User login
Intermittent explosive disorder: Taming temper tantrums in the volatile, impulsive adult
Mr. P, age 41, has a “problem with anger.” Since age 17, he has had sudden outbursts of screaming and shouting, with occasional minor damage to objects. These outbursts—including episodes of “road rage”—occur once or more per week and almost daily for months at a time.
Mr. P has also had more violent episodes— sometimes every 2 to 3 months—in which he has punched holes in walls, destroyed a computer with a hammer, and assaulted other people with his fists. These events are not premeditated and are typically triggered by Mr. P’s frustration at not being “perfect” or by others breaking what he considers “general rules of conduct.”
The day before his initial visit, while he was stuck in traffic, Mr. P saw a car speeding down the shoulder. Enraged, he pulled in front of the car so that the driver had to slam on the brakes. He jumped out of his car and approached the other driver, shouting obscenities. The other driver locked her door and tried to ignore Mr. P until he returned to his car. Mr. P noted that this episode “ruined” his day because of his lingering anger and irritability.
Intermittent explosive disorder (IED) is more common and complex than was once thought, based on recent evidence. Recurrent, problematic, impulsive aggression is highly comorbid with other psychiatric conditions—including mood and personality disorders—and undermines social relationships and job performance. Typical characteristics of IED are outlined in Table 1.1-3
Table 1
Typical characteristics of intermittent explosive disorder
| Onset in childhood or adolescence (mean age 15), with average duration ±20 years |
| Aggressive outbursts: |
|
| Some episodes may appear without identifiable provocation |
| Male to female ratio 3:1, although some data suggest gender parity |
| Source: Adapted from references 1-3 |
This article offers updated diagnostic criteria and a two-pronged algorithm that can help you diagnose and treat this aggression disorder.
HOW COMMON IS IED?
DSM-IV states that IED is “apparently rare.” This statement is far from surprising, given the limitations of DSM criteria. Surveys of hospitalized patients in the 1980s found that only 1.1% met DSM-III criteria for IED.4 In another study of more than 400 patients seeking treatment for aggression, only 1.8% met DSM-III criteria for IED (although far more would likely have met DSM-IV criteria).5
A more recent survey of 411 psychiatric outpatients6 found that 3.8% met current and 6.2% met lifetime DSM-IV criteria for IED, using the Structured Clinical Interview for DSM-IV Diagnoses (SCID). Reanalysis of a threefold larger data set from the same study site (Coccaro and Zimmerman, unpublished) yielded the same result.
Far from rare. More recently, our findings from a small sample suggested that the community rate of lifetime IED is about 4% by DSM-IV criteria and 5% by research criteria. In the United States, we estimate that the lifetime rate of IED could be 4.5 to 18 million persons using DSM-IV criteria or 6.7 to 22.2 million using IED research criteria. If so, IED is at least as common as other major psychiatric disorders, including schizophrenia or bipolar illness. The ongoing National Comorbidity Study is expected to produce more definitive community data.
PSYCHIATRIC COMORBIDITY
Axis I disorders. IED is highly comorbid with mood, anxiety, and substance use disorders,3,7,8 although no causal relationship has been shown
Mood and substance abuse disorders. IED’s age of onset may precede that of mood and substance use disorders, according to analysis of our unpublished data. If so, comorbid IED may not occur in the context of mood or substance use disorders.
Anxiety disorders. We have noted a similar pattern with IED and anxiety disorders, although phobic anxiety disorders (simple or social phobia) tend to manifest earlier than IED. This suggests that early-onset phobic anxiety might be associated with an increased risk of IED in adolescence or young adulthood.
Bipolar disorder. McElroy9 has suggested a relationship between IED and bipolar disorder. In some samples, as many as one-half of IED patients (56%) have comorbid bipolar disorder when one includes bipolar II and cyclothymia.3 Moreover, some subjects’ aggressive episodes appear to resemble “microdysphoric manic episodes.”9 Other studies,8 however, find a much lower rate (10% or less) of IED comorbidity with bipolar illness.
Bipolar disorder overall may not be highly comorbid with IED, although rates may be higher in specialty clinic samples. In individuals with any kind of bipolar disorder, mood stabilizers— rather than selective serotonin reuptake inhibitors (SSRIs)—are probably the better choice as first-line treatment of IED.9
Axis II disorders. DSM-IV allows IED diagnosis in individuals with borderline or antisocial personality disorder, as long as these cluster B disorders do not better explain the aggressive behavior. How a clinician makes this distinction is not clear; in fact, most clinicians do not diagnose IED in patients with personality disorders, regardless of the clinical picture.
IED comorbidity with borderline or antisocial personality disorders varies with the sample. Persons with personality disorders who seek treatment of aggressive behavior are more likely to have comorbid IED (90%) than those not seeking treatment who are outpatients (50%) or in the community (25%).1,7
Individuals with personality disorders and IED score higher in aggression and lower in psychosocial function than do similar individuals without IED,7 indicating that the additional diagnosis is relevant.
Case report continued.
Mr. P’s outbursts have cost him several friendships, including romantic relationships. He has never advanced at work because he is seen as too volatile to supervise subordinates. Though some of Mr. P’s aggressive outbursts have occurred under the influence of alcohol, most are not related to alcohol or drug use. He has no medical problems and no other psychiatric history.
A full diagnostic evaluation uncovers a personality disorder, not otherwise specified (eight scattered traits from obsessive-compulsive personality disorder and from each of the cluster B personality disorders), and no Axis I condition other than intermittent explosive disorder.
PROBLEMS DEFINING IED
Intermittent explosive disorder is the only DSM diagnosis that applies to persons with histories of recurrent, problematic aggression not caused by another mental or physical disorder. Even so, little research on IED is available. DSM criteria for IED are poorly operationalized and have improved only modestly since the diagnosis was first included in DSM-III. In that revision, IED had four criteria.
“A” criteria specified recurrent outbursts of “seriously assaultive or destructive behavior,” but left unanswered important questions such as:
- What behavior crosses the threshold for seriously” assaultive or destructive?
- Does any physical assault qualify, or only those that cause physical injury (or stigmata)?
- How often or within what time must the behavior occur?
The phrase “recurrent acts of aggression” suggested that at least three acts of aggression were required to reach the threshold, but DSM-III provided no guidelines.
“B” criteria stated that the aggression should be out of proportion to the provocation. But how should one judge this criterion, when provocative stimuli sometimes are clearly sufficient to prompt a justifiably aggressive act?
“C” criteria excluded persons who are aggressive or impulsive between ill-defined “aggressive episodes.” This exclusion was especially limiting because individuals with recurrent, problematic, aggressive behaviors generally are impulsive and aggressive between more-severe outbursts. Excluding those who otherwise met diagnostic criteria for IED led to a spuriously low prevalence rate and limited the number of research subjects. DSM-IV eliminated this criterion but made no other notable changes in IED criteria.
“D” criteria in DSM-III and III-R further restricted the number of individuals who could meet this diagnosis:
- In DSM-III, antisocial personality disorder excluded the diagnosis of IED.
- In DSM-III-R, borderline personality disorder was added as an exclusionary factor.
Because of these restrictions, very few clinically valid cases of IED (individuals meeting A and B criteria) could receive an IED diagnosis.10
EVOLVING DIAGNOSTIC CRITERIA
By the early 1990s, DSM diagnostic criteria clearly severely restricted the study of recurrent, problematic aggression, even though research since DSM-III had greatly advanced our understanding of human aggression. For example, data linked impulsive aggression to deficits in central serotonergic function and suggested that agents that enhance serotonergic activity could modify this behavior.
Some investigators proposed research criteria for IED (IED-R) so that individuals with recurrent, problematic, impulsive aggression could be identified and studied. Research criteria first published in 19987 proposed six changes/clarifications in IED diagnostic criteria:
Lower-intensity aggression. The scope of aggressive behavior was expanded to include verbal and indirect physical aggression, provided that these behaviors are associated with distress and/or impairment. Data from double-blind, placebo-controlled trials indicated that these lower-intensity (although usually higher frequency) behaviors respond well to treatment with SSRIs.11,12
Impulsivity. The aggression was specified as impulsive. This change identified individuals with greater liability for deficits in central serotonergic function and excluded individuals with premeditated or criminal aggression.
A minimal frequency of aggression over time was proposed to make the IED diagnosis more reliable and to ensure that persons with only occasional impulsive aggressive outbursts (especially of low severity) were given this diagnosis.
Subjective distress (in the individual) and/or social or occupational dysfunction was proposed so that putatively aggressive individuals are not diagnosed for manifesting behaviors that are not functionally severe.
Diagnostic exclusionary criteria were modified so that individuals with:
- antisocial or borderline personality disorder could be diagnosed with IED if otherwise warranted
- aggressive behaviors confined within major depression episodes could not be diagnosed with IED.
This last change recognized that impulsive, aggressive outbursts could point to major depressive disorder.
When the revised criteria were tested in patients seeking treatment for aggression, those who met IED-R criteria were found to exhibit significantly greater aggression and impulsivity (using validated scales) and lower global functioning than those who did not.7 Statistical adjustments made to account for aggression score differences eliminated the difference in global functioning, which suggested a direct link between aggression and global function in individuals with IED-R.
Two patterns. Later research uncovered at least patterns of aggressive outbursts:
- low intensity at high frequency (such as verbal arguments or door slamming approximately twice weekly)
- high intensity at low frequency (such as physical aggression resulting in injury or destruction of nontrivial property at least three times per year).
Data revealed that 69% of individuals with IED-like histories displayed both aggression patterns, 20% displayed only the high-intensity/low-frequency pattern, and 11% displayed only the low-intensity/high-frequency pattern.
Because further analysis revealed no important differences between these groups in measures of aggression and impulsivity, IED-R criteria were revised to include both patterns in the “A” criteria. This revision integrated the essences of IED-R and DSM criteria into one diagnostic set (Table 2).
INFLUENCE OF HEREDITY
No twin or adoption studies of IED have been performed. However, family history data suggest that IED (or IED-type behavior) is familial. I recently conducted a blinded, controlled, family history study using IED-R criteria and found a significantly elevated risk for IED (p < 0.01) in relatives of persons with a history of IED (26%), compared with non-IED controls (8%). Comorbid conditions did not affect the risk among the IED subjects or their relatives, suggesting that IED is familial and independent of other conditions.13
Nearly all studies of aggression’s biology and treatment have measured aggression as a dimensional variable along a continuous scale from low to high.14 Our studies have allowed us to explore biological and treatment response correlates. In preliminary analyses, we have found that the maximal prolactin response to d-fenfluramine challenge and the number of platelet serotonin transporter binding sites are:
- reduced in subjects meeting research criteria for IED
- inversely correlated with dimensional measures of impulsive aggression.
Table 2
Updated diagnostic criteria for intermittent explosive disorder
| A. Recurrent incidents of aggression manifest as either: |
| 1. Verbal or physical aggression towards other people, animals, or property occurring twice weekly on average for 1 month |
| OR |
| 2. Three episodes involving physical assault against other people or destruction of property over a 1-year period |
| B. The degree of aggressiveness expressed is grossly out of proportion to the provocation or any precipitating psychosocial stressors |
| C. The aggressive behavior is generally not premeditated (ie, is impulsive) and is not committed to achieve a tangible objective (such as money, power, intimidation, etc.) |
| D. The aggressive behavior causes marked distress in the individual or impairs occupational or interpersonal functioning |
| E. The aggressive behavior is not better explained by another mental disorder (such as a major depressive/manic/psychotic disorder, attention-deficit/hyperactivity disorder, general medical condition [head trauma, Alzheimer’s disease], or due to the direct physiologic effects of a substance) |
| Source: Adapted from reference 7 |
Earlier, Virkkunen et al15 reported reduced cerebrospinal fluid 5-hydroxyindoleacetic acid concentrations in persons diagnosed with IED based on DSM-III criteria, compared with persons who were not diagnosed with IED and those who demonstrated nonimpulsive aggression.
TREATING IED
Cognitive therapy. Few double-blind, randomized, placebo-controlled trials of any treatments for IED have been published. Trials using cognitive-behavioral approaches have reduced self-rated anger and its expression in young adults with anger disorders.16 Although many of these subjects may have had IED, it is not known if this approach works in IED.
Table 3
Characteristic behaviors of aggressive individuals*
| Severity | Behaviors |
|---|---|
| Mildly aggressive | Occasional verbal arguments and/or temper tantrums |
| Moderately aggressive | Frequent verbal arguments and temper tantrums (about twice weekly on average), occasional destruction of property, rare or occasional physical assault against others (usually without injury) |
| Highly aggressive | Frequent verbal arguments and temper tantrums (about twice weekly) and/or more than occasional destruction of property or physical assault against others, sometimes with injury |
| * Characteristics given are descriptive and not based on data. | |
Drug therapy. SSRIs. A trial by this author using fluoxetine showed that impulsive aggressive behavior responds to treatment that targets the central serotonergic system.12 Forty subjects with personality disorders and histories of impulsive aggression received fluoxetine, 20 to 60 mg qd, or placebo for 12 weeks. Fluoxetine reduced overt aggression and irritability about 67% more than placebo, as assessed by the Overt Aggression Scale Modified for Outpatients (OAS-M).
All subjects met research criteria for IED. A reanalysis suggests that SSRIs may be most effective in moderately aggressive patients (Table 3),17 whose serotonergic system may be less impaired than that of highly aggressive patients.18
Mood stabilizers. Impulsively aggressive subjects who do not respond to an SSRI may respond to a mood stabilizer.19 An antiaggressive response in IED-like subjects has been reported for lithium,20 carbamazepine,21 and diphenylhydantoin.22
Recently, Hollander et al23 reported greater reduction in overt aggression scores in IED subjects with a DSM cluster B personality disorder who were treated with divalproex, compared with placebo. This study used the same design and outcome measure as our study12 and included subjects who met both DSM-IV and research criteria for IED.
For unknown reasons, divalproex was no more effective than placebo in IED subjects without cluster B personality disorder. More research is needed to uncover predictors of antiaggressive response in IED subjects.
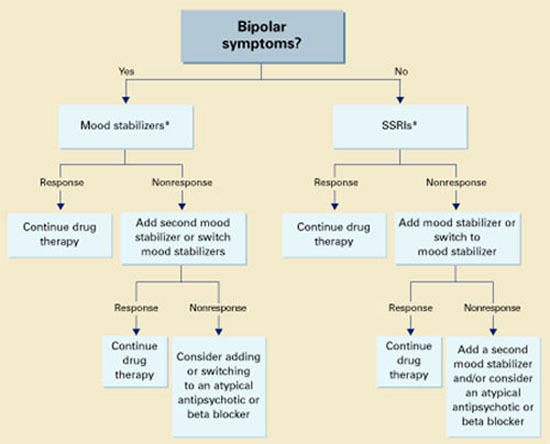
Unipolar vs. bipolar. McElroy9 has suggested using SSRIs (or other antidepressants) as first-line treatment for IED subjects with unipolar affective symptoms and mood stabilizers for those with bipolar affective symptoms. IED subjects without bipolar affective symptoms should be treated first with SSRIs (Algorithm). Preliminary data suggest a role for atypical antipsychotics to treat aggressive behavior in patients with schizophrenia or bipolar disorder, but no empiric data exist.
Beta blockers such as propranolol also may be considered.2 However, beta blockers are more difficult to dose and are associated with more burdensome side effects, compared with SSRIs.
Algorithm Suggested 2-pronged approach for treating intermittent explosive disorder
* With or without an anger management program, which may precede drug interventionThe full effects of antiaggressive treatment with an SSRI (E. Coccaro, unpublished observations) or a mood stabilizer19 may take 3 months to observe12,20,22,23 and tend to disappear soon after treatment is discontinued.
Therefore, an adequate trial of SSRIs or mood stabilizers is no less than 3 months. If improvement is seen, continue drug treatment indefinitely.
Case report continued.
Mr. P was started on an SSRI. His aggressive outbursts decreased in intensity and frequency over 3 months but were not eliminated. After 6 months he dropped out of treatment, but returned 5 weeks later because his aggressive outbursts had resumed their pre-treatment level.
SSRI treatment was restarted, and Mr. P began a 12-week anger management course of relaxation training, cognitive restructuring, and coping skills training. He gained greater control over his aggressive outbursts and continues monthly medication checks and anger management “booster sessions.”
Related resources
- Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
- Olvera RL. Intermittent explosive disorder: epidemiology, diagnosis and management. CNS Drugs 2002;16:517-26.
Drug brand names
- Carbamazepine • Tegretol
- Diphenylhydantoin • Dilantin
- Divalproex • Depakote
- Fluoxetine • Prozac
- Lithium • Lithobid
- Propanolol • Inderal
Disclosure
Dr. Coccaro reports that he receives research grants and serves on the speaker’s bureau or as a consultant to Eli Lilly and Co., Abbott Laboratories, GlaxoSmithKline, and Forrest Laboratories.
1. Coccaro EF, Schimdt CA, Samuels JF, et al. Lifetime rates of intermittent explosive disorder in a community sample (abstract). Philadelphia: American Psychiatric Association annual meeting, 2002.
2. Mattes JA. Comparative effectiveness of carbamazepine and propranolol for rage outbursts. J Neuropsychiatry Clin Neurosci 1990;2:159-64.
3. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry 1998;59:203-10.
4. Monopolis S, Lion JR. Problems in the diagnosis of intermittent explosive disorder. Am J Psychiatry 1983;140:1200-2.
5. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (abstract 265). Washington, DC: American Psychiatric Association annual meeting, 1998.
6. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (APA new research abstracts #265). Washington, DC: American Psychiatric Publishing, Inc., 1998.
7. Coccaro EF, Kavoussi RJ, Berman ME, Lish JD. Intermittent explosive disorder-revised: development, reliability and validity of research criteria. Compr Psychiatry 1998;39:368-76.
8. Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
9. McElroy SL. Recognition and treatment of DSM-IV intermittent explosive disorder. J Clin Psychiatry 1999;60(suppl 15):12-16.
10. Felthous AR, Bryant G, Wingerter CB, Barratt E. The diagnosis of intermittent explosive disorder in violent men. Bull Am Acad Psychiatry Law 1991;19:71-9.
11. Salzman C, Wolfson AN, Schatzberg A, et al. Effect of fluoxetine on anger in symptomatic volunteers with borderline personality disorder. J Clin Psychopharmacology 1995;15:23-9.
12. Coccaro EF, Kavoussi RJ. Fluoxetine and impulsive aggressive behavior in personality disordered subjects. Arch Gen Psychiatry 1997;54:1081-8.
13. Coccaro EF. Family history study of intermittent explosive disorder (abstract). Washington, DC: American Psychiatric Association annual meeting, 1999.
14. Coccaro EF, Siever LJ. Pathophysiology and treatment of aggression. In: Davis KL, Charney D, Coyle JT, Nemeroff D (eds). Psychopharmacology: the fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002;1709-24
15. Virkkunen M, Rawlings R, Tokola R, et al. CSF biochemistries, glucose metabolism, and diurnal activity rhythms in alcoholic, violent offenders, fire setters, and healthy volunteers. Arch Gen Psychiatry 1994;51:20-7.
16. Deffenbacher JL. Psychosocial interventions: anger disorders. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
17. Lee R, Coccaro EF. Treatment of aggression: serotonergic agents. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
18. Coccaro EF, Kavoussi RJ, Hauger RL. Serotonin function and antiaggressive responses to fluoxetine: a pilot study. Biol Psychiatry 1997;42:546-52.
19. Kavoussi RJ, Coccaro EF. Divalproex sodium for impulsive aggressive behavior in patients with personality disorder. J Clin Psychiatry 1998;59:676-80.
20. Sheard MH, Marini J, Bridges CI, Wagner E. The effect of lithium on impulsive aggressive behavior in man. Am J Psychiatry 1976;133:1409-13.
21. Cowdry RW, Gardner DL. Pharmacotherapy of borderline personality disorder: alprazolam, carbamazepine, trifluroperazine, and tranylcypromine. Arch Gen Psychiatry 1988;45:111-19.
22. Barratt ES, Stanford MS, Felthous AR, Kent TA. The effects of phenytoin on impulsive and premeditated aggression: a controlled study. J Clin Psychopharmacology 1997;17:341-9.
23. Hollander E, Tracy KA, Swann AC, et al. Divalproex sodium is superior to placebo for impulsive aggression in Cluster B personality disorders. Neuropsychopharmacology 2003;28:1186-97.
Mr. P, age 41, has a “problem with anger.” Since age 17, he has had sudden outbursts of screaming and shouting, with occasional minor damage to objects. These outbursts—including episodes of “road rage”—occur once or more per week and almost daily for months at a time.
Mr. P has also had more violent episodes— sometimes every 2 to 3 months—in which he has punched holes in walls, destroyed a computer with a hammer, and assaulted other people with his fists. These events are not premeditated and are typically triggered by Mr. P’s frustration at not being “perfect” or by others breaking what he considers “general rules of conduct.”
The day before his initial visit, while he was stuck in traffic, Mr. P saw a car speeding down the shoulder. Enraged, he pulled in front of the car so that the driver had to slam on the brakes. He jumped out of his car and approached the other driver, shouting obscenities. The other driver locked her door and tried to ignore Mr. P until he returned to his car. Mr. P noted that this episode “ruined” his day because of his lingering anger and irritability.
Intermittent explosive disorder (IED) is more common and complex than was once thought, based on recent evidence. Recurrent, problematic, impulsive aggression is highly comorbid with other psychiatric conditions—including mood and personality disorders—and undermines social relationships and job performance. Typical characteristics of IED are outlined in Table 1.1-3
Table 1
Typical characteristics of intermittent explosive disorder
| Onset in childhood or adolescence (mean age 15), with average duration ±20 years |
| Aggressive outbursts: |
|
| Some episodes may appear without identifiable provocation |
| Male to female ratio 3:1, although some data suggest gender parity |
| Source: Adapted from references 1-3 |
This article offers updated diagnostic criteria and a two-pronged algorithm that can help you diagnose and treat this aggression disorder.
HOW COMMON IS IED?
DSM-IV states that IED is “apparently rare.” This statement is far from surprising, given the limitations of DSM criteria. Surveys of hospitalized patients in the 1980s found that only 1.1% met DSM-III criteria for IED.4 In another study of more than 400 patients seeking treatment for aggression, only 1.8% met DSM-III criteria for IED (although far more would likely have met DSM-IV criteria).5
A more recent survey of 411 psychiatric outpatients6 found that 3.8% met current and 6.2% met lifetime DSM-IV criteria for IED, using the Structured Clinical Interview for DSM-IV Diagnoses (SCID). Reanalysis of a threefold larger data set from the same study site (Coccaro and Zimmerman, unpublished) yielded the same result.
Far from rare. More recently, our findings from a small sample suggested that the community rate of lifetime IED is about 4% by DSM-IV criteria and 5% by research criteria. In the United States, we estimate that the lifetime rate of IED could be 4.5 to 18 million persons using DSM-IV criteria or 6.7 to 22.2 million using IED research criteria. If so, IED is at least as common as other major psychiatric disorders, including schizophrenia or bipolar illness. The ongoing National Comorbidity Study is expected to produce more definitive community data.
PSYCHIATRIC COMORBIDITY
Axis I disorders. IED is highly comorbid with mood, anxiety, and substance use disorders,3,7,8 although no causal relationship has been shown
Mood and substance abuse disorders. IED’s age of onset may precede that of mood and substance use disorders, according to analysis of our unpublished data. If so, comorbid IED may not occur in the context of mood or substance use disorders.
Anxiety disorders. We have noted a similar pattern with IED and anxiety disorders, although phobic anxiety disorders (simple or social phobia) tend to manifest earlier than IED. This suggests that early-onset phobic anxiety might be associated with an increased risk of IED in adolescence or young adulthood.
Bipolar disorder. McElroy9 has suggested a relationship between IED and bipolar disorder. In some samples, as many as one-half of IED patients (56%) have comorbid bipolar disorder when one includes bipolar II and cyclothymia.3 Moreover, some subjects’ aggressive episodes appear to resemble “microdysphoric manic episodes.”9 Other studies,8 however, find a much lower rate (10% or less) of IED comorbidity with bipolar illness.
Bipolar disorder overall may not be highly comorbid with IED, although rates may be higher in specialty clinic samples. In individuals with any kind of bipolar disorder, mood stabilizers— rather than selective serotonin reuptake inhibitors (SSRIs)—are probably the better choice as first-line treatment of IED.9
Axis II disorders. DSM-IV allows IED diagnosis in individuals with borderline or antisocial personality disorder, as long as these cluster B disorders do not better explain the aggressive behavior. How a clinician makes this distinction is not clear; in fact, most clinicians do not diagnose IED in patients with personality disorders, regardless of the clinical picture.
IED comorbidity with borderline or antisocial personality disorders varies with the sample. Persons with personality disorders who seek treatment of aggressive behavior are more likely to have comorbid IED (90%) than those not seeking treatment who are outpatients (50%) or in the community (25%).1,7
Individuals with personality disorders and IED score higher in aggression and lower in psychosocial function than do similar individuals without IED,7 indicating that the additional diagnosis is relevant.
Case report continued.
Mr. P’s outbursts have cost him several friendships, including romantic relationships. He has never advanced at work because he is seen as too volatile to supervise subordinates. Though some of Mr. P’s aggressive outbursts have occurred under the influence of alcohol, most are not related to alcohol or drug use. He has no medical problems and no other psychiatric history.
A full diagnostic evaluation uncovers a personality disorder, not otherwise specified (eight scattered traits from obsessive-compulsive personality disorder and from each of the cluster B personality disorders), and no Axis I condition other than intermittent explosive disorder.
PROBLEMS DEFINING IED
Intermittent explosive disorder is the only DSM diagnosis that applies to persons with histories of recurrent, problematic aggression not caused by another mental or physical disorder. Even so, little research on IED is available. DSM criteria for IED are poorly operationalized and have improved only modestly since the diagnosis was first included in DSM-III. In that revision, IED had four criteria.
“A” criteria specified recurrent outbursts of “seriously assaultive or destructive behavior,” but left unanswered important questions such as:
- What behavior crosses the threshold for seriously” assaultive or destructive?
- Does any physical assault qualify, or only those that cause physical injury (or stigmata)?
- How often or within what time must the behavior occur?
The phrase “recurrent acts of aggression” suggested that at least three acts of aggression were required to reach the threshold, but DSM-III provided no guidelines.
“B” criteria stated that the aggression should be out of proportion to the provocation. But how should one judge this criterion, when provocative stimuli sometimes are clearly sufficient to prompt a justifiably aggressive act?
“C” criteria excluded persons who are aggressive or impulsive between ill-defined “aggressive episodes.” This exclusion was especially limiting because individuals with recurrent, problematic, aggressive behaviors generally are impulsive and aggressive between more-severe outbursts. Excluding those who otherwise met diagnostic criteria for IED led to a spuriously low prevalence rate and limited the number of research subjects. DSM-IV eliminated this criterion but made no other notable changes in IED criteria.
“D” criteria in DSM-III and III-R further restricted the number of individuals who could meet this diagnosis:
- In DSM-III, antisocial personality disorder excluded the diagnosis of IED.
- In DSM-III-R, borderline personality disorder was added as an exclusionary factor.
Because of these restrictions, very few clinically valid cases of IED (individuals meeting A and B criteria) could receive an IED diagnosis.10
EVOLVING DIAGNOSTIC CRITERIA
By the early 1990s, DSM diagnostic criteria clearly severely restricted the study of recurrent, problematic aggression, even though research since DSM-III had greatly advanced our understanding of human aggression. For example, data linked impulsive aggression to deficits in central serotonergic function and suggested that agents that enhance serotonergic activity could modify this behavior.
Some investigators proposed research criteria for IED (IED-R) so that individuals with recurrent, problematic, impulsive aggression could be identified and studied. Research criteria first published in 19987 proposed six changes/clarifications in IED diagnostic criteria:
Lower-intensity aggression. The scope of aggressive behavior was expanded to include verbal and indirect physical aggression, provided that these behaviors are associated with distress and/or impairment. Data from double-blind, placebo-controlled trials indicated that these lower-intensity (although usually higher frequency) behaviors respond well to treatment with SSRIs.11,12
Impulsivity. The aggression was specified as impulsive. This change identified individuals with greater liability for deficits in central serotonergic function and excluded individuals with premeditated or criminal aggression.
A minimal frequency of aggression over time was proposed to make the IED diagnosis more reliable and to ensure that persons with only occasional impulsive aggressive outbursts (especially of low severity) were given this diagnosis.
Subjective distress (in the individual) and/or social or occupational dysfunction was proposed so that putatively aggressive individuals are not diagnosed for manifesting behaviors that are not functionally severe.
Diagnostic exclusionary criteria were modified so that individuals with:
- antisocial or borderline personality disorder could be diagnosed with IED if otherwise warranted
- aggressive behaviors confined within major depression episodes could not be diagnosed with IED.
This last change recognized that impulsive, aggressive outbursts could point to major depressive disorder.
When the revised criteria were tested in patients seeking treatment for aggression, those who met IED-R criteria were found to exhibit significantly greater aggression and impulsivity (using validated scales) and lower global functioning than those who did not.7 Statistical adjustments made to account for aggression score differences eliminated the difference in global functioning, which suggested a direct link between aggression and global function in individuals with IED-R.
Two patterns. Later research uncovered at least patterns of aggressive outbursts:
- low intensity at high frequency (such as verbal arguments or door slamming approximately twice weekly)
- high intensity at low frequency (such as physical aggression resulting in injury or destruction of nontrivial property at least three times per year).
Data revealed that 69% of individuals with IED-like histories displayed both aggression patterns, 20% displayed only the high-intensity/low-frequency pattern, and 11% displayed only the low-intensity/high-frequency pattern.
Because further analysis revealed no important differences between these groups in measures of aggression and impulsivity, IED-R criteria were revised to include both patterns in the “A” criteria. This revision integrated the essences of IED-R and DSM criteria into one diagnostic set (Table 2).
INFLUENCE OF HEREDITY
No twin or adoption studies of IED have been performed. However, family history data suggest that IED (or IED-type behavior) is familial. I recently conducted a blinded, controlled, family history study using IED-R criteria and found a significantly elevated risk for IED (p < 0.01) in relatives of persons with a history of IED (26%), compared with non-IED controls (8%). Comorbid conditions did not affect the risk among the IED subjects or their relatives, suggesting that IED is familial and independent of other conditions.13
Nearly all studies of aggression’s biology and treatment have measured aggression as a dimensional variable along a continuous scale from low to high.14 Our studies have allowed us to explore biological and treatment response correlates. In preliminary analyses, we have found that the maximal prolactin response to d-fenfluramine challenge and the number of platelet serotonin transporter binding sites are:
- reduced in subjects meeting research criteria for IED
- inversely correlated with dimensional measures of impulsive aggression.
Table 2
Updated diagnostic criteria for intermittent explosive disorder
| A. Recurrent incidents of aggression manifest as either: |
| 1. Verbal or physical aggression towards other people, animals, or property occurring twice weekly on average for 1 month |
| OR |
| 2. Three episodes involving physical assault against other people or destruction of property over a 1-year period |
| B. The degree of aggressiveness expressed is grossly out of proportion to the provocation or any precipitating psychosocial stressors |
| C. The aggressive behavior is generally not premeditated (ie, is impulsive) and is not committed to achieve a tangible objective (such as money, power, intimidation, etc.) |
| D. The aggressive behavior causes marked distress in the individual or impairs occupational or interpersonal functioning |
| E. The aggressive behavior is not better explained by another mental disorder (such as a major depressive/manic/psychotic disorder, attention-deficit/hyperactivity disorder, general medical condition [head trauma, Alzheimer’s disease], or due to the direct physiologic effects of a substance) |
| Source: Adapted from reference 7 |
Earlier, Virkkunen et al15 reported reduced cerebrospinal fluid 5-hydroxyindoleacetic acid concentrations in persons diagnosed with IED based on DSM-III criteria, compared with persons who were not diagnosed with IED and those who demonstrated nonimpulsive aggression.
TREATING IED
Cognitive therapy. Few double-blind, randomized, placebo-controlled trials of any treatments for IED have been published. Trials using cognitive-behavioral approaches have reduced self-rated anger and its expression in young adults with anger disorders.16 Although many of these subjects may have had IED, it is not known if this approach works in IED.
Table 3
Characteristic behaviors of aggressive individuals*
| Severity | Behaviors |
|---|---|
| Mildly aggressive | Occasional verbal arguments and/or temper tantrums |
| Moderately aggressive | Frequent verbal arguments and temper tantrums (about twice weekly on average), occasional destruction of property, rare or occasional physical assault against others (usually without injury) |
| Highly aggressive | Frequent verbal arguments and temper tantrums (about twice weekly) and/or more than occasional destruction of property or physical assault against others, sometimes with injury |
| * Characteristics given are descriptive and not based on data. | |
Drug therapy. SSRIs. A trial by this author using fluoxetine showed that impulsive aggressive behavior responds to treatment that targets the central serotonergic system.12 Forty subjects with personality disorders and histories of impulsive aggression received fluoxetine, 20 to 60 mg qd, or placebo for 12 weeks. Fluoxetine reduced overt aggression and irritability about 67% more than placebo, as assessed by the Overt Aggression Scale Modified for Outpatients (OAS-M).
All subjects met research criteria for IED. A reanalysis suggests that SSRIs may be most effective in moderately aggressive patients (Table 3),17 whose serotonergic system may be less impaired than that of highly aggressive patients.18
Mood stabilizers. Impulsively aggressive subjects who do not respond to an SSRI may respond to a mood stabilizer.19 An antiaggressive response in IED-like subjects has been reported for lithium,20 carbamazepine,21 and diphenylhydantoin.22
Recently, Hollander et al23 reported greater reduction in overt aggression scores in IED subjects with a DSM cluster B personality disorder who were treated with divalproex, compared with placebo. This study used the same design and outcome measure as our study12 and included subjects who met both DSM-IV and research criteria for IED.
For unknown reasons, divalproex was no more effective than placebo in IED subjects without cluster B personality disorder. More research is needed to uncover predictors of antiaggressive response in IED subjects.
Unipolar vs. bipolar. McElroy9 has suggested using SSRIs (or other antidepressants) as first-line treatment for IED subjects with unipolar affective symptoms and mood stabilizers for those with bipolar affective symptoms. IED subjects without bipolar affective symptoms should be treated first with SSRIs (Algorithm). Preliminary data suggest a role for atypical antipsychotics to treat aggressive behavior in patients with schizophrenia or bipolar disorder, but no empiric data exist.
Beta blockers such as propranolol also may be considered.2 However, beta blockers are more difficult to dose and are associated with more burdensome side effects, compared with SSRIs.
Algorithm Suggested 2-pronged approach for treating intermittent explosive disorder
* With or without an anger management program, which may precede drug interventionThe full effects of antiaggressive treatment with an SSRI (E. Coccaro, unpublished observations) or a mood stabilizer19 may take 3 months to observe12,20,22,23 and tend to disappear soon after treatment is discontinued.
Therefore, an adequate trial of SSRIs or mood stabilizers is no less than 3 months. If improvement is seen, continue drug treatment indefinitely.
Case report continued.
Mr. P was started on an SSRI. His aggressive outbursts decreased in intensity and frequency over 3 months but were not eliminated. After 6 months he dropped out of treatment, but returned 5 weeks later because his aggressive outbursts had resumed their pre-treatment level.
SSRI treatment was restarted, and Mr. P began a 12-week anger management course of relaxation training, cognitive restructuring, and coping skills training. He gained greater control over his aggressive outbursts and continues monthly medication checks and anger management “booster sessions.”
Related resources
- Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
- Olvera RL. Intermittent explosive disorder: epidemiology, diagnosis and management. CNS Drugs 2002;16:517-26.
Drug brand names
- Carbamazepine • Tegretol
- Diphenylhydantoin • Dilantin
- Divalproex • Depakote
- Fluoxetine • Prozac
- Lithium • Lithobid
- Propanolol • Inderal
Disclosure
Dr. Coccaro reports that he receives research grants and serves on the speaker’s bureau or as a consultant to Eli Lilly and Co., Abbott Laboratories, GlaxoSmithKline, and Forrest Laboratories.
Mr. P, age 41, has a “problem with anger.” Since age 17, he has had sudden outbursts of screaming and shouting, with occasional minor damage to objects. These outbursts—including episodes of “road rage”—occur once or more per week and almost daily for months at a time.
Mr. P has also had more violent episodes— sometimes every 2 to 3 months—in which he has punched holes in walls, destroyed a computer with a hammer, and assaulted other people with his fists. These events are not premeditated and are typically triggered by Mr. P’s frustration at not being “perfect” or by others breaking what he considers “general rules of conduct.”
The day before his initial visit, while he was stuck in traffic, Mr. P saw a car speeding down the shoulder. Enraged, he pulled in front of the car so that the driver had to slam on the brakes. He jumped out of his car and approached the other driver, shouting obscenities. The other driver locked her door and tried to ignore Mr. P until he returned to his car. Mr. P noted that this episode “ruined” his day because of his lingering anger and irritability.
Intermittent explosive disorder (IED) is more common and complex than was once thought, based on recent evidence. Recurrent, problematic, impulsive aggression is highly comorbid with other psychiatric conditions—including mood and personality disorders—and undermines social relationships and job performance. Typical characteristics of IED are outlined in Table 1.1-3
Table 1
Typical characteristics of intermittent explosive disorder
| Onset in childhood or adolescence (mean age 15), with average duration ±20 years |
| Aggressive outbursts: |
|
| Some episodes may appear without identifiable provocation |
| Male to female ratio 3:1, although some data suggest gender parity |
| Source: Adapted from references 1-3 |
This article offers updated diagnostic criteria and a two-pronged algorithm that can help you diagnose and treat this aggression disorder.
HOW COMMON IS IED?
DSM-IV states that IED is “apparently rare.” This statement is far from surprising, given the limitations of DSM criteria. Surveys of hospitalized patients in the 1980s found that only 1.1% met DSM-III criteria for IED.4 In another study of more than 400 patients seeking treatment for aggression, only 1.8% met DSM-III criteria for IED (although far more would likely have met DSM-IV criteria).5
A more recent survey of 411 psychiatric outpatients6 found that 3.8% met current and 6.2% met lifetime DSM-IV criteria for IED, using the Structured Clinical Interview for DSM-IV Diagnoses (SCID). Reanalysis of a threefold larger data set from the same study site (Coccaro and Zimmerman, unpublished) yielded the same result.
Far from rare. More recently, our findings from a small sample suggested that the community rate of lifetime IED is about 4% by DSM-IV criteria and 5% by research criteria. In the United States, we estimate that the lifetime rate of IED could be 4.5 to 18 million persons using DSM-IV criteria or 6.7 to 22.2 million using IED research criteria. If so, IED is at least as common as other major psychiatric disorders, including schizophrenia or bipolar illness. The ongoing National Comorbidity Study is expected to produce more definitive community data.
PSYCHIATRIC COMORBIDITY
Axis I disorders. IED is highly comorbid with mood, anxiety, and substance use disorders,3,7,8 although no causal relationship has been shown
Mood and substance abuse disorders. IED’s age of onset may precede that of mood and substance use disorders, according to analysis of our unpublished data. If so, comorbid IED may not occur in the context of mood or substance use disorders.
Anxiety disorders. We have noted a similar pattern with IED and anxiety disorders, although phobic anxiety disorders (simple or social phobia) tend to manifest earlier than IED. This suggests that early-onset phobic anxiety might be associated with an increased risk of IED in adolescence or young adulthood.
Bipolar disorder. McElroy9 has suggested a relationship between IED and bipolar disorder. In some samples, as many as one-half of IED patients (56%) have comorbid bipolar disorder when one includes bipolar II and cyclothymia.3 Moreover, some subjects’ aggressive episodes appear to resemble “microdysphoric manic episodes.”9 Other studies,8 however, find a much lower rate (10% or less) of IED comorbidity with bipolar illness.
Bipolar disorder overall may not be highly comorbid with IED, although rates may be higher in specialty clinic samples. In individuals with any kind of bipolar disorder, mood stabilizers— rather than selective serotonin reuptake inhibitors (SSRIs)—are probably the better choice as first-line treatment of IED.9
Axis II disorders. DSM-IV allows IED diagnosis in individuals with borderline or antisocial personality disorder, as long as these cluster B disorders do not better explain the aggressive behavior. How a clinician makes this distinction is not clear; in fact, most clinicians do not diagnose IED in patients with personality disorders, regardless of the clinical picture.
IED comorbidity with borderline or antisocial personality disorders varies with the sample. Persons with personality disorders who seek treatment of aggressive behavior are more likely to have comorbid IED (90%) than those not seeking treatment who are outpatients (50%) or in the community (25%).1,7
Individuals with personality disorders and IED score higher in aggression and lower in psychosocial function than do similar individuals without IED,7 indicating that the additional diagnosis is relevant.
Case report continued.
Mr. P’s outbursts have cost him several friendships, including romantic relationships. He has never advanced at work because he is seen as too volatile to supervise subordinates. Though some of Mr. P’s aggressive outbursts have occurred under the influence of alcohol, most are not related to alcohol or drug use. He has no medical problems and no other psychiatric history.
A full diagnostic evaluation uncovers a personality disorder, not otherwise specified (eight scattered traits from obsessive-compulsive personality disorder and from each of the cluster B personality disorders), and no Axis I condition other than intermittent explosive disorder.
PROBLEMS DEFINING IED
Intermittent explosive disorder is the only DSM diagnosis that applies to persons with histories of recurrent, problematic aggression not caused by another mental or physical disorder. Even so, little research on IED is available. DSM criteria for IED are poorly operationalized and have improved only modestly since the diagnosis was first included in DSM-III. In that revision, IED had four criteria.
“A” criteria specified recurrent outbursts of “seriously assaultive or destructive behavior,” but left unanswered important questions such as:
- What behavior crosses the threshold for seriously” assaultive or destructive?
- Does any physical assault qualify, or only those that cause physical injury (or stigmata)?
- How often or within what time must the behavior occur?
The phrase “recurrent acts of aggression” suggested that at least three acts of aggression were required to reach the threshold, but DSM-III provided no guidelines.
“B” criteria stated that the aggression should be out of proportion to the provocation. But how should one judge this criterion, when provocative stimuli sometimes are clearly sufficient to prompt a justifiably aggressive act?
“C” criteria excluded persons who are aggressive or impulsive between ill-defined “aggressive episodes.” This exclusion was especially limiting because individuals with recurrent, problematic, aggressive behaviors generally are impulsive and aggressive between more-severe outbursts. Excluding those who otherwise met diagnostic criteria for IED led to a spuriously low prevalence rate and limited the number of research subjects. DSM-IV eliminated this criterion but made no other notable changes in IED criteria.
“D” criteria in DSM-III and III-R further restricted the number of individuals who could meet this diagnosis:
- In DSM-III, antisocial personality disorder excluded the diagnosis of IED.
- In DSM-III-R, borderline personality disorder was added as an exclusionary factor.
Because of these restrictions, very few clinically valid cases of IED (individuals meeting A and B criteria) could receive an IED diagnosis.10
EVOLVING DIAGNOSTIC CRITERIA
By the early 1990s, DSM diagnostic criteria clearly severely restricted the study of recurrent, problematic aggression, even though research since DSM-III had greatly advanced our understanding of human aggression. For example, data linked impulsive aggression to deficits in central serotonergic function and suggested that agents that enhance serotonergic activity could modify this behavior.
Some investigators proposed research criteria for IED (IED-R) so that individuals with recurrent, problematic, impulsive aggression could be identified and studied. Research criteria first published in 19987 proposed six changes/clarifications in IED diagnostic criteria:
Lower-intensity aggression. The scope of aggressive behavior was expanded to include verbal and indirect physical aggression, provided that these behaviors are associated with distress and/or impairment. Data from double-blind, placebo-controlled trials indicated that these lower-intensity (although usually higher frequency) behaviors respond well to treatment with SSRIs.11,12
Impulsivity. The aggression was specified as impulsive. This change identified individuals with greater liability for deficits in central serotonergic function and excluded individuals with premeditated or criminal aggression.
A minimal frequency of aggression over time was proposed to make the IED diagnosis more reliable and to ensure that persons with only occasional impulsive aggressive outbursts (especially of low severity) were given this diagnosis.
Subjective distress (in the individual) and/or social or occupational dysfunction was proposed so that putatively aggressive individuals are not diagnosed for manifesting behaviors that are not functionally severe.
Diagnostic exclusionary criteria were modified so that individuals with:
- antisocial or borderline personality disorder could be diagnosed with IED if otherwise warranted
- aggressive behaviors confined within major depression episodes could not be diagnosed with IED.
This last change recognized that impulsive, aggressive outbursts could point to major depressive disorder.
When the revised criteria were tested in patients seeking treatment for aggression, those who met IED-R criteria were found to exhibit significantly greater aggression and impulsivity (using validated scales) and lower global functioning than those who did not.7 Statistical adjustments made to account for aggression score differences eliminated the difference in global functioning, which suggested a direct link between aggression and global function in individuals with IED-R.
Two patterns. Later research uncovered at least patterns of aggressive outbursts:
- low intensity at high frequency (such as verbal arguments or door slamming approximately twice weekly)
- high intensity at low frequency (such as physical aggression resulting in injury or destruction of nontrivial property at least three times per year).
Data revealed that 69% of individuals with IED-like histories displayed both aggression patterns, 20% displayed only the high-intensity/low-frequency pattern, and 11% displayed only the low-intensity/high-frequency pattern.
Because further analysis revealed no important differences between these groups in measures of aggression and impulsivity, IED-R criteria were revised to include both patterns in the “A” criteria. This revision integrated the essences of IED-R and DSM criteria into one diagnostic set (Table 2).
INFLUENCE OF HEREDITY
No twin or adoption studies of IED have been performed. However, family history data suggest that IED (or IED-type behavior) is familial. I recently conducted a blinded, controlled, family history study using IED-R criteria and found a significantly elevated risk for IED (p < 0.01) in relatives of persons with a history of IED (26%), compared with non-IED controls (8%). Comorbid conditions did not affect the risk among the IED subjects or their relatives, suggesting that IED is familial and independent of other conditions.13
Nearly all studies of aggression’s biology and treatment have measured aggression as a dimensional variable along a continuous scale from low to high.14 Our studies have allowed us to explore biological and treatment response correlates. In preliminary analyses, we have found that the maximal prolactin response to d-fenfluramine challenge and the number of platelet serotonin transporter binding sites are:
- reduced in subjects meeting research criteria for IED
- inversely correlated with dimensional measures of impulsive aggression.
Table 2
Updated diagnostic criteria for intermittent explosive disorder
| A. Recurrent incidents of aggression manifest as either: |
| 1. Verbal or physical aggression towards other people, animals, or property occurring twice weekly on average for 1 month |
| OR |
| 2. Three episodes involving physical assault against other people or destruction of property over a 1-year period |
| B. The degree of aggressiveness expressed is grossly out of proportion to the provocation or any precipitating psychosocial stressors |
| C. The aggressive behavior is generally not premeditated (ie, is impulsive) and is not committed to achieve a tangible objective (such as money, power, intimidation, etc.) |
| D. The aggressive behavior causes marked distress in the individual or impairs occupational or interpersonal functioning |
| E. The aggressive behavior is not better explained by another mental disorder (such as a major depressive/manic/psychotic disorder, attention-deficit/hyperactivity disorder, general medical condition [head trauma, Alzheimer’s disease], or due to the direct physiologic effects of a substance) |
| Source: Adapted from reference 7 |
Earlier, Virkkunen et al15 reported reduced cerebrospinal fluid 5-hydroxyindoleacetic acid concentrations in persons diagnosed with IED based on DSM-III criteria, compared with persons who were not diagnosed with IED and those who demonstrated nonimpulsive aggression.
TREATING IED
Cognitive therapy. Few double-blind, randomized, placebo-controlled trials of any treatments for IED have been published. Trials using cognitive-behavioral approaches have reduced self-rated anger and its expression in young adults with anger disorders.16 Although many of these subjects may have had IED, it is not known if this approach works in IED.
Table 3
Characteristic behaviors of aggressive individuals*
| Severity | Behaviors |
|---|---|
| Mildly aggressive | Occasional verbal arguments and/or temper tantrums |
| Moderately aggressive | Frequent verbal arguments and temper tantrums (about twice weekly on average), occasional destruction of property, rare or occasional physical assault against others (usually without injury) |
| Highly aggressive | Frequent verbal arguments and temper tantrums (about twice weekly) and/or more than occasional destruction of property or physical assault against others, sometimes with injury |
| * Characteristics given are descriptive and not based on data. | |
Drug therapy. SSRIs. A trial by this author using fluoxetine showed that impulsive aggressive behavior responds to treatment that targets the central serotonergic system.12 Forty subjects with personality disorders and histories of impulsive aggression received fluoxetine, 20 to 60 mg qd, or placebo for 12 weeks. Fluoxetine reduced overt aggression and irritability about 67% more than placebo, as assessed by the Overt Aggression Scale Modified for Outpatients (OAS-M).
All subjects met research criteria for IED. A reanalysis suggests that SSRIs may be most effective in moderately aggressive patients (Table 3),17 whose serotonergic system may be less impaired than that of highly aggressive patients.18
Mood stabilizers. Impulsively aggressive subjects who do not respond to an SSRI may respond to a mood stabilizer.19 An antiaggressive response in IED-like subjects has been reported for lithium,20 carbamazepine,21 and diphenylhydantoin.22
Recently, Hollander et al23 reported greater reduction in overt aggression scores in IED subjects with a DSM cluster B personality disorder who were treated with divalproex, compared with placebo. This study used the same design and outcome measure as our study12 and included subjects who met both DSM-IV and research criteria for IED.
For unknown reasons, divalproex was no more effective than placebo in IED subjects without cluster B personality disorder. More research is needed to uncover predictors of antiaggressive response in IED subjects.
Unipolar vs. bipolar. McElroy9 has suggested using SSRIs (or other antidepressants) as first-line treatment for IED subjects with unipolar affective symptoms and mood stabilizers for those with bipolar affective symptoms. IED subjects without bipolar affective symptoms should be treated first with SSRIs (Algorithm). Preliminary data suggest a role for atypical antipsychotics to treat aggressive behavior in patients with schizophrenia or bipolar disorder, but no empiric data exist.
Beta blockers such as propranolol also may be considered.2 However, beta blockers are more difficult to dose and are associated with more burdensome side effects, compared with SSRIs.
Algorithm Suggested 2-pronged approach for treating intermittent explosive disorder
* With or without an anger management program, which may precede drug interventionThe full effects of antiaggressive treatment with an SSRI (E. Coccaro, unpublished observations) or a mood stabilizer19 may take 3 months to observe12,20,22,23 and tend to disappear soon after treatment is discontinued.
Therefore, an adequate trial of SSRIs or mood stabilizers is no less than 3 months. If improvement is seen, continue drug treatment indefinitely.
Case report continued.
Mr. P was started on an SSRI. His aggressive outbursts decreased in intensity and frequency over 3 months but were not eliminated. After 6 months he dropped out of treatment, but returned 5 weeks later because his aggressive outbursts had resumed their pre-treatment level.
SSRI treatment was restarted, and Mr. P began a 12-week anger management course of relaxation training, cognitive restructuring, and coping skills training. He gained greater control over his aggressive outbursts and continues monthly medication checks and anger management “booster sessions.”
Related resources
- Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
- Olvera RL. Intermittent explosive disorder: epidemiology, diagnosis and management. CNS Drugs 2002;16:517-26.
Drug brand names
- Carbamazepine • Tegretol
- Diphenylhydantoin • Dilantin
- Divalproex • Depakote
- Fluoxetine • Prozac
- Lithium • Lithobid
- Propanolol • Inderal
Disclosure
Dr. Coccaro reports that he receives research grants and serves on the speaker’s bureau or as a consultant to Eli Lilly and Co., Abbott Laboratories, GlaxoSmithKline, and Forrest Laboratories.
1. Coccaro EF, Schimdt CA, Samuels JF, et al. Lifetime rates of intermittent explosive disorder in a community sample (abstract). Philadelphia: American Psychiatric Association annual meeting, 2002.
2. Mattes JA. Comparative effectiveness of carbamazepine and propranolol for rage outbursts. J Neuropsychiatry Clin Neurosci 1990;2:159-64.
3. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry 1998;59:203-10.
4. Monopolis S, Lion JR. Problems in the diagnosis of intermittent explosive disorder. Am J Psychiatry 1983;140:1200-2.
5. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (abstract 265). Washington, DC: American Psychiatric Association annual meeting, 1998.
6. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (APA new research abstracts #265). Washington, DC: American Psychiatric Publishing, Inc., 1998.
7. Coccaro EF, Kavoussi RJ, Berman ME, Lish JD. Intermittent explosive disorder-revised: development, reliability and validity of research criteria. Compr Psychiatry 1998;39:368-76.
8. Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
9. McElroy SL. Recognition and treatment of DSM-IV intermittent explosive disorder. J Clin Psychiatry 1999;60(suppl 15):12-16.
10. Felthous AR, Bryant G, Wingerter CB, Barratt E. The diagnosis of intermittent explosive disorder in violent men. Bull Am Acad Psychiatry Law 1991;19:71-9.
11. Salzman C, Wolfson AN, Schatzberg A, et al. Effect of fluoxetine on anger in symptomatic volunteers with borderline personality disorder. J Clin Psychopharmacology 1995;15:23-9.
12. Coccaro EF, Kavoussi RJ. Fluoxetine and impulsive aggressive behavior in personality disordered subjects. Arch Gen Psychiatry 1997;54:1081-8.
13. Coccaro EF. Family history study of intermittent explosive disorder (abstract). Washington, DC: American Psychiatric Association annual meeting, 1999.
14. Coccaro EF, Siever LJ. Pathophysiology and treatment of aggression. In: Davis KL, Charney D, Coyle JT, Nemeroff D (eds). Psychopharmacology: the fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002;1709-24
15. Virkkunen M, Rawlings R, Tokola R, et al. CSF biochemistries, glucose metabolism, and diurnal activity rhythms in alcoholic, violent offenders, fire setters, and healthy volunteers. Arch Gen Psychiatry 1994;51:20-7.
16. Deffenbacher JL. Psychosocial interventions: anger disorders. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
17. Lee R, Coccaro EF. Treatment of aggression: serotonergic agents. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
18. Coccaro EF, Kavoussi RJ, Hauger RL. Serotonin function and antiaggressive responses to fluoxetine: a pilot study. Biol Psychiatry 1997;42:546-52.
19. Kavoussi RJ, Coccaro EF. Divalproex sodium for impulsive aggressive behavior in patients with personality disorder. J Clin Psychiatry 1998;59:676-80.
20. Sheard MH, Marini J, Bridges CI, Wagner E. The effect of lithium on impulsive aggressive behavior in man. Am J Psychiatry 1976;133:1409-13.
21. Cowdry RW, Gardner DL. Pharmacotherapy of borderline personality disorder: alprazolam, carbamazepine, trifluroperazine, and tranylcypromine. Arch Gen Psychiatry 1988;45:111-19.
22. Barratt ES, Stanford MS, Felthous AR, Kent TA. The effects of phenytoin on impulsive and premeditated aggression: a controlled study. J Clin Psychopharmacology 1997;17:341-9.
23. Hollander E, Tracy KA, Swann AC, et al. Divalproex sodium is superior to placebo for impulsive aggression in Cluster B personality disorders. Neuropsychopharmacology 2003;28:1186-97.
1. Coccaro EF, Schimdt CA, Samuels JF, et al. Lifetime rates of intermittent explosive disorder in a community sample (abstract). Philadelphia: American Psychiatric Association annual meeting, 2002.
2. Mattes JA. Comparative effectiveness of carbamazepine and propranolol for rage outbursts. J Neuropsychiatry Clin Neurosci 1990;2:159-64.
3. McElroy SL, Soutullo CA, Beckman DA, et al. DSM-IV intermittent explosive disorder: a report of 27 cases. J Clin Psychiatry 1998;59:203-10.
4. Monopolis S, Lion JR. Problems in the diagnosis of intermittent explosive disorder. Am J Psychiatry 1983;140:1200-2.
5. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (abstract 265). Washington, DC: American Psychiatric Association annual meeting, 1998.
6. Zimmerman M, Mattia J, Younken S, Torres M. The prevalence of DSM-IV impulse control disorders in psychiatric outpatients (APA new research abstracts #265). Washington, DC: American Psychiatric Publishing, Inc., 1998.
7. Coccaro EF, Kavoussi RJ, Berman ME, Lish JD. Intermittent explosive disorder-revised: development, reliability and validity of research criteria. Compr Psychiatry 1998;39:368-76.
8. Galovski T, Blanchard EB, Veazey C. Intermittent explosive disorder and other psychiatric comorbidity among court-referred and self-referred aggressive drivers. Behav Res Ther 2002;40:641-51.
9. McElroy SL. Recognition and treatment of DSM-IV intermittent explosive disorder. J Clin Psychiatry 1999;60(suppl 15):12-16.
10. Felthous AR, Bryant G, Wingerter CB, Barratt E. The diagnosis of intermittent explosive disorder in violent men. Bull Am Acad Psychiatry Law 1991;19:71-9.
11. Salzman C, Wolfson AN, Schatzberg A, et al. Effect of fluoxetine on anger in symptomatic volunteers with borderline personality disorder. J Clin Psychopharmacology 1995;15:23-9.
12. Coccaro EF, Kavoussi RJ. Fluoxetine and impulsive aggressive behavior in personality disordered subjects. Arch Gen Psychiatry 1997;54:1081-8.
13. Coccaro EF. Family history study of intermittent explosive disorder (abstract). Washington, DC: American Psychiatric Association annual meeting, 1999.
14. Coccaro EF, Siever LJ. Pathophysiology and treatment of aggression. In: Davis KL, Charney D, Coyle JT, Nemeroff D (eds). Psychopharmacology: the fifth generation of progress. Philadelphia: Lippincott Williams & Wilkins, 2002;1709-24
15. Virkkunen M, Rawlings R, Tokola R, et al. CSF biochemistries, glucose metabolism, and diurnal activity rhythms in alcoholic, violent offenders, fire setters, and healthy volunteers. Arch Gen Psychiatry 1994;51:20-7.
16. Deffenbacher JL. Psychosocial interventions: anger disorders. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
17. Lee R, Coccaro EF. Treatment of aggression: serotonergic agents. In: Coccaro EF (ed). Aggression: assessment and treatment. New York: Marcel Dekker (in press).
18. Coccaro EF, Kavoussi RJ, Hauger RL. Serotonin function and antiaggressive responses to fluoxetine: a pilot study. Biol Psychiatry 1997;42:546-52.
19. Kavoussi RJ, Coccaro EF. Divalproex sodium for impulsive aggressive behavior in patients with personality disorder. J Clin Psychiatry 1998;59:676-80.
20. Sheard MH, Marini J, Bridges CI, Wagner E. The effect of lithium on impulsive aggressive behavior in man. Am J Psychiatry 1976;133:1409-13.
21. Cowdry RW, Gardner DL. Pharmacotherapy of borderline personality disorder: alprazolam, carbamazepine, trifluroperazine, and tranylcypromine. Arch Gen Psychiatry 1988;45:111-19.
22. Barratt ES, Stanford MS, Felthous AR, Kent TA. The effects of phenytoin on impulsive and premeditated aggression: a controlled study. J Clin Psychopharmacology 1997;17:341-9.
23. Hollander E, Tracy KA, Swann AC, et al. Divalproex sodium is superior to placebo for impulsive aggression in Cluster B personality disorders. Neuropsychopharmacology 2003;28:1186-97.
Therapy-resistant major depression The attraction of magnetism: How effective—and safe—is rTMS?
Using magnets to improve health is sometimes hawked in dubious classified ads and “infomercials.” However, a legitimate use of magnetism—repetitive transcranial magnetic stimulation (rTMS)—is showing promise in treating severe depression (Box) 1-4 and other psychiatric disorders.
Patients or their families are likely to ask psychiatrists about rTMS as more becomes known about this investigational technology. Drawing from our experience and the evidence, we offer an update on whether rTMS may be an alternative for treating depression and address issues that must be resolved before it could be used in clinical practice.
WHAT IS RTMS?
rTMS consists of a series of magnetic pulses produced by a stimulator, which can be adjusted for:
- coil type and placement
- stimulation site, intensity, frequency, and number
- amount of time between stimulations
- treatment duration.
In 1985, Barker and colleagues developed single-pulse transcranial magnetic stimulation to examine motor cortex function.1 The single-pulse mechanism they discovered was subsequently adapted to deliver repetitive pulses and is referred to as repetitive transcranial magnetic stimulation (rTMS).
How rTMS works. Transcranial magnetic stimulation uses an electromagnetic coil applied to the head to produce an intense, localized, fluctuating magnetic field that passes unimpeded into a small area of the brain, inducing an electrical current. This results in neuronal depolarization in a localized area under the coil, and possibly distal effects as well.2 During the neurophysiological studies, it was discovered that subjects also experienced a change in mood.
Antidepressant effects. Similar physiologic effects induced by rTMS, electroconvulsive therapy (ECT), and antidepressants on the endocrine system, sleep parameters, and biochemical measures suggest antidepressant properties.3 In 1993, the first published study examining rTMS in psychiatric patients reported reduced depressive symptoms in two subjects.4 Since then, several clinical trials have examined rTMS’ antidepressive effects. In 2001, Canada’s Health Ministry approved rTMS for treating major depression. In the United States, rTMS remains investigational and is FDA-approved only for clinical trials.
Coil type and placement. Initial studies involved stimulation—typically low-frequency—over the vertex, but most subsequent rTMS trials in depression have stimulated the left dorsolateral prefrontal cortex. Neuroimaging studies have shown prefrontal functioning abnormalities in depressed subjects, and it is hypothesized that stimulating this area (plus possible distal effects) may produce an antidepressant effect.5
Various configurations have been used, but circular and figure-eight-shaped coils are most common. These flat coils are made of tightly wound ferromagnetic material such as copper, enclosed in a heavy plastic cover. With the figure-eight coil, the intersection of the two loops produces the strongest magnetic field.
Stimulation site. Stimulation intensity depends on the individual’s motor threshold, and the site can be determined visually or electrophysiologically.
- With the visual method, the motor threshold over the left primary motor cortex site for the first dorsal interosseous muscle (FDI) or the abductor pollius brevis (APB) is determined by iteration. This involves placing the coil at a progression of sites and increasing stimulation intensity until reliable (in 5 of 10 stimulations) contractions are seen in the right FDI or APB.
- Similarly, the electrophysiologic method uses 5 of 10 motorevoked potentials of 50 microvolts to locate the site.
The only small trial that compared visual and electrophysiologic site determination showed similar results with both methods.6 The most common stimulation site is the left dorsolateral prefrontal cortex, 5 cm anterior and parasagittal to the FDI or APB motor cortex. Alternately, frameless stereotactic systems or the international 10-20 proportional system used in EEG labs have been recommended to target sites more accurately.
Stimulus intensity. Each individual’s motor threshold determines stimulus intensity. Using functional MRI studies, researchers from the Medical University of South Carolina concluded that higher stimulation intensity relative to the motor threshold may have a more robust effect, as the magnetic field declines with distance from the coil.7 However, intensities >120% of the motor threshold are generally avoided because of possible increased seizure risk.9
Frequency of stimulation. Most researchers apply frequencies of 1 to 20 Hz over the left dorsolateral prefrontal cortex, but also use lower frequencies (<1 Hz) over the right dorsolateral prefrontal cortex. Using higher frequencies in major depression is attractive in theory because of:
- the reported association of decreased regional cerebral blood flow with hypometabolism in the left dorsolateral prefrontal cortex
- higher-frequency stimulation’s ability to produce temporary excitation and neuronal depolarization.
Number of stimulations. The number of stimulations is determined by frequency (Hz) and stimulation train duration (for example, 10 Hz for 5 seconds equals 50 stimulations). A typical treatment session incorporates 10 to 30 stimulation trains several seconds apart (the inter-train interval). Thus, a typical session delivers 1,000 to 1,200 stimulations. In studies of unmedicated depressed patients, the total number of stimulations has varied from 8,000 to 32,000 per treatment course.
Duration between two stimulation trains. Chen et al have demonstrated that shorter (<1 second) inter-train intervals increase seizure risk with higher frequencies (such as 20 Hz) and intensities (>100% of motor threshold) of stimulation.9 Based on their studies with healthy volunteers, they recommended several “safe” ranges (such as 5 seconds at 110% of motor threshold). Most trials use 30- to 60-second inter-train intervals.
Most treatments continue 2 to 4 weeks, Monday through Friday, although more frequent treatments are being studied.
EFFICACY FOR DEPRESSION
Most studies of rTMS in depression have compared real rTMS to a sham control or electroconvulsive therapy (ECT).
In earlier studies, the sham procedure typically involved tilting the coil away from the skull. This method has been questioned, however, because of evidence of neuronal depolarization.10
More recent sham coils mimic the real coils’ sound and sensation, without magnetic stimulation.
Despite these methodologic problems and some mixed results, depressed patients receiving rTMS show more favorable results than those receiving sham rTMS.11,12 Several meta-analyses have attempted to quantify rTMS’ efficacy for depression:
- Holtzheimer et al concluded that rTMS was statistically superior to sham rTMS, but the clinical significance of these findings was modest in a population of mostly outpatients with less-severe depression.13
- Burt et al found a statistically strong antidepressant effect, but its magnitude varied and few of the studies yielded a substantial clinical response or remission. The team also noted that rTMS’ long-term efficacy or adverse effects are unknown.14
- Kozel et al concluded that left prefrontal rTMS rendered a significant antidepressant effect with measurable clinical improvement.15
- Gershon et al16 supported an antidepressant effect for rTMS when compared with sham rTMS or ECT.
Ongoing rTMS research includes subjects with many types of mild to severe psychiatric illnesses, including major depression, obsessive-compulsive disorder, and psychosis. Typically, patients referred for experimental approaches have not responded to or tolerated available treatments. Exclusion criteria used by most rTMS studies are listed in the Table.
Table
Medical conditions that preclude use of rTMS
| Serious medical conditions History of seizures Increased intracranial pressure Serious head trauma |
| Myocardial infarction within the past 6 months |
| Pregnancy or childbearing potential (unless reliable contraception is being used) |
| Intracranial metallic implants |
| Pacemakers or other implanted devices |
rTMS vs. ECT. Four randomized, controlled trials have compared rTMS with ECT for treating severely ill, often medication-resistant patients.17-20 Although their methodologies differed, all four studies concluded that rTMS and ECT offer similar efficacy, except that rTMS may be less effective for treating psychotic depression.
One study found ECT more effective than rTMS for psychotic depression, although the patients who received ECT were also treated with antipsychotics and/or antidepressants.17 Our study,19 which did not use these agents, has not corroborated this observation. Preliminary data also indicate comparable relapse rates following acute ECT and rTMS when subjects are followed on maintenance medication.21
ADVERSE EFFECTS
The potential adverse effects of new treatments must always be considered. Thus far, rTMS appears to produce minimal, relatively benign complications, including:
- mild discomfort at the stimulation site
- localized muscle twitching during stimulation
- mild post-treatment headaches—believed caused by muscle contractions—which usually respond to aspirin or acetaminophen
- treatment stimulation-related seizures (rarely).8
The rTMS device makes a loud clicking noise, and subjects wear protective ear plugs during treatment.
Patient experience. The first rTMS session—during which the patient’s motor threshold is determined—can last up to 45 minutes. Subsequent sessions are usually 15 to 20 minutes. Patients are typically apprehensive before the first session but become more relaxed with experience and tolerate the treatments easily.
During the procedure, many patients describe a tapping sensation on the forehead, and some experience slight muscle twitching around the eye or corner of the mouth. As the coil warms, the skin it touches sometimes flushes pink, although this does not seem to bother our patients. They can return to their daily routines immediately after a session.
rTMS for major depression. In our experience, rTMS may help patients with major depression. For example, one patient diagnosed with a major depressive episode with psychotic features was referred to our study comparing rTMS with ECT.19 Her depression had lasted several months, with partial response to ECT treatments. She signed informed consent and was randomly assigned to receive rTMS treatment.
At study admission, the patient’s Hamilton Depression Rating Scale (HDRS) score was 48, indicating moderate to severe depression. Following 10 rTMS sessions, her HDRS score had dropped to 2, with remission of symptoms. No follow-up results were documented.
Cognitive effects. Whereas mood disorders are associated with medication-independent neuropsychological deficits, most studies have found no adverse cognitive effects with rTMS.22 Indeed, some of our rTMS patients have improved in certain cognitive tests, although this may be explained by test-retest effects or better attention and concentration associated with mood improvement.
Figure Potential roles for rTMS in treating major depression
Solid lines represent current standards of practice. Dotted lines represent hypothetical roles for rTMS.
Source: Adapted and reprinted with permission from Dowd et al. Is repetitive transcranial magnetic stimulation an alternative to ECTfor the treatment of depression? Contemp Psychiatry 2002;1:1-10.
POTENTIAL ROLE FOR rTMS
Today’s standard treatment of major depressive episodes begins with an antidepressant (plus an antipsychotic, if necessary) and proceeds to augmentation strategies if response is insufficient. rTMS may one day become an augmentation or monotherapy option for patients who do not respond sufficiently to standard treatments (Figure).
ECT treatment may be initiated if a patient has had a prior good response to ECT, is intolerant to medication, or prefers ECT. In that case, rTMS may be used as an alternate initial treatment or with ECT. Thus, rTMS may be used:
- to augment antidepressants
- as an alternative to antidepressants or ECT
- or sequentially with ECT.
Before that can happen, however, optimal treatment parameters need to be clarified by larger, well-designed, controlled studies comparing rTMS to a valid sham treatment, antidepressants, and ECT.
Related resources
- International Society for Transcranial Stimulation. www.ists.unibe.ch/
- Repetitive Transcranial Magnetic Stimulation Research Clinic at Yale-New Haven Psychiatric Hospital.
Disclosure
The authors report that they have no proprietary interest in the technology discussed in this article.
1. Barker A, Jalinous R, Freeston I. Non-invasive magnetic stimulation of human motor cortex. Lancet 1985;1:1106-7.
2. Lisanby SH, Datto CJ, Szuba MP. ECT and rTMS: past, present, and future. Depress Anxiety 2000;12:115-17.
3. Post A, Keck PE, Jr. Transcranial magnetic stimulation as a therapeutic tool in psychiatry: what do we know about the neurobiological mechanisms? J Psychiatr Res 2001;35:193-215.
4. Holfich G, Kasper S, Hufnagel A, et al. Application of transcranial magnetic stimulation in treatment of drug resistant major depression—a report of two cases. Human Psychopharmacol 1993;8:361-5.
5. George MS, Nahas Z, Speer AM, et al. Transcranial magnetic stimulation—a new method for investigating the neuroanatomy of depression. In: Ebert D, Ebmeier K (eds). New models for depression. New York: Karger, 1998;94-122.
6. Pridmore A, Americo Fernandes Filho J, Nahas Z, et al. Motor threshold in transcranial magnetic stimulation: a comparison of a neurophysiological method and a visualization of movement method. J ECT 1998;14(1):25-7.
7. Kozel FA, Nahas Z, deBrux C, et al. How coil-cortex distance relates to age, motor threshold, and antidepressant response to repetitive transcranial magnetic stimulation. J Neuropsychiatry Clin Neurosci 2000;13:376-84.
8. Wassermann EM. Risk and safety of repetitive transcranial magnetic stimulation: report and suggested guidelines from the International Workshop on the Safety of Repetitive Transcranial Magnetic Stimulation, 1996. Electroencephalogr Clin Neurophysiol 1998;108:1-16.
9. Chen R, Gerloff C, Classen J, et al. Safety of different inter-train intervals for repetitive transcranial magnetic stimulation and recommendations for safe ranges of stimulation parameters. Electroencephalogr Clin Neurophysiol 1997;105:415-21.
10. Loo CK, Taylor JL, Gandevia SC, et al. Transcranial magnetic stimulation in controlled treatment studies: Are some “sham” forms active? Biol Psychiatry. 2000;47:325-31.
11. George MS, Nahas Z, Molloy M, et al. A controlled trial of daily left prefrontal cortex TMS for treating depression. Biol Psychiatry 2000;48:962-70.
12. Berman RM, Narasimhan M, Sanacora G, et al. A randomized clinical trial of repetitive transcranial magnetic stimulation in the treatment of major depression. Biol Psychiatry 2000;47:332-7.
13. Holtzheimer PE, Russo J, Avery D. A meta-analysis of repetitive transcranial magnetic stimulation in the treatment of depression. Psychopharmacol Bull 2001;35:149-69.
14. Burt T, Lisanby SH, Sackeim HA. Neuropsychiatric applications of transcranial magnetic stimulation: a meta-analysis. Int J Neuropsychopharmacol 2002;5:73-103.
15. Kozel FE, George MS. Meta-analysis of left prefrontal repetitive transcranial magnetic stimulation (rTMS) to treat depression. J Psychiatr Pract 2002;8:270-5.
16. Gershon AA, Dannon PN, Grunhaus L. Transcranial magnetic stimulation in the treatment of depression. Am JPsychiatry 2003;160(5):835-45.
17. Grunhaus L, Dannon PN, Schreiber S, et al. Repetitive transcranial magnetic stimulation is as effective as electroconvulsive therapy in the treatment of nondelusional major depressive disorder: an open study. Biol Psychiatry 2000;47:314-24.
18. Pridmore S, Bruno R, Turnier-Shea Y, et al. Comparison of unlimited numbers of rapid transcranial magnetic stimulation and ECT treatment sessions in major depression episodes. Int J Neuropsychopharmacol 2000;3:129-34.
19. Janicak PG, Dowd SM, Martis B, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: preliminary results of a randomized trial. Biol Psychiatry 2002;51:659-67
20. Grunhaus L, Schreiber S, Dolberg OT, et al. A randomized controlled comparison of electroconvulsive therapy and repetitive transcranial magnetic stimulation in severe and resistant nonpsychotic major depression. Biol Psychiatry 2003;53:324-31.
21. Dannon PH, Dolberg OT, Schreiber S, Grunhaus L. Three and six month outcome following courses of either ECT or rTMS in a population of severely depressed individuals—preliminary report. Biol Psychiatry 2002;15:687-90.
22. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiology (in press).
Using magnets to improve health is sometimes hawked in dubious classified ads and “infomercials.” However, a legitimate use of magnetism—repetitive transcranial magnetic stimulation (rTMS)—is showing promise in treating severe depression (Box) 1-4 and other psychiatric disorders.
Patients or their families are likely to ask psychiatrists about rTMS as more becomes known about this investigational technology. Drawing from our experience and the evidence, we offer an update on whether rTMS may be an alternative for treating depression and address issues that must be resolved before it could be used in clinical practice.
WHAT IS RTMS?
rTMS consists of a series of magnetic pulses produced by a stimulator, which can be adjusted for:
- coil type and placement
- stimulation site, intensity, frequency, and number
- amount of time between stimulations
- treatment duration.
In 1985, Barker and colleagues developed single-pulse transcranial magnetic stimulation to examine motor cortex function.1 The single-pulse mechanism they discovered was subsequently adapted to deliver repetitive pulses and is referred to as repetitive transcranial magnetic stimulation (rTMS).
How rTMS works. Transcranial magnetic stimulation uses an electromagnetic coil applied to the head to produce an intense, localized, fluctuating magnetic field that passes unimpeded into a small area of the brain, inducing an electrical current. This results in neuronal depolarization in a localized area under the coil, and possibly distal effects as well.2 During the neurophysiological studies, it was discovered that subjects also experienced a change in mood.
Antidepressant effects. Similar physiologic effects induced by rTMS, electroconvulsive therapy (ECT), and antidepressants on the endocrine system, sleep parameters, and biochemical measures suggest antidepressant properties.3 In 1993, the first published study examining rTMS in psychiatric patients reported reduced depressive symptoms in two subjects.4 Since then, several clinical trials have examined rTMS’ antidepressive effects. In 2001, Canada’s Health Ministry approved rTMS for treating major depression. In the United States, rTMS remains investigational and is FDA-approved only for clinical trials.
Coil type and placement. Initial studies involved stimulation—typically low-frequency—over the vertex, but most subsequent rTMS trials in depression have stimulated the left dorsolateral prefrontal cortex. Neuroimaging studies have shown prefrontal functioning abnormalities in depressed subjects, and it is hypothesized that stimulating this area (plus possible distal effects) may produce an antidepressant effect.5
Various configurations have been used, but circular and figure-eight-shaped coils are most common. These flat coils are made of tightly wound ferromagnetic material such as copper, enclosed in a heavy plastic cover. With the figure-eight coil, the intersection of the two loops produces the strongest magnetic field.
Stimulation site. Stimulation intensity depends on the individual’s motor threshold, and the site can be determined visually or electrophysiologically.
- With the visual method, the motor threshold over the left primary motor cortex site for the first dorsal interosseous muscle (FDI) or the abductor pollius brevis (APB) is determined by iteration. This involves placing the coil at a progression of sites and increasing stimulation intensity until reliable (in 5 of 10 stimulations) contractions are seen in the right FDI or APB.
- Similarly, the electrophysiologic method uses 5 of 10 motorevoked potentials of 50 microvolts to locate the site.
The only small trial that compared visual and electrophysiologic site determination showed similar results with both methods.6 The most common stimulation site is the left dorsolateral prefrontal cortex, 5 cm anterior and parasagittal to the FDI or APB motor cortex. Alternately, frameless stereotactic systems or the international 10-20 proportional system used in EEG labs have been recommended to target sites more accurately.
Stimulus intensity. Each individual’s motor threshold determines stimulus intensity. Using functional MRI studies, researchers from the Medical University of South Carolina concluded that higher stimulation intensity relative to the motor threshold may have a more robust effect, as the magnetic field declines with distance from the coil.7 However, intensities >120% of the motor threshold are generally avoided because of possible increased seizure risk.9
Frequency of stimulation. Most researchers apply frequencies of 1 to 20 Hz over the left dorsolateral prefrontal cortex, but also use lower frequencies (<1 Hz) over the right dorsolateral prefrontal cortex. Using higher frequencies in major depression is attractive in theory because of:
- the reported association of decreased regional cerebral blood flow with hypometabolism in the left dorsolateral prefrontal cortex
- higher-frequency stimulation’s ability to produce temporary excitation and neuronal depolarization.
Number of stimulations. The number of stimulations is determined by frequency (Hz) and stimulation train duration (for example, 10 Hz for 5 seconds equals 50 stimulations). A typical treatment session incorporates 10 to 30 stimulation trains several seconds apart (the inter-train interval). Thus, a typical session delivers 1,000 to 1,200 stimulations. In studies of unmedicated depressed patients, the total number of stimulations has varied from 8,000 to 32,000 per treatment course.
Duration between two stimulation trains. Chen et al have demonstrated that shorter (<1 second) inter-train intervals increase seizure risk with higher frequencies (such as 20 Hz) and intensities (>100% of motor threshold) of stimulation.9 Based on their studies with healthy volunteers, they recommended several “safe” ranges (such as 5 seconds at 110% of motor threshold). Most trials use 30- to 60-second inter-train intervals.
Most treatments continue 2 to 4 weeks, Monday through Friday, although more frequent treatments are being studied.
EFFICACY FOR DEPRESSION
Most studies of rTMS in depression have compared real rTMS to a sham control or electroconvulsive therapy (ECT).
In earlier studies, the sham procedure typically involved tilting the coil away from the skull. This method has been questioned, however, because of evidence of neuronal depolarization.10
More recent sham coils mimic the real coils’ sound and sensation, without magnetic stimulation.
Despite these methodologic problems and some mixed results, depressed patients receiving rTMS show more favorable results than those receiving sham rTMS.11,12 Several meta-analyses have attempted to quantify rTMS’ efficacy for depression:
- Holtzheimer et al concluded that rTMS was statistically superior to sham rTMS, but the clinical significance of these findings was modest in a population of mostly outpatients with less-severe depression.13
- Burt et al found a statistically strong antidepressant effect, but its magnitude varied and few of the studies yielded a substantial clinical response or remission. The team also noted that rTMS’ long-term efficacy or adverse effects are unknown.14
- Kozel et al concluded that left prefrontal rTMS rendered a significant antidepressant effect with measurable clinical improvement.15
- Gershon et al16 supported an antidepressant effect for rTMS when compared with sham rTMS or ECT.
Ongoing rTMS research includes subjects with many types of mild to severe psychiatric illnesses, including major depression, obsessive-compulsive disorder, and psychosis. Typically, patients referred for experimental approaches have not responded to or tolerated available treatments. Exclusion criteria used by most rTMS studies are listed in the Table.
Table
Medical conditions that preclude use of rTMS
| Serious medical conditions History of seizures Increased intracranial pressure Serious head trauma |
| Myocardial infarction within the past 6 months |
| Pregnancy or childbearing potential (unless reliable contraception is being used) |
| Intracranial metallic implants |
| Pacemakers or other implanted devices |
rTMS vs. ECT. Four randomized, controlled trials have compared rTMS with ECT for treating severely ill, often medication-resistant patients.17-20 Although their methodologies differed, all four studies concluded that rTMS and ECT offer similar efficacy, except that rTMS may be less effective for treating psychotic depression.
One study found ECT more effective than rTMS for psychotic depression, although the patients who received ECT were also treated with antipsychotics and/or antidepressants.17 Our study,19 which did not use these agents, has not corroborated this observation. Preliminary data also indicate comparable relapse rates following acute ECT and rTMS when subjects are followed on maintenance medication.21
ADVERSE EFFECTS
The potential adverse effects of new treatments must always be considered. Thus far, rTMS appears to produce minimal, relatively benign complications, including:
- mild discomfort at the stimulation site
- localized muscle twitching during stimulation
- mild post-treatment headaches—believed caused by muscle contractions—which usually respond to aspirin or acetaminophen
- treatment stimulation-related seizures (rarely).8
The rTMS device makes a loud clicking noise, and subjects wear protective ear plugs during treatment.
Patient experience. The first rTMS session—during which the patient’s motor threshold is determined—can last up to 45 minutes. Subsequent sessions are usually 15 to 20 minutes. Patients are typically apprehensive before the first session but become more relaxed with experience and tolerate the treatments easily.
During the procedure, many patients describe a tapping sensation on the forehead, and some experience slight muscle twitching around the eye or corner of the mouth. As the coil warms, the skin it touches sometimes flushes pink, although this does not seem to bother our patients. They can return to their daily routines immediately after a session.
rTMS for major depression. In our experience, rTMS may help patients with major depression. For example, one patient diagnosed with a major depressive episode with psychotic features was referred to our study comparing rTMS with ECT.19 Her depression had lasted several months, with partial response to ECT treatments. She signed informed consent and was randomly assigned to receive rTMS treatment.
At study admission, the patient’s Hamilton Depression Rating Scale (HDRS) score was 48, indicating moderate to severe depression. Following 10 rTMS sessions, her HDRS score had dropped to 2, with remission of symptoms. No follow-up results were documented.
Cognitive effects. Whereas mood disorders are associated with medication-independent neuropsychological deficits, most studies have found no adverse cognitive effects with rTMS.22 Indeed, some of our rTMS patients have improved in certain cognitive tests, although this may be explained by test-retest effects or better attention and concentration associated with mood improvement.
Figure Potential roles for rTMS in treating major depression
Solid lines represent current standards of practice. Dotted lines represent hypothetical roles for rTMS.
Source: Adapted and reprinted with permission from Dowd et al. Is repetitive transcranial magnetic stimulation an alternative to ECTfor the treatment of depression? Contemp Psychiatry 2002;1:1-10.
POTENTIAL ROLE FOR rTMS
Today’s standard treatment of major depressive episodes begins with an antidepressant (plus an antipsychotic, if necessary) and proceeds to augmentation strategies if response is insufficient. rTMS may one day become an augmentation or monotherapy option for patients who do not respond sufficiently to standard treatments (Figure).
ECT treatment may be initiated if a patient has had a prior good response to ECT, is intolerant to medication, or prefers ECT. In that case, rTMS may be used as an alternate initial treatment or with ECT. Thus, rTMS may be used:
- to augment antidepressants
- as an alternative to antidepressants or ECT
- or sequentially with ECT.
Before that can happen, however, optimal treatment parameters need to be clarified by larger, well-designed, controlled studies comparing rTMS to a valid sham treatment, antidepressants, and ECT.
Related resources
- International Society for Transcranial Stimulation. www.ists.unibe.ch/
- Repetitive Transcranial Magnetic Stimulation Research Clinic at Yale-New Haven Psychiatric Hospital.
Disclosure
The authors report that they have no proprietary interest in the technology discussed in this article.
Using magnets to improve health is sometimes hawked in dubious classified ads and “infomercials.” However, a legitimate use of magnetism—repetitive transcranial magnetic stimulation (rTMS)—is showing promise in treating severe depression (Box) 1-4 and other psychiatric disorders.
Patients or their families are likely to ask psychiatrists about rTMS as more becomes known about this investigational technology. Drawing from our experience and the evidence, we offer an update on whether rTMS may be an alternative for treating depression and address issues that must be resolved before it could be used in clinical practice.
WHAT IS RTMS?
rTMS consists of a series of magnetic pulses produced by a stimulator, which can be adjusted for:
- coil type and placement
- stimulation site, intensity, frequency, and number
- amount of time between stimulations
- treatment duration.
In 1985, Barker and colleagues developed single-pulse transcranial magnetic stimulation to examine motor cortex function.1 The single-pulse mechanism they discovered was subsequently adapted to deliver repetitive pulses and is referred to as repetitive transcranial magnetic stimulation (rTMS).
How rTMS works. Transcranial magnetic stimulation uses an electromagnetic coil applied to the head to produce an intense, localized, fluctuating magnetic field that passes unimpeded into a small area of the brain, inducing an electrical current. This results in neuronal depolarization in a localized area under the coil, and possibly distal effects as well.2 During the neurophysiological studies, it was discovered that subjects also experienced a change in mood.
Antidepressant effects. Similar physiologic effects induced by rTMS, electroconvulsive therapy (ECT), and antidepressants on the endocrine system, sleep parameters, and biochemical measures suggest antidepressant properties.3 In 1993, the first published study examining rTMS in psychiatric patients reported reduced depressive symptoms in two subjects.4 Since then, several clinical trials have examined rTMS’ antidepressive effects. In 2001, Canada’s Health Ministry approved rTMS for treating major depression. In the United States, rTMS remains investigational and is FDA-approved only for clinical trials.
Coil type and placement. Initial studies involved stimulation—typically low-frequency—over the vertex, but most subsequent rTMS trials in depression have stimulated the left dorsolateral prefrontal cortex. Neuroimaging studies have shown prefrontal functioning abnormalities in depressed subjects, and it is hypothesized that stimulating this area (plus possible distal effects) may produce an antidepressant effect.5
Various configurations have been used, but circular and figure-eight-shaped coils are most common. These flat coils are made of tightly wound ferromagnetic material such as copper, enclosed in a heavy plastic cover. With the figure-eight coil, the intersection of the two loops produces the strongest magnetic field.
Stimulation site. Stimulation intensity depends on the individual’s motor threshold, and the site can be determined visually or electrophysiologically.
- With the visual method, the motor threshold over the left primary motor cortex site for the first dorsal interosseous muscle (FDI) or the abductor pollius brevis (APB) is determined by iteration. This involves placing the coil at a progression of sites and increasing stimulation intensity until reliable (in 5 of 10 stimulations) contractions are seen in the right FDI or APB.
- Similarly, the electrophysiologic method uses 5 of 10 motorevoked potentials of 50 microvolts to locate the site.
The only small trial that compared visual and electrophysiologic site determination showed similar results with both methods.6 The most common stimulation site is the left dorsolateral prefrontal cortex, 5 cm anterior and parasagittal to the FDI or APB motor cortex. Alternately, frameless stereotactic systems or the international 10-20 proportional system used in EEG labs have been recommended to target sites more accurately.
Stimulus intensity. Each individual’s motor threshold determines stimulus intensity. Using functional MRI studies, researchers from the Medical University of South Carolina concluded that higher stimulation intensity relative to the motor threshold may have a more robust effect, as the magnetic field declines with distance from the coil.7 However, intensities >120% of the motor threshold are generally avoided because of possible increased seizure risk.9
Frequency of stimulation. Most researchers apply frequencies of 1 to 20 Hz over the left dorsolateral prefrontal cortex, but also use lower frequencies (<1 Hz) over the right dorsolateral prefrontal cortex. Using higher frequencies in major depression is attractive in theory because of:
- the reported association of decreased regional cerebral blood flow with hypometabolism in the left dorsolateral prefrontal cortex
- higher-frequency stimulation’s ability to produce temporary excitation and neuronal depolarization.
Number of stimulations. The number of stimulations is determined by frequency (Hz) and stimulation train duration (for example, 10 Hz for 5 seconds equals 50 stimulations). A typical treatment session incorporates 10 to 30 stimulation trains several seconds apart (the inter-train interval). Thus, a typical session delivers 1,000 to 1,200 stimulations. In studies of unmedicated depressed patients, the total number of stimulations has varied from 8,000 to 32,000 per treatment course.
Duration between two stimulation trains. Chen et al have demonstrated that shorter (<1 second) inter-train intervals increase seizure risk with higher frequencies (such as 20 Hz) and intensities (>100% of motor threshold) of stimulation.9 Based on their studies with healthy volunteers, they recommended several “safe” ranges (such as 5 seconds at 110% of motor threshold). Most trials use 30- to 60-second inter-train intervals.
Most treatments continue 2 to 4 weeks, Monday through Friday, although more frequent treatments are being studied.
EFFICACY FOR DEPRESSION
Most studies of rTMS in depression have compared real rTMS to a sham control or electroconvulsive therapy (ECT).
In earlier studies, the sham procedure typically involved tilting the coil away from the skull. This method has been questioned, however, because of evidence of neuronal depolarization.10
More recent sham coils mimic the real coils’ sound and sensation, without magnetic stimulation.
Despite these methodologic problems and some mixed results, depressed patients receiving rTMS show more favorable results than those receiving sham rTMS.11,12 Several meta-analyses have attempted to quantify rTMS’ efficacy for depression:
- Holtzheimer et al concluded that rTMS was statistically superior to sham rTMS, but the clinical significance of these findings was modest in a population of mostly outpatients with less-severe depression.13
- Burt et al found a statistically strong antidepressant effect, but its magnitude varied and few of the studies yielded a substantial clinical response or remission. The team also noted that rTMS’ long-term efficacy or adverse effects are unknown.14
- Kozel et al concluded that left prefrontal rTMS rendered a significant antidepressant effect with measurable clinical improvement.15
- Gershon et al16 supported an antidepressant effect for rTMS when compared with sham rTMS or ECT.
Ongoing rTMS research includes subjects with many types of mild to severe psychiatric illnesses, including major depression, obsessive-compulsive disorder, and psychosis. Typically, patients referred for experimental approaches have not responded to or tolerated available treatments. Exclusion criteria used by most rTMS studies are listed in the Table.
Table
Medical conditions that preclude use of rTMS
| Serious medical conditions History of seizures Increased intracranial pressure Serious head trauma |
| Myocardial infarction within the past 6 months |
| Pregnancy or childbearing potential (unless reliable contraception is being used) |
| Intracranial metallic implants |
| Pacemakers or other implanted devices |
rTMS vs. ECT. Four randomized, controlled trials have compared rTMS with ECT for treating severely ill, often medication-resistant patients.17-20 Although their methodologies differed, all four studies concluded that rTMS and ECT offer similar efficacy, except that rTMS may be less effective for treating psychotic depression.
One study found ECT more effective than rTMS for psychotic depression, although the patients who received ECT were also treated with antipsychotics and/or antidepressants.17 Our study,19 which did not use these agents, has not corroborated this observation. Preliminary data also indicate comparable relapse rates following acute ECT and rTMS when subjects are followed on maintenance medication.21
ADVERSE EFFECTS
The potential adverse effects of new treatments must always be considered. Thus far, rTMS appears to produce minimal, relatively benign complications, including:
- mild discomfort at the stimulation site
- localized muscle twitching during stimulation
- mild post-treatment headaches—believed caused by muscle contractions—which usually respond to aspirin or acetaminophen
- treatment stimulation-related seizures (rarely).8
The rTMS device makes a loud clicking noise, and subjects wear protective ear plugs during treatment.
Patient experience. The first rTMS session—during which the patient’s motor threshold is determined—can last up to 45 minutes. Subsequent sessions are usually 15 to 20 minutes. Patients are typically apprehensive before the first session but become more relaxed with experience and tolerate the treatments easily.
During the procedure, many patients describe a tapping sensation on the forehead, and some experience slight muscle twitching around the eye or corner of the mouth. As the coil warms, the skin it touches sometimes flushes pink, although this does not seem to bother our patients. They can return to their daily routines immediately after a session.
rTMS for major depression. In our experience, rTMS may help patients with major depression. For example, one patient diagnosed with a major depressive episode with psychotic features was referred to our study comparing rTMS with ECT.19 Her depression had lasted several months, with partial response to ECT treatments. She signed informed consent and was randomly assigned to receive rTMS treatment.
At study admission, the patient’s Hamilton Depression Rating Scale (HDRS) score was 48, indicating moderate to severe depression. Following 10 rTMS sessions, her HDRS score had dropped to 2, with remission of symptoms. No follow-up results were documented.
Cognitive effects. Whereas mood disorders are associated with medication-independent neuropsychological deficits, most studies have found no adverse cognitive effects with rTMS.22 Indeed, some of our rTMS patients have improved in certain cognitive tests, although this may be explained by test-retest effects or better attention and concentration associated with mood improvement.
Figure Potential roles for rTMS in treating major depression
Solid lines represent current standards of practice. Dotted lines represent hypothetical roles for rTMS.
Source: Adapted and reprinted with permission from Dowd et al. Is repetitive transcranial magnetic stimulation an alternative to ECTfor the treatment of depression? Contemp Psychiatry 2002;1:1-10.
POTENTIAL ROLE FOR rTMS
Today’s standard treatment of major depressive episodes begins with an antidepressant (plus an antipsychotic, if necessary) and proceeds to augmentation strategies if response is insufficient. rTMS may one day become an augmentation or monotherapy option for patients who do not respond sufficiently to standard treatments (Figure).
ECT treatment may be initiated if a patient has had a prior good response to ECT, is intolerant to medication, or prefers ECT. In that case, rTMS may be used as an alternate initial treatment or with ECT. Thus, rTMS may be used:
- to augment antidepressants
- as an alternative to antidepressants or ECT
- or sequentially with ECT.
Before that can happen, however, optimal treatment parameters need to be clarified by larger, well-designed, controlled studies comparing rTMS to a valid sham treatment, antidepressants, and ECT.
Related resources
- International Society for Transcranial Stimulation. www.ists.unibe.ch/
- Repetitive Transcranial Magnetic Stimulation Research Clinic at Yale-New Haven Psychiatric Hospital.
Disclosure
The authors report that they have no proprietary interest in the technology discussed in this article.
1. Barker A, Jalinous R, Freeston I. Non-invasive magnetic stimulation of human motor cortex. Lancet 1985;1:1106-7.
2. Lisanby SH, Datto CJ, Szuba MP. ECT and rTMS: past, present, and future. Depress Anxiety 2000;12:115-17.
3. Post A, Keck PE, Jr. Transcranial magnetic stimulation as a therapeutic tool in psychiatry: what do we know about the neurobiological mechanisms? J Psychiatr Res 2001;35:193-215.
4. Holfich G, Kasper S, Hufnagel A, et al. Application of transcranial magnetic stimulation in treatment of drug resistant major depression—a report of two cases. Human Psychopharmacol 1993;8:361-5.
5. George MS, Nahas Z, Speer AM, et al. Transcranial magnetic stimulation—a new method for investigating the neuroanatomy of depression. In: Ebert D, Ebmeier K (eds). New models for depression. New York: Karger, 1998;94-122.
6. Pridmore A, Americo Fernandes Filho J, Nahas Z, et al. Motor threshold in transcranial magnetic stimulation: a comparison of a neurophysiological method and a visualization of movement method. J ECT 1998;14(1):25-7.
7. Kozel FA, Nahas Z, deBrux C, et al. How coil-cortex distance relates to age, motor threshold, and antidepressant response to repetitive transcranial magnetic stimulation. J Neuropsychiatry Clin Neurosci 2000;13:376-84.
8. Wassermann EM. Risk and safety of repetitive transcranial magnetic stimulation: report and suggested guidelines from the International Workshop on the Safety of Repetitive Transcranial Magnetic Stimulation, 1996. Electroencephalogr Clin Neurophysiol 1998;108:1-16.
9. Chen R, Gerloff C, Classen J, et al. Safety of different inter-train intervals for repetitive transcranial magnetic stimulation and recommendations for safe ranges of stimulation parameters. Electroencephalogr Clin Neurophysiol 1997;105:415-21.
10. Loo CK, Taylor JL, Gandevia SC, et al. Transcranial magnetic stimulation in controlled treatment studies: Are some “sham” forms active? Biol Psychiatry. 2000;47:325-31.
11. George MS, Nahas Z, Molloy M, et al. A controlled trial of daily left prefrontal cortex TMS for treating depression. Biol Psychiatry 2000;48:962-70.
12. Berman RM, Narasimhan M, Sanacora G, et al. A randomized clinical trial of repetitive transcranial magnetic stimulation in the treatment of major depression. Biol Psychiatry 2000;47:332-7.
13. Holtzheimer PE, Russo J, Avery D. A meta-analysis of repetitive transcranial magnetic stimulation in the treatment of depression. Psychopharmacol Bull 2001;35:149-69.
14. Burt T, Lisanby SH, Sackeim HA. Neuropsychiatric applications of transcranial magnetic stimulation: a meta-analysis. Int J Neuropsychopharmacol 2002;5:73-103.
15. Kozel FE, George MS. Meta-analysis of left prefrontal repetitive transcranial magnetic stimulation (rTMS) to treat depression. J Psychiatr Pract 2002;8:270-5.
16. Gershon AA, Dannon PN, Grunhaus L. Transcranial magnetic stimulation in the treatment of depression. Am JPsychiatry 2003;160(5):835-45.
17. Grunhaus L, Dannon PN, Schreiber S, et al. Repetitive transcranial magnetic stimulation is as effective as electroconvulsive therapy in the treatment of nondelusional major depressive disorder: an open study. Biol Psychiatry 2000;47:314-24.
18. Pridmore S, Bruno R, Turnier-Shea Y, et al. Comparison of unlimited numbers of rapid transcranial magnetic stimulation and ECT treatment sessions in major depression episodes. Int J Neuropsychopharmacol 2000;3:129-34.
19. Janicak PG, Dowd SM, Martis B, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: preliminary results of a randomized trial. Biol Psychiatry 2002;51:659-67
20. Grunhaus L, Schreiber S, Dolberg OT, et al. A randomized controlled comparison of electroconvulsive therapy and repetitive transcranial magnetic stimulation in severe and resistant nonpsychotic major depression. Biol Psychiatry 2003;53:324-31.
21. Dannon PH, Dolberg OT, Schreiber S, Grunhaus L. Three and six month outcome following courses of either ECT or rTMS in a population of severely depressed individuals—preliminary report. Biol Psychiatry 2002;15:687-90.
22. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiology (in press).
1. Barker A, Jalinous R, Freeston I. Non-invasive magnetic stimulation of human motor cortex. Lancet 1985;1:1106-7.
2. Lisanby SH, Datto CJ, Szuba MP. ECT and rTMS: past, present, and future. Depress Anxiety 2000;12:115-17.
3. Post A, Keck PE, Jr. Transcranial magnetic stimulation as a therapeutic tool in psychiatry: what do we know about the neurobiological mechanisms? J Psychiatr Res 2001;35:193-215.
4. Holfich G, Kasper S, Hufnagel A, et al. Application of transcranial magnetic stimulation in treatment of drug resistant major depression—a report of two cases. Human Psychopharmacol 1993;8:361-5.
5. George MS, Nahas Z, Speer AM, et al. Transcranial magnetic stimulation—a new method for investigating the neuroanatomy of depression. In: Ebert D, Ebmeier K (eds). New models for depression. New York: Karger, 1998;94-122.
6. Pridmore A, Americo Fernandes Filho J, Nahas Z, et al. Motor threshold in transcranial magnetic stimulation: a comparison of a neurophysiological method and a visualization of movement method. J ECT 1998;14(1):25-7.
7. Kozel FA, Nahas Z, deBrux C, et al. How coil-cortex distance relates to age, motor threshold, and antidepressant response to repetitive transcranial magnetic stimulation. J Neuropsychiatry Clin Neurosci 2000;13:376-84.
8. Wassermann EM. Risk and safety of repetitive transcranial magnetic stimulation: report and suggested guidelines from the International Workshop on the Safety of Repetitive Transcranial Magnetic Stimulation, 1996. Electroencephalogr Clin Neurophysiol 1998;108:1-16.
9. Chen R, Gerloff C, Classen J, et al. Safety of different inter-train intervals for repetitive transcranial magnetic stimulation and recommendations for safe ranges of stimulation parameters. Electroencephalogr Clin Neurophysiol 1997;105:415-21.
10. Loo CK, Taylor JL, Gandevia SC, et al. Transcranial magnetic stimulation in controlled treatment studies: Are some “sham” forms active? Biol Psychiatry. 2000;47:325-31.
11. George MS, Nahas Z, Molloy M, et al. A controlled trial of daily left prefrontal cortex TMS for treating depression. Biol Psychiatry 2000;48:962-70.
12. Berman RM, Narasimhan M, Sanacora G, et al. A randomized clinical trial of repetitive transcranial magnetic stimulation in the treatment of major depression. Biol Psychiatry 2000;47:332-7.
13. Holtzheimer PE, Russo J, Avery D. A meta-analysis of repetitive transcranial magnetic stimulation in the treatment of depression. Psychopharmacol Bull 2001;35:149-69.
14. Burt T, Lisanby SH, Sackeim HA. Neuropsychiatric applications of transcranial magnetic stimulation: a meta-analysis. Int J Neuropsychopharmacol 2002;5:73-103.
15. Kozel FE, George MS. Meta-analysis of left prefrontal repetitive transcranial magnetic stimulation (rTMS) to treat depression. J Psychiatr Pract 2002;8:270-5.
16. Gershon AA, Dannon PN, Grunhaus L. Transcranial magnetic stimulation in the treatment of depression. Am JPsychiatry 2003;160(5):835-45.
17. Grunhaus L, Dannon PN, Schreiber S, et al. Repetitive transcranial magnetic stimulation is as effective as electroconvulsive therapy in the treatment of nondelusional major depressive disorder: an open study. Biol Psychiatry 2000;47:314-24.
18. Pridmore S, Bruno R, Turnier-Shea Y, et al. Comparison of unlimited numbers of rapid transcranial magnetic stimulation and ECT treatment sessions in major depression episodes. Int J Neuropsychopharmacol 2000;3:129-34.
19. Janicak PG, Dowd SM, Martis B, et al. Repetitive transcranial magnetic stimulation versus electroconvulsive therapy for major depression: preliminary results of a randomized trial. Biol Psychiatry 2002;51:659-67
20. Grunhaus L, Schreiber S, Dolberg OT, et al. A randomized controlled comparison of electroconvulsive therapy and repetitive transcranial magnetic stimulation in severe and resistant nonpsychotic major depression. Biol Psychiatry 2003;53:324-31.
21. Dannon PH, Dolberg OT, Schreiber S, Grunhaus L. Three and six month outcome following courses of either ECT or rTMS in a population of severely depressed individuals—preliminary report. Biol Psychiatry 2002;15:687-90.
22. Martis B, Alam D, Dowd SM, et al. Neurocognitive effects of repetitive transcranial magnetic stimulation in severe major depression. Clin Neurophysiology (in press).
Irritable bowel syndrome and psychiatric illness: Three clinical challenges
Psychiatrists often treat patients with irritable bowel syndrome (IBS) and an accompanying mental illness. Knowledge of available treatments and communication with the referring doctor are crucial to treating both the IBS symptoms and the comorbidity.
This article presents three cases that illustrate the challenges of identifying target symptoms, avoiding drug-drug interactions, ruling out serious underlying medical problems, and formulating treatment.
WHO GETS IBS?
Approximately 12% of the United States population reports IBS symptoms (abdominal pain, bloating, altered bowel habits).1 These symptoms begin before age 35 in most patients and during childhood in some. Onset after age 65 is rare.
IBS is common among patients with alcohol abuse disorder (32%),2 chronic fatigue syndrome (92%), fibromyalgia (77%), or temporomandibular joint syndrome (64%).3 Seventy percent of patients with IBS are women.4 Chronic pelvic pain, dyspareunia, dysmenorrhea, or a history of abdominal surgeries are risk factors for IBS in women.
LINK BETWEEN IBS AND MENTAL ILLNESS
Although mental illness often coexists with IBS, no cause-effect relationship has been shown.5
IBS is often preceded by stressful life events, such as family death or divorce,3 and some believe IBS is a precursor to numerous psychiatric disorders. Generalized anxiety disorder, major depression, panic disorder, social phobia, somatization disorder, or dysthymia have been diagnosed in most IBS patients.2
CASE 1: IBS AND DEPRESSION
Ms. R, age 55, has had IBS for 10 years. She has occasional diarrhea and abdominal cramps relieved by bowel movements. She is taking a bulking agent but still sometimes suffers abdominal pain.
She is referred to a psychiatrist after complaining of fatigue, loss of interest in hobbies, and crying spells for 2 months. She denies suicidal ideations. Her referring physician reports that she is taking conjugated estrogens to manage menopause symptoms. She denies any recent stressful life events. Thyroid function, glucose, and CBC are normal.
The challenge: Deciding which to treat first—the IBS symptoms or the depression—and how.
Discussion: The predominant symptom (in Ms. R’s case, abdominal pain) can help determine choice of medication. Bulk-forming agents, antispasmodics, barbiturates, benzodiazepines, and serotonin reuptake inhibitors have historically been used to treat IBS,6 but scant evidence supports their use.
Obtaining a thorough prescription history from the primary care physician, OB/GYN, and other treatment team members is critical before formulating a treatment plan. Ms. R’s estrogen use will not affect the choice of psychotropic or IBS medication because there are no significant interactions between estrogen and these classes of drugs.
Ms. R’s abdominal pain and depression can be treated simultaneously. Randomized, controlled trials have demonstrated that tricyclic antidepressants reduce abdominal pain and that behavioral therapy (relaxation therapy, hypnotherapy, and cognitive-behavioral therapy) may relieve individual IBS symptoms.7
Case 1 concluded: After reviewing Ms. R’s medications, the psychiatrist starts:
- desipramine, 50 mg at bedtime, to minimize anticholinergic side effects
- and short-term psychotherapy, which helped her identify support mechanisms and ways to better balance her life stresses.
After 6 weeks, her Beck Depression Inventory score improved from 30 at baseline to 8. She reports her abdominal pain is “the best it has been in 10 years.” Six months after diagnosis, she continues to take desipramine and is doing well.
CASE 2: IBS, DEPRESSION, AND PSYCHOSIS
Ms. H, age 32, is referred to a psychiatrist for treatment of depression with paranoid features.
Four years ago, a gastroenterologist diagnosed her as having IBS. She experiences frequent diarrhea and lower abdominal cramping. For 2 years she has been taking the antimuscarinic dicyclomine, 10 mg tid, which has provided some relief from her cramps. An estimated 20 diarrhea attacks per day leaves her housebound much of the time, however.
She reports fatigue, loss of interest in hobbies across 2 months, and paranoid thinking. She denies hallucinations or delusions but believes that her teenage children are discussing her “sickness” and plotting to “drive her crazy.” She is not suicidal.
The challenge: Treating Ms. H’s depression and paranoia while avoiding drug-drug interactions.
Discussion: Adverse drug-drug interactions can occur when prescribing psychotropics to patients with IBS (Table 1). Additive constipation, diarrhea, abdominal pain, and sedation are common interactions between psychotropics and the 5-HT3 antagonists and 5HT4 agonists commonly prescribed for IBS.
Table 1
Interactions between psychotropics and agents prescribed for IBS
| Antispasmodics | Benzodiazepines | SSRIs | Tricyclics | |
|---|---|---|---|---|
| MAOIs | Additive sedation | Additive dizziness, sedation, dry mouth, | Contraindicated–hyperpyrexia and severe neurologic effects | Contraindicated–hyperpyrexia, seizures, and death |
| SSRIs | Additive sedation | Additive sedation | —- | Increased tricyclic levels with concurrent use |
| Tricyclics | Additive sedation, dry mouth | Additive sedation | Additive sedation, dry mouth, increased tricyclic levels | —- |
| Anticonvulsants | Additive sedation | Additive sedation | Increased levels of anticonvulsants | Additive sedation, dry mouth, constipation |
| Benzodiazepines | Additive sedation | —- | Additive sedation and dry mouth | Additive sedation |
| Buspirone | Additive sedation, dizziness | Additive sedation | Additive sedation, dizziness, nausea | Additive sedation, dry mouth, constipation, increased tricyclic level |
| Traditional antipsychotics | Additive sedation, CNS effects | Additive sedation, CNS effects | Additive sedation, dizziness | Additive sedation and anticholinergic effects; increased tricyclic level |
| Atypical antipsychotics | Additive sedation, CNS effects | Contraindicated–respiratory and cardiovascular collapse | Elevated antipsychotic levels | Levels of both drugs increased |
| Aripiprazole | Somnolence,constipation | Additive sedation | Increased blood levels of aripiprazole | Increased sedation and anticholinergic effects |
| Psychotropics and 5-HT3 antagonists taken concomitantly typically lead to additive constipation and abdominal pain. | ||||
| Psychotropics and 5-HT4 agonists taken concomitantly typically lead to additive diarrhea and/or abdominal pain. | ||||
| Source: Physician’s Desk Reference. Mobile PDR release version 32. Database version 437. Montvale, NJ: Thomson Healthcare 2003. | ||||
- Hematochezia
- Weight loss < 10 pounds
- Family history of colon cancer
- Recurrent fever
- Anemia
- Chronic severe diarrhea
Source: American College of Gastroenterology Functional Gastrointestinal Disorders Task Force. Am J Gastroenterol. 2002;97:S1-S5.
Other than fiber supplements, most traditional IBS medications are sedating and are associated with anticholinergic side effects. In Ms. H’s case, extreme caution is necessary before prescribing an antidepressant or antipsychotic because of dicyclomine’s additive sedating effects.
Case 2 concluded: After a thorough initial patient interview, the psychiatrist elects to treat Ms. H’s major depression with an antidepressant but delays the use of an antipsychotic to avoid additive sedation.
After talking with Ms. H’s family physician, the psychiatrist stops her dicyclomine and starts sertraline, 100 mg/d. She tolerates the sertraline well and the dosage is titrated across 1 month to 200 mg/d.
Four weeks later, Ms. H’s Beck Depression Inventory score has improved from 26 at baseline to 5, but her paranoid thoughts and frequent diarrhea persist. The psychiatrist adds low-dose olanzapine (5 mg at bedtime) to minimize extrapyramidal side effects. One month later, her depression and paranoia have resolved.
Ms. H’s gastroenterologist instructs her to begin taking alosetron, 1 mg bid, for her continued frequent diarrhea. Adding this agent to her sertraline/olanzapine regimen can lead to additive constipation and abdominal pain, so the psychiatrist monitors her psychiatric medications. One month later, she reports that her affect is much improved and her diarrhea is “gone.”
CASE 3: DEPRESSION AND ABDOMINAL PAIN
Mr. J, age 52, has had depression for 1 year. His depressive symptoms have improved significantly on fluoxetine, 20 mg/d; he once again enjoys life and has a more positive outlook.
The patient was in reasonably good health until about 1 month ago, when he began to experience abdominal pain. He has lost 14 lbs over the past month. He is not taking other medications.
The challenge: Find the cause of Mr. J’s persistent abdominal pain without undermining depression therapy.
Discussion: Although Mr. J’s symptoms might be side effects of fluoxetine, his abdominal pain and weight loss >10 lbs within 1 month are cause for concern. The American College of Gastroenterology has identified six alarm symptoms that could point to a serious medical problem in patients with severe abdominal pain (Box).7
Patients who exhibit any of these symptoms should be referred for endoscopic and stool studies. Colon cancer screening should be considered for all patients age 50 and older.
Patients with IBS usually present first to their primary care physicians with abdominal pain and altered bowel habits. These symptoms can occur in many gastrointestinal and systemic illnesses (Table 2).8
Table 2
Diagnosing irritable bowel syndrome: What to rule out
| Differential diagnosis | Examples |
|---|---|
| Inflammatory bowel disease | Crohn’s disease, ulcerative colitis |
| Medication effects | Laxatives, constipating agents |
| Infections | Parasitic, bacterial, viral, opportunistic |
| Malabsorption syndromes | Celiac disease, pancreatic insufficiency |
| Endocrine disorders | Hypothyroidism, hyperthyroidism, diabetes, Addison’s disease |
| Endocrine tumors (extremely uncommon) | Gastrinoma, carcinoid |
| Colorectal carcinoma | Adenocarcinoma, villous adenoma |
| Intestinal pseudo-obstruction | Diabetes, scleroderma |
| Lactose intolerance | —- |
| Psychiatric disorders | Depression, anxiety, somatization disorders |
| Source: Dalton CB, Drossman D. Am Fam Physician. 1997;55(3):875-80. | |
Case 3 concluded: The psychiatrist and primary care physician consult a gastroenterologist, who performs a colonoscopy and identifies a resectable Duke’s Class B adenocarcinoma in the transverse colon. A partial colectomy is performed.
Three years later, Mr. J is cancer-free and his depression is stable. The psychiatrist advises him to keep taking fluoxetine, 20 mg/d, because the stress of his cancer therapy increases the risk of depression recurrence.
Related resources
- National Institute of Diabetes and Digestive and Kidney Diseases —Irritable Bowel Syndrome www.niddk.nih.gov/health/digest/pubs/irrbowel/irrbowel.htm
Drug brand names
- Alosetron • Lotronex
- Aripiprazole • Abilify
- Buspirone • BuSpar
- Desipramine • Norpramin
- Dicyclomine • Bentyl
- Fluoxetine • Prozac
- Olanzapine • Zyprexa
- Sertraline • Zoloft
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Locke GR, 3rd. The epidemiology of functional gastrointestinal disorders in North America. Gastroenterol Clin North Am. 1996;25:1-19.
2. Goldberg J, Davidson P. A biopsychosocial understanding of the irritable bowel syndrome: a review. Can J Psychiatry. 1997;42:835-40.
3. Aaron LA, Burke MM, Buchwald D. Overlapping conditions among chronic fatigue syndrome, fibromyalgia, and temporomandibular disorder. Arch Intern Med. 2000;160:221-7.
4. Smith RP. Lower gastrointestinal disease in women. Obstet Gynecol Clin North Am. 2001;28:351-62.
5. Olden KW, Drossman DA. Psychologic and psychiatric aspects of gastrointestinal disease. Med Clin North Am. 2000;84:1313-276.
6. Mobile PDR Release Version 32. Database Version 437. An abbreviated, up-to-date version of the PDR onto computing devices. Thomson Healthcare, Ortho-Biotech Oncology, 2003.
7. American College of Gastroenterology Functional Gastrointestinal Disorders Task Force. Evidence-based position statement on the management of irritable bowel syndrome in North America. Am J Gastroenterol. 2002;97:S1-S5.
8. Dalton CB, Drossman D. Diagnosis and treatment of irritable bowel syndrome. Am Fam Physician. 1997;55(3):875-80.
Psychiatrists often treat patients with irritable bowel syndrome (IBS) and an accompanying mental illness. Knowledge of available treatments and communication with the referring doctor are crucial to treating both the IBS symptoms and the comorbidity.
This article presents three cases that illustrate the challenges of identifying target symptoms, avoiding drug-drug interactions, ruling out serious underlying medical problems, and formulating treatment.
WHO GETS IBS?
Approximately 12% of the United States population reports IBS symptoms (abdominal pain, bloating, altered bowel habits).1 These symptoms begin before age 35 in most patients and during childhood in some. Onset after age 65 is rare.
IBS is common among patients with alcohol abuse disorder (32%),2 chronic fatigue syndrome (92%), fibromyalgia (77%), or temporomandibular joint syndrome (64%).3 Seventy percent of patients with IBS are women.4 Chronic pelvic pain, dyspareunia, dysmenorrhea, or a history of abdominal surgeries are risk factors for IBS in women.
LINK BETWEEN IBS AND MENTAL ILLNESS
Although mental illness often coexists with IBS, no cause-effect relationship has been shown.5
IBS is often preceded by stressful life events, such as family death or divorce,3 and some believe IBS is a precursor to numerous psychiatric disorders. Generalized anxiety disorder, major depression, panic disorder, social phobia, somatization disorder, or dysthymia have been diagnosed in most IBS patients.2
CASE 1: IBS AND DEPRESSION
Ms. R, age 55, has had IBS for 10 years. She has occasional diarrhea and abdominal cramps relieved by bowel movements. She is taking a bulking agent but still sometimes suffers abdominal pain.
She is referred to a psychiatrist after complaining of fatigue, loss of interest in hobbies, and crying spells for 2 months. She denies suicidal ideations. Her referring physician reports that she is taking conjugated estrogens to manage menopause symptoms. She denies any recent stressful life events. Thyroid function, glucose, and CBC are normal.
The challenge: Deciding which to treat first—the IBS symptoms or the depression—and how.
Discussion: The predominant symptom (in Ms. R’s case, abdominal pain) can help determine choice of medication. Bulk-forming agents, antispasmodics, barbiturates, benzodiazepines, and serotonin reuptake inhibitors have historically been used to treat IBS,6 but scant evidence supports their use.
Obtaining a thorough prescription history from the primary care physician, OB/GYN, and other treatment team members is critical before formulating a treatment plan. Ms. R’s estrogen use will not affect the choice of psychotropic or IBS medication because there are no significant interactions between estrogen and these classes of drugs.
Ms. R’s abdominal pain and depression can be treated simultaneously. Randomized, controlled trials have demonstrated that tricyclic antidepressants reduce abdominal pain and that behavioral therapy (relaxation therapy, hypnotherapy, and cognitive-behavioral therapy) may relieve individual IBS symptoms.7
Case 1 concluded: After reviewing Ms. R’s medications, the psychiatrist starts:
- desipramine, 50 mg at bedtime, to minimize anticholinergic side effects
- and short-term psychotherapy, which helped her identify support mechanisms and ways to better balance her life stresses.
After 6 weeks, her Beck Depression Inventory score improved from 30 at baseline to 8. She reports her abdominal pain is “the best it has been in 10 years.” Six months after diagnosis, she continues to take desipramine and is doing well.
CASE 2: IBS, DEPRESSION, AND PSYCHOSIS
Ms. H, age 32, is referred to a psychiatrist for treatment of depression with paranoid features.
Four years ago, a gastroenterologist diagnosed her as having IBS. She experiences frequent diarrhea and lower abdominal cramping. For 2 years she has been taking the antimuscarinic dicyclomine, 10 mg tid, which has provided some relief from her cramps. An estimated 20 diarrhea attacks per day leaves her housebound much of the time, however.
She reports fatigue, loss of interest in hobbies across 2 months, and paranoid thinking. She denies hallucinations or delusions but believes that her teenage children are discussing her “sickness” and plotting to “drive her crazy.” She is not suicidal.
The challenge: Treating Ms. H’s depression and paranoia while avoiding drug-drug interactions.
Discussion: Adverse drug-drug interactions can occur when prescribing psychotropics to patients with IBS (Table 1). Additive constipation, diarrhea, abdominal pain, and sedation are common interactions between psychotropics and the 5-HT3 antagonists and 5HT4 agonists commonly prescribed for IBS.
Table 1
Interactions between psychotropics and agents prescribed for IBS
| Antispasmodics | Benzodiazepines | SSRIs | Tricyclics | |
|---|---|---|---|---|
| MAOIs | Additive sedation | Additive dizziness, sedation, dry mouth, | Contraindicated–hyperpyrexia and severe neurologic effects | Contraindicated–hyperpyrexia, seizures, and death |
| SSRIs | Additive sedation | Additive sedation | —- | Increased tricyclic levels with concurrent use |
| Tricyclics | Additive sedation, dry mouth | Additive sedation | Additive sedation, dry mouth, increased tricyclic levels | —- |
| Anticonvulsants | Additive sedation | Additive sedation | Increased levels of anticonvulsants | Additive sedation, dry mouth, constipation |
| Benzodiazepines | Additive sedation | —- | Additive sedation and dry mouth | Additive sedation |
| Buspirone | Additive sedation, dizziness | Additive sedation | Additive sedation, dizziness, nausea | Additive sedation, dry mouth, constipation, increased tricyclic level |
| Traditional antipsychotics | Additive sedation, CNS effects | Additive sedation, CNS effects | Additive sedation, dizziness | Additive sedation and anticholinergic effects; increased tricyclic level |
| Atypical antipsychotics | Additive sedation, CNS effects | Contraindicated–respiratory and cardiovascular collapse | Elevated antipsychotic levels | Levels of both drugs increased |
| Aripiprazole | Somnolence,constipation | Additive sedation | Increased blood levels of aripiprazole | Increased sedation and anticholinergic effects |
| Psychotropics and 5-HT3 antagonists taken concomitantly typically lead to additive constipation and abdominal pain. | ||||
| Psychotropics and 5-HT4 agonists taken concomitantly typically lead to additive diarrhea and/or abdominal pain. | ||||
| Source: Physician’s Desk Reference. Mobile PDR release version 32. Database version 437. Montvale, NJ: Thomson Healthcare 2003. | ||||
- Hematochezia
- Weight loss < 10 pounds
- Family history of colon cancer
- Recurrent fever
- Anemia
- Chronic severe diarrhea
Source: American College of Gastroenterology Functional Gastrointestinal Disorders Task Force. Am J Gastroenterol. 2002;97:S1-S5.
Other than fiber supplements, most traditional IBS medications are sedating and are associated with anticholinergic side effects. In Ms. H’s case, extreme caution is necessary before prescribing an antidepressant or antipsychotic because of dicyclomine’s additive sedating effects.
Case 2 concluded: After a thorough initial patient interview, the psychiatrist elects to treat Ms. H’s major depression with an antidepressant but delays the use of an antipsychotic to avoid additive sedation.
After talking with Ms. H’s family physician, the psychiatrist stops her dicyclomine and starts sertraline, 100 mg/d. She tolerates the sertraline well and the dosage is titrated across 1 month to 200 mg/d.
Four weeks later, Ms. H’s Beck Depression Inventory score has improved from 26 at baseline to 5, but her paranoid thoughts and frequent diarrhea persist. The psychiatrist adds low-dose olanzapine (5 mg at bedtime) to minimize extrapyramidal side effects. One month later, her depression and paranoia have resolved.
Ms. H’s gastroenterologist instructs her to begin taking alosetron, 1 mg bid, for her continued frequent diarrhea. Adding this agent to her sertraline/olanzapine regimen can lead to additive constipation and abdominal pain, so the psychiatrist monitors her psychiatric medications. One month later, she reports that her affect is much improved and her diarrhea is “gone.”
CASE 3: DEPRESSION AND ABDOMINAL PAIN
Mr. J, age 52, has had depression for 1 year. His depressive symptoms have improved significantly on fluoxetine, 20 mg/d; he once again enjoys life and has a more positive outlook.
The patient was in reasonably good health until about 1 month ago, when he began to experience abdominal pain. He has lost 14 lbs over the past month. He is not taking other medications.
The challenge: Find the cause of Mr. J’s persistent abdominal pain without undermining depression therapy.
Discussion: Although Mr. J’s symptoms might be side effects of fluoxetine, his abdominal pain and weight loss >10 lbs within 1 month are cause for concern. The American College of Gastroenterology has identified six alarm symptoms that could point to a serious medical problem in patients with severe abdominal pain (Box).7
Patients who exhibit any of these symptoms should be referred for endoscopic and stool studies. Colon cancer screening should be considered for all patients age 50 and older.
Patients with IBS usually present first to their primary care physicians with abdominal pain and altered bowel habits. These symptoms can occur in many gastrointestinal and systemic illnesses (Table 2).8
Table 2
Diagnosing irritable bowel syndrome: What to rule out
| Differential diagnosis | Examples |
|---|---|
| Inflammatory bowel disease | Crohn’s disease, ulcerative colitis |
| Medication effects | Laxatives, constipating agents |
| Infections | Parasitic, bacterial, viral, opportunistic |
| Malabsorption syndromes | Celiac disease, pancreatic insufficiency |
| Endocrine disorders | Hypothyroidism, hyperthyroidism, diabetes, Addison’s disease |
| Endocrine tumors (extremely uncommon) | Gastrinoma, carcinoid |
| Colorectal carcinoma | Adenocarcinoma, villous adenoma |
| Intestinal pseudo-obstruction | Diabetes, scleroderma |
| Lactose intolerance | —- |
| Psychiatric disorders | Depression, anxiety, somatization disorders |
| Source: Dalton CB, Drossman D. Am Fam Physician. 1997;55(3):875-80. | |
Case 3 concluded: The psychiatrist and primary care physician consult a gastroenterologist, who performs a colonoscopy and identifies a resectable Duke’s Class B adenocarcinoma in the transverse colon. A partial colectomy is performed.
Three years later, Mr. J is cancer-free and his depression is stable. The psychiatrist advises him to keep taking fluoxetine, 20 mg/d, because the stress of his cancer therapy increases the risk of depression recurrence.
Related resources
- National Institute of Diabetes and Digestive and Kidney Diseases —Irritable Bowel Syndrome www.niddk.nih.gov/health/digest/pubs/irrbowel/irrbowel.htm
Drug brand names
- Alosetron • Lotronex
- Aripiprazole • Abilify
- Buspirone • BuSpar
- Desipramine • Norpramin
- Dicyclomine • Bentyl
- Fluoxetine • Prozac
- Olanzapine • Zyprexa
- Sertraline • Zoloft
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Psychiatrists often treat patients with irritable bowel syndrome (IBS) and an accompanying mental illness. Knowledge of available treatments and communication with the referring doctor are crucial to treating both the IBS symptoms and the comorbidity.
This article presents three cases that illustrate the challenges of identifying target symptoms, avoiding drug-drug interactions, ruling out serious underlying medical problems, and formulating treatment.
WHO GETS IBS?
Approximately 12% of the United States population reports IBS symptoms (abdominal pain, bloating, altered bowel habits).1 These symptoms begin before age 35 in most patients and during childhood in some. Onset after age 65 is rare.
IBS is common among patients with alcohol abuse disorder (32%),2 chronic fatigue syndrome (92%), fibromyalgia (77%), or temporomandibular joint syndrome (64%).3 Seventy percent of patients with IBS are women.4 Chronic pelvic pain, dyspareunia, dysmenorrhea, or a history of abdominal surgeries are risk factors for IBS in women.
LINK BETWEEN IBS AND MENTAL ILLNESS
Although mental illness often coexists with IBS, no cause-effect relationship has been shown.5
IBS is often preceded by stressful life events, such as family death or divorce,3 and some believe IBS is a precursor to numerous psychiatric disorders. Generalized anxiety disorder, major depression, panic disorder, social phobia, somatization disorder, or dysthymia have been diagnosed in most IBS patients.2
CASE 1: IBS AND DEPRESSION
Ms. R, age 55, has had IBS for 10 years. She has occasional diarrhea and abdominal cramps relieved by bowel movements. She is taking a bulking agent but still sometimes suffers abdominal pain.
She is referred to a psychiatrist after complaining of fatigue, loss of interest in hobbies, and crying spells for 2 months. She denies suicidal ideations. Her referring physician reports that she is taking conjugated estrogens to manage menopause symptoms. She denies any recent stressful life events. Thyroid function, glucose, and CBC are normal.
The challenge: Deciding which to treat first—the IBS symptoms or the depression—and how.
Discussion: The predominant symptom (in Ms. R’s case, abdominal pain) can help determine choice of medication. Bulk-forming agents, antispasmodics, barbiturates, benzodiazepines, and serotonin reuptake inhibitors have historically been used to treat IBS,6 but scant evidence supports their use.
Obtaining a thorough prescription history from the primary care physician, OB/GYN, and other treatment team members is critical before formulating a treatment plan. Ms. R’s estrogen use will not affect the choice of psychotropic or IBS medication because there are no significant interactions between estrogen and these classes of drugs.
Ms. R’s abdominal pain and depression can be treated simultaneously. Randomized, controlled trials have demonstrated that tricyclic antidepressants reduce abdominal pain and that behavioral therapy (relaxation therapy, hypnotherapy, and cognitive-behavioral therapy) may relieve individual IBS symptoms.7
Case 1 concluded: After reviewing Ms. R’s medications, the psychiatrist starts:
- desipramine, 50 mg at bedtime, to minimize anticholinergic side effects
- and short-term psychotherapy, which helped her identify support mechanisms and ways to better balance her life stresses.
After 6 weeks, her Beck Depression Inventory score improved from 30 at baseline to 8. She reports her abdominal pain is “the best it has been in 10 years.” Six months after diagnosis, she continues to take desipramine and is doing well.
CASE 2: IBS, DEPRESSION, AND PSYCHOSIS
Ms. H, age 32, is referred to a psychiatrist for treatment of depression with paranoid features.
Four years ago, a gastroenterologist diagnosed her as having IBS. She experiences frequent diarrhea and lower abdominal cramping. For 2 years she has been taking the antimuscarinic dicyclomine, 10 mg tid, which has provided some relief from her cramps. An estimated 20 diarrhea attacks per day leaves her housebound much of the time, however.
She reports fatigue, loss of interest in hobbies across 2 months, and paranoid thinking. She denies hallucinations or delusions but believes that her teenage children are discussing her “sickness” and plotting to “drive her crazy.” She is not suicidal.
The challenge: Treating Ms. H’s depression and paranoia while avoiding drug-drug interactions.
Discussion: Adverse drug-drug interactions can occur when prescribing psychotropics to patients with IBS (Table 1). Additive constipation, diarrhea, abdominal pain, and sedation are common interactions between psychotropics and the 5-HT3 antagonists and 5HT4 agonists commonly prescribed for IBS.
Table 1
Interactions between psychotropics and agents prescribed for IBS
| Antispasmodics | Benzodiazepines | SSRIs | Tricyclics | |
|---|---|---|---|---|
| MAOIs | Additive sedation | Additive dizziness, sedation, dry mouth, | Contraindicated–hyperpyrexia and severe neurologic effects | Contraindicated–hyperpyrexia, seizures, and death |
| SSRIs | Additive sedation | Additive sedation | —- | Increased tricyclic levels with concurrent use |
| Tricyclics | Additive sedation, dry mouth | Additive sedation | Additive sedation, dry mouth, increased tricyclic levels | —- |
| Anticonvulsants | Additive sedation | Additive sedation | Increased levels of anticonvulsants | Additive sedation, dry mouth, constipation |
| Benzodiazepines | Additive sedation | —- | Additive sedation and dry mouth | Additive sedation |
| Buspirone | Additive sedation, dizziness | Additive sedation | Additive sedation, dizziness, nausea | Additive sedation, dry mouth, constipation, increased tricyclic level |
| Traditional antipsychotics | Additive sedation, CNS effects | Additive sedation, CNS effects | Additive sedation, dizziness | Additive sedation and anticholinergic effects; increased tricyclic level |
| Atypical antipsychotics | Additive sedation, CNS effects | Contraindicated–respiratory and cardiovascular collapse | Elevated antipsychotic levels | Levels of both drugs increased |
| Aripiprazole | Somnolence,constipation | Additive sedation | Increased blood levels of aripiprazole | Increased sedation and anticholinergic effects |
| Psychotropics and 5-HT3 antagonists taken concomitantly typically lead to additive constipation and abdominal pain. | ||||
| Psychotropics and 5-HT4 agonists taken concomitantly typically lead to additive diarrhea and/or abdominal pain. | ||||
| Source: Physician’s Desk Reference. Mobile PDR release version 32. Database version 437. Montvale, NJ: Thomson Healthcare 2003. | ||||
- Hematochezia
- Weight loss < 10 pounds
- Family history of colon cancer
- Recurrent fever
- Anemia
- Chronic severe diarrhea
Source: American College of Gastroenterology Functional Gastrointestinal Disorders Task Force. Am J Gastroenterol. 2002;97:S1-S5.
Other than fiber supplements, most traditional IBS medications are sedating and are associated with anticholinergic side effects. In Ms. H’s case, extreme caution is necessary before prescribing an antidepressant or antipsychotic because of dicyclomine’s additive sedating effects.
Case 2 concluded: After a thorough initial patient interview, the psychiatrist elects to treat Ms. H’s major depression with an antidepressant but delays the use of an antipsychotic to avoid additive sedation.
After talking with Ms. H’s family physician, the psychiatrist stops her dicyclomine and starts sertraline, 100 mg/d. She tolerates the sertraline well and the dosage is titrated across 1 month to 200 mg/d.
Four weeks later, Ms. H’s Beck Depression Inventory score has improved from 26 at baseline to 5, but her paranoid thoughts and frequent diarrhea persist. The psychiatrist adds low-dose olanzapine (5 mg at bedtime) to minimize extrapyramidal side effects. One month later, her depression and paranoia have resolved.
Ms. H’s gastroenterologist instructs her to begin taking alosetron, 1 mg bid, for her continued frequent diarrhea. Adding this agent to her sertraline/olanzapine regimen can lead to additive constipation and abdominal pain, so the psychiatrist monitors her psychiatric medications. One month later, she reports that her affect is much improved and her diarrhea is “gone.”
CASE 3: DEPRESSION AND ABDOMINAL PAIN
Mr. J, age 52, has had depression for 1 year. His depressive symptoms have improved significantly on fluoxetine, 20 mg/d; he once again enjoys life and has a more positive outlook.
The patient was in reasonably good health until about 1 month ago, when he began to experience abdominal pain. He has lost 14 lbs over the past month. He is not taking other medications.
The challenge: Find the cause of Mr. J’s persistent abdominal pain without undermining depression therapy.
Discussion: Although Mr. J’s symptoms might be side effects of fluoxetine, his abdominal pain and weight loss >10 lbs within 1 month are cause for concern. The American College of Gastroenterology has identified six alarm symptoms that could point to a serious medical problem in patients with severe abdominal pain (Box).7
Patients who exhibit any of these symptoms should be referred for endoscopic and stool studies. Colon cancer screening should be considered for all patients age 50 and older.
Patients with IBS usually present first to their primary care physicians with abdominal pain and altered bowel habits. These symptoms can occur in many gastrointestinal and systemic illnesses (Table 2).8
Table 2
Diagnosing irritable bowel syndrome: What to rule out
| Differential diagnosis | Examples |
|---|---|
| Inflammatory bowel disease | Crohn’s disease, ulcerative colitis |
| Medication effects | Laxatives, constipating agents |
| Infections | Parasitic, bacterial, viral, opportunistic |
| Malabsorption syndromes | Celiac disease, pancreatic insufficiency |
| Endocrine disorders | Hypothyroidism, hyperthyroidism, diabetes, Addison’s disease |
| Endocrine tumors (extremely uncommon) | Gastrinoma, carcinoid |
| Colorectal carcinoma | Adenocarcinoma, villous adenoma |
| Intestinal pseudo-obstruction | Diabetes, scleroderma |
| Lactose intolerance | —- |
| Psychiatric disorders | Depression, anxiety, somatization disorders |
| Source: Dalton CB, Drossman D. Am Fam Physician. 1997;55(3):875-80. | |
Case 3 concluded: The psychiatrist and primary care physician consult a gastroenterologist, who performs a colonoscopy and identifies a resectable Duke’s Class B adenocarcinoma in the transverse colon. A partial colectomy is performed.
Three years later, Mr. J is cancer-free and his depression is stable. The psychiatrist advises him to keep taking fluoxetine, 20 mg/d, because the stress of his cancer therapy increases the risk of depression recurrence.
Related resources
- National Institute of Diabetes and Digestive and Kidney Diseases —Irritable Bowel Syndrome www.niddk.nih.gov/health/digest/pubs/irrbowel/irrbowel.htm
Drug brand names
- Alosetron • Lotronex
- Aripiprazole • Abilify
- Buspirone • BuSpar
- Desipramine • Norpramin
- Dicyclomine • Bentyl
- Fluoxetine • Prozac
- Olanzapine • Zyprexa
- Sertraline • Zoloft
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Locke GR, 3rd. The epidemiology of functional gastrointestinal disorders in North America. Gastroenterol Clin North Am. 1996;25:1-19.
2. Goldberg J, Davidson P. A biopsychosocial understanding of the irritable bowel syndrome: a review. Can J Psychiatry. 1997;42:835-40.
3. Aaron LA, Burke MM, Buchwald D. Overlapping conditions among chronic fatigue syndrome, fibromyalgia, and temporomandibular disorder. Arch Intern Med. 2000;160:221-7.
4. Smith RP. Lower gastrointestinal disease in women. Obstet Gynecol Clin North Am. 2001;28:351-62.
5. Olden KW, Drossman DA. Psychologic and psychiatric aspects of gastrointestinal disease. Med Clin North Am. 2000;84:1313-276.
6. Mobile PDR Release Version 32. Database Version 437. An abbreviated, up-to-date version of the PDR onto computing devices. Thomson Healthcare, Ortho-Biotech Oncology, 2003.
7. American College of Gastroenterology Functional Gastrointestinal Disorders Task Force. Evidence-based position statement on the management of irritable bowel syndrome in North America. Am J Gastroenterol. 2002;97:S1-S5.
8. Dalton CB, Drossman D. Diagnosis and treatment of irritable bowel syndrome. Am Fam Physician. 1997;55(3):875-80.
1. Locke GR, 3rd. The epidemiology of functional gastrointestinal disorders in North America. Gastroenterol Clin North Am. 1996;25:1-19.
2. Goldberg J, Davidson P. A biopsychosocial understanding of the irritable bowel syndrome: a review. Can J Psychiatry. 1997;42:835-40.
3. Aaron LA, Burke MM, Buchwald D. Overlapping conditions among chronic fatigue syndrome, fibromyalgia, and temporomandibular disorder. Arch Intern Med. 2000;160:221-7.
4. Smith RP. Lower gastrointestinal disease in women. Obstet Gynecol Clin North Am. 2001;28:351-62.
5. Olden KW, Drossman DA. Psychologic and psychiatric aspects of gastrointestinal disease. Med Clin North Am. 2000;84:1313-276.
6. Mobile PDR Release Version 32. Database Version 437. An abbreviated, up-to-date version of the PDR onto computing devices. Thomson Healthcare, Ortho-Biotech Oncology, 2003.
7. American College of Gastroenterology Functional Gastrointestinal Disorders Task Force. Evidence-based position statement on the management of irritable bowel syndrome in North America. Am J Gastroenterol. 2002;97:S1-S5.
8. Dalton CB, Drossman D. Diagnosis and treatment of irritable bowel syndrome. Am Fam Physician. 1997;55(3):875-80.
How to avoid ethnic bias when diagnosing schizophrenia
In patients with psychotic symptoms, why are African-Americans more likely than whites to be diagnosed with schizophrenia? After more than 30 years of debate, some answers—and remedies for the problem—are becoming clear.
In psychiatry, where interpersonal interactions are key to eliciting diagnostic symptoms and signs, there is an intrinsic risk of misinterpretation when clinician and patient are of different cultural, ethnic, or socioeconomic backgrounds. This article analyzes four factors that contribute to misinterpretation and to ethnic misdiagnosis of schizophrenia. Culturally sensitive strategies are offered to avoid diagnostic bias in clinical practice.
SCHIZOPHRENIA MISDIAGNOSIS
Large epidemiologic studies report similar rates of schizophrenia and bipolar disorder in African-American and white populations.1 Although patients of both races have been wrongly diagnosed with schizophrenia, the pattern is stronger and more persistent in African-Americans.
Two (or more) of the following, each present for a significant portion of time during a 1-month period (or less if successfully treated):
- Delusions
- Hallucinations
- Disorganized speech (eg, frequent derailment or incoherence)
- Grossly disorganized or catatonic behavior
- Negative symptoms (ie, affective flattening, alogia, or avolition)
Note: Only one Criterion A symptom is required if delusions are bizarre or hallucinations consist of a voice keeping up a running commentary on the person’s behavior or thoughts, or two or more voices conversing with each other.
295.30 Paranoid type
A type of schizophrenia in which the following criteria are met:
- Preoccupation with one or more delusions or frequent auditory hallucinations
- None of the following is prominent: disorganized speech, disorganized or catatonic behavior, or flat or inappropriate affect.
Source: DSM-IV-TR
In the 1970s, Simon et al2 studied 192 hospitalized patients, of whom all African-Americans and 85% of whites had been identified clinically as having schizophrenia. Using a structured interview, the researchers found that only 40% of the African Americans and 50% of the whites met diagnostic criteria for schizophrenia. African-Americans with mood disorders were found to be at particular risk of schizophrenia misdiagnosis.
In the 1980s, among 76 patients with a clinical diagnosis of schizophrenia, Mukherjee et al3 diagnosed one-half (52%) with bipolar disorder using a structured clinical interview. Schizophrenia misdiagnoses were more common in African-Americans (86%) and Hispanics (83%) than in whites (51%). In particular, African-Americans were most likely to be misdiagnosed with paranoid schizophrenia. African-Americans complained more commonly than whites of auditory hallucinations, which may represent an ethnic difference in symptomatic presentation of psychotic mood disorders.
In the 1990s, colleagues and I conducted two studies—one of 173 patients in a Tennessee psychiatric hospital4 and the other of 490 patients in an Ohio psychiatric emergency service5—and found yet again that African-Americans were more likely than whites to be diagnosed with schizophrenia. In the hospital study, higher rates of schizophrenia diagnosis were associated with lower rates of mood disorder diagnosis. This inverse relationship implied that African-Americans with mood disorders were being misdiagnosed with schizophrenia.
Men were more likely than women to be diagnosed with schizophrenia, suggesting that African-American men were most likely to be misdiagnosed. When adjustments were made for gender, black women were found to be at higher risk for misdiagnosis than white women.
Lawson et al6 extended this research in a population-based study of African-Americans living in Tennessee. They found that African-Americans constituted 16% of the state’s population but 48% of psychiatric inpatients diagnosed with schizophrenia and 37% of psychiatric outpatients.
CONSEQUENCES OF INACCURATE DIAGNOSIS
Differentiating between schizophrenia (Box 1) and a psychotic mood disorder (Box 2) is more than a semantic exercise. Schizophrenia implies a chronic, unremitting, debilitating illness that worsens over time. Though this perception of schizophrenia is not entirely accurate, in clinical practice its diagnosis imparts a bleak prognosis that may lower the clinician’s expectations for the patient.7
Schizophrenia misdiagnosis also may lead the psychiatrist to rely excessively on antipsychotics, rather than attempting thymoleptic and psychotherapy trials. Studies suggest that African-American patients are more likely than similar white patients to receive antipsychotics4,8,9 and less likely to receive psychotherapy.5,10
Reasons why African-Americans are often misdiagnosed with schizophrenia remain unclear but probably include four contributing factors:
- differences in symptom presentation compared with whites
- failure by clinicians to identify affective symptoms in African-Americans
- minority patients’ wariness when dealing with health services
- and racial stereotyping.
DIFFERENCES IN SYMPTOM EXPRESSION
African-American patients with mood disorders or schizophrenia are more likely than are similar white patients to complain of auditory hallucinations.11-13 For example, Strakowski et al14 examined 330 patients with nonaffective and psychotic diagnoses in a study that was used to develop DSM-IV criteria for schizophrenia. Auditory hallucinations were rated as more severe in African-American than in similar white patients.
MAJOR DEPRESSIVE EPISODE
Five or more of the following symptoms present during the same 2-week period and representing a change from previous functioning; must include either depressed mood or loss of interest or pleasure.
- Depressed mood
- Markedly diminished interest or pleasure
- Significant weight loss
- Insomnia or hypersomnia
- Psychomotor agitation or retardation
- Fatigue
- Feelings of worthlessness or excessive guilt
- Diminished ability to concentrate
SEVERE MAJOR DEPRESSION WITH PSYCHOTIC FEATURES
Mood-congruent
Delusions or hallucinations whose content is entirely consistent with the typical depressive themes of personal inadequacy, guilt, disease, death, nihilism, or deserved punishment
Mood-incongruent
Delusions or hallucinations whose content does not involve typical depressive themes. Includes symptoms such as persecutory delusions, thought insertion, thought broadcasting, and delusions of control
Source: DSM-IV-TR
African-American patients also are more likely than whites to exhibit so-called Schneiderian first-rank symptoms of schizophrenia,15 including:
- delusions of thought broadcasting or insertion
- auditory hallucinations of voices conversing about the patient in the third person.
These symptoms were once used to diagnose schizophrenia, but their lack of specificity has been well documented.2,16 First-rank symptoms of schizophrenia depend on the specific form of the hallucination or delusion, are likely to be influenced by a patient’s culture, and may be misleading in multicultural populations. Though first-rank symptoms now occupy a minor role in U.S. diagnostic systems, they might continue to sway clinicians—even when using structured diagnostic interviews—to inappropriately diagnose schizophrenia in lieu of affective disorders in minority patients.15
To extend this finding, our group16 studied rates and severity of affective and psychotic symptoms—particularly first-rank symptoms—in 100 patients with psychotic mania who met DSM-III-R criteria for bipolar disorder (80%) or schizoaffective disorder, bipolar type (20%) as determined by structured diagnostic interview. No differences in affective symptoms between African-American and white patients were seen. African-Americans were more likely to endorse auditory hallucinations and to report severe auditory hallucinations of voices commenting on their behavior—the only first-rank symptom on which they differed from whites.
Though their affective symptoms were similar, African-Americans were significantly more likely than whites to have been diagnosed with a schizophrenia-spectrum disorder. Because misdiagnosis of African-Americans could not be explained by psychotic symptoms—which were as severe as those of white patients—these findings suggest other mechanisms were at work.
UNIDENTIFIED AFFECTIVE SYMPTOMS
Underidentification of mood disorders in African-American patients may also lead to over-diagnosis of schizophrenia. In a sample of 99 patients, colleagues and I17 compared clinical diagnoses made in a psychiatric emergency service with those by research investigators using a structured clinical interview. Reasons for diagnostic differences were identified and divided into two categories:
- the same symptoms were recorded but applied differently to diagnostic criteria (criterion variance)
- different information was recorded, which led to diagnostic discrepancies (information variance).
Differences occurred significantly more often in African-American than in white patients, but only information variance was associated with ethnicity. This suggests that clinicians are less likely to elicit appropriate information from African-American than from white psychiatric patients. The fact that researchers obtained this information during diagnostic interviews suggests that the patients could provide it when given appropriate prompts. Specifically, affective symptoms were less likely to be elicited by clinicians than by researchers.
PATIENT WARINESS
Minority patients, when interacting with clinicians of the majority population, may project “protective wariness.”18 Specific behaviors include hesitancy or reluctance to fully engage with the care provider as a precaution against being exploited or harmed. Cultural misunderstandings19 and patient concerns about past reports of minorities receiving substandard or unethical health care20 may contribute to this behavior.
Whaley21 compared nonpathologic distrust and paranoia in 404 community-living African-Americans and whites. Some were healthy, and some had diagnoses of schizophrenia or depression. African-Americans—particularly those with psychiatric disorders—showed higher levels of distrust than whites. Distrust was also associated with depression in African-Americans but not in whites. Whaley concluded that:
- depressed African-Americans may exhibit more distrust toward clinicians than do whites
- this distrust puts African-Americans at risk of being perceived as paranoid and being misdiagnosed with paranoid schizophrenia.
Table
Remedial actions to avoid ethnic bias in diagnosing schizophrenia
| Problem | Remedies |
|---|---|
| Failure to recognize differences in symptom expression | Become familiar with ethnic differences in how patients describe symptoms Incorporate structured interviews or rating scales into the clinical assessment |
| Failure to elicit affective symptoms | Incorporate structured interviews or rating scales into the clinical assessment Maintain a high index of suspicion for affective symptoms (see Box 2) |
| Misinterpreted protective wariness | Clarify the patient’s degree of suspicion; consider this in the historical context of abuses toward minorities by majority populations Become familiar with ethnic differences in how symptoms are described |
| Covert and overt stereotyping and cultural insensitivity | Review practice patterns Consult with culturally sensitive clinicians as necessary |
Though Whaley did not report differences in distrust between African-American men and women, others have noted that distrust of health providers may be more common in minority men.18
RACIAL STEREOTYPING
Compared with similar white men, African-American men with mental disorders are more likely to be:
- referred for mental health care through social and legal—rather than medical—systems and to be involuntarily committed
- perceived as violent—even though controlled research suggests they are not. This misperception can lead to excessive medication and restraints.22
Differential treatment of African-American men may create a cycle of distrust, hostility, and additional inappropriate treatment. Together, these factors may increase the risk that African-American men will be misdiagnosed with schizophrenia.
Past racism in biomedical and psychiatric practice and research has been documented23,24 and more recently reviewed by Lawson.19 Historically, African-Americans were perceived to have a “primitive psychic” nature that was thought to be more susceptible to schizophrenia than depression.19 Whether these or similar racist stereotypes continue to inject ethnic bias into clinical assessment requires further study.
WHERE DO WE GO FROM HERE?
Although research into methods to eliminate ethnicity bias is sparse, the work reviewed in this article suggests ways that psychiatrists can minimize this problem (Table).
Obtain comprehensive information. Use structured interviews, such as the Structured Clinical Interview for DSM-IV (SCID), and rating scales, such as the Hamilton Depression Scale, which require clinicians to ask about all types of symptoms, particularly affective symptoms.
Review treatment records. Review your practice patterns for evidence of schizophrenia over-diagnosis in African-Americans or other ethnic groups. Examine ethnic differences in legal referrals or use of restraints or seclusion, which may indicate an ethnic bias in how threats are perceived. Only by being aware of bias can one correct it.
Become familiar with cultural and ethnic differences in idioms of distress. Specifically, review research in cultural psychiatry to identify potential differences among cultural groups in how they describe psychiatric symptoms. Talk with colleagues or friends from other cultural groups, and read literature from different ethnic perspectives to increase your cultural sensitivity.
Consult with psychiatrists with expertise in cultural variability of clinical presentation when the diagnosis or threat assessment seems unclear. Consultation is recommended if a patient’s diagnosis is uncertain or if you detect bias in your practice.
These interventions require clinicians to become familiar with psychosocial differences in how patients of various cultural and ethnic groups express psychiatric symptoms. With this understanding, we can better engage wary patients, obtain valid information, and improve clinical practice and patient outcomes.
Finally, psychiatry’s diagnostic systems need to continually address how patient assessment is influenced by ethnicity, culture, gender, and other socio-demographic factors. Studies are needed to examine the contributions of multiple factors—such as symptom differences and stereotyping—that contribute to ethnic-related diagnostic disparities.
Related resources
- Paul AM. Painting insanity black: Why are there more black schizophrenics? Salon.com Dec. 1, 1999. http://www.salon.com/books/it/1999/12/01/schizo/index.html
- Alarcon RD, Westermeyer J, Foulks EF, Ruiz P. Clinical relevance of contemporary cultural psychiatry. J Nerv Ment Dis 1999;187: 465-71.
- Williams DR, Neighbors HW, Jackson JS. Racial/ethnic discrimination and health: findings from community studies. Am J Public Health 2003;93:200-8.
- Lin KM, Smith MW, Ortiz V. Culture and psychopharmacology. Psychiatr Clin North Am 2001; 24:523-38.
Acknowledgement
Preparation of this manuscript was supported in part by National Institutes of Health grant MH56352.
1. Robins LN, Regier DA (eds). Psychiatric disorders in America: the Epidemiologic Catchment Area study. New York: The Free Press. 1991.
2. Simon RJ, Fleiss JL, Gurland BJ, et al. Depression and schizophrenia in hospitalized black and white mental patients. Arch Gen Psychiatry 1973;28:509-12.
3. Mukherjee S, Shukla S, Woodle J, et al. Misdiagnosis of schizophrenia in bipolar patients: a multiethnic comparison. Am J Psychiatry 1983;140:1571-4.
4. Strakowski SM, Shelton RC, Kolbrener ML. The effects of race and comorbidity on clinical diagnosis in patients with psychosis. J Clin Psychiatry 1993;54:96-102.
5. Strakowski SM, Lonczak HS, Sax KW, et al. The effects of race on diagnosis and disposition from a psychiatric emergency service. J Clin Psychiatry 1995;56:101-7.
6. Lawson WB, Hepler N, Holladay J, Cuffel B. Race as a factor in inpatient and outpatient admissions and diagnosis. Hosp Comm Psychiatry 1994;45:72-4.
7. Hoffman H, Kupper Z, Kunz B. Hopelessness and its impact on rehabilitation outcome in schizophrenia—an exploratory study. Schizophr Res 2000;43:147-58.
8. Walkup JT, McAlpine DD, Olfson M, et al. Patients with schizophrenia at risk for excessive antipsychotic dosing. J Clin Psychiatry 2000;61:344-8.
9. Segal SP, Bola JR, Watson MA. Race, quality of care, and antipsychotic prescribing practices in psychiatric emergency services. Psychiatr Serv 1996;47:282-6.
10. Flaskerud JH, Hu L. Racial/ethnic identity and amount and type of psychiatric treatment. Am J Psychiatry 1992;149:379-84.
11. Adebimpe VR, Klein HE, Fried J. Hallucinations and delusions in black psychiatric patients. J Natl Med Assoc 1981;73:517-20.
12. Adebimpe VR, Chu CC, Klein HE, Lange MH. Racial and geographic differences in the psychopathology of schizophrenia. Am J Psychiatry 1982;139:888-91.
13. Fabrega H, Jr, Mezzich J, Ulrich RF. Black-white differences in psychopathology in an urban psychiatric population. Compr Psychiatry 1988;29:285-97.
14. Strakowski SM, Flaum M, Amador X, et al. Racial differences in the diagnosis of psychosis. Schizophr Res 1996;21:117-24.
15. Schneider K. Clinical psychopathology (translated by Hamilton MW). New York: Grune and Stratton, 1959.
16. Strakowski SM, McElroy SL, Keck PE, Jr, West SA. Racial influence on diagnosis in psychotic mania. J Aff Disord. 1996;39:157-62.
17. Strakowski SM, Hawkins JM, Keck PE, Jr, et al. The effects of race and information variance on disagreement between psychiatric emergency service and research diagnoses in first-episode psychosis. J Clin Psychiatry 1997;58:457-63.
18. Jones BE, Gray BA. Problems in diagnosing schizophrenia and affective disorders among blacks. Hosp Comm Psychiatry 1986;37:61-5.
19. Neighbors HW, Jackson JS, Campbell L, Williams D. The influence of racial factors on psychiatric diagnosis: A review and suggestions for research. Comm Ment Health J 1989;25:301-11.
20. Lawson WB. Racial and ethnic factors in psychiatric research. Hosp Comm Psychiatry 1986;37:50-4.
21. Whaley AL. Ethnicity/race, paranoia, and psychiatric diagnoses: Clinician bias versus sociocultural differences. J Psychopathol Behav Assess 1997;19:1-20.
22. Lawson WB, Yesavage JA, Werner RD. Race, violence, and psychopathology. J Clin Psychiatry 1984;45:294-7.
23. Spurlock J. Psychiatric states. In: Williams RA (ed). Textbook of black-related diseases. New York: McGraw-Hill, 1975.
24. Thomas A, Sillen S. Racism and psychiatry. New York: Brunner/Mazel, 1972.
In patients with psychotic symptoms, why are African-Americans more likely than whites to be diagnosed with schizophrenia? After more than 30 years of debate, some answers—and remedies for the problem—are becoming clear.
In psychiatry, where interpersonal interactions are key to eliciting diagnostic symptoms and signs, there is an intrinsic risk of misinterpretation when clinician and patient are of different cultural, ethnic, or socioeconomic backgrounds. This article analyzes four factors that contribute to misinterpretation and to ethnic misdiagnosis of schizophrenia. Culturally sensitive strategies are offered to avoid diagnostic bias in clinical practice.
SCHIZOPHRENIA MISDIAGNOSIS
Large epidemiologic studies report similar rates of schizophrenia and bipolar disorder in African-American and white populations.1 Although patients of both races have been wrongly diagnosed with schizophrenia, the pattern is stronger and more persistent in African-Americans.
Two (or more) of the following, each present for a significant portion of time during a 1-month period (or less if successfully treated):
- Delusions
- Hallucinations
- Disorganized speech (eg, frequent derailment or incoherence)
- Grossly disorganized or catatonic behavior
- Negative symptoms (ie, affective flattening, alogia, or avolition)
Note: Only one Criterion A symptom is required if delusions are bizarre or hallucinations consist of a voice keeping up a running commentary on the person’s behavior or thoughts, or two or more voices conversing with each other.
295.30 Paranoid type
A type of schizophrenia in which the following criteria are met:
- Preoccupation with one or more delusions or frequent auditory hallucinations
- None of the following is prominent: disorganized speech, disorganized or catatonic behavior, or flat or inappropriate affect.
Source: DSM-IV-TR
In the 1970s, Simon et al2 studied 192 hospitalized patients, of whom all African-Americans and 85% of whites had been identified clinically as having schizophrenia. Using a structured interview, the researchers found that only 40% of the African Americans and 50% of the whites met diagnostic criteria for schizophrenia. African-Americans with mood disorders were found to be at particular risk of schizophrenia misdiagnosis.
In the 1980s, among 76 patients with a clinical diagnosis of schizophrenia, Mukherjee et al3 diagnosed one-half (52%) with bipolar disorder using a structured clinical interview. Schizophrenia misdiagnoses were more common in African-Americans (86%) and Hispanics (83%) than in whites (51%). In particular, African-Americans were most likely to be misdiagnosed with paranoid schizophrenia. African-Americans complained more commonly than whites of auditory hallucinations, which may represent an ethnic difference in symptomatic presentation of psychotic mood disorders.
In the 1990s, colleagues and I conducted two studies—one of 173 patients in a Tennessee psychiatric hospital4 and the other of 490 patients in an Ohio psychiatric emergency service5—and found yet again that African-Americans were more likely than whites to be diagnosed with schizophrenia. In the hospital study, higher rates of schizophrenia diagnosis were associated with lower rates of mood disorder diagnosis. This inverse relationship implied that African-Americans with mood disorders were being misdiagnosed with schizophrenia.
Men were more likely than women to be diagnosed with schizophrenia, suggesting that African-American men were most likely to be misdiagnosed. When adjustments were made for gender, black women were found to be at higher risk for misdiagnosis than white women.
Lawson et al6 extended this research in a population-based study of African-Americans living in Tennessee. They found that African-Americans constituted 16% of the state’s population but 48% of psychiatric inpatients diagnosed with schizophrenia and 37% of psychiatric outpatients.
CONSEQUENCES OF INACCURATE DIAGNOSIS
Differentiating between schizophrenia (Box 1) and a psychotic mood disorder (Box 2) is more than a semantic exercise. Schizophrenia implies a chronic, unremitting, debilitating illness that worsens over time. Though this perception of schizophrenia is not entirely accurate, in clinical practice its diagnosis imparts a bleak prognosis that may lower the clinician’s expectations for the patient.7
Schizophrenia misdiagnosis also may lead the psychiatrist to rely excessively on antipsychotics, rather than attempting thymoleptic and psychotherapy trials. Studies suggest that African-American patients are more likely than similar white patients to receive antipsychotics4,8,9 and less likely to receive psychotherapy.5,10
Reasons why African-Americans are often misdiagnosed with schizophrenia remain unclear but probably include four contributing factors:
- differences in symptom presentation compared with whites
- failure by clinicians to identify affective symptoms in African-Americans
- minority patients’ wariness when dealing with health services
- and racial stereotyping.
DIFFERENCES IN SYMPTOM EXPRESSION
African-American patients with mood disorders or schizophrenia are more likely than are similar white patients to complain of auditory hallucinations.11-13 For example, Strakowski et al14 examined 330 patients with nonaffective and psychotic diagnoses in a study that was used to develop DSM-IV criteria for schizophrenia. Auditory hallucinations were rated as more severe in African-American than in similar white patients.
MAJOR DEPRESSIVE EPISODE
Five or more of the following symptoms present during the same 2-week period and representing a change from previous functioning; must include either depressed mood or loss of interest or pleasure.
- Depressed mood
- Markedly diminished interest or pleasure
- Significant weight loss
- Insomnia or hypersomnia
- Psychomotor agitation or retardation
- Fatigue
- Feelings of worthlessness or excessive guilt
- Diminished ability to concentrate
SEVERE MAJOR DEPRESSION WITH PSYCHOTIC FEATURES
Mood-congruent
Delusions or hallucinations whose content is entirely consistent with the typical depressive themes of personal inadequacy, guilt, disease, death, nihilism, or deserved punishment
Mood-incongruent
Delusions or hallucinations whose content does not involve typical depressive themes. Includes symptoms such as persecutory delusions, thought insertion, thought broadcasting, and delusions of control
Source: DSM-IV-TR
African-American patients also are more likely than whites to exhibit so-called Schneiderian first-rank symptoms of schizophrenia,15 including:
- delusions of thought broadcasting or insertion
- auditory hallucinations of voices conversing about the patient in the third person.
These symptoms were once used to diagnose schizophrenia, but their lack of specificity has been well documented.2,16 First-rank symptoms of schizophrenia depend on the specific form of the hallucination or delusion, are likely to be influenced by a patient’s culture, and may be misleading in multicultural populations. Though first-rank symptoms now occupy a minor role in U.S. diagnostic systems, they might continue to sway clinicians—even when using structured diagnostic interviews—to inappropriately diagnose schizophrenia in lieu of affective disorders in minority patients.15
To extend this finding, our group16 studied rates and severity of affective and psychotic symptoms—particularly first-rank symptoms—in 100 patients with psychotic mania who met DSM-III-R criteria for bipolar disorder (80%) or schizoaffective disorder, bipolar type (20%) as determined by structured diagnostic interview. No differences in affective symptoms between African-American and white patients were seen. African-Americans were more likely to endorse auditory hallucinations and to report severe auditory hallucinations of voices commenting on their behavior—the only first-rank symptom on which they differed from whites.
Though their affective symptoms were similar, African-Americans were significantly more likely than whites to have been diagnosed with a schizophrenia-spectrum disorder. Because misdiagnosis of African-Americans could not be explained by psychotic symptoms—which were as severe as those of white patients—these findings suggest other mechanisms were at work.
UNIDENTIFIED AFFECTIVE SYMPTOMS
Underidentification of mood disorders in African-American patients may also lead to over-diagnosis of schizophrenia. In a sample of 99 patients, colleagues and I17 compared clinical diagnoses made in a psychiatric emergency service with those by research investigators using a structured clinical interview. Reasons for diagnostic differences were identified and divided into two categories:
- the same symptoms were recorded but applied differently to diagnostic criteria (criterion variance)
- different information was recorded, which led to diagnostic discrepancies (information variance).
Differences occurred significantly more often in African-American than in white patients, but only information variance was associated with ethnicity. This suggests that clinicians are less likely to elicit appropriate information from African-American than from white psychiatric patients. The fact that researchers obtained this information during diagnostic interviews suggests that the patients could provide it when given appropriate prompts. Specifically, affective symptoms were less likely to be elicited by clinicians than by researchers.
PATIENT WARINESS
Minority patients, when interacting with clinicians of the majority population, may project “protective wariness.”18 Specific behaviors include hesitancy or reluctance to fully engage with the care provider as a precaution against being exploited or harmed. Cultural misunderstandings19 and patient concerns about past reports of minorities receiving substandard or unethical health care20 may contribute to this behavior.
Whaley21 compared nonpathologic distrust and paranoia in 404 community-living African-Americans and whites. Some were healthy, and some had diagnoses of schizophrenia or depression. African-Americans—particularly those with psychiatric disorders—showed higher levels of distrust than whites. Distrust was also associated with depression in African-Americans but not in whites. Whaley concluded that:
- depressed African-Americans may exhibit more distrust toward clinicians than do whites
- this distrust puts African-Americans at risk of being perceived as paranoid and being misdiagnosed with paranoid schizophrenia.
Table
Remedial actions to avoid ethnic bias in diagnosing schizophrenia
| Problem | Remedies |
|---|---|
| Failure to recognize differences in symptom expression | Become familiar with ethnic differences in how patients describe symptoms Incorporate structured interviews or rating scales into the clinical assessment |
| Failure to elicit affective symptoms | Incorporate structured interviews or rating scales into the clinical assessment Maintain a high index of suspicion for affective symptoms (see Box 2) |
| Misinterpreted protective wariness | Clarify the patient’s degree of suspicion; consider this in the historical context of abuses toward minorities by majority populations Become familiar with ethnic differences in how symptoms are described |
| Covert and overt stereotyping and cultural insensitivity | Review practice patterns Consult with culturally sensitive clinicians as necessary |
Though Whaley did not report differences in distrust between African-American men and women, others have noted that distrust of health providers may be more common in minority men.18
RACIAL STEREOTYPING
Compared with similar white men, African-American men with mental disorders are more likely to be:
- referred for mental health care through social and legal—rather than medical—systems and to be involuntarily committed
- perceived as violent—even though controlled research suggests they are not. This misperception can lead to excessive medication and restraints.22
Differential treatment of African-American men may create a cycle of distrust, hostility, and additional inappropriate treatment. Together, these factors may increase the risk that African-American men will be misdiagnosed with schizophrenia.
Past racism in biomedical and psychiatric practice and research has been documented23,24 and more recently reviewed by Lawson.19 Historically, African-Americans were perceived to have a “primitive psychic” nature that was thought to be more susceptible to schizophrenia than depression.19 Whether these or similar racist stereotypes continue to inject ethnic bias into clinical assessment requires further study.
WHERE DO WE GO FROM HERE?
Although research into methods to eliminate ethnicity bias is sparse, the work reviewed in this article suggests ways that psychiatrists can minimize this problem (Table).
Obtain comprehensive information. Use structured interviews, such as the Structured Clinical Interview for DSM-IV (SCID), and rating scales, such as the Hamilton Depression Scale, which require clinicians to ask about all types of symptoms, particularly affective symptoms.
Review treatment records. Review your practice patterns for evidence of schizophrenia over-diagnosis in African-Americans or other ethnic groups. Examine ethnic differences in legal referrals or use of restraints or seclusion, which may indicate an ethnic bias in how threats are perceived. Only by being aware of bias can one correct it.
Become familiar with cultural and ethnic differences in idioms of distress. Specifically, review research in cultural psychiatry to identify potential differences among cultural groups in how they describe psychiatric symptoms. Talk with colleagues or friends from other cultural groups, and read literature from different ethnic perspectives to increase your cultural sensitivity.
Consult with psychiatrists with expertise in cultural variability of clinical presentation when the diagnosis or threat assessment seems unclear. Consultation is recommended if a patient’s diagnosis is uncertain or if you detect bias in your practice.
These interventions require clinicians to become familiar with psychosocial differences in how patients of various cultural and ethnic groups express psychiatric symptoms. With this understanding, we can better engage wary patients, obtain valid information, and improve clinical practice and patient outcomes.
Finally, psychiatry’s diagnostic systems need to continually address how patient assessment is influenced by ethnicity, culture, gender, and other socio-demographic factors. Studies are needed to examine the contributions of multiple factors—such as symptom differences and stereotyping—that contribute to ethnic-related diagnostic disparities.
Related resources
- Paul AM. Painting insanity black: Why are there more black schizophrenics? Salon.com Dec. 1, 1999. http://www.salon.com/books/it/1999/12/01/schizo/index.html
- Alarcon RD, Westermeyer J, Foulks EF, Ruiz P. Clinical relevance of contemporary cultural psychiatry. J Nerv Ment Dis 1999;187: 465-71.
- Williams DR, Neighbors HW, Jackson JS. Racial/ethnic discrimination and health: findings from community studies. Am J Public Health 2003;93:200-8.
- Lin KM, Smith MW, Ortiz V. Culture and psychopharmacology. Psychiatr Clin North Am 2001; 24:523-38.
Acknowledgement
Preparation of this manuscript was supported in part by National Institutes of Health grant MH56352.
In patients with psychotic symptoms, why are African-Americans more likely than whites to be diagnosed with schizophrenia? After more than 30 years of debate, some answers—and remedies for the problem—are becoming clear.
In psychiatry, where interpersonal interactions are key to eliciting diagnostic symptoms and signs, there is an intrinsic risk of misinterpretation when clinician and patient are of different cultural, ethnic, or socioeconomic backgrounds. This article analyzes four factors that contribute to misinterpretation and to ethnic misdiagnosis of schizophrenia. Culturally sensitive strategies are offered to avoid diagnostic bias in clinical practice.
SCHIZOPHRENIA MISDIAGNOSIS
Large epidemiologic studies report similar rates of schizophrenia and bipolar disorder in African-American and white populations.1 Although patients of both races have been wrongly diagnosed with schizophrenia, the pattern is stronger and more persistent in African-Americans.
Two (or more) of the following, each present for a significant portion of time during a 1-month period (or less if successfully treated):
- Delusions
- Hallucinations
- Disorganized speech (eg, frequent derailment or incoherence)
- Grossly disorganized or catatonic behavior
- Negative symptoms (ie, affective flattening, alogia, or avolition)
Note: Only one Criterion A symptom is required if delusions are bizarre or hallucinations consist of a voice keeping up a running commentary on the person’s behavior or thoughts, or two or more voices conversing with each other.
295.30 Paranoid type
A type of schizophrenia in which the following criteria are met:
- Preoccupation with one or more delusions or frequent auditory hallucinations
- None of the following is prominent: disorganized speech, disorganized or catatonic behavior, or flat or inappropriate affect.
Source: DSM-IV-TR
In the 1970s, Simon et al2 studied 192 hospitalized patients, of whom all African-Americans and 85% of whites had been identified clinically as having schizophrenia. Using a structured interview, the researchers found that only 40% of the African Americans and 50% of the whites met diagnostic criteria for schizophrenia. African-Americans with mood disorders were found to be at particular risk of schizophrenia misdiagnosis.
In the 1980s, among 76 patients with a clinical diagnosis of schizophrenia, Mukherjee et al3 diagnosed one-half (52%) with bipolar disorder using a structured clinical interview. Schizophrenia misdiagnoses were more common in African-Americans (86%) and Hispanics (83%) than in whites (51%). In particular, African-Americans were most likely to be misdiagnosed with paranoid schizophrenia. African-Americans complained more commonly than whites of auditory hallucinations, which may represent an ethnic difference in symptomatic presentation of psychotic mood disorders.
In the 1990s, colleagues and I conducted two studies—one of 173 patients in a Tennessee psychiatric hospital4 and the other of 490 patients in an Ohio psychiatric emergency service5—and found yet again that African-Americans were more likely than whites to be diagnosed with schizophrenia. In the hospital study, higher rates of schizophrenia diagnosis were associated with lower rates of mood disorder diagnosis. This inverse relationship implied that African-Americans with mood disorders were being misdiagnosed with schizophrenia.
Men were more likely than women to be diagnosed with schizophrenia, suggesting that African-American men were most likely to be misdiagnosed. When adjustments were made for gender, black women were found to be at higher risk for misdiagnosis than white women.
Lawson et al6 extended this research in a population-based study of African-Americans living in Tennessee. They found that African-Americans constituted 16% of the state’s population but 48% of psychiatric inpatients diagnosed with schizophrenia and 37% of psychiatric outpatients.
CONSEQUENCES OF INACCURATE DIAGNOSIS
Differentiating between schizophrenia (Box 1) and a psychotic mood disorder (Box 2) is more than a semantic exercise. Schizophrenia implies a chronic, unremitting, debilitating illness that worsens over time. Though this perception of schizophrenia is not entirely accurate, in clinical practice its diagnosis imparts a bleak prognosis that may lower the clinician’s expectations for the patient.7
Schizophrenia misdiagnosis also may lead the psychiatrist to rely excessively on antipsychotics, rather than attempting thymoleptic and psychotherapy trials. Studies suggest that African-American patients are more likely than similar white patients to receive antipsychotics4,8,9 and less likely to receive psychotherapy.5,10
Reasons why African-Americans are often misdiagnosed with schizophrenia remain unclear but probably include four contributing factors:
- differences in symptom presentation compared with whites
- failure by clinicians to identify affective symptoms in African-Americans
- minority patients’ wariness when dealing with health services
- and racial stereotyping.
DIFFERENCES IN SYMPTOM EXPRESSION
African-American patients with mood disorders or schizophrenia are more likely than are similar white patients to complain of auditory hallucinations.11-13 For example, Strakowski et al14 examined 330 patients with nonaffective and psychotic diagnoses in a study that was used to develop DSM-IV criteria for schizophrenia. Auditory hallucinations were rated as more severe in African-American than in similar white patients.
MAJOR DEPRESSIVE EPISODE
Five or more of the following symptoms present during the same 2-week period and representing a change from previous functioning; must include either depressed mood or loss of interest or pleasure.
- Depressed mood
- Markedly diminished interest or pleasure
- Significant weight loss
- Insomnia or hypersomnia
- Psychomotor agitation or retardation
- Fatigue
- Feelings of worthlessness or excessive guilt
- Diminished ability to concentrate
SEVERE MAJOR DEPRESSION WITH PSYCHOTIC FEATURES
Mood-congruent
Delusions or hallucinations whose content is entirely consistent with the typical depressive themes of personal inadequacy, guilt, disease, death, nihilism, or deserved punishment
Mood-incongruent
Delusions or hallucinations whose content does not involve typical depressive themes. Includes symptoms such as persecutory delusions, thought insertion, thought broadcasting, and delusions of control
Source: DSM-IV-TR
African-American patients also are more likely than whites to exhibit so-called Schneiderian first-rank symptoms of schizophrenia,15 including:
- delusions of thought broadcasting or insertion
- auditory hallucinations of voices conversing about the patient in the third person.
These symptoms were once used to diagnose schizophrenia, but their lack of specificity has been well documented.2,16 First-rank symptoms of schizophrenia depend on the specific form of the hallucination or delusion, are likely to be influenced by a patient’s culture, and may be misleading in multicultural populations. Though first-rank symptoms now occupy a minor role in U.S. diagnostic systems, they might continue to sway clinicians—even when using structured diagnostic interviews—to inappropriately diagnose schizophrenia in lieu of affective disorders in minority patients.15
To extend this finding, our group16 studied rates and severity of affective and psychotic symptoms—particularly first-rank symptoms—in 100 patients with psychotic mania who met DSM-III-R criteria for bipolar disorder (80%) or schizoaffective disorder, bipolar type (20%) as determined by structured diagnostic interview. No differences in affective symptoms between African-American and white patients were seen. African-Americans were more likely to endorse auditory hallucinations and to report severe auditory hallucinations of voices commenting on their behavior—the only first-rank symptom on which they differed from whites.
Though their affective symptoms were similar, African-Americans were significantly more likely than whites to have been diagnosed with a schizophrenia-spectrum disorder. Because misdiagnosis of African-Americans could not be explained by psychotic symptoms—which were as severe as those of white patients—these findings suggest other mechanisms were at work.
UNIDENTIFIED AFFECTIVE SYMPTOMS
Underidentification of mood disorders in African-American patients may also lead to over-diagnosis of schizophrenia. In a sample of 99 patients, colleagues and I17 compared clinical diagnoses made in a psychiatric emergency service with those by research investigators using a structured clinical interview. Reasons for diagnostic differences were identified and divided into two categories:
- the same symptoms were recorded but applied differently to diagnostic criteria (criterion variance)
- different information was recorded, which led to diagnostic discrepancies (information variance).
Differences occurred significantly more often in African-American than in white patients, but only information variance was associated with ethnicity. This suggests that clinicians are less likely to elicit appropriate information from African-American than from white psychiatric patients. The fact that researchers obtained this information during diagnostic interviews suggests that the patients could provide it when given appropriate prompts. Specifically, affective symptoms were less likely to be elicited by clinicians than by researchers.
PATIENT WARINESS
Minority patients, when interacting with clinicians of the majority population, may project “protective wariness.”18 Specific behaviors include hesitancy or reluctance to fully engage with the care provider as a precaution against being exploited or harmed. Cultural misunderstandings19 and patient concerns about past reports of minorities receiving substandard or unethical health care20 may contribute to this behavior.
Whaley21 compared nonpathologic distrust and paranoia in 404 community-living African-Americans and whites. Some were healthy, and some had diagnoses of schizophrenia or depression. African-Americans—particularly those with psychiatric disorders—showed higher levels of distrust than whites. Distrust was also associated with depression in African-Americans but not in whites. Whaley concluded that:
- depressed African-Americans may exhibit more distrust toward clinicians than do whites
- this distrust puts African-Americans at risk of being perceived as paranoid and being misdiagnosed with paranoid schizophrenia.
Table
Remedial actions to avoid ethnic bias in diagnosing schizophrenia
| Problem | Remedies |
|---|---|
| Failure to recognize differences in symptom expression | Become familiar with ethnic differences in how patients describe symptoms Incorporate structured interviews or rating scales into the clinical assessment |
| Failure to elicit affective symptoms | Incorporate structured interviews or rating scales into the clinical assessment Maintain a high index of suspicion for affective symptoms (see Box 2) |
| Misinterpreted protective wariness | Clarify the patient’s degree of suspicion; consider this in the historical context of abuses toward minorities by majority populations Become familiar with ethnic differences in how symptoms are described |
| Covert and overt stereotyping and cultural insensitivity | Review practice patterns Consult with culturally sensitive clinicians as necessary |
Though Whaley did not report differences in distrust between African-American men and women, others have noted that distrust of health providers may be more common in minority men.18
RACIAL STEREOTYPING
Compared with similar white men, African-American men with mental disorders are more likely to be:
- referred for mental health care through social and legal—rather than medical—systems and to be involuntarily committed
- perceived as violent—even though controlled research suggests they are not. This misperception can lead to excessive medication and restraints.22
Differential treatment of African-American men may create a cycle of distrust, hostility, and additional inappropriate treatment. Together, these factors may increase the risk that African-American men will be misdiagnosed with schizophrenia.
Past racism in biomedical and psychiatric practice and research has been documented23,24 and more recently reviewed by Lawson.19 Historically, African-Americans were perceived to have a “primitive psychic” nature that was thought to be more susceptible to schizophrenia than depression.19 Whether these or similar racist stereotypes continue to inject ethnic bias into clinical assessment requires further study.
WHERE DO WE GO FROM HERE?
Although research into methods to eliminate ethnicity bias is sparse, the work reviewed in this article suggests ways that psychiatrists can minimize this problem (Table).
Obtain comprehensive information. Use structured interviews, such as the Structured Clinical Interview for DSM-IV (SCID), and rating scales, such as the Hamilton Depression Scale, which require clinicians to ask about all types of symptoms, particularly affective symptoms.
Review treatment records. Review your practice patterns for evidence of schizophrenia over-diagnosis in African-Americans or other ethnic groups. Examine ethnic differences in legal referrals or use of restraints or seclusion, which may indicate an ethnic bias in how threats are perceived. Only by being aware of bias can one correct it.
Become familiar with cultural and ethnic differences in idioms of distress. Specifically, review research in cultural psychiatry to identify potential differences among cultural groups in how they describe psychiatric symptoms. Talk with colleagues or friends from other cultural groups, and read literature from different ethnic perspectives to increase your cultural sensitivity.
Consult with psychiatrists with expertise in cultural variability of clinical presentation when the diagnosis or threat assessment seems unclear. Consultation is recommended if a patient’s diagnosis is uncertain or if you detect bias in your practice.
These interventions require clinicians to become familiar with psychosocial differences in how patients of various cultural and ethnic groups express psychiatric symptoms. With this understanding, we can better engage wary patients, obtain valid information, and improve clinical practice and patient outcomes.
Finally, psychiatry’s diagnostic systems need to continually address how patient assessment is influenced by ethnicity, culture, gender, and other socio-demographic factors. Studies are needed to examine the contributions of multiple factors—such as symptom differences and stereotyping—that contribute to ethnic-related diagnostic disparities.
Related resources
- Paul AM. Painting insanity black: Why are there more black schizophrenics? Salon.com Dec. 1, 1999. http://www.salon.com/books/it/1999/12/01/schizo/index.html
- Alarcon RD, Westermeyer J, Foulks EF, Ruiz P. Clinical relevance of contemporary cultural psychiatry. J Nerv Ment Dis 1999;187: 465-71.
- Williams DR, Neighbors HW, Jackson JS. Racial/ethnic discrimination and health: findings from community studies. Am J Public Health 2003;93:200-8.
- Lin KM, Smith MW, Ortiz V. Culture and psychopharmacology. Psychiatr Clin North Am 2001; 24:523-38.
Acknowledgement
Preparation of this manuscript was supported in part by National Institutes of Health grant MH56352.
1. Robins LN, Regier DA (eds). Psychiatric disorders in America: the Epidemiologic Catchment Area study. New York: The Free Press. 1991.
2. Simon RJ, Fleiss JL, Gurland BJ, et al. Depression and schizophrenia in hospitalized black and white mental patients. Arch Gen Psychiatry 1973;28:509-12.
3. Mukherjee S, Shukla S, Woodle J, et al. Misdiagnosis of schizophrenia in bipolar patients: a multiethnic comparison. Am J Psychiatry 1983;140:1571-4.
4. Strakowski SM, Shelton RC, Kolbrener ML. The effects of race and comorbidity on clinical diagnosis in patients with psychosis. J Clin Psychiatry 1993;54:96-102.
5. Strakowski SM, Lonczak HS, Sax KW, et al. The effects of race on diagnosis and disposition from a psychiatric emergency service. J Clin Psychiatry 1995;56:101-7.
6. Lawson WB, Hepler N, Holladay J, Cuffel B. Race as a factor in inpatient and outpatient admissions and diagnosis. Hosp Comm Psychiatry 1994;45:72-4.
7. Hoffman H, Kupper Z, Kunz B. Hopelessness and its impact on rehabilitation outcome in schizophrenia—an exploratory study. Schizophr Res 2000;43:147-58.
8. Walkup JT, McAlpine DD, Olfson M, et al. Patients with schizophrenia at risk for excessive antipsychotic dosing. J Clin Psychiatry 2000;61:344-8.
9. Segal SP, Bola JR, Watson MA. Race, quality of care, and antipsychotic prescribing practices in psychiatric emergency services. Psychiatr Serv 1996;47:282-6.
10. Flaskerud JH, Hu L. Racial/ethnic identity and amount and type of psychiatric treatment. Am J Psychiatry 1992;149:379-84.
11. Adebimpe VR, Klein HE, Fried J. Hallucinations and delusions in black psychiatric patients. J Natl Med Assoc 1981;73:517-20.
12. Adebimpe VR, Chu CC, Klein HE, Lange MH. Racial and geographic differences in the psychopathology of schizophrenia. Am J Psychiatry 1982;139:888-91.
13. Fabrega H, Jr, Mezzich J, Ulrich RF. Black-white differences in psychopathology in an urban psychiatric population. Compr Psychiatry 1988;29:285-97.
14. Strakowski SM, Flaum M, Amador X, et al. Racial differences in the diagnosis of psychosis. Schizophr Res 1996;21:117-24.
15. Schneider K. Clinical psychopathology (translated by Hamilton MW). New York: Grune and Stratton, 1959.
16. Strakowski SM, McElroy SL, Keck PE, Jr, West SA. Racial influence on diagnosis in psychotic mania. J Aff Disord. 1996;39:157-62.
17. Strakowski SM, Hawkins JM, Keck PE, Jr, et al. The effects of race and information variance on disagreement between psychiatric emergency service and research diagnoses in first-episode psychosis. J Clin Psychiatry 1997;58:457-63.
18. Jones BE, Gray BA. Problems in diagnosing schizophrenia and affective disorders among blacks. Hosp Comm Psychiatry 1986;37:61-5.
19. Neighbors HW, Jackson JS, Campbell L, Williams D. The influence of racial factors on psychiatric diagnosis: A review and suggestions for research. Comm Ment Health J 1989;25:301-11.
20. Lawson WB. Racial and ethnic factors in psychiatric research. Hosp Comm Psychiatry 1986;37:50-4.
21. Whaley AL. Ethnicity/race, paranoia, and psychiatric diagnoses: Clinician bias versus sociocultural differences. J Psychopathol Behav Assess 1997;19:1-20.
22. Lawson WB, Yesavage JA, Werner RD. Race, violence, and psychopathology. J Clin Psychiatry 1984;45:294-7.
23. Spurlock J. Psychiatric states. In: Williams RA (ed). Textbook of black-related diseases. New York: McGraw-Hill, 1975.
24. Thomas A, Sillen S. Racism and psychiatry. New York: Brunner/Mazel, 1972.
1. Robins LN, Regier DA (eds). Psychiatric disorders in America: the Epidemiologic Catchment Area study. New York: The Free Press. 1991.
2. Simon RJ, Fleiss JL, Gurland BJ, et al. Depression and schizophrenia in hospitalized black and white mental patients. Arch Gen Psychiatry 1973;28:509-12.
3. Mukherjee S, Shukla S, Woodle J, et al. Misdiagnosis of schizophrenia in bipolar patients: a multiethnic comparison. Am J Psychiatry 1983;140:1571-4.
4. Strakowski SM, Shelton RC, Kolbrener ML. The effects of race and comorbidity on clinical diagnosis in patients with psychosis. J Clin Psychiatry 1993;54:96-102.
5. Strakowski SM, Lonczak HS, Sax KW, et al. The effects of race on diagnosis and disposition from a psychiatric emergency service. J Clin Psychiatry 1995;56:101-7.
6. Lawson WB, Hepler N, Holladay J, Cuffel B. Race as a factor in inpatient and outpatient admissions and diagnosis. Hosp Comm Psychiatry 1994;45:72-4.
7. Hoffman H, Kupper Z, Kunz B. Hopelessness and its impact on rehabilitation outcome in schizophrenia—an exploratory study. Schizophr Res 2000;43:147-58.
8. Walkup JT, McAlpine DD, Olfson M, et al. Patients with schizophrenia at risk for excessive antipsychotic dosing. J Clin Psychiatry 2000;61:344-8.
9. Segal SP, Bola JR, Watson MA. Race, quality of care, and antipsychotic prescribing practices in psychiatric emergency services. Psychiatr Serv 1996;47:282-6.
10. Flaskerud JH, Hu L. Racial/ethnic identity and amount and type of psychiatric treatment. Am J Psychiatry 1992;149:379-84.
11. Adebimpe VR, Klein HE, Fried J. Hallucinations and delusions in black psychiatric patients. J Natl Med Assoc 1981;73:517-20.
12. Adebimpe VR, Chu CC, Klein HE, Lange MH. Racial and geographic differences in the psychopathology of schizophrenia. Am J Psychiatry 1982;139:888-91.
13. Fabrega H, Jr, Mezzich J, Ulrich RF. Black-white differences in psychopathology in an urban psychiatric population. Compr Psychiatry 1988;29:285-97.
14. Strakowski SM, Flaum M, Amador X, et al. Racial differences in the diagnosis of psychosis. Schizophr Res 1996;21:117-24.
15. Schneider K. Clinical psychopathology (translated by Hamilton MW). New York: Grune and Stratton, 1959.
16. Strakowski SM, McElroy SL, Keck PE, Jr, West SA. Racial influence on diagnosis in psychotic mania. J Aff Disord. 1996;39:157-62.
17. Strakowski SM, Hawkins JM, Keck PE, Jr, et al. The effects of race and information variance on disagreement between psychiatric emergency service and research diagnoses in first-episode psychosis. J Clin Psychiatry 1997;58:457-63.
18. Jones BE, Gray BA. Problems in diagnosing schizophrenia and affective disorders among blacks. Hosp Comm Psychiatry 1986;37:61-5.
19. Neighbors HW, Jackson JS, Campbell L, Williams D. The influence of racial factors on psychiatric diagnosis: A review and suggestions for research. Comm Ment Health J 1989;25:301-11.
20. Lawson WB. Racial and ethnic factors in psychiatric research. Hosp Comm Psychiatry 1986;37:50-4.
21. Whaley AL. Ethnicity/race, paranoia, and psychiatric diagnoses: Clinician bias versus sociocultural differences. J Psychopathol Behav Assess 1997;19:1-20.
22. Lawson WB, Yesavage JA, Werner RD. Race, violence, and psychopathology. J Clin Psychiatry 1984;45:294-7.
23. Spurlock J. Psychiatric states. In: Williams RA (ed). Textbook of black-related diseases. New York: McGraw-Hill, 1975.
24. Thomas A, Sillen S. Racism and psychiatry. New York: Brunner/Mazel, 1972.
Therapy-resistant major depression When to consider ECT: Algorithm seeks respect for neglected therapy
Patients with what’s called “therapy-resistant” depression (TRD)—with subtherapeutic response to medications and psychotherapy—are often actually suffering from unrecognized, inadequately treated psychotic depression. Psychiatrists could greatly diminish the clinical challenge of TRD by recognizing psychotic depression and treating it more effectively.1 And the most effective treatment for psychotic depression is neither antidepressants nor antipsychotic drugs but electroconvulsive therapy (ECT).
Despite ECT’s superior efficacy in TRD, however, algorithms for treating major depression relegate ECT to an option of last resort. By not considering ECT sooner, we consign many severely depressed patients to less-effective treatments and the risk of chronic illness.
Table
Diagnostic signs of psychosis in patients with major depression
| Sign | Example |
|---|---|
| Somatic concern | Delusions of fatal illness |
| Grandiosity | Special relation to God or royalty |
| Suspiciousness | Delusions of spousal infidelity |
| Hallucinations | Foul body odor |
| Unusual thought | Bizarre, confused ideation |
| Depressive delusion | Worthlessness, guilt, feelings of deserving death or punishment |
| Source: Based on the Brief Psychiatric Rating Scale.14 | |
It is time for a more realistic algorithm that recommends ECT earlier for major depressive episodes, with or without psychotic features. This article proposes such an algorithm and discusses the supporting evidence.
TREATING PSYCHOTIC DEPRESSION
Patients with delusions or hallucinations were classified as suffering from schizophrenia until the mid-1970s. Researchers then found that depressed patients with psychotic features responded well to ECT but poorly to adequate serum levels of imipramine.2
These observations were confirmed by a large Italian study, in which 437 depressed patients were treated with high-dose imipramine (200 to 350 mg/d). Depression remitted in 244 patients (56%). Those who remained depressed were then treated with ECT, and depression remitted in 136 of 190 (72%). Psychosis was the marker of poor response to imipramine.3 DSM-III codified these findings by separating the syndrome of “major depression with psychosis” (296.34) from “major depression without psychosis” (296.33).
As psychiatry recognized psychotic depression as a distinct form of depression, it became clear that drugs often could not adequately treat it. Less than one-third of patients with psychotic depression respond to tricyclics alone.4-6
Response to antipsychotic monotherapy averaged 50% and increased to 75% with combined antipsychotics and antidepressants. However, high daily dosages —at least 32 mg of perphenazine and 225 mg of amitriptyline—were required for an adequate response,7 and side effects made sustaining such heroic dosing was difficult. The greatest improvement rates were seen with ECT.
Few other drug combinations have been reported to be effective in psychotic depression, but we lack proper studies. Schatzberg8 addressed the use of newer antidepressants and atypical antipsychotics without offering an algorithm based on the data. Evidence on combination therapies consists mainly of case reports, with few designed studies.
EFFICACY OF ECT
ECT is the most effective treatment for psychotic major depression—achieving remission rates >80% within 3 weeks—as demonstrated by the ongoing, four-hospital Consortium for Research in ECT (CORE), supported by the National Institute of Mental Health.
CORE researchers are studying the efficacy of bilateral ECT in treating severe unipolar depression in patients ages 18 to 85 and of continuation treatments with ECT or lithium plus nortriptyline.9 Under the CORE protocol, diagnoses are made by structured clinical interview using DSM-III-R criteria, and remission is defined as >60% reduction in Hamilton Rating Scale for Depression scores, with final scores 10 sustained for 1 week.
In the first 253 CORE patients treated with ECT, depression remitted in 75% and did not remit in 11%; 14% dropped out. Psychotic depression was identified in 30% (77 of 253), and the remission rate among these patients was 83%.
Among patients who completed the full ECT course (at least 12 sessions), remission rates were 96% for psychotic depression and 83% for nonpsychotic depression. The overall remission rate was 87%.
Treatments were given three times per week. Among patients who completed treatment in weeks 1 through 4, remission rates were 5%, 45%, 81%, and 100%, respectively. Psychotic depression remitted more rapidly than nonpsychotic depression.
Suicide risk. CORE findings suggest that ECT also may reduce suicide risk. In item 3 of the Hamilton Rating Scale for Depression, scores of 2 to 4 indicate preoccupation with death or suicide, or a recent suicide attempt. Nearly 60% of 404 patients (237) reported baseline scores of 2 to 4, but their scores dropped rapidly with ECT. Scores of 0 were reported in 68% after 1 week of ECT, in 87% after 2 weeks, and in 93% after 3 weeks.10
Summary. In patients with severe depressive illness, CORE’s remission rates of 95% for psychotic depression and 83% for nonpsychotic depression are remarkable. Another group is independently reporting a 92% remission rate for psychotic depression treated with ECT.11
Psychotic depression is difficult to recognize and treat, even for clinicians with advanced training. For example:
- only 2 of 52 psychotic depressed patients were determined to have been adequately treated before referral to a National Institute of Mental Health-supported study of ECT15
- only 3 of 46 psychotic depressed patients had been adequately treated prior to enrollment in the Consortium for Research in ECT (CORE) study.9
The three most useful diagnostic criteria are spelled out in the Brief Psychiatric Rating Scale:
- any sign of psychosis is sufficient for designating a major depression as “psychotic”
- one well-developed sign is sufficient to prescribe treatment for psychotic depression
- well-developed vegetative signs also indicate the need to treat psychotic depression.14
ROADBLOCKS TO WIDER USE OF ECT
Many eligible patients never receive ECT, despite its track record of producing high remission rates in psychotic depression. Reasons include:
Limited access. Few psychiatrists—less than 8%—offer ECT as a treatment option, and most who do offer it practice in private hospitals.12-14
Academic low regard. Psychiatry’s academic lecturers largely ignore ECT’s efficacy in psychotic depression and therapy-resistant depression. This low regard for ECT is codified in expert algorithms, which cite ECT as an option of last resort.
Social stigma. In a recent essay summarizing medication’s weak effect in treating psychotic depression, Schatzberg states, “While ECT is a remarkably effective treatment for psychotic depression, requirements for its use are stringent, and public perception about the overall appropriateness of shock treatment is negative.”8
Algorithm Treatment of major depression, with or without psychotic features

ALGORITHM FOR MAJOR DEPRESSION
Because patients with psychotic and nonpsychotic major depression clearly require different treatments, differentiating between these two types is critical. Although psychotic depression can be difficult to diagnose,15,16 commonly recognized criteria are listed in the Table
Assessment. Assess each severely depressed patient for psychotic features, such as delusions and hallucinations. A useful assessment guide is the Brief Psychiatric Rating Scale (Box).17 Also treat those with melancholia, inanition, severe weight loss and insomnia, concentration and memory difficulty, stupor, or suicidal ideation as if they had psychotic depression. These symptoms and signs are evidence that the patient’s neuroendocrine system is disturbed, an indication of severe depression that responds poorly to antidepressant drugs alone.
Treatment. Nonpsychotic depressed patients are best offered antidepressants—tricyclics or selective serotonin reuptake inhibitors (SSRIs)—as recommended by conventional guidelines. Insufficient response to two adequate trials calls for a careful assessment for psychosis and, if found, treatment with effective drug combinations or ECT (Algorithm).18 For patients with psychotic depression—especially those who fail medication trials or whose severe symptoms would likely respond to ECT as a primary treatment—bilateral ECT is the effective standard.19
ECT is the appropriate first option for hospitalized patients with psychotic depression—especially those who are suicidal or require supplementary feeding and sedation. It may also be considered the first option in patients who have:
- attempted suicide
- lost more than 10% of body weight (approximately 15 lbs for adults) in the weeks of their illness
- or show signs of severe melancholia, such as catatonia or pseudodementia.
TREATING NONPSYCHOTIC DEPRESSION
When medications are first-line treatment for nonpsychotic depression, how long should a trial be continued before taking another tack? How many courses should you try before you declare therapy resistance and consider ECT?
Studies of clozapine provide a useful model.20 Because of clozapine’s association with agranulocytosis and induced seizures, patients with schizophrenia usually do not receive this antipsychotic unless two 4- to 6-week trials of other neuroleptics have proven ineffective. Ethicists have deemed two failed medication trials to be sufficient before a more hazardous treatment is offered.
Figure Remission of major depression with ECT
We can apply a similar standard when considering ECT in patients first treated with medication.18 A patient’s depression could be defined as therapy-resistant after an inadequate response to 4-week courses (in either order) of:
- an SSRI at dosages equivalent to fluoxetine, 40 mg/d
- a tricyclic at 200 mg/d.
In depressed patients with bipolar disorder, a trial of lithium or an anticonvulsant may replace an antidepressant.
ECT is appropriate when debilitating depression persists after two adequate medication trials. Only after an adequate ECT trial has failed—and such failure is infrequent—is it reasonable to offer poorly tested augmentation and combination strategies.
What is ‘adequate’ ECT? For patients with major depression, the definition of an adequate ECT trial is complex. Although many doctors expect six ECT sessions to be sufficient, the CORE studies are finding that only 45% of patients remitted with six ECT, 81% with nine ECT, and almost all with 12 ECT sessions (Figure).9 These patients were treated with bitemporal electrode placement, the more effective form of ECT. When unilateral electrode placements are used, ECT courses may be inadequate, as this form requires special attention to electrical dosing.
Further, the quality of the seizure—like the dosing of medications—directly influences outcome. Seizure monitoring is essential for assessing the adequacy of each treatment and the treatment course.21,22
Related resources
- Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus non-psychotic depressed patients: a report from CORE. J ECT 2001;17:244-53.
Drug brand names
- Amitriptyline • Elavil
- Clozapine • Clozaril
- Imipramine • Tofranil
- Nortriptyline • Pamelor
- Perphenazine • Trilafon
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Preparation of this manuscript was aided by grants from the Scion Natural Science Association, Inc., St. James, NY.
1. Thase M. New approaches to managing difficult-to-treat depressions. J Clin Psychiatry 2003;64[suppl1]:3-4.
2. Kantor SJ, Glassman AH. Delusional depressions: natural history and response to treatment. Br J Psychiatry 1977;131:351-6.
3. Avery D, Lubrano A. Depression treated with imipramine and ECT: the deCarolis study reconsidered. Am J Psychiatry 1979;136:559-62.
4. Kroessler D. Relative efficacy rates for therapies of delusional depression. Convuls Ther 1985;1:173-82.
5. Parker G, Roy K, Hadzi-Pavlovic D, Pedic F. Psychotic (delusional) depression: a meta-analysis of physical treatments. J Affect Dis 1992;24:17-24.
6. Wheeler Vega JA, Mortimer AM, Tyson PJ. Somatic treatment of psychotic depression: review and recommendations for practice. J Clin Psychopharmacol 2000;20:504-19.
7. Spiker DG, Weiss JC, Dealy RS, et al. The pharmacologic treatment of delusional depression. Am J Psychiatry 1985;142:430-6.
8. Schatzberg AF. New approaches to managing psychotic depression. J Clin Psychiatry 2003;64[suppl1]:19-23.
9. Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus non-psychotic depressed patients: a report from CORE. J ECT 2001;17:244-53.
10. Kellner CH, Fink M, Knapp R, et al. Bilateral ECT rapidly relieves suicidality: findings from phase I of the CORE ECT study. Am J Psychiatry (submitted).
11. Birkenhäger TK, Pluijms EM, Lucius SAP. ECT response in delusional versus non-delusional depressed inpatients. J Affect Dis (in press).
12. Hermann RC, Ettner SL, Dorwart RA, et al. Characteristics of psychiatrists who perform ECT. Am J Psychiatry 1998;155:889-94.
13. Thompson JW, Weiner RD, Myers CP. Use of ECT in the United States in 1975, 1980, and 1986. Am J Psychiatry 1994;151:1657-61.
14. Kramer BA. Use of ECT in California revisited: 1984-1994. J ECT 1999;15:245-51.
15. Prudic J, Sackeim HA, Devanand DP. Medication resistance and clinical response to electroconvulsive therapy. Psychiatry Res 1990;31:287-96.
16. Mulsant BH, Haskett RF, Prudic J, et al. Low use of neuroleptic drugs in the treatment of psychotic major depression. Am J Psychiatry 1997;154:559-61.
17. Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychol Rep 1962;10:799-812.
18. Fink M. Electroconvulsive therapy in medication-resistant depression. In: Amsterdam J, Hornig-Rohan M, Nierenberg A (eds.): Treatment-resistant mood disorders. Cambridge, UK: Cambridge University Press, 2001;223-38.
19. Fink M. The efficacy of ECT and “treatment resistance.” J ECT 2002;18:1-2.
20. Lieberman JA, Kane JM, Johns CA. Clozapine: guidelines for clinical management. J Clin Psychiatry 1989;50:329-38.
21. Abrams R. Electroconvulsive therapy. (4th ed). New York: Oxford University Press, 2002.
22. Fink M. Optimizing ECT. Encephale 1994;20:297-302.
Patients with what’s called “therapy-resistant” depression (TRD)—with subtherapeutic response to medications and psychotherapy—are often actually suffering from unrecognized, inadequately treated psychotic depression. Psychiatrists could greatly diminish the clinical challenge of TRD by recognizing psychotic depression and treating it more effectively.1 And the most effective treatment for psychotic depression is neither antidepressants nor antipsychotic drugs but electroconvulsive therapy (ECT).
Despite ECT’s superior efficacy in TRD, however, algorithms for treating major depression relegate ECT to an option of last resort. By not considering ECT sooner, we consign many severely depressed patients to less-effective treatments and the risk of chronic illness.
Table
Diagnostic signs of psychosis in patients with major depression
| Sign | Example |
|---|---|
| Somatic concern | Delusions of fatal illness |
| Grandiosity | Special relation to God or royalty |
| Suspiciousness | Delusions of spousal infidelity |
| Hallucinations | Foul body odor |
| Unusual thought | Bizarre, confused ideation |
| Depressive delusion | Worthlessness, guilt, feelings of deserving death or punishment |
| Source: Based on the Brief Psychiatric Rating Scale.14 | |
It is time for a more realistic algorithm that recommends ECT earlier for major depressive episodes, with or without psychotic features. This article proposes such an algorithm and discusses the supporting evidence.
TREATING PSYCHOTIC DEPRESSION
Patients with delusions or hallucinations were classified as suffering from schizophrenia until the mid-1970s. Researchers then found that depressed patients with psychotic features responded well to ECT but poorly to adequate serum levels of imipramine.2
These observations were confirmed by a large Italian study, in which 437 depressed patients were treated with high-dose imipramine (200 to 350 mg/d). Depression remitted in 244 patients (56%). Those who remained depressed were then treated with ECT, and depression remitted in 136 of 190 (72%). Psychosis was the marker of poor response to imipramine.3 DSM-III codified these findings by separating the syndrome of “major depression with psychosis” (296.34) from “major depression without psychosis” (296.33).
As psychiatry recognized psychotic depression as a distinct form of depression, it became clear that drugs often could not adequately treat it. Less than one-third of patients with psychotic depression respond to tricyclics alone.4-6
Response to antipsychotic monotherapy averaged 50% and increased to 75% with combined antipsychotics and antidepressants. However, high daily dosages —at least 32 mg of perphenazine and 225 mg of amitriptyline—were required for an adequate response,7 and side effects made sustaining such heroic dosing was difficult. The greatest improvement rates were seen with ECT.
Few other drug combinations have been reported to be effective in psychotic depression, but we lack proper studies. Schatzberg8 addressed the use of newer antidepressants and atypical antipsychotics without offering an algorithm based on the data. Evidence on combination therapies consists mainly of case reports, with few designed studies.
EFFICACY OF ECT
ECT is the most effective treatment for psychotic major depression—achieving remission rates >80% within 3 weeks—as demonstrated by the ongoing, four-hospital Consortium for Research in ECT (CORE), supported by the National Institute of Mental Health.
CORE researchers are studying the efficacy of bilateral ECT in treating severe unipolar depression in patients ages 18 to 85 and of continuation treatments with ECT or lithium plus nortriptyline.9 Under the CORE protocol, diagnoses are made by structured clinical interview using DSM-III-R criteria, and remission is defined as >60% reduction in Hamilton Rating Scale for Depression scores, with final scores 10 sustained for 1 week.
In the first 253 CORE patients treated with ECT, depression remitted in 75% and did not remit in 11%; 14% dropped out. Psychotic depression was identified in 30% (77 of 253), and the remission rate among these patients was 83%.
Among patients who completed the full ECT course (at least 12 sessions), remission rates were 96% for psychotic depression and 83% for nonpsychotic depression. The overall remission rate was 87%.
Treatments were given three times per week. Among patients who completed treatment in weeks 1 through 4, remission rates were 5%, 45%, 81%, and 100%, respectively. Psychotic depression remitted more rapidly than nonpsychotic depression.
Suicide risk. CORE findings suggest that ECT also may reduce suicide risk. In item 3 of the Hamilton Rating Scale for Depression, scores of 2 to 4 indicate preoccupation with death or suicide, or a recent suicide attempt. Nearly 60% of 404 patients (237) reported baseline scores of 2 to 4, but their scores dropped rapidly with ECT. Scores of 0 were reported in 68% after 1 week of ECT, in 87% after 2 weeks, and in 93% after 3 weeks.10
Summary. In patients with severe depressive illness, CORE’s remission rates of 95% for psychotic depression and 83% for nonpsychotic depression are remarkable. Another group is independently reporting a 92% remission rate for psychotic depression treated with ECT.11
Psychotic depression is difficult to recognize and treat, even for clinicians with advanced training. For example:
- only 2 of 52 psychotic depressed patients were determined to have been adequately treated before referral to a National Institute of Mental Health-supported study of ECT15
- only 3 of 46 psychotic depressed patients had been adequately treated prior to enrollment in the Consortium for Research in ECT (CORE) study.9
The three most useful diagnostic criteria are spelled out in the Brief Psychiatric Rating Scale:
- any sign of psychosis is sufficient for designating a major depression as “psychotic”
- one well-developed sign is sufficient to prescribe treatment for psychotic depression
- well-developed vegetative signs also indicate the need to treat psychotic depression.14
ROADBLOCKS TO WIDER USE OF ECT
Many eligible patients never receive ECT, despite its track record of producing high remission rates in psychotic depression. Reasons include:
Limited access. Few psychiatrists—less than 8%—offer ECT as a treatment option, and most who do offer it practice in private hospitals.12-14
Academic low regard. Psychiatry’s academic lecturers largely ignore ECT’s efficacy in psychotic depression and therapy-resistant depression. This low regard for ECT is codified in expert algorithms, which cite ECT as an option of last resort.
Social stigma. In a recent essay summarizing medication’s weak effect in treating psychotic depression, Schatzberg states, “While ECT is a remarkably effective treatment for psychotic depression, requirements for its use are stringent, and public perception about the overall appropriateness of shock treatment is negative.”8
Algorithm Treatment of major depression, with or without psychotic features
ALGORITHM FOR MAJOR DEPRESSION
Because patients with psychotic and nonpsychotic major depression clearly require different treatments, differentiating between these two types is critical. Although psychotic depression can be difficult to diagnose,15,16 commonly recognized criteria are listed in the Table
Assessment. Assess each severely depressed patient for psychotic features, such as delusions and hallucinations. A useful assessment guide is the Brief Psychiatric Rating Scale (Box).17 Also treat those with melancholia, inanition, severe weight loss and insomnia, concentration and memory difficulty, stupor, or suicidal ideation as if they had psychotic depression. These symptoms and signs are evidence that the patient’s neuroendocrine system is disturbed, an indication of severe depression that responds poorly to antidepressant drugs alone.
Treatment. Nonpsychotic depressed patients are best offered antidepressants—tricyclics or selective serotonin reuptake inhibitors (SSRIs)—as recommended by conventional guidelines. Insufficient response to two adequate trials calls for a careful assessment for psychosis and, if found, treatment with effective drug combinations or ECT (Algorithm).18 For patients with psychotic depression—especially those who fail medication trials or whose severe symptoms would likely respond to ECT as a primary treatment—bilateral ECT is the effective standard.19
ECT is the appropriate first option for hospitalized patients with psychotic depression—especially those who are suicidal or require supplementary feeding and sedation. It may also be considered the first option in patients who have:
- attempted suicide
- lost more than 10% of body weight (approximately 15 lbs for adults) in the weeks of their illness
- or show signs of severe melancholia, such as catatonia or pseudodementia.
TREATING NONPSYCHOTIC DEPRESSION
When medications are first-line treatment for nonpsychotic depression, how long should a trial be continued before taking another tack? How many courses should you try before you declare therapy resistance and consider ECT?
Studies of clozapine provide a useful model.20 Because of clozapine’s association with agranulocytosis and induced seizures, patients with schizophrenia usually do not receive this antipsychotic unless two 4- to 6-week trials of other neuroleptics have proven ineffective. Ethicists have deemed two failed medication trials to be sufficient before a more hazardous treatment is offered.
Figure Remission of major depression with ECT
We can apply a similar standard when considering ECT in patients first treated with medication.18 A patient’s depression could be defined as therapy-resistant after an inadequate response to 4-week courses (in either order) of:
- an SSRI at dosages equivalent to fluoxetine, 40 mg/d
- a tricyclic at 200 mg/d.
In depressed patients with bipolar disorder, a trial of lithium or an anticonvulsant may replace an antidepressant.
ECT is appropriate when debilitating depression persists after two adequate medication trials. Only after an adequate ECT trial has failed—and such failure is infrequent—is it reasonable to offer poorly tested augmentation and combination strategies.
What is ‘adequate’ ECT? For patients with major depression, the definition of an adequate ECT trial is complex. Although many doctors expect six ECT sessions to be sufficient, the CORE studies are finding that only 45% of patients remitted with six ECT, 81% with nine ECT, and almost all with 12 ECT sessions (Figure).9 These patients were treated with bitemporal electrode placement, the more effective form of ECT. When unilateral electrode placements are used, ECT courses may be inadequate, as this form requires special attention to electrical dosing.
Further, the quality of the seizure—like the dosing of medications—directly influences outcome. Seizure monitoring is essential for assessing the adequacy of each treatment and the treatment course.21,22
Related resources
- Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus non-psychotic depressed patients: a report from CORE. J ECT 2001;17:244-53.
Drug brand names
- Amitriptyline • Elavil
- Clozapine • Clozaril
- Imipramine • Tofranil
- Nortriptyline • Pamelor
- Perphenazine • Trilafon
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Preparation of this manuscript was aided by grants from the Scion Natural Science Association, Inc., St. James, NY.
Patients with what’s called “therapy-resistant” depression (TRD)—with subtherapeutic response to medications and psychotherapy—are often actually suffering from unrecognized, inadequately treated psychotic depression. Psychiatrists could greatly diminish the clinical challenge of TRD by recognizing psychotic depression and treating it more effectively.1 And the most effective treatment for psychotic depression is neither antidepressants nor antipsychotic drugs but electroconvulsive therapy (ECT).
Despite ECT’s superior efficacy in TRD, however, algorithms for treating major depression relegate ECT to an option of last resort. By not considering ECT sooner, we consign many severely depressed patients to less-effective treatments and the risk of chronic illness.
Table
Diagnostic signs of psychosis in patients with major depression
| Sign | Example |
|---|---|
| Somatic concern | Delusions of fatal illness |
| Grandiosity | Special relation to God or royalty |
| Suspiciousness | Delusions of spousal infidelity |
| Hallucinations | Foul body odor |
| Unusual thought | Bizarre, confused ideation |
| Depressive delusion | Worthlessness, guilt, feelings of deserving death or punishment |
| Source: Based on the Brief Psychiatric Rating Scale.14 | |
It is time for a more realistic algorithm that recommends ECT earlier for major depressive episodes, with or without psychotic features. This article proposes such an algorithm and discusses the supporting evidence.
TREATING PSYCHOTIC DEPRESSION
Patients with delusions or hallucinations were classified as suffering from schizophrenia until the mid-1970s. Researchers then found that depressed patients with psychotic features responded well to ECT but poorly to adequate serum levels of imipramine.2
These observations were confirmed by a large Italian study, in which 437 depressed patients were treated with high-dose imipramine (200 to 350 mg/d). Depression remitted in 244 patients (56%). Those who remained depressed were then treated with ECT, and depression remitted in 136 of 190 (72%). Psychosis was the marker of poor response to imipramine.3 DSM-III codified these findings by separating the syndrome of “major depression with psychosis” (296.34) from “major depression without psychosis” (296.33).
As psychiatry recognized psychotic depression as a distinct form of depression, it became clear that drugs often could not adequately treat it. Less than one-third of patients with psychotic depression respond to tricyclics alone.4-6
Response to antipsychotic monotherapy averaged 50% and increased to 75% with combined antipsychotics and antidepressants. However, high daily dosages —at least 32 mg of perphenazine and 225 mg of amitriptyline—were required for an adequate response,7 and side effects made sustaining such heroic dosing was difficult. The greatest improvement rates were seen with ECT.
Few other drug combinations have been reported to be effective in psychotic depression, but we lack proper studies. Schatzberg8 addressed the use of newer antidepressants and atypical antipsychotics without offering an algorithm based on the data. Evidence on combination therapies consists mainly of case reports, with few designed studies.
EFFICACY OF ECT
ECT is the most effective treatment for psychotic major depression—achieving remission rates >80% within 3 weeks—as demonstrated by the ongoing, four-hospital Consortium for Research in ECT (CORE), supported by the National Institute of Mental Health.
CORE researchers are studying the efficacy of bilateral ECT in treating severe unipolar depression in patients ages 18 to 85 and of continuation treatments with ECT or lithium plus nortriptyline.9 Under the CORE protocol, diagnoses are made by structured clinical interview using DSM-III-R criteria, and remission is defined as >60% reduction in Hamilton Rating Scale for Depression scores, with final scores 10 sustained for 1 week.
In the first 253 CORE patients treated with ECT, depression remitted in 75% and did not remit in 11%; 14% dropped out. Psychotic depression was identified in 30% (77 of 253), and the remission rate among these patients was 83%.
Among patients who completed the full ECT course (at least 12 sessions), remission rates were 96% for psychotic depression and 83% for nonpsychotic depression. The overall remission rate was 87%.
Treatments were given three times per week. Among patients who completed treatment in weeks 1 through 4, remission rates were 5%, 45%, 81%, and 100%, respectively. Psychotic depression remitted more rapidly than nonpsychotic depression.
Suicide risk. CORE findings suggest that ECT also may reduce suicide risk. In item 3 of the Hamilton Rating Scale for Depression, scores of 2 to 4 indicate preoccupation with death or suicide, or a recent suicide attempt. Nearly 60% of 404 patients (237) reported baseline scores of 2 to 4, but their scores dropped rapidly with ECT. Scores of 0 were reported in 68% after 1 week of ECT, in 87% after 2 weeks, and in 93% after 3 weeks.10
Summary. In patients with severe depressive illness, CORE’s remission rates of 95% for psychotic depression and 83% for nonpsychotic depression are remarkable. Another group is independently reporting a 92% remission rate for psychotic depression treated with ECT.11
Psychotic depression is difficult to recognize and treat, even for clinicians with advanced training. For example:
- only 2 of 52 psychotic depressed patients were determined to have been adequately treated before referral to a National Institute of Mental Health-supported study of ECT15
- only 3 of 46 psychotic depressed patients had been adequately treated prior to enrollment in the Consortium for Research in ECT (CORE) study.9
The three most useful diagnostic criteria are spelled out in the Brief Psychiatric Rating Scale:
- any sign of psychosis is sufficient for designating a major depression as “psychotic”
- one well-developed sign is sufficient to prescribe treatment for psychotic depression
- well-developed vegetative signs also indicate the need to treat psychotic depression.14
ROADBLOCKS TO WIDER USE OF ECT
Many eligible patients never receive ECT, despite its track record of producing high remission rates in psychotic depression. Reasons include:
Limited access. Few psychiatrists—less than 8%—offer ECT as a treatment option, and most who do offer it practice in private hospitals.12-14
Academic low regard. Psychiatry’s academic lecturers largely ignore ECT’s efficacy in psychotic depression and therapy-resistant depression. This low regard for ECT is codified in expert algorithms, which cite ECT as an option of last resort.
Social stigma. In a recent essay summarizing medication’s weak effect in treating psychotic depression, Schatzberg states, “While ECT is a remarkably effective treatment for psychotic depression, requirements for its use are stringent, and public perception about the overall appropriateness of shock treatment is negative.”8
Algorithm Treatment of major depression, with or without psychotic features
ALGORITHM FOR MAJOR DEPRESSION
Because patients with psychotic and nonpsychotic major depression clearly require different treatments, differentiating between these two types is critical. Although psychotic depression can be difficult to diagnose,15,16 commonly recognized criteria are listed in the Table
Assessment. Assess each severely depressed patient for psychotic features, such as delusions and hallucinations. A useful assessment guide is the Brief Psychiatric Rating Scale (Box).17 Also treat those with melancholia, inanition, severe weight loss and insomnia, concentration and memory difficulty, stupor, or suicidal ideation as if they had psychotic depression. These symptoms and signs are evidence that the patient’s neuroendocrine system is disturbed, an indication of severe depression that responds poorly to antidepressant drugs alone.
Treatment. Nonpsychotic depressed patients are best offered antidepressants—tricyclics or selective serotonin reuptake inhibitors (SSRIs)—as recommended by conventional guidelines. Insufficient response to two adequate trials calls for a careful assessment for psychosis and, if found, treatment with effective drug combinations or ECT (Algorithm).18 For patients with psychotic depression—especially those who fail medication trials or whose severe symptoms would likely respond to ECT as a primary treatment—bilateral ECT is the effective standard.19
ECT is the appropriate first option for hospitalized patients with psychotic depression—especially those who are suicidal or require supplementary feeding and sedation. It may also be considered the first option in patients who have:
- attempted suicide
- lost more than 10% of body weight (approximately 15 lbs for adults) in the weeks of their illness
- or show signs of severe melancholia, such as catatonia or pseudodementia.
TREATING NONPSYCHOTIC DEPRESSION
When medications are first-line treatment for nonpsychotic depression, how long should a trial be continued before taking another tack? How many courses should you try before you declare therapy resistance and consider ECT?
Studies of clozapine provide a useful model.20 Because of clozapine’s association with agranulocytosis and induced seizures, patients with schizophrenia usually do not receive this antipsychotic unless two 4- to 6-week trials of other neuroleptics have proven ineffective. Ethicists have deemed two failed medication trials to be sufficient before a more hazardous treatment is offered.
Figure Remission of major depression with ECT
We can apply a similar standard when considering ECT in patients first treated with medication.18 A patient’s depression could be defined as therapy-resistant after an inadequate response to 4-week courses (in either order) of:
- an SSRI at dosages equivalent to fluoxetine, 40 mg/d
- a tricyclic at 200 mg/d.
In depressed patients with bipolar disorder, a trial of lithium or an anticonvulsant may replace an antidepressant.
ECT is appropriate when debilitating depression persists after two adequate medication trials. Only after an adequate ECT trial has failed—and such failure is infrequent—is it reasonable to offer poorly tested augmentation and combination strategies.
What is ‘adequate’ ECT? For patients with major depression, the definition of an adequate ECT trial is complex. Although many doctors expect six ECT sessions to be sufficient, the CORE studies are finding that only 45% of patients remitted with six ECT, 81% with nine ECT, and almost all with 12 ECT sessions (Figure).9 These patients were treated with bitemporal electrode placement, the more effective form of ECT. When unilateral electrode placements are used, ECT courses may be inadequate, as this form requires special attention to electrical dosing.
Further, the quality of the seizure—like the dosing of medications—directly influences outcome. Seizure monitoring is essential for assessing the adequacy of each treatment and the treatment course.21,22
Related resources
- Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus non-psychotic depressed patients: a report from CORE. J ECT 2001;17:244-53.
Drug brand names
- Amitriptyline • Elavil
- Clozapine • Clozaril
- Imipramine • Tofranil
- Nortriptyline • Pamelor
- Perphenazine • Trilafon
Disclosure
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgment
Preparation of this manuscript was aided by grants from the Scion Natural Science Association, Inc., St. James, NY.
1. Thase M. New approaches to managing difficult-to-treat depressions. J Clin Psychiatry 2003;64[suppl1]:3-4.
2. Kantor SJ, Glassman AH. Delusional depressions: natural history and response to treatment. Br J Psychiatry 1977;131:351-6.
3. Avery D, Lubrano A. Depression treated with imipramine and ECT: the deCarolis study reconsidered. Am J Psychiatry 1979;136:559-62.
4. Kroessler D. Relative efficacy rates for therapies of delusional depression. Convuls Ther 1985;1:173-82.
5. Parker G, Roy K, Hadzi-Pavlovic D, Pedic F. Psychotic (delusional) depression: a meta-analysis of physical treatments. J Affect Dis 1992;24:17-24.
6. Wheeler Vega JA, Mortimer AM, Tyson PJ. Somatic treatment of psychotic depression: review and recommendations for practice. J Clin Psychopharmacol 2000;20:504-19.
7. Spiker DG, Weiss JC, Dealy RS, et al. The pharmacologic treatment of delusional depression. Am J Psychiatry 1985;142:430-6.
8. Schatzberg AF. New approaches to managing psychotic depression. J Clin Psychiatry 2003;64[suppl1]:19-23.
9. Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus non-psychotic depressed patients: a report from CORE. J ECT 2001;17:244-53.
10. Kellner CH, Fink M, Knapp R, et al. Bilateral ECT rapidly relieves suicidality: findings from phase I of the CORE ECT study. Am J Psychiatry (submitted).
11. Birkenhäger TK, Pluijms EM, Lucius SAP. ECT response in delusional versus non-delusional depressed inpatients. J Affect Dis (in press).
12. Hermann RC, Ettner SL, Dorwart RA, et al. Characteristics of psychiatrists who perform ECT. Am J Psychiatry 1998;155:889-94.
13. Thompson JW, Weiner RD, Myers CP. Use of ECT in the United States in 1975, 1980, and 1986. Am J Psychiatry 1994;151:1657-61.
14. Kramer BA. Use of ECT in California revisited: 1984-1994. J ECT 1999;15:245-51.
15. Prudic J, Sackeim HA, Devanand DP. Medication resistance and clinical response to electroconvulsive therapy. Psychiatry Res 1990;31:287-96.
16. Mulsant BH, Haskett RF, Prudic J, et al. Low use of neuroleptic drugs in the treatment of psychotic major depression. Am J Psychiatry 1997;154:559-61.
17. Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychol Rep 1962;10:799-812.
18. Fink M. Electroconvulsive therapy in medication-resistant depression. In: Amsterdam J, Hornig-Rohan M, Nierenberg A (eds.): Treatment-resistant mood disorders. Cambridge, UK: Cambridge University Press, 2001;223-38.
19. Fink M. The efficacy of ECT and “treatment resistance.” J ECT 2002;18:1-2.
20. Lieberman JA, Kane JM, Johns CA. Clozapine: guidelines for clinical management. J Clin Psychiatry 1989;50:329-38.
21. Abrams R. Electroconvulsive therapy. (4th ed). New York: Oxford University Press, 2002.
22. Fink M. Optimizing ECT. Encephale 1994;20:297-302.
1. Thase M. New approaches to managing difficult-to-treat depressions. J Clin Psychiatry 2003;64[suppl1]:3-4.
2. Kantor SJ, Glassman AH. Delusional depressions: natural history and response to treatment. Br J Psychiatry 1977;131:351-6.
3. Avery D, Lubrano A. Depression treated with imipramine and ECT: the deCarolis study reconsidered. Am J Psychiatry 1979;136:559-62.
4. Kroessler D. Relative efficacy rates for therapies of delusional depression. Convuls Ther 1985;1:173-82.
5. Parker G, Roy K, Hadzi-Pavlovic D, Pedic F. Psychotic (delusional) depression: a meta-analysis of physical treatments. J Affect Dis 1992;24:17-24.
6. Wheeler Vega JA, Mortimer AM, Tyson PJ. Somatic treatment of psychotic depression: review and recommendations for practice. J Clin Psychopharmacol 2000;20:504-19.
7. Spiker DG, Weiss JC, Dealy RS, et al. The pharmacologic treatment of delusional depression. Am J Psychiatry 1985;142:430-6.
8. Schatzberg AF. New approaches to managing psychotic depression. J Clin Psychiatry 2003;64[suppl1]:19-23.
9. Petrides G, Fink M, Husain MM, et al. ECT remission rates in psychotic versus non-psychotic depressed patients: a report from CORE. J ECT 2001;17:244-53.
10. Kellner CH, Fink M, Knapp R, et al. Bilateral ECT rapidly relieves suicidality: findings from phase I of the CORE ECT study. Am J Psychiatry (submitted).
11. Birkenhäger TK, Pluijms EM, Lucius SAP. ECT response in delusional versus non-delusional depressed inpatients. J Affect Dis (in press).
12. Hermann RC, Ettner SL, Dorwart RA, et al. Characteristics of psychiatrists who perform ECT. Am J Psychiatry 1998;155:889-94.
13. Thompson JW, Weiner RD, Myers CP. Use of ECT in the United States in 1975, 1980, and 1986. Am J Psychiatry 1994;151:1657-61.
14. Kramer BA. Use of ECT in California revisited: 1984-1994. J ECT 1999;15:245-51.
15. Prudic J, Sackeim HA, Devanand DP. Medication resistance and clinical response to electroconvulsive therapy. Psychiatry Res 1990;31:287-96.
16. Mulsant BH, Haskett RF, Prudic J, et al. Low use of neuroleptic drugs in the treatment of psychotic major depression. Am J Psychiatry 1997;154:559-61.
17. Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychol Rep 1962;10:799-812.
18. Fink M. Electroconvulsive therapy in medication-resistant depression. In: Amsterdam J, Hornig-Rohan M, Nierenberg A (eds.): Treatment-resistant mood disorders. Cambridge, UK: Cambridge University Press, 2001;223-38.
19. Fink M. The efficacy of ECT and “treatment resistance.” J ECT 2002;18:1-2.
20. Lieberman JA, Kane JM, Johns CA. Clozapine: guidelines for clinical management. J Clin Psychiatry 1989;50:329-38.
21. Abrams R. Electroconvulsive therapy. (4th ed). New York: Oxford University Press, 2002.
22. Fink M. Optimizing ECT. Encephale 1994;20:297-302.
Getting to the bottom of problem drinking: The case for routine screening
Do you know which of your patients have alcohol problems? Though alcohol use disorders may be difficult to detect, self-report and biochemical measures followed by a thorough face-to-face assessment improve diagnostic accuracy. New tools—such as the serum carbohydrate-deficient transferrin (CDT) test—are changing how psychiatrists screen for alcohol problems, provide motivational feedback, and monitor patients for relapse.
FOUR REASONS TO SCREEN
Screening for excessive alcohol consumption is important in psychiatric practice because:
- Alcohol use disorders coexist with many psychiatric problems, most notably affective and anxiety disorders and—not surprisingly—other substance abuse disorders (Table 1).1,2
- Patients with psychiatric comorbidity who abuse alcohol have poorer prognoses, are less adherent to treatment, and are more likely to drop out of treatment than are psychiatric patients who do not have alcohol problems.3
- Alcohol interacts with many psychotropics, and chronic heavy drinking can cause pharmacokinetic changes that affect a patient’s response to medications.
- Alcohol-dependent patients are more likely than nondrinkers to become dependent on anxiolytics and sedative-hypnotics.
Table 1
Overlap of alcohol problems with common psychiatric disorders
| Disorder | Risk of alcohol use disorder (odds ratio) | Source of data (population survey) |
|---|---|---|
| Drug use disorder | 25.1 | NLAES |
| Mania | 5.6 | NCS |
| Major depression | 3.7 | NLAES |
| Obsessive-compulsive disorder | 3.4 | ECA |
| Generalized anxiety disorder | 2.7 | NCS |
| Phobia | 2.3 | NCS |
| Posttraumatic stress disorder | 2.2 | NCS |
| Panic disorder | 1.4 | NCS |
| NLAES: National Longitudinal Alcohol Epidemiological Survey | ||
| NCS: National Comorbidity Survey | ||
| ECA: Epidemiologic Catchment Area | ||
Because alcohol problems are common in psychiatric patients, routine screening for alcohol abuse and dependence at the onset of any treatment can be very useful. Thereafter, screening can be done periodically—perhaps annually or more often if the patient’s functioning declines.
CHOOSING A SELF-REPORT MEASURE
Many self-report alcohol screening scales are available,4 the most popular being the CAGE5 and the Michigan Alcoholism Screening Test (MAST).6 Though both instruments can help identify alcohol problems, each has shortcomings:
- The CAGE performs less reliably in women and adolescents than in men, and its validity depends on the patient’s sensitivity to the emotional impacts of alcohol dependence.
- The MAST is long (25 items), concentrates on late-stage alcoholism symptoms, and uses differential weighting—not validated in subsequent studies—of particular items in deriving the score.
Neither addresses drinking behavior or when symptoms occurred and thus may misclassify recovered alcoholics or former problem drinkers.
AUDIT. A more reliable choice is the Alcohol Use Disorders Identification Test (AUDIT).7 It was designed by the World Health Organization (WHO) to be valid across gender and culture and to identify even early stage problem drinking. The AUDIT’s 10 items deal with drinking behavior, dependence on alcohol, and adverse consequences of drinking during the past year (Box). The survey takes less than 5 minutes; can be administered orally, in writing, or online; and it retains its validity when given as part of a comprehensive health risk appraisal.8
The WHO offers an excellent manual detailing how to administer and interpret the AUDIT (see Related resources). A patient’s score is computed by summing the values associated with his or her responses to each item. A score of 8 or greater indicates excessive alcohol consumption, although some researchers have argued that for women a more accurate threshold might be 6 or 7 points.
Standardized for adults, the AUDIT also appears to accurately gauge drinking behavior in adolescents9 and in psychiatric patients, although only three studies have explored its use in the latter population.10-12 Abbreviated AUDIT versions have been found to be psychometrically sound8 and may be useful in an emergency room or busy primary care clinic. In comparisons with other screening tools, the AUDIT almost always has been found to be more valid.9,13
USING BIOCHEMICAL MEASURES
Self-report screens for alcohol problems, especially the AUDIT, are highly sensitive and specific, though their accuracy depends on the patient's memory, understanding of the questions, and candor. In chronic heavy drinkers, biochemical measures (Table 2) can augment self-reports.14
Self-report and biochemical screens have different strengths and weaknesses (Table 3). It is important to see them as complementary because each contributes to accurate screening.
CDT. Most biomarkers screen indirectly for alcohol problems by measuring damage to an end organ-typically the liver-caused by chronic excessive alcohol consumption. False positive results are common because of nonalcohol-related organ damage, medications, smoking, obesity, and other confounding factors. An exception appears to be the serum test for carbohydrate-deficient transferrin (CDT), a biomarker for heavy drinking approved in kit form 3 years ago by the Food and Drug Administration.
The value of measuring CDT levels is that few conditions other than excessive alcohol consumption elevate them. For unclear reasons,15 average daily consumption of >60 grams of alcohol (about five standard drinks) during the previous 2 weeks causes a higher percent of transferrin—a glycoenzyme that transports iron in the body—to lack its usual carbohydrate content.
Bio-Rad Laboratories (www.bio-rad.com) offers a reagent kit (%CDT Turbidimetric Immunoassay). It quantifies CDT as a percent of total serum transferrin, rather than total CDT, thus correcting for individual variations in transferrin levels. CDT values are obtained from a 100-microliter serum sample. The blood is clotted and the serum separated. The sample may be stored at 2 to 8 °C if the test is to be run within 1 week. Samples must be tested at a reference lab (Bio-Rad offers a list of labs). Results are available in a few days.
Patients who deny problem drinking may need convincing to submit to a blood draw. It may help to explain that alcohol use can exacerbate emotional problems and that the test can provide information on possible risky alcohol use.
GGT. Using a second biochemical marker may improve the sensitivity of CDT to detect heavy drinking.16-18
The most-researched choice for a second marker is gamma glutamyltransferase (GGT). Patients are considered to have tested positive for an alcohol problem if either CDT or GGT levels are elevated. Combining these tests may be especially useful in alcohol-dependent women, in whom the reliability of CDT testing alone has been questioned.
Recommendation. Start with a self-report screening measure. If the patient scores slightly below the threshold for an alcohol problem, follow up with the more costly CDT and GGT tests.
For example, biomarkers might be useful for follow-up in men with AUDIT screening scores of 6 or 7 or women with scores of 5 to 7. Biomarkers also are recommended when you suspect an alcohol problem for another reason or question whether the patient responded accurately to the self-report measure.
- How often do you have a drink containing alcohol?
- How many drinks containing alcohol do you have on a typical day when you are drinking?
- How often do you have 6 or more drinks on one occasion?
- How often during the last year have you found that you were not able to stop drinking once you had started?
- How often during the last year have you failed to do what was normally expected from you because of drinking?
- How often during the last year have you needed a first drink in the morning to get yourself going after a heavy drinking session?
- How often during the last year have you had a feeling of guilt or remorse after drinking?
- How often during the last year have you been unable to remember what happened the night before because you had been drinking?
- Have you or someone else been injured as a result of your drinking?
- Has a relative, friend, doctor, or other health worker been concerned about your drinking or suggested that you cut down?
The World Health Organization offers a manual on how to administer and interpret AUDIT (http://www.who.int/substance_abuse/pubs_alcohol.htm).
Source: World Health Organization
Table 2
Biochemical markers of heavy drinking
| Marker | Time needed for return to normal limits | Level of drinking characterized | Comments |
|---|---|---|---|
| Gamma glutamyltransferase (GGT) | 2 to 6 weeks of abstinence | ~70 drinks/wk for several weeks | Most common and reliable of the traditional markers of heavy drinking; many sources of false positives |
| Aspartate aminotransferase (AST) (formerly SGOT) | 7 days, but much variability in declines with abstinence | Unknown, but heavy | Present in many organs; many sources of false positives; moderate correlations with GGT |
| Alanine aminotransferase (ALT) (formerly SGPT) | Unknown | Unknown, but heavy | Many sources of false positives and less sensitive than AST; ratio of AST to ALT may be more accurate |
| Macrocytic volume (MCV) | Unknown; half-life ~40 days | Unknown, but regular and heavy | Poor sensitivity and specificity; even with abstinence, very slow return to normal limits and may increase at first; little, if any, gender effect |
| Carbohydrate-deficient transferrin (CDT) | 2 to 4 weeks of abstinence | >60 grams/day for approximately 2 weeks | Few sources of false positives; excellent indicator of relapse |
MONITORING PATIENTS IN TREATMENT
MET. Motivational enhancement therapy (MET) has gained popularity as a means of changing problematic drinking.19 Project MATCH—a multi-site trial on alcohol abuse treatment—studied MET and two other interventions. MET required fewer sessions but equaled the other interventions in reducing drinking days and aver-age amount of alcohol consumed.20
A key component of MET—and other brief interventions—is to provide patients with empathetic, nonjudgmental feedback.19-21 Responses to the first three AUDIT items can provide such feedback to patients with drinking problems. Amazingly, heavy drinkers and alcoholics often do not realize how much more they drink than other people. To help them develop this insight, show them their self-report responses in contrast with national normative data.
Biomarker results can be used similarly, in this case comparing the patient’s score with the test’s reference range values. Kristenson et al22 showed that giving men with elevated scores recurrent biomarker information and advice significantly reduced morbidity and mortality and improved their work performance.
Using visual aids can deepen patients’ under-standing of motivational feedback. For example, displaying sequential test results on a timeline can reinforce motivation by showing how their drinking behavior has improved with continuing treatment and sustained effort.
Table 3
Self-report and biochemical measures of drinking: Pros and cons
| Measure | Strengths | Weaknesses |
|---|---|---|
| Self-report | Noninvasive Inexpensive High validity Flexible window of assessment Immediate results | Easily feigned Accuracy depends on patient’s verbal skills and memory |
| Biochemical | Objective Results may be more compelling to patients than self-reports May reflect organ damage Useful in tracking treatment progress | Window of assessment is limited to recent past Results often not immediately available May be more costly than self-report measures |
DETECTING RELAPSE
Although treatment for alcohol problems is often successful,23 relapse to some level of drinking is not uncommon, especially during the first 3 or 4 months after patients complete treatment.20 Recognizing relapse quickly can help you:
- decrease risk of harm from resumed alcohol use
- reduce the potential for drinking to again become habitual
- identify circumstances and cues that may have triggered the drinking episode, for use in tailoring future interventions.
In clinical practice, relapse is most often revealed via comments from the family, direct observation by the clinician, or voluntary acknowledgment by the patient. Interestingly, CDT has demonstrated a relapse “heralding effect,”24 meaning that it tends to rise well before a patient will admit he or she resumed drinking.24-26 Although other markers may also rise following relapse, their elevation tends to be delayed and less dramatic.
The sensitivity and specificity of CDT alone and with GGT in identifying relapse have been evaluated. Across male-only studies, CDT’s median sensitivity (percent of relapsed patients with elevated scores) was 0.73, with a specificity (percent of those not relapsed who had low scores) of 0.91. In the two female-only studies, median sensitivity and specificity for CDT were 0.32 and 0.86, respectively. For women, using CDT and GGT in combination substantially raised median sensitivity to 0.62, although specificity fell slightly to 0.80.27
Recommendation. When using biomarkers to identify relapse, examine the temporal pattern of test results to date. Assume that an increase of 30% or more above the lowest observed lab value indicates a relapse.28
Frequent testing—probably biweekly—is recommended during the first 3 or 4 months after patients complete treatment. If there is no indication of relapse, testing frequency could be tapered down.
- AUDIT. The Alcohol Use Disorders Identification Test. Guidelines for primary care use. Available at: http://www.who.int/substance_abuse/pubs_alcohol.htm
- Allen JP, Litten RZ. Psychometric and laboratory measures to assist in the treatment of alcoholism. Clin Psychol Rev 1993;13(3):223-39.
- Salaspuro M. Carbohydrate-deficient transferrin as compared to other markers of alcoholism: a systematic review. Alcohol 1999; 19(3):261-71.
Disclosure
Dr. Allen reports that he serves as a consultant to Axis-Shield ASA and Bio-Rad Laboratories, patent holder and U.S. distributor, respectively, of the carbohydrate-deficient transferrin (%CDT) reagent kit.
Dr. Anthenelli reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hall W. What have population surveys revealed about substance use disorders and their co-morbidity with other mental disorders? Drug Alcohol Rev 1996;15(2):157-70.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: Results of a national survey. Drug Alcohol Depend 1995;39(3):197-206.
3. Shivani R, Goldsmith RJ, Anthenelli RM. Alcoholism and psychiatric disorders: diagnostic challenges. Alcohol Res Health 2002;26(2):90-8.
4. Allen JP, Columbus M (eds). Assessing alcohol problems: a guide for clinicians and researchers. NIAAA Treatment Handbook, Series 4. Washington, DC: U.S. Department of Health and Human Services, 1995 [NIH publication no. 95-3745].
5. Mayfield D, McLeod G, Hall P. CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry 1974;131(10):1121-3.
6. Seltzer ML. The Michigan Alcoholism Screening Test: the quest for a new diagnostic instrument. Am J Psychiatry 1971;127:1653-8.
7. Saunders JB, Aasland OG, Babor TF, et al. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO collaborative project on early detection of persons with harmful alcohol consumption—II. Addiction 1993;88(6):791-804.
8. Daeppen J, Yersin B, Landry U, et al. Reliability and validity of the Alcohol Use Disorders Identification Test (AUDIT) imbedded within a general health risk screening questionnaire: results of a survey in 332 primary care patients. Alcohol Clin Exp Res 2000;24:659-65.
9. Reinert DF, Allen JP. The Alcohol Use Disorders Identification Test: a review of recent research. Alcohol Clin Exp Res 2002;26(2):272-9.
10. Maisto SA, Carey MP, Carey KB, et al. Use of the AUDIT and the DAST-10 to identify alcohol and drug use disorders among adults with a severe and persistent mental illness. Psychol Assess 2002;12:186-92.
11. Hulse GK, Saunders JB, Roydhouse RM, et al. Screening for hazardous alcohol use and dependence in psychiatric inpatients using the AUDIT questionnaire. Drug Alcohol Rev 2000;19:291-8.
12. Dawe S, Seinen A, Kavanagh D. An examination of the utility of the AUDIT in people with schizophrenia. J Stud Alcohol 2000;61:744-75.
13. Allen JP, Litten RZ, Fertig JB, Babor T. A review of research on the Alcohol Use Disorders Identification Test (AUDIT). Alcohol Clin Exp Res 1997;21:613-19.
14. Hermansson U, Helander A, Huss A, et al. The Alcohol Use Disorders Identification Test (AUDIT) and carbohydrate-deficient transferrin (CDT) in a routine workplace health examination. Alcohol Clin Exp Res 2000;24:180-7.
15. Sillanaukee P, Strid N, Allen JP, Litten RZ. Possible reasons why heavy drinking increases carbohydrate-deficient transferrin. Alcohol Clin Exp Res 2001;25(1):34-40.
16. Litten RZ, Allen JP, Fertig JB. Gamma-glutamyltranspeptidase and carbohydrate deficient transferrin: alternative measures of excessive alcohol consumption. Alcohol Clin Exp Res 1995;19(6):1541-6.
17. Allen JP, Litten RZ, Fertig JB, Sillanaukee P. Carbohydrate-deficient transferrin, gamma glutamyl transferase and macrocytic volume as biomarkers of alcohol problems in women. Alcohol Clin Exp Res 2000;24(4):492-6.
18. Sillanaukee P, Strid N, Allen JP, Litten RZ. Combining biomarkers to screen for alcohol problems (manuscript submitted for publication).
19. Miller WR, Zweben A, DiClemente CC, et al. Motivational enhancement therapy manual: A clinical research guide for therapists treating individuals with alcohol abuse and dependence. Rockville, MD: U.S. Department of Health and Human Services, 1995.
20. Project MATCH research group. Matching alcoholism treatments to client heterogeneity: Project MATCH post-treatment drinking outcomes. J Stud Alcohol 1997;58(1):7-29.
21. Bien TH, Miller WR, Tonigan S. Brief intervention for alcohol problems: a review. Addiction 1993;88:315-36.
22. Kristenson H, Hood B, Peterson B, et al. Prevention of alcohol-related problems in urban middle-aged males. Alcohol 1985;2(3):545-9.
23. Miller WR, Walter ST, Bennett ME. How effective is alcoholism treatment in the United States? J Stud Alcohol. 2001;62:211-20.
24. Mitchell C, Simpson D, Chick J. Carbohydrate-deficient transferrin in detecting relapse in alcohol dependence. Drug Alcohol Depend 1997;48:97-103.
25. Borg S, Helander A, Voltaire Carlsson A, Hogstrom Brandt AM. Detection of relapses in alcohol-dependent patients using carbohydrate-deficient transferrin: improvement with individualized reference levels during long-term monitoring. Alcohol Clin Exp Res 1995;19(4):961-3.
26. Schmidt LG, Schmidt K, Dufeu P, et al. Superiority of carbohydrate-deficient transferrin to gamma-glutamyltransferase in detecting relapse in alcoholism. Am J Psychiatry 1997;154(1):75-80.
27. Allen JP, Anton R. Biomarkers as aids to identification of relapse in alcoholic patients. In: Galanter M (ed). Recent developments in alcoholism: research on alcoholism treatment (vol. XVI). New York: Plenum Press (in press).
28. Anton RF, Lieber C, Tabakoff B, et al. Carbohydrate-deficient transferrin and gamma-glutamyltransferase for the detection and monitoring of alcohol use: results from a multi-site study. Alcohol Clin Exp Res 2002;26(8):1215-22.
Do you know which of your patients have alcohol problems? Though alcohol use disorders may be difficult to detect, self-report and biochemical measures followed by a thorough face-to-face assessment improve diagnostic accuracy. New tools—such as the serum carbohydrate-deficient transferrin (CDT) test—are changing how psychiatrists screen for alcohol problems, provide motivational feedback, and monitor patients for relapse.
FOUR REASONS TO SCREEN
Screening for excessive alcohol consumption is important in psychiatric practice because:
- Alcohol use disorders coexist with many psychiatric problems, most notably affective and anxiety disorders and—not surprisingly—other substance abuse disorders (Table 1).1,2
- Patients with psychiatric comorbidity who abuse alcohol have poorer prognoses, are less adherent to treatment, and are more likely to drop out of treatment than are psychiatric patients who do not have alcohol problems.3
- Alcohol interacts with many psychotropics, and chronic heavy drinking can cause pharmacokinetic changes that affect a patient’s response to medications.
- Alcohol-dependent patients are more likely than nondrinkers to become dependent on anxiolytics and sedative-hypnotics.
Table 1
Overlap of alcohol problems with common psychiatric disorders
| Disorder | Risk of alcohol use disorder (odds ratio) | Source of data (population survey) |
|---|---|---|
| Drug use disorder | 25.1 | NLAES |
| Mania | 5.6 | NCS |
| Major depression | 3.7 | NLAES |
| Obsessive-compulsive disorder | 3.4 | ECA |
| Generalized anxiety disorder | 2.7 | NCS |
| Phobia | 2.3 | NCS |
| Posttraumatic stress disorder | 2.2 | NCS |
| Panic disorder | 1.4 | NCS |
| NLAES: National Longitudinal Alcohol Epidemiological Survey | ||
| NCS: National Comorbidity Survey | ||
| ECA: Epidemiologic Catchment Area | ||
Because alcohol problems are common in psychiatric patients, routine screening for alcohol abuse and dependence at the onset of any treatment can be very useful. Thereafter, screening can be done periodically—perhaps annually or more often if the patient’s functioning declines.
CHOOSING A SELF-REPORT MEASURE
Many self-report alcohol screening scales are available,4 the most popular being the CAGE5 and the Michigan Alcoholism Screening Test (MAST).6 Though both instruments can help identify alcohol problems, each has shortcomings:
- The CAGE performs less reliably in women and adolescents than in men, and its validity depends on the patient’s sensitivity to the emotional impacts of alcohol dependence.
- The MAST is long (25 items), concentrates on late-stage alcoholism symptoms, and uses differential weighting—not validated in subsequent studies—of particular items in deriving the score.
Neither addresses drinking behavior or when symptoms occurred and thus may misclassify recovered alcoholics or former problem drinkers.
AUDIT. A more reliable choice is the Alcohol Use Disorders Identification Test (AUDIT).7 It was designed by the World Health Organization (WHO) to be valid across gender and culture and to identify even early stage problem drinking. The AUDIT’s 10 items deal with drinking behavior, dependence on alcohol, and adverse consequences of drinking during the past year (Box). The survey takes less than 5 minutes; can be administered orally, in writing, or online; and it retains its validity when given as part of a comprehensive health risk appraisal.8
The WHO offers an excellent manual detailing how to administer and interpret the AUDIT (see Related resources). A patient’s score is computed by summing the values associated with his or her responses to each item. A score of 8 or greater indicates excessive alcohol consumption, although some researchers have argued that for women a more accurate threshold might be 6 or 7 points.
Standardized for adults, the AUDIT also appears to accurately gauge drinking behavior in adolescents9 and in psychiatric patients, although only three studies have explored its use in the latter population.10-12 Abbreviated AUDIT versions have been found to be psychometrically sound8 and may be useful in an emergency room or busy primary care clinic. In comparisons with other screening tools, the AUDIT almost always has been found to be more valid.9,13
USING BIOCHEMICAL MEASURES
Self-report screens for alcohol problems, especially the AUDIT, are highly sensitive and specific, though their accuracy depends on the patient's memory, understanding of the questions, and candor. In chronic heavy drinkers, biochemical measures (Table 2) can augment self-reports.14
Self-report and biochemical screens have different strengths and weaknesses (Table 3). It is important to see them as complementary because each contributes to accurate screening.
CDT. Most biomarkers screen indirectly for alcohol problems by measuring damage to an end organ-typically the liver-caused by chronic excessive alcohol consumption. False positive results are common because of nonalcohol-related organ damage, medications, smoking, obesity, and other confounding factors. An exception appears to be the serum test for carbohydrate-deficient transferrin (CDT), a biomarker for heavy drinking approved in kit form 3 years ago by the Food and Drug Administration.
The value of measuring CDT levels is that few conditions other than excessive alcohol consumption elevate them. For unclear reasons,15 average daily consumption of >60 grams of alcohol (about five standard drinks) during the previous 2 weeks causes a higher percent of transferrin—a glycoenzyme that transports iron in the body—to lack its usual carbohydrate content.
Bio-Rad Laboratories (www.bio-rad.com) offers a reagent kit (%CDT Turbidimetric Immunoassay). It quantifies CDT as a percent of total serum transferrin, rather than total CDT, thus correcting for individual variations in transferrin levels. CDT values are obtained from a 100-microliter serum sample. The blood is clotted and the serum separated. The sample may be stored at 2 to 8 °C if the test is to be run within 1 week. Samples must be tested at a reference lab (Bio-Rad offers a list of labs). Results are available in a few days.
Patients who deny problem drinking may need convincing to submit to a blood draw. It may help to explain that alcohol use can exacerbate emotional problems and that the test can provide information on possible risky alcohol use.
GGT. Using a second biochemical marker may improve the sensitivity of CDT to detect heavy drinking.16-18
The most-researched choice for a second marker is gamma glutamyltransferase (GGT). Patients are considered to have tested positive for an alcohol problem if either CDT or GGT levels are elevated. Combining these tests may be especially useful in alcohol-dependent women, in whom the reliability of CDT testing alone has been questioned.
Recommendation. Start with a self-report screening measure. If the patient scores slightly below the threshold for an alcohol problem, follow up with the more costly CDT and GGT tests.
For example, biomarkers might be useful for follow-up in men with AUDIT screening scores of 6 or 7 or women with scores of 5 to 7. Biomarkers also are recommended when you suspect an alcohol problem for another reason or question whether the patient responded accurately to the self-report measure.
- How often do you have a drink containing alcohol?
- How many drinks containing alcohol do you have on a typical day when you are drinking?
- How often do you have 6 or more drinks on one occasion?
- How often during the last year have you found that you were not able to stop drinking once you had started?
- How often during the last year have you failed to do what was normally expected from you because of drinking?
- How often during the last year have you needed a first drink in the morning to get yourself going after a heavy drinking session?
- How often during the last year have you had a feeling of guilt or remorse after drinking?
- How often during the last year have you been unable to remember what happened the night before because you had been drinking?
- Have you or someone else been injured as a result of your drinking?
- Has a relative, friend, doctor, or other health worker been concerned about your drinking or suggested that you cut down?
The World Health Organization offers a manual on how to administer and interpret AUDIT (http://www.who.int/substance_abuse/pubs_alcohol.htm).
Source: World Health Organization
Table 2
Biochemical markers of heavy drinking
| Marker | Time needed for return to normal limits | Level of drinking characterized | Comments |
|---|---|---|---|
| Gamma glutamyltransferase (GGT) | 2 to 6 weeks of abstinence | ~70 drinks/wk for several weeks | Most common and reliable of the traditional markers of heavy drinking; many sources of false positives |
| Aspartate aminotransferase (AST) (formerly SGOT) | 7 days, but much variability in declines with abstinence | Unknown, but heavy | Present in many organs; many sources of false positives; moderate correlations with GGT |
| Alanine aminotransferase (ALT) (formerly SGPT) | Unknown | Unknown, but heavy | Many sources of false positives and less sensitive than AST; ratio of AST to ALT may be more accurate |
| Macrocytic volume (MCV) | Unknown; half-life ~40 days | Unknown, but regular and heavy | Poor sensitivity and specificity; even with abstinence, very slow return to normal limits and may increase at first; little, if any, gender effect |
| Carbohydrate-deficient transferrin (CDT) | 2 to 4 weeks of abstinence | >60 grams/day for approximately 2 weeks | Few sources of false positives; excellent indicator of relapse |
MONITORING PATIENTS IN TREATMENT
MET. Motivational enhancement therapy (MET) has gained popularity as a means of changing problematic drinking.19 Project MATCH—a multi-site trial on alcohol abuse treatment—studied MET and two other interventions. MET required fewer sessions but equaled the other interventions in reducing drinking days and aver-age amount of alcohol consumed.20
A key component of MET—and other brief interventions—is to provide patients with empathetic, nonjudgmental feedback.19-21 Responses to the first three AUDIT items can provide such feedback to patients with drinking problems. Amazingly, heavy drinkers and alcoholics often do not realize how much more they drink than other people. To help them develop this insight, show them their self-report responses in contrast with national normative data.
Biomarker results can be used similarly, in this case comparing the patient’s score with the test’s reference range values. Kristenson et al22 showed that giving men with elevated scores recurrent biomarker information and advice significantly reduced morbidity and mortality and improved their work performance.
Using visual aids can deepen patients’ under-standing of motivational feedback. For example, displaying sequential test results on a timeline can reinforce motivation by showing how their drinking behavior has improved with continuing treatment and sustained effort.
Table 3
Self-report and biochemical measures of drinking: Pros and cons
| Measure | Strengths | Weaknesses |
|---|---|---|
| Self-report | Noninvasive Inexpensive High validity Flexible window of assessment Immediate results | Easily feigned Accuracy depends on patient’s verbal skills and memory |
| Biochemical | Objective Results may be more compelling to patients than self-reports May reflect organ damage Useful in tracking treatment progress | Window of assessment is limited to recent past Results often not immediately available May be more costly than self-report measures |
DETECTING RELAPSE
Although treatment for alcohol problems is often successful,23 relapse to some level of drinking is not uncommon, especially during the first 3 or 4 months after patients complete treatment.20 Recognizing relapse quickly can help you:
- decrease risk of harm from resumed alcohol use
- reduce the potential for drinking to again become habitual
- identify circumstances and cues that may have triggered the drinking episode, for use in tailoring future interventions.
In clinical practice, relapse is most often revealed via comments from the family, direct observation by the clinician, or voluntary acknowledgment by the patient. Interestingly, CDT has demonstrated a relapse “heralding effect,”24 meaning that it tends to rise well before a patient will admit he or she resumed drinking.24-26 Although other markers may also rise following relapse, their elevation tends to be delayed and less dramatic.
The sensitivity and specificity of CDT alone and with GGT in identifying relapse have been evaluated. Across male-only studies, CDT’s median sensitivity (percent of relapsed patients with elevated scores) was 0.73, with a specificity (percent of those not relapsed who had low scores) of 0.91. In the two female-only studies, median sensitivity and specificity for CDT were 0.32 and 0.86, respectively. For women, using CDT and GGT in combination substantially raised median sensitivity to 0.62, although specificity fell slightly to 0.80.27
Recommendation. When using biomarkers to identify relapse, examine the temporal pattern of test results to date. Assume that an increase of 30% or more above the lowest observed lab value indicates a relapse.28
Frequent testing—probably biweekly—is recommended during the first 3 or 4 months after patients complete treatment. If there is no indication of relapse, testing frequency could be tapered down.
- AUDIT. The Alcohol Use Disorders Identification Test. Guidelines for primary care use. Available at: http://www.who.int/substance_abuse/pubs_alcohol.htm
- Allen JP, Litten RZ. Psychometric and laboratory measures to assist in the treatment of alcoholism. Clin Psychol Rev 1993;13(3):223-39.
- Salaspuro M. Carbohydrate-deficient transferrin as compared to other markers of alcoholism: a systematic review. Alcohol 1999; 19(3):261-71.
Disclosure
Dr. Allen reports that he serves as a consultant to Axis-Shield ASA and Bio-Rad Laboratories, patent holder and U.S. distributor, respectively, of the carbohydrate-deficient transferrin (%CDT) reagent kit.
Dr. Anthenelli reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Do you know which of your patients have alcohol problems? Though alcohol use disorders may be difficult to detect, self-report and biochemical measures followed by a thorough face-to-face assessment improve diagnostic accuracy. New tools—such as the serum carbohydrate-deficient transferrin (CDT) test—are changing how psychiatrists screen for alcohol problems, provide motivational feedback, and monitor patients for relapse.
FOUR REASONS TO SCREEN
Screening for excessive alcohol consumption is important in psychiatric practice because:
- Alcohol use disorders coexist with many psychiatric problems, most notably affective and anxiety disorders and—not surprisingly—other substance abuse disorders (Table 1).1,2
- Patients with psychiatric comorbidity who abuse alcohol have poorer prognoses, are less adherent to treatment, and are more likely to drop out of treatment than are psychiatric patients who do not have alcohol problems.3
- Alcohol interacts with many psychotropics, and chronic heavy drinking can cause pharmacokinetic changes that affect a patient’s response to medications.
- Alcohol-dependent patients are more likely than nondrinkers to become dependent on anxiolytics and sedative-hypnotics.
Table 1
Overlap of alcohol problems with common psychiatric disorders
| Disorder | Risk of alcohol use disorder (odds ratio) | Source of data (population survey) |
|---|---|---|
| Drug use disorder | 25.1 | NLAES |
| Mania | 5.6 | NCS |
| Major depression | 3.7 | NLAES |
| Obsessive-compulsive disorder | 3.4 | ECA |
| Generalized anxiety disorder | 2.7 | NCS |
| Phobia | 2.3 | NCS |
| Posttraumatic stress disorder | 2.2 | NCS |
| Panic disorder | 1.4 | NCS |
| NLAES: National Longitudinal Alcohol Epidemiological Survey | ||
| NCS: National Comorbidity Survey | ||
| ECA: Epidemiologic Catchment Area | ||
Because alcohol problems are common in psychiatric patients, routine screening for alcohol abuse and dependence at the onset of any treatment can be very useful. Thereafter, screening can be done periodically—perhaps annually or more often if the patient’s functioning declines.
CHOOSING A SELF-REPORT MEASURE
Many self-report alcohol screening scales are available,4 the most popular being the CAGE5 and the Michigan Alcoholism Screening Test (MAST).6 Though both instruments can help identify alcohol problems, each has shortcomings:
- The CAGE performs less reliably in women and adolescents than in men, and its validity depends on the patient’s sensitivity to the emotional impacts of alcohol dependence.
- The MAST is long (25 items), concentrates on late-stage alcoholism symptoms, and uses differential weighting—not validated in subsequent studies—of particular items in deriving the score.
Neither addresses drinking behavior or when symptoms occurred and thus may misclassify recovered alcoholics or former problem drinkers.
AUDIT. A more reliable choice is the Alcohol Use Disorders Identification Test (AUDIT).7 It was designed by the World Health Organization (WHO) to be valid across gender and culture and to identify even early stage problem drinking. The AUDIT’s 10 items deal with drinking behavior, dependence on alcohol, and adverse consequences of drinking during the past year (Box). The survey takes less than 5 minutes; can be administered orally, in writing, or online; and it retains its validity when given as part of a comprehensive health risk appraisal.8
The WHO offers an excellent manual detailing how to administer and interpret the AUDIT (see Related resources). A patient’s score is computed by summing the values associated with his or her responses to each item. A score of 8 or greater indicates excessive alcohol consumption, although some researchers have argued that for women a more accurate threshold might be 6 or 7 points.
Standardized for adults, the AUDIT also appears to accurately gauge drinking behavior in adolescents9 and in psychiatric patients, although only three studies have explored its use in the latter population.10-12 Abbreviated AUDIT versions have been found to be psychometrically sound8 and may be useful in an emergency room or busy primary care clinic. In comparisons with other screening tools, the AUDIT almost always has been found to be more valid.9,13
USING BIOCHEMICAL MEASURES
Self-report screens for alcohol problems, especially the AUDIT, are highly sensitive and specific, though their accuracy depends on the patient's memory, understanding of the questions, and candor. In chronic heavy drinkers, biochemical measures (Table 2) can augment self-reports.14
Self-report and biochemical screens have different strengths and weaknesses (Table 3). It is important to see them as complementary because each contributes to accurate screening.
CDT. Most biomarkers screen indirectly for alcohol problems by measuring damage to an end organ-typically the liver-caused by chronic excessive alcohol consumption. False positive results are common because of nonalcohol-related organ damage, medications, smoking, obesity, and other confounding factors. An exception appears to be the serum test for carbohydrate-deficient transferrin (CDT), a biomarker for heavy drinking approved in kit form 3 years ago by the Food and Drug Administration.
The value of measuring CDT levels is that few conditions other than excessive alcohol consumption elevate them. For unclear reasons,15 average daily consumption of >60 grams of alcohol (about five standard drinks) during the previous 2 weeks causes a higher percent of transferrin—a glycoenzyme that transports iron in the body—to lack its usual carbohydrate content.
Bio-Rad Laboratories (www.bio-rad.com) offers a reagent kit (%CDT Turbidimetric Immunoassay). It quantifies CDT as a percent of total serum transferrin, rather than total CDT, thus correcting for individual variations in transferrin levels. CDT values are obtained from a 100-microliter serum sample. The blood is clotted and the serum separated. The sample may be stored at 2 to 8 °C if the test is to be run within 1 week. Samples must be tested at a reference lab (Bio-Rad offers a list of labs). Results are available in a few days.
Patients who deny problem drinking may need convincing to submit to a blood draw. It may help to explain that alcohol use can exacerbate emotional problems and that the test can provide information on possible risky alcohol use.
GGT. Using a second biochemical marker may improve the sensitivity of CDT to detect heavy drinking.16-18
The most-researched choice for a second marker is gamma glutamyltransferase (GGT). Patients are considered to have tested positive for an alcohol problem if either CDT or GGT levels are elevated. Combining these tests may be especially useful in alcohol-dependent women, in whom the reliability of CDT testing alone has been questioned.
Recommendation. Start with a self-report screening measure. If the patient scores slightly below the threshold for an alcohol problem, follow up with the more costly CDT and GGT tests.
For example, biomarkers might be useful for follow-up in men with AUDIT screening scores of 6 or 7 or women with scores of 5 to 7. Biomarkers also are recommended when you suspect an alcohol problem for another reason or question whether the patient responded accurately to the self-report measure.
- How often do you have a drink containing alcohol?
- How many drinks containing alcohol do you have on a typical day when you are drinking?
- How often do you have 6 or more drinks on one occasion?
- How often during the last year have you found that you were not able to stop drinking once you had started?
- How often during the last year have you failed to do what was normally expected from you because of drinking?
- How often during the last year have you needed a first drink in the morning to get yourself going after a heavy drinking session?
- How often during the last year have you had a feeling of guilt or remorse after drinking?
- How often during the last year have you been unable to remember what happened the night before because you had been drinking?
- Have you or someone else been injured as a result of your drinking?
- Has a relative, friend, doctor, or other health worker been concerned about your drinking or suggested that you cut down?
The World Health Organization offers a manual on how to administer and interpret AUDIT (http://www.who.int/substance_abuse/pubs_alcohol.htm).
Source: World Health Organization
Table 2
Biochemical markers of heavy drinking
| Marker | Time needed for return to normal limits | Level of drinking characterized | Comments |
|---|---|---|---|
| Gamma glutamyltransferase (GGT) | 2 to 6 weeks of abstinence | ~70 drinks/wk for several weeks | Most common and reliable of the traditional markers of heavy drinking; many sources of false positives |
| Aspartate aminotransferase (AST) (formerly SGOT) | 7 days, but much variability in declines with abstinence | Unknown, but heavy | Present in many organs; many sources of false positives; moderate correlations with GGT |
| Alanine aminotransferase (ALT) (formerly SGPT) | Unknown | Unknown, but heavy | Many sources of false positives and less sensitive than AST; ratio of AST to ALT may be more accurate |
| Macrocytic volume (MCV) | Unknown; half-life ~40 days | Unknown, but regular and heavy | Poor sensitivity and specificity; even with abstinence, very slow return to normal limits and may increase at first; little, if any, gender effect |
| Carbohydrate-deficient transferrin (CDT) | 2 to 4 weeks of abstinence | >60 grams/day for approximately 2 weeks | Few sources of false positives; excellent indicator of relapse |
MONITORING PATIENTS IN TREATMENT
MET. Motivational enhancement therapy (MET) has gained popularity as a means of changing problematic drinking.19 Project MATCH—a multi-site trial on alcohol abuse treatment—studied MET and two other interventions. MET required fewer sessions but equaled the other interventions in reducing drinking days and aver-age amount of alcohol consumed.20
A key component of MET—and other brief interventions—is to provide patients with empathetic, nonjudgmental feedback.19-21 Responses to the first three AUDIT items can provide such feedback to patients with drinking problems. Amazingly, heavy drinkers and alcoholics often do not realize how much more they drink than other people. To help them develop this insight, show them their self-report responses in contrast with national normative data.
Biomarker results can be used similarly, in this case comparing the patient’s score with the test’s reference range values. Kristenson et al22 showed that giving men with elevated scores recurrent biomarker information and advice significantly reduced morbidity and mortality and improved their work performance.
Using visual aids can deepen patients’ under-standing of motivational feedback. For example, displaying sequential test results on a timeline can reinforce motivation by showing how their drinking behavior has improved with continuing treatment and sustained effort.
Table 3
Self-report and biochemical measures of drinking: Pros and cons
| Measure | Strengths | Weaknesses |
|---|---|---|
| Self-report | Noninvasive Inexpensive High validity Flexible window of assessment Immediate results | Easily feigned Accuracy depends on patient’s verbal skills and memory |
| Biochemical | Objective Results may be more compelling to patients than self-reports May reflect organ damage Useful in tracking treatment progress | Window of assessment is limited to recent past Results often not immediately available May be more costly than self-report measures |
DETECTING RELAPSE
Although treatment for alcohol problems is often successful,23 relapse to some level of drinking is not uncommon, especially during the first 3 or 4 months after patients complete treatment.20 Recognizing relapse quickly can help you:
- decrease risk of harm from resumed alcohol use
- reduce the potential for drinking to again become habitual
- identify circumstances and cues that may have triggered the drinking episode, for use in tailoring future interventions.
In clinical practice, relapse is most often revealed via comments from the family, direct observation by the clinician, or voluntary acknowledgment by the patient. Interestingly, CDT has demonstrated a relapse “heralding effect,”24 meaning that it tends to rise well before a patient will admit he or she resumed drinking.24-26 Although other markers may also rise following relapse, their elevation tends to be delayed and less dramatic.
The sensitivity and specificity of CDT alone and with GGT in identifying relapse have been evaluated. Across male-only studies, CDT’s median sensitivity (percent of relapsed patients with elevated scores) was 0.73, with a specificity (percent of those not relapsed who had low scores) of 0.91. In the two female-only studies, median sensitivity and specificity for CDT were 0.32 and 0.86, respectively. For women, using CDT and GGT in combination substantially raised median sensitivity to 0.62, although specificity fell slightly to 0.80.27
Recommendation. When using biomarkers to identify relapse, examine the temporal pattern of test results to date. Assume that an increase of 30% or more above the lowest observed lab value indicates a relapse.28
Frequent testing—probably biweekly—is recommended during the first 3 or 4 months after patients complete treatment. If there is no indication of relapse, testing frequency could be tapered down.
- AUDIT. The Alcohol Use Disorders Identification Test. Guidelines for primary care use. Available at: http://www.who.int/substance_abuse/pubs_alcohol.htm
- Allen JP, Litten RZ. Psychometric and laboratory measures to assist in the treatment of alcoholism. Clin Psychol Rev 1993;13(3):223-39.
- Salaspuro M. Carbohydrate-deficient transferrin as compared to other markers of alcoholism: a systematic review. Alcohol 1999; 19(3):261-71.
Disclosure
Dr. Allen reports that he serves as a consultant to Axis-Shield ASA and Bio-Rad Laboratories, patent holder and U.S. distributor, respectively, of the carbohydrate-deficient transferrin (%CDT) reagent kit.
Dr. Anthenelli reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hall W. What have population surveys revealed about substance use disorders and their co-morbidity with other mental disorders? Drug Alcohol Rev 1996;15(2):157-70.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: Results of a national survey. Drug Alcohol Depend 1995;39(3):197-206.
3. Shivani R, Goldsmith RJ, Anthenelli RM. Alcoholism and psychiatric disorders: diagnostic challenges. Alcohol Res Health 2002;26(2):90-8.
4. Allen JP, Columbus M (eds). Assessing alcohol problems: a guide for clinicians and researchers. NIAAA Treatment Handbook, Series 4. Washington, DC: U.S. Department of Health and Human Services, 1995 [NIH publication no. 95-3745].
5. Mayfield D, McLeod G, Hall P. CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry 1974;131(10):1121-3.
6. Seltzer ML. The Michigan Alcoholism Screening Test: the quest for a new diagnostic instrument. Am J Psychiatry 1971;127:1653-8.
7. Saunders JB, Aasland OG, Babor TF, et al. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO collaborative project on early detection of persons with harmful alcohol consumption—II. Addiction 1993;88(6):791-804.
8. Daeppen J, Yersin B, Landry U, et al. Reliability and validity of the Alcohol Use Disorders Identification Test (AUDIT) imbedded within a general health risk screening questionnaire: results of a survey in 332 primary care patients. Alcohol Clin Exp Res 2000;24:659-65.
9. Reinert DF, Allen JP. The Alcohol Use Disorders Identification Test: a review of recent research. Alcohol Clin Exp Res 2002;26(2):272-9.
10. Maisto SA, Carey MP, Carey KB, et al. Use of the AUDIT and the DAST-10 to identify alcohol and drug use disorders among adults with a severe and persistent mental illness. Psychol Assess 2002;12:186-92.
11. Hulse GK, Saunders JB, Roydhouse RM, et al. Screening for hazardous alcohol use and dependence in psychiatric inpatients using the AUDIT questionnaire. Drug Alcohol Rev 2000;19:291-8.
12. Dawe S, Seinen A, Kavanagh D. An examination of the utility of the AUDIT in people with schizophrenia. J Stud Alcohol 2000;61:744-75.
13. Allen JP, Litten RZ, Fertig JB, Babor T. A review of research on the Alcohol Use Disorders Identification Test (AUDIT). Alcohol Clin Exp Res 1997;21:613-19.
14. Hermansson U, Helander A, Huss A, et al. The Alcohol Use Disorders Identification Test (AUDIT) and carbohydrate-deficient transferrin (CDT) in a routine workplace health examination. Alcohol Clin Exp Res 2000;24:180-7.
15. Sillanaukee P, Strid N, Allen JP, Litten RZ. Possible reasons why heavy drinking increases carbohydrate-deficient transferrin. Alcohol Clin Exp Res 2001;25(1):34-40.
16. Litten RZ, Allen JP, Fertig JB. Gamma-glutamyltranspeptidase and carbohydrate deficient transferrin: alternative measures of excessive alcohol consumption. Alcohol Clin Exp Res 1995;19(6):1541-6.
17. Allen JP, Litten RZ, Fertig JB, Sillanaukee P. Carbohydrate-deficient transferrin, gamma glutamyl transferase and macrocytic volume as biomarkers of alcohol problems in women. Alcohol Clin Exp Res 2000;24(4):492-6.
18. Sillanaukee P, Strid N, Allen JP, Litten RZ. Combining biomarkers to screen for alcohol problems (manuscript submitted for publication).
19. Miller WR, Zweben A, DiClemente CC, et al. Motivational enhancement therapy manual: A clinical research guide for therapists treating individuals with alcohol abuse and dependence. Rockville, MD: U.S. Department of Health and Human Services, 1995.
20. Project MATCH research group. Matching alcoholism treatments to client heterogeneity: Project MATCH post-treatment drinking outcomes. J Stud Alcohol 1997;58(1):7-29.
21. Bien TH, Miller WR, Tonigan S. Brief intervention for alcohol problems: a review. Addiction 1993;88:315-36.
22. Kristenson H, Hood B, Peterson B, et al. Prevention of alcohol-related problems in urban middle-aged males. Alcohol 1985;2(3):545-9.
23. Miller WR, Walter ST, Bennett ME. How effective is alcoholism treatment in the United States? J Stud Alcohol. 2001;62:211-20.
24. Mitchell C, Simpson D, Chick J. Carbohydrate-deficient transferrin in detecting relapse in alcohol dependence. Drug Alcohol Depend 1997;48:97-103.
25. Borg S, Helander A, Voltaire Carlsson A, Hogstrom Brandt AM. Detection of relapses in alcohol-dependent patients using carbohydrate-deficient transferrin: improvement with individualized reference levels during long-term monitoring. Alcohol Clin Exp Res 1995;19(4):961-3.
26. Schmidt LG, Schmidt K, Dufeu P, et al. Superiority of carbohydrate-deficient transferrin to gamma-glutamyltransferase in detecting relapse in alcoholism. Am J Psychiatry 1997;154(1):75-80.
27. Allen JP, Anton R. Biomarkers as aids to identification of relapse in alcoholic patients. In: Galanter M (ed). Recent developments in alcoholism: research on alcoholism treatment (vol. XVI). New York: Plenum Press (in press).
28. Anton RF, Lieber C, Tabakoff B, et al. Carbohydrate-deficient transferrin and gamma-glutamyltransferase for the detection and monitoring of alcohol use: results from a multi-site study. Alcohol Clin Exp Res 2002;26(8):1215-22.
1. Hall W. What have population surveys revealed about substance use disorders and their co-morbidity with other mental disorders? Drug Alcohol Rev 1996;15(2):157-70.
2. Grant BF, Harford TC. Comorbidity between DSM-IV alcohol use disorders and major depression: Results of a national survey. Drug Alcohol Depend 1995;39(3):197-206.
3. Shivani R, Goldsmith RJ, Anthenelli RM. Alcoholism and psychiatric disorders: diagnostic challenges. Alcohol Res Health 2002;26(2):90-8.
4. Allen JP, Columbus M (eds). Assessing alcohol problems: a guide for clinicians and researchers. NIAAA Treatment Handbook, Series 4. Washington, DC: U.S. Department of Health and Human Services, 1995 [NIH publication no. 95-3745].
5. Mayfield D, McLeod G, Hall P. CAGE questionnaire: validation of a new alcoholism screening instrument. Am J Psychiatry 1974;131(10):1121-3.
6. Seltzer ML. The Michigan Alcoholism Screening Test: the quest for a new diagnostic instrument. Am J Psychiatry 1971;127:1653-8.
7. Saunders JB, Aasland OG, Babor TF, et al. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO collaborative project on early detection of persons with harmful alcohol consumption—II. Addiction 1993;88(6):791-804.
8. Daeppen J, Yersin B, Landry U, et al. Reliability and validity of the Alcohol Use Disorders Identification Test (AUDIT) imbedded within a general health risk screening questionnaire: results of a survey in 332 primary care patients. Alcohol Clin Exp Res 2000;24:659-65.
9. Reinert DF, Allen JP. The Alcohol Use Disorders Identification Test: a review of recent research. Alcohol Clin Exp Res 2002;26(2):272-9.
10. Maisto SA, Carey MP, Carey KB, et al. Use of the AUDIT and the DAST-10 to identify alcohol and drug use disorders among adults with a severe and persistent mental illness. Psychol Assess 2002;12:186-92.
11. Hulse GK, Saunders JB, Roydhouse RM, et al. Screening for hazardous alcohol use and dependence in psychiatric inpatients using the AUDIT questionnaire. Drug Alcohol Rev 2000;19:291-8.
12. Dawe S, Seinen A, Kavanagh D. An examination of the utility of the AUDIT in people with schizophrenia. J Stud Alcohol 2000;61:744-75.
13. Allen JP, Litten RZ, Fertig JB, Babor T. A review of research on the Alcohol Use Disorders Identification Test (AUDIT). Alcohol Clin Exp Res 1997;21:613-19.
14. Hermansson U, Helander A, Huss A, et al. The Alcohol Use Disorders Identification Test (AUDIT) and carbohydrate-deficient transferrin (CDT) in a routine workplace health examination. Alcohol Clin Exp Res 2000;24:180-7.
15. Sillanaukee P, Strid N, Allen JP, Litten RZ. Possible reasons why heavy drinking increases carbohydrate-deficient transferrin. Alcohol Clin Exp Res 2001;25(1):34-40.
16. Litten RZ, Allen JP, Fertig JB. Gamma-glutamyltranspeptidase and carbohydrate deficient transferrin: alternative measures of excessive alcohol consumption. Alcohol Clin Exp Res 1995;19(6):1541-6.
17. Allen JP, Litten RZ, Fertig JB, Sillanaukee P. Carbohydrate-deficient transferrin, gamma glutamyl transferase and macrocytic volume as biomarkers of alcohol problems in women. Alcohol Clin Exp Res 2000;24(4):492-6.
18. Sillanaukee P, Strid N, Allen JP, Litten RZ. Combining biomarkers to screen for alcohol problems (manuscript submitted for publication).
19. Miller WR, Zweben A, DiClemente CC, et al. Motivational enhancement therapy manual: A clinical research guide for therapists treating individuals with alcohol abuse and dependence. Rockville, MD: U.S. Department of Health and Human Services, 1995.
20. Project MATCH research group. Matching alcoholism treatments to client heterogeneity: Project MATCH post-treatment drinking outcomes. J Stud Alcohol 1997;58(1):7-29.
21. Bien TH, Miller WR, Tonigan S. Brief intervention for alcohol problems: a review. Addiction 1993;88:315-36.
22. Kristenson H, Hood B, Peterson B, et al. Prevention of alcohol-related problems in urban middle-aged males. Alcohol 1985;2(3):545-9.
23. Miller WR, Walter ST, Bennett ME. How effective is alcoholism treatment in the United States? J Stud Alcohol. 2001;62:211-20.
24. Mitchell C, Simpson D, Chick J. Carbohydrate-deficient transferrin in detecting relapse in alcohol dependence. Drug Alcohol Depend 1997;48:97-103.
25. Borg S, Helander A, Voltaire Carlsson A, Hogstrom Brandt AM. Detection of relapses in alcohol-dependent patients using carbohydrate-deficient transferrin: improvement with individualized reference levels during long-term monitoring. Alcohol Clin Exp Res 1995;19(4):961-3.
26. Schmidt LG, Schmidt K, Dufeu P, et al. Superiority of carbohydrate-deficient transferrin to gamma-glutamyltransferase in detecting relapse in alcoholism. Am J Psychiatry 1997;154(1):75-80.
27. Allen JP, Anton R. Biomarkers as aids to identification of relapse in alcoholic patients. In: Galanter M (ed). Recent developments in alcoholism: research on alcoholism treatment (vol. XVI). New York: Plenum Press (in press).
28. Anton RF, Lieber C, Tabakoff B, et al. Carbohydrate-deficient transferrin and gamma-glutamyltransferase for the detection and monitoring of alcohol use: results from a multi-site study. Alcohol Clin Exp Res 2002;26(8):1215-22.
Serotonin syndrome: How to avoid, identify, & treat dangerous drug interactions
Promptly identifying serotonin syndrome and acting decisively can keep side effects at the mild end of the spectrum. Symptoms of this potentially dangerous syndrome range from minimal in patients starting selective serotonin reuptake inhibitors (SSRIs) to fatal in those combining monoamine oxidase inhibitors (MAOIs) with serotonergic agents.
This article presents the latest evidence on how to:
- reduce the risk of serotonin syndrome
- recognize its symptoms
- and treat patients with mild to life-threatening symptoms.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome is characterized by changes in autonomic, neuromotor, and cognitive-behavioral function (Table 1) triggered by increased serotonergic stimulation. It typically results from pharmacodynamic and/or pharmacokinetic interactions between drugs that increase serotonin activity.1,2
Table 1
How to recognize serotonin syndrome
| System | Clinical signs and symptoms |
|---|---|
| Autonomic | Diaphoresis, hyperthermia, hypertension, tachycardia, pupillary dilatation, nausea, diarrhea, shivering |
| Neuromotor | Hyperreflexia, myoclonus, restlessness, tremor, incoordination, rigidity, clonus, teeth chattering, trismus, seizures |
| Cognitive-behavioral | Confusion, agitation, anxiety, hypomania, insomnia, hallucinations, headache |
The syndrome was first identified in animal studies, followed by case reports in humans. The first review—with suggested diagnostic criteria— was published in 1991.1
Since then, case reports have described serotonin syndrome with many drug combinations, including nonpsychotropics and illicit drugs. Using an irreversible MAOI with a serotonergic agent is the most toxic reported combination, but any drug or combination that increases serotonin can, in theory, cause serotonin syndrome (Table 2). A clinical scale3 is being developed to define and identify this potentially dangerous state, but no consensus has emerged on diagnostic criteria.
Pathophysiology. Serotonin syndrome’s symptoms and signs appear to result from stimulation of specific central and peripheral serotonin receptors, especially 5HT1a and 5HT2. Others—such as 5HT3 and 5HT4—may also be involved in causing GI symptoms and may affect dopaminergic transmission.
Damaged vascular or pulmonary endothelium, atherosclerosis, hypertension, or hypercholesterolemia may increase the risk for serotonin syndrome. In patients with these common medical conditions, reduced endothelial MAO-A activity or reduced ability to secrete endothelium-derived nitric oxide may diminish the ability to metabolize serotonin.2
POTENTIALLY DANGEROUS COMBINATIONS
MAOIs. Serotonin syndrome has been reported as a result of interactions between MAOIs— including selegiline and reversible MAO-A inhibitors (RIMAs)—and various serotonergic compounds. These reports have included fatalities,4 some of which were preceded by severe hyperthermia with complications such as disseminated intravascular coagulation, rhabdomyolysis, and renal failure. Some cases resulted from overdoses, but others did not.
Most disturbingly, some cases occurred after patients had undergone the traditional 2-week washout from the MAOI and then took a serotonergic agent.5-7 In one instance,8 a patient who had discontinued fluoxetine for 6 weeks developed serotonin syndrome after starting tranylcypromine. These cases remind us to be vigilant when switching patients from irreversible MAOIs to serotonergic antidepressants or vice versa—even when recommended wash-out times are observed—and not to combine these agents acutely.
Selegiline is a relatively selective MAO-B inhibitor when used at 5 to 10 mg/d to treat Parkinson’s disease, though it loses MAO-B selectivity when used at higher dosages to treat depression. In a study9 of 4,568 patients with Parkinson’s disease who received selegiline (in dosages selective for MAO-B) plus an antidepressant:
- 11 (0.24%) experienced symptoms “possibly” consistent with serotonin syndrome
- 2 others (0.04%) experienced serious serotonin syndrome symptoms.9
Serotonin syndrome has been reported when MAO-B-selective doses of selegiline were combined with meperidine10 and nortriptyline.11 This underscores the need for caution when combining these agents, especially if transdermal selegiline— which would not be MAO-B-selective—becomes available for treating depression.
Moclobemide is a RIMA used in treating depression and anxiety, with a purported reduced risk of drug and food interactions compared with other MAOIs. Moclobemide is not approved in the United States, but some patients obtain it elsewhere.
Joffe and Bakish reported on safely combining moclobemide with SSRIs,12 and a review of MAOIs—including RIMAs—indicated that moclobemide was involved in only 9 of 226 cases of adverse effects and 3 of 105 cases of defined serotonin syndrome.13 Most moclobemide-SSRI interactions—including fatalities—involved overdoses in suicide attempts, although toxic symptoms have been reported with clomipramine or meperidine taken at normal dosages.14,15
In one study,16 18 healthy controls received fluoxetine, 20 to 40 mg/d, for 23 days, then were given moclobemide, up to 600 mg/d, or placebo and observed for adverse effects. No indication of serotonin syndrome was observed.
Linezolid is an oxazolidinone antibiotic with relatively weak, nonspecific, but reversible MAO inhibition. Cases of potential serotonin syndrome have been reported with linezolid plus paroxetine17 or sertraline.18 Patients in each case were medically ill and taking several other medications, which complicates interpretation of these reports. Nonetheless, physicians should be aware of the potential risk of serotonin syndrome if this antibiotic is combined with serotonergic agents.
Table 2
Serotonergic agents and their actions
| Actions | Agents |
|---|---|
| Inhibit serotonin reuptake | Fluoxetine, sertraline, citalopram, escitalopram, paroxetine, clomipramine, venlafaxine, fluvoxamine, tramadol, trazodone, nefazodone, tricyclic antidepressants, amphetamine, cocaine, dextromethorphan, meperidine, St. John’s wort |
| Increases serotonin synthesis | Tryptophan |
| Inhibit serotonin metabolism | Phenelzine, tranylcypromine, isocarboxazid, selegiline (deprenyl), linezolid, moclobemide |
| Increase serotonin release | MDMA (“Ecstasy”), amphetamine, cocaine, fenfluramine |
| Increase serotonin activity | Lithium, ECT |
| Serotonin receptor agonists | Buspirone, sumatriptan and other “triptans” used for migraine |
Atypical antipsychotics. Original diagnostic criteria for serotonin syndrome excluded the addition of, or increase in, an antipsychotic prior to the syndrome’s onset.1 However, serotonin syndrome has been reported with combinations of risperidone with paroxetine,19 olanzapine with mirtazapine and tramadol,20 and olanzapine with lithium and citalopram.21 The 5HT2 antagonist effect of these atypical antipsychotics may have led indirectly to overactivation of 5HT1a receptors and serotonin syndrome. In each case, neuroleptic malignant syndrome was ruled out.
Table 3
Signs and symptoms that differentiate 5 hyperthermic states
| Hyperthermic state | Symptoms/signs | Lab findings | Cause |
|---|---|---|---|
| Serotonin syndrome | Typically rapid onset with hyperreflexia, tremors, myoclonus, diaphoresis, confusion, agitation, or shivering; muscular rigidity not invariably present | Nonspecific | Increased serotonergic tone |
| Neuroleptic malignant syndrome | Variable rapidity of onset; severe muscular rigidity, diaphoresis, delirium, fluctuating blood pressure, tachycardia, extrapyramidal symptoms | Elevated CPK, leukocytosis | Blockade of dopamine receptors or abrupt withdrawal of a dopamine agonist |
| Lethal catatonia | Muscular rigidity, diaphoresis, delirium, alternating extreme excitement and stupor, tremors, hypertension | Nonspecific | Evidence of pre-existing psychosis (bipolar disorder, schizophrenia) |
| Anticholinergic toxicity | Hot, dry skin, pupillary dilatation, tachycardia, constipation, urinary retention, confusion, hallucinations, muscular relaxation | Nonspecific | Agents that block central and peripheral muscarinic cholinergic receptors |
| Malignant hyperthermia | Rapid onset, severe muscular rigidity, ischemia, hypotension | Elevated CPK, potassium, magnesium; DIC; acidosis; rhabdomyolysis | Inherited disorder with onset after exposure to anesthetic agents that block the neuromuscular junction |
| CPK: creatine phosphokinase | |||
| DIC: disseminated intravascular coagulation | |||
Tramadol is an analgesic with opioid and serotonin-reuptake inhibiting properties that is metabolized by the cytochrome P (CYP)-450 isoenzyme 2D6. Serotonin syndrome has been reported from interactions between tramadol and sertraline22 and fluoxetine.23 Possible causes include SSRI inhibition of CYP 2D6 metabolism of tramadol, tramadol abuse,23 and multiple coadministered medications.22
Sumatriptan is one of the selective 5HT1D agonists used in treating migraine. Gardner and Lynd24 concluded that most patients tolerate sumatriptan with SSRIs or lithium. They felt they could not ensure the safety of sumatriptan with MAOIs, however, because sumatriptan elimination depends on hepatic MAO activity.
Among the 5HT1D agonists, using sumatriptan, zolmitriptan, rizatriptan, or almotriptan with an MAOI or within 2 weeks of discontinuing an MAOI is contraindicated. Naratriptan and frovatriptan appear less likely to interact with MAOIs, based on FDA-approved labeling.
MDMA. 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) is widely used as a recreational drug, especially at crowded dances (“raves”) and with other drugs.25 This illicit amphetamine derivative stimulates the release of serotonin and inhibits its reuptake.
Kaskey reported the rapid onset of serotonin syndrome when a patient taking lithium and phenelzine ingested MDMA.26 Signs and symptoms of serotonin syndrome also may develop when MDMA is used alone, facilitated by the high ambient temperatures on crowded dance floors and the dancers’ relative dehydration.
Fatalities have been blamed on complications including disseminated intravascular coagulation, rhabdomyolysis, and acute hepatic, renal, or cardiac failure.25 Cases are difficult to interpret because of uncertainty about whether the victim ingested MDMA or another agent or combination.
St. John’s wort (Hypericum perforatum) contains numerous constituents, including hypericin and hyperforin, which have been found to inhibit the synaptic uptake of monoamines, including serotonin.27 Which constituents are responsible for its clinical effect is unclear. Adverse effects from monotherapy include GI symptoms, confusion, dry mouth, dizziness, headache, fatigue, allergic skin reactions, photosensitivity, and urinary frequency.27
Several cases of purported serotonin syndrome have been associated with St. John’s wort alone28 or in combination with SSRIs, nefazodone, or fenfluramine.29,30 GI symptoms and anxiety were the primary complaints and resolved without complications (adjunctive cyproheptadine was prescribed in two cases, though it is not clear that this agent contributed to resolution).
MISCELLANEOUS COMBINATIONS
Antiretroviral therapy. Five cases of serotonin syndrome were reported in HIV-infected patients taking fluoxetine with antiretroviral therapy.31 In particular, the use or addition of ritonavir—a potent CYP 2D6 inhibitor—was implicated, though saquinavir, efavirenz, or grapefruit juice (all primarily CYP 3A4 inhibitors) were also used, suggesting that pharmacokinetic interactions increased serotonergic stimulation. All five patients were taking multiple additional medications and had complex medical and/or psychiatric histories. Reducing SSRI dosages by one-half when used with ritonavir has been recommended to minimize adverse effects from a pharmacokinetic interaction.
Erythromycin was reported to induce serotonin syndrome in a 12-year-old boy when added to ongoing treatment with sertraline, an effect believed to be secondary to CYP 3A4 inhibition of sertraline metabolism.32
Mirtazapine was reported to induce serotonin syndrome in an elderly man 8 days after it was added to a regimen he had been taking for several years to treat chronic obstructive pulmonary disease.33 Serotonin syndrome also developed in a 12-year-old boy with Ewing’s sarcoma when the 5HT3 antagonist ondansetron was added to mirtazapine and morphine34 and in an 11-year-old girl with acute lymphoblastic leukemia when ondansetron was added to fentanyl. Interestingly, another report35 suggested using mirtazapine to treat serotonin syndrome caused by serotonergic antagonist effects.
Reports have associated the following combinations with serotonin syndrome, perhaps as the result of pharmacodynamic and/or pharmacokinetic interactions:
- paroxetine plus dextromethorphan and pseudoephedrine
- paroxetine plus nefazodone
- fluoxetine plus clomipramine and buspirone
- fluvoxamine plus buspirone
- fluoxetine plus buspirone
- amitriptyline plus meperidine and venlafaxine
- venlafaxine and dextroamphetamine
- fluoxetine plus clomipramine.
Table 4
Clinical signs that distinguish hyperthermic states
| Signs | Possible diagnosis |
|---|---|
| Prominent muscular rigidity | Neuroleptic malignant syndrome, malignant hyperthermia, catatonia |
| Myoclonus/hyperreflexia | Serotonin syndrome |
| Diaphoresis | Serotonin syndrome, neuroleptic malignant syndrome, catatonia |
| Hot dry skin | Anticholinergic toxicity |
| Elevated creatine phosphokinase | Neuroleptic malignant syndrome, malignant hyperthermia |
| Family history of anesthetic-induced hyperthermia | Malignant hyperthermia |
HOW TO RECOGNIZE SEROTONIN SYNDROME
Signs and symptoms of serotonin syndrome can overlap with those seen in neuroleptic malignant syndrome, lethal catatonia, malignant hyperthermia, and anticholinergic toxicity (Table 3),1,36,37 particularly with fever or hyperthermia (>40.5 °C, 105 °F). Fink37 has opined that acute neurotoxic syndromes such as serotonin syndrome and neuroleptic malignant syndrome also meet criteria for catatonia and are therefore subtypes of catatonia. The types of drugs involved and clinical findings can help distinguish the various hyperthermic states (Table 4).
As mentioned above, original diagnostic criteria for serotonin syndrome excluded the addition of, or increase in, an antipsychotic agent. This exclusion was intended to avoid confusion between serotonin syndrome and neuroleptic malignant syndrome. Co-administering antipsychotic and serotonergic agents requires heightened awareness for both neurotoxic syndromes.
TREATING MILD TO SEVERE CASES
If a patient develops serotonin syndrome, immediately discontinue the suspected agent(s) and observe carefully. In most cases, serotonin syndrome will resolve within 24 hours.
In mild cases, lorazepam, 1 to 2 mg slow IV push every 30 minutes until excessive sedation develops, may help. In moderate to severe cases, agents that block serotonin’s action are recommended,2 including:
- cyproheptadine (4 mg po every 4 hours as needed, up to 20 mg in 24 hours)
- propranolol (1 to 3 mg IV every 5 minutes, up to 0.1 mg/kg).
Case reports attest to these agents’ potential benefit. Other clinicians have reported using mirtazapine,35 nitroglycerin,38 and chlorpromazine.1
Serotonin syndrome symptoms resolved within minutes when IV nitroglycerin was used in a patient with serotonin syndrome and cardiac ischemia. The authors hypothesized that nitroglycerin, via nitric acid, provided an “off” signal for serotonin, though they did not advocate this as a routine treatment.38
The rationale for using chlorpromazine is its potential to block serotonin receptors. I would avoid the routine use of any antipsychotic agent in this setting, however, to minimize the risk of neuroleptic malignant syndrome.
Severe cases. Intensive care observation and treatment is required for patients with severe serotonin syndrome, including evidence of hyperthermia, DIC, rhabdomyolysis, renal failure, or aspiration. In cases of hyperthermia, supportive measures and standard treatments include muscle relaxants, cooling, and endotracheal intubation.
Severe complications are most likely with interactions between MAOIs and serotonergic agents, especially in overdose. Therefore, using such combinations requires close observation.
Related resources
- Di Rosa AE, Morgante L, Spina E et al. Epidemiology and pathoetiology of neurological syndromes with hyperthermia. Funct Neurol 1995;10:111-19.
- Radomski, JW, Dursun SM, Reveley MA, et al. An exploratory approach to the serotonin syndrome: an update of clinical phenomenology and revised diagnostic criteria. Med Hypothesis 2000;55: 218-24.
- Lane R, Baldwin D. Selective serotonin reuptake inhibitor-induced serotonin syndrome: review. J Clin Psychopharmacol 1997;17:208-21.
Drug brand names
- Almotriptan • Axert
- Amitriptyline • Elavil
- Buspirone • Buspar
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Cyproheptadine • Periactin
- Dextroamphetamine • Dexedrine
- Dextromethorphan • Delsym
- Efavirenz • Sustiva
- Escitalopram • Lexapro
- Fenfluramine • Pondimin
- Fentanyl • Sublimaze
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Isocarboxazid • Marplan
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Moclobemide • Aurorix
- Nortriptyline • Pamelor
- Naratriptan, • Amerge
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Ondansetron • Zofran
- Paroxetine • Paxil
- Phenelzine • Nardil
- Propranolol • Inderal
- Risperidone • Risperdal
- Ritonavir • Norvir
- Rizatriptan • Maxalt
- Saquinavir • Invirase
- Selegiline • Eldepryl
- Sertraline • Zoloft
- Sumatriptan • Imitrex
- Tramadol • Ultram
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Zolmitriptan • Zomig
Disclosure
Dr. Sternbach receives research grants from Otsuka America Pharmaceuticals and Eli Lilly and Co. and owns stock in Merck & Co., Pfizer Inc., and Johnson & Johnson.
1. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148:705-13.
2. Brown TM, Skop BP, Mareth TR. Pathophysiology and management of the serotonin syndrome. Ann Pharmacother 1996;30:527-33.
3. Hegerl U, Bottlender R, Gallinat J, et al. The serotonin syndrome scale: first results on validity. Eur Arch Psychiatry Clin Neurosci 1998;248:96-103.
4. Beasley CM, Jr, Masica DN, Heiligenstein JH, et al. Possible monoamine-oxidase inhibitor-serotonin uptake inhibitor interaction: fluoxetine clinical data and pre-clinical findings. J Clin Psychopharmacol 1993;13:312-20.
5. Ruiz F. Fluoxetine and the serotonin syndrome. Ann Emerg Med 1994;34:983-5.
6. Gitlin MJ. Venlafaxine, monoamine oxidase inhibitors and the serotonin syndrome. J Clin Psychopharmacol 1997;17:66-7.
7. Kolecki P. Venlafaxine induced serotonin syndrome occurring after abstinence from phenelzine for more than two weeks. Clin Toxicol 1997;35:211-12.
8. Coplan JD, Gorman JM. Detectable levels of fluoxetine metabolites after discontinuation: an unexpected serotonin syndrome. Am J Psychiatry 1993;15:837.-
9. Richard IH, Kurlan R, Tanner C, et al. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson’s disease. Neurology 1997;48:1070-7.
10. Zornberg GL, Bodkin JA, Cohen BM. Severe adverse interaction between pethidine and selegiline. Lancet 1991;337:246.-
11. Hinds NP, Hillier CE, Wiles CM. Possible serotonin syndrome arising from an interaction between nortriptyline and selegiline in a lady with parkinsonism. J Neurol 2000;247:811.-
12. Joffe RT, Bakish D. Combined SSRI-moclobemide treatment of psychiatric illness. J Clin Psychiatry 1994;55:24-5.
13. Hilton SE, Maradit H, Moller HJ. Serotonin syndrome and drug combinations: focus on MAOI and RIMA. Eur Arch Psychiatry Clin Neurosci 1997;247:113-19.
14. Dardennes RM, Even C, Ballon N, et al. Serotonin syndrome caused by a clomipramine-moclobemide interaction. J Clin Psychiatry 1998;59:382-3.
15. Gillman PK. Possible serotonin syndrome with moclobemide and pethidine. Med J Aust 1995;162:554.-
16. Dingemanse J, Wallnofer A, Gieschke R, et al. Pharmacokinetic and pharmacodynamic interaction between fluoxetine and moclobemide in the investigation of development of the “serotonin syndrome.” Clin Pharmacol Ther 1998;63:403-13.
17. Wigen CL, Goetz MB. Serotonin syndrome and linezolid. Clin Infect Dis 2002;34:1651-2.
18. Lavery S, Ravi H, McDaniel WW, et al. Linezolid and serotonin syndrome. Psychosomatics 2001;42:432-4.
19. Hamilton S, Malone K. Serotonin syndrome during treatment with paroxetine and risperidone. J Clin Psychopharmacol 2000;20:103-5.
20. Duggal HS, Fetchko J. Serotonin syndrome and atypical antipsychotics. Am J Psychiatry 2002;159:672-3.
21. Haslett CD, Kumar S. Can olanzapine be implicated in causing serotonin syndrome? Psychiatry Clin Neurosci 2002;56:533-6.
22. Mason BJ, Blackburn KH. Possible serotonin syndrome associated with tramadol and sertraline coadministration. Ann Pharmacother 1997;31:175-7.
23. Lange-Asschenfeldt C, Weigmann H, Hiemke C, et al. Serotonin syndrome as a result of fluoxetine in a patient with tramadol abuse: plasma level-correlated symptomatology. J Clin Psychopharmacol 2002;22:440-1.
24. Gardner DM, Lynd LD. Sumatriptan contraindications and the serotonin syndrome. Ann Pharmacother 1998;32:33-8.
25. Parrott AC. Recreational Ecstasy/MDMA, the serotonin syndrome and serotonergic neurotoxicity. Pharmacol Biochem Behav 2002;71:837-44.
26. Kaskey GB. Possible interaction between an MAOI and “Ecstasy.” Am J Psychiatry 1992;149:411-12.
27. De Smet PA. Herbal remedies. N Engl J Med 2002;347:2046-56.
28. Parker V, Wong AH, Boon HS, et al. Adverse reactions to St. John’s wort. Can J Psychiatry 2001;46:77-9.
29. Lantz MS, Buchalter E, Giambanco V. St. John’s wort and antidepressant drug interactions in the elderly. J Geriatr Psychiatry Neurol 1999;12:7-10.
30. Beckman SE, Sommi RW, Switzer J. Consumer use of St. John’s wort: a survey of effectiveness, safety, and tolerability. Pharmacotherapy 2000;20:568-74.
31. De Silva KE, Le Flore DB, Marston BJ, et al. Serotonin syndrome in HIV-infected individuals receiving antiretroviral therapy and fluoxetine. AIDS 2001;15:1281-5.
32. Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999;19:894-6.
33. Hernandez JL, Ramos FJ, Infante J, et al. Severe serotonin syndrome induced by mirtazapine monotherapy. Ann Pharmacother 2002;36:641-3.
34. Turkel SB, Nadala JGB, Wincor MZ. Possible serotonin syndrome in association with 5HT3 antagonist agents. Psychosomatics 2001;42:258-60.
35. Hoes MJ, Zeijpveld JH. Mirtazapine as treatment for serotonin syndrome. Pharmacopsychiatry 1996;29:81.-
36. Theoharides TC, Harris RS, Weckstein D. Neuroleptic malignant-like syndrome due to cyclobenzaprine? J Clin Psychopharmacol 1995;15:79-81.
37. Fink M. Toxic serotonin syndrome or neuroleptic malignant syndrome? Pharmacopsychiatry 1996;29:159-61.
38. Brown TM, Skop BP. Nitroglycerin in the treatment of the serotonin syndrome. Ann Pharmacother 1996;30:191-2.
Promptly identifying serotonin syndrome and acting decisively can keep side effects at the mild end of the spectrum. Symptoms of this potentially dangerous syndrome range from minimal in patients starting selective serotonin reuptake inhibitors (SSRIs) to fatal in those combining monoamine oxidase inhibitors (MAOIs) with serotonergic agents.
This article presents the latest evidence on how to:
- reduce the risk of serotonin syndrome
- recognize its symptoms
- and treat patients with mild to life-threatening symptoms.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome is characterized by changes in autonomic, neuromotor, and cognitive-behavioral function (Table 1) triggered by increased serotonergic stimulation. It typically results from pharmacodynamic and/or pharmacokinetic interactions between drugs that increase serotonin activity.1,2
Table 1
How to recognize serotonin syndrome
| System | Clinical signs and symptoms |
|---|---|
| Autonomic | Diaphoresis, hyperthermia, hypertension, tachycardia, pupillary dilatation, nausea, diarrhea, shivering |
| Neuromotor | Hyperreflexia, myoclonus, restlessness, tremor, incoordination, rigidity, clonus, teeth chattering, trismus, seizures |
| Cognitive-behavioral | Confusion, agitation, anxiety, hypomania, insomnia, hallucinations, headache |
The syndrome was first identified in animal studies, followed by case reports in humans. The first review—with suggested diagnostic criteria— was published in 1991.1
Since then, case reports have described serotonin syndrome with many drug combinations, including nonpsychotropics and illicit drugs. Using an irreversible MAOI with a serotonergic agent is the most toxic reported combination, but any drug or combination that increases serotonin can, in theory, cause serotonin syndrome (Table 2). A clinical scale3 is being developed to define and identify this potentially dangerous state, but no consensus has emerged on diagnostic criteria.
Pathophysiology. Serotonin syndrome’s symptoms and signs appear to result from stimulation of specific central and peripheral serotonin receptors, especially 5HT1a and 5HT2. Others—such as 5HT3 and 5HT4—may also be involved in causing GI symptoms and may affect dopaminergic transmission.
Damaged vascular or pulmonary endothelium, atherosclerosis, hypertension, or hypercholesterolemia may increase the risk for serotonin syndrome. In patients with these common medical conditions, reduced endothelial MAO-A activity or reduced ability to secrete endothelium-derived nitric oxide may diminish the ability to metabolize serotonin.2
POTENTIALLY DANGEROUS COMBINATIONS
MAOIs. Serotonin syndrome has been reported as a result of interactions between MAOIs— including selegiline and reversible MAO-A inhibitors (RIMAs)—and various serotonergic compounds. These reports have included fatalities,4 some of which were preceded by severe hyperthermia with complications such as disseminated intravascular coagulation, rhabdomyolysis, and renal failure. Some cases resulted from overdoses, but others did not.
Most disturbingly, some cases occurred after patients had undergone the traditional 2-week washout from the MAOI and then took a serotonergic agent.5-7 In one instance,8 a patient who had discontinued fluoxetine for 6 weeks developed serotonin syndrome after starting tranylcypromine. These cases remind us to be vigilant when switching patients from irreversible MAOIs to serotonergic antidepressants or vice versa—even when recommended wash-out times are observed—and not to combine these agents acutely.
Selegiline is a relatively selective MAO-B inhibitor when used at 5 to 10 mg/d to treat Parkinson’s disease, though it loses MAO-B selectivity when used at higher dosages to treat depression. In a study9 of 4,568 patients with Parkinson’s disease who received selegiline (in dosages selective for MAO-B) plus an antidepressant:
- 11 (0.24%) experienced symptoms “possibly” consistent with serotonin syndrome
- 2 others (0.04%) experienced serious serotonin syndrome symptoms.9
Serotonin syndrome has been reported when MAO-B-selective doses of selegiline were combined with meperidine10 and nortriptyline.11 This underscores the need for caution when combining these agents, especially if transdermal selegiline— which would not be MAO-B-selective—becomes available for treating depression.
Moclobemide is a RIMA used in treating depression and anxiety, with a purported reduced risk of drug and food interactions compared with other MAOIs. Moclobemide is not approved in the United States, but some patients obtain it elsewhere.
Joffe and Bakish reported on safely combining moclobemide with SSRIs,12 and a review of MAOIs—including RIMAs—indicated that moclobemide was involved in only 9 of 226 cases of adverse effects and 3 of 105 cases of defined serotonin syndrome.13 Most moclobemide-SSRI interactions—including fatalities—involved overdoses in suicide attempts, although toxic symptoms have been reported with clomipramine or meperidine taken at normal dosages.14,15
In one study,16 18 healthy controls received fluoxetine, 20 to 40 mg/d, for 23 days, then were given moclobemide, up to 600 mg/d, or placebo and observed for adverse effects. No indication of serotonin syndrome was observed.
Linezolid is an oxazolidinone antibiotic with relatively weak, nonspecific, but reversible MAO inhibition. Cases of potential serotonin syndrome have been reported with linezolid plus paroxetine17 or sertraline.18 Patients in each case were medically ill and taking several other medications, which complicates interpretation of these reports. Nonetheless, physicians should be aware of the potential risk of serotonin syndrome if this antibiotic is combined with serotonergic agents.
Table 2
Serotonergic agents and their actions
| Actions | Agents |
|---|---|
| Inhibit serotonin reuptake | Fluoxetine, sertraline, citalopram, escitalopram, paroxetine, clomipramine, venlafaxine, fluvoxamine, tramadol, trazodone, nefazodone, tricyclic antidepressants, amphetamine, cocaine, dextromethorphan, meperidine, St. John’s wort |
| Increases serotonin synthesis | Tryptophan |
| Inhibit serotonin metabolism | Phenelzine, tranylcypromine, isocarboxazid, selegiline (deprenyl), linezolid, moclobemide |
| Increase serotonin release | MDMA (“Ecstasy”), amphetamine, cocaine, fenfluramine |
| Increase serotonin activity | Lithium, ECT |
| Serotonin receptor agonists | Buspirone, sumatriptan and other “triptans” used for migraine |
Atypical antipsychotics. Original diagnostic criteria for serotonin syndrome excluded the addition of, or increase in, an antipsychotic prior to the syndrome’s onset.1 However, serotonin syndrome has been reported with combinations of risperidone with paroxetine,19 olanzapine with mirtazapine and tramadol,20 and olanzapine with lithium and citalopram.21 The 5HT2 antagonist effect of these atypical antipsychotics may have led indirectly to overactivation of 5HT1a receptors and serotonin syndrome. In each case, neuroleptic malignant syndrome was ruled out.
Table 3
Signs and symptoms that differentiate 5 hyperthermic states
| Hyperthermic state | Symptoms/signs | Lab findings | Cause |
|---|---|---|---|
| Serotonin syndrome | Typically rapid onset with hyperreflexia, tremors, myoclonus, diaphoresis, confusion, agitation, or shivering; muscular rigidity not invariably present | Nonspecific | Increased serotonergic tone |
| Neuroleptic malignant syndrome | Variable rapidity of onset; severe muscular rigidity, diaphoresis, delirium, fluctuating blood pressure, tachycardia, extrapyramidal symptoms | Elevated CPK, leukocytosis | Blockade of dopamine receptors or abrupt withdrawal of a dopamine agonist |
| Lethal catatonia | Muscular rigidity, diaphoresis, delirium, alternating extreme excitement and stupor, tremors, hypertension | Nonspecific | Evidence of pre-existing psychosis (bipolar disorder, schizophrenia) |
| Anticholinergic toxicity | Hot, dry skin, pupillary dilatation, tachycardia, constipation, urinary retention, confusion, hallucinations, muscular relaxation | Nonspecific | Agents that block central and peripheral muscarinic cholinergic receptors |
| Malignant hyperthermia | Rapid onset, severe muscular rigidity, ischemia, hypotension | Elevated CPK, potassium, magnesium; DIC; acidosis; rhabdomyolysis | Inherited disorder with onset after exposure to anesthetic agents that block the neuromuscular junction |
| CPK: creatine phosphokinase | |||
| DIC: disseminated intravascular coagulation | |||
Tramadol is an analgesic with opioid and serotonin-reuptake inhibiting properties that is metabolized by the cytochrome P (CYP)-450 isoenzyme 2D6. Serotonin syndrome has been reported from interactions between tramadol and sertraline22 and fluoxetine.23 Possible causes include SSRI inhibition of CYP 2D6 metabolism of tramadol, tramadol abuse,23 and multiple coadministered medications.22
Sumatriptan is one of the selective 5HT1D agonists used in treating migraine. Gardner and Lynd24 concluded that most patients tolerate sumatriptan with SSRIs or lithium. They felt they could not ensure the safety of sumatriptan with MAOIs, however, because sumatriptan elimination depends on hepatic MAO activity.
Among the 5HT1D agonists, using sumatriptan, zolmitriptan, rizatriptan, or almotriptan with an MAOI or within 2 weeks of discontinuing an MAOI is contraindicated. Naratriptan and frovatriptan appear less likely to interact with MAOIs, based on FDA-approved labeling.
MDMA. 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) is widely used as a recreational drug, especially at crowded dances (“raves”) and with other drugs.25 This illicit amphetamine derivative stimulates the release of serotonin and inhibits its reuptake.
Kaskey reported the rapid onset of serotonin syndrome when a patient taking lithium and phenelzine ingested MDMA.26 Signs and symptoms of serotonin syndrome also may develop when MDMA is used alone, facilitated by the high ambient temperatures on crowded dance floors and the dancers’ relative dehydration.
Fatalities have been blamed on complications including disseminated intravascular coagulation, rhabdomyolysis, and acute hepatic, renal, or cardiac failure.25 Cases are difficult to interpret because of uncertainty about whether the victim ingested MDMA or another agent or combination.
St. John’s wort (Hypericum perforatum) contains numerous constituents, including hypericin and hyperforin, which have been found to inhibit the synaptic uptake of monoamines, including serotonin.27 Which constituents are responsible for its clinical effect is unclear. Adverse effects from monotherapy include GI symptoms, confusion, dry mouth, dizziness, headache, fatigue, allergic skin reactions, photosensitivity, and urinary frequency.27
Several cases of purported serotonin syndrome have been associated with St. John’s wort alone28 or in combination with SSRIs, nefazodone, or fenfluramine.29,30 GI symptoms and anxiety were the primary complaints and resolved without complications (adjunctive cyproheptadine was prescribed in two cases, though it is not clear that this agent contributed to resolution).
MISCELLANEOUS COMBINATIONS
Antiretroviral therapy. Five cases of serotonin syndrome were reported in HIV-infected patients taking fluoxetine with antiretroviral therapy.31 In particular, the use or addition of ritonavir—a potent CYP 2D6 inhibitor—was implicated, though saquinavir, efavirenz, or grapefruit juice (all primarily CYP 3A4 inhibitors) were also used, suggesting that pharmacokinetic interactions increased serotonergic stimulation. All five patients were taking multiple additional medications and had complex medical and/or psychiatric histories. Reducing SSRI dosages by one-half when used with ritonavir has been recommended to minimize adverse effects from a pharmacokinetic interaction.
Erythromycin was reported to induce serotonin syndrome in a 12-year-old boy when added to ongoing treatment with sertraline, an effect believed to be secondary to CYP 3A4 inhibition of sertraline metabolism.32
Mirtazapine was reported to induce serotonin syndrome in an elderly man 8 days after it was added to a regimen he had been taking for several years to treat chronic obstructive pulmonary disease.33 Serotonin syndrome also developed in a 12-year-old boy with Ewing’s sarcoma when the 5HT3 antagonist ondansetron was added to mirtazapine and morphine34 and in an 11-year-old girl with acute lymphoblastic leukemia when ondansetron was added to fentanyl. Interestingly, another report35 suggested using mirtazapine to treat serotonin syndrome caused by serotonergic antagonist effects.
Reports have associated the following combinations with serotonin syndrome, perhaps as the result of pharmacodynamic and/or pharmacokinetic interactions:
- paroxetine plus dextromethorphan and pseudoephedrine
- paroxetine plus nefazodone
- fluoxetine plus clomipramine and buspirone
- fluvoxamine plus buspirone
- fluoxetine plus buspirone
- amitriptyline plus meperidine and venlafaxine
- venlafaxine and dextroamphetamine
- fluoxetine plus clomipramine.
Table 4
Clinical signs that distinguish hyperthermic states
| Signs | Possible diagnosis |
|---|---|
| Prominent muscular rigidity | Neuroleptic malignant syndrome, malignant hyperthermia, catatonia |
| Myoclonus/hyperreflexia | Serotonin syndrome |
| Diaphoresis | Serotonin syndrome, neuroleptic malignant syndrome, catatonia |
| Hot dry skin | Anticholinergic toxicity |
| Elevated creatine phosphokinase | Neuroleptic malignant syndrome, malignant hyperthermia |
| Family history of anesthetic-induced hyperthermia | Malignant hyperthermia |
HOW TO RECOGNIZE SEROTONIN SYNDROME
Signs and symptoms of serotonin syndrome can overlap with those seen in neuroleptic malignant syndrome, lethal catatonia, malignant hyperthermia, and anticholinergic toxicity (Table 3),1,36,37 particularly with fever or hyperthermia (>40.5 °C, 105 °F). Fink37 has opined that acute neurotoxic syndromes such as serotonin syndrome and neuroleptic malignant syndrome also meet criteria for catatonia and are therefore subtypes of catatonia. The types of drugs involved and clinical findings can help distinguish the various hyperthermic states (Table 4).
As mentioned above, original diagnostic criteria for serotonin syndrome excluded the addition of, or increase in, an antipsychotic agent. This exclusion was intended to avoid confusion between serotonin syndrome and neuroleptic malignant syndrome. Co-administering antipsychotic and serotonergic agents requires heightened awareness for both neurotoxic syndromes.
TREATING MILD TO SEVERE CASES
If a patient develops serotonin syndrome, immediately discontinue the suspected agent(s) and observe carefully. In most cases, serotonin syndrome will resolve within 24 hours.
In mild cases, lorazepam, 1 to 2 mg slow IV push every 30 minutes until excessive sedation develops, may help. In moderate to severe cases, agents that block serotonin’s action are recommended,2 including:
- cyproheptadine (4 mg po every 4 hours as needed, up to 20 mg in 24 hours)
- propranolol (1 to 3 mg IV every 5 minutes, up to 0.1 mg/kg).
Case reports attest to these agents’ potential benefit. Other clinicians have reported using mirtazapine,35 nitroglycerin,38 and chlorpromazine.1
Serotonin syndrome symptoms resolved within minutes when IV nitroglycerin was used in a patient with serotonin syndrome and cardiac ischemia. The authors hypothesized that nitroglycerin, via nitric acid, provided an “off” signal for serotonin, though they did not advocate this as a routine treatment.38
The rationale for using chlorpromazine is its potential to block serotonin receptors. I would avoid the routine use of any antipsychotic agent in this setting, however, to minimize the risk of neuroleptic malignant syndrome.
Severe cases. Intensive care observation and treatment is required for patients with severe serotonin syndrome, including evidence of hyperthermia, DIC, rhabdomyolysis, renal failure, or aspiration. In cases of hyperthermia, supportive measures and standard treatments include muscle relaxants, cooling, and endotracheal intubation.
Severe complications are most likely with interactions between MAOIs and serotonergic agents, especially in overdose. Therefore, using such combinations requires close observation.
Related resources
- Di Rosa AE, Morgante L, Spina E et al. Epidemiology and pathoetiology of neurological syndromes with hyperthermia. Funct Neurol 1995;10:111-19.
- Radomski, JW, Dursun SM, Reveley MA, et al. An exploratory approach to the serotonin syndrome: an update of clinical phenomenology and revised diagnostic criteria. Med Hypothesis 2000;55: 218-24.
- Lane R, Baldwin D. Selective serotonin reuptake inhibitor-induced serotonin syndrome: review. J Clin Psychopharmacol 1997;17:208-21.
Drug brand names
- Almotriptan • Axert
- Amitriptyline • Elavil
- Buspirone • Buspar
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Cyproheptadine • Periactin
- Dextroamphetamine • Dexedrine
- Dextromethorphan • Delsym
- Efavirenz • Sustiva
- Escitalopram • Lexapro
- Fenfluramine • Pondimin
- Fentanyl • Sublimaze
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Isocarboxazid • Marplan
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Moclobemide • Aurorix
- Nortriptyline • Pamelor
- Naratriptan, • Amerge
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Ondansetron • Zofran
- Paroxetine • Paxil
- Phenelzine • Nardil
- Propranolol • Inderal
- Risperidone • Risperdal
- Ritonavir • Norvir
- Rizatriptan • Maxalt
- Saquinavir • Invirase
- Selegiline • Eldepryl
- Sertraline • Zoloft
- Sumatriptan • Imitrex
- Tramadol • Ultram
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Zolmitriptan • Zomig
Disclosure
Dr. Sternbach receives research grants from Otsuka America Pharmaceuticals and Eli Lilly and Co. and owns stock in Merck & Co., Pfizer Inc., and Johnson & Johnson.
Promptly identifying serotonin syndrome and acting decisively can keep side effects at the mild end of the spectrum. Symptoms of this potentially dangerous syndrome range from minimal in patients starting selective serotonin reuptake inhibitors (SSRIs) to fatal in those combining monoamine oxidase inhibitors (MAOIs) with serotonergic agents.
This article presents the latest evidence on how to:
- reduce the risk of serotonin syndrome
- recognize its symptoms
- and treat patients with mild to life-threatening symptoms.
WHAT IS SEROTONIN SYNDROME?
Serotonin syndrome is characterized by changes in autonomic, neuromotor, and cognitive-behavioral function (Table 1) triggered by increased serotonergic stimulation. It typically results from pharmacodynamic and/or pharmacokinetic interactions between drugs that increase serotonin activity.1,2
Table 1
How to recognize serotonin syndrome
| System | Clinical signs and symptoms |
|---|---|
| Autonomic | Diaphoresis, hyperthermia, hypertension, tachycardia, pupillary dilatation, nausea, diarrhea, shivering |
| Neuromotor | Hyperreflexia, myoclonus, restlessness, tremor, incoordination, rigidity, clonus, teeth chattering, trismus, seizures |
| Cognitive-behavioral | Confusion, agitation, anxiety, hypomania, insomnia, hallucinations, headache |
The syndrome was first identified in animal studies, followed by case reports in humans. The first review—with suggested diagnostic criteria— was published in 1991.1
Since then, case reports have described serotonin syndrome with many drug combinations, including nonpsychotropics and illicit drugs. Using an irreversible MAOI with a serotonergic agent is the most toxic reported combination, but any drug or combination that increases serotonin can, in theory, cause serotonin syndrome (Table 2). A clinical scale3 is being developed to define and identify this potentially dangerous state, but no consensus has emerged on diagnostic criteria.
Pathophysiology. Serotonin syndrome’s symptoms and signs appear to result from stimulation of specific central and peripheral serotonin receptors, especially 5HT1a and 5HT2. Others—such as 5HT3 and 5HT4—may also be involved in causing GI symptoms and may affect dopaminergic transmission.
Damaged vascular or pulmonary endothelium, atherosclerosis, hypertension, or hypercholesterolemia may increase the risk for serotonin syndrome. In patients with these common medical conditions, reduced endothelial MAO-A activity or reduced ability to secrete endothelium-derived nitric oxide may diminish the ability to metabolize serotonin.2
POTENTIALLY DANGEROUS COMBINATIONS
MAOIs. Serotonin syndrome has been reported as a result of interactions between MAOIs— including selegiline and reversible MAO-A inhibitors (RIMAs)—and various serotonergic compounds. These reports have included fatalities,4 some of which were preceded by severe hyperthermia with complications such as disseminated intravascular coagulation, rhabdomyolysis, and renal failure. Some cases resulted from overdoses, but others did not.
Most disturbingly, some cases occurred after patients had undergone the traditional 2-week washout from the MAOI and then took a serotonergic agent.5-7 In one instance,8 a patient who had discontinued fluoxetine for 6 weeks developed serotonin syndrome after starting tranylcypromine. These cases remind us to be vigilant when switching patients from irreversible MAOIs to serotonergic antidepressants or vice versa—even when recommended wash-out times are observed—and not to combine these agents acutely.
Selegiline is a relatively selective MAO-B inhibitor when used at 5 to 10 mg/d to treat Parkinson’s disease, though it loses MAO-B selectivity when used at higher dosages to treat depression. In a study9 of 4,568 patients with Parkinson’s disease who received selegiline (in dosages selective for MAO-B) plus an antidepressant:
- 11 (0.24%) experienced symptoms “possibly” consistent with serotonin syndrome
- 2 others (0.04%) experienced serious serotonin syndrome symptoms.9
Serotonin syndrome has been reported when MAO-B-selective doses of selegiline were combined with meperidine10 and nortriptyline.11 This underscores the need for caution when combining these agents, especially if transdermal selegiline— which would not be MAO-B-selective—becomes available for treating depression.
Moclobemide is a RIMA used in treating depression and anxiety, with a purported reduced risk of drug and food interactions compared with other MAOIs. Moclobemide is not approved in the United States, but some patients obtain it elsewhere.
Joffe and Bakish reported on safely combining moclobemide with SSRIs,12 and a review of MAOIs—including RIMAs—indicated that moclobemide was involved in only 9 of 226 cases of adverse effects and 3 of 105 cases of defined serotonin syndrome.13 Most moclobemide-SSRI interactions—including fatalities—involved overdoses in suicide attempts, although toxic symptoms have been reported with clomipramine or meperidine taken at normal dosages.14,15
In one study,16 18 healthy controls received fluoxetine, 20 to 40 mg/d, for 23 days, then were given moclobemide, up to 600 mg/d, or placebo and observed for adverse effects. No indication of serotonin syndrome was observed.
Linezolid is an oxazolidinone antibiotic with relatively weak, nonspecific, but reversible MAO inhibition. Cases of potential serotonin syndrome have been reported with linezolid plus paroxetine17 or sertraline.18 Patients in each case were medically ill and taking several other medications, which complicates interpretation of these reports. Nonetheless, physicians should be aware of the potential risk of serotonin syndrome if this antibiotic is combined with serotonergic agents.
Table 2
Serotonergic agents and their actions
| Actions | Agents |
|---|---|
| Inhibit serotonin reuptake | Fluoxetine, sertraline, citalopram, escitalopram, paroxetine, clomipramine, venlafaxine, fluvoxamine, tramadol, trazodone, nefazodone, tricyclic antidepressants, amphetamine, cocaine, dextromethorphan, meperidine, St. John’s wort |
| Increases serotonin synthesis | Tryptophan |
| Inhibit serotonin metabolism | Phenelzine, tranylcypromine, isocarboxazid, selegiline (deprenyl), linezolid, moclobemide |
| Increase serotonin release | MDMA (“Ecstasy”), amphetamine, cocaine, fenfluramine |
| Increase serotonin activity | Lithium, ECT |
| Serotonin receptor agonists | Buspirone, sumatriptan and other “triptans” used for migraine |
Atypical antipsychotics. Original diagnostic criteria for serotonin syndrome excluded the addition of, or increase in, an antipsychotic prior to the syndrome’s onset.1 However, serotonin syndrome has been reported with combinations of risperidone with paroxetine,19 olanzapine with mirtazapine and tramadol,20 and olanzapine with lithium and citalopram.21 The 5HT2 antagonist effect of these atypical antipsychotics may have led indirectly to overactivation of 5HT1a receptors and serotonin syndrome. In each case, neuroleptic malignant syndrome was ruled out.
Table 3
Signs and symptoms that differentiate 5 hyperthermic states
| Hyperthermic state | Symptoms/signs | Lab findings | Cause |
|---|---|---|---|
| Serotonin syndrome | Typically rapid onset with hyperreflexia, tremors, myoclonus, diaphoresis, confusion, agitation, or shivering; muscular rigidity not invariably present | Nonspecific | Increased serotonergic tone |
| Neuroleptic malignant syndrome | Variable rapidity of onset; severe muscular rigidity, diaphoresis, delirium, fluctuating blood pressure, tachycardia, extrapyramidal symptoms | Elevated CPK, leukocytosis | Blockade of dopamine receptors or abrupt withdrawal of a dopamine agonist |
| Lethal catatonia | Muscular rigidity, diaphoresis, delirium, alternating extreme excitement and stupor, tremors, hypertension | Nonspecific | Evidence of pre-existing psychosis (bipolar disorder, schizophrenia) |
| Anticholinergic toxicity | Hot, dry skin, pupillary dilatation, tachycardia, constipation, urinary retention, confusion, hallucinations, muscular relaxation | Nonspecific | Agents that block central and peripheral muscarinic cholinergic receptors |
| Malignant hyperthermia | Rapid onset, severe muscular rigidity, ischemia, hypotension | Elevated CPK, potassium, magnesium; DIC; acidosis; rhabdomyolysis | Inherited disorder with onset after exposure to anesthetic agents that block the neuromuscular junction |
| CPK: creatine phosphokinase | |||
| DIC: disseminated intravascular coagulation | |||
Tramadol is an analgesic with opioid and serotonin-reuptake inhibiting properties that is metabolized by the cytochrome P (CYP)-450 isoenzyme 2D6. Serotonin syndrome has been reported from interactions between tramadol and sertraline22 and fluoxetine.23 Possible causes include SSRI inhibition of CYP 2D6 metabolism of tramadol, tramadol abuse,23 and multiple coadministered medications.22
Sumatriptan is one of the selective 5HT1D agonists used in treating migraine. Gardner and Lynd24 concluded that most patients tolerate sumatriptan with SSRIs or lithium. They felt they could not ensure the safety of sumatriptan with MAOIs, however, because sumatriptan elimination depends on hepatic MAO activity.
Among the 5HT1D agonists, using sumatriptan, zolmitriptan, rizatriptan, or almotriptan with an MAOI or within 2 weeks of discontinuing an MAOI is contraindicated. Naratriptan and frovatriptan appear less likely to interact with MAOIs, based on FDA-approved labeling.
MDMA. 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) is widely used as a recreational drug, especially at crowded dances (“raves”) and with other drugs.25 This illicit amphetamine derivative stimulates the release of serotonin and inhibits its reuptake.
Kaskey reported the rapid onset of serotonin syndrome when a patient taking lithium and phenelzine ingested MDMA.26 Signs and symptoms of serotonin syndrome also may develop when MDMA is used alone, facilitated by the high ambient temperatures on crowded dance floors and the dancers’ relative dehydration.
Fatalities have been blamed on complications including disseminated intravascular coagulation, rhabdomyolysis, and acute hepatic, renal, or cardiac failure.25 Cases are difficult to interpret because of uncertainty about whether the victim ingested MDMA or another agent or combination.
St. John’s wort (Hypericum perforatum) contains numerous constituents, including hypericin and hyperforin, which have been found to inhibit the synaptic uptake of monoamines, including serotonin.27 Which constituents are responsible for its clinical effect is unclear. Adverse effects from monotherapy include GI symptoms, confusion, dry mouth, dizziness, headache, fatigue, allergic skin reactions, photosensitivity, and urinary frequency.27
Several cases of purported serotonin syndrome have been associated with St. John’s wort alone28 or in combination with SSRIs, nefazodone, or fenfluramine.29,30 GI symptoms and anxiety were the primary complaints and resolved without complications (adjunctive cyproheptadine was prescribed in two cases, though it is not clear that this agent contributed to resolution).
MISCELLANEOUS COMBINATIONS
Antiretroviral therapy. Five cases of serotonin syndrome were reported in HIV-infected patients taking fluoxetine with antiretroviral therapy.31 In particular, the use or addition of ritonavir—a potent CYP 2D6 inhibitor—was implicated, though saquinavir, efavirenz, or grapefruit juice (all primarily CYP 3A4 inhibitors) were also used, suggesting that pharmacokinetic interactions increased serotonergic stimulation. All five patients were taking multiple additional medications and had complex medical and/or psychiatric histories. Reducing SSRI dosages by one-half when used with ritonavir has been recommended to minimize adverse effects from a pharmacokinetic interaction.
Erythromycin was reported to induce serotonin syndrome in a 12-year-old boy when added to ongoing treatment with sertraline, an effect believed to be secondary to CYP 3A4 inhibition of sertraline metabolism.32
Mirtazapine was reported to induce serotonin syndrome in an elderly man 8 days after it was added to a regimen he had been taking for several years to treat chronic obstructive pulmonary disease.33 Serotonin syndrome also developed in a 12-year-old boy with Ewing’s sarcoma when the 5HT3 antagonist ondansetron was added to mirtazapine and morphine34 and in an 11-year-old girl with acute lymphoblastic leukemia when ondansetron was added to fentanyl. Interestingly, another report35 suggested using mirtazapine to treat serotonin syndrome caused by serotonergic antagonist effects.
Reports have associated the following combinations with serotonin syndrome, perhaps as the result of pharmacodynamic and/or pharmacokinetic interactions:
- paroxetine plus dextromethorphan and pseudoephedrine
- paroxetine plus nefazodone
- fluoxetine plus clomipramine and buspirone
- fluvoxamine plus buspirone
- fluoxetine plus buspirone
- amitriptyline plus meperidine and venlafaxine
- venlafaxine and dextroamphetamine
- fluoxetine plus clomipramine.
Table 4
Clinical signs that distinguish hyperthermic states
| Signs | Possible diagnosis |
|---|---|
| Prominent muscular rigidity | Neuroleptic malignant syndrome, malignant hyperthermia, catatonia |
| Myoclonus/hyperreflexia | Serotonin syndrome |
| Diaphoresis | Serotonin syndrome, neuroleptic malignant syndrome, catatonia |
| Hot dry skin | Anticholinergic toxicity |
| Elevated creatine phosphokinase | Neuroleptic malignant syndrome, malignant hyperthermia |
| Family history of anesthetic-induced hyperthermia | Malignant hyperthermia |
HOW TO RECOGNIZE SEROTONIN SYNDROME
Signs and symptoms of serotonin syndrome can overlap with those seen in neuroleptic malignant syndrome, lethal catatonia, malignant hyperthermia, and anticholinergic toxicity (Table 3),1,36,37 particularly with fever or hyperthermia (>40.5 °C, 105 °F). Fink37 has opined that acute neurotoxic syndromes such as serotonin syndrome and neuroleptic malignant syndrome also meet criteria for catatonia and are therefore subtypes of catatonia. The types of drugs involved and clinical findings can help distinguish the various hyperthermic states (Table 4).
As mentioned above, original diagnostic criteria for serotonin syndrome excluded the addition of, or increase in, an antipsychotic agent. This exclusion was intended to avoid confusion between serotonin syndrome and neuroleptic malignant syndrome. Co-administering antipsychotic and serotonergic agents requires heightened awareness for both neurotoxic syndromes.
TREATING MILD TO SEVERE CASES
If a patient develops serotonin syndrome, immediately discontinue the suspected agent(s) and observe carefully. In most cases, serotonin syndrome will resolve within 24 hours.
In mild cases, lorazepam, 1 to 2 mg slow IV push every 30 minutes until excessive sedation develops, may help. In moderate to severe cases, agents that block serotonin’s action are recommended,2 including:
- cyproheptadine (4 mg po every 4 hours as needed, up to 20 mg in 24 hours)
- propranolol (1 to 3 mg IV every 5 minutes, up to 0.1 mg/kg).
Case reports attest to these agents’ potential benefit. Other clinicians have reported using mirtazapine,35 nitroglycerin,38 and chlorpromazine.1
Serotonin syndrome symptoms resolved within minutes when IV nitroglycerin was used in a patient with serotonin syndrome and cardiac ischemia. The authors hypothesized that nitroglycerin, via nitric acid, provided an “off” signal for serotonin, though they did not advocate this as a routine treatment.38
The rationale for using chlorpromazine is its potential to block serotonin receptors. I would avoid the routine use of any antipsychotic agent in this setting, however, to minimize the risk of neuroleptic malignant syndrome.
Severe cases. Intensive care observation and treatment is required for patients with severe serotonin syndrome, including evidence of hyperthermia, DIC, rhabdomyolysis, renal failure, or aspiration. In cases of hyperthermia, supportive measures and standard treatments include muscle relaxants, cooling, and endotracheal intubation.
Severe complications are most likely with interactions between MAOIs and serotonergic agents, especially in overdose. Therefore, using such combinations requires close observation.
Related resources
- Di Rosa AE, Morgante L, Spina E et al. Epidemiology and pathoetiology of neurological syndromes with hyperthermia. Funct Neurol 1995;10:111-19.
- Radomski, JW, Dursun SM, Reveley MA, et al. An exploratory approach to the serotonin syndrome: an update of clinical phenomenology and revised diagnostic criteria. Med Hypothesis 2000;55: 218-24.
- Lane R, Baldwin D. Selective serotonin reuptake inhibitor-induced serotonin syndrome: review. J Clin Psychopharmacol 1997;17:208-21.
Drug brand names
- Almotriptan • Axert
- Amitriptyline • Elavil
- Buspirone • Buspar
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clomipramine • Anafranil
- Cyproheptadine • Periactin
- Dextroamphetamine • Dexedrine
- Dextromethorphan • Delsym
- Efavirenz • Sustiva
- Escitalopram • Lexapro
- Fenfluramine • Pondimin
- Fentanyl • Sublimaze
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Isocarboxazid • Marplan
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Moclobemide • Aurorix
- Nortriptyline • Pamelor
- Naratriptan, • Amerge
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Ondansetron • Zofran
- Paroxetine • Paxil
- Phenelzine • Nardil
- Propranolol • Inderal
- Risperidone • Risperdal
- Ritonavir • Norvir
- Rizatriptan • Maxalt
- Saquinavir • Invirase
- Selegiline • Eldepryl
- Sertraline • Zoloft
- Sumatriptan • Imitrex
- Tramadol • Ultram
- Tranylcypromine • Parnate
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Zolmitriptan • Zomig
Disclosure
Dr. Sternbach receives research grants from Otsuka America Pharmaceuticals and Eli Lilly and Co. and owns stock in Merck & Co., Pfizer Inc., and Johnson & Johnson.
1. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148:705-13.
2. Brown TM, Skop BP, Mareth TR. Pathophysiology and management of the serotonin syndrome. Ann Pharmacother 1996;30:527-33.
3. Hegerl U, Bottlender R, Gallinat J, et al. The serotonin syndrome scale: first results on validity. Eur Arch Psychiatry Clin Neurosci 1998;248:96-103.
4. Beasley CM, Jr, Masica DN, Heiligenstein JH, et al. Possible monoamine-oxidase inhibitor-serotonin uptake inhibitor interaction: fluoxetine clinical data and pre-clinical findings. J Clin Psychopharmacol 1993;13:312-20.
5. Ruiz F. Fluoxetine and the serotonin syndrome. Ann Emerg Med 1994;34:983-5.
6. Gitlin MJ. Venlafaxine, monoamine oxidase inhibitors and the serotonin syndrome. J Clin Psychopharmacol 1997;17:66-7.
7. Kolecki P. Venlafaxine induced serotonin syndrome occurring after abstinence from phenelzine for more than two weeks. Clin Toxicol 1997;35:211-12.
8. Coplan JD, Gorman JM. Detectable levels of fluoxetine metabolites after discontinuation: an unexpected serotonin syndrome. Am J Psychiatry 1993;15:837.-
9. Richard IH, Kurlan R, Tanner C, et al. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson’s disease. Neurology 1997;48:1070-7.
10. Zornberg GL, Bodkin JA, Cohen BM. Severe adverse interaction between pethidine and selegiline. Lancet 1991;337:246.-
11. Hinds NP, Hillier CE, Wiles CM. Possible serotonin syndrome arising from an interaction between nortriptyline and selegiline in a lady with parkinsonism. J Neurol 2000;247:811.-
12. Joffe RT, Bakish D. Combined SSRI-moclobemide treatment of psychiatric illness. J Clin Psychiatry 1994;55:24-5.
13. Hilton SE, Maradit H, Moller HJ. Serotonin syndrome and drug combinations: focus on MAOI and RIMA. Eur Arch Psychiatry Clin Neurosci 1997;247:113-19.
14. Dardennes RM, Even C, Ballon N, et al. Serotonin syndrome caused by a clomipramine-moclobemide interaction. J Clin Psychiatry 1998;59:382-3.
15. Gillman PK. Possible serotonin syndrome with moclobemide and pethidine. Med J Aust 1995;162:554.-
16. Dingemanse J, Wallnofer A, Gieschke R, et al. Pharmacokinetic and pharmacodynamic interaction between fluoxetine and moclobemide in the investigation of development of the “serotonin syndrome.” Clin Pharmacol Ther 1998;63:403-13.
17. Wigen CL, Goetz MB. Serotonin syndrome and linezolid. Clin Infect Dis 2002;34:1651-2.
18. Lavery S, Ravi H, McDaniel WW, et al. Linezolid and serotonin syndrome. Psychosomatics 2001;42:432-4.
19. Hamilton S, Malone K. Serotonin syndrome during treatment with paroxetine and risperidone. J Clin Psychopharmacol 2000;20:103-5.
20. Duggal HS, Fetchko J. Serotonin syndrome and atypical antipsychotics. Am J Psychiatry 2002;159:672-3.
21. Haslett CD, Kumar S. Can olanzapine be implicated in causing serotonin syndrome? Psychiatry Clin Neurosci 2002;56:533-6.
22. Mason BJ, Blackburn KH. Possible serotonin syndrome associated with tramadol and sertraline coadministration. Ann Pharmacother 1997;31:175-7.
23. Lange-Asschenfeldt C, Weigmann H, Hiemke C, et al. Serotonin syndrome as a result of fluoxetine in a patient with tramadol abuse: plasma level-correlated symptomatology. J Clin Psychopharmacol 2002;22:440-1.
24. Gardner DM, Lynd LD. Sumatriptan contraindications and the serotonin syndrome. Ann Pharmacother 1998;32:33-8.
25. Parrott AC. Recreational Ecstasy/MDMA, the serotonin syndrome and serotonergic neurotoxicity. Pharmacol Biochem Behav 2002;71:837-44.
26. Kaskey GB. Possible interaction between an MAOI and “Ecstasy.” Am J Psychiatry 1992;149:411-12.
27. De Smet PA. Herbal remedies. N Engl J Med 2002;347:2046-56.
28. Parker V, Wong AH, Boon HS, et al. Adverse reactions to St. John’s wort. Can J Psychiatry 2001;46:77-9.
29. Lantz MS, Buchalter E, Giambanco V. St. John’s wort and antidepressant drug interactions in the elderly. J Geriatr Psychiatry Neurol 1999;12:7-10.
30. Beckman SE, Sommi RW, Switzer J. Consumer use of St. John’s wort: a survey of effectiveness, safety, and tolerability. Pharmacotherapy 2000;20:568-74.
31. De Silva KE, Le Flore DB, Marston BJ, et al. Serotonin syndrome in HIV-infected individuals receiving antiretroviral therapy and fluoxetine. AIDS 2001;15:1281-5.
32. Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999;19:894-6.
33. Hernandez JL, Ramos FJ, Infante J, et al. Severe serotonin syndrome induced by mirtazapine monotherapy. Ann Pharmacother 2002;36:641-3.
34. Turkel SB, Nadala JGB, Wincor MZ. Possible serotonin syndrome in association with 5HT3 antagonist agents. Psychosomatics 2001;42:258-60.
35. Hoes MJ, Zeijpveld JH. Mirtazapine as treatment for serotonin syndrome. Pharmacopsychiatry 1996;29:81.-
36. Theoharides TC, Harris RS, Weckstein D. Neuroleptic malignant-like syndrome due to cyclobenzaprine? J Clin Psychopharmacol 1995;15:79-81.
37. Fink M. Toxic serotonin syndrome or neuroleptic malignant syndrome? Pharmacopsychiatry 1996;29:159-61.
38. Brown TM, Skop BP. Nitroglycerin in the treatment of the serotonin syndrome. Ann Pharmacother 1996;30:191-2.
1. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148:705-13.
2. Brown TM, Skop BP, Mareth TR. Pathophysiology and management of the serotonin syndrome. Ann Pharmacother 1996;30:527-33.
3. Hegerl U, Bottlender R, Gallinat J, et al. The serotonin syndrome scale: first results on validity. Eur Arch Psychiatry Clin Neurosci 1998;248:96-103.
4. Beasley CM, Jr, Masica DN, Heiligenstein JH, et al. Possible monoamine-oxidase inhibitor-serotonin uptake inhibitor interaction: fluoxetine clinical data and pre-clinical findings. J Clin Psychopharmacol 1993;13:312-20.
5. Ruiz F. Fluoxetine and the serotonin syndrome. Ann Emerg Med 1994;34:983-5.
6. Gitlin MJ. Venlafaxine, monoamine oxidase inhibitors and the serotonin syndrome. J Clin Psychopharmacol 1997;17:66-7.
7. Kolecki P. Venlafaxine induced serotonin syndrome occurring after abstinence from phenelzine for more than two weeks. Clin Toxicol 1997;35:211-12.
8. Coplan JD, Gorman JM. Detectable levels of fluoxetine metabolites after discontinuation: an unexpected serotonin syndrome. Am J Psychiatry 1993;15:837.-
9. Richard IH, Kurlan R, Tanner C, et al. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson’s disease. Neurology 1997;48:1070-7.
10. Zornberg GL, Bodkin JA, Cohen BM. Severe adverse interaction between pethidine and selegiline. Lancet 1991;337:246.-
11. Hinds NP, Hillier CE, Wiles CM. Possible serotonin syndrome arising from an interaction between nortriptyline and selegiline in a lady with parkinsonism. J Neurol 2000;247:811.-
12. Joffe RT, Bakish D. Combined SSRI-moclobemide treatment of psychiatric illness. J Clin Psychiatry 1994;55:24-5.
13. Hilton SE, Maradit H, Moller HJ. Serotonin syndrome and drug combinations: focus on MAOI and RIMA. Eur Arch Psychiatry Clin Neurosci 1997;247:113-19.
14. Dardennes RM, Even C, Ballon N, et al. Serotonin syndrome caused by a clomipramine-moclobemide interaction. J Clin Psychiatry 1998;59:382-3.
15. Gillman PK. Possible serotonin syndrome with moclobemide and pethidine. Med J Aust 1995;162:554.-
16. Dingemanse J, Wallnofer A, Gieschke R, et al. Pharmacokinetic and pharmacodynamic interaction between fluoxetine and moclobemide in the investigation of development of the “serotonin syndrome.” Clin Pharmacol Ther 1998;63:403-13.
17. Wigen CL, Goetz MB. Serotonin syndrome and linezolid. Clin Infect Dis 2002;34:1651-2.
18. Lavery S, Ravi H, McDaniel WW, et al. Linezolid and serotonin syndrome. Psychosomatics 2001;42:432-4.
19. Hamilton S, Malone K. Serotonin syndrome during treatment with paroxetine and risperidone. J Clin Psychopharmacol 2000;20:103-5.
20. Duggal HS, Fetchko J. Serotonin syndrome and atypical antipsychotics. Am J Psychiatry 2002;159:672-3.
21. Haslett CD, Kumar S. Can olanzapine be implicated in causing serotonin syndrome? Psychiatry Clin Neurosci 2002;56:533-6.
22. Mason BJ, Blackburn KH. Possible serotonin syndrome associated with tramadol and sertraline coadministration. Ann Pharmacother 1997;31:175-7.
23. Lange-Asschenfeldt C, Weigmann H, Hiemke C, et al. Serotonin syndrome as a result of fluoxetine in a patient with tramadol abuse: plasma level-correlated symptomatology. J Clin Psychopharmacol 2002;22:440-1.
24. Gardner DM, Lynd LD. Sumatriptan contraindications and the serotonin syndrome. Ann Pharmacother 1998;32:33-8.
25. Parrott AC. Recreational Ecstasy/MDMA, the serotonin syndrome and serotonergic neurotoxicity. Pharmacol Biochem Behav 2002;71:837-44.
26. Kaskey GB. Possible interaction between an MAOI and “Ecstasy.” Am J Psychiatry 1992;149:411-12.
27. De Smet PA. Herbal remedies. N Engl J Med 2002;347:2046-56.
28. Parker V, Wong AH, Boon HS, et al. Adverse reactions to St. John’s wort. Can J Psychiatry 2001;46:77-9.
29. Lantz MS, Buchalter E, Giambanco V. St. John’s wort and antidepressant drug interactions in the elderly. J Geriatr Psychiatry Neurol 1999;12:7-10.
30. Beckman SE, Sommi RW, Switzer J. Consumer use of St. John’s wort: a survey of effectiveness, safety, and tolerability. Pharmacotherapy 2000;20:568-74.
31. De Silva KE, Le Flore DB, Marston BJ, et al. Serotonin syndrome in HIV-infected individuals receiving antiretroviral therapy and fluoxetine. AIDS 2001;15:1281-5.
32. Lee DO, Lee CD. Serotonin syndrome in a child associated with erythromycin and sertraline. Pharmacotherapy 1999;19:894-6.
33. Hernandez JL, Ramos FJ, Infante J, et al. Severe serotonin syndrome induced by mirtazapine monotherapy. Ann Pharmacother 2002;36:641-3.
34. Turkel SB, Nadala JGB, Wincor MZ. Possible serotonin syndrome in association with 5HT3 antagonist agents. Psychosomatics 2001;42:258-60.
35. Hoes MJ, Zeijpveld JH. Mirtazapine as treatment for serotonin syndrome. Pharmacopsychiatry 1996;29:81.-
36. Theoharides TC, Harris RS, Weckstein D. Neuroleptic malignant-like syndrome due to cyclobenzaprine? J Clin Psychopharmacol 1995;15:79-81.
37. Fink M. Toxic serotonin syndrome or neuroleptic malignant syndrome? Pharmacopsychiatry 1996;29:159-61.
38. Brown TM, Skop BP. Nitroglycerin in the treatment of the serotonin syndrome. Ann Pharmacother 1996;30:191-2.
When patients can’t sleep: Practical guide to using and choosing hypnotic therapy
Careful investigation can often reveal insomnia’s cause1—whether a psychiatric or medical condition or poor sleep habits. Understanding why patients can’t sleep is key to effective therapy.
Acute and chronic sleep deprivation is associated with measurable declines in daytime performance (Box). Some data even suggest that long-term sleeplessness increases the risk of new psychiatric disorders—most notably major depression.3
PSYCHIATRIC DISORDERS AND INSOMNIA
Depression. Many depressed persons—up to 80%—experience insomnia, although no one sleep pattern seems typical.2 Depression may be associated with:
- difficulties in falling asleep
- interrupted nocturnal sleep
- and early morning awakening.
Anxiety disorders. Generalized anxiety disorder (GAD), social phobia, panic attacks, and posttraumatic stress disorder (PTSD) are all associated with disrupted sleep. Patients with GAD experience prolonged sleep latency (time needed to fall asleep after lights out) and fragmented sleep, similar to those with primary insomnia.
One-half of adult Americans experience insomnia during their lives, and 10% report persistent sleep difficulties (longer than 2 weeks). Individuals who complain of insomnia report:
- daytime drowsiness
- diminished memory and concentration
- depression
- strained relationships
- increased risk of accidents
- impaired job performance.
Despite these complaints, a surprising 70% of those with insomnia never seek medical help. Only 6% visit their physicians specifically for insomnia, and 24% address sleep difficulty as a secondary complaint. Many (40%) self-medicate with over-the-counter sleep aids or alcohol.2
Insomnia becomes more frequent with aging, associated with increased rates of medical and psychiatric illness and an age-related deterioration in the brain’s sleep-generating processes.3
Subjective sleep quality may be impaired in patients with social phobia. Some patients experience panic symptoms while sleeping, possibly in association with mild hypercapnia. Patients with sleep panic attacks tend to have earlier onset of panic disorder and a higher likelihood of comorbid mood and other anxiety disorders.4
In patients with PTSD, disturbed sleep continuity and increased REM phasic activity—such as eye movements—are directly correlated with severity of PTSD symptoms. Nightmares and disturbed REM sleep are hypothesized hallmarks of PTSD.5
Schizophrenia. Patients with schizophrenia often have disrupted sleep patterns. These include prolonged sleep latency, fragmented sleep with frequent arousals, decreased slow-wave sleep, variable REM latency, and decreased REM rebound after sleep deprivation. Despite investigations going back to the 1950s, no specific link between REM sleep and psychosis has been found.6 Interestingly, increases in REM sleep time and REM activity have been associated with an increased risk of suicide in patients with schizophrenia.7
Adjustment sleep disorder. Acute emotional stressors—such as bereavement, job loss, or hospitalization—often cause adjustment sleep disorder. Symptoms typically remit soon after the stressors abate, so this transient insomnia usually lasts a few days to a few weeks. Treatment with behavioral therapies and hypnotics8 is warranted if:
- sleepiness and fatigue interfere with daytime functioning
- a pattern of recurring episodes develops.9
Psychophysiologic insomnia. Once initiated—regardless of cause—insomnia may persist well after its precipitating factors resolve. Thus, short-term insomnia may develop into long-term, chronic difficulty with recurring episodes or a constant, daily pattern of insomnia. Sufferers often spend hours in bed awake focused upon—and brooding over—their sleeplessness. which in turn further aggravates their insomnia.
Adjustment sleep disorder and psychophysiologic insomnia are included within DSM-IV’s term “primary insomnia.”
OTHER CAUSES OF INSOMNIA
Medications that may affect sleep quality include antidepressants (Table 1),10,11 antihypertensives, antineoplastic agents, bronchodilators, stimulants, corticosteroids, decongestants, diuretics, histamine-2 receptor blockers, and smoking cessation aids.
Recreational drugs, such as cocaine, often cause insomnia. Hypnotics and anxiolytics can cause insomnia following long-term use and during withdrawal.
Other disorders known to disturb sleep include periodic limb movement disorder (PLMD), restless legs syndrome (RLS), sleep apnea syndrome, disrupted circadian rhythms (as with travel or shift work), cardiopulmonary disorders, chronic pain, diabetes, hyperthyroidism, hot flashes associated with menopause, seizures, dementia, and Parkinson’s disease, to name a few.
WORKUP OF SLEEP COMPLAINTS
Acute. Most short-term insomnias—lasting a few weeks or less—are caused by situational stressors, circadian rhythm alterations, and sleep hygiene violations. A logical initial approach, therefore, is to combine sleep hygiene measures with supportive psychotherapy. Hypnotic agents may be considered for apparent daytime consequences—such as sleepiness and occupational impairment—or if the insomnia seems to be escalating.
Chronic. For longer-term insomnias—lasting more than a few weeks—consider a more thorough evaluation, including medical and psychiatric history, physical examination, and mental status examination. Inquire about cardinal symptoms of disorders associated with insomnia, including:
- snoring or breathing pauses during sleep (sleep apnea syndrome)
- restlessness or twitching in the lower extremities (PLMS/RLS).
Question the bed partner, who may be more aware of such symptoms than the patient. Carefully review sleep patterns on weekdays and weekends, bedtime habits, sleep hygiene habits, and substance and medication use.
Table 1
Antidepressants’ effects on sleep and wakefulness
| Activating agents | Bupropion, protriptyline, most selective serotonin reuptake inhibitors, venlafaxine, monoamine oxidase inhibitors |
| Sedating agents | Amitriptyline, doxepin, trimipramine, nefazodone, trazodone, mirtazapine |
| Neutral agents | Citalopram, escitalopram |
Sleep clinic referrals. Consider an evaluation by a sleep disorders center when:
- the diagnosis remains unclear
- or treatment of the presumed conditions fails after a reasonable time
BEHAVIORAL TREATMENTS
Behavioral treatments—with or without hypnotics—are appropriate for a wide variety of insomnia complaints, including adjustment sleep disorder, psychophysiologic insomnia, and depression. Behavioral measures may take longer to implement than drug therapy, but their effects have been shown to last longer in patients with primary insomnia. In many cases, it may be useful to start with both hypnotic and behavioral treatments and withdraw the hypnotic after behavioral measures take effect.
Sleep hygiene. Many individuals unknowingly engage in habitual behaviors that impair sleep. Those with insomnia, for example, often try to compensate for lost sleep by staying in bed later in the morning or by napping, which further fragment nocturnal sleep. Advise these patients to adhere to a regular awakening time—regardless of how long they slept the night before—and to avoid naps. Other tips for getting a good night’s sleep are outlined in Table 2.12
Caffeine has a plasma half-life of 3 to 7 hours, although individual sensitivity varies widely and caffeine’s erratic absorption can prolong its effects. Advise patients with insomnia to avoid caffeine-containing beverages—including coffee, tea, and soft drinks—after noon.
Relaxation training. Muscle tension can be reduced through electromyography (EMG) biofeedback, abdominal breathing exercises, or progressive muscle relaxation techniques, among others. Relaxation training is usually effective within a few weeks.
Psychotherapy. Cognitive-behavioral therapy can help identify and dispel tension-producing thoughts that are disrupting sleep, such as preoccupation with unpleasant work experiences or school examinations. Reassurance may help patients overcome fears about sleeplessness; suggest that patients deal with anxiety-producing thoughts during therapy sessions and at times other than bedtime.
Insight-oriented psychotherapy may enhance patients’ awareness of psychological conflicts from their past that may be producing anxiety and contributing to sleeplessness.
PRESCRIBING HYPNOTICS
Sedative-hypnotics are indicated primarily for short-term management of insomnia. Most are used prophylactically at bedtime until insomnia dissipates or the physician advises the patient to take a break.
Treatment principles. Because many insomnias are recurrent, prolonged hypnotic treatment given in short bouts is often optimal. Longer treatment—months to years—is not recommended by standard textbooks but is clearly needed by a small number of patients with chronic insomnia. In these cases, carefully monitor for tolerance, as manifested by dosage escalation. Long-term hypnotic treatment is not suitable for patients with drug abuse or dependence histories.

Table 2
How to get a good night’s sleep
|
Although chloral hydrate and barbiturates are effective hypnotics, adverse effects limit their safety and usefulness. Benzodiazepines and more recently introduced agents have milder side effect profiles (Table 3). Choose agents based on the patient’s situation, preferences, and effects of prior trials with similar agents. Guidelines for hypnotics discourage chronic use to minimize abuse, misuse, and habituation (Table 4).
Elimination half-life is the primary pharmacokinetic property that differentiates the hypnotics from each other:13
- longer half-life: flurazepam, quazepam
- intermediate half-life: estazolam, temazepam
- short half-life: triazolam, zolpidem, zaleplon (Table 3).
Table 3
Actions and available doses of common hypnotics
| Class/drug | Onset of action | Half-life (hrs) | Active metabolites | Doses (mg) |
|---|---|---|---|---|
| Benzodiazepines | ||||
| Flurazepam | Rapid | 40 to 250 | Yes | 15, 30 |
| Quazepam | Rapid | 40 to 250 | Yes | 7.5, 15 |
| Estazolam | Rapid | 10 to 24 | Yes | 0.5, 1, 2 |
| Temazepam | Intermediate | 8 to 22 | No | 7.5, 15 |
| Triazolam | Rapid | <6 | No | 0.125, 0.25, 0.5 |
| Imidazopyridine | ||||
| Zolpidem | Rapid | 2.5 | No | 5, 10 |
| Pyrazolopyrimidine | ||||
| Zaleplon | Rapid | 1 | No | 5, 10, 20 |
Whereas benzodiazepines bind to benzodiazepine receptor types 1 and 2, zolpidem and zaleplon (and possibly quazepam) bind selectively to type 1. This selectivity may explain why zolpidem and zaleplon are more easily tolerated.
Hypnotic agents with relatively longer half-lives tend to be associated with greater potential for residual daytime effects such as sedation, motor incoordination, amnesia, and slowed reflexes. These effects may impair performance and increase the risk of auto accidents and injuries, especially hip fractures in the elderly.
Nonbenzodiazepines. Because of its ultra-short half-life, zaleplon is least likely to cause residual daytime effects when administered at bedtime. At 10-mg doses, its side effects seem to last no more than 4 hours following administration. Zaleplon can be safely taken after nocturnal awakenings if the patient remains in bed 4 hours or longer after taking it.14
Some patients feel that taking zaleplon only when needed allows them to use hypnotics more sparingly. On the other hand, zaleplon’s ultrashort half-life makes it less useful for patients who have frequent episodes of sleep-interruption insomnia every night. For them, a longer elimination half-life agent such as zolpidem may be more predictably effective for the entire night.15 Short half-life hypnotics have many advantages, but they do not offer anxiolysis for patients with daytime anxiety, as the longer half-life agents do.
Tolerance and rebound. Tolerance can develop following repeated dosing with benzodiazepines—primarily triazolam—and rebound insomnia can follow abrupt discontinuation. Despite widespread concerns, neither tolerance nor rebound insomnia has been well documented. Nevertheless, both can be minimized by using benzodiazepines at the lowest effective dosages and for brief periods. Gradual tapering when discontinuing the drug can help control rebound.
Tolerance and rebound seem to be less of a concern with the newer hypnotics than with benzodiazepines. In preliminary uncontrolled trials, zolpidem and zaleplon did not show evidence of these problems in 1 year of continued use.
NONHYPNOTIC SLEEP AIDS
Sedating antidepressants. Physicians often prescribe low doses of sedating antidepressants to control insomnia, a practice supported by some controlled clinical trials. For example, polysomnography showed that patients who took doxepin, 25 to 50 mg at bedtime, had enhanced sleep efficiency (ratio of time slept to time spent in bed) yet no change in sleep latency. Liver abnormalities, leukopenia, and thrombopenia developed in a few patients.16 Controlled studies have also shown subjective efficacy of trazodone17 and trimipramine18 in treating insomnia.
Some physicians advocate using the more sedating antidepressants—at dosages needed to treat depression—to control insomnia in depressed patients. Evening dosing can minimize daytime sedation. If you choose an activating antidepressant, the potential side effect of insomnia can be managed by judicious use of hypnotic agents. Little is known about antidepressants’ effects on sleep quality after the first 6 to 8 weeks of treatment.19
Although possibly helpful as sleep aids, antidepressants are also associated with side effects. Trazodone, for example, may cause daytime sedation, orthostatic hypotension, and priapism. As a class, the tricyclics are associated with anticholinergic effects such as dry mouth, urinary flow difficulties, and cardiac dysrhythmias.
Table 4
Guidelines for safe use of hypnotics
|
Alcohol. Patients with insomnia often self-medicate with agents that are not specifically indicated to induce sleep. Alcohol is widely used at bedtime because it enhances sleepiness and induces a more rapid sleep onset.20 Drinking a “nightcap” is a poor choice, however, because alcohol—especially after prolonged use—can impair sleep quality, resulting in daytime somnolence. Alcohol is also associated with rapid development of tolerance.
Patients who use alcohol report unrefreshing and disturbed sleep, with frequent nocturnal awakenings even after prolonged abstinence. Alcohol also can further impair sleep-related respiration in patients with obstructive sleep apnea syndrome.
Antihistamines and over-the-counter products whose main active ingredients are antihistamines—such as doxylamine and diphenhydramine—can cause unpredictable efficacy and side effects such as daytime sedation, confusion, and systemic anticholinergic effects.21
Melatonin is a dietary supplement used in dosages of 0.5 to 3,000 mg. Anecdotal reports indicate it may be efficacious in certain subtypes of insomnia—such as shift work, jetlag, blindness, delayed sleep phase syndrome—and in the elderly. However, melatonin’s efficacy has not been established conclusively and is in doubt. Concerns have been expressed regarding the purity of available preparations and possible coronary artery tissue stimulation, as observed in animal studies of melatonin.
Related resources
- American Academy of Sleep Medicine. Sleep logs, patient education materials. www.aasmnet.org
- American Sleep Apnea Association. www.sleepapnea.org
- National Sleep Foundation. www.sleepfoundation.org
Drug brand names
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Doxepin • Sinequan
- Escitalopram • Lexapro
- Estazolam • Prosom
- Flurazepam • Dalmane
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Protriptyline • Vivactil
- Quazepam • Doral
- Temazepam • Restoril
- Trazodone • Desyrel
- Triazolam • Halcion
- Trimipramine • Surmontil
- Venlafaxine • Effexor
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosure
Dr. Doghramji receives research grant support from Cephalon Inc., GlaxoSmithKline, Merck & Co., and Sanofi-Synthelabo.
1. Sateia MJ, Doghramji K, Hauri PJ, Morin CM. Evaluation of chronic insomnia. Sleep 2000;23:243-81.
2. Reynolds CF III, Kupfer DJ. Sleep research in affective illness: state of the art circa 1987. Sleep 1987;10:199-215.
3. Ford DE, Kamerow DB. Epidemiologic study of sleep disturbances and psychiatric disorders. JAMA 1989;262:1479-84.
4. Labbate LA, Pollack MH, Otto MW, et al. Sleep panic attacks: an association with childhood anxiety and adult psychopathology. Biol Psychiatry 1994;43:840-2.
5. Ross RJ, Ball WA, Sullivan KA, et al. Sleep disturbance as the hallmark of posttraumatic stress disorder. Am J Psychiatry 1989;146:697-707.
6. Neylan TC, Reynolds CF III, Kupfer DJ. Sleep disorders. In: Hales RE, Yudofsky SC (eds). Textbook of clinical psychiatry(4th ed). Washington, DC: American Psychiatric Publishing, 2003;978-90.
7. Lewis CF, Tandon R, Shipley JE, et al. Biological predictors of suicidality in schizophrenia. Acta Psychiatr Scand 1996;94:416-20.
8. Spielman AJ, Glovinsky P. The varied nature of insomnia. In: Hauri P (ed). Case studies in insomnia. New York: Plenum Press, 1991;1-15.
9. American Sleep Disorders Association International classification of sleep disorders (rev). Diagnostic and coding manual. Rochester: American Sleep Disorders Association, 1997.
10. Winokur A, Reynolds CF. The effects of antidepressants on sleep physiology. Primary Psychiatry 1994;6:22-7.
11. Gillin JC, Rapaport M, Erman MK, Winokur A, Albala BJ. A comparison of nefazodone and fluoxetine on mood and on objective, subjective, and clinician-rated measures of sleep in depressed patients: a double-blind, 8-week clinical trial. J Clin Psychiatry 1997;58:185-92.
12. Doghramji K. The evaluation and management of sleep disorders. In: Stoudemire A (ed). Clinical psychiatry for medical students (3rd ed). Philadelphia: J.B. Lippincott Co., 1998;783-818.
13. Gillin JC. The long and short of sleeping pills. N Engl J Med 1991;324:1735-7.
14. Corser B, Mayleben D, Doghramji K, et al. No next-day residual sedation four hours after middle-of-the-night treatment with zaleplon. Sleep 2000;23 (S2):A309.-
15. Holm KJ, Goa KL. Zolpidem: an update of its pharmacology, therapeutic efficacy and tolerability in the treatment of insomnia. Drugs 2000;59:865-89.
16. Hajak G, Rodenbeck A, Voderholzer U, et al. Doxepin in the treatment of primary insomnia: a placebo-controlled, double-blind, polysomnographic study. J Clin Psych 2001;62:453-63.
17. Walsh JK, Erman M, Erwin CE, et al. Subjective hypnotic efficacy of trazodone and zolpidem in DSM-III-R primary insomnia. Hum Psychopharmacol 1998;13(3):191-8.
18. Hohagen F, Monero RF, Weiss E, et al. Treatment of primary insomnia with trimipramine: an alternative to benzodiazepine hypnotics? Eur Arch Psychiatry Clin Neurosci 1994;244(2):65-72.
19. Thase ME. Antidepressant treatment of the depressed patient with insomnia. J Clin Psychiatry 1999;60(suppl 17):28-31.
20. Johnson EO, Roehrs T, Roth T, Breslau N. Epidemiology of alcohol and medication as aids to sleep in early adulthood. Sleep 1998;21:178-86.
21. Gengo F, Gabos C, Miller JK. The pharmacodynamics of diphenhydramine-induced drowsiness and changes in mental performance. Clin Pharmacol Ther 1989;45:15-21.
Careful investigation can often reveal insomnia’s cause1—whether a psychiatric or medical condition or poor sleep habits. Understanding why patients can’t sleep is key to effective therapy.
Acute and chronic sleep deprivation is associated with measurable declines in daytime performance (Box). Some data even suggest that long-term sleeplessness increases the risk of new psychiatric disorders—most notably major depression.3
PSYCHIATRIC DISORDERS AND INSOMNIA
Depression. Many depressed persons—up to 80%—experience insomnia, although no one sleep pattern seems typical.2 Depression may be associated with:
- difficulties in falling asleep
- interrupted nocturnal sleep
- and early morning awakening.
Anxiety disorders. Generalized anxiety disorder (GAD), social phobia, panic attacks, and posttraumatic stress disorder (PTSD) are all associated with disrupted sleep. Patients with GAD experience prolonged sleep latency (time needed to fall asleep after lights out) and fragmented sleep, similar to those with primary insomnia.
One-half of adult Americans experience insomnia during their lives, and 10% report persistent sleep difficulties (longer than 2 weeks). Individuals who complain of insomnia report:
- daytime drowsiness
- diminished memory and concentration
- depression
- strained relationships
- increased risk of accidents
- impaired job performance.
Despite these complaints, a surprising 70% of those with insomnia never seek medical help. Only 6% visit their physicians specifically for insomnia, and 24% address sleep difficulty as a secondary complaint. Many (40%) self-medicate with over-the-counter sleep aids or alcohol.2
Insomnia becomes more frequent with aging, associated with increased rates of medical and psychiatric illness and an age-related deterioration in the brain’s sleep-generating processes.3
Subjective sleep quality may be impaired in patients with social phobia. Some patients experience panic symptoms while sleeping, possibly in association with mild hypercapnia. Patients with sleep panic attacks tend to have earlier onset of panic disorder and a higher likelihood of comorbid mood and other anxiety disorders.4
In patients with PTSD, disturbed sleep continuity and increased REM phasic activity—such as eye movements—are directly correlated with severity of PTSD symptoms. Nightmares and disturbed REM sleep are hypothesized hallmarks of PTSD.5
Schizophrenia. Patients with schizophrenia often have disrupted sleep patterns. These include prolonged sleep latency, fragmented sleep with frequent arousals, decreased slow-wave sleep, variable REM latency, and decreased REM rebound after sleep deprivation. Despite investigations going back to the 1950s, no specific link between REM sleep and psychosis has been found.6 Interestingly, increases in REM sleep time and REM activity have been associated with an increased risk of suicide in patients with schizophrenia.7
Adjustment sleep disorder. Acute emotional stressors—such as bereavement, job loss, or hospitalization—often cause adjustment sleep disorder. Symptoms typically remit soon after the stressors abate, so this transient insomnia usually lasts a few days to a few weeks. Treatment with behavioral therapies and hypnotics8 is warranted if:
- sleepiness and fatigue interfere with daytime functioning
- a pattern of recurring episodes develops.9
Psychophysiologic insomnia. Once initiated—regardless of cause—insomnia may persist well after its precipitating factors resolve. Thus, short-term insomnia may develop into long-term, chronic difficulty with recurring episodes or a constant, daily pattern of insomnia. Sufferers often spend hours in bed awake focused upon—and brooding over—their sleeplessness. which in turn further aggravates their insomnia.
Adjustment sleep disorder and psychophysiologic insomnia are included within DSM-IV’s term “primary insomnia.”
OTHER CAUSES OF INSOMNIA
Medications that may affect sleep quality include antidepressants (Table 1),10,11 antihypertensives, antineoplastic agents, bronchodilators, stimulants, corticosteroids, decongestants, diuretics, histamine-2 receptor blockers, and smoking cessation aids.
Recreational drugs, such as cocaine, often cause insomnia. Hypnotics and anxiolytics can cause insomnia following long-term use and during withdrawal.
Other disorders known to disturb sleep include periodic limb movement disorder (PLMD), restless legs syndrome (RLS), sleep apnea syndrome, disrupted circadian rhythms (as with travel or shift work), cardiopulmonary disorders, chronic pain, diabetes, hyperthyroidism, hot flashes associated with menopause, seizures, dementia, and Parkinson’s disease, to name a few.
WORKUP OF SLEEP COMPLAINTS
Acute. Most short-term insomnias—lasting a few weeks or less—are caused by situational stressors, circadian rhythm alterations, and sleep hygiene violations. A logical initial approach, therefore, is to combine sleep hygiene measures with supportive psychotherapy. Hypnotic agents may be considered for apparent daytime consequences—such as sleepiness and occupational impairment—or if the insomnia seems to be escalating.
Chronic. For longer-term insomnias—lasting more than a few weeks—consider a more thorough evaluation, including medical and psychiatric history, physical examination, and mental status examination. Inquire about cardinal symptoms of disorders associated with insomnia, including:
- snoring or breathing pauses during sleep (sleep apnea syndrome)
- restlessness or twitching in the lower extremities (PLMS/RLS).
Question the bed partner, who may be more aware of such symptoms than the patient. Carefully review sleep patterns on weekdays and weekends, bedtime habits, sleep hygiene habits, and substance and medication use.
Table 1
Antidepressants’ effects on sleep and wakefulness
| Activating agents | Bupropion, protriptyline, most selective serotonin reuptake inhibitors, venlafaxine, monoamine oxidase inhibitors |
| Sedating agents | Amitriptyline, doxepin, trimipramine, nefazodone, trazodone, mirtazapine |
| Neutral agents | Citalopram, escitalopram |
Sleep clinic referrals. Consider an evaluation by a sleep disorders center when:
- the diagnosis remains unclear
- or treatment of the presumed conditions fails after a reasonable time
BEHAVIORAL TREATMENTS
Behavioral treatments—with or without hypnotics—are appropriate for a wide variety of insomnia complaints, including adjustment sleep disorder, psychophysiologic insomnia, and depression. Behavioral measures may take longer to implement than drug therapy, but their effects have been shown to last longer in patients with primary insomnia. In many cases, it may be useful to start with both hypnotic and behavioral treatments and withdraw the hypnotic after behavioral measures take effect.
Sleep hygiene. Many individuals unknowingly engage in habitual behaviors that impair sleep. Those with insomnia, for example, often try to compensate for lost sleep by staying in bed later in the morning or by napping, which further fragment nocturnal sleep. Advise these patients to adhere to a regular awakening time—regardless of how long they slept the night before—and to avoid naps. Other tips for getting a good night’s sleep are outlined in Table 2.12
Caffeine has a plasma half-life of 3 to 7 hours, although individual sensitivity varies widely and caffeine’s erratic absorption can prolong its effects. Advise patients with insomnia to avoid caffeine-containing beverages—including coffee, tea, and soft drinks—after noon.
Relaxation training. Muscle tension can be reduced through electromyography (EMG) biofeedback, abdominal breathing exercises, or progressive muscle relaxation techniques, among others. Relaxation training is usually effective within a few weeks.
Psychotherapy. Cognitive-behavioral therapy can help identify and dispel tension-producing thoughts that are disrupting sleep, such as preoccupation with unpleasant work experiences or school examinations. Reassurance may help patients overcome fears about sleeplessness; suggest that patients deal with anxiety-producing thoughts during therapy sessions and at times other than bedtime.
Insight-oriented psychotherapy may enhance patients’ awareness of psychological conflicts from their past that may be producing anxiety and contributing to sleeplessness.
PRESCRIBING HYPNOTICS
Sedative-hypnotics are indicated primarily for short-term management of insomnia. Most are used prophylactically at bedtime until insomnia dissipates or the physician advises the patient to take a break.
Treatment principles. Because many insomnias are recurrent, prolonged hypnotic treatment given in short bouts is often optimal. Longer treatment—months to years—is not recommended by standard textbooks but is clearly needed by a small number of patients with chronic insomnia. In these cases, carefully monitor for tolerance, as manifested by dosage escalation. Long-term hypnotic treatment is not suitable for patients with drug abuse or dependence histories.
Table 2
How to get a good night’s sleep
|
Although chloral hydrate and barbiturates are effective hypnotics, adverse effects limit their safety and usefulness. Benzodiazepines and more recently introduced agents have milder side effect profiles (Table 3). Choose agents based on the patient’s situation, preferences, and effects of prior trials with similar agents. Guidelines for hypnotics discourage chronic use to minimize abuse, misuse, and habituation (Table 4).
Elimination half-life is the primary pharmacokinetic property that differentiates the hypnotics from each other:13
- longer half-life: flurazepam, quazepam
- intermediate half-life: estazolam, temazepam
- short half-life: triazolam, zolpidem, zaleplon (Table 3).
Table 3
Actions and available doses of common hypnotics
| Class/drug | Onset of action | Half-life (hrs) | Active metabolites | Doses (mg) |
|---|---|---|---|---|
| Benzodiazepines | ||||
| Flurazepam | Rapid | 40 to 250 | Yes | 15, 30 |
| Quazepam | Rapid | 40 to 250 | Yes | 7.5, 15 |
| Estazolam | Rapid | 10 to 24 | Yes | 0.5, 1, 2 |
| Temazepam | Intermediate | 8 to 22 | No | 7.5, 15 |
| Triazolam | Rapid | <6 | No | 0.125, 0.25, 0.5 |
| Imidazopyridine | ||||
| Zolpidem | Rapid | 2.5 | No | 5, 10 |
| Pyrazolopyrimidine | ||||
| Zaleplon | Rapid | 1 | No | 5, 10, 20 |
Whereas benzodiazepines bind to benzodiazepine receptor types 1 and 2, zolpidem and zaleplon (and possibly quazepam) bind selectively to type 1. This selectivity may explain why zolpidem and zaleplon are more easily tolerated.
Hypnotic agents with relatively longer half-lives tend to be associated with greater potential for residual daytime effects such as sedation, motor incoordination, amnesia, and slowed reflexes. These effects may impair performance and increase the risk of auto accidents and injuries, especially hip fractures in the elderly.
Nonbenzodiazepines. Because of its ultra-short half-life, zaleplon is least likely to cause residual daytime effects when administered at bedtime. At 10-mg doses, its side effects seem to last no more than 4 hours following administration. Zaleplon can be safely taken after nocturnal awakenings if the patient remains in bed 4 hours or longer after taking it.14
Some patients feel that taking zaleplon only when needed allows them to use hypnotics more sparingly. On the other hand, zaleplon’s ultrashort half-life makes it less useful for patients who have frequent episodes of sleep-interruption insomnia every night. For them, a longer elimination half-life agent such as zolpidem may be more predictably effective for the entire night.15 Short half-life hypnotics have many advantages, but they do not offer anxiolysis for patients with daytime anxiety, as the longer half-life agents do.
Tolerance and rebound. Tolerance can develop following repeated dosing with benzodiazepines—primarily triazolam—and rebound insomnia can follow abrupt discontinuation. Despite widespread concerns, neither tolerance nor rebound insomnia has been well documented. Nevertheless, both can be minimized by using benzodiazepines at the lowest effective dosages and for brief periods. Gradual tapering when discontinuing the drug can help control rebound.
Tolerance and rebound seem to be less of a concern with the newer hypnotics than with benzodiazepines. In preliminary uncontrolled trials, zolpidem and zaleplon did not show evidence of these problems in 1 year of continued use.
NONHYPNOTIC SLEEP AIDS
Sedating antidepressants. Physicians often prescribe low doses of sedating antidepressants to control insomnia, a practice supported by some controlled clinical trials. For example, polysomnography showed that patients who took doxepin, 25 to 50 mg at bedtime, had enhanced sleep efficiency (ratio of time slept to time spent in bed) yet no change in sleep latency. Liver abnormalities, leukopenia, and thrombopenia developed in a few patients.16 Controlled studies have also shown subjective efficacy of trazodone17 and trimipramine18 in treating insomnia.
Some physicians advocate using the more sedating antidepressants—at dosages needed to treat depression—to control insomnia in depressed patients. Evening dosing can minimize daytime sedation. If you choose an activating antidepressant, the potential side effect of insomnia can be managed by judicious use of hypnotic agents. Little is known about antidepressants’ effects on sleep quality after the first 6 to 8 weeks of treatment.19
Although possibly helpful as sleep aids, antidepressants are also associated with side effects. Trazodone, for example, may cause daytime sedation, orthostatic hypotension, and priapism. As a class, the tricyclics are associated with anticholinergic effects such as dry mouth, urinary flow difficulties, and cardiac dysrhythmias.
Table 4
Guidelines for safe use of hypnotics
|
Alcohol. Patients with insomnia often self-medicate with agents that are not specifically indicated to induce sleep. Alcohol is widely used at bedtime because it enhances sleepiness and induces a more rapid sleep onset.20 Drinking a “nightcap” is a poor choice, however, because alcohol—especially after prolonged use—can impair sleep quality, resulting in daytime somnolence. Alcohol is also associated with rapid development of tolerance.
Patients who use alcohol report unrefreshing and disturbed sleep, with frequent nocturnal awakenings even after prolonged abstinence. Alcohol also can further impair sleep-related respiration in patients with obstructive sleep apnea syndrome.
Antihistamines and over-the-counter products whose main active ingredients are antihistamines—such as doxylamine and diphenhydramine—can cause unpredictable efficacy and side effects such as daytime sedation, confusion, and systemic anticholinergic effects.21
Melatonin is a dietary supplement used in dosages of 0.5 to 3,000 mg. Anecdotal reports indicate it may be efficacious in certain subtypes of insomnia—such as shift work, jetlag, blindness, delayed sleep phase syndrome—and in the elderly. However, melatonin’s efficacy has not been established conclusively and is in doubt. Concerns have been expressed regarding the purity of available preparations and possible coronary artery tissue stimulation, as observed in animal studies of melatonin.
Related resources
- American Academy of Sleep Medicine. Sleep logs, patient education materials. www.aasmnet.org
- American Sleep Apnea Association. www.sleepapnea.org
- National Sleep Foundation. www.sleepfoundation.org
Drug brand names
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Doxepin • Sinequan
- Escitalopram • Lexapro
- Estazolam • Prosom
- Flurazepam • Dalmane
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Protriptyline • Vivactil
- Quazepam • Doral
- Temazepam • Restoril
- Trazodone • Desyrel
- Triazolam • Halcion
- Trimipramine • Surmontil
- Venlafaxine • Effexor
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosure
Dr. Doghramji receives research grant support from Cephalon Inc., GlaxoSmithKline, Merck & Co., and Sanofi-Synthelabo.
Careful investigation can often reveal insomnia’s cause1—whether a psychiatric or medical condition or poor sleep habits. Understanding why patients can’t sleep is key to effective therapy.
Acute and chronic sleep deprivation is associated with measurable declines in daytime performance (Box). Some data even suggest that long-term sleeplessness increases the risk of new psychiatric disorders—most notably major depression.3
PSYCHIATRIC DISORDERS AND INSOMNIA
Depression. Many depressed persons—up to 80%—experience insomnia, although no one sleep pattern seems typical.2 Depression may be associated with:
- difficulties in falling asleep
- interrupted nocturnal sleep
- and early morning awakening.
Anxiety disorders. Generalized anxiety disorder (GAD), social phobia, panic attacks, and posttraumatic stress disorder (PTSD) are all associated with disrupted sleep. Patients with GAD experience prolonged sleep latency (time needed to fall asleep after lights out) and fragmented sleep, similar to those with primary insomnia.
One-half of adult Americans experience insomnia during their lives, and 10% report persistent sleep difficulties (longer than 2 weeks). Individuals who complain of insomnia report:
- daytime drowsiness
- diminished memory and concentration
- depression
- strained relationships
- increased risk of accidents
- impaired job performance.
Despite these complaints, a surprising 70% of those with insomnia never seek medical help. Only 6% visit their physicians specifically for insomnia, and 24% address sleep difficulty as a secondary complaint. Many (40%) self-medicate with over-the-counter sleep aids or alcohol.2
Insomnia becomes more frequent with aging, associated with increased rates of medical and psychiatric illness and an age-related deterioration in the brain’s sleep-generating processes.3
Subjective sleep quality may be impaired in patients with social phobia. Some patients experience panic symptoms while sleeping, possibly in association with mild hypercapnia. Patients with sleep panic attacks tend to have earlier onset of panic disorder and a higher likelihood of comorbid mood and other anxiety disorders.4
In patients with PTSD, disturbed sleep continuity and increased REM phasic activity—such as eye movements—are directly correlated with severity of PTSD symptoms. Nightmares and disturbed REM sleep are hypothesized hallmarks of PTSD.5
Schizophrenia. Patients with schizophrenia often have disrupted sleep patterns. These include prolonged sleep latency, fragmented sleep with frequent arousals, decreased slow-wave sleep, variable REM latency, and decreased REM rebound after sleep deprivation. Despite investigations going back to the 1950s, no specific link between REM sleep and psychosis has been found.6 Interestingly, increases in REM sleep time and REM activity have been associated with an increased risk of suicide in patients with schizophrenia.7
Adjustment sleep disorder. Acute emotional stressors—such as bereavement, job loss, or hospitalization—often cause adjustment sleep disorder. Symptoms typically remit soon after the stressors abate, so this transient insomnia usually lasts a few days to a few weeks. Treatment with behavioral therapies and hypnotics8 is warranted if:
- sleepiness and fatigue interfere with daytime functioning
- a pattern of recurring episodes develops.9
Psychophysiologic insomnia. Once initiated—regardless of cause—insomnia may persist well after its precipitating factors resolve. Thus, short-term insomnia may develop into long-term, chronic difficulty with recurring episodes or a constant, daily pattern of insomnia. Sufferers often spend hours in bed awake focused upon—and brooding over—their sleeplessness. which in turn further aggravates their insomnia.
Adjustment sleep disorder and psychophysiologic insomnia are included within DSM-IV’s term “primary insomnia.”
OTHER CAUSES OF INSOMNIA
Medications that may affect sleep quality include antidepressants (Table 1),10,11 antihypertensives, antineoplastic agents, bronchodilators, stimulants, corticosteroids, decongestants, diuretics, histamine-2 receptor blockers, and smoking cessation aids.
Recreational drugs, such as cocaine, often cause insomnia. Hypnotics and anxiolytics can cause insomnia following long-term use and during withdrawal.
Other disorders known to disturb sleep include periodic limb movement disorder (PLMD), restless legs syndrome (RLS), sleep apnea syndrome, disrupted circadian rhythms (as with travel or shift work), cardiopulmonary disorders, chronic pain, diabetes, hyperthyroidism, hot flashes associated with menopause, seizures, dementia, and Parkinson’s disease, to name a few.
WORKUP OF SLEEP COMPLAINTS
Acute. Most short-term insomnias—lasting a few weeks or less—are caused by situational stressors, circadian rhythm alterations, and sleep hygiene violations. A logical initial approach, therefore, is to combine sleep hygiene measures with supportive psychotherapy. Hypnotic agents may be considered for apparent daytime consequences—such as sleepiness and occupational impairment—or if the insomnia seems to be escalating.
Chronic. For longer-term insomnias—lasting more than a few weeks—consider a more thorough evaluation, including medical and psychiatric history, physical examination, and mental status examination. Inquire about cardinal symptoms of disorders associated with insomnia, including:
- snoring or breathing pauses during sleep (sleep apnea syndrome)
- restlessness or twitching in the lower extremities (PLMS/RLS).
Question the bed partner, who may be more aware of such symptoms than the patient. Carefully review sleep patterns on weekdays and weekends, bedtime habits, sleep hygiene habits, and substance and medication use.
Table 1
Antidepressants’ effects on sleep and wakefulness
| Activating agents | Bupropion, protriptyline, most selective serotonin reuptake inhibitors, venlafaxine, monoamine oxidase inhibitors |
| Sedating agents | Amitriptyline, doxepin, trimipramine, nefazodone, trazodone, mirtazapine |
| Neutral agents | Citalopram, escitalopram |
Sleep clinic referrals. Consider an evaluation by a sleep disorders center when:
- the diagnosis remains unclear
- or treatment of the presumed conditions fails after a reasonable time
BEHAVIORAL TREATMENTS
Behavioral treatments—with or without hypnotics—are appropriate for a wide variety of insomnia complaints, including adjustment sleep disorder, psychophysiologic insomnia, and depression. Behavioral measures may take longer to implement than drug therapy, but their effects have been shown to last longer in patients with primary insomnia. In many cases, it may be useful to start with both hypnotic and behavioral treatments and withdraw the hypnotic after behavioral measures take effect.
Sleep hygiene. Many individuals unknowingly engage in habitual behaviors that impair sleep. Those with insomnia, for example, often try to compensate for lost sleep by staying in bed later in the morning or by napping, which further fragment nocturnal sleep. Advise these patients to adhere to a regular awakening time—regardless of how long they slept the night before—and to avoid naps. Other tips for getting a good night’s sleep are outlined in Table 2.12
Caffeine has a plasma half-life of 3 to 7 hours, although individual sensitivity varies widely and caffeine’s erratic absorption can prolong its effects. Advise patients with insomnia to avoid caffeine-containing beverages—including coffee, tea, and soft drinks—after noon.
Relaxation training. Muscle tension can be reduced through electromyography (EMG) biofeedback, abdominal breathing exercises, or progressive muscle relaxation techniques, among others. Relaxation training is usually effective within a few weeks.
Psychotherapy. Cognitive-behavioral therapy can help identify and dispel tension-producing thoughts that are disrupting sleep, such as preoccupation with unpleasant work experiences or school examinations. Reassurance may help patients overcome fears about sleeplessness; suggest that patients deal with anxiety-producing thoughts during therapy sessions and at times other than bedtime.
Insight-oriented psychotherapy may enhance patients’ awareness of psychological conflicts from their past that may be producing anxiety and contributing to sleeplessness.
PRESCRIBING HYPNOTICS
Sedative-hypnotics are indicated primarily for short-term management of insomnia. Most are used prophylactically at bedtime until insomnia dissipates or the physician advises the patient to take a break.
Treatment principles. Because many insomnias are recurrent, prolonged hypnotic treatment given in short bouts is often optimal. Longer treatment—months to years—is not recommended by standard textbooks but is clearly needed by a small number of patients with chronic insomnia. In these cases, carefully monitor for tolerance, as manifested by dosage escalation. Long-term hypnotic treatment is not suitable for patients with drug abuse or dependence histories.
Table 2
How to get a good night’s sleep
|
Although chloral hydrate and barbiturates are effective hypnotics, adverse effects limit their safety and usefulness. Benzodiazepines and more recently introduced agents have milder side effect profiles (Table 3). Choose agents based on the patient’s situation, preferences, and effects of prior trials with similar agents. Guidelines for hypnotics discourage chronic use to minimize abuse, misuse, and habituation (Table 4).
Elimination half-life is the primary pharmacokinetic property that differentiates the hypnotics from each other:13
- longer half-life: flurazepam, quazepam
- intermediate half-life: estazolam, temazepam
- short half-life: triazolam, zolpidem, zaleplon (Table 3).
Table 3
Actions and available doses of common hypnotics
| Class/drug | Onset of action | Half-life (hrs) | Active metabolites | Doses (mg) |
|---|---|---|---|---|
| Benzodiazepines | ||||
| Flurazepam | Rapid | 40 to 250 | Yes | 15, 30 |
| Quazepam | Rapid | 40 to 250 | Yes | 7.5, 15 |
| Estazolam | Rapid | 10 to 24 | Yes | 0.5, 1, 2 |
| Temazepam | Intermediate | 8 to 22 | No | 7.5, 15 |
| Triazolam | Rapid | <6 | No | 0.125, 0.25, 0.5 |
| Imidazopyridine | ||||
| Zolpidem | Rapid | 2.5 | No | 5, 10 |
| Pyrazolopyrimidine | ||||
| Zaleplon | Rapid | 1 | No | 5, 10, 20 |
Whereas benzodiazepines bind to benzodiazepine receptor types 1 and 2, zolpidem and zaleplon (and possibly quazepam) bind selectively to type 1. This selectivity may explain why zolpidem and zaleplon are more easily tolerated.
Hypnotic agents with relatively longer half-lives tend to be associated with greater potential for residual daytime effects such as sedation, motor incoordination, amnesia, and slowed reflexes. These effects may impair performance and increase the risk of auto accidents and injuries, especially hip fractures in the elderly.
Nonbenzodiazepines. Because of its ultra-short half-life, zaleplon is least likely to cause residual daytime effects when administered at bedtime. At 10-mg doses, its side effects seem to last no more than 4 hours following administration. Zaleplon can be safely taken after nocturnal awakenings if the patient remains in bed 4 hours or longer after taking it.14
Some patients feel that taking zaleplon only when needed allows them to use hypnotics more sparingly. On the other hand, zaleplon’s ultrashort half-life makes it less useful for patients who have frequent episodes of sleep-interruption insomnia every night. For them, a longer elimination half-life agent such as zolpidem may be more predictably effective for the entire night.15 Short half-life hypnotics have many advantages, but they do not offer anxiolysis for patients with daytime anxiety, as the longer half-life agents do.
Tolerance and rebound. Tolerance can develop following repeated dosing with benzodiazepines—primarily triazolam—and rebound insomnia can follow abrupt discontinuation. Despite widespread concerns, neither tolerance nor rebound insomnia has been well documented. Nevertheless, both can be minimized by using benzodiazepines at the lowest effective dosages and for brief periods. Gradual tapering when discontinuing the drug can help control rebound.
Tolerance and rebound seem to be less of a concern with the newer hypnotics than with benzodiazepines. In preliminary uncontrolled trials, zolpidem and zaleplon did not show evidence of these problems in 1 year of continued use.
NONHYPNOTIC SLEEP AIDS
Sedating antidepressants. Physicians often prescribe low doses of sedating antidepressants to control insomnia, a practice supported by some controlled clinical trials. For example, polysomnography showed that patients who took doxepin, 25 to 50 mg at bedtime, had enhanced sleep efficiency (ratio of time slept to time spent in bed) yet no change in sleep latency. Liver abnormalities, leukopenia, and thrombopenia developed in a few patients.16 Controlled studies have also shown subjective efficacy of trazodone17 and trimipramine18 in treating insomnia.
Some physicians advocate using the more sedating antidepressants—at dosages needed to treat depression—to control insomnia in depressed patients. Evening dosing can minimize daytime sedation. If you choose an activating antidepressant, the potential side effect of insomnia can be managed by judicious use of hypnotic agents. Little is known about antidepressants’ effects on sleep quality after the first 6 to 8 weeks of treatment.19
Although possibly helpful as sleep aids, antidepressants are also associated with side effects. Trazodone, for example, may cause daytime sedation, orthostatic hypotension, and priapism. As a class, the tricyclics are associated with anticholinergic effects such as dry mouth, urinary flow difficulties, and cardiac dysrhythmias.
Table 4
Guidelines for safe use of hypnotics
|
Alcohol. Patients with insomnia often self-medicate with agents that are not specifically indicated to induce sleep. Alcohol is widely used at bedtime because it enhances sleepiness and induces a more rapid sleep onset.20 Drinking a “nightcap” is a poor choice, however, because alcohol—especially after prolonged use—can impair sleep quality, resulting in daytime somnolence. Alcohol is also associated with rapid development of tolerance.
Patients who use alcohol report unrefreshing and disturbed sleep, with frequent nocturnal awakenings even after prolonged abstinence. Alcohol also can further impair sleep-related respiration in patients with obstructive sleep apnea syndrome.
Antihistamines and over-the-counter products whose main active ingredients are antihistamines—such as doxylamine and diphenhydramine—can cause unpredictable efficacy and side effects such as daytime sedation, confusion, and systemic anticholinergic effects.21
Melatonin is a dietary supplement used in dosages of 0.5 to 3,000 mg. Anecdotal reports indicate it may be efficacious in certain subtypes of insomnia—such as shift work, jetlag, blindness, delayed sleep phase syndrome—and in the elderly. However, melatonin’s efficacy has not been established conclusively and is in doubt. Concerns have been expressed regarding the purity of available preparations and possible coronary artery tissue stimulation, as observed in animal studies of melatonin.
Related resources
- American Academy of Sleep Medicine. Sleep logs, patient education materials. www.aasmnet.org
- American Sleep Apnea Association. www.sleepapnea.org
- National Sleep Foundation. www.sleepfoundation.org
Drug brand names
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Doxepin • Sinequan
- Escitalopram • Lexapro
- Estazolam • Prosom
- Flurazepam • Dalmane
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Protriptyline • Vivactil
- Quazepam • Doral
- Temazepam • Restoril
- Trazodone • Desyrel
- Triazolam • Halcion
- Trimipramine • Surmontil
- Venlafaxine • Effexor
- Zaleplon • Sonata
- Zolpidem • Ambien
Disclosure
Dr. Doghramji receives research grant support from Cephalon Inc., GlaxoSmithKline, Merck & Co., and Sanofi-Synthelabo.
1. Sateia MJ, Doghramji K, Hauri PJ, Morin CM. Evaluation of chronic insomnia. Sleep 2000;23:243-81.
2. Reynolds CF III, Kupfer DJ. Sleep research in affective illness: state of the art circa 1987. Sleep 1987;10:199-215.
3. Ford DE, Kamerow DB. Epidemiologic study of sleep disturbances and psychiatric disorders. JAMA 1989;262:1479-84.
4. Labbate LA, Pollack MH, Otto MW, et al. Sleep panic attacks: an association with childhood anxiety and adult psychopathology. Biol Psychiatry 1994;43:840-2.
5. Ross RJ, Ball WA, Sullivan KA, et al. Sleep disturbance as the hallmark of posttraumatic stress disorder. Am J Psychiatry 1989;146:697-707.
6. Neylan TC, Reynolds CF III, Kupfer DJ. Sleep disorders. In: Hales RE, Yudofsky SC (eds). Textbook of clinical psychiatry(4th ed). Washington, DC: American Psychiatric Publishing, 2003;978-90.
7. Lewis CF, Tandon R, Shipley JE, et al. Biological predictors of suicidality in schizophrenia. Acta Psychiatr Scand 1996;94:416-20.
8. Spielman AJ, Glovinsky P. The varied nature of insomnia. In: Hauri P (ed). Case studies in insomnia. New York: Plenum Press, 1991;1-15.
9. American Sleep Disorders Association International classification of sleep disorders (rev). Diagnostic and coding manual. Rochester: American Sleep Disorders Association, 1997.
10. Winokur A, Reynolds CF. The effects of antidepressants on sleep physiology. Primary Psychiatry 1994;6:22-7.
11. Gillin JC, Rapaport M, Erman MK, Winokur A, Albala BJ. A comparison of nefazodone and fluoxetine on mood and on objective, subjective, and clinician-rated measures of sleep in depressed patients: a double-blind, 8-week clinical trial. J Clin Psychiatry 1997;58:185-92.
12. Doghramji K. The evaluation and management of sleep disorders. In: Stoudemire A (ed). Clinical psychiatry for medical students (3rd ed). Philadelphia: J.B. Lippincott Co., 1998;783-818.
13. Gillin JC. The long and short of sleeping pills. N Engl J Med 1991;324:1735-7.
14. Corser B, Mayleben D, Doghramji K, et al. No next-day residual sedation four hours after middle-of-the-night treatment with zaleplon. Sleep 2000;23 (S2):A309.-
15. Holm KJ, Goa KL. Zolpidem: an update of its pharmacology, therapeutic efficacy and tolerability in the treatment of insomnia. Drugs 2000;59:865-89.
16. Hajak G, Rodenbeck A, Voderholzer U, et al. Doxepin in the treatment of primary insomnia: a placebo-controlled, double-blind, polysomnographic study. J Clin Psych 2001;62:453-63.
17. Walsh JK, Erman M, Erwin CE, et al. Subjective hypnotic efficacy of trazodone and zolpidem in DSM-III-R primary insomnia. Hum Psychopharmacol 1998;13(3):191-8.
18. Hohagen F, Monero RF, Weiss E, et al. Treatment of primary insomnia with trimipramine: an alternative to benzodiazepine hypnotics? Eur Arch Psychiatry Clin Neurosci 1994;244(2):65-72.
19. Thase ME. Antidepressant treatment of the depressed patient with insomnia. J Clin Psychiatry 1999;60(suppl 17):28-31.
20. Johnson EO, Roehrs T, Roth T, Breslau N. Epidemiology of alcohol and medication as aids to sleep in early adulthood. Sleep 1998;21:178-86.
21. Gengo F, Gabos C, Miller JK. The pharmacodynamics of diphenhydramine-induced drowsiness and changes in mental performance. Clin Pharmacol Ther 1989;45:15-21.
1. Sateia MJ, Doghramji K, Hauri PJ, Morin CM. Evaluation of chronic insomnia. Sleep 2000;23:243-81.
2. Reynolds CF III, Kupfer DJ. Sleep research in affective illness: state of the art circa 1987. Sleep 1987;10:199-215.
3. Ford DE, Kamerow DB. Epidemiologic study of sleep disturbances and psychiatric disorders. JAMA 1989;262:1479-84.
4. Labbate LA, Pollack MH, Otto MW, et al. Sleep panic attacks: an association with childhood anxiety and adult psychopathology. Biol Psychiatry 1994;43:840-2.
5. Ross RJ, Ball WA, Sullivan KA, et al. Sleep disturbance as the hallmark of posttraumatic stress disorder. Am J Psychiatry 1989;146:697-707.
6. Neylan TC, Reynolds CF III, Kupfer DJ. Sleep disorders. In: Hales RE, Yudofsky SC (eds). Textbook of clinical psychiatry(4th ed). Washington, DC: American Psychiatric Publishing, 2003;978-90.
7. Lewis CF, Tandon R, Shipley JE, et al. Biological predictors of suicidality in schizophrenia. Acta Psychiatr Scand 1996;94:416-20.
8. Spielman AJ, Glovinsky P. The varied nature of insomnia. In: Hauri P (ed). Case studies in insomnia. New York: Plenum Press, 1991;1-15.
9. American Sleep Disorders Association International classification of sleep disorders (rev). Diagnostic and coding manual. Rochester: American Sleep Disorders Association, 1997.
10. Winokur A, Reynolds CF. The effects of antidepressants on sleep physiology. Primary Psychiatry 1994;6:22-7.
11. Gillin JC, Rapaport M, Erman MK, Winokur A, Albala BJ. A comparison of nefazodone and fluoxetine on mood and on objective, subjective, and clinician-rated measures of sleep in depressed patients: a double-blind, 8-week clinical trial. J Clin Psychiatry 1997;58:185-92.
12. Doghramji K. The evaluation and management of sleep disorders. In: Stoudemire A (ed). Clinical psychiatry for medical students (3rd ed). Philadelphia: J.B. Lippincott Co., 1998;783-818.
13. Gillin JC. The long and short of sleeping pills. N Engl J Med 1991;324:1735-7.
14. Corser B, Mayleben D, Doghramji K, et al. No next-day residual sedation four hours after middle-of-the-night treatment with zaleplon. Sleep 2000;23 (S2):A309.-
15. Holm KJ, Goa KL. Zolpidem: an update of its pharmacology, therapeutic efficacy and tolerability in the treatment of insomnia. Drugs 2000;59:865-89.
16. Hajak G, Rodenbeck A, Voderholzer U, et al. Doxepin in the treatment of primary insomnia: a placebo-controlled, double-blind, polysomnographic study. J Clin Psych 2001;62:453-63.
17. Walsh JK, Erman M, Erwin CE, et al. Subjective hypnotic efficacy of trazodone and zolpidem in DSM-III-R primary insomnia. Hum Psychopharmacol 1998;13(3):191-8.
18. Hohagen F, Monero RF, Weiss E, et al. Treatment of primary insomnia with trimipramine: an alternative to benzodiazepine hypnotics? Eur Arch Psychiatry Clin Neurosci 1994;244(2):65-72.
19. Thase ME. Antidepressant treatment of the depressed patient with insomnia. J Clin Psychiatry 1999;60(suppl 17):28-31.
20. Johnson EO, Roehrs T, Roth T, Breslau N. Epidemiology of alcohol and medication as aids to sleep in early adulthood. Sleep 1998;21:178-86.
21. Gengo F, Gabos C, Miller JK. The pharmacodynamics of diphenhydramine-induced drowsiness and changes in mental performance. Clin Pharmacol Ther 1989;45:15-21.
Benzodiazepines for substance abusers
How should you treat anxiety in substance-abusing patients: deny them benzodiazepines and risk under-treatment, or prescribe benzodiazepines for the anxiolytic effect and risk contributing to addiction?
There is no definitive answer, but one thing is clear: Among psychiatric patients, substance abusers are most likely to abuse benzodiazepines and become addicted to them.
Some argue that the abuse potential is overstated, but only limited data suggest that benzodiazepines can be safely prescribed to patients who are abusing alcohol or drugs. In this article, we discuss benzodiazepine use in these patients and offer a sobriety-based treatment approach.
Table 1
Benzodiazepines’ potency and half-lives, including half-lives of active metabolites
| Potency | Shorter half-life (hr) | Longer half-life (hr) |
|---|---|---|
| High | Alprazolam (6 to 12) Lorazepam (10 to 20) Triazolam (2) | Clonazepam (18 to 50) |
| Low | Oxazepam (4 to 15) Temazepam (8 to 22) | Chlordiazepoxide (5 to 30) [36 to 200]* Clorazepate [36 to 200]* Diazepam (20 to 100) [36 to 200]* Flurazepam [40 to 250]* |
| * [active metabolite] | ||
| Source: Ashton CH. Benzodiazepine equivalence table. Available at www.benzo.org.uk | ||
BENEFITS AND RISKS
Considered a safe substitute for barbiturates, benzodiazepines were heralded as wonder drugs when they were introduced in the 1950s. Reports of their addictive potential surfaced in the 1970s, and since then researchers have disagreed on whether benzodiazepines should be prescribed to substance-abusing or -dependent patients.
Clinical utility. Benzodiazepines are used in many clinical situations because of their:
- anxiolytic, hypnotic, anticonvulsant, antipanic, antidepressant, amnestic, anesthetic, and antispastic effects
- relatively mild side effects, when compared with alternatives such as barbiturates.
In psychiatry, benzodiazepines are used to treat anxiety disorders, agitation, and insomnia. Because of cross-tolerance with alcohol and barbiturates, benzodiazepines also are used to manage alcohol or barbiturate withdrawal.
Interactions. Benzodiazepines can interact with other psychotropics, including lithium, antipsychotics, and selective serotonin reuptake inhibitors (SSRIs). Respiratory arrest has been reported in patients taking both a high-potency benzodiazepine and clozapine.1
Overdose and withdrawal symptoms. Benzodiazepine overdose is characterized by slurred speech, sedation, memory impairment, incoordination, respiratory depression, hypotension, stupor, and coma. Abrupt withdrawal may produce life-threatening delirium, hallucinations, grand mal seizures, and symptoms similar to those of alcohol withdrawal (insomnia, anxiety, tremor, hyperactivity, nausea, vomiting, and psychomotor agitation).1
ABUSE PATTERNS
The few empiric studies examining benzodiazepines’ abuse potential in substance abusers have shown inconsistent results. However, it is generally accepted that:
- long-term benzodiazepine use may lead to tolerance and physiologic dependence
- withdrawal symptoms can occur if benzodiazepines are stopped suddenly, especially after long-term (months to years) use.
Even though most benzodiazepine prescriptions are not abused,2 a history of alcohol and drug abuse suggests high potential for benzodiazepine abuse. Also, long-term users of prescribed benzodiazepines often develop tolerance and may escalate their doses to get the same desired effects. If their supply is threatened, these patients may seek benzodiazepines illicitly.
Benzodiazepines may enhance or prolong the elation (“high”) associated with other drugs or mitigate the depression (“crash”) that follows a stimulant “high.” Sometimes benzodiazepines are the drug of choice, as high doses of potent, short-acting agents may provide a stimulant “high.”
WHO ABUSES BENZODIAZEPINES?
Alcohol and substance abusers tend to ingest benzodiazepines for recreational purposes. Thirty to 50% of alcoholics undergoing detoxification and 44% of IV drug abusers also may be abusing benzodiazepines.3
Benzodiazepines are cross-tolerant with alcohol, and alcoholics may use them with alcohol or as a substitute when alcohol is unavailable. They also may self-medicate with benzodiazepines to ease alcohol’s withdrawal symptoms. Opiate, amphetamine, and cocaine abusers may use benzodiazepines with their drugs of choice, as may younger abusers of MDMA (“Ecstasy”) and LSD.
Even patients who begin taking benzodiazepines for legitimate reasons may end up abusing them. In one study of 2,600 patients prescribed diazepam, up to 60% had abused and/or become dependent on it.4
Benzodiazepine abuse may start with other sedative/hypnotic abuse or as experimentation with drugs or alcohol, typically around age 13 or 14.5 The average benzodiazepine abuser is age 19 to 31, and the male-to-female ratio is about 2:1.6
Multi-drug abuse. Benzodiazepines are usually not the preferred or sole drug of abuse. Roughly 80% of benzodiazepine abuse may be a component of poly-drug abuse, most commonly with opioid addiction.7 A 2-year National Institute on Drug Abuse study of heroin abusers suggested that 15% also abused benzodiazepines daily for more than 1 year, and 73% had abused benzodiazepines several times during the previous week.8 Other studies suggest that up to 90% of methadone users regularly abuse benzodiazepines, often at high doses.9
Table 2
Should benzodiazepines be prescribed to substance abusers?
YES: Arguments for
|
NO: Arguments against
|
ILLICIT USE POTENTIAL
Prescriptions are the primary source of supply for benzodiazepine abusers. These patients are doctor shoppers and often change pharmacies. They visit emergency rooms frequently and may feign symptoms to obtain benzodiazepine prescriptions. They fill prescriptions for personal use or sell the drugs to illicit sources to support their addictions.
On the street, brand-name benzodiazepines are worth much more than generics because they can be identified by photographs of brand-name benzodiazepines on the Internet or in reference books. In many cities, the street value of the Xanax or Klonopin brands may be $5 to $10 per pill. A 5mg tablet of Valium-brand diazepam may sell for $5, and 10-mg tablets are worth up to $10.
Higher abuse potential. All benzodiazepines have abuse potential, and most have been reported in the literature as being abused. Those most likely to be abused have a short half-life10 (Table 1) or rapidly cross the blood brain barrier, such as alprazolam.11
Alprazolam and lorazepam are popular among benzodiazepine abusers. In experienced but nondependent users, 1 mg of alprazolam produces a sense of elation and carries an abuse potential similar to that of 10 mg of dextroamphetamine.12 Lipophilic agents such as diazepam also have a high abuse and addiction potential.
In the United States, diazepam and alprazolam appear to be the most abused benzodi-azepines.13 Flunitrazepam has become popular among high school students and drug addicts, particularly in the south and southwest. This potent benzodiazepine is not approved for use in the United States but is diverted from Latin America or Europe in the illegal drug trade.
Lower abuse potential. Benzodiazepines with longer half-lives generally are less likely to be abused, although diazepam—with a half-life of up to 100 hours—is the exception. Chlordiazepoxide has been reported to produce a lower “high” than other benzodiazepines.14 Among the short half-life benzodiazepines, oxazepam may have a relatively low abuse potential.14 Clonazepam—a high-potency benzodiazepine with a long half-life—is generally safe and may have a lower abuse or addiction potential, although its abuse has been report-ed.15 Similarly, oxazepam, clorazepate, and chlordiazepoxide may be less reinforcing than other benzodiazepines, although reports have linked these agents to abuse as well.16
TO PRESCRIBE OR NOT TO PRESCRIBE
Opponents blame benzodiazepines for promoting the drug culture and argue that prescribing benzodiazepines promotes drug abuse. Advocates of benzodiazepine therapy contend that restricting an effective and safe medication is unethical, even in substance abusers. Arguments from each perspective are summarized in Table 2.17-27
In 1990, an American Psychiatric Association task force concluded that alcohol and substance abusers could be prescribed benzodiazepines with very close monitoring but did not recommend specific standards.28
RECOMMENDATIONS
Prescribing benzodiazepines to substance abusers is not absolutely contraindicated, despite an elevated relative risk of abuse or dependence. In the absence of convincing data, physicians must decide on their own—usually case by case—the merits of using benzodiazepines to treat anxiety in substance abusers.
A sobriety-based approach. Our group at the Substance Abuse Treatment Center, VA Medical Center, Omaha, Nebraska, has developed a treatment algorithm for substance abusers presenting with anxiety (see Algorithm). It is based on clinical experience, more than 200 relevant articles, and the consensus of psychiatrists trained and certified by the American Board of Psychiatry and Neurology and the American Society of Addiction Medicine.
Algorithm Sobriety-based protocol for treating anxiety in substance abusers
Precautions for prescribing benzodiazepines
- Inform patient of planned duration of therapy
- Prescribe for brief periods (weeks to months), with follow-up at least monthly
- No refills without follow-up, and no refills over the phone
- Use random urine toxicology screening every 1 to 3 months to monitor for relapse
- Attempt to taper dosage after 3 to 6 months—even if patient resists—and monitor for objective withdrawal
- If no objective withdrawal, terminate benzodiazepine; continue other medications
- If objective withdrawal, continue benzodiazepine and reattempt taper in 3 to 6 months; continue Alcoholics/Narcotics Anonymous
We suggest that you begin by encouraging sobriety and referring willing patients to detoxification. Because most addicts deny or greatly minimize their substance abuse, investigate all potential drug or alcohol abuse thoroughly and address it appropriately.
If anxiety persists after detoxification, begin drug therapy with nonbenzodiazepines:
- Diphenhydramine, 50 to 100 mg/d, may reduce anxiety and often improves sleep, but consider its anticholinergic side effects before prescribing.
- Some SSRIs and venlafaxine are used to treat anxiety, but they generally take weeks to produce a therapeutic effect and some patients cannot wait that long.
- Mirtazapine, 15 to 30 mg/d, provides relatively rapid sedation and helps with sleep and anxiety.
- Buspirone may reduce anxiety, especially when given at 30 to 60 mg/d.
- Gabapentin, 100 to 300 mg tid or higher, may reduce anxiety and help with sleep.
- Tricyclic antidepressants may be considered, but watch for cardiac and anticholinergic side effects and overdose risks.
If anxiety does not improve with an adequate trial of first-line agents, consider adding long-acting benzodiazepines at sufficient dosages, such as clonazepam, 0.5 to 1 mg bid to tid. Prescribe scheduled doses, rather than “as needed.” Continue the first-line antianxiety agent, and reiterate to the patient that benzodiazepine therapy will be short-term.
Observe prescribing precautions (see Algorithm), and screen patients’ urine randomly every 1 to 3 months to monitor their adherence to substance abuse treatment.
Keep patient records current, with attention to dates of visits and prescriptions and quantity of benzodiazepines prescribed. To ensure proper continued use of benzodiazepines, consider consulting with physicians who have expertise in treating similar patients. Watch for possible signs of benzodiazepine dependence and abuse, such as requests for dose increases or early refills.
NONDRUG TREATMENTS
Nondrug treatments have been shown to reduce substance use and control anxiety in some studies. These include cognitive-behavioral therapy, motivational enhancement therapy, interpersonal therapy, and brief dynamic therapy, among others. Their use requires specific training or referral to more experienced colleagues. For information on these treatments, consult the Web sites of the National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism (Related resources).
Group and self-help therapies such as Alcoholics Anonymous or Narcotics Anonymous also have been shown to reduce substance use.
- Parran TV. Prescription drug abuse. A question of balance. Med Clin North Am 1997;81:967-78.
- Lader M, Russell J. Guidelines for the prevention and treatment of benzodiazepine dependence: summary of a report from the Mental Health Foundation. Addiction 1993;88(12):1707-8.
- National Institute on Drug Abuse. www.nida.nih.gov
- National Institute on Alcohol Abuse and Addiction. www.niaaa.nih.gov
Drug brand names
- Alprazolam • Xanax
- Buspirone • BuSpar
- Chlordiazepoxide • Librium
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Flurazepam • Dalmane
- Gabapentin • Neurontin
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Temazepam • Restoril
- Triazolam • Halcion
- Venlafaxine • Effexor
Disclosure
This work was supported by the Attorney General’s Office, Commonwealth of Massachusetts and the United States Department of Veterans Affairs.
Dr. Sattar has received grant funding from Abbott Laboratories and is a speaker for AstraZeneca and Eli Lilly and Co.
Dr. Bhatia is a speaker for AstraZeneca, Eli Lilly and Co., Janssen Pharmaceutica, and Bristol-Myers Squibb Co.
Acknowledgment
The authors wish to thank Jennifer Hong, second-year medical student, Creighton University School of Medicine, Omaha, NE, for her assistance in preparing this article for publication.
1. Hales RE, Yudofsky SC. Textbook of clinical psychiatry, 4th ed. Washington, DC: American Psychiatric Publishing, 2003:318-19,493,501,1098.
2. Woods JH, Katz JL, Winger G. Use and abuse of benzodiazepines. Issues relevant to prescribing. JAMA 1988;260(23):3476-80.
3. Shaw M, Brabbins C, Ruben S. Misuse of benzodiazepines. Specify the formulation when prescribing. BMJ 1994;308(6945):1709.-
4. Woody GE, O’Brien CP, Greenstein R. Misuse and abuse of diazepam: an increasingly common medical problem. Int J Addict 1975;10(5):843-8.
5. Pedersen W, Lavik NJ. Adolescents and benzodiazepines: prescribed use, self-medication and intoxication. Acta Psychiatrica Scand 1991;84:94-8.
6. Ruben SM, Morrison CL. Temazepam misuse in a group of injecting drug users. Br J Addict 1992;87:1387-92,
7. Gold MS, Miller NS, Stennie K, Populla-Vardi C. Epidemiology of benzodiazepine use and dependence. Psychiatr Annals 1995;25:146-8.
8. Dumont RL. Abuse of benzodiazepines—the problems and the solutions. A report of a Committee of the Institute for Behavior and Health, Inc. Am J Drug Alcohol Abuse. 1988;14(suppl 1):1-69.
9. Iguchi MY, Griffiths RR, Bickel WK, et al. Relative abuse liability of benzodiazepines in methadone-maintained populations in three cities. In: Harris LS (ed). Problems of drug dependence, 1988. Proceedings of the 50th annual scientific meeting, the Committee on Problems of Drug Dependence, Inc. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute on Drug Abuse, Office of Science, 1989. DHHS publication no. (ADM) 89-1605.
10. Longo LP. Non-benzodiazepine pharmacotherapy of anxiety and panic in substance abusing patients. Psychiatr Annals 1998;28(3):142-53.
11. Roache JD, Meisch RA. Findings from self-administration research on the addiction potential of benzodiazepines. Psychiatr Annals 1995;25(3):153-7.
12. Zawertailo LA, Busto U, Kaplan HL, Sellers EM. Comparative abuse liability of sertraline, alprazolam, and dextroamphetamine in humans. J Clin Psychopharmacol 1995;15(2):117-24.
13. Griffiths RR, McLeod DR, Bigelow GE, et al. Comparison of diazepam and oxazepam: preference, liking and extent of abuse. J Pharmacol Exp Ther 1984;229(2):501-8.
14. Griffiths RR, Wolf B. Relative abuse liability of different benzodiazepines in drug abusers. J Clin Psychopharmacol 1990;10(4):237-43.
15. Albeck JH. Withdrawal and detoxification from benzodiazepine dependence: a potential role for clonazepam. J Clin Psychiatry 1987;48(suppl):43-9.
16. Woods JH, Katz JL, Winger G. Use and abuse of benzodiazepines: issues relevant to prescribing. JAMA 1988;260(23):3476-80.
17. Maletzky BM, Klotter J. Addiction to diazepam. Int J Addict 1976;II(1):95-115.
18. Lader M. Short-term versus long-term benzodiazepine therapy. Curr Med Res Opin 1984(8, suppl 4);120-6.
19. Ciraulo DA, Sands BK, Shader RI. Critical review of liability for benzodiazepine abuse among alcoholics. Am J Psychiatry 1988;145(12):1501-6.
20. Berner R. The patient’s perspective. NYS J Med 1991;91(11, suppl):37S-39S.
21. Schatzberg AF. Benzodiazepines: therapeutic, biological and psychosocial issues. J Psychiatr Res 1990;24(2):1-2.
22. Lader M. Drug development optimization—benzodiazepines. Agents Actions 1990;29:59-69.
23. Sellers EM, Marshman JA, Kaplan Hl, et al. Acute and chronic drug abuse emergencies in metropolitan Toronto. Int J Addict 1981;16(2):283-303.
24. Hamlin M. Guidelines for benzodiazepine prescribing. Br J Hosp Med 1989;42(1):82.-
25. Piesiur Strehlow B, Strehlow U, Poser W. Mortality of patients dependent on benzodiazepines. Acta Psychiatr Scand 1986;73:330-5.
26. Bendtsen P, Hensing G, McKenzie L, Stardsman AK. Prescribing benzodiazepines—a critical incident study of a physician dilemma. Soc Sci Med 1999;49:459-67.
27. Linnoila MI. Benzodiazepines and alcohol. J Psychiatric Res 1990;24(2, suppl):121-7.
28. Benzodiazepine dependence, toxicity and abuse. A task force report. Washington, DC: American Psychiatric Association, 1990.
How should you treat anxiety in substance-abusing patients: deny them benzodiazepines and risk under-treatment, or prescribe benzodiazepines for the anxiolytic effect and risk contributing to addiction?
There is no definitive answer, but one thing is clear: Among psychiatric patients, substance abusers are most likely to abuse benzodiazepines and become addicted to them.
Some argue that the abuse potential is overstated, but only limited data suggest that benzodiazepines can be safely prescribed to patients who are abusing alcohol or drugs. In this article, we discuss benzodiazepine use in these patients and offer a sobriety-based treatment approach.
Table 1
Benzodiazepines’ potency and half-lives, including half-lives of active metabolites
| Potency | Shorter half-life (hr) | Longer half-life (hr) |
|---|---|---|
| High | Alprazolam (6 to 12) Lorazepam (10 to 20) Triazolam (2) | Clonazepam (18 to 50) |
| Low | Oxazepam (4 to 15) Temazepam (8 to 22) | Chlordiazepoxide (5 to 30) [36 to 200]* Clorazepate [36 to 200]* Diazepam (20 to 100) [36 to 200]* Flurazepam [40 to 250]* |
| * [active metabolite] | ||
| Source: Ashton CH. Benzodiazepine equivalence table. Available at www.benzo.org.uk | ||
BENEFITS AND RISKS
Considered a safe substitute for barbiturates, benzodiazepines were heralded as wonder drugs when they were introduced in the 1950s. Reports of their addictive potential surfaced in the 1970s, and since then researchers have disagreed on whether benzodiazepines should be prescribed to substance-abusing or -dependent patients.
Clinical utility. Benzodiazepines are used in many clinical situations because of their:
- anxiolytic, hypnotic, anticonvulsant, antipanic, antidepressant, amnestic, anesthetic, and antispastic effects
- relatively mild side effects, when compared with alternatives such as barbiturates.
In psychiatry, benzodiazepines are used to treat anxiety disorders, agitation, and insomnia. Because of cross-tolerance with alcohol and barbiturates, benzodiazepines also are used to manage alcohol or barbiturate withdrawal.
Interactions. Benzodiazepines can interact with other psychotropics, including lithium, antipsychotics, and selective serotonin reuptake inhibitors (SSRIs). Respiratory arrest has been reported in patients taking both a high-potency benzodiazepine and clozapine.1
Overdose and withdrawal symptoms. Benzodiazepine overdose is characterized by slurred speech, sedation, memory impairment, incoordination, respiratory depression, hypotension, stupor, and coma. Abrupt withdrawal may produce life-threatening delirium, hallucinations, grand mal seizures, and symptoms similar to those of alcohol withdrawal (insomnia, anxiety, tremor, hyperactivity, nausea, vomiting, and psychomotor agitation).1
ABUSE PATTERNS
The few empiric studies examining benzodiazepines’ abuse potential in substance abusers have shown inconsistent results. However, it is generally accepted that:
- long-term benzodiazepine use may lead to tolerance and physiologic dependence
- withdrawal symptoms can occur if benzodiazepines are stopped suddenly, especially after long-term (months to years) use.
Even though most benzodiazepine prescriptions are not abused,2 a history of alcohol and drug abuse suggests high potential for benzodiazepine abuse. Also, long-term users of prescribed benzodiazepines often develop tolerance and may escalate their doses to get the same desired effects. If their supply is threatened, these patients may seek benzodiazepines illicitly.
Benzodiazepines may enhance or prolong the elation (“high”) associated with other drugs or mitigate the depression (“crash”) that follows a stimulant “high.” Sometimes benzodiazepines are the drug of choice, as high doses of potent, short-acting agents may provide a stimulant “high.”
WHO ABUSES BENZODIAZEPINES?
Alcohol and substance abusers tend to ingest benzodiazepines for recreational purposes. Thirty to 50% of alcoholics undergoing detoxification and 44% of IV drug abusers also may be abusing benzodiazepines.3
Benzodiazepines are cross-tolerant with alcohol, and alcoholics may use them with alcohol or as a substitute when alcohol is unavailable. They also may self-medicate with benzodiazepines to ease alcohol’s withdrawal symptoms. Opiate, amphetamine, and cocaine abusers may use benzodiazepines with their drugs of choice, as may younger abusers of MDMA (“Ecstasy”) and LSD.
Even patients who begin taking benzodiazepines for legitimate reasons may end up abusing them. In one study of 2,600 patients prescribed diazepam, up to 60% had abused and/or become dependent on it.4
Benzodiazepine abuse may start with other sedative/hypnotic abuse or as experimentation with drugs or alcohol, typically around age 13 or 14.5 The average benzodiazepine abuser is age 19 to 31, and the male-to-female ratio is about 2:1.6
Multi-drug abuse. Benzodiazepines are usually not the preferred or sole drug of abuse. Roughly 80% of benzodiazepine abuse may be a component of poly-drug abuse, most commonly with opioid addiction.7 A 2-year National Institute on Drug Abuse study of heroin abusers suggested that 15% also abused benzodiazepines daily for more than 1 year, and 73% had abused benzodiazepines several times during the previous week.8 Other studies suggest that up to 90% of methadone users regularly abuse benzodiazepines, often at high doses.9
Table 2
Should benzodiazepines be prescribed to substance abusers?
YES: Arguments for
|
NO: Arguments against
|
ILLICIT USE POTENTIAL
Prescriptions are the primary source of supply for benzodiazepine abusers. These patients are doctor shoppers and often change pharmacies. They visit emergency rooms frequently and may feign symptoms to obtain benzodiazepine prescriptions. They fill prescriptions for personal use or sell the drugs to illicit sources to support their addictions.
On the street, brand-name benzodiazepines are worth much more than generics because they can be identified by photographs of brand-name benzodiazepines on the Internet or in reference books. In many cities, the street value of the Xanax or Klonopin brands may be $5 to $10 per pill. A 5mg tablet of Valium-brand diazepam may sell for $5, and 10-mg tablets are worth up to $10.
Higher abuse potential. All benzodiazepines have abuse potential, and most have been reported in the literature as being abused. Those most likely to be abused have a short half-life10 (Table 1) or rapidly cross the blood brain barrier, such as alprazolam.11
Alprazolam and lorazepam are popular among benzodiazepine abusers. In experienced but nondependent users, 1 mg of alprazolam produces a sense of elation and carries an abuse potential similar to that of 10 mg of dextroamphetamine.12 Lipophilic agents such as diazepam also have a high abuse and addiction potential.
In the United States, diazepam and alprazolam appear to be the most abused benzodi-azepines.13 Flunitrazepam has become popular among high school students and drug addicts, particularly in the south and southwest. This potent benzodiazepine is not approved for use in the United States but is diverted from Latin America or Europe in the illegal drug trade.
Lower abuse potential. Benzodiazepines with longer half-lives generally are less likely to be abused, although diazepam—with a half-life of up to 100 hours—is the exception. Chlordiazepoxide has been reported to produce a lower “high” than other benzodiazepines.14 Among the short half-life benzodiazepines, oxazepam may have a relatively low abuse potential.14 Clonazepam—a high-potency benzodiazepine with a long half-life—is generally safe and may have a lower abuse or addiction potential, although its abuse has been report-ed.15 Similarly, oxazepam, clorazepate, and chlordiazepoxide may be less reinforcing than other benzodiazepines, although reports have linked these agents to abuse as well.16
TO PRESCRIBE OR NOT TO PRESCRIBE
Opponents blame benzodiazepines for promoting the drug culture and argue that prescribing benzodiazepines promotes drug abuse. Advocates of benzodiazepine therapy contend that restricting an effective and safe medication is unethical, even in substance abusers. Arguments from each perspective are summarized in Table 2.17-27
In 1990, an American Psychiatric Association task force concluded that alcohol and substance abusers could be prescribed benzodiazepines with very close monitoring but did not recommend specific standards.28
RECOMMENDATIONS
Prescribing benzodiazepines to substance abusers is not absolutely contraindicated, despite an elevated relative risk of abuse or dependence. In the absence of convincing data, physicians must decide on their own—usually case by case—the merits of using benzodiazepines to treat anxiety in substance abusers.
A sobriety-based approach. Our group at the Substance Abuse Treatment Center, VA Medical Center, Omaha, Nebraska, has developed a treatment algorithm for substance abusers presenting with anxiety (see Algorithm). It is based on clinical experience, more than 200 relevant articles, and the consensus of psychiatrists trained and certified by the American Board of Psychiatry and Neurology and the American Society of Addiction Medicine.
Algorithm Sobriety-based protocol for treating anxiety in substance abusers
Precautions for prescribing benzodiazepines
- Inform patient of planned duration of therapy
- Prescribe for brief periods (weeks to months), with follow-up at least monthly
- No refills without follow-up, and no refills over the phone
- Use random urine toxicology screening every 1 to 3 months to monitor for relapse
- Attempt to taper dosage after 3 to 6 months—even if patient resists—and monitor for objective withdrawal
- If no objective withdrawal, terminate benzodiazepine; continue other medications
- If objective withdrawal, continue benzodiazepine and reattempt taper in 3 to 6 months; continue Alcoholics/Narcotics Anonymous
We suggest that you begin by encouraging sobriety and referring willing patients to detoxification. Because most addicts deny or greatly minimize their substance abuse, investigate all potential drug or alcohol abuse thoroughly and address it appropriately.
If anxiety persists after detoxification, begin drug therapy with nonbenzodiazepines:
- Diphenhydramine, 50 to 100 mg/d, may reduce anxiety and often improves sleep, but consider its anticholinergic side effects before prescribing.
- Some SSRIs and venlafaxine are used to treat anxiety, but they generally take weeks to produce a therapeutic effect and some patients cannot wait that long.
- Mirtazapine, 15 to 30 mg/d, provides relatively rapid sedation and helps with sleep and anxiety.
- Buspirone may reduce anxiety, especially when given at 30 to 60 mg/d.
- Gabapentin, 100 to 300 mg tid or higher, may reduce anxiety and help with sleep.
- Tricyclic antidepressants may be considered, but watch for cardiac and anticholinergic side effects and overdose risks.
If anxiety does not improve with an adequate trial of first-line agents, consider adding long-acting benzodiazepines at sufficient dosages, such as clonazepam, 0.5 to 1 mg bid to tid. Prescribe scheduled doses, rather than “as needed.” Continue the first-line antianxiety agent, and reiterate to the patient that benzodiazepine therapy will be short-term.
Observe prescribing precautions (see Algorithm), and screen patients’ urine randomly every 1 to 3 months to monitor their adherence to substance abuse treatment.
Keep patient records current, with attention to dates of visits and prescriptions and quantity of benzodiazepines prescribed. To ensure proper continued use of benzodiazepines, consider consulting with physicians who have expertise in treating similar patients. Watch for possible signs of benzodiazepine dependence and abuse, such as requests for dose increases or early refills.
NONDRUG TREATMENTS
Nondrug treatments have been shown to reduce substance use and control anxiety in some studies. These include cognitive-behavioral therapy, motivational enhancement therapy, interpersonal therapy, and brief dynamic therapy, among others. Their use requires specific training or referral to more experienced colleagues. For information on these treatments, consult the Web sites of the National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism (Related resources).
Group and self-help therapies such as Alcoholics Anonymous or Narcotics Anonymous also have been shown to reduce substance use.
- Parran TV. Prescription drug abuse. A question of balance. Med Clin North Am 1997;81:967-78.
- Lader M, Russell J. Guidelines for the prevention and treatment of benzodiazepine dependence: summary of a report from the Mental Health Foundation. Addiction 1993;88(12):1707-8.
- National Institute on Drug Abuse. www.nida.nih.gov
- National Institute on Alcohol Abuse and Addiction. www.niaaa.nih.gov
Drug brand names
- Alprazolam • Xanax
- Buspirone • BuSpar
- Chlordiazepoxide • Librium
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Flurazepam • Dalmane
- Gabapentin • Neurontin
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Temazepam • Restoril
- Triazolam • Halcion
- Venlafaxine • Effexor
Disclosure
This work was supported by the Attorney General’s Office, Commonwealth of Massachusetts and the United States Department of Veterans Affairs.
Dr. Sattar has received grant funding from Abbott Laboratories and is a speaker for AstraZeneca and Eli Lilly and Co.
Dr. Bhatia is a speaker for AstraZeneca, Eli Lilly and Co., Janssen Pharmaceutica, and Bristol-Myers Squibb Co.
Acknowledgment
The authors wish to thank Jennifer Hong, second-year medical student, Creighton University School of Medicine, Omaha, NE, for her assistance in preparing this article for publication.
How should you treat anxiety in substance-abusing patients: deny them benzodiazepines and risk under-treatment, or prescribe benzodiazepines for the anxiolytic effect and risk contributing to addiction?
There is no definitive answer, but one thing is clear: Among psychiatric patients, substance abusers are most likely to abuse benzodiazepines and become addicted to them.
Some argue that the abuse potential is overstated, but only limited data suggest that benzodiazepines can be safely prescribed to patients who are abusing alcohol or drugs. In this article, we discuss benzodiazepine use in these patients and offer a sobriety-based treatment approach.
Table 1
Benzodiazepines’ potency and half-lives, including half-lives of active metabolites
| Potency | Shorter half-life (hr) | Longer half-life (hr) |
|---|---|---|
| High | Alprazolam (6 to 12) Lorazepam (10 to 20) Triazolam (2) | Clonazepam (18 to 50) |
| Low | Oxazepam (4 to 15) Temazepam (8 to 22) | Chlordiazepoxide (5 to 30) [36 to 200]* Clorazepate [36 to 200]* Diazepam (20 to 100) [36 to 200]* Flurazepam [40 to 250]* |
| * [active metabolite] | ||
| Source: Ashton CH. Benzodiazepine equivalence table. Available at www.benzo.org.uk | ||
BENEFITS AND RISKS
Considered a safe substitute for barbiturates, benzodiazepines were heralded as wonder drugs when they were introduced in the 1950s. Reports of their addictive potential surfaced in the 1970s, and since then researchers have disagreed on whether benzodiazepines should be prescribed to substance-abusing or -dependent patients.
Clinical utility. Benzodiazepines are used in many clinical situations because of their:
- anxiolytic, hypnotic, anticonvulsant, antipanic, antidepressant, amnestic, anesthetic, and antispastic effects
- relatively mild side effects, when compared with alternatives such as barbiturates.
In psychiatry, benzodiazepines are used to treat anxiety disorders, agitation, and insomnia. Because of cross-tolerance with alcohol and barbiturates, benzodiazepines also are used to manage alcohol or barbiturate withdrawal.
Interactions. Benzodiazepines can interact with other psychotropics, including lithium, antipsychotics, and selective serotonin reuptake inhibitors (SSRIs). Respiratory arrest has been reported in patients taking both a high-potency benzodiazepine and clozapine.1
Overdose and withdrawal symptoms. Benzodiazepine overdose is characterized by slurred speech, sedation, memory impairment, incoordination, respiratory depression, hypotension, stupor, and coma. Abrupt withdrawal may produce life-threatening delirium, hallucinations, grand mal seizures, and symptoms similar to those of alcohol withdrawal (insomnia, anxiety, tremor, hyperactivity, nausea, vomiting, and psychomotor agitation).1
ABUSE PATTERNS
The few empiric studies examining benzodiazepines’ abuse potential in substance abusers have shown inconsistent results. However, it is generally accepted that:
- long-term benzodiazepine use may lead to tolerance and physiologic dependence
- withdrawal symptoms can occur if benzodiazepines are stopped suddenly, especially after long-term (months to years) use.
Even though most benzodiazepine prescriptions are not abused,2 a history of alcohol and drug abuse suggests high potential for benzodiazepine abuse. Also, long-term users of prescribed benzodiazepines often develop tolerance and may escalate their doses to get the same desired effects. If their supply is threatened, these patients may seek benzodiazepines illicitly.
Benzodiazepines may enhance or prolong the elation (“high”) associated with other drugs or mitigate the depression (“crash”) that follows a stimulant “high.” Sometimes benzodiazepines are the drug of choice, as high doses of potent, short-acting agents may provide a stimulant “high.”
WHO ABUSES BENZODIAZEPINES?
Alcohol and substance abusers tend to ingest benzodiazepines for recreational purposes. Thirty to 50% of alcoholics undergoing detoxification and 44% of IV drug abusers also may be abusing benzodiazepines.3
Benzodiazepines are cross-tolerant with alcohol, and alcoholics may use them with alcohol or as a substitute when alcohol is unavailable. They also may self-medicate with benzodiazepines to ease alcohol’s withdrawal symptoms. Opiate, amphetamine, and cocaine abusers may use benzodiazepines with their drugs of choice, as may younger abusers of MDMA (“Ecstasy”) and LSD.
Even patients who begin taking benzodiazepines for legitimate reasons may end up abusing them. In one study of 2,600 patients prescribed diazepam, up to 60% had abused and/or become dependent on it.4
Benzodiazepine abuse may start with other sedative/hypnotic abuse or as experimentation with drugs or alcohol, typically around age 13 or 14.5 The average benzodiazepine abuser is age 19 to 31, and the male-to-female ratio is about 2:1.6
Multi-drug abuse. Benzodiazepines are usually not the preferred or sole drug of abuse. Roughly 80% of benzodiazepine abuse may be a component of poly-drug abuse, most commonly with opioid addiction.7 A 2-year National Institute on Drug Abuse study of heroin abusers suggested that 15% also abused benzodiazepines daily for more than 1 year, and 73% had abused benzodiazepines several times during the previous week.8 Other studies suggest that up to 90% of methadone users regularly abuse benzodiazepines, often at high doses.9
Table 2
Should benzodiazepines be prescribed to substance abusers?
YES: Arguments for
|
NO: Arguments against
|
ILLICIT USE POTENTIAL
Prescriptions are the primary source of supply for benzodiazepine abusers. These patients are doctor shoppers and often change pharmacies. They visit emergency rooms frequently and may feign symptoms to obtain benzodiazepine prescriptions. They fill prescriptions for personal use or sell the drugs to illicit sources to support their addictions.
On the street, brand-name benzodiazepines are worth much more than generics because they can be identified by photographs of brand-name benzodiazepines on the Internet or in reference books. In many cities, the street value of the Xanax or Klonopin brands may be $5 to $10 per pill. A 5mg tablet of Valium-brand diazepam may sell for $5, and 10-mg tablets are worth up to $10.
Higher abuse potential. All benzodiazepines have abuse potential, and most have been reported in the literature as being abused. Those most likely to be abused have a short half-life10 (Table 1) or rapidly cross the blood brain barrier, such as alprazolam.11
Alprazolam and lorazepam are popular among benzodiazepine abusers. In experienced but nondependent users, 1 mg of alprazolam produces a sense of elation and carries an abuse potential similar to that of 10 mg of dextroamphetamine.12 Lipophilic agents such as diazepam also have a high abuse and addiction potential.
In the United States, diazepam and alprazolam appear to be the most abused benzodi-azepines.13 Flunitrazepam has become popular among high school students and drug addicts, particularly in the south and southwest. This potent benzodiazepine is not approved for use in the United States but is diverted from Latin America or Europe in the illegal drug trade.
Lower abuse potential. Benzodiazepines with longer half-lives generally are less likely to be abused, although diazepam—with a half-life of up to 100 hours—is the exception. Chlordiazepoxide has been reported to produce a lower “high” than other benzodiazepines.14 Among the short half-life benzodiazepines, oxazepam may have a relatively low abuse potential.14 Clonazepam—a high-potency benzodiazepine with a long half-life—is generally safe and may have a lower abuse or addiction potential, although its abuse has been report-ed.15 Similarly, oxazepam, clorazepate, and chlordiazepoxide may be less reinforcing than other benzodiazepines, although reports have linked these agents to abuse as well.16
TO PRESCRIBE OR NOT TO PRESCRIBE
Opponents blame benzodiazepines for promoting the drug culture and argue that prescribing benzodiazepines promotes drug abuse. Advocates of benzodiazepine therapy contend that restricting an effective and safe medication is unethical, even in substance abusers. Arguments from each perspective are summarized in Table 2.17-27
In 1990, an American Psychiatric Association task force concluded that alcohol and substance abusers could be prescribed benzodiazepines with very close monitoring but did not recommend specific standards.28
RECOMMENDATIONS
Prescribing benzodiazepines to substance abusers is not absolutely contraindicated, despite an elevated relative risk of abuse or dependence. In the absence of convincing data, physicians must decide on their own—usually case by case—the merits of using benzodiazepines to treat anxiety in substance abusers.
A sobriety-based approach. Our group at the Substance Abuse Treatment Center, VA Medical Center, Omaha, Nebraska, has developed a treatment algorithm for substance abusers presenting with anxiety (see Algorithm). It is based on clinical experience, more than 200 relevant articles, and the consensus of psychiatrists trained and certified by the American Board of Psychiatry and Neurology and the American Society of Addiction Medicine.
Algorithm Sobriety-based protocol for treating anxiety in substance abusers
Precautions for prescribing benzodiazepines
- Inform patient of planned duration of therapy
- Prescribe for brief periods (weeks to months), with follow-up at least monthly
- No refills without follow-up, and no refills over the phone
- Use random urine toxicology screening every 1 to 3 months to monitor for relapse
- Attempt to taper dosage after 3 to 6 months—even if patient resists—and monitor for objective withdrawal
- If no objective withdrawal, terminate benzodiazepine; continue other medications
- If objective withdrawal, continue benzodiazepine and reattempt taper in 3 to 6 months; continue Alcoholics/Narcotics Anonymous
We suggest that you begin by encouraging sobriety and referring willing patients to detoxification. Because most addicts deny or greatly minimize their substance abuse, investigate all potential drug or alcohol abuse thoroughly and address it appropriately.
If anxiety persists after detoxification, begin drug therapy with nonbenzodiazepines:
- Diphenhydramine, 50 to 100 mg/d, may reduce anxiety and often improves sleep, but consider its anticholinergic side effects before prescribing.
- Some SSRIs and venlafaxine are used to treat anxiety, but they generally take weeks to produce a therapeutic effect and some patients cannot wait that long.
- Mirtazapine, 15 to 30 mg/d, provides relatively rapid sedation and helps with sleep and anxiety.
- Buspirone may reduce anxiety, especially when given at 30 to 60 mg/d.
- Gabapentin, 100 to 300 mg tid or higher, may reduce anxiety and help with sleep.
- Tricyclic antidepressants may be considered, but watch for cardiac and anticholinergic side effects and overdose risks.
If anxiety does not improve with an adequate trial of first-line agents, consider adding long-acting benzodiazepines at sufficient dosages, such as clonazepam, 0.5 to 1 mg bid to tid. Prescribe scheduled doses, rather than “as needed.” Continue the first-line antianxiety agent, and reiterate to the patient that benzodiazepine therapy will be short-term.
Observe prescribing precautions (see Algorithm), and screen patients’ urine randomly every 1 to 3 months to monitor their adherence to substance abuse treatment.
Keep patient records current, with attention to dates of visits and prescriptions and quantity of benzodiazepines prescribed. To ensure proper continued use of benzodiazepines, consider consulting with physicians who have expertise in treating similar patients. Watch for possible signs of benzodiazepine dependence and abuse, such as requests for dose increases or early refills.
NONDRUG TREATMENTS
Nondrug treatments have been shown to reduce substance use and control anxiety in some studies. These include cognitive-behavioral therapy, motivational enhancement therapy, interpersonal therapy, and brief dynamic therapy, among others. Their use requires specific training or referral to more experienced colleagues. For information on these treatments, consult the Web sites of the National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism (Related resources).
Group and self-help therapies such as Alcoholics Anonymous or Narcotics Anonymous also have been shown to reduce substance use.
- Parran TV. Prescription drug abuse. A question of balance. Med Clin North Am 1997;81:967-78.
- Lader M, Russell J. Guidelines for the prevention and treatment of benzodiazepine dependence: summary of a report from the Mental Health Foundation. Addiction 1993;88(12):1707-8.
- National Institute on Drug Abuse. www.nida.nih.gov
- National Institute on Alcohol Abuse and Addiction. www.niaaa.nih.gov
Drug brand names
- Alprazolam • Xanax
- Buspirone • BuSpar
- Chlordiazepoxide • Librium
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Diazepam • Valium
- Flurazepam • Dalmane
- Gabapentin • Neurontin
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Oxazepam • Serax
- Temazepam • Restoril
- Triazolam • Halcion
- Venlafaxine • Effexor
Disclosure
This work was supported by the Attorney General’s Office, Commonwealth of Massachusetts and the United States Department of Veterans Affairs.
Dr. Sattar has received grant funding from Abbott Laboratories and is a speaker for AstraZeneca and Eli Lilly and Co.
Dr. Bhatia is a speaker for AstraZeneca, Eli Lilly and Co., Janssen Pharmaceutica, and Bristol-Myers Squibb Co.
Acknowledgment
The authors wish to thank Jennifer Hong, second-year medical student, Creighton University School of Medicine, Omaha, NE, for her assistance in preparing this article for publication.
1. Hales RE, Yudofsky SC. Textbook of clinical psychiatry, 4th ed. Washington, DC: American Psychiatric Publishing, 2003:318-19,493,501,1098.
2. Woods JH, Katz JL, Winger G. Use and abuse of benzodiazepines. Issues relevant to prescribing. JAMA 1988;260(23):3476-80.
3. Shaw M, Brabbins C, Ruben S. Misuse of benzodiazepines. Specify the formulation when prescribing. BMJ 1994;308(6945):1709.-
4. Woody GE, O’Brien CP, Greenstein R. Misuse and abuse of diazepam: an increasingly common medical problem. Int J Addict 1975;10(5):843-8.
5. Pedersen W, Lavik NJ. Adolescents and benzodiazepines: prescribed use, self-medication and intoxication. Acta Psychiatrica Scand 1991;84:94-8.
6. Ruben SM, Morrison CL. Temazepam misuse in a group of injecting drug users. Br J Addict 1992;87:1387-92,
7. Gold MS, Miller NS, Stennie K, Populla-Vardi C. Epidemiology of benzodiazepine use and dependence. Psychiatr Annals 1995;25:146-8.
8. Dumont RL. Abuse of benzodiazepines—the problems and the solutions. A report of a Committee of the Institute for Behavior and Health, Inc. Am J Drug Alcohol Abuse. 1988;14(suppl 1):1-69.
9. Iguchi MY, Griffiths RR, Bickel WK, et al. Relative abuse liability of benzodiazepines in methadone-maintained populations in three cities. In: Harris LS (ed). Problems of drug dependence, 1988. Proceedings of the 50th annual scientific meeting, the Committee on Problems of Drug Dependence, Inc. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute on Drug Abuse, Office of Science, 1989. DHHS publication no. (ADM) 89-1605.
10. Longo LP. Non-benzodiazepine pharmacotherapy of anxiety and panic in substance abusing patients. Psychiatr Annals 1998;28(3):142-53.
11. Roache JD, Meisch RA. Findings from self-administration research on the addiction potential of benzodiazepines. Psychiatr Annals 1995;25(3):153-7.
12. Zawertailo LA, Busto U, Kaplan HL, Sellers EM. Comparative abuse liability of sertraline, alprazolam, and dextroamphetamine in humans. J Clin Psychopharmacol 1995;15(2):117-24.
13. Griffiths RR, McLeod DR, Bigelow GE, et al. Comparison of diazepam and oxazepam: preference, liking and extent of abuse. J Pharmacol Exp Ther 1984;229(2):501-8.
14. Griffiths RR, Wolf B. Relative abuse liability of different benzodiazepines in drug abusers. J Clin Psychopharmacol 1990;10(4):237-43.
15. Albeck JH. Withdrawal and detoxification from benzodiazepine dependence: a potential role for clonazepam. J Clin Psychiatry 1987;48(suppl):43-9.
16. Woods JH, Katz JL, Winger G. Use and abuse of benzodiazepines: issues relevant to prescribing. JAMA 1988;260(23):3476-80.
17. Maletzky BM, Klotter J. Addiction to diazepam. Int J Addict 1976;II(1):95-115.
18. Lader M. Short-term versus long-term benzodiazepine therapy. Curr Med Res Opin 1984(8, suppl 4);120-6.
19. Ciraulo DA, Sands BK, Shader RI. Critical review of liability for benzodiazepine abuse among alcoholics. Am J Psychiatry 1988;145(12):1501-6.
20. Berner R. The patient’s perspective. NYS J Med 1991;91(11, suppl):37S-39S.
21. Schatzberg AF. Benzodiazepines: therapeutic, biological and psychosocial issues. J Psychiatr Res 1990;24(2):1-2.
22. Lader M. Drug development optimization—benzodiazepines. Agents Actions 1990;29:59-69.
23. Sellers EM, Marshman JA, Kaplan Hl, et al. Acute and chronic drug abuse emergencies in metropolitan Toronto. Int J Addict 1981;16(2):283-303.
24. Hamlin M. Guidelines for benzodiazepine prescribing. Br J Hosp Med 1989;42(1):82.-
25. Piesiur Strehlow B, Strehlow U, Poser W. Mortality of patients dependent on benzodiazepines. Acta Psychiatr Scand 1986;73:330-5.
26. Bendtsen P, Hensing G, McKenzie L, Stardsman AK. Prescribing benzodiazepines—a critical incident study of a physician dilemma. Soc Sci Med 1999;49:459-67.
27. Linnoila MI. Benzodiazepines and alcohol. J Psychiatric Res 1990;24(2, suppl):121-7.
28. Benzodiazepine dependence, toxicity and abuse. A task force report. Washington, DC: American Psychiatric Association, 1990.
1. Hales RE, Yudofsky SC. Textbook of clinical psychiatry, 4th ed. Washington, DC: American Psychiatric Publishing, 2003:318-19,493,501,1098.
2. Woods JH, Katz JL, Winger G. Use and abuse of benzodiazepines. Issues relevant to prescribing. JAMA 1988;260(23):3476-80.
3. Shaw M, Brabbins C, Ruben S. Misuse of benzodiazepines. Specify the formulation when prescribing. BMJ 1994;308(6945):1709.-
4. Woody GE, O’Brien CP, Greenstein R. Misuse and abuse of diazepam: an increasingly common medical problem. Int J Addict 1975;10(5):843-8.
5. Pedersen W, Lavik NJ. Adolescents and benzodiazepines: prescribed use, self-medication and intoxication. Acta Psychiatrica Scand 1991;84:94-8.
6. Ruben SM, Morrison CL. Temazepam misuse in a group of injecting drug users. Br J Addict 1992;87:1387-92,
7. Gold MS, Miller NS, Stennie K, Populla-Vardi C. Epidemiology of benzodiazepine use and dependence. Psychiatr Annals 1995;25:146-8.
8. Dumont RL. Abuse of benzodiazepines—the problems and the solutions. A report of a Committee of the Institute for Behavior and Health, Inc. Am J Drug Alcohol Abuse. 1988;14(suppl 1):1-69.
9. Iguchi MY, Griffiths RR, Bickel WK, et al. Relative abuse liability of benzodiazepines in methadone-maintained populations in three cities. In: Harris LS (ed). Problems of drug dependence, 1988. Proceedings of the 50th annual scientific meeting, the Committee on Problems of Drug Dependence, Inc. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute on Drug Abuse, Office of Science, 1989. DHHS publication no. (ADM) 89-1605.
10. Longo LP. Non-benzodiazepine pharmacotherapy of anxiety and panic in substance abusing patients. Psychiatr Annals 1998;28(3):142-53.
11. Roache JD, Meisch RA. Findings from self-administration research on the addiction potential of benzodiazepines. Psychiatr Annals 1995;25(3):153-7.
12. Zawertailo LA, Busto U, Kaplan HL, Sellers EM. Comparative abuse liability of sertraline, alprazolam, and dextroamphetamine in humans. J Clin Psychopharmacol 1995;15(2):117-24.
13. Griffiths RR, McLeod DR, Bigelow GE, et al. Comparison of diazepam and oxazepam: preference, liking and extent of abuse. J Pharmacol Exp Ther 1984;229(2):501-8.
14. Griffiths RR, Wolf B. Relative abuse liability of different benzodiazepines in drug abusers. J Clin Psychopharmacol 1990;10(4):237-43.
15. Albeck JH. Withdrawal and detoxification from benzodiazepine dependence: a potential role for clonazepam. J Clin Psychiatry 1987;48(suppl):43-9.
16. Woods JH, Katz JL, Winger G. Use and abuse of benzodiazepines: issues relevant to prescribing. JAMA 1988;260(23):3476-80.
17. Maletzky BM, Klotter J. Addiction to diazepam. Int J Addict 1976;II(1):95-115.
18. Lader M. Short-term versus long-term benzodiazepine therapy. Curr Med Res Opin 1984(8, suppl 4);120-6.
19. Ciraulo DA, Sands BK, Shader RI. Critical review of liability for benzodiazepine abuse among alcoholics. Am J Psychiatry 1988;145(12):1501-6.
20. Berner R. The patient’s perspective. NYS J Med 1991;91(11, suppl):37S-39S.
21. Schatzberg AF. Benzodiazepines: therapeutic, biological and psychosocial issues. J Psychiatr Res 1990;24(2):1-2.
22. Lader M. Drug development optimization—benzodiazepines. Agents Actions 1990;29:59-69.
23. Sellers EM, Marshman JA, Kaplan Hl, et al. Acute and chronic drug abuse emergencies in metropolitan Toronto. Int J Addict 1981;16(2):283-303.
24. Hamlin M. Guidelines for benzodiazepine prescribing. Br J Hosp Med 1989;42(1):82.-
25. Piesiur Strehlow B, Strehlow U, Poser W. Mortality of patients dependent on benzodiazepines. Acta Psychiatr Scand 1986;73:330-5.
26. Bendtsen P, Hensing G, McKenzie L, Stardsman AK. Prescribing benzodiazepines—a critical incident study of a physician dilemma. Soc Sci Med 1999;49:459-67.
27. Linnoila MI. Benzodiazepines and alcohol. J Psychiatric Res 1990;24(2, suppl):121-7.
28. Benzodiazepine dependence, toxicity and abuse. A task force report. Washington, DC: American Psychiatric Association, 1990.
First psychotic episode—a window of opportunity: Seize the moment to build a therapeutic alliance
A first psychotic episode offers the opportunity to build a therapeutic alliance at a teachable moment—while patients and their families are dealing with a devastating diagnosis. With a proactive approach, you can influence how patients view themselves and their experience, including psychotic illness, your efforts to treat its symptoms, and the costs and benefits of interventions.
Unfortunately, the typical first psychotic episode goes undiagnosed and untreated for 1 to 2 years, which some studies suggest may allow schizophrenia to progress. Although controversial, evidence links a prolonged duration of untreated psychosis to poorer outcome.1 Interventions during a prodromal (ie, pre-psychotic but already symptomatic) phase of schizophrenia also is being investigated, with the goal of attenuating—or perhaps even preventing—progression to frank psychosis.2-6
The implication for clinicians: timely identification and treatment may improve response, reduce relapse rates, and ultimately improve schizophrenic patients’ quality of life.
High rates of response—and relapse
Patients with a first psychotic episode show a higher response rate to antipsychotics—up to 87% within 1 year7 —and are more sensitive to side effects than are multi-episode patients.8 Yet despite their high response rate, new-onset patients often suffer from residual symptoms, even when treated in controlled settings. They also have a high rate of relapse—82% within 5 years.9
The strongest modifiable predictor of relapse is medication non-adherence, which has been shown to increase the risk of relapse five-fold.7 The first treatment experience provides a window of opportunity to help the patient accept taking medications as a normal part of life.
Mr. C, a 19-year-old college student, was brought for psychiatric admission after he told his roommates he was a new messiah who “needed to starve himself during the sunlight to enhance his holiness.” Approximately 7 months earlier he had become socially withdrawn and less able to do his college work. Two months later, he started using cannabis frequently. About 5 weeks prior to admission, he developed paranoid ideas involving his roommates and immersed himself in Eastern religions.
History and work-up. Mr. C was overweight and presented with mild dehydration. He did not report relevant signs of depression or mania and had no history of medical or psychiatric problems. Admission work-up included physical and neurologic exams, head CT, and blood work, which were unremarkable except for a positive cannabis toxicology. Family history was significant for one grandfather with alcohol abuse and one uncle who required psychiatric hospitalization in his 20s and never recovered functionally.
Family concerns. Mr. C’s parents were convinced a new diet was causing his symptoms and demanded that he be admitted to a medical ward. His brother insisted the symptoms were secondary to some “bad weed” and that everything would clear up in a few days. Although a brief medication-free observation period was considered to rule out substance-induced psychosis, the prodromal pattern of functional decline for more than 6 months and the bizarre quality of his delusions led to the diagnosis of a first episode of schizophrenia.
Treatment strategy. The treatment team met with Mr. C and his family to educate them about psychotic illness, the risks and benefits of novel antipsychotics, and the need to begin immediate treatment. With the patient’s and family’s consent, risperidone was initiated at 0.5 mg at bedtime and slowly increased over 1 week to 3 mg/d, with only mild and transient sedation. Within 3 weeks, Mr. C responded robustly and was discharged back to his family. Over the next 7 months, he continued taking risperidone, 3 mg/d, with some residual negative symptoms (social isolation without depression) and full remission of positive symptoms, which enabled him to return to college.
Therapeutic alliance. Your approach is key to building a therapeutic alliance with a person whose reality often is clouded by paranoia and referential thinking. Trust begins with the first clinical contact—during history-taking, ordering of tests, answering questions about the diagnosis, and discussing treatment options. Patients and their families must be informed about:
- target symptoms
- medication side effects
- predictors of response and relapse
- lack of certainty about how or when a patient will respond to any antipsychotic
- and the importance of rapid and uninterrupted treatment.
Supportive therapy. Support groups for the patient and family can help destigmatize the illness and reduce stress. Information about schizophrenia’s nature and course is available from the National Alliance for the Mentally Ill, National Mental Health Association, and other sources (see “Related resources”).10
CBT. Adjunctive cognitive-behavioral therapy (CBT) may speed up acute symptom response,11,12 reduce rates of nonresponse, and shorten hospital stays13 by helping patients deal with uncertainty about outer and inner realities. CBT approaches are understudied but so far have not been found to reduce relapse rates.
A moving target. As treatment moves from acute to consolidation and maintenance, target symptoms may change, side effects can limit the preferred approach, and partial or nonresponse may require drug or dosing adjustments. It is prudent to be prepared to re-evaluate the initial diagnosis as new symptoms emerge, response patterns develop, additional test or historical data become available, or as the illness’ course becomes more clear. To improve outcome, address comorbid or concurrent diseases—such as substance abuse or dependence, mood disorders, anxiety and obsessive-compulsive symptoms, or eating disorders.
Diagnostic work-up
As in Mr. C’s case (Box), a first psychotic episode is characterized by DSM-IV diagnostic criteria for schizophrenia, including hallucinations, delusions, disorganized thoughts or speech, disorganized behavior(s), or negative symptoms (such as anhedonia, amotivation, asociality, alogia, or affective flattening). The work-up is more comprehensive than that for subsequent episodes and includes a thorough history, complete physical examination, and brain imaging (Table 1) to explore other possible medical and psychiatric diagnoses (Table 2).
Table 1
WORK-UP OF PATIENTS PRESENTING WITHA FIRST EPISODE OF PSYCHOSIS
| Priority | Mode of evaluation |
|---|---|
| Routine | History Symptoms, time course, medical conditions, current/previous medications, herbs, drugs Medical and neurologic exam Blood work: CBC with differential, complete metabolic panel, thyroid and liver function tests, syphilis serology, pregnancy test, toxicology Urinalysis, toxicology ECG |
| Recommended | Fasting glucose and lipid profile (ideally before starting atypical antipsychotic) Head CT (especially if history of recent trauma) or brain MRI |
| Optional | Erythrocyte sedimentation rate, antinuclear antibodies, lumbar puncture, sleep-deprived EEG |
The history—ideally gathered from the patient and others—includes:
- medical and psychiatric diagnoses
- medications (prescribed and over-the-counter remedies)
- presence of stressors/triggers
- chronology of symptoms
- potential for the episode to endanger the patient or others.
Imaging. Despite a relatively low yield, we recommend that every patient with a first psychotic episode undergo a brain CT or MRI to rule out a potentially treatable organic cause for the psychosis.14
Other tests. Because of the increased risk of hyperglycemia, dyslipidemia, and possible cardiac conduction abnormalities with atypical antipsychotics, obtain a baseline fasting blood glucose, lipid profile, and ECG. A sleep-deprived EEG is recommended for patients with unclear motor movements or family history of epilepsy.
Choosing medications
Medication choices for the patient with first-episode schizophrenia are influenced by:
- target symptoms
- whether the symptoms endanger the patient or others
- the patient’s personal or family history of medication response or side effects
- a generally increased sensitivity to side effects in patients who have never been exposed to antipsychotics
- concurrent medical and/or psychiatric disorders
- prescriber, patient, and family preferences.
Psychiatrists generally select psychotropic classes by symptom domains (Table 3) and individual agents in each class by side effect profile. Except for clozapine’s superior effectiveness in patients with refractory psychosis, controlled studies have shown no clinically significant differences in efficacy among the drugs in each class—including the antipsychotics. Individual patients, however, may respond differently to different agents.
Principles of prescribing antipsychotics
Antipsychotics are effective in treating most psychotic core symptoms, such as hallucinations, delusions, agitation, aggression, and disorganized thinking and behavior. Other medications can be added to speed up or enhance treatment response or to target other domains.
Dosages. First-episode patients often require lower dosages and slower titration than multi-episode patients. As a rule, antipsychotics are started at about one-half the dosage given to patients with a chronic treatment history, although symptom severity and absence of side effects at lower dosages can help individualize titration.
Side effects. Atypical antipsychotics are preferred because of their reduced risk of extrapyramidal symptoms (EPS), positive effects on depressive and cognitive symptoms, and improved patient satisfaction and adherence, compared with the older antipsychotics.15-17 Atypicals’ potential side effects include weight gain, hyperglycemia, and dyslipidemia,18 as well as often-overlooked sexual side effects.19
Table 2
DIFFERENTIAL DIAGNOSIS OF FIRST-EPISODE PSYCHOSIS
| Possible diagnosis | Key points for differentiation |
|---|---|
| Schizophrenia | 6 months of psychosis* (including prodromal symptoms); total duration of mood episodes brief relative to active and residual psychotic phases; not directly caused by medical condition or substance |
| Schizophreniform disorder | Same as above, except symptoms are present 1 to 6 months |
| Brief psychotic disorder | Same as above, except symptoms are present 1 day to 1 month |
| Delusional disorder | Apart from non-bizarre delusions, functioning not markedly impaired; total duration of mood episodes brief relative to active and residual psychotic phases; not caused by direct physiologic effects of medical condition or substance |
| Psychotic disorder NOS | Psychotic symptoms insufficient to make a specific diagnosis |
| Schizoaffective disorder | Like schizophrenia for at least 2 weeks, but with mania or major depression present for much of the active and residual psychotic periods |
| Mood disorder with psychosis | Psychotic symptoms occur exclusively during mood disorder episodes |
| Psychosis due to general medical condition | Psychotic symptoms caused by direct physiologic effects of a general medical condition |
| Delirium due to general medical condition | Psychotic symptoms associated with a disturbance in consciousness and other cognitive deficits; characterized by a fluctuating course |
| Dementia due to general medical condition | Psychotic symptoms associated with memory impairment and other cognitive deficits |
| Substance-induced psychotic disorder | Psychotic symptoms caused by direct physiologic effects of a substance; reaction exceeds that usually encountered with intoxication or withdrawal |
| Substance-induced psychotic delirium | Similar to above, but associated with a disturbance in consciousness and other cognitive deficits; characterized by a fluctuating course |
| Substance intoxication or withdrawal | Caused by direct physiologic effects of a substance; reaction is typically encountered with intoxication or withdrawal |
| Conversion disorder | Contradictory and inconsistent history and presentation; secondary gain |
| Malingering | Contradictory and inconsistent history and presentation; primary gain |
| * Psychotic symptoms must interfere with functioning, and at least two of the following are required: delusions, hallucinations, disorganized thoughts or speech, disorganized behavior, or negative symptoms (avolition, alogia, affective flattening, asociality, or anhedonia), unless delusions are bizarre (impossible), or hallucinations consist of a running commentary or of two or more voices conversing with each other. | |
| Source: Adapted from DSM-IV handbook of differential diagnosis. Washington, DC: American Psychiatric Press, 1995. | |
Consider the patient’s risk for side effects when choosing an atypical antipsychotic, as each drug has strengths and weaknesses. For example, it may be reasonable to consider:
- risperidone, ziprasidone, or aripiprazole—rather than olanzapine or quetiapine—for patients at high risk for weight gain or with a family history of diabetes
- avoiding risperidone for patients with an early indication of sensitivity to EPS or prolactin-related side effects
- an agent that can be loaded rapidly, such as olanzapine or aripiprazole—rather than an agent that requires titration, such as quetiapine—for a patient presenting with severe agitation.
Clozapine—because of its side effect potential—is generally reserved for patients who have not responded to at least two antipsychotic trials of at least 4 to 6 weeks duration and have a steady-state clozapine level >350 ng/dl.
Acute agitation. Short-acting IM formulations can be used effectively for acute agitation and impulsivity or for patients who refuse oral antipsychotics. Until recently, only first-generation antipsychotics such as haloperidol and fluphenazine were available in IM formulations. Injectable ziprasidone mesylate is now available for acute agitation and has been found to be as safe and effective as haloperidol. Lorazepam may be used to treat agitation or as an adjunct to a patient’s antipsychotic agent.
Adjunctive therapies
When antipsychotic monotherapy is inadequate, adjunctive medications may be considered:
- to treat catatonia, obsessive-compulsive disorder, or depression
- to resolve agitation or mania more quickly
- to control agitation or anxiety while an antipsychotic dosage is being titrated
- when side effects emerge or residual symptoms remain despite adequate dosage and duration of antipsychotic treatment.
Deciding if and when to add another drug depends on the nature and severity of target symptoms, the degree and time course of response, and whether side effects appear. If you use adjunctive therapies during acute stabilization, attempt to taper and discontinue them after the patient’s symptoms have improved. Avoid combining antipsychotics, as no clinical data support the effectiveness and safety of this practice.20
Benzodiazepines are often used adjunctively for agitation, anxiety, or temporary insomnia in patients with schizophrenia. Common dosages are:
- for agitation or anxiety, lorazepam, 0.5 to 2 mg bid or tid, or clonazepam, 0.5 mg bid to 2 mg tid
- for insomnia, lorazepam or clonazepam, 1 to 2 mg at bedtime, or zolpidem, 5 to 10 mg at bedtime.
Benzodiazepines also are effective for catatonia, which may be misdiagnosed as negative symptoms or depression when it presents as marked psychomotor retardation, staring, selective mutism, negativism, mild posturing, or stereotypies. If undiagnosed, catatonia may worsen during antipsychotic titration. Symptoms usually respond to high-dose lorazepam, 1 mg bid or tid, with increases up to a maximum dosage of 12 mg/d.
Mood stabilizers are often used adjunctively to treat acute agitation and disinhibition21 or as add-on agents for residual psychotic or affective symptoms. Recommended dosages and blood levels, as tolerated, are:
- valproic acid, starting at 10 to 20 mg/kg bid, with a target serum level of 60 to 120 μg/ml. A once-daily, extended-release formulation may improve compliance.
- lithium, starting at 300 mg bid to tid, aiming for a serum level of 0.8 to 1.2 mEq/L.
Manic symptoms in a schizoaffective presentation may require one or even two mood stabilizers.
Lamotrigine may be the treatment of choice for depressed patients with schizoaffective disorder, bipolar type.22 However, it must be started at 25 mg/d and titrated extremely slowly—by 25 mg every other week, up to a target 200 to 400 mg/d—to avoid the risk of potentially fatal Stevens-Johnson syndrome.
Gabapentin, 300 mg tid to 1,500 mg tid, may help treat anxiety—particularly in patients with comorbid substance abuse, in whom benzodiazepines should be used sparingly after stabilization.
Table 3
SYMPTOM-BASED DRUG TREATMENT OF FIRST-EPISODE SCHIZOPHRENIA
| Target symptom | Medication choices | Dosage range | Comments |
|---|---|---|---|
| Agitation | Atypical antipsychotic Mood stabilizer Benzodiazepine ECT | Depends on level of agitation and individual agent | Ziprasidone IM may be useful for acute agitation |
| Psychosis | Atypical antipsychotic ECT | =50% of dosage used for multiple episode patients | Start low/go slow, monitor side effects |
| Catatonia | Lorazepam ECT | 1 mg/d bid to4 mg/d tid | Use sedation, symptom resolution as threshold |
| Negative symptoms | Atypical antipsychotic Glycine, cycloserine may help | Dictated by positive symptom response | Effect on functioning, quality of life unclear |
| Cognitive symptoms | Atypical antipsychotic | Same as above for negative symptoms | Same as above |
| Insomnia | Lorazepam Zolpidem Trazodone Mirtazapine | 1 to 2 mg HS 5 to 10 mg HS 50 to 200 mg HS 7.5 to 15 mg HS | Short-term treatment preferred |
| Depression, obsessive-compulsive symptoms, anxiety/panic | SSRI, SNDRI, NDRI, SARI, NASA | As per individual agent Possible interference with | antipsychotic blood levels* |
| Parkinsonism | Benztropine Trihexyphenidyl | 0.5 to 2 mg/d 1 to 15 mg/d | Possible effect of decreased cognition |
| Akathisia | Propranolol | 20 to 160 mg/d | Monitor blood pressure |
| Weight gain | Ziprasidone Aripiprazole | Dictated by positive symptom response | Prevention more effective than remediation |
| Non-adherence | Olanzapine oral disintegrating tablets Risperidone liquid Risperidone microspheres | 5 to 20 mg/d 1 to 4 mg/d IM injections every 2 weeks | Dissolves instantly Can be mixed with water, coffee, orange juice, or low-fat milk Not yet available |
| * Fluoxetine and paroxetine increase risperidone levels by CYP-P450 2D6 inhibition; fluvoxamine increases clozapine and olanzapine levels by CYP-P450 1A2 inhibition; fluvoxamine and nefazodone increase quetiapine and may increase ziprasidone levels by CYP-P450 3A4 inhibition; all three CYP-P450 inhibitors may increase aripiprazole levels, but the extent is not known. | |||
| ECT: electroconvulsive therapy; SSRI: selective serotonin reuptake inhibitor; SNDRI: serotonin-norepinephrine-dopamine reuptake inhibitor; NDRI: norepinephrinedopamine reuptake inhibitor; SARI: serotonin antagonist and reuptake inhibitor; NASA: norepinephrine-antagonist and serotonin antagonist. | |||
Antidepressants. Patients with schizophrenia can develop depression, even if they do not meet diagnostic criteria for schizoaffective disorder. Untreated depression can lead to non-adherence, self-medication with alcohol or illicit substances, and increased risk of suicide.
Differentiating depression from negative symptoms may be difficult, but there are subtle distinctions:
- Patients with negative symptoms appear more emotionally flat and unconcerned about their lack of motivation and diminished social and role functioning.
- Depressed persons often verbalize their demoralization, hopelessness, and desire to feel and behave differently.
Treat depression with any selective serotonin reuptake inhibitor or other newer-generation antidepressant such as mirtazapine, nefazodone, or venlafaxine at usual doses, as tolerated.
Miscellaneous medications. Use anticholinergic medications such as benztropine, 0.5 to 2 mg bid, or trihexyphenidyl, 1 to 5 mg bid, if parkinsonian symptoms occur and changing to an antipsychotic with a lower EPS potential is not feasible.
For akathisia, propranolol (10 mg bid or tid; titrate up to 160 mg/d if pulse rate and blood pressure remain stable) or benzodiazepines may be useful. Amantadine may also be used at dosages between 50 and 150 mg bid.
Insomnia may be treated with low dosages of sedating antidepressants, such as trazodone, 50 to 200 mg HS, or mirtazapine, 7.5 to 15 mg HS.
Preventing relapse during maintenance
Medication adherence depends on patient insight and attitude towards medications.23 Once you start a first-episode patient on drug therapy, encourage adherence by monitoring symptoms and anticipating side effects. Every 3 months after the acute phase:
- Use a structured evaluation, such as the Brief Psychiatric Rating Scale,24 to plot symptom severity, response, and risk for relapse.
- Rate EPS with the Simpson-Angus Scale25 and tardive dyskinesia (TD) with the Abnormal Involuntary Movement Scale26 because EPS and TD are associated with poor symptom response, adherence, and outcome.7
Reinforce information about the chronic nature of schizophrenia, especially when the patient or family question why treatment is needed if symptoms have resolved. Continue to counsel them about the patient’s need for:
- regular sleep of sufficient duration and without sleep-wake reversal
- gradual return to premorbid social, educational, and vocational activities/responsibilities
- ongoing treatment.
Encourage vigilance for relapse warning signs, including insomnia, social withdrawal, anxiety, refusal to eat or take medications, suspiciousness, agitation, disorganization, preoccupation with overvalued ideas, or responses to internal stimuli.
If the patient is noncompliant with antipsychotics in tablets or capsules, options include:
- liquid risperidone or olanzapine in a rapidly dissolving form that the patient cannot hide and spit out later
- long-acting depot formulations if the patient cannot be supervised and monitored daily. Older antipsychotics (such as haloperidol decanoate and fluphenazine decanoate) are available in depot formulations, and the FDA is considering risperidone in a microsphere formulation that would allow biweekly injections.
Medication withdrawal
Although the ideal duration of maintenance treatment after a first psychotic episode is debatable, we recommend that antipsychotics be continued at the full dosage that achieved symptom remission for at least 1 year.27 Then, if the patient has returned to the premorbid baseline, you can attempt a gradual medication withdrawal across 2 to 4 months, ideally when the patient’s environment is stable.
Be cautious when withdrawing antipsychotics from patients with a family history of psychosis. Consider a more gradual dose reduction, ongoing group and/or individual psychotherapy, and at least monthly monitoring. When possible, involve people who are significant in the patient’s life and educate them to look for deterioration’s warning signs, such as insomnia, irritability, anxiety, social withdrawal, preoccupation with overvalued ideas, or pacing.
If relapse occurs, carefully assess how well the patient has adhered to medication. Once a second psychotic episode occurs, his or her medication probably should be continued indefinitely.
- National Alliance for the Mentally Ill (800) 950-NAMI (6264); www.nami.org
- National Mental Health Association (800) 969-NMHA (6642); www.nmha.org
- National Alliance for Research on Schizophrenia and Depression (516) 829-0091; www.narsad.org/index.html
- Miller R, Mason SE. Diagnosis schizophrenia. A comprehensive resource. New York: Columbia University Press, 2002.
Drug brand names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
- Zolpidem • Ambien
Disclosure
Dr. Correll reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Mendelowitz receives grant/research support from, is a consultant to, and/or is a speaker for Pfizer Inc., Bristol-Myers Squibb Co.; and AstraZeneca Pharmaceuticals.
Acknowledgments
Research for this article was supported by grant 5P30MH60575 to The Zucker Hillside Hospital Intervention Research Center for Schizophrenia from the National Institute of Mental Health, Bethesda, MD.
1. Norman RM, Malla AK. Duration of untreated psychosis: a critical examination of the concept and its importance. Psychol Med 2001;31:381-400.
2. Cornblatt B, Lencz T, Obuchowski M. The schizophrenia prodrome: treatment and high-risk perspectives. Schizophr Res 2002;54(1-2):177-86.
3. McGorry PD, Yung AR, Phillips LJ, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry 2002;59(10):921-8.
4. Heinssen RK, Perkins DO, Appelbaum PS, et al. Informed consent in early psychosis research: National Institute of Mental Health workshop, November 15, 2000. Schizophr Bull 2001;27(4):571-83.
5. Miller TJ, McGlashan TH, Rosen JL, et al. Prospective diagnosis of the initial prodrome for schizophrenia based on the Structured Interview for Prodromal Syndromes: preliminary evidence of interrater reliability and predictive validity. Am J Psychiatry 2002;159(5):863-5.
6. Young LT, Bakish D, Beaulieu S. The neurobiology of treatment response to antidepressants and mood stabilizing medications. J Psychiatry Neurosci 2002;27(4):260-5.
7. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry 1999;56(3):241-7.
8. Tauscher J, Kapur S. Choosing the right dose of antipsychotics in schizophrenia: lessons from neuroimaging studies. CNS Drugs 2001;15:671-8.
9. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry 1999;56(3):241-7.
10. Miller R, Mason SE. Diagnosis schizophrenia. A comprehensive resource, New York: Columbia University Press, 2002.
11. Lewis S, Tarrier N, Haddock G, et al. Randomised controlled trial of cognitivebehavioural therapy in early schizophrenia: acute-phase outcomes. Br J Psychiatry Suppl 2002;43:S91-7.
12. Turkington D, Kingdon D, Turner T. Effectiveness of a brief cognitive-behavioural therapy intervention in the treatment of schizophrenia. Br J Psychiatry 2002;180:523-7.
13. Cormac I, Jones C, Campbell C. Cognitive behaviour therapy for schizophrenia. Cochrane Database Syst Rev 2002;(1):CD000524.-
14. Smith GN, Flynn SW, Kopala LC, et al. A comprehensive method of assessing routine CT scans in schizophrenia. Acta Psychiatr Scand 1997;96:395-401.
15. Leucht S, Pitschel-Walz G, Abraham D, et al. Efficacy and extrapyramidal sideeffects of the new antipsychotics olanzapine, quetiapine, risperidone, and sertindole compared to conventional antipsychotics and placebo. A meta-analysis of randomized controlled trials. Schizophr Res 1999;35(1):51-68.
16. Keefe RS, Silva SG, Perkins DO, Lieberman JA. The effects of atypical antipsychotic drugs on neurocognitive impairment in schizophrenia: A review and meta-analysis. Schizophr Bull 1999;25(2):201-22.
17. Dolder CR, Lacro JP, Dunn LB, Jeste DV. Antipsychotic medication adherence: is there a difference between typical and atypical agents? Am J Psychiatry 2002;159(1):103-8.
18. Vieweg WVR, Adler RA, Fernandez A. Weight control and antipsychotics: How to tip the scale away from diabetes and heart disease. Current Psychiatry 2002;1(5):10-19.
19. Compton MT, Miller AH. Sexual side effects associated with conventional and atypical antipsychotics. Psychopharmacol Bull 2001;35:89-108.
20. Stahl SM. Antipsychotic polypharmacy: squandering precious resources? J Clin Psychiatry 2002;63(2):93-4.
21. Casey DE, Daniel DG, Wassef AA, Tracy KA, Wozniak P, Sommerville KW. Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology 2003;28(1):182-92.
22. Calabrese JR, Shelton MD, Rapport DJ, Kimmel SE. Bipolar disorders and the effectiveness of novel anticonvulsants. J Clin Psychiatry. 2002;63(Suppl 3):5-9.
23. Kampman O, Laippala P, Vaananen J, et al. Indicators of medication compliance in first-episode psychosis. Psychiatry Res 2002;110:39-48.
24. Overall JE, Gorham DR. Brief psychiatric rating scale. Psychol Rep 1962;10:799-812.
25. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand 1970(suppl);212:11-19.
26. Guy W (ed). ECDEU assessment manual for psychopharmacology. Publication ABM 76-338. Washington, DC: U.S. Department of Health, Education, and Welfare, 1976;534-7.
27. Bosveld-van Haandel LJ, Slooff CJ, van den Bosch RJ. Reasoning about the optimal duration of prophylactic antipsychotic medication in schizophrenia: evidence and arguments. Acta Psychiatr Scand 2001;103(5):335-46.
A first psychotic episode offers the opportunity to build a therapeutic alliance at a teachable moment—while patients and their families are dealing with a devastating diagnosis. With a proactive approach, you can influence how patients view themselves and their experience, including psychotic illness, your efforts to treat its symptoms, and the costs and benefits of interventions.
Unfortunately, the typical first psychotic episode goes undiagnosed and untreated for 1 to 2 years, which some studies suggest may allow schizophrenia to progress. Although controversial, evidence links a prolonged duration of untreated psychosis to poorer outcome.1 Interventions during a prodromal (ie, pre-psychotic but already symptomatic) phase of schizophrenia also is being investigated, with the goal of attenuating—or perhaps even preventing—progression to frank psychosis.2-6
The implication for clinicians: timely identification and treatment may improve response, reduce relapse rates, and ultimately improve schizophrenic patients’ quality of life.
High rates of response—and relapse
Patients with a first psychotic episode show a higher response rate to antipsychotics—up to 87% within 1 year7 —and are more sensitive to side effects than are multi-episode patients.8 Yet despite their high response rate, new-onset patients often suffer from residual symptoms, even when treated in controlled settings. They also have a high rate of relapse—82% within 5 years.9
The strongest modifiable predictor of relapse is medication non-adherence, which has been shown to increase the risk of relapse five-fold.7 The first treatment experience provides a window of opportunity to help the patient accept taking medications as a normal part of life.
Mr. C, a 19-year-old college student, was brought for psychiatric admission after he told his roommates he was a new messiah who “needed to starve himself during the sunlight to enhance his holiness.” Approximately 7 months earlier he had become socially withdrawn and less able to do his college work. Two months later, he started using cannabis frequently. About 5 weeks prior to admission, he developed paranoid ideas involving his roommates and immersed himself in Eastern religions.
History and work-up. Mr. C was overweight and presented with mild dehydration. He did not report relevant signs of depression or mania and had no history of medical or psychiatric problems. Admission work-up included physical and neurologic exams, head CT, and blood work, which were unremarkable except for a positive cannabis toxicology. Family history was significant for one grandfather with alcohol abuse and one uncle who required psychiatric hospitalization in his 20s and never recovered functionally.
Family concerns. Mr. C’s parents were convinced a new diet was causing his symptoms and demanded that he be admitted to a medical ward. His brother insisted the symptoms were secondary to some “bad weed” and that everything would clear up in a few days. Although a brief medication-free observation period was considered to rule out substance-induced psychosis, the prodromal pattern of functional decline for more than 6 months and the bizarre quality of his delusions led to the diagnosis of a first episode of schizophrenia.
Treatment strategy. The treatment team met with Mr. C and his family to educate them about psychotic illness, the risks and benefits of novel antipsychotics, and the need to begin immediate treatment. With the patient’s and family’s consent, risperidone was initiated at 0.5 mg at bedtime and slowly increased over 1 week to 3 mg/d, with only mild and transient sedation. Within 3 weeks, Mr. C responded robustly and was discharged back to his family. Over the next 7 months, he continued taking risperidone, 3 mg/d, with some residual negative symptoms (social isolation without depression) and full remission of positive symptoms, which enabled him to return to college.
Therapeutic alliance. Your approach is key to building a therapeutic alliance with a person whose reality often is clouded by paranoia and referential thinking. Trust begins with the first clinical contact—during history-taking, ordering of tests, answering questions about the diagnosis, and discussing treatment options. Patients and their families must be informed about:
- target symptoms
- medication side effects
- predictors of response and relapse
- lack of certainty about how or when a patient will respond to any antipsychotic
- and the importance of rapid and uninterrupted treatment.
Supportive therapy. Support groups for the patient and family can help destigmatize the illness and reduce stress. Information about schizophrenia’s nature and course is available from the National Alliance for the Mentally Ill, National Mental Health Association, and other sources (see “Related resources”).10
CBT. Adjunctive cognitive-behavioral therapy (CBT) may speed up acute symptom response,11,12 reduce rates of nonresponse, and shorten hospital stays13 by helping patients deal with uncertainty about outer and inner realities. CBT approaches are understudied but so far have not been found to reduce relapse rates.
A moving target. As treatment moves from acute to consolidation and maintenance, target symptoms may change, side effects can limit the preferred approach, and partial or nonresponse may require drug or dosing adjustments. It is prudent to be prepared to re-evaluate the initial diagnosis as new symptoms emerge, response patterns develop, additional test or historical data become available, or as the illness’ course becomes more clear. To improve outcome, address comorbid or concurrent diseases—such as substance abuse or dependence, mood disorders, anxiety and obsessive-compulsive symptoms, or eating disorders.
Diagnostic work-up
As in Mr. C’s case (Box), a first psychotic episode is characterized by DSM-IV diagnostic criteria for schizophrenia, including hallucinations, delusions, disorganized thoughts or speech, disorganized behavior(s), or negative symptoms (such as anhedonia, amotivation, asociality, alogia, or affective flattening). The work-up is more comprehensive than that for subsequent episodes and includes a thorough history, complete physical examination, and brain imaging (Table 1) to explore other possible medical and psychiatric diagnoses (Table 2).
Table 1
WORK-UP OF PATIENTS PRESENTING WITHA FIRST EPISODE OF PSYCHOSIS
| Priority | Mode of evaluation |
|---|---|
| Routine | History Symptoms, time course, medical conditions, current/previous medications, herbs, drugs Medical and neurologic exam Blood work: CBC with differential, complete metabolic panel, thyroid and liver function tests, syphilis serology, pregnancy test, toxicology Urinalysis, toxicology ECG |
| Recommended | Fasting glucose and lipid profile (ideally before starting atypical antipsychotic) Head CT (especially if history of recent trauma) or brain MRI |
| Optional | Erythrocyte sedimentation rate, antinuclear antibodies, lumbar puncture, sleep-deprived EEG |
The history—ideally gathered from the patient and others—includes:
- medical and psychiatric diagnoses
- medications (prescribed and over-the-counter remedies)
- presence of stressors/triggers
- chronology of symptoms
- potential for the episode to endanger the patient or others.
Imaging. Despite a relatively low yield, we recommend that every patient with a first psychotic episode undergo a brain CT or MRI to rule out a potentially treatable organic cause for the psychosis.14
Other tests. Because of the increased risk of hyperglycemia, dyslipidemia, and possible cardiac conduction abnormalities with atypical antipsychotics, obtain a baseline fasting blood glucose, lipid profile, and ECG. A sleep-deprived EEG is recommended for patients with unclear motor movements or family history of epilepsy.
Choosing medications
Medication choices for the patient with first-episode schizophrenia are influenced by:
- target symptoms
- whether the symptoms endanger the patient or others
- the patient’s personal or family history of medication response or side effects
- a generally increased sensitivity to side effects in patients who have never been exposed to antipsychotics
- concurrent medical and/or psychiatric disorders
- prescriber, patient, and family preferences.
Psychiatrists generally select psychotropic classes by symptom domains (Table 3) and individual agents in each class by side effect profile. Except for clozapine’s superior effectiveness in patients with refractory psychosis, controlled studies have shown no clinically significant differences in efficacy among the drugs in each class—including the antipsychotics. Individual patients, however, may respond differently to different agents.
Principles of prescribing antipsychotics
Antipsychotics are effective in treating most psychotic core symptoms, such as hallucinations, delusions, agitation, aggression, and disorganized thinking and behavior. Other medications can be added to speed up or enhance treatment response or to target other domains.
Dosages. First-episode patients often require lower dosages and slower titration than multi-episode patients. As a rule, antipsychotics are started at about one-half the dosage given to patients with a chronic treatment history, although symptom severity and absence of side effects at lower dosages can help individualize titration.
Side effects. Atypical antipsychotics are preferred because of their reduced risk of extrapyramidal symptoms (EPS), positive effects on depressive and cognitive symptoms, and improved patient satisfaction and adherence, compared with the older antipsychotics.15-17 Atypicals’ potential side effects include weight gain, hyperglycemia, and dyslipidemia,18 as well as often-overlooked sexual side effects.19
Table 2
DIFFERENTIAL DIAGNOSIS OF FIRST-EPISODE PSYCHOSIS
| Possible diagnosis | Key points for differentiation |
|---|---|
| Schizophrenia | 6 months of psychosis* (including prodromal symptoms); total duration of mood episodes brief relative to active and residual psychotic phases; not directly caused by medical condition or substance |
| Schizophreniform disorder | Same as above, except symptoms are present 1 to 6 months |
| Brief psychotic disorder | Same as above, except symptoms are present 1 day to 1 month |
| Delusional disorder | Apart from non-bizarre delusions, functioning not markedly impaired; total duration of mood episodes brief relative to active and residual psychotic phases; not caused by direct physiologic effects of medical condition or substance |
| Psychotic disorder NOS | Psychotic symptoms insufficient to make a specific diagnosis |
| Schizoaffective disorder | Like schizophrenia for at least 2 weeks, but with mania or major depression present for much of the active and residual psychotic periods |
| Mood disorder with psychosis | Psychotic symptoms occur exclusively during mood disorder episodes |
| Psychosis due to general medical condition | Psychotic symptoms caused by direct physiologic effects of a general medical condition |
| Delirium due to general medical condition | Psychotic symptoms associated with a disturbance in consciousness and other cognitive deficits; characterized by a fluctuating course |
| Dementia due to general medical condition | Psychotic symptoms associated with memory impairment and other cognitive deficits |
| Substance-induced psychotic disorder | Psychotic symptoms caused by direct physiologic effects of a substance; reaction exceeds that usually encountered with intoxication or withdrawal |
| Substance-induced psychotic delirium | Similar to above, but associated with a disturbance in consciousness and other cognitive deficits; characterized by a fluctuating course |
| Substance intoxication or withdrawal | Caused by direct physiologic effects of a substance; reaction is typically encountered with intoxication or withdrawal |
| Conversion disorder | Contradictory and inconsistent history and presentation; secondary gain |
| Malingering | Contradictory and inconsistent history and presentation; primary gain |
| * Psychotic symptoms must interfere with functioning, and at least two of the following are required: delusions, hallucinations, disorganized thoughts or speech, disorganized behavior, or negative symptoms (avolition, alogia, affective flattening, asociality, or anhedonia), unless delusions are bizarre (impossible), or hallucinations consist of a running commentary or of two or more voices conversing with each other. | |
| Source: Adapted from DSM-IV handbook of differential diagnosis. Washington, DC: American Psychiatric Press, 1995. | |
Consider the patient’s risk for side effects when choosing an atypical antipsychotic, as each drug has strengths and weaknesses. For example, it may be reasonable to consider:
- risperidone, ziprasidone, or aripiprazole—rather than olanzapine or quetiapine—for patients at high risk for weight gain or with a family history of diabetes
- avoiding risperidone for patients with an early indication of sensitivity to EPS or prolactin-related side effects
- an agent that can be loaded rapidly, such as olanzapine or aripiprazole—rather than an agent that requires titration, such as quetiapine—for a patient presenting with severe agitation.
Clozapine—because of its side effect potential—is generally reserved for patients who have not responded to at least two antipsychotic trials of at least 4 to 6 weeks duration and have a steady-state clozapine level >350 ng/dl.
Acute agitation. Short-acting IM formulations can be used effectively for acute agitation and impulsivity or for patients who refuse oral antipsychotics. Until recently, only first-generation antipsychotics such as haloperidol and fluphenazine were available in IM formulations. Injectable ziprasidone mesylate is now available for acute agitation and has been found to be as safe and effective as haloperidol. Lorazepam may be used to treat agitation or as an adjunct to a patient’s antipsychotic agent.
Adjunctive therapies
When antipsychotic monotherapy is inadequate, adjunctive medications may be considered:
- to treat catatonia, obsessive-compulsive disorder, or depression
- to resolve agitation or mania more quickly
- to control agitation or anxiety while an antipsychotic dosage is being titrated
- when side effects emerge or residual symptoms remain despite adequate dosage and duration of antipsychotic treatment.
Deciding if and when to add another drug depends on the nature and severity of target symptoms, the degree and time course of response, and whether side effects appear. If you use adjunctive therapies during acute stabilization, attempt to taper and discontinue them after the patient’s symptoms have improved. Avoid combining antipsychotics, as no clinical data support the effectiveness and safety of this practice.20
Benzodiazepines are often used adjunctively for agitation, anxiety, or temporary insomnia in patients with schizophrenia. Common dosages are:
- for agitation or anxiety, lorazepam, 0.5 to 2 mg bid or tid, or clonazepam, 0.5 mg bid to 2 mg tid
- for insomnia, lorazepam or clonazepam, 1 to 2 mg at bedtime, or zolpidem, 5 to 10 mg at bedtime.
Benzodiazepines also are effective for catatonia, which may be misdiagnosed as negative symptoms or depression when it presents as marked psychomotor retardation, staring, selective mutism, negativism, mild posturing, or stereotypies. If undiagnosed, catatonia may worsen during antipsychotic titration. Symptoms usually respond to high-dose lorazepam, 1 mg bid or tid, with increases up to a maximum dosage of 12 mg/d.
Mood stabilizers are often used adjunctively to treat acute agitation and disinhibition21 or as add-on agents for residual psychotic or affective symptoms. Recommended dosages and blood levels, as tolerated, are:
- valproic acid, starting at 10 to 20 mg/kg bid, with a target serum level of 60 to 120 μg/ml. A once-daily, extended-release formulation may improve compliance.
- lithium, starting at 300 mg bid to tid, aiming for a serum level of 0.8 to 1.2 mEq/L.
Manic symptoms in a schizoaffective presentation may require one or even two mood stabilizers.
Lamotrigine may be the treatment of choice for depressed patients with schizoaffective disorder, bipolar type.22 However, it must be started at 25 mg/d and titrated extremely slowly—by 25 mg every other week, up to a target 200 to 400 mg/d—to avoid the risk of potentially fatal Stevens-Johnson syndrome.
Gabapentin, 300 mg tid to 1,500 mg tid, may help treat anxiety—particularly in patients with comorbid substance abuse, in whom benzodiazepines should be used sparingly after stabilization.
Table 3
SYMPTOM-BASED DRUG TREATMENT OF FIRST-EPISODE SCHIZOPHRENIA
| Target symptom | Medication choices | Dosage range | Comments |
|---|---|---|---|
| Agitation | Atypical antipsychotic Mood stabilizer Benzodiazepine ECT | Depends on level of agitation and individual agent | Ziprasidone IM may be useful for acute agitation |
| Psychosis | Atypical antipsychotic ECT | =50% of dosage used for multiple episode patients | Start low/go slow, monitor side effects |
| Catatonia | Lorazepam ECT | 1 mg/d bid to4 mg/d tid | Use sedation, symptom resolution as threshold |
| Negative symptoms | Atypical antipsychotic Glycine, cycloserine may help | Dictated by positive symptom response | Effect on functioning, quality of life unclear |
| Cognitive symptoms | Atypical antipsychotic | Same as above for negative symptoms | Same as above |
| Insomnia | Lorazepam Zolpidem Trazodone Mirtazapine | 1 to 2 mg HS 5 to 10 mg HS 50 to 200 mg HS 7.5 to 15 mg HS | Short-term treatment preferred |
| Depression, obsessive-compulsive symptoms, anxiety/panic | SSRI, SNDRI, NDRI, SARI, NASA | As per individual agent Possible interference with | antipsychotic blood levels* |
| Parkinsonism | Benztropine Trihexyphenidyl | 0.5 to 2 mg/d 1 to 15 mg/d | Possible effect of decreased cognition |
| Akathisia | Propranolol | 20 to 160 mg/d | Monitor blood pressure |
| Weight gain | Ziprasidone Aripiprazole | Dictated by positive symptom response | Prevention more effective than remediation |
| Non-adherence | Olanzapine oral disintegrating tablets Risperidone liquid Risperidone microspheres | 5 to 20 mg/d 1 to 4 mg/d IM injections every 2 weeks | Dissolves instantly Can be mixed with water, coffee, orange juice, or low-fat milk Not yet available |
| * Fluoxetine and paroxetine increase risperidone levels by CYP-P450 2D6 inhibition; fluvoxamine increases clozapine and olanzapine levels by CYP-P450 1A2 inhibition; fluvoxamine and nefazodone increase quetiapine and may increase ziprasidone levels by CYP-P450 3A4 inhibition; all three CYP-P450 inhibitors may increase aripiprazole levels, but the extent is not known. | |||
| ECT: electroconvulsive therapy; SSRI: selective serotonin reuptake inhibitor; SNDRI: serotonin-norepinephrine-dopamine reuptake inhibitor; NDRI: norepinephrinedopamine reuptake inhibitor; SARI: serotonin antagonist and reuptake inhibitor; NASA: norepinephrine-antagonist and serotonin antagonist. | |||
Antidepressants. Patients with schizophrenia can develop depression, even if they do not meet diagnostic criteria for schizoaffective disorder. Untreated depression can lead to non-adherence, self-medication with alcohol or illicit substances, and increased risk of suicide.
Differentiating depression from negative symptoms may be difficult, but there are subtle distinctions:
- Patients with negative symptoms appear more emotionally flat and unconcerned about their lack of motivation and diminished social and role functioning.
- Depressed persons often verbalize their demoralization, hopelessness, and desire to feel and behave differently.
Treat depression with any selective serotonin reuptake inhibitor or other newer-generation antidepressant such as mirtazapine, nefazodone, or venlafaxine at usual doses, as tolerated.
Miscellaneous medications. Use anticholinergic medications such as benztropine, 0.5 to 2 mg bid, or trihexyphenidyl, 1 to 5 mg bid, if parkinsonian symptoms occur and changing to an antipsychotic with a lower EPS potential is not feasible.
For akathisia, propranolol (10 mg bid or tid; titrate up to 160 mg/d if pulse rate and blood pressure remain stable) or benzodiazepines may be useful. Amantadine may also be used at dosages between 50 and 150 mg bid.
Insomnia may be treated with low dosages of sedating antidepressants, such as trazodone, 50 to 200 mg HS, or mirtazapine, 7.5 to 15 mg HS.
Preventing relapse during maintenance
Medication adherence depends on patient insight and attitude towards medications.23 Once you start a first-episode patient on drug therapy, encourage adherence by monitoring symptoms and anticipating side effects. Every 3 months after the acute phase:
- Use a structured evaluation, such as the Brief Psychiatric Rating Scale,24 to plot symptom severity, response, and risk for relapse.
- Rate EPS with the Simpson-Angus Scale25 and tardive dyskinesia (TD) with the Abnormal Involuntary Movement Scale26 because EPS and TD are associated with poor symptom response, adherence, and outcome.7
Reinforce information about the chronic nature of schizophrenia, especially when the patient or family question why treatment is needed if symptoms have resolved. Continue to counsel them about the patient’s need for:
- regular sleep of sufficient duration and without sleep-wake reversal
- gradual return to premorbid social, educational, and vocational activities/responsibilities
- ongoing treatment.
Encourage vigilance for relapse warning signs, including insomnia, social withdrawal, anxiety, refusal to eat or take medications, suspiciousness, agitation, disorganization, preoccupation with overvalued ideas, or responses to internal stimuli.
If the patient is noncompliant with antipsychotics in tablets or capsules, options include:
- liquid risperidone or olanzapine in a rapidly dissolving form that the patient cannot hide and spit out later
- long-acting depot formulations if the patient cannot be supervised and monitored daily. Older antipsychotics (such as haloperidol decanoate and fluphenazine decanoate) are available in depot formulations, and the FDA is considering risperidone in a microsphere formulation that would allow biweekly injections.
Medication withdrawal
Although the ideal duration of maintenance treatment after a first psychotic episode is debatable, we recommend that antipsychotics be continued at the full dosage that achieved symptom remission for at least 1 year.27 Then, if the patient has returned to the premorbid baseline, you can attempt a gradual medication withdrawal across 2 to 4 months, ideally when the patient’s environment is stable.
Be cautious when withdrawing antipsychotics from patients with a family history of psychosis. Consider a more gradual dose reduction, ongoing group and/or individual psychotherapy, and at least monthly monitoring. When possible, involve people who are significant in the patient’s life and educate them to look for deterioration’s warning signs, such as insomnia, irritability, anxiety, social withdrawal, preoccupation with overvalued ideas, or pacing.
If relapse occurs, carefully assess how well the patient has adhered to medication. Once a second psychotic episode occurs, his or her medication probably should be continued indefinitely.
- National Alliance for the Mentally Ill (800) 950-NAMI (6264); www.nami.org
- National Mental Health Association (800) 969-NMHA (6642); www.nmha.org
- National Alliance for Research on Schizophrenia and Depression (516) 829-0091; www.narsad.org/index.html
- Miller R, Mason SE. Diagnosis schizophrenia. A comprehensive resource. New York: Columbia University Press, 2002.
Drug brand names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
- Zolpidem • Ambien
Disclosure
Dr. Correll reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Mendelowitz receives grant/research support from, is a consultant to, and/or is a speaker for Pfizer Inc., Bristol-Myers Squibb Co.; and AstraZeneca Pharmaceuticals.
Acknowledgments
Research for this article was supported by grant 5P30MH60575 to The Zucker Hillside Hospital Intervention Research Center for Schizophrenia from the National Institute of Mental Health, Bethesda, MD.
A first psychotic episode offers the opportunity to build a therapeutic alliance at a teachable moment—while patients and their families are dealing with a devastating diagnosis. With a proactive approach, you can influence how patients view themselves and their experience, including psychotic illness, your efforts to treat its symptoms, and the costs and benefits of interventions.
Unfortunately, the typical first psychotic episode goes undiagnosed and untreated for 1 to 2 years, which some studies suggest may allow schizophrenia to progress. Although controversial, evidence links a prolonged duration of untreated psychosis to poorer outcome.1 Interventions during a prodromal (ie, pre-psychotic but already symptomatic) phase of schizophrenia also is being investigated, with the goal of attenuating—or perhaps even preventing—progression to frank psychosis.2-6
The implication for clinicians: timely identification and treatment may improve response, reduce relapse rates, and ultimately improve schizophrenic patients’ quality of life.
High rates of response—and relapse
Patients with a first psychotic episode show a higher response rate to antipsychotics—up to 87% within 1 year7 —and are more sensitive to side effects than are multi-episode patients.8 Yet despite their high response rate, new-onset patients often suffer from residual symptoms, even when treated in controlled settings. They also have a high rate of relapse—82% within 5 years.9
The strongest modifiable predictor of relapse is medication non-adherence, which has been shown to increase the risk of relapse five-fold.7 The first treatment experience provides a window of opportunity to help the patient accept taking medications as a normal part of life.
Mr. C, a 19-year-old college student, was brought for psychiatric admission after he told his roommates he was a new messiah who “needed to starve himself during the sunlight to enhance his holiness.” Approximately 7 months earlier he had become socially withdrawn and less able to do his college work. Two months later, he started using cannabis frequently. About 5 weeks prior to admission, he developed paranoid ideas involving his roommates and immersed himself in Eastern religions.
History and work-up. Mr. C was overweight and presented with mild dehydration. He did not report relevant signs of depression or mania and had no history of medical or psychiatric problems. Admission work-up included physical and neurologic exams, head CT, and blood work, which were unremarkable except for a positive cannabis toxicology. Family history was significant for one grandfather with alcohol abuse and one uncle who required psychiatric hospitalization in his 20s and never recovered functionally.
Family concerns. Mr. C’s parents were convinced a new diet was causing his symptoms and demanded that he be admitted to a medical ward. His brother insisted the symptoms were secondary to some “bad weed” and that everything would clear up in a few days. Although a brief medication-free observation period was considered to rule out substance-induced psychosis, the prodromal pattern of functional decline for more than 6 months and the bizarre quality of his delusions led to the diagnosis of a first episode of schizophrenia.
Treatment strategy. The treatment team met with Mr. C and his family to educate them about psychotic illness, the risks and benefits of novel antipsychotics, and the need to begin immediate treatment. With the patient’s and family’s consent, risperidone was initiated at 0.5 mg at bedtime and slowly increased over 1 week to 3 mg/d, with only mild and transient sedation. Within 3 weeks, Mr. C responded robustly and was discharged back to his family. Over the next 7 months, he continued taking risperidone, 3 mg/d, with some residual negative symptoms (social isolation without depression) and full remission of positive symptoms, which enabled him to return to college.
Therapeutic alliance. Your approach is key to building a therapeutic alliance with a person whose reality often is clouded by paranoia and referential thinking. Trust begins with the first clinical contact—during history-taking, ordering of tests, answering questions about the diagnosis, and discussing treatment options. Patients and their families must be informed about:
- target symptoms
- medication side effects
- predictors of response and relapse
- lack of certainty about how or when a patient will respond to any antipsychotic
- and the importance of rapid and uninterrupted treatment.
Supportive therapy. Support groups for the patient and family can help destigmatize the illness and reduce stress. Information about schizophrenia’s nature and course is available from the National Alliance for the Mentally Ill, National Mental Health Association, and other sources (see “Related resources”).10
CBT. Adjunctive cognitive-behavioral therapy (CBT) may speed up acute symptom response,11,12 reduce rates of nonresponse, and shorten hospital stays13 by helping patients deal with uncertainty about outer and inner realities. CBT approaches are understudied but so far have not been found to reduce relapse rates.
A moving target. As treatment moves from acute to consolidation and maintenance, target symptoms may change, side effects can limit the preferred approach, and partial or nonresponse may require drug or dosing adjustments. It is prudent to be prepared to re-evaluate the initial diagnosis as new symptoms emerge, response patterns develop, additional test or historical data become available, or as the illness’ course becomes more clear. To improve outcome, address comorbid or concurrent diseases—such as substance abuse or dependence, mood disorders, anxiety and obsessive-compulsive symptoms, or eating disorders.
Diagnostic work-up
As in Mr. C’s case (Box), a first psychotic episode is characterized by DSM-IV diagnostic criteria for schizophrenia, including hallucinations, delusions, disorganized thoughts or speech, disorganized behavior(s), or negative symptoms (such as anhedonia, amotivation, asociality, alogia, or affective flattening). The work-up is more comprehensive than that for subsequent episodes and includes a thorough history, complete physical examination, and brain imaging (Table 1) to explore other possible medical and psychiatric diagnoses (Table 2).
Table 1
WORK-UP OF PATIENTS PRESENTING WITHA FIRST EPISODE OF PSYCHOSIS
| Priority | Mode of evaluation |
|---|---|
| Routine | History Symptoms, time course, medical conditions, current/previous medications, herbs, drugs Medical and neurologic exam Blood work: CBC with differential, complete metabolic panel, thyroid and liver function tests, syphilis serology, pregnancy test, toxicology Urinalysis, toxicology ECG |
| Recommended | Fasting glucose and lipid profile (ideally before starting atypical antipsychotic) Head CT (especially if history of recent trauma) or brain MRI |
| Optional | Erythrocyte sedimentation rate, antinuclear antibodies, lumbar puncture, sleep-deprived EEG |
The history—ideally gathered from the patient and others—includes:
- medical and psychiatric diagnoses
- medications (prescribed and over-the-counter remedies)
- presence of stressors/triggers
- chronology of symptoms
- potential for the episode to endanger the patient or others.
Imaging. Despite a relatively low yield, we recommend that every patient with a first psychotic episode undergo a brain CT or MRI to rule out a potentially treatable organic cause for the psychosis.14
Other tests. Because of the increased risk of hyperglycemia, dyslipidemia, and possible cardiac conduction abnormalities with atypical antipsychotics, obtain a baseline fasting blood glucose, lipid profile, and ECG. A sleep-deprived EEG is recommended for patients with unclear motor movements or family history of epilepsy.
Choosing medications
Medication choices for the patient with first-episode schizophrenia are influenced by:
- target symptoms
- whether the symptoms endanger the patient or others
- the patient’s personal or family history of medication response or side effects
- a generally increased sensitivity to side effects in patients who have never been exposed to antipsychotics
- concurrent medical and/or psychiatric disorders
- prescriber, patient, and family preferences.
Psychiatrists generally select psychotropic classes by symptom domains (Table 3) and individual agents in each class by side effect profile. Except for clozapine’s superior effectiveness in patients with refractory psychosis, controlled studies have shown no clinically significant differences in efficacy among the drugs in each class—including the antipsychotics. Individual patients, however, may respond differently to different agents.
Principles of prescribing antipsychotics
Antipsychotics are effective in treating most psychotic core symptoms, such as hallucinations, delusions, agitation, aggression, and disorganized thinking and behavior. Other medications can be added to speed up or enhance treatment response or to target other domains.
Dosages. First-episode patients often require lower dosages and slower titration than multi-episode patients. As a rule, antipsychotics are started at about one-half the dosage given to patients with a chronic treatment history, although symptom severity and absence of side effects at lower dosages can help individualize titration.
Side effects. Atypical antipsychotics are preferred because of their reduced risk of extrapyramidal symptoms (EPS), positive effects on depressive and cognitive symptoms, and improved patient satisfaction and adherence, compared with the older antipsychotics.15-17 Atypicals’ potential side effects include weight gain, hyperglycemia, and dyslipidemia,18 as well as often-overlooked sexual side effects.19
Table 2
DIFFERENTIAL DIAGNOSIS OF FIRST-EPISODE PSYCHOSIS
| Possible diagnosis | Key points for differentiation |
|---|---|
| Schizophrenia | 6 months of psychosis* (including prodromal symptoms); total duration of mood episodes brief relative to active and residual psychotic phases; not directly caused by medical condition or substance |
| Schizophreniform disorder | Same as above, except symptoms are present 1 to 6 months |
| Brief psychotic disorder | Same as above, except symptoms are present 1 day to 1 month |
| Delusional disorder | Apart from non-bizarre delusions, functioning not markedly impaired; total duration of mood episodes brief relative to active and residual psychotic phases; not caused by direct physiologic effects of medical condition or substance |
| Psychotic disorder NOS | Psychotic symptoms insufficient to make a specific diagnosis |
| Schizoaffective disorder | Like schizophrenia for at least 2 weeks, but with mania or major depression present for much of the active and residual psychotic periods |
| Mood disorder with psychosis | Psychotic symptoms occur exclusively during mood disorder episodes |
| Psychosis due to general medical condition | Psychotic symptoms caused by direct physiologic effects of a general medical condition |
| Delirium due to general medical condition | Psychotic symptoms associated with a disturbance in consciousness and other cognitive deficits; characterized by a fluctuating course |
| Dementia due to general medical condition | Psychotic symptoms associated with memory impairment and other cognitive deficits |
| Substance-induced psychotic disorder | Psychotic symptoms caused by direct physiologic effects of a substance; reaction exceeds that usually encountered with intoxication or withdrawal |
| Substance-induced psychotic delirium | Similar to above, but associated with a disturbance in consciousness and other cognitive deficits; characterized by a fluctuating course |
| Substance intoxication or withdrawal | Caused by direct physiologic effects of a substance; reaction is typically encountered with intoxication or withdrawal |
| Conversion disorder | Contradictory and inconsistent history and presentation; secondary gain |
| Malingering | Contradictory and inconsistent history and presentation; primary gain |
| * Psychotic symptoms must interfere with functioning, and at least two of the following are required: delusions, hallucinations, disorganized thoughts or speech, disorganized behavior, or negative symptoms (avolition, alogia, affective flattening, asociality, or anhedonia), unless delusions are bizarre (impossible), or hallucinations consist of a running commentary or of two or more voices conversing with each other. | |
| Source: Adapted from DSM-IV handbook of differential diagnosis. Washington, DC: American Psychiatric Press, 1995. | |
Consider the patient’s risk for side effects when choosing an atypical antipsychotic, as each drug has strengths and weaknesses. For example, it may be reasonable to consider:
- risperidone, ziprasidone, or aripiprazole—rather than olanzapine or quetiapine—for patients at high risk for weight gain or with a family history of diabetes
- avoiding risperidone for patients with an early indication of sensitivity to EPS or prolactin-related side effects
- an agent that can be loaded rapidly, such as olanzapine or aripiprazole—rather than an agent that requires titration, such as quetiapine—for a patient presenting with severe agitation.
Clozapine—because of its side effect potential—is generally reserved for patients who have not responded to at least two antipsychotic trials of at least 4 to 6 weeks duration and have a steady-state clozapine level >350 ng/dl.
Acute agitation. Short-acting IM formulations can be used effectively for acute agitation and impulsivity or for patients who refuse oral antipsychotics. Until recently, only first-generation antipsychotics such as haloperidol and fluphenazine were available in IM formulations. Injectable ziprasidone mesylate is now available for acute agitation and has been found to be as safe and effective as haloperidol. Lorazepam may be used to treat agitation or as an adjunct to a patient’s antipsychotic agent.
Adjunctive therapies
When antipsychotic monotherapy is inadequate, adjunctive medications may be considered:
- to treat catatonia, obsessive-compulsive disorder, or depression
- to resolve agitation or mania more quickly
- to control agitation or anxiety while an antipsychotic dosage is being titrated
- when side effects emerge or residual symptoms remain despite adequate dosage and duration of antipsychotic treatment.
Deciding if and when to add another drug depends on the nature and severity of target symptoms, the degree and time course of response, and whether side effects appear. If you use adjunctive therapies during acute stabilization, attempt to taper and discontinue them after the patient’s symptoms have improved. Avoid combining antipsychotics, as no clinical data support the effectiveness and safety of this practice.20
Benzodiazepines are often used adjunctively for agitation, anxiety, or temporary insomnia in patients with schizophrenia. Common dosages are:
- for agitation or anxiety, lorazepam, 0.5 to 2 mg bid or tid, or clonazepam, 0.5 mg bid to 2 mg tid
- for insomnia, lorazepam or clonazepam, 1 to 2 mg at bedtime, or zolpidem, 5 to 10 mg at bedtime.
Benzodiazepines also are effective for catatonia, which may be misdiagnosed as negative symptoms or depression when it presents as marked psychomotor retardation, staring, selective mutism, negativism, mild posturing, or stereotypies. If undiagnosed, catatonia may worsen during antipsychotic titration. Symptoms usually respond to high-dose lorazepam, 1 mg bid or tid, with increases up to a maximum dosage of 12 mg/d.
Mood stabilizers are often used adjunctively to treat acute agitation and disinhibition21 or as add-on agents for residual psychotic or affective symptoms. Recommended dosages and blood levels, as tolerated, are:
- valproic acid, starting at 10 to 20 mg/kg bid, with a target serum level of 60 to 120 μg/ml. A once-daily, extended-release formulation may improve compliance.
- lithium, starting at 300 mg bid to tid, aiming for a serum level of 0.8 to 1.2 mEq/L.
Manic symptoms in a schizoaffective presentation may require one or even two mood stabilizers.
Lamotrigine may be the treatment of choice for depressed patients with schizoaffective disorder, bipolar type.22 However, it must be started at 25 mg/d and titrated extremely slowly—by 25 mg every other week, up to a target 200 to 400 mg/d—to avoid the risk of potentially fatal Stevens-Johnson syndrome.
Gabapentin, 300 mg tid to 1,500 mg tid, may help treat anxiety—particularly in patients with comorbid substance abuse, in whom benzodiazepines should be used sparingly after stabilization.
Table 3
SYMPTOM-BASED DRUG TREATMENT OF FIRST-EPISODE SCHIZOPHRENIA
| Target symptom | Medication choices | Dosage range | Comments |
|---|---|---|---|
| Agitation | Atypical antipsychotic Mood stabilizer Benzodiazepine ECT | Depends on level of agitation and individual agent | Ziprasidone IM may be useful for acute agitation |
| Psychosis | Atypical antipsychotic ECT | =50% of dosage used for multiple episode patients | Start low/go slow, monitor side effects |
| Catatonia | Lorazepam ECT | 1 mg/d bid to4 mg/d tid | Use sedation, symptom resolution as threshold |
| Negative symptoms | Atypical antipsychotic Glycine, cycloserine may help | Dictated by positive symptom response | Effect on functioning, quality of life unclear |
| Cognitive symptoms | Atypical antipsychotic | Same as above for negative symptoms | Same as above |
| Insomnia | Lorazepam Zolpidem Trazodone Mirtazapine | 1 to 2 mg HS 5 to 10 mg HS 50 to 200 mg HS 7.5 to 15 mg HS | Short-term treatment preferred |
| Depression, obsessive-compulsive symptoms, anxiety/panic | SSRI, SNDRI, NDRI, SARI, NASA | As per individual agent Possible interference with | antipsychotic blood levels* |
| Parkinsonism | Benztropine Trihexyphenidyl | 0.5 to 2 mg/d 1 to 15 mg/d | Possible effect of decreased cognition |
| Akathisia | Propranolol | 20 to 160 mg/d | Monitor blood pressure |
| Weight gain | Ziprasidone Aripiprazole | Dictated by positive symptom response | Prevention more effective than remediation |
| Non-adherence | Olanzapine oral disintegrating tablets Risperidone liquid Risperidone microspheres | 5 to 20 mg/d 1 to 4 mg/d IM injections every 2 weeks | Dissolves instantly Can be mixed with water, coffee, orange juice, or low-fat milk Not yet available |
| * Fluoxetine and paroxetine increase risperidone levels by CYP-P450 2D6 inhibition; fluvoxamine increases clozapine and olanzapine levels by CYP-P450 1A2 inhibition; fluvoxamine and nefazodone increase quetiapine and may increase ziprasidone levels by CYP-P450 3A4 inhibition; all three CYP-P450 inhibitors may increase aripiprazole levels, but the extent is not known. | |||
| ECT: electroconvulsive therapy; SSRI: selective serotonin reuptake inhibitor; SNDRI: serotonin-norepinephrine-dopamine reuptake inhibitor; NDRI: norepinephrinedopamine reuptake inhibitor; SARI: serotonin antagonist and reuptake inhibitor; NASA: norepinephrine-antagonist and serotonin antagonist. | |||
Antidepressants. Patients with schizophrenia can develop depression, even if they do not meet diagnostic criteria for schizoaffective disorder. Untreated depression can lead to non-adherence, self-medication with alcohol or illicit substances, and increased risk of suicide.
Differentiating depression from negative symptoms may be difficult, but there are subtle distinctions:
- Patients with negative symptoms appear more emotionally flat and unconcerned about their lack of motivation and diminished social and role functioning.
- Depressed persons often verbalize their demoralization, hopelessness, and desire to feel and behave differently.
Treat depression with any selective serotonin reuptake inhibitor or other newer-generation antidepressant such as mirtazapine, nefazodone, or venlafaxine at usual doses, as tolerated.
Miscellaneous medications. Use anticholinergic medications such as benztropine, 0.5 to 2 mg bid, or trihexyphenidyl, 1 to 5 mg bid, if parkinsonian symptoms occur and changing to an antipsychotic with a lower EPS potential is not feasible.
For akathisia, propranolol (10 mg bid or tid; titrate up to 160 mg/d if pulse rate and blood pressure remain stable) or benzodiazepines may be useful. Amantadine may also be used at dosages between 50 and 150 mg bid.
Insomnia may be treated with low dosages of sedating antidepressants, such as trazodone, 50 to 200 mg HS, or mirtazapine, 7.5 to 15 mg HS.
Preventing relapse during maintenance
Medication adherence depends on patient insight and attitude towards medications.23 Once you start a first-episode patient on drug therapy, encourage adherence by monitoring symptoms and anticipating side effects. Every 3 months after the acute phase:
- Use a structured evaluation, such as the Brief Psychiatric Rating Scale,24 to plot symptom severity, response, and risk for relapse.
- Rate EPS with the Simpson-Angus Scale25 and tardive dyskinesia (TD) with the Abnormal Involuntary Movement Scale26 because EPS and TD are associated with poor symptom response, adherence, and outcome.7
Reinforce information about the chronic nature of schizophrenia, especially when the patient or family question why treatment is needed if symptoms have resolved. Continue to counsel them about the patient’s need for:
- regular sleep of sufficient duration and without sleep-wake reversal
- gradual return to premorbid social, educational, and vocational activities/responsibilities
- ongoing treatment.
Encourage vigilance for relapse warning signs, including insomnia, social withdrawal, anxiety, refusal to eat or take medications, suspiciousness, agitation, disorganization, preoccupation with overvalued ideas, or responses to internal stimuli.
If the patient is noncompliant with antipsychotics in tablets or capsules, options include:
- liquid risperidone or olanzapine in a rapidly dissolving form that the patient cannot hide and spit out later
- long-acting depot formulations if the patient cannot be supervised and monitored daily. Older antipsychotics (such as haloperidol decanoate and fluphenazine decanoate) are available in depot formulations, and the FDA is considering risperidone in a microsphere formulation that would allow biweekly injections.
Medication withdrawal
Although the ideal duration of maintenance treatment after a first psychotic episode is debatable, we recommend that antipsychotics be continued at the full dosage that achieved symptom remission for at least 1 year.27 Then, if the patient has returned to the premorbid baseline, you can attempt a gradual medication withdrawal across 2 to 4 months, ideally when the patient’s environment is stable.
Be cautious when withdrawing antipsychotics from patients with a family history of psychosis. Consider a more gradual dose reduction, ongoing group and/or individual psychotherapy, and at least monthly monitoring. When possible, involve people who are significant in the patient’s life and educate them to look for deterioration’s warning signs, such as insomnia, irritability, anxiety, social withdrawal, preoccupation with overvalued ideas, or pacing.
If relapse occurs, carefully assess how well the patient has adhered to medication. Once a second psychotic episode occurs, his or her medication probably should be continued indefinitely.
- National Alliance for the Mentally Ill (800) 950-NAMI (6264); www.nami.org
- National Mental Health Association (800) 969-NMHA (6642); www.nmha.org
- National Alliance for Research on Schizophrenia and Depression (516) 829-0091; www.narsad.org/index.html
- Miller R, Mason SE. Diagnosis schizophrenia. A comprehensive resource. New York: Columbia University Press, 2002.
Drug brand names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Citalopram • Celexa
- Clozapine • Clozaril
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Gabapentin • Neurontin
- Lamotrigine • Lamictal
- Lorazepam • Ativan
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Sertraline • Zoloft
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
- Zolpidem • Ambien
Disclosure
Dr. Correll reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Mendelowitz receives grant/research support from, is a consultant to, and/or is a speaker for Pfizer Inc., Bristol-Myers Squibb Co.; and AstraZeneca Pharmaceuticals.
Acknowledgments
Research for this article was supported by grant 5P30MH60575 to The Zucker Hillside Hospital Intervention Research Center for Schizophrenia from the National Institute of Mental Health, Bethesda, MD.
1. Norman RM, Malla AK. Duration of untreated psychosis: a critical examination of the concept and its importance. Psychol Med 2001;31:381-400.
2. Cornblatt B, Lencz T, Obuchowski M. The schizophrenia prodrome: treatment and high-risk perspectives. Schizophr Res 2002;54(1-2):177-86.
3. McGorry PD, Yung AR, Phillips LJ, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry 2002;59(10):921-8.
4. Heinssen RK, Perkins DO, Appelbaum PS, et al. Informed consent in early psychosis research: National Institute of Mental Health workshop, November 15, 2000. Schizophr Bull 2001;27(4):571-83.
5. Miller TJ, McGlashan TH, Rosen JL, et al. Prospective diagnosis of the initial prodrome for schizophrenia based on the Structured Interview for Prodromal Syndromes: preliminary evidence of interrater reliability and predictive validity. Am J Psychiatry 2002;159(5):863-5.
6. Young LT, Bakish D, Beaulieu S. The neurobiology of treatment response to antidepressants and mood stabilizing medications. J Psychiatry Neurosci 2002;27(4):260-5.
7. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry 1999;56(3):241-7.
8. Tauscher J, Kapur S. Choosing the right dose of antipsychotics in schizophrenia: lessons from neuroimaging studies. CNS Drugs 2001;15:671-8.
9. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry 1999;56(3):241-7.
10. Miller R, Mason SE. Diagnosis schizophrenia. A comprehensive resource, New York: Columbia University Press, 2002.
11. Lewis S, Tarrier N, Haddock G, et al. Randomised controlled trial of cognitivebehavioural therapy in early schizophrenia: acute-phase outcomes. Br J Psychiatry Suppl 2002;43:S91-7.
12. Turkington D, Kingdon D, Turner T. Effectiveness of a brief cognitive-behavioural therapy intervention in the treatment of schizophrenia. Br J Psychiatry 2002;180:523-7.
13. Cormac I, Jones C, Campbell C. Cognitive behaviour therapy for schizophrenia. Cochrane Database Syst Rev 2002;(1):CD000524.-
14. Smith GN, Flynn SW, Kopala LC, et al. A comprehensive method of assessing routine CT scans in schizophrenia. Acta Psychiatr Scand 1997;96:395-401.
15. Leucht S, Pitschel-Walz G, Abraham D, et al. Efficacy and extrapyramidal sideeffects of the new antipsychotics olanzapine, quetiapine, risperidone, and sertindole compared to conventional antipsychotics and placebo. A meta-analysis of randomized controlled trials. Schizophr Res 1999;35(1):51-68.
16. Keefe RS, Silva SG, Perkins DO, Lieberman JA. The effects of atypical antipsychotic drugs on neurocognitive impairment in schizophrenia: A review and meta-analysis. Schizophr Bull 1999;25(2):201-22.
17. Dolder CR, Lacro JP, Dunn LB, Jeste DV. Antipsychotic medication adherence: is there a difference between typical and atypical agents? Am J Psychiatry 2002;159(1):103-8.
18. Vieweg WVR, Adler RA, Fernandez A. Weight control and antipsychotics: How to tip the scale away from diabetes and heart disease. Current Psychiatry 2002;1(5):10-19.
19. Compton MT, Miller AH. Sexual side effects associated with conventional and atypical antipsychotics. Psychopharmacol Bull 2001;35:89-108.
20. Stahl SM. Antipsychotic polypharmacy: squandering precious resources? J Clin Psychiatry 2002;63(2):93-4.
21. Casey DE, Daniel DG, Wassef AA, Tracy KA, Wozniak P, Sommerville KW. Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology 2003;28(1):182-92.
22. Calabrese JR, Shelton MD, Rapport DJ, Kimmel SE. Bipolar disorders and the effectiveness of novel anticonvulsants. J Clin Psychiatry. 2002;63(Suppl 3):5-9.
23. Kampman O, Laippala P, Vaananen J, et al. Indicators of medication compliance in first-episode psychosis. Psychiatry Res 2002;110:39-48.
24. Overall JE, Gorham DR. Brief psychiatric rating scale. Psychol Rep 1962;10:799-812.
25. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand 1970(suppl);212:11-19.
26. Guy W (ed). ECDEU assessment manual for psychopharmacology. Publication ABM 76-338. Washington, DC: U.S. Department of Health, Education, and Welfare, 1976;534-7.
27. Bosveld-van Haandel LJ, Slooff CJ, van den Bosch RJ. Reasoning about the optimal duration of prophylactic antipsychotic medication in schizophrenia: evidence and arguments. Acta Psychiatr Scand 2001;103(5):335-46.
1. Norman RM, Malla AK. Duration of untreated psychosis: a critical examination of the concept and its importance. Psychol Med 2001;31:381-400.
2. Cornblatt B, Lencz T, Obuchowski M. The schizophrenia prodrome: treatment and high-risk perspectives. Schizophr Res 2002;54(1-2):177-86.
3. McGorry PD, Yung AR, Phillips LJ, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to first-episode psychosis in a clinical sample with subthreshold symptoms. Arch Gen Psychiatry 2002;59(10):921-8.
4. Heinssen RK, Perkins DO, Appelbaum PS, et al. Informed consent in early psychosis research: National Institute of Mental Health workshop, November 15, 2000. Schizophr Bull 2001;27(4):571-83.
5. Miller TJ, McGlashan TH, Rosen JL, et al. Prospective diagnosis of the initial prodrome for schizophrenia based on the Structured Interview for Prodromal Syndromes: preliminary evidence of interrater reliability and predictive validity. Am J Psychiatry 2002;159(5):863-5.
6. Young LT, Bakish D, Beaulieu S. The neurobiology of treatment response to antidepressants and mood stabilizing medications. J Psychiatry Neurosci 2002;27(4):260-5.
7. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry 1999;56(3):241-7.
8. Tauscher J, Kapur S. Choosing the right dose of antipsychotics in schizophrenia: lessons from neuroimaging studies. CNS Drugs 2001;15:671-8.
9. Robinson D, Woerner MG, Alvir JM, et al. Predictors of relapse following response from a first episode of schizophrenia or schizoaffective disorder. Arch Gen Psychiatry 1999;56(3):241-7.
10. Miller R, Mason SE. Diagnosis schizophrenia. A comprehensive resource, New York: Columbia University Press, 2002.
11. Lewis S, Tarrier N, Haddock G, et al. Randomised controlled trial of cognitivebehavioural therapy in early schizophrenia: acute-phase outcomes. Br J Psychiatry Suppl 2002;43:S91-7.
12. Turkington D, Kingdon D, Turner T. Effectiveness of a brief cognitive-behavioural therapy intervention in the treatment of schizophrenia. Br J Psychiatry 2002;180:523-7.
13. Cormac I, Jones C, Campbell C. Cognitive behaviour therapy for schizophrenia. Cochrane Database Syst Rev 2002;(1):CD000524.-
14. Smith GN, Flynn SW, Kopala LC, et al. A comprehensive method of assessing routine CT scans in schizophrenia. Acta Psychiatr Scand 1997;96:395-401.
15. Leucht S, Pitschel-Walz G, Abraham D, et al. Efficacy and extrapyramidal sideeffects of the new antipsychotics olanzapine, quetiapine, risperidone, and sertindole compared to conventional antipsychotics and placebo. A meta-analysis of randomized controlled trials. Schizophr Res 1999;35(1):51-68.
16. Keefe RS, Silva SG, Perkins DO, Lieberman JA. The effects of atypical antipsychotic drugs on neurocognitive impairment in schizophrenia: A review and meta-analysis. Schizophr Bull 1999;25(2):201-22.
17. Dolder CR, Lacro JP, Dunn LB, Jeste DV. Antipsychotic medication adherence: is there a difference between typical and atypical agents? Am J Psychiatry 2002;159(1):103-8.
18. Vieweg WVR, Adler RA, Fernandez A. Weight control and antipsychotics: How to tip the scale away from diabetes and heart disease. Current Psychiatry 2002;1(5):10-19.
19. Compton MT, Miller AH. Sexual side effects associated with conventional and atypical antipsychotics. Psychopharmacol Bull 2001;35:89-108.
20. Stahl SM. Antipsychotic polypharmacy: squandering precious resources? J Clin Psychiatry 2002;63(2):93-4.
21. Casey DE, Daniel DG, Wassef AA, Tracy KA, Wozniak P, Sommerville KW. Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology 2003;28(1):182-92.
22. Calabrese JR, Shelton MD, Rapport DJ, Kimmel SE. Bipolar disorders and the effectiveness of novel anticonvulsants. J Clin Psychiatry. 2002;63(Suppl 3):5-9.
23. Kampman O, Laippala P, Vaananen J, et al. Indicators of medication compliance in first-episode psychosis. Psychiatry Res 2002;110:39-48.
24. Overall JE, Gorham DR. Brief psychiatric rating scale. Psychol Rep 1962;10:799-812.
25. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand 1970(suppl);212:11-19.
26. Guy W (ed). ECDEU assessment manual for psychopharmacology. Publication ABM 76-338. Washington, DC: U.S. Department of Health, Education, and Welfare, 1976;534-7.
27. Bosveld-van Haandel LJ, Slooff CJ, van den Bosch RJ. Reasoning about the optimal duration of prophylactic antipsychotic medication in schizophrenia: evidence and arguments. Acta Psychiatr Scand 2001;103(5):335-46.