history, I would focus my physical exam on looking for evidence of parenchymal lung disease, signs of pulmonary hypertension, and pneumonia.
Her surgical history includes a previous hysterectomy, cholecystectomy, hernia repair, and left hepatic lobectomy for a benign mass. Her outpatient medications were ibuprofen, bupropion, fluvastatin, atenolol, potassium, aspirin, clopidogrel, albuterol inhaler, fluticasone/salmeterol inhaler, and omeprazole. She reports an allergy to penicillin and to sulfa drugs. Her mother died of an unknown cancer at age 77 years. She denied any international travel and she has always lived in Georgia.
The patient has been retired since 1992, having previously worked for the U.S. Postal Service. She admits to occasional alcohol intake (2 to 3 drinks a month). No recent travel, surgery, or prolonged immobilization was noted.
On initial examination she was alert and mentally appropriate, but appeared to be in mild respiratory distress with a respiratory rate of 28 breaths/minute. Her blood pressure (BP) was 99/70, heart rate 102, temperature of 38.2C, and oxygen saturation of 93% on room air and 97% on 2 L of oxygen via nasal cannula. Auscultation of her lungs revealed crackles over her left anterior lung field, bronchial breath sounds in the left posterior midlung, and bibasilar crackles. No wheezing was noted. Her cardiovascular exam and the remainder of her physical exam were unremarkable except for morbid obesity.
While my initial thoughts were leaning toward an exacerbation of chronic lung disease or possibly a new PE, at this moment, infection seems more likely. Indeed, her pulmonary findings suggest a left‐sided inflammatory process, and her vital signs meet criteria for systemic inflammatory response syndrome (SIRS). My primary concern is sepsis due to a drug‐resistant bacterial infection, including Staphylococcus aureus or gram‐negative bacteria or possibly more unusual organisms such as Nocardia or fungi, due to her recent use of antibiotics and chronic inhaled steroid use and recent course of oral glucocorticoids.
Conversely, the SIRS could be a manifestation of a noninfectious lung process such as acute interstitial pneumonia or an eosinophilic pneumonia. Given the diagnostic complexity, I would strongly consider consulting a pulmonologist if the patient did not improve quickly. At this point, I would like to review a posterior‐anterior (PA) and lateral chest radiograph, and room air arterial blood gas (ABG) in addition to basic laboratory test values.
Laboratory data obtained on admission was remarkable for a white blood cell (WBC) count of 26,500/L with 75% neutrophils and 6% eosinophils. Hemoglobin was 14.4 gm/dL. Platelet count was 454,000/L. Serum chemistries showed a sodium of 137 mEq/dL, potassium 4.3 mEq/dL, Cl 108 mEq/dL, bicarbonate 19 mEq/dL, blood urea nitrogen (BUN) 8 mg/dL, creatinine 1.0 mg/dL, and glucose 137 mg/dL. Cardiac enzymes were normal. Calcium was 9.8 mg/dL, albumin 2.7 gm/dL, total protein 6.9 gm/dL, AST 36 U/L, ALT 54 U/L and the bilirubin was normal. Chest radiograph (Figure 1) demonstrated a left perihilar infiltrate with air bronchograms and marked right hemidiaphragm elevation as seen on previous films. Unchanged increased interstitial markings were also present. Her electrocardiogram (ECG) showed normal sinus rhythm, normal axis, and QRS duration with nonspecific diffuse T‐wave abnormalities.
Given her presentation, I am worried about how well she is oxygenating and ventilating. An ABG should be done to assess her status more accurately. An albumin of 2.7 gm/dL indicates that she is fairly sick. I would not hesitate to consider testing the patient for human immunodeficiency virus (HIV) given how this information would dramatically change the differential diagnoses of her pulmonary process.
I am still most concerned about sepsis secondary to pneumonia in this patient with multiple chronic comorbidities, underlying chronic lung disease, receiving chronic inhaled glucocorticoids and a recent course of oral glucocorticoids and antibiotics. While I would initiate hydration I do not see a clear indication for early goal‐directed therapy for severe sepsis. In addition to obtaining an ABG and starting intravenous fluids, I would also draw blood cultures, send sputum for gram stain, culture, and sensitivity, and perform a urinalysis. I would also administer empiric antibiotics as quickly as possible based on a number of pneumonia clinical studies suggesting improved outcomes with early antibiotic administration. Because of her use of antibiotics and both inhaled and oral glucocorticoids, she is at higher risk for potentially multidrug‐resistant bacterial pathogens, including Staphyloccocus aureus and gram‐negative bacteria such as Pseudomonas and Klebsiella (Table 1). Therefore, I would initially cover her broadly for these organisms.
| Meets Any of the Following |
|---|
| Antimicrobial therapy in the preceding 90 days |
| Current hospitalization of 5 days or more |
| High frequency of antibiotic resistance in the community or in the specific hospital unit |
| Presence of risk factors for healthcare‐associated pneumonia (HCAP) |
| Hospitalization for >2 days in the preceding 90 days |
| Residence in nursing home or long‐term care facility (LTAC) for at least 5 days in last 90 days |
| Home infusion therapy including intravenous antibiotics within 30 days |
| Home wound care within 30 days |
| Chronic hemodialysis in hospital or clinic within 30 days |
| Family member with multidrug‐resistant pathogen |
| Immunosuppressive disease and/or therapy |
In addition to initial treatment choice, the inpatient triage decision is another important issue, especially at a community hospital where intensive care unit (ICU) resources are rare and often the admission decision is between sending a moderately sick patient to a regular floor bed or the medical ICU. Both the American Thoracic Society and Infectious Diseases Society of America support an ICU triage protocol in their guidelines for the management of community‐acquired pneumonia in adults that utilizes the following 9 minor criteria, of which the presence of at least 3 should support ICU admission: respiratory rate 30 breaths/minute; oxygenation index (pressure of oxygen [PaO2]/fraction of inspired oxygen [FiO2] ratio) 250; multilobar infiltrates; confusion/disorientation; uremia (BUN level 20 mg/dL); leukopenia (WBC count <4,000 cells/mm3); thrombocytopenia (platelet count <100,000 cells/mm3); hypothermia (core temperature <36C); and hypotension requiring aggressive fluid. Despite the absence of these criteria in this patient, it is important to note that no triage protocol has been adequately prospectively validated. Retrospective study of the minor criteria has found that the presence of at least 2 of the following 3 clinical criteria to have the highest specificity for predicting cardiopulmonary decompensation and subsequent need for ICU care: (1) initial hypotension (BP <90/60) on presentation with response to initial intravenous fluids to a BP >90/60; (2) oxygenation failure as indicated by PaO2/FiO2 ratio less than 250; or (3) the presence of multilobar or bilateral infiltrates on chest radiography.
I also want to comment on the relative elevation of her calcium, especially given the low albumin. This may simply be due to volume depletion, as many older patients have asymptomatic mild primary hyperparathyroidism. However, this elevated calcium may be a clue to the underlying lung process. Granulomatous lung disease due to tuberculosis or fungal infection could yield elevated calcium levels via increases in macrophage production of the active vitamin D metabolite calcitriol. This will need to be followed and a parathormone (PTH) level would be the best first test to request if the calcium level remains elevated. If the PTH level is suppressed, granulomatous disease or malignancy would be the more likely cause.
The patient was admitted with a presumptive diagnosis of community‐acquired pneumonia, was started on ceftriaxone and azithromycin, and given intravenous fluids, oxygen, and continued on inhaled salmeterol/fluticasone. Sputum was ordered for gram stain, culture, and sensitivity, and blood cultures were obtained. Urinalysis showed 1‐5 WBCs/high‐power field. Venous thromboembolism prophylaxis was initiated with subcutaneous heparin 5,000 units 8 hours. Her blood pressure normalized rapidly and during the next few days she stated she was feeling better. Despite continued significant wheezing her oxygen saturation remained at 98% on 2 L of oxygen via nasal cannula and she was less tachypneic. Attempts at obtaining an ABG were unsuccessful, and the patient subsequently refused additional attempts. Over the first few days her WBC count remained elevated above 20,000/L, with worsening bandemia (11%), and fever ranging from 38C to 39C. Sputum analysis was initially unsuccessful and blood cultures remained negative.
I am concerned about the persistent fever and elevated WBC count, and want to emphasize that I might have treated her with broader spectrum antibiotics to cover additional multidrug‐resistant bacterial organisms. I would have initially ordered vancomycin to cover methicillin resistant Staphylococcus aureus (MRSA) plus 2 additional antibiotics that cover multidrug‐resistant gram negative pathogens including Pseudomonas aeruginosa.
On the fifth hospital day, her WBC count dropped to 13,400/L and she defervesced. However, her respiratory status worsened during that same day with increased tachypnea. Of note, no results were reported from the initial sputum cultures and they were reordered and a noncontrast chest computed tomography (CT) was also ordered.
I think at this point, even though she has remained stable hemodynamically and oxygenating easily with supplemental oxygen, the question of whether or not her primary process is infectious or noninfectious lingers. I agree with obtaining a chest CT scan.
I am not surprised that sputum was not evaluated despite the orders. Among hospitalized patients with pneumonia, we frequently find that about a third of the time sputum cannot be obtained, about a third of the time it is obtained but the quality is unsatisfactory, and only a third of the time does the sputum sample meet criteria (less than 5 squamous epithelial cells per high‐power field) for adequate interpretation of the gram‐stain and culture result. Unfortunately, no one has developed a better way to improve this process. Nonetheless, I believe we do not try hard enough to obtain sputum in the first hours of evaluating our patients. I joke with our internal medicine residents that they should carry a sputum cup with them when they evaluate a patient with possible pneumonia. One recent prospective study of the value of sputum gram‐staining in community‐acquired pneumonia has found it to be highly specific for identifying Streptococcus pneumoniae or Haemophilus influenzae pneumonia.
The CT scan (Figure 2) performed on hospital day 6 demonstrated consolidation in the left upper lobe with areas of cavitation. There was also interstitial infiltrate extending into the lingula. Elevation of the right hemidiaphragm with atelectasis in both lung bases was also noted. A small effusion was present on the left and possibly a minimal effusion on the right as well. There was no pericardial effusion and only a few small pretracheal and periaortic lymph nodes were noted.
Given her failure to improve significantly after 6 days of antibiotic treatment, and her recent use of glucocorticoids, I would expand my diagnostic considerations to include other necrotizing bacterial infections, tuberculosis, fungus, and Nocardia.
Given the results of the CT scan she was placed in respiratory isolation to rule out active pulmonary tuberculosis. Though tachypneic, her blood pressure and pulse remained stable. However, her oxygen saturation deteriorated, declining to 92% on 2 L of oxygen via nasal cannula during hospital days 6 and 7. Subsequent successful attempts at collecting sputum yielded rapid growth of yeast (not Cryptococcus spp.). Pulmonary and infectious disease consultations were obtained and vancomycin was added to her regimen. The patient subsequently agreed to undergo diagnostic bronchoscopy.
I agree with obtaining input from expert consultants. I think we too often underutilize consultation in patients that are better but not completely better when we are not entirely sure what is going on. Evidence of noncryptococcus yeast in sputum may sometimes indicate colonization with Candida spp. without any significant clinical consequence. This finding may alternatively suggest the possibility of a true fungal pneumonia caused by 1 of the dimorphic fungi, including Histoplasma capsulatum, Paracoccidioides brasiliensis, Blastomyces dermatitides, or Coccidioides immitis. However, in this case there is not a strong epidemiologic patient history of exposure to any of these types of fungi.
Three sputum smears were negative for acid fast bacilli (AFB). Bronchoscopy revealed grossly abnormal mucosa in the left upper lobe and bronchomalacia, but no obstructive lesions. A transthoracic echocardiogram was ordered to evaluate her degree of pulmonary hypertension.
The 3 sputum specimens that were negative for AFB despite cavitary lung disease have high sensitivity for ruling out pulmonary tuberculosis. In addition, given the absence of any bacterial pathogen isolated from these specimens, I would pursue the possibility of other potential fungal pathogens given the patient's subacute course, history of using inhaled and oral corticosteroids, sputum results, and the presence of a cavitary lesion on her CT scan images.
Cytologic examination of the bronchoalveolar lavage (BAL) sample showed a cell differential of 1% bands, 58% neutrophils, 9% lymphs, and 27% eosinophils. The routine postbronchoscopy chest radiograph showed complete opacification of the left lung. The patient's WBC count rose to 26,000/L but she remained afebrile. Echocardiogram was reported to be of very poor quality due to her obesity. The cardiologist reviewing the echocardiogram called the attending physicians and stated there was possibly something in the left pulmonary artery and aortic dissection could not be ruled out.
The presence of eosinophilia on BAL may be a very important clue as to what lung pathology she has. In fact, eosinophilia in this setting may indicate the possibility of parasitic or fungal infection of the lung, or inflammation of the airway associated to drug toxicity, asthma, or environmental toxin exposure. With this additional information, I am concerned that she may be harboring an atypical infection such as an invasive fungus. The echocardiogram results are unclear to me but will need to be clarified with additional testing.
The interpretation of the transbronchial biopsy specimen was limited but suggested invasive pseudomembranous tracheal bronchitis due to aspergillosis. The routine hematoxylin and eosin stain showed portions of alveolar lung tissue and some collapsed submucosal bronchial glands with relatively normal‐looking lung tissue but along the edge of the spaces were obvious fungal organisms. The Gomori's methenamine silver (GMS) stain suggested the presence of Aspergillus organisms (Figure 3). Fungal cultures were also negative for any of the other dimorphic fungi or for molds.
Despite the negative culture results, the overall clinical picture suggests a necrotizing pneumonia caused by an invasive Aspergillus affecting both the bronchial tree and the lower respiratory tract. Generally, necrotizing pneumonias usually have a slow response to antimicrobial therapy. Given the inherent difficulty in differentiating clearly between invasive and noninvasive disease based on a transbronchial biopsy specimen, initiating antifungal therapy for invasive aspergillosis is appropriate in this patient. This patient's recent use of oral glucocorticoids and chronic use of inhaled glucocorticoids are both potential risk factors that predisposed this patient to develop invasive aspergillosis.
Many times we simply follow treatment guidelines for different categories of pneumonia, and have limited or inadequate clinical information to make more definitive diagnoses. While we need these treatment protocols, physicians must avoid falling into the trap that antibiotics treat all infectious etiologies in the lung and we should make reasonable efforts to pin down the etiology. All of us have been fooled by atypical presentations of tuberculosis, fungus, and noninfectious diseases of the lung. I think it behooves us to be vigilant about alternative diagnoses and consider pursuing additional studies whenever the clinical response to initial treatment does not meet our expectations.
Subsequently, the patient's additional cultures remained negative. The official echocardiogram report was read as questionable PE in the pulmonary artery. A spiral CT angiogram revealed a pulmonary artery embolus in the left upper lobe and she was treated with anticoagulation. Her shortness of breath improved steadily and she was successfully discharged after receiving 9 days of oral voriconazole. Outpatient pulmonary function testing documented the presence of chronic obstructive lung disease. She completed a 5‐month course of voriconazole therapy with significant clinical and radiologic improvement of her pulmonary infiltrate. She also completed a 12‐month treatment with warfarin for the concomitant pulmonary embolism. On follow‐up at 12 months she was doing well.
COMMENTARY
Aspergillosis caused particularly by Aspergillus fumigatus is considered an emerging infectious disease that frequently produces significant morbidity and mortality among immunocompromised patients.1, 2 The most frequently‐affected organs by this fungal pathogen include the lung and the central nervous system. There are 3 pathogenic mechanisms of Aspergillus infection of the lung: colonization, hypersensitivity reaction, and invasive aspergillosis.1
Invasive pulmonary aspergillosis is predominantly seen among individuals with severe degrees of immunosuppression as a result of solid‐organ transplantation, immunosuppressive therapies for autoimmune diseases, systemic glucocorticoids, and chemotherapy for hematologic malignancies. Mortality due to invasive aspergillosis continues to be very high (>58%) despite our improved ability to diagnose this condition and newer therapies to treat immunocompromised individuals.1 Invasive aspergillosis can manifest clinically in multiple ways. These include: (1) an invasive vascular process in which fungal organisms invade blood vessels, causing a rapidly progressive and often fatal illness; (2) necrotizing pseudomembranous tracheal bronchitis; (3) chronic necrotizing aspergillosis; (4) bronchopleural fistula; or (5) empyema.35 In our case, while the pathologic findings were most suggestive of an invasive pseudomembranous tracheal bronchitis, the overall clinical picture was most compatible with a necrotizing pneumonia due to invasive aspergillosis.
In addition to the traditional identified risk factors for invasive pulmonary aspergillosis, a number of reports during the last decade have demonstrated the occurrence of invasive aspergillosis in patients with COPD.14 A systematic review of the literature demonstrated that among 1,941 patients with invasive aspergillosis, 26 (1.3%) had evidence of COPD as the main risk factor for developing invasive aspergillosis.1 A single report has associated the potential use of inhaled steroids with the occurrence of invasive aspergillosis in this patient population.2 However, other factors that may promote increased susceptibility to invasive fungal infection among patients with COPD include the use of long‐term or repeated short‐term glucocorticoid treatments, and the presence of multiple additional comorbidities, which may be found in this same population such as diabetes, malnutrition, or end‐stage renal disease.3, 4 Most reported series have demonstrated a high mortality rate of invasive pulmonary aspergillosis in patients with COPD.14
The diagnosis of invasive pulmonary aspergillosis represents a significant clinical challenge. Diagnostic algorithms incorporating CT, antigen detection testing (for serum galactomannan and ‐glucan) as well as polymerase chain reaction diagnostic testing appear to be beneficial in the early diagnosis of invasive aspergillosis in particular settings such as in allogeneic hematopoietic stem cell transplantation.5 The role of antigen testing to identify early invasive aspergillosis in patients with COPD remains uncertain since it has been evaluated in a limited number of patients and therefore clinical suspicion is critical to push clinicians to pursue invasive tissue biopsy and cultures to confirm the diagnosis.3, 4
Based on the available clinical case series and in our case, invasive pulmonary aspergillosis should be suspected in COPD patients with rapidly progressive pneumonia not responding to antibacterial therapy and who have received oral or inhaled glucocorticoids in the recent past. In addition, this case also illustrates that occasionally, patients present with more than 1 life‐threatening diagnosis. This patient was also diagnosed with PE despite adequate prophylaxis. In addition to the well‐known clinical risk factors of obesity and lung disease, the underlying infection may have contributed to a systemic or local hypercoagulable condition that further increased her risk for venous thromboembolism.
KEY TEACHING POINTS
-
Clinicians should remember to consider a broad differential in patients presenting with pneumonia, including the possibility of fungal pathogens in patients with known risk factors and in patients with multiple, potentially immunosuppressive comorbidities, or in patients who do not improve on standard antibiotic therapy.
-
There is some evidence of an association between COPD and invasive aspergillosis, likely due to the frequent use of oral corticosteroids and/or chronic inhaled steroids in this population.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 71‐year‐old African‐American woman presented to the emergency department with chest pain, shortness of breath, and cough. She had initially presented to her primary care physician 2 weeks previously complaining of worsening cough and shortness of breath and was told to continue her inhaled albuterol and glucocorticoids and was prescribed a prednisone taper and an unknown course of antibiotics. She noted no improvement in her symptoms despite compliance with this treatment. Three days prior to admission she described the gradual onset of left‐sided pleuritic chest pain with continued cough, associated with yellow sputum and worsening dyspnea. Review of systems was remarkable for generalized weakness and malaise. She denied fever, chills, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, diarrhea, nausea, vomiting, or abdominal pain.
Her past medical history included a diagnosis of chronic obstructive pulmonary disease (COPD) but pulmonary function tests 7 years prior to admission showed an forced expiratory volume in the first second (FEV1)/forced vital capacity (FVC) ratio of 81%. She had a 30 pack‐year history of smoking, but quit 35 years ago. The patient also carried a diagnosis of heart failure, but an echocardiogram done 1 year ago demonstrated a left ventricular ejection fraction of 65% to 70% without diastolic dysfunction but mild right ventricular dilation and hypertrophy. Additionally, she had known nonobstructive coronary atherosclerotic heart disease, dyslipidemia, hypertension, morbid obesity, depression, and a documented chronic right hemidiaphragm elevation.
At this point the history suggests that the patient does not have a clear diagnosis of COPD. The lack of definitive spirometry evidence of chronic airway obstruction concerns me; I think that she may have been mistakenly treated with chronic inhaled steroids and doses of antibiotics for an acute exacerbation of chronic lung disease. Additional review of her history gives some indication of advanced lung disease, with her recent echocardiogram showing strain on the right ventricle with right ventricular hypertrophy and dilation, but there is no mention of the presence or severity of pulmonary hypertension. Nonetheless, I would be concerned that she probably has underlying significant cor pulmonale.
The patient now re‐presents with a worsening of her pulmonary symptoms. Her left‐sided pleuritic pain would make me concerned that she had a pulmonary embolus (PE). This morbidly obese patient with new pulmonary symptoms, right ventricular strain on her previous echocardiogram, and a persistent elevated right hemidiaphragm suggests a presentation of another PE.
At this time I cannot rule out other common possibilities such as infectious pneumonia. If she does have pneumonia, I would be concerned she could be harboring a multidrug‐resistant bacterial infection given her recent course of antibiotics in addition to her use of both chronic inhaled and intermittent oral glucocorticoids.
After gathering the rest of her full medical history, I would focus my physical exam on looking for evidence of parenchymal lung disease, signs of pulmonary hypertension, and pneumonia.
Her surgical history includes a previous hysterectomy, cholecystectomy, hernia repair, and left hepatic lobectomy for a benign mass. Her outpatient medications were ibuprofen, bupropion, fluvastatin, atenolol, potassium, aspirin, clopidogrel, albuterol inhaler, fluticasone/salmeterol inhaler, and omeprazole. She reports an allergy to penicillin and to sulfa drugs. Her mother died of an unknown cancer at age 77 years. She denied any international travel and she has always lived in Georgia.
The patient has been retired since 1992, having previously worked for the U.S. Postal Service. She admits to occasional alcohol intake (2 to 3 drinks a month). No recent travel, surgery, or prolonged immobilization was noted.
On initial examination she was alert and mentally appropriate, but appeared to be in mild respiratory distress with a respiratory rate of 28 breaths/minute. Her blood pressure (BP) was 99/70, heart rate 102, temperature of 38.2C, and oxygen saturation of 93% on room air and 97% on 2 L of oxygen via nasal cannula. Auscultation of her lungs revealed crackles over her left anterior lung field, bronchial breath sounds in the left posterior midlung, and bibasilar crackles. No wheezing was noted. Her cardiovascular exam and the remainder of her physical exam were unremarkable except for morbid obesity.
While my initial thoughts were leaning toward an exacerbation of chronic lung disease or possibly a new PE, at this moment, infection seems more likely. Indeed, her pulmonary findings suggest a left‐sided inflammatory process, and her vital signs meet criteria for systemic inflammatory response syndrome (SIRS). My primary concern is sepsis due to a drug‐resistant bacterial infection, including Staphylococcus aureus or gram‐negative bacteria or possibly more unusual organisms such as Nocardia or fungi, due to her recent use of antibiotics and chronic inhaled steroid use and recent course of oral glucocorticoids.
Conversely, the SIRS could be a manifestation of a noninfectious lung process such as acute interstitial pneumonia or an eosinophilic pneumonia. Given the diagnostic complexity, I would strongly consider consulting a pulmonologist if the patient did not improve quickly. At this point, I would like to review a posterior‐anterior (PA) and lateral chest radiograph, and room air arterial blood gas (ABG) in addition to basic laboratory test values.
Laboratory data obtained on admission was remarkable for a white blood cell (WBC) count of 26,500/L with 75% neutrophils and 6% eosinophils. Hemoglobin was 14.4 gm/dL. Platelet count was 454,000/L. Serum chemistries showed a sodium of 137 mEq/dL, potassium 4.3 mEq/dL, Cl 108 mEq/dL, bicarbonate 19 mEq/dL, blood urea nitrogen (BUN) 8 mg/dL, creatinine 1.0 mg/dL, and glucose 137 mg/dL. Cardiac enzymes were normal. Calcium was 9.8 mg/dL, albumin 2.7 gm/dL, total protein 6.9 gm/dL, AST 36 U/L, ALT 54 U/L and the bilirubin was normal. Chest radiograph (Figure 1) demonstrated a left perihilar infiltrate with air bronchograms and marked right hemidiaphragm elevation as seen on previous films. Unchanged increased interstitial markings were also present. Her electrocardiogram (ECG) showed normal sinus rhythm, normal axis, and QRS duration with nonspecific diffuse T‐wave abnormalities.
Given her presentation, I am worried about how well she is oxygenating and ventilating. An ABG should be done to assess her status more accurately. An albumin of 2.7 gm/dL indicates that she is fairly sick. I would not hesitate to consider testing the patient for human immunodeficiency virus (HIV) given how this information would dramatically change the differential diagnoses of her pulmonary process.
I am still most concerned about sepsis secondary to pneumonia in this patient with multiple chronic comorbidities, underlying chronic lung disease, receiving chronic inhaled glucocorticoids and a recent course of oral glucocorticoids and antibiotics. While I would initiate hydration I do not see a clear indication for early goal‐directed therapy for severe sepsis. In addition to obtaining an ABG and starting intravenous fluids, I would also draw blood cultures, send sputum for gram stain, culture, and sensitivity, and perform a urinalysis. I would also administer empiric antibiotics as quickly as possible based on a number of pneumonia clinical studies suggesting improved outcomes with early antibiotic administration. Because of her use of antibiotics and both inhaled and oral glucocorticoids, she is at higher risk for potentially multidrug‐resistant bacterial pathogens, including Staphyloccocus aureus and gram‐negative bacteria such as Pseudomonas and Klebsiella (Table 1). Therefore, I would initially cover her broadly for these organisms.
| Meets Any of the Following |
|---|
| Antimicrobial therapy in the preceding 90 days |
| Current hospitalization of 5 days or more |
| High frequency of antibiotic resistance in the community or in the specific hospital unit |
| Presence of risk factors for healthcare‐associated pneumonia (HCAP) |
| Hospitalization for >2 days in the preceding 90 days |
| Residence in nursing home or long‐term care facility (LTAC) for at least 5 days in last 90 days |
| Home infusion therapy including intravenous antibiotics within 30 days |
| Home wound care within 30 days |
| Chronic hemodialysis in hospital or clinic within 30 days |
| Family member with multidrug‐resistant pathogen |
| Immunosuppressive disease and/or therapy |
In addition to initial treatment choice, the inpatient triage decision is another important issue, especially at a community hospital where intensive care unit (ICU) resources are rare and often the admission decision is between sending a moderately sick patient to a regular floor bed or the medical ICU. Both the American Thoracic Society and Infectious Diseases Society of America support an ICU triage protocol in their guidelines for the management of community‐acquired pneumonia in adults that utilizes the following 9 minor criteria, of which the presence of at least 3 should support ICU admission: respiratory rate 30 breaths/minute; oxygenation index (pressure of oxygen [PaO2]/fraction of inspired oxygen [FiO2] ratio) 250; multilobar infiltrates; confusion/disorientation; uremia (BUN level 20 mg/dL); leukopenia (WBC count <4,000 cells/mm3); thrombocytopenia (platelet count <100,000 cells/mm3); hypothermia (core temperature <36C); and hypotension requiring aggressive fluid. Despite the absence of these criteria in this patient, it is important to note that no triage protocol has been adequately prospectively validated. Retrospective study of the minor criteria has found that the presence of at least 2 of the following 3 clinical criteria to have the highest specificity for predicting cardiopulmonary decompensation and subsequent need for ICU care: (1) initial hypotension (BP <90/60) on presentation with response to initial intravenous fluids to a BP >90/60; (2) oxygenation failure as indicated by PaO2/FiO2 ratio less than 250; or (3) the presence of multilobar or bilateral infiltrates on chest radiography.
I also want to comment on the relative elevation of her calcium, especially given the low albumin. This may simply be due to volume depletion, as many older patients have asymptomatic mild primary hyperparathyroidism. However, this elevated calcium may be a clue to the underlying lung process. Granulomatous lung disease due to tuberculosis or fungal infection could yield elevated calcium levels via increases in macrophage production of the active vitamin D metabolite calcitriol. This will need to be followed and a parathormone (PTH) level would be the best first test to request if the calcium level remains elevated. If the PTH level is suppressed, granulomatous disease or malignancy would be the more likely cause.
The patient was admitted with a presumptive diagnosis of community‐acquired pneumonia, was started on ceftriaxone and azithromycin, and given intravenous fluids, oxygen, and continued on inhaled salmeterol/fluticasone. Sputum was ordered for gram stain, culture, and sensitivity, and blood cultures were obtained. Urinalysis showed 1‐5 WBCs/high‐power field. Venous thromboembolism prophylaxis was initiated with subcutaneous heparin 5,000 units 8 hours. Her blood pressure normalized rapidly and during the next few days she stated she was feeling better. Despite continued significant wheezing her oxygen saturation remained at 98% on 2 L of oxygen via nasal cannula and she was less tachypneic. Attempts at obtaining an ABG were unsuccessful, and the patient subsequently refused additional attempts. Over the first few days her WBC count remained elevated above 20,000/L, with worsening bandemia (11%), and fever ranging from 38C to 39C. Sputum analysis was initially unsuccessful and blood cultures remained negative.
I am concerned about the persistent fever and elevated WBC count, and want to emphasize that I might have treated her with broader spectrum antibiotics to cover additional multidrug‐resistant bacterial organisms. I would have initially ordered vancomycin to cover methicillin resistant Staphylococcus aureus (MRSA) plus 2 additional antibiotics that cover multidrug‐resistant gram negative pathogens including Pseudomonas aeruginosa.
On the fifth hospital day, her WBC count dropped to 13,400/L and she defervesced. However, her respiratory status worsened during that same day with increased tachypnea. Of note, no results were reported from the initial sputum cultures and they were reordered and a noncontrast chest computed tomography (CT) was also ordered.
I think at this point, even though she has remained stable hemodynamically and oxygenating easily with supplemental oxygen, the question of whether or not her primary process is infectious or noninfectious lingers. I agree with obtaining a chest CT scan.
I am not surprised that sputum was not evaluated despite the orders. Among hospitalized patients with pneumonia, we frequently find that about a third of the time sputum cannot be obtained, about a third of the time it is obtained but the quality is unsatisfactory, and only a third of the time does the sputum sample meet criteria (less than 5 squamous epithelial cells per high‐power field) for adequate interpretation of the gram‐stain and culture result. Unfortunately, no one has developed a better way to improve this process. Nonetheless, I believe we do not try hard enough to obtain sputum in the first hours of evaluating our patients. I joke with our internal medicine residents that they should carry a sputum cup with them when they evaluate a patient with possible pneumonia. One recent prospective study of the value of sputum gram‐staining in community‐acquired pneumonia has found it to be highly specific for identifying Streptococcus pneumoniae or Haemophilus influenzae pneumonia.
The CT scan (Figure 2) performed on hospital day 6 demonstrated consolidation in the left upper lobe with areas of cavitation. There was also interstitial infiltrate extending into the lingula. Elevation of the right hemidiaphragm with atelectasis in both lung bases was also noted. A small effusion was present on the left and possibly a minimal effusion on the right as well. There was no pericardial effusion and only a few small pretracheal and periaortic lymph nodes were noted.
Given her failure to improve significantly after 6 days of antibiotic treatment, and her recent use of glucocorticoids, I would expand my diagnostic considerations to include other necrotizing bacterial infections, tuberculosis, fungus, and Nocardia.
Given the results of the CT scan she was placed in respiratory isolation to rule out active pulmonary tuberculosis. Though tachypneic, her blood pressure and pulse remained stable. However, her oxygen saturation deteriorated, declining to 92% on 2 L of oxygen via nasal cannula during hospital days 6 and 7. Subsequent successful attempts at collecting sputum yielded rapid growth of yeast (not Cryptococcus spp.). Pulmonary and infectious disease consultations were obtained and vancomycin was added to her regimen. The patient subsequently agreed to undergo diagnostic bronchoscopy.
I agree with obtaining input from expert consultants. I think we too often underutilize consultation in patients that are better but not completely better when we are not entirely sure what is going on. Evidence of noncryptococcus yeast in sputum may sometimes indicate colonization with Candida spp. without any significant clinical consequence. This finding may alternatively suggest the possibility of a true fungal pneumonia caused by 1 of the dimorphic fungi, including Histoplasma capsulatum, Paracoccidioides brasiliensis, Blastomyces dermatitides, or Coccidioides immitis. However, in this case there is not a strong epidemiologic patient history of exposure to any of these types of fungi.
Three sputum smears were negative for acid fast bacilli (AFB). Bronchoscopy revealed grossly abnormal mucosa in the left upper lobe and bronchomalacia, but no obstructive lesions. A transthoracic echocardiogram was ordered to evaluate her degree of pulmonary hypertension.
The 3 sputum specimens that were negative for AFB despite cavitary lung disease have high sensitivity for ruling out pulmonary tuberculosis. In addition, given the absence of any bacterial pathogen isolated from these specimens, I would pursue the possibility of other potential fungal pathogens given the patient's subacute course, history of using inhaled and oral corticosteroids, sputum results, and the presence of a cavitary lesion on her CT scan images.
Cytologic examination of the bronchoalveolar lavage (BAL) sample showed a cell differential of 1% bands, 58% neutrophils, 9% lymphs, and 27% eosinophils. The routine postbronchoscopy chest radiograph showed complete opacification of the left lung. The patient's WBC count rose to 26,000/L but she remained afebrile. Echocardiogram was reported to be of very poor quality due to her obesity. The cardiologist reviewing the echocardiogram called the attending physicians and stated there was possibly something in the left pulmonary artery and aortic dissection could not be ruled out.
The presence of eosinophilia on BAL may be a very important clue as to what lung pathology she has. In fact, eosinophilia in this setting may indicate the possibility of parasitic or fungal infection of the lung, or inflammation of the airway associated to drug toxicity, asthma, or environmental toxin exposure. With this additional information, I am concerned that she may be harboring an atypical infection such as an invasive fungus. The echocardiogram results are unclear to me but will need to be clarified with additional testing.
The interpretation of the transbronchial biopsy specimen was limited but suggested invasive pseudomembranous tracheal bronchitis due to aspergillosis. The routine hematoxylin and eosin stain showed portions of alveolar lung tissue and some collapsed submucosal bronchial glands with relatively normal‐looking lung tissue but along the edge of the spaces were obvious fungal organisms. The Gomori's methenamine silver (GMS) stain suggested the presence of Aspergillus organisms (Figure 3). Fungal cultures were also negative for any of the other dimorphic fungi or for molds.
Despite the negative culture results, the overall clinical picture suggests a necrotizing pneumonia caused by an invasive Aspergillus affecting both the bronchial tree and the lower respiratory tract. Generally, necrotizing pneumonias usually have a slow response to antimicrobial therapy. Given the inherent difficulty in differentiating clearly between invasive and noninvasive disease based on a transbronchial biopsy specimen, initiating antifungal therapy for invasive aspergillosis is appropriate in this patient. This patient's recent use of oral glucocorticoids and chronic use of inhaled glucocorticoids are both potential risk factors that predisposed this patient to develop invasive aspergillosis.
Many times we simply follow treatment guidelines for different categories of pneumonia, and have limited or inadequate clinical information to make more definitive diagnoses. While we need these treatment protocols, physicians must avoid falling into the trap that antibiotics treat all infectious etiologies in the lung and we should make reasonable efforts to pin down the etiology. All of us have been fooled by atypical presentations of tuberculosis, fungus, and noninfectious diseases of the lung. I think it behooves us to be vigilant about alternative diagnoses and consider pursuing additional studies whenever the clinical response to initial treatment does not meet our expectations.
Subsequently, the patient's additional cultures remained negative. The official echocardiogram report was read as questionable PE in the pulmonary artery. A spiral CT angiogram revealed a pulmonary artery embolus in the left upper lobe and she was treated with anticoagulation. Her shortness of breath improved steadily and she was successfully discharged after receiving 9 days of oral voriconazole. Outpatient pulmonary function testing documented the presence of chronic obstructive lung disease. She completed a 5‐month course of voriconazole therapy with significant clinical and radiologic improvement of her pulmonary infiltrate. She also completed a 12‐month treatment with warfarin for the concomitant pulmonary embolism. On follow‐up at 12 months she was doing well.
COMMENTARY
Aspergillosis caused particularly by Aspergillus fumigatus is considered an emerging infectious disease that frequently produces significant morbidity and mortality among immunocompromised patients.1, 2 The most frequently‐affected organs by this fungal pathogen include the lung and the central nervous system. There are 3 pathogenic mechanisms of Aspergillus infection of the lung: colonization, hypersensitivity reaction, and invasive aspergillosis.1
Invasive pulmonary aspergillosis is predominantly seen among individuals with severe degrees of immunosuppression as a result of solid‐organ transplantation, immunosuppressive therapies for autoimmune diseases, systemic glucocorticoids, and chemotherapy for hematologic malignancies. Mortality due to invasive aspergillosis continues to be very high (>58%) despite our improved ability to diagnose this condition and newer therapies to treat immunocompromised individuals.1 Invasive aspergillosis can manifest clinically in multiple ways. These include: (1) an invasive vascular process in which fungal organisms invade blood vessels, causing a rapidly progressive and often fatal illness; (2) necrotizing pseudomembranous tracheal bronchitis; (3) chronic necrotizing aspergillosis; (4) bronchopleural fistula; or (5) empyema.35 In our case, while the pathologic findings were most suggestive of an invasive pseudomembranous tracheal bronchitis, the overall clinical picture was most compatible with a necrotizing pneumonia due to invasive aspergillosis.
In addition to the traditional identified risk factors for invasive pulmonary aspergillosis, a number of reports during the last decade have demonstrated the occurrence of invasive aspergillosis in patients with COPD.14 A systematic review of the literature demonstrated that among 1,941 patients with invasive aspergillosis, 26 (1.3%) had evidence of COPD as the main risk factor for developing invasive aspergillosis.1 A single report has associated the potential use of inhaled steroids with the occurrence of invasive aspergillosis in this patient population.2 However, other factors that may promote increased susceptibility to invasive fungal infection among patients with COPD include the use of long‐term or repeated short‐term glucocorticoid treatments, and the presence of multiple additional comorbidities, which may be found in this same population such as diabetes, malnutrition, or end‐stage renal disease.3, 4 Most reported series have demonstrated a high mortality rate of invasive pulmonary aspergillosis in patients with COPD.14
The diagnosis of invasive pulmonary aspergillosis represents a significant clinical challenge. Diagnostic algorithms incorporating CT, antigen detection testing (for serum galactomannan and ‐glucan) as well as polymerase chain reaction diagnostic testing appear to be beneficial in the early diagnosis of invasive aspergillosis in particular settings such as in allogeneic hematopoietic stem cell transplantation.5 The role of antigen testing to identify early invasive aspergillosis in patients with COPD remains uncertain since it has been evaluated in a limited number of patients and therefore clinical suspicion is critical to push clinicians to pursue invasive tissue biopsy and cultures to confirm the diagnosis.3, 4
Based on the available clinical case series and in our case, invasive pulmonary aspergillosis should be suspected in COPD patients with rapidly progressive pneumonia not responding to antibacterial therapy and who have received oral or inhaled glucocorticoids in the recent past. In addition, this case also illustrates that occasionally, patients present with more than 1 life‐threatening diagnosis. This patient was also diagnosed with PE despite adequate prophylaxis. In addition to the well‐known clinical risk factors of obesity and lung disease, the underlying infection may have contributed to a systemic or local hypercoagulable condition that further increased her risk for venous thromboembolism.
KEY TEACHING POINTS
-
Clinicians should remember to consider a broad differential in patients presenting with pneumonia, including the possibility of fungal pathogens in patients with known risk factors and in patients with multiple, potentially immunosuppressive comorbidities, or in patients who do not improve on standard antibiotic therapy.
-
There is some evidence of an association between COPD and invasive aspergillosis, likely due to the frequent use of oral corticosteroids and/or chronic inhaled steroids in this population.
- ,,.Aspergillosis case‐fatality rate: systematic review of the literature.Clin Infect Dis.2001;32:358–366.
- ,,,,.Invasive pulmonary filamentous fungal infection in a patient receiving inhaled corticosteroid therapy.Clin Infect Dis.2002;35:e54–e56.
- ,,, et al.Invasive pulmonary aspergillosis in chronic obstructive pulmonary disease: an emerging fungal pathogen.Clin Microbiol Infect.2005;11:427–429.
- ,,,,,.Invasive pulmonary aspergillosis in patients with chronic obstructive pulmonary disease: report of eight cases and review.Clin Infect Dis.1998;26:1473–1475.
- ,.Current approaches to diagnosis and treatment to invasive aspergillosis.Am J Respir Crit Care Med.2006;173:707–717.
- ,,.Aspergillosis case‐fatality rate: systematic review of the literature.Clin Infect Dis.2001;32:358–366.
- ,,,,.Invasive pulmonary filamentous fungal infection in a patient receiving inhaled corticosteroid therapy.Clin Infect Dis.2002;35:e54–e56.
- ,,, et al.Invasive pulmonary aspergillosis in chronic obstructive pulmonary disease: an emerging fungal pathogen.Clin Microbiol Infect.2005;11:427–429.
- ,,,,,.Invasive pulmonary aspergillosis in patients with chronic obstructive pulmonary disease: report of eight cases and review.Clin Infect Dis.1998;26:1473–1475.
- ,.Current approaches to diagnosis and treatment to invasive aspergillosis.Am J Respir Crit Care Med.2006;173:707–717.
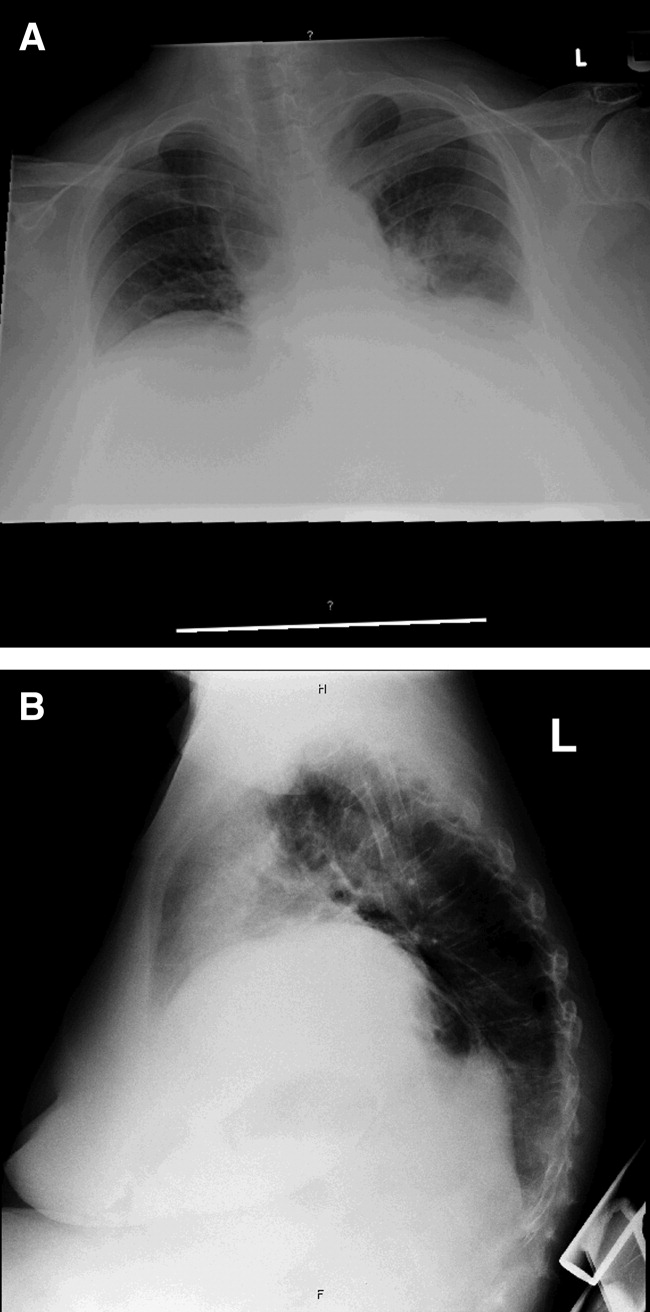
One Hundred Years Later
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 36 year‐old male physician was admitted to a Baltimore hospital in April 1907 with weight loss, weakness, arthralgias, and abdominal distension that had progressed over 5 years.
In 1907, major causes of unexplained weight loss included tuberculosis, hyperthyroidism, cancer, and diabetes. Arthralgias and weakness are not specific. The insidious progression over 5 years narrows the infectious possibilities; tuberculosis and syphilis are important considerations. Since surgical removal was the main treatment for malignancy in 1907, a history of prior surgery might point to a previously diagnosed malignancy that is now progressing.
Five years earlier, while visiting Turkey as a medical missionary, he first noted the onset of arthralgias that lasted 6 to 8 hours and occurred 3 to 4 times per week. Over time, these attacks lasted up to 24 hours and became associated with warmth, swelling, and tenderness of both small and large joints. He gradually lost weight and strength. One year prior to arrival in the hospital, he developed a cough productive of yellow sputum. Seven months prior, he returned from Turkey to Atlanta and noticed an increase in his cough, along with fevers of 100 Fahrenheit and night sweats.
The primacy of the arthralgias in this illness lead me to consider primary rheumatic diseases, and multisystem diseases (including infections) with a predominant skeletal component. In 1907, tests for lupus and the rheumatoid factor were not available. Neither skeletal remains nor works of art provide evidence that rheumatoid arthritis existed until the 19th century, whereas ankylosing spondylitis, gout, and rickets were present by then.
As a medical missionary, he might have acquired a disease endemic to the areas he visited, or the travel history may be a red herring. Familial Mediterranean fever, though prevalent in Turkey and a cause of arthralgias accompanied by recurrent attacks of abdominal pain and fever, is not an acquired disease. Behcet's disease, also known as Silk Trader's Route disease, is found in descendents of the countries that comprised the ancient Silk Route from Japan to the Middle East and may cause arthritis along with oral ulcers, genital lesions, pathergy or uveitis. I would inquire about his ancestry and fevers before dismissing these possibilities.
Although 5 years would be unusually long for tuberculosis to go unrecognized, a physician in the first half of the 20th century would place tuberculosis near the top of possible diagnoses. In 1930, a time when the population of the United States was considerably less, there were over 300,000 cases of tuberculosis. Physicians, and in particular pathologists since autopsies were more commonly performed, often died from tuberculosis since streptomycin, the first antituberculous medication, did not arrive until 1944. At the turn of the 20th century, the ability to detect tubercle bacilli was quite good. Thus, I would include tuberculous peritonitis as a cause of the progressive abdominal symptoms in this physician. In approximately one‐third of patients with tuberculous peritonitis there is evidence of pulmonary disease, and I would try to culture tuberculosis in samples of sputum, a test then so common it probably rivaled our frequent complete blood counts in popularity.
Six months prior, examinations of sputum were negative for tubercle bacilli. Four months prior to arrival, the patient moved to New Mexico. His cough improved but he continued to lose weight and had diarrhea consisting of 3 to 4 loose or semiformed bowel movements per day. Three months prior to admission, he noted an increase in abdominal girth along with right lower quadrant fullness. One month prior, he noted painful swelling and warmth in both ankles as well as dyspnea with exertion.
The increased abdominal girth in the context of chronic illness might be due to ascites, adenopathy, visceromegaly, or mass lesions such as a neoplasm or abscess. If ascites is the cause, one would need to consider primary hepatic disorders, as well as extrahepatic diseases that could progress over years. Infection with hepatitis A virus does not cause chronic liver disease. Hepatitis B, in those days, was referred to as serum hepatitis, and a serum marker for the B virusthe Australia antigenwas not identified until 1967. Cardiac causes of ascites include congestive heart failure and constrictive pericarditis, the latter an important consideration because it is potentially curable. Also, constrictive pericarditis can present as an indolent weight‐losing disease because of chronic visceral congestion. Other considerations include nephrotic syndrome, infection, and neoplasm, including mesothelioma.
Abdominal distention might also be seen with a smoldering abscess. In addition to an appendiceal process, the travel and right lower quadrant localization reminds us to consider ameboma. This patient surely was in an area where amebiasis was endemic, and amebomaa chronic inflammatory form of infection with E. histolytica not associated with diarrhea or liver cystsmay mimic cecal carcinoma. Exertional dyspnea suggests at least the possibility of cardiac disease. Despite the negative sputum cultures, tuberculosis remains high on the list as a cause of constrictive pericarditis or peritonitis, either of which may occur in the absence of active pulmonary disease.
Past medical history included measles and whooping cough as a child, mild pleurisy at age 14, mild influenza 7 years previously. The patient had a tonsillectomy as a child and had a portion of his inferior turbinate bone removed in an attempt to relieve a nasal condition.
On physical exam, the patient was thin and the skin over his face and hands was deep brown. His temperature was 101.5 Fahrenheit, the heart rate was 100, and the respiratory rate was 24. Small lymph nodes were palpable in the axillary and epitrochlear areas. His thorax moved asymmetrically, with less movement on the left apex and slight dullness to percussion in that area. The pulmonic component of the second heart sound was mildly accentuated. The abdomen displayed fullness and tympany, most pronounced in the right lower quadrant without hepatosplenomegaly. The left ankle was swollen, and the overlying skin was tense, shiny, and hot. On both lower legs, areas of discoloration and slight induration were observed, felt to be consistent with faded erythema nodosum.
Though pleurisy has numerous causes, its presence raises the specter of tuberculosis again. The nasal condition triggers thoughts of Wegener's granulomatosis or lethal midline granuloma, both unlikely diagnoses here. The pulmonary exam suggests an apical process, such as tuberculosis, and the accentuated pulmonic heart sound implies pulmonary hypertension, which could be due to a number of chronic pulmonary diseases. The epitrochlear nodes are of interest since lymphoma and Hodgkin's disease rarely involve this area; syphilis and human immunodeficiency virus (HIV) are a few of the chronic diseases that may involve this lymph node region. More helpful is the absence of hepatosplenomegaly, since many indolent malignancies and infections would be expected to enlarge these organs by this point.
Monoarticular arthritis is often due to infection, and less likely due to rheumatoid disease. When rheumatoid arthritis flares, the entire skeleton flares, not single joints. Given the indolence and this single joint involvement, tuberculosis again comes to mind.
I would next want to obtain a plain chest radiograph, looking for evidence of tuberculosis. As with any test, one should ask how this will change management. In 1907, antituberculous medications were not available, so therapy was directed at lowering oxygen tension in the primary site of infection; for example, pulmonary disease was addressed via pneumothorax. If the chest radiograph provides little hint of tuberculosis, then consideration must be given to exploratory surgery of the abdomen given the focal abnormality in the right lower quadrant.
A peripheral blood smear revealed a hypochromic microcytic anemia. The total red blood cell count was 4.468 million/mm3 (normal range for men is 4.52‐5.90 million/mm3), white blood cell count was 8180/mm3, including 80% granulocytes and 9% eosinophils. On gross inspection, the stool was clay‐colored, and stool microscopy demonstrated large numbers of neutral fat droplets, but no ova, parasites, or tubercle bacilli. Urinalysis revealed no albumin or casts, and the bones were normal on ankle radiographs. Another sample of sputum revealed no tubercle bacilli, and intradermal placement of tuberculin provoked no reaction.
His negative tuberculin skin reaction is unusual for that era, because of the prevalence of tuberculosis. Most likely, he is anergic because of his severe underlying illness, and the absent reaction is thus not all that helpful a clue. Multiple negative sputum examinations lower the possibility of pulmonary, but not extrapulmonary, tuberculosis. The absence of bony destruction on ankle radiographs lowers my suspicion for tuberculous arthritis.
The excess stool fat implies steatogenic diarrhea from malabsorption, and 2 categories here are pancreatic and luminal diseases. Of these 2, pancreatic etiologies produce more severe malabsorption. We do not hear mention of jaundice, however, and I cannot see how to link the pancreas to the arthritis. A chronic infection which may produce malabsorption and eosinophilia is strongyloidiasis, endemic in the southeastern United States. However, this patient did not manifest the most common finding of chronic strongyloidiasis, namely asthma. Adrenal insufficiency, as might result from disseminated tuberculosis, is associated with increased skin pigmentation, diarrhea, and eosinophilia. However, the diarrhea of adrenal insufficiency is not malabsorptive, and serum electrolytes and cortisol tests were not available then to confirm this diagnosis antemortem.
In an attempt to identify a unifying cause of chronic arthritis, malabsorption, and increased skin pigmentation, I must consider Whipple's disease first and foremost. Physicians then were strapped and observation was often the default mode of the day. Given the abdominal findings, an exploratory laparotomy would be warranted if his condition deteriorated.
Despite forced oral feedings, the patient continued to lose weight, from his normal of 175 pounds to a nadir of 145 pounds. Because of worsening abdominal distention, the patient underwent exploratory abdominal surgery on the twenty‐first hospital day. Intraoperatively, no ascites was seen, but his mesenteric lymph nodes were hard and markedly enlarged. The abdomen was closed without further intervention. Two days after the surgery, the patient abruptly developed dyspnea. His respirations were 40 per minute, heart rate was 120, and he had minimal rales at the lung bases without findings of consolidation. He died 2 hours later, on the twenty‐third hospital day, and an autopsy was performed.
The final event may have been a pulmonary embolism. As for the adenopathy, lymphoma and tuberculosis are possible. Heavy chain disease, an unusual lymphoproliferative disorder found in persons from the old Silk Trader's Route from the Middle East to the Orient, is a remote prospect. However, 5 years is just too indolent for most cancers and would be very unusual for tuberculosis. I think the findings support Whipple's disease, and I wonder if this was the first reported case.
On postmortem examination, the abdominal adenopathy was striking. The small intestine contained enlarged villi with thickened submucosa, and the mesenteric nodes were enlarged with fat deposits and abnormal foamy cells. Within these foamy cells, microscopy revealed numerous rod‐shaped organisms. All studies were negative for tuberculosis, and although the pathologist, Dr. George Hoyt Whipple, suspected an infectious etiology, he offered the name intestinal lipodystrophy to emphasize the striking small intestinal changes he witnessed at autopsy, and which are the hallmarks of the disease that now bears his name. Whipple also shared the 1934 Nobel Prize in Physiology or Medicine with Minot and Murphy for their discovery that a nutritional substance in liver, now known as vitamin B12, was beneficial in treating pernicious anemia.
COMMENTARY
This is the index case of Whipple's disease, summarized from the original 1907 description.1 George Hoyt Whipple, then a pathologist at Johns Hopkins, highlights the value of keen observation and a well‐done case report in describing a new disease entity. One of the roles of case reports is to detail the features of an unknown disease. In this capacity, Whipple's summary is exemplary. His achievement was having the openness of mind to realize he was witnessing something novel, and to take the first step on the road to discovery. Although Whipple suspected he was staring at a unique disease, he could not pinpoint the culprit bacteria and he had trouble squaring the extraintestinal findings with the marked intestinal anomalies. It was left to decades of input from others to confirm the association of arthralgias, eosinophilia, skin hyperpigmentation, and cardiac valve abnormalities with intestinal malabsorption, and to culture the infectious agent.
In his discussion, Whipple recognized he was confronted with a novel clinical entity. Prior to surgery, pulmonary and mesenteric tuberculosis were suspected, based on the fevers, weight loss, cough, fat malabsorption, and lymphadenopathy. However, he felt the left apical exam was more representative of retraction from prior disease than active infection. He was also bothered by the negative skin reaction and sputum tests. At surgery, the pronounced adenopathy suggested sarcoma or Hodgkin's disease but postmortem examination eliminated these possibilities. At autopsy, the abdominal findings were most striking. The small intestine demonstrated enlarged villi with thickened submucosa and markedly enlarged mesenteric lymph glands containing large fat deposits and distinctly abnormal foamy cells. These foamy macrophages contained great numbers of rod‐shaped organisms resembling the tubercle bacillus. However, all tests were negative for tuberculosis, and the lungs contained no active disease. Though he suspected an infectious etiology, Whipple offered the name intestinal lipodystrophy to emphasize the striking small intestinal pathology.
Although Whipple had surmised a novel infectious agent in 1907, it took almost a century to isolate the causative microbe. Granules within foamy macrophages of the small intestine were detected on periodic acid‐Schiff (PAS) staining in 1949.2 Similar PAS‐positive granules were soon discovered in other tissues and fluid, providing a plausible explanation for the systemic features of the disease.3 Electron microscopy confirmed the presence of infectious bacilli in 1961,4 ushering in the era of antimicrobial treatment for this disease. More recently, using polymerase chain reaction (PCR), a unique bacterial 16S ribosomal RNA gene was isolated in patients with Whipple's disease.5, 6 Phylogenetically classified with the actinobacteria, Tropheryma whipplei (fom the Greek trophe, nourishment, and eryma, barrier) was ultimately subcultured in 2000,7 and immunodetection testing became possible. Using this technique, the archived pathology specimens from the 1907 index case demonstrated numerous intracellular bacteria in the lamina propria, closing the loop started by Whipple nearly a century earlier.8
Whipple's index case report described most of the manifestations of the disease we are familiar with today. As in the original description, arthralgias are the most common initial symptom and may precede diagnosis by a mean of 8 years. Other cardinal features include weight loss, abdominal pain and steatorrhea due to small intestinal involvement. Table 1 summarizes the important signs and symptoms of Whipple's disease.9, 10 One notable manifestation missing in Whipple's report is central nervous system involvement. Central nervous system (CNS) disease ranges from cognitive deficits to encephalitis and focal defects, and may occur years after treatment and without concomitant intestinal symptoms.
| Clinical Feature | Comment |
|---|---|
| |
| Cardinal features (present in 60% to 90%) | |
| Arthropathy | Most common initial symptom, preceding diagnosis by a mean of 8 years. Migratory, nonerosive, mainly in the peripheral joints. |
| Weight loss | |
| Diarrhea | Usually steatorrhea, may be associated with pain or occult blood in the stool |
| Other common features (present in 20% to 45%) | |
| Fever | |
| Lymphadenopathy | May present as a palpable mass |
| Increased skin pigmentation | Mechanism unknown (evidence of adrenal insufficiency has not been found in Whipple's) |
| Cardiac disease | Culture‐negative endocarditis |
| Hypotension | |
| Peripheral edema | |
| Uncommon clinical features | |
| Central nervous system involvement | May be global (dementia, personality change, sleep disturbance) or focal (cranial neuropathy, nystagmus) |
| Eye disease | Uveitis, retinitis |
| Hepatosplenomegaly | |
| Polyserositis | |
| Ascites | |
A remaining mystery is why this pathogen results only rarely in clinical disease. Caucasians comprise the majority of infected patients, and men are affected 8 times more often than women. An overrepresentation of HLA‐B27 suggests a genetic predisposition, though its role in pathogenesis is unclear. T. whipplei has been identified by PCR methods in asymptomatic individuals, implying additional abnormalities must be present in susceptible hosts for symptoms to occur following colonization.11 The exact immune defects are speculative, and immunodeficiency states (such as HIV) have not been consistently identified in patients with Whipple's disease.
The cornerstone of diagnosing Whipple's disease is upper endoscopy with duodenal biopsy. Flattening of the villi and markedly increased PAS‐positive staining of lamina propria macrophages are strongly suggestive of the diagnosis. PAS‐positive staining is not unique to T. whipplei, however. In patients with profound immunodeficiency, Mycobacterium avium intracellulare may stain positive with PAS. Since Whipple's disease is only rarely associated with HIV, a negative HIV test would favor a diagnosis of Whipple's disease. Electron microscopy may distinguish T. whipplei from its mimickers by morphology. For extraintestinal disease, PCR testing on samples from infected tissue has been found to be a reliable diagnostic aid.9
Given the rarity of the disease, controlled clinical trials addressing optimal treatment are lacking. Current recommendations include initial therapy for 14 days with an agent that crosses the blood‐brain barrier (eg, ceftriaxone) to reduce the incidence of CNS disease. This is then followed by a year or more of oral antimicrobial therapy with trimethoprim‐sulfamethoxazole or a tetracycline.9 While most patients respond within 2 to 3 weeks, relapse may occur in as many as one‐third of patients.
Historical case reports reinforce the case‐based learning paradigm. As the discussant remarks, observation was all too often the only recourse for physicians a century ago. In recounting the 7‐year progression of disease in 1 individual, Whipple provides a unique window into the natural evolution of the key features of this systemic disease. Viewed through the prism of Whipple's eyes, we can recall the striking lymphoid hyperplasia and unusual organisms in the small intestine, cementing our understanding of the pathogenesis of this disorder. Revisiting past cases allows us to learn of and learn from the past.
Teaching Points
-
Whipple's disease should be considered in patients with unexplained arthralgias accompanied by weight loss, malabsorption, and abdominal pain.
-
For suspected intestinal Whipple's disease, diagnosis is best made by duodenal biopsy demonstrating PAS‐positive staining in lamina propria macrophages.
-
Systemic manifestations of Whipple's disease include culture‐negative endocarditis and CNS disease. PCR testing of involved sites for T. whipplei is recommended to confirm extraintestinal disease.
- .A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues.Bull Johns Hopkins Hosp.1907;18:382–391.
- .The tinctoral demonstration of a glycoprotein in Whipple's disease.Proc Soc Exp Biol Med.1949;72:225–227.
- ,,.Whipple's disease: clinical, biochemical, and histopathologic features and assessment of treatment in 29 patients.Mayo Clin Proc.1988;63:539–551.
- ,.Combined electron and light microscopy in Whipple's disease: demonstration of “bacillary bodies” in the intestine.Bull Johns Hopkins Hosp.1961;109:80–98.
- ,,,.Phylogeny of the Whipple's disease‐associated bacterium.Lancet.1991;338:474–475.
- ,,,.Identification of the uncultured bacillus of Whipple's disease.N Engl J Med.1992;327:293–301.
- ,,, et al.Cultivation of the bacillus of Whipple's disease.N Engl J Med.2000;342:620–625.
- ,,,.Immunodetection of Tropheryma whipplei in intestinal tissue from Dr. Whipple's 1907 patient.N Engl J Med.2003;348:1411– 1412.
- ,.Whipple's disease.Lancet.2003;361:239–246.
- ,,, et al.Diagnostic guidelines in central nervous system Whipple's disease.Ann Neurol.1996;40:561–568.
- ,,, et al.PCR‐positive tests for Tropheryma whippleii in patients without Whipple's disease.Lancet.1999;353:2214.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 36 year‐old male physician was admitted to a Baltimore hospital in April 1907 with weight loss, weakness, arthralgias, and abdominal distension that had progressed over 5 years.
In 1907, major causes of unexplained weight loss included tuberculosis, hyperthyroidism, cancer, and diabetes. Arthralgias and weakness are not specific. The insidious progression over 5 years narrows the infectious possibilities; tuberculosis and syphilis are important considerations. Since surgical removal was the main treatment for malignancy in 1907, a history of prior surgery might point to a previously diagnosed malignancy that is now progressing.
Five years earlier, while visiting Turkey as a medical missionary, he first noted the onset of arthralgias that lasted 6 to 8 hours and occurred 3 to 4 times per week. Over time, these attacks lasted up to 24 hours and became associated with warmth, swelling, and tenderness of both small and large joints. He gradually lost weight and strength. One year prior to arrival in the hospital, he developed a cough productive of yellow sputum. Seven months prior, he returned from Turkey to Atlanta and noticed an increase in his cough, along with fevers of 100 Fahrenheit and night sweats.
The primacy of the arthralgias in this illness lead me to consider primary rheumatic diseases, and multisystem diseases (including infections) with a predominant skeletal component. In 1907, tests for lupus and the rheumatoid factor were not available. Neither skeletal remains nor works of art provide evidence that rheumatoid arthritis existed until the 19th century, whereas ankylosing spondylitis, gout, and rickets were present by then.
As a medical missionary, he might have acquired a disease endemic to the areas he visited, or the travel history may be a red herring. Familial Mediterranean fever, though prevalent in Turkey and a cause of arthralgias accompanied by recurrent attacks of abdominal pain and fever, is not an acquired disease. Behcet's disease, also known as Silk Trader's Route disease, is found in descendents of the countries that comprised the ancient Silk Route from Japan to the Middle East and may cause arthritis along with oral ulcers, genital lesions, pathergy or uveitis. I would inquire about his ancestry and fevers before dismissing these possibilities.
Although 5 years would be unusually long for tuberculosis to go unrecognized, a physician in the first half of the 20th century would place tuberculosis near the top of possible diagnoses. In 1930, a time when the population of the United States was considerably less, there were over 300,000 cases of tuberculosis. Physicians, and in particular pathologists since autopsies were more commonly performed, often died from tuberculosis since streptomycin, the first antituberculous medication, did not arrive until 1944. At the turn of the 20th century, the ability to detect tubercle bacilli was quite good. Thus, I would include tuberculous peritonitis as a cause of the progressive abdominal symptoms in this physician. In approximately one‐third of patients with tuberculous peritonitis there is evidence of pulmonary disease, and I would try to culture tuberculosis in samples of sputum, a test then so common it probably rivaled our frequent complete blood counts in popularity.
Six months prior, examinations of sputum were negative for tubercle bacilli. Four months prior to arrival, the patient moved to New Mexico. His cough improved but he continued to lose weight and had diarrhea consisting of 3 to 4 loose or semiformed bowel movements per day. Three months prior to admission, he noted an increase in abdominal girth along with right lower quadrant fullness. One month prior, he noted painful swelling and warmth in both ankles as well as dyspnea with exertion.
The increased abdominal girth in the context of chronic illness might be due to ascites, adenopathy, visceromegaly, or mass lesions such as a neoplasm or abscess. If ascites is the cause, one would need to consider primary hepatic disorders, as well as extrahepatic diseases that could progress over years. Infection with hepatitis A virus does not cause chronic liver disease. Hepatitis B, in those days, was referred to as serum hepatitis, and a serum marker for the B virusthe Australia antigenwas not identified until 1967. Cardiac causes of ascites include congestive heart failure and constrictive pericarditis, the latter an important consideration because it is potentially curable. Also, constrictive pericarditis can present as an indolent weight‐losing disease because of chronic visceral congestion. Other considerations include nephrotic syndrome, infection, and neoplasm, including mesothelioma.
Abdominal distention might also be seen with a smoldering abscess. In addition to an appendiceal process, the travel and right lower quadrant localization reminds us to consider ameboma. This patient surely was in an area where amebiasis was endemic, and amebomaa chronic inflammatory form of infection with E. histolytica not associated with diarrhea or liver cystsmay mimic cecal carcinoma. Exertional dyspnea suggests at least the possibility of cardiac disease. Despite the negative sputum cultures, tuberculosis remains high on the list as a cause of constrictive pericarditis or peritonitis, either of which may occur in the absence of active pulmonary disease.
Past medical history included measles and whooping cough as a child, mild pleurisy at age 14, mild influenza 7 years previously. The patient had a tonsillectomy as a child and had a portion of his inferior turbinate bone removed in an attempt to relieve a nasal condition.
On physical exam, the patient was thin and the skin over his face and hands was deep brown. His temperature was 101.5 Fahrenheit, the heart rate was 100, and the respiratory rate was 24. Small lymph nodes were palpable in the axillary and epitrochlear areas. His thorax moved asymmetrically, with less movement on the left apex and slight dullness to percussion in that area. The pulmonic component of the second heart sound was mildly accentuated. The abdomen displayed fullness and tympany, most pronounced in the right lower quadrant without hepatosplenomegaly. The left ankle was swollen, and the overlying skin was tense, shiny, and hot. On both lower legs, areas of discoloration and slight induration were observed, felt to be consistent with faded erythema nodosum.
Though pleurisy has numerous causes, its presence raises the specter of tuberculosis again. The nasal condition triggers thoughts of Wegener's granulomatosis or lethal midline granuloma, both unlikely diagnoses here. The pulmonary exam suggests an apical process, such as tuberculosis, and the accentuated pulmonic heart sound implies pulmonary hypertension, which could be due to a number of chronic pulmonary diseases. The epitrochlear nodes are of interest since lymphoma and Hodgkin's disease rarely involve this area; syphilis and human immunodeficiency virus (HIV) are a few of the chronic diseases that may involve this lymph node region. More helpful is the absence of hepatosplenomegaly, since many indolent malignancies and infections would be expected to enlarge these organs by this point.
Monoarticular arthritis is often due to infection, and less likely due to rheumatoid disease. When rheumatoid arthritis flares, the entire skeleton flares, not single joints. Given the indolence and this single joint involvement, tuberculosis again comes to mind.
I would next want to obtain a plain chest radiograph, looking for evidence of tuberculosis. As with any test, one should ask how this will change management. In 1907, antituberculous medications were not available, so therapy was directed at lowering oxygen tension in the primary site of infection; for example, pulmonary disease was addressed via pneumothorax. If the chest radiograph provides little hint of tuberculosis, then consideration must be given to exploratory surgery of the abdomen given the focal abnormality in the right lower quadrant.
A peripheral blood smear revealed a hypochromic microcytic anemia. The total red blood cell count was 4.468 million/mm3 (normal range for men is 4.52‐5.90 million/mm3), white blood cell count was 8180/mm3, including 80% granulocytes and 9% eosinophils. On gross inspection, the stool was clay‐colored, and stool microscopy demonstrated large numbers of neutral fat droplets, but no ova, parasites, or tubercle bacilli. Urinalysis revealed no albumin or casts, and the bones were normal on ankle radiographs. Another sample of sputum revealed no tubercle bacilli, and intradermal placement of tuberculin provoked no reaction.
His negative tuberculin skin reaction is unusual for that era, because of the prevalence of tuberculosis. Most likely, he is anergic because of his severe underlying illness, and the absent reaction is thus not all that helpful a clue. Multiple negative sputum examinations lower the possibility of pulmonary, but not extrapulmonary, tuberculosis. The absence of bony destruction on ankle radiographs lowers my suspicion for tuberculous arthritis.
The excess stool fat implies steatogenic diarrhea from malabsorption, and 2 categories here are pancreatic and luminal diseases. Of these 2, pancreatic etiologies produce more severe malabsorption. We do not hear mention of jaundice, however, and I cannot see how to link the pancreas to the arthritis. A chronic infection which may produce malabsorption and eosinophilia is strongyloidiasis, endemic in the southeastern United States. However, this patient did not manifest the most common finding of chronic strongyloidiasis, namely asthma. Adrenal insufficiency, as might result from disseminated tuberculosis, is associated with increased skin pigmentation, diarrhea, and eosinophilia. However, the diarrhea of adrenal insufficiency is not malabsorptive, and serum electrolytes and cortisol tests were not available then to confirm this diagnosis antemortem.
In an attempt to identify a unifying cause of chronic arthritis, malabsorption, and increased skin pigmentation, I must consider Whipple's disease first and foremost. Physicians then were strapped and observation was often the default mode of the day. Given the abdominal findings, an exploratory laparotomy would be warranted if his condition deteriorated.
Despite forced oral feedings, the patient continued to lose weight, from his normal of 175 pounds to a nadir of 145 pounds. Because of worsening abdominal distention, the patient underwent exploratory abdominal surgery on the twenty‐first hospital day. Intraoperatively, no ascites was seen, but his mesenteric lymph nodes were hard and markedly enlarged. The abdomen was closed without further intervention. Two days after the surgery, the patient abruptly developed dyspnea. His respirations were 40 per minute, heart rate was 120, and he had minimal rales at the lung bases without findings of consolidation. He died 2 hours later, on the twenty‐third hospital day, and an autopsy was performed.
The final event may have been a pulmonary embolism. As for the adenopathy, lymphoma and tuberculosis are possible. Heavy chain disease, an unusual lymphoproliferative disorder found in persons from the old Silk Trader's Route from the Middle East to the Orient, is a remote prospect. However, 5 years is just too indolent for most cancers and would be very unusual for tuberculosis. I think the findings support Whipple's disease, and I wonder if this was the first reported case.
On postmortem examination, the abdominal adenopathy was striking. The small intestine contained enlarged villi with thickened submucosa, and the mesenteric nodes were enlarged with fat deposits and abnormal foamy cells. Within these foamy cells, microscopy revealed numerous rod‐shaped organisms. All studies were negative for tuberculosis, and although the pathologist, Dr. George Hoyt Whipple, suspected an infectious etiology, he offered the name intestinal lipodystrophy to emphasize the striking small intestinal changes he witnessed at autopsy, and which are the hallmarks of the disease that now bears his name. Whipple also shared the 1934 Nobel Prize in Physiology or Medicine with Minot and Murphy for their discovery that a nutritional substance in liver, now known as vitamin B12, was beneficial in treating pernicious anemia.
COMMENTARY
This is the index case of Whipple's disease, summarized from the original 1907 description.1 George Hoyt Whipple, then a pathologist at Johns Hopkins, highlights the value of keen observation and a well‐done case report in describing a new disease entity. One of the roles of case reports is to detail the features of an unknown disease. In this capacity, Whipple's summary is exemplary. His achievement was having the openness of mind to realize he was witnessing something novel, and to take the first step on the road to discovery. Although Whipple suspected he was staring at a unique disease, he could not pinpoint the culprit bacteria and he had trouble squaring the extraintestinal findings with the marked intestinal anomalies. It was left to decades of input from others to confirm the association of arthralgias, eosinophilia, skin hyperpigmentation, and cardiac valve abnormalities with intestinal malabsorption, and to culture the infectious agent.
In his discussion, Whipple recognized he was confronted with a novel clinical entity. Prior to surgery, pulmonary and mesenteric tuberculosis were suspected, based on the fevers, weight loss, cough, fat malabsorption, and lymphadenopathy. However, he felt the left apical exam was more representative of retraction from prior disease than active infection. He was also bothered by the negative skin reaction and sputum tests. At surgery, the pronounced adenopathy suggested sarcoma or Hodgkin's disease but postmortem examination eliminated these possibilities. At autopsy, the abdominal findings were most striking. The small intestine demonstrated enlarged villi with thickened submucosa and markedly enlarged mesenteric lymph glands containing large fat deposits and distinctly abnormal foamy cells. These foamy macrophages contained great numbers of rod‐shaped organisms resembling the tubercle bacillus. However, all tests were negative for tuberculosis, and the lungs contained no active disease. Though he suspected an infectious etiology, Whipple offered the name intestinal lipodystrophy to emphasize the striking small intestinal pathology.
Although Whipple had surmised a novel infectious agent in 1907, it took almost a century to isolate the causative microbe. Granules within foamy macrophages of the small intestine were detected on periodic acid‐Schiff (PAS) staining in 1949.2 Similar PAS‐positive granules were soon discovered in other tissues and fluid, providing a plausible explanation for the systemic features of the disease.3 Electron microscopy confirmed the presence of infectious bacilli in 1961,4 ushering in the era of antimicrobial treatment for this disease. More recently, using polymerase chain reaction (PCR), a unique bacterial 16S ribosomal RNA gene was isolated in patients with Whipple's disease.5, 6 Phylogenetically classified with the actinobacteria, Tropheryma whipplei (fom the Greek trophe, nourishment, and eryma, barrier) was ultimately subcultured in 2000,7 and immunodetection testing became possible. Using this technique, the archived pathology specimens from the 1907 index case demonstrated numerous intracellular bacteria in the lamina propria, closing the loop started by Whipple nearly a century earlier.8
Whipple's index case report described most of the manifestations of the disease we are familiar with today. As in the original description, arthralgias are the most common initial symptom and may precede diagnosis by a mean of 8 years. Other cardinal features include weight loss, abdominal pain and steatorrhea due to small intestinal involvement. Table 1 summarizes the important signs and symptoms of Whipple's disease.9, 10 One notable manifestation missing in Whipple's report is central nervous system involvement. Central nervous system (CNS) disease ranges from cognitive deficits to encephalitis and focal defects, and may occur years after treatment and without concomitant intestinal symptoms.
| Clinical Feature | Comment |
|---|---|
| |
| Cardinal features (present in 60% to 90%) | |
| Arthropathy | Most common initial symptom, preceding diagnosis by a mean of 8 years. Migratory, nonerosive, mainly in the peripheral joints. |
| Weight loss | |
| Diarrhea | Usually steatorrhea, may be associated with pain or occult blood in the stool |
| Other common features (present in 20% to 45%) | |
| Fever | |
| Lymphadenopathy | May present as a palpable mass |
| Increased skin pigmentation | Mechanism unknown (evidence of adrenal insufficiency has not been found in Whipple's) |
| Cardiac disease | Culture‐negative endocarditis |
| Hypotension | |
| Peripheral edema | |
| Uncommon clinical features | |
| Central nervous system involvement | May be global (dementia, personality change, sleep disturbance) or focal (cranial neuropathy, nystagmus) |
| Eye disease | Uveitis, retinitis |
| Hepatosplenomegaly | |
| Polyserositis | |
| Ascites | |
A remaining mystery is why this pathogen results only rarely in clinical disease. Caucasians comprise the majority of infected patients, and men are affected 8 times more often than women. An overrepresentation of HLA‐B27 suggests a genetic predisposition, though its role in pathogenesis is unclear. T. whipplei has been identified by PCR methods in asymptomatic individuals, implying additional abnormalities must be present in susceptible hosts for symptoms to occur following colonization.11 The exact immune defects are speculative, and immunodeficiency states (such as HIV) have not been consistently identified in patients with Whipple's disease.
The cornerstone of diagnosing Whipple's disease is upper endoscopy with duodenal biopsy. Flattening of the villi and markedly increased PAS‐positive staining of lamina propria macrophages are strongly suggestive of the diagnosis. PAS‐positive staining is not unique to T. whipplei, however. In patients with profound immunodeficiency, Mycobacterium avium intracellulare may stain positive with PAS. Since Whipple's disease is only rarely associated with HIV, a negative HIV test would favor a diagnosis of Whipple's disease. Electron microscopy may distinguish T. whipplei from its mimickers by morphology. For extraintestinal disease, PCR testing on samples from infected tissue has been found to be a reliable diagnostic aid.9
Given the rarity of the disease, controlled clinical trials addressing optimal treatment are lacking. Current recommendations include initial therapy for 14 days with an agent that crosses the blood‐brain barrier (eg, ceftriaxone) to reduce the incidence of CNS disease. This is then followed by a year or more of oral antimicrobial therapy with trimethoprim‐sulfamethoxazole or a tetracycline.9 While most patients respond within 2 to 3 weeks, relapse may occur in as many as one‐third of patients.
Historical case reports reinforce the case‐based learning paradigm. As the discussant remarks, observation was all too often the only recourse for physicians a century ago. In recounting the 7‐year progression of disease in 1 individual, Whipple provides a unique window into the natural evolution of the key features of this systemic disease. Viewed through the prism of Whipple's eyes, we can recall the striking lymphoid hyperplasia and unusual organisms in the small intestine, cementing our understanding of the pathogenesis of this disorder. Revisiting past cases allows us to learn of and learn from the past.
Teaching Points
-
Whipple's disease should be considered in patients with unexplained arthralgias accompanied by weight loss, malabsorption, and abdominal pain.
-
For suspected intestinal Whipple's disease, diagnosis is best made by duodenal biopsy demonstrating PAS‐positive staining in lamina propria macrophages.
-
Systemic manifestations of Whipple's disease include culture‐negative endocarditis and CNS disease. PCR testing of involved sites for T. whipplei is recommended to confirm extraintestinal disease.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 36 year‐old male physician was admitted to a Baltimore hospital in April 1907 with weight loss, weakness, arthralgias, and abdominal distension that had progressed over 5 years.
In 1907, major causes of unexplained weight loss included tuberculosis, hyperthyroidism, cancer, and diabetes. Arthralgias and weakness are not specific. The insidious progression over 5 years narrows the infectious possibilities; tuberculosis and syphilis are important considerations. Since surgical removal was the main treatment for malignancy in 1907, a history of prior surgery might point to a previously diagnosed malignancy that is now progressing.
Five years earlier, while visiting Turkey as a medical missionary, he first noted the onset of arthralgias that lasted 6 to 8 hours and occurred 3 to 4 times per week. Over time, these attacks lasted up to 24 hours and became associated with warmth, swelling, and tenderness of both small and large joints. He gradually lost weight and strength. One year prior to arrival in the hospital, he developed a cough productive of yellow sputum. Seven months prior, he returned from Turkey to Atlanta and noticed an increase in his cough, along with fevers of 100 Fahrenheit and night sweats.
The primacy of the arthralgias in this illness lead me to consider primary rheumatic diseases, and multisystem diseases (including infections) with a predominant skeletal component. In 1907, tests for lupus and the rheumatoid factor were not available. Neither skeletal remains nor works of art provide evidence that rheumatoid arthritis existed until the 19th century, whereas ankylosing spondylitis, gout, and rickets were present by then.
As a medical missionary, he might have acquired a disease endemic to the areas he visited, or the travel history may be a red herring. Familial Mediterranean fever, though prevalent in Turkey and a cause of arthralgias accompanied by recurrent attacks of abdominal pain and fever, is not an acquired disease. Behcet's disease, also known as Silk Trader's Route disease, is found in descendents of the countries that comprised the ancient Silk Route from Japan to the Middle East and may cause arthritis along with oral ulcers, genital lesions, pathergy or uveitis. I would inquire about his ancestry and fevers before dismissing these possibilities.
Although 5 years would be unusually long for tuberculosis to go unrecognized, a physician in the first half of the 20th century would place tuberculosis near the top of possible diagnoses. In 1930, a time when the population of the United States was considerably less, there were over 300,000 cases of tuberculosis. Physicians, and in particular pathologists since autopsies were more commonly performed, often died from tuberculosis since streptomycin, the first antituberculous medication, did not arrive until 1944. At the turn of the 20th century, the ability to detect tubercle bacilli was quite good. Thus, I would include tuberculous peritonitis as a cause of the progressive abdominal symptoms in this physician. In approximately one‐third of patients with tuberculous peritonitis there is evidence of pulmonary disease, and I would try to culture tuberculosis in samples of sputum, a test then so common it probably rivaled our frequent complete blood counts in popularity.
Six months prior, examinations of sputum were negative for tubercle bacilli. Four months prior to arrival, the patient moved to New Mexico. His cough improved but he continued to lose weight and had diarrhea consisting of 3 to 4 loose or semiformed bowel movements per day. Three months prior to admission, he noted an increase in abdominal girth along with right lower quadrant fullness. One month prior, he noted painful swelling and warmth in both ankles as well as dyspnea with exertion.
The increased abdominal girth in the context of chronic illness might be due to ascites, adenopathy, visceromegaly, or mass lesions such as a neoplasm or abscess. If ascites is the cause, one would need to consider primary hepatic disorders, as well as extrahepatic diseases that could progress over years. Infection with hepatitis A virus does not cause chronic liver disease. Hepatitis B, in those days, was referred to as serum hepatitis, and a serum marker for the B virusthe Australia antigenwas not identified until 1967. Cardiac causes of ascites include congestive heart failure and constrictive pericarditis, the latter an important consideration because it is potentially curable. Also, constrictive pericarditis can present as an indolent weight‐losing disease because of chronic visceral congestion. Other considerations include nephrotic syndrome, infection, and neoplasm, including mesothelioma.
Abdominal distention might also be seen with a smoldering abscess. In addition to an appendiceal process, the travel and right lower quadrant localization reminds us to consider ameboma. This patient surely was in an area where amebiasis was endemic, and amebomaa chronic inflammatory form of infection with E. histolytica not associated with diarrhea or liver cystsmay mimic cecal carcinoma. Exertional dyspnea suggests at least the possibility of cardiac disease. Despite the negative sputum cultures, tuberculosis remains high on the list as a cause of constrictive pericarditis or peritonitis, either of which may occur in the absence of active pulmonary disease.
Past medical history included measles and whooping cough as a child, mild pleurisy at age 14, mild influenza 7 years previously. The patient had a tonsillectomy as a child and had a portion of his inferior turbinate bone removed in an attempt to relieve a nasal condition.
On physical exam, the patient was thin and the skin over his face and hands was deep brown. His temperature was 101.5 Fahrenheit, the heart rate was 100, and the respiratory rate was 24. Small lymph nodes were palpable in the axillary and epitrochlear areas. His thorax moved asymmetrically, with less movement on the left apex and slight dullness to percussion in that area. The pulmonic component of the second heart sound was mildly accentuated. The abdomen displayed fullness and tympany, most pronounced in the right lower quadrant without hepatosplenomegaly. The left ankle was swollen, and the overlying skin was tense, shiny, and hot. On both lower legs, areas of discoloration and slight induration were observed, felt to be consistent with faded erythema nodosum.
Though pleurisy has numerous causes, its presence raises the specter of tuberculosis again. The nasal condition triggers thoughts of Wegener's granulomatosis or lethal midline granuloma, both unlikely diagnoses here. The pulmonary exam suggests an apical process, such as tuberculosis, and the accentuated pulmonic heart sound implies pulmonary hypertension, which could be due to a number of chronic pulmonary diseases. The epitrochlear nodes are of interest since lymphoma and Hodgkin's disease rarely involve this area; syphilis and human immunodeficiency virus (HIV) are a few of the chronic diseases that may involve this lymph node region. More helpful is the absence of hepatosplenomegaly, since many indolent malignancies and infections would be expected to enlarge these organs by this point.
Monoarticular arthritis is often due to infection, and less likely due to rheumatoid disease. When rheumatoid arthritis flares, the entire skeleton flares, not single joints. Given the indolence and this single joint involvement, tuberculosis again comes to mind.
I would next want to obtain a plain chest radiograph, looking for evidence of tuberculosis. As with any test, one should ask how this will change management. In 1907, antituberculous medications were not available, so therapy was directed at lowering oxygen tension in the primary site of infection; for example, pulmonary disease was addressed via pneumothorax. If the chest radiograph provides little hint of tuberculosis, then consideration must be given to exploratory surgery of the abdomen given the focal abnormality in the right lower quadrant.
A peripheral blood smear revealed a hypochromic microcytic anemia. The total red blood cell count was 4.468 million/mm3 (normal range for men is 4.52‐5.90 million/mm3), white blood cell count was 8180/mm3, including 80% granulocytes and 9% eosinophils. On gross inspection, the stool was clay‐colored, and stool microscopy demonstrated large numbers of neutral fat droplets, but no ova, parasites, or tubercle bacilli. Urinalysis revealed no albumin or casts, and the bones were normal on ankle radiographs. Another sample of sputum revealed no tubercle bacilli, and intradermal placement of tuberculin provoked no reaction.
His negative tuberculin skin reaction is unusual for that era, because of the prevalence of tuberculosis. Most likely, he is anergic because of his severe underlying illness, and the absent reaction is thus not all that helpful a clue. Multiple negative sputum examinations lower the possibility of pulmonary, but not extrapulmonary, tuberculosis. The absence of bony destruction on ankle radiographs lowers my suspicion for tuberculous arthritis.
The excess stool fat implies steatogenic diarrhea from malabsorption, and 2 categories here are pancreatic and luminal diseases. Of these 2, pancreatic etiologies produce more severe malabsorption. We do not hear mention of jaundice, however, and I cannot see how to link the pancreas to the arthritis. A chronic infection which may produce malabsorption and eosinophilia is strongyloidiasis, endemic in the southeastern United States. However, this patient did not manifest the most common finding of chronic strongyloidiasis, namely asthma. Adrenal insufficiency, as might result from disseminated tuberculosis, is associated with increased skin pigmentation, diarrhea, and eosinophilia. However, the diarrhea of adrenal insufficiency is not malabsorptive, and serum electrolytes and cortisol tests were not available then to confirm this diagnosis antemortem.
In an attempt to identify a unifying cause of chronic arthritis, malabsorption, and increased skin pigmentation, I must consider Whipple's disease first and foremost. Physicians then were strapped and observation was often the default mode of the day. Given the abdominal findings, an exploratory laparotomy would be warranted if his condition deteriorated.
Despite forced oral feedings, the patient continued to lose weight, from his normal of 175 pounds to a nadir of 145 pounds. Because of worsening abdominal distention, the patient underwent exploratory abdominal surgery on the twenty‐first hospital day. Intraoperatively, no ascites was seen, but his mesenteric lymph nodes were hard and markedly enlarged. The abdomen was closed without further intervention. Two days after the surgery, the patient abruptly developed dyspnea. His respirations were 40 per minute, heart rate was 120, and he had minimal rales at the lung bases without findings of consolidation. He died 2 hours later, on the twenty‐third hospital day, and an autopsy was performed.
The final event may have been a pulmonary embolism. As for the adenopathy, lymphoma and tuberculosis are possible. Heavy chain disease, an unusual lymphoproliferative disorder found in persons from the old Silk Trader's Route from the Middle East to the Orient, is a remote prospect. However, 5 years is just too indolent for most cancers and would be very unusual for tuberculosis. I think the findings support Whipple's disease, and I wonder if this was the first reported case.
On postmortem examination, the abdominal adenopathy was striking. The small intestine contained enlarged villi with thickened submucosa, and the mesenteric nodes were enlarged with fat deposits and abnormal foamy cells. Within these foamy cells, microscopy revealed numerous rod‐shaped organisms. All studies were negative for tuberculosis, and although the pathologist, Dr. George Hoyt Whipple, suspected an infectious etiology, he offered the name intestinal lipodystrophy to emphasize the striking small intestinal changes he witnessed at autopsy, and which are the hallmarks of the disease that now bears his name. Whipple also shared the 1934 Nobel Prize in Physiology or Medicine with Minot and Murphy for their discovery that a nutritional substance in liver, now known as vitamin B12, was beneficial in treating pernicious anemia.
COMMENTARY
This is the index case of Whipple's disease, summarized from the original 1907 description.1 George Hoyt Whipple, then a pathologist at Johns Hopkins, highlights the value of keen observation and a well‐done case report in describing a new disease entity. One of the roles of case reports is to detail the features of an unknown disease. In this capacity, Whipple's summary is exemplary. His achievement was having the openness of mind to realize he was witnessing something novel, and to take the first step on the road to discovery. Although Whipple suspected he was staring at a unique disease, he could not pinpoint the culprit bacteria and he had trouble squaring the extraintestinal findings with the marked intestinal anomalies. It was left to decades of input from others to confirm the association of arthralgias, eosinophilia, skin hyperpigmentation, and cardiac valve abnormalities with intestinal malabsorption, and to culture the infectious agent.
In his discussion, Whipple recognized he was confronted with a novel clinical entity. Prior to surgery, pulmonary and mesenteric tuberculosis were suspected, based on the fevers, weight loss, cough, fat malabsorption, and lymphadenopathy. However, he felt the left apical exam was more representative of retraction from prior disease than active infection. He was also bothered by the negative skin reaction and sputum tests. At surgery, the pronounced adenopathy suggested sarcoma or Hodgkin's disease but postmortem examination eliminated these possibilities. At autopsy, the abdominal findings were most striking. The small intestine demonstrated enlarged villi with thickened submucosa and markedly enlarged mesenteric lymph glands containing large fat deposits and distinctly abnormal foamy cells. These foamy macrophages contained great numbers of rod‐shaped organisms resembling the tubercle bacillus. However, all tests were negative for tuberculosis, and the lungs contained no active disease. Though he suspected an infectious etiology, Whipple offered the name intestinal lipodystrophy to emphasize the striking small intestinal pathology.
Although Whipple had surmised a novel infectious agent in 1907, it took almost a century to isolate the causative microbe. Granules within foamy macrophages of the small intestine were detected on periodic acid‐Schiff (PAS) staining in 1949.2 Similar PAS‐positive granules were soon discovered in other tissues and fluid, providing a plausible explanation for the systemic features of the disease.3 Electron microscopy confirmed the presence of infectious bacilli in 1961,4 ushering in the era of antimicrobial treatment for this disease. More recently, using polymerase chain reaction (PCR), a unique bacterial 16S ribosomal RNA gene was isolated in patients with Whipple's disease.5, 6 Phylogenetically classified with the actinobacteria, Tropheryma whipplei (fom the Greek trophe, nourishment, and eryma, barrier) was ultimately subcultured in 2000,7 and immunodetection testing became possible. Using this technique, the archived pathology specimens from the 1907 index case demonstrated numerous intracellular bacteria in the lamina propria, closing the loop started by Whipple nearly a century earlier.8
Whipple's index case report described most of the manifestations of the disease we are familiar with today. As in the original description, arthralgias are the most common initial symptom and may precede diagnosis by a mean of 8 years. Other cardinal features include weight loss, abdominal pain and steatorrhea due to small intestinal involvement. Table 1 summarizes the important signs and symptoms of Whipple's disease.9, 10 One notable manifestation missing in Whipple's report is central nervous system involvement. Central nervous system (CNS) disease ranges from cognitive deficits to encephalitis and focal defects, and may occur years after treatment and without concomitant intestinal symptoms.
| Clinical Feature | Comment |
|---|---|
| |
| Cardinal features (present in 60% to 90%) | |
| Arthropathy | Most common initial symptom, preceding diagnosis by a mean of 8 years. Migratory, nonerosive, mainly in the peripheral joints. |
| Weight loss | |
| Diarrhea | Usually steatorrhea, may be associated with pain or occult blood in the stool |
| Other common features (present in 20% to 45%) | |
| Fever | |
| Lymphadenopathy | May present as a palpable mass |
| Increased skin pigmentation | Mechanism unknown (evidence of adrenal insufficiency has not been found in Whipple's) |
| Cardiac disease | Culture‐negative endocarditis |
| Hypotension | |
| Peripheral edema | |
| Uncommon clinical features | |
| Central nervous system involvement | May be global (dementia, personality change, sleep disturbance) or focal (cranial neuropathy, nystagmus) |
| Eye disease | Uveitis, retinitis |
| Hepatosplenomegaly | |
| Polyserositis | |
| Ascites | |
A remaining mystery is why this pathogen results only rarely in clinical disease. Caucasians comprise the majority of infected patients, and men are affected 8 times more often than women. An overrepresentation of HLA‐B27 suggests a genetic predisposition, though its role in pathogenesis is unclear. T. whipplei has been identified by PCR methods in asymptomatic individuals, implying additional abnormalities must be present in susceptible hosts for symptoms to occur following colonization.11 The exact immune defects are speculative, and immunodeficiency states (such as HIV) have not been consistently identified in patients with Whipple's disease.
The cornerstone of diagnosing Whipple's disease is upper endoscopy with duodenal biopsy. Flattening of the villi and markedly increased PAS‐positive staining of lamina propria macrophages are strongly suggestive of the diagnosis. PAS‐positive staining is not unique to T. whipplei, however. In patients with profound immunodeficiency, Mycobacterium avium intracellulare may stain positive with PAS. Since Whipple's disease is only rarely associated with HIV, a negative HIV test would favor a diagnosis of Whipple's disease. Electron microscopy may distinguish T. whipplei from its mimickers by morphology. For extraintestinal disease, PCR testing on samples from infected tissue has been found to be a reliable diagnostic aid.9
Given the rarity of the disease, controlled clinical trials addressing optimal treatment are lacking. Current recommendations include initial therapy for 14 days with an agent that crosses the blood‐brain barrier (eg, ceftriaxone) to reduce the incidence of CNS disease. This is then followed by a year or more of oral antimicrobial therapy with trimethoprim‐sulfamethoxazole or a tetracycline.9 While most patients respond within 2 to 3 weeks, relapse may occur in as many as one‐third of patients.
Historical case reports reinforce the case‐based learning paradigm. As the discussant remarks, observation was all too often the only recourse for physicians a century ago. In recounting the 7‐year progression of disease in 1 individual, Whipple provides a unique window into the natural evolution of the key features of this systemic disease. Viewed through the prism of Whipple's eyes, we can recall the striking lymphoid hyperplasia and unusual organisms in the small intestine, cementing our understanding of the pathogenesis of this disorder. Revisiting past cases allows us to learn of and learn from the past.
Teaching Points
-
Whipple's disease should be considered in patients with unexplained arthralgias accompanied by weight loss, malabsorption, and abdominal pain.
-
For suspected intestinal Whipple's disease, diagnosis is best made by duodenal biopsy demonstrating PAS‐positive staining in lamina propria macrophages.
-
Systemic manifestations of Whipple's disease include culture‐negative endocarditis and CNS disease. PCR testing of involved sites for T. whipplei is recommended to confirm extraintestinal disease.
- .A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues.Bull Johns Hopkins Hosp.1907;18:382–391.
- .The tinctoral demonstration of a glycoprotein in Whipple's disease.Proc Soc Exp Biol Med.1949;72:225–227.
- ,,.Whipple's disease: clinical, biochemical, and histopathologic features and assessment of treatment in 29 patients.Mayo Clin Proc.1988;63:539–551.
- ,.Combined electron and light microscopy in Whipple's disease: demonstration of “bacillary bodies” in the intestine.Bull Johns Hopkins Hosp.1961;109:80–98.
- ,,,.Phylogeny of the Whipple's disease‐associated bacterium.Lancet.1991;338:474–475.
- ,,,.Identification of the uncultured bacillus of Whipple's disease.N Engl J Med.1992;327:293–301.
- ,,, et al.Cultivation of the bacillus of Whipple's disease.N Engl J Med.2000;342:620–625.
- ,,,.Immunodetection of Tropheryma whipplei in intestinal tissue from Dr. Whipple's 1907 patient.N Engl J Med.2003;348:1411– 1412.
- ,.Whipple's disease.Lancet.2003;361:239–246.
- ,,, et al.Diagnostic guidelines in central nervous system Whipple's disease.Ann Neurol.1996;40:561–568.
- ,,, et al.PCR‐positive tests for Tropheryma whippleii in patients without Whipple's disease.Lancet.1999;353:2214.
- .A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues.Bull Johns Hopkins Hosp.1907;18:382–391.
- .The tinctoral demonstration of a glycoprotein in Whipple's disease.Proc Soc Exp Biol Med.1949;72:225–227.
- ,,.Whipple's disease: clinical, biochemical, and histopathologic features and assessment of treatment in 29 patients.Mayo Clin Proc.1988;63:539–551.
- ,.Combined electron and light microscopy in Whipple's disease: demonstration of “bacillary bodies” in the intestine.Bull Johns Hopkins Hosp.1961;109:80–98.
- ,,,.Phylogeny of the Whipple's disease‐associated bacterium.Lancet.1991;338:474–475.
- ,,,.Identification of the uncultured bacillus of Whipple's disease.N Engl J Med.1992;327:293–301.
- ,,, et al.Cultivation of the bacillus of Whipple's disease.N Engl J Med.2000;342:620–625.
- ,,,.Immunodetection of Tropheryma whipplei in intestinal tissue from Dr. Whipple's 1907 patient.N Engl J Med.2003;348:1411– 1412.
- ,.Whipple's disease.Lancet.2003;361:239–246.
- ,,, et al.Diagnostic guidelines in central nervous system Whipple's disease.Ann Neurol.1996;40:561–568.
- ,,, et al.PCR‐positive tests for Tropheryma whippleii in patients without Whipple's disease.Lancet.1999;353:2214.
The Irritable Heart
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A30‐year‐old woman was referred for evaluation of chest pain, palpitations, and exercise intolerance. She had been previously healthy, active, and physically fit. Five months prior to our evaluation, she had an elective C5C6 cervical spine discectomy with interbody allograft fusion for a chronic neck injury that occurred 11 years ago during gymnastics. Two weeks after spine surgery, the patient developed numbness and tingling of her left thumb and palm that occurred with exertion or exposure to cold and subsided with rest. These episodes increased in frequency and intensity and after 1 week became associated with sharp, occasionally stabbing chest pain that radiated to the left arm. On one occasion, the patient had an episode of exertional chest pain with prolonged left arm cyanosis. Emergent left upper extremity angiography revealed normal great vessel anatomy with spasm of the radial artery and collateral ulnar flow. The patient was diagnosed with Raynaud's phenomenon and was started on nifedipine. A subsequent rheumatologic evaluation was unrevealing, and the patient was empirically switched to amlodipine with no improvement in symptoms.
This otherwise very healthy 30‐year‐old developed a multitude of symptoms. The patient's chest pain is atypical and in a young woman is unlikely to signify atherosclerotic coronary disease, but it should not be entirely disregarded. Vasospasm triggered by exposure to cold does raise suspicion for Raynaud's phenomenon, which is not uncommon in this demographic. However, this presentation is quite unusual because the vasospasm was limited to one vascular distribution of one extremity. Associated coronary vasospasm could explain the other symptoms, although coronary spasm is generally not associated with Raynaud's phenomenon. Vasculitis may also affect the pulmonary vasculature, leading to pulmonary hypertension and exercise intolerance. The temporal association with her spine surgery is intriguing but of unclear significance.
The patient continued to have frequent exertional episodes of sharp precordial chest pain radiating to her left arm that were accompanied by dyspnea and left upper extremity symptoms despite amlodipine therapy. These now occurred with limited activity when she walked 1 to 2 blocks uphill. Over the previous 2 months, she had also noticed palpitations occurring reliably with exercise that were relieved with 15 to 20 min of rest. With prolonged episodes, she reported dizziness, nausea, and blurry vision that improved with lying down. She twice had syncope with these symptoms. She noted lower extremity edema while taking calcium channel blockers, but this had resolved after discontinuation of the drugs.
The patient's past medical history included several high‐school orthopedic injuries. She had 2 kidney stones at ages 18 and 23 and had an appendectomy at age 28. Her only medication was an oral contraceptive, and she had discontinued the amlodipine. She denied the use of tobacco, alcohol, herbal medications, or illicit substances. There was no family history of sudden death or heart disease.
Palpitations in a 30‐year‐old woman may signify a cardiac arrhythmia. Paroxysmal supraventricular arrhythmias, such as atrioventricular nodal reentrant tachycardia, atrial tachycardia, and atrial fibrillation, are well described in the young. Ventricular tachycardia (VT) is another possible cause and could be idiopathic or related to occult structural heart disease. Young patients typically tolerate lone arrhythmias quite well, and her failure to do so raises suspicion for concomitant structural heart disease. Her palpitations may be from appropriate sinus tachycardia, which could be compensatory because of inadequate cardiac output reserve, which in turn could be caused by valvular disease, congenital heart disease, or ventricular dysfunction. The exertional chest pain is worrisome for ischemia. Pulmonary hypertension, severe ventricular hypertrophy, or congenital anomalies of the coronary circulation could lead to subendocardial myocardial ischemia with exertion, resulting in angina, dyspnea, and arrhythmias. However, the patient also experiences exertional palpitations without chest pain, which may signify an exertional tachyarrhythmia possibly mediated by catecholamines. Based solely on the history, the differential diagnosis remains broad.
On physical examination, the patient was a fit, thin, healthy woman. Her blood pressure was 120/70 mm Hg supine in both arms and 115/75 mm Hg standing; her pulse was 85 supine and 110 standing, Oxygen saturation was 100% on room air. A cardiac exam revealed a normal jugular venous pressure, normal point of maximal impulse, regular rhythm with occasional ectopy, normal S1, and physiologically split S2 without extra heart sounds or murmurs. The right ventricular impulse was faintly palpable at the left sternal border. Head, neck, chest, abdominal, musculoskeletal, neurologic, extremity, and peripheral pulse examinations were normal.
Laboratory data showed a normal complete blood count and normal chemistries. Serum tests for hepatitis C antibody, cardiolipin antibody, rheumatoid factor, cryoglobulins, and anti‐nuclear antibody were negative. The erythrocyte sedimentation rate and thyroid stimulating hormone levels were within normal limits. An electrocardiogram (ECG) demonstrated a normal sinus rhythm with frequent premature ventricular complexes (PVCs) and normal axis and intervals. (Figure 1). The PR segment was normal and without preexcitation. A prior ECG from 3 months ago was similar with ventricular trigeminy.
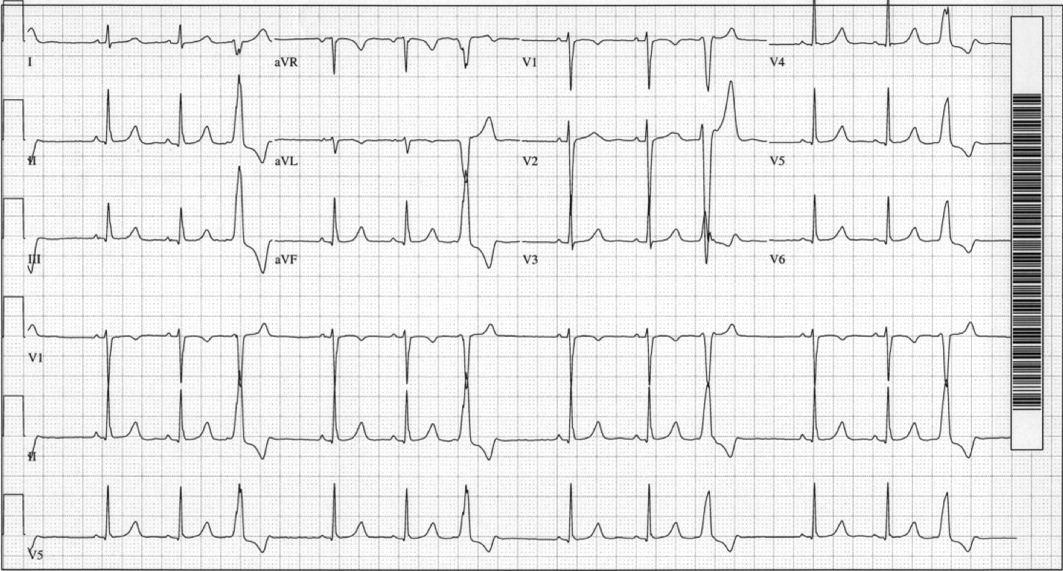
Her unremarkable cardiac examination does not favor structural or valvular heart disease, and there are no obvious stigmata of vasculitis. She did become mildly tachycardic upon standing, and this raises the possibility of orthostatic tachycardia. A comprehensive rheumatologic panel revealed no evidence of autoimmune disease or vasculitis, and the clinical constellation is not consistent with primary or secondary Raynaud's disease. The ECG demonstrates frequent monomorphic PVCs complexes with a left bundle branch block pattern and an inferior axis. This pattern suggests that the PVCs arise from the right ventricular outflow tract. Idiopathic right ventricular outflow tract VT and arrhythmogenic right ventricular dysplasia must be considered as a cause of exertional or catecholamine‐mediated tachycardia. The normal ECG argues against arrhythmogenic right ventricular dysplasia, in which patients typically have incomplete or complete right bundle branch block, right precordial T wave abnormalities, and occasionally epsilon waves. Her QT interval is normal, but excluding long‐QT syndrome with a single ECG has poor sensitivity. The next critical step is to document her cardiac rhythm during symptoms and to exclude malignant arrhythmias.
An event recorder and exercise echocardiogram were ordered. While the patient was wearing her event recorder, she had 4 episodes of exertional syncope while hiking and successfully triggered event recording before losing consciousness. She had chest pain and left arm pain after regaining consciousness. The patient came to the emergency room for evaluation. Her blood pressure was 116/80 mm Hg supine and 112/70 mm Hg seated. Her heart rate increased from 82 supine to 132 seated. The physical examination was unremarkable. ECG showed sinus rhythm with frequent PVCs. Troponin‐I measurements 10 hours apart were 0.7 and 0.3 g/L (normal < 1.1), with normal creatinine kinase and creatinine kinase MB fractions. Interrogation of the event recorder revealed multiple episodes of a narrow complex tachycardia with rates up to 180 bpm that correlated with symptoms (Figure 2). There were no episodes of wide complex tachycardia.
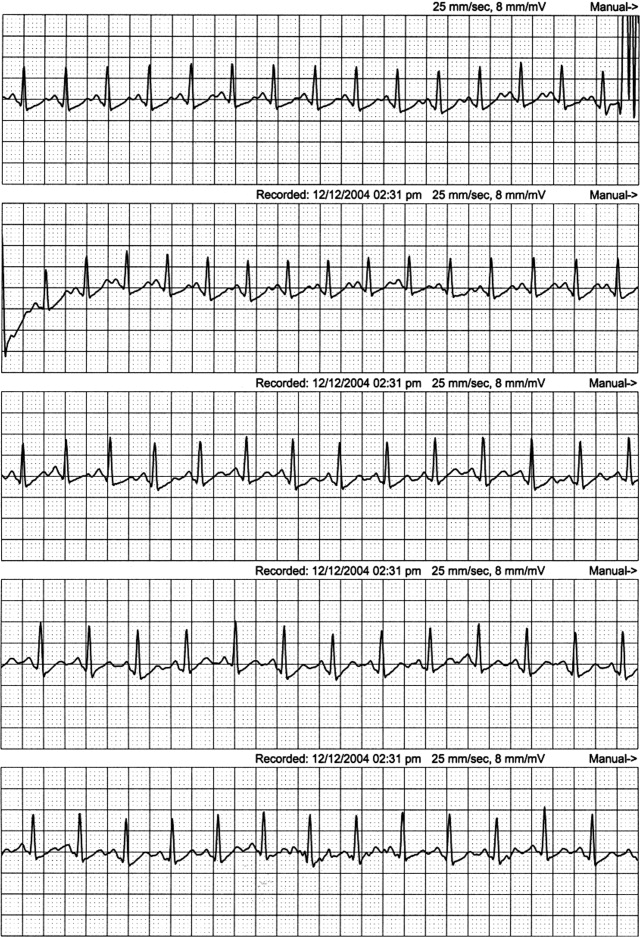
The patient was not hypotensive in the emergency room, but she had evidence of a marked orthostatic tachycardia. The minimal but significant troponin elevations are also troubling. Although her clinical picture is not consistent with an acute coronary syndrome, I am concerned about other mechanisms of myocardial ischemia or injury, such as a coronary anomaly or subendocardial ischemia from globally reduced myocardial perfusion. The presence of event recorder data from her syncopal events was fortuitous and revealed a supraventricular tachycardia. The arrhythmia was gradual in onset and resolution and had no triggers, such as premature atrial or ventricular complexes, which could suggest reentrant arrhythmias. The P wave morphology was also unchanged, and this argues against an atrial tachycardia. These findings are consistent with sinus tachycardia, which was notably out of proportion to her workload. This arrhythmia may be the primary cause of syncope, such as in inappropriate sinus tachycardia, or it may be a compensatory mechanism. Tachycardia from coronary vasospasm is often preceded by ST segment changes, which are not seen here. Although the event recorder had no episodes of VT, the patient's persistent frequent PVCs are still of concern. I would obtain an echocardiogram to exclude structural heart disease and an exercise test to exclude exertional VT. Finally, coronary angiography may be helpful in excluding congenital anomalies.
The patient was admitted for evaluation. An exercise treadmill test was performed, and the patient exercised 20 min on the standard Bruce protocol with a peak heart rate of 180 bpm. The test was notable for a premature rise in heart rate (in stage 1) without a rise in blood pressure. There were no symptoms or ST/T wave changes. Transthoracic echocardiogram showed normal left ventricular size and function with normal anatomy, valves, and hemodynamics. Coronary angiography showed a right dominant system with normal anatomy and no atherosclerotic disease.
Ventricular arrhythmias could not be elicited with exercise. Her high exercise tolerance virtually excluded hemodynamically significant structural or valvular disease, and this was confirmed by the echocardiogram. Coronary angiography excluded coronary anomalies and myocardial bridging. The most intriguing finding is the rise in the patient's heart rate out of proportion to the workload. This, along with her orthostatic tachycardia, raises the issue of inappropriate sinus tachycardia or postural orthostatic tachycardia syndrome (POTS). Carotid hypersensitivity is also a possibility. The patient was hiking when she fainted, and even light pressure on the patient's neck with head turning or from a camera strap, for example, could produce syncope. Although carotid hypersensitivity usually results in sinus bradycardia and AV block, it may be followed by reflex tachycardia, which was seen in this patient's event recordings. I would perform a tilt‐table test with carotid massage to make the diagnosis.
Tilt‐table testing was performed (Figure 3). Her supine blood pressure was 128/68 mm Hg, and her heart rate was 72 bpm with no change during the 10‐min supine period. Upon elevation to a 70‐degree tilt, the patient had an immediate increase in her heart rate to 160 bpm with a blood pressure nadir of 109/58 mm Hg and symptoms of palpitations, dizziness, dyspnea, chest pain, blurry vision, and nausea. Her peak heart rate was 172 bpm, and her peak blood pressure was 122/72 mm Hg. Vital signs did not change in response to carotid sinus massage in the supine or upright positions.
The tilt‐table test has 3 notable findings. First, her heart rate increased rapidly with tilt and decreased rapidly in supine recovery. Second, her usual symptoms started immediately after tilt and quickly resolved in recovery when vital signs returned to baseline. Finally, there was only a modest drop in blood pressure. These findings are classic for POTS. POTS is defined as symptomatic orthostasis with a heart rate increase of 30 bpm or a heart rate of 120 bpm. The physiologic lesions found in the syndrome are heterogeneous, but they all lead to a failure of orthostatic compensation. In POTS, the tachycardia is a reflex secondary to hypotension (baroreceptor reflex) or reduced preload (cardiac mechanoreceptors), in contrast to inappropriate sinus tachycardia. Interestingly, blood pressure is usually preserved until the final moments preceding syncope, when venous return further declines, tachycardia decreases the diastolic filling time and stroke volume, and mean arterial pressure sharply falls.
The patient was started on labetalol (200 mg 3 times daily), and her symptoms worsened. She also developed nausea and constipation. Midodrine and pindolol were also tried without success. She was then switched to fludrocortisone, salt supplementation, and leg support stockings with dramatic improvement.
COMMENTARY
In 1871, DeCosta1 published a report on the irritable heart, noting an affliction of extreme fatigue and exercise intolerance that occurred suddenly and without obvious cause. Subsequently, the terms vasoregulatory asthenia and neurocirculatory asthenia were used to link cardiovascular symptoms to impaired regulation of peripheral blood flow.2, 3 The term POTS was first used in 1982 to describe a single patient with postural tachycardia without hypotension and palpitations, weakness, abdominal pain, and presyncope.4
POTS is one of several disorders of autonomic control associated with orthostatic intolerance. The criteria for diagnosis are listed in Table 1. POTS typically occurs in women between the ages of 15 and 50 but tends to present during adolescence or young adulthood. The physiology has only recently been elucidated. When a person stands, 500 cc of the total blood volume is displaced to the dependent extremities and inferior mesenteric vessels.5 Normally, orthostatic stabilization occurs in less than 1 minute via 3 mechanisms: baroreceptor input, sympathetic reflex tachycardia and vasoconstriction, and enhanced venous return via the pumping action of skeletal muscles and venoconstriction. In POTS, there is a failure of at least one of these mechanisms, leading to decreased venous return, a 40% reduction in stroke volume, and cerebral hypoperfusion.6
| 1. Consistent symptoms of orthostatic intolerance [may include excessive fatigue, exercise intolerance, recurrent syncope or near syncope, dizziness, nausea, tachycardia, palpitations, visual disturbances, blurred vision, tunnel vision, tremulousness, weakness (most noticeable in the legs), chest discomfort, shortness of breath, mood swings, and gastrointestinal complaints] |
| 2. Heart rate increase 30 bpm or heart rate 120 bpm within 10 min of standing or head‐up tilt |
| 3. Absence of a known cause of autonomic neuropathy |
POTS is divided into 2 major subtypes on the basis of pathophysiology.5, 7 The partial dysautonomic form is the most common and the type that this patient most likely had. In this form, the development of an acquired peripheral autonomic neuropathy results in a failure of sympathetic venoconstriction, which leads to excessive venous pooling in the lower extremities and splanchnic circulation.8, 9 Failure to mobilize this venous reservoir upon standing leads to excessive orthostatic tachycardia secondary to a marked reduction in stroke volume. Peripheral arterial vasoconstriction is generally preserved, which is why midodrine, an arterial vasoconstrictor, did not improve symptoms. The labetalol may have further exacerbated peripheral pooling because of its alpha‐adrenergic blocking properties. Because total plasma volume is decreased and plasma renin activity is inappropriately low,10 volume expanders, including salt, low‐dose steroids, and fluids, can attenuate symptoms.11 The extrinsic venous compression from leg and abdominal support stockings may also dramatically reduce venous pooling.
In the less common hyperadrenergic form of POTS, patients may have orthostatic hypertension, tremulousness, cold, sweaty extremities, and anxiety due to an exaggerated response to beta‐adrenergic stimulation.7 The excessive sympathetic activity, which is poorly modulated by baroreflex activity, may be due to impaired mechanisms of norepinephrine reuptake by sympathetic ganglia.12 Consequently, serum norepinephrine levels are markedly elevated (>600 pg/mL).5
In adults, the presence of a POTS trigger is common and is usually an antecedent viral illness. Antibodies to the ganglionic acetylcholine receptor have been found in a subset of POTS patients,13 and this may suggest an idiopathic or postinflammatory autoimmune mechanism.14 This patient's presentation is unique because her symptoms developed after C5C6 spine surgery. The cervical spinal cord and sympathetic ganglia are dense with nerves involved in autonomic cardiovascular control, and damage to these fibers could explain the patient's physiology and symptoms. Among these, the descending vasomotor pathways traverse through the C5C8 area to innervate the splanchnic and leg venous circulation, receiving input from the heart along the way.15 The pattern of numbness and tingling fits the C5/C6 dermatomal distribution, as does the innervation of the radial artery. The frequent PVCs with a left bundle branch block pattern and inferior axis appear to arise from the right ventricular outflow tract and may be associated with regional sympathetic denervation, which has been described in idiopathic ventricular arrhythmias.16 POTS has been anecdotally reported after neck injury from motor vehicle accidents (whiplash), which is also thought to be related to cervical sympathetic nerve damage (B.P. Grubb, personal communication, 2005). Most cases of triggered POTS improve spontaneously after months to years, but this patient's prognosis remains uncertain because of the presumed mechanical disruption of the autonomic nerve fibers at the time of surgery.
This case demonstrates the complexities of arriving at a unifying diagnosis in the setting of a constellation of nonspecific symptoms and findings, some of which even suggest life‐threatening conditions. Because young women are primarily affected, symptoms of POTS can be mistakenly attributed to anxiety or other nonphysiological factors. A systematic approach excluded life‐threatening causes, including primary ventricular arrhythmias, coronary vasospasm, and coronary anomalies. The investigations narrowed the differential diagnosis, and the tilt‐table test confirmed POTS. Because the cardiac and circulatory dysautonomias encompass an array of distinct physiologic processes, understanding the patient's mechanism is critical to her management. The only effective therapies were those that counteracted venous pooling and improved venous return.
Teaching Points
-
The differential diagnosis of exertional syncope is extremely broad, ranging from benign to malignant conditions, and requires a systematic evaluation of the heart and circulatory system.
-
The diagnosis of POTS is elusive and frequently missed. Referral for tilt‐table testing is useful in identifying the mechanism of sinus tachycardia and syncope. Marked orthostatic tachycardia and symptoms of cerebral hypoperfusion out of proportion to the degree of hypotension strongly suggest POTS.
-
Cardiac and circulatory dysautonomias have distinct and varied mechanisms. Therapies, including beta‐blockers, vasoconstrictors, and volume expanders, must be directed at the underlying physiological defect.
- .An irritable heart.Am J Med Sci.1871;27:145–161.
- ,,,,,.Low physical working capacity in suspected heart cases due to inadequate adjustment of peripheral blood flow (vasoregulatory asthenia).Acta Med Scand.1957;158(6):413–436.
- ,,.Orthostatic tachycardia and orthostatic hypotension: defects in the return of venous blood to the heart.Am Heart J.1944;27:145–163.
- ,.Postural tachycardia syndrome. Reversal of sympathetic hyperresponsiveness and clinical improvement during sodium loading.Am J Med.1982;72(5):847–850.
- ,,.The postural orthostatic tachycardia syndrome: definitions, diagnosis, and management.Pacing Clin Electrophysiol.2003;26(8):1747– 1757.
- ,.Clinical disorders of the autonomic nervous system associated with orthostatic intolerance: an overview of classification, clinical evaluation, and management.Pacing Clin Electrophysiol.1999;22(5):798–810.
- ,.Idiopathic orthostatic intolerance and postural tachycardia syndromes.Am J Med Sci.1999;317(2):88–101.
- ,,, et al.Splanchnic‐mesenteric capacitance bed in the postural tachycardia syndrome (POTS).Auton Neurosci.2000;86(1–2):107–113.
- ,,,.Abnormal orthostatic changes in blood pressure and heart rate in subjects with intact sympathetic nervous function: evidence for excessive venous pooling.J Lab Clin Med.1988;111(3):326–335.
- ,,, et al.Renin‐aldosterone paradox and perturbed blood volume regulation underlying postural tachycardia syndrome.Circulation.2005;111(13):1574–1582.
- .Clinical practice. Neurocardiogenic syncope.N Engl J Med.2005;352(10):1004–1010.
- ,,, et al.Orthostatic intolerance and tachycardia associated with norepinephrine‐transporter deficiency.N Engl J Med.2000;342(8):541–549.
- ,,,,,.Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies.N Engl J Med.2000;343(12):847–855.
- ,,.The postural tachycardia syndrome: a concise guide to diagnosis and management.J Cardiovasc Electrophysiol.2006;17(1):108–112.
- ,,.Neurovegetative regulation of the vascular system. In:Lanzer P,Topol EJ, eds.Panvascular Medicine.Berlin, Germany:Springer‐Verlag;2002:175–187.
- ,,, et al.Regional cardiac sympathetic denervation in patients with ventricular tachycardia in the absence of coronary artery disease.J Am Coll Cardiol.1993;22(5):1344–1353.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A30‐year‐old woman was referred for evaluation of chest pain, palpitations, and exercise intolerance. She had been previously healthy, active, and physically fit. Five months prior to our evaluation, she had an elective C5C6 cervical spine discectomy with interbody allograft fusion for a chronic neck injury that occurred 11 years ago during gymnastics. Two weeks after spine surgery, the patient developed numbness and tingling of her left thumb and palm that occurred with exertion or exposure to cold and subsided with rest. These episodes increased in frequency and intensity and after 1 week became associated with sharp, occasionally stabbing chest pain that radiated to the left arm. On one occasion, the patient had an episode of exertional chest pain with prolonged left arm cyanosis. Emergent left upper extremity angiography revealed normal great vessel anatomy with spasm of the radial artery and collateral ulnar flow. The patient was diagnosed with Raynaud's phenomenon and was started on nifedipine. A subsequent rheumatologic evaluation was unrevealing, and the patient was empirically switched to amlodipine with no improvement in symptoms.
This otherwise very healthy 30‐year‐old developed a multitude of symptoms. The patient's chest pain is atypical and in a young woman is unlikely to signify atherosclerotic coronary disease, but it should not be entirely disregarded. Vasospasm triggered by exposure to cold does raise suspicion for Raynaud's phenomenon, which is not uncommon in this demographic. However, this presentation is quite unusual because the vasospasm was limited to one vascular distribution of one extremity. Associated coronary vasospasm could explain the other symptoms, although coronary spasm is generally not associated with Raynaud's phenomenon. Vasculitis may also affect the pulmonary vasculature, leading to pulmonary hypertension and exercise intolerance. The temporal association with her spine surgery is intriguing but of unclear significance.
The patient continued to have frequent exertional episodes of sharp precordial chest pain radiating to her left arm that were accompanied by dyspnea and left upper extremity symptoms despite amlodipine therapy. These now occurred with limited activity when she walked 1 to 2 blocks uphill. Over the previous 2 months, she had also noticed palpitations occurring reliably with exercise that were relieved with 15 to 20 min of rest. With prolonged episodes, she reported dizziness, nausea, and blurry vision that improved with lying down. She twice had syncope with these symptoms. She noted lower extremity edema while taking calcium channel blockers, but this had resolved after discontinuation of the drugs.
The patient's past medical history included several high‐school orthopedic injuries. She had 2 kidney stones at ages 18 and 23 and had an appendectomy at age 28. Her only medication was an oral contraceptive, and she had discontinued the amlodipine. She denied the use of tobacco, alcohol, herbal medications, or illicit substances. There was no family history of sudden death or heart disease.
Palpitations in a 30‐year‐old woman may signify a cardiac arrhythmia. Paroxysmal supraventricular arrhythmias, such as atrioventricular nodal reentrant tachycardia, atrial tachycardia, and atrial fibrillation, are well described in the young. Ventricular tachycardia (VT) is another possible cause and could be idiopathic or related to occult structural heart disease. Young patients typically tolerate lone arrhythmias quite well, and her failure to do so raises suspicion for concomitant structural heart disease. Her palpitations may be from appropriate sinus tachycardia, which could be compensatory because of inadequate cardiac output reserve, which in turn could be caused by valvular disease, congenital heart disease, or ventricular dysfunction. The exertional chest pain is worrisome for ischemia. Pulmonary hypertension, severe ventricular hypertrophy, or congenital anomalies of the coronary circulation could lead to subendocardial myocardial ischemia with exertion, resulting in angina, dyspnea, and arrhythmias. However, the patient also experiences exertional palpitations without chest pain, which may signify an exertional tachyarrhythmia possibly mediated by catecholamines. Based solely on the history, the differential diagnosis remains broad.
On physical examination, the patient was a fit, thin, healthy woman. Her blood pressure was 120/70 mm Hg supine in both arms and 115/75 mm Hg standing; her pulse was 85 supine and 110 standing, Oxygen saturation was 100% on room air. A cardiac exam revealed a normal jugular venous pressure, normal point of maximal impulse, regular rhythm with occasional ectopy, normal S1, and physiologically split S2 without extra heart sounds or murmurs. The right ventricular impulse was faintly palpable at the left sternal border. Head, neck, chest, abdominal, musculoskeletal, neurologic, extremity, and peripheral pulse examinations were normal.
Laboratory data showed a normal complete blood count and normal chemistries. Serum tests for hepatitis C antibody, cardiolipin antibody, rheumatoid factor, cryoglobulins, and anti‐nuclear antibody were negative. The erythrocyte sedimentation rate and thyroid stimulating hormone levels were within normal limits. An electrocardiogram (ECG) demonstrated a normal sinus rhythm with frequent premature ventricular complexes (PVCs) and normal axis and intervals. (Figure 1). The PR segment was normal and without preexcitation. A prior ECG from 3 months ago was similar with ventricular trigeminy.
Her unremarkable cardiac examination does not favor structural or valvular heart disease, and there are no obvious stigmata of vasculitis. She did become mildly tachycardic upon standing, and this raises the possibility of orthostatic tachycardia. A comprehensive rheumatologic panel revealed no evidence of autoimmune disease or vasculitis, and the clinical constellation is not consistent with primary or secondary Raynaud's disease. The ECG demonstrates frequent monomorphic PVCs complexes with a left bundle branch block pattern and an inferior axis. This pattern suggests that the PVCs arise from the right ventricular outflow tract. Idiopathic right ventricular outflow tract VT and arrhythmogenic right ventricular dysplasia must be considered as a cause of exertional or catecholamine‐mediated tachycardia. The normal ECG argues against arrhythmogenic right ventricular dysplasia, in which patients typically have incomplete or complete right bundle branch block, right precordial T wave abnormalities, and occasionally epsilon waves. Her QT interval is normal, but excluding long‐QT syndrome with a single ECG has poor sensitivity. The next critical step is to document her cardiac rhythm during symptoms and to exclude malignant arrhythmias.
An event recorder and exercise echocardiogram were ordered. While the patient was wearing her event recorder, she had 4 episodes of exertional syncope while hiking and successfully triggered event recording before losing consciousness. She had chest pain and left arm pain after regaining consciousness. The patient came to the emergency room for evaluation. Her blood pressure was 116/80 mm Hg supine and 112/70 mm Hg seated. Her heart rate increased from 82 supine to 132 seated. The physical examination was unremarkable. ECG showed sinus rhythm with frequent PVCs. Troponin‐I measurements 10 hours apart were 0.7 and 0.3 g/L (normal < 1.1), with normal creatinine kinase and creatinine kinase MB fractions. Interrogation of the event recorder revealed multiple episodes of a narrow complex tachycardia with rates up to 180 bpm that correlated with symptoms (Figure 2). There were no episodes of wide complex tachycardia.
The patient was not hypotensive in the emergency room, but she had evidence of a marked orthostatic tachycardia. The minimal but significant troponin elevations are also troubling. Although her clinical picture is not consistent with an acute coronary syndrome, I am concerned about other mechanisms of myocardial ischemia or injury, such as a coronary anomaly or subendocardial ischemia from globally reduced myocardial perfusion. The presence of event recorder data from her syncopal events was fortuitous and revealed a supraventricular tachycardia. The arrhythmia was gradual in onset and resolution and had no triggers, such as premature atrial or ventricular complexes, which could suggest reentrant arrhythmias. The P wave morphology was also unchanged, and this argues against an atrial tachycardia. These findings are consistent with sinus tachycardia, which was notably out of proportion to her workload. This arrhythmia may be the primary cause of syncope, such as in inappropriate sinus tachycardia, or it may be a compensatory mechanism. Tachycardia from coronary vasospasm is often preceded by ST segment changes, which are not seen here. Although the event recorder had no episodes of VT, the patient's persistent frequent PVCs are still of concern. I would obtain an echocardiogram to exclude structural heart disease and an exercise test to exclude exertional VT. Finally, coronary angiography may be helpful in excluding congenital anomalies.
The patient was admitted for evaluation. An exercise treadmill test was performed, and the patient exercised 20 min on the standard Bruce protocol with a peak heart rate of 180 bpm. The test was notable for a premature rise in heart rate (in stage 1) without a rise in blood pressure. There were no symptoms or ST/T wave changes. Transthoracic echocardiogram showed normal left ventricular size and function with normal anatomy, valves, and hemodynamics. Coronary angiography showed a right dominant system with normal anatomy and no atherosclerotic disease.
Ventricular arrhythmias could not be elicited with exercise. Her high exercise tolerance virtually excluded hemodynamically significant structural or valvular disease, and this was confirmed by the echocardiogram. Coronary angiography excluded coronary anomalies and myocardial bridging. The most intriguing finding is the rise in the patient's heart rate out of proportion to the workload. This, along with her orthostatic tachycardia, raises the issue of inappropriate sinus tachycardia or postural orthostatic tachycardia syndrome (POTS). Carotid hypersensitivity is also a possibility. The patient was hiking when she fainted, and even light pressure on the patient's neck with head turning or from a camera strap, for example, could produce syncope. Although carotid hypersensitivity usually results in sinus bradycardia and AV block, it may be followed by reflex tachycardia, which was seen in this patient's event recordings. I would perform a tilt‐table test with carotid massage to make the diagnosis.
Tilt‐table testing was performed (Figure 3). Her supine blood pressure was 128/68 mm Hg, and her heart rate was 72 bpm with no change during the 10‐min supine period. Upon elevation to a 70‐degree tilt, the patient had an immediate increase in her heart rate to 160 bpm with a blood pressure nadir of 109/58 mm Hg and symptoms of palpitations, dizziness, dyspnea, chest pain, blurry vision, and nausea. Her peak heart rate was 172 bpm, and her peak blood pressure was 122/72 mm Hg. Vital signs did not change in response to carotid sinus massage in the supine or upright positions.
The tilt‐table test has 3 notable findings. First, her heart rate increased rapidly with tilt and decreased rapidly in supine recovery. Second, her usual symptoms started immediately after tilt and quickly resolved in recovery when vital signs returned to baseline. Finally, there was only a modest drop in blood pressure. These findings are classic for POTS. POTS is defined as symptomatic orthostasis with a heart rate increase of 30 bpm or a heart rate of 120 bpm. The physiologic lesions found in the syndrome are heterogeneous, but they all lead to a failure of orthostatic compensation. In POTS, the tachycardia is a reflex secondary to hypotension (baroreceptor reflex) or reduced preload (cardiac mechanoreceptors), in contrast to inappropriate sinus tachycardia. Interestingly, blood pressure is usually preserved until the final moments preceding syncope, when venous return further declines, tachycardia decreases the diastolic filling time and stroke volume, and mean arterial pressure sharply falls.
The patient was started on labetalol (200 mg 3 times daily), and her symptoms worsened. She also developed nausea and constipation. Midodrine and pindolol were also tried without success. She was then switched to fludrocortisone, salt supplementation, and leg support stockings with dramatic improvement.
COMMENTARY
In 1871, DeCosta1 published a report on the irritable heart, noting an affliction of extreme fatigue and exercise intolerance that occurred suddenly and without obvious cause. Subsequently, the terms vasoregulatory asthenia and neurocirculatory asthenia were used to link cardiovascular symptoms to impaired regulation of peripheral blood flow.2, 3 The term POTS was first used in 1982 to describe a single patient with postural tachycardia without hypotension and palpitations, weakness, abdominal pain, and presyncope.4
POTS is one of several disorders of autonomic control associated with orthostatic intolerance. The criteria for diagnosis are listed in Table 1. POTS typically occurs in women between the ages of 15 and 50 but tends to present during adolescence or young adulthood. The physiology has only recently been elucidated. When a person stands, 500 cc of the total blood volume is displaced to the dependent extremities and inferior mesenteric vessels.5 Normally, orthostatic stabilization occurs in less than 1 minute via 3 mechanisms: baroreceptor input, sympathetic reflex tachycardia and vasoconstriction, and enhanced venous return via the pumping action of skeletal muscles and venoconstriction. In POTS, there is a failure of at least one of these mechanisms, leading to decreased venous return, a 40% reduction in stroke volume, and cerebral hypoperfusion.6
| 1. Consistent symptoms of orthostatic intolerance [may include excessive fatigue, exercise intolerance, recurrent syncope or near syncope, dizziness, nausea, tachycardia, palpitations, visual disturbances, blurred vision, tunnel vision, tremulousness, weakness (most noticeable in the legs), chest discomfort, shortness of breath, mood swings, and gastrointestinal complaints] |
| 2. Heart rate increase 30 bpm or heart rate 120 bpm within 10 min of standing or head‐up tilt |
| 3. Absence of a known cause of autonomic neuropathy |
POTS is divided into 2 major subtypes on the basis of pathophysiology.5, 7 The partial dysautonomic form is the most common and the type that this patient most likely had. In this form, the development of an acquired peripheral autonomic neuropathy results in a failure of sympathetic venoconstriction, which leads to excessive venous pooling in the lower extremities and splanchnic circulation.8, 9 Failure to mobilize this venous reservoir upon standing leads to excessive orthostatic tachycardia secondary to a marked reduction in stroke volume. Peripheral arterial vasoconstriction is generally preserved, which is why midodrine, an arterial vasoconstrictor, did not improve symptoms. The labetalol may have further exacerbated peripheral pooling because of its alpha‐adrenergic blocking properties. Because total plasma volume is decreased and plasma renin activity is inappropriately low,10 volume expanders, including salt, low‐dose steroids, and fluids, can attenuate symptoms.11 The extrinsic venous compression from leg and abdominal support stockings may also dramatically reduce venous pooling.
In the less common hyperadrenergic form of POTS, patients may have orthostatic hypertension, tremulousness, cold, sweaty extremities, and anxiety due to an exaggerated response to beta‐adrenergic stimulation.7 The excessive sympathetic activity, which is poorly modulated by baroreflex activity, may be due to impaired mechanisms of norepinephrine reuptake by sympathetic ganglia.12 Consequently, serum norepinephrine levels are markedly elevated (>600 pg/mL).5
In adults, the presence of a POTS trigger is common and is usually an antecedent viral illness. Antibodies to the ganglionic acetylcholine receptor have been found in a subset of POTS patients,13 and this may suggest an idiopathic or postinflammatory autoimmune mechanism.14 This patient's presentation is unique because her symptoms developed after C5C6 spine surgery. The cervical spinal cord and sympathetic ganglia are dense with nerves involved in autonomic cardiovascular control, and damage to these fibers could explain the patient's physiology and symptoms. Among these, the descending vasomotor pathways traverse through the C5C8 area to innervate the splanchnic and leg venous circulation, receiving input from the heart along the way.15 The pattern of numbness and tingling fits the C5/C6 dermatomal distribution, as does the innervation of the radial artery. The frequent PVCs with a left bundle branch block pattern and inferior axis appear to arise from the right ventricular outflow tract and may be associated with regional sympathetic denervation, which has been described in idiopathic ventricular arrhythmias.16 POTS has been anecdotally reported after neck injury from motor vehicle accidents (whiplash), which is also thought to be related to cervical sympathetic nerve damage (B.P. Grubb, personal communication, 2005). Most cases of triggered POTS improve spontaneously after months to years, but this patient's prognosis remains uncertain because of the presumed mechanical disruption of the autonomic nerve fibers at the time of surgery.
This case demonstrates the complexities of arriving at a unifying diagnosis in the setting of a constellation of nonspecific symptoms and findings, some of which even suggest life‐threatening conditions. Because young women are primarily affected, symptoms of POTS can be mistakenly attributed to anxiety or other nonphysiological factors. A systematic approach excluded life‐threatening causes, including primary ventricular arrhythmias, coronary vasospasm, and coronary anomalies. The investigations narrowed the differential diagnosis, and the tilt‐table test confirmed POTS. Because the cardiac and circulatory dysautonomias encompass an array of distinct physiologic processes, understanding the patient's mechanism is critical to her management. The only effective therapies were those that counteracted venous pooling and improved venous return.
Teaching Points
-
The differential diagnosis of exertional syncope is extremely broad, ranging from benign to malignant conditions, and requires a systematic evaluation of the heart and circulatory system.
-
The diagnosis of POTS is elusive and frequently missed. Referral for tilt‐table testing is useful in identifying the mechanism of sinus tachycardia and syncope. Marked orthostatic tachycardia and symptoms of cerebral hypoperfusion out of proportion to the degree of hypotension strongly suggest POTS.
-
Cardiac and circulatory dysautonomias have distinct and varied mechanisms. Therapies, including beta‐blockers, vasoconstrictors, and volume expanders, must be directed at the underlying physiological defect.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A30‐year‐old woman was referred for evaluation of chest pain, palpitations, and exercise intolerance. She had been previously healthy, active, and physically fit. Five months prior to our evaluation, she had an elective C5C6 cervical spine discectomy with interbody allograft fusion for a chronic neck injury that occurred 11 years ago during gymnastics. Two weeks after spine surgery, the patient developed numbness and tingling of her left thumb and palm that occurred with exertion or exposure to cold and subsided with rest. These episodes increased in frequency and intensity and after 1 week became associated with sharp, occasionally stabbing chest pain that radiated to the left arm. On one occasion, the patient had an episode of exertional chest pain with prolonged left arm cyanosis. Emergent left upper extremity angiography revealed normal great vessel anatomy with spasm of the radial artery and collateral ulnar flow. The patient was diagnosed with Raynaud's phenomenon and was started on nifedipine. A subsequent rheumatologic evaluation was unrevealing, and the patient was empirically switched to amlodipine with no improvement in symptoms.
This otherwise very healthy 30‐year‐old developed a multitude of symptoms. The patient's chest pain is atypical and in a young woman is unlikely to signify atherosclerotic coronary disease, but it should not be entirely disregarded. Vasospasm triggered by exposure to cold does raise suspicion for Raynaud's phenomenon, which is not uncommon in this demographic. However, this presentation is quite unusual because the vasospasm was limited to one vascular distribution of one extremity. Associated coronary vasospasm could explain the other symptoms, although coronary spasm is generally not associated with Raynaud's phenomenon. Vasculitis may also affect the pulmonary vasculature, leading to pulmonary hypertension and exercise intolerance. The temporal association with her spine surgery is intriguing but of unclear significance.
The patient continued to have frequent exertional episodes of sharp precordial chest pain radiating to her left arm that were accompanied by dyspnea and left upper extremity symptoms despite amlodipine therapy. These now occurred with limited activity when she walked 1 to 2 blocks uphill. Over the previous 2 months, she had also noticed palpitations occurring reliably with exercise that were relieved with 15 to 20 min of rest. With prolonged episodes, she reported dizziness, nausea, and blurry vision that improved with lying down. She twice had syncope with these symptoms. She noted lower extremity edema while taking calcium channel blockers, but this had resolved after discontinuation of the drugs.
The patient's past medical history included several high‐school orthopedic injuries. She had 2 kidney stones at ages 18 and 23 and had an appendectomy at age 28. Her only medication was an oral contraceptive, and she had discontinued the amlodipine. She denied the use of tobacco, alcohol, herbal medications, or illicit substances. There was no family history of sudden death or heart disease.
Palpitations in a 30‐year‐old woman may signify a cardiac arrhythmia. Paroxysmal supraventricular arrhythmias, such as atrioventricular nodal reentrant tachycardia, atrial tachycardia, and atrial fibrillation, are well described in the young. Ventricular tachycardia (VT) is another possible cause and could be idiopathic or related to occult structural heart disease. Young patients typically tolerate lone arrhythmias quite well, and her failure to do so raises suspicion for concomitant structural heart disease. Her palpitations may be from appropriate sinus tachycardia, which could be compensatory because of inadequate cardiac output reserve, which in turn could be caused by valvular disease, congenital heart disease, or ventricular dysfunction. The exertional chest pain is worrisome for ischemia. Pulmonary hypertension, severe ventricular hypertrophy, or congenital anomalies of the coronary circulation could lead to subendocardial myocardial ischemia with exertion, resulting in angina, dyspnea, and arrhythmias. However, the patient also experiences exertional palpitations without chest pain, which may signify an exertional tachyarrhythmia possibly mediated by catecholamines. Based solely on the history, the differential diagnosis remains broad.
On physical examination, the patient was a fit, thin, healthy woman. Her blood pressure was 120/70 mm Hg supine in both arms and 115/75 mm Hg standing; her pulse was 85 supine and 110 standing, Oxygen saturation was 100% on room air. A cardiac exam revealed a normal jugular venous pressure, normal point of maximal impulse, regular rhythm with occasional ectopy, normal S1, and physiologically split S2 without extra heart sounds or murmurs. The right ventricular impulse was faintly palpable at the left sternal border. Head, neck, chest, abdominal, musculoskeletal, neurologic, extremity, and peripheral pulse examinations were normal.
Laboratory data showed a normal complete blood count and normal chemistries. Serum tests for hepatitis C antibody, cardiolipin antibody, rheumatoid factor, cryoglobulins, and anti‐nuclear antibody were negative. The erythrocyte sedimentation rate and thyroid stimulating hormone levels were within normal limits. An electrocardiogram (ECG) demonstrated a normal sinus rhythm with frequent premature ventricular complexes (PVCs) and normal axis and intervals. (Figure 1). The PR segment was normal and without preexcitation. A prior ECG from 3 months ago was similar with ventricular trigeminy.
Her unremarkable cardiac examination does not favor structural or valvular heart disease, and there are no obvious stigmata of vasculitis. She did become mildly tachycardic upon standing, and this raises the possibility of orthostatic tachycardia. A comprehensive rheumatologic panel revealed no evidence of autoimmune disease or vasculitis, and the clinical constellation is not consistent with primary or secondary Raynaud's disease. The ECG demonstrates frequent monomorphic PVCs complexes with a left bundle branch block pattern and an inferior axis. This pattern suggests that the PVCs arise from the right ventricular outflow tract. Idiopathic right ventricular outflow tract VT and arrhythmogenic right ventricular dysplasia must be considered as a cause of exertional or catecholamine‐mediated tachycardia. The normal ECG argues against arrhythmogenic right ventricular dysplasia, in which patients typically have incomplete or complete right bundle branch block, right precordial T wave abnormalities, and occasionally epsilon waves. Her QT interval is normal, but excluding long‐QT syndrome with a single ECG has poor sensitivity. The next critical step is to document her cardiac rhythm during symptoms and to exclude malignant arrhythmias.
An event recorder and exercise echocardiogram were ordered. While the patient was wearing her event recorder, she had 4 episodes of exertional syncope while hiking and successfully triggered event recording before losing consciousness. She had chest pain and left arm pain after regaining consciousness. The patient came to the emergency room for evaluation. Her blood pressure was 116/80 mm Hg supine and 112/70 mm Hg seated. Her heart rate increased from 82 supine to 132 seated. The physical examination was unremarkable. ECG showed sinus rhythm with frequent PVCs. Troponin‐I measurements 10 hours apart were 0.7 and 0.3 g/L (normal < 1.1), with normal creatinine kinase and creatinine kinase MB fractions. Interrogation of the event recorder revealed multiple episodes of a narrow complex tachycardia with rates up to 180 bpm that correlated with symptoms (Figure 2). There were no episodes of wide complex tachycardia.
The patient was not hypotensive in the emergency room, but she had evidence of a marked orthostatic tachycardia. The minimal but significant troponin elevations are also troubling. Although her clinical picture is not consistent with an acute coronary syndrome, I am concerned about other mechanisms of myocardial ischemia or injury, such as a coronary anomaly or subendocardial ischemia from globally reduced myocardial perfusion. The presence of event recorder data from her syncopal events was fortuitous and revealed a supraventricular tachycardia. The arrhythmia was gradual in onset and resolution and had no triggers, such as premature atrial or ventricular complexes, which could suggest reentrant arrhythmias. The P wave morphology was also unchanged, and this argues against an atrial tachycardia. These findings are consistent with sinus tachycardia, which was notably out of proportion to her workload. This arrhythmia may be the primary cause of syncope, such as in inappropriate sinus tachycardia, or it may be a compensatory mechanism. Tachycardia from coronary vasospasm is often preceded by ST segment changes, which are not seen here. Although the event recorder had no episodes of VT, the patient's persistent frequent PVCs are still of concern. I would obtain an echocardiogram to exclude structural heart disease and an exercise test to exclude exertional VT. Finally, coronary angiography may be helpful in excluding congenital anomalies.
The patient was admitted for evaluation. An exercise treadmill test was performed, and the patient exercised 20 min on the standard Bruce protocol with a peak heart rate of 180 bpm. The test was notable for a premature rise in heart rate (in stage 1) without a rise in blood pressure. There were no symptoms or ST/T wave changes. Transthoracic echocardiogram showed normal left ventricular size and function with normal anatomy, valves, and hemodynamics. Coronary angiography showed a right dominant system with normal anatomy and no atherosclerotic disease.
Ventricular arrhythmias could not be elicited with exercise. Her high exercise tolerance virtually excluded hemodynamically significant structural or valvular disease, and this was confirmed by the echocardiogram. Coronary angiography excluded coronary anomalies and myocardial bridging. The most intriguing finding is the rise in the patient's heart rate out of proportion to the workload. This, along with her orthostatic tachycardia, raises the issue of inappropriate sinus tachycardia or postural orthostatic tachycardia syndrome (POTS). Carotid hypersensitivity is also a possibility. The patient was hiking when she fainted, and even light pressure on the patient's neck with head turning or from a camera strap, for example, could produce syncope. Although carotid hypersensitivity usually results in sinus bradycardia and AV block, it may be followed by reflex tachycardia, which was seen in this patient's event recordings. I would perform a tilt‐table test with carotid massage to make the diagnosis.
Tilt‐table testing was performed (Figure 3). Her supine blood pressure was 128/68 mm Hg, and her heart rate was 72 bpm with no change during the 10‐min supine period. Upon elevation to a 70‐degree tilt, the patient had an immediate increase in her heart rate to 160 bpm with a blood pressure nadir of 109/58 mm Hg and symptoms of palpitations, dizziness, dyspnea, chest pain, blurry vision, and nausea. Her peak heart rate was 172 bpm, and her peak blood pressure was 122/72 mm Hg. Vital signs did not change in response to carotid sinus massage in the supine or upright positions.
The tilt‐table test has 3 notable findings. First, her heart rate increased rapidly with tilt and decreased rapidly in supine recovery. Second, her usual symptoms started immediately after tilt and quickly resolved in recovery when vital signs returned to baseline. Finally, there was only a modest drop in blood pressure. These findings are classic for POTS. POTS is defined as symptomatic orthostasis with a heart rate increase of 30 bpm or a heart rate of 120 bpm. The physiologic lesions found in the syndrome are heterogeneous, but they all lead to a failure of orthostatic compensation. In POTS, the tachycardia is a reflex secondary to hypotension (baroreceptor reflex) or reduced preload (cardiac mechanoreceptors), in contrast to inappropriate sinus tachycardia. Interestingly, blood pressure is usually preserved until the final moments preceding syncope, when venous return further declines, tachycardia decreases the diastolic filling time and stroke volume, and mean arterial pressure sharply falls.
The patient was started on labetalol (200 mg 3 times daily), and her symptoms worsened. She also developed nausea and constipation. Midodrine and pindolol were also tried without success. She was then switched to fludrocortisone, salt supplementation, and leg support stockings with dramatic improvement.
COMMENTARY
In 1871, DeCosta1 published a report on the irritable heart, noting an affliction of extreme fatigue and exercise intolerance that occurred suddenly and without obvious cause. Subsequently, the terms vasoregulatory asthenia and neurocirculatory asthenia were used to link cardiovascular symptoms to impaired regulation of peripheral blood flow.2, 3 The term POTS was first used in 1982 to describe a single patient with postural tachycardia without hypotension and palpitations, weakness, abdominal pain, and presyncope.4
POTS is one of several disorders of autonomic control associated with orthostatic intolerance. The criteria for diagnosis are listed in Table 1. POTS typically occurs in women between the ages of 15 and 50 but tends to present during adolescence or young adulthood. The physiology has only recently been elucidated. When a person stands, 500 cc of the total blood volume is displaced to the dependent extremities and inferior mesenteric vessels.5 Normally, orthostatic stabilization occurs in less than 1 minute via 3 mechanisms: baroreceptor input, sympathetic reflex tachycardia and vasoconstriction, and enhanced venous return via the pumping action of skeletal muscles and venoconstriction. In POTS, there is a failure of at least one of these mechanisms, leading to decreased venous return, a 40% reduction in stroke volume, and cerebral hypoperfusion.6
| 1. Consistent symptoms of orthostatic intolerance [may include excessive fatigue, exercise intolerance, recurrent syncope or near syncope, dizziness, nausea, tachycardia, palpitations, visual disturbances, blurred vision, tunnel vision, tremulousness, weakness (most noticeable in the legs), chest discomfort, shortness of breath, mood swings, and gastrointestinal complaints] |
| 2. Heart rate increase 30 bpm or heart rate 120 bpm within 10 min of standing or head‐up tilt |
| 3. Absence of a known cause of autonomic neuropathy |
POTS is divided into 2 major subtypes on the basis of pathophysiology.5, 7 The partial dysautonomic form is the most common and the type that this patient most likely had. In this form, the development of an acquired peripheral autonomic neuropathy results in a failure of sympathetic venoconstriction, which leads to excessive venous pooling in the lower extremities and splanchnic circulation.8, 9 Failure to mobilize this venous reservoir upon standing leads to excessive orthostatic tachycardia secondary to a marked reduction in stroke volume. Peripheral arterial vasoconstriction is generally preserved, which is why midodrine, an arterial vasoconstrictor, did not improve symptoms. The labetalol may have further exacerbated peripheral pooling because of its alpha‐adrenergic blocking properties. Because total plasma volume is decreased and plasma renin activity is inappropriately low,10 volume expanders, including salt, low‐dose steroids, and fluids, can attenuate symptoms.11 The extrinsic venous compression from leg and abdominal support stockings may also dramatically reduce venous pooling.
In the less common hyperadrenergic form of POTS, patients may have orthostatic hypertension, tremulousness, cold, sweaty extremities, and anxiety due to an exaggerated response to beta‐adrenergic stimulation.7 The excessive sympathetic activity, which is poorly modulated by baroreflex activity, may be due to impaired mechanisms of norepinephrine reuptake by sympathetic ganglia.12 Consequently, serum norepinephrine levels are markedly elevated (>600 pg/mL).5
In adults, the presence of a POTS trigger is common and is usually an antecedent viral illness. Antibodies to the ganglionic acetylcholine receptor have been found in a subset of POTS patients,13 and this may suggest an idiopathic or postinflammatory autoimmune mechanism.14 This patient's presentation is unique because her symptoms developed after C5C6 spine surgery. The cervical spinal cord and sympathetic ganglia are dense with nerves involved in autonomic cardiovascular control, and damage to these fibers could explain the patient's physiology and symptoms. Among these, the descending vasomotor pathways traverse through the C5C8 area to innervate the splanchnic and leg venous circulation, receiving input from the heart along the way.15 The pattern of numbness and tingling fits the C5/C6 dermatomal distribution, as does the innervation of the radial artery. The frequent PVCs with a left bundle branch block pattern and inferior axis appear to arise from the right ventricular outflow tract and may be associated with regional sympathetic denervation, which has been described in idiopathic ventricular arrhythmias.16 POTS has been anecdotally reported after neck injury from motor vehicle accidents (whiplash), which is also thought to be related to cervical sympathetic nerve damage (B.P. Grubb, personal communication, 2005). Most cases of triggered POTS improve spontaneously after months to years, but this patient's prognosis remains uncertain because of the presumed mechanical disruption of the autonomic nerve fibers at the time of surgery.
This case demonstrates the complexities of arriving at a unifying diagnosis in the setting of a constellation of nonspecific symptoms and findings, some of which even suggest life‐threatening conditions. Because young women are primarily affected, symptoms of POTS can be mistakenly attributed to anxiety or other nonphysiological factors. A systematic approach excluded life‐threatening causes, including primary ventricular arrhythmias, coronary vasospasm, and coronary anomalies. The investigations narrowed the differential diagnosis, and the tilt‐table test confirmed POTS. Because the cardiac and circulatory dysautonomias encompass an array of distinct physiologic processes, understanding the patient's mechanism is critical to her management. The only effective therapies were those that counteracted venous pooling and improved venous return.
Teaching Points
-
The differential diagnosis of exertional syncope is extremely broad, ranging from benign to malignant conditions, and requires a systematic evaluation of the heart and circulatory system.
-
The diagnosis of POTS is elusive and frequently missed. Referral for tilt‐table testing is useful in identifying the mechanism of sinus tachycardia and syncope. Marked orthostatic tachycardia and symptoms of cerebral hypoperfusion out of proportion to the degree of hypotension strongly suggest POTS.
-
Cardiac and circulatory dysautonomias have distinct and varied mechanisms. Therapies, including beta‐blockers, vasoconstrictors, and volume expanders, must be directed at the underlying physiological defect.
- .An irritable heart.Am J Med Sci.1871;27:145–161.
- ,,,,,.Low physical working capacity in suspected heart cases due to inadequate adjustment of peripheral blood flow (vasoregulatory asthenia).Acta Med Scand.1957;158(6):413–436.
- ,,.Orthostatic tachycardia and orthostatic hypotension: defects in the return of venous blood to the heart.Am Heart J.1944;27:145–163.
- ,.Postural tachycardia syndrome. Reversal of sympathetic hyperresponsiveness and clinical improvement during sodium loading.Am J Med.1982;72(5):847–850.
- ,,.The postural orthostatic tachycardia syndrome: definitions, diagnosis, and management.Pacing Clin Electrophysiol.2003;26(8):1747– 1757.
- ,.Clinical disorders of the autonomic nervous system associated with orthostatic intolerance: an overview of classification, clinical evaluation, and management.Pacing Clin Electrophysiol.1999;22(5):798–810.
- ,.Idiopathic orthostatic intolerance and postural tachycardia syndromes.Am J Med Sci.1999;317(2):88–101.
- ,,, et al.Splanchnic‐mesenteric capacitance bed in the postural tachycardia syndrome (POTS).Auton Neurosci.2000;86(1–2):107–113.
- ,,,.Abnormal orthostatic changes in blood pressure and heart rate in subjects with intact sympathetic nervous function: evidence for excessive venous pooling.J Lab Clin Med.1988;111(3):326–335.
- ,,, et al.Renin‐aldosterone paradox and perturbed blood volume regulation underlying postural tachycardia syndrome.Circulation.2005;111(13):1574–1582.
- .Clinical practice. Neurocardiogenic syncope.N Engl J Med.2005;352(10):1004–1010.
- ,,, et al.Orthostatic intolerance and tachycardia associated with norepinephrine‐transporter deficiency.N Engl J Med.2000;342(8):541–549.
- ,,,,,.Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies.N Engl J Med.2000;343(12):847–855.
- ,,.The postural tachycardia syndrome: a concise guide to diagnosis and management.J Cardiovasc Electrophysiol.2006;17(1):108–112.
- ,,.Neurovegetative regulation of the vascular system. In:Lanzer P,Topol EJ, eds.Panvascular Medicine.Berlin, Germany:Springer‐Verlag;2002:175–187.
- ,,, et al.Regional cardiac sympathetic denervation in patients with ventricular tachycardia in the absence of coronary artery disease.J Am Coll Cardiol.1993;22(5):1344–1353.
- .An irritable heart.Am J Med Sci.1871;27:145–161.
- ,,,,,.Low physical working capacity in suspected heart cases due to inadequate adjustment of peripheral blood flow (vasoregulatory asthenia).Acta Med Scand.1957;158(6):413–436.
- ,,.Orthostatic tachycardia and orthostatic hypotension: defects in the return of venous blood to the heart.Am Heart J.1944;27:145–163.
- ,.Postural tachycardia syndrome. Reversal of sympathetic hyperresponsiveness and clinical improvement during sodium loading.Am J Med.1982;72(5):847–850.
- ,,.The postural orthostatic tachycardia syndrome: definitions, diagnosis, and management.Pacing Clin Electrophysiol.2003;26(8):1747– 1757.
- ,.Clinical disorders of the autonomic nervous system associated with orthostatic intolerance: an overview of classification, clinical evaluation, and management.Pacing Clin Electrophysiol.1999;22(5):798–810.
- ,.Idiopathic orthostatic intolerance and postural tachycardia syndromes.Am J Med Sci.1999;317(2):88–101.
- ,,, et al.Splanchnic‐mesenteric capacitance bed in the postural tachycardia syndrome (POTS).Auton Neurosci.2000;86(1–2):107–113.
- ,,,.Abnormal orthostatic changes in blood pressure and heart rate in subjects with intact sympathetic nervous function: evidence for excessive venous pooling.J Lab Clin Med.1988;111(3):326–335.
- ,,, et al.Renin‐aldosterone paradox and perturbed blood volume regulation underlying postural tachycardia syndrome.Circulation.2005;111(13):1574–1582.
- .Clinical practice. Neurocardiogenic syncope.N Engl J Med.2005;352(10):1004–1010.
- ,,, et al.Orthostatic intolerance and tachycardia associated with norepinephrine‐transporter deficiency.N Engl J Med.2000;342(8):541–549.
- ,,,,,.Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies.N Engl J Med.2000;343(12):847–855.
- ,,.The postural tachycardia syndrome: a concise guide to diagnosis and management.J Cardiovasc Electrophysiol.2006;17(1):108–112.
- ,,.Neurovegetative regulation of the vascular system. In:Lanzer P,Topol EJ, eds.Panvascular Medicine.Berlin, Germany:Springer‐Verlag;2002:175–187.
- ,,, et al.Regional cardiac sympathetic denervation in patients with ventricular tachycardia in the absence of coronary artery disease.J Am Coll Cardiol.1993;22(5):1344–1353.
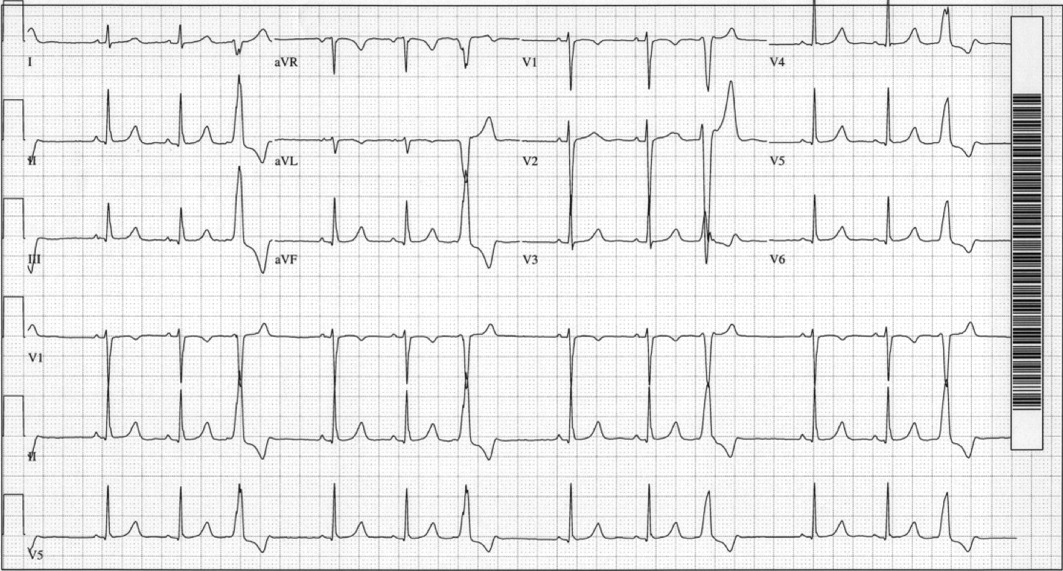
Nerves of Steal
A 60 year‐old woman with advanced kidney disease presented with one month of progressively worsening, sharp burning pain and decreased sensation in her left hand. Cold air exacerbated the pain. She noted decreasing ability to utilize her left fingers, a weakened grip and that the muscles in her hand looked smaller.
Localized sensory and motor symptoms in a discrete region of a single limb suggest neuropathy. The lack of symptoms in the face or ipsilateral lower extremity would dissuade a clinician from considering central etiologies; the presence of neuropathic pain is uncommon for cortical lesions. The involvement of motor and sensory nerves indicates peripheral nerve involvement.
The general approach to patients with peripheral neuropathy begins with identifying the neuropathy as a mononeuropathy (involving a single nerve), a polyneuropathy (symmetric involvement of multiple nerves) or a mononeuropathy multiplex (asymmetric involvement of multiple nerves). The patient, in this case, described subacute neuropathic pain, sensory loss, and weakness in her left hand in a distribution consistent with mononeuropathy or mononeuropathy multiplex.
This patient could have carpal tunnel syndrome given its prevalence in patients with advanced renal disease. The differential diagnosis is broad, however, and includes ulnar mononeuropathy, nerve ischemia due to vasculitis or vasculopathy, lower cervical radiculopathy (though the patient does not describe neck or radicular pain), lower brachial plexopathy, and complex regional pain syndrome.
The patient was diagnosed with advanced kidney disease one year ago when biopsy revealed focal segmental glomerulosclerosis secondary to lithium. Since her diagnosis, two grafts were placed in the left upper arm in anticipation of dialysis: the first, placed seven months prior to this admission, failed to mature; the second, placed one month prior to this admission, was complicated by bleeding at the fistula site and was not yet mature. Prior to this admission she had not required hemodialysis. Her past history included hypertension, dyslipidemia, hypothyroidism, secondary hyperparathyroidism, a remote history of cervical cancer (stage unknown, recent PAP smear negative), microcytosis and schizoaffective disorder. Her medications were furosemide, amlodipine, lisinopril, atenolol, atorvastatin, pantoprazole, olanzapine, levothyroxine, iron, darbepoetin, sevelamer, multivitamin and docusate.
Given the history of procedures in the left arm one should consider ischemic injury to the left median nerve. Other local complications could include compressive lesions such as an abscess or hematoma or direct nerve injury from the procedure. Carpal tunnel syndrome remains high on the differential due to its prevalence and because renal failure and hypothyroidism increase the risk of carpal tunnel syndrome.
A careful physical examination would localize the nerve or nerves involved. Examination findings that would be consistent with carpal tunnel syndrome include sensory loss in the distribution of the left median nerve, weakness of muscles innervated by the median nerve, including the abductor pollicus brevis and opponens muscles, and a Tinels and Phalens sign of the left wrist. A proximal median neuropathy resulting from ischemia or compression might also involve median‐innervated forearm muscle such as the pronator teres (forearm pronation) and flexor carpi radialis (hand flexion and abduction) muscles. Complex regional pain syndrome can be seen after a traumatic injury or surgery and is typified by severe neuropathic pain in a limb, often in combination with trophic changes in the affected extremity. A cervical radiculopathy would affect muscles supplied by the injured nerve root. For example, a C8 radiculopathy would affect all of the intrinsic hand muscles, the wrist and finger extensors, and the triceps brachii. A lower brachial plexopathy would present similarly to a lower cervical radiculopathy on clinical examination; electrodiagnostic evaluation would be necessary to distinguish these two disorders.
The patient appeared fatigued and her left hand was wrapped in blankets. Her vital signs were stable. There was no thyroid enlargement or lymphadenopathy. There was marked thenar, hypothenar and forearm atrophy on the left. Strength testing of her left hand demonstrated a grip strength of 2/5, finger extension and interosseous strength of 1/5, left wrist flexion and extension of 3/5; left biceps and triceps were 5/5. Sensation was mildly decreased to light touch, temperature, pain and proprioception throughout the left hand. Strength and sensation were intact in the right upper and bilateral lower extremities. Reflexes were 2+ throughout. The left radial pulse was diminished compared to the left ulnar that was 1+. The left hand was dry and cool. Laboratory evaluation revealed an elevated white blood cell count of 15,300/mm3, a hematocrit of 33 percent, mean corpuscular volume of 73 fL, and a normal platelet count. Electrolytes were consistent with advanced renal disease. The thyroid stimulating hormone level was normal.
The patient's examination reveals an injury to the sensory and motor components of the left ulnar, median, and distal radial nerves. The volar forearm wasting suggests proximal median motor nerve injury in the forearm. Because the triceps is spared, the weakness of finger and wrist extension implies distal radial motor nerve injury. The interosseous weakness is consistent with injury to the ulnar motor nerve. Weakness of grip and wrist flexion is less specific and it may be explained by injury to either the median or ulnar motor nerves. The diffuse sensory loss over the palmar and dorsal aspect of the left hand is consistent with neuropathic injury to the left median, ulnar, and radial sensory nerves. The preserved deep tendon reflexes are controlled by the musculocutaneous nerve (the biceps reflex) and branches arising from the proximal radial nerve (the triceps and brachioradialis reflexes), both of which are proximal to the apparent level of neuropathic insult.
Carpal tunnel syndrome is excluded since abnormalities extend beyond the median nerve distribution. The presentation is consistent with a mononeuropathy multiplex. Axonal etiologies of mononeuropathy multiplexincluding vasculitis, ischemia, neoplastic infiltration, and infectious etiologies such as Lyme diseaseare more common than demyelinating causes. Vasculitic neuropathy commonly involves the lower extremities and may have systemic symptoms, not present in this case. Neoplasm or other compressive lesions such as hematoma could explain these findings. Abscess must be considered given the leukocytosis. A lower brachial plexopathy, technically a mononeuropathy multiplex involving the proximal arm at the level of the brachial plexus, is also in the differential diagnosis. Another axonal disorder to consider would be neuralgic amyotrophy, an idiopathic form of acute brachial plexopathy associated with pain that is a complication of surgery that typically presents within a few hours to weeks of the procedure.
Demyelinating causes of mononeuropathy multiplex are less likely and include a variant of chronic inflammatory demyelinating polyneuropathy (Lewis‐Sumner syndrome) and hereditary neuropathy with liability to pressure palsies. Both processes are typically indolent and usually not painful, though the latter may present with fulminant numbness and weakness.
Nerve conduction and needle electromyography of the arm and cervical paraspinal muscles would differentiate axonal degeneration from demyelination. It would identify the affected nerves and any nerve root involvement. Given the concern for abscess or hematoma, MR neurography focused on the surgical site would also be important.
Electromyography and nerve conduction velocities demonstrated severe axonal loss of the left median, ulnar and distal radial sensory nerves consistent with acute denervation. A magnetic resonance neurogram following the course of these nerves revealed enlarged ulnar and median nerves with abnormal signal, but no compressive lesion (Fig. 1).
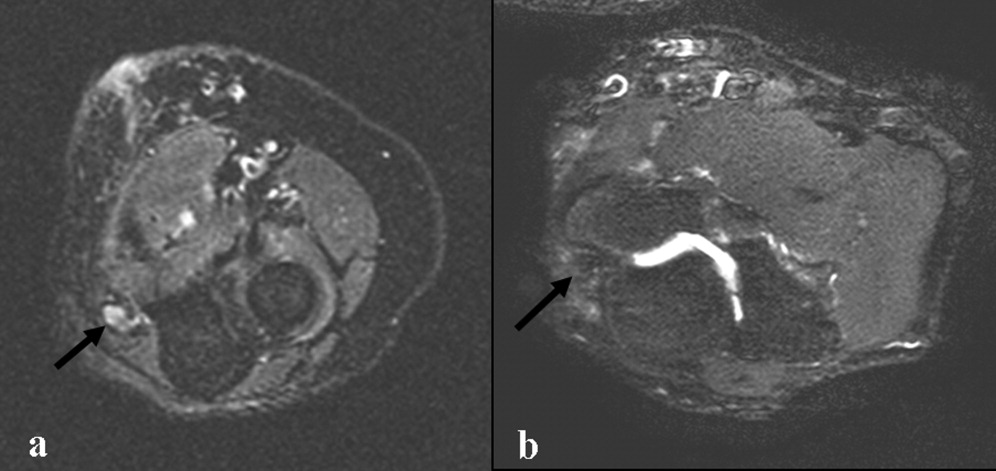
The electrodiagnostic testing is consistent with severe acute axonal injury to the left ulnar, median and radial nerves. This supports a diagnosis of mononeuropathy multiplex with axonal injury. Demyelinating causes are excluded at this point.
The MR neurogram demonstrates nonspecific nerve enlargement, which may be seen in ischemia, neoplastic processes (primary or metastatic), demyelinating disease, or, rarely, amyloidosis. Neoplastic involvement is unlikely in this case given the absence of a compressive mass lesion and the long segmental involvement of both the median and ulnar nerves. Compression from an abscess or hematoma is excluded. Neuralgic amyotrophy does not typically cause nerve enlargement.
Ischemia is the most likely diagnosis. Laboratory evaluation for vasculitis would be reasonable. Vasculitides that could present in this fashion include: polyarteritis nodosa, mixed connective tissue disease, Wegner's granulomatosis, Churg‐Strauss angiitis, Sjogren's, hepatitis C with serum cryoglobulinemia and possibly rheumatoid arthritis. Given the history of fistula placement in the affected limb, vascular sufficiency must be assessed.
Anti‐nuclear antibodies and anti‐neutrophilic cytoplasmic antibodies were negative, and a C‐ reactive protein was 5.9 mg/L (normal range, 0 to 10 mg/L). The erythrocyte sedimentation rate was 32mm/hr (normal range, 0 to 20) and serologies for hepatitis B and C were negative. There was no evidence of serum cryoglobulins.
A modestly elevated sedimentation rate and normal C‐reactive protein argue against a diagnosis of vasculitis. The negative ANA, ANCA, hepatitis serologies and cryoglobulin tests render unlikely the diagnoses of polyarteritis nodosa, Wegner's granulomatosis, Churg‐Strauss angiitis, or hepatitis related cryoglobulinemia. Eosinophilia (present in Churg‐Strauss), ENA (positive in mixed connective tissue disease), anti‐SSA and SSB (positive in Sjogren's) and a rheumatoid factor would round out this evaluation for vasculitis. A left radial sensory nerve biopsy could also be of value in diagnosing vasculitic neuropathy in this patient.
Given the evidence against vasculitis, the possibility of ischemia due to vascular insufficiency is concerning. Two ischemic complications of hemodialysis are known to cause distal multiple mononeuropathies. The first, ischemic monomelic neuropathy syndrome is seen almost exclusively in diabetics. It is characterized by the development of acute pain, weakness of the forearm and hand muscles, and sensory loss within minutes or hours of AV graft placement. Transient occlusion of the blood supply to the nerves of the forearm and hand induces nerve ischemia, but does not cause necrosis of other tissues. The nerve conduction findings in this patient are consistent with ischemic monomelic neuropathy syndrome. The delayed onset of her symptoms, however, makes this diagnosis unlikely.
The second ischemic complication of hemodialysis, and the likelier diagnosis, is vascular steal syndrome. This has a similar clinical and electrodiagnostic presentation to ischemic monomelic neuropathy syndrome, but has a latency period after surgery of days to months. Vascular steal occurs when a reversal of blood flow into the fistula steals flow from the palmar arch arteries and induces ischemia of the vasa nervorum. Vascular studies should be obtained urgently when this diagnosis is considered.
Evaluation of the arteriovenous graft and vascular surgery consultation were sought. Digital photoplethysmography revealed diminished waveforms in all fingers of the left hand. Arterial Doppler evaluation of the left upper extremity confirmed low‐velocity flow in the radial and ulnar arteries and failed to confirm flow in the brachial artery distal to the arteriovenous fistula. The patient underwent an angiogram of the left axillary and brachial arteries. There was normal flow until the level of the arteriovenous fistula but minimal flow distal to the fistula (Fig. 2).
The diminished waveforms on digital photoplethysmography are consistent with poor perfusion distally. The angiogram suggests that the multiple mononeuropathies are a consequence of ischemia from impaired blood flow.
Consulting the vascular surgeons in this setting is essential because restoring adequate blood flow to the affected nerves can prevent further loss of function. Prognosis is dependent on many factors, including the severity of the functional loss and the duration of the symptoms prior to the restoration of blood flow. The patient's severe weakness and substantial muscle atrophy, manifestations of axonal degeneration, imply a poorer prognosis for recovery of function.
Embolization of the arteriovenous fistula was performed by interventional radiology. Post embolization angiograms demonstrated improved peripheral arterial flow (Fig. 3). One day later, the patient's finger flexion and extension improved. She reported mildly decreased dysesthesias and on examination her fingers were warmer to the touch. One month after discharge, her strength continued to be impaired, though improved and she still experienced pain.
COMMENTARY
Hospitalists must be equipped to recognize urgent and potentially reversible causes of neuropathy. The hospitalist should maintain a high index of suspicion for ischemia (either due to vasculitis or vascular compromise), traumatic nerve injury, nerve compression or entrapment, lymphoma or metastatic infiltration, hepatitis C with cryoglobulinemia, Guillain‐Barre syndrome and toxic exposures. Table 1 highlights important causes of mononeuropathy multiplex and summarizes associated findings and indicated diagnostic tests for specific evaluation.
| Diagnosis | Associated features | Specific evaluation |
|---|---|---|
| Axonal neuropathies | ||
| Ischemia (including vascular steal) | Poor arterial pulses, history of vascular surgery | Digital photoplethysmography, Doppler, angiography |
| Nerve compression and trauma | History of traumatic injury, mass, infection/abscess | MR neurography |
| Lymphoma or metastatic infiltration | History of known cancer, weight loss | PET, whole body CT, bone marrow biopsy |
| Vasculitis | Waxing and waning symptoms, association with connective tissue diseases, painful | CRP, ESR, Hepatitis C, cryoglobulins, ANA, ANCA, antibodies to SSA/SSB, ENA, eosinophil count, serum complement, SPEP/UPEP, RF, nerve biopsy |
| Neurosarcoidosis | Hilar lymphadenopathy, chronic cough | Chest CT, ACE, nerve biopsy |
| Lyme | Tick bite, erythema chronicum migrans | Lyme serology |
| Leprosy | Resident of southeast Asia, skin lesions | Skin smear for acid fast bacilli (mycobacterium), nerve biopsy |
| Demyelinating neuropathies | ||
| Lewis‐Sumner syndrome (i.e. asymmetric CIDP) | Relapsing remitting or chronic progressive course, areflexia | Lumbar puncture (increased spinal fluid protein common) |
| Hereditary neuropathy with liability to pressure palsy | Family history, recurrent episodes of entrapment/compression neuropathies | Genetic testing (deletion in the gene for peripheral myelin protein‐22) |
| Multifocal motor neuropathy with conduction block | Multifocal weakness in the distal arms/legs without sensory symptoms | Only motor abnormalities on nerve conduction including conduction block |
Another challenge for hospitalists is efficient evaluation of neuropathy. A systematic framework for creating a differential diagnosis and familiarity with available diagnostic tests is crucial. Hospitalists should be aware of three broad categories of neuropathy: mononeuropathy, polyneuropathy and mononeuropathy multiplex. Electrodiagnostic testing is essential to confirm the involved nerves and distinguishes axonal from demyelinating etiologies. Ultrasound, MR neurogram and, when indicated, nerve biopsy may be useful. Table 2 reviews these diagnostic tools as well as their indications and limitations.
| Test | Indications | Limitations |
|---|---|---|
| Electrodiagnostic Testing7 | Any peripheral neuropathy, muscle or neuromuscular junction disorder | Concomitant disease can reduce accuracy |
| Detects severity, chronicity, axonal v. demyelinating, diffuse v. focal, asymmetric v. symmetric | ||
| Electromyography (EMG) | EMG | EMG |
| Monopolar/concentric needle electrode inserted into the muscle belly | Differentiates axonal v. muscle damage; sensitive for even mild axon degeneration; localizes lesions. | Patient discomfort |
| Evaluates only motor fibers | ||
| Measures action potential at rest vs. during voluntary activation | Does not detect demyelination | |
| Might not be positive in first 21 days of symptoms | ||
| Nerve Conduction Studies (NCS) | NCS | NCS |
| Sensory | High sensitivity to differentiate axon loss from demyelination; localizes lesions. | Certain sensory responses lost with aging |
| Recording electrode placed over sensory nerve | ||
| Sensory nerve stimulated distally | Sensory localizes lesion to proximal vs. distal or to dorsal root ganglion | Less sensitive for mild axonal loss |
| Measures stimulus at proximal site | ||
| Motor | Motor amount of axonal loss | |
| Recording electrode placed over muscle belly | ||
| Motor nerve stimulated proximally | ||
| Measures stimulus at muscle | ||
| Specialized NCS tests | Specialized testing can identify radiculopathy, peripheral neuropathy, myasthenia gravis | |
| Ultrasound8 | ||
| Performed with typical ultrasound equipment | Suspected nerve entrapment | Doesn't show pathologic changes within nerves |
| Clinician must localize lesion for technician and explicitly guide test process | Evidence strongest for evaluation of median and ulnar nerves and Morton's neuroma | Difficult to visualize deep nerves or nerves surrounded by fat |
| Normal nerves appear tubular with linear echoes on a longitudinal scan; honeycomb on transverse scan | Detects lesions, nerve thickening, decreased echogenicity | Small field of view unless reconstructed |
| Results operator dependent | ||
| Less accurate than MRI for tumors | ||
| MRI9, 10 MR neurography | ||
| Standard MRI equipment | Concern regarding entrapment, trauma or mass lesions | Expense |
| Optimizes nerve resolution compared with surrounding tissues | To narrow differential when clinical and electrodiagnostic studies are inconclusive | Time (1560 minutes depending on scan requested) |
| When carpal tunnel syndrome does not respond to conservative management | ||
| Detects mass lesions compressing nerves, nerve enlargement and abnormal signal (neuritis, infiltration), increased signal in denervated muscle groups (once strength is 3 of 5). These changes can be seen as early at 4 days post trauma compared to 23 weeks on EMG. | ||
| Nerve Biopsy11 | ||
| Biopsy a nerve in the region of sensory loss or of a sensory nerve demonstrating electrophysiological abnormalities (decrease risk of adverse effects and to increase the likelihood of diagnosis | Rarely necessary | Painful, often for months |
| Concomitant muscle biopsy increases likelihood of diagnosing vasculitis or sarcoidosis | Use as last resort when evaluation not definitive | Risk of bleeding and infection |
| Greatest yield in multifocal neuropathies, or suspected amyloidotic polyneuropathy, vasculitis, sarcoidosis, lepromatous neuropathy, or rare hereditary disease where no genetic testing exists | ||
| Detects inflammation, amyloid deposits, tumor infiltration | ||
| Commonly targeted nerves include: LE sural, superficial peroneal, UE superficial radial |
Ischemic steal syndrome should be considered when neuropathy develops in a limb subsequent to arterio‐venous access procedures. Any vascular network, including the vertebral, carotid and coronary arteries, is at risk for steal. A feature common to all steal syndromes is the diversion of blood away from its original destination toward a lower pressure alternative. In some cases, this leads to a reversal of arterial flow and ischemia. Ischemic complications from AV access occur in 1‐9% of patients.1 Symptoms of steal can be mild, such as self‐limited dialysis induced pain, coldness and numbness, or severe, including severe pain, sensory and motor loss.2 If vascular compromise is sufficient, gangrene can ensue. Sensory deficits usually precede motor loss and the radial pulse is commonly absent or diminished. Other findings can include pallor of the fingers, muscle atrophy, resorption of the nail bed, and gangrene or ulcerations of the fingers. Risk factors for steal include atherosclerotic disease, female gender, age greater than 60 years, diabetes mellitus, previous surgery on the same arm, and use of the brachial artery as a donor.3 Symptoms of ischemic steal typically present within the first month after surgery, but can also be delayed; there is one report of a patient presenting one year postoperatively.4
Imaging studies such as doppler and angiography can be helpful in diagnosing ischemic steal syndrome. Fistulagrams may reveal a reversal of blood flow in the distal arm and hand, but these are reserved for cases with suspected proximal obstructive arterial disease.5 Vascular imaging studies can be misleading, however, as many patients will have physiologic but asymptomatic reversal of flow. Thus, a functional assessment such as digital plethysmography is recommended, especially in cases where clinical symptoms are vague. Digital pressures less than 60mmHg demonstrated 100% sensitivity and 87% specificity in one case control study of 40 patients.6 Treatment of ischemic steal syndrome is aimed at decreasing flow through the access shunt.
In conclusion, this case highlights the importance of timely and systematic evaluation of peripheral neuropathy in the hospital setting. Neuropathy with rapid progression and high potential for permanent damage necessitates early neurologic, or in this case, vascular consultation. Hospitalists should be facile in evaluating peripheral neuropathies and recognizing the appropriate indications for diagnostic tests and procedures.
- .Upper limb ischemia after vascular access surgery: differential diagnosis and management.Sem Dial2000;13:312–315.
- ,,,,,.Steal syndrome complicating hemodialysis access.Cardiovascular Surg (London, England)1997;5:648–653.
- ,,,,,.Onset of arterial ‘steal’ following proximal angioaccess: immediate and delayed types.Nephrol Dial Transplant2003;18:2387–2390.
- ,,,,.Incidence and characteristics of patients with hand ischemia after hemodialysis access procedure.J Surg Res1998;74:8–10
- ,,,,,.Ischemic steal syndrome: a case series and review of current management.Curr Surg2006;63:130–135.
- ,,,.Use of digital pressure measurements for the diagnosis of AV access‐induced hand ischemia.Vasc Med2006;11:227–231.
- ,.Electrodiagnostic testing of nerves and muscles: when, why, and how to order.Cleve Clin J Med2005;72:37–48.
- ,.High‐resolution sonography of the peripheral nervous system—a review of the literature.Eur J Neurol2004;11:305–314.
- ,,, et al.Role of magnetic resonance imaging in entrapment and compressive neuropathy‐what, where, and how to see the peripheral nerves on the musculoskeletal magnetic resonance image: part 2. Upper extremity.Eur Radiol2007;17:509–522.
- ,,,,,.The utility of magnetic resonance imaging in evaluating peripheral nerve disorders.Muscle Nerve2002;25:314–331.
- .Indications and usefulness of nerve biopsy.Arch Neurol2002;59:1532–1535.
A 60 year‐old woman with advanced kidney disease presented with one month of progressively worsening, sharp burning pain and decreased sensation in her left hand. Cold air exacerbated the pain. She noted decreasing ability to utilize her left fingers, a weakened grip and that the muscles in her hand looked smaller.
Localized sensory and motor symptoms in a discrete region of a single limb suggest neuropathy. The lack of symptoms in the face or ipsilateral lower extremity would dissuade a clinician from considering central etiologies; the presence of neuropathic pain is uncommon for cortical lesions. The involvement of motor and sensory nerves indicates peripheral nerve involvement.
The general approach to patients with peripheral neuropathy begins with identifying the neuropathy as a mononeuropathy (involving a single nerve), a polyneuropathy (symmetric involvement of multiple nerves) or a mononeuropathy multiplex (asymmetric involvement of multiple nerves). The patient, in this case, described subacute neuropathic pain, sensory loss, and weakness in her left hand in a distribution consistent with mononeuropathy or mononeuropathy multiplex.
This patient could have carpal tunnel syndrome given its prevalence in patients with advanced renal disease. The differential diagnosis is broad, however, and includes ulnar mononeuropathy, nerve ischemia due to vasculitis or vasculopathy, lower cervical radiculopathy (though the patient does not describe neck or radicular pain), lower brachial plexopathy, and complex regional pain syndrome.
The patient was diagnosed with advanced kidney disease one year ago when biopsy revealed focal segmental glomerulosclerosis secondary to lithium. Since her diagnosis, two grafts were placed in the left upper arm in anticipation of dialysis: the first, placed seven months prior to this admission, failed to mature; the second, placed one month prior to this admission, was complicated by bleeding at the fistula site and was not yet mature. Prior to this admission she had not required hemodialysis. Her past history included hypertension, dyslipidemia, hypothyroidism, secondary hyperparathyroidism, a remote history of cervical cancer (stage unknown, recent PAP smear negative), microcytosis and schizoaffective disorder. Her medications were furosemide, amlodipine, lisinopril, atenolol, atorvastatin, pantoprazole, olanzapine, levothyroxine, iron, darbepoetin, sevelamer, multivitamin and docusate.
Given the history of procedures in the left arm one should consider ischemic injury to the left median nerve. Other local complications could include compressive lesions such as an abscess or hematoma or direct nerve injury from the procedure. Carpal tunnel syndrome remains high on the differential due to its prevalence and because renal failure and hypothyroidism increase the risk of carpal tunnel syndrome.
A careful physical examination would localize the nerve or nerves involved. Examination findings that would be consistent with carpal tunnel syndrome include sensory loss in the distribution of the left median nerve, weakness of muscles innervated by the median nerve, including the abductor pollicus brevis and opponens muscles, and a Tinels and Phalens sign of the left wrist. A proximal median neuropathy resulting from ischemia or compression might also involve median‐innervated forearm muscle such as the pronator teres (forearm pronation) and flexor carpi radialis (hand flexion and abduction) muscles. Complex regional pain syndrome can be seen after a traumatic injury or surgery and is typified by severe neuropathic pain in a limb, often in combination with trophic changes in the affected extremity. A cervical radiculopathy would affect muscles supplied by the injured nerve root. For example, a C8 radiculopathy would affect all of the intrinsic hand muscles, the wrist and finger extensors, and the triceps brachii. A lower brachial plexopathy would present similarly to a lower cervical radiculopathy on clinical examination; electrodiagnostic evaluation would be necessary to distinguish these two disorders.
The patient appeared fatigued and her left hand was wrapped in blankets. Her vital signs were stable. There was no thyroid enlargement or lymphadenopathy. There was marked thenar, hypothenar and forearm atrophy on the left. Strength testing of her left hand demonstrated a grip strength of 2/5, finger extension and interosseous strength of 1/5, left wrist flexion and extension of 3/5; left biceps and triceps were 5/5. Sensation was mildly decreased to light touch, temperature, pain and proprioception throughout the left hand. Strength and sensation were intact in the right upper and bilateral lower extremities. Reflexes were 2+ throughout. The left radial pulse was diminished compared to the left ulnar that was 1+. The left hand was dry and cool. Laboratory evaluation revealed an elevated white blood cell count of 15,300/mm3, a hematocrit of 33 percent, mean corpuscular volume of 73 fL, and a normal platelet count. Electrolytes were consistent with advanced renal disease. The thyroid stimulating hormone level was normal.
The patient's examination reveals an injury to the sensory and motor components of the left ulnar, median, and distal radial nerves. The volar forearm wasting suggests proximal median motor nerve injury in the forearm. Because the triceps is spared, the weakness of finger and wrist extension implies distal radial motor nerve injury. The interosseous weakness is consistent with injury to the ulnar motor nerve. Weakness of grip and wrist flexion is less specific and it may be explained by injury to either the median or ulnar motor nerves. The diffuse sensory loss over the palmar and dorsal aspect of the left hand is consistent with neuropathic injury to the left median, ulnar, and radial sensory nerves. The preserved deep tendon reflexes are controlled by the musculocutaneous nerve (the biceps reflex) and branches arising from the proximal radial nerve (the triceps and brachioradialis reflexes), both of which are proximal to the apparent level of neuropathic insult.
Carpal tunnel syndrome is excluded since abnormalities extend beyond the median nerve distribution. The presentation is consistent with a mononeuropathy multiplex. Axonal etiologies of mononeuropathy multiplexincluding vasculitis, ischemia, neoplastic infiltration, and infectious etiologies such as Lyme diseaseare more common than demyelinating causes. Vasculitic neuropathy commonly involves the lower extremities and may have systemic symptoms, not present in this case. Neoplasm or other compressive lesions such as hematoma could explain these findings. Abscess must be considered given the leukocytosis. A lower brachial plexopathy, technically a mononeuropathy multiplex involving the proximal arm at the level of the brachial plexus, is also in the differential diagnosis. Another axonal disorder to consider would be neuralgic amyotrophy, an idiopathic form of acute brachial plexopathy associated with pain that is a complication of surgery that typically presents within a few hours to weeks of the procedure.
Demyelinating causes of mononeuropathy multiplex are less likely and include a variant of chronic inflammatory demyelinating polyneuropathy (Lewis‐Sumner syndrome) and hereditary neuropathy with liability to pressure palsies. Both processes are typically indolent and usually not painful, though the latter may present with fulminant numbness and weakness.
Nerve conduction and needle electromyography of the arm and cervical paraspinal muscles would differentiate axonal degeneration from demyelination. It would identify the affected nerves and any nerve root involvement. Given the concern for abscess or hematoma, MR neurography focused on the surgical site would also be important.
Electromyography and nerve conduction velocities demonstrated severe axonal loss of the left median, ulnar and distal radial sensory nerves consistent with acute denervation. A magnetic resonance neurogram following the course of these nerves revealed enlarged ulnar and median nerves with abnormal signal, but no compressive lesion (Fig. 1).
The electrodiagnostic testing is consistent with severe acute axonal injury to the left ulnar, median and radial nerves. This supports a diagnosis of mononeuropathy multiplex with axonal injury. Demyelinating causes are excluded at this point.
The MR neurogram demonstrates nonspecific nerve enlargement, which may be seen in ischemia, neoplastic processes (primary or metastatic), demyelinating disease, or, rarely, amyloidosis. Neoplastic involvement is unlikely in this case given the absence of a compressive mass lesion and the long segmental involvement of both the median and ulnar nerves. Compression from an abscess or hematoma is excluded. Neuralgic amyotrophy does not typically cause nerve enlargement.
Ischemia is the most likely diagnosis. Laboratory evaluation for vasculitis would be reasonable. Vasculitides that could present in this fashion include: polyarteritis nodosa, mixed connective tissue disease, Wegner's granulomatosis, Churg‐Strauss angiitis, Sjogren's, hepatitis C with serum cryoglobulinemia and possibly rheumatoid arthritis. Given the history of fistula placement in the affected limb, vascular sufficiency must be assessed.
Anti‐nuclear antibodies and anti‐neutrophilic cytoplasmic antibodies were negative, and a C‐ reactive protein was 5.9 mg/L (normal range, 0 to 10 mg/L). The erythrocyte sedimentation rate was 32mm/hr (normal range, 0 to 20) and serologies for hepatitis B and C were negative. There was no evidence of serum cryoglobulins.
A modestly elevated sedimentation rate and normal C‐reactive protein argue against a diagnosis of vasculitis. The negative ANA, ANCA, hepatitis serologies and cryoglobulin tests render unlikely the diagnoses of polyarteritis nodosa, Wegner's granulomatosis, Churg‐Strauss angiitis, or hepatitis related cryoglobulinemia. Eosinophilia (present in Churg‐Strauss), ENA (positive in mixed connective tissue disease), anti‐SSA and SSB (positive in Sjogren's) and a rheumatoid factor would round out this evaluation for vasculitis. A left radial sensory nerve biopsy could also be of value in diagnosing vasculitic neuropathy in this patient.
Given the evidence against vasculitis, the possibility of ischemia due to vascular insufficiency is concerning. Two ischemic complications of hemodialysis are known to cause distal multiple mononeuropathies. The first, ischemic monomelic neuropathy syndrome is seen almost exclusively in diabetics. It is characterized by the development of acute pain, weakness of the forearm and hand muscles, and sensory loss within minutes or hours of AV graft placement. Transient occlusion of the blood supply to the nerves of the forearm and hand induces nerve ischemia, but does not cause necrosis of other tissues. The nerve conduction findings in this patient are consistent with ischemic monomelic neuropathy syndrome. The delayed onset of her symptoms, however, makes this diagnosis unlikely.
The second ischemic complication of hemodialysis, and the likelier diagnosis, is vascular steal syndrome. This has a similar clinical and electrodiagnostic presentation to ischemic monomelic neuropathy syndrome, but has a latency period after surgery of days to months. Vascular steal occurs when a reversal of blood flow into the fistula steals flow from the palmar arch arteries and induces ischemia of the vasa nervorum. Vascular studies should be obtained urgently when this diagnosis is considered.
Evaluation of the arteriovenous graft and vascular surgery consultation were sought. Digital photoplethysmography revealed diminished waveforms in all fingers of the left hand. Arterial Doppler evaluation of the left upper extremity confirmed low‐velocity flow in the radial and ulnar arteries and failed to confirm flow in the brachial artery distal to the arteriovenous fistula. The patient underwent an angiogram of the left axillary and brachial arteries. There was normal flow until the level of the arteriovenous fistula but minimal flow distal to the fistula (Fig. 2).
The diminished waveforms on digital photoplethysmography are consistent with poor perfusion distally. The angiogram suggests that the multiple mononeuropathies are a consequence of ischemia from impaired blood flow.
Consulting the vascular surgeons in this setting is essential because restoring adequate blood flow to the affected nerves can prevent further loss of function. Prognosis is dependent on many factors, including the severity of the functional loss and the duration of the symptoms prior to the restoration of blood flow. The patient's severe weakness and substantial muscle atrophy, manifestations of axonal degeneration, imply a poorer prognosis for recovery of function.
Embolization of the arteriovenous fistula was performed by interventional radiology. Post embolization angiograms demonstrated improved peripheral arterial flow (Fig. 3). One day later, the patient's finger flexion and extension improved. She reported mildly decreased dysesthesias and on examination her fingers were warmer to the touch. One month after discharge, her strength continued to be impaired, though improved and she still experienced pain.
COMMENTARY
Hospitalists must be equipped to recognize urgent and potentially reversible causes of neuropathy. The hospitalist should maintain a high index of suspicion for ischemia (either due to vasculitis or vascular compromise), traumatic nerve injury, nerve compression or entrapment, lymphoma or metastatic infiltration, hepatitis C with cryoglobulinemia, Guillain‐Barre syndrome and toxic exposures. Table 1 highlights important causes of mononeuropathy multiplex and summarizes associated findings and indicated diagnostic tests for specific evaluation.
| Diagnosis | Associated features | Specific evaluation |
|---|---|---|
| Axonal neuropathies | ||
| Ischemia (including vascular steal) | Poor arterial pulses, history of vascular surgery | Digital photoplethysmography, Doppler, angiography |
| Nerve compression and trauma | History of traumatic injury, mass, infection/abscess | MR neurography |
| Lymphoma or metastatic infiltration | History of known cancer, weight loss | PET, whole body CT, bone marrow biopsy |
| Vasculitis | Waxing and waning symptoms, association with connective tissue diseases, painful | CRP, ESR, Hepatitis C, cryoglobulins, ANA, ANCA, antibodies to SSA/SSB, ENA, eosinophil count, serum complement, SPEP/UPEP, RF, nerve biopsy |
| Neurosarcoidosis | Hilar lymphadenopathy, chronic cough | Chest CT, ACE, nerve biopsy |
| Lyme | Tick bite, erythema chronicum migrans | Lyme serology |
| Leprosy | Resident of southeast Asia, skin lesions | Skin smear for acid fast bacilli (mycobacterium), nerve biopsy |
| Demyelinating neuropathies | ||
| Lewis‐Sumner syndrome (i.e. asymmetric CIDP) | Relapsing remitting or chronic progressive course, areflexia | Lumbar puncture (increased spinal fluid protein common) |
| Hereditary neuropathy with liability to pressure palsy | Family history, recurrent episodes of entrapment/compression neuropathies | Genetic testing (deletion in the gene for peripheral myelin protein‐22) |
| Multifocal motor neuropathy with conduction block | Multifocal weakness in the distal arms/legs without sensory symptoms | Only motor abnormalities on nerve conduction including conduction block |
Another challenge for hospitalists is efficient evaluation of neuropathy. A systematic framework for creating a differential diagnosis and familiarity with available diagnostic tests is crucial. Hospitalists should be aware of three broad categories of neuropathy: mononeuropathy, polyneuropathy and mononeuropathy multiplex. Electrodiagnostic testing is essential to confirm the involved nerves and distinguishes axonal from demyelinating etiologies. Ultrasound, MR neurogram and, when indicated, nerve biopsy may be useful. Table 2 reviews these diagnostic tools as well as their indications and limitations.
| Test | Indications | Limitations |
|---|---|---|
| Electrodiagnostic Testing7 | Any peripheral neuropathy, muscle or neuromuscular junction disorder | Concomitant disease can reduce accuracy |
| Detects severity, chronicity, axonal v. demyelinating, diffuse v. focal, asymmetric v. symmetric | ||
| Electromyography (EMG) | EMG | EMG |
| Monopolar/concentric needle electrode inserted into the muscle belly | Differentiates axonal v. muscle damage; sensitive for even mild axon degeneration; localizes lesions. | Patient discomfort |
| Evaluates only motor fibers | ||
| Measures action potential at rest vs. during voluntary activation | Does not detect demyelination | |
| Might not be positive in first 21 days of symptoms | ||
| Nerve Conduction Studies (NCS) | NCS | NCS |
| Sensory | High sensitivity to differentiate axon loss from demyelination; localizes lesions. | Certain sensory responses lost with aging |
| Recording electrode placed over sensory nerve | ||
| Sensory nerve stimulated distally | Sensory localizes lesion to proximal vs. distal or to dorsal root ganglion | Less sensitive for mild axonal loss |
| Measures stimulus at proximal site | ||
| Motor | Motor amount of axonal loss | |
| Recording electrode placed over muscle belly | ||
| Motor nerve stimulated proximally | ||
| Measures stimulus at muscle | ||
| Specialized NCS tests | Specialized testing can identify radiculopathy, peripheral neuropathy, myasthenia gravis | |
| Ultrasound8 | ||
| Performed with typical ultrasound equipment | Suspected nerve entrapment | Doesn't show pathologic changes within nerves |
| Clinician must localize lesion for technician and explicitly guide test process | Evidence strongest for evaluation of median and ulnar nerves and Morton's neuroma | Difficult to visualize deep nerves or nerves surrounded by fat |
| Normal nerves appear tubular with linear echoes on a longitudinal scan; honeycomb on transverse scan | Detects lesions, nerve thickening, decreased echogenicity | Small field of view unless reconstructed |
| Results operator dependent | ||
| Less accurate than MRI for tumors | ||
| MRI9, 10 MR neurography | ||
| Standard MRI equipment | Concern regarding entrapment, trauma or mass lesions | Expense |
| Optimizes nerve resolution compared with surrounding tissues | To narrow differential when clinical and electrodiagnostic studies are inconclusive | Time (1560 minutes depending on scan requested) |
| When carpal tunnel syndrome does not respond to conservative management | ||
| Detects mass lesions compressing nerves, nerve enlargement and abnormal signal (neuritis, infiltration), increased signal in denervated muscle groups (once strength is 3 of 5). These changes can be seen as early at 4 days post trauma compared to 23 weeks on EMG. | ||
| Nerve Biopsy11 | ||
| Biopsy a nerve in the region of sensory loss or of a sensory nerve demonstrating electrophysiological abnormalities (decrease risk of adverse effects and to increase the likelihood of diagnosis | Rarely necessary | Painful, often for months |
| Concomitant muscle biopsy increases likelihood of diagnosing vasculitis or sarcoidosis | Use as last resort when evaluation not definitive | Risk of bleeding and infection |
| Greatest yield in multifocal neuropathies, or suspected amyloidotic polyneuropathy, vasculitis, sarcoidosis, lepromatous neuropathy, or rare hereditary disease where no genetic testing exists | ||
| Detects inflammation, amyloid deposits, tumor infiltration | ||
| Commonly targeted nerves include: LE sural, superficial peroneal, UE superficial radial |
Ischemic steal syndrome should be considered when neuropathy develops in a limb subsequent to arterio‐venous access procedures. Any vascular network, including the vertebral, carotid and coronary arteries, is at risk for steal. A feature common to all steal syndromes is the diversion of blood away from its original destination toward a lower pressure alternative. In some cases, this leads to a reversal of arterial flow and ischemia. Ischemic complications from AV access occur in 1‐9% of patients.1 Symptoms of steal can be mild, such as self‐limited dialysis induced pain, coldness and numbness, or severe, including severe pain, sensory and motor loss.2 If vascular compromise is sufficient, gangrene can ensue. Sensory deficits usually precede motor loss and the radial pulse is commonly absent or diminished. Other findings can include pallor of the fingers, muscle atrophy, resorption of the nail bed, and gangrene or ulcerations of the fingers. Risk factors for steal include atherosclerotic disease, female gender, age greater than 60 years, diabetes mellitus, previous surgery on the same arm, and use of the brachial artery as a donor.3 Symptoms of ischemic steal typically present within the first month after surgery, but can also be delayed; there is one report of a patient presenting one year postoperatively.4
Imaging studies such as doppler and angiography can be helpful in diagnosing ischemic steal syndrome. Fistulagrams may reveal a reversal of blood flow in the distal arm and hand, but these are reserved for cases with suspected proximal obstructive arterial disease.5 Vascular imaging studies can be misleading, however, as many patients will have physiologic but asymptomatic reversal of flow. Thus, a functional assessment such as digital plethysmography is recommended, especially in cases where clinical symptoms are vague. Digital pressures less than 60mmHg demonstrated 100% sensitivity and 87% specificity in one case control study of 40 patients.6 Treatment of ischemic steal syndrome is aimed at decreasing flow through the access shunt.
In conclusion, this case highlights the importance of timely and systematic evaluation of peripheral neuropathy in the hospital setting. Neuropathy with rapid progression and high potential for permanent damage necessitates early neurologic, or in this case, vascular consultation. Hospitalists should be facile in evaluating peripheral neuropathies and recognizing the appropriate indications for diagnostic tests and procedures.
A 60 year‐old woman with advanced kidney disease presented with one month of progressively worsening, sharp burning pain and decreased sensation in her left hand. Cold air exacerbated the pain. She noted decreasing ability to utilize her left fingers, a weakened grip and that the muscles in her hand looked smaller.
Localized sensory and motor symptoms in a discrete region of a single limb suggest neuropathy. The lack of symptoms in the face or ipsilateral lower extremity would dissuade a clinician from considering central etiologies; the presence of neuropathic pain is uncommon for cortical lesions. The involvement of motor and sensory nerves indicates peripheral nerve involvement.
The general approach to patients with peripheral neuropathy begins with identifying the neuropathy as a mononeuropathy (involving a single nerve), a polyneuropathy (symmetric involvement of multiple nerves) or a mononeuropathy multiplex (asymmetric involvement of multiple nerves). The patient, in this case, described subacute neuropathic pain, sensory loss, and weakness in her left hand in a distribution consistent with mononeuropathy or mononeuropathy multiplex.
This patient could have carpal tunnel syndrome given its prevalence in patients with advanced renal disease. The differential diagnosis is broad, however, and includes ulnar mononeuropathy, nerve ischemia due to vasculitis or vasculopathy, lower cervical radiculopathy (though the patient does not describe neck or radicular pain), lower brachial plexopathy, and complex regional pain syndrome.
The patient was diagnosed with advanced kidney disease one year ago when biopsy revealed focal segmental glomerulosclerosis secondary to lithium. Since her diagnosis, two grafts were placed in the left upper arm in anticipation of dialysis: the first, placed seven months prior to this admission, failed to mature; the second, placed one month prior to this admission, was complicated by bleeding at the fistula site and was not yet mature. Prior to this admission she had not required hemodialysis. Her past history included hypertension, dyslipidemia, hypothyroidism, secondary hyperparathyroidism, a remote history of cervical cancer (stage unknown, recent PAP smear negative), microcytosis and schizoaffective disorder. Her medications were furosemide, amlodipine, lisinopril, atenolol, atorvastatin, pantoprazole, olanzapine, levothyroxine, iron, darbepoetin, sevelamer, multivitamin and docusate.
Given the history of procedures in the left arm one should consider ischemic injury to the left median nerve. Other local complications could include compressive lesions such as an abscess or hematoma or direct nerve injury from the procedure. Carpal tunnel syndrome remains high on the differential due to its prevalence and because renal failure and hypothyroidism increase the risk of carpal tunnel syndrome.
A careful physical examination would localize the nerve or nerves involved. Examination findings that would be consistent with carpal tunnel syndrome include sensory loss in the distribution of the left median nerve, weakness of muscles innervated by the median nerve, including the abductor pollicus brevis and opponens muscles, and a Tinels and Phalens sign of the left wrist. A proximal median neuropathy resulting from ischemia or compression might also involve median‐innervated forearm muscle such as the pronator teres (forearm pronation) and flexor carpi radialis (hand flexion and abduction) muscles. Complex regional pain syndrome can be seen after a traumatic injury or surgery and is typified by severe neuropathic pain in a limb, often in combination with trophic changes in the affected extremity. A cervical radiculopathy would affect muscles supplied by the injured nerve root. For example, a C8 radiculopathy would affect all of the intrinsic hand muscles, the wrist and finger extensors, and the triceps brachii. A lower brachial plexopathy would present similarly to a lower cervical radiculopathy on clinical examination; electrodiagnostic evaluation would be necessary to distinguish these two disorders.
The patient appeared fatigued and her left hand was wrapped in blankets. Her vital signs were stable. There was no thyroid enlargement or lymphadenopathy. There was marked thenar, hypothenar and forearm atrophy on the left. Strength testing of her left hand demonstrated a grip strength of 2/5, finger extension and interosseous strength of 1/5, left wrist flexion and extension of 3/5; left biceps and triceps were 5/5. Sensation was mildly decreased to light touch, temperature, pain and proprioception throughout the left hand. Strength and sensation were intact in the right upper and bilateral lower extremities. Reflexes were 2+ throughout. The left radial pulse was diminished compared to the left ulnar that was 1+. The left hand was dry and cool. Laboratory evaluation revealed an elevated white blood cell count of 15,300/mm3, a hematocrit of 33 percent, mean corpuscular volume of 73 fL, and a normal platelet count. Electrolytes were consistent with advanced renal disease. The thyroid stimulating hormone level was normal.
The patient's examination reveals an injury to the sensory and motor components of the left ulnar, median, and distal radial nerves. The volar forearm wasting suggests proximal median motor nerve injury in the forearm. Because the triceps is spared, the weakness of finger and wrist extension implies distal radial motor nerve injury. The interosseous weakness is consistent with injury to the ulnar motor nerve. Weakness of grip and wrist flexion is less specific and it may be explained by injury to either the median or ulnar motor nerves. The diffuse sensory loss over the palmar and dorsal aspect of the left hand is consistent with neuropathic injury to the left median, ulnar, and radial sensory nerves. The preserved deep tendon reflexes are controlled by the musculocutaneous nerve (the biceps reflex) and branches arising from the proximal radial nerve (the triceps and brachioradialis reflexes), both of which are proximal to the apparent level of neuropathic insult.
Carpal tunnel syndrome is excluded since abnormalities extend beyond the median nerve distribution. The presentation is consistent with a mononeuropathy multiplex. Axonal etiologies of mononeuropathy multiplexincluding vasculitis, ischemia, neoplastic infiltration, and infectious etiologies such as Lyme diseaseare more common than demyelinating causes. Vasculitic neuropathy commonly involves the lower extremities and may have systemic symptoms, not present in this case. Neoplasm or other compressive lesions such as hematoma could explain these findings. Abscess must be considered given the leukocytosis. A lower brachial plexopathy, technically a mononeuropathy multiplex involving the proximal arm at the level of the brachial plexus, is also in the differential diagnosis. Another axonal disorder to consider would be neuralgic amyotrophy, an idiopathic form of acute brachial plexopathy associated with pain that is a complication of surgery that typically presents within a few hours to weeks of the procedure.
Demyelinating causes of mononeuropathy multiplex are less likely and include a variant of chronic inflammatory demyelinating polyneuropathy (Lewis‐Sumner syndrome) and hereditary neuropathy with liability to pressure palsies. Both processes are typically indolent and usually not painful, though the latter may present with fulminant numbness and weakness.
Nerve conduction and needle electromyography of the arm and cervical paraspinal muscles would differentiate axonal degeneration from demyelination. It would identify the affected nerves and any nerve root involvement. Given the concern for abscess or hematoma, MR neurography focused on the surgical site would also be important.
Electromyography and nerve conduction velocities demonstrated severe axonal loss of the left median, ulnar and distal radial sensory nerves consistent with acute denervation. A magnetic resonance neurogram following the course of these nerves revealed enlarged ulnar and median nerves with abnormal signal, but no compressive lesion (Fig. 1).
The electrodiagnostic testing is consistent with severe acute axonal injury to the left ulnar, median and radial nerves. This supports a diagnosis of mononeuropathy multiplex with axonal injury. Demyelinating causes are excluded at this point.
The MR neurogram demonstrates nonspecific nerve enlargement, which may be seen in ischemia, neoplastic processes (primary or metastatic), demyelinating disease, or, rarely, amyloidosis. Neoplastic involvement is unlikely in this case given the absence of a compressive mass lesion and the long segmental involvement of both the median and ulnar nerves. Compression from an abscess or hematoma is excluded. Neuralgic amyotrophy does not typically cause nerve enlargement.
Ischemia is the most likely diagnosis. Laboratory evaluation for vasculitis would be reasonable. Vasculitides that could present in this fashion include: polyarteritis nodosa, mixed connective tissue disease, Wegner's granulomatosis, Churg‐Strauss angiitis, Sjogren's, hepatitis C with serum cryoglobulinemia and possibly rheumatoid arthritis. Given the history of fistula placement in the affected limb, vascular sufficiency must be assessed.
Anti‐nuclear antibodies and anti‐neutrophilic cytoplasmic antibodies were negative, and a C‐ reactive protein was 5.9 mg/L (normal range, 0 to 10 mg/L). The erythrocyte sedimentation rate was 32mm/hr (normal range, 0 to 20) and serologies for hepatitis B and C were negative. There was no evidence of serum cryoglobulins.
A modestly elevated sedimentation rate and normal C‐reactive protein argue against a diagnosis of vasculitis. The negative ANA, ANCA, hepatitis serologies and cryoglobulin tests render unlikely the diagnoses of polyarteritis nodosa, Wegner's granulomatosis, Churg‐Strauss angiitis, or hepatitis related cryoglobulinemia. Eosinophilia (present in Churg‐Strauss), ENA (positive in mixed connective tissue disease), anti‐SSA and SSB (positive in Sjogren's) and a rheumatoid factor would round out this evaluation for vasculitis. A left radial sensory nerve biopsy could also be of value in diagnosing vasculitic neuropathy in this patient.
Given the evidence against vasculitis, the possibility of ischemia due to vascular insufficiency is concerning. Two ischemic complications of hemodialysis are known to cause distal multiple mononeuropathies. The first, ischemic monomelic neuropathy syndrome is seen almost exclusively in diabetics. It is characterized by the development of acute pain, weakness of the forearm and hand muscles, and sensory loss within minutes or hours of AV graft placement. Transient occlusion of the blood supply to the nerves of the forearm and hand induces nerve ischemia, but does not cause necrosis of other tissues. The nerve conduction findings in this patient are consistent with ischemic monomelic neuropathy syndrome. The delayed onset of her symptoms, however, makes this diagnosis unlikely.
The second ischemic complication of hemodialysis, and the likelier diagnosis, is vascular steal syndrome. This has a similar clinical and electrodiagnostic presentation to ischemic monomelic neuropathy syndrome, but has a latency period after surgery of days to months. Vascular steal occurs when a reversal of blood flow into the fistula steals flow from the palmar arch arteries and induces ischemia of the vasa nervorum. Vascular studies should be obtained urgently when this diagnosis is considered.
Evaluation of the arteriovenous graft and vascular surgery consultation were sought. Digital photoplethysmography revealed diminished waveforms in all fingers of the left hand. Arterial Doppler evaluation of the left upper extremity confirmed low‐velocity flow in the radial and ulnar arteries and failed to confirm flow in the brachial artery distal to the arteriovenous fistula. The patient underwent an angiogram of the left axillary and brachial arteries. There was normal flow until the level of the arteriovenous fistula but minimal flow distal to the fistula (Fig. 2).
The diminished waveforms on digital photoplethysmography are consistent with poor perfusion distally. The angiogram suggests that the multiple mononeuropathies are a consequence of ischemia from impaired blood flow.
Consulting the vascular surgeons in this setting is essential because restoring adequate blood flow to the affected nerves can prevent further loss of function. Prognosis is dependent on many factors, including the severity of the functional loss and the duration of the symptoms prior to the restoration of blood flow. The patient's severe weakness and substantial muscle atrophy, manifestations of axonal degeneration, imply a poorer prognosis for recovery of function.
Embolization of the arteriovenous fistula was performed by interventional radiology. Post embolization angiograms demonstrated improved peripheral arterial flow (Fig. 3). One day later, the patient's finger flexion and extension improved. She reported mildly decreased dysesthesias and on examination her fingers were warmer to the touch. One month after discharge, her strength continued to be impaired, though improved and she still experienced pain.
COMMENTARY
Hospitalists must be equipped to recognize urgent and potentially reversible causes of neuropathy. The hospitalist should maintain a high index of suspicion for ischemia (either due to vasculitis or vascular compromise), traumatic nerve injury, nerve compression or entrapment, lymphoma or metastatic infiltration, hepatitis C with cryoglobulinemia, Guillain‐Barre syndrome and toxic exposures. Table 1 highlights important causes of mononeuropathy multiplex and summarizes associated findings and indicated diagnostic tests for specific evaluation.
| Diagnosis | Associated features | Specific evaluation |
|---|---|---|
| Axonal neuropathies | ||
| Ischemia (including vascular steal) | Poor arterial pulses, history of vascular surgery | Digital photoplethysmography, Doppler, angiography |
| Nerve compression and trauma | History of traumatic injury, mass, infection/abscess | MR neurography |
| Lymphoma or metastatic infiltration | History of known cancer, weight loss | PET, whole body CT, bone marrow biopsy |
| Vasculitis | Waxing and waning symptoms, association with connective tissue diseases, painful | CRP, ESR, Hepatitis C, cryoglobulins, ANA, ANCA, antibodies to SSA/SSB, ENA, eosinophil count, serum complement, SPEP/UPEP, RF, nerve biopsy |
| Neurosarcoidosis | Hilar lymphadenopathy, chronic cough | Chest CT, ACE, nerve biopsy |
| Lyme | Tick bite, erythema chronicum migrans | Lyme serology |
| Leprosy | Resident of southeast Asia, skin lesions | Skin smear for acid fast bacilli (mycobacterium), nerve biopsy |
| Demyelinating neuropathies | ||
| Lewis‐Sumner syndrome (i.e. asymmetric CIDP) | Relapsing remitting or chronic progressive course, areflexia | Lumbar puncture (increased spinal fluid protein common) |
| Hereditary neuropathy with liability to pressure palsy | Family history, recurrent episodes of entrapment/compression neuropathies | Genetic testing (deletion in the gene for peripheral myelin protein‐22) |
| Multifocal motor neuropathy with conduction block | Multifocal weakness in the distal arms/legs without sensory symptoms | Only motor abnormalities on nerve conduction including conduction block |
Another challenge for hospitalists is efficient evaluation of neuropathy. A systematic framework for creating a differential diagnosis and familiarity with available diagnostic tests is crucial. Hospitalists should be aware of three broad categories of neuropathy: mononeuropathy, polyneuropathy and mononeuropathy multiplex. Electrodiagnostic testing is essential to confirm the involved nerves and distinguishes axonal from demyelinating etiologies. Ultrasound, MR neurogram and, when indicated, nerve biopsy may be useful. Table 2 reviews these diagnostic tools as well as their indications and limitations.
| Test | Indications | Limitations |
|---|---|---|
| Electrodiagnostic Testing7 | Any peripheral neuropathy, muscle or neuromuscular junction disorder | Concomitant disease can reduce accuracy |
| Detects severity, chronicity, axonal v. demyelinating, diffuse v. focal, asymmetric v. symmetric | ||
| Electromyography (EMG) | EMG | EMG |
| Monopolar/concentric needle electrode inserted into the muscle belly | Differentiates axonal v. muscle damage; sensitive for even mild axon degeneration; localizes lesions. | Patient discomfort |
| Evaluates only motor fibers | ||
| Measures action potential at rest vs. during voluntary activation | Does not detect demyelination | |
| Might not be positive in first 21 days of symptoms | ||
| Nerve Conduction Studies (NCS) | NCS | NCS |
| Sensory | High sensitivity to differentiate axon loss from demyelination; localizes lesions. | Certain sensory responses lost with aging |
| Recording electrode placed over sensory nerve | ||
| Sensory nerve stimulated distally | Sensory localizes lesion to proximal vs. distal or to dorsal root ganglion | Less sensitive for mild axonal loss |
| Measures stimulus at proximal site | ||
| Motor | Motor amount of axonal loss | |
| Recording electrode placed over muscle belly | ||
| Motor nerve stimulated proximally | ||
| Measures stimulus at muscle | ||
| Specialized NCS tests | Specialized testing can identify radiculopathy, peripheral neuropathy, myasthenia gravis | |
| Ultrasound8 | ||
| Performed with typical ultrasound equipment | Suspected nerve entrapment | Doesn't show pathologic changes within nerves |
| Clinician must localize lesion for technician and explicitly guide test process | Evidence strongest for evaluation of median and ulnar nerves and Morton's neuroma | Difficult to visualize deep nerves or nerves surrounded by fat |
| Normal nerves appear tubular with linear echoes on a longitudinal scan; honeycomb on transverse scan | Detects lesions, nerve thickening, decreased echogenicity | Small field of view unless reconstructed |
| Results operator dependent | ||
| Less accurate than MRI for tumors | ||
| MRI9, 10 MR neurography | ||
| Standard MRI equipment | Concern regarding entrapment, trauma or mass lesions | Expense |
| Optimizes nerve resolution compared with surrounding tissues | To narrow differential when clinical and electrodiagnostic studies are inconclusive | Time (1560 minutes depending on scan requested) |
| When carpal tunnel syndrome does not respond to conservative management | ||
| Detects mass lesions compressing nerves, nerve enlargement and abnormal signal (neuritis, infiltration), increased signal in denervated muscle groups (once strength is 3 of 5). These changes can be seen as early at 4 days post trauma compared to 23 weeks on EMG. | ||
| Nerve Biopsy11 | ||
| Biopsy a nerve in the region of sensory loss or of a sensory nerve demonstrating electrophysiological abnormalities (decrease risk of adverse effects and to increase the likelihood of diagnosis | Rarely necessary | Painful, often for months |
| Concomitant muscle biopsy increases likelihood of diagnosing vasculitis or sarcoidosis | Use as last resort when evaluation not definitive | Risk of bleeding and infection |
| Greatest yield in multifocal neuropathies, or suspected amyloidotic polyneuropathy, vasculitis, sarcoidosis, lepromatous neuropathy, or rare hereditary disease where no genetic testing exists | ||
| Detects inflammation, amyloid deposits, tumor infiltration | ||
| Commonly targeted nerves include: LE sural, superficial peroneal, UE superficial radial |
Ischemic steal syndrome should be considered when neuropathy develops in a limb subsequent to arterio‐venous access procedures. Any vascular network, including the vertebral, carotid and coronary arteries, is at risk for steal. A feature common to all steal syndromes is the diversion of blood away from its original destination toward a lower pressure alternative. In some cases, this leads to a reversal of arterial flow and ischemia. Ischemic complications from AV access occur in 1‐9% of patients.1 Symptoms of steal can be mild, such as self‐limited dialysis induced pain, coldness and numbness, or severe, including severe pain, sensory and motor loss.2 If vascular compromise is sufficient, gangrene can ensue. Sensory deficits usually precede motor loss and the radial pulse is commonly absent or diminished. Other findings can include pallor of the fingers, muscle atrophy, resorption of the nail bed, and gangrene or ulcerations of the fingers. Risk factors for steal include atherosclerotic disease, female gender, age greater than 60 years, diabetes mellitus, previous surgery on the same arm, and use of the brachial artery as a donor.3 Symptoms of ischemic steal typically present within the first month after surgery, but can also be delayed; there is one report of a patient presenting one year postoperatively.4
Imaging studies such as doppler and angiography can be helpful in diagnosing ischemic steal syndrome. Fistulagrams may reveal a reversal of blood flow in the distal arm and hand, but these are reserved for cases with suspected proximal obstructive arterial disease.5 Vascular imaging studies can be misleading, however, as many patients will have physiologic but asymptomatic reversal of flow. Thus, a functional assessment such as digital plethysmography is recommended, especially in cases where clinical symptoms are vague. Digital pressures less than 60mmHg demonstrated 100% sensitivity and 87% specificity in one case control study of 40 patients.6 Treatment of ischemic steal syndrome is aimed at decreasing flow through the access shunt.
In conclusion, this case highlights the importance of timely and systematic evaluation of peripheral neuropathy in the hospital setting. Neuropathy with rapid progression and high potential for permanent damage necessitates early neurologic, or in this case, vascular consultation. Hospitalists should be facile in evaluating peripheral neuropathies and recognizing the appropriate indications for diagnostic tests and procedures.
- .Upper limb ischemia after vascular access surgery: differential diagnosis and management.Sem Dial2000;13:312–315.
- ,,,,,.Steal syndrome complicating hemodialysis access.Cardiovascular Surg (London, England)1997;5:648–653.
- ,,,,,.Onset of arterial ‘steal’ following proximal angioaccess: immediate and delayed types.Nephrol Dial Transplant2003;18:2387–2390.
- ,,,,.Incidence and characteristics of patients with hand ischemia after hemodialysis access procedure.J Surg Res1998;74:8–10
- ,,,,,.Ischemic steal syndrome: a case series and review of current management.Curr Surg2006;63:130–135.
- ,,,.Use of digital pressure measurements for the diagnosis of AV access‐induced hand ischemia.Vasc Med2006;11:227–231.
- ,.Electrodiagnostic testing of nerves and muscles: when, why, and how to order.Cleve Clin J Med2005;72:37–48.
- ,.High‐resolution sonography of the peripheral nervous system—a review of the literature.Eur J Neurol2004;11:305–314.
- ,,, et al.Role of magnetic resonance imaging in entrapment and compressive neuropathy‐what, where, and how to see the peripheral nerves on the musculoskeletal magnetic resonance image: part 2. Upper extremity.Eur Radiol2007;17:509–522.
- ,,,,,.The utility of magnetic resonance imaging in evaluating peripheral nerve disorders.Muscle Nerve2002;25:314–331.
- .Indications and usefulness of nerve biopsy.Arch Neurol2002;59:1532–1535.
- .Upper limb ischemia after vascular access surgery: differential diagnosis and management.Sem Dial2000;13:312–315.
- ,,,,,.Steal syndrome complicating hemodialysis access.Cardiovascular Surg (London, England)1997;5:648–653.
- ,,,,,.Onset of arterial ‘steal’ following proximal angioaccess: immediate and delayed types.Nephrol Dial Transplant2003;18:2387–2390.
- ,,,,.Incidence and characteristics of patients with hand ischemia after hemodialysis access procedure.J Surg Res1998;74:8–10
- ,,,,,.Ischemic steal syndrome: a case series and review of current management.Curr Surg2006;63:130–135.
- ,,,.Use of digital pressure measurements for the diagnosis of AV access‐induced hand ischemia.Vasc Med2006;11:227–231.
- ,.Electrodiagnostic testing of nerves and muscles: when, why, and how to order.Cleve Clin J Med2005;72:37–48.
- ,.High‐resolution sonography of the peripheral nervous system—a review of the literature.Eur J Neurol2004;11:305–314.
- ,,, et al.Role of magnetic resonance imaging in entrapment and compressive neuropathy‐what, where, and how to see the peripheral nerves on the musculoskeletal magnetic resonance image: part 2. Upper extremity.Eur Radiol2007;17:509–522.
- ,,,,,.The utility of magnetic resonance imaging in evaluating peripheral nerve disorders.Muscle Nerve2002;25:314–331.
- .Indications and usefulness of nerve biopsy.Arch Neurol2002;59:1532–1535.
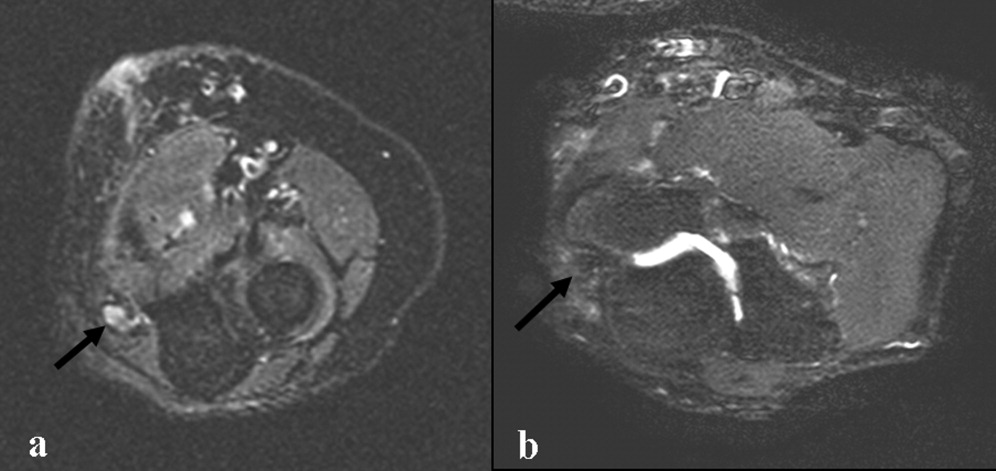
Diagnosis of Exclusion
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
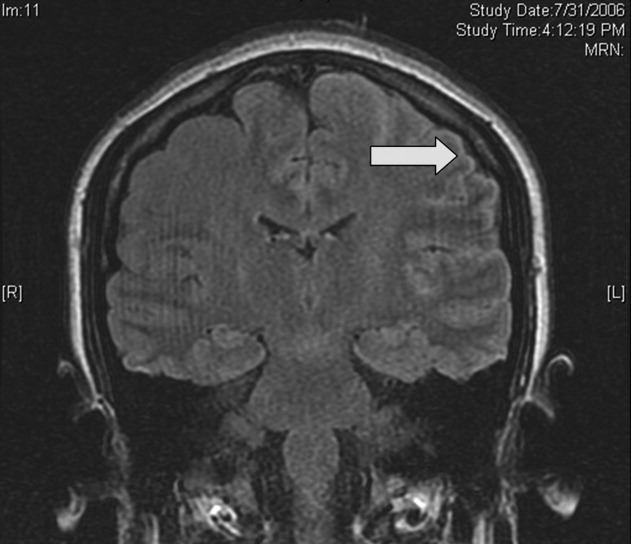
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
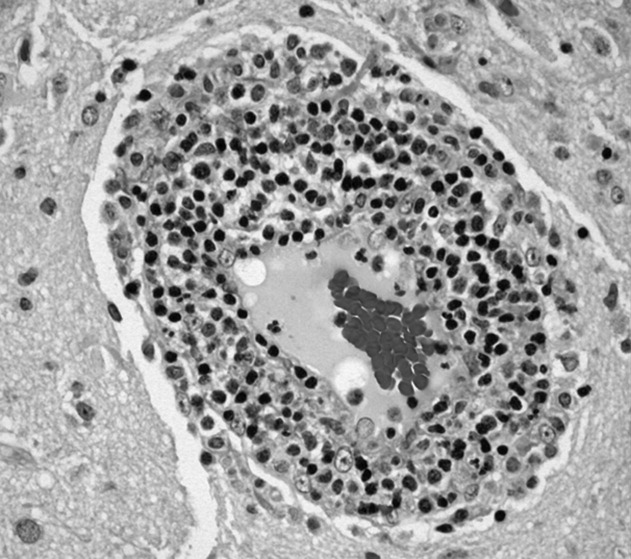
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
A 26‐year‐old woman was brought to the emergency department following several episodes of seizures. The patient's friend witnessed several 15‐minute episodes of sudden jerks and tremors of her right arm during which the patient bit her tongue, had word‐finding difficulty, had horizontal eye deviation, and was incontinent of urine. She became unresponsive during the episodes, with incomplete recovery of consciousness between attacks. She was afebrile. Her neurologic exam 4 hours after several seizures revealed word‐finding difficulty and right arm weakness. A complete blood count, chemistry panel including renal and liver function tests, urine toxicology screen, and computed tomography (CT) of the head were normal. After a loading dose of fosphenytoin, the patient did not experience further seizures and was discharged on a maintenance dose of phenytoin.
Over the next week, the patient continued to note a sensation of heaviness in her right arm and felt fatigued. The patient's mother brought her back to the emergency department after witnessing a similar seizure episode that persisted for an hour. On arrival, the patient was no longer seizing.
Although it can sometimes be difficult to differentiate between seizure, stroke, syncope, and other causes of transient loss of consciousness, this constellation of symptoms strongly points to a seizure. I would classify the patient's focal arm movements associated with impaired consciousness as partial complex seizures. One of the first considerations is determining whether the seizure is caused by a systemic process or by an intrinsic central nervous system disorder. Common systemic illnesses include infections, metabolic disturbances, toxins, and malignancies, none of which are evident on the preliminary evaluation. The absence of fever is important as is the time frame (now extending over 1 week) in excluding acute bacterial meningitis. A negative urine toxicology is very helpful but does not exclude the possibility that the seizure is from unmeasured drug intoxication, for example, tricyclic antidepressants, or from drug withdrawal, for example, benzodiazepines, barbiturates, ethanol, and antiepileptic drugs. The persistent right arm heaviness and right arm jerking during the seizures suggest a left cortical focus that the CT scan did not detect. Without a clear diagnosis and with recurrent seizures despite antiepileptic drugs, hospitalization is warranted.
The patient experienced migraine headaches each month during menses. There was no family history of seizures. Her only medication was phenytoin. The mother was unaware of any use of tobacco, alcohol, or recreational drugs. The patient was raised in New Jersey and moved to the San Francisco Bay area 9 months ago. She had no pets and had traveled to Florida and Montreal in the past 6 months. She was a graduate student in performing arts. During the preceding 2 weeks she had been under significant stress and had not slept much in preparation for an upcoming production. The patient's mother was not aware of any head trauma, recent illness, fevers, chills, weight loss, photosensitivity, arthralgias, nausea, vomiting, or diarrhea.
Recent sleep deprivation could provoke seizures in a patient with a latent anatomic focus or metabolic predisposition. Nonadherence to antiepileptic drug therapy is the most common reason for patients to present to the ED with seizures; therefore, I would check a phenytoin level to assess whether she is at a therapeutic level and would consider administering another loading dose. In the absence of immunocompromise or unusual activities or exposures, North American travel does not bring to mind additional etiologies at this time.
On exam, temperature was 37.3C, blood pressure was 148/84 mm Hg, heart rate was 120 per minute, and respiratory rate was 16 per minute. The patient was stuporous and withdrew from painful stimuli. She was unable to speak. Pupils were 4 mm in diameter and reacted to light. No gaze preference or nystagmus was present. There was no meningismus. Deep tendon reflexes were 1+ and symmetrical in both upper and lower extremities. Plantar reflexes were extensor bilaterally. The tone in the right upper extremity was mildly increased compared to the left. The patient demonstrated semipurposeful movement of the limbs, such as reaching for the bed rails with her arms. Examination of the heart, lungs, abdomen, skin, and oropharynx was normal.
The white blood cell count was 22,300/mm3 with 50% neutrophils, 40% lymphocytes, 7% monocytes, and 3% eosinophils. Results of the chemistry panel including electrolytes, glucose, creatinine, and liver enzymes, urinalysis, and thyroid‐stimulating hormone were normal. Serum phenytoin level was 8.1 g/mL. Urine toxicology screen, obtained after the patient had received lorazepam, was positive only for benzodiazepines. A chest radiograph was normal.
The cerebrospinal fluid (CSF) was colorless, containing 35 white blood cells/mm3 (48% lymphocytes, 30% neutrophils, 22% monocytes), 3 red blood cells/mm3, 62 mg/dL protein, and 50 mg/dL glucose. There was no xanthochromia. The CSF was negative for cryptococcal antigen, antibodies to West Nile virus, PCR for herpes simplex viruses‐1 and ‐2, and PCR for Borrelia burgdorferi. CSF bacterial culture, cryptococcal antigen, and AFB stain were negative. The serum antinuclear antibody, rheumatoid factor, and rapid plasma reagin were negative. Serum antibodies to human immunodeficiency virus, hepatitis B and C viruses, Borrelia burgdorferi, and herpes simplex viruses were negative. The erythrocyte sedimentation rate was 25 mm/hr. There was no growth in her blood cultures.
These CSF findings have to be interpreted in light of her clinical picture, as they are congruent with both an aseptic meningitis and encephalitis. In practice, these can be hard to distinguish, but the early and dominant cortical findings (focal neurologic deficits, prominent altered mental status, bilateral extensor plantar reflexes) and absence of meningeal signs favor encephalitis. This CSF profile can be seen in a variety of disease processes causing a meningoencephalitis, including partially treated bacterial meningitis; meningitis due to viruses, fungi, mycobacteria, or atypical bacteria (eg, Listeria); neurosarcoidosis; carcinomatous meningitis; and infection or inflammation from a parameningeal focus in the sinuses, epidural space, or brain parenchyma. Seizure itself can lead to a postictal pleocytosis in the CSF, although this degree of inflammation would be unusual. Many tests can be sent, and the clinicians appropriately focused on some of the most treatable and serious etiologies first. The negative HIV test limits the list of opportunistic pathogens. The negative ANA substantially lowers the likelihood of systemic lupus, an important consideration in a young woman with an inflammatory disorder involving the central nervous system.
Magnetic resonance imaging (MRI) of the brain showed cortical T2 prolongation with significant enhancement with gadolinium in the cortex and leptomeninges of the left parietal and posterotemporal lobes and right cingulate gyrus region (Fig. 1). The patient was admitted to the intensive care unit, and phenytoin and levetiracetam were administered. Over the next several days, she remained afebrile, and her leukocytosis resolved. She continued to have seizures every day despite receiving phenytoin, levetiracetam, and lamotrigine. She was alert and complained about persistent right arm weakness and word‐finding difficulties. Posterior cervical lymphadenopathy at the base of her left occiput was detected on subsequent exam.
An excisional lymph node biopsy demonstrated extensive necrosis without evidence of granulomata, malignancy, or lymphoproliferative disease. Stains and cultures for bacteria, fungi, and mycobacteria were negative. The patient's electroencephalogram captured epileptiform activity over the left hemisphere 2 hours after a cluster of seizures. MR angiography and cerebral angiography demonstrated no abnormalities.
Despite this additional information, there is no distinguishing clue that points to a single diagnosis. This is a 26‐year‐old healthy, seemingly immunocompetent woman who has had a 2‐week progressive and refractory seizure disorder secondary to a multifocal neuroinvasive process with a CSF pleocytosis. She does not have evidence of a systemic underlying disorder, save for nonspecific localized lymphadenopathy and a transient episode of leukocytosis on admission, and has no distinguishing epidemiological factors or exposures.
Despite my initial concerns for infectious meningoencephalitis, the negative stains, serologies, and cultures of the blood, CSF, and lymph nodes in the setting of a normal immune system and no suspect exposure substantially lower this probability. Arthropod‐borne viruses are still possible, especially West Nile virus, because the serological tests are less sensitive early in the illness, acknowledging that the absence of fever, weakness, and known mosquito bites detracts from this diagnosis. Pathogens that cause regional lymphadenopathy and encephalitis such as Bartonella remain possibilities, as the history of exposure to a kitten can be easily overlooked.
Rheumatologic disorders merit close attention in a young woman, but the negative ANA makes lupus cerebritis unlikely, and the 2 angiograms did not detect evidence of vasculitis. Finally, there is the question of malignancy and other miscellaneous infiltrative disorders (such as sarcoid), which are of importance here because of the multifocal cortical involvement on imaging.
At this point, I would resample the CSF for viral etiologies (eg, West Nile virus) and cytology and would send serum Bartonella serologies. If these studies were negative, a brain biopsy, primarily to exclude malignancy but also to uncover an unsuspected process, would be indicated. I cannot make a definitive diagnosis or find a perfect fit here, but in the absence of strong evidence of an infection, I am concerned about a malignancy, perhaps a low‐grade primary brain tumor.
Brain biopsy of the leptomeninges and cortex of the left parietal lobe showed multiple blood vessels infiltrated by lymphocytes, neutrophils, and eosinophils (Fig. 2). The pattern of inflammation was consistent with primary angiitis of the central nervous system (PACNS). The patient received 1 g of intravenous methylprednisolone on 3 consecutive days, followed by oral prednisone and cyclophosphamide. The seizures ceased, and she made steady progress with rehabilitation therapy. Four months after discharge a cerebral angiogram (done to ensure there was no interval evidence of vasculitis prior to tapering therapy) demonstrated patency of all major intracranial arteries and venous sinuses.
COMMENTARY
When a patient presents with symptoms or signs referable to the central nervous system (CNS), hospitalists must simultaneously consider primary neurologic disorders and systemic diseases that involve the CNS. Initial evaluation includes a thorough history and physical examination, basic lab studies, routine CSF analysis, and neuroimaging (often a CT scan of the head). Complicated neurologic cases may warrant more elaborate testing including EEG, brain MRI, cerebral angiography, and specialized blood and CSF studies. Clinicians may still find themselves faced with a patient who has clear CNS dysfunction but no obvious diagnosis despite an exhaustive and expensive evaluation. Several disorders match this profile including intravascular lymphoma, prion diseases, paraneoplastic syndromes, and cerebritis. Primary angiitis of the central nervous system (PACNS), a rare disorder characterized by inflammation of the medium‐sized and small arteries of the CNS, is among these disorders. Although the aforementioned diseases may sometimes have suggestive or even pathognomonic features (eg, the string of beads angiographic appearance in vasculitides), they are challenging to diagnose when such findings are absent.
Like any vasculitis of the CNS, PACNS may present with a wide spectrum of clinical features.12 Although headache and altered mental status are the most common complaints, paresis, seizures, ataxia, visual changes, and aphasia have all been described. The onset of symptoms ranges from acute to chronic, and neurologic deficits can be focal or diffuse. Systemic manifestations such as fever and weight loss are rare. The average age of onset is 42 years, with no significant sex preponderance. The histopathology of PACNS is granulomatous inflammation of arteries in the parenchyma and leptomeninges of the brain and less commonly in the spinal cord. The narrowing of the affected vessels causes cerebral ischemia and the associated neurologic deficits. The trigger for this focal inflammation is unknown.
After common disorders have been excluded in cases of CNS dysfunction, compatible CSF findings and imaging results may prompt consideration of PACNS. CSF analysis in patients with PACNS typically demonstrates a lymphocytic pleocytosis. MRI abnormalities in PACNS include multiple infarcts in the cortex, deep white matter, or leptomeninges.34 Less specific findings are contrast enhancement in the leptomeninges and white matter disease, both of which may direct the site for meningeal and brain biopsy.
Both brain MRA and cerebral angiography have a limited role in the diagnosis of vasculitis within the CNS. In 18 patients with CNS vasculitis due to autoimmune disease, all had parenchymal abnormalities on MRA but only 65% had evidence of vasculitis on angiography. In 2 retrospective studies of patients with suspected PACNS, abnormal angiograms had a specificity less than 30% for PACNS, whereas brain biopsies had a negative predictive value of 70%.57 Although in practice patients with compatible clinical features are sometimes diagnosed with CNS vasculitis on the basis of angiographic findings, brain biopsy is necessary to differentiate vasculitis from other vasculopathies and to establish a definitive diagnosis.
Before a diagnosis of PACNS is made, care must be taken to exclude infections, neoplasms, and autoimmune processes that cause angiitis of the CNS (Table 1). The presence of any extracranial abnormalities (which were not present in this case) should prompt consideration of an underlying systemic disorder causing a secondary CNS vasculitis and should cast doubt on the diagnosis of PACNS. Meningovascular syphilis and tuberculosis are among the long list of infections that may cause inflammation of the CNS vasculature. Autoimmune disorders that may cause vasculitis inside the brain include polyarteritis nodosa and Wegener's granulomatosis. Reversible cerebral vasoconstrictive disease, which is most commonly seen in women ages 20 to 50, and sympathomimetic toxins such as cocaine and amphetamine may exhibit clinical and angiographic abnormalities indistinguishable from PACNS.89
| Infection: Viruses (HIV, varicella‐zoster virus, hepatitis C virus), syphilis, Borrelia burgdorferi, Bartonella, Mycobacterium tuberculosis, fungi (Aspergillus, Coccidioides), bacteria. |
| Autoimmune: Polyarteritis nodosa, Wegener's granulomatosis, temporal arteritis, cryoglobulinemic vasculitis, lupus vasculitis, rheumatoid vasculitis. |
| Toxins: Amphetamine, cocaine, ephedrine, heroin. |
| Malignancy: Primary CNS lymphoma, angioimmunoproliferative disorders, infiltrating glioma. |
There are no prospective trials investigating PACNS treatment. Aggressive immunosuppression with cyclophosphamide and glucocorticoids is the mainstay of treatment. The duration of treatment varies with the severity of the disease and response to therapy. One study suggests that treatment should be continued for 6 to 12 months.10 Neurologic deficits may remain irreversible because of scarring of the affected vessels. Serial brain MRI examinations are often used to follow radiographic resolution during and after the therapy, although radiographic changes do not predict clinical response.11 New abnormalities on MRI, however, delay any tapering of treatment. The availability of neuroimaging studies and immunosuppressive therapy has improved the prognosis of PACNS. One study reported a favorable outcome with a 29% relapse rate and a 10% mortality rate in 54 patients over a mean follow‐up period of 35 months.12
PACNS remains a challenging diagnosis because of its rarity, the wide range of neurologic manifestations, and the difficulty in establishing a diagnosis noninvasively. It is an extremely uncommon disease but should be considered in patients with unexplained neurologic deficits referable to the CNS alone after an exhaustive workup. Ultimately, the diagnosis is made by a thorough history and physical examination, exclusion of underlying conditions (particularly systemic vasculitides and infections), and histological confirmation.
Key Points for Hospitalists
-
Serious disorders that may present with CNS abnormalities and nondiagnostic abnormal findings on lumbar puncture, brain MRI, and cerebral angiography include intravascular lymphoma, prion diseases, cerebritis, paraneoplastic syndromes, and CNS vasculitis.
-
PACNS is a challenging diagnosis with varied clinical features and often normal angiographic findings. In particular, the specificity of brain MRA and cerebral angiography is low. Although PACNS is rare, it should be on the differential diagnosis, as the condition is fatal without prompt treatment.
-
A diagnosis of PACNS is made only after excluding secondary causes of CNS vasculitis such as infections, malignancies, autoimmune conditions, reversible cerebral vasoconstrictive disease, and medications. The diagnosis is confirmed with a biopsy of the brain and meninges.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
- ,.Medical progress: small‐vessel vasculitis.N Engl J Med.1997;337:1512–1523.
- ,.Arthritis and Allied Conditions: A Textbook of Rheumatology.15th ed.Philadelphia:Lippincott Williams 2005.
- ,,,.Primary angiitis of the central nervous system: unusual MR appearance.Am J Neuroradiol.1994;15:331–334.
- ,,,.Radiographic features of central nervous system vasculitis.Neurol Clin.1997;15:779–804.
- ,,,,,.Primary angiitis of the central nervous system at conventional angiography.Radiology.2004;233:878–882.
- ,,,,.CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.Am J Neuroradiol.1999;20:75–85.
- ,.Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system.J Rheumatol.1995;22:662–667.
- ,.Amphetamine abuse and intracranial haemorrhage.J R Soc Med.2000;93:472–477.
- ,.Drug‐induced vasculitis.Curr Opin Rheumatol.1996;8:34–40.
- ,,.Vasculitis in the central nervous system.Arthritis Rheum.1997;40:1189–1201.
- .Therapy of systemic vasculitis.Neurol Clin.1997;15:973–991.
- ,,, et al.Long‐term outcomes of patients with primary angiitis of the central nervous system.Arthritis Rheum.2000;43:S162.
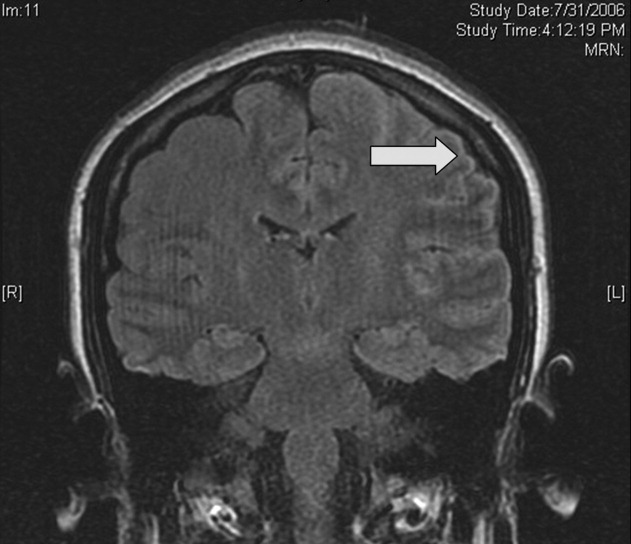
Thinking Inside the Box
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
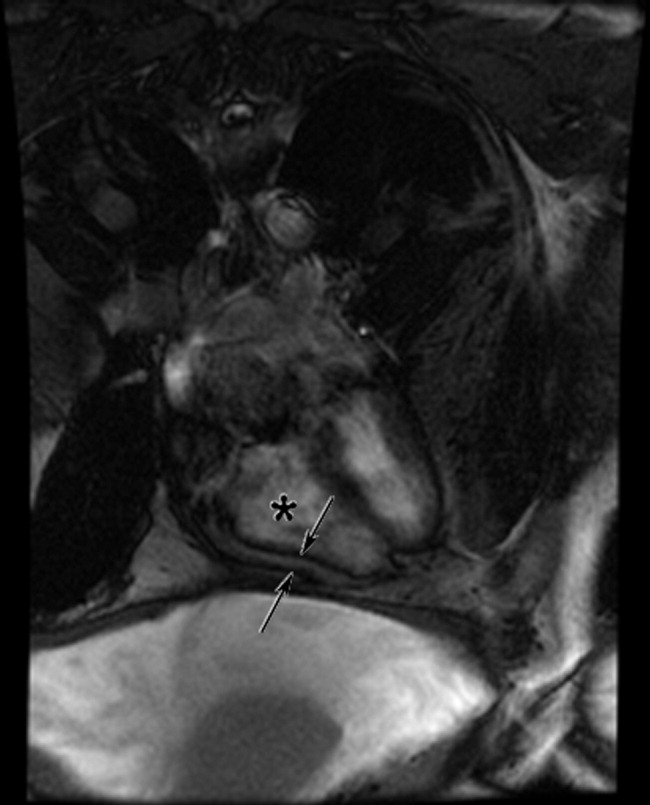
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
A 65‐year‐old man was referred for evaluation of worsening ascites and end‐stage liver disease. The patient had been well until 1 year ago, when he developed lower extremity edema and abdominal distention. After evaluation by his primary care physician, he was given a diagnosis of cryptogenic cirrhosis. He underwent several paracenteses and was placed on furosemide and spironolactone. The patient had been stable on his diuretic regimen until 2 weeks previously, when he suddenly developed worsening edema and ascites, along with dizziness, nausea, and hypotension. His physician stopped the diuretics and referred him to the hospital.
Before diagnosing a patient with cryptogenic cirrhosis, it is necessary to exclude common etiologies of cirrhosis such as alcohol, viral hepatitis, and non‐alcoholic fatty liver disease and numerous uncommon causes, including Wilson's disease, hemochromatosis, Budd‐Chiari, and biliary cirrhosis. It is also important to remember that patients with liver disease are not immune to extrahepatic causes of ascites, such as peritoneal carcinomatosis and tuberculous ascites. Simultaneously, reasons for chronic liver disease decompensating acutely must be considered: medication nonadherence, excess salt intake, hepatotoxicity from acetaminophen or alcohol, and other acute insults, such as hepatocellular carcinoma, an intervening infection (especially spontaneous bacterial peritonitis), ascending cholangitis, or a flare of chronic viral hepatitis.
Past medical and surgical history included diabetes mellitus (diagnosed 10 years previously), obstructive sleep apnea, hypertension, hypothyroidism, and mild chronic kidney disease. Medications included levothyroxine, lactulose, sulfamethoxazole, pioglitazone (started 4 months prior), and ibuprofen. Furosemide and spironolactone had been discontinued 2 weeks previously. He currently resided in the Central Valley of California. He had lived in Thailand from age 7 to 17 and traveled to India more than 1 year ago. He did not smoke and had never used intravenous drugs or received a blood transfusion. He rarely drank alcohol. He worked as a chemist. There was no family history of liver disease.
There is no obvious explanation for the underlying liver disease or the acute decompensation. Sulfamethoxazole is a rare cause of allergic or granulomatous hepatitis. Pioglitazone is a thiazolinedione which in earlier formulations was linked to hepatitis but can be excluded as a cause of this patient's cirrhosis because it was started after liver disease was detected. As a chemist, he might have been exposed to carbon tetrachloride, a known hepatotoxin. Obstructive sleep apnea causes pulmonary hypertension, but severe ascites and acute hepatic decompensation would be unusual. Ibuprofen might precipitate worsening renal function and fluid accumulation. Time in Thailand and India raises the possibility of tuberculous ascites.
The patient had no headache, vision changes, abdominal pain, emesis, melena, hematochezia, chest pain, palpitations, dysuria, polyuria, pruritus, dark urine, or rashes. He reported difficulty with concentration when lactulose was decreased. He noted worsening exercise tolerance with dyspnea after 10 steps and reported a weight gain of 12 pounds in the past 2 weeks.
On examination, temperature was 36.8C; blood pressure, 129/87 mm Hg; heart rate, 85 beats per minute; respirations, 20 per minute; and oxygen saturation, 94% on room air. He was uncomfortable but alert. There was no scleral icterus or conjunctival pallor. Jugular venous pressure was elevated. The lungs were clear, and the heart was regular, with no murmur, rub, or gallops. The abdomen was massively distended with a fluid wave; the liver and spleen could not be palpated. There was pitting edema of the sacrum and lower extremities. There was no asterixis, palmar erythema, spider angiomata, or skin discoloration.
The additional history and physical exam suggest that the primary problem may lie outside the liver, especially as signs of advanced liver disease (other than ascites) are absent. Dyspnea on exertion is consistent with the physical stress of a large volume of ascites or could be secondary to several pulmonary complications associated with liver disease, including portopulmonary hypertension, hepatopulmonary syndrome, or hepatic hydrothorax. Alternatively, the dyspnea raises the possibility that the ascites is not related to a primary liver disorder but rather to anemia or to a cardiac disorder, such as chronic left ventricular failure, isolated right‐sided heart failure, or constrictive pericarditis. These diagnoses are suggested by the elevated jugular venous pressure, which is atypical in cirrhosis.
Although portal hypertension accounts for most cases of ascites, peritoneal fluid should be examined to exclude peritoneal carcinomatosis and tuberculous ascites. I am interested in the results of an echocardiogram.
Initial laboratory studies demonstrated a sodium concentration of 136 mEq/dL; potassium, 4.7 mEq/dL; chloride, 99 mEq/dL; bicarbonate, 24 mEq/dL; blood urea nitrogen, 54 mg/dL; creatinine, 3.3 mg/dL (increased from baseline of 1.6 mg/dL 4 months previously); white cell count, 7000/mm3; hemoglobin, 10.5 g/dL; MCV, 89 fL; platelet count, 205,000/mm3; bilirubin, 0.6 mg/dL; aspartate aminotransferase, 15 U/L; alanine aminotransferase, 8 U/L; alkaline phosphatase, 102 U/L; albumin, 4.2 g/dL; total protein, 8.2 g/dL; international normalized ratio, 1.2; and partial thromboplastin time, 31.8 seconds. A urine dipstick demonstrated 1+ protein. The chest radiograph was normal. Electrocardiogram had borderline low voltage with nonspecific T‐wave abnormalities. Additional studies showed a serum iron concentration of 49 mg/dL, transferrin saturation of 16%, total iron binding capacity of 310 mg/dL, and ferritin of 247 mg/mL. Hemoglobin A1c was 7.0%. Acute and chronic antibodies to hepatitis A, B, and C viruses were negative. The following study results were normal or negative: antinuclear antibody, alpha‐1‐antitrypsin, ceruloplasmin, alpha‐fetoprotein, carcinoembryonic antigen, and 24‐hour urinary copper. The thyroid function studies were normal. A purified protein derivative (PPD) skin test was nonreactive.
There continues to be a paucity of evidence of a primary liver disorder. The hepatic enzymes and tests of liver synthetic function are normal, and there is no pancytopenia, as might result from hypersplenism. I remain most suspicious of either a primary cardiac or pericardial disorder with secondary hepatic congestion or a disease that simultaneously affects the heart and liver.
The reasons for the low voltage on the electrocardiogram include processes that infiltrate the myocardium (amyloidosis, sarcoidosis, hemochromatosis, and myxedema fluid) and processes that increase the distance between the myocardium and surface electrodes, such as adipose tissue, air (from emphysema or pneumothorax), or pericardial effusion. Pericardial effusion may present subacutely with predominant features of right ventricular failure. Low voltage, liver disease, and possible heart failure raise the possibility of amyloidosis or hemochromatosis. The low transferrin saturation renders hemochromatosis unlikely. Although normal alkaline phosphatase and serum albumin are not characteristic when AL amyloid affects the liver and kidneys, serum and urine protein electrophoresis and immunofixation should be considered.
With paracentesis 3.5 L of ascitic fluid was removed. The red cell count was 4000/mm3, and white blood cell count was 505/mm3, of which 25% were polymorphonuclear cells, 22% were lymphocytes, and 53% were monocytes. Additional peritoneal fluid chemistries included albumin of 3.0 g/dL and total protein of 5.3 g/dL. Abdominal ultrasound with Doppler demonstrated a liver of normal size and echogenicity with patent hepatic arteries, hepatic veins, and portal vein. There was mild splenomegaly with normal kidneys. Evaluation for a possible liver transplant was initiated. Blood, urine, and peritoneal fluid cultures demonstrated no growth. Echocardiography demonstrated borderline concentric left ventricular hypertrophy, normal right and left ventricular function, dilated superior and inferior vena cavae, and no pericardial effusion or thickening.
The serum‐ascites albumin gradient (SAAG) of 1.2 is consistent with portal hypertension as the cause of the ascites. The Doppler findings exclude postsinusoidal causes of portal hypertension from hepatic vein obstruction or thrombosis. The combination of the elevated SAAG, elevated jugular venous pressure, borderline low voltage on ECG, and elevated peritoneal total protein make cardiac and pericardial disease the leading considerations. Given the normal ventricular function, I am concerned about elevated intracardiac pressures resulting from pericardial disease or restrictive cardiomyopathy. At this point, right heart catheterization would be useful for assessing intracardiac pressures.
On the fourth hospital day, paracentesis was repeated, and 15 L of fluid was removed. A transjugular liver biopsy demonstrated diffuse patchy fibrosis consistent with early cirrhosis and minor intralobular changes with minimal ballooning. There was no steatosis, active inflammation, granulomata, iron deposition, or evidence of viral hepatitis. Right heart catheterization revealed a right atrial pressure of 18 cm H20, right ventricular pressure of 34/20 cm H20, pulmonary artery pressure of 34/18 cm H20 (mean 25), pulmonary capillary wedge pressure of 20 cm H20, cardiac output of 5.8 L/min, and cardiac index of 2.5 L/min/m2.
The mild hepatic histologic abnormalities do not support an intrinsic liver disease as the cause of his massive ascites and end‐stage liver disease physiology. Cardiac catheterization demonstrates equalization of diastolic pressures, which suggests constrictive pericarditis or restrictive cardiomyopathy. Despite the normal chest radiograph and nonreactive PPD, tuberculosis would be my leading explanation for constrictive pericarditis given the time spent in areas endemic with TB. Although lateral chest radiography may demonstrate pericardial calcifications, magnetic resonance imaging (MRI) is the best imaging modality to detect constrictive pericarditis. Alternately, cardiac amyloidosis could cause restrictive cardiomyopathy and has not been definitively excluded. A cardiac MRI to assess the pericardium would be my next test, and I would request Congo red stains of the liver biopsy. If these tests are unrevealing, endomyocardial biopsy may be necessary.
The cardiac MRI revealed a severely thickened 7‐mm pericardium (normal < 3 mm) most prominent over the right atrium and ventricle. The right ventricle was described as bullet‐shaped, suggesting constrictive pericardial disease (Fig. 1). Left heart catheterization to evaluate coronary anatomy and left ventricular pressures revealed no significant coronary arterial disease and demonstrated an elevated left ventricular end‐diastolic pressure consistent with constrictive pericarditis. Endomyocardial biopsy showed no evidence of infiltrative disease, granulomata, or other significant abnormality. The following day the patient underwent pericardiectomy. Postoperatively, his ascites was easily managed with low doses of diuretics. The pericardial tissue revealed chronic inflammatory cells and dense collagenous fibrosis characteristic of constrictive pericarditis without evidence of malignancy or granulomatous disease. Pericardial cultures were negative for bacteria, viruses, fungi, and mycobacteria.
DISCUSSION
Constrictive pericarditis is characterized by chronic fibrous thickening of the once‐elastic pericardial sac and can occur following any disease process that affects the pericardium (Table 1).1, 2 The challenge in the diagnosis of constrictive pericarditis lies in the recognition of this slowly progressive and uncommon disease. In many cases, nonspecific symptoms of reduced cardiac output and insidious right‐sided heart failure are present for 12 months or longer before a diagnosis is established.1, 3 A typical presentation of constrictive pericarditis is peripheral edema, ascites, and hepatomegaly, a combination that may understandably lead to a misdiagnosis of chronic liver disease and even subject a patient to the unnecessary risk of a liver biopsy, as in this case.
|
Cryptogenic cirrhosis, the initial diagnosis of this patient, is a term used only after excluding the common and uncommon causes of cirrhosis (Table 2).46 With expanded knowledge of the causes of cirrhosis, especially nonalcoholic fatty liver disease, the number of cases of cirrhosis considered to be cryptogenic has decreased from nearly one‐third of all cases in 1960 to approximately 5% in a modern series.7, 8 Chronic or repetitive heart failure can lead to progressive hepatic fibrosis and cirrhosis. Distinguishing features compared to other causes of cirrhosis include an ascitic protein concentration greater than 2.5 g/dL, relatively preserved synthetic function, and infrequent stigmata of end‐stage liver disease such as spider angiomata or pronounced jaundice.9, 10
|
Most common
|
Less common
|
A key exam feature that distinguishes cardiac cirrhosis from other causes of liver failure is an elevated jugular venous pressure. Hepatic causes of cirrhosis induce increased nitric oxide production, which leads to splanchnic and peripheral arterial vasodilatation with a reduced effective circulating volume and normal or low jugular venous pressure.11, 12 Therefore, a patient with cirrhosis and ascites having an elevated jugular venous pressure should prompt echocardiographic evaluation.13 When echocardiography excludes ventricular dysfunction, valvular abnormalities, and pulmonary hypertension, constrictive pericarditis and restrictive cardiomyopathy remain important diagnostic considerations.
In both constrictive pericarditis and restrictive cardiomyopathy, ventricular filling is limited. Pressures in the chambers rise abruptly and rapidly during ventricular filling until equilibrium is reached in early diastole. This can be conceptualized as the cardiac chambers being constrained by the limitations of a rigid external box. In constrictive pericarditis, the rigid external box is the fibrosed and thickened pericardial sac, which loses its elasticity and impairs filling of the ventricles. In restrictive cardiomyopathy, the stiff myocardium limits ventricular filling.
There is considerable overlap in the clinical, echocardiographic, and hemodynamic findings of constrictive pericarditis and restrictive cardiomyopathy.14 Both may present insidiously with progressive heart failure. Echocardiography demonstrates impaired diastolic function. Cardiac hemodynamics demonstrate abrupt and rapid early diastolic filling, elevated and equal ventricular end‐diastolic pressures, and reduced stroke volume and cardiac output. A diagnosis of constrictive pericarditis is favored when a marked inspiratory increase in right ventricular pressures and decrease in left ventricular pressures are seen on heart catheterization or a similar inspiratory increase in transvalvular flow velocities across the tricuspid valve compared with the mitral valve is shown by echocardiography. This finding results from normal inspiratory increases in intrathoracic pressures, which are unable to be transmitted through the rigid pericardium but continue to augment venous return to the right side of the heart. As many as one‐third of patients with pericardial constriction lack these characteristic findings on echocardiogram.14
The results of pericardial imaging may suggest a diagnosis of constrictive pericarditis. Lateral chest radiography demonstrates pericardial calcifications in less than 30% of cases.15 Cardiac computed tomography (CT) and MRI are the best imaging modalities for detecting an increase in pericardial thickness (3 mm or greater).16 However, in as many as 20% of patients with surgically confirmed constrictive pericarditis, CT and MRI will demonstrate a pericardium of normal thickness.17
When faced with the diagnostic conundrum of constrictive pericarditis versus restrictive cardiomyopathy, strong clinical suspicion, thorough echocardiography, careful hemodynamic assessment with right and left heart catheterization,14, 18 pericardial imaging, and sometimes endomyocardial biopsy to exclude restrictive cardiomyopathy are often needed before proceeding to pericardiectomy, which carries a significant surgical risk but can also be curative.
This case highlights many of the features of constrictive pericarditis, the challenges and delay in its diagnosis, and its occasional misdiagnosis as chronic liver disease. Clinicians may recognize the typical combination of cirrhosis (or suspected cirrhosis), high SAAG ascites, and edema as characteristic of advanced intrinsic liver disease. However, they must not be seduced into immediate pattern recognition when contrary evidencesuch as elevated neck veins, elevated ascitic total protein, or relatively preserved hepatic synthetic functionaccompanies that picture. Under such circumstances, they must remember to think outside the box and bear in mind that the heart may be trapped inside a box.
Take‐Home Points
-
Constrictive pericarditis is often unrecognized initially, resulting in delayed diagnosis. Patients typically present with nonspecific signs and symptoms of low cardiac output and progressive right‐sided heart failure. Clinical suspicion is key to prompt diagnosis and pericardiectomy, which may be curative.
-
Distinguishing features in the presentation of cardiac or pericardial etiologies of ascites and cirrhosis include elevated neck veins, elevated ascitic protein content, relatively preserved hepatic synthetic function, and absence of the stigmata of end‐stage liver disease.
-
Constrictive pericarditis and restrictive cardiomyopathy can present with a similar clinical picture and hemodynamics showing impaired ventricular filling. Right and left heart catheterization, pericardial imaging, and endomyocardial biopsy may differentiate the 2 conditions. For constrictive pericarditis, surgical and pathological confirmation is the gold standard for diagnosis and the only definitive treatment.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
- ,,, et al.Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy.Circulation.1999;100:1380–1386.
- ,,, et al.Constrictive pericarditis: etiology and cause‐specific survival after pericardiectomy.J Am Coll Cardiol.2004;43:1445–1452.
- .Chronic constrictive pericarditis.Am J Cardiol.1961;7:48–61.
- American Gastroenterological Association.AGA technical review on the evaluation of liver chemistry tests.Gastroenterology.2002;123:1367–1384.
- ,.AASLD practice guidelines: evaluation of the patient for liver transplantation.Hepatology.2005;41:1–26.
- Feldman M,Friedman LS,Brandt LJ, eds.Sleisenger and Fordtran's Gastrointestinal and Liver Disease: Pathophysiology, Diagnosis, Management.Philadelphia:Saunders Elsevier;2006.
- ,,, et al.Cirrhosis of the liver: a study of alcoholic and nonalcoholic patients in Boston and London.N Engl J Med.1960;261:1–9.
- ,,, et al.Liver transplantation for cryptogenic cirrhosis.Liver Transpl Surg.1997;3:359–364.
- ,,, et al.Heart diseases affecting the liver and liver disease affecting the heart.Am Heart J.2000;140:111–120.
- ,,.The liver in heart failure.Clin Liver Dis.2002;6:947–967.
- ,,, et al.Portal hypertension: from pathophysiology to clinical practice.Liver Int.2005;25:1079–1090.
- .Portal hypertension.Curr Opin Gastroenterol.2006;22:254–262.
- ,,, et al.Negative influences of ascites on the cardiac function of cirrhotic patients.Am J Med.1975;59:165–170.
- .Constrictive pericarditis in the modern era: a diagnostic dilemma.Heart.2001;86:619–623.
- ,,, et al.Calcific constrictive pericarditis: is it still with us?Ann Intern Med.2000;132:444–450.
- ,,,,,.CT and MR imaging of pericardial disease.Radiographics.2003;23:S167–S180.
- ,,, et al.Constrictive pericarditis in 26 patients with histologically normal pericardial thickness.Circulation.2003;108:1852–1857.
- ,,, et al.Value of dynamic respiratory changes in left and right ventricular pressures for the diagnosis of constrictive pericarditis.Circulation.1996;93:2007–2013.
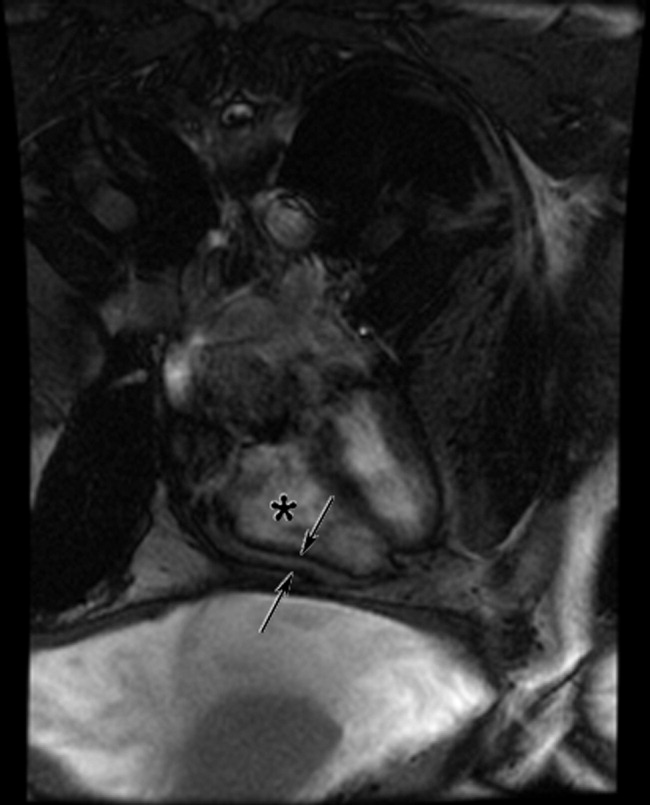
A Rash Decision
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
A 38‐year‐old HIV+ Ohio man with a recent CD4+ count of 534 cells/mL presented to his physician with 3 weeks of fever as high as 102F. He noted mild myalgias, pruritus, and an occasional cough but no headache, sore throat, dyspnea, rash, or gastrointestinal or genitourinary complaints. He had been seen elsewhere 2 weeks previously, when he had reported a single episode of receptive oral sex with a male partner several weeks earlier. He had been prescribed ciprofloxacin and azithromycin, but a throat swab came back negative for Chlamydia and Neisseria gonorrhoeae, and he reported no change in his symptoms after the course of antibiotics. He denied smoking or using street drugs. His only medications were citalopram and trazodone for depression.
This is a HIV+ man with a mild degree of immunosuppression with a fever of unknown origin (FUO). It is not yet known if the requisite basic infectious evaluation has been completed to meet this definition, but the duration certainly qualifies, and regardless of semantics, the FUO framework is a helpful starting point. The primary considerations in FUO are infections, neoplasms, and autoimmune illnesses. Autoimmune diseases are relatively less common in HIV patients. Although pruritis is quite common in HIV alone, it may also herald renal failure, cholestasis, or a malignancy (usually hematologic). Drugs must also be considered as a cause of unexplained fever; the pruritis might suggest an allergic reaction, although I do not think of citalopram or trazodone as having this effect. The failure to respond to broad‐spectrum antimicrobials (along with the duration) lowers my suspicion for common infections such as pneumonia, urinary tract infection, or cellulitis. Among sexually transmitted diseases, syphilis can be protean and merits consideration.
On examination he appeared well. His temperature was 102.4F, pulse 111 beats/min, blood pressure 138/78 mm Hg. The head, neck, cardiovascular system, and lungs appeared normal on examination. The abdomen was soft and nontender without organomegaly; skin, extremities, and neurological system were unremarkable. Rectal examination showed small anal condylomata. Hemoglobin was 14.3 g/dL, white blood cell count 6200/cm3, and platelet count 230,000/cm3. Serum electrolytes and lactate dehydrogenase were normal. The results of his liver function tests (LFTs) demonstrated a serum aspartate transaminase of 60 U/L (normal, 7‐40 U/L), alanine transaminase of 125 U/L (normal, 5‐50 U/L), alkaline phosphatase 218 U/L (normal, 40‐150 U/L), and total bilirubin 2.1 mg/dL (normal, 0.0‐1.5 mg/dL). Urinalysis demonstrated 2+ bilirubin and was otherwise normal. His erythrocyte sedimentation rate was 32 mm/hr (normal, 0‐15 mm/hr).
After 3 weeks of illness, his CBC demonstrates no signs of chronic illness (such as anemia of a chronic disease or a reactive leukocytosis or thrombocytosis). The results of his liver function tests showed moderate elevation, slightly more cholestatic than hepatocellular. This finding may reflect a disease process involving the liver, but such abnormal findings are often nonspecific in acute and chronic illnesses. With an unremitting fever, infectious complications in the liver merit early consideration. The time course rules out common biliary disorders such as cholangitis or cholecystitis. Pyogenic or amoebic liver abscesses are possible (homosexual men are at increased risk for the latter), but the absence of pain or abdominal tenderness is atypical. This biochemical profile can also be seen in chronic (but not acute) viral infections of the liver. Chronic hepatitis B and C predispose to hepatocellular carcinoma (HCC), which can be associated with fever. Cancers that infiltrate the liver, such as lymphoma or carcinoma, could also account for this picture. Indolent infections such as tuberculosis (TB) and syphilis are also possible, so associated signs of these systemic diseases should be sought. I do not believe either of his antibiotics is commonly associated with LFT abnormalities, and his CD4 count is too high for HIV cholangiopathy. In sum, a host of liver diseases are possible, but an extrahepatic systemic disease deserves equal attention.
His CD4+ count was 537 cells/mL, and his HIV RNA viral load was 44,300 copies/mL. Radiographs of the chest were normal. Two sets of blood cultures were negative. The rapid plasma reagin (RPR) was nonreactive. The results of serologies for acute hepatitis A, B, C, and E, chronic hepatitis B and C, and toxoplasmosis were negative. Testing for both Epstein‐Barr virus and cytomegalovirus showed evidence of remote infection. Results of serologies for bartonella species, human herpesviruses 6 and 7, and parvovirus B19 were negative.
The negative RPR makes disseminated (secondary) syphilis improbable, provided the prozone phenomenon has been excluded. An extensive serological workup is common in the evaluation of fever of unknown origin, although the threat of false‐positive results always looms when many studies are sent simultaneously. This must be considered in advance here, as his relatively preserved CD4 count affords him significant protection against many opportunistic infections. His HIV infection, however, regardless of CD4 count, increases his risk for TB and lymphoma, which remain high on my list. Both may be residing primarily in the liver. In FUO, the abdominal CT is frequently a high‐yield test (primarily by demonstrating unsuspected tumors and abscesses), even in the absence of symptoms, and would certainly be of interest here given the liver function test results. Imaging could diagnose febrile tumors such as lymphoma, HCC, or renal cell carcinoma. In the event that imaging is unrevealing, causes of granulomatous hepatitis should be entertained. The constellation of cough, LFT abnormalities, and fever is compatible with Q fever. As with any FUO case, I would also carefully revisit this patient's history to discern where he was born, where he has been, and what activities or exposures he is engaged in.
He was seen 2 days later with fever of 104F and new papules over his sternal area. Over the next week, he had intermittent fevers and severe fatigue. The rash progressed, predominantly involving his chest and back, but also his legs, arms, and face (see Fig. 1). The lesions spared his palms and soles. The exanthem was intensely pruritic and maculopapular, consisting of lesions with a diameter of 0.5 cm or less, with some scaling. There were no vesicles or pustular lesions. There were no other new findings on examination. His transaminase and bilirubin had normalized, and his CBC and electrolytes were unchanged. Repeat blood cultures held for extended incubation were negative. Computerized tomography of the chest, abdomen, and pelvis demonstrated mild lymphadenopathy at the porta hepatis with increased portocaval and periaortic lymphadenopathy.
The only LFT abnormality that persists is the elevated alkaline phosphatase, which suggests (1) that liver involvement was not specific and that there is a disease process involving the bone, (2) that there is a persistent infiltrative disorder of the liver such as infection or malignancy or, less likely, amyloidosis or sarcoidosis, or (3) that the porta hepatis lymphadenopathy is causing biliary obstruction. The underlying diagnosis must explain the rash, intraabdominal lymphadenopathy, and fever. The time course does somewhat limit the extensive differential of fever and rash. After 3 weeks of illness, some of the most life‐threatening entities such as meningococcal disease, Rocky Mountain spotted fever, and toxic shock syndrome are unlikely. Concern remains for infections that are more indolent, such as mycobacteria, fungi, or spirochetes. The most striking elements of the rash are the extensive distribution, rapid progression, large number, and discreteness of the lesions, which collectively point more toward disseminated fungal (eg, histoplasmosis, as he lives in Ohio), spirochetal, rickettsial, or viral etiologies, rather than bacterial or mycobacterial entities. The absence of vesicles detracts from the diagnosis of a disseminated herpes virus such as herpes simplex or varicella. I believe that this rash is too disseminated to be caused by a common mycobacterial illness. This extent of cutaneous metastases would usually accompany a far more ill patient with an obvious primary cancer (none is seen on imaging, including the liver), and it appears too extensive to be caused by a paraneoplastic phenomenon such as Sweet's syndrome. A systemic vasculitis or another autoimmune disease remains possible, but there is minimal evidence of visceral organ involvement. All the aforementioned diseases could explain the intraabdominal lymphadenopathy, but my suspicion is highest for infection. I would biopsy and culture the skin lesions, repeat the RPR and/or send a treponemal‐specific test, place a PPD skin test, and send fungal studies (serum serologies and urine antigens) for evaluation. If the results of these noninvasive studies are unrevealing, I would consider a liver biopsy.
The patient's medications were discontinued, and a skin biopsy of the rash from his chest showed atypical lymphohistiocytic infiltrates without acute inflammatory cells and with negative Gomori methenamine silver (GMS), acid‐fast bacilli (AFB), and Fite (for Nocardia) stains. The infiltrates were predominantly T cells with a 1:1 CD4:CD8 ratio. This was read as suspicious for cytotoxic (CD8) mycosis fungoides.
I do not have reason to doubt the pathologist's impression of mycosis fungoides on histopathologic grounds, but from a clinical standpoint, I do not think mycosis fungoides is a disease that has a prolonged febrile prodrome or an explosive cutaneous onset. Rather, it is frequently preceded by nonspecific skin findings over a long period. Thinking broadly and pathophysiologically and noting that T cells are the predominant lymphocytes in skin, I wonder if they could represent a nonmalignant, immunological reaction in the skin. The stains, although not perfectly sensitive, make mycobacterial and fungal diseases less likely, although incubation of cultures is necessary.
Over the next 10 days (bringing the total duration of the patient's illness to 6 weeks), the skin lesions increased in number. In the physician's office at his next follow‐up, the patient had a temperature of 104.1F, was uncomfortable, shivering, and ill‐appearing. His blood pressure was 108/66 mm Hg, and his pulse 114 beats/min. He complained of severe shooting pains, predominantly in his pretibial regions and arms. Examination showed no other new findings, including no focal neurological findings. The results of the T‐cell rearrangement study from the skin biopsy showed evidence of a monoclonal T‐cell population. He was admitted to the hospital for further evaluation and treatment.
The extremity dysesthesias could represent a lesion of the spinal cord (including the CSF/meninges), a polyradiculopathy, or a polyneuropathy. Unfortunately, this does not add a tremendous amount of diagnostic resolution, as infection, malignancy, and autoimmune syndromes, such as vasculitis, may all involve the nervous system in these ways. In general, I associate monoclonal lymphocyte responses with hematological malignancies and polyclonal responses with the less specific inflammation that could accompany infection, autoimmunity, or solid malignancies. His age, fever, and rapid progression seem atypical for mycosis fungoides, but given the monoclonal T cells, this must now be considered. Adult T‐cell leukemia/lymphoma, with its prominent skin manifestations and its association with HLTV‐1, is an alternative T‐cell malignancy that could explain the fever, neurological symptoms, and possible visceral involvement (elevated alkaline phosphatase, which could reflect liver or bone). In cases that are diagnostic challenges, one of the highest‐yield maneuvers is to repeat the preceding evaluation, starting with the history, exam, and basic labs, and if necessary, to review or repeat the imaging or skin biopsy. Given the elevated alkaline phosphatase, disseminated rash, new neurological symptoms, and his HIV status, I remain particularly concerned about syphilis and would do further testing (accounting for the prozone phenomenon) before proceeding with the malignancy evaluation.
A lumbar puncture demonstrated clear cerebrospinal fluid, with 2 leukocytes and 195 erythrocytes/cm3, protein of 26 mg/dL, and glucose of 52 mg/dL. Bacterial and fungal cultures of the fluid were negative. The results of colonoscopy were normal. A bone marrow biopsy demonstrated ring granulomas. GMS, AFB, Fite, and Steiner (for spirochetes) stains were negative, cultures of the aspirate were negative for bacteria, and smears were negative for fungi and mycobacteria. Antibody tests for human T‐cell lymphotropic virus types I and II, Coxiella burnetii, and Bartonella henselae were negative. The dermatology consultant believed the absence of lymphadenopathy and the pruritic nature of the lesions was atypical for cytotoxic T‐cell lymphoma (CTCL). Before initiating therapy for CTCL, she suggested repeating the skin biopsy and RPR.
The repeat RPR was positive at 1:64 dilutions, and a confirmatory fluorescent treponemal antibody absorption test showed a positive result. He was prescribed intramuscular benzathine pencillin 2.4 million units weekly for 3 weeks, with almost immediate defervescence and slower resolution of his rash and shooting pains in his limbs. The repeat skin biopsy done during the hospitalization demonstrated lichenoid‐type dermatitis with interstitial and perivascular lymphohistiocytic infiltrates and granulomas. Steiner stains for spirochetes were positive. Immunohistochemical stains ruled out a lymphoproliferative process. One year later his RPR was nonreactive.
COMMENTARY
Fever of unknown origin (FUO) was first defined by Petersdorf and Beeson in 1961 as a temperature higher than 38.3C on several occasions lasting longer than 3 weeks and defying diagnosis despite 1 week of inpatient investigation.1 Dramatic changes in medical practice have rendered this definition outdated, with more recent proposals allowing thoughtful outpatient investigation to serve as a surrogate for hospitalization. Some have proposed that HIV‐associated FUO be considered a distinct entity, with the most complete North American series finding the etiology of the HIV‐associated FUO in 56 of 70 patients.2 The mean CD4+ count in this series was 58/cm3. Disseminated M. avium was the most frequently diagnosed cause, followed by P. jirovecii pneumonia, cytomegalovirus infection, disseminated histoplasmosis, and lymphoma. Of 14 patients with fever of no definable etiology, 12 eventually proved to have self‐limiting illness.
Despite numerous attempts to reduce the investigation of the patient with FUO to an algorithm, the approach must be individualized. A thorough history and careful, serial physical examinations are frequently and appropriately stressed as the foundation, followed by thoughtful selection of laboratory and imaging studies. Although FUO has a lengthy differential diagnosis, it often proves to be, as Mackowiak and Durack stress, an unusual manifestation of a common disease, rather than a typical presentation of a rare disease.3 A relatively uncommon disease in conjunction with an initially negative diagnostic test result, as was the case with this patient, may lead to a protracted diagnostic puzzle.
Syphilis is a rare cause of FUO. In 6 large studies of a total of 947 patients published over a 40‐year period, only 2 cases of syphilis (1 secondary and 1 neurosyphilis) were reported.1, 48 Syphilis as a cause of prolonged cryptic fever appears to have been seen with greater frequency in the preantibiotic era.9 In the first half of the 20th century, syphilis was known as the great imitator, with its unusual manifestations recognized and indeed expected. As a result of the dramatically lower incidence of syphilis in recent decades, these lessons have largely been forgotten, however, which may lead to diagnostic confusion when syphilis presents atypically. The manifestations of secondary syphilis are protean, including a variety of rashes, aphthous ulcers, arthralgias, pharyngitis, weight loss, fever, meningitis, ocular symptoms, cranial nerve palsies, glomerulonephritis, hepatitis, and periostitis (which afflicted this patient, who complained of severe shooting pains in his arms and shins).
After declining in the last decade of the 20th century, the rates of primary and secondary syphilis are rising in the United States.10 Oral sex is a clear risk factor for syphilis transmission, particularly for men who have sex with men.11 Because of the patient's exposure history and clinical picture, his outpatient physician considered the diagnosis of secondary syphilis early in the course of his illness. The diagnosis was not entertained further when an RPR test, highly sensitive at this stage of the disease, returned nonreactive. Likewise, when a rash subsequently appeared, the lack of palm and sole involvement dissuaded multiple clinicians from reconsidering the diagnosis of syphilis. A skin biopsy that appeared to lead in a distinctly different direction understandably confused the picture still further. Even at the time of the lumbar puncture, VDRL of the CSF was not ordered.
In retrospect, the chief confounder in the case was the false‐negative RPR test, as the discussant suspected early on. Although nontreponemal tests are generally accurate in individuals with HIV, delayed seropositivity and false‐negatives have been reported in this population.12 The false‐negative could have also been a result of the prozone phenomenon, an unusual event, occurring in fewer than 2% of cases of secondary syphilis and attributed to a mismatch between antibody and very high antigen level. The prozone reaction can be corrected for by requesting dilution of the serum prior to repeating the test. Simple lab error must be considered as well, but without access to this patient's serum from his original testing, the cause of his initial false‐negative test cannot be known with certainty.
An unusual presentation in conjunction with failure to recognize the causes of rare false‐negative testing for secondary syphilis led to a delayed diagnosis in this patient. Although syphilis and mycosis fungoides have previously been reported to mimic one another both clinically and histopathologically, the potential for secondary syphilis to be misdiagnosed in this fashion is not generally appreciated.1315 Recognition of the possibility of secondary syphilis occurred just in time to spare this patient the rash decision of treating him with cytotoxic therapy directed against CTCL.
Teaching Points
-
HIV‐associated FUO can be a diagnostic challenge, but an etiology can be found in most cases.
-
Syphilis continues to be an unusual cause of FUO and can have protean manifestations affecting nearly every organ system
-
The sensitivity of RPR is extremely high in secondary syphilis, but false‐negative tests can be seen in HIV because of both the prozone phenomenon and a delayed rise in antibodies.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
- ,.Fever of unexplained origin: Report on 100 cases.Medicine.1961;40:1–30.
- ,,.Human immunodeficiency virus‐associated fever of unknown origin: A study of 70 patients in the United States and review.Clin Infect Dis.1999;28:341–345.
- ,.Fever of unknown origin. In:Mandell GL,Bennett JE,Dolin R, eds.Principles and Practice of Infectious Diseases.6th ed.Philadelphia:Elsevier Churchill Livingstone;2005:718–729.
- ,,.Fever of unknown origin: Diagnosis and follow‐up of 105 cases, 1970‐1980.Medicine.1982;61:269–292.
- ,,,.Fever of unknown origin in the 1980s: An update of the diagnostic spectrum.Arch Intern Med.1992;152:51–55.
- .Fever of unknown origin: Review of 86 patients treated in community hospitals.Clin Infect Dis.1992;15:968–973.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO study group.Medicine.1997;76:392–400.
- ,,, et al.From prolonged febrile illness to fever of unknown origin: The challenge continues.Arch Intern Med.2003;163:1033–1041.
- ,.The diagnosis of obscure fever. II. The diagnosis of unexplained high fever.Bull Johns Hopkins Hosp.1936;58:307–331.
- Centers for Disease Control and Prevention.Primary and secondary syphilis—United States, 2003–2004.MMWR.2006;55:269–273.
- Transmission of primary and secondary syphilis by oral sex—Chicago, Illinois, 1998‐2202.MMWR.2004;53:966–968.
- ,,, et al.Seronegative secondary syphilis in 2 patients coinfected with human immunodeficiency virus.Arch Dermatol.2005;141:431–433.
- ,,,.Secondary syphilis mimicking mycosis fungoides.J Am Acad Dermatol.1980;3:92–94
- ,.A case of lues maligna in a patient with acquired immunodeficiency syndrome (AIDS).Scand J Infect Dis.2005;37:697–700.
- ,,,,.Unusual presentation of secondary syphilis in 2 HIV‐1 positive patients.Cutis.2000;66:383–389.
Fishing for a Diagnosis
A 54‐year‐old man with hypertension and type 2 diabetes mellitus entered the Chest Pain Evaluation Unit of a teaching hospital after 12 hours of intermittent thoracic discomfort. The pain began during dinner and was sharp, bandlike, and located beneath the sternum and across the entire chest. He had dyspnea but no diaphoresis or nausea. A recumbent position relieved the pain after dinner, but it recurred during the night and again the following morning. He did not smoke and had no family history of coronary artery disease.
A useful approach in evaluating acute chest pain is to employ a hierarchical differential diagnosis that emphasizes life‐threatening disorders requiring prompt recognition and intervention. Most prominent are cardiac ischemia, pericardial tamponade, pneumothorax, pulmonary embolus, esophageal rupture, and aortic dissection. The concurrent dyspnea and retrosternal location and intermittent nature of the pain that this patient has are consistent with myocardial ischemia, but the sharp quality of the pain and the relief gained by being recumbent are atypical. Pain with pericarditis is characteristically pleuritic and often worse when lying down. The pain of pneumothorax is typically unilateral, not intermittent, and unlikely to improve with recumbency. Although pain during eating suggests the possibility of an esophageal source, spontaneous rupture usually follows vomiting. The pain is typically continuous and severe. The pain of pulmonary embolism may be unilateral and pleuritic but often is more diffuse. Relief by recumbency is unusual, but the intermittent nature could suggest recurrent emboli. The patient has a history of hypertension, which predisposes him to aortic dissection, in which the pain is typically sharp, continuous, and severe but occasionally intermittent. Among numerous less urgent diagnoses are esophagitis and thoracic diabetic radiculopathy.
Important features to look for during this patient's examination include: disappearance of the radial pulse during inhalation, a simple screening test that is insensitive but very specific for pericardial tamponade; elevated neck veins, which can occur with tension pneumothorax, massive pulmonary embolism, and pericardial tamponade; pericardial and pleural friction rubs; discrepant blood pressures in the 2 arms, sometimes a sign of aortic dissection; local thoracic tenderness from chest wall disorders; and sensory examination of the chest surface, which is often abnormal in diabetic thoracic radiculopathy. Given this patient's age and history of diabetes, I am most concerned about myocardial ischemia. The most appropriate diagnostic tests include an electrocardiogram and a chest radiograph.
The patient appeared apprehensive but reported no pain. He had a temperature of 36.0 C, heart rate of 95 beats/minute, blood pressure of 138/77 mm Hg, respiratory rate of 16/minute, and oxygen saturation of 99% while breathing ambient air. Blood pressures were equal in both arms. Jugular venous distention was absent, and the lung and cardiac examinations had normal results. His pain did not increase on chest wall palpation. Examination of the abdomen, extremities, and the neurologic system showed normal results.
The results of laboratory tests showed a leukocyte count of 12,700/cm3, with 85% neutrophils, 8% lymphocytes, 6% monocytes, and 1% eosinophils. The hematocrit was 45%, and the platelet count was 172,000/cm3. The results of his chemistry panel were remarkable only for a glucose of 225 mg/dL. An electrocardiogram (ECG) showed a normal sinus rhythm and left anterior fascicular block without acute ST‐ or T‐wave changes. Prior ECGs were unavailable. An anteroposterior radiograph disclosed low lung volumes and bibasilar opacities. No pleural effusion was noted. Serial serum troponin and creatine kinase levels were normal. A Tc‐99m tetrofosmin cardiac nuclear perfusion test performed at rest demonstrated a moderate area of mildly decreased uptake along the inferior wall extending to the apex. An exercise treadmill test, terminated after 1 minute, 45 seconds because of chest pain, provoked no ECG changes diagnostic of ischemic disease.
The absence of elevated jugular venous pressure virtually eliminates pericardial tamponade as a diagnosis, and the chest film excludes pneumothorax. The intermittent nature of the chest pain, the absence on the chest radiograph of such findings as mediastinal gas, left pneumothorax, or hydropneumothorax, and the lack of a predisposing cause make esophageal rupture unlikely. Pulmonary emboli remain a consideration despite the normal oxygen saturation because there is no hypoxemia in a substantial minority of such cases. The normal cardiac enzyme levels and the lack of significant changes on the ECG exclude that a myocardial infarction has recently occurred, but cardiac ischemia remains a possibility, especially because the patient had chest pain on exercise and the nuclear scan indicated diminished blood flow to the inferior left ventricle. Aortic dissection still lurks as a possibility. The inferior wall abnormalities seen on the scan could result from dissection into the right coronary artery, which is more frequently involved than the left, or compression of it by an enlarged aorta, but they also may be artifacts. The leukocytosis may be a nonspecific response to stress but could indicate, although unlikely, infections such as mediastinitis from esophageal rupture or bacterial aortitis.
A conscientious clinician would repeat the history, reexamine the patient, and scrutinize the chest film to determine what the bilateral opacities represent. Given the story so far, however, I might consider a thoracic computed tomography (CT) angiogram because I am most concerned about pulmonary emboli and aortic dissection.
On hospital day 3, the patient had worsening dyspnea and persistent chest pain. His temperature was 39.3C, and his oxygen saturation decreased to 89% while breathing room air. Repeat chest radiography showed new bilateral pleural effusions and increased bibasilar opacification (Fig. 1). His leukocyte count was 19,000/cm3, with 88% neutrophils, 6% lymphocytes, 5% monocytes, and 1% eosinophils. Care was transferred from the chest pain team to an inpatient general medicine ward team. A pulmonary CT angiogram showed no large central clots but suggested emboli in the right superior subsegmental artery and a right upper lobe subsegmental artery. Bilateral pleural effusions were observed, as were bilateral pleural‐based atelectasis or infiltrates in the lower lungs. A hiatal hernia was noted, but no aortic dissection. The patient received supplemental oxygen, intravenous levofloxacin, and unfractionated heparin by continuous infusion.
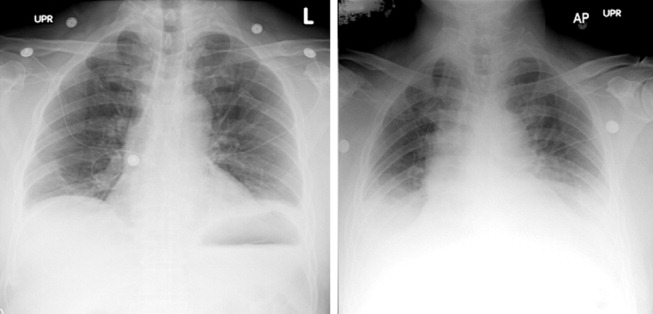
Without other information, I will assume that the fever is part of the patient's original disease and not a nosocomial infection or drug fever. At this point, a crucial part of the evaluation is examining the CT scan with experienced radiologists to determine whether the abnormalities noted are genuinely convincing for pulmonary emboli. If the findings are equivocal, the next step might be a pulmonary angiogram or the indirect approach of evaluating the leg veins with ultrasound, reasoning that the presence of proximal leg vein thromboses would require anticoagulation in any event.
The patient's worsening chest pain and hypoxemia are consistent with multiple pulmonary emboli. Bilateral pleural effusions and leukocytosis can occur but are uncommon. Because of the fever, another possibility is septic pulmonary emboli, but he has no evidence of suppurative thrombophlebitis of the peripheral veins, apparent infection elsewhere, or previous intravenous drug abuse causing right‐sided endocarditis. An alternative diagnosis is infection of an initially bland pulmonary infarct.
An important consideration is a thoracentesis, depending on how persuasive the CT diagnosis of pulmonary embolism is and the size of the pleural effusions. It should be done before instituting antimicrobial therapy, which may decrease the yield of the cultures, and before starting heparin, which increases the risk of bleeding and occasionally causes a substantial, even fatal hemothorax.
The patient's oxygenation and dyspnea did not improve. Over the next day, he repeatedly mentioned that swallowing, particularly solid foods, worsened his chest pain. He had a temperature of 39.9C, a heart rate of 121 beats/minute, blood pressure of 149/94 mm Hg, a respiratory rate of 28/minute, and oxygen saturation of 93% while breathing 40% oxygen. He had inspiratory splinting, percussive dullness at both lung bases, and distant heart sounds. A contrast esophagogram showed distal narrowing that prevented solid contrast from passing, but no hiatal hernia. Blood and urine cultures obtained before antibiotic therapy were sterile. Duplex ultrasonography of bilateral lower extremities showed no evidence of deep venous thrombosis. A pulmonary angiogram revealed no emboli, and heparin was discontinued. Bilateral thoracentesis yielded grossly bloody fluid. Repeat chest CT (Fig. 2) demonstrated large bilateral effusions, a new large pericardial effusion, and a prominence at the gastroesophageal junction more concerning for a soft‐tissue mass than for a hiatal hernia, although the quality of the study was suboptimal because of an absence of oral contrast.
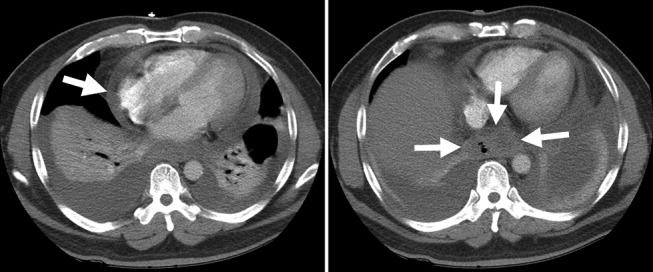
The CT scan suggests a paraesophageal abscess from an esophageal rupture. As mentioned earlier, if rupture occurs spontaneously, it typically follows retching or vomiting and is called Boerhaave's syndrome. Another consideration is a rupture secondary to an external insult, such as trauma or ingestion of a caustic substance. In evaluating these possibilities, the patient should have been asked 4 questions at the initial interview that I neglected to explicitly highlight earlier. First, what was he eating when he developed the chest pain? Second, did the pain begin during swallowing? Third, did he have previous symptoms suggesting an esophageal disorder such as dysphagia, odynophagia, or heartburn? These might indicate a cancer that could perforate or another problem such as a stricture or disordered esophageal motility that might have caused a swallowed item to lodge in the esophagus. Finally, did he have retching or vomiting? Though not routinely part of the review of systems, the former 2 questions are an appropriate history‐prompted line of questioning of a patient with onset of chest pain while eating.
At this point, a reasonable approach would be an esophagoscopy to delineate any intraluminal problems, such as a cancer or a foreign body. The apparent obstruction seen on barium swallow may be from extrinsic pressure from a paraesophageal abscess. The patient should receive broad‐spectrum antimicrobial therapy effective against oral anaerobes. Although occasionally patients recover with antibiotics alone, surgery is usually required. I am surprised that the original CT scan did not show evidence of an esophageal perforation. Possibly, the hiatal hernia was a paraesophageal abscess poorly characterized because of the lack of oral contrast.
The team, concerned about esophageal perforation, began the patient on intravenous clindamycin. The patient underwent video‐assisted thoracoscopic drainage, which yielded a moderate amount of turbid, bloody fluid from each hemithorax. The pericardium contained approximately 500 cm3 of turbid fluid. Gram stain and culture of these fluids were negative. No esophageal or mediastinal mass was noted during surgery. Intraoperative esophagogastroduodenoscopy with endoscopic ultrasound showed a healing linear mucosal tear in the distal esophagus (Fig. 3) as well as air/fluid collection in the esophageal soft tissue (not shown).
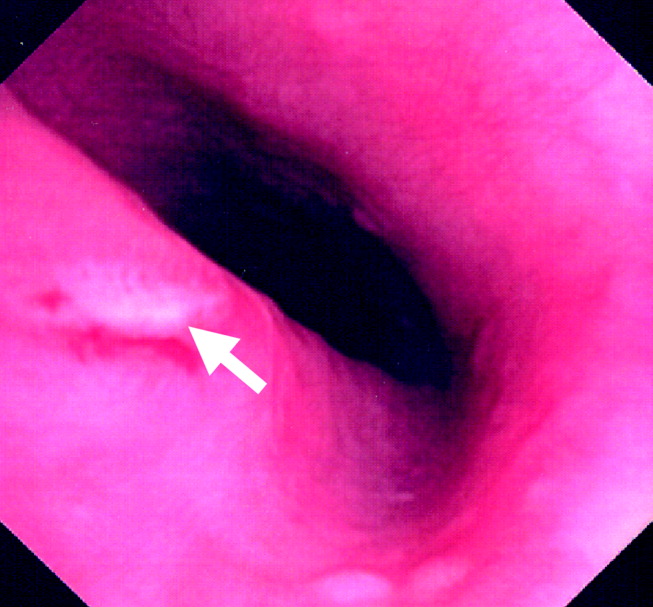
On further questioning postoperatively, the patient reported eating bony fish during the dinner when he first experienced chest pain. The patient received a 21‐day course of oral clindamycin and completely recovered. Five weeks later, a chest CT showed decreased distal esophageal thickening and no mediastinal air.
COMMENTARY
Esophageal perforation is an uncommon but life‐threatening cause of chest pain. In most series iatrogenic injury accounts for more than 70% of cases, whereas most of the other cases have spontaneous (5%20%) or traumatic (4%10%) causes (Table 1).14 Perforation as a complication of ingesting fish bones, although rare, is well described and continues to be reported.57
| Etiology | Percent |
|---|---|
| |
| Iatrogenic | 45%77% |
| Rigid or flexible endoscopy, balloon dilation, Blakemore tube, sclerotherapy, operative injury | |
| Increased intraesophageal pressure (Boerhaave's syndrome) | 5%20% |
| Vomiting or retching, weightlifting, childbirth | |
| Traumatic | 4%10% |
| Penetrating or blunt injury to neck or chest | |
| Ingestion | 0%12% |
| Foreign body, toxic or caustic substance | |
| Miscellaneous | 0%5% |
| Malignancy, Barrett's esophagus, infection, aortic dissection | |
Diagnosis of esophageal perforation secondary to a foreign body may be difficult because of the considerable overlap of symptoms with other causes of chest pain and failure to consider this infrequent condition in the absence of a classic history of retching. To diagnose such a disease, physicians must gather data from various sourcesespecially the history, physical examination, and medical recordformulate hypotheses, integrate results from diagnostic tests, and then assess the importance of the available information in the context of a differential diagnosis. Incorrectly evaluating or failing to obtain essential data can lead to incorrect or delayed diagnoses.
In This Patient's Evaluation, What Prevented Prompt Recognition of Esophageal Perforation?
The critical misstep was an incomplete history, both on arrival and when the patient was transferred to a second team. The presence of risk factors for coronary artery disease led the providers to first consider myocardial ischemia. They failed to ask crucial questions about the onset of the painwhen it occurred during the meal and what he was eatingeven when the patient later complained of odynophagia. As a result of the incomplete history, the providers, puzzled by the patient's ongoing and evolving symptoms, ordered numerous unnecessary diagnostic tests that gave false‐positive results, leading to potentially harmful treatment including anticoagulation. The discussant mentions that the preferred response to a puzzling clinical situation is to return to the bedside and repeat the history, reexamine the patient, and reevaluate available informationsimple steps that can often resolve diagnostic dilemmas.
There is ongoing concern that the history‐taking and physical examination skills of clinicians are in decline.814 Many speculate this is in part due to reliance on increasingly sophisticated diagnostic tests. Providers may overly rely on modern diagnostic tests because of their familiarity with the sensitivity and specificity of such tests, fear of malpractice litigation, diminishing opportunity to elucidate the complete history and physical exam, or lack of confidence in their history‐taking and examination skills.814 Although the rapid development and implementation of advanced diagnostic technologies have had a significant impact on diagnostic accuracy, the estimated rate of disease misdiagnosis remains elevated at 24%.1518 In contrast to technology‐based testing, the history and physical provide an inexpensive, safe, and effective means of at arriving at a correct diagnosis. In outpatient medical visits the history and physical, when completely elicited, result in a correct diagnosis of up to 70%90% of patients.8, 19, 20 Even for illnesses whose diagnosis requires confirmation by a diagnostic test, the definitive test can only be selected after a sufficient history and exam provide an assessment of the pretest probability of disease.
In evaluating chest pain there is an additional potential factor that diminishes reliance on bedside assessment. Modern quality assurance measures and chest pain units encourage clinicians to evaluate patients with chest pain quickly because any delay diminishes the benefits of therapies for acute coronary syndromes. In the emergency room, these patients find themselves on a rapidly moving diagnostic conveyor belt, an approach that is efficient and appropriate given the high prevalence of coronary disease but that also contributes to inattentiveness and error for patients with unusual diagnoses.
How Could Clinicians in Our Case Use Bedside Evidence to Help Differentiate Our Patient?
For most patients with chest pain there is no finding that would change diagnostic probabilities enough to take them off the diagnostic conveyor belt. Nevertheless, several bedside findings can help providers to rank‐order a differential diagnosis, thereby improving the sequence in which diagnostic testing is done. For patients with chest pain the ECG has the highest predictive ability of all studied history, physical exam, and ECG findings (Table 2).21 A history of sharp and positional pain descriptors diminishes the probability of myocardial ischemia.21 Unfortunately, no history, exam, or ECG feature is sensitive enough, either alone or in combination, to effectively rule out myocardial ischemia.
| Finding | Positive LR* |
|---|---|
| Myocardial ischemia | |
| ST segment elevation or Q wave | 22 |
| S3 gallop, blood pressure < 100 mm Hg, or ST segment depression | 3.0 |
| Sharp or positional pain | 0.3 |
| Pulmonary embolism | |
| Low clinical probability | 0.2 |
| Medium clinical probability | 1.8 |
| High clinical probability | 17.1 |
| Aortic dissection | |
| Tearing or ripping pain | 10.8 |
| Focal neurologic deficits | 6.633 |
| Ipsilateral versus contralateral pulse deficit | 5.7 |
| Cardiac tamponade | |
| Pulsus paradoxus > 12 mm Hg | 5.9 |
| Esophageal perforation | |
| Dysphagia, odynophagia, retching, vomiting, or subcutaneous emphysema | ? |
The history and exam can also facilitate differentiation of noncoronary causes of life‐threatening chest pain. The dismal performance of individual bedside findings for pulmonary embolism is what led to development of quantitative D‐dimer assays and objective methods based on bedside evaluation, including the widely used Wells Score.22 This score can be used to classify patients as having low, medium, and high risk of pulmonary embolism, facilitating management decisions after diagnostic imaging is obtained.23 Fewer than half of all patients with thoracic aortic dissection have classic exam findings; however, when present, they can appropriately raise the probability of dissection higher on the differential diagnosis.24 Importantly, no history or exam finding argues against dissection.24 Most patients with cardiac tamponade will have elevated jugular venous pressure (76%100%); however, poor interobserver agreement about this finding may decrease its detection.11, 25, 26 As the discussant notes, total paradox, defined as the palpable pulse disappearing with inspiration, is an insensitive test for tamponade, present in only 23% of patients with the disorder. In contrast, an inspiratory drop in systolic blood pressure of more than 12 mm Hg should prompt consideration for tamponade.11, 26 Commonly taught features of esophageal perforation, including chest pain, dysphagia, odynophagia, prior retching or vomiting, subcutaneous emphysema, dyspnea, and pleural effusions, vary in their reported sensitivity, but their specificity is virtually never reported.27
Like most patients with chest pain, our patient lacked all these symptoms and signs, arguing for myocardial ischemia, although he had a few signs that argued against it (sharp and positional chest pain). After the initial CXR and ECG, further testing with cardiac biomarkers was appropriate, but a fundamental error was made in not returning to the patient's bedside to repeat the interview and examination after the cardiac biomarkers were found to be normal. Had this been done, several cluesdysphagia, onset of pain with eating bony fish, and feverwould have pushed esophageal perforation to the top of the differential diagnosis. Subsequent testing would have led to the correct diagnosis and avoided a potentially harmful diagnostic fishing expedition.
Take‐Home Points
-
Esophageal perforation is an uncommon but life‐threatening cause of chest pain that is difficult to diagnose because of its nonspecific symptoms.
-
An accurate and complete history and exam can reveal signs and symptoms that influence the likelihood of each life‐threatening cause of chest pain. Evaluating patients for these features is vital to the rank ordering of a differential diagnosis and the selection of appropriate diagnostic tests.
-
There is no substitute for repeating the history, reexamining the patient, and reevaluating available information when confronted with a confusing constellation of symptoms.
Acknowledgements
The authors thank Steve McGee for his thoughtful review and comments on the manuscript.
- ,.Esophageal perforations: a 15 year experience.Am J Surg.1982;143:495–503.
- ,,.Esophageal perforation: emphasis on management.Ann Thorac Surg.1996;61:1447–1451; discussion1451–1452.
- ,,,,,.Evolving options in the management of esophageal perforation.Ann Thorac Surg.2004;77:1475–1783.
- ,.Personal management of 57 consecutive patients with esophageal perforation.Am J Surg.2004;187:58–63.
- ,,,,.Perforation of the oesophagus and aorta after eating fish: an unusual cause of chest pain.Emerg Med J.2003;20:385–386.
- ,,,.Esophageal perforation and mediastinitis from fish bone ingestion.South Med J.2003;96:516–520.
- ,,,.A 73‐year‐old man with chest pain 4 days after a fish dinner.Chest.2004;126:294–297.
- ,.The science of the art of the clinical examination.JAMA.1992;267:2650–2652.
- .Clinical skills in the 21st century.Arch Intern Med.1994;154:22–24.
- ,.Pulmonary auscultatory skills during training in internal medicine and family practice.Am J Respir Crit Care Med.1999;159:1119–1124.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:Saunders;2001.
- .Simple is beautiful: the neglected power of simple tests.Arch Intern Med.2004;164:2198–2200.
- ,.Pearls and pitfalls in patient care: need to revive traditional clinical values.Am J Med Sci.2004;327:79–85.
- ,. Physical diagnosis: a lost art? Agency for Health Research and Quality. WebM75:29–40.
- .Low‐tech autopsies in the era of high‐tech medicine: continued value for quality assurance and patient safety.JAMA.1998;280:1273–1274.
- ,,,.Has misdiagnosis of appendicitis decreased over time? A population‐based analysis.JAMA.2001;286:1748–1753.
- ,,,.Changes in rates of autopsy‐detected diagnostic errors over time: a systematic review.JAMA.2003;289:2849–2856.
- .Diagnostic Process.J Coll Gen Pract.1963;54:579–589.
- .The importance of the history in the medical clinic and the cost of unnecessary tests.Am Heart J.1980;100:928–931.
- ,.Bedside diagnosis of coronary artery disease: a systematic review.Am J Med.2004;117:334–343.
- ,,, et al.Use of a clinical model for safe management of patients with suspected pulmonary embolism.Ann Intern Med.1998;129:997–1005.
- ,,, et al.Diagnostic pathways in acute pulmonary embolism: recommendations of the PIOPED II investigators.Am J Med.2006;119:1048–1055.
- .Does this patient have an acute thoracic aortic dissection?JAMA.2002;287:2262–2272.
- ,.The rational clinical examination. Does this patient have abnormal central venous pressure?JAMA.1996;275:630–634.
- ,,,.Does this patient with a pericardial effusion have cardiac tamponade?JAMA.2007;297:1810–1818.
- ,.Spontaneous esophageal rupture: a frequently missed diagnosis.Am Surg.1999;65:449–452.
A 54‐year‐old man with hypertension and type 2 diabetes mellitus entered the Chest Pain Evaluation Unit of a teaching hospital after 12 hours of intermittent thoracic discomfort. The pain began during dinner and was sharp, bandlike, and located beneath the sternum and across the entire chest. He had dyspnea but no diaphoresis or nausea. A recumbent position relieved the pain after dinner, but it recurred during the night and again the following morning. He did not smoke and had no family history of coronary artery disease.
A useful approach in evaluating acute chest pain is to employ a hierarchical differential diagnosis that emphasizes life‐threatening disorders requiring prompt recognition and intervention. Most prominent are cardiac ischemia, pericardial tamponade, pneumothorax, pulmonary embolus, esophageal rupture, and aortic dissection. The concurrent dyspnea and retrosternal location and intermittent nature of the pain that this patient has are consistent with myocardial ischemia, but the sharp quality of the pain and the relief gained by being recumbent are atypical. Pain with pericarditis is characteristically pleuritic and often worse when lying down. The pain of pneumothorax is typically unilateral, not intermittent, and unlikely to improve with recumbency. Although pain during eating suggests the possibility of an esophageal source, spontaneous rupture usually follows vomiting. The pain is typically continuous and severe. The pain of pulmonary embolism may be unilateral and pleuritic but often is more diffuse. Relief by recumbency is unusual, but the intermittent nature could suggest recurrent emboli. The patient has a history of hypertension, which predisposes him to aortic dissection, in which the pain is typically sharp, continuous, and severe but occasionally intermittent. Among numerous less urgent diagnoses are esophagitis and thoracic diabetic radiculopathy.
Important features to look for during this patient's examination include: disappearance of the radial pulse during inhalation, a simple screening test that is insensitive but very specific for pericardial tamponade; elevated neck veins, which can occur with tension pneumothorax, massive pulmonary embolism, and pericardial tamponade; pericardial and pleural friction rubs; discrepant blood pressures in the 2 arms, sometimes a sign of aortic dissection; local thoracic tenderness from chest wall disorders; and sensory examination of the chest surface, which is often abnormal in diabetic thoracic radiculopathy. Given this patient's age and history of diabetes, I am most concerned about myocardial ischemia. The most appropriate diagnostic tests include an electrocardiogram and a chest radiograph.
The patient appeared apprehensive but reported no pain. He had a temperature of 36.0 C, heart rate of 95 beats/minute, blood pressure of 138/77 mm Hg, respiratory rate of 16/minute, and oxygen saturation of 99% while breathing ambient air. Blood pressures were equal in both arms. Jugular venous distention was absent, and the lung and cardiac examinations had normal results. His pain did not increase on chest wall palpation. Examination of the abdomen, extremities, and the neurologic system showed normal results.
The results of laboratory tests showed a leukocyte count of 12,700/cm3, with 85% neutrophils, 8% lymphocytes, 6% monocytes, and 1% eosinophils. The hematocrit was 45%, and the platelet count was 172,000/cm3. The results of his chemistry panel were remarkable only for a glucose of 225 mg/dL. An electrocardiogram (ECG) showed a normal sinus rhythm and left anterior fascicular block without acute ST‐ or T‐wave changes. Prior ECGs were unavailable. An anteroposterior radiograph disclosed low lung volumes and bibasilar opacities. No pleural effusion was noted. Serial serum troponin and creatine kinase levels were normal. A Tc‐99m tetrofosmin cardiac nuclear perfusion test performed at rest demonstrated a moderate area of mildly decreased uptake along the inferior wall extending to the apex. An exercise treadmill test, terminated after 1 minute, 45 seconds because of chest pain, provoked no ECG changes diagnostic of ischemic disease.
The absence of elevated jugular venous pressure virtually eliminates pericardial tamponade as a diagnosis, and the chest film excludes pneumothorax. The intermittent nature of the chest pain, the absence on the chest radiograph of such findings as mediastinal gas, left pneumothorax, or hydropneumothorax, and the lack of a predisposing cause make esophageal rupture unlikely. Pulmonary emboli remain a consideration despite the normal oxygen saturation because there is no hypoxemia in a substantial minority of such cases. The normal cardiac enzyme levels and the lack of significant changes on the ECG exclude that a myocardial infarction has recently occurred, but cardiac ischemia remains a possibility, especially because the patient had chest pain on exercise and the nuclear scan indicated diminished blood flow to the inferior left ventricle. Aortic dissection still lurks as a possibility. The inferior wall abnormalities seen on the scan could result from dissection into the right coronary artery, which is more frequently involved than the left, or compression of it by an enlarged aorta, but they also may be artifacts. The leukocytosis may be a nonspecific response to stress but could indicate, although unlikely, infections such as mediastinitis from esophageal rupture or bacterial aortitis.
A conscientious clinician would repeat the history, reexamine the patient, and scrutinize the chest film to determine what the bilateral opacities represent. Given the story so far, however, I might consider a thoracic computed tomography (CT) angiogram because I am most concerned about pulmonary emboli and aortic dissection.
On hospital day 3, the patient had worsening dyspnea and persistent chest pain. His temperature was 39.3C, and his oxygen saturation decreased to 89% while breathing room air. Repeat chest radiography showed new bilateral pleural effusions and increased bibasilar opacification (Fig. 1). His leukocyte count was 19,000/cm3, with 88% neutrophils, 6% lymphocytes, 5% monocytes, and 1% eosinophils. Care was transferred from the chest pain team to an inpatient general medicine ward team. A pulmonary CT angiogram showed no large central clots but suggested emboli in the right superior subsegmental artery and a right upper lobe subsegmental artery. Bilateral pleural effusions were observed, as were bilateral pleural‐based atelectasis or infiltrates in the lower lungs. A hiatal hernia was noted, but no aortic dissection. The patient received supplemental oxygen, intravenous levofloxacin, and unfractionated heparin by continuous infusion.
Without other information, I will assume that the fever is part of the patient's original disease and not a nosocomial infection or drug fever. At this point, a crucial part of the evaluation is examining the CT scan with experienced radiologists to determine whether the abnormalities noted are genuinely convincing for pulmonary emboli. If the findings are equivocal, the next step might be a pulmonary angiogram or the indirect approach of evaluating the leg veins with ultrasound, reasoning that the presence of proximal leg vein thromboses would require anticoagulation in any event.
The patient's worsening chest pain and hypoxemia are consistent with multiple pulmonary emboli. Bilateral pleural effusions and leukocytosis can occur but are uncommon. Because of the fever, another possibility is septic pulmonary emboli, but he has no evidence of suppurative thrombophlebitis of the peripheral veins, apparent infection elsewhere, or previous intravenous drug abuse causing right‐sided endocarditis. An alternative diagnosis is infection of an initially bland pulmonary infarct.
An important consideration is a thoracentesis, depending on how persuasive the CT diagnosis of pulmonary embolism is and the size of the pleural effusions. It should be done before instituting antimicrobial therapy, which may decrease the yield of the cultures, and before starting heparin, which increases the risk of bleeding and occasionally causes a substantial, even fatal hemothorax.
The patient's oxygenation and dyspnea did not improve. Over the next day, he repeatedly mentioned that swallowing, particularly solid foods, worsened his chest pain. He had a temperature of 39.9C, a heart rate of 121 beats/minute, blood pressure of 149/94 mm Hg, a respiratory rate of 28/minute, and oxygen saturation of 93% while breathing 40% oxygen. He had inspiratory splinting, percussive dullness at both lung bases, and distant heart sounds. A contrast esophagogram showed distal narrowing that prevented solid contrast from passing, but no hiatal hernia. Blood and urine cultures obtained before antibiotic therapy were sterile. Duplex ultrasonography of bilateral lower extremities showed no evidence of deep venous thrombosis. A pulmonary angiogram revealed no emboli, and heparin was discontinued. Bilateral thoracentesis yielded grossly bloody fluid. Repeat chest CT (Fig. 2) demonstrated large bilateral effusions, a new large pericardial effusion, and a prominence at the gastroesophageal junction more concerning for a soft‐tissue mass than for a hiatal hernia, although the quality of the study was suboptimal because of an absence of oral contrast.
The CT scan suggests a paraesophageal abscess from an esophageal rupture. As mentioned earlier, if rupture occurs spontaneously, it typically follows retching or vomiting and is called Boerhaave's syndrome. Another consideration is a rupture secondary to an external insult, such as trauma or ingestion of a caustic substance. In evaluating these possibilities, the patient should have been asked 4 questions at the initial interview that I neglected to explicitly highlight earlier. First, what was he eating when he developed the chest pain? Second, did the pain begin during swallowing? Third, did he have previous symptoms suggesting an esophageal disorder such as dysphagia, odynophagia, or heartburn? These might indicate a cancer that could perforate or another problem such as a stricture or disordered esophageal motility that might have caused a swallowed item to lodge in the esophagus. Finally, did he have retching or vomiting? Though not routinely part of the review of systems, the former 2 questions are an appropriate history‐prompted line of questioning of a patient with onset of chest pain while eating.
At this point, a reasonable approach would be an esophagoscopy to delineate any intraluminal problems, such as a cancer or a foreign body. The apparent obstruction seen on barium swallow may be from extrinsic pressure from a paraesophageal abscess. The patient should receive broad‐spectrum antimicrobial therapy effective against oral anaerobes. Although occasionally patients recover with antibiotics alone, surgery is usually required. I am surprised that the original CT scan did not show evidence of an esophageal perforation. Possibly, the hiatal hernia was a paraesophageal abscess poorly characterized because of the lack of oral contrast.
The team, concerned about esophageal perforation, began the patient on intravenous clindamycin. The patient underwent video‐assisted thoracoscopic drainage, which yielded a moderate amount of turbid, bloody fluid from each hemithorax. The pericardium contained approximately 500 cm3 of turbid fluid. Gram stain and culture of these fluids were negative. No esophageal or mediastinal mass was noted during surgery. Intraoperative esophagogastroduodenoscopy with endoscopic ultrasound showed a healing linear mucosal tear in the distal esophagus (Fig. 3) as well as air/fluid collection in the esophageal soft tissue (not shown).
On further questioning postoperatively, the patient reported eating bony fish during the dinner when he first experienced chest pain. The patient received a 21‐day course of oral clindamycin and completely recovered. Five weeks later, a chest CT showed decreased distal esophageal thickening and no mediastinal air.
COMMENTARY
Esophageal perforation is an uncommon but life‐threatening cause of chest pain. In most series iatrogenic injury accounts for more than 70% of cases, whereas most of the other cases have spontaneous (5%20%) or traumatic (4%10%) causes (Table 1).14 Perforation as a complication of ingesting fish bones, although rare, is well described and continues to be reported.57
| Etiology | Percent |
|---|---|
| |
| Iatrogenic | 45%77% |
| Rigid or flexible endoscopy, balloon dilation, Blakemore tube, sclerotherapy, operative injury | |
| Increased intraesophageal pressure (Boerhaave's syndrome) | 5%20% |
| Vomiting or retching, weightlifting, childbirth | |
| Traumatic | 4%10% |
| Penetrating or blunt injury to neck or chest | |
| Ingestion | 0%12% |
| Foreign body, toxic or caustic substance | |
| Miscellaneous | 0%5% |
| Malignancy, Barrett's esophagus, infection, aortic dissection | |
Diagnosis of esophageal perforation secondary to a foreign body may be difficult because of the considerable overlap of symptoms with other causes of chest pain and failure to consider this infrequent condition in the absence of a classic history of retching. To diagnose such a disease, physicians must gather data from various sourcesespecially the history, physical examination, and medical recordformulate hypotheses, integrate results from diagnostic tests, and then assess the importance of the available information in the context of a differential diagnosis. Incorrectly evaluating or failing to obtain essential data can lead to incorrect or delayed diagnoses.
In This Patient's Evaluation, What Prevented Prompt Recognition of Esophageal Perforation?
The critical misstep was an incomplete history, both on arrival and when the patient was transferred to a second team. The presence of risk factors for coronary artery disease led the providers to first consider myocardial ischemia. They failed to ask crucial questions about the onset of the painwhen it occurred during the meal and what he was eatingeven when the patient later complained of odynophagia. As a result of the incomplete history, the providers, puzzled by the patient's ongoing and evolving symptoms, ordered numerous unnecessary diagnostic tests that gave false‐positive results, leading to potentially harmful treatment including anticoagulation. The discussant mentions that the preferred response to a puzzling clinical situation is to return to the bedside and repeat the history, reexamine the patient, and reevaluate available informationsimple steps that can often resolve diagnostic dilemmas.
There is ongoing concern that the history‐taking and physical examination skills of clinicians are in decline.814 Many speculate this is in part due to reliance on increasingly sophisticated diagnostic tests. Providers may overly rely on modern diagnostic tests because of their familiarity with the sensitivity and specificity of such tests, fear of malpractice litigation, diminishing opportunity to elucidate the complete history and physical exam, or lack of confidence in their history‐taking and examination skills.814 Although the rapid development and implementation of advanced diagnostic technologies have had a significant impact on diagnostic accuracy, the estimated rate of disease misdiagnosis remains elevated at 24%.1518 In contrast to technology‐based testing, the history and physical provide an inexpensive, safe, and effective means of at arriving at a correct diagnosis. In outpatient medical visits the history and physical, when completely elicited, result in a correct diagnosis of up to 70%90% of patients.8, 19, 20 Even for illnesses whose diagnosis requires confirmation by a diagnostic test, the definitive test can only be selected after a sufficient history and exam provide an assessment of the pretest probability of disease.
In evaluating chest pain there is an additional potential factor that diminishes reliance on bedside assessment. Modern quality assurance measures and chest pain units encourage clinicians to evaluate patients with chest pain quickly because any delay diminishes the benefits of therapies for acute coronary syndromes. In the emergency room, these patients find themselves on a rapidly moving diagnostic conveyor belt, an approach that is efficient and appropriate given the high prevalence of coronary disease but that also contributes to inattentiveness and error for patients with unusual diagnoses.
How Could Clinicians in Our Case Use Bedside Evidence to Help Differentiate Our Patient?
For most patients with chest pain there is no finding that would change diagnostic probabilities enough to take them off the diagnostic conveyor belt. Nevertheless, several bedside findings can help providers to rank‐order a differential diagnosis, thereby improving the sequence in which diagnostic testing is done. For patients with chest pain the ECG has the highest predictive ability of all studied history, physical exam, and ECG findings (Table 2).21 A history of sharp and positional pain descriptors diminishes the probability of myocardial ischemia.21 Unfortunately, no history, exam, or ECG feature is sensitive enough, either alone or in combination, to effectively rule out myocardial ischemia.
| Finding | Positive LR* |
|---|---|
| Myocardial ischemia | |
| ST segment elevation or Q wave | 22 |
| S3 gallop, blood pressure < 100 mm Hg, or ST segment depression | 3.0 |
| Sharp or positional pain | 0.3 |
| Pulmonary embolism | |
| Low clinical probability | 0.2 |
| Medium clinical probability | 1.8 |
| High clinical probability | 17.1 |
| Aortic dissection | |
| Tearing or ripping pain | 10.8 |
| Focal neurologic deficits | 6.633 |
| Ipsilateral versus contralateral pulse deficit | 5.7 |
| Cardiac tamponade | |
| Pulsus paradoxus > 12 mm Hg | 5.9 |
| Esophageal perforation | |
| Dysphagia, odynophagia, retching, vomiting, or subcutaneous emphysema | ? |
The history and exam can also facilitate differentiation of noncoronary causes of life‐threatening chest pain. The dismal performance of individual bedside findings for pulmonary embolism is what led to development of quantitative D‐dimer assays and objective methods based on bedside evaluation, including the widely used Wells Score.22 This score can be used to classify patients as having low, medium, and high risk of pulmonary embolism, facilitating management decisions after diagnostic imaging is obtained.23 Fewer than half of all patients with thoracic aortic dissection have classic exam findings; however, when present, they can appropriately raise the probability of dissection higher on the differential diagnosis.24 Importantly, no history or exam finding argues against dissection.24 Most patients with cardiac tamponade will have elevated jugular venous pressure (76%100%); however, poor interobserver agreement about this finding may decrease its detection.11, 25, 26 As the discussant notes, total paradox, defined as the palpable pulse disappearing with inspiration, is an insensitive test for tamponade, present in only 23% of patients with the disorder. In contrast, an inspiratory drop in systolic blood pressure of more than 12 mm Hg should prompt consideration for tamponade.11, 26 Commonly taught features of esophageal perforation, including chest pain, dysphagia, odynophagia, prior retching or vomiting, subcutaneous emphysema, dyspnea, and pleural effusions, vary in their reported sensitivity, but their specificity is virtually never reported.27
Like most patients with chest pain, our patient lacked all these symptoms and signs, arguing for myocardial ischemia, although he had a few signs that argued against it (sharp and positional chest pain). After the initial CXR and ECG, further testing with cardiac biomarkers was appropriate, but a fundamental error was made in not returning to the patient's bedside to repeat the interview and examination after the cardiac biomarkers were found to be normal. Had this been done, several cluesdysphagia, onset of pain with eating bony fish, and feverwould have pushed esophageal perforation to the top of the differential diagnosis. Subsequent testing would have led to the correct diagnosis and avoided a potentially harmful diagnostic fishing expedition.
Take‐Home Points
-
Esophageal perforation is an uncommon but life‐threatening cause of chest pain that is difficult to diagnose because of its nonspecific symptoms.
-
An accurate and complete history and exam can reveal signs and symptoms that influence the likelihood of each life‐threatening cause of chest pain. Evaluating patients for these features is vital to the rank ordering of a differential diagnosis and the selection of appropriate diagnostic tests.
-
There is no substitute for repeating the history, reexamining the patient, and reevaluating available information when confronted with a confusing constellation of symptoms.
Acknowledgements
The authors thank Steve McGee for his thoughtful review and comments on the manuscript.
A 54‐year‐old man with hypertension and type 2 diabetes mellitus entered the Chest Pain Evaluation Unit of a teaching hospital after 12 hours of intermittent thoracic discomfort. The pain began during dinner and was sharp, bandlike, and located beneath the sternum and across the entire chest. He had dyspnea but no diaphoresis or nausea. A recumbent position relieved the pain after dinner, but it recurred during the night and again the following morning. He did not smoke and had no family history of coronary artery disease.
A useful approach in evaluating acute chest pain is to employ a hierarchical differential diagnosis that emphasizes life‐threatening disorders requiring prompt recognition and intervention. Most prominent are cardiac ischemia, pericardial tamponade, pneumothorax, pulmonary embolus, esophageal rupture, and aortic dissection. The concurrent dyspnea and retrosternal location and intermittent nature of the pain that this patient has are consistent with myocardial ischemia, but the sharp quality of the pain and the relief gained by being recumbent are atypical. Pain with pericarditis is characteristically pleuritic and often worse when lying down. The pain of pneumothorax is typically unilateral, not intermittent, and unlikely to improve with recumbency. Although pain during eating suggests the possibility of an esophageal source, spontaneous rupture usually follows vomiting. The pain is typically continuous and severe. The pain of pulmonary embolism may be unilateral and pleuritic but often is more diffuse. Relief by recumbency is unusual, but the intermittent nature could suggest recurrent emboli. The patient has a history of hypertension, which predisposes him to aortic dissection, in which the pain is typically sharp, continuous, and severe but occasionally intermittent. Among numerous less urgent diagnoses are esophagitis and thoracic diabetic radiculopathy.
Important features to look for during this patient's examination include: disappearance of the radial pulse during inhalation, a simple screening test that is insensitive but very specific for pericardial tamponade; elevated neck veins, which can occur with tension pneumothorax, massive pulmonary embolism, and pericardial tamponade; pericardial and pleural friction rubs; discrepant blood pressures in the 2 arms, sometimes a sign of aortic dissection; local thoracic tenderness from chest wall disorders; and sensory examination of the chest surface, which is often abnormal in diabetic thoracic radiculopathy. Given this patient's age and history of diabetes, I am most concerned about myocardial ischemia. The most appropriate diagnostic tests include an electrocardiogram and a chest radiograph.
The patient appeared apprehensive but reported no pain. He had a temperature of 36.0 C, heart rate of 95 beats/minute, blood pressure of 138/77 mm Hg, respiratory rate of 16/minute, and oxygen saturation of 99% while breathing ambient air. Blood pressures were equal in both arms. Jugular venous distention was absent, and the lung and cardiac examinations had normal results. His pain did not increase on chest wall palpation. Examination of the abdomen, extremities, and the neurologic system showed normal results.
The results of laboratory tests showed a leukocyte count of 12,700/cm3, with 85% neutrophils, 8% lymphocytes, 6% monocytes, and 1% eosinophils. The hematocrit was 45%, and the platelet count was 172,000/cm3. The results of his chemistry panel were remarkable only for a glucose of 225 mg/dL. An electrocardiogram (ECG) showed a normal sinus rhythm and left anterior fascicular block without acute ST‐ or T‐wave changes. Prior ECGs were unavailable. An anteroposterior radiograph disclosed low lung volumes and bibasilar opacities. No pleural effusion was noted. Serial serum troponin and creatine kinase levels were normal. A Tc‐99m tetrofosmin cardiac nuclear perfusion test performed at rest demonstrated a moderate area of mildly decreased uptake along the inferior wall extending to the apex. An exercise treadmill test, terminated after 1 minute, 45 seconds because of chest pain, provoked no ECG changes diagnostic of ischemic disease.
The absence of elevated jugular venous pressure virtually eliminates pericardial tamponade as a diagnosis, and the chest film excludes pneumothorax. The intermittent nature of the chest pain, the absence on the chest radiograph of such findings as mediastinal gas, left pneumothorax, or hydropneumothorax, and the lack of a predisposing cause make esophageal rupture unlikely. Pulmonary emboli remain a consideration despite the normal oxygen saturation because there is no hypoxemia in a substantial minority of such cases. The normal cardiac enzyme levels and the lack of significant changes on the ECG exclude that a myocardial infarction has recently occurred, but cardiac ischemia remains a possibility, especially because the patient had chest pain on exercise and the nuclear scan indicated diminished blood flow to the inferior left ventricle. Aortic dissection still lurks as a possibility. The inferior wall abnormalities seen on the scan could result from dissection into the right coronary artery, which is more frequently involved than the left, or compression of it by an enlarged aorta, but they also may be artifacts. The leukocytosis may be a nonspecific response to stress but could indicate, although unlikely, infections such as mediastinitis from esophageal rupture or bacterial aortitis.
A conscientious clinician would repeat the history, reexamine the patient, and scrutinize the chest film to determine what the bilateral opacities represent. Given the story so far, however, I might consider a thoracic computed tomography (CT) angiogram because I am most concerned about pulmonary emboli and aortic dissection.
On hospital day 3, the patient had worsening dyspnea and persistent chest pain. His temperature was 39.3C, and his oxygen saturation decreased to 89% while breathing room air. Repeat chest radiography showed new bilateral pleural effusions and increased bibasilar opacification (Fig. 1). His leukocyte count was 19,000/cm3, with 88% neutrophils, 6% lymphocytes, 5% monocytes, and 1% eosinophils. Care was transferred from the chest pain team to an inpatient general medicine ward team. A pulmonary CT angiogram showed no large central clots but suggested emboli in the right superior subsegmental artery and a right upper lobe subsegmental artery. Bilateral pleural effusions were observed, as were bilateral pleural‐based atelectasis or infiltrates in the lower lungs. A hiatal hernia was noted, but no aortic dissection. The patient received supplemental oxygen, intravenous levofloxacin, and unfractionated heparin by continuous infusion.
Without other information, I will assume that the fever is part of the patient's original disease and not a nosocomial infection or drug fever. At this point, a crucial part of the evaluation is examining the CT scan with experienced radiologists to determine whether the abnormalities noted are genuinely convincing for pulmonary emboli. If the findings are equivocal, the next step might be a pulmonary angiogram or the indirect approach of evaluating the leg veins with ultrasound, reasoning that the presence of proximal leg vein thromboses would require anticoagulation in any event.
The patient's worsening chest pain and hypoxemia are consistent with multiple pulmonary emboli. Bilateral pleural effusions and leukocytosis can occur but are uncommon. Because of the fever, another possibility is septic pulmonary emboli, but he has no evidence of suppurative thrombophlebitis of the peripheral veins, apparent infection elsewhere, or previous intravenous drug abuse causing right‐sided endocarditis. An alternative diagnosis is infection of an initially bland pulmonary infarct.
An important consideration is a thoracentesis, depending on how persuasive the CT diagnosis of pulmonary embolism is and the size of the pleural effusions. It should be done before instituting antimicrobial therapy, which may decrease the yield of the cultures, and before starting heparin, which increases the risk of bleeding and occasionally causes a substantial, even fatal hemothorax.
The patient's oxygenation and dyspnea did not improve. Over the next day, he repeatedly mentioned that swallowing, particularly solid foods, worsened his chest pain. He had a temperature of 39.9C, a heart rate of 121 beats/minute, blood pressure of 149/94 mm Hg, a respiratory rate of 28/minute, and oxygen saturation of 93% while breathing 40% oxygen. He had inspiratory splinting, percussive dullness at both lung bases, and distant heart sounds. A contrast esophagogram showed distal narrowing that prevented solid contrast from passing, but no hiatal hernia. Blood and urine cultures obtained before antibiotic therapy were sterile. Duplex ultrasonography of bilateral lower extremities showed no evidence of deep venous thrombosis. A pulmonary angiogram revealed no emboli, and heparin was discontinued. Bilateral thoracentesis yielded grossly bloody fluid. Repeat chest CT (Fig. 2) demonstrated large bilateral effusions, a new large pericardial effusion, and a prominence at the gastroesophageal junction more concerning for a soft‐tissue mass than for a hiatal hernia, although the quality of the study was suboptimal because of an absence of oral contrast.
The CT scan suggests a paraesophageal abscess from an esophageal rupture. As mentioned earlier, if rupture occurs spontaneously, it typically follows retching or vomiting and is called Boerhaave's syndrome. Another consideration is a rupture secondary to an external insult, such as trauma or ingestion of a caustic substance. In evaluating these possibilities, the patient should have been asked 4 questions at the initial interview that I neglected to explicitly highlight earlier. First, what was he eating when he developed the chest pain? Second, did the pain begin during swallowing? Third, did he have previous symptoms suggesting an esophageal disorder such as dysphagia, odynophagia, or heartburn? These might indicate a cancer that could perforate or another problem such as a stricture or disordered esophageal motility that might have caused a swallowed item to lodge in the esophagus. Finally, did he have retching or vomiting? Though not routinely part of the review of systems, the former 2 questions are an appropriate history‐prompted line of questioning of a patient with onset of chest pain while eating.
At this point, a reasonable approach would be an esophagoscopy to delineate any intraluminal problems, such as a cancer or a foreign body. The apparent obstruction seen on barium swallow may be from extrinsic pressure from a paraesophageal abscess. The patient should receive broad‐spectrum antimicrobial therapy effective against oral anaerobes. Although occasionally patients recover with antibiotics alone, surgery is usually required. I am surprised that the original CT scan did not show evidence of an esophageal perforation. Possibly, the hiatal hernia was a paraesophageal abscess poorly characterized because of the lack of oral contrast.
The team, concerned about esophageal perforation, began the patient on intravenous clindamycin. The patient underwent video‐assisted thoracoscopic drainage, which yielded a moderate amount of turbid, bloody fluid from each hemithorax. The pericardium contained approximately 500 cm3 of turbid fluid. Gram stain and culture of these fluids were negative. No esophageal or mediastinal mass was noted during surgery. Intraoperative esophagogastroduodenoscopy with endoscopic ultrasound showed a healing linear mucosal tear in the distal esophagus (Fig. 3) as well as air/fluid collection in the esophageal soft tissue (not shown).
On further questioning postoperatively, the patient reported eating bony fish during the dinner when he first experienced chest pain. The patient received a 21‐day course of oral clindamycin and completely recovered. Five weeks later, a chest CT showed decreased distal esophageal thickening and no mediastinal air.
COMMENTARY
Esophageal perforation is an uncommon but life‐threatening cause of chest pain. In most series iatrogenic injury accounts for more than 70% of cases, whereas most of the other cases have spontaneous (5%20%) or traumatic (4%10%) causes (Table 1).14 Perforation as a complication of ingesting fish bones, although rare, is well described and continues to be reported.57
| Etiology | Percent |
|---|---|
| |
| Iatrogenic | 45%77% |
| Rigid or flexible endoscopy, balloon dilation, Blakemore tube, sclerotherapy, operative injury | |
| Increased intraesophageal pressure (Boerhaave's syndrome) | 5%20% |
| Vomiting or retching, weightlifting, childbirth | |
| Traumatic | 4%10% |
| Penetrating or blunt injury to neck or chest | |
| Ingestion | 0%12% |
| Foreign body, toxic or caustic substance | |
| Miscellaneous | 0%5% |
| Malignancy, Barrett's esophagus, infection, aortic dissection | |
Diagnosis of esophageal perforation secondary to a foreign body may be difficult because of the considerable overlap of symptoms with other causes of chest pain and failure to consider this infrequent condition in the absence of a classic history of retching. To diagnose such a disease, physicians must gather data from various sourcesespecially the history, physical examination, and medical recordformulate hypotheses, integrate results from diagnostic tests, and then assess the importance of the available information in the context of a differential diagnosis. Incorrectly evaluating or failing to obtain essential data can lead to incorrect or delayed diagnoses.
In This Patient's Evaluation, What Prevented Prompt Recognition of Esophageal Perforation?
The critical misstep was an incomplete history, both on arrival and when the patient was transferred to a second team. The presence of risk factors for coronary artery disease led the providers to first consider myocardial ischemia. They failed to ask crucial questions about the onset of the painwhen it occurred during the meal and what he was eatingeven when the patient later complained of odynophagia. As a result of the incomplete history, the providers, puzzled by the patient's ongoing and evolving symptoms, ordered numerous unnecessary diagnostic tests that gave false‐positive results, leading to potentially harmful treatment including anticoagulation. The discussant mentions that the preferred response to a puzzling clinical situation is to return to the bedside and repeat the history, reexamine the patient, and reevaluate available informationsimple steps that can often resolve diagnostic dilemmas.
There is ongoing concern that the history‐taking and physical examination skills of clinicians are in decline.814 Many speculate this is in part due to reliance on increasingly sophisticated diagnostic tests. Providers may overly rely on modern diagnostic tests because of their familiarity with the sensitivity and specificity of such tests, fear of malpractice litigation, diminishing opportunity to elucidate the complete history and physical exam, or lack of confidence in their history‐taking and examination skills.814 Although the rapid development and implementation of advanced diagnostic technologies have had a significant impact on diagnostic accuracy, the estimated rate of disease misdiagnosis remains elevated at 24%.1518 In contrast to technology‐based testing, the history and physical provide an inexpensive, safe, and effective means of at arriving at a correct diagnosis. In outpatient medical visits the history and physical, when completely elicited, result in a correct diagnosis of up to 70%90% of patients.8, 19, 20 Even for illnesses whose diagnosis requires confirmation by a diagnostic test, the definitive test can only be selected after a sufficient history and exam provide an assessment of the pretest probability of disease.
In evaluating chest pain there is an additional potential factor that diminishes reliance on bedside assessment. Modern quality assurance measures and chest pain units encourage clinicians to evaluate patients with chest pain quickly because any delay diminishes the benefits of therapies for acute coronary syndromes. In the emergency room, these patients find themselves on a rapidly moving diagnostic conveyor belt, an approach that is efficient and appropriate given the high prevalence of coronary disease but that also contributes to inattentiveness and error for patients with unusual diagnoses.
How Could Clinicians in Our Case Use Bedside Evidence to Help Differentiate Our Patient?
For most patients with chest pain there is no finding that would change diagnostic probabilities enough to take them off the diagnostic conveyor belt. Nevertheless, several bedside findings can help providers to rank‐order a differential diagnosis, thereby improving the sequence in which diagnostic testing is done. For patients with chest pain the ECG has the highest predictive ability of all studied history, physical exam, and ECG findings (Table 2).21 A history of sharp and positional pain descriptors diminishes the probability of myocardial ischemia.21 Unfortunately, no history, exam, or ECG feature is sensitive enough, either alone or in combination, to effectively rule out myocardial ischemia.
| Finding | Positive LR* |
|---|---|
| Myocardial ischemia | |
| ST segment elevation or Q wave | 22 |
| S3 gallop, blood pressure < 100 mm Hg, or ST segment depression | 3.0 |
| Sharp or positional pain | 0.3 |
| Pulmonary embolism | |
| Low clinical probability | 0.2 |
| Medium clinical probability | 1.8 |
| High clinical probability | 17.1 |
| Aortic dissection | |
| Tearing or ripping pain | 10.8 |
| Focal neurologic deficits | 6.633 |
| Ipsilateral versus contralateral pulse deficit | 5.7 |
| Cardiac tamponade | |
| Pulsus paradoxus > 12 mm Hg | 5.9 |
| Esophageal perforation | |
| Dysphagia, odynophagia, retching, vomiting, or subcutaneous emphysema | ? |
The history and exam can also facilitate differentiation of noncoronary causes of life‐threatening chest pain. The dismal performance of individual bedside findings for pulmonary embolism is what led to development of quantitative D‐dimer assays and objective methods based on bedside evaluation, including the widely used Wells Score.22 This score can be used to classify patients as having low, medium, and high risk of pulmonary embolism, facilitating management decisions after diagnostic imaging is obtained.23 Fewer than half of all patients with thoracic aortic dissection have classic exam findings; however, when present, they can appropriately raise the probability of dissection higher on the differential diagnosis.24 Importantly, no history or exam finding argues against dissection.24 Most patients with cardiac tamponade will have elevated jugular venous pressure (76%100%); however, poor interobserver agreement about this finding may decrease its detection.11, 25, 26 As the discussant notes, total paradox, defined as the palpable pulse disappearing with inspiration, is an insensitive test for tamponade, present in only 23% of patients with the disorder. In contrast, an inspiratory drop in systolic blood pressure of more than 12 mm Hg should prompt consideration for tamponade.11, 26 Commonly taught features of esophageal perforation, including chest pain, dysphagia, odynophagia, prior retching or vomiting, subcutaneous emphysema, dyspnea, and pleural effusions, vary in their reported sensitivity, but their specificity is virtually never reported.27
Like most patients with chest pain, our patient lacked all these symptoms and signs, arguing for myocardial ischemia, although he had a few signs that argued against it (sharp and positional chest pain). After the initial CXR and ECG, further testing with cardiac biomarkers was appropriate, but a fundamental error was made in not returning to the patient's bedside to repeat the interview and examination after the cardiac biomarkers were found to be normal. Had this been done, several cluesdysphagia, onset of pain with eating bony fish, and feverwould have pushed esophageal perforation to the top of the differential diagnosis. Subsequent testing would have led to the correct diagnosis and avoided a potentially harmful diagnostic fishing expedition.
Take‐Home Points
-
Esophageal perforation is an uncommon but life‐threatening cause of chest pain that is difficult to diagnose because of its nonspecific symptoms.
-
An accurate and complete history and exam can reveal signs and symptoms that influence the likelihood of each life‐threatening cause of chest pain. Evaluating patients for these features is vital to the rank ordering of a differential diagnosis and the selection of appropriate diagnostic tests.
-
There is no substitute for repeating the history, reexamining the patient, and reevaluating available information when confronted with a confusing constellation of symptoms.
Acknowledgements
The authors thank Steve McGee for his thoughtful review and comments on the manuscript.
- ,.Esophageal perforations: a 15 year experience.Am J Surg.1982;143:495–503.
- ,,.Esophageal perforation: emphasis on management.Ann Thorac Surg.1996;61:1447–1451; discussion1451–1452.
- ,,,,,.Evolving options in the management of esophageal perforation.Ann Thorac Surg.2004;77:1475–1783.
- ,.Personal management of 57 consecutive patients with esophageal perforation.Am J Surg.2004;187:58–63.
- ,,,,.Perforation of the oesophagus and aorta after eating fish: an unusual cause of chest pain.Emerg Med J.2003;20:385–386.
- ,,,.Esophageal perforation and mediastinitis from fish bone ingestion.South Med J.2003;96:516–520.
- ,,,.A 73‐year‐old man with chest pain 4 days after a fish dinner.Chest.2004;126:294–297.
- ,.The science of the art of the clinical examination.JAMA.1992;267:2650–2652.
- .Clinical skills in the 21st century.Arch Intern Med.1994;154:22–24.
- ,.Pulmonary auscultatory skills during training in internal medicine and family practice.Am J Respir Crit Care Med.1999;159:1119–1124.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:Saunders;2001.
- .Simple is beautiful: the neglected power of simple tests.Arch Intern Med.2004;164:2198–2200.
- ,.Pearls and pitfalls in patient care: need to revive traditional clinical values.Am J Med Sci.2004;327:79–85.
- ,. Physical diagnosis: a lost art? Agency for Health Research and Quality. WebM75:29–40.
- .Low‐tech autopsies in the era of high‐tech medicine: continued value for quality assurance and patient safety.JAMA.1998;280:1273–1274.
- ,,,.Has misdiagnosis of appendicitis decreased over time? A population‐based analysis.JAMA.2001;286:1748–1753.
- ,,,.Changes in rates of autopsy‐detected diagnostic errors over time: a systematic review.JAMA.2003;289:2849–2856.
- .Diagnostic Process.J Coll Gen Pract.1963;54:579–589.
- .The importance of the history in the medical clinic and the cost of unnecessary tests.Am Heart J.1980;100:928–931.
- ,.Bedside diagnosis of coronary artery disease: a systematic review.Am J Med.2004;117:334–343.
- ,,, et al.Use of a clinical model for safe management of patients with suspected pulmonary embolism.Ann Intern Med.1998;129:997–1005.
- ,,, et al.Diagnostic pathways in acute pulmonary embolism: recommendations of the PIOPED II investigators.Am J Med.2006;119:1048–1055.
- .Does this patient have an acute thoracic aortic dissection?JAMA.2002;287:2262–2272.
- ,.The rational clinical examination. Does this patient have abnormal central venous pressure?JAMA.1996;275:630–634.
- ,,,.Does this patient with a pericardial effusion have cardiac tamponade?JAMA.2007;297:1810–1818.
- ,.Spontaneous esophageal rupture: a frequently missed diagnosis.Am Surg.1999;65:449–452.
- ,.Esophageal perforations: a 15 year experience.Am J Surg.1982;143:495–503.
- ,,.Esophageal perforation: emphasis on management.Ann Thorac Surg.1996;61:1447–1451; discussion1451–1452.
- ,,,,,.Evolving options in the management of esophageal perforation.Ann Thorac Surg.2004;77:1475–1783.
- ,.Personal management of 57 consecutive patients with esophageal perforation.Am J Surg.2004;187:58–63.
- ,,,,.Perforation of the oesophagus and aorta after eating fish: an unusual cause of chest pain.Emerg Med J.2003;20:385–386.
- ,,,.Esophageal perforation and mediastinitis from fish bone ingestion.South Med J.2003;96:516–520.
- ,,,.A 73‐year‐old man with chest pain 4 days after a fish dinner.Chest.2004;126:294–297.
- ,.The science of the art of the clinical examination.JAMA.1992;267:2650–2652.
- .Clinical skills in the 21st century.Arch Intern Med.1994;154:22–24.
- ,.Pulmonary auscultatory skills during training in internal medicine and family practice.Am J Respir Crit Care Med.1999;159:1119–1124.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:Saunders;2001.
- .Simple is beautiful: the neglected power of simple tests.Arch Intern Med.2004;164:2198–2200.
- ,.Pearls and pitfalls in patient care: need to revive traditional clinical values.Am J Med Sci.2004;327:79–85.
- ,. Physical diagnosis: a lost art? Agency for Health Research and Quality. WebM75:29–40.
- .Low‐tech autopsies in the era of high‐tech medicine: continued value for quality assurance and patient safety.JAMA.1998;280:1273–1274.
- ,,,.Has misdiagnosis of appendicitis decreased over time? A population‐based analysis.JAMA.2001;286:1748–1753.
- ,,,.Changes in rates of autopsy‐detected diagnostic errors over time: a systematic review.JAMA.2003;289:2849–2856.
- .Diagnostic Process.J Coll Gen Pract.1963;54:579–589.
- .The importance of the history in the medical clinic and the cost of unnecessary tests.Am Heart J.1980;100:928–931.
- ,.Bedside diagnosis of coronary artery disease: a systematic review.Am J Med.2004;117:334–343.
- ,,, et al.Use of a clinical model for safe management of patients with suspected pulmonary embolism.Ann Intern Med.1998;129:997–1005.
- ,,, et al.Diagnostic pathways in acute pulmonary embolism: recommendations of the PIOPED II investigators.Am J Med.2006;119:1048–1055.
- .Does this patient have an acute thoracic aortic dissection?JAMA.2002;287:2262–2272.
- ,.The rational clinical examination. Does this patient have abnormal central venous pressure?JAMA.1996;275:630–634.
- ,,,.Does this patient with a pericardial effusion have cardiac tamponade?JAMA.2007;297:1810–1818.
- ,.Spontaneous esophageal rupture: a frequently missed diagnosis.Am Surg.1999;65:449–452.
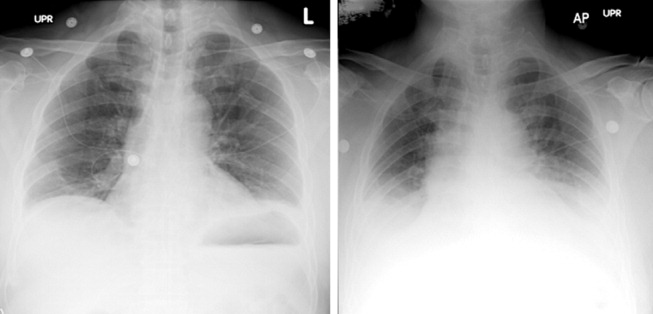
Consequences of Missed Opportunities
A 58‐year‐old man was evaluated for 3 weeks of leg numbness and weakness. His symptoms began with numbness and tingling in the distal left leg that progressed to weakness that impaired his ability to walk. He had no history of trauma or incontinence but endorsed several months of back pain that worsened when lying flat. He had a history of type 2 diabetes mellitus, hepatitis C infection, hypertension, and posttraumatic stress disorder. He had a remote history of intravenous drug use and had quit tobacco 9 years earlier. Medications he was taking included hydrochlorothiazide, rosiglitazone, oxycodone/acetaminophen, baclofen, ibuprofen, and gabapentin.
Internists see this constellation of complaints frequently in an acute care setting. Finding a unifying diagnosis may be difficult initially, so thinking of the symptoms in series is helpful. The complaint of leg weakness and the pattern of numbness should be further elucidated. Is this true weakness, or is it a feeling of instability because of foot numbness? What is the pattern of the numbness? Peripheral neuropathy typically begins in a symmetric stocking pattern (involving the plantar surface of the feet), then progresses to a glove distribution (involving the hands from the fingers distally to the wrist proximally). Such a pattern in a patient with diabetes would be consistent with distal polyneuropathy, a mixed sensory and motor process. Other possible causes of peripheral neuropathy in this patient include HIV, B12 deficiency, and syphilis. These symptoms could be tied to the back pain if this were intervertebral disk disease, a compression fracture, or a lytic lesion in the vertebrae with resulting nerve impingement or if it were epidural spinal cord compression. The lack of bowel or bladder dysfunction speaks against a cauda equina syndrome but does not rule out more cepahalad spinal pathology.
On neurological examination, I would concentrate on differentiating weakness from pain. I would attempt to determine whether the weakness was of central or peripheral nerve etiology. Helpful findings would include increased tone with upper motor lesions and flaccid tone with lower motor lesions, hyperreflexia with upper motor lesions and hyporeflexia with lower motor lesions, a Babinski sign, muscle atrophy or fasciculations, and gait. A rectal examination would also be helpful to assess for deficits in rectal tone, wink reflex, or saddle anesthesia.
All patients with low back pain who have alarm signs of age older than 50, pain duration of more than 1 month, known cancer, lack of relief with conservative measures, or systemic B symptoms should have imaging of the spine. Although plain films may reveal bony abnormalities, computed tomography (CT) is better for evaluating osseous structures and magnetic resonance imaging (MRI) for evaluating pathology in patients suspected of having an infection or a malignancy. I would obtain imaging of the spine in this patient.
The patient was receiving care at an outside clinic for 2 liver lesions discovered on abdominal ultrasound 19 months prior to admission. CT showed that the lesions were 4.0 and 2.3 cm in diameter 17 months prior to admission and 5.0 and 3.0 cm in diameter 5 months prior to admission. No cirrhosis was appreciated on the ultrasound or CT. The patient was referred for CT‐guided biopsy of the larger mass after the second CT, but he became anxious and left before the biopsy was obtained.
This piece of the history is ominous, as it increases the possibility of cancer in our differential. Metastatic disease could provide a unifying diagnosis, explaining the constellation of back pain, leg weakness, and liver lesions. Lung cancer commonly metastasizes to the liver and to bone, so I would obtain a chest x‐ray. Other possible types of cancer in this situation include cancer of the prostate, colon, or thyroid and melanoma. In this patient, who has hepatitis C, hepatocellular carcinoma (HCC) could be the primary etiology, although cirrhosis was not seen on CT and HCC metastasizes to the spine less commonly than do other primary cancers (eg, lung, breast, prostate). Nonetheless, I would obtain an alpha‐fetoprotein level, which would confirm HCC in a patient with liver lesions if it was greater than 200 g/L. Pancreatic cancer has been associated with both type 2 diabetes and liver lesions and could explain his abdominal pain.
There is no comment on the arterial‐phase CT imaging of the liver lesions. Dual‐phase CT scans examine the hepatic arterial and portal vein phases of contrast filling. Triple‐phase CT scans also examine the portal vein influx phase. Both hemangiomas and hypervascular HCCs enhance on the arterial phase, as they derive their blood flow from the hepatic artery. Therefore, arterial‐phase imaging can help to distinguish vascular tumors that flush with contrast, such as hemangiomas, melanoma, and HCC, from less vascular tumors such as pancreatic and colon cancer. Other liver lesions such as focal nodular hyperplasia and adenomas cannot be excluded in this situation as they also may enhance during the arterial phase and can grow over time, as this patient's repeat imaging documented. It seems unlikely that this patient has a liver abscess because he has a paucity of constitutional symptoms and no travel history. The liver lesions seen on initial imaging were larger than 1.0 cm, so I would have favored an earlier biopsy to obtain a tissue diagnosis.
The patient was afebrile, and all other vital signs were normal. He appeared well nourished and anicteric. There was no lymphadenopathy. Cardiac auscultation was regular without murmurs. The lungs were clear. The abdomen was without fluid wave or hepatosplenomegaly and was tender to palpation in the right upper and lower quadrants. There was no midline tenderness to palpation of the spine.
Cranial nerves II‐XII were intact. Lower extremity muscle tone could not be accurately assessed due to splinting from back pain. Strength was 3 of 5 in the left hip extensors and left knee flexors and extensors, and 1 of 5 in the left hip flexors. He had no motor strength in the distal left lower extremity extensors. Bilateral upper extremity and right leg strength were normal. Sensation to light touch, temperature, and pain was decreased circumferentially below the xiphoid. The patient had hyperesthesia in a band around the thorax just above the xiphoid and paresthesia of the perineal area. Left patellar tendon reflexes were brisk, and left ankle jerk was absent, but other reflexes were normal. Toes were down‐going bilaterally. The anal wink was absent, and rectal tone was decreased. Results of the cerebellar exam were normal. Gait could not be assessed.
The results of the exam are notable for not showing the stigmata of end‐stage liver disease. The results of the neurological exam are concerning, with decreased sensation at approximately the T7 level that is almost certainly a result of epidural compression of the spinal cord. Hematogenous metastasis to the vertebrae from one of the tumors mentioned above, with spread into the thecal sac, is the most likely culprit. An epidural abscess is possible because the patient has diabetes and a history of injection drug use.
The thoracic spine is involved in 60% of spinal cord metastases. This patient's left‐sided distal leg weakness is consistent with having corticospinal tract compression and indicates thoracic spine involvement. Flaccid paralysis is classically found in lower motor neuron weakness, but is also seen in the early stages of upper motor neuron pathology. Lesions found above the cauda equina often spare the perineal area, but low thoracic lesions involving the conus medullaris (from T10 to L1) could explain both his loss of anal wink and his decreased rectal tone.
This patient's presentation is unfortunately classic for epidural spinal cord compression. Because the onset of compression is insidious, the diagnosis is often delayed, even in patients with known cancer. Urgent imaging is imperative to evaluate this possibility, as having any meaningful chance of recovery of function depends on rapid relief of the spinal cord compression. I would obtain an emergent MRI of the thoracic and lumbosacral spine.
Laboratory studies showed the following: hemoglobin, 13.1 g/dL; mean corpuscular volume, 80 m3; platelet count, 149,000/L; creatinine, 1.9 mg/dL; aspartate aminotransferase, 66 U/L (5‐35 U/L); alanine aminotransferase, 66 U/L (7‐56 U/L); alkaline phosphatase, 87 U/L (40‐125 U/L); total bilirubin, 1.3 mg/dL; prostate specific antigen (PSA), 1.6 g/dL; and alpha‐fetoprotein (AFP), 10.3 g/L. White cell count, sodium, glucose, calcium, and albumin levels, and prothrombin and partial‐thromboplastin times were within normal ranges.
His liver function tests likely reflect chronic hepatitis C infection. His renal insufficiency could be a result of hypertension, diabetes, or dehydration given that he has been bed‐bound.
Most intriguing are the normal PSA level and only slightly elevated AFP level. PSA is useful for detecting recurrence of prostate cancer or following response of therapy, but the utility of PSA as a screening tool remains controversial in part because of its low specificity. Prostate cancer is the most commonly diagnosed cancer among men and cannot be ruled out by a normal PSA. In a patient with hepatitis C, cirrhosis (which we have not conclusively diagnosed), and a radiologically suspicious liver lesion, an AFP > 200 g/L would be diagnostic of HCC. In this case, however, mildly elevated AFP does not help us to either diagnose or exclude HCC.
The chest x‐ray showed no abnormalities. MRI of the spine revealed lytic lesions in the T7‐T10 vertebral bodies with spinal cord compression at the T7 level (Fig. 1).
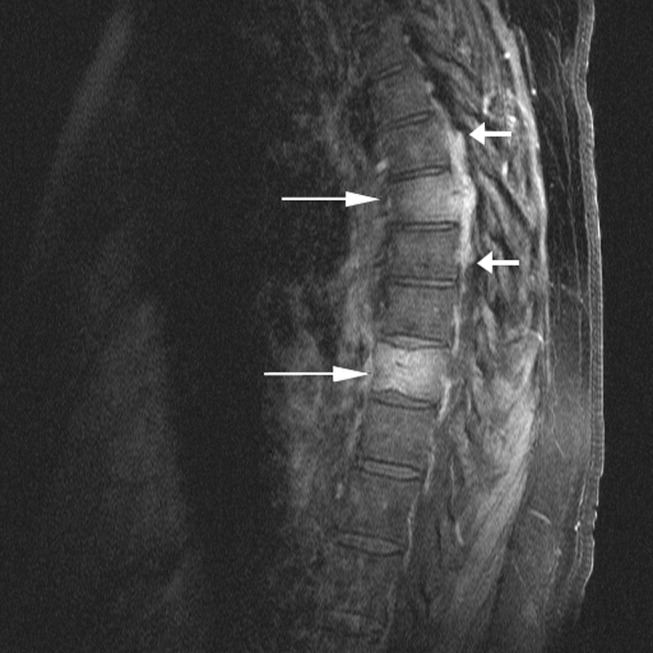
A repeat CT scan of the abdomen showed a coarse, nodular liver with 2 heterogeneous, early‐enhancing masses (4.7 4.2 and 3.4 2.4 cm in diameter) with surrounding satellite lesions (Fig. 2).
The enhancement pattern on dual‐phase liver protocol CT was not characteristic of HCC. The left portal vein was not visualized. Splenomegaly and esophageal varices were observed. The adrenal glands showed bilateral, heterogeneous enhancing masses. The epiphrenic, retroperitoneal, and periportal lymph nodes were enlarged. Lytic lesions were seen in the sacrum, left iliac wing, and T7‐T10 vertebral bodies.
Intravenous high‐dose steroids were started. The neurosurgery team advised that no surgical interventions were appropriate because of the patient's poor functional status and the extent of his disease.
It is unfortunate that no neurosurgical interventions could help this patient, especially because we are not yet sure of the final diagnosis. Standard indications for neurosurgical decompression include compression from bone fragments, spinal instability requiring fixation, and lack of response to radiation therapy. Patients must also be able to tolerate surgery. Although evidence supports the use of corticosteroids in reducing edema, inflammation, and neurological deficits in malignant spinal cord compression, there is not consensus on what the optimal dose is. Doses of 16‐100 mg of dexamethasone per day appear to be beneficial, as long as higher doses are rapidly tapered to avoid toxic effects. High‐dose steroids minimize the initial edema but are unlikely to change the long‐term outcome of patients who are nonambulatory on arrival.
The CT scan does not help us distinguish between metastatic cancer and primary HCC. Adrenal metastases are very uncommon in HCC. Lung cancer, however, metastasizes to the liver, adrenal glands, and spine, even without significant pulmonary symptoms. HCC may be seen on CT as a solitary mass, a dominant mass with surrounding satellite lesions, multifocal lesions, or a diffusely infiltrating tumor. This diagnosis now seems more likely given the finding of cirrhosis, which increases the risk of HCC in individuals with hepatitis C infection.
We need to obtain tissue for diagnosis and prognosis and to guide therapy. I would consult with radiology and gastroenterology colleagues about the best location to biopsy, but a bone biopsy should be avoided because the pathologic yield is lower.
The radiology and gastroenterology consultants recommended adrenal biopsy because there was easier posterior access for tissue. A liver biopsy was avoided because of the risk of bleeding with hypervascular masses. Fine‐needle aspiration of the mass in the right adrenal gland was performed. The pathology demonstrated bile production and hexagonal arrangement of cells with endothelial cuffing consistent with hepatocellular carcinoma. The oncology staff was consulted about palliative chemotherapy options. The patient began radiation therapy directed at the T7 lesion compressing the spinal cord. He regained minimal movement of his foot. After discussing treatment options with the oncology staff, the patient declined chemotherapy and was transitioned to hospice, where he died 3 weeks later.
COMMENTARY
Hepatocellular carcinoma (HCC) is the third‐leading cause of cancer death and the fifth‐leading cause of cancer worldwide. It causes nearly 1 million deaths annually, and unlike many other cancers, its incidence and mortality rate are rising. Most cases of HCC in Africa and Asia are a result of chronic hepatitis B infection, but in the United States HCC is primarily attributable to hepatitis C infection.1 The annual incidence of HCC in the U.S. population, now about 4 cases per 100,000 people,2 is rising because of the increased prevalence of hepatitis C. Other causes of HCC, such as alcoholic liver disease, hepatitis B infection, and hemochromatosis, have remained stable and have not contributed as significantly to the rising incidence of HCC. For the individual patient, hepatitis C infection conveys a 20‐fold increase in the risk for HCC (2%‐8% risk/year).1 Eighty percent of cases of HCC develop in patients with cirrhosis.3 Unlike patients with hepatitis B infection, persons chronically infected with hepatitis C rarely develop HCC unless they have cirrhosis.
The American Association for the Study of Liver Disease recommends that hepatitis Binfected individuals at high risk for HCC (eg, men older than 40 years and persons with cirrhosis or a family history of HCC) and hepatitis Cinfected individuals with cirrhosis4 be periodically screened for HCC with alpha‐fetoprotein (AFP) and ultrasonography (every 6 months to approximate the doubling time of the tumor5). Using the most commonly reported cutoff for a positive test result for hepatocellular carcinoma (AFP level > 20 g/L) resulted in the following test characteristics: sensitivity, 41%‐65%; specificity, 80%‐94%; positive likelihood ratio, 3.1‐6.8; and negative likelihood ratio, 0.4‐0.6.6 AFP alone is therefore a poor screening test for HCC, and as shown in this case, AFP levels can be normal or only minimally elevated in the setting of diffusely metastatic disease. Ultrasonography alone is only 35%‐87% sensitive in detecting HCC,79 but the combination of AFP and ultrasonography identified 100% of the HCC cases in one small case series.10
For the patient in this case, the optimal clinical pathway would have been to transition from screening to diagnostic measures in a timely manner. Consensus guidelines from the European Association for Study of the Liver in 2001 recommend biopsy of all focal liver lesions that are between 1 and 2 cm.11 The American Association for the Study of Liver Diseases (AASLD) recommends that focal liver lesions between 1 and 2 cm found on ultrasound in cirrhotic livers be followed by 2 dynamic studies: CT, MRI, or contrast ultrasound. If 2 separate studies reveal typical characteristics of HCC, then the lesion should be treated as HCC, and if not typical, then the lesion should be biopsied.4 Although no studies were available to support the recommendations, both the EASL and AASLD advise that lesions greater than 2 cm with demonstrated vascularity on both ultrasonography and CT can be diagnosed as HCC without biopsy and that lesions smaller than 1 cm be monitored.4, 11
Hepatocellular carcinoma can metastasize to almost anywhere in the body by hematologic or lymphatic spread or by direct extension. The most common site for metastases of HCC is the lung. Metastases to the lung arise primarily from arterial emboli and therefore are most common in the lower lobes, where there is greater perfusion.12 The second most common site is intraabdominal lymph nodes. The axial skeleton is the third most common site of metastases and, as in this case, primarily involves the spine.13 Other sites of metastases include the peritoneum, the inferior vena cava and right atrium by direct extension, and, less commonly, the gallbladder and spleen. Autopsy studies of patients with HCC found that 8% had metastases to the adrenal glands, as did this patient.13 Metastasis to the central nervous system is rare.
There were several challenging aspects of this case, including atypical radiologic appearance, an unusual metastatic pattern, and minimally elevated AFP level. This case raises 3 key points that we must remember as clinicians:
-
Patients infected with hepatitis C who are found to have suspicious hepatic lesions should be aggressively evaluated for HCC.
-
Using an AFP level < 20 g/L as a screening test is not helpful because this level can be seen even with widely metastatic disease.
-
Knowledge of available screening tests as well as the many possible manifestations of HCC helps clinicians to diagnose HCC earlier, when the disease is potentially curable.
Acknowledgements
The authors thank Gurpreet Dhaliwal, MD, for reviewing an early version of this manuscript.
- .Hepatocellular carcinoma: epidemiology, risk factors, and screening.Semin Liver Dis.2005;25:143–154.
- American Cancer Society. Cancer Facts and Figures 2005. Atlanta, GA: American Cancer Society, 2005. Available at: http://www.cancer.org/docroot/STT/stt_0.asp. Accessed October 17,2005.
- ,,.Hepatocellular carcinoma.Lancet.2003;362:1907–1917.
- ,.Management of hepatocellular carcinoma. AASLD Practice Guideline.Hepatology.2005;42:1208–1236.
- ,,, et al.Growth rate of asymptomatic hepatocellular carcinoma and its clinical implications.Gastroenterology.1985;89:259–266.
- ,,.Test characteristics of alpha‐fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C.Ann Intern Med.2003;139:46–50.
- ,,,.Sonographic screening for hepatocellular carcinoma in patients with chronic hepatitis or cirrhosis: an evaluation.Am J Roentgenol.1998;171:433–435.
- ,,,,.Detection of malignant tumors in end‐stage cirrhotic livers: efficacy of sonography as a screening technique.Am J Roentgenol.1992;159:727–733.
- ,,, et al.The diagnosis of small hepatocellular carcinomas: efficacy of various imaging procedures in 100 patients.Am J Roentgenol.1990;155:49–54
- ,,,,,.Outcome of 67 patients with hepatocellular cancer detected during screening of 1125 patients with chronic hepatitis.Ann Surg.1998;277:513–518.
- ,,, et al.;EASL Panel of Experts on HCC.Clinical management of hepatocellular carcinoma: conclusions of the Barcelona‐2000 EASL conference: European Association for the Study of the Liver.J Hepatol.2001;35:421–430.
- ,,, et al.Extrahepatic spread of hepatocellular carcinoma: a pictorial review.Eur Radiol.2003;13:874–882.
- ,,,,,.Extrahepatic metastases of hepatocellular carcinoma.Radiology.2000;216:698–703.
A 58‐year‐old man was evaluated for 3 weeks of leg numbness and weakness. His symptoms began with numbness and tingling in the distal left leg that progressed to weakness that impaired his ability to walk. He had no history of trauma or incontinence but endorsed several months of back pain that worsened when lying flat. He had a history of type 2 diabetes mellitus, hepatitis C infection, hypertension, and posttraumatic stress disorder. He had a remote history of intravenous drug use and had quit tobacco 9 years earlier. Medications he was taking included hydrochlorothiazide, rosiglitazone, oxycodone/acetaminophen, baclofen, ibuprofen, and gabapentin.
Internists see this constellation of complaints frequently in an acute care setting. Finding a unifying diagnosis may be difficult initially, so thinking of the symptoms in series is helpful. The complaint of leg weakness and the pattern of numbness should be further elucidated. Is this true weakness, or is it a feeling of instability because of foot numbness? What is the pattern of the numbness? Peripheral neuropathy typically begins in a symmetric stocking pattern (involving the plantar surface of the feet), then progresses to a glove distribution (involving the hands from the fingers distally to the wrist proximally). Such a pattern in a patient with diabetes would be consistent with distal polyneuropathy, a mixed sensory and motor process. Other possible causes of peripheral neuropathy in this patient include HIV, B12 deficiency, and syphilis. These symptoms could be tied to the back pain if this were intervertebral disk disease, a compression fracture, or a lytic lesion in the vertebrae with resulting nerve impingement or if it were epidural spinal cord compression. The lack of bowel or bladder dysfunction speaks against a cauda equina syndrome but does not rule out more cepahalad spinal pathology.
On neurological examination, I would concentrate on differentiating weakness from pain. I would attempt to determine whether the weakness was of central or peripheral nerve etiology. Helpful findings would include increased tone with upper motor lesions and flaccid tone with lower motor lesions, hyperreflexia with upper motor lesions and hyporeflexia with lower motor lesions, a Babinski sign, muscle atrophy or fasciculations, and gait. A rectal examination would also be helpful to assess for deficits in rectal tone, wink reflex, or saddle anesthesia.
All patients with low back pain who have alarm signs of age older than 50, pain duration of more than 1 month, known cancer, lack of relief with conservative measures, or systemic B symptoms should have imaging of the spine. Although plain films may reveal bony abnormalities, computed tomography (CT) is better for evaluating osseous structures and magnetic resonance imaging (MRI) for evaluating pathology in patients suspected of having an infection or a malignancy. I would obtain imaging of the spine in this patient.
The patient was receiving care at an outside clinic for 2 liver lesions discovered on abdominal ultrasound 19 months prior to admission. CT showed that the lesions were 4.0 and 2.3 cm in diameter 17 months prior to admission and 5.0 and 3.0 cm in diameter 5 months prior to admission. No cirrhosis was appreciated on the ultrasound or CT. The patient was referred for CT‐guided biopsy of the larger mass after the second CT, but he became anxious and left before the biopsy was obtained.
This piece of the history is ominous, as it increases the possibility of cancer in our differential. Metastatic disease could provide a unifying diagnosis, explaining the constellation of back pain, leg weakness, and liver lesions. Lung cancer commonly metastasizes to the liver and to bone, so I would obtain a chest x‐ray. Other possible types of cancer in this situation include cancer of the prostate, colon, or thyroid and melanoma. In this patient, who has hepatitis C, hepatocellular carcinoma (HCC) could be the primary etiology, although cirrhosis was not seen on CT and HCC metastasizes to the spine less commonly than do other primary cancers (eg, lung, breast, prostate). Nonetheless, I would obtain an alpha‐fetoprotein level, which would confirm HCC in a patient with liver lesions if it was greater than 200 g/L. Pancreatic cancer has been associated with both type 2 diabetes and liver lesions and could explain his abdominal pain.
There is no comment on the arterial‐phase CT imaging of the liver lesions. Dual‐phase CT scans examine the hepatic arterial and portal vein phases of contrast filling. Triple‐phase CT scans also examine the portal vein influx phase. Both hemangiomas and hypervascular HCCs enhance on the arterial phase, as they derive their blood flow from the hepatic artery. Therefore, arterial‐phase imaging can help to distinguish vascular tumors that flush with contrast, such as hemangiomas, melanoma, and HCC, from less vascular tumors such as pancreatic and colon cancer. Other liver lesions such as focal nodular hyperplasia and adenomas cannot be excluded in this situation as they also may enhance during the arterial phase and can grow over time, as this patient's repeat imaging documented. It seems unlikely that this patient has a liver abscess because he has a paucity of constitutional symptoms and no travel history. The liver lesions seen on initial imaging were larger than 1.0 cm, so I would have favored an earlier biopsy to obtain a tissue diagnosis.
The patient was afebrile, and all other vital signs were normal. He appeared well nourished and anicteric. There was no lymphadenopathy. Cardiac auscultation was regular without murmurs. The lungs were clear. The abdomen was without fluid wave or hepatosplenomegaly and was tender to palpation in the right upper and lower quadrants. There was no midline tenderness to palpation of the spine.
Cranial nerves II‐XII were intact. Lower extremity muscle tone could not be accurately assessed due to splinting from back pain. Strength was 3 of 5 in the left hip extensors and left knee flexors and extensors, and 1 of 5 in the left hip flexors. He had no motor strength in the distal left lower extremity extensors. Bilateral upper extremity and right leg strength were normal. Sensation to light touch, temperature, and pain was decreased circumferentially below the xiphoid. The patient had hyperesthesia in a band around the thorax just above the xiphoid and paresthesia of the perineal area. Left patellar tendon reflexes were brisk, and left ankle jerk was absent, but other reflexes were normal. Toes were down‐going bilaterally. The anal wink was absent, and rectal tone was decreased. Results of the cerebellar exam were normal. Gait could not be assessed.
The results of the exam are notable for not showing the stigmata of end‐stage liver disease. The results of the neurological exam are concerning, with decreased sensation at approximately the T7 level that is almost certainly a result of epidural compression of the spinal cord. Hematogenous metastasis to the vertebrae from one of the tumors mentioned above, with spread into the thecal sac, is the most likely culprit. An epidural abscess is possible because the patient has diabetes and a history of injection drug use.
The thoracic spine is involved in 60% of spinal cord metastases. This patient's left‐sided distal leg weakness is consistent with having corticospinal tract compression and indicates thoracic spine involvement. Flaccid paralysis is classically found in lower motor neuron weakness, but is also seen in the early stages of upper motor neuron pathology. Lesions found above the cauda equina often spare the perineal area, but low thoracic lesions involving the conus medullaris (from T10 to L1) could explain both his loss of anal wink and his decreased rectal tone.
This patient's presentation is unfortunately classic for epidural spinal cord compression. Because the onset of compression is insidious, the diagnosis is often delayed, even in patients with known cancer. Urgent imaging is imperative to evaluate this possibility, as having any meaningful chance of recovery of function depends on rapid relief of the spinal cord compression. I would obtain an emergent MRI of the thoracic and lumbosacral spine.
Laboratory studies showed the following: hemoglobin, 13.1 g/dL; mean corpuscular volume, 80 m3; platelet count, 149,000/L; creatinine, 1.9 mg/dL; aspartate aminotransferase, 66 U/L (5‐35 U/L); alanine aminotransferase, 66 U/L (7‐56 U/L); alkaline phosphatase, 87 U/L (40‐125 U/L); total bilirubin, 1.3 mg/dL; prostate specific antigen (PSA), 1.6 g/dL; and alpha‐fetoprotein (AFP), 10.3 g/L. White cell count, sodium, glucose, calcium, and albumin levels, and prothrombin and partial‐thromboplastin times were within normal ranges.
His liver function tests likely reflect chronic hepatitis C infection. His renal insufficiency could be a result of hypertension, diabetes, or dehydration given that he has been bed‐bound.
Most intriguing are the normal PSA level and only slightly elevated AFP level. PSA is useful for detecting recurrence of prostate cancer or following response of therapy, but the utility of PSA as a screening tool remains controversial in part because of its low specificity. Prostate cancer is the most commonly diagnosed cancer among men and cannot be ruled out by a normal PSA. In a patient with hepatitis C, cirrhosis (which we have not conclusively diagnosed), and a radiologically suspicious liver lesion, an AFP > 200 g/L would be diagnostic of HCC. In this case, however, mildly elevated AFP does not help us to either diagnose or exclude HCC.
The chest x‐ray showed no abnormalities. MRI of the spine revealed lytic lesions in the T7‐T10 vertebral bodies with spinal cord compression at the T7 level (Fig. 1).
A repeat CT scan of the abdomen showed a coarse, nodular liver with 2 heterogeneous, early‐enhancing masses (4.7 4.2 and 3.4 2.4 cm in diameter) with surrounding satellite lesions (Fig. 2).
The enhancement pattern on dual‐phase liver protocol CT was not characteristic of HCC. The left portal vein was not visualized. Splenomegaly and esophageal varices were observed. The adrenal glands showed bilateral, heterogeneous enhancing masses. The epiphrenic, retroperitoneal, and periportal lymph nodes were enlarged. Lytic lesions were seen in the sacrum, left iliac wing, and T7‐T10 vertebral bodies.
Intravenous high‐dose steroids were started. The neurosurgery team advised that no surgical interventions were appropriate because of the patient's poor functional status and the extent of his disease.
It is unfortunate that no neurosurgical interventions could help this patient, especially because we are not yet sure of the final diagnosis. Standard indications for neurosurgical decompression include compression from bone fragments, spinal instability requiring fixation, and lack of response to radiation therapy. Patients must also be able to tolerate surgery. Although evidence supports the use of corticosteroids in reducing edema, inflammation, and neurological deficits in malignant spinal cord compression, there is not consensus on what the optimal dose is. Doses of 16‐100 mg of dexamethasone per day appear to be beneficial, as long as higher doses are rapidly tapered to avoid toxic effects. High‐dose steroids minimize the initial edema but are unlikely to change the long‐term outcome of patients who are nonambulatory on arrival.
The CT scan does not help us distinguish between metastatic cancer and primary HCC. Adrenal metastases are very uncommon in HCC. Lung cancer, however, metastasizes to the liver, adrenal glands, and spine, even without significant pulmonary symptoms. HCC may be seen on CT as a solitary mass, a dominant mass with surrounding satellite lesions, multifocal lesions, or a diffusely infiltrating tumor. This diagnosis now seems more likely given the finding of cirrhosis, which increases the risk of HCC in individuals with hepatitis C infection.
We need to obtain tissue for diagnosis and prognosis and to guide therapy. I would consult with radiology and gastroenterology colleagues about the best location to biopsy, but a bone biopsy should be avoided because the pathologic yield is lower.
The radiology and gastroenterology consultants recommended adrenal biopsy because there was easier posterior access for tissue. A liver biopsy was avoided because of the risk of bleeding with hypervascular masses. Fine‐needle aspiration of the mass in the right adrenal gland was performed. The pathology demonstrated bile production and hexagonal arrangement of cells with endothelial cuffing consistent with hepatocellular carcinoma. The oncology staff was consulted about palliative chemotherapy options. The patient began radiation therapy directed at the T7 lesion compressing the spinal cord. He regained minimal movement of his foot. After discussing treatment options with the oncology staff, the patient declined chemotherapy and was transitioned to hospice, where he died 3 weeks later.
COMMENTARY
Hepatocellular carcinoma (HCC) is the third‐leading cause of cancer death and the fifth‐leading cause of cancer worldwide. It causes nearly 1 million deaths annually, and unlike many other cancers, its incidence and mortality rate are rising. Most cases of HCC in Africa and Asia are a result of chronic hepatitis B infection, but in the United States HCC is primarily attributable to hepatitis C infection.1 The annual incidence of HCC in the U.S. population, now about 4 cases per 100,000 people,2 is rising because of the increased prevalence of hepatitis C. Other causes of HCC, such as alcoholic liver disease, hepatitis B infection, and hemochromatosis, have remained stable and have not contributed as significantly to the rising incidence of HCC. For the individual patient, hepatitis C infection conveys a 20‐fold increase in the risk for HCC (2%‐8% risk/year).1 Eighty percent of cases of HCC develop in patients with cirrhosis.3 Unlike patients with hepatitis B infection, persons chronically infected with hepatitis C rarely develop HCC unless they have cirrhosis.
The American Association for the Study of Liver Disease recommends that hepatitis Binfected individuals at high risk for HCC (eg, men older than 40 years and persons with cirrhosis or a family history of HCC) and hepatitis Cinfected individuals with cirrhosis4 be periodically screened for HCC with alpha‐fetoprotein (AFP) and ultrasonography (every 6 months to approximate the doubling time of the tumor5). Using the most commonly reported cutoff for a positive test result for hepatocellular carcinoma (AFP level > 20 g/L) resulted in the following test characteristics: sensitivity, 41%‐65%; specificity, 80%‐94%; positive likelihood ratio, 3.1‐6.8; and negative likelihood ratio, 0.4‐0.6.6 AFP alone is therefore a poor screening test for HCC, and as shown in this case, AFP levels can be normal or only minimally elevated in the setting of diffusely metastatic disease. Ultrasonography alone is only 35%‐87% sensitive in detecting HCC,79 but the combination of AFP and ultrasonography identified 100% of the HCC cases in one small case series.10
For the patient in this case, the optimal clinical pathway would have been to transition from screening to diagnostic measures in a timely manner. Consensus guidelines from the European Association for Study of the Liver in 2001 recommend biopsy of all focal liver lesions that are between 1 and 2 cm.11 The American Association for the Study of Liver Diseases (AASLD) recommends that focal liver lesions between 1 and 2 cm found on ultrasound in cirrhotic livers be followed by 2 dynamic studies: CT, MRI, or contrast ultrasound. If 2 separate studies reveal typical characteristics of HCC, then the lesion should be treated as HCC, and if not typical, then the lesion should be biopsied.4 Although no studies were available to support the recommendations, both the EASL and AASLD advise that lesions greater than 2 cm with demonstrated vascularity on both ultrasonography and CT can be diagnosed as HCC without biopsy and that lesions smaller than 1 cm be monitored.4, 11
Hepatocellular carcinoma can metastasize to almost anywhere in the body by hematologic or lymphatic spread or by direct extension. The most common site for metastases of HCC is the lung. Metastases to the lung arise primarily from arterial emboli and therefore are most common in the lower lobes, where there is greater perfusion.12 The second most common site is intraabdominal lymph nodes. The axial skeleton is the third most common site of metastases and, as in this case, primarily involves the spine.13 Other sites of metastases include the peritoneum, the inferior vena cava and right atrium by direct extension, and, less commonly, the gallbladder and spleen. Autopsy studies of patients with HCC found that 8% had metastases to the adrenal glands, as did this patient.13 Metastasis to the central nervous system is rare.
There were several challenging aspects of this case, including atypical radiologic appearance, an unusual metastatic pattern, and minimally elevated AFP level. This case raises 3 key points that we must remember as clinicians:
-
Patients infected with hepatitis C who are found to have suspicious hepatic lesions should be aggressively evaluated for HCC.
-
Using an AFP level < 20 g/L as a screening test is not helpful because this level can be seen even with widely metastatic disease.
-
Knowledge of available screening tests as well as the many possible manifestations of HCC helps clinicians to diagnose HCC earlier, when the disease is potentially curable.
Acknowledgements
The authors thank Gurpreet Dhaliwal, MD, for reviewing an early version of this manuscript.
A 58‐year‐old man was evaluated for 3 weeks of leg numbness and weakness. His symptoms began with numbness and tingling in the distal left leg that progressed to weakness that impaired his ability to walk. He had no history of trauma or incontinence but endorsed several months of back pain that worsened when lying flat. He had a history of type 2 diabetes mellitus, hepatitis C infection, hypertension, and posttraumatic stress disorder. He had a remote history of intravenous drug use and had quit tobacco 9 years earlier. Medications he was taking included hydrochlorothiazide, rosiglitazone, oxycodone/acetaminophen, baclofen, ibuprofen, and gabapentin.
Internists see this constellation of complaints frequently in an acute care setting. Finding a unifying diagnosis may be difficult initially, so thinking of the symptoms in series is helpful. The complaint of leg weakness and the pattern of numbness should be further elucidated. Is this true weakness, or is it a feeling of instability because of foot numbness? What is the pattern of the numbness? Peripheral neuropathy typically begins in a symmetric stocking pattern (involving the plantar surface of the feet), then progresses to a glove distribution (involving the hands from the fingers distally to the wrist proximally). Such a pattern in a patient with diabetes would be consistent with distal polyneuropathy, a mixed sensory and motor process. Other possible causes of peripheral neuropathy in this patient include HIV, B12 deficiency, and syphilis. These symptoms could be tied to the back pain if this were intervertebral disk disease, a compression fracture, or a lytic lesion in the vertebrae with resulting nerve impingement or if it were epidural spinal cord compression. The lack of bowel or bladder dysfunction speaks against a cauda equina syndrome but does not rule out more cepahalad spinal pathology.
On neurological examination, I would concentrate on differentiating weakness from pain. I would attempt to determine whether the weakness was of central or peripheral nerve etiology. Helpful findings would include increased tone with upper motor lesions and flaccid tone with lower motor lesions, hyperreflexia with upper motor lesions and hyporeflexia with lower motor lesions, a Babinski sign, muscle atrophy or fasciculations, and gait. A rectal examination would also be helpful to assess for deficits in rectal tone, wink reflex, or saddle anesthesia.
All patients with low back pain who have alarm signs of age older than 50, pain duration of more than 1 month, known cancer, lack of relief with conservative measures, or systemic B symptoms should have imaging of the spine. Although plain films may reveal bony abnormalities, computed tomography (CT) is better for evaluating osseous structures and magnetic resonance imaging (MRI) for evaluating pathology in patients suspected of having an infection or a malignancy. I would obtain imaging of the spine in this patient.
The patient was receiving care at an outside clinic for 2 liver lesions discovered on abdominal ultrasound 19 months prior to admission. CT showed that the lesions were 4.0 and 2.3 cm in diameter 17 months prior to admission and 5.0 and 3.0 cm in diameter 5 months prior to admission. No cirrhosis was appreciated on the ultrasound or CT. The patient was referred for CT‐guided biopsy of the larger mass after the second CT, but he became anxious and left before the biopsy was obtained.
This piece of the history is ominous, as it increases the possibility of cancer in our differential. Metastatic disease could provide a unifying diagnosis, explaining the constellation of back pain, leg weakness, and liver lesions. Lung cancer commonly metastasizes to the liver and to bone, so I would obtain a chest x‐ray. Other possible types of cancer in this situation include cancer of the prostate, colon, or thyroid and melanoma. In this patient, who has hepatitis C, hepatocellular carcinoma (HCC) could be the primary etiology, although cirrhosis was not seen on CT and HCC metastasizes to the spine less commonly than do other primary cancers (eg, lung, breast, prostate). Nonetheless, I would obtain an alpha‐fetoprotein level, which would confirm HCC in a patient with liver lesions if it was greater than 200 g/L. Pancreatic cancer has been associated with both type 2 diabetes and liver lesions and could explain his abdominal pain.
There is no comment on the arterial‐phase CT imaging of the liver lesions. Dual‐phase CT scans examine the hepatic arterial and portal vein phases of contrast filling. Triple‐phase CT scans also examine the portal vein influx phase. Both hemangiomas and hypervascular HCCs enhance on the arterial phase, as they derive their blood flow from the hepatic artery. Therefore, arterial‐phase imaging can help to distinguish vascular tumors that flush with contrast, such as hemangiomas, melanoma, and HCC, from less vascular tumors such as pancreatic and colon cancer. Other liver lesions such as focal nodular hyperplasia and adenomas cannot be excluded in this situation as they also may enhance during the arterial phase and can grow over time, as this patient's repeat imaging documented. It seems unlikely that this patient has a liver abscess because he has a paucity of constitutional symptoms and no travel history. The liver lesions seen on initial imaging were larger than 1.0 cm, so I would have favored an earlier biopsy to obtain a tissue diagnosis.
The patient was afebrile, and all other vital signs were normal. He appeared well nourished and anicteric. There was no lymphadenopathy. Cardiac auscultation was regular without murmurs. The lungs were clear. The abdomen was without fluid wave or hepatosplenomegaly and was tender to palpation in the right upper and lower quadrants. There was no midline tenderness to palpation of the spine.
Cranial nerves II‐XII were intact. Lower extremity muscle tone could not be accurately assessed due to splinting from back pain. Strength was 3 of 5 in the left hip extensors and left knee flexors and extensors, and 1 of 5 in the left hip flexors. He had no motor strength in the distal left lower extremity extensors. Bilateral upper extremity and right leg strength were normal. Sensation to light touch, temperature, and pain was decreased circumferentially below the xiphoid. The patient had hyperesthesia in a band around the thorax just above the xiphoid and paresthesia of the perineal area. Left patellar tendon reflexes were brisk, and left ankle jerk was absent, but other reflexes were normal. Toes were down‐going bilaterally. The anal wink was absent, and rectal tone was decreased. Results of the cerebellar exam were normal. Gait could not be assessed.
The results of the exam are notable for not showing the stigmata of end‐stage liver disease. The results of the neurological exam are concerning, with decreased sensation at approximately the T7 level that is almost certainly a result of epidural compression of the spinal cord. Hematogenous metastasis to the vertebrae from one of the tumors mentioned above, with spread into the thecal sac, is the most likely culprit. An epidural abscess is possible because the patient has diabetes and a history of injection drug use.
The thoracic spine is involved in 60% of spinal cord metastases. This patient's left‐sided distal leg weakness is consistent with having corticospinal tract compression and indicates thoracic spine involvement. Flaccid paralysis is classically found in lower motor neuron weakness, but is also seen in the early stages of upper motor neuron pathology. Lesions found above the cauda equina often spare the perineal area, but low thoracic lesions involving the conus medullaris (from T10 to L1) could explain both his loss of anal wink and his decreased rectal tone.
This patient's presentation is unfortunately classic for epidural spinal cord compression. Because the onset of compression is insidious, the diagnosis is often delayed, even in patients with known cancer. Urgent imaging is imperative to evaluate this possibility, as having any meaningful chance of recovery of function depends on rapid relief of the spinal cord compression. I would obtain an emergent MRI of the thoracic and lumbosacral spine.
Laboratory studies showed the following: hemoglobin, 13.1 g/dL; mean corpuscular volume, 80 m3; platelet count, 149,000/L; creatinine, 1.9 mg/dL; aspartate aminotransferase, 66 U/L (5‐35 U/L); alanine aminotransferase, 66 U/L (7‐56 U/L); alkaline phosphatase, 87 U/L (40‐125 U/L); total bilirubin, 1.3 mg/dL; prostate specific antigen (PSA), 1.6 g/dL; and alpha‐fetoprotein (AFP), 10.3 g/L. White cell count, sodium, glucose, calcium, and albumin levels, and prothrombin and partial‐thromboplastin times were within normal ranges.
His liver function tests likely reflect chronic hepatitis C infection. His renal insufficiency could be a result of hypertension, diabetes, or dehydration given that he has been bed‐bound.
Most intriguing are the normal PSA level and only slightly elevated AFP level. PSA is useful for detecting recurrence of prostate cancer or following response of therapy, but the utility of PSA as a screening tool remains controversial in part because of its low specificity. Prostate cancer is the most commonly diagnosed cancer among men and cannot be ruled out by a normal PSA. In a patient with hepatitis C, cirrhosis (which we have not conclusively diagnosed), and a radiologically suspicious liver lesion, an AFP > 200 g/L would be diagnostic of HCC. In this case, however, mildly elevated AFP does not help us to either diagnose or exclude HCC.
The chest x‐ray showed no abnormalities. MRI of the spine revealed lytic lesions in the T7‐T10 vertebral bodies with spinal cord compression at the T7 level (Fig. 1).
A repeat CT scan of the abdomen showed a coarse, nodular liver with 2 heterogeneous, early‐enhancing masses (4.7 4.2 and 3.4 2.4 cm in diameter) with surrounding satellite lesions (Fig. 2).
The enhancement pattern on dual‐phase liver protocol CT was not characteristic of HCC. The left portal vein was not visualized. Splenomegaly and esophageal varices were observed. The adrenal glands showed bilateral, heterogeneous enhancing masses. The epiphrenic, retroperitoneal, and periportal lymph nodes were enlarged. Lytic lesions were seen in the sacrum, left iliac wing, and T7‐T10 vertebral bodies.
Intravenous high‐dose steroids were started. The neurosurgery team advised that no surgical interventions were appropriate because of the patient's poor functional status and the extent of his disease.
It is unfortunate that no neurosurgical interventions could help this patient, especially because we are not yet sure of the final diagnosis. Standard indications for neurosurgical decompression include compression from bone fragments, spinal instability requiring fixation, and lack of response to radiation therapy. Patients must also be able to tolerate surgery. Although evidence supports the use of corticosteroids in reducing edema, inflammation, and neurological deficits in malignant spinal cord compression, there is not consensus on what the optimal dose is. Doses of 16‐100 mg of dexamethasone per day appear to be beneficial, as long as higher doses are rapidly tapered to avoid toxic effects. High‐dose steroids minimize the initial edema but are unlikely to change the long‐term outcome of patients who are nonambulatory on arrival.
The CT scan does not help us distinguish between metastatic cancer and primary HCC. Adrenal metastases are very uncommon in HCC. Lung cancer, however, metastasizes to the liver, adrenal glands, and spine, even without significant pulmonary symptoms. HCC may be seen on CT as a solitary mass, a dominant mass with surrounding satellite lesions, multifocal lesions, or a diffusely infiltrating tumor. This diagnosis now seems more likely given the finding of cirrhosis, which increases the risk of HCC in individuals with hepatitis C infection.
We need to obtain tissue for diagnosis and prognosis and to guide therapy. I would consult with radiology and gastroenterology colleagues about the best location to biopsy, but a bone biopsy should be avoided because the pathologic yield is lower.
The radiology and gastroenterology consultants recommended adrenal biopsy because there was easier posterior access for tissue. A liver biopsy was avoided because of the risk of bleeding with hypervascular masses. Fine‐needle aspiration of the mass in the right adrenal gland was performed. The pathology demonstrated bile production and hexagonal arrangement of cells with endothelial cuffing consistent with hepatocellular carcinoma. The oncology staff was consulted about palliative chemotherapy options. The patient began radiation therapy directed at the T7 lesion compressing the spinal cord. He regained minimal movement of his foot. After discussing treatment options with the oncology staff, the patient declined chemotherapy and was transitioned to hospice, where he died 3 weeks later.
COMMENTARY
Hepatocellular carcinoma (HCC) is the third‐leading cause of cancer death and the fifth‐leading cause of cancer worldwide. It causes nearly 1 million deaths annually, and unlike many other cancers, its incidence and mortality rate are rising. Most cases of HCC in Africa and Asia are a result of chronic hepatitis B infection, but in the United States HCC is primarily attributable to hepatitis C infection.1 The annual incidence of HCC in the U.S. population, now about 4 cases per 100,000 people,2 is rising because of the increased prevalence of hepatitis C. Other causes of HCC, such as alcoholic liver disease, hepatitis B infection, and hemochromatosis, have remained stable and have not contributed as significantly to the rising incidence of HCC. For the individual patient, hepatitis C infection conveys a 20‐fold increase in the risk for HCC (2%‐8% risk/year).1 Eighty percent of cases of HCC develop in patients with cirrhosis.3 Unlike patients with hepatitis B infection, persons chronically infected with hepatitis C rarely develop HCC unless they have cirrhosis.
The American Association for the Study of Liver Disease recommends that hepatitis Binfected individuals at high risk for HCC (eg, men older than 40 years and persons with cirrhosis or a family history of HCC) and hepatitis Cinfected individuals with cirrhosis4 be periodically screened for HCC with alpha‐fetoprotein (AFP) and ultrasonography (every 6 months to approximate the doubling time of the tumor5). Using the most commonly reported cutoff for a positive test result for hepatocellular carcinoma (AFP level > 20 g/L) resulted in the following test characteristics: sensitivity, 41%‐65%; specificity, 80%‐94%; positive likelihood ratio, 3.1‐6.8; and negative likelihood ratio, 0.4‐0.6.6 AFP alone is therefore a poor screening test for HCC, and as shown in this case, AFP levels can be normal or only minimally elevated in the setting of diffusely metastatic disease. Ultrasonography alone is only 35%‐87% sensitive in detecting HCC,79 but the combination of AFP and ultrasonography identified 100% of the HCC cases in one small case series.10
For the patient in this case, the optimal clinical pathway would have been to transition from screening to diagnostic measures in a timely manner. Consensus guidelines from the European Association for Study of the Liver in 2001 recommend biopsy of all focal liver lesions that are between 1 and 2 cm.11 The American Association for the Study of Liver Diseases (AASLD) recommends that focal liver lesions between 1 and 2 cm found on ultrasound in cirrhotic livers be followed by 2 dynamic studies: CT, MRI, or contrast ultrasound. If 2 separate studies reveal typical characteristics of HCC, then the lesion should be treated as HCC, and if not typical, then the lesion should be biopsied.4 Although no studies were available to support the recommendations, both the EASL and AASLD advise that lesions greater than 2 cm with demonstrated vascularity on both ultrasonography and CT can be diagnosed as HCC without biopsy and that lesions smaller than 1 cm be monitored.4, 11
Hepatocellular carcinoma can metastasize to almost anywhere in the body by hematologic or lymphatic spread or by direct extension. The most common site for metastases of HCC is the lung. Metastases to the lung arise primarily from arterial emboli and therefore are most common in the lower lobes, where there is greater perfusion.12 The second most common site is intraabdominal lymph nodes. The axial skeleton is the third most common site of metastases and, as in this case, primarily involves the spine.13 Other sites of metastases include the peritoneum, the inferior vena cava and right atrium by direct extension, and, less commonly, the gallbladder and spleen. Autopsy studies of patients with HCC found that 8% had metastases to the adrenal glands, as did this patient.13 Metastasis to the central nervous system is rare.
There were several challenging aspects of this case, including atypical radiologic appearance, an unusual metastatic pattern, and minimally elevated AFP level. This case raises 3 key points that we must remember as clinicians:
-
Patients infected with hepatitis C who are found to have suspicious hepatic lesions should be aggressively evaluated for HCC.
-
Using an AFP level < 20 g/L as a screening test is not helpful because this level can be seen even with widely metastatic disease.
-
Knowledge of available screening tests as well as the many possible manifestations of HCC helps clinicians to diagnose HCC earlier, when the disease is potentially curable.
Acknowledgements
The authors thank Gurpreet Dhaliwal, MD, for reviewing an early version of this manuscript.
- .Hepatocellular carcinoma: epidemiology, risk factors, and screening.Semin Liver Dis.2005;25:143–154.
- American Cancer Society. Cancer Facts and Figures 2005. Atlanta, GA: American Cancer Society, 2005. Available at: http://www.cancer.org/docroot/STT/stt_0.asp. Accessed October 17,2005.
- ,,.Hepatocellular carcinoma.Lancet.2003;362:1907–1917.
- ,.Management of hepatocellular carcinoma. AASLD Practice Guideline.Hepatology.2005;42:1208–1236.
- ,,, et al.Growth rate of asymptomatic hepatocellular carcinoma and its clinical implications.Gastroenterology.1985;89:259–266.
- ,,.Test characteristics of alpha‐fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C.Ann Intern Med.2003;139:46–50.
- ,,,.Sonographic screening for hepatocellular carcinoma in patients with chronic hepatitis or cirrhosis: an evaluation.Am J Roentgenol.1998;171:433–435.
- ,,,,.Detection of malignant tumors in end‐stage cirrhotic livers: efficacy of sonography as a screening technique.Am J Roentgenol.1992;159:727–733.
- ,,, et al.The diagnosis of small hepatocellular carcinomas: efficacy of various imaging procedures in 100 patients.Am J Roentgenol.1990;155:49–54
- ,,,,,.Outcome of 67 patients with hepatocellular cancer detected during screening of 1125 patients with chronic hepatitis.Ann Surg.1998;277:513–518.
- ,,, et al.;EASL Panel of Experts on HCC.Clinical management of hepatocellular carcinoma: conclusions of the Barcelona‐2000 EASL conference: European Association for the Study of the Liver.J Hepatol.2001;35:421–430.
- ,,, et al.Extrahepatic spread of hepatocellular carcinoma: a pictorial review.Eur Radiol.2003;13:874–882.
- ,,,,,.Extrahepatic metastases of hepatocellular carcinoma.Radiology.2000;216:698–703.
- .Hepatocellular carcinoma: epidemiology, risk factors, and screening.Semin Liver Dis.2005;25:143–154.
- American Cancer Society. Cancer Facts and Figures 2005. Atlanta, GA: American Cancer Society, 2005. Available at: http://www.cancer.org/docroot/STT/stt_0.asp. Accessed October 17,2005.
- ,,.Hepatocellular carcinoma.Lancet.2003;362:1907–1917.
- ,.Management of hepatocellular carcinoma. AASLD Practice Guideline.Hepatology.2005;42:1208–1236.
- ,,, et al.Growth rate of asymptomatic hepatocellular carcinoma and its clinical implications.Gastroenterology.1985;89:259–266.
- ,,.Test characteristics of alpha‐fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C.Ann Intern Med.2003;139:46–50.
- ,,,.Sonographic screening for hepatocellular carcinoma in patients with chronic hepatitis or cirrhosis: an evaluation.Am J Roentgenol.1998;171:433–435.
- ,,,,.Detection of malignant tumors in end‐stage cirrhotic livers: efficacy of sonography as a screening technique.Am J Roentgenol.1992;159:727–733.
- ,,, et al.The diagnosis of small hepatocellular carcinomas: efficacy of various imaging procedures in 100 patients.Am J Roentgenol.1990;155:49–54
- ,,,,,.Outcome of 67 patients with hepatocellular cancer detected during screening of 1125 patients with chronic hepatitis.Ann Surg.1998;277:513–518.
- ,,, et al.;EASL Panel of Experts on HCC.Clinical management of hepatocellular carcinoma: conclusions of the Barcelona‐2000 EASL conference: European Association for the Study of the Liver.J Hepatol.2001;35:421–430.
- ,,, et al.Extrahepatic spread of hepatocellular carcinoma: a pictorial review.Eur Radiol.2003;13:874–882.
- ,,,,,.Extrahepatic metastases of hepatocellular carcinoma.Radiology.2000;216:698–703.