User login
Tips to manage and prevent discontinuation syndromes
Abruptly stopping common psychotropics—particularly antidepressants, benzodiazepines, or atypical antipsychotics—can trigger a discontinuation syndrome, with:
- rebound or relapse of original symptoms
- uncomfortable new physical and psychological symptoms
- physiologic withdrawal at times.
To increase health professionals’ awareness of the risk of these adverse effects,1 this article describes discontinuation syndromes associated with various psychotropics and offers strategies to anticipate, recognize, and manage them.
Antidepressant Discontinuation Syndromes
Discontinuation syndromes can occur with tricyclic and tetracyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), and other newer antidepressants. Symptoms usually start within a few days of stopping a drug—or less commonly, reducing its dosage—and are usually mild and self-limited. Serious outcomes have been reported.
Distinguishing antidepressant discontinuation symptoms from depression recurrence is important. Discontinuation symptoms emerge within 1 to 3 days, whereas depressive symptoms usually occur 2 to 3 weeks after an antidepressant is stopped. Discontinuation reactions remit within a few days, especially if the antidepressant is re-instituted.
TCAs block serotonin and norepinephrine reuptake, increasing the availability of these biogenic amines at receptor sites in the brain and other tissues. Abrupt discontinuation can cause physical symptoms—such as lethargy, headache, and tremor—and psychological symptoms including irritability, anxiety, agitation, and low mood (Table 1).2
Long-term use of TCAs with potent anticholinergic properties leads to upregulation of postsynaptic muscarinic receptors, creating a “supersensitive” state. Abrupt discontinuation can cause cholinergic rebound, with symptoms emerging as soon as 12 hours—but typically 24 to 48 hours—after the last dose.
Table 1
Discontinuation symptoms seen with TCAs
| Physical symptoms | Lethargy, headache, tremor, sweating, anorexia, insomnia, nausea, vomiting, diarrhea, akathisia (rare), parkinsonism (rare) |
| Psychological symptoms | Irritability, anxiety/agitation, low mood, excessive dreaming, nightmares, paradoxical activation resulting in manic/hypomanic symptoms (rare) |
| TCA: Tricyclic antidepressants | |
| Source: Reference 2 | |
MAOIs such as phenelzine and tranylcypromine inhibit the enzyme monoamine oxidase, which is responsible for monoamine degradation and increases synaptic monoamine concentrations. Discontinuation syndromes may include acute confusional states, paranoid delusions, hallucinations, or worsening of depressive symptoms.3 These problems rarely occur in clinical practice, however, because MAOIs’ serious side effects discourage doctors from prescribing them.
SSRIs and other agents. SSRIs block synaptic reuptake of serotonin. Serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine and duloxetine inhibit both serotonin and norepinephrine reuptake. Mirtazapine—an alpha2-adrenergic and heteroreceptor antagonist—increases serotonin and norepinephrine at the synapse. Bupropion increases dopamine and norepinephrine turnover in the CNS and also blocks serotonin.
Up to 30% of patients who stop taking SSRIs develop discontinuation symptoms.4 Six symptom clusters—disequilibrium, sensory symptoms, general somatic symptoms, sleep disturbance, GI symptoms, and affective symptoms—characterize the SSRI discontinuation syndrome (Table 2).5 The four most common symptoms—in decreasing order of frequency—are dizziness, nausea, lethargy, and headache.6 Ataxia, sensory abnormalities, and possibly aggressive and impulsive behavior differentiate this discontinuation syndrome from that of the TCAs.
Table 2
Discontinuation symptoms seen with SSRIs
| Type | Symptoms |
|---|---|
| Disequilibrium | Lightheadedness/dizziness, vertigo, ataxia |
| Sensory symptoms | Paraesthesia, numbness, electric shock-like sensations |
| General somatic symptoms | Lethargy, headache, tremor, sweating, anorexia |
| Sleep disturbance | Insomnia, nightmares, excessive dreaming |
| GI symptoms | Nausea, vomiting, diarrhea |
| Affective symptoms | Irritability, anxiety/agitation, low mood |
| SSRIs: Selective serotonin reuptake inhibitors | |
| Source: Reference 5 | |
Risk factors. Risk factors for SSRI discontinuation syndrome have been identified (Table 3).7 Symptoms usually begin 1 to 3 days after an SSRI is abruptly stopped and are usually mild. However, some patients report falls, inability to work, and difficulty walking and driving. Untreated symptoms are short-lived and remit within 1 to 2 weeks. They also remit if the original antidepressant is reintroduced or a pharmacologically similar agent is substituted.
Discontinuation syndrome risk among SSRIs is highest for paroxetine, intermediate for sertraline and fluvoxamine, and lowest for fluoxetine.4 Citalopram may cause a mild and transient discontinuation syndrome.8 Citalopram’s long elimination half-life (30 to 35 hours) and fewer and much less-potent active metabolites9 may explain its relatively low risk of discontinuation symptoms.
Discontinuation reactions have been reported to occur 100 times more frequently with paroxetine than with fluoxetine.10 Fluoxetine’s lower rate could be explained by its 2- to 3-day half-life, compared with half-lives of 33 hours or less for paroxetine, sertraline, citalopram, and fluvoxamine. A longer half-life might protect against a discontinuation syndrome.
Among other newer antidepressants:
- venlafaxine’s discontinuation syndrome is similar to the SSRI syndrome11
- no discontinuation symptoms have been reported with mirtazapine, bupropion, or duloxetine.
Table 3
SSRI discontinuation syndrome: The patient at risk…
| Is taking an SSRI with a relatively short half-life |
| Has received antidepressant treatment > 4 weeks |
| Has history of treatment-emergent anxiety, discontinuation symptoms, nonadherence |
| SSRI: Selective serotonin reuptake inhibitor |
| Source: Reference 7 |
Causes. Theories to explain SSRI discontinuation syndrome include cholinergic rebound,12 as described with TCAs, or a decrease in available synaptic serotonin coinciding with down-regulated serotonin receptors.13 Paroxetine’s pharmacologic properties—cholinergic effects, short halflife, and high potency of serotonin reuptake blockade—may explain its relatively high frequency of discontinuation symptoms.
Atypical Antipsychotic Discontinuation Syndromes
Except for aripiprazole—which is a partial dopamine receptor agonist—most atypical antipsychotics are serotonin-dopamine antagonists. Discontinuation syndrome occurs most commonly with clozapine.
Clozapine. Abruptly stopping clozapine can exacerbate psychosis or cause delirium, agitation, confusion, and diaphoresis. Less-common symptoms may include extrapyramidal effects, nausea, diarrhea, headache, or restlessness.14 Clozapine is a weak dopamine D2 antagonist and a potent antagonist at the serotonin 5HT2, alpha adrenergic, histaminergic, and anticholinergic receptors. Thus, rebound from cholinergic, serotonin, dopamine and/or adrenergic receptor supersensitivity is thought to cause its discontinuation syndrome.15
Other atypicals. Case reports describe tics and withdrawal-emergent dyskinesia with risperidone16 and supersensitivity psychosis and a cholinergic/serotonergic syndrome with olanzapine.17,18 Anecdotal reports suggest that abruptly discontinuing quetiapine can cause nausea, emesis, lightheadedness, diaphoresis, orthostasis, tachycardia, and nervousness.19,20 Although discontinuation syndromes have not been reported with ziprasidone or aripiprazole, tapering any atypical antipsychotic during discontinuation is prudent.
Benzodiazepine Discontinuation Syndromes
Benzodiazepines modulate the neurotransmitter activity of gamma-aminobutyric acid (GABA). They differ in their pharmacokinetic properties and have varying half-lives:
- chlordiazepoxide and diazepam have long half-lives (48 hours)
- clonazepam has an intermediate half-life (10 to 24 hours)
- alprazolam, lorazepam, and oxazepam have short half-lives (10 hours).
Abruptly discontinuing benzodiazepines can cause relapse or rebound of pretreatment symptoms. Rebound—with symptoms exceeding pretreatment levels—sometimes occurs after 4 weeks of therapy. The syndrome may last 1 to 3 weeks and is more common with agents having relatively short half-lives.21
Withdrawal. During benzodiazepine withdrawal, new symptoms emerge and pre-existing symptoms worsen. An autonomic component differentiates withdrawal from relapse or rebound. Prominent symptoms include excess sensitivity to light and sound, insomnia, tachycardia, mild systolic hypertension, anxiety, nausea, irritability, tremors, sweating, and abdominal distress. Less-common but serious symptoms include confusion, paranoid delusions, hallucinations, and seizures.22
Withdrawal symptoms are more likely to occur after 6 months of benzodiazepine therapy, when physical dependence also can develop. More-severe benzodiazepine discontinuation syndrome is associated with higher dosages, longer duration of therapy, shorter half-lives, and rapid tapers. Patient factors associated with withdrawal symptoms include:
- personality traits such as dependency and neuroticism
- high pretreatment anxious and depressive symptoms
- history of substance abuse or dependence.23
Preventing discontinuation syndromes
Antidepressants. For TCAs, no discontinuation protocols exist, although some experts suggest tapering regimens over 4 weeks to 3 months. For MAOIs, reducing dosages 10% per week has been suggested.24 The SSRI taper rate depends on the drug’s pharmacologic profile, how long the patient has been taking the SSRI, and the dosage.25
With paroxetine, for example, a gradual reduction of 10 mg/d per week is recommended, based on clinical trial experience. When you reach 20 mg/d, continue this dosage for 1 week before stopping treatment. If reducing a dosage or discontinuing paroxetine causes intolerable symptoms, consider resuming the previously prescribed dosage and then taper more gradually.26
Also gradually taper other SSRIs with short half-lives. Suggested taper regimens for sertraline and fluvoxamine call for weekly reductions of 50 mg/d until you reach 25 to 50 mg. It is not unusual for this final dosage to be lower than the starting dosage.25 Substituting fluoxetine—with its longer half-life—for other SSRIs at the end of treatment has been suggested to suppress withdrawal symptoms,27 although the safety and efficacy of this approach is unknown.5 With venlafaxine, taper over a minimum of 2 to 4 weeks.28
Antipsychotics. To prevent psychotic relapse when discontinuing clozapine, some experts advocate starting a new antipsychotic in a therapeutic dosage before stopping clozapine. When highdose clozapine must be withdrawn immediately, hospitalize the patient and consider using cholinergics to prevent cholinergic rebound.15
Data on managing discontinuation syndromes associated with risperidone, olanzapine, or quetiapine are limited. In some cases, reinstituting the original drug, gradually tapering the antipsychotic,18,19 or using prochlorperazine20 have been useful.
Benzodiazepines. Taper oral benzodiazepines if a patient has taken them >4 to 6 weeks. Also taper IV midazolam used >7 days to sedate a critically ill patient. For the elderly, an 8- to 10-week taper may be required to discontinue benzodiazepines used >3 months for insomnia.
The American Psychiatric Association practice guideline for patients with panic disorder29 recommends tapering benzodiazepines across 2 to 4 months, reducing dosages not more than 10% weekly. Another option is to reduce the daily dosage by 25% per week, but close monitoring and flexibility are required during this taper.
Outcomes when tapering benzodiazepines, according to Rickels et al,23 depend less on pharmacologic adjuvant treatment than on benzodiazepine dosage before the taper, initial psychopathology severity, and patient personality traits (such as passivity/dependency). Before tapering, those authors recommend that you:
- establish a stable patient-physician relationship
- aggressively treat clinically significant anxiety and depression symptoms with medication or other means while the patient continues the established benzodiazepine dosage.
When the taper is nearly complete, maintain the reduced benzodiazepine dosage several months before the final taper.23 Carbamazepine, imipramine, valproate, or trazodone may help alleviate benzodiazepine discontinuation symptoms in select patients.21
When discontinuation occurs
Medical comorbidity. Common medical conditions, including pregnancy or acute surgical procedures, may necessitate abrupt psychotropic discontinuation (Table 4).
Because up to 30% of medical patients have a psychiatric disorder,30 primary care physicians often start psychotropics to manage anxiety and depressive symptoms and may seek psychiatric advice when switching or stopping medications. Moreover, 10% to 15% of hospitalized medically ill patients require dosage reduction or discontinuation of psychotropics that are contributing to the clinical presentation.31
Table 4
Common conditions requiring abrupt psychotropic discontinuation
|
Switching. When switching psychotropics, effects from the first psychotropic may appear to be adverse effects of the new psychotropic. Thus, unrecognized discontinuation syndromes may lead to unnecessary treatment changes.
In our experience, a general rule is to cross-taper and decrease the psychotropic being discontinued by 10% every 1 to 2 weeks. Prescribe adequate dosages of the new psychotropic, closely monitor vital signs, and watch for emerging discontinuation symptoms.
Pregnancy. For women who become pregnant while taking psychotropics, consider the patient’s psychiatric stability, week of pregnancy, psychotropic agent, and treatment preferences when adjusting the treatment plan. In one study of 34 women who stopped psychotropics abruptly for fear of harming the fetus:
- 26 (70%) reported physical and psychological adverse effects
- 11 (30%) reported suicidal ideation, and 4 were hospitalized.32
Patient education. In the study described above, some of the pregnant women’s physicians were unaware of the risks associated with abrupt psychotropic discontinuation and others were aware but failed to inform their patients.32 Thus, patient and family/caregiver education is important. When stopping psychotropics, discuss their risks/benefits, address unrealistic expectations, and individualize therapy by tapering and providing adequate dosing. Watch for suicidality; a weekly telephone call might be useful.
Related resource
- Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics (10th ed). New York: McGraw-Hill, 2001.
Drug Brand Names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Equetro, Tegretol
- Chlordiazepoxide • Librium
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Diazepam • Valium
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Sertraline • Zoloft
- Valproate • Depakene
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Young AH, Currie A. Physicians’ knowledge of antidepressant withdrawal effects: a survey. J Clin Psychiatry 1997;58(7):28-30.
2. Dilsaver SC, Greden JF, Snider RM. Antidepressant withdrawal syndromes: phenomenology and pathophysiology. Int Clin Psychopharmacol 1987;2(1):1-19.
3. Liskin B, Roose S, Walsh T. Acute psychosis following phenelzine discontinuation. J Clin Psychopharmacol 1985;5:46-7.
4. Coupland NJ, Bell CJ, Potokar JP. Serotonin reuptake inhibitor withdrawal. J Clin Psychopharmacol 1996;16(5):356-62.
5. Haddad PM. Antidepressant discontinuation syndromes. Drug Safety 2001;24(3):183-97.
6. Haddad P. The SSRI discontinuation syndrome. J Psychopharmacol 1998;2(3):305-13.
7. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Serotonin reuptake inhibitor discontinuation syndrome: a hypothetical definition. J Clin Psychiatry 1997;58(S7):5-10.
8. Markowitz JS, DeVane CL, Liston HL, et al. An assessment of selective serotonin reuptake inhibitor discontinuation symptoms with citalopram. Int Clin Psychopharmacol 2000;15(6):329-33.
9. Bezchlibnyk-Butler K, Aleksic I, Kennedy SH. Citalopram—a review of pharmacological and clinical effects. J Psychiatry Neurosci 2000;25(3):241-54.
10. Price JS, Waller PC, Wood SM, et al. A comparison of the post-marketing safety of four selective serotonin reuptake inhibitors, including the investigation of symptoms occurring on withdrawal. Br J Clin Pharmacol 1996;42:757-63.
11. Fava M, Mulroy R, Alpert J, et al. Emergence of adverse events following discontinuation of treatment with extended-release venlafaxine. Am J Psychiatry 1997;154(12):1760-2.
12. Barr LC, Goodman WK, Price LH. Physical symptoms associated with paroxetine discontinuation. Am J Psychiatry 1994;151(2):289.-
13. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Possible biological mechanisms of the serotonin reuptake inhibitor discontinuation syndrome. J Clin Psychiatry 1997;58(S7):23-7.
14. Shore D. Clinical implications of clozapine discontinuation: report of an NIMH workshop. Schizophr Bull 1995;21(2):333-8.
15. de Leon J, Stanilla JK, White AO, Simpson GM. Anticholinergics to treat clozapine withdrawal. J Clin Psychiatry 1994;55(3):119-20.
16. Rosebush PI, Kennedy K, Dalton B, Mazurek MF. Protracted akathisia after risperidone withdrawal. Am J Psychiatry 1997;154(3):437-8.
17. Llorca PM, Vaiva G, Lancon C. Supersensitivity psychosis in patients with schizophrenia after sudden olanzapine withdrawal. Can J Psychiatry 2001;46(1):87-8.
18. Nayudu SK, Scheftner WA. Case report of withdrawal syndrome after olanzapine discontinuation. J Clin Psychopharmacol 2000;20:489-90.
19. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry 2000;61:602-3.
20. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry 2005 May;162(5):1020.-
21. McLean W, Ariano R. Benzodiazepine withdrawal schedule and symptoms In: Klasco RK (ed). DRUGDEX® System (vol. 124). Greenwood Village, CO: Thomson Micromedex, 2005.
22. Greenblatt DJ, Miller LG, Shader RI. Benzodiazepine discontinuation syndromes. J Psychiatr Res 1990;24(S2):73-9.
23. Rickels K, Schweizer E, Case WG, Greenblatt DJ. Long-term therapeutic use of benzodiazepines. I. Effects of abrupt discontinuation. Arch Gen Psychiatry 1990;47(10):899-907.
24. Lejoyeux M, Ades J, Mourad I, et al. Antidepressant withdrawal syndrome: recognition, prevalence and management. CNS Drugs 1996;5:278-92.
25. Rosenbaum JF, Zajecka J. Clinical management of antidepressant discontinuation. J Clin Psychiatry 1997;58(S7):37-40.
26. Paxil (paroxetine) package labeling GlaxoSmithKline, 2002.
27. Keuthen NJ, Cyr P, Ricciardi JA, et al. Medication withdrawal symptoms in obsessive-compulsive disorder patients treated with paroxetine. J Clin Psychopharmacol 1994;14(3):206-7.
28. Dallal A, Chouinard G. Withdrawal and rebound symptoms associated with abrupt discontinuation of venlafaxine. J Clin Psychopharmacol 1998;18(4):343-4.
29. American Psychiatric Association Work Group on Panic Disorder Practice guideline for the treatment of patients with panic disorder. Am J Psychiatry 1998;155(S5):1-34.
30. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994;272(22):1749-56.
31. Bronheim HE, Fulop G, Kunkel EJ, et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. Psychosomatics 1998;39(4):S8-30.
32. Einarson A, Selby P, Koren G. Abrupt discontinuation of psychotropic drugs during pregnancy: fear of teratogenic risk and impact of counseling. J Psychiatry Neurosci 2001;26(1):44-8.
Abruptly stopping common psychotropics—particularly antidepressants, benzodiazepines, or atypical antipsychotics—can trigger a discontinuation syndrome, with:
- rebound or relapse of original symptoms
- uncomfortable new physical and psychological symptoms
- physiologic withdrawal at times.
To increase health professionals’ awareness of the risk of these adverse effects,1 this article describes discontinuation syndromes associated with various psychotropics and offers strategies to anticipate, recognize, and manage them.
Antidepressant Discontinuation Syndromes
Discontinuation syndromes can occur with tricyclic and tetracyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), and other newer antidepressants. Symptoms usually start within a few days of stopping a drug—or less commonly, reducing its dosage—and are usually mild and self-limited. Serious outcomes have been reported.
Distinguishing antidepressant discontinuation symptoms from depression recurrence is important. Discontinuation symptoms emerge within 1 to 3 days, whereas depressive symptoms usually occur 2 to 3 weeks after an antidepressant is stopped. Discontinuation reactions remit within a few days, especially if the antidepressant is re-instituted.
TCAs block serotonin and norepinephrine reuptake, increasing the availability of these biogenic amines at receptor sites in the brain and other tissues. Abrupt discontinuation can cause physical symptoms—such as lethargy, headache, and tremor—and psychological symptoms including irritability, anxiety, agitation, and low mood (Table 1).2
Long-term use of TCAs with potent anticholinergic properties leads to upregulation of postsynaptic muscarinic receptors, creating a “supersensitive” state. Abrupt discontinuation can cause cholinergic rebound, with symptoms emerging as soon as 12 hours—but typically 24 to 48 hours—after the last dose.
Table 1
Discontinuation symptoms seen with TCAs
| Physical symptoms | Lethargy, headache, tremor, sweating, anorexia, insomnia, nausea, vomiting, diarrhea, akathisia (rare), parkinsonism (rare) |
| Psychological symptoms | Irritability, anxiety/agitation, low mood, excessive dreaming, nightmares, paradoxical activation resulting in manic/hypomanic symptoms (rare) |
| TCA: Tricyclic antidepressants | |
| Source: Reference 2 | |
MAOIs such as phenelzine and tranylcypromine inhibit the enzyme monoamine oxidase, which is responsible for monoamine degradation and increases synaptic monoamine concentrations. Discontinuation syndromes may include acute confusional states, paranoid delusions, hallucinations, or worsening of depressive symptoms.3 These problems rarely occur in clinical practice, however, because MAOIs’ serious side effects discourage doctors from prescribing them.
SSRIs and other agents. SSRIs block synaptic reuptake of serotonin. Serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine and duloxetine inhibit both serotonin and norepinephrine reuptake. Mirtazapine—an alpha2-adrenergic and heteroreceptor antagonist—increases serotonin and norepinephrine at the synapse. Bupropion increases dopamine and norepinephrine turnover in the CNS and also blocks serotonin.
Up to 30% of patients who stop taking SSRIs develop discontinuation symptoms.4 Six symptom clusters—disequilibrium, sensory symptoms, general somatic symptoms, sleep disturbance, GI symptoms, and affective symptoms—characterize the SSRI discontinuation syndrome (Table 2).5 The four most common symptoms—in decreasing order of frequency—are dizziness, nausea, lethargy, and headache.6 Ataxia, sensory abnormalities, and possibly aggressive and impulsive behavior differentiate this discontinuation syndrome from that of the TCAs.
Table 2
Discontinuation symptoms seen with SSRIs
| Type | Symptoms |
|---|---|
| Disequilibrium | Lightheadedness/dizziness, vertigo, ataxia |
| Sensory symptoms | Paraesthesia, numbness, electric shock-like sensations |
| General somatic symptoms | Lethargy, headache, tremor, sweating, anorexia |
| Sleep disturbance | Insomnia, nightmares, excessive dreaming |
| GI symptoms | Nausea, vomiting, diarrhea |
| Affective symptoms | Irritability, anxiety/agitation, low mood |
| SSRIs: Selective serotonin reuptake inhibitors | |
| Source: Reference 5 | |
Risk factors. Risk factors for SSRI discontinuation syndrome have been identified (Table 3).7 Symptoms usually begin 1 to 3 days after an SSRI is abruptly stopped and are usually mild. However, some patients report falls, inability to work, and difficulty walking and driving. Untreated symptoms are short-lived and remit within 1 to 2 weeks. They also remit if the original antidepressant is reintroduced or a pharmacologically similar agent is substituted.
Discontinuation syndrome risk among SSRIs is highest for paroxetine, intermediate for sertraline and fluvoxamine, and lowest for fluoxetine.4 Citalopram may cause a mild and transient discontinuation syndrome.8 Citalopram’s long elimination half-life (30 to 35 hours) and fewer and much less-potent active metabolites9 may explain its relatively low risk of discontinuation symptoms.
Discontinuation reactions have been reported to occur 100 times more frequently with paroxetine than with fluoxetine.10 Fluoxetine’s lower rate could be explained by its 2- to 3-day half-life, compared with half-lives of 33 hours or less for paroxetine, sertraline, citalopram, and fluvoxamine. A longer half-life might protect against a discontinuation syndrome.
Among other newer antidepressants:
- venlafaxine’s discontinuation syndrome is similar to the SSRI syndrome11
- no discontinuation symptoms have been reported with mirtazapine, bupropion, or duloxetine.
Table 3
SSRI discontinuation syndrome: The patient at risk…
| Is taking an SSRI with a relatively short half-life |
| Has received antidepressant treatment > 4 weeks |
| Has history of treatment-emergent anxiety, discontinuation symptoms, nonadherence |
| SSRI: Selective serotonin reuptake inhibitor |
| Source: Reference 7 |
Causes. Theories to explain SSRI discontinuation syndrome include cholinergic rebound,12 as described with TCAs, or a decrease in available synaptic serotonin coinciding with down-regulated serotonin receptors.13 Paroxetine’s pharmacologic properties—cholinergic effects, short halflife, and high potency of serotonin reuptake blockade—may explain its relatively high frequency of discontinuation symptoms.
Atypical Antipsychotic Discontinuation Syndromes
Except for aripiprazole—which is a partial dopamine receptor agonist—most atypical antipsychotics are serotonin-dopamine antagonists. Discontinuation syndrome occurs most commonly with clozapine.
Clozapine. Abruptly stopping clozapine can exacerbate psychosis or cause delirium, agitation, confusion, and diaphoresis. Less-common symptoms may include extrapyramidal effects, nausea, diarrhea, headache, or restlessness.14 Clozapine is a weak dopamine D2 antagonist and a potent antagonist at the serotonin 5HT2, alpha adrenergic, histaminergic, and anticholinergic receptors. Thus, rebound from cholinergic, serotonin, dopamine and/or adrenergic receptor supersensitivity is thought to cause its discontinuation syndrome.15
Other atypicals. Case reports describe tics and withdrawal-emergent dyskinesia with risperidone16 and supersensitivity psychosis and a cholinergic/serotonergic syndrome with olanzapine.17,18 Anecdotal reports suggest that abruptly discontinuing quetiapine can cause nausea, emesis, lightheadedness, diaphoresis, orthostasis, tachycardia, and nervousness.19,20 Although discontinuation syndromes have not been reported with ziprasidone or aripiprazole, tapering any atypical antipsychotic during discontinuation is prudent.
Benzodiazepine Discontinuation Syndromes
Benzodiazepines modulate the neurotransmitter activity of gamma-aminobutyric acid (GABA). They differ in their pharmacokinetic properties and have varying half-lives:
- chlordiazepoxide and diazepam have long half-lives (48 hours)
- clonazepam has an intermediate half-life (10 to 24 hours)
- alprazolam, lorazepam, and oxazepam have short half-lives (10 hours).
Abruptly discontinuing benzodiazepines can cause relapse or rebound of pretreatment symptoms. Rebound—with symptoms exceeding pretreatment levels—sometimes occurs after 4 weeks of therapy. The syndrome may last 1 to 3 weeks and is more common with agents having relatively short half-lives.21
Withdrawal. During benzodiazepine withdrawal, new symptoms emerge and pre-existing symptoms worsen. An autonomic component differentiates withdrawal from relapse or rebound. Prominent symptoms include excess sensitivity to light and sound, insomnia, tachycardia, mild systolic hypertension, anxiety, nausea, irritability, tremors, sweating, and abdominal distress. Less-common but serious symptoms include confusion, paranoid delusions, hallucinations, and seizures.22
Withdrawal symptoms are more likely to occur after 6 months of benzodiazepine therapy, when physical dependence also can develop. More-severe benzodiazepine discontinuation syndrome is associated with higher dosages, longer duration of therapy, shorter half-lives, and rapid tapers. Patient factors associated with withdrawal symptoms include:
- personality traits such as dependency and neuroticism
- high pretreatment anxious and depressive symptoms
- history of substance abuse or dependence.23
Preventing discontinuation syndromes
Antidepressants. For TCAs, no discontinuation protocols exist, although some experts suggest tapering regimens over 4 weeks to 3 months. For MAOIs, reducing dosages 10% per week has been suggested.24 The SSRI taper rate depends on the drug’s pharmacologic profile, how long the patient has been taking the SSRI, and the dosage.25
With paroxetine, for example, a gradual reduction of 10 mg/d per week is recommended, based on clinical trial experience. When you reach 20 mg/d, continue this dosage for 1 week before stopping treatment. If reducing a dosage or discontinuing paroxetine causes intolerable symptoms, consider resuming the previously prescribed dosage and then taper more gradually.26
Also gradually taper other SSRIs with short half-lives. Suggested taper regimens for sertraline and fluvoxamine call for weekly reductions of 50 mg/d until you reach 25 to 50 mg. It is not unusual for this final dosage to be lower than the starting dosage.25 Substituting fluoxetine—with its longer half-life—for other SSRIs at the end of treatment has been suggested to suppress withdrawal symptoms,27 although the safety and efficacy of this approach is unknown.5 With venlafaxine, taper over a minimum of 2 to 4 weeks.28
Antipsychotics. To prevent psychotic relapse when discontinuing clozapine, some experts advocate starting a new antipsychotic in a therapeutic dosage before stopping clozapine. When highdose clozapine must be withdrawn immediately, hospitalize the patient and consider using cholinergics to prevent cholinergic rebound.15
Data on managing discontinuation syndromes associated with risperidone, olanzapine, or quetiapine are limited. In some cases, reinstituting the original drug, gradually tapering the antipsychotic,18,19 or using prochlorperazine20 have been useful.
Benzodiazepines. Taper oral benzodiazepines if a patient has taken them >4 to 6 weeks. Also taper IV midazolam used >7 days to sedate a critically ill patient. For the elderly, an 8- to 10-week taper may be required to discontinue benzodiazepines used >3 months for insomnia.
The American Psychiatric Association practice guideline for patients with panic disorder29 recommends tapering benzodiazepines across 2 to 4 months, reducing dosages not more than 10% weekly. Another option is to reduce the daily dosage by 25% per week, but close monitoring and flexibility are required during this taper.
Outcomes when tapering benzodiazepines, according to Rickels et al,23 depend less on pharmacologic adjuvant treatment than on benzodiazepine dosage before the taper, initial psychopathology severity, and patient personality traits (such as passivity/dependency). Before tapering, those authors recommend that you:
- establish a stable patient-physician relationship
- aggressively treat clinically significant anxiety and depression symptoms with medication or other means while the patient continues the established benzodiazepine dosage.
When the taper is nearly complete, maintain the reduced benzodiazepine dosage several months before the final taper.23 Carbamazepine, imipramine, valproate, or trazodone may help alleviate benzodiazepine discontinuation symptoms in select patients.21
When discontinuation occurs
Medical comorbidity. Common medical conditions, including pregnancy or acute surgical procedures, may necessitate abrupt psychotropic discontinuation (Table 4).
Because up to 30% of medical patients have a psychiatric disorder,30 primary care physicians often start psychotropics to manage anxiety and depressive symptoms and may seek psychiatric advice when switching or stopping medications. Moreover, 10% to 15% of hospitalized medically ill patients require dosage reduction or discontinuation of psychotropics that are contributing to the clinical presentation.31
Table 4
Common conditions requiring abrupt psychotropic discontinuation
|
Switching. When switching psychotropics, effects from the first psychotropic may appear to be adverse effects of the new psychotropic. Thus, unrecognized discontinuation syndromes may lead to unnecessary treatment changes.
In our experience, a general rule is to cross-taper and decrease the psychotropic being discontinued by 10% every 1 to 2 weeks. Prescribe adequate dosages of the new psychotropic, closely monitor vital signs, and watch for emerging discontinuation symptoms.
Pregnancy. For women who become pregnant while taking psychotropics, consider the patient’s psychiatric stability, week of pregnancy, psychotropic agent, and treatment preferences when adjusting the treatment plan. In one study of 34 women who stopped psychotropics abruptly for fear of harming the fetus:
- 26 (70%) reported physical and psychological adverse effects
- 11 (30%) reported suicidal ideation, and 4 were hospitalized.32
Patient education. In the study described above, some of the pregnant women’s physicians were unaware of the risks associated with abrupt psychotropic discontinuation and others were aware but failed to inform their patients.32 Thus, patient and family/caregiver education is important. When stopping psychotropics, discuss their risks/benefits, address unrealistic expectations, and individualize therapy by tapering and providing adequate dosing. Watch for suicidality; a weekly telephone call might be useful.
Related resource
- Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics (10th ed). New York: McGraw-Hill, 2001.
Drug Brand Names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Equetro, Tegretol
- Chlordiazepoxide • Librium
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Diazepam • Valium
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Sertraline • Zoloft
- Valproate • Depakene
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Abruptly stopping common psychotropics—particularly antidepressants, benzodiazepines, or atypical antipsychotics—can trigger a discontinuation syndrome, with:
- rebound or relapse of original symptoms
- uncomfortable new physical and psychological symptoms
- physiologic withdrawal at times.
To increase health professionals’ awareness of the risk of these adverse effects,1 this article describes discontinuation syndromes associated with various psychotropics and offers strategies to anticipate, recognize, and manage them.
Antidepressant Discontinuation Syndromes
Discontinuation syndromes can occur with tricyclic and tetracyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), and other newer antidepressants. Symptoms usually start within a few days of stopping a drug—or less commonly, reducing its dosage—and are usually mild and self-limited. Serious outcomes have been reported.
Distinguishing antidepressant discontinuation symptoms from depression recurrence is important. Discontinuation symptoms emerge within 1 to 3 days, whereas depressive symptoms usually occur 2 to 3 weeks after an antidepressant is stopped. Discontinuation reactions remit within a few days, especially if the antidepressant is re-instituted.
TCAs block serotonin and norepinephrine reuptake, increasing the availability of these biogenic amines at receptor sites in the brain and other tissues. Abrupt discontinuation can cause physical symptoms—such as lethargy, headache, and tremor—and psychological symptoms including irritability, anxiety, agitation, and low mood (Table 1).2
Long-term use of TCAs with potent anticholinergic properties leads to upregulation of postsynaptic muscarinic receptors, creating a “supersensitive” state. Abrupt discontinuation can cause cholinergic rebound, with symptoms emerging as soon as 12 hours—but typically 24 to 48 hours—after the last dose.
Table 1
Discontinuation symptoms seen with TCAs
| Physical symptoms | Lethargy, headache, tremor, sweating, anorexia, insomnia, nausea, vomiting, diarrhea, akathisia (rare), parkinsonism (rare) |
| Psychological symptoms | Irritability, anxiety/agitation, low mood, excessive dreaming, nightmares, paradoxical activation resulting in manic/hypomanic symptoms (rare) |
| TCA: Tricyclic antidepressants | |
| Source: Reference 2 | |
MAOIs such as phenelzine and tranylcypromine inhibit the enzyme monoamine oxidase, which is responsible for monoamine degradation and increases synaptic monoamine concentrations. Discontinuation syndromes may include acute confusional states, paranoid delusions, hallucinations, or worsening of depressive symptoms.3 These problems rarely occur in clinical practice, however, because MAOIs’ serious side effects discourage doctors from prescribing them.
SSRIs and other agents. SSRIs block synaptic reuptake of serotonin. Serotonin-norepinephrine reuptake inhibitors (SNRIs) such as venlafaxine and duloxetine inhibit both serotonin and norepinephrine reuptake. Mirtazapine—an alpha2-adrenergic and heteroreceptor antagonist—increases serotonin and norepinephrine at the synapse. Bupropion increases dopamine and norepinephrine turnover in the CNS and also blocks serotonin.
Up to 30% of patients who stop taking SSRIs develop discontinuation symptoms.4 Six symptom clusters—disequilibrium, sensory symptoms, general somatic symptoms, sleep disturbance, GI symptoms, and affective symptoms—characterize the SSRI discontinuation syndrome (Table 2).5 The four most common symptoms—in decreasing order of frequency—are dizziness, nausea, lethargy, and headache.6 Ataxia, sensory abnormalities, and possibly aggressive and impulsive behavior differentiate this discontinuation syndrome from that of the TCAs.
Table 2
Discontinuation symptoms seen with SSRIs
| Type | Symptoms |
|---|---|
| Disequilibrium | Lightheadedness/dizziness, vertigo, ataxia |
| Sensory symptoms | Paraesthesia, numbness, electric shock-like sensations |
| General somatic symptoms | Lethargy, headache, tremor, sweating, anorexia |
| Sleep disturbance | Insomnia, nightmares, excessive dreaming |
| GI symptoms | Nausea, vomiting, diarrhea |
| Affective symptoms | Irritability, anxiety/agitation, low mood |
| SSRIs: Selective serotonin reuptake inhibitors | |
| Source: Reference 5 | |
Risk factors. Risk factors for SSRI discontinuation syndrome have been identified (Table 3).7 Symptoms usually begin 1 to 3 days after an SSRI is abruptly stopped and are usually mild. However, some patients report falls, inability to work, and difficulty walking and driving. Untreated symptoms are short-lived and remit within 1 to 2 weeks. They also remit if the original antidepressant is reintroduced or a pharmacologically similar agent is substituted.
Discontinuation syndrome risk among SSRIs is highest for paroxetine, intermediate for sertraline and fluvoxamine, and lowest for fluoxetine.4 Citalopram may cause a mild and transient discontinuation syndrome.8 Citalopram’s long elimination half-life (30 to 35 hours) and fewer and much less-potent active metabolites9 may explain its relatively low risk of discontinuation symptoms.
Discontinuation reactions have been reported to occur 100 times more frequently with paroxetine than with fluoxetine.10 Fluoxetine’s lower rate could be explained by its 2- to 3-day half-life, compared with half-lives of 33 hours or less for paroxetine, sertraline, citalopram, and fluvoxamine. A longer half-life might protect against a discontinuation syndrome.
Among other newer antidepressants:
- venlafaxine’s discontinuation syndrome is similar to the SSRI syndrome11
- no discontinuation symptoms have been reported with mirtazapine, bupropion, or duloxetine.
Table 3
SSRI discontinuation syndrome: The patient at risk…
| Is taking an SSRI with a relatively short half-life |
| Has received antidepressant treatment > 4 weeks |
| Has history of treatment-emergent anxiety, discontinuation symptoms, nonadherence |
| SSRI: Selective serotonin reuptake inhibitor |
| Source: Reference 7 |
Causes. Theories to explain SSRI discontinuation syndrome include cholinergic rebound,12 as described with TCAs, or a decrease in available synaptic serotonin coinciding with down-regulated serotonin receptors.13 Paroxetine’s pharmacologic properties—cholinergic effects, short halflife, and high potency of serotonin reuptake blockade—may explain its relatively high frequency of discontinuation symptoms.
Atypical Antipsychotic Discontinuation Syndromes
Except for aripiprazole—which is a partial dopamine receptor agonist—most atypical antipsychotics are serotonin-dopamine antagonists. Discontinuation syndrome occurs most commonly with clozapine.
Clozapine. Abruptly stopping clozapine can exacerbate psychosis or cause delirium, agitation, confusion, and diaphoresis. Less-common symptoms may include extrapyramidal effects, nausea, diarrhea, headache, or restlessness.14 Clozapine is a weak dopamine D2 antagonist and a potent antagonist at the serotonin 5HT2, alpha adrenergic, histaminergic, and anticholinergic receptors. Thus, rebound from cholinergic, serotonin, dopamine and/or adrenergic receptor supersensitivity is thought to cause its discontinuation syndrome.15
Other atypicals. Case reports describe tics and withdrawal-emergent dyskinesia with risperidone16 and supersensitivity psychosis and a cholinergic/serotonergic syndrome with olanzapine.17,18 Anecdotal reports suggest that abruptly discontinuing quetiapine can cause nausea, emesis, lightheadedness, diaphoresis, orthostasis, tachycardia, and nervousness.19,20 Although discontinuation syndromes have not been reported with ziprasidone or aripiprazole, tapering any atypical antipsychotic during discontinuation is prudent.
Benzodiazepine Discontinuation Syndromes
Benzodiazepines modulate the neurotransmitter activity of gamma-aminobutyric acid (GABA). They differ in their pharmacokinetic properties and have varying half-lives:
- chlordiazepoxide and diazepam have long half-lives (48 hours)
- clonazepam has an intermediate half-life (10 to 24 hours)
- alprazolam, lorazepam, and oxazepam have short half-lives (10 hours).
Abruptly discontinuing benzodiazepines can cause relapse or rebound of pretreatment symptoms. Rebound—with symptoms exceeding pretreatment levels—sometimes occurs after 4 weeks of therapy. The syndrome may last 1 to 3 weeks and is more common with agents having relatively short half-lives.21
Withdrawal. During benzodiazepine withdrawal, new symptoms emerge and pre-existing symptoms worsen. An autonomic component differentiates withdrawal from relapse or rebound. Prominent symptoms include excess sensitivity to light and sound, insomnia, tachycardia, mild systolic hypertension, anxiety, nausea, irritability, tremors, sweating, and abdominal distress. Less-common but serious symptoms include confusion, paranoid delusions, hallucinations, and seizures.22
Withdrawal symptoms are more likely to occur after 6 months of benzodiazepine therapy, when physical dependence also can develop. More-severe benzodiazepine discontinuation syndrome is associated with higher dosages, longer duration of therapy, shorter half-lives, and rapid tapers. Patient factors associated with withdrawal symptoms include:
- personality traits such as dependency and neuroticism
- high pretreatment anxious and depressive symptoms
- history of substance abuse or dependence.23
Preventing discontinuation syndromes
Antidepressants. For TCAs, no discontinuation protocols exist, although some experts suggest tapering regimens over 4 weeks to 3 months. For MAOIs, reducing dosages 10% per week has been suggested.24 The SSRI taper rate depends on the drug’s pharmacologic profile, how long the patient has been taking the SSRI, and the dosage.25
With paroxetine, for example, a gradual reduction of 10 mg/d per week is recommended, based on clinical trial experience. When you reach 20 mg/d, continue this dosage for 1 week before stopping treatment. If reducing a dosage or discontinuing paroxetine causes intolerable symptoms, consider resuming the previously prescribed dosage and then taper more gradually.26
Also gradually taper other SSRIs with short half-lives. Suggested taper regimens for sertraline and fluvoxamine call for weekly reductions of 50 mg/d until you reach 25 to 50 mg. It is not unusual for this final dosage to be lower than the starting dosage.25 Substituting fluoxetine—with its longer half-life—for other SSRIs at the end of treatment has been suggested to suppress withdrawal symptoms,27 although the safety and efficacy of this approach is unknown.5 With venlafaxine, taper over a minimum of 2 to 4 weeks.28
Antipsychotics. To prevent psychotic relapse when discontinuing clozapine, some experts advocate starting a new antipsychotic in a therapeutic dosage before stopping clozapine. When highdose clozapine must be withdrawn immediately, hospitalize the patient and consider using cholinergics to prevent cholinergic rebound.15
Data on managing discontinuation syndromes associated with risperidone, olanzapine, or quetiapine are limited. In some cases, reinstituting the original drug, gradually tapering the antipsychotic,18,19 or using prochlorperazine20 have been useful.
Benzodiazepines. Taper oral benzodiazepines if a patient has taken them >4 to 6 weeks. Also taper IV midazolam used >7 days to sedate a critically ill patient. For the elderly, an 8- to 10-week taper may be required to discontinue benzodiazepines used >3 months for insomnia.
The American Psychiatric Association practice guideline for patients with panic disorder29 recommends tapering benzodiazepines across 2 to 4 months, reducing dosages not more than 10% weekly. Another option is to reduce the daily dosage by 25% per week, but close monitoring and flexibility are required during this taper.
Outcomes when tapering benzodiazepines, according to Rickels et al,23 depend less on pharmacologic adjuvant treatment than on benzodiazepine dosage before the taper, initial psychopathology severity, and patient personality traits (such as passivity/dependency). Before tapering, those authors recommend that you:
- establish a stable patient-physician relationship
- aggressively treat clinically significant anxiety and depression symptoms with medication or other means while the patient continues the established benzodiazepine dosage.
When the taper is nearly complete, maintain the reduced benzodiazepine dosage several months before the final taper.23 Carbamazepine, imipramine, valproate, or trazodone may help alleviate benzodiazepine discontinuation symptoms in select patients.21
When discontinuation occurs
Medical comorbidity. Common medical conditions, including pregnancy or acute surgical procedures, may necessitate abrupt psychotropic discontinuation (Table 4).
Because up to 30% of medical patients have a psychiatric disorder,30 primary care physicians often start psychotropics to manage anxiety and depressive symptoms and may seek psychiatric advice when switching or stopping medications. Moreover, 10% to 15% of hospitalized medically ill patients require dosage reduction or discontinuation of psychotropics that are contributing to the clinical presentation.31
Table 4
Common conditions requiring abrupt psychotropic discontinuation
|
Switching. When switching psychotropics, effects from the first psychotropic may appear to be adverse effects of the new psychotropic. Thus, unrecognized discontinuation syndromes may lead to unnecessary treatment changes.
In our experience, a general rule is to cross-taper and decrease the psychotropic being discontinued by 10% every 1 to 2 weeks. Prescribe adequate dosages of the new psychotropic, closely monitor vital signs, and watch for emerging discontinuation symptoms.
Pregnancy. For women who become pregnant while taking psychotropics, consider the patient’s psychiatric stability, week of pregnancy, psychotropic agent, and treatment preferences when adjusting the treatment plan. In one study of 34 women who stopped psychotropics abruptly for fear of harming the fetus:
- 26 (70%) reported physical and psychological adverse effects
- 11 (30%) reported suicidal ideation, and 4 were hospitalized.32
Patient education. In the study described above, some of the pregnant women’s physicians were unaware of the risks associated with abrupt psychotropic discontinuation and others were aware but failed to inform their patients.32 Thus, patient and family/caregiver education is important. When stopping psychotropics, discuss their risks/benefits, address unrealistic expectations, and individualize therapy by tapering and providing adequate dosing. Watch for suicidality; a weekly telephone call might be useful.
Related resource
- Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman’s the pharmacological basis of therapeutics (10th ed). New York: McGraw-Hill, 2001.
Drug Brand Names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Equetro, Tegretol
- Chlordiazepoxide • Librium
- Citalopram • Celexa
- Clonazepam • Klonopin
- Clozapine • Clozaril
- Diazepam • Valium
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenelzine • Nardil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Sertraline • Zoloft
- Valproate • Depakene
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Young AH, Currie A. Physicians’ knowledge of antidepressant withdrawal effects: a survey. J Clin Psychiatry 1997;58(7):28-30.
2. Dilsaver SC, Greden JF, Snider RM. Antidepressant withdrawal syndromes: phenomenology and pathophysiology. Int Clin Psychopharmacol 1987;2(1):1-19.
3. Liskin B, Roose S, Walsh T. Acute psychosis following phenelzine discontinuation. J Clin Psychopharmacol 1985;5:46-7.
4. Coupland NJ, Bell CJ, Potokar JP. Serotonin reuptake inhibitor withdrawal. J Clin Psychopharmacol 1996;16(5):356-62.
5. Haddad PM. Antidepressant discontinuation syndromes. Drug Safety 2001;24(3):183-97.
6. Haddad P. The SSRI discontinuation syndrome. J Psychopharmacol 1998;2(3):305-13.
7. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Serotonin reuptake inhibitor discontinuation syndrome: a hypothetical definition. J Clin Psychiatry 1997;58(S7):5-10.
8. Markowitz JS, DeVane CL, Liston HL, et al. An assessment of selective serotonin reuptake inhibitor discontinuation symptoms with citalopram. Int Clin Psychopharmacol 2000;15(6):329-33.
9. Bezchlibnyk-Butler K, Aleksic I, Kennedy SH. Citalopram—a review of pharmacological and clinical effects. J Psychiatry Neurosci 2000;25(3):241-54.
10. Price JS, Waller PC, Wood SM, et al. A comparison of the post-marketing safety of four selective serotonin reuptake inhibitors, including the investigation of symptoms occurring on withdrawal. Br J Clin Pharmacol 1996;42:757-63.
11. Fava M, Mulroy R, Alpert J, et al. Emergence of adverse events following discontinuation of treatment with extended-release venlafaxine. Am J Psychiatry 1997;154(12):1760-2.
12. Barr LC, Goodman WK, Price LH. Physical symptoms associated with paroxetine discontinuation. Am J Psychiatry 1994;151(2):289.-
13. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Possible biological mechanisms of the serotonin reuptake inhibitor discontinuation syndrome. J Clin Psychiatry 1997;58(S7):23-7.
14. Shore D. Clinical implications of clozapine discontinuation: report of an NIMH workshop. Schizophr Bull 1995;21(2):333-8.
15. de Leon J, Stanilla JK, White AO, Simpson GM. Anticholinergics to treat clozapine withdrawal. J Clin Psychiatry 1994;55(3):119-20.
16. Rosebush PI, Kennedy K, Dalton B, Mazurek MF. Protracted akathisia after risperidone withdrawal. Am J Psychiatry 1997;154(3):437-8.
17. Llorca PM, Vaiva G, Lancon C. Supersensitivity psychosis in patients with schizophrenia after sudden olanzapine withdrawal. Can J Psychiatry 2001;46(1):87-8.
18. Nayudu SK, Scheftner WA. Case report of withdrawal syndrome after olanzapine discontinuation. J Clin Psychopharmacol 2000;20:489-90.
19. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry 2000;61:602-3.
20. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry 2005 May;162(5):1020.-
21. McLean W, Ariano R. Benzodiazepine withdrawal schedule and symptoms In: Klasco RK (ed). DRUGDEX® System (vol. 124). Greenwood Village, CO: Thomson Micromedex, 2005.
22. Greenblatt DJ, Miller LG, Shader RI. Benzodiazepine discontinuation syndromes. J Psychiatr Res 1990;24(S2):73-9.
23. Rickels K, Schweizer E, Case WG, Greenblatt DJ. Long-term therapeutic use of benzodiazepines. I. Effects of abrupt discontinuation. Arch Gen Psychiatry 1990;47(10):899-907.
24. Lejoyeux M, Ades J, Mourad I, et al. Antidepressant withdrawal syndrome: recognition, prevalence and management. CNS Drugs 1996;5:278-92.
25. Rosenbaum JF, Zajecka J. Clinical management of antidepressant discontinuation. J Clin Psychiatry 1997;58(S7):37-40.
26. Paxil (paroxetine) package labeling GlaxoSmithKline, 2002.
27. Keuthen NJ, Cyr P, Ricciardi JA, et al. Medication withdrawal symptoms in obsessive-compulsive disorder patients treated with paroxetine. J Clin Psychopharmacol 1994;14(3):206-7.
28. Dallal A, Chouinard G. Withdrawal and rebound symptoms associated with abrupt discontinuation of venlafaxine. J Clin Psychopharmacol 1998;18(4):343-4.
29. American Psychiatric Association Work Group on Panic Disorder Practice guideline for the treatment of patients with panic disorder. Am J Psychiatry 1998;155(S5):1-34.
30. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994;272(22):1749-56.
31. Bronheim HE, Fulop G, Kunkel EJ, et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. Psychosomatics 1998;39(4):S8-30.
32. Einarson A, Selby P, Koren G. Abrupt discontinuation of psychotropic drugs during pregnancy: fear of teratogenic risk and impact of counseling. J Psychiatry Neurosci 2001;26(1):44-8.
1. Young AH, Currie A. Physicians’ knowledge of antidepressant withdrawal effects: a survey. J Clin Psychiatry 1997;58(7):28-30.
2. Dilsaver SC, Greden JF, Snider RM. Antidepressant withdrawal syndromes: phenomenology and pathophysiology. Int Clin Psychopharmacol 1987;2(1):1-19.
3. Liskin B, Roose S, Walsh T. Acute psychosis following phenelzine discontinuation. J Clin Psychopharmacol 1985;5:46-7.
4. Coupland NJ, Bell CJ, Potokar JP. Serotonin reuptake inhibitor withdrawal. J Clin Psychopharmacol 1996;16(5):356-62.
5. Haddad PM. Antidepressant discontinuation syndromes. Drug Safety 2001;24(3):183-97.
6. Haddad P. The SSRI discontinuation syndrome. J Psychopharmacol 1998;2(3):305-13.
7. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Serotonin reuptake inhibitor discontinuation syndrome: a hypothetical definition. J Clin Psychiatry 1997;58(S7):5-10.
8. Markowitz JS, DeVane CL, Liston HL, et al. An assessment of selective serotonin reuptake inhibitor discontinuation symptoms with citalopram. Int Clin Psychopharmacol 2000;15(6):329-33.
9. Bezchlibnyk-Butler K, Aleksic I, Kennedy SH. Citalopram—a review of pharmacological and clinical effects. J Psychiatry Neurosci 2000;25(3):241-54.
10. Price JS, Waller PC, Wood SM, et al. A comparison of the post-marketing safety of four selective serotonin reuptake inhibitors, including the investigation of symptoms occurring on withdrawal. Br J Clin Pharmacol 1996;42:757-63.
11. Fava M, Mulroy R, Alpert J, et al. Emergence of adverse events following discontinuation of treatment with extended-release venlafaxine. Am J Psychiatry 1997;154(12):1760-2.
12. Barr LC, Goodman WK, Price LH. Physical symptoms associated with paroxetine discontinuation. Am J Psychiatry 1994;151(2):289.-
13. Schatzberg AF, Haddad P, Kaplan EM, et al. for the Discontinuation Consensus Panel Possible biological mechanisms of the serotonin reuptake inhibitor discontinuation syndrome. J Clin Psychiatry 1997;58(S7):23-7.
14. Shore D. Clinical implications of clozapine discontinuation: report of an NIMH workshop. Schizophr Bull 1995;21(2):333-8.
15. de Leon J, Stanilla JK, White AO, Simpson GM. Anticholinergics to treat clozapine withdrawal. J Clin Psychiatry 1994;55(3):119-20.
16. Rosebush PI, Kennedy K, Dalton B, Mazurek MF. Protracted akathisia after risperidone withdrawal. Am J Psychiatry 1997;154(3):437-8.
17. Llorca PM, Vaiva G, Lancon C. Supersensitivity psychosis in patients with schizophrenia after sudden olanzapine withdrawal. Can J Psychiatry 2001;46(1):87-8.
18. Nayudu SK, Scheftner WA. Case report of withdrawal syndrome after olanzapine discontinuation. J Clin Psychopharmacol 2000;20:489-90.
19. Thurstone CC, Alahi P. A possible case of quetiapine withdrawal syndrome. J Clin Psychiatry 2000;61:602-3.
20. Kim DR, Staab JP. Quetiapine discontinuation syndrome. Am J Psychiatry 2005 May;162(5):1020.-
21. McLean W, Ariano R. Benzodiazepine withdrawal schedule and symptoms In: Klasco RK (ed). DRUGDEX® System (vol. 124). Greenwood Village, CO: Thomson Micromedex, 2005.
22. Greenblatt DJ, Miller LG, Shader RI. Benzodiazepine discontinuation syndromes. J Psychiatr Res 1990;24(S2):73-9.
23. Rickels K, Schweizer E, Case WG, Greenblatt DJ. Long-term therapeutic use of benzodiazepines. I. Effects of abrupt discontinuation. Arch Gen Psychiatry 1990;47(10):899-907.
24. Lejoyeux M, Ades J, Mourad I, et al. Antidepressant withdrawal syndrome: recognition, prevalence and management. CNS Drugs 1996;5:278-92.
25. Rosenbaum JF, Zajecka J. Clinical management of antidepressant discontinuation. J Clin Psychiatry 1997;58(S7):37-40.
26. Paxil (paroxetine) package labeling GlaxoSmithKline, 2002.
27. Keuthen NJ, Cyr P, Ricciardi JA, et al. Medication withdrawal symptoms in obsessive-compulsive disorder patients treated with paroxetine. J Clin Psychopharmacol 1994;14(3):206-7.
28. Dallal A, Chouinard G. Withdrawal and rebound symptoms associated with abrupt discontinuation of venlafaxine. J Clin Psychopharmacol 1998;18(4):343-4.
29. American Psychiatric Association Work Group on Panic Disorder Practice guideline for the treatment of patients with panic disorder. Am J Psychiatry 1998;155(S5):1-34.
30. Spitzer RL, Williams JB, Kroenke K, et al. Utility of a new procedure for diagnosing mental disorders in primary care. The PRIME-MD 1000 study. JAMA 1994;272(22):1749-56.
31. Bronheim HE, Fulop G, Kunkel EJ, et al. The Academy of Psychosomatic Medicine practice guidelines for psychiatric consultation in the general medical setting. Psychosomatics 1998;39(4):S8-30.
32. Einarson A, Selby P, Koren G. Abrupt discontinuation of psychotropic drugs during pregnancy: fear of teratogenic risk and impact of counseling. J Psychiatry Neurosci 2001;26(1):44-8.
Treating depression to remission: Target recovery, and give patients back their lives
Remission is considered the standard of treatment for major depression,1-4 but many patients fall short of this goal:
- 25% to 50% of those who respond to treatment have residual symptoms5
- 60% to 70% respond to treatment, but only 20% to 40% achieve remission.6
We offer practical, evidence-based suggestions and resources to help you take more of your patients beyond response to remission and then to recovery (Table 1).7,8
Table 1
Outcomes in depression: Defining the 4 ‘R’s
| Outcome | Definition | Comment |
|---|---|---|
| Response | Clinically significant reduction of symptoms | 50% reduction in symptoms on psychometric scales may leave severely depressed patients with disabling symptoms |
| Remission | Depression resolves completely or nearly completely, with return to baseline function | Score of ≤7 on the HRSD used in many studies; ACNP Task Force defines remission by 9 core depression symptoms in DSM-IV-TR |
| Recovery | Remission lasts for extended time; signifies end of a major depressive episode | ACNP Task Force defines recovery as 4 months of remission |
| Relapse | Return to full symptoms during remission but before recovery; signifies re-emergence of current depressive episode | Residual symptoms during response or remission greatly increase chances of relapse |
| HRSD: Hamilton Rating Scale for Depression | ||
| ACNP: American College of Neuropsychopharmacology | ||
| DSM-IV-TR: Diagnostic and statistical manual of mental disorders, 4th ed., text rev. | ||
| Source: References 7, 8 | ||
Case report: missing the target
Ms. M, age 32, develops depressive symptoms after taking on several new projects in her work as an accountant. At first she notices difficulty falling asleep at night and that she seems tired all day. Typically efficient and neat, she finds herself absently staring at her computer screen while a messy pile of unfinished paperwork accumulates on her desk.
She begins chastising herself for falling behind, yet she feels she will never catch up. When she unexpectedly bursts out sobbing in a board meeting, she knows she needs help.
Ms. M reports that she has been taking fluoxetine, 20 mg/d, for 4 weeks as her primary care physician prescribed, with no improvement. She has no history of depression or other psychiatric illness, is taking no other medication, and has no medical illnesses. Her brother has a history of bipolar disorder.
Initial diagnostic workup includes laboratory tests such as thyroid stimulating hormone, vitamin B12, and folate. All values are within normal limits. Her Hamilton Rating Scale for Depression (HRSD) score is 17, indicating moderate depression.
The psychiatrist increases fluoxetine to 40 mg/d, and after about 3 weeks Ms. M starts feeling better. Her hopelessness lifts, she is more engaged, and her sleep improves, yet she continues to feel sluggish and dazed. Her financial reports contain uncharacteristic errors, and her pace is noticeably slow. Twice her supervisor approaches her about substandard work, then a week later warns that she will lose her job unless she improves.
Barriers to remission. Patient, provider, and health care system barriers prevent patients with major depression from achieving remission (Table 2).3 Patients may feel better with antidepressant therapy but do not recognize and report residual depressive symptoms, such as Ms. M’s fatigue and substandard job performance.
Clinicians also play a role in depression undertreatment. For example, in a study of 239 patients with ≥ 5 depression symptoms,9 28% did not receive treatment consistent with depression management guidelines.
Fava et al10 suggested the following reasons for depression undertreatment: “… clinicians have partial therapeutic targets, neglect residual symptoms, and equate therapeutic response with full remission.” Others have found that physicians may underdose medications or fail to plan treatment in clear phases.11
Table 2
Barriers to remission during depression treatment
| Who and what | Behavior and system problems |
|---|---|
| Patients | Nonadherence to treatment, underreporting of symptoms, satisfaction with suboptimal outcomes, failure to recognize depressive symptoms, underestimating depression severity, limited access to care, reluctance to see a mental health specialist |
| Providers | Medication under-dosing, inadequate treatment duration, inaccurate diagnosis, failure to recognize residual symptoms, limited training in interpersonal skills, inadequate time to evaluate and treat depression, failure to consider psychotherapeutic approaches |
| Health care systems | Limited therapeutic choices, limited number of mental health care visits, restricted access to providers |
| Source: Reference 3 | |
Relapse risk
Increased risk of relapse is perhaps the greatest cost of undertreated major depression. Patients with residual subsyndromal depressive symptoms relapse five times faster than patients in full remission.12
Residual symptoms may be a more powerful relapse predictor than number of past depressive episodes:13
- Chronic mood symptoms for ≥ 2 years double the relapse risk.14
- 50% to 80% of patients in partial remission relapse.15
In a study of patients in recovery from a major depressive episode, 76% (13 of 17) with residual symptoms relapsed within 15 months, compared with 25% (10 of 40) who completely recovered.16
Illness course. After a first major depressive episode, 26 patients with residual subsyndromal symptoms showed a more-severe, chronic illness compared with 70 asymptomatic patients:
- those with residual symptoms had more depression recurrences, with faster relapse and shorter intervals when they felt well
- subsequent depressive episodes occurred >3 times sooner
- well intervals between depressive episodes were 7 times shorter.17
The authors noted that “patients recovering from major depressive episodes with residual subsyndromal depression experience very rapid episode relapse and have strikingly more chronic future courses of illness that are characterized by early and more frequent episode relapses and recurrences.”18 Each major depression recurrence increases the risk of a successive episode.13,19,20
Treatment resistance. Over time, incomplete remission may contribute to treatment resistance,21 although this theory remains untested.
Social health costs
Residual depressive symptoms and impaired psychosocial, interpersonal, and occupational functioning are strongly correlated. In a study of patients who recovered from a major depressive episode, those with residual symptoms scored significantly worse on markers of social performance, relationships, and occupational functioning.6
Psychosocial costs. Compared with the general population, persons with subsyndromal depression have poorer health status and job functioning (as did Ms. M), and greater familial discord, financial strain, irritability, restricted activity, and number of days in bed.22 In 635 patients with chronic depression treated for 12 weeks with imipramine or sertraline:
- nonresponders scored lowest in psychosocial functioning
- responders scored in a mid-range
- remitters scored highest—as well or nearly as well as healthy controls did.23
Occupational costs. Depression leads to workplace absenteeism—twice the rate of nondepressed workers—and less effectiveness. Depressed persons are seven times more likely to be unemployed, employed part-time, or in jobs below their education levels, compared with community samples.24
Medical costs of depressed persons average twice those of the nondepressed,25 and depressed persons use three times as many health care services.26 Depression may increase the risk of:
- cardiac morbidity and mortality
- poor glycemic control in diabetic patients
- HIV progression
- cerebrovascular events
- and overall mortality.
Depression is associated with worse outcomes after myocardial infarction and among nursing home patients, stroke patients, and those with cancer or HIV infection.21
How to improve remission rates
To improve remission rates, we recommend that you follow a rational treatment progression and observe established guidelines, as described in the follow-up report on Ms. M:
Case continued: Part way there. Back at the psychiatrist’s office for 30-day medication monitoring, Ms. M reports that increasing her antidepressant has worked—no more crying in meetings or feeling down on herself. She even sleeps better. Her HRSD score is now 10, indicating improvement, though with some residual symptoms.
When the psychiatrist asks about her job performance, Ms. M is surprised to learn that her fatigue and disorganized thoughts might be lingering features of depression. She said she thought she just wasn’t trying hard enough.
Following practice guidelines,27 the psychiatrist increases fluoxetine to 60 mg/d. This higher dosage remains less than the maximum recommended 80 mg/d, and Ms. M has shown partial improvement with fluoxetine.
Patients being treated for depression need adequate follow-up to ensure they are improving. As with Ms. M, encourage patients to describe residual symptoms and functional domains that remain suboptimal. Educate them about the importance of taking antidepressants as prescribed ( Box). If poor response continues, address possible nonadherence.
Use objective assessments. Global, haphazard, or subjective assessments of patient progress can miss important ongoing depressive symptoms. We recommend using patient- or clinician-rated symptom scales to rapidly identify and monitor residual symptoms ( Table 3).
You may wish to design your own questionnaires to elicit easily missed data or symptoms particular to a certain treatment—such as common side effects of the medication a patient is taking. Nurses or other providers in a busy practice can help assess patients between or before doctor visits.
Keep in mind that the common practice of defining treatment response as a 50% reduction in HRSD scores7,8 leaves many patients with residual depressive symptoms. For example, an HRSD score reduction from 32 to 16 would signify treatment response, but this patient would remain quite depressed.
- Full remission from depression is the treatment goal, and any lesser outcome requires further attention
- Finding the proper medication may require trial and error, and several weeks may pass before a drug’s therapeutic effect occurs
- Continuing to take the medication as prescribed is important to achieving remission
- Medication may have predictable side effects
Table 3
Useful scales to identify and monitor depressive symptoms
| Scale | Administration | Features |
|---|---|---|
| Hamilton Rating Scale for Depression (21- or 17-item HRSD versions) | 15 to 20 minutes, clinician-rated | Focuses on somatic symptoms, excellent reliability, often used to evaluate response to medications |
| Beck Depression Inventory (BDI versions I or II) | 5 to 10 minutes, self-administered | Focuses on behavioral and cognitive elements (somatic symptoms added to BDI-II), good for measuring depression severity, not for depression screening |
| Zung Self-Rating Depression Scale | 5 to 10 minutes, self-administered | Good for screening, not studied as extensively as Hamilton and Beck scales |
| Montgomery-Asberg Depression Rating Scale | 5 to 10 minutes, clinician-rated | 10 items, often used in research |
A Stepwise approach
An expert panel recommends a stepwise approach for patients who respond inadequately to initial antidepressant therapy (Algorithm).27
Re-evaluate the diagnosis. Patients with bipolar disorder or comorbid medical or psychiatric disorders may need medications other than antidepressants. Address concomitant substance abuse, which may interfere with depression treatment. Also exclude or appropriately treat depressive symptoms associated with general medical conditions, such as hypothyroidism.
Optimize dosages. Consider increasing medication dosages as needed until limited by side effects or the drug’s safety profile. Before exceeding an FDA-recommended dosage (Table 4), obtain the patient’s informed consent and document this discussion in the chart.
Consider augmenting or switching. For patients who continue to show partial response, consider combining the initial medication with another antidepressant or augment with another agent, such as lithium, stimulants, thyroid hormone, or even atypical antipsychotics. For patients with no response to optimal dosages of the initial medication after 3 to 4 weeks, try switching to another antidepressant—not necessarily in a different class. One switch within the same class is reasonable.
Some authors emphasize the choice of antidepressant in attaining remission. Although no antidepressant is clearly more efficacious than another, those with fewer side effects (such as selective serotonin reuptake inhibitors vs. tricyclics) may improve adherence.
Numerous trials have shown higher remission rates with serotonin/norepinephrine reuptake inhibitors such as venlafaxine or duloxetine than with other antidepressants.28-30 This evidence is not universally accepted, however.31 Depressive illness probably has a heterogeneous biology, and with greater understanding we may eventually tailor treatment to individual patients’ needs.
Consider psychotherapy or ECT. Patients who do not achieve remission with medication may be candidates for combined treatment with psychotherapy or electroconvulsive therapy (ECT). Life issues—such as family or work stressors—may need to be addressed along with depressive symptoms.
Algorithm A stepwise approach to remission when antidepressant therapy falls short
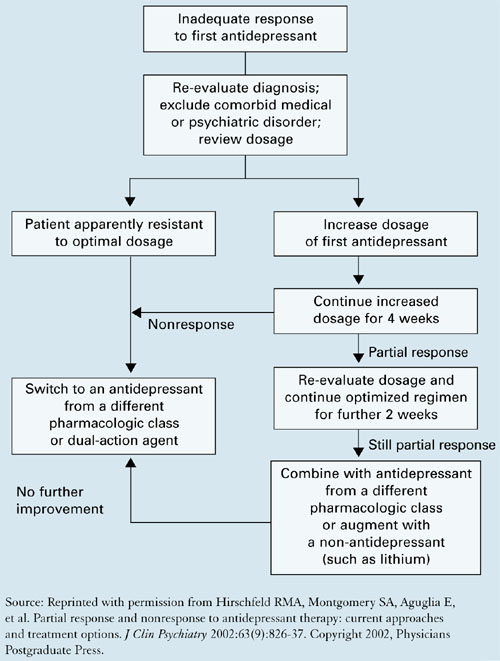
Table 4
Using common antidepressants for adults with major depression
| FDA-approved maximum dosage* | |||
|---|---|---|---|
| Antidepressant mg/d | mg/d | (mg/kg/d) | Common side effects at maximum dosage* |
| SSRIs | |||
| Citalopram | 80 | (1.0) | Nausea, dry mouth, somnolence |
| Escitalopram | 20 | Nausea, delayed ejaculation, insomnia | |
| Fluoxetine | 80 | (1.33) | Nausea, headache, insomnia |
| Paroxetine | 50 | (0.83) | Nausea, somnolence, headache |
| Sertraline | 200 | (3.33) | Nausea, headache, insomnia |
| SNRIs | |||
| Duloxetine | 120 | Nausea, dry mouth, fatigue | |
| Venlafaxine | 375 | (6.25) | Nausea, somnolence, dry mouth |
| Tricyclics | |||
| Amitriptyline | 300 | (5.0) | Drowsiness, dry mouth, dizziness |
| Desipramine | 300 | (5.0) | Same as above |
| Imipramine | 300 | Same as above | |
| Nortriptyline | 200 | (1.67) | Same as above |
| Others | |||
| Bupropion | 450 | (7.5) | Insomnia, dry mouth, headache |
| Mirtazapine | 45 | (0.75) | Somnolence, dry mouth, increased appetite |
| SSRIs: selective serotonin reuptake inhibitors | |||
| SNRIs: serotonin-norepinephrine reuptake inhibitors | |||
| * Informed consent discussion and documentation is recommended for dosages that exceed FDA-approved maximums. | |||
| Sources: Food and Drug Administration, and Kaplan HI, Sadock B. Kaplan & Sadock’s synopsis of psychiatry (9th ed). Philadelphia: Lippincott Williams & Wilkins, 2002. | |||
Case: monitoring after remission
Ms. M feels back to normal 2 weeks after starting fluoxetine at 60 mg/d. She experienced some transient nausea and headache at this dosage but did not stop the medication because her psychiatrist had told her these side effects might occur.
Ms. M also agrees to short-term psychotherapy to address self-esteem issues that may have contributed to her depressive episode. She soon files the mountain of papers on her desk and corrects erroneous financial statements she has made. Her supervisor is relieved—and so is she.
The psychiatrist schedules monthly medication monitoring and plans to gradually reduce the fluoxetine dosage if depressive symptoms remain in remission for 6 months. Because Ms. M had no past depressive episodes, the medication trial may not need to be extended past 6 months.
Related resources
- McManamy J. McMan’s Depression and Bipolar Web. Treating to remission. http://www.mcmanweb.com/article-181.htm.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (2nd ed.). http://www.psych.org/psych_pract/treatg/pg/Depression2e.book.cfm.
- Stahl S. Essential psychopharmacology: Neuroscientific basis and practical applications (2nd ed.). Cambridge, UK: Cambridge University Presss, 2004, chapters 5-7.
Drug brand names
- Amitriptyline • Elavil, Endep
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Van Rhoads reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Gelenberg receives research grants from Pfizer Inc. and Novartis Pharmaceuticals Corp. He is a consultant or a speaker for Eli Lilly and Co., Pfizer Inc., Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Wyeth, GlaxoSmithKline, and Cyberonics.
1. Agency for Health Care Policy and Research. Clinical practice guideline, number 5: Depression in primary care: vol. 2. Treatment of major depression. AHCPR publication 93-0551. Rockville, MD: US Department of Health and Human Services, 1993.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. Am J Psychiatry 2000;157(suppl 4):1-45.
3. Hirschfeld RM, Keller MB, Panico BS, et al. The National Depressive and Manic-Depressive Association consensus statement on the undertreatment of depression. JAMA 1997;277:333-40.
4. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: Report of the Texas Consensus Conference Panel on medication treatment of major depressive disorder. J Clin Psychiatry 1999;60:142-56.
5. Nierenberg AA, Keefe BR, Leslie VA, et al. Residual symptoms in depressed patients who respond acutely to fluoxetine. J Clin Psychiatry 1999;60:221-5.
6. Kennedy N, Paykel ES. Residual symptoms at remission from depression: impact on long-term outcome. J Affect Disord 2004;80(2-3):135-44.
7. Rush AJ, Kraemer HC, Sackeim HA, et al. Report by the ACNP Task Force on Response and Remission in Major Depressive Disorder. Submitted for publication.
8. Keller M. Past, present, and future directions for defining optimal treatment outcome in depression: remission and beyond. JAMA 2003;289(23):3152-60.
9. Nutting P, Rost K, Diceinson M, et al. Barriers to initiating depression treatment in primary care practice. J Gen Intern Med 2002;17:103-11.
10. Fava GA, Rafanelli C, Grandi S, et al. Letter to the editor. Arch Gen Psychiatry 1999;56:765.-
11. Ramana R, Paykel ES, Cooper Z, et al. Remission and relapse in major depression: a two year prospective follow-up study. Psychol Med 1995;25(6):1171-80.
12. Judd LL, Akiskal HS, Maser JD, et al. Major depressive disorder: a prospective study of residual subthreshold depressive symptoms as a predictor of rapid relapse. J Affect Disord 1999;50:97-108.
13. Pintor L, Gastó G, Navarro V, et al. Relapse of major depression after complete and partial remission during a 2-year follow-up. J Affect Disord 2003;73:237-44.
14. Lin E, Katon W, Von Korff M, et al. Relapse of depression in primary care: rate and clinical predictors. Arch Fam Med 1998;7(5):443-9.
15. Cornwall P, Scott J. Partial remission in depressive disorders. Acta Psychiatr Scand 1997;95(4):265-71.
16. Paykel ES, Ramana R, Cooper Z, et al. Residual symptoms after partial remission: an important outcome in depression. Psychol Med 1995;25(6):1171-80.
17. Judd LL, Paulus MJ, Schettler PJ, et al. Does incomplete recovery from first lifetime major depressive episode herald a chronic course of illness? Am J Psychiatry 2000;157(9):1501-4.
18. Judd LL, Paulus MP, Zeller P. The role of residual subthreshold depressive symptoms in early episode relapse in unipolar major depressive disorder. Arch Gen Psychiatry 1999;56(8):764-5.
19. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry 2000;157(2):229-33.
20. Kennedy N, Paykel ES. Residual symptoms at remission from depression: impact on long-term outcome. J Affect Disord 2004;80(2-3):135-44.
21. Zajecka JM. Treating depression to remission. J Clin Psychiatry 2003;64(suppl 15):7-12.
22. Judd LL, Paulus MP, Wells KB, Rapaport MH. Socioeconomic burden of subsyndromal depressive symptoms and major depressionin a sample of the general population. Am J Psychiatry 1996;153(11):1411-7.
23. Miller IW, Keitner GI, Schatzberg AF, et al. The treatment of chronic depression, part 3: psychosocial functioning before and after treatment with sertraline or imipramine. J Clin Psychiatry 1998;59(11):608-19.
24. Druss B, Schlesinger M, Allen H. Depressive symptoms, satisfaction with health care, and 2-year work outcomes in an employed population. Am J Psychiatry 2001;158(5):731-4.
25. Simon G, Ormel J, VonKorff M, et al. Health care costs associated with depressive and anxiety disorders in primary care. Am J Psychiatry 1995;152(3):352-7.
26. Katon W, Schulberg H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14(4):237-47.
27. Hirschfeld RMA, Montgomery SA, Aguglia E, et al. Partial response and nonresponse to antidepressant therapy: current approaches and treatment options. J Clin Psychiatry 2002;63(9):826-37.
28. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
29. Kelsey J, Entsuah R. Venlafaxine offers significant therapeutic benefits over existing SSRI treatments irrespective of the patient’s depressive duration (poster presentation). Montreal: Collegium Internationale Neuropsychopharmacologicum, June 2002.
30. Thase ME. Effectiveness of antidepressants: comparative remission rates. J Clin Psychiatry 2003;64(suppl 2):3-7.
31. Shelton C. Long-term management of major depressive disorder: are differences among antidepressant treatments meaningful? J Clin Psychiatry 2004;65(suppl 17):29-33.
Remission is considered the standard of treatment for major depression,1-4 but many patients fall short of this goal:
- 25% to 50% of those who respond to treatment have residual symptoms5
- 60% to 70% respond to treatment, but only 20% to 40% achieve remission.6
We offer practical, evidence-based suggestions and resources to help you take more of your patients beyond response to remission and then to recovery (Table 1).7,8
Table 1
Outcomes in depression: Defining the 4 ‘R’s
| Outcome | Definition | Comment |
|---|---|---|
| Response | Clinically significant reduction of symptoms | 50% reduction in symptoms on psychometric scales may leave severely depressed patients with disabling symptoms |
| Remission | Depression resolves completely or nearly completely, with return to baseline function | Score of ≤7 on the HRSD used in many studies; ACNP Task Force defines remission by 9 core depression symptoms in DSM-IV-TR |
| Recovery | Remission lasts for extended time; signifies end of a major depressive episode | ACNP Task Force defines recovery as 4 months of remission |
| Relapse | Return to full symptoms during remission but before recovery; signifies re-emergence of current depressive episode | Residual symptoms during response or remission greatly increase chances of relapse |
| HRSD: Hamilton Rating Scale for Depression | ||
| ACNP: American College of Neuropsychopharmacology | ||
| DSM-IV-TR: Diagnostic and statistical manual of mental disorders, 4th ed., text rev. | ||
| Source: References 7, 8 | ||
Case report: missing the target
Ms. M, age 32, develops depressive symptoms after taking on several new projects in her work as an accountant. At first she notices difficulty falling asleep at night and that she seems tired all day. Typically efficient and neat, she finds herself absently staring at her computer screen while a messy pile of unfinished paperwork accumulates on her desk.
She begins chastising herself for falling behind, yet she feels she will never catch up. When she unexpectedly bursts out sobbing in a board meeting, she knows she needs help.
Ms. M reports that she has been taking fluoxetine, 20 mg/d, for 4 weeks as her primary care physician prescribed, with no improvement. She has no history of depression or other psychiatric illness, is taking no other medication, and has no medical illnesses. Her brother has a history of bipolar disorder.
Initial diagnostic workup includes laboratory tests such as thyroid stimulating hormone, vitamin B12, and folate. All values are within normal limits. Her Hamilton Rating Scale for Depression (HRSD) score is 17, indicating moderate depression.
The psychiatrist increases fluoxetine to 40 mg/d, and after about 3 weeks Ms. M starts feeling better. Her hopelessness lifts, she is more engaged, and her sleep improves, yet she continues to feel sluggish and dazed. Her financial reports contain uncharacteristic errors, and her pace is noticeably slow. Twice her supervisor approaches her about substandard work, then a week later warns that she will lose her job unless she improves.
Barriers to remission. Patient, provider, and health care system barriers prevent patients with major depression from achieving remission (Table 2).3 Patients may feel better with antidepressant therapy but do not recognize and report residual depressive symptoms, such as Ms. M’s fatigue and substandard job performance.
Clinicians also play a role in depression undertreatment. For example, in a study of 239 patients with ≥ 5 depression symptoms,9 28% did not receive treatment consistent with depression management guidelines.
Fava et al10 suggested the following reasons for depression undertreatment: “… clinicians have partial therapeutic targets, neglect residual symptoms, and equate therapeutic response with full remission.” Others have found that physicians may underdose medications or fail to plan treatment in clear phases.11
Table 2
Barriers to remission during depression treatment
| Who and what | Behavior and system problems |
|---|---|
| Patients | Nonadherence to treatment, underreporting of symptoms, satisfaction with suboptimal outcomes, failure to recognize depressive symptoms, underestimating depression severity, limited access to care, reluctance to see a mental health specialist |
| Providers | Medication under-dosing, inadequate treatment duration, inaccurate diagnosis, failure to recognize residual symptoms, limited training in interpersonal skills, inadequate time to evaluate and treat depression, failure to consider psychotherapeutic approaches |
| Health care systems | Limited therapeutic choices, limited number of mental health care visits, restricted access to providers |
| Source: Reference 3 | |
Relapse risk
Increased risk of relapse is perhaps the greatest cost of undertreated major depression. Patients with residual subsyndromal depressive symptoms relapse five times faster than patients in full remission.12
Residual symptoms may be a more powerful relapse predictor than number of past depressive episodes:13
- Chronic mood symptoms for ≥ 2 years double the relapse risk.14
- 50% to 80% of patients in partial remission relapse.15
In a study of patients in recovery from a major depressive episode, 76% (13 of 17) with residual symptoms relapsed within 15 months, compared with 25% (10 of 40) who completely recovered.16
Illness course. After a first major depressive episode, 26 patients with residual subsyndromal symptoms showed a more-severe, chronic illness compared with 70 asymptomatic patients:
- those with residual symptoms had more depression recurrences, with faster relapse and shorter intervals when they felt well
- subsequent depressive episodes occurred >3 times sooner
- well intervals between depressive episodes were 7 times shorter.17
The authors noted that “patients recovering from major depressive episodes with residual subsyndromal depression experience very rapid episode relapse and have strikingly more chronic future courses of illness that are characterized by early and more frequent episode relapses and recurrences.”18 Each major depression recurrence increases the risk of a successive episode.13,19,20
Treatment resistance. Over time, incomplete remission may contribute to treatment resistance,21 although this theory remains untested.
Social health costs
Residual depressive symptoms and impaired psychosocial, interpersonal, and occupational functioning are strongly correlated. In a study of patients who recovered from a major depressive episode, those with residual symptoms scored significantly worse on markers of social performance, relationships, and occupational functioning.6
Psychosocial costs. Compared with the general population, persons with subsyndromal depression have poorer health status and job functioning (as did Ms. M), and greater familial discord, financial strain, irritability, restricted activity, and number of days in bed.22 In 635 patients with chronic depression treated for 12 weeks with imipramine or sertraline:
- nonresponders scored lowest in psychosocial functioning
- responders scored in a mid-range
- remitters scored highest—as well or nearly as well as healthy controls did.23
Occupational costs. Depression leads to workplace absenteeism—twice the rate of nondepressed workers—and less effectiveness. Depressed persons are seven times more likely to be unemployed, employed part-time, or in jobs below their education levels, compared with community samples.24
Medical costs of depressed persons average twice those of the nondepressed,25 and depressed persons use three times as many health care services.26 Depression may increase the risk of:
- cardiac morbidity and mortality
- poor glycemic control in diabetic patients
- HIV progression
- cerebrovascular events
- and overall mortality.
Depression is associated with worse outcomes after myocardial infarction and among nursing home patients, stroke patients, and those with cancer or HIV infection.21
How to improve remission rates
To improve remission rates, we recommend that you follow a rational treatment progression and observe established guidelines, as described in the follow-up report on Ms. M:
Case continued: Part way there. Back at the psychiatrist’s office for 30-day medication monitoring, Ms. M reports that increasing her antidepressant has worked—no more crying in meetings or feeling down on herself. She even sleeps better. Her HRSD score is now 10, indicating improvement, though with some residual symptoms.
When the psychiatrist asks about her job performance, Ms. M is surprised to learn that her fatigue and disorganized thoughts might be lingering features of depression. She said she thought she just wasn’t trying hard enough.
Following practice guidelines,27 the psychiatrist increases fluoxetine to 60 mg/d. This higher dosage remains less than the maximum recommended 80 mg/d, and Ms. M has shown partial improvement with fluoxetine.
Patients being treated for depression need adequate follow-up to ensure they are improving. As with Ms. M, encourage patients to describe residual symptoms and functional domains that remain suboptimal. Educate them about the importance of taking antidepressants as prescribed ( Box). If poor response continues, address possible nonadherence.
Use objective assessments. Global, haphazard, or subjective assessments of patient progress can miss important ongoing depressive symptoms. We recommend using patient- or clinician-rated symptom scales to rapidly identify and monitor residual symptoms ( Table 3).
You may wish to design your own questionnaires to elicit easily missed data or symptoms particular to a certain treatment—such as common side effects of the medication a patient is taking. Nurses or other providers in a busy practice can help assess patients between or before doctor visits.
Keep in mind that the common practice of defining treatment response as a 50% reduction in HRSD scores7,8 leaves many patients with residual depressive symptoms. For example, an HRSD score reduction from 32 to 16 would signify treatment response, but this patient would remain quite depressed.
- Full remission from depression is the treatment goal, and any lesser outcome requires further attention
- Finding the proper medication may require trial and error, and several weeks may pass before a drug’s therapeutic effect occurs
- Continuing to take the medication as prescribed is important to achieving remission
- Medication may have predictable side effects
Table 3
Useful scales to identify and monitor depressive symptoms
| Scale | Administration | Features |
|---|---|---|
| Hamilton Rating Scale for Depression (21- or 17-item HRSD versions) | 15 to 20 minutes, clinician-rated | Focuses on somatic symptoms, excellent reliability, often used to evaluate response to medications |
| Beck Depression Inventory (BDI versions I or II) | 5 to 10 minutes, self-administered | Focuses on behavioral and cognitive elements (somatic symptoms added to BDI-II), good for measuring depression severity, not for depression screening |
| Zung Self-Rating Depression Scale | 5 to 10 minutes, self-administered | Good for screening, not studied as extensively as Hamilton and Beck scales |
| Montgomery-Asberg Depression Rating Scale | 5 to 10 minutes, clinician-rated | 10 items, often used in research |
A Stepwise approach
An expert panel recommends a stepwise approach for patients who respond inadequately to initial antidepressant therapy (Algorithm).27
Re-evaluate the diagnosis. Patients with bipolar disorder or comorbid medical or psychiatric disorders may need medications other than antidepressants. Address concomitant substance abuse, which may interfere with depression treatment. Also exclude or appropriately treat depressive symptoms associated with general medical conditions, such as hypothyroidism.
Optimize dosages. Consider increasing medication dosages as needed until limited by side effects or the drug’s safety profile. Before exceeding an FDA-recommended dosage (Table 4), obtain the patient’s informed consent and document this discussion in the chart.
Consider augmenting or switching. For patients who continue to show partial response, consider combining the initial medication with another antidepressant or augment with another agent, such as lithium, stimulants, thyroid hormone, or even atypical antipsychotics. For patients with no response to optimal dosages of the initial medication after 3 to 4 weeks, try switching to another antidepressant—not necessarily in a different class. One switch within the same class is reasonable.
Some authors emphasize the choice of antidepressant in attaining remission. Although no antidepressant is clearly more efficacious than another, those with fewer side effects (such as selective serotonin reuptake inhibitors vs. tricyclics) may improve adherence.
Numerous trials have shown higher remission rates with serotonin/norepinephrine reuptake inhibitors such as venlafaxine or duloxetine than with other antidepressants.28-30 This evidence is not universally accepted, however.31 Depressive illness probably has a heterogeneous biology, and with greater understanding we may eventually tailor treatment to individual patients’ needs.
Consider psychotherapy or ECT. Patients who do not achieve remission with medication may be candidates for combined treatment with psychotherapy or electroconvulsive therapy (ECT). Life issues—such as family or work stressors—may need to be addressed along with depressive symptoms.
Algorithm A stepwise approach to remission when antidepressant therapy falls short
Table 4
Using common antidepressants for adults with major depression
| FDA-approved maximum dosage* | |||
|---|---|---|---|
| Antidepressant mg/d | mg/d | (mg/kg/d) | Common side effects at maximum dosage* |
| SSRIs | |||
| Citalopram | 80 | (1.0) | Nausea, dry mouth, somnolence |
| Escitalopram | 20 | Nausea, delayed ejaculation, insomnia | |
| Fluoxetine | 80 | (1.33) | Nausea, headache, insomnia |
| Paroxetine | 50 | (0.83) | Nausea, somnolence, headache |
| Sertraline | 200 | (3.33) | Nausea, headache, insomnia |
| SNRIs | |||
| Duloxetine | 120 | Nausea, dry mouth, fatigue | |
| Venlafaxine | 375 | (6.25) | Nausea, somnolence, dry mouth |
| Tricyclics | |||
| Amitriptyline | 300 | (5.0) | Drowsiness, dry mouth, dizziness |
| Desipramine | 300 | (5.0) | Same as above |
| Imipramine | 300 | Same as above | |
| Nortriptyline | 200 | (1.67) | Same as above |
| Others | |||
| Bupropion | 450 | (7.5) | Insomnia, dry mouth, headache |
| Mirtazapine | 45 | (0.75) | Somnolence, dry mouth, increased appetite |
| SSRIs: selective serotonin reuptake inhibitors | |||
| SNRIs: serotonin-norepinephrine reuptake inhibitors | |||
| * Informed consent discussion and documentation is recommended for dosages that exceed FDA-approved maximums. | |||
| Sources: Food and Drug Administration, and Kaplan HI, Sadock B. Kaplan & Sadock’s synopsis of psychiatry (9th ed). Philadelphia: Lippincott Williams & Wilkins, 2002. | |||
Case: monitoring after remission
Ms. M feels back to normal 2 weeks after starting fluoxetine at 60 mg/d. She experienced some transient nausea and headache at this dosage but did not stop the medication because her psychiatrist had told her these side effects might occur.
Ms. M also agrees to short-term psychotherapy to address self-esteem issues that may have contributed to her depressive episode. She soon files the mountain of papers on her desk and corrects erroneous financial statements she has made. Her supervisor is relieved—and so is she.
The psychiatrist schedules monthly medication monitoring and plans to gradually reduce the fluoxetine dosage if depressive symptoms remain in remission for 6 months. Because Ms. M had no past depressive episodes, the medication trial may not need to be extended past 6 months.
Related resources
- McManamy J. McMan’s Depression and Bipolar Web. Treating to remission. http://www.mcmanweb.com/article-181.htm.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (2nd ed.). http://www.psych.org/psych_pract/treatg/pg/Depression2e.book.cfm.
- Stahl S. Essential psychopharmacology: Neuroscientific basis and practical applications (2nd ed.). Cambridge, UK: Cambridge University Presss, 2004, chapters 5-7.
Drug brand names
- Amitriptyline • Elavil, Endep
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Van Rhoads reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Gelenberg receives research grants from Pfizer Inc. and Novartis Pharmaceuticals Corp. He is a consultant or a speaker for Eli Lilly and Co., Pfizer Inc., Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Wyeth, GlaxoSmithKline, and Cyberonics.
Remission is considered the standard of treatment for major depression,1-4 but many patients fall short of this goal:
- 25% to 50% of those who respond to treatment have residual symptoms5
- 60% to 70% respond to treatment, but only 20% to 40% achieve remission.6
We offer practical, evidence-based suggestions and resources to help you take more of your patients beyond response to remission and then to recovery (Table 1).7,8
Table 1
Outcomes in depression: Defining the 4 ‘R’s
| Outcome | Definition | Comment |
|---|---|---|
| Response | Clinically significant reduction of symptoms | 50% reduction in symptoms on psychometric scales may leave severely depressed patients with disabling symptoms |
| Remission | Depression resolves completely or nearly completely, with return to baseline function | Score of ≤7 on the HRSD used in many studies; ACNP Task Force defines remission by 9 core depression symptoms in DSM-IV-TR |
| Recovery | Remission lasts for extended time; signifies end of a major depressive episode | ACNP Task Force defines recovery as 4 months of remission |
| Relapse | Return to full symptoms during remission but before recovery; signifies re-emergence of current depressive episode | Residual symptoms during response or remission greatly increase chances of relapse |
| HRSD: Hamilton Rating Scale for Depression | ||
| ACNP: American College of Neuropsychopharmacology | ||
| DSM-IV-TR: Diagnostic and statistical manual of mental disorders, 4th ed., text rev. | ||
| Source: References 7, 8 | ||
Case report: missing the target
Ms. M, age 32, develops depressive symptoms after taking on several new projects in her work as an accountant. At first she notices difficulty falling asleep at night and that she seems tired all day. Typically efficient and neat, she finds herself absently staring at her computer screen while a messy pile of unfinished paperwork accumulates on her desk.
She begins chastising herself for falling behind, yet she feels she will never catch up. When she unexpectedly bursts out sobbing in a board meeting, she knows she needs help.
Ms. M reports that she has been taking fluoxetine, 20 mg/d, for 4 weeks as her primary care physician prescribed, with no improvement. She has no history of depression or other psychiatric illness, is taking no other medication, and has no medical illnesses. Her brother has a history of bipolar disorder.
Initial diagnostic workup includes laboratory tests such as thyroid stimulating hormone, vitamin B12, and folate. All values are within normal limits. Her Hamilton Rating Scale for Depression (HRSD) score is 17, indicating moderate depression.
The psychiatrist increases fluoxetine to 40 mg/d, and after about 3 weeks Ms. M starts feeling better. Her hopelessness lifts, she is more engaged, and her sleep improves, yet she continues to feel sluggish and dazed. Her financial reports contain uncharacteristic errors, and her pace is noticeably slow. Twice her supervisor approaches her about substandard work, then a week later warns that she will lose her job unless she improves.
Barriers to remission. Patient, provider, and health care system barriers prevent patients with major depression from achieving remission (Table 2).3 Patients may feel better with antidepressant therapy but do not recognize and report residual depressive symptoms, such as Ms. M’s fatigue and substandard job performance.
Clinicians also play a role in depression undertreatment. For example, in a study of 239 patients with ≥ 5 depression symptoms,9 28% did not receive treatment consistent with depression management guidelines.
Fava et al10 suggested the following reasons for depression undertreatment: “… clinicians have partial therapeutic targets, neglect residual symptoms, and equate therapeutic response with full remission.” Others have found that physicians may underdose medications or fail to plan treatment in clear phases.11
Table 2
Barriers to remission during depression treatment
| Who and what | Behavior and system problems |
|---|---|
| Patients | Nonadherence to treatment, underreporting of symptoms, satisfaction with suboptimal outcomes, failure to recognize depressive symptoms, underestimating depression severity, limited access to care, reluctance to see a mental health specialist |
| Providers | Medication under-dosing, inadequate treatment duration, inaccurate diagnosis, failure to recognize residual symptoms, limited training in interpersonal skills, inadequate time to evaluate and treat depression, failure to consider psychotherapeutic approaches |
| Health care systems | Limited therapeutic choices, limited number of mental health care visits, restricted access to providers |
| Source: Reference 3 | |
Relapse risk
Increased risk of relapse is perhaps the greatest cost of undertreated major depression. Patients with residual subsyndromal depressive symptoms relapse five times faster than patients in full remission.12
Residual symptoms may be a more powerful relapse predictor than number of past depressive episodes:13
- Chronic mood symptoms for ≥ 2 years double the relapse risk.14
- 50% to 80% of patients in partial remission relapse.15
In a study of patients in recovery from a major depressive episode, 76% (13 of 17) with residual symptoms relapsed within 15 months, compared with 25% (10 of 40) who completely recovered.16
Illness course. After a first major depressive episode, 26 patients with residual subsyndromal symptoms showed a more-severe, chronic illness compared with 70 asymptomatic patients:
- those with residual symptoms had more depression recurrences, with faster relapse and shorter intervals when they felt well
- subsequent depressive episodes occurred >3 times sooner
- well intervals between depressive episodes were 7 times shorter.17
The authors noted that “patients recovering from major depressive episodes with residual subsyndromal depression experience very rapid episode relapse and have strikingly more chronic future courses of illness that are characterized by early and more frequent episode relapses and recurrences.”18 Each major depression recurrence increases the risk of a successive episode.13,19,20
Treatment resistance. Over time, incomplete remission may contribute to treatment resistance,21 although this theory remains untested.
Social health costs
Residual depressive symptoms and impaired psychosocial, interpersonal, and occupational functioning are strongly correlated. In a study of patients who recovered from a major depressive episode, those with residual symptoms scored significantly worse on markers of social performance, relationships, and occupational functioning.6
Psychosocial costs. Compared with the general population, persons with subsyndromal depression have poorer health status and job functioning (as did Ms. M), and greater familial discord, financial strain, irritability, restricted activity, and number of days in bed.22 In 635 patients with chronic depression treated for 12 weeks with imipramine or sertraline:
- nonresponders scored lowest in psychosocial functioning
- responders scored in a mid-range
- remitters scored highest—as well or nearly as well as healthy controls did.23
Occupational costs. Depression leads to workplace absenteeism—twice the rate of nondepressed workers—and less effectiveness. Depressed persons are seven times more likely to be unemployed, employed part-time, or in jobs below their education levels, compared with community samples.24
Medical costs of depressed persons average twice those of the nondepressed,25 and depressed persons use three times as many health care services.26 Depression may increase the risk of:
- cardiac morbidity and mortality
- poor glycemic control in diabetic patients
- HIV progression
- cerebrovascular events
- and overall mortality.
Depression is associated with worse outcomes after myocardial infarction and among nursing home patients, stroke patients, and those with cancer or HIV infection.21
How to improve remission rates
To improve remission rates, we recommend that you follow a rational treatment progression and observe established guidelines, as described in the follow-up report on Ms. M:
Case continued: Part way there. Back at the psychiatrist’s office for 30-day medication monitoring, Ms. M reports that increasing her antidepressant has worked—no more crying in meetings or feeling down on herself. She even sleeps better. Her HRSD score is now 10, indicating improvement, though with some residual symptoms.
When the psychiatrist asks about her job performance, Ms. M is surprised to learn that her fatigue and disorganized thoughts might be lingering features of depression. She said she thought she just wasn’t trying hard enough.
Following practice guidelines,27 the psychiatrist increases fluoxetine to 60 mg/d. This higher dosage remains less than the maximum recommended 80 mg/d, and Ms. M has shown partial improvement with fluoxetine.
Patients being treated for depression need adequate follow-up to ensure they are improving. As with Ms. M, encourage patients to describe residual symptoms and functional domains that remain suboptimal. Educate them about the importance of taking antidepressants as prescribed ( Box). If poor response continues, address possible nonadherence.
Use objective assessments. Global, haphazard, or subjective assessments of patient progress can miss important ongoing depressive symptoms. We recommend using patient- or clinician-rated symptom scales to rapidly identify and monitor residual symptoms ( Table 3).
You may wish to design your own questionnaires to elicit easily missed data or symptoms particular to a certain treatment—such as common side effects of the medication a patient is taking. Nurses or other providers in a busy practice can help assess patients between or before doctor visits.
Keep in mind that the common practice of defining treatment response as a 50% reduction in HRSD scores7,8 leaves many patients with residual depressive symptoms. For example, an HRSD score reduction from 32 to 16 would signify treatment response, but this patient would remain quite depressed.
- Full remission from depression is the treatment goal, and any lesser outcome requires further attention
- Finding the proper medication may require trial and error, and several weeks may pass before a drug’s therapeutic effect occurs
- Continuing to take the medication as prescribed is important to achieving remission
- Medication may have predictable side effects
Table 3
Useful scales to identify and monitor depressive symptoms
| Scale | Administration | Features |
|---|---|---|
| Hamilton Rating Scale for Depression (21- or 17-item HRSD versions) | 15 to 20 minutes, clinician-rated | Focuses on somatic symptoms, excellent reliability, often used to evaluate response to medications |
| Beck Depression Inventory (BDI versions I or II) | 5 to 10 minutes, self-administered | Focuses on behavioral and cognitive elements (somatic symptoms added to BDI-II), good for measuring depression severity, not for depression screening |
| Zung Self-Rating Depression Scale | 5 to 10 minutes, self-administered | Good for screening, not studied as extensively as Hamilton and Beck scales |
| Montgomery-Asberg Depression Rating Scale | 5 to 10 minutes, clinician-rated | 10 items, often used in research |
A Stepwise approach
An expert panel recommends a stepwise approach for patients who respond inadequately to initial antidepressant therapy (Algorithm).27
Re-evaluate the diagnosis. Patients with bipolar disorder or comorbid medical or psychiatric disorders may need medications other than antidepressants. Address concomitant substance abuse, which may interfere with depression treatment. Also exclude or appropriately treat depressive symptoms associated with general medical conditions, such as hypothyroidism.
Optimize dosages. Consider increasing medication dosages as needed until limited by side effects or the drug’s safety profile. Before exceeding an FDA-recommended dosage (Table 4), obtain the patient’s informed consent and document this discussion in the chart.
Consider augmenting or switching. For patients who continue to show partial response, consider combining the initial medication with another antidepressant or augment with another agent, such as lithium, stimulants, thyroid hormone, or even atypical antipsychotics. For patients with no response to optimal dosages of the initial medication after 3 to 4 weeks, try switching to another antidepressant—not necessarily in a different class. One switch within the same class is reasonable.
Some authors emphasize the choice of antidepressant in attaining remission. Although no antidepressant is clearly more efficacious than another, those with fewer side effects (such as selective serotonin reuptake inhibitors vs. tricyclics) may improve adherence.
Numerous trials have shown higher remission rates with serotonin/norepinephrine reuptake inhibitors such as venlafaxine or duloxetine than with other antidepressants.28-30 This evidence is not universally accepted, however.31 Depressive illness probably has a heterogeneous biology, and with greater understanding we may eventually tailor treatment to individual patients’ needs.
Consider psychotherapy or ECT. Patients who do not achieve remission with medication may be candidates for combined treatment with psychotherapy or electroconvulsive therapy (ECT). Life issues—such as family or work stressors—may need to be addressed along with depressive symptoms.
Algorithm A stepwise approach to remission when antidepressant therapy falls short
Table 4
Using common antidepressants for adults with major depression
| FDA-approved maximum dosage* | |||
|---|---|---|---|
| Antidepressant mg/d | mg/d | (mg/kg/d) | Common side effects at maximum dosage* |
| SSRIs | |||
| Citalopram | 80 | (1.0) | Nausea, dry mouth, somnolence |
| Escitalopram | 20 | Nausea, delayed ejaculation, insomnia | |
| Fluoxetine | 80 | (1.33) | Nausea, headache, insomnia |
| Paroxetine | 50 | (0.83) | Nausea, somnolence, headache |
| Sertraline | 200 | (3.33) | Nausea, headache, insomnia |
| SNRIs | |||
| Duloxetine | 120 | Nausea, dry mouth, fatigue | |
| Venlafaxine | 375 | (6.25) | Nausea, somnolence, dry mouth |
| Tricyclics | |||
| Amitriptyline | 300 | (5.0) | Drowsiness, dry mouth, dizziness |
| Desipramine | 300 | (5.0) | Same as above |
| Imipramine | 300 | Same as above | |
| Nortriptyline | 200 | (1.67) | Same as above |
| Others | |||
| Bupropion | 450 | (7.5) | Insomnia, dry mouth, headache |
| Mirtazapine | 45 | (0.75) | Somnolence, dry mouth, increased appetite |
| SSRIs: selective serotonin reuptake inhibitors | |||
| SNRIs: serotonin-norepinephrine reuptake inhibitors | |||
| * Informed consent discussion and documentation is recommended for dosages that exceed FDA-approved maximums. | |||
| Sources: Food and Drug Administration, and Kaplan HI, Sadock B. Kaplan & Sadock’s synopsis of psychiatry (9th ed). Philadelphia: Lippincott Williams & Wilkins, 2002. | |||
Case: monitoring after remission
Ms. M feels back to normal 2 weeks after starting fluoxetine at 60 mg/d. She experienced some transient nausea and headache at this dosage but did not stop the medication because her psychiatrist had told her these side effects might occur.
Ms. M also agrees to short-term psychotherapy to address self-esteem issues that may have contributed to her depressive episode. She soon files the mountain of papers on her desk and corrects erroneous financial statements she has made. Her supervisor is relieved—and so is she.
The psychiatrist schedules monthly medication monitoring and plans to gradually reduce the fluoxetine dosage if depressive symptoms remain in remission for 6 months. Because Ms. M had no past depressive episodes, the medication trial may not need to be extended past 6 months.
Related resources
- McManamy J. McMan’s Depression and Bipolar Web. Treating to remission. http://www.mcmanweb.com/article-181.htm.
- American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder (2nd ed.). http://www.psych.org/psych_pract/treatg/pg/Depression2e.book.cfm.
- Stahl S. Essential psychopharmacology: Neuroscientific basis and practical applications (2nd ed.). Cambridge, UK: Cambridge University Presss, 2004, chapters 5-7.
Drug brand names
- Amitriptyline • Elavil, Endep
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Desipramine • Norpramin
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Imipramine • Tofranil
- Mirtazapine • Remeron
- Nortriptyline • Pamelor
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Van Rhoads reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Gelenberg receives research grants from Pfizer Inc. and Novartis Pharmaceuticals Corp. He is a consultant or a speaker for Eli Lilly and Co., Pfizer Inc., Bristol-Myers Squibb Co., AstraZeneca Pharmaceuticals, Wyeth, GlaxoSmithKline, and Cyberonics.
1. Agency for Health Care Policy and Research. Clinical practice guideline, number 5: Depression in primary care: vol. 2. Treatment of major depression. AHCPR publication 93-0551. Rockville, MD: US Department of Health and Human Services, 1993.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. Am J Psychiatry 2000;157(suppl 4):1-45.
3. Hirschfeld RM, Keller MB, Panico BS, et al. The National Depressive and Manic-Depressive Association consensus statement on the undertreatment of depression. JAMA 1997;277:333-40.
4. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: Report of the Texas Consensus Conference Panel on medication treatment of major depressive disorder. J Clin Psychiatry 1999;60:142-56.
5. Nierenberg AA, Keefe BR, Leslie VA, et al. Residual symptoms in depressed patients who respond acutely to fluoxetine. J Clin Psychiatry 1999;60:221-5.
6. Kennedy N, Paykel ES. Residual symptoms at remission from depression: impact on long-term outcome. J Affect Disord 2004;80(2-3):135-44.
7. Rush AJ, Kraemer HC, Sackeim HA, et al. Report by the ACNP Task Force on Response and Remission in Major Depressive Disorder. Submitted for publication.
8. Keller M. Past, present, and future directions for defining optimal treatment outcome in depression: remission and beyond. JAMA 2003;289(23):3152-60.
9. Nutting P, Rost K, Diceinson M, et al. Barriers to initiating depression treatment in primary care practice. J Gen Intern Med 2002;17:103-11.
10. Fava GA, Rafanelli C, Grandi S, et al. Letter to the editor. Arch Gen Psychiatry 1999;56:765.-
11. Ramana R, Paykel ES, Cooper Z, et al. Remission and relapse in major depression: a two year prospective follow-up study. Psychol Med 1995;25(6):1171-80.
12. Judd LL, Akiskal HS, Maser JD, et al. Major depressive disorder: a prospective study of residual subthreshold depressive symptoms as a predictor of rapid relapse. J Affect Disord 1999;50:97-108.
13. Pintor L, Gastó G, Navarro V, et al. Relapse of major depression after complete and partial remission during a 2-year follow-up. J Affect Disord 2003;73:237-44.
14. Lin E, Katon W, Von Korff M, et al. Relapse of depression in primary care: rate and clinical predictors. Arch Fam Med 1998;7(5):443-9.
15. Cornwall P, Scott J. Partial remission in depressive disorders. Acta Psychiatr Scand 1997;95(4):265-71.
16. Paykel ES, Ramana R, Cooper Z, et al. Residual symptoms after partial remission: an important outcome in depression. Psychol Med 1995;25(6):1171-80.
17. Judd LL, Paulus MJ, Schettler PJ, et al. Does incomplete recovery from first lifetime major depressive episode herald a chronic course of illness? Am J Psychiatry 2000;157(9):1501-4.
18. Judd LL, Paulus MP, Zeller P. The role of residual subthreshold depressive symptoms in early episode relapse in unipolar major depressive disorder. Arch Gen Psychiatry 1999;56(8):764-5.
19. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry 2000;157(2):229-33.
20. Kennedy N, Paykel ES. Residual symptoms at remission from depression: impact on long-term outcome. J Affect Disord 2004;80(2-3):135-44.
21. Zajecka JM. Treating depression to remission. J Clin Psychiatry 2003;64(suppl 15):7-12.
22. Judd LL, Paulus MP, Wells KB, Rapaport MH. Socioeconomic burden of subsyndromal depressive symptoms and major depressionin a sample of the general population. Am J Psychiatry 1996;153(11):1411-7.
23. Miller IW, Keitner GI, Schatzberg AF, et al. The treatment of chronic depression, part 3: psychosocial functioning before and after treatment with sertraline or imipramine. J Clin Psychiatry 1998;59(11):608-19.
24. Druss B, Schlesinger M, Allen H. Depressive symptoms, satisfaction with health care, and 2-year work outcomes in an employed population. Am J Psychiatry 2001;158(5):731-4.
25. Simon G, Ormel J, VonKorff M, et al. Health care costs associated with depressive and anxiety disorders in primary care. Am J Psychiatry 1995;152(3):352-7.
26. Katon W, Schulberg H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14(4):237-47.
27. Hirschfeld RMA, Montgomery SA, Aguglia E, et al. Partial response and nonresponse to antidepressant therapy: current approaches and treatment options. J Clin Psychiatry 2002;63(9):826-37.
28. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
29. Kelsey J, Entsuah R. Venlafaxine offers significant therapeutic benefits over existing SSRI treatments irrespective of the patient’s depressive duration (poster presentation). Montreal: Collegium Internationale Neuropsychopharmacologicum, June 2002.
30. Thase ME. Effectiveness of antidepressants: comparative remission rates. J Clin Psychiatry 2003;64(suppl 2):3-7.
31. Shelton C. Long-term management of major depressive disorder: are differences among antidepressant treatments meaningful? J Clin Psychiatry 2004;65(suppl 17):29-33.
1. Agency for Health Care Policy and Research. Clinical practice guideline, number 5: Depression in primary care: vol. 2. Treatment of major depression. AHCPR publication 93-0551. Rockville, MD: US Department of Health and Human Services, 1993.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder. Am J Psychiatry 2000;157(suppl 4):1-45.
3. Hirschfeld RM, Keller MB, Panico BS, et al. The National Depressive and Manic-Depressive Association consensus statement on the undertreatment of depression. JAMA 1997;277:333-40.
4. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: Report of the Texas Consensus Conference Panel on medication treatment of major depressive disorder. J Clin Psychiatry 1999;60:142-56.
5. Nierenberg AA, Keefe BR, Leslie VA, et al. Residual symptoms in depressed patients who respond acutely to fluoxetine. J Clin Psychiatry 1999;60:221-5.
6. Kennedy N, Paykel ES. Residual symptoms at remission from depression: impact on long-term outcome. J Affect Disord 2004;80(2-3):135-44.
7. Rush AJ, Kraemer HC, Sackeim HA, et al. Report by the ACNP Task Force on Response and Remission in Major Depressive Disorder. Submitted for publication.
8. Keller M. Past, present, and future directions for defining optimal treatment outcome in depression: remission and beyond. JAMA 2003;289(23):3152-60.
9. Nutting P, Rost K, Diceinson M, et al. Barriers to initiating depression treatment in primary care practice. J Gen Intern Med 2002;17:103-11.
10. Fava GA, Rafanelli C, Grandi S, et al. Letter to the editor. Arch Gen Psychiatry 1999;56:765.-
11. Ramana R, Paykel ES, Cooper Z, et al. Remission and relapse in major depression: a two year prospective follow-up study. Psychol Med 1995;25(6):1171-80.
12. Judd LL, Akiskal HS, Maser JD, et al. Major depressive disorder: a prospective study of residual subthreshold depressive symptoms as a predictor of rapid relapse. J Affect Disord 1999;50:97-108.
13. Pintor L, Gastó G, Navarro V, et al. Relapse of major depression after complete and partial remission during a 2-year follow-up. J Affect Disord 2003;73:237-44.
14. Lin E, Katon W, Von Korff M, et al. Relapse of depression in primary care: rate and clinical predictors. Arch Fam Med 1998;7(5):443-9.
15. Cornwall P, Scott J. Partial remission in depressive disorders. Acta Psychiatr Scand 1997;95(4):265-71.
16. Paykel ES, Ramana R, Cooper Z, et al. Residual symptoms after partial remission: an important outcome in depression. Psychol Med 1995;25(6):1171-80.
17. Judd LL, Paulus MJ, Schettler PJ, et al. Does incomplete recovery from first lifetime major depressive episode herald a chronic course of illness? Am J Psychiatry 2000;157(9):1501-4.
18. Judd LL, Paulus MP, Zeller P. The role of residual subthreshold depressive symptoms in early episode relapse in unipolar major depressive disorder. Arch Gen Psychiatry 1999;56(8):764-5.
19. Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry 2000;157(2):229-33.
20. Kennedy N, Paykel ES. Residual symptoms at remission from depression: impact on long-term outcome. J Affect Disord 2004;80(2-3):135-44.
21. Zajecka JM. Treating depression to remission. J Clin Psychiatry 2003;64(suppl 15):7-12.
22. Judd LL, Paulus MP, Wells KB, Rapaport MH. Socioeconomic burden of subsyndromal depressive symptoms and major depressionin a sample of the general population. Am J Psychiatry 1996;153(11):1411-7.
23. Miller IW, Keitner GI, Schatzberg AF, et al. The treatment of chronic depression, part 3: psychosocial functioning before and after treatment with sertraline or imipramine. J Clin Psychiatry 1998;59(11):608-19.
24. Druss B, Schlesinger M, Allen H. Depressive symptoms, satisfaction with health care, and 2-year work outcomes in an employed population. Am J Psychiatry 2001;158(5):731-4.
25. Simon G, Ormel J, VonKorff M, et al. Health care costs associated with depressive and anxiety disorders in primary care. Am J Psychiatry 1995;152(3):352-7.
26. Katon W, Schulberg H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14(4):237-47.
27. Hirschfeld RMA, Montgomery SA, Aguglia E, et al. Partial response and nonresponse to antidepressant therapy: current approaches and treatment options. J Clin Psychiatry 2002;63(9):826-37.
28. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry 2001;178:234-41.
29. Kelsey J, Entsuah R. Venlafaxine offers significant therapeutic benefits over existing SSRI treatments irrespective of the patient’s depressive duration (poster presentation). Montreal: Collegium Internationale Neuropsychopharmacologicum, June 2002.
30. Thase ME. Effectiveness of antidepressants: comparative remission rates. J Clin Psychiatry 2003;64(suppl 2):3-7.
31. Shelton C. Long-term management of major depressive disorder: are differences among antidepressant treatments meaningful? J Clin Psychiatry 2004;65(suppl 17):29-33.
Counseling trauma victims: 4 brief therapies meet the test
Therapists once believed trauma survivors required years of treatment, yet we now know that relatively brief cognitive-behavioral interventions can yield long-term gains in psychosocial and psychological function.1 Many psychiatric patients meet diagnostic criteria for posttraumatic stress disorder (PTSD), including:
- 33% of women experiencing sexual assault2
- 30% of male war veterans3
- 30% of the 5 million U.S. children exposed to trauma each year4(Box).5
We offer recommendations on how to prepare traumatized adults and children for cognitive-behavioral therapy (CBT) and discuss four tested models—prolonged exposure (PE), cognitive processing therapy (CPT), eye movement desensitization and reprocessing (EMDR), and stress inoculation training (SIT)—that psychiatrists may find effective when treating PTSD.
Exposure therapy with children is usually more gradual than with adults, and the child is first taught relaxation techniques to use while recalling traumatic experiences. Although re-exposing children to traumatic events may seem harsh, exposure-based cognitive-behavioral therapy (CBT) appears to be most effective when trauma memories or reminders are most distressing to the child.
As with adults, CBT with children typically includes:
- exposure
- identifying and challenging unhealthy or distorted trauma-related thoughts
- teaching anxiety management techniques such as relaxation or assertiveness training.
In initial studies, CBT has been found safe and effective for treating posttraumatic stress disorder (PTSD) in children and adolescents.17 Through therapy, they can learn not to be afraid of their memories and can develop healthier, more-appropriate thoughts about the trauma. Children with uncomplicated PTSD—without severe, long-term physical injury—typically receive 12 to 20 CBT sessions. More sessions are needed for complex cases, such as when the trauma perpetrator was an integral family member.
Comorbid conditions—such as conduct disorder, attention-deficit/hyperactivity disorder, or depression—may need to be treated before PTSD or concurrently, using medication or other interventions.
Preparing trauma patients for CBT
Before starting CBT, evaluate patients thoroughly to determine if they meet DSM-IV-TR full or subthreshold criteria (
Not all patients are ready to confront their traumas when they arrive for psychiatric evaluation. For example:
- For a domestic violence victim, the therapist’s priority is to help begin safety planning and to address trauma after the patient is out of danger.
- Patients with poor coping skills and little social support often find it difficult to begin trauma treatment. For them, focus on building skills to offset the distress that accompanies trauma therapy.
- Patients with PTSD and substance abuse may benefit more from CBT if the therapist first addresses the substance dependence.
CBT core concepts
CBT therapists typically help patients identify and evaluate disruptive cognitions, which helps them challenge and modify emotions, thoughts, and behaviors related to traumatic experience(s). Other CBT components include:
- educating patients about PTSD
- exposing them to the traumatic material
- challenging and modifying their disruptive thoughts.
International Society for Traumatic Stress Studies (ISTSS) practice guidelines for PTSD8 include assessment and treatment suggestions (see Related resources). Whatever the model, CBT appears help patients manage their distress, not only during treatment but up to 5 years after completing therapy.9
Which CBT? Comparison studies have shown all four CBT interventions to be effective in treating PTSD, although initial trend data suggest that patients with:
- fear-based PTSD may do better with PE or EMDR
- PTSD-related guilt, anger, or other cognitive distortions may benefit more from CPT.
If you refer a patient, make sure the therapist is trained in CBT interventions and in working with trauma patients. To be effective, the therapist must be skilled in handling trauma processing work, suicidal thoughts/intent, and comorbid personality disorders.
Prolonged exposure
PE (Table 1) is typically conducted in 9 to 12 sessions lasting 90 minutes each and has been used to treat PTSD after sexual assault, combat, sexual abuse, and natural disasters. Although frequently offered in individual sessions, group PE has also been found to be effective.10
After educating the patient about PTSD and the treatment rationale, the therapist repeatedly asks the patient to describe the traumatic event as if it were occurring. During 45 to 60 minutes of this exposure, the therapist frequently asks the patient to rate his or her distress. This identifies “hot spots” in the account that need to be repeated. The therapist does not necessarily challenge distorted cognitions about the event (such as “I am to blame for the rape” or “No one can be trusted”).
Researchers hypothesize that exposing a PTSD patient to traumatic memories engages his or her brain’s pathologic “fear network,” which triggers an excessive fear response to non-threatening stimuli. Continued exposure allows the patient to habituate to this network, with subsequent extinction of fear and anxiety reactions. Foa et al11 found that mentally re-experiencing a traumatic event helps patients organize memory cues about it, which encourages cognitive restructuring of the trauma.
PE has been shown to enhance the trauma survivor’s self-control and personal competence and to decrease generalization of fear to non-assault stimuli.12 For example, many combat veterans report fear of situations—such as going to the beach or into the woods—that bring back memories of traumatic events. Their fears may keep them from enjoying a walk in the park or family vacations.
Through in vivo exposure, these patients can face associations between environmental cues and their trauma. As they learn to modify the fears associated with these cues, their personal and social functioning improves.
PE can be successful for those who complete therapy, but it has a relatively high drop-out rate, reported as 8%13 to 41%.14 The pain of continually reliving a traumatic event probably causes many patients to quit. To reduce drop-out rates, many therapists combine PE with cognitive restructuring or other techniques that help build patients’ coping skills.
Table 1
Using prolonged exposure therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Treatment rationale | |
| Review of PTSD symptom response | |
| Introduce breathing retraining | |
| 2 | Review handout, ‘Common reactions to trauma’ |
| Introduce Subjective Units of Distress | |
| Create fear hierarchy for in vivo exposures | |
| 3 | Provide rationale for imaginal exposure |
| Conduct imaginal exposure | |
| Assign in vivo exposure homework | |
| 4 to 8 | Conduct imaginal exposure |
| Discuss in vivo exposures | |
| 9 or 9 to 12 | Conduct imaginal exposure |
| Suggest continued in vivo exercises | |
| Termination | |
| Source: Foa EB, Rothbaum BO. Treating the trauma of rape: cognitive behavioral therapy for PTSD. New York: Guilford Press, 1998. | |
Cognitive processing therapy
CPT (Table 2) was created as a protocol to treat PTSD and related symptoms in rape survivors.7 Sessions can be group, individual, or combined, depending on the needs and resources of the patients and clinic.
Originally, CPT contained 12 weekly sessions, although versions up to 17 weeks have been developed for adult survivors of child sexual abuse, domestic violence survivors, and war veterans.15 Sessions can be added or adapted to address each population’s type of traumatic experience (such as developmental impairment of sexual abuse survivors).
CPT is based on information processing theory, which suggests that as people access a traumatic memory, they experience and extinguish emotions attached to the event. Guided by the therapist, the patient identifies and challenges distortions the trauma created in three cognition domains: the self, others, and the world. Patients learn to change or replace these cognitive distortions—which therapists often call “stuck points” or “rules”—with more-adaptive, healthier beliefs.
Common byproducts of trauma are feeling out of control or hopeless. Thus, CPT focuses on personal safety, trust, power/control, esteem, and intimacy within each of the three domains. Modules on assertiveness, communication, and social support can also be added.
Although CPT is being adapted for populations other than rape survivors, comparison studies are needed to determine if it is as effective as other CBT therapies for these groups.
Table 2
Using cognitive processing therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Review of symptoms | |
| Introduce ‘stuck points’/rules | |
| Write impact of event statement (IES) | |
| 2 | Review IES |
| Identify stuck points | |
| Introduce A-B-C sheets | |
| 3 | Review A-B-C sheets |
| Assign writing of traumatic account | |
| 4 | Read traumatic account |
| Identify stuck points | |
| Rewrite the account | |
| 5 | Read rewritten account |
| Identify stuck points | |
| Introduce challenging questions sheet (CQS) | |
| Assign writing of next-most traumatic incident and CQS | |
| 6 | Review CQS |
| Assign review of faulty thinking patterns (FTP) | |
| 7 | Review FTP |
| Assign safety module and challenging beliefs worksheets (CBW) on safety | |
| 8 | Review CBWs on safety |
| Assign module on trust | |
| 9 | Review CBWs on trust |
| Assign module on power/control | |
| 10 | Review CBWs on power/control |
| Assign module on esteem | |
| 11 | Review CBWs on esteem |
| Assign module on intimacy | |
| Rewrite IES | |
| 12 | Review CBWs on intimacy |
| Read both impact statements | |
| Address remaining areas of concern | |
| Termination | |
| Source: Resick PA, Schnicke MK Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage, 1993. | |
Eye movement desensitization and reprocessing
Like other PTSD treatments, EMDR is based on an “accelerated information-processing” model.16 Because it also incorporates dissociation and nonverbal representation of traumas (such as visual memories), EMDR is often classified as a cognitive treatment, although ISTSS practice guidelines8 present it as a separate category.
EMDR protocols call for the trauma patient to watch rapid, rhythmic movements of the therapist’s hand or a set of lights to distract attention from the stress he or she feels when visualizing the traumatic event. The original technique—developed by Francine Shapiro, PhD—is based on the observation that persons with PTSD often have disrupted rapid eye-movement sleep. In theory, inducing eye movements inhibits stress, allowing patients to more freely access their memory networks and process disturbances. Subsequently, Dr. Shapiro has suggested that using other auditory cues or hand taps may be as effective as eye movements.16
EMDR is often conducted in 12 to 15 sessions, although some studies report positive changes after 3 to 6 sessions. After obtaining a patient history, establishing rapport, and explaining the treatment, the therapist asks the patient to identify:
- visual images of the trauma
- his or her affective and physiologic responses to the trauma
- negative self-representations the trauma created
- positive, alternate self-representations.
EMDR has been effective in treating male war veterans, rape victims, and other trauma groups.17 Initial dismantling studies suggest that eye movements (or other distracting cues) might not be essential for trauma reprocessing, calling into question the mechanisms thought to create change in EMDR. Studies with larger samples comparing EMDR with other CBT models are needed to assess EMDR’s efficacy for trauma survivors.17
Stress inoculation training
SIT was designed by Meichenbaum18 (Table 3) to treat anxiety and stress and was adapted for use with trauma survivors. It appears most effective in relieving fear, anxiety, and depressive symptoms associated with traumatic experiences. SIT includes education, muscle relaxation training, breathing retraining, covert modeling, role-playing, guided self-dialog, and thought stopping. Therapists often teach these skills to patients in modules that build on each other.
For example, a patient might receive relaxation training while role-playing a difficult scenario she may face in the future. This helps her learn to remain calm in anxiety-provoking situations.
Unlike PE, SIT does not directly ask patients to recount their traumatic memories, although exposure may be indirect (such as during role-playing exercises). Its purpose is to give patients new skills to manage their anxiety, which in turn decreases PTSD symptoms.
Studies suggest that PE is more effective than SIT alone or SIT/PE combined.13 Thus, instead of using SIT as a trauma-focused treatment, some therapists find it useful to help patients gain coping skills before beginning other trauma treatments.
Table 3
Where to learn more about cognitive therapies for PTSD
| CBT model | PTSD related to… | Resources |
|---|---|---|
| Prolonged exposure | Combat experience, sexual assault, childhood abuse, motor vehicle accidents | Foa EB, Rothbaum BO. Treating the trauma of rape: Cognitive-behavioral therapy for PTSD. New York: Guilford Press; 1998 |
| Cognitive processing | Sexual assault, childhood abuse, incarceration (of adolescents) | Resick P, Schnicke M. Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage Publications; 1996 |
| EMDR | Combat experience, sexual assault, civilian disasters (for children or adults) | Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols, and procedures (2nd ed). New York: Guilford Press; 2001 |
| EMDR Institute, Inc. Available at: http://www.emdr.com | ||
| Stress inoculation training | Sexual and physical assault, motor vehicle accidents | Meichenbaum D. Stress inoculation training for coping with stressors. Available at: http://www.apa.org/divisions/div12/rev_est/sit_stress.html |
| EMDR: Eye movement desensitization and reprocessing | ||
- International Society for Traumatic Stress Studies. www.istss.org.
- Foa EB, Keane TM, Friedman MJ. Effective treatments for PTSD. New York: Guilford Press; 2000.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rothbaum BO, Meadows EA, Resick PA, Foy DW. Cognitive-behavioral therapy. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
2. Kilpatrick D, Edmonds CN, Seymour AK. Rape in America: A report to the nation. Arlington, VA: National Victims Center; 1992.
3. Kulka RA, Schlenger WE, Fairbank JA, et al. Trauma and the Vietnam War generation: report of findings from the National Vietnam Veterans Readjustment Study. New York: Brunner/Mazel; 1990.
4. Pfefferbaum B. Posttraumatic stress disorder in children: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry 1997;36(11):1503-11.
5. Deblinger E, McLeer SV, Henry D. Cognitive behavioral treatment for sexually abused children suffering post-traumatic stress. J Am Acad Child Adolesc Psychiatry 1990;5:747-52.
6. Najavits LM. Seeking Safety: a treatment manual for PTSD and substance abuse. New York: Guilford Press; 2002.
7. Resick PA, Nishith P, Weaver TL, et al. A comparison of cognitive processing therapy, prolonged exposure and a waiting condition for the treatment of posttraumatic stress disorder in female rape victims. J Consult Clin Psychol 2002;70:867-79.
8. Foa EB, Keane TM. Friedman MJ (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
9. Tarrier N, Sommerfield C. Treatment of chronic PTSD by cognitive therapy and exposure: 5-year follow-up. Behavior Ther 2004;35(2):231-46.
10. Foa EB, Rauch SA. Cognitive changes during prolonged exposure versus prolonged exposure plus cognitive restructuring in female assault survivors with posttraumatic stress disorder. J Consult Clin Psychol 2004;72(5):879-84.
11. Foa EB, Riggs DS, Massie ED, Yarczower M. The impact of fear activation and anger on the efficacy of exposure treatment for PTSD. Behav Ther 1995;26:487-99.
12. Foa EB, Rothbaum EO, Riggs D, Murdock T. Treatment of PTSD in rape victims: a comparison between cognitive-behavioral procedures and counseling. J Consult Clin Psychol 1991;59:715-23.
13. Foa EB, Dancu CV, Hembree EA, Jaycox LH, et al. A comparison of exposure therapy, stress inoculation training, and their combination for reducing posttraumatic stress disorder in female assault victims. J Consult Clin Psychol 1999;67:194-200.
14. McDonagh A, Friedman M, McHugo G, et al. Randomized trial of cognitive-behavioral therapy for chronic posttraumatic stress disorder in adult female survivors of childhood sexual abuse. J Consult Clin Psychol 2005;73:515-24.
15. Chard K. An evaluation of cognitive processing therapy for the treatment of posttraumatic stress disorder related to childhood sexual abuse. J Consult Clin Psychol. (in press).
16. Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols and procedures (2nd ed). New York: Guilford Press; 2001.
17. Chemtob CM, Tolin DF, van der Kolk BA, Pitman RK. Eye movement desensitization and reprocessing. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
18. Meichenbaum D. Cognitive-behavior modification: An integrative approach. New York: Plenum Press; 1977.
Therapists once believed trauma survivors required years of treatment, yet we now know that relatively brief cognitive-behavioral interventions can yield long-term gains in psychosocial and psychological function.1 Many psychiatric patients meet diagnostic criteria for posttraumatic stress disorder (PTSD), including:
- 33% of women experiencing sexual assault2
- 30% of male war veterans3
- 30% of the 5 million U.S. children exposed to trauma each year4(Box).5
We offer recommendations on how to prepare traumatized adults and children for cognitive-behavioral therapy (CBT) and discuss four tested models—prolonged exposure (PE), cognitive processing therapy (CPT), eye movement desensitization and reprocessing (EMDR), and stress inoculation training (SIT)—that psychiatrists may find effective when treating PTSD.
Exposure therapy with children is usually more gradual than with adults, and the child is first taught relaxation techniques to use while recalling traumatic experiences. Although re-exposing children to traumatic events may seem harsh, exposure-based cognitive-behavioral therapy (CBT) appears to be most effective when trauma memories or reminders are most distressing to the child.
As with adults, CBT with children typically includes:
- exposure
- identifying and challenging unhealthy or distorted trauma-related thoughts
- teaching anxiety management techniques such as relaxation or assertiveness training.
In initial studies, CBT has been found safe and effective for treating posttraumatic stress disorder (PTSD) in children and adolescents.17 Through therapy, they can learn not to be afraid of their memories and can develop healthier, more-appropriate thoughts about the trauma. Children with uncomplicated PTSD—without severe, long-term physical injury—typically receive 12 to 20 CBT sessions. More sessions are needed for complex cases, such as when the trauma perpetrator was an integral family member.
Comorbid conditions—such as conduct disorder, attention-deficit/hyperactivity disorder, or depression—may need to be treated before PTSD or concurrently, using medication or other interventions.
Preparing trauma patients for CBT
Before starting CBT, evaluate patients thoroughly to determine if they meet DSM-IV-TR full or subthreshold criteria (
Not all patients are ready to confront their traumas when they arrive for psychiatric evaluation. For example:
- For a domestic violence victim, the therapist’s priority is to help begin safety planning and to address trauma after the patient is out of danger.
- Patients with poor coping skills and little social support often find it difficult to begin trauma treatment. For them, focus on building skills to offset the distress that accompanies trauma therapy.
- Patients with PTSD and substance abuse may benefit more from CBT if the therapist first addresses the substance dependence.
CBT core concepts
CBT therapists typically help patients identify and evaluate disruptive cognitions, which helps them challenge and modify emotions, thoughts, and behaviors related to traumatic experience(s). Other CBT components include:
- educating patients about PTSD
- exposing them to the traumatic material
- challenging and modifying their disruptive thoughts.
International Society for Traumatic Stress Studies (ISTSS) practice guidelines for PTSD8 include assessment and treatment suggestions (see Related resources). Whatever the model, CBT appears help patients manage their distress, not only during treatment but up to 5 years after completing therapy.9
Which CBT? Comparison studies have shown all four CBT interventions to be effective in treating PTSD, although initial trend data suggest that patients with:
- fear-based PTSD may do better with PE or EMDR
- PTSD-related guilt, anger, or other cognitive distortions may benefit more from CPT.
If you refer a patient, make sure the therapist is trained in CBT interventions and in working with trauma patients. To be effective, the therapist must be skilled in handling trauma processing work, suicidal thoughts/intent, and comorbid personality disorders.
Prolonged exposure
PE (Table 1) is typically conducted in 9 to 12 sessions lasting 90 minutes each and has been used to treat PTSD after sexual assault, combat, sexual abuse, and natural disasters. Although frequently offered in individual sessions, group PE has also been found to be effective.10
After educating the patient about PTSD and the treatment rationale, the therapist repeatedly asks the patient to describe the traumatic event as if it were occurring. During 45 to 60 minutes of this exposure, the therapist frequently asks the patient to rate his or her distress. This identifies “hot spots” in the account that need to be repeated. The therapist does not necessarily challenge distorted cognitions about the event (such as “I am to blame for the rape” or “No one can be trusted”).
Researchers hypothesize that exposing a PTSD patient to traumatic memories engages his or her brain’s pathologic “fear network,” which triggers an excessive fear response to non-threatening stimuli. Continued exposure allows the patient to habituate to this network, with subsequent extinction of fear and anxiety reactions. Foa et al11 found that mentally re-experiencing a traumatic event helps patients organize memory cues about it, which encourages cognitive restructuring of the trauma.
PE has been shown to enhance the trauma survivor’s self-control and personal competence and to decrease generalization of fear to non-assault stimuli.12 For example, many combat veterans report fear of situations—such as going to the beach or into the woods—that bring back memories of traumatic events. Their fears may keep them from enjoying a walk in the park or family vacations.
Through in vivo exposure, these patients can face associations between environmental cues and their trauma. As they learn to modify the fears associated with these cues, their personal and social functioning improves.
PE can be successful for those who complete therapy, but it has a relatively high drop-out rate, reported as 8%13 to 41%.14 The pain of continually reliving a traumatic event probably causes many patients to quit. To reduce drop-out rates, many therapists combine PE with cognitive restructuring or other techniques that help build patients’ coping skills.
Table 1
Using prolonged exposure therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Treatment rationale | |
| Review of PTSD symptom response | |
| Introduce breathing retraining | |
| 2 | Review handout, ‘Common reactions to trauma’ |
| Introduce Subjective Units of Distress | |
| Create fear hierarchy for in vivo exposures | |
| 3 | Provide rationale for imaginal exposure |
| Conduct imaginal exposure | |
| Assign in vivo exposure homework | |
| 4 to 8 | Conduct imaginal exposure |
| Discuss in vivo exposures | |
| 9 or 9 to 12 | Conduct imaginal exposure |
| Suggest continued in vivo exercises | |
| Termination | |
| Source: Foa EB, Rothbaum BO. Treating the trauma of rape: cognitive behavioral therapy for PTSD. New York: Guilford Press, 1998. | |
Cognitive processing therapy
CPT (Table 2) was created as a protocol to treat PTSD and related symptoms in rape survivors.7 Sessions can be group, individual, or combined, depending on the needs and resources of the patients and clinic.
Originally, CPT contained 12 weekly sessions, although versions up to 17 weeks have been developed for adult survivors of child sexual abuse, domestic violence survivors, and war veterans.15 Sessions can be added or adapted to address each population’s type of traumatic experience (such as developmental impairment of sexual abuse survivors).
CPT is based on information processing theory, which suggests that as people access a traumatic memory, they experience and extinguish emotions attached to the event. Guided by the therapist, the patient identifies and challenges distortions the trauma created in three cognition domains: the self, others, and the world. Patients learn to change or replace these cognitive distortions—which therapists often call “stuck points” or “rules”—with more-adaptive, healthier beliefs.
Common byproducts of trauma are feeling out of control or hopeless. Thus, CPT focuses on personal safety, trust, power/control, esteem, and intimacy within each of the three domains. Modules on assertiveness, communication, and social support can also be added.
Although CPT is being adapted for populations other than rape survivors, comparison studies are needed to determine if it is as effective as other CBT therapies for these groups.
Table 2
Using cognitive processing therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Review of symptoms | |
| Introduce ‘stuck points’/rules | |
| Write impact of event statement (IES) | |
| 2 | Review IES |
| Identify stuck points | |
| Introduce A-B-C sheets | |
| 3 | Review A-B-C sheets |
| Assign writing of traumatic account | |
| 4 | Read traumatic account |
| Identify stuck points | |
| Rewrite the account | |
| 5 | Read rewritten account |
| Identify stuck points | |
| Introduce challenging questions sheet (CQS) | |
| Assign writing of next-most traumatic incident and CQS | |
| 6 | Review CQS |
| Assign review of faulty thinking patterns (FTP) | |
| 7 | Review FTP |
| Assign safety module and challenging beliefs worksheets (CBW) on safety | |
| 8 | Review CBWs on safety |
| Assign module on trust | |
| 9 | Review CBWs on trust |
| Assign module on power/control | |
| 10 | Review CBWs on power/control |
| Assign module on esteem | |
| 11 | Review CBWs on esteem |
| Assign module on intimacy | |
| Rewrite IES | |
| 12 | Review CBWs on intimacy |
| Read both impact statements | |
| Address remaining areas of concern | |
| Termination | |
| Source: Resick PA, Schnicke MK Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage, 1993. | |
Eye movement desensitization and reprocessing
Like other PTSD treatments, EMDR is based on an “accelerated information-processing” model.16 Because it also incorporates dissociation and nonverbal representation of traumas (such as visual memories), EMDR is often classified as a cognitive treatment, although ISTSS practice guidelines8 present it as a separate category.
EMDR protocols call for the trauma patient to watch rapid, rhythmic movements of the therapist’s hand or a set of lights to distract attention from the stress he or she feels when visualizing the traumatic event. The original technique—developed by Francine Shapiro, PhD—is based on the observation that persons with PTSD often have disrupted rapid eye-movement sleep. In theory, inducing eye movements inhibits stress, allowing patients to more freely access their memory networks and process disturbances. Subsequently, Dr. Shapiro has suggested that using other auditory cues or hand taps may be as effective as eye movements.16
EMDR is often conducted in 12 to 15 sessions, although some studies report positive changes after 3 to 6 sessions. After obtaining a patient history, establishing rapport, and explaining the treatment, the therapist asks the patient to identify:
- visual images of the trauma
- his or her affective and physiologic responses to the trauma
- negative self-representations the trauma created
- positive, alternate self-representations.
EMDR has been effective in treating male war veterans, rape victims, and other trauma groups.17 Initial dismantling studies suggest that eye movements (or other distracting cues) might not be essential for trauma reprocessing, calling into question the mechanisms thought to create change in EMDR. Studies with larger samples comparing EMDR with other CBT models are needed to assess EMDR’s efficacy for trauma survivors.17
Stress inoculation training
SIT was designed by Meichenbaum18 (Table 3) to treat anxiety and stress and was adapted for use with trauma survivors. It appears most effective in relieving fear, anxiety, and depressive symptoms associated with traumatic experiences. SIT includes education, muscle relaxation training, breathing retraining, covert modeling, role-playing, guided self-dialog, and thought stopping. Therapists often teach these skills to patients in modules that build on each other.
For example, a patient might receive relaxation training while role-playing a difficult scenario she may face in the future. This helps her learn to remain calm in anxiety-provoking situations.
Unlike PE, SIT does not directly ask patients to recount their traumatic memories, although exposure may be indirect (such as during role-playing exercises). Its purpose is to give patients new skills to manage their anxiety, which in turn decreases PTSD symptoms.
Studies suggest that PE is more effective than SIT alone or SIT/PE combined.13 Thus, instead of using SIT as a trauma-focused treatment, some therapists find it useful to help patients gain coping skills before beginning other trauma treatments.
Table 3
Where to learn more about cognitive therapies for PTSD
| CBT model | PTSD related to… | Resources |
|---|---|---|
| Prolonged exposure | Combat experience, sexual assault, childhood abuse, motor vehicle accidents | Foa EB, Rothbaum BO. Treating the trauma of rape: Cognitive-behavioral therapy for PTSD. New York: Guilford Press; 1998 |
| Cognitive processing | Sexual assault, childhood abuse, incarceration (of adolescents) | Resick P, Schnicke M. Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage Publications; 1996 |
| EMDR | Combat experience, sexual assault, civilian disasters (for children or adults) | Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols, and procedures (2nd ed). New York: Guilford Press; 2001 |
| EMDR Institute, Inc. Available at: http://www.emdr.com | ||
| Stress inoculation training | Sexual and physical assault, motor vehicle accidents | Meichenbaum D. Stress inoculation training for coping with stressors. Available at: http://www.apa.org/divisions/div12/rev_est/sit_stress.html |
| EMDR: Eye movement desensitization and reprocessing | ||
- International Society for Traumatic Stress Studies. www.istss.org.
- Foa EB, Keane TM, Friedman MJ. Effective treatments for PTSD. New York: Guilford Press; 2000.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Therapists once believed trauma survivors required years of treatment, yet we now know that relatively brief cognitive-behavioral interventions can yield long-term gains in psychosocial and psychological function.1 Many psychiatric patients meet diagnostic criteria for posttraumatic stress disorder (PTSD), including:
- 33% of women experiencing sexual assault2
- 30% of male war veterans3
- 30% of the 5 million U.S. children exposed to trauma each year4(Box).5
We offer recommendations on how to prepare traumatized adults and children for cognitive-behavioral therapy (CBT) and discuss four tested models—prolonged exposure (PE), cognitive processing therapy (CPT), eye movement desensitization and reprocessing (EMDR), and stress inoculation training (SIT)—that psychiatrists may find effective when treating PTSD.
Exposure therapy with children is usually more gradual than with adults, and the child is first taught relaxation techniques to use while recalling traumatic experiences. Although re-exposing children to traumatic events may seem harsh, exposure-based cognitive-behavioral therapy (CBT) appears to be most effective when trauma memories or reminders are most distressing to the child.
As with adults, CBT with children typically includes:
- exposure
- identifying and challenging unhealthy or distorted trauma-related thoughts
- teaching anxiety management techniques such as relaxation or assertiveness training.
In initial studies, CBT has been found safe and effective for treating posttraumatic stress disorder (PTSD) in children and adolescents.17 Through therapy, they can learn not to be afraid of their memories and can develop healthier, more-appropriate thoughts about the trauma. Children with uncomplicated PTSD—without severe, long-term physical injury—typically receive 12 to 20 CBT sessions. More sessions are needed for complex cases, such as when the trauma perpetrator was an integral family member.
Comorbid conditions—such as conduct disorder, attention-deficit/hyperactivity disorder, or depression—may need to be treated before PTSD or concurrently, using medication or other interventions.
Preparing trauma patients for CBT
Before starting CBT, evaluate patients thoroughly to determine if they meet DSM-IV-TR full or subthreshold criteria (
Not all patients are ready to confront their traumas when they arrive for psychiatric evaluation. For example:
- For a domestic violence victim, the therapist’s priority is to help begin safety planning and to address trauma after the patient is out of danger.
- Patients with poor coping skills and little social support often find it difficult to begin trauma treatment. For them, focus on building skills to offset the distress that accompanies trauma therapy.
- Patients with PTSD and substance abuse may benefit more from CBT if the therapist first addresses the substance dependence.
CBT core concepts
CBT therapists typically help patients identify and evaluate disruptive cognitions, which helps them challenge and modify emotions, thoughts, and behaviors related to traumatic experience(s). Other CBT components include:
- educating patients about PTSD
- exposing them to the traumatic material
- challenging and modifying their disruptive thoughts.
International Society for Traumatic Stress Studies (ISTSS) practice guidelines for PTSD8 include assessment and treatment suggestions (see Related resources). Whatever the model, CBT appears help patients manage their distress, not only during treatment but up to 5 years after completing therapy.9
Which CBT? Comparison studies have shown all four CBT interventions to be effective in treating PTSD, although initial trend data suggest that patients with:
- fear-based PTSD may do better with PE or EMDR
- PTSD-related guilt, anger, or other cognitive distortions may benefit more from CPT.
If you refer a patient, make sure the therapist is trained in CBT interventions and in working with trauma patients. To be effective, the therapist must be skilled in handling trauma processing work, suicidal thoughts/intent, and comorbid personality disorders.
Prolonged exposure
PE (Table 1) is typically conducted in 9 to 12 sessions lasting 90 minutes each and has been used to treat PTSD after sexual assault, combat, sexual abuse, and natural disasters. Although frequently offered in individual sessions, group PE has also been found to be effective.10
After educating the patient about PTSD and the treatment rationale, the therapist repeatedly asks the patient to describe the traumatic event as if it were occurring. During 45 to 60 minutes of this exposure, the therapist frequently asks the patient to rate his or her distress. This identifies “hot spots” in the account that need to be repeated. The therapist does not necessarily challenge distorted cognitions about the event (such as “I am to blame for the rape” or “No one can be trusted”).
Researchers hypothesize that exposing a PTSD patient to traumatic memories engages his or her brain’s pathologic “fear network,” which triggers an excessive fear response to non-threatening stimuli. Continued exposure allows the patient to habituate to this network, with subsequent extinction of fear and anxiety reactions. Foa et al11 found that mentally re-experiencing a traumatic event helps patients organize memory cues about it, which encourages cognitive restructuring of the trauma.
PE has been shown to enhance the trauma survivor’s self-control and personal competence and to decrease generalization of fear to non-assault stimuli.12 For example, many combat veterans report fear of situations—such as going to the beach or into the woods—that bring back memories of traumatic events. Their fears may keep them from enjoying a walk in the park or family vacations.
Through in vivo exposure, these patients can face associations between environmental cues and their trauma. As they learn to modify the fears associated with these cues, their personal and social functioning improves.
PE can be successful for those who complete therapy, but it has a relatively high drop-out rate, reported as 8%13 to 41%.14 The pain of continually reliving a traumatic event probably causes many patients to quit. To reduce drop-out rates, many therapists combine PE with cognitive restructuring or other techniques that help build patients’ coping skills.
Table 1
Using prolonged exposure therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Treatment rationale | |
| Review of PTSD symptom response | |
| Introduce breathing retraining | |
| 2 | Review handout, ‘Common reactions to trauma’ |
| Introduce Subjective Units of Distress | |
| Create fear hierarchy for in vivo exposures | |
| 3 | Provide rationale for imaginal exposure |
| Conduct imaginal exposure | |
| Assign in vivo exposure homework | |
| 4 to 8 | Conduct imaginal exposure |
| Discuss in vivo exposures | |
| 9 or 9 to 12 | Conduct imaginal exposure |
| Suggest continued in vivo exercises | |
| Termination | |
| Source: Foa EB, Rothbaum BO. Treating the trauma of rape: cognitive behavioral therapy for PTSD. New York: Guilford Press, 1998. | |
Cognitive processing therapy
CPT (Table 2) was created as a protocol to treat PTSD and related symptoms in rape survivors.7 Sessions can be group, individual, or combined, depending on the needs and resources of the patients and clinic.
Originally, CPT contained 12 weekly sessions, although versions up to 17 weeks have been developed for adult survivors of child sexual abuse, domestic violence survivors, and war veterans.15 Sessions can be added or adapted to address each population’s type of traumatic experience (such as developmental impairment of sexual abuse survivors).
CPT is based on information processing theory, which suggests that as people access a traumatic memory, they experience and extinguish emotions attached to the event. Guided by the therapist, the patient identifies and challenges distortions the trauma created in three cognition domains: the self, others, and the world. Patients learn to change or replace these cognitive distortions—which therapists often call “stuck points” or “rules”—with more-adaptive, healthier beliefs.
Common byproducts of trauma are feeling out of control or hopeless. Thus, CPT focuses on personal safety, trust, power/control, esteem, and intimacy within each of the three domains. Modules on assertiveness, communication, and social support can also be added.
Although CPT is being adapted for populations other than rape survivors, comparison studies are needed to determine if it is as effective as other CBT therapies for these groups.
Table 2
Using cognitive processing therapy to treat PTSD, session by session
| Session | Content |
|---|---|
| 1 | Education |
| Review of symptoms | |
| Introduce ‘stuck points’/rules | |
| Write impact of event statement (IES) | |
| 2 | Review IES |
| Identify stuck points | |
| Introduce A-B-C sheets | |
| 3 | Review A-B-C sheets |
| Assign writing of traumatic account | |
| 4 | Read traumatic account |
| Identify stuck points | |
| Rewrite the account | |
| 5 | Read rewritten account |
| Identify stuck points | |
| Introduce challenging questions sheet (CQS) | |
| Assign writing of next-most traumatic incident and CQS | |
| 6 | Review CQS |
| Assign review of faulty thinking patterns (FTP) | |
| 7 | Review FTP |
| Assign safety module and challenging beliefs worksheets (CBW) on safety | |
| 8 | Review CBWs on safety |
| Assign module on trust | |
| 9 | Review CBWs on trust |
| Assign module on power/control | |
| 10 | Review CBWs on power/control |
| Assign module on esteem | |
| 11 | Review CBWs on esteem |
| Assign module on intimacy | |
| Rewrite IES | |
| 12 | Review CBWs on intimacy |
| Read both impact statements | |
| Address remaining areas of concern | |
| Termination | |
| Source: Resick PA, Schnicke MK Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage, 1993. | |
Eye movement desensitization and reprocessing
Like other PTSD treatments, EMDR is based on an “accelerated information-processing” model.16 Because it also incorporates dissociation and nonverbal representation of traumas (such as visual memories), EMDR is often classified as a cognitive treatment, although ISTSS practice guidelines8 present it as a separate category.
EMDR protocols call for the trauma patient to watch rapid, rhythmic movements of the therapist’s hand or a set of lights to distract attention from the stress he or she feels when visualizing the traumatic event. The original technique—developed by Francine Shapiro, PhD—is based on the observation that persons with PTSD often have disrupted rapid eye-movement sleep. In theory, inducing eye movements inhibits stress, allowing patients to more freely access their memory networks and process disturbances. Subsequently, Dr. Shapiro has suggested that using other auditory cues or hand taps may be as effective as eye movements.16
EMDR is often conducted in 12 to 15 sessions, although some studies report positive changes after 3 to 6 sessions. After obtaining a patient history, establishing rapport, and explaining the treatment, the therapist asks the patient to identify:
- visual images of the trauma
- his or her affective and physiologic responses to the trauma
- negative self-representations the trauma created
- positive, alternate self-representations.
EMDR has been effective in treating male war veterans, rape victims, and other trauma groups.17 Initial dismantling studies suggest that eye movements (or other distracting cues) might not be essential for trauma reprocessing, calling into question the mechanisms thought to create change in EMDR. Studies with larger samples comparing EMDR with other CBT models are needed to assess EMDR’s efficacy for trauma survivors.17
Stress inoculation training
SIT was designed by Meichenbaum18 (Table 3) to treat anxiety and stress and was adapted for use with trauma survivors. It appears most effective in relieving fear, anxiety, and depressive symptoms associated with traumatic experiences. SIT includes education, muscle relaxation training, breathing retraining, covert modeling, role-playing, guided self-dialog, and thought stopping. Therapists often teach these skills to patients in modules that build on each other.
For example, a patient might receive relaxation training while role-playing a difficult scenario she may face in the future. This helps her learn to remain calm in anxiety-provoking situations.
Unlike PE, SIT does not directly ask patients to recount their traumatic memories, although exposure may be indirect (such as during role-playing exercises). Its purpose is to give patients new skills to manage their anxiety, which in turn decreases PTSD symptoms.
Studies suggest that PE is more effective than SIT alone or SIT/PE combined.13 Thus, instead of using SIT as a trauma-focused treatment, some therapists find it useful to help patients gain coping skills before beginning other trauma treatments.
Table 3
Where to learn more about cognitive therapies for PTSD
| CBT model | PTSD related to… | Resources |
|---|---|---|
| Prolonged exposure | Combat experience, sexual assault, childhood abuse, motor vehicle accidents | Foa EB, Rothbaum BO. Treating the trauma of rape: Cognitive-behavioral therapy for PTSD. New York: Guilford Press; 1998 |
| Cognitive processing | Sexual assault, childhood abuse, incarceration (of adolescents) | Resick P, Schnicke M. Cognitive processing therapy for rape victims: a treatment manual. Newbury Park, CA: Sage Publications; 1996 |
| EMDR | Combat experience, sexual assault, civilian disasters (for children or adults) | Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols, and procedures (2nd ed). New York: Guilford Press; 2001 |
| EMDR Institute, Inc. Available at: http://www.emdr.com | ||
| Stress inoculation training | Sexual and physical assault, motor vehicle accidents | Meichenbaum D. Stress inoculation training for coping with stressors. Available at: http://www.apa.org/divisions/div12/rev_est/sit_stress.html |
| EMDR: Eye movement desensitization and reprocessing | ||
- International Society for Traumatic Stress Studies. www.istss.org.
- Foa EB, Keane TM, Friedman MJ. Effective treatments for PTSD. New York: Guilford Press; 2000.
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Rothbaum BO, Meadows EA, Resick PA, Foy DW. Cognitive-behavioral therapy. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
2. Kilpatrick D, Edmonds CN, Seymour AK. Rape in America: A report to the nation. Arlington, VA: National Victims Center; 1992.
3. Kulka RA, Schlenger WE, Fairbank JA, et al. Trauma and the Vietnam War generation: report of findings from the National Vietnam Veterans Readjustment Study. New York: Brunner/Mazel; 1990.
4. Pfefferbaum B. Posttraumatic stress disorder in children: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry 1997;36(11):1503-11.
5. Deblinger E, McLeer SV, Henry D. Cognitive behavioral treatment for sexually abused children suffering post-traumatic stress. J Am Acad Child Adolesc Psychiatry 1990;5:747-52.
6. Najavits LM. Seeking Safety: a treatment manual for PTSD and substance abuse. New York: Guilford Press; 2002.
7. Resick PA, Nishith P, Weaver TL, et al. A comparison of cognitive processing therapy, prolonged exposure and a waiting condition for the treatment of posttraumatic stress disorder in female rape victims. J Consult Clin Psychol 2002;70:867-79.
8. Foa EB, Keane TM. Friedman MJ (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
9. Tarrier N, Sommerfield C. Treatment of chronic PTSD by cognitive therapy and exposure: 5-year follow-up. Behavior Ther 2004;35(2):231-46.
10. Foa EB, Rauch SA. Cognitive changes during prolonged exposure versus prolonged exposure plus cognitive restructuring in female assault survivors with posttraumatic stress disorder. J Consult Clin Psychol 2004;72(5):879-84.
11. Foa EB, Riggs DS, Massie ED, Yarczower M. The impact of fear activation and anger on the efficacy of exposure treatment for PTSD. Behav Ther 1995;26:487-99.
12. Foa EB, Rothbaum EO, Riggs D, Murdock T. Treatment of PTSD in rape victims: a comparison between cognitive-behavioral procedures and counseling. J Consult Clin Psychol 1991;59:715-23.
13. Foa EB, Dancu CV, Hembree EA, Jaycox LH, et al. A comparison of exposure therapy, stress inoculation training, and their combination for reducing posttraumatic stress disorder in female assault victims. J Consult Clin Psychol 1999;67:194-200.
14. McDonagh A, Friedman M, McHugo G, et al. Randomized trial of cognitive-behavioral therapy for chronic posttraumatic stress disorder in adult female survivors of childhood sexual abuse. J Consult Clin Psychol 2005;73:515-24.
15. Chard K. An evaluation of cognitive processing therapy for the treatment of posttraumatic stress disorder related to childhood sexual abuse. J Consult Clin Psychol. (in press).
16. Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols and procedures (2nd ed). New York: Guilford Press; 2001.
17. Chemtob CM, Tolin DF, van der Kolk BA, Pitman RK. Eye movement desensitization and reprocessing. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
18. Meichenbaum D. Cognitive-behavior modification: An integrative approach. New York: Plenum Press; 1977.
1. Rothbaum BO, Meadows EA, Resick PA, Foy DW. Cognitive-behavioral therapy. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
2. Kilpatrick D, Edmonds CN, Seymour AK. Rape in America: A report to the nation. Arlington, VA: National Victims Center; 1992.
3. Kulka RA, Schlenger WE, Fairbank JA, et al. Trauma and the Vietnam War generation: report of findings from the National Vietnam Veterans Readjustment Study. New York: Brunner/Mazel; 1990.
4. Pfefferbaum B. Posttraumatic stress disorder in children: a review of the past 10 years. J Am Acad Child Adolesc Psychiatry 1997;36(11):1503-11.
5. Deblinger E, McLeer SV, Henry D. Cognitive behavioral treatment for sexually abused children suffering post-traumatic stress. J Am Acad Child Adolesc Psychiatry 1990;5:747-52.
6. Najavits LM. Seeking Safety: a treatment manual for PTSD and substance abuse. New York: Guilford Press; 2002.
7. Resick PA, Nishith P, Weaver TL, et al. A comparison of cognitive processing therapy, prolonged exposure and a waiting condition for the treatment of posttraumatic stress disorder in female rape victims. J Consult Clin Psychol 2002;70:867-79.
8. Foa EB, Keane TM. Friedman MJ (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
9. Tarrier N, Sommerfield C. Treatment of chronic PTSD by cognitive therapy and exposure: 5-year follow-up. Behavior Ther 2004;35(2):231-46.
10. Foa EB, Rauch SA. Cognitive changes during prolonged exposure versus prolonged exposure plus cognitive restructuring in female assault survivors with posttraumatic stress disorder. J Consult Clin Psychol 2004;72(5):879-84.
11. Foa EB, Riggs DS, Massie ED, Yarczower M. The impact of fear activation and anger on the efficacy of exposure treatment for PTSD. Behav Ther 1995;26:487-99.
12. Foa EB, Rothbaum EO, Riggs D, Murdock T. Treatment of PTSD in rape victims: a comparison between cognitive-behavioral procedures and counseling. J Consult Clin Psychol 1991;59:715-23.
13. Foa EB, Dancu CV, Hembree EA, Jaycox LH, et al. A comparison of exposure therapy, stress inoculation training, and their combination for reducing posttraumatic stress disorder in female assault victims. J Consult Clin Psychol 1999;67:194-200.
14. McDonagh A, Friedman M, McHugo G, et al. Randomized trial of cognitive-behavioral therapy for chronic posttraumatic stress disorder in adult female survivors of childhood sexual abuse. J Consult Clin Psychol 2005;73:515-24.
15. Chard K. An evaluation of cognitive processing therapy for the treatment of posttraumatic stress disorder related to childhood sexual abuse. J Consult Clin Psychol. (in press).
16. Shapiro F. Eye movement desensitization and reprocessing: basic principles, protocols and procedures (2nd ed). New York: Guilford Press; 2001.
17. Chemtob CM, Tolin DF, van der Kolk BA, Pitman RK. Eye movement desensitization and reprocessing. In: Foa E, Keane T, Friedman M (eds). Effective treatments for PTSD. New York: Guilford Press; 2000.
18. Meichenbaum D. Cognitive-behavior modification: An integrative approach. New York: Plenum Press; 1977.
Are anticonvulsants safe for pediatric bipolar disorder?
Are anticonvulsants safe and effective mood stabilizers for children and adolescents with bipolar disorder? The answer is unclear because most bipolar disorder treatment trials have included adults only, and clinicians are desperate for data.1
To help you care for young patients, we report what is known about the potential benefits and risks of using mood stabilizers and anticonvulsants in bipolar youth. We base our dosing, target serum level, and monitoring recommendations on clinical experience and the limited published evidence.
AGENTS OF CHOICE?
Bipolar disorder’s “atypical” presentation in children—often more irritability and explosiveness than euphoria—can complicate diagnosis. Bipolar children and adolescents often have comorbid attention-deficit/hyperactivity disorder (ADHD), other disruptive behavior disorders, or anxiety disorders. Thus, comorbidities and presenting symptoms often dictate medication choice.
An expert consensus guideline acknowledges that more evidence on pediatric bipolar disorder is needed. In the meantime, the guideline suggests trying valproate or lithium first to treat nonpsychotic mania in pediatric bipolar patients.1 It also recommends three atypical antipsychotics— olanzapine, quetiapine, and risperidone—as potential first-line treatments. Valproate and lithium may be preferred because of atypicals’ risk of weight gain and metabolic syndrome.
Trying other anticonvulsants may be justified for bipolar youths who are not functioning well with first-line agents. Lamotrigine, for example, has antidepressant and antimanic effects.2 When you try anticonvulsants that lack double-blind, placebo-controlled trials, we recommend that you:
- obtain consent from the parents and child
- monitor carefully for side effects.
LITHIUM: STRONGEST EVIDENCE
Lithium is one of the most well-studied medications for pediatric bipolar disorder and the only mood stabilizer FDA-approved for children and adolescents (Table 1).3 Although approved for ages 12 and older, lithium has been used in younger children in practice and in clinical trials.
Table 1
FDA-approval status of medications used to treat bipolar disorder
| Medication | Indications for adults | Indications for children |
|---|---|---|
| Carbamazepine | Acute manic episode and acute mixed episode | Not approved |
| Lamotrigine | Maintenance therapy | Not approved |
| Lithium | Acute manic episode and maintenance therapy | Age ≥ 12 years |
| Oxcarbazepine | Not approved | Not approved |
| Topiramate | Not approved | Not approved |
| Valproate | Acute manic episode | Not approved |
| Source: Reference 3 | ||
In the only double-blind, placebo-controlled trial of lithium in adolescents with bipolar disorder, some subjects had secondary substance dependency disorders.7 For 6 weeks, 25 outpatient adolescents received lithium (13 patients) or placebo (12 patients). Lithium was effective in treating bipolar and substance dependency symptoms, with significantly improved clinical global assessment scores and decreased positive urine assays for drugs. Little difference was seen in mood item scores on the Schedule for Affective Disorders and Schizophrenia, child version (KSADS-1986), whether patients were taking lithium or placebo.
Pediatric dosing. For bipolar patients ages 6 to 12, use the child’s weight to determine lithium dosage (Table 2).8 Maintain serum levels between 0.8 and 1.2 mEq/L,9 and check them frequently when starting therapy.10 After mood stabilization, check levels every 1 to 3 months or when you suspect noncompliance. Obtain renal and thyroid function values at baseline and every 4 to 6 months.
Table 2
Guide to dosing lithium for prepubertal school-aged children*
| Doses (mg) | ||||
|---|---|---|---|---|
| Child’s weight (kg) | 8 AM | 12 PM | 6 PM | Total daily |
| 150 | 150 | 300 | 600 | |
| 25 to 40 | 300 | 300 | 300 | 900 |
| 40 to 50 | 300 | 300 | 600 | 1,200 |
| 50 to 60 | 600 | 300 | 600 | 1,500 |
| * Maintain specified dose at least 5 days, drawing serum levels 12 hrs after the last lithium dose until two consecutive levels appear in the therapeutic range (0.6 to 1.2 mEq/L). Dose may then be adjusted based on serum level, side effects, or clinical response. Do not exceed 1.4 mEq/L. | ||||
| Source: Reference 8 | ||||
Consider teratogenicity when choosing mood stabilizers for bipolar adolescent girls who may be sexually active. Lithium, valproate, and carbamazepine are labeled pregnancy category D because of their potential to cause birth defects.
Lithium treatment has been associated with increased risk of cardiac defects, specifically Ebstein’s anomaly (malformation of the tricuspid valve). Its incidence in children of women who used lithium during pregnancy is estimated to be 1:1,000 (0.10%) to 2:1,000 (0.05%)— 20 to 40 times the rate in the general population.12
Valproate.Results from the North American Antiepileptic Drug (AED) Pregnancy Registry showed a 10.7% rate of major congenital malformations (MCM)— including neural tube defects (spina bifida) and cardiac defects (pulmonary atresia)—in children of women who used valproate during pregnancy. The rate of births with MCMs in the general population is 2.9%.13
Carbamazepine.Data from the Australian Pregnancy Registry showed no significant increase in malformation rates in infants of carbamazepine users compared with those of women receiving no antiepileptics.14 Other studies, however, have linked carbamazepine with an increased risk of craniofacial defects (11%), neural tube defects (0.5 to 1%), and cardiac malformations.12
Lamotrigine.The teratogenic effects of the newer anticonvulsants are unclear. An 11-year study of lamotrigine15 found MCM risk after first-trimester exposure to lamotrigine to be similar to the general population’s MCM risk.
Combination therapy.Teratogenic risk appears to increase when multiple antiepileptic drugs are used (9.9% risk in polytherapy vs 6.2% in monotherapy).16
VALPROATE: OPEN-LABEL TRIALS ONLY
Efficacy. No double-blind, placebo-controlled study has shown valproate to be effective in treating bipolar disorder in children and adolescents. When used as monotherapy in open-label studies, valproate has produced response rates of:
- 53% in a 6-week, randomized, open-label trial in which 42 outpatients (mean age 11.4 years) with bipolar disorder type I or II received lithium, divalproex sodium, or carbamazepine9
- 61% in an open-label study of 40 patients ages 7 to 19 with a manic, hypomanic, or mixed episode who received divalproex for 2 to 8 weeks17
- 80% in an 8-week open-label trial of 40 patients ages 6 to 17 with bipolar disorder type I (77.5%) or type II (22.5%) and a Young Mania Rating Scale (YMRS)score ≥ 14.18
Safety: Black-box warnings. Valproate therapy carries risks of hepatic failure, pancreatitis, and birth defects. Monitor blood counts and hepatic enzymes throughout therapy (Table 3).3 Rare yet potentially fatal hepatic toxicity appears to occur most often in children age 21 Other studies suggest:
- an association with congenital malformations, including spina bifida and pulmonary atresia, in children exposed to valproate in utero6
- a link between valproate and hyperammonemic encephalopathy, especially in patients with urea cycle disorders22
- potential for benign thrombocytopenia23
- increased incidence of polycystic ovary syndrome—ovarian cysts, hyperandrogenism, chronic anovulation—in peripubertal mentally retarded women treated with valproate for seizure disorders.24
Table 3
Mood stabilizers’ side effects and recommended monitoring
| Medication | Major side effects | Monitoring |
|---|---|---|
| Carbamazepine | Allergic skin rash, drowsiness, blood dyscrasias, diplopia | CBC with reticulocytes, iron, LFTs, urinalysis, BUN, TFTs, sodium, serum carbamazepine levels |
| Lamotrigine | Stevens-Johnson syndrome, headache, dizziness, ataxia, somnolence, nausea, diplopia, blurred vision, rhinitis | No serum monitoring recommended |
| Lithium | Polyuria, polydipsia, nausea, diarrhea, tininecleatremor, enuresis, fatigue, ataxia, leukocytosis, malaise, cardiac arrhythmias, weight gain | BUN/creatinine, crearance, TFTs, calcium/phosphorus, ECG, serum lithium levels every 1 to 3 months once stabilized |
| Oxcarbazepine | Dizziness, somnolence/fatigue, ataxia/gait disturbance, vertigo, headache, tremor, rash, hyponatremia, hypersensitivity reaction, GI symptoms, diplopia | Sodium levels (particularly ifirst 3 months) |
| Topiramate | Hyperchloremic metabolic acidosis, oligohydrosis and hyperthermia, acute myopia, somnolence/fatigue, nausea, anorexia/weight loss, paresthesia, tremor, difficulty concentrating | BUN/creatinine, sodium bicarbonate |
| Valproate | Irritability/restlessness, ataxia, headache, weight gain, hyperammonemic encephalopathy, alopecia, GI upset, pancreatitis,sedation, thrombocytopenia, liver failure, polycystic ovaries/hyperandrogenism, teratogenic effects,rash | Ammonia, LFTs, bilirubin, CBC with platelets, serum valproate levels |
| BUN: blood urea nitrogen; CBC: complete blood count; ECG: electrocardiography; LFT: liver function tests; TFTs: thyroid function tests | ||
| Note: Bolded items included in black-box warnings | ||
| Source: Reference 3 | ||
CARBAMAZEPINE: DRUG INTERACTION RISK
Carbamazepine is used less often than lithium or divalproex for bipolar disorder. It tends to be used adjunctively when lithium alone is ineffective.
Efficacy. In an open-label study,9 42 patients ages 8 to 18 with bipolar disorder type I or II were randomly assigned to lithium, divalproex sodium, or carbamazepine monotherapy for 6 weeks. Response rates—measured as a ≥ 50% change from baseline in YMRS scores—were 53% with divalproex, 38% with lithium, and 38% with carbamazepine.
A retrospective review of 44 hospitalized bipolar patients ages 5 to 12 treated for at least 7 days with lithium, valproate, or carbamazepine reported higher (ie, worse) Clinical Global Impression of Improvement scores with carbamazepine.27 Small sample sizes, particularly in the carbamazepine group, limited this naturalistic study.
Safety: Black-box warnings. Carbamazepine’s hematologic “black box” warns of increased risk of aplastic anemia, agranulocytosis, leukopenia, and thrombocytopenia. Risks associated with carbamazepine have been estimated at:
- aplastic anemia: 5.1/million patient years
- agranulocytosis: 1.4/million patient years.28
Body weight. Carbamazepine is not associated with significant weight gain, which could be clinically important for some patients.
Drug interactions. Carbamazepine activates the cytochrome P-450 liver enzyme system, increasing the metabolism of many medications and decreasing their blood levels. Consider monitoring serum levels when using carbamazepine with valproate, imipramine, corticosteroids, warfarin, oral contraceptives, and some antibiotics. Because carbamazepine induces its own metabolism, you might need to increase its dosage if its effects appear to be waning.3
Carbamazepine and tricyclic antidepressants may show cross-sensitivity because of structural similarity. Do not use monoamine oxidase inhibitors with carbamazepine; discontinue them at least 14 days before starting carbamazepine.3
OXCARBAZEPINE: FEWER INTERACTIONS
Oxcarbazepine has similar efficacy to carbamazepine but less side effect risk and does not require plasma level monitoring. A weaker inducer of CYP-450, it causes fewer clinically important drug-drug interactions and may be useful for patients who respond to carbamazepine but cannot tolerate its side effects.30
Efficacy. Case studies31,32 have been encouraging, but no published, double-blind, placebo-controlled studies support using oxcarbazepine in bipolar children and adolescents.
Safety. Oxcarbazepine appears to be generally well-tolerated but can cause potentially serious reactions—including hyponatremia.33 Somnolence, emesis, and ataxia are the most common side effects in pediatric patients.3
Hyponatremia —plasma sodium 125 mEq/L—occurs in 2.5% of adults taking oxcarbazepine3 and has been reported in a similar percentage of children.34 This potentially severe reaction—characterized by nausea, lethargy, malaise, headache, confusion, decreased seizure threshold, or simply decreased serum sodium35—is usually noted within the first 12 weeks of therapy. The risk increases with concomitant use of other sodium-altering drugs, such as antidepressants or antipsychotics.36
Evaluate serum sodium when starting oxcarbazepine, periodically in the first 3 months, and if symptoms occur.34,36 For sodium levels of 125 to 130 mEq/L, obtain repeat measurements to confirm that hyponatremia is not worsening. Intervention is often required when levels fall below 125 mEq/L.36
Other serious adverse reactions include Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions; 25% to 30% of patients with hypersensitivity to carbamazepine also will react to oxcarbazepine.33
Contraceptive concerns. Oxcarbazepine may reduce contraceptive efficacy by altering estrogen and progesterone plasma concentrations.37 Consider other birth control methods for sexuallyactive bipolar adolescent girls.
LAMOTRIGINE
Neurologists often use lamotrigine for children with atypical seizure disorders, but no controlled data exist on the drug’s efficacy and safety in youths with bipolar disorder.
Efficacy. In a prospective, open-label study,38 13 adolescents with type I bipolar disorder received lamotrigine, 200 to 400 mg/d. After 12 weeks (mean dosage 241 mg/d), their symptoms had improved as shown by these mean scores:
- Montgomery-Asberg Depression Rating Scale: from 21 at baseline to 4 at endpoint
- Clinical Global Impressions–Severity of Illness scale: from 4 to 1
- Children’s Depression Rating Scale (CDRS-R): from 74 to 40
- YMRS: from 20 to 6.
Safety: Severe rash. An age-related association with Stevens-Johnson syndrome may limit pediatric use of lamotrigine. Severe and potentially life-threatening rashes have been reported in 0.8% of children treated with lamotrigine.40 Discontinue lamotrigine if a rash develops, unless it clearly is not drug-related. Three factors that increase rash risk include:
- co-administering lamotrigine with valproate
- higher-than-recommended initial dosages
- rapid dose titration.41
Pediatric dosing. We find no published studies of efficacious dosages and plasma levels of lamotrigine in pediatric bipolar disorder (Table 4).3,9,17 Based on our clinical experience, we recommend starting lamotrigine at 1 to 5 mg/kg/day (1 to 3 mg/kg/day if given with valproate) divided into two daily doses. Watch for rash or skin disorders. Do not exceed the recommended daily dosage by 200 mg in children age
Table 4
Using anticonvulsants in pediatric bipolar disorder patients
| Drug | Recommended dosage | Target serum level |
|---|---|---|
| Carbamazepine | Age 6 to 12: 20 to 30 mg/kg/d | ≥ 7.0 μg/L |
| Age >12: 400 to 1,200 mg/d | ||
| Lamotrigine* | Unknown | Unknown |
| Oxcarbazepine | 11 to 16 mg/kg/d (as adjunct) | Unknown |
| Topiramate* | Unknown | Unknown |
| Valproate | 15 to 20 mg/kg/d | 45 to 125 μg/mL (trough) |
| 85 to 110 μg/mL (target) | ||
| * No published studies found for efficacious dosage and plasma levels in pediatric bipolar disorder. | ||
| Dosage supported by case reports only; no studies found examining efficacious plasma levels. | ||
| Source: References 3,9, and 17. | ||
TOPIRAMATE: LIMITED INFORMATION
Efficacy. Little is known about using topiramate in children and adolescents. A retrospective chart review42 of 26 patients with bipolar disorder type I (n=23) or II (n=3) showed adjunctive topiramate to be effective, with response rates of 73% for mania and 62% overall. Topiramate was well tolerated, and no serious events were reported.
A randomized, controlled trial of topiramate for acute mania in youths with type I bipolar disorder43 was recently halted because of lack of efficacy in adul trials. Preliminary data from 56 of the pediatric patients—analyzed before the study was halted—showed improved YMRS scores. Although results were not statistically significant, the authors suggest topiramate might be effective in treating children and adolescents with bipolar disorder.
Safety: FDA warning. Decreased sodium bicarbonate leading to hyperchloremic metabolic acidosis has been reported in youths treated with topiramate for seizure disorder,44 leading to an FDA warning to prescribers.3 Although no monitoring guidelines exist, we recommend baseline and periodic serum bicarbonate measurements and acidbase evaluations during topiramate treatment, especially when adding other antiepileptics.44
Other rare but serious reactions include:
- impaired sweat production and resultant hyperthermia45
- ophthalmologic symptoms characterized by secondary acute angle closure glaucoma and acute myacute myopia (usually within 1 month of starting treatment)46
- sedation and cognitive difficulties.47
Cognitive effects? Reports of “word finding difficulties” with topiramate47 may suggest cognitive effects. Thus, be very cautious about using this medication in children and adolescents.
Related resources
- Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(3):213-35.
- Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
- American Academy of Child and Adolescent Psychiatry. Treatment guidelines for childhood psychiatric disorders. www.aacap.org
Dr. Kloos, Dr. Hitchcock, and Dr. Ronald Weller report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Elizabeth Weller has been a consultant to or received research grants from GlaxoSmithKline, Johnson & Johnson, Novartis Pharmaceuticals Corp., Abbott Laboratories, and Shire Pharmaceuticals.
Drug brand names
- Carbamazepine • Tegretol
- Divalproex • Depakote
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Oxcarbazepine • Trileptal
- Topiramate • Topamax
1. Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44(3):213-35
2. Wang P, Ketter T, Becker O, et al. New anticonvulsant medication uses in bipolar disorder. CNS Spectrums 2003;8(12):930-47.
3. Prescribing information. Physicians’ Desk Reference (59th ed.) Montvale, NJ: Thomson Healthcare, 2005.
4. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a large open trial. J Am Acad Child Adolesc Psychiatry 2003;42(9):1038-45.
5. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a placebo-controlled discontinuation study. J Am Acad Child Adolesc Psychiatry 2004;43(8):984-93.
6. Strober M, DeAntonio M, Schmidt-Lackner S, et al. Early childhood attention deficit hyperactivity disorder predicts poorer response to acute lithium therapy in adolescent mania. J Affect Disord 1998;51(2):145-51.
7. Geller B, Cooper T, Zimerman B, et al. Lithium for prepubertal depressed children with family history predictors of future bipolarity: a double-blind, placebo-controlled study. J Affect Disord 1998;51(2):165-75.
8. Weller E, Weller R, Fristad M. Lithium dosage guide for prepubertal children: a preliminary report. J Am Acad Child Adolesc Psychiatry 1986;25(1):92-5.
9. Kowatch R, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamazepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39(6):713-20.
10. Hagino O, Weller E, Weller R, et al. Untoward effects of lithium treatment in children aged four through six years. J Am Acad Child Adolesc Psychiatry 1995;34(12):1584-90.
11. Hagino O, Weller E, Weller R, Fristad M. Comparison of lithium dosage methods for preschool- and early school-age children. J Am Acad Child Adolesc Psychiatry 1995;37(1):60-5.
12. Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
13. Holmes L. The North American antiepileptic drug pregnancy registry: a seven-year experience. (paper presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
14. Vajda F, Lander C, Cook M, et al. Antiepileptic medication in pregnancy: the Australian Pregnancy Register: 52 months data (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
15. Messenheimer J, Tennis P, Cunnington M. Eleven-year interim results of an international observational study of pregnancy outcomes following exposure to lamotrigine (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
16. Torbjorn T, Battino D, Bonizzoni E, et al. Eurap: an international registry of antiepileptic drugs and pregnancy (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
17. Wagner K, Weller E, Carlson G, et al. An open-label trial of divalproex in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2002;41(10):1224-30.
18. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
19. Findling R, McNamara N, Gracious B, et al. Combination lithium and divalproex sodium in pediatric bipolarity. J Am Acad Child Adolesc Psychiatry 2003;42(8):895-901.
20. DelBello M, Adler C, Strakowski S. Divalproex for the treatment of aggression associated with adolescent mania. J Child Adolesc Psychopharmacol 2004;14(2):325-8.
21. Anderson G. Children versus adults: pharmokinetic and adverseeffect differences. Epilepsia 2002;43(suppl 3):53-9.
22. Yehya N, Saldarini C, Koski M, et al. Valproate-induced hyperammonemic encephalopathy. J Am Acad Child Adolesc Psychiatry 2004;43(8):926-7.
23. Verrotti A, Greco R, Matera V, et al. Platelet count and function in children receiving sodium valproate. Pediatr Neurol 1999;21(3):611-14.
24. Isojarvi J, Tauboll E, Pakarinen A, et al. Altered ovarian function and cardiovascular risk factors in valproate-treated women. Am J Med 2001;111(4):290-6.
25. Bowden C, Calabrese J, McElroy S, et al. A randomized, placebocontrolled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Arch Gen Psychiatry 2000;57(5):481-9.
26. Findling R, Gracious B, McNamara N, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3(4):202-10.
27. Davanzo P, Gunderson B, Belin T, et al. Mood stabilizers in hospitalized children with bipolar disorder: a retrospective review. Psychiatry Clin Neurosci 2003;57(5):504-10.
28. Pellock J. Carbamazepine side effects in children and adults. Epilepsia 1987;28(suppl 3):64-70.
29. Sobotka J, Alexander B, Cook B. A review of carbamazepine’s hematologic reactions and monitoring. DICP 1990;24(12):1214-19.
30. Hellewell J. Oxcarbazepine (Trileptal) in the treatment of bipolar disorders: a review of efficacy and tolerability. J Affect Disord 2002;72(supp 1):23-34.
31. Davanzo P, Nikore V, Yehya N, Stevenson L. Oxcarbazepine treatment of juvenile-onset bipolar disorder. J Child Adolesc Psychopharmacol 2004;14(3):344-5.
32. Teitelbaum M. Oxcarbazepine in bipolar disorder. J Am Acad Child Adolesc Psychiatry 2001;40(9):993-4.
33. Dietrich D, Kropp S, Emrich H. Oxcarbazepine in affective and schizoaffective disorders. Pharmacopsychiatry 2001;34(6):242-50.
34. Holtmann M, Krause M, Opp J, et al. Oxcarbazepine-induced hyponatremia and the regulation of serum sodium after replacing carbamazepine with oxcarbazepine in children. Neuropediatrics 2002;33(6):298-300.
35. Prescribing information for oxcarbazepine (Trileptal) Novartis Pharmaceuticals Corp. 2005. Available at: http://www.pharma.us. novartis.com/product/pi/pdf/trileptal.pdf. Accessed June 26, 2005.
36. Asconape J. Some common issues in the use of antiepileptic drugs. Semin Neurol 2002;22(1):27-39.
37. Fattore C, Cipolla G, Gatti G, et al. Induction of ethinylestradiol and levonorgestrel metabolism by oxcarbazepine in healthy women. Epilepsia 1999;40(6):783-7.
38. Swope G, Hoopes S, Amy L, et al. An open-label study of lamotrigine in adolescents with bipolar mood disorder (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
39. Saxena K, Howe M, Chang K. Lamotrigine adjunct or monotherapy for adolescent bipolar depression or mixed mania (poster presentation). Washington, DC: American Academy of Child and Adolescent Psychiatry annual meeting, 2004.
40. Prescribing information for lamotrigine (Lamictal). GlaxoSmith Kline 2004. Available at: http://us.gsk.com/products/assets/us_ lamictal.pdf. Accessed June 2, 2005.
41. Messenheimer J, Mullens E, Giorgi L, Young F. Safety review of adult clinical trial experience with lamotrigine. Drug Safety 1998;18(4):281-96.
42. DelBello M, Kowatch R, Warner J, et al. Adjunctive topiramate treatment for pediatric bipolar disorder: a retrospective chart review. J Child Adolesc Psychopharmacol 2002;12(4):323-30.
43. DelBello M, Kushner S, Wang D, et al. Topiramate for acute mania in children and adolescents with bipolar I disorder (abstract). New York: American Psychiatric Association annual meeting, 2004.
44. Philippi H, Boor R, Reitter B. Topiramate and metabolic acidosis in infants and toddlers. Epilepsia 2002;43(7):744-7.
45. Arcas J, Ferrer T, Roche M, et al. Hypohidrosis related to the administration of topiramate to children. Epilepsia 2001;42(10):1363-5.
46. Davanzo P, Cantwell E, Kleiner J, et al. Cognitive changes during topiramate therapy. J Am Acad Child Adolesc Psychiatry 2001;40(3):262-3.
47. McIntyre R, Mancini D, McCann S, et al. Topiramate versus bupropion SR when added to mood stabilizer therapy for the depressive phase of bipolar disorder: a preliminary single-blind study. Bipolar Disord 2002;4(3):207-13.
Are anticonvulsants safe and effective mood stabilizers for children and adolescents with bipolar disorder? The answer is unclear because most bipolar disorder treatment trials have included adults only, and clinicians are desperate for data.1
To help you care for young patients, we report what is known about the potential benefits and risks of using mood stabilizers and anticonvulsants in bipolar youth. We base our dosing, target serum level, and monitoring recommendations on clinical experience and the limited published evidence.
AGENTS OF CHOICE?
Bipolar disorder’s “atypical” presentation in children—often more irritability and explosiveness than euphoria—can complicate diagnosis. Bipolar children and adolescents often have comorbid attention-deficit/hyperactivity disorder (ADHD), other disruptive behavior disorders, or anxiety disorders. Thus, comorbidities and presenting symptoms often dictate medication choice.
An expert consensus guideline acknowledges that more evidence on pediatric bipolar disorder is needed. In the meantime, the guideline suggests trying valproate or lithium first to treat nonpsychotic mania in pediatric bipolar patients.1 It also recommends three atypical antipsychotics— olanzapine, quetiapine, and risperidone—as potential first-line treatments. Valproate and lithium may be preferred because of atypicals’ risk of weight gain and metabolic syndrome.
Trying other anticonvulsants may be justified for bipolar youths who are not functioning well with first-line agents. Lamotrigine, for example, has antidepressant and antimanic effects.2 When you try anticonvulsants that lack double-blind, placebo-controlled trials, we recommend that you:
- obtain consent from the parents and child
- monitor carefully for side effects.
LITHIUM: STRONGEST EVIDENCE
Lithium is one of the most well-studied medications for pediatric bipolar disorder and the only mood stabilizer FDA-approved for children and adolescents (Table 1).3 Although approved for ages 12 and older, lithium has been used in younger children in practice and in clinical trials.
Table 1
FDA-approval status of medications used to treat bipolar disorder
| Medication | Indications for adults | Indications for children |
|---|---|---|
| Carbamazepine | Acute manic episode and acute mixed episode | Not approved |
| Lamotrigine | Maintenance therapy | Not approved |
| Lithium | Acute manic episode and maintenance therapy | Age ≥ 12 years |
| Oxcarbazepine | Not approved | Not approved |
| Topiramate | Not approved | Not approved |
| Valproate | Acute manic episode | Not approved |
| Source: Reference 3 | ||
In the only double-blind, placebo-controlled trial of lithium in adolescents with bipolar disorder, some subjects had secondary substance dependency disorders.7 For 6 weeks, 25 outpatient adolescents received lithium (13 patients) or placebo (12 patients). Lithium was effective in treating bipolar and substance dependency symptoms, with significantly improved clinical global assessment scores and decreased positive urine assays for drugs. Little difference was seen in mood item scores on the Schedule for Affective Disorders and Schizophrenia, child version (KSADS-1986), whether patients were taking lithium or placebo.
Pediatric dosing. For bipolar patients ages 6 to 12, use the child’s weight to determine lithium dosage (Table 2).8 Maintain serum levels between 0.8 and 1.2 mEq/L,9 and check them frequently when starting therapy.10 After mood stabilization, check levels every 1 to 3 months or when you suspect noncompliance. Obtain renal and thyroid function values at baseline and every 4 to 6 months.
Table 2
Guide to dosing lithium for prepubertal school-aged children*
| Doses (mg) | ||||
|---|---|---|---|---|
| Child’s weight (kg) | 8 AM | 12 PM | 6 PM | Total daily |
| 150 | 150 | 300 | 600 | |
| 25 to 40 | 300 | 300 | 300 | 900 |
| 40 to 50 | 300 | 300 | 600 | 1,200 |
| 50 to 60 | 600 | 300 | 600 | 1,500 |
| * Maintain specified dose at least 5 days, drawing serum levels 12 hrs after the last lithium dose until two consecutive levels appear in the therapeutic range (0.6 to 1.2 mEq/L). Dose may then be adjusted based on serum level, side effects, or clinical response. Do not exceed 1.4 mEq/L. | ||||
| Source: Reference 8 | ||||
Consider teratogenicity when choosing mood stabilizers for bipolar adolescent girls who may be sexually active. Lithium, valproate, and carbamazepine are labeled pregnancy category D because of their potential to cause birth defects.
Lithium treatment has been associated with increased risk of cardiac defects, specifically Ebstein’s anomaly (malformation of the tricuspid valve). Its incidence in children of women who used lithium during pregnancy is estimated to be 1:1,000 (0.10%) to 2:1,000 (0.05%)— 20 to 40 times the rate in the general population.12
Valproate.Results from the North American Antiepileptic Drug (AED) Pregnancy Registry showed a 10.7% rate of major congenital malformations (MCM)— including neural tube defects (spina bifida) and cardiac defects (pulmonary atresia)—in children of women who used valproate during pregnancy. The rate of births with MCMs in the general population is 2.9%.13
Carbamazepine.Data from the Australian Pregnancy Registry showed no significant increase in malformation rates in infants of carbamazepine users compared with those of women receiving no antiepileptics.14 Other studies, however, have linked carbamazepine with an increased risk of craniofacial defects (11%), neural tube defects (0.5 to 1%), and cardiac malformations.12
Lamotrigine.The teratogenic effects of the newer anticonvulsants are unclear. An 11-year study of lamotrigine15 found MCM risk after first-trimester exposure to lamotrigine to be similar to the general population’s MCM risk.
Combination therapy.Teratogenic risk appears to increase when multiple antiepileptic drugs are used (9.9% risk in polytherapy vs 6.2% in monotherapy).16
VALPROATE: OPEN-LABEL TRIALS ONLY
Efficacy. No double-blind, placebo-controlled study has shown valproate to be effective in treating bipolar disorder in children and adolescents. When used as monotherapy in open-label studies, valproate has produced response rates of:
- 53% in a 6-week, randomized, open-label trial in which 42 outpatients (mean age 11.4 years) with bipolar disorder type I or II received lithium, divalproex sodium, or carbamazepine9
- 61% in an open-label study of 40 patients ages 7 to 19 with a manic, hypomanic, or mixed episode who received divalproex for 2 to 8 weeks17
- 80% in an 8-week open-label trial of 40 patients ages 6 to 17 with bipolar disorder type I (77.5%) or type II (22.5%) and a Young Mania Rating Scale (YMRS)score ≥ 14.18
Safety: Black-box warnings. Valproate therapy carries risks of hepatic failure, pancreatitis, and birth defects. Monitor blood counts and hepatic enzymes throughout therapy (Table 3).3 Rare yet potentially fatal hepatic toxicity appears to occur most often in children age 21 Other studies suggest:
- an association with congenital malformations, including spina bifida and pulmonary atresia, in children exposed to valproate in utero6
- a link between valproate and hyperammonemic encephalopathy, especially in patients with urea cycle disorders22
- potential for benign thrombocytopenia23
- increased incidence of polycystic ovary syndrome—ovarian cysts, hyperandrogenism, chronic anovulation—in peripubertal mentally retarded women treated with valproate for seizure disorders.24
Table 3
Mood stabilizers’ side effects and recommended monitoring
| Medication | Major side effects | Monitoring |
|---|---|---|
| Carbamazepine | Allergic skin rash, drowsiness, blood dyscrasias, diplopia | CBC with reticulocytes, iron, LFTs, urinalysis, BUN, TFTs, sodium, serum carbamazepine levels |
| Lamotrigine | Stevens-Johnson syndrome, headache, dizziness, ataxia, somnolence, nausea, diplopia, blurred vision, rhinitis | No serum monitoring recommended |
| Lithium | Polyuria, polydipsia, nausea, diarrhea, tininecleatremor, enuresis, fatigue, ataxia, leukocytosis, malaise, cardiac arrhythmias, weight gain | BUN/creatinine, crearance, TFTs, calcium/phosphorus, ECG, serum lithium levels every 1 to 3 months once stabilized |
| Oxcarbazepine | Dizziness, somnolence/fatigue, ataxia/gait disturbance, vertigo, headache, tremor, rash, hyponatremia, hypersensitivity reaction, GI symptoms, diplopia | Sodium levels (particularly ifirst 3 months) |
| Topiramate | Hyperchloremic metabolic acidosis, oligohydrosis and hyperthermia, acute myopia, somnolence/fatigue, nausea, anorexia/weight loss, paresthesia, tremor, difficulty concentrating | BUN/creatinine, sodium bicarbonate |
| Valproate | Irritability/restlessness, ataxia, headache, weight gain, hyperammonemic encephalopathy, alopecia, GI upset, pancreatitis,sedation, thrombocytopenia, liver failure, polycystic ovaries/hyperandrogenism, teratogenic effects,rash | Ammonia, LFTs, bilirubin, CBC with platelets, serum valproate levels |
| BUN: blood urea nitrogen; CBC: complete blood count; ECG: electrocardiography; LFT: liver function tests; TFTs: thyroid function tests | ||
| Note: Bolded items included in black-box warnings | ||
| Source: Reference 3 | ||
CARBAMAZEPINE: DRUG INTERACTION RISK
Carbamazepine is used less often than lithium or divalproex for bipolar disorder. It tends to be used adjunctively when lithium alone is ineffective.
Efficacy. In an open-label study,9 42 patients ages 8 to 18 with bipolar disorder type I or II were randomly assigned to lithium, divalproex sodium, or carbamazepine monotherapy for 6 weeks. Response rates—measured as a ≥ 50% change from baseline in YMRS scores—were 53% with divalproex, 38% with lithium, and 38% with carbamazepine.
A retrospective review of 44 hospitalized bipolar patients ages 5 to 12 treated for at least 7 days with lithium, valproate, or carbamazepine reported higher (ie, worse) Clinical Global Impression of Improvement scores with carbamazepine.27 Small sample sizes, particularly in the carbamazepine group, limited this naturalistic study.
Safety: Black-box warnings. Carbamazepine’s hematologic “black box” warns of increased risk of aplastic anemia, agranulocytosis, leukopenia, and thrombocytopenia. Risks associated with carbamazepine have been estimated at:
- aplastic anemia: 5.1/million patient years
- agranulocytosis: 1.4/million patient years.28
Body weight. Carbamazepine is not associated with significant weight gain, which could be clinically important for some patients.
Drug interactions. Carbamazepine activates the cytochrome P-450 liver enzyme system, increasing the metabolism of many medications and decreasing their blood levels. Consider monitoring serum levels when using carbamazepine with valproate, imipramine, corticosteroids, warfarin, oral contraceptives, and some antibiotics. Because carbamazepine induces its own metabolism, you might need to increase its dosage if its effects appear to be waning.3
Carbamazepine and tricyclic antidepressants may show cross-sensitivity because of structural similarity. Do not use monoamine oxidase inhibitors with carbamazepine; discontinue them at least 14 days before starting carbamazepine.3
OXCARBAZEPINE: FEWER INTERACTIONS
Oxcarbazepine has similar efficacy to carbamazepine but less side effect risk and does not require plasma level monitoring. A weaker inducer of CYP-450, it causes fewer clinically important drug-drug interactions and may be useful for patients who respond to carbamazepine but cannot tolerate its side effects.30
Efficacy. Case studies31,32 have been encouraging, but no published, double-blind, placebo-controlled studies support using oxcarbazepine in bipolar children and adolescents.
Safety. Oxcarbazepine appears to be generally well-tolerated but can cause potentially serious reactions—including hyponatremia.33 Somnolence, emesis, and ataxia are the most common side effects in pediatric patients.3
Hyponatremia —plasma sodium 125 mEq/L—occurs in 2.5% of adults taking oxcarbazepine3 and has been reported in a similar percentage of children.34 This potentially severe reaction—characterized by nausea, lethargy, malaise, headache, confusion, decreased seizure threshold, or simply decreased serum sodium35—is usually noted within the first 12 weeks of therapy. The risk increases with concomitant use of other sodium-altering drugs, such as antidepressants or antipsychotics.36
Evaluate serum sodium when starting oxcarbazepine, periodically in the first 3 months, and if symptoms occur.34,36 For sodium levels of 125 to 130 mEq/L, obtain repeat measurements to confirm that hyponatremia is not worsening. Intervention is often required when levels fall below 125 mEq/L.36
Other serious adverse reactions include Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions; 25% to 30% of patients with hypersensitivity to carbamazepine also will react to oxcarbazepine.33
Contraceptive concerns. Oxcarbazepine may reduce contraceptive efficacy by altering estrogen and progesterone plasma concentrations.37 Consider other birth control methods for sexuallyactive bipolar adolescent girls.
LAMOTRIGINE
Neurologists often use lamotrigine for children with atypical seizure disorders, but no controlled data exist on the drug’s efficacy and safety in youths with bipolar disorder.
Efficacy. In a prospective, open-label study,38 13 adolescents with type I bipolar disorder received lamotrigine, 200 to 400 mg/d. After 12 weeks (mean dosage 241 mg/d), their symptoms had improved as shown by these mean scores:
- Montgomery-Asberg Depression Rating Scale: from 21 at baseline to 4 at endpoint
- Clinical Global Impressions–Severity of Illness scale: from 4 to 1
- Children’s Depression Rating Scale (CDRS-R): from 74 to 40
- YMRS: from 20 to 6.
Safety: Severe rash. An age-related association with Stevens-Johnson syndrome may limit pediatric use of lamotrigine. Severe and potentially life-threatening rashes have been reported in 0.8% of children treated with lamotrigine.40 Discontinue lamotrigine if a rash develops, unless it clearly is not drug-related. Three factors that increase rash risk include:
- co-administering lamotrigine with valproate
- higher-than-recommended initial dosages
- rapid dose titration.41
Pediatric dosing. We find no published studies of efficacious dosages and plasma levels of lamotrigine in pediatric bipolar disorder (Table 4).3,9,17 Based on our clinical experience, we recommend starting lamotrigine at 1 to 5 mg/kg/day (1 to 3 mg/kg/day if given with valproate) divided into two daily doses. Watch for rash or skin disorders. Do not exceed the recommended daily dosage by 200 mg in children age
Table 4
Using anticonvulsants in pediatric bipolar disorder patients
| Drug | Recommended dosage | Target serum level |
|---|---|---|
| Carbamazepine | Age 6 to 12: 20 to 30 mg/kg/d | ≥ 7.0 μg/L |
| Age >12: 400 to 1,200 mg/d | ||
| Lamotrigine* | Unknown | Unknown |
| Oxcarbazepine | 11 to 16 mg/kg/d (as adjunct) | Unknown |
| Topiramate* | Unknown | Unknown |
| Valproate | 15 to 20 mg/kg/d | 45 to 125 μg/mL (trough) |
| 85 to 110 μg/mL (target) | ||
| * No published studies found for efficacious dosage and plasma levels in pediatric bipolar disorder. | ||
| Dosage supported by case reports only; no studies found examining efficacious plasma levels. | ||
| Source: References 3,9, and 17. | ||
TOPIRAMATE: LIMITED INFORMATION
Efficacy. Little is known about using topiramate in children and adolescents. A retrospective chart review42 of 26 patients with bipolar disorder type I (n=23) or II (n=3) showed adjunctive topiramate to be effective, with response rates of 73% for mania and 62% overall. Topiramate was well tolerated, and no serious events were reported.
A randomized, controlled trial of topiramate for acute mania in youths with type I bipolar disorder43 was recently halted because of lack of efficacy in adul trials. Preliminary data from 56 of the pediatric patients—analyzed before the study was halted—showed improved YMRS scores. Although results were not statistically significant, the authors suggest topiramate might be effective in treating children and adolescents with bipolar disorder.
Safety: FDA warning. Decreased sodium bicarbonate leading to hyperchloremic metabolic acidosis has been reported in youths treated with topiramate for seizure disorder,44 leading to an FDA warning to prescribers.3 Although no monitoring guidelines exist, we recommend baseline and periodic serum bicarbonate measurements and acidbase evaluations during topiramate treatment, especially when adding other antiepileptics.44
Other rare but serious reactions include:
- impaired sweat production and resultant hyperthermia45
- ophthalmologic symptoms characterized by secondary acute angle closure glaucoma and acute myacute myopia (usually within 1 month of starting treatment)46
- sedation and cognitive difficulties.47
Cognitive effects? Reports of “word finding difficulties” with topiramate47 may suggest cognitive effects. Thus, be very cautious about using this medication in children and adolescents.
Related resources
- Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(3):213-35.
- Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
- American Academy of Child and Adolescent Psychiatry. Treatment guidelines for childhood psychiatric disorders. www.aacap.org
Dr. Kloos, Dr. Hitchcock, and Dr. Ronald Weller report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Elizabeth Weller has been a consultant to or received research grants from GlaxoSmithKline, Johnson & Johnson, Novartis Pharmaceuticals Corp., Abbott Laboratories, and Shire Pharmaceuticals.
Drug brand names
- Carbamazepine • Tegretol
- Divalproex • Depakote
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Oxcarbazepine • Trileptal
- Topiramate • Topamax
Are anticonvulsants safe and effective mood stabilizers for children and adolescents with bipolar disorder? The answer is unclear because most bipolar disorder treatment trials have included adults only, and clinicians are desperate for data.1
To help you care for young patients, we report what is known about the potential benefits and risks of using mood stabilizers and anticonvulsants in bipolar youth. We base our dosing, target serum level, and monitoring recommendations on clinical experience and the limited published evidence.
AGENTS OF CHOICE?
Bipolar disorder’s “atypical” presentation in children—often more irritability and explosiveness than euphoria—can complicate diagnosis. Bipolar children and adolescents often have comorbid attention-deficit/hyperactivity disorder (ADHD), other disruptive behavior disorders, or anxiety disorders. Thus, comorbidities and presenting symptoms often dictate medication choice.
An expert consensus guideline acknowledges that more evidence on pediatric bipolar disorder is needed. In the meantime, the guideline suggests trying valproate or lithium first to treat nonpsychotic mania in pediatric bipolar patients.1 It also recommends three atypical antipsychotics— olanzapine, quetiapine, and risperidone—as potential first-line treatments. Valproate and lithium may be preferred because of atypicals’ risk of weight gain and metabolic syndrome.
Trying other anticonvulsants may be justified for bipolar youths who are not functioning well with first-line agents. Lamotrigine, for example, has antidepressant and antimanic effects.2 When you try anticonvulsants that lack double-blind, placebo-controlled trials, we recommend that you:
- obtain consent from the parents and child
- monitor carefully for side effects.
LITHIUM: STRONGEST EVIDENCE
Lithium is one of the most well-studied medications for pediatric bipolar disorder and the only mood stabilizer FDA-approved for children and adolescents (Table 1).3 Although approved for ages 12 and older, lithium has been used in younger children in practice and in clinical trials.
Table 1
FDA-approval status of medications used to treat bipolar disorder
| Medication | Indications for adults | Indications for children |
|---|---|---|
| Carbamazepine | Acute manic episode and acute mixed episode | Not approved |
| Lamotrigine | Maintenance therapy | Not approved |
| Lithium | Acute manic episode and maintenance therapy | Age ≥ 12 years |
| Oxcarbazepine | Not approved | Not approved |
| Topiramate | Not approved | Not approved |
| Valproate | Acute manic episode | Not approved |
| Source: Reference 3 | ||
In the only double-blind, placebo-controlled trial of lithium in adolescents with bipolar disorder, some subjects had secondary substance dependency disorders.7 For 6 weeks, 25 outpatient adolescents received lithium (13 patients) or placebo (12 patients). Lithium was effective in treating bipolar and substance dependency symptoms, with significantly improved clinical global assessment scores and decreased positive urine assays for drugs. Little difference was seen in mood item scores on the Schedule for Affective Disorders and Schizophrenia, child version (KSADS-1986), whether patients were taking lithium or placebo.
Pediatric dosing. For bipolar patients ages 6 to 12, use the child’s weight to determine lithium dosage (Table 2).8 Maintain serum levels between 0.8 and 1.2 mEq/L,9 and check them frequently when starting therapy.10 After mood stabilization, check levels every 1 to 3 months or when you suspect noncompliance. Obtain renal and thyroid function values at baseline and every 4 to 6 months.
Table 2
Guide to dosing lithium for prepubertal school-aged children*
| Doses (mg) | ||||
|---|---|---|---|---|
| Child’s weight (kg) | 8 AM | 12 PM | 6 PM | Total daily |
| 150 | 150 | 300 | 600 | |
| 25 to 40 | 300 | 300 | 300 | 900 |
| 40 to 50 | 300 | 300 | 600 | 1,200 |
| 50 to 60 | 600 | 300 | 600 | 1,500 |
| * Maintain specified dose at least 5 days, drawing serum levels 12 hrs after the last lithium dose until two consecutive levels appear in the therapeutic range (0.6 to 1.2 mEq/L). Dose may then be adjusted based on serum level, side effects, or clinical response. Do not exceed 1.4 mEq/L. | ||||
| Source: Reference 8 | ||||
Consider teratogenicity when choosing mood stabilizers for bipolar adolescent girls who may be sexually active. Lithium, valproate, and carbamazepine are labeled pregnancy category D because of their potential to cause birth defects.
Lithium treatment has been associated with increased risk of cardiac defects, specifically Ebstein’s anomaly (malformation of the tricuspid valve). Its incidence in children of women who used lithium during pregnancy is estimated to be 1:1,000 (0.10%) to 2:1,000 (0.05%)— 20 to 40 times the rate in the general population.12
Valproate.Results from the North American Antiepileptic Drug (AED) Pregnancy Registry showed a 10.7% rate of major congenital malformations (MCM)— including neural tube defects (spina bifida) and cardiac defects (pulmonary atresia)—in children of women who used valproate during pregnancy. The rate of births with MCMs in the general population is 2.9%.13
Carbamazepine.Data from the Australian Pregnancy Registry showed no significant increase in malformation rates in infants of carbamazepine users compared with those of women receiving no antiepileptics.14 Other studies, however, have linked carbamazepine with an increased risk of craniofacial defects (11%), neural tube defects (0.5 to 1%), and cardiac malformations.12
Lamotrigine.The teratogenic effects of the newer anticonvulsants are unclear. An 11-year study of lamotrigine15 found MCM risk after first-trimester exposure to lamotrigine to be similar to the general population’s MCM risk.
Combination therapy.Teratogenic risk appears to increase when multiple antiepileptic drugs are used (9.9% risk in polytherapy vs 6.2% in monotherapy).16
VALPROATE: OPEN-LABEL TRIALS ONLY
Efficacy. No double-blind, placebo-controlled study has shown valproate to be effective in treating bipolar disorder in children and adolescents. When used as monotherapy in open-label studies, valproate has produced response rates of:
- 53% in a 6-week, randomized, open-label trial in which 42 outpatients (mean age 11.4 years) with bipolar disorder type I or II received lithium, divalproex sodium, or carbamazepine9
- 61% in an open-label study of 40 patients ages 7 to 19 with a manic, hypomanic, or mixed episode who received divalproex for 2 to 8 weeks17
- 80% in an 8-week open-label trial of 40 patients ages 6 to 17 with bipolar disorder type I (77.5%) or type II (22.5%) and a Young Mania Rating Scale (YMRS)score ≥ 14.18
Safety: Black-box warnings. Valproate therapy carries risks of hepatic failure, pancreatitis, and birth defects. Monitor blood counts and hepatic enzymes throughout therapy (Table 3).3 Rare yet potentially fatal hepatic toxicity appears to occur most often in children age 21 Other studies suggest:
- an association with congenital malformations, including spina bifida and pulmonary atresia, in children exposed to valproate in utero6
- a link between valproate and hyperammonemic encephalopathy, especially in patients with urea cycle disorders22
- potential for benign thrombocytopenia23
- increased incidence of polycystic ovary syndrome—ovarian cysts, hyperandrogenism, chronic anovulation—in peripubertal mentally retarded women treated with valproate for seizure disorders.24
Table 3
Mood stabilizers’ side effects and recommended monitoring
| Medication | Major side effects | Monitoring |
|---|---|---|
| Carbamazepine | Allergic skin rash, drowsiness, blood dyscrasias, diplopia | CBC with reticulocytes, iron, LFTs, urinalysis, BUN, TFTs, sodium, serum carbamazepine levels |
| Lamotrigine | Stevens-Johnson syndrome, headache, dizziness, ataxia, somnolence, nausea, diplopia, blurred vision, rhinitis | No serum monitoring recommended |
| Lithium | Polyuria, polydipsia, nausea, diarrhea, tininecleatremor, enuresis, fatigue, ataxia, leukocytosis, malaise, cardiac arrhythmias, weight gain | BUN/creatinine, crearance, TFTs, calcium/phosphorus, ECG, serum lithium levels every 1 to 3 months once stabilized |
| Oxcarbazepine | Dizziness, somnolence/fatigue, ataxia/gait disturbance, vertigo, headache, tremor, rash, hyponatremia, hypersensitivity reaction, GI symptoms, diplopia | Sodium levels (particularly ifirst 3 months) |
| Topiramate | Hyperchloremic metabolic acidosis, oligohydrosis and hyperthermia, acute myopia, somnolence/fatigue, nausea, anorexia/weight loss, paresthesia, tremor, difficulty concentrating | BUN/creatinine, sodium bicarbonate |
| Valproate | Irritability/restlessness, ataxia, headache, weight gain, hyperammonemic encephalopathy, alopecia, GI upset, pancreatitis,sedation, thrombocytopenia, liver failure, polycystic ovaries/hyperandrogenism, teratogenic effects,rash | Ammonia, LFTs, bilirubin, CBC with platelets, serum valproate levels |
| BUN: blood urea nitrogen; CBC: complete blood count; ECG: electrocardiography; LFT: liver function tests; TFTs: thyroid function tests | ||
| Note: Bolded items included in black-box warnings | ||
| Source: Reference 3 | ||
CARBAMAZEPINE: DRUG INTERACTION RISK
Carbamazepine is used less often than lithium or divalproex for bipolar disorder. It tends to be used adjunctively when lithium alone is ineffective.
Efficacy. In an open-label study,9 42 patients ages 8 to 18 with bipolar disorder type I or II were randomly assigned to lithium, divalproex sodium, or carbamazepine monotherapy for 6 weeks. Response rates—measured as a ≥ 50% change from baseline in YMRS scores—were 53% with divalproex, 38% with lithium, and 38% with carbamazepine.
A retrospective review of 44 hospitalized bipolar patients ages 5 to 12 treated for at least 7 days with lithium, valproate, or carbamazepine reported higher (ie, worse) Clinical Global Impression of Improvement scores with carbamazepine.27 Small sample sizes, particularly in the carbamazepine group, limited this naturalistic study.
Safety: Black-box warnings. Carbamazepine’s hematologic “black box” warns of increased risk of aplastic anemia, agranulocytosis, leukopenia, and thrombocytopenia. Risks associated with carbamazepine have been estimated at:
- aplastic anemia: 5.1/million patient years
- agranulocytosis: 1.4/million patient years.28
Body weight. Carbamazepine is not associated with significant weight gain, which could be clinically important for some patients.
Drug interactions. Carbamazepine activates the cytochrome P-450 liver enzyme system, increasing the metabolism of many medications and decreasing their blood levels. Consider monitoring serum levels when using carbamazepine with valproate, imipramine, corticosteroids, warfarin, oral contraceptives, and some antibiotics. Because carbamazepine induces its own metabolism, you might need to increase its dosage if its effects appear to be waning.3
Carbamazepine and tricyclic antidepressants may show cross-sensitivity because of structural similarity. Do not use monoamine oxidase inhibitors with carbamazepine; discontinue them at least 14 days before starting carbamazepine.3
OXCARBAZEPINE: FEWER INTERACTIONS
Oxcarbazepine has similar efficacy to carbamazepine but less side effect risk and does not require plasma level monitoring. A weaker inducer of CYP-450, it causes fewer clinically important drug-drug interactions and may be useful for patients who respond to carbamazepine but cannot tolerate its side effects.30
Efficacy. Case studies31,32 have been encouraging, but no published, double-blind, placebo-controlled studies support using oxcarbazepine in bipolar children and adolescents.
Safety. Oxcarbazepine appears to be generally well-tolerated but can cause potentially serious reactions—including hyponatremia.33 Somnolence, emesis, and ataxia are the most common side effects in pediatric patients.3
Hyponatremia —plasma sodium 125 mEq/L—occurs in 2.5% of adults taking oxcarbazepine3 and has been reported in a similar percentage of children.34 This potentially severe reaction—characterized by nausea, lethargy, malaise, headache, confusion, decreased seizure threshold, or simply decreased serum sodium35—is usually noted within the first 12 weeks of therapy. The risk increases with concomitant use of other sodium-altering drugs, such as antidepressants or antipsychotics.36
Evaluate serum sodium when starting oxcarbazepine, periodically in the first 3 months, and if symptoms occur.34,36 For sodium levels of 125 to 130 mEq/L, obtain repeat measurements to confirm that hyponatremia is not worsening. Intervention is often required when levels fall below 125 mEq/L.36
Other serious adverse reactions include Stevens-Johnson syndrome, toxic epidermal necrolysis, and hypersensitivity reactions; 25% to 30% of patients with hypersensitivity to carbamazepine also will react to oxcarbazepine.33
Contraceptive concerns. Oxcarbazepine may reduce contraceptive efficacy by altering estrogen and progesterone plasma concentrations.37 Consider other birth control methods for sexuallyactive bipolar adolescent girls.
LAMOTRIGINE
Neurologists often use lamotrigine for children with atypical seizure disorders, but no controlled data exist on the drug’s efficacy and safety in youths with bipolar disorder.
Efficacy. In a prospective, open-label study,38 13 adolescents with type I bipolar disorder received lamotrigine, 200 to 400 mg/d. After 12 weeks (mean dosage 241 mg/d), their symptoms had improved as shown by these mean scores:
- Montgomery-Asberg Depression Rating Scale: from 21 at baseline to 4 at endpoint
- Clinical Global Impressions–Severity of Illness scale: from 4 to 1
- Children’s Depression Rating Scale (CDRS-R): from 74 to 40
- YMRS: from 20 to 6.
Safety: Severe rash. An age-related association with Stevens-Johnson syndrome may limit pediatric use of lamotrigine. Severe and potentially life-threatening rashes have been reported in 0.8% of children treated with lamotrigine.40 Discontinue lamotrigine if a rash develops, unless it clearly is not drug-related. Three factors that increase rash risk include:
- co-administering lamotrigine with valproate
- higher-than-recommended initial dosages
- rapid dose titration.41
Pediatric dosing. We find no published studies of efficacious dosages and plasma levels of lamotrigine in pediatric bipolar disorder (Table 4).3,9,17 Based on our clinical experience, we recommend starting lamotrigine at 1 to 5 mg/kg/day (1 to 3 mg/kg/day if given with valproate) divided into two daily doses. Watch for rash or skin disorders. Do not exceed the recommended daily dosage by 200 mg in children age
Table 4
Using anticonvulsants in pediatric bipolar disorder patients
| Drug | Recommended dosage | Target serum level |
|---|---|---|
| Carbamazepine | Age 6 to 12: 20 to 30 mg/kg/d | ≥ 7.0 μg/L |
| Age >12: 400 to 1,200 mg/d | ||
| Lamotrigine* | Unknown | Unknown |
| Oxcarbazepine | 11 to 16 mg/kg/d (as adjunct) | Unknown |
| Topiramate* | Unknown | Unknown |
| Valproate | 15 to 20 mg/kg/d | 45 to 125 μg/mL (trough) |
| 85 to 110 μg/mL (target) | ||
| * No published studies found for efficacious dosage and plasma levels in pediatric bipolar disorder. | ||
| Dosage supported by case reports only; no studies found examining efficacious plasma levels. | ||
| Source: References 3,9, and 17. | ||
TOPIRAMATE: LIMITED INFORMATION
Efficacy. Little is known about using topiramate in children and adolescents. A retrospective chart review42 of 26 patients with bipolar disorder type I (n=23) or II (n=3) showed adjunctive topiramate to be effective, with response rates of 73% for mania and 62% overall. Topiramate was well tolerated, and no serious events were reported.
A randomized, controlled trial of topiramate for acute mania in youths with type I bipolar disorder43 was recently halted because of lack of efficacy in adul trials. Preliminary data from 56 of the pediatric patients—analyzed before the study was halted—showed improved YMRS scores. Although results were not statistically significant, the authors suggest topiramate might be effective in treating children and adolescents with bipolar disorder.
Safety: FDA warning. Decreased sodium bicarbonate leading to hyperchloremic metabolic acidosis has been reported in youths treated with topiramate for seizure disorder,44 leading to an FDA warning to prescribers.3 Although no monitoring guidelines exist, we recommend baseline and periodic serum bicarbonate measurements and acidbase evaluations during topiramate treatment, especially when adding other antiepileptics.44
Other rare but serious reactions include:
- impaired sweat production and resultant hyperthermia45
- ophthalmologic symptoms characterized by secondary acute angle closure glaucoma and acute myacute myopia (usually within 1 month of starting treatment)46
- sedation and cognitive difficulties.47
Cognitive effects? Reports of “word finding difficulties” with topiramate47 may suggest cognitive effects. Thus, be very cautious about using this medication in children and adolescents.
Related resources
- Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2005;44(3):213-35.
- Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
- American Academy of Child and Adolescent Psychiatry. Treatment guidelines for childhood psychiatric disorders. www.aacap.org
Dr. Kloos, Dr. Hitchcock, and Dr. Ronald Weller report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Elizabeth Weller has been a consultant to or received research grants from GlaxoSmithKline, Johnson & Johnson, Novartis Pharmaceuticals Corp., Abbott Laboratories, and Shire Pharmaceuticals.
Drug brand names
- Carbamazepine • Tegretol
- Divalproex • Depakote
- Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid, others
- Oxcarbazepine • Trileptal
- Topiramate • Topamax
1. Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44(3):213-35
2. Wang P, Ketter T, Becker O, et al. New anticonvulsant medication uses in bipolar disorder. CNS Spectrums 2003;8(12):930-47.
3. Prescribing information. Physicians’ Desk Reference (59th ed.) Montvale, NJ: Thomson Healthcare, 2005.
4. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a large open trial. J Am Acad Child Adolesc Psychiatry 2003;42(9):1038-45.
5. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a placebo-controlled discontinuation study. J Am Acad Child Adolesc Psychiatry 2004;43(8):984-93.
6. Strober M, DeAntonio M, Schmidt-Lackner S, et al. Early childhood attention deficit hyperactivity disorder predicts poorer response to acute lithium therapy in adolescent mania. J Affect Disord 1998;51(2):145-51.
7. Geller B, Cooper T, Zimerman B, et al. Lithium for prepubertal depressed children with family history predictors of future bipolarity: a double-blind, placebo-controlled study. J Affect Disord 1998;51(2):165-75.
8. Weller E, Weller R, Fristad M. Lithium dosage guide for prepubertal children: a preliminary report. J Am Acad Child Adolesc Psychiatry 1986;25(1):92-5.
9. Kowatch R, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamazepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39(6):713-20.
10. Hagino O, Weller E, Weller R, et al. Untoward effects of lithium treatment in children aged four through six years. J Am Acad Child Adolesc Psychiatry 1995;34(12):1584-90.
11. Hagino O, Weller E, Weller R, Fristad M. Comparison of lithium dosage methods for preschool- and early school-age children. J Am Acad Child Adolesc Psychiatry 1995;37(1):60-5.
12. Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
13. Holmes L. The North American antiepileptic drug pregnancy registry: a seven-year experience. (paper presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
14. Vajda F, Lander C, Cook M, et al. Antiepileptic medication in pregnancy: the Australian Pregnancy Register: 52 months data (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
15. Messenheimer J, Tennis P, Cunnington M. Eleven-year interim results of an international observational study of pregnancy outcomes following exposure to lamotrigine (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
16. Torbjorn T, Battino D, Bonizzoni E, et al. Eurap: an international registry of antiepileptic drugs and pregnancy (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
17. Wagner K, Weller E, Carlson G, et al. An open-label trial of divalproex in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2002;41(10):1224-30.
18. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
19. Findling R, McNamara N, Gracious B, et al. Combination lithium and divalproex sodium in pediatric bipolarity. J Am Acad Child Adolesc Psychiatry 2003;42(8):895-901.
20. DelBello M, Adler C, Strakowski S. Divalproex for the treatment of aggression associated with adolescent mania. J Child Adolesc Psychopharmacol 2004;14(2):325-8.
21. Anderson G. Children versus adults: pharmokinetic and adverseeffect differences. Epilepsia 2002;43(suppl 3):53-9.
22. Yehya N, Saldarini C, Koski M, et al. Valproate-induced hyperammonemic encephalopathy. J Am Acad Child Adolesc Psychiatry 2004;43(8):926-7.
23. Verrotti A, Greco R, Matera V, et al. Platelet count and function in children receiving sodium valproate. Pediatr Neurol 1999;21(3):611-14.
24. Isojarvi J, Tauboll E, Pakarinen A, et al. Altered ovarian function and cardiovascular risk factors in valproate-treated women. Am J Med 2001;111(4):290-6.
25. Bowden C, Calabrese J, McElroy S, et al. A randomized, placebocontrolled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Arch Gen Psychiatry 2000;57(5):481-9.
26. Findling R, Gracious B, McNamara N, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3(4):202-10.
27. Davanzo P, Gunderson B, Belin T, et al. Mood stabilizers in hospitalized children with bipolar disorder: a retrospective review. Psychiatry Clin Neurosci 2003;57(5):504-10.
28. Pellock J. Carbamazepine side effects in children and adults. Epilepsia 1987;28(suppl 3):64-70.
29. Sobotka J, Alexander B, Cook B. A review of carbamazepine’s hematologic reactions and monitoring. DICP 1990;24(12):1214-19.
30. Hellewell J. Oxcarbazepine (Trileptal) in the treatment of bipolar disorders: a review of efficacy and tolerability. J Affect Disord 2002;72(supp 1):23-34.
31. Davanzo P, Nikore V, Yehya N, Stevenson L. Oxcarbazepine treatment of juvenile-onset bipolar disorder. J Child Adolesc Psychopharmacol 2004;14(3):344-5.
32. Teitelbaum M. Oxcarbazepine in bipolar disorder. J Am Acad Child Adolesc Psychiatry 2001;40(9):993-4.
33. Dietrich D, Kropp S, Emrich H. Oxcarbazepine in affective and schizoaffective disorders. Pharmacopsychiatry 2001;34(6):242-50.
34. Holtmann M, Krause M, Opp J, et al. Oxcarbazepine-induced hyponatremia and the regulation of serum sodium after replacing carbamazepine with oxcarbazepine in children. Neuropediatrics 2002;33(6):298-300.
35. Prescribing information for oxcarbazepine (Trileptal) Novartis Pharmaceuticals Corp. 2005. Available at: http://www.pharma.us. novartis.com/product/pi/pdf/trileptal.pdf. Accessed June 26, 2005.
36. Asconape J. Some common issues in the use of antiepileptic drugs. Semin Neurol 2002;22(1):27-39.
37. Fattore C, Cipolla G, Gatti G, et al. Induction of ethinylestradiol and levonorgestrel metabolism by oxcarbazepine in healthy women. Epilepsia 1999;40(6):783-7.
38. Swope G, Hoopes S, Amy L, et al. An open-label study of lamotrigine in adolescents with bipolar mood disorder (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
39. Saxena K, Howe M, Chang K. Lamotrigine adjunct or monotherapy for adolescent bipolar depression or mixed mania (poster presentation). Washington, DC: American Academy of Child and Adolescent Psychiatry annual meeting, 2004.
40. Prescribing information for lamotrigine (Lamictal). GlaxoSmith Kline 2004. Available at: http://us.gsk.com/products/assets/us_ lamictal.pdf. Accessed June 2, 2005.
41. Messenheimer J, Mullens E, Giorgi L, Young F. Safety review of adult clinical trial experience with lamotrigine. Drug Safety 1998;18(4):281-96.
42. DelBello M, Kowatch R, Warner J, et al. Adjunctive topiramate treatment for pediatric bipolar disorder: a retrospective chart review. J Child Adolesc Psychopharmacol 2002;12(4):323-30.
43. DelBello M, Kushner S, Wang D, et al. Topiramate for acute mania in children and adolescents with bipolar I disorder (abstract). New York: American Psychiatric Association annual meeting, 2004.
44. Philippi H, Boor R, Reitter B. Topiramate and metabolic acidosis in infants and toddlers. Epilepsia 2002;43(7):744-7.
45. Arcas J, Ferrer T, Roche M, et al. Hypohidrosis related to the administration of topiramate to children. Epilepsia 2001;42(10):1363-5.
46. Davanzo P, Cantwell E, Kleiner J, et al. Cognitive changes during topiramate therapy. J Am Acad Child Adolesc Psychiatry 2001;40(3):262-3.
47. McIntyre R, Mancini D, McCann S, et al. Topiramate versus bupropion SR when added to mood stabilizer therapy for the depressive phase of bipolar disorder: a preliminary single-blind study. Bipolar Disord 2002;4(3):207-13.
1. Kowatch R, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44(3):213-35
2. Wang P, Ketter T, Becker O, et al. New anticonvulsant medication uses in bipolar disorder. CNS Spectrums 2003;8(12):930-47.
3. Prescribing information. Physicians’ Desk Reference (59th ed.) Montvale, NJ: Thomson Healthcare, 2005.
4. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a large open trial. J Am Acad Child Adolesc Psychiatry 2003;42(9):1038-45.
5. Kafantaris V, Coletti D, Dicker R, et al. Lithium treatment of acute mania in adolescents: a placebo-controlled discontinuation study. J Am Acad Child Adolesc Psychiatry 2004;43(8):984-93.
6. Strober M, DeAntonio M, Schmidt-Lackner S, et al. Early childhood attention deficit hyperactivity disorder predicts poorer response to acute lithium therapy in adolescent mania. J Affect Disord 1998;51(2):145-51.
7. Geller B, Cooper T, Zimerman B, et al. Lithium for prepubertal depressed children with family history predictors of future bipolarity: a double-blind, placebo-controlled study. J Affect Disord 1998;51(2):165-75.
8. Weller E, Weller R, Fristad M. Lithium dosage guide for prepubertal children: a preliminary report. J Am Acad Child Adolesc Psychiatry 1986;25(1):92-5.
9. Kowatch R, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamazepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39(6):713-20.
10. Hagino O, Weller E, Weller R, et al. Untoward effects of lithium treatment in children aged four through six years. J Am Acad Child Adolesc Psychiatry 1995;34(12):1584-90.
11. Hagino O, Weller E, Weller R, Fristad M. Comparison of lithium dosage methods for preschool- and early school-age children. J Am Acad Child Adolesc Psychiatry 1995;37(1):60-5.
12. Yonkers K, Wisner K, Stowe Z, et al. Management of bipolar disorder during pregnancy and the postpartum period. Am J Psychiatry 2004;161(4):608-20.
13. Holmes L. The North American antiepileptic drug pregnancy registry: a seven-year experience. (paper presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
14. Vajda F, Lander C, Cook M, et al. Antiepileptic medication in pregnancy: the Australian Pregnancy Register: 52 months data (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
15. Messenheimer J, Tennis P, Cunnington M. Eleven-year interim results of an international observational study of pregnancy outcomes following exposure to lamotrigine (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
16. Torbjorn T, Battino D, Bonizzoni E, et al. Eurap: an international registry of antiepileptic drugs and pregnancy (poster presentation). New Orleans: American Epilepsy Society annual meeting, 2004.
17. Wagner K, Weller E, Carlson G, et al. An open-label trial of divalproex in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2002;41(10):1224-30.
18. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162(1):58-64.
19. Findling R, McNamara N, Gracious B, et al. Combination lithium and divalproex sodium in pediatric bipolarity. J Am Acad Child Adolesc Psychiatry 2003;42(8):895-901.
20. DelBello M, Adler C, Strakowski S. Divalproex for the treatment of aggression associated with adolescent mania. J Child Adolesc Psychopharmacol 2004;14(2):325-8.
21. Anderson G. Children versus adults: pharmokinetic and adverseeffect differences. Epilepsia 2002;43(suppl 3):53-9.
22. Yehya N, Saldarini C, Koski M, et al. Valproate-induced hyperammonemic encephalopathy. J Am Acad Child Adolesc Psychiatry 2004;43(8):926-7.
23. Verrotti A, Greco R, Matera V, et al. Platelet count and function in children receiving sodium valproate. Pediatr Neurol 1999;21(3):611-14.
24. Isojarvi J, Tauboll E, Pakarinen A, et al. Altered ovarian function and cardiovascular risk factors in valproate-treated women. Am J Med 2001;111(4):290-6.
25. Bowden C, Calabrese J, McElroy S, et al. A randomized, placebocontrolled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Arch Gen Psychiatry 2000;57(5):481-9.
26. Findling R, Gracious B, McNamara N, et al. Rapid, continuous cycling and psychiatric co-morbidity in pediatric bipolar I disorder. Bipolar Disord 2001;3(4):202-10.
27. Davanzo P, Gunderson B, Belin T, et al. Mood stabilizers in hospitalized children with bipolar disorder: a retrospective review. Psychiatry Clin Neurosci 2003;57(5):504-10.
28. Pellock J. Carbamazepine side effects in children and adults. Epilepsia 1987;28(suppl 3):64-70.
29. Sobotka J, Alexander B, Cook B. A review of carbamazepine’s hematologic reactions and monitoring. DICP 1990;24(12):1214-19.
30. Hellewell J. Oxcarbazepine (Trileptal) in the treatment of bipolar disorders: a review of efficacy and tolerability. J Affect Disord 2002;72(supp 1):23-34.
31. Davanzo P, Nikore V, Yehya N, Stevenson L. Oxcarbazepine treatment of juvenile-onset bipolar disorder. J Child Adolesc Psychopharmacol 2004;14(3):344-5.
32. Teitelbaum M. Oxcarbazepine in bipolar disorder. J Am Acad Child Adolesc Psychiatry 2001;40(9):993-4.
33. Dietrich D, Kropp S, Emrich H. Oxcarbazepine in affective and schizoaffective disorders. Pharmacopsychiatry 2001;34(6):242-50.
34. Holtmann M, Krause M, Opp J, et al. Oxcarbazepine-induced hyponatremia and the regulation of serum sodium after replacing carbamazepine with oxcarbazepine in children. Neuropediatrics 2002;33(6):298-300.
35. Prescribing information for oxcarbazepine (Trileptal) Novartis Pharmaceuticals Corp. 2005. Available at: http://www.pharma.us. novartis.com/product/pi/pdf/trileptal.pdf. Accessed June 26, 2005.
36. Asconape J. Some common issues in the use of antiepileptic drugs. Semin Neurol 2002;22(1):27-39.
37. Fattore C, Cipolla G, Gatti G, et al. Induction of ethinylestradiol and levonorgestrel metabolism by oxcarbazepine in healthy women. Epilepsia 1999;40(6):783-7.
38. Swope G, Hoopes S, Amy L, et al. An open-label study of lamotrigine in adolescents with bipolar mood disorder (poster presentation). New York: American Psychiatric Association annual meeting, 2004.
39. Saxena K, Howe M, Chang K. Lamotrigine adjunct or monotherapy for adolescent bipolar depression or mixed mania (poster presentation). Washington, DC: American Academy of Child and Adolescent Psychiatry annual meeting, 2004.
40. Prescribing information for lamotrigine (Lamictal). GlaxoSmith Kline 2004. Available at: http://us.gsk.com/products/assets/us_ lamictal.pdf. Accessed June 2, 2005.
41. Messenheimer J, Mullens E, Giorgi L, Young F. Safety review of adult clinical trial experience with lamotrigine. Drug Safety 1998;18(4):281-96.
42. DelBello M, Kowatch R, Warner J, et al. Adjunctive topiramate treatment for pediatric bipolar disorder: a retrospective chart review. J Child Adolesc Psychopharmacol 2002;12(4):323-30.
43. DelBello M, Kushner S, Wang D, et al. Topiramate for acute mania in children and adolescents with bipolar I disorder (abstract). New York: American Psychiatric Association annual meeting, 2004.
44. Philippi H, Boor R, Reitter B. Topiramate and metabolic acidosis in infants and toddlers. Epilepsia 2002;43(7):744-7.
45. Arcas J, Ferrer T, Roche M, et al. Hypohidrosis related to the administration of topiramate to children. Epilepsia 2001;42(10):1363-5.
46. Davanzo P, Cantwell E, Kleiner J, et al. Cognitive changes during topiramate therapy. J Am Acad Child Adolesc Psychiatry 2001;40(3):262-3.
47. McIntyre R, Mancini D, McCann S, et al. Topiramate versus bupropion SR when added to mood stabilizer therapy for the depressive phase of bipolar disorder: a preliminary single-blind study. Bipolar Disord 2002;4(3):207-13.
Antidepressants for bipolar depression: Tips to stay out of trouble
In clinical practice, 50% to 80% of bipolar patients receive long-term antidepressants,1 although potential benefits probably outweigh risks in 20% to 40%. This gap suggests that psychiatrists could do more to stay out of trouble when prescribing antidepressants for patients with bipolar depression.
Antidepressants have not shown efficacy in long-term treatment, and evidence of their effectiveness in acute bipolar depression is limited. They appear to pose greater risk of switching and mood destabilization for some patients and certain types of bipolar illness, and some antidepressant classes are more worrisome than others.
Because carefully analyzing risks and benefits is essential when considering antidepressants for a patient with bipolar illness, this article clarifies that delicate balance and offers evidence-based recommendations for using antidepressants in bipolar depression.
ACUTE THERAPY
Clinical trials support antidepressants as the treatment of choice for unipolar depression, but less evidence supports efficacy and safety in acute bipolar depression. Depressive episodes predominate in bipolar disorder, with chronic subsyndromal symptoms being most characteristic.2,3 Compared with mania or hypomania, depressive episodes:
- last longer and are more frequent
- contribute to greater morbidity and mortality
- pose a greater treatment challenge.
Antidepressants have shown benefit in multiple double-blind, bipolar depression trials and were as effective as mood stabilizers in one small study.4 Even so, no trials have found them more effective than mood stabilizers in acute bipolar depression.
Controlled trials. Two randomized, double-blind, placebo-controlled trials have examined antidepressant use in bipolar depression.5,6 The larger and better designed—a prospective 10-week study by Nemeroff et al6—examined 117 outpatients with type I bipolar disorder.
Subjects who had been taking lithium (serum levels 0.5 to 1.2 mEq/L) for ≥6 weeks and were experiencing moderate breakthrough depression then received paroxetine (mean dosage 32.6 mg/d), imipramine (mean dosage 166.7 mg/d), or placebo. Therapeutic response was defined as ≤7 on the Hamilton Rating Scale for Depression (HRSD) or ≤2 on the Clinical Global Impression (CGI) scale—normally considered criteria for depressive remission.
The authors hoped to show a statistically significant medication-placebo difference, but the antidepressants’ effects were similar to those of placebo. Thus, adding antidepressants to lithium conferred no added benefit, though the small sample size may have created a false negative.
Interestingly, a post-hoc analysis found different treatment outcomes when patients were separated into two groups by lithium serum levels:
- low therapeutic (≤0.8 mEq/L)
- high therapeutic (>0.8 mEq/L).
Adding antidepressants significantly reduced HRSD scores compared with placebo in the low lithium group but not in the high lithium group. Thus, therapeutic lithium levels may have moderate antidepressant effects, and adding antidepressants may help patients who cannot tolerate therapeutic lithium levels.
MAINTENANCE THERAPY
Antidepressants may have modest efficacy in acute bipolar depression, but they have not shown benefit—with or without mood stabilizers—in 7 studies of bipolar depression maintenance therapy. Most were double-blind, long-term trials comparing tricyclic antidepressants (TCAs) with lithium or adding TCAs to lithium; 3 were placebo-controlled.7 Antidepressants were not more effective than mood stabilizers such as lithium or lamotrigine in preventing bipolar depression.
Type II patients. For depression in type II bipolar disorder, the only data on using antidepressants as acute or maintenance therapy come from post-hoc analyses of unipolar depression trials and retrospective assessments of “manic switches.” No specific mania rating scales have been used.8,9
Long-term antidepressants. Two naturalistic studies by Altshuler et al10,11 explored continuing antidepressants as bipolar depression maintenance treatment. The larger trial11 included 84 patients (most with type I bipolar disorder) who experienced breakthrough depression while taking a mood stabilizer. This subset (15%) of the Stanley Foundation Bipolar Network had tolerated antidepressants at least 2 months without switching into hypomania/mania and remained in remission at least 6 weeks. None were rapid cyclers.
With counseling from clinicians, patients chose to continue or discontinue taking antidepressants. Relapse rates after 1 year were 70% in patients who stopped antidepressants after <6 months, compared with 24% in those who continued taking them for 1 year. The authors concluded that bipolar patients may benefit from staying on antidepressants at least 6 months and perhaps 12 or more months after depressive remission.
Keep in mind, however, that these findings may not apply to all bipolar patients. This study pertains to a minority of robust responders—none of whom were rapid cyclers—who tolerated the medication well and were not randomly assigned to continue or discontinue antidepressants. Other evidence suggests that depressed bipolar patients are three times more likely than unipolar patients (54% vs 16%) to develop tolerance to antidepressants.12
ANTIDEPRESSANT RISKS
Risks of using antidepressants in bipolar patients include acute switches into hypo/mania, usually within 8 weeks of starting an antidepressant, and new-onset mood destabilization—with cycle acceleration or rapid cycling—or worsening of pre-existing rapid cycling (Table 1).1
Table 1
Switches vs destabilization: Defining antidepressant risks
| Risk | Definition |
|---|---|
| Acute switches to hypomania/mania | ≤8 weeks by convention, unless dosage is increased |
| Mood destabilization | |
| Cycle acceleration | Increase of ≥2 mood episodes while taking antidepressants, compared with a similar exposure time before treatment |
| Rapid cycling | ≥4mood episodes in previous 12 months (new-onset or exacerbation of baseline pattern), according to DSM-IV-TR |
| Source: Reference 1 | |
Switching risk. Some researchers have reported antidepressant-induced switches to be milder and more brief than spontaneous hypo/manias,13 whereas others have observed more-severe mixed14 and even psychotic episodes. Risk factors that may predispose patients to switching include:
- personal or family history of switches or mood destabilization
- family history of bipolar disorder
- exposure to multiple antidepressant trials
- history of substance abuse or dependence
- early onset (age <25) and/or treatment of mood symptoms.15,16
True switch rates are difficult to estimate because clinical trials have used different switching definitions, durations, antidepressants (with or without mood stabilizers, and with different mood stabilizers), and cohorts (often excluding rapid cyclers). Except for the Nemeroff et al study,6 no prospective, double-blind, placebo-controlled studies have examined switch rates, and even this study was not large enough to detect statistically significant differences.
Thus we must rely on naturalistic evidence that is less rigorous but more applicable to clinical practice. This literature reveals switch rates of:
- 30% to 60% with TCAs and monoamine oxidase inhibitors (MAOIs)
- 15% to 27% with selective serotonin reuptake inhibitors (SSRIs), bupropion, and venlafaxine.
Average switch rates are thought to be approximately 40% with TCAs/MAOIs and 20% with the newer antidepressants.1 Preliminary data associate venlafaxine with higher switch rates than SSRIs or bupropion, so perhaps antidepressants with some noradrenergic effects (including TCAs) facilitate the switching phenomenon.17
Mood destabilization. Three randomized, controlled trials suggest that antidepressants—especially TCAs—increase the risk of cycle acceleration or rapid cycling in bipolar patients. The best designed study—sponsored by the National Institute of Mental Health—was a 10-year, prospective, double-blind trial of 51 rapid-cycling patients. The trial’s on-off-on design showed that 20% of these patients developed rapid cycling as a direct result of taking TCAs.18
Unfortunately, most randomized, controlled trials are not designed to show a relationship between antidepressants and mood destabilization. Observational literature is mixed but suggests that antidepressant use is associated with rapid cycling. Most evidence supports a relationship between antidepressants and long-term mood destabilization—especially cycle acceleration, which is believed to occur in approximately 20% of patients using TCAs or SSRIs.1
Are mood stabilizers protective? Some studies suggest that mood stabilizers may help protect against switches. Most of the evidence—using lithium and TCAs—suggests a 50% drop in switch rates when patients receive mood stabilizers with antidepressants. In one study, lithium was more protective than anticonvulsants for SSRI-induced mania, but the difference was not statistically significant.19
Because study data variability, we don’t know if some mood stabilizers are more effective than others in preventing antidepressant-related switching. This variability is likely caused by:
- medication-specific factors (such as higher switch rates with TCAs and possibly dual-reuptake inhibitors than with SSRIs)
- illness-specific factors (such as rapid cycling and cycle pattern)
- patient-specific factors, already described. Mood stabilizers appear to be more protective against switching than against mood destabilization, in which their effects are less clear (Table 2).15
Table 2
Frequency of switching or mood destabilization with antidepressants
| Bipolar risk | Causative agents | Frequency | Mood stabilizer effect |
|---|---|---|---|
| Acute switch | TCAs, MAOIs | ~ 40% | Variably protective; apparent partial risk reduction ~ 50% |
| SSRIs, bupropion, venlafaxine | ~ 20% | ||
| Mood destabilization | TCAs, SSRIs | ~ 20% | Not as clearly protective against mood destabilization as against acute switching |
| TCAs: tricyclic antidepressants; MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors | |||
| Source: References 1, 15 | |||
TREATMENT RECOMMENDATIONS
How does a clinician decide which bipolar depressed patients should receive antidepressants?
The first step in treating bipolar depression (Algorithm) is to provide optimal dosages of the patient’s mood stabilizers. Consensus guidelines20 suggest lithium or lamotrigine as first-line treatments for bipolar depression. Evidence also shows efficacy for atypical antipsychotics, including the olanzapine/fluoxetine combination (OFC)—FDA-approved for acute bipolar depression21—and quetiapine monotherapy.22 Dosages vary, but suggested ranges include:
- lithium: 0.6 to 1.2 mEq/L; aim for approximately 0.8 mEq/L, but some data suggest 0.6 to 0.7 mEq/L may be sufficient
- lamotrigine: 50 to 250 mg/d (the higher dosage is based on maintenance studies)
- OFC: 6 to 12 mg olanzapine/25 to 50 mg fluoxetine
- quetiapine: 300 to 600 mg/d.
The next step is an antidepressant risk/benefit analysis, weighing the considerable risks of switching/mood destabilization with the patient’s depressive illness severity, type of bipolar disorder (such as rapid cycling), and cycle pattern.
Algorithm Recommended treatment of bipolar depression
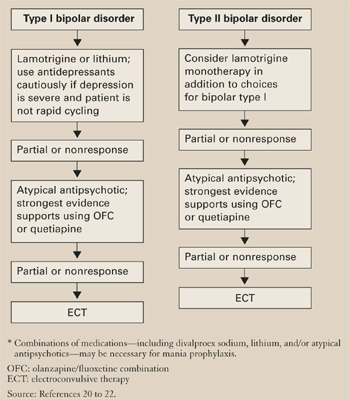
Cycle patterns. In a naturalistic study, Macqueen et al23 used life chart data for 42 bipolar patients to assess how the mood state preceding a prospectively observed depressive episode affected treatment response:
- A euthymic mood state in the previous 2 months represented a uniphasic pattern and an isolated depressive episode.
- A preceding hypomanic/manic mood state indicated a biphasic pattern.
Approximately 60% of bipolar patients show a biphasic pattern, although the episode sequence is usually depression-hypomania/mania rather than hypomania/mania-depression. These authors included patients whose breakthrough depressive episodes were treated with an antidepressant or a putative mood stabilizer but not an atypical antipsychotic.
In patients treated with an antidepressant, the response-to-switch ratio was 10:1 for those previously euthymic, compared with a less beneficial 0.75:1 in previously hypomanic/manic patients. This small study suggests that a patient’s cycle pattern may help you decide whether to use an antidepressant for bipolar depression.
How to use antidepressants. As described, some depressed bipolar patients are better candidates for antidepressant therapy than others (Table 3).
Table 3
Antidepressants for bipolar depression? Consider ‘ideal patient’ traits
| Severe depression refractory to optimal doses of ≥1 mood stabilizers |
| Uniphasic cycle pattern |
| Not rapid cycling |
| No history of switching or mood destabilization |
| No comorbid substance abuse |
Use antidepressants cautiously and conservatively in a minority of bipolar patients (approximately 20% to 40%) and usually for short periods (discussed below). SSRIs or bupropion are first-line agents because:
- they appear to be relatively less likely to cause switching than other antidepressant classes
- controlled trials have examined these antidepressants in bipolar depression.
Depressed patients with very mild, nonrapid-cycling, bipolar II disorder and no more than three previous hypomanic episodes might be candidates for antidepressant monotherapy. In other bipolar patients, always use at least one mood stabilizer if you decide to use an antidepressant.
TREATMENT DURATION
No randomized, controlled trial has examined what duration of antidepressant treatment may be optimum for bipolar depression, but consensus guidelines recommend:
- approximately 3 to 7 months, depending on depression severity
- approximately one-half that duration (2 to 4 months) for rapid-cycling bipolar disorder.20
Because of the switching risk, one could also argue for a shorter treatment duration in patients with a biphasic cycle pattern—especially with an episode sequence of depression to hypomania/mania to euthymia.
Ideally, patients would stay on antidepressants no longer than the natural course of their depression (usually 2 to 6 months in bipolar depression), although it could be shorter in rapid cyclers. Approximately 15% to 20% of patients may have a robust initial response to antidepressants and need to be maintained on these medications, especially after several tapers and relapses have failed.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
Drug brand names
- Bupropion • Wellbutrin
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid, others
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Venlafaxine • Effexor
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
1. Ghaemi SN, Hsu DJ, Soldani F, et al. Antidepressants in bipolar disorder: the case for caution. Bipolar Disorders 2003;5:421-33.
2. Judd LL. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
3. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry 2003;60(3):261-9.
4. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry 2000;157:124-6.
5. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmacol 1989;4(4):313-22.
6. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158(6):906-12.
7. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry 2001;62(7):565-9.
8. Amsterdam JD, Garcia-Espana F, Fawcett J, et al. Efficacy and safety of fluoxetine in treating bipolar II major depressive episode. J Clin Psychopharmacol 1998;18:435-40.
9. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
10. Altshuler L, Kiriakos L, Calcagno J, et al. Impact of antidepressant discontinuation versus antidepressant continuation at 1-year risk for relapse of bipolar depression: a retrospective chart review. J Clin Psychiatry 2001;62:612-16.
11. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry 2003;160:1252-62.
12. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry 2004;161:163-5.
13. Stoll AL, Mayer PV, Kolbrener M, et al. Antidepressant-associated mania: a controlled comparison with spontaneous mania. Am J Psychiatry 1994;151:1642-5.
14. Zubieta JK, Demitrack MA. Possible bupropion precipitation of mania and mixed affective state. J Clin Psychopharmacol 1991;11(5):327-8.
15. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disorders 2003;5:407-20.
16. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry 2002;63:791-5.
17. Post RM, Leverich GS, Nolen WA, et al. A re-evaluation of the role of antidepressants in the treatment of bipolar depression: data from the Stanley Foundation Bipolar Network. Bipolar Disorders 2003;5:396-406.
18. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
19. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry 2001;62:249-55.
20. Keck PE, Jr, Perlis RH, Otto MW, et al. The Expert Consensus Guideline Series: Treatment of bipolar disorder 2004. Postgrad Med 2004;1-120.
21. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
22. Calabrese JR. Quetiapine BOLDER study [presentation]. New York: American Psychiatric Association annual meeting, 2004.
23. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand 2002;105:414-18.
In clinical practice, 50% to 80% of bipolar patients receive long-term antidepressants,1 although potential benefits probably outweigh risks in 20% to 40%. This gap suggests that psychiatrists could do more to stay out of trouble when prescribing antidepressants for patients with bipolar depression.
Antidepressants have not shown efficacy in long-term treatment, and evidence of their effectiveness in acute bipolar depression is limited. They appear to pose greater risk of switching and mood destabilization for some patients and certain types of bipolar illness, and some antidepressant classes are more worrisome than others.
Because carefully analyzing risks and benefits is essential when considering antidepressants for a patient with bipolar illness, this article clarifies that delicate balance and offers evidence-based recommendations for using antidepressants in bipolar depression.
ACUTE THERAPY
Clinical trials support antidepressants as the treatment of choice for unipolar depression, but less evidence supports efficacy and safety in acute bipolar depression. Depressive episodes predominate in bipolar disorder, with chronic subsyndromal symptoms being most characteristic.2,3 Compared with mania or hypomania, depressive episodes:
- last longer and are more frequent
- contribute to greater morbidity and mortality
- pose a greater treatment challenge.
Antidepressants have shown benefit in multiple double-blind, bipolar depression trials and were as effective as mood stabilizers in one small study.4 Even so, no trials have found them more effective than mood stabilizers in acute bipolar depression.
Controlled trials. Two randomized, double-blind, placebo-controlled trials have examined antidepressant use in bipolar depression.5,6 The larger and better designed—a prospective 10-week study by Nemeroff et al6—examined 117 outpatients with type I bipolar disorder.
Subjects who had been taking lithium (serum levels 0.5 to 1.2 mEq/L) for ≥6 weeks and were experiencing moderate breakthrough depression then received paroxetine (mean dosage 32.6 mg/d), imipramine (mean dosage 166.7 mg/d), or placebo. Therapeutic response was defined as ≤7 on the Hamilton Rating Scale for Depression (HRSD) or ≤2 on the Clinical Global Impression (CGI) scale—normally considered criteria for depressive remission.
The authors hoped to show a statistically significant medication-placebo difference, but the antidepressants’ effects were similar to those of placebo. Thus, adding antidepressants to lithium conferred no added benefit, though the small sample size may have created a false negative.
Interestingly, a post-hoc analysis found different treatment outcomes when patients were separated into two groups by lithium serum levels:
- low therapeutic (≤0.8 mEq/L)
- high therapeutic (>0.8 mEq/L).
Adding antidepressants significantly reduced HRSD scores compared with placebo in the low lithium group but not in the high lithium group. Thus, therapeutic lithium levels may have moderate antidepressant effects, and adding antidepressants may help patients who cannot tolerate therapeutic lithium levels.
MAINTENANCE THERAPY
Antidepressants may have modest efficacy in acute bipolar depression, but they have not shown benefit—with or without mood stabilizers—in 7 studies of bipolar depression maintenance therapy. Most were double-blind, long-term trials comparing tricyclic antidepressants (TCAs) with lithium or adding TCAs to lithium; 3 were placebo-controlled.7 Antidepressants were not more effective than mood stabilizers such as lithium or lamotrigine in preventing bipolar depression.
Type II patients. For depression in type II bipolar disorder, the only data on using antidepressants as acute or maintenance therapy come from post-hoc analyses of unipolar depression trials and retrospective assessments of “manic switches.” No specific mania rating scales have been used.8,9
Long-term antidepressants. Two naturalistic studies by Altshuler et al10,11 explored continuing antidepressants as bipolar depression maintenance treatment. The larger trial11 included 84 patients (most with type I bipolar disorder) who experienced breakthrough depression while taking a mood stabilizer. This subset (15%) of the Stanley Foundation Bipolar Network had tolerated antidepressants at least 2 months without switching into hypomania/mania and remained in remission at least 6 weeks. None were rapid cyclers.
With counseling from clinicians, patients chose to continue or discontinue taking antidepressants. Relapse rates after 1 year were 70% in patients who stopped antidepressants after <6 months, compared with 24% in those who continued taking them for 1 year. The authors concluded that bipolar patients may benefit from staying on antidepressants at least 6 months and perhaps 12 or more months after depressive remission.
Keep in mind, however, that these findings may not apply to all bipolar patients. This study pertains to a minority of robust responders—none of whom were rapid cyclers—who tolerated the medication well and were not randomly assigned to continue or discontinue antidepressants. Other evidence suggests that depressed bipolar patients are three times more likely than unipolar patients (54% vs 16%) to develop tolerance to antidepressants.12
ANTIDEPRESSANT RISKS
Risks of using antidepressants in bipolar patients include acute switches into hypo/mania, usually within 8 weeks of starting an antidepressant, and new-onset mood destabilization—with cycle acceleration or rapid cycling—or worsening of pre-existing rapid cycling (Table 1).1
Table 1
Switches vs destabilization: Defining antidepressant risks
| Risk | Definition |
|---|---|
| Acute switches to hypomania/mania | ≤8 weeks by convention, unless dosage is increased |
| Mood destabilization | |
| Cycle acceleration | Increase of ≥2 mood episodes while taking antidepressants, compared with a similar exposure time before treatment |
| Rapid cycling | ≥4mood episodes in previous 12 months (new-onset or exacerbation of baseline pattern), according to DSM-IV-TR |
| Source: Reference 1 | |
Switching risk. Some researchers have reported antidepressant-induced switches to be milder and more brief than spontaneous hypo/manias,13 whereas others have observed more-severe mixed14 and even psychotic episodes. Risk factors that may predispose patients to switching include:
- personal or family history of switches or mood destabilization
- family history of bipolar disorder
- exposure to multiple antidepressant trials
- history of substance abuse or dependence
- early onset (age <25) and/or treatment of mood symptoms.15,16
True switch rates are difficult to estimate because clinical trials have used different switching definitions, durations, antidepressants (with or without mood stabilizers, and with different mood stabilizers), and cohorts (often excluding rapid cyclers). Except for the Nemeroff et al study,6 no prospective, double-blind, placebo-controlled studies have examined switch rates, and even this study was not large enough to detect statistically significant differences.
Thus we must rely on naturalistic evidence that is less rigorous but more applicable to clinical practice. This literature reveals switch rates of:
- 30% to 60% with TCAs and monoamine oxidase inhibitors (MAOIs)
- 15% to 27% with selective serotonin reuptake inhibitors (SSRIs), bupropion, and venlafaxine.
Average switch rates are thought to be approximately 40% with TCAs/MAOIs and 20% with the newer antidepressants.1 Preliminary data associate venlafaxine with higher switch rates than SSRIs or bupropion, so perhaps antidepressants with some noradrenergic effects (including TCAs) facilitate the switching phenomenon.17
Mood destabilization. Three randomized, controlled trials suggest that antidepressants—especially TCAs—increase the risk of cycle acceleration or rapid cycling in bipolar patients. The best designed study—sponsored by the National Institute of Mental Health—was a 10-year, prospective, double-blind trial of 51 rapid-cycling patients. The trial’s on-off-on design showed that 20% of these patients developed rapid cycling as a direct result of taking TCAs.18
Unfortunately, most randomized, controlled trials are not designed to show a relationship between antidepressants and mood destabilization. Observational literature is mixed but suggests that antidepressant use is associated with rapid cycling. Most evidence supports a relationship between antidepressants and long-term mood destabilization—especially cycle acceleration, which is believed to occur in approximately 20% of patients using TCAs or SSRIs.1
Are mood stabilizers protective? Some studies suggest that mood stabilizers may help protect against switches. Most of the evidence—using lithium and TCAs—suggests a 50% drop in switch rates when patients receive mood stabilizers with antidepressants. In one study, lithium was more protective than anticonvulsants for SSRI-induced mania, but the difference was not statistically significant.19
Because study data variability, we don’t know if some mood stabilizers are more effective than others in preventing antidepressant-related switching. This variability is likely caused by:
- medication-specific factors (such as higher switch rates with TCAs and possibly dual-reuptake inhibitors than with SSRIs)
- illness-specific factors (such as rapid cycling and cycle pattern)
- patient-specific factors, already described. Mood stabilizers appear to be more protective against switching than against mood destabilization, in which their effects are less clear (Table 2).15
Table 2
Frequency of switching or mood destabilization with antidepressants
| Bipolar risk | Causative agents | Frequency | Mood stabilizer effect |
|---|---|---|---|
| Acute switch | TCAs, MAOIs | ~ 40% | Variably protective; apparent partial risk reduction ~ 50% |
| SSRIs, bupropion, venlafaxine | ~ 20% | ||
| Mood destabilization | TCAs, SSRIs | ~ 20% | Not as clearly protective against mood destabilization as against acute switching |
| TCAs: tricyclic antidepressants; MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors | |||
| Source: References 1, 15 | |||
TREATMENT RECOMMENDATIONS
How does a clinician decide which bipolar depressed patients should receive antidepressants?
The first step in treating bipolar depression (Algorithm) is to provide optimal dosages of the patient’s mood stabilizers. Consensus guidelines20 suggest lithium or lamotrigine as first-line treatments for bipolar depression. Evidence also shows efficacy for atypical antipsychotics, including the olanzapine/fluoxetine combination (OFC)—FDA-approved for acute bipolar depression21—and quetiapine monotherapy.22 Dosages vary, but suggested ranges include:
- lithium: 0.6 to 1.2 mEq/L; aim for approximately 0.8 mEq/L, but some data suggest 0.6 to 0.7 mEq/L may be sufficient
- lamotrigine: 50 to 250 mg/d (the higher dosage is based on maintenance studies)
- OFC: 6 to 12 mg olanzapine/25 to 50 mg fluoxetine
- quetiapine: 300 to 600 mg/d.
The next step is an antidepressant risk/benefit analysis, weighing the considerable risks of switching/mood destabilization with the patient’s depressive illness severity, type of bipolar disorder (such as rapid cycling), and cycle pattern.
Algorithm Recommended treatment of bipolar depression
Cycle patterns. In a naturalistic study, Macqueen et al23 used life chart data for 42 bipolar patients to assess how the mood state preceding a prospectively observed depressive episode affected treatment response:
- A euthymic mood state in the previous 2 months represented a uniphasic pattern and an isolated depressive episode.
- A preceding hypomanic/manic mood state indicated a biphasic pattern.
Approximately 60% of bipolar patients show a biphasic pattern, although the episode sequence is usually depression-hypomania/mania rather than hypomania/mania-depression. These authors included patients whose breakthrough depressive episodes were treated with an antidepressant or a putative mood stabilizer but not an atypical antipsychotic.
In patients treated with an antidepressant, the response-to-switch ratio was 10:1 for those previously euthymic, compared with a less beneficial 0.75:1 in previously hypomanic/manic patients. This small study suggests that a patient’s cycle pattern may help you decide whether to use an antidepressant for bipolar depression.
How to use antidepressants. As described, some depressed bipolar patients are better candidates for antidepressant therapy than others (Table 3).
Table 3
Antidepressants for bipolar depression? Consider ‘ideal patient’ traits
| Severe depression refractory to optimal doses of ≥1 mood stabilizers |
| Uniphasic cycle pattern |
| Not rapid cycling |
| No history of switching or mood destabilization |
| No comorbid substance abuse |
Use antidepressants cautiously and conservatively in a minority of bipolar patients (approximately 20% to 40%) and usually for short periods (discussed below). SSRIs or bupropion are first-line agents because:
- they appear to be relatively less likely to cause switching than other antidepressant classes
- controlled trials have examined these antidepressants in bipolar depression.
Depressed patients with very mild, nonrapid-cycling, bipolar II disorder and no more than three previous hypomanic episodes might be candidates for antidepressant monotherapy. In other bipolar patients, always use at least one mood stabilizer if you decide to use an antidepressant.
TREATMENT DURATION
No randomized, controlled trial has examined what duration of antidepressant treatment may be optimum for bipolar depression, but consensus guidelines recommend:
- approximately 3 to 7 months, depending on depression severity
- approximately one-half that duration (2 to 4 months) for rapid-cycling bipolar disorder.20
Because of the switching risk, one could also argue for a shorter treatment duration in patients with a biphasic cycle pattern—especially with an episode sequence of depression to hypomania/mania to euthymia.
Ideally, patients would stay on antidepressants no longer than the natural course of their depression (usually 2 to 6 months in bipolar depression), although it could be shorter in rapid cyclers. Approximately 15% to 20% of patients may have a robust initial response to antidepressants and need to be maintained on these medications, especially after several tapers and relapses have failed.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
Drug brand names
- Bupropion • Wellbutrin
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid, others
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Venlafaxine • Effexor
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
In clinical practice, 50% to 80% of bipolar patients receive long-term antidepressants,1 although potential benefits probably outweigh risks in 20% to 40%. This gap suggests that psychiatrists could do more to stay out of trouble when prescribing antidepressants for patients with bipolar depression.
Antidepressants have not shown efficacy in long-term treatment, and evidence of their effectiveness in acute bipolar depression is limited. They appear to pose greater risk of switching and mood destabilization for some patients and certain types of bipolar illness, and some antidepressant classes are more worrisome than others.
Because carefully analyzing risks and benefits is essential when considering antidepressants for a patient with bipolar illness, this article clarifies that delicate balance and offers evidence-based recommendations for using antidepressants in bipolar depression.
ACUTE THERAPY
Clinical trials support antidepressants as the treatment of choice for unipolar depression, but less evidence supports efficacy and safety in acute bipolar depression. Depressive episodes predominate in bipolar disorder, with chronic subsyndromal symptoms being most characteristic.2,3 Compared with mania or hypomania, depressive episodes:
- last longer and are more frequent
- contribute to greater morbidity and mortality
- pose a greater treatment challenge.
Antidepressants have shown benefit in multiple double-blind, bipolar depression trials and were as effective as mood stabilizers in one small study.4 Even so, no trials have found them more effective than mood stabilizers in acute bipolar depression.
Controlled trials. Two randomized, double-blind, placebo-controlled trials have examined antidepressant use in bipolar depression.5,6 The larger and better designed—a prospective 10-week study by Nemeroff et al6—examined 117 outpatients with type I bipolar disorder.
Subjects who had been taking lithium (serum levels 0.5 to 1.2 mEq/L) for ≥6 weeks and were experiencing moderate breakthrough depression then received paroxetine (mean dosage 32.6 mg/d), imipramine (mean dosage 166.7 mg/d), or placebo. Therapeutic response was defined as ≤7 on the Hamilton Rating Scale for Depression (HRSD) or ≤2 on the Clinical Global Impression (CGI) scale—normally considered criteria for depressive remission.
The authors hoped to show a statistically significant medication-placebo difference, but the antidepressants’ effects were similar to those of placebo. Thus, adding antidepressants to lithium conferred no added benefit, though the small sample size may have created a false negative.
Interestingly, a post-hoc analysis found different treatment outcomes when patients were separated into two groups by lithium serum levels:
- low therapeutic (≤0.8 mEq/L)
- high therapeutic (>0.8 mEq/L).
Adding antidepressants significantly reduced HRSD scores compared with placebo in the low lithium group but not in the high lithium group. Thus, therapeutic lithium levels may have moderate antidepressant effects, and adding antidepressants may help patients who cannot tolerate therapeutic lithium levels.
MAINTENANCE THERAPY
Antidepressants may have modest efficacy in acute bipolar depression, but they have not shown benefit—with or without mood stabilizers—in 7 studies of bipolar depression maintenance therapy. Most were double-blind, long-term trials comparing tricyclic antidepressants (TCAs) with lithium or adding TCAs to lithium; 3 were placebo-controlled.7 Antidepressants were not more effective than mood stabilizers such as lithium or lamotrigine in preventing bipolar depression.
Type II patients. For depression in type II bipolar disorder, the only data on using antidepressants as acute or maintenance therapy come from post-hoc analyses of unipolar depression trials and retrospective assessments of “manic switches.” No specific mania rating scales have been used.8,9
Long-term antidepressants. Two naturalistic studies by Altshuler et al10,11 explored continuing antidepressants as bipolar depression maintenance treatment. The larger trial11 included 84 patients (most with type I bipolar disorder) who experienced breakthrough depression while taking a mood stabilizer. This subset (15%) of the Stanley Foundation Bipolar Network had tolerated antidepressants at least 2 months without switching into hypomania/mania and remained in remission at least 6 weeks. None were rapid cyclers.
With counseling from clinicians, patients chose to continue or discontinue taking antidepressants. Relapse rates after 1 year were 70% in patients who stopped antidepressants after <6 months, compared with 24% in those who continued taking them for 1 year. The authors concluded that bipolar patients may benefit from staying on antidepressants at least 6 months and perhaps 12 or more months after depressive remission.
Keep in mind, however, that these findings may not apply to all bipolar patients. This study pertains to a minority of robust responders—none of whom were rapid cyclers—who tolerated the medication well and were not randomly assigned to continue or discontinue antidepressants. Other evidence suggests that depressed bipolar patients are three times more likely than unipolar patients (54% vs 16%) to develop tolerance to antidepressants.12
ANTIDEPRESSANT RISKS
Risks of using antidepressants in bipolar patients include acute switches into hypo/mania, usually within 8 weeks of starting an antidepressant, and new-onset mood destabilization—with cycle acceleration or rapid cycling—or worsening of pre-existing rapid cycling (Table 1).1
Table 1
Switches vs destabilization: Defining antidepressant risks
| Risk | Definition |
|---|---|
| Acute switches to hypomania/mania | ≤8 weeks by convention, unless dosage is increased |
| Mood destabilization | |
| Cycle acceleration | Increase of ≥2 mood episodes while taking antidepressants, compared with a similar exposure time before treatment |
| Rapid cycling | ≥4mood episodes in previous 12 months (new-onset or exacerbation of baseline pattern), according to DSM-IV-TR |
| Source: Reference 1 | |
Switching risk. Some researchers have reported antidepressant-induced switches to be milder and more brief than spontaneous hypo/manias,13 whereas others have observed more-severe mixed14 and even psychotic episodes. Risk factors that may predispose patients to switching include:
- personal or family history of switches or mood destabilization
- family history of bipolar disorder
- exposure to multiple antidepressant trials
- history of substance abuse or dependence
- early onset (age <25) and/or treatment of mood symptoms.15,16
True switch rates are difficult to estimate because clinical trials have used different switching definitions, durations, antidepressants (with or without mood stabilizers, and with different mood stabilizers), and cohorts (often excluding rapid cyclers). Except for the Nemeroff et al study,6 no prospective, double-blind, placebo-controlled studies have examined switch rates, and even this study was not large enough to detect statistically significant differences.
Thus we must rely on naturalistic evidence that is less rigorous but more applicable to clinical practice. This literature reveals switch rates of:
- 30% to 60% with TCAs and monoamine oxidase inhibitors (MAOIs)
- 15% to 27% with selective serotonin reuptake inhibitors (SSRIs), bupropion, and venlafaxine.
Average switch rates are thought to be approximately 40% with TCAs/MAOIs and 20% with the newer antidepressants.1 Preliminary data associate venlafaxine with higher switch rates than SSRIs or bupropion, so perhaps antidepressants with some noradrenergic effects (including TCAs) facilitate the switching phenomenon.17
Mood destabilization. Three randomized, controlled trials suggest that antidepressants—especially TCAs—increase the risk of cycle acceleration or rapid cycling in bipolar patients. The best designed study—sponsored by the National Institute of Mental Health—was a 10-year, prospective, double-blind trial of 51 rapid-cycling patients. The trial’s on-off-on design showed that 20% of these patients developed rapid cycling as a direct result of taking TCAs.18
Unfortunately, most randomized, controlled trials are not designed to show a relationship between antidepressants and mood destabilization. Observational literature is mixed but suggests that antidepressant use is associated with rapid cycling. Most evidence supports a relationship between antidepressants and long-term mood destabilization—especially cycle acceleration, which is believed to occur in approximately 20% of patients using TCAs or SSRIs.1
Are mood stabilizers protective? Some studies suggest that mood stabilizers may help protect against switches. Most of the evidence—using lithium and TCAs—suggests a 50% drop in switch rates when patients receive mood stabilizers with antidepressants. In one study, lithium was more protective than anticonvulsants for SSRI-induced mania, but the difference was not statistically significant.19
Because study data variability, we don’t know if some mood stabilizers are more effective than others in preventing antidepressant-related switching. This variability is likely caused by:
- medication-specific factors (such as higher switch rates with TCAs and possibly dual-reuptake inhibitors than with SSRIs)
- illness-specific factors (such as rapid cycling and cycle pattern)
- patient-specific factors, already described. Mood stabilizers appear to be more protective against switching than against mood destabilization, in which their effects are less clear (Table 2).15
Table 2
Frequency of switching or mood destabilization with antidepressants
| Bipolar risk | Causative agents | Frequency | Mood stabilizer effect |
|---|---|---|---|
| Acute switch | TCAs, MAOIs | ~ 40% | Variably protective; apparent partial risk reduction ~ 50% |
| SSRIs, bupropion, venlafaxine | ~ 20% | ||
| Mood destabilization | TCAs, SSRIs | ~ 20% | Not as clearly protective against mood destabilization as against acute switching |
| TCAs: tricyclic antidepressants; MAOIs: monoamine oxidase inhibitors; SSRIs: selective serotonin reuptake inhibitors | |||
| Source: References 1, 15 | |||
TREATMENT RECOMMENDATIONS
How does a clinician decide which bipolar depressed patients should receive antidepressants?
The first step in treating bipolar depression (Algorithm) is to provide optimal dosages of the patient’s mood stabilizers. Consensus guidelines20 suggest lithium or lamotrigine as first-line treatments for bipolar depression. Evidence also shows efficacy for atypical antipsychotics, including the olanzapine/fluoxetine combination (OFC)—FDA-approved for acute bipolar depression21—and quetiapine monotherapy.22 Dosages vary, but suggested ranges include:
- lithium: 0.6 to 1.2 mEq/L; aim for approximately 0.8 mEq/L, but some data suggest 0.6 to 0.7 mEq/L may be sufficient
- lamotrigine: 50 to 250 mg/d (the higher dosage is based on maintenance studies)
- OFC: 6 to 12 mg olanzapine/25 to 50 mg fluoxetine
- quetiapine: 300 to 600 mg/d.
The next step is an antidepressant risk/benefit analysis, weighing the considerable risks of switching/mood destabilization with the patient’s depressive illness severity, type of bipolar disorder (such as rapid cycling), and cycle pattern.
Algorithm Recommended treatment of bipolar depression
Cycle patterns. In a naturalistic study, Macqueen et al23 used life chart data for 42 bipolar patients to assess how the mood state preceding a prospectively observed depressive episode affected treatment response:
- A euthymic mood state in the previous 2 months represented a uniphasic pattern and an isolated depressive episode.
- A preceding hypomanic/manic mood state indicated a biphasic pattern.
Approximately 60% of bipolar patients show a biphasic pattern, although the episode sequence is usually depression-hypomania/mania rather than hypomania/mania-depression. These authors included patients whose breakthrough depressive episodes were treated with an antidepressant or a putative mood stabilizer but not an atypical antipsychotic.
In patients treated with an antidepressant, the response-to-switch ratio was 10:1 for those previously euthymic, compared with a less beneficial 0.75:1 in previously hypomanic/manic patients. This small study suggests that a patient’s cycle pattern may help you decide whether to use an antidepressant for bipolar depression.
How to use antidepressants. As described, some depressed bipolar patients are better candidates for antidepressant therapy than others (Table 3).
Table 3
Antidepressants for bipolar depression? Consider ‘ideal patient’ traits
| Severe depression refractory to optimal doses of ≥1 mood stabilizers |
| Uniphasic cycle pattern |
| Not rapid cycling |
| No history of switching or mood destabilization |
| No comorbid substance abuse |
Use antidepressants cautiously and conservatively in a minority of bipolar patients (approximately 20% to 40%) and usually for short periods (discussed below). SSRIs or bupropion are first-line agents because:
- they appear to be relatively less likely to cause switching than other antidepressant classes
- controlled trials have examined these antidepressants in bipolar depression.
Depressed patients with very mild, nonrapid-cycling, bipolar II disorder and no more than three previous hypomanic episodes might be candidates for antidepressant monotherapy. In other bipolar patients, always use at least one mood stabilizer if you decide to use an antidepressant.
TREATMENT DURATION
No randomized, controlled trial has examined what duration of antidepressant treatment may be optimum for bipolar depression, but consensus guidelines recommend:
- approximately 3 to 7 months, depending on depression severity
- approximately one-half that duration (2 to 4 months) for rapid-cycling bipolar disorder.20
Because of the switching risk, one could also argue for a shorter treatment duration in patients with a biphasic cycle pattern—especially with an episode sequence of depression to hypomania/mania to euthymia.
Ideally, patients would stay on antidepressants no longer than the natural course of their depression (usually 2 to 6 months in bipolar depression), although it could be shorter in rapid cyclers. Approximately 15% to 20% of patients may have a robust initial response to antidepressants and need to be maintained on these medications, especially after several tapers and relapses have failed.
Related resources
- Bipolar Clinic and Research Program. Massachusetts General Hospital. Includes tools for clinicians and the clinical site for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD). www.manicdepressive.org.
- Goodwin FK, Jamison KR. Manic-depressive illness. New York: Oxford University Press, 1990.
Drug brand names
- Bupropion • Wellbutrin
- Imipramine • Tofranil
- Lamotrigine • Lamictal
- Lithium • Lithobid, others
- Olanzapine/fluoxetine • Symbyax
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Venlafaxine • Effexor
Disclosures
Dr. Altman is a speaker for Forest Pharmaceuticals, Janssen Pharmaceutica, AstraZeneca Pharmaceuticals, and Abbott Laboratories.
1. Ghaemi SN, Hsu DJ, Soldani F, et al. Antidepressants in bipolar disorder: the case for caution. Bipolar Disorders 2003;5:421-33.
2. Judd LL. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
3. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry 2003;60(3):261-9.
4. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry 2000;157:124-6.
5. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmacol 1989;4(4):313-22.
6. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158(6):906-12.
7. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry 2001;62(7):565-9.
8. Amsterdam JD, Garcia-Espana F, Fawcett J, et al. Efficacy and safety of fluoxetine in treating bipolar II major depressive episode. J Clin Psychopharmacol 1998;18:435-40.
9. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
10. Altshuler L, Kiriakos L, Calcagno J, et al. Impact of antidepressant discontinuation versus antidepressant continuation at 1-year risk for relapse of bipolar depression: a retrospective chart review. J Clin Psychiatry 2001;62:612-16.
11. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry 2003;160:1252-62.
12. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry 2004;161:163-5.
13. Stoll AL, Mayer PV, Kolbrener M, et al. Antidepressant-associated mania: a controlled comparison with spontaneous mania. Am J Psychiatry 1994;151:1642-5.
14. Zubieta JK, Demitrack MA. Possible bupropion precipitation of mania and mixed affective state. J Clin Psychopharmacol 1991;11(5):327-8.
15. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disorders 2003;5:407-20.
16. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry 2002;63:791-5.
17. Post RM, Leverich GS, Nolen WA, et al. A re-evaluation of the role of antidepressants in the treatment of bipolar depression: data from the Stanley Foundation Bipolar Network. Bipolar Disorders 2003;5:396-406.
18. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
19. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry 2001;62:249-55.
20. Keck PE, Jr, Perlis RH, Otto MW, et al. The Expert Consensus Guideline Series: Treatment of bipolar disorder 2004. Postgrad Med 2004;1-120.
21. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
22. Calabrese JR. Quetiapine BOLDER study [presentation]. New York: American Psychiatric Association annual meeting, 2004.
23. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand 2002;105:414-18.
1. Ghaemi SN, Hsu DJ, Soldani F, et al. Antidepressants in bipolar disorder: the case for caution. Bipolar Disorders 2003;5:421-33.
2. Judd LL. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry 2002;59:530-7.
3. Judd LL, Akiskal HS, Schettler PJ, et al. A prospective investigation of the natural history of the long-term weekly symptomatic status of bipolar II disorder. Arch Gen Psychiatry 2003;60(3):261-9.
4. Young LT, Joffe RT, Robb JC, et al. Double-blind comparison of addition of a second mood stabilizer versus an antidepressant to an initial mood stabilizer for treatment of patients with bipolar depression. Am J Psychiatry 2000;157:124-6.
5. Cohn JB, Collins G, Ashbrook E, et al. A comparison of fluoxetine, imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmacol 1989;4(4):313-22.
6. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry 2001;158(6):906-12.
7. Ghaemi SN, Lenox MS, Baldessarini RJ. Effectiveness and safety of long-term antidepressant treatment in bipolar disorder. J Clin Psychiatry 2001;62(7):565-9.
8. Amsterdam JD, Garcia-Espana F, Fawcett J, et al. Efficacy and safety of fluoxetine in treating bipolar II major depressive episode. J Clin Psychopharmacol 1998;18:435-40.
9. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
10. Altshuler L, Kiriakos L, Calcagno J, et al. Impact of antidepressant discontinuation versus antidepressant continuation at 1-year risk for relapse of bipolar depression: a retrospective chart review. J Clin Psychiatry 2001;62:612-16.
11. Altshuler L, Suppes T, Black D, et al. Impact of antidepressant discontinuation after acute bipolar depression remission on rates of depressive relapse at 1-year follow-up. Am J Psychiatry 2003;160:1252-62.
12. Ghaemi SN, Rosenquist KJ, Ko JY, et al. Antidepressant treatment in bipolar versus unipolar depression. Am J Psychiatry 2004;161:163-5.
13. Stoll AL, Mayer PV, Kolbrener M, et al. Antidepressant-associated mania: a controlled comparison with spontaneous mania. Am J Psychiatry 1994;151:1642-5.
14. Zubieta JK, Demitrack MA. Possible bupropion precipitation of mania and mixed affective state. J Clin Psychopharmacol 1991;11(5):327-8.
15. Goldberg JF, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disorders 2003;5:407-20.
16. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry 2002;63:791-5.
17. Post RM, Leverich GS, Nolen WA, et al. A re-evaluation of the role of antidepressants in the treatment of bipolar depression: data from the Stanley Foundation Bipolar Network. Bipolar Disorders 2003;5:396-406.
18. Wehr TA, Sack DA, Rosenthal NE, et al. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
19. Henry C, Sorbara F, Lacoste J, et al. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry 2001;62:249-55.
20. Keck PE, Jr, Perlis RH, Otto MW, et al. The Expert Consensus Guideline Series: Treatment of bipolar disorder 2004. Postgrad Med 2004;1-120.
21. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 2003;60:1079-88.
22. Calabrese JR. Quetiapine BOLDER study [presentation]. New York: American Psychiatric Association annual meeting, 2004.
23. MacQueen GM, Young LT, Marriott M, et al. Previous mood state predicts response and switch rates in patients with bipolar depression. Acta Psychiatr Scand 2002;105:414-18.
New Investigators: What makes aripiprazole the ‘different’ antipsychotic
Aripiprazole, the first FDA-approved partial dopamine agonist, causes few side effects when used to treat schizophrenia or acute bipolar mania. This relatively safe profile in approved uses has led clinicians to try aripiprazole for off-label uses as well, though evidence of the drug’s efficacy and safety in other psychiatric conditions is limited (Box 1).1-7 Because this practice may involve unknown risks, this article:
- reviews aripiprazole’s role in correcting dopaminergic dysfunction in patients with schizophrenia
- cites adverse effects reported with aripiprazole use and discusses concerns about its off-label use.
Aripiprazole shows clear efficacy for treating acute mania and schizophrenia’s negative and positive symptoms, but its effectiveness and safety in other psychiatric illnesses is unknown. Even so, it is being used to treat psychotic unipolar and bipolar depression, attention-deficit/hyperactivity disorder, oppositional defiant disorder, and pervasive developmental disorders in childrenand adults.
Dopamine differences. Do not assume that aripiprazole’s safety and effectiveness in treating schizophrenia translates to other psychiatric conditions. Dopamine dysfunction patterns differ in persons with and without schizophrenia; therefore, aripiprazole’s regional and functional selectivity at dopamine receptors—and treatment response and tolerability—is also likely to differ.
Antipsychotic differences. Aripiprazole is unlike other psychotropics and cannot be assumed to have similar therapeutic effects. The agent’s dopamine agonist and antagonist properties hinge on regional dopamine concentrations as well as the drug itself.
Dopaminergic risks in combination. Numerous drugs modulate dopamine function, and switching and combining drugs places patients at unknown risk of dopaminergic side effects with aripiprazole. Adding an antagonist or a low-dose antagonist combination with aripiprazole may increase competition for dopamine receptors and modify its intrinsic activity, depending on the antagonist dosage and regional dopamine concentrations.
Source: References 1-7
DOPAMINE AND SCHIZOPHRENIA
Dopamine neurons arise from two major nuclei in the mesencephalon (midbrain): the substantia nigra and ventral tegmental area (VTA). Neurons from the substantia nigra extend to the basal ganglia via the mesostriatal (nigrostriatal) pathway, which influences extrapyramidal motor function. The VTA sends dopaminergic neurons through mesolimbic and mesocortical pathways.
The basic limbic system includes the cingulate and orbitofrontal gyri, hippocampus, hypothalamus, thalamus, amygdala, medial temporal cortex, and the periaqueductal gray. This system controls emotion, episodic memory, pain, and primitive behaviors such as eating, fighting, sexual desire, and grooming.8 The limbic system is surrounded by cortex, where higher-order sensory, cognitive, and motor processes occur. The VTA links limbic and cortical functions via dopamine.
The tuberoinfundibular pathway, another clinically important dopaminergic route, projects from the hypothalamus to the anterior pituitary gland and regulates prolactin secretion.
Functional dopamine neurotransmission abnormalities in schizophrenia are generally characterized by region, with:
- excessive mesolimbic pathway activity resulting in positive symptoms such as delusions and hallucinations
- mesocortical projection deficits resulting in cognitive and negative symptoms such as impaired memory and attention, emotional blunting, alogia, avolition, and anhedonia.
Dopamine function abnormalities in schizophrenia occur in:
- projections from the substantia nigra to the caudate and putamen in the basal ganglia
- mesolimbic connections to the anterior cingulate, hippocampus, and parahippocampus
- mesocortical projections to the prefrontal cortex.
These dysfunctions contribute to abnormal motor function, perception, attention, memory, volition, emotion, and executive function.9
WHY ARIPIPRAZOLE WAS CREATED
Differences in regional dopamine function and observations of dopamine agonists’ therapeutic effects in schizophrenia10 led to development of partial dopamine agonists such as aripiprazole (Box 2).11,12
Aripiprazole and the investigational agent bifeprunox have complex pharmacologic actions involving numerous neurotransmitters—dopamine, serotonin, and histamine—but are believed to be principally partial agonists at pre- and postsynaptic dopamine receptors. Specifically, aripiprazole decreases hyperdopaminergic states while preserving dopamine function by partial agonism and decreased stimulation of presynaptic regulatory autoreceptors.5
The FDA approved aripiprazole for treating schizophrenia in 2002 and acute bipolar mania in 2004. A dihydroquinolone unrelated to other antipsychotics, it has an active partial agonist metabolite (dehydro-aripiprazole), high affinity for D2 receptors (Figure 1), and partial agonism at dopamine and serotonin receptors.13
Phase III trials are in progress for bifeprunox, a partial dopamine agonist/antagonist and serotonin receptor agonist being investigated for schizophrenia. An FDA decision on its approvability is expected in 2007.
Aripiprazole has approximately 30% intrinsic dopaminergic activity, estimated from studies showing a low incidence of extrapyramidal symptoms (EPS) with dosages up to 30 mg and PET studies showing a therapeutic range of postsynaptic D2 receptor occupancy of 80% to 95%.14 This small intrinsic activity limits excessive stimulation and dopamine receptor blockade.11 It also limits down-regulation of regulatory dopamine autoreceptors and preserves dopaminergic function in pre- and postsynaptic neurons.
The therapeutic window for dopamine receptor agonists (DA) and serotonin-dopamine receptor agonists (SDA) used in schizophrenia is 60% to 80% D2 receptor occupancy in mesostriatal neurons. D2 receptor occupancy and antagonism >80% significantly increases risk of EPS.15 Excessive dosing of high-potency antipsychotics with strong postsynaptic dopamine receptor affinity carries the highest risk of hypodopaminergic side effects, such as cognitive impairment, worsening of negative symptoms, akathisia, Parkinsonism, and hyperprolactinemia.
Aripiprazole’s intrinsic activity allows for higher D2 receptor occupancy with fewer side effects associated with full dopamine receptor antagonism. Clinical implications of this profile are unknown. Dopamine’s role in schizophrenia’s pathophysiology is inferred from knowledge that all effective antipsychotics offer some postsynaptic D2 receptor blockade.
Conventional dopamine receptor agonists (DAs) such as haloperidol and the newer serotonin-dopamine receptor agonists (SDAs) modeled after clozapine are thought to exert clinical effect by blocking D2 and D3 receptors. By contrast, partial dopamine agonists are agonists or antagonists, depending on cell-specific, synaptic dopamine concentrations. DAs and SDAs are competitive, full antagonists at pre- and postsynaptic dopamine receptors, whereas partial dopamine agonists are:
- agonists at presynaptic regulatory autoreceptors and in hypodopaminergic (mesocortical) synapses
- antagonists with small intrinsic activity in hyperdopaminergic (mesolimbic) synapses. Intrinsic activity is a compound’s degree of agonism in proportion to full agonism.
‘Dopamine stabilizers.’ Because of this combination of high-affinity receptor antagonism with preserved intrinsic activity, some call partial dopamine agonists “dopamine stabilizers.”
Source: References 11, 12
FigureAntipsychotics’ D2 receptor binding affinities
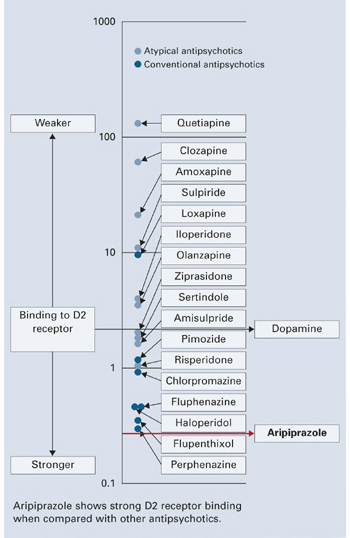
SIDE EFFECTS
Evidence shows fewer side effects with aripiprazole when used to treat schizophrenia and schizoaffective disorder compared with DAs such as haloperidol and SDAs such as risperidone.1,2,4
Dosage-dependent somnolence occurs with aripiprazole; the most common side effects include headache, agitation, anxiety, and insomnia (24.5%, 34.6%, 24%, and 18.6%, respectively).2 continued on page 60 A 10-week, placebo-controlled study using aripiprazole, 2 to 15 mg/d, to treat psychosis in Alzheimer’s dementia showed significantly increased risk of somnolence, accidental injury, and bronchitis (likely caused by aspiration).16
In placebo-controlled trials, aripiprazole, 2 to 30 mg/d, did not increase risk of cardiac, lipid, or prolactin-related side effects2 but showed increased risk of:
- tremor when used at 15 mg/d for up to 26 weeks in chronic schizophrenia, (9% incidence vs 1% with placebo)17
- akathisia when used at mean dosages of 27.9 mg/d to treat acute bipolar mania (11% incidence vs 2%with placebo).3 Aripiprazole also increased akathisia incidence in normal subjects.18
Risk of tardive dyskinesia or hyperglycemia-related adverse events with aripiprazole are unknown. Studies report weight gain of <1 kg (<2.2 lbs) in patients taking aripiprazole, 2 to 30 mg/d. One study found that patients switched from DA and SDA antipsychotics to aripiprazole, 30 mg/d, lost on average 1.5 kg (3.2 lbs) across 8weeks.19
Determining neuroleptic malignant syndrome risk with aripiprazole is difficult; two cases were reported in the premarketing sample.16 One animal study showed diminished catalepsy with chronic aripiprazole use, in contrast to persistent catalepsy with haloperidol.20
A Medline search for aripiprazole in February 2005 found several reports of treatment-emergent side effects, including:
- 3 reports of worsening agitation or psychosis21-23
- 2 reports of EPS24,25
- 1 report of excessive somnolence in a child.26
Adverse effects are probably underrepresented. Clinical deterioration and adverse effects were reported after starting, switching to, or combining aripiprazole with other antipsychotics or serotonergic agents (trazodone, sertraline, or venlafaxine).27,28
This paper by Dr. Mahendra T. Bhati was entered in the 2005 Promising New Investigators competition sponsored by the Neuroleptic Malignant Syndrome Information Service (NMSIS). The theme of this year’s scholarly papers was “New insights on psychotropic drug safety and side effects.”
Current Psychiatry is honored to publish this peer-reviewed, evidence-based article on a clinically important topic for practicing psychiatrists.
NMSIS is dedicated to reducing morbidity and mortality of NMS by improving medical and psychiatric care of patients with heat-related disorders; providing support information for medical professionals, patients and families; and improving scientific understanding of these conditions through research.
Related resources
- Lieberman, J. Aripiprazole. In: Schatzberg, A, Nemeroff, C (eds.) Textbook of Psychopharmacology. Washington DC: American Psychiatric Publishing, 2004:487-494.
- FDA Center for Drug Evaluation and Research. http://www.fda.gov/cder/foi/nda/2002/21-436_Abilify.htm
- Medline Plus Drug Information. http://www.nlm.nih.gov/medlineplus/druginfo/medmaster/a603012.html
Drug brand names
- Aripiprazole •Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Risperidone • Risperdal
- Trazodone • Desyrel
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
This author is supported by grant 2R25MH060490-06 from the National Institute of Mental Health.
1. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
2. Marder SR, McQuade RD, Stock E, et al. Aripiprazole in the treatment of schizophrenia: safety and tolerability in short-term, placebo-controlled trials. Schizophr Res 2003;61:123-36.
3. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry 2003;160:1651-8.
4. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry 2003;60:681-90.
5. Grady MA, Gasperoni TL, Kirkpatrick P. Aripiprazole. Nat Rev Drug Discov 2003;2:427-8.
6. Stigler KA, Posey DJ, McDougle CJ. Aripiprazole for maladaptive behavior in pervasive developmental disorders. J Child Adolesc Psychopharmacol 2004;14:455-63.
7. Tamminga CA. Partial dopamine agonists in the treatment of psychosis. J Neural Transm 2002;109:411-20.
8. Arciniegas DB, Beresford TP. Neuropsychiatry: an Introductory Approach .Cambridge, UK: Cambridge University Press; 2001.
9. Stahl SM. Psychopharmacology of Antipsychotics .London: Martin Dunitz Ltd.; 1999.
10. Tamminga CA, Gotts MD, Thaker GK, et al. Dopamine agonist treatment of schizophrenia with N-propylnorapomorphine. Arch Gen Psychiatry 1986;43:398-402.
11. Grunder G, Carlsson A, Wong D. Mechanism of new antipsychotic medications: occupancy is not just antagonism. Arch Gen Psychiatry 2003;60:974-7.
12. Burris K, Molski T, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
13. Lieberman J. Dopamine partial agonists: a new class of antipsychotic. CNS Drugs 2004;18:251-67.
14. Yokoi F, Grunder G, Biziere K, et al. Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]raclopride. Neuropsychopharmacology 2002;27:248-59.
15. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 2000;157:514-20.
16. U.S. Food and Drug Administration. Medwatch online. Available at: http://www.fda.gov/medwatch/SAFETY/2004/abilify_pi.pdf. Accessed June 7, 2004.
17. Pigott T, Carson W, Saha A, et al. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry 2003;64:1048-56.
18. Mallikaarjun S, Salazar D, Bramer S. Pharmacokinetics, tolerability, and safety of aripiprazole following multiple oral dosing in normal healthy volunteers. J Clin Pharmacol 2004;44:179-87.
19. Casey D, Carson W, Saha A, et al. Switching patients to aripiprazole from other antipsychotic agents: a multicenter randomized study. Psychopharmacology 2003;166:391-9.
20. Nakai S, Hirose T, Uwahodo Y, et al. Diminished catalepsy and dopamine metabolism distinguish aripiprazole from haloperidol or risperidone. Eur J Pharmacol 2003;472:89-97.
21. Sajbel T, Cheney E, DeQuardo J. Aripiprazole-associated dyskinesia. Ann Pharmacother 2005;39:200-1.
22. Reeves R, Mack J. Worsening schizoaffective disorder with aripiprazole. Am J Psychiatry 2004;161:1308.-
23. Ramaswamy S, Vijay D, William M, et al. Aripiprazole possibly worsens psychosis. Int Clin Psychopharmacol 2004;19:45-8.
24. Mendhekar D. Aripiprazole-induced rabbit syndrome. Aust N Z J Psychiatry 2004;38:561.-
25. Lindsey R, Kaplan D, Koliatsos V, et al. Aripiprazole and extrapyramidal symptoms. J Am Acad Child Adolesc Psychiatry 2003;42:1268-9.
26. Davenport J, McCarthy M, Buck M. Excessive somnolence from aripiprazole in a child. Pharmacotherapy 2004;24:522-5.
27. DeQuardo J. Worsened agitation with aripiprazole: adverse effect of dopamine partial agonism? J Clin Psychiatry 2004;65:132-3.
28. Cohen S, Rulf D, Pies R. Extrapyramidal side effects associated with aripiprazole coprescription in 2 patients. J Clin Psychiatry 2005;66:135-6.
Aripiprazole, the first FDA-approved partial dopamine agonist, causes few side effects when used to treat schizophrenia or acute bipolar mania. This relatively safe profile in approved uses has led clinicians to try aripiprazole for off-label uses as well, though evidence of the drug’s efficacy and safety in other psychiatric conditions is limited (Box 1).1-7 Because this practice may involve unknown risks, this article:
- reviews aripiprazole’s role in correcting dopaminergic dysfunction in patients with schizophrenia
- cites adverse effects reported with aripiprazole use and discusses concerns about its off-label use.
Aripiprazole shows clear efficacy for treating acute mania and schizophrenia’s negative and positive symptoms, but its effectiveness and safety in other psychiatric illnesses is unknown. Even so, it is being used to treat psychotic unipolar and bipolar depression, attention-deficit/hyperactivity disorder, oppositional defiant disorder, and pervasive developmental disorders in childrenand adults.
Dopamine differences. Do not assume that aripiprazole’s safety and effectiveness in treating schizophrenia translates to other psychiatric conditions. Dopamine dysfunction patterns differ in persons with and without schizophrenia; therefore, aripiprazole’s regional and functional selectivity at dopamine receptors—and treatment response and tolerability—is also likely to differ.
Antipsychotic differences. Aripiprazole is unlike other psychotropics and cannot be assumed to have similar therapeutic effects. The agent’s dopamine agonist and antagonist properties hinge on regional dopamine concentrations as well as the drug itself.
Dopaminergic risks in combination. Numerous drugs modulate dopamine function, and switching and combining drugs places patients at unknown risk of dopaminergic side effects with aripiprazole. Adding an antagonist or a low-dose antagonist combination with aripiprazole may increase competition for dopamine receptors and modify its intrinsic activity, depending on the antagonist dosage and regional dopamine concentrations.
Source: References 1-7
DOPAMINE AND SCHIZOPHRENIA
Dopamine neurons arise from two major nuclei in the mesencephalon (midbrain): the substantia nigra and ventral tegmental area (VTA). Neurons from the substantia nigra extend to the basal ganglia via the mesostriatal (nigrostriatal) pathway, which influences extrapyramidal motor function. The VTA sends dopaminergic neurons through mesolimbic and mesocortical pathways.
The basic limbic system includes the cingulate and orbitofrontal gyri, hippocampus, hypothalamus, thalamus, amygdala, medial temporal cortex, and the periaqueductal gray. This system controls emotion, episodic memory, pain, and primitive behaviors such as eating, fighting, sexual desire, and grooming.8 The limbic system is surrounded by cortex, where higher-order sensory, cognitive, and motor processes occur. The VTA links limbic and cortical functions via dopamine.
The tuberoinfundibular pathway, another clinically important dopaminergic route, projects from the hypothalamus to the anterior pituitary gland and regulates prolactin secretion.
Functional dopamine neurotransmission abnormalities in schizophrenia are generally characterized by region, with:
- excessive mesolimbic pathway activity resulting in positive symptoms such as delusions and hallucinations
- mesocortical projection deficits resulting in cognitive and negative symptoms such as impaired memory and attention, emotional blunting, alogia, avolition, and anhedonia.
Dopamine function abnormalities in schizophrenia occur in:
- projections from the substantia nigra to the caudate and putamen in the basal ganglia
- mesolimbic connections to the anterior cingulate, hippocampus, and parahippocampus
- mesocortical projections to the prefrontal cortex.
These dysfunctions contribute to abnormal motor function, perception, attention, memory, volition, emotion, and executive function.9
WHY ARIPIPRAZOLE WAS CREATED
Differences in regional dopamine function and observations of dopamine agonists’ therapeutic effects in schizophrenia10 led to development of partial dopamine agonists such as aripiprazole (Box 2).11,12
Aripiprazole and the investigational agent bifeprunox have complex pharmacologic actions involving numerous neurotransmitters—dopamine, serotonin, and histamine—but are believed to be principally partial agonists at pre- and postsynaptic dopamine receptors. Specifically, aripiprazole decreases hyperdopaminergic states while preserving dopamine function by partial agonism and decreased stimulation of presynaptic regulatory autoreceptors.5
The FDA approved aripiprazole for treating schizophrenia in 2002 and acute bipolar mania in 2004. A dihydroquinolone unrelated to other antipsychotics, it has an active partial agonist metabolite (dehydro-aripiprazole), high affinity for D2 receptors (Figure 1), and partial agonism at dopamine and serotonin receptors.13
Phase III trials are in progress for bifeprunox, a partial dopamine agonist/antagonist and serotonin receptor agonist being investigated for schizophrenia. An FDA decision on its approvability is expected in 2007.
Aripiprazole has approximately 30% intrinsic dopaminergic activity, estimated from studies showing a low incidence of extrapyramidal symptoms (EPS) with dosages up to 30 mg and PET studies showing a therapeutic range of postsynaptic D2 receptor occupancy of 80% to 95%.14 This small intrinsic activity limits excessive stimulation and dopamine receptor blockade.11 It also limits down-regulation of regulatory dopamine autoreceptors and preserves dopaminergic function in pre- and postsynaptic neurons.
The therapeutic window for dopamine receptor agonists (DA) and serotonin-dopamine receptor agonists (SDA) used in schizophrenia is 60% to 80% D2 receptor occupancy in mesostriatal neurons. D2 receptor occupancy and antagonism >80% significantly increases risk of EPS.15 Excessive dosing of high-potency antipsychotics with strong postsynaptic dopamine receptor affinity carries the highest risk of hypodopaminergic side effects, such as cognitive impairment, worsening of negative symptoms, akathisia, Parkinsonism, and hyperprolactinemia.
Aripiprazole’s intrinsic activity allows for higher D2 receptor occupancy with fewer side effects associated with full dopamine receptor antagonism. Clinical implications of this profile are unknown. Dopamine’s role in schizophrenia’s pathophysiology is inferred from knowledge that all effective antipsychotics offer some postsynaptic D2 receptor blockade.
Conventional dopamine receptor agonists (DAs) such as haloperidol and the newer serotonin-dopamine receptor agonists (SDAs) modeled after clozapine are thought to exert clinical effect by blocking D2 and D3 receptors. By contrast, partial dopamine agonists are agonists or antagonists, depending on cell-specific, synaptic dopamine concentrations. DAs and SDAs are competitive, full antagonists at pre- and postsynaptic dopamine receptors, whereas partial dopamine agonists are:
- agonists at presynaptic regulatory autoreceptors and in hypodopaminergic (mesocortical) synapses
- antagonists with small intrinsic activity in hyperdopaminergic (mesolimbic) synapses. Intrinsic activity is a compound’s degree of agonism in proportion to full agonism.
‘Dopamine stabilizers.’ Because of this combination of high-affinity receptor antagonism with preserved intrinsic activity, some call partial dopamine agonists “dopamine stabilizers.”
Source: References 11, 12
FigureAntipsychotics’ D2 receptor binding affinities
SIDE EFFECTS
Evidence shows fewer side effects with aripiprazole when used to treat schizophrenia and schizoaffective disorder compared with DAs such as haloperidol and SDAs such as risperidone.1,2,4
Dosage-dependent somnolence occurs with aripiprazole; the most common side effects include headache, agitation, anxiety, and insomnia (24.5%, 34.6%, 24%, and 18.6%, respectively).2 continued on page 60 A 10-week, placebo-controlled study using aripiprazole, 2 to 15 mg/d, to treat psychosis in Alzheimer’s dementia showed significantly increased risk of somnolence, accidental injury, and bronchitis (likely caused by aspiration).16
In placebo-controlled trials, aripiprazole, 2 to 30 mg/d, did not increase risk of cardiac, lipid, or prolactin-related side effects2 but showed increased risk of:
- tremor when used at 15 mg/d for up to 26 weeks in chronic schizophrenia, (9% incidence vs 1% with placebo)17
- akathisia when used at mean dosages of 27.9 mg/d to treat acute bipolar mania (11% incidence vs 2%with placebo).3 Aripiprazole also increased akathisia incidence in normal subjects.18
Risk of tardive dyskinesia or hyperglycemia-related adverse events with aripiprazole are unknown. Studies report weight gain of <1 kg (<2.2 lbs) in patients taking aripiprazole, 2 to 30 mg/d. One study found that patients switched from DA and SDA antipsychotics to aripiprazole, 30 mg/d, lost on average 1.5 kg (3.2 lbs) across 8weeks.19
Determining neuroleptic malignant syndrome risk with aripiprazole is difficult; two cases were reported in the premarketing sample.16 One animal study showed diminished catalepsy with chronic aripiprazole use, in contrast to persistent catalepsy with haloperidol.20
A Medline search for aripiprazole in February 2005 found several reports of treatment-emergent side effects, including:
- 3 reports of worsening agitation or psychosis21-23
- 2 reports of EPS24,25
- 1 report of excessive somnolence in a child.26
Adverse effects are probably underrepresented. Clinical deterioration and adverse effects were reported after starting, switching to, or combining aripiprazole with other antipsychotics or serotonergic agents (trazodone, sertraline, or venlafaxine).27,28
This paper by Dr. Mahendra T. Bhati was entered in the 2005 Promising New Investigators competition sponsored by the Neuroleptic Malignant Syndrome Information Service (NMSIS). The theme of this year’s scholarly papers was “New insights on psychotropic drug safety and side effects.”
Current Psychiatry is honored to publish this peer-reviewed, evidence-based article on a clinically important topic for practicing psychiatrists.
NMSIS is dedicated to reducing morbidity and mortality of NMS by improving medical and psychiatric care of patients with heat-related disorders; providing support information for medical professionals, patients and families; and improving scientific understanding of these conditions through research.
Related resources
- Lieberman, J. Aripiprazole. In: Schatzberg, A, Nemeroff, C (eds.) Textbook of Psychopharmacology. Washington DC: American Psychiatric Publishing, 2004:487-494.
- FDA Center for Drug Evaluation and Research. http://www.fda.gov/cder/foi/nda/2002/21-436_Abilify.htm
- Medline Plus Drug Information. http://www.nlm.nih.gov/medlineplus/druginfo/medmaster/a603012.html
Drug brand names
- Aripiprazole •Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Risperidone • Risperdal
- Trazodone • Desyrel
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
This author is supported by grant 2R25MH060490-06 from the National Institute of Mental Health.
Aripiprazole, the first FDA-approved partial dopamine agonist, causes few side effects when used to treat schizophrenia or acute bipolar mania. This relatively safe profile in approved uses has led clinicians to try aripiprazole for off-label uses as well, though evidence of the drug’s efficacy and safety in other psychiatric conditions is limited (Box 1).1-7 Because this practice may involve unknown risks, this article:
- reviews aripiprazole’s role in correcting dopaminergic dysfunction in patients with schizophrenia
- cites adverse effects reported with aripiprazole use and discusses concerns about its off-label use.
Aripiprazole shows clear efficacy for treating acute mania and schizophrenia’s negative and positive symptoms, but its effectiveness and safety in other psychiatric illnesses is unknown. Even so, it is being used to treat psychotic unipolar and bipolar depression, attention-deficit/hyperactivity disorder, oppositional defiant disorder, and pervasive developmental disorders in childrenand adults.
Dopamine differences. Do not assume that aripiprazole’s safety and effectiveness in treating schizophrenia translates to other psychiatric conditions. Dopamine dysfunction patterns differ in persons with and without schizophrenia; therefore, aripiprazole’s regional and functional selectivity at dopamine receptors—and treatment response and tolerability—is also likely to differ.
Antipsychotic differences. Aripiprazole is unlike other psychotropics and cannot be assumed to have similar therapeutic effects. The agent’s dopamine agonist and antagonist properties hinge on regional dopamine concentrations as well as the drug itself.
Dopaminergic risks in combination. Numerous drugs modulate dopamine function, and switching and combining drugs places patients at unknown risk of dopaminergic side effects with aripiprazole. Adding an antagonist or a low-dose antagonist combination with aripiprazole may increase competition for dopamine receptors and modify its intrinsic activity, depending on the antagonist dosage and regional dopamine concentrations.
Source: References 1-7
DOPAMINE AND SCHIZOPHRENIA
Dopamine neurons arise from two major nuclei in the mesencephalon (midbrain): the substantia nigra and ventral tegmental area (VTA). Neurons from the substantia nigra extend to the basal ganglia via the mesostriatal (nigrostriatal) pathway, which influences extrapyramidal motor function. The VTA sends dopaminergic neurons through mesolimbic and mesocortical pathways.
The basic limbic system includes the cingulate and orbitofrontal gyri, hippocampus, hypothalamus, thalamus, amygdala, medial temporal cortex, and the periaqueductal gray. This system controls emotion, episodic memory, pain, and primitive behaviors such as eating, fighting, sexual desire, and grooming.8 The limbic system is surrounded by cortex, where higher-order sensory, cognitive, and motor processes occur. The VTA links limbic and cortical functions via dopamine.
The tuberoinfundibular pathway, another clinically important dopaminergic route, projects from the hypothalamus to the anterior pituitary gland and regulates prolactin secretion.
Functional dopamine neurotransmission abnormalities in schizophrenia are generally characterized by region, with:
- excessive mesolimbic pathway activity resulting in positive symptoms such as delusions and hallucinations
- mesocortical projection deficits resulting in cognitive and negative symptoms such as impaired memory and attention, emotional blunting, alogia, avolition, and anhedonia.
Dopamine function abnormalities in schizophrenia occur in:
- projections from the substantia nigra to the caudate and putamen in the basal ganglia
- mesolimbic connections to the anterior cingulate, hippocampus, and parahippocampus
- mesocortical projections to the prefrontal cortex.
These dysfunctions contribute to abnormal motor function, perception, attention, memory, volition, emotion, and executive function.9
WHY ARIPIPRAZOLE WAS CREATED
Differences in regional dopamine function and observations of dopamine agonists’ therapeutic effects in schizophrenia10 led to development of partial dopamine agonists such as aripiprazole (Box 2).11,12
Aripiprazole and the investigational agent bifeprunox have complex pharmacologic actions involving numerous neurotransmitters—dopamine, serotonin, and histamine—but are believed to be principally partial agonists at pre- and postsynaptic dopamine receptors. Specifically, aripiprazole decreases hyperdopaminergic states while preserving dopamine function by partial agonism and decreased stimulation of presynaptic regulatory autoreceptors.5
The FDA approved aripiprazole for treating schizophrenia in 2002 and acute bipolar mania in 2004. A dihydroquinolone unrelated to other antipsychotics, it has an active partial agonist metabolite (dehydro-aripiprazole), high affinity for D2 receptors (Figure 1), and partial agonism at dopamine and serotonin receptors.13
Phase III trials are in progress for bifeprunox, a partial dopamine agonist/antagonist and serotonin receptor agonist being investigated for schizophrenia. An FDA decision on its approvability is expected in 2007.
Aripiprazole has approximately 30% intrinsic dopaminergic activity, estimated from studies showing a low incidence of extrapyramidal symptoms (EPS) with dosages up to 30 mg and PET studies showing a therapeutic range of postsynaptic D2 receptor occupancy of 80% to 95%.14 This small intrinsic activity limits excessive stimulation and dopamine receptor blockade.11 It also limits down-regulation of regulatory dopamine autoreceptors and preserves dopaminergic function in pre- and postsynaptic neurons.
The therapeutic window for dopamine receptor agonists (DA) and serotonin-dopamine receptor agonists (SDA) used in schizophrenia is 60% to 80% D2 receptor occupancy in mesostriatal neurons. D2 receptor occupancy and antagonism >80% significantly increases risk of EPS.15 Excessive dosing of high-potency antipsychotics with strong postsynaptic dopamine receptor affinity carries the highest risk of hypodopaminergic side effects, such as cognitive impairment, worsening of negative symptoms, akathisia, Parkinsonism, and hyperprolactinemia.
Aripiprazole’s intrinsic activity allows for higher D2 receptor occupancy with fewer side effects associated with full dopamine receptor antagonism. Clinical implications of this profile are unknown. Dopamine’s role in schizophrenia’s pathophysiology is inferred from knowledge that all effective antipsychotics offer some postsynaptic D2 receptor blockade.
Conventional dopamine receptor agonists (DAs) such as haloperidol and the newer serotonin-dopamine receptor agonists (SDAs) modeled after clozapine are thought to exert clinical effect by blocking D2 and D3 receptors. By contrast, partial dopamine agonists are agonists or antagonists, depending on cell-specific, synaptic dopamine concentrations. DAs and SDAs are competitive, full antagonists at pre- and postsynaptic dopamine receptors, whereas partial dopamine agonists are:
- agonists at presynaptic regulatory autoreceptors and in hypodopaminergic (mesocortical) synapses
- antagonists with small intrinsic activity in hyperdopaminergic (mesolimbic) synapses. Intrinsic activity is a compound’s degree of agonism in proportion to full agonism.
‘Dopamine stabilizers.’ Because of this combination of high-affinity receptor antagonism with preserved intrinsic activity, some call partial dopamine agonists “dopamine stabilizers.”
Source: References 11, 12
FigureAntipsychotics’ D2 receptor binding affinities
SIDE EFFECTS
Evidence shows fewer side effects with aripiprazole when used to treat schizophrenia and schizoaffective disorder compared with DAs such as haloperidol and SDAs such as risperidone.1,2,4
Dosage-dependent somnolence occurs with aripiprazole; the most common side effects include headache, agitation, anxiety, and insomnia (24.5%, 34.6%, 24%, and 18.6%, respectively).2 continued on page 60 A 10-week, placebo-controlled study using aripiprazole, 2 to 15 mg/d, to treat psychosis in Alzheimer’s dementia showed significantly increased risk of somnolence, accidental injury, and bronchitis (likely caused by aspiration).16
In placebo-controlled trials, aripiprazole, 2 to 30 mg/d, did not increase risk of cardiac, lipid, or prolactin-related side effects2 but showed increased risk of:
- tremor when used at 15 mg/d for up to 26 weeks in chronic schizophrenia, (9% incidence vs 1% with placebo)17
- akathisia when used at mean dosages of 27.9 mg/d to treat acute bipolar mania (11% incidence vs 2%with placebo).3 Aripiprazole also increased akathisia incidence in normal subjects.18
Risk of tardive dyskinesia or hyperglycemia-related adverse events with aripiprazole are unknown. Studies report weight gain of <1 kg (<2.2 lbs) in patients taking aripiprazole, 2 to 30 mg/d. One study found that patients switched from DA and SDA antipsychotics to aripiprazole, 30 mg/d, lost on average 1.5 kg (3.2 lbs) across 8weeks.19
Determining neuroleptic malignant syndrome risk with aripiprazole is difficult; two cases were reported in the premarketing sample.16 One animal study showed diminished catalepsy with chronic aripiprazole use, in contrast to persistent catalepsy with haloperidol.20
A Medline search for aripiprazole in February 2005 found several reports of treatment-emergent side effects, including:
- 3 reports of worsening agitation or psychosis21-23
- 2 reports of EPS24,25
- 1 report of excessive somnolence in a child.26
Adverse effects are probably underrepresented. Clinical deterioration and adverse effects were reported after starting, switching to, or combining aripiprazole with other antipsychotics or serotonergic agents (trazodone, sertraline, or venlafaxine).27,28
This paper by Dr. Mahendra T. Bhati was entered in the 2005 Promising New Investigators competition sponsored by the Neuroleptic Malignant Syndrome Information Service (NMSIS). The theme of this year’s scholarly papers was “New insights on psychotropic drug safety and side effects.”
Current Psychiatry is honored to publish this peer-reviewed, evidence-based article on a clinically important topic for practicing psychiatrists.
NMSIS is dedicated to reducing morbidity and mortality of NMS by improving medical and psychiatric care of patients with heat-related disorders; providing support information for medical professionals, patients and families; and improving scientific understanding of these conditions through research.
Related resources
- Lieberman, J. Aripiprazole. In: Schatzberg, A, Nemeroff, C (eds.) Textbook of Psychopharmacology. Washington DC: American Psychiatric Publishing, 2004:487-494.
- FDA Center for Drug Evaluation and Research. http://www.fda.gov/cder/foi/nda/2002/21-436_Abilify.htm
- Medline Plus Drug Information. http://www.nlm.nih.gov/medlineplus/druginfo/medmaster/a603012.html
Drug brand names
- Aripiprazole •Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Risperidone • Risperdal
- Trazodone • Desyrel
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosure
This author is supported by grant 2R25MH060490-06 from the National Institute of Mental Health.
1. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
2. Marder SR, McQuade RD, Stock E, et al. Aripiprazole in the treatment of schizophrenia: safety and tolerability in short-term, placebo-controlled trials. Schizophr Res 2003;61:123-36.
3. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry 2003;160:1651-8.
4. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry 2003;60:681-90.
5. Grady MA, Gasperoni TL, Kirkpatrick P. Aripiprazole. Nat Rev Drug Discov 2003;2:427-8.
6. Stigler KA, Posey DJ, McDougle CJ. Aripiprazole for maladaptive behavior in pervasive developmental disorders. J Child Adolesc Psychopharmacol 2004;14:455-63.
7. Tamminga CA. Partial dopamine agonists in the treatment of psychosis. J Neural Transm 2002;109:411-20.
8. Arciniegas DB, Beresford TP. Neuropsychiatry: an Introductory Approach .Cambridge, UK: Cambridge University Press; 2001.
9. Stahl SM. Psychopharmacology of Antipsychotics .London: Martin Dunitz Ltd.; 1999.
10. Tamminga CA, Gotts MD, Thaker GK, et al. Dopamine agonist treatment of schizophrenia with N-propylnorapomorphine. Arch Gen Psychiatry 1986;43:398-402.
11. Grunder G, Carlsson A, Wong D. Mechanism of new antipsychotic medications: occupancy is not just antagonism. Arch Gen Psychiatry 2003;60:974-7.
12. Burris K, Molski T, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
13. Lieberman J. Dopamine partial agonists: a new class of antipsychotic. CNS Drugs 2004;18:251-67.
14. Yokoi F, Grunder G, Biziere K, et al. Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]raclopride. Neuropsychopharmacology 2002;27:248-59.
15. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 2000;157:514-20.
16. U.S. Food and Drug Administration. Medwatch online. Available at: http://www.fda.gov/medwatch/SAFETY/2004/abilify_pi.pdf. Accessed June 7, 2004.
17. Pigott T, Carson W, Saha A, et al. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry 2003;64:1048-56.
18. Mallikaarjun S, Salazar D, Bramer S. Pharmacokinetics, tolerability, and safety of aripiprazole following multiple oral dosing in normal healthy volunteers. J Clin Pharmacol 2004;44:179-87.
19. Casey D, Carson W, Saha A, et al. Switching patients to aripiprazole from other antipsychotic agents: a multicenter randomized study. Psychopharmacology 2003;166:391-9.
20. Nakai S, Hirose T, Uwahodo Y, et al. Diminished catalepsy and dopamine metabolism distinguish aripiprazole from haloperidol or risperidone. Eur J Pharmacol 2003;472:89-97.
21. Sajbel T, Cheney E, DeQuardo J. Aripiprazole-associated dyskinesia. Ann Pharmacother 2005;39:200-1.
22. Reeves R, Mack J. Worsening schizoaffective disorder with aripiprazole. Am J Psychiatry 2004;161:1308.-
23. Ramaswamy S, Vijay D, William M, et al. Aripiprazole possibly worsens psychosis. Int Clin Psychopharmacol 2004;19:45-8.
24. Mendhekar D. Aripiprazole-induced rabbit syndrome. Aust N Z J Psychiatry 2004;38:561.-
25. Lindsey R, Kaplan D, Koliatsos V, et al. Aripiprazole and extrapyramidal symptoms. J Am Acad Child Adolesc Psychiatry 2003;42:1268-9.
26. Davenport J, McCarthy M, Buck M. Excessive somnolence from aripiprazole in a child. Pharmacotherapy 2004;24:522-5.
27. DeQuardo J. Worsened agitation with aripiprazole: adverse effect of dopamine partial agonism? J Clin Psychiatry 2004;65:132-3.
28. Cohen S, Rulf D, Pies R. Extrapyramidal side effects associated with aripiprazole coprescription in 2 patients. J Clin Psychiatry 2005;66:135-6.
1. Kane JM, Carson WH, Saha AR, et al. Efficacy and safety of aripiprazole and haloperidol versus placebo in patients with schizophrenia and schizoaffective disorder. J Clin Psychiatry 2002;63:763-71.
2. Marder SR, McQuade RD, Stock E, et al. Aripiprazole in the treatment of schizophrenia: safety and tolerability in short-term, placebo-controlled trials. Schizophr Res 2003;61:123-36.
3. Keck PE, Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry 2003;160:1651-8.
4. Potkin SG, Saha AR, Kujawa MJ, et al. Aripiprazole, an antipsychotic with a novel mechanism of action, and risperidone vs placebo in patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry 2003;60:681-90.
5. Grady MA, Gasperoni TL, Kirkpatrick P. Aripiprazole. Nat Rev Drug Discov 2003;2:427-8.
6. Stigler KA, Posey DJ, McDougle CJ. Aripiprazole for maladaptive behavior in pervasive developmental disorders. J Child Adolesc Psychopharmacol 2004;14:455-63.
7. Tamminga CA. Partial dopamine agonists in the treatment of psychosis. J Neural Transm 2002;109:411-20.
8. Arciniegas DB, Beresford TP. Neuropsychiatry: an Introductory Approach .Cambridge, UK: Cambridge University Press; 2001.
9. Stahl SM. Psychopharmacology of Antipsychotics .London: Martin Dunitz Ltd.; 1999.
10. Tamminga CA, Gotts MD, Thaker GK, et al. Dopamine agonist treatment of schizophrenia with N-propylnorapomorphine. Arch Gen Psychiatry 1986;43:398-402.
11. Grunder G, Carlsson A, Wong D. Mechanism of new antipsychotic medications: occupancy is not just antagonism. Arch Gen Psychiatry 2003;60:974-7.
12. Burris K, Molski T, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
13. Lieberman J. Dopamine partial agonists: a new class of antipsychotic. CNS Drugs 2004;18:251-67.
14. Yokoi F, Grunder G, Biziere K, et al. Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]raclopride. Neuropsychopharmacology 2002;27:248-59.
15. Kapur S, Zipursky R, Jones C, et al. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry 2000;157:514-20.
16. U.S. Food and Drug Administration. Medwatch online. Available at: http://www.fda.gov/medwatch/SAFETY/2004/abilify_pi.pdf. Accessed June 7, 2004.
17. Pigott T, Carson W, Saha A, et al. Aripiprazole for the prevention of relapse in stabilized patients with chronic schizophrenia: a placebo-controlled 26-week study. J Clin Psychiatry 2003;64:1048-56.
18. Mallikaarjun S, Salazar D, Bramer S. Pharmacokinetics, tolerability, and safety of aripiprazole following multiple oral dosing in normal healthy volunteers. J Clin Pharmacol 2004;44:179-87.
19. Casey D, Carson W, Saha A, et al. Switching patients to aripiprazole from other antipsychotic agents: a multicenter randomized study. Psychopharmacology 2003;166:391-9.
20. Nakai S, Hirose T, Uwahodo Y, et al. Diminished catalepsy and dopamine metabolism distinguish aripiprazole from haloperidol or risperidone. Eur J Pharmacol 2003;472:89-97.
21. Sajbel T, Cheney E, DeQuardo J. Aripiprazole-associated dyskinesia. Ann Pharmacother 2005;39:200-1.
22. Reeves R, Mack J. Worsening schizoaffective disorder with aripiprazole. Am J Psychiatry 2004;161:1308.-
23. Ramaswamy S, Vijay D, William M, et al. Aripiprazole possibly worsens psychosis. Int Clin Psychopharmacol 2004;19:45-8.
24. Mendhekar D. Aripiprazole-induced rabbit syndrome. Aust N Z J Psychiatry 2004;38:561.-
25. Lindsey R, Kaplan D, Koliatsos V, et al. Aripiprazole and extrapyramidal symptoms. J Am Acad Child Adolesc Psychiatry 2003;42:1268-9.
26. Davenport J, McCarthy M, Buck M. Excessive somnolence from aripiprazole in a child. Pharmacotherapy 2004;24:522-5.
27. DeQuardo J. Worsened agitation with aripiprazole: adverse effect of dopamine partial agonism? J Clin Psychiatry 2004;65:132-3.
28. Cohen S, Rulf D, Pies R. Extrapyramidal side effects associated with aripiprazole coprescription in 2 patients. J Clin Psychiatry 2005;66:135-6.
Is it PANDAS? How to confirm the sore throat/OCD connection
John, age 6, presented for psychiatric evaluation with acute, incapacitating obsessive-compulsive symptoms. For 4 weeks he washed his hands compulsively and had pervasive obsessions about death by choking.
These symptoms had suddenly worsened over 2 days. At first, he washed his hands more than 35 times per day in rituals lasting several minutes each. Then, within 2 weeks, John’s handwashing spontaneously decreased, but his choking fears dramatically increased. He refused all solid foods and continuously sought reassurance from his parents that he would not choke or die.
Approximately 1 week before these symptoms began, John had a sore throat and tested positive via throat culture for group A beta-hemolytic streptococcal infection (GABHS).
Sore throat followed by sudden-onset obsessive-compulsive symptoms or tics in a child such as John suggests a pediatric autoimmune neuropsychiatric disorder associated with streptococcal infection (PANDAS). The association between GABHS infection and these symptoms remains uncertain, as the mechanism by which GABHS infection may cause obsessive-compulsive symptoms and other childhood-onset neuropsychiatric disorders is largely unknown.
Since PANDAS was recognized (Box 1),1-6 some data have emerged on the disorder’s symptoms, course, and prognosis. However:
- diagnostic criteria are not well-defined
- few controlled studies have examined treatment response
- using antibiotics and immunotherapies to treat or prevent PANDAS symptoms remains controversial because of unproven efficacy and potential adverse effects.
To help you diagnose and treat patients with suspected PANDAS, this article examines the limited evidence for the disorder, discusses diagnostic guidelines, and reviews preliminary indications for behavioral and medical treatments.
PANDAS stands for pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. It describes childhoodonset OCD or tic cases whose onset or worsening appears to be linked to group A beta-hemolytic streptococcal (GABHS) infection.
GABHS antibodies cross-react with the cellular components of the basal ganglia, particularly in the caudate nucleus.1 PANDAS was first recognized in 1987 during an unexpected resurgence of rheumatic fever in the United States.2 Sydenham chorea is thought to occur when GABHS antibodies undergo molecular mimicry and cross-react with epitopes on neurons in the basal ganglia and other brain areas, causing motor and behavioral disruptions.3 Rheumatic fever patients with Sydenham chorea show a high prevalence (up to 70%) of obsessive-compulsive disorder (OCD).4,5
Although individuals with Sydenham chorea appear to be at greatest risk for OCD after GABHS infection, rheumatic fever patients without chorea also appear to be at increased OCD risk.6
CASE REPORT CONTINUED: PANDAS CLUES
John’s sudden-onset compulsive behaviors and obsessive thoughts exemplify the rapid symptom onset often seen in children with PANDAS. His medical records showed a temporal relationship between his streptococcal infection and symptom exacerbations, which his parents confirmed. On examination, we noted choreiform movements when we asked John to extend his hands in a supinated position.
Because this was John’s first documented presentation of PANDAS-like symptoms, an additional episode would provide more convincing support for classifying his OCD as the PANDAS subtype.
DIAGNOSTIC CRITERIA
National Institute of Mental Health (NIMH) diagnostic guidelines for PANDAS,7 initially proposed as working guidelines by Swedo and colleagues,8 are listed in Table 1.
Time between GABHS infection and symptom onset varies, but post-streptococcal diseases generally emerge after a few days to several weeks.9 Because this latent period makes retrospective assessment difficult,10 NIMH guidelines require a prospective link between GABHS infection and at least two OCD/tic symptom episodes.7,8,11 These additional criteria are necessary to avoid misdiagnosing PANDAS in cases when the GABHS infection/OCD connection is spurious.
Table 1
Guidelines for PANDAS diagnosis
| Presence of obsessive-compulsive disorder and/or tic disorder |
| Pediatric symptom onset (age 3 years to puberty) |
| Episodic course of symptom severity |
| Prospectively established association between group A beta-hemolytic streptococcal infection (GABHS)—as shown by positive throat culture and/or elevated anti-GABHS antibody titers and at least 2 separate OCD/tic symptom episodes |
| Association with neurologic abnormalities (motoric hyperactivity or adventitious movements, such as choreiform movements) |
| PANDAS: pediatric autoimmune neuropsychiatric disorders associated with streptococcus |
| Source: References 7, 8, and 11 |
PROSPECTIVE DIAGNOSIS
Neuropsychiatric symptoms. Early PANDAS symptoms are often similar to those of pediatric OCD and tic disorders (Table 2). Notable differences include:
- Sudden onset of obsessive-compulsive or tic behaviors shortly after GABHS infection, as opposed to OCD’s typical insidious course.
- Prepubertal onset (average age 7, as with Tourette’s syndrome7,8), compared with average age 10 of childhood OCD.12
Compulsions reported in PANDAS include germ-related behaviors such as hand washing, hoarding, and excessive toilet hygiene rituals. Most studies show consistent gender differences, with more washing behaviors by girls and more checking behaviors, aggression, and tics among boys.13
Recurrences. PANDAS has an episodic course, and approximately 50% of patients experience recurrences.13 Whether PANDAS remits completely, becomes dormant when neuropsychiatric symptoms are waning, or consistently progresses to a more chronic illness is unclear.
Because young children diagnosed with PANDAS often have repeated, frequent GABHS infections,8 give careful attention to:
- unexplained abdominal pain accompanied by fever
- history of scarlet fever
- brief episodes of tics, OCD, or compulsive urination that remitted
- illness accompanied by sudden onset of OCD or tic-like behaviors
- history of sore throats not severe enough to seek medical attention
- dramatic improvement in behavior/neuropsychiatric symptoms following standard antibiotictherapy for unrelated infection.
Differential diagnosis of OCD, tic disorders, and PANDAS
| Characteristic | OCD | Tourette’s/tic disorders | PANDAS |
|---|---|---|---|
| Typical age of onset | 10 years | 7 years | 7 years |
| Gender relatedness | Slightly higher prevalence in boys than girls before age 15; female-to-male ratio increases after puberty | 2:1 male-to-female ratio | 5:1 male-to-female ratio before age 8; thereafter, boys slightly outnumber girls |
| Course | Typically unremitting, though some episodic cases reported | Peak severity at age 10; 50% of cases remit by late teens | Episodic or sawtooth course; long-term prognosis unknown |
| Involvement of basal ganglia | Strong evidence | Strong evidence | Good evidence |
| GABHS trigger | Reported; cause uncertain | Reported in some cases; cause uncertain | Proposed association |
| Neurologic findings | Increased findings of NSS, including choreiform movements | Increased findings of NSS, including choreiform movements | Choreiform movements |
| GABHS: group A beta-hemolytic streptococcal infection | |||
| NSS: neurologic soft signs | |||
| OCD: obsessive-compulsive disorder | |||
| PANDAS: pediatric autoimmune neuropsychiatric disorders associated with streptococcus | |||
WEIGHING TREATMENT OPTIONS
Antibiotics. Antibiotic treatment of GABHS infection has been thoroughly studied among patients with rheumatic fever. American Heart Association guidelines for preventing rheumatic fever after GABHS infection recommend oral penicillin, 250 mg bid.14 Studies also indicate that using azithromycin, 500 mg once weekly, can protect against GABHS infection but may also increase resistance to macrolide antibiotics.15
Because antibiotic prophylaxis for GABHS infection is effective for rheumatic fever, some researchers have hypothesized that similar treatment would reduce neuropsychiatric symptoms in PANDAS patients.
In a double-blind, randomized, controlled trial, Snider et al16 found significant decreases in GABHS infection and neuropsychiatric symptoms in 23 PANDAS patients who took penicillin (250 mg bid) or azithromycin (250 mg bid on one day of the week) for 12 months.
An earlier study using penicillin for PANDAS prophylaxis was inconclusive. Its design limited more-definitive conclusions by allowing a high rate of antibiotic use during the placebo phase.17
An uncontrolled prospective study by Murphy et al13 documented rapid resolution of primary OCD, tic, and anxiety symptoms after appropriate antibiotic treatment in 12 children with PANDAS. Obsessive-compulsive symptoms remitted 5 to 21 days after patients received penicillin, amoxicillin/clavulanate potassium, or a cephalosporin. Symptoms resolved much more quickly than nonPANDAS obsessive-compulsive and tic disorders usually remit with cognitive-behavioral, habit reversal, and/or drug treatment.18 One-half of patients had at least one OCD recurrence, all documented as GABHS-positive with throat culture or rapid antigen-detection assay.
Recommendation. Obtain a GABHS culture if a child presents with sudden-onset OCD. If positive, treat with a standard course of antibiotics.19 Caution is strongly recommended when using antibiotics in children, as antibiotic-resistant organisms may develop. Collaborate with the child’s pediatrician to ensure that strep infections are treated consistently.
CASE CONTINUED: USING CBT FOR PANDAS
Giving John antibiotics when he had the sore throat might have been a rational choice to manage acute OCD symptoms. However, the scant literature on antibiotic prophylaxis for PANDAS subtype OCD led us to also consider cognitive-behavioral therapy (CBT).
CBT alone or with a selective serotonin reuptake inhibitor (SSRI) is first-line therapy for pediatric OCD.18,20 We hypothesized, therefore, that CBT might also be useful in PANDAS and provided John with five CBT sessions within 1 week, without giving an antibiotic or other medication. [See our study21 for therapy details.]
At baseline, John’s score on the Children’s Yale-Brown Obsessive-Compulsive Scale (CY-BOCS) was 34, indicating severe OCD symptoms, and his score on the Anxiety/Depression subscale of the Child Behavior Checklist (CBCL) was elevated (t = 66). After five CBT sessions, John’s CY-BOCS score decreased by 75% to 8 and his CBCL Anxiety/Depression score decreased into the average range (t = 50).21
Given PANDAS’ fluctuating course, his symptoms could have remitted spontaneously. His symptoms remained in remission 6 months later.
We believe John’s case is the only published description of using CBT alone to treat a patient with PANDAS. Since then, our team has successfully treated several other PANDAS patients using CBT. Based on our experience with trained clinicians, CBT provided an appropriate treatment option for this handful of cases. Controlled trials are needed to establish CBT’s efficacy for treating documented PANDAS.
SSRIs. As stated, CBT alone or with an SSRI is first-line therapy for pediatric OCD, and CBT alone or with an SSRI reduces pediatric OCD symptoms more effectively than antidepressants alone.18 Because no published reports of SSRI use in PANDAS exist, we recommend treating a child with PANDAS as you would any child presenting with OCD and tics:
- For milder cases with recent onset, begin with clinical monitoring for GABHS, without using SSRIs or antibiotics. Early CBT may prevent symptom worsening.
- For more severecases of longer duration, continue with CBT, then consider adding an SSRI.
Immunomodulatory therapies? Immunomodulatory therapies such as IV immunoglobulin (IVIG) and plasma exchange are not appropriate for refractory OCD or tic cases that have no clear GABHS association and a relapsing/remitting course. No studies support using immunomodulatory agents in disorders without an immune-mediated cause.
You might consider these therapies for severe, clearly established PANDAS only when less-invasive treatments (antibiotics, standard OCD therapies) have been ineffective and then only under research protocols and by physicians experienced in giving them.
Immunomodulatory therapies interrupt autoantibodies’ actions on the CNS and have shown moderate (40% to 50%) symptom reduction in some CNS diseases. In the NIMH trial, plasma exchange was better tolerated than IVIG and provided greater symptom relief.22 However, at least one study has shown plasma exchange to be ineffective for chronic OCD.23
USING ANTIBIOTICS FOR PANDAS
Snider and Swedo24 recommend guidelines for treating PANDAS, based on risks of using antibiotics in children, research and clinical experience, and American Academy of Child and Adolescent Psychiatry practice parameters (Box 2).
Elevated streptococcal titers are common in the community population25 and are not necessarily diagnostic of PANDAS. Thus, it is important to demonstrate a change in titer levels (such as a 4-fold dilution rise in antistreptococcal antibody titers 4 to 6 weeks after infection).
In patients with new-onset OCD/tics or recent symptom exacerbation, a positive throat culture provides support that symptoms were triggered by subclinical GABHS infection but does not rule out the possibility that the child is a GABHS carrier.
After streptococcal infections, titers may remain elevated for 6 months to 1 year. Murphy et al25 found persistent elevations in one or more strep titers in patients with dramatically fluctuating neuropsychiatric symptoms, compared with those whose course was inconsistent with PANDAS. It is unclear if these children had undetected, frequent GABHS infections or the elevated titers reflect a chronic immune activation to GABHS.
Assess for GABHS infectionin young children with abrupt-onset, obsessive-compulsive/tic-like behaviors (suspected PANDAS), using a 48-hour throat culture. If positive, promptly give a 10-day course of antibiotics effective for acute GABHS treatment (penicillins, cephalosporins, azithromycin).
Attempt to documenta preceding GABHS infection if neuropsychiatric symptoms began abruptly 4 to 6 weeks ago. Perform a 48-hour throat culture and a blood test for antistreptococcal antibody titers (ASO and anti-DNaseB). Do not give antibiotics unless GABHS culture is positive.
A rising titer 4 to 6 weeks later would suggest a recent infection. A single elevated titer does not adequately support a recent strep infection, as some individuals have elevated titers 6 months or longer after GABHS infection.
Consider prospective assessmentfor GABHS infections in children with episodic symptoms. Obtain throat cultures when neuropsychiatric symptoms return/exacerbate, as even untreated strep infections are usually self-limited.
Reserve antibiotic prophylaxisfor use under research protocols and based on solid evidence of PANDAS diagnosis.
Use immunomodulatory therapiesunder research protocols and only for children with acute, severe symptoms who fit the PANDAS designation.
Source: Reference 24.
- National Institute of Mental Health. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS).http://intramural.nimh.nih.gov/pdn/web.htm.
- Murphy TK, Herbstman, DM, Edge PJ. Infectious trigger in obsessive compulsive and tic disorders. In: Fatemi SH (ed). Infectious etiologies of neuropsychiatric disorders. New York: Taylor & Francis (in press).
- Amoxicillin/potassium clavulanate • Augmentin
- Azithromycin • Zithromax
Michael Larson reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Storch receives grant support from the National Institute of Health and Genentech Inc.
Dr. Murphy receives grant support from the National Institute of Mental Health, Bristol-Myers Squibb Co., and the Tourette Syndrome Association (TSA). She is a speaker for Pfizer, Inc.
1. Snider LA, Swedo SE. PANDAS: current status and directions for research. Mol Psychiatry 2004;9(10):900-7.
2. Hosier DM, Craenen JM, Teske DW, Wheller JJ. Resurgence of acute rheumatic fever. Am J Dis Child 1987;141:730-3.
3. Stollerman GH. Rheumatic fever. Lancet 1997;349:935-42.
4. Swedo S, Leonard HL, Schapiro MB, et al. Sydenham’s chorea: Physical and psychological symptoms of St.Vitus’ dance. Pediatrics 1993;91:706-13.
5. Asbahr FR, Negrao AB, Gentil V, et al. Obsessive-compulsive and related symptoms in children and adolescents with rheumatic fever with and without chorea: A prospective 6-month study. Am J Psychiatry 1998;155:1122-4.
6. Mercadante MT, Busatto GF, Lombroso PJ, et al. The psychiatric symptoms of rheumatic fever. Am J Psychiatry 2000;157:2036-8.
7. National Institute of Mental Health. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. Available at: http://intramural.nimh.nih.gov/pdn/web.htm. Accessed June 9, 2005.
8. Swedo SE, Leonard HL, Garvey M, et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. Am J Psychiatry 1998;155(2):264-71.
9. Kim SW, Grant JE, Kim SI, et al. A possible association of recurrent streptococcal infections and acute onset of obsessive-compulsive disorder. J Neuropsychiatry Clin Neurosci 2004;16(3):252-60.
10. Garvey MA, Giedd J, Swedo SE. PANDAS: the search for environmental triggers of pediatric neuropsychiatric disorders. Lessons from rheumatic fever. J Child Neurol 1998;13(9):413-23.
11. March JS. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS): implications for clinical practice. Arch Pediatr Adolesc Med 2004;158:927-9.
12. Zohar AH. The epidemiology of obsessive-compulsive disorder in children and adolescents. Child Adolesc Psychiatr Clin N Am 1999;8:445-59.
13. Murphy ML, Pichichero ME. Prospective identification and treatment of children with pediatric autoimmune neuropsychiatric disorder associated with group A streptococcal infection (PANDAS). Arch Pediatr Adolesc Med 2002;156(4):356-61.
14. Dajani A, Taubert K, Ferrieri P, et al. Treatment of acute streptococcal pharyngitis and prevention of rheumatic fever: a statement for health professionals. Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, the American Heart Association. Pediatrics 1995;96:758-764.
15. Gray GC, McPhate DC, Leinonen M, et al. Weekly oral azithromycin as prophylaxis for agents causing acute respiratory disease. Clin Infect Dis 1998;26:103-10.
16. Snider LA, Lougee L, Slattery M, et al. Antibiotic prophylaxis with azithromycin or penicillin for childhood-onset neuropsychiatric disorders. Biol Psychiatry 2005;57:788-92.
17. Garvey MA, Perlmutter SJ, Allen AJ, et al. A pilot study of penicillin prophylaxis for neuropsychiatric exacerbations triggered by streptococcal infections. Biol Psychiatry 1999;45(12):1564-71.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA 2004;292(16):1969-76.
19. Leonard HL, Swedo SE. Paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS). Int J Neuropsychopharmacol 2001;4(2):191-8.
20. Snider LA, Swedo SE. Childhood-onset obsessive-compulsive disorder and tic disorders: case report and literature review. J Child Adolesc Psychopharmacol 2003;13(suppl 1):S81-S88.
21. Storch EA, Gerdes AC, Adkins JW, et al. Behavioral treatment of a child with PANDAS. J Am Acad Child Adolesc Psychiatry 2004;43(5):510-11.
22. Nicolson R, Swedo SE, Lenane M, et al. An open trial of plasma exchange in childhood-onset obsessive-compulsive disorder without poststreptococcal exacerbations. J Am Acad Child Adolesc Psychiatry 2000;39(10):1313-5.
23. Perlmutter SJ, Leitman SF, Garvey MA, et al. Therapeutic plasma exchange and intravenous immunoglobulin for obsessive-compulsive disorder and tic disorders in childhood. Lancet 1999;354 (9185):1153-8.
24. Snider LA, Swedo SE. Post-streptococcal autoimmune disorders of the central nervous system. Curr Opin Neurol 2003;16:359-65.
25. Murphy TK, Sajid M, Soto O, et al. Detecting pediatric autoimmune neuropsychiatric disorders associated with streptococcus in children with obsessive-compulsive disorder and tics. Biol Psychiatry 2004;55(1):61-8.
John, age 6, presented for psychiatric evaluation with acute, incapacitating obsessive-compulsive symptoms. For 4 weeks he washed his hands compulsively and had pervasive obsessions about death by choking.
These symptoms had suddenly worsened over 2 days. At first, he washed his hands more than 35 times per day in rituals lasting several minutes each. Then, within 2 weeks, John’s handwashing spontaneously decreased, but his choking fears dramatically increased. He refused all solid foods and continuously sought reassurance from his parents that he would not choke or die.
Approximately 1 week before these symptoms began, John had a sore throat and tested positive via throat culture for group A beta-hemolytic streptococcal infection (GABHS).
Sore throat followed by sudden-onset obsessive-compulsive symptoms or tics in a child such as John suggests a pediatric autoimmune neuropsychiatric disorder associated with streptococcal infection (PANDAS). The association between GABHS infection and these symptoms remains uncertain, as the mechanism by which GABHS infection may cause obsessive-compulsive symptoms and other childhood-onset neuropsychiatric disorders is largely unknown.
Since PANDAS was recognized (Box 1),1-6 some data have emerged on the disorder’s symptoms, course, and prognosis. However:
- diagnostic criteria are not well-defined
- few controlled studies have examined treatment response
- using antibiotics and immunotherapies to treat or prevent PANDAS symptoms remains controversial because of unproven efficacy and potential adverse effects.
To help you diagnose and treat patients with suspected PANDAS, this article examines the limited evidence for the disorder, discusses diagnostic guidelines, and reviews preliminary indications for behavioral and medical treatments.
PANDAS stands for pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. It describes childhoodonset OCD or tic cases whose onset or worsening appears to be linked to group A beta-hemolytic streptococcal (GABHS) infection.
GABHS antibodies cross-react with the cellular components of the basal ganglia, particularly in the caudate nucleus.1 PANDAS was first recognized in 1987 during an unexpected resurgence of rheumatic fever in the United States.2 Sydenham chorea is thought to occur when GABHS antibodies undergo molecular mimicry and cross-react with epitopes on neurons in the basal ganglia and other brain areas, causing motor and behavioral disruptions.3 Rheumatic fever patients with Sydenham chorea show a high prevalence (up to 70%) of obsessive-compulsive disorder (OCD).4,5
Although individuals with Sydenham chorea appear to be at greatest risk for OCD after GABHS infection, rheumatic fever patients without chorea also appear to be at increased OCD risk.6
CASE REPORT CONTINUED: PANDAS CLUES
John’s sudden-onset compulsive behaviors and obsessive thoughts exemplify the rapid symptom onset often seen in children with PANDAS. His medical records showed a temporal relationship between his streptococcal infection and symptom exacerbations, which his parents confirmed. On examination, we noted choreiform movements when we asked John to extend his hands in a supinated position.
Because this was John’s first documented presentation of PANDAS-like symptoms, an additional episode would provide more convincing support for classifying his OCD as the PANDAS subtype.
DIAGNOSTIC CRITERIA
National Institute of Mental Health (NIMH) diagnostic guidelines for PANDAS,7 initially proposed as working guidelines by Swedo and colleagues,8 are listed in Table 1.
Time between GABHS infection and symptom onset varies, but post-streptococcal diseases generally emerge after a few days to several weeks.9 Because this latent period makes retrospective assessment difficult,10 NIMH guidelines require a prospective link between GABHS infection and at least two OCD/tic symptom episodes.7,8,11 These additional criteria are necessary to avoid misdiagnosing PANDAS in cases when the GABHS infection/OCD connection is spurious.
Table 1
Guidelines for PANDAS diagnosis
| Presence of obsessive-compulsive disorder and/or tic disorder |
| Pediatric symptom onset (age 3 years to puberty) |
| Episodic course of symptom severity |
| Prospectively established association between group A beta-hemolytic streptococcal infection (GABHS)—as shown by positive throat culture and/or elevated anti-GABHS antibody titers and at least 2 separate OCD/tic symptom episodes |
| Association with neurologic abnormalities (motoric hyperactivity or adventitious movements, such as choreiform movements) |
| PANDAS: pediatric autoimmune neuropsychiatric disorders associated with streptococcus |
| Source: References 7, 8, and 11 |
PROSPECTIVE DIAGNOSIS
Neuropsychiatric symptoms. Early PANDAS symptoms are often similar to those of pediatric OCD and tic disorders (Table 2). Notable differences include:
- Sudden onset of obsessive-compulsive or tic behaviors shortly after GABHS infection, as opposed to OCD’s typical insidious course.
- Prepubertal onset (average age 7, as with Tourette’s syndrome7,8), compared with average age 10 of childhood OCD.12
Compulsions reported in PANDAS include germ-related behaviors such as hand washing, hoarding, and excessive toilet hygiene rituals. Most studies show consistent gender differences, with more washing behaviors by girls and more checking behaviors, aggression, and tics among boys.13
Recurrences. PANDAS has an episodic course, and approximately 50% of patients experience recurrences.13 Whether PANDAS remits completely, becomes dormant when neuropsychiatric symptoms are waning, or consistently progresses to a more chronic illness is unclear.
Because young children diagnosed with PANDAS often have repeated, frequent GABHS infections,8 give careful attention to:
- unexplained abdominal pain accompanied by fever
- history of scarlet fever
- brief episodes of tics, OCD, or compulsive urination that remitted
- illness accompanied by sudden onset of OCD or tic-like behaviors
- history of sore throats not severe enough to seek medical attention
- dramatic improvement in behavior/neuropsychiatric symptoms following standard antibiotictherapy for unrelated infection.
Differential diagnosis of OCD, tic disorders, and PANDAS
| Characteristic | OCD | Tourette’s/tic disorders | PANDAS |
|---|---|---|---|
| Typical age of onset | 10 years | 7 years | 7 years |
| Gender relatedness | Slightly higher prevalence in boys than girls before age 15; female-to-male ratio increases after puberty | 2:1 male-to-female ratio | 5:1 male-to-female ratio before age 8; thereafter, boys slightly outnumber girls |
| Course | Typically unremitting, though some episodic cases reported | Peak severity at age 10; 50% of cases remit by late teens | Episodic or sawtooth course; long-term prognosis unknown |
| Involvement of basal ganglia | Strong evidence | Strong evidence | Good evidence |
| GABHS trigger | Reported; cause uncertain | Reported in some cases; cause uncertain | Proposed association |
| Neurologic findings | Increased findings of NSS, including choreiform movements | Increased findings of NSS, including choreiform movements | Choreiform movements |
| GABHS: group A beta-hemolytic streptococcal infection | |||
| NSS: neurologic soft signs | |||
| OCD: obsessive-compulsive disorder | |||
| PANDAS: pediatric autoimmune neuropsychiatric disorders associated with streptococcus | |||
WEIGHING TREATMENT OPTIONS
Antibiotics. Antibiotic treatment of GABHS infection has been thoroughly studied among patients with rheumatic fever. American Heart Association guidelines for preventing rheumatic fever after GABHS infection recommend oral penicillin, 250 mg bid.14 Studies also indicate that using azithromycin, 500 mg once weekly, can protect against GABHS infection but may also increase resistance to macrolide antibiotics.15
Because antibiotic prophylaxis for GABHS infection is effective for rheumatic fever, some researchers have hypothesized that similar treatment would reduce neuropsychiatric symptoms in PANDAS patients.
In a double-blind, randomized, controlled trial, Snider et al16 found significant decreases in GABHS infection and neuropsychiatric symptoms in 23 PANDAS patients who took penicillin (250 mg bid) or azithromycin (250 mg bid on one day of the week) for 12 months.
An earlier study using penicillin for PANDAS prophylaxis was inconclusive. Its design limited more-definitive conclusions by allowing a high rate of antibiotic use during the placebo phase.17
An uncontrolled prospective study by Murphy et al13 documented rapid resolution of primary OCD, tic, and anxiety symptoms after appropriate antibiotic treatment in 12 children with PANDAS. Obsessive-compulsive symptoms remitted 5 to 21 days after patients received penicillin, amoxicillin/clavulanate potassium, or a cephalosporin. Symptoms resolved much more quickly than nonPANDAS obsessive-compulsive and tic disorders usually remit with cognitive-behavioral, habit reversal, and/or drug treatment.18 One-half of patients had at least one OCD recurrence, all documented as GABHS-positive with throat culture or rapid antigen-detection assay.
Recommendation. Obtain a GABHS culture if a child presents with sudden-onset OCD. If positive, treat with a standard course of antibiotics.19 Caution is strongly recommended when using antibiotics in children, as antibiotic-resistant organisms may develop. Collaborate with the child’s pediatrician to ensure that strep infections are treated consistently.
CASE CONTINUED: USING CBT FOR PANDAS
Giving John antibiotics when he had the sore throat might have been a rational choice to manage acute OCD symptoms. However, the scant literature on antibiotic prophylaxis for PANDAS subtype OCD led us to also consider cognitive-behavioral therapy (CBT).
CBT alone or with a selective serotonin reuptake inhibitor (SSRI) is first-line therapy for pediatric OCD.18,20 We hypothesized, therefore, that CBT might also be useful in PANDAS and provided John with five CBT sessions within 1 week, without giving an antibiotic or other medication. [See our study21 for therapy details.]
At baseline, John’s score on the Children’s Yale-Brown Obsessive-Compulsive Scale (CY-BOCS) was 34, indicating severe OCD symptoms, and his score on the Anxiety/Depression subscale of the Child Behavior Checklist (CBCL) was elevated (t = 66). After five CBT sessions, John’s CY-BOCS score decreased by 75% to 8 and his CBCL Anxiety/Depression score decreased into the average range (t = 50).21
Given PANDAS’ fluctuating course, his symptoms could have remitted spontaneously. His symptoms remained in remission 6 months later.
We believe John’s case is the only published description of using CBT alone to treat a patient with PANDAS. Since then, our team has successfully treated several other PANDAS patients using CBT. Based on our experience with trained clinicians, CBT provided an appropriate treatment option for this handful of cases. Controlled trials are needed to establish CBT’s efficacy for treating documented PANDAS.
SSRIs. As stated, CBT alone or with an SSRI is first-line therapy for pediatric OCD, and CBT alone or with an SSRI reduces pediatric OCD symptoms more effectively than antidepressants alone.18 Because no published reports of SSRI use in PANDAS exist, we recommend treating a child with PANDAS as you would any child presenting with OCD and tics:
- For milder cases with recent onset, begin with clinical monitoring for GABHS, without using SSRIs or antibiotics. Early CBT may prevent symptom worsening.
- For more severecases of longer duration, continue with CBT, then consider adding an SSRI.
Immunomodulatory therapies? Immunomodulatory therapies such as IV immunoglobulin (IVIG) and plasma exchange are not appropriate for refractory OCD or tic cases that have no clear GABHS association and a relapsing/remitting course. No studies support using immunomodulatory agents in disorders without an immune-mediated cause.
You might consider these therapies for severe, clearly established PANDAS only when less-invasive treatments (antibiotics, standard OCD therapies) have been ineffective and then only under research protocols and by physicians experienced in giving them.
Immunomodulatory therapies interrupt autoantibodies’ actions on the CNS and have shown moderate (40% to 50%) symptom reduction in some CNS diseases. In the NIMH trial, plasma exchange was better tolerated than IVIG and provided greater symptom relief.22 However, at least one study has shown plasma exchange to be ineffective for chronic OCD.23
USING ANTIBIOTICS FOR PANDAS
Snider and Swedo24 recommend guidelines for treating PANDAS, based on risks of using antibiotics in children, research and clinical experience, and American Academy of Child and Adolescent Psychiatry practice parameters (Box 2).
Elevated streptococcal titers are common in the community population25 and are not necessarily diagnostic of PANDAS. Thus, it is important to demonstrate a change in titer levels (such as a 4-fold dilution rise in antistreptococcal antibody titers 4 to 6 weeks after infection).
In patients with new-onset OCD/tics or recent symptom exacerbation, a positive throat culture provides support that symptoms were triggered by subclinical GABHS infection but does not rule out the possibility that the child is a GABHS carrier.
After streptococcal infections, titers may remain elevated for 6 months to 1 year. Murphy et al25 found persistent elevations in one or more strep titers in patients with dramatically fluctuating neuropsychiatric symptoms, compared with those whose course was inconsistent with PANDAS. It is unclear if these children had undetected, frequent GABHS infections or the elevated titers reflect a chronic immune activation to GABHS.
Assess for GABHS infectionin young children with abrupt-onset, obsessive-compulsive/tic-like behaviors (suspected PANDAS), using a 48-hour throat culture. If positive, promptly give a 10-day course of antibiotics effective for acute GABHS treatment (penicillins, cephalosporins, azithromycin).
Attempt to documenta preceding GABHS infection if neuropsychiatric symptoms began abruptly 4 to 6 weeks ago. Perform a 48-hour throat culture and a blood test for antistreptococcal antibody titers (ASO and anti-DNaseB). Do not give antibiotics unless GABHS culture is positive.
A rising titer 4 to 6 weeks later would suggest a recent infection. A single elevated titer does not adequately support a recent strep infection, as some individuals have elevated titers 6 months or longer after GABHS infection.
Consider prospective assessmentfor GABHS infections in children with episodic symptoms. Obtain throat cultures when neuropsychiatric symptoms return/exacerbate, as even untreated strep infections are usually self-limited.
Reserve antibiotic prophylaxisfor use under research protocols and based on solid evidence of PANDAS diagnosis.
Use immunomodulatory therapiesunder research protocols and only for children with acute, severe symptoms who fit the PANDAS designation.
Source: Reference 24.
- National Institute of Mental Health. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS).http://intramural.nimh.nih.gov/pdn/web.htm.
- Murphy TK, Herbstman, DM, Edge PJ. Infectious trigger in obsessive compulsive and tic disorders. In: Fatemi SH (ed). Infectious etiologies of neuropsychiatric disorders. New York: Taylor & Francis (in press).
- Amoxicillin/potassium clavulanate • Augmentin
- Azithromycin • Zithromax
Michael Larson reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Storch receives grant support from the National Institute of Health and Genentech Inc.
Dr. Murphy receives grant support from the National Institute of Mental Health, Bristol-Myers Squibb Co., and the Tourette Syndrome Association (TSA). She is a speaker for Pfizer, Inc.
John, age 6, presented for psychiatric evaluation with acute, incapacitating obsessive-compulsive symptoms. For 4 weeks he washed his hands compulsively and had pervasive obsessions about death by choking.
These symptoms had suddenly worsened over 2 days. At first, he washed his hands more than 35 times per day in rituals lasting several minutes each. Then, within 2 weeks, John’s handwashing spontaneously decreased, but his choking fears dramatically increased. He refused all solid foods and continuously sought reassurance from his parents that he would not choke or die.
Approximately 1 week before these symptoms began, John had a sore throat and tested positive via throat culture for group A beta-hemolytic streptococcal infection (GABHS).
Sore throat followed by sudden-onset obsessive-compulsive symptoms or tics in a child such as John suggests a pediatric autoimmune neuropsychiatric disorder associated with streptococcal infection (PANDAS). The association between GABHS infection and these symptoms remains uncertain, as the mechanism by which GABHS infection may cause obsessive-compulsive symptoms and other childhood-onset neuropsychiatric disorders is largely unknown.
Since PANDAS was recognized (Box 1),1-6 some data have emerged on the disorder’s symptoms, course, and prognosis. However:
- diagnostic criteria are not well-defined
- few controlled studies have examined treatment response
- using antibiotics and immunotherapies to treat or prevent PANDAS symptoms remains controversial because of unproven efficacy and potential adverse effects.
To help you diagnose and treat patients with suspected PANDAS, this article examines the limited evidence for the disorder, discusses diagnostic guidelines, and reviews preliminary indications for behavioral and medical treatments.
PANDAS stands for pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. It describes childhoodonset OCD or tic cases whose onset or worsening appears to be linked to group A beta-hemolytic streptococcal (GABHS) infection.
GABHS antibodies cross-react with the cellular components of the basal ganglia, particularly in the caudate nucleus.1 PANDAS was first recognized in 1987 during an unexpected resurgence of rheumatic fever in the United States.2 Sydenham chorea is thought to occur when GABHS antibodies undergo molecular mimicry and cross-react with epitopes on neurons in the basal ganglia and other brain areas, causing motor and behavioral disruptions.3 Rheumatic fever patients with Sydenham chorea show a high prevalence (up to 70%) of obsessive-compulsive disorder (OCD).4,5
Although individuals with Sydenham chorea appear to be at greatest risk for OCD after GABHS infection, rheumatic fever patients without chorea also appear to be at increased OCD risk.6
CASE REPORT CONTINUED: PANDAS CLUES
John’s sudden-onset compulsive behaviors and obsessive thoughts exemplify the rapid symptom onset often seen in children with PANDAS. His medical records showed a temporal relationship between his streptococcal infection and symptom exacerbations, which his parents confirmed. On examination, we noted choreiform movements when we asked John to extend his hands in a supinated position.
Because this was John’s first documented presentation of PANDAS-like symptoms, an additional episode would provide more convincing support for classifying his OCD as the PANDAS subtype.
DIAGNOSTIC CRITERIA
National Institute of Mental Health (NIMH) diagnostic guidelines for PANDAS,7 initially proposed as working guidelines by Swedo and colleagues,8 are listed in Table 1.
Time between GABHS infection and symptom onset varies, but post-streptococcal diseases generally emerge after a few days to several weeks.9 Because this latent period makes retrospective assessment difficult,10 NIMH guidelines require a prospective link between GABHS infection and at least two OCD/tic symptom episodes.7,8,11 These additional criteria are necessary to avoid misdiagnosing PANDAS in cases when the GABHS infection/OCD connection is spurious.
Table 1
Guidelines for PANDAS diagnosis
| Presence of obsessive-compulsive disorder and/or tic disorder |
| Pediatric symptom onset (age 3 years to puberty) |
| Episodic course of symptom severity |
| Prospectively established association between group A beta-hemolytic streptococcal infection (GABHS)—as shown by positive throat culture and/or elevated anti-GABHS antibody titers and at least 2 separate OCD/tic symptom episodes |
| Association with neurologic abnormalities (motoric hyperactivity or adventitious movements, such as choreiform movements) |
| PANDAS: pediatric autoimmune neuropsychiatric disorders associated with streptococcus |
| Source: References 7, 8, and 11 |
PROSPECTIVE DIAGNOSIS
Neuropsychiatric symptoms. Early PANDAS symptoms are often similar to those of pediatric OCD and tic disorders (Table 2). Notable differences include:
- Sudden onset of obsessive-compulsive or tic behaviors shortly after GABHS infection, as opposed to OCD’s typical insidious course.
- Prepubertal onset (average age 7, as with Tourette’s syndrome7,8), compared with average age 10 of childhood OCD.12
Compulsions reported in PANDAS include germ-related behaviors such as hand washing, hoarding, and excessive toilet hygiene rituals. Most studies show consistent gender differences, with more washing behaviors by girls and more checking behaviors, aggression, and tics among boys.13
Recurrences. PANDAS has an episodic course, and approximately 50% of patients experience recurrences.13 Whether PANDAS remits completely, becomes dormant when neuropsychiatric symptoms are waning, or consistently progresses to a more chronic illness is unclear.
Because young children diagnosed with PANDAS often have repeated, frequent GABHS infections,8 give careful attention to:
- unexplained abdominal pain accompanied by fever
- history of scarlet fever
- brief episodes of tics, OCD, or compulsive urination that remitted
- illness accompanied by sudden onset of OCD or tic-like behaviors
- history of sore throats not severe enough to seek medical attention
- dramatic improvement in behavior/neuropsychiatric symptoms following standard antibiotictherapy for unrelated infection.
Differential diagnosis of OCD, tic disorders, and PANDAS
| Characteristic | OCD | Tourette’s/tic disorders | PANDAS |
|---|---|---|---|
| Typical age of onset | 10 years | 7 years | 7 years |
| Gender relatedness | Slightly higher prevalence in boys than girls before age 15; female-to-male ratio increases after puberty | 2:1 male-to-female ratio | 5:1 male-to-female ratio before age 8; thereafter, boys slightly outnumber girls |
| Course | Typically unremitting, though some episodic cases reported | Peak severity at age 10; 50% of cases remit by late teens | Episodic or sawtooth course; long-term prognosis unknown |
| Involvement of basal ganglia | Strong evidence | Strong evidence | Good evidence |
| GABHS trigger | Reported; cause uncertain | Reported in some cases; cause uncertain | Proposed association |
| Neurologic findings | Increased findings of NSS, including choreiform movements | Increased findings of NSS, including choreiform movements | Choreiform movements |
| GABHS: group A beta-hemolytic streptococcal infection | |||
| NSS: neurologic soft signs | |||
| OCD: obsessive-compulsive disorder | |||
| PANDAS: pediatric autoimmune neuropsychiatric disorders associated with streptococcus | |||
WEIGHING TREATMENT OPTIONS
Antibiotics. Antibiotic treatment of GABHS infection has been thoroughly studied among patients with rheumatic fever. American Heart Association guidelines for preventing rheumatic fever after GABHS infection recommend oral penicillin, 250 mg bid.14 Studies also indicate that using azithromycin, 500 mg once weekly, can protect against GABHS infection but may also increase resistance to macrolide antibiotics.15
Because antibiotic prophylaxis for GABHS infection is effective for rheumatic fever, some researchers have hypothesized that similar treatment would reduce neuropsychiatric symptoms in PANDAS patients.
In a double-blind, randomized, controlled trial, Snider et al16 found significant decreases in GABHS infection and neuropsychiatric symptoms in 23 PANDAS patients who took penicillin (250 mg bid) or azithromycin (250 mg bid on one day of the week) for 12 months.
An earlier study using penicillin for PANDAS prophylaxis was inconclusive. Its design limited more-definitive conclusions by allowing a high rate of antibiotic use during the placebo phase.17
An uncontrolled prospective study by Murphy et al13 documented rapid resolution of primary OCD, tic, and anxiety symptoms after appropriate antibiotic treatment in 12 children with PANDAS. Obsessive-compulsive symptoms remitted 5 to 21 days after patients received penicillin, amoxicillin/clavulanate potassium, or a cephalosporin. Symptoms resolved much more quickly than nonPANDAS obsessive-compulsive and tic disorders usually remit with cognitive-behavioral, habit reversal, and/or drug treatment.18 One-half of patients had at least one OCD recurrence, all documented as GABHS-positive with throat culture or rapid antigen-detection assay.
Recommendation. Obtain a GABHS culture if a child presents with sudden-onset OCD. If positive, treat with a standard course of antibiotics.19 Caution is strongly recommended when using antibiotics in children, as antibiotic-resistant organisms may develop. Collaborate with the child’s pediatrician to ensure that strep infections are treated consistently.
CASE CONTINUED: USING CBT FOR PANDAS
Giving John antibiotics when he had the sore throat might have been a rational choice to manage acute OCD symptoms. However, the scant literature on antibiotic prophylaxis for PANDAS subtype OCD led us to also consider cognitive-behavioral therapy (CBT).
CBT alone or with a selective serotonin reuptake inhibitor (SSRI) is first-line therapy for pediatric OCD.18,20 We hypothesized, therefore, that CBT might also be useful in PANDAS and provided John with five CBT sessions within 1 week, without giving an antibiotic or other medication. [See our study21 for therapy details.]
At baseline, John’s score on the Children’s Yale-Brown Obsessive-Compulsive Scale (CY-BOCS) was 34, indicating severe OCD symptoms, and his score on the Anxiety/Depression subscale of the Child Behavior Checklist (CBCL) was elevated (t = 66). After five CBT sessions, John’s CY-BOCS score decreased by 75% to 8 and his CBCL Anxiety/Depression score decreased into the average range (t = 50).21
Given PANDAS’ fluctuating course, his symptoms could have remitted spontaneously. His symptoms remained in remission 6 months later.
We believe John’s case is the only published description of using CBT alone to treat a patient with PANDAS. Since then, our team has successfully treated several other PANDAS patients using CBT. Based on our experience with trained clinicians, CBT provided an appropriate treatment option for this handful of cases. Controlled trials are needed to establish CBT’s efficacy for treating documented PANDAS.
SSRIs. As stated, CBT alone or with an SSRI is first-line therapy for pediatric OCD, and CBT alone or with an SSRI reduces pediatric OCD symptoms more effectively than antidepressants alone.18 Because no published reports of SSRI use in PANDAS exist, we recommend treating a child with PANDAS as you would any child presenting with OCD and tics:
- For milder cases with recent onset, begin with clinical monitoring for GABHS, without using SSRIs or antibiotics. Early CBT may prevent symptom worsening.
- For more severecases of longer duration, continue with CBT, then consider adding an SSRI.
Immunomodulatory therapies? Immunomodulatory therapies such as IV immunoglobulin (IVIG) and plasma exchange are not appropriate for refractory OCD or tic cases that have no clear GABHS association and a relapsing/remitting course. No studies support using immunomodulatory agents in disorders without an immune-mediated cause.
You might consider these therapies for severe, clearly established PANDAS only when less-invasive treatments (antibiotics, standard OCD therapies) have been ineffective and then only under research protocols and by physicians experienced in giving them.
Immunomodulatory therapies interrupt autoantibodies’ actions on the CNS and have shown moderate (40% to 50%) symptom reduction in some CNS diseases. In the NIMH trial, plasma exchange was better tolerated than IVIG and provided greater symptom relief.22 However, at least one study has shown plasma exchange to be ineffective for chronic OCD.23
USING ANTIBIOTICS FOR PANDAS
Snider and Swedo24 recommend guidelines for treating PANDAS, based on risks of using antibiotics in children, research and clinical experience, and American Academy of Child and Adolescent Psychiatry practice parameters (Box 2).
Elevated streptococcal titers are common in the community population25 and are not necessarily diagnostic of PANDAS. Thus, it is important to demonstrate a change in titer levels (such as a 4-fold dilution rise in antistreptococcal antibody titers 4 to 6 weeks after infection).
In patients with new-onset OCD/tics or recent symptom exacerbation, a positive throat culture provides support that symptoms were triggered by subclinical GABHS infection but does not rule out the possibility that the child is a GABHS carrier.
After streptococcal infections, titers may remain elevated for 6 months to 1 year. Murphy et al25 found persistent elevations in one or more strep titers in patients with dramatically fluctuating neuropsychiatric symptoms, compared with those whose course was inconsistent with PANDAS. It is unclear if these children had undetected, frequent GABHS infections or the elevated titers reflect a chronic immune activation to GABHS.
Assess for GABHS infectionin young children with abrupt-onset, obsessive-compulsive/tic-like behaviors (suspected PANDAS), using a 48-hour throat culture. If positive, promptly give a 10-day course of antibiotics effective for acute GABHS treatment (penicillins, cephalosporins, azithromycin).
Attempt to documenta preceding GABHS infection if neuropsychiatric symptoms began abruptly 4 to 6 weeks ago. Perform a 48-hour throat culture and a blood test for antistreptococcal antibody titers (ASO and anti-DNaseB). Do not give antibiotics unless GABHS culture is positive.
A rising titer 4 to 6 weeks later would suggest a recent infection. A single elevated titer does not adequately support a recent strep infection, as some individuals have elevated titers 6 months or longer after GABHS infection.
Consider prospective assessmentfor GABHS infections in children with episodic symptoms. Obtain throat cultures when neuropsychiatric symptoms return/exacerbate, as even untreated strep infections are usually self-limited.
Reserve antibiotic prophylaxisfor use under research protocols and based on solid evidence of PANDAS diagnosis.
Use immunomodulatory therapiesunder research protocols and only for children with acute, severe symptoms who fit the PANDAS designation.
Source: Reference 24.
- National Institute of Mental Health. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS).http://intramural.nimh.nih.gov/pdn/web.htm.
- Murphy TK, Herbstman, DM, Edge PJ. Infectious trigger in obsessive compulsive and tic disorders. In: Fatemi SH (ed). Infectious etiologies of neuropsychiatric disorders. New York: Taylor & Francis (in press).
- Amoxicillin/potassium clavulanate • Augmentin
- Azithromycin • Zithromax
Michael Larson reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Storch receives grant support from the National Institute of Health and Genentech Inc.
Dr. Murphy receives grant support from the National Institute of Mental Health, Bristol-Myers Squibb Co., and the Tourette Syndrome Association (TSA). She is a speaker for Pfizer, Inc.
1. Snider LA, Swedo SE. PANDAS: current status and directions for research. Mol Psychiatry 2004;9(10):900-7.
2. Hosier DM, Craenen JM, Teske DW, Wheller JJ. Resurgence of acute rheumatic fever. Am J Dis Child 1987;141:730-3.
3. Stollerman GH. Rheumatic fever. Lancet 1997;349:935-42.
4. Swedo S, Leonard HL, Schapiro MB, et al. Sydenham’s chorea: Physical and psychological symptoms of St.Vitus’ dance. Pediatrics 1993;91:706-13.
5. Asbahr FR, Negrao AB, Gentil V, et al. Obsessive-compulsive and related symptoms in children and adolescents with rheumatic fever with and without chorea: A prospective 6-month study. Am J Psychiatry 1998;155:1122-4.
6. Mercadante MT, Busatto GF, Lombroso PJ, et al. The psychiatric symptoms of rheumatic fever. Am J Psychiatry 2000;157:2036-8.
7. National Institute of Mental Health. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. Available at: http://intramural.nimh.nih.gov/pdn/web.htm. Accessed June 9, 2005.
8. Swedo SE, Leonard HL, Garvey M, et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. Am J Psychiatry 1998;155(2):264-71.
9. Kim SW, Grant JE, Kim SI, et al. A possible association of recurrent streptococcal infections and acute onset of obsessive-compulsive disorder. J Neuropsychiatry Clin Neurosci 2004;16(3):252-60.
10. Garvey MA, Giedd J, Swedo SE. PANDAS: the search for environmental triggers of pediatric neuropsychiatric disorders. Lessons from rheumatic fever. J Child Neurol 1998;13(9):413-23.
11. March JS. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS): implications for clinical practice. Arch Pediatr Adolesc Med 2004;158:927-9.
12. Zohar AH. The epidemiology of obsessive-compulsive disorder in children and adolescents. Child Adolesc Psychiatr Clin N Am 1999;8:445-59.
13. Murphy ML, Pichichero ME. Prospective identification and treatment of children with pediatric autoimmune neuropsychiatric disorder associated with group A streptococcal infection (PANDAS). Arch Pediatr Adolesc Med 2002;156(4):356-61.
14. Dajani A, Taubert K, Ferrieri P, et al. Treatment of acute streptococcal pharyngitis and prevention of rheumatic fever: a statement for health professionals. Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, the American Heart Association. Pediatrics 1995;96:758-764.
15. Gray GC, McPhate DC, Leinonen M, et al. Weekly oral azithromycin as prophylaxis for agents causing acute respiratory disease. Clin Infect Dis 1998;26:103-10.
16. Snider LA, Lougee L, Slattery M, et al. Antibiotic prophylaxis with azithromycin or penicillin for childhood-onset neuropsychiatric disorders. Biol Psychiatry 2005;57:788-92.
17. Garvey MA, Perlmutter SJ, Allen AJ, et al. A pilot study of penicillin prophylaxis for neuropsychiatric exacerbations triggered by streptococcal infections. Biol Psychiatry 1999;45(12):1564-71.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA 2004;292(16):1969-76.
19. Leonard HL, Swedo SE. Paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS). Int J Neuropsychopharmacol 2001;4(2):191-8.
20. Snider LA, Swedo SE. Childhood-onset obsessive-compulsive disorder and tic disorders: case report and literature review. J Child Adolesc Psychopharmacol 2003;13(suppl 1):S81-S88.
21. Storch EA, Gerdes AC, Adkins JW, et al. Behavioral treatment of a child with PANDAS. J Am Acad Child Adolesc Psychiatry 2004;43(5):510-11.
22. Nicolson R, Swedo SE, Lenane M, et al. An open trial of plasma exchange in childhood-onset obsessive-compulsive disorder without poststreptococcal exacerbations. J Am Acad Child Adolesc Psychiatry 2000;39(10):1313-5.
23. Perlmutter SJ, Leitman SF, Garvey MA, et al. Therapeutic plasma exchange and intravenous immunoglobulin for obsessive-compulsive disorder and tic disorders in childhood. Lancet 1999;354 (9185):1153-8.
24. Snider LA, Swedo SE. Post-streptococcal autoimmune disorders of the central nervous system. Curr Opin Neurol 2003;16:359-65.
25. Murphy TK, Sajid M, Soto O, et al. Detecting pediatric autoimmune neuropsychiatric disorders associated with streptococcus in children with obsessive-compulsive disorder and tics. Biol Psychiatry 2004;55(1):61-8.
1. Snider LA, Swedo SE. PANDAS: current status and directions for research. Mol Psychiatry 2004;9(10):900-7.
2. Hosier DM, Craenen JM, Teske DW, Wheller JJ. Resurgence of acute rheumatic fever. Am J Dis Child 1987;141:730-3.
3. Stollerman GH. Rheumatic fever. Lancet 1997;349:935-42.
4. Swedo S, Leonard HL, Schapiro MB, et al. Sydenham’s chorea: Physical and psychological symptoms of St.Vitus’ dance. Pediatrics 1993;91:706-13.
5. Asbahr FR, Negrao AB, Gentil V, et al. Obsessive-compulsive and related symptoms in children and adolescents with rheumatic fever with and without chorea: A prospective 6-month study. Am J Psychiatry 1998;155:1122-4.
6. Mercadante MT, Busatto GF, Lombroso PJ, et al. The psychiatric symptoms of rheumatic fever. Am J Psychiatry 2000;157:2036-8.
7. National Institute of Mental Health. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. Available at: http://intramural.nimh.nih.gov/pdn/web.htm. Accessed June 9, 2005.
8. Swedo SE, Leonard HL, Garvey M, et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. Am J Psychiatry 1998;155(2):264-71.
9. Kim SW, Grant JE, Kim SI, et al. A possible association of recurrent streptococcal infections and acute onset of obsessive-compulsive disorder. J Neuropsychiatry Clin Neurosci 2004;16(3):252-60.
10. Garvey MA, Giedd J, Swedo SE. PANDAS: the search for environmental triggers of pediatric neuropsychiatric disorders. Lessons from rheumatic fever. J Child Neurol 1998;13(9):413-23.
11. March JS. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS): implications for clinical practice. Arch Pediatr Adolesc Med 2004;158:927-9.
12. Zohar AH. The epidemiology of obsessive-compulsive disorder in children and adolescents. Child Adolesc Psychiatr Clin N Am 1999;8:445-59.
13. Murphy ML, Pichichero ME. Prospective identification and treatment of children with pediatric autoimmune neuropsychiatric disorder associated with group A streptococcal infection (PANDAS). Arch Pediatr Adolesc Med 2002;156(4):356-61.
14. Dajani A, Taubert K, Ferrieri P, et al. Treatment of acute streptococcal pharyngitis and prevention of rheumatic fever: a statement for health professionals. Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, the American Heart Association. Pediatrics 1995;96:758-764.
15. Gray GC, McPhate DC, Leinonen M, et al. Weekly oral azithromycin as prophylaxis for agents causing acute respiratory disease. Clin Infect Dis 1998;26:103-10.
16. Snider LA, Lougee L, Slattery M, et al. Antibiotic prophylaxis with azithromycin or penicillin for childhood-onset neuropsychiatric disorders. Biol Psychiatry 2005;57:788-92.
17. Garvey MA, Perlmutter SJ, Allen AJ, et al. A pilot study of penicillin prophylaxis for neuropsychiatric exacerbations triggered by streptococcal infections. Biol Psychiatry 1999;45(12):1564-71.
18. Pediatric OCD Treatment Study (POTS) Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA 2004;292(16):1969-76.
19. Leonard HL, Swedo SE. Paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS). Int J Neuropsychopharmacol 2001;4(2):191-8.
20. Snider LA, Swedo SE. Childhood-onset obsessive-compulsive disorder and tic disorders: case report and literature review. J Child Adolesc Psychopharmacol 2003;13(suppl 1):S81-S88.
21. Storch EA, Gerdes AC, Adkins JW, et al. Behavioral treatment of a child with PANDAS. J Am Acad Child Adolesc Psychiatry 2004;43(5):510-11.
22. Nicolson R, Swedo SE, Lenane M, et al. An open trial of plasma exchange in childhood-onset obsessive-compulsive disorder without poststreptococcal exacerbations. J Am Acad Child Adolesc Psychiatry 2000;39(10):1313-5.
23. Perlmutter SJ, Leitman SF, Garvey MA, et al. Therapeutic plasma exchange and intravenous immunoglobulin for obsessive-compulsive disorder and tic disorders in childhood. Lancet 1999;354 (9185):1153-8.
24. Snider LA, Swedo SE. Post-streptococcal autoimmune disorders of the central nervous system. Curr Opin Neurol 2003;16:359-65.
25. Murphy TK, Sajid M, Soto O, et al. Detecting pediatric autoimmune neuropsychiatric disorders associated with streptococcus in children with obsessive-compulsive disorder and tics. Biol Psychiatry 2004;55(1):61-8.
Treatment-resistant psychosis: Are 2 antipsychotics more effective than 1?
Off-label prescribing of two or more antipsychotics for treatment-resistant psychosis carries inherent risks for both schizophrenia patients and their psychiatrists. You can reduce these risks by demonstrating that your patient will benefit more from combining antipsychotics than from monotherapy alternatives.
Until more empiric data become available, clinicians carry the burden of documenting individual patient response to justify combining antipsychotics. This article can help you identify and counsel possible candidates for this therapy, assess the risks and benefits, and defend your treatment choicesif necessary.
What Does ‘Combining’ Mean?
For this discussion, “combining antipsychotics” does not mean simultaneously using two or more antipsychotics to treat psychosis. Rather, it refers to using two or more antipsychotics for long-term control of psychotic symptoms. It does not apply to short-term tactics, such as overlapping antipsychotics when switching between them or combining a parenteral and an oral antipsychotic when treating an acute episode.
How long must a combination be used to qualify as long-term treatment? No standard exists, but a period >6 weeks exceeds normal short-term treatment.
How Safe Are Combinations?
Patient risk. Combination antipsychotics are used to treat 10% to 20% of schizophrenia patients in this country and >90% of those in some Asian countries, such as Japan.1 Statistics on the prevalence of combining antipsychotics seldom:
- distinguish between short- and long-term use
- identify when practitioners use combinations to treat co-existing symptoms such as insomnia, rather than for psychosis.
Clinicians, however, must define safety in terms of risks and benefits. Given that every medication has risks—some (such as allergic reactions) unique to the individual patient—adding another drug to a treatment regimen will always add risk. When considering more than one antipsychotic, ask yourself:
- Do benefits outweigh risks?
- Is a combination the least risky way to achieve these benefits?
How much does a practitioner’s perception of medicolegal risk influence treatment selection? No doubt, comfort level varies greatly. I have frequently met prescribers who use antipsychotic combinations instead of clozapine for fear of being sued should agranulocytosis occur.
Most trials have used criteriafrom the seminal clozapine study to define “treatment resistant” schizophrenia:
- History of poor response to one or more antipsychotics.
- Demonstrated nonresponse to adequate dosage and duration of a nonclozapine antipsychotic before starting clozapine.2
This work defines treatment resistance as persistent positive symptoms despite treatment with ≥2 different antipsychotics other than clozapine.
Newer antipsychotics are different. The original clozapine studies found little likelihood that patients who had not responded to haloperidol would respond to chlorpromazine. Newer-generation antipsychotics are pharmacologically more heterogeneous than the first-generation agents, however, and failure to respond to one might not mean that none of the others will help.
When to start clozapine? Solid evidence is limited, but a consensus panel has recommended starting clozapine after two unsuccessful trials of newer-generation antipsychotics, with the option to try a third antipsychotic (first- or newer-generation) if clinically warranted.3
Symptoms vs function. In clinical practice, defining treatment resistance as persistent positive symptoms may exclude patients impaired by severe negative symptoms and/or disorganized thought processes. The treatment goal is to restore function, not just to reduce psychosis. As treatments evolve, we can expect more emphasis on a recovery model that addresses total patient well-being and combines psychosocial and pharmacologic interventions.
A Practical View
Medical risks. Treating patients with antipsychotic combinations may be associated with medical risks (Table 1). Patients are less likely to adhere to complex medication regimens than to simple ones. Thus, adding a second antipsychotic may decrease adherence to the first antipsychotic and to other medications, such as for hypertension, diabetes, etc.
Increased side-effect risk has been reported with antipsychotic combinations.4 Side effects include those expected from one or both drugs in the combination, especially extrapyramidal effects when conventional antipsychotics are included.4,5 Because the total antipsychotic dosage can be considerably higher than usual in patients receiving combinations,5 some of the increased side effect burden is probably related to high dosing, rather than to the combinations.
Pharmacodynamic or pharmacokinetic interactions or the loss of an agent’s advantages are infrequently reported, perhaps because prescribers are not looking for these effects or do not consider them worth reporting. Of particular concern is tardive dyskinesia (TD), which may develop when conventional antipsychotics are added to agents with very low propensity to cause TD—such as clozapine.
Table 1
Potential risks and benefits of combining antipsychotics
| Medical risks |
| Decreased adherence to multiple medications |
| Increased and/or unexpected side effects |
| Increased potential for undesirable pharmacokinetic or pharmacodynamic interactions |
| Difficulties in making rational dose adjustments |
| Loss of advantages of one of the medications |
| Nonmedical risks |
| Lack of evidence to defend practice in medicolegal cases |
| Increased costs if combining second-generation antipsychotics |
| Increased quality assurance scrutiny and paperwork |
| Medical benefits |
| Reduced symptoms |
| Reduced metabolic side effects (such as partially substituting another atypical antipsychotic for a high clozapine dosage) |
litigation. As noted, some clinicians prefer combining antipsychotics instead of using clozapine because they fear legal action should a patient develop agranulocytosis. In the author’s view, this fear is not well-grounded. Successful lawsuits typically find evidence that the clinician committed errors of omission or commission. In the case of blood monitoring for clozapine-induced neutropenia, the parameters are clear and enforced. You must follow community standards of practice, which provide a strong legal defense.
Administrative scrutiny. Quality assurance programs are increasingly identifying and monitoring antipsychotic combinations—an obvious target by being frequent, lacking a strong evidence base, increasing costs, and raising liability concerns. Typically, such programs discourage antipsychotic combinations and impose administrative hurdles to starting or continuing them.
Gathering data and documenting it in the patient’s medical record—to be discussed later—is key to demonstrating that a combination’s superior efficacy for the individual patient justifies its use.
Medical benefits. Guidelines and algorithms for drug treatment of schizophrenia either omit combination antipsychotics or suggest this strategy when all else has failed.6,7 Lack of evidence for a practice is not the same as evidence against it, however. A combination may be better for some patients than any available antipsychotic monotherapy.
Antipsychotic combinations have been examined in more literature reviews than randomized controlled trials—all of which have addressed augmenting clozapine with another antipsychotic (Table 2).8-12 Augmentation with psychotropics other than a second antipsychotic has most often been tested for negative and cognitive symptoms,4 but some evidence has shown adjunctive anticonvulsants and cognition-enhancing agents to improve positive symptoms.6
Reducing side effects is not directly related to treatment-resistant psychosis, but some articles describe managing clozapine’s metabolic side effects by partially substituting another atypical antipsychotic.11,13
Because clozapine is the treatment of choice for treatment-resistant psychosis, consider tactics that maintain its benefits while reducing its metabolic liabilities. Reducing the clozapine dosage (if feasible) is the first-line approach, but partially substituting another antipsychotic might help if psychotic symptoms return.
Table 2
Results of studies using combination antipsychotic therapy
| Combination | Type of trial | First author | Outcome measures | Results |
|---|---|---|---|---|
| Clozapine/sulpiride* | RCT | Shiloh8 | Positive, negative, and depressive symptoms | Positive |
| Clozapine/risperidone | RCT | Josiassen9 | Positive and negative symptoms | Positive |
| Clozapine/risperidone | RCT | Yagcioglu10 | Positive and negative symptoms | Negative |
| Clozapine/quetiapine | LCS | Reinstein11 | Body weight, serum glucose | Positive |
| Clozapine/amisulpride* | LCS | Agelink12 | Positive and negative symptoms | Positive |
| RCT: randomized controlled trial; LCS: large case series | ||||
| *Sulpiride and amisulpride are not available in the United States. | ||||
Clinical Management Hints
Document, document, document. Crucial to “non-standard” treatments such as combining antipsychotics is using and documenting good medical practices. These include:
- accurately assessing patients
- carefully weighing illness risks vs treatment risks
- talking regularly to patients about their treatment and therapeutic options (Table 3).
Use objective measures. Brief scales to measure psychotic symptoms are being increasingly used in public mental health settings.6 For example, four psychosis items from the Brief Psychiatric Rating Scale (hallucinations, unusual thought content, paranoia, and disorganized thought) can quickly assess and capture much of the variance of the full scale in patients with schizophrenia.14
When properly used, these objective and reliable scales can document clinical change and track illness course across time and providers. Other objective outcome measures can contribute to quality of care and its documentation. For more information, visit the Substance Abuse and Mental Health Services Administration Web site (see Related Resources).
Although it seems obvious that persistent illness is not a good thing, the risks of persistent psychosis vary from patient to patient. Some function relatively well despite ongoing psychotic thoughts, whereas others are terribly impaired, demoralized, and/or suicidal. The rationale for nonstandard treatment such as an antipsychotic combination is to reduce patient suffering and risk and/or to increase function.
Discuss options with patients. Finally, schizophrenia patients (and involved significant others) need to understand and participate in treatment. Ongoing discussion of treatment options is an important part of this process. For example, initially reluctant patients often come to accept clozapine treatment after discussing its risks and benefits with clinicians and other patients who are taking clozapine. Documenting these discussions is as beneficial to the patient and clinician as is documenting symptoms and treatment effects.
Table 3
3 tips for managing risk and medications
| Use objective, quantifiable symptom measures at regular intervals |
| Document patient’s risk of persistent psychosis, based on history |
| Discuss risks and benefits of other treatment options with the patient and/or family at regular intervals |
- How to use medication in a systematic and effective way, as part of the overall treatment for severe mental illness. In: Evidence-based practices: Shaping mental health services toward recovery. Medication management approaches in psychiatry. Substance Abuse and Mental Health Services Administration. U.S. Department of Health and Human Services. http://mentalhealth.samhsa.gov/cmhs/communitysupport/toolkits/medication/.
- Texas Medication Algorithm Project (TMAP). http://www.dshs.state.tx.us/mhprograms/TIMA.shtm.
- Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, 2nd ed. Am J Psychiatry 2004;161(suppl):1–56.
- Clozapine • Clozaril
- Haloperidol • Haldol
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Miller receives research support from or is a consultant or speaker for Abbott Laboratories, Almirall, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Pfizer Inc., and InforMedix.
1. Miller A, Craig CS. Combination antipsychotics: pros, cons, and questions. Schizophr Bull 2002;28(1):105-9.
2. Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry 1988;45(9):789-96.
3. Miller AL, Hall CS, Buchanan RW, et al. The Texas Medication Algorithm Project antipsychotic algorithm for schizophrenia: 2003 update. J Clin Psychiatry 2004;65(4):500-8.
4. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: Comparing monotherapy with polypharmacy and augmentation. Curr Med Chem 2004;11(3):313-27.
5. Centorrino F, Goren JL, Hennen J, et al. Multiple versus single antipsychotic agents for hospitalized psychiatric patients: Case-control study of risks versus benefits. Am J Psychiatry 2004;161(4):700-6.
6. Miller AL. Polypharmacy in schizophrenia: science, art, and fiction. Curr Psychosis Ther Rep (in press).
7. Miller AL, Chiles JA, Chiles JK, et al. The Texas Medication Algorithm Project (TMAP) schizophrenia algorithms. J Clin Psychiatry 1999;60(10):649-57.
8. Shiloh R, Zemishlany Z, Aizenberg D, et al. Sulpiride augmentation in people with schizophrenia partially responsive to clozapine. A double-blind, placebo-controlled study. Br J Psychiatry 1997;171:569-73.
9. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry 2005;162(1):130-6.
10. Anil Yagcioglu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry 2005;66(1):63-72.
11. Reinstein MJ, Sirotovskaya LA, Jones LE, et al. Effect of clozapine-quetiapine combination therapy on weight and glycaemic control. Clin Drug Invest 1999;18(2):99-104.
12. Agelink MW, Kavuk I, Ak I. Clozapine with amisulpride for refractory schizophrenia. Am J Psychiatry 2004;161:924-5.
13. Kaye NS. Ziprasidone augmentation of clozapine in 11 patients. J Clin Psychiatry 2003;64(2):215-16.
14. Shores-Wilson K, Biggs MM, Miller AL, et al. Itemized clinician ratings versus global ratings of symptom severity in patients with schizophrenia. Int J Methods Psychiatr Res 2002;11(1):45-53.
Off-label prescribing of two or more antipsychotics for treatment-resistant psychosis carries inherent risks for both schizophrenia patients and their psychiatrists. You can reduce these risks by demonstrating that your patient will benefit more from combining antipsychotics than from monotherapy alternatives.
Until more empiric data become available, clinicians carry the burden of documenting individual patient response to justify combining antipsychotics. This article can help you identify and counsel possible candidates for this therapy, assess the risks and benefits, and defend your treatment choicesif necessary.
What Does ‘Combining’ Mean?
For this discussion, “combining antipsychotics” does not mean simultaneously using two or more antipsychotics to treat psychosis. Rather, it refers to using two or more antipsychotics for long-term control of psychotic symptoms. It does not apply to short-term tactics, such as overlapping antipsychotics when switching between them or combining a parenteral and an oral antipsychotic when treating an acute episode.
How long must a combination be used to qualify as long-term treatment? No standard exists, but a period >6 weeks exceeds normal short-term treatment.
How Safe Are Combinations?
Patient risk. Combination antipsychotics are used to treat 10% to 20% of schizophrenia patients in this country and >90% of those in some Asian countries, such as Japan.1 Statistics on the prevalence of combining antipsychotics seldom:
- distinguish between short- and long-term use
- identify when practitioners use combinations to treat co-existing symptoms such as insomnia, rather than for psychosis.
Clinicians, however, must define safety in terms of risks and benefits. Given that every medication has risks—some (such as allergic reactions) unique to the individual patient—adding another drug to a treatment regimen will always add risk. When considering more than one antipsychotic, ask yourself:
- Do benefits outweigh risks?
- Is a combination the least risky way to achieve these benefits?
How much does a practitioner’s perception of medicolegal risk influence treatment selection? No doubt, comfort level varies greatly. I have frequently met prescribers who use antipsychotic combinations instead of clozapine for fear of being sued should agranulocytosis occur.
Most trials have used criteriafrom the seminal clozapine study to define “treatment resistant” schizophrenia:
- History of poor response to one or more antipsychotics.
- Demonstrated nonresponse to adequate dosage and duration of a nonclozapine antipsychotic before starting clozapine.2
This work defines treatment resistance as persistent positive symptoms despite treatment with ≥2 different antipsychotics other than clozapine.
Newer antipsychotics are different. The original clozapine studies found little likelihood that patients who had not responded to haloperidol would respond to chlorpromazine. Newer-generation antipsychotics are pharmacologically more heterogeneous than the first-generation agents, however, and failure to respond to one might not mean that none of the others will help.
When to start clozapine? Solid evidence is limited, but a consensus panel has recommended starting clozapine after two unsuccessful trials of newer-generation antipsychotics, with the option to try a third antipsychotic (first- or newer-generation) if clinically warranted.3
Symptoms vs function. In clinical practice, defining treatment resistance as persistent positive symptoms may exclude patients impaired by severe negative symptoms and/or disorganized thought processes. The treatment goal is to restore function, not just to reduce psychosis. As treatments evolve, we can expect more emphasis on a recovery model that addresses total patient well-being and combines psychosocial and pharmacologic interventions.
A Practical View
Medical risks. Treating patients with antipsychotic combinations may be associated with medical risks (Table 1). Patients are less likely to adhere to complex medication regimens than to simple ones. Thus, adding a second antipsychotic may decrease adherence to the first antipsychotic and to other medications, such as for hypertension, diabetes, etc.
Increased side-effect risk has been reported with antipsychotic combinations.4 Side effects include those expected from one or both drugs in the combination, especially extrapyramidal effects when conventional antipsychotics are included.4,5 Because the total antipsychotic dosage can be considerably higher than usual in patients receiving combinations,5 some of the increased side effect burden is probably related to high dosing, rather than to the combinations.
Pharmacodynamic or pharmacokinetic interactions or the loss of an agent’s advantages are infrequently reported, perhaps because prescribers are not looking for these effects or do not consider them worth reporting. Of particular concern is tardive dyskinesia (TD), which may develop when conventional antipsychotics are added to agents with very low propensity to cause TD—such as clozapine.
Table 1
Potential risks and benefits of combining antipsychotics
| Medical risks |
| Decreased adherence to multiple medications |
| Increased and/or unexpected side effects |
| Increased potential for undesirable pharmacokinetic or pharmacodynamic interactions |
| Difficulties in making rational dose adjustments |
| Loss of advantages of one of the medications |
| Nonmedical risks |
| Lack of evidence to defend practice in medicolegal cases |
| Increased costs if combining second-generation antipsychotics |
| Increased quality assurance scrutiny and paperwork |
| Medical benefits |
| Reduced symptoms |
| Reduced metabolic side effects (such as partially substituting another atypical antipsychotic for a high clozapine dosage) |
litigation. As noted, some clinicians prefer combining antipsychotics instead of using clozapine because they fear legal action should a patient develop agranulocytosis. In the author’s view, this fear is not well-grounded. Successful lawsuits typically find evidence that the clinician committed errors of omission or commission. In the case of blood monitoring for clozapine-induced neutropenia, the parameters are clear and enforced. You must follow community standards of practice, which provide a strong legal defense.
Administrative scrutiny. Quality assurance programs are increasingly identifying and monitoring antipsychotic combinations—an obvious target by being frequent, lacking a strong evidence base, increasing costs, and raising liability concerns. Typically, such programs discourage antipsychotic combinations and impose administrative hurdles to starting or continuing them.
Gathering data and documenting it in the patient’s medical record—to be discussed later—is key to demonstrating that a combination’s superior efficacy for the individual patient justifies its use.
Medical benefits. Guidelines and algorithms for drug treatment of schizophrenia either omit combination antipsychotics or suggest this strategy when all else has failed.6,7 Lack of evidence for a practice is not the same as evidence against it, however. A combination may be better for some patients than any available antipsychotic monotherapy.
Antipsychotic combinations have been examined in more literature reviews than randomized controlled trials—all of which have addressed augmenting clozapine with another antipsychotic (Table 2).8-12 Augmentation with psychotropics other than a second antipsychotic has most often been tested for negative and cognitive symptoms,4 but some evidence has shown adjunctive anticonvulsants and cognition-enhancing agents to improve positive symptoms.6
Reducing side effects is not directly related to treatment-resistant psychosis, but some articles describe managing clozapine’s metabolic side effects by partially substituting another atypical antipsychotic.11,13
Because clozapine is the treatment of choice for treatment-resistant psychosis, consider tactics that maintain its benefits while reducing its metabolic liabilities. Reducing the clozapine dosage (if feasible) is the first-line approach, but partially substituting another antipsychotic might help if psychotic symptoms return.
Table 2
Results of studies using combination antipsychotic therapy
| Combination | Type of trial | First author | Outcome measures | Results |
|---|---|---|---|---|
| Clozapine/sulpiride* | RCT | Shiloh8 | Positive, negative, and depressive symptoms | Positive |
| Clozapine/risperidone | RCT | Josiassen9 | Positive and negative symptoms | Positive |
| Clozapine/risperidone | RCT | Yagcioglu10 | Positive and negative symptoms | Negative |
| Clozapine/quetiapine | LCS | Reinstein11 | Body weight, serum glucose | Positive |
| Clozapine/amisulpride* | LCS | Agelink12 | Positive and negative symptoms | Positive |
| RCT: randomized controlled trial; LCS: large case series | ||||
| *Sulpiride and amisulpride are not available in the United States. | ||||
Clinical Management Hints
Document, document, document. Crucial to “non-standard” treatments such as combining antipsychotics is using and documenting good medical practices. These include:
- accurately assessing patients
- carefully weighing illness risks vs treatment risks
- talking regularly to patients about their treatment and therapeutic options (Table 3).
Use objective measures. Brief scales to measure psychotic symptoms are being increasingly used in public mental health settings.6 For example, four psychosis items from the Brief Psychiatric Rating Scale (hallucinations, unusual thought content, paranoia, and disorganized thought) can quickly assess and capture much of the variance of the full scale in patients with schizophrenia.14
When properly used, these objective and reliable scales can document clinical change and track illness course across time and providers. Other objective outcome measures can contribute to quality of care and its documentation. For more information, visit the Substance Abuse and Mental Health Services Administration Web site (see Related Resources).
Although it seems obvious that persistent illness is not a good thing, the risks of persistent psychosis vary from patient to patient. Some function relatively well despite ongoing psychotic thoughts, whereas others are terribly impaired, demoralized, and/or suicidal. The rationale for nonstandard treatment such as an antipsychotic combination is to reduce patient suffering and risk and/or to increase function.
Discuss options with patients. Finally, schizophrenia patients (and involved significant others) need to understand and participate in treatment. Ongoing discussion of treatment options is an important part of this process. For example, initially reluctant patients often come to accept clozapine treatment after discussing its risks and benefits with clinicians and other patients who are taking clozapine. Documenting these discussions is as beneficial to the patient and clinician as is documenting symptoms and treatment effects.
Table 3
3 tips for managing risk and medications
| Use objective, quantifiable symptom measures at regular intervals |
| Document patient’s risk of persistent psychosis, based on history |
| Discuss risks and benefits of other treatment options with the patient and/or family at regular intervals |
- How to use medication in a systematic and effective way, as part of the overall treatment for severe mental illness. In: Evidence-based practices: Shaping mental health services toward recovery. Medication management approaches in psychiatry. Substance Abuse and Mental Health Services Administration. U.S. Department of Health and Human Services. http://mentalhealth.samhsa.gov/cmhs/communitysupport/toolkits/medication/.
- Texas Medication Algorithm Project (TMAP). http://www.dshs.state.tx.us/mhprograms/TIMA.shtm.
- Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, 2nd ed. Am J Psychiatry 2004;161(suppl):1–56.
- Clozapine • Clozaril
- Haloperidol • Haldol
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Miller receives research support from or is a consultant or speaker for Abbott Laboratories, Almirall, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Pfizer Inc., and InforMedix.
Off-label prescribing of two or more antipsychotics for treatment-resistant psychosis carries inherent risks for both schizophrenia patients and their psychiatrists. You can reduce these risks by demonstrating that your patient will benefit more from combining antipsychotics than from monotherapy alternatives.
Until more empiric data become available, clinicians carry the burden of documenting individual patient response to justify combining antipsychotics. This article can help you identify and counsel possible candidates for this therapy, assess the risks and benefits, and defend your treatment choicesif necessary.
What Does ‘Combining’ Mean?
For this discussion, “combining antipsychotics” does not mean simultaneously using two or more antipsychotics to treat psychosis. Rather, it refers to using two or more antipsychotics for long-term control of psychotic symptoms. It does not apply to short-term tactics, such as overlapping antipsychotics when switching between them or combining a parenteral and an oral antipsychotic when treating an acute episode.
How long must a combination be used to qualify as long-term treatment? No standard exists, but a period >6 weeks exceeds normal short-term treatment.
How Safe Are Combinations?
Patient risk. Combination antipsychotics are used to treat 10% to 20% of schizophrenia patients in this country and >90% of those in some Asian countries, such as Japan.1 Statistics on the prevalence of combining antipsychotics seldom:
- distinguish between short- and long-term use
- identify when practitioners use combinations to treat co-existing symptoms such as insomnia, rather than for psychosis.
Clinicians, however, must define safety in terms of risks and benefits. Given that every medication has risks—some (such as allergic reactions) unique to the individual patient—adding another drug to a treatment regimen will always add risk. When considering more than one antipsychotic, ask yourself:
- Do benefits outweigh risks?
- Is a combination the least risky way to achieve these benefits?
How much does a practitioner’s perception of medicolegal risk influence treatment selection? No doubt, comfort level varies greatly. I have frequently met prescribers who use antipsychotic combinations instead of clozapine for fear of being sued should agranulocytosis occur.
Most trials have used criteriafrom the seminal clozapine study to define “treatment resistant” schizophrenia:
- History of poor response to one or more antipsychotics.
- Demonstrated nonresponse to adequate dosage and duration of a nonclozapine antipsychotic before starting clozapine.2
This work defines treatment resistance as persistent positive symptoms despite treatment with ≥2 different antipsychotics other than clozapine.
Newer antipsychotics are different. The original clozapine studies found little likelihood that patients who had not responded to haloperidol would respond to chlorpromazine. Newer-generation antipsychotics are pharmacologically more heterogeneous than the first-generation agents, however, and failure to respond to one might not mean that none of the others will help.
When to start clozapine? Solid evidence is limited, but a consensus panel has recommended starting clozapine after two unsuccessful trials of newer-generation antipsychotics, with the option to try a third antipsychotic (first- or newer-generation) if clinically warranted.3
Symptoms vs function. In clinical practice, defining treatment resistance as persistent positive symptoms may exclude patients impaired by severe negative symptoms and/or disorganized thought processes. The treatment goal is to restore function, not just to reduce psychosis. As treatments evolve, we can expect more emphasis on a recovery model that addresses total patient well-being and combines psychosocial and pharmacologic interventions.
A Practical View
Medical risks. Treating patients with antipsychotic combinations may be associated with medical risks (Table 1). Patients are less likely to adhere to complex medication regimens than to simple ones. Thus, adding a second antipsychotic may decrease adherence to the first antipsychotic and to other medications, such as for hypertension, diabetes, etc.
Increased side-effect risk has been reported with antipsychotic combinations.4 Side effects include those expected from one or both drugs in the combination, especially extrapyramidal effects when conventional antipsychotics are included.4,5 Because the total antipsychotic dosage can be considerably higher than usual in patients receiving combinations,5 some of the increased side effect burden is probably related to high dosing, rather than to the combinations.
Pharmacodynamic or pharmacokinetic interactions or the loss of an agent’s advantages are infrequently reported, perhaps because prescribers are not looking for these effects or do not consider them worth reporting. Of particular concern is tardive dyskinesia (TD), which may develop when conventional antipsychotics are added to agents with very low propensity to cause TD—such as clozapine.
Table 1
Potential risks and benefits of combining antipsychotics
| Medical risks |
| Decreased adherence to multiple medications |
| Increased and/or unexpected side effects |
| Increased potential for undesirable pharmacokinetic or pharmacodynamic interactions |
| Difficulties in making rational dose adjustments |
| Loss of advantages of one of the medications |
| Nonmedical risks |
| Lack of evidence to defend practice in medicolegal cases |
| Increased costs if combining second-generation antipsychotics |
| Increased quality assurance scrutiny and paperwork |
| Medical benefits |
| Reduced symptoms |
| Reduced metabolic side effects (such as partially substituting another atypical antipsychotic for a high clozapine dosage) |
litigation. As noted, some clinicians prefer combining antipsychotics instead of using clozapine because they fear legal action should a patient develop agranulocytosis. In the author’s view, this fear is not well-grounded. Successful lawsuits typically find evidence that the clinician committed errors of omission or commission. In the case of blood monitoring for clozapine-induced neutropenia, the parameters are clear and enforced. You must follow community standards of practice, which provide a strong legal defense.
Administrative scrutiny. Quality assurance programs are increasingly identifying and monitoring antipsychotic combinations—an obvious target by being frequent, lacking a strong evidence base, increasing costs, and raising liability concerns. Typically, such programs discourage antipsychotic combinations and impose administrative hurdles to starting or continuing them.
Gathering data and documenting it in the patient’s medical record—to be discussed later—is key to demonstrating that a combination’s superior efficacy for the individual patient justifies its use.
Medical benefits. Guidelines and algorithms for drug treatment of schizophrenia either omit combination antipsychotics or suggest this strategy when all else has failed.6,7 Lack of evidence for a practice is not the same as evidence against it, however. A combination may be better for some patients than any available antipsychotic monotherapy.
Antipsychotic combinations have been examined in more literature reviews than randomized controlled trials—all of which have addressed augmenting clozapine with another antipsychotic (Table 2).8-12 Augmentation with psychotropics other than a second antipsychotic has most often been tested for negative and cognitive symptoms,4 but some evidence has shown adjunctive anticonvulsants and cognition-enhancing agents to improve positive symptoms.6
Reducing side effects is not directly related to treatment-resistant psychosis, but some articles describe managing clozapine’s metabolic side effects by partially substituting another atypical antipsychotic.11,13
Because clozapine is the treatment of choice for treatment-resistant psychosis, consider tactics that maintain its benefits while reducing its metabolic liabilities. Reducing the clozapine dosage (if feasible) is the first-line approach, but partially substituting another antipsychotic might help if psychotic symptoms return.
Table 2
Results of studies using combination antipsychotic therapy
| Combination | Type of trial | First author | Outcome measures | Results |
|---|---|---|---|---|
| Clozapine/sulpiride* | RCT | Shiloh8 | Positive, negative, and depressive symptoms | Positive |
| Clozapine/risperidone | RCT | Josiassen9 | Positive and negative symptoms | Positive |
| Clozapine/risperidone | RCT | Yagcioglu10 | Positive and negative symptoms | Negative |
| Clozapine/quetiapine | LCS | Reinstein11 | Body weight, serum glucose | Positive |
| Clozapine/amisulpride* | LCS | Agelink12 | Positive and negative symptoms | Positive |
| RCT: randomized controlled trial; LCS: large case series | ||||
| *Sulpiride and amisulpride are not available in the United States. | ||||
Clinical Management Hints
Document, document, document. Crucial to “non-standard” treatments such as combining antipsychotics is using and documenting good medical practices. These include:
- accurately assessing patients
- carefully weighing illness risks vs treatment risks
- talking regularly to patients about their treatment and therapeutic options (Table 3).
Use objective measures. Brief scales to measure psychotic symptoms are being increasingly used in public mental health settings.6 For example, four psychosis items from the Brief Psychiatric Rating Scale (hallucinations, unusual thought content, paranoia, and disorganized thought) can quickly assess and capture much of the variance of the full scale in patients with schizophrenia.14
When properly used, these objective and reliable scales can document clinical change and track illness course across time and providers. Other objective outcome measures can contribute to quality of care and its documentation. For more information, visit the Substance Abuse and Mental Health Services Administration Web site (see Related Resources).
Although it seems obvious that persistent illness is not a good thing, the risks of persistent psychosis vary from patient to patient. Some function relatively well despite ongoing psychotic thoughts, whereas others are terribly impaired, demoralized, and/or suicidal. The rationale for nonstandard treatment such as an antipsychotic combination is to reduce patient suffering and risk and/or to increase function.
Discuss options with patients. Finally, schizophrenia patients (and involved significant others) need to understand and participate in treatment. Ongoing discussion of treatment options is an important part of this process. For example, initially reluctant patients often come to accept clozapine treatment after discussing its risks and benefits with clinicians and other patients who are taking clozapine. Documenting these discussions is as beneficial to the patient and clinician as is documenting symptoms and treatment effects.
Table 3
3 tips for managing risk and medications
| Use objective, quantifiable symptom measures at regular intervals |
| Document patient’s risk of persistent psychosis, based on history |
| Discuss risks and benefits of other treatment options with the patient and/or family at regular intervals |
- How to use medication in a systematic and effective way, as part of the overall treatment for severe mental illness. In: Evidence-based practices: Shaping mental health services toward recovery. Medication management approaches in psychiatry. Substance Abuse and Mental Health Services Administration. U.S. Department of Health and Human Services. http://mentalhealth.samhsa.gov/cmhs/communitysupport/toolkits/medication/.
- Texas Medication Algorithm Project (TMAP). http://www.dshs.state.tx.us/mhprograms/TIMA.shtm.
- Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, 2nd ed. Am J Psychiatry 2004;161(suppl):1–56.
- Clozapine • Clozaril
- Haloperidol • Haldol
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Miller receives research support from or is a consultant or speaker for Abbott Laboratories, Almirall, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Eli Lilly and Co., Janssen Pharmaceutica, Pfizer Inc., and InforMedix.
1. Miller A, Craig CS. Combination antipsychotics: pros, cons, and questions. Schizophr Bull 2002;28(1):105-9.
2. Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry 1988;45(9):789-96.
3. Miller AL, Hall CS, Buchanan RW, et al. The Texas Medication Algorithm Project antipsychotic algorithm for schizophrenia: 2003 update. J Clin Psychiatry 2004;65(4):500-8.
4. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: Comparing monotherapy with polypharmacy and augmentation. Curr Med Chem 2004;11(3):313-27.
5. Centorrino F, Goren JL, Hennen J, et al. Multiple versus single antipsychotic agents for hospitalized psychiatric patients: Case-control study of risks versus benefits. Am J Psychiatry 2004;161(4):700-6.
6. Miller AL. Polypharmacy in schizophrenia: science, art, and fiction. Curr Psychosis Ther Rep (in press).
7. Miller AL, Chiles JA, Chiles JK, et al. The Texas Medication Algorithm Project (TMAP) schizophrenia algorithms. J Clin Psychiatry 1999;60(10):649-57.
8. Shiloh R, Zemishlany Z, Aizenberg D, et al. Sulpiride augmentation in people with schizophrenia partially responsive to clozapine. A double-blind, placebo-controlled study. Br J Psychiatry 1997;171:569-73.
9. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry 2005;162(1):130-6.
10. Anil Yagcioglu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry 2005;66(1):63-72.
11. Reinstein MJ, Sirotovskaya LA, Jones LE, et al. Effect of clozapine-quetiapine combination therapy on weight and glycaemic control. Clin Drug Invest 1999;18(2):99-104.
12. Agelink MW, Kavuk I, Ak I. Clozapine with amisulpride for refractory schizophrenia. Am J Psychiatry 2004;161:924-5.
13. Kaye NS. Ziprasidone augmentation of clozapine in 11 patients. J Clin Psychiatry 2003;64(2):215-16.
14. Shores-Wilson K, Biggs MM, Miller AL, et al. Itemized clinician ratings versus global ratings of symptom severity in patients with schizophrenia. Int J Methods Psychiatr Res 2002;11(1):45-53.
1. Miller A, Craig CS. Combination antipsychotics: pros, cons, and questions. Schizophr Bull 2002;28(1):105-9.
2. Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry 1988;45(9):789-96.
3. Miller AL, Hall CS, Buchanan RW, et al. The Texas Medication Algorithm Project antipsychotic algorithm for schizophrenia: 2003 update. J Clin Psychiatry 2004;65(4):500-8.
4. Stahl SM, Grady MM. A critical review of atypical antipsychotic utilization: Comparing monotherapy with polypharmacy and augmentation. Curr Med Chem 2004;11(3):313-27.
5. Centorrino F, Goren JL, Hennen J, et al. Multiple versus single antipsychotic agents for hospitalized psychiatric patients: Case-control study of risks versus benefits. Am J Psychiatry 2004;161(4):700-6.
6. Miller AL. Polypharmacy in schizophrenia: science, art, and fiction. Curr Psychosis Ther Rep (in press).
7. Miller AL, Chiles JA, Chiles JK, et al. The Texas Medication Algorithm Project (TMAP) schizophrenia algorithms. J Clin Psychiatry 1999;60(10):649-57.
8. Shiloh R, Zemishlany Z, Aizenberg D, et al. Sulpiride augmentation in people with schizophrenia partially responsive to clozapine. A double-blind, placebo-controlled study. Br J Psychiatry 1997;171:569-73.
9. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry 2005;162(1):130-6.
10. Anil Yagcioglu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry 2005;66(1):63-72.
11. Reinstein MJ, Sirotovskaya LA, Jones LE, et al. Effect of clozapine-quetiapine combination therapy on weight and glycaemic control. Clin Drug Invest 1999;18(2):99-104.
12. Agelink MW, Kavuk I, Ak I. Clozapine with amisulpride for refractory schizophrenia. Am J Psychiatry 2004;161:924-5.
13. Kaye NS. Ziprasidone augmentation of clozapine in 11 patients. J Clin Psychiatry 2003;64(2):215-16.
14. Shores-Wilson K, Biggs MM, Miller AL, et al. Itemized clinician ratings versus global ratings of symptom severity in patients with schizophrenia. Int J Methods Psychiatr Res 2002;11(1):45-53.
Psychological testing: Use do-it-yourself tools or refer?
Mr. A, age 38, presents with severe anxiety symptoms that suggest generalized anxiety disorder (GAD). You wish to confirm the diagnosis before starting medication, measure treatment response, and provide documentation to Mr. A’s managed care company.
Miss B, age 73, complains of memory and organization problems. Her history of transient ischemic attacks suggests vascular dementia, but the gradual symptom onset suggests Alzheimer’s dementia. You need to clarify the diagnosis.
Informed use of psychological testing can help you plan treatment by clarifying the causes, diagnosis, and prognosis of patients’ symptoms. With hundreds of instruments available, we offer an overview to help you quickly choose appropriate in-office tools or refer for more-intensive testing.
QUICK BUT IMPERFECT
Checklists and rating scales can quickly gauge a personality trait such as impulsivity or target symptom such as anxiety, using a numerical list of words or statements:
- A checklist’s response format is dichotomous (typically yes/no).
- Rating scales offer greater options, such as a 4-point scale for measuring symptoms as 0 (not present), 1 (mild), 2 (moderate), 3 (severe).
Some checklists/rating scales can assess more than one disorder or target symptom. These wide-band instruments—often called inventories or schedules—tend to be lengthy (1 to 2 hours), often require an interview, and generally require specialized training to administer.1-4
Pros. Two attributes make checklist/rating scales popular in clinical practice: their convenience, and managed care’s quest for documentation of service need, quality of care, cost-effectiveness, and symptom reduction.5 Brief, accurate, efficient checklists/rating scales can help you give managed care firms the documentation they require to authorize continued treatment—whether psychotherapy or medication monitoring.
Cons. Many checklists/rating scales are psychometrically weak, with low reliability and unproven validity. Some are lengthy or have other traits that diminish their clinical value (Table 1).
Table 1
Pros and cons of checklists/rating scales
| Pros |
|
| Cons |
|
LONGER AND MORE DETAILED
Objective tests typically contain true/false questions for which responses are reported as percentiles or standard scores. Examples are the Minnesota Multiphasic Personality Inventory (MMPI-2), used to clarify axis I diagnoses, and Millon Clinical Multiaxial Inventory (MCMI-III), chiefly used to assess personality disorders. Objective tests’ ability to assess a wide band of psychopathology can help you evaluate patients with complex differential diagnoses.6
Projective tests are unstructured instruments developed to detect covert psychosis and pathologic conflicts/impulses. Patients respond to ambiguous stimuli (inkblots, pictures, incomplete sentences) that are assumed to function as a screen onto which a person projects his or her conflicts and issues.3
Useful projective tests include the Rorschach ink blot test, Thematic Apperception Test (TAT) of interpersonal relationships, and several sentence-completion tests. The Rorschach can take 1 to 2 hours to administer and score and requires years to master. The Rotter Incomplete Sentences Blank (2nd ed) (RISB) is well-constructed; available in high school, college, and adult forms; and can help clarify major conflicts.3
Projective tests’ psychometric properties have been questioned, but the Rorschach is considered useful in detecting subtle psychoses.6
Neuropsychological tests can identify and localize brain injury. Board-certified neuropsychologists (with 2 years’ postdoctoral training) use them to assess traumatic brain injury, evaluate post-stroke syndromes or early dementia, and differentiate dementia and depression.7 These tests also have litigation and forensic applications, such as assessing competence or malingering.
Some neuropsychologists use a comprehensive instrument such as the Halstead-Reitan Neuropsychological Test Battery, which evaluates memory, abstract thought, language, sensory-motor integration, imperception, and motor dexterity. Others may select specific instruments to answer a referring psychiatrist’s question.
CHOOSING AN INSTRUMENT
Medical reference librarians can help research specific instruments and choose useful testing tools. We also recommend Corcoran and Fischer’s Measures for Clinical Practice,8 which provides practical information on administration, advantages, and disadvantages of instruments that:
- are used in clinical practice
- provide data on psychometric properties
- take
- are rapidly scored
- provide information on symptom severity
- can be used to document change.
Table 2
Commonly used checklists/rating scales for adult assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Anger | Anger, Irritability and Assault Questionnaire (AIAQ) |
| Anxiety | |
| Phobias | Fear Questionnaire (FQ) |
| GAD | Beck Anxiety Inventory (BAI) |
| OCD | Yale-Brown Obsessive Compulsive Scale (Y-BOCS) |
| PTSD | Posttraumatic Stress Diagnostic Scale (PDS) |
| Bipolar disorder | Young Mania Rating Scale (YMRS) |
| Depression | Beck Depression Inventory (BDI) |
| Zung Self-rating Depression Scale (SDS) | |
| Eating disorders | Eating Disorders Inventory-2 |
| Family issues | Family Assessment Device (FAD) |
| Impulse control | Barratt Impulsiveness Scale, Version II (BIS-II) |
| Pain | McGill Pain Questionnaire (MPQ) |
| Personality disorders | Millon Clinical Multiaxial Inventory (MCMI-III) |
| Psychosis | Brief Psychiatric Rating Scale (BPRS) |
| Manchester Scale | |
| Sexuality | Sexual Interaction Inventory (SII) |
| Sleep | Sleep Disorder Questionnaire (SDQ) |
| Suicide risk | Beck Scale for Suicide Ideation (BSS) |
| GAD: Generalized anxiety disorder | |
| OCD: Obsessive-compulsive disorder | |
| PTSD: Posttraumatic stress disorder | |
Common checklists/rating scales for geriatric assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Cognitive status | Mini-Mental State Examination (MMSE) |
| Neurobehavioral Cognitive Status Examination (Cognistat) | |
| Dementia Rating Scale | |
| Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) | |
| Caregiver stress | Caregiver’s Burden Scale (CBS) |
| Death concerns | Concern About Death-Dying (CADD) and Coping (C) checklist |
| Depression | Geriatric Depression Scale (GDS) |
Common checklists/rating scales for child and adolescent assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Anxiety | Multidimensional Anxiety Scale for Children (MASC) |
| Assertiveness | Assertiveness Scale for Adolescents (ASA) |
| Conduct | Child Behavior Checklist (CBCL) |
| Depression | Children’s Depression Self Rating Scale (CDRS) |
| Drug/alcohol risk | CAGE Questionnaire |
| Michigan Alcohol Screening Test (MAST) | |
| Impulsivity | Impulsivity Scale (IS) |
| PTSD | Child Report of Posttraumatic Symptoms (CROPS) |
| Reaction to divorce | Children’s Belief About Parental Divorce Scale (CBAPS) |
| Self-esteem | Rosenberg Self-Esteem Scale (RSE) |
| Suicide risk | Multi-Attitude Suicide Tendency Scale (MAST) |
| Test anxiety | Children’s Cognitive Assessment Questionnaire (CCAQ) |
| PTSD: Posttraumatic stress disorder | |
IN-OFFICE TESTING VS REFERRAL
You could use in-office testing to diagnose Mr. A’s anxiety symptoms and provide documentation to his managed care company. For Miss B’s memory problems, we recommend referral for psychological testing.
Mr. A completes the 21-item Beck Anxiety Inventory (BAI) in your office. You select the BAI because it is psychometrically sound, brief (about 10 minutes to complete and score), and easily understood. Results can be readily used for feedback to patients or third-party payers.
Mr. A’s score of 19 is consistent with GAD and justifies a medication trial. The BAI provides information about his experience of anxiety (subjective vs. somatic) that can guide psychotherapy. You plan to repeat the BAI over time to monitor treatment.
Miss B would benefit from referral to a neuropsychologist, as screening tools do not reliably differentiate among the dementias. The neuropsychologist will likely use all or part of the Halstead-Reitan Neuropsychological Test Battery to localize any ischemic-related brain injury and clarify the diagnosis. This test also can provide data to stage her dementia and help you and her family with care decisions.
PSYCHOLOGIST REFERRAL
When referring patients for psychological testing, we recommend that you tell the psychologist what information you need and let him or her select the tests. Relying on their expertise can save time and yield a report that targets the referral question.
Three cases follow that illustrate types of referral questions doctoral-level psychologists can help answer with appropriately chosen tests:
WHAT EXPLAINS TREATMENT RESISTANCE?
Mr. C, age 43, presents with mixed anxiety and depression. He complains of insomnia, fatigue, tightness in the chest, and trembling hands. You give him the Beck Depression Inventory (BDI) and Beck Anxiety Inventory (BAI), which show mild depression/anxiety. You prescribe fluoxetine, 20 mg/d, and 8 weeks later his symptoms are unchanged. The patient is demanding, critical, and has a pattern of interpersonal difficulty. You suspect a personality disorder is complicating treatment.
In this case, the MMPI-2 and MCMI-III would be useful to clarify diagnosis. The MMPI-2 gauges anxiety (state anxiety, phobias, social anxiety and posttraumatic stress disorder), and depression. The MCMI-III was developed to assess axis II diagnoses and has scales to assess each personality disorder. These tests provide information about psychological-mindedness, treatment resistance, and characteristics that can guide psychotherapy.
DRUGS, PSYCHOSIS, OR BIPOLAR DISORDER?
Mr. D’s parents report that their 20-year-old is isolating himself in his room, is not sleeping, and has grandiose beliefs of special powers and knowledge. He has no psychiatric history. Because these symptoms could suggest numerous psychopathologies, you would like help with the differential diagnosis.
Mr. D’s symptoms could suggest drug abuse, schizophrenia, psychotic depression, or bipolar disorder. The psychologist might use the MMPI-2 to assess drug abuse, depression, mania, and psychosis. The relative elevation of each scale could be clinically useful; if scales gauging psychosis and depression are both elevated, psychotic depression is likely, whereas an elevation chiefly on the mania scale would point to bipolar disorder.
The Rorschach test could assess psychotic process. The MMPI-2 could be repeated in a few months to gauge treatment response.
IS THIS EARLY ALZHEIMER’S DISEASE?
Mr. E, age 78, presents with mild memory and word-finding deficits and complains of fatigue, loss of appetite, and anhedonia. Physical exam and lab tests are unremarkable, and you suspect early Alzheimer’s dementia and depression. You wish to confirm the diagnosis to decide whether to start a cholinesterase inhibitor, antidepressant, or other medication. You also wish to document change over time.
An in-office depression checklist would be appropriate for Mr. E. The 30-item, self-rated Geriatric Depression Scale is psychometrically sound and can be completed in 15 to 20 minutes.
Referral is recommended for dementia screening with an tool such as the Neurobehavioral Cognitive Status Examination (Cognistat) or Repeatable Battery for the Assessment of Neuropsychological Status (RBANS). The Mini-Mental State Examination (MMSE) is used for in-office screening of cognitive deficits but lacks sensitivity to detect mild decline. Cognistat or RBANS are less influenced by the patient’s education level and are more sensitive than the MMSE to early dementia.
All three instruments are brief enough to repeat as needed to document change.
Related resources
- Corcoran K, Fischer J. Measures for clinical practice, vols. 1 and 2. New York: Free Press; 2000.
- Maruish ME (ed). The use of psychological testing for treatment planning and outcomes assessment. Mahwah, NJ: Lawrence Erlbaum Associates; 1999.
- Rush AJ, Pincus HJ, First MB, et al. (eds). Handbook of psychiatric measures. Washington, DC: American Psychiatric Association; 2000.
- American Psychological Association. FAQ/Finding information about psychological tests. http://www.apa.org/science/faq-findtests.html.
- Fluoxetine • Prozac
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Maruish ME (ed) The use of psychological testing for treatment planning and outcomes assessment. Mahwah, NJ: Lawrence Erlbaum Associates; 1999.
2. Sajatovic M, Ramirez LF. Rating scales in mental health. Hudson, OH: Lexi-Comp; 2003.
3. Aiken LR. Assessment of adult personality. New York: Springer; 1997.
4. Rush AJ, Pincus HJ, First MB, et al (eds). Handbook of Psychiatric Measures. Washington, DC: American Psychiatric Association; 2000.
5. Belar CD. Psychological assessment in capitated care. In: Butcher JN (ed). Personality assessment in managed care: Using the MMPI-2 in treatment planning. New York: Oxford Press; 1997;13:80.-
6. Adams RL, Culbertson JL. Personality assessment: adults and children. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;702:21.-
7. Swanda RM, Haaland KY, La Rue A. Clinical neuropsychology and intellectual assessment of adults. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;689:701.-
8. Corcoran K, Fischer J. Measures for clinical practice, vols 1 and 2. New York: Free Press; 2000.
9. Blacker D. Psychiatric rating scales. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;755-83.
Mr. A, age 38, presents with severe anxiety symptoms that suggest generalized anxiety disorder (GAD). You wish to confirm the diagnosis before starting medication, measure treatment response, and provide documentation to Mr. A’s managed care company.
Miss B, age 73, complains of memory and organization problems. Her history of transient ischemic attacks suggests vascular dementia, but the gradual symptom onset suggests Alzheimer’s dementia. You need to clarify the diagnosis.
Informed use of psychological testing can help you plan treatment by clarifying the causes, diagnosis, and prognosis of patients’ symptoms. With hundreds of instruments available, we offer an overview to help you quickly choose appropriate in-office tools or refer for more-intensive testing.
QUICK BUT IMPERFECT
Checklists and rating scales can quickly gauge a personality trait such as impulsivity or target symptom such as anxiety, using a numerical list of words or statements:
- A checklist’s response format is dichotomous (typically yes/no).
- Rating scales offer greater options, such as a 4-point scale for measuring symptoms as 0 (not present), 1 (mild), 2 (moderate), 3 (severe).
Some checklists/rating scales can assess more than one disorder or target symptom. These wide-band instruments—often called inventories or schedules—tend to be lengthy (1 to 2 hours), often require an interview, and generally require specialized training to administer.1-4
Pros. Two attributes make checklist/rating scales popular in clinical practice: their convenience, and managed care’s quest for documentation of service need, quality of care, cost-effectiveness, and symptom reduction.5 Brief, accurate, efficient checklists/rating scales can help you give managed care firms the documentation they require to authorize continued treatment—whether psychotherapy or medication monitoring.
Cons. Many checklists/rating scales are psychometrically weak, with low reliability and unproven validity. Some are lengthy or have other traits that diminish their clinical value (Table 1).
Table 1
Pros and cons of checklists/rating scales
| Pros |
|
| Cons |
|
LONGER AND MORE DETAILED
Objective tests typically contain true/false questions for which responses are reported as percentiles or standard scores. Examples are the Minnesota Multiphasic Personality Inventory (MMPI-2), used to clarify axis I diagnoses, and Millon Clinical Multiaxial Inventory (MCMI-III), chiefly used to assess personality disorders. Objective tests’ ability to assess a wide band of psychopathology can help you evaluate patients with complex differential diagnoses.6
Projective tests are unstructured instruments developed to detect covert psychosis and pathologic conflicts/impulses. Patients respond to ambiguous stimuli (inkblots, pictures, incomplete sentences) that are assumed to function as a screen onto which a person projects his or her conflicts and issues.3
Useful projective tests include the Rorschach ink blot test, Thematic Apperception Test (TAT) of interpersonal relationships, and several sentence-completion tests. The Rorschach can take 1 to 2 hours to administer and score and requires years to master. The Rotter Incomplete Sentences Blank (2nd ed) (RISB) is well-constructed; available in high school, college, and adult forms; and can help clarify major conflicts.3
Projective tests’ psychometric properties have been questioned, but the Rorschach is considered useful in detecting subtle psychoses.6
Neuropsychological tests can identify and localize brain injury. Board-certified neuropsychologists (with 2 years’ postdoctoral training) use them to assess traumatic brain injury, evaluate post-stroke syndromes or early dementia, and differentiate dementia and depression.7 These tests also have litigation and forensic applications, such as assessing competence or malingering.
Some neuropsychologists use a comprehensive instrument such as the Halstead-Reitan Neuropsychological Test Battery, which evaluates memory, abstract thought, language, sensory-motor integration, imperception, and motor dexterity. Others may select specific instruments to answer a referring psychiatrist’s question.
CHOOSING AN INSTRUMENT
Medical reference librarians can help research specific instruments and choose useful testing tools. We also recommend Corcoran and Fischer’s Measures for Clinical Practice,8 which provides practical information on administration, advantages, and disadvantages of instruments that:
- are used in clinical practice
- provide data on psychometric properties
- take
- are rapidly scored
- provide information on symptom severity
- can be used to document change.
Table 2
Commonly used checklists/rating scales for adult assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Anger | Anger, Irritability and Assault Questionnaire (AIAQ) |
| Anxiety | |
| Phobias | Fear Questionnaire (FQ) |
| GAD | Beck Anxiety Inventory (BAI) |
| OCD | Yale-Brown Obsessive Compulsive Scale (Y-BOCS) |
| PTSD | Posttraumatic Stress Diagnostic Scale (PDS) |
| Bipolar disorder | Young Mania Rating Scale (YMRS) |
| Depression | Beck Depression Inventory (BDI) |
| Zung Self-rating Depression Scale (SDS) | |
| Eating disorders | Eating Disorders Inventory-2 |
| Family issues | Family Assessment Device (FAD) |
| Impulse control | Barratt Impulsiveness Scale, Version II (BIS-II) |
| Pain | McGill Pain Questionnaire (MPQ) |
| Personality disorders | Millon Clinical Multiaxial Inventory (MCMI-III) |
| Psychosis | Brief Psychiatric Rating Scale (BPRS) |
| Manchester Scale | |
| Sexuality | Sexual Interaction Inventory (SII) |
| Sleep | Sleep Disorder Questionnaire (SDQ) |
| Suicide risk | Beck Scale for Suicide Ideation (BSS) |
| GAD: Generalized anxiety disorder | |
| OCD: Obsessive-compulsive disorder | |
| PTSD: Posttraumatic stress disorder | |
Common checklists/rating scales for geriatric assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Cognitive status | Mini-Mental State Examination (MMSE) |
| Neurobehavioral Cognitive Status Examination (Cognistat) | |
| Dementia Rating Scale | |
| Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) | |
| Caregiver stress | Caregiver’s Burden Scale (CBS) |
| Death concerns | Concern About Death-Dying (CADD) and Coping (C) checklist |
| Depression | Geriatric Depression Scale (GDS) |
Common checklists/rating scales for child and adolescent assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Anxiety | Multidimensional Anxiety Scale for Children (MASC) |
| Assertiveness | Assertiveness Scale for Adolescents (ASA) |
| Conduct | Child Behavior Checklist (CBCL) |
| Depression | Children’s Depression Self Rating Scale (CDRS) |
| Drug/alcohol risk | CAGE Questionnaire |
| Michigan Alcohol Screening Test (MAST) | |
| Impulsivity | Impulsivity Scale (IS) |
| PTSD | Child Report of Posttraumatic Symptoms (CROPS) |
| Reaction to divorce | Children’s Belief About Parental Divorce Scale (CBAPS) |
| Self-esteem | Rosenberg Self-Esteem Scale (RSE) |
| Suicide risk | Multi-Attitude Suicide Tendency Scale (MAST) |
| Test anxiety | Children’s Cognitive Assessment Questionnaire (CCAQ) |
| PTSD: Posttraumatic stress disorder | |
IN-OFFICE TESTING VS REFERRAL
You could use in-office testing to diagnose Mr. A’s anxiety symptoms and provide documentation to his managed care company. For Miss B’s memory problems, we recommend referral for psychological testing.
Mr. A completes the 21-item Beck Anxiety Inventory (BAI) in your office. You select the BAI because it is psychometrically sound, brief (about 10 minutes to complete and score), and easily understood. Results can be readily used for feedback to patients or third-party payers.
Mr. A’s score of 19 is consistent with GAD and justifies a medication trial. The BAI provides information about his experience of anxiety (subjective vs. somatic) that can guide psychotherapy. You plan to repeat the BAI over time to monitor treatment.
Miss B would benefit from referral to a neuropsychologist, as screening tools do not reliably differentiate among the dementias. The neuropsychologist will likely use all or part of the Halstead-Reitan Neuropsychological Test Battery to localize any ischemic-related brain injury and clarify the diagnosis. This test also can provide data to stage her dementia and help you and her family with care decisions.
PSYCHOLOGIST REFERRAL
When referring patients for psychological testing, we recommend that you tell the psychologist what information you need and let him or her select the tests. Relying on their expertise can save time and yield a report that targets the referral question.
Three cases follow that illustrate types of referral questions doctoral-level psychologists can help answer with appropriately chosen tests:
WHAT EXPLAINS TREATMENT RESISTANCE?
Mr. C, age 43, presents with mixed anxiety and depression. He complains of insomnia, fatigue, tightness in the chest, and trembling hands. You give him the Beck Depression Inventory (BDI) and Beck Anxiety Inventory (BAI), which show mild depression/anxiety. You prescribe fluoxetine, 20 mg/d, and 8 weeks later his symptoms are unchanged. The patient is demanding, critical, and has a pattern of interpersonal difficulty. You suspect a personality disorder is complicating treatment.
In this case, the MMPI-2 and MCMI-III would be useful to clarify diagnosis. The MMPI-2 gauges anxiety (state anxiety, phobias, social anxiety and posttraumatic stress disorder), and depression. The MCMI-III was developed to assess axis II diagnoses and has scales to assess each personality disorder. These tests provide information about psychological-mindedness, treatment resistance, and characteristics that can guide psychotherapy.
DRUGS, PSYCHOSIS, OR BIPOLAR DISORDER?
Mr. D’s parents report that their 20-year-old is isolating himself in his room, is not sleeping, and has grandiose beliefs of special powers and knowledge. He has no psychiatric history. Because these symptoms could suggest numerous psychopathologies, you would like help with the differential diagnosis.
Mr. D’s symptoms could suggest drug abuse, schizophrenia, psychotic depression, or bipolar disorder. The psychologist might use the MMPI-2 to assess drug abuse, depression, mania, and psychosis. The relative elevation of each scale could be clinically useful; if scales gauging psychosis and depression are both elevated, psychotic depression is likely, whereas an elevation chiefly on the mania scale would point to bipolar disorder.
The Rorschach test could assess psychotic process. The MMPI-2 could be repeated in a few months to gauge treatment response.
IS THIS EARLY ALZHEIMER’S DISEASE?
Mr. E, age 78, presents with mild memory and word-finding deficits and complains of fatigue, loss of appetite, and anhedonia. Physical exam and lab tests are unremarkable, and you suspect early Alzheimer’s dementia and depression. You wish to confirm the diagnosis to decide whether to start a cholinesterase inhibitor, antidepressant, or other medication. You also wish to document change over time.
An in-office depression checklist would be appropriate for Mr. E. The 30-item, self-rated Geriatric Depression Scale is psychometrically sound and can be completed in 15 to 20 minutes.
Referral is recommended for dementia screening with an tool such as the Neurobehavioral Cognitive Status Examination (Cognistat) or Repeatable Battery for the Assessment of Neuropsychological Status (RBANS). The Mini-Mental State Examination (MMSE) is used for in-office screening of cognitive deficits but lacks sensitivity to detect mild decline. Cognistat or RBANS are less influenced by the patient’s education level and are more sensitive than the MMSE to early dementia.
All three instruments are brief enough to repeat as needed to document change.
Related resources
- Corcoran K, Fischer J. Measures for clinical practice, vols. 1 and 2. New York: Free Press; 2000.
- Maruish ME (ed). The use of psychological testing for treatment planning and outcomes assessment. Mahwah, NJ: Lawrence Erlbaum Associates; 1999.
- Rush AJ, Pincus HJ, First MB, et al. (eds). Handbook of psychiatric measures. Washington, DC: American Psychiatric Association; 2000.
- American Psychological Association. FAQ/Finding information about psychological tests. http://www.apa.org/science/faq-findtests.html.
- Fluoxetine • Prozac
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. A, age 38, presents with severe anxiety symptoms that suggest generalized anxiety disorder (GAD). You wish to confirm the diagnosis before starting medication, measure treatment response, and provide documentation to Mr. A’s managed care company.
Miss B, age 73, complains of memory and organization problems. Her history of transient ischemic attacks suggests vascular dementia, but the gradual symptom onset suggests Alzheimer’s dementia. You need to clarify the diagnosis.
Informed use of psychological testing can help you plan treatment by clarifying the causes, diagnosis, and prognosis of patients’ symptoms. With hundreds of instruments available, we offer an overview to help you quickly choose appropriate in-office tools or refer for more-intensive testing.
QUICK BUT IMPERFECT
Checklists and rating scales can quickly gauge a personality trait such as impulsivity or target symptom such as anxiety, using a numerical list of words or statements:
- A checklist’s response format is dichotomous (typically yes/no).
- Rating scales offer greater options, such as a 4-point scale for measuring symptoms as 0 (not present), 1 (mild), 2 (moderate), 3 (severe).
Some checklists/rating scales can assess more than one disorder or target symptom. These wide-band instruments—often called inventories or schedules—tend to be lengthy (1 to 2 hours), often require an interview, and generally require specialized training to administer.1-4
Pros. Two attributes make checklist/rating scales popular in clinical practice: their convenience, and managed care’s quest for documentation of service need, quality of care, cost-effectiveness, and symptom reduction.5 Brief, accurate, efficient checklists/rating scales can help you give managed care firms the documentation they require to authorize continued treatment—whether psychotherapy or medication monitoring.
Cons. Many checklists/rating scales are psychometrically weak, with low reliability and unproven validity. Some are lengthy or have other traits that diminish their clinical value (Table 1).
Table 1
Pros and cons of checklists/rating scales
| Pros |
|
| Cons |
|
LONGER AND MORE DETAILED
Objective tests typically contain true/false questions for which responses are reported as percentiles or standard scores. Examples are the Minnesota Multiphasic Personality Inventory (MMPI-2), used to clarify axis I diagnoses, and Millon Clinical Multiaxial Inventory (MCMI-III), chiefly used to assess personality disorders. Objective tests’ ability to assess a wide band of psychopathology can help you evaluate patients with complex differential diagnoses.6
Projective tests are unstructured instruments developed to detect covert psychosis and pathologic conflicts/impulses. Patients respond to ambiguous stimuli (inkblots, pictures, incomplete sentences) that are assumed to function as a screen onto which a person projects his or her conflicts and issues.3
Useful projective tests include the Rorschach ink blot test, Thematic Apperception Test (TAT) of interpersonal relationships, and several sentence-completion tests. The Rorschach can take 1 to 2 hours to administer and score and requires years to master. The Rotter Incomplete Sentences Blank (2nd ed) (RISB) is well-constructed; available in high school, college, and adult forms; and can help clarify major conflicts.3
Projective tests’ psychometric properties have been questioned, but the Rorschach is considered useful in detecting subtle psychoses.6
Neuropsychological tests can identify and localize brain injury. Board-certified neuropsychologists (with 2 years’ postdoctoral training) use them to assess traumatic brain injury, evaluate post-stroke syndromes or early dementia, and differentiate dementia and depression.7 These tests also have litigation and forensic applications, such as assessing competence or malingering.
Some neuropsychologists use a comprehensive instrument such as the Halstead-Reitan Neuropsychological Test Battery, which evaluates memory, abstract thought, language, sensory-motor integration, imperception, and motor dexterity. Others may select specific instruments to answer a referring psychiatrist’s question.
CHOOSING AN INSTRUMENT
Medical reference librarians can help research specific instruments and choose useful testing tools. We also recommend Corcoran and Fischer’s Measures for Clinical Practice,8 which provides practical information on administration, advantages, and disadvantages of instruments that:
- are used in clinical practice
- provide data on psychometric properties
- take
- are rapidly scored
- provide information on symptom severity
- can be used to document change.
Table 2
Commonly used checklists/rating scales for adult assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Anger | Anger, Irritability and Assault Questionnaire (AIAQ) |
| Anxiety | |
| Phobias | Fear Questionnaire (FQ) |
| GAD | Beck Anxiety Inventory (BAI) |
| OCD | Yale-Brown Obsessive Compulsive Scale (Y-BOCS) |
| PTSD | Posttraumatic Stress Diagnostic Scale (PDS) |
| Bipolar disorder | Young Mania Rating Scale (YMRS) |
| Depression | Beck Depression Inventory (BDI) |
| Zung Self-rating Depression Scale (SDS) | |
| Eating disorders | Eating Disorders Inventory-2 |
| Family issues | Family Assessment Device (FAD) |
| Impulse control | Barratt Impulsiveness Scale, Version II (BIS-II) |
| Pain | McGill Pain Questionnaire (MPQ) |
| Personality disorders | Millon Clinical Multiaxial Inventory (MCMI-III) |
| Psychosis | Brief Psychiatric Rating Scale (BPRS) |
| Manchester Scale | |
| Sexuality | Sexual Interaction Inventory (SII) |
| Sleep | Sleep Disorder Questionnaire (SDQ) |
| Suicide risk | Beck Scale for Suicide Ideation (BSS) |
| GAD: Generalized anxiety disorder | |
| OCD: Obsessive-compulsive disorder | |
| PTSD: Posttraumatic stress disorder | |
Common checklists/rating scales for geriatric assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Cognitive status | Mini-Mental State Examination (MMSE) |
| Neurobehavioral Cognitive Status Examination (Cognistat) | |
| Dementia Rating Scale | |
| Repeatable Battery for the Assessment of Neuropsychological Status (RBANS) | |
| Caregiver stress | Caregiver’s Burden Scale (CBS) |
| Death concerns | Concern About Death-Dying (CADD) and Coping (C) checklist |
| Depression | Geriatric Depression Scale (GDS) |
Common checklists/rating scales for child and adolescent assessment
| Disorder/target symptom | Commonly used scales |
|---|---|
| Anxiety | Multidimensional Anxiety Scale for Children (MASC) |
| Assertiveness | Assertiveness Scale for Adolescents (ASA) |
| Conduct | Child Behavior Checklist (CBCL) |
| Depression | Children’s Depression Self Rating Scale (CDRS) |
| Drug/alcohol risk | CAGE Questionnaire |
| Michigan Alcohol Screening Test (MAST) | |
| Impulsivity | Impulsivity Scale (IS) |
| PTSD | Child Report of Posttraumatic Symptoms (CROPS) |
| Reaction to divorce | Children’s Belief About Parental Divorce Scale (CBAPS) |
| Self-esteem | Rosenberg Self-Esteem Scale (RSE) |
| Suicide risk | Multi-Attitude Suicide Tendency Scale (MAST) |
| Test anxiety | Children’s Cognitive Assessment Questionnaire (CCAQ) |
| PTSD: Posttraumatic stress disorder | |
IN-OFFICE TESTING VS REFERRAL
You could use in-office testing to diagnose Mr. A’s anxiety symptoms and provide documentation to his managed care company. For Miss B’s memory problems, we recommend referral for psychological testing.
Mr. A completes the 21-item Beck Anxiety Inventory (BAI) in your office. You select the BAI because it is psychometrically sound, brief (about 10 minutes to complete and score), and easily understood. Results can be readily used for feedback to patients or third-party payers.
Mr. A’s score of 19 is consistent with GAD and justifies a medication trial. The BAI provides information about his experience of anxiety (subjective vs. somatic) that can guide psychotherapy. You plan to repeat the BAI over time to monitor treatment.
Miss B would benefit from referral to a neuropsychologist, as screening tools do not reliably differentiate among the dementias. The neuropsychologist will likely use all or part of the Halstead-Reitan Neuropsychological Test Battery to localize any ischemic-related brain injury and clarify the diagnosis. This test also can provide data to stage her dementia and help you and her family with care decisions.
PSYCHOLOGIST REFERRAL
When referring patients for psychological testing, we recommend that you tell the psychologist what information you need and let him or her select the tests. Relying on their expertise can save time and yield a report that targets the referral question.
Three cases follow that illustrate types of referral questions doctoral-level psychologists can help answer with appropriately chosen tests:
WHAT EXPLAINS TREATMENT RESISTANCE?
Mr. C, age 43, presents with mixed anxiety and depression. He complains of insomnia, fatigue, tightness in the chest, and trembling hands. You give him the Beck Depression Inventory (BDI) and Beck Anxiety Inventory (BAI), which show mild depression/anxiety. You prescribe fluoxetine, 20 mg/d, and 8 weeks later his symptoms are unchanged. The patient is demanding, critical, and has a pattern of interpersonal difficulty. You suspect a personality disorder is complicating treatment.
In this case, the MMPI-2 and MCMI-III would be useful to clarify diagnosis. The MMPI-2 gauges anxiety (state anxiety, phobias, social anxiety and posttraumatic stress disorder), and depression. The MCMI-III was developed to assess axis II diagnoses and has scales to assess each personality disorder. These tests provide information about psychological-mindedness, treatment resistance, and characteristics that can guide psychotherapy.
DRUGS, PSYCHOSIS, OR BIPOLAR DISORDER?
Mr. D’s parents report that their 20-year-old is isolating himself in his room, is not sleeping, and has grandiose beliefs of special powers and knowledge. He has no psychiatric history. Because these symptoms could suggest numerous psychopathologies, you would like help with the differential diagnosis.
Mr. D’s symptoms could suggest drug abuse, schizophrenia, psychotic depression, or bipolar disorder. The psychologist might use the MMPI-2 to assess drug abuse, depression, mania, and psychosis. The relative elevation of each scale could be clinically useful; if scales gauging psychosis and depression are both elevated, psychotic depression is likely, whereas an elevation chiefly on the mania scale would point to bipolar disorder.
The Rorschach test could assess psychotic process. The MMPI-2 could be repeated in a few months to gauge treatment response.
IS THIS EARLY ALZHEIMER’S DISEASE?
Mr. E, age 78, presents with mild memory and word-finding deficits and complains of fatigue, loss of appetite, and anhedonia. Physical exam and lab tests are unremarkable, and you suspect early Alzheimer’s dementia and depression. You wish to confirm the diagnosis to decide whether to start a cholinesterase inhibitor, antidepressant, or other medication. You also wish to document change over time.
An in-office depression checklist would be appropriate for Mr. E. The 30-item, self-rated Geriatric Depression Scale is psychometrically sound and can be completed in 15 to 20 minutes.
Referral is recommended for dementia screening with an tool such as the Neurobehavioral Cognitive Status Examination (Cognistat) or Repeatable Battery for the Assessment of Neuropsychological Status (RBANS). The Mini-Mental State Examination (MMSE) is used for in-office screening of cognitive deficits but lacks sensitivity to detect mild decline. Cognistat or RBANS are less influenced by the patient’s education level and are more sensitive than the MMSE to early dementia.
All three instruments are brief enough to repeat as needed to document change.
Related resources
- Corcoran K, Fischer J. Measures for clinical practice, vols. 1 and 2. New York: Free Press; 2000.
- Maruish ME (ed). The use of psychological testing for treatment planning and outcomes assessment. Mahwah, NJ: Lawrence Erlbaum Associates; 1999.
- Rush AJ, Pincus HJ, First MB, et al. (eds). Handbook of psychiatric measures. Washington, DC: American Psychiatric Association; 2000.
- American Psychological Association. FAQ/Finding information about psychological tests. http://www.apa.org/science/faq-findtests.html.
- Fluoxetine • Prozac
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Maruish ME (ed) The use of psychological testing for treatment planning and outcomes assessment. Mahwah, NJ: Lawrence Erlbaum Associates; 1999.
2. Sajatovic M, Ramirez LF. Rating scales in mental health. Hudson, OH: Lexi-Comp; 2003.
3. Aiken LR. Assessment of adult personality. New York: Springer; 1997.
4. Rush AJ, Pincus HJ, First MB, et al (eds). Handbook of Psychiatric Measures. Washington, DC: American Psychiatric Association; 2000.
5. Belar CD. Psychological assessment in capitated care. In: Butcher JN (ed). Personality assessment in managed care: Using the MMPI-2 in treatment planning. New York: Oxford Press; 1997;13:80.-
6. Adams RL, Culbertson JL. Personality assessment: adults and children. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;702:21.-
7. Swanda RM, Haaland KY, La Rue A. Clinical neuropsychology and intellectual assessment of adults. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;689:701.-
8. Corcoran K, Fischer J. Measures for clinical practice, vols 1 and 2. New York: Free Press; 2000.
9. Blacker D. Psychiatric rating scales. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;755-83.
1. Maruish ME (ed) The use of psychological testing for treatment planning and outcomes assessment. Mahwah, NJ: Lawrence Erlbaum Associates; 1999.
2. Sajatovic M, Ramirez LF. Rating scales in mental health. Hudson, OH: Lexi-Comp; 2003.
3. Aiken LR. Assessment of adult personality. New York: Springer; 1997.
4. Rush AJ, Pincus HJ, First MB, et al (eds). Handbook of Psychiatric Measures. Washington, DC: American Psychiatric Association; 2000.
5. Belar CD. Psychological assessment in capitated care. In: Butcher JN (ed). Personality assessment in managed care: Using the MMPI-2 in treatment planning. New York: Oxford Press; 1997;13:80.-
6. Adams RL, Culbertson JL. Personality assessment: adults and children. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;702:21.-
7. Swanda RM, Haaland KY, La Rue A. Clinical neuropsychology and intellectual assessment of adults. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;689:701.-
8. Corcoran K, Fischer J. Measures for clinical practice, vols 1 and 2. New York: Free Press; 2000.
9. Blacker D. Psychiatric rating scales. In: Sadock BJ, Sadock VA (eds). Comprehensive textbook of psychiatry, vol. 1 (7th ed). Baltimore: Lippincott Williams & Wilkins; 2000;755-83.
Perimenopausal depression? Ask how she’s sleeping
Trying to treat depression or anxiety in a midlife woman without asking how she’s sleeping may doom your treatment plan. Asking about sleep addresses issues that affect her quality of life and can provide valuable insight into effective interventions.
Psychiatric, psychosocial, and medical problems can disturb sleep during perimenopause.1 To help you diagnose and treat both mood disorders and insomnia, this article:
- describes how irregular hormone levels and psychosocial changes are linked to perimenopausal mood and sleep disorders
- offers evidence-based strategies for hormone replacement therapy (HRT), antidepressants, hypnotics, and psychotherapy.
DEPRESSION AND INSOMNIA AT MIDLIFE
Sixty-five percent of women seeking outpatient treatment for depression report disturbed sleep.2 Even mild anxiety and depression can undermine sleep quality, whereas insomnia can precede other symptoms of an evolving major depression.
Depressive disorders affect up to 29% of perimenopausal women (depending on the assessment tool used), compared with 8% to 12% of premenopausal women. Menopausal symptoms—hot flashes, poor sleep, memory problems—and not using HRT are associated with depression.3
Causes of midlife depression. Gonadal hormone changes have been implicated as a cause of increased depression in midlife women; declines in serum estradiol and testosterone are inversely associated with depression.4 The natural menopause transition (perimenopause) begins during the mid-40s, persists to the early 50s, and lasts an average 2 to 9 years. Estradiol produced by the ovary becomes erratic then decreases. Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) serum levels increase, then plateau and serve as laboratory markers of menopause.5
Sociodemographic factors also may contribute to depression, anxiety, and insomnia. A midlife woman may experience role transitions—such as children leaving home and aging parents needing care. She may be adapting to her or her spouse’s retirement or to the loss of her partner by divorce or death. She may be grappling with her own aging and questions about mortality and life purpose.
In the workup, consider medical factors that may worsen sleep problems, such as hot flashes, sleep apnea, thyroid disease, urinary frequency, chronic pain, restless leg syndrome, caffeine use, sedentary lifestyle, and primary insomnia. Some women lose sleep from a bed partner’s snoring or movement (“spousal arousal”). Stimulating drugs such as theophylline can also play a role.
SLEEP CHANGES AT PERIMENOPAUSE
Sleep changes are among the most common physical and psychological experiences healthy women describe during perimenopause:
- 100 consecutive women surveyed at a menopause clinic reported fatigue (91%), hot flashes (80%), insomnia and early awakenings (77%), and depression (65%).6
- Sleep problems were reported by >50% of 203 women interviewed for the Decisions at Menopause Study (DAMES).7
- Difficulty sleeping across 2 weeks was reported by 38% of a multiethnic population of 12,603 women ages 40 to 55.8
Sleep problems occur more often during perimenopause than earlier in life. In a clinic sample of 521 women, Owens et al1 found insomnia in 33% to 36% of those in premenopause and in 44% to 61% of women during perimenopause. In the total sample of healthy middle-aged women, 42% had sleep complaints, including:
- initial insomnia: 49%
- middle insomnia: 92%
- early morning awakening: 59%.
No association? Individuals experience sleep quality subjectively, and these assessments may not match those obtained objectively. The Wisconsin Sleep Cohort Study,9 for example, found no association between menopause and diminished sleep quality in polysomnographic studies of 589 community-dwelling women. Even so, the peri- and postmenopausal women in the study reported less sleep satisfaction than premenopausal women did.
Most clinicians agree that a woman’s subjective experience of sleep is clinically relevant. Thus, rule out underlying sleep disorders before you attribute a midlife woman’s depressive signs and symptoms primarily to menopause.10
Treatment. Combination therapy may be useful, depending on the patient’s psychiatric and medical comorbidities (Algorithm).
TREATING PERIMENOPAUSAL DEPRESSION
HRT. Before the Women’s Health Initiative (WHI),10 guidelines recommended HRT for a first depressive episode during perimenopause and antidepressants for severe depressive symptoms and for women with a history of depression.11 This practice changed when the WHI found risks of thromboembolism, breast cancer, stroke, and coronary artery disease that increased over time with HRT.
HRT remains a short-term treatment option but is no longer considered the first or only approach to mood symptoms at perimenopause. Discuss with your patient potential benefits of short-term HRT for a first episode of depression—especially if she has vasomotor symptoms—versus potential risks.
Antidepressants can improve perimenopausal depression, but few studies have tested these agents’ effects on sleep. To reduce treatment-associated insomnia:
- select a relatively sedating antidepressant such as mirtazapine
- accept some insomnia for 3 to 4 weeks, until a stimulating antidepressant has had a full ffect on mood and its associated side effects would be expected to resolve
- or augment the antidepressant with a hypnotic such as zolpidem, zaleplon, eszopiclone, or trazodone.
When choosing therapy, consider patient factors and insomnia severity. For example, mirtazapine is typically associated with weight gain, so consider other options for overweight patients. Those with severe insomnia may prefer not to wait 3 to 4 weeks for improved sleep. With hypnotics, consider cost, any coexisting chemical dependency, and potential for morning hangover.
Psychotherapy can help perimenopausal patients accept aging, evaluate relationships, and examine their roles in the lives of more-dependent parents and less-dependent children.
HOT FLASHES AND INSOMNIA
Persistent hot flashes that disturb sleep may cause depression.12 They can wake a perimenopausal woman repeatedly (Figure 1). The awakenings may be brief—90% last <3 minutes—but a severely affected woman can lose an hour of sleep in a night.13 Even after a hot flash resolves, other factors such as anxiety may keep her awake.
Up to 85% of perimenopausal women experience hot flashes, especially during the first year after menses cease. Hot flashes persist for 5 years after menopause in 25% of women and indefinitely in a minority (Box ).14
Estrogen deficiency is thought to cause hot flashes via decreased serotonin synthesis and up-regulated 5HT2A receptors—the mediators of heat loss. As a result, a woman’s thermoregulatory zone narrows during perimenopause, reducing her tolerance for core body temperature changes. The thermoregulatory nucleus resides in the medial preoptic area of the anterior hypothalamus.
A hot flash begins with facial warmth when core temperatures exceed the thermoregulatory line. Heat spreads to the chest, often accompanied by flushing, diaphoresis, and headache. A woman may feel agitated, irritable, and distressed.
CNS noradrenergic activity may initiate hot flashes. Freedman et al13 compared the effects of IV clonidine (an alpha2 adrenergic agonist) plus yohimbine (an alpha2 adrenergic antagonist) or placebo in menopausal women with or without vasomotor symptoms. Among 9 symptomatic women, 6 experienced hot flashes when given yohimbine, and none did with placebo. No hot flashes occurred in asymptomatic women. Clonidine increased the duration of peripheral heating needed to trigger a hot flash and reduced the number of hot flashes in symptomatic women, compared with baseline.
Risk factors for nocturnal hot flashes include surgical menopause, Caucasian versus Asian ethnicity, lack of exercise, and nicotine use.8 Women suffering anxiety and stress also are at increased risk.15
HOT-FLASH THERAPIES
Placebo-controlled trials of hot flash therapies have found efficacies from 85% for HRT to 25% for placebo, vitamin E, black cohosh, soy, and behavioral therapy (Figure 2).16 Most trials were not designed to test the link between hot flashes and sleep, and many enrolled cancer patients not experiencing natural menopause. With the 25% placeboresponse rate, some therapies’ efficacy is unclear.
HRT can reduce nocturnal hot flash frequency. In a polysomnographic study,17 21 postmenopausal women received 6 months of conjugated estrogens, 0.625 mg/d, with medroxyprogesterone, 5 mg/d, or micronized progesterone, 200 mg/d. Sleep efficiency improved by 8% in women receiving micronized progesterone but was unchanged with medroxyprogesterone. Even so, both groups reported improved sleep quality and duration, with decreased awakenings.
The Wisconsin Sleep Cohort Study9 found that HRT was not associated with improved sleep, as measured by polysomnography. Even so, the women in that study noted subjective sleep improvement with HRT.
Antidepressants. Venlafaxine, 75 mg/d, and fluoxetine, 20 mg/d, have shown benefit in reducing hot flashes,18 presumably by increasing CNS serotonin. As mentioned, however, many antidepressants can cause insomnia, and few studies have examined this problem.
Gabapentin has been effective for patients with hot flashes.19 This agent, which increases GABA levels and may modestly increase slow-wave sleep—can improve conditions that disrupt sleep, including restless legs syndrome and chronic pain. It is well-tolerated, even at 900 mg/d, and is more-sedating than most serotonergic antidepressants.
Hypnotics. Surprisingly little evidence addresses hypnotics’ role in managing insomnia caused by hot flashes. No data have been published on the role of benzodiazepines or the benzodiazepine receptor agonists (zolpidem, zaleplon, and eszopiclone). In my experience, benzodiazepine receptor agonists improve sleep quality compromised by multiple factors, including hot flashes.
Soy and black cohosh. Isoflavones in soy may be estrogen receptor modulators. Twelve randomized, controlled trials of soy or soy extracts have shown a modest benefit for hot flashes.20
Black cohosh extracts, 8 mg/d, were given to 80 postmenopausal women in a randomized, double-blind, placebo-controlled trial (RCT). Hot flashes in those receiving black cohosh decreased from 4.9 to 0.7 daily, compared with reductions of 5.2 to 3.2 in women receiving estrogen and 5.1 to 3.1 in those receiving placebo.21 As a result, the National Institutes of Health is funding a 12-month, RCT to determine whether black cohosh reduces hot flash frequency and intensity.
Alternative agents are widely used and warrant study. Those shown to be safe can be used alone or with other therapies, but advise the patient that these agents may not be effective. Relaxation and exercise may decrease hot flashes,22 although some outcomes have been similar to a placebo response.
SLEEP APNEA AT PERIMENOPAUSE
Obstructive sleep apnea (OSA), although more common in men than women, appears to increase during perimenopause. Women with untreated OSA are twice as likely as men to be treated for depression, less likely to report excessive daytime sleepiness and snoring, and more likely to present with depression, anxiety, and morning headache.
Bixler et al23 interviewed 12,219 women and 4,364 men ages 20 to 100 and conducted 1-night sleep studies in 1,000 women and 741 men. OSA rates were 3.9% in men, 0.6% in premenopausal women, 2.7% in postmenopausal women not taking HRT, and 0.5% in postmenopausal women taking HRT.
The risk of sleep-disordered breathing is lower during early menopause and peaks at approximately age 65. Declining hormones likely play a role; progesterone increases ventilatory drive, and estrogen increases ventilatory centers’ sensitivity to progesterone’s stimulant effect. In small studies, exogenous progesterone has shown a slight effect in improving OSA.24
OSA’s transient, repetitive upper airway collapse increases inspiratory effort and may cause hypoxemia. Repeated arousals can lead to prolonged awakenings and unrefreshing sleep. Snoring and increased body mass index are strongly associated factors, although the Wisconsin Sleep Cohort Study10 showed an increase in sleep apnea in perimenopausal women that was unrelated to increased body mass index.
Obesity may not explain the increase in obstructive sleep apnea at perimenopause (Figure 3),28 although body fat distribution does change with aging. Women at perimenopause are likely to develop abdominal weight distribution.
Figure 3 Increased OSA in postmenopausal women is unrelated to obesity (BMI >32)
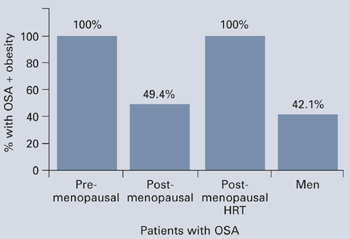
Obstructive sleep apnea (OSA) in 1,000 women and 741 men was associated exclusively with obesity in premenopausal women and postmenopausal women using HRT, but nearly one-half of the postmenopausal women with OSA were not obese.
Source: Adapted from reference 23.Treatment. In the Sleep Heart Health Study25 of 2,852 women age 50 or older, HRT users had one-half the apnea prevalence of nonusers (6% vs 14%). HRT users were less likely to awaken at night and to get inadequate sleep. Snoring rates were similar (25% for HRT users, 23% for nonusers).
Nasal continuous positive airway pressure (CPAP) is the mainstay of apnea treatment, although some women appear to have difficulty accepting CPAP.26 Weight loss and moderate exercise can help manage weight and improve sleep quality by increasing slow-wave sleep. Regular exercise also may improve depressed mood.
Related resources
- American College of Obstetricians and Gynecologists. www.acog.org
- The North American Menopause Society. www.menopause.org
Drug brand names
- Conjugated estrogens • Premarin
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Medroxyprogesterone • Provera
- Micronized progesterone • Prometrium
- Mirtazapine • Remeron
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosures
Dr. Krahn reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Owens JF, Matthews KA. Sleep disturbance in healthy middle-aged women. Maturitas 1998;30:41-50.
2. Perlis ML, Giles DE, Buysse DJ, et al. Self-reported sleep disturbance as a prodromal symptom in recurrent depression. J Affect Disord 1997;42(2-3):209-12.
3. Bromberger JT, Assmann SF, Avis NE, et al. Persistent mood symptoms in a multiethnic community cohort of pre- and perimenopausal women. Am J Epidemiol 2003;158:347-56.
4. Sherwin BB. Changes in sexual behavior as a function of plasma sex steroid levels in post-menopausal women. Maturitas 1985;7(3):225-33.
5. Brizendine L. Minding menopause. Psychotropics vs estrogen: What you need to know now. Current Psychiatry 2003;2(10):12-31.
6. Anderson E, Hamburger S, Liu JH, Rebar RW. Characteristics of menopausal women seeking assistance. Am J Obstet Gynecol 1987;156(2):428-33.
7. Obermeyer CM, Reynolds RF, Price K, Abraham A. Therapeutic decisions for menopause: results of the DAMES project in central Massachusetts. Menopause 2004;11(4):456-65.
8. Kravitz HM, Ganz PA, Bromberger J, et al. Sleep difficulty in women at midlife: a community survey of sleep and the menopausal transition. Menopause 2003;10(1):19-28.
9. Young T, Rabago D, Zgierska A, et al. Objective and subjective sleep quality in premenopausal, perimenopausal, and postmenopausal women in the Wisconsin Sleep Cohort Study. Sleep 2003;26(6):667-72.
10. Wassertheil-Smoller S, Shumaker S, Ockene J, et al. Depression and cardiovascular sequelae in postmenopausal women. Arch Intern Med 2004;164:289-98.
11. Altshuler LL, Cohen LS, Moline ML, et al. The expert consensus guideline series. Treatment of depression in women. Postgrad Med 2001;March:1-107.
12. Krystal AD. Insomnia in women. Clin Cornerstone 2003;5(3):41-50.
13. Freedman RR. Physiology of hot flashes. Am J Hum Biol 2001;13(4):453-64.
14. Freedman RR, Woodward S, Sabharwal SC. A2-adrenergic mechanism in menopausal hot flushes. Obstet Gynecol 1990;76(4):573-8.
15. Miller AG, Li RM. Measuring hot flashes: summary of a NIH workshop. Mayo Clin Proc 2004;79:777-81.
16. Joffe H, Soares CN, Cohen LS. Assessment and treatment of hot flushes and menopausal mood disturbance. Psychiatr Clin North Am 2003 Sep;26(3):563-80.
17. Montplaisir J, Lorrain J, Denesle R, Petit D. Sleep in menopause: differential effects of two forms of hormone replacement therapy. Menopause 2001;8(1):10-16.
18. Loprinzi CL, Sloan JA, Perez EA, et al. Phase III evaluation of fluoxetine for treatment of hot flashes. J Clin Oncol 2002;20:1578-83.
19. Guttoso T, Jr, Kurlan R, McDermott MP, Kieburtz K. Gabapentin’s effects on hot flashes in postmenopausal women: a randomized controlled trial. Obstet Gynecol 2003;101:337-45.
20. Kessel B, Kronenberg F. The role of complementary and alternative medicine in management of menopausal symptoms. Endocrinol Metab Clin North Am 2004;33:717-39.
21. National Institutes of Health. National Center for Complementary and Alternative Medicine. Office of Dietary Supplements. Questions and answers about black cohosh and the symptoms of menopause. Available at: http://ods.od.nih.gov/factsheets/blackcohosh.asp. Accessed May 9, 2005.
22. Ivarsson T, Spetz AC, Hammar M. Physical exercise and vasomotor symptoms in postmenopausal women. Mauritas 1998;29:139-46.
23. Bixler EO, Vgontzas AN, Lin HM, et al. Prevalence of sleep-disordered breathing in women: effects of gender. Am J Respir Crit Care Med 2001;163:608-13.
24. Block AJ, Wynne JW, Boysen PG, et al. Menopause, medroxyprogesterone and breathing during sleep. Am J Med 1981;70:506-10.
25. Shahar E, Redline S, Young T, et al. Hormone replacement therapy and sleep-disordered breathing. Am J Respir Crit Care Med 2003;167:1186-92.
26. McArdle N, Devereux G, Heidarnejad H, et al. Long-term use of CPAP therapy for sleep apnea/hypopnea syndrome. Am J Respir Crit. Care Med 1999;159:1108-14.
Trying to treat depression or anxiety in a midlife woman without asking how she’s sleeping may doom your treatment plan. Asking about sleep addresses issues that affect her quality of life and can provide valuable insight into effective interventions.
Psychiatric, psychosocial, and medical problems can disturb sleep during perimenopause.1 To help you diagnose and treat both mood disorders and insomnia, this article:
- describes how irregular hormone levels and psychosocial changes are linked to perimenopausal mood and sleep disorders
- offers evidence-based strategies for hormone replacement therapy (HRT), antidepressants, hypnotics, and psychotherapy.
DEPRESSION AND INSOMNIA AT MIDLIFE
Sixty-five percent of women seeking outpatient treatment for depression report disturbed sleep.2 Even mild anxiety and depression can undermine sleep quality, whereas insomnia can precede other symptoms of an evolving major depression.
Depressive disorders affect up to 29% of perimenopausal women (depending on the assessment tool used), compared with 8% to 12% of premenopausal women. Menopausal symptoms—hot flashes, poor sleep, memory problems—and not using HRT are associated with depression.3
Causes of midlife depression. Gonadal hormone changes have been implicated as a cause of increased depression in midlife women; declines in serum estradiol and testosterone are inversely associated with depression.4 The natural menopause transition (perimenopause) begins during the mid-40s, persists to the early 50s, and lasts an average 2 to 9 years. Estradiol produced by the ovary becomes erratic then decreases. Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) serum levels increase, then plateau and serve as laboratory markers of menopause.5
Sociodemographic factors also may contribute to depression, anxiety, and insomnia. A midlife woman may experience role transitions—such as children leaving home and aging parents needing care. She may be adapting to her or her spouse’s retirement or to the loss of her partner by divorce or death. She may be grappling with her own aging and questions about mortality and life purpose.
In the workup, consider medical factors that may worsen sleep problems, such as hot flashes, sleep apnea, thyroid disease, urinary frequency, chronic pain, restless leg syndrome, caffeine use, sedentary lifestyle, and primary insomnia. Some women lose sleep from a bed partner’s snoring or movement (“spousal arousal”). Stimulating drugs such as theophylline can also play a role.
SLEEP CHANGES AT PERIMENOPAUSE
Sleep changes are among the most common physical and psychological experiences healthy women describe during perimenopause:
- 100 consecutive women surveyed at a menopause clinic reported fatigue (91%), hot flashes (80%), insomnia and early awakenings (77%), and depression (65%).6
- Sleep problems were reported by >50% of 203 women interviewed for the Decisions at Menopause Study (DAMES).7
- Difficulty sleeping across 2 weeks was reported by 38% of a multiethnic population of 12,603 women ages 40 to 55.8
Sleep problems occur more often during perimenopause than earlier in life. In a clinic sample of 521 women, Owens et al1 found insomnia in 33% to 36% of those in premenopause and in 44% to 61% of women during perimenopause. In the total sample of healthy middle-aged women, 42% had sleep complaints, including:
- initial insomnia: 49%
- middle insomnia: 92%
- early morning awakening: 59%.
No association? Individuals experience sleep quality subjectively, and these assessments may not match those obtained objectively. The Wisconsin Sleep Cohort Study,9 for example, found no association between menopause and diminished sleep quality in polysomnographic studies of 589 community-dwelling women. Even so, the peri- and postmenopausal women in the study reported less sleep satisfaction than premenopausal women did.
Most clinicians agree that a woman’s subjective experience of sleep is clinically relevant. Thus, rule out underlying sleep disorders before you attribute a midlife woman’s depressive signs and symptoms primarily to menopause.10
Treatment. Combination therapy may be useful, depending on the patient’s psychiatric and medical comorbidities (Algorithm).
TREATING PERIMENOPAUSAL DEPRESSION
HRT. Before the Women’s Health Initiative (WHI),10 guidelines recommended HRT for a first depressive episode during perimenopause and antidepressants for severe depressive symptoms and for women with a history of depression.11 This practice changed when the WHI found risks of thromboembolism, breast cancer, stroke, and coronary artery disease that increased over time with HRT.
HRT remains a short-term treatment option but is no longer considered the first or only approach to mood symptoms at perimenopause. Discuss with your patient potential benefits of short-term HRT for a first episode of depression—especially if she has vasomotor symptoms—versus potential risks.
Antidepressants can improve perimenopausal depression, but few studies have tested these agents’ effects on sleep. To reduce treatment-associated insomnia:
- select a relatively sedating antidepressant such as mirtazapine
- accept some insomnia for 3 to 4 weeks, until a stimulating antidepressant has had a full ffect on mood and its associated side effects would be expected to resolve
- or augment the antidepressant with a hypnotic such as zolpidem, zaleplon, eszopiclone, or trazodone.
When choosing therapy, consider patient factors and insomnia severity. For example, mirtazapine is typically associated with weight gain, so consider other options for overweight patients. Those with severe insomnia may prefer not to wait 3 to 4 weeks for improved sleep. With hypnotics, consider cost, any coexisting chemical dependency, and potential for morning hangover.
Psychotherapy can help perimenopausal patients accept aging, evaluate relationships, and examine their roles in the lives of more-dependent parents and less-dependent children.
HOT FLASHES AND INSOMNIA
Persistent hot flashes that disturb sleep may cause depression.12 They can wake a perimenopausal woman repeatedly (Figure 1). The awakenings may be brief—90% last <3 minutes—but a severely affected woman can lose an hour of sleep in a night.13 Even after a hot flash resolves, other factors such as anxiety may keep her awake.
Up to 85% of perimenopausal women experience hot flashes, especially during the first year after menses cease. Hot flashes persist for 5 years after menopause in 25% of women and indefinitely in a minority (Box ).14
Estrogen deficiency is thought to cause hot flashes via decreased serotonin synthesis and up-regulated 5HT2A receptors—the mediators of heat loss. As a result, a woman’s thermoregulatory zone narrows during perimenopause, reducing her tolerance for core body temperature changes. The thermoregulatory nucleus resides in the medial preoptic area of the anterior hypothalamus.
A hot flash begins with facial warmth when core temperatures exceed the thermoregulatory line. Heat spreads to the chest, often accompanied by flushing, diaphoresis, and headache. A woman may feel agitated, irritable, and distressed.
CNS noradrenergic activity may initiate hot flashes. Freedman et al13 compared the effects of IV clonidine (an alpha2 adrenergic agonist) plus yohimbine (an alpha2 adrenergic antagonist) or placebo in menopausal women with or without vasomotor symptoms. Among 9 symptomatic women, 6 experienced hot flashes when given yohimbine, and none did with placebo. No hot flashes occurred in asymptomatic women. Clonidine increased the duration of peripheral heating needed to trigger a hot flash and reduced the number of hot flashes in symptomatic women, compared with baseline.
Risk factors for nocturnal hot flashes include surgical menopause, Caucasian versus Asian ethnicity, lack of exercise, and nicotine use.8 Women suffering anxiety and stress also are at increased risk.15
HOT-FLASH THERAPIES
Placebo-controlled trials of hot flash therapies have found efficacies from 85% for HRT to 25% for placebo, vitamin E, black cohosh, soy, and behavioral therapy (Figure 2).16 Most trials were not designed to test the link between hot flashes and sleep, and many enrolled cancer patients not experiencing natural menopause. With the 25% placeboresponse rate, some therapies’ efficacy is unclear.
HRT can reduce nocturnal hot flash frequency. In a polysomnographic study,17 21 postmenopausal women received 6 months of conjugated estrogens, 0.625 mg/d, with medroxyprogesterone, 5 mg/d, or micronized progesterone, 200 mg/d. Sleep efficiency improved by 8% in women receiving micronized progesterone but was unchanged with medroxyprogesterone. Even so, both groups reported improved sleep quality and duration, with decreased awakenings.
The Wisconsin Sleep Cohort Study9 found that HRT was not associated with improved sleep, as measured by polysomnography. Even so, the women in that study noted subjective sleep improvement with HRT.
Antidepressants. Venlafaxine, 75 mg/d, and fluoxetine, 20 mg/d, have shown benefit in reducing hot flashes,18 presumably by increasing CNS serotonin. As mentioned, however, many antidepressants can cause insomnia, and few studies have examined this problem.
Gabapentin has been effective for patients with hot flashes.19 This agent, which increases GABA levels and may modestly increase slow-wave sleep—can improve conditions that disrupt sleep, including restless legs syndrome and chronic pain. It is well-tolerated, even at 900 mg/d, and is more-sedating than most serotonergic antidepressants.
Hypnotics. Surprisingly little evidence addresses hypnotics’ role in managing insomnia caused by hot flashes. No data have been published on the role of benzodiazepines or the benzodiazepine receptor agonists (zolpidem, zaleplon, and eszopiclone). In my experience, benzodiazepine receptor agonists improve sleep quality compromised by multiple factors, including hot flashes.
Soy and black cohosh. Isoflavones in soy may be estrogen receptor modulators. Twelve randomized, controlled trials of soy or soy extracts have shown a modest benefit for hot flashes.20
Black cohosh extracts, 8 mg/d, were given to 80 postmenopausal women in a randomized, double-blind, placebo-controlled trial (RCT). Hot flashes in those receiving black cohosh decreased from 4.9 to 0.7 daily, compared with reductions of 5.2 to 3.2 in women receiving estrogen and 5.1 to 3.1 in those receiving placebo.21 As a result, the National Institutes of Health is funding a 12-month, RCT to determine whether black cohosh reduces hot flash frequency and intensity.
Alternative agents are widely used and warrant study. Those shown to be safe can be used alone or with other therapies, but advise the patient that these agents may not be effective. Relaxation and exercise may decrease hot flashes,22 although some outcomes have been similar to a placebo response.
SLEEP APNEA AT PERIMENOPAUSE
Obstructive sleep apnea (OSA), although more common in men than women, appears to increase during perimenopause. Women with untreated OSA are twice as likely as men to be treated for depression, less likely to report excessive daytime sleepiness and snoring, and more likely to present with depression, anxiety, and morning headache.
Bixler et al23 interviewed 12,219 women and 4,364 men ages 20 to 100 and conducted 1-night sleep studies in 1,000 women and 741 men. OSA rates were 3.9% in men, 0.6% in premenopausal women, 2.7% in postmenopausal women not taking HRT, and 0.5% in postmenopausal women taking HRT.
The risk of sleep-disordered breathing is lower during early menopause and peaks at approximately age 65. Declining hormones likely play a role; progesterone increases ventilatory drive, and estrogen increases ventilatory centers’ sensitivity to progesterone’s stimulant effect. In small studies, exogenous progesterone has shown a slight effect in improving OSA.24
OSA’s transient, repetitive upper airway collapse increases inspiratory effort and may cause hypoxemia. Repeated arousals can lead to prolonged awakenings and unrefreshing sleep. Snoring and increased body mass index are strongly associated factors, although the Wisconsin Sleep Cohort Study10 showed an increase in sleep apnea in perimenopausal women that was unrelated to increased body mass index.
Obesity may not explain the increase in obstructive sleep apnea at perimenopause (Figure 3),28 although body fat distribution does change with aging. Women at perimenopause are likely to develop abdominal weight distribution.
Figure 3 Increased OSA in postmenopausal women is unrelated to obesity (BMI >32)
Obstructive sleep apnea (OSA) in 1,000 women and 741 men was associated exclusively with obesity in premenopausal women and postmenopausal women using HRT, but nearly one-half of the postmenopausal women with OSA were not obese.
Source: Adapted from reference 23.Treatment. In the Sleep Heart Health Study25 of 2,852 women age 50 or older, HRT users had one-half the apnea prevalence of nonusers (6% vs 14%). HRT users were less likely to awaken at night and to get inadequate sleep. Snoring rates were similar (25% for HRT users, 23% for nonusers).
Nasal continuous positive airway pressure (CPAP) is the mainstay of apnea treatment, although some women appear to have difficulty accepting CPAP.26 Weight loss and moderate exercise can help manage weight and improve sleep quality by increasing slow-wave sleep. Regular exercise also may improve depressed mood.
Related resources
- American College of Obstetricians and Gynecologists. www.acog.org
- The North American Menopause Society. www.menopause.org
Drug brand names
- Conjugated estrogens • Premarin
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Medroxyprogesterone • Provera
- Micronized progesterone • Prometrium
- Mirtazapine • Remeron
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosures
Dr. Krahn reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Trying to treat depression or anxiety in a midlife woman without asking how she’s sleeping may doom your treatment plan. Asking about sleep addresses issues that affect her quality of life and can provide valuable insight into effective interventions.
Psychiatric, psychosocial, and medical problems can disturb sleep during perimenopause.1 To help you diagnose and treat both mood disorders and insomnia, this article:
- describes how irregular hormone levels and psychosocial changes are linked to perimenopausal mood and sleep disorders
- offers evidence-based strategies for hormone replacement therapy (HRT), antidepressants, hypnotics, and psychotherapy.
DEPRESSION AND INSOMNIA AT MIDLIFE
Sixty-five percent of women seeking outpatient treatment for depression report disturbed sleep.2 Even mild anxiety and depression can undermine sleep quality, whereas insomnia can precede other symptoms of an evolving major depression.
Depressive disorders affect up to 29% of perimenopausal women (depending on the assessment tool used), compared with 8% to 12% of premenopausal women. Menopausal symptoms—hot flashes, poor sleep, memory problems—and not using HRT are associated with depression.3
Causes of midlife depression. Gonadal hormone changes have been implicated as a cause of increased depression in midlife women; declines in serum estradiol and testosterone are inversely associated with depression.4 The natural menopause transition (perimenopause) begins during the mid-40s, persists to the early 50s, and lasts an average 2 to 9 years. Estradiol produced by the ovary becomes erratic then decreases. Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) serum levels increase, then plateau and serve as laboratory markers of menopause.5
Sociodemographic factors also may contribute to depression, anxiety, and insomnia. A midlife woman may experience role transitions—such as children leaving home and aging parents needing care. She may be adapting to her or her spouse’s retirement or to the loss of her partner by divorce or death. She may be grappling with her own aging and questions about mortality and life purpose.
In the workup, consider medical factors that may worsen sleep problems, such as hot flashes, sleep apnea, thyroid disease, urinary frequency, chronic pain, restless leg syndrome, caffeine use, sedentary lifestyle, and primary insomnia. Some women lose sleep from a bed partner’s snoring or movement (“spousal arousal”). Stimulating drugs such as theophylline can also play a role.
SLEEP CHANGES AT PERIMENOPAUSE
Sleep changes are among the most common physical and psychological experiences healthy women describe during perimenopause:
- 100 consecutive women surveyed at a menopause clinic reported fatigue (91%), hot flashes (80%), insomnia and early awakenings (77%), and depression (65%).6
- Sleep problems were reported by >50% of 203 women interviewed for the Decisions at Menopause Study (DAMES).7
- Difficulty sleeping across 2 weeks was reported by 38% of a multiethnic population of 12,603 women ages 40 to 55.8
Sleep problems occur more often during perimenopause than earlier in life. In a clinic sample of 521 women, Owens et al1 found insomnia in 33% to 36% of those in premenopause and in 44% to 61% of women during perimenopause. In the total sample of healthy middle-aged women, 42% had sleep complaints, including:
- initial insomnia: 49%
- middle insomnia: 92%
- early morning awakening: 59%.
No association? Individuals experience sleep quality subjectively, and these assessments may not match those obtained objectively. The Wisconsin Sleep Cohort Study,9 for example, found no association between menopause and diminished sleep quality in polysomnographic studies of 589 community-dwelling women. Even so, the peri- and postmenopausal women in the study reported less sleep satisfaction than premenopausal women did.
Most clinicians agree that a woman’s subjective experience of sleep is clinically relevant. Thus, rule out underlying sleep disorders before you attribute a midlife woman’s depressive signs and symptoms primarily to menopause.10
Treatment. Combination therapy may be useful, depending on the patient’s psychiatric and medical comorbidities (Algorithm).
TREATING PERIMENOPAUSAL DEPRESSION
HRT. Before the Women’s Health Initiative (WHI),10 guidelines recommended HRT for a first depressive episode during perimenopause and antidepressants for severe depressive symptoms and for women with a history of depression.11 This practice changed when the WHI found risks of thromboembolism, breast cancer, stroke, and coronary artery disease that increased over time with HRT.
HRT remains a short-term treatment option but is no longer considered the first or only approach to mood symptoms at perimenopause. Discuss with your patient potential benefits of short-term HRT for a first episode of depression—especially if she has vasomotor symptoms—versus potential risks.
Antidepressants can improve perimenopausal depression, but few studies have tested these agents’ effects on sleep. To reduce treatment-associated insomnia:
- select a relatively sedating antidepressant such as mirtazapine
- accept some insomnia for 3 to 4 weeks, until a stimulating antidepressant has had a full ffect on mood and its associated side effects would be expected to resolve
- or augment the antidepressant with a hypnotic such as zolpidem, zaleplon, eszopiclone, or trazodone.
When choosing therapy, consider patient factors and insomnia severity. For example, mirtazapine is typically associated with weight gain, so consider other options for overweight patients. Those with severe insomnia may prefer not to wait 3 to 4 weeks for improved sleep. With hypnotics, consider cost, any coexisting chemical dependency, and potential for morning hangover.
Psychotherapy can help perimenopausal patients accept aging, evaluate relationships, and examine their roles in the lives of more-dependent parents and less-dependent children.
HOT FLASHES AND INSOMNIA
Persistent hot flashes that disturb sleep may cause depression.12 They can wake a perimenopausal woman repeatedly (Figure 1). The awakenings may be brief—90% last <3 minutes—but a severely affected woman can lose an hour of sleep in a night.13 Even after a hot flash resolves, other factors such as anxiety may keep her awake.
Up to 85% of perimenopausal women experience hot flashes, especially during the first year after menses cease. Hot flashes persist for 5 years after menopause in 25% of women and indefinitely in a minority (Box ).14
Estrogen deficiency is thought to cause hot flashes via decreased serotonin synthesis and up-regulated 5HT2A receptors—the mediators of heat loss. As a result, a woman’s thermoregulatory zone narrows during perimenopause, reducing her tolerance for core body temperature changes. The thermoregulatory nucleus resides in the medial preoptic area of the anterior hypothalamus.
A hot flash begins with facial warmth when core temperatures exceed the thermoregulatory line. Heat spreads to the chest, often accompanied by flushing, diaphoresis, and headache. A woman may feel agitated, irritable, and distressed.
CNS noradrenergic activity may initiate hot flashes. Freedman et al13 compared the effects of IV clonidine (an alpha2 adrenergic agonist) plus yohimbine (an alpha2 adrenergic antagonist) or placebo in menopausal women with or without vasomotor symptoms. Among 9 symptomatic women, 6 experienced hot flashes when given yohimbine, and none did with placebo. No hot flashes occurred in asymptomatic women. Clonidine increased the duration of peripheral heating needed to trigger a hot flash and reduced the number of hot flashes in symptomatic women, compared with baseline.
Risk factors for nocturnal hot flashes include surgical menopause, Caucasian versus Asian ethnicity, lack of exercise, and nicotine use.8 Women suffering anxiety and stress also are at increased risk.15
HOT-FLASH THERAPIES
Placebo-controlled trials of hot flash therapies have found efficacies from 85% for HRT to 25% for placebo, vitamin E, black cohosh, soy, and behavioral therapy (Figure 2).16 Most trials were not designed to test the link between hot flashes and sleep, and many enrolled cancer patients not experiencing natural menopause. With the 25% placeboresponse rate, some therapies’ efficacy is unclear.
HRT can reduce nocturnal hot flash frequency. In a polysomnographic study,17 21 postmenopausal women received 6 months of conjugated estrogens, 0.625 mg/d, with medroxyprogesterone, 5 mg/d, or micronized progesterone, 200 mg/d. Sleep efficiency improved by 8% in women receiving micronized progesterone but was unchanged with medroxyprogesterone. Even so, both groups reported improved sleep quality and duration, with decreased awakenings.
The Wisconsin Sleep Cohort Study9 found that HRT was not associated with improved sleep, as measured by polysomnography. Even so, the women in that study noted subjective sleep improvement with HRT.
Antidepressants. Venlafaxine, 75 mg/d, and fluoxetine, 20 mg/d, have shown benefit in reducing hot flashes,18 presumably by increasing CNS serotonin. As mentioned, however, many antidepressants can cause insomnia, and few studies have examined this problem.
Gabapentin has been effective for patients with hot flashes.19 This agent, which increases GABA levels and may modestly increase slow-wave sleep—can improve conditions that disrupt sleep, including restless legs syndrome and chronic pain. It is well-tolerated, even at 900 mg/d, and is more-sedating than most serotonergic antidepressants.
Hypnotics. Surprisingly little evidence addresses hypnotics’ role in managing insomnia caused by hot flashes. No data have been published on the role of benzodiazepines or the benzodiazepine receptor agonists (zolpidem, zaleplon, and eszopiclone). In my experience, benzodiazepine receptor agonists improve sleep quality compromised by multiple factors, including hot flashes.
Soy and black cohosh. Isoflavones in soy may be estrogen receptor modulators. Twelve randomized, controlled trials of soy or soy extracts have shown a modest benefit for hot flashes.20
Black cohosh extracts, 8 mg/d, were given to 80 postmenopausal women in a randomized, double-blind, placebo-controlled trial (RCT). Hot flashes in those receiving black cohosh decreased from 4.9 to 0.7 daily, compared with reductions of 5.2 to 3.2 in women receiving estrogen and 5.1 to 3.1 in those receiving placebo.21 As a result, the National Institutes of Health is funding a 12-month, RCT to determine whether black cohosh reduces hot flash frequency and intensity.
Alternative agents are widely used and warrant study. Those shown to be safe can be used alone or with other therapies, but advise the patient that these agents may not be effective. Relaxation and exercise may decrease hot flashes,22 although some outcomes have been similar to a placebo response.
SLEEP APNEA AT PERIMENOPAUSE
Obstructive sleep apnea (OSA), although more common in men than women, appears to increase during perimenopause. Women with untreated OSA are twice as likely as men to be treated for depression, less likely to report excessive daytime sleepiness and snoring, and more likely to present with depression, anxiety, and morning headache.
Bixler et al23 interviewed 12,219 women and 4,364 men ages 20 to 100 and conducted 1-night sleep studies in 1,000 women and 741 men. OSA rates were 3.9% in men, 0.6% in premenopausal women, 2.7% in postmenopausal women not taking HRT, and 0.5% in postmenopausal women taking HRT.
The risk of sleep-disordered breathing is lower during early menopause and peaks at approximately age 65. Declining hormones likely play a role; progesterone increases ventilatory drive, and estrogen increases ventilatory centers’ sensitivity to progesterone’s stimulant effect. In small studies, exogenous progesterone has shown a slight effect in improving OSA.24
OSA’s transient, repetitive upper airway collapse increases inspiratory effort and may cause hypoxemia. Repeated arousals can lead to prolonged awakenings and unrefreshing sleep. Snoring and increased body mass index are strongly associated factors, although the Wisconsin Sleep Cohort Study10 showed an increase in sleep apnea in perimenopausal women that was unrelated to increased body mass index.
Obesity may not explain the increase in obstructive sleep apnea at perimenopause (Figure 3),28 although body fat distribution does change with aging. Women at perimenopause are likely to develop abdominal weight distribution.
Figure 3 Increased OSA in postmenopausal women is unrelated to obesity (BMI >32)
Obstructive sleep apnea (OSA) in 1,000 women and 741 men was associated exclusively with obesity in premenopausal women and postmenopausal women using HRT, but nearly one-half of the postmenopausal women with OSA were not obese.
Source: Adapted from reference 23.Treatment. In the Sleep Heart Health Study25 of 2,852 women age 50 or older, HRT users had one-half the apnea prevalence of nonusers (6% vs 14%). HRT users were less likely to awaken at night and to get inadequate sleep. Snoring rates were similar (25% for HRT users, 23% for nonusers).
Nasal continuous positive airway pressure (CPAP) is the mainstay of apnea treatment, although some women appear to have difficulty accepting CPAP.26 Weight loss and moderate exercise can help manage weight and improve sleep quality by increasing slow-wave sleep. Regular exercise also may improve depressed mood.
Related resources
- American College of Obstetricians and Gynecologists. www.acog.org
- The North American Menopause Society. www.menopause.org
Drug brand names
- Conjugated estrogens • Premarin
- Eszopiclone • Lunesta
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Medroxyprogesterone • Provera
- Micronized progesterone • Prometrium
- Mirtazapine • Remeron
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosures
Dr. Krahn reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Owens JF, Matthews KA. Sleep disturbance in healthy middle-aged women. Maturitas 1998;30:41-50.
2. Perlis ML, Giles DE, Buysse DJ, et al. Self-reported sleep disturbance as a prodromal symptom in recurrent depression. J Affect Disord 1997;42(2-3):209-12.
3. Bromberger JT, Assmann SF, Avis NE, et al. Persistent mood symptoms in a multiethnic community cohort of pre- and perimenopausal women. Am J Epidemiol 2003;158:347-56.
4. Sherwin BB. Changes in sexual behavior as a function of plasma sex steroid levels in post-menopausal women. Maturitas 1985;7(3):225-33.
5. Brizendine L. Minding menopause. Psychotropics vs estrogen: What you need to know now. Current Psychiatry 2003;2(10):12-31.
6. Anderson E, Hamburger S, Liu JH, Rebar RW. Characteristics of menopausal women seeking assistance. Am J Obstet Gynecol 1987;156(2):428-33.
7. Obermeyer CM, Reynolds RF, Price K, Abraham A. Therapeutic decisions for menopause: results of the DAMES project in central Massachusetts. Menopause 2004;11(4):456-65.
8. Kravitz HM, Ganz PA, Bromberger J, et al. Sleep difficulty in women at midlife: a community survey of sleep and the menopausal transition. Menopause 2003;10(1):19-28.
9. Young T, Rabago D, Zgierska A, et al. Objective and subjective sleep quality in premenopausal, perimenopausal, and postmenopausal women in the Wisconsin Sleep Cohort Study. Sleep 2003;26(6):667-72.
10. Wassertheil-Smoller S, Shumaker S, Ockene J, et al. Depression and cardiovascular sequelae in postmenopausal women. Arch Intern Med 2004;164:289-98.
11. Altshuler LL, Cohen LS, Moline ML, et al. The expert consensus guideline series. Treatment of depression in women. Postgrad Med 2001;March:1-107.
12. Krystal AD. Insomnia in women. Clin Cornerstone 2003;5(3):41-50.
13. Freedman RR. Physiology of hot flashes. Am J Hum Biol 2001;13(4):453-64.
14. Freedman RR, Woodward S, Sabharwal SC. A2-adrenergic mechanism in menopausal hot flushes. Obstet Gynecol 1990;76(4):573-8.
15. Miller AG, Li RM. Measuring hot flashes: summary of a NIH workshop. Mayo Clin Proc 2004;79:777-81.
16. Joffe H, Soares CN, Cohen LS. Assessment and treatment of hot flushes and menopausal mood disturbance. Psychiatr Clin North Am 2003 Sep;26(3):563-80.
17. Montplaisir J, Lorrain J, Denesle R, Petit D. Sleep in menopause: differential effects of two forms of hormone replacement therapy. Menopause 2001;8(1):10-16.
18. Loprinzi CL, Sloan JA, Perez EA, et al. Phase III evaluation of fluoxetine for treatment of hot flashes. J Clin Oncol 2002;20:1578-83.
19. Guttoso T, Jr, Kurlan R, McDermott MP, Kieburtz K. Gabapentin’s effects on hot flashes in postmenopausal women: a randomized controlled trial. Obstet Gynecol 2003;101:337-45.
20. Kessel B, Kronenberg F. The role of complementary and alternative medicine in management of menopausal symptoms. Endocrinol Metab Clin North Am 2004;33:717-39.
21. National Institutes of Health. National Center for Complementary and Alternative Medicine. Office of Dietary Supplements. Questions and answers about black cohosh and the symptoms of menopause. Available at: http://ods.od.nih.gov/factsheets/blackcohosh.asp. Accessed May 9, 2005.
22. Ivarsson T, Spetz AC, Hammar M. Physical exercise and vasomotor symptoms in postmenopausal women. Mauritas 1998;29:139-46.
23. Bixler EO, Vgontzas AN, Lin HM, et al. Prevalence of sleep-disordered breathing in women: effects of gender. Am J Respir Crit Care Med 2001;163:608-13.
24. Block AJ, Wynne JW, Boysen PG, et al. Menopause, medroxyprogesterone and breathing during sleep. Am J Med 1981;70:506-10.
25. Shahar E, Redline S, Young T, et al. Hormone replacement therapy and sleep-disordered breathing. Am J Respir Crit Care Med 2003;167:1186-92.
26. McArdle N, Devereux G, Heidarnejad H, et al. Long-term use of CPAP therapy for sleep apnea/hypopnea syndrome. Am J Respir Crit. Care Med 1999;159:1108-14.
1. Owens JF, Matthews KA. Sleep disturbance in healthy middle-aged women. Maturitas 1998;30:41-50.
2. Perlis ML, Giles DE, Buysse DJ, et al. Self-reported sleep disturbance as a prodromal symptom in recurrent depression. J Affect Disord 1997;42(2-3):209-12.
3. Bromberger JT, Assmann SF, Avis NE, et al. Persistent mood symptoms in a multiethnic community cohort of pre- and perimenopausal women. Am J Epidemiol 2003;158:347-56.
4. Sherwin BB. Changes in sexual behavior as a function of plasma sex steroid levels in post-menopausal women. Maturitas 1985;7(3):225-33.
5. Brizendine L. Minding menopause. Psychotropics vs estrogen: What you need to know now. Current Psychiatry 2003;2(10):12-31.
6. Anderson E, Hamburger S, Liu JH, Rebar RW. Characteristics of menopausal women seeking assistance. Am J Obstet Gynecol 1987;156(2):428-33.
7. Obermeyer CM, Reynolds RF, Price K, Abraham A. Therapeutic decisions for menopause: results of the DAMES project in central Massachusetts. Menopause 2004;11(4):456-65.
8. Kravitz HM, Ganz PA, Bromberger J, et al. Sleep difficulty in women at midlife: a community survey of sleep and the menopausal transition. Menopause 2003;10(1):19-28.
9. Young T, Rabago D, Zgierska A, et al. Objective and subjective sleep quality in premenopausal, perimenopausal, and postmenopausal women in the Wisconsin Sleep Cohort Study. Sleep 2003;26(6):667-72.
10. Wassertheil-Smoller S, Shumaker S, Ockene J, et al. Depression and cardiovascular sequelae in postmenopausal women. Arch Intern Med 2004;164:289-98.
11. Altshuler LL, Cohen LS, Moline ML, et al. The expert consensus guideline series. Treatment of depression in women. Postgrad Med 2001;March:1-107.
12. Krystal AD. Insomnia in women. Clin Cornerstone 2003;5(3):41-50.
13. Freedman RR. Physiology of hot flashes. Am J Hum Biol 2001;13(4):453-64.
14. Freedman RR, Woodward S, Sabharwal SC. A2-adrenergic mechanism in menopausal hot flushes. Obstet Gynecol 1990;76(4):573-8.
15. Miller AG, Li RM. Measuring hot flashes: summary of a NIH workshop. Mayo Clin Proc 2004;79:777-81.
16. Joffe H, Soares CN, Cohen LS. Assessment and treatment of hot flushes and menopausal mood disturbance. Psychiatr Clin North Am 2003 Sep;26(3):563-80.
17. Montplaisir J, Lorrain J, Denesle R, Petit D. Sleep in menopause: differential effects of two forms of hormone replacement therapy. Menopause 2001;8(1):10-16.
18. Loprinzi CL, Sloan JA, Perez EA, et al. Phase III evaluation of fluoxetine for treatment of hot flashes. J Clin Oncol 2002;20:1578-83.
19. Guttoso T, Jr, Kurlan R, McDermott MP, Kieburtz K. Gabapentin’s effects on hot flashes in postmenopausal women: a randomized controlled trial. Obstet Gynecol 2003;101:337-45.
20. Kessel B, Kronenberg F. The role of complementary and alternative medicine in management of menopausal symptoms. Endocrinol Metab Clin North Am 2004;33:717-39.
21. National Institutes of Health. National Center for Complementary and Alternative Medicine. Office of Dietary Supplements. Questions and answers about black cohosh and the symptoms of menopause. Available at: http://ods.od.nih.gov/factsheets/blackcohosh.asp. Accessed May 9, 2005.
22. Ivarsson T, Spetz AC, Hammar M. Physical exercise and vasomotor symptoms in postmenopausal women. Mauritas 1998;29:139-46.
23. Bixler EO, Vgontzas AN, Lin HM, et al. Prevalence of sleep-disordered breathing in women: effects of gender. Am J Respir Crit Care Med 2001;163:608-13.
24. Block AJ, Wynne JW, Boysen PG, et al. Menopause, medroxyprogesterone and breathing during sleep. Am J Med 1981;70:506-10.
25. Shahar E, Redline S, Young T, et al. Hormone replacement therapy and sleep-disordered breathing. Am J Respir Crit Care Med 2003;167:1186-92.
26. McArdle N, Devereux G, Heidarnejad H, et al. Long-term use of CPAP therapy for sleep apnea/hypopnea syndrome. Am J Respir Crit. Care Med 1999;159:1108-14.