User login
When Clozapine is not enough: Augment with lamotrigine?
Current antipsychotics are reasonably effective in treating positive symptoms, but they do less to improve the negative and cognitive symptoms1 that contribute to patients’ long-term poor functional capacity and quality of life.2 So what do psychiatrists do in clinical practice to mitigate antipsychotics’ limitations? We augment.
Schizophrenia patients routinely are treated with polypharmacy—often with antidepressants or anticonvulsants—in attempts to improve negative symptoms, aggression, and impulsivity.3 Most adjuncts, however—including divalproex, antidepressants, and lithium—have shown very small, inconsistent, or no effects.4,5 The only agent with a recent meta-analysis supporting its use as augmentation in treatment-resistant schizophrenia is lamotrigine,6 an anticonvulsant approved for use in epilepsy.7
This article examines the evidence supporting off-label use of lamotrigine as an augmenting agent in schizophrenia and explains the rationale, based on lamotrigine’s probable mechanism of action as a stabilizer of glutamate neurotransmission.
Is lamotrigine worth trying?
Some 20% of schizophrenia patients are considered treatment-resistant, with persistent positive symptoms despite having undergone ≥2 adequate antipsychotic trials.8 Evidence suggests clozapine then should be tried,4 but approximately one-half of treatment-resistant patients do not respond to clozapine. Treatment guidelines are limited for these 10% of schizophrenia patients with an inadequate response to available therapies, including clozapine.4
In a meta-analysis of 5 controlled trials in patients with treatment-resistant schizophrenia, adjunctive lamotrigine was shown to significantly reduce Positive and Negative Syndrome Scale (PANSS) total scores, positive symptom subscores, and negative symptom subscores.6 In these trials, lamotrigine was added to various antipsychotics, including clozapine. Based on the results—as outlined below—we suggest:
- In treatment-resistant patients with residual symptoms while taking clozapine, lamotrigine given in dosages ≥200 mg/d could be a first-line adjunct (Figure 1).
- Lamotrigine augmentation also might help patients whose positive symptoms are adequately controlled but who have persistent negative and/or cognitive symptoms.
- Evidence does not support routine use of lamotrigine in patients taking antipsychotics other than clozapine.
Managing side effects. Lamotrigine is generally well tolerated; in the meta-analysis, nausea was the only side effect more common with lamotrigine (9%) than with placebo (3.9%).6 Close follow-up is required, however, as a few case reports have noted worsening positive symptoms when lamotrigine was added to antipsychotics.9,10
Lamotrigine produces a skin rash in approximately 10% of patients; the rash usually is benign but may be severe, including the potentially fatal Stevens-Johnson syndrome.11 In the meta-analysis, rash was no more likely in patients receiving placebo (3%) than those receiving lamotrigine (2.2%), and no serious rashes were reported.6 Even so, lamotrigine needs to be titrated upwards very slowly over weeks, and patients must be able to monitor for rash.
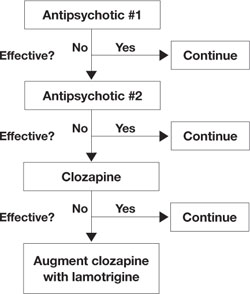
Figure 1 An evidence-based approach to treatment-resistant schizophrenia
Treatment-resistant schizophrenia is defined as residual positive symptoms after ≥2 adequate antipsychotic trials. Evidence supports trying clozapine as the next step.4 When patients show an inadequate response to clozapine, a meta-analysis of 5 controlled trials6 indicates that lamotrigine may be a useful first-line adjunct.
Why consider lamotrigine?
During clinical trials of lamotrigine for epilepsy, patients showed improved mood12 as is seen with other anticonvulsants such as valproate and carbamazepine.13 A series of randomized trials then demonstrated lamotrigine’s effectiveness in treating patients with bipolar I disorder, especially during depressive episodes,14,15 and the FDA approved lamotrigine for maintenance treatment of bipolar I disorder.16 In those early studies, lamotrigine also improved bipolar patients’ quality of life and cognitive function in addition to showing mood-stabilizing properties.12
The glutamate hypothesis. Lamotrigine is an inhibitor of voltage-gated sodium channels and has been shown to inhibit the excessive synaptic release of glutamate.17 Glutamate is the primary excitatory neurotransmitter for at least 60% of neurons in the brain, including all cortical pyramidal neurons. A large body of evidence implicates dysfunctional glutamate signaling in the pathophysiology of schizophrenia.18
For example, phencyclidine (PCP) and ketamine—antagonists of one subtype of glutamate receptor, the N-methyl-D-aspartate (NMDA) receptor—are well known to produce positive psychotic symptoms, negative symptoms, and cognitive dysfunction.19 This led to a long-held hypothesis that schizophrenia is caused by too little glutamate. However, ketamine and PCP also increase the release of glutamate at synapses that then can act on glutamate receptors other than the NMDA receptor, which suggests that too much glutamate also may be involved in schizophrenia.
Too little or too much glutamate? These competing hypotheses could both be at least partially true, suggesting an “inverted-U” pattern of glutamate signaling (Figure 2). Because glutamate is involved in most cortical functions, too little glutamate can cause cognitive and processing deficits such as those seen in schizophrenia. On the other hand, too much glutamate can be toxic to neurons and may be a factor in neurodegeneration, such as in Alzheimer’s disease.20 Indeed, schizophrenia may be associated with gradual neurodegeneration.21
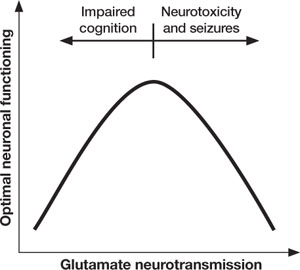
Figure 2 Inverted U-curve may explain dysfunctional glutamate signaling in schizophrenia
Both too little or too much glutamate may play a role in schizophrenia’s pathophysiology. Glutamate, the major excitatory neurotransmitter of the cerebral cortex, is involved in most cognitive functions. Too little (or glutamate inhibition) can impair cognition, whereas too much can lead to seizures, neurotoxicity, and cell death.
Glutamate stabilization?
Because lamotrigine prevents excessive glutamate release at synapses, it stabilizes neuronal membranes by preventing toxicity from too much glutamate without interfering with glutamate’s normal functions.22 Thus, lamotrigine may have potential to maintain optimal glutamate signaling in patients with schizophrenia.
In 16 healthy volunteers, a 300-mg dose of lamotrigine was significantly more effective than placebo in reducing ketamine-induced positive symptoms, as assessed by the Brief Psychiatric Rating Scale positive symptoms subscale (P < .001). Lamotrigine pretreatment also reduced negative symptoms and improved learning and memory.23
More recently, lamotrigine pretreatment was shown to prevent many ketamine-induced changes on functional MRI.24 Few antipsychotics have clinically significant effects on ketamine-induced symptoms—especially in a single dose—although repeated dosing with clozapine attenuates some ketamine-induced effects.25
Given the limitations of available antipsychotics, adding a drug such as lamotrigine—which may modulate and stabilize the glutamate system—could be effective in treatment-resistant schizophrenia.
What is the evidence?
Case reports and open-label case series first showed that lamotrigine augmentation could be effective in treatment-resistant schizophrenia patients receiving clozapine.26–28 One naturalistic case series also included patients receiving olanzapine or risperidone and suggested greater improvement with lamotrigine augmentation in patients on clozapine.26
Controlled trials. In a placebo-controlled trial, Tiihonen et al29 reported significantly lower ratings of positive symptoms—but not negative symptoms—after 38 treatment-resistant schizophrenia patients on clozapine received adjunctive lamotrigine, 200 mg/d, for 14 weeks (Table 1).
A subsequent controlled trial in which Kremer et al30 added lamotrigine, ≤400 mg/d, showed significant improvements in positive and negative symptoms among 31 treatment-resistant schizophrenia patients who completed the 10-week study. Patients were taking conventional and atypical antipsychotics, including clozapine. All groups improved, but the study was not powered to detect differences among the groups.
Table 1
Lamotrigine augmentation: 5 double-blind, placebo-controlled trials
| Trial duration | Patient diagnosis (number) | Antipsychotic(s) | Lamotrigine (mg/d) | Results |
|---|---|---|---|---|
| 14 weeks (Tiihonen et al, 200329) | Treatment-resistant schizophrenia (n=34) | Clozapine | 200 | Significantly reduced psychosis ratings, with no significant improvement in negative symptoms |
| 10 weeks (Kremer et al, 200430) | Treatment-resistant schizophrenia (n=38) | Conventional and atypical, including clozapine | ≤400 | Significant improvements with all antipsychotics, especially clozapine, in positive and negative symptoms* |
| 8 weeks (Akhondzadeh et al, 200531) | Schizophrenia (n=36) | Risperidone | 150 | Significant improvement in negative symptoms and cognition; less improvement in positive symptoms |
| 12 weeks, multicenter (Goff et al, 200732) | Schizophrenia, schizoaffective patients with residual symptoms (n=217+212) | Conventional and atypical, including clozapine | 100 to 400 | No significant improvement in any symptom domain; improved negative symptoms only in study 1 and cognitive symptoms only in study 2 |
| 24 weeks (Zoccali et al, 200733) | Treatment-resistant schizophrenia (n=51) | Clozapine | ≤200 | Significant improvement in positive and negative symptoms as well as some cognitive symptoms |
| * Significance achieved only in study completers, not in the last-observation-carried-forward analysis | ||||
A third trial by Akhondzadeh et al,31 augmenting risperidone with lamotrigine, 150 mg/d, resulted in modest improvements in negative and cognitive symptoms and slight improvement in positive symptoms.
Multicenter trials. Preliminary trials led to 2 randomized, double-blind, multicenter studies. In a total of 429 schizophrenia outpatients with residual psychotic symptoms on atypical antipsychotics, lamotrigine, 100 to 400 mg/d, or placebo was added for 12 weeks.32 The combined results failed to show significant improvement with adjunctive lamotrigine in any symptom domain compared with placebo. One study showed some improved negative symptoms, and the other showed improved cognitive symptoms.
Possible reasons for these negative results were unclear, although:
- a relatively large placebo response, compared with other studies, suggests a “failed” clinical trial
- the small number of patients receiving clozapine in this study suggests that they may have been less treatment-resistant than those enrolled in prior studies.
Meta-analysis. A meta-analysis of data from these 5 randomized, controlled trials found the “positive, negative, and general psychopathology subscale scores as measured with the PANSS … showed significant difference favoring adjuvant lamotrigine” (Table 2).6 As for study limitations, the authors noted that effectiveness data could be usefully analyzed in <70 of the 537 patients from the controlled trials, and “the small mean decrease in scores may not be really clinically relevant.”6 Thus, they said, caution is warranted in translating these results to clinical practice.
One more trial. Since the meta-analysis, an additional placebo-controlled trial has been reported.33 In this 24-week trial, lamotrigine augmentation, ≤200 mg/d, was statistically more effective than placebo in reducing positive and negative symptoms in 51 stable treatment-resistant patients on clozapine. Cognitive function also improved.
Table 2
How symptom scores changed with add-on lamotrigine in the meta-analysis of controlled trials
| PANSS subscales: Individual items scored 1 to 7, with 1=absent and 7=extreme | Change [95% CI]* |
|---|---|
| Positive symptom subscale (max 49) Delusions, conceptual disorganization, hallucinatory behavior, excitement, grandiosity, suspiciousness, hostility | -5.10 [-8.86, -1.34] |
| Negative symptom subscale (max 49) Blunted affect, emotional withdrawal, poor rapport, passive-apathetic social withdrawal, difficulty in abstract thinking, lack of spontaneity and flow of conversation, stereotyped thinking | -5.25 [-7.07, -3.43] |
| General psychopathology subscale (max 112) Somatic concern, anxiety, guilt feelings, tension, mannerisms and posturing, depression, motor retardation, uncooperativeness, unusual thought content, disorientation, poor attention, lack of judgment and insight, disturbance of volition, poor impulse control, preoccupation, active social avoidance | -10.74 [-16.53, -4.96] |
| * See text for limitations of the meta-analysis | |
| CI: confidence interval; PANSS: Positive and Negative Syndrome Scale | |
| Source: Reference 6 | |
Only treatment-resistant patients?
In controlled trials, lamotrigine augmentation has had the greatest effect on positive and negative symptoms in treatment-resistant schizophrenia patients, especially those on clozapine. Could lamotrigine augmentation be of benefit only in treatment-resistant schizophrenia?
Analysis of trial findings. As mentioned, outpatients who comprised the majority of subjects in the 2 large “negative” (or possibly failed) trials32 might have been less treatment-resistant than subjects in the other trials. Lower mean lamotrigine dosages (205 mg/d and 241 mg/d) also were used in the 2 negative trials and in the trial by Akhondzadeh et al (150 mg/d)31—compared with up to 400 mg/d in the trial by Kremer et al.30 This suggests that insufficient dosing might have caused the nonsignificant findings.
Given schizophrenia’s heterogeneity, treatment-resistant patients may represent a subgroup that has greater glutamatergic dysfunction, whereas patients who respond more completely to antipsychotics may have greater dopaminergic dysfunction. Thus, lamotrigine augmentation might be more beneficial in the subset of treatment-resistant patients. Lamotrigine or other glutamate stabilizers have been proposed to act as neuroprotective agents, slowing functional decline in chronic schizophrenia34 (although long-term studies needed to test this hypothesis are unlikely to occur because of cost and time constraints).
Another hypothetical, yet intriguing, explanation for the greater effects of lamotrigine augmentation in patients on clozapine is a pharmacodynamic interaction between these 2 drugs. Clozapine (and possibly olanzapine) have been shown to enhance cortical glutamatergic transmission.25 We propose that clozapine-induced boosting of glutamate in concert with stabilization of the glutamate system by lamotrigine improves neuronal functioning. Clinical trial data regarding lamotrigine augmentation of antipsychotics other than clozapine are needed to determine if the relationship between clozapine and lamotrigine is unique.
Related resources
- Lamotrigine prescribing information and patient handout. www.lamictal.com/bipolar/hcp/prescibing_information. html.
- Augmentation strategies for schizophrenia. IPAP Schizophrenia algorithm flowchart (online interactive version), node 11. www.ipap.org/algorithms.php.
Drug brand names
- Carbamazepine • Carbatrol, Equetro, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Valproate • Depacon, Depakene
Disclosures
Dr. Gray reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Risch receives research support from the National Institute of Mental Health and is a speaker for AstraZeneca and Pfizer Inc.
1. Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12(10):904-922.
2. Agid Y, Buzsaki G, Diamond DM, et al. How can drug discovery for psychiatric disorders be improved? Nat Rev Drug Discov. 2007;6(3):189-201.
3. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: comparing monotherapy with polypharmacy and augmentation. Curr Med Chem. 2004;11(3):313-327.
4. Miller AL, McEvoy SP, Jeste DV, et al. Treatment of chronic schizophrenia. In: Lieberman JA, Stroup TS, Perkins DO, eds. Textbook of schizophrenia. Arlington, VA: American Psychiatric Publishing; 2006:365-381.
5. Miller AL. Combination treatments for schizophrenia. CNS Spectr. 2004;9(9 suppl 9):19-23.
6. Premkumar TS, Pick J. Lamotrigine for schizophrenia. Cochrane Database Syst Rev. 2006;(4):CD005962.-
7. Brodie MJ, Richens A, Yuen AW. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK lamotrigine/carbamazepine monotherapy trial group. Lancet. 1995;345(8948):476-479.
8. Buckley P, Miller A, Olsen J, et al. When symptoms persist: clozapine augmentation strategies. Schizophr Bull. 2001;27(4):615-628.
9. Chan YC, Miller KM, Shaheen N, et al. Worsening of psychotic symptoms in schizophrenia with addition of lamotrigine: a case report. Schizophr Res. 2005;78(2-3):343-345.
10. Konstantakopoulos G, Oulis P, Koulouris GC, et al. Lamotrigine-associated exacerbation of positive symptoms in paranoid schizophrenia. Schizophr Res. 2008;98(1-3):325-326.
11. Messenheimer J, Mullens EL, Giorgi L, et al. Safety review of adult clinical trial experience with lamotrigine. Drug Saf. 1998;18(4):281-296.
12. Smith D, Baker G, Davies G, et al. Outcomes of add-on treatment with lamotrigine in partial epilepsy. Epilepsia. 1993;34(2):312-322.
13. Post RM, Ketter TA, Denicoff K, et al. The place of anticonvulsant therapy in bipolar illness. Psychopharmacology (Berl). 1996;128(2):115-129.
14. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 study group. J Clin Psychiatry. 1999;60(2):79-88.
15. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. Lamictal 614 study group. J Clin Psychiatry. 2000;61(11):841-850.
16. Bowden CL, Calabrese JR, Sachs G, et al. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
17. Large CH, Webster EL, Goff DC. The potential role of lamotrigine in schizophrenia. Psychopharmacology (Berl). 2005;181(3):415-436.
18. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9):1367-1377.
19. Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry. 2004;9(11):984-997.
20. Chohan MO, Iqbal K. From tau to toxicity: emerging roles of NMDA receptor in Alzheimer’s disease. J Alzheimers Dis. 2006;10(1):81-87.
21. Konradi C, Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther. 2003;97(2):153-179.
22. Leach MJ, Baxter MG, Critchley MA. Neurochemical and behavioral aspects of lamotrigine. Epilepsia. 1991;32(suppl 2):S4-S8.
23. Anand A, Charney DS, Oren DA, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of n-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57(3):270-276.
24. Deakin JF, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65(2):154-164.
25. Large CH. Do NMDA receptor antagonist models of schizophrenia predict the clinical efficacy of antipsychotic drugs? J Psychopharmacol. 2007;21(3):283-301.
26. Dursun SM, Deakin JF. Augmenting antipsychotic treatment with lamotrigine or topiramate in patients with treatment-resistant schizophrenia: a naturalistic case-series outcome study. J Psychopharmacol. 2001;15(4):297-301.
27. Dursun SM, McIntosh D, Milliken H. Clozapine plus lamotrigine in treatment-resistant schizophrenia. Arch Gen Psychiatry. 1999;56(10):950.-
28. Saba G, Dumortier G, Kalalou K, et al. Lamotrigine-clozapine combination in refractory schizophrenia: three cases. J Neuropsychiatry Clin Neurosci. 2002;14(1):86.-
29. Tiihonen J, Hallikainen T, Ryynanen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry. 2003;54(11):1241-1248.
30. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):441-446.
31. Akhondzadeh S, Mackinejad K, Ahmadi-Abhari SA, et al. Does the addition of lamotrigine to risperidone improve psychotic symptoms and cognitive impairments in chronic schizophrenia? Therapy. 2005;2(3):399-406.
32. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol. 2007;27(6):582-589.
33. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res. 2007;93(1-3):109-116.
34. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(3 suppl 4):1-13.
Current antipsychotics are reasonably effective in treating positive symptoms, but they do less to improve the negative and cognitive symptoms1 that contribute to patients’ long-term poor functional capacity and quality of life.2 So what do psychiatrists do in clinical practice to mitigate antipsychotics’ limitations? We augment.
Schizophrenia patients routinely are treated with polypharmacy—often with antidepressants or anticonvulsants—in attempts to improve negative symptoms, aggression, and impulsivity.3 Most adjuncts, however—including divalproex, antidepressants, and lithium—have shown very small, inconsistent, or no effects.4,5 The only agent with a recent meta-analysis supporting its use as augmentation in treatment-resistant schizophrenia is lamotrigine,6 an anticonvulsant approved for use in epilepsy.7
This article examines the evidence supporting off-label use of lamotrigine as an augmenting agent in schizophrenia and explains the rationale, based on lamotrigine’s probable mechanism of action as a stabilizer of glutamate neurotransmission.
Is lamotrigine worth trying?
Some 20% of schizophrenia patients are considered treatment-resistant, with persistent positive symptoms despite having undergone ≥2 adequate antipsychotic trials.8 Evidence suggests clozapine then should be tried,4 but approximately one-half of treatment-resistant patients do not respond to clozapine. Treatment guidelines are limited for these 10% of schizophrenia patients with an inadequate response to available therapies, including clozapine.4
In a meta-analysis of 5 controlled trials in patients with treatment-resistant schizophrenia, adjunctive lamotrigine was shown to significantly reduce Positive and Negative Syndrome Scale (PANSS) total scores, positive symptom subscores, and negative symptom subscores.6 In these trials, lamotrigine was added to various antipsychotics, including clozapine. Based on the results—as outlined below—we suggest:
- In treatment-resistant patients with residual symptoms while taking clozapine, lamotrigine given in dosages ≥200 mg/d could be a first-line adjunct (Figure 1).
- Lamotrigine augmentation also might help patients whose positive symptoms are adequately controlled but who have persistent negative and/or cognitive symptoms.
- Evidence does not support routine use of lamotrigine in patients taking antipsychotics other than clozapine.
Managing side effects. Lamotrigine is generally well tolerated; in the meta-analysis, nausea was the only side effect more common with lamotrigine (9%) than with placebo (3.9%).6 Close follow-up is required, however, as a few case reports have noted worsening positive symptoms when lamotrigine was added to antipsychotics.9,10
Lamotrigine produces a skin rash in approximately 10% of patients; the rash usually is benign but may be severe, including the potentially fatal Stevens-Johnson syndrome.11 In the meta-analysis, rash was no more likely in patients receiving placebo (3%) than those receiving lamotrigine (2.2%), and no serious rashes were reported.6 Even so, lamotrigine needs to be titrated upwards very slowly over weeks, and patients must be able to monitor for rash.
Figure 1 An evidence-based approach to treatment-resistant schizophrenia
Treatment-resistant schizophrenia is defined as residual positive symptoms after ≥2 adequate antipsychotic trials. Evidence supports trying clozapine as the next step.4 When patients show an inadequate response to clozapine, a meta-analysis of 5 controlled trials6 indicates that lamotrigine may be a useful first-line adjunct.
Why consider lamotrigine?
During clinical trials of lamotrigine for epilepsy, patients showed improved mood12 as is seen with other anticonvulsants such as valproate and carbamazepine.13 A series of randomized trials then demonstrated lamotrigine’s effectiveness in treating patients with bipolar I disorder, especially during depressive episodes,14,15 and the FDA approved lamotrigine for maintenance treatment of bipolar I disorder.16 In those early studies, lamotrigine also improved bipolar patients’ quality of life and cognitive function in addition to showing mood-stabilizing properties.12
The glutamate hypothesis. Lamotrigine is an inhibitor of voltage-gated sodium channels and has been shown to inhibit the excessive synaptic release of glutamate.17 Glutamate is the primary excitatory neurotransmitter for at least 60% of neurons in the brain, including all cortical pyramidal neurons. A large body of evidence implicates dysfunctional glutamate signaling in the pathophysiology of schizophrenia.18
For example, phencyclidine (PCP) and ketamine—antagonists of one subtype of glutamate receptor, the N-methyl-D-aspartate (NMDA) receptor—are well known to produce positive psychotic symptoms, negative symptoms, and cognitive dysfunction.19 This led to a long-held hypothesis that schizophrenia is caused by too little glutamate. However, ketamine and PCP also increase the release of glutamate at synapses that then can act on glutamate receptors other than the NMDA receptor, which suggests that too much glutamate also may be involved in schizophrenia.
Too little or too much glutamate? These competing hypotheses could both be at least partially true, suggesting an “inverted-U” pattern of glutamate signaling (Figure 2). Because glutamate is involved in most cortical functions, too little glutamate can cause cognitive and processing deficits such as those seen in schizophrenia. On the other hand, too much glutamate can be toxic to neurons and may be a factor in neurodegeneration, such as in Alzheimer’s disease.20 Indeed, schizophrenia may be associated with gradual neurodegeneration.21
Figure 2 Inverted U-curve may explain dysfunctional glutamate signaling in schizophrenia
Both too little or too much glutamate may play a role in schizophrenia’s pathophysiology. Glutamate, the major excitatory neurotransmitter of the cerebral cortex, is involved in most cognitive functions. Too little (or glutamate inhibition) can impair cognition, whereas too much can lead to seizures, neurotoxicity, and cell death.
Glutamate stabilization?
Because lamotrigine prevents excessive glutamate release at synapses, it stabilizes neuronal membranes by preventing toxicity from too much glutamate without interfering with glutamate’s normal functions.22 Thus, lamotrigine may have potential to maintain optimal glutamate signaling in patients with schizophrenia.
In 16 healthy volunteers, a 300-mg dose of lamotrigine was significantly more effective than placebo in reducing ketamine-induced positive symptoms, as assessed by the Brief Psychiatric Rating Scale positive symptoms subscale (P < .001). Lamotrigine pretreatment also reduced negative symptoms and improved learning and memory.23
More recently, lamotrigine pretreatment was shown to prevent many ketamine-induced changes on functional MRI.24 Few antipsychotics have clinically significant effects on ketamine-induced symptoms—especially in a single dose—although repeated dosing with clozapine attenuates some ketamine-induced effects.25
Given the limitations of available antipsychotics, adding a drug such as lamotrigine—which may modulate and stabilize the glutamate system—could be effective in treatment-resistant schizophrenia.
What is the evidence?
Case reports and open-label case series first showed that lamotrigine augmentation could be effective in treatment-resistant schizophrenia patients receiving clozapine.26–28 One naturalistic case series also included patients receiving olanzapine or risperidone and suggested greater improvement with lamotrigine augmentation in patients on clozapine.26
Controlled trials. In a placebo-controlled trial, Tiihonen et al29 reported significantly lower ratings of positive symptoms—but not negative symptoms—after 38 treatment-resistant schizophrenia patients on clozapine received adjunctive lamotrigine, 200 mg/d, for 14 weeks (Table 1).
A subsequent controlled trial in which Kremer et al30 added lamotrigine, ≤400 mg/d, showed significant improvements in positive and negative symptoms among 31 treatment-resistant schizophrenia patients who completed the 10-week study. Patients were taking conventional and atypical antipsychotics, including clozapine. All groups improved, but the study was not powered to detect differences among the groups.
Table 1
Lamotrigine augmentation: 5 double-blind, placebo-controlled trials
| Trial duration | Patient diagnosis (number) | Antipsychotic(s) | Lamotrigine (mg/d) | Results |
|---|---|---|---|---|
| 14 weeks (Tiihonen et al, 200329) | Treatment-resistant schizophrenia (n=34) | Clozapine | 200 | Significantly reduced psychosis ratings, with no significant improvement in negative symptoms |
| 10 weeks (Kremer et al, 200430) | Treatment-resistant schizophrenia (n=38) | Conventional and atypical, including clozapine | ≤400 | Significant improvements with all antipsychotics, especially clozapine, in positive and negative symptoms* |
| 8 weeks (Akhondzadeh et al, 200531) | Schizophrenia (n=36) | Risperidone | 150 | Significant improvement in negative symptoms and cognition; less improvement in positive symptoms |
| 12 weeks, multicenter (Goff et al, 200732) | Schizophrenia, schizoaffective patients with residual symptoms (n=217+212) | Conventional and atypical, including clozapine | 100 to 400 | No significant improvement in any symptom domain; improved negative symptoms only in study 1 and cognitive symptoms only in study 2 |
| 24 weeks (Zoccali et al, 200733) | Treatment-resistant schizophrenia (n=51) | Clozapine | ≤200 | Significant improvement in positive and negative symptoms as well as some cognitive symptoms |
| * Significance achieved only in study completers, not in the last-observation-carried-forward analysis | ||||
A third trial by Akhondzadeh et al,31 augmenting risperidone with lamotrigine, 150 mg/d, resulted in modest improvements in negative and cognitive symptoms and slight improvement in positive symptoms.
Multicenter trials. Preliminary trials led to 2 randomized, double-blind, multicenter studies. In a total of 429 schizophrenia outpatients with residual psychotic symptoms on atypical antipsychotics, lamotrigine, 100 to 400 mg/d, or placebo was added for 12 weeks.32 The combined results failed to show significant improvement with adjunctive lamotrigine in any symptom domain compared with placebo. One study showed some improved negative symptoms, and the other showed improved cognitive symptoms.
Possible reasons for these negative results were unclear, although:
- a relatively large placebo response, compared with other studies, suggests a “failed” clinical trial
- the small number of patients receiving clozapine in this study suggests that they may have been less treatment-resistant than those enrolled in prior studies.
Meta-analysis. A meta-analysis of data from these 5 randomized, controlled trials found the “positive, negative, and general psychopathology subscale scores as measured with the PANSS … showed significant difference favoring adjuvant lamotrigine” (Table 2).6 As for study limitations, the authors noted that effectiveness data could be usefully analyzed in <70 of the 537 patients from the controlled trials, and “the small mean decrease in scores may not be really clinically relevant.”6 Thus, they said, caution is warranted in translating these results to clinical practice.
One more trial. Since the meta-analysis, an additional placebo-controlled trial has been reported.33 In this 24-week trial, lamotrigine augmentation, ≤200 mg/d, was statistically more effective than placebo in reducing positive and negative symptoms in 51 stable treatment-resistant patients on clozapine. Cognitive function also improved.
Table 2
How symptom scores changed with add-on lamotrigine in the meta-analysis of controlled trials
| PANSS subscales: Individual items scored 1 to 7, with 1=absent and 7=extreme | Change [95% CI]* |
|---|---|
| Positive symptom subscale (max 49) Delusions, conceptual disorganization, hallucinatory behavior, excitement, grandiosity, suspiciousness, hostility | -5.10 [-8.86, -1.34] |
| Negative symptom subscale (max 49) Blunted affect, emotional withdrawal, poor rapport, passive-apathetic social withdrawal, difficulty in abstract thinking, lack of spontaneity and flow of conversation, stereotyped thinking | -5.25 [-7.07, -3.43] |
| General psychopathology subscale (max 112) Somatic concern, anxiety, guilt feelings, tension, mannerisms and posturing, depression, motor retardation, uncooperativeness, unusual thought content, disorientation, poor attention, lack of judgment and insight, disturbance of volition, poor impulse control, preoccupation, active social avoidance | -10.74 [-16.53, -4.96] |
| * See text for limitations of the meta-analysis | |
| CI: confidence interval; PANSS: Positive and Negative Syndrome Scale | |
| Source: Reference 6 | |
Only treatment-resistant patients?
In controlled trials, lamotrigine augmentation has had the greatest effect on positive and negative symptoms in treatment-resistant schizophrenia patients, especially those on clozapine. Could lamotrigine augmentation be of benefit only in treatment-resistant schizophrenia?
Analysis of trial findings. As mentioned, outpatients who comprised the majority of subjects in the 2 large “negative” (or possibly failed) trials32 might have been less treatment-resistant than subjects in the other trials. Lower mean lamotrigine dosages (205 mg/d and 241 mg/d) also were used in the 2 negative trials and in the trial by Akhondzadeh et al (150 mg/d)31—compared with up to 400 mg/d in the trial by Kremer et al.30 This suggests that insufficient dosing might have caused the nonsignificant findings.
Given schizophrenia’s heterogeneity, treatment-resistant patients may represent a subgroup that has greater glutamatergic dysfunction, whereas patients who respond more completely to antipsychotics may have greater dopaminergic dysfunction. Thus, lamotrigine augmentation might be more beneficial in the subset of treatment-resistant patients. Lamotrigine or other glutamate stabilizers have been proposed to act as neuroprotective agents, slowing functional decline in chronic schizophrenia34 (although long-term studies needed to test this hypothesis are unlikely to occur because of cost and time constraints).
Another hypothetical, yet intriguing, explanation for the greater effects of lamotrigine augmentation in patients on clozapine is a pharmacodynamic interaction between these 2 drugs. Clozapine (and possibly olanzapine) have been shown to enhance cortical glutamatergic transmission.25 We propose that clozapine-induced boosting of glutamate in concert with stabilization of the glutamate system by lamotrigine improves neuronal functioning. Clinical trial data regarding lamotrigine augmentation of antipsychotics other than clozapine are needed to determine if the relationship between clozapine and lamotrigine is unique.
Related resources
- Lamotrigine prescribing information and patient handout. www.lamictal.com/bipolar/hcp/prescibing_information. html.
- Augmentation strategies for schizophrenia. IPAP Schizophrenia algorithm flowchart (online interactive version), node 11. www.ipap.org/algorithms.php.
Drug brand names
- Carbamazepine • Carbatrol, Equetro, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Valproate • Depacon, Depakene
Disclosures
Dr. Gray reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Risch receives research support from the National Institute of Mental Health and is a speaker for AstraZeneca and Pfizer Inc.
Current antipsychotics are reasonably effective in treating positive symptoms, but they do less to improve the negative and cognitive symptoms1 that contribute to patients’ long-term poor functional capacity and quality of life.2 So what do psychiatrists do in clinical practice to mitigate antipsychotics’ limitations? We augment.
Schizophrenia patients routinely are treated with polypharmacy—often with antidepressants or anticonvulsants—in attempts to improve negative symptoms, aggression, and impulsivity.3 Most adjuncts, however—including divalproex, antidepressants, and lithium—have shown very small, inconsistent, or no effects.4,5 The only agent with a recent meta-analysis supporting its use as augmentation in treatment-resistant schizophrenia is lamotrigine,6 an anticonvulsant approved for use in epilepsy.7
This article examines the evidence supporting off-label use of lamotrigine as an augmenting agent in schizophrenia and explains the rationale, based on lamotrigine’s probable mechanism of action as a stabilizer of glutamate neurotransmission.
Is lamotrigine worth trying?
Some 20% of schizophrenia patients are considered treatment-resistant, with persistent positive symptoms despite having undergone ≥2 adequate antipsychotic trials.8 Evidence suggests clozapine then should be tried,4 but approximately one-half of treatment-resistant patients do not respond to clozapine. Treatment guidelines are limited for these 10% of schizophrenia patients with an inadequate response to available therapies, including clozapine.4
In a meta-analysis of 5 controlled trials in patients with treatment-resistant schizophrenia, adjunctive lamotrigine was shown to significantly reduce Positive and Negative Syndrome Scale (PANSS) total scores, positive symptom subscores, and negative symptom subscores.6 In these trials, lamotrigine was added to various antipsychotics, including clozapine. Based on the results—as outlined below—we suggest:
- In treatment-resistant patients with residual symptoms while taking clozapine, lamotrigine given in dosages ≥200 mg/d could be a first-line adjunct (Figure 1).
- Lamotrigine augmentation also might help patients whose positive symptoms are adequately controlled but who have persistent negative and/or cognitive symptoms.
- Evidence does not support routine use of lamotrigine in patients taking antipsychotics other than clozapine.
Managing side effects. Lamotrigine is generally well tolerated; in the meta-analysis, nausea was the only side effect more common with lamotrigine (9%) than with placebo (3.9%).6 Close follow-up is required, however, as a few case reports have noted worsening positive symptoms when lamotrigine was added to antipsychotics.9,10
Lamotrigine produces a skin rash in approximately 10% of patients; the rash usually is benign but may be severe, including the potentially fatal Stevens-Johnson syndrome.11 In the meta-analysis, rash was no more likely in patients receiving placebo (3%) than those receiving lamotrigine (2.2%), and no serious rashes were reported.6 Even so, lamotrigine needs to be titrated upwards very slowly over weeks, and patients must be able to monitor for rash.
Figure 1 An evidence-based approach to treatment-resistant schizophrenia
Treatment-resistant schizophrenia is defined as residual positive symptoms after ≥2 adequate antipsychotic trials. Evidence supports trying clozapine as the next step.4 When patients show an inadequate response to clozapine, a meta-analysis of 5 controlled trials6 indicates that lamotrigine may be a useful first-line adjunct.
Why consider lamotrigine?
During clinical trials of lamotrigine for epilepsy, patients showed improved mood12 as is seen with other anticonvulsants such as valproate and carbamazepine.13 A series of randomized trials then demonstrated lamotrigine’s effectiveness in treating patients with bipolar I disorder, especially during depressive episodes,14,15 and the FDA approved lamotrigine for maintenance treatment of bipolar I disorder.16 In those early studies, lamotrigine also improved bipolar patients’ quality of life and cognitive function in addition to showing mood-stabilizing properties.12
The glutamate hypothesis. Lamotrigine is an inhibitor of voltage-gated sodium channels and has been shown to inhibit the excessive synaptic release of glutamate.17 Glutamate is the primary excitatory neurotransmitter for at least 60% of neurons in the brain, including all cortical pyramidal neurons. A large body of evidence implicates dysfunctional glutamate signaling in the pathophysiology of schizophrenia.18
For example, phencyclidine (PCP) and ketamine—antagonists of one subtype of glutamate receptor, the N-methyl-D-aspartate (NMDA) receptor—are well known to produce positive psychotic symptoms, negative symptoms, and cognitive dysfunction.19 This led to a long-held hypothesis that schizophrenia is caused by too little glutamate. However, ketamine and PCP also increase the release of glutamate at synapses that then can act on glutamate receptors other than the NMDA receptor, which suggests that too much glutamate also may be involved in schizophrenia.
Too little or too much glutamate? These competing hypotheses could both be at least partially true, suggesting an “inverted-U” pattern of glutamate signaling (Figure 2). Because glutamate is involved in most cortical functions, too little glutamate can cause cognitive and processing deficits such as those seen in schizophrenia. On the other hand, too much glutamate can be toxic to neurons and may be a factor in neurodegeneration, such as in Alzheimer’s disease.20 Indeed, schizophrenia may be associated with gradual neurodegeneration.21
Figure 2 Inverted U-curve may explain dysfunctional glutamate signaling in schizophrenia
Both too little or too much glutamate may play a role in schizophrenia’s pathophysiology. Glutamate, the major excitatory neurotransmitter of the cerebral cortex, is involved in most cognitive functions. Too little (or glutamate inhibition) can impair cognition, whereas too much can lead to seizures, neurotoxicity, and cell death.
Glutamate stabilization?
Because lamotrigine prevents excessive glutamate release at synapses, it stabilizes neuronal membranes by preventing toxicity from too much glutamate without interfering with glutamate’s normal functions.22 Thus, lamotrigine may have potential to maintain optimal glutamate signaling in patients with schizophrenia.
In 16 healthy volunteers, a 300-mg dose of lamotrigine was significantly more effective than placebo in reducing ketamine-induced positive symptoms, as assessed by the Brief Psychiatric Rating Scale positive symptoms subscale (P < .001). Lamotrigine pretreatment also reduced negative symptoms and improved learning and memory.23
More recently, lamotrigine pretreatment was shown to prevent many ketamine-induced changes on functional MRI.24 Few antipsychotics have clinically significant effects on ketamine-induced symptoms—especially in a single dose—although repeated dosing with clozapine attenuates some ketamine-induced effects.25
Given the limitations of available antipsychotics, adding a drug such as lamotrigine—which may modulate and stabilize the glutamate system—could be effective in treatment-resistant schizophrenia.
What is the evidence?
Case reports and open-label case series first showed that lamotrigine augmentation could be effective in treatment-resistant schizophrenia patients receiving clozapine.26–28 One naturalistic case series also included patients receiving olanzapine or risperidone and suggested greater improvement with lamotrigine augmentation in patients on clozapine.26
Controlled trials. In a placebo-controlled trial, Tiihonen et al29 reported significantly lower ratings of positive symptoms—but not negative symptoms—after 38 treatment-resistant schizophrenia patients on clozapine received adjunctive lamotrigine, 200 mg/d, for 14 weeks (Table 1).
A subsequent controlled trial in which Kremer et al30 added lamotrigine, ≤400 mg/d, showed significant improvements in positive and negative symptoms among 31 treatment-resistant schizophrenia patients who completed the 10-week study. Patients were taking conventional and atypical antipsychotics, including clozapine. All groups improved, but the study was not powered to detect differences among the groups.
Table 1
Lamotrigine augmentation: 5 double-blind, placebo-controlled trials
| Trial duration | Patient diagnosis (number) | Antipsychotic(s) | Lamotrigine (mg/d) | Results |
|---|---|---|---|---|
| 14 weeks (Tiihonen et al, 200329) | Treatment-resistant schizophrenia (n=34) | Clozapine | 200 | Significantly reduced psychosis ratings, with no significant improvement in negative symptoms |
| 10 weeks (Kremer et al, 200430) | Treatment-resistant schizophrenia (n=38) | Conventional and atypical, including clozapine | ≤400 | Significant improvements with all antipsychotics, especially clozapine, in positive and negative symptoms* |
| 8 weeks (Akhondzadeh et al, 200531) | Schizophrenia (n=36) | Risperidone | 150 | Significant improvement in negative symptoms and cognition; less improvement in positive symptoms |
| 12 weeks, multicenter (Goff et al, 200732) | Schizophrenia, schizoaffective patients with residual symptoms (n=217+212) | Conventional and atypical, including clozapine | 100 to 400 | No significant improvement in any symptom domain; improved negative symptoms only in study 1 and cognitive symptoms only in study 2 |
| 24 weeks (Zoccali et al, 200733) | Treatment-resistant schizophrenia (n=51) | Clozapine | ≤200 | Significant improvement in positive and negative symptoms as well as some cognitive symptoms |
| * Significance achieved only in study completers, not in the last-observation-carried-forward analysis | ||||
A third trial by Akhondzadeh et al,31 augmenting risperidone with lamotrigine, 150 mg/d, resulted in modest improvements in negative and cognitive symptoms and slight improvement in positive symptoms.
Multicenter trials. Preliminary trials led to 2 randomized, double-blind, multicenter studies. In a total of 429 schizophrenia outpatients with residual psychotic symptoms on atypical antipsychotics, lamotrigine, 100 to 400 mg/d, or placebo was added for 12 weeks.32 The combined results failed to show significant improvement with adjunctive lamotrigine in any symptom domain compared with placebo. One study showed some improved negative symptoms, and the other showed improved cognitive symptoms.
Possible reasons for these negative results were unclear, although:
- a relatively large placebo response, compared with other studies, suggests a “failed” clinical trial
- the small number of patients receiving clozapine in this study suggests that they may have been less treatment-resistant than those enrolled in prior studies.
Meta-analysis. A meta-analysis of data from these 5 randomized, controlled trials found the “positive, negative, and general psychopathology subscale scores as measured with the PANSS … showed significant difference favoring adjuvant lamotrigine” (Table 2).6 As for study limitations, the authors noted that effectiveness data could be usefully analyzed in <70 of the 537 patients from the controlled trials, and “the small mean decrease in scores may not be really clinically relevant.”6 Thus, they said, caution is warranted in translating these results to clinical practice.
One more trial. Since the meta-analysis, an additional placebo-controlled trial has been reported.33 In this 24-week trial, lamotrigine augmentation, ≤200 mg/d, was statistically more effective than placebo in reducing positive and negative symptoms in 51 stable treatment-resistant patients on clozapine. Cognitive function also improved.
Table 2
How symptom scores changed with add-on lamotrigine in the meta-analysis of controlled trials
| PANSS subscales: Individual items scored 1 to 7, with 1=absent and 7=extreme | Change [95% CI]* |
|---|---|
| Positive symptom subscale (max 49) Delusions, conceptual disorganization, hallucinatory behavior, excitement, grandiosity, suspiciousness, hostility | -5.10 [-8.86, -1.34] |
| Negative symptom subscale (max 49) Blunted affect, emotional withdrawal, poor rapport, passive-apathetic social withdrawal, difficulty in abstract thinking, lack of spontaneity and flow of conversation, stereotyped thinking | -5.25 [-7.07, -3.43] |
| General psychopathology subscale (max 112) Somatic concern, anxiety, guilt feelings, tension, mannerisms and posturing, depression, motor retardation, uncooperativeness, unusual thought content, disorientation, poor attention, lack of judgment and insight, disturbance of volition, poor impulse control, preoccupation, active social avoidance | -10.74 [-16.53, -4.96] |
| * See text for limitations of the meta-analysis | |
| CI: confidence interval; PANSS: Positive and Negative Syndrome Scale | |
| Source: Reference 6 | |
Only treatment-resistant patients?
In controlled trials, lamotrigine augmentation has had the greatest effect on positive and negative symptoms in treatment-resistant schizophrenia patients, especially those on clozapine. Could lamotrigine augmentation be of benefit only in treatment-resistant schizophrenia?
Analysis of trial findings. As mentioned, outpatients who comprised the majority of subjects in the 2 large “negative” (or possibly failed) trials32 might have been less treatment-resistant than subjects in the other trials. Lower mean lamotrigine dosages (205 mg/d and 241 mg/d) also were used in the 2 negative trials and in the trial by Akhondzadeh et al (150 mg/d)31—compared with up to 400 mg/d in the trial by Kremer et al.30 This suggests that insufficient dosing might have caused the nonsignificant findings.
Given schizophrenia’s heterogeneity, treatment-resistant patients may represent a subgroup that has greater glutamatergic dysfunction, whereas patients who respond more completely to antipsychotics may have greater dopaminergic dysfunction. Thus, lamotrigine augmentation might be more beneficial in the subset of treatment-resistant patients. Lamotrigine or other glutamate stabilizers have been proposed to act as neuroprotective agents, slowing functional decline in chronic schizophrenia34 (although long-term studies needed to test this hypothesis are unlikely to occur because of cost and time constraints).
Another hypothetical, yet intriguing, explanation for the greater effects of lamotrigine augmentation in patients on clozapine is a pharmacodynamic interaction between these 2 drugs. Clozapine (and possibly olanzapine) have been shown to enhance cortical glutamatergic transmission.25 We propose that clozapine-induced boosting of glutamate in concert with stabilization of the glutamate system by lamotrigine improves neuronal functioning. Clinical trial data regarding lamotrigine augmentation of antipsychotics other than clozapine are needed to determine if the relationship between clozapine and lamotrigine is unique.
Related resources
- Lamotrigine prescribing information and patient handout. www.lamictal.com/bipolar/hcp/prescibing_information. html.
- Augmentation strategies for schizophrenia. IPAP Schizophrenia algorithm flowchart (online interactive version), node 11. www.ipap.org/algorithms.php.
Drug brand names
- Carbamazepine • Carbatrol, Equetro, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Olanzapine • Zyprexa
- Risperidone • Risperdal
- Valproate • Depacon, Depakene
Disclosures
Dr. Gray reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Risch receives research support from the National Institute of Mental Health and is a speaker for AstraZeneca and Pfizer Inc.
1. Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12(10):904-922.
2. Agid Y, Buzsaki G, Diamond DM, et al. How can drug discovery for psychiatric disorders be improved? Nat Rev Drug Discov. 2007;6(3):189-201.
3. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: comparing monotherapy with polypharmacy and augmentation. Curr Med Chem. 2004;11(3):313-327.
4. Miller AL, McEvoy SP, Jeste DV, et al. Treatment of chronic schizophrenia. In: Lieberman JA, Stroup TS, Perkins DO, eds. Textbook of schizophrenia. Arlington, VA: American Psychiatric Publishing; 2006:365-381.
5. Miller AL. Combination treatments for schizophrenia. CNS Spectr. 2004;9(9 suppl 9):19-23.
6. Premkumar TS, Pick J. Lamotrigine for schizophrenia. Cochrane Database Syst Rev. 2006;(4):CD005962.-
7. Brodie MJ, Richens A, Yuen AW. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK lamotrigine/carbamazepine monotherapy trial group. Lancet. 1995;345(8948):476-479.
8. Buckley P, Miller A, Olsen J, et al. When symptoms persist: clozapine augmentation strategies. Schizophr Bull. 2001;27(4):615-628.
9. Chan YC, Miller KM, Shaheen N, et al. Worsening of psychotic symptoms in schizophrenia with addition of lamotrigine: a case report. Schizophr Res. 2005;78(2-3):343-345.
10. Konstantakopoulos G, Oulis P, Koulouris GC, et al. Lamotrigine-associated exacerbation of positive symptoms in paranoid schizophrenia. Schizophr Res. 2008;98(1-3):325-326.
11. Messenheimer J, Mullens EL, Giorgi L, et al. Safety review of adult clinical trial experience with lamotrigine. Drug Saf. 1998;18(4):281-296.
12. Smith D, Baker G, Davies G, et al. Outcomes of add-on treatment with lamotrigine in partial epilepsy. Epilepsia. 1993;34(2):312-322.
13. Post RM, Ketter TA, Denicoff K, et al. The place of anticonvulsant therapy in bipolar illness. Psychopharmacology (Berl). 1996;128(2):115-129.
14. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 study group. J Clin Psychiatry. 1999;60(2):79-88.
15. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. Lamictal 614 study group. J Clin Psychiatry. 2000;61(11):841-850.
16. Bowden CL, Calabrese JR, Sachs G, et al. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
17. Large CH, Webster EL, Goff DC. The potential role of lamotrigine in schizophrenia. Psychopharmacology (Berl). 2005;181(3):415-436.
18. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9):1367-1377.
19. Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry. 2004;9(11):984-997.
20. Chohan MO, Iqbal K. From tau to toxicity: emerging roles of NMDA receptor in Alzheimer’s disease. J Alzheimers Dis. 2006;10(1):81-87.
21. Konradi C, Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther. 2003;97(2):153-179.
22. Leach MJ, Baxter MG, Critchley MA. Neurochemical and behavioral aspects of lamotrigine. Epilepsia. 1991;32(suppl 2):S4-S8.
23. Anand A, Charney DS, Oren DA, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of n-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57(3):270-276.
24. Deakin JF, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65(2):154-164.
25. Large CH. Do NMDA receptor antagonist models of schizophrenia predict the clinical efficacy of antipsychotic drugs? J Psychopharmacol. 2007;21(3):283-301.
26. Dursun SM, Deakin JF. Augmenting antipsychotic treatment with lamotrigine or topiramate in patients with treatment-resistant schizophrenia: a naturalistic case-series outcome study. J Psychopharmacol. 2001;15(4):297-301.
27. Dursun SM, McIntosh D, Milliken H. Clozapine plus lamotrigine in treatment-resistant schizophrenia. Arch Gen Psychiatry. 1999;56(10):950.-
28. Saba G, Dumortier G, Kalalou K, et al. Lamotrigine-clozapine combination in refractory schizophrenia: three cases. J Neuropsychiatry Clin Neurosci. 2002;14(1):86.-
29. Tiihonen J, Hallikainen T, Ryynanen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry. 2003;54(11):1241-1248.
30. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):441-446.
31. Akhondzadeh S, Mackinejad K, Ahmadi-Abhari SA, et al. Does the addition of lamotrigine to risperidone improve psychotic symptoms and cognitive impairments in chronic schizophrenia? Therapy. 2005;2(3):399-406.
32. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol. 2007;27(6):582-589.
33. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res. 2007;93(1-3):109-116.
34. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(3 suppl 4):1-13.
1. Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12(10):904-922.
2. Agid Y, Buzsaki G, Diamond DM, et al. How can drug discovery for psychiatric disorders be improved? Nat Rev Drug Discov. 2007;6(3):189-201.
3. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: comparing monotherapy with polypharmacy and augmentation. Curr Med Chem. 2004;11(3):313-327.
4. Miller AL, McEvoy SP, Jeste DV, et al. Treatment of chronic schizophrenia. In: Lieberman JA, Stroup TS, Perkins DO, eds. Textbook of schizophrenia. Arlington, VA: American Psychiatric Publishing; 2006:365-381.
5. Miller AL. Combination treatments for schizophrenia. CNS Spectr. 2004;9(9 suppl 9):19-23.
6. Premkumar TS, Pick J. Lamotrigine for schizophrenia. Cochrane Database Syst Rev. 2006;(4):CD005962.-
7. Brodie MJ, Richens A, Yuen AW. Double-blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK lamotrigine/carbamazepine monotherapy trial group. Lancet. 1995;345(8948):476-479.
8. Buckley P, Miller A, Olsen J, et al. When symptoms persist: clozapine augmentation strategies. Schizophr Bull. 2001;27(4):615-628.
9. Chan YC, Miller KM, Shaheen N, et al. Worsening of psychotic symptoms in schizophrenia with addition of lamotrigine: a case report. Schizophr Res. 2005;78(2-3):343-345.
10. Konstantakopoulos G, Oulis P, Koulouris GC, et al. Lamotrigine-associated exacerbation of positive symptoms in paranoid schizophrenia. Schizophr Res. 2008;98(1-3):325-326.
11. Messenheimer J, Mullens EL, Giorgi L, et al. Safety review of adult clinical trial experience with lamotrigine. Drug Saf. 1998;18(4):281-296.
12. Smith D, Baker G, Davies G, et al. Outcomes of add-on treatment with lamotrigine in partial epilepsy. Epilepsia. 1993;34(2):312-322.
13. Post RM, Ketter TA, Denicoff K, et al. The place of anticonvulsant therapy in bipolar illness. Psychopharmacology (Berl). 1996;128(2):115-129.
14. Calabrese JR, Bowden CL, Sachs GS, et al. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 study group. J Clin Psychiatry. 1999;60(2):79-88.
15. Calabrese JR, Suppes T, Bowden CL, et al. A double-blind, placebo-controlled, prophylaxis study of lamotrigine in rapid-cycling bipolar disorder. Lamictal 614 study group. J Clin Psychiatry. 2000;61(11):841-850.
16. Bowden CL, Calabrese JR, Sachs G, et al. A placebo-controlled 18-month trial of lamotrigine and lithium maintenance treatment in recently manic or hypomanic patients with bipolar I disorder. Arch Gen Psychiatry. 2003;60(4):392-400.
17. Large CH, Webster EL, Goff DC. The potential role of lamotrigine in schizophrenia. Psychopharmacology (Berl). 2005;181(3):415-436.
18. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry. 2001;158(9):1367-1377.
19. Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry. 2004;9(11):984-997.
20. Chohan MO, Iqbal K. From tau to toxicity: emerging roles of NMDA receptor in Alzheimer’s disease. J Alzheimers Dis. 2006;10(1):81-87.
21. Konradi C, Heckers S. Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol Ther. 2003;97(2):153-179.
22. Leach MJ, Baxter MG, Critchley MA. Neurochemical and behavioral aspects of lamotrigine. Epilepsia. 1991;32(suppl 2):S4-S8.
23. Anand A, Charney DS, Oren DA, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of n-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57(3):270-276.
24. Deakin JF, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65(2):154-164.
25. Large CH. Do NMDA receptor antagonist models of schizophrenia predict the clinical efficacy of antipsychotic drugs? J Psychopharmacol. 2007;21(3):283-301.
26. Dursun SM, Deakin JF. Augmenting antipsychotic treatment with lamotrigine or topiramate in patients with treatment-resistant schizophrenia: a naturalistic case-series outcome study. J Psychopharmacol. 2001;15(4):297-301.
27. Dursun SM, McIntosh D, Milliken H. Clozapine plus lamotrigine in treatment-resistant schizophrenia. Arch Gen Psychiatry. 1999;56(10):950.-
28. Saba G, Dumortier G, Kalalou K, et al. Lamotrigine-clozapine combination in refractory schizophrenia: three cases. J Neuropsychiatry Clin Neurosci. 2002;14(1):86.-
29. Tiihonen J, Hallikainen T, Ryynanen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry. 2003;54(11):1241-1248.
30. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry. 2004;56(6):441-446.
31. Akhondzadeh S, Mackinejad K, Ahmadi-Abhari SA, et al. Does the addition of lamotrigine to risperidone improve psychotic symptoms and cognitive impairments in chronic schizophrenia? Therapy. 2005;2(3):399-406.
32. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol. 2007;27(6):582-589.
33. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res. 2007;93(1-3):109-116.
34. Lieberman JA, Perkins DO, Jarskog LF. Neuroprotection: a therapeutic strategy to prevent deterioration associated with schizophrenia. CNS Spectr. 2007;12(3 suppl 4):1-13.
Emerging clues: Is this teen at risk for substance abuse?
Traditionally, clinicians have identified children at high risk for substance abuse disorders (SUDs) by their family history—such as “children of alcoholics.” Advances in etiology research, however, have led to the identification of other risks for SUDs seen during childhood (Table). The clustering of these SUD risk factors—genetic influences, family characteristics, and predictive phenotypes—makes it feasible to identify children and adolescents who are very likely to develop problematic substance use.
Table
Risk factors for substance abuse in children and adolescents
| Genetic predisposition |
| Parental substance use |
| Maltreatment |
| Inadequate supervision |
| Impulsive behavior, inattention, irritability |
| Substance availability |
| Early substance use |
Nature vs nurture
Genetic influences. Heritable risk accounts for a substantial proportion of the variation in SUDs, as multiple genes differentially influence substance initiation, metabolism, and reinforcing properties.1 For example, well-characterized genetic variations determine individual differences in alcohol dehydrogenase and aldehyde dehydrogenase—the enzymes involved in alcohol metabolism—and influence liability to alcohol use disorders (AUDs).2,3 Researchers are exploring ways in which genes might impact SUD risk (Box 1).1,4,5
Genetic influences on substance use may be less important during adolescence than adulthood. In a study of 1,796 male twins’ alcohol, nicotine, and cannabis use from early adolescence to middle adulthood, genetic variations had little or no influence on substance use in early adolescence. The influence of genetic factors gradually increased with age.6
Familial environmental factors, by contrast, were important in early adolescence and gradually decreased in effect with increasing age. During adolescence, the family’s influence on substance use apparently operates more through environmental characteristics than through heritable factors.6
Familial influences. Parents with ongoing SUDs model problematic substance use and create environments of child maltreatment and inadequate supervision.
Child maltreatment. Children of parents with SUDs are more likely to suffer sexual abuse, physical abuse, or neglect.7 The effects of sexual abuse on the child may vary by abuse severity and the child’s gender, developmental stage, and relationship to the perpetrator. Maltreatment may cause the child difficulties in psychological regulation and social development, leading to related psychopathology; these characteristics may contribute to later SUDs.8
Inadequate supervision. Adolescents who report that their parents do not effectively monitor their activities have an increased likelihood of developing SUDs. However, children/adolescents who exhibit difficulties with psychological regulation—such as impulsive behavior and irritability—are difficult to parent, and adolescents with early substance involvement may subvert parental supervision efforts.9,10
Recent investigations have examined genes that might confer risk across substance types. Promising research has focused on:
- genes that influence functional variations in neurotransmitter systems
- gene-environment interactions
- the search for neurobiological endophenotypes—characteristics that cannot be observed by conventional means, such as brain development characteristics that are seen through neuroimaging.1,4,5
Specific molecular-level genetic variations can be measured in individual patients but cannot yet validly quantify risk.
Predictive phenotypes
Predictive phenotypes—measurable individual characteristics that predict SUDs—may be considered risk factors but should not be viewed as causal influences akin to genetic and familial/environmental factors. Rather, predictive phenotypes may reflect propensities that are manifested by specific behaviors and other features according to developmental stage and environmental facilitation.
- specific psychiatric disorders
- specific personality traits that collectively are called psychological dysregulation
- early substance use.11
Recent studies have demonstrated that this clustering of problems—including impulsive behavior, inattention, and negative affect—represents a single continuous dimension termed psychological dysregulation.12 The construct of psychological dysregulation has origins in neuropathology and provides a conceptual link between childhood psychopathological characteristics known to predict SUD and neurobiological deficits.5 Childhood indices of psychological dysregulation—such as the Behavior Rating Inventory of Executive Function (BRIEF)14—complement other risk factors, such as parental SUDs and early substance use, in predicting accelerated substance use and SUDs.15
Neurobiological characteristics. Recent investigations have focused on relationships between variations in normal brain development and differences in psychological regulation.5 Several brain structures thought relevant to the development of psychological regulation—including the prefrontal cortex, limbic structures, and reward circuits—develop during adolescence. Delays or deficits in the development of these structures are called neurodevelopmental dysmaturation.5
Variation in genes that influence these brain areas may interact with environmental influences—including child maltreatment and early substance use—to produce neurodevelopmental dysmaturation that manifests as psychological dysregulation. Thus, genetic and environmental causes are hypothesized to lead to an endophenotype (neurodevelopmental dysmaturation) and developmentally specific phenotypes, such as:
- ADHD in childhood
- CD and accelerated substance use initiation in early adolescence
- SUDs involving alcohol and cannabis in late adolescence.5
Consuming small quantities of alcohol under parental supervision is culturally normative and does not predict problematic drinking.17 On the other hand, regularly consuming “standard drink” quantities of alcohol in late childhood typically occurs in unsupervised settings and predicts adolescent-onset AUDs.18
Problem-focused interview methods—including CAGE, TWEAK, and CRAFFT—have been developed and tested to screen adolescents for AUDs. None has been as consistently successful as the World Health Organization’s Alcohol Use Disorders Identification Test (AUDIT) questionnaire18 (see Related Resources).
Childhood cigarette smoking also predicts accelerated substance use and SUDs.15 Marijuana use predicts both cannabis use disorders and other illicit drug use.1 This observation supports the controversial “gateway hypothesis,” which proposes that marijuana use accelerates the onset of other illicit drug use.15,19,20 An alternate hypothesis proposes that use of marijuana and other illicit drugs is a developmentally specific manifestation of a more general liability for SUDs.1
Identifying those at high risk
Screening for SUD risk factors makes it possible to identify children and adolescents who are very likely to develop problematic substance use. For example, in a study of 560 children age 10 to 12 at recruitment, this author (DBC) identified subjects as high risk if they had 2 parents with SUDs, tobacco or alcohol use by age 12, and high psychological dysregulation as measured by combined assessments of cognitive, emotional, and behavioral regulation. By age 18:
- three-quarters of these adolescents used tobacco daily
- more than one-half had alcohol problems
- nearly one-half had cannabis abuse or dependence.15
Recommendations. Children and adolescents receiving health care services—including primary care, ongoing treatment for chronic conditions, and treatment for psychiatric disorders—should be evaluated for SUD risk. Screening ideally occurs at the initial evaluation or early in the course of treatment. Family history determines genetic risk.
Direct questioning is needed because unstructured evaluations often fail to reveal the presence of important SUD risks.21 Explore possible child maltreatment by questioning the parent and child about physical abuse, sexual abuse, and neglect. Key mental disorders include CD, ADHD, and PTSD. Ask about use of tobacco, alcohol, cannabis, and other drugs. Follow acknowledgement of use with inquiries on frequency, quantity, and problems.
Prevention and early intervention
By identifying characteristics that confer risk for SUDs, you can target these characteristics in prevention and early treatment efforts. These efforts may involve parents as well as children. Many promising approaches have been developed, including universal or selective interventions based on family, school, community, or multi-component approaches.22
Because parental SUDs are a prominent risk factor for children, interventions to reduce or eliminate parental substance use may be helpful, particularly for diminishing childhood psychological dysregulation.23 Early treatment of childhood predictive phenotypes, including CD and ADHD, is another promising approach.12 Community efforts to limit adolescents’ access to addictive substances have met with some success.22
Parents, teachers, and children and adolescents can obtain a wealth of information from the Web sites of the National Institute on Alcohol Abuse and Alcoholism and the National Institute on Drug Abuse (Box 2). The Centers for Disease Control and Prevention offers infomation about preventing smoking (see Related Resources.)
Parents/teachers
The National Institute on Drug Abuse (NIDA) provides a Web site for parents and teachers at www.drugabuse.gov/parent-teacher.html. Parents also can find resources at NIDA for teens at http://teens.drugabuse.gov.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) provides several free booklets for parents at www.niaaa.nih.gov/Publications/PamphletsBrochuresPosters/English.
My Brain, My Body (www.mybrainmybody.com) is an NIAAA-supported educational tool for teaching middle school students. Developmentally specific NIDA Junior Scientist Programs have been developed for kindergarten through grade 5. The Office of Safe and Drug-Free Schools, part of the U.S. Department of Education, also provides relevant resources.
Children and adolescents
An NIAAA-sponsored educational resource (www.thecoolspot.gov) provides education on alcohol designed for young teens (age 11 to 13). The site includes quizzes, suggestions for resisting peer pressure, and activities that encourage refusing drinking opportunities.
NIDA for Teens (http://teens.drugabuse.gov) provides information and activities designed for those age 11 to 15.
Related resources
- Alcohol Use Disorders Identification Test (AUDIT). World Health Organization. http://whqlibdoc.who.int/hq/2001/WHO_MSD_MSB_01.6a.pdf.
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov.
- National Institute on Drug Abuse. www.nida.nih.gov.
- Centers for Disease Control and Prevention. Youth Tobacco Prevention. www.cdc.gov/tobacco/youth.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vanyukov MM, Tarter RE, Kirisci L, et al. Liability to substance use disorders: 1. Common mechanisms and manifestations. Neurosci Biobehav Rev. 2003;27(6):507-515.
2. Karpyak VM, Hall-Flavin DK, Mrazek DA. Can genetics predict risk for alcohol dependence? Current Psychiatry. 2008;7(3):57-73.
3. Kuo PH, Kalsi G, Prescott CA, et al. Association of ADH and ALDH genes with alcohol dependence in the Irish Affected Sib Pair Study of alcohol dependence (IASPSAD) sample. Alcohol Clin Exp Res. 2008;32(5):785-795.
4. Vanyukov MM, Maher BS, Devlin B, et al. The MAOA promoter polymorphism, disruptive behavior disorders, and early onset substance use disorder: gene-environment interaction. Psychiatr Genet. 2007;17(6):323-332.
5. Clark DB, Thatcher DL, Tapert S. Alcohol, psychological dysregulation and adolescent brain development. Alcohol Clin Exp Res. 2008;32(3):375-385.
6. Kendler KS, Schmitt E, Aggen SH, et al. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry. 2008;65(6):674-682.
7. Sher KJ, Gershuny BS, Peterson L, et al. The role of childhood stressors in the intergenerational transmission of alcohol use disorders. J Stud Alcohol. 1997;58:414-427.
8. Clark DB, De Bellis MD, Lynch KG, et al. Physical and sexual abuse, depression and alcohol use disorders in adolescents. Drug Alcohol Depend. 2003;69(1):51-60.
9. Clark DB, Thatcher DL, Maisto S. Adolescent neglect and alcohol use disorders in two-parent families. Child Maltreat. 2004;9(4):357-370.
10. Clark DB, Kirisci L, Mezzich A, et al. Parental supervision and alcohol use in early adolescence: developmentally specific interactions. J Dev Behav Pediatr. 2008;29(4):285-292.
11. Clark DB, Kirisci L, Moss HB. Early adolescent gateway drug use in sons of fathers with substance use disorders. Addict Behav. 1998;23:561-566.
12. Clark DB, Winters KC. Measuring risks and outcomes in substance use disorders prevention research. J Consult Clin Psychol. 2002;70(6):1207-1223.
13. Clark DB, Cornelius JR, Wood DS, et al. Psychopathology risk transmission in children of parents with substance use disorders. Am J Psychiatry. 2004;161(4):685-691.
14. Guy SC, Isquith PK, Gioia GA. Behavior rating inventory of executive function—self-report version professional manual. Lutz, FL: Psychological Assessment Resources; 2004.
15. Clark DB, Cornelius JR, Kirisci L, et al. Childhood risk categories for adolescent substance involvement: a general liability typology. Drug Alcohol Depend. 2005;77(1):13-21.
16. Clark DB. The natural history of adolescent alcohol use disorders. Addiction. 2004;99(suppl 2):5-22.
17. Donovan JE, Molina BS. Children’s introduction to alcohol use: sips and tastes. Alcohol Clin Exp Res. 2008;32(1):108-119.
18. Clark DB, Chung T, Martin CS. Alcohol use frequency as a screen for alcohol use disorders in adolescents. Int J Adolesc Med Health. 2006;18(1):181-187.
19. Tarter RE, Kirisci L, Mezzich A, et al. Neurobehavioral disinhibition in childhood predicts early age onset substance use disorder. Am J Psychiatry. 2003;160(6):1078-1085.
20. Tarter RE, Vanyukov M, Kirisci L, et al. Predictors of marijuana use in adolescents before and after licit drug use: examination of the gateway hypothesis. Am J Psychiatry. 2006;163(12):2134-2140.
21. Clark DB, Bukstein OG, Smith MG, et al. Identifying anxiety disorders in adolescents hospitalized for alcohol abuse or dependence. Psychiatr Serv. 1995;46:618-620.
22. Spoth R, Greenberg M, Turrisi R. Preventive interventions addressing underage drinking: state of the evidence and steps toward public health impact. Pediatrics. 2008;121(suppl 4):S311-336.
23. Kelley ML, Fals-Stewart W. Treating paternal drug abuse using Learning Sobriety Together: effects on adolescents versus children. Drug Alcohol Depend. 2008;92(1-3):228-238.
Traditionally, clinicians have identified children at high risk for substance abuse disorders (SUDs) by their family history—such as “children of alcoholics.” Advances in etiology research, however, have led to the identification of other risks for SUDs seen during childhood (Table). The clustering of these SUD risk factors—genetic influences, family characteristics, and predictive phenotypes—makes it feasible to identify children and adolescents who are very likely to develop problematic substance use.
Table
Risk factors for substance abuse in children and adolescents
| Genetic predisposition |
| Parental substance use |
| Maltreatment |
| Inadequate supervision |
| Impulsive behavior, inattention, irritability |
| Substance availability |
| Early substance use |
Nature vs nurture
Genetic influences. Heritable risk accounts for a substantial proportion of the variation in SUDs, as multiple genes differentially influence substance initiation, metabolism, and reinforcing properties.1 For example, well-characterized genetic variations determine individual differences in alcohol dehydrogenase and aldehyde dehydrogenase—the enzymes involved in alcohol metabolism—and influence liability to alcohol use disorders (AUDs).2,3 Researchers are exploring ways in which genes might impact SUD risk (Box 1).1,4,5
Genetic influences on substance use may be less important during adolescence than adulthood. In a study of 1,796 male twins’ alcohol, nicotine, and cannabis use from early adolescence to middle adulthood, genetic variations had little or no influence on substance use in early adolescence. The influence of genetic factors gradually increased with age.6
Familial environmental factors, by contrast, were important in early adolescence and gradually decreased in effect with increasing age. During adolescence, the family’s influence on substance use apparently operates more through environmental characteristics than through heritable factors.6
Familial influences. Parents with ongoing SUDs model problematic substance use and create environments of child maltreatment and inadequate supervision.
Child maltreatment. Children of parents with SUDs are more likely to suffer sexual abuse, physical abuse, or neglect.7 The effects of sexual abuse on the child may vary by abuse severity and the child’s gender, developmental stage, and relationship to the perpetrator. Maltreatment may cause the child difficulties in psychological regulation and social development, leading to related psychopathology; these characteristics may contribute to later SUDs.8
Inadequate supervision. Adolescents who report that their parents do not effectively monitor their activities have an increased likelihood of developing SUDs. However, children/adolescents who exhibit difficulties with psychological regulation—such as impulsive behavior and irritability—are difficult to parent, and adolescents with early substance involvement may subvert parental supervision efforts.9,10
Recent investigations have examined genes that might confer risk across substance types. Promising research has focused on:
- genes that influence functional variations in neurotransmitter systems
- gene-environment interactions
- the search for neurobiological endophenotypes—characteristics that cannot be observed by conventional means, such as brain development characteristics that are seen through neuroimaging.1,4,5
Specific molecular-level genetic variations can be measured in individual patients but cannot yet validly quantify risk.
Predictive phenotypes
Predictive phenotypes—measurable individual characteristics that predict SUDs—may be considered risk factors but should not be viewed as causal influences akin to genetic and familial/environmental factors. Rather, predictive phenotypes may reflect propensities that are manifested by specific behaviors and other features according to developmental stage and environmental facilitation.
- specific psychiatric disorders
- specific personality traits that collectively are called psychological dysregulation
- early substance use.11
Recent studies have demonstrated that this clustering of problems—including impulsive behavior, inattention, and negative affect—represents a single continuous dimension termed psychological dysregulation.12 The construct of psychological dysregulation has origins in neuropathology and provides a conceptual link between childhood psychopathological characteristics known to predict SUD and neurobiological deficits.5 Childhood indices of psychological dysregulation—such as the Behavior Rating Inventory of Executive Function (BRIEF)14—complement other risk factors, such as parental SUDs and early substance use, in predicting accelerated substance use and SUDs.15
Neurobiological characteristics. Recent investigations have focused on relationships between variations in normal brain development and differences in psychological regulation.5 Several brain structures thought relevant to the development of psychological regulation—including the prefrontal cortex, limbic structures, and reward circuits—develop during adolescence. Delays or deficits in the development of these structures are called neurodevelopmental dysmaturation.5
Variation in genes that influence these brain areas may interact with environmental influences—including child maltreatment and early substance use—to produce neurodevelopmental dysmaturation that manifests as psychological dysregulation. Thus, genetic and environmental causes are hypothesized to lead to an endophenotype (neurodevelopmental dysmaturation) and developmentally specific phenotypes, such as:
- ADHD in childhood
- CD and accelerated substance use initiation in early adolescence
- SUDs involving alcohol and cannabis in late adolescence.5
Consuming small quantities of alcohol under parental supervision is culturally normative and does not predict problematic drinking.17 On the other hand, regularly consuming “standard drink” quantities of alcohol in late childhood typically occurs in unsupervised settings and predicts adolescent-onset AUDs.18
Problem-focused interview methods—including CAGE, TWEAK, and CRAFFT—have been developed and tested to screen adolescents for AUDs. None has been as consistently successful as the World Health Organization’s Alcohol Use Disorders Identification Test (AUDIT) questionnaire18 (see Related Resources).
Childhood cigarette smoking also predicts accelerated substance use and SUDs.15 Marijuana use predicts both cannabis use disorders and other illicit drug use.1 This observation supports the controversial “gateway hypothesis,” which proposes that marijuana use accelerates the onset of other illicit drug use.15,19,20 An alternate hypothesis proposes that use of marijuana and other illicit drugs is a developmentally specific manifestation of a more general liability for SUDs.1
Identifying those at high risk
Screening for SUD risk factors makes it possible to identify children and adolescents who are very likely to develop problematic substance use. For example, in a study of 560 children age 10 to 12 at recruitment, this author (DBC) identified subjects as high risk if they had 2 parents with SUDs, tobacco or alcohol use by age 12, and high psychological dysregulation as measured by combined assessments of cognitive, emotional, and behavioral regulation. By age 18:
- three-quarters of these adolescents used tobacco daily
- more than one-half had alcohol problems
- nearly one-half had cannabis abuse or dependence.15
Recommendations. Children and adolescents receiving health care services—including primary care, ongoing treatment for chronic conditions, and treatment for psychiatric disorders—should be evaluated for SUD risk. Screening ideally occurs at the initial evaluation or early in the course of treatment. Family history determines genetic risk.
Direct questioning is needed because unstructured evaluations often fail to reveal the presence of important SUD risks.21 Explore possible child maltreatment by questioning the parent and child about physical abuse, sexual abuse, and neglect. Key mental disorders include CD, ADHD, and PTSD. Ask about use of tobacco, alcohol, cannabis, and other drugs. Follow acknowledgement of use with inquiries on frequency, quantity, and problems.
Prevention and early intervention
By identifying characteristics that confer risk for SUDs, you can target these characteristics in prevention and early treatment efforts. These efforts may involve parents as well as children. Many promising approaches have been developed, including universal or selective interventions based on family, school, community, or multi-component approaches.22
Because parental SUDs are a prominent risk factor for children, interventions to reduce or eliminate parental substance use may be helpful, particularly for diminishing childhood psychological dysregulation.23 Early treatment of childhood predictive phenotypes, including CD and ADHD, is another promising approach.12 Community efforts to limit adolescents’ access to addictive substances have met with some success.22
Parents, teachers, and children and adolescents can obtain a wealth of information from the Web sites of the National Institute on Alcohol Abuse and Alcoholism and the National Institute on Drug Abuse (Box 2). The Centers for Disease Control and Prevention offers infomation about preventing smoking (see Related Resources.)
Parents/teachers
The National Institute on Drug Abuse (NIDA) provides a Web site for parents and teachers at www.drugabuse.gov/parent-teacher.html. Parents also can find resources at NIDA for teens at http://teens.drugabuse.gov.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) provides several free booklets for parents at www.niaaa.nih.gov/Publications/PamphletsBrochuresPosters/English.
My Brain, My Body (www.mybrainmybody.com) is an NIAAA-supported educational tool for teaching middle school students. Developmentally specific NIDA Junior Scientist Programs have been developed for kindergarten through grade 5. The Office of Safe and Drug-Free Schools, part of the U.S. Department of Education, also provides relevant resources.
Children and adolescents
An NIAAA-sponsored educational resource (www.thecoolspot.gov) provides education on alcohol designed for young teens (age 11 to 13). The site includes quizzes, suggestions for resisting peer pressure, and activities that encourage refusing drinking opportunities.
NIDA for Teens (http://teens.drugabuse.gov) provides information and activities designed for those age 11 to 15.
Related resources
- Alcohol Use Disorders Identification Test (AUDIT). World Health Organization. http://whqlibdoc.who.int/hq/2001/WHO_MSD_MSB_01.6a.pdf.
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov.
- National Institute on Drug Abuse. www.nida.nih.gov.
- Centers for Disease Control and Prevention. Youth Tobacco Prevention. www.cdc.gov/tobacco/youth.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Traditionally, clinicians have identified children at high risk for substance abuse disorders (SUDs) by their family history—such as “children of alcoholics.” Advances in etiology research, however, have led to the identification of other risks for SUDs seen during childhood (Table). The clustering of these SUD risk factors—genetic influences, family characteristics, and predictive phenotypes—makes it feasible to identify children and adolescents who are very likely to develop problematic substance use.
Table
Risk factors for substance abuse in children and adolescents
| Genetic predisposition |
| Parental substance use |
| Maltreatment |
| Inadequate supervision |
| Impulsive behavior, inattention, irritability |
| Substance availability |
| Early substance use |
Nature vs nurture
Genetic influences. Heritable risk accounts for a substantial proportion of the variation in SUDs, as multiple genes differentially influence substance initiation, metabolism, and reinforcing properties.1 For example, well-characterized genetic variations determine individual differences in alcohol dehydrogenase and aldehyde dehydrogenase—the enzymes involved in alcohol metabolism—and influence liability to alcohol use disorders (AUDs).2,3 Researchers are exploring ways in which genes might impact SUD risk (Box 1).1,4,5
Genetic influences on substance use may be less important during adolescence than adulthood. In a study of 1,796 male twins’ alcohol, nicotine, and cannabis use from early adolescence to middle adulthood, genetic variations had little or no influence on substance use in early adolescence. The influence of genetic factors gradually increased with age.6
Familial environmental factors, by contrast, were important in early adolescence and gradually decreased in effect with increasing age. During adolescence, the family’s influence on substance use apparently operates more through environmental characteristics than through heritable factors.6
Familial influences. Parents with ongoing SUDs model problematic substance use and create environments of child maltreatment and inadequate supervision.
Child maltreatment. Children of parents with SUDs are more likely to suffer sexual abuse, physical abuse, or neglect.7 The effects of sexual abuse on the child may vary by abuse severity and the child’s gender, developmental stage, and relationship to the perpetrator. Maltreatment may cause the child difficulties in psychological regulation and social development, leading to related psychopathology; these characteristics may contribute to later SUDs.8
Inadequate supervision. Adolescents who report that their parents do not effectively monitor their activities have an increased likelihood of developing SUDs. However, children/adolescents who exhibit difficulties with psychological regulation—such as impulsive behavior and irritability—are difficult to parent, and adolescents with early substance involvement may subvert parental supervision efforts.9,10
Recent investigations have examined genes that might confer risk across substance types. Promising research has focused on:
- genes that influence functional variations in neurotransmitter systems
- gene-environment interactions
- the search for neurobiological endophenotypes—characteristics that cannot be observed by conventional means, such as brain development characteristics that are seen through neuroimaging.1,4,5
Specific molecular-level genetic variations can be measured in individual patients but cannot yet validly quantify risk.
Predictive phenotypes
Predictive phenotypes—measurable individual characteristics that predict SUDs—may be considered risk factors but should not be viewed as causal influences akin to genetic and familial/environmental factors. Rather, predictive phenotypes may reflect propensities that are manifested by specific behaviors and other features according to developmental stage and environmental facilitation.
- specific psychiatric disorders
- specific personality traits that collectively are called psychological dysregulation
- early substance use.11
Recent studies have demonstrated that this clustering of problems—including impulsive behavior, inattention, and negative affect—represents a single continuous dimension termed psychological dysregulation.12 The construct of psychological dysregulation has origins in neuropathology and provides a conceptual link between childhood psychopathological characteristics known to predict SUD and neurobiological deficits.5 Childhood indices of psychological dysregulation—such as the Behavior Rating Inventory of Executive Function (BRIEF)14—complement other risk factors, such as parental SUDs and early substance use, in predicting accelerated substance use and SUDs.15
Neurobiological characteristics. Recent investigations have focused on relationships between variations in normal brain development and differences in psychological regulation.5 Several brain structures thought relevant to the development of psychological regulation—including the prefrontal cortex, limbic structures, and reward circuits—develop during adolescence. Delays or deficits in the development of these structures are called neurodevelopmental dysmaturation.5
Variation in genes that influence these brain areas may interact with environmental influences—including child maltreatment and early substance use—to produce neurodevelopmental dysmaturation that manifests as psychological dysregulation. Thus, genetic and environmental causes are hypothesized to lead to an endophenotype (neurodevelopmental dysmaturation) and developmentally specific phenotypes, such as:
- ADHD in childhood
- CD and accelerated substance use initiation in early adolescence
- SUDs involving alcohol and cannabis in late adolescence.5
Consuming small quantities of alcohol under parental supervision is culturally normative and does not predict problematic drinking.17 On the other hand, regularly consuming “standard drink” quantities of alcohol in late childhood typically occurs in unsupervised settings and predicts adolescent-onset AUDs.18
Problem-focused interview methods—including CAGE, TWEAK, and CRAFFT—have been developed and tested to screen adolescents for AUDs. None has been as consistently successful as the World Health Organization’s Alcohol Use Disorders Identification Test (AUDIT) questionnaire18 (see Related Resources).
Childhood cigarette smoking also predicts accelerated substance use and SUDs.15 Marijuana use predicts both cannabis use disorders and other illicit drug use.1 This observation supports the controversial “gateway hypothesis,” which proposes that marijuana use accelerates the onset of other illicit drug use.15,19,20 An alternate hypothesis proposes that use of marijuana and other illicit drugs is a developmentally specific manifestation of a more general liability for SUDs.1
Identifying those at high risk
Screening for SUD risk factors makes it possible to identify children and adolescents who are very likely to develop problematic substance use. For example, in a study of 560 children age 10 to 12 at recruitment, this author (DBC) identified subjects as high risk if they had 2 parents with SUDs, tobacco or alcohol use by age 12, and high psychological dysregulation as measured by combined assessments of cognitive, emotional, and behavioral regulation. By age 18:
- three-quarters of these adolescents used tobacco daily
- more than one-half had alcohol problems
- nearly one-half had cannabis abuse or dependence.15
Recommendations. Children and adolescents receiving health care services—including primary care, ongoing treatment for chronic conditions, and treatment for psychiatric disorders—should be evaluated for SUD risk. Screening ideally occurs at the initial evaluation or early in the course of treatment. Family history determines genetic risk.
Direct questioning is needed because unstructured evaluations often fail to reveal the presence of important SUD risks.21 Explore possible child maltreatment by questioning the parent and child about physical abuse, sexual abuse, and neglect. Key mental disorders include CD, ADHD, and PTSD. Ask about use of tobacco, alcohol, cannabis, and other drugs. Follow acknowledgement of use with inquiries on frequency, quantity, and problems.
Prevention and early intervention
By identifying characteristics that confer risk for SUDs, you can target these characteristics in prevention and early treatment efforts. These efforts may involve parents as well as children. Many promising approaches have been developed, including universal or selective interventions based on family, school, community, or multi-component approaches.22
Because parental SUDs are a prominent risk factor for children, interventions to reduce or eliminate parental substance use may be helpful, particularly for diminishing childhood psychological dysregulation.23 Early treatment of childhood predictive phenotypes, including CD and ADHD, is another promising approach.12 Community efforts to limit adolescents’ access to addictive substances have met with some success.22
Parents, teachers, and children and adolescents can obtain a wealth of information from the Web sites of the National Institute on Alcohol Abuse and Alcoholism and the National Institute on Drug Abuse (Box 2). The Centers for Disease Control and Prevention offers infomation about preventing smoking (see Related Resources.)
Parents/teachers
The National Institute on Drug Abuse (NIDA) provides a Web site for parents and teachers at www.drugabuse.gov/parent-teacher.html. Parents also can find resources at NIDA for teens at http://teens.drugabuse.gov.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) provides several free booklets for parents at www.niaaa.nih.gov/Publications/PamphletsBrochuresPosters/English.
My Brain, My Body (www.mybrainmybody.com) is an NIAAA-supported educational tool for teaching middle school students. Developmentally specific NIDA Junior Scientist Programs have been developed for kindergarten through grade 5. The Office of Safe and Drug-Free Schools, part of the U.S. Department of Education, also provides relevant resources.
Children and adolescents
An NIAAA-sponsored educational resource (www.thecoolspot.gov) provides education on alcohol designed for young teens (age 11 to 13). The site includes quizzes, suggestions for resisting peer pressure, and activities that encourage refusing drinking opportunities.
NIDA for Teens (http://teens.drugabuse.gov) provides information and activities designed for those age 11 to 15.
Related resources
- Alcohol Use Disorders Identification Test (AUDIT). World Health Organization. http://whqlibdoc.who.int/hq/2001/WHO_MSD_MSB_01.6a.pdf.
- National Institute on Alcohol Abuse and Alcoholism. www.niaaa.nih.gov.
- National Institute on Drug Abuse. www.nida.nih.gov.
- Centers for Disease Control and Prevention. Youth Tobacco Prevention. www.cdc.gov/tobacco/youth.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Vanyukov MM, Tarter RE, Kirisci L, et al. Liability to substance use disorders: 1. Common mechanisms and manifestations. Neurosci Biobehav Rev. 2003;27(6):507-515.
2. Karpyak VM, Hall-Flavin DK, Mrazek DA. Can genetics predict risk for alcohol dependence? Current Psychiatry. 2008;7(3):57-73.
3. Kuo PH, Kalsi G, Prescott CA, et al. Association of ADH and ALDH genes with alcohol dependence in the Irish Affected Sib Pair Study of alcohol dependence (IASPSAD) sample. Alcohol Clin Exp Res. 2008;32(5):785-795.
4. Vanyukov MM, Maher BS, Devlin B, et al. The MAOA promoter polymorphism, disruptive behavior disorders, and early onset substance use disorder: gene-environment interaction. Psychiatr Genet. 2007;17(6):323-332.
5. Clark DB, Thatcher DL, Tapert S. Alcohol, psychological dysregulation and adolescent brain development. Alcohol Clin Exp Res. 2008;32(3):375-385.
6. Kendler KS, Schmitt E, Aggen SH, et al. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry. 2008;65(6):674-682.
7. Sher KJ, Gershuny BS, Peterson L, et al. The role of childhood stressors in the intergenerational transmission of alcohol use disorders. J Stud Alcohol. 1997;58:414-427.
8. Clark DB, De Bellis MD, Lynch KG, et al. Physical and sexual abuse, depression and alcohol use disorders in adolescents. Drug Alcohol Depend. 2003;69(1):51-60.
9. Clark DB, Thatcher DL, Maisto S. Adolescent neglect and alcohol use disorders in two-parent families. Child Maltreat. 2004;9(4):357-370.
10. Clark DB, Kirisci L, Mezzich A, et al. Parental supervision and alcohol use in early adolescence: developmentally specific interactions. J Dev Behav Pediatr. 2008;29(4):285-292.
11. Clark DB, Kirisci L, Moss HB. Early adolescent gateway drug use in sons of fathers with substance use disorders. Addict Behav. 1998;23:561-566.
12. Clark DB, Winters KC. Measuring risks and outcomes in substance use disorders prevention research. J Consult Clin Psychol. 2002;70(6):1207-1223.
13. Clark DB, Cornelius JR, Wood DS, et al. Psychopathology risk transmission in children of parents with substance use disorders. Am J Psychiatry. 2004;161(4):685-691.
14. Guy SC, Isquith PK, Gioia GA. Behavior rating inventory of executive function—self-report version professional manual. Lutz, FL: Psychological Assessment Resources; 2004.
15. Clark DB, Cornelius JR, Kirisci L, et al. Childhood risk categories for adolescent substance involvement: a general liability typology. Drug Alcohol Depend. 2005;77(1):13-21.
16. Clark DB. The natural history of adolescent alcohol use disorders. Addiction. 2004;99(suppl 2):5-22.
17. Donovan JE, Molina BS. Children’s introduction to alcohol use: sips and tastes. Alcohol Clin Exp Res. 2008;32(1):108-119.
18. Clark DB, Chung T, Martin CS. Alcohol use frequency as a screen for alcohol use disorders in adolescents. Int J Adolesc Med Health. 2006;18(1):181-187.
19. Tarter RE, Kirisci L, Mezzich A, et al. Neurobehavioral disinhibition in childhood predicts early age onset substance use disorder. Am J Psychiatry. 2003;160(6):1078-1085.
20. Tarter RE, Vanyukov M, Kirisci L, et al. Predictors of marijuana use in adolescents before and after licit drug use: examination of the gateway hypothesis. Am J Psychiatry. 2006;163(12):2134-2140.
21. Clark DB, Bukstein OG, Smith MG, et al. Identifying anxiety disorders in adolescents hospitalized for alcohol abuse or dependence. Psychiatr Serv. 1995;46:618-620.
22. Spoth R, Greenberg M, Turrisi R. Preventive interventions addressing underage drinking: state of the evidence and steps toward public health impact. Pediatrics. 2008;121(suppl 4):S311-336.
23. Kelley ML, Fals-Stewart W. Treating paternal drug abuse using Learning Sobriety Together: effects on adolescents versus children. Drug Alcohol Depend. 2008;92(1-3):228-238.
1. Vanyukov MM, Tarter RE, Kirisci L, et al. Liability to substance use disorders: 1. Common mechanisms and manifestations. Neurosci Biobehav Rev. 2003;27(6):507-515.
2. Karpyak VM, Hall-Flavin DK, Mrazek DA. Can genetics predict risk for alcohol dependence? Current Psychiatry. 2008;7(3):57-73.
3. Kuo PH, Kalsi G, Prescott CA, et al. Association of ADH and ALDH genes with alcohol dependence in the Irish Affected Sib Pair Study of alcohol dependence (IASPSAD) sample. Alcohol Clin Exp Res. 2008;32(5):785-795.
4. Vanyukov MM, Maher BS, Devlin B, et al. The MAOA promoter polymorphism, disruptive behavior disorders, and early onset substance use disorder: gene-environment interaction. Psychiatr Genet. 2007;17(6):323-332.
5. Clark DB, Thatcher DL, Tapert S. Alcohol, psychological dysregulation and adolescent brain development. Alcohol Clin Exp Res. 2008;32(3):375-385.
6. Kendler KS, Schmitt E, Aggen SH, et al. Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch Gen Psychiatry. 2008;65(6):674-682.
7. Sher KJ, Gershuny BS, Peterson L, et al. The role of childhood stressors in the intergenerational transmission of alcohol use disorders. J Stud Alcohol. 1997;58:414-427.
8. Clark DB, De Bellis MD, Lynch KG, et al. Physical and sexual abuse, depression and alcohol use disorders in adolescents. Drug Alcohol Depend. 2003;69(1):51-60.
9. Clark DB, Thatcher DL, Maisto S. Adolescent neglect and alcohol use disorders in two-parent families. Child Maltreat. 2004;9(4):357-370.
10. Clark DB, Kirisci L, Mezzich A, et al. Parental supervision and alcohol use in early adolescence: developmentally specific interactions. J Dev Behav Pediatr. 2008;29(4):285-292.
11. Clark DB, Kirisci L, Moss HB. Early adolescent gateway drug use in sons of fathers with substance use disorders. Addict Behav. 1998;23:561-566.
12. Clark DB, Winters KC. Measuring risks and outcomes in substance use disorders prevention research. J Consult Clin Psychol. 2002;70(6):1207-1223.
13. Clark DB, Cornelius JR, Wood DS, et al. Psychopathology risk transmission in children of parents with substance use disorders. Am J Psychiatry. 2004;161(4):685-691.
14. Guy SC, Isquith PK, Gioia GA. Behavior rating inventory of executive function—self-report version professional manual. Lutz, FL: Psychological Assessment Resources; 2004.
15. Clark DB, Cornelius JR, Kirisci L, et al. Childhood risk categories for adolescent substance involvement: a general liability typology. Drug Alcohol Depend. 2005;77(1):13-21.
16. Clark DB. The natural history of adolescent alcohol use disorders. Addiction. 2004;99(suppl 2):5-22.
17. Donovan JE, Molina BS. Children’s introduction to alcohol use: sips and tastes. Alcohol Clin Exp Res. 2008;32(1):108-119.
18. Clark DB, Chung T, Martin CS. Alcohol use frequency as a screen for alcohol use disorders in adolescents. Int J Adolesc Med Health. 2006;18(1):181-187.
19. Tarter RE, Kirisci L, Mezzich A, et al. Neurobehavioral disinhibition in childhood predicts early age onset substance use disorder. Am J Psychiatry. 2003;160(6):1078-1085.
20. Tarter RE, Vanyukov M, Kirisci L, et al. Predictors of marijuana use in adolescents before and after licit drug use: examination of the gateway hypothesis. Am J Psychiatry. 2006;163(12):2134-2140.
21. Clark DB, Bukstein OG, Smith MG, et al. Identifying anxiety disorders in adolescents hospitalized for alcohol abuse or dependence. Psychiatr Serv. 1995;46:618-620.
22. Spoth R, Greenberg M, Turrisi R. Preventive interventions addressing underage drinking: state of the evidence and steps toward public health impact. Pediatrics. 2008;121(suppl 4):S311-336.
23. Kelley ML, Fals-Stewart W. Treating paternal drug abuse using Learning Sobriety Together: effects on adolescents versus children. Drug Alcohol Depend. 2008;92(1-3):228-238.
Is dialectical behavior therapy the right ‘fit’ for your patient?
Strong evidence for the efficacy of dialectical behavior therapy (DBT) for patients with borderline personality disorder (BPD) has brought hope to clinicians and patients alike. By including cognitive therapy, behavioral strategies, skills training, and exposure therapy, DBT addresses more than just self-harm and suicidal behavior (Box 1).1-13 In fact, DBT’s primary interventions—such as skills training in emotion regulation and a straightforward approach to dysfunctional behaviors—could help many people.
Because DBT is so comprehensive and practical, clinicians might be tempted to refer almost anyone who seems even slightly “borderline” for DBT. But some patients—particularly those with mood and anxiety disorders—might benefit more from other treatments. To help you make appropriate evidence-based referrals for DBT and other psychological treatments, this article recommends 4 steps:
- Know what the treatment involves.
- Consider the evidence for the treatment in patients similar to yours.
- Consider why your patient—with unique characteristics and problems—would benefit from these specific interventions.
- Communicate to the patient your reasons for the referral.
Marsha Linehan, PhD, developed dialectical behavior therapy (DBT) in an attempt to devise an effective protocol for highly suicidal women. Over time, she realized that many of these women met criteria for borderline personality disorder (BPD), and DBT evolved to address their emotional, interpersonal, and mental health issues.1
Linehan et al2 published results from the first randomized clinical trial (RCT) of any psychological treatment for BPD. In this study, chronically parasuicidal women who met criteria for BPD received 1 year of DBT or “treatment as usual” in community settings. Those treated with DBT experienced fewer and less severe parasuicidal events, were more likely to remain in treatment, and required fewer days of inpatient care.
Findings from 9 additional RCTs have supported the efficacy of DBT for women with BPD and other populations.3 These RCTs have examined DBT (or adapted versions of DBT) for treating:
- women with BPD and substance dependence4,5
- men and women with BPD in a community setting6
- women veterans with BPD7
- non-BPD women with bulimia8 or binge-eating disorder9
- women with BPD in the Netherlands (53% of study subjects had a substance use disorder)10,11
- depressed older adults12
- suicidal women with BPD.13
Step 1. What does DBT involve?
Difficulty with emotion regulation. DBT is based on a biosocial theory of BPD.1 Within this framework, BPD is caused by the transaction (mutual interplay) of a biologically based vulnerability to emotions with an invalidating rearing environment. The patient with BPD typically experiences strong and long-lasting emotional reactions, often to seemingly small or insignificant events such as a slight look of disappointment on someone’s face or a minor daily hassle. Patients with BPD often are especially attuned to emotional reactions, particularly signs of rejection or disapproval.
Caregivers in an invalidating environment fail to provide the support a highly emotional child needs to learn to manage intense emotions. An invalidating environment:
- indiscriminately rejects the child’s communication of emotions and thoughts as invalid, independent of the validity of the child’s experience
- punishes emotional displays, then intermittently reinforces emotional escalation
- oversimplifies the ease of problem solving or coping.1
As a result, the fledgling BPD individual learns to mistrust and fear emotions and does not learn how to manage them. A patient with BPD is like a car with a powerful “emotional engine” but lacking brakes.
Team treatment. The standard DBT treatment package is an outpatient program run by a team.1 Therapists meet weekly for consultation to help them execute DBT according to the manual, prevent burn-out, and improve skills and motivation to treat patients with multiple, severe problems. The team also maintains the DBT program’s integrity and functioning and ensures that all treatment components—including individual therapy and skills training—are in place.
In individual therapy, the therapist and client collaborate to help the client reduce dysfunctional behaviors, increase motivation, and work toward goals. Because persons with BPD often present with many serious life problems, the therapist organizes session time to address 3 main priorities:
- Life-threatening behavior (intentional self-injury or imminent threat of intentional self-injury, including suicidal crises or threats, severe suicidal ideation or urges, suicide attempts, nonsuicidal self-injury or self-injury urges, or similar behaviors).
- Therapy-interfering behaviors (actions by the therapist or patient that interfere with progress, such as angry outbursts, missed sessions, or tardiness).
- Quality-of-life-interfering behavior (behaviors or problems—such as depression, eating disorders, or substance use disorders; homelessness or financial difficulties; or serious interpersonal discord—that make it hard for the patient to establish a reasonable quality of life).
Additional priorities include skills deficits and secondary targets.1 Each week, the client monitors his or her behaviors, emotions, and actions using a diary card. The therapist uses this information to collaboratively prioritize the focus of each individual therapy session.
Skills training typically occurs weekly in group sessions of 1.5 to 2.5 hours with 1 or 2 therapists. This structured, psycho-educational training focuses on skills that persons with BPD often lack:
- mindfulness (paying attention to the experience of the present moment)
- emotion regulation (regulating or managing distressing emotions)
- distress tolerance (averting crises, tolerating or accepting distressing situations or emotions)
- interpersonal effectiveness (maintaining relationships and asserting needs or wishes).
Therapists often use the first half of group sessions to review each patient’s homework and to provide feedback and coaching on effective skill use. The remaining time is spent teaching new skills. The therapist then assigns homework to practice new skills and closes with a wind-down exercise, often involving relaxation training.
Step 2. Consider the evidence
Before you make a referral for DBT (or any psychological treatment), know what the research says about who is likely to benefit from it. For women with BPD, DBT is the only treatment that can be considered “well-established.”3,14 The literature on DBT includes 10 randomized controlled trials (as well as many uncontrolled trials), and the strongest research supports its use in women with BPD.2,4-13
Based on a detailed review of the literature on DBT, I recommend a basic, evidence-based priority list for referrals (Table 1).3,12,13 Patient groups at the top are most likely to benefit from DBT—according to the most solid, rigorous research—and deserve your strongest consideration for referral. Patient groups further down the list—with fewer rigorous studies of DBT—merit less consideration of DBT as the treatment of choice. Of course to use this list, an accurate diagnosis of your patient’s problems is essential.
DBT’s treatment strategies—exposure therapy, skills training, cognitive therapy, emotion regulation training, and mindfulness—can work for other types of patients. I have noticed, however, that some clinicians refer patients with depression, anxiety disorders, or even bipolar disorder for DBT. Despite DBT’s intuitive appeal, sufficient evidence does not yet support its use in patients with these disorders. Other evidence-based treatments may be more suitable for patients with uncomplicated mood and anxiety disorders (Table 2).3
Table 1
Candidates for DBT: An evidence-based referral priority list*
 | Women with BPD who are suicidal or who self-harm (without bipolar disorder, a psychotic disorder, or mental retardation). One randomized clinical trial with suicidal individuals with BPD included men. Two studies excluded participants with substance dependence, but the most recent, largest study13 did not. |
| Women with BPD and substance use problems (without bipolar disorder, a psychotic disorder, or cognitive impairment) | |
| Women with bulimia nervosa or binge-eating disorder (without substance abuse, psychotic disorder, or suicidal ideation). Other empirically supported treatments exist for these patients (Table 2). | |
| Depressed older adults (age ≥60, without bipolar disorder, a psychotic disorder, or cognitive impairment). Investigated treatments included group DBT skills training, telephone consultation, selective serotonin reuptake inhibitor medications, and psychiatric clinical management.12 | |
| Suicidal and nonsuicidal adolescentswith oppositional defiant disorder or bipolar disorder | |
| Incarcerated men and womenwith or without BPD, in high- and low-security forensic settings | |
| Couples and families where 1 member has BPDor where domestic violence occurs in an intimate relationship | |
| * Persons at the top of the list are the ones for whom the most solid, rigorous research has demonstrated the efficacy of DBT. Fewer rigorous studies of DBT have been conducted in persons further down the list. | |
| BPD: borderline personality disorder; DBT: dialectical behavior therapy | |
| Source: References 3,12,13 | |
Table 2
When not to refer a patient for DBT: Evidence is stronger for alternate treatments
| Diagnosis | Treatments with empirical support |
|---|---|
| Major depressive disorder | CBT, behavioral activation, interpersonal therapy, antidepressant medication, mindfulness-based cognitive therapy for depressive relapse |
| Panic disorder/panic disorder with agoraphobia | CBT involving cognitive therapy and exposure-based with agoraphobia interventions |
| Posttraumatic stress disorder | Prolonged exposure therapy, cognitive therapy, EMDR |
| Bulimia nervosa | CBT, interpersonal therapy |
| Primary substance use disorders | CBT, motivational enhancement/motivational interviewing, community reinforcement approach |
| Psychotic disorders | Medication management, social skills training, family-based interventions |
| CBT: cognitive-behavioral therapy; DBT: dialectical behavior therapy; EMDR: eye movement desensitization and reprocessing therapy | |
| Source: Reference 3 | |
Step 3: Would this patient benefit?
Would your patient, with unique struggles and characteristics, benefit from DBT? Consider to what degree DBT’s interventions would solve some of your patient’s problems and whether DBT fits your patient’s needs.
DBT’s target problems. In controlled trials, DBT’s pragmatic approach outperforms comparator treatments in reducing suicidal behaviors and self-injury, and DBT therapists monitor and target these behaviors. Thus, because few treatments reduce self-injury,15,16 you might consider DBT for patients who self-injure even if they do not have BPD.
DBT also includes a strong focus on emotions and emotion regulation. Therefore, if difficulty managing emotions is among your patient’s primary problems, DBT may offer some benefit. DBT also includes structured interpersonal skills training that might be useful for patients who lack assertiveness.
Finally, if you have a patient with multiple diagnoses and severe problems—but not psychosis—the DBT approach to organizing and prioritizing treatment targets may be beneficial. Some multi-diagnosis patients may struggle with aspects of DBT (such as learning new skills), but DBT is set up to incorporate other empirically supported treatment protocols for co-occurring Axis I and II disorders.
Does DBT ‘fit’ your patient? DBT is very structured and involves direct discussions of maladaptive behaviors. If your patient prefers or would benefit from a structured approach, you might consider a referral for DBT.
DBT is an outpatient behavioral treatment that focuses on the here and now. DBT might not be the best fit if your patient:
- views his or her problems as resulting primarily from childhood experiences or relationships with parents
- would prefer insight-oriented therapy.
If, however, your patient would like a practical approach focused on problem-solving, DBT could be an effective choice.
DBT is based in part on a dialectical philosophy, and DBT therapists often seek to bring together or synthesize polarized thinking. If your patient struggles with “black or white” thinking, this dialectical philosophy might be helpful. On the other hand, DBT might not be the best fit if your patient is particularly rigid in thinking or seems to require cognitive therapy to address his or her thought patterns.
DBT is not the treatment of choice for all personality disorders. Most of the evidence examines its use for BPD, and few studies have looked at any other personality disorder. Also, keep in mind that being interpersonally “difficult” does not mean that a patient is “borderline” or needs DBT.
Step 4: Communicate reasons for referral to your patient
Finally, communicate to your patient the reasons you are referring him or her for DBT. Patients often walk into my office for DBT, confused about why they are there. If patients understand why they have been referred for DBT and how it may help them, they may be more likely to follow through and realize its benefits.
A sample explanation of referral that I offer to guide this communication (Box 2) includes 3 main points:
- my diagnosis or conceptualization of the patient’s clinical problems
- a brief description of DBT
- a rationale for why DBT would be a good fit, and what kinds of benefits the patient might receive.
Based on my initial assessment, you seem to meet criteria for a diagnosis of borderline personality disorder, or BPD. A diagnosis is a category for different symptoms or experiences. To receive a BPD diagnosis, a person has to have at least 5 of 9 symptoms, and you seem to have about 6 of them. From what you have said, the main problems you struggle with are roller-coaster emotions and moods, problems with relationships with other people, and self-harm.
A lot of people recover from BPD, and there’s no reason to think you will have these problems for the rest of your life. In fact, there is a very effective treatment for BPD. This treatment is called dialectical behavior therapy, or DBT. I think you’re a great candidate for DBT. Of course, there’s no guarantee that DBT is the ideal treatment for you, but several studies have shown that DBT helps people learn how to manage their emotions, reduce self-harm, and improve their functioning in life.
DBT includes a couple of different things: meeting once a week with a therapist on an individual basis, then meeting once a week with a group. In the group, you will learn how to manage your emotions, pay attention to the present moment, deal with other people, and tolerate being upset without getting into a crisis.
I know some people in town who provide DBT. Is this something you think you might be interested in? If so, what questions do you have?
Related Resources
- Chapman AL, Gratz KL. The borderline personality disorder survival guide: everything you need to know about living with BPD. Oakland, CA: New Harbinger Publications; 2007.
- National Education Alliance for Borderline Personality Disorder. Information for professionals, patients, and families. www.neabpd.org.
- Behavioral Tech, LLC, founded by Marsha Linehan, PhD. DBT training and resources, including a directory of DBT therapists. www.behavioraltech.org.
- Dialectical Behaviour Therapy Centre of Vancouver. www.dbtvancouver.com.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Linehan MM. Cognitive behavior treatment of borderline personality disorder. New York, NY: The Guilford Press; 1993.
2. Linehan MM, Armstrong HE, Suarez A, et al. Cognitive-behavioral treatment of chronically parasuicidal borderline patients. Arch Gen Psychiatry 1991;48:1060-4.
3. Robins CJ, Chapman AL. Dialectical behavior therapy: current status, recent developments, and future directions. J Personal Disord 2004;18:73-9.
4. Linehan MM, Schmidt HI, Dimeff LA, et al. Dialectical behavior therapy for patients with borderline personality disorder and drug-dependence. Am J Addictions 1999;8:279-92.
5. Turner RM. Naturalistic evaluation of dialectical behavior therapy-oriented treatment for borderline personality disorder. Cognit Behav Pract 2000;7:413-9.
6. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectical behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend 2002;67:13-26.
7. Koons C, Robins CJ, Tweed JL, et al. Efficacy of dialectical behavior therapy in women veterans with borderline personality disorder. Behav Ther 2001;32:371-90.
8. Safer DL, Telch CF, Agras WS. Dialectical behavior therapy for bulimia nervosa. Am J Psychiatry 2001;158:632-4.
9. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061-5.
10. van den Bosch LMC, Verheul R, Schippers GM, van den Brink W. Dialectical behavior therapy of borderline patients with and without substance abuse problems: implementation and long-term effects. Addict Behav 2002;27:911-23.
11. Verheul R, van den Bosch LMC, Koeter MWJ, et al. Dialectical behavior therapy for women with borderline personality disorder. Br J Psychiatry 2003;182:135-40.
12. Lynch TR, Morse JQ, Mendelson T, Robins CJ. Dialectical behavior therapy for depressed older adults: a randomized pilot study. Am J Geriatr Psychiatry 2003;11:33-45.
13. Linehan MM, Comtois KA, Murray AM, et al. Two-year randomized controlled trial and follow-up of dialectical behavior therapy vs. therapy by experts for suicidal behaviors and borderline personality disorder. Arch Gen Psychiatry 2006;63:757-66.
14. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol 2001;52:685-716.
15. Hawton K, Arensman E, Townsend E, et al. Deliberate self-harm: systematic review of efficacy of psychosocial and pharmacological treatments in preventing repetition. BMJ 1998;317:441-7.
16. Tyrer P, Tom B, Byford S, et al. Differential effects of manual-assisted cognitive behavior therapy in the treatment of recurrent deliberate self-harm and personality disturbance: the POPMACT study. J Personal Disord 2004;18:102-16.
Strong evidence for the efficacy of dialectical behavior therapy (DBT) for patients with borderline personality disorder (BPD) has brought hope to clinicians and patients alike. By including cognitive therapy, behavioral strategies, skills training, and exposure therapy, DBT addresses more than just self-harm and suicidal behavior (Box 1).1-13 In fact, DBT’s primary interventions—such as skills training in emotion regulation and a straightforward approach to dysfunctional behaviors—could help many people.
Because DBT is so comprehensive and practical, clinicians might be tempted to refer almost anyone who seems even slightly “borderline” for DBT. But some patients—particularly those with mood and anxiety disorders—might benefit more from other treatments. To help you make appropriate evidence-based referrals for DBT and other psychological treatments, this article recommends 4 steps:
- Know what the treatment involves.
- Consider the evidence for the treatment in patients similar to yours.
- Consider why your patient—with unique characteristics and problems—would benefit from these specific interventions.
- Communicate to the patient your reasons for the referral.
Marsha Linehan, PhD, developed dialectical behavior therapy (DBT) in an attempt to devise an effective protocol for highly suicidal women. Over time, she realized that many of these women met criteria for borderline personality disorder (BPD), and DBT evolved to address their emotional, interpersonal, and mental health issues.1
Linehan et al2 published results from the first randomized clinical trial (RCT) of any psychological treatment for BPD. In this study, chronically parasuicidal women who met criteria for BPD received 1 year of DBT or “treatment as usual” in community settings. Those treated with DBT experienced fewer and less severe parasuicidal events, were more likely to remain in treatment, and required fewer days of inpatient care.
Findings from 9 additional RCTs have supported the efficacy of DBT for women with BPD and other populations.3 These RCTs have examined DBT (or adapted versions of DBT) for treating:
- women with BPD and substance dependence4,5
- men and women with BPD in a community setting6
- women veterans with BPD7
- non-BPD women with bulimia8 or binge-eating disorder9
- women with BPD in the Netherlands (53% of study subjects had a substance use disorder)10,11
- depressed older adults12
- suicidal women with BPD.13
Step 1. What does DBT involve?
Difficulty with emotion regulation. DBT is based on a biosocial theory of BPD.1 Within this framework, BPD is caused by the transaction (mutual interplay) of a biologically based vulnerability to emotions with an invalidating rearing environment. The patient with BPD typically experiences strong and long-lasting emotional reactions, often to seemingly small or insignificant events such as a slight look of disappointment on someone’s face or a minor daily hassle. Patients with BPD often are especially attuned to emotional reactions, particularly signs of rejection or disapproval.
Caregivers in an invalidating environment fail to provide the support a highly emotional child needs to learn to manage intense emotions. An invalidating environment:
- indiscriminately rejects the child’s communication of emotions and thoughts as invalid, independent of the validity of the child’s experience
- punishes emotional displays, then intermittently reinforces emotional escalation
- oversimplifies the ease of problem solving or coping.1
As a result, the fledgling BPD individual learns to mistrust and fear emotions and does not learn how to manage them. A patient with BPD is like a car with a powerful “emotional engine” but lacking brakes.
Team treatment. The standard DBT treatment package is an outpatient program run by a team.1 Therapists meet weekly for consultation to help them execute DBT according to the manual, prevent burn-out, and improve skills and motivation to treat patients with multiple, severe problems. The team also maintains the DBT program’s integrity and functioning and ensures that all treatment components—including individual therapy and skills training—are in place.
In individual therapy, the therapist and client collaborate to help the client reduce dysfunctional behaviors, increase motivation, and work toward goals. Because persons with BPD often present with many serious life problems, the therapist organizes session time to address 3 main priorities:
- Life-threatening behavior (intentional self-injury or imminent threat of intentional self-injury, including suicidal crises or threats, severe suicidal ideation or urges, suicide attempts, nonsuicidal self-injury or self-injury urges, or similar behaviors).
- Therapy-interfering behaviors (actions by the therapist or patient that interfere with progress, such as angry outbursts, missed sessions, or tardiness).
- Quality-of-life-interfering behavior (behaviors or problems—such as depression, eating disorders, or substance use disorders; homelessness or financial difficulties; or serious interpersonal discord—that make it hard for the patient to establish a reasonable quality of life).
Additional priorities include skills deficits and secondary targets.1 Each week, the client monitors his or her behaviors, emotions, and actions using a diary card. The therapist uses this information to collaboratively prioritize the focus of each individual therapy session.
Skills training typically occurs weekly in group sessions of 1.5 to 2.5 hours with 1 or 2 therapists. This structured, psycho-educational training focuses on skills that persons with BPD often lack:
- mindfulness (paying attention to the experience of the present moment)
- emotion regulation (regulating or managing distressing emotions)
- distress tolerance (averting crises, tolerating or accepting distressing situations or emotions)
- interpersonal effectiveness (maintaining relationships and asserting needs or wishes).
Therapists often use the first half of group sessions to review each patient’s homework and to provide feedback and coaching on effective skill use. The remaining time is spent teaching new skills. The therapist then assigns homework to practice new skills and closes with a wind-down exercise, often involving relaxation training.
Step 2. Consider the evidence
Before you make a referral for DBT (or any psychological treatment), know what the research says about who is likely to benefit from it. For women with BPD, DBT is the only treatment that can be considered “well-established.”3,14 The literature on DBT includes 10 randomized controlled trials (as well as many uncontrolled trials), and the strongest research supports its use in women with BPD.2,4-13
Based on a detailed review of the literature on DBT, I recommend a basic, evidence-based priority list for referrals (Table 1).3,12,13 Patient groups at the top are most likely to benefit from DBT—according to the most solid, rigorous research—and deserve your strongest consideration for referral. Patient groups further down the list—with fewer rigorous studies of DBT—merit less consideration of DBT as the treatment of choice. Of course to use this list, an accurate diagnosis of your patient’s problems is essential.
DBT’s treatment strategies—exposure therapy, skills training, cognitive therapy, emotion regulation training, and mindfulness—can work for other types of patients. I have noticed, however, that some clinicians refer patients with depression, anxiety disorders, or even bipolar disorder for DBT. Despite DBT’s intuitive appeal, sufficient evidence does not yet support its use in patients with these disorders. Other evidence-based treatments may be more suitable for patients with uncomplicated mood and anxiety disorders (Table 2).3
Table 1
Candidates for DBT: An evidence-based referral priority list*
| Women with BPD who are suicidal or who self-harm (without bipolar disorder, a psychotic disorder, or mental retardation). One randomized clinical trial with suicidal individuals with BPD included men. Two studies excluded participants with substance dependence, but the most recent, largest study13 did not. |
| Women with BPD and substance use problems (without bipolar disorder, a psychotic disorder, or cognitive impairment) | |
| Women with bulimia nervosa or binge-eating disorder (without substance abuse, psychotic disorder, or suicidal ideation). Other empirically supported treatments exist for these patients (Table 2). | |
| Depressed older adults (age ≥60, without bipolar disorder, a psychotic disorder, or cognitive impairment). Investigated treatments included group DBT skills training, telephone consultation, selective serotonin reuptake inhibitor medications, and psychiatric clinical management.12 | |
| Suicidal and nonsuicidal adolescentswith oppositional defiant disorder or bipolar disorder | |
| Incarcerated men and womenwith or without BPD, in high- and low-security forensic settings | |
| Couples and families where 1 member has BPDor where domestic violence occurs in an intimate relationship | |
| * Persons at the top of the list are the ones for whom the most solid, rigorous research has demonstrated the efficacy of DBT. Fewer rigorous studies of DBT have been conducted in persons further down the list. | |
| BPD: borderline personality disorder; DBT: dialectical behavior therapy | |
| Source: References 3,12,13 | |
Table 2
When not to refer a patient for DBT: Evidence is stronger for alternate treatments
| Diagnosis | Treatments with empirical support |
|---|---|
| Major depressive disorder | CBT, behavioral activation, interpersonal therapy, antidepressant medication, mindfulness-based cognitive therapy for depressive relapse |
| Panic disorder/panic disorder with agoraphobia | CBT involving cognitive therapy and exposure-based with agoraphobia interventions |
| Posttraumatic stress disorder | Prolonged exposure therapy, cognitive therapy, EMDR |
| Bulimia nervosa | CBT, interpersonal therapy |
| Primary substance use disorders | CBT, motivational enhancement/motivational interviewing, community reinforcement approach |
| Psychotic disorders | Medication management, social skills training, family-based interventions |
| CBT: cognitive-behavioral therapy; DBT: dialectical behavior therapy; EMDR: eye movement desensitization and reprocessing therapy | |
| Source: Reference 3 | |
Step 3: Would this patient benefit?
Would your patient, with unique struggles and characteristics, benefit from DBT? Consider to what degree DBT’s interventions would solve some of your patient’s problems and whether DBT fits your patient’s needs.
DBT’s target problems. In controlled trials, DBT’s pragmatic approach outperforms comparator treatments in reducing suicidal behaviors and self-injury, and DBT therapists monitor and target these behaviors. Thus, because few treatments reduce self-injury,15,16 you might consider DBT for patients who self-injure even if they do not have BPD.
DBT also includes a strong focus on emotions and emotion regulation. Therefore, if difficulty managing emotions is among your patient’s primary problems, DBT may offer some benefit. DBT also includes structured interpersonal skills training that might be useful for patients who lack assertiveness.
Finally, if you have a patient with multiple diagnoses and severe problems—but not psychosis—the DBT approach to organizing and prioritizing treatment targets may be beneficial. Some multi-diagnosis patients may struggle with aspects of DBT (such as learning new skills), but DBT is set up to incorporate other empirically supported treatment protocols for co-occurring Axis I and II disorders.
Does DBT ‘fit’ your patient? DBT is very structured and involves direct discussions of maladaptive behaviors. If your patient prefers or would benefit from a structured approach, you might consider a referral for DBT.
DBT is an outpatient behavioral treatment that focuses on the here and now. DBT might not be the best fit if your patient:
- views his or her problems as resulting primarily from childhood experiences or relationships with parents
- would prefer insight-oriented therapy.
If, however, your patient would like a practical approach focused on problem-solving, DBT could be an effective choice.
DBT is based in part on a dialectical philosophy, and DBT therapists often seek to bring together or synthesize polarized thinking. If your patient struggles with “black or white” thinking, this dialectical philosophy might be helpful. On the other hand, DBT might not be the best fit if your patient is particularly rigid in thinking or seems to require cognitive therapy to address his or her thought patterns.
DBT is not the treatment of choice for all personality disorders. Most of the evidence examines its use for BPD, and few studies have looked at any other personality disorder. Also, keep in mind that being interpersonally “difficult” does not mean that a patient is “borderline” or needs DBT.
Step 4: Communicate reasons for referral to your patient
Finally, communicate to your patient the reasons you are referring him or her for DBT. Patients often walk into my office for DBT, confused about why they are there. If patients understand why they have been referred for DBT and how it may help them, they may be more likely to follow through and realize its benefits.
A sample explanation of referral that I offer to guide this communication (Box 2) includes 3 main points:
- my diagnosis or conceptualization of the patient’s clinical problems
- a brief description of DBT
- a rationale for why DBT would be a good fit, and what kinds of benefits the patient might receive.
Based on my initial assessment, you seem to meet criteria for a diagnosis of borderline personality disorder, or BPD. A diagnosis is a category for different symptoms or experiences. To receive a BPD diagnosis, a person has to have at least 5 of 9 symptoms, and you seem to have about 6 of them. From what you have said, the main problems you struggle with are roller-coaster emotions and moods, problems with relationships with other people, and self-harm.
A lot of people recover from BPD, and there’s no reason to think you will have these problems for the rest of your life. In fact, there is a very effective treatment for BPD. This treatment is called dialectical behavior therapy, or DBT. I think you’re a great candidate for DBT. Of course, there’s no guarantee that DBT is the ideal treatment for you, but several studies have shown that DBT helps people learn how to manage their emotions, reduce self-harm, and improve their functioning in life.
DBT includes a couple of different things: meeting once a week with a therapist on an individual basis, then meeting once a week with a group. In the group, you will learn how to manage your emotions, pay attention to the present moment, deal with other people, and tolerate being upset without getting into a crisis.
I know some people in town who provide DBT. Is this something you think you might be interested in? If so, what questions do you have?
Related Resources
- Chapman AL, Gratz KL. The borderline personality disorder survival guide: everything you need to know about living with BPD. Oakland, CA: New Harbinger Publications; 2007.
- National Education Alliance for Borderline Personality Disorder. Information for professionals, patients, and families. www.neabpd.org.
- Behavioral Tech, LLC, founded by Marsha Linehan, PhD. DBT training and resources, including a directory of DBT therapists. www.behavioraltech.org.
- Dialectical Behaviour Therapy Centre of Vancouver. www.dbtvancouver.com.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Strong evidence for the efficacy of dialectical behavior therapy (DBT) for patients with borderline personality disorder (BPD) has brought hope to clinicians and patients alike. By including cognitive therapy, behavioral strategies, skills training, and exposure therapy, DBT addresses more than just self-harm and suicidal behavior (Box 1).1-13 In fact, DBT’s primary interventions—such as skills training in emotion regulation and a straightforward approach to dysfunctional behaviors—could help many people.
Because DBT is so comprehensive and practical, clinicians might be tempted to refer almost anyone who seems even slightly “borderline” for DBT. But some patients—particularly those with mood and anxiety disorders—might benefit more from other treatments. To help you make appropriate evidence-based referrals for DBT and other psychological treatments, this article recommends 4 steps:
- Know what the treatment involves.
- Consider the evidence for the treatment in patients similar to yours.
- Consider why your patient—with unique characteristics and problems—would benefit from these specific interventions.
- Communicate to the patient your reasons for the referral.
Marsha Linehan, PhD, developed dialectical behavior therapy (DBT) in an attempt to devise an effective protocol for highly suicidal women. Over time, she realized that many of these women met criteria for borderline personality disorder (BPD), and DBT evolved to address their emotional, interpersonal, and mental health issues.1
Linehan et al2 published results from the first randomized clinical trial (RCT) of any psychological treatment for BPD. In this study, chronically parasuicidal women who met criteria for BPD received 1 year of DBT or “treatment as usual” in community settings. Those treated with DBT experienced fewer and less severe parasuicidal events, were more likely to remain in treatment, and required fewer days of inpatient care.
Findings from 9 additional RCTs have supported the efficacy of DBT for women with BPD and other populations.3 These RCTs have examined DBT (or adapted versions of DBT) for treating:
- women with BPD and substance dependence4,5
- men and women with BPD in a community setting6
- women veterans with BPD7
- non-BPD women with bulimia8 or binge-eating disorder9
- women with BPD in the Netherlands (53% of study subjects had a substance use disorder)10,11
- depressed older adults12
- suicidal women with BPD.13
Step 1. What does DBT involve?
Difficulty with emotion regulation. DBT is based on a biosocial theory of BPD.1 Within this framework, BPD is caused by the transaction (mutual interplay) of a biologically based vulnerability to emotions with an invalidating rearing environment. The patient with BPD typically experiences strong and long-lasting emotional reactions, often to seemingly small or insignificant events such as a slight look of disappointment on someone’s face or a minor daily hassle. Patients with BPD often are especially attuned to emotional reactions, particularly signs of rejection or disapproval.
Caregivers in an invalidating environment fail to provide the support a highly emotional child needs to learn to manage intense emotions. An invalidating environment:
- indiscriminately rejects the child’s communication of emotions and thoughts as invalid, independent of the validity of the child’s experience
- punishes emotional displays, then intermittently reinforces emotional escalation
- oversimplifies the ease of problem solving or coping.1
As a result, the fledgling BPD individual learns to mistrust and fear emotions and does not learn how to manage them. A patient with BPD is like a car with a powerful “emotional engine” but lacking brakes.
Team treatment. The standard DBT treatment package is an outpatient program run by a team.1 Therapists meet weekly for consultation to help them execute DBT according to the manual, prevent burn-out, and improve skills and motivation to treat patients with multiple, severe problems. The team also maintains the DBT program’s integrity and functioning and ensures that all treatment components—including individual therapy and skills training—are in place.
In individual therapy, the therapist and client collaborate to help the client reduce dysfunctional behaviors, increase motivation, and work toward goals. Because persons with BPD often present with many serious life problems, the therapist organizes session time to address 3 main priorities:
- Life-threatening behavior (intentional self-injury or imminent threat of intentional self-injury, including suicidal crises or threats, severe suicidal ideation or urges, suicide attempts, nonsuicidal self-injury or self-injury urges, or similar behaviors).
- Therapy-interfering behaviors (actions by the therapist or patient that interfere with progress, such as angry outbursts, missed sessions, or tardiness).
- Quality-of-life-interfering behavior (behaviors or problems—such as depression, eating disorders, or substance use disorders; homelessness or financial difficulties; or serious interpersonal discord—that make it hard for the patient to establish a reasonable quality of life).
Additional priorities include skills deficits and secondary targets.1 Each week, the client monitors his or her behaviors, emotions, and actions using a diary card. The therapist uses this information to collaboratively prioritize the focus of each individual therapy session.
Skills training typically occurs weekly in group sessions of 1.5 to 2.5 hours with 1 or 2 therapists. This structured, psycho-educational training focuses on skills that persons with BPD often lack:
- mindfulness (paying attention to the experience of the present moment)
- emotion regulation (regulating or managing distressing emotions)
- distress tolerance (averting crises, tolerating or accepting distressing situations or emotions)
- interpersonal effectiveness (maintaining relationships and asserting needs or wishes).
Therapists often use the first half of group sessions to review each patient’s homework and to provide feedback and coaching on effective skill use. The remaining time is spent teaching new skills. The therapist then assigns homework to practice new skills and closes with a wind-down exercise, often involving relaxation training.
Step 2. Consider the evidence
Before you make a referral for DBT (or any psychological treatment), know what the research says about who is likely to benefit from it. For women with BPD, DBT is the only treatment that can be considered “well-established.”3,14 The literature on DBT includes 10 randomized controlled trials (as well as many uncontrolled trials), and the strongest research supports its use in women with BPD.2,4-13
Based on a detailed review of the literature on DBT, I recommend a basic, evidence-based priority list for referrals (Table 1).3,12,13 Patient groups at the top are most likely to benefit from DBT—according to the most solid, rigorous research—and deserve your strongest consideration for referral. Patient groups further down the list—with fewer rigorous studies of DBT—merit less consideration of DBT as the treatment of choice. Of course to use this list, an accurate diagnosis of your patient’s problems is essential.
DBT’s treatment strategies—exposure therapy, skills training, cognitive therapy, emotion regulation training, and mindfulness—can work for other types of patients. I have noticed, however, that some clinicians refer patients with depression, anxiety disorders, or even bipolar disorder for DBT. Despite DBT’s intuitive appeal, sufficient evidence does not yet support its use in patients with these disorders. Other evidence-based treatments may be more suitable for patients with uncomplicated mood and anxiety disorders (Table 2).3
Table 1
Candidates for DBT: An evidence-based referral priority list*
| Women with BPD who are suicidal or who self-harm (without bipolar disorder, a psychotic disorder, or mental retardation). One randomized clinical trial with suicidal individuals with BPD included men. Two studies excluded participants with substance dependence, but the most recent, largest study13 did not. |
| Women with BPD and substance use problems (without bipolar disorder, a psychotic disorder, or cognitive impairment) | |
| Women with bulimia nervosa or binge-eating disorder (without substance abuse, psychotic disorder, or suicidal ideation). Other empirically supported treatments exist for these patients (Table 2). | |
| Depressed older adults (age ≥60, without bipolar disorder, a psychotic disorder, or cognitive impairment). Investigated treatments included group DBT skills training, telephone consultation, selective serotonin reuptake inhibitor medications, and psychiatric clinical management.12 | |
| Suicidal and nonsuicidal adolescentswith oppositional defiant disorder or bipolar disorder | |
| Incarcerated men and womenwith or without BPD, in high- and low-security forensic settings | |
| Couples and families where 1 member has BPDor where domestic violence occurs in an intimate relationship | |
| * Persons at the top of the list are the ones for whom the most solid, rigorous research has demonstrated the efficacy of DBT. Fewer rigorous studies of DBT have been conducted in persons further down the list. | |
| BPD: borderline personality disorder; DBT: dialectical behavior therapy | |
| Source: References 3,12,13 | |
Table 2
When not to refer a patient for DBT: Evidence is stronger for alternate treatments
| Diagnosis | Treatments with empirical support |
|---|---|
| Major depressive disorder | CBT, behavioral activation, interpersonal therapy, antidepressant medication, mindfulness-based cognitive therapy for depressive relapse |
| Panic disorder/panic disorder with agoraphobia | CBT involving cognitive therapy and exposure-based with agoraphobia interventions |
| Posttraumatic stress disorder | Prolonged exposure therapy, cognitive therapy, EMDR |
| Bulimia nervosa | CBT, interpersonal therapy |
| Primary substance use disorders | CBT, motivational enhancement/motivational interviewing, community reinforcement approach |
| Psychotic disorders | Medication management, social skills training, family-based interventions |
| CBT: cognitive-behavioral therapy; DBT: dialectical behavior therapy; EMDR: eye movement desensitization and reprocessing therapy | |
| Source: Reference 3 | |
Step 3: Would this patient benefit?
Would your patient, with unique struggles and characteristics, benefit from DBT? Consider to what degree DBT’s interventions would solve some of your patient’s problems and whether DBT fits your patient’s needs.
DBT’s target problems. In controlled trials, DBT’s pragmatic approach outperforms comparator treatments in reducing suicidal behaviors and self-injury, and DBT therapists monitor and target these behaviors. Thus, because few treatments reduce self-injury,15,16 you might consider DBT for patients who self-injure even if they do not have BPD.
DBT also includes a strong focus on emotions and emotion regulation. Therefore, if difficulty managing emotions is among your patient’s primary problems, DBT may offer some benefit. DBT also includes structured interpersonal skills training that might be useful for patients who lack assertiveness.
Finally, if you have a patient with multiple diagnoses and severe problems—but not psychosis—the DBT approach to organizing and prioritizing treatment targets may be beneficial. Some multi-diagnosis patients may struggle with aspects of DBT (such as learning new skills), but DBT is set up to incorporate other empirically supported treatment protocols for co-occurring Axis I and II disorders.
Does DBT ‘fit’ your patient? DBT is very structured and involves direct discussions of maladaptive behaviors. If your patient prefers or would benefit from a structured approach, you might consider a referral for DBT.
DBT is an outpatient behavioral treatment that focuses on the here and now. DBT might not be the best fit if your patient:
- views his or her problems as resulting primarily from childhood experiences or relationships with parents
- would prefer insight-oriented therapy.
If, however, your patient would like a practical approach focused on problem-solving, DBT could be an effective choice.
DBT is based in part on a dialectical philosophy, and DBT therapists often seek to bring together or synthesize polarized thinking. If your patient struggles with “black or white” thinking, this dialectical philosophy might be helpful. On the other hand, DBT might not be the best fit if your patient is particularly rigid in thinking or seems to require cognitive therapy to address his or her thought patterns.
DBT is not the treatment of choice for all personality disorders. Most of the evidence examines its use for BPD, and few studies have looked at any other personality disorder. Also, keep in mind that being interpersonally “difficult” does not mean that a patient is “borderline” or needs DBT.
Step 4: Communicate reasons for referral to your patient
Finally, communicate to your patient the reasons you are referring him or her for DBT. Patients often walk into my office for DBT, confused about why they are there. If patients understand why they have been referred for DBT and how it may help them, they may be more likely to follow through and realize its benefits.
A sample explanation of referral that I offer to guide this communication (Box 2) includes 3 main points:
- my diagnosis or conceptualization of the patient’s clinical problems
- a brief description of DBT
- a rationale for why DBT would be a good fit, and what kinds of benefits the patient might receive.
Based on my initial assessment, you seem to meet criteria for a diagnosis of borderline personality disorder, or BPD. A diagnosis is a category for different symptoms or experiences. To receive a BPD diagnosis, a person has to have at least 5 of 9 symptoms, and you seem to have about 6 of them. From what you have said, the main problems you struggle with are roller-coaster emotions and moods, problems with relationships with other people, and self-harm.
A lot of people recover from BPD, and there’s no reason to think you will have these problems for the rest of your life. In fact, there is a very effective treatment for BPD. This treatment is called dialectical behavior therapy, or DBT. I think you’re a great candidate for DBT. Of course, there’s no guarantee that DBT is the ideal treatment for you, but several studies have shown that DBT helps people learn how to manage their emotions, reduce self-harm, and improve their functioning in life.
DBT includes a couple of different things: meeting once a week with a therapist on an individual basis, then meeting once a week with a group. In the group, you will learn how to manage your emotions, pay attention to the present moment, deal with other people, and tolerate being upset without getting into a crisis.
I know some people in town who provide DBT. Is this something you think you might be interested in? If so, what questions do you have?
Related Resources
- Chapman AL, Gratz KL. The borderline personality disorder survival guide: everything you need to know about living with BPD. Oakland, CA: New Harbinger Publications; 2007.
- National Education Alliance for Borderline Personality Disorder. Information for professionals, patients, and families. www.neabpd.org.
- Behavioral Tech, LLC, founded by Marsha Linehan, PhD. DBT training and resources, including a directory of DBT therapists. www.behavioraltech.org.
- Dialectical Behaviour Therapy Centre of Vancouver. www.dbtvancouver.com.
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Linehan MM. Cognitive behavior treatment of borderline personality disorder. New York, NY: The Guilford Press; 1993.
2. Linehan MM, Armstrong HE, Suarez A, et al. Cognitive-behavioral treatment of chronically parasuicidal borderline patients. Arch Gen Psychiatry 1991;48:1060-4.
3. Robins CJ, Chapman AL. Dialectical behavior therapy: current status, recent developments, and future directions. J Personal Disord 2004;18:73-9.
4. Linehan MM, Schmidt HI, Dimeff LA, et al. Dialectical behavior therapy for patients with borderline personality disorder and drug-dependence. Am J Addictions 1999;8:279-92.
5. Turner RM. Naturalistic evaluation of dialectical behavior therapy-oriented treatment for borderline personality disorder. Cognit Behav Pract 2000;7:413-9.
6. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectical behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend 2002;67:13-26.
7. Koons C, Robins CJ, Tweed JL, et al. Efficacy of dialectical behavior therapy in women veterans with borderline personality disorder. Behav Ther 2001;32:371-90.
8. Safer DL, Telch CF, Agras WS. Dialectical behavior therapy for bulimia nervosa. Am J Psychiatry 2001;158:632-4.
9. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061-5.
10. van den Bosch LMC, Verheul R, Schippers GM, van den Brink W. Dialectical behavior therapy of borderline patients with and without substance abuse problems: implementation and long-term effects. Addict Behav 2002;27:911-23.
11. Verheul R, van den Bosch LMC, Koeter MWJ, et al. Dialectical behavior therapy for women with borderline personality disorder. Br J Psychiatry 2003;182:135-40.
12. Lynch TR, Morse JQ, Mendelson T, Robins CJ. Dialectical behavior therapy for depressed older adults: a randomized pilot study. Am J Geriatr Psychiatry 2003;11:33-45.
13. Linehan MM, Comtois KA, Murray AM, et al. Two-year randomized controlled trial and follow-up of dialectical behavior therapy vs. therapy by experts for suicidal behaviors and borderline personality disorder. Arch Gen Psychiatry 2006;63:757-66.
14. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol 2001;52:685-716.
15. Hawton K, Arensman E, Townsend E, et al. Deliberate self-harm: systematic review of efficacy of psychosocial and pharmacological treatments in preventing repetition. BMJ 1998;317:441-7.
16. Tyrer P, Tom B, Byford S, et al. Differential effects of manual-assisted cognitive behavior therapy in the treatment of recurrent deliberate self-harm and personality disturbance: the POPMACT study. J Personal Disord 2004;18:102-16.
1. Linehan MM. Cognitive behavior treatment of borderline personality disorder. New York, NY: The Guilford Press; 1993.
2. Linehan MM, Armstrong HE, Suarez A, et al. Cognitive-behavioral treatment of chronically parasuicidal borderline patients. Arch Gen Psychiatry 1991;48:1060-4.
3. Robins CJ, Chapman AL. Dialectical behavior therapy: current status, recent developments, and future directions. J Personal Disord 2004;18:73-9.
4. Linehan MM, Schmidt HI, Dimeff LA, et al. Dialectical behavior therapy for patients with borderline personality disorder and drug-dependence. Am J Addictions 1999;8:279-92.
5. Turner RM. Naturalistic evaluation of dialectical behavior therapy-oriented treatment for borderline personality disorder. Cognit Behav Pract 2000;7:413-9.
6. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectical behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend 2002;67:13-26.
7. Koons C, Robins CJ, Tweed JL, et al. Efficacy of dialectical behavior therapy in women veterans with borderline personality disorder. Behav Ther 2001;32:371-90.
8. Safer DL, Telch CF, Agras WS. Dialectical behavior therapy for bulimia nervosa. Am J Psychiatry 2001;158:632-4.
9. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061-5.
10. van den Bosch LMC, Verheul R, Schippers GM, van den Brink W. Dialectical behavior therapy of borderline patients with and without substance abuse problems: implementation and long-term effects. Addict Behav 2002;27:911-23.
11. Verheul R, van den Bosch LMC, Koeter MWJ, et al. Dialectical behavior therapy for women with borderline personality disorder. Br J Psychiatry 2003;182:135-40.
12. Lynch TR, Morse JQ, Mendelson T, Robins CJ. Dialectical behavior therapy for depressed older adults: a randomized pilot study. Am J Geriatr Psychiatry 2003;11:33-45.
13. Linehan MM, Comtois KA, Murray AM, et al. Two-year randomized controlled trial and follow-up of dialectical behavior therapy vs. therapy by experts for suicidal behaviors and borderline personality disorder. Arch Gen Psychiatry 2006;63:757-66.
14. Chambless DL, Ollendick TH. Empirically supported psychological interventions: controversies and evidence. Annu Rev Psychol 2001;52:685-716.
15. Hawton K, Arensman E, Townsend E, et al. Deliberate self-harm: systematic review of efficacy of psychosocial and pharmacological treatments in preventing repetition. BMJ 1998;317:441-7.
16. Tyrer P, Tom B, Byford S, et al. Differential effects of manual-assisted cognitive behavior therapy in the treatment of recurrent deliberate self-harm and personality disturbance: the POPMACT study. J Personal Disord 2004;18:102-16.
Driving with dementia: How to assess safety behind the wheel
Mr. D, age 75, presents to your office with a 5-year history of gradually declining memory. His wife reports he is having difficulty with word finding, managing his finances, and shopping, and he needs supervision when using the stove. Nonetheless, he enjoys playing golf and drives himself to the golf course 3 times a week. He is compliant with his chronic medical therapy for hypertension, hypercholesterolemia, and asthma.
Patients with dementia who continue to drive pose a potential danger on the road, worry their families, and present challenges to clinicians. Most people would agree that patients with moderate or severe dementia should not drive, but a careful evaluation is required to assess whether a patient such as Mr. D with mild dementia remains fit to drive.
This article explores how dementia exacerbates age-related changes in driving ability and discusses how to assess driving in patients with dementia. Our goal is to help clinicians sort through data from in-office physical and cognitive assessments, family caregivers/informants’ reports, and (when available) on-road testing. We also discuss:
- guidelines for assessing older drivers that can help balance patients’ need for autonomy with public safety
- strategies for discussing driving cessation with patients and their families.
Driving: A privilege, not a right
Driving is central to older adults’ autonomy, and >75% of persons age ≥75 rely on driving as their primary mode of transportation.1 Driving cessation in this population has been associated with a 3-fold decrease in out-of-home activity2 and a 2.5-fold increase in depressive symptoms.3 Nonetheless, some 4.5 million Americans have Alzheimer’s disease (AD),4 and dementia poses a substantial risk to safe driving.
Although driving must be sacrificed when personal and public safety is at risk, most physicians perceive an uncomfortable conflict of interest between patient confidentiality and public safety.5 Assessing driving safety of patients with dementia can undermine the doctor-patient relationship and pose hardships for patients.
Mr. D has a 5-year history of memory problems that affect his daily functioning, yet he continues to drive. A longitudinal study of persons with dementia found that among the 29% who were driving at baseline, more than one-half were still behind the wheel 2 years later.6
Age and driving safety. Even in the absence of dementia, driving ability declines with aging (Tables 1 and 2).7,8 Older persons may self-regulate and restrict their driving to shorter distances, with fewer trips at night, on high-speed roads, or in unfamiliar situations. Their driving is rarely aggressive and they are unlikely to speed, but they may drive more slowly than other traffic.7,8 Although the overall rate of motor vehicle collisions declines with age:
- the rate of collisions per mile driven increases after age 659
- drivers age >65 have the highest fatality rate per mile driven among adults age ≥25.10
A dementia diagnosis is not sufficient to withdraw driving privileges, according to American Medical Association (AMA)/National Highway Traffic Safety Administration (NHTSA) guidelines. These recommend that you base decisions on the individual’s driving ability, and—when you have concerns—factor in a focused medical assessment and formal assessment of driving skills.10
Table 1
Age-related changes that may affect driving fitness
| Decreased physical capabilities, including declining muscle tone, flexibility, and reaction time |
| Decreased hearing and visual acuity |
| Increased fragility, resulting in longer time to heal should injuries occur |
| Increased medication use with possible side effect of drowsiness |
| Source: References 7,8 |
Table 2
Older drivers’ common traffic violations leading to crashes*
| Failure to obey traffic signals, including stop signs and red lights |
| Unsafe left turns (driver may inaccurately judge speed of oncoming vehicle) |
| Inappropriate turns (such as difficulty judging distance from oncoming cars, wide or narrow turns, or not timing the turn correctly with traffic lights) |
| Unsafe passing |
| Failure to yield |
| * These errors often lead to multivehicle accidents Source: References 7,8 |
CASE CONTINUED: Cognitive deficits quantified
You perform a Mini-Mental State Examination (MMSE). Mr. D scores 24/30, losing 1 point for orientation, 2 points for attention, 2 points for recall, and 1 point for copying. This score, along with his history, indicates mild dementia, although he claims he is a safe driver. On further cognitive testing, Mr. D completes the Trails A test in 90 seconds and Trails B test in 250 seconds (well below 1.5 standard deviations of the norm for his age and education).11 On the clock-drawing task, he drew a poorly organized clock, with unequal spaces between numbers and hands pointing to “10” and “11” instead of properly indicating “10 after 11.”
Mr. D and his wife live in a rural area, 5 miles from the nearest grocery store. His wife never drove, and she relies on him for weekly shopping trips and to drive her to her bridge club. She denies any problems with his driving but states, “Other drivers have become so aggressive; they’re always honking at him.” Their daughter denies that Mr. D has driving problems but admits that for the last 2 years she has refused to allow her child to ride in his car.
Focused in-office assessment
Information to assess driving ability can come from the patient, family caregiver/informant, and clinical judgment. Patients with dementia are notoriously inaccurate in self-reported driving ability, either for lack of insight or as a testament to the importance of driving to their autonomy. Caregivers often are more accurate in describing a patient’s driving, but other agendas may color their responses.
In a study of patients with very mild or mild AD, 94% reported themselves as safe drivers, whereas on-road driving instructors rated <50% of drivers in these groups as safe. Caregivers were better able to classify driving performance, but 36% of their ratings were incorrect.12
Cognitive assessment. To assess older drivers’ cognition, AMA/NHTSA’s Guide to Assessing and Counseling Older Drivers recommends the Trail-Making Test, Part B and the clock-drawing test.10 The Canadian Medical Association suggests the MMSE.13 Both guides say that abnormalities in these tests indicate a need for more detailed testing, including referral to specialized driving assessment and retesting at regular intervals (Algorithm). Retest patients with mild dementia at least every 6 months or sooner when dementia severity increases noticeably14 (Box 1).6,15
The MMSE is widely used to screen for cognitive impairment and identify dementia or delirium, but it is not a diagnostic tool or proxy driving test. A patient with dementia may produce a high MMSE score and yet be an unsafe driver. For example, well-educated patients or those with vascular or frontotemporal dementia may retain cognitive abilities as measured by the MMSE until later in the disease.
Considerable effort has been put into developing tools to help clinicians quickly and accurately differentiate safe from unsafe drivers by assessing cognition. Unfortunately, no consistent link has been found between cognitive test results and driving outcome measures. A systematic review of office-based predictors of fitness to drive in dementia found 5 studies showing an association between MMSE scores and driving and 5 studies showing no such association.16 Thus, although the AMA/NHTSA guide recommends the MMSE, Trails B, and clock-drawing tests, cognitive tests—including these—are not sufficient to assess driving ability.
Severity of dementia. International consensus groups have attempted to create guidelines for patients with dementia who drive. American, Canadian, and Australian groups suggest that a diagnosis of moderate to severe dementia precludes driving, and the driver’s licenses of persons with these conditions should be revoked.17
In general, AD is considered severe when the MMSE score is <10 or the patient becomes dependent on a caregiver for survival.18 AD of moderate severity is more difficult to define, but a Canadian consensus conference suggested a practical approach: Patients with AD would be considered to have moderate to severe dementia and should not drive when they cannot independently perform multiple instrumental activities of daily living or any of the basic activities of daily living.19
Some dementias may impair driving more quickly than AD does. For example, hallucinations may occur early in Lewy body dementia, as may impulsivity in frontotemporal dementia and motor impairment in vascular dementia.
Mrs. Y visits your office for a follow-up regarding mild Alzheimer’s disease (AD), which was diagnosed 2 years ago. She passed an on-road test 3 months ago and has an Mini-Mental State Examination score of 24/30. Over the last month she has become depressed, with insomnia and mild psychomotor retardation. She occasionally has hallucinations.
Behavioral and psychological symptoms such as agitation, aggression, hallucinations, apathy, depression, and anxiety are common neuropsychiatric sequelae of AD. Little is known about the risks these symptoms pose to road safety, but we recommend that clinicians strongly consider the potential for impaired driving.
In a longitudinal study, cognitive impairment and behavioral disturbances—especially agitation, apathy, and hallucinations—were strong predictors of driving cessation among patients with dementia.6 Furthermore, a case crossover study of patients with dementia found a 54% increase in risk of motor vehicle collisions associated with the use of psychotropic medications.15
Consider all aspects of the patient’s clinical status, including neuropsychiatric symptoms, psychotropic medications, comorbid medical conditions (including hearing and vision impairment), and concomitant therapy for medical conditions. Any could change a safe driver with mild dementia into an unsafe driver.
Algorithm: 3 options for drivers with dementia, based on in-office assessment
* Observe legislation or statutes that address reporting unsafe drivers to the department of motor vehicles or ministry of transportation
On-road driving tests
Because some individuals with mild dementia can drive safely for extended periods, international recommendations for assessing the driver with dementia emphasize on-road driving tests.10,13,20–22 American10 and Canadian guidelines13 suggest that a dementia diagnosis is not sufficient to withdraw licensure.
A formal driving assessment is necessary to establish road safety for patients with mild dementia except when the need for license withdrawal is evident, such as when the patient has:
- a history of major driving problems (such as crashes or driving the wrong way on a highway)
- significant contraindications to driving on the history or physical examination (such as severe inattention or psychosis).
Challenges of on-road testing. On-road tests may be the gold standard, but they are not without clinical problems.
Need to retest. Because almost all dementias are progressive and driving skills deteriorate over time, most guidelines recommend periodic retesting. For patients with dementia who pass on-road evaluations, limited evidence supports retesting every 6 months.14 Take an individual approach, however, because of the various rates at which the dementias progress.
Testing vs real world conditions. Structured on-road testing is not equivalent to unstructured real-world driving, in which the patient often must navigate without instruction or assistance.
Rural vs urban driving. Road tests conducted in urban areas assess skills associated with complex conditions and the need to respond quickly to crises. They might not assess as well rural driving, which requires sustained attention on monotonous roads.
Inaccessibility. Cost and lack of availability of on-road tests, particularly in rural areas, limit the number of patients whose performance can be evaluated.
CASE CONTINUED: Distressing results
Mr. D has a history of decline in cognition and function, objective cognitive difficulties, and a subtle history of driving problems. You refer him for a specialized on-road test, and the report indicates that he failed. Errors included wide turns, driving too slowly, getting caught in an intersection twice during red lights while attempting to turn left, driving on the shoulder, and failing to signal lane changes. You review the results with Mr. D and his wife and recommend that he cease driving immediately.
Mr. D is furious, and his wife is dismayed. He demands to know how he can continue to play golf, which is his only form of exercise and recreation. Will she have to give up her bridge club? How will they shop for food? They request permission to at least to drive to the grocery store during the daytime.
You explain that no system allows individuals to drive only at certain times, and for the sake of safety you cannot grant them special permission. You discuss alternatives, such as asking their daughter for assistance with grocery shopping and taking taxis or ride-sharing with friends who play golf and bridge.
Remain firm, but ease the blow
Driving cessation orders distress patients, families, and clinicians. A failed road test clearly indicates unsafe driving, and driving cessation is critical to public safety.
A review by Man-Son-Hing et al23 found that drivers with dementia performed worse than nondemented controls in all studies that examined driving performance (on-road, simulator, or caregiver report). Simulators showed problems such as off-road driving, deviation from posted speed, and more time to negotiate left turns.24
By comparison, only 1 of 3 studies using state crash records showed an increased risk of collisions in persons with dementia compared with controls.23 From a research perspective, however, studies that use state-reported collisions to assess driving risk are confounded by driving restrictions on persons with dementia.
Mr. D wants to continue driving with restrictions. No studies have shown reduced crash rates when drivers with dementia used compensatory strategies such as restrictions, retraining/education, having a passenger “co-pilot,” on-board navigation, or cognitive enhancers.23
If Mr. D had passed the road test, the situation would have been more ambiguous. Two studies have examined on-road driving performance over time in patients with early-stage dementia.25,26 Both studies followed drivers prospectively for 2 years, and those with mild dementia (vs very mild or no dementia) were most likely to show a decline in driving skills:
- All participants with mild dementia were rated as “not safe” by the end of 2 years by Duchek et al.25
- Median time to “failure” (or a rating of unsafe) was 324 days for drivers with mild dementia vs 605 days for those with very mild dementia, as reported by Ott et al.26
Mr. D’s passionate plea for reconsideration highlights the need for communities to develop alternate transportation for seniors whose driving becomes unsafe (Box 2).
Legal liability? Physicians often are concerned about legal responsibilities and risks involved in reporting unsafe drivers. Be aware of local statutes or legislations regarding mandatory reporting of patients you deem unsafe to drive.17 These laws usually protect physicians from lawsuits related to violating patient confidentiality. Civil lawsuits remain possible, however, if clinicians fail to report an unsafe driver who subsequently is involved in a motor vehicle collision.27
1. Meet with family first. Help them assume a positive and supportive role. Explain concretely and empathically your concern for the safety of the patient and others. Clearly outline your findings that the patient is not fit to drive, and explain that the law requires you to report the patient to the authorities.
Remind family members that the goal of driving assessment is to prevent a collision, and they carry some responsibility because they are aware of the potential risk of letting their family member continue to drive. If necessary, have family members witness a repeat performance by the patient on the most revealing test. Discuss the importance of finding alternate transportation to reduce the risk of isolation and depression that can follow driving cessation.
2. Meet with patient. Having the family present can be helpful, but ask them to assume a supportive role. Give the patient a positive role by recognizing that he or she has been a responsible driver, and part of this responsibility is to stop driving before an accident occurs. Acknowledge that it is normal to be unhappy upon learning that one’s driving privileges are being revoked.
Sometimes it helps to give the patient a prescription in their name that says, “Do not drive.” Families who receive a copy may find this very helpful, too, for reminding the patient later about what you said.
If your patient argues with your position, remain firm and do not argue. Indicate that you have made notes on the meeting and are notifying the authorities about the patient’s unsafe driving. You can add that your chart could be subpoenaed and the patient may be legally liable and financially responsible should he or she continue to drive and have a collision.
3. Talk about transportation options. Family members could share driving responsibilities. Taxi rides can cost less than maintaining a car if the patient drives <4,000 km (2,500 miles) per year. Suggest that patients or families find volunteer drivers or contact helpful taxi drivers a day before an outing is planned.
4. If patient refuses to comply, meet with the family again and encourage them to remove the patient’s opportunity to drive (confiscate the keys, disable the car, or remove the car altogether).
Provide a written statement to the patient and family outlining why the patient can no longer drive. Indicate that it is your legal responsibility to report unsafe drivers, and you intend to notify the authorities regarding the patient’s driving status. If the patient remains noncompliant, continue to encourage family to remove the opportunity to drive.
Related resources
- Physician’s guide to assessing and counseling older drivers. American Medical Association and the National Highway Traffic Safety Administration, 2003. www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook.
- Determining medical fitness to operate motor vehicles. Canadian Medical Association. 2006. www.cma.ca/index.cfm/ci_id/18223/la_id/1.htm.
- Canadian Driving Research Initiative for Vehicular Safety in the Elderly (CanDRIVE). www.candrive.ca.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rapoport receives grant/research support from the Canadian Institute of Health Research and the Ontario Neurotrauma Foundation.
1. Stowell-Ritter A, Straight A, Evans EL. Understanding senior transportation: report and analysis of a survey of consumers age 50+. Washington, DC: American Association of Retired Persons; 2002.
2. Marottoli RA, de Leon CFM, Glass TA, et al. Consequences of driving cessation: decreased out-of-home activity levels. J Gerontol B Psychol Sci Soc Sci 2000;55(6):S334-40.
3. Marottoli RA, Mendes de Leon CF, Glass TA, et al. Driving cessation and increased depressive symptoms: prospective evidence from the New Haven EPESE. Established Populations for Epidemiologic Studies of the Elderly. J Am Geriatr Soc 1997;45(2):202-6.
4. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003;60(8):1119-22.
5. Jang RW, Man-Son-Hing M, Molnar FJ, et al. Family physicians’ attitudes and practices regarding assessments of medical fitness to drive in older persons. J Gen Intern Med 2007;22(4):531-43.
6. Herrmann N, Rapoport MJ, Sambrook R, et al. Predictors of driving cessation in mild-to-moderate dementia. CMAJ 2006;175(6):591-5.
7. Hedlund J. Countermeasures that work: a highway safety countermeasure guide for state highway safety offices. Washington, DC: National Highway Traffic Safety Administration; 2006.
8. Dobbs B. Medical conditions and driving: a review of the literature (1960-2000). Washington, DC: National Highway Traffic Safety Administration; 2005. Available at: http://www.nhtsa.dot.gov/people/injury/research/Medical_Condition_Driving/pages/TRD.html. Accessed September 29, 2008.
9. Li G, Braver ER, Chen LH. Fragility versus excessive crash involvement as determinants of high death rates per vehicle-mile of travel among older drivers. Accid Anal Prev 2003;35(2):227-35.
10. Physician’s guide to assessing and counseling older drivers. Washington, DC: National Highway Traffic Safety Administration; 2003. Available at: http://www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook. Accessed September 29, 2008.
11. Yeudall LT, Reddon JR, Gill DM, et al. Normative data for the Halstead-Reitan neuropsychological tests stratified by age and sex. J Clin Psychol 1987;43(3):346-67.
12. Brown LB, Ott BR, Papandonatos GD, et al. Prediction of on-road driving performance in patients with early Alzheimer’s disease. J Am Geriatr Soc 2005;53(1):94-8.
13. Determining medical fitness to operate motor vehicles. CMA driver’s guide. 7th ed. Ottawa, Ontario, Canada: Canadian Medical Association; 2006.
14. Molnar FJ, Patel A, Marshall SC, et al. Systematic review of the optimal frequency of follow-up in persons with mild dementia who continue to drive. Alzheimer Dis Assoc Disord 2006;20(4):295-7.
15. Rapoport MJ, Herrmann N, Molnar FJ, et al. Psychotropic medications and motor vehicle collisions in patients with dementia (letter). J Am Geriatr Soc 2008;56(10):1968-70.
16. Molnar FJ, Patel A, Marshall SC, et al. Clinical utility of office-based cognitive predictors of fitness to drive in persons with dementia: a systematic review. J Am Geriatr Soc 2006;54(12):1809-24.
17. Rapoport MJ, Herrmann N, Molnar FJ, et al. Sharing the responsibility for assessing the risk of the driver with dementia. CMAJ. 2007;177(6):599-601.
18. Herrmann N, Gauthier S, Lysy PG. Clinical practice guidelines for severe Alzheimer’s disease. Alzheimers Dement 2007;3(4):385-97.
19. Hogan DB, Bailey P, Carswell A, et al. Management of mild to moderate Alzheimer’s disease and dementia. Alzheimers Dement 2007;3(4):355-84.
19. Assessing fitness to drive for commercial and private vehicle drivers. Sydney, Australia: National Library of Australia; 2006. Available at: http://www.austroads.com.au/aftd/index.html. Accessed November 4, 2008.
21. Medical aspects of fitness to drive. A guide for medical practitioners. Wellington, New Zealand: Land Transport Safety Authority; 2002. Available at: http://www.transfund.govt.nz/licensing/docs/ltsa-medical-aspects.pdf. Accessed September 29, 2008.
22. At a glance guide to the current medical standards of fitness to drive. Swansea, UK: Drivers Medical Group, Driver and Vehicle Licensing Agency; 2008. Available at: http://www.dvla.gov.uk/medical/ataglance.aspx. Accessed September 29, 2008.
23. Man-Son-Hing M, Marshall SC, Molnar FJ, Wilson KG. Systematic review of driving risk and the efficacy of compensatory strategies in persons with dementia. J Am Geriatr Soc 2007;55(6):878-84.
24. Cox DJ, Quillian WC, Thorndike FP, et al. Evaluating driving performance of outpatients with Alzheimer disease. J Am Board Fam Pract 1998;11(4):264-71.
25. Duchek JM, Carr DB, Hunt L, et al. Longitudinal driving performance in early-stage dementia of the Alzheimer type. J Am Geriatr Soc 2003;51(10):1342-7.
26. Ott BR, Heindel WC, Papandonatos GD, et al. A longitudinal study of drivers with Alzheimer disease. Neurology 2008;70(14):1171-8.
27. Molnar FJ, Byszewski AM, Marshall SC, Man-Son-Hing M. In-office evaluation of medical fitness to drive: practical approaches for assessing older people. Can Fam Physician 2005;51:372-9.
Mr. D, age 75, presents to your office with a 5-year history of gradually declining memory. His wife reports he is having difficulty with word finding, managing his finances, and shopping, and he needs supervision when using the stove. Nonetheless, he enjoys playing golf and drives himself to the golf course 3 times a week. He is compliant with his chronic medical therapy for hypertension, hypercholesterolemia, and asthma.
Patients with dementia who continue to drive pose a potential danger on the road, worry their families, and present challenges to clinicians. Most people would agree that patients with moderate or severe dementia should not drive, but a careful evaluation is required to assess whether a patient such as Mr. D with mild dementia remains fit to drive.
This article explores how dementia exacerbates age-related changes in driving ability and discusses how to assess driving in patients with dementia. Our goal is to help clinicians sort through data from in-office physical and cognitive assessments, family caregivers/informants’ reports, and (when available) on-road testing. We also discuss:
- guidelines for assessing older drivers that can help balance patients’ need for autonomy with public safety
- strategies for discussing driving cessation with patients and their families.
Driving: A privilege, not a right
Driving is central to older adults’ autonomy, and >75% of persons age ≥75 rely on driving as their primary mode of transportation.1 Driving cessation in this population has been associated with a 3-fold decrease in out-of-home activity2 and a 2.5-fold increase in depressive symptoms.3 Nonetheless, some 4.5 million Americans have Alzheimer’s disease (AD),4 and dementia poses a substantial risk to safe driving.
Although driving must be sacrificed when personal and public safety is at risk, most physicians perceive an uncomfortable conflict of interest between patient confidentiality and public safety.5 Assessing driving safety of patients with dementia can undermine the doctor-patient relationship and pose hardships for patients.
Mr. D has a 5-year history of memory problems that affect his daily functioning, yet he continues to drive. A longitudinal study of persons with dementia found that among the 29% who were driving at baseline, more than one-half were still behind the wheel 2 years later.6
Age and driving safety. Even in the absence of dementia, driving ability declines with aging (Tables 1 and 2).7,8 Older persons may self-regulate and restrict their driving to shorter distances, with fewer trips at night, on high-speed roads, or in unfamiliar situations. Their driving is rarely aggressive and they are unlikely to speed, but they may drive more slowly than other traffic.7,8 Although the overall rate of motor vehicle collisions declines with age:
- the rate of collisions per mile driven increases after age 659
- drivers age >65 have the highest fatality rate per mile driven among adults age ≥25.10
A dementia diagnosis is not sufficient to withdraw driving privileges, according to American Medical Association (AMA)/National Highway Traffic Safety Administration (NHTSA) guidelines. These recommend that you base decisions on the individual’s driving ability, and—when you have concerns—factor in a focused medical assessment and formal assessment of driving skills.10
Table 1
Age-related changes that may affect driving fitness
| Decreased physical capabilities, including declining muscle tone, flexibility, and reaction time |
| Decreased hearing and visual acuity |
| Increased fragility, resulting in longer time to heal should injuries occur |
| Increased medication use with possible side effect of drowsiness |
| Source: References 7,8 |
Table 2
Older drivers’ common traffic violations leading to crashes*
| Failure to obey traffic signals, including stop signs and red lights |
| Unsafe left turns (driver may inaccurately judge speed of oncoming vehicle) |
| Inappropriate turns (such as difficulty judging distance from oncoming cars, wide or narrow turns, or not timing the turn correctly with traffic lights) |
| Unsafe passing |
| Failure to yield |
| * These errors often lead to multivehicle accidents Source: References 7,8 |
CASE CONTINUED: Cognitive deficits quantified
You perform a Mini-Mental State Examination (MMSE). Mr. D scores 24/30, losing 1 point for orientation, 2 points for attention, 2 points for recall, and 1 point for copying. This score, along with his history, indicates mild dementia, although he claims he is a safe driver. On further cognitive testing, Mr. D completes the Trails A test in 90 seconds and Trails B test in 250 seconds (well below 1.5 standard deviations of the norm for his age and education).11 On the clock-drawing task, he drew a poorly organized clock, with unequal spaces between numbers and hands pointing to “10” and “11” instead of properly indicating “10 after 11.”
Mr. D and his wife live in a rural area, 5 miles from the nearest grocery store. His wife never drove, and she relies on him for weekly shopping trips and to drive her to her bridge club. She denies any problems with his driving but states, “Other drivers have become so aggressive; they’re always honking at him.” Their daughter denies that Mr. D has driving problems but admits that for the last 2 years she has refused to allow her child to ride in his car.
Focused in-office assessment
Information to assess driving ability can come from the patient, family caregiver/informant, and clinical judgment. Patients with dementia are notoriously inaccurate in self-reported driving ability, either for lack of insight or as a testament to the importance of driving to their autonomy. Caregivers often are more accurate in describing a patient’s driving, but other agendas may color their responses.
In a study of patients with very mild or mild AD, 94% reported themselves as safe drivers, whereas on-road driving instructors rated <50% of drivers in these groups as safe. Caregivers were better able to classify driving performance, but 36% of their ratings were incorrect.12
Cognitive assessment. To assess older drivers’ cognition, AMA/NHTSA’s Guide to Assessing and Counseling Older Drivers recommends the Trail-Making Test, Part B and the clock-drawing test.10 The Canadian Medical Association suggests the MMSE.13 Both guides say that abnormalities in these tests indicate a need for more detailed testing, including referral to specialized driving assessment and retesting at regular intervals (Algorithm). Retest patients with mild dementia at least every 6 months or sooner when dementia severity increases noticeably14 (Box 1).6,15
The MMSE is widely used to screen for cognitive impairment and identify dementia or delirium, but it is not a diagnostic tool or proxy driving test. A patient with dementia may produce a high MMSE score and yet be an unsafe driver. For example, well-educated patients or those with vascular or frontotemporal dementia may retain cognitive abilities as measured by the MMSE until later in the disease.
Considerable effort has been put into developing tools to help clinicians quickly and accurately differentiate safe from unsafe drivers by assessing cognition. Unfortunately, no consistent link has been found between cognitive test results and driving outcome measures. A systematic review of office-based predictors of fitness to drive in dementia found 5 studies showing an association between MMSE scores and driving and 5 studies showing no such association.16 Thus, although the AMA/NHTSA guide recommends the MMSE, Trails B, and clock-drawing tests, cognitive tests—including these—are not sufficient to assess driving ability.
Severity of dementia. International consensus groups have attempted to create guidelines for patients with dementia who drive. American, Canadian, and Australian groups suggest that a diagnosis of moderate to severe dementia precludes driving, and the driver’s licenses of persons with these conditions should be revoked.17
In general, AD is considered severe when the MMSE score is <10 or the patient becomes dependent on a caregiver for survival.18 AD of moderate severity is more difficult to define, but a Canadian consensus conference suggested a practical approach: Patients with AD would be considered to have moderate to severe dementia and should not drive when they cannot independently perform multiple instrumental activities of daily living or any of the basic activities of daily living.19
Some dementias may impair driving more quickly than AD does. For example, hallucinations may occur early in Lewy body dementia, as may impulsivity in frontotemporal dementia and motor impairment in vascular dementia.
Mrs. Y visits your office for a follow-up regarding mild Alzheimer’s disease (AD), which was diagnosed 2 years ago. She passed an on-road test 3 months ago and has an Mini-Mental State Examination score of 24/30. Over the last month she has become depressed, with insomnia and mild psychomotor retardation. She occasionally has hallucinations.
Behavioral and psychological symptoms such as agitation, aggression, hallucinations, apathy, depression, and anxiety are common neuropsychiatric sequelae of AD. Little is known about the risks these symptoms pose to road safety, but we recommend that clinicians strongly consider the potential for impaired driving.
In a longitudinal study, cognitive impairment and behavioral disturbances—especially agitation, apathy, and hallucinations—were strong predictors of driving cessation among patients with dementia.6 Furthermore, a case crossover study of patients with dementia found a 54% increase in risk of motor vehicle collisions associated with the use of psychotropic medications.15
Consider all aspects of the patient’s clinical status, including neuropsychiatric symptoms, psychotropic medications, comorbid medical conditions (including hearing and vision impairment), and concomitant therapy for medical conditions. Any could change a safe driver with mild dementia into an unsafe driver.
Algorithm: 3 options for drivers with dementia, based on in-office assessment
* Observe legislation or statutes that address reporting unsafe drivers to the department of motor vehicles or ministry of transportation
On-road driving tests
Because some individuals with mild dementia can drive safely for extended periods, international recommendations for assessing the driver with dementia emphasize on-road driving tests.10,13,20–22 American10 and Canadian guidelines13 suggest that a dementia diagnosis is not sufficient to withdraw licensure.
A formal driving assessment is necessary to establish road safety for patients with mild dementia except when the need for license withdrawal is evident, such as when the patient has:
- a history of major driving problems (such as crashes or driving the wrong way on a highway)
- significant contraindications to driving on the history or physical examination (such as severe inattention or psychosis).
Challenges of on-road testing. On-road tests may be the gold standard, but they are not without clinical problems.
Need to retest. Because almost all dementias are progressive and driving skills deteriorate over time, most guidelines recommend periodic retesting. For patients with dementia who pass on-road evaluations, limited evidence supports retesting every 6 months.14 Take an individual approach, however, because of the various rates at which the dementias progress.
Testing vs real world conditions. Structured on-road testing is not equivalent to unstructured real-world driving, in which the patient often must navigate without instruction or assistance.
Rural vs urban driving. Road tests conducted in urban areas assess skills associated with complex conditions and the need to respond quickly to crises. They might not assess as well rural driving, which requires sustained attention on monotonous roads.
Inaccessibility. Cost and lack of availability of on-road tests, particularly in rural areas, limit the number of patients whose performance can be evaluated.
CASE CONTINUED: Distressing results
Mr. D has a history of decline in cognition and function, objective cognitive difficulties, and a subtle history of driving problems. You refer him for a specialized on-road test, and the report indicates that he failed. Errors included wide turns, driving too slowly, getting caught in an intersection twice during red lights while attempting to turn left, driving on the shoulder, and failing to signal lane changes. You review the results with Mr. D and his wife and recommend that he cease driving immediately.
Mr. D is furious, and his wife is dismayed. He demands to know how he can continue to play golf, which is his only form of exercise and recreation. Will she have to give up her bridge club? How will they shop for food? They request permission to at least to drive to the grocery store during the daytime.
You explain that no system allows individuals to drive only at certain times, and for the sake of safety you cannot grant them special permission. You discuss alternatives, such as asking their daughter for assistance with grocery shopping and taking taxis or ride-sharing with friends who play golf and bridge.
Remain firm, but ease the blow
Driving cessation orders distress patients, families, and clinicians. A failed road test clearly indicates unsafe driving, and driving cessation is critical to public safety.
A review by Man-Son-Hing et al23 found that drivers with dementia performed worse than nondemented controls in all studies that examined driving performance (on-road, simulator, or caregiver report). Simulators showed problems such as off-road driving, deviation from posted speed, and more time to negotiate left turns.24
By comparison, only 1 of 3 studies using state crash records showed an increased risk of collisions in persons with dementia compared with controls.23 From a research perspective, however, studies that use state-reported collisions to assess driving risk are confounded by driving restrictions on persons with dementia.
Mr. D wants to continue driving with restrictions. No studies have shown reduced crash rates when drivers with dementia used compensatory strategies such as restrictions, retraining/education, having a passenger “co-pilot,” on-board navigation, or cognitive enhancers.23
If Mr. D had passed the road test, the situation would have been more ambiguous. Two studies have examined on-road driving performance over time in patients with early-stage dementia.25,26 Both studies followed drivers prospectively for 2 years, and those with mild dementia (vs very mild or no dementia) were most likely to show a decline in driving skills:
- All participants with mild dementia were rated as “not safe” by the end of 2 years by Duchek et al.25
- Median time to “failure” (or a rating of unsafe) was 324 days for drivers with mild dementia vs 605 days for those with very mild dementia, as reported by Ott et al.26
Mr. D’s passionate plea for reconsideration highlights the need for communities to develop alternate transportation for seniors whose driving becomes unsafe (Box 2).
Legal liability? Physicians often are concerned about legal responsibilities and risks involved in reporting unsafe drivers. Be aware of local statutes or legislations regarding mandatory reporting of patients you deem unsafe to drive.17 These laws usually protect physicians from lawsuits related to violating patient confidentiality. Civil lawsuits remain possible, however, if clinicians fail to report an unsafe driver who subsequently is involved in a motor vehicle collision.27
1. Meet with family first. Help them assume a positive and supportive role. Explain concretely and empathically your concern for the safety of the patient and others. Clearly outline your findings that the patient is not fit to drive, and explain that the law requires you to report the patient to the authorities.
Remind family members that the goal of driving assessment is to prevent a collision, and they carry some responsibility because they are aware of the potential risk of letting their family member continue to drive. If necessary, have family members witness a repeat performance by the patient on the most revealing test. Discuss the importance of finding alternate transportation to reduce the risk of isolation and depression that can follow driving cessation.
2. Meet with patient. Having the family present can be helpful, but ask them to assume a supportive role. Give the patient a positive role by recognizing that he or she has been a responsible driver, and part of this responsibility is to stop driving before an accident occurs. Acknowledge that it is normal to be unhappy upon learning that one’s driving privileges are being revoked.
Sometimes it helps to give the patient a prescription in their name that says, “Do not drive.” Families who receive a copy may find this very helpful, too, for reminding the patient later about what you said.
If your patient argues with your position, remain firm and do not argue. Indicate that you have made notes on the meeting and are notifying the authorities about the patient’s unsafe driving. You can add that your chart could be subpoenaed and the patient may be legally liable and financially responsible should he or she continue to drive and have a collision.
3. Talk about transportation options. Family members could share driving responsibilities. Taxi rides can cost less than maintaining a car if the patient drives <4,000 km (2,500 miles) per year. Suggest that patients or families find volunteer drivers or contact helpful taxi drivers a day before an outing is planned.
4. If patient refuses to comply, meet with the family again and encourage them to remove the patient’s opportunity to drive (confiscate the keys, disable the car, or remove the car altogether).
Provide a written statement to the patient and family outlining why the patient can no longer drive. Indicate that it is your legal responsibility to report unsafe drivers, and you intend to notify the authorities regarding the patient’s driving status. If the patient remains noncompliant, continue to encourage family to remove the opportunity to drive.
Related resources
- Physician’s guide to assessing and counseling older drivers. American Medical Association and the National Highway Traffic Safety Administration, 2003. www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook.
- Determining medical fitness to operate motor vehicles. Canadian Medical Association. 2006. www.cma.ca/index.cfm/ci_id/18223/la_id/1.htm.
- Canadian Driving Research Initiative for Vehicular Safety in the Elderly (CanDRIVE). www.candrive.ca.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rapoport receives grant/research support from the Canadian Institute of Health Research and the Ontario Neurotrauma Foundation.
Mr. D, age 75, presents to your office with a 5-year history of gradually declining memory. His wife reports he is having difficulty with word finding, managing his finances, and shopping, and he needs supervision when using the stove. Nonetheless, he enjoys playing golf and drives himself to the golf course 3 times a week. He is compliant with his chronic medical therapy for hypertension, hypercholesterolemia, and asthma.
Patients with dementia who continue to drive pose a potential danger on the road, worry their families, and present challenges to clinicians. Most people would agree that patients with moderate or severe dementia should not drive, but a careful evaluation is required to assess whether a patient such as Mr. D with mild dementia remains fit to drive.
This article explores how dementia exacerbates age-related changes in driving ability and discusses how to assess driving in patients with dementia. Our goal is to help clinicians sort through data from in-office physical and cognitive assessments, family caregivers/informants’ reports, and (when available) on-road testing. We also discuss:
- guidelines for assessing older drivers that can help balance patients’ need for autonomy with public safety
- strategies for discussing driving cessation with patients and their families.
Driving: A privilege, not a right
Driving is central to older adults’ autonomy, and >75% of persons age ≥75 rely on driving as their primary mode of transportation.1 Driving cessation in this population has been associated with a 3-fold decrease in out-of-home activity2 and a 2.5-fold increase in depressive symptoms.3 Nonetheless, some 4.5 million Americans have Alzheimer’s disease (AD),4 and dementia poses a substantial risk to safe driving.
Although driving must be sacrificed when personal and public safety is at risk, most physicians perceive an uncomfortable conflict of interest between patient confidentiality and public safety.5 Assessing driving safety of patients with dementia can undermine the doctor-patient relationship and pose hardships for patients.
Mr. D has a 5-year history of memory problems that affect his daily functioning, yet he continues to drive. A longitudinal study of persons with dementia found that among the 29% who were driving at baseline, more than one-half were still behind the wheel 2 years later.6
Age and driving safety. Even in the absence of dementia, driving ability declines with aging (Tables 1 and 2).7,8 Older persons may self-regulate and restrict their driving to shorter distances, with fewer trips at night, on high-speed roads, or in unfamiliar situations. Their driving is rarely aggressive and they are unlikely to speed, but they may drive more slowly than other traffic.7,8 Although the overall rate of motor vehicle collisions declines with age:
- the rate of collisions per mile driven increases after age 659
- drivers age >65 have the highest fatality rate per mile driven among adults age ≥25.10
A dementia diagnosis is not sufficient to withdraw driving privileges, according to American Medical Association (AMA)/National Highway Traffic Safety Administration (NHTSA) guidelines. These recommend that you base decisions on the individual’s driving ability, and—when you have concerns—factor in a focused medical assessment and formal assessment of driving skills.10
Table 1
Age-related changes that may affect driving fitness
| Decreased physical capabilities, including declining muscle tone, flexibility, and reaction time |
| Decreased hearing and visual acuity |
| Increased fragility, resulting in longer time to heal should injuries occur |
| Increased medication use with possible side effect of drowsiness |
| Source: References 7,8 |
Table 2
Older drivers’ common traffic violations leading to crashes*
| Failure to obey traffic signals, including stop signs and red lights |
| Unsafe left turns (driver may inaccurately judge speed of oncoming vehicle) |
| Inappropriate turns (such as difficulty judging distance from oncoming cars, wide or narrow turns, or not timing the turn correctly with traffic lights) |
| Unsafe passing |
| Failure to yield |
| * These errors often lead to multivehicle accidents Source: References 7,8 |
CASE CONTINUED: Cognitive deficits quantified
You perform a Mini-Mental State Examination (MMSE). Mr. D scores 24/30, losing 1 point for orientation, 2 points for attention, 2 points for recall, and 1 point for copying. This score, along with his history, indicates mild dementia, although he claims he is a safe driver. On further cognitive testing, Mr. D completes the Trails A test in 90 seconds and Trails B test in 250 seconds (well below 1.5 standard deviations of the norm for his age and education).11 On the clock-drawing task, he drew a poorly organized clock, with unequal spaces between numbers and hands pointing to “10” and “11” instead of properly indicating “10 after 11.”
Mr. D and his wife live in a rural area, 5 miles from the nearest grocery store. His wife never drove, and she relies on him for weekly shopping trips and to drive her to her bridge club. She denies any problems with his driving but states, “Other drivers have become so aggressive; they’re always honking at him.” Their daughter denies that Mr. D has driving problems but admits that for the last 2 years she has refused to allow her child to ride in his car.
Focused in-office assessment
Information to assess driving ability can come from the patient, family caregiver/informant, and clinical judgment. Patients with dementia are notoriously inaccurate in self-reported driving ability, either for lack of insight or as a testament to the importance of driving to their autonomy. Caregivers often are more accurate in describing a patient’s driving, but other agendas may color their responses.
In a study of patients with very mild or mild AD, 94% reported themselves as safe drivers, whereas on-road driving instructors rated <50% of drivers in these groups as safe. Caregivers were better able to classify driving performance, but 36% of their ratings were incorrect.12
Cognitive assessment. To assess older drivers’ cognition, AMA/NHTSA’s Guide to Assessing and Counseling Older Drivers recommends the Trail-Making Test, Part B and the clock-drawing test.10 The Canadian Medical Association suggests the MMSE.13 Both guides say that abnormalities in these tests indicate a need for more detailed testing, including referral to specialized driving assessment and retesting at regular intervals (Algorithm). Retest patients with mild dementia at least every 6 months or sooner when dementia severity increases noticeably14 (Box 1).6,15
The MMSE is widely used to screen for cognitive impairment and identify dementia or delirium, but it is not a diagnostic tool or proxy driving test. A patient with dementia may produce a high MMSE score and yet be an unsafe driver. For example, well-educated patients or those with vascular or frontotemporal dementia may retain cognitive abilities as measured by the MMSE until later in the disease.
Considerable effort has been put into developing tools to help clinicians quickly and accurately differentiate safe from unsafe drivers by assessing cognition. Unfortunately, no consistent link has been found between cognitive test results and driving outcome measures. A systematic review of office-based predictors of fitness to drive in dementia found 5 studies showing an association between MMSE scores and driving and 5 studies showing no such association.16 Thus, although the AMA/NHTSA guide recommends the MMSE, Trails B, and clock-drawing tests, cognitive tests—including these—are not sufficient to assess driving ability.
Severity of dementia. International consensus groups have attempted to create guidelines for patients with dementia who drive. American, Canadian, and Australian groups suggest that a diagnosis of moderate to severe dementia precludes driving, and the driver’s licenses of persons with these conditions should be revoked.17
In general, AD is considered severe when the MMSE score is <10 or the patient becomes dependent on a caregiver for survival.18 AD of moderate severity is more difficult to define, but a Canadian consensus conference suggested a practical approach: Patients with AD would be considered to have moderate to severe dementia and should not drive when they cannot independently perform multiple instrumental activities of daily living or any of the basic activities of daily living.19
Some dementias may impair driving more quickly than AD does. For example, hallucinations may occur early in Lewy body dementia, as may impulsivity in frontotemporal dementia and motor impairment in vascular dementia.
Mrs. Y visits your office for a follow-up regarding mild Alzheimer’s disease (AD), which was diagnosed 2 years ago. She passed an on-road test 3 months ago and has an Mini-Mental State Examination score of 24/30. Over the last month she has become depressed, with insomnia and mild psychomotor retardation. She occasionally has hallucinations.
Behavioral and psychological symptoms such as agitation, aggression, hallucinations, apathy, depression, and anxiety are common neuropsychiatric sequelae of AD. Little is known about the risks these symptoms pose to road safety, but we recommend that clinicians strongly consider the potential for impaired driving.
In a longitudinal study, cognitive impairment and behavioral disturbances—especially agitation, apathy, and hallucinations—were strong predictors of driving cessation among patients with dementia.6 Furthermore, a case crossover study of patients with dementia found a 54% increase in risk of motor vehicle collisions associated with the use of psychotropic medications.15
Consider all aspects of the patient’s clinical status, including neuropsychiatric symptoms, psychotropic medications, comorbid medical conditions (including hearing and vision impairment), and concomitant therapy for medical conditions. Any could change a safe driver with mild dementia into an unsafe driver.
Algorithm: 3 options for drivers with dementia, based on in-office assessment
* Observe legislation or statutes that address reporting unsafe drivers to the department of motor vehicles or ministry of transportation
On-road driving tests
Because some individuals with mild dementia can drive safely for extended periods, international recommendations for assessing the driver with dementia emphasize on-road driving tests.10,13,20–22 American10 and Canadian guidelines13 suggest that a dementia diagnosis is not sufficient to withdraw licensure.
A formal driving assessment is necessary to establish road safety for patients with mild dementia except when the need for license withdrawal is evident, such as when the patient has:
- a history of major driving problems (such as crashes or driving the wrong way on a highway)
- significant contraindications to driving on the history or physical examination (such as severe inattention or psychosis).
Challenges of on-road testing. On-road tests may be the gold standard, but they are not without clinical problems.
Need to retest. Because almost all dementias are progressive and driving skills deteriorate over time, most guidelines recommend periodic retesting. For patients with dementia who pass on-road evaluations, limited evidence supports retesting every 6 months.14 Take an individual approach, however, because of the various rates at which the dementias progress.
Testing vs real world conditions. Structured on-road testing is not equivalent to unstructured real-world driving, in which the patient often must navigate without instruction or assistance.
Rural vs urban driving. Road tests conducted in urban areas assess skills associated with complex conditions and the need to respond quickly to crises. They might not assess as well rural driving, which requires sustained attention on monotonous roads.
Inaccessibility. Cost and lack of availability of on-road tests, particularly in rural areas, limit the number of patients whose performance can be evaluated.
CASE CONTINUED: Distressing results
Mr. D has a history of decline in cognition and function, objective cognitive difficulties, and a subtle history of driving problems. You refer him for a specialized on-road test, and the report indicates that he failed. Errors included wide turns, driving too slowly, getting caught in an intersection twice during red lights while attempting to turn left, driving on the shoulder, and failing to signal lane changes. You review the results with Mr. D and his wife and recommend that he cease driving immediately.
Mr. D is furious, and his wife is dismayed. He demands to know how he can continue to play golf, which is his only form of exercise and recreation. Will she have to give up her bridge club? How will they shop for food? They request permission to at least to drive to the grocery store during the daytime.
You explain that no system allows individuals to drive only at certain times, and for the sake of safety you cannot grant them special permission. You discuss alternatives, such as asking their daughter for assistance with grocery shopping and taking taxis or ride-sharing with friends who play golf and bridge.
Remain firm, but ease the blow
Driving cessation orders distress patients, families, and clinicians. A failed road test clearly indicates unsafe driving, and driving cessation is critical to public safety.
A review by Man-Son-Hing et al23 found that drivers with dementia performed worse than nondemented controls in all studies that examined driving performance (on-road, simulator, or caregiver report). Simulators showed problems such as off-road driving, deviation from posted speed, and more time to negotiate left turns.24
By comparison, only 1 of 3 studies using state crash records showed an increased risk of collisions in persons with dementia compared with controls.23 From a research perspective, however, studies that use state-reported collisions to assess driving risk are confounded by driving restrictions on persons with dementia.
Mr. D wants to continue driving with restrictions. No studies have shown reduced crash rates when drivers with dementia used compensatory strategies such as restrictions, retraining/education, having a passenger “co-pilot,” on-board navigation, or cognitive enhancers.23
If Mr. D had passed the road test, the situation would have been more ambiguous. Two studies have examined on-road driving performance over time in patients with early-stage dementia.25,26 Both studies followed drivers prospectively for 2 years, and those with mild dementia (vs very mild or no dementia) were most likely to show a decline in driving skills:
- All participants with mild dementia were rated as “not safe” by the end of 2 years by Duchek et al.25
- Median time to “failure” (or a rating of unsafe) was 324 days for drivers with mild dementia vs 605 days for those with very mild dementia, as reported by Ott et al.26
Mr. D’s passionate plea for reconsideration highlights the need for communities to develop alternate transportation for seniors whose driving becomes unsafe (Box 2).
Legal liability? Physicians often are concerned about legal responsibilities and risks involved in reporting unsafe drivers. Be aware of local statutes or legislations regarding mandatory reporting of patients you deem unsafe to drive.17 These laws usually protect physicians from lawsuits related to violating patient confidentiality. Civil lawsuits remain possible, however, if clinicians fail to report an unsafe driver who subsequently is involved in a motor vehicle collision.27
1. Meet with family first. Help them assume a positive and supportive role. Explain concretely and empathically your concern for the safety of the patient and others. Clearly outline your findings that the patient is not fit to drive, and explain that the law requires you to report the patient to the authorities.
Remind family members that the goal of driving assessment is to prevent a collision, and they carry some responsibility because they are aware of the potential risk of letting their family member continue to drive. If necessary, have family members witness a repeat performance by the patient on the most revealing test. Discuss the importance of finding alternate transportation to reduce the risk of isolation and depression that can follow driving cessation.
2. Meet with patient. Having the family present can be helpful, but ask them to assume a supportive role. Give the patient a positive role by recognizing that he or she has been a responsible driver, and part of this responsibility is to stop driving before an accident occurs. Acknowledge that it is normal to be unhappy upon learning that one’s driving privileges are being revoked.
Sometimes it helps to give the patient a prescription in their name that says, “Do not drive.” Families who receive a copy may find this very helpful, too, for reminding the patient later about what you said.
If your patient argues with your position, remain firm and do not argue. Indicate that you have made notes on the meeting and are notifying the authorities about the patient’s unsafe driving. You can add that your chart could be subpoenaed and the patient may be legally liable and financially responsible should he or she continue to drive and have a collision.
3. Talk about transportation options. Family members could share driving responsibilities. Taxi rides can cost less than maintaining a car if the patient drives <4,000 km (2,500 miles) per year. Suggest that patients or families find volunteer drivers or contact helpful taxi drivers a day before an outing is planned.
4. If patient refuses to comply, meet with the family again and encourage them to remove the patient’s opportunity to drive (confiscate the keys, disable the car, or remove the car altogether).
Provide a written statement to the patient and family outlining why the patient can no longer drive. Indicate that it is your legal responsibility to report unsafe drivers, and you intend to notify the authorities regarding the patient’s driving status. If the patient remains noncompliant, continue to encourage family to remove the opportunity to drive.
Related resources
- Physician’s guide to assessing and counseling older drivers. American Medical Association and the National Highway Traffic Safety Administration, 2003. www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook.
- Determining medical fitness to operate motor vehicles. Canadian Medical Association. 2006. www.cma.ca/index.cfm/ci_id/18223/la_id/1.htm.
- Canadian Driving Research Initiative for Vehicular Safety in the Elderly (CanDRIVE). www.candrive.ca.
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Rapoport receives grant/research support from the Canadian Institute of Health Research and the Ontario Neurotrauma Foundation.
1. Stowell-Ritter A, Straight A, Evans EL. Understanding senior transportation: report and analysis of a survey of consumers age 50+. Washington, DC: American Association of Retired Persons; 2002.
2. Marottoli RA, de Leon CFM, Glass TA, et al. Consequences of driving cessation: decreased out-of-home activity levels. J Gerontol B Psychol Sci Soc Sci 2000;55(6):S334-40.
3. Marottoli RA, Mendes de Leon CF, Glass TA, et al. Driving cessation and increased depressive symptoms: prospective evidence from the New Haven EPESE. Established Populations for Epidemiologic Studies of the Elderly. J Am Geriatr Soc 1997;45(2):202-6.
4. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003;60(8):1119-22.
5. Jang RW, Man-Son-Hing M, Molnar FJ, et al. Family physicians’ attitudes and practices regarding assessments of medical fitness to drive in older persons. J Gen Intern Med 2007;22(4):531-43.
6. Herrmann N, Rapoport MJ, Sambrook R, et al. Predictors of driving cessation in mild-to-moderate dementia. CMAJ 2006;175(6):591-5.
7. Hedlund J. Countermeasures that work: a highway safety countermeasure guide for state highway safety offices. Washington, DC: National Highway Traffic Safety Administration; 2006.
8. Dobbs B. Medical conditions and driving: a review of the literature (1960-2000). Washington, DC: National Highway Traffic Safety Administration; 2005. Available at: http://www.nhtsa.dot.gov/people/injury/research/Medical_Condition_Driving/pages/TRD.html. Accessed September 29, 2008.
9. Li G, Braver ER, Chen LH. Fragility versus excessive crash involvement as determinants of high death rates per vehicle-mile of travel among older drivers. Accid Anal Prev 2003;35(2):227-35.
10. Physician’s guide to assessing and counseling older drivers. Washington, DC: National Highway Traffic Safety Administration; 2003. Available at: http://www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook. Accessed September 29, 2008.
11. Yeudall LT, Reddon JR, Gill DM, et al. Normative data for the Halstead-Reitan neuropsychological tests stratified by age and sex. J Clin Psychol 1987;43(3):346-67.
12. Brown LB, Ott BR, Papandonatos GD, et al. Prediction of on-road driving performance in patients with early Alzheimer’s disease. J Am Geriatr Soc 2005;53(1):94-8.
13. Determining medical fitness to operate motor vehicles. CMA driver’s guide. 7th ed. Ottawa, Ontario, Canada: Canadian Medical Association; 2006.
14. Molnar FJ, Patel A, Marshall SC, et al. Systematic review of the optimal frequency of follow-up in persons with mild dementia who continue to drive. Alzheimer Dis Assoc Disord 2006;20(4):295-7.
15. Rapoport MJ, Herrmann N, Molnar FJ, et al. Psychotropic medications and motor vehicle collisions in patients with dementia (letter). J Am Geriatr Soc 2008;56(10):1968-70.
16. Molnar FJ, Patel A, Marshall SC, et al. Clinical utility of office-based cognitive predictors of fitness to drive in persons with dementia: a systematic review. J Am Geriatr Soc 2006;54(12):1809-24.
17. Rapoport MJ, Herrmann N, Molnar FJ, et al. Sharing the responsibility for assessing the risk of the driver with dementia. CMAJ. 2007;177(6):599-601.
18. Herrmann N, Gauthier S, Lysy PG. Clinical practice guidelines for severe Alzheimer’s disease. Alzheimers Dement 2007;3(4):385-97.
19. Hogan DB, Bailey P, Carswell A, et al. Management of mild to moderate Alzheimer’s disease and dementia. Alzheimers Dement 2007;3(4):355-84.
19. Assessing fitness to drive for commercial and private vehicle drivers. Sydney, Australia: National Library of Australia; 2006. Available at: http://www.austroads.com.au/aftd/index.html. Accessed November 4, 2008.
21. Medical aspects of fitness to drive. A guide for medical practitioners. Wellington, New Zealand: Land Transport Safety Authority; 2002. Available at: http://www.transfund.govt.nz/licensing/docs/ltsa-medical-aspects.pdf. Accessed September 29, 2008.
22. At a glance guide to the current medical standards of fitness to drive. Swansea, UK: Drivers Medical Group, Driver and Vehicle Licensing Agency; 2008. Available at: http://www.dvla.gov.uk/medical/ataglance.aspx. Accessed September 29, 2008.
23. Man-Son-Hing M, Marshall SC, Molnar FJ, Wilson KG. Systematic review of driving risk and the efficacy of compensatory strategies in persons with dementia. J Am Geriatr Soc 2007;55(6):878-84.
24. Cox DJ, Quillian WC, Thorndike FP, et al. Evaluating driving performance of outpatients with Alzheimer disease. J Am Board Fam Pract 1998;11(4):264-71.
25. Duchek JM, Carr DB, Hunt L, et al. Longitudinal driving performance in early-stage dementia of the Alzheimer type. J Am Geriatr Soc 2003;51(10):1342-7.
26. Ott BR, Heindel WC, Papandonatos GD, et al. A longitudinal study of drivers with Alzheimer disease. Neurology 2008;70(14):1171-8.
27. Molnar FJ, Byszewski AM, Marshall SC, Man-Son-Hing M. In-office evaluation of medical fitness to drive: practical approaches for assessing older people. Can Fam Physician 2005;51:372-9.
1. Stowell-Ritter A, Straight A, Evans EL. Understanding senior transportation: report and analysis of a survey of consumers age 50+. Washington, DC: American Association of Retired Persons; 2002.
2. Marottoli RA, de Leon CFM, Glass TA, et al. Consequences of driving cessation: decreased out-of-home activity levels. J Gerontol B Psychol Sci Soc Sci 2000;55(6):S334-40.
3. Marottoli RA, Mendes de Leon CF, Glass TA, et al. Driving cessation and increased depressive symptoms: prospective evidence from the New Haven EPESE. Established Populations for Epidemiologic Studies of the Elderly. J Am Geriatr Soc 1997;45(2):202-6.
4. Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 2003;60(8):1119-22.
5. Jang RW, Man-Son-Hing M, Molnar FJ, et al. Family physicians’ attitudes and practices regarding assessments of medical fitness to drive in older persons. J Gen Intern Med 2007;22(4):531-43.
6. Herrmann N, Rapoport MJ, Sambrook R, et al. Predictors of driving cessation in mild-to-moderate dementia. CMAJ 2006;175(6):591-5.
7. Hedlund J. Countermeasures that work: a highway safety countermeasure guide for state highway safety offices. Washington, DC: National Highway Traffic Safety Administration; 2006.
8. Dobbs B. Medical conditions and driving: a review of the literature (1960-2000). Washington, DC: National Highway Traffic Safety Administration; 2005. Available at: http://www.nhtsa.dot.gov/people/injury/research/Medical_Condition_Driving/pages/TRD.html. Accessed September 29, 2008.
9. Li G, Braver ER, Chen LH. Fragility versus excessive crash involvement as determinants of high death rates per vehicle-mile of travel among older drivers. Accid Anal Prev 2003;35(2):227-35.
10. Physician’s guide to assessing and counseling older drivers. Washington, DC: National Highway Traffic Safety Administration; 2003. Available at: http://www.nhtsa.dot.gov/people/injury/olddrive/OlderDriversBook. Accessed September 29, 2008.
11. Yeudall LT, Reddon JR, Gill DM, et al. Normative data for the Halstead-Reitan neuropsychological tests stratified by age and sex. J Clin Psychol 1987;43(3):346-67.
12. Brown LB, Ott BR, Papandonatos GD, et al. Prediction of on-road driving performance in patients with early Alzheimer’s disease. J Am Geriatr Soc 2005;53(1):94-8.
13. Determining medical fitness to operate motor vehicles. CMA driver’s guide. 7th ed. Ottawa, Ontario, Canada: Canadian Medical Association; 2006.
14. Molnar FJ, Patel A, Marshall SC, et al. Systematic review of the optimal frequency of follow-up in persons with mild dementia who continue to drive. Alzheimer Dis Assoc Disord 2006;20(4):295-7.
15. Rapoport MJ, Herrmann N, Molnar FJ, et al. Psychotropic medications and motor vehicle collisions in patients with dementia (letter). J Am Geriatr Soc 2008;56(10):1968-70.
16. Molnar FJ, Patel A, Marshall SC, et al. Clinical utility of office-based cognitive predictors of fitness to drive in persons with dementia: a systematic review. J Am Geriatr Soc 2006;54(12):1809-24.
17. Rapoport MJ, Herrmann N, Molnar FJ, et al. Sharing the responsibility for assessing the risk of the driver with dementia. CMAJ. 2007;177(6):599-601.
18. Herrmann N, Gauthier S, Lysy PG. Clinical practice guidelines for severe Alzheimer’s disease. Alzheimers Dement 2007;3(4):385-97.
19. Hogan DB, Bailey P, Carswell A, et al. Management of mild to moderate Alzheimer’s disease and dementia. Alzheimers Dement 2007;3(4):355-84.
19. Assessing fitness to drive for commercial and private vehicle drivers. Sydney, Australia: National Library of Australia; 2006. Available at: http://www.austroads.com.au/aftd/index.html. Accessed November 4, 2008.
21. Medical aspects of fitness to drive. A guide for medical practitioners. Wellington, New Zealand: Land Transport Safety Authority; 2002. Available at: http://www.transfund.govt.nz/licensing/docs/ltsa-medical-aspects.pdf. Accessed September 29, 2008.
22. At a glance guide to the current medical standards of fitness to drive. Swansea, UK: Drivers Medical Group, Driver and Vehicle Licensing Agency; 2008. Available at: http://www.dvla.gov.uk/medical/ataglance.aspx. Accessed September 29, 2008.
23. Man-Son-Hing M, Marshall SC, Molnar FJ, Wilson KG. Systematic review of driving risk and the efficacy of compensatory strategies in persons with dementia. J Am Geriatr Soc 2007;55(6):878-84.
24. Cox DJ, Quillian WC, Thorndike FP, et al. Evaluating driving performance of outpatients with Alzheimer disease. J Am Board Fam Pract 1998;11(4):264-71.
25. Duchek JM, Carr DB, Hunt L, et al. Longitudinal driving performance in early-stage dementia of the Alzheimer type. J Am Geriatr Soc 2003;51(10):1342-7.
26. Ott BR, Heindel WC, Papandonatos GD, et al. A longitudinal study of drivers with Alzheimer disease. Neurology 2008;70(14):1171-8.
27. Molnar FJ, Byszewski AM, Marshall SC, Man-Son-Hing M. In-office evaluation of medical fitness to drive: practical approaches for assessing older people. Can Fam Physician 2005;51:372-9.
Words to the wise: 4 secrets of successful pharmacotherapy
Any medication’s therapeutic success depends on the interaction between its specific biochemical effects and nonspecific factors.1 Thus, clinical trial designers may view the placebo effect as undesirable, but it can be a valuable response that improves treatment outcomes in clinical practice. As Freud stated, “Expectation colored by hope and faith is an effective force with which we have to reckon…in all our attempts at treatment and cure.”2
This article describes how experienced clinicians make use of the placebo effect and 3 other powerful, nonspecific elements of successful pharmacotherapy.
The placebo effect
The placebo effect is any effect attributable to a pill or potion that does not originate from its specific pharmacologic properties.3 Its clinical value has been trivialized, in part because of misconceptions (Table 1). For example, the placebo effect is commonly believed to be short-lived, whereas in fact it can last a long time.4
In clinical practice, our goal is to enhance the placebo effect to maximize a desirable therapeutic outcome (Table 2).5 Therefore, before I prescribe a medication, I tell my patient that I have selected a particular medication because I have had good results with it in many other patients and I believe it will work well for him or her, too.
Too often, doctors feel pessimistic about a medication’s potential therapeutic result and communicate this pessimism. What the patient hears is, “There’s nothing else I can do for you; why not try this medication, even though I don’t believe it’s going to work.” This may create a negative placebo effect6—termed the “nocebo” effect—which gives the patient a negative expectation about the treatment’s outcome. The patient internalizes the doctor’s words and lives out this negative expectation.
Table 1
Correcting misconceptions about the placebo effect
| Misconception | What the evidence shows |
|---|---|
| Placebo effects are short-lived | The placebo effect has been documented to last for a long time |
| Only complaints that are psychologically originated respond to placebo | Changes after placebo have been documented for most symptoms, including those originating from somatic diseases |
| Placebo responders are distinctly different from nonresponders | There is no difference between placebo responders and nonresponders |
| The placebo effect is only about one-third of the total therapeutic effect | The placebo effect can be up to 100% of the total therapeutic effect |
| Only about one-third of the population responds to placebo | The placebo response is context-dependent and may include >90% of the patient population |
| Source: Reference 4 | |
Clinical strategies to enhance the placebo effect
| |
| Source: Reference 5 | |
CASE REPORT: Predicting positive results
Mr. B, age 42, has a history of recurrent depression associated with severe insomnia, poor appetite, significant weight loss, and psychosocial withdrawal with feelings of hopelessness. After I take a detailed history and do a mental status examination, I suggest that he be treated with cognitive-behavioral therapy (CBT) and mirtazapine.
Even though studies of antidepressants rarely show mood improvements within the first 7 days, it is not unusual to hear patients report feeling less depressed within days after they start a new antidepressant. Although the drug’s specific chemical effects on the brain may not be sufficient to explain this phenomenon, the explanation probably lies in nonspecific effects—such as the patient expecting that this medication will make him feel better.
The placebo effect can occur as soon as a patient starts a medication. Experienced clinicians understand the placebo effect’s power and harness it to benefit their patients.
Conditioned responses
Many biological responses can be associated with visual, auditory, tactile, olfactory, or gustatory stimuli. Nonconditioned physiologic responses paired with conditioned stimuli induce the same biological effects of a drug. Evidence supporting this phenomenon includes successful conditioning of the immune system.7-10 Conditioned responses—as demonstrated in glycemia regulation10 and with psychopharmacology11—also can enhance the desirable results of pharmacotherapy.
CASE REPORT: A soothing drink
Ms. L, a 22-year-old college student, suffers from obsessive-compulsive disorder associated with anxiety and depression. She arrives at the appointment hurried and worried that she might be late. She is short of breath and looks stressed. The nurse offers Ms. L a cup of tea or water. She chooses a glass of water and is asked to bring it into her session.
Following a comprehensive interview and mental status examination, I recommend CBT plus medication. Considering Ms. L’s medication history, we agree to start treatment with sertraline. We review its potential benefits and expectations that it will reduce her anxiety, alleviate her ruminating obsessive worries, and improve her mood. I give her a 50-mg sample and inform her that some patients experience positive effects soon after taking the medication. I then ask her to take the first pill, using her glass of water. She does so and thanks me for being attentive to her needs.
I instruct her to call within 1 week and report on her condition, even if she feels better. Seven days later she reports that she is feeling better and is looking forward to her next appointment. She reports no side effects.
Often patients come to my office feeling thirsty. My staff or I offer them a glass of water or a cup of tea. As patients sip from the cup, they swallow and incorporate the liquid into their bodies. At the same time, I use verbal interventions to make them feel listened to and understood. They internalize this emotional experience in connection with swallowing the liquid.
Later, when swallowing the new medication as instructed, the patient re-experiences the positive therapeutic effect that was internalized in the doctor’s office.
The power of suggestion
The power of suggestion has been shown to positively or negatively affect treatment outcomes.12,13 In practice, most clinicians give unintentional suggestions by how and what they communicate to the patient.
CASE REPORT: Predicting improvement
Mrs. J, age 48, has had dysthymic disorder and fibromyalgia for many years. She describes how various specialists have tried to alleviate her depression and chronic pain. Follow-up questions reveal that whenever she received a new prescription the physician would alert her to all the possible side effects and instruct her to call the office if she developed a problem with the new medication.
Invariably, Mrs. J would call as instructed and describe side effects she developed with the new medication. Often the doctor would discontinue the medication, depriving Mrs. J of benefits she might have derived later.
When Mrs. J calls to report on her status, she mentions that she is sleeping better and has begun to feel better during the day. She says that her husband told her she has started to smile again.
This vignette illustrates the importance of suggesting to the patient a positive outcome of pharmacotherapy associated with a particular action (calling the doctor’s office to report results). When the patient promised to call, she internalized the suggestion that calling would be associated with feeling better—and that is what happened. This intervention contrasts with saying to the patient, “Call me if you have a problem with any of these side effects,” which gives the patient a suggestion to call and report a problem.
The suggestion effect also can be used to reframe a predictable side effect as a positive sign that indicates the beginning of change leading to recovery (Box).
Ms. M, age 32 and single, has an anxiety disorder associated with bipolar depression. She has discontinued several psychotropics because of uncomfortable side effects, such as constipation.
After taking a detailed history, I decide to prescribe quetiapine. I tell Ms. M about this medication’s potential benefits and side effects. One common side effect is dry mouth, which often occurs before patients experience therapeutic effects.
I inform Ms. M that a dry mouth will be her sign that the medication has begun to work, and beneficial effects—such as improved sleep, reduced anxiety, and improved mood—will soon follow. I then instruct her to call my office and report when she experiences a dry mouth.
Discussion. In pharmacotherapy, side effects may appear before patients experience a medication’s beneficial/therapeutic effects. Patients’ initial experience often determines whether or not they will continue taking a prescribed medication. I know Ms. M may stop taking quetiapine—as she has done with other medications—if she initially has uncomfortable side effects.
Instructing patients to expect a specific side effect (such as a dry mouth with quetiapine) and associating it with a future therapeutic benefit sets up a road map of expectations. They know their experience is compatible with the doctor’s predictions. For Ms. M, I reframed the side effect as a positive sign that recovery has begun, with more positive changes to come.
Participatory pharmacotherapy
Many patients seek ownership in making decisions about their treatment and medications. In participatory pharmacotherapy, patients provide you with data and valuable information—such as family history, personal medical history, and experience with treatment—and inform you about which medications worked best and which did not work. You invite patients to predict how they see themselves getting better and into recovery.
CASE REPORT: All in the family
Mr. A, age 28 and single, has been diagnosed with a bipolar mood disorder. As part of a detailed family history, he reports that his maternal grandfather, mother, and a maternal uncle were diagnosed with mood swings and were successfully treated with medications, specifically lithium. He states that he believes he has the same condition.
I compliment Mr. A for being so well informed about his grandfather and uncle and educate him about mood stabilizers’ benefits in bipolar disorder. I tell him about the finding that if lithium has helped his relatives, it will probably help him as well.
I also reassure Mr. A that, in deciding what medications to avoid and what medications to use, I will consider his experience with specific antidepressants that did not help him. He thanks me for considering his suggestion about what medication to use for him.
- a successful therapeutic alliance
- adherence with prescribed medications
- the best possible outcome with pharmacotherapy.
In patients with a defiant-oppositional personality, consider framing the treatment decision as a choice between 2 equally efficacious medications. This gives the patient the sense of control in choosing his or her own medication, which is jointly monitored.
Table 3
Choosing patients for participatory pharmacotherapy
| Good candidates | Exclusionary qualities |
|---|---|
| Adults | Children, adolescents, and prison inmates |
| No history of alcoholism or drug addiction | Alcohol dependence or drug addiction |
| Average and above intelligence | Below-average intelligence |
| Intact cognitive function | Cognitive deficits, such as dementia |
| Not psychotic | Actively psychotic |
| Good comprehension of diagnosis and treatment | Poor comprehension of diagnosis and treatment |
| Therapeutic alliance is present | Therapeutic alliance is absent |
| Personality style or disorder with a need to be in control of treatment, such as obsessive-compulsive personality | Passive, dependent personality style or disorder; these patients may view a participatory approach as the doctor’s lack of confidence |
- Brody H. The placebo response: how you can release the body’s inner pharmacy for better health. New York, NY: HarperCollins Publishers; 2000.
- Spiro H. The power of hope: a doctor’s perspective. New Haven, CT: Yale University Press; 1998.
- Ernst E. Placebo: new insights into an old enigma. Drug Discov Today 2007;12:413-8.
- Lithium • Eskalith, Lithobid
- Mirtazapine • Remeron
- Quetiapine • Seroquel
- Sertraline • Zoloft
Dr. Torem reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Frank JD, Frank JB. Persuasion and healing. Baltimore, MD: The Johns Hopkins University Press; 1991.
2. Freud S. The complete psychological works of Sigmund Freud. Strachey J, trans-ed. Toronto, Ontario, Canada: Hogarth Press; 1953.
3. Wolf S. The pharmacology of placebos. Pharmacol Rev 1959;11:689-704.
4. Ernst E. Placebo: new insights into an old enigma. Drug Discov Today 2007;12:413-8.
5. Brody H. The placebo response: recent research and implications for family medicine. J Fam Pract 2000;49:649-54.
6. Spiegel H. Nocebo, the power of suggestibility. Prev Med 1997;26:616-21.
7. Ader R, Cohen N. Behaviorally conditioned immunosuppression and murine systemic lupus erythematosus. Science 1982;215:1534-6.
8. Ader R. The role of conditioning in pharmacotherapy. In: Harrington A, ed. The placebo effect: an interdisciplinary exploration. Cambridge, MA: Harvard University Press; 1997;138-65.
9. Olness K, Ader R. Conditioning as an adjunct in the pharmacotherapy of lupus erythematosus. J Dev Behav Pediatr 1992;13:124-5.
10. Stockhorst U, Mahl N, Krueger M, et al. Classical conditioning and conditionability of insulin and glucose effect in healthy humans. Physiol Behav 2004;81:375-88.
11. Wolf S. Effect of suggestion and conditioning on the action of chemical agents in human subjects—the pharmacology of placebos. J Clin Invest 1950;29:100-9.
12. Lown B. The verbal conditioning of angina pectoris during exercise testing. Am J Cardiol 1977;40:630-4.
13. Lown B. Introduction. In: Cousins N. The healing heart. New York, NY: W.W. Norton; 1983:11-28.
14. Watzlawick P. If you desire to see, learn how to act. In: Nardone G, Watzlawick P, eds. The art of change San Francisco, CA: Jossey-Bass; 1993:1-16.
Any medication’s therapeutic success depends on the interaction between its specific biochemical effects and nonspecific factors.1 Thus, clinical trial designers may view the placebo effect as undesirable, but it can be a valuable response that improves treatment outcomes in clinical practice. As Freud stated, “Expectation colored by hope and faith is an effective force with which we have to reckon…in all our attempts at treatment and cure.”2
This article describes how experienced clinicians make use of the placebo effect and 3 other powerful, nonspecific elements of successful pharmacotherapy.
The placebo effect
The placebo effect is any effect attributable to a pill or potion that does not originate from its specific pharmacologic properties.3 Its clinical value has been trivialized, in part because of misconceptions (Table 1). For example, the placebo effect is commonly believed to be short-lived, whereas in fact it can last a long time.4
In clinical practice, our goal is to enhance the placebo effect to maximize a desirable therapeutic outcome (Table 2).5 Therefore, before I prescribe a medication, I tell my patient that I have selected a particular medication because I have had good results with it in many other patients and I believe it will work well for him or her, too.
Too often, doctors feel pessimistic about a medication’s potential therapeutic result and communicate this pessimism. What the patient hears is, “There’s nothing else I can do for you; why not try this medication, even though I don’t believe it’s going to work.” This may create a negative placebo effect6—termed the “nocebo” effect—which gives the patient a negative expectation about the treatment’s outcome. The patient internalizes the doctor’s words and lives out this negative expectation.
Table 1
Correcting misconceptions about the placebo effect
| Misconception | What the evidence shows |
|---|---|
| Placebo effects are short-lived | The placebo effect has been documented to last for a long time |
| Only complaints that are psychologically originated respond to placebo | Changes after placebo have been documented for most symptoms, including those originating from somatic diseases |
| Placebo responders are distinctly different from nonresponders | There is no difference between placebo responders and nonresponders |
| The placebo effect is only about one-third of the total therapeutic effect | The placebo effect can be up to 100% of the total therapeutic effect |
| Only about one-third of the population responds to placebo | The placebo response is context-dependent and may include >90% of the patient population |
| Source: Reference 4 | |
Clinical strategies to enhance the placebo effect
| |
| Source: Reference 5 | |
CASE REPORT: Predicting positive results
Mr. B, age 42, has a history of recurrent depression associated with severe insomnia, poor appetite, significant weight loss, and psychosocial withdrawal with feelings of hopelessness. After I take a detailed history and do a mental status examination, I suggest that he be treated with cognitive-behavioral therapy (CBT) and mirtazapine.
Even though studies of antidepressants rarely show mood improvements within the first 7 days, it is not unusual to hear patients report feeling less depressed within days after they start a new antidepressant. Although the drug’s specific chemical effects on the brain may not be sufficient to explain this phenomenon, the explanation probably lies in nonspecific effects—such as the patient expecting that this medication will make him feel better.
The placebo effect can occur as soon as a patient starts a medication. Experienced clinicians understand the placebo effect’s power and harness it to benefit their patients.
Conditioned responses
Many biological responses can be associated with visual, auditory, tactile, olfactory, or gustatory stimuli. Nonconditioned physiologic responses paired with conditioned stimuli induce the same biological effects of a drug. Evidence supporting this phenomenon includes successful conditioning of the immune system.7-10 Conditioned responses—as demonstrated in glycemia regulation10 and with psychopharmacology11—also can enhance the desirable results of pharmacotherapy.
CASE REPORT: A soothing drink
Ms. L, a 22-year-old college student, suffers from obsessive-compulsive disorder associated with anxiety and depression. She arrives at the appointment hurried and worried that she might be late. She is short of breath and looks stressed. The nurse offers Ms. L a cup of tea or water. She chooses a glass of water and is asked to bring it into her session.
Following a comprehensive interview and mental status examination, I recommend CBT plus medication. Considering Ms. L’s medication history, we agree to start treatment with sertraline. We review its potential benefits and expectations that it will reduce her anxiety, alleviate her ruminating obsessive worries, and improve her mood. I give her a 50-mg sample and inform her that some patients experience positive effects soon after taking the medication. I then ask her to take the first pill, using her glass of water. She does so and thanks me for being attentive to her needs.
I instruct her to call within 1 week and report on her condition, even if she feels better. Seven days later she reports that she is feeling better and is looking forward to her next appointment. She reports no side effects.
Often patients come to my office feeling thirsty. My staff or I offer them a glass of water or a cup of tea. As patients sip from the cup, they swallow and incorporate the liquid into their bodies. At the same time, I use verbal interventions to make them feel listened to and understood. They internalize this emotional experience in connection with swallowing the liquid.
Later, when swallowing the new medication as instructed, the patient re-experiences the positive therapeutic effect that was internalized in the doctor’s office.
The power of suggestion
The power of suggestion has been shown to positively or negatively affect treatment outcomes.12,13 In practice, most clinicians give unintentional suggestions by how and what they communicate to the patient.
CASE REPORT: Predicting improvement
Mrs. J, age 48, has had dysthymic disorder and fibromyalgia for many years. She describes how various specialists have tried to alleviate her depression and chronic pain. Follow-up questions reveal that whenever she received a new prescription the physician would alert her to all the possible side effects and instruct her to call the office if she developed a problem with the new medication.
Invariably, Mrs. J would call as instructed and describe side effects she developed with the new medication. Often the doctor would discontinue the medication, depriving Mrs. J of benefits she might have derived later.
When Mrs. J calls to report on her status, she mentions that she is sleeping better and has begun to feel better during the day. She says that her husband told her she has started to smile again.
This vignette illustrates the importance of suggesting to the patient a positive outcome of pharmacotherapy associated with a particular action (calling the doctor’s office to report results). When the patient promised to call, she internalized the suggestion that calling would be associated with feeling better—and that is what happened. This intervention contrasts with saying to the patient, “Call me if you have a problem with any of these side effects,” which gives the patient a suggestion to call and report a problem.
The suggestion effect also can be used to reframe a predictable side effect as a positive sign that indicates the beginning of change leading to recovery (Box).
Ms. M, age 32 and single, has an anxiety disorder associated with bipolar depression. She has discontinued several psychotropics because of uncomfortable side effects, such as constipation.
After taking a detailed history, I decide to prescribe quetiapine. I tell Ms. M about this medication’s potential benefits and side effects. One common side effect is dry mouth, which often occurs before patients experience therapeutic effects.
I inform Ms. M that a dry mouth will be her sign that the medication has begun to work, and beneficial effects—such as improved sleep, reduced anxiety, and improved mood—will soon follow. I then instruct her to call my office and report when she experiences a dry mouth.
Discussion. In pharmacotherapy, side effects may appear before patients experience a medication’s beneficial/therapeutic effects. Patients’ initial experience often determines whether or not they will continue taking a prescribed medication. I know Ms. M may stop taking quetiapine—as she has done with other medications—if she initially has uncomfortable side effects.
Instructing patients to expect a specific side effect (such as a dry mouth with quetiapine) and associating it with a future therapeutic benefit sets up a road map of expectations. They know their experience is compatible with the doctor’s predictions. For Ms. M, I reframed the side effect as a positive sign that recovery has begun, with more positive changes to come.
Participatory pharmacotherapy
Many patients seek ownership in making decisions about their treatment and medications. In participatory pharmacotherapy, patients provide you with data and valuable information—such as family history, personal medical history, and experience with treatment—and inform you about which medications worked best and which did not work. You invite patients to predict how they see themselves getting better and into recovery.
CASE REPORT: All in the family
Mr. A, age 28 and single, has been diagnosed with a bipolar mood disorder. As part of a detailed family history, he reports that his maternal grandfather, mother, and a maternal uncle were diagnosed with mood swings and were successfully treated with medications, specifically lithium. He states that he believes he has the same condition.
I compliment Mr. A for being so well informed about his grandfather and uncle and educate him about mood stabilizers’ benefits in bipolar disorder. I tell him about the finding that if lithium has helped his relatives, it will probably help him as well.
I also reassure Mr. A that, in deciding what medications to avoid and what medications to use, I will consider his experience with specific antidepressants that did not help him. He thanks me for considering his suggestion about what medication to use for him.
- a successful therapeutic alliance
- adherence with prescribed medications
- the best possible outcome with pharmacotherapy.
In patients with a defiant-oppositional personality, consider framing the treatment decision as a choice between 2 equally efficacious medications. This gives the patient the sense of control in choosing his or her own medication, which is jointly monitored.
Table 3
Choosing patients for participatory pharmacotherapy
| Good candidates | Exclusionary qualities |
|---|---|
| Adults | Children, adolescents, and prison inmates |
| No history of alcoholism or drug addiction | Alcohol dependence or drug addiction |
| Average and above intelligence | Below-average intelligence |
| Intact cognitive function | Cognitive deficits, such as dementia |
| Not psychotic | Actively psychotic |
| Good comprehension of diagnosis and treatment | Poor comprehension of diagnosis and treatment |
| Therapeutic alliance is present | Therapeutic alliance is absent |
| Personality style or disorder with a need to be in control of treatment, such as obsessive-compulsive personality | Passive, dependent personality style or disorder; these patients may view a participatory approach as the doctor’s lack of confidence |
- Brody H. The placebo response: how you can release the body’s inner pharmacy for better health. New York, NY: HarperCollins Publishers; 2000.
- Spiro H. The power of hope: a doctor’s perspective. New Haven, CT: Yale University Press; 1998.
- Ernst E. Placebo: new insights into an old enigma. Drug Discov Today 2007;12:413-8.
- Lithium • Eskalith, Lithobid
- Mirtazapine • Remeron
- Quetiapine • Seroquel
- Sertraline • Zoloft
Dr. Torem reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Any medication’s therapeutic success depends on the interaction between its specific biochemical effects and nonspecific factors.1 Thus, clinical trial designers may view the placebo effect as undesirable, but it can be a valuable response that improves treatment outcomes in clinical practice. As Freud stated, “Expectation colored by hope and faith is an effective force with which we have to reckon…in all our attempts at treatment and cure.”2
This article describes how experienced clinicians make use of the placebo effect and 3 other powerful, nonspecific elements of successful pharmacotherapy.
The placebo effect
The placebo effect is any effect attributable to a pill or potion that does not originate from its specific pharmacologic properties.3 Its clinical value has been trivialized, in part because of misconceptions (Table 1). For example, the placebo effect is commonly believed to be short-lived, whereas in fact it can last a long time.4
In clinical practice, our goal is to enhance the placebo effect to maximize a desirable therapeutic outcome (Table 2).5 Therefore, before I prescribe a medication, I tell my patient that I have selected a particular medication because I have had good results with it in many other patients and I believe it will work well for him or her, too.
Too often, doctors feel pessimistic about a medication’s potential therapeutic result and communicate this pessimism. What the patient hears is, “There’s nothing else I can do for you; why not try this medication, even though I don’t believe it’s going to work.” This may create a negative placebo effect6—termed the “nocebo” effect—which gives the patient a negative expectation about the treatment’s outcome. The patient internalizes the doctor’s words and lives out this negative expectation.
Table 1
Correcting misconceptions about the placebo effect
| Misconception | What the evidence shows |
|---|---|
| Placebo effects are short-lived | The placebo effect has been documented to last for a long time |
| Only complaints that are psychologically originated respond to placebo | Changes after placebo have been documented for most symptoms, including those originating from somatic diseases |
| Placebo responders are distinctly different from nonresponders | There is no difference between placebo responders and nonresponders |
| The placebo effect is only about one-third of the total therapeutic effect | The placebo effect can be up to 100% of the total therapeutic effect |
| Only about one-third of the population responds to placebo | The placebo response is context-dependent and may include >90% of the patient population |
| Source: Reference 4 | |
Clinical strategies to enhance the placebo effect
| |
| Source: Reference 5 | |
CASE REPORT: Predicting positive results
Mr. B, age 42, has a history of recurrent depression associated with severe insomnia, poor appetite, significant weight loss, and psychosocial withdrawal with feelings of hopelessness. After I take a detailed history and do a mental status examination, I suggest that he be treated with cognitive-behavioral therapy (CBT) and mirtazapine.
Even though studies of antidepressants rarely show mood improvements within the first 7 days, it is not unusual to hear patients report feeling less depressed within days after they start a new antidepressant. Although the drug’s specific chemical effects on the brain may not be sufficient to explain this phenomenon, the explanation probably lies in nonspecific effects—such as the patient expecting that this medication will make him feel better.
The placebo effect can occur as soon as a patient starts a medication. Experienced clinicians understand the placebo effect’s power and harness it to benefit their patients.
Conditioned responses
Many biological responses can be associated with visual, auditory, tactile, olfactory, or gustatory stimuli. Nonconditioned physiologic responses paired with conditioned stimuli induce the same biological effects of a drug. Evidence supporting this phenomenon includes successful conditioning of the immune system.7-10 Conditioned responses—as demonstrated in glycemia regulation10 and with psychopharmacology11—also can enhance the desirable results of pharmacotherapy.
CASE REPORT: A soothing drink
Ms. L, a 22-year-old college student, suffers from obsessive-compulsive disorder associated with anxiety and depression. She arrives at the appointment hurried and worried that she might be late. She is short of breath and looks stressed. The nurse offers Ms. L a cup of tea or water. She chooses a glass of water and is asked to bring it into her session.
Following a comprehensive interview and mental status examination, I recommend CBT plus medication. Considering Ms. L’s medication history, we agree to start treatment with sertraline. We review its potential benefits and expectations that it will reduce her anxiety, alleviate her ruminating obsessive worries, and improve her mood. I give her a 50-mg sample and inform her that some patients experience positive effects soon after taking the medication. I then ask her to take the first pill, using her glass of water. She does so and thanks me for being attentive to her needs.
I instruct her to call within 1 week and report on her condition, even if she feels better. Seven days later she reports that she is feeling better and is looking forward to her next appointment. She reports no side effects.
Often patients come to my office feeling thirsty. My staff or I offer them a glass of water or a cup of tea. As patients sip from the cup, they swallow and incorporate the liquid into their bodies. At the same time, I use verbal interventions to make them feel listened to and understood. They internalize this emotional experience in connection with swallowing the liquid.
Later, when swallowing the new medication as instructed, the patient re-experiences the positive therapeutic effect that was internalized in the doctor’s office.
The power of suggestion
The power of suggestion has been shown to positively or negatively affect treatment outcomes.12,13 In practice, most clinicians give unintentional suggestions by how and what they communicate to the patient.
CASE REPORT: Predicting improvement
Mrs. J, age 48, has had dysthymic disorder and fibromyalgia for many years. She describes how various specialists have tried to alleviate her depression and chronic pain. Follow-up questions reveal that whenever she received a new prescription the physician would alert her to all the possible side effects and instruct her to call the office if she developed a problem with the new medication.
Invariably, Mrs. J would call as instructed and describe side effects she developed with the new medication. Often the doctor would discontinue the medication, depriving Mrs. J of benefits she might have derived later.
When Mrs. J calls to report on her status, she mentions that she is sleeping better and has begun to feel better during the day. She says that her husband told her she has started to smile again.
This vignette illustrates the importance of suggesting to the patient a positive outcome of pharmacotherapy associated with a particular action (calling the doctor’s office to report results). When the patient promised to call, she internalized the suggestion that calling would be associated with feeling better—and that is what happened. This intervention contrasts with saying to the patient, “Call me if you have a problem with any of these side effects,” which gives the patient a suggestion to call and report a problem.
The suggestion effect also can be used to reframe a predictable side effect as a positive sign that indicates the beginning of change leading to recovery (Box).
Ms. M, age 32 and single, has an anxiety disorder associated with bipolar depression. She has discontinued several psychotropics because of uncomfortable side effects, such as constipation.
After taking a detailed history, I decide to prescribe quetiapine. I tell Ms. M about this medication’s potential benefits and side effects. One common side effect is dry mouth, which often occurs before patients experience therapeutic effects.
I inform Ms. M that a dry mouth will be her sign that the medication has begun to work, and beneficial effects—such as improved sleep, reduced anxiety, and improved mood—will soon follow. I then instruct her to call my office and report when she experiences a dry mouth.
Discussion. In pharmacotherapy, side effects may appear before patients experience a medication’s beneficial/therapeutic effects. Patients’ initial experience often determines whether or not they will continue taking a prescribed medication. I know Ms. M may stop taking quetiapine—as she has done with other medications—if she initially has uncomfortable side effects.
Instructing patients to expect a specific side effect (such as a dry mouth with quetiapine) and associating it with a future therapeutic benefit sets up a road map of expectations. They know their experience is compatible with the doctor’s predictions. For Ms. M, I reframed the side effect as a positive sign that recovery has begun, with more positive changes to come.
Participatory pharmacotherapy
Many patients seek ownership in making decisions about their treatment and medications. In participatory pharmacotherapy, patients provide you with data and valuable information—such as family history, personal medical history, and experience with treatment—and inform you about which medications worked best and which did not work. You invite patients to predict how they see themselves getting better and into recovery.
CASE REPORT: All in the family
Mr. A, age 28 and single, has been diagnosed with a bipolar mood disorder. As part of a detailed family history, he reports that his maternal grandfather, mother, and a maternal uncle were diagnosed with mood swings and were successfully treated with medications, specifically lithium. He states that he believes he has the same condition.
I compliment Mr. A for being so well informed about his grandfather and uncle and educate him about mood stabilizers’ benefits in bipolar disorder. I tell him about the finding that if lithium has helped his relatives, it will probably help him as well.
I also reassure Mr. A that, in deciding what medications to avoid and what medications to use, I will consider his experience with specific antidepressants that did not help him. He thanks me for considering his suggestion about what medication to use for him.
- a successful therapeutic alliance
- adherence with prescribed medications
- the best possible outcome with pharmacotherapy.
In patients with a defiant-oppositional personality, consider framing the treatment decision as a choice between 2 equally efficacious medications. This gives the patient the sense of control in choosing his or her own medication, which is jointly monitored.
Table 3
Choosing patients for participatory pharmacotherapy
| Good candidates | Exclusionary qualities |
|---|---|
| Adults | Children, adolescents, and prison inmates |
| No history of alcoholism or drug addiction | Alcohol dependence or drug addiction |
| Average and above intelligence | Below-average intelligence |
| Intact cognitive function | Cognitive deficits, such as dementia |
| Not psychotic | Actively psychotic |
| Good comprehension of diagnosis and treatment | Poor comprehension of diagnosis and treatment |
| Therapeutic alliance is present | Therapeutic alliance is absent |
| Personality style or disorder with a need to be in control of treatment, such as obsessive-compulsive personality | Passive, dependent personality style or disorder; these patients may view a participatory approach as the doctor’s lack of confidence |
- Brody H. The placebo response: how you can release the body’s inner pharmacy for better health. New York, NY: HarperCollins Publishers; 2000.
- Spiro H. The power of hope: a doctor’s perspective. New Haven, CT: Yale University Press; 1998.
- Ernst E. Placebo: new insights into an old enigma. Drug Discov Today 2007;12:413-8.
- Lithium • Eskalith, Lithobid
- Mirtazapine • Remeron
- Quetiapine • Seroquel
- Sertraline • Zoloft
Dr. Torem reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Frank JD, Frank JB. Persuasion and healing. Baltimore, MD: The Johns Hopkins University Press; 1991.
2. Freud S. The complete psychological works of Sigmund Freud. Strachey J, trans-ed. Toronto, Ontario, Canada: Hogarth Press; 1953.
3. Wolf S. The pharmacology of placebos. Pharmacol Rev 1959;11:689-704.
4. Ernst E. Placebo: new insights into an old enigma. Drug Discov Today 2007;12:413-8.
5. Brody H. The placebo response: recent research and implications for family medicine. J Fam Pract 2000;49:649-54.
6. Spiegel H. Nocebo, the power of suggestibility. Prev Med 1997;26:616-21.
7. Ader R, Cohen N. Behaviorally conditioned immunosuppression and murine systemic lupus erythematosus. Science 1982;215:1534-6.
8. Ader R. The role of conditioning in pharmacotherapy. In: Harrington A, ed. The placebo effect: an interdisciplinary exploration. Cambridge, MA: Harvard University Press; 1997;138-65.
9. Olness K, Ader R. Conditioning as an adjunct in the pharmacotherapy of lupus erythematosus. J Dev Behav Pediatr 1992;13:124-5.
10. Stockhorst U, Mahl N, Krueger M, et al. Classical conditioning and conditionability of insulin and glucose effect in healthy humans. Physiol Behav 2004;81:375-88.
11. Wolf S. Effect of suggestion and conditioning on the action of chemical agents in human subjects—the pharmacology of placebos. J Clin Invest 1950;29:100-9.
12. Lown B. The verbal conditioning of angina pectoris during exercise testing. Am J Cardiol 1977;40:630-4.
13. Lown B. Introduction. In: Cousins N. The healing heart. New York, NY: W.W. Norton; 1983:11-28.
14. Watzlawick P. If you desire to see, learn how to act. In: Nardone G, Watzlawick P, eds. The art of change San Francisco, CA: Jossey-Bass; 1993:1-16.
1. Frank JD, Frank JB. Persuasion and healing. Baltimore, MD: The Johns Hopkins University Press; 1991.
2. Freud S. The complete psychological works of Sigmund Freud. Strachey J, trans-ed. Toronto, Ontario, Canada: Hogarth Press; 1953.
3. Wolf S. The pharmacology of placebos. Pharmacol Rev 1959;11:689-704.
4. Ernst E. Placebo: new insights into an old enigma. Drug Discov Today 2007;12:413-8.
5. Brody H. The placebo response: recent research and implications for family medicine. J Fam Pract 2000;49:649-54.
6. Spiegel H. Nocebo, the power of suggestibility. Prev Med 1997;26:616-21.
7. Ader R, Cohen N. Behaviorally conditioned immunosuppression and murine systemic lupus erythematosus. Science 1982;215:1534-6.
8. Ader R. The role of conditioning in pharmacotherapy. In: Harrington A, ed. The placebo effect: an interdisciplinary exploration. Cambridge, MA: Harvard University Press; 1997;138-65.
9. Olness K, Ader R. Conditioning as an adjunct in the pharmacotherapy of lupus erythematosus. J Dev Behav Pediatr 1992;13:124-5.
10. Stockhorst U, Mahl N, Krueger M, et al. Classical conditioning and conditionability of insulin and glucose effect in healthy humans. Physiol Behav 2004;81:375-88.
11. Wolf S. Effect of suggestion and conditioning on the action of chemical agents in human subjects—the pharmacology of placebos. J Clin Invest 1950;29:100-9.
12. Lown B. The verbal conditioning of angina pectoris during exercise testing. Am J Cardiol 1977;40:630-4.
13. Lown B. Introduction. In: Cousins N. The healing heart. New York, NY: W.W. Norton; 1983:11-28.
14. Watzlawick P. If you desire to see, learn how to act. In: Nardone G, Watzlawick P, eds. The art of change San Francisco, CA: Jossey-Bass; 1993:1-16.
Screening for Chlamydia? New advice for sexually active women
- Ask female psychiatric patients about high-risk sexual behaviors, and recommend Chlamydia screening when appropriate.
- Recommend Chlamydia screening for all sexually active women age <25 years.
- Chlamydia screening decreases incidence of pelvic inflammatory disease, improves pregnancy outcomes, and lowers risk of other sexually transmitted infections.
- Urine nucleic acid amplification tests minimize patient discomfort and remove logistical barriers of speculum or urethra specimens.
- Evidence is insufficient to recommend screening men.
Chlamydia trachomatis is the most common bacterial sexually transmitted infection (STI), with nearly 3 million new cases diagnosed annually in the United States.1 In July 2007 the U.S. Preventive Services Task Force (USPSTF) updated its recommendation on Chlamydia screening of sexually active female adolescents and adults ( Table 1 ).2 Routine screening is not recommended for men because there is not enough data to determine the benefits and risks of screening.
Although the USPSTF recommendation targets the general population, it is important to assess each patient’s sexual behavior. Women exhibiting impulsivity caused by bipolar mania, substance abuse, or personality disorders or who exchange sex for food, shelter, substances, or money are at high risk of infection ( Table 2 ).1,2
The new guideline introduces a screening cutoff at age 25 because women age <25 years are 5 times more likely than women age >30 to have chlamydial infection.2 In women age ≥25, yearly screening is recommended only for those at high risk as indicated by:
- previous chlamydial infection or other STIs
- new or multiple sexual partners
- inconsistent condom use
- being a sex worker ( Table 2 ).2
Chlamydia screening reduces the incidence of pelvic inflammatory disease (PID) in nonpregnant adolescent and adult women, which can cause infertility, ectopic pregnancy, and chronic pelvic pain.2 If untreated, the risk of PID approaches 40%.1 In pregnant women, Chlamydia treatment significantly improves birth outcomes. Pregnant women should be screened during the first prenatal visit and in the third trimester if at continued risk.
Discussant: Glen L. Xiong, MD
Psychological implications of screening. Address possible themes of guilt and shame with patients to clarify and alleviate the burden they may feel about STI screening and treatment. Compared with men, women experience more stigmatization, blame, and denial about the source of infection. Women also report being more concerned about potential threats to their relationships.1
Table 1
U.S. Preventive Services Task Force
screening recommendations for chlamydial infection
| Nonpregnant women* | Pregnant women† | |
|---|---|---|
| Age <25 | Screen | Screen |
| Age ≥25 | Screen those at increased risk | Screen those at increased risk |
| Interval‡ | At least annually | First prenatal visit, third trimester if at continued risk |
| * Grade A recommendation: high quality evidence | ||
| † Grade B recommendation: moderate quality evidence | ||
| ‡ The optimal screening interval remains to be determined | ||
| Source: Reference 2 | ||
Table 2
Risks for chlamydial infection
| • Sexually active women age <25 years |
| • History of chlamydial or other sexually transmitted infection |
| • New or multiple sexual partners |
| • Inconsistent condom use |
| • Exchange of sex for money, drugs, or shelter |
| Other demographic groups at high risk |
| • African-American and Hispanic women |
| • Incarcerated men and women |
| • Military recruits |
| Source: References 1,2 |
Screening test. The Centers for Disease Control and Prevention (CDC) recommends screening with nucleic acid amplification tests (NAATs), which have high specificity (>95%) and sensitivity (80% to 93%) for chlamydial infections. Urine specimens are comparable to cervical and urethral specimens and avert the cost and patient discomfort associated with speculum exams.3 NAATs do not exclude other infections, such as trichomonas, however, and are not sufficient in patients with active urinary or vaginal symptoms.
Clinical presentation and treatment. Most persons with Chlamydia are asymptomatic and may infect new sexual partners. In women, chlamydial infection may cause cervicitis, urethritis, PID, chronic pelvic pain, ectopic pregnancy, miscarriage, preterm labor, and infertility. In men, chlamydial infection may cause urethritis, urethral strictures, and epididymis. In both genders, chlamydial infection increases the risk of acquiring other STIs, such as human immunodeficiency virus.4
The CDC recommends treating chlamydial infection with azithromycin, 1 g/d PO for pregnant and nonpregnant women. Alternatives include amoxicillin, 500 mg tid for 7 days for pregnant women, or doxycycline, 100 mg bid for 7 days for nonpregnant women. Sexual partners of an infected individual should be treated presumptively or tested and then treated. Because Chlamydia NAAT is highly sensitive, patients with a negative test do not need treatment. Patients who test positive for gonorrhea and receive a negative non-NAAT (antigen-based tests that are less sensitive than NAATs) for Chlamydia should be treated for both.5
Related resources
- Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines. www.cdc.gov/std/treatment.
- U.S. Department of Health and Human Services. Agency for Healthcare Research and Quality. Preventive Services. www.preventiveservices.ahrq.gov.
Drug brand names
- Amoxicillin • Amoxil, others
- Azithromycin • Zithromax
- Doxycycline • Vibramycin
Disclosure
Dr. Xiong reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyers DS, Halvorson H, Luckhaupt S. Screening for chlamydial infection: an evidence update for the U.S. Preventive Services Task Force. Ann Intern Med 2007;147(2):135-42.
2. U.S. Preventive Services Task Force. Screening for chlamydial infection: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2007;147(2):128-34.
3. Cook RI, Hutchison SL, Østergaard L, et al. Systematic review: noninvasive testing for Chlamydia trachomatis and Neisseria gonorrhoea. Ann Intern Med 2005;142(11):914-25.
4. Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex Transm Infect 1999;75(1):3-17.
5. Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines 2006: dual therapy for gonococcal and chlamydial infections. Available at: http://www.cdc.gov/std/treatment/2006/urethritis-and-cervicitis.htm#dualtherapy. Accessed June 16, 2008.
Dr. Xiong is assistant clinical professor, departments of internal medicine and psychiatry, University of California, Davis.

Robert M. McCarron, DO
Series Editor
Robert M. McCarron, DO
Series Editor
Robert M. McCarron, DO
Series Editor
- Ask female psychiatric patients about high-risk sexual behaviors, and recommend Chlamydia screening when appropriate.
- Recommend Chlamydia screening for all sexually active women age <25 years.
- Chlamydia screening decreases incidence of pelvic inflammatory disease, improves pregnancy outcomes, and lowers risk of other sexually transmitted infections.
- Urine nucleic acid amplification tests minimize patient discomfort and remove logistical barriers of speculum or urethra specimens.
- Evidence is insufficient to recommend screening men.
Chlamydia trachomatis is the most common bacterial sexually transmitted infection (STI), with nearly 3 million new cases diagnosed annually in the United States.1 In July 2007 the U.S. Preventive Services Task Force (USPSTF) updated its recommendation on Chlamydia screening of sexually active female adolescents and adults ( Table 1 ).2 Routine screening is not recommended for men because there is not enough data to determine the benefits and risks of screening.
Although the USPSTF recommendation targets the general population, it is important to assess each patient’s sexual behavior. Women exhibiting impulsivity caused by bipolar mania, substance abuse, or personality disorders or who exchange sex for food, shelter, substances, or money are at high risk of infection ( Table 2 ).1,2
The new guideline introduces a screening cutoff at age 25 because women age <25 years are 5 times more likely than women age >30 to have chlamydial infection.2 In women age ≥25, yearly screening is recommended only for those at high risk as indicated by:
- previous chlamydial infection or other STIs
- new or multiple sexual partners
- inconsistent condom use
- being a sex worker ( Table 2 ).2
Chlamydia screening reduces the incidence of pelvic inflammatory disease (PID) in nonpregnant adolescent and adult women, which can cause infertility, ectopic pregnancy, and chronic pelvic pain.2 If untreated, the risk of PID approaches 40%.1 In pregnant women, Chlamydia treatment significantly improves birth outcomes. Pregnant women should be screened during the first prenatal visit and in the third trimester if at continued risk.
Discussant: Glen L. Xiong, MD
Psychological implications of screening. Address possible themes of guilt and shame with patients to clarify and alleviate the burden they may feel about STI screening and treatment. Compared with men, women experience more stigmatization, blame, and denial about the source of infection. Women also report being more concerned about potential threats to their relationships.1
Table 1
U.S. Preventive Services Task Force
screening recommendations for chlamydial infection
| Nonpregnant women* | Pregnant women† | |
|---|---|---|
| Age <25 | Screen | Screen |
| Age ≥25 | Screen those at increased risk | Screen those at increased risk |
| Interval‡ | At least annually | First prenatal visit, third trimester if at continued risk |
| * Grade A recommendation: high quality evidence | ||
| † Grade B recommendation: moderate quality evidence | ||
| ‡ The optimal screening interval remains to be determined | ||
| Source: Reference 2 | ||
Table 2
Risks for chlamydial infection
| • Sexually active women age <25 years |
| • History of chlamydial or other sexually transmitted infection |
| • New or multiple sexual partners |
| • Inconsistent condom use |
| • Exchange of sex for money, drugs, or shelter |
| Other demographic groups at high risk |
| • African-American and Hispanic women |
| • Incarcerated men and women |
| • Military recruits |
| Source: References 1,2 |
Screening test. The Centers for Disease Control and Prevention (CDC) recommends screening with nucleic acid amplification tests (NAATs), which have high specificity (>95%) and sensitivity (80% to 93%) for chlamydial infections. Urine specimens are comparable to cervical and urethral specimens and avert the cost and patient discomfort associated with speculum exams.3 NAATs do not exclude other infections, such as trichomonas, however, and are not sufficient in patients with active urinary or vaginal symptoms.
Clinical presentation and treatment. Most persons with Chlamydia are asymptomatic and may infect new sexual partners. In women, chlamydial infection may cause cervicitis, urethritis, PID, chronic pelvic pain, ectopic pregnancy, miscarriage, preterm labor, and infertility. In men, chlamydial infection may cause urethritis, urethral strictures, and epididymis. In both genders, chlamydial infection increases the risk of acquiring other STIs, such as human immunodeficiency virus.4
The CDC recommends treating chlamydial infection with azithromycin, 1 g/d PO for pregnant and nonpregnant women. Alternatives include amoxicillin, 500 mg tid for 7 days for pregnant women, or doxycycline, 100 mg bid for 7 days for nonpregnant women. Sexual partners of an infected individual should be treated presumptively or tested and then treated. Because Chlamydia NAAT is highly sensitive, patients with a negative test do not need treatment. Patients who test positive for gonorrhea and receive a negative non-NAAT (antigen-based tests that are less sensitive than NAATs) for Chlamydia should be treated for both.5
Related resources
- Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines. www.cdc.gov/std/treatment.
- U.S. Department of Health and Human Services. Agency for Healthcare Research and Quality. Preventive Services. www.preventiveservices.ahrq.gov.
Drug brand names
- Amoxicillin • Amoxil, others
- Azithromycin • Zithromax
- Doxycycline • Vibramycin
Disclosure
Dr. Xiong reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
- Ask female psychiatric patients about high-risk sexual behaviors, and recommend Chlamydia screening when appropriate.
- Recommend Chlamydia screening for all sexually active women age <25 years.
- Chlamydia screening decreases incidence of pelvic inflammatory disease, improves pregnancy outcomes, and lowers risk of other sexually transmitted infections.
- Urine nucleic acid amplification tests minimize patient discomfort and remove logistical barriers of speculum or urethra specimens.
- Evidence is insufficient to recommend screening men.
Chlamydia trachomatis is the most common bacterial sexually transmitted infection (STI), with nearly 3 million new cases diagnosed annually in the United States.1 In July 2007 the U.S. Preventive Services Task Force (USPSTF) updated its recommendation on Chlamydia screening of sexually active female adolescents and adults ( Table 1 ).2 Routine screening is not recommended for men because there is not enough data to determine the benefits and risks of screening.
Although the USPSTF recommendation targets the general population, it is important to assess each patient’s sexual behavior. Women exhibiting impulsivity caused by bipolar mania, substance abuse, or personality disorders or who exchange sex for food, shelter, substances, or money are at high risk of infection ( Table 2 ).1,2
The new guideline introduces a screening cutoff at age 25 because women age <25 years are 5 times more likely than women age >30 to have chlamydial infection.2 In women age ≥25, yearly screening is recommended only for those at high risk as indicated by:
- previous chlamydial infection or other STIs
- new or multiple sexual partners
- inconsistent condom use
- being a sex worker ( Table 2 ).2
Chlamydia screening reduces the incidence of pelvic inflammatory disease (PID) in nonpregnant adolescent and adult women, which can cause infertility, ectopic pregnancy, and chronic pelvic pain.2 If untreated, the risk of PID approaches 40%.1 In pregnant women, Chlamydia treatment significantly improves birth outcomes. Pregnant women should be screened during the first prenatal visit and in the third trimester if at continued risk.
Discussant: Glen L. Xiong, MD
Psychological implications of screening. Address possible themes of guilt and shame with patients to clarify and alleviate the burden they may feel about STI screening and treatment. Compared with men, women experience more stigmatization, blame, and denial about the source of infection. Women also report being more concerned about potential threats to their relationships.1
Table 1
U.S. Preventive Services Task Force
screening recommendations for chlamydial infection
| Nonpregnant women* | Pregnant women† | |
|---|---|---|
| Age <25 | Screen | Screen |
| Age ≥25 | Screen those at increased risk | Screen those at increased risk |
| Interval‡ | At least annually | First prenatal visit, third trimester if at continued risk |
| * Grade A recommendation: high quality evidence | ||
| † Grade B recommendation: moderate quality evidence | ||
| ‡ The optimal screening interval remains to be determined | ||
| Source: Reference 2 | ||
Table 2
Risks for chlamydial infection
| • Sexually active women age <25 years |
| • History of chlamydial or other sexually transmitted infection |
| • New or multiple sexual partners |
| • Inconsistent condom use |
| • Exchange of sex for money, drugs, or shelter |
| Other demographic groups at high risk |
| • African-American and Hispanic women |
| • Incarcerated men and women |
| • Military recruits |
| Source: References 1,2 |
Screening test. The Centers for Disease Control and Prevention (CDC) recommends screening with nucleic acid amplification tests (NAATs), which have high specificity (>95%) and sensitivity (80% to 93%) for chlamydial infections. Urine specimens are comparable to cervical and urethral specimens and avert the cost and patient discomfort associated with speculum exams.3 NAATs do not exclude other infections, such as trichomonas, however, and are not sufficient in patients with active urinary or vaginal symptoms.
Clinical presentation and treatment. Most persons with Chlamydia are asymptomatic and may infect new sexual partners. In women, chlamydial infection may cause cervicitis, urethritis, PID, chronic pelvic pain, ectopic pregnancy, miscarriage, preterm labor, and infertility. In men, chlamydial infection may cause urethritis, urethral strictures, and epididymis. In both genders, chlamydial infection increases the risk of acquiring other STIs, such as human immunodeficiency virus.4
The CDC recommends treating chlamydial infection with azithromycin, 1 g/d PO for pregnant and nonpregnant women. Alternatives include amoxicillin, 500 mg tid for 7 days for pregnant women, or doxycycline, 100 mg bid for 7 days for nonpregnant women. Sexual partners of an infected individual should be treated presumptively or tested and then treated. Because Chlamydia NAAT is highly sensitive, patients with a negative test do not need treatment. Patients who test positive for gonorrhea and receive a negative non-NAAT (antigen-based tests that are less sensitive than NAATs) for Chlamydia should be treated for both.5
Related resources
- Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines. www.cdc.gov/std/treatment.
- U.S. Department of Health and Human Services. Agency for Healthcare Research and Quality. Preventive Services. www.preventiveservices.ahrq.gov.
Drug brand names
- Amoxicillin • Amoxil, others
- Azithromycin • Zithromax
- Doxycycline • Vibramycin
Disclosure
Dr. Xiong reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyers DS, Halvorson H, Luckhaupt S. Screening for chlamydial infection: an evidence update for the U.S. Preventive Services Task Force. Ann Intern Med 2007;147(2):135-42.
2. U.S. Preventive Services Task Force. Screening for chlamydial infection: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2007;147(2):128-34.
3. Cook RI, Hutchison SL, Østergaard L, et al. Systematic review: noninvasive testing for Chlamydia trachomatis and Neisseria gonorrhoea. Ann Intern Med 2005;142(11):914-25.
4. Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex Transm Infect 1999;75(1):3-17.
5. Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines 2006: dual therapy for gonococcal and chlamydial infections. Available at: http://www.cdc.gov/std/treatment/2006/urethritis-and-cervicitis.htm#dualtherapy. Accessed June 16, 2008.
Dr. Xiong is assistant clinical professor, departments of internal medicine and psychiatry, University of California, Davis.
1. Meyers DS, Halvorson H, Luckhaupt S. Screening for chlamydial infection: an evidence update for the U.S. Preventive Services Task Force. Ann Intern Med 2007;147(2):135-42.
2. U.S. Preventive Services Task Force. Screening for chlamydial infection: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 2007;147(2):128-34.
3. Cook RI, Hutchison SL, Østergaard L, et al. Systematic review: noninvasive testing for Chlamydia trachomatis and Neisseria gonorrhoea. Ann Intern Med 2005;142(11):914-25.
4. Fleming DT, Wasserheit JN. From epidemiological synergy to public health policy and practice: the contribution of other sexually transmitted diseases to sexual transmission of HIV infection. Sex Transm Infect 1999;75(1):3-17.
5. Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines 2006: dual therapy for gonococcal and chlamydial infections. Available at: http://www.cdc.gov/std/treatment/2006/urethritis-and-cervicitis.htm#dualtherapy. Accessed June 16, 2008.
Dr. Xiong is assistant clinical professor, departments of internal medicine and psychiatry, University of California, Davis.
Hyperprolactinemia: Monitoring children on long-term risperidone
Serum prolactin increases in children and adolescents when risperidone therapy begins, then decreases over time in many patients. When prolactin levels remain elevated, evidence suggests that children may experience adverse effects such as delayed sexual maturation or reduced bone growth because of hypothalamic-pituitary-gonadal axis (HPG) dysfunction.
To help you make informed prescribing decisions, we discuss what the evidence says about the effects of elevated prolactin in children and adolescents. We then suggest clinical steps to help you manage hyperprolactinemia when prescribing risperidone.
Pediatric indications
Based on short-term clinical trials of efficacy and tolerability, risperidone is FDA-approved for 3 pediatric indications:
- short-term treatment of acute mania or mixed episodes associated with bipolar I disorder in patients age 10 to 17
- schizophrenia treatment in patients age 13 to 17
- treatment of irritability (including aggression, self-injury, temper tantrums, and mood swings) associated with autistic disorder in patients age 5 to 16.
Recommended risperidone dosages are lower for children and adolescents than for adults (Table 1). Off-label pediatric uses described in case reports include psychotic, mood, disruptive, movement, and pervasive developmental disorders.
Table 1
Recommended risperidone dosing for pediatric indications*
| Indication | Starting dose | Maximum dose |
|---|---|---|
| Acute mania or mixed episodes | 0.5 mg once daily in morning or evening | 2.5 mg/d |
| Irritability in autism | 0.25 mg/d for patients weighing <20 kg 0.5 mg/d for patients weighing ≥20 kg | 0.5 mg/d for patients weighing <20 kg 1 mg/d for patients weighing ≥20 kg |
| Schizophrenia | 0.5 mg once daily in morning or evening | 3 mg/d |
| * FDA-approved dosages; individualize based on response and tolerability | ||
| Source: Drug facts and comparisons. St. Louis, MO: Wolters Kluwer Health; 2008:949-50 | ||
Prolactin physiology
Prolactin’s primary physiologic function is to cause breast enlargement during pregnancy and milk secretion during lactation.1 A polypeptide hormone, prolactin is secreted by lactotroph cells in the anterior pituitary, under the complex control of stimulatory and inhibitory factors (Table 2). Its pulsatile secretion peaks 13 to 14 times daily, with approximately 95 minutes between pulses.
Serum prolactin levels show marked circadian variation.2 The reference value for serum prolactin is 1 to 25 ng/mL for women and 1 to 20 ng/mL for men. The higher prolactin levels seen in women begin after puberty and presumably are caused by estrogen’s stimulatory effect.3 Age- and sex-specific normal prolactin ranges vary widely and from lab to lab (Table 3).
Risperidone is a strong dopamine D2 and serotonin 5HT-2A antagonist with low affinity for alpha-1 and alpha-2 adrenergic receptors and histamine H1 receptors.4 Antagonism of these receptors is thought to explain the drug’s therapeutic effects and many of its side effects, including hyperprolactinemia.5 Prolactin release is also influenced by thyrotropin-releasing hormone.6 A rare association between pituitary tumors and atypical antipsychotics has been proposed as a probable cause of sustained prolactin elevation.7
Pituitary prolactin secretion is regulated by neuroendocrine neurons in the hypothalamus, specifically in the tuberoinfundibular tract that extends from the arcuate nucleus of the mediobasal hypothalamus (tuberal region) and projects to the median eminence (infundibular region). Neurosecretory dopamine neurons of the arcuate nucleus inhibit prolactin secretion. Hence, prolactin secretion increases when antipsychotic therapy results in dopamine receptor blockade.
Antipsychotics vary in affinity for the D2 dopamine receptor, rate of dissociation from the receptor, and ability to act on the receptor as both a dopamine agonist (which lowers serum prolactin) and a dopamine antagonist (which increases serum prolactin). Based on adult and pediatric data, the relative potency of antipsychotic drugs in inducing hyperprolactinemia is roughly risperidone > haloperidol > olanzapine > ziprasidone > quetiapine > clozapine > aripiprazole.8 Even though risperidone ranks highest in the hierarchy to cause hyperprolactinemia, it is accepted as the first-line antipsychotic in children and adolescents. This is probably because risperidone:
- has been in clinical use longer than other atypical antipsychotics except clozapine
- has received FDA approval for 3 pediatric indications.
Table 2
Factors that regulate prolactin secretion
| Effect | Factors | Mechanism |
|---|---|---|
| Inhibitory | Dopamine, gonadotropin-associated protein, acetylcholine | D2 receptor stimulation of lactotroph cells |
| Stimulatory | Serotonin, thyrotropin-releasing hormone, cholecystokinin | Through 5-HT1A and 5-HT2 |
Table 3
Sample age- and sex-specific reference ranges for serum prolactin (ng/mL)*
| Age | Males | Females |
|---|---|---|
| 0 to 1 month | 3.7 to 81.2 | 0.3 to 95.0 |
| 1 to 12 months | 0.3 to 28.9 | 0.2 to 29.9 |
| 1 to 3 years | 2.3 to 13.2 | 1.0 to 17.0 |
| 4 to 6 years | 0.8 to 16.9 | 1.6 to 13.1 |
| 7 to 9 years | 1.9 to 11.6 | 0.3 to 12.9 |
| 10 to 12 years | 0.9 to 12.9 | 1.9 to 9.6 |
| 13 to 15 years | 1.6 to 16.6 | 3.0 to 14.4 |
| Adult | 2.1 to 17.7 | 2.8 to 29.2 |
| Female: nonpregnant | 2.8 to 29.2 | |
| Female: pregnant | 9.7 to 208.5 | |
| Postmenopausal | 1.8 to 20.3 | |
| * Reference values may vary from lab to lab | ||
| Source: LabCorp, Birmingham, AL | ||
Prolactin and the HPG axis
Elevated serum prolactin inhibits the hypothalamus’ pulsatile release of gonadotrophin-releasing hormone (GnRH), which in turn decreases the pituitary’s secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). In women, prolactin also blocks the feedback effect of estradiol on LH secretion (Figure). The prolactin level that triggers gonadal hypofunction appears to vary substantially among individuals.9
Symptoms of elevated prolactin can occur as a direct result of prolactin’s physiologic effect on breast tissue or indirectly through hypogonadism related to decreased FSH and LH. Symptoms of hyperprolactinemia—which can be seen more readily in sexually mature adolescents than in children—include:
- amenorrhea or oligorrhea
- breast enlargement or engorgement in females and males
- galactorrhea (females > males)
- decreased libido
- erectile dysfunction.
Although evidence is inconclusive, other problems may be associated with increased prolactin in children and adolescents. These include failure to enter or progress through puberty,8 increased risk of benign breast tumors,22 and reduced bone density.10
Bone changes. Decreased estrogen related to hyperprolactinemia may inhibit bone mineralization, causing osteopenia, osteoporosis, and increased fracture risk.10 The mechanism of bone density loss may be estrogen’s osteoclast activating and osteoblast inhibiting action. The level and duration of prolactin elevation that can hamper bone growth has not been defined, although evidence suggests a pervasive effect:
- 65% of a group of 38 premenstrual patients developed osteoporosis or osteopenia when taking risperidone or typical antipsychotics for schizophrenia for a mean of 8 years.11
- Bone loss has persisted 2 years after prolactin normalized in adolescents with prolactinomas.12
Prolactin secretion is controlled by stimulatory and inhibitory influences (A). Antipsychotic blockade of dopamine’s inhibitory influence (B) increases serum prolactin and its effect on mammary tissue (C). In the brain, hyperprolactinemia inhibits the release of gonadotropin-releasing hormone (GnRH) by the hypothalamus, which results in decreased follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secretion by the pituitary (D). FSH and LH are important determinants of male and female gonadal maturation by their direct action on testes and ovaries within the hypothalamic-pituitary-gonadal axis.
Source: Developed by Manpreet Khemka, MBBS, and Jeffrey Ali, MD, MSc. Current Psychiatry Illustration by Rob Flewell
Hyperprolactinemia in children and adolescents
We suggest that children and adolescents receiving prolonged risperidone treatment can present with symptoms similar to those associated with hyperprolactinemia secondary to other causes, including:
- prolactinomas (the most common cause)13
- thyrotropin-releasing hormone stimulation in primary hypothyroidism
- hypoglycemia
- inherited endocrine syndromes
- physiologic stress
- medications.
The most common presenting symptoms of prolactinomas are headache, amenorrhea, and galactorrhea. A few patients have delayed puberty.13 In a review of hyperprolactinemia in children, Massart and Saggese14 proposed a correlation between elevated serum prolactin and underlying pathology:
- >100 ng/mL usually suggests organic pathology and requires MRI or CT confirmation
- <100 ng/mL usually indicates functional pathology.
What the evidence says. Using the key words “risperidone and hyperprolactinemia in children and adolescents” in a PubMed search, we identified 7 prospective, cross-sectional, and retrospective studies.14-20 We then analyzed these studies in terms of subjects’ age, sex, primary psychiatric disorder, dosage of risperidone used, prolactin elevation pattern, reported clinical consequences, and interventions used to ameliorate asymptomatic or symptomatic hyperprolactinemia.
Prolactin and antipsychotics. In a cross-sectional study, Staller15 compared serum prolactin at baseline and after 6 months in 50 children treated with atypical antipsychotics. Patients taking risperidone showed greater increases in prolactin than those taking quetiapine or olanzapine. Saito et al16 reached a similar conclusion in a prospective study of 40 children treated with atypical antipsychotics for 4 to 15 weeks.
Ups and downs. Prolactin levels increase sharply in the first weeks of risperidone treatment, peak at around 6 to 8 weeks, and then trend downward toward normal.17 In a post hoc analysis of pooled data from 5 clinical trials totaling 700 patients age 5 to 15, Findling et al17 reported that mean serum prolactin:
- peaked in the first 1 to 2 months of patients’ starting risperidone, 0.02 to 0.06 mg/kg/d
- returned to within or close to normal range by 3 to 5 months.
No correlation was seen between prolactin elevation and side effects that could be attributed to prolactin.
In a 2-part study, Anderson et al18 examined the short- and long-term effects of risperidone treatment on prolactin in children age 5 to 17 with autism. In the initial double-blind, placebo-controlled trial, 101 children were randomly assigned to risperidone 1.8 mg/d, or placebo. After 8 weeks, 63 children continued with open-label risperidone, mean dose 1.96 mg/d, for up to a total 22 months. Serum prolactin was measured at baseline (9.3±7.5 ng/ mL) and then at 2, 6, and 22 months.
Serum prolactin increased sharply in children treated with risperidone in the placebo-controlled trial. After 8 weeks, prolactin levels were 4 times higher with risperidone treatment than with placebo (39.0±19.2 ng/mL vs 10.1±8.8 ng/mL [P< 0.0001]). In the open-label risperidone continuation trial, prolactin levels remained significantly higher than at baseline but decreased over time to:
- 32.4±17.8 ng/mL in 43 children who remained in the study at 6 months (P< 0.0001)
- 25.3±15.6 ng/mL in 30 children who remained at 22 months (P< 0.0001).
In this study, a sharp rise in serum prolactin in the first 2 months trended down to the upper limit of normal at 22 months. None of the children showed known clinical manifestations of elevated prolactin, including gynecomastia, galactorrhea, or menstrual disturbance.
A double-blind, placebo-controlled trial by Hellings et al18 examined risperidone’s effect on aggression and self-injury in children, adolescents, and adults with mental retardation and pervasive developmental disorders. In a subset of 10 children and adolescents whose serum prolactin was measured during the trial, prolactin remained elevated during at least 26 weeks of risperidone treatment. Mean prolactin levels were:
- 13.2±8.6 ng/mL at baseline
- 31.0±11.6 ng/mL during acute risperidone therapy
- 37.9±10.4 ng/mL during maintenance therapy.
Clinical features. Higher risperidone dosages—rather than longer duration of use—appear more likely to cause symptomatic elevated serum prolactin. A case series of 3 adolescents with symptomatic prolactin elevation showed:
- gynecomastia and galactorrhea in 2 adolescent males age 17 and 18, receiving risperidone, 4 mg/d and 5 mg/d, respectively
- amenorrhea within 2 to 6 weeks of starting treatment in a female patient age 15 receiving risperidone, 6 mg/d.20
Holzer et al21 described 5 adolescents who showed symptoms of elevated prolactin after 3 to 15 months while taking risperidone, 2 to 6 mg/d, as treatment for psychosis.
Long-term health risks?
Some evidence suggests an association between elevated serum prolactin and carcinogenesis and infertility in adults. No studies have examined these long-term risks in children and adolescents who develop hyperprolactinemia from risperidone treatment. A link may be possible, however, if prolactin elevation affects postpubertal and HPG axis development.
Breast cancer. Halbriech et al22 reviewed mammograms and charts of 275 female psychiatric hospital patients age >40 and 928 women of similar age at a hospital radiology clinic. The incidence of breast cancer among psychiatric patients was:
- >3.5 times higher than among radiology clinic patients
- 9.5 times higher than in the general population.
The authors speculated that the observed increased breast cancer incidence in psychiatric patients could be associated with medications, although high rates of cigarette smoking and alcohol consumption also might have played a role.
A case-control study by Hankinson et al23 of blood samples collected from women in the Nurses’ Health Study found a statistically significant association between hyperprolactinemia and breast cancer. This analysis included 306 postmenopausal women diagnosed with breast cancer and 448 controls matched for age, postmenopausal hormone use, and time of day and month when blood samples were drawn.
Several putative mechanisms have been proposed to explain a possible role of prolactin in breast carcinoma. Breast tissue—whether normal or cancerous—expresses the prolactin receptor, but the density of prolactin receptors is higher in tumor tissue. In several mouse models, prolactin induces tumor formation and increases tumor growth rates.23,24
Infertility. As noted, hyperprolactinemia can cause HPG axis dysfunction.9 A retrospective review by Sigman and Jarow25 linked endocrine disorders with infertility in 10% of 1,035 consecutive men attending 2 infertility centers. Hyperprolactinemia accounted for infertility in 0.4% of that population. No studies have associated hyperprolactinemia with female infertility.
Targeting hyperprolactinemia
When prescribing risperidone, consider obtaining a baseline serum prolactin level, especially in sexually mature patients. Repeat after 2 months, and ask the patient about menstruation, nipple discharge, sexual functioning, and pubertal development. Sexual side effects may be difficult to ascertain in patients receiving antipsychotics because of psychiatric comorbidities in this population.
Elevation without symptoms. If serum prolactin is elevated after 2 months but the patient has no clinical symptoms, repeat evaluation after another 2 months without altering the risperidone dosage. As discussed, serum prolactin tends to decline and may normalize with continued antipsychotic therapy in adults and children. A reasonable approach may be to wait 6 to 12 months for symptoms to resolve and hyperprolactinemia to diminish in patients who benefit from risperidone and have no or mild prolactin-related symptoms.8
Elevation with symptoms. Intervene if serum prolactin is elevated and your patient has clinical symptoms of hyperprolactinemia. Consider gradually tapering risperidone over 2 weeks and switching to a prolactin-sparing antipsychotic such as aripiprazole. If serum prolactin is >200 ng/mL or is persistently elevated despite switching to a prolactin-sparing antipsychotic, obtain an MRI of the sella turcica to look for a pituitary adenoma or parasellar tumor.
Use of dopamine agonists. Few studies have evaluated the safety and efficacy of using dopamine agonists such as cabergoline or amantadine to resolve the effects of hyperprolactinemia.26,27 Further research is warranted before this approach can be recommended.
Related resources
- Masi G, Cosenza A. Prolactin levels in young children with pervasive developmental disorders during risperidone treatment. J Child Adolesc Psychopharmacol 2001;11:389-94.
- Cheng-Shannon J, McGough JJ, Pataki C, McCracken JT. Second-generation antipsychotic medications in children and adolescents. J Child Adolesc Psychopharmacol 2004;14:372-94.
Drug brand names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Cabergoline • Dostinex
- Clozapine • Clozaril
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Freeman ME, Kanyicska B, Lerant A. Prolactin: structure, function, and regulation of secretion. Physiol Rev 2000;80:1523-1631.
2. Frantz AG. Prolactin. N Engl J Med 1978;198:201-7.
3. Guber HA, Farag AF. Evaluation of endocrine function. In: McPherson RA, Pincus MR, eds. Henry’s clinical diagnosis and management by laboratory methods. 21st ed. Philadelphia, PA: WB Saunders; 2006.
4. Melmed S, Jameson JL. Endocrinology and metabolism. In: Kasper DL, Braunwald E, Fauci AS, et al. eds. Harrison’s principles of internal medicine. 16th ed. New York, NY: McGraw-Hill; 2007.
5. Malone RP, Maislin G, Choudhary MS, et al. Risperidone treatment in children and adolescents with autism—short-and long-term safety and effectiveness. J Am Acad Child Adolesc Psychiatry 2002;41:140-7.
6. Colao A, Loche S, Cappabianca P. Pituitary adenomas in children and adolescents. Endocrinologist 2000;10:314-27.
7. Szarfman A, Tonning JM, Levine JG, et al. Atypical antipsychotics and pituitary tumors—a pharmacovigilance study. Pharmacotherapy 2006;26:748-58.
8. Correll CU, Carlson HE. Endocrine and metabolic adverse effects of psychotropic medications in children and adolescents. J Am Acad Child Adolesc Psychiatry 2006;45(7):771-91.
9. Haddad PM, Wieck A. Antipsychotic induced hyperprolactinemia: mechanisms, clinical features and management. Drugs 2004;64:2291-314.
10. Naidoo U, Goff DC, Klibanski A. Hyperprolactinemia and bone mineral density—the potential impact of antipsychotic agents. Psychoneuroendocrinol 2003;28:97-108.
11. O’Keane V, Meaney AM. Antipsychotic drugs: a new risk factor for osteoporosis in young women with schizophrenia? J Clin Psychopharmacol 2005;25:26-31.
12. Colao A, Di Somma C, Loche S, et al. Prolactinomas in adolescents: persistent bone loss after 2 years of prolactin normalization. Clin Endocrinol (Oxf) 2000;52:319-27.
13. Parks JS, Felner EI. Hormones of the hypothalamus and pituitary gland. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson textbook of pediatrics. 17th ed. Philadelphia, PA: Elsevier Science; 2003.
14. Massart F, Saggese G. Hyperprolactinaemia in children—a common diagnostic dilemma. Eur Endocrine Rev January 2006. Available at: http://www.touchbriefings.com/download.cfm?fileID=7546. Accessed August 26, 2008.
15. Staller J. The effect of long-term antipsychotic treatment on prolactin. J Child Adolesc Psychopharmacol 2006;16:317-26.
16. Saito E, Correll CU, Gallelli K, et al. A prospective study of hyperprolactinemia in children and adolescents treated with atypical antipsychotic agents. J Child Adolesc Psychopharmacol 2004;14:350-8.
17. Findling RL, Kusumakar V, Daneman D, et al. Prolactin levels during long-term risperidone treatment in children and adolescents. J Clin Psychiatry 2003;64:1362-9.
18. Anderson GM, Scahill L, McCracken JT, et al. Effects of short-and long-term risperidone treatment on prolactin levels in children with autism. Biol Psychiatry 2007;61:545-50.
19. Hellings JA, Zarcone JR, Valdovinos MG, et al. Risperidone-induced prolactin elevation in a prospective study of children, adolescents, and adults with mental retardation and pervasive developmental disorders. J Child Adolesc Psychopharmacol 2005;15:885-92.
20. Madhusoodanan S, Moise D. Risperidone induced hyperprolactinemia in adolescents: a case series. J Clin Psychiatry 2006;67:1110-3.
21. Holzer L, Eap CB. Risperidone-induced symptomatic hyperprolactinaemia in adolescents. J Clin Psychopharmacol 2006;26:167-71.
22. Halbreich U, Shen J, Panaro V. Are chronic psychiatric patients at increased risk for developing breast cancer? Am J Psychiatry 1996;153:59-60.
23. Hankinson SE, Willett WC, Michaud DS, et al. Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst 1999;91:629-34.
24. Ginsburg E, Vonderhann B. Prolactin synthesis and secretion by human breast cells. Cancer Res 1995;55:2591-5.
25. Sigman M, Jarow JP. Endocrine evaluation of infertile men. Urology 1997;50:659-64.
26. Cavallaro R, Cocchi F, Angelone SM, et al. Cabergoline treatment of risperidone-induced hyperprolactinemia: a pilot study. J Clin Psychiatry 2004;65(2):187-90.
27. Cohen LG, Biederman J. Treatment of risperidone-induced hyperprolactinemia with a dopamine agonist in children. J Child Adolesc Psychopharmacol 2001;11:435-40.
Serum prolactin increases in children and adolescents when risperidone therapy begins, then decreases over time in many patients. When prolactin levels remain elevated, evidence suggests that children may experience adverse effects such as delayed sexual maturation or reduced bone growth because of hypothalamic-pituitary-gonadal axis (HPG) dysfunction.
To help you make informed prescribing decisions, we discuss what the evidence says about the effects of elevated prolactin in children and adolescents. We then suggest clinical steps to help you manage hyperprolactinemia when prescribing risperidone.
Pediatric indications
Based on short-term clinical trials of efficacy and tolerability, risperidone is FDA-approved for 3 pediatric indications:
- short-term treatment of acute mania or mixed episodes associated with bipolar I disorder in patients age 10 to 17
- schizophrenia treatment in patients age 13 to 17
- treatment of irritability (including aggression, self-injury, temper tantrums, and mood swings) associated with autistic disorder in patients age 5 to 16.
Recommended risperidone dosages are lower for children and adolescents than for adults (Table 1). Off-label pediatric uses described in case reports include psychotic, mood, disruptive, movement, and pervasive developmental disorders.
Table 1
Recommended risperidone dosing for pediatric indications*
| Indication | Starting dose | Maximum dose |
|---|---|---|
| Acute mania or mixed episodes | 0.5 mg once daily in morning or evening | 2.5 mg/d |
| Irritability in autism | 0.25 mg/d for patients weighing <20 kg 0.5 mg/d for patients weighing ≥20 kg | 0.5 mg/d for patients weighing <20 kg 1 mg/d for patients weighing ≥20 kg |
| Schizophrenia | 0.5 mg once daily in morning or evening | 3 mg/d |
| * FDA-approved dosages; individualize based on response and tolerability | ||
| Source: Drug facts and comparisons. St. Louis, MO: Wolters Kluwer Health; 2008:949-50 | ||
Prolactin physiology
Prolactin’s primary physiologic function is to cause breast enlargement during pregnancy and milk secretion during lactation.1 A polypeptide hormone, prolactin is secreted by lactotroph cells in the anterior pituitary, under the complex control of stimulatory and inhibitory factors (Table 2). Its pulsatile secretion peaks 13 to 14 times daily, with approximately 95 minutes between pulses.
Serum prolactin levels show marked circadian variation.2 The reference value for serum prolactin is 1 to 25 ng/mL for women and 1 to 20 ng/mL for men. The higher prolactin levels seen in women begin after puberty and presumably are caused by estrogen’s stimulatory effect.3 Age- and sex-specific normal prolactin ranges vary widely and from lab to lab (Table 3).
Risperidone is a strong dopamine D2 and serotonin 5HT-2A antagonist with low affinity for alpha-1 and alpha-2 adrenergic receptors and histamine H1 receptors.4 Antagonism of these receptors is thought to explain the drug’s therapeutic effects and many of its side effects, including hyperprolactinemia.5 Prolactin release is also influenced by thyrotropin-releasing hormone.6 A rare association between pituitary tumors and atypical antipsychotics has been proposed as a probable cause of sustained prolactin elevation.7
Pituitary prolactin secretion is regulated by neuroendocrine neurons in the hypothalamus, specifically in the tuberoinfundibular tract that extends from the arcuate nucleus of the mediobasal hypothalamus (tuberal region) and projects to the median eminence (infundibular region). Neurosecretory dopamine neurons of the arcuate nucleus inhibit prolactin secretion. Hence, prolactin secretion increases when antipsychotic therapy results in dopamine receptor blockade.
Antipsychotics vary in affinity for the D2 dopamine receptor, rate of dissociation from the receptor, and ability to act on the receptor as both a dopamine agonist (which lowers serum prolactin) and a dopamine antagonist (which increases serum prolactin). Based on adult and pediatric data, the relative potency of antipsychotic drugs in inducing hyperprolactinemia is roughly risperidone > haloperidol > olanzapine > ziprasidone > quetiapine > clozapine > aripiprazole.8 Even though risperidone ranks highest in the hierarchy to cause hyperprolactinemia, it is accepted as the first-line antipsychotic in children and adolescents. This is probably because risperidone:
- has been in clinical use longer than other atypical antipsychotics except clozapine
- has received FDA approval for 3 pediatric indications.
Table 2
Factors that regulate prolactin secretion
| Effect | Factors | Mechanism |
|---|---|---|
| Inhibitory | Dopamine, gonadotropin-associated protein, acetylcholine | D2 receptor stimulation of lactotroph cells |
| Stimulatory | Serotonin, thyrotropin-releasing hormone, cholecystokinin | Through 5-HT1A and 5-HT2 |
Table 3
Sample age- and sex-specific reference ranges for serum prolactin (ng/mL)*
| Age | Males | Females |
|---|---|---|
| 0 to 1 month | 3.7 to 81.2 | 0.3 to 95.0 |
| 1 to 12 months | 0.3 to 28.9 | 0.2 to 29.9 |
| 1 to 3 years | 2.3 to 13.2 | 1.0 to 17.0 |
| 4 to 6 years | 0.8 to 16.9 | 1.6 to 13.1 |
| 7 to 9 years | 1.9 to 11.6 | 0.3 to 12.9 |
| 10 to 12 years | 0.9 to 12.9 | 1.9 to 9.6 |
| 13 to 15 years | 1.6 to 16.6 | 3.0 to 14.4 |
| Adult | 2.1 to 17.7 | 2.8 to 29.2 |
| Female: nonpregnant | 2.8 to 29.2 | |
| Female: pregnant | 9.7 to 208.5 | |
| Postmenopausal | 1.8 to 20.3 | |
| * Reference values may vary from lab to lab | ||
| Source: LabCorp, Birmingham, AL | ||
Prolactin and the HPG axis
Elevated serum prolactin inhibits the hypothalamus’ pulsatile release of gonadotrophin-releasing hormone (GnRH), which in turn decreases the pituitary’s secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). In women, prolactin also blocks the feedback effect of estradiol on LH secretion (Figure). The prolactin level that triggers gonadal hypofunction appears to vary substantially among individuals.9
Symptoms of elevated prolactin can occur as a direct result of prolactin’s physiologic effect on breast tissue or indirectly through hypogonadism related to decreased FSH and LH. Symptoms of hyperprolactinemia—which can be seen more readily in sexually mature adolescents than in children—include:
- amenorrhea or oligorrhea
- breast enlargement or engorgement in females and males
- galactorrhea (females > males)
- decreased libido
- erectile dysfunction.
Although evidence is inconclusive, other problems may be associated with increased prolactin in children and adolescents. These include failure to enter or progress through puberty,8 increased risk of benign breast tumors,22 and reduced bone density.10
Bone changes. Decreased estrogen related to hyperprolactinemia may inhibit bone mineralization, causing osteopenia, osteoporosis, and increased fracture risk.10 The mechanism of bone density loss may be estrogen’s osteoclast activating and osteoblast inhibiting action. The level and duration of prolactin elevation that can hamper bone growth has not been defined, although evidence suggests a pervasive effect:
- 65% of a group of 38 premenstrual patients developed osteoporosis or osteopenia when taking risperidone or typical antipsychotics for schizophrenia for a mean of 8 years.11
- Bone loss has persisted 2 years after prolactin normalized in adolescents with prolactinomas.12
Prolactin secretion is controlled by stimulatory and inhibitory influences (A). Antipsychotic blockade of dopamine’s inhibitory influence (B) increases serum prolactin and its effect on mammary tissue (C). In the brain, hyperprolactinemia inhibits the release of gonadotropin-releasing hormone (GnRH) by the hypothalamus, which results in decreased follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secretion by the pituitary (D). FSH and LH are important determinants of male and female gonadal maturation by their direct action on testes and ovaries within the hypothalamic-pituitary-gonadal axis.
Source: Developed by Manpreet Khemka, MBBS, and Jeffrey Ali, MD, MSc. Current Psychiatry Illustration by Rob Flewell
Hyperprolactinemia in children and adolescents
We suggest that children and adolescents receiving prolonged risperidone treatment can present with symptoms similar to those associated with hyperprolactinemia secondary to other causes, including:
- prolactinomas (the most common cause)13
- thyrotropin-releasing hormone stimulation in primary hypothyroidism
- hypoglycemia
- inherited endocrine syndromes
- physiologic stress
- medications.
The most common presenting symptoms of prolactinomas are headache, amenorrhea, and galactorrhea. A few patients have delayed puberty.13 In a review of hyperprolactinemia in children, Massart and Saggese14 proposed a correlation between elevated serum prolactin and underlying pathology:
- >100 ng/mL usually suggests organic pathology and requires MRI or CT confirmation
- <100 ng/mL usually indicates functional pathology.
What the evidence says. Using the key words “risperidone and hyperprolactinemia in children and adolescents” in a PubMed search, we identified 7 prospective, cross-sectional, and retrospective studies.14-20 We then analyzed these studies in terms of subjects’ age, sex, primary psychiatric disorder, dosage of risperidone used, prolactin elevation pattern, reported clinical consequences, and interventions used to ameliorate asymptomatic or symptomatic hyperprolactinemia.
Prolactin and antipsychotics. In a cross-sectional study, Staller15 compared serum prolactin at baseline and after 6 months in 50 children treated with atypical antipsychotics. Patients taking risperidone showed greater increases in prolactin than those taking quetiapine or olanzapine. Saito et al16 reached a similar conclusion in a prospective study of 40 children treated with atypical antipsychotics for 4 to 15 weeks.
Ups and downs. Prolactin levels increase sharply in the first weeks of risperidone treatment, peak at around 6 to 8 weeks, and then trend downward toward normal.17 In a post hoc analysis of pooled data from 5 clinical trials totaling 700 patients age 5 to 15, Findling et al17 reported that mean serum prolactin:
- peaked in the first 1 to 2 months of patients’ starting risperidone, 0.02 to 0.06 mg/kg/d
- returned to within or close to normal range by 3 to 5 months.
No correlation was seen between prolactin elevation and side effects that could be attributed to prolactin.
In a 2-part study, Anderson et al18 examined the short- and long-term effects of risperidone treatment on prolactin in children age 5 to 17 with autism. In the initial double-blind, placebo-controlled trial, 101 children were randomly assigned to risperidone 1.8 mg/d, or placebo. After 8 weeks, 63 children continued with open-label risperidone, mean dose 1.96 mg/d, for up to a total 22 months. Serum prolactin was measured at baseline (9.3±7.5 ng/ mL) and then at 2, 6, and 22 months.
Serum prolactin increased sharply in children treated with risperidone in the placebo-controlled trial. After 8 weeks, prolactin levels were 4 times higher with risperidone treatment than with placebo (39.0±19.2 ng/mL vs 10.1±8.8 ng/mL [P< 0.0001]). In the open-label risperidone continuation trial, prolactin levels remained significantly higher than at baseline but decreased over time to:
- 32.4±17.8 ng/mL in 43 children who remained in the study at 6 months (P< 0.0001)
- 25.3±15.6 ng/mL in 30 children who remained at 22 months (P< 0.0001).
In this study, a sharp rise in serum prolactin in the first 2 months trended down to the upper limit of normal at 22 months. None of the children showed known clinical manifestations of elevated prolactin, including gynecomastia, galactorrhea, or menstrual disturbance.
A double-blind, placebo-controlled trial by Hellings et al18 examined risperidone’s effect on aggression and self-injury in children, adolescents, and adults with mental retardation and pervasive developmental disorders. In a subset of 10 children and adolescents whose serum prolactin was measured during the trial, prolactin remained elevated during at least 26 weeks of risperidone treatment. Mean prolactin levels were:
- 13.2±8.6 ng/mL at baseline
- 31.0±11.6 ng/mL during acute risperidone therapy
- 37.9±10.4 ng/mL during maintenance therapy.
Clinical features. Higher risperidone dosages—rather than longer duration of use—appear more likely to cause symptomatic elevated serum prolactin. A case series of 3 adolescents with symptomatic prolactin elevation showed:
- gynecomastia and galactorrhea in 2 adolescent males age 17 and 18, receiving risperidone, 4 mg/d and 5 mg/d, respectively
- amenorrhea within 2 to 6 weeks of starting treatment in a female patient age 15 receiving risperidone, 6 mg/d.20
Holzer et al21 described 5 adolescents who showed symptoms of elevated prolactin after 3 to 15 months while taking risperidone, 2 to 6 mg/d, as treatment for psychosis.
Long-term health risks?
Some evidence suggests an association between elevated serum prolactin and carcinogenesis and infertility in adults. No studies have examined these long-term risks in children and adolescents who develop hyperprolactinemia from risperidone treatment. A link may be possible, however, if prolactin elevation affects postpubertal and HPG axis development.
Breast cancer. Halbriech et al22 reviewed mammograms and charts of 275 female psychiatric hospital patients age >40 and 928 women of similar age at a hospital radiology clinic. The incidence of breast cancer among psychiatric patients was:
- >3.5 times higher than among radiology clinic patients
- 9.5 times higher than in the general population.
The authors speculated that the observed increased breast cancer incidence in psychiatric patients could be associated with medications, although high rates of cigarette smoking and alcohol consumption also might have played a role.
A case-control study by Hankinson et al23 of blood samples collected from women in the Nurses’ Health Study found a statistically significant association between hyperprolactinemia and breast cancer. This analysis included 306 postmenopausal women diagnosed with breast cancer and 448 controls matched for age, postmenopausal hormone use, and time of day and month when blood samples were drawn.
Several putative mechanisms have been proposed to explain a possible role of prolactin in breast carcinoma. Breast tissue—whether normal or cancerous—expresses the prolactin receptor, but the density of prolactin receptors is higher in tumor tissue. In several mouse models, prolactin induces tumor formation and increases tumor growth rates.23,24
Infertility. As noted, hyperprolactinemia can cause HPG axis dysfunction.9 A retrospective review by Sigman and Jarow25 linked endocrine disorders with infertility in 10% of 1,035 consecutive men attending 2 infertility centers. Hyperprolactinemia accounted for infertility in 0.4% of that population. No studies have associated hyperprolactinemia with female infertility.
Targeting hyperprolactinemia
When prescribing risperidone, consider obtaining a baseline serum prolactin level, especially in sexually mature patients. Repeat after 2 months, and ask the patient about menstruation, nipple discharge, sexual functioning, and pubertal development. Sexual side effects may be difficult to ascertain in patients receiving antipsychotics because of psychiatric comorbidities in this population.
Elevation without symptoms. If serum prolactin is elevated after 2 months but the patient has no clinical symptoms, repeat evaluation after another 2 months without altering the risperidone dosage. As discussed, serum prolactin tends to decline and may normalize with continued antipsychotic therapy in adults and children. A reasonable approach may be to wait 6 to 12 months for symptoms to resolve and hyperprolactinemia to diminish in patients who benefit from risperidone and have no or mild prolactin-related symptoms.8
Elevation with symptoms. Intervene if serum prolactin is elevated and your patient has clinical symptoms of hyperprolactinemia. Consider gradually tapering risperidone over 2 weeks and switching to a prolactin-sparing antipsychotic such as aripiprazole. If serum prolactin is >200 ng/mL or is persistently elevated despite switching to a prolactin-sparing antipsychotic, obtain an MRI of the sella turcica to look for a pituitary adenoma or parasellar tumor.
Use of dopamine agonists. Few studies have evaluated the safety and efficacy of using dopamine agonists such as cabergoline or amantadine to resolve the effects of hyperprolactinemia.26,27 Further research is warranted before this approach can be recommended.
Related resources
- Masi G, Cosenza A. Prolactin levels in young children with pervasive developmental disorders during risperidone treatment. J Child Adolesc Psychopharmacol 2001;11:389-94.
- Cheng-Shannon J, McGough JJ, Pataki C, McCracken JT. Second-generation antipsychotic medications in children and adolescents. J Child Adolesc Psychopharmacol 2004;14:372-94.
Drug brand names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Cabergoline • Dostinex
- Clozapine • Clozaril
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Serum prolactin increases in children and adolescents when risperidone therapy begins, then decreases over time in many patients. When prolactin levels remain elevated, evidence suggests that children may experience adverse effects such as delayed sexual maturation or reduced bone growth because of hypothalamic-pituitary-gonadal axis (HPG) dysfunction.
To help you make informed prescribing decisions, we discuss what the evidence says about the effects of elevated prolactin in children and adolescents. We then suggest clinical steps to help you manage hyperprolactinemia when prescribing risperidone.
Pediatric indications
Based on short-term clinical trials of efficacy and tolerability, risperidone is FDA-approved for 3 pediatric indications:
- short-term treatment of acute mania or mixed episodes associated with bipolar I disorder in patients age 10 to 17
- schizophrenia treatment in patients age 13 to 17
- treatment of irritability (including aggression, self-injury, temper tantrums, and mood swings) associated with autistic disorder in patients age 5 to 16.
Recommended risperidone dosages are lower for children and adolescents than for adults (Table 1). Off-label pediatric uses described in case reports include psychotic, mood, disruptive, movement, and pervasive developmental disorders.
Table 1
Recommended risperidone dosing for pediatric indications*
| Indication | Starting dose | Maximum dose |
|---|---|---|
| Acute mania or mixed episodes | 0.5 mg once daily in morning or evening | 2.5 mg/d |
| Irritability in autism | 0.25 mg/d for patients weighing <20 kg 0.5 mg/d for patients weighing ≥20 kg | 0.5 mg/d for patients weighing <20 kg 1 mg/d for patients weighing ≥20 kg |
| Schizophrenia | 0.5 mg once daily in morning or evening | 3 mg/d |
| * FDA-approved dosages; individualize based on response and tolerability | ||
| Source: Drug facts and comparisons. St. Louis, MO: Wolters Kluwer Health; 2008:949-50 | ||
Prolactin physiology
Prolactin’s primary physiologic function is to cause breast enlargement during pregnancy and milk secretion during lactation.1 A polypeptide hormone, prolactin is secreted by lactotroph cells in the anterior pituitary, under the complex control of stimulatory and inhibitory factors (Table 2). Its pulsatile secretion peaks 13 to 14 times daily, with approximately 95 minutes between pulses.
Serum prolactin levels show marked circadian variation.2 The reference value for serum prolactin is 1 to 25 ng/mL for women and 1 to 20 ng/mL for men. The higher prolactin levels seen in women begin after puberty and presumably are caused by estrogen’s stimulatory effect.3 Age- and sex-specific normal prolactin ranges vary widely and from lab to lab (Table 3).
Risperidone is a strong dopamine D2 and serotonin 5HT-2A antagonist with low affinity for alpha-1 and alpha-2 adrenergic receptors and histamine H1 receptors.4 Antagonism of these receptors is thought to explain the drug’s therapeutic effects and many of its side effects, including hyperprolactinemia.5 Prolactin release is also influenced by thyrotropin-releasing hormone.6 A rare association between pituitary tumors and atypical antipsychotics has been proposed as a probable cause of sustained prolactin elevation.7
Pituitary prolactin secretion is regulated by neuroendocrine neurons in the hypothalamus, specifically in the tuberoinfundibular tract that extends from the arcuate nucleus of the mediobasal hypothalamus (tuberal region) and projects to the median eminence (infundibular region). Neurosecretory dopamine neurons of the arcuate nucleus inhibit prolactin secretion. Hence, prolactin secretion increases when antipsychotic therapy results in dopamine receptor blockade.
Antipsychotics vary in affinity for the D2 dopamine receptor, rate of dissociation from the receptor, and ability to act on the receptor as both a dopamine agonist (which lowers serum prolactin) and a dopamine antagonist (which increases serum prolactin). Based on adult and pediatric data, the relative potency of antipsychotic drugs in inducing hyperprolactinemia is roughly risperidone > haloperidol > olanzapine > ziprasidone > quetiapine > clozapine > aripiprazole.8 Even though risperidone ranks highest in the hierarchy to cause hyperprolactinemia, it is accepted as the first-line antipsychotic in children and adolescents. This is probably because risperidone:
- has been in clinical use longer than other atypical antipsychotics except clozapine
- has received FDA approval for 3 pediatric indications.
Table 2
Factors that regulate prolactin secretion
| Effect | Factors | Mechanism |
|---|---|---|
| Inhibitory | Dopamine, gonadotropin-associated protein, acetylcholine | D2 receptor stimulation of lactotroph cells |
| Stimulatory | Serotonin, thyrotropin-releasing hormone, cholecystokinin | Through 5-HT1A and 5-HT2 |
Table 3
Sample age- and sex-specific reference ranges for serum prolactin (ng/mL)*
| Age | Males | Females |
|---|---|---|
| 0 to 1 month | 3.7 to 81.2 | 0.3 to 95.0 |
| 1 to 12 months | 0.3 to 28.9 | 0.2 to 29.9 |
| 1 to 3 years | 2.3 to 13.2 | 1.0 to 17.0 |
| 4 to 6 years | 0.8 to 16.9 | 1.6 to 13.1 |
| 7 to 9 years | 1.9 to 11.6 | 0.3 to 12.9 |
| 10 to 12 years | 0.9 to 12.9 | 1.9 to 9.6 |
| 13 to 15 years | 1.6 to 16.6 | 3.0 to 14.4 |
| Adult | 2.1 to 17.7 | 2.8 to 29.2 |
| Female: nonpregnant | 2.8 to 29.2 | |
| Female: pregnant | 9.7 to 208.5 | |
| Postmenopausal | 1.8 to 20.3 | |
| * Reference values may vary from lab to lab | ||
| Source: LabCorp, Birmingham, AL | ||
Prolactin and the HPG axis
Elevated serum prolactin inhibits the hypothalamus’ pulsatile release of gonadotrophin-releasing hormone (GnRH), which in turn decreases the pituitary’s secretion of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). In women, prolactin also blocks the feedback effect of estradiol on LH secretion (Figure). The prolactin level that triggers gonadal hypofunction appears to vary substantially among individuals.9
Symptoms of elevated prolactin can occur as a direct result of prolactin’s physiologic effect on breast tissue or indirectly through hypogonadism related to decreased FSH and LH. Symptoms of hyperprolactinemia—which can be seen more readily in sexually mature adolescents than in children—include:
- amenorrhea or oligorrhea
- breast enlargement or engorgement in females and males
- galactorrhea (females > males)
- decreased libido
- erectile dysfunction.
Although evidence is inconclusive, other problems may be associated with increased prolactin in children and adolescents. These include failure to enter or progress through puberty,8 increased risk of benign breast tumors,22 and reduced bone density.10
Bone changes. Decreased estrogen related to hyperprolactinemia may inhibit bone mineralization, causing osteopenia, osteoporosis, and increased fracture risk.10 The mechanism of bone density loss may be estrogen’s osteoclast activating and osteoblast inhibiting action. The level and duration of prolactin elevation that can hamper bone growth has not been defined, although evidence suggests a pervasive effect:
- 65% of a group of 38 premenstrual patients developed osteoporosis or osteopenia when taking risperidone or typical antipsychotics for schizophrenia for a mean of 8 years.11
- Bone loss has persisted 2 years after prolactin normalized in adolescents with prolactinomas.12
Prolactin secretion is controlled by stimulatory and inhibitory influences (A). Antipsychotic blockade of dopamine’s inhibitory influence (B) increases serum prolactin and its effect on mammary tissue (C). In the brain, hyperprolactinemia inhibits the release of gonadotropin-releasing hormone (GnRH) by the hypothalamus, which results in decreased follicle-stimulating hormone (FSH) and luteinizing hormone (LH) secretion by the pituitary (D). FSH and LH are important determinants of male and female gonadal maturation by their direct action on testes and ovaries within the hypothalamic-pituitary-gonadal axis.
Source: Developed by Manpreet Khemka, MBBS, and Jeffrey Ali, MD, MSc. Current Psychiatry Illustration by Rob Flewell
Hyperprolactinemia in children and adolescents
We suggest that children and adolescents receiving prolonged risperidone treatment can present with symptoms similar to those associated with hyperprolactinemia secondary to other causes, including:
- prolactinomas (the most common cause)13
- thyrotropin-releasing hormone stimulation in primary hypothyroidism
- hypoglycemia
- inherited endocrine syndromes
- physiologic stress
- medications.
The most common presenting symptoms of prolactinomas are headache, amenorrhea, and galactorrhea. A few patients have delayed puberty.13 In a review of hyperprolactinemia in children, Massart and Saggese14 proposed a correlation between elevated serum prolactin and underlying pathology:
- >100 ng/mL usually suggests organic pathology and requires MRI or CT confirmation
- <100 ng/mL usually indicates functional pathology.
What the evidence says. Using the key words “risperidone and hyperprolactinemia in children and adolescents” in a PubMed search, we identified 7 prospective, cross-sectional, and retrospective studies.14-20 We then analyzed these studies in terms of subjects’ age, sex, primary psychiatric disorder, dosage of risperidone used, prolactin elevation pattern, reported clinical consequences, and interventions used to ameliorate asymptomatic or symptomatic hyperprolactinemia.
Prolactin and antipsychotics. In a cross-sectional study, Staller15 compared serum prolactin at baseline and after 6 months in 50 children treated with atypical antipsychotics. Patients taking risperidone showed greater increases in prolactin than those taking quetiapine or olanzapine. Saito et al16 reached a similar conclusion in a prospective study of 40 children treated with atypical antipsychotics for 4 to 15 weeks.
Ups and downs. Prolactin levels increase sharply in the first weeks of risperidone treatment, peak at around 6 to 8 weeks, and then trend downward toward normal.17 In a post hoc analysis of pooled data from 5 clinical trials totaling 700 patients age 5 to 15, Findling et al17 reported that mean serum prolactin:
- peaked in the first 1 to 2 months of patients’ starting risperidone, 0.02 to 0.06 mg/kg/d
- returned to within or close to normal range by 3 to 5 months.
No correlation was seen between prolactin elevation and side effects that could be attributed to prolactin.
In a 2-part study, Anderson et al18 examined the short- and long-term effects of risperidone treatment on prolactin in children age 5 to 17 with autism. In the initial double-blind, placebo-controlled trial, 101 children were randomly assigned to risperidone 1.8 mg/d, or placebo. After 8 weeks, 63 children continued with open-label risperidone, mean dose 1.96 mg/d, for up to a total 22 months. Serum prolactin was measured at baseline (9.3±7.5 ng/ mL) and then at 2, 6, and 22 months.
Serum prolactin increased sharply in children treated with risperidone in the placebo-controlled trial. After 8 weeks, prolactin levels were 4 times higher with risperidone treatment than with placebo (39.0±19.2 ng/mL vs 10.1±8.8 ng/mL [P< 0.0001]). In the open-label risperidone continuation trial, prolactin levels remained significantly higher than at baseline but decreased over time to:
- 32.4±17.8 ng/mL in 43 children who remained in the study at 6 months (P< 0.0001)
- 25.3±15.6 ng/mL in 30 children who remained at 22 months (P< 0.0001).
In this study, a sharp rise in serum prolactin in the first 2 months trended down to the upper limit of normal at 22 months. None of the children showed known clinical manifestations of elevated prolactin, including gynecomastia, galactorrhea, or menstrual disturbance.
A double-blind, placebo-controlled trial by Hellings et al18 examined risperidone’s effect on aggression and self-injury in children, adolescents, and adults with mental retardation and pervasive developmental disorders. In a subset of 10 children and adolescents whose serum prolactin was measured during the trial, prolactin remained elevated during at least 26 weeks of risperidone treatment. Mean prolactin levels were:
- 13.2±8.6 ng/mL at baseline
- 31.0±11.6 ng/mL during acute risperidone therapy
- 37.9±10.4 ng/mL during maintenance therapy.
Clinical features. Higher risperidone dosages—rather than longer duration of use—appear more likely to cause symptomatic elevated serum prolactin. A case series of 3 adolescents with symptomatic prolactin elevation showed:
- gynecomastia and galactorrhea in 2 adolescent males age 17 and 18, receiving risperidone, 4 mg/d and 5 mg/d, respectively
- amenorrhea within 2 to 6 weeks of starting treatment in a female patient age 15 receiving risperidone, 6 mg/d.20
Holzer et al21 described 5 adolescents who showed symptoms of elevated prolactin after 3 to 15 months while taking risperidone, 2 to 6 mg/d, as treatment for psychosis.
Long-term health risks?
Some evidence suggests an association between elevated serum prolactin and carcinogenesis and infertility in adults. No studies have examined these long-term risks in children and adolescents who develop hyperprolactinemia from risperidone treatment. A link may be possible, however, if prolactin elevation affects postpubertal and HPG axis development.
Breast cancer. Halbriech et al22 reviewed mammograms and charts of 275 female psychiatric hospital patients age >40 and 928 women of similar age at a hospital radiology clinic. The incidence of breast cancer among psychiatric patients was:
- >3.5 times higher than among radiology clinic patients
- 9.5 times higher than in the general population.
The authors speculated that the observed increased breast cancer incidence in psychiatric patients could be associated with medications, although high rates of cigarette smoking and alcohol consumption also might have played a role.
A case-control study by Hankinson et al23 of blood samples collected from women in the Nurses’ Health Study found a statistically significant association between hyperprolactinemia and breast cancer. This analysis included 306 postmenopausal women diagnosed with breast cancer and 448 controls matched for age, postmenopausal hormone use, and time of day and month when blood samples were drawn.
Several putative mechanisms have been proposed to explain a possible role of prolactin in breast carcinoma. Breast tissue—whether normal or cancerous—expresses the prolactin receptor, but the density of prolactin receptors is higher in tumor tissue. In several mouse models, prolactin induces tumor formation and increases tumor growth rates.23,24
Infertility. As noted, hyperprolactinemia can cause HPG axis dysfunction.9 A retrospective review by Sigman and Jarow25 linked endocrine disorders with infertility in 10% of 1,035 consecutive men attending 2 infertility centers. Hyperprolactinemia accounted for infertility in 0.4% of that population. No studies have associated hyperprolactinemia with female infertility.
Targeting hyperprolactinemia
When prescribing risperidone, consider obtaining a baseline serum prolactin level, especially in sexually mature patients. Repeat after 2 months, and ask the patient about menstruation, nipple discharge, sexual functioning, and pubertal development. Sexual side effects may be difficult to ascertain in patients receiving antipsychotics because of psychiatric comorbidities in this population.
Elevation without symptoms. If serum prolactin is elevated after 2 months but the patient has no clinical symptoms, repeat evaluation after another 2 months without altering the risperidone dosage. As discussed, serum prolactin tends to decline and may normalize with continued antipsychotic therapy in adults and children. A reasonable approach may be to wait 6 to 12 months for symptoms to resolve and hyperprolactinemia to diminish in patients who benefit from risperidone and have no or mild prolactin-related symptoms.8
Elevation with symptoms. Intervene if serum prolactin is elevated and your patient has clinical symptoms of hyperprolactinemia. Consider gradually tapering risperidone over 2 weeks and switching to a prolactin-sparing antipsychotic such as aripiprazole. If serum prolactin is >200 ng/mL or is persistently elevated despite switching to a prolactin-sparing antipsychotic, obtain an MRI of the sella turcica to look for a pituitary adenoma or parasellar tumor.
Use of dopamine agonists. Few studies have evaluated the safety and efficacy of using dopamine agonists such as cabergoline or amantadine to resolve the effects of hyperprolactinemia.26,27 Further research is warranted before this approach can be recommended.
Related resources
- Masi G, Cosenza A. Prolactin levels in young children with pervasive developmental disorders during risperidone treatment. J Child Adolesc Psychopharmacol 2001;11:389-94.
- Cheng-Shannon J, McGough JJ, Pataki C, McCracken JT. Second-generation antipsychotic medications in children and adolescents. J Child Adolesc Psychopharmacol 2004;14:372-94.
Drug brand names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Cabergoline • Dostinex
- Clozapine • Clozaril
- Haloperidol • Haldol
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Freeman ME, Kanyicska B, Lerant A. Prolactin: structure, function, and regulation of secretion. Physiol Rev 2000;80:1523-1631.
2. Frantz AG. Prolactin. N Engl J Med 1978;198:201-7.
3. Guber HA, Farag AF. Evaluation of endocrine function. In: McPherson RA, Pincus MR, eds. Henry’s clinical diagnosis and management by laboratory methods. 21st ed. Philadelphia, PA: WB Saunders; 2006.
4. Melmed S, Jameson JL. Endocrinology and metabolism. In: Kasper DL, Braunwald E, Fauci AS, et al. eds. Harrison’s principles of internal medicine. 16th ed. New York, NY: McGraw-Hill; 2007.
5. Malone RP, Maislin G, Choudhary MS, et al. Risperidone treatment in children and adolescents with autism—short-and long-term safety and effectiveness. J Am Acad Child Adolesc Psychiatry 2002;41:140-7.
6. Colao A, Loche S, Cappabianca P. Pituitary adenomas in children and adolescents. Endocrinologist 2000;10:314-27.
7. Szarfman A, Tonning JM, Levine JG, et al. Atypical antipsychotics and pituitary tumors—a pharmacovigilance study. Pharmacotherapy 2006;26:748-58.
8. Correll CU, Carlson HE. Endocrine and metabolic adverse effects of psychotropic medications in children and adolescents. J Am Acad Child Adolesc Psychiatry 2006;45(7):771-91.
9. Haddad PM, Wieck A. Antipsychotic induced hyperprolactinemia: mechanisms, clinical features and management. Drugs 2004;64:2291-314.
10. Naidoo U, Goff DC, Klibanski A. Hyperprolactinemia and bone mineral density—the potential impact of antipsychotic agents. Psychoneuroendocrinol 2003;28:97-108.
11. O’Keane V, Meaney AM. Antipsychotic drugs: a new risk factor for osteoporosis in young women with schizophrenia? J Clin Psychopharmacol 2005;25:26-31.
12. Colao A, Di Somma C, Loche S, et al. Prolactinomas in adolescents: persistent bone loss after 2 years of prolactin normalization. Clin Endocrinol (Oxf) 2000;52:319-27.
13. Parks JS, Felner EI. Hormones of the hypothalamus and pituitary gland. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson textbook of pediatrics. 17th ed. Philadelphia, PA: Elsevier Science; 2003.
14. Massart F, Saggese G. Hyperprolactinaemia in children—a common diagnostic dilemma. Eur Endocrine Rev January 2006. Available at: http://www.touchbriefings.com/download.cfm?fileID=7546. Accessed August 26, 2008.
15. Staller J. The effect of long-term antipsychotic treatment on prolactin. J Child Adolesc Psychopharmacol 2006;16:317-26.
16. Saito E, Correll CU, Gallelli K, et al. A prospective study of hyperprolactinemia in children and adolescents treated with atypical antipsychotic agents. J Child Adolesc Psychopharmacol 2004;14:350-8.
17. Findling RL, Kusumakar V, Daneman D, et al. Prolactin levels during long-term risperidone treatment in children and adolescents. J Clin Psychiatry 2003;64:1362-9.
18. Anderson GM, Scahill L, McCracken JT, et al. Effects of short-and long-term risperidone treatment on prolactin levels in children with autism. Biol Psychiatry 2007;61:545-50.
19. Hellings JA, Zarcone JR, Valdovinos MG, et al. Risperidone-induced prolactin elevation in a prospective study of children, adolescents, and adults with mental retardation and pervasive developmental disorders. J Child Adolesc Psychopharmacol 2005;15:885-92.
20. Madhusoodanan S, Moise D. Risperidone induced hyperprolactinemia in adolescents: a case series. J Clin Psychiatry 2006;67:1110-3.
21. Holzer L, Eap CB. Risperidone-induced symptomatic hyperprolactinaemia in adolescents. J Clin Psychopharmacol 2006;26:167-71.
22. Halbreich U, Shen J, Panaro V. Are chronic psychiatric patients at increased risk for developing breast cancer? Am J Psychiatry 1996;153:59-60.
23. Hankinson SE, Willett WC, Michaud DS, et al. Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst 1999;91:629-34.
24. Ginsburg E, Vonderhann B. Prolactin synthesis and secretion by human breast cells. Cancer Res 1995;55:2591-5.
25. Sigman M, Jarow JP. Endocrine evaluation of infertile men. Urology 1997;50:659-64.
26. Cavallaro R, Cocchi F, Angelone SM, et al. Cabergoline treatment of risperidone-induced hyperprolactinemia: a pilot study. J Clin Psychiatry 2004;65(2):187-90.
27. Cohen LG, Biederman J. Treatment of risperidone-induced hyperprolactinemia with a dopamine agonist in children. J Child Adolesc Psychopharmacol 2001;11:435-40.
1. Freeman ME, Kanyicska B, Lerant A. Prolactin: structure, function, and regulation of secretion. Physiol Rev 2000;80:1523-1631.
2. Frantz AG. Prolactin. N Engl J Med 1978;198:201-7.
3. Guber HA, Farag AF. Evaluation of endocrine function. In: McPherson RA, Pincus MR, eds. Henry’s clinical diagnosis and management by laboratory methods. 21st ed. Philadelphia, PA: WB Saunders; 2006.
4. Melmed S, Jameson JL. Endocrinology and metabolism. In: Kasper DL, Braunwald E, Fauci AS, et al. eds. Harrison’s principles of internal medicine. 16th ed. New York, NY: McGraw-Hill; 2007.
5. Malone RP, Maislin G, Choudhary MS, et al. Risperidone treatment in children and adolescents with autism—short-and long-term safety and effectiveness. J Am Acad Child Adolesc Psychiatry 2002;41:140-7.
6. Colao A, Loche S, Cappabianca P. Pituitary adenomas in children and adolescents. Endocrinologist 2000;10:314-27.
7. Szarfman A, Tonning JM, Levine JG, et al. Atypical antipsychotics and pituitary tumors—a pharmacovigilance study. Pharmacotherapy 2006;26:748-58.
8. Correll CU, Carlson HE. Endocrine and metabolic adverse effects of psychotropic medications in children and adolescents. J Am Acad Child Adolesc Psychiatry 2006;45(7):771-91.
9. Haddad PM, Wieck A. Antipsychotic induced hyperprolactinemia: mechanisms, clinical features and management. Drugs 2004;64:2291-314.
10. Naidoo U, Goff DC, Klibanski A. Hyperprolactinemia and bone mineral density—the potential impact of antipsychotic agents. Psychoneuroendocrinol 2003;28:97-108.
11. O’Keane V, Meaney AM. Antipsychotic drugs: a new risk factor for osteoporosis in young women with schizophrenia? J Clin Psychopharmacol 2005;25:26-31.
12. Colao A, Di Somma C, Loche S, et al. Prolactinomas in adolescents: persistent bone loss after 2 years of prolactin normalization. Clin Endocrinol (Oxf) 2000;52:319-27.
13. Parks JS, Felner EI. Hormones of the hypothalamus and pituitary gland. In: Behrman RE, Kliegman RM, Jenson HB, eds. Nelson textbook of pediatrics. 17th ed. Philadelphia, PA: Elsevier Science; 2003.
14. Massart F, Saggese G. Hyperprolactinaemia in children—a common diagnostic dilemma. Eur Endocrine Rev January 2006. Available at: http://www.touchbriefings.com/download.cfm?fileID=7546. Accessed August 26, 2008.
15. Staller J. The effect of long-term antipsychotic treatment on prolactin. J Child Adolesc Psychopharmacol 2006;16:317-26.
16. Saito E, Correll CU, Gallelli K, et al. A prospective study of hyperprolactinemia in children and adolescents treated with atypical antipsychotic agents. J Child Adolesc Psychopharmacol 2004;14:350-8.
17. Findling RL, Kusumakar V, Daneman D, et al. Prolactin levels during long-term risperidone treatment in children and adolescents. J Clin Psychiatry 2003;64:1362-9.
18. Anderson GM, Scahill L, McCracken JT, et al. Effects of short-and long-term risperidone treatment on prolactin levels in children with autism. Biol Psychiatry 2007;61:545-50.
19. Hellings JA, Zarcone JR, Valdovinos MG, et al. Risperidone-induced prolactin elevation in a prospective study of children, adolescents, and adults with mental retardation and pervasive developmental disorders. J Child Adolesc Psychopharmacol 2005;15:885-92.
20. Madhusoodanan S, Moise D. Risperidone induced hyperprolactinemia in adolescents: a case series. J Clin Psychiatry 2006;67:1110-3.
21. Holzer L, Eap CB. Risperidone-induced symptomatic hyperprolactinaemia in adolescents. J Clin Psychopharmacol 2006;26:167-71.
22. Halbreich U, Shen J, Panaro V. Are chronic psychiatric patients at increased risk for developing breast cancer? Am J Psychiatry 1996;153:59-60.
23. Hankinson SE, Willett WC, Michaud DS, et al. Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst 1999;91:629-34.
24. Ginsburg E, Vonderhann B. Prolactin synthesis and secretion by human breast cells. Cancer Res 1995;55:2591-5.
25. Sigman M, Jarow JP. Endocrine evaluation of infertile men. Urology 1997;50:659-64.
26. Cavallaro R, Cocchi F, Angelone SM, et al. Cabergoline treatment of risperidone-induced hyperprolactinemia: a pilot study. J Clin Psychiatry 2004;65(2):187-90.
27. Cohen LG, Biederman J. Treatment of risperidone-induced hyperprolactinemia with a dopamine agonist in children. J Child Adolesc Psychopharmacol 2001;11:435-40.
Stimulants for adult bipolar disorder?
Patients with bipolar disorder show an unpredictable range of responses to stimulants, from virtually no ill effects to emerging manic-like symptoms.1 Thus, although stimulants may be beneficial to some bipolar patients, there is a great deal of concern about using stimulants in this population. Even so, stimulants may be a rational adjunct for treating certain aspects of bipolar illness, particularly resistant depression, iatrogenic sedation, and comorbid attention-deficit/hyperactivity disorder (ADHD).
To help you decide if and when your patient might be a candidate for stimulant therapy, this article:
- reviews the evidence on stimulants’ safety and tolerability for patients with bipolar disorder
- weighs potential benefits and risks of using stimulants in this population
- addresses stimulants’ possible adverse effects on illness course and from interactions with other psychotropics
- discusses treatment options based on the limited evidence and our clinical experience.
Limited support
We are aware that using stimulants to treat patients with bipolar disorder is not an uncommon clinical practice, but supportive evidence is limited (Table 1). In searching the literature, we found only 2 randomized controlled studies—Frye et al2 and Scheffer et al3—that addressed this practice. (One author of this review [TS] participated as a coinvestigator with Frye et al.2) Other evidence that suggests a role for stimulants in bipolar disorder comes from case reports, retrospective case series, and open-label studies.4-11
- “traditional” stimulants (including amphetamine-based compounds such as dextroamphetamine, methylphenidate, dexmethylphenidate, and lisdexamfetamine) thought to affect the dopamine transporter, resulting in increased dopamine in nerve terminals
- the “novel” psychostimulant modafinil, thought to affect multiple neurotransmitter systems (dopamine, GABA, serotonin, histamine, and glutamate), although its mechanism of action is unclear.
Table 1
Clinical studies of stimulant use in patients with bipolar disorder
| Stimulant(s) studied | Study design | Patients studied | Clinical outcomes |
|---|---|---|---|
| Traditional stimulants | |||
| Adjunctive methylphenidate | Chart review, naturalistic12 | 16 adults (5 with comorbid ADHD, 11 with bipolar depression) | Improvements in depression, overall functioning, and ability to concentrate; sleep disturbance, irritability/agitation reported |
| Adjunctive methylphenidate or racemic mixture of AMPH salts | Chart review of sedation and depressive symptoms13 | 8 adults (BD II) | Improved clinical impression of bipolar illness; no manic switches, changes in cycling patterns, or substance abuse noted |
| Adjunctive methylphenidate | 12-week open study, bipolar depression14 | 12 adults (10 BD I, 2 BD II) | Significant clinical improvements in depressive symptoms; no change in manic symptoms; anxiety, agitation, and hypomania reported |
| Multiple stimulants | Chart review, history of stimulants and bipolar illness course25 | 34 hospitalized adolescents | Prior stimulant treatment associated with earlier age of illness onset |
| Adjunctive mixed amphetamine salts | Randomized, placebo-controlled; comorbid BD and ADHD3 | 30 children with ADHD symptoms stabilized on divalproex sodium | Decrease in ADHD symptoms with adjunctive amphetamine treatment but not with divalproex sodium alone; 1 case of mania |
| Novel stimulant | |||
| Adjunctive modafinil | Case series15 | Mixed sample of depressed adults (4 unipolar, 3 bipolar) | Significant improvement in depressive symptoms |
| Adjunctive modafinil | Randomized, double-blind, placebo-controlled2 | 85 adults with bipolar depression | Treatment group showed greater response and remission of depressive symptoms compared with placebo group; no difference in development of manic symptoms |
| ADHD: attention-deficit/hyperactivity disorder; AMPH: amphetamine; BD: bipolar disorder; NOS: not otherwise specified | |||
Depression and iatrogenic sedation
Small, uncontrolled trials have reported some benefit and tolerability in bipolar disorder patients when stimulants are used to treat residual depressive symptoms or iatrogenic sedation associated with mood stabilizers.
Traditional stimulants. A retrospective chart review of 16 patients treated with adjunctive methylphenidate noted improved functioning, as measured by the Global Assessment of Functioning scale. Some patients’ depressive symptoms and concentration also appeared to improve, but how these parameters were assessed is not clear. Some patients tolerated stimulants well, whereas others experienced irritability, agitation, and sleep disturbances.12
Another retrospective chart review described 8 patients with iatrogenic sedation or depression who received adjunctive methylphenidate, mean 20 to 40 mg/d, or a racemic mixture of amphetamine salts, mean 20 to 40 mg/d. Overall bipolar symptoms decreased in severity, as measured by Clinical Global Impression (CGI) scores, but the authors did not directly measure sedation or depression. The stimulants were well-tolerated, with no evidence of stimulant-induced mania.13
In a 12-week open-label trial of methylphenidate in 14 patients with bipolar disorder, depressive symptoms improved as measured by the Hamilton Depression Rating Scale (HAM-D). Mean doses were 10 mg/d for the 3 patients who discontinued because of anxiety, agitation, or hypomania and 16.6 mg/d for those who completed the trial.14
Modafinil may have some efficacy in treating bipolar depression. In a case series of 7 depressed patients (4 unipolar and 3 bipolar), 5 patients showed a 50% decrease in HAM-D scores with adjunctive modafinil. Dosages ranged from 100 to 200 mg/d, although most patients took 200 mg/d. In this series, modafinil was added to a variety of treatments, including bupropion, nefazodone, paroxetine, venlafaxine, an unspecified tricyclic antidepressant (TCA), divalproex sodium, lamotrigine, lithium, electroconvulsive therapy, olanzapine, and gabapentin.15
The only randomized, double-blind, placebo-controlled trial of adjunctive modafinil for bipolar depression enrolled 85 patients with moderate or more severe depression. In this 6-week trial by Frye et al,2 41 patients received modafinil, 100 to 200 mg/d (mean dose 174.2 mg/d), and 44 received placebo.
Bipolar disorder plus ADHD
An estimated 10% to 21% of bipolar patients meet criteria for ADHD,16-19 although at times the line differentiating these 2 disorders is unclear. Co-occurring ADHD worsens the course of bipolar illness,20-22 and data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD) trial suggest that only 2% of dual-diagnosis patients are receiving treatment specifically for ADHD symptoms.23
Theoretically, overlapping symptoms such as talkativeness, distractibility, and physical activity remain relatively constant in ADHD but wax and wane with bipolar disorder’s manic and depressive phases. Recent evidence suggests, however, that many bipolar patients experience prodromal symptoms that may resemble ADHD, including cognitive impairment, distractibility, and increased psychomotor activity.24 In addition, medications used to treat bipolar disorder may impair cognitive function, making ADHD diagnosis difficult in this population.
We are not aware of any clinical trials that examined stimulants’ safety and efficacy in adult bipolar patients with co-occurring ADHD. One of the only studies to examine stimulant treatment of ADHD symptoms in a bipolar population was a retrospective chart review of 34 adolescents hospitalized with bipolar mania. An earlier age of bipolar illness onset was reported in adolescents who had been exposed to stimulants, whether or not they also had ADHD.25
Possible adverse events
Some bipolar disorder patients tolerate stimulants well, whereas others experience serious side effects, toxicities, and illness destabilization (Table 2). Because mood-stabilizer treatment may attenuate stimulants’ undesirable effects in bipolar disorder patients,26,27 be sure to use adequate dosing of a mood stabilizer if you determine a stimulant trial is warranted in your patient.
Destabilization. Stimulants can have a direct negative effect on mood; they can cause restlessness, irritability, anxiety, and mood lability. Some bipolar patients may be more sensitive to these adverse effects than others. Particularly concerning is the possibility of switching to mania or worsening of manic symptoms.28,29 Other potential destabilizing effects include:
- changing cycling patterns, such as inducing rapid cycling
- sleep disturbance because stimulants promote wakefulness.
If you are considering stimulant treatment for a bipolar disorder patient in whom substance abuse is a concern, modafinil or lisdexamfetamine may have a lower abuse potential compared with immediate-release psychostimulants. Lisdexamfetamine is metabolized in the GI tract and does not produce high d-amphetamine blood levels or cause reinforcing effects if injected or snorted.34
Table 2
Possible stimulant side effects, signs of toxicity, and contraindications
| Stimulant class | Possible side effects | Signs of toxicity/overdose | Contraindications/cautions |
|---|---|---|---|
| Traditional (amphetamine mixtures, dexmethylphenidate, dextroamphetamine, lisdexamfetamine methylphenidate)* | Restlessness, insomnia, mood lability, anxiety | Agitation, confusion, tremor, tachycardia, hyperreflexia, hypertension, sweating, psychomotor agitation, seizure, arrhythmia, coma, psychosis | Cardiovascular disease, hypertension, hyperthyroidism, glaucoma, Tourette’s syndrome/motor tics, history of seizure disorder, hypersensitivity to medication class |
| Novel (modafinil) | Restlessness, insomnia, mood lability, anxiety | Agitation, tremor, nausea, diarrhea, confusion | Cardiovascular disease, hepatic impairment, psychosis |
| * Amphetamines and dextroamphetamine (Adderall, Adderall XR); dexmethylphenidate (Focalin, Focalin XR), dextroamphetamine (Dexedrine, DextroStat); lisdexamfetamine (Vyvanse); methylphenidate (Concerta, Daytrana, Metadate CD, Methylin, Methylin ER, Ritalin, Ritalin LA, Ritalin SR) | |||
Drug-drug interactions
Polypharmacy is the rule in treating bipolar disorder, and stimulants can interact with many other psychotropics (Table 3).
Antidepressants. Never use traditional stimulants with monoamine oxidase inhibitors, as this combination may precipitate a hypertensive crisis. Coadministered stimulants also may decrease the metabolism of serotonergic agents—such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs)—and cause side effects associated with increased serotonin neurotransmission, including serotonin syndrome.
Combining traditional stimulants with TCAs may increase TCA concentrations. When coadministered with bupropion, stimulants can increase the risk of seizures.
Modafinil is both an inducer and inhibitor of cytochrome P450 isoenzymes. Because it induces CYP3A4 and inhibits CYP2C19 and CYP2C9, modafinil interacts with many other psychopharmacologic agents:
- Its induction of CYP3A4 may increase the metabolism of commonly used medications such as carbamazepine, aripiprazole, and triazolam.
- Its inhibition of CYP2C19 may decrease the metabolism of many SSRIs, TCAs, diazepam, and clozapine, increasing these drugs’ effects and adverse events.
Possible stimulant interactions with other psychotropics
| Stimulant class | Psychotropic medication | Possible adverse effects |
|---|---|---|
| Traditional (amphetamine mixtures, dexmethylphenidate, dextroamphetamine, lisdexamfetamine methylphenidate)* | MAOIs | Hypertensive crisis |
| CBZ | Reduced methylphenidate levels; abruptly stopping CBZ increases methylphenidate’s effect | |
| TCAs | Increased TCA concentration | |
| SSRIs, SNRIs | Possible decreased metabolism of antidepressants; potential for serotonin syndrome or NMS-like syndrome | |
| Typical and atypical antipsychotics | Each may interfere with the other’s therapeutic action | |
| Novel (modafinil) | CBZ | Decreased modafinil efficacy; decreased CBZ levels |
| Triazolam | Decreased triazolam efficacy; increased effects of triazolam with modafinil discontinuation | |
| Fluoxetine, fluvoxamine | Decreased modafinil clearance | |
| Citalopram, escitalopram, sertraline | Prolonged elimination and increased levels of antidepressant | |
| MAOIs | Hypertensive crisis(?); not recommended | |
| Diazepam | Prolonged elimination and increased levels of diazepam | |
| TCAs | Prolonged elimination and increased levels of TCAs | |
| Clozapine | Increased clozapine concentration (case report) | |
| Aripiprazole | Decreased levels of aripiprazole | |
| * Amphetamines and dextroamphetamine (Adderall, Adderall XR); dexmethylphenidate (Focalin, Focalin XR), dextroamphetamine (Dexedrine, DextroStat); lisdexamfetamine (Vyvanse); methylphenidate (Concerta, Daytrana, Metadate CD, Methylin, Methylin ER, Ritalin, Ritalin LA, Ritalin SR) | ||
| CBZ: carbamazepine; MAOIs: monoamine oxidase inhibitors; NMS: neuroleptic malignant syndrome; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants | ||
Treatment considerations
Without evidence to support stimulants’ safety and efficacy in patients with bipolar disorder, we cannot make specific recommendations. We would, however, like to offer some general recommendations if you decide to use stimulants when treating patients with bipolar disorder (Table 4).
Carefully assess and—in many cases—reassess the patient’s symptoms to clarify the diagnosis. As mentioned, ADHD and bipolar disorder share many symptoms, particularly in the manic phase of bipolar illness. Overlapping symptoms include decreased ability to concentrate and focus, distractibility, hyperactivity and psychomotor agitation, racing thoughts, and impulsivity.
Substance abuse can negatively impact bipolar illness and present as clinical scenarios in which stimulants are used (such as treatment-resistant depression, impulsivity, somnolence, or fatigue).
Treat medical conditions such as thyroid disease, diabetes, and sleep apnea, which may worsen depression, cause somnolence and sedation, and present with symptoms similar to those of ADHD.
When possible, use lifestyle techniques to help patients manage the course of bipolar illness. Encourage good sleep hygiene, exercise, stable social rhythms, and limited use of alcohol and caffeine (both of which can impair sleep quality, which affects illness stability).
The next step. When you have explored all medication options and ruled out all other causes for the patient’s symptoms, stimulant treatment may be an appropriate next step. In these cases:
Encourage patients to participate in treatment, particularly in monitoring mood changes (as with life charts), symptoms associated with mood episodes, and emergence of side effects. When possible, involve family members in monitoring for adverse events.
Administration. Start stimulants only when bipolar illness is well-stabilized, especially regarding manic symptoms. We highly caution against using stimulants in patients with manic or hypomanic symptoms, including mixed states. We recommend not using stimulants in patients with:
- clinically significant insomnia or sleep fragmentation
- active suicidal ideation or psychotic symptoms, particularly if associated with manic symptoms.
Schedule frequent office visits when prescribing stimulants. At least initially, see patients every other week to assess for the emergence of adverse events.
Table 4
6 recommendations when using stimulants in bipolar disorder
| Carefully assess patient’s symptoms | Manic symptoms vs ADHD; medical conditions such as thyroid disorders, diabetes, or sleep apnea |
| Review possible iatrogenic causes of symptoms | Somnolence, decreased energy/fatigue, sedation, difficulty with concentration/focus |
| Engage patient in the therapeutic process | Discuss risks and benefits; monitor mood with life charts; enlist help of family, significant others when appropriate |
| Use caution in clinical scenarios that may herald adverse response to stimulants | Manic/hypomanic symptoms; sleep disturbances; psychosis; history of substance abuse |
| Administer stimulants with caution | Start low and go slow; always use stimulants in conjunction with a mood-stabilizing agent; be aware of possible interactions with patient’s other medications; schedule more frequent visits when starting stimulants |
| Monitor for adverse events associated with stimulant administration | Manic symptoms, changes in cycling patterns, sleep disturbances, substance abuse |
| ADHD: attention-deficit/hyperactivity disorder | |
- The Texas Medication Algorithm Project. Texas Department of State Health Services. www.dshs.state.tx.us/mhprograms/tmapover.shtm.
- The Cochrane Collaboration. www.cochrane.org.
- Amphetamine and dextroamphetamine • Adderall
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clozapine • Clozaril
- Dexmethylphenidate • Focalin
- Dextroamphetamine • Dexedrine, DextroStat
- Diazepam • Valium
- Divalproex sodium • Depakote
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Lisdexamfetamine • Vyvanse
- Lithium • various
- Methylphenidate • Ritalin, Concerta, others
- Modafinil • Provigil
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Triazolam • Halcion
- Valproic acid • Depakene
- Venlafaxine • Effexor
Dr. Gonzalez reports no financial relationship with any company whose products are mentioned in the article or with manufacturers of competing products. He is a recipient of a T32 Ruth L. Kirschstein National Research Service Awards training fellowship sponsored by the National Institutes of Health.
Dr. Suppes receives grants/research support from Abbott Laboratories, AstraZeneca, GlaxoSmithKline, JDS Pharmaceuticals, Janssen Pharmaceutica, National Institute of Mental Health, Novartis, Pfizer Inc., and the Stanley Medical Research Institute.
1. Silberman EK, Reus VI, Jimerson DC, et al. Heterogeneity of amphetamine response in depressed patients. Am J Psychiatry 1981;138(10):1302-7.
2. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry 2007;164(8):1242-9.
3. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
4. Meyers B. Treatment of imipramine-resistant depression and lithium-refractory mania through drug interactions. Am J Psychiatry 1978;135(11):1420-1.
5. Bannet J, Ebstein RP, Belmaker RH. Clinical aspects of the interaction of lithium and stimulants. Br J Psychiatry 1980;136:204.-
6. Drimmer EJ, Gitlin MJ, Gwirtsman HE. Desipramine and methylphenidate combination treatment for depression: case report. Am J Psychiatry 1983;140(2):241-2.
7. Fernandes PP, Petty F. Modafinil for remitted bipolar depression with hypersomnia. Ann Pharmacother 2003;37(12):1807-9.
8. Berigan TR. Augmentation with modafinil to achieve remission in depression: a case report. Prim Care Companion J Clin Psychiatry 2001;3(1):32.-
9. Berigan TR. Modafinil treatment of excessive daytime sedation and fatigue associated with topiramate. Prim Care Companion J Clin Psychiatry 2002;4(6):249-50.
10. Berigan T. Modafinil treatment of excessive sedation associated with divalproex sodium. Can J Psychiatry 2004;49(1):72-3.
11. Even C, Thuile J, Santos J, Bourgin P. Modafinil as an adjunctive treatment to sleep deprivation in depression. J Psychiatry Neurosci 2005;30(6):432-3.
12. Lydon E, El-Mallakh RS. Naturalistic long-term use of methylphenidate in bipolar disorder. J Clin Psychopharmacol 2006;26(5):516-8.
13. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: treatment of residual depression and sedation. Bipolar Disord 2004;6(5):416-20.
14. El-Mallakh RS. An open study of methylphenidate in bipolar depression. Bipolar Disord 2000;2(1):56-9.
15. Menza MA, Kaufman KR, Castellanos A. Modafinil augmentation of antidepressant treatment in depression. J Clin Psychiatry 2000;61(5):378-81.
16. Wingo AP, Ghaemi SN. A systematic review of rates and diagnostic validity of comorbid adult attention-deficit/hyperactivity disorder and bipolar disorder. J Clin Psychiatry 2007;68(11):1776-84.
17. Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry 2006;163(4):716-23.
18. Nierenberg AA, Miyahara S, Spencer T, et al. Clinical and diagnostic implications of lifetime attention-deficit/hyperactivity disorder comorbidity in adults with bipolar disorder: data from the first 1000 STEP-BD participants. Biol Psychiatry 2005;57(11):1467-73.
19. Tamam L, Tuglu C, Karatas G, Ozcan S. Adult attention-deficit hyperactivity disorder in patients with bipolar I disorder in remission: preliminary study. Psychiatry Clin Neurosci 2006;60(4):480-5.
20. Faraone SV, Biederman J, Mennin D, et al. Attention-deficit hyperactivity disorder with bipolar disorder: a familial subtype? J Am Acad Child Adolesc Psychiatry 1997;36(10):1378-87; discussion 1387-90.
21. Faraone SV, Biederman J, Monuteaux MC. Attention deficit hyperactivity disorder with bipolar disorder in girls: further evidence for a familial subtype? J Affect Disord 2001;64(1):19-26.
22. Faraone SV, Glatt SJ, Tsuang MT. The genetics of pediatriconset bipolar disorder. Biol Psychiatry 2003;53(11):970-7.
23. Simon NM, Otto MW, Weiss RD, et al. Pharmacotherapy for bipolar disorder and comorbid conditions: baseline data from STEP-BD. J Clin Psychopharmacol 2004;24(5):512-20.
24. Calabrese JR. Overview of patient care issues and treatment in bipolar spectrum and bipolar II disorder. J Clin Psychiatry 2008;69(6):e18.-
25. DelBello MP, Soutullo CA, Hendricks W, et al. Prior stimulant treatment in adolescents with bipolar disorder: association with age at onset. Bipolar Disord 2001;3(2):53-7.
26. Van Kammen DP, Murphy DL. Attenuation of the euphoriant and activating effects of d- and l-amphetamine by lithium carbonate treatment. Psychopharmacologia 1975;44(3):215-24.
27. Huey LY, Janowsky DS, Judd LL, et al. Effects of lithium carbonate on methylphenidate-induced mood, behavior, and cognitive processes. Psychopharmacology (Berl) 1981;73(2):161-4.
28. Gerner RH, Post RM, Bunney WE, Jr. A dopaminergic mechanism in mania. Am J Psychiatry 1976;133(10):1177-80.
29. Koehler-Troy C, Strober M, Malenbaum R. Methylphenidateinduced mania in a prepubertal child. J Clin Psychiatry 1986;47(11):566-7.
30. Brady KT, Sonne SC. The relationship between substance abuse and bipolar disorder. J Clin Psychiatry 1995;56(suppl 3):19-24.
31. Sonne SC, Brady KT. Substance abuse and bipolar comorbidity. Psychiatr Clin North Am 1999;22(3):609-27,ix.
32. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA 1990;264(19):2511-8.
33. Estroff TW, Dackis CA, Gold MS, Pottash AL. Drug abuse and bipolar disorders. Int J Psychiatry Med 1985-1986;15(1):37-40.
34. Faraone SV. Lisdexamfetamine dimesylate: the first longacting prodrug stimulant treatment for attention deficit/hyperactivity disorder. Expert Opin Pharmacother 2008;9(9):1565-74.
Patients with bipolar disorder show an unpredictable range of responses to stimulants, from virtually no ill effects to emerging manic-like symptoms.1 Thus, although stimulants may be beneficial to some bipolar patients, there is a great deal of concern about using stimulants in this population. Even so, stimulants may be a rational adjunct for treating certain aspects of bipolar illness, particularly resistant depression, iatrogenic sedation, and comorbid attention-deficit/hyperactivity disorder (ADHD).
To help you decide if and when your patient might be a candidate for stimulant therapy, this article:
- reviews the evidence on stimulants’ safety and tolerability for patients with bipolar disorder
- weighs potential benefits and risks of using stimulants in this population
- addresses stimulants’ possible adverse effects on illness course and from interactions with other psychotropics
- discusses treatment options based on the limited evidence and our clinical experience.
Limited support
We are aware that using stimulants to treat patients with bipolar disorder is not an uncommon clinical practice, but supportive evidence is limited (Table 1). In searching the literature, we found only 2 randomized controlled studies—Frye et al2 and Scheffer et al3—that addressed this practice. (One author of this review [TS] participated as a coinvestigator with Frye et al.2) Other evidence that suggests a role for stimulants in bipolar disorder comes from case reports, retrospective case series, and open-label studies.4-11
- “traditional” stimulants (including amphetamine-based compounds such as dextroamphetamine, methylphenidate, dexmethylphenidate, and lisdexamfetamine) thought to affect the dopamine transporter, resulting in increased dopamine in nerve terminals
- the “novel” psychostimulant modafinil, thought to affect multiple neurotransmitter systems (dopamine, GABA, serotonin, histamine, and glutamate), although its mechanism of action is unclear.
Table 1
Clinical studies of stimulant use in patients with bipolar disorder
| Stimulant(s) studied | Study design | Patients studied | Clinical outcomes |
|---|---|---|---|
| Traditional stimulants | |||
| Adjunctive methylphenidate | Chart review, naturalistic12 | 16 adults (5 with comorbid ADHD, 11 with bipolar depression) | Improvements in depression, overall functioning, and ability to concentrate; sleep disturbance, irritability/agitation reported |
| Adjunctive methylphenidate or racemic mixture of AMPH salts | Chart review of sedation and depressive symptoms13 | 8 adults (BD II) | Improved clinical impression of bipolar illness; no manic switches, changes in cycling patterns, or substance abuse noted |
| Adjunctive methylphenidate | 12-week open study, bipolar depression14 | 12 adults (10 BD I, 2 BD II) | Significant clinical improvements in depressive symptoms; no change in manic symptoms; anxiety, agitation, and hypomania reported |
| Multiple stimulants | Chart review, history of stimulants and bipolar illness course25 | 34 hospitalized adolescents | Prior stimulant treatment associated with earlier age of illness onset |
| Adjunctive mixed amphetamine salts | Randomized, placebo-controlled; comorbid BD and ADHD3 | 30 children with ADHD symptoms stabilized on divalproex sodium | Decrease in ADHD symptoms with adjunctive amphetamine treatment but not with divalproex sodium alone; 1 case of mania |
| Novel stimulant | |||
| Adjunctive modafinil | Case series15 | Mixed sample of depressed adults (4 unipolar, 3 bipolar) | Significant improvement in depressive symptoms |
| Adjunctive modafinil | Randomized, double-blind, placebo-controlled2 | 85 adults with bipolar depression | Treatment group showed greater response and remission of depressive symptoms compared with placebo group; no difference in development of manic symptoms |
| ADHD: attention-deficit/hyperactivity disorder; AMPH: amphetamine; BD: bipolar disorder; NOS: not otherwise specified | |||
Depression and iatrogenic sedation
Small, uncontrolled trials have reported some benefit and tolerability in bipolar disorder patients when stimulants are used to treat residual depressive symptoms or iatrogenic sedation associated with mood stabilizers.
Traditional stimulants. A retrospective chart review of 16 patients treated with adjunctive methylphenidate noted improved functioning, as measured by the Global Assessment of Functioning scale. Some patients’ depressive symptoms and concentration also appeared to improve, but how these parameters were assessed is not clear. Some patients tolerated stimulants well, whereas others experienced irritability, agitation, and sleep disturbances.12
Another retrospective chart review described 8 patients with iatrogenic sedation or depression who received adjunctive methylphenidate, mean 20 to 40 mg/d, or a racemic mixture of amphetamine salts, mean 20 to 40 mg/d. Overall bipolar symptoms decreased in severity, as measured by Clinical Global Impression (CGI) scores, but the authors did not directly measure sedation or depression. The stimulants were well-tolerated, with no evidence of stimulant-induced mania.13
In a 12-week open-label trial of methylphenidate in 14 patients with bipolar disorder, depressive symptoms improved as measured by the Hamilton Depression Rating Scale (HAM-D). Mean doses were 10 mg/d for the 3 patients who discontinued because of anxiety, agitation, or hypomania and 16.6 mg/d for those who completed the trial.14
Modafinil may have some efficacy in treating bipolar depression. In a case series of 7 depressed patients (4 unipolar and 3 bipolar), 5 patients showed a 50% decrease in HAM-D scores with adjunctive modafinil. Dosages ranged from 100 to 200 mg/d, although most patients took 200 mg/d. In this series, modafinil was added to a variety of treatments, including bupropion, nefazodone, paroxetine, venlafaxine, an unspecified tricyclic antidepressant (TCA), divalproex sodium, lamotrigine, lithium, electroconvulsive therapy, olanzapine, and gabapentin.15
The only randomized, double-blind, placebo-controlled trial of adjunctive modafinil for bipolar depression enrolled 85 patients with moderate or more severe depression. In this 6-week trial by Frye et al,2 41 patients received modafinil, 100 to 200 mg/d (mean dose 174.2 mg/d), and 44 received placebo.
Bipolar disorder plus ADHD
An estimated 10% to 21% of bipolar patients meet criteria for ADHD,16-19 although at times the line differentiating these 2 disorders is unclear. Co-occurring ADHD worsens the course of bipolar illness,20-22 and data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD) trial suggest that only 2% of dual-diagnosis patients are receiving treatment specifically for ADHD symptoms.23
Theoretically, overlapping symptoms such as talkativeness, distractibility, and physical activity remain relatively constant in ADHD but wax and wane with bipolar disorder’s manic and depressive phases. Recent evidence suggests, however, that many bipolar patients experience prodromal symptoms that may resemble ADHD, including cognitive impairment, distractibility, and increased psychomotor activity.24 In addition, medications used to treat bipolar disorder may impair cognitive function, making ADHD diagnosis difficult in this population.
We are not aware of any clinical trials that examined stimulants’ safety and efficacy in adult bipolar patients with co-occurring ADHD. One of the only studies to examine stimulant treatment of ADHD symptoms in a bipolar population was a retrospective chart review of 34 adolescents hospitalized with bipolar mania. An earlier age of bipolar illness onset was reported in adolescents who had been exposed to stimulants, whether or not they also had ADHD.25
Possible adverse events
Some bipolar disorder patients tolerate stimulants well, whereas others experience serious side effects, toxicities, and illness destabilization (Table 2). Because mood-stabilizer treatment may attenuate stimulants’ undesirable effects in bipolar disorder patients,26,27 be sure to use adequate dosing of a mood stabilizer if you determine a stimulant trial is warranted in your patient.
Destabilization. Stimulants can have a direct negative effect on mood; they can cause restlessness, irritability, anxiety, and mood lability. Some bipolar patients may be more sensitive to these adverse effects than others. Particularly concerning is the possibility of switching to mania or worsening of manic symptoms.28,29 Other potential destabilizing effects include:
- changing cycling patterns, such as inducing rapid cycling
- sleep disturbance because stimulants promote wakefulness.
If you are considering stimulant treatment for a bipolar disorder patient in whom substance abuse is a concern, modafinil or lisdexamfetamine may have a lower abuse potential compared with immediate-release psychostimulants. Lisdexamfetamine is metabolized in the GI tract and does not produce high d-amphetamine blood levels or cause reinforcing effects if injected or snorted.34
Table 2
Possible stimulant side effects, signs of toxicity, and contraindications
| Stimulant class | Possible side effects | Signs of toxicity/overdose | Contraindications/cautions |
|---|---|---|---|
| Traditional (amphetamine mixtures, dexmethylphenidate, dextroamphetamine, lisdexamfetamine methylphenidate)* | Restlessness, insomnia, mood lability, anxiety | Agitation, confusion, tremor, tachycardia, hyperreflexia, hypertension, sweating, psychomotor agitation, seizure, arrhythmia, coma, psychosis | Cardiovascular disease, hypertension, hyperthyroidism, glaucoma, Tourette’s syndrome/motor tics, history of seizure disorder, hypersensitivity to medication class |
| Novel (modafinil) | Restlessness, insomnia, mood lability, anxiety | Agitation, tremor, nausea, diarrhea, confusion | Cardiovascular disease, hepatic impairment, psychosis |
| * Amphetamines and dextroamphetamine (Adderall, Adderall XR); dexmethylphenidate (Focalin, Focalin XR), dextroamphetamine (Dexedrine, DextroStat); lisdexamfetamine (Vyvanse); methylphenidate (Concerta, Daytrana, Metadate CD, Methylin, Methylin ER, Ritalin, Ritalin LA, Ritalin SR) | |||
Drug-drug interactions
Polypharmacy is the rule in treating bipolar disorder, and stimulants can interact with many other psychotropics (Table 3).
Antidepressants. Never use traditional stimulants with monoamine oxidase inhibitors, as this combination may precipitate a hypertensive crisis. Coadministered stimulants also may decrease the metabolism of serotonergic agents—such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs)—and cause side effects associated with increased serotonin neurotransmission, including serotonin syndrome.
Combining traditional stimulants with TCAs may increase TCA concentrations. When coadministered with bupropion, stimulants can increase the risk of seizures.
Modafinil is both an inducer and inhibitor of cytochrome P450 isoenzymes. Because it induces CYP3A4 and inhibits CYP2C19 and CYP2C9, modafinil interacts with many other psychopharmacologic agents:
- Its induction of CYP3A4 may increase the metabolism of commonly used medications such as carbamazepine, aripiprazole, and triazolam.
- Its inhibition of CYP2C19 may decrease the metabolism of many SSRIs, TCAs, diazepam, and clozapine, increasing these drugs’ effects and adverse events.
Possible stimulant interactions with other psychotropics
| Stimulant class | Psychotropic medication | Possible adverse effects |
|---|---|---|
| Traditional (amphetamine mixtures, dexmethylphenidate, dextroamphetamine, lisdexamfetamine methylphenidate)* | MAOIs | Hypertensive crisis |
| CBZ | Reduced methylphenidate levels; abruptly stopping CBZ increases methylphenidate’s effect | |
| TCAs | Increased TCA concentration | |
| SSRIs, SNRIs | Possible decreased metabolism of antidepressants; potential for serotonin syndrome or NMS-like syndrome | |
| Typical and atypical antipsychotics | Each may interfere with the other’s therapeutic action | |
| Novel (modafinil) | CBZ | Decreased modafinil efficacy; decreased CBZ levels |
| Triazolam | Decreased triazolam efficacy; increased effects of triazolam with modafinil discontinuation | |
| Fluoxetine, fluvoxamine | Decreased modafinil clearance | |
| Citalopram, escitalopram, sertraline | Prolonged elimination and increased levels of antidepressant | |
| MAOIs | Hypertensive crisis(?); not recommended | |
| Diazepam | Prolonged elimination and increased levels of diazepam | |
| TCAs | Prolonged elimination and increased levels of TCAs | |
| Clozapine | Increased clozapine concentration (case report) | |
| Aripiprazole | Decreased levels of aripiprazole | |
| * Amphetamines and dextroamphetamine (Adderall, Adderall XR); dexmethylphenidate (Focalin, Focalin XR), dextroamphetamine (Dexedrine, DextroStat); lisdexamfetamine (Vyvanse); methylphenidate (Concerta, Daytrana, Metadate CD, Methylin, Methylin ER, Ritalin, Ritalin LA, Ritalin SR) | ||
| CBZ: carbamazepine; MAOIs: monoamine oxidase inhibitors; NMS: neuroleptic malignant syndrome; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants | ||
Treatment considerations
Without evidence to support stimulants’ safety and efficacy in patients with bipolar disorder, we cannot make specific recommendations. We would, however, like to offer some general recommendations if you decide to use stimulants when treating patients with bipolar disorder (Table 4).
Carefully assess and—in many cases—reassess the patient’s symptoms to clarify the diagnosis. As mentioned, ADHD and bipolar disorder share many symptoms, particularly in the manic phase of bipolar illness. Overlapping symptoms include decreased ability to concentrate and focus, distractibility, hyperactivity and psychomotor agitation, racing thoughts, and impulsivity.
Substance abuse can negatively impact bipolar illness and present as clinical scenarios in which stimulants are used (such as treatment-resistant depression, impulsivity, somnolence, or fatigue).
Treat medical conditions such as thyroid disease, diabetes, and sleep apnea, which may worsen depression, cause somnolence and sedation, and present with symptoms similar to those of ADHD.
When possible, use lifestyle techniques to help patients manage the course of bipolar illness. Encourage good sleep hygiene, exercise, stable social rhythms, and limited use of alcohol and caffeine (both of which can impair sleep quality, which affects illness stability).
The next step. When you have explored all medication options and ruled out all other causes for the patient’s symptoms, stimulant treatment may be an appropriate next step. In these cases:
Encourage patients to participate in treatment, particularly in monitoring mood changes (as with life charts), symptoms associated with mood episodes, and emergence of side effects. When possible, involve family members in monitoring for adverse events.
Administration. Start stimulants only when bipolar illness is well-stabilized, especially regarding manic symptoms. We highly caution against using stimulants in patients with manic or hypomanic symptoms, including mixed states. We recommend not using stimulants in patients with:
- clinically significant insomnia or sleep fragmentation
- active suicidal ideation or psychotic symptoms, particularly if associated with manic symptoms.
Schedule frequent office visits when prescribing stimulants. At least initially, see patients every other week to assess for the emergence of adverse events.
Table 4
6 recommendations when using stimulants in bipolar disorder
| Carefully assess patient’s symptoms | Manic symptoms vs ADHD; medical conditions such as thyroid disorders, diabetes, or sleep apnea |
| Review possible iatrogenic causes of symptoms | Somnolence, decreased energy/fatigue, sedation, difficulty with concentration/focus |
| Engage patient in the therapeutic process | Discuss risks and benefits; monitor mood with life charts; enlist help of family, significant others when appropriate |
| Use caution in clinical scenarios that may herald adverse response to stimulants | Manic/hypomanic symptoms; sleep disturbances; psychosis; history of substance abuse |
| Administer stimulants with caution | Start low and go slow; always use stimulants in conjunction with a mood-stabilizing agent; be aware of possible interactions with patient’s other medications; schedule more frequent visits when starting stimulants |
| Monitor for adverse events associated with stimulant administration | Manic symptoms, changes in cycling patterns, sleep disturbances, substance abuse |
| ADHD: attention-deficit/hyperactivity disorder | |
- The Texas Medication Algorithm Project. Texas Department of State Health Services. www.dshs.state.tx.us/mhprograms/tmapover.shtm.
- The Cochrane Collaboration. www.cochrane.org.
- Amphetamine and dextroamphetamine • Adderall
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clozapine • Clozaril
- Dexmethylphenidate • Focalin
- Dextroamphetamine • Dexedrine, DextroStat
- Diazepam • Valium
- Divalproex sodium • Depakote
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Lisdexamfetamine • Vyvanse
- Lithium • various
- Methylphenidate • Ritalin, Concerta, others
- Modafinil • Provigil
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Triazolam • Halcion
- Valproic acid • Depakene
- Venlafaxine • Effexor
Dr. Gonzalez reports no financial relationship with any company whose products are mentioned in the article or with manufacturers of competing products. He is a recipient of a T32 Ruth L. Kirschstein National Research Service Awards training fellowship sponsored by the National Institutes of Health.
Dr. Suppes receives grants/research support from Abbott Laboratories, AstraZeneca, GlaxoSmithKline, JDS Pharmaceuticals, Janssen Pharmaceutica, National Institute of Mental Health, Novartis, Pfizer Inc., and the Stanley Medical Research Institute.
Patients with bipolar disorder show an unpredictable range of responses to stimulants, from virtually no ill effects to emerging manic-like symptoms.1 Thus, although stimulants may be beneficial to some bipolar patients, there is a great deal of concern about using stimulants in this population. Even so, stimulants may be a rational adjunct for treating certain aspects of bipolar illness, particularly resistant depression, iatrogenic sedation, and comorbid attention-deficit/hyperactivity disorder (ADHD).
To help you decide if and when your patient might be a candidate for stimulant therapy, this article:
- reviews the evidence on stimulants’ safety and tolerability for patients with bipolar disorder
- weighs potential benefits and risks of using stimulants in this population
- addresses stimulants’ possible adverse effects on illness course and from interactions with other psychotropics
- discusses treatment options based on the limited evidence and our clinical experience.
Limited support
We are aware that using stimulants to treat patients with bipolar disorder is not an uncommon clinical practice, but supportive evidence is limited (Table 1). In searching the literature, we found only 2 randomized controlled studies—Frye et al2 and Scheffer et al3—that addressed this practice. (One author of this review [TS] participated as a coinvestigator with Frye et al.2) Other evidence that suggests a role for stimulants in bipolar disorder comes from case reports, retrospective case series, and open-label studies.4-11
- “traditional” stimulants (including amphetamine-based compounds such as dextroamphetamine, methylphenidate, dexmethylphenidate, and lisdexamfetamine) thought to affect the dopamine transporter, resulting in increased dopamine in nerve terminals
- the “novel” psychostimulant modafinil, thought to affect multiple neurotransmitter systems (dopamine, GABA, serotonin, histamine, and glutamate), although its mechanism of action is unclear.
Table 1
Clinical studies of stimulant use in patients with bipolar disorder
| Stimulant(s) studied | Study design | Patients studied | Clinical outcomes |
|---|---|---|---|
| Traditional stimulants | |||
| Adjunctive methylphenidate | Chart review, naturalistic12 | 16 adults (5 with comorbid ADHD, 11 with bipolar depression) | Improvements in depression, overall functioning, and ability to concentrate; sleep disturbance, irritability/agitation reported |
| Adjunctive methylphenidate or racemic mixture of AMPH salts | Chart review of sedation and depressive symptoms13 | 8 adults (BD II) | Improved clinical impression of bipolar illness; no manic switches, changes in cycling patterns, or substance abuse noted |
| Adjunctive methylphenidate | 12-week open study, bipolar depression14 | 12 adults (10 BD I, 2 BD II) | Significant clinical improvements in depressive symptoms; no change in manic symptoms; anxiety, agitation, and hypomania reported |
| Multiple stimulants | Chart review, history of stimulants and bipolar illness course25 | 34 hospitalized adolescents | Prior stimulant treatment associated with earlier age of illness onset |
| Adjunctive mixed amphetamine salts | Randomized, placebo-controlled; comorbid BD and ADHD3 | 30 children with ADHD symptoms stabilized on divalproex sodium | Decrease in ADHD symptoms with adjunctive amphetamine treatment but not with divalproex sodium alone; 1 case of mania |
| Novel stimulant | |||
| Adjunctive modafinil | Case series15 | Mixed sample of depressed adults (4 unipolar, 3 bipolar) | Significant improvement in depressive symptoms |
| Adjunctive modafinil | Randomized, double-blind, placebo-controlled2 | 85 adults with bipolar depression | Treatment group showed greater response and remission of depressive symptoms compared with placebo group; no difference in development of manic symptoms |
| ADHD: attention-deficit/hyperactivity disorder; AMPH: amphetamine; BD: bipolar disorder; NOS: not otherwise specified | |||
Depression and iatrogenic sedation
Small, uncontrolled trials have reported some benefit and tolerability in bipolar disorder patients when stimulants are used to treat residual depressive symptoms or iatrogenic sedation associated with mood stabilizers.
Traditional stimulants. A retrospective chart review of 16 patients treated with adjunctive methylphenidate noted improved functioning, as measured by the Global Assessment of Functioning scale. Some patients’ depressive symptoms and concentration also appeared to improve, but how these parameters were assessed is not clear. Some patients tolerated stimulants well, whereas others experienced irritability, agitation, and sleep disturbances.12
Another retrospective chart review described 8 patients with iatrogenic sedation or depression who received adjunctive methylphenidate, mean 20 to 40 mg/d, or a racemic mixture of amphetamine salts, mean 20 to 40 mg/d. Overall bipolar symptoms decreased in severity, as measured by Clinical Global Impression (CGI) scores, but the authors did not directly measure sedation or depression. The stimulants were well-tolerated, with no evidence of stimulant-induced mania.13
In a 12-week open-label trial of methylphenidate in 14 patients with bipolar disorder, depressive symptoms improved as measured by the Hamilton Depression Rating Scale (HAM-D). Mean doses were 10 mg/d for the 3 patients who discontinued because of anxiety, agitation, or hypomania and 16.6 mg/d for those who completed the trial.14
Modafinil may have some efficacy in treating bipolar depression. In a case series of 7 depressed patients (4 unipolar and 3 bipolar), 5 patients showed a 50% decrease in HAM-D scores with adjunctive modafinil. Dosages ranged from 100 to 200 mg/d, although most patients took 200 mg/d. In this series, modafinil was added to a variety of treatments, including bupropion, nefazodone, paroxetine, venlafaxine, an unspecified tricyclic antidepressant (TCA), divalproex sodium, lamotrigine, lithium, electroconvulsive therapy, olanzapine, and gabapentin.15
The only randomized, double-blind, placebo-controlled trial of adjunctive modafinil for bipolar depression enrolled 85 patients with moderate or more severe depression. In this 6-week trial by Frye et al,2 41 patients received modafinil, 100 to 200 mg/d (mean dose 174.2 mg/d), and 44 received placebo.
Bipolar disorder plus ADHD
An estimated 10% to 21% of bipolar patients meet criteria for ADHD,16-19 although at times the line differentiating these 2 disorders is unclear. Co-occurring ADHD worsens the course of bipolar illness,20-22 and data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD) trial suggest that only 2% of dual-diagnosis patients are receiving treatment specifically for ADHD symptoms.23
Theoretically, overlapping symptoms such as talkativeness, distractibility, and physical activity remain relatively constant in ADHD but wax and wane with bipolar disorder’s manic and depressive phases. Recent evidence suggests, however, that many bipolar patients experience prodromal symptoms that may resemble ADHD, including cognitive impairment, distractibility, and increased psychomotor activity.24 In addition, medications used to treat bipolar disorder may impair cognitive function, making ADHD diagnosis difficult in this population.
We are not aware of any clinical trials that examined stimulants’ safety and efficacy in adult bipolar patients with co-occurring ADHD. One of the only studies to examine stimulant treatment of ADHD symptoms in a bipolar population was a retrospective chart review of 34 adolescents hospitalized with bipolar mania. An earlier age of bipolar illness onset was reported in adolescents who had been exposed to stimulants, whether or not they also had ADHD.25
Possible adverse events
Some bipolar disorder patients tolerate stimulants well, whereas others experience serious side effects, toxicities, and illness destabilization (Table 2). Because mood-stabilizer treatment may attenuate stimulants’ undesirable effects in bipolar disorder patients,26,27 be sure to use adequate dosing of a mood stabilizer if you determine a stimulant trial is warranted in your patient.
Destabilization. Stimulants can have a direct negative effect on mood; they can cause restlessness, irritability, anxiety, and mood lability. Some bipolar patients may be more sensitive to these adverse effects than others. Particularly concerning is the possibility of switching to mania or worsening of manic symptoms.28,29 Other potential destabilizing effects include:
- changing cycling patterns, such as inducing rapid cycling
- sleep disturbance because stimulants promote wakefulness.
If you are considering stimulant treatment for a bipolar disorder patient in whom substance abuse is a concern, modafinil or lisdexamfetamine may have a lower abuse potential compared with immediate-release psychostimulants. Lisdexamfetamine is metabolized in the GI tract and does not produce high d-amphetamine blood levels or cause reinforcing effects if injected or snorted.34
Table 2
Possible stimulant side effects, signs of toxicity, and contraindications
| Stimulant class | Possible side effects | Signs of toxicity/overdose | Contraindications/cautions |
|---|---|---|---|
| Traditional (amphetamine mixtures, dexmethylphenidate, dextroamphetamine, lisdexamfetamine methylphenidate)* | Restlessness, insomnia, mood lability, anxiety | Agitation, confusion, tremor, tachycardia, hyperreflexia, hypertension, sweating, psychomotor agitation, seizure, arrhythmia, coma, psychosis | Cardiovascular disease, hypertension, hyperthyroidism, glaucoma, Tourette’s syndrome/motor tics, history of seizure disorder, hypersensitivity to medication class |
| Novel (modafinil) | Restlessness, insomnia, mood lability, anxiety | Agitation, tremor, nausea, diarrhea, confusion | Cardiovascular disease, hepatic impairment, psychosis |
| * Amphetamines and dextroamphetamine (Adderall, Adderall XR); dexmethylphenidate (Focalin, Focalin XR), dextroamphetamine (Dexedrine, DextroStat); lisdexamfetamine (Vyvanse); methylphenidate (Concerta, Daytrana, Metadate CD, Methylin, Methylin ER, Ritalin, Ritalin LA, Ritalin SR) | |||
Drug-drug interactions
Polypharmacy is the rule in treating bipolar disorder, and stimulants can interact with many other psychotropics (Table 3).
Antidepressants. Never use traditional stimulants with monoamine oxidase inhibitors, as this combination may precipitate a hypertensive crisis. Coadministered stimulants also may decrease the metabolism of serotonergic agents—such as selective serotonin reuptake inhibitors (SSRIs) or serotonin-norepinephrine reuptake inhibitors (SNRIs)—and cause side effects associated with increased serotonin neurotransmission, including serotonin syndrome.
Combining traditional stimulants with TCAs may increase TCA concentrations. When coadministered with bupropion, stimulants can increase the risk of seizures.
Modafinil is both an inducer and inhibitor of cytochrome P450 isoenzymes. Because it induces CYP3A4 and inhibits CYP2C19 and CYP2C9, modafinil interacts with many other psychopharmacologic agents:
- Its induction of CYP3A4 may increase the metabolism of commonly used medications such as carbamazepine, aripiprazole, and triazolam.
- Its inhibition of CYP2C19 may decrease the metabolism of many SSRIs, TCAs, diazepam, and clozapine, increasing these drugs’ effects and adverse events.
Possible stimulant interactions with other psychotropics
| Stimulant class | Psychotropic medication | Possible adverse effects |
|---|---|---|
| Traditional (amphetamine mixtures, dexmethylphenidate, dextroamphetamine, lisdexamfetamine methylphenidate)* | MAOIs | Hypertensive crisis |
| CBZ | Reduced methylphenidate levels; abruptly stopping CBZ increases methylphenidate’s effect | |
| TCAs | Increased TCA concentration | |
| SSRIs, SNRIs | Possible decreased metabolism of antidepressants; potential for serotonin syndrome or NMS-like syndrome | |
| Typical and atypical antipsychotics | Each may interfere with the other’s therapeutic action | |
| Novel (modafinil) | CBZ | Decreased modafinil efficacy; decreased CBZ levels |
| Triazolam | Decreased triazolam efficacy; increased effects of triazolam with modafinil discontinuation | |
| Fluoxetine, fluvoxamine | Decreased modafinil clearance | |
| Citalopram, escitalopram, sertraline | Prolonged elimination and increased levels of antidepressant | |
| MAOIs | Hypertensive crisis(?); not recommended | |
| Diazepam | Prolonged elimination and increased levels of diazepam | |
| TCAs | Prolonged elimination and increased levels of TCAs | |
| Clozapine | Increased clozapine concentration (case report) | |
| Aripiprazole | Decreased levels of aripiprazole | |
| * Amphetamines and dextroamphetamine (Adderall, Adderall XR); dexmethylphenidate (Focalin, Focalin XR), dextroamphetamine (Dexedrine, DextroStat); lisdexamfetamine (Vyvanse); methylphenidate (Concerta, Daytrana, Metadate CD, Methylin, Methylin ER, Ritalin, Ritalin LA, Ritalin SR) | ||
| CBZ: carbamazepine; MAOIs: monoamine oxidase inhibitors; NMS: neuroleptic malignant syndrome; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants | ||
Treatment considerations
Without evidence to support stimulants’ safety and efficacy in patients with bipolar disorder, we cannot make specific recommendations. We would, however, like to offer some general recommendations if you decide to use stimulants when treating patients with bipolar disorder (Table 4).
Carefully assess and—in many cases—reassess the patient’s symptoms to clarify the diagnosis. As mentioned, ADHD and bipolar disorder share many symptoms, particularly in the manic phase of bipolar illness. Overlapping symptoms include decreased ability to concentrate and focus, distractibility, hyperactivity and psychomotor agitation, racing thoughts, and impulsivity.
Substance abuse can negatively impact bipolar illness and present as clinical scenarios in which stimulants are used (such as treatment-resistant depression, impulsivity, somnolence, or fatigue).
Treat medical conditions such as thyroid disease, diabetes, and sleep apnea, which may worsen depression, cause somnolence and sedation, and present with symptoms similar to those of ADHD.
When possible, use lifestyle techniques to help patients manage the course of bipolar illness. Encourage good sleep hygiene, exercise, stable social rhythms, and limited use of alcohol and caffeine (both of which can impair sleep quality, which affects illness stability).
The next step. When you have explored all medication options and ruled out all other causes for the patient’s symptoms, stimulant treatment may be an appropriate next step. In these cases:
Encourage patients to participate in treatment, particularly in monitoring mood changes (as with life charts), symptoms associated with mood episodes, and emergence of side effects. When possible, involve family members in monitoring for adverse events.
Administration. Start stimulants only when bipolar illness is well-stabilized, especially regarding manic symptoms. We highly caution against using stimulants in patients with manic or hypomanic symptoms, including mixed states. We recommend not using stimulants in patients with:
- clinically significant insomnia or sleep fragmentation
- active suicidal ideation or psychotic symptoms, particularly if associated with manic symptoms.
Schedule frequent office visits when prescribing stimulants. At least initially, see patients every other week to assess for the emergence of adverse events.
Table 4
6 recommendations when using stimulants in bipolar disorder
| Carefully assess patient’s symptoms | Manic symptoms vs ADHD; medical conditions such as thyroid disorders, diabetes, or sleep apnea |
| Review possible iatrogenic causes of symptoms | Somnolence, decreased energy/fatigue, sedation, difficulty with concentration/focus |
| Engage patient in the therapeutic process | Discuss risks and benefits; monitor mood with life charts; enlist help of family, significant others when appropriate |
| Use caution in clinical scenarios that may herald adverse response to stimulants | Manic/hypomanic symptoms; sleep disturbances; psychosis; history of substance abuse |
| Administer stimulants with caution | Start low and go slow; always use stimulants in conjunction with a mood-stabilizing agent; be aware of possible interactions with patient’s other medications; schedule more frequent visits when starting stimulants |
| Monitor for adverse events associated with stimulant administration | Manic symptoms, changes in cycling patterns, sleep disturbances, substance abuse |
| ADHD: attention-deficit/hyperactivity disorder | |
- The Texas Medication Algorithm Project. Texas Department of State Health Services. www.dshs.state.tx.us/mhprograms/tmapover.shtm.
- The Cochrane Collaboration. www.cochrane.org.
- Amphetamine and dextroamphetamine • Adderall
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clozapine • Clozaril
- Dexmethylphenidate • Focalin
- Dextroamphetamine • Dexedrine, DextroStat
- Diazepam • Valium
- Divalproex sodium • Depakote
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Lisdexamfetamine • Vyvanse
- Lithium • various
- Methylphenidate • Ritalin, Concerta, others
- Modafinil • Provigil
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Triazolam • Halcion
- Valproic acid • Depakene
- Venlafaxine • Effexor
Dr. Gonzalez reports no financial relationship with any company whose products are mentioned in the article or with manufacturers of competing products. He is a recipient of a T32 Ruth L. Kirschstein National Research Service Awards training fellowship sponsored by the National Institutes of Health.
Dr. Suppes receives grants/research support from Abbott Laboratories, AstraZeneca, GlaxoSmithKline, JDS Pharmaceuticals, Janssen Pharmaceutica, National Institute of Mental Health, Novartis, Pfizer Inc., and the Stanley Medical Research Institute.
1. Silberman EK, Reus VI, Jimerson DC, et al. Heterogeneity of amphetamine response in depressed patients. Am J Psychiatry 1981;138(10):1302-7.
2. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry 2007;164(8):1242-9.
3. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
4. Meyers B. Treatment of imipramine-resistant depression and lithium-refractory mania through drug interactions. Am J Psychiatry 1978;135(11):1420-1.
5. Bannet J, Ebstein RP, Belmaker RH. Clinical aspects of the interaction of lithium and stimulants. Br J Psychiatry 1980;136:204.-
6. Drimmer EJ, Gitlin MJ, Gwirtsman HE. Desipramine and methylphenidate combination treatment for depression: case report. Am J Psychiatry 1983;140(2):241-2.
7. Fernandes PP, Petty F. Modafinil for remitted bipolar depression with hypersomnia. Ann Pharmacother 2003;37(12):1807-9.
8. Berigan TR. Augmentation with modafinil to achieve remission in depression: a case report. Prim Care Companion J Clin Psychiatry 2001;3(1):32.-
9. Berigan TR. Modafinil treatment of excessive daytime sedation and fatigue associated with topiramate. Prim Care Companion J Clin Psychiatry 2002;4(6):249-50.
10. Berigan T. Modafinil treatment of excessive sedation associated with divalproex sodium. Can J Psychiatry 2004;49(1):72-3.
11. Even C, Thuile J, Santos J, Bourgin P. Modafinil as an adjunctive treatment to sleep deprivation in depression. J Psychiatry Neurosci 2005;30(6):432-3.
12. Lydon E, El-Mallakh RS. Naturalistic long-term use of methylphenidate in bipolar disorder. J Clin Psychopharmacol 2006;26(5):516-8.
13. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: treatment of residual depression and sedation. Bipolar Disord 2004;6(5):416-20.
14. El-Mallakh RS. An open study of methylphenidate in bipolar depression. Bipolar Disord 2000;2(1):56-9.
15. Menza MA, Kaufman KR, Castellanos A. Modafinil augmentation of antidepressant treatment in depression. J Clin Psychiatry 2000;61(5):378-81.
16. Wingo AP, Ghaemi SN. A systematic review of rates and diagnostic validity of comorbid adult attention-deficit/hyperactivity disorder and bipolar disorder. J Clin Psychiatry 2007;68(11):1776-84.
17. Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry 2006;163(4):716-23.
18. Nierenberg AA, Miyahara S, Spencer T, et al. Clinical and diagnostic implications of lifetime attention-deficit/hyperactivity disorder comorbidity in adults with bipolar disorder: data from the first 1000 STEP-BD participants. Biol Psychiatry 2005;57(11):1467-73.
19. Tamam L, Tuglu C, Karatas G, Ozcan S. Adult attention-deficit hyperactivity disorder in patients with bipolar I disorder in remission: preliminary study. Psychiatry Clin Neurosci 2006;60(4):480-5.
20. Faraone SV, Biederman J, Mennin D, et al. Attention-deficit hyperactivity disorder with bipolar disorder: a familial subtype? J Am Acad Child Adolesc Psychiatry 1997;36(10):1378-87; discussion 1387-90.
21. Faraone SV, Biederman J, Monuteaux MC. Attention deficit hyperactivity disorder with bipolar disorder in girls: further evidence for a familial subtype? J Affect Disord 2001;64(1):19-26.
22. Faraone SV, Glatt SJ, Tsuang MT. The genetics of pediatriconset bipolar disorder. Biol Psychiatry 2003;53(11):970-7.
23. Simon NM, Otto MW, Weiss RD, et al. Pharmacotherapy for bipolar disorder and comorbid conditions: baseline data from STEP-BD. J Clin Psychopharmacol 2004;24(5):512-20.
24. Calabrese JR. Overview of patient care issues and treatment in bipolar spectrum and bipolar II disorder. J Clin Psychiatry 2008;69(6):e18.-
25. DelBello MP, Soutullo CA, Hendricks W, et al. Prior stimulant treatment in adolescents with bipolar disorder: association with age at onset. Bipolar Disord 2001;3(2):53-7.
26. Van Kammen DP, Murphy DL. Attenuation of the euphoriant and activating effects of d- and l-amphetamine by lithium carbonate treatment. Psychopharmacologia 1975;44(3):215-24.
27. Huey LY, Janowsky DS, Judd LL, et al. Effects of lithium carbonate on methylphenidate-induced mood, behavior, and cognitive processes. Psychopharmacology (Berl) 1981;73(2):161-4.
28. Gerner RH, Post RM, Bunney WE, Jr. A dopaminergic mechanism in mania. Am J Psychiatry 1976;133(10):1177-80.
29. Koehler-Troy C, Strober M, Malenbaum R. Methylphenidateinduced mania in a prepubertal child. J Clin Psychiatry 1986;47(11):566-7.
30. Brady KT, Sonne SC. The relationship between substance abuse and bipolar disorder. J Clin Psychiatry 1995;56(suppl 3):19-24.
31. Sonne SC, Brady KT. Substance abuse and bipolar comorbidity. Psychiatr Clin North Am 1999;22(3):609-27,ix.
32. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA 1990;264(19):2511-8.
33. Estroff TW, Dackis CA, Gold MS, Pottash AL. Drug abuse and bipolar disorders. Int J Psychiatry Med 1985-1986;15(1):37-40.
34. Faraone SV. Lisdexamfetamine dimesylate: the first longacting prodrug stimulant treatment for attention deficit/hyperactivity disorder. Expert Opin Pharmacother 2008;9(9):1565-74.
1. Silberman EK, Reus VI, Jimerson DC, et al. Heterogeneity of amphetamine response in depressed patients. Am J Psychiatry 1981;138(10):1302-7.
2. Frye MA, Grunze H, Suppes T, et al. A placebo-controlled evaluation of adjunctive modafinil in the treatment of bipolar depression. Am J Psychiatry 2007;164(8):1242-9.
3. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
4. Meyers B. Treatment of imipramine-resistant depression and lithium-refractory mania through drug interactions. Am J Psychiatry 1978;135(11):1420-1.
5. Bannet J, Ebstein RP, Belmaker RH. Clinical aspects of the interaction of lithium and stimulants. Br J Psychiatry 1980;136:204.-
6. Drimmer EJ, Gitlin MJ, Gwirtsman HE. Desipramine and methylphenidate combination treatment for depression: case report. Am J Psychiatry 1983;140(2):241-2.
7. Fernandes PP, Petty F. Modafinil for remitted bipolar depression with hypersomnia. Ann Pharmacother 2003;37(12):1807-9.
8. Berigan TR. Augmentation with modafinil to achieve remission in depression: a case report. Prim Care Companion J Clin Psychiatry 2001;3(1):32.-
9. Berigan TR. Modafinil treatment of excessive daytime sedation and fatigue associated with topiramate. Prim Care Companion J Clin Psychiatry 2002;4(6):249-50.
10. Berigan T. Modafinil treatment of excessive sedation associated with divalproex sodium. Can J Psychiatry 2004;49(1):72-3.
11. Even C, Thuile J, Santos J, Bourgin P. Modafinil as an adjunctive treatment to sleep deprivation in depression. J Psychiatry Neurosci 2005;30(6):432-3.
12. Lydon E, El-Mallakh RS. Naturalistic long-term use of methylphenidate in bipolar disorder. J Clin Psychopharmacol 2006;26(5):516-8.
13. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: treatment of residual depression and sedation. Bipolar Disord 2004;6(5):416-20.
14. El-Mallakh RS. An open study of methylphenidate in bipolar depression. Bipolar Disord 2000;2(1):56-9.
15. Menza MA, Kaufman KR, Castellanos A. Modafinil augmentation of antidepressant treatment in depression. J Clin Psychiatry 2000;61(5):378-81.
16. Wingo AP, Ghaemi SN. A systematic review of rates and diagnostic validity of comorbid adult attention-deficit/hyperactivity disorder and bipolar disorder. J Clin Psychiatry 2007;68(11):1776-84.
17. Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry 2006;163(4):716-23.
18. Nierenberg AA, Miyahara S, Spencer T, et al. Clinical and diagnostic implications of lifetime attention-deficit/hyperactivity disorder comorbidity in adults with bipolar disorder: data from the first 1000 STEP-BD participants. Biol Psychiatry 2005;57(11):1467-73.
19. Tamam L, Tuglu C, Karatas G, Ozcan S. Adult attention-deficit hyperactivity disorder in patients with bipolar I disorder in remission: preliminary study. Psychiatry Clin Neurosci 2006;60(4):480-5.
20. Faraone SV, Biederman J, Mennin D, et al. Attention-deficit hyperactivity disorder with bipolar disorder: a familial subtype? J Am Acad Child Adolesc Psychiatry 1997;36(10):1378-87; discussion 1387-90.
21. Faraone SV, Biederman J, Monuteaux MC. Attention deficit hyperactivity disorder with bipolar disorder in girls: further evidence for a familial subtype? J Affect Disord 2001;64(1):19-26.
22. Faraone SV, Glatt SJ, Tsuang MT. The genetics of pediatriconset bipolar disorder. Biol Psychiatry 2003;53(11):970-7.
23. Simon NM, Otto MW, Weiss RD, et al. Pharmacotherapy for bipolar disorder and comorbid conditions: baseline data from STEP-BD. J Clin Psychopharmacol 2004;24(5):512-20.
24. Calabrese JR. Overview of patient care issues and treatment in bipolar spectrum and bipolar II disorder. J Clin Psychiatry 2008;69(6):e18.-
25. DelBello MP, Soutullo CA, Hendricks W, et al. Prior stimulant treatment in adolescents with bipolar disorder: association with age at onset. Bipolar Disord 2001;3(2):53-7.
26. Van Kammen DP, Murphy DL. Attenuation of the euphoriant and activating effects of d- and l-amphetamine by lithium carbonate treatment. Psychopharmacologia 1975;44(3):215-24.
27. Huey LY, Janowsky DS, Judd LL, et al. Effects of lithium carbonate on methylphenidate-induced mood, behavior, and cognitive processes. Psychopharmacology (Berl) 1981;73(2):161-4.
28. Gerner RH, Post RM, Bunney WE, Jr. A dopaminergic mechanism in mania. Am J Psychiatry 1976;133(10):1177-80.
29. Koehler-Troy C, Strober M, Malenbaum R. Methylphenidateinduced mania in a prepubertal child. J Clin Psychiatry 1986;47(11):566-7.
30. Brady KT, Sonne SC. The relationship between substance abuse and bipolar disorder. J Clin Psychiatry 1995;56(suppl 3):19-24.
31. Sonne SC, Brady KT. Substance abuse and bipolar comorbidity. Psychiatr Clin North Am 1999;22(3):609-27,ix.
32. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA 1990;264(19):2511-8.
33. Estroff TW, Dackis CA, Gold MS, Pottash AL. Drug abuse and bipolar disorders. Int J Psychiatry Med 1985-1986;15(1):37-40.
34. Faraone SV. Lisdexamfetamine dimesylate: the first longacting prodrug stimulant treatment for attention deficit/hyperactivity disorder. Expert Opin Pharmacother 2008;9(9):1565-74.
Will my patient attempt suicide again?
Ms. J, age 32, comes to our mental health clinic seeking treatment for depression and anxiety. She reports she has attempted suicide 3 times. Ms. J describes the first 2 attempts—both of which occurred when she was in her 20s after the end of a relationship—as “cries for attention” that were relatively innocuous. Her third suicide attempt, however, was an acetaminophen overdose approximately 1 year ago that resulted in hospitalization and irreversible liver damage.
Ms. J acknowledges that over the last several weeks she has been thinking about suicide almost constantly, especially as the anniversary of her fiancé’s death approaches. She says she has a nearly full bottle of zolpidem in her medicine cabinet and fantasizes about taking all of them and just “going to sleep.”
Many patients—especially those with depression—experience recurrent thoughts of death or a wish to die, but only about 10% attempt suicide.1 To identify those who are at highest risk and warrant hospitalization, it is vital to assess how a history of suicidal behavior and other factors impact the risk for future suicide attempts. This article:
- examines research on patients who have attempted suicide and risk factors for repeat suicide attempts
- describes characteristics of patients with multiple attempts
- explores the link between a history of self-injurious behavior and suicide attempts.
A strong predictor
A previous suicide attempt is among the strongest predictors of future suicide attempts.2-4 In a sample of clinically referred European adolescents, those who had attempted suicide were 3 times more likely to try again during the 1-year follow-up compared with those who had never attempted suicide.5 In addition, Harris et al6 found that patients with a previous suicide attempt were 38 times more likely to eventually commit suicide than those with no past attempts.
Other risk factors
Other factors might help predict which individuals will continue to engage in suicidal behavior after a first attempt (Table 1).7,8 Spirito et al7 followed 58 adolescent suicide attempters for 3 months after their initial attempt. Seven (12%) made a subsequent attempt, and 26 (45%) reported continued suicidal ideation. Depressed mood was the strongest predictor of subsequent suicidal behavior, followed by poor family functioning, affect regulation difficulty, and hopelessness.
Hopelessness. Beck et al9 found that patients who scored ≥9 on the Beck Hopelessness Scale (BHS)—the most common self-report measure of hopelessness—were approximately 11 times more likely to commit suicide than patients who scored ≤8. A study of hospitalized suicide attempters found that BHS scores were unique predictors of future suicide attempts.10 Several studies have found that persons who remain consistently hopeless are more likely to kill themselves compared with those who have fluctuating hopelessness levels.11,12
History of abuse—specifically sexual abuse—is associated with suicidal behavior. A study of depressed women age >50 found that among those who were sexually abused before age 18, 83% reported 1 suicide attempt and 67% made multiple attempts.13 Among women who had not experienced childhood sexual abuse, 58% reported a past suicide attempt and 27% made multiple attempts.13
In a separate study of psychiatric inpatients, those who had been physically or sexually abused were more likely to have made a suicide attempt than patients with no such history.14 This study did not find a difference in reported abuse between single and multiple suicide attempters.
Stressors. In many cases suicide attempts are precipitated by acute or chronic stressors, including:
- job stress
- chronic illness
- financial problems
- relationship discord
- retirement and declining physical health (especially for older men)
- death of a loved one.15
Risk is not necessarily cumulative—and not all risk factors are weighted equally. In general, however, the more risk factors a patient has, the greater the likelihood that he or she may attempt suicide.17
Table 1
Repeated suicidal behavior: Factors that increase risk
| History of ≥1 suicide attempts |
| Feelings of hopelessness |
| Presence of an Axis I or II disorder |
| High levels of perceived stress |
| History of physical or sexual abuse |
| Source: References 7,8 |
Red flag: Multiple attempts
When assessing a patient’s suicide history, ask about the number of attempts. A person who makes >1 suicide attempt—a multiple attempter—has a significantly higher chance of making subsequent attempts compared with those with 1 or no attempts.18,19
Persons who make multiple attempts share certain characteristics (Table 2).19-21 Rudd et al19 compared 68 multiple attempters with 128 single attempters and found that multiple attempters had higher levels of:
- suicide ideation
- depression
- hopelessness
- perceived stress.
Similarly, Foreman et al20 found that compared with single suicide attempters, multiple attempters had higher levels of depression, hopelessness, and suicidal ideation and met criteria for more Axis I diagnoses. Multiple attempters also were more likely to be:
- diagnosed with substance use disorders, psychotic disorder, or borderline personality disorder
- unemployed and have relationship difficulties, a history of emotional abuse, and a family history of psychiatric problems and suicide.
Among 326 individuals in a military medical setting treated for suicidal behavior or severe suicidal ideation, multiple suicide attempters reported higher levels of ongoing distress that was unrelated to specific life stressors.22 This suggests these patients may not respond well to psychological interventions that focus on problem-solving.
Table 2
Common characteristics of multiple suicide attempters
| History of Axis I disorder (major depressive disorder, bipolar disorder, schizophrenia, substance use disorders, eating disorders) |
| High levels of perceived stress |
| High levels of depression |
| Symptoms of borderline personality disorder |
| Poor problem-solving skills |
| Family history of psychiatric illness |
| Source: References 19-21 |
Self-harm and suicidal behavior
Patients who engage in nonsuicidal self harm—also called self-injurious behavior (SIB)—may be mistaken for suicide attempters. Although differences exist between suicide attempters and those who engage in SIB, evidence suggests that a history of SIB increases risk for suicidal behavior.23,24 In a retrospective study of 4,167 self-harmers, females who engaged in ≥4 acts of SIB were more likely to die from suicide than those who engaged in ≤3 acts.25 A cross-sectional analysis of data from 3,069 students responding to a random Web-based survey found that an increased incidence of SIB significantly increased the odds of suicidal behavior.26
Although the link between SIB and suicide attempts remains unclear, evidence suggests SIB is a risk factor for suicidal behavior and therefore should be assessed when evaluating a patient’s suicide risk.
CASE CONTINUED: At high risk
Ms. J has several risk factors for making another suicide attempt. She has 3 previous attempts, and because her last attempt caused liver damage we know she is capable of lethal behavior. In addition, the anniversary of the death of her fiancé is approaching. Ms. J also reports almost constant suicidal ideation, with a specific plan (to overdose). Her fantasies of taking pills could be interpreted as mental rehearsal and desensitization to the behavior.
Because we believe Ms. J is at high risk for a serious, if not lethal, suicide attempt we conduct a 4-question suicide inquiry. It is clear that Ms. J had suicidal thoughts and a plan. Her answer to “How likely is it that once you leave my office you will do something to hurt yourself?” is the key to determining whether or not she requires hospitalization. Ms. J states that she is “pretty certain she will hurt herself” once she leaves the office, so we hospitalize her.
To determine if a patient requires immediate hospitalization, perform a specific suicide inquiry. Although there is no surefire way to determine if a patient will kill himself or herself, asking specific questions can help you gauge risk. Based on evidence28 and my clinical experience, I focus on patients’ answers to 4 questions (Table 3). Affirmative answers to these questions are a strong indication that a patient requires hospitalization.
Occasionally, patients are not truthful when asked about their suicidal intent. If you suspect a patient is lying, clinical judgment and the patient’s history guide the decision on hospitalization.
Table 3
Hospitalize? 4 questions to guide your decision
| Are you having thoughts of hurting or killing yourself? If yes: What are you thinking/planning to do? |
| Do you have access to lethal means? |
| What is the likelihood that you will hurt yourself? |
| Have you ever done something to hurt yourself (either suicide attempt or self-injurious behavior)? If yes: How many times? |
- Joiner TE. Why people die by suicide. Cambridge, MA: Harvard University Press; 2005:46-93,203-22.
- American Foundation for Suicide Prevention. www.afsp.org.
- SAVE: Suicide Awareness Voices of Education. www.save.org.
- Zolpidem • Ambien
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Office of Applied Studies, Substance Abuse and Mental Health Services Administration. Suicidal thoughts, suicide attempts, major depressive episode, and substance use among adults. Rockville, MD: US Department of Health and Human Services; 2006.
2. Pfeffer CR, Klerman GL, Hurt SW, et al. Suicidal children grow up: rates and psychosocial risk factors for suicide attempts during follow-up. J Am Acad Child Adolesc Psychiatry 1993;32:106-13.
3. Lewinsohn PM, Rohde P, Seeley JR. Psychosocial risk factors for future adolescent suicide attempts. J Consult Clin Psychol 1994;62:297-305.
4. Brown GK, Beck AT, Steer RA, et al. Risk factors for suicide in psychiatric outpatients: a 20-year prospective study. J Consult Clin Psychol 2000;68:371-7.
5. Hultén A, Jiang GX, Wasserman D, et al. Repetition of attempted suicide among teenagers in Europe: frequency, timing, and risk factors. Eur Child Adolesc Psychiatry 2001;10:161-9.
6. Harris EC, Barraclough B. Suicide as an outcome for mental disorders: a metaanalysis. Br J Psychiatr 1997;170:205-28.
7. Spirito A, Valeri S, Boergers J, et al. Predictors of continued suicidal behavior in adolescents following a suicide attempt. J Clin Child Adolesc Psychol 2003;32(2):284-9.
8. Moscicki EK. Epidemiology of completed and attempted suicide: toward a framework for prevention. Clin Neurosci Res 2001;1:310-23.
9. Beck AT, Steer RA. Clinical predictors of eventual suicide: a 5- to 10-year prospective study of suicide attempters. J Affec Disord 1989;17:203-9.
10. Petrie K, Chamberlain K, Clarke D. Psychological predictors of future suicidal behaviour in hospitalized suicide attempters. Br J Clin Psychol 1988;27:247-57.
11. Dahlsgaard KK, Beck AT, Brown GK. Inadequate response to therapy as a predictor of suicide. Suicide Life Threat Behav 1998;28:197-204.
12. Young MA, Fogg LF, Scheftner W, et al. Stable trait components of hopelessness: baseline and sensitivity to depression. J Abnorm Psychol 1996;105(2):155-65.
13. Talbot NL, Duberstein PR, Cox C, et al. Preliminary report on childhood sexual abuse, suicidal ideation, and suicide attempts among middle-aged and older depressed women. Am J Geriatr Psychiatry 2004;12:536-8.
14. Andover MS, Zlotnick C, Miller IW. Childhood physical and sexual abuse in depressed patients with single and multiple suicide attempts. Suicide Life Threat Behav 2007;37(4):467-74.
15. Heikkinen M, Aro H, Lönnqvist J. The partners’ views on precipitant stressors in suicide. Acta Psychiatr Scand 1992;85(5):380-4.
16. Bunch J, Barraclough B. The influence of parental death anniversaries upon suicide dates. Br J Psychiatry 1971;118:621-6.
17. Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Survey. Arch Gen Psychiatry 1999;56(7):617-26.
18. Goldston DB, Daniel SS, Reboussin DM, et al. Suicide attempts among formerly hospitalized adolescents: a prospective naturalistic study of risk during the first 5 years after discharge. J Am Acad Child Adolesc Psychiatry 1999;38:660-71.
19. Rudd MD, Joiner T, Rajab MH. Relationships among suicide ideators, attempters, and multiple attempters in a young-adult sample. J Abnorm Psychol 1996;105(4):541-50.
20. Foreman EM, Berk MS, Henriques GR, et al. History of multiple suicide attempts as a behavioral marker of severe psychopathology. Am J Psychiatry 2004;161(3):437-43.
21. Miranda R, Scott M, Roger H, et al. Suicide attempt characteristics, diagnoses, and future attempts: comparing multiple attempters to single attempters and ideators. J Am Acad Child Adolesc Psychiatry 2008;47:32-40.
22. Joiner TE, Rudd MD. Intensity and duration of suicidal crises vary as a function of previous suicide attempts and negative life events. J Consult Clin Psychol 2000;68(5):909-16.
23. Suominen K, Isometsä E, Suokas J, et al. Completed suicide after a suicide attempt: a 37-year follow-up study. Am J Psychiatry 2004;161:562-3.
24. Owens D, Wood C, Greenwood D, et al. Mortality and suicide after non-fatal self-poisoning: a 16-year outcome study of patients attending accident and emergency. Br J Psychiatry 2005;187:470-5.
25. Haw C, Bergen H, Casey D, Hawton K. Repetition of deliberate self-harm: a study of the characteristics and subsequent deaths in patients presenting to a general hospital according to extent of repetition. Suicide Life Threat Behav 2007;37(4):379-96.
26. Whitlock J, Knox KL. The relationship between self-injurious behavior and suicide in a young adult population. Arch Pediatr Adolesc Med 2007;161:634-40.
27. Joiner TE. Why people die by suicide. Cambridge, MA: Harvard University Press; 2005:46-93,203-22.
28. Gliatto MF, Rai AK. Evaluation and treatment of patients with suicidal ideation. Am Fam Physician. 1999;59(6):1500-6.
Ms. J, age 32, comes to our mental health clinic seeking treatment for depression and anxiety. She reports she has attempted suicide 3 times. Ms. J describes the first 2 attempts—both of which occurred when she was in her 20s after the end of a relationship—as “cries for attention” that were relatively innocuous. Her third suicide attempt, however, was an acetaminophen overdose approximately 1 year ago that resulted in hospitalization and irreversible liver damage.
Ms. J acknowledges that over the last several weeks she has been thinking about suicide almost constantly, especially as the anniversary of her fiancé’s death approaches. She says she has a nearly full bottle of zolpidem in her medicine cabinet and fantasizes about taking all of them and just “going to sleep.”
Many patients—especially those with depression—experience recurrent thoughts of death or a wish to die, but only about 10% attempt suicide.1 To identify those who are at highest risk and warrant hospitalization, it is vital to assess how a history of suicidal behavior and other factors impact the risk for future suicide attempts. This article:
- examines research on patients who have attempted suicide and risk factors for repeat suicide attempts
- describes characteristics of patients with multiple attempts
- explores the link between a history of self-injurious behavior and suicide attempts.
A strong predictor
A previous suicide attempt is among the strongest predictors of future suicide attempts.2-4 In a sample of clinically referred European adolescents, those who had attempted suicide were 3 times more likely to try again during the 1-year follow-up compared with those who had never attempted suicide.5 In addition, Harris et al6 found that patients with a previous suicide attempt were 38 times more likely to eventually commit suicide than those with no past attempts.
Other risk factors
Other factors might help predict which individuals will continue to engage in suicidal behavior after a first attempt (Table 1).7,8 Spirito et al7 followed 58 adolescent suicide attempters for 3 months after their initial attempt. Seven (12%) made a subsequent attempt, and 26 (45%) reported continued suicidal ideation. Depressed mood was the strongest predictor of subsequent suicidal behavior, followed by poor family functioning, affect regulation difficulty, and hopelessness.
Hopelessness. Beck et al9 found that patients who scored ≥9 on the Beck Hopelessness Scale (BHS)—the most common self-report measure of hopelessness—were approximately 11 times more likely to commit suicide than patients who scored ≤8. A study of hospitalized suicide attempters found that BHS scores were unique predictors of future suicide attempts.10 Several studies have found that persons who remain consistently hopeless are more likely to kill themselves compared with those who have fluctuating hopelessness levels.11,12
History of abuse—specifically sexual abuse—is associated with suicidal behavior. A study of depressed women age >50 found that among those who were sexually abused before age 18, 83% reported 1 suicide attempt and 67% made multiple attempts.13 Among women who had not experienced childhood sexual abuse, 58% reported a past suicide attempt and 27% made multiple attempts.13
In a separate study of psychiatric inpatients, those who had been physically or sexually abused were more likely to have made a suicide attempt than patients with no such history.14 This study did not find a difference in reported abuse between single and multiple suicide attempters.
Stressors. In many cases suicide attempts are precipitated by acute or chronic stressors, including:
- job stress
- chronic illness
- financial problems
- relationship discord
- retirement and declining physical health (especially for older men)
- death of a loved one.15
Risk is not necessarily cumulative—and not all risk factors are weighted equally. In general, however, the more risk factors a patient has, the greater the likelihood that he or she may attempt suicide.17
Table 1
Repeated suicidal behavior: Factors that increase risk
| History of ≥1 suicide attempts |
| Feelings of hopelessness |
| Presence of an Axis I or II disorder |
| High levels of perceived stress |
| History of physical or sexual abuse |
| Source: References 7,8 |
Red flag: Multiple attempts
When assessing a patient’s suicide history, ask about the number of attempts. A person who makes >1 suicide attempt—a multiple attempter—has a significantly higher chance of making subsequent attempts compared with those with 1 or no attempts.18,19
Persons who make multiple attempts share certain characteristics (Table 2).19-21 Rudd et al19 compared 68 multiple attempters with 128 single attempters and found that multiple attempters had higher levels of:
- suicide ideation
- depression
- hopelessness
- perceived stress.
Similarly, Foreman et al20 found that compared with single suicide attempters, multiple attempters had higher levels of depression, hopelessness, and suicidal ideation and met criteria for more Axis I diagnoses. Multiple attempters also were more likely to be:
- diagnosed with substance use disorders, psychotic disorder, or borderline personality disorder
- unemployed and have relationship difficulties, a history of emotional abuse, and a family history of psychiatric problems and suicide.
Among 326 individuals in a military medical setting treated for suicidal behavior or severe suicidal ideation, multiple suicide attempters reported higher levels of ongoing distress that was unrelated to specific life stressors.22 This suggests these patients may not respond well to psychological interventions that focus on problem-solving.
Table 2
Common characteristics of multiple suicide attempters
| History of Axis I disorder (major depressive disorder, bipolar disorder, schizophrenia, substance use disorders, eating disorders) |
| High levels of perceived stress |
| High levels of depression |
| Symptoms of borderline personality disorder |
| Poor problem-solving skills |
| Family history of psychiatric illness |
| Source: References 19-21 |
Self-harm and suicidal behavior
Patients who engage in nonsuicidal self harm—also called self-injurious behavior (SIB)—may be mistaken for suicide attempters. Although differences exist between suicide attempters and those who engage in SIB, evidence suggests that a history of SIB increases risk for suicidal behavior.23,24 In a retrospective study of 4,167 self-harmers, females who engaged in ≥4 acts of SIB were more likely to die from suicide than those who engaged in ≤3 acts.25 A cross-sectional analysis of data from 3,069 students responding to a random Web-based survey found that an increased incidence of SIB significantly increased the odds of suicidal behavior.26
Although the link between SIB and suicide attempts remains unclear, evidence suggests SIB is a risk factor for suicidal behavior and therefore should be assessed when evaluating a patient’s suicide risk.
CASE CONTINUED: At high risk
Ms. J has several risk factors for making another suicide attempt. She has 3 previous attempts, and because her last attempt caused liver damage we know she is capable of lethal behavior. In addition, the anniversary of the death of her fiancé is approaching. Ms. J also reports almost constant suicidal ideation, with a specific plan (to overdose). Her fantasies of taking pills could be interpreted as mental rehearsal and desensitization to the behavior.
Because we believe Ms. J is at high risk for a serious, if not lethal, suicide attempt we conduct a 4-question suicide inquiry. It is clear that Ms. J had suicidal thoughts and a plan. Her answer to “How likely is it that once you leave my office you will do something to hurt yourself?” is the key to determining whether or not she requires hospitalization. Ms. J states that she is “pretty certain she will hurt herself” once she leaves the office, so we hospitalize her.
To determine if a patient requires immediate hospitalization, perform a specific suicide inquiry. Although there is no surefire way to determine if a patient will kill himself or herself, asking specific questions can help you gauge risk. Based on evidence28 and my clinical experience, I focus on patients’ answers to 4 questions (Table 3). Affirmative answers to these questions are a strong indication that a patient requires hospitalization.
Occasionally, patients are not truthful when asked about their suicidal intent. If you suspect a patient is lying, clinical judgment and the patient’s history guide the decision on hospitalization.
Table 3
Hospitalize? 4 questions to guide your decision
| Are you having thoughts of hurting or killing yourself? If yes: What are you thinking/planning to do? |
| Do you have access to lethal means? |
| What is the likelihood that you will hurt yourself? |
| Have you ever done something to hurt yourself (either suicide attempt or self-injurious behavior)? If yes: How many times? |
- Joiner TE. Why people die by suicide. Cambridge, MA: Harvard University Press; 2005:46-93,203-22.
- American Foundation for Suicide Prevention. www.afsp.org.
- SAVE: Suicide Awareness Voices of Education. www.save.org.
- Zolpidem • Ambien
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. J, age 32, comes to our mental health clinic seeking treatment for depression and anxiety. She reports she has attempted suicide 3 times. Ms. J describes the first 2 attempts—both of which occurred when she was in her 20s after the end of a relationship—as “cries for attention” that were relatively innocuous. Her third suicide attempt, however, was an acetaminophen overdose approximately 1 year ago that resulted in hospitalization and irreversible liver damage.
Ms. J acknowledges that over the last several weeks she has been thinking about suicide almost constantly, especially as the anniversary of her fiancé’s death approaches. She says she has a nearly full bottle of zolpidem in her medicine cabinet and fantasizes about taking all of them and just “going to sleep.”
Many patients—especially those with depression—experience recurrent thoughts of death or a wish to die, but only about 10% attempt suicide.1 To identify those who are at highest risk and warrant hospitalization, it is vital to assess how a history of suicidal behavior and other factors impact the risk for future suicide attempts. This article:
- examines research on patients who have attempted suicide and risk factors for repeat suicide attempts
- describes characteristics of patients with multiple attempts
- explores the link between a history of self-injurious behavior and suicide attempts.
A strong predictor
A previous suicide attempt is among the strongest predictors of future suicide attempts.2-4 In a sample of clinically referred European adolescents, those who had attempted suicide were 3 times more likely to try again during the 1-year follow-up compared with those who had never attempted suicide.5 In addition, Harris et al6 found that patients with a previous suicide attempt were 38 times more likely to eventually commit suicide than those with no past attempts.
Other risk factors
Other factors might help predict which individuals will continue to engage in suicidal behavior after a first attempt (Table 1).7,8 Spirito et al7 followed 58 adolescent suicide attempters for 3 months after their initial attempt. Seven (12%) made a subsequent attempt, and 26 (45%) reported continued suicidal ideation. Depressed mood was the strongest predictor of subsequent suicidal behavior, followed by poor family functioning, affect regulation difficulty, and hopelessness.
Hopelessness. Beck et al9 found that patients who scored ≥9 on the Beck Hopelessness Scale (BHS)—the most common self-report measure of hopelessness—were approximately 11 times more likely to commit suicide than patients who scored ≤8. A study of hospitalized suicide attempters found that BHS scores were unique predictors of future suicide attempts.10 Several studies have found that persons who remain consistently hopeless are more likely to kill themselves compared with those who have fluctuating hopelessness levels.11,12
History of abuse—specifically sexual abuse—is associated with suicidal behavior. A study of depressed women age >50 found that among those who were sexually abused before age 18, 83% reported 1 suicide attempt and 67% made multiple attempts.13 Among women who had not experienced childhood sexual abuse, 58% reported a past suicide attempt and 27% made multiple attempts.13
In a separate study of psychiatric inpatients, those who had been physically or sexually abused were more likely to have made a suicide attempt than patients with no such history.14 This study did not find a difference in reported abuse between single and multiple suicide attempters.
Stressors. In many cases suicide attempts are precipitated by acute or chronic stressors, including:
- job stress
- chronic illness
- financial problems
- relationship discord
- retirement and declining physical health (especially for older men)
- death of a loved one.15
Risk is not necessarily cumulative—and not all risk factors are weighted equally. In general, however, the more risk factors a patient has, the greater the likelihood that he or she may attempt suicide.17
Table 1
Repeated suicidal behavior: Factors that increase risk
| History of ≥1 suicide attempts |
| Feelings of hopelessness |
| Presence of an Axis I or II disorder |
| High levels of perceived stress |
| History of physical or sexual abuse |
| Source: References 7,8 |
Red flag: Multiple attempts
When assessing a patient’s suicide history, ask about the number of attempts. A person who makes >1 suicide attempt—a multiple attempter—has a significantly higher chance of making subsequent attempts compared with those with 1 or no attempts.18,19
Persons who make multiple attempts share certain characteristics (Table 2).19-21 Rudd et al19 compared 68 multiple attempters with 128 single attempters and found that multiple attempters had higher levels of:
- suicide ideation
- depression
- hopelessness
- perceived stress.
Similarly, Foreman et al20 found that compared with single suicide attempters, multiple attempters had higher levels of depression, hopelessness, and suicidal ideation and met criteria for more Axis I diagnoses. Multiple attempters also were more likely to be:
- diagnosed with substance use disorders, psychotic disorder, or borderline personality disorder
- unemployed and have relationship difficulties, a history of emotional abuse, and a family history of psychiatric problems and suicide.
Among 326 individuals in a military medical setting treated for suicidal behavior or severe suicidal ideation, multiple suicide attempters reported higher levels of ongoing distress that was unrelated to specific life stressors.22 This suggests these patients may not respond well to psychological interventions that focus on problem-solving.
Table 2
Common characteristics of multiple suicide attempters
| History of Axis I disorder (major depressive disorder, bipolar disorder, schizophrenia, substance use disorders, eating disorders) |
| High levels of perceived stress |
| High levels of depression |
| Symptoms of borderline personality disorder |
| Poor problem-solving skills |
| Family history of psychiatric illness |
| Source: References 19-21 |
Self-harm and suicidal behavior
Patients who engage in nonsuicidal self harm—also called self-injurious behavior (SIB)—may be mistaken for suicide attempters. Although differences exist between suicide attempters and those who engage in SIB, evidence suggests that a history of SIB increases risk for suicidal behavior.23,24 In a retrospective study of 4,167 self-harmers, females who engaged in ≥4 acts of SIB were more likely to die from suicide than those who engaged in ≤3 acts.25 A cross-sectional analysis of data from 3,069 students responding to a random Web-based survey found that an increased incidence of SIB significantly increased the odds of suicidal behavior.26
Although the link between SIB and suicide attempts remains unclear, evidence suggests SIB is a risk factor for suicidal behavior and therefore should be assessed when evaluating a patient’s suicide risk.
CASE CONTINUED: At high risk
Ms. J has several risk factors for making another suicide attempt. She has 3 previous attempts, and because her last attempt caused liver damage we know she is capable of lethal behavior. In addition, the anniversary of the death of her fiancé is approaching. Ms. J also reports almost constant suicidal ideation, with a specific plan (to overdose). Her fantasies of taking pills could be interpreted as mental rehearsal and desensitization to the behavior.
Because we believe Ms. J is at high risk for a serious, if not lethal, suicide attempt we conduct a 4-question suicide inquiry. It is clear that Ms. J had suicidal thoughts and a plan. Her answer to “How likely is it that once you leave my office you will do something to hurt yourself?” is the key to determining whether or not she requires hospitalization. Ms. J states that she is “pretty certain she will hurt herself” once she leaves the office, so we hospitalize her.
To determine if a patient requires immediate hospitalization, perform a specific suicide inquiry. Although there is no surefire way to determine if a patient will kill himself or herself, asking specific questions can help you gauge risk. Based on evidence28 and my clinical experience, I focus on patients’ answers to 4 questions (Table 3). Affirmative answers to these questions are a strong indication that a patient requires hospitalization.
Occasionally, patients are not truthful when asked about their suicidal intent. If you suspect a patient is lying, clinical judgment and the patient’s history guide the decision on hospitalization.
Table 3
Hospitalize? 4 questions to guide your decision
| Are you having thoughts of hurting or killing yourself? If yes: What are you thinking/planning to do? |
| Do you have access to lethal means? |
| What is the likelihood that you will hurt yourself? |
| Have you ever done something to hurt yourself (either suicide attempt or self-injurious behavior)? If yes: How many times? |
- Joiner TE. Why people die by suicide. Cambridge, MA: Harvard University Press; 2005:46-93,203-22.
- American Foundation for Suicide Prevention. www.afsp.org.
- SAVE: Suicide Awareness Voices of Education. www.save.org.
- Zolpidem • Ambien
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Office of Applied Studies, Substance Abuse and Mental Health Services Administration. Suicidal thoughts, suicide attempts, major depressive episode, and substance use among adults. Rockville, MD: US Department of Health and Human Services; 2006.
2. Pfeffer CR, Klerman GL, Hurt SW, et al. Suicidal children grow up: rates and psychosocial risk factors for suicide attempts during follow-up. J Am Acad Child Adolesc Psychiatry 1993;32:106-13.
3. Lewinsohn PM, Rohde P, Seeley JR. Psychosocial risk factors for future adolescent suicide attempts. J Consult Clin Psychol 1994;62:297-305.
4. Brown GK, Beck AT, Steer RA, et al. Risk factors for suicide in psychiatric outpatients: a 20-year prospective study. J Consult Clin Psychol 2000;68:371-7.
5. Hultén A, Jiang GX, Wasserman D, et al. Repetition of attempted suicide among teenagers in Europe: frequency, timing, and risk factors. Eur Child Adolesc Psychiatry 2001;10:161-9.
6. Harris EC, Barraclough B. Suicide as an outcome for mental disorders: a metaanalysis. Br J Psychiatr 1997;170:205-28.
7. Spirito A, Valeri S, Boergers J, et al. Predictors of continued suicidal behavior in adolescents following a suicide attempt. J Clin Child Adolesc Psychol 2003;32(2):284-9.
8. Moscicki EK. Epidemiology of completed and attempted suicide: toward a framework for prevention. Clin Neurosci Res 2001;1:310-23.
9. Beck AT, Steer RA. Clinical predictors of eventual suicide: a 5- to 10-year prospective study of suicide attempters. J Affec Disord 1989;17:203-9.
10. Petrie K, Chamberlain K, Clarke D. Psychological predictors of future suicidal behaviour in hospitalized suicide attempters. Br J Clin Psychol 1988;27:247-57.
11. Dahlsgaard KK, Beck AT, Brown GK. Inadequate response to therapy as a predictor of suicide. Suicide Life Threat Behav 1998;28:197-204.
12. Young MA, Fogg LF, Scheftner W, et al. Stable trait components of hopelessness: baseline and sensitivity to depression. J Abnorm Psychol 1996;105(2):155-65.
13. Talbot NL, Duberstein PR, Cox C, et al. Preliminary report on childhood sexual abuse, suicidal ideation, and suicide attempts among middle-aged and older depressed women. Am J Geriatr Psychiatry 2004;12:536-8.
14. Andover MS, Zlotnick C, Miller IW. Childhood physical and sexual abuse in depressed patients with single and multiple suicide attempts. Suicide Life Threat Behav 2007;37(4):467-74.
15. Heikkinen M, Aro H, Lönnqvist J. The partners’ views on precipitant stressors in suicide. Acta Psychiatr Scand 1992;85(5):380-4.
16. Bunch J, Barraclough B. The influence of parental death anniversaries upon suicide dates. Br J Psychiatry 1971;118:621-6.
17. Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Survey. Arch Gen Psychiatry 1999;56(7):617-26.
18. Goldston DB, Daniel SS, Reboussin DM, et al. Suicide attempts among formerly hospitalized adolescents: a prospective naturalistic study of risk during the first 5 years after discharge. J Am Acad Child Adolesc Psychiatry 1999;38:660-71.
19. Rudd MD, Joiner T, Rajab MH. Relationships among suicide ideators, attempters, and multiple attempters in a young-adult sample. J Abnorm Psychol 1996;105(4):541-50.
20. Foreman EM, Berk MS, Henriques GR, et al. History of multiple suicide attempts as a behavioral marker of severe psychopathology. Am J Psychiatry 2004;161(3):437-43.
21. Miranda R, Scott M, Roger H, et al. Suicide attempt characteristics, diagnoses, and future attempts: comparing multiple attempters to single attempters and ideators. J Am Acad Child Adolesc Psychiatry 2008;47:32-40.
22. Joiner TE, Rudd MD. Intensity and duration of suicidal crises vary as a function of previous suicide attempts and negative life events. J Consult Clin Psychol 2000;68(5):909-16.
23. Suominen K, Isometsä E, Suokas J, et al. Completed suicide after a suicide attempt: a 37-year follow-up study. Am J Psychiatry 2004;161:562-3.
24. Owens D, Wood C, Greenwood D, et al. Mortality and suicide after non-fatal self-poisoning: a 16-year outcome study of patients attending accident and emergency. Br J Psychiatry 2005;187:470-5.
25. Haw C, Bergen H, Casey D, Hawton K. Repetition of deliberate self-harm: a study of the characteristics and subsequent deaths in patients presenting to a general hospital according to extent of repetition. Suicide Life Threat Behav 2007;37(4):379-96.
26. Whitlock J, Knox KL. The relationship between self-injurious behavior and suicide in a young adult population. Arch Pediatr Adolesc Med 2007;161:634-40.
27. Joiner TE. Why people die by suicide. Cambridge, MA: Harvard University Press; 2005:46-93,203-22.
28. Gliatto MF, Rai AK. Evaluation and treatment of patients with suicidal ideation. Am Fam Physician. 1999;59(6):1500-6.
1. Office of Applied Studies, Substance Abuse and Mental Health Services Administration. Suicidal thoughts, suicide attempts, major depressive episode, and substance use among adults. Rockville, MD: US Department of Health and Human Services; 2006.
2. Pfeffer CR, Klerman GL, Hurt SW, et al. Suicidal children grow up: rates and psychosocial risk factors for suicide attempts during follow-up. J Am Acad Child Adolesc Psychiatry 1993;32:106-13.
3. Lewinsohn PM, Rohde P, Seeley JR. Psychosocial risk factors for future adolescent suicide attempts. J Consult Clin Psychol 1994;62:297-305.
4. Brown GK, Beck AT, Steer RA, et al. Risk factors for suicide in psychiatric outpatients: a 20-year prospective study. J Consult Clin Psychol 2000;68:371-7.
5. Hultén A, Jiang GX, Wasserman D, et al. Repetition of attempted suicide among teenagers in Europe: frequency, timing, and risk factors. Eur Child Adolesc Psychiatry 2001;10:161-9.
6. Harris EC, Barraclough B. Suicide as an outcome for mental disorders: a metaanalysis. Br J Psychiatr 1997;170:205-28.
7. Spirito A, Valeri S, Boergers J, et al. Predictors of continued suicidal behavior in adolescents following a suicide attempt. J Clin Child Adolesc Psychol 2003;32(2):284-9.
8. Moscicki EK. Epidemiology of completed and attempted suicide: toward a framework for prevention. Clin Neurosci Res 2001;1:310-23.
9. Beck AT, Steer RA. Clinical predictors of eventual suicide: a 5- to 10-year prospective study of suicide attempters. J Affec Disord 1989;17:203-9.
10. Petrie K, Chamberlain K, Clarke D. Psychological predictors of future suicidal behaviour in hospitalized suicide attempters. Br J Clin Psychol 1988;27:247-57.
11. Dahlsgaard KK, Beck AT, Brown GK. Inadequate response to therapy as a predictor of suicide. Suicide Life Threat Behav 1998;28:197-204.
12. Young MA, Fogg LF, Scheftner W, et al. Stable trait components of hopelessness: baseline and sensitivity to depression. J Abnorm Psychol 1996;105(2):155-65.
13. Talbot NL, Duberstein PR, Cox C, et al. Preliminary report on childhood sexual abuse, suicidal ideation, and suicide attempts among middle-aged and older depressed women. Am J Geriatr Psychiatry 2004;12:536-8.
14. Andover MS, Zlotnick C, Miller IW. Childhood physical and sexual abuse in depressed patients with single and multiple suicide attempts. Suicide Life Threat Behav 2007;37(4):467-74.
15. Heikkinen M, Aro H, Lönnqvist J. The partners’ views on precipitant stressors in suicide. Acta Psychiatr Scand 1992;85(5):380-4.
16. Bunch J, Barraclough B. The influence of parental death anniversaries upon suicide dates. Br J Psychiatry 1971;118:621-6.
17. Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Survey. Arch Gen Psychiatry 1999;56(7):617-26.
18. Goldston DB, Daniel SS, Reboussin DM, et al. Suicide attempts among formerly hospitalized adolescents: a prospective naturalistic study of risk during the first 5 years after discharge. J Am Acad Child Adolesc Psychiatry 1999;38:660-71.
19. Rudd MD, Joiner T, Rajab MH. Relationships among suicide ideators, attempters, and multiple attempters in a young-adult sample. J Abnorm Psychol 1996;105(4):541-50.
20. Foreman EM, Berk MS, Henriques GR, et al. History of multiple suicide attempts as a behavioral marker of severe psychopathology. Am J Psychiatry 2004;161(3):437-43.
21. Miranda R, Scott M, Roger H, et al. Suicide attempt characteristics, diagnoses, and future attempts: comparing multiple attempters to single attempters and ideators. J Am Acad Child Adolesc Psychiatry 2008;47:32-40.
22. Joiner TE, Rudd MD. Intensity and duration of suicidal crises vary as a function of previous suicide attempts and negative life events. J Consult Clin Psychol 2000;68(5):909-16.
23. Suominen K, Isometsä E, Suokas J, et al. Completed suicide after a suicide attempt: a 37-year follow-up study. Am J Psychiatry 2004;161:562-3.
24. Owens D, Wood C, Greenwood D, et al. Mortality and suicide after non-fatal self-poisoning: a 16-year outcome study of patients attending accident and emergency. Br J Psychiatry 2005;187:470-5.
25. Haw C, Bergen H, Casey D, Hawton K. Repetition of deliberate self-harm: a study of the characteristics and subsequent deaths in patients presenting to a general hospital according to extent of repetition. Suicide Life Threat Behav 2007;37(4):379-96.
26. Whitlock J, Knox KL. The relationship between self-injurious behavior and suicide in a young adult population. Arch Pediatr Adolesc Med 2007;161:634-40.
27. Joiner TE. Why people die by suicide. Cambridge, MA: Harvard University Press; 2005:46-93,203-22.
28. Gliatto MF, Rai AK. Evaluation and treatment of patients with suicidal ideation. Am Fam Physician. 1999;59(6):1500-6.
Perimenopausal depression: Covering mood and vasomotor symptoms
Symptoms of perimenopausal depression are not inherently different from those of depression diagnosed at any other time in life, but they present in a unique context:
- Hormonal fluctuations may persist for a long duration.
- Women experiencing hormonal fluctuations may be vulnerable to mood problems.
- Psychosocial/psychodynamic stressors often complicate this life transition.
Managing perimenopausal depression has become more complicated since the Women’s Health Initiative (WHI) studies found fewer benefits and greater risks with hormone replacement therapy (HRT) than had been perceived. This article discusses the clinical presentation of perimenopausal depression, its risk factors, and treatment options in post-WHI psychiatric practice.
Who is at risk?
Perimenopausal depression is diagnosed when onset of major depressive disorder (MDD) is associated with menstrual cycle irregularity and/or somatic symptoms of the menopausal transition.1 Diagnosis is based on the overall clinical picture, and treatment requires a thoughtful exploration of the complex relationship between hormonal function and mood regulation.
Presentation. For many women, perimenopause is characterized by mild to severe vasomotor, cognitive, and mood symptoms (Table 1). Thus, in your workup of depression in midlife women, document somatic symptoms—such as hot flushes, vaginal dryness, and incontinence—and affective/behavioral symp toms such as mood and sleep disturbances.
Table 1
Vasomotor, cognitive, and mood symptoms of perimenopause
| Vasomotor | Cognitive and mood |
|---|---|
| Hot flushes | Decreased concentration |
| Sweating | Anxiety |
| Heart palpitations | Irritability |
| Painful intercourse | Mood lability |
| Vaginal dryness and discomfort | Memory difficulty |
| Sleep disruption | |
| Headache |
Explore psychiatric and medical histories of your patient and her close relatives. Ask about depression, dysthymia, hypomania, or mood fluctuations around hormonal events such as menses, pregnancy, postpartum, or starting/stopping oral contraceptives. In the differential diagnosis, consider:
- Is low mood temporally connected with hot flushes and disturbed sleep?
- Is low mood secondary to stressful life events?
- Does the patient have another medical illness (such as thyroid disorder) with symptoms similar to depression?
- Is low mood secondary to anxiety or another psychiatric disorder?
Screening. Menopause is considered to have been reached after 12 months of amenorrhea not due to another cause. Median ages for this transition in the United States are 47.5 for perimenopause and 51 for menopause, with an average of 8 years between regular cycles and amenorrhea.2 Therefore, begin talking with women about perimenopausal symptoms when they turn 40.
Evidence supports screening perimenopausal women for depressive symptoms even when their primary complaints are vasomotor. The Greene Climacteric Scale3 is convenient for quantifying and monitoring perimenopausal symptoms. It includes depressive symptoms plus physical and cognitive markers. The Quick Inventory of Depressive Symptomatology—Self Report (QIDS-SR)4 questionnaire:
- takes minutes to complete
- is easy to score
- quantitates the number and severity of depressive symptoms (see Related Resources).
Psychosocial factors can predict depression at any time in life, but some are specific to the menopausal transition (Table 2).5 The “empty nest syndrome,” for example, is often used to explain depressive symptoms in midlife mothers, but no evidence links mood lability with the maturation and departure of children. What may be more stressing for women is supporting adolescents/young adults in their exit to independence while caring for aging parents.
Table 2
Risk factors for depression in women
| Predictive over lifetime | High risk during menopausal transition |
|---|---|
| History of depression | History of PMS, perinatal depression, mood symptoms associated with contraceptives |
| Family history of affective disorders | Premature or surgical menopause |
| Insomnia | Lengthy menopausal transition (≥27 months) |
| Reduced physical activities | Persistent and/or severe vasomotor symptoms |
| Weight gain | Negative attitudes toward menopause and aging |
| Less education | |
| Perceived lower economic status | |
| Perceived lower social support | |
| Perceived lower health status | |
| Smoking | |
| Stressful life events | |
| History of trauma | |
| Marital dissatisfaction | |
| PMS: Premenstrual syndrome | |
Sociocultural beliefs about sexuality and menopause may play a role in how your patient experiences and reports her symptoms. In some cultures, menopause elevates a woman’s social status and is associated with increased respect and authority. In others, such as Western societies that emphasize youth and beauty, women may view menopause and its physical changes in a negative light.6
Therefore, give careful attention to the psychosocial context of menopause to your patient and the social resources available to her. Questions to ask include:
- Has your lifestyle changed recently?
- Have your husband, family members, or close friends noticed any changes in your functioning?
- Is there anyone in your life that you feel comfortable confiding in?
Explaining the complexity of this life transition may ease her anxiety by normalizing her experience, helping her understand her symptoms, and validating her distress.
What might be the cause?
Although the exact pathophysiology of perimenopausal depression is unknown, hormonal changes,7 general health, the experience of menopause,8 and the psychosocial context2 likely work together to increase vulnerability for depressive symptoms (Figure).
Figure Biopsychosocial milieu of depression during perimenopauseHormonal fluctuation. The estrogen withdrawal theory7 explains depressive symptoms as resulting from a sustained decline in ovarian estrogen in tandem with spiking secretions of follicle-stimulating hormone by the pituitary. The finding that women with surgical menopause have a higher incidence of depressive symptoms than women with natural menopause supports this hypothesis.
Mood disorders occur across various female reproductive events, and increased risk appears to be associated with fluctuating gonadal hormones. Thus, declining estrogen may be less causative of perimenopausal depression than extreme fluctuations in estradiol activity.9,10
Estrogen interacts with dopamine, norepinephrine, beta-endorphin, and serotonin metabolism. In particular, estrogen facilitates serotonin delivery to neurons across the brain. These findings—and the success of selective serotonin reuptake inhibitors (SSRIs) in treating mood disorders—support the theory that fluctuating estrogen affects the serotonergic system and may cause depressive symptoms.
‘Domino theory.’ Others have hypothesized that depressive symptoms are the secondhand result of somatic symptoms of perimenopause. In a “domino effect,” hot flushes and night sweats disrupt women’s sleep, bringing fatigue and impaired daytime concentration, which lead to irritability and feelings of being overwhelmed.8
This theory, which incorporates perimenopausal hormone changes, is supported by elevated levels of depression in women who report frequent and intense vasomotor symptoms persisting >27 months.2
The psychosocial theory suggests that depression results from increased stress or adverse events.2 Midlife women with depressive symptoms report many possible sources of stress:
- demanding jobs
- family responsibilities
- dual demands of career and family
- little time for self
- poverty or employment stressors
- not enough sleep
- changing social relationships.
Negative interpretations of aging or the menopausal transition also have been implicated in cross-cultural studies.6 The predictive nature of psychosocial issues for depression during perimenopause supports this theory.
Evidence-based treatment
HRT. Research and clinical reports suggest that estrogen may have antidepressant effects, either alone or as an adjunct to antidepressant medication.11 Before the WHI studies, expert consensus guidelines on treating depression in women recommended HRT as first-line treatment for patients experiencing a first lifetime onset of mild to moderate depression during perimenopause.12 WHI findings since 2002 that associated HRT with increased risk of stroke, deep vein thrombosis, and pulmonary embolism—without clear protection against coronary heart disease or cognitive decline—have left HRT a controversial option for treating perimenopausal depression. In the WHI trials:
- 10,739 postmenopausal women age 50 to 79 without a uterus received unopposed conjugated equine estrogens, 0.625 mg/d, or placebo for an average 6.8 years.13
- 16,608 postmenopausal women age 50 to 79 with an intact uterus received combination HRT (conjugated equine estrogens, 0.625 mg/d, plus 2.5 mg of medroxyprogesterone), or placebo for an average 5.6 years.14
The study using combination HRT found increased risks of breast cancer, ischemic stroke, blood clots, and coronary heart disease.15 A follow-up study showed that vasomotor symptoms returned in more than one-half the women after they stopped using combination HRT.15
A companion WHI trial found that estrogen, 0.625 mg/d—given unopposed or with a progestin—did not prevent cognitive decline in women age 65 to 79 and may have been associated with a slightly greater risk of probable dementia.16,17
The FDA recommends that women who want to use HRT to control menopausal symptoms use the lowest effective dose for the shortest time necessary.18
Antidepressants. SSRIs may be more useful than estrogen for producing MDD remission in perimenopausal women.19 SSRIs and other psychotropics may reduce perimenopausal vasomotor symptoms in addition to addressing depressive symptoms (Table 3). When choosing antidepressant therapy, consider the patient’s dominant presenting perimenopausal symptoms and side effects associated with treatment.20
Table 3
Nonhormone medications for perimenopausal depression: Evidence-based dosages and target symptoms
| Medication | Dosage effective for perimenopausal depression | Symptoms assessed |
|---|---|---|
| SSRIs | ||
| Citaloprama | 40 to 60 mg | Depressive and vasomotor |
| Escitalopramb,c | 5 to 20 mg | Depressive and vasomotor |
| Fluoxetined | 20 to 40 mg | Depressive and vasomotor |
| Paroxetinee,f | 12.5 or 25 mg | Depressive and vasomotor |
| Sertralineg | 100 mg | Depressive and vasomotor |
| Other antidepressants | ||
| Duloxetineh | 60 to 120 mg | Depressive and vasomotor |
| Venlafaxinei | 75 to 225 mg | Depressive and vasomotor |
| Mirtazapinej | 30 to 60 mg | Severe depressive symptoms; used as an adjunct to estrogen |
| Hypnotics | ||
| Eszopiclonek | 3 mg | Depressive and vasomotor; insomnia |
| Zolpideml | 5 to 10 mg | Insomnia |
| Anticonvulsant | ||
| Gabapentinm | 300 to 900 mg | Vasomotor |
| SSRIs: selective serotonin reuptake inhibitors | ||
| Source: Reference Citations | ||
Nonpharmacologic interventions are viable options for women who are reluctant to begin HRT or psychotropics.
Psychotherapy. Interpersonal psychotherapy (IPT) and cognitive-behavioral therapy (CBT) have been recommended to address psychosocial elements of perimenopausal mood lability.21 For women with climacteric depression, IPT focuses on role transitions, loss, and interpersonal support, whereas CBT focuses on identifying and altering negative thoughts and beliefs.
Although no randomized trials have examined psychotherapies for perimenopausal depression, a pilot open trial provided group CBT—psychoeducation, group discussion, and coping skills training—to 30 women with climacteric symptoms. Anxiety, depression, partnership relations, overall sexuality, hot flushes, and cardiac complaints improved significantly, based on pre- and post-intervention surveys. Sexual satisfaction and the stressfulness of menopausal symptoms did not change.22
Integrative medicine. Plant-based substances and herbal remedies such as phytoestrogens, red-clover isoflavones, black cohosh, and evening primrose oil have been included in a few research investigations, and the evidence is equivocal. Because of potential interactions between alternative therapies and medications, inquire about their use. Although a comprehensive review of integrative medicine for perimenopausal symptoms is beyond the scope of this article, see suggested readings (Box).
- Albertazzi P. Non-estrogenic approaches for the treatment of climacteric symptoms. Climacteric 2007;10(suppl 2):115-20.
- Blair YA, Gold EB, Zhang G, et al. Use of complementary and alternative medicine during the menopause transition: longitudinal results from the Study of Women’s Health Across the Nation. Menopause 2008;15:32-43.
- Freeman MP, Helgason C, Hill RA. Selected integrative medicine treatments for depression: considerations for women. J Am Med Womens Assoc 2004;59(3):216-24.
- Mischoulon D. Update and critique of natural remedies as antidepressant treatments. Psychiatr Clin North Am 2007;30:51-68.
- Thachil AF, Mohan R, Bhugra D. The evidence base of complementary and alternative therapies in depression. J Affect Disord 2007;97:23-35.
- Tremblay A, Sheeran L, Aranda SK. Psychoeducational interventions to alleviate hot flashes: a systematic review. J North Am Menopause Soc 2008;15:193-202.
Clinical recommendations
Explore options with your patient; discuss side effects, risks, and expected minimum duration of treatment. Antidepressants, hormonal therapies, psychotherapy, and complementary and alternative treatments each might have a role in managing perimenopausal depression. A patient’s preferences, psychiatric history, and depression severity help determine which options to consider and in what order. How she responded to past treatments also can help you individualize a plan.
HRT may be appropriate for women who express a preference for HRT, have responded well to past hormone therapy, and have no personal history or high-risk factors for breast cancer. Based on the WHI findings, we consider a history of breast cancer in the patient or a first- or second-degree relative a contraindication to HRT.
Estrogen can be used alone or with an antidepressant. Studies support 17β-estradiol, 0.1 to 0.3 mg/d, for 8 to 12 weeks.11,23 Concomitant progesterone may be indicated to offset the effects of unopposed estrogen in women with an intact uterus. This option calls for an informed discussion with the patient about risks and benefits.
No data support long-term use of estrogen for recurrent or chronic depression. Because HRT’s risks and benefits vary with the length of exposure, individualize the extended use of estrogen solely to augment treatment for depression. Because vasomotor symptoms may recur when HRT is discontinued,15 we recommend that women make an informed decision in consultation with a gynecologist or primary care physician.
Antidepressants that have serotonergic activity—such as SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs)—appear most promising for treating comorbid depressive and vasomotor symptoms. If a patient has had a good response to an antidepressant in the past, consider starting with that medication.
Common antidepressant side effects are difficult to assess in perimenopausal patients with MDD because the symptoms attributed to antidepressant side effects—such as low libido, sleep disturbance, and weight changes—also can be caused by mood disorders and hormonal changes. Therefore, inquire about these symptoms when you initiate antidepressant therapy and at follow-up assessments.
Psychotherapy. We recommend that all women who present with perimenopausal depression receive information about psychotherapy. Psychotherapy alone often is adequate for mild depression, and adding psychotherapy to antidepressant treatment usually enhances recovery from moderate and severe depression episodes. In addition, patients who engage in psychotherapy for depression may have a lower rate of relapse.24
Individual psychotherapy can help patients with perimenopausal depression:
- accept this life transition
- recognize the benefits of menopause, such as no need for contraception
- develop awareness of personal potential in the years ahead.
Because depression often occurs in an interpersonal context, consider including family members in psychotherapy to improve the patient’s interpersonal support.
Integrative therapies. A full evaluation and consideration of standard treatment options is indicated for all women with MDD. Integrative medicine appeals to many patients but has not been sufficiently studied for perimenopausal depression. Supplemental omega-3 fatty acids and folate are reasonable adjuncts to the treatment of MDD25-27 and deserve study in perimenopausal MDD.
- Greene Climacteric Scale. www.menopausematters.co.uk/greenescale.php.
- Quick Inventory of Depressive Symptomatology. www.idsqids.org.
- National Center for Complementary and Alternative Medicine. National Institutes of Health. http://nccam.nih.gov.
Drug brand names
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Estradiol • various
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Medroxyprogesterone • Provera
- Mirtazapine • Remeron
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
- Zolpidem • Ambien
Disclosures
Dr. Brandon and Dr. Shivakumar report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Freeman receives research support from GlaxoSmithKline, Forest Pharmaceuticals, and Eli Lilly and Company. She was associate professor of psychiatry at the University of Texas Southwestern Medical Center at Dallas when this article was written and is now on the faculty at Harvard Medical School and Massachusetts General Hospital, Boston.
1. Schmidt PJ, Rubinow DR. Reproductive ageing, sex steroids and depression. J Br Menopause Soc 2006;12(4):178-85.
2. Rasgon N, Shelton S, Halbreich U. Perimenopausal mental disorders: epidemiology and phenomenology. CNS Spectr 2005;10(6):471-8.
3. Greene JG. Constructing a standard climacteric scale. Maturitas 1998;29(1):25-31.
4. Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry 2003;54(5):573-83.Erratum in: Biol Psychiatry. 2003;54(5):585.
5. Feld J, Halbreich U, Karkun S. The association of perimenopausal mood disorders with other reproductive-related disorders. CNS Spectr 2005;10(6):461-70.
6. Avis NE, Stellato R, Crawford S, et al. Is there a menopausal syndrome? Menopausal status and symptoms across racial/ethnic groups. Soc Sci Med 2001;52:345-56.
7. Campbell S, Whitehead M. Oestrogen therapy and the menopause syndrome. Clin Obstet Gynecol 1977;4:31-47.
8. Schmidt PJ, Rubinow DR. Menopause-related affective disorders: a justification for further study. Am J Psychiatry 1991;48:844-52.
9. Soares CN. Menopausal transition and depression: who is at risk and how to treat it? Expert Rev Neurother 2007;7(10):1285-93.
10. Prior JC. The complex endocrinology of menopausal transition. Endocrinol Rev 1998;19:397-428.
11. Rasgon N, Altshuler LL, Fairbanks LA, et al. Estrogen replacement therapy in the treatment of major depressive disorder in perimenopausal women. J Clin Psychiatry 2002;63(suppl):45-8.
12. Altshuler LL, Cohen LS, Moline ML, et al. The Expert Consensus Guideline Series. Treatment of depression in women. Postgrad Med 2001 Mar;(Spec No):1-107.
13. The Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 2004;291:1701-12.
14. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002;288:321-33.
15. Ockene JK, Barad DH, Cochrane BB, et al. Symptom experience after discontinuing use of estrogen plus progestin. JAMA 2005;294:183-93.
16. Shumaker SA, Legault C, Kuller L, et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004;291:2947-58.
17. Shumaker SA, Legault C, Rapp SR. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women. The Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003;289:2651-62.
18. FDA approves new labeling and provides new advice to postmenopausal women who use or who are considering using estrogen and estrogen with progestin [FDA Fact Sheet January 8, 2003]. Available at: http://www.fda.gov/oc/factsheets/WHI.html. Accessed September 11, 2008.
19. Soares CN, Arsenio H, Joffe H, et al. Escitalopram versus ethinyl estradiol and norethindrone acetate for symptomatic peri- and postmenopausal women: impact on depression, vasomotor symptoms, sleep, and quality of life. Menopause 2006;13(5):780-6.
20. Cohen LS, Soares CN, Joffe H. Diagnosis and management of mood disorders during the menopausal transition. Am J Med 2005;118(suppl 12B):93-7.
21. Kahn DA, Moline ML, Ross RW, et al. Depression during the transition to menopause: a guide for patients and families. Postgrad Med 2001 Mar;(Spec No):110-1.
22. Alder J, Besken KE, Armbruster U, et al. Cognitive-behavioural group intervention for climacteric syndrome. Psychother Psychosom 2006;75(5):298-303.
23. Soares CN, Almeida OP, Joffe H, Cohen LS. Efficacy of estradiol for the treatment of depressive disorders in perimenopausal women: a double-blind, randomized, placebo-controlled trial. Arch Gen Psychiatry 2001;58(6):529-34.
24. Otto MW, Smits JA, Reese HE. Combined psychotherapy and pharmacotherapy for mood and anxiety disorders in adults: review and analysis. Clinical Psychology: Science and Practice 2005;12:72-86.
25. Coppen A, Bailey J. Enhancement of the antidepressant action of fluoxetine by folic acid: a randomized, placebo controlled trial. J Affect Disord 2000;60:121-30.
26. Freeman MP, Hibbeln JR, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry [American Psychiatric Association subcommittee report]. J Clin Psychiatry 2006;67:1954-67.
27. Otto MW, Church TS, Craft LL, et al. Exercise for mood and anxiety disorders. J Clin Psychiatry 2007;68:669-76.
Symptoms of perimenopausal depression are not inherently different from those of depression diagnosed at any other time in life, but they present in a unique context:
- Hormonal fluctuations may persist for a long duration.
- Women experiencing hormonal fluctuations may be vulnerable to mood problems.
- Psychosocial/psychodynamic stressors often complicate this life transition.
Managing perimenopausal depression has become more complicated since the Women’s Health Initiative (WHI) studies found fewer benefits and greater risks with hormone replacement therapy (HRT) than had been perceived. This article discusses the clinical presentation of perimenopausal depression, its risk factors, and treatment options in post-WHI psychiatric practice.
Who is at risk?
Perimenopausal depression is diagnosed when onset of major depressive disorder (MDD) is associated with menstrual cycle irregularity and/or somatic symptoms of the menopausal transition.1 Diagnosis is based on the overall clinical picture, and treatment requires a thoughtful exploration of the complex relationship between hormonal function and mood regulation.
Presentation. For many women, perimenopause is characterized by mild to severe vasomotor, cognitive, and mood symptoms (Table 1). Thus, in your workup of depression in midlife women, document somatic symptoms—such as hot flushes, vaginal dryness, and incontinence—and affective/behavioral symp toms such as mood and sleep disturbances.
Table 1
Vasomotor, cognitive, and mood symptoms of perimenopause
| Vasomotor | Cognitive and mood |
|---|---|
| Hot flushes | Decreased concentration |
| Sweating | Anxiety |
| Heart palpitations | Irritability |
| Painful intercourse | Mood lability |
| Vaginal dryness and discomfort | Memory difficulty |
| Sleep disruption | |
| Headache |
Explore psychiatric and medical histories of your patient and her close relatives. Ask about depression, dysthymia, hypomania, or mood fluctuations around hormonal events such as menses, pregnancy, postpartum, or starting/stopping oral contraceptives. In the differential diagnosis, consider:
- Is low mood temporally connected with hot flushes and disturbed sleep?
- Is low mood secondary to stressful life events?
- Does the patient have another medical illness (such as thyroid disorder) with symptoms similar to depression?
- Is low mood secondary to anxiety or another psychiatric disorder?
Screening. Menopause is considered to have been reached after 12 months of amenorrhea not due to another cause. Median ages for this transition in the United States are 47.5 for perimenopause and 51 for menopause, with an average of 8 years between regular cycles and amenorrhea.2 Therefore, begin talking with women about perimenopausal symptoms when they turn 40.
Evidence supports screening perimenopausal women for depressive symptoms even when their primary complaints are vasomotor. The Greene Climacteric Scale3 is convenient for quantifying and monitoring perimenopausal symptoms. It includes depressive symptoms plus physical and cognitive markers. The Quick Inventory of Depressive Symptomatology—Self Report (QIDS-SR)4 questionnaire:
- takes minutes to complete
- is easy to score
- quantitates the number and severity of depressive symptoms (see Related Resources).
Psychosocial factors can predict depression at any time in life, but some are specific to the menopausal transition (Table 2).5 The “empty nest syndrome,” for example, is often used to explain depressive symptoms in midlife mothers, but no evidence links mood lability with the maturation and departure of children. What may be more stressing for women is supporting adolescents/young adults in their exit to independence while caring for aging parents.
Table 2
Risk factors for depression in women
| Predictive over lifetime | High risk during menopausal transition |
|---|---|
| History of depression | History of PMS, perinatal depression, mood symptoms associated with contraceptives |
| Family history of affective disorders | Premature or surgical menopause |
| Insomnia | Lengthy menopausal transition (≥27 months) |
| Reduced physical activities | Persistent and/or severe vasomotor symptoms |
| Weight gain | Negative attitudes toward menopause and aging |
| Less education | |
| Perceived lower economic status | |
| Perceived lower social support | |
| Perceived lower health status | |
| Smoking | |
| Stressful life events | |
| History of trauma | |
| Marital dissatisfaction | |
| PMS: Premenstrual syndrome | |
Sociocultural beliefs about sexuality and menopause may play a role in how your patient experiences and reports her symptoms. In some cultures, menopause elevates a woman’s social status and is associated with increased respect and authority. In others, such as Western societies that emphasize youth and beauty, women may view menopause and its physical changes in a negative light.6
Therefore, give careful attention to the psychosocial context of menopause to your patient and the social resources available to her. Questions to ask include:
- Has your lifestyle changed recently?
- Have your husband, family members, or close friends noticed any changes in your functioning?
- Is there anyone in your life that you feel comfortable confiding in?
Explaining the complexity of this life transition may ease her anxiety by normalizing her experience, helping her understand her symptoms, and validating her distress.
What might be the cause?
Although the exact pathophysiology of perimenopausal depression is unknown, hormonal changes,7 general health, the experience of menopause,8 and the psychosocial context2 likely work together to increase vulnerability for depressive symptoms (Figure).
Figure Biopsychosocial milieu of depression during perimenopauseHormonal fluctuation. The estrogen withdrawal theory7 explains depressive symptoms as resulting from a sustained decline in ovarian estrogen in tandem with spiking secretions of follicle-stimulating hormone by the pituitary. The finding that women with surgical menopause have a higher incidence of depressive symptoms than women with natural menopause supports this hypothesis.
Mood disorders occur across various female reproductive events, and increased risk appears to be associated with fluctuating gonadal hormones. Thus, declining estrogen may be less causative of perimenopausal depression than extreme fluctuations in estradiol activity.9,10
Estrogen interacts with dopamine, norepinephrine, beta-endorphin, and serotonin metabolism. In particular, estrogen facilitates serotonin delivery to neurons across the brain. These findings—and the success of selective serotonin reuptake inhibitors (SSRIs) in treating mood disorders—support the theory that fluctuating estrogen affects the serotonergic system and may cause depressive symptoms.
‘Domino theory.’ Others have hypothesized that depressive symptoms are the secondhand result of somatic symptoms of perimenopause. In a “domino effect,” hot flushes and night sweats disrupt women’s sleep, bringing fatigue and impaired daytime concentration, which lead to irritability and feelings of being overwhelmed.8
This theory, which incorporates perimenopausal hormone changes, is supported by elevated levels of depression in women who report frequent and intense vasomotor symptoms persisting >27 months.2
The psychosocial theory suggests that depression results from increased stress or adverse events.2 Midlife women with depressive symptoms report many possible sources of stress:
- demanding jobs
- family responsibilities
- dual demands of career and family
- little time for self
- poverty or employment stressors
- not enough sleep
- changing social relationships.
Negative interpretations of aging or the menopausal transition also have been implicated in cross-cultural studies.6 The predictive nature of psychosocial issues for depression during perimenopause supports this theory.
Evidence-based treatment
HRT. Research and clinical reports suggest that estrogen may have antidepressant effects, either alone or as an adjunct to antidepressant medication.11 Before the WHI studies, expert consensus guidelines on treating depression in women recommended HRT as first-line treatment for patients experiencing a first lifetime onset of mild to moderate depression during perimenopause.12 WHI findings since 2002 that associated HRT with increased risk of stroke, deep vein thrombosis, and pulmonary embolism—without clear protection against coronary heart disease or cognitive decline—have left HRT a controversial option for treating perimenopausal depression. In the WHI trials:
- 10,739 postmenopausal women age 50 to 79 without a uterus received unopposed conjugated equine estrogens, 0.625 mg/d, or placebo for an average 6.8 years.13
- 16,608 postmenopausal women age 50 to 79 with an intact uterus received combination HRT (conjugated equine estrogens, 0.625 mg/d, plus 2.5 mg of medroxyprogesterone), or placebo for an average 5.6 years.14
The study using combination HRT found increased risks of breast cancer, ischemic stroke, blood clots, and coronary heart disease.15 A follow-up study showed that vasomotor symptoms returned in more than one-half the women after they stopped using combination HRT.15
A companion WHI trial found that estrogen, 0.625 mg/d—given unopposed or with a progestin—did not prevent cognitive decline in women age 65 to 79 and may have been associated with a slightly greater risk of probable dementia.16,17
The FDA recommends that women who want to use HRT to control menopausal symptoms use the lowest effective dose for the shortest time necessary.18
Antidepressants. SSRIs may be more useful than estrogen for producing MDD remission in perimenopausal women.19 SSRIs and other psychotropics may reduce perimenopausal vasomotor symptoms in addition to addressing depressive symptoms (Table 3). When choosing antidepressant therapy, consider the patient’s dominant presenting perimenopausal symptoms and side effects associated with treatment.20
Table 3
Nonhormone medications for perimenopausal depression: Evidence-based dosages and target symptoms
| Medication | Dosage effective for perimenopausal depression | Symptoms assessed |
|---|---|---|
| SSRIs | ||
| Citaloprama | 40 to 60 mg | Depressive and vasomotor |
| Escitalopramb,c | 5 to 20 mg | Depressive and vasomotor |
| Fluoxetined | 20 to 40 mg | Depressive and vasomotor |
| Paroxetinee,f | 12.5 or 25 mg | Depressive and vasomotor |
| Sertralineg | 100 mg | Depressive and vasomotor |
| Other antidepressants | ||
| Duloxetineh | 60 to 120 mg | Depressive and vasomotor |
| Venlafaxinei | 75 to 225 mg | Depressive and vasomotor |
| Mirtazapinej | 30 to 60 mg | Severe depressive symptoms; used as an adjunct to estrogen |
| Hypnotics | ||
| Eszopiclonek | 3 mg | Depressive and vasomotor; insomnia |
| Zolpideml | 5 to 10 mg | Insomnia |
| Anticonvulsant | ||
| Gabapentinm | 300 to 900 mg | Vasomotor |
| SSRIs: selective serotonin reuptake inhibitors | ||
| Source: Reference Citations | ||
Nonpharmacologic interventions are viable options for women who are reluctant to begin HRT or psychotropics.
Psychotherapy. Interpersonal psychotherapy (IPT) and cognitive-behavioral therapy (CBT) have been recommended to address psychosocial elements of perimenopausal mood lability.21 For women with climacteric depression, IPT focuses on role transitions, loss, and interpersonal support, whereas CBT focuses on identifying and altering negative thoughts and beliefs.
Although no randomized trials have examined psychotherapies for perimenopausal depression, a pilot open trial provided group CBT—psychoeducation, group discussion, and coping skills training—to 30 women with climacteric symptoms. Anxiety, depression, partnership relations, overall sexuality, hot flushes, and cardiac complaints improved significantly, based on pre- and post-intervention surveys. Sexual satisfaction and the stressfulness of menopausal symptoms did not change.22
Integrative medicine. Plant-based substances and herbal remedies such as phytoestrogens, red-clover isoflavones, black cohosh, and evening primrose oil have been included in a few research investigations, and the evidence is equivocal. Because of potential interactions between alternative therapies and medications, inquire about their use. Although a comprehensive review of integrative medicine for perimenopausal symptoms is beyond the scope of this article, see suggested readings (Box).
- Albertazzi P. Non-estrogenic approaches for the treatment of climacteric symptoms. Climacteric 2007;10(suppl 2):115-20.
- Blair YA, Gold EB, Zhang G, et al. Use of complementary and alternative medicine during the menopause transition: longitudinal results from the Study of Women’s Health Across the Nation. Menopause 2008;15:32-43.
- Freeman MP, Helgason C, Hill RA. Selected integrative medicine treatments for depression: considerations for women. J Am Med Womens Assoc 2004;59(3):216-24.
- Mischoulon D. Update and critique of natural remedies as antidepressant treatments. Psychiatr Clin North Am 2007;30:51-68.
- Thachil AF, Mohan R, Bhugra D. The evidence base of complementary and alternative therapies in depression. J Affect Disord 2007;97:23-35.
- Tremblay A, Sheeran L, Aranda SK. Psychoeducational interventions to alleviate hot flashes: a systematic review. J North Am Menopause Soc 2008;15:193-202.
Clinical recommendations
Explore options with your patient; discuss side effects, risks, and expected minimum duration of treatment. Antidepressants, hormonal therapies, psychotherapy, and complementary and alternative treatments each might have a role in managing perimenopausal depression. A patient’s preferences, psychiatric history, and depression severity help determine which options to consider and in what order. How she responded to past treatments also can help you individualize a plan.
HRT may be appropriate for women who express a preference for HRT, have responded well to past hormone therapy, and have no personal history or high-risk factors for breast cancer. Based on the WHI findings, we consider a history of breast cancer in the patient or a first- or second-degree relative a contraindication to HRT.
Estrogen can be used alone or with an antidepressant. Studies support 17β-estradiol, 0.1 to 0.3 mg/d, for 8 to 12 weeks.11,23 Concomitant progesterone may be indicated to offset the effects of unopposed estrogen in women with an intact uterus. This option calls for an informed discussion with the patient about risks and benefits.
No data support long-term use of estrogen for recurrent or chronic depression. Because HRT’s risks and benefits vary with the length of exposure, individualize the extended use of estrogen solely to augment treatment for depression. Because vasomotor symptoms may recur when HRT is discontinued,15 we recommend that women make an informed decision in consultation with a gynecologist or primary care physician.
Antidepressants that have serotonergic activity—such as SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs)—appear most promising for treating comorbid depressive and vasomotor symptoms. If a patient has had a good response to an antidepressant in the past, consider starting with that medication.
Common antidepressant side effects are difficult to assess in perimenopausal patients with MDD because the symptoms attributed to antidepressant side effects—such as low libido, sleep disturbance, and weight changes—also can be caused by mood disorders and hormonal changes. Therefore, inquire about these symptoms when you initiate antidepressant therapy and at follow-up assessments.
Psychotherapy. We recommend that all women who present with perimenopausal depression receive information about psychotherapy. Psychotherapy alone often is adequate for mild depression, and adding psychotherapy to antidepressant treatment usually enhances recovery from moderate and severe depression episodes. In addition, patients who engage in psychotherapy for depression may have a lower rate of relapse.24
Individual psychotherapy can help patients with perimenopausal depression:
- accept this life transition
- recognize the benefits of menopause, such as no need for contraception
- develop awareness of personal potential in the years ahead.
Because depression often occurs in an interpersonal context, consider including family members in psychotherapy to improve the patient’s interpersonal support.
Integrative therapies. A full evaluation and consideration of standard treatment options is indicated for all women with MDD. Integrative medicine appeals to many patients but has not been sufficiently studied for perimenopausal depression. Supplemental omega-3 fatty acids and folate are reasonable adjuncts to the treatment of MDD25-27 and deserve study in perimenopausal MDD.
- Greene Climacteric Scale. www.menopausematters.co.uk/greenescale.php.
- Quick Inventory of Depressive Symptomatology. www.idsqids.org.
- National Center for Complementary and Alternative Medicine. National Institutes of Health. http://nccam.nih.gov.
Drug brand names
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Estradiol • various
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Medroxyprogesterone • Provera
- Mirtazapine • Remeron
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
- Zolpidem • Ambien
Disclosures
Dr. Brandon and Dr. Shivakumar report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Freeman receives research support from GlaxoSmithKline, Forest Pharmaceuticals, and Eli Lilly and Company. She was associate professor of psychiatry at the University of Texas Southwestern Medical Center at Dallas when this article was written and is now on the faculty at Harvard Medical School and Massachusetts General Hospital, Boston.
Symptoms of perimenopausal depression are not inherently different from those of depression diagnosed at any other time in life, but they present in a unique context:
- Hormonal fluctuations may persist for a long duration.
- Women experiencing hormonal fluctuations may be vulnerable to mood problems.
- Psychosocial/psychodynamic stressors often complicate this life transition.
Managing perimenopausal depression has become more complicated since the Women’s Health Initiative (WHI) studies found fewer benefits and greater risks with hormone replacement therapy (HRT) than had been perceived. This article discusses the clinical presentation of perimenopausal depression, its risk factors, and treatment options in post-WHI psychiatric practice.
Who is at risk?
Perimenopausal depression is diagnosed when onset of major depressive disorder (MDD) is associated with menstrual cycle irregularity and/or somatic symptoms of the menopausal transition.1 Diagnosis is based on the overall clinical picture, and treatment requires a thoughtful exploration of the complex relationship between hormonal function and mood regulation.
Presentation. For many women, perimenopause is characterized by mild to severe vasomotor, cognitive, and mood symptoms (Table 1). Thus, in your workup of depression in midlife women, document somatic symptoms—such as hot flushes, vaginal dryness, and incontinence—and affective/behavioral symp toms such as mood and sleep disturbances.
Table 1
Vasomotor, cognitive, and mood symptoms of perimenopause
| Vasomotor | Cognitive and mood |
|---|---|
| Hot flushes | Decreased concentration |
| Sweating | Anxiety |
| Heart palpitations | Irritability |
| Painful intercourse | Mood lability |
| Vaginal dryness and discomfort | Memory difficulty |
| Sleep disruption | |
| Headache |
Explore psychiatric and medical histories of your patient and her close relatives. Ask about depression, dysthymia, hypomania, or mood fluctuations around hormonal events such as menses, pregnancy, postpartum, or starting/stopping oral contraceptives. In the differential diagnosis, consider:
- Is low mood temporally connected with hot flushes and disturbed sleep?
- Is low mood secondary to stressful life events?
- Does the patient have another medical illness (such as thyroid disorder) with symptoms similar to depression?
- Is low mood secondary to anxiety or another psychiatric disorder?
Screening. Menopause is considered to have been reached after 12 months of amenorrhea not due to another cause. Median ages for this transition in the United States are 47.5 for perimenopause and 51 for menopause, with an average of 8 years between regular cycles and amenorrhea.2 Therefore, begin talking with women about perimenopausal symptoms when they turn 40.
Evidence supports screening perimenopausal women for depressive symptoms even when their primary complaints are vasomotor. The Greene Climacteric Scale3 is convenient for quantifying and monitoring perimenopausal symptoms. It includes depressive symptoms plus physical and cognitive markers. The Quick Inventory of Depressive Symptomatology—Self Report (QIDS-SR)4 questionnaire:
- takes minutes to complete
- is easy to score
- quantitates the number and severity of depressive symptoms (see Related Resources).
Psychosocial factors can predict depression at any time in life, but some are specific to the menopausal transition (Table 2).5 The “empty nest syndrome,” for example, is often used to explain depressive symptoms in midlife mothers, but no evidence links mood lability with the maturation and departure of children. What may be more stressing for women is supporting adolescents/young adults in their exit to independence while caring for aging parents.
Table 2
Risk factors for depression in women
| Predictive over lifetime | High risk during menopausal transition |
|---|---|
| History of depression | History of PMS, perinatal depression, mood symptoms associated with contraceptives |
| Family history of affective disorders | Premature or surgical menopause |
| Insomnia | Lengthy menopausal transition (≥27 months) |
| Reduced physical activities | Persistent and/or severe vasomotor symptoms |
| Weight gain | Negative attitudes toward menopause and aging |
| Less education | |
| Perceived lower economic status | |
| Perceived lower social support | |
| Perceived lower health status | |
| Smoking | |
| Stressful life events | |
| History of trauma | |
| Marital dissatisfaction | |
| PMS: Premenstrual syndrome | |
Sociocultural beliefs about sexuality and menopause may play a role in how your patient experiences and reports her symptoms. In some cultures, menopause elevates a woman’s social status and is associated with increased respect and authority. In others, such as Western societies that emphasize youth and beauty, women may view menopause and its physical changes in a negative light.6
Therefore, give careful attention to the psychosocial context of menopause to your patient and the social resources available to her. Questions to ask include:
- Has your lifestyle changed recently?
- Have your husband, family members, or close friends noticed any changes in your functioning?
- Is there anyone in your life that you feel comfortable confiding in?
Explaining the complexity of this life transition may ease her anxiety by normalizing her experience, helping her understand her symptoms, and validating her distress.
What might be the cause?
Although the exact pathophysiology of perimenopausal depression is unknown, hormonal changes,7 general health, the experience of menopause,8 and the psychosocial context2 likely work together to increase vulnerability for depressive symptoms (Figure).
Figure Biopsychosocial milieu of depression during perimenopauseHormonal fluctuation. The estrogen withdrawal theory7 explains depressive symptoms as resulting from a sustained decline in ovarian estrogen in tandem with spiking secretions of follicle-stimulating hormone by the pituitary. The finding that women with surgical menopause have a higher incidence of depressive symptoms than women with natural menopause supports this hypothesis.
Mood disorders occur across various female reproductive events, and increased risk appears to be associated with fluctuating gonadal hormones. Thus, declining estrogen may be less causative of perimenopausal depression than extreme fluctuations in estradiol activity.9,10
Estrogen interacts with dopamine, norepinephrine, beta-endorphin, and serotonin metabolism. In particular, estrogen facilitates serotonin delivery to neurons across the brain. These findings—and the success of selective serotonin reuptake inhibitors (SSRIs) in treating mood disorders—support the theory that fluctuating estrogen affects the serotonergic system and may cause depressive symptoms.
‘Domino theory.’ Others have hypothesized that depressive symptoms are the secondhand result of somatic symptoms of perimenopause. In a “domino effect,” hot flushes and night sweats disrupt women’s sleep, bringing fatigue and impaired daytime concentration, which lead to irritability and feelings of being overwhelmed.8
This theory, which incorporates perimenopausal hormone changes, is supported by elevated levels of depression in women who report frequent and intense vasomotor symptoms persisting >27 months.2
The psychosocial theory suggests that depression results from increased stress or adverse events.2 Midlife women with depressive symptoms report many possible sources of stress:
- demanding jobs
- family responsibilities
- dual demands of career and family
- little time for self
- poverty or employment stressors
- not enough sleep
- changing social relationships.
Negative interpretations of aging or the menopausal transition also have been implicated in cross-cultural studies.6 The predictive nature of psychosocial issues for depression during perimenopause supports this theory.
Evidence-based treatment
HRT. Research and clinical reports suggest that estrogen may have antidepressant effects, either alone or as an adjunct to antidepressant medication.11 Before the WHI studies, expert consensus guidelines on treating depression in women recommended HRT as first-line treatment for patients experiencing a first lifetime onset of mild to moderate depression during perimenopause.12 WHI findings since 2002 that associated HRT with increased risk of stroke, deep vein thrombosis, and pulmonary embolism—without clear protection against coronary heart disease or cognitive decline—have left HRT a controversial option for treating perimenopausal depression. In the WHI trials:
- 10,739 postmenopausal women age 50 to 79 without a uterus received unopposed conjugated equine estrogens, 0.625 mg/d, or placebo for an average 6.8 years.13
- 16,608 postmenopausal women age 50 to 79 with an intact uterus received combination HRT (conjugated equine estrogens, 0.625 mg/d, plus 2.5 mg of medroxyprogesterone), or placebo for an average 5.6 years.14
The study using combination HRT found increased risks of breast cancer, ischemic stroke, blood clots, and coronary heart disease.15 A follow-up study showed that vasomotor symptoms returned in more than one-half the women after they stopped using combination HRT.15
A companion WHI trial found that estrogen, 0.625 mg/d—given unopposed or with a progestin—did not prevent cognitive decline in women age 65 to 79 and may have been associated with a slightly greater risk of probable dementia.16,17
The FDA recommends that women who want to use HRT to control menopausal symptoms use the lowest effective dose for the shortest time necessary.18
Antidepressants. SSRIs may be more useful than estrogen for producing MDD remission in perimenopausal women.19 SSRIs and other psychotropics may reduce perimenopausal vasomotor symptoms in addition to addressing depressive symptoms (Table 3). When choosing antidepressant therapy, consider the patient’s dominant presenting perimenopausal symptoms and side effects associated with treatment.20
Table 3
Nonhormone medications for perimenopausal depression: Evidence-based dosages and target symptoms
| Medication | Dosage effective for perimenopausal depression | Symptoms assessed |
|---|---|---|
| SSRIs | ||
| Citaloprama | 40 to 60 mg | Depressive and vasomotor |
| Escitalopramb,c | 5 to 20 mg | Depressive and vasomotor |
| Fluoxetined | 20 to 40 mg | Depressive and vasomotor |
| Paroxetinee,f | 12.5 or 25 mg | Depressive and vasomotor |
| Sertralineg | 100 mg | Depressive and vasomotor |
| Other antidepressants | ||
| Duloxetineh | 60 to 120 mg | Depressive and vasomotor |
| Venlafaxinei | 75 to 225 mg | Depressive and vasomotor |
| Mirtazapinej | 30 to 60 mg | Severe depressive symptoms; used as an adjunct to estrogen |
| Hypnotics | ||
| Eszopiclonek | 3 mg | Depressive and vasomotor; insomnia |
| Zolpideml | 5 to 10 mg | Insomnia |
| Anticonvulsant | ||
| Gabapentinm | 300 to 900 mg | Vasomotor |
| SSRIs: selective serotonin reuptake inhibitors | ||
| Source: Reference Citations | ||
Nonpharmacologic interventions are viable options for women who are reluctant to begin HRT or psychotropics.
Psychotherapy. Interpersonal psychotherapy (IPT) and cognitive-behavioral therapy (CBT) have been recommended to address psychosocial elements of perimenopausal mood lability.21 For women with climacteric depression, IPT focuses on role transitions, loss, and interpersonal support, whereas CBT focuses on identifying and altering negative thoughts and beliefs.
Although no randomized trials have examined psychotherapies for perimenopausal depression, a pilot open trial provided group CBT—psychoeducation, group discussion, and coping skills training—to 30 women with climacteric symptoms. Anxiety, depression, partnership relations, overall sexuality, hot flushes, and cardiac complaints improved significantly, based on pre- and post-intervention surveys. Sexual satisfaction and the stressfulness of menopausal symptoms did not change.22
Integrative medicine. Plant-based substances and herbal remedies such as phytoestrogens, red-clover isoflavones, black cohosh, and evening primrose oil have been included in a few research investigations, and the evidence is equivocal. Because of potential interactions between alternative therapies and medications, inquire about their use. Although a comprehensive review of integrative medicine for perimenopausal symptoms is beyond the scope of this article, see suggested readings (Box).
- Albertazzi P. Non-estrogenic approaches for the treatment of climacteric symptoms. Climacteric 2007;10(suppl 2):115-20.
- Blair YA, Gold EB, Zhang G, et al. Use of complementary and alternative medicine during the menopause transition: longitudinal results from the Study of Women’s Health Across the Nation. Menopause 2008;15:32-43.
- Freeman MP, Helgason C, Hill RA. Selected integrative medicine treatments for depression: considerations for women. J Am Med Womens Assoc 2004;59(3):216-24.
- Mischoulon D. Update and critique of natural remedies as antidepressant treatments. Psychiatr Clin North Am 2007;30:51-68.
- Thachil AF, Mohan R, Bhugra D. The evidence base of complementary and alternative therapies in depression. J Affect Disord 2007;97:23-35.
- Tremblay A, Sheeran L, Aranda SK. Psychoeducational interventions to alleviate hot flashes: a systematic review. J North Am Menopause Soc 2008;15:193-202.
Clinical recommendations
Explore options with your patient; discuss side effects, risks, and expected minimum duration of treatment. Antidepressants, hormonal therapies, psychotherapy, and complementary and alternative treatments each might have a role in managing perimenopausal depression. A patient’s preferences, psychiatric history, and depression severity help determine which options to consider and in what order. How she responded to past treatments also can help you individualize a plan.
HRT may be appropriate for women who express a preference for HRT, have responded well to past hormone therapy, and have no personal history or high-risk factors for breast cancer. Based on the WHI findings, we consider a history of breast cancer in the patient or a first- or second-degree relative a contraindication to HRT.
Estrogen can be used alone or with an antidepressant. Studies support 17β-estradiol, 0.1 to 0.3 mg/d, for 8 to 12 weeks.11,23 Concomitant progesterone may be indicated to offset the effects of unopposed estrogen in women with an intact uterus. This option calls for an informed discussion with the patient about risks and benefits.
No data support long-term use of estrogen for recurrent or chronic depression. Because HRT’s risks and benefits vary with the length of exposure, individualize the extended use of estrogen solely to augment treatment for depression. Because vasomotor symptoms may recur when HRT is discontinued,15 we recommend that women make an informed decision in consultation with a gynecologist or primary care physician.
Antidepressants that have serotonergic activity—such as SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs)—appear most promising for treating comorbid depressive and vasomotor symptoms. If a patient has had a good response to an antidepressant in the past, consider starting with that medication.
Common antidepressant side effects are difficult to assess in perimenopausal patients with MDD because the symptoms attributed to antidepressant side effects—such as low libido, sleep disturbance, and weight changes—also can be caused by mood disorders and hormonal changes. Therefore, inquire about these symptoms when you initiate antidepressant therapy and at follow-up assessments.
Psychotherapy. We recommend that all women who present with perimenopausal depression receive information about psychotherapy. Psychotherapy alone often is adequate for mild depression, and adding psychotherapy to antidepressant treatment usually enhances recovery from moderate and severe depression episodes. In addition, patients who engage in psychotherapy for depression may have a lower rate of relapse.24
Individual psychotherapy can help patients with perimenopausal depression:
- accept this life transition
- recognize the benefits of menopause, such as no need for contraception
- develop awareness of personal potential in the years ahead.
Because depression often occurs in an interpersonal context, consider including family members in psychotherapy to improve the patient’s interpersonal support.
Integrative therapies. A full evaluation and consideration of standard treatment options is indicated for all women with MDD. Integrative medicine appeals to many patients but has not been sufficiently studied for perimenopausal depression. Supplemental omega-3 fatty acids and folate are reasonable adjuncts to the treatment of MDD25-27 and deserve study in perimenopausal MDD.
- Greene Climacteric Scale. www.menopausematters.co.uk/greenescale.php.
- Quick Inventory of Depressive Symptomatology. www.idsqids.org.
- National Center for Complementary and Alternative Medicine. National Institutes of Health. http://nccam.nih.gov.
Drug brand names
- Citalopram • Celexa
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Estradiol • various
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Medroxyprogesterone • Provera
- Mirtazapine • Remeron
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
- Zolpidem • Ambien
Disclosures
Dr. Brandon and Dr. Shivakumar report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Freeman receives research support from GlaxoSmithKline, Forest Pharmaceuticals, and Eli Lilly and Company. She was associate professor of psychiatry at the University of Texas Southwestern Medical Center at Dallas when this article was written and is now on the faculty at Harvard Medical School and Massachusetts General Hospital, Boston.
1. Schmidt PJ, Rubinow DR. Reproductive ageing, sex steroids and depression. J Br Menopause Soc 2006;12(4):178-85.
2. Rasgon N, Shelton S, Halbreich U. Perimenopausal mental disorders: epidemiology and phenomenology. CNS Spectr 2005;10(6):471-8.
3. Greene JG. Constructing a standard climacteric scale. Maturitas 1998;29(1):25-31.
4. Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry 2003;54(5):573-83.Erratum in: Biol Psychiatry. 2003;54(5):585.
5. Feld J, Halbreich U, Karkun S. The association of perimenopausal mood disorders with other reproductive-related disorders. CNS Spectr 2005;10(6):461-70.
6. Avis NE, Stellato R, Crawford S, et al. Is there a menopausal syndrome? Menopausal status and symptoms across racial/ethnic groups. Soc Sci Med 2001;52:345-56.
7. Campbell S, Whitehead M. Oestrogen therapy and the menopause syndrome. Clin Obstet Gynecol 1977;4:31-47.
8. Schmidt PJ, Rubinow DR. Menopause-related affective disorders: a justification for further study. Am J Psychiatry 1991;48:844-52.
9. Soares CN. Menopausal transition and depression: who is at risk and how to treat it? Expert Rev Neurother 2007;7(10):1285-93.
10. Prior JC. The complex endocrinology of menopausal transition. Endocrinol Rev 1998;19:397-428.
11. Rasgon N, Altshuler LL, Fairbanks LA, et al. Estrogen replacement therapy in the treatment of major depressive disorder in perimenopausal women. J Clin Psychiatry 2002;63(suppl):45-8.
12. Altshuler LL, Cohen LS, Moline ML, et al. The Expert Consensus Guideline Series. Treatment of depression in women. Postgrad Med 2001 Mar;(Spec No):1-107.
13. The Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 2004;291:1701-12.
14. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002;288:321-33.
15. Ockene JK, Barad DH, Cochrane BB, et al. Symptom experience after discontinuing use of estrogen plus progestin. JAMA 2005;294:183-93.
16. Shumaker SA, Legault C, Kuller L, et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004;291:2947-58.
17. Shumaker SA, Legault C, Rapp SR. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women. The Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003;289:2651-62.
18. FDA approves new labeling and provides new advice to postmenopausal women who use or who are considering using estrogen and estrogen with progestin [FDA Fact Sheet January 8, 2003]. Available at: http://www.fda.gov/oc/factsheets/WHI.html. Accessed September 11, 2008.
19. Soares CN, Arsenio H, Joffe H, et al. Escitalopram versus ethinyl estradiol and norethindrone acetate for symptomatic peri- and postmenopausal women: impact on depression, vasomotor symptoms, sleep, and quality of life. Menopause 2006;13(5):780-6.
20. Cohen LS, Soares CN, Joffe H. Diagnosis and management of mood disorders during the menopausal transition. Am J Med 2005;118(suppl 12B):93-7.
21. Kahn DA, Moline ML, Ross RW, et al. Depression during the transition to menopause: a guide for patients and families. Postgrad Med 2001 Mar;(Spec No):110-1.
22. Alder J, Besken KE, Armbruster U, et al. Cognitive-behavioural group intervention for climacteric syndrome. Psychother Psychosom 2006;75(5):298-303.
23. Soares CN, Almeida OP, Joffe H, Cohen LS. Efficacy of estradiol for the treatment of depressive disorders in perimenopausal women: a double-blind, randomized, placebo-controlled trial. Arch Gen Psychiatry 2001;58(6):529-34.
24. Otto MW, Smits JA, Reese HE. Combined psychotherapy and pharmacotherapy for mood and anxiety disorders in adults: review and analysis. Clinical Psychology: Science and Practice 2005;12:72-86.
25. Coppen A, Bailey J. Enhancement of the antidepressant action of fluoxetine by folic acid: a randomized, placebo controlled trial. J Affect Disord 2000;60:121-30.
26. Freeman MP, Hibbeln JR, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry [American Psychiatric Association subcommittee report]. J Clin Psychiatry 2006;67:1954-67.
27. Otto MW, Church TS, Craft LL, et al. Exercise for mood and anxiety disorders. J Clin Psychiatry 2007;68:669-76.
1. Schmidt PJ, Rubinow DR. Reproductive ageing, sex steroids and depression. J Br Menopause Soc 2006;12(4):178-85.
2. Rasgon N, Shelton S, Halbreich U. Perimenopausal mental disorders: epidemiology and phenomenology. CNS Spectr 2005;10(6):471-8.
3. Greene JG. Constructing a standard climacteric scale. Maturitas 1998;29(1):25-31.
4. Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry 2003;54(5):573-83.Erratum in: Biol Psychiatry. 2003;54(5):585.
5. Feld J, Halbreich U, Karkun S. The association of perimenopausal mood disorders with other reproductive-related disorders. CNS Spectr 2005;10(6):461-70.
6. Avis NE, Stellato R, Crawford S, et al. Is there a menopausal syndrome? Menopausal status and symptoms across racial/ethnic groups. Soc Sci Med 2001;52:345-56.
7. Campbell S, Whitehead M. Oestrogen therapy and the menopause syndrome. Clin Obstet Gynecol 1977;4:31-47.
8. Schmidt PJ, Rubinow DR. Menopause-related affective disorders: a justification for further study. Am J Psychiatry 1991;48:844-52.
9. Soares CN. Menopausal transition and depression: who is at risk and how to treat it? Expert Rev Neurother 2007;7(10):1285-93.
10. Prior JC. The complex endocrinology of menopausal transition. Endocrinol Rev 1998;19:397-428.
11. Rasgon N, Altshuler LL, Fairbanks LA, et al. Estrogen replacement therapy in the treatment of major depressive disorder in perimenopausal women. J Clin Psychiatry 2002;63(suppl):45-8.
12. Altshuler LL, Cohen LS, Moline ML, et al. The Expert Consensus Guideline Series. Treatment of depression in women. Postgrad Med 2001 Mar;(Spec No):1-107.
13. The Women’s Health Initiative Steering Committee. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 2004;291:1701-12.
14. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002;288:321-33.
15. Ockene JK, Barad DH, Cochrane BB, et al. Symptom experience after discontinuing use of estrogen plus progestin. JAMA 2005;294:183-93.
16. Shumaker SA, Legault C, Kuller L, et al. Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 2004;291:2947-58.
17. Shumaker SA, Legault C, Rapp SR. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women. The Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003;289:2651-62.
18. FDA approves new labeling and provides new advice to postmenopausal women who use or who are considering using estrogen and estrogen with progestin [FDA Fact Sheet January 8, 2003]. Available at: http://www.fda.gov/oc/factsheets/WHI.html. Accessed September 11, 2008.
19. Soares CN, Arsenio H, Joffe H, et al. Escitalopram versus ethinyl estradiol and norethindrone acetate for symptomatic peri- and postmenopausal women: impact on depression, vasomotor symptoms, sleep, and quality of life. Menopause 2006;13(5):780-6.
20. Cohen LS, Soares CN, Joffe H. Diagnosis and management of mood disorders during the menopausal transition. Am J Med 2005;118(suppl 12B):93-7.
21. Kahn DA, Moline ML, Ross RW, et al. Depression during the transition to menopause: a guide for patients and families. Postgrad Med 2001 Mar;(Spec No):110-1.
22. Alder J, Besken KE, Armbruster U, et al. Cognitive-behavioural group intervention for climacteric syndrome. Psychother Psychosom 2006;75(5):298-303.
23. Soares CN, Almeida OP, Joffe H, Cohen LS. Efficacy of estradiol for the treatment of depressive disorders in perimenopausal women: a double-blind, randomized, placebo-controlled trial. Arch Gen Psychiatry 2001;58(6):529-34.
24. Otto MW, Smits JA, Reese HE. Combined psychotherapy and pharmacotherapy for mood and anxiety disorders in adults: review and analysis. Clinical Psychology: Science and Practice 2005;12:72-86.
25. Coppen A, Bailey J. Enhancement of the antidepressant action of fluoxetine by folic acid: a randomized, placebo controlled trial. J Affect Disord 2000;60:121-30.
26. Freeman MP, Hibbeln JR, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry [American Psychiatric Association subcommittee report]. J Clin Psychiatry 2006;67:1954-67.
27. Otto MW, Church TS, Craft LL, et al. Exercise for mood and anxiety disorders. J Clin Psychiatry 2007;68:669-76.