User login
Panic attacks: Help sufferers recover with cognitive-behavioral therapy
With panic attacks, alarming physiologic symptoms mount swiftly—tachycardia, chest pain, sweating, trembling, smothering or choking, dizziness, fear of losing control or going crazy—even fear of dying.1 Patients constantly fear the next attack, worry about its consequences, and change their behaviors to avoid or withdraw from anxiety-provoking situations.
To relieve their suffering, cognitive-behavioral therapy (CBT) may offer benefits you would not realize with medication alone. CBT can:
- improve long-term patient outcomes
- enhance medication management
- boost treatment response when medication alone is inadequate
- ease drug discontinuation.2
Whether you or a CBT-trained psychotherapist guides the sessions, you can achieve optimal results for your patients with panic disorder.
How Effective is CBT?
Panic disorder is chronic, often disabling, and characterized by spontaneous, unpredictable panic attacks (Boxes 1 and 23-11). When treated with CBT, about three-quarters of patients become panic-free and maintain treatment gains at follow-up, and one-half become both panic-free and free of excess anxiety.9
Typical therapy is 12 individual, once-weekly visits for psycho-education, relaxation, and breathing training; cognitive restructuring; and exposure therapies.
Briefer protocols, “reduced therapist contact,”12 and group therapy13 also can help patients and in some studies have been as beneficial as 12 weeks of individual therapy. Although trained psychotherapists have higher success rates than nonbehaviorists when treating panic patients, nonbehaviorists also can provide effective therapy after relatively brief training.14
American Psychiatric Association15 treatment guidelines recommend medications—such as selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), and benzodiazepines—as well as CBT as first-line therapies for panic disorder. Other treatment guidelines concur16 and note that CBT is more cost-effective than medications.
In comparison studies, CBT has been at least as effective for panic symptoms as SSRIs,17,18 TCAs,19 and alprazolam.20 Antidepressants are the preferred drug for panic disorder16 because they lack benzodiazepines’ dependence and abuse potential.
Providing medication during CBT may maintain patients’ therapeutic gains better than CBT alone if the medication is continued after CBT is completed. Interestingly, patients who use benzodiazepines during CBT may have higher relapse rates than those who do not use benzodiazepines, particularly when the benzodiazepines are withdrawn.9
CBT produces improvement rates similar to those of pharmacologic treatment at one-quarter to one-half the cost in the first year. Patients also appear to have better clinical outcomes if they receive CBT while SSRIs or benzodiazepines are being discontinued, compared with simply stopping the medications.8
Panic attacks typically begin between ages 10 and 40. The cause is unknown, but evidence points to multiple factors, including heredity, neurobiology, provocations, and psychological conditioning (Box 2).3-9 prevalence is approximately 5%,10 and about three-fourths of panic disorder patients are female.11
Comorbidity. Up to 50% of persons with panic disorder also experience agoraphobia.1 Depression, other anxiety disorders, and substance abuse may complicate the clinical picture.
BIOLOGICAL THEORIES
Genetics. About 10% of persons who experience panic attacks have first-degree relatives with panic disorder. Twin studies suggest heritability of up to 43%
Neurobiology. Anxiety responses appear to be organized at different neuroanatomic levels:
- automatic responses by periaqueductal grey matter or locus coeruleus
- practiced responses by the amygdala and septohippocampal regions
- cognitively complex responses by higher cortical regions.
The hypothalamus mediates neurohormonal responses. Panic disorder patients’ response to SSRIs, tricyclic antidepressants, and benzodiazepines suggest a link with neurotransmitters serotonin, norepinephrine, and GABA. Adenosine, cannabinoids, neuropeptides, hormones, neurotrophins, cytokines, and cellular mediators may also be involved.
Provocation. Panic disorder may have a physiologic mechanism. When exposed in the laboratory to panicogenic substances (such as carbon dioxide, sodium lactate, yohimbine, and caffeine), persons with panic disorders experience greater numbers of panic attacks than do those without panic disorders. These laboratory-induced panic attacks resemble real attacks, and anti-panic medications block the induced panic attacks.
PSYCHOLOGICAL THEORIES
The cognitive-behavioral model postulates that panic disorder patients:
- have a predisposed vulnerability to respond with physiologic arousal to negative stressors
- tend to see anxiety symptoms as harmful
- have negative and catastrophizing cognitions about those symptoms.
With conditioning, patients associate early physiologic arousal with other panic symptoms as the arousal progresses. Ultimately, they become hypervigilant for symptoms and develop a learned escalation of anxiety and apprehension (with accompanying negative cognitions) when the early symptoms re-occur.
Source: References 3-9
CBT Candidates
To diagnose panic disorder, conduct a thorough psychiatric evaluation that includes assessing for comorbid mental and substance use disorders. The history and physical exam are essential to rule out medical causes of the patient’s symptoms, such as heart disease causing dizziness or palpitations. Asking patients to keep panic attack records can help you identify panic symptoms’ frequency and triggers.9
An assessment tool such as the Albany Panic and Phobia Questionnaire (Figure) can be a useful starting point. It has 27 items and three subscales to quantify a patient’s fear of agoraphobic situations, social phobia situations, and situations that produce bodily sensations (interoceptive symptoms). Items on the interoceptive subscale include activities such as exercising vigorously, ingesting caffeine, and experiencing intense emotion.21 Using the Anxiety Sensitivity Index is another assessment option.22
Not all patients with panic attacks respond well to CBT; predictors of poor response in clinical trials have included:
- severe baseline panic symptoms, personality disorders, and possibly depressed mood
- marital dissatisfaction
- low motivation for treatment.2,9
Figure Patient assessment: Albany Panic and Phobia Questionnaire
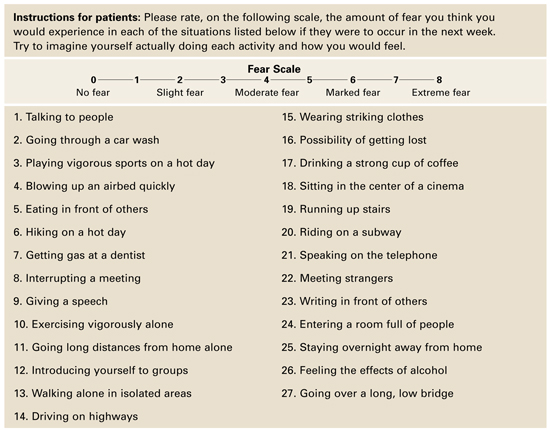
Source: Reprinted with permission from Rapee RM, Craske MG, Barlow DH. Assessment instrument for panic disorder that includes fear of sensation-producing activities: The Albany Panic and Phobia Questionnaire. Anxiety 1995;1:114-22. Copyright 1995, Wiley-Liss, Inc.
Cognitive Therapies for Panic Disorder
Psychoeducation. Begin by defining and explaining anxiety, panic attacks, panic disorder, and any comorbid psychopathology the patient may have (agoraphobia, depression). Explain panic symptoms as physiologic and psychological responses to stressors.
Address the patient’s fears that anxiety’s physiologic symptoms represent a serious or undiagnosed medical disorder or that a panic attack could cause serious harm. Assign self-help and reading materials to reinforce this discussion (see Related resources). Finally, explain the rationale for using CBT to treat panic symptoms.
Use cognitive restructuring to address faulty or irrational information-processing patterns that underlie pathologic anxiety. Identify automatic thought patterns (such as catastrophizing, overgeneralization, all-or-nothing thinking, and personalization), then provide a careful “reality check,” in which you systematically substitute a more-rational thought process.
Have the patient keep a self-monitoring diary to help you assess thought patterns and re-direct irrational thoughts. A diary may identify anxiety-provoking scenarios on which to focus therapy. Encourage patients to document anxiety events using the “triple-column” technique:23
- column 1: circumstances of the anxiety or panic
- column 2: their emotional state at the time
- column 3: any thoughts they can identify.
Instruct them to log this data while experiencing symptoms or immediately afterward. Later, during the intervention phase, patients can record how they tried to restructure their thoughts and any consequent mood changes. Review diary entries with them during subsequent sessions.
Exposure Therapy
Graduated exposure and response prevention (ERP) is the core component of CBT for panic disorder. ERP exercises require the patient to confront anxiety-producing stimuli while agreeing not to engage in maladaptive behavior that avoids, prematurely reduces, or prevents the anxiety. The stimuli may be external cues—such as bridges, stores, or heights—or interoceptive cues such as dizziness, tachycardia, or tachypnea.
Creating fear hierarchies. To begin, we recommend that you work with the patient to create lists of all external situations and interoceptive stimuli that cause him or her anxiety. Separating the stimuli into two lists helps patients recognize that their bodily stimuli are at least as important as environmental stimuli in promoting a panic attack.
The patient then rates each stimulus using a Subjective Units of Distress scale (SUDS)—assigning 0 to 100 points from no anxiety to overwhelming anxiety—and ranks items on the lists from mildest to worst anxiety. Instruct patients to rate the distress they would feel if they could not escape from the stimuli.
First experience. After the hierarchies are created, the therapist introduces the patient to exposure therapy by choosing an item that causes mild to moderate anxiety. Starting at this anxiety level, patients are likely to succeed with their first exercise without feeling overwhelmed. The therapist teaches the patient about the process, then begins the exposure by helping the patient create and confront the very scenario (or a representation of that scenario) that causes anxiety.
When working on interoceptive cues, various exercises can be used to reproduce bother-some bodily symptoms, such as:
- running up a flight of stairs or running in place to generate tachycardia
- purposefully hyperventilating to produce lightheadedness.
- spinning in place to create dizziness.
The patient agrees not to actively attempt to escape the scenario but to tolerate and perhaps even focus on the anxiety (Box 3). Using “safety cues”—such as leaning against a wall or keeping eyes closed—is also forbidden. Patients soon see that the anxiety does not last indefinitely but begins to diminish fairly rapidly.
Reaching the goal. After repeated exposure sessions, anxiety associated with a stimulus begins to extinguish. Having experienced the success of tolerating a previously difficult stimuli and feeling much less anxious, the patient is ready to take on increasingly difficult tasks. The therapist also assigns the patient “homework” to practice exposure exercises already mastered during sessions. As exposure therapy progresses, the patient takes a larger role in designing and executing sessions. The goal is for the patient to learn to become his or her own behaviorist and to intervene early when panic symptoms begin.
Resist temptation to rescue patients from their anxiety during exposure sessions, such as by chatting about the weather or current events or providing other distractions. To extinguish the link between the stimuli and anxiety, the patient must experience anxiety all the way through the exercise—preferably giving ongoing Subjective Units of Distress (SUDS) ratings—until symptoms inevitably wane and cease.
Similarly, avoid assigning exposure homework to be done “until you can’t stand it anymore, then take a rest.” Although well-intentioned, allowing the patient to escape the exposure when anxiety peaks increases conditioned anxiety and strongly reinforces avoidance behaviors.
Other Behavioral Techniques
Imaginal exposure sessions can be created using visualizations of feared stimuli, gradually presented as with in vivo exposure. For example, as you recount a target scenario, ask the patient to imagine a progression of events or bodily cues that have led to panic attacks. The patient supplies SUDS ratings and refrains from imagining an avoidance or maladaptive response. You can tape-record the session for homework and assign the patient to listen to it and participate daily.
Imaginal exposure may help treat phobic avoidance (such as agoraphobic symptoms), but study results have been disappointing in panic symptoms.24 However, this approach may help reluctant patients initiate in vivo exposure therapy.
Relaxation training—such as progressive muscle relaxation, visual imagery, or autogenic protocols—has shown mixed results in treating panic.25,26 Relaxation may help patients cope with panic’s physiologic arousal, but it is not suitable as a singular intervention.
Breathing retraining. Because hyperventilation and panic symptoms are related, instruction and practice in slow, diaphragmatic breathing has long been a component of CBT for panic symptoms. Little evidence supports breathing retraining,9 although Meuret et al27,28 have described a respiratory feedback paradigm that may reduce panic symptoms in appropriately selected patients.
Many studies that have assessed breathing retraining as monotherapy for panic have had methodologic flaws.27
Building a Therapeutic Alliance
Successful therapists have been found to use empathic listening more than directives and explanations in the first therapy session.9 They understand the suffering from panic disorder and the value of listening as patients explain their symptoms, thoughts, and feelings. The rapport built during this initial interaction can help sustain motivation as the therapist then takes charge of subsequent sessions.
Among important skills for CBT therapists, Seligman29 includes empathy, caring, warmth, and active listening, as well as the ability to:
- be a teacher, scientist, and co-investigator
- demystify treatment
- engage clients as “active, knowledgeable, and responsible partners” in their therapy.
Finally, although CBT clinicians suggest tasks and interventions for this “shared endeavor,” patients are primarily responsible for change.
- Anxiety Disorders Association of America. www.adaa.org.
- Craske M, Barlow D, Cary NC. Mastery of your anxiety and panic, (3rd ed). Therapist guide and client workbook. Oxford, UK: Oxford University Press; 2000.
- Otto MW. Stopping anxiety medication: panic control therapy for benzodiazepine discontinuation. Therapist guide and patient workbook. Oxford, UK: Oxford University Press; 2004.
- The Panic Center. Patient diaries and other self-help resources. www.paniccenter.net
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association; 1994.
2. Otto MW, Deckersbach T. Cognitive-behavioral therapy for panic disorder: Theory, strategies, and outcome. In: Rosenbaum JF, Pollack M (eds). Panic disorder and its treatment. New York: Marcel Dekker; 1998.
3. Hettema, JM, Neale, MC, Kendler, KS. A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry 2001;158(10):1568-78.
4. Kendler KS, Gardner CO, Prescott CA. Panic syndromes in a population-based sample of male and female twins. Psychol Med 2001;31:989-1000.
5. Sandford JJ, Argyropoulos SV, Nutt DJ. The psychobiology of anxiolytic drugs. Part I: basic neurobiology. Pharmacol Ther 2000;88:197-212.
6. Millan MJ. The neurobiology and control of anxious states. Prog Neurobiol 2003;70:83-244.
7. Sanderson WC, Rego SA. Empirically supported psychological treatment of panic disorder and agoraphobia. Medscape. Available at www.medscape.com/viewprogram/350_pnt. Accessed Nov. 8, 2005.
8. Rayburn NR, Otto MW. Cognitive-behavioral therapy for panic disorder: a review of treatment elements, strategies, and outcomes. CNS Spectr 2003;8(5):356-62.
9. Craske MG, Barlow DH. Panic disorder and agoraphobia. In: Barlow DH (ed). Clinical handbook of psychological disorders: A step-by-step treatment manual. New York: Guilford Press; 2001;1-59.
10. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey replication. Arch Gen Psychiatry 2005;62:593-602.
11. Rapee RM, Barlow DH. Generalized anxiety disorders, panic disorders, and phobias. In: Sutker PB, Adams HE (eds). Comprehensive handbook of psychopathology (3rd ed). New York: Kluwer Academic/Plenum; 2001.
12. Cote G, Gauthier JG, Laberge B, et al. Reduced therapist contact in the cognitive behavioral treatment of panic disorder. Behav Ther 1994;25:123-45.
13. Telch MJ, Lucas JA, Schmidt NB, et al. Group cognitive-behavioral treatment of panic disorder. Behav Res Ther 1993;31:279-28.
14. Welkowitz LA, Papp LA, Cloitre M, et al. Cognitive-behavioral therapy for panic disorder delivered by psychopharmacologically oriented clinicians. J Nerv Ment Dis 1991;179:473-77.
15. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. Washington, DC: American Psychiatric Association; 1998.
16. Royal Australian and New Zealand College of Psychiatrists. Australian and New Zealand clinical practice guidelines for the treatment of panic disorder and agoraphobia. Aust NZ J Psychiatry 2003;37:641-56.Available at: www.ranzcp.org/publicarea/cpg.asp. Accessed Aug. 24, 2005.
17. Black DW, Wesner R, Bowers W, Gabel J. A comparison of fluvoxamine, cognitive therapy, and placebo in the treatment of panic disorder. Arch Gen Psychiatry 1993;31:383-94.
18. Dannon PN, Gon-Usishkin M, Gelbert A, et al. Cognitive behavioral group therapy in panic disorder patients: The efficacy of CBGT versus drug treatment. Ann Clin Psychiatry 2004;16:41-6.
19. Clark DM, Salkovskis PM, Hackmann A, et al. A comparison of cognitive therapy, applied relaxation, and imipramine in the treatment of panic disorder. Br J Psychiatry 1994;164:759-69.
20. Klosko JS, Barlow DH, Tassinari R, Cerny JA. A comparison of alprazolam and behavior therapy in treatment of panic disorder. J Consult Clin Psychol 1990;58:77-84.
21. Rapee RM, Craske MG, Barlow DH. Assessment instrument for panic disorder that includes fear of sensation-producing activities: The Albany Panic and Phobia Questionnaire. Anxiety 1995;1:114-22.
22. Reiss S, Peterson R, Gursky D, McNally R. Anxiety sensitivity, anxiety frequency, and the prediction of fearfulness. Behav Res Ther 1986;24:1-8.
23. Burns DD. Feeling good: The new mood therapy. New York: William Morrow and Co.; 1980.
24. Clum GA, Watkins PL, Borden JW, et al. A comparison of guided imaginal coping and imaginal exposure in the treatment of panic disorder. J Rational-Emotive & Cognitive Behavior Therapy 1993;11(4):179-93.
25. Craske MG, Brown TA, Barlow DH. Behavioral treatment of panic disorder: A two year follow-up. Behav Ther 1991;22:289-304.
26. Ost LG, Westling BE. Applied relaxation vs cognitive behavior therapy in the treatment of panic disorder. Behav Res Ther 1995;33:145-58.
27. Meuret AE, Wilhelm FH, Ritz T, Roth WT. Breathing training for treating panic disorder: useful intervention or impediment. Behav Mod 2003;27(5):731-54.
28. Meuret AE, Wilhelm FH, Roth WT. Respiratory feedback for treating panic disorder. J Clin Psychol 2004;60:197-207.
29. Seligman L. Systems, strategies, and skills of counseling and psychotherapy. Saddle River, NJ: Prentice-Hall; 2001.
With panic attacks, alarming physiologic symptoms mount swiftly—tachycardia, chest pain, sweating, trembling, smothering or choking, dizziness, fear of losing control or going crazy—even fear of dying.1 Patients constantly fear the next attack, worry about its consequences, and change their behaviors to avoid or withdraw from anxiety-provoking situations.
To relieve their suffering, cognitive-behavioral therapy (CBT) may offer benefits you would not realize with medication alone. CBT can:
- improve long-term patient outcomes
- enhance medication management
- boost treatment response when medication alone is inadequate
- ease drug discontinuation.2
Whether you or a CBT-trained psychotherapist guides the sessions, you can achieve optimal results for your patients with panic disorder.
How Effective is CBT?
Panic disorder is chronic, often disabling, and characterized by spontaneous, unpredictable panic attacks (Boxes 1 and 23-11). When treated with CBT, about three-quarters of patients become panic-free and maintain treatment gains at follow-up, and one-half become both panic-free and free of excess anxiety.9
Typical therapy is 12 individual, once-weekly visits for psycho-education, relaxation, and breathing training; cognitive restructuring; and exposure therapies.
Briefer protocols, “reduced therapist contact,”12 and group therapy13 also can help patients and in some studies have been as beneficial as 12 weeks of individual therapy. Although trained psychotherapists have higher success rates than nonbehaviorists when treating panic patients, nonbehaviorists also can provide effective therapy after relatively brief training.14
American Psychiatric Association15 treatment guidelines recommend medications—such as selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), and benzodiazepines—as well as CBT as first-line therapies for panic disorder. Other treatment guidelines concur16 and note that CBT is more cost-effective than medications.
In comparison studies, CBT has been at least as effective for panic symptoms as SSRIs,17,18 TCAs,19 and alprazolam.20 Antidepressants are the preferred drug for panic disorder16 because they lack benzodiazepines’ dependence and abuse potential.
Providing medication during CBT may maintain patients’ therapeutic gains better than CBT alone if the medication is continued after CBT is completed. Interestingly, patients who use benzodiazepines during CBT may have higher relapse rates than those who do not use benzodiazepines, particularly when the benzodiazepines are withdrawn.9
CBT produces improvement rates similar to those of pharmacologic treatment at one-quarter to one-half the cost in the first year. Patients also appear to have better clinical outcomes if they receive CBT while SSRIs or benzodiazepines are being discontinued, compared with simply stopping the medications.8
Panic attacks typically begin between ages 10 and 40. The cause is unknown, but evidence points to multiple factors, including heredity, neurobiology, provocations, and psychological conditioning (Box 2).3-9 prevalence is approximately 5%,10 and about three-fourths of panic disorder patients are female.11
Comorbidity. Up to 50% of persons with panic disorder also experience agoraphobia.1 Depression, other anxiety disorders, and substance abuse may complicate the clinical picture.
BIOLOGICAL THEORIES
Genetics. About 10% of persons who experience panic attacks have first-degree relatives with panic disorder. Twin studies suggest heritability of up to 43%
Neurobiology. Anxiety responses appear to be organized at different neuroanatomic levels:
- automatic responses by periaqueductal grey matter or locus coeruleus
- practiced responses by the amygdala and septohippocampal regions
- cognitively complex responses by higher cortical regions.
The hypothalamus mediates neurohormonal responses. Panic disorder patients’ response to SSRIs, tricyclic antidepressants, and benzodiazepines suggest a link with neurotransmitters serotonin, norepinephrine, and GABA. Adenosine, cannabinoids, neuropeptides, hormones, neurotrophins, cytokines, and cellular mediators may also be involved.
Provocation. Panic disorder may have a physiologic mechanism. When exposed in the laboratory to panicogenic substances (such as carbon dioxide, sodium lactate, yohimbine, and caffeine), persons with panic disorders experience greater numbers of panic attacks than do those without panic disorders. These laboratory-induced panic attacks resemble real attacks, and anti-panic medications block the induced panic attacks.
PSYCHOLOGICAL THEORIES
The cognitive-behavioral model postulates that panic disorder patients:
- have a predisposed vulnerability to respond with physiologic arousal to negative stressors
- tend to see anxiety symptoms as harmful
- have negative and catastrophizing cognitions about those symptoms.
With conditioning, patients associate early physiologic arousal with other panic symptoms as the arousal progresses. Ultimately, they become hypervigilant for symptoms and develop a learned escalation of anxiety and apprehension (with accompanying negative cognitions) when the early symptoms re-occur.
Source: References 3-9
CBT Candidates
To diagnose panic disorder, conduct a thorough psychiatric evaluation that includes assessing for comorbid mental and substance use disorders. The history and physical exam are essential to rule out medical causes of the patient’s symptoms, such as heart disease causing dizziness or palpitations. Asking patients to keep panic attack records can help you identify panic symptoms’ frequency and triggers.9
An assessment tool such as the Albany Panic and Phobia Questionnaire (Figure) can be a useful starting point. It has 27 items and three subscales to quantify a patient’s fear of agoraphobic situations, social phobia situations, and situations that produce bodily sensations (interoceptive symptoms). Items on the interoceptive subscale include activities such as exercising vigorously, ingesting caffeine, and experiencing intense emotion.21 Using the Anxiety Sensitivity Index is another assessment option.22
Not all patients with panic attacks respond well to CBT; predictors of poor response in clinical trials have included:
- severe baseline panic symptoms, personality disorders, and possibly depressed mood
- marital dissatisfaction
- low motivation for treatment.2,9
Figure Patient assessment: Albany Panic and Phobia Questionnaire
Source: Reprinted with permission from Rapee RM, Craske MG, Barlow DH. Assessment instrument for panic disorder that includes fear of sensation-producing activities: The Albany Panic and Phobia Questionnaire. Anxiety 1995;1:114-22. Copyright 1995, Wiley-Liss, Inc.
Cognitive Therapies for Panic Disorder
Psychoeducation. Begin by defining and explaining anxiety, panic attacks, panic disorder, and any comorbid psychopathology the patient may have (agoraphobia, depression). Explain panic symptoms as physiologic and psychological responses to stressors.
Address the patient’s fears that anxiety’s physiologic symptoms represent a serious or undiagnosed medical disorder or that a panic attack could cause serious harm. Assign self-help and reading materials to reinforce this discussion (see Related resources). Finally, explain the rationale for using CBT to treat panic symptoms.
Use cognitive restructuring to address faulty or irrational information-processing patterns that underlie pathologic anxiety. Identify automatic thought patterns (such as catastrophizing, overgeneralization, all-or-nothing thinking, and personalization), then provide a careful “reality check,” in which you systematically substitute a more-rational thought process.
Have the patient keep a self-monitoring diary to help you assess thought patterns and re-direct irrational thoughts. A diary may identify anxiety-provoking scenarios on which to focus therapy. Encourage patients to document anxiety events using the “triple-column” technique:23
- column 1: circumstances of the anxiety or panic
- column 2: their emotional state at the time
- column 3: any thoughts they can identify.
Instruct them to log this data while experiencing symptoms or immediately afterward. Later, during the intervention phase, patients can record how they tried to restructure their thoughts and any consequent mood changes. Review diary entries with them during subsequent sessions.
Exposure Therapy
Graduated exposure and response prevention (ERP) is the core component of CBT for panic disorder. ERP exercises require the patient to confront anxiety-producing stimuli while agreeing not to engage in maladaptive behavior that avoids, prematurely reduces, or prevents the anxiety. The stimuli may be external cues—such as bridges, stores, or heights—or interoceptive cues such as dizziness, tachycardia, or tachypnea.
Creating fear hierarchies. To begin, we recommend that you work with the patient to create lists of all external situations and interoceptive stimuli that cause him or her anxiety. Separating the stimuli into two lists helps patients recognize that their bodily stimuli are at least as important as environmental stimuli in promoting a panic attack.
The patient then rates each stimulus using a Subjective Units of Distress scale (SUDS)—assigning 0 to 100 points from no anxiety to overwhelming anxiety—and ranks items on the lists from mildest to worst anxiety. Instruct patients to rate the distress they would feel if they could not escape from the stimuli.
First experience. After the hierarchies are created, the therapist introduces the patient to exposure therapy by choosing an item that causes mild to moderate anxiety. Starting at this anxiety level, patients are likely to succeed with their first exercise without feeling overwhelmed. The therapist teaches the patient about the process, then begins the exposure by helping the patient create and confront the very scenario (or a representation of that scenario) that causes anxiety.
When working on interoceptive cues, various exercises can be used to reproduce bother-some bodily symptoms, such as:
- running up a flight of stairs or running in place to generate tachycardia
- purposefully hyperventilating to produce lightheadedness.
- spinning in place to create dizziness.
The patient agrees not to actively attempt to escape the scenario but to tolerate and perhaps even focus on the anxiety (Box 3). Using “safety cues”—such as leaning against a wall or keeping eyes closed—is also forbidden. Patients soon see that the anxiety does not last indefinitely but begins to diminish fairly rapidly.
Reaching the goal. After repeated exposure sessions, anxiety associated with a stimulus begins to extinguish. Having experienced the success of tolerating a previously difficult stimuli and feeling much less anxious, the patient is ready to take on increasingly difficult tasks. The therapist also assigns the patient “homework” to practice exposure exercises already mastered during sessions. As exposure therapy progresses, the patient takes a larger role in designing and executing sessions. The goal is for the patient to learn to become his or her own behaviorist and to intervene early when panic symptoms begin.
Resist temptation to rescue patients from their anxiety during exposure sessions, such as by chatting about the weather or current events or providing other distractions. To extinguish the link between the stimuli and anxiety, the patient must experience anxiety all the way through the exercise—preferably giving ongoing Subjective Units of Distress (SUDS) ratings—until symptoms inevitably wane and cease.
Similarly, avoid assigning exposure homework to be done “until you can’t stand it anymore, then take a rest.” Although well-intentioned, allowing the patient to escape the exposure when anxiety peaks increases conditioned anxiety and strongly reinforces avoidance behaviors.
Other Behavioral Techniques
Imaginal exposure sessions can be created using visualizations of feared stimuli, gradually presented as with in vivo exposure. For example, as you recount a target scenario, ask the patient to imagine a progression of events or bodily cues that have led to panic attacks. The patient supplies SUDS ratings and refrains from imagining an avoidance or maladaptive response. You can tape-record the session for homework and assign the patient to listen to it and participate daily.
Imaginal exposure may help treat phobic avoidance (such as agoraphobic symptoms), but study results have been disappointing in panic symptoms.24 However, this approach may help reluctant patients initiate in vivo exposure therapy.
Relaxation training—such as progressive muscle relaxation, visual imagery, or autogenic protocols—has shown mixed results in treating panic.25,26 Relaxation may help patients cope with panic’s physiologic arousal, but it is not suitable as a singular intervention.
Breathing retraining. Because hyperventilation and panic symptoms are related, instruction and practice in slow, diaphragmatic breathing has long been a component of CBT for panic symptoms. Little evidence supports breathing retraining,9 although Meuret et al27,28 have described a respiratory feedback paradigm that may reduce panic symptoms in appropriately selected patients.
Many studies that have assessed breathing retraining as monotherapy for panic have had methodologic flaws.27
Building a Therapeutic Alliance
Successful therapists have been found to use empathic listening more than directives and explanations in the first therapy session.9 They understand the suffering from panic disorder and the value of listening as patients explain their symptoms, thoughts, and feelings. The rapport built during this initial interaction can help sustain motivation as the therapist then takes charge of subsequent sessions.
Among important skills for CBT therapists, Seligman29 includes empathy, caring, warmth, and active listening, as well as the ability to:
- be a teacher, scientist, and co-investigator
- demystify treatment
- engage clients as “active, knowledgeable, and responsible partners” in their therapy.
Finally, although CBT clinicians suggest tasks and interventions for this “shared endeavor,” patients are primarily responsible for change.
- Anxiety Disorders Association of America. www.adaa.org.
- Craske M, Barlow D, Cary NC. Mastery of your anxiety and panic, (3rd ed). Therapist guide and client workbook. Oxford, UK: Oxford University Press; 2000.
- Otto MW. Stopping anxiety medication: panic control therapy for benzodiazepine discontinuation. Therapist guide and patient workbook. Oxford, UK: Oxford University Press; 2004.
- The Panic Center. Patient diaries and other self-help resources. www.paniccenter.net
With panic attacks, alarming physiologic symptoms mount swiftly—tachycardia, chest pain, sweating, trembling, smothering or choking, dizziness, fear of losing control or going crazy—even fear of dying.1 Patients constantly fear the next attack, worry about its consequences, and change their behaviors to avoid or withdraw from anxiety-provoking situations.
To relieve their suffering, cognitive-behavioral therapy (CBT) may offer benefits you would not realize with medication alone. CBT can:
- improve long-term patient outcomes
- enhance medication management
- boost treatment response when medication alone is inadequate
- ease drug discontinuation.2
Whether you or a CBT-trained psychotherapist guides the sessions, you can achieve optimal results for your patients with panic disorder.
How Effective is CBT?
Panic disorder is chronic, often disabling, and characterized by spontaneous, unpredictable panic attacks (Boxes 1 and 23-11). When treated with CBT, about three-quarters of patients become panic-free and maintain treatment gains at follow-up, and one-half become both panic-free and free of excess anxiety.9
Typical therapy is 12 individual, once-weekly visits for psycho-education, relaxation, and breathing training; cognitive restructuring; and exposure therapies.
Briefer protocols, “reduced therapist contact,”12 and group therapy13 also can help patients and in some studies have been as beneficial as 12 weeks of individual therapy. Although trained psychotherapists have higher success rates than nonbehaviorists when treating panic patients, nonbehaviorists also can provide effective therapy after relatively brief training.14
American Psychiatric Association15 treatment guidelines recommend medications—such as selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), and benzodiazepines—as well as CBT as first-line therapies for panic disorder. Other treatment guidelines concur16 and note that CBT is more cost-effective than medications.
In comparison studies, CBT has been at least as effective for panic symptoms as SSRIs,17,18 TCAs,19 and alprazolam.20 Antidepressants are the preferred drug for panic disorder16 because they lack benzodiazepines’ dependence and abuse potential.
Providing medication during CBT may maintain patients’ therapeutic gains better than CBT alone if the medication is continued after CBT is completed. Interestingly, patients who use benzodiazepines during CBT may have higher relapse rates than those who do not use benzodiazepines, particularly when the benzodiazepines are withdrawn.9
CBT produces improvement rates similar to those of pharmacologic treatment at one-quarter to one-half the cost in the first year. Patients also appear to have better clinical outcomes if they receive CBT while SSRIs or benzodiazepines are being discontinued, compared with simply stopping the medications.8
Panic attacks typically begin between ages 10 and 40. The cause is unknown, but evidence points to multiple factors, including heredity, neurobiology, provocations, and psychological conditioning (Box 2).3-9 prevalence is approximately 5%,10 and about three-fourths of panic disorder patients are female.11
Comorbidity. Up to 50% of persons with panic disorder also experience agoraphobia.1 Depression, other anxiety disorders, and substance abuse may complicate the clinical picture.
BIOLOGICAL THEORIES
Genetics. About 10% of persons who experience panic attacks have first-degree relatives with panic disorder. Twin studies suggest heritability of up to 43%
Neurobiology. Anxiety responses appear to be organized at different neuroanatomic levels:
- automatic responses by periaqueductal grey matter or locus coeruleus
- practiced responses by the amygdala and septohippocampal regions
- cognitively complex responses by higher cortical regions.
The hypothalamus mediates neurohormonal responses. Panic disorder patients’ response to SSRIs, tricyclic antidepressants, and benzodiazepines suggest a link with neurotransmitters serotonin, norepinephrine, and GABA. Adenosine, cannabinoids, neuropeptides, hormones, neurotrophins, cytokines, and cellular mediators may also be involved.
Provocation. Panic disorder may have a physiologic mechanism. When exposed in the laboratory to panicogenic substances (such as carbon dioxide, sodium lactate, yohimbine, and caffeine), persons with panic disorders experience greater numbers of panic attacks than do those without panic disorders. These laboratory-induced panic attacks resemble real attacks, and anti-panic medications block the induced panic attacks.
PSYCHOLOGICAL THEORIES
The cognitive-behavioral model postulates that panic disorder patients:
- have a predisposed vulnerability to respond with physiologic arousal to negative stressors
- tend to see anxiety symptoms as harmful
- have negative and catastrophizing cognitions about those symptoms.
With conditioning, patients associate early physiologic arousal with other panic symptoms as the arousal progresses. Ultimately, they become hypervigilant for symptoms and develop a learned escalation of anxiety and apprehension (with accompanying negative cognitions) when the early symptoms re-occur.
Source: References 3-9
CBT Candidates
To diagnose panic disorder, conduct a thorough psychiatric evaluation that includes assessing for comorbid mental and substance use disorders. The history and physical exam are essential to rule out medical causes of the patient’s symptoms, such as heart disease causing dizziness or palpitations. Asking patients to keep panic attack records can help you identify panic symptoms’ frequency and triggers.9
An assessment tool such as the Albany Panic and Phobia Questionnaire (Figure) can be a useful starting point. It has 27 items and three subscales to quantify a patient’s fear of agoraphobic situations, social phobia situations, and situations that produce bodily sensations (interoceptive symptoms). Items on the interoceptive subscale include activities such as exercising vigorously, ingesting caffeine, and experiencing intense emotion.21 Using the Anxiety Sensitivity Index is another assessment option.22
Not all patients with panic attacks respond well to CBT; predictors of poor response in clinical trials have included:
- severe baseline panic symptoms, personality disorders, and possibly depressed mood
- marital dissatisfaction
- low motivation for treatment.2,9
Figure Patient assessment: Albany Panic and Phobia Questionnaire
Source: Reprinted with permission from Rapee RM, Craske MG, Barlow DH. Assessment instrument for panic disorder that includes fear of sensation-producing activities: The Albany Panic and Phobia Questionnaire. Anxiety 1995;1:114-22. Copyright 1995, Wiley-Liss, Inc.
Cognitive Therapies for Panic Disorder
Psychoeducation. Begin by defining and explaining anxiety, panic attacks, panic disorder, and any comorbid psychopathology the patient may have (agoraphobia, depression). Explain panic symptoms as physiologic and psychological responses to stressors.
Address the patient’s fears that anxiety’s physiologic symptoms represent a serious or undiagnosed medical disorder or that a panic attack could cause serious harm. Assign self-help and reading materials to reinforce this discussion (see Related resources). Finally, explain the rationale for using CBT to treat panic symptoms.
Use cognitive restructuring to address faulty or irrational information-processing patterns that underlie pathologic anxiety. Identify automatic thought patterns (such as catastrophizing, overgeneralization, all-or-nothing thinking, and personalization), then provide a careful “reality check,” in which you systematically substitute a more-rational thought process.
Have the patient keep a self-monitoring diary to help you assess thought patterns and re-direct irrational thoughts. A diary may identify anxiety-provoking scenarios on which to focus therapy. Encourage patients to document anxiety events using the “triple-column” technique:23
- column 1: circumstances of the anxiety or panic
- column 2: their emotional state at the time
- column 3: any thoughts they can identify.
Instruct them to log this data while experiencing symptoms or immediately afterward. Later, during the intervention phase, patients can record how they tried to restructure their thoughts and any consequent mood changes. Review diary entries with them during subsequent sessions.
Exposure Therapy
Graduated exposure and response prevention (ERP) is the core component of CBT for panic disorder. ERP exercises require the patient to confront anxiety-producing stimuli while agreeing not to engage in maladaptive behavior that avoids, prematurely reduces, or prevents the anxiety. The stimuli may be external cues—such as bridges, stores, or heights—or interoceptive cues such as dizziness, tachycardia, or tachypnea.
Creating fear hierarchies. To begin, we recommend that you work with the patient to create lists of all external situations and interoceptive stimuli that cause him or her anxiety. Separating the stimuli into two lists helps patients recognize that their bodily stimuli are at least as important as environmental stimuli in promoting a panic attack.
The patient then rates each stimulus using a Subjective Units of Distress scale (SUDS)—assigning 0 to 100 points from no anxiety to overwhelming anxiety—and ranks items on the lists from mildest to worst anxiety. Instruct patients to rate the distress they would feel if they could not escape from the stimuli.
First experience. After the hierarchies are created, the therapist introduces the patient to exposure therapy by choosing an item that causes mild to moderate anxiety. Starting at this anxiety level, patients are likely to succeed with their first exercise without feeling overwhelmed. The therapist teaches the patient about the process, then begins the exposure by helping the patient create and confront the very scenario (or a representation of that scenario) that causes anxiety.
When working on interoceptive cues, various exercises can be used to reproduce bother-some bodily symptoms, such as:
- running up a flight of stairs or running in place to generate tachycardia
- purposefully hyperventilating to produce lightheadedness.
- spinning in place to create dizziness.
The patient agrees not to actively attempt to escape the scenario but to tolerate and perhaps even focus on the anxiety (Box 3). Using “safety cues”—such as leaning against a wall or keeping eyes closed—is also forbidden. Patients soon see that the anxiety does not last indefinitely but begins to diminish fairly rapidly.
Reaching the goal. After repeated exposure sessions, anxiety associated with a stimulus begins to extinguish. Having experienced the success of tolerating a previously difficult stimuli and feeling much less anxious, the patient is ready to take on increasingly difficult tasks. The therapist also assigns the patient “homework” to practice exposure exercises already mastered during sessions. As exposure therapy progresses, the patient takes a larger role in designing and executing sessions. The goal is for the patient to learn to become his or her own behaviorist and to intervene early when panic symptoms begin.
Resist temptation to rescue patients from their anxiety during exposure sessions, such as by chatting about the weather or current events or providing other distractions. To extinguish the link between the stimuli and anxiety, the patient must experience anxiety all the way through the exercise—preferably giving ongoing Subjective Units of Distress (SUDS) ratings—until symptoms inevitably wane and cease.
Similarly, avoid assigning exposure homework to be done “until you can’t stand it anymore, then take a rest.” Although well-intentioned, allowing the patient to escape the exposure when anxiety peaks increases conditioned anxiety and strongly reinforces avoidance behaviors.
Other Behavioral Techniques
Imaginal exposure sessions can be created using visualizations of feared stimuli, gradually presented as with in vivo exposure. For example, as you recount a target scenario, ask the patient to imagine a progression of events or bodily cues that have led to panic attacks. The patient supplies SUDS ratings and refrains from imagining an avoidance or maladaptive response. You can tape-record the session for homework and assign the patient to listen to it and participate daily.
Imaginal exposure may help treat phobic avoidance (such as agoraphobic symptoms), but study results have been disappointing in panic symptoms.24 However, this approach may help reluctant patients initiate in vivo exposure therapy.
Relaxation training—such as progressive muscle relaxation, visual imagery, or autogenic protocols—has shown mixed results in treating panic.25,26 Relaxation may help patients cope with panic’s physiologic arousal, but it is not suitable as a singular intervention.
Breathing retraining. Because hyperventilation and panic symptoms are related, instruction and practice in slow, diaphragmatic breathing has long been a component of CBT for panic symptoms. Little evidence supports breathing retraining,9 although Meuret et al27,28 have described a respiratory feedback paradigm that may reduce panic symptoms in appropriately selected patients.
Many studies that have assessed breathing retraining as monotherapy for panic have had methodologic flaws.27
Building a Therapeutic Alliance
Successful therapists have been found to use empathic listening more than directives and explanations in the first therapy session.9 They understand the suffering from panic disorder and the value of listening as patients explain their symptoms, thoughts, and feelings. The rapport built during this initial interaction can help sustain motivation as the therapist then takes charge of subsequent sessions.
Among important skills for CBT therapists, Seligman29 includes empathy, caring, warmth, and active listening, as well as the ability to:
- be a teacher, scientist, and co-investigator
- demystify treatment
- engage clients as “active, knowledgeable, and responsible partners” in their therapy.
Finally, although CBT clinicians suggest tasks and interventions for this “shared endeavor,” patients are primarily responsible for change.
- Anxiety Disorders Association of America. www.adaa.org.
- Craske M, Barlow D, Cary NC. Mastery of your anxiety and panic, (3rd ed). Therapist guide and client workbook. Oxford, UK: Oxford University Press; 2000.
- Otto MW. Stopping anxiety medication: panic control therapy for benzodiazepine discontinuation. Therapist guide and patient workbook. Oxford, UK: Oxford University Press; 2004.
- The Panic Center. Patient diaries and other self-help resources. www.paniccenter.net
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association; 1994.
2. Otto MW, Deckersbach T. Cognitive-behavioral therapy for panic disorder: Theory, strategies, and outcome. In: Rosenbaum JF, Pollack M (eds). Panic disorder and its treatment. New York: Marcel Dekker; 1998.
3. Hettema, JM, Neale, MC, Kendler, KS. A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry 2001;158(10):1568-78.
4. Kendler KS, Gardner CO, Prescott CA. Panic syndromes in a population-based sample of male and female twins. Psychol Med 2001;31:989-1000.
5. Sandford JJ, Argyropoulos SV, Nutt DJ. The psychobiology of anxiolytic drugs. Part I: basic neurobiology. Pharmacol Ther 2000;88:197-212.
6. Millan MJ. The neurobiology and control of anxious states. Prog Neurobiol 2003;70:83-244.
7. Sanderson WC, Rego SA. Empirically supported psychological treatment of panic disorder and agoraphobia. Medscape. Available at www.medscape.com/viewprogram/350_pnt. Accessed Nov. 8, 2005.
8. Rayburn NR, Otto MW. Cognitive-behavioral therapy for panic disorder: a review of treatment elements, strategies, and outcomes. CNS Spectr 2003;8(5):356-62.
9. Craske MG, Barlow DH. Panic disorder and agoraphobia. In: Barlow DH (ed). Clinical handbook of psychological disorders: A step-by-step treatment manual. New York: Guilford Press; 2001;1-59.
10. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey replication. Arch Gen Psychiatry 2005;62:593-602.
11. Rapee RM, Barlow DH. Generalized anxiety disorders, panic disorders, and phobias. In: Sutker PB, Adams HE (eds). Comprehensive handbook of psychopathology (3rd ed). New York: Kluwer Academic/Plenum; 2001.
12. Cote G, Gauthier JG, Laberge B, et al. Reduced therapist contact in the cognitive behavioral treatment of panic disorder. Behav Ther 1994;25:123-45.
13. Telch MJ, Lucas JA, Schmidt NB, et al. Group cognitive-behavioral treatment of panic disorder. Behav Res Ther 1993;31:279-28.
14. Welkowitz LA, Papp LA, Cloitre M, et al. Cognitive-behavioral therapy for panic disorder delivered by psychopharmacologically oriented clinicians. J Nerv Ment Dis 1991;179:473-77.
15. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. Washington, DC: American Psychiatric Association; 1998.
16. Royal Australian and New Zealand College of Psychiatrists. Australian and New Zealand clinical practice guidelines for the treatment of panic disorder and agoraphobia. Aust NZ J Psychiatry 2003;37:641-56.Available at: www.ranzcp.org/publicarea/cpg.asp. Accessed Aug. 24, 2005.
17. Black DW, Wesner R, Bowers W, Gabel J. A comparison of fluvoxamine, cognitive therapy, and placebo in the treatment of panic disorder. Arch Gen Psychiatry 1993;31:383-94.
18. Dannon PN, Gon-Usishkin M, Gelbert A, et al. Cognitive behavioral group therapy in panic disorder patients: The efficacy of CBGT versus drug treatment. Ann Clin Psychiatry 2004;16:41-6.
19. Clark DM, Salkovskis PM, Hackmann A, et al. A comparison of cognitive therapy, applied relaxation, and imipramine in the treatment of panic disorder. Br J Psychiatry 1994;164:759-69.
20. Klosko JS, Barlow DH, Tassinari R, Cerny JA. A comparison of alprazolam and behavior therapy in treatment of panic disorder. J Consult Clin Psychol 1990;58:77-84.
21. Rapee RM, Craske MG, Barlow DH. Assessment instrument for panic disorder that includes fear of sensation-producing activities: The Albany Panic and Phobia Questionnaire. Anxiety 1995;1:114-22.
22. Reiss S, Peterson R, Gursky D, McNally R. Anxiety sensitivity, anxiety frequency, and the prediction of fearfulness. Behav Res Ther 1986;24:1-8.
23. Burns DD. Feeling good: The new mood therapy. New York: William Morrow and Co.; 1980.
24. Clum GA, Watkins PL, Borden JW, et al. A comparison of guided imaginal coping and imaginal exposure in the treatment of panic disorder. J Rational-Emotive & Cognitive Behavior Therapy 1993;11(4):179-93.
25. Craske MG, Brown TA, Barlow DH. Behavioral treatment of panic disorder: A two year follow-up. Behav Ther 1991;22:289-304.
26. Ost LG, Westling BE. Applied relaxation vs cognitive behavior therapy in the treatment of panic disorder. Behav Res Ther 1995;33:145-58.
27. Meuret AE, Wilhelm FH, Ritz T, Roth WT. Breathing training for treating panic disorder: useful intervention or impediment. Behav Mod 2003;27(5):731-54.
28. Meuret AE, Wilhelm FH, Roth WT. Respiratory feedback for treating panic disorder. J Clin Psychol 2004;60:197-207.
29. Seligman L. Systems, strategies, and skills of counseling and psychotherapy. Saddle River, NJ: Prentice-Hall; 2001.
1. American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association; 1994.
2. Otto MW, Deckersbach T. Cognitive-behavioral therapy for panic disorder: Theory, strategies, and outcome. In: Rosenbaum JF, Pollack M (eds). Panic disorder and its treatment. New York: Marcel Dekker; 1998.
3. Hettema, JM, Neale, MC, Kendler, KS. A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry 2001;158(10):1568-78.
4. Kendler KS, Gardner CO, Prescott CA. Panic syndromes in a population-based sample of male and female twins. Psychol Med 2001;31:989-1000.
5. Sandford JJ, Argyropoulos SV, Nutt DJ. The psychobiology of anxiolytic drugs. Part I: basic neurobiology. Pharmacol Ther 2000;88:197-212.
6. Millan MJ. The neurobiology and control of anxious states. Prog Neurobiol 2003;70:83-244.
7. Sanderson WC, Rego SA. Empirically supported psychological treatment of panic disorder and agoraphobia. Medscape. Available at www.medscape.com/viewprogram/350_pnt. Accessed Nov. 8, 2005.
8. Rayburn NR, Otto MW. Cognitive-behavioral therapy for panic disorder: a review of treatment elements, strategies, and outcomes. CNS Spectr 2003;8(5):356-62.
9. Craske MG, Barlow DH. Panic disorder and agoraphobia. In: Barlow DH (ed). Clinical handbook of psychological disorders: A step-by-step treatment manual. New York: Guilford Press; 2001;1-59.
10. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey replication. Arch Gen Psychiatry 2005;62:593-602.
11. Rapee RM, Barlow DH. Generalized anxiety disorders, panic disorders, and phobias. In: Sutker PB, Adams HE (eds). Comprehensive handbook of psychopathology (3rd ed). New York: Kluwer Academic/Plenum; 2001.
12. Cote G, Gauthier JG, Laberge B, et al. Reduced therapist contact in the cognitive behavioral treatment of panic disorder. Behav Ther 1994;25:123-45.
13. Telch MJ, Lucas JA, Schmidt NB, et al. Group cognitive-behavioral treatment of panic disorder. Behav Res Ther 1993;31:279-28.
14. Welkowitz LA, Papp LA, Cloitre M, et al. Cognitive-behavioral therapy for panic disorder delivered by psychopharmacologically oriented clinicians. J Nerv Ment Dis 1991;179:473-77.
15. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. Washington, DC: American Psychiatric Association; 1998.
16. Royal Australian and New Zealand College of Psychiatrists. Australian and New Zealand clinical practice guidelines for the treatment of panic disorder and agoraphobia. Aust NZ J Psychiatry 2003;37:641-56.Available at: www.ranzcp.org/publicarea/cpg.asp. Accessed Aug. 24, 2005.
17. Black DW, Wesner R, Bowers W, Gabel J. A comparison of fluvoxamine, cognitive therapy, and placebo in the treatment of panic disorder. Arch Gen Psychiatry 1993;31:383-94.
18. Dannon PN, Gon-Usishkin M, Gelbert A, et al. Cognitive behavioral group therapy in panic disorder patients: The efficacy of CBGT versus drug treatment. Ann Clin Psychiatry 2004;16:41-6.
19. Clark DM, Salkovskis PM, Hackmann A, et al. A comparison of cognitive therapy, applied relaxation, and imipramine in the treatment of panic disorder. Br J Psychiatry 1994;164:759-69.
20. Klosko JS, Barlow DH, Tassinari R, Cerny JA. A comparison of alprazolam and behavior therapy in treatment of panic disorder. J Consult Clin Psychol 1990;58:77-84.
21. Rapee RM, Craske MG, Barlow DH. Assessment instrument for panic disorder that includes fear of sensation-producing activities: The Albany Panic and Phobia Questionnaire. Anxiety 1995;1:114-22.
22. Reiss S, Peterson R, Gursky D, McNally R. Anxiety sensitivity, anxiety frequency, and the prediction of fearfulness. Behav Res Ther 1986;24:1-8.
23. Burns DD. Feeling good: The new mood therapy. New York: William Morrow and Co.; 1980.
24. Clum GA, Watkins PL, Borden JW, et al. A comparison of guided imaginal coping and imaginal exposure in the treatment of panic disorder. J Rational-Emotive & Cognitive Behavior Therapy 1993;11(4):179-93.
25. Craske MG, Brown TA, Barlow DH. Behavioral treatment of panic disorder: A two year follow-up. Behav Ther 1991;22:289-304.
26. Ost LG, Westling BE. Applied relaxation vs cognitive behavior therapy in the treatment of panic disorder. Behav Res Ther 1995;33:145-58.
27. Meuret AE, Wilhelm FH, Ritz T, Roth WT. Breathing training for treating panic disorder: useful intervention or impediment. Behav Mod 2003;27(5):731-54.
28. Meuret AE, Wilhelm FH, Roth WT. Respiratory feedback for treating panic disorder. J Clin Psychol 2004;60:197-207.
29. Seligman L. Systems, strategies, and skills of counseling and psychotherapy. Saddle River, NJ: Prentice-Hall; 2001.
Bipolar disorder: New strategy for checking serum valproate
Valproate’s well-accepted therapeutic range for treating epilepsy—50 to 100 mcg/mL—was adopted for bipolar disorder treatment without rigorous evaluation of serum levels and response relationships. Because most literature on monitoring serum valproate refers to its use as an anticonvulsant, you may wonder:
- When should I measure serum valproate in bipolar patients?
- What do serum valproate levels mean in their clinical care?
To answer these questions, we discuss when to monitor serum valproate, whether routinely or in specific situations. We then review studies that show how serum levels affect valproate’s efficacy and safety in three phases of bipolar disorder management: acute mania, maintenance therapy, and acute depression.
Is monitoring overused?
Some neurologists consider serum levels nonessential—and, in some cases, overused—when valproate is used as an anticonvulsant for healthy patients.1,2 A multicenter, randomized controlled trial evaluating the impact of antiepileptic drug monitoring on patient outcomes3 supports this notion, at least in part. Serum monitoring did not improve therapeutic outcome, suggesting that patients with epilepsy could be satisfactorily treated by adjusting dosages based on clinical response.
On the other hand, American Psychiatric Association (APA) guidelines for bipolar disorder suggest routine serum monitoring every 6 months along with other hematologic and hepatic assessments, or more frequently if necessary. The APA recommends maintaining serum valproate levels of 50 to 125 mcg/mL when treating:
- acutely manic patients
- outpatients
- the elderly
- patients who are hypomanic or euthymic.4
Table 1
4 situations where serum valproate monitoring may be clinically useful
| To establish a baseline effective level in individual patients |
| To assess lack or loss of efficacy, including patient adherence |
| When drug-drug interactions increase or decrease valproate clearance (such as with aspirin, carbamazepine, felbamate, or phenytoin)5 |
| When dose-dependent side effects occur (such as alopecia, elevated liver function, thrombocytopenia, or pancreatitis) |
Effective levels in acute mania
In one of the first randomized, double-blind, placebo-controlled trials to examine valproate use in adults with acute mania, Pope et al6 used the epilepsy reference range to adjust dosages. Patients (n=17) initially received valproate, 750 mg/d, and dosages were then adjusted to serum levels of 50 to 100 mcg/mL. Nineteen patients received placebo. Mean (SD) baseline Young Mania Rating Scale (YMRS) scores for the valproate and placebo groups were 28.2 (5.8) and 28.6 (6.9), respectively.
Patients receiving valproate showed the greatest symptomatic improvement—as indicated by YMRS scores—within 1 to 4 days of achieving a serum level ≥50 mcg/mL. Serum valproate for all patients was maintained at >50 mcg/mL, which limits our ability to draw conclusions about a minimum level associated with efficacy.
Minimum threshold for efficacy. In another randomized, double-blind, placebo-controlled study of acute mania, Bowden et al7 compared the efficacy of divalproex (n=69) versus lithium (n=36) or placebo (n=74) given for 3 weeks. Patients met criteria for manic disorder using the Schedule for Affective Disorders and Schizophrenia (SADS) and had Mania Rating Scale scores (derived from the SADS) of at least 14.
Those in the divalproex group received 750 mg/d for 2 days, then 1,000 mg/d for 3 days. Dosages were then adjusted to target a serum level of 150 mcg/mL, unless limited by side effects. Mean serum valproate levels on days 8 and 21 were 77 and 93.2 mcg/mL, respectively. Marked improvement, defined as ≥50% reduction in Mania Rating Scale scores, was seen in 48% of the divalproex group, compared with 25% of the placebo group.
The authors then analyzed the relationship between serum valproate levels and clinical response and tolerability.8 At day 5, patients with serum valproate ≥45 mcg/mL were 2 to 7 times more likely to show 20% or greater improvement in SADS mania subscales (manic syndrome, and behavior and ideation).
This study provided a minimum threshold for valproate efficacy in bipolar mania—45 to 50 mcg/mL—but not a level above which further clinical benefit would not be gained.
Optimum serum ranges. Allen et al9 recently conducted a post hoc analysis of pooled intent-to-treat data from three randomized, fixed dose, placebo-controlled studies of divalproex for acute mania. Subjects were stratified into a placebo group (n=171) and six serum valproate ranges:
- ≤55 mcg/mL (n=35)
- >55 to 71.3 mcg/mL (n=32)
- >71.3 to 85 mcg/mL (n=36)
- >85 to 94 mcg/mL (n=34)
- >94 to 107 mcg/mL (n=33)
- >107 mcg/mL (n=33).
Loading for rapid response. Patients with acute mania may respond sooner when loading doses are used to attain therapeutic serum valproate levels.
Keck et al10 examined time to onset of improvement in adults with acute mania (N=19) receiving oral loading doses of valproate (20 mg/kg/d in divided doses for 5 days) to rapidly attain valproate levels ≥50 mcg/mL. Ten (53%) patients who received at least 1 loading dose showed a ≥50% reduction in MRS scores and the greatest improvement across the first 3 days.
Hirschfeld et al11 also reported that patients’ symptoms began to improve sooner when divalproex was given at 30 mg/kg/d on days 1 and 2, and 20 mg/kg/d on days 3 to 10 (n=20), compared with standard titration (750 mg/d on days 1 and 2, and gradual dose titration on days 3 to 10 [n=20]).
Discussion. In acute mania, evidence suggests that patients with serum valproate ≥45 to 50 mcg/mL may show greater clinical improvement than patients with lower serum levels. Loading doses may achieve a minimum therapeutic serum level more quickly, yielding faster clinical improvement. A serum level >90 mcg/mL may confer additional benefit.
Although a minimum serum level has been recommended, no data have established a maximum level beyond which further clinical improvement would not be observed.
In maintenance therapy
What serum valproate levels are most effective for bipolar maintenance therapy? Some evidence is emerging.
Bowden et al12 compared divalproex (n=187), lithium (n=90), and placebo (n=92) in a 52-week, double-blind, parallel-group study of bipolar adult outpatients who met recovery criteria 3 months after an index manic episode. Divalproex dosages were adjusted to achieve trough serum concentrations between 71 and 125 mcg/mL. Mean (SD) and median serum valproate levels were 84.8 (29.9) mcg/mL and 83.9 mcg/mL, respectively. Serum valproate levels significantly correlated with Mania Rating Scale scores. No minimum threshold for efficacy was reported.
Thirteen subjects in the divalproex group were then stratified into 4 categories:
- nontherapeutic (
- low therapeutic (50 to 74.9 mcg/mL)
- medium therapeutic (75 to 99.9 mcg/mL)
- high therapeutic (>100 mcg/mL).
Discussion. Serum valproate levels of 75 to 100 mcg/mL may be most effective in preventing subsequent mood episodes with acceptable tolerability. Prospective, longitudinal studies are needed to better establish a therapeutic range for valproate in bipolar maintenance therapy.
In bipolar depression
Little evidence supports a therapeutic serum valproate range for treating acute bipolar depression.
In an 8-week, double-blind study, Davis et al14 randomly assigned adults with bipolar depression to divalproex (n=13) or placebo (n=12). Bipolar depression diagnoses were confirmed using the Structured Clinical Interview for DSM-IV, and patients were required to have a Hamilton Rating Scale for Depression (HRSD) score ≥16.
Valproate was started at 500 mg/d and titrated to serum levels of 50 to 150 mcg/mL. Mean (SD) serum valproate levels at weeks 4 and 8 were 80 (9.3) mcg/mL and 81 (19.2) mcg/mL, respectively. Remission rate (defined a priori as a >50% improvement and total HRSD score 15 In Sachs’ 8-week study, the mean (SD) valproate level was 61.5 (42.8) mcg/mL.
Discussion. The relationship between serum valproate and therapeutic efficacy in acute bipolar depression—and the range of levels considered therapeutic—are undefined. For now we recommend that individual patients’ clinical response and tolerability guide optimum serum valproate in acute bipolar depression (Box).16
When evaluating serum valproate levels–especially for assessing adherence–be careful to:
- obtain blood samples 12 hours after the most recent dose to accurately assess serum trough concentrations
- account for valproate’s saturation of protein binding sites and increased free fraction with increased serum concentration.16
Valproate clearance is increased when more free drug is available for metabolism, and this may result in disproportionately lower steady-state serum concentrations. Smaller increases in total valproate after dosage increases may be misinterpreted as medication nonadherence.
High levels and safety
High serum valproate levels may increase the risk and frequency of side effects. For example, serum levels >125 mcg/mL have been associated with:
- increased nausea, vomiting, dizziness, and sedation in acutely manic patients8
- weight gain and reduced platelets and white blood cells in patients receiving valproate as maintenance treatment.12
In the loading dose study by Hirschfeld et al,11 patients receiving divalproex, 20 to 30 mg/kg/d, did not experience a higher frequency or severity of side effects compared with patients receiving standard titration. Keck et al10 also reported minimal valproate-related side effects in their open-label study. Neither study suggested an upper-limit valproate level associated with increased side effects.
Discussion. Serum valproate >125 mcg/mL has been associated with increased side effects (Table 2), but more studies are needed.
Table 2
For bipolar disorder, suggested serum valproate therapeutic ranges*
| Serum valproate (mcg/mL) | |||
|---|---|---|---|
| Lower level | Upper level | Comments | |
| Acute mania | 45 to 50 | 125 | Upper level based on tolerability, not efficacy |
| Maintenance | 75 | 100 | Levels based primarily on retrospective analysis |
| Acute bipolar | Not established | Not established | |
| * Based on available data | |||
Clinical recommendations
Carefully consider when to monitor serum valproate levels in your patients with bipolar disorder:
- Obtaining routine serum levels can be expensive, and no data support the cost-effectiveness of this approach in bipolar disorder.
- Individualize valproate dosing; a specific patient’s therapeutic range may differ from another’s or from those published in the literature or used by a clinical laboratory.
- Monitoring serum valproate levels does not replace the need to adjust dosages based on patients’ therapeutic response and tolerance.
- American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry2002; 159(suppl 4):1–50.
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- Carbamazepine • Tegretol, Equetro
- Divalproex sodium • Depakote
- Felbamate • Felbatol
- Phenytoin • Dilantin
Dr. Kaneria reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Patel is a consultant to and speaker for Eli Lilly and Co. and a speaker for Pfizer.
Dr. Keck receives research support from or is a consultant to or advisor for Abbott Laboratories, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., GlaxoSmithKline, Janssen Pharmaceutica, Eli Lilly and Co., Organon, Ortho-McNeil Pharmaceutical, Merck & Co., Pfizer, Shire, and UCB Pharma.
1. Glauser TA, Pippenger CE. Controversies in blood-level monitoring: reexamining its role in the treatment of epilepsy. Epilepsia 2000;41(suppl 8):S6-S15.
2. Pellock JM, Willmore LJ. A rational guide to routine blood monitoring in patients receiving antiepileptic drugs. Neurology 1991;41:961-4.
3. Jannuzzi G, Cian P, Fattore C, et al. A multicenter randomized controlled trial on the clinical impact of therapeutic drug monitoring in patients with newly diagnosed epilepsy. The Italian TDM Study Group in Epilepsy. Epilepsia 2000;41:222-30.
4. AmericanPsychiatric Association Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry 2002;159(suppl 4):1-50.
5. Depakote (divalproex sodium) package insert Abbott Park, IL: Abbott Laboratories; October 2005.
6. Pope HG, Jr, McElroy SL, Keck PE, Jr, Hudson JI. Valproate in the treatment of acute mania. A placebo-controlled study. Arch Gen Psychiatry 1991;48:62-8.
7. Bowden CL, Brugger AM, Swann AC, et al. Efficacy of divalproex vs lithium and placebo in the treatment of mania. The Depakote Mania Study Group. JAMA 1994;271:918-24.
8. Bowden CL, Janicak PG, Orsulak P, et al. Relation of serum valproate concentration to response in mania. Am J Psychiatry 1996;153:765-70.
9. Allen MH, Baker J, Wozniak PJ. Relationship of serum valproate level to response in mania (abstract presentation). New York: American Psychiatric Association annual meeting, 2004.
10. Keck PE, Jr, McElroy SL, Tugrul KC, Bennett JA. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry 1993;54:305-8.
11. Hirschfeld RM, Allen MH, McEvoy JP, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry 1999;60:815-18.
12. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex Maintenance Study Group. Arch Gen Psychiatry 2000;57:481-9.
13. Keck PE, Jr, Bowden CL, Meinhold JM, et al. Relationship between serum valproate and lithium levels and efficacy and tolerability in bipolar maintenance therapy. Int J Psychiatry Clin Pract (in press).
14. Davis LL, Bartolucci A, Petty F. Divalproex in the treatment of bipolar depression: a placebo-controlled study. J Affect Disord 2005;85:259-66.
15. Sachs GS, Collins MA, Altshuler LL, et al. Divalproex sodium versus placebo for the treatment of bipolar depression (abstract presentation). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2001.
16. Wilder BJ. Pharmacokinetics of valproate and carbamazepine. J Clin Psychopharmacol 1992;12(suppl 1):64S-68S.
Valproate’s well-accepted therapeutic range for treating epilepsy—50 to 100 mcg/mL—was adopted for bipolar disorder treatment without rigorous evaluation of serum levels and response relationships. Because most literature on monitoring serum valproate refers to its use as an anticonvulsant, you may wonder:
- When should I measure serum valproate in bipolar patients?
- What do serum valproate levels mean in their clinical care?
To answer these questions, we discuss when to monitor serum valproate, whether routinely or in specific situations. We then review studies that show how serum levels affect valproate’s efficacy and safety in three phases of bipolar disorder management: acute mania, maintenance therapy, and acute depression.
Is monitoring overused?
Some neurologists consider serum levels nonessential—and, in some cases, overused—when valproate is used as an anticonvulsant for healthy patients.1,2 A multicenter, randomized controlled trial evaluating the impact of antiepileptic drug monitoring on patient outcomes3 supports this notion, at least in part. Serum monitoring did not improve therapeutic outcome, suggesting that patients with epilepsy could be satisfactorily treated by adjusting dosages based on clinical response.
On the other hand, American Psychiatric Association (APA) guidelines for bipolar disorder suggest routine serum monitoring every 6 months along with other hematologic and hepatic assessments, or more frequently if necessary. The APA recommends maintaining serum valproate levels of 50 to 125 mcg/mL when treating:
- acutely manic patients
- outpatients
- the elderly
- patients who are hypomanic or euthymic.4
Table 1
4 situations where serum valproate monitoring may be clinically useful
| To establish a baseline effective level in individual patients |
| To assess lack or loss of efficacy, including patient adherence |
| When drug-drug interactions increase or decrease valproate clearance (such as with aspirin, carbamazepine, felbamate, or phenytoin)5 |
| When dose-dependent side effects occur (such as alopecia, elevated liver function, thrombocytopenia, or pancreatitis) |
Effective levels in acute mania
In one of the first randomized, double-blind, placebo-controlled trials to examine valproate use in adults with acute mania, Pope et al6 used the epilepsy reference range to adjust dosages. Patients (n=17) initially received valproate, 750 mg/d, and dosages were then adjusted to serum levels of 50 to 100 mcg/mL. Nineteen patients received placebo. Mean (SD) baseline Young Mania Rating Scale (YMRS) scores for the valproate and placebo groups were 28.2 (5.8) and 28.6 (6.9), respectively.
Patients receiving valproate showed the greatest symptomatic improvement—as indicated by YMRS scores—within 1 to 4 days of achieving a serum level ≥50 mcg/mL. Serum valproate for all patients was maintained at >50 mcg/mL, which limits our ability to draw conclusions about a minimum level associated with efficacy.
Minimum threshold for efficacy. In another randomized, double-blind, placebo-controlled study of acute mania, Bowden et al7 compared the efficacy of divalproex (n=69) versus lithium (n=36) or placebo (n=74) given for 3 weeks. Patients met criteria for manic disorder using the Schedule for Affective Disorders and Schizophrenia (SADS) and had Mania Rating Scale scores (derived from the SADS) of at least 14.
Those in the divalproex group received 750 mg/d for 2 days, then 1,000 mg/d for 3 days. Dosages were then adjusted to target a serum level of 150 mcg/mL, unless limited by side effects. Mean serum valproate levels on days 8 and 21 were 77 and 93.2 mcg/mL, respectively. Marked improvement, defined as ≥50% reduction in Mania Rating Scale scores, was seen in 48% of the divalproex group, compared with 25% of the placebo group.
The authors then analyzed the relationship between serum valproate levels and clinical response and tolerability.8 At day 5, patients with serum valproate ≥45 mcg/mL were 2 to 7 times more likely to show 20% or greater improvement in SADS mania subscales (manic syndrome, and behavior and ideation).
This study provided a minimum threshold for valproate efficacy in bipolar mania—45 to 50 mcg/mL—but not a level above which further clinical benefit would not be gained.
Optimum serum ranges. Allen et al9 recently conducted a post hoc analysis of pooled intent-to-treat data from three randomized, fixed dose, placebo-controlled studies of divalproex for acute mania. Subjects were stratified into a placebo group (n=171) and six serum valproate ranges:
- ≤55 mcg/mL (n=35)
- >55 to 71.3 mcg/mL (n=32)
- >71.3 to 85 mcg/mL (n=36)
- >85 to 94 mcg/mL (n=34)
- >94 to 107 mcg/mL (n=33)
- >107 mcg/mL (n=33).
Loading for rapid response. Patients with acute mania may respond sooner when loading doses are used to attain therapeutic serum valproate levels.
Keck et al10 examined time to onset of improvement in adults with acute mania (N=19) receiving oral loading doses of valproate (20 mg/kg/d in divided doses for 5 days) to rapidly attain valproate levels ≥50 mcg/mL. Ten (53%) patients who received at least 1 loading dose showed a ≥50% reduction in MRS scores and the greatest improvement across the first 3 days.
Hirschfeld et al11 also reported that patients’ symptoms began to improve sooner when divalproex was given at 30 mg/kg/d on days 1 and 2, and 20 mg/kg/d on days 3 to 10 (n=20), compared with standard titration (750 mg/d on days 1 and 2, and gradual dose titration on days 3 to 10 [n=20]).
Discussion. In acute mania, evidence suggests that patients with serum valproate ≥45 to 50 mcg/mL may show greater clinical improvement than patients with lower serum levels. Loading doses may achieve a minimum therapeutic serum level more quickly, yielding faster clinical improvement. A serum level >90 mcg/mL may confer additional benefit.
Although a minimum serum level has been recommended, no data have established a maximum level beyond which further clinical improvement would not be observed.
In maintenance therapy
What serum valproate levels are most effective for bipolar maintenance therapy? Some evidence is emerging.
Bowden et al12 compared divalproex (n=187), lithium (n=90), and placebo (n=92) in a 52-week, double-blind, parallel-group study of bipolar adult outpatients who met recovery criteria 3 months after an index manic episode. Divalproex dosages were adjusted to achieve trough serum concentrations between 71 and 125 mcg/mL. Mean (SD) and median serum valproate levels were 84.8 (29.9) mcg/mL and 83.9 mcg/mL, respectively. Serum valproate levels significantly correlated with Mania Rating Scale scores. No minimum threshold for efficacy was reported.
Thirteen subjects in the divalproex group were then stratified into 4 categories:
- nontherapeutic (
- low therapeutic (50 to 74.9 mcg/mL)
- medium therapeutic (75 to 99.9 mcg/mL)
- high therapeutic (>100 mcg/mL).
Discussion. Serum valproate levels of 75 to 100 mcg/mL may be most effective in preventing subsequent mood episodes with acceptable tolerability. Prospective, longitudinal studies are needed to better establish a therapeutic range for valproate in bipolar maintenance therapy.
In bipolar depression
Little evidence supports a therapeutic serum valproate range for treating acute bipolar depression.
In an 8-week, double-blind study, Davis et al14 randomly assigned adults with bipolar depression to divalproex (n=13) or placebo (n=12). Bipolar depression diagnoses were confirmed using the Structured Clinical Interview for DSM-IV, and patients were required to have a Hamilton Rating Scale for Depression (HRSD) score ≥16.
Valproate was started at 500 mg/d and titrated to serum levels of 50 to 150 mcg/mL. Mean (SD) serum valproate levels at weeks 4 and 8 were 80 (9.3) mcg/mL and 81 (19.2) mcg/mL, respectively. Remission rate (defined a priori as a >50% improvement and total HRSD score 15 In Sachs’ 8-week study, the mean (SD) valproate level was 61.5 (42.8) mcg/mL.
Discussion. The relationship between serum valproate and therapeutic efficacy in acute bipolar depression—and the range of levels considered therapeutic—are undefined. For now we recommend that individual patients’ clinical response and tolerability guide optimum serum valproate in acute bipolar depression (Box).16
When evaluating serum valproate levels–especially for assessing adherence–be careful to:
- obtain blood samples 12 hours after the most recent dose to accurately assess serum trough concentrations
- account for valproate’s saturation of protein binding sites and increased free fraction with increased serum concentration.16
Valproate clearance is increased when more free drug is available for metabolism, and this may result in disproportionately lower steady-state serum concentrations. Smaller increases in total valproate after dosage increases may be misinterpreted as medication nonadherence.
High levels and safety
High serum valproate levels may increase the risk and frequency of side effects. For example, serum levels >125 mcg/mL have been associated with:
- increased nausea, vomiting, dizziness, and sedation in acutely manic patients8
- weight gain and reduced platelets and white blood cells in patients receiving valproate as maintenance treatment.12
In the loading dose study by Hirschfeld et al,11 patients receiving divalproex, 20 to 30 mg/kg/d, did not experience a higher frequency or severity of side effects compared with patients receiving standard titration. Keck et al10 also reported minimal valproate-related side effects in their open-label study. Neither study suggested an upper-limit valproate level associated with increased side effects.
Discussion. Serum valproate >125 mcg/mL has been associated with increased side effects (Table 2), but more studies are needed.
Table 2
For bipolar disorder, suggested serum valproate therapeutic ranges*
| Serum valproate (mcg/mL) | |||
|---|---|---|---|
| Lower level | Upper level | Comments | |
| Acute mania | 45 to 50 | 125 | Upper level based on tolerability, not efficacy |
| Maintenance | 75 | 100 | Levels based primarily on retrospective analysis |
| Acute bipolar | Not established | Not established | |
| * Based on available data | |||
Clinical recommendations
Carefully consider when to monitor serum valproate levels in your patients with bipolar disorder:
- Obtaining routine serum levels can be expensive, and no data support the cost-effectiveness of this approach in bipolar disorder.
- Individualize valproate dosing; a specific patient’s therapeutic range may differ from another’s or from those published in the literature or used by a clinical laboratory.
- Monitoring serum valproate levels does not replace the need to adjust dosages based on patients’ therapeutic response and tolerance.
- American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry2002; 159(suppl 4):1–50.
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- Carbamazepine • Tegretol, Equetro
- Divalproex sodium • Depakote
- Felbamate • Felbatol
- Phenytoin • Dilantin
Dr. Kaneria reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Patel is a consultant to and speaker for Eli Lilly and Co. and a speaker for Pfizer.
Dr. Keck receives research support from or is a consultant to or advisor for Abbott Laboratories, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., GlaxoSmithKline, Janssen Pharmaceutica, Eli Lilly and Co., Organon, Ortho-McNeil Pharmaceutical, Merck & Co., Pfizer, Shire, and UCB Pharma.
Valproate’s well-accepted therapeutic range for treating epilepsy—50 to 100 mcg/mL—was adopted for bipolar disorder treatment without rigorous evaluation of serum levels and response relationships. Because most literature on monitoring serum valproate refers to its use as an anticonvulsant, you may wonder:
- When should I measure serum valproate in bipolar patients?
- What do serum valproate levels mean in their clinical care?
To answer these questions, we discuss when to monitor serum valproate, whether routinely or in specific situations. We then review studies that show how serum levels affect valproate’s efficacy and safety in three phases of bipolar disorder management: acute mania, maintenance therapy, and acute depression.
Is monitoring overused?
Some neurologists consider serum levels nonessential—and, in some cases, overused—when valproate is used as an anticonvulsant for healthy patients.1,2 A multicenter, randomized controlled trial evaluating the impact of antiepileptic drug monitoring on patient outcomes3 supports this notion, at least in part. Serum monitoring did not improve therapeutic outcome, suggesting that patients with epilepsy could be satisfactorily treated by adjusting dosages based on clinical response.
On the other hand, American Psychiatric Association (APA) guidelines for bipolar disorder suggest routine serum monitoring every 6 months along with other hematologic and hepatic assessments, or more frequently if necessary. The APA recommends maintaining serum valproate levels of 50 to 125 mcg/mL when treating:
- acutely manic patients
- outpatients
- the elderly
- patients who are hypomanic or euthymic.4
Table 1
4 situations where serum valproate monitoring may be clinically useful
| To establish a baseline effective level in individual patients |
| To assess lack or loss of efficacy, including patient adherence |
| When drug-drug interactions increase or decrease valproate clearance (such as with aspirin, carbamazepine, felbamate, or phenytoin)5 |
| When dose-dependent side effects occur (such as alopecia, elevated liver function, thrombocytopenia, or pancreatitis) |
Effective levels in acute mania
In one of the first randomized, double-blind, placebo-controlled trials to examine valproate use in adults with acute mania, Pope et al6 used the epilepsy reference range to adjust dosages. Patients (n=17) initially received valproate, 750 mg/d, and dosages were then adjusted to serum levels of 50 to 100 mcg/mL. Nineteen patients received placebo. Mean (SD) baseline Young Mania Rating Scale (YMRS) scores for the valproate and placebo groups were 28.2 (5.8) and 28.6 (6.9), respectively.
Patients receiving valproate showed the greatest symptomatic improvement—as indicated by YMRS scores—within 1 to 4 days of achieving a serum level ≥50 mcg/mL. Serum valproate for all patients was maintained at >50 mcg/mL, which limits our ability to draw conclusions about a minimum level associated with efficacy.
Minimum threshold for efficacy. In another randomized, double-blind, placebo-controlled study of acute mania, Bowden et al7 compared the efficacy of divalproex (n=69) versus lithium (n=36) or placebo (n=74) given for 3 weeks. Patients met criteria for manic disorder using the Schedule for Affective Disorders and Schizophrenia (SADS) and had Mania Rating Scale scores (derived from the SADS) of at least 14.
Those in the divalproex group received 750 mg/d for 2 days, then 1,000 mg/d for 3 days. Dosages were then adjusted to target a serum level of 150 mcg/mL, unless limited by side effects. Mean serum valproate levels on days 8 and 21 were 77 and 93.2 mcg/mL, respectively. Marked improvement, defined as ≥50% reduction in Mania Rating Scale scores, was seen in 48% of the divalproex group, compared with 25% of the placebo group.
The authors then analyzed the relationship between serum valproate levels and clinical response and tolerability.8 At day 5, patients with serum valproate ≥45 mcg/mL were 2 to 7 times more likely to show 20% or greater improvement in SADS mania subscales (manic syndrome, and behavior and ideation).
This study provided a minimum threshold for valproate efficacy in bipolar mania—45 to 50 mcg/mL—but not a level above which further clinical benefit would not be gained.
Optimum serum ranges. Allen et al9 recently conducted a post hoc analysis of pooled intent-to-treat data from three randomized, fixed dose, placebo-controlled studies of divalproex for acute mania. Subjects were stratified into a placebo group (n=171) and six serum valproate ranges:
- ≤55 mcg/mL (n=35)
- >55 to 71.3 mcg/mL (n=32)
- >71.3 to 85 mcg/mL (n=36)
- >85 to 94 mcg/mL (n=34)
- >94 to 107 mcg/mL (n=33)
- >107 mcg/mL (n=33).
Loading for rapid response. Patients with acute mania may respond sooner when loading doses are used to attain therapeutic serum valproate levels.
Keck et al10 examined time to onset of improvement in adults with acute mania (N=19) receiving oral loading doses of valproate (20 mg/kg/d in divided doses for 5 days) to rapidly attain valproate levels ≥50 mcg/mL. Ten (53%) patients who received at least 1 loading dose showed a ≥50% reduction in MRS scores and the greatest improvement across the first 3 days.
Hirschfeld et al11 also reported that patients’ symptoms began to improve sooner when divalproex was given at 30 mg/kg/d on days 1 and 2, and 20 mg/kg/d on days 3 to 10 (n=20), compared with standard titration (750 mg/d on days 1 and 2, and gradual dose titration on days 3 to 10 [n=20]).
Discussion. In acute mania, evidence suggests that patients with serum valproate ≥45 to 50 mcg/mL may show greater clinical improvement than patients with lower serum levels. Loading doses may achieve a minimum therapeutic serum level more quickly, yielding faster clinical improvement. A serum level >90 mcg/mL may confer additional benefit.
Although a minimum serum level has been recommended, no data have established a maximum level beyond which further clinical improvement would not be observed.
In maintenance therapy
What serum valproate levels are most effective for bipolar maintenance therapy? Some evidence is emerging.
Bowden et al12 compared divalproex (n=187), lithium (n=90), and placebo (n=92) in a 52-week, double-blind, parallel-group study of bipolar adult outpatients who met recovery criteria 3 months after an index manic episode. Divalproex dosages were adjusted to achieve trough serum concentrations between 71 and 125 mcg/mL. Mean (SD) and median serum valproate levels were 84.8 (29.9) mcg/mL and 83.9 mcg/mL, respectively. Serum valproate levels significantly correlated with Mania Rating Scale scores. No minimum threshold for efficacy was reported.
Thirteen subjects in the divalproex group were then stratified into 4 categories:
- nontherapeutic (
- low therapeutic (50 to 74.9 mcg/mL)
- medium therapeutic (75 to 99.9 mcg/mL)
- high therapeutic (>100 mcg/mL).
Discussion. Serum valproate levels of 75 to 100 mcg/mL may be most effective in preventing subsequent mood episodes with acceptable tolerability. Prospective, longitudinal studies are needed to better establish a therapeutic range for valproate in bipolar maintenance therapy.
In bipolar depression
Little evidence supports a therapeutic serum valproate range for treating acute bipolar depression.
In an 8-week, double-blind study, Davis et al14 randomly assigned adults with bipolar depression to divalproex (n=13) or placebo (n=12). Bipolar depression diagnoses were confirmed using the Structured Clinical Interview for DSM-IV, and patients were required to have a Hamilton Rating Scale for Depression (HRSD) score ≥16.
Valproate was started at 500 mg/d and titrated to serum levels of 50 to 150 mcg/mL. Mean (SD) serum valproate levels at weeks 4 and 8 were 80 (9.3) mcg/mL and 81 (19.2) mcg/mL, respectively. Remission rate (defined a priori as a >50% improvement and total HRSD score 15 In Sachs’ 8-week study, the mean (SD) valproate level was 61.5 (42.8) mcg/mL.
Discussion. The relationship between serum valproate and therapeutic efficacy in acute bipolar depression—and the range of levels considered therapeutic—are undefined. For now we recommend that individual patients’ clinical response and tolerability guide optimum serum valproate in acute bipolar depression (Box).16
When evaluating serum valproate levels–especially for assessing adherence–be careful to:
- obtain blood samples 12 hours after the most recent dose to accurately assess serum trough concentrations
- account for valproate’s saturation of protein binding sites and increased free fraction with increased serum concentration.16
Valproate clearance is increased when more free drug is available for metabolism, and this may result in disproportionately lower steady-state serum concentrations. Smaller increases in total valproate after dosage increases may be misinterpreted as medication nonadherence.
High levels and safety
High serum valproate levels may increase the risk and frequency of side effects. For example, serum levels >125 mcg/mL have been associated with:
- increased nausea, vomiting, dizziness, and sedation in acutely manic patients8
- weight gain and reduced platelets and white blood cells in patients receiving valproate as maintenance treatment.12
In the loading dose study by Hirschfeld et al,11 patients receiving divalproex, 20 to 30 mg/kg/d, did not experience a higher frequency or severity of side effects compared with patients receiving standard titration. Keck et al10 also reported minimal valproate-related side effects in their open-label study. Neither study suggested an upper-limit valproate level associated with increased side effects.
Discussion. Serum valproate >125 mcg/mL has been associated with increased side effects (Table 2), but more studies are needed.
Table 2
For bipolar disorder, suggested serum valproate therapeutic ranges*
| Serum valproate (mcg/mL) | |||
|---|---|---|---|
| Lower level | Upper level | Comments | |
| Acute mania | 45 to 50 | 125 | Upper level based on tolerability, not efficacy |
| Maintenance | 75 | 100 | Levels based primarily on retrospective analysis |
| Acute bipolar | Not established | Not established | |
| * Based on available data | |||
Clinical recommendations
Carefully consider when to monitor serum valproate levels in your patients with bipolar disorder:
- Obtaining routine serum levels can be expensive, and no data support the cost-effectiveness of this approach in bipolar disorder.
- Individualize valproate dosing; a specific patient’s therapeutic range may differ from another’s or from those published in the literature or used by a clinical laboratory.
- Monitoring serum valproate levels does not replace the need to adjust dosages based on patients’ therapeutic response and tolerance.
- American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry2002; 159(suppl 4):1–50.
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- Carbamazepine • Tegretol, Equetro
- Divalproex sodium • Depakote
- Felbamate • Felbatol
- Phenytoin • Dilantin
Dr. Kaneria reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Patel is a consultant to and speaker for Eli Lilly and Co. and a speaker for Pfizer.
Dr. Keck receives research support from or is a consultant to or advisor for Abbott Laboratories, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., GlaxoSmithKline, Janssen Pharmaceutica, Eli Lilly and Co., Organon, Ortho-McNeil Pharmaceutical, Merck & Co., Pfizer, Shire, and UCB Pharma.
1. Glauser TA, Pippenger CE. Controversies in blood-level monitoring: reexamining its role in the treatment of epilepsy. Epilepsia 2000;41(suppl 8):S6-S15.
2. Pellock JM, Willmore LJ. A rational guide to routine blood monitoring in patients receiving antiepileptic drugs. Neurology 1991;41:961-4.
3. Jannuzzi G, Cian P, Fattore C, et al. A multicenter randomized controlled trial on the clinical impact of therapeutic drug monitoring in patients with newly diagnosed epilepsy. The Italian TDM Study Group in Epilepsy. Epilepsia 2000;41:222-30.
4. AmericanPsychiatric Association Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry 2002;159(suppl 4):1-50.
5. Depakote (divalproex sodium) package insert Abbott Park, IL: Abbott Laboratories; October 2005.
6. Pope HG, Jr, McElroy SL, Keck PE, Jr, Hudson JI. Valproate in the treatment of acute mania. A placebo-controlled study. Arch Gen Psychiatry 1991;48:62-8.
7. Bowden CL, Brugger AM, Swann AC, et al. Efficacy of divalproex vs lithium and placebo in the treatment of mania. The Depakote Mania Study Group. JAMA 1994;271:918-24.
8. Bowden CL, Janicak PG, Orsulak P, et al. Relation of serum valproate concentration to response in mania. Am J Psychiatry 1996;153:765-70.
9. Allen MH, Baker J, Wozniak PJ. Relationship of serum valproate level to response in mania (abstract presentation). New York: American Psychiatric Association annual meeting, 2004.
10. Keck PE, Jr, McElroy SL, Tugrul KC, Bennett JA. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry 1993;54:305-8.
11. Hirschfeld RM, Allen MH, McEvoy JP, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry 1999;60:815-18.
12. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex Maintenance Study Group. Arch Gen Psychiatry 2000;57:481-9.
13. Keck PE, Jr, Bowden CL, Meinhold JM, et al. Relationship between serum valproate and lithium levels and efficacy and tolerability in bipolar maintenance therapy. Int J Psychiatry Clin Pract (in press).
14. Davis LL, Bartolucci A, Petty F. Divalproex in the treatment of bipolar depression: a placebo-controlled study. J Affect Disord 2005;85:259-66.
15. Sachs GS, Collins MA, Altshuler LL, et al. Divalproex sodium versus placebo for the treatment of bipolar depression (abstract presentation). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2001.
16. Wilder BJ. Pharmacokinetics of valproate and carbamazepine. J Clin Psychopharmacol 1992;12(suppl 1):64S-68S.
1. Glauser TA, Pippenger CE. Controversies in blood-level monitoring: reexamining its role in the treatment of epilepsy. Epilepsia 2000;41(suppl 8):S6-S15.
2. Pellock JM, Willmore LJ. A rational guide to routine blood monitoring in patients receiving antiepileptic drugs. Neurology 1991;41:961-4.
3. Jannuzzi G, Cian P, Fattore C, et al. A multicenter randomized controlled trial on the clinical impact of therapeutic drug monitoring in patients with newly diagnosed epilepsy. The Italian TDM Study Group in Epilepsy. Epilepsia 2000;41:222-30.
4. AmericanPsychiatric Association Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry 2002;159(suppl 4):1-50.
5. Depakote (divalproex sodium) package insert Abbott Park, IL: Abbott Laboratories; October 2005.
6. Pope HG, Jr, McElroy SL, Keck PE, Jr, Hudson JI. Valproate in the treatment of acute mania. A placebo-controlled study. Arch Gen Psychiatry 1991;48:62-8.
7. Bowden CL, Brugger AM, Swann AC, et al. Efficacy of divalproex vs lithium and placebo in the treatment of mania. The Depakote Mania Study Group. JAMA 1994;271:918-24.
8. Bowden CL, Janicak PG, Orsulak P, et al. Relation of serum valproate concentration to response in mania. Am J Psychiatry 1996;153:765-70.
9. Allen MH, Baker J, Wozniak PJ. Relationship of serum valproate level to response in mania (abstract presentation). New York: American Psychiatric Association annual meeting, 2004.
10. Keck PE, Jr, McElroy SL, Tugrul KC, Bennett JA. Valproate oral loading in the treatment of acute mania. J Clin Psychiatry 1993;54:305-8.
11. Hirschfeld RM, Allen MH, McEvoy JP, et al. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. J Clin Psychiatry 1999;60:815-18.
12. Bowden CL, Calabrese JR, McElroy SL, et al. A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Divalproex Maintenance Study Group. Arch Gen Psychiatry 2000;57:481-9.
13. Keck PE, Jr, Bowden CL, Meinhold JM, et al. Relationship between serum valproate and lithium levels and efficacy and tolerability in bipolar maintenance therapy. Int J Psychiatry Clin Pract (in press).
14. Davis LL, Bartolucci A, Petty F. Divalproex in the treatment of bipolar depression: a placebo-controlled study. J Affect Disord 2005;85:259-66.
15. Sachs GS, Collins MA, Altshuler LL, et al. Divalproex sodium versus placebo for the treatment of bipolar depression (abstract presentation). San Juan, PR: American College of Neuropsychopharmacology annual meeting, 2001.
16. Wilder BJ. Pharmacokinetics of valproate and carbamazepine. J Clin Psychopharmacol 1992;12(suppl 1):64S-68S.
When every minute counts: What workup is sufficient for diagnosis under pressure?
Police officers bring Mr. A, age 25, to the emergency department (ED) in handcuffs after an alleged assault at work. He is calm but will provide no information about himself. ED staff don’t know if he has been using illicit substances, is on medications, or has any medical conditions.
Mr. A says the FBI is after him, but he makes no threats to ED staff. He talks about milking cows on a farm and of hearing animal sounds, though he lives in the city. After about 30 minutes, he consents to a lab draw and provides a urine sample.
Because no charges are pending and Mr. A is semi-cooperative, police remove his handcuffs and leave him in the care of two ED security officers. He is given something to eat and drink and seems fairly content. He asks how long he will need to stay in the room but does not demand to leave.
In a fast-paced ED, physicians might not notice signs of psychiatric illness, such as Mr. A’s paranoid and delusional thinking. By being familiar with techniques to manage patients’ psychiatric emergencies, you can help your ED colleagues:
- establish working psychiatric diagnoses and medical causes of psychiatric symptoms in the fast-paced ED
- maintain a safe ED environment for patients and clinicians.
What ed patients want
To understand how ED patients feel, put yourself in Mr. A’s shoes. You were at work and began to hallucinate. You believed your boss was out to harm you, and in fear you made comments perceived as threatening.
The next thing you know, you’re in a police car with handcuffs on. All of your coworkers witnessed your embarrassment. Now you are in a small ED room, wondering what’s going to happen next. Are you going to be put in a straight jacket and a padded room?
Patients may experience anxiety-provoking thoughts whether they come to the ED voluntarily or involuntarily. Fear and confusion can affect their behavior in the ED, and how providers respond to patients in crisis can escalate or de-escalate an already-difficult situation.
Psychiatric illness in the ed
Mr. A may have a psychiatric disorder, as do at least 3% of patients seen in EDs.1 This figure may be low, however:
- Kunen et al2 asserted that EDs are underdiagnosing psychiatric disorders, given a U.S. Department of Health and Human Services 1999 estimate that 20% to 28% of Americans have psychiatric illnesses. Using ED discharge records across 6 months in three emergency departments, the authors found the psychiatric diagnosis rate to be 5.27% in 33,000 ED visits.
- Another study, done in a university teaching hospital ED, showed that ED physicians trained to focus on patients’ presenting problems often missed comorbid medical or psychiatric illnesses.
In the randomized, controlled trial by Schriger et al,3 218 patients with nonspecific complaints suggesting occult psychiatric illness (such as chronic headache, abdominal pain, or back pain) completed the Primary Care Evaluation of Mental Disorders (PRIME-MD) questionnaire. This 27-item self-report asks questions about mood, alcohol use, obsessive-compulsive symptoms, phobias, and somatoform symptoms.
Participants were then randomly assigned to “report” or “nonreport” groups, depending on whether or not ED physicians received their PRIME-MD scores. Even when informed of patients’ psychiatric symptoms, ED physicians rarely diagnosed or treated psychiatric disorders. Lack of mental status documentation and psychiatric interviews was apparent, the authors noted.
Case continued: a toxic cocktail
Mr. A’s urine drug screen and lab results are positive for benzodiazepines, methamphetamines, and cannabis. The staff decide Mr. A will require further observation and detoxification, and he is told this. A bed is not available at the hospital, however, and calls to nearby facilities find no empty beds.
As time passes, Mr. A shows signs of agitation and arousal. He paces the examination room—his jaw clenched and his face flushed—and begins raising his voice, asking to be discharged.
Recommendations. Unpleasantness is sometimes unavoidable, but no one in the ED has tried to create an alliance with Mr. A (Box). Try to make patients’ ED experiences as positive as possible. Make it clear that you share a common goal: to help the patient feel better. In fact, psychiatric patients and emergency psychiatrists have similar ideas about what constitutes quality ED care. When surveyed,4 ED patients said they preferred:
- verbal interventions compared with medications
- a collaborative approach with ED physicians
- having medications selected for their specific problems, medication experiences, and choices
- benzodiazepines rather than conventional antipsychotics such as haloperidol.
Treat patients with respect, and preserve their sense of dignity
Offer patients choices when reasonable to help them feel they have some control
Strongly (and early) encourage smokers to accept nicotine replacement to avoid withdrawal and heightened arousal
Offer food, beverages, a blanket, or other comfort measures that would not compromise safety (do not give hot coffee, in case the patient throws the cup at someone)
Allow patients to call a loved one, friend, or pastor (offer a cordless phone to avoid strangulation attempts)
Allow relatives or friends to sit and talk with the patient if this would not compromise safety
Keep patients informed on what is going on and why
Answer questions asked by the patient and family or friends
Offer oral medications first
Get to know your security staff well
A pragmatic workup
Medical illnesses such as delirium, stroke, drug toxicity, or urinary tract infections can trigger or worsen psychiatric illness (Table 1).5 Comorbidities such as diabetes, hypertension, obesity, and heart disease are common in patients with psychiatric disorders, and psychotropics can cause or exacerbate these conditions.
In the high-pressure ED, a sufficient workup for complicated medical conditions lies somewhere between extensive/unnecessary and inadequate. Thus, determining an exact diagnosis is not as important as establishing a diagnostic category to guide emergency treatment.
Think of psychiatric disorders as they are organized in DSM-IV-TR—mood, anxiety, psychotic, substance use/withdrawal/intoxication, cognitive, adjustment, somatoform, and personality disorders—and whether they are primary or secondary to a general medical condition or substance use. For example:
- Anxiety disorder secondary to a general medical condition means the history, physical exam, or lab reports suggest a medical condition is the direct physiologic cause of the mood disturbance.
- Methamphetamine-induced psychotic disorder would be the diagnosis if methamphetamines are presumed to be causing a patient’s psychotic symptoms.
Hospitalization. ED staff often develop a treatment plan based on a patient’s clinical picture and a working diagnosis. The plan hinges on deciding if the patient needs to be admitted to the hospital. Admission may be warranted for life-threatening medical conditions or safety issues, such as threats to self or others or inability to care for oneself at home. Other issues come into play—such as starting or changing medications and follow-up to ensure continuity of care—if you decide to discharge the patient.
Even after medical clearance, patients in the psychiatric emergency service may have underlying medical illnesses (Table 2).6
Table 1
Medical disorders that can cause psychiatric symptoms
| Medical/toxic disorders | Examples |
|---|---|
| Alcohol and drugs of abuse | Amphetamines (including methamphetamine), cocaine, heroin, Jimson weed, ketamine, marijuana, MDMA (‘Ecstasy’), LSD, PCP |
| Prescription drugs | Antibiotics, anticholinergics, anticonvulsants, antihypertensives, benzodiazepines, chemotherapeutic agents, cimetidine, corticosteroids, digitalis, narcotics, propranolol, sleep medications, tricyclic antidepressants |
| CNS disease | Hypertensive encephalopathy, intracranial aneurysm, metastases, normal pressure hydrocephalus, postictal nonconvulsive status, primary CNS infection, seizure disorders, stroke, subdural hematoma, tumor |
| Infections | Acute rheumatic fever, diphtheria, malaria, Legionnaires’ disease, pneumonia, Rocky Mountain spotted fever, sepsis, syphilis, typhoid fever, urinary tract infection |
| Metabolic/endocrine disorders | Adrenal disease, diabetic ketoacidosis, hepatic encephalopathy, hypoglycemia, pituitary dysfunction, renal disease, serum electrolyte imbalances (sodium, potassium, calcium), thyroid disease, vitamin deficiencies, Wilson’s disease |
| Cardiopulmonary disease | Arrhythmias, congestive heart failure, COPD/asthma, myocardial infarction, pulmonary embolism |
| Miscellaneous | Anemia, lupus, multiple sclerosis, temporal arteritis, vasculitis |
| Source: Reprinted with permission from Williams ER, Shepherd SM. Medical clearance of psychiatric patients. Emerg Med Clin North Am 2000;18(2):185-90. Copyright 2000, Elsevier. | |
Table 2
Reasonable medical assessment in psychiatric emergencies
| DO |
| Obtain a medical history, the best determinant of medical need |
| Listen to patients. If they say they have a medical condition, believe them; if they say they don’t, try to believe them |
| Thoroughly check vital signs |
| Conduct a focused physical examination |
| Maintain a high index of suspicion |
Be selective with laboratory testing. Check:
|
| DON’T |
| Order blanket laboratory screening |
| Order an ECG in healthy young patients in the absence of clinical findings |
| Order chest radiography in the absence of known disease/exposure/symptoms |
| Source: Reprinted from Currier GW. Medical assessment on the psychiatric emergency service. Psychiatric Issues in Emergency Care Settings 2004;3(July):17, with permission from Cliggott Publishing Group of CMP Healthcare Media. Copyright 2004. |
Overwhelming demand
In the study of ED patient preferences,4 one-fifth of patients said they went to the ED because they lacked access to routine mental health care. Therefore, besides psychiatric conditions caused by medical illnesses, ED physicians can see patients with any primary psychiatric diagnosis, including mood and anxiety disorders and psychosis.
Under pressures of time and limited collateral information, ED staff must:
- individualize psychiatric treatment
- consider use of medications and/or restraints
- rule out life-threatening causes for psychiatric symptoms
- stabilize patients and prevent injury to self and others.
These tasks are becoming increasingly difficult as more and more patients present to emergency rooms. Nationally, ED visits increased from 19 million in 1992 to 108 million in 2000, according to the U.S. Department of Health and Human Services.1
Psychiatric patients are seeking ED care in greater numbers, and the number of those staying longer than anticipated (“boarding”) also has increased, according to a 2004 survey of 340 physicians by the American College of Emergency Physicians, American Psychiatric Association, National Mental Health Association, and National Alliance for the Mentally Ill. Surveyed physicians blamed inadequate Medicaid funding and bed shortages for the increasing ED visits.7
In crowded emergency rooms, where patients wait longer and longer to be seen, the influx of acutely ill psychiatric patients increases the risks of agitation, violence, and injury, as well as litigation.8
Case continued: going up in smoke
Recognizing Mr. A’s arousal, ED staff tries to reassure him and offers him food, something to drink, a phone Call, and a magazine. When these attempts fail to de-escalate his agitation, staff offers to make him more comfortable by giving oral lorazepam, which he adamantly refuses. He is told again that he must stay until a transfer facility is found for him.
Mr. A then demands to go outside “for a smoke.” When he is told ED patients cannot leave to smoke and is offered nicotine replacement, he begins to scream and lunges at one of the security officers. He is extremely strong, and additional officers are summoned. He retreats inside the room, slams the door, shatters the door window with a chair, and begins punching the broken glass. He slides to the floor in a vasovagal reaction at the sight of his bleeding hands but soon becomes combative again.
Staff give Mr. A IM haloperidol, 10 mg, and lorazepam, 2 mg, to manage his extreme agitation and place him in physical restraints to protect him and others. Within 25 to 30 minutes he is calm, and a safe environment has been re-established. The lacerations on his hands are sutured, and he is admitted to an inpatient psychiatric hospital for further stabilization and treatment.
No place for complacency
Mr. A’s experience illustrates how situations can become dangerous when precautions are not taken. Five steps can help you prepare and protect yourself when evaluating patients in the ED:
- seek the patient history
- evaluate the context in which the patient is being assessed
- identify arousal states (fear, anger, confusion, and humiliation)
- structure the interview for safety
- keep your guard up during the clinical encounter.9
Risk is high when law enforcement officers bring a patient to the ED. Be on guard, even if the patient is 80 years old and in a wheelchair. Complacency has no place in the ED; prepare as much as you can before interviewing the patient.
When restraints are needed. Involuntary medication and/or restraints may be necessary when reasonable interventions have failed, the patient will not cooperate, and he or she is exhibiting behavior/symptoms that could result in injury. Approximately 10% to 20% of psychiatric patients require physical or chemical restraint in the ED.10
Expert consensus guidelines suggest starting with verbal intervention, voluntary medication, and show of force, although emergency medication may be a reasonable first treatment (Algorithm).11 Offer oral medication first; IM medications carry risks including acute dystonia and akathisia, although these can be treated.
Lorazepam, 1 to 2 mg oral/IM, combined with haloperidol, 2 to 5 mg oral/IM, is a reasonable start in most cases. If the patient remains extremely agitated, the same medications and dosages can be repeated 30 to 60 minutes after the initial administration.12
Conventional oral/IM agents are usually more readily available in the ED than atypical antipsychotics, which must be ordered from the pharmacy. Recent FDA black-box warnings also emphasize that atypical antipsychotics are approved only for treating schizophrenia, acute manic and mixed episodes of bipolar I disorder, and for maintenance treatment in bipolar disorder. When compared with placebo, atypical antipsychotics have been associated with:
- increased risk for cerebrovascular events in elderly patients with dementia
- death in elderly patients with dementia-related psychosis.
Atypicals may be more appropriate than conventional antipsychotics for emergency treatment of agitation and aggression in some patients with complicating medical conditions or histories. For example, avoid high-potency conventional antipsychotics in patients with a history of extrapyramidal side effects and in those with mental retardation/developmental delay.11 Similarly, avoid benzodiazepines in patients with chronic obstructive pulmonary disease (COPD) or a history of drug-seeking behavior or drug abuse.
Of course not all psychiatric interventions in the ED are involuntary. For example, the ED physician may start an antidepressant for a patient diagnosed with mild to moderate depression for whom hospitalization is not indicated. Characteristics of patients who may be good candidates for starting antidepressants in the ED include a clear diagnosis, no substance abuse, low suicide risk, no psychosis or agitation, available social supports, clear follow-up plan, desire to begin treatment, and ability to pay for and obtain medications.13
Algorithm Consensus guideline for treating a behavioral emergency
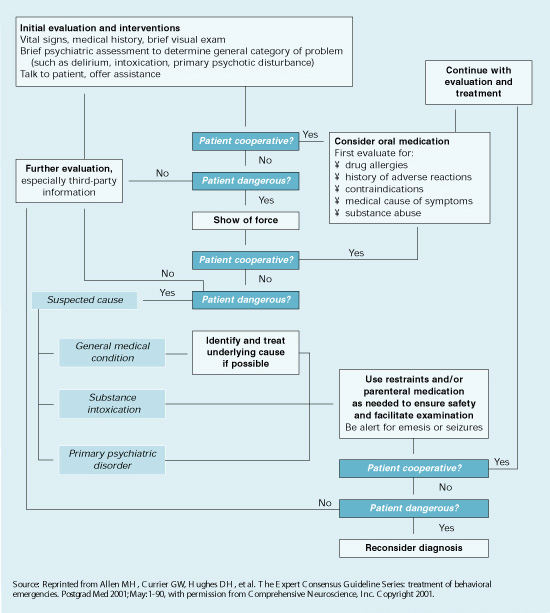
Related resources
- Allen MH, Currier GW, Hughes DH, et al. The Expert Consensus Guideline Series: Treatment of behavioral emergencies. Postgrad Med 2001;May(Spec No):1-88.
- American College of Emergency Physicians. www.acep.org
- National Alliance on Mental Illness. www.nami.org
Drug brand names
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. McCaig LF, Ly N. National Hospital Ambulatory Medical Care Survey: 2000 emergency department summary. Adv Data 2002;327:1-27.
2. Kunen S, Niederhauser R, Smith PO, et al. Race disparities in psychiatric rates in emergency departments. J Consult Clin Psychol 2005;73(1):116-126.
3. Schriger DL, Gibbons PS, Langone CA, et al. Enabling the diagnosis of occult psychiatric illness in the emergency department: a randomized, controlled trial of the computerized, self-administered PRIME-MD diagnostic system. Ann Emerg Med 2001;37(2):132-40.
4. Allen M, Carpenter D, Sheets JL, et al. What do consumers say they want and need during a psychiatric emergency? J Psychiatr Pract 2003;9(1):39-58.
5. Williams ER, Shepherd SM. Medical clearance of psychiatric patients. Emerg Med Clin North Am 2000;18(2):185-98.
6. Allen MH, Currier GW. Medical assessment on the psychiatric emergency service. New Dir Ment Health Serv 1999;82:21-8.
7. Mulligan K. ER docs report large increase in psychiatric patients. Psychiatr News 2004;39(12):10.-
8. Karcz A, Holbrook J, Auerbach BS, et al. Preventability of malpractice claims in emergency medicine: a closed claims study. Ann Emerg Med 1990;19(8):865-73.
9. Battaglia J. Is this patient dangerous? 5 steps to assess risk for violence. Current Psychiatry 2004;3(2):14-21.
10. De Fruyt J, Demyttenaere K. Rapid tranquilization: new approaches in the emergency treatment of behavioral disturbances. Eur Psychiatry 2004;19:243-9.
11. Allen MH, Currier GW, Hughes DH, et al. The Expert Consensus Guideline Series: Treatment of behavioral emergencies. Postgrad Med 2001;May(Spec No):1-88.
12. Hughes DH. Acute psychopharmacological management of the aggressive psychotic patient. Psychiatr Serv 1999;50(9):1135-7.
13. Glick RL. Starting antidepressant treatment in the emergency setting. Psychiatric Issues in Emergency Care Settings 2004;3(2):6-10.
Police officers bring Mr. A, age 25, to the emergency department (ED) in handcuffs after an alleged assault at work. He is calm but will provide no information about himself. ED staff don’t know if he has been using illicit substances, is on medications, or has any medical conditions.
Mr. A says the FBI is after him, but he makes no threats to ED staff. He talks about milking cows on a farm and of hearing animal sounds, though he lives in the city. After about 30 minutes, he consents to a lab draw and provides a urine sample.
Because no charges are pending and Mr. A is semi-cooperative, police remove his handcuffs and leave him in the care of two ED security officers. He is given something to eat and drink and seems fairly content. He asks how long he will need to stay in the room but does not demand to leave.
In a fast-paced ED, physicians might not notice signs of psychiatric illness, such as Mr. A’s paranoid and delusional thinking. By being familiar with techniques to manage patients’ psychiatric emergencies, you can help your ED colleagues:
- establish working psychiatric diagnoses and medical causes of psychiatric symptoms in the fast-paced ED
- maintain a safe ED environment for patients and clinicians.
What ed patients want
To understand how ED patients feel, put yourself in Mr. A’s shoes. You were at work and began to hallucinate. You believed your boss was out to harm you, and in fear you made comments perceived as threatening.
The next thing you know, you’re in a police car with handcuffs on. All of your coworkers witnessed your embarrassment. Now you are in a small ED room, wondering what’s going to happen next. Are you going to be put in a straight jacket and a padded room?
Patients may experience anxiety-provoking thoughts whether they come to the ED voluntarily or involuntarily. Fear and confusion can affect their behavior in the ED, and how providers respond to patients in crisis can escalate or de-escalate an already-difficult situation.
Psychiatric illness in the ed
Mr. A may have a psychiatric disorder, as do at least 3% of patients seen in EDs.1 This figure may be low, however:
- Kunen et al2 asserted that EDs are underdiagnosing psychiatric disorders, given a U.S. Department of Health and Human Services 1999 estimate that 20% to 28% of Americans have psychiatric illnesses. Using ED discharge records across 6 months in three emergency departments, the authors found the psychiatric diagnosis rate to be 5.27% in 33,000 ED visits.
- Another study, done in a university teaching hospital ED, showed that ED physicians trained to focus on patients’ presenting problems often missed comorbid medical or psychiatric illnesses.
In the randomized, controlled trial by Schriger et al,3 218 patients with nonspecific complaints suggesting occult psychiatric illness (such as chronic headache, abdominal pain, or back pain) completed the Primary Care Evaluation of Mental Disorders (PRIME-MD) questionnaire. This 27-item self-report asks questions about mood, alcohol use, obsessive-compulsive symptoms, phobias, and somatoform symptoms.
Participants were then randomly assigned to “report” or “nonreport” groups, depending on whether or not ED physicians received their PRIME-MD scores. Even when informed of patients’ psychiatric symptoms, ED physicians rarely diagnosed or treated psychiatric disorders. Lack of mental status documentation and psychiatric interviews was apparent, the authors noted.
Case continued: a toxic cocktail
Mr. A’s urine drug screen and lab results are positive for benzodiazepines, methamphetamines, and cannabis. The staff decide Mr. A will require further observation and detoxification, and he is told this. A bed is not available at the hospital, however, and calls to nearby facilities find no empty beds.
As time passes, Mr. A shows signs of agitation and arousal. He paces the examination room—his jaw clenched and his face flushed—and begins raising his voice, asking to be discharged.
Recommendations. Unpleasantness is sometimes unavoidable, but no one in the ED has tried to create an alliance with Mr. A (Box). Try to make patients’ ED experiences as positive as possible. Make it clear that you share a common goal: to help the patient feel better. In fact, psychiatric patients and emergency psychiatrists have similar ideas about what constitutes quality ED care. When surveyed,4 ED patients said they preferred:
- verbal interventions compared with medications
- a collaborative approach with ED physicians
- having medications selected for their specific problems, medication experiences, and choices
- benzodiazepines rather than conventional antipsychotics such as haloperidol.
Treat patients with respect, and preserve their sense of dignity
Offer patients choices when reasonable to help them feel they have some control
Strongly (and early) encourage smokers to accept nicotine replacement to avoid withdrawal and heightened arousal
Offer food, beverages, a blanket, or other comfort measures that would not compromise safety (do not give hot coffee, in case the patient throws the cup at someone)
Allow patients to call a loved one, friend, or pastor (offer a cordless phone to avoid strangulation attempts)
Allow relatives or friends to sit and talk with the patient if this would not compromise safety
Keep patients informed on what is going on and why
Answer questions asked by the patient and family or friends
Offer oral medications first
Get to know your security staff well
A pragmatic workup
Medical illnesses such as delirium, stroke, drug toxicity, or urinary tract infections can trigger or worsen psychiatric illness (Table 1).5 Comorbidities such as diabetes, hypertension, obesity, and heart disease are common in patients with psychiatric disorders, and psychotropics can cause or exacerbate these conditions.
In the high-pressure ED, a sufficient workup for complicated medical conditions lies somewhere between extensive/unnecessary and inadequate. Thus, determining an exact diagnosis is not as important as establishing a diagnostic category to guide emergency treatment.
Think of psychiatric disorders as they are organized in DSM-IV-TR—mood, anxiety, psychotic, substance use/withdrawal/intoxication, cognitive, adjustment, somatoform, and personality disorders—and whether they are primary or secondary to a general medical condition or substance use. For example:
- Anxiety disorder secondary to a general medical condition means the history, physical exam, or lab reports suggest a medical condition is the direct physiologic cause of the mood disturbance.
- Methamphetamine-induced psychotic disorder would be the diagnosis if methamphetamines are presumed to be causing a patient’s psychotic symptoms.
Hospitalization. ED staff often develop a treatment plan based on a patient’s clinical picture and a working diagnosis. The plan hinges on deciding if the patient needs to be admitted to the hospital. Admission may be warranted for life-threatening medical conditions or safety issues, such as threats to self or others or inability to care for oneself at home. Other issues come into play—such as starting or changing medications and follow-up to ensure continuity of care—if you decide to discharge the patient.
Even after medical clearance, patients in the psychiatric emergency service may have underlying medical illnesses (Table 2).6
Table 1
Medical disorders that can cause psychiatric symptoms
| Medical/toxic disorders | Examples |
|---|---|
| Alcohol and drugs of abuse | Amphetamines (including methamphetamine), cocaine, heroin, Jimson weed, ketamine, marijuana, MDMA (‘Ecstasy’), LSD, PCP |
| Prescription drugs | Antibiotics, anticholinergics, anticonvulsants, antihypertensives, benzodiazepines, chemotherapeutic agents, cimetidine, corticosteroids, digitalis, narcotics, propranolol, sleep medications, tricyclic antidepressants |
| CNS disease | Hypertensive encephalopathy, intracranial aneurysm, metastases, normal pressure hydrocephalus, postictal nonconvulsive status, primary CNS infection, seizure disorders, stroke, subdural hematoma, tumor |
| Infections | Acute rheumatic fever, diphtheria, malaria, Legionnaires’ disease, pneumonia, Rocky Mountain spotted fever, sepsis, syphilis, typhoid fever, urinary tract infection |
| Metabolic/endocrine disorders | Adrenal disease, diabetic ketoacidosis, hepatic encephalopathy, hypoglycemia, pituitary dysfunction, renal disease, serum electrolyte imbalances (sodium, potassium, calcium), thyroid disease, vitamin deficiencies, Wilson’s disease |
| Cardiopulmonary disease | Arrhythmias, congestive heart failure, COPD/asthma, myocardial infarction, pulmonary embolism |
| Miscellaneous | Anemia, lupus, multiple sclerosis, temporal arteritis, vasculitis |
| Source: Reprinted with permission from Williams ER, Shepherd SM. Medical clearance of psychiatric patients. Emerg Med Clin North Am 2000;18(2):185-90. Copyright 2000, Elsevier. | |
Table 2
Reasonable medical assessment in psychiatric emergencies
| DO |
| Obtain a medical history, the best determinant of medical need |
| Listen to patients. If they say they have a medical condition, believe them; if they say they don’t, try to believe them |
| Thoroughly check vital signs |
| Conduct a focused physical examination |
| Maintain a high index of suspicion |
Be selective with laboratory testing. Check:
|
| DON’T |
| Order blanket laboratory screening |
| Order an ECG in healthy young patients in the absence of clinical findings |
| Order chest radiography in the absence of known disease/exposure/symptoms |
| Source: Reprinted from Currier GW. Medical assessment on the psychiatric emergency service. Psychiatric Issues in Emergency Care Settings 2004;3(July):17, with permission from Cliggott Publishing Group of CMP Healthcare Media. Copyright 2004. |
Overwhelming demand
In the study of ED patient preferences,4 one-fifth of patients said they went to the ED because they lacked access to routine mental health care. Therefore, besides psychiatric conditions caused by medical illnesses, ED physicians can see patients with any primary psychiatric diagnosis, including mood and anxiety disorders and psychosis.
Under pressures of time and limited collateral information, ED staff must:
- individualize psychiatric treatment
- consider use of medications and/or restraints
- rule out life-threatening causes for psychiatric symptoms
- stabilize patients and prevent injury to self and others.
These tasks are becoming increasingly difficult as more and more patients present to emergency rooms. Nationally, ED visits increased from 19 million in 1992 to 108 million in 2000, according to the U.S. Department of Health and Human Services.1
Psychiatric patients are seeking ED care in greater numbers, and the number of those staying longer than anticipated (“boarding”) also has increased, according to a 2004 survey of 340 physicians by the American College of Emergency Physicians, American Psychiatric Association, National Mental Health Association, and National Alliance for the Mentally Ill. Surveyed physicians blamed inadequate Medicaid funding and bed shortages for the increasing ED visits.7
In crowded emergency rooms, where patients wait longer and longer to be seen, the influx of acutely ill psychiatric patients increases the risks of agitation, violence, and injury, as well as litigation.8
Case continued: going up in smoke
Recognizing Mr. A’s arousal, ED staff tries to reassure him and offers him food, something to drink, a phone Call, and a magazine. When these attempts fail to de-escalate his agitation, staff offers to make him more comfortable by giving oral lorazepam, which he adamantly refuses. He is told again that he must stay until a transfer facility is found for him.
Mr. A then demands to go outside “for a smoke.” When he is told ED patients cannot leave to smoke and is offered nicotine replacement, he begins to scream and lunges at one of the security officers. He is extremely strong, and additional officers are summoned. He retreats inside the room, slams the door, shatters the door window with a chair, and begins punching the broken glass. He slides to the floor in a vasovagal reaction at the sight of his bleeding hands but soon becomes combative again.
Staff give Mr. A IM haloperidol, 10 mg, and lorazepam, 2 mg, to manage his extreme agitation and place him in physical restraints to protect him and others. Within 25 to 30 minutes he is calm, and a safe environment has been re-established. The lacerations on his hands are sutured, and he is admitted to an inpatient psychiatric hospital for further stabilization and treatment.
No place for complacency
Mr. A’s experience illustrates how situations can become dangerous when precautions are not taken. Five steps can help you prepare and protect yourself when evaluating patients in the ED:
- seek the patient history
- evaluate the context in which the patient is being assessed
- identify arousal states (fear, anger, confusion, and humiliation)
- structure the interview for safety
- keep your guard up during the clinical encounter.9
Risk is high when law enforcement officers bring a patient to the ED. Be on guard, even if the patient is 80 years old and in a wheelchair. Complacency has no place in the ED; prepare as much as you can before interviewing the patient.
When restraints are needed. Involuntary medication and/or restraints may be necessary when reasonable interventions have failed, the patient will not cooperate, and he or she is exhibiting behavior/symptoms that could result in injury. Approximately 10% to 20% of psychiatric patients require physical or chemical restraint in the ED.10
Expert consensus guidelines suggest starting with verbal intervention, voluntary medication, and show of force, although emergency medication may be a reasonable first treatment (Algorithm).11 Offer oral medication first; IM medications carry risks including acute dystonia and akathisia, although these can be treated.
Lorazepam, 1 to 2 mg oral/IM, combined with haloperidol, 2 to 5 mg oral/IM, is a reasonable start in most cases. If the patient remains extremely agitated, the same medications and dosages can be repeated 30 to 60 minutes after the initial administration.12
Conventional oral/IM agents are usually more readily available in the ED than atypical antipsychotics, which must be ordered from the pharmacy. Recent FDA black-box warnings also emphasize that atypical antipsychotics are approved only for treating schizophrenia, acute manic and mixed episodes of bipolar I disorder, and for maintenance treatment in bipolar disorder. When compared with placebo, atypical antipsychotics have been associated with:
- increased risk for cerebrovascular events in elderly patients with dementia
- death in elderly patients with dementia-related psychosis.
Atypicals may be more appropriate than conventional antipsychotics for emergency treatment of agitation and aggression in some patients with complicating medical conditions or histories. For example, avoid high-potency conventional antipsychotics in patients with a history of extrapyramidal side effects and in those with mental retardation/developmental delay.11 Similarly, avoid benzodiazepines in patients with chronic obstructive pulmonary disease (COPD) or a history of drug-seeking behavior or drug abuse.
Of course not all psychiatric interventions in the ED are involuntary. For example, the ED physician may start an antidepressant for a patient diagnosed with mild to moderate depression for whom hospitalization is not indicated. Characteristics of patients who may be good candidates for starting antidepressants in the ED include a clear diagnosis, no substance abuse, low suicide risk, no psychosis or agitation, available social supports, clear follow-up plan, desire to begin treatment, and ability to pay for and obtain medications.13
Algorithm Consensus guideline for treating a behavioral emergency
Related resources
- Allen MH, Currier GW, Hughes DH, et al. The Expert Consensus Guideline Series: Treatment of behavioral emergencies. Postgrad Med 2001;May(Spec No):1-88.
- American College of Emergency Physicians. www.acep.org
- National Alliance on Mental Illness. www.nami.org
Drug brand names
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Police officers bring Mr. A, age 25, to the emergency department (ED) in handcuffs after an alleged assault at work. He is calm but will provide no information about himself. ED staff don’t know if he has been using illicit substances, is on medications, or has any medical conditions.
Mr. A says the FBI is after him, but he makes no threats to ED staff. He talks about milking cows on a farm and of hearing animal sounds, though he lives in the city. After about 30 minutes, he consents to a lab draw and provides a urine sample.
Because no charges are pending and Mr. A is semi-cooperative, police remove his handcuffs and leave him in the care of two ED security officers. He is given something to eat and drink and seems fairly content. He asks how long he will need to stay in the room but does not demand to leave.
In a fast-paced ED, physicians might not notice signs of psychiatric illness, such as Mr. A’s paranoid and delusional thinking. By being familiar with techniques to manage patients’ psychiatric emergencies, you can help your ED colleagues:
- establish working psychiatric diagnoses and medical causes of psychiatric symptoms in the fast-paced ED
- maintain a safe ED environment for patients and clinicians.
What ed patients want
To understand how ED patients feel, put yourself in Mr. A’s shoes. You were at work and began to hallucinate. You believed your boss was out to harm you, and in fear you made comments perceived as threatening.
The next thing you know, you’re in a police car with handcuffs on. All of your coworkers witnessed your embarrassment. Now you are in a small ED room, wondering what’s going to happen next. Are you going to be put in a straight jacket and a padded room?
Patients may experience anxiety-provoking thoughts whether they come to the ED voluntarily or involuntarily. Fear and confusion can affect their behavior in the ED, and how providers respond to patients in crisis can escalate or de-escalate an already-difficult situation.
Psychiatric illness in the ed
Mr. A may have a psychiatric disorder, as do at least 3% of patients seen in EDs.1 This figure may be low, however:
- Kunen et al2 asserted that EDs are underdiagnosing psychiatric disorders, given a U.S. Department of Health and Human Services 1999 estimate that 20% to 28% of Americans have psychiatric illnesses. Using ED discharge records across 6 months in three emergency departments, the authors found the psychiatric diagnosis rate to be 5.27% in 33,000 ED visits.
- Another study, done in a university teaching hospital ED, showed that ED physicians trained to focus on patients’ presenting problems often missed comorbid medical or psychiatric illnesses.
In the randomized, controlled trial by Schriger et al,3 218 patients with nonspecific complaints suggesting occult psychiatric illness (such as chronic headache, abdominal pain, or back pain) completed the Primary Care Evaluation of Mental Disorders (PRIME-MD) questionnaire. This 27-item self-report asks questions about mood, alcohol use, obsessive-compulsive symptoms, phobias, and somatoform symptoms.
Participants were then randomly assigned to “report” or “nonreport” groups, depending on whether or not ED physicians received their PRIME-MD scores. Even when informed of patients’ psychiatric symptoms, ED physicians rarely diagnosed or treated psychiatric disorders. Lack of mental status documentation and psychiatric interviews was apparent, the authors noted.
Case continued: a toxic cocktail
Mr. A’s urine drug screen and lab results are positive for benzodiazepines, methamphetamines, and cannabis. The staff decide Mr. A will require further observation and detoxification, and he is told this. A bed is not available at the hospital, however, and calls to nearby facilities find no empty beds.
As time passes, Mr. A shows signs of agitation and arousal. He paces the examination room—his jaw clenched and his face flushed—and begins raising his voice, asking to be discharged.
Recommendations. Unpleasantness is sometimes unavoidable, but no one in the ED has tried to create an alliance with Mr. A (Box). Try to make patients’ ED experiences as positive as possible. Make it clear that you share a common goal: to help the patient feel better. In fact, psychiatric patients and emergency psychiatrists have similar ideas about what constitutes quality ED care. When surveyed,4 ED patients said they preferred:
- verbal interventions compared with medications
- a collaborative approach with ED physicians
- having medications selected for their specific problems, medication experiences, and choices
- benzodiazepines rather than conventional antipsychotics such as haloperidol.
Treat patients with respect, and preserve their sense of dignity
Offer patients choices when reasonable to help them feel they have some control
Strongly (and early) encourage smokers to accept nicotine replacement to avoid withdrawal and heightened arousal
Offer food, beverages, a blanket, or other comfort measures that would not compromise safety (do not give hot coffee, in case the patient throws the cup at someone)
Allow patients to call a loved one, friend, or pastor (offer a cordless phone to avoid strangulation attempts)
Allow relatives or friends to sit and talk with the patient if this would not compromise safety
Keep patients informed on what is going on and why
Answer questions asked by the patient and family or friends
Offer oral medications first
Get to know your security staff well
A pragmatic workup
Medical illnesses such as delirium, stroke, drug toxicity, or urinary tract infections can trigger or worsen psychiatric illness (Table 1).5 Comorbidities such as diabetes, hypertension, obesity, and heart disease are common in patients with psychiatric disorders, and psychotropics can cause or exacerbate these conditions.
In the high-pressure ED, a sufficient workup for complicated medical conditions lies somewhere between extensive/unnecessary and inadequate. Thus, determining an exact diagnosis is not as important as establishing a diagnostic category to guide emergency treatment.
Think of psychiatric disorders as they are organized in DSM-IV-TR—mood, anxiety, psychotic, substance use/withdrawal/intoxication, cognitive, adjustment, somatoform, and personality disorders—and whether they are primary or secondary to a general medical condition or substance use. For example:
- Anxiety disorder secondary to a general medical condition means the history, physical exam, or lab reports suggest a medical condition is the direct physiologic cause of the mood disturbance.
- Methamphetamine-induced psychotic disorder would be the diagnosis if methamphetamines are presumed to be causing a patient’s psychotic symptoms.
Hospitalization. ED staff often develop a treatment plan based on a patient’s clinical picture and a working diagnosis. The plan hinges on deciding if the patient needs to be admitted to the hospital. Admission may be warranted for life-threatening medical conditions or safety issues, such as threats to self or others or inability to care for oneself at home. Other issues come into play—such as starting or changing medications and follow-up to ensure continuity of care—if you decide to discharge the patient.
Even after medical clearance, patients in the psychiatric emergency service may have underlying medical illnesses (Table 2).6
Table 1
Medical disorders that can cause psychiatric symptoms
| Medical/toxic disorders | Examples |
|---|---|
| Alcohol and drugs of abuse | Amphetamines (including methamphetamine), cocaine, heroin, Jimson weed, ketamine, marijuana, MDMA (‘Ecstasy’), LSD, PCP |
| Prescription drugs | Antibiotics, anticholinergics, anticonvulsants, antihypertensives, benzodiazepines, chemotherapeutic agents, cimetidine, corticosteroids, digitalis, narcotics, propranolol, sleep medications, tricyclic antidepressants |
| CNS disease | Hypertensive encephalopathy, intracranial aneurysm, metastases, normal pressure hydrocephalus, postictal nonconvulsive status, primary CNS infection, seizure disorders, stroke, subdural hematoma, tumor |
| Infections | Acute rheumatic fever, diphtheria, malaria, Legionnaires’ disease, pneumonia, Rocky Mountain spotted fever, sepsis, syphilis, typhoid fever, urinary tract infection |
| Metabolic/endocrine disorders | Adrenal disease, diabetic ketoacidosis, hepatic encephalopathy, hypoglycemia, pituitary dysfunction, renal disease, serum electrolyte imbalances (sodium, potassium, calcium), thyroid disease, vitamin deficiencies, Wilson’s disease |
| Cardiopulmonary disease | Arrhythmias, congestive heart failure, COPD/asthma, myocardial infarction, pulmonary embolism |
| Miscellaneous | Anemia, lupus, multiple sclerosis, temporal arteritis, vasculitis |
| Source: Reprinted with permission from Williams ER, Shepherd SM. Medical clearance of psychiatric patients. Emerg Med Clin North Am 2000;18(2):185-90. Copyright 2000, Elsevier. | |
Table 2
Reasonable medical assessment in psychiatric emergencies
| DO |
| Obtain a medical history, the best determinant of medical need |
| Listen to patients. If they say they have a medical condition, believe them; if they say they don’t, try to believe them |
| Thoroughly check vital signs |
| Conduct a focused physical examination |
| Maintain a high index of suspicion |
Be selective with laboratory testing. Check:
|
| DON’T |
| Order blanket laboratory screening |
| Order an ECG in healthy young patients in the absence of clinical findings |
| Order chest radiography in the absence of known disease/exposure/symptoms |
| Source: Reprinted from Currier GW. Medical assessment on the psychiatric emergency service. Psychiatric Issues in Emergency Care Settings 2004;3(July):17, with permission from Cliggott Publishing Group of CMP Healthcare Media. Copyright 2004. |
Overwhelming demand
In the study of ED patient preferences,4 one-fifth of patients said they went to the ED because they lacked access to routine mental health care. Therefore, besides psychiatric conditions caused by medical illnesses, ED physicians can see patients with any primary psychiatric diagnosis, including mood and anxiety disorders and psychosis.
Under pressures of time and limited collateral information, ED staff must:
- individualize psychiatric treatment
- consider use of medications and/or restraints
- rule out life-threatening causes for psychiatric symptoms
- stabilize patients and prevent injury to self and others.
These tasks are becoming increasingly difficult as more and more patients present to emergency rooms. Nationally, ED visits increased from 19 million in 1992 to 108 million in 2000, according to the U.S. Department of Health and Human Services.1
Psychiatric patients are seeking ED care in greater numbers, and the number of those staying longer than anticipated (“boarding”) also has increased, according to a 2004 survey of 340 physicians by the American College of Emergency Physicians, American Psychiatric Association, National Mental Health Association, and National Alliance for the Mentally Ill. Surveyed physicians blamed inadequate Medicaid funding and bed shortages for the increasing ED visits.7
In crowded emergency rooms, where patients wait longer and longer to be seen, the influx of acutely ill psychiatric patients increases the risks of agitation, violence, and injury, as well as litigation.8
Case continued: going up in smoke
Recognizing Mr. A’s arousal, ED staff tries to reassure him and offers him food, something to drink, a phone Call, and a magazine. When these attempts fail to de-escalate his agitation, staff offers to make him more comfortable by giving oral lorazepam, which he adamantly refuses. He is told again that he must stay until a transfer facility is found for him.
Mr. A then demands to go outside “for a smoke.” When he is told ED patients cannot leave to smoke and is offered nicotine replacement, he begins to scream and lunges at one of the security officers. He is extremely strong, and additional officers are summoned. He retreats inside the room, slams the door, shatters the door window with a chair, and begins punching the broken glass. He slides to the floor in a vasovagal reaction at the sight of his bleeding hands but soon becomes combative again.
Staff give Mr. A IM haloperidol, 10 mg, and lorazepam, 2 mg, to manage his extreme agitation and place him in physical restraints to protect him and others. Within 25 to 30 minutes he is calm, and a safe environment has been re-established. The lacerations on his hands are sutured, and he is admitted to an inpatient psychiatric hospital for further stabilization and treatment.
No place for complacency
Mr. A’s experience illustrates how situations can become dangerous when precautions are not taken. Five steps can help you prepare and protect yourself when evaluating patients in the ED:
- seek the patient history
- evaluate the context in which the patient is being assessed
- identify arousal states (fear, anger, confusion, and humiliation)
- structure the interview for safety
- keep your guard up during the clinical encounter.9
Risk is high when law enforcement officers bring a patient to the ED. Be on guard, even if the patient is 80 years old and in a wheelchair. Complacency has no place in the ED; prepare as much as you can before interviewing the patient.
When restraints are needed. Involuntary medication and/or restraints may be necessary when reasonable interventions have failed, the patient will not cooperate, and he or she is exhibiting behavior/symptoms that could result in injury. Approximately 10% to 20% of psychiatric patients require physical or chemical restraint in the ED.10
Expert consensus guidelines suggest starting with verbal intervention, voluntary medication, and show of force, although emergency medication may be a reasonable first treatment (Algorithm).11 Offer oral medication first; IM medications carry risks including acute dystonia and akathisia, although these can be treated.
Lorazepam, 1 to 2 mg oral/IM, combined with haloperidol, 2 to 5 mg oral/IM, is a reasonable start in most cases. If the patient remains extremely agitated, the same medications and dosages can be repeated 30 to 60 minutes after the initial administration.12
Conventional oral/IM agents are usually more readily available in the ED than atypical antipsychotics, which must be ordered from the pharmacy. Recent FDA black-box warnings also emphasize that atypical antipsychotics are approved only for treating schizophrenia, acute manic and mixed episodes of bipolar I disorder, and for maintenance treatment in bipolar disorder. When compared with placebo, atypical antipsychotics have been associated with:
- increased risk for cerebrovascular events in elderly patients with dementia
- death in elderly patients with dementia-related psychosis.
Atypicals may be more appropriate than conventional antipsychotics for emergency treatment of agitation and aggression in some patients with complicating medical conditions or histories. For example, avoid high-potency conventional antipsychotics in patients with a history of extrapyramidal side effects and in those with mental retardation/developmental delay.11 Similarly, avoid benzodiazepines in patients with chronic obstructive pulmonary disease (COPD) or a history of drug-seeking behavior or drug abuse.
Of course not all psychiatric interventions in the ED are involuntary. For example, the ED physician may start an antidepressant for a patient diagnosed with mild to moderate depression for whom hospitalization is not indicated. Characteristics of patients who may be good candidates for starting antidepressants in the ED include a clear diagnosis, no substance abuse, low suicide risk, no psychosis or agitation, available social supports, clear follow-up plan, desire to begin treatment, and ability to pay for and obtain medications.13
Algorithm Consensus guideline for treating a behavioral emergency
Related resources
- Allen MH, Currier GW, Hughes DH, et al. The Expert Consensus Guideline Series: Treatment of behavioral emergencies. Postgrad Med 2001;May(Spec No):1-88.
- American College of Emergency Physicians. www.acep.org
- National Alliance on Mental Illness. www.nami.org
Drug brand names
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Lorazepam • Ativan
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. McCaig LF, Ly N. National Hospital Ambulatory Medical Care Survey: 2000 emergency department summary. Adv Data 2002;327:1-27.
2. Kunen S, Niederhauser R, Smith PO, et al. Race disparities in psychiatric rates in emergency departments. J Consult Clin Psychol 2005;73(1):116-126.
3. Schriger DL, Gibbons PS, Langone CA, et al. Enabling the diagnosis of occult psychiatric illness in the emergency department: a randomized, controlled trial of the computerized, self-administered PRIME-MD diagnostic system. Ann Emerg Med 2001;37(2):132-40.
4. Allen M, Carpenter D, Sheets JL, et al. What do consumers say they want and need during a psychiatric emergency? J Psychiatr Pract 2003;9(1):39-58.
5. Williams ER, Shepherd SM. Medical clearance of psychiatric patients. Emerg Med Clin North Am 2000;18(2):185-98.
6. Allen MH, Currier GW. Medical assessment on the psychiatric emergency service. New Dir Ment Health Serv 1999;82:21-8.
7. Mulligan K. ER docs report large increase in psychiatric patients. Psychiatr News 2004;39(12):10.-
8. Karcz A, Holbrook J, Auerbach BS, et al. Preventability of malpractice claims in emergency medicine: a closed claims study. Ann Emerg Med 1990;19(8):865-73.
9. Battaglia J. Is this patient dangerous? 5 steps to assess risk for violence. Current Psychiatry 2004;3(2):14-21.
10. De Fruyt J, Demyttenaere K. Rapid tranquilization: new approaches in the emergency treatment of behavioral disturbances. Eur Psychiatry 2004;19:243-9.
11. Allen MH, Currier GW, Hughes DH, et al. The Expert Consensus Guideline Series: Treatment of behavioral emergencies. Postgrad Med 2001;May(Spec No):1-88.
12. Hughes DH. Acute psychopharmacological management of the aggressive psychotic patient. Psychiatr Serv 1999;50(9):1135-7.
13. Glick RL. Starting antidepressant treatment in the emergency setting. Psychiatric Issues in Emergency Care Settings 2004;3(2):6-10.
1. McCaig LF, Ly N. National Hospital Ambulatory Medical Care Survey: 2000 emergency department summary. Adv Data 2002;327:1-27.
2. Kunen S, Niederhauser R, Smith PO, et al. Race disparities in psychiatric rates in emergency departments. J Consult Clin Psychol 2005;73(1):116-126.
3. Schriger DL, Gibbons PS, Langone CA, et al. Enabling the diagnosis of occult psychiatric illness in the emergency department: a randomized, controlled trial of the computerized, self-administered PRIME-MD diagnostic system. Ann Emerg Med 2001;37(2):132-40.
4. Allen M, Carpenter D, Sheets JL, et al. What do consumers say they want and need during a psychiatric emergency? J Psychiatr Pract 2003;9(1):39-58.
5. Williams ER, Shepherd SM. Medical clearance of psychiatric patients. Emerg Med Clin North Am 2000;18(2):185-98.
6. Allen MH, Currier GW. Medical assessment on the psychiatric emergency service. New Dir Ment Health Serv 1999;82:21-8.
7. Mulligan K. ER docs report large increase in psychiatric patients. Psychiatr News 2004;39(12):10.-
8. Karcz A, Holbrook J, Auerbach BS, et al. Preventability of malpractice claims in emergency medicine: a closed claims study. Ann Emerg Med 1990;19(8):865-73.
9. Battaglia J. Is this patient dangerous? 5 steps to assess risk for violence. Current Psychiatry 2004;3(2):14-21.
10. De Fruyt J, Demyttenaere K. Rapid tranquilization: new approaches in the emergency treatment of behavioral disturbances. Eur Psychiatry 2004;19:243-9.
11. Allen MH, Currier GW, Hughes DH, et al. The Expert Consensus Guideline Series: Treatment of behavioral emergencies. Postgrad Med 2001;May(Spec No):1-88.
12. Hughes DH. Acute psychopharmacological management of the aggressive psychotic patient. Psychiatr Serv 1999;50(9):1135-7.
13. Glick RL. Starting antidepressant treatment in the emergency setting. Psychiatric Issues in Emergency Care Settings 2004;3(2):6-10.
Which cholinesterase inhibitor for early dementia?
Using a cholinesterase inhibitor (ChEI) makes sense for any disorder with a significant cholinergic deficit, such as Alzheimer’s disease (AD) and other forms of mild-to-moderate dementia (Box 1).1-3 Yet the ChEIs tacrine, donepezil, rivastigmine, and galantamine have pharmacologic differences, and individual patients respond differently to them.
To help you choose the safest, most effective treatment for each patient, we discuss:
- three cases that show how ChEIs differ in mechanism of action, administration, and side effects
- evidence of ChEIs’ efficacy in AD—for which they are approved—and in other dementias for which they have been tried
- when to switch agents, and how long to continue treatment.
Probable Alzheimer’s disease (AD) accounts for 64% of all dementias in the United States. Less-common causes include:
- vascular dementia (5%)
- combined vascular dementia and AD (10%)
- probable dementia with Lewy bodies, Parkinson’s dementia, or diffuse Lewy body disease (9%)
- Lewy body variant of AD, or AD and dementia with Lewy bodies (6%)
- frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, or Creutzfeldt-Jakob disease (6%).2,3
In our experience, many primary care physicians choose to follow their patients with dementia, even when clinical features are atypical or suggest unusual causes. Psychiatrists are asked most often to assist in diagnosis and management of patients with:
- uncommon dementias, including frontotemporal dementia or dementia with Lewy bodies
- rapidly progressive dementia
- dementia in a patient age
- dementia with psychiatric comorbidities or severe behavior disturbances.4
How Cheis Differ
Although dementia remains incurable, recognizing cognitive decline early allows you to start ChEI therapy before substantial neuronal loss occurs (Box 2).3,4 The goal of early treatment is to improve or stabilize cognition, behavior, and activities of daily living for as long as possible.
In comparison studies,5,6 ChEIs have shown differences in tolerability but not consistent differences in efficacy for mild to moderate AD—though these studies had methodologic limitations. Because the agents appear similarly effective, the initial ChEI choice often depends on how their differences might benefit your patient (Table 1). Consider the following cases:
An early dementia diagnosis enables you educate the patient and family (Box 3) and begin the most effective treatment for the person with cognitive decline. Although dementia remains incurable, early recognition presents the opportunity to start cholinesterase inhibitors before substantial neuronal loss occurs.3,4
Patient workup. The Alzheimer’s Association offers online information for health care professionals on AD diagnosis and treatment protocols (see Related resources). A detailed history, physical examination, and Mini-Mental State Examination (MMSE) are necessary if you suspect Alzheimer’s or a related dementia.
Also recommended are a comprehensive metabolic screen, complete blood counts with differential, urine analysis, serum B12 and folate studies, homocysteine levels, thyroid studies, chest radiography, ECG, lipid profile, and brain scan (MRI or CT). Perform studies such as the rapid plasma reagin test for syphilis and HIV testing as appropriate.
Similarities and differences among cholinesterase inhibitors
| Tacrine | Donepezil | Rivastigmine | Galantamine | |
|---|---|---|---|---|
| Administration | Four times daily | Once daily | Twice daily with full meals | Once daily (extended-release formulation) |
| AChE inhibitor | Yes | Yes | Yes | Yes |
| BuChE inhibitor | Yes | No | Yes | No |
| Allosteric modulation of nicotinic receptor | No | Yes | No | Yes |
| Pharmacodynamic nicotinic/muscarinic effect | Yes | Yes | Yes | Yes |
| GI side effects | Present | Present | Present | Present |
| Hepatotoxicity | Present | Absent | Absent | Absent |
| Metabolism | CYP-450 | CYP-450 | Autohydrolysis | CYP-450 |
| Drug–drug interactions | Yes | Yes | None reported | Yes |
| AChE: acetylcholinesterase | ||||
| BuChE: butyrylcholinesterase | ||||
| CYP-450: cytochrome P-450 hepatic isoenzymes | ||||
Case 1: Gradual Memory Loss
Mrs. J, age 76, has experienced a slow, insidious memory decline across 5 years. She has become socially withdrawn and shows some language difficulties. She has had peptic ulcer disease and often does not take medications as prescribed.
Her psychiatrist diagnoses probable AD and chooses donepezil with its easy dosing schedule because of Mrs. J’s history of nonadherence. Donepezil’s GI tolerability is also a factor in this choice because of the patient’s peptic ulcer disease.
Case 2: Dementia And Motor Deficits
Mr. L, age 82, has gradually developed memory loss and parkinsonian symptoms, including slowness of movement and shuffling gait. He has visual hallucinations of people and episodic confusion. His medications include warfarin and digoxin for atrial fibrillation and congestive heart failure.
Mr. L is diagnosed with probable dementia with Lewy bodies. His psychiatrist chooses rivastigmine because it has shown efficacy in this type of dementia and is not known to interact significantly with cardiovascular medications.
Case 3: Stroke, Then Rapid Decline
Mrs. D, age 68, has a history of hypertension and suffered a stroke in the past. Her family says her memory and behavior—anger outbursts and excessive irritability—have worsened rapidly across 2 years. Examination reveals some focal neurologic deficits.
Her psychiatrist diagnoses probable vascular dementia and chooses galantamine for its efficacy in patients with this dementia type. Mrs. D has no history of GI illness and will likely tolerate the drug’s GI side effects. Follow-up care will include monitoring for tolerability.
Mechanism. Donepezil inhibits the enzyme acetylcholinesterase, and rivastigmine inhibits acetylcholinesterase and butyrylcholinesterase. Galantamine inhibits acetylcholinesterase and shows allosteric modulation of the presynaptic nicotinic receptor.
Data indicating that rivastigmine is particularly effective in patients with rapidly progressive illness is consistent with the possible advantage of inhibiting both butyrylcholinesterase and acetylcholinesterase. It has been argued that galantamine’s binding to nicotinic receptors modulates their function, which may enhance acetylcholine release.
Among the three agents, only rivastigmine shows a consistent, linear dose-response relationship. It is rapidly and extensively metabolized, primarily via cholinesterase-mediated hydrolysis to the decarbamylated metabolite (autohydrolysis). Minimal metabolism occurs via the major cytochrome P (CYP)-450 isoenzymes. Donepezil and galantamine are metabolized by isoenzymes 2D6 and 3A4 and undergo glucuronidation.7
Drug interactions. Because rivastigmine avoids hepatic metabolism, interactions with drugs metabolized by CYP-450 isoenzymes have not been reported.8
Donepezil interacts with ketoconazole and quinidine, which inhibit donepezil metabolism and increase mean donepezil concentrations. Galantamine interacts with ketoconazole, paroxetine, and erythromycin, which increase mean galantamine concentrations.9
Administration. Donepezil and extended-release galantamine are given once daily because of their long half-lives, whereas regular galantamine and rivastigmine are taken twice daily with meals to minimize GI effects (Table 2). Nausea and vomiting can occur with any of the ChEIs but are more common and troublesome with rivastigmine and galantamine.
Table 2
How to use cholinesterase inhibitors for patients with dementia
| Drug | Recommended dosing | Possible side effects | Titration | Administration |
|---|---|---|---|---|
| Tacrine | Initial: 40 mg/d Maximum: 160 mg/d | Liver damage causing increase in ALT levels, GI effects (nausea, indigestion, vomiting, diarrhea, abdominal pain), skin rash | Dosage can be increased every 4 weeks | Divide into four doses; take on empty stomach |
| Donepezil | Initial: 5 mg/d Maximum: 10 mg/d | GI effects (nausea, diarrhea, vomiting, loss of appetite), insomnia, muscle cramps, fatigue | Increase dosage after 4 weeks | Once daily in morning or at bedtime |
| Rivastigmine | Initial: 3 mg/d Maximum: 12 mg/d | GI effects (nausea, vomiting, loss of appetite, weight loss, diarrhea, heartburn) | Increase dosage every 4 weeks | Twice daily after meals |
| Galantamine (regular, ER) | Initial: 8 mg/d Maximum: 24 mg/d | GI effects (nausea, vomiting, diarrhea, weight loss), possible increased mortality risk in patients with MCI | Increase dosage every 4 weeks | Regular: Twice daily after meals ER: Once daily after a meal |
| ALT: alanine transferase | ||||
| ER: extended-release formulation | ||||
| MCI: mild cognitive impairment | ||||
Efficacy In Early AD
In controlled clinical trials, all four ChEIs have significantly improved cognition, behavior, and activities of daily living in patients with mild-to-moderate AD.10-12 Tacrine—the first FDA-approved ChEI—is rarely used because its associated hepatoxicity requires ongoing liver enzyme monitoring.13 Among the other three:
Donepezil. A review of 16 trials involving 4,365 participants10 found significant benefits in cognitive functioning, activities of daily living, and behavior in persons with mild, moderate, or severe AD who were treated with donepezil for 12, 24, or 52 weeks.
Rivastigmine improved or maintained cognitive function, activities of daily living, and behavior for up to 52 weeks in patients with mild to moderate AD, according to a review of studies from 1995 to 2002.11 GI irritation was the most common adverse effect. Giving rivastigmine for up to 2 years may reduce the cost of caring for patients with AD, mostly by delaying nursing home placement.
Galantamine has beneficial effects on cognition, global function, activities of daily living, and behavior in patients with AD, vascular dementia, and AD with cerebrovascular components, according to a review of clinical studies.12 Adverse events are generally mild to moderate, transient, and gastrointestinal.
Efficacy In Other Dementias
In addition to their FDA-approved use for mildto-moderate AD, ChEIs also have been studied in persons with other types of dementia and mild cognitive impairment (MCI).
Dementia with Lewy bodies. Rivastigmine given with flexible titration from 6 to 12 mg/d improved behavior in 120 patients with Lewy body dementia.14 In the double-blind, multicenter study, patients taking rivastigmine, mean 9.7 mg/d for 20 weeks, were less apathetic and anxious and had fewer delusions and hallucinations than did those taking placebo. The drug was judged to be safe and well tolerated.
Vascular dementia. Patients with vascular dementia showed improved cognition and global function when treated with donepezil, 5 or 10 mg/d, for up to 24 weeks. Donepezil was well tolerated in this combined analysis of two randomized, placebo-controlled trials.15
Kumar et al16 compared two rivastigmine dosages in patients with mild-to-moderate AD, some of whom also had vascular dementia risk factors. Patients were randomly assigned to placebo, low-dose rivastigmine (1 to 4 mg/d), or high-dose rivastigmine (6 to 12 mg/d) for 26 weeks. Cognition, activities of daily living, and disease severity improved with rivastigmine in patients with or without vascular risk factors. Greater benefit was seen with high-dose than low-dose rivastigmine and in patients with AD plus vascular risk factors than in those with AD alone.
In a multicenter, double-blind trial,17 patients with vascular dementia or AD with vascular risk factors received galantamine, up to 24 mg/d, or placebo for 6 months. Compared with controls, those taking galantamine showed improved cognition, behavior, and function. The drug overall was well tolerated, with nausea and vomiting the most common side effects.
Parkinson’s dementia. Emre et al18 evaluated rivastigmine’s efficacy and safety in patients whose mild-to-moderate dementia developed at least 2 years after a clinical diagnosis of Parkinson’s disease (PD). Patients were randomly assigned to placebo or rivastigmine, 3 to 12 mg/d, for 24 weeks, and 410 of 541 enrollees completed the study. Compared with placebo, rivastigmine was associated with statistically significant improvements in cognition and global measures in dementia associated with PD but also with higher rates of nausea, vomiting, and tremor. PD’s motor symptoms did not change significantly in either group.
Mixed dementia states. As mentioned, galantamine improved cognitive and noncognitive abilities in patients with vascular dementia or AD with vascular risk factors in a 6-month, double-blind trial.17 Patients who received galantamine or placebo could then continue open-label galantamine, 24 mg/d, for another 6 months. In patients treated the full 12 months, galantamine continued to improve or maintain:
- cognition, based on Alzheimer’s Disease Assessment Scale-cognitive subscale scores
- functional ability, measured by the 40-item Disability Assessment for Dementia
- behavior, measured by the Neuro-psychiatric Inventory.19
After 12 months, the rivastigmine-treated patients were less behaviorally impaired than the matched patients, and their caregivers reported reduced stress. Rivastigmine did not prevent cognitive deterioration, as assessed with the Mini-Mental State Examination (MMSE).
Mild cognitive impairment. Persons with MCI have objective psychometric evidence of memory loss compared with their peers, but they are not significantly impaired in activities of daily living or other cognitive functions (language, abstract thinking, or problem-solving).
At this time, we do not recommend using ChEIs to treat MCI. These agents have shown little benefit and potential risk in patients who do not meet diagnostic criteria for dementia:
- Salloway et al21 tested donepezil’s efficacy and safety in 270 patients with MCI in a 24-week, double-blind, placebo-controlled trial. Donepezil was started at 5 mg/d for 42 days, then escalated to 10 mg/d. Compared with placebo, donepezil showed no significant effects on recall, but some improvements were seen in attention and psychomotor speed.
- In two unpublished placebo-controlled trials, galantamine did not improve memory when given for 2 years to elderly patients with MCI. A precaution was added to the drug’s prescribing information because 13 of the 1,026 patients taking galantamine died, compared with 1 of 1,022 taking placebo. Vascular disease caused one-half of the galantamine group deaths. No evidence of increased mortality risk has been seen in studies of galantamine in patients with mild-to-moderate AD, for which it is indicated.
Getting The Greatest Response
To gauge response to ChEI therapy, family reports about the patient are helpful—such as that cognition has improved or cognitive decline has not progressed as rapidly as before. Assessment tools such as the MMSE can document improvement or stabilization.
We recommend trying an initial ChEI for at least 6 months to determine its efficacy. If your patient cannot tolerate one ChEI or fails to respond to initial treatment, two consensus panels22,23 recommend that you consider changing ChEIs:
- If switching because of intolerable side effects, wait at least 2 to 3 days after stopping the first ChEI before starting another.
- If switching because of poor response, you can start a different ChEI immediately after the first one is stopped.
- Cholinesterase inhibitors may help improve or stabilize cognition, behavior, and/or activities of daily living
- Persons receiving these agents may decline more slowly than those who have not been treated
- Common side effects include nausea, vomiting, diarrhea, and loss of appetite
- Other less-common side effects are muscle cramps, slowed heart rate, dizziness, and fainting
- Because of differences in these agents, it may make sense to switch to another cholinesterase inhibitor if the patient has intolerable side effects or does not improve with the first one tried
Related resources
- Alzheimer’s Association. Information for health care professionals:
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- • Treating cognitive symptoms www.alz.org/Health/Treating/symptoms.asp
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- American Association for Geriatric Psychiatry. Information for older patients and their families on Alzheimer’s disease, other dementias. www.gmhfonline.org/gmhf/consumer/alzheimers.html.
- Tacrine • Cognex
- Donepezil • Aricept
- Rivastigmine • Exelon
- Galantamine • Razadyne (was Reminyl)
Drs. Kamat and LeFevre report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Boehringer Ingelheim Pharmaceuticals, Cyberonics, Eli Lilly and Co., Eunoe, Forest Pharmaceuticals, Novartis Pharmaceuticals Corp., Pfizer, and Wyeth Pharmaceuticals. He is a consultant to AstraZeneca Pharmaceuticals, Forest Pharmaceuticals, Janssen Pharmaceutica, KV Pharma, Novartis Pharmaceuticals Corp., Organon International, and Sanofi-Synthelabo.
Acknowledgment
The authors thank Anjali Baliga, MD, for her contribution and help in preparing this article
1. Small GW, Rabins PV, Barry PP, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA 1997;278(16):1363-71.
2. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 suppl 5):S4-S9.
3. Grossberg GT, Lake JT. The role of the psychiatrist in Alzheimer’s disease. J Clin Psychiatry 1998;59(suppl 9):3-6.
4. Doraiswamy PM, Steffens DC, Pitchumoni S, Tabrizi S. Early recognition of Alzheimer’s disease: what is consensual? What is controversial? What is practical? J Clin Psychiatry 1998;59(suppl 13):6-18.
5. Wilkinson DG, Passmore AP, Bullock R, et al. A multinational, randomised, 12-week, comparative study of donepezil and rivastigmine in patients with mild to moderate Alzheimer’s disease. Int J Clin Pract 2002;56(6):441-6
6. Jones RW, Soininen H, Hager K, et al. A multinational, randomised, 12-week study comparing the effects of donepezil and galantamine in patients with mild to moderate Alzheimer’s disease. Int J Geriatr Psychiatry 2004;19(1):58-67.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twentytwo classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. U. S. Bureau of the Census. 2004 International database: Midyear population, by age and sex. Table 094. U.S. Bureau of the Census; 2004.
9. Reminyl (galantamine HBr). Physicians’ desk reference (59th ed). Montvale, NJ: Thomson PDR; 2005:1739.
10. Birks JS, Harvey R. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev 2003;(3):CD001190.-
11. Williams BR, Nazarians A, Gill MA. A review of rivastigmine: a reversible cholinesterase inhibitor. Clin Ther 2003;25(6):1634-53.
12. Corey-Bloom J. Galantamine: a review of its use in Alzheimer’s disease and vascular dementia. Int J Clin Pract 2003;57(3):219-23.
13. Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994;271(13):992-8.
14. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000;356(9247):2031-6.
15. Passmore AP, Bayer AJ, Steinhagen-Thiessen E. Cognitive, global, and functional benefits of donepezil in Alzheimer’s disease and vascular dementia: results from large-scale clinical trials. J Neurol Sci 2005;229-30:141-6.
16. Kumar V, Anand R, Messina J, et al. An efficacy and safety analysis of rivastigmine in Alzheimer’s disease patients with concurrent vascular risk factors. Eur J Neurol 2000;7(2):159-69.
17. Kurz AF, Erkinjuntti T, Gauthier S, et al. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002;359(9314):1283-90.
18. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351(24):2509-18.
19. Erkinjuntti T, Kurz A, Small GW, et al. An open-label extension trial of galantamine in patients with probable vascular dementia and mixed dementia. Clin Ther 2003;25(6):1765-82.
20. Moretti R, Torre P, Antonello RM, et al. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004;21(14):931-7.
21. Salloway S, Ferris S, Kluger A, et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology 2004;63(4):651-7.
22. Emre M. Switching cholinesterase inhibitors in patients with Alzheimer’s disease. Int J Clin Pract Suppl 2002;(127):64-72.
23. Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int J Clin Pract Suppl 2002;(127):45-63.
Using a cholinesterase inhibitor (ChEI) makes sense for any disorder with a significant cholinergic deficit, such as Alzheimer’s disease (AD) and other forms of mild-to-moderate dementia (Box 1).1-3 Yet the ChEIs tacrine, donepezil, rivastigmine, and galantamine have pharmacologic differences, and individual patients respond differently to them.
To help you choose the safest, most effective treatment for each patient, we discuss:
- three cases that show how ChEIs differ in mechanism of action, administration, and side effects
- evidence of ChEIs’ efficacy in AD—for which they are approved—and in other dementias for which they have been tried
- when to switch agents, and how long to continue treatment.
Probable Alzheimer’s disease (AD) accounts for 64% of all dementias in the United States. Less-common causes include:
- vascular dementia (5%)
- combined vascular dementia and AD (10%)
- probable dementia with Lewy bodies, Parkinson’s dementia, or diffuse Lewy body disease (9%)
- Lewy body variant of AD, or AD and dementia with Lewy bodies (6%)
- frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, or Creutzfeldt-Jakob disease (6%).2,3
In our experience, many primary care physicians choose to follow their patients with dementia, even when clinical features are atypical or suggest unusual causes. Psychiatrists are asked most often to assist in diagnosis and management of patients with:
- uncommon dementias, including frontotemporal dementia or dementia with Lewy bodies
- rapidly progressive dementia
- dementia in a patient age
- dementia with psychiatric comorbidities or severe behavior disturbances.4
How Cheis Differ
Although dementia remains incurable, recognizing cognitive decline early allows you to start ChEI therapy before substantial neuronal loss occurs (Box 2).3,4 The goal of early treatment is to improve or stabilize cognition, behavior, and activities of daily living for as long as possible.
In comparison studies,5,6 ChEIs have shown differences in tolerability but not consistent differences in efficacy for mild to moderate AD—though these studies had methodologic limitations. Because the agents appear similarly effective, the initial ChEI choice often depends on how their differences might benefit your patient (Table 1). Consider the following cases:
An early dementia diagnosis enables you educate the patient and family (Box 3) and begin the most effective treatment for the person with cognitive decline. Although dementia remains incurable, early recognition presents the opportunity to start cholinesterase inhibitors before substantial neuronal loss occurs.3,4
Patient workup. The Alzheimer’s Association offers online information for health care professionals on AD diagnosis and treatment protocols (see Related resources). A detailed history, physical examination, and Mini-Mental State Examination (MMSE) are necessary if you suspect Alzheimer’s or a related dementia.
Also recommended are a comprehensive metabolic screen, complete blood counts with differential, urine analysis, serum B12 and folate studies, homocysteine levels, thyroid studies, chest radiography, ECG, lipid profile, and brain scan (MRI or CT). Perform studies such as the rapid plasma reagin test for syphilis and HIV testing as appropriate.
Similarities and differences among cholinesterase inhibitors
| Tacrine | Donepezil | Rivastigmine | Galantamine | |
|---|---|---|---|---|
| Administration | Four times daily | Once daily | Twice daily with full meals | Once daily (extended-release formulation) |
| AChE inhibitor | Yes | Yes | Yes | Yes |
| BuChE inhibitor | Yes | No | Yes | No |
| Allosteric modulation of nicotinic receptor | No | Yes | No | Yes |
| Pharmacodynamic nicotinic/muscarinic effect | Yes | Yes | Yes | Yes |
| GI side effects | Present | Present | Present | Present |
| Hepatotoxicity | Present | Absent | Absent | Absent |
| Metabolism | CYP-450 | CYP-450 | Autohydrolysis | CYP-450 |
| Drug–drug interactions | Yes | Yes | None reported | Yes |
| AChE: acetylcholinesterase | ||||
| BuChE: butyrylcholinesterase | ||||
| CYP-450: cytochrome P-450 hepatic isoenzymes | ||||
Case 1: Gradual Memory Loss
Mrs. J, age 76, has experienced a slow, insidious memory decline across 5 years. She has become socially withdrawn and shows some language difficulties. She has had peptic ulcer disease and often does not take medications as prescribed.
Her psychiatrist diagnoses probable AD and chooses donepezil with its easy dosing schedule because of Mrs. J’s history of nonadherence. Donepezil’s GI tolerability is also a factor in this choice because of the patient’s peptic ulcer disease.
Case 2: Dementia And Motor Deficits
Mr. L, age 82, has gradually developed memory loss and parkinsonian symptoms, including slowness of movement and shuffling gait. He has visual hallucinations of people and episodic confusion. His medications include warfarin and digoxin for atrial fibrillation and congestive heart failure.
Mr. L is diagnosed with probable dementia with Lewy bodies. His psychiatrist chooses rivastigmine because it has shown efficacy in this type of dementia and is not known to interact significantly with cardiovascular medications.
Case 3: Stroke, Then Rapid Decline
Mrs. D, age 68, has a history of hypertension and suffered a stroke in the past. Her family says her memory and behavior—anger outbursts and excessive irritability—have worsened rapidly across 2 years. Examination reveals some focal neurologic deficits.
Her psychiatrist diagnoses probable vascular dementia and chooses galantamine for its efficacy in patients with this dementia type. Mrs. D has no history of GI illness and will likely tolerate the drug’s GI side effects. Follow-up care will include monitoring for tolerability.
Mechanism. Donepezil inhibits the enzyme acetylcholinesterase, and rivastigmine inhibits acetylcholinesterase and butyrylcholinesterase. Galantamine inhibits acetylcholinesterase and shows allosteric modulation of the presynaptic nicotinic receptor.
Data indicating that rivastigmine is particularly effective in patients with rapidly progressive illness is consistent with the possible advantage of inhibiting both butyrylcholinesterase and acetylcholinesterase. It has been argued that galantamine’s binding to nicotinic receptors modulates their function, which may enhance acetylcholine release.
Among the three agents, only rivastigmine shows a consistent, linear dose-response relationship. It is rapidly and extensively metabolized, primarily via cholinesterase-mediated hydrolysis to the decarbamylated metabolite (autohydrolysis). Minimal metabolism occurs via the major cytochrome P (CYP)-450 isoenzymes. Donepezil and galantamine are metabolized by isoenzymes 2D6 and 3A4 and undergo glucuronidation.7
Drug interactions. Because rivastigmine avoids hepatic metabolism, interactions with drugs metabolized by CYP-450 isoenzymes have not been reported.8
Donepezil interacts with ketoconazole and quinidine, which inhibit donepezil metabolism and increase mean donepezil concentrations. Galantamine interacts with ketoconazole, paroxetine, and erythromycin, which increase mean galantamine concentrations.9
Administration. Donepezil and extended-release galantamine are given once daily because of their long half-lives, whereas regular galantamine and rivastigmine are taken twice daily with meals to minimize GI effects (Table 2). Nausea and vomiting can occur with any of the ChEIs but are more common and troublesome with rivastigmine and galantamine.
Table 2
How to use cholinesterase inhibitors for patients with dementia
| Drug | Recommended dosing | Possible side effects | Titration | Administration |
|---|---|---|---|---|
| Tacrine | Initial: 40 mg/d Maximum: 160 mg/d | Liver damage causing increase in ALT levels, GI effects (nausea, indigestion, vomiting, diarrhea, abdominal pain), skin rash | Dosage can be increased every 4 weeks | Divide into four doses; take on empty stomach |
| Donepezil | Initial: 5 mg/d Maximum: 10 mg/d | GI effects (nausea, diarrhea, vomiting, loss of appetite), insomnia, muscle cramps, fatigue | Increase dosage after 4 weeks | Once daily in morning or at bedtime |
| Rivastigmine | Initial: 3 mg/d Maximum: 12 mg/d | GI effects (nausea, vomiting, loss of appetite, weight loss, diarrhea, heartburn) | Increase dosage every 4 weeks | Twice daily after meals |
| Galantamine (regular, ER) | Initial: 8 mg/d Maximum: 24 mg/d | GI effects (nausea, vomiting, diarrhea, weight loss), possible increased mortality risk in patients with MCI | Increase dosage every 4 weeks | Regular: Twice daily after meals ER: Once daily after a meal |
| ALT: alanine transferase | ||||
| ER: extended-release formulation | ||||
| MCI: mild cognitive impairment | ||||
Efficacy In Early AD
In controlled clinical trials, all four ChEIs have significantly improved cognition, behavior, and activities of daily living in patients with mild-to-moderate AD.10-12 Tacrine—the first FDA-approved ChEI—is rarely used because its associated hepatoxicity requires ongoing liver enzyme monitoring.13 Among the other three:
Donepezil. A review of 16 trials involving 4,365 participants10 found significant benefits in cognitive functioning, activities of daily living, and behavior in persons with mild, moderate, or severe AD who were treated with donepezil for 12, 24, or 52 weeks.
Rivastigmine improved or maintained cognitive function, activities of daily living, and behavior for up to 52 weeks in patients with mild to moderate AD, according to a review of studies from 1995 to 2002.11 GI irritation was the most common adverse effect. Giving rivastigmine for up to 2 years may reduce the cost of caring for patients with AD, mostly by delaying nursing home placement.
Galantamine has beneficial effects on cognition, global function, activities of daily living, and behavior in patients with AD, vascular dementia, and AD with cerebrovascular components, according to a review of clinical studies.12 Adverse events are generally mild to moderate, transient, and gastrointestinal.
Efficacy In Other Dementias
In addition to their FDA-approved use for mildto-moderate AD, ChEIs also have been studied in persons with other types of dementia and mild cognitive impairment (MCI).
Dementia with Lewy bodies. Rivastigmine given with flexible titration from 6 to 12 mg/d improved behavior in 120 patients with Lewy body dementia.14 In the double-blind, multicenter study, patients taking rivastigmine, mean 9.7 mg/d for 20 weeks, were less apathetic and anxious and had fewer delusions and hallucinations than did those taking placebo. The drug was judged to be safe and well tolerated.
Vascular dementia. Patients with vascular dementia showed improved cognition and global function when treated with donepezil, 5 or 10 mg/d, for up to 24 weeks. Donepezil was well tolerated in this combined analysis of two randomized, placebo-controlled trials.15
Kumar et al16 compared two rivastigmine dosages in patients with mild-to-moderate AD, some of whom also had vascular dementia risk factors. Patients were randomly assigned to placebo, low-dose rivastigmine (1 to 4 mg/d), or high-dose rivastigmine (6 to 12 mg/d) for 26 weeks. Cognition, activities of daily living, and disease severity improved with rivastigmine in patients with or without vascular risk factors. Greater benefit was seen with high-dose than low-dose rivastigmine and in patients with AD plus vascular risk factors than in those with AD alone.
In a multicenter, double-blind trial,17 patients with vascular dementia or AD with vascular risk factors received galantamine, up to 24 mg/d, or placebo for 6 months. Compared with controls, those taking galantamine showed improved cognition, behavior, and function. The drug overall was well tolerated, with nausea and vomiting the most common side effects.
Parkinson’s dementia. Emre et al18 evaluated rivastigmine’s efficacy and safety in patients whose mild-to-moderate dementia developed at least 2 years after a clinical diagnosis of Parkinson’s disease (PD). Patients were randomly assigned to placebo or rivastigmine, 3 to 12 mg/d, for 24 weeks, and 410 of 541 enrollees completed the study. Compared with placebo, rivastigmine was associated with statistically significant improvements in cognition and global measures in dementia associated with PD but also with higher rates of nausea, vomiting, and tremor. PD’s motor symptoms did not change significantly in either group.
Mixed dementia states. As mentioned, galantamine improved cognitive and noncognitive abilities in patients with vascular dementia or AD with vascular risk factors in a 6-month, double-blind trial.17 Patients who received galantamine or placebo could then continue open-label galantamine, 24 mg/d, for another 6 months. In patients treated the full 12 months, galantamine continued to improve or maintain:
- cognition, based on Alzheimer’s Disease Assessment Scale-cognitive subscale scores
- functional ability, measured by the 40-item Disability Assessment for Dementia
- behavior, measured by the Neuro-psychiatric Inventory.19
After 12 months, the rivastigmine-treated patients were less behaviorally impaired than the matched patients, and their caregivers reported reduced stress. Rivastigmine did not prevent cognitive deterioration, as assessed with the Mini-Mental State Examination (MMSE).
Mild cognitive impairment. Persons with MCI have objective psychometric evidence of memory loss compared with their peers, but they are not significantly impaired in activities of daily living or other cognitive functions (language, abstract thinking, or problem-solving).
At this time, we do not recommend using ChEIs to treat MCI. These agents have shown little benefit and potential risk in patients who do not meet diagnostic criteria for dementia:
- Salloway et al21 tested donepezil’s efficacy and safety in 270 patients with MCI in a 24-week, double-blind, placebo-controlled trial. Donepezil was started at 5 mg/d for 42 days, then escalated to 10 mg/d. Compared with placebo, donepezil showed no significant effects on recall, but some improvements were seen in attention and psychomotor speed.
- In two unpublished placebo-controlled trials, galantamine did not improve memory when given for 2 years to elderly patients with MCI. A precaution was added to the drug’s prescribing information because 13 of the 1,026 patients taking galantamine died, compared with 1 of 1,022 taking placebo. Vascular disease caused one-half of the galantamine group deaths. No evidence of increased mortality risk has been seen in studies of galantamine in patients with mild-to-moderate AD, for which it is indicated.
Getting The Greatest Response
To gauge response to ChEI therapy, family reports about the patient are helpful—such as that cognition has improved or cognitive decline has not progressed as rapidly as before. Assessment tools such as the MMSE can document improvement or stabilization.
We recommend trying an initial ChEI for at least 6 months to determine its efficacy. If your patient cannot tolerate one ChEI or fails to respond to initial treatment, two consensus panels22,23 recommend that you consider changing ChEIs:
- If switching because of intolerable side effects, wait at least 2 to 3 days after stopping the first ChEI before starting another.
- If switching because of poor response, you can start a different ChEI immediately after the first one is stopped.
- Cholinesterase inhibitors may help improve or stabilize cognition, behavior, and/or activities of daily living
- Persons receiving these agents may decline more slowly than those who have not been treated
- Common side effects include nausea, vomiting, diarrhea, and loss of appetite
- Other less-common side effects are muscle cramps, slowed heart rate, dizziness, and fainting
- Because of differences in these agents, it may make sense to switch to another cholinesterase inhibitor if the patient has intolerable side effects or does not improve with the first one tried
Related resources
- Alzheimer’s Association. Information for health care professionals:
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- • Treating cognitive symptoms www.alz.org/Health/Treating/symptoms.asp
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- American Association for Geriatric Psychiatry. Information for older patients and their families on Alzheimer’s disease, other dementias. www.gmhfonline.org/gmhf/consumer/alzheimers.html.
- Tacrine • Cognex
- Donepezil • Aricept
- Rivastigmine • Exelon
- Galantamine • Razadyne (was Reminyl)
Drs. Kamat and LeFevre report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Boehringer Ingelheim Pharmaceuticals, Cyberonics, Eli Lilly and Co., Eunoe, Forest Pharmaceuticals, Novartis Pharmaceuticals Corp., Pfizer, and Wyeth Pharmaceuticals. He is a consultant to AstraZeneca Pharmaceuticals, Forest Pharmaceuticals, Janssen Pharmaceutica, KV Pharma, Novartis Pharmaceuticals Corp., Organon International, and Sanofi-Synthelabo.
Acknowledgment
The authors thank Anjali Baliga, MD, for her contribution and help in preparing this article
Using a cholinesterase inhibitor (ChEI) makes sense for any disorder with a significant cholinergic deficit, such as Alzheimer’s disease (AD) and other forms of mild-to-moderate dementia (Box 1).1-3 Yet the ChEIs tacrine, donepezil, rivastigmine, and galantamine have pharmacologic differences, and individual patients respond differently to them.
To help you choose the safest, most effective treatment for each patient, we discuss:
- three cases that show how ChEIs differ in mechanism of action, administration, and side effects
- evidence of ChEIs’ efficacy in AD—for which they are approved—and in other dementias for which they have been tried
- when to switch agents, and how long to continue treatment.
Probable Alzheimer’s disease (AD) accounts for 64% of all dementias in the United States. Less-common causes include:
- vascular dementia (5%)
- combined vascular dementia and AD (10%)
- probable dementia with Lewy bodies, Parkinson’s dementia, or diffuse Lewy body disease (9%)
- Lewy body variant of AD, or AD and dementia with Lewy bodies (6%)
- frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, or Creutzfeldt-Jakob disease (6%).2,3
In our experience, many primary care physicians choose to follow their patients with dementia, even when clinical features are atypical or suggest unusual causes. Psychiatrists are asked most often to assist in diagnosis and management of patients with:
- uncommon dementias, including frontotemporal dementia or dementia with Lewy bodies
- rapidly progressive dementia
- dementia in a patient age
- dementia with psychiatric comorbidities or severe behavior disturbances.4
How Cheis Differ
Although dementia remains incurable, recognizing cognitive decline early allows you to start ChEI therapy before substantial neuronal loss occurs (Box 2).3,4 The goal of early treatment is to improve or stabilize cognition, behavior, and activities of daily living for as long as possible.
In comparison studies,5,6 ChEIs have shown differences in tolerability but not consistent differences in efficacy for mild to moderate AD—though these studies had methodologic limitations. Because the agents appear similarly effective, the initial ChEI choice often depends on how their differences might benefit your patient (Table 1). Consider the following cases:
An early dementia diagnosis enables you educate the patient and family (Box 3) and begin the most effective treatment for the person with cognitive decline. Although dementia remains incurable, early recognition presents the opportunity to start cholinesterase inhibitors before substantial neuronal loss occurs.3,4
Patient workup. The Alzheimer’s Association offers online information for health care professionals on AD diagnosis and treatment protocols (see Related resources). A detailed history, physical examination, and Mini-Mental State Examination (MMSE) are necessary if you suspect Alzheimer’s or a related dementia.
Also recommended are a comprehensive metabolic screen, complete blood counts with differential, urine analysis, serum B12 and folate studies, homocysteine levels, thyroid studies, chest radiography, ECG, lipid profile, and brain scan (MRI or CT). Perform studies such as the rapid plasma reagin test for syphilis and HIV testing as appropriate.
Similarities and differences among cholinesterase inhibitors
| Tacrine | Donepezil | Rivastigmine | Galantamine | |
|---|---|---|---|---|
| Administration | Four times daily | Once daily | Twice daily with full meals | Once daily (extended-release formulation) |
| AChE inhibitor | Yes | Yes | Yes | Yes |
| BuChE inhibitor | Yes | No | Yes | No |
| Allosteric modulation of nicotinic receptor | No | Yes | No | Yes |
| Pharmacodynamic nicotinic/muscarinic effect | Yes | Yes | Yes | Yes |
| GI side effects | Present | Present | Present | Present |
| Hepatotoxicity | Present | Absent | Absent | Absent |
| Metabolism | CYP-450 | CYP-450 | Autohydrolysis | CYP-450 |
| Drug–drug interactions | Yes | Yes | None reported | Yes |
| AChE: acetylcholinesterase | ||||
| BuChE: butyrylcholinesterase | ||||
| CYP-450: cytochrome P-450 hepatic isoenzymes | ||||
Case 1: Gradual Memory Loss
Mrs. J, age 76, has experienced a slow, insidious memory decline across 5 years. She has become socially withdrawn and shows some language difficulties. She has had peptic ulcer disease and often does not take medications as prescribed.
Her psychiatrist diagnoses probable AD and chooses donepezil with its easy dosing schedule because of Mrs. J’s history of nonadherence. Donepezil’s GI tolerability is also a factor in this choice because of the patient’s peptic ulcer disease.
Case 2: Dementia And Motor Deficits
Mr. L, age 82, has gradually developed memory loss and parkinsonian symptoms, including slowness of movement and shuffling gait. He has visual hallucinations of people and episodic confusion. His medications include warfarin and digoxin for atrial fibrillation and congestive heart failure.
Mr. L is diagnosed with probable dementia with Lewy bodies. His psychiatrist chooses rivastigmine because it has shown efficacy in this type of dementia and is not known to interact significantly with cardiovascular medications.
Case 3: Stroke, Then Rapid Decline
Mrs. D, age 68, has a history of hypertension and suffered a stroke in the past. Her family says her memory and behavior—anger outbursts and excessive irritability—have worsened rapidly across 2 years. Examination reveals some focal neurologic deficits.
Her psychiatrist diagnoses probable vascular dementia and chooses galantamine for its efficacy in patients with this dementia type. Mrs. D has no history of GI illness and will likely tolerate the drug’s GI side effects. Follow-up care will include monitoring for tolerability.
Mechanism. Donepezil inhibits the enzyme acetylcholinesterase, and rivastigmine inhibits acetylcholinesterase and butyrylcholinesterase. Galantamine inhibits acetylcholinesterase and shows allosteric modulation of the presynaptic nicotinic receptor.
Data indicating that rivastigmine is particularly effective in patients with rapidly progressive illness is consistent with the possible advantage of inhibiting both butyrylcholinesterase and acetylcholinesterase. It has been argued that galantamine’s binding to nicotinic receptors modulates their function, which may enhance acetylcholine release.
Among the three agents, only rivastigmine shows a consistent, linear dose-response relationship. It is rapidly and extensively metabolized, primarily via cholinesterase-mediated hydrolysis to the decarbamylated metabolite (autohydrolysis). Minimal metabolism occurs via the major cytochrome P (CYP)-450 isoenzymes. Donepezil and galantamine are metabolized by isoenzymes 2D6 and 3A4 and undergo glucuronidation.7
Drug interactions. Because rivastigmine avoids hepatic metabolism, interactions with drugs metabolized by CYP-450 isoenzymes have not been reported.8
Donepezil interacts with ketoconazole and quinidine, which inhibit donepezil metabolism and increase mean donepezil concentrations. Galantamine interacts with ketoconazole, paroxetine, and erythromycin, which increase mean galantamine concentrations.9
Administration. Donepezil and extended-release galantamine are given once daily because of their long half-lives, whereas regular galantamine and rivastigmine are taken twice daily with meals to minimize GI effects (Table 2). Nausea and vomiting can occur with any of the ChEIs but are more common and troublesome with rivastigmine and galantamine.
Table 2
How to use cholinesterase inhibitors for patients with dementia
| Drug | Recommended dosing | Possible side effects | Titration | Administration |
|---|---|---|---|---|
| Tacrine | Initial: 40 mg/d Maximum: 160 mg/d | Liver damage causing increase in ALT levels, GI effects (nausea, indigestion, vomiting, diarrhea, abdominal pain), skin rash | Dosage can be increased every 4 weeks | Divide into four doses; take on empty stomach |
| Donepezil | Initial: 5 mg/d Maximum: 10 mg/d | GI effects (nausea, diarrhea, vomiting, loss of appetite), insomnia, muscle cramps, fatigue | Increase dosage after 4 weeks | Once daily in morning or at bedtime |
| Rivastigmine | Initial: 3 mg/d Maximum: 12 mg/d | GI effects (nausea, vomiting, loss of appetite, weight loss, diarrhea, heartburn) | Increase dosage every 4 weeks | Twice daily after meals |
| Galantamine (regular, ER) | Initial: 8 mg/d Maximum: 24 mg/d | GI effects (nausea, vomiting, diarrhea, weight loss), possible increased mortality risk in patients with MCI | Increase dosage every 4 weeks | Regular: Twice daily after meals ER: Once daily after a meal |
| ALT: alanine transferase | ||||
| ER: extended-release formulation | ||||
| MCI: mild cognitive impairment | ||||
Efficacy In Early AD
In controlled clinical trials, all four ChEIs have significantly improved cognition, behavior, and activities of daily living in patients with mild-to-moderate AD.10-12 Tacrine—the first FDA-approved ChEI—is rarely used because its associated hepatoxicity requires ongoing liver enzyme monitoring.13 Among the other three:
Donepezil. A review of 16 trials involving 4,365 participants10 found significant benefits in cognitive functioning, activities of daily living, and behavior in persons with mild, moderate, or severe AD who were treated with donepezil for 12, 24, or 52 weeks.
Rivastigmine improved or maintained cognitive function, activities of daily living, and behavior for up to 52 weeks in patients with mild to moderate AD, according to a review of studies from 1995 to 2002.11 GI irritation was the most common adverse effect. Giving rivastigmine for up to 2 years may reduce the cost of caring for patients with AD, mostly by delaying nursing home placement.
Galantamine has beneficial effects on cognition, global function, activities of daily living, and behavior in patients with AD, vascular dementia, and AD with cerebrovascular components, according to a review of clinical studies.12 Adverse events are generally mild to moderate, transient, and gastrointestinal.
Efficacy In Other Dementias
In addition to their FDA-approved use for mildto-moderate AD, ChEIs also have been studied in persons with other types of dementia and mild cognitive impairment (MCI).
Dementia with Lewy bodies. Rivastigmine given with flexible titration from 6 to 12 mg/d improved behavior in 120 patients with Lewy body dementia.14 In the double-blind, multicenter study, patients taking rivastigmine, mean 9.7 mg/d for 20 weeks, were less apathetic and anxious and had fewer delusions and hallucinations than did those taking placebo. The drug was judged to be safe and well tolerated.
Vascular dementia. Patients with vascular dementia showed improved cognition and global function when treated with donepezil, 5 or 10 mg/d, for up to 24 weeks. Donepezil was well tolerated in this combined analysis of two randomized, placebo-controlled trials.15
Kumar et al16 compared two rivastigmine dosages in patients with mild-to-moderate AD, some of whom also had vascular dementia risk factors. Patients were randomly assigned to placebo, low-dose rivastigmine (1 to 4 mg/d), or high-dose rivastigmine (6 to 12 mg/d) for 26 weeks. Cognition, activities of daily living, and disease severity improved with rivastigmine in patients with or without vascular risk factors. Greater benefit was seen with high-dose than low-dose rivastigmine and in patients with AD plus vascular risk factors than in those with AD alone.
In a multicenter, double-blind trial,17 patients with vascular dementia or AD with vascular risk factors received galantamine, up to 24 mg/d, or placebo for 6 months. Compared with controls, those taking galantamine showed improved cognition, behavior, and function. The drug overall was well tolerated, with nausea and vomiting the most common side effects.
Parkinson’s dementia. Emre et al18 evaluated rivastigmine’s efficacy and safety in patients whose mild-to-moderate dementia developed at least 2 years after a clinical diagnosis of Parkinson’s disease (PD). Patients were randomly assigned to placebo or rivastigmine, 3 to 12 mg/d, for 24 weeks, and 410 of 541 enrollees completed the study. Compared with placebo, rivastigmine was associated with statistically significant improvements in cognition and global measures in dementia associated with PD but also with higher rates of nausea, vomiting, and tremor. PD’s motor symptoms did not change significantly in either group.
Mixed dementia states. As mentioned, galantamine improved cognitive and noncognitive abilities in patients with vascular dementia or AD with vascular risk factors in a 6-month, double-blind trial.17 Patients who received galantamine or placebo could then continue open-label galantamine, 24 mg/d, for another 6 months. In patients treated the full 12 months, galantamine continued to improve or maintain:
- cognition, based on Alzheimer’s Disease Assessment Scale-cognitive subscale scores
- functional ability, measured by the 40-item Disability Assessment for Dementia
- behavior, measured by the Neuro-psychiatric Inventory.19
After 12 months, the rivastigmine-treated patients were less behaviorally impaired than the matched patients, and their caregivers reported reduced stress. Rivastigmine did not prevent cognitive deterioration, as assessed with the Mini-Mental State Examination (MMSE).
Mild cognitive impairment. Persons with MCI have objective psychometric evidence of memory loss compared with their peers, but they are not significantly impaired in activities of daily living or other cognitive functions (language, abstract thinking, or problem-solving).
At this time, we do not recommend using ChEIs to treat MCI. These agents have shown little benefit and potential risk in patients who do not meet diagnostic criteria for dementia:
- Salloway et al21 tested donepezil’s efficacy and safety in 270 patients with MCI in a 24-week, double-blind, placebo-controlled trial. Donepezil was started at 5 mg/d for 42 days, then escalated to 10 mg/d. Compared with placebo, donepezil showed no significant effects on recall, but some improvements were seen in attention and psychomotor speed.
- In two unpublished placebo-controlled trials, galantamine did not improve memory when given for 2 years to elderly patients with MCI. A precaution was added to the drug’s prescribing information because 13 of the 1,026 patients taking galantamine died, compared with 1 of 1,022 taking placebo. Vascular disease caused one-half of the galantamine group deaths. No evidence of increased mortality risk has been seen in studies of galantamine in patients with mild-to-moderate AD, for which it is indicated.
Getting The Greatest Response
To gauge response to ChEI therapy, family reports about the patient are helpful—such as that cognition has improved or cognitive decline has not progressed as rapidly as before. Assessment tools such as the MMSE can document improvement or stabilization.
We recommend trying an initial ChEI for at least 6 months to determine its efficacy. If your patient cannot tolerate one ChEI or fails to respond to initial treatment, two consensus panels22,23 recommend that you consider changing ChEIs:
- If switching because of intolerable side effects, wait at least 2 to 3 days after stopping the first ChEI before starting another.
- If switching because of poor response, you can start a different ChEI immediately after the first one is stopped.
- Cholinesterase inhibitors may help improve or stabilize cognition, behavior, and/or activities of daily living
- Persons receiving these agents may decline more slowly than those who have not been treated
- Common side effects include nausea, vomiting, diarrhea, and loss of appetite
- Other less-common side effects are muscle cramps, slowed heart rate, dizziness, and fainting
- Because of differences in these agents, it may make sense to switch to another cholinesterase inhibitor if the patient has intolerable side effects or does not improve with the first one tried
Related resources
- Alzheimer’s Association. Information for health care professionals:
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- • Treating cognitive symptoms www.alz.org/Health/Treating/symptoms.asp
- • Diagnosing Alzheimer’s www.alz.org/Health/Diagnose/overview.asp
- American Association for Geriatric Psychiatry. Information for older patients and their families on Alzheimer’s disease, other dementias. www.gmhfonline.org/gmhf/consumer/alzheimers.html.
- Tacrine • Cognex
- Donepezil • Aricept
- Rivastigmine • Exelon
- Galantamine • Razadyne (was Reminyl)
Drs. Kamat and LeFevre report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg receives grant/research support from Abbott Laboratories, Boehringer Ingelheim Pharmaceuticals, Cyberonics, Eli Lilly and Co., Eunoe, Forest Pharmaceuticals, Novartis Pharmaceuticals Corp., Pfizer, and Wyeth Pharmaceuticals. He is a consultant to AstraZeneca Pharmaceuticals, Forest Pharmaceuticals, Janssen Pharmaceutica, KV Pharma, Novartis Pharmaceuticals Corp., Organon International, and Sanofi-Synthelabo.
Acknowledgment
The authors thank Anjali Baliga, MD, for her contribution and help in preparing this article
1. Small GW, Rabins PV, Barry PP, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA 1997;278(16):1363-71.
2. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 suppl 5):S4-S9.
3. Grossberg GT, Lake JT. The role of the psychiatrist in Alzheimer’s disease. J Clin Psychiatry 1998;59(suppl 9):3-6.
4. Doraiswamy PM, Steffens DC, Pitchumoni S, Tabrizi S. Early recognition of Alzheimer’s disease: what is consensual? What is controversial? What is practical? J Clin Psychiatry 1998;59(suppl 13):6-18.
5. Wilkinson DG, Passmore AP, Bullock R, et al. A multinational, randomised, 12-week, comparative study of donepezil and rivastigmine in patients with mild to moderate Alzheimer’s disease. Int J Clin Pract 2002;56(6):441-6
6. Jones RW, Soininen H, Hager K, et al. A multinational, randomised, 12-week study comparing the effects of donepezil and galantamine in patients with mild to moderate Alzheimer’s disease. Int J Geriatr Psychiatry 2004;19(1):58-67.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twentytwo classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. U. S. Bureau of the Census. 2004 International database: Midyear population, by age and sex. Table 094. U.S. Bureau of the Census; 2004.
9. Reminyl (galantamine HBr). Physicians’ desk reference (59th ed). Montvale, NJ: Thomson PDR; 2005:1739.
10. Birks JS, Harvey R. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev 2003;(3):CD001190.-
11. Williams BR, Nazarians A, Gill MA. A review of rivastigmine: a reversible cholinesterase inhibitor. Clin Ther 2003;25(6):1634-53.
12. Corey-Bloom J. Galantamine: a review of its use in Alzheimer’s disease and vascular dementia. Int J Clin Pract 2003;57(3):219-23.
13. Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994;271(13):992-8.
14. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000;356(9247):2031-6.
15. Passmore AP, Bayer AJ, Steinhagen-Thiessen E. Cognitive, global, and functional benefits of donepezil in Alzheimer’s disease and vascular dementia: results from large-scale clinical trials. J Neurol Sci 2005;229-30:141-6.
16. Kumar V, Anand R, Messina J, et al. An efficacy and safety analysis of rivastigmine in Alzheimer’s disease patients with concurrent vascular risk factors. Eur J Neurol 2000;7(2):159-69.
17. Kurz AF, Erkinjuntti T, Gauthier S, et al. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002;359(9314):1283-90.
18. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351(24):2509-18.
19. Erkinjuntti T, Kurz A, Small GW, et al. An open-label extension trial of galantamine in patients with probable vascular dementia and mixed dementia. Clin Ther 2003;25(6):1765-82.
20. Moretti R, Torre P, Antonello RM, et al. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004;21(14):931-7.
21. Salloway S, Ferris S, Kluger A, et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology 2004;63(4):651-7.
22. Emre M. Switching cholinesterase inhibitors in patients with Alzheimer’s disease. Int J Clin Pract Suppl 2002;(127):64-72.
23. Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int J Clin Pract Suppl 2002;(127):45-63.
1. Small GW, Rabins PV, Barry PP, et al. Diagnosis and treatment of Alzheimer disease and related disorders. Consensus statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA 1997;278(16):1363-71.
2. Lobo A, Launer LJ, Fratiglioni L, et al. Prevalence of dementia and major subtypes in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 suppl 5):S4-S9.
3. Grossberg GT, Lake JT. The role of the psychiatrist in Alzheimer’s disease. J Clin Psychiatry 1998;59(suppl 9):3-6.
4. Doraiswamy PM, Steffens DC, Pitchumoni S, Tabrizi S. Early recognition of Alzheimer’s disease: what is consensual? What is controversial? What is practical? J Clin Psychiatry 1998;59(suppl 13):6-18.
5. Wilkinson DG, Passmore AP, Bullock R, et al. A multinational, randomised, 12-week, comparative study of donepezil and rivastigmine in patients with mild to moderate Alzheimer’s disease. Int J Clin Pract 2002;56(6):441-6
6. Jones RW, Soininen H, Hager K, et al. A multinational, randomised, 12-week study comparing the effects of donepezil and galantamine in patients with mild to moderate Alzheimer’s disease. Int J Geriatr Psychiatry 2004;19(1):58-67.
7. Grossberg GT, Stahelin HB, Messina JC, et al. Lack of adverse pharmacodynamic drug interactions with rivastigmine and twentytwo classes of medications. Int J Geriatr Psychiatry 2000;15(3):242-7.
8. U. S. Bureau of the Census. 2004 International database: Midyear population, by age and sex. Table 094. U.S. Bureau of the Census; 2004.
9. Reminyl (galantamine HBr). Physicians’ desk reference (59th ed). Montvale, NJ: Thomson PDR; 2005:1739.
10. Birks JS, Harvey R. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev 2003;(3):CD001190.-
11. Williams BR, Nazarians A, Gill MA. A review of rivastigmine: a reversible cholinesterase inhibitor. Clin Ther 2003;25(6):1634-53.
12. Corey-Bloom J. Galantamine: a review of its use in Alzheimer’s disease and vascular dementia. Int J Clin Pract 2003;57(3):219-23.
13. Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 1994;271(13):992-8.
14. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000;356(9247):2031-6.
15. Passmore AP, Bayer AJ, Steinhagen-Thiessen E. Cognitive, global, and functional benefits of donepezil in Alzheimer’s disease and vascular dementia: results from large-scale clinical trials. J Neurol Sci 2005;229-30:141-6.
16. Kumar V, Anand R, Messina J, et al. An efficacy and safety analysis of rivastigmine in Alzheimer’s disease patients with concurrent vascular risk factors. Eur J Neurol 2000;7(2):159-69.
17. Kurz AF, Erkinjuntti T, Gauthier S, et al. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002;359(9314):1283-90.
18. Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med 2004;351(24):2509-18.
19. Erkinjuntti T, Kurz A, Small GW, et al. An open-label extension trial of galantamine in patients with probable vascular dementia and mixed dementia. Clin Ther 2003;25(6):1765-82.
20. Moretti R, Torre P, Antonello RM, et al. Rivastigmine in frontotemporal dementia: an open-label study. Drugs Aging 2004;21(14):931-7.
21. Salloway S, Ferris S, Kluger A, et al. Efficacy of donepezil in mild cognitive impairment: a randomized placebo-controlled trial. Neurology 2004;63(4):651-7.
22. Emre M. Switching cholinesterase inhibitors in patients with Alzheimer’s disease. Int J Clin Pract Suppl 2002;(127):64-72.
23. Inglis F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int J Clin Pract Suppl 2002;(127):45-63.
Late-life depression: Focused IPT eases loss and role changes
Mrs. E, age 74, has been distraught for 6 months since the death of her husband of 45 years. She is brought for evaluation by her daughter, who is exasperated and worried about her mother’s sad mood, frequent tearfulness, weight loss (11 pounds), and social isolation.
Mrs. E says she feels lost and paralyzed, that she “sticks out like a sore thumb” when among couples “that still have each other.” She refuses to go to church, though she attended regularly in the past.
Unresolved grief appears to be linked to the onset and persistence of Mrs. E’s depressive symptoms. After a thorough evaluation confirms major depression, the psychiatrist explains the diagnosis to Mrs. E. She agrees to begin an antidepressant and interpersonal psychotherapy (IPT).
IPT is easy to use and well-suited to address abnormal grieving, role transitions, and role disputes in depressed older patients. In controlled trials, IPT has been shown effective as acute1 and maintenance treatment2,3 of depression. This article describes how IPT can work effectively for depressed older adults and their clinicians.
IPT and elder depression
New-onset or recurrent depression is common in older patients experiencing retirement, relocation, disabilities, or loss of important persons in their lives (Box 1) 4 IPT recognizes that depression, regardless of psychosocial stress or biologic vulnerability, is expressed in an interpersonal environment (Box 2).5,6 The environment may have contributed to the depression, but it also can be a platform for intervention.
Depressed older adults who are verbal, nondemented, and engageable are candidates for IPT, with or without adjunctive antidepressant therapy. Psychotherapy is not indicated for patients with severe dementia, but this article will describe how IPT is being adapted for those with early dementia or mild cognitive impairment.
Biologic insults. Any brain injury that is more common in late life or that accumulates with age (such as cerebrovascular, Alzheimer’s, or Parkinson’s disease) increases the risk of damage to the neural circuitry that maintains mood.4 Common metabolic abnormalities such as hypothyroidism and vitamin B12 deficiency also can contribute to late-life depression, which is why routine blood screening is recommended.
Older patients often take multiple medications, increasing the risk for interactions and adverse events. Drugs with depression as a potential side effect include antipsychotics, antihypertensives, and corticosteroids.
Losses in later life can include bereavement for departed family and friends; changes in ego support and financial security with retirement; lack of transportation to sustain hobbies and interests; and declining vision, hearing, and physical function such as urinary continence or ambulation.
Role disputes and interpersonal conflicts. Marriages may be strained by role changes related to retirement or to caring for a physically frail or cognitively impaired partner. Problems of adult children or grandchildren—illnesses, substance abuse, unemployment—can burden elders, especially if families expect financial support, child-care help, or cohabitation. Elder abuse or neglect may also add to late-life stress.
Death and dying issues. Older persons may worry about dying, experiencing pain, being a burden to their families, and whether their lives have been meaningful. Moving to a long-term care facility can demoralize those who view this transition as “the last abode before the grave.”
Klerman et al5 developed interpersonal psychotherapy (IPT) in the 1970s while working with depressed adults. These authors adopted an empiric approach, reviewing the literature for evidence-based outcomes from various schools of thought to pull together elements that proved to be effective in treating depression.
Social workers on the team reported that interpersonal themes—such as family disputes, life changes, and grief reactions—seemed to trigger or perpetuate many patients’ depressions. Using these observations and the literature review, the group developed IPT as a practica psychotherapy to address depression in an interpersonal environment. IPT’s case discussions and guidelines are designed to help health professionals learn the approach quickly, use it with broad populations, and complete therapy within weeks rather than years.5,6
Case continued: ‘he made all the decisions’
At Mrs. E’s first IPT session, the therapist assigns her the “sick role.” They contract to meet 12 to 16 weeks, and Mrs. E’s daughter agrees to drive her to sessions.
In the next few weeks, the therapist explains depression’s biopsychosocial model and explores dual strategies with Mrs. E: to ease her mourning and explore new interests or relationships. The therapist encourages her to express her feelings and seeks to understand the dynamics of her marriage.
Mrs. E said she was raised by nurturing parents and married soon after high school. She depended on her husband for almost every decision, including their social calendar. She describes their relationship as mutually loving. As part of an interpersonal inventory, her therapist encourages her to describe in detail all the ways she misses him.
The ‘sick role.’ The therapist assigned Mrs. E the “sick role” to emphasize that major depression can be a severe illness. A therapist might say: “If you had pneumonia, you wouldn’t think of trying to rake leaves. You would rest and take care of yourself to speed the healing. Persons with major depression should do the same.”
The contract. The therapist also explained to Mrs. E that contracting to meet weekly for 12 to 16 weeks is part of the treatment. A contract:
- encourages patients to commit to an IPT trial for a reasonable time
- presses patients to achieve adequate progress by the deadline
- discourages digression, avoidance of painful subjects, and dependence on the therapist.
IPT begins with a complete psychiatric evaluation, including the patient’s past, family, and social histories; alcohol and drug use; medical comorbidities; mental status exam; and sometimes blood screening to rule out metabolic abnormalities. Antidepressants are prescribed as needed to relieve vegetative symptoms and are used during IPT when indicated.
Interpersonal inventory. Each of the patient’s interpersonal relationships is then systematically reviewed. This inventory sets the stage for exploring relationships that may be linked to the depressive symptoms or offer opportunities for trying alternate coping trategies, such as learning to seek social support (Table 2).
Table 1
IPT’s understanding of depression comprises 3 component processes
| Component | Description |
|---|---|
| Symptom function | Biological or psychological causes may trigger neurovegetative signs and symptoms |
| Interpersonal and social relations | Influenced by childhood learning, social reinforcement, personal mastery, and competence |
| Personality and character problems | Enduring traits such as excess anger, guilt, impaired communication, or low self-esteem may impair patient’s ability to maintain satisfying interpersonal relationships |
| Source: References 5 and 6 | |
‘How-to’ checklist of IPT procedures
|
IPT’S FOUR FOCI
IPT’s goal is to relieve depressive symptoms by identifying and focusing on problems that may have caused or are perpetuating those symptoms. Most of the reasons depressed patients give for seeking help fall into four foci: unresolved grief, role transition, role dispute, and interpersonal deficit (Table 3).5,6 The therapist uses clarification, interpretation, confrontation, and testing of perceptions and performance to address each focus, as detailed in the IPT manual.6
The therapist acts as the patient’s advocate, and focuses treatment on interpersonal relationships in the “here and now,” not past traumas, childhood conflicts, cognitive-behavioral interventions, or intrapsychic themes. No attempt is made to restructure personality.
Progress in relieving depressive symptoms is reviewed regularly, and treatment ends within the contract’s time limits in many cases. Older patients may need additional sessions because they often take longer to respond to antidepressant trials (6 to 8 weeks, compared with 3 to 4 weeks for younger adults). We allow older patients to “catch up” with additional sessions if illness, lack of transportation, or other problems prevent them from receiving the “full dose” of IPT.
IPT does not work for all patients. Consider other types of treatment if a patient shows no discernable benefit.
Table 3
IPT’s 4 foci, specific to late-life depression
| Focus | Description |
|---|---|
| Unresolved grief | Emotional reactions to the death of another person (not the loss of a job or one’s health) |
| Role transition | Difficulty adjusting to life change (such as retirement, ceasing to drive, or moving to an apartment) |
| Role dispute | Nonreciprocal expectations between two or more persons that predispose or perpetuate depressive symptoms |
| Interpersonal deficit | History of social impoverishment or inadequate or nonsustaining interpersonal relationships |
| Source: References 5 and 6 | |
Case continued: stalled in grief
As the weeks pass, Mrs. E improves but remains hypoactive and reclusive. She seems afraid to take any action without her late husband’s approval. Thinking about making independent decisions overwhelms her, and she withdraws to her couch to hide.
Her therapist discerns that Mrs. E needs more-active confrontation to accept that her new life requires her to make choices, even though decision-making is difficult for her. They develop a game, hronicling all decisions Mrs. E has made for the first time, such as calling a repairman and planting the summer vegetable garden by herself.
The therapist applauds these “firsts” and points out that Mrs. E’s depressive symptoms have improved as her list of completed decisions has grown. Mrs. E holds the power to make decisions, the therapist stresses, and bears the consequences of not taking action.
Applying ipt to late-life depression
Our group has used IPT in research protocols for 15 years. We and others7-11 have found that IPT is well-suited for treating late-life depression because:
- Older patients without psychotherapy experience or psychological sophistication can easily participate.
- Persons with limited education can understand IPT’s informal explanations of depression.
- Two foci of IPT—grief and role transition—address common themes of aging, such as spousal role disputes after retirement or caregiver stress when one partner becomes ill or shows signs of dementia.
Only minor IPT adaptations were required for older patients, such as:
- shorter sessions for those who reported physical discomfort
- accommodating for hearing loss, arranging transportation, and conducting sessions by telephone when patients were ill or shut in by inclement weather.
Case continued: more ‘firsts’ build confidence
Mrs. E makes slow, sometimes painful, but steady progress. Her therapist encourages her to keep trying more “firsts,”such as going back to church and attending her first social event alone, and to review her emotional reactions.
Mrs. E’s depressive symptoms wane as her confidence builds, and she readjusts her self-image to that of a widow who enjoyed a good marriage with a benevolent but overprotective husband. Her therapist links her progress to her string of successful “firsts” and to the contributing benefit of anti-depressant medication.
IPT As maintenance therapy
In the Maintenance Therapies for Late Life Depression (MTLLD) study—a randomized, double-blind, placebo-controlled trial12—we showed IPT to be effective as maintenance therapy for recurrent depression in patients age 60 and older. The 187 patients (mean age 67, one-third age ≥70) with nonpsychotic unipolar major depression were first treated to remission with IPT plus nortriptyline (80 to 120 ng/mL).
We then randomly assigned the 107 who achieved recovery to one of four maintenance therapies. After 3 years of monthly follow-up, relapse rates were:
- 20% with nortriptyline plus maintenance IPT
- 43% with nortriptyline plus medication clinic visits
- 64% with maintenance IPT plus placebo
- 90% with medication clinic plus placebo.
Further analysis showed that patients age ≥70 required combined treatment with nortriptyline and IPT to stay well, whereas those ages 60 to 69 stayed well with drug therapy alone. Patients age ≥70 also had a higher and more rapid relapse rate.
Recurrence by therapy focus. In patients who received placebo instead of nortriptyline:
- Time without a new depressive episode was similar for patients with a focus on grief or role transition, whether they received IPT or medication checkups.
- Recurrence rates were clearly lower in patients whose initial focus was role dispute if they received monthly maintenance IPT sessions instead of medication check visits.
Case continued: looking ahead
As the 12- to 16-week contracted period winds down, Mrs. E admits she still longs for her husband’s protection. She said she would gladly give up her independence to have that “safe” feeling back.
The therapist acknowledges that feeling but gently reminds her that she has the tools to face her new life realistically. During therapy, Mrs. E has shown she can assess life’s many decisions, make rational choices, and live with the consequences.
Their final discussion touches on the notion that Mrs. E could imagine having some kind of friendship with another man in the future.
Wrapping up. The last IPT sessions focus on reviewing any decline in depressive symptoms that may be linked to having learned new coping skills. With successful IPT, patients learn to appraise their strengths and remaining vulnerabilities and gain skills, self-confidence, and understanding to confront remaining obstacles after therapy ends.
Adapting ipt for special populations
Resistant depression. Researchers at the University of Pittsburgh are investigating whether adding IPT can achieve remission in depressed older patients who show partial response to a 6-week trial of escitalopram, 10 mg/d. In this ongoing trial, patients with Hamilton Depression Rating Scale scores of 11 to 14 after 6 weeks receive an increased escitalopram dosage (20 mg/d) and are randomly assigned to medication alone or medication plus 16 weeks of IPT.
Cognitive impairment. An unpublished follow-up to the MTLLD study enrolled 116 patients aged ≥70 and used a similar design, except that:
- patients were not required to have had recurrent depressive episodes
- paroxetine was used instead of nortriptyline
- patients with cognitive impairment (Mini-Mental Status Examination scores ?18/30) were included.
Cognitive impairment may have interfered with patients’ ability to benefit from traditional IPT. Thus, to improve the quality of life of depressed, cognitively-impaired elders, researchers are involving caregivers (usually a spouse or adult child) in modified forms of IPT couples therapy.6,13-18 A team at the University of Pittsburgh is developing a flexible approach that includes meetings with the patient, the caregiver, or both. Two papers on IPT-CI (for cognitive impairment) are under review.
Related resources
- International Society for Interpersonal Psychotherapy. Accreditation, training, and research information. www.interpersonalpsychotherapy.org.
- Stuart S, Robertson M. Interpersonal psychotherapy: a clinician’s guide. London: Edward Arnold Ltd.; 2002.
- Miller MD, Reynolds CF. Interpersonal psychotherapy. In: Hepple J, Pierce J, Wilkinson P (eds). Psychological therapies with older people. East Sussex, UK: Brunner-Routledge; 2002:103-27.
- Miller MD, Reynolds CF. Living longer depression-free: a family guide to the recognition, treatment and prevention of depression in later life. Baltimore: Johns Hopkins University Press; 2002.
- Escitalopram • Lexapro
- Nortriptyline • Pamelor
- Paroxetine • Paxil
Dr. Miller is a consultant to Forest Laboratories and GlaxoSmithKline and is a speaker for Forest Laboratories, GlaxoSmithKline, and Wyeth Pharmaceuticals.
1. Elkin I, Shea MT, Watkins JT, et al. National Institute of Mental Health treatment of depression collaborative research program: general effectiveness of treatments. Arch Gen Psychiatry 1989;46:971-82.
2. Frank E, Kupfer DJ, Perel JM, et al. Three-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry 1990;47;1093-9.
3. Kupfer DJ, Frank E, Perel JM, et al. Five-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry 1992;49:769-73.
4. Miller MD, Reynolds CF. Living longer depression free: A family guide to the recognition, treatment and prevention of depression in later life. Baltimore: Johns Hopkins University Press; 2002.
5. Klerman GL, Weissman MM, Rounsaville BJ, Chevron E. Interpersonal psychotherapy of depression. New York: Academic Press; 1984.
6. Weissman M, Markowitz JC, Klerman GL. Comprehensive guide to interpersonal psychotherapy. New York: Basic Books; 2000.
7. Miller MD, Cornes C, Frank E, et al. Interpersonal psychotherapy for late-life depression: Past, present and future. J Psychother Pract Res 2001;10(4):231-8.
8. Wolfson LK, Miller M, Houck PR, et al. Foci of interpersonal psychotherapy (IPT) in depressed elders: clinical and outcome correlates in a combined IPT/nortriptyline protocol. Psychother Res 1997;7(1):45-55.
9. Miller MD, Frank E, Cornes C, et al. Value of maintenance Interpersonal Psychotherapy (IPT) in elder adults with different IPT foci. Am J Geriatr Psychiatry 2003;11(1):97-102.-
10. Joiner T, Coyne JC. The interactional nature of depression: advances in interpersonal approaches. Washington, DC: American Psychological Association; 1999.
11. Sherrill JT, Frank E, Geary M, et al. An extension of psychoeducational family workshops to elderly patients with recurrent major depression: Description and evaluation. Psychiatr Serv 1997;48(1):76-81.
12. Reynolds CF, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression: a randomized controlled trial in patients older than 59 years. JAMA 1999;281(1):39-45.
13. Alexopoulos GS, Meyers BS, Young RC, et al. Executive dysfunction and long-term outcomes of geriatric depression. Arch Gen Psychiatry 2000;57(3):285-90.
14. Bozoki A, Giordani B, Heidebrink JL, et al. Mild cognitive impairments predict dementia in nondemented elderly patients with memory loss. Arch Neurol 2001;58(3):411-6.
15. Hinrichsen GA, Zweig R. Family issues in late-life depression. J Long Term Home Health Care 1994;13:4-15.
16. Reischies FM, Neu P. Comorbidity of mild cognitive disorder and depression—a neuropsychological analysis. Eur Arch Psychiatry Clin Neurosci 2000;250(4):186-93.
17. Teri L. Behavior and caregiver burden: behavioral problems in patients with Alzheimer disease and its association with caregiver distress. Alzheimer Dis Assoc Disord 1997;11(suppl 4):S35-S38.
18. Wright LK, Clipp EC, George LK. Health consequences of caregiver stress. Medicine, Exercise, Nutrition, and Health 1993;2:181-95.
Mrs. E, age 74, has been distraught for 6 months since the death of her husband of 45 years. She is brought for evaluation by her daughter, who is exasperated and worried about her mother’s sad mood, frequent tearfulness, weight loss (11 pounds), and social isolation.
Mrs. E says she feels lost and paralyzed, that she “sticks out like a sore thumb” when among couples “that still have each other.” She refuses to go to church, though she attended regularly in the past.
Unresolved grief appears to be linked to the onset and persistence of Mrs. E’s depressive symptoms. After a thorough evaluation confirms major depression, the psychiatrist explains the diagnosis to Mrs. E. She agrees to begin an antidepressant and interpersonal psychotherapy (IPT).
IPT is easy to use and well-suited to address abnormal grieving, role transitions, and role disputes in depressed older patients. In controlled trials, IPT has been shown effective as acute1 and maintenance treatment2,3 of depression. This article describes how IPT can work effectively for depressed older adults and their clinicians.
IPT and elder depression
New-onset or recurrent depression is common in older patients experiencing retirement, relocation, disabilities, or loss of important persons in their lives (Box 1) 4 IPT recognizes that depression, regardless of psychosocial stress or biologic vulnerability, is expressed in an interpersonal environment (Box 2).5,6 The environment may have contributed to the depression, but it also can be a platform for intervention.
Depressed older adults who are verbal, nondemented, and engageable are candidates for IPT, with or without adjunctive antidepressant therapy. Psychotherapy is not indicated for patients with severe dementia, but this article will describe how IPT is being adapted for those with early dementia or mild cognitive impairment.
Biologic insults. Any brain injury that is more common in late life or that accumulates with age (such as cerebrovascular, Alzheimer’s, or Parkinson’s disease) increases the risk of damage to the neural circuitry that maintains mood.4 Common metabolic abnormalities such as hypothyroidism and vitamin B12 deficiency also can contribute to late-life depression, which is why routine blood screening is recommended.
Older patients often take multiple medications, increasing the risk for interactions and adverse events. Drugs with depression as a potential side effect include antipsychotics, antihypertensives, and corticosteroids.
Losses in later life can include bereavement for departed family and friends; changes in ego support and financial security with retirement; lack of transportation to sustain hobbies and interests; and declining vision, hearing, and physical function such as urinary continence or ambulation.
Role disputes and interpersonal conflicts. Marriages may be strained by role changes related to retirement or to caring for a physically frail or cognitively impaired partner. Problems of adult children or grandchildren—illnesses, substance abuse, unemployment—can burden elders, especially if families expect financial support, child-care help, or cohabitation. Elder abuse or neglect may also add to late-life stress.
Death and dying issues. Older persons may worry about dying, experiencing pain, being a burden to their families, and whether their lives have been meaningful. Moving to a long-term care facility can demoralize those who view this transition as “the last abode before the grave.”
Klerman et al5 developed interpersonal psychotherapy (IPT) in the 1970s while working with depressed adults. These authors adopted an empiric approach, reviewing the literature for evidence-based outcomes from various schools of thought to pull together elements that proved to be effective in treating depression.
Social workers on the team reported that interpersonal themes—such as family disputes, life changes, and grief reactions—seemed to trigger or perpetuate many patients’ depressions. Using these observations and the literature review, the group developed IPT as a practica psychotherapy to address depression in an interpersonal environment. IPT’s case discussions and guidelines are designed to help health professionals learn the approach quickly, use it with broad populations, and complete therapy within weeks rather than years.5,6
Case continued: ‘he made all the decisions’
At Mrs. E’s first IPT session, the therapist assigns her the “sick role.” They contract to meet 12 to 16 weeks, and Mrs. E’s daughter agrees to drive her to sessions.
In the next few weeks, the therapist explains depression’s biopsychosocial model and explores dual strategies with Mrs. E: to ease her mourning and explore new interests or relationships. The therapist encourages her to express her feelings and seeks to understand the dynamics of her marriage.
Mrs. E said she was raised by nurturing parents and married soon after high school. She depended on her husband for almost every decision, including their social calendar. She describes their relationship as mutually loving. As part of an interpersonal inventory, her therapist encourages her to describe in detail all the ways she misses him.
The ‘sick role.’ The therapist assigned Mrs. E the “sick role” to emphasize that major depression can be a severe illness. A therapist might say: “If you had pneumonia, you wouldn’t think of trying to rake leaves. You would rest and take care of yourself to speed the healing. Persons with major depression should do the same.”
The contract. The therapist also explained to Mrs. E that contracting to meet weekly for 12 to 16 weeks is part of the treatment. A contract:
- encourages patients to commit to an IPT trial for a reasonable time
- presses patients to achieve adequate progress by the deadline
- discourages digression, avoidance of painful subjects, and dependence on the therapist.
IPT begins with a complete psychiatric evaluation, including the patient’s past, family, and social histories; alcohol and drug use; medical comorbidities; mental status exam; and sometimes blood screening to rule out metabolic abnormalities. Antidepressants are prescribed as needed to relieve vegetative symptoms and are used during IPT when indicated.
Interpersonal inventory. Each of the patient’s interpersonal relationships is then systematically reviewed. This inventory sets the stage for exploring relationships that may be linked to the depressive symptoms or offer opportunities for trying alternate coping trategies, such as learning to seek social support (Table 2).
Table 1
IPT’s understanding of depression comprises 3 component processes
| Component | Description |
|---|---|
| Symptom function | Biological or psychological causes may trigger neurovegetative signs and symptoms |
| Interpersonal and social relations | Influenced by childhood learning, social reinforcement, personal mastery, and competence |
| Personality and character problems | Enduring traits such as excess anger, guilt, impaired communication, or low self-esteem may impair patient’s ability to maintain satisfying interpersonal relationships |
| Source: References 5 and 6 | |
‘How-to’ checklist of IPT procedures
|
IPT’S FOUR FOCI
IPT’s goal is to relieve depressive symptoms by identifying and focusing on problems that may have caused or are perpetuating those symptoms. Most of the reasons depressed patients give for seeking help fall into four foci: unresolved grief, role transition, role dispute, and interpersonal deficit (Table 3).5,6 The therapist uses clarification, interpretation, confrontation, and testing of perceptions and performance to address each focus, as detailed in the IPT manual.6
The therapist acts as the patient’s advocate, and focuses treatment on interpersonal relationships in the “here and now,” not past traumas, childhood conflicts, cognitive-behavioral interventions, or intrapsychic themes. No attempt is made to restructure personality.
Progress in relieving depressive symptoms is reviewed regularly, and treatment ends within the contract’s time limits in many cases. Older patients may need additional sessions because they often take longer to respond to antidepressant trials (6 to 8 weeks, compared with 3 to 4 weeks for younger adults). We allow older patients to “catch up” with additional sessions if illness, lack of transportation, or other problems prevent them from receiving the “full dose” of IPT.
IPT does not work for all patients. Consider other types of treatment if a patient shows no discernable benefit.
Table 3
IPT’s 4 foci, specific to late-life depression
| Focus | Description |
|---|---|
| Unresolved grief | Emotional reactions to the death of another person (not the loss of a job or one’s health) |
| Role transition | Difficulty adjusting to life change (such as retirement, ceasing to drive, or moving to an apartment) |
| Role dispute | Nonreciprocal expectations between two or more persons that predispose or perpetuate depressive symptoms |
| Interpersonal deficit | History of social impoverishment or inadequate or nonsustaining interpersonal relationships |
| Source: References 5 and 6 | |
Case continued: stalled in grief
As the weeks pass, Mrs. E improves but remains hypoactive and reclusive. She seems afraid to take any action without her late husband’s approval. Thinking about making independent decisions overwhelms her, and she withdraws to her couch to hide.
Her therapist discerns that Mrs. E needs more-active confrontation to accept that her new life requires her to make choices, even though decision-making is difficult for her. They develop a game, hronicling all decisions Mrs. E has made for the first time, such as calling a repairman and planting the summer vegetable garden by herself.
The therapist applauds these “firsts” and points out that Mrs. E’s depressive symptoms have improved as her list of completed decisions has grown. Mrs. E holds the power to make decisions, the therapist stresses, and bears the consequences of not taking action.
Applying ipt to late-life depression
Our group has used IPT in research protocols for 15 years. We and others7-11 have found that IPT is well-suited for treating late-life depression because:
- Older patients without psychotherapy experience or psychological sophistication can easily participate.
- Persons with limited education can understand IPT’s informal explanations of depression.
- Two foci of IPT—grief and role transition—address common themes of aging, such as spousal role disputes after retirement or caregiver stress when one partner becomes ill or shows signs of dementia.
Only minor IPT adaptations were required for older patients, such as:
- shorter sessions for those who reported physical discomfort
- accommodating for hearing loss, arranging transportation, and conducting sessions by telephone when patients were ill or shut in by inclement weather.
Case continued: more ‘firsts’ build confidence
Mrs. E makes slow, sometimes painful, but steady progress. Her therapist encourages her to keep trying more “firsts,”such as going back to church and attending her first social event alone, and to review her emotional reactions.
Mrs. E’s depressive symptoms wane as her confidence builds, and she readjusts her self-image to that of a widow who enjoyed a good marriage with a benevolent but overprotective husband. Her therapist links her progress to her string of successful “firsts” and to the contributing benefit of anti-depressant medication.
IPT As maintenance therapy
In the Maintenance Therapies for Late Life Depression (MTLLD) study—a randomized, double-blind, placebo-controlled trial12—we showed IPT to be effective as maintenance therapy for recurrent depression in patients age 60 and older. The 187 patients (mean age 67, one-third age ≥70) with nonpsychotic unipolar major depression were first treated to remission with IPT plus nortriptyline (80 to 120 ng/mL).
We then randomly assigned the 107 who achieved recovery to one of four maintenance therapies. After 3 years of monthly follow-up, relapse rates were:
- 20% with nortriptyline plus maintenance IPT
- 43% with nortriptyline plus medication clinic visits
- 64% with maintenance IPT plus placebo
- 90% with medication clinic plus placebo.
Further analysis showed that patients age ≥70 required combined treatment with nortriptyline and IPT to stay well, whereas those ages 60 to 69 stayed well with drug therapy alone. Patients age ≥70 also had a higher and more rapid relapse rate.
Recurrence by therapy focus. In patients who received placebo instead of nortriptyline:
- Time without a new depressive episode was similar for patients with a focus on grief or role transition, whether they received IPT or medication checkups.
- Recurrence rates were clearly lower in patients whose initial focus was role dispute if they received monthly maintenance IPT sessions instead of medication check visits.
Case continued: looking ahead
As the 12- to 16-week contracted period winds down, Mrs. E admits she still longs for her husband’s protection. She said she would gladly give up her independence to have that “safe” feeling back.
The therapist acknowledges that feeling but gently reminds her that she has the tools to face her new life realistically. During therapy, Mrs. E has shown she can assess life’s many decisions, make rational choices, and live with the consequences.
Their final discussion touches on the notion that Mrs. E could imagine having some kind of friendship with another man in the future.
Wrapping up. The last IPT sessions focus on reviewing any decline in depressive symptoms that may be linked to having learned new coping skills. With successful IPT, patients learn to appraise their strengths and remaining vulnerabilities and gain skills, self-confidence, and understanding to confront remaining obstacles after therapy ends.
Adapting ipt for special populations
Resistant depression. Researchers at the University of Pittsburgh are investigating whether adding IPT can achieve remission in depressed older patients who show partial response to a 6-week trial of escitalopram, 10 mg/d. In this ongoing trial, patients with Hamilton Depression Rating Scale scores of 11 to 14 after 6 weeks receive an increased escitalopram dosage (20 mg/d) and are randomly assigned to medication alone or medication plus 16 weeks of IPT.
Cognitive impairment. An unpublished follow-up to the MTLLD study enrolled 116 patients aged ≥70 and used a similar design, except that:
- patients were not required to have had recurrent depressive episodes
- paroxetine was used instead of nortriptyline
- patients with cognitive impairment (Mini-Mental Status Examination scores ?18/30) were included.
Cognitive impairment may have interfered with patients’ ability to benefit from traditional IPT. Thus, to improve the quality of life of depressed, cognitively-impaired elders, researchers are involving caregivers (usually a spouse or adult child) in modified forms of IPT couples therapy.6,13-18 A team at the University of Pittsburgh is developing a flexible approach that includes meetings with the patient, the caregiver, or both. Two papers on IPT-CI (for cognitive impairment) are under review.
Related resources
- International Society for Interpersonal Psychotherapy. Accreditation, training, and research information. www.interpersonalpsychotherapy.org.
- Stuart S, Robertson M. Interpersonal psychotherapy: a clinician’s guide. London: Edward Arnold Ltd.; 2002.
- Miller MD, Reynolds CF. Interpersonal psychotherapy. In: Hepple J, Pierce J, Wilkinson P (eds). Psychological therapies with older people. East Sussex, UK: Brunner-Routledge; 2002:103-27.
- Miller MD, Reynolds CF. Living longer depression-free: a family guide to the recognition, treatment and prevention of depression in later life. Baltimore: Johns Hopkins University Press; 2002.
- Escitalopram • Lexapro
- Nortriptyline • Pamelor
- Paroxetine • Paxil
Dr. Miller is a consultant to Forest Laboratories and GlaxoSmithKline and is a speaker for Forest Laboratories, GlaxoSmithKline, and Wyeth Pharmaceuticals.
Mrs. E, age 74, has been distraught for 6 months since the death of her husband of 45 years. She is brought for evaluation by her daughter, who is exasperated and worried about her mother’s sad mood, frequent tearfulness, weight loss (11 pounds), and social isolation.
Mrs. E says she feels lost and paralyzed, that she “sticks out like a sore thumb” when among couples “that still have each other.” She refuses to go to church, though she attended regularly in the past.
Unresolved grief appears to be linked to the onset and persistence of Mrs. E’s depressive symptoms. After a thorough evaluation confirms major depression, the psychiatrist explains the diagnosis to Mrs. E. She agrees to begin an antidepressant and interpersonal psychotherapy (IPT).
IPT is easy to use and well-suited to address abnormal grieving, role transitions, and role disputes in depressed older patients. In controlled trials, IPT has been shown effective as acute1 and maintenance treatment2,3 of depression. This article describes how IPT can work effectively for depressed older adults and their clinicians.
IPT and elder depression
New-onset or recurrent depression is common in older patients experiencing retirement, relocation, disabilities, or loss of important persons in their lives (Box 1) 4 IPT recognizes that depression, regardless of psychosocial stress or biologic vulnerability, is expressed in an interpersonal environment (Box 2).5,6 The environment may have contributed to the depression, but it also can be a platform for intervention.
Depressed older adults who are verbal, nondemented, and engageable are candidates for IPT, with or without adjunctive antidepressant therapy. Psychotherapy is not indicated for patients with severe dementia, but this article will describe how IPT is being adapted for those with early dementia or mild cognitive impairment.
Biologic insults. Any brain injury that is more common in late life or that accumulates with age (such as cerebrovascular, Alzheimer’s, or Parkinson’s disease) increases the risk of damage to the neural circuitry that maintains mood.4 Common metabolic abnormalities such as hypothyroidism and vitamin B12 deficiency also can contribute to late-life depression, which is why routine blood screening is recommended.
Older patients often take multiple medications, increasing the risk for interactions and adverse events. Drugs with depression as a potential side effect include antipsychotics, antihypertensives, and corticosteroids.
Losses in later life can include bereavement for departed family and friends; changes in ego support and financial security with retirement; lack of transportation to sustain hobbies and interests; and declining vision, hearing, and physical function such as urinary continence or ambulation.
Role disputes and interpersonal conflicts. Marriages may be strained by role changes related to retirement or to caring for a physically frail or cognitively impaired partner. Problems of adult children or grandchildren—illnesses, substance abuse, unemployment—can burden elders, especially if families expect financial support, child-care help, or cohabitation. Elder abuse or neglect may also add to late-life stress.
Death and dying issues. Older persons may worry about dying, experiencing pain, being a burden to their families, and whether their lives have been meaningful. Moving to a long-term care facility can demoralize those who view this transition as “the last abode before the grave.”
Klerman et al5 developed interpersonal psychotherapy (IPT) in the 1970s while working with depressed adults. These authors adopted an empiric approach, reviewing the literature for evidence-based outcomes from various schools of thought to pull together elements that proved to be effective in treating depression.
Social workers on the team reported that interpersonal themes—such as family disputes, life changes, and grief reactions—seemed to trigger or perpetuate many patients’ depressions. Using these observations and the literature review, the group developed IPT as a practica psychotherapy to address depression in an interpersonal environment. IPT’s case discussions and guidelines are designed to help health professionals learn the approach quickly, use it with broad populations, and complete therapy within weeks rather than years.5,6
Case continued: ‘he made all the decisions’
At Mrs. E’s first IPT session, the therapist assigns her the “sick role.” They contract to meet 12 to 16 weeks, and Mrs. E’s daughter agrees to drive her to sessions.
In the next few weeks, the therapist explains depression’s biopsychosocial model and explores dual strategies with Mrs. E: to ease her mourning and explore new interests or relationships. The therapist encourages her to express her feelings and seeks to understand the dynamics of her marriage.
Mrs. E said she was raised by nurturing parents and married soon after high school. She depended on her husband for almost every decision, including their social calendar. She describes their relationship as mutually loving. As part of an interpersonal inventory, her therapist encourages her to describe in detail all the ways she misses him.
The ‘sick role.’ The therapist assigned Mrs. E the “sick role” to emphasize that major depression can be a severe illness. A therapist might say: “If you had pneumonia, you wouldn’t think of trying to rake leaves. You would rest and take care of yourself to speed the healing. Persons with major depression should do the same.”
The contract. The therapist also explained to Mrs. E that contracting to meet weekly for 12 to 16 weeks is part of the treatment. A contract:
- encourages patients to commit to an IPT trial for a reasonable time
- presses patients to achieve adequate progress by the deadline
- discourages digression, avoidance of painful subjects, and dependence on the therapist.
IPT begins with a complete psychiatric evaluation, including the patient’s past, family, and social histories; alcohol and drug use; medical comorbidities; mental status exam; and sometimes blood screening to rule out metabolic abnormalities. Antidepressants are prescribed as needed to relieve vegetative symptoms and are used during IPT when indicated.
Interpersonal inventory. Each of the patient’s interpersonal relationships is then systematically reviewed. This inventory sets the stage for exploring relationships that may be linked to the depressive symptoms or offer opportunities for trying alternate coping trategies, such as learning to seek social support (Table 2).
Table 1
IPT’s understanding of depression comprises 3 component processes
| Component | Description |
|---|---|
| Symptom function | Biological or psychological causes may trigger neurovegetative signs and symptoms |
| Interpersonal and social relations | Influenced by childhood learning, social reinforcement, personal mastery, and competence |
| Personality and character problems | Enduring traits such as excess anger, guilt, impaired communication, or low self-esteem may impair patient’s ability to maintain satisfying interpersonal relationships |
| Source: References 5 and 6 | |
‘How-to’ checklist of IPT procedures
|
IPT’S FOUR FOCI
IPT’s goal is to relieve depressive symptoms by identifying and focusing on problems that may have caused or are perpetuating those symptoms. Most of the reasons depressed patients give for seeking help fall into four foci: unresolved grief, role transition, role dispute, and interpersonal deficit (Table 3).5,6 The therapist uses clarification, interpretation, confrontation, and testing of perceptions and performance to address each focus, as detailed in the IPT manual.6
The therapist acts as the patient’s advocate, and focuses treatment on interpersonal relationships in the “here and now,” not past traumas, childhood conflicts, cognitive-behavioral interventions, or intrapsychic themes. No attempt is made to restructure personality.
Progress in relieving depressive symptoms is reviewed regularly, and treatment ends within the contract’s time limits in many cases. Older patients may need additional sessions because they often take longer to respond to antidepressant trials (6 to 8 weeks, compared with 3 to 4 weeks for younger adults). We allow older patients to “catch up” with additional sessions if illness, lack of transportation, or other problems prevent them from receiving the “full dose” of IPT.
IPT does not work for all patients. Consider other types of treatment if a patient shows no discernable benefit.
Table 3
IPT’s 4 foci, specific to late-life depression
| Focus | Description |
|---|---|
| Unresolved grief | Emotional reactions to the death of another person (not the loss of a job or one’s health) |
| Role transition | Difficulty adjusting to life change (such as retirement, ceasing to drive, or moving to an apartment) |
| Role dispute | Nonreciprocal expectations between two or more persons that predispose or perpetuate depressive symptoms |
| Interpersonal deficit | History of social impoverishment or inadequate or nonsustaining interpersonal relationships |
| Source: References 5 and 6 | |
Case continued: stalled in grief
As the weeks pass, Mrs. E improves but remains hypoactive and reclusive. She seems afraid to take any action without her late husband’s approval. Thinking about making independent decisions overwhelms her, and she withdraws to her couch to hide.
Her therapist discerns that Mrs. E needs more-active confrontation to accept that her new life requires her to make choices, even though decision-making is difficult for her. They develop a game, hronicling all decisions Mrs. E has made for the first time, such as calling a repairman and planting the summer vegetable garden by herself.
The therapist applauds these “firsts” and points out that Mrs. E’s depressive symptoms have improved as her list of completed decisions has grown. Mrs. E holds the power to make decisions, the therapist stresses, and bears the consequences of not taking action.
Applying ipt to late-life depression
Our group has used IPT in research protocols for 15 years. We and others7-11 have found that IPT is well-suited for treating late-life depression because:
- Older patients without psychotherapy experience or psychological sophistication can easily participate.
- Persons with limited education can understand IPT’s informal explanations of depression.
- Two foci of IPT—grief and role transition—address common themes of aging, such as spousal role disputes after retirement or caregiver stress when one partner becomes ill or shows signs of dementia.
Only minor IPT adaptations were required for older patients, such as:
- shorter sessions for those who reported physical discomfort
- accommodating for hearing loss, arranging transportation, and conducting sessions by telephone when patients were ill or shut in by inclement weather.
Case continued: more ‘firsts’ build confidence
Mrs. E makes slow, sometimes painful, but steady progress. Her therapist encourages her to keep trying more “firsts,”such as going back to church and attending her first social event alone, and to review her emotional reactions.
Mrs. E’s depressive symptoms wane as her confidence builds, and she readjusts her self-image to that of a widow who enjoyed a good marriage with a benevolent but overprotective husband. Her therapist links her progress to her string of successful “firsts” and to the contributing benefit of anti-depressant medication.
IPT As maintenance therapy
In the Maintenance Therapies for Late Life Depression (MTLLD) study—a randomized, double-blind, placebo-controlled trial12—we showed IPT to be effective as maintenance therapy for recurrent depression in patients age 60 and older. The 187 patients (mean age 67, one-third age ≥70) with nonpsychotic unipolar major depression were first treated to remission with IPT plus nortriptyline (80 to 120 ng/mL).
We then randomly assigned the 107 who achieved recovery to one of four maintenance therapies. After 3 years of monthly follow-up, relapse rates were:
- 20% with nortriptyline plus maintenance IPT
- 43% with nortriptyline plus medication clinic visits
- 64% with maintenance IPT plus placebo
- 90% with medication clinic plus placebo.
Further analysis showed that patients age ≥70 required combined treatment with nortriptyline and IPT to stay well, whereas those ages 60 to 69 stayed well with drug therapy alone. Patients age ≥70 also had a higher and more rapid relapse rate.
Recurrence by therapy focus. In patients who received placebo instead of nortriptyline:
- Time without a new depressive episode was similar for patients with a focus on grief or role transition, whether they received IPT or medication checkups.
- Recurrence rates were clearly lower in patients whose initial focus was role dispute if they received monthly maintenance IPT sessions instead of medication check visits.
Case continued: looking ahead
As the 12- to 16-week contracted period winds down, Mrs. E admits she still longs for her husband’s protection. She said she would gladly give up her independence to have that “safe” feeling back.
The therapist acknowledges that feeling but gently reminds her that she has the tools to face her new life realistically. During therapy, Mrs. E has shown she can assess life’s many decisions, make rational choices, and live with the consequences.
Their final discussion touches on the notion that Mrs. E could imagine having some kind of friendship with another man in the future.
Wrapping up. The last IPT sessions focus on reviewing any decline in depressive symptoms that may be linked to having learned new coping skills. With successful IPT, patients learn to appraise their strengths and remaining vulnerabilities and gain skills, self-confidence, and understanding to confront remaining obstacles after therapy ends.
Adapting ipt for special populations
Resistant depression. Researchers at the University of Pittsburgh are investigating whether adding IPT can achieve remission in depressed older patients who show partial response to a 6-week trial of escitalopram, 10 mg/d. In this ongoing trial, patients with Hamilton Depression Rating Scale scores of 11 to 14 after 6 weeks receive an increased escitalopram dosage (20 mg/d) and are randomly assigned to medication alone or medication plus 16 weeks of IPT.
Cognitive impairment. An unpublished follow-up to the MTLLD study enrolled 116 patients aged ≥70 and used a similar design, except that:
- patients were not required to have had recurrent depressive episodes
- paroxetine was used instead of nortriptyline
- patients with cognitive impairment (Mini-Mental Status Examination scores ?18/30) were included.
Cognitive impairment may have interfered with patients’ ability to benefit from traditional IPT. Thus, to improve the quality of life of depressed, cognitively-impaired elders, researchers are involving caregivers (usually a spouse or adult child) in modified forms of IPT couples therapy.6,13-18 A team at the University of Pittsburgh is developing a flexible approach that includes meetings with the patient, the caregiver, or both. Two papers on IPT-CI (for cognitive impairment) are under review.
Related resources
- International Society for Interpersonal Psychotherapy. Accreditation, training, and research information. www.interpersonalpsychotherapy.org.
- Stuart S, Robertson M. Interpersonal psychotherapy: a clinician’s guide. London: Edward Arnold Ltd.; 2002.
- Miller MD, Reynolds CF. Interpersonal psychotherapy. In: Hepple J, Pierce J, Wilkinson P (eds). Psychological therapies with older people. East Sussex, UK: Brunner-Routledge; 2002:103-27.
- Miller MD, Reynolds CF. Living longer depression-free: a family guide to the recognition, treatment and prevention of depression in later life. Baltimore: Johns Hopkins University Press; 2002.
- Escitalopram • Lexapro
- Nortriptyline • Pamelor
- Paroxetine • Paxil
Dr. Miller is a consultant to Forest Laboratories and GlaxoSmithKline and is a speaker for Forest Laboratories, GlaxoSmithKline, and Wyeth Pharmaceuticals.
1. Elkin I, Shea MT, Watkins JT, et al. National Institute of Mental Health treatment of depression collaborative research program: general effectiveness of treatments. Arch Gen Psychiatry 1989;46:971-82.
2. Frank E, Kupfer DJ, Perel JM, et al. Three-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry 1990;47;1093-9.
3. Kupfer DJ, Frank E, Perel JM, et al. Five-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry 1992;49:769-73.
4. Miller MD, Reynolds CF. Living longer depression free: A family guide to the recognition, treatment and prevention of depression in later life. Baltimore: Johns Hopkins University Press; 2002.
5. Klerman GL, Weissman MM, Rounsaville BJ, Chevron E. Interpersonal psychotherapy of depression. New York: Academic Press; 1984.
6. Weissman M, Markowitz JC, Klerman GL. Comprehensive guide to interpersonal psychotherapy. New York: Basic Books; 2000.
7. Miller MD, Cornes C, Frank E, et al. Interpersonal psychotherapy for late-life depression: Past, present and future. J Psychother Pract Res 2001;10(4):231-8.
8. Wolfson LK, Miller M, Houck PR, et al. Foci of interpersonal psychotherapy (IPT) in depressed elders: clinical and outcome correlates in a combined IPT/nortriptyline protocol. Psychother Res 1997;7(1):45-55.
9. Miller MD, Frank E, Cornes C, et al. Value of maintenance Interpersonal Psychotherapy (IPT) in elder adults with different IPT foci. Am J Geriatr Psychiatry 2003;11(1):97-102.-
10. Joiner T, Coyne JC. The interactional nature of depression: advances in interpersonal approaches. Washington, DC: American Psychological Association; 1999.
11. Sherrill JT, Frank E, Geary M, et al. An extension of psychoeducational family workshops to elderly patients with recurrent major depression: Description and evaluation. Psychiatr Serv 1997;48(1):76-81.
12. Reynolds CF, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression: a randomized controlled trial in patients older than 59 years. JAMA 1999;281(1):39-45.
13. Alexopoulos GS, Meyers BS, Young RC, et al. Executive dysfunction and long-term outcomes of geriatric depression. Arch Gen Psychiatry 2000;57(3):285-90.
14. Bozoki A, Giordani B, Heidebrink JL, et al. Mild cognitive impairments predict dementia in nondemented elderly patients with memory loss. Arch Neurol 2001;58(3):411-6.
15. Hinrichsen GA, Zweig R. Family issues in late-life depression. J Long Term Home Health Care 1994;13:4-15.
16. Reischies FM, Neu P. Comorbidity of mild cognitive disorder and depression—a neuropsychological analysis. Eur Arch Psychiatry Clin Neurosci 2000;250(4):186-93.
17. Teri L. Behavior and caregiver burden: behavioral problems in patients with Alzheimer disease and its association with caregiver distress. Alzheimer Dis Assoc Disord 1997;11(suppl 4):S35-S38.
18. Wright LK, Clipp EC, George LK. Health consequences of caregiver stress. Medicine, Exercise, Nutrition, and Health 1993;2:181-95.
1. Elkin I, Shea MT, Watkins JT, et al. National Institute of Mental Health treatment of depression collaborative research program: general effectiveness of treatments. Arch Gen Psychiatry 1989;46:971-82.
2. Frank E, Kupfer DJ, Perel JM, et al. Three-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry 1990;47;1093-9.
3. Kupfer DJ, Frank E, Perel JM, et al. Five-year outcomes for maintenance therapies in recurrent depression. Arch Gen Psychiatry 1992;49:769-73.
4. Miller MD, Reynolds CF. Living longer depression free: A family guide to the recognition, treatment and prevention of depression in later life. Baltimore: Johns Hopkins University Press; 2002.
5. Klerman GL, Weissman MM, Rounsaville BJ, Chevron E. Interpersonal psychotherapy of depression. New York: Academic Press; 1984.
6. Weissman M, Markowitz JC, Klerman GL. Comprehensive guide to interpersonal psychotherapy. New York: Basic Books; 2000.
7. Miller MD, Cornes C, Frank E, et al. Interpersonal psychotherapy for late-life depression: Past, present and future. J Psychother Pract Res 2001;10(4):231-8.
8. Wolfson LK, Miller M, Houck PR, et al. Foci of interpersonal psychotherapy (IPT) in depressed elders: clinical and outcome correlates in a combined IPT/nortriptyline protocol. Psychother Res 1997;7(1):45-55.
9. Miller MD, Frank E, Cornes C, et al. Value of maintenance Interpersonal Psychotherapy (IPT) in elder adults with different IPT foci. Am J Geriatr Psychiatry 2003;11(1):97-102.-
10. Joiner T, Coyne JC. The interactional nature of depression: advances in interpersonal approaches. Washington, DC: American Psychological Association; 1999.
11. Sherrill JT, Frank E, Geary M, et al. An extension of psychoeducational family workshops to elderly patients with recurrent major depression: Description and evaluation. Psychiatr Serv 1997;48(1):76-81.
12. Reynolds CF, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression: a randomized controlled trial in patients older than 59 years. JAMA 1999;281(1):39-45.
13. Alexopoulos GS, Meyers BS, Young RC, et al. Executive dysfunction and long-term outcomes of geriatric depression. Arch Gen Psychiatry 2000;57(3):285-90.
14. Bozoki A, Giordani B, Heidebrink JL, et al. Mild cognitive impairments predict dementia in nondemented elderly patients with memory loss. Arch Neurol 2001;58(3):411-6.
15. Hinrichsen GA, Zweig R. Family issues in late-life depression. J Long Term Home Health Care 1994;13:4-15.
16. Reischies FM, Neu P. Comorbidity of mild cognitive disorder and depression—a neuropsychological analysis. Eur Arch Psychiatry Clin Neurosci 2000;250(4):186-93.
17. Teri L. Behavior and caregiver burden: behavioral problems in patients with Alzheimer disease and its association with caregiver distress. Alzheimer Dis Assoc Disord 1997;11(suppl 4):S35-S38.
18. Wright LK, Clipp EC, George LK. Health consequences of caregiver stress. Medicine, Exercise, Nutrition, and Health 1993;2:181-95.
Faking it: How to detect malingered psychosis
Reputed Cosa Nostra boss Vincent “The Chin” Gigante deceived “the most respected minds in forensic psychiatry” for years by malingering schizophrenia.1 Ultimately, he admitted to maintaining his charade from 1990 to 1997 during evaluations of his competency to stand trial for racketeering.
A lesson from this case—said a psychiatrist who concluded Gigante was malingering—is, “When feigning is a consideration, we must be more critical and less accepting of our impressions when we conduct and interpret a psychiatric examination…than might be the case in a typical clinical situation.”2
Even in typical clinical situations, however, psychiatrists may be reluctant to diagnose malingering3 for fear of being sued, assaulted—or wrong. An inaccurate diagnosis of malingering may unjustly stigmatize a patient and deny him needed care.4
Because psychiatrists need a systematized approach to detect malingering,5 we offer specific clinical factors and approaches to help you recognize malingered psychosis.
What is Malingering?
No other syndrome is as easy to define yet so difficult to diagnose as malingering. Reliably diagnosing malingered mental illness is complex, requiring the psychiatrist to consider collateral data beyond the patient interview.
Malingering is the intentional production of false or grossly exaggerated physical or psychological symptoms, motivated by external incentives.6 In practice, malingering commonly must be differentiated from factitious disorder, which also involves intentional production of symptoms. In factitious disorders, the patient’s motivation is to assume the sick role, which can be thought of as an internal or psychological incentive.
Three categories of malingering include:
- pure malingering (feigning a nonexistent disorder)
- partial malingering (consciously exaggerating real symptoms)
- false imputation (ascribing real symptoms to a cause the individual knows is unrelated to the symptoms).7
Motivations. Individuals usually malinger to avoid pain (such as difficult situations or punishment) or to seek pleasure (such as to obtain compensation or medications) (Table 1). In correctional settings, for example, inmates may malinger mental illness to do “easier time” or to obtain drugs. On the other hand, malingering in prison also may be an adaptive response by a mentally ill inmate to relatively sparse and difficult-to-obtain mental health resources.8
Table 1
Common motives of malingerers
| Motives | Examples |
|---|---|
| To avoid pain | To avoid: |
| Arrest | |
| Criminal prosecution | |
| Conscription into the military | |
| To seek pleasure | To obtain: |
| Controlled substances | |
| Free room and board | |
| Workers’ compensation or disability benefits for alleged psychological injury |
Interview Style
When you suspect a patient is malingering, keep your suspicions to yourself and conduct an objective evaluation. Patients are likely to become defensive if you show annoyance or incredulity, and putting them on guard decreases your ability to uncover evidence of malingering.9
Begin by asking open-ended questions, which allow patients to report symptoms in their own words. To avoid hinting at correct responses, carefully phrase initial inquiries about symptoms. Later in the interview, you can proceed to more-detailed questions of specific symptoms, as discussed below.
If possible, review collateral data before the interview, when it is most helpful. Consider information that would support or refute the alleged symptoms, such as treatment and insurance records, police reports, and interviews of close friends or family.
The patient interview may be prolonged because fatigue may diminish a malingerer’s ability to maintain fake symptoms. In very difficult cases, consider monitoring during inpatient assessment because feigned psychosis is extremely difficult to maintain 24 hours a day.
Watch for individuals who endorse rare or improbable symptoms. Rare symptoms—by definition—occur very infrequently, and even severely disturbed patients almost never report improbable symptoms.10 Consider asking malingerers about improbable symptoms to see if they will endorse them. For example:
- “When people talk to you, do you see the words they speak spelled out?”11
- “Have you ever believed that automobiles are members of an organized religion?”12
Watch closely for internal or external inconsistency in the suspected malingerer’s presentation (Table 2).
Table 2
Clues to identify malingering during patient evaluation
| Internal inconsistencies | Example |
| In subject’s report of symptoms | Gives a clear and articulate explanation of being confused |
| In subject’s own reported history | Gives conflicting versions |
| External inconsistencies | Example |
| Between reported and observed symptoms | Alleges having active auditory and visual hallucinations yet shows no evidence of being distracted |
| Between reported and observed level of functioning | Behaves in disorganized or confused manner around psychiatrist, yet plays excellent chess on ward with other patients |
| Between reported symptoms and nature of genuine symptoms | Reports seeing visual hallucinations in black and white, whereas genuine visual hallucinations are seen in color |
| Between reported symptoms and psychological test results | Alleges genuine psychotic symptoms, yet testing suggests faking or exaggeration |
Malingered Psychotic Symptoms
Detecting malingered mental illness is considered an advanced psychiatric skill, partly because you must understand thoroughly how genuine psychotic symptoms manifest.
Hallucinations. If a patient alleges atypical hallucinations, ask about them in detail. Hallucinations are usually (88%) associated with delusions.13 Genuine hallucinations are typically intermittent rather than continuous.
Continue to: Auditory hallucinations
Auditory hallucinations are usually clear, not vague (7%) or inaudible. Both male and female voices are commonly heard (75%), and voices are usually perceived as originating outside the head (88%).14 In schizophrenia, the major themes are persecutory or instructive.15
Command auditory hallucinations are easy to fabricate. Persons experiencing genuine command hallucinations:
- do not always obey the voices, especially if doing so would be dangerous16
- usually present with noncommand hallucinations (85%) and delusions (75%) as well17
Thus, view with suspicion someone who alleges an isolated command hallucination without other psychotic symptoms.
Genuine schizophrenic hallucinations tend to diminish when patients are involved in activities. Thus, to deal with their hallucinations, persons with schizophrenia typically cope by:
- engaging in activities (working, listening to a radio, watching TV)
- changing posture (lying down, walking)
- seeking interpersonal contact
- taking medications.
If you suspect a person of malingered auditory hallucinations, ask what he or she does to make the voices go away or diminish in intensity. Patients with genuine schizophrenia often can stop their auditory hallucinations while in remission but not during acute illness.
Malingerers may report auditory hallucinations of stilted or implausible language. For example, we have evaluated:
- an individual charged with attempted rape who alleged that voices said, “Go commit a sex offense.”
- a bank robber who alleged that voices kept screaming, “Stick up, stick up, stick up!”
Both examples contain language that is very questionable for genuine hallucinations, while providing the patient with “psychotic justification” for an illegal act that has a rational alternative motive.
Visual hallucinations are experienced by an estimated 24% to 30% of psychotic individuals but are reported much more often by malingerers (46%) than by persons with genuine psychosis (4%).18
Genuine visual hallucinations are usually of normalsized people and are seen in color.14 On rare occasions, genuine visual hallucinations of small people (Lilliputian hallucinations) may be associated with alcohol use, organic disease, or toxic psychosis (such as anticholinergic toxicity) but are rarely seen by persons with schizophrenia.
Psychotic visual hallucinations do not typically change if the eyes are closed or open, whereas drug-induced hallucinations are more readily seen with eyes closed or in the dark. Unformed hallucinations—such as flashes of light, shadows, or moving objects—are typically associated with neurologic disease and substance use.19
Suspect malingering if the patient reports dramatic or atypical visual hallucinations. For example, one defendant charged with bank robbery calmly reported seeing “a 30-foot tall, red giant smashing down the walls” of the interview room. When he was asked detailed questions, he frequently replied, “I don’t know.” He eventually admitted to malingering.
Delusions. Genuine delusions vary in content, theme, degree of systemization, and relevance to the person’s life. The complexity and sophistication of delusional systems usually reflect the individual’s intelligence. Persecutory delusions are more likely to be acted upon than are other types of delusions.20
Malingerers may claim that a delusion began or disappeared suddenly. In reality, systematized delusions usually take weeks to develop and much longer to disappear. Typically, the delusion will become somewhat less relevant, and the individual will gradually relinquish its importance over time after adequate treatment. In general, the more bizarre the delusion’s content, the more disorganized the individual’s thinking is likely to be (Table 3).
With genuine delusions, the individual’s behavior usually conforms to the delusions’ content. For example, Russell Weston—who suffered from schizophrenia—made a deadly assault on the U.S. Capitol in 1998 because he held a delusional belief that cannibalism was destroying Washington, DC. Before he shot and killed two U.S. Capitol security officers, he had gone to the Central Intelligence Agency several years before and voiced the same delusional concerns.
Suspect malingering if a patient alleges persecutory delusions without engaging in corresponding paranoid behaviors. One exception is the person with long-standing schizophrenia who has grown accustomed to the delusion and whose behavior is no longer consistent with it.
Table 3
Uncommon psychosis presentations that suggest malingering
| Hallucinations |
|
| Delusions |
|
Where Malingerers Trip Up
Malingerers may have inadequate or incomplete knowledge of the mental illness they are faking. Indeed, malingerers are like actors who can portray a role only as well as they understand it. They often overact their part or mistakenly believe the more bizarre their behavior, the more convincing they will be. Conversely, “successful” malingerers are more likely to endorse fewer symptoms and avoid endorsing overly bizarre or unusual symptoms.21
Continue to: Numerous clinical factors suggest malingering...
Numerous clinical factors suggest malingering (Table 4). Malingerers are more likely to eagerly “thrust forward” their illness, whereas patients with genuine schizophrenia are often reluctant to discuss their symptoms.22
Malingerers may attempt to take control of the interview and behave in an intimidating or hostile manner. They may accuse the psychiatrist of inferring that they are faking. Such behavior is rare in genuinely psychotic individuals. Although DSM-IV-TR states that antisocial personality disorder should arouse suspicions of malingering, some studies have failed to show a relationship. One study has associated psychopathic traits with malingering.23
Malingerers often believe that faking intellectual deficits, in addition to psychotic symptoms, will make them more believable. For example, a man who had completed several years of college alleged that he did not know the colors of the American flag.
Malingerers are more likely to give vague or hedging answers to straightforward questions. For example, when asked whether an alleged voice was male or female, one malingerer replied, “It was probably a man’s voice.” Malingerers may also answer, “I don’t know” to detailed questions about psychotic symptoms. Whereas a person with genuine psychotic symptoms could easily give an answer, the malingerer may have never experienced the symptoms and consequently “doesn’t know” the correct answer.
Psychotic symptoms such as derailment, neologisms, loose associations, and word salad are rarely simulated. This is because it is much more difficult for a malingerer to successfully imitate psychotic thought processes than psychotic thought content. Similarly, it is unusual for a malingerer to fake schizophrenia’s subtle signs, such as negative symptoms.
Table 4
Clinical factors that suggest malingering
| Absence of active or subtle signs of psychosis |
| Marked inconsistencies, contradictions |
Patient endorses improbable psychiatric symptoms
|
Patient is evasive or uncooperative
|
| Psychological testing indicates malingering (SIRS, M-FAST, MMPI-2) |
| SIRS: Structured Interview of Reported Symptoms |
| M-FAST: Miller Forensic Assessment of Symptoms Test |
| MMPI-2: Minnesota Multiphasic Personality Inventory, Revised |
Psychological Testing
Although many psychometric tests are available for detecting malingered psychosis, few have been validated. Among the more reliable are:
- Structured Interview of Reported Symptoms (SIRS)
- Minnesota Multiphasic Personality Inventory, Revised (MMPI-2)
- Miller Forensic Assessment of Symptoms Test (M-FAST).11
SIRS includes questions about rare symptoms, uncommon symptom pairing, atypical symptoms, and other indices involving excessive symptom reporting. It takes 30 to 60 minutes to administer. Tested in inpatient, forensic, and correctional populations, the SIRS has shown consistently high accuracy in detecting malingered psychiatric illness.24
Two MMPI-2 scales—F-scale and F-K Index—are the most frequently used test for evaluating suspected malingering. When using the MMPI-2 in this manner, consult the literature for appropriate cutoff scores (see Related resources). Although the MMPI-2 is the most validated psychometric method to detect malingering, a malingerer with high intelligence and previous knowledge of the test could evade detection.25
M-FAST was developed to provide a brief, reliable screen for malingered mental illness. This test takes 10 to 15 minutes to administer and measures rare symptom combinations, excessive reporting, and atypical symptoms.11 It has shown good validity and high correlation with the SIRS and MMPI-2.26,27
Confronting the Malingerer
If a thorough investigation indicates that a patient is malingering psychosis, you may decide to confront the evaluee. Avoid direct accusations of lying,10 and give the suspected malingerer every opportunity to save face. For example, it is preferable to say, “You haven’t told me the whole truth.”
A thoughtful approach that asks the evaluee to clarify inconsistencies is more likely to be productive and safer for the examiner. When confronting individuals with a history of violence and aggression, have adequate security personnel with you.
- Structured Interview of Reported Symptoms (SIRS). Available for purchase from Psychological Assessment Resources at www3.parinc.com (enter “SIRS” in search field).
- Graham JR. MMPI-2: Assessing personality and psychopathology. New York: Oxford Press; 2000. (Source of cutoff scores to use MMPI-2 scales [F-scale and F-K Index] to evaluate suspected malingering).
- Psychological Assessment Resources, Inc. Miller Forensic Assessment of Symptoms Test (M-FAST). Available at: www3.parinc.com (enter “M-FAST” in search field).
1. Newman A. Analyze this: Vincent Gigante, not crazy after all those years. New York Times, April 13, 2003.
2. Brodie JD. Personal communication, 2005.
3. Yates BD, Nordquist CR, Schultz-Ross RA. Feigned psychiatric symptoms in the emergency room. Psychiatr Serv 1996;47:998-1000.
4. Kropp PR, Rogers R. Understanding malingering: motivation, method, and detection. In: Lewis M, Saarini C (eds). Lying and deception. New York: Guilford Press; 1993.
5. Kucharski LT, et al. Clinical symptom presentation in suspected malingerers: an empirical investigation. Bull Am Acad Psychiatry Law. 1998;26:579-85.
6. Diagnostic and statistical manual of mental disorders (4th ed., text rev.). Washington, DC: American Psychiatric Association; 2000.
7. Resnick PJ. Malingering of posttraumatic stress disorders. In: Rogers R (ed). Clinical assessment of malingering and deception (2nd ed.). New York: Guilford Press; 1997;130-52.
8. Kupers TA. Malingering in correctional settings. Correctional Ment Health Rep. 2004;5(6):81-95.
9. Sadock BJ, Sadock VA. Kaplan & Sadock’s synopsis of psychiatry, 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2003;898.-
10. Thompson JW, LeBourgeois HW, Black FW. Malingering. In: Simon R, Gold L (eds). Textbook of forensic psychiatry. Washington, DC: American Psychiatric Publishing; 2004.
11. Miller HA. M.-FAST interview booklet. Lutz, FL: Psychological Assessment Resources; 2001.
12. Rogers R. Assessment of malingering within a forensic context. In Weisstub DW (ed.). Law and psychiatry: international perspectives. New York: Plenum Press; 1987;3:209-37.
13. Lewinsohn PM. An empirical test of several popular notions about hallucinations in schizophrenic patients. In: Keup W (ed.). Origin and mechanisms of hallucinations. New York: Plenum Press; 1970;401-3.
14. Goodwin DW, Anderson P, Rosenthal R. Clinical significance of hallucinations in psychiatric disorders: a study of 116 hallucinatory patients. Arch Gen Psychiatry 1971;24:76-80.
15. Small IF, Small JG, Andersen JM. Clinical characteristics of hallucinations of schizophrenia. Dis Nerv Sys 1966;27:349-53.
16. Kasper ME, Rogers R, Adams PA. Dangerousness and command hallucinations: an investigation of psychotic inpatients. Bull Am Acad Psychiatry Law 1996;24:219-24.
17. Thompson JS, Stuart GL, Holden CE. Command hallucinations and legal insanity. Forensic Rep 1992;5:29-43.
18. Cornell DG, Hawk GL. Clinical presentation of malingerers diagnosed by experienced forensic psychologists. Law Hum Behav 1989;13:375-83.
19. Cummings JL, Miller BL. Visual hallucinations: clinical occurrence and use in differential diagnosis. West J Med 1987;146:46-51.
20. Wessely S, Buchanan A, Reed A, et al. Acting on delusions: I. Prevalence. Br J Psychiatry 1993;163:69-76.
21. Edens JF, Guy LS, Otto RK, et al. Factors differentiating successful versus unsuccessful malingerers. J Pers Assess 2001;77(2):333-8.
22. Ritson B, Forest A. The simulation of psychosis: a contemporary presentation. Br J Psychol 1970;43:31-7.
23. Edens JF, Buffington JK, Tomicic TL. An investigation of the relationship between psychopathic traits and malingering on the Psychopathic Personality Inventory. Assessment 2000;7:281-96.
24. Rogers R. Structured interviews and dissimulation. In: Rogers R (ed). Clinical assessment of malingering and deception. New York: Guilford Press; 1997.
25. Pelfrey WV. The relationship between malingerers’ intelligence and MMPI-2 knowledge and their ability to avoid detection. Int J Offender Ther Comp Criminol. 2004;48(6):649-63.
26. Jackson RL, Rogers R, Sewell KW. Forensic applications of the Miller Forensic Assessment of Symptoms Test (MFAST): screening for feigned disorders in competency to stand trial evaluations. Law Human Behav 2005;29(2):199-210.
27. Miller HA. Examining the use of the M-FAST with criminal defendants incompetent to stand trial. Int J Offender Ther Comp Criminol 2004;48(3):268-80.
Reputed Cosa Nostra boss Vincent “The Chin” Gigante deceived “the most respected minds in forensic psychiatry” for years by malingering schizophrenia.1 Ultimately, he admitted to maintaining his charade from 1990 to 1997 during evaluations of his competency to stand trial for racketeering.
A lesson from this case—said a psychiatrist who concluded Gigante was malingering—is, “When feigning is a consideration, we must be more critical and less accepting of our impressions when we conduct and interpret a psychiatric examination…than might be the case in a typical clinical situation.”2
Even in typical clinical situations, however, psychiatrists may be reluctant to diagnose malingering3 for fear of being sued, assaulted—or wrong. An inaccurate diagnosis of malingering may unjustly stigmatize a patient and deny him needed care.4
Because psychiatrists need a systematized approach to detect malingering,5 we offer specific clinical factors and approaches to help you recognize malingered psychosis.
What is Malingering?
No other syndrome is as easy to define yet so difficult to diagnose as malingering. Reliably diagnosing malingered mental illness is complex, requiring the psychiatrist to consider collateral data beyond the patient interview.
Malingering is the intentional production of false or grossly exaggerated physical or psychological symptoms, motivated by external incentives.6 In practice, malingering commonly must be differentiated from factitious disorder, which also involves intentional production of symptoms. In factitious disorders, the patient’s motivation is to assume the sick role, which can be thought of as an internal or psychological incentive.
Three categories of malingering include:
- pure malingering (feigning a nonexistent disorder)
- partial malingering (consciously exaggerating real symptoms)
- false imputation (ascribing real symptoms to a cause the individual knows is unrelated to the symptoms).7
Motivations. Individuals usually malinger to avoid pain (such as difficult situations or punishment) or to seek pleasure (such as to obtain compensation or medications) (Table 1). In correctional settings, for example, inmates may malinger mental illness to do “easier time” or to obtain drugs. On the other hand, malingering in prison also may be an adaptive response by a mentally ill inmate to relatively sparse and difficult-to-obtain mental health resources.8
Table 1
Common motives of malingerers
| Motives | Examples |
|---|---|
| To avoid pain | To avoid: |
| Arrest | |
| Criminal prosecution | |
| Conscription into the military | |
| To seek pleasure | To obtain: |
| Controlled substances | |
| Free room and board | |
| Workers’ compensation or disability benefits for alleged psychological injury |
Interview Style
When you suspect a patient is malingering, keep your suspicions to yourself and conduct an objective evaluation. Patients are likely to become defensive if you show annoyance or incredulity, and putting them on guard decreases your ability to uncover evidence of malingering.9
Begin by asking open-ended questions, which allow patients to report symptoms in their own words. To avoid hinting at correct responses, carefully phrase initial inquiries about symptoms. Later in the interview, you can proceed to more-detailed questions of specific symptoms, as discussed below.
If possible, review collateral data before the interview, when it is most helpful. Consider information that would support or refute the alleged symptoms, such as treatment and insurance records, police reports, and interviews of close friends or family.
The patient interview may be prolonged because fatigue may diminish a malingerer’s ability to maintain fake symptoms. In very difficult cases, consider monitoring during inpatient assessment because feigned psychosis is extremely difficult to maintain 24 hours a day.
Watch for individuals who endorse rare or improbable symptoms. Rare symptoms—by definition—occur very infrequently, and even severely disturbed patients almost never report improbable symptoms.10 Consider asking malingerers about improbable symptoms to see if they will endorse them. For example:
- “When people talk to you, do you see the words they speak spelled out?”11
- “Have you ever believed that automobiles are members of an organized religion?”12
Watch closely for internal or external inconsistency in the suspected malingerer’s presentation (Table 2).
Table 2
Clues to identify malingering during patient evaluation
| Internal inconsistencies | Example |
| In subject’s report of symptoms | Gives a clear and articulate explanation of being confused |
| In subject’s own reported history | Gives conflicting versions |
| External inconsistencies | Example |
| Between reported and observed symptoms | Alleges having active auditory and visual hallucinations yet shows no evidence of being distracted |
| Between reported and observed level of functioning | Behaves in disorganized or confused manner around psychiatrist, yet plays excellent chess on ward with other patients |
| Between reported symptoms and nature of genuine symptoms | Reports seeing visual hallucinations in black and white, whereas genuine visual hallucinations are seen in color |
| Between reported symptoms and psychological test results | Alleges genuine psychotic symptoms, yet testing suggests faking or exaggeration |
Malingered Psychotic Symptoms
Detecting malingered mental illness is considered an advanced psychiatric skill, partly because you must understand thoroughly how genuine psychotic symptoms manifest.
Hallucinations. If a patient alleges atypical hallucinations, ask about them in detail. Hallucinations are usually (88%) associated with delusions.13 Genuine hallucinations are typically intermittent rather than continuous.
Continue to: Auditory hallucinations
Auditory hallucinations are usually clear, not vague (7%) or inaudible. Both male and female voices are commonly heard (75%), and voices are usually perceived as originating outside the head (88%).14 In schizophrenia, the major themes are persecutory or instructive.15
Command auditory hallucinations are easy to fabricate. Persons experiencing genuine command hallucinations:
- do not always obey the voices, especially if doing so would be dangerous16
- usually present with noncommand hallucinations (85%) and delusions (75%) as well17
Thus, view with suspicion someone who alleges an isolated command hallucination without other psychotic symptoms.
Genuine schizophrenic hallucinations tend to diminish when patients are involved in activities. Thus, to deal with their hallucinations, persons with schizophrenia typically cope by:
- engaging in activities (working, listening to a radio, watching TV)
- changing posture (lying down, walking)
- seeking interpersonal contact
- taking medications.
If you suspect a person of malingered auditory hallucinations, ask what he or she does to make the voices go away or diminish in intensity. Patients with genuine schizophrenia often can stop their auditory hallucinations while in remission but not during acute illness.
Malingerers may report auditory hallucinations of stilted or implausible language. For example, we have evaluated:
- an individual charged with attempted rape who alleged that voices said, “Go commit a sex offense.”
- a bank robber who alleged that voices kept screaming, “Stick up, stick up, stick up!”
Both examples contain language that is very questionable for genuine hallucinations, while providing the patient with “psychotic justification” for an illegal act that has a rational alternative motive.
Visual hallucinations are experienced by an estimated 24% to 30% of psychotic individuals but are reported much more often by malingerers (46%) than by persons with genuine psychosis (4%).18
Genuine visual hallucinations are usually of normalsized people and are seen in color.14 On rare occasions, genuine visual hallucinations of small people (Lilliputian hallucinations) may be associated with alcohol use, organic disease, or toxic psychosis (such as anticholinergic toxicity) but are rarely seen by persons with schizophrenia.
Psychotic visual hallucinations do not typically change if the eyes are closed or open, whereas drug-induced hallucinations are more readily seen with eyes closed or in the dark. Unformed hallucinations—such as flashes of light, shadows, or moving objects—are typically associated with neurologic disease and substance use.19
Suspect malingering if the patient reports dramatic or atypical visual hallucinations. For example, one defendant charged with bank robbery calmly reported seeing “a 30-foot tall, red giant smashing down the walls” of the interview room. When he was asked detailed questions, he frequently replied, “I don’t know.” He eventually admitted to malingering.
Delusions. Genuine delusions vary in content, theme, degree of systemization, and relevance to the person’s life. The complexity and sophistication of delusional systems usually reflect the individual’s intelligence. Persecutory delusions are more likely to be acted upon than are other types of delusions.20
Malingerers may claim that a delusion began or disappeared suddenly. In reality, systematized delusions usually take weeks to develop and much longer to disappear. Typically, the delusion will become somewhat less relevant, and the individual will gradually relinquish its importance over time after adequate treatment. In general, the more bizarre the delusion’s content, the more disorganized the individual’s thinking is likely to be (Table 3).
With genuine delusions, the individual’s behavior usually conforms to the delusions’ content. For example, Russell Weston—who suffered from schizophrenia—made a deadly assault on the U.S. Capitol in 1998 because he held a delusional belief that cannibalism was destroying Washington, DC. Before he shot and killed two U.S. Capitol security officers, he had gone to the Central Intelligence Agency several years before and voiced the same delusional concerns.
Suspect malingering if a patient alleges persecutory delusions without engaging in corresponding paranoid behaviors. One exception is the person with long-standing schizophrenia who has grown accustomed to the delusion and whose behavior is no longer consistent with it.
Table 3
Uncommon psychosis presentations that suggest malingering
| Hallucinations |
|
| Delusions |
|
Where Malingerers Trip Up
Malingerers may have inadequate or incomplete knowledge of the mental illness they are faking. Indeed, malingerers are like actors who can portray a role only as well as they understand it. They often overact their part or mistakenly believe the more bizarre their behavior, the more convincing they will be. Conversely, “successful” malingerers are more likely to endorse fewer symptoms and avoid endorsing overly bizarre or unusual symptoms.21
Continue to: Numerous clinical factors suggest malingering...
Numerous clinical factors suggest malingering (Table 4). Malingerers are more likely to eagerly “thrust forward” their illness, whereas patients with genuine schizophrenia are often reluctant to discuss their symptoms.22
Malingerers may attempt to take control of the interview and behave in an intimidating or hostile manner. They may accuse the psychiatrist of inferring that they are faking. Such behavior is rare in genuinely psychotic individuals. Although DSM-IV-TR states that antisocial personality disorder should arouse suspicions of malingering, some studies have failed to show a relationship. One study has associated psychopathic traits with malingering.23
Malingerers often believe that faking intellectual deficits, in addition to psychotic symptoms, will make them more believable. For example, a man who had completed several years of college alleged that he did not know the colors of the American flag.
Malingerers are more likely to give vague or hedging answers to straightforward questions. For example, when asked whether an alleged voice was male or female, one malingerer replied, “It was probably a man’s voice.” Malingerers may also answer, “I don’t know” to detailed questions about psychotic symptoms. Whereas a person with genuine psychotic symptoms could easily give an answer, the malingerer may have never experienced the symptoms and consequently “doesn’t know” the correct answer.
Psychotic symptoms such as derailment, neologisms, loose associations, and word salad are rarely simulated. This is because it is much more difficult for a malingerer to successfully imitate psychotic thought processes than psychotic thought content. Similarly, it is unusual for a malingerer to fake schizophrenia’s subtle signs, such as negative symptoms.
Table 4
Clinical factors that suggest malingering
| Absence of active or subtle signs of psychosis |
| Marked inconsistencies, contradictions |
Patient endorses improbable psychiatric symptoms
|
Patient is evasive or uncooperative
|
| Psychological testing indicates malingering (SIRS, M-FAST, MMPI-2) |
| SIRS: Structured Interview of Reported Symptoms |
| M-FAST: Miller Forensic Assessment of Symptoms Test |
| MMPI-2: Minnesota Multiphasic Personality Inventory, Revised |
Psychological Testing
Although many psychometric tests are available for detecting malingered psychosis, few have been validated. Among the more reliable are:
- Structured Interview of Reported Symptoms (SIRS)
- Minnesota Multiphasic Personality Inventory, Revised (MMPI-2)
- Miller Forensic Assessment of Symptoms Test (M-FAST).11
SIRS includes questions about rare symptoms, uncommon symptom pairing, atypical symptoms, and other indices involving excessive symptom reporting. It takes 30 to 60 minutes to administer. Tested in inpatient, forensic, and correctional populations, the SIRS has shown consistently high accuracy in detecting malingered psychiatric illness.24
Two MMPI-2 scales—F-scale and F-K Index—are the most frequently used test for evaluating suspected malingering. When using the MMPI-2 in this manner, consult the literature for appropriate cutoff scores (see Related resources). Although the MMPI-2 is the most validated psychometric method to detect malingering, a malingerer with high intelligence and previous knowledge of the test could evade detection.25
M-FAST was developed to provide a brief, reliable screen for malingered mental illness. This test takes 10 to 15 minutes to administer and measures rare symptom combinations, excessive reporting, and atypical symptoms.11 It has shown good validity and high correlation with the SIRS and MMPI-2.26,27
Confronting the Malingerer
If a thorough investigation indicates that a patient is malingering psychosis, you may decide to confront the evaluee. Avoid direct accusations of lying,10 and give the suspected malingerer every opportunity to save face. For example, it is preferable to say, “You haven’t told me the whole truth.”
A thoughtful approach that asks the evaluee to clarify inconsistencies is more likely to be productive and safer for the examiner. When confronting individuals with a history of violence and aggression, have adequate security personnel with you.
- Structured Interview of Reported Symptoms (SIRS). Available for purchase from Psychological Assessment Resources at www3.parinc.com (enter “SIRS” in search field).
- Graham JR. MMPI-2: Assessing personality and psychopathology. New York: Oxford Press; 2000. (Source of cutoff scores to use MMPI-2 scales [F-scale and F-K Index] to evaluate suspected malingering).
- Psychological Assessment Resources, Inc. Miller Forensic Assessment of Symptoms Test (M-FAST). Available at: www3.parinc.com (enter “M-FAST” in search field).
Reputed Cosa Nostra boss Vincent “The Chin” Gigante deceived “the most respected minds in forensic psychiatry” for years by malingering schizophrenia.1 Ultimately, he admitted to maintaining his charade from 1990 to 1997 during evaluations of his competency to stand trial for racketeering.
A lesson from this case—said a psychiatrist who concluded Gigante was malingering—is, “When feigning is a consideration, we must be more critical and less accepting of our impressions when we conduct and interpret a psychiatric examination…than might be the case in a typical clinical situation.”2
Even in typical clinical situations, however, psychiatrists may be reluctant to diagnose malingering3 for fear of being sued, assaulted—or wrong. An inaccurate diagnosis of malingering may unjustly stigmatize a patient and deny him needed care.4
Because psychiatrists need a systematized approach to detect malingering,5 we offer specific clinical factors and approaches to help you recognize malingered psychosis.
What is Malingering?
No other syndrome is as easy to define yet so difficult to diagnose as malingering. Reliably diagnosing malingered mental illness is complex, requiring the psychiatrist to consider collateral data beyond the patient interview.
Malingering is the intentional production of false or grossly exaggerated physical or psychological symptoms, motivated by external incentives.6 In practice, malingering commonly must be differentiated from factitious disorder, which also involves intentional production of symptoms. In factitious disorders, the patient’s motivation is to assume the sick role, which can be thought of as an internal or psychological incentive.
Three categories of malingering include:
- pure malingering (feigning a nonexistent disorder)
- partial malingering (consciously exaggerating real symptoms)
- false imputation (ascribing real symptoms to a cause the individual knows is unrelated to the symptoms).7
Motivations. Individuals usually malinger to avoid pain (such as difficult situations or punishment) or to seek pleasure (such as to obtain compensation or medications) (Table 1). In correctional settings, for example, inmates may malinger mental illness to do “easier time” or to obtain drugs. On the other hand, malingering in prison also may be an adaptive response by a mentally ill inmate to relatively sparse and difficult-to-obtain mental health resources.8
Table 1
Common motives of malingerers
| Motives | Examples |
|---|---|
| To avoid pain | To avoid: |
| Arrest | |
| Criminal prosecution | |
| Conscription into the military | |
| To seek pleasure | To obtain: |
| Controlled substances | |
| Free room and board | |
| Workers’ compensation or disability benefits for alleged psychological injury |
Interview Style
When you suspect a patient is malingering, keep your suspicions to yourself and conduct an objective evaluation. Patients are likely to become defensive if you show annoyance or incredulity, and putting them on guard decreases your ability to uncover evidence of malingering.9
Begin by asking open-ended questions, which allow patients to report symptoms in their own words. To avoid hinting at correct responses, carefully phrase initial inquiries about symptoms. Later in the interview, you can proceed to more-detailed questions of specific symptoms, as discussed below.
If possible, review collateral data before the interview, when it is most helpful. Consider information that would support or refute the alleged symptoms, such as treatment and insurance records, police reports, and interviews of close friends or family.
The patient interview may be prolonged because fatigue may diminish a malingerer’s ability to maintain fake symptoms. In very difficult cases, consider monitoring during inpatient assessment because feigned psychosis is extremely difficult to maintain 24 hours a day.
Watch for individuals who endorse rare or improbable symptoms. Rare symptoms—by definition—occur very infrequently, and even severely disturbed patients almost never report improbable symptoms.10 Consider asking malingerers about improbable symptoms to see if they will endorse them. For example:
- “When people talk to you, do you see the words they speak spelled out?”11
- “Have you ever believed that automobiles are members of an organized religion?”12
Watch closely for internal or external inconsistency in the suspected malingerer’s presentation (Table 2).
Table 2
Clues to identify malingering during patient evaluation
| Internal inconsistencies | Example |
| In subject’s report of symptoms | Gives a clear and articulate explanation of being confused |
| In subject’s own reported history | Gives conflicting versions |
| External inconsistencies | Example |
| Between reported and observed symptoms | Alleges having active auditory and visual hallucinations yet shows no evidence of being distracted |
| Between reported and observed level of functioning | Behaves in disorganized or confused manner around psychiatrist, yet plays excellent chess on ward with other patients |
| Between reported symptoms and nature of genuine symptoms | Reports seeing visual hallucinations in black and white, whereas genuine visual hallucinations are seen in color |
| Between reported symptoms and psychological test results | Alleges genuine psychotic symptoms, yet testing suggests faking or exaggeration |
Malingered Psychotic Symptoms
Detecting malingered mental illness is considered an advanced psychiatric skill, partly because you must understand thoroughly how genuine psychotic symptoms manifest.
Hallucinations. If a patient alleges atypical hallucinations, ask about them in detail. Hallucinations are usually (88%) associated with delusions.13 Genuine hallucinations are typically intermittent rather than continuous.
Continue to: Auditory hallucinations
Auditory hallucinations are usually clear, not vague (7%) or inaudible. Both male and female voices are commonly heard (75%), and voices are usually perceived as originating outside the head (88%).14 In schizophrenia, the major themes are persecutory or instructive.15
Command auditory hallucinations are easy to fabricate. Persons experiencing genuine command hallucinations:
- do not always obey the voices, especially if doing so would be dangerous16
- usually present with noncommand hallucinations (85%) and delusions (75%) as well17
Thus, view with suspicion someone who alleges an isolated command hallucination without other psychotic symptoms.
Genuine schizophrenic hallucinations tend to diminish when patients are involved in activities. Thus, to deal with their hallucinations, persons with schizophrenia typically cope by:
- engaging in activities (working, listening to a radio, watching TV)
- changing posture (lying down, walking)
- seeking interpersonal contact
- taking medications.
If you suspect a person of malingered auditory hallucinations, ask what he or she does to make the voices go away or diminish in intensity. Patients with genuine schizophrenia often can stop their auditory hallucinations while in remission but not during acute illness.
Malingerers may report auditory hallucinations of stilted or implausible language. For example, we have evaluated:
- an individual charged with attempted rape who alleged that voices said, “Go commit a sex offense.”
- a bank robber who alleged that voices kept screaming, “Stick up, stick up, stick up!”
Both examples contain language that is very questionable for genuine hallucinations, while providing the patient with “psychotic justification” for an illegal act that has a rational alternative motive.
Visual hallucinations are experienced by an estimated 24% to 30% of psychotic individuals but are reported much more often by malingerers (46%) than by persons with genuine psychosis (4%).18
Genuine visual hallucinations are usually of normalsized people and are seen in color.14 On rare occasions, genuine visual hallucinations of small people (Lilliputian hallucinations) may be associated with alcohol use, organic disease, or toxic psychosis (such as anticholinergic toxicity) but are rarely seen by persons with schizophrenia.
Psychotic visual hallucinations do not typically change if the eyes are closed or open, whereas drug-induced hallucinations are more readily seen with eyes closed or in the dark. Unformed hallucinations—such as flashes of light, shadows, or moving objects—are typically associated with neurologic disease and substance use.19
Suspect malingering if the patient reports dramatic or atypical visual hallucinations. For example, one defendant charged with bank robbery calmly reported seeing “a 30-foot tall, red giant smashing down the walls” of the interview room. When he was asked detailed questions, he frequently replied, “I don’t know.” He eventually admitted to malingering.
Delusions. Genuine delusions vary in content, theme, degree of systemization, and relevance to the person’s life. The complexity and sophistication of delusional systems usually reflect the individual’s intelligence. Persecutory delusions are more likely to be acted upon than are other types of delusions.20
Malingerers may claim that a delusion began or disappeared suddenly. In reality, systematized delusions usually take weeks to develop and much longer to disappear. Typically, the delusion will become somewhat less relevant, and the individual will gradually relinquish its importance over time after adequate treatment. In general, the more bizarre the delusion’s content, the more disorganized the individual’s thinking is likely to be (Table 3).
With genuine delusions, the individual’s behavior usually conforms to the delusions’ content. For example, Russell Weston—who suffered from schizophrenia—made a deadly assault on the U.S. Capitol in 1998 because he held a delusional belief that cannibalism was destroying Washington, DC. Before he shot and killed two U.S. Capitol security officers, he had gone to the Central Intelligence Agency several years before and voiced the same delusional concerns.
Suspect malingering if a patient alleges persecutory delusions without engaging in corresponding paranoid behaviors. One exception is the person with long-standing schizophrenia who has grown accustomed to the delusion and whose behavior is no longer consistent with it.
Table 3
Uncommon psychosis presentations that suggest malingering
| Hallucinations |
|
| Delusions |
|
Where Malingerers Trip Up
Malingerers may have inadequate or incomplete knowledge of the mental illness they are faking. Indeed, malingerers are like actors who can portray a role only as well as they understand it. They often overact their part or mistakenly believe the more bizarre their behavior, the more convincing they will be. Conversely, “successful” malingerers are more likely to endorse fewer symptoms and avoid endorsing overly bizarre or unusual symptoms.21
Continue to: Numerous clinical factors suggest malingering...
Numerous clinical factors suggest malingering (Table 4). Malingerers are more likely to eagerly “thrust forward” their illness, whereas patients with genuine schizophrenia are often reluctant to discuss their symptoms.22
Malingerers may attempt to take control of the interview and behave in an intimidating or hostile manner. They may accuse the psychiatrist of inferring that they are faking. Such behavior is rare in genuinely psychotic individuals. Although DSM-IV-TR states that antisocial personality disorder should arouse suspicions of malingering, some studies have failed to show a relationship. One study has associated psychopathic traits with malingering.23
Malingerers often believe that faking intellectual deficits, in addition to psychotic symptoms, will make them more believable. For example, a man who had completed several years of college alleged that he did not know the colors of the American flag.
Malingerers are more likely to give vague or hedging answers to straightforward questions. For example, when asked whether an alleged voice was male or female, one malingerer replied, “It was probably a man’s voice.” Malingerers may also answer, “I don’t know” to detailed questions about psychotic symptoms. Whereas a person with genuine psychotic symptoms could easily give an answer, the malingerer may have never experienced the symptoms and consequently “doesn’t know” the correct answer.
Psychotic symptoms such as derailment, neologisms, loose associations, and word salad are rarely simulated. This is because it is much more difficult for a malingerer to successfully imitate psychotic thought processes than psychotic thought content. Similarly, it is unusual for a malingerer to fake schizophrenia’s subtle signs, such as negative symptoms.
Table 4
Clinical factors that suggest malingering
| Absence of active or subtle signs of psychosis |
| Marked inconsistencies, contradictions |
Patient endorses improbable psychiatric symptoms
|
Patient is evasive or uncooperative
|
| Psychological testing indicates malingering (SIRS, M-FAST, MMPI-2) |
| SIRS: Structured Interview of Reported Symptoms |
| M-FAST: Miller Forensic Assessment of Symptoms Test |
| MMPI-2: Minnesota Multiphasic Personality Inventory, Revised |
Psychological Testing
Although many psychometric tests are available for detecting malingered psychosis, few have been validated. Among the more reliable are:
- Structured Interview of Reported Symptoms (SIRS)
- Minnesota Multiphasic Personality Inventory, Revised (MMPI-2)
- Miller Forensic Assessment of Symptoms Test (M-FAST).11
SIRS includes questions about rare symptoms, uncommon symptom pairing, atypical symptoms, and other indices involving excessive symptom reporting. It takes 30 to 60 minutes to administer. Tested in inpatient, forensic, and correctional populations, the SIRS has shown consistently high accuracy in detecting malingered psychiatric illness.24
Two MMPI-2 scales—F-scale and F-K Index—are the most frequently used test for evaluating suspected malingering. When using the MMPI-2 in this manner, consult the literature for appropriate cutoff scores (see Related resources). Although the MMPI-2 is the most validated psychometric method to detect malingering, a malingerer with high intelligence and previous knowledge of the test could evade detection.25
M-FAST was developed to provide a brief, reliable screen for malingered mental illness. This test takes 10 to 15 minutes to administer and measures rare symptom combinations, excessive reporting, and atypical symptoms.11 It has shown good validity and high correlation with the SIRS and MMPI-2.26,27
Confronting the Malingerer
If a thorough investigation indicates that a patient is malingering psychosis, you may decide to confront the evaluee. Avoid direct accusations of lying,10 and give the suspected malingerer every opportunity to save face. For example, it is preferable to say, “You haven’t told me the whole truth.”
A thoughtful approach that asks the evaluee to clarify inconsistencies is more likely to be productive and safer for the examiner. When confronting individuals with a history of violence and aggression, have adequate security personnel with you.
- Structured Interview of Reported Symptoms (SIRS). Available for purchase from Psychological Assessment Resources at www3.parinc.com (enter “SIRS” in search field).
- Graham JR. MMPI-2: Assessing personality and psychopathology. New York: Oxford Press; 2000. (Source of cutoff scores to use MMPI-2 scales [F-scale and F-K Index] to evaluate suspected malingering).
- Psychological Assessment Resources, Inc. Miller Forensic Assessment of Symptoms Test (M-FAST). Available at: www3.parinc.com (enter “M-FAST” in search field).
1. Newman A. Analyze this: Vincent Gigante, not crazy after all those years. New York Times, April 13, 2003.
2. Brodie JD. Personal communication, 2005.
3. Yates BD, Nordquist CR, Schultz-Ross RA. Feigned psychiatric symptoms in the emergency room. Psychiatr Serv 1996;47:998-1000.
4. Kropp PR, Rogers R. Understanding malingering: motivation, method, and detection. In: Lewis M, Saarini C (eds). Lying and deception. New York: Guilford Press; 1993.
5. Kucharski LT, et al. Clinical symptom presentation in suspected malingerers: an empirical investigation. Bull Am Acad Psychiatry Law. 1998;26:579-85.
6. Diagnostic and statistical manual of mental disorders (4th ed., text rev.). Washington, DC: American Psychiatric Association; 2000.
7. Resnick PJ. Malingering of posttraumatic stress disorders. In: Rogers R (ed). Clinical assessment of malingering and deception (2nd ed.). New York: Guilford Press; 1997;130-52.
8. Kupers TA. Malingering in correctional settings. Correctional Ment Health Rep. 2004;5(6):81-95.
9. Sadock BJ, Sadock VA. Kaplan & Sadock’s synopsis of psychiatry, 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2003;898.-
10. Thompson JW, LeBourgeois HW, Black FW. Malingering. In: Simon R, Gold L (eds). Textbook of forensic psychiatry. Washington, DC: American Psychiatric Publishing; 2004.
11. Miller HA. M.-FAST interview booklet. Lutz, FL: Psychological Assessment Resources; 2001.
12. Rogers R. Assessment of malingering within a forensic context. In Weisstub DW (ed.). Law and psychiatry: international perspectives. New York: Plenum Press; 1987;3:209-37.
13. Lewinsohn PM. An empirical test of several popular notions about hallucinations in schizophrenic patients. In: Keup W (ed.). Origin and mechanisms of hallucinations. New York: Plenum Press; 1970;401-3.
14. Goodwin DW, Anderson P, Rosenthal R. Clinical significance of hallucinations in psychiatric disorders: a study of 116 hallucinatory patients. Arch Gen Psychiatry 1971;24:76-80.
15. Small IF, Small JG, Andersen JM. Clinical characteristics of hallucinations of schizophrenia. Dis Nerv Sys 1966;27:349-53.
16. Kasper ME, Rogers R, Adams PA. Dangerousness and command hallucinations: an investigation of psychotic inpatients. Bull Am Acad Psychiatry Law 1996;24:219-24.
17. Thompson JS, Stuart GL, Holden CE. Command hallucinations and legal insanity. Forensic Rep 1992;5:29-43.
18. Cornell DG, Hawk GL. Clinical presentation of malingerers diagnosed by experienced forensic psychologists. Law Hum Behav 1989;13:375-83.
19. Cummings JL, Miller BL. Visual hallucinations: clinical occurrence and use in differential diagnosis. West J Med 1987;146:46-51.
20. Wessely S, Buchanan A, Reed A, et al. Acting on delusions: I. Prevalence. Br J Psychiatry 1993;163:69-76.
21. Edens JF, Guy LS, Otto RK, et al. Factors differentiating successful versus unsuccessful malingerers. J Pers Assess 2001;77(2):333-8.
22. Ritson B, Forest A. The simulation of psychosis: a contemporary presentation. Br J Psychol 1970;43:31-7.
23. Edens JF, Buffington JK, Tomicic TL. An investigation of the relationship between psychopathic traits and malingering on the Psychopathic Personality Inventory. Assessment 2000;7:281-96.
24. Rogers R. Structured interviews and dissimulation. In: Rogers R (ed). Clinical assessment of malingering and deception. New York: Guilford Press; 1997.
25. Pelfrey WV. The relationship between malingerers’ intelligence and MMPI-2 knowledge and their ability to avoid detection. Int J Offender Ther Comp Criminol. 2004;48(6):649-63.
26. Jackson RL, Rogers R, Sewell KW. Forensic applications of the Miller Forensic Assessment of Symptoms Test (MFAST): screening for feigned disorders in competency to stand trial evaluations. Law Human Behav 2005;29(2):199-210.
27. Miller HA. Examining the use of the M-FAST with criminal defendants incompetent to stand trial. Int J Offender Ther Comp Criminol 2004;48(3):268-80.
1. Newman A. Analyze this: Vincent Gigante, not crazy after all those years. New York Times, April 13, 2003.
2. Brodie JD. Personal communication, 2005.
3. Yates BD, Nordquist CR, Schultz-Ross RA. Feigned psychiatric symptoms in the emergency room. Psychiatr Serv 1996;47:998-1000.
4. Kropp PR, Rogers R. Understanding malingering: motivation, method, and detection. In: Lewis M, Saarini C (eds). Lying and deception. New York: Guilford Press; 1993.
5. Kucharski LT, et al. Clinical symptom presentation in suspected malingerers: an empirical investigation. Bull Am Acad Psychiatry Law. 1998;26:579-85.
6. Diagnostic and statistical manual of mental disorders (4th ed., text rev.). Washington, DC: American Psychiatric Association; 2000.
7. Resnick PJ. Malingering of posttraumatic stress disorders. In: Rogers R (ed). Clinical assessment of malingering and deception (2nd ed.). New York: Guilford Press; 1997;130-52.
8. Kupers TA. Malingering in correctional settings. Correctional Ment Health Rep. 2004;5(6):81-95.
9. Sadock BJ, Sadock VA. Kaplan & Sadock’s synopsis of psychiatry, 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2003;898.-
10. Thompson JW, LeBourgeois HW, Black FW. Malingering. In: Simon R, Gold L (eds). Textbook of forensic psychiatry. Washington, DC: American Psychiatric Publishing; 2004.
11. Miller HA. M.-FAST interview booklet. Lutz, FL: Psychological Assessment Resources; 2001.
12. Rogers R. Assessment of malingering within a forensic context. In Weisstub DW (ed.). Law and psychiatry: international perspectives. New York: Plenum Press; 1987;3:209-37.
13. Lewinsohn PM. An empirical test of several popular notions about hallucinations in schizophrenic patients. In: Keup W (ed.). Origin and mechanisms of hallucinations. New York: Plenum Press; 1970;401-3.
14. Goodwin DW, Anderson P, Rosenthal R. Clinical significance of hallucinations in psychiatric disorders: a study of 116 hallucinatory patients. Arch Gen Psychiatry 1971;24:76-80.
15. Small IF, Small JG, Andersen JM. Clinical characteristics of hallucinations of schizophrenia. Dis Nerv Sys 1966;27:349-53.
16. Kasper ME, Rogers R, Adams PA. Dangerousness and command hallucinations: an investigation of psychotic inpatients. Bull Am Acad Psychiatry Law 1996;24:219-24.
17. Thompson JS, Stuart GL, Holden CE. Command hallucinations and legal insanity. Forensic Rep 1992;5:29-43.
18. Cornell DG, Hawk GL. Clinical presentation of malingerers diagnosed by experienced forensic psychologists. Law Hum Behav 1989;13:375-83.
19. Cummings JL, Miller BL. Visual hallucinations: clinical occurrence and use in differential diagnosis. West J Med 1987;146:46-51.
20. Wessely S, Buchanan A, Reed A, et al. Acting on delusions: I. Prevalence. Br J Psychiatry 1993;163:69-76.
21. Edens JF, Guy LS, Otto RK, et al. Factors differentiating successful versus unsuccessful malingerers. J Pers Assess 2001;77(2):333-8.
22. Ritson B, Forest A. The simulation of psychosis: a contemporary presentation. Br J Psychol 1970;43:31-7.
23. Edens JF, Buffington JK, Tomicic TL. An investigation of the relationship between psychopathic traits and malingering on the Psychopathic Personality Inventory. Assessment 2000;7:281-96.
24. Rogers R. Structured interviews and dissimulation. In: Rogers R (ed). Clinical assessment of malingering and deception. New York: Guilford Press; 1997.
25. Pelfrey WV. The relationship between malingerers’ intelligence and MMPI-2 knowledge and their ability to avoid detection. Int J Offender Ther Comp Criminol. 2004;48(6):649-63.
26. Jackson RL, Rogers R, Sewell KW. Forensic applications of the Miller Forensic Assessment of Symptoms Test (MFAST): screening for feigned disorders in competency to stand trial evaluations. Law Human Behav 2005;29(2):199-210.
27. Miller HA. Examining the use of the M-FAST with criminal defendants incompetent to stand trial. Int J Offender Ther Comp Criminol 2004;48(3):268-80.
Beyond the mirror: Treating body dysmorphic disorder
Identifying which came first—body dysmorphic disorder (BDD) or comorbid anxiety or depressive disorders—can be as complex as treating the disorder’s delusional thinking and high suicide risk. To help you when working alone or with a psychotherapist, we offer strategies we have found useful for:
- diagnosing BDD
- educating patients and families about it
- choosing and dosing medications
- addressing inaccurate perceptions with targeted cognitive-behavioral therapies.
Though many recommendations are based on published data, we also draw on our clinical experience because research on effective BDD treatments is limited.
Body dysmorphic disorder (BDD) is preoccupation with an imagined defect in physical appearance or excessive concern about a slight physical anomaly that causes significant distress or impairs social, occupational, or other functioning.1 BDD patients have obsessive thoughts about their “flaws” and engage in compulsive behaviors and avoidances related to how they perceive their appearance, similar to behavior seen in obsessive-compulsive disorder. BDD causes great distress and disability, often accompanied by depression and suicidality.2
BDD occurs in an estimated 0.7% of the general population3 and in 6 to 14% of persons receiving treatment for anxiety or depressive disorders.4,5 These estimates may be low, however, as persons with BDD often do not seek treatment. Men and women are equally affected.6 Average age of onset is 16, although diagnosis often doesn’t occur for another 10 to 15 years.7
Assessment
BDD causes patients great distress and disability—often accompanied by major depression—but is easy to miss or misdiagnose (Box).1-7 Even when suicidal, BDD patients often do not reveal their symptoms to clinicians,2 probably because of poor insight or shame about their appearance. When a patient describes being unable to stop thinking about specific aspects of his or her appearance, assess further for BDD.
BDD patients’ conviction that their appearance is defective ranges from good insight to mildly overvalued ideation to frankly delusional.8 They often have ideas of reference (such as thinking others may be looking at their “defective” body part) and delusions of reference (such as being convinced others are talking about their “defective” body part). Asking a patient the questions in Table 1 can help establish the diagnosis. BDD also is included in the Structured Clinical Interview for DSM-IV (SCID). Useful assessment tools include:
- Body Dysmorphic Disorder Questionnaire,9 a 5-minute, patient-rated scale for screening
- Body Dysmorphic Disorder Examination,10 to diagnose BDD, survey BDD symptoms, and measure severity
- Yale-Brown Obsessive-Compulsive Scale modified for Body Dysmorphic Disorder (BDD-YBOCS),11 for measuring symptom severity and changes over time.
Comorbidity. Psychiatric comorbidity is common in BDD (Table 2),6,7,12-14 and deciding which disorder to address first can be difficult. If there is acute mania or non-BDD psychosis, we suggest that you stabilize these before treating BDD. Suicidality or severe substance dependence or abuse may result from BDD and therefore needs to be treated in conjunction with BDD.
If comorbid obsessive-compulsive disorder (OCD) or social phobia symptoms are interconnected with the patient’s BDD, treat concurrently; if not, address sequentially, starting with the more-severe symptoms. For example, symptoms that suggest social phobia (such as fear of public speaking) may be related to BDD, and treatment should focus on BDD. A patient with obsessive fears about how “contaminants” will affect her skin’s appearance may need to have the OCD and BDD addressed concurrently.
For other comorbidities, the treatment hierarchy is less clear. Major depression, for example, may be caused by severe BDD and might not improve until BDD improves. Even when a patient has several concurrent Axis I disorders, don’t over-look treating BDD; otherwise, the patient may remain quite impaired.
Assess suicide risk, as ≥ 25% of BDD patients may attempt suicide in their lifetimes.2 Safety measures include frequent monitoring, medication, family involvement, and—if necessary—hospitalization.
Table 1
Patient interview: Questions to help diagnose BDD
| Are you concerned about specific parts of your appearance that you believe are ugly or defective? |
| Do you find it difficult to stop thinking about parts of your appearance? |
| Do you avoid certain situations, places, or being seen in general because of your appearance? |
| Do you feel anxious, ashamed, disgusted, or depressed by specific aspects of your appearance? |
| Are any of your behaviors influenced by your appearance, such as trying to hide parts of your appearance or taking a long time getting ready to leave your residence? |
| Does your preoccupation cause you a lot of distress, anxiety, disgust, and/or shame? |
| Is preoccupation with your appearance interfering with your social life, ability to work, job performance, or other important areas of your life? |
| Do you tend to use mirrors very often or avoid them? |
| Does what you see in the mirror determine your mood that day? |
| How important do you think appearance is in life? |
| Do you use any oral or topical medications for dermatologic reasons or to prevent hair loss? |
| Have you ever had cosmetic surgery? If so, how satisfied were you with the outcome? Did you have any revisions? |
Table 2
Lifetime prevalence (%) of comorbid Axis I disorders in BDD
| Study | N | Major depression | Social phobia | OCD | Substance use disorders |
|---|---|---|---|---|---|
| Gunstad and Phillips (2003)*12 | 175 | 75 | 37 | 30 | 30 |
| Zimmerman and Mattia (1998)14 | 16 | 69 | 69 | 38 | 6 |
| Perugi et al (1997)13 | 58 | 41 | 12 | 41 | † |
| Veale et al (1996)7 | 50 | 8 | 16 | 6 | 2 |
| Hollander et al (1993)6 | 50 | 68 | 12 | 78 | 22 |
| N: number of study subjects | |||||
| OCD: obsessive-compulsive disorder | |||||
| * Phenomenology group | |||||
| † not reported | |||||
| Source: Adapted and reprinted with permission from reference 12. | |||||
Patient education
Improving insight. Educate patients that BDD is a brain disorder that creates faulty, inaccurate thoughts and perceptions about appearance. Many patients initially resist a BDD diagnosis; delusional thinking and poor insight lead them to assume the “flaw” they see is an accurate perception. They may need to hear about other persons with similar concerns to realize that a psychiatric disorder is causing their distress.
Other helpful resources for improving insight include:
- group therapy
- The Broken Mirror, by Katharine A. Phillips, MD,15 which contains case examples to which BDD sufferers may relate
- Websites and online forums (see Related resources).
Explaining BDD. Discuss possible causes of BDD, giving patients alternate explanations for the physical defects they perceive. Contributing factors may include:
- neurobiological abnormalities and genetic factors16
- a history since childhood of shyness, perfectionism, or anxious temperament
- being teased, abused, or in poor family and peer relationships.17
Emphasize that multiple, different, converging factors cause BDD for each individual.
The obsessive-compulsive cycle. Explain that thoughts create distressing emotions, and that persons with BDD try to relieve or prevent these emotions by performing compulsive behaviors. Compulsions then strengthen the association between intrusive thoughts about appearance “defects” and negative feelings about appearance.
Review a list of common compulsions (Table 3) with BDD patients, as many have engaged in these behaviors for years without realizing they are compulsions.
Table 3
Common BDD compulsions and avoidances
| Excessive grooming |
| Excessive checking or avoidance of mirrors and other reflective surfaces |
| Asking for reassurance about appearance |
| “Camouflaging” (hiding or covering up) supposed defects |
| Scrutinizing the appearance of other people and comparing to oneself |
| Avoiding social interactions |
| Avoidance of certain lighting conditions |
| Skin-picking to “fix” perceived flaws |
| Having repeated cosmetic or dermatological procedures, such as dermabrasion, cosmetic surgery, etc. |
Pharmacotherapy
BDD is a severe and complex disorder that often requires multimodal treatment using cognitive-behavioral therapy (CBT) and medication (algorithm).18 In our experience, most BDD patients need medication for the disorder and for common comorbidities. We recommend starting medications before or when beginning CBT for patients with moderate to severe BDD (BDD YBOCs ≥ 20).
Serotonin reuptake inhibitors (SRIs) have reduced BDD symptoms in open-label19,20 and controlled trials.21,22 As first-line treatments, SRIs decrease distress, compulsions, and frequency and intensity of obsessions about perceived defects; they also can improve insight.21-24 SRIs appear equally effective for delusional and nondelusional patients;21,23 whether CBT is similarly effective is unclear.
Relatively high dosages are usually necessary, according to published flexible-dosing trials in BDD,19-23 a retrospective chart review24 and our experience. Try dosages similar to those used for OCD (Table 4) as tolerated, and monitor for side effects. Twelve to 16 weeks of treatment are often needed for a full therapeutic effect.20-21
Augmentation. Consider adding another agent if a full SRI trial achieves partial symptom relief. One open-label trial of 13 BDD patients found that 6 (46%) improved when buspirone (mean dosage 48.3 mg/d) was added to SRI therapy.25 In a chart review, Phillips et al24 reported variable response rates of BDD patients treated with augmentation trials of clomipramine (4/9), buspirone (12/36), lithium (1/5), methylphenidate (1/6), and antipsychotics (2/13).
Very few studies have examined antipsychotic use in BDD. Placebo-controlled data are available only for pimozide.27 Conventional antipsychotics are unlikely to be effective, either as monotherapy26 or augmentation.27 As for the atypicals, olanzapine augmentation showed little to no efficacy in one small trial, although the average dosage used was low (4.6 mg/d).28 In our experience, atypicals—such as aripiprazole, 5 to 30 mg/d; quetiapine 100 to 300 mg/d; olanzapine, 7.5 to 15 mg/d; or risperidone, 1 to 3 mg/d—can improve BDD core symptoms and improve insight.
Benzodiazepines can be useful for acute anxiety or agitation. Carefully monitor benzodiazepine use, however, as substance abuse is relatively common in BDD patients.29
Table 4
Recommended SRI dosages for treating BDD*†
| Drug | Dosage range (mg/d) |
|---|---|
| Citalopram | 40 to 100 |
| Clomipramine | 150 to 250 |
| Escitalopram | 20 to 50 |
| Fluoxetine | 40 to 100 |
| Fluvoxamine | 200 to 400 |
| Paroxetine | 40 to 100 |
| Sertraline | 150 to 400 |
| * Off-label use. | |
| † May exceed FDA-recommended maximum dosages. | |
Specialized cbt techniques
Cognitive restructuring. Trying to convince BDD patients there is nothing wrong with their appearance will not be successful. Instead, we use cognitive restructuring to challenge the rationality of their thoughts and beliefs and to find alternate, more rational ones:
Therapist: “I know I cannot convince you that your (body area) is not defective, but can you give me evidence of how this ‘defect’ has affected your life?”
BDD patient: “Well, I haven’t had a date for a long time. I think this is evidence that my (body part) must be ugly, and that no one wants to date me because of it.”
Therapist: “What are some other possible reasons why you haven’t had a date in a long time? You admitted that you have barely left your house for many months. Is it possible that you have not had a date for a long time because you rarely go outside?”
With cognitive restructuring, patients learn to:
- identify automatic thoughts and beliefs that provoke distress
- examine evidence supporting or refuting these beliefs
- de-catastrophize (such as “What is the worst thing that could happen if you left the house today without checking your [body part]? Do you think you would eventually be able to cope with that?”)
- learn to more accurately assess the probability of feared negative consequences
- arrive at rational responses.
In our experience—which is supported by OCD literature—participating in CBT is very hard for patients with frank delusions, and insight determines how effective cognitive restructuring can be.30 If a patient is convinced a body part is defective, she is unlikely to stay in treatment—much less be open to restructuring her thoughts. Even unsuccessful attempts can help you gauge the intensity of patients’ beliefs, however.
During cognitive restructuring, it is important to uncover patients’ core beliefs (underlying, organizing principles they hold about themselves, others, and the world). BDD patients commonly believe that appearance is of utmost importance and that no one could love them because of their “defect.” The therapist can then help the patient challenge the rationality of those core beliefs.
Behavioral therapy. Basic behavioral therapy attempts to normalize excessive response to appearance concerns and to prepare patients for exposure and response prevention therapy (ERP). Having identified their compulsions, the next step is to guide patients in changing these behaviors, such as by:
- decreasing reassurance-seeking
- reducing avoidance of social situations
- decreasing opportunities to use the mirror
- reducing time spent on the Internet seeking cosmetic solutions
- increasing eye contact in social situations
- decreasing scanning of others’ physical features.
For example, suggest that BDD patients stand at least an arm’s length away when using a mirror for normal grooming. Then, instead of focusing on their body part, they will view it within the context of their entire face and body.
Exposure and response prevention
ERP exposes the patient to situations that evoke negative emotions—primarily shame and anxiety in BDD—so that they gradually habituate to these feelings. Individualize exposure exercises, targeting the body parts each person believes are defective. Because these exercises are intended to induce the discomfort patients usually experience, do not attempt ERP until the patient has had extensive education, developed insight, and has consented to treatment.
Create a hierarchy of ERP tasks (Table 5), ranking situations from low- to high-distress. Address items lower on the hierarchy first, and progress to higher items as the lower ones become easier to perform. Do not attempt the highest-distress items until the patient has improved insight and is not severely ill and suicidal.
During exposures, patients must remain in distress-provoking situations—without performing compulsive behaviors—until their negative feelings decrease by at least 50% of the initial subjective, self-rated distress level. Leaving the situation before stress diminishes may reinforce shame and discomfort. Performing compulsive behaviors during or after an exposure will negate the exposure’s effect.
Mirrors and ERP. Some therapists use mirrors for exposure exercises, but this is a complex issue. Mirror-checking is a common BDD compulsion that provides temporary relief but ultimately reinforces negative, intrusive thoughts about the disliked body area. How BDD patients perceive themselves changes from moment to moment; they may stare at and analyze any reflective surface in hopes that their “defect” will not appear as deformed or ugly that day. Thus, one cannot predict whether looking in the mirror at any one time is an exposure or a compulsion.
ERP exercises for BDD need to emphasize behaviors that involve interactions with the outside world, rather than reinforcing the importance of appearance. Using the mirror for ERP could promote checking compulsions and may send the message that appearance is the focal point of treatment. On the other hand, for patients with persistent mirror avoidance, gradual mirror exposures may be useful. A technique called mirror retraining helps patients objectively view their appearance and has been used with success in some individuals.
Table 5
Exposure and response therapy: a BDD patient’s sample hierarchy
| High-distress tasks | Subjective distress rating (scale of 0 to 100) |
| 1. Purposely creating the appearance of acne/skin defects | 100 |
| 2. Intentionally messing up my hair before going in public | 100 |
| 3. Standing under bright or fluorescent lighting in public | 90 |
| 4. Sitting in a position where others can directly see my face for an extended period | 85 |
| 5. Highlighting my face with a flashlight or bright light, while sitting in front of my therapist or another person. | 80 |
| Lower-distress tasks | |
| 6. Intentionally going outside in daylight hours, instead of only after dark | 70 |
| 7. Not turning away from others in an attempt to prevent them from seeing my face | 65 |
| 8. Standing close to people when talking to them, rather than standing at a distance | 50 |
| 9. Going out in public without camouflaging my hair with hats or scarves | 40 |
Psychosocial development
BDD therapy challenges the disorder’s core theme—that appearance is one’s only important attribute—and helps patients identify and develop qualities not related to appearance. Through social interactions, the BDD patient can:
- develop a multidimensional sense of self
- receive nonappearance-related positive feedback from the outside world.
Explore psychosocial development during the assessment phase and when a patient shows little progress in CBT. In some patients, for example, BDD onset in childhood or adolescence interferes with developmental transition to adulthood.
In our experience, some patients may resist treatment because of conscious and unconscious fears of adult responsibilities and relationships. We focus therapy on making them aware of these phenomena, exploring fears of development, and encouraging them to seek new relationships and responsibilities.
Because a BDD patient’s symptoms often create conflict and distress at home, offer the family support and education about the disorder. Occasionally, forces within the family seem to be working against the individual’s recovery and/or independence.
In some families, an individual with BDD may become the “identified patient,” diverting attention from other dysfunctional family members or relationships. During therapy, the BDD patient’s goal to develop a sense of self that is not appearance-based may run counter to the family’s need to keep him or her in the “sick” role.
If therapy is to succeed, talk to the patient about these dynamics. Consider family therapy if resistance to change is strong. When a patient is not progressing well with CBT, we find understanding the family system can be useful to comprehensive BDD treatment, although this observation remains to be validated.
Preventing and treating relapse
Educate patients that BDD is usually chronic, even when treated with psychotherapy and medication.31 Relapse often occurs, especially when patients discontinue medications on their own24 or drop out of therapy early. No guidelines exist, but based on our experience:
- we continue medication for at least 1 year after a patient improves
- psychotherapy is more variable but may need to last 6 to 12 months or more.
When therapy ends, we encourage patients to practice and reinforce everything they learned during treatment. Casting BDD resurgence as normal—and not as failure—will help patients who relapse to resist the downward spiral of low self-esteem, shame, and turning to the mirror for reassurance. Identifying BDD symptom triggers and developing plans to cope with them may also prevent relapse. CBT “booster sessions” scheduled monthly for 3 to 6 months may help patients who have completed therapy.
FOR CLINICIANS:
- Phillips KA. “I’m as ugly as the elephant man:” How to recognize and treat body dysmorphic disorder. Current Psychiatry. 2002;1(1):58-65.
- Cororve MB, Gleaves DH. Body dysmorphic disorder: a review of conceptualizations, assessment, and treatment strategies. Clin Psychol Rev. 2001;21(6):949-70.
FOR PATIENTS AND FAMILIES:
- Phillips KA. The broken mirror. New York: Oxford University Press; 2005.
- BDD and body image program, Butler Hospital, Providence, RI. BDD education and support. www.BDDcentral.com.
- Winograd A. Director, Accurate Reflections, Los Angeles, CA. Support group and information on BDD and obsessive compulsive spectrum disorders. www.AccurateReflections.com
Drug brand names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Buspirone • BuSpar
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Lithium • Lithobid, others
- Methylphenidate • Ritalin, Concerta
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders (4th ed. text rev.). Washington, DC: American Psychiatric Association; 2000.
2. Phillips KA, Coles ME, Menard W, et al. Suicidal ideation and suicide attempts in body dysmorphic disorder. J Clin Psychiatry 2005;66(6):717-25.
3. Otto MW, Wilhelm S, Cohen LS, Harlow BL. Prevalence of body dysmorphic disorder in a community sample of women. Am J Psychiatry 2001;158(12):2061-3.
4. Wilhelm S, Otto MW, Zucker BG, Pollack MH. Prevalence of body dysmorphic disorder in patients with anxiety disorders. J Anxiety Disord 1997;11(5):499-502.
5. Phillips KA, Nierenberg AA, Brendel G, Fava M. Prevalence and clinical features of body dysmorphic disorder in atypical major depression. J Nerv Ment Dis 1996;184(2):125-9.
6. Hollander E, Cohen L, Simeon D. Body dysmorphic disorder. Psychiatr Ann 1993;23:359-64.
7. Veale D, Boocock A, Gournay K, et al. Body dysmorphic disorder. survey of fifty cases. Br J Psychiatry 1996;169(2):196-201.
8. Phillips KA. Psychosis in body dysmorphic disorder. J Psychiatr Res 2004;38(1):63-72.
9. Dufresne RG, Phillips KA, Vittorio CC, Wilkel CS. A screening questionnaire for body dysmorphic disorder in a cosmetic dermatologic surgery practice. Dermatol Surg 2001;27(5):457-62.
10. Rosen JC, Reiter J. Development of the body dysmorphic disorder examination. Behav Res Ther 1996;34(9):755-66.
11. Phillips KA, Hollander E, Rasmussen SA, et al. A severity rating scale for body dysmorphic disorder: development, reliability, and validity of a modified version of the Yale-Brown Obsessive Compulsive Scale. Psychopharmacol Bull 1997;33(1):17-22.
12. Gunstad J, Phillips KA. Axis I comorbidity in body dysmorphic disorder. Compr Psychiatry 2003;44(4):270-6.
13. Perugi G, Akiskal HS, Giannotti D, et al. Gender-related differences in body dysmorphic disorder (dysmorphophobia). J Nerv Ment Dis 1997;185(9):578-82.
14. Zimmerman M, Mattia JI. Body dysmorphic disorder in psychiatric outpatients: recognition, prevalence, comorbidity, demographic, and clinical correlates. Compr Psychiatry 1998;39(5):265-70.
15. Phillips KA. The broken mirror. New York: Oxford University Press; 2005.
16. Rauch SL, Phillips KA, Segal E, et al. A preliminary morphometric magnetic resonance imaging study of regional brain volumes in body dysmorphic disorder. Psychiatry Res 2003;122(1):13-19.
17. Veale D. Body dysmorphic disorder. Postgrad Med J 2004;80(940):67-71.
18. Saxena S, Winograd A, Dunkin JJ, et al. A retrospective review of clinical characteristics and treatment response in body dysmorphic disorder versus obsessive-compulsive disorder. J Clin Psychiatry 2001;62:67-72.
19. Phillips KA, Najjar F. An open-label study of citalopram in body dysmorphic disorder. J Clin Psychiatry 2003;64(6):715-20.
20. Phillips KA, Dwight MM, McElroy SL. Efficacy and safety of fluvoxamine in body dysmorphic disorder. J Clin Psychiatry 1998;59(4):165-71.
21. Phillips KA, Albertini RS, Rasmussen SA. A randomized placebo-controlled trial of fluoxetine in body dysmorphic disorder. Arch Gen Psychiatry 2002;59(4):381-8.
22. Hollander E, Allen A, Kwon J, et al. Clomipramine vs desipramine crossover trial in body dysmorphic disorder: Selective efficacy of a serotonin reuptake inhibitor in imagined ugliness. Arch Gen Psychiatry 1999;56(11):1033-9.
23. Phillips KA, McElroy SL, Dwight MM, et al. Delusionality and response to open-label fluvoxamine in body dysmorphic disorder. J Clin Psychiatry 2001;62(2):87-91.
24. Phillips KA, Albertini RS, Siniscalchi JM, et al. Effectiveness of pharmacotherapy for body dysmorphic disorder: a chart-review study. J Clin Psychiatry 2001;62(9):721-7.
25. Phillips KA. An open study of buspirone augmentation of serotonin-reuptake inhibitors in body dysmorphic disorder. Psychopharmacol Bull 1996;32(1):175-80.
26. Phillips KA, McElroy SL, Keck PE, Jr, et al. A comparison of delusional and nondelusional body dysmorphic disorder in 100 cases. Psychopharmacol Bull 1994;30(2):179-86.
27. Phillips KA. Placebo-controlled study of pimozide augmentation of fluoxetine in body dysmorphic disorder. Am J Psychiatry 2005;162(2):377-9.
28. Phillips KA. Olanzapine augmentation of fluoxetine in body dysmorphic disorder. Am J Psychiatry 2005;162(5):1022-3.
29. Grant JE, Menard W, Pagano ME, et al. Substance use disorders in individuals with body dysmorphic disorder. J Clin Psychiatry 2005;66(3):309-16.
30. Foa EB. Failures in treating obsessive-compulsives. Behav Res Ther 1979;17:169-76.
31. Phillips KA, McElroy SL, Keck PE, Jr, et al. Body dysmorphic disorder: 30 cases of imagined ugliness. Am J Psychiatry 1993;150(2):302-8.
Identifying which came first—body dysmorphic disorder (BDD) or comorbid anxiety or depressive disorders—can be as complex as treating the disorder’s delusional thinking and high suicide risk. To help you when working alone or with a psychotherapist, we offer strategies we have found useful for:
- diagnosing BDD
- educating patients and families about it
- choosing and dosing medications
- addressing inaccurate perceptions with targeted cognitive-behavioral therapies.
Though many recommendations are based on published data, we also draw on our clinical experience because research on effective BDD treatments is limited.
Body dysmorphic disorder (BDD) is preoccupation with an imagined defect in physical appearance or excessive concern about a slight physical anomaly that causes significant distress or impairs social, occupational, or other functioning.1 BDD patients have obsessive thoughts about their “flaws” and engage in compulsive behaviors and avoidances related to how they perceive their appearance, similar to behavior seen in obsessive-compulsive disorder. BDD causes great distress and disability, often accompanied by depression and suicidality.2
BDD occurs in an estimated 0.7% of the general population3 and in 6 to 14% of persons receiving treatment for anxiety or depressive disorders.4,5 These estimates may be low, however, as persons with BDD often do not seek treatment. Men and women are equally affected.6 Average age of onset is 16, although diagnosis often doesn’t occur for another 10 to 15 years.7
Assessment
BDD causes patients great distress and disability—often accompanied by major depression—but is easy to miss or misdiagnose (Box).1-7 Even when suicidal, BDD patients often do not reveal their symptoms to clinicians,2 probably because of poor insight or shame about their appearance. When a patient describes being unable to stop thinking about specific aspects of his or her appearance, assess further for BDD.
BDD patients’ conviction that their appearance is defective ranges from good insight to mildly overvalued ideation to frankly delusional.8 They often have ideas of reference (such as thinking others may be looking at their “defective” body part) and delusions of reference (such as being convinced others are talking about their “defective” body part). Asking a patient the questions in Table 1 can help establish the diagnosis. BDD also is included in the Structured Clinical Interview for DSM-IV (SCID). Useful assessment tools include:
- Body Dysmorphic Disorder Questionnaire,9 a 5-minute, patient-rated scale for screening
- Body Dysmorphic Disorder Examination,10 to diagnose BDD, survey BDD symptoms, and measure severity
- Yale-Brown Obsessive-Compulsive Scale modified for Body Dysmorphic Disorder (BDD-YBOCS),11 for measuring symptom severity and changes over time.
Comorbidity. Psychiatric comorbidity is common in BDD (Table 2),6,7,12-14 and deciding which disorder to address first can be difficult. If there is acute mania or non-BDD psychosis, we suggest that you stabilize these before treating BDD. Suicidality or severe substance dependence or abuse may result from BDD and therefore needs to be treated in conjunction with BDD.
If comorbid obsessive-compulsive disorder (OCD) or social phobia symptoms are interconnected with the patient’s BDD, treat concurrently; if not, address sequentially, starting with the more-severe symptoms. For example, symptoms that suggest social phobia (such as fear of public speaking) may be related to BDD, and treatment should focus on BDD. A patient with obsessive fears about how “contaminants” will affect her skin’s appearance may need to have the OCD and BDD addressed concurrently.
For other comorbidities, the treatment hierarchy is less clear. Major depression, for example, may be caused by severe BDD and might not improve until BDD improves. Even when a patient has several concurrent Axis I disorders, don’t over-look treating BDD; otherwise, the patient may remain quite impaired.
Assess suicide risk, as ≥ 25% of BDD patients may attempt suicide in their lifetimes.2 Safety measures include frequent monitoring, medication, family involvement, and—if necessary—hospitalization.
Table 1
Patient interview: Questions to help diagnose BDD
| Are you concerned about specific parts of your appearance that you believe are ugly or defective? |
| Do you find it difficult to stop thinking about parts of your appearance? |
| Do you avoid certain situations, places, or being seen in general because of your appearance? |
| Do you feel anxious, ashamed, disgusted, or depressed by specific aspects of your appearance? |
| Are any of your behaviors influenced by your appearance, such as trying to hide parts of your appearance or taking a long time getting ready to leave your residence? |
| Does your preoccupation cause you a lot of distress, anxiety, disgust, and/or shame? |
| Is preoccupation with your appearance interfering with your social life, ability to work, job performance, or other important areas of your life? |
| Do you tend to use mirrors very often or avoid them? |
| Does what you see in the mirror determine your mood that day? |
| How important do you think appearance is in life? |
| Do you use any oral or topical medications for dermatologic reasons or to prevent hair loss? |
| Have you ever had cosmetic surgery? If so, how satisfied were you with the outcome? Did you have any revisions? |
Table 2
Lifetime prevalence (%) of comorbid Axis I disorders in BDD
| Study | N | Major depression | Social phobia | OCD | Substance use disorders |
|---|---|---|---|---|---|
| Gunstad and Phillips (2003)*12 | 175 | 75 | 37 | 30 | 30 |
| Zimmerman and Mattia (1998)14 | 16 | 69 | 69 | 38 | 6 |
| Perugi et al (1997)13 | 58 | 41 | 12 | 41 | † |
| Veale et al (1996)7 | 50 | 8 | 16 | 6 | 2 |
| Hollander et al (1993)6 | 50 | 68 | 12 | 78 | 22 |
| N: number of study subjects | |||||
| OCD: obsessive-compulsive disorder | |||||
| * Phenomenology group | |||||
| † not reported | |||||
| Source: Adapted and reprinted with permission from reference 12. | |||||
Patient education
Improving insight. Educate patients that BDD is a brain disorder that creates faulty, inaccurate thoughts and perceptions about appearance. Many patients initially resist a BDD diagnosis; delusional thinking and poor insight lead them to assume the “flaw” they see is an accurate perception. They may need to hear about other persons with similar concerns to realize that a psychiatric disorder is causing their distress.
Other helpful resources for improving insight include:
- group therapy
- The Broken Mirror, by Katharine A. Phillips, MD,15 which contains case examples to which BDD sufferers may relate
- Websites and online forums (see Related resources).
Explaining BDD. Discuss possible causes of BDD, giving patients alternate explanations for the physical defects they perceive. Contributing factors may include:
- neurobiological abnormalities and genetic factors16
- a history since childhood of shyness, perfectionism, or anxious temperament
- being teased, abused, or in poor family and peer relationships.17
Emphasize that multiple, different, converging factors cause BDD for each individual.
The obsessive-compulsive cycle. Explain that thoughts create distressing emotions, and that persons with BDD try to relieve or prevent these emotions by performing compulsive behaviors. Compulsions then strengthen the association between intrusive thoughts about appearance “defects” and negative feelings about appearance.
Review a list of common compulsions (Table 3) with BDD patients, as many have engaged in these behaviors for years without realizing they are compulsions.
Table 3
Common BDD compulsions and avoidances
| Excessive grooming |
| Excessive checking or avoidance of mirrors and other reflective surfaces |
| Asking for reassurance about appearance |
| “Camouflaging” (hiding or covering up) supposed defects |
| Scrutinizing the appearance of other people and comparing to oneself |
| Avoiding social interactions |
| Avoidance of certain lighting conditions |
| Skin-picking to “fix” perceived flaws |
| Having repeated cosmetic or dermatological procedures, such as dermabrasion, cosmetic surgery, etc. |
Pharmacotherapy
BDD is a severe and complex disorder that often requires multimodal treatment using cognitive-behavioral therapy (CBT) and medication (algorithm).18 In our experience, most BDD patients need medication for the disorder and for common comorbidities. We recommend starting medications before or when beginning CBT for patients with moderate to severe BDD (BDD YBOCs ≥ 20).
Serotonin reuptake inhibitors (SRIs) have reduced BDD symptoms in open-label19,20 and controlled trials.21,22 As first-line treatments, SRIs decrease distress, compulsions, and frequency and intensity of obsessions about perceived defects; they also can improve insight.21-24 SRIs appear equally effective for delusional and nondelusional patients;21,23 whether CBT is similarly effective is unclear.
Relatively high dosages are usually necessary, according to published flexible-dosing trials in BDD,19-23 a retrospective chart review24 and our experience. Try dosages similar to those used for OCD (Table 4) as tolerated, and monitor for side effects. Twelve to 16 weeks of treatment are often needed for a full therapeutic effect.20-21
Augmentation. Consider adding another agent if a full SRI trial achieves partial symptom relief. One open-label trial of 13 BDD patients found that 6 (46%) improved when buspirone (mean dosage 48.3 mg/d) was added to SRI therapy.25 In a chart review, Phillips et al24 reported variable response rates of BDD patients treated with augmentation trials of clomipramine (4/9), buspirone (12/36), lithium (1/5), methylphenidate (1/6), and antipsychotics (2/13).
Very few studies have examined antipsychotic use in BDD. Placebo-controlled data are available only for pimozide.27 Conventional antipsychotics are unlikely to be effective, either as monotherapy26 or augmentation.27 As for the atypicals, olanzapine augmentation showed little to no efficacy in one small trial, although the average dosage used was low (4.6 mg/d).28 In our experience, atypicals—such as aripiprazole, 5 to 30 mg/d; quetiapine 100 to 300 mg/d; olanzapine, 7.5 to 15 mg/d; or risperidone, 1 to 3 mg/d—can improve BDD core symptoms and improve insight.
Benzodiazepines can be useful for acute anxiety or agitation. Carefully monitor benzodiazepine use, however, as substance abuse is relatively common in BDD patients.29
Table 4
Recommended SRI dosages for treating BDD*†
| Drug | Dosage range (mg/d) |
|---|---|
| Citalopram | 40 to 100 |
| Clomipramine | 150 to 250 |
| Escitalopram | 20 to 50 |
| Fluoxetine | 40 to 100 |
| Fluvoxamine | 200 to 400 |
| Paroxetine | 40 to 100 |
| Sertraline | 150 to 400 |
| * Off-label use. | |
| † May exceed FDA-recommended maximum dosages. | |
Specialized cbt techniques
Cognitive restructuring. Trying to convince BDD patients there is nothing wrong with their appearance will not be successful. Instead, we use cognitive restructuring to challenge the rationality of their thoughts and beliefs and to find alternate, more rational ones:
Therapist: “I know I cannot convince you that your (body area) is not defective, but can you give me evidence of how this ‘defect’ has affected your life?”
BDD patient: “Well, I haven’t had a date for a long time. I think this is evidence that my (body part) must be ugly, and that no one wants to date me because of it.”
Therapist: “What are some other possible reasons why you haven’t had a date in a long time? You admitted that you have barely left your house for many months. Is it possible that you have not had a date for a long time because you rarely go outside?”
With cognitive restructuring, patients learn to:
- identify automatic thoughts and beliefs that provoke distress
- examine evidence supporting or refuting these beliefs
- de-catastrophize (such as “What is the worst thing that could happen if you left the house today without checking your [body part]? Do you think you would eventually be able to cope with that?”)
- learn to more accurately assess the probability of feared negative consequences
- arrive at rational responses.
In our experience—which is supported by OCD literature—participating in CBT is very hard for patients with frank delusions, and insight determines how effective cognitive restructuring can be.30 If a patient is convinced a body part is defective, she is unlikely to stay in treatment—much less be open to restructuring her thoughts. Even unsuccessful attempts can help you gauge the intensity of patients’ beliefs, however.
During cognitive restructuring, it is important to uncover patients’ core beliefs (underlying, organizing principles they hold about themselves, others, and the world). BDD patients commonly believe that appearance is of utmost importance and that no one could love them because of their “defect.” The therapist can then help the patient challenge the rationality of those core beliefs.
Behavioral therapy. Basic behavioral therapy attempts to normalize excessive response to appearance concerns and to prepare patients for exposure and response prevention therapy (ERP). Having identified their compulsions, the next step is to guide patients in changing these behaviors, such as by:
- decreasing reassurance-seeking
- reducing avoidance of social situations
- decreasing opportunities to use the mirror
- reducing time spent on the Internet seeking cosmetic solutions
- increasing eye contact in social situations
- decreasing scanning of others’ physical features.
For example, suggest that BDD patients stand at least an arm’s length away when using a mirror for normal grooming. Then, instead of focusing on their body part, they will view it within the context of their entire face and body.
Exposure and response prevention
ERP exposes the patient to situations that evoke negative emotions—primarily shame and anxiety in BDD—so that they gradually habituate to these feelings. Individualize exposure exercises, targeting the body parts each person believes are defective. Because these exercises are intended to induce the discomfort patients usually experience, do not attempt ERP until the patient has had extensive education, developed insight, and has consented to treatment.
Create a hierarchy of ERP tasks (Table 5), ranking situations from low- to high-distress. Address items lower on the hierarchy first, and progress to higher items as the lower ones become easier to perform. Do not attempt the highest-distress items until the patient has improved insight and is not severely ill and suicidal.
During exposures, patients must remain in distress-provoking situations—without performing compulsive behaviors—until their negative feelings decrease by at least 50% of the initial subjective, self-rated distress level. Leaving the situation before stress diminishes may reinforce shame and discomfort. Performing compulsive behaviors during or after an exposure will negate the exposure’s effect.
Mirrors and ERP. Some therapists use mirrors for exposure exercises, but this is a complex issue. Mirror-checking is a common BDD compulsion that provides temporary relief but ultimately reinforces negative, intrusive thoughts about the disliked body area. How BDD patients perceive themselves changes from moment to moment; they may stare at and analyze any reflective surface in hopes that their “defect” will not appear as deformed or ugly that day. Thus, one cannot predict whether looking in the mirror at any one time is an exposure or a compulsion.
ERP exercises for BDD need to emphasize behaviors that involve interactions with the outside world, rather than reinforcing the importance of appearance. Using the mirror for ERP could promote checking compulsions and may send the message that appearance is the focal point of treatment. On the other hand, for patients with persistent mirror avoidance, gradual mirror exposures may be useful. A technique called mirror retraining helps patients objectively view their appearance and has been used with success in some individuals.
Table 5
Exposure and response therapy: a BDD patient’s sample hierarchy
| High-distress tasks | Subjective distress rating (scale of 0 to 100) |
| 1. Purposely creating the appearance of acne/skin defects | 100 |
| 2. Intentionally messing up my hair before going in public | 100 |
| 3. Standing under bright or fluorescent lighting in public | 90 |
| 4. Sitting in a position where others can directly see my face for an extended period | 85 |
| 5. Highlighting my face with a flashlight or bright light, while sitting in front of my therapist or another person. | 80 |
| Lower-distress tasks | |
| 6. Intentionally going outside in daylight hours, instead of only after dark | 70 |
| 7. Not turning away from others in an attempt to prevent them from seeing my face | 65 |
| 8. Standing close to people when talking to them, rather than standing at a distance | 50 |
| 9. Going out in public without camouflaging my hair with hats or scarves | 40 |
Psychosocial development
BDD therapy challenges the disorder’s core theme—that appearance is one’s only important attribute—and helps patients identify and develop qualities not related to appearance. Through social interactions, the BDD patient can:
- develop a multidimensional sense of self
- receive nonappearance-related positive feedback from the outside world.
Explore psychosocial development during the assessment phase and when a patient shows little progress in CBT. In some patients, for example, BDD onset in childhood or adolescence interferes with developmental transition to adulthood.
In our experience, some patients may resist treatment because of conscious and unconscious fears of adult responsibilities and relationships. We focus therapy on making them aware of these phenomena, exploring fears of development, and encouraging them to seek new relationships and responsibilities.
Because a BDD patient’s symptoms often create conflict and distress at home, offer the family support and education about the disorder. Occasionally, forces within the family seem to be working against the individual’s recovery and/or independence.
In some families, an individual with BDD may become the “identified patient,” diverting attention from other dysfunctional family members or relationships. During therapy, the BDD patient’s goal to develop a sense of self that is not appearance-based may run counter to the family’s need to keep him or her in the “sick” role.
If therapy is to succeed, talk to the patient about these dynamics. Consider family therapy if resistance to change is strong. When a patient is not progressing well with CBT, we find understanding the family system can be useful to comprehensive BDD treatment, although this observation remains to be validated.
Preventing and treating relapse
Educate patients that BDD is usually chronic, even when treated with psychotherapy and medication.31 Relapse often occurs, especially when patients discontinue medications on their own24 or drop out of therapy early. No guidelines exist, but based on our experience:
- we continue medication for at least 1 year after a patient improves
- psychotherapy is more variable but may need to last 6 to 12 months or more.
When therapy ends, we encourage patients to practice and reinforce everything they learned during treatment. Casting BDD resurgence as normal—and not as failure—will help patients who relapse to resist the downward spiral of low self-esteem, shame, and turning to the mirror for reassurance. Identifying BDD symptom triggers and developing plans to cope with them may also prevent relapse. CBT “booster sessions” scheduled monthly for 3 to 6 months may help patients who have completed therapy.
FOR CLINICIANS:
- Phillips KA. “I’m as ugly as the elephant man:” How to recognize and treat body dysmorphic disorder. Current Psychiatry. 2002;1(1):58-65.
- Cororve MB, Gleaves DH. Body dysmorphic disorder: a review of conceptualizations, assessment, and treatment strategies. Clin Psychol Rev. 2001;21(6):949-70.
FOR PATIENTS AND FAMILIES:
- Phillips KA. The broken mirror. New York: Oxford University Press; 2005.
- BDD and body image program, Butler Hospital, Providence, RI. BDD education and support. www.BDDcentral.com.
- Winograd A. Director, Accurate Reflections, Los Angeles, CA. Support group and information on BDD and obsessive compulsive spectrum disorders. www.AccurateReflections.com
Drug brand names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Buspirone • BuSpar
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Lithium • Lithobid, others
- Methylphenidate • Ritalin, Concerta
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Identifying which came first—body dysmorphic disorder (BDD) or comorbid anxiety or depressive disorders—can be as complex as treating the disorder’s delusional thinking and high suicide risk. To help you when working alone or with a psychotherapist, we offer strategies we have found useful for:
- diagnosing BDD
- educating patients and families about it
- choosing and dosing medications
- addressing inaccurate perceptions with targeted cognitive-behavioral therapies.
Though many recommendations are based on published data, we also draw on our clinical experience because research on effective BDD treatments is limited.
Body dysmorphic disorder (BDD) is preoccupation with an imagined defect in physical appearance or excessive concern about a slight physical anomaly that causes significant distress or impairs social, occupational, or other functioning.1 BDD patients have obsessive thoughts about their “flaws” and engage in compulsive behaviors and avoidances related to how they perceive their appearance, similar to behavior seen in obsessive-compulsive disorder. BDD causes great distress and disability, often accompanied by depression and suicidality.2
BDD occurs in an estimated 0.7% of the general population3 and in 6 to 14% of persons receiving treatment for anxiety or depressive disorders.4,5 These estimates may be low, however, as persons with BDD often do not seek treatment. Men and women are equally affected.6 Average age of onset is 16, although diagnosis often doesn’t occur for another 10 to 15 years.7
Assessment
BDD causes patients great distress and disability—often accompanied by major depression—but is easy to miss or misdiagnose (Box).1-7 Even when suicidal, BDD patients often do not reveal their symptoms to clinicians,2 probably because of poor insight or shame about their appearance. When a patient describes being unable to stop thinking about specific aspects of his or her appearance, assess further for BDD.
BDD patients’ conviction that their appearance is defective ranges from good insight to mildly overvalued ideation to frankly delusional.8 They often have ideas of reference (such as thinking others may be looking at their “defective” body part) and delusions of reference (such as being convinced others are talking about their “defective” body part). Asking a patient the questions in Table 1 can help establish the diagnosis. BDD also is included in the Structured Clinical Interview for DSM-IV (SCID). Useful assessment tools include:
- Body Dysmorphic Disorder Questionnaire,9 a 5-minute, patient-rated scale for screening
- Body Dysmorphic Disorder Examination,10 to diagnose BDD, survey BDD symptoms, and measure severity
- Yale-Brown Obsessive-Compulsive Scale modified for Body Dysmorphic Disorder (BDD-YBOCS),11 for measuring symptom severity and changes over time.
Comorbidity. Psychiatric comorbidity is common in BDD (Table 2),6,7,12-14 and deciding which disorder to address first can be difficult. If there is acute mania or non-BDD psychosis, we suggest that you stabilize these before treating BDD. Suicidality or severe substance dependence or abuse may result from BDD and therefore needs to be treated in conjunction with BDD.
If comorbid obsessive-compulsive disorder (OCD) or social phobia symptoms are interconnected with the patient’s BDD, treat concurrently; if not, address sequentially, starting with the more-severe symptoms. For example, symptoms that suggest social phobia (such as fear of public speaking) may be related to BDD, and treatment should focus on BDD. A patient with obsessive fears about how “contaminants” will affect her skin’s appearance may need to have the OCD and BDD addressed concurrently.
For other comorbidities, the treatment hierarchy is less clear. Major depression, for example, may be caused by severe BDD and might not improve until BDD improves. Even when a patient has several concurrent Axis I disorders, don’t over-look treating BDD; otherwise, the patient may remain quite impaired.
Assess suicide risk, as ≥ 25% of BDD patients may attempt suicide in their lifetimes.2 Safety measures include frequent monitoring, medication, family involvement, and—if necessary—hospitalization.
Table 1
Patient interview: Questions to help diagnose BDD
| Are you concerned about specific parts of your appearance that you believe are ugly or defective? |
| Do you find it difficult to stop thinking about parts of your appearance? |
| Do you avoid certain situations, places, or being seen in general because of your appearance? |
| Do you feel anxious, ashamed, disgusted, or depressed by specific aspects of your appearance? |
| Are any of your behaviors influenced by your appearance, such as trying to hide parts of your appearance or taking a long time getting ready to leave your residence? |
| Does your preoccupation cause you a lot of distress, anxiety, disgust, and/or shame? |
| Is preoccupation with your appearance interfering with your social life, ability to work, job performance, or other important areas of your life? |
| Do you tend to use mirrors very often or avoid them? |
| Does what you see in the mirror determine your mood that day? |
| How important do you think appearance is in life? |
| Do you use any oral or topical medications for dermatologic reasons or to prevent hair loss? |
| Have you ever had cosmetic surgery? If so, how satisfied were you with the outcome? Did you have any revisions? |
Table 2
Lifetime prevalence (%) of comorbid Axis I disorders in BDD
| Study | N | Major depression | Social phobia | OCD | Substance use disorders |
|---|---|---|---|---|---|
| Gunstad and Phillips (2003)*12 | 175 | 75 | 37 | 30 | 30 |
| Zimmerman and Mattia (1998)14 | 16 | 69 | 69 | 38 | 6 |
| Perugi et al (1997)13 | 58 | 41 | 12 | 41 | † |
| Veale et al (1996)7 | 50 | 8 | 16 | 6 | 2 |
| Hollander et al (1993)6 | 50 | 68 | 12 | 78 | 22 |
| N: number of study subjects | |||||
| OCD: obsessive-compulsive disorder | |||||
| * Phenomenology group | |||||
| † not reported | |||||
| Source: Adapted and reprinted with permission from reference 12. | |||||
Patient education
Improving insight. Educate patients that BDD is a brain disorder that creates faulty, inaccurate thoughts and perceptions about appearance. Many patients initially resist a BDD diagnosis; delusional thinking and poor insight lead them to assume the “flaw” they see is an accurate perception. They may need to hear about other persons with similar concerns to realize that a psychiatric disorder is causing their distress.
Other helpful resources for improving insight include:
- group therapy
- The Broken Mirror, by Katharine A. Phillips, MD,15 which contains case examples to which BDD sufferers may relate
- Websites and online forums (see Related resources).
Explaining BDD. Discuss possible causes of BDD, giving patients alternate explanations for the physical defects they perceive. Contributing factors may include:
- neurobiological abnormalities and genetic factors16
- a history since childhood of shyness, perfectionism, or anxious temperament
- being teased, abused, or in poor family and peer relationships.17
Emphasize that multiple, different, converging factors cause BDD for each individual.
The obsessive-compulsive cycle. Explain that thoughts create distressing emotions, and that persons with BDD try to relieve or prevent these emotions by performing compulsive behaviors. Compulsions then strengthen the association between intrusive thoughts about appearance “defects” and negative feelings about appearance.
Review a list of common compulsions (Table 3) with BDD patients, as many have engaged in these behaviors for years without realizing they are compulsions.
Table 3
Common BDD compulsions and avoidances
| Excessive grooming |
| Excessive checking or avoidance of mirrors and other reflective surfaces |
| Asking for reassurance about appearance |
| “Camouflaging” (hiding or covering up) supposed defects |
| Scrutinizing the appearance of other people and comparing to oneself |
| Avoiding social interactions |
| Avoidance of certain lighting conditions |
| Skin-picking to “fix” perceived flaws |
| Having repeated cosmetic or dermatological procedures, such as dermabrasion, cosmetic surgery, etc. |
Pharmacotherapy
BDD is a severe and complex disorder that often requires multimodal treatment using cognitive-behavioral therapy (CBT) and medication (algorithm).18 In our experience, most BDD patients need medication for the disorder and for common comorbidities. We recommend starting medications before or when beginning CBT for patients with moderate to severe BDD (BDD YBOCs ≥ 20).
Serotonin reuptake inhibitors (SRIs) have reduced BDD symptoms in open-label19,20 and controlled trials.21,22 As first-line treatments, SRIs decrease distress, compulsions, and frequency and intensity of obsessions about perceived defects; they also can improve insight.21-24 SRIs appear equally effective for delusional and nondelusional patients;21,23 whether CBT is similarly effective is unclear.
Relatively high dosages are usually necessary, according to published flexible-dosing trials in BDD,19-23 a retrospective chart review24 and our experience. Try dosages similar to those used for OCD (Table 4) as tolerated, and monitor for side effects. Twelve to 16 weeks of treatment are often needed for a full therapeutic effect.20-21
Augmentation. Consider adding another agent if a full SRI trial achieves partial symptom relief. One open-label trial of 13 BDD patients found that 6 (46%) improved when buspirone (mean dosage 48.3 mg/d) was added to SRI therapy.25 In a chart review, Phillips et al24 reported variable response rates of BDD patients treated with augmentation trials of clomipramine (4/9), buspirone (12/36), lithium (1/5), methylphenidate (1/6), and antipsychotics (2/13).
Very few studies have examined antipsychotic use in BDD. Placebo-controlled data are available only for pimozide.27 Conventional antipsychotics are unlikely to be effective, either as monotherapy26 or augmentation.27 As for the atypicals, olanzapine augmentation showed little to no efficacy in one small trial, although the average dosage used was low (4.6 mg/d).28 In our experience, atypicals—such as aripiprazole, 5 to 30 mg/d; quetiapine 100 to 300 mg/d; olanzapine, 7.5 to 15 mg/d; or risperidone, 1 to 3 mg/d—can improve BDD core symptoms and improve insight.
Benzodiazepines can be useful for acute anxiety or agitation. Carefully monitor benzodiazepine use, however, as substance abuse is relatively common in BDD patients.29
Table 4
Recommended SRI dosages for treating BDD*†
| Drug | Dosage range (mg/d) |
|---|---|
| Citalopram | 40 to 100 |
| Clomipramine | 150 to 250 |
| Escitalopram | 20 to 50 |
| Fluoxetine | 40 to 100 |
| Fluvoxamine | 200 to 400 |
| Paroxetine | 40 to 100 |
| Sertraline | 150 to 400 |
| * Off-label use. | |
| † May exceed FDA-recommended maximum dosages. | |
Specialized cbt techniques
Cognitive restructuring. Trying to convince BDD patients there is nothing wrong with their appearance will not be successful. Instead, we use cognitive restructuring to challenge the rationality of their thoughts and beliefs and to find alternate, more rational ones:
Therapist: “I know I cannot convince you that your (body area) is not defective, but can you give me evidence of how this ‘defect’ has affected your life?”
BDD patient: “Well, I haven’t had a date for a long time. I think this is evidence that my (body part) must be ugly, and that no one wants to date me because of it.”
Therapist: “What are some other possible reasons why you haven’t had a date in a long time? You admitted that you have barely left your house for many months. Is it possible that you have not had a date for a long time because you rarely go outside?”
With cognitive restructuring, patients learn to:
- identify automatic thoughts and beliefs that provoke distress
- examine evidence supporting or refuting these beliefs
- de-catastrophize (such as “What is the worst thing that could happen if you left the house today without checking your [body part]? Do you think you would eventually be able to cope with that?”)
- learn to more accurately assess the probability of feared negative consequences
- arrive at rational responses.
In our experience—which is supported by OCD literature—participating in CBT is very hard for patients with frank delusions, and insight determines how effective cognitive restructuring can be.30 If a patient is convinced a body part is defective, she is unlikely to stay in treatment—much less be open to restructuring her thoughts. Even unsuccessful attempts can help you gauge the intensity of patients’ beliefs, however.
During cognitive restructuring, it is important to uncover patients’ core beliefs (underlying, organizing principles they hold about themselves, others, and the world). BDD patients commonly believe that appearance is of utmost importance and that no one could love them because of their “defect.” The therapist can then help the patient challenge the rationality of those core beliefs.
Behavioral therapy. Basic behavioral therapy attempts to normalize excessive response to appearance concerns and to prepare patients for exposure and response prevention therapy (ERP). Having identified their compulsions, the next step is to guide patients in changing these behaviors, such as by:
- decreasing reassurance-seeking
- reducing avoidance of social situations
- decreasing opportunities to use the mirror
- reducing time spent on the Internet seeking cosmetic solutions
- increasing eye contact in social situations
- decreasing scanning of others’ physical features.
For example, suggest that BDD patients stand at least an arm’s length away when using a mirror for normal grooming. Then, instead of focusing on their body part, they will view it within the context of their entire face and body.
Exposure and response prevention
ERP exposes the patient to situations that evoke negative emotions—primarily shame and anxiety in BDD—so that they gradually habituate to these feelings. Individualize exposure exercises, targeting the body parts each person believes are defective. Because these exercises are intended to induce the discomfort patients usually experience, do not attempt ERP until the patient has had extensive education, developed insight, and has consented to treatment.
Create a hierarchy of ERP tasks (Table 5), ranking situations from low- to high-distress. Address items lower on the hierarchy first, and progress to higher items as the lower ones become easier to perform. Do not attempt the highest-distress items until the patient has improved insight and is not severely ill and suicidal.
During exposures, patients must remain in distress-provoking situations—without performing compulsive behaviors—until their negative feelings decrease by at least 50% of the initial subjective, self-rated distress level. Leaving the situation before stress diminishes may reinforce shame and discomfort. Performing compulsive behaviors during or after an exposure will negate the exposure’s effect.
Mirrors and ERP. Some therapists use mirrors for exposure exercises, but this is a complex issue. Mirror-checking is a common BDD compulsion that provides temporary relief but ultimately reinforces negative, intrusive thoughts about the disliked body area. How BDD patients perceive themselves changes from moment to moment; they may stare at and analyze any reflective surface in hopes that their “defect” will not appear as deformed or ugly that day. Thus, one cannot predict whether looking in the mirror at any one time is an exposure or a compulsion.
ERP exercises for BDD need to emphasize behaviors that involve interactions with the outside world, rather than reinforcing the importance of appearance. Using the mirror for ERP could promote checking compulsions and may send the message that appearance is the focal point of treatment. On the other hand, for patients with persistent mirror avoidance, gradual mirror exposures may be useful. A technique called mirror retraining helps patients objectively view their appearance and has been used with success in some individuals.
Table 5
Exposure and response therapy: a BDD patient’s sample hierarchy
| High-distress tasks | Subjective distress rating (scale of 0 to 100) |
| 1. Purposely creating the appearance of acne/skin defects | 100 |
| 2. Intentionally messing up my hair before going in public | 100 |
| 3. Standing under bright or fluorescent lighting in public | 90 |
| 4. Sitting in a position where others can directly see my face for an extended period | 85 |
| 5. Highlighting my face with a flashlight or bright light, while sitting in front of my therapist or another person. | 80 |
| Lower-distress tasks | |
| 6. Intentionally going outside in daylight hours, instead of only after dark | 70 |
| 7. Not turning away from others in an attempt to prevent them from seeing my face | 65 |
| 8. Standing close to people when talking to them, rather than standing at a distance | 50 |
| 9. Going out in public without camouflaging my hair with hats or scarves | 40 |
Psychosocial development
BDD therapy challenges the disorder’s core theme—that appearance is one’s only important attribute—and helps patients identify and develop qualities not related to appearance. Through social interactions, the BDD patient can:
- develop a multidimensional sense of self
- receive nonappearance-related positive feedback from the outside world.
Explore psychosocial development during the assessment phase and when a patient shows little progress in CBT. In some patients, for example, BDD onset in childhood or adolescence interferes with developmental transition to adulthood.
In our experience, some patients may resist treatment because of conscious and unconscious fears of adult responsibilities and relationships. We focus therapy on making them aware of these phenomena, exploring fears of development, and encouraging them to seek new relationships and responsibilities.
Because a BDD patient’s symptoms often create conflict and distress at home, offer the family support and education about the disorder. Occasionally, forces within the family seem to be working against the individual’s recovery and/or independence.
In some families, an individual with BDD may become the “identified patient,” diverting attention from other dysfunctional family members or relationships. During therapy, the BDD patient’s goal to develop a sense of self that is not appearance-based may run counter to the family’s need to keep him or her in the “sick” role.
If therapy is to succeed, talk to the patient about these dynamics. Consider family therapy if resistance to change is strong. When a patient is not progressing well with CBT, we find understanding the family system can be useful to comprehensive BDD treatment, although this observation remains to be validated.
Preventing and treating relapse
Educate patients that BDD is usually chronic, even when treated with psychotherapy and medication.31 Relapse often occurs, especially when patients discontinue medications on their own24 or drop out of therapy early. No guidelines exist, but based on our experience:
- we continue medication for at least 1 year after a patient improves
- psychotherapy is more variable but may need to last 6 to 12 months or more.
When therapy ends, we encourage patients to practice and reinforce everything they learned during treatment. Casting BDD resurgence as normal—and not as failure—will help patients who relapse to resist the downward spiral of low self-esteem, shame, and turning to the mirror for reassurance. Identifying BDD symptom triggers and developing plans to cope with them may also prevent relapse. CBT “booster sessions” scheduled monthly for 3 to 6 months may help patients who have completed therapy.
FOR CLINICIANS:
- Phillips KA. “I’m as ugly as the elephant man:” How to recognize and treat body dysmorphic disorder. Current Psychiatry. 2002;1(1):58-65.
- Cororve MB, Gleaves DH. Body dysmorphic disorder: a review of conceptualizations, assessment, and treatment strategies. Clin Psychol Rev. 2001;21(6):949-70.
FOR PATIENTS AND FAMILIES:
- Phillips KA. The broken mirror. New York: Oxford University Press; 2005.
- BDD and body image program, Butler Hospital, Providence, RI. BDD education and support. www.BDDcentral.com.
- Winograd A. Director, Accurate Reflections, Los Angeles, CA. Support group and information on BDD and obsessive compulsive spectrum disorders. www.AccurateReflections.com
Drug brand names
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Buspirone • BuSpar
- Citalopram • Celexa
- Clomipramine • Anafranil
- Desipramine • Norpramin
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Lithium • Lithobid, others
- Methylphenidate • Ritalin, Concerta
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders (4th ed. text rev.). Washington, DC: American Psychiatric Association; 2000.
2. Phillips KA, Coles ME, Menard W, et al. Suicidal ideation and suicide attempts in body dysmorphic disorder. J Clin Psychiatry 2005;66(6):717-25.
3. Otto MW, Wilhelm S, Cohen LS, Harlow BL. Prevalence of body dysmorphic disorder in a community sample of women. Am J Psychiatry 2001;158(12):2061-3.
4. Wilhelm S, Otto MW, Zucker BG, Pollack MH. Prevalence of body dysmorphic disorder in patients with anxiety disorders. J Anxiety Disord 1997;11(5):499-502.
5. Phillips KA, Nierenberg AA, Brendel G, Fava M. Prevalence and clinical features of body dysmorphic disorder in atypical major depression. J Nerv Ment Dis 1996;184(2):125-9.
6. Hollander E, Cohen L, Simeon D. Body dysmorphic disorder. Psychiatr Ann 1993;23:359-64.
7. Veale D, Boocock A, Gournay K, et al. Body dysmorphic disorder. survey of fifty cases. Br J Psychiatry 1996;169(2):196-201.
8. Phillips KA. Psychosis in body dysmorphic disorder. J Psychiatr Res 2004;38(1):63-72.
9. Dufresne RG, Phillips KA, Vittorio CC, Wilkel CS. A screening questionnaire for body dysmorphic disorder in a cosmetic dermatologic surgery practice. Dermatol Surg 2001;27(5):457-62.
10. Rosen JC, Reiter J. Development of the body dysmorphic disorder examination. Behav Res Ther 1996;34(9):755-66.
11. Phillips KA, Hollander E, Rasmussen SA, et al. A severity rating scale for body dysmorphic disorder: development, reliability, and validity of a modified version of the Yale-Brown Obsessive Compulsive Scale. Psychopharmacol Bull 1997;33(1):17-22.
12. Gunstad J, Phillips KA. Axis I comorbidity in body dysmorphic disorder. Compr Psychiatry 2003;44(4):270-6.
13. Perugi G, Akiskal HS, Giannotti D, et al. Gender-related differences in body dysmorphic disorder (dysmorphophobia). J Nerv Ment Dis 1997;185(9):578-82.
14. Zimmerman M, Mattia JI. Body dysmorphic disorder in psychiatric outpatients: recognition, prevalence, comorbidity, demographic, and clinical correlates. Compr Psychiatry 1998;39(5):265-70.
15. Phillips KA. The broken mirror. New York: Oxford University Press; 2005.
16. Rauch SL, Phillips KA, Segal E, et al. A preliminary morphometric magnetic resonance imaging study of regional brain volumes in body dysmorphic disorder. Psychiatry Res 2003;122(1):13-19.
17. Veale D. Body dysmorphic disorder. Postgrad Med J 2004;80(940):67-71.
18. Saxena S, Winograd A, Dunkin JJ, et al. A retrospective review of clinical characteristics and treatment response in body dysmorphic disorder versus obsessive-compulsive disorder. J Clin Psychiatry 2001;62:67-72.
19. Phillips KA, Najjar F. An open-label study of citalopram in body dysmorphic disorder. J Clin Psychiatry 2003;64(6):715-20.
20. Phillips KA, Dwight MM, McElroy SL. Efficacy and safety of fluvoxamine in body dysmorphic disorder. J Clin Psychiatry 1998;59(4):165-71.
21. Phillips KA, Albertini RS, Rasmussen SA. A randomized placebo-controlled trial of fluoxetine in body dysmorphic disorder. Arch Gen Psychiatry 2002;59(4):381-8.
22. Hollander E, Allen A, Kwon J, et al. Clomipramine vs desipramine crossover trial in body dysmorphic disorder: Selective efficacy of a serotonin reuptake inhibitor in imagined ugliness. Arch Gen Psychiatry 1999;56(11):1033-9.
23. Phillips KA, McElroy SL, Dwight MM, et al. Delusionality and response to open-label fluvoxamine in body dysmorphic disorder. J Clin Psychiatry 2001;62(2):87-91.
24. Phillips KA, Albertini RS, Siniscalchi JM, et al. Effectiveness of pharmacotherapy for body dysmorphic disorder: a chart-review study. J Clin Psychiatry 2001;62(9):721-7.
25. Phillips KA. An open study of buspirone augmentation of serotonin-reuptake inhibitors in body dysmorphic disorder. Psychopharmacol Bull 1996;32(1):175-80.
26. Phillips KA, McElroy SL, Keck PE, Jr, et al. A comparison of delusional and nondelusional body dysmorphic disorder in 100 cases. Psychopharmacol Bull 1994;30(2):179-86.
27. Phillips KA. Placebo-controlled study of pimozide augmentation of fluoxetine in body dysmorphic disorder. Am J Psychiatry 2005;162(2):377-9.
28. Phillips KA. Olanzapine augmentation of fluoxetine in body dysmorphic disorder. Am J Psychiatry 2005;162(5):1022-3.
29. Grant JE, Menard W, Pagano ME, et al. Substance use disorders in individuals with body dysmorphic disorder. J Clin Psychiatry 2005;66(3):309-16.
30. Foa EB. Failures in treating obsessive-compulsives. Behav Res Ther 1979;17:169-76.
31. Phillips KA, McElroy SL, Keck PE, Jr, et al. Body dysmorphic disorder: 30 cases of imagined ugliness. Am J Psychiatry 1993;150(2):302-8.
1. Diagnostic and statistical manual of mental disorders (4th ed. text rev.). Washington, DC: American Psychiatric Association; 2000.
2. Phillips KA, Coles ME, Menard W, et al. Suicidal ideation and suicide attempts in body dysmorphic disorder. J Clin Psychiatry 2005;66(6):717-25.
3. Otto MW, Wilhelm S, Cohen LS, Harlow BL. Prevalence of body dysmorphic disorder in a community sample of women. Am J Psychiatry 2001;158(12):2061-3.
4. Wilhelm S, Otto MW, Zucker BG, Pollack MH. Prevalence of body dysmorphic disorder in patients with anxiety disorders. J Anxiety Disord 1997;11(5):499-502.
5. Phillips KA, Nierenberg AA, Brendel G, Fava M. Prevalence and clinical features of body dysmorphic disorder in atypical major depression. J Nerv Ment Dis 1996;184(2):125-9.
6. Hollander E, Cohen L, Simeon D. Body dysmorphic disorder. Psychiatr Ann 1993;23:359-64.
7. Veale D, Boocock A, Gournay K, et al. Body dysmorphic disorder. survey of fifty cases. Br J Psychiatry 1996;169(2):196-201.
8. Phillips KA. Psychosis in body dysmorphic disorder. J Psychiatr Res 2004;38(1):63-72.
9. Dufresne RG, Phillips KA, Vittorio CC, Wilkel CS. A screening questionnaire for body dysmorphic disorder in a cosmetic dermatologic surgery practice. Dermatol Surg 2001;27(5):457-62.
10. Rosen JC, Reiter J. Development of the body dysmorphic disorder examination. Behav Res Ther 1996;34(9):755-66.
11. Phillips KA, Hollander E, Rasmussen SA, et al. A severity rating scale for body dysmorphic disorder: development, reliability, and validity of a modified version of the Yale-Brown Obsessive Compulsive Scale. Psychopharmacol Bull 1997;33(1):17-22.
12. Gunstad J, Phillips KA. Axis I comorbidity in body dysmorphic disorder. Compr Psychiatry 2003;44(4):270-6.
13. Perugi G, Akiskal HS, Giannotti D, et al. Gender-related differences in body dysmorphic disorder (dysmorphophobia). J Nerv Ment Dis 1997;185(9):578-82.
14. Zimmerman M, Mattia JI. Body dysmorphic disorder in psychiatric outpatients: recognition, prevalence, comorbidity, demographic, and clinical correlates. Compr Psychiatry 1998;39(5):265-70.
15. Phillips KA. The broken mirror. New York: Oxford University Press; 2005.
16. Rauch SL, Phillips KA, Segal E, et al. A preliminary morphometric magnetic resonance imaging study of regional brain volumes in body dysmorphic disorder. Psychiatry Res 2003;122(1):13-19.
17. Veale D. Body dysmorphic disorder. Postgrad Med J 2004;80(940):67-71.
18. Saxena S, Winograd A, Dunkin JJ, et al. A retrospective review of clinical characteristics and treatment response in body dysmorphic disorder versus obsessive-compulsive disorder. J Clin Psychiatry 2001;62:67-72.
19. Phillips KA, Najjar F. An open-label study of citalopram in body dysmorphic disorder. J Clin Psychiatry 2003;64(6):715-20.
20. Phillips KA, Dwight MM, McElroy SL. Efficacy and safety of fluvoxamine in body dysmorphic disorder. J Clin Psychiatry 1998;59(4):165-71.
21. Phillips KA, Albertini RS, Rasmussen SA. A randomized placebo-controlled trial of fluoxetine in body dysmorphic disorder. Arch Gen Psychiatry 2002;59(4):381-8.
22. Hollander E, Allen A, Kwon J, et al. Clomipramine vs desipramine crossover trial in body dysmorphic disorder: Selective efficacy of a serotonin reuptake inhibitor in imagined ugliness. Arch Gen Psychiatry 1999;56(11):1033-9.
23. Phillips KA, McElroy SL, Dwight MM, et al. Delusionality and response to open-label fluvoxamine in body dysmorphic disorder. J Clin Psychiatry 2001;62(2):87-91.
24. Phillips KA, Albertini RS, Siniscalchi JM, et al. Effectiveness of pharmacotherapy for body dysmorphic disorder: a chart-review study. J Clin Psychiatry 2001;62(9):721-7.
25. Phillips KA. An open study of buspirone augmentation of serotonin-reuptake inhibitors in body dysmorphic disorder. Psychopharmacol Bull 1996;32(1):175-80.
26. Phillips KA, McElroy SL, Keck PE, Jr, et al. A comparison of delusional and nondelusional body dysmorphic disorder in 100 cases. Psychopharmacol Bull 1994;30(2):179-86.
27. Phillips KA. Placebo-controlled study of pimozide augmentation of fluoxetine in body dysmorphic disorder. Am J Psychiatry 2005;162(2):377-9.
28. Phillips KA. Olanzapine augmentation of fluoxetine in body dysmorphic disorder. Am J Psychiatry 2005;162(5):1022-3.
29. Grant JE, Menard W, Pagano ME, et al. Substance use disorders in individuals with body dysmorphic disorder. J Clin Psychiatry 2005;66(3):309-16.
30. Foa EB. Failures in treating obsessive-compulsives. Behav Res Ther 1979;17:169-76.
31. Phillips KA, McElroy SL, Keck PE, Jr, et al. Body dysmorphic disorder: 30 cases of imagined ugliness. Am J Psychiatry 1993;150(2):302-8.
How to reduce mania risk when prescribing stimulants
Stimulants are most effective for childhood attention-deficit/hyperactivity disorder (ADHD),1 but they may induce mania or trigger a treatment-resistant course in children with comorbid bipolar disorder. To help you safely manage these complicated symptoms, this article offers a treatment algorithm and tips to:
- differentiate bipolar and ADHD symptoms
- identify patients at risk for stimulantinduced mania
- choose medications by a hierarchythat may reduce the risk of mood destabilization.
Bipolar mood symptoms emerge before age 20 in about 25% of persons with bipolar disorder (BP).3 Early-onset BP may be more severe than the adult-onset form, with more-affected family members and greater comorbidity with other disorders, especially ADHD.4
In one study, 91% of children with BP also met criteria for ADHD, and 19% of patients with ADHD also received a diagnosis of BP.5 Among 31 children ages 2 to 5 with BP, 80% met criteria for concurrent ADHD.6
Of 40 children age <5 presenting consecutively to a mental health clinic, 11 (28%) met criteria for mania, which was usually associated with euphoria.7 These 11 children also met criteria for ADHD.
A comparison study8 of children (mean age 12) found greater impairment, suicidality, irritability, and sadness in 43 with ADHD plus bipolar depression than in:
- 109 with ADHD plus major depressive disorder
- 128 without depression or mania.
Family prevalence of bipolar disorder and major depression was highest in the bipolar-ADHD group, which also had the highest rates of comorbid conduct disorder, oppositional defiant disorder, alcohol abuse, and agoraphobia. Average age of bipolar diagnosis was 6.3 years.
Adhd and/or bipolar disorder?
Some 70% to 90% of bipolar children and at least 30% to 40% of bipolar adolescents also have ADHD.2 This high comorbidity (Box 1)3-8 might mean that:
- one disorder predisposes to the other
- one is a precursor of the other
- they share common vulnerabilities or causes
- their symptoms overlap so much that patients with one disorder appear to meet criteria for the other.
Some experts contend that bipolar disorder and ADHD usually can be differentiated. Bipolar children score higher than those with ADHD on measures of anxiety/depression, aggression, and attention problems on the Child Behavior Checklist.9 Others believe ADHD symptoms that occur with bipolar disorder are a dimension of bipolar illness rather than a separate disorder.10
For every DSM-IV-TR diagnostic criterion for ADHD, a corresponding diagnostic criterion or common feature of bipolar disorder can be identified (Table 1). Mania and hypomania are obviously associated with hyperactivity and impulsivity, and tangential thinking and distractibility interfere with attention in many patients with bipolar disorder.
Though most ADHD symptoms can occur in bipolar patients, some features of bipolar illness are not characteristic of ADHD (Table 2). Children with ADHD can become hyper-focused on video games and television, for example, but they usually do not become engrossed in long, complicated books or preoccupied with other people, as can occur in bipolar disorder.
Table 1
How ADHD, bipolar symptoms overlap in three domains
| ADHD | Bipolar disorder |
|---|---|
| Inattention | |
| Fails to pay attention | Racing and tangential thoughts |
| Difficulty sustaining attention | Attention driven by racing thoughts, affective themes, and psychosis |
| Does not follow through | Direction of activity shifts with shifting mood |
| Difficulty organizing tasks | Disorganization, psychosis, excessive energy |
| Easily distracted | Distractibility |
| Hyperactivity | |
| Fidgets or squirms | Increased energy and activity |
| Runs about or climbs excessively | Hyperactivity, thrill-seeking |
| Difficulty engaging quietly in leisure activities | Increased energy, boredom |
| Often on the go | Increased energy, hyperactivity |
| Talks excessively | Rapid, pressured speech |
| Impulsivity | |
| Blurts out answers | Rapid, pressured, impulsive speech |
| Difficulty awaiting turn | Hyperactivity, increased energy, impatience, grandiosity |
| Interrupts or intrudes on others | Grandiosity, impatience, pressured speech, increased mental content |
Table 2
Bipolar features not seen in ADHD
|
A treatment hierarchy
Whether a bipolar patient’s attention problems are features of the primary condition or caused by comorbid ADHD may be unclear, but the treatment implications are important. All antidepressants can induce mania/hypomania and increase the risk of mixed states and mood cycling. Because stimulants have antidepressant properties and because some antidepressants are used to treat ADHD, a systematic approach is necessary when treating inattention in juvenile bipolar disorder.
A treatment hierarchy developed by the American Academy of Child and Adolescent Psychiatry Workgroup on Bipolar Disorder recommends beginning psychosocial approaches, such as training parents in behavior management techniques, and:
- treating bipolar disorder first in children who clearly have both ADHD and bipolar disorder
- adding ADHD treatment if ADHD symptoms persist and impair functioning.2
Who’s at risk for mood destabilization?
No data address differences between bipolar patients whose mood disorders deteriorate with stimulant use and those who remain stable. However, risk factors for mood destabilization that have been reported with antidepressants likely also apply to stimulants (Table 3) because stimulants’ adverse effects in bipolar disorder are probably related to their antidepressant properties.
For example, depressed patients who report that an antidepressant worked within hours to days may have bipolar disorder and be at risk for mood destabilization leading to treatment resistance.11 Antidepressant-induced mania also may be more likely:
- when depression is mixed with hypomanic symptoms such as racing thoughts, excessive talkativeness, aggression, irritability, distractibility, and increased drive12
- in patients with a history of antidepressant-induced mania, family history of bipolar disorder, or multiple antidepressant trials.13
Similarly, patients who report feeling better immediately after starting a stimulant—especially if they have evidence of elation, increased irritability, more aggression or impulsivity, decreased sleep, or related symptoms—may be developing stimulant-induced hypomania.
Table 3
Risk factors that may increase risk of stimulant-induced mania
|
| Source: Reference 25 |
Antidepressant-induced mania
Most studies of antidepressant-induced mania have examined outright mania, but hypomania and subsyndromal hypomanic syndromes also may cause significant morbidity and may worsen bipolar disorder’s course. A change in polarity may worsen a patient’s prognosis, but how do we know that antidepressants (or stimulants) caused it?
One suggested criterion is that mania or hypomania develops within 8 weeks of starting an antidepressant for the first time. A chart review of 51 bipolar patients who had extensive life charting found that 82% developed mania while taking an antidepressant—35% of them within 8 weeks.14 The authors attributed 50% of the risk of a first manic episode and/or cycle acceleration to antidepressants and 50% to spontaneous mood swings. They also noted that:
- an initial manic episode appeared to sensitize patients to subsequent manic episodes and rapid cycling
- mood stabilizers did not seem to prevent these outcomes.
A meta-analysis of 12 randomized, controlled, 4-to 12-week trials among 1,088 patients found antidepressants no more likely than placebo to induce mania in the short term.15 These trials did not, however, consider less-severe forms of overstimulation and were not designed to determine mania risk in bipolar depressed patients.
Post-mania cycling. Rapid and ultradian cycling and other forms of deterioration are more likely to occur after a manic or hypomanic episode than after a depressive episode.16
A longitudinal study17 indicated that antidepressant use did not predictably predate rapid cycling when depression was controlled. The authors, however, looked at the correlation between taking an antidepressant at study entry and rapid cycling over 1 year but did not examine whether antidepressants were started or stopped during the study.18 Rapid cycling prevalence declined from 19% to 5% during the study, but they did not determine whether withdrawing antidepressants was associated with this change.
In an earlier prospective study, rapid cycling was more severe while patients were taking antidepressants—despite the use of mood stabilizers—and cycling duration decreased when antidepressants were withdrawn.19
TCAs vs. newer agents. Tricyclic antidepressants (TCAs) are perceived as more likely to induce mania than are selective serotonin reuptake inhibitors (SSRIs) or bupropion. Comparing TCAs’ and newer antidepressants’ switch rates is difficult, however. Most antidepressant trials were designed to show efficacy and safety in unipolar, not bipolar, depression. Moreover, as exclusion criteria have improved with greater awareness of bipolar illness’ polymorphic manifestations, recent studies likely have enrolled fewer bipolar patients—who are most at risk to develop a manic switch—than did earlier TCA trials.
Bupropion, which has been used to treat ADHD, has been thought to have a low risk of inducing mania. In open observation, however, >50% of 11 patients with a history of developing mania with other antidepressants also had a manic switch on bupropion, even though they were taking mood stabilizers.20
Analysis of 155 antidepressant trials in 41 depressed patients found mania risk to be similar with bupropion, SSRIs, TCAs, monoamine oxidase inhibitors (MAOIs), and other newer antidepressants.21 Mania risk doubled when patients were not also taking mood stabilizers.
Going without mood stabilizers. Reports have emerged of patients with bipolar depression taking antidepressants such as fluoxetine and venlafaxine without a mood stabilizer for extended periods, without high rates of mania or mood cycling.22-24 These reports suggest that some bipolar depressed patients can tolerate antidepressants without a mood stabilizer, although we have no way to identify such patients in advance.
Cycle acceleration and treatment resistance may follow antidepressant-induced mania.25 In DSM-IV field trials, antidepressants appeared to have triggered rapid cycling in some 20% of bipolar patients.26 Mood stabilizers were not particularly effective in patients with treatment-resistant ultradian cycling, but withdrawing antidepressants improved outcome.27
Stimulant-induced mania
Compared with antidepressants, less information is available about stimulant-induced mania and rapid cycling.
Some carefully selected bipolar patients may tolerate ongoing stimulant treatment. For example, in 2 years of open experience with 5 bipolar type I and 3 bipolar type II adults, adding methylphenidate or amphetamine for residual depression or sedation was moderately helpful and did not lead to manic switching or drug misuse.28
On the other hand, affective symptoms worsened in nearly two-thirds of 31 children ages 2 to 5 when treated with stimulants or antidepressants without mood stabilizers. Most of the children also had ADHD, and valproate usually helped.6
In 40 patients, mean age 10, who entered the open-label phase of an 8-week trial of divalproex for manic and ADHD symptoms:
- Young Mania Rating Scale (YMRS) scores declined by≥50% in 32 (80%) by week 8, a greater initial response than usually reported in pediatric bipolar disorder with comorbid ADHD.
- ADHD symptoms, measured by Clinical Global Impressions (CGI) scores, did not change significantly.29
Thirty divalproex responders then received mixed amphetamine salts, 10 mg/d, or placebo plus divalproex, crossing over to the other treatment in a 4-week, double-blind trial. ADHD symptoms improved twice as much with the stimulant as with placebo, as measured by CGI scores, whereas YMRS scores did not differ significantly. Among 23 patients who continued the stimulant and divalproex for 12 more weeks, 45% required an increase in stimulant dosage and 1 relapsed into mania.
In this study, ADHD symptoms did not respond to mania treatment but did improve when a stimulant was added. This suggests either that patients had two disorders or that not all bipolar features remit at the same time. The trial’s low stimulant dosage and short duration provide insufficient evidence to support using stimulants over long periods in bipolar children.
LOng-term stimulant effects
Without long-term observations, some investigators have inferred stimulants’ impact on bipolar disorder. A poll of pediatric psychiatrists in the Netherlands, for example, found bipolar disorder in 39 children ages <13 (0.001%) in the previous year, compared with a prevalence of at least 1% in the United States.3 The authors concluded:
- Bipolar disorder emerges at younger ages in the United States than in the Netherlands.
- One reason may be that U.S. psychiatrists have a lower threshold for treating pediatric depression and hyperactivity with antidepressants and stimulants than Dutch psychiatrists do, evoking more-obvious bipolar symptoms at an earlier age.
Observations of 30 U.S. children with a manic episode and ADHD suggested that stimulants can induce manic symptoms:
- Mean age of ADHD onset was 5.5 years.
- Mean age of starting stimulants was 6.9 years.
- Mean age of hypomanic or manic symptom onset was 7.1 years.30
Similarly, in a survey of 34 adolescent manic inpatients, those who had taken stimulants had earlier mania onset (mean age 10.7) than did those who had not taken stimulants (mean age 13.9). Exposure to two stimulants was associated with earlier onset than exposure to one, but comorbid ADHD alone did not affect age of bipolar disorder onset.31
The same group10 reviewed charts of 80 consecutively hospitalized adolescents with a manic or mixed bipolar episode and found stimulant exposure was associated with relatively worse inpatient course, longer length of stay, more emergency medications, and more seclusion and restraint orders. Comorbid ADHD, mixed versus manic episode, and prior antidepressant exposure did not worsen the inpatient course.
A chart review by El-Mallakh et al32 found bipolar disorder was diagnosed at mean age 10.7 in 49 children exposed to antidepressants or stimulants, compared with mean age 12.7 in 44 unexposed children. The exposed group appeared to have tolerated stimulants longer than antidepressants before mania or hypomania emerged.33
In contrast, a retrospective review by Carlson et al34 of data from a longitudinal study of 75 boys with “hyperkinetic reaction of childhood” found that methylphenidate treatment did not appear more common in boys later diagnosed with bipolar disorder than in those without a bipolar diagnosis. This study had obvious methodologic limitations, lacking a hypothesis and focusing on a population with “minimal brain dysfunction.”
In a reanalysis of data from a 1-month methylphenidate titration trial, Galanter et al35 examined whether some 300 children ages 5 to 12 experienced manic symptoms, using the Diagnostic Interview Schedule for Children or the Child Behavior Checklist. At least during this brief trial, patients with and without manic symptoms showed no differences in response rates or adverse effects with stimulant therapy.
Drug treatment hierarchy
Mood stabilizers. Evidence supports starting all bipolar children with a mood stabilizer such as lithium or valproate (Algorithm). A few patients may tolerate stimulants without mood stabilizers, but the risk is high of inducing mania and precipitating a more complex and treatment-resistant disorder.
Carbamazepine can be effective, but it makes some youths aggressive or disorganized. Antipsychotics have not been tested in controlled trials in bipolar children and are not considered first-line treatments, especially as mood stabilizers. They can be effective for childhood mania, but outpatients needing ADHD treatment usually do not have severe manic syndromes.
Algorithm Reducing mania risk: Using stimulants in children with bipolar disorder
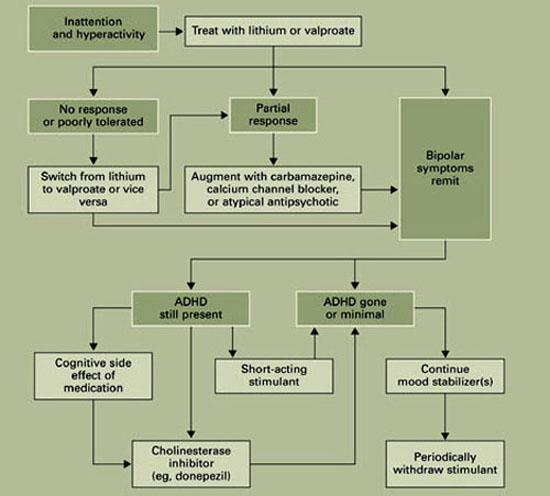
Combination therapy. Like many adults, bipolar children often require combinations of mood stabilizers. Kowatch et al36 found that 16 of 20 acutely ill bipolar children (mean age 11) responded to a combination of mood stabilizers after not responding to 8 weeks of a single mood stabilizer. Because bipolar disorder with comorbid ADHD suggests a complex pathophysiology, patients with both disorders may be more likely to require mood-stabilizer combinations than those with bipolar disorder alone.
The goal in treating bipolar disorder is to eliminate symptoms as completely as possible. In bipolar children with comorbid ADHD, be certain that subtle hypomanic symptoms—irritability, decreased sleep, hypersensitivity to interactions, psychosis—have remitted, as they could account for continued inattention. Persistent mood lability may indicate incomplete treatment of the mood disorder, which can increase sensitivity to destabilization by a stimulant.
If a child remains inattentive after the mood disorder is controlled, consider whether medications for the mood disorder are to blame. If medications are working well but causing cognitive side effects, a cholinesterase inhibitor may help.
Adding stimulants. If attention problems persist, consider cautiously adding a stimulant. Informed consent includes telling patients and families about the risks of mood destabilization with stimulants, even when used with mood stabilizers.
Increase stimulant dosage very slowly, and monitor the patient closely for emerging mood instability or subtle evidence of dysphoric hypomania. Address hypersensitivity to sounds, increased irritability, or other signs of activation with more-aggressive mood stabilization before assuming that these are ADHD symptoms that require a higher stimulant dosage.
Sustained-release stimulant preparations are probably second-line choices in patients with concomitant bipolar disorder. With long-acting stimulants, any worsening of the mood disorder will take longer to wear off. Antidepressants such as bupropion are potential alternatives to stimulants but are as likely to induce hypomania and mood cycling and may not be as effective.
Compared with stimulants, atomoxetine has a less-potent antidepressant effect and may be somewhat safer, but it is not as effective for ADHD and is longer-acting. Thus, atomoxetine could be a first-line alternative for comorbid ADHD, with stimulants being added if it is not effective. Clonidine can reduce hyperactivity but does not stabilize mood or improve attention.
When an antidepressant has brought bipolar depression into remission, discontinue it slowly to reduce the risk of rebound while continuing mood stabilizers to prevent recurrence. Because ADHD is not cyclical like bipolar depression, inattention returns for many patients when stimulants are withdrawn.
We do not yet know whether the risk of mood destabilization increases with long-term stimulant use, but discontinuation-induced refractoriness has not been reported with stimulants as it has with mood stabilizers and antidepressants. Thus, trying to withdraw stimulants once ADHD symptoms have remitted is prudent, while supplementing the regimen with behavioral treatments. If managing ADHD symptoms requires continued stimulant treatment, monitor the patient closely for mood destabilization.
Related resources
- American Academy of Child and Adolescent Psychiatry. Facts for families: Bipolar disorder in children and teens.
www.aacap.org/publications/factsFam/bipolar.htm. - National Institute of Mental Health. Database on ADHD.
www.nimh.nih.gov/publicat/adhd.cfm.
Drug brand names
- Amphetamine salts • Adderall
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, others
- Clonidine • Catapres
- Dexmethylphenidate • Focalin
- Fluoxetine • Prozac
- Lithium • Lithobid, others
- Methylphenidate • Concerta,
- Ritalin, others
- Valproate • Depakene, Depakote
- Venlafaxine • Effexor
Disclosures
Dr. Dubovsky receives research support from UCB Pharma, Forest Laboratories, and Solvay Pharmaceuticals, and is a speaker for Janssen Pharmaceutica and Forest Laboratories.
1. Greenhill LL, Pliszka S, Dulcan MK, et al. AACAP. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41(suppl 2):26S-49S.
2. Kowatch RA, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder: child psychiatric workgroup on bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44:213-35.
3. Reichart CG, Nolen W. Earlier onset of bipolar disorder in children by antidepressants or stimulants? An hypothesis. J Affect Disord 2004;78:81-4.
4. Faraone SV, Glatt SJ, Tsuang MT. The genetics of pediatric-onset bipolar disorder. Biol Psychiatry 2003;53:970-7.
5. Geller B, Zimmerman B, Williams MB, et al. Bipolar disorder at prospective follow-up of adults who had prepubertal major depressive disorder. Am J Psychiatry 2001;158:125-7.
6. Scheffer RE, Niskala Apps JA. The diagnosis of preschool bipolar disorder presenting with mania: open pharmacological treatment. J Affect Disord. 2004;82(suppl 1):S25-S34.
7. Dilsaver SC, Akiskal HS. Preschool-onset mania: incidence, phenomenology and family history. J Affect Disord 2004;82(suppl 1):S35-S43.
8. Wozniak J, Spencer T, Biederman J, et al. The clinical characteristics of unipolar vs. bipolar major depression in ADHD youth. J Affect Disord 2004;82(suppl 1):S59-S69.
9. Mick E, Biederman J, Pandina G, Faraone SV. A preliminary meta-analysis of the Child Behavior Checklist in pediatric bipolar disorder. Biol Psychiatry 2003;53:1021-7.
10. Soutullo CA, DelBello MP, Ochsner JE, et al. Severity of bipolarity in hospitalized manic adolescents with history of stimulant or antidepressant treatment. J Affect Disord 2002;70:323-7.
11. Piver A. Ultrarapid response to an antidepressant: A clue to bipolarity? Can J Psychiatry 2003;48:427-8.
12. Bottlender R, Sato T, Kleindienst N, et al. Mixed depressive features predict maniform switch during treatment of depression in bipolar I disorder. J Affect Disord 2004;78:149-52.
13. Goldberg J, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord 2003;5:407-20.
14. Altshuler LL, Post RM, Leverich GS, et al. Antidepressant-induced mania and cycle acceleration: a controversy revisited. Am J Psychiatry 1995;152(8):1130-8.
15. Gijsman HJ, Geddes J, Rendell J, et al. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry 2004;161:1537-47.
16. Post RM, Roy-Byrne PP, Uhde TW. Graphic representation of the life course of illness in patients with affective disorder. Am J Psychiatry 1988;145:844-8.
17. Coryell W, Endicott J, Keller M. Rapidly cycling affective disorder: demographics, diagnosis, family history and course. Arch Gen Psychiatry 1992;49:126-31.
18. Wehr TA. Can antidepressants induce rapid cycling? Arch Gen Psychiatry 1993;50(6):495-6.
19. Wehr TA, Sack DA, Rosenthal NE, Cowdry RW. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
20. Fogelson DL, Bystritsky A, Pasnau R. Bupropion in the treatment of bipolar disorders: the same old story. J Clin Psychiatry 1992;53:443-6.
21. Goldberg J, Ernst CL. Features associated with the delayed initiation of mood stabilizers at illness onset in bipolar disorder. J Clin Psychiatry 2002;63:985-91.
22. Amsterdam JD, Shults J, Brunswick DJ, Hundert M. Short-term fluoxetine monotherapy for bipolar type II or bipolar NOS major depression—low manic switch rate. Bipolar Disord 2004;6:75-81.
23. Simpson SG, DePaulo JR. Fluoxetine treatment of bipolar II depression. J Clin Psychopharmacol 1991;11:52-4.
24. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
25. Goldberg J. When do antidepressants worsen the course of bipolar disorder? J Psychiatr Pract 2003;9:181-94.
26. Bauer M, Calabrese JR, Dunner DL. Multisite data reanalysis of the validity of rapid cycling as a course modifier for bipolar disorder in DSM-IV. Am J Psychiatry 1994;151:506-15.
27. Prien RF, Kupfer DJ, Mansky PA. Drug therapy in the prevention of recurrences in unipolar and bipolar affective disorders: Report of the NIMH Collaborative Study Group comparing lithium carbonate, imipramine, and a lithium carbonate-imipramine combination. Arch Gen Psychiatry 1984;41:1096-1104.
28. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: treatment of residual depression and sedation. Bipolar Disord 2004;6:416-20.
29. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162:58-64.
30. Kowatch RA, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamezepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39:713-20.
31. DelBello MP, Soutullo CA, Hendricks W, et al. Prior stimulant treatment in adolescents with bipolar disorder: association with age at onset. Bipolar Disord 2001;3:53-7.
32. El-Mallakh RS, Cicero D, Holman J, Robertson J. Antidepressant exposure in children diagnosed with bipolar disorder. Bipolar Disord 2001;3(suppl 1):35-9.
33. Cicero D, El-Mallakh RS, Holman J, Robertson J. Antidepressant exposure in bipolar children. Psychiatry 2003;66:317-22.
34. Carlson G, Loney J, Salisbury H, et al. Stimulant treatment in young boys with symptoms suggesting childhood mania: A report from a longitudinal study. J Child Adolesc Psychopharmacol 2000;10:175-84.
35. Galanter CA, Carlson GA, Jensen PS, et al. Response to methylphenidate in children with attention deficit hyperactivity disorder and manic symptoms in the multimodal treatment study of children with attention deficit hyperactivity disorder titration trial. J Child Adolesc Psychopharmacol 2003;13:123-36.
36. Kowatch RA, Sethuraman G, Hume JH, Kromelis M, Weinberg WA. Combination pharmacotherapy in children and adolescents with bipolar disorder. Biol Psychiatry. 2003;53:978-84.
Stimulants are most effective for childhood attention-deficit/hyperactivity disorder (ADHD),1 but they may induce mania or trigger a treatment-resistant course in children with comorbid bipolar disorder. To help you safely manage these complicated symptoms, this article offers a treatment algorithm and tips to:
- differentiate bipolar and ADHD symptoms
- identify patients at risk for stimulantinduced mania
- choose medications by a hierarchythat may reduce the risk of mood destabilization.
Bipolar mood symptoms emerge before age 20 in about 25% of persons with bipolar disorder (BP).3 Early-onset BP may be more severe than the adult-onset form, with more-affected family members and greater comorbidity with other disorders, especially ADHD.4
In one study, 91% of children with BP also met criteria for ADHD, and 19% of patients with ADHD also received a diagnosis of BP.5 Among 31 children ages 2 to 5 with BP, 80% met criteria for concurrent ADHD.6
Of 40 children age <5 presenting consecutively to a mental health clinic, 11 (28%) met criteria for mania, which was usually associated with euphoria.7 These 11 children also met criteria for ADHD.
A comparison study8 of children (mean age 12) found greater impairment, suicidality, irritability, and sadness in 43 with ADHD plus bipolar depression than in:
- 109 with ADHD plus major depressive disorder
- 128 without depression or mania.
Family prevalence of bipolar disorder and major depression was highest in the bipolar-ADHD group, which also had the highest rates of comorbid conduct disorder, oppositional defiant disorder, alcohol abuse, and agoraphobia. Average age of bipolar diagnosis was 6.3 years.
Adhd and/or bipolar disorder?
Some 70% to 90% of bipolar children and at least 30% to 40% of bipolar adolescents also have ADHD.2 This high comorbidity (Box 1)3-8 might mean that:
- one disorder predisposes to the other
- one is a precursor of the other
- they share common vulnerabilities or causes
- their symptoms overlap so much that patients with one disorder appear to meet criteria for the other.
Some experts contend that bipolar disorder and ADHD usually can be differentiated. Bipolar children score higher than those with ADHD on measures of anxiety/depression, aggression, and attention problems on the Child Behavior Checklist.9 Others believe ADHD symptoms that occur with bipolar disorder are a dimension of bipolar illness rather than a separate disorder.10
For every DSM-IV-TR diagnostic criterion for ADHD, a corresponding diagnostic criterion or common feature of bipolar disorder can be identified (Table 1). Mania and hypomania are obviously associated with hyperactivity and impulsivity, and tangential thinking and distractibility interfere with attention in many patients with bipolar disorder.
Though most ADHD symptoms can occur in bipolar patients, some features of bipolar illness are not characteristic of ADHD (Table 2). Children with ADHD can become hyper-focused on video games and television, for example, but they usually do not become engrossed in long, complicated books or preoccupied with other people, as can occur in bipolar disorder.
Table 1
How ADHD, bipolar symptoms overlap in three domains
| ADHD | Bipolar disorder |
|---|---|
| Inattention | |
| Fails to pay attention | Racing and tangential thoughts |
| Difficulty sustaining attention | Attention driven by racing thoughts, affective themes, and psychosis |
| Does not follow through | Direction of activity shifts with shifting mood |
| Difficulty organizing tasks | Disorganization, psychosis, excessive energy |
| Easily distracted | Distractibility |
| Hyperactivity | |
| Fidgets or squirms | Increased energy and activity |
| Runs about or climbs excessively | Hyperactivity, thrill-seeking |
| Difficulty engaging quietly in leisure activities | Increased energy, boredom |
| Often on the go | Increased energy, hyperactivity |
| Talks excessively | Rapid, pressured speech |
| Impulsivity | |
| Blurts out answers | Rapid, pressured, impulsive speech |
| Difficulty awaiting turn | Hyperactivity, increased energy, impatience, grandiosity |
| Interrupts or intrudes on others | Grandiosity, impatience, pressured speech, increased mental content |
Table 2
Bipolar features not seen in ADHD
|
A treatment hierarchy
Whether a bipolar patient’s attention problems are features of the primary condition or caused by comorbid ADHD may be unclear, but the treatment implications are important. All antidepressants can induce mania/hypomania and increase the risk of mixed states and mood cycling. Because stimulants have antidepressant properties and because some antidepressants are used to treat ADHD, a systematic approach is necessary when treating inattention in juvenile bipolar disorder.
A treatment hierarchy developed by the American Academy of Child and Adolescent Psychiatry Workgroup on Bipolar Disorder recommends beginning psychosocial approaches, such as training parents in behavior management techniques, and:
- treating bipolar disorder first in children who clearly have both ADHD and bipolar disorder
- adding ADHD treatment if ADHD symptoms persist and impair functioning.2
Who’s at risk for mood destabilization?
No data address differences between bipolar patients whose mood disorders deteriorate with stimulant use and those who remain stable. However, risk factors for mood destabilization that have been reported with antidepressants likely also apply to stimulants (Table 3) because stimulants’ adverse effects in bipolar disorder are probably related to their antidepressant properties.
For example, depressed patients who report that an antidepressant worked within hours to days may have bipolar disorder and be at risk for mood destabilization leading to treatment resistance.11 Antidepressant-induced mania also may be more likely:
- when depression is mixed with hypomanic symptoms such as racing thoughts, excessive talkativeness, aggression, irritability, distractibility, and increased drive12
- in patients with a history of antidepressant-induced mania, family history of bipolar disorder, or multiple antidepressant trials.13
Similarly, patients who report feeling better immediately after starting a stimulant—especially if they have evidence of elation, increased irritability, more aggression or impulsivity, decreased sleep, or related symptoms—may be developing stimulant-induced hypomania.
Table 3
Risk factors that may increase risk of stimulant-induced mania
|
| Source: Reference 25 |
Antidepressant-induced mania
Most studies of antidepressant-induced mania have examined outright mania, but hypomania and subsyndromal hypomanic syndromes also may cause significant morbidity and may worsen bipolar disorder’s course. A change in polarity may worsen a patient’s prognosis, but how do we know that antidepressants (or stimulants) caused it?
One suggested criterion is that mania or hypomania develops within 8 weeks of starting an antidepressant for the first time. A chart review of 51 bipolar patients who had extensive life charting found that 82% developed mania while taking an antidepressant—35% of them within 8 weeks.14 The authors attributed 50% of the risk of a first manic episode and/or cycle acceleration to antidepressants and 50% to spontaneous mood swings. They also noted that:
- an initial manic episode appeared to sensitize patients to subsequent manic episodes and rapid cycling
- mood stabilizers did not seem to prevent these outcomes.
A meta-analysis of 12 randomized, controlled, 4-to 12-week trials among 1,088 patients found antidepressants no more likely than placebo to induce mania in the short term.15 These trials did not, however, consider less-severe forms of overstimulation and were not designed to determine mania risk in bipolar depressed patients.
Post-mania cycling. Rapid and ultradian cycling and other forms of deterioration are more likely to occur after a manic or hypomanic episode than after a depressive episode.16
A longitudinal study17 indicated that antidepressant use did not predictably predate rapid cycling when depression was controlled. The authors, however, looked at the correlation between taking an antidepressant at study entry and rapid cycling over 1 year but did not examine whether antidepressants were started or stopped during the study.18 Rapid cycling prevalence declined from 19% to 5% during the study, but they did not determine whether withdrawing antidepressants was associated with this change.
In an earlier prospective study, rapid cycling was more severe while patients were taking antidepressants—despite the use of mood stabilizers—and cycling duration decreased when antidepressants were withdrawn.19
TCAs vs. newer agents. Tricyclic antidepressants (TCAs) are perceived as more likely to induce mania than are selective serotonin reuptake inhibitors (SSRIs) or bupropion. Comparing TCAs’ and newer antidepressants’ switch rates is difficult, however. Most antidepressant trials were designed to show efficacy and safety in unipolar, not bipolar, depression. Moreover, as exclusion criteria have improved with greater awareness of bipolar illness’ polymorphic manifestations, recent studies likely have enrolled fewer bipolar patients—who are most at risk to develop a manic switch—than did earlier TCA trials.
Bupropion, which has been used to treat ADHD, has been thought to have a low risk of inducing mania. In open observation, however, >50% of 11 patients with a history of developing mania with other antidepressants also had a manic switch on bupropion, even though they were taking mood stabilizers.20
Analysis of 155 antidepressant trials in 41 depressed patients found mania risk to be similar with bupropion, SSRIs, TCAs, monoamine oxidase inhibitors (MAOIs), and other newer antidepressants.21 Mania risk doubled when patients were not also taking mood stabilizers.
Going without mood stabilizers. Reports have emerged of patients with bipolar depression taking antidepressants such as fluoxetine and venlafaxine without a mood stabilizer for extended periods, without high rates of mania or mood cycling.22-24 These reports suggest that some bipolar depressed patients can tolerate antidepressants without a mood stabilizer, although we have no way to identify such patients in advance.
Cycle acceleration and treatment resistance may follow antidepressant-induced mania.25 In DSM-IV field trials, antidepressants appeared to have triggered rapid cycling in some 20% of bipolar patients.26 Mood stabilizers were not particularly effective in patients with treatment-resistant ultradian cycling, but withdrawing antidepressants improved outcome.27
Stimulant-induced mania
Compared with antidepressants, less information is available about stimulant-induced mania and rapid cycling.
Some carefully selected bipolar patients may tolerate ongoing stimulant treatment. For example, in 2 years of open experience with 5 bipolar type I and 3 bipolar type II adults, adding methylphenidate or amphetamine for residual depression or sedation was moderately helpful and did not lead to manic switching or drug misuse.28
On the other hand, affective symptoms worsened in nearly two-thirds of 31 children ages 2 to 5 when treated with stimulants or antidepressants without mood stabilizers. Most of the children also had ADHD, and valproate usually helped.6
In 40 patients, mean age 10, who entered the open-label phase of an 8-week trial of divalproex for manic and ADHD symptoms:
- Young Mania Rating Scale (YMRS) scores declined by≥50% in 32 (80%) by week 8, a greater initial response than usually reported in pediatric bipolar disorder with comorbid ADHD.
- ADHD symptoms, measured by Clinical Global Impressions (CGI) scores, did not change significantly.29
Thirty divalproex responders then received mixed amphetamine salts, 10 mg/d, or placebo plus divalproex, crossing over to the other treatment in a 4-week, double-blind trial. ADHD symptoms improved twice as much with the stimulant as with placebo, as measured by CGI scores, whereas YMRS scores did not differ significantly. Among 23 patients who continued the stimulant and divalproex for 12 more weeks, 45% required an increase in stimulant dosage and 1 relapsed into mania.
In this study, ADHD symptoms did not respond to mania treatment but did improve when a stimulant was added. This suggests either that patients had two disorders or that not all bipolar features remit at the same time. The trial’s low stimulant dosage and short duration provide insufficient evidence to support using stimulants over long periods in bipolar children.
LOng-term stimulant effects
Without long-term observations, some investigators have inferred stimulants’ impact on bipolar disorder. A poll of pediatric psychiatrists in the Netherlands, for example, found bipolar disorder in 39 children ages <13 (0.001%) in the previous year, compared with a prevalence of at least 1% in the United States.3 The authors concluded:
- Bipolar disorder emerges at younger ages in the United States than in the Netherlands.
- One reason may be that U.S. psychiatrists have a lower threshold for treating pediatric depression and hyperactivity with antidepressants and stimulants than Dutch psychiatrists do, evoking more-obvious bipolar symptoms at an earlier age.
Observations of 30 U.S. children with a manic episode and ADHD suggested that stimulants can induce manic symptoms:
- Mean age of ADHD onset was 5.5 years.
- Mean age of starting stimulants was 6.9 years.
- Mean age of hypomanic or manic symptom onset was 7.1 years.30
Similarly, in a survey of 34 adolescent manic inpatients, those who had taken stimulants had earlier mania onset (mean age 10.7) than did those who had not taken stimulants (mean age 13.9). Exposure to two stimulants was associated with earlier onset than exposure to one, but comorbid ADHD alone did not affect age of bipolar disorder onset.31
The same group10 reviewed charts of 80 consecutively hospitalized adolescents with a manic or mixed bipolar episode and found stimulant exposure was associated with relatively worse inpatient course, longer length of stay, more emergency medications, and more seclusion and restraint orders. Comorbid ADHD, mixed versus manic episode, and prior antidepressant exposure did not worsen the inpatient course.
A chart review by El-Mallakh et al32 found bipolar disorder was diagnosed at mean age 10.7 in 49 children exposed to antidepressants or stimulants, compared with mean age 12.7 in 44 unexposed children. The exposed group appeared to have tolerated stimulants longer than antidepressants before mania or hypomania emerged.33
In contrast, a retrospective review by Carlson et al34 of data from a longitudinal study of 75 boys with “hyperkinetic reaction of childhood” found that methylphenidate treatment did not appear more common in boys later diagnosed with bipolar disorder than in those without a bipolar diagnosis. This study had obvious methodologic limitations, lacking a hypothesis and focusing on a population with “minimal brain dysfunction.”
In a reanalysis of data from a 1-month methylphenidate titration trial, Galanter et al35 examined whether some 300 children ages 5 to 12 experienced manic symptoms, using the Diagnostic Interview Schedule for Children or the Child Behavior Checklist. At least during this brief trial, patients with and without manic symptoms showed no differences in response rates or adverse effects with stimulant therapy.
Drug treatment hierarchy
Mood stabilizers. Evidence supports starting all bipolar children with a mood stabilizer such as lithium or valproate (Algorithm). A few patients may tolerate stimulants without mood stabilizers, but the risk is high of inducing mania and precipitating a more complex and treatment-resistant disorder.
Carbamazepine can be effective, but it makes some youths aggressive or disorganized. Antipsychotics have not been tested in controlled trials in bipolar children and are not considered first-line treatments, especially as mood stabilizers. They can be effective for childhood mania, but outpatients needing ADHD treatment usually do not have severe manic syndromes.
Algorithm Reducing mania risk: Using stimulants in children with bipolar disorder
Combination therapy. Like many adults, bipolar children often require combinations of mood stabilizers. Kowatch et al36 found that 16 of 20 acutely ill bipolar children (mean age 11) responded to a combination of mood stabilizers after not responding to 8 weeks of a single mood stabilizer. Because bipolar disorder with comorbid ADHD suggests a complex pathophysiology, patients with both disorders may be more likely to require mood-stabilizer combinations than those with bipolar disorder alone.
The goal in treating bipolar disorder is to eliminate symptoms as completely as possible. In bipolar children with comorbid ADHD, be certain that subtle hypomanic symptoms—irritability, decreased sleep, hypersensitivity to interactions, psychosis—have remitted, as they could account for continued inattention. Persistent mood lability may indicate incomplete treatment of the mood disorder, which can increase sensitivity to destabilization by a stimulant.
If a child remains inattentive after the mood disorder is controlled, consider whether medications for the mood disorder are to blame. If medications are working well but causing cognitive side effects, a cholinesterase inhibitor may help.
Adding stimulants. If attention problems persist, consider cautiously adding a stimulant. Informed consent includes telling patients and families about the risks of mood destabilization with stimulants, even when used with mood stabilizers.
Increase stimulant dosage very slowly, and monitor the patient closely for emerging mood instability or subtle evidence of dysphoric hypomania. Address hypersensitivity to sounds, increased irritability, or other signs of activation with more-aggressive mood stabilization before assuming that these are ADHD symptoms that require a higher stimulant dosage.
Sustained-release stimulant preparations are probably second-line choices in patients with concomitant bipolar disorder. With long-acting stimulants, any worsening of the mood disorder will take longer to wear off. Antidepressants such as bupropion are potential alternatives to stimulants but are as likely to induce hypomania and mood cycling and may not be as effective.
Compared with stimulants, atomoxetine has a less-potent antidepressant effect and may be somewhat safer, but it is not as effective for ADHD and is longer-acting. Thus, atomoxetine could be a first-line alternative for comorbid ADHD, with stimulants being added if it is not effective. Clonidine can reduce hyperactivity but does not stabilize mood or improve attention.
When an antidepressant has brought bipolar depression into remission, discontinue it slowly to reduce the risk of rebound while continuing mood stabilizers to prevent recurrence. Because ADHD is not cyclical like bipolar depression, inattention returns for many patients when stimulants are withdrawn.
We do not yet know whether the risk of mood destabilization increases with long-term stimulant use, but discontinuation-induced refractoriness has not been reported with stimulants as it has with mood stabilizers and antidepressants. Thus, trying to withdraw stimulants once ADHD symptoms have remitted is prudent, while supplementing the regimen with behavioral treatments. If managing ADHD symptoms requires continued stimulant treatment, monitor the patient closely for mood destabilization.
Related resources
- American Academy of Child and Adolescent Psychiatry. Facts for families: Bipolar disorder in children and teens.
www.aacap.org/publications/factsFam/bipolar.htm. - National Institute of Mental Health. Database on ADHD.
www.nimh.nih.gov/publicat/adhd.cfm.
Drug brand names
- Amphetamine salts • Adderall
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, others
- Clonidine • Catapres
- Dexmethylphenidate • Focalin
- Fluoxetine • Prozac
- Lithium • Lithobid, others
- Methylphenidate • Concerta,
- Ritalin, others
- Valproate • Depakene, Depakote
- Venlafaxine • Effexor
Disclosures
Dr. Dubovsky receives research support from UCB Pharma, Forest Laboratories, and Solvay Pharmaceuticals, and is a speaker for Janssen Pharmaceutica and Forest Laboratories.
Stimulants are most effective for childhood attention-deficit/hyperactivity disorder (ADHD),1 but they may induce mania or trigger a treatment-resistant course in children with comorbid bipolar disorder. To help you safely manage these complicated symptoms, this article offers a treatment algorithm and tips to:
- differentiate bipolar and ADHD symptoms
- identify patients at risk for stimulantinduced mania
- choose medications by a hierarchythat may reduce the risk of mood destabilization.
Bipolar mood symptoms emerge before age 20 in about 25% of persons with bipolar disorder (BP).3 Early-onset BP may be more severe than the adult-onset form, with more-affected family members and greater comorbidity with other disorders, especially ADHD.4
In one study, 91% of children with BP also met criteria for ADHD, and 19% of patients with ADHD also received a diagnosis of BP.5 Among 31 children ages 2 to 5 with BP, 80% met criteria for concurrent ADHD.6
Of 40 children age <5 presenting consecutively to a mental health clinic, 11 (28%) met criteria for mania, which was usually associated with euphoria.7 These 11 children also met criteria for ADHD.
A comparison study8 of children (mean age 12) found greater impairment, suicidality, irritability, and sadness in 43 with ADHD plus bipolar depression than in:
- 109 with ADHD plus major depressive disorder
- 128 without depression or mania.
Family prevalence of bipolar disorder and major depression was highest in the bipolar-ADHD group, which also had the highest rates of comorbid conduct disorder, oppositional defiant disorder, alcohol abuse, and agoraphobia. Average age of bipolar diagnosis was 6.3 years.
Adhd and/or bipolar disorder?
Some 70% to 90% of bipolar children and at least 30% to 40% of bipolar adolescents also have ADHD.2 This high comorbidity (Box 1)3-8 might mean that:
- one disorder predisposes to the other
- one is a precursor of the other
- they share common vulnerabilities or causes
- their symptoms overlap so much that patients with one disorder appear to meet criteria for the other.
Some experts contend that bipolar disorder and ADHD usually can be differentiated. Bipolar children score higher than those with ADHD on measures of anxiety/depression, aggression, and attention problems on the Child Behavior Checklist.9 Others believe ADHD symptoms that occur with bipolar disorder are a dimension of bipolar illness rather than a separate disorder.10
For every DSM-IV-TR diagnostic criterion for ADHD, a corresponding diagnostic criterion or common feature of bipolar disorder can be identified (Table 1). Mania and hypomania are obviously associated with hyperactivity and impulsivity, and tangential thinking and distractibility interfere with attention in many patients with bipolar disorder.
Though most ADHD symptoms can occur in bipolar patients, some features of bipolar illness are not characteristic of ADHD (Table 2). Children with ADHD can become hyper-focused on video games and television, for example, but they usually do not become engrossed in long, complicated books or preoccupied with other people, as can occur in bipolar disorder.
Table 1
How ADHD, bipolar symptoms overlap in three domains
| ADHD | Bipolar disorder |
|---|---|
| Inattention | |
| Fails to pay attention | Racing and tangential thoughts |
| Difficulty sustaining attention | Attention driven by racing thoughts, affective themes, and psychosis |
| Does not follow through | Direction of activity shifts with shifting mood |
| Difficulty organizing tasks | Disorganization, psychosis, excessive energy |
| Easily distracted | Distractibility |
| Hyperactivity | |
| Fidgets or squirms | Increased energy and activity |
| Runs about or climbs excessively | Hyperactivity, thrill-seeking |
| Difficulty engaging quietly in leisure activities | Increased energy, boredom |
| Often on the go | Increased energy, hyperactivity |
| Talks excessively | Rapid, pressured speech |
| Impulsivity | |
| Blurts out answers | Rapid, pressured, impulsive speech |
| Difficulty awaiting turn | Hyperactivity, increased energy, impatience, grandiosity |
| Interrupts or intrudes on others | Grandiosity, impatience, pressured speech, increased mental content |
Table 2
Bipolar features not seen in ADHD
|
A treatment hierarchy
Whether a bipolar patient’s attention problems are features of the primary condition or caused by comorbid ADHD may be unclear, but the treatment implications are important. All antidepressants can induce mania/hypomania and increase the risk of mixed states and mood cycling. Because stimulants have antidepressant properties and because some antidepressants are used to treat ADHD, a systematic approach is necessary when treating inattention in juvenile bipolar disorder.
A treatment hierarchy developed by the American Academy of Child and Adolescent Psychiatry Workgroup on Bipolar Disorder recommends beginning psychosocial approaches, such as training parents in behavior management techniques, and:
- treating bipolar disorder first in children who clearly have both ADHD and bipolar disorder
- adding ADHD treatment if ADHD symptoms persist and impair functioning.2
Who’s at risk for mood destabilization?
No data address differences between bipolar patients whose mood disorders deteriorate with stimulant use and those who remain stable. However, risk factors for mood destabilization that have been reported with antidepressants likely also apply to stimulants (Table 3) because stimulants’ adverse effects in bipolar disorder are probably related to their antidepressant properties.
For example, depressed patients who report that an antidepressant worked within hours to days may have bipolar disorder and be at risk for mood destabilization leading to treatment resistance.11 Antidepressant-induced mania also may be more likely:
- when depression is mixed with hypomanic symptoms such as racing thoughts, excessive talkativeness, aggression, irritability, distractibility, and increased drive12
- in patients with a history of antidepressant-induced mania, family history of bipolar disorder, or multiple antidepressant trials.13
Similarly, patients who report feeling better immediately after starting a stimulant—especially if they have evidence of elation, increased irritability, more aggression or impulsivity, decreased sleep, or related symptoms—may be developing stimulant-induced hypomania.
Table 3
Risk factors that may increase risk of stimulant-induced mania
|
| Source: Reference 25 |
Antidepressant-induced mania
Most studies of antidepressant-induced mania have examined outright mania, but hypomania and subsyndromal hypomanic syndromes also may cause significant morbidity and may worsen bipolar disorder’s course. A change in polarity may worsen a patient’s prognosis, but how do we know that antidepressants (or stimulants) caused it?
One suggested criterion is that mania or hypomania develops within 8 weeks of starting an antidepressant for the first time. A chart review of 51 bipolar patients who had extensive life charting found that 82% developed mania while taking an antidepressant—35% of them within 8 weeks.14 The authors attributed 50% of the risk of a first manic episode and/or cycle acceleration to antidepressants and 50% to spontaneous mood swings. They also noted that:
- an initial manic episode appeared to sensitize patients to subsequent manic episodes and rapid cycling
- mood stabilizers did not seem to prevent these outcomes.
A meta-analysis of 12 randomized, controlled, 4-to 12-week trials among 1,088 patients found antidepressants no more likely than placebo to induce mania in the short term.15 These trials did not, however, consider less-severe forms of overstimulation and were not designed to determine mania risk in bipolar depressed patients.
Post-mania cycling. Rapid and ultradian cycling and other forms of deterioration are more likely to occur after a manic or hypomanic episode than after a depressive episode.16
A longitudinal study17 indicated that antidepressant use did not predictably predate rapid cycling when depression was controlled. The authors, however, looked at the correlation between taking an antidepressant at study entry and rapid cycling over 1 year but did not examine whether antidepressants were started or stopped during the study.18 Rapid cycling prevalence declined from 19% to 5% during the study, but they did not determine whether withdrawing antidepressants was associated with this change.
In an earlier prospective study, rapid cycling was more severe while patients were taking antidepressants—despite the use of mood stabilizers—and cycling duration decreased when antidepressants were withdrawn.19
TCAs vs. newer agents. Tricyclic antidepressants (TCAs) are perceived as more likely to induce mania than are selective serotonin reuptake inhibitors (SSRIs) or bupropion. Comparing TCAs’ and newer antidepressants’ switch rates is difficult, however. Most antidepressant trials were designed to show efficacy and safety in unipolar, not bipolar, depression. Moreover, as exclusion criteria have improved with greater awareness of bipolar illness’ polymorphic manifestations, recent studies likely have enrolled fewer bipolar patients—who are most at risk to develop a manic switch—than did earlier TCA trials.
Bupropion, which has been used to treat ADHD, has been thought to have a low risk of inducing mania. In open observation, however, >50% of 11 patients with a history of developing mania with other antidepressants also had a manic switch on bupropion, even though they were taking mood stabilizers.20
Analysis of 155 antidepressant trials in 41 depressed patients found mania risk to be similar with bupropion, SSRIs, TCAs, monoamine oxidase inhibitors (MAOIs), and other newer antidepressants.21 Mania risk doubled when patients were not also taking mood stabilizers.
Going without mood stabilizers. Reports have emerged of patients with bipolar depression taking antidepressants such as fluoxetine and venlafaxine without a mood stabilizer for extended periods, without high rates of mania or mood cycling.22-24 These reports suggest that some bipolar depressed patients can tolerate antidepressants without a mood stabilizer, although we have no way to identify such patients in advance.
Cycle acceleration and treatment resistance may follow antidepressant-induced mania.25 In DSM-IV field trials, antidepressants appeared to have triggered rapid cycling in some 20% of bipolar patients.26 Mood stabilizers were not particularly effective in patients with treatment-resistant ultradian cycling, but withdrawing antidepressants improved outcome.27
Stimulant-induced mania
Compared with antidepressants, less information is available about stimulant-induced mania and rapid cycling.
Some carefully selected bipolar patients may tolerate ongoing stimulant treatment. For example, in 2 years of open experience with 5 bipolar type I and 3 bipolar type II adults, adding methylphenidate or amphetamine for residual depression or sedation was moderately helpful and did not lead to manic switching or drug misuse.28
On the other hand, affective symptoms worsened in nearly two-thirds of 31 children ages 2 to 5 when treated with stimulants or antidepressants without mood stabilizers. Most of the children also had ADHD, and valproate usually helped.6
In 40 patients, mean age 10, who entered the open-label phase of an 8-week trial of divalproex for manic and ADHD symptoms:
- Young Mania Rating Scale (YMRS) scores declined by≥50% in 32 (80%) by week 8, a greater initial response than usually reported in pediatric bipolar disorder with comorbid ADHD.
- ADHD symptoms, measured by Clinical Global Impressions (CGI) scores, did not change significantly.29
Thirty divalproex responders then received mixed amphetamine salts, 10 mg/d, or placebo plus divalproex, crossing over to the other treatment in a 4-week, double-blind trial. ADHD symptoms improved twice as much with the stimulant as with placebo, as measured by CGI scores, whereas YMRS scores did not differ significantly. Among 23 patients who continued the stimulant and divalproex for 12 more weeks, 45% required an increase in stimulant dosage and 1 relapsed into mania.
In this study, ADHD symptoms did not respond to mania treatment but did improve when a stimulant was added. This suggests either that patients had two disorders or that not all bipolar features remit at the same time. The trial’s low stimulant dosage and short duration provide insufficient evidence to support using stimulants over long periods in bipolar children.
LOng-term stimulant effects
Without long-term observations, some investigators have inferred stimulants’ impact on bipolar disorder. A poll of pediatric psychiatrists in the Netherlands, for example, found bipolar disorder in 39 children ages <13 (0.001%) in the previous year, compared with a prevalence of at least 1% in the United States.3 The authors concluded:
- Bipolar disorder emerges at younger ages in the United States than in the Netherlands.
- One reason may be that U.S. psychiatrists have a lower threshold for treating pediatric depression and hyperactivity with antidepressants and stimulants than Dutch psychiatrists do, evoking more-obvious bipolar symptoms at an earlier age.
Observations of 30 U.S. children with a manic episode and ADHD suggested that stimulants can induce manic symptoms:
- Mean age of ADHD onset was 5.5 years.
- Mean age of starting stimulants was 6.9 years.
- Mean age of hypomanic or manic symptom onset was 7.1 years.30
Similarly, in a survey of 34 adolescent manic inpatients, those who had taken stimulants had earlier mania onset (mean age 10.7) than did those who had not taken stimulants (mean age 13.9). Exposure to two stimulants was associated with earlier onset than exposure to one, but comorbid ADHD alone did not affect age of bipolar disorder onset.31
The same group10 reviewed charts of 80 consecutively hospitalized adolescents with a manic or mixed bipolar episode and found stimulant exposure was associated with relatively worse inpatient course, longer length of stay, more emergency medications, and more seclusion and restraint orders. Comorbid ADHD, mixed versus manic episode, and prior antidepressant exposure did not worsen the inpatient course.
A chart review by El-Mallakh et al32 found bipolar disorder was diagnosed at mean age 10.7 in 49 children exposed to antidepressants or stimulants, compared with mean age 12.7 in 44 unexposed children. The exposed group appeared to have tolerated stimulants longer than antidepressants before mania or hypomania emerged.33
In contrast, a retrospective review by Carlson et al34 of data from a longitudinal study of 75 boys with “hyperkinetic reaction of childhood” found that methylphenidate treatment did not appear more common in boys later diagnosed with bipolar disorder than in those without a bipolar diagnosis. This study had obvious methodologic limitations, lacking a hypothesis and focusing on a population with “minimal brain dysfunction.”
In a reanalysis of data from a 1-month methylphenidate titration trial, Galanter et al35 examined whether some 300 children ages 5 to 12 experienced manic symptoms, using the Diagnostic Interview Schedule for Children or the Child Behavior Checklist. At least during this brief trial, patients with and without manic symptoms showed no differences in response rates or adverse effects with stimulant therapy.
Drug treatment hierarchy
Mood stabilizers. Evidence supports starting all bipolar children with a mood stabilizer such as lithium or valproate (Algorithm). A few patients may tolerate stimulants without mood stabilizers, but the risk is high of inducing mania and precipitating a more complex and treatment-resistant disorder.
Carbamazepine can be effective, but it makes some youths aggressive or disorganized. Antipsychotics have not been tested in controlled trials in bipolar children and are not considered first-line treatments, especially as mood stabilizers. They can be effective for childhood mania, but outpatients needing ADHD treatment usually do not have severe manic syndromes.
Algorithm Reducing mania risk: Using stimulants in children with bipolar disorder
Combination therapy. Like many adults, bipolar children often require combinations of mood stabilizers. Kowatch et al36 found that 16 of 20 acutely ill bipolar children (mean age 11) responded to a combination of mood stabilizers after not responding to 8 weeks of a single mood stabilizer. Because bipolar disorder with comorbid ADHD suggests a complex pathophysiology, patients with both disorders may be more likely to require mood-stabilizer combinations than those with bipolar disorder alone.
The goal in treating bipolar disorder is to eliminate symptoms as completely as possible. In bipolar children with comorbid ADHD, be certain that subtle hypomanic symptoms—irritability, decreased sleep, hypersensitivity to interactions, psychosis—have remitted, as they could account for continued inattention. Persistent mood lability may indicate incomplete treatment of the mood disorder, which can increase sensitivity to destabilization by a stimulant.
If a child remains inattentive after the mood disorder is controlled, consider whether medications for the mood disorder are to blame. If medications are working well but causing cognitive side effects, a cholinesterase inhibitor may help.
Adding stimulants. If attention problems persist, consider cautiously adding a stimulant. Informed consent includes telling patients and families about the risks of mood destabilization with stimulants, even when used with mood stabilizers.
Increase stimulant dosage very slowly, and monitor the patient closely for emerging mood instability or subtle evidence of dysphoric hypomania. Address hypersensitivity to sounds, increased irritability, or other signs of activation with more-aggressive mood stabilization before assuming that these are ADHD symptoms that require a higher stimulant dosage.
Sustained-release stimulant preparations are probably second-line choices in patients with concomitant bipolar disorder. With long-acting stimulants, any worsening of the mood disorder will take longer to wear off. Antidepressants such as bupropion are potential alternatives to stimulants but are as likely to induce hypomania and mood cycling and may not be as effective.
Compared with stimulants, atomoxetine has a less-potent antidepressant effect and may be somewhat safer, but it is not as effective for ADHD and is longer-acting. Thus, atomoxetine could be a first-line alternative for comorbid ADHD, with stimulants being added if it is not effective. Clonidine can reduce hyperactivity but does not stabilize mood or improve attention.
When an antidepressant has brought bipolar depression into remission, discontinue it slowly to reduce the risk of rebound while continuing mood stabilizers to prevent recurrence. Because ADHD is not cyclical like bipolar depression, inattention returns for many patients when stimulants are withdrawn.
We do not yet know whether the risk of mood destabilization increases with long-term stimulant use, but discontinuation-induced refractoriness has not been reported with stimulants as it has with mood stabilizers and antidepressants. Thus, trying to withdraw stimulants once ADHD symptoms have remitted is prudent, while supplementing the regimen with behavioral treatments. If managing ADHD symptoms requires continued stimulant treatment, monitor the patient closely for mood destabilization.
Related resources
- American Academy of Child and Adolescent Psychiatry. Facts for families: Bipolar disorder in children and teens.
www.aacap.org/publications/factsFam/bipolar.htm. - National Institute of Mental Health. Database on ADHD.
www.nimh.nih.gov/publicat/adhd.cfm.
Drug brand names
- Amphetamine salts • Adderall
- Atomoxetine • Strattera
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol, others
- Clonidine • Catapres
- Dexmethylphenidate • Focalin
- Fluoxetine • Prozac
- Lithium • Lithobid, others
- Methylphenidate • Concerta,
- Ritalin, others
- Valproate • Depakene, Depakote
- Venlafaxine • Effexor
Disclosures
Dr. Dubovsky receives research support from UCB Pharma, Forest Laboratories, and Solvay Pharmaceuticals, and is a speaker for Janssen Pharmaceutica and Forest Laboratories.
1. Greenhill LL, Pliszka S, Dulcan MK, et al. AACAP. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41(suppl 2):26S-49S.
2. Kowatch RA, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder: child psychiatric workgroup on bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44:213-35.
3. Reichart CG, Nolen W. Earlier onset of bipolar disorder in children by antidepressants or stimulants? An hypothesis. J Affect Disord 2004;78:81-4.
4. Faraone SV, Glatt SJ, Tsuang MT. The genetics of pediatric-onset bipolar disorder. Biol Psychiatry 2003;53:970-7.
5. Geller B, Zimmerman B, Williams MB, et al. Bipolar disorder at prospective follow-up of adults who had prepubertal major depressive disorder. Am J Psychiatry 2001;158:125-7.
6. Scheffer RE, Niskala Apps JA. The diagnosis of preschool bipolar disorder presenting with mania: open pharmacological treatment. J Affect Disord. 2004;82(suppl 1):S25-S34.
7. Dilsaver SC, Akiskal HS. Preschool-onset mania: incidence, phenomenology and family history. J Affect Disord 2004;82(suppl 1):S35-S43.
8. Wozniak J, Spencer T, Biederman J, et al. The clinical characteristics of unipolar vs. bipolar major depression in ADHD youth. J Affect Disord 2004;82(suppl 1):S59-S69.
9. Mick E, Biederman J, Pandina G, Faraone SV. A preliminary meta-analysis of the Child Behavior Checklist in pediatric bipolar disorder. Biol Psychiatry 2003;53:1021-7.
10. Soutullo CA, DelBello MP, Ochsner JE, et al. Severity of bipolarity in hospitalized manic adolescents with history of stimulant or antidepressant treatment. J Affect Disord 2002;70:323-7.
11. Piver A. Ultrarapid response to an antidepressant: A clue to bipolarity? Can J Psychiatry 2003;48:427-8.
12. Bottlender R, Sato T, Kleindienst N, et al. Mixed depressive features predict maniform switch during treatment of depression in bipolar I disorder. J Affect Disord 2004;78:149-52.
13. Goldberg J, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord 2003;5:407-20.
14. Altshuler LL, Post RM, Leverich GS, et al. Antidepressant-induced mania and cycle acceleration: a controversy revisited. Am J Psychiatry 1995;152(8):1130-8.
15. Gijsman HJ, Geddes J, Rendell J, et al. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry 2004;161:1537-47.
16. Post RM, Roy-Byrne PP, Uhde TW. Graphic representation of the life course of illness in patients with affective disorder. Am J Psychiatry 1988;145:844-8.
17. Coryell W, Endicott J, Keller M. Rapidly cycling affective disorder: demographics, diagnosis, family history and course. Arch Gen Psychiatry 1992;49:126-31.
18. Wehr TA. Can antidepressants induce rapid cycling? Arch Gen Psychiatry 1993;50(6):495-6.
19. Wehr TA, Sack DA, Rosenthal NE, Cowdry RW. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
20. Fogelson DL, Bystritsky A, Pasnau R. Bupropion in the treatment of bipolar disorders: the same old story. J Clin Psychiatry 1992;53:443-6.
21. Goldberg J, Ernst CL. Features associated with the delayed initiation of mood stabilizers at illness onset in bipolar disorder. J Clin Psychiatry 2002;63:985-91.
22. Amsterdam JD, Shults J, Brunswick DJ, Hundert M. Short-term fluoxetine monotherapy for bipolar type II or bipolar NOS major depression—low manic switch rate. Bipolar Disord 2004;6:75-81.
23. Simpson SG, DePaulo JR. Fluoxetine treatment of bipolar II depression. J Clin Psychopharmacol 1991;11:52-4.
24. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
25. Goldberg J. When do antidepressants worsen the course of bipolar disorder? J Psychiatr Pract 2003;9:181-94.
26. Bauer M, Calabrese JR, Dunner DL. Multisite data reanalysis of the validity of rapid cycling as a course modifier for bipolar disorder in DSM-IV. Am J Psychiatry 1994;151:506-15.
27. Prien RF, Kupfer DJ, Mansky PA. Drug therapy in the prevention of recurrences in unipolar and bipolar affective disorders: Report of the NIMH Collaborative Study Group comparing lithium carbonate, imipramine, and a lithium carbonate-imipramine combination. Arch Gen Psychiatry 1984;41:1096-1104.
28. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: treatment of residual depression and sedation. Bipolar Disord 2004;6:416-20.
29. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162:58-64.
30. Kowatch RA, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamezepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39:713-20.
31. DelBello MP, Soutullo CA, Hendricks W, et al. Prior stimulant treatment in adolescents with bipolar disorder: association with age at onset. Bipolar Disord 2001;3:53-7.
32. El-Mallakh RS, Cicero D, Holman J, Robertson J. Antidepressant exposure in children diagnosed with bipolar disorder. Bipolar Disord 2001;3(suppl 1):35-9.
33. Cicero D, El-Mallakh RS, Holman J, Robertson J. Antidepressant exposure in bipolar children. Psychiatry 2003;66:317-22.
34. Carlson G, Loney J, Salisbury H, et al. Stimulant treatment in young boys with symptoms suggesting childhood mania: A report from a longitudinal study. J Child Adolesc Psychopharmacol 2000;10:175-84.
35. Galanter CA, Carlson GA, Jensen PS, et al. Response to methylphenidate in children with attention deficit hyperactivity disorder and manic symptoms in the multimodal treatment study of children with attention deficit hyperactivity disorder titration trial. J Child Adolesc Psychopharmacol 2003;13:123-36.
36. Kowatch RA, Sethuraman G, Hume JH, Kromelis M, Weinberg WA. Combination pharmacotherapy in children and adolescents with bipolar disorder. Biol Psychiatry. 2003;53:978-84.
1. Greenhill LL, Pliszka S, Dulcan MK, et al. AACAP. Practice parameter for the use of stimulant medications in the treatment of children, adolescents, and adults. J Am Acad Child Adolesc Psychiatry 2002;41(suppl 2):26S-49S.
2. Kowatch RA, Fristad M, Birmaher B, et al. Treatment guidelines for children and adolescents with bipolar disorder: child psychiatric workgroup on bipolar disorder. J Am Acad Child Adolesc Psychiatry 2005;44:213-35.
3. Reichart CG, Nolen W. Earlier onset of bipolar disorder in children by antidepressants or stimulants? An hypothesis. J Affect Disord 2004;78:81-4.
4. Faraone SV, Glatt SJ, Tsuang MT. The genetics of pediatric-onset bipolar disorder. Biol Psychiatry 2003;53:970-7.
5. Geller B, Zimmerman B, Williams MB, et al. Bipolar disorder at prospective follow-up of adults who had prepubertal major depressive disorder. Am J Psychiatry 2001;158:125-7.
6. Scheffer RE, Niskala Apps JA. The diagnosis of preschool bipolar disorder presenting with mania: open pharmacological treatment. J Affect Disord. 2004;82(suppl 1):S25-S34.
7. Dilsaver SC, Akiskal HS. Preschool-onset mania: incidence, phenomenology and family history. J Affect Disord 2004;82(suppl 1):S35-S43.
8. Wozniak J, Spencer T, Biederman J, et al. The clinical characteristics of unipolar vs. bipolar major depression in ADHD youth. J Affect Disord 2004;82(suppl 1):S59-S69.
9. Mick E, Biederman J, Pandina G, Faraone SV. A preliminary meta-analysis of the Child Behavior Checklist in pediatric bipolar disorder. Biol Psychiatry 2003;53:1021-7.
10. Soutullo CA, DelBello MP, Ochsner JE, et al. Severity of bipolarity in hospitalized manic adolescents with history of stimulant or antidepressant treatment. J Affect Disord 2002;70:323-7.
11. Piver A. Ultrarapid response to an antidepressant: A clue to bipolarity? Can J Psychiatry 2003;48:427-8.
12. Bottlender R, Sato T, Kleindienst N, et al. Mixed depressive features predict maniform switch during treatment of depression in bipolar I disorder. J Affect Disord 2004;78:149-52.
13. Goldberg J, Truman CJ. Antidepressant-induced mania: an overview of current controversies. Bipolar Disord 2003;5:407-20.
14. Altshuler LL, Post RM, Leverich GS, et al. Antidepressant-induced mania and cycle acceleration: a controversy revisited. Am J Psychiatry 1995;152(8):1130-8.
15. Gijsman HJ, Geddes J, Rendell J, et al. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry 2004;161:1537-47.
16. Post RM, Roy-Byrne PP, Uhde TW. Graphic representation of the life course of illness in patients with affective disorder. Am J Psychiatry 1988;145:844-8.
17. Coryell W, Endicott J, Keller M. Rapidly cycling affective disorder: demographics, diagnosis, family history and course. Arch Gen Psychiatry 1992;49:126-31.
18. Wehr TA. Can antidepressants induce rapid cycling? Arch Gen Psychiatry 1993;50(6):495-6.
19. Wehr TA, Sack DA, Rosenthal NE, Cowdry RW. Rapid cycling affective disorder: contributing factors and treatment responses in 51 patients. Am J Psychiatry 1988;145:179-84.
20. Fogelson DL, Bystritsky A, Pasnau R. Bupropion in the treatment of bipolar disorders: the same old story. J Clin Psychiatry 1992;53:443-6.
21. Goldberg J, Ernst CL. Features associated with the delayed initiation of mood stabilizers at illness onset in bipolar disorder. J Clin Psychiatry 2002;63:985-91.
22. Amsterdam JD, Shults J, Brunswick DJ, Hundert M. Short-term fluoxetine monotherapy for bipolar type II or bipolar NOS major depression—low manic switch rate. Bipolar Disord 2004;6:75-81.
23. Simpson SG, DePaulo JR. Fluoxetine treatment of bipolar II depression. J Clin Psychopharmacol 1991;11:52-4.
24. Amsterdam JD, Garcia-Espana F. Venlafaxine monotherapy in women with bipolar II and unipolar major depression. J Affect Disord 2000;59:225-9.
25. Goldberg J. When do antidepressants worsen the course of bipolar disorder? J Psychiatr Pract 2003;9:181-94.
26. Bauer M, Calabrese JR, Dunner DL. Multisite data reanalysis of the validity of rapid cycling as a course modifier for bipolar disorder in DSM-IV. Am J Psychiatry 1994;151:506-15.
27. Prien RF, Kupfer DJ, Mansky PA. Drug therapy in the prevention of recurrences in unipolar and bipolar affective disorders: Report of the NIMH Collaborative Study Group comparing lithium carbonate, imipramine, and a lithium carbonate-imipramine combination. Arch Gen Psychiatry 1984;41:1096-1104.
28. Carlson PJ, Merlock MC, Suppes T. Adjunctive stimulant use in patients with bipolar disorder: treatment of residual depression and sedation. Bipolar Disord 2004;6:416-20.
29. Scheffer RE, Kowatch RA, Carmody T, Rush AJ. Randomized, placebo-controlled trial of mixed amphetamine salts for symptoms of comorbid ADHD in pediatric bipolar disorder after mood stabilization with divalproex sodium. Am J Psychiatry 2005;162:58-64.
30. Kowatch RA, Suppes T, Carmody T, et al. Effect size of lithium, divalproex sodium, and carbamezepine in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry 2000;39:713-20.
31. DelBello MP, Soutullo CA, Hendricks W, et al. Prior stimulant treatment in adolescents with bipolar disorder: association with age at onset. Bipolar Disord 2001;3:53-7.
32. El-Mallakh RS, Cicero D, Holman J, Robertson J. Antidepressant exposure in children diagnosed with bipolar disorder. Bipolar Disord 2001;3(suppl 1):35-9.
33. Cicero D, El-Mallakh RS, Holman J, Robertson J. Antidepressant exposure in bipolar children. Psychiatry 2003;66:317-22.
34. Carlson G, Loney J, Salisbury H, et al. Stimulant treatment in young boys with symptoms suggesting childhood mania: A report from a longitudinal study. J Child Adolesc Psychopharmacol 2000;10:175-84.
35. Galanter CA, Carlson GA, Jensen PS, et al. Response to methylphenidate in children with attention deficit hyperactivity disorder and manic symptoms in the multimodal treatment study of children with attention deficit hyperactivity disorder titration trial. J Child Adolesc Psychopharmacol 2003;13:123-36.
36. Kowatch RA, Sethuraman G, Hume JH, Kromelis M, Weinberg WA. Combination pharmacotherapy in children and adolescents with bipolar disorder. Biol Psychiatry. 2003;53:978-84.
EARLY LIFE STRESS AND DEPRESSION Childhood trauma may lead to neurobiologically unique mood disorders
Sadly, parental neglect and child abuse are very common in the United States and worldwide. Patients abused or exploited in childhood, who experience neglect or the loss of a parent during childhood, are haunted by these experiences.
Considerable evidence from laboratory animal and clinical studies indicate that stressful or traumatic events early in development have long-lasting effects on brain development. In particular, the neural and endocrine systems mediating the response to stress exhibit persistent alterations after adverse childhood events.
Clinically, patients with a history of childhood trauma often struggle with variable symptom complexes including both depression and anxiety. In this article, we review evidence that depression in patients with a history of early life stress (ELS) is biologically and clinically distinct from depression in patients without childhood abuse or neglect.
Data linking ELS to depression
Conservative estimates suggest that every year in the United States more than 1 million children are exposed to sexual or physical abuse or severe neglect.1 Unfortunately, this is only the tip of the iceberg. Emotional abuse is by definition comorbid with sexual and physical abuse and may occur alone at even higher rates.
Psychiatric sequelae of child abuse have been studied in adult survivors in considerable detail (Table). Women abused as children report greater numbers of depression, anxiety, somatic, and substance abuse symptoms compared with women without such a history.2 Not only are these women at increased risk for attempted suicide, but they attempt suicide at a rate that is proportional to the number of early life traumatic events that occurred during childhood.3 Men also are at increased risk for depression in the wake of child abuse.4
Sexual abuse in particular is a marker of especially severe childhood trauma. Depressed women who were sexually abused as children report more childhood physical abuse, childhood emotional abuse, parental conflict, and an earlier onset of depression than depressed women without a history of sexual abuse.5
Finally, recent data drawn from the National Comorbidity Survey suggest that child abuse and neglect may independently elevate risk for several stress-related diseases including cardiac disease, peptic ulcer, autoimmune disease, diabetes mellitus, and lung disease.6
Table
Early life stress as a risk factor for mood and anxiety disorders
| Child abuse and neglect are predictors of episodes of major depression in identical twins |
| Women with a history of childhood abuse are more than twice as likely to develop depression as non-abused women |
| Childhood abuse is related to the development of anxiety disorders in adulthood |
| Childhood physical abuse predisposes for combat-related posttraumatic stress disorder (PTSD) |
| Stress early in life may induce a vulnerability to stress later in life, resulting in an increased risk for stress-related disorders |
Depression and the biology of stress
Preclinical research using laboratory animals and clinical research with humans has provided significant insight into the relationship between the pathophysiology of depression and the neurobiology of stress. A burgeoning database suggests that disruption of the neural systems mediating the stress response plays a significant role in the etiology of certain forms of depression and anxiety.7 Much of this work has focused on the preeminent role of corticotropin-releasing factor (CRF) in this process (Figure).
CRF is one of the principal mediators of the mammalian stress response. One CRF system is composed of neurons of the paraventricular nuclei of the hypothalamus that project nerve terminals to the median eminence, where they secrete CRF into the hypophyseal portal system. CRF is then transported within the portal system to the anterior pituitary where it acts on corticotrophs to increase adrenocorticotrophic hormone (ACTH) secretion, thereby controlling hypothalamic-pituitary-adrenal (HPA) axis activity.8 CRF is also found in extrahypothalamic brain areas where it functions, in concert with the hypothalamic CRF system, as a neurotransmitter in coordinating the behavioral, autonomic, and immune responses to stress.9
Direct central nervous system (CNS) administration of CRF in laboratory animals, typically rodents or nonhuman primates, results in activation of the autonomic nervous system leading to elevation of peripheral catecholamines, modification of gastrointestinal activity, increased heart rate and increased blood pressure. In addition, changes in behavior similar to those observed in human depression occur, including disturbed sleep patterns, reduced food intake, decreased reproductive behavior, and enhanced fear conditioning.10,11
In humans, elevated CRF concentrations are found in the cerebrospinal fluid (CSF) of patients with depression12,13 and of combat veterans with posttraumatic stress disorder (PTSD).14,15 Further, postmortem studies of suicide victims have revealed decreased density of CRF receptors in the frontal cortex,16 decreased expression of CRF receptor mRNA and increased CRF concentrations in the frontal cortex when compared with controls,17 and increased concentrations of cisternal CSF CRF.18 Collectively, these clinical data are consistent with the hypothesis that CRF is chronically hypersecreted in patients with depression or PTSD.
Figure Biological and behavioral effects of chronic CRF hypersecretion
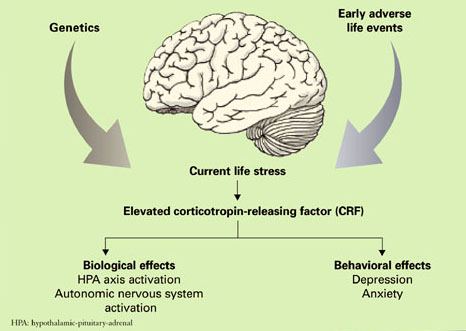
A distinct ‘ELS depression’?
Depression has a complex etiology based on interacting contributions from genes and the environment19 that may ultimately result in biologically and clinically distinct forms of depression.20 Exposure to stress, particularly during neurobiologically vulnerable periods of development, may be one means whereby the environment influences the development of depression in genetically susceptible individuals.21
Heredity. Kendler and colleagues22 studied 1,404 female adult twins and observed that childhood sexual abuse was associated with both an increased risk for major depression and a marked increased sensitivity to the depressogenic effects of stressful life events. Moreover, research in human gene-environment interactions has identified a functional polymorphism in the promoter region of the gene for the serotonin transporter which appears to moderate the influence of stressful life events on the development of depression and potential for suicide.23,24
Environment. Similarly, a key variable in determining the clinical outcome of childhood trauma may be the developmental timing of the abuse. Women abused before age 13 are at equivalent risk for developing PTSD or major depressive disorder (MDD), whereas women abused after age 13 are more likely to develop PTSD.25
Thus a major challenge in depression research is to understand the biological mechanisms that mediate the effects of trauma during development through the genetic windows of vulnerability and resilience.
Animal models of ELS have been studied to elucidate the neurobiological consequences of early life trauma in adult humans. This work has largely been performed in rodents and nonhuman primates using a variety of experimental paradigms.
Although a comprehensive review of these data is beyond the scope of this article, ELS in laboratory animals has consistently been found to produce both short- and long-term adverse neurobiological and endocrine effects as well as cognitive dysfunction and abnormal behavior.21 One possible mechanism mediating these effects is a persistent hyperresponsiveness of different components of the HPA axis following exposure to stress.
Studies in adult women have sought to uncover the long-term effects of ELS (prepubertal physical or sexual abuse) on reactivity of the HPA axis in response to the Trier Social Stress Test, a standardized psychosocial stress test.26 It consists of the subject giving a 10-minute speech and performing a mental arithmetic task in front of a panel of stern-appearing evaluators. Variables measured include heart rate, plasma ACTH, and cortisol concentration at intervals before and after the performance component of the test. The four groups in this study included:
- women without psychiatric illness or history of ELS serving as a control group (CON)
- depressed women without a history of ELS (non-ELS/MDD)
- depressed women with a history of ELS (ELS/MDD)
- non-depressed women with a history of ELS (ELS/non-MDD).
The largest ACTH and cortisol responses and increases in heart rate following this stress exposure were seen in the ELS/MDD group. In fact, the ACTH response of these women was more than 6 times greater than that observed in the control group. The ELS/MDD group of women also had greater rates of comorbid PTSD (85%) in comparison to the other experimental groups as well.
These data are consistent with the hypothesis that ELS produces enduring sensitization of the HPA axis and autonomic nervous system response to stress. This phenomenon may constitute an important etiological element in the development of stress-related adult psychiatric illnesses such as depression or PTSD.
To further explore the hypothesis that ELS alters set points of the HPA axis, we sought to characterize the effects of standard HPA axis challenge tests (CRF stimulation test and ACTH1-24 stimulation test) in a similar population of women.27 Depressed women with a history of ELS and depressed women without a history of ELS both exhibited a blunted ACTH response to infusion of exogenous CRF. Conversely, women with a history of ELS but without current depression had an increased ACTH response following CRF infusion.
With respect to the ACTH1-24 stimulation test, abused women who were not depressed had lower plasma cortisol levels at baseline and after administration of ACTH1-24. Similar to the findings of our previous study,26 women with MDD and a history of ELS were more likely to report current life stress and to also have comorbid PTSD than women with ELS who were not depressed. Blunting of the ACTH response to exogenous CRF in depressed women with a history of ELS may in part be secondary to acute downregulation of pituitary CRF receptors as a result of chronic CRF hypersecretion.
More recently, Carpenter and colleagues28 evaluated the relationship between the perception of ELS and CSF CRF in patients with depression and healthy control subjects. The perception of ELS predicted CSF CRF concentration independent of the presence or absence of depression. Further, and most interestingly, the developmental timing of the stress exposure was predictive of either relatively increased or decreased CSF CRF. ELS before age 6 was associated with elevated CSF CRF, whereas perinatal and preteen exposure to stressful events was associated with decreased CSF CRF.
Brain structure changes? In addition to the neuroendocrine changes observed in patients with ELS, there is evidence that ELS may also alter brain structure. Reduced hippocampal volume is found in some but not all patients with unipolar depression.29 In patients with a history of depression who also have hippocampal atrophy, the extent of atrophy is greater in patients with higher total lifetime duration of depression.30,31
Patients with ELS also have been found to have decreased hippocampal volume.32,33 However, previous structural imaging studies have not controlled for the presence of ELS when attempting to determine the relationship between depression and structural changes in the hippocampus, and this methodologic confound may explain in part the inconsistent relationship between altered hippocampal volume and depression.
To evaluate this hypothesis, hippocampal volume was measured in depressed women with and without a history of ELS and in a control group of women. Reduced hippocampal volume was found to occur solely in depressed women with a history of ELS. Depressed women without ELS and women from the control group had similar hippocampal volumes.34 These data suggest that previous reports of reduced hippocampal size in patients with depression may in fact be related to a history of ELS rather than depression.
Treatment implications
The data discussed in this paper indicate that patients with depression and a history of ELS may constitute a unique subgroup among depressed patients as a whole. A growing body of evidence suggests that depressed patients with ELS may also be unique with respect to their response to treatment.
ELS has been found to impact the clinical response of patients to pharmacotherapy with either dysthymia or depression.35,36 Further, patients with depression and a history of ELS have been reported to exhibit increased rates of relapse following treatment of depression.37 The course of depression in individuals with ELS is often characterized by chronicity.
ELS and therapeutic response, Recently, our group has sought to determine whether ELS in patients with chronic depression moderates their response to pharmacotherapy or psychotherapy.38 In this study, data from a large multicenter trial39 originally designed to compare the relative efficacy of pharmacotherapy (nefazodone), psychotherapy (Cognitive Behavioral Analysis System of Psychotherapy), or their combination in the treatment of chronic depression was reanalyzed by stratifying patients based on the presence or absence of ELS. In the overall sample of patients with chronic depression, psychotherapy and pharmacotherapy were comparable in efficacy but significantly less effective than their combination.
ELS in chronically depressed patients was highly prevalent. Approximately one-third experienced loss of a parent before age 15, 45% experienced childhood physical abuse, 16% experienced childhood sexual abuse, and 10% experienced neglect. Most significantly, depressed patients with a history of ELS had a superior response to psychotherapy alone compared with antidepressant monotherapy. In addition, combination therapy was only slightly more effective than psychotherapy alone in the group of depressed patients with ELS.
These data suggest that ELS is common in the population of patients with chronic depression and that psychotherapy is a critical element in the treatment of depressed patients with ELS40 (Box). However, it will be important in future studies to ascertain whether the differences in treatment response for psychotherapy compared with antidepressant in patients with ELS and depression are able to be replicated with the SSRI class of antidepressants.
Assessment of trauma and neglect should be a standard component of the diagnostic interview. Patients with a history of early life stress (ELS) may present for treatment with complaints that represent depression, anxiety, or substance abuse, but they may also have complicated presentations involving psychotic or dissociative symptoms, reflecting the diagnostic comorbidity in this population.
How to identify ELS. No standardized office-based screening tools exist for ELS, and clinical interviewing is the primary means of assessing exposure to ELS. A common error in history-taking with this population, particularly in high-volume settings, is to merely ask patients whether they were abused or neglected as children or to elaborate only very slightly on this aspect of the history. We risk not finding information that is critical to understanding our patients if we assume they share a common definition of abuse and neglect with us, can recognize such events in their personal history, and are willing to share that information with us.
When framing questions about abuse or neglect, it is important to remember that our own sense of what constitutes neglect or abuse may be very different from what a patient thinks of as neglect or abuse. For example, some patients may not consider their experience as a child abusive because of a distorted sense of responsibility, possibly further exaggerated by comorbid depression (ie, “My parents locked me in the closet overnight all the time when I was a child because I deserved it.”)
Other patients may try to minimize the impact of the experience or the responsibility of the perpetrator and attempt to normalize it (ie, “My uncle used to touch me between my legs in the swimming pool but he didn’t mean anything by it; he did it to everybody.”)
Avoiding ‘false memories.’ As important as it is to identify abuse or neglect when it has occurred, it is equally important to avoid intensifying the impact of an incident of abuse or, worse, creating a “false memory” of abuse in suggestible patients (bearing in mind that there is no definitive way to exclude the presence of “suggestibility” in patients). Our task as clinicians is to help patients correctly identify experiences of abuse and neglect and understand their response to these experiences clinically to facilitate case formulation and a treatment plan.
Creating a therapeutic alliance. Abuse and neglect during early life fundamentally alter the core assumptions that patients have about trust and safety in their relationships with others. Not only does this potentially impact the disclosure by patients of the nature and extent of trauma they have experienced, but it can also slow the formation of an effective therapeutic alliance.
To that end, asking open-ended questions about neglect and specific forms of abuse, creating an atmosphere of safety and trust, and a warm, empathic, nonjudgmental manner are central to the accurate assessment of ELS and provide the foundation for treatment by establishing an effective therapeutic alliance.
Optimal treatment. No published clinical trials have specifically compared the relative efficacy of particular forms of psychotherapy or pharmacotherapy for depressed patients with a history of ELS. However, it is clear from the available data that psychotherapy should be considered a core component of treatment for these patients.
Because psychiatric comorbidity is common in these patients, their treatment should be individualized in a manner that accounts for and addresses depression as well as associated diagnoses such as panic disorder or posttraumatic stress disorder (PTSD) with disorder-specific psychotherapy. Pharmacotherapy in combination with psychotherapy may also be helpful to patients with ELS and depression, though definitive data are lacking.
Judicious combination of medications such as antidepressants and benzodiazepines, particularly in patients with comorbid panic or PTSD, in concert with psychotherapy probably constitutes optimal treatment.
Related resources
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- National Clearinghouse on Child Abuse and Neglect. http://nccanch.acf.hhs.gov.
- Charney DS, Nemeroff CB. The peace of mind prescription: An authoritative guide to finding the most effective treatment for anxiety and depression. Boston: Houghton Mifflin, 2004.
Drug brand name
- Nefazodone • Serzone
Disclosure
Dr. Gillespie reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Nemeroff receives research grants from or is a consultant/speaker for Abbott Laboratories, Acadia Pharmaceuticals, American Foundation for Suicide Prevention, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Corcept Therapeutics, Cyberonics, Cypress Biosciences, Eli Lilly and Co., Forest Laboratories, GlaxoSmithKline, Janssen Pharmaceutica, Merck & Co., Neurocrine Biosciences, Otsuka Inc., Pfizer Inc., Sanofi Aventis, and Wyeth.
Acknowledgement
Supported by NIH MH-42088, MH-52899 (CBN), and NIH NCRRM01-RR00039 to Emory University.
1. Sedlack AJ, Broadhurst DD. Third National Incidence Study of Child Abuse and Neglect. Washington, DC: US Department of Health and Human Services; 1996.
2. McCauley J, Kern DE, Kolodner K, et al. Clinical characteristics of women with a history of child abuse: unhealed wounds. JAMA 1997;277(17):1362-8.
3. Dube SR, Anda RF, Felitti VJ, et al. Childhood abuse, household dysfunction, and the risk of attempted suicide throughout the life span: findings from the adverse childhood experiences study. JAMA 2001;286(24):3089-96.
4. Chapman DP, Whitfield CL, Felitti VJ, et al. Adverse childhood experiences and the risk of depressive disorders in adulthood. J Affect Disord 2004;82(2):217-25.
5. Gladstone GL, Parker GB, Mitchell PB, et al. Implications of childhood trauma for depressed women: an analysis of pathways from childhood sexual abuse to deliberate self-harm and revictimization. Am J Psychiatry 2004;161(8):1417-25.
6. Goodwin RD, Stein MB. Association between childhood trauma and physical disorders among adults in the United States. Psychol Med 2004;34(3):509-20.
7. Heim C, Plotsky PM, Nemeroff CB. Importance of studying the contributions of early adverse experience to neurobiological findings in depression. Neuropsychopharmacology 2004;29(4):641-8.
8. Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 1983;36(3):165-86.
9. Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol 1999;160(1):1-12.
10. Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotrophin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev 1990;15(2):71-100.
11. Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotrophin-releasing factor. Pharmacol Rev 1991;43(4):425-73.
12. Nemeroff CB, Widerlov E, Bissette G, et al. Elevated concentrations of CSF corticotrophin-releasing factor-like immunoreactivity in depressed patients. Science 1984;226(4680):1342-4.
13. Hartline KM, Owens MJ, Nemeroff CB. Postmortem and cerebrospinal fluid studies of corticotropin-releasing factor in humans. Ann NY Acad Sci 1996;780:96-105.
14. Bremner JD, Licinio J, Darnell A, et al. Elevated CSF corticotrophin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 1997;154(5):624-9.
15. Baker DG, West SA, Nicholson WE, et al. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 1999;156(4):585-8.
16. Nemeroff CB, Owens MJ, Bissette G, et al. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry 1988;45(6):577-9.
17. Merali Z, Du L, Hrdina P, et al. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci 2004;24(6):1478-85.
18. Arato M, Banki CM, Bissette G, Nemeroff CB. Elevated CSF CRF in suicide victims. Biol Psychiatry 1989;25(3):355-9.
19. Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000;157(10):1552-62.
20. Hasler G, Drevets WC, Manji HK, Charney DS. Discovering endophenotypes for major depression. Neuropsychopharmacology 2004;29(10):1765-81.
21. Gutman DA, Nemeroff CB. Persistent central nervous system effects of an adverse early environment: clinical and preclinical studies. Physiol Behav 2003;79(3):471-8.
22. Kendler KS, Kuhn JW, Prescott CA. Childhood sexual abuse, stressful life events and risk for major depression in women. Psychol Med 2004;34(8):1475-82.
23. Caspi A, Sugden K, Moffitt TE. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003;301(5631):386-9.
24. Kaufman J, Yang BZ, Douglas-Palumberi H, et al. Social supports and serotonin transporter gene moderate depression in maltreated children. Proc Natl Acad Sci USA 2004;101(49):17316-21.
25. Maercker A, Michael T, Fehm L, et al. Age of traumatisation as a predictor of post-traumatic stress disorder or major depression in young women. Br J Psychiatry 2004;184:482-7.
26. Heim C, Newport DJ, Heit S, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA 2000;284(5):592-7.
27. Heim C, Newport DJ, Bonsall R, et al. Altered pituitary-adrenal axis responses to provocative challenge tests in adult survivors of childhood abuse. Am J Psychiatry 2001;158(4):575-81.
28. Carpenter LL, Tyrka AR, McDougle CJ, et al. Cerebrospinal fluid corticotropin-releasing factor and perceived early-life stress in depressed patients and healthy control subjects. Neuropsychopharmacology 2004;29(4):777-84.
29. Campbell S, Macqueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci 2004;29(6):417-26.
30. Sheline YI, Wang PW, Gado MH, et al. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 1996;93(9):3908-13.
31. Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci 1999;19(12):5034-43.
32. Stein MB, Koverola C, Hanna C, et al. Hippocampal volume in women victimized by childhood sexual abuse. Psychol Med 1997;27(4):951-9.
33. Driessen M, Herrmann J, Stahl K, et al. Magnetic resonance imaging volumes of the hippocampus and the amygdala in women with borderline personality disorder and early traumatization. Arch Gen Psychiatry 2000;57(12):1115-22.
34. Vythilingam M, Heim C, Newport DJ, et al. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry 2002;159(12):2072-80.
35. Hayden EP, Klein DM. Outcome of dysthymic disorder at 5-year follow-up: the effect of familial psychopathology, early adversity, personality, comorbidity, and chronic stress. Am J Psychiatry 2001;158(11):1864-70.
36. Kaplan MJ, Klinetob NA. Childhood emotional trauma and chronic posttraumatic stress disorder in adult outpatients with treatment-resistant depression. J Nerv Ment Dis 2000;188(9):596-601.
37. Lara ME, Klein DN, Kasch KL. Psychosocial predictors of the short-term course and outcome of major depression: a longitudinal study of a nonclinical sample with recent-onset episodes. J Abnorm Psychol 2000;109(4):644-50.
38. Nemeroff CB, Heim CM, Thase ME, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci U S A 2003;100(24):14293-6.
39. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342(20):1462-70.
40. Craighead WE, Nemeroff CB. The impact of early trauma on response to psychotherapy. Clin Neurosci Res 2005;4(5-6):405-11.
Sadly, parental neglect and child abuse are very common in the United States and worldwide. Patients abused or exploited in childhood, who experience neglect or the loss of a parent during childhood, are haunted by these experiences.
Considerable evidence from laboratory animal and clinical studies indicate that stressful or traumatic events early in development have long-lasting effects on brain development. In particular, the neural and endocrine systems mediating the response to stress exhibit persistent alterations after adverse childhood events.
Clinically, patients with a history of childhood trauma often struggle with variable symptom complexes including both depression and anxiety. In this article, we review evidence that depression in patients with a history of early life stress (ELS) is biologically and clinically distinct from depression in patients without childhood abuse or neglect.
Data linking ELS to depression
Conservative estimates suggest that every year in the United States more than 1 million children are exposed to sexual or physical abuse or severe neglect.1 Unfortunately, this is only the tip of the iceberg. Emotional abuse is by definition comorbid with sexual and physical abuse and may occur alone at even higher rates.
Psychiatric sequelae of child abuse have been studied in adult survivors in considerable detail (Table). Women abused as children report greater numbers of depression, anxiety, somatic, and substance abuse symptoms compared with women without such a history.2 Not only are these women at increased risk for attempted suicide, but they attempt suicide at a rate that is proportional to the number of early life traumatic events that occurred during childhood.3 Men also are at increased risk for depression in the wake of child abuse.4
Sexual abuse in particular is a marker of especially severe childhood trauma. Depressed women who were sexually abused as children report more childhood physical abuse, childhood emotional abuse, parental conflict, and an earlier onset of depression than depressed women without a history of sexual abuse.5
Finally, recent data drawn from the National Comorbidity Survey suggest that child abuse and neglect may independently elevate risk for several stress-related diseases including cardiac disease, peptic ulcer, autoimmune disease, diabetes mellitus, and lung disease.6
Table
Early life stress as a risk factor for mood and anxiety disorders
| Child abuse and neglect are predictors of episodes of major depression in identical twins |
| Women with a history of childhood abuse are more than twice as likely to develop depression as non-abused women |
| Childhood abuse is related to the development of anxiety disorders in adulthood |
| Childhood physical abuse predisposes for combat-related posttraumatic stress disorder (PTSD) |
| Stress early in life may induce a vulnerability to stress later in life, resulting in an increased risk for stress-related disorders |
Depression and the biology of stress
Preclinical research using laboratory animals and clinical research with humans has provided significant insight into the relationship between the pathophysiology of depression and the neurobiology of stress. A burgeoning database suggests that disruption of the neural systems mediating the stress response plays a significant role in the etiology of certain forms of depression and anxiety.7 Much of this work has focused on the preeminent role of corticotropin-releasing factor (CRF) in this process (Figure).
CRF is one of the principal mediators of the mammalian stress response. One CRF system is composed of neurons of the paraventricular nuclei of the hypothalamus that project nerve terminals to the median eminence, where they secrete CRF into the hypophyseal portal system. CRF is then transported within the portal system to the anterior pituitary where it acts on corticotrophs to increase adrenocorticotrophic hormone (ACTH) secretion, thereby controlling hypothalamic-pituitary-adrenal (HPA) axis activity.8 CRF is also found in extrahypothalamic brain areas where it functions, in concert with the hypothalamic CRF system, as a neurotransmitter in coordinating the behavioral, autonomic, and immune responses to stress.9
Direct central nervous system (CNS) administration of CRF in laboratory animals, typically rodents or nonhuman primates, results in activation of the autonomic nervous system leading to elevation of peripheral catecholamines, modification of gastrointestinal activity, increased heart rate and increased blood pressure. In addition, changes in behavior similar to those observed in human depression occur, including disturbed sleep patterns, reduced food intake, decreased reproductive behavior, and enhanced fear conditioning.10,11
In humans, elevated CRF concentrations are found in the cerebrospinal fluid (CSF) of patients with depression12,13 and of combat veterans with posttraumatic stress disorder (PTSD).14,15 Further, postmortem studies of suicide victims have revealed decreased density of CRF receptors in the frontal cortex,16 decreased expression of CRF receptor mRNA and increased CRF concentrations in the frontal cortex when compared with controls,17 and increased concentrations of cisternal CSF CRF.18 Collectively, these clinical data are consistent with the hypothesis that CRF is chronically hypersecreted in patients with depression or PTSD.
Figure Biological and behavioral effects of chronic CRF hypersecretion
A distinct ‘ELS depression’?
Depression has a complex etiology based on interacting contributions from genes and the environment19 that may ultimately result in biologically and clinically distinct forms of depression.20 Exposure to stress, particularly during neurobiologically vulnerable periods of development, may be one means whereby the environment influences the development of depression in genetically susceptible individuals.21
Heredity. Kendler and colleagues22 studied 1,404 female adult twins and observed that childhood sexual abuse was associated with both an increased risk for major depression and a marked increased sensitivity to the depressogenic effects of stressful life events. Moreover, research in human gene-environment interactions has identified a functional polymorphism in the promoter region of the gene for the serotonin transporter which appears to moderate the influence of stressful life events on the development of depression and potential for suicide.23,24
Environment. Similarly, a key variable in determining the clinical outcome of childhood trauma may be the developmental timing of the abuse. Women abused before age 13 are at equivalent risk for developing PTSD or major depressive disorder (MDD), whereas women abused after age 13 are more likely to develop PTSD.25
Thus a major challenge in depression research is to understand the biological mechanisms that mediate the effects of trauma during development through the genetic windows of vulnerability and resilience.
Animal models of ELS have been studied to elucidate the neurobiological consequences of early life trauma in adult humans. This work has largely been performed in rodents and nonhuman primates using a variety of experimental paradigms.
Although a comprehensive review of these data is beyond the scope of this article, ELS in laboratory animals has consistently been found to produce both short- and long-term adverse neurobiological and endocrine effects as well as cognitive dysfunction and abnormal behavior.21 One possible mechanism mediating these effects is a persistent hyperresponsiveness of different components of the HPA axis following exposure to stress.
Studies in adult women have sought to uncover the long-term effects of ELS (prepubertal physical or sexual abuse) on reactivity of the HPA axis in response to the Trier Social Stress Test, a standardized psychosocial stress test.26 It consists of the subject giving a 10-minute speech and performing a mental arithmetic task in front of a panel of stern-appearing evaluators. Variables measured include heart rate, plasma ACTH, and cortisol concentration at intervals before and after the performance component of the test. The four groups in this study included:
- women without psychiatric illness or history of ELS serving as a control group (CON)
- depressed women without a history of ELS (non-ELS/MDD)
- depressed women with a history of ELS (ELS/MDD)
- non-depressed women with a history of ELS (ELS/non-MDD).
The largest ACTH and cortisol responses and increases in heart rate following this stress exposure were seen in the ELS/MDD group. In fact, the ACTH response of these women was more than 6 times greater than that observed in the control group. The ELS/MDD group of women also had greater rates of comorbid PTSD (85%) in comparison to the other experimental groups as well.
These data are consistent with the hypothesis that ELS produces enduring sensitization of the HPA axis and autonomic nervous system response to stress. This phenomenon may constitute an important etiological element in the development of stress-related adult psychiatric illnesses such as depression or PTSD.
To further explore the hypothesis that ELS alters set points of the HPA axis, we sought to characterize the effects of standard HPA axis challenge tests (CRF stimulation test and ACTH1-24 stimulation test) in a similar population of women.27 Depressed women with a history of ELS and depressed women without a history of ELS both exhibited a blunted ACTH response to infusion of exogenous CRF. Conversely, women with a history of ELS but without current depression had an increased ACTH response following CRF infusion.
With respect to the ACTH1-24 stimulation test, abused women who were not depressed had lower plasma cortisol levels at baseline and after administration of ACTH1-24. Similar to the findings of our previous study,26 women with MDD and a history of ELS were more likely to report current life stress and to also have comorbid PTSD than women with ELS who were not depressed. Blunting of the ACTH response to exogenous CRF in depressed women with a history of ELS may in part be secondary to acute downregulation of pituitary CRF receptors as a result of chronic CRF hypersecretion.
More recently, Carpenter and colleagues28 evaluated the relationship between the perception of ELS and CSF CRF in patients with depression and healthy control subjects. The perception of ELS predicted CSF CRF concentration independent of the presence or absence of depression. Further, and most interestingly, the developmental timing of the stress exposure was predictive of either relatively increased or decreased CSF CRF. ELS before age 6 was associated with elevated CSF CRF, whereas perinatal and preteen exposure to stressful events was associated with decreased CSF CRF.
Brain structure changes? In addition to the neuroendocrine changes observed in patients with ELS, there is evidence that ELS may also alter brain structure. Reduced hippocampal volume is found in some but not all patients with unipolar depression.29 In patients with a history of depression who also have hippocampal atrophy, the extent of atrophy is greater in patients with higher total lifetime duration of depression.30,31
Patients with ELS also have been found to have decreased hippocampal volume.32,33 However, previous structural imaging studies have not controlled for the presence of ELS when attempting to determine the relationship between depression and structural changes in the hippocampus, and this methodologic confound may explain in part the inconsistent relationship between altered hippocampal volume and depression.
To evaluate this hypothesis, hippocampal volume was measured in depressed women with and without a history of ELS and in a control group of women. Reduced hippocampal volume was found to occur solely in depressed women with a history of ELS. Depressed women without ELS and women from the control group had similar hippocampal volumes.34 These data suggest that previous reports of reduced hippocampal size in patients with depression may in fact be related to a history of ELS rather than depression.
Treatment implications
The data discussed in this paper indicate that patients with depression and a history of ELS may constitute a unique subgroup among depressed patients as a whole. A growing body of evidence suggests that depressed patients with ELS may also be unique with respect to their response to treatment.
ELS has been found to impact the clinical response of patients to pharmacotherapy with either dysthymia or depression.35,36 Further, patients with depression and a history of ELS have been reported to exhibit increased rates of relapse following treatment of depression.37 The course of depression in individuals with ELS is often characterized by chronicity.
ELS and therapeutic response, Recently, our group has sought to determine whether ELS in patients with chronic depression moderates their response to pharmacotherapy or psychotherapy.38 In this study, data from a large multicenter trial39 originally designed to compare the relative efficacy of pharmacotherapy (nefazodone), psychotherapy (Cognitive Behavioral Analysis System of Psychotherapy), or their combination in the treatment of chronic depression was reanalyzed by stratifying patients based on the presence or absence of ELS. In the overall sample of patients with chronic depression, psychotherapy and pharmacotherapy were comparable in efficacy but significantly less effective than their combination.
ELS in chronically depressed patients was highly prevalent. Approximately one-third experienced loss of a parent before age 15, 45% experienced childhood physical abuse, 16% experienced childhood sexual abuse, and 10% experienced neglect. Most significantly, depressed patients with a history of ELS had a superior response to psychotherapy alone compared with antidepressant monotherapy. In addition, combination therapy was only slightly more effective than psychotherapy alone in the group of depressed patients with ELS.
These data suggest that ELS is common in the population of patients with chronic depression and that psychotherapy is a critical element in the treatment of depressed patients with ELS40 (Box). However, it will be important in future studies to ascertain whether the differences in treatment response for psychotherapy compared with antidepressant in patients with ELS and depression are able to be replicated with the SSRI class of antidepressants.
Assessment of trauma and neglect should be a standard component of the diagnostic interview. Patients with a history of early life stress (ELS) may present for treatment with complaints that represent depression, anxiety, or substance abuse, but they may also have complicated presentations involving psychotic or dissociative symptoms, reflecting the diagnostic comorbidity in this population.
How to identify ELS. No standardized office-based screening tools exist for ELS, and clinical interviewing is the primary means of assessing exposure to ELS. A common error in history-taking with this population, particularly in high-volume settings, is to merely ask patients whether they were abused or neglected as children or to elaborate only very slightly on this aspect of the history. We risk not finding information that is critical to understanding our patients if we assume they share a common definition of abuse and neglect with us, can recognize such events in their personal history, and are willing to share that information with us.
When framing questions about abuse or neglect, it is important to remember that our own sense of what constitutes neglect or abuse may be very different from what a patient thinks of as neglect or abuse. For example, some patients may not consider their experience as a child abusive because of a distorted sense of responsibility, possibly further exaggerated by comorbid depression (ie, “My parents locked me in the closet overnight all the time when I was a child because I deserved it.”)
Other patients may try to minimize the impact of the experience or the responsibility of the perpetrator and attempt to normalize it (ie, “My uncle used to touch me between my legs in the swimming pool but he didn’t mean anything by it; he did it to everybody.”)
Avoiding ‘false memories.’ As important as it is to identify abuse or neglect when it has occurred, it is equally important to avoid intensifying the impact of an incident of abuse or, worse, creating a “false memory” of abuse in suggestible patients (bearing in mind that there is no definitive way to exclude the presence of “suggestibility” in patients). Our task as clinicians is to help patients correctly identify experiences of abuse and neglect and understand their response to these experiences clinically to facilitate case formulation and a treatment plan.
Creating a therapeutic alliance. Abuse and neglect during early life fundamentally alter the core assumptions that patients have about trust and safety in their relationships with others. Not only does this potentially impact the disclosure by patients of the nature and extent of trauma they have experienced, but it can also slow the formation of an effective therapeutic alliance.
To that end, asking open-ended questions about neglect and specific forms of abuse, creating an atmosphere of safety and trust, and a warm, empathic, nonjudgmental manner are central to the accurate assessment of ELS and provide the foundation for treatment by establishing an effective therapeutic alliance.
Optimal treatment. No published clinical trials have specifically compared the relative efficacy of particular forms of psychotherapy or pharmacotherapy for depressed patients with a history of ELS. However, it is clear from the available data that psychotherapy should be considered a core component of treatment for these patients.
Because psychiatric comorbidity is common in these patients, their treatment should be individualized in a manner that accounts for and addresses depression as well as associated diagnoses such as panic disorder or posttraumatic stress disorder (PTSD) with disorder-specific psychotherapy. Pharmacotherapy in combination with psychotherapy may also be helpful to patients with ELS and depression, though definitive data are lacking.
Judicious combination of medications such as antidepressants and benzodiazepines, particularly in patients with comorbid panic or PTSD, in concert with psychotherapy probably constitutes optimal treatment.
Related resources
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- National Clearinghouse on Child Abuse and Neglect. http://nccanch.acf.hhs.gov.
- Charney DS, Nemeroff CB. The peace of mind prescription: An authoritative guide to finding the most effective treatment for anxiety and depression. Boston: Houghton Mifflin, 2004.
Drug brand name
- Nefazodone • Serzone
Disclosure
Dr. Gillespie reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Nemeroff receives research grants from or is a consultant/speaker for Abbott Laboratories, Acadia Pharmaceuticals, American Foundation for Suicide Prevention, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Corcept Therapeutics, Cyberonics, Cypress Biosciences, Eli Lilly and Co., Forest Laboratories, GlaxoSmithKline, Janssen Pharmaceutica, Merck & Co., Neurocrine Biosciences, Otsuka Inc., Pfizer Inc., Sanofi Aventis, and Wyeth.
Acknowledgement
Supported by NIH MH-42088, MH-52899 (CBN), and NIH NCRRM01-RR00039 to Emory University.
Sadly, parental neglect and child abuse are very common in the United States and worldwide. Patients abused or exploited in childhood, who experience neglect or the loss of a parent during childhood, are haunted by these experiences.
Considerable evidence from laboratory animal and clinical studies indicate that stressful or traumatic events early in development have long-lasting effects on brain development. In particular, the neural and endocrine systems mediating the response to stress exhibit persistent alterations after adverse childhood events.
Clinically, patients with a history of childhood trauma often struggle with variable symptom complexes including both depression and anxiety. In this article, we review evidence that depression in patients with a history of early life stress (ELS) is biologically and clinically distinct from depression in patients without childhood abuse or neglect.
Data linking ELS to depression
Conservative estimates suggest that every year in the United States more than 1 million children are exposed to sexual or physical abuse or severe neglect.1 Unfortunately, this is only the tip of the iceberg. Emotional abuse is by definition comorbid with sexual and physical abuse and may occur alone at even higher rates.
Psychiatric sequelae of child abuse have been studied in adult survivors in considerable detail (Table). Women abused as children report greater numbers of depression, anxiety, somatic, and substance abuse symptoms compared with women without such a history.2 Not only are these women at increased risk for attempted suicide, but they attempt suicide at a rate that is proportional to the number of early life traumatic events that occurred during childhood.3 Men also are at increased risk for depression in the wake of child abuse.4
Sexual abuse in particular is a marker of especially severe childhood trauma. Depressed women who were sexually abused as children report more childhood physical abuse, childhood emotional abuse, parental conflict, and an earlier onset of depression than depressed women without a history of sexual abuse.5
Finally, recent data drawn from the National Comorbidity Survey suggest that child abuse and neglect may independently elevate risk for several stress-related diseases including cardiac disease, peptic ulcer, autoimmune disease, diabetes mellitus, and lung disease.6
Table
Early life stress as a risk factor for mood and anxiety disorders
| Child abuse and neglect are predictors of episodes of major depression in identical twins |
| Women with a history of childhood abuse are more than twice as likely to develop depression as non-abused women |
| Childhood abuse is related to the development of anxiety disorders in adulthood |
| Childhood physical abuse predisposes for combat-related posttraumatic stress disorder (PTSD) |
| Stress early in life may induce a vulnerability to stress later in life, resulting in an increased risk for stress-related disorders |
Depression and the biology of stress
Preclinical research using laboratory animals and clinical research with humans has provided significant insight into the relationship between the pathophysiology of depression and the neurobiology of stress. A burgeoning database suggests that disruption of the neural systems mediating the stress response plays a significant role in the etiology of certain forms of depression and anxiety.7 Much of this work has focused on the preeminent role of corticotropin-releasing factor (CRF) in this process (Figure).
CRF is one of the principal mediators of the mammalian stress response. One CRF system is composed of neurons of the paraventricular nuclei of the hypothalamus that project nerve terminals to the median eminence, where they secrete CRF into the hypophyseal portal system. CRF is then transported within the portal system to the anterior pituitary where it acts on corticotrophs to increase adrenocorticotrophic hormone (ACTH) secretion, thereby controlling hypothalamic-pituitary-adrenal (HPA) axis activity.8 CRF is also found in extrahypothalamic brain areas where it functions, in concert with the hypothalamic CRF system, as a neurotransmitter in coordinating the behavioral, autonomic, and immune responses to stress.9
Direct central nervous system (CNS) administration of CRF in laboratory animals, typically rodents or nonhuman primates, results in activation of the autonomic nervous system leading to elevation of peripheral catecholamines, modification of gastrointestinal activity, increased heart rate and increased blood pressure. In addition, changes in behavior similar to those observed in human depression occur, including disturbed sleep patterns, reduced food intake, decreased reproductive behavior, and enhanced fear conditioning.10,11
In humans, elevated CRF concentrations are found in the cerebrospinal fluid (CSF) of patients with depression12,13 and of combat veterans with posttraumatic stress disorder (PTSD).14,15 Further, postmortem studies of suicide victims have revealed decreased density of CRF receptors in the frontal cortex,16 decreased expression of CRF receptor mRNA and increased CRF concentrations in the frontal cortex when compared with controls,17 and increased concentrations of cisternal CSF CRF.18 Collectively, these clinical data are consistent with the hypothesis that CRF is chronically hypersecreted in patients with depression or PTSD.
Figure Biological and behavioral effects of chronic CRF hypersecretion
A distinct ‘ELS depression’?
Depression has a complex etiology based on interacting contributions from genes and the environment19 that may ultimately result in biologically and clinically distinct forms of depression.20 Exposure to stress, particularly during neurobiologically vulnerable periods of development, may be one means whereby the environment influences the development of depression in genetically susceptible individuals.21
Heredity. Kendler and colleagues22 studied 1,404 female adult twins and observed that childhood sexual abuse was associated with both an increased risk for major depression and a marked increased sensitivity to the depressogenic effects of stressful life events. Moreover, research in human gene-environment interactions has identified a functional polymorphism in the promoter region of the gene for the serotonin transporter which appears to moderate the influence of stressful life events on the development of depression and potential for suicide.23,24
Environment. Similarly, a key variable in determining the clinical outcome of childhood trauma may be the developmental timing of the abuse. Women abused before age 13 are at equivalent risk for developing PTSD or major depressive disorder (MDD), whereas women abused after age 13 are more likely to develop PTSD.25
Thus a major challenge in depression research is to understand the biological mechanisms that mediate the effects of trauma during development through the genetic windows of vulnerability and resilience.
Animal models of ELS have been studied to elucidate the neurobiological consequences of early life trauma in adult humans. This work has largely been performed in rodents and nonhuman primates using a variety of experimental paradigms.
Although a comprehensive review of these data is beyond the scope of this article, ELS in laboratory animals has consistently been found to produce both short- and long-term adverse neurobiological and endocrine effects as well as cognitive dysfunction and abnormal behavior.21 One possible mechanism mediating these effects is a persistent hyperresponsiveness of different components of the HPA axis following exposure to stress.
Studies in adult women have sought to uncover the long-term effects of ELS (prepubertal physical or sexual abuse) on reactivity of the HPA axis in response to the Trier Social Stress Test, a standardized psychosocial stress test.26 It consists of the subject giving a 10-minute speech and performing a mental arithmetic task in front of a panel of stern-appearing evaluators. Variables measured include heart rate, plasma ACTH, and cortisol concentration at intervals before and after the performance component of the test. The four groups in this study included:
- women without psychiatric illness or history of ELS serving as a control group (CON)
- depressed women without a history of ELS (non-ELS/MDD)
- depressed women with a history of ELS (ELS/MDD)
- non-depressed women with a history of ELS (ELS/non-MDD).
The largest ACTH and cortisol responses and increases in heart rate following this stress exposure were seen in the ELS/MDD group. In fact, the ACTH response of these women was more than 6 times greater than that observed in the control group. The ELS/MDD group of women also had greater rates of comorbid PTSD (85%) in comparison to the other experimental groups as well.
These data are consistent with the hypothesis that ELS produces enduring sensitization of the HPA axis and autonomic nervous system response to stress. This phenomenon may constitute an important etiological element in the development of stress-related adult psychiatric illnesses such as depression or PTSD.
To further explore the hypothesis that ELS alters set points of the HPA axis, we sought to characterize the effects of standard HPA axis challenge tests (CRF stimulation test and ACTH1-24 stimulation test) in a similar population of women.27 Depressed women with a history of ELS and depressed women without a history of ELS both exhibited a blunted ACTH response to infusion of exogenous CRF. Conversely, women with a history of ELS but without current depression had an increased ACTH response following CRF infusion.
With respect to the ACTH1-24 stimulation test, abused women who were not depressed had lower plasma cortisol levels at baseline and after administration of ACTH1-24. Similar to the findings of our previous study,26 women with MDD and a history of ELS were more likely to report current life stress and to also have comorbid PTSD than women with ELS who were not depressed. Blunting of the ACTH response to exogenous CRF in depressed women with a history of ELS may in part be secondary to acute downregulation of pituitary CRF receptors as a result of chronic CRF hypersecretion.
More recently, Carpenter and colleagues28 evaluated the relationship between the perception of ELS and CSF CRF in patients with depression and healthy control subjects. The perception of ELS predicted CSF CRF concentration independent of the presence or absence of depression. Further, and most interestingly, the developmental timing of the stress exposure was predictive of either relatively increased or decreased CSF CRF. ELS before age 6 was associated with elevated CSF CRF, whereas perinatal and preteen exposure to stressful events was associated with decreased CSF CRF.
Brain structure changes? In addition to the neuroendocrine changes observed in patients with ELS, there is evidence that ELS may also alter brain structure. Reduced hippocampal volume is found in some but not all patients with unipolar depression.29 In patients with a history of depression who also have hippocampal atrophy, the extent of atrophy is greater in patients with higher total lifetime duration of depression.30,31
Patients with ELS also have been found to have decreased hippocampal volume.32,33 However, previous structural imaging studies have not controlled for the presence of ELS when attempting to determine the relationship between depression and structural changes in the hippocampus, and this methodologic confound may explain in part the inconsistent relationship between altered hippocampal volume and depression.
To evaluate this hypothesis, hippocampal volume was measured in depressed women with and without a history of ELS and in a control group of women. Reduced hippocampal volume was found to occur solely in depressed women with a history of ELS. Depressed women without ELS and women from the control group had similar hippocampal volumes.34 These data suggest that previous reports of reduced hippocampal size in patients with depression may in fact be related to a history of ELS rather than depression.
Treatment implications
The data discussed in this paper indicate that patients with depression and a history of ELS may constitute a unique subgroup among depressed patients as a whole. A growing body of evidence suggests that depressed patients with ELS may also be unique with respect to their response to treatment.
ELS has been found to impact the clinical response of patients to pharmacotherapy with either dysthymia or depression.35,36 Further, patients with depression and a history of ELS have been reported to exhibit increased rates of relapse following treatment of depression.37 The course of depression in individuals with ELS is often characterized by chronicity.
ELS and therapeutic response, Recently, our group has sought to determine whether ELS in patients with chronic depression moderates their response to pharmacotherapy or psychotherapy.38 In this study, data from a large multicenter trial39 originally designed to compare the relative efficacy of pharmacotherapy (nefazodone), psychotherapy (Cognitive Behavioral Analysis System of Psychotherapy), or their combination in the treatment of chronic depression was reanalyzed by stratifying patients based on the presence or absence of ELS. In the overall sample of patients with chronic depression, psychotherapy and pharmacotherapy were comparable in efficacy but significantly less effective than their combination.
ELS in chronically depressed patients was highly prevalent. Approximately one-third experienced loss of a parent before age 15, 45% experienced childhood physical abuse, 16% experienced childhood sexual abuse, and 10% experienced neglect. Most significantly, depressed patients with a history of ELS had a superior response to psychotherapy alone compared with antidepressant monotherapy. In addition, combination therapy was only slightly more effective than psychotherapy alone in the group of depressed patients with ELS.
These data suggest that ELS is common in the population of patients with chronic depression and that psychotherapy is a critical element in the treatment of depressed patients with ELS40 (Box). However, it will be important in future studies to ascertain whether the differences in treatment response for psychotherapy compared with antidepressant in patients with ELS and depression are able to be replicated with the SSRI class of antidepressants.
Assessment of trauma and neglect should be a standard component of the diagnostic interview. Patients with a history of early life stress (ELS) may present for treatment with complaints that represent depression, anxiety, or substance abuse, but they may also have complicated presentations involving psychotic or dissociative symptoms, reflecting the diagnostic comorbidity in this population.
How to identify ELS. No standardized office-based screening tools exist for ELS, and clinical interviewing is the primary means of assessing exposure to ELS. A common error in history-taking with this population, particularly in high-volume settings, is to merely ask patients whether they were abused or neglected as children or to elaborate only very slightly on this aspect of the history. We risk not finding information that is critical to understanding our patients if we assume they share a common definition of abuse and neglect with us, can recognize such events in their personal history, and are willing to share that information with us.
When framing questions about abuse or neglect, it is important to remember that our own sense of what constitutes neglect or abuse may be very different from what a patient thinks of as neglect or abuse. For example, some patients may not consider their experience as a child abusive because of a distorted sense of responsibility, possibly further exaggerated by comorbid depression (ie, “My parents locked me in the closet overnight all the time when I was a child because I deserved it.”)
Other patients may try to minimize the impact of the experience or the responsibility of the perpetrator and attempt to normalize it (ie, “My uncle used to touch me between my legs in the swimming pool but he didn’t mean anything by it; he did it to everybody.”)
Avoiding ‘false memories.’ As important as it is to identify abuse or neglect when it has occurred, it is equally important to avoid intensifying the impact of an incident of abuse or, worse, creating a “false memory” of abuse in suggestible patients (bearing in mind that there is no definitive way to exclude the presence of “suggestibility” in patients). Our task as clinicians is to help patients correctly identify experiences of abuse and neglect and understand their response to these experiences clinically to facilitate case formulation and a treatment plan.
Creating a therapeutic alliance. Abuse and neglect during early life fundamentally alter the core assumptions that patients have about trust and safety in their relationships with others. Not only does this potentially impact the disclosure by patients of the nature and extent of trauma they have experienced, but it can also slow the formation of an effective therapeutic alliance.
To that end, asking open-ended questions about neglect and specific forms of abuse, creating an atmosphere of safety and trust, and a warm, empathic, nonjudgmental manner are central to the accurate assessment of ELS and provide the foundation for treatment by establishing an effective therapeutic alliance.
Optimal treatment. No published clinical trials have specifically compared the relative efficacy of particular forms of psychotherapy or pharmacotherapy for depressed patients with a history of ELS. However, it is clear from the available data that psychotherapy should be considered a core component of treatment for these patients.
Because psychiatric comorbidity is common in these patients, their treatment should be individualized in a manner that accounts for and addresses depression as well as associated diagnoses such as panic disorder or posttraumatic stress disorder (PTSD) with disorder-specific psychotherapy. Pharmacotherapy in combination with psychotherapy may also be helpful to patients with ELS and depression, though definitive data are lacking.
Judicious combination of medications such as antidepressants and benzodiazepines, particularly in patients with comorbid panic or PTSD, in concert with psychotherapy probably constitutes optimal treatment.
Related resources
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- National Clearinghouse on Child Abuse and Neglect. http://nccanch.acf.hhs.gov.
- Charney DS, Nemeroff CB. The peace of mind prescription: An authoritative guide to finding the most effective treatment for anxiety and depression. Boston: Houghton Mifflin, 2004.
Drug brand name
- Nefazodone • Serzone
Disclosure
Dr. Gillespie reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Nemeroff receives research grants from or is a consultant/speaker for Abbott Laboratories, Acadia Pharmaceuticals, American Foundation for Suicide Prevention, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb Co., Corcept Therapeutics, Cyberonics, Cypress Biosciences, Eli Lilly and Co., Forest Laboratories, GlaxoSmithKline, Janssen Pharmaceutica, Merck & Co., Neurocrine Biosciences, Otsuka Inc., Pfizer Inc., Sanofi Aventis, and Wyeth.
Acknowledgement
Supported by NIH MH-42088, MH-52899 (CBN), and NIH NCRRM01-RR00039 to Emory University.
1. Sedlack AJ, Broadhurst DD. Third National Incidence Study of Child Abuse and Neglect. Washington, DC: US Department of Health and Human Services; 1996.
2. McCauley J, Kern DE, Kolodner K, et al. Clinical characteristics of women with a history of child abuse: unhealed wounds. JAMA 1997;277(17):1362-8.
3. Dube SR, Anda RF, Felitti VJ, et al. Childhood abuse, household dysfunction, and the risk of attempted suicide throughout the life span: findings from the adverse childhood experiences study. JAMA 2001;286(24):3089-96.
4. Chapman DP, Whitfield CL, Felitti VJ, et al. Adverse childhood experiences and the risk of depressive disorders in adulthood. J Affect Disord 2004;82(2):217-25.
5. Gladstone GL, Parker GB, Mitchell PB, et al. Implications of childhood trauma for depressed women: an analysis of pathways from childhood sexual abuse to deliberate self-harm and revictimization. Am J Psychiatry 2004;161(8):1417-25.
6. Goodwin RD, Stein MB. Association between childhood trauma and physical disorders among adults in the United States. Psychol Med 2004;34(3):509-20.
7. Heim C, Plotsky PM, Nemeroff CB. Importance of studying the contributions of early adverse experience to neurobiological findings in depression. Neuropsychopharmacology 2004;29(4):641-8.
8. Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 1983;36(3):165-86.
9. Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol 1999;160(1):1-12.
10. Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotrophin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev 1990;15(2):71-100.
11. Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotrophin-releasing factor. Pharmacol Rev 1991;43(4):425-73.
12. Nemeroff CB, Widerlov E, Bissette G, et al. Elevated concentrations of CSF corticotrophin-releasing factor-like immunoreactivity in depressed patients. Science 1984;226(4680):1342-4.
13. Hartline KM, Owens MJ, Nemeroff CB. Postmortem and cerebrospinal fluid studies of corticotropin-releasing factor in humans. Ann NY Acad Sci 1996;780:96-105.
14. Bremner JD, Licinio J, Darnell A, et al. Elevated CSF corticotrophin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 1997;154(5):624-9.
15. Baker DG, West SA, Nicholson WE, et al. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 1999;156(4):585-8.
16. Nemeroff CB, Owens MJ, Bissette G, et al. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry 1988;45(6):577-9.
17. Merali Z, Du L, Hrdina P, et al. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci 2004;24(6):1478-85.
18. Arato M, Banki CM, Bissette G, Nemeroff CB. Elevated CSF CRF in suicide victims. Biol Psychiatry 1989;25(3):355-9.
19. Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000;157(10):1552-62.
20. Hasler G, Drevets WC, Manji HK, Charney DS. Discovering endophenotypes for major depression. Neuropsychopharmacology 2004;29(10):1765-81.
21. Gutman DA, Nemeroff CB. Persistent central nervous system effects of an adverse early environment: clinical and preclinical studies. Physiol Behav 2003;79(3):471-8.
22. Kendler KS, Kuhn JW, Prescott CA. Childhood sexual abuse, stressful life events and risk for major depression in women. Psychol Med 2004;34(8):1475-82.
23. Caspi A, Sugden K, Moffitt TE. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003;301(5631):386-9.
24. Kaufman J, Yang BZ, Douglas-Palumberi H, et al. Social supports and serotonin transporter gene moderate depression in maltreated children. Proc Natl Acad Sci USA 2004;101(49):17316-21.
25. Maercker A, Michael T, Fehm L, et al. Age of traumatisation as a predictor of post-traumatic stress disorder or major depression in young women. Br J Psychiatry 2004;184:482-7.
26. Heim C, Newport DJ, Heit S, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA 2000;284(5):592-7.
27. Heim C, Newport DJ, Bonsall R, et al. Altered pituitary-adrenal axis responses to provocative challenge tests in adult survivors of childhood abuse. Am J Psychiatry 2001;158(4):575-81.
28. Carpenter LL, Tyrka AR, McDougle CJ, et al. Cerebrospinal fluid corticotropin-releasing factor and perceived early-life stress in depressed patients and healthy control subjects. Neuropsychopharmacology 2004;29(4):777-84.
29. Campbell S, Macqueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci 2004;29(6):417-26.
30. Sheline YI, Wang PW, Gado MH, et al. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 1996;93(9):3908-13.
31. Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci 1999;19(12):5034-43.
32. Stein MB, Koverola C, Hanna C, et al. Hippocampal volume in women victimized by childhood sexual abuse. Psychol Med 1997;27(4):951-9.
33. Driessen M, Herrmann J, Stahl K, et al. Magnetic resonance imaging volumes of the hippocampus and the amygdala in women with borderline personality disorder and early traumatization. Arch Gen Psychiatry 2000;57(12):1115-22.
34. Vythilingam M, Heim C, Newport DJ, et al. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry 2002;159(12):2072-80.
35. Hayden EP, Klein DM. Outcome of dysthymic disorder at 5-year follow-up: the effect of familial psychopathology, early adversity, personality, comorbidity, and chronic stress. Am J Psychiatry 2001;158(11):1864-70.
36. Kaplan MJ, Klinetob NA. Childhood emotional trauma and chronic posttraumatic stress disorder in adult outpatients with treatment-resistant depression. J Nerv Ment Dis 2000;188(9):596-601.
37. Lara ME, Klein DN, Kasch KL. Psychosocial predictors of the short-term course and outcome of major depression: a longitudinal study of a nonclinical sample with recent-onset episodes. J Abnorm Psychol 2000;109(4):644-50.
38. Nemeroff CB, Heim CM, Thase ME, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci U S A 2003;100(24):14293-6.
39. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342(20):1462-70.
40. Craighead WE, Nemeroff CB. The impact of early trauma on response to psychotherapy. Clin Neurosci Res 2005;4(5-6):405-11.
1. Sedlack AJ, Broadhurst DD. Third National Incidence Study of Child Abuse and Neglect. Washington, DC: US Department of Health and Human Services; 1996.
2. McCauley J, Kern DE, Kolodner K, et al. Clinical characteristics of women with a history of child abuse: unhealed wounds. JAMA 1997;277(17):1362-8.
3. Dube SR, Anda RF, Felitti VJ, et al. Childhood abuse, household dysfunction, and the risk of attempted suicide throughout the life span: findings from the adverse childhood experiences study. JAMA 2001;286(24):3089-96.
4. Chapman DP, Whitfield CL, Felitti VJ, et al. Adverse childhood experiences and the risk of depressive disorders in adulthood. J Affect Disord 2004;82(2):217-25.
5. Gladstone GL, Parker GB, Mitchell PB, et al. Implications of childhood trauma for depressed women: an analysis of pathways from childhood sexual abuse to deliberate self-harm and revictimization. Am J Psychiatry 2004;161(8):1417-25.
6. Goodwin RD, Stein MB. Association between childhood trauma and physical disorders among adults in the United States. Psychol Med 2004;34(3):509-20.
7. Heim C, Plotsky PM, Nemeroff CB. Importance of studying the contributions of early adverse experience to neurobiological findings in depression. Neuropsychopharmacology 2004;29(4):641-8.
8. Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 1983;36(3):165-86.
9. Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol 1999;160(1):1-12.
10. Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotrophin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev 1990;15(2):71-100.
11. Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotrophin-releasing factor. Pharmacol Rev 1991;43(4):425-73.
12. Nemeroff CB, Widerlov E, Bissette G, et al. Elevated concentrations of CSF corticotrophin-releasing factor-like immunoreactivity in depressed patients. Science 1984;226(4680):1342-4.
13. Hartline KM, Owens MJ, Nemeroff CB. Postmortem and cerebrospinal fluid studies of corticotropin-releasing factor in humans. Ann NY Acad Sci 1996;780:96-105.
14. Bremner JD, Licinio J, Darnell A, et al. Elevated CSF corticotrophin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry 1997;154(5):624-9.
15. Baker DG, West SA, Nicholson WE, et al. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry 1999;156(4):585-8.
16. Nemeroff CB, Owens MJ, Bissette G, et al. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry 1988;45(6):577-9.
17. Merali Z, Du L, Hrdina P, et al. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci 2004;24(6):1478-85.
18. Arato M, Banki CM, Bissette G, Nemeroff CB. Elevated CSF CRF in suicide victims. Biol Psychiatry 1989;25(3):355-9.
19. Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 2000;157(10):1552-62.
20. Hasler G, Drevets WC, Manji HK, Charney DS. Discovering endophenotypes for major depression. Neuropsychopharmacology 2004;29(10):1765-81.
21. Gutman DA, Nemeroff CB. Persistent central nervous system effects of an adverse early environment: clinical and preclinical studies. Physiol Behav 2003;79(3):471-8.
22. Kendler KS, Kuhn JW, Prescott CA. Childhood sexual abuse, stressful life events and risk for major depression in women. Psychol Med 2004;34(8):1475-82.
23. Caspi A, Sugden K, Moffitt TE. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 2003;301(5631):386-9.
24. Kaufman J, Yang BZ, Douglas-Palumberi H, et al. Social supports and serotonin transporter gene moderate depression in maltreated children. Proc Natl Acad Sci USA 2004;101(49):17316-21.
25. Maercker A, Michael T, Fehm L, et al. Age of traumatisation as a predictor of post-traumatic stress disorder or major depression in young women. Br J Psychiatry 2004;184:482-7.
26. Heim C, Newport DJ, Heit S, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA 2000;284(5):592-7.
27. Heim C, Newport DJ, Bonsall R, et al. Altered pituitary-adrenal axis responses to provocative challenge tests in adult survivors of childhood abuse. Am J Psychiatry 2001;158(4):575-81.
28. Carpenter LL, Tyrka AR, McDougle CJ, et al. Cerebrospinal fluid corticotropin-releasing factor and perceived early-life stress in depressed patients and healthy control subjects. Neuropsychopharmacology 2004;29(4):777-84.
29. Campbell S, Macqueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci 2004;29(6):417-26.
30. Sheline YI, Wang PW, Gado MH, et al. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA 1996;93(9):3908-13.
31. Sheline YI, Sanghavi M, Mintun MA, Gado MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci 1999;19(12):5034-43.
32. Stein MB, Koverola C, Hanna C, et al. Hippocampal volume in women victimized by childhood sexual abuse. Psychol Med 1997;27(4):951-9.
33. Driessen M, Herrmann J, Stahl K, et al. Magnetic resonance imaging volumes of the hippocampus and the amygdala in women with borderline personality disorder and early traumatization. Arch Gen Psychiatry 2000;57(12):1115-22.
34. Vythilingam M, Heim C, Newport DJ, et al. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am J Psychiatry 2002;159(12):2072-80.
35. Hayden EP, Klein DM. Outcome of dysthymic disorder at 5-year follow-up: the effect of familial psychopathology, early adversity, personality, comorbidity, and chronic stress. Am J Psychiatry 2001;158(11):1864-70.
36. Kaplan MJ, Klinetob NA. Childhood emotional trauma and chronic posttraumatic stress disorder in adult outpatients with treatment-resistant depression. J Nerv Ment Dis 2000;188(9):596-601.
37. Lara ME, Klein DN, Kasch KL. Psychosocial predictors of the short-term course and outcome of major depression: a longitudinal study of a nonclinical sample with recent-onset episodes. J Abnorm Psychol 2000;109(4):644-50.
38. Nemeroff CB, Heim CM, Thase ME, et al. Differential responses to psychotherapy versus pharmacotherapy in patients with chronic forms of major depression and childhood trauma. Proc Natl Acad Sci U S A 2003;100(24):14293-6.
39. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342(20):1462-70.
40. Craighead WE, Nemeroff CB. The impact of early trauma on response to psychotherapy. Clin Neurosci Res 2005;4(5-6):405-11.
Acute MI risk? Protecting your patients’ heart health
One in five of your patients could suffer a heart attack in the near future—unless you take steps to ensure their heart health.
Psychiatric patients have more modifiable risk factors for coronary artery disease (CAD) compared with the general population.When depression treatment goes nowhere,” Current Psychiatry, August 2005.)
To help keep you abreast of constantly changing guidelines and strategies for recognizing and minimizing CAD risk, this article discusses:
- preventive and diagnostic guidelines for managing hypertension, diabetes, and dyslipidemia
- practical advice on convincing at-risk patients to adopt a healthier lifestyle and have a primary care doctor monitor their health.
Case: cigarettes and supersizing
Mr. H, age 54, is receiving cognitive-behavioral therapy for mild depression. He has been smoking one pack of cigarettes per day for 20 years and has never seriously considered quitting.
The patient, a school teacher, says his “busy schedule” keeps him from exercising and eating properly; he eats fast-food hamburgers and fries approximately five times per week. His father had a heart attack at age 52 and died in his sleep 10 years later.
Mr. H says he feels fine and has never seen a physician other than his psychiatrist. He is reluctant to see a primary care physician for a check-up and, because he is asymptomatic, has no incentive to do so. The psychiatrist thus decides to do a routine examination.
Blood pressure is 148/86; other vital signs are normal. Mr. H’s waist size is 42 inches, he weighs 242 lbs, and his body mass index (BMI) is 34 kg/m2, indicating clinical obesity. Cardiovascular, pulmonary, and abdominal exams are unremarkable.
Discussion. Mr. H is at high risk of a myocardial ischemic event in the near future. He has six risk factors for CAD (Table 1)—four of which are modifiable:
- family history
- age
- current cigarette use
- provisional hypertension diagnosis
- obesity
- physical inactivity.
Table 1
Risk factors for coronary artery disease
| Core risk factors |
| Age ≥45 for men* |
| Age ≥55 for women or premature menopause without estrogen-replacement therapy* |
Family history: premature coronary artery disease with myocardial infarction or sudden death before:
|
| Current cigarette smoking |
| Hypertension or antihypertensive treatment* |
| Elevated LDL cholesterol (>130 mg/dL in patients with low cardiac risk) |
| HDL cholesterol |
| Triglycerides >150 mg/dL |
| Total cholesterol >200 mg/dL* |
| Obesity (BMI >30 kg/m2)† |
| Sedentary lifestyle |
| Other risk factors |
| Elevated C-reactive protein |
| Elevated homocysteine |
| Chronic renal failure |
| Depression |
| Negative (cardio-protective) risk factors |
| HDL >60 mg/dL |
| Moderate alcohol use—no more than 1 to 2 drinks per day (1 drink = 12 oz beer or 5 oz of wine) |
| If >1 risk factor, refer to primary care doctor or quantify 10-year risk by using the Framingham/ATP III point system scale (www.nhlbi.nih.gov). |
| * Framingham/ATP III point system scale variables |
| † Use BMI calculator (http://www.nhlbisupport.com/bmi/bmicalc.htm) to determine body mass index. |
| HDL: High-density lipoprotein |
| LDL: Low-density lipoprotein |
| Source: References 9,10 |
Tools for assessing risk
The lifetime risk at age 40 for developing CAD is 49% and 32% in men and women, respectively.6
The National Cholesterol Education Program Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel) has focused on decreasing heart disease incidence by educating patients and providers. Preventive strategies and standards of care have changed several times over the past decade; the Adult Treatment Panel (ATP III) was last revised in July 2004.7
The American College of Cardiology and American Heart Association both endorse the modified Framingham/ATP III scale to measure CAD risk (see Related resources). Although this somewhat tedious point system has limitations, it can precisely calculate coronary risk across 10 years.8 Variables not included in the scoring system—such as C-reactive protein, homocysteine, and postmenopausal state—may be clinically significant and should be gauged separately.
An easier-to-use alternative, the ATP III “core risk factors” scale, estimates hypertension, hypercholesterolemia, family history, current cigarette smoking, and age as low, intermediate, or high (“risk equivalent”) risks (Table 1).8 Psychiatrists can quickly obtain this information from a brief history, blood pressure assessment, and relatively inexpensive lab studies.
Generally, the more risk factors present, the higher the risk of having a major coronary event. Presence of ≥ 2 risk factors signals intermediate or high risk and necessitates referral to a primary care doctor for monitoring.
Patients with a cardiac “risk equivalent” face a >20% risk of having a cardiac ischemic event within 10 years8 (Table 2). Examples of risk equivalents include diabetes or significant vascular disease in any artery.
“Non-core” variables. Also consider certain “non-core” variables—such as pre-existing psychiatric illness—when estimating clinical risk for heart disease. Depression, anxiety, and stress are correlated with an increase in pro-inflammatory markers such as C-reactive protein and predispose patients to CAD.11,12 Depression has repeatedly been shown to increase morbidity and mortality two- to four-fold after myocardial infarction (MI).9,13,14 Interestingly, however, depression treatment after an acute coronary event does not clearly decrease mortality.15 Although prospective, randomized studies are lacking, mood and anxiety disorder treatment is presumed to help prevent CAD development.16
Table 2
Risk equivalents for CAD*
| Established coronary artery disease |
| Symptomatic carotid artery disease |
| Peripheral vascular disease |
| Abdominal aortic aneurism |
| Diabetes mellitus |
| *Risk equivalent: Patient is assumed to have coronary artery disease (CAD). |
Recognizing cad risk
At what point do hypertension and dyslipidemia become risk factors for CAD? When and how often should patients be screened for diabetes mellitus?
Hypertension is one of the most common and deadly CAD risk factors, affecting 50 million Americans.10 Although hypertension awareness and treatment have improved, only 35% of adults have “controlled” blood pressure (
According to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7), normalizing blood pressure can reduce stroke incidence by 35% and MI by 25%, respectively. JNC 7, however, also found that 90% of persons who are normotensive at age 55 eventually develop hypertension.10
Based on these findings, JNC 7 in 2003 drastically changed the standard of care for diagnosing hypertension. JNC 7 defines normal blood pressure as
- systolic blood pressure 120 to 139 mm Hg
- diastolic blood pressure 80 to 89 mm Hg (Table 3).
Patients with diabetes mellitus or chronic kidney disease are considered hypertensive with blood pressure >130 mm Hg systolic and/or >80 mm Hg diastolic.
As with Mr. H, a blood pressure check is imperative for patients who have rarely or never seen a primary care physician in recent years. The U.S. Preventive Services Task Force strongly recommends measuring blood pressure during a routine medical evaluation at least every 2 years. A second abnormal reading at a separate visit at any time should prompt a hypertension diagnosis. Once diagnosed with hypertension, patients should be treated and checked monthly until stable, then monitored every 3 to 6 months indefinitely.10
If you cannot measure blood pressure in the office, urge patients to use an over-the-counter blood pressure measuring device and refer them to a primary care physician. Check the patient’s self-test reading for accuracy against a clinician’s measurement.
Diabetes is now considered a risk equivalent for CAD development.8 Patients diagnosed with diabetes are extremely likely to have established vascular disease,8 which predisposes them to MI, stroke, kidney disease, blindness, and lower-extremity amputations.17 Those with type 1 diabetes usually present with acute symptoms—including polyuria, polydipsia, weight loss, malaise, dry mouth, and blurred vision—and are readily diagnosed with elevated plasma glucose.
Screening for diabetes is critical because one-third of patients with the disease are undiagnosed. Also, more than 90% of patients with diabetes are non-insulin-dependent (type 2) and are asymptomatic early in the disease course.
No data definitively show benefits from screening asymptomatic adults. Recently revised diagnostic criteria for diabetes, however, call for re-testing asymptomatic patients who were found to have normal fasting plasma glucose (FPG) levels and were considered “free” of diabetes. The American Diabetes Association recommends measuring FPG after no caloric intake for ≥ 8 hours for asymptomatic patients.
FPG measurement is cost-effective and generally more convenient than other diabetes tests.17 Expert consensus strongly suggests checking FPG every 3 years beginning at age 45:17
- FPG
- FPG 100 to 125 mg/dL suggests prediabetes or impaired fasting glucose
- FPG ≥ 126 mg/dL demands a provisional diabetes diagnosis and a follow-up test on another day to confirm the diagnosis.
- comorbid cardiac risk factors
- history of polycystic ovary disease
- a first-degree relative with diabetes
- habitual inactivity
- or FPG 100 to 125 mg/dL.
Do not base diabetes diagnosis on glycosylated hemoglobin measurements, as this test can produce false-negative results in patients with new-onset diabetes.
Dyslipidemia. Every 10% reduction in serum cholesterol reduces cardiovascular mortality by 10% to 15%.19 Data from the large, prospective Framingham heart study show a 25% increase in MIs with each 5-mg/dL decrease in high-density lipoprotein cholesterol (HDL) below the age-based median for men and women.20 Serum triglycerides >150 mg/dL clearly predict future CAD and increase the likelihood of abnormally low HDL.
Every 30-mg/dL increase in low-density lipoprotein cholesterol (LDL) raises the relative risk for CAD by 30%.7 ATP III classifies LDL as the “primary target of cholesterol-lowering therapy.”8Table 4 lists LDL target levels based on other CAD risk factors.
Check fasting lipid profile or serum cholesterol, LDL, HDL, and triglycerides beginning at age 20 and about every 5 years thereafter.8 Total cholesterol
Table 3
JNC 7: What blood pressure readings mean
| Category | Systolic BP (mm Hg) | Diastolic BP (mm Hg) |
|---|---|---|
| Normal | and | |
| Prehypertension | 120-139 | or 80-89 |
| Stage 1 hypertension* | 140-159 | or 90-99 |
| Stage 2 hypertension | 160 | or ≥100 |
| JNC 7: Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure | ||
| *Patients with diabetes mellitus or chronic kidney disease have stage 1 hypertension at >130/80 mm Hg. | ||
| Source: Reference 10 | ||
Acceptable LDL cholesterol levels for adults based on CAD risk
| Risk category | Existing CAD risk factors | LDL goal |
|---|---|---|
| High risk (10-year risk > 20%) | History of diabetes, CAD, symptomatic carotid artery disease, peripheral vascular disease, or abdominal aortic aneurysm | |
| Moderate high risk* (10-year risk 10 to 20%) | >2 risk factors | |
| Moderate risk* (10-year risk | >2 risk factors | |
| Low risk | 0 to 1 risk factor | ≤160 mg/dL |
| CAD: Coronary artery disease | ||
| *Same goals apply to managing moderate high and moderate risk. Find 10-year risk calculations at nhlbi.nih.gov/guidelines/cholesterol. | ||
| Source: Reference 7 | ||
Addressing smoking, obesity
Smoking. Before trying nicotine patches or bupropion, Mr. H should realistically contemplate his risks with continued smoking; if he doesn’t want to stop, periodically encourage him to reconsider.10 Most people know the dangers of smoking but few understand that complete cessation for 1 to 2 years often nearly reverses cardiovascular disease.21
Obesity and lack of exercise go hand in hand. Reducing Mr. H’s waist size to 2 is a reasonable short-term goal. To that end, encourage him to:
- decrease his number of weekly fast-food meals from five to three, with an eventual goal of one per week. As an alternative, microwaveable, low-calorie meals—each with at least two servings of fruits or vegetables—can be prepared at home or work.
- walk 30 minutes three times weekly and progress to 1 hour five times weekly over 6 months. As with any exercise program, remind Mr. H to “start low and go slow.”
The patient’s role in treatment. Patients often feel overwhelmed after getting large amounts of information on CAD risk and may feel hopeless and unenthusiastic about improving their physical health. Work with the primary care doctor to emphasize a patient care plan that clearly defines easily attainable, step-by-step goals. Make sure the patient agrees to these goals.
Case continued: no more supersizing
Mr. H now understands the importance of minimizing his CAD risk and realizes that CAD and many associated risk factors are asymptomatic in the early stages of development.
With help from his doctors, Mr. H quit smoking. He also became more mindful of his caloric intake and the types of foods he was eating. He advanced from briskly walking 30 minutes three times per week to slow jogging 40 minutes five times weekly. He still eats at fast-food restaurants but usually orders broiled chicken, salads, or the occasional burger.
- National Cholesterol Education Program. CAD risk assessment tool and ATP III guidelines. www.nhlbi.nih.gov/guidelines/cholesterol/.
- U.S. Preventive Services Task Force preventive guidelines. www.ahrq.gov/clinic/uspstfix.htm.
- National Heart, Lung, and Blood Institute. Calculate your body mass index. http://nhlbisupport.com/bmi/.
- American Heart Association. www.americanheart.org.
1. Holt RI, Peveler RC, Byrne CD. Schizophrenia, the metabolic syndrome and diabetes. Diabetes Med 2004;21:515-23.
2. Carney RM, Freedland KE, Miller GE, Jaffe AS. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
3. Weiser M, Reichenberg A, Grotto I, et al. Higher rates of cigarette smoking in male adolescents before the onset of schizophrenia: a historical-prospective cohort study. Am J Psychiatry 2004;161:1219-23.
4. Druss B, Rosenheck R. Mental disorders and access to medical care in the United States. Am J Psychiatry 1998;155:1775-7.
5. Druss B, Rosenheck R, Desai MM, Perlin JB. Quality of preventive medical care for patients with mental disorders. Med Care 2002;40:129-36.
6. Lloyd-Jones DM, Larson MG, Beiser A, Levy D. Lifetime risk of developing coronary heart disease. Lancet 1999;353(9147):89-92.
7. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004;110:227-9.
8. Expert Panel on Detection. Evaluation and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001;285:2486-97.
9. Frasure-Smith N, Lesperance F, Talajic M, et al. Depression and 18-month prognosis after myocardial infarction. Circulation 1995;91:999-1005.
10. Chobanian AV, Bakris GL, Black HR, et al. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003;289:2560-72.
11. Ford DE, Erlinger TP. Depression and C-reactive protein in U.S. adults: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med 2004;164:1010-14.
12. Panagiotakos DB, Pitsavos C, Chrysohoou C, et al. Inflammation, coagulation and depressive symptomatology in cardiovascular disease-free people: the ATTICA study. Eur Heart J 2004;25:492-9.
13. Frasure-Smith N, Lesperance F, Talajic M. Depression following myocardial infarction. Impact on 6-month survival. JAMA 1993;270:1819-25.
14. Ladwig KH, Kieser M, Konig J, et al. Affective disorders and survival after acute myocardial infarction: results from the post-infarction late potential study. Eur Heart J 1991;12:959-64.
15. Berkman LF, Blumenthal J, Burg M, et al. Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Artery Heart Disease Patients (ENRICHD) randomized trial. JAMA 2003;289:3106-16.
16. Rosengren A, Hawken S, Ounpuu S, et al. Association of psychological risk factors with risk of acute myocardial infarction in 11,119 cases and 13,648 controls from 52 countries (the INTERHEART study). Lancet 2004;364:953-62.
17. American Diabetes Association. Screening for type 2 diabetes. Diabetes Care 2004;27(suppl 1):11-14.
18. American Diabetes Association; American Psychiatric Association. American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 2004;27:596-601.
19. Gould AL, Rossouw JE, Santanello NC, et al. Cholesterol reduction yields clinical benefit; impact of statin trials. Circulation 1998;97:946-52.
20. Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary artery disease. The Framingham Study. Am J Med 1977;62:707-14.
21. Rigotti N, Pasternak L. Changing the natural history of coronary artery disease. Cardiology Clinics 1996;14:51-68.
One in five of your patients could suffer a heart attack in the near future—unless you take steps to ensure their heart health.
Psychiatric patients have more modifiable risk factors for coronary artery disease (CAD) compared with the general population.When depression treatment goes nowhere,” Current Psychiatry, August 2005.)
To help keep you abreast of constantly changing guidelines and strategies for recognizing and minimizing CAD risk, this article discusses:
- preventive and diagnostic guidelines for managing hypertension, diabetes, and dyslipidemia
- practical advice on convincing at-risk patients to adopt a healthier lifestyle and have a primary care doctor monitor their health.
Case: cigarettes and supersizing
Mr. H, age 54, is receiving cognitive-behavioral therapy for mild depression. He has been smoking one pack of cigarettes per day for 20 years and has never seriously considered quitting.
The patient, a school teacher, says his “busy schedule” keeps him from exercising and eating properly; he eats fast-food hamburgers and fries approximately five times per week. His father had a heart attack at age 52 and died in his sleep 10 years later.
Mr. H says he feels fine and has never seen a physician other than his psychiatrist. He is reluctant to see a primary care physician for a check-up and, because he is asymptomatic, has no incentive to do so. The psychiatrist thus decides to do a routine examination.
Blood pressure is 148/86; other vital signs are normal. Mr. H’s waist size is 42 inches, he weighs 242 lbs, and his body mass index (BMI) is 34 kg/m2, indicating clinical obesity. Cardiovascular, pulmonary, and abdominal exams are unremarkable.
Discussion. Mr. H is at high risk of a myocardial ischemic event in the near future. He has six risk factors for CAD (Table 1)—four of which are modifiable:
- family history
- age
- current cigarette use
- provisional hypertension diagnosis
- obesity
- physical inactivity.
Table 1
Risk factors for coronary artery disease
| Core risk factors |
| Age ≥45 for men* |
| Age ≥55 for women or premature menopause without estrogen-replacement therapy* |
Family history: premature coronary artery disease with myocardial infarction or sudden death before:
|
| Current cigarette smoking |
| Hypertension or antihypertensive treatment* |
| Elevated LDL cholesterol (>130 mg/dL in patients with low cardiac risk) |
| HDL cholesterol |
| Triglycerides >150 mg/dL |
| Total cholesterol >200 mg/dL* |
| Obesity (BMI >30 kg/m2)† |
| Sedentary lifestyle |
| Other risk factors |
| Elevated C-reactive protein |
| Elevated homocysteine |
| Chronic renal failure |
| Depression |
| Negative (cardio-protective) risk factors |
| HDL >60 mg/dL |
| Moderate alcohol use—no more than 1 to 2 drinks per day (1 drink = 12 oz beer or 5 oz of wine) |
| If >1 risk factor, refer to primary care doctor or quantify 10-year risk by using the Framingham/ATP III point system scale (www.nhlbi.nih.gov). |
| * Framingham/ATP III point system scale variables |
| † Use BMI calculator (http://www.nhlbisupport.com/bmi/bmicalc.htm) to determine body mass index. |
| HDL: High-density lipoprotein |
| LDL: Low-density lipoprotein |
| Source: References 9,10 |
Tools for assessing risk
The lifetime risk at age 40 for developing CAD is 49% and 32% in men and women, respectively.6
The National Cholesterol Education Program Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel) has focused on decreasing heart disease incidence by educating patients and providers. Preventive strategies and standards of care have changed several times over the past decade; the Adult Treatment Panel (ATP III) was last revised in July 2004.7
The American College of Cardiology and American Heart Association both endorse the modified Framingham/ATP III scale to measure CAD risk (see Related resources). Although this somewhat tedious point system has limitations, it can precisely calculate coronary risk across 10 years.8 Variables not included in the scoring system—such as C-reactive protein, homocysteine, and postmenopausal state—may be clinically significant and should be gauged separately.
An easier-to-use alternative, the ATP III “core risk factors” scale, estimates hypertension, hypercholesterolemia, family history, current cigarette smoking, and age as low, intermediate, or high (“risk equivalent”) risks (Table 1).8 Psychiatrists can quickly obtain this information from a brief history, blood pressure assessment, and relatively inexpensive lab studies.
Generally, the more risk factors present, the higher the risk of having a major coronary event. Presence of ≥ 2 risk factors signals intermediate or high risk and necessitates referral to a primary care doctor for monitoring.
Patients with a cardiac “risk equivalent” face a >20% risk of having a cardiac ischemic event within 10 years8 (Table 2). Examples of risk equivalents include diabetes or significant vascular disease in any artery.
“Non-core” variables. Also consider certain “non-core” variables—such as pre-existing psychiatric illness—when estimating clinical risk for heart disease. Depression, anxiety, and stress are correlated with an increase in pro-inflammatory markers such as C-reactive protein and predispose patients to CAD.11,12 Depression has repeatedly been shown to increase morbidity and mortality two- to four-fold after myocardial infarction (MI).9,13,14 Interestingly, however, depression treatment after an acute coronary event does not clearly decrease mortality.15 Although prospective, randomized studies are lacking, mood and anxiety disorder treatment is presumed to help prevent CAD development.16
Table 2
Risk equivalents for CAD*
| Established coronary artery disease |
| Symptomatic carotid artery disease |
| Peripheral vascular disease |
| Abdominal aortic aneurism |
| Diabetes mellitus |
| *Risk equivalent: Patient is assumed to have coronary artery disease (CAD). |
Recognizing cad risk
At what point do hypertension and dyslipidemia become risk factors for CAD? When and how often should patients be screened for diabetes mellitus?
Hypertension is one of the most common and deadly CAD risk factors, affecting 50 million Americans.10 Although hypertension awareness and treatment have improved, only 35% of adults have “controlled” blood pressure (
According to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7), normalizing blood pressure can reduce stroke incidence by 35% and MI by 25%, respectively. JNC 7, however, also found that 90% of persons who are normotensive at age 55 eventually develop hypertension.10
Based on these findings, JNC 7 in 2003 drastically changed the standard of care for diagnosing hypertension. JNC 7 defines normal blood pressure as
- systolic blood pressure 120 to 139 mm Hg
- diastolic blood pressure 80 to 89 mm Hg (Table 3).
Patients with diabetes mellitus or chronic kidney disease are considered hypertensive with blood pressure >130 mm Hg systolic and/or >80 mm Hg diastolic.
As with Mr. H, a blood pressure check is imperative for patients who have rarely or never seen a primary care physician in recent years. The U.S. Preventive Services Task Force strongly recommends measuring blood pressure during a routine medical evaluation at least every 2 years. A second abnormal reading at a separate visit at any time should prompt a hypertension diagnosis. Once diagnosed with hypertension, patients should be treated and checked monthly until stable, then monitored every 3 to 6 months indefinitely.10
If you cannot measure blood pressure in the office, urge patients to use an over-the-counter blood pressure measuring device and refer them to a primary care physician. Check the patient’s self-test reading for accuracy against a clinician’s measurement.
Diabetes is now considered a risk equivalent for CAD development.8 Patients diagnosed with diabetes are extremely likely to have established vascular disease,8 which predisposes them to MI, stroke, kidney disease, blindness, and lower-extremity amputations.17 Those with type 1 diabetes usually present with acute symptoms—including polyuria, polydipsia, weight loss, malaise, dry mouth, and blurred vision—and are readily diagnosed with elevated plasma glucose.
Screening for diabetes is critical because one-third of patients with the disease are undiagnosed. Also, more than 90% of patients with diabetes are non-insulin-dependent (type 2) and are asymptomatic early in the disease course.
No data definitively show benefits from screening asymptomatic adults. Recently revised diagnostic criteria for diabetes, however, call for re-testing asymptomatic patients who were found to have normal fasting plasma glucose (FPG) levels and were considered “free” of diabetes. The American Diabetes Association recommends measuring FPG after no caloric intake for ≥ 8 hours for asymptomatic patients.
FPG measurement is cost-effective and generally more convenient than other diabetes tests.17 Expert consensus strongly suggests checking FPG every 3 years beginning at age 45:17
- FPG
- FPG 100 to 125 mg/dL suggests prediabetes or impaired fasting glucose
- FPG ≥ 126 mg/dL demands a provisional diabetes diagnosis and a follow-up test on another day to confirm the diagnosis.
- comorbid cardiac risk factors
- history of polycystic ovary disease
- a first-degree relative with diabetes
- habitual inactivity
- or FPG 100 to 125 mg/dL.
Do not base diabetes diagnosis on glycosylated hemoglobin measurements, as this test can produce false-negative results in patients with new-onset diabetes.
Dyslipidemia. Every 10% reduction in serum cholesterol reduces cardiovascular mortality by 10% to 15%.19 Data from the large, prospective Framingham heart study show a 25% increase in MIs with each 5-mg/dL decrease in high-density lipoprotein cholesterol (HDL) below the age-based median for men and women.20 Serum triglycerides >150 mg/dL clearly predict future CAD and increase the likelihood of abnormally low HDL.
Every 30-mg/dL increase in low-density lipoprotein cholesterol (LDL) raises the relative risk for CAD by 30%.7 ATP III classifies LDL as the “primary target of cholesterol-lowering therapy.”8Table 4 lists LDL target levels based on other CAD risk factors.
Check fasting lipid profile or serum cholesterol, LDL, HDL, and triglycerides beginning at age 20 and about every 5 years thereafter.8 Total cholesterol
Table 3
JNC 7: What blood pressure readings mean
| Category | Systolic BP (mm Hg) | Diastolic BP (mm Hg) |
|---|---|---|
| Normal | and | |
| Prehypertension | 120-139 | or 80-89 |
| Stage 1 hypertension* | 140-159 | or 90-99 |
| Stage 2 hypertension | 160 | or ≥100 |
| JNC 7: Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure | ||
| *Patients with diabetes mellitus or chronic kidney disease have stage 1 hypertension at >130/80 mm Hg. | ||
| Source: Reference 10 | ||
Acceptable LDL cholesterol levels for adults based on CAD risk
| Risk category | Existing CAD risk factors | LDL goal |
|---|---|---|
| High risk (10-year risk > 20%) | History of diabetes, CAD, symptomatic carotid artery disease, peripheral vascular disease, or abdominal aortic aneurysm | |
| Moderate high risk* (10-year risk 10 to 20%) | >2 risk factors | |
| Moderate risk* (10-year risk | >2 risk factors | |
| Low risk | 0 to 1 risk factor | ≤160 mg/dL |
| CAD: Coronary artery disease | ||
| *Same goals apply to managing moderate high and moderate risk. Find 10-year risk calculations at nhlbi.nih.gov/guidelines/cholesterol. | ||
| Source: Reference 7 | ||
Addressing smoking, obesity
Smoking. Before trying nicotine patches or bupropion, Mr. H should realistically contemplate his risks with continued smoking; if he doesn’t want to stop, periodically encourage him to reconsider.10 Most people know the dangers of smoking but few understand that complete cessation for 1 to 2 years often nearly reverses cardiovascular disease.21
Obesity and lack of exercise go hand in hand. Reducing Mr. H’s waist size to 2 is a reasonable short-term goal. To that end, encourage him to:
- decrease his number of weekly fast-food meals from five to three, with an eventual goal of one per week. As an alternative, microwaveable, low-calorie meals—each with at least two servings of fruits or vegetables—can be prepared at home or work.
- walk 30 minutes three times weekly and progress to 1 hour five times weekly over 6 months. As with any exercise program, remind Mr. H to “start low and go slow.”
The patient’s role in treatment. Patients often feel overwhelmed after getting large amounts of information on CAD risk and may feel hopeless and unenthusiastic about improving their physical health. Work with the primary care doctor to emphasize a patient care plan that clearly defines easily attainable, step-by-step goals. Make sure the patient agrees to these goals.
Case continued: no more supersizing
Mr. H now understands the importance of minimizing his CAD risk and realizes that CAD and many associated risk factors are asymptomatic in the early stages of development.
With help from his doctors, Mr. H quit smoking. He also became more mindful of his caloric intake and the types of foods he was eating. He advanced from briskly walking 30 minutes three times per week to slow jogging 40 minutes five times weekly. He still eats at fast-food restaurants but usually orders broiled chicken, salads, or the occasional burger.
- National Cholesterol Education Program. CAD risk assessment tool and ATP III guidelines. www.nhlbi.nih.gov/guidelines/cholesterol/.
- U.S. Preventive Services Task Force preventive guidelines. www.ahrq.gov/clinic/uspstfix.htm.
- National Heart, Lung, and Blood Institute. Calculate your body mass index. http://nhlbisupport.com/bmi/.
- American Heart Association. www.americanheart.org.
One in five of your patients could suffer a heart attack in the near future—unless you take steps to ensure their heart health.
Psychiatric patients have more modifiable risk factors for coronary artery disease (CAD) compared with the general population.When depression treatment goes nowhere,” Current Psychiatry, August 2005.)
To help keep you abreast of constantly changing guidelines and strategies for recognizing and minimizing CAD risk, this article discusses:
- preventive and diagnostic guidelines for managing hypertension, diabetes, and dyslipidemia
- practical advice on convincing at-risk patients to adopt a healthier lifestyle and have a primary care doctor monitor their health.
Case: cigarettes and supersizing
Mr. H, age 54, is receiving cognitive-behavioral therapy for mild depression. He has been smoking one pack of cigarettes per day for 20 years and has never seriously considered quitting.
The patient, a school teacher, says his “busy schedule” keeps him from exercising and eating properly; he eats fast-food hamburgers and fries approximately five times per week. His father had a heart attack at age 52 and died in his sleep 10 years later.
Mr. H says he feels fine and has never seen a physician other than his psychiatrist. He is reluctant to see a primary care physician for a check-up and, because he is asymptomatic, has no incentive to do so. The psychiatrist thus decides to do a routine examination.
Blood pressure is 148/86; other vital signs are normal. Mr. H’s waist size is 42 inches, he weighs 242 lbs, and his body mass index (BMI) is 34 kg/m2, indicating clinical obesity. Cardiovascular, pulmonary, and abdominal exams are unremarkable.
Discussion. Mr. H is at high risk of a myocardial ischemic event in the near future. He has six risk factors for CAD (Table 1)—four of which are modifiable:
- family history
- age
- current cigarette use
- provisional hypertension diagnosis
- obesity
- physical inactivity.
Table 1
Risk factors for coronary artery disease
| Core risk factors |
| Age ≥45 for men* |
| Age ≥55 for women or premature menopause without estrogen-replacement therapy* |
Family history: premature coronary artery disease with myocardial infarction or sudden death before:
|
| Current cigarette smoking |
| Hypertension or antihypertensive treatment* |
| Elevated LDL cholesterol (>130 mg/dL in patients with low cardiac risk) |
| HDL cholesterol |
| Triglycerides >150 mg/dL |
| Total cholesterol >200 mg/dL* |
| Obesity (BMI >30 kg/m2)† |
| Sedentary lifestyle |
| Other risk factors |
| Elevated C-reactive protein |
| Elevated homocysteine |
| Chronic renal failure |
| Depression |
| Negative (cardio-protective) risk factors |
| HDL >60 mg/dL |
| Moderate alcohol use—no more than 1 to 2 drinks per day (1 drink = 12 oz beer or 5 oz of wine) |
| If >1 risk factor, refer to primary care doctor or quantify 10-year risk by using the Framingham/ATP III point system scale (www.nhlbi.nih.gov). |
| * Framingham/ATP III point system scale variables |
| † Use BMI calculator (http://www.nhlbisupport.com/bmi/bmicalc.htm) to determine body mass index. |
| HDL: High-density lipoprotein |
| LDL: Low-density lipoprotein |
| Source: References 9,10 |
Tools for assessing risk
The lifetime risk at age 40 for developing CAD is 49% and 32% in men and women, respectively.6
The National Cholesterol Education Program Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel) has focused on decreasing heart disease incidence by educating patients and providers. Preventive strategies and standards of care have changed several times over the past decade; the Adult Treatment Panel (ATP III) was last revised in July 2004.7
The American College of Cardiology and American Heart Association both endorse the modified Framingham/ATP III scale to measure CAD risk (see Related resources). Although this somewhat tedious point system has limitations, it can precisely calculate coronary risk across 10 years.8 Variables not included in the scoring system—such as C-reactive protein, homocysteine, and postmenopausal state—may be clinically significant and should be gauged separately.
An easier-to-use alternative, the ATP III “core risk factors” scale, estimates hypertension, hypercholesterolemia, family history, current cigarette smoking, and age as low, intermediate, or high (“risk equivalent”) risks (Table 1).8 Psychiatrists can quickly obtain this information from a brief history, blood pressure assessment, and relatively inexpensive lab studies.
Generally, the more risk factors present, the higher the risk of having a major coronary event. Presence of ≥ 2 risk factors signals intermediate or high risk and necessitates referral to a primary care doctor for monitoring.
Patients with a cardiac “risk equivalent” face a >20% risk of having a cardiac ischemic event within 10 years8 (Table 2). Examples of risk equivalents include diabetes or significant vascular disease in any artery.
“Non-core” variables. Also consider certain “non-core” variables—such as pre-existing psychiatric illness—when estimating clinical risk for heart disease. Depression, anxiety, and stress are correlated with an increase in pro-inflammatory markers such as C-reactive protein and predispose patients to CAD.11,12 Depression has repeatedly been shown to increase morbidity and mortality two- to four-fold after myocardial infarction (MI).9,13,14 Interestingly, however, depression treatment after an acute coronary event does not clearly decrease mortality.15 Although prospective, randomized studies are lacking, mood and anxiety disorder treatment is presumed to help prevent CAD development.16
Table 2
Risk equivalents for CAD*
| Established coronary artery disease |
| Symptomatic carotid artery disease |
| Peripheral vascular disease |
| Abdominal aortic aneurism |
| Diabetes mellitus |
| *Risk equivalent: Patient is assumed to have coronary artery disease (CAD). |
Recognizing cad risk
At what point do hypertension and dyslipidemia become risk factors for CAD? When and how often should patients be screened for diabetes mellitus?
Hypertension is one of the most common and deadly CAD risk factors, affecting 50 million Americans.10 Although hypertension awareness and treatment have improved, only 35% of adults have “controlled” blood pressure (
According to the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC 7), normalizing blood pressure can reduce stroke incidence by 35% and MI by 25%, respectively. JNC 7, however, also found that 90% of persons who are normotensive at age 55 eventually develop hypertension.10
Based on these findings, JNC 7 in 2003 drastically changed the standard of care for diagnosing hypertension. JNC 7 defines normal blood pressure as
- systolic blood pressure 120 to 139 mm Hg
- diastolic blood pressure 80 to 89 mm Hg (Table 3).
Patients with diabetes mellitus or chronic kidney disease are considered hypertensive with blood pressure >130 mm Hg systolic and/or >80 mm Hg diastolic.
As with Mr. H, a blood pressure check is imperative for patients who have rarely or never seen a primary care physician in recent years. The U.S. Preventive Services Task Force strongly recommends measuring blood pressure during a routine medical evaluation at least every 2 years. A second abnormal reading at a separate visit at any time should prompt a hypertension diagnosis. Once diagnosed with hypertension, patients should be treated and checked monthly until stable, then monitored every 3 to 6 months indefinitely.10
If you cannot measure blood pressure in the office, urge patients to use an over-the-counter blood pressure measuring device and refer them to a primary care physician. Check the patient’s self-test reading for accuracy against a clinician’s measurement.
Diabetes is now considered a risk equivalent for CAD development.8 Patients diagnosed with diabetes are extremely likely to have established vascular disease,8 which predisposes them to MI, stroke, kidney disease, blindness, and lower-extremity amputations.17 Those with type 1 diabetes usually present with acute symptoms—including polyuria, polydipsia, weight loss, malaise, dry mouth, and blurred vision—and are readily diagnosed with elevated plasma glucose.
Screening for diabetes is critical because one-third of patients with the disease are undiagnosed. Also, more than 90% of patients with diabetes are non-insulin-dependent (type 2) and are asymptomatic early in the disease course.
No data definitively show benefits from screening asymptomatic adults. Recently revised diagnostic criteria for diabetes, however, call for re-testing asymptomatic patients who were found to have normal fasting plasma glucose (FPG) levels and were considered “free” of diabetes. The American Diabetes Association recommends measuring FPG after no caloric intake for ≥ 8 hours for asymptomatic patients.
FPG measurement is cost-effective and generally more convenient than other diabetes tests.17 Expert consensus strongly suggests checking FPG every 3 years beginning at age 45:17
- FPG
- FPG 100 to 125 mg/dL suggests prediabetes or impaired fasting glucose
- FPG ≥ 126 mg/dL demands a provisional diabetes diagnosis and a follow-up test on another day to confirm the diagnosis.
- comorbid cardiac risk factors
- history of polycystic ovary disease
- a first-degree relative with diabetes
- habitual inactivity
- or FPG 100 to 125 mg/dL.
Do not base diabetes diagnosis on glycosylated hemoglobin measurements, as this test can produce false-negative results in patients with new-onset diabetes.
Dyslipidemia. Every 10% reduction in serum cholesterol reduces cardiovascular mortality by 10% to 15%.19 Data from the large, prospective Framingham heart study show a 25% increase in MIs with each 5-mg/dL decrease in high-density lipoprotein cholesterol (HDL) below the age-based median for men and women.20 Serum triglycerides >150 mg/dL clearly predict future CAD and increase the likelihood of abnormally low HDL.
Every 30-mg/dL increase in low-density lipoprotein cholesterol (LDL) raises the relative risk for CAD by 30%.7 ATP III classifies LDL as the “primary target of cholesterol-lowering therapy.”8Table 4 lists LDL target levels based on other CAD risk factors.
Check fasting lipid profile or serum cholesterol, LDL, HDL, and triglycerides beginning at age 20 and about every 5 years thereafter.8 Total cholesterol
Table 3
JNC 7: What blood pressure readings mean
| Category | Systolic BP (mm Hg) | Diastolic BP (mm Hg) |
|---|---|---|
| Normal | and | |
| Prehypertension | 120-139 | or 80-89 |
| Stage 1 hypertension* | 140-159 | or 90-99 |
| Stage 2 hypertension | 160 | or ≥100 |
| JNC 7: Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure | ||
| *Patients with diabetes mellitus or chronic kidney disease have stage 1 hypertension at >130/80 mm Hg. | ||
| Source: Reference 10 | ||
Acceptable LDL cholesterol levels for adults based on CAD risk
| Risk category | Existing CAD risk factors | LDL goal |
|---|---|---|
| High risk (10-year risk > 20%) | History of diabetes, CAD, symptomatic carotid artery disease, peripheral vascular disease, or abdominal aortic aneurysm | |
| Moderate high risk* (10-year risk 10 to 20%) | >2 risk factors | |
| Moderate risk* (10-year risk | >2 risk factors | |
| Low risk | 0 to 1 risk factor | ≤160 mg/dL |
| CAD: Coronary artery disease | ||
| *Same goals apply to managing moderate high and moderate risk. Find 10-year risk calculations at nhlbi.nih.gov/guidelines/cholesterol. | ||
| Source: Reference 7 | ||
Addressing smoking, obesity
Smoking. Before trying nicotine patches or bupropion, Mr. H should realistically contemplate his risks with continued smoking; if he doesn’t want to stop, periodically encourage him to reconsider.10 Most people know the dangers of smoking but few understand that complete cessation for 1 to 2 years often nearly reverses cardiovascular disease.21
Obesity and lack of exercise go hand in hand. Reducing Mr. H’s waist size to 2 is a reasonable short-term goal. To that end, encourage him to:
- decrease his number of weekly fast-food meals from five to three, with an eventual goal of one per week. As an alternative, microwaveable, low-calorie meals—each with at least two servings of fruits or vegetables—can be prepared at home or work.
- walk 30 minutes three times weekly and progress to 1 hour five times weekly over 6 months. As with any exercise program, remind Mr. H to “start low and go slow.”
The patient’s role in treatment. Patients often feel overwhelmed after getting large amounts of information on CAD risk and may feel hopeless and unenthusiastic about improving their physical health. Work with the primary care doctor to emphasize a patient care plan that clearly defines easily attainable, step-by-step goals. Make sure the patient agrees to these goals.
Case continued: no more supersizing
Mr. H now understands the importance of minimizing his CAD risk and realizes that CAD and many associated risk factors are asymptomatic in the early stages of development.
With help from his doctors, Mr. H quit smoking. He also became more mindful of his caloric intake and the types of foods he was eating. He advanced from briskly walking 30 minutes three times per week to slow jogging 40 minutes five times weekly. He still eats at fast-food restaurants but usually orders broiled chicken, salads, or the occasional burger.
- National Cholesterol Education Program. CAD risk assessment tool and ATP III guidelines. www.nhlbi.nih.gov/guidelines/cholesterol/.
- U.S. Preventive Services Task Force preventive guidelines. www.ahrq.gov/clinic/uspstfix.htm.
- National Heart, Lung, and Blood Institute. Calculate your body mass index. http://nhlbisupport.com/bmi/.
- American Heart Association. www.americanheart.org.
1. Holt RI, Peveler RC, Byrne CD. Schizophrenia, the metabolic syndrome and diabetes. Diabetes Med 2004;21:515-23.
2. Carney RM, Freedland KE, Miller GE, Jaffe AS. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
3. Weiser M, Reichenberg A, Grotto I, et al. Higher rates of cigarette smoking in male adolescents before the onset of schizophrenia: a historical-prospective cohort study. Am J Psychiatry 2004;161:1219-23.
4. Druss B, Rosenheck R. Mental disorders and access to medical care in the United States. Am J Psychiatry 1998;155:1775-7.
5. Druss B, Rosenheck R, Desai MM, Perlin JB. Quality of preventive medical care for patients with mental disorders. Med Care 2002;40:129-36.
6. Lloyd-Jones DM, Larson MG, Beiser A, Levy D. Lifetime risk of developing coronary heart disease. Lancet 1999;353(9147):89-92.
7. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004;110:227-9.
8. Expert Panel on Detection. Evaluation and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001;285:2486-97.
9. Frasure-Smith N, Lesperance F, Talajic M, et al. Depression and 18-month prognosis after myocardial infarction. Circulation 1995;91:999-1005.
10. Chobanian AV, Bakris GL, Black HR, et al. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003;289:2560-72.
11. Ford DE, Erlinger TP. Depression and C-reactive protein in U.S. adults: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med 2004;164:1010-14.
12. Panagiotakos DB, Pitsavos C, Chrysohoou C, et al. Inflammation, coagulation and depressive symptomatology in cardiovascular disease-free people: the ATTICA study. Eur Heart J 2004;25:492-9.
13. Frasure-Smith N, Lesperance F, Talajic M. Depression following myocardial infarction. Impact on 6-month survival. JAMA 1993;270:1819-25.
14. Ladwig KH, Kieser M, Konig J, et al. Affective disorders and survival after acute myocardial infarction: results from the post-infarction late potential study. Eur Heart J 1991;12:959-64.
15. Berkman LF, Blumenthal J, Burg M, et al. Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Artery Heart Disease Patients (ENRICHD) randomized trial. JAMA 2003;289:3106-16.
16. Rosengren A, Hawken S, Ounpuu S, et al. Association of psychological risk factors with risk of acute myocardial infarction in 11,119 cases and 13,648 controls from 52 countries (the INTERHEART study). Lancet 2004;364:953-62.
17. American Diabetes Association. Screening for type 2 diabetes. Diabetes Care 2004;27(suppl 1):11-14.
18. American Diabetes Association; American Psychiatric Association. American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 2004;27:596-601.
19. Gould AL, Rossouw JE, Santanello NC, et al. Cholesterol reduction yields clinical benefit; impact of statin trials. Circulation 1998;97:946-52.
20. Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary artery disease. The Framingham Study. Am J Med 1977;62:707-14.
21. Rigotti N, Pasternak L. Changing the natural history of coronary artery disease. Cardiology Clinics 1996;14:51-68.
1. Holt RI, Peveler RC, Byrne CD. Schizophrenia, the metabolic syndrome and diabetes. Diabetes Med 2004;21:515-23.
2. Carney RM, Freedland KE, Miller GE, Jaffe AS. Depression as a risk factor for cardiac mortality and morbidity: a review of potential mechanisms. J Psychosom Res 2002;53:897-902.
3. Weiser M, Reichenberg A, Grotto I, et al. Higher rates of cigarette smoking in male adolescents before the onset of schizophrenia: a historical-prospective cohort study. Am J Psychiatry 2004;161:1219-23.
4. Druss B, Rosenheck R. Mental disorders and access to medical care in the United States. Am J Psychiatry 1998;155:1775-7.
5. Druss B, Rosenheck R, Desai MM, Perlin JB. Quality of preventive medical care for patients with mental disorders. Med Care 2002;40:129-36.
6. Lloyd-Jones DM, Larson MG, Beiser A, Levy D. Lifetime risk of developing coronary heart disease. Lancet 1999;353(9147):89-92.
7. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004;110:227-9.
8. Expert Panel on Detection. Evaluation and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001;285:2486-97.
9. Frasure-Smith N, Lesperance F, Talajic M, et al. Depression and 18-month prognosis after myocardial infarction. Circulation 1995;91:999-1005.
10. Chobanian AV, Bakris GL, Black HR, et al. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003;289:2560-72.
11. Ford DE, Erlinger TP. Depression and C-reactive protein in U.S. adults: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med 2004;164:1010-14.
12. Panagiotakos DB, Pitsavos C, Chrysohoou C, et al. Inflammation, coagulation and depressive symptomatology in cardiovascular disease-free people: the ATTICA study. Eur Heart J 2004;25:492-9.
13. Frasure-Smith N, Lesperance F, Talajic M. Depression following myocardial infarction. Impact on 6-month survival. JAMA 1993;270:1819-25.
14. Ladwig KH, Kieser M, Konig J, et al. Affective disorders and survival after acute myocardial infarction: results from the post-infarction late potential study. Eur Heart J 1991;12:959-64.
15. Berkman LF, Blumenthal J, Burg M, et al. Effects of treating depression and low perceived social support on clinical events after myocardial infarction: the Enhancing Recovery in Coronary Artery Heart Disease Patients (ENRICHD) randomized trial. JAMA 2003;289:3106-16.
16. Rosengren A, Hawken S, Ounpuu S, et al. Association of psychological risk factors with risk of acute myocardial infarction in 11,119 cases and 13,648 controls from 52 countries (the INTERHEART study). Lancet 2004;364:953-62.
17. American Diabetes Association. Screening for type 2 diabetes. Diabetes Care 2004;27(suppl 1):11-14.
18. American Diabetes Association; American Psychiatric Association. American Association of Clinical Endocrinologists; North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 2004;27:596-601.
19. Gould AL, Rossouw JE, Santanello NC, et al. Cholesterol reduction yields clinical benefit; impact of statin trials. Circulation 1998;97:946-52.
20. Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary artery disease. The Framingham Study. Am J Med 1977;62:707-14.
21. Rigotti N, Pasternak L. Changing the natural history of coronary artery disease. Cardiology Clinics 1996;14:51-68.