User login
Detecting and managing hereditary colorectal cancer syndromes in your practice
Hereditary colorectal cancer syndromes account for 5% to 10% of cases of colorectal cancer.
Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history. An accurate and comprehensive family history should cover three generations and include ethnic background, ages and causes of death of relatives, and any diagnosis of cancer, including age at onset and history of polyps.

Red flags for a hereditary colorectal cancer syndrome in the personal or family history are:
- Early age of onset of cancer (eg, colorectal cancer before age 50)
- More than 10 colorectal adenomas
- Synchronous (ie, occurring at the same time) or metachronous (occurring at different times) primary cancers
- Multiple relatives in successive generations with the same or related cancers (eg, colon or endometrial cancer)
- A family member with a known hereditary colorectal cancer syndrome (Table 1).
Any of these red flags should prompt a referral for genetic counseling.
SYNDROMES ARE CLASSIFIED AS WITH OR WITHOUT POLYPOSIS

Many hereditary syndromes are associated with a higher risk of colorectal cancer. Generally, they can be divided into two categories (Table 2): polyposis syndromes (in which patients have numerous colorectal polyps) and nonpolyposis syndromes (with few or no polyps).
These two main types are subclassified on the basis of the histology of most of the polyps detected: adenomatous, hamartomatous, serrated, or mixed types.
In this review, we will address the three most common of these syndromes: Lynch syndrome (hereditary nonpolyposis colorectal cancer), familial adenomatous polyposis, and MYH-associated polyposis. However, as noted in Table 2, other hereditary colorectal cancer syndromes exist, and suspicion of these conditions should prompt a referral for further evaluation.
LYNCH SYNDROME (HEREDITARY NONPOLYPOSIS COLORECTAL CANCER)
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer, predisposes people to a variety of cancers.
Colorectal cancer is the most common type of cancer associated with Lynch syndrome. Recent research suggests that the cumulative risk of developing colorectal cancer by age 80 is 42% for all patients with Lynch syndrome.1 The median age at onset is 45 years.1 For patients who undergo segmental resection of their initial cancer, the cumulative risk of metachronous colorectal cancer (ie, a new tumor arising later) is 16% at 10 years, 41% at 20 years, and up to 62% after 30 years.2
Endometrial cancer occurs in 17% to 57% of women with Lynch syndrome by age 70, with a median age at onset of 49 years.1
Other extracolonic cancers in Lynch syndrome include cancers of the:
- Stomach (1%–10% risk by age 70 years)
- Ovaries (1%–20% risk)
- Hepatobiliary tract (1%–2% risk)
- Urinary tract (1%–12% risk)
- Small bowel (1%–2% risk)
- Brain (1%–8% risk)
- Skin (sebaceous adenomas, adenocarcinomas, and keratoacanthomas).1,3,4
Earlier studies reported higher rates of associated cancer than those shown here. However, their data were largely derived from registries and may be overestimates. The numbers shown above are from population-based studies.
Genetics of Lynch syndrome
Lynch syndrome is caused by a germline mutation in the MLH1, MSH2, MSH6, PMS2, or EPCAM genes.5 These genes code for proteins that are responsible DNA mismatch repair—one of the cell’s proofreading mechanisms during DNA replication.
These mutations are inherited in an autosomal dominant manner. Though de novo mutations in these genes have been reported, they are rare and the exact frequency with which they occur is unknown.6
In whom should Lynch syndrome be suspected?
Lynch syndrome can be suspected on the basis of family history and clinical criteria.
In 1991, the same group of experts who coined the term “hereditary nonpolyposis colorectal cancer” developed family history criteria for it1:
- At least three relatives with histologically confirmed colorectal cancer, one of whom is a first-degree relative of the other two
- At least two successive generations involved
- At least one of the cancers diagnosed before age 50
- Familial adenomatous polyposis is excluded.

Known as the Amsterdam criteria, these were to be used in collaborative studies of families with hereditary colorectal cancer.7 In 1999, these criteria were broadened to include extracolonic cancers and became known as the Amsterdam II criteria (Table 3).8
Patients whose families meet the Amsterdam II criteria or who have molecular pathologic evidence of Lynch syndrome (see below) are appropriate candidates for genetic counseling and testing.
Diagnosis of Lynch syndrome
The diagnosis of Lynch syndrome is based on molecular pathologic analysis (performed on tumor samples) and confirmed by genetic testing.
Molecular pathologic evidence of Lynch syndrome includes microsatellite instability and loss of expression of one or more of the DNA mismatch repair proteins (detected using immunohistochemistry) (more on these below). The revised Bethesda guidelines (TABLE 3) were intended to identify individuals whose tumors should be tested for one or both of these phenomena.9
In 2009, the Evaluation of Genomic Applications in Practice and Prevention working group recommended that all patients with newly diagnosed colorectal cancer undergo microsatellite instability analysis, immunohistochemistry testing, or both, regardless of whether they meet the Amsterdam II or the Bethesda guideline criteria.10
Microsatellite instability analysis. Microsatellites are short sequences of repeated DNA. The tumor cells of patients who carry defective mismatch repair genes have microsatellites that are longer or shorter than in normal cells, a condition called microsatellite instability (ie, “MSI-high”).
Microsatellite instability testing, using a standardized panel of five DNA markers, is performed on normal and tumor tissue. If more than two of the five microsatellite markers in the tumor show instability, the lesion is considered to have a high level of microsatellite instability. About 15% of colorectal cancers have this high level, although most are not associated with Lynch syndrome and lose MLH1 expression by promoter methylation.11,12
While only 2% of patients with colorectal cancer have Lynch syndrome, from 90% to 95% of colorectal cancers from patients with Lynch syndrome have high levels of microsatellite instability.10 The presence of MLH1 promoter hypermethylation, the BRAF mutation V600E, or both within the tumor suggests that the cancer is not associated with Lynch syndrome.
Some families that meet the Amsterdam I criteria have microsatellite-stable tumors: their condition has been called familial colorectal cancer type X.13 This condition is associated with a higher risk of colorectal cancer but not the other malignancies observed in Lynch syndrome.
Immunohistochemistry is performed to assess for expression of the mismatch repair proteins MSH2, MSH6, MLH1, and PMS2. Absence of expression of the specific protein within tumor cells compared with normal cells within the specimen suggests dysfunction of the specific gene and guides germline mutation testing (Figure 1). For example, a patient who lacks expression of the MSH2 protein in his or her colon cancer most likely has a mutation in the MSH2 gene. Therefore, germ-line genetic testing should initially target the MSH2 gene. Approximately 88% of Lynch syndrome-associated colorectal cancers have abnormal immunohistochemical staining.10
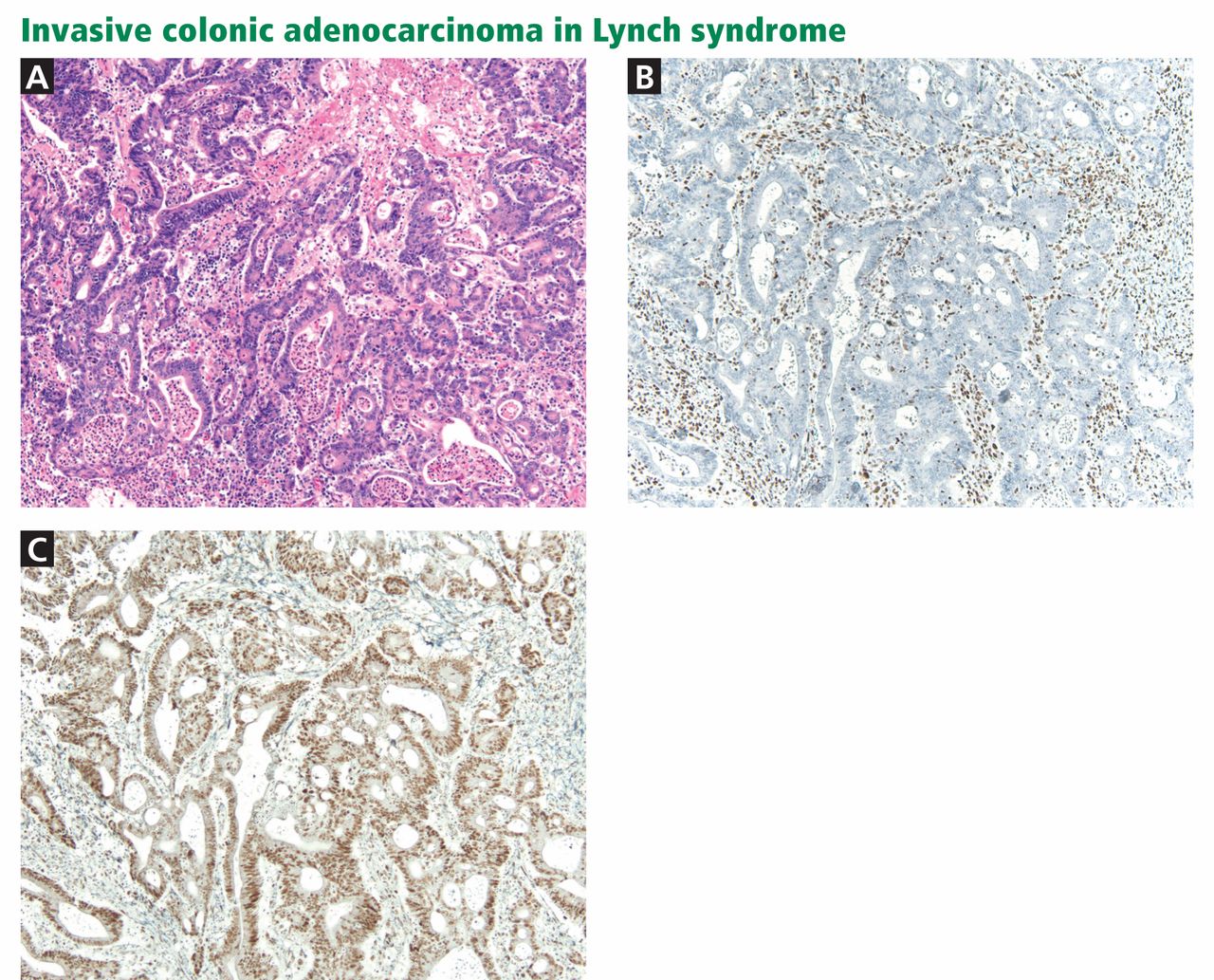
Testing for microsatellite instability and mismatch repair gene expression ideally precedes germline genetic testing and helps to guide which gene or genes should be tested.9,14
Genetic testing for Lynch syndrome is routinely performed on a blood or saliva sample, using DNA from white blood cells and sequencing the gene or genes involved to look for mutations. Positive results from a germline genetic test confirm the diagnosis of Lynch syndrome and allow for predictive testing for relatives at risk. The term Lynch syndrome is used exclusively to describe individuals with evidence of a mutation in one of the mismatch repair genes.15
If a patient’s results are positive, genetic counseling and genetic testing should be offered to at-risk relatives age 18 and over.
Management of Lynch syndrome
Aggressive cancer surveillance is essential for people with Lynch syndrome and for those who are considered at risk but have not pursued genetic testing, such as a sibling of a person with Lynch syndrome.
Colorectal cancer. Colonoscopy is recommended every 1 to 2 years beginning at the age of 20 to 25 years, or 2 to 5 years earlier than the age of the youngest relative affected with colorectal cancer if the initial diagnosis was before age 25. When patients turn 40 years old, colonoscopy is done annually.16–18 A significant reduction in cancer incidence and in the mortality rate has been shown with colonoscopic surveillance.19–21
Chemoprevention may also have a role. Patients with Lynch syndrome who took aspirin 600 mg per day for an average of 25 months had a significantly lower incidence of colorectal cancer during a 55-month follow-up period compared with patients randomized to placebo.22
For patients with Lynch syndrome who are diagnosed with colorectal cancer, the high risk of metachronous cancers after standard segmental colectomy calls for a more extended resection. Retrospective analysis of 382 Lynch syndrome patients found that none of the 50 who underwent total or subtotal colectomy were diagnosed with metachronous colorectal cancer, whereas a metachronous cancer developed in 74 (22%) of the 332 patients who had had segmented resection.2 Annual surveillance of the remaining colon, rectum, or both is indicated postoperatively.
Gynecologic cancers. Women with Lynch syndrome should also consider gynecologic surveillance and risk-reducing surgery. This includes annual gynecologic examination, transvaginal ultrasonography, and endometrial aspiration, beginning at age 30 to 35 years. Although this surveillance does detect premalignant lesions and early symptomatic cancers, its effect on the mortality rate is unknown. Hysterectomy with bilateral salpingo-oophorectomy has been shown to significantly reduce endometrial and ovarian cancers in women with Lynch syndrome.23,24
Urothelial cancers. Carriers of MSH2 mutations have a significantly higher risk of urothelial cancers.4 Therefore, MSH2 carriers should consider ultrasonography of the urinary tract, urinary cytology, and urinalysis every 1 to 2 years beginning at age 40.4
Other extracolonic cancers. Poor evidence exists for systematic screening for the other extracolonic tumors associated with Lynch syndrome. However, the National Comprehensive Cancer Network advises considering esophagogastroduodenoscopy with extended duodenoscopy as well as capsule endoscopy every 2 to 3 years beginning at age 30 to 35.14
ADENOMATOUS POLYPOSIS SYNDROMES
Familial adenomatous polyposis and MYH-associated polyposis are the next most common hereditary colorectal cancer syndromes. Each of these accounts for about 1% of cases of colorectal cancer. Clinically, these two syndromes can be challenging to distinguish because they overlap phenotypically to a significant degree.
FAMILIAL ADENOMATOUS POLYPOSIS
Familial adenomatous polyposis is caused by mutations in the APC gene. Its prevalence is 2.29 to 3.2 per 100,000 individuals.25,26
Genetics of familial adenomatous polyposis
APC is the only gene known to cause familial adenomatous polyposis. Mutations in APC are inherited in an autosomal dominant manner. Approximately 25% of cases of familial adenomatous polyposis are due to a de novo mutation in APC.27
Clinical presentation of familial adenomatous polyposis
Familial adenomatous polyposis is classified by the burden of colorectal adenomas.
Patients who have fewer than 100 adenomas have an attenuated form of the disease. In this group, polyps usually begin to form in the late teenage years or early 20s and tend to develop in the proximal colon. The attenuated form is associated with an approximately 70% lifetime risk of colorectal cancer.28
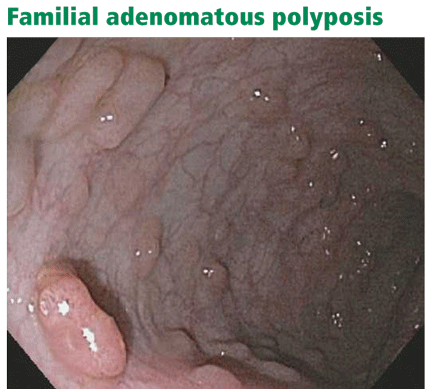
Patients who have more than 100 polyps are considered to have the classic form of the disease, and those with more than 1,000 polyps have profuse familial adenomatous polyposis (Figure 2). In these groups, polyps typically begin to develop in the preteenage to mid-teenage years. Without surgery, there is nearly a 100% risk of colorectal cancer. The average age at diagnosis of colorectal cancer is 39 years for patients with classic disease.
Upper gastrointestinal polyps are common in familial adenomatous polyposis. Nearly 90% of patients develop duodenal adenomas by a mean age of 44, with a cumulative lifetime risk of nearly 100%.29 Fundic gland polyposis occurs in nearly 90% of patients,30 while gastric adenomas are reported in fewer than 15% of patients.
Duodenal and periampullary cancer is the second most common malignancy in familial adenomatous polyposis. The lifetime risk ranges from 2% to 36%, depending on the Spigelman stage. People with Spigelman stage I, II, or III have a 2.5% risk of duodenal cancer, while those with stage IV disease have up to a 36% lifetime risk.
Gastric cancer, arising from fundic gland polyps, has been reported but is rare in Western populations.
In familial adenomatous polyposis, the incidence of jejunal adenomas and cancer is less than 10%, and the risk of ileal adenomas and cancer is less than 1%.31
Familial adenomatous polyposis is also associated with a higher risk of other malignancies, including:
- Pancreatic cancer (2% lifetime risk)
- Thyroid cancer (2% to 3% lifetime risk, typically papillary carcinoma)32
- Hepatoblastoma (1% to 2% lifetime risk)
- Brain tumors (< 1% lifetime risk)
- Biliary cancer (higher risk than in the general population).33
Benign extracolonic manifestations that have been observed include osteomas, dental abnormalities (supernumerary teeth, unerupted or absent teeth, odontomas), congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions (epidermoid cysts and fibromas), and desmoid tumors.33 The term “Gardner syndrome” has been used to describe patients who have familial adenomatous polyposis but also have osteomas and soft-tissue tumors.34 These patients carry the same risk of colorectal cancer as other patients with familial adenomatous polyposis.
Diagnosing familial adenomatous polyposis
The diagnosis of familial adenomatous polyposis is suspected when a patient has more than 10 adenomatous polyps.
Seventy-five percent of patients with familial adenomatous polyposis have a family history of the condition. Therefore, most cases are identified at a young age on screening sigmoidoscopy or colonoscopy or by predictive gene testing. Patients rarely have cancer at the time of diagnosis.
The other 25% of patients typically are diagnosed when symptoms develop from the polyps or cancer. Over 50% of these symptomatic patients have cancer at the time of diagnosis.
It is recommended that people who have more than 10 adenomas detected on a single colonoscopy or who are first-degree relatives of patients with familial adenomatous polyposis undergo a genetic evaluation and testing for mutations in the APC gene.14 Once an APC mutation is identified in the family, at-risk relatives should be offered testing around age 10 years for families with classic familial adenomatous polyposis or in the mid to late teenage years for those with the attenuated form. It also appropriate to refer patients with desmoid tumors, duodenal adenomas, and bilateral or multifocal congenital hypertrophy of the retinal pigment epithelium for a genetic evaluation.
Management of familial adenomatous polyposis
Flexible sigmoidoscopy every 1 to 2 years beginning at age 10 to 12 years is recommended for individuals and families who have been phenotypically or genetically diagnosed with familial adenomatous polyposis.35–37 If colorectal adenomas are found, surgical options should be discussed and annual colonoscopic surveillance should commence.
For people with the attenuated form, because of the later age of disease onset and the tendency for right-sided disease, colonoscopy every 1 to 2 years should commence at about age 18.35–37 If polyps are found, colonoscopy should be performed every year.
The decision of when to offer colectomy is based on polyp burden (taking into account the number, pathologic appearance, and size of the polyps) and psychosocial factors such as patient maturity. Surgical options include total colectomy and ileorectal anastomosis or total proctocolectomy and ileal pouch anal anastomosis.38 Colonic and extracolonic phenotype as well as genotype should factor into the type of operation recommended. After colectomy, annual endoscopic surveillance of the rectum or ileal pouch is indicated to screen for recurrent polyposis and cancer.
Chemoprevention with sulindac (Clinoril) 150 mg or celecoxib (Celebrex) 400 mg twice a day causes regression of colorectal adenomas in familial adenomatous polyposis and may be useful as an adjunct to endoscopy in managing the colorectal polyp burden.39,40
Forward and side-viewing upper endoscopy should commence at age 20. This should include visualization and biopsy of the papilla and periampulllary region.29 The frequency of endoscopic surveillance depends on the Spigelman stage, which reflects the duodenal polyp burden. It is recommended that patients with Spigelman stage IV duodenal polyposis be seen in consultation with an experienced gastrointestinal surgeon for consideration of a prophylactic, pylorus-preserving, pancreas-sparing duodenectomy. This procedure has been shown to be more effective in polyp control and cancer prevention than endoscopic polyp ablation and local surgical resection.41
Some evidence for the utility of celecoxib 400 mg twice daily for the regression of duodenal polyposis was noted in a 6-month placebo-controlled trial.42 Some experts recommend removal of large duodenal adenomas, with adjunctive celecoxib therapy to control polyposis burden.30
People with familial adenomatous polyposis have been shown to have a 2.6% risk of thyroid cancer, and ultrasonography of the neck with attention to the thyroid is recommended for them.32
MYH-ASSOCIATED POLYPOSIS
Biallelic mutations in the MYH gene result in an adenomatous polyposis syndrome that may be indistinguishable from the attenuated or classic forms of familial adenomatous polyposis. A characteristic autosomal recessive pattern of inheritance in the family can be useful for identifying these patients in the clinic.
Genetics of MYH-associated polyposis
MYH-associated polyposis is the only known autosomal recessive hereditary colorectal cancer syndrome. In white populations, the most commonly reported mutations in MYH are Y179C (previously called Y165C) and G396D (previously called G382D), which account for up to 80% of cases.43 These two mutations are estimated to occur in 1% to 2% of the general population.44
Clinical presentation of MYH-associated polyposis
MYH-associated polyposis typically presents as multiple adenomatous polyps and is diagnosed at a mean age of 47 years. Eleven percent to 42% of affected individuals are reported to have fewer than 100 adenomas, while a minority (7.5% to 29%) of patients present with classic polyposis.45–47 In one study, an estimated 19% of patients presented with colorectal cancer and reported no history of colorectal polyps.48 Synchronous colorectal cancer is seen in more than 60% of patients with biallelic MYH mutations.49 Patients with monoallelic (heterozygous) MYH mutations appear to have the same risk of developing colorectal adenomas and cancer as the general population.49
Upper-gastrointestinal polyps have been reported in MYH-associated polyposis; as many as 17% to 25% of patients have duodenal adenomas.50,51
Diagnosis of MYH-associated polyposis
Genetic testing for biallelic MYH mutations should be performed in patients who test negative for an APC mutation but who have clinical features of familial adenomatous polyposis, a personal history of more than 10 colorectal adenomas, or a recessive family history of polyposis. 14 It has been shown that up to 29% of patients with familial adenomatous polyposis who are APC-negative will have biallelic mutations in the MYH gene.52 The siblings of a patient with biallelic MYH mutations should be offered genetic counseling and testing in their late teens or early 20s. All children of an individual with MYH-associated polyposis will carry one MYH mutation and are only at risk of having the syndrome if the other parent is also a MYH carrier and passed on his or her mutation.
Management of MYH-associated polyposis
The management of patients with MYH-associated polyposis is similar to that recommended for attenuated and classic familial adenomatous polyposis.14 Genetic counseling and testing and colonic and extracolonic surveillance are warranted. There are no data on the use of chemoprevention in MYH-associated polyposis. Surgery should be considered early because of the high risk of colorectal cancer, even in individuals with very few adenomas. Patients with monoallelic MYH mutations should follow the general population screening guidelines for colorectal cancer.49
GENETIC COUNSELING AND GENETIC TESTING
The American College of Gastroenterology advises that patients suspected of having hereditary colorectal cancer syndromes be advised to pursue genetic counseling and, if appropriate, genetic testing.16 They further recommend genetic counseling and informed consent before genetic testing.16
Genetic counseling is a process of working with patients and families whereby:
- A detailed medical and family history is obtained
- A formal risk assessment is performed
- Education about the disease in question and about genetic testing is provided
- Psychosocial concerns are assessed
- Informed consent is obtained when genetic testing is recommended.53
This process is important for helping patients better understand their cancer risks, the benefits and limitations of genetic testing, and the protections that are in place for people who undergo genetic testing, including the Genetic Information Non-Discrimination Act.
In 1996 the American Society of Clinical Oncology issued a policy statement highlighting the essential elements of informed consent for genetic testing for cancer susceptibility, and this was updated in 2003.54 In particular, it notes that patients should be informed of the implications of positive and negative results and of the possibility that the test may be uninformative.
When a hereditary colorectal cancer syndrome is suspected, a positive genetic test result confirms the diagnosis and allows for predictive testing of the patient’s relatives. However, no genetic test for a hereditary colorectal cancer syndrome is 100% sensitive. Therefore, a negative result does not rule out the syndrome in question.
Further, all cancer susceptibility genes have variants of uncertain significance, which are genetic alterations for which there are insufficient data to determine if the mutation is disease-causing or polymorphic (benign). Both negative and uninformative results can be confusing for patients and providers and can lead to false reassurance or undue worry when patients are not properly educated about these potential outcomes of testing.
Genetic testing is an evolving field, and with additional research and improved testing technologies, appropriate diagnoses can be made over time. That is why it is important for the genetic counseling relationship to continue over time.
- Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011; 305:2304–2310.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011; 60:950–957.
- Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009; 75:141–149.
- van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 2010; 47:464–470.
- Wijnen JT, Vasen HF, Khan PM, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 1998; 339:511–518.
- Bisgaard ML, Bernstein I. HNPCC mutation rate. Familial Cancer 2003; 2.
- Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34:424–425.
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116:1453–1456.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96:261–268.
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med 2009; 11:15–20.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260:812–816.
- Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994; 145:148–156.
- Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293:1979–1985.
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines) colorectal cancer screening version 2.2011. www.nccn.org. Accessed October 2, 2012.
- Jass JR. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol 2006; 12:4943–4950.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Winawer S, Fletcher R, Rex D, et al; Gastrointestinal Consortium Panel. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 2003; 124:544–560.
- Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006; 296:1507–1517.
- de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006; 130:665–671.
- Mecklin JP, Aarnio M, Läärä E, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007; 133:1093–1098.
- Engel C, Rahner N, Schulmann K, et al; German HNPCC Consortium. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 2010; 8:174–182.
- Burn J, Gerdes AM, Macrae F, et al; CAPP2 Investigators. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011; 378:2081–2087.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44:353–362.
- Manchanda R, Menon U, Michaelson-Cohen R, Beller U, Jacobs I. Hereditary non-polyposis colorectal cancer or Lynch syndrome: the gynaecological perspective. Curr Opin Obstet Gynecol 2009; 21:31–38.
- Burn J, Chapman P, Delhanty J, et al. The UK Northern region genetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of the retinal pigment epithelium, and DNA markers in risk calculations. J Med Genet 1991; 28:289–296.
- Järvinen HJ. Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut 1992; 33:357–360.
- Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3:121–125.
- Neklason DW, Stevens J, Boucher KM, et al. American founder mutation for attenuated familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:46–52.
- Burke CA, Beck GJ, Church JM, van Stolk RU. The natural history of untreated duodenal and ampullary adenomas in patients with familial adenomatous polyposis followed in an endoscopic surveillance program. Gastrointest Endosc 1999; 49:358–364.
- Bianchi LK, Burke CA, Bennett AE, Lopez R, Hasson H, Church JM. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:180–185.
- Kadmon M, Tandara A, Herfarth C. Duodenal adenomatosis in familial adenomatous polyposis coli. A review of the literature and results from the Heidelberg Polyposis Register. Int J Colorectal Dis 2001; 16:63–75.
- Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg 2011; 253:515–521.
- Jasperson KW, Burt RW. APC-associated polyposis conditions. In:Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews (Internet). Seattle, WA: University of Washington; 2011.
- Gardner EJ, Richards RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5:139–147.
- Dunlop MG; British Society for Gastroenterology. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polyposis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 2002; 51(suppl 5):V21–V27.
- Burke W, Petersen G, Lynch P, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. Cancer Genetics Studies Consortium. JAMA 1997; 277:915–919.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57:704–713.
- Church J. Familial adenomatous polyposis. Surg Oncol Clin N Am 2009; 18:585–598.
- Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993; 328:1313–1316.
- Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000; 342:1946–1952.
- Johnson MD, Mackey R, Brown N, Church J, Burke C, Walsh RM. Outcome based on management for duodenal adenomas: sporadic versus familial disease. J Gastrointest Surg 2010; 14:229–235.
- Phillips RK, Wallace MH, Lynch PM, et al; FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002; 50:857–860.
- Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer 2006; 95:239–242.
- Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004; 96:1631–1634.
- Croitoru ME, Cleary SP, Berk T, et al. Germline MYH mutations in a clinic-based series of Canadian multiple colorectal adenoma patients. J Surg Oncol 2007; 95:499–506.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362:39–41.
- Nielsen M, Franken PF, Reinards TH, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet 2005; 42:e54.
- Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 2009; 136:1251–1260.
- Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol 2009; 27:3975–3980.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006; 119:807–814.
- Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009; 137:1976–1985.e1–e10.
- Gismondi V, Meta M, Bonelli L, et al. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer 2004; 109:680–684.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors. J Genet Couns 2004; 13:83–114.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
Hereditary colorectal cancer syndromes account for 5% to 10% of cases of colorectal cancer.
Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history. An accurate and comprehensive family history should cover three generations and include ethnic background, ages and causes of death of relatives, and any diagnosis of cancer, including age at onset and history of polyps.
Red flags for a hereditary colorectal cancer syndrome in the personal or family history are:
- Early age of onset of cancer (eg, colorectal cancer before age 50)
- More than 10 colorectal adenomas
- Synchronous (ie, occurring at the same time) or metachronous (occurring at different times) primary cancers
- Multiple relatives in successive generations with the same or related cancers (eg, colon or endometrial cancer)
- A family member with a known hereditary colorectal cancer syndrome (Table 1).
Any of these red flags should prompt a referral for genetic counseling.
SYNDROMES ARE CLASSIFIED AS WITH OR WITHOUT POLYPOSIS
Many hereditary syndromes are associated with a higher risk of colorectal cancer. Generally, they can be divided into two categories (Table 2): polyposis syndromes (in which patients have numerous colorectal polyps) and nonpolyposis syndromes (with few or no polyps).
These two main types are subclassified on the basis of the histology of most of the polyps detected: adenomatous, hamartomatous, serrated, or mixed types.
In this review, we will address the three most common of these syndromes: Lynch syndrome (hereditary nonpolyposis colorectal cancer), familial adenomatous polyposis, and MYH-associated polyposis. However, as noted in Table 2, other hereditary colorectal cancer syndromes exist, and suspicion of these conditions should prompt a referral for further evaluation.
LYNCH SYNDROME (HEREDITARY NONPOLYPOSIS COLORECTAL CANCER)
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer, predisposes people to a variety of cancers.
Colorectal cancer is the most common type of cancer associated with Lynch syndrome. Recent research suggests that the cumulative risk of developing colorectal cancer by age 80 is 42% for all patients with Lynch syndrome.1 The median age at onset is 45 years.1 For patients who undergo segmental resection of their initial cancer, the cumulative risk of metachronous colorectal cancer (ie, a new tumor arising later) is 16% at 10 years, 41% at 20 years, and up to 62% after 30 years.2
Endometrial cancer occurs in 17% to 57% of women with Lynch syndrome by age 70, with a median age at onset of 49 years.1
Other extracolonic cancers in Lynch syndrome include cancers of the:
- Stomach (1%–10% risk by age 70 years)
- Ovaries (1%–20% risk)
- Hepatobiliary tract (1%–2% risk)
- Urinary tract (1%–12% risk)
- Small bowel (1%–2% risk)
- Brain (1%–8% risk)
- Skin (sebaceous adenomas, adenocarcinomas, and keratoacanthomas).1,3,4
Earlier studies reported higher rates of associated cancer than those shown here. However, their data were largely derived from registries and may be overestimates. The numbers shown above are from population-based studies.
Genetics of Lynch syndrome
Lynch syndrome is caused by a germline mutation in the MLH1, MSH2, MSH6, PMS2, or EPCAM genes.5 These genes code for proteins that are responsible DNA mismatch repair—one of the cell’s proofreading mechanisms during DNA replication.
These mutations are inherited in an autosomal dominant manner. Though de novo mutations in these genes have been reported, they are rare and the exact frequency with which they occur is unknown.6
In whom should Lynch syndrome be suspected?
Lynch syndrome can be suspected on the basis of family history and clinical criteria.
In 1991, the same group of experts who coined the term “hereditary nonpolyposis colorectal cancer” developed family history criteria for it1:
- At least three relatives with histologically confirmed colorectal cancer, one of whom is a first-degree relative of the other two
- At least two successive generations involved
- At least one of the cancers diagnosed before age 50
- Familial adenomatous polyposis is excluded.
Known as the Amsterdam criteria, these were to be used in collaborative studies of families with hereditary colorectal cancer.7 In 1999, these criteria were broadened to include extracolonic cancers and became known as the Amsterdam II criteria (Table 3).8
Patients whose families meet the Amsterdam II criteria or who have molecular pathologic evidence of Lynch syndrome (see below) are appropriate candidates for genetic counseling and testing.
Diagnosis of Lynch syndrome
The diagnosis of Lynch syndrome is based on molecular pathologic analysis (performed on tumor samples) and confirmed by genetic testing.
Molecular pathologic evidence of Lynch syndrome includes microsatellite instability and loss of expression of one or more of the DNA mismatch repair proteins (detected using immunohistochemistry) (more on these below). The revised Bethesda guidelines (TABLE 3) were intended to identify individuals whose tumors should be tested for one or both of these phenomena.9
In 2009, the Evaluation of Genomic Applications in Practice and Prevention working group recommended that all patients with newly diagnosed colorectal cancer undergo microsatellite instability analysis, immunohistochemistry testing, or both, regardless of whether they meet the Amsterdam II or the Bethesda guideline criteria.10
Microsatellite instability analysis. Microsatellites are short sequences of repeated DNA. The tumor cells of patients who carry defective mismatch repair genes have microsatellites that are longer or shorter than in normal cells, a condition called microsatellite instability (ie, “MSI-high”).
Microsatellite instability testing, using a standardized panel of five DNA markers, is performed on normal and tumor tissue. If more than two of the five microsatellite markers in the tumor show instability, the lesion is considered to have a high level of microsatellite instability. About 15% of colorectal cancers have this high level, although most are not associated with Lynch syndrome and lose MLH1 expression by promoter methylation.11,12
While only 2% of patients with colorectal cancer have Lynch syndrome, from 90% to 95% of colorectal cancers from patients with Lynch syndrome have high levels of microsatellite instability.10 The presence of MLH1 promoter hypermethylation, the BRAF mutation V600E, or both within the tumor suggests that the cancer is not associated with Lynch syndrome.
Some families that meet the Amsterdam I criteria have microsatellite-stable tumors: their condition has been called familial colorectal cancer type X.13 This condition is associated with a higher risk of colorectal cancer but not the other malignancies observed in Lynch syndrome.
Immunohistochemistry is performed to assess for expression of the mismatch repair proteins MSH2, MSH6, MLH1, and PMS2. Absence of expression of the specific protein within tumor cells compared with normal cells within the specimen suggests dysfunction of the specific gene and guides germline mutation testing (Figure 1). For example, a patient who lacks expression of the MSH2 protein in his or her colon cancer most likely has a mutation in the MSH2 gene. Therefore, germ-line genetic testing should initially target the MSH2 gene. Approximately 88% of Lynch syndrome-associated colorectal cancers have abnormal immunohistochemical staining.10
Testing for microsatellite instability and mismatch repair gene expression ideally precedes germline genetic testing and helps to guide which gene or genes should be tested.9,14
Genetic testing for Lynch syndrome is routinely performed on a blood or saliva sample, using DNA from white blood cells and sequencing the gene or genes involved to look for mutations. Positive results from a germline genetic test confirm the diagnosis of Lynch syndrome and allow for predictive testing for relatives at risk. The term Lynch syndrome is used exclusively to describe individuals with evidence of a mutation in one of the mismatch repair genes.15
If a patient’s results are positive, genetic counseling and genetic testing should be offered to at-risk relatives age 18 and over.
Management of Lynch syndrome
Aggressive cancer surveillance is essential for people with Lynch syndrome and for those who are considered at risk but have not pursued genetic testing, such as a sibling of a person with Lynch syndrome.
Colorectal cancer. Colonoscopy is recommended every 1 to 2 years beginning at the age of 20 to 25 years, or 2 to 5 years earlier than the age of the youngest relative affected with colorectal cancer if the initial diagnosis was before age 25. When patients turn 40 years old, colonoscopy is done annually.16–18 A significant reduction in cancer incidence and in the mortality rate has been shown with colonoscopic surveillance.19–21
Chemoprevention may also have a role. Patients with Lynch syndrome who took aspirin 600 mg per day for an average of 25 months had a significantly lower incidence of colorectal cancer during a 55-month follow-up period compared with patients randomized to placebo.22
For patients with Lynch syndrome who are diagnosed with colorectal cancer, the high risk of metachronous cancers after standard segmental colectomy calls for a more extended resection. Retrospective analysis of 382 Lynch syndrome patients found that none of the 50 who underwent total or subtotal colectomy were diagnosed with metachronous colorectal cancer, whereas a metachronous cancer developed in 74 (22%) of the 332 patients who had had segmented resection.2 Annual surveillance of the remaining colon, rectum, or both is indicated postoperatively.
Gynecologic cancers. Women with Lynch syndrome should also consider gynecologic surveillance and risk-reducing surgery. This includes annual gynecologic examination, transvaginal ultrasonography, and endometrial aspiration, beginning at age 30 to 35 years. Although this surveillance does detect premalignant lesions and early symptomatic cancers, its effect on the mortality rate is unknown. Hysterectomy with bilateral salpingo-oophorectomy has been shown to significantly reduce endometrial and ovarian cancers in women with Lynch syndrome.23,24
Urothelial cancers. Carriers of MSH2 mutations have a significantly higher risk of urothelial cancers.4 Therefore, MSH2 carriers should consider ultrasonography of the urinary tract, urinary cytology, and urinalysis every 1 to 2 years beginning at age 40.4
Other extracolonic cancers. Poor evidence exists for systematic screening for the other extracolonic tumors associated with Lynch syndrome. However, the National Comprehensive Cancer Network advises considering esophagogastroduodenoscopy with extended duodenoscopy as well as capsule endoscopy every 2 to 3 years beginning at age 30 to 35.14
ADENOMATOUS POLYPOSIS SYNDROMES
Familial adenomatous polyposis and MYH-associated polyposis are the next most common hereditary colorectal cancer syndromes. Each of these accounts for about 1% of cases of colorectal cancer. Clinically, these two syndromes can be challenging to distinguish because they overlap phenotypically to a significant degree.
FAMILIAL ADENOMATOUS POLYPOSIS
Familial adenomatous polyposis is caused by mutations in the APC gene. Its prevalence is 2.29 to 3.2 per 100,000 individuals.25,26
Genetics of familial adenomatous polyposis
APC is the only gene known to cause familial adenomatous polyposis. Mutations in APC are inherited in an autosomal dominant manner. Approximately 25% of cases of familial adenomatous polyposis are due to a de novo mutation in APC.27
Clinical presentation of familial adenomatous polyposis
Familial adenomatous polyposis is classified by the burden of colorectal adenomas.
Patients who have fewer than 100 adenomas have an attenuated form of the disease. In this group, polyps usually begin to form in the late teenage years or early 20s and tend to develop in the proximal colon. The attenuated form is associated with an approximately 70% lifetime risk of colorectal cancer.28
Patients who have more than 100 polyps are considered to have the classic form of the disease, and those with more than 1,000 polyps have profuse familial adenomatous polyposis (Figure 2). In these groups, polyps typically begin to develop in the preteenage to mid-teenage years. Without surgery, there is nearly a 100% risk of colorectal cancer. The average age at diagnosis of colorectal cancer is 39 years for patients with classic disease.
Upper gastrointestinal polyps are common in familial adenomatous polyposis. Nearly 90% of patients develop duodenal adenomas by a mean age of 44, with a cumulative lifetime risk of nearly 100%.29 Fundic gland polyposis occurs in nearly 90% of patients,30 while gastric adenomas are reported in fewer than 15% of patients.
Duodenal and periampullary cancer is the second most common malignancy in familial adenomatous polyposis. The lifetime risk ranges from 2% to 36%, depending on the Spigelman stage. People with Spigelman stage I, II, or III have a 2.5% risk of duodenal cancer, while those with stage IV disease have up to a 36% lifetime risk.
Gastric cancer, arising from fundic gland polyps, has been reported but is rare in Western populations.
In familial adenomatous polyposis, the incidence of jejunal adenomas and cancer is less than 10%, and the risk of ileal adenomas and cancer is less than 1%.31
Familial adenomatous polyposis is also associated with a higher risk of other malignancies, including:
- Pancreatic cancer (2% lifetime risk)
- Thyroid cancer (2% to 3% lifetime risk, typically papillary carcinoma)32
- Hepatoblastoma (1% to 2% lifetime risk)
- Brain tumors (< 1% lifetime risk)
- Biliary cancer (higher risk than in the general population).33
Benign extracolonic manifestations that have been observed include osteomas, dental abnormalities (supernumerary teeth, unerupted or absent teeth, odontomas), congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions (epidermoid cysts and fibromas), and desmoid tumors.33 The term “Gardner syndrome” has been used to describe patients who have familial adenomatous polyposis but also have osteomas and soft-tissue tumors.34 These patients carry the same risk of colorectal cancer as other patients with familial adenomatous polyposis.
Diagnosing familial adenomatous polyposis
The diagnosis of familial adenomatous polyposis is suspected when a patient has more than 10 adenomatous polyps.
Seventy-five percent of patients with familial adenomatous polyposis have a family history of the condition. Therefore, most cases are identified at a young age on screening sigmoidoscopy or colonoscopy or by predictive gene testing. Patients rarely have cancer at the time of diagnosis.
The other 25% of patients typically are diagnosed when symptoms develop from the polyps or cancer. Over 50% of these symptomatic patients have cancer at the time of diagnosis.
It is recommended that people who have more than 10 adenomas detected on a single colonoscopy or who are first-degree relatives of patients with familial adenomatous polyposis undergo a genetic evaluation and testing for mutations in the APC gene.14 Once an APC mutation is identified in the family, at-risk relatives should be offered testing around age 10 years for families with classic familial adenomatous polyposis or in the mid to late teenage years for those with the attenuated form. It also appropriate to refer patients with desmoid tumors, duodenal adenomas, and bilateral or multifocal congenital hypertrophy of the retinal pigment epithelium for a genetic evaluation.
Management of familial adenomatous polyposis
Flexible sigmoidoscopy every 1 to 2 years beginning at age 10 to 12 years is recommended for individuals and families who have been phenotypically or genetically diagnosed with familial adenomatous polyposis.35–37 If colorectal adenomas are found, surgical options should be discussed and annual colonoscopic surveillance should commence.
For people with the attenuated form, because of the later age of disease onset and the tendency for right-sided disease, colonoscopy every 1 to 2 years should commence at about age 18.35–37 If polyps are found, colonoscopy should be performed every year.
The decision of when to offer colectomy is based on polyp burden (taking into account the number, pathologic appearance, and size of the polyps) and psychosocial factors such as patient maturity. Surgical options include total colectomy and ileorectal anastomosis or total proctocolectomy and ileal pouch anal anastomosis.38 Colonic and extracolonic phenotype as well as genotype should factor into the type of operation recommended. After colectomy, annual endoscopic surveillance of the rectum or ileal pouch is indicated to screen for recurrent polyposis and cancer.
Chemoprevention with sulindac (Clinoril) 150 mg or celecoxib (Celebrex) 400 mg twice a day causes regression of colorectal adenomas in familial adenomatous polyposis and may be useful as an adjunct to endoscopy in managing the colorectal polyp burden.39,40
Forward and side-viewing upper endoscopy should commence at age 20. This should include visualization and biopsy of the papilla and periampulllary region.29 The frequency of endoscopic surveillance depends on the Spigelman stage, which reflects the duodenal polyp burden. It is recommended that patients with Spigelman stage IV duodenal polyposis be seen in consultation with an experienced gastrointestinal surgeon for consideration of a prophylactic, pylorus-preserving, pancreas-sparing duodenectomy. This procedure has been shown to be more effective in polyp control and cancer prevention than endoscopic polyp ablation and local surgical resection.41
Some evidence for the utility of celecoxib 400 mg twice daily for the regression of duodenal polyposis was noted in a 6-month placebo-controlled trial.42 Some experts recommend removal of large duodenal adenomas, with adjunctive celecoxib therapy to control polyposis burden.30
People with familial adenomatous polyposis have been shown to have a 2.6% risk of thyroid cancer, and ultrasonography of the neck with attention to the thyroid is recommended for them.32
MYH-ASSOCIATED POLYPOSIS
Biallelic mutations in the MYH gene result in an adenomatous polyposis syndrome that may be indistinguishable from the attenuated or classic forms of familial adenomatous polyposis. A characteristic autosomal recessive pattern of inheritance in the family can be useful for identifying these patients in the clinic.
Genetics of MYH-associated polyposis
MYH-associated polyposis is the only known autosomal recessive hereditary colorectal cancer syndrome. In white populations, the most commonly reported mutations in MYH are Y179C (previously called Y165C) and G396D (previously called G382D), which account for up to 80% of cases.43 These two mutations are estimated to occur in 1% to 2% of the general population.44
Clinical presentation of MYH-associated polyposis
MYH-associated polyposis typically presents as multiple adenomatous polyps and is diagnosed at a mean age of 47 years. Eleven percent to 42% of affected individuals are reported to have fewer than 100 adenomas, while a minority (7.5% to 29%) of patients present with classic polyposis.45–47 In one study, an estimated 19% of patients presented with colorectal cancer and reported no history of colorectal polyps.48 Synchronous colorectal cancer is seen in more than 60% of patients with biallelic MYH mutations.49 Patients with monoallelic (heterozygous) MYH mutations appear to have the same risk of developing colorectal adenomas and cancer as the general population.49
Upper-gastrointestinal polyps have been reported in MYH-associated polyposis; as many as 17% to 25% of patients have duodenal adenomas.50,51
Diagnosis of MYH-associated polyposis
Genetic testing for biallelic MYH mutations should be performed in patients who test negative for an APC mutation but who have clinical features of familial adenomatous polyposis, a personal history of more than 10 colorectal adenomas, or a recessive family history of polyposis. 14 It has been shown that up to 29% of patients with familial adenomatous polyposis who are APC-negative will have biallelic mutations in the MYH gene.52 The siblings of a patient with biallelic MYH mutations should be offered genetic counseling and testing in their late teens or early 20s. All children of an individual with MYH-associated polyposis will carry one MYH mutation and are only at risk of having the syndrome if the other parent is also a MYH carrier and passed on his or her mutation.
Management of MYH-associated polyposis
The management of patients with MYH-associated polyposis is similar to that recommended for attenuated and classic familial adenomatous polyposis.14 Genetic counseling and testing and colonic and extracolonic surveillance are warranted. There are no data on the use of chemoprevention in MYH-associated polyposis. Surgery should be considered early because of the high risk of colorectal cancer, even in individuals with very few adenomas. Patients with monoallelic MYH mutations should follow the general population screening guidelines for colorectal cancer.49
GENETIC COUNSELING AND GENETIC TESTING
The American College of Gastroenterology advises that patients suspected of having hereditary colorectal cancer syndromes be advised to pursue genetic counseling and, if appropriate, genetic testing.16 They further recommend genetic counseling and informed consent before genetic testing.16
Genetic counseling is a process of working with patients and families whereby:
- A detailed medical and family history is obtained
- A formal risk assessment is performed
- Education about the disease in question and about genetic testing is provided
- Psychosocial concerns are assessed
- Informed consent is obtained when genetic testing is recommended.53
This process is important for helping patients better understand their cancer risks, the benefits and limitations of genetic testing, and the protections that are in place for people who undergo genetic testing, including the Genetic Information Non-Discrimination Act.
In 1996 the American Society of Clinical Oncology issued a policy statement highlighting the essential elements of informed consent for genetic testing for cancer susceptibility, and this was updated in 2003.54 In particular, it notes that patients should be informed of the implications of positive and negative results and of the possibility that the test may be uninformative.
When a hereditary colorectal cancer syndrome is suspected, a positive genetic test result confirms the diagnosis and allows for predictive testing of the patient’s relatives. However, no genetic test for a hereditary colorectal cancer syndrome is 100% sensitive. Therefore, a negative result does not rule out the syndrome in question.
Further, all cancer susceptibility genes have variants of uncertain significance, which are genetic alterations for which there are insufficient data to determine if the mutation is disease-causing or polymorphic (benign). Both negative and uninformative results can be confusing for patients and providers and can lead to false reassurance or undue worry when patients are not properly educated about these potential outcomes of testing.
Genetic testing is an evolving field, and with additional research and improved testing technologies, appropriate diagnoses can be made over time. That is why it is important for the genetic counseling relationship to continue over time.
Hereditary colorectal cancer syndromes account for 5% to 10% of cases of colorectal cancer.
Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history. An accurate and comprehensive family history should cover three generations and include ethnic background, ages and causes of death of relatives, and any diagnosis of cancer, including age at onset and history of polyps.
Red flags for a hereditary colorectal cancer syndrome in the personal or family history are:
- Early age of onset of cancer (eg, colorectal cancer before age 50)
- More than 10 colorectal adenomas
- Synchronous (ie, occurring at the same time) or metachronous (occurring at different times) primary cancers
- Multiple relatives in successive generations with the same or related cancers (eg, colon or endometrial cancer)
- A family member with a known hereditary colorectal cancer syndrome (Table 1).
Any of these red flags should prompt a referral for genetic counseling.
SYNDROMES ARE CLASSIFIED AS WITH OR WITHOUT POLYPOSIS
Many hereditary syndromes are associated with a higher risk of colorectal cancer. Generally, they can be divided into two categories (Table 2): polyposis syndromes (in which patients have numerous colorectal polyps) and nonpolyposis syndromes (with few or no polyps).
These two main types are subclassified on the basis of the histology of most of the polyps detected: adenomatous, hamartomatous, serrated, or mixed types.
In this review, we will address the three most common of these syndromes: Lynch syndrome (hereditary nonpolyposis colorectal cancer), familial adenomatous polyposis, and MYH-associated polyposis. However, as noted in Table 2, other hereditary colorectal cancer syndromes exist, and suspicion of these conditions should prompt a referral for further evaluation.
LYNCH SYNDROME (HEREDITARY NONPOLYPOSIS COLORECTAL CANCER)
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer, predisposes people to a variety of cancers.
Colorectal cancer is the most common type of cancer associated with Lynch syndrome. Recent research suggests that the cumulative risk of developing colorectal cancer by age 80 is 42% for all patients with Lynch syndrome.1 The median age at onset is 45 years.1 For patients who undergo segmental resection of their initial cancer, the cumulative risk of metachronous colorectal cancer (ie, a new tumor arising later) is 16% at 10 years, 41% at 20 years, and up to 62% after 30 years.2
Endometrial cancer occurs in 17% to 57% of women with Lynch syndrome by age 70, with a median age at onset of 49 years.1
Other extracolonic cancers in Lynch syndrome include cancers of the:
- Stomach (1%–10% risk by age 70 years)
- Ovaries (1%–20% risk)
- Hepatobiliary tract (1%–2% risk)
- Urinary tract (1%–12% risk)
- Small bowel (1%–2% risk)
- Brain (1%–8% risk)
- Skin (sebaceous adenomas, adenocarcinomas, and keratoacanthomas).1,3,4
Earlier studies reported higher rates of associated cancer than those shown here. However, their data were largely derived from registries and may be overestimates. The numbers shown above are from population-based studies.
Genetics of Lynch syndrome
Lynch syndrome is caused by a germline mutation in the MLH1, MSH2, MSH6, PMS2, or EPCAM genes.5 These genes code for proteins that are responsible DNA mismatch repair—one of the cell’s proofreading mechanisms during DNA replication.
These mutations are inherited in an autosomal dominant manner. Though de novo mutations in these genes have been reported, they are rare and the exact frequency with which they occur is unknown.6
In whom should Lynch syndrome be suspected?
Lynch syndrome can be suspected on the basis of family history and clinical criteria.
In 1991, the same group of experts who coined the term “hereditary nonpolyposis colorectal cancer” developed family history criteria for it1:
- At least three relatives with histologically confirmed colorectal cancer, one of whom is a first-degree relative of the other two
- At least two successive generations involved
- At least one of the cancers diagnosed before age 50
- Familial adenomatous polyposis is excluded.
Known as the Amsterdam criteria, these were to be used in collaborative studies of families with hereditary colorectal cancer.7 In 1999, these criteria were broadened to include extracolonic cancers and became known as the Amsterdam II criteria (Table 3).8
Patients whose families meet the Amsterdam II criteria or who have molecular pathologic evidence of Lynch syndrome (see below) are appropriate candidates for genetic counseling and testing.
Diagnosis of Lynch syndrome
The diagnosis of Lynch syndrome is based on molecular pathologic analysis (performed on tumor samples) and confirmed by genetic testing.
Molecular pathologic evidence of Lynch syndrome includes microsatellite instability and loss of expression of one or more of the DNA mismatch repair proteins (detected using immunohistochemistry) (more on these below). The revised Bethesda guidelines (TABLE 3) were intended to identify individuals whose tumors should be tested for one or both of these phenomena.9
In 2009, the Evaluation of Genomic Applications in Practice and Prevention working group recommended that all patients with newly diagnosed colorectal cancer undergo microsatellite instability analysis, immunohistochemistry testing, or both, regardless of whether they meet the Amsterdam II or the Bethesda guideline criteria.10
Microsatellite instability analysis. Microsatellites are short sequences of repeated DNA. The tumor cells of patients who carry defective mismatch repair genes have microsatellites that are longer or shorter than in normal cells, a condition called microsatellite instability (ie, “MSI-high”).
Microsatellite instability testing, using a standardized panel of five DNA markers, is performed on normal and tumor tissue. If more than two of the five microsatellite markers in the tumor show instability, the lesion is considered to have a high level of microsatellite instability. About 15% of colorectal cancers have this high level, although most are not associated with Lynch syndrome and lose MLH1 expression by promoter methylation.11,12
While only 2% of patients with colorectal cancer have Lynch syndrome, from 90% to 95% of colorectal cancers from patients with Lynch syndrome have high levels of microsatellite instability.10 The presence of MLH1 promoter hypermethylation, the BRAF mutation V600E, or both within the tumor suggests that the cancer is not associated with Lynch syndrome.
Some families that meet the Amsterdam I criteria have microsatellite-stable tumors: their condition has been called familial colorectal cancer type X.13 This condition is associated with a higher risk of colorectal cancer but not the other malignancies observed in Lynch syndrome.
Immunohistochemistry is performed to assess for expression of the mismatch repair proteins MSH2, MSH6, MLH1, and PMS2. Absence of expression of the specific protein within tumor cells compared with normal cells within the specimen suggests dysfunction of the specific gene and guides germline mutation testing (Figure 1). For example, a patient who lacks expression of the MSH2 protein in his or her colon cancer most likely has a mutation in the MSH2 gene. Therefore, germ-line genetic testing should initially target the MSH2 gene. Approximately 88% of Lynch syndrome-associated colorectal cancers have abnormal immunohistochemical staining.10
Testing for microsatellite instability and mismatch repair gene expression ideally precedes germline genetic testing and helps to guide which gene or genes should be tested.9,14
Genetic testing for Lynch syndrome is routinely performed on a blood or saliva sample, using DNA from white blood cells and sequencing the gene or genes involved to look for mutations. Positive results from a germline genetic test confirm the diagnosis of Lynch syndrome and allow for predictive testing for relatives at risk. The term Lynch syndrome is used exclusively to describe individuals with evidence of a mutation in one of the mismatch repair genes.15
If a patient’s results are positive, genetic counseling and genetic testing should be offered to at-risk relatives age 18 and over.
Management of Lynch syndrome
Aggressive cancer surveillance is essential for people with Lynch syndrome and for those who are considered at risk but have not pursued genetic testing, such as a sibling of a person with Lynch syndrome.
Colorectal cancer. Colonoscopy is recommended every 1 to 2 years beginning at the age of 20 to 25 years, or 2 to 5 years earlier than the age of the youngest relative affected with colorectal cancer if the initial diagnosis was before age 25. When patients turn 40 years old, colonoscopy is done annually.16–18 A significant reduction in cancer incidence and in the mortality rate has been shown with colonoscopic surveillance.19–21
Chemoprevention may also have a role. Patients with Lynch syndrome who took aspirin 600 mg per day for an average of 25 months had a significantly lower incidence of colorectal cancer during a 55-month follow-up period compared with patients randomized to placebo.22
For patients with Lynch syndrome who are diagnosed with colorectal cancer, the high risk of metachronous cancers after standard segmental colectomy calls for a more extended resection. Retrospective analysis of 382 Lynch syndrome patients found that none of the 50 who underwent total or subtotal colectomy were diagnosed with metachronous colorectal cancer, whereas a metachronous cancer developed in 74 (22%) of the 332 patients who had had segmented resection.2 Annual surveillance of the remaining colon, rectum, or both is indicated postoperatively.
Gynecologic cancers. Women with Lynch syndrome should also consider gynecologic surveillance and risk-reducing surgery. This includes annual gynecologic examination, transvaginal ultrasonography, and endometrial aspiration, beginning at age 30 to 35 years. Although this surveillance does detect premalignant lesions and early symptomatic cancers, its effect on the mortality rate is unknown. Hysterectomy with bilateral salpingo-oophorectomy has been shown to significantly reduce endometrial and ovarian cancers in women with Lynch syndrome.23,24
Urothelial cancers. Carriers of MSH2 mutations have a significantly higher risk of urothelial cancers.4 Therefore, MSH2 carriers should consider ultrasonography of the urinary tract, urinary cytology, and urinalysis every 1 to 2 years beginning at age 40.4
Other extracolonic cancers. Poor evidence exists for systematic screening for the other extracolonic tumors associated with Lynch syndrome. However, the National Comprehensive Cancer Network advises considering esophagogastroduodenoscopy with extended duodenoscopy as well as capsule endoscopy every 2 to 3 years beginning at age 30 to 35.14
ADENOMATOUS POLYPOSIS SYNDROMES
Familial adenomatous polyposis and MYH-associated polyposis are the next most common hereditary colorectal cancer syndromes. Each of these accounts for about 1% of cases of colorectal cancer. Clinically, these two syndromes can be challenging to distinguish because they overlap phenotypically to a significant degree.
FAMILIAL ADENOMATOUS POLYPOSIS
Familial adenomatous polyposis is caused by mutations in the APC gene. Its prevalence is 2.29 to 3.2 per 100,000 individuals.25,26
Genetics of familial adenomatous polyposis
APC is the only gene known to cause familial adenomatous polyposis. Mutations in APC are inherited in an autosomal dominant manner. Approximately 25% of cases of familial adenomatous polyposis are due to a de novo mutation in APC.27
Clinical presentation of familial adenomatous polyposis
Familial adenomatous polyposis is classified by the burden of colorectal adenomas.
Patients who have fewer than 100 adenomas have an attenuated form of the disease. In this group, polyps usually begin to form in the late teenage years or early 20s and tend to develop in the proximal colon. The attenuated form is associated with an approximately 70% lifetime risk of colorectal cancer.28
Patients who have more than 100 polyps are considered to have the classic form of the disease, and those with more than 1,000 polyps have profuse familial adenomatous polyposis (Figure 2). In these groups, polyps typically begin to develop in the preteenage to mid-teenage years. Without surgery, there is nearly a 100% risk of colorectal cancer. The average age at diagnosis of colorectal cancer is 39 years for patients with classic disease.
Upper gastrointestinal polyps are common in familial adenomatous polyposis. Nearly 90% of patients develop duodenal adenomas by a mean age of 44, with a cumulative lifetime risk of nearly 100%.29 Fundic gland polyposis occurs in nearly 90% of patients,30 while gastric adenomas are reported in fewer than 15% of patients.
Duodenal and periampullary cancer is the second most common malignancy in familial adenomatous polyposis. The lifetime risk ranges from 2% to 36%, depending on the Spigelman stage. People with Spigelman stage I, II, or III have a 2.5% risk of duodenal cancer, while those with stage IV disease have up to a 36% lifetime risk.
Gastric cancer, arising from fundic gland polyps, has been reported but is rare in Western populations.
In familial adenomatous polyposis, the incidence of jejunal adenomas and cancer is less than 10%, and the risk of ileal adenomas and cancer is less than 1%.31
Familial adenomatous polyposis is also associated with a higher risk of other malignancies, including:
- Pancreatic cancer (2% lifetime risk)
- Thyroid cancer (2% to 3% lifetime risk, typically papillary carcinoma)32
- Hepatoblastoma (1% to 2% lifetime risk)
- Brain tumors (< 1% lifetime risk)
- Biliary cancer (higher risk than in the general population).33
Benign extracolonic manifestations that have been observed include osteomas, dental abnormalities (supernumerary teeth, unerupted or absent teeth, odontomas), congenital hypertrophy of the retinal pigment epithelium, benign cutaneous lesions (epidermoid cysts and fibromas), and desmoid tumors.33 The term “Gardner syndrome” has been used to describe patients who have familial adenomatous polyposis but also have osteomas and soft-tissue tumors.34 These patients carry the same risk of colorectal cancer as other patients with familial adenomatous polyposis.
Diagnosing familial adenomatous polyposis
The diagnosis of familial adenomatous polyposis is suspected when a patient has more than 10 adenomatous polyps.
Seventy-five percent of patients with familial adenomatous polyposis have a family history of the condition. Therefore, most cases are identified at a young age on screening sigmoidoscopy or colonoscopy or by predictive gene testing. Patients rarely have cancer at the time of diagnosis.
The other 25% of patients typically are diagnosed when symptoms develop from the polyps or cancer. Over 50% of these symptomatic patients have cancer at the time of diagnosis.
It is recommended that people who have more than 10 adenomas detected on a single colonoscopy or who are first-degree relatives of patients with familial adenomatous polyposis undergo a genetic evaluation and testing for mutations in the APC gene.14 Once an APC mutation is identified in the family, at-risk relatives should be offered testing around age 10 years for families with classic familial adenomatous polyposis or in the mid to late teenage years for those with the attenuated form. It also appropriate to refer patients with desmoid tumors, duodenal adenomas, and bilateral or multifocal congenital hypertrophy of the retinal pigment epithelium for a genetic evaluation.
Management of familial adenomatous polyposis
Flexible sigmoidoscopy every 1 to 2 years beginning at age 10 to 12 years is recommended for individuals and families who have been phenotypically or genetically diagnosed with familial adenomatous polyposis.35–37 If colorectal adenomas are found, surgical options should be discussed and annual colonoscopic surveillance should commence.
For people with the attenuated form, because of the later age of disease onset and the tendency for right-sided disease, colonoscopy every 1 to 2 years should commence at about age 18.35–37 If polyps are found, colonoscopy should be performed every year.
The decision of when to offer colectomy is based on polyp burden (taking into account the number, pathologic appearance, and size of the polyps) and psychosocial factors such as patient maturity. Surgical options include total colectomy and ileorectal anastomosis or total proctocolectomy and ileal pouch anal anastomosis.38 Colonic and extracolonic phenotype as well as genotype should factor into the type of operation recommended. After colectomy, annual endoscopic surveillance of the rectum or ileal pouch is indicated to screen for recurrent polyposis and cancer.
Chemoprevention with sulindac (Clinoril) 150 mg or celecoxib (Celebrex) 400 mg twice a day causes regression of colorectal adenomas in familial adenomatous polyposis and may be useful as an adjunct to endoscopy in managing the colorectal polyp burden.39,40
Forward and side-viewing upper endoscopy should commence at age 20. This should include visualization and biopsy of the papilla and periampulllary region.29 The frequency of endoscopic surveillance depends on the Spigelman stage, which reflects the duodenal polyp burden. It is recommended that patients with Spigelman stage IV duodenal polyposis be seen in consultation with an experienced gastrointestinal surgeon for consideration of a prophylactic, pylorus-preserving, pancreas-sparing duodenectomy. This procedure has been shown to be more effective in polyp control and cancer prevention than endoscopic polyp ablation and local surgical resection.41
Some evidence for the utility of celecoxib 400 mg twice daily for the regression of duodenal polyposis was noted in a 6-month placebo-controlled trial.42 Some experts recommend removal of large duodenal adenomas, with adjunctive celecoxib therapy to control polyposis burden.30
People with familial adenomatous polyposis have been shown to have a 2.6% risk of thyroid cancer, and ultrasonography of the neck with attention to the thyroid is recommended for them.32
MYH-ASSOCIATED POLYPOSIS
Biallelic mutations in the MYH gene result in an adenomatous polyposis syndrome that may be indistinguishable from the attenuated or classic forms of familial adenomatous polyposis. A characteristic autosomal recessive pattern of inheritance in the family can be useful for identifying these patients in the clinic.
Genetics of MYH-associated polyposis
MYH-associated polyposis is the only known autosomal recessive hereditary colorectal cancer syndrome. In white populations, the most commonly reported mutations in MYH are Y179C (previously called Y165C) and G396D (previously called G382D), which account for up to 80% of cases.43 These two mutations are estimated to occur in 1% to 2% of the general population.44
Clinical presentation of MYH-associated polyposis
MYH-associated polyposis typically presents as multiple adenomatous polyps and is diagnosed at a mean age of 47 years. Eleven percent to 42% of affected individuals are reported to have fewer than 100 adenomas, while a minority (7.5% to 29%) of patients present with classic polyposis.45–47 In one study, an estimated 19% of patients presented with colorectal cancer and reported no history of colorectal polyps.48 Synchronous colorectal cancer is seen in more than 60% of patients with biallelic MYH mutations.49 Patients with monoallelic (heterozygous) MYH mutations appear to have the same risk of developing colorectal adenomas and cancer as the general population.49
Upper-gastrointestinal polyps have been reported in MYH-associated polyposis; as many as 17% to 25% of patients have duodenal adenomas.50,51
Diagnosis of MYH-associated polyposis
Genetic testing for biallelic MYH mutations should be performed in patients who test negative for an APC mutation but who have clinical features of familial adenomatous polyposis, a personal history of more than 10 colorectal adenomas, or a recessive family history of polyposis. 14 It has been shown that up to 29% of patients with familial adenomatous polyposis who are APC-negative will have biallelic mutations in the MYH gene.52 The siblings of a patient with biallelic MYH mutations should be offered genetic counseling and testing in their late teens or early 20s. All children of an individual with MYH-associated polyposis will carry one MYH mutation and are only at risk of having the syndrome if the other parent is also a MYH carrier and passed on his or her mutation.
Management of MYH-associated polyposis
The management of patients with MYH-associated polyposis is similar to that recommended for attenuated and classic familial adenomatous polyposis.14 Genetic counseling and testing and colonic and extracolonic surveillance are warranted. There are no data on the use of chemoprevention in MYH-associated polyposis. Surgery should be considered early because of the high risk of colorectal cancer, even in individuals with very few adenomas. Patients with monoallelic MYH mutations should follow the general population screening guidelines for colorectal cancer.49
GENETIC COUNSELING AND GENETIC TESTING
The American College of Gastroenterology advises that patients suspected of having hereditary colorectal cancer syndromes be advised to pursue genetic counseling and, if appropriate, genetic testing.16 They further recommend genetic counseling and informed consent before genetic testing.16
Genetic counseling is a process of working with patients and families whereby:
- A detailed medical and family history is obtained
- A formal risk assessment is performed
- Education about the disease in question and about genetic testing is provided
- Psychosocial concerns are assessed
- Informed consent is obtained when genetic testing is recommended.53
This process is important for helping patients better understand their cancer risks, the benefits and limitations of genetic testing, and the protections that are in place for people who undergo genetic testing, including the Genetic Information Non-Discrimination Act.
In 1996 the American Society of Clinical Oncology issued a policy statement highlighting the essential elements of informed consent for genetic testing for cancer susceptibility, and this was updated in 2003.54 In particular, it notes that patients should be informed of the implications of positive and negative results and of the possibility that the test may be uninformative.
When a hereditary colorectal cancer syndrome is suspected, a positive genetic test result confirms the diagnosis and allows for predictive testing of the patient’s relatives. However, no genetic test for a hereditary colorectal cancer syndrome is 100% sensitive. Therefore, a negative result does not rule out the syndrome in question.
Further, all cancer susceptibility genes have variants of uncertain significance, which are genetic alterations for which there are insufficient data to determine if the mutation is disease-causing or polymorphic (benign). Both negative and uninformative results can be confusing for patients and providers and can lead to false reassurance or undue worry when patients are not properly educated about these potential outcomes of testing.
Genetic testing is an evolving field, and with additional research and improved testing technologies, appropriate diagnoses can be made over time. That is why it is important for the genetic counseling relationship to continue over time.
- Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011; 305:2304–2310.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011; 60:950–957.
- Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009; 75:141–149.
- van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 2010; 47:464–470.
- Wijnen JT, Vasen HF, Khan PM, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 1998; 339:511–518.
- Bisgaard ML, Bernstein I. HNPCC mutation rate. Familial Cancer 2003; 2.
- Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34:424–425.
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116:1453–1456.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96:261–268.
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med 2009; 11:15–20.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260:812–816.
- Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994; 145:148–156.
- Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293:1979–1985.
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines) colorectal cancer screening version 2.2011. www.nccn.org. Accessed October 2, 2012.
- Jass JR. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol 2006; 12:4943–4950.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Winawer S, Fletcher R, Rex D, et al; Gastrointestinal Consortium Panel. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 2003; 124:544–560.
- Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006; 296:1507–1517.
- de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006; 130:665–671.
- Mecklin JP, Aarnio M, Läärä E, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007; 133:1093–1098.
- Engel C, Rahner N, Schulmann K, et al; German HNPCC Consortium. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 2010; 8:174–182.
- Burn J, Gerdes AM, Macrae F, et al; CAPP2 Investigators. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011; 378:2081–2087.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44:353–362.
- Manchanda R, Menon U, Michaelson-Cohen R, Beller U, Jacobs I. Hereditary non-polyposis colorectal cancer or Lynch syndrome: the gynaecological perspective. Curr Opin Obstet Gynecol 2009; 21:31–38.
- Burn J, Chapman P, Delhanty J, et al. The UK Northern region genetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of the retinal pigment epithelium, and DNA markers in risk calculations. J Med Genet 1991; 28:289–296.
- Järvinen HJ. Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut 1992; 33:357–360.
- Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3:121–125.
- Neklason DW, Stevens J, Boucher KM, et al. American founder mutation for attenuated familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:46–52.
- Burke CA, Beck GJ, Church JM, van Stolk RU. The natural history of untreated duodenal and ampullary adenomas in patients with familial adenomatous polyposis followed in an endoscopic surveillance program. Gastrointest Endosc 1999; 49:358–364.
- Bianchi LK, Burke CA, Bennett AE, Lopez R, Hasson H, Church JM. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:180–185.
- Kadmon M, Tandara A, Herfarth C. Duodenal adenomatosis in familial adenomatous polyposis coli. A review of the literature and results from the Heidelberg Polyposis Register. Int J Colorectal Dis 2001; 16:63–75.
- Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg 2011; 253:515–521.
- Jasperson KW, Burt RW. APC-associated polyposis conditions. In:Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews (Internet). Seattle, WA: University of Washington; 2011.
- Gardner EJ, Richards RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5:139–147.
- Dunlop MG; British Society for Gastroenterology. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polyposis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 2002; 51(suppl 5):V21–V27.
- Burke W, Petersen G, Lynch P, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. Cancer Genetics Studies Consortium. JAMA 1997; 277:915–919.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57:704–713.
- Church J. Familial adenomatous polyposis. Surg Oncol Clin N Am 2009; 18:585–598.
- Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993; 328:1313–1316.
- Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000; 342:1946–1952.
- Johnson MD, Mackey R, Brown N, Church J, Burke C, Walsh RM. Outcome based on management for duodenal adenomas: sporadic versus familial disease. J Gastrointest Surg 2010; 14:229–235.
- Phillips RK, Wallace MH, Lynch PM, et al; FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002; 50:857–860.
- Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer 2006; 95:239–242.
- Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004; 96:1631–1634.
- Croitoru ME, Cleary SP, Berk T, et al. Germline MYH mutations in a clinic-based series of Canadian multiple colorectal adenoma patients. J Surg Oncol 2007; 95:499–506.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362:39–41.
- Nielsen M, Franken PF, Reinards TH, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet 2005; 42:e54.
- Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 2009; 136:1251–1260.
- Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol 2009; 27:3975–3980.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006; 119:807–814.
- Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009; 137:1976–1985.e1–e10.
- Gismondi V, Meta M, Bonelli L, et al. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer 2004; 109:680–684.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors. J Genet Couns 2004; 13:83–114.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
- Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011; 305:2304–2310.
- Parry S, Win AK, Parry B, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011; 60:950–957.
- Barrow E, Robinson L, Alduaij W, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009; 75:141–149.
- van der Post RS, Kiemeney LA, Ligtenberg MJ, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet 2010; 47:464–470.
- Wijnen JT, Vasen HF, Khan PM, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med 1998; 339:511–518.
- Bisgaard ML, Bernstein I. HNPCC mutation rate. Familial Cancer 2003; 2.
- Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34:424–425.
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116:1453–1456.
- Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96:261–268.
- Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: can UGT1A1 genotyping reduce morbidity and mortality in patients with metastatic colorectal cancer treated with irinotecan? Genet Med 2009; 11:15–20.
- Aaltonen LA, Peltomäki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993; 260:812–816.
- Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 1994; 145:148–156.
- Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293:1979–1985.
- National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology (NCCN guidelines) colorectal cancer screening version 2.2011. www.nccn.org. Accessed October 2, 2012.
- Jass JR. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol 2006; 12:4943–4950.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 [corrected]. Am J Gastroenterol 2009; 104:739–750.
- Winawer S, Fletcher R, Rex D, et al; Gastrointestinal Consortium Panel. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 2003; 124:544–560.
- Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA 2006; 296:1507–1517.
- de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006; 130:665–671.
- Mecklin JP, Aarnio M, Läärä E, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007; 133:1093–1098.
- Engel C, Rahner N, Schulmann K, et al; German HNPCC Consortium. Efficacy of annual colonoscopic surveillance in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 2010; 8:174–182.
- Burn J, Gerdes AM, Macrae F, et al; CAPP2 Investigators. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011; 378:2081–2087.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44:353–362.
- Manchanda R, Menon U, Michaelson-Cohen R, Beller U, Jacobs I. Hereditary non-polyposis colorectal cancer or Lynch syndrome: the gynaecological perspective. Curr Opin Obstet Gynecol 2009; 21:31–38.
- Burn J, Chapman P, Delhanty J, et al. The UK Northern region genetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of the retinal pigment epithelium, and DNA markers in risk calculations. J Med Genet 1991; 28:289–296.
- Järvinen HJ. Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut 1992; 33:357–360.
- Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3:121–125.
- Neklason DW, Stevens J, Boucher KM, et al. American founder mutation for attenuated familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:46–52.
- Burke CA, Beck GJ, Church JM, van Stolk RU. The natural history of untreated duodenal and ampullary adenomas in patients with familial adenomatous polyposis followed in an endoscopic surveillance program. Gastrointest Endosc 1999; 49:358–364.
- Bianchi LK, Burke CA, Bennett AE, Lopez R, Hasson H, Church JM. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008; 6:180–185.
- Kadmon M, Tandara A, Herfarth C. Duodenal adenomatosis in familial adenomatous polyposis coli. A review of the literature and results from the Heidelberg Polyposis Register. Int J Colorectal Dis 2001; 16:63–75.
- Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg 2011; 253:515–521.
- Jasperson KW, Burt RW. APC-associated polyposis conditions. In:Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews (Internet). Seattle, WA: University of Washington; 2011.
- Gardner EJ, Richards RC. Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5:139–147.
- Dunlop MG; British Society for Gastroenterology. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polyposis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 2002; 51(suppl 5):V21–V27.
- Burke W, Petersen G, Lynch P, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. I. Hereditary nonpolyposis colon cancer. Cancer Genetics Studies Consortium. JAMA 1997; 277:915–919.
- Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57:704–713.
- Church J. Familial adenomatous polyposis. Surg Oncol Clin N Am 2009; 18:585–598.
- Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993; 328:1313–1316.
- Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000; 342:1946–1952.
- Johnson MD, Mackey R, Brown N, Church J, Burke C, Walsh RM. Outcome based on management for duodenal adenomas: sporadic versus familial disease. J Gastrointest Surg 2010; 14:229–235.
- Phillips RK, Wallace MH, Lynch PM, et al; FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002; 50:857–860.
- Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer 2006; 95:239–242.
- Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004; 96:1631–1634.
- Croitoru ME, Cleary SP, Berk T, et al. Germline MYH mutations in a clinic-based series of Canadian multiple colorectal adenoma patients. J Surg Oncol 2007; 95:499–506.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362:39–41.
- Nielsen M, Franken PF, Reinards TH, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet 2005; 42:e54.
- Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 2009; 136:1251–1260.
- Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol 2009; 27:3975–3980.
- Aretz S, Uhlhaas S, Goergens H, et al. MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006; 119:807–814.
- Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009; 137:1976–1985.e1–e10.
- Gismondi V, Meta M, Bonelli L, et al. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer 2004; 109:680–684.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors. J Genet Couns 2004; 13:83–114.
- American Society of Clinical Oncology. American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 2003; 21:2397–2406.
KEY POINTS
- Hereditary colorectal cancer syndromes carry a substantial risk of intestinal and extraintestinal tumors.
- Affected patients need increased cancer surveillance and may benefit from prophylactic surgery.
- Identifying these patients in clinical practice begins by assessing a patient’s personal and family health history.
- Patients suspected of having hereditary colorectal cancer syndromes should be referred for genetic counseling and, if appropriate, for genetic testing.

Genetic counselors: Your partners in clinical practice
Suppose a new patient walks into your office for a routine physical examination. As part of your discussion, you ask about her family history. She relates that her grandmother and uncle had colon cancer.
You know that colon cancer can be hereditary, but you are unsure whether this patient’s family history is significant. You know genetic testing can be ordered, but you only have 15 minutes with the patient and you are unsure which test is appropriate and how it can be ordered. What should you do next?
With advances in genetics and genomics have come expectations that health care providers understand and apply these discoveries to patient care. Identification of a genetic diagnosis can lead to personalized treatment and intensive screening, which can reduce the patient’s risk of contracting the disease in question or dying of it.1,2 But genetic testing may also take patients on an emotional journey as they adjust to learning new information about themselves and the health care implications such a diagnosis may have for themselves and their family members.
Genetic counseling is an important component of risk assessment and testing. With increasing demands and shorter appointment times, physicians are finding it harder to provide comprehensive risk assessment and genetic counseling.3–5 Just as “physician extenders” have helped streamline various aspects of health care, genetic counselors can serve as partners to physicians, from helping determine which testing to consider to helping guide follow-up care after test results are received.
Genetic counselors can help not only patients who have a personal or family history of a hereditary condition, but also their physicians and family members. This article will explain the process of genetic counseling and testing, highlight complexities through case examples, and provide a brief review outlining which patients should be referred for genetic counseling.
WHAT IS GENETIC COUNSELING?
The National Society of Genetic Counselors defines genetic counseling as “the process of helping people understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease.”6 The process includes:
- Interpretation of family and medical histories to assess the chance of disease occurrence or recurrence
- Education about inheritance, testing, management, prevention, resources, and research
- Counseling to promote informed choices and adaptation to the risk or condition.6
WHAT HAPPENS DURING A COUNSELING SESSION?

The goals and outcomes of a successful genetic counseling session (Table 1) reflect the need for genetic counselors to not only give patients enough information to understand what is being discussed, but also to monitor their emotional responses and respond to their needs for support.7 The components of a typical genetic counseling session include:
- Contracting (reviewing why the patient is here)
- Reviewing the patient’s personal medical history
- Documenting relevant diagnoses in the family history
- Educating about the condition in question and relevant basic information about genetics
- If testing is indicated, educating about what the test will and will not tell the patient
- If test results are being discussed, discussing the implications of the results for the patient’s management and the utility of testing for relatives
- Identifying additional sources of support and education for patients, such as disease-specific support groups
- Making sure the patient understands the information provided
- Monitoring the patient’s emotional and psychological reactions and responding appropriately.
Before the visit, which may last from 30 minutes to several hours, the genetic counselor reviews the patient’s available medical information, performs a literature search covering relevant topics, and prepares supporting educational resources such as visual aids. After the visit, the genetic counselor contacts the patient to discuss the results of any tests ordered, makes sure the follow-up plan is clear, and arranges return visits if these are indicated. Studies have shown that these nonbillable patient-related activities take at least as much time as the actual patient visit.8,9
EVIDENCE THAT GENETIC COUNSELING IS BENEFICIAL
Although genetic counseling may be time-consuming, its benefits to patients have been proven in a number of studies.
Improved patient knowledge. Three controlled trials found a significant increase in knowledge about cancer genetics in patients who received genetic counseling as part of their clinical services.10–12 Additionally, a large prospective multicenter study found a continued significant increase in cancer genetics knowledge in women who had received genetic counseling for inherited breast cancer risk 1 year earlier.13
More accurate perception of risk. A meta-analysis of three studies found a significant increase in the accuracy of breast cancer risk perceptions among women who had received genetic counseling.14
Improved psychosocial outcomes. Anxiety was reduced in 82% of parents who received genetic counseling after screening of their newborn was positive for hemoglobinopathy trait.15 And 1 year after genetic counseling, parents of patients with psychotic disorders reported reduced anxiety as a result of an increased understanding of accurate recurrence risks.16
Improved risk-reducing behaviors. Increased genetic counseling support led to improved communication and increased contact with genetics services for at-risk family members.17 Genetic counseling also led to higher rates of mammography, clinical breast examination, and breast self-examination.18
WHO ARE GENETIC COUNSELORS?
Genetic counselors are allied health professionals with a master’s degree and with specific expertise in identifying and educating patients at risk for inherited conditions. They are certified through the American Board of Genetic Counseling. Genetic counseling is a licensed profession in many states,19 and licensure legislation is pending in several others.
HOW GENETIC COUNSELORS FACILITATE DIFFICULT COMPONENTS OF GENETIC TESTING
Genetic counselors can serve as complementary practitioners who possess the time and expertise to discuss some of the more complex components of the genetic testing process, further discussed here.
Making sure that testing is appropriate and that the right test is ordered
Let us revisit our introductory scenario—a patient presents to your office and relates a family history of colon cancer. What would you do if she then says, “I know there’s a gene for colon cancer; I want that test today so I can know if I’m at risk.” You get the sense that the patient is anxious and determined to get this testing done today. Which of the following would you do?
- Say “OK,” enter “colon cancer gene” in your hospital’s laboratory ordering system, and pray that the results are normal.
- Remember that a representative from a genetic testing company came by your office and left sample collection kits. Say “OK,” draw the patient’s blood in the tubes provided, check off testing for “comprehensive colorectal genetics panel,” and pray the results are normal.
- Tell the patient: “Most colon cancers are not necessarily caused by an inherited syndrome. However, a detailed analysis of your family history seems warranted. There are many genes that can play a role in inherited colon cancer risk, and I want to make sure the right test is done for the right person in your family. I’m going to refer you to a genetic counselor who can take a detailed family history and discuss the risks and benefits of genetic testing with you.” You make the referral and within 1 or 2 weeks, your patient is seen for genetic counseling.
If you chose ‘colon cancer gene’ testing
The phlebotomy and laboratory personnel at your facility are likely unsure what kind of sample to draw and where it should be sent. As of this writing, at least 14 genes have been associated with a risk of colorectal cancer, and testing for these genes is available through dozens of laboratories across the country.
In this scenario, your hospital does not have sufficient information to follow through on your orders, and someone pages you to discuss it. However, you are in the midst of a busy clinic and are not able to return the page promptly, so the laboratory informs the patient that it cannot draw her blood for testing today. The patient leaves feeling angry and upset.
If you chose commercial genetic testing
You may have just ordered testing for four of the genes known to cause Lynch syndrome, an inherited condition predisposing to colon, uterine, and a few other cancer types. While testing like this may be labeled as “comprehensive,” it may not include all disorders associated with colon cancer. Such shotgun approaches to patient care without consideration of family history can often lead to ordering genetic testing that may be not only medically unnecessary, but also not reimbursable by insurance companies.
Continuing with the case above, the patient’s insurance company determines that testing is not medically necessary, and she is billed for the entire cost of more than $4,400. Her results are normal, and she feels reassured that she is not at increased risk of colon cancer.
A year later, the patient phones you to say that her uncle had genetic testing with positive results. She sends you the letter she received along with the genetic counselor’s clinic note—the uncle’s mutation is in a completely different gene from the ones you tested. While she was previously told she was at low risk, the appropriate site-specific genetic test (average cost range $185–$450) to target the specific mutation is positive, and she is at increased risk of colon cancer, but is now able to pursue increased screening to reduce her risks of developing and dying from this disease.
If the patient had not been made aware of her uncle’s results, she may not have received this screening. If she were diagnosed with later-stage colon cancer after developing symptoms, she may feel you are liable for this diagnosis based on her perception that she was not at risk according to the previously negative genetic testing results ordered by you. After learning about her family history and that the right test was not ordered for her, the patient pursues legal action.
If you chose genetic counseling
If you chose to refer the patient for genetic counseling, congratulations! Your patient is seen for risk assessment and genetic counseling.
As part of the genetic counseling session, a comprehensive family history identifies the patient’s uncle who was diagnosed with colon cancer. He was previously seen for genetics assessment and was found to have a mutation in the APC gene, predisposing him to familial adenomatous polyposis. Site-specific testing, which the genetic counselor is able to get covered by the patient’s insurance through a letter of medical necessity, reveals that your patient shares her uncle’s mutation. As indicated by national guidelines, you refer the patient to a gastroenterologist for medical management, which will significantly reduce her chances of developing and dying of colorectal cancer.
It is preferable to see the family member at highest risk for an inherited condition—usually, but not always the affected relative—for genetic consultation first. During the consultation the genetic counselor would decide which syndrome, if any, is the best fit for the family.
If the affected relative tests positive, targeted and less costly testing for the specific mutation identified (ie, site-specific testing) can then be offered to family members to provide a yes-or-no answer as to their risk status.
If the relative most likely to be gene-positive tests negative, no genetic testing would be recommended for family members, as the genetic cause of the cancer in the family is unknown. In this situation, family members may be advised to pursue the same screening measures as those with a positive gene test due to their strong family history.
INFORMED CONSENT FOR GENETIC TESTING
Genetic testing consists of much more than a simple blood draw. Obtaining informed consent for genetic testing is a crucial step in the testing process, as the results can be complex and often affect multiple family members. When predictive genetic testing is being discussed, special conversations need to take place to make sure that decisions are well informed. Genetic counselors can facilitate these discussions and guide patients and families through the decision-making process.
Example: Huntington disease
The need for genetic counseling before predictive testing is best illustrated by Huntington disease, a progressive neurodegenerative disorder with typical onset in the third or fourth decade of life. Over the disease course, patients experience decreases in motor control (leading to the aptly named “Huntington chorea”), cognitive decline, and changes in psychiatric state. Ultimately, most patients die 15 to 20 years after the onset of symptoms. Treatment is palliative and symptom-based.
Huntington disease is inherited in an autosomal dominant manner, meaning that each child of an affected person has a 50% risk of inheriting the gene change responsible for this condition and of eventually developing the disease. It is caused by an expansion within the HD gene; this expansion may grow with successive generations, leading to earlier onset of symptoms.20
The availability of predictive testing—which enables people who are at risk but who are without symptoms to find out their genetic status—ultimately leads each at-risk person to ask herself or himself, Do I want to know? Studies have found that only 15% to 67% of offspring of parents with Huntington disease (offspring are at 50% risk of the disease) elected to be tested, and in one longitudinal study, this rate of “uptake” decreased over time.21,22 However, any estimates of uptake may be falsely elevated, given the likelihood that those not wishing to consider testing may not feel the need for a clinical visit focused on this subject.
After predictive testing became available, an increased risk of suicide in persons at risk of Huntington disease was documented.23,24 In view of this risk and the careful decision-making support that people at risk need, predictive testing guidelines were developed by a committee of medical experts and members of Huntington disease family organizations.25 As part of the guidelines, a multivisit pretesting process was established that includes extensive education and counseling. Delay of testing is recommended when contraindications are identified, such as evidence of coercion or a serious psychiatric condition. Most genetic testing companies offering predictive testing require a signature from the ordering clinician verifying that pretest counseling has been completed; some also include a provision that the ordering clinician will relay results to the patient in person.
More than 15 years after these guidelines were adopted, a study of suicide risk in at-risk persons continued to find rates higher than in the general population, but lower than in earlier studies.26 Whether this careful pretest counseling protocol is directly related to a possible decrease in suicide risk has yet to be established, but its successful use in patients undergoing predictive Huntington disease testing has led to its adoption in other neurodegenerative diseases such as Alzheimer disease and Parkinson disease.
EXPLAINING POSITIVE GENETIC TESTING RESULTS
If genetic testing identifies a mutation, genetic counselors can help patients understand the implications of the results for themselves and for their relatives. Some patients become quite inquisitive, and the genetic counseling session morphs into a graduate-level discussion of genes, DNA, disease pathways, genetic-environmental interactions, availability of gene therapy, and clinical trials. The genetic counselor also makes the patient aware of other resources, such as disease-specific support groups, which may be developed by patients and families to provide support and practical knowledge.
In some cases, attention turns to at-risk relatives, and the genetic counselor may role-play with the patient to rehearse ways to share information with them. Genetic counselors may give patients a letter to distribute to family members with a copy of the patient’s test results, briefly explaining the condition identified and how relatives may find a genetic counselor in their area for their own risk assessment.
WHAT ABOUT GENETIC DISCRIMINATION?
Genetic discrimination is addressed in many genetic counseling sessions.
As defined by the National Human Genome Research Institute, genetic discrimination is “prejudice directed against people who have or may have a genetic disease.”27
In May 2008, the Genetic Information Nondiscrimination Act (GINA) was signed into law, providing some legal protections against genetic discrimination for patients undergoing predictive genetic testing. The law applies to most employers and health insurers but does not protect against discrimination by life or disability insurers. When discussing genetic testing, genetic counselors ensure that patients are aware of their rights and protections.
GINA would not be relevant for a patient who has a medical condition that may affect his or her insurability. For example, someone with thyroid cancer who is found to have an underlying gene mutation may still be denied any type of insurance coverage on the basis of his or her personal cancer diagnosis. However, should that person’s son who has not been diagnosed with cancer opt to undergo predictive testing, GINA would provide protection against employment and health insurance discrimination, as described above.
DIRECT-TO-CONSUMER GENETIC TESTING
As DNA technology has become increasingly complex, so has the task of understanding new tests and their clinical relevance to patients.
In the last several years, more companies have begun to offer direct-to-consumer genetic testing, which may be ordered without the involvement of a health care professional. While some companies hire or work closely with genetic counselors to conduct pretest and posttest genetic counseling, others do not, and preliminary research has found that only a minority of primary care physicians feel prepared to answer patients’ questions about direct-to-consumer genetic testing.28
Genetic counselors stay abreast of emerging technologies and are prepared to answer questions from patients who are considering or have already undergone such testing and from physicians who may wonder if a patient’s direct-to-consumer genetic testing results should affect his or her management.
Direct-to-consumer genetic testing will be discussed in depth in a future article in this series.
EXPLAINING ‘NORMAL’ (NEGATIVE) GENETIC TEST RESULTS
When testing results are normal, patients are educated about the meaning of “normal” results, the residual risk, and screening that might be appropriate in each person’s situation.
Sometimes a normal result does not mean the patient is not at risk for disease—for most diseases, genetic testing is not perfect and cannot identify a mutation in every at-risk family. Patients who have a family history of certain conditions may still face a higher risk despite normal test results. In these situations it is imperative that the family continue to adhere to follow-up recommendations even with normal test results.
Example: Marfan syndrome
Marfan syndrome is an autosomal dominant connective tissue disorder that, if unrecognized, is associated with significant morbidity and mortality. People with Marfan syndrome are at increased risk of aortic aneurysms, which can rupture spontaneously, leading to sudden death.
Although at least 70% of patients with Marfan syndrome have a mutation in FBN1, other patients meeting the clinical diagnostic criteria do not. Despite a normal genetic test result, they should adhere to the same screening guidelines as a person who tests positive.29
This concept—that screening should still be done despite a normal “Marfan test”—may be difficult for patients to grasp without a discussion of the imperfect sensitivity of genetic testing and of their real ongoing risks. Even more difficult to understand is the idea that the patient’s family members should also be screened as though they have the disease, given that the family’s mutation is unknown and predictive testing cannot be conducted.
Further complicating matters, other disorders such as Loeys-Dietz and vascular Ehlers-Danlos syndrome can mimic Marfan syndrome by causing aortic aneurysms, but management recommendations for them are very different.30,31
The appropriate genetic diagnosis for patients with aortic aneurysms can be facilitated by referring them to genetic counselors, who can identify appropriate testing. In this way, physicians can personalize medical management and give screening recommendations specific to the genetic disorder present.
EXPLAINING UNCERTAIN RESULTS
There are three possible results for most genetic tests—positive (a pathogenic or disease-causing mutation was found), negative (normal), and the frustrating “variant of uncertain significance” (VUS).
A VUS result means that an abnormality was detected in the gene, but that there are insufficient data about whether the abnormality alters the function of the gene in question and, thus, leads to disease. Since some gene variants are known to be common in the general population and not linked to disease and others are known to definitely alter a gene’s function and cause disease, a VUS that is clearly unknown poses a challenge not only to patient management, but also to family members seeking personal risk assessments.
Knowledge of how or if specific variants relate to disease is emerging. In time, some variants become reclassified as either disease-causing mutations or benign polymorphisms. However, careful consideration needs to be given to how to explain the abnormal result to the patient and to at-risk family members, as well as to how to explain the clinical implications of the VUS.
Example: Hereditary breast and ovarian cancer syndrome
People with hereditary breast and ovarian cancer syndrome face a lifetime risk of breast cancer of up to 87% and a risk of ovarian cancer of up to 44%. Most families with this syndrome have an inherited change in either the BRCA1 or BRCA2 gene.32,33 Given these risks, prophylactic mastectomy and oophorectomy are among the management options for mutation-positive patients. In the absence of clear genetic counseling, a patient with a VUS might see the “abnormal” test result and believe herself to be mutation-positive and thus at very high cancer risk.
An important role for the genetic counselor is to clarify the pathogenicity of a particular VUS. When a VUS is found, genetic counselors search for information about the variant by reviewing the medical literature, discussing it with the testing laboratory, arranging for family studies when appropriate, and contacting researchers whose work focuses on the gene in question.
Failure to properly research a particular VUS can lead to unnecessary and risky surgical procedures, as well as to falsely labelling relatives as being at risk. Until a VUS is reclassified as a disease-causing mutation, testing for it should not be offered to family members (unless through a research study), nor should medical management be based solely on the results of a particular VUS. In time, a VUS may be reclassified as either a benign polymorphism or a disease-causing mutation, and the genetic counselor will recontact the patient and physician with updated information and recommendations.
WHOM SHOULD I REFER?
Genetic counseling is available for patients and families in diverse settings within health systems. The six primary areas of practice are general, cardiovascular, cancer, preconception, prenatal, and pediatrics.
Patients with a personal or family history of a hereditary condition can benefit from genetic counseling regardless of whether they would be considered appropriate for genetic testing.34

At current count, there are 4,424 genetic disorders for which the underlying cause has been identified.35 Individually, each disorder is rare, but when they are considered as a whole, they affect a significant minority of the general population. It is estimated that before age 25 years, 53 (5.3%) of every 1,000 people will be diagnosed with a disease that has an important genetic component.36 From 20% to 30% of infant deaths are related to a genetic disorder,37,38 and 22% of unaffected adults have a family history of cancer significant enough to warrant a genetics referral.39 See Table 2 for a list of common indications for referral.
HOW CAN I FIND GENETIC COUNSELING SERVICES?
The National Society of Genetic Counselors (www.nsgc.org) and American Board of Genetic Counseling (www.abgc.net) both provide searchable databases of registered genetic counselors.
KNOWLEDGE CONTINUES TO EXPAND
Genetic knowledge continues to expand, and testing is becoming available for a growing number of medical conditions. Appropriate identification of individuals with and at risk for genetic disorders through the use of genetic testing and screening is a cornerstone of personalized medicine, with the ultimate goal of improving patient outcomes. However, in this era of value-based medicine and fewer health care dollars, genetic testing must be used in a way that maximizes its clinical impact with a careful fiscal approach.
Genetic counselors are specially trained health care professionals with expertise in genetic and genomic medicine who work in collaboration with physicians to guide patients through the complexities of heritable conditions and emerging technologies. They are trained to personalize, interpret, and communicate complex science into data that will assure best outcomes for patients and their families. Developing a partnership with the genetic counselors in your area can provide multiple benefits to your patients as well as to your own practice.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Hunt SC, Gwinn M, Adams TD. Family history assessment: strategies for prevention of cardiovascular disease. Am J Prev Med 2003; 24:136–142.
- Wood ME, Stockdale A, Flynn BS. Interviews with primary care physicians regarding taking and interpreting the cancer family history. Fam Pract 2008; 25:334–340.
- Bellcross CA, Kolor K, Goddard KA, Coates RJ, Reyes M, Khoury MJ. Awareness and utilization of BRCA1/2 testing among U.S. primary care physicians. Am J Prev Med 2011; 40:61–66.
- Hindorff LA, Burke W, Laberge AM, et al. Motivating factors for physician ordering of factor V Leiden genetic tests. Arch Intern Med 2009; 169:68–74.
- National Society of Genetic Counselors. Definition of genetic counseling. www.nsgc.org/About/FAQsDefinitions/tabid/97/Default.aspx. Accessed June 4, 2012.
- Bernhardt BA, Biesecker BB, Mastromarino CL. Goals, benefits, and outcomes of genetic counseling: client and genetic counselor assessment. Am J Med Genet 2000; 94:189–197.
- Bernhardt BA, Pyeritz RE. The economics of clinical genetics services. III. Cognitive genetics services are not self-supporting. Am J Hum Genet 1989; 44:288–293.
- McPherson E, Zaleski C, Benishek K, et al. Clinical genetics provider real-time workflow study. Genet Med 2008; 10:699–706.
- Brain K, Gray J, Norman P, et al. Randomized trial of a specialist genetic assessment service for familial breast cancer. J Natl Cancer Inst 2000; 92:1345–1351.
- Lerman C, Biesecker B, Benkendorf JL, et al. Controlled trial of pretest education approaches to enhance informed decision-making for BRCA1 gene testing. J Natl Cancer Inst 1997; 89:148–157.
- Randall J, Butow P, Kirk J, Tucker K. Psychological impact of genetic counselling and testing in women previously diagnosed with breast cancer. Intern Med J 2001; 31:397–405.
- Meiser B, Butow PN, Barratt AL, et al; Psychological Impact Collaborative Group. Long-term outcomes of genetic counseling in women at increased risk of developing hereditary breast cancer. Patient Educ Couns 2001; 44:215–225.
- Meiser B, Halliday JL. What is the impact of genetic counselling in women at increased risk of developing hereditary breast cancer? A meta-analytic review. Soc Sci Med 2002; 54:1463–1470.
- Kladny B, Williams A, Gupta A, Gettig EA, Krishnamurti L. Genetic counseling following the detection of hemoglobinopathy trait on the newborn screen is well received, improves knowledge, and relieves anxiety. Genet Med 2011; 13:658–661.
- Austin JC, Honer WG. Psychiatric genetic counselling for parents of individuals affected with psychotic disorders: a pilot study. Early Interv Psychiatry 2008; 2:80–89.
- Forrest LE, Burke J, Bacic S, Amor DJ. Increased genetic counseling support improves communication of genetic information in families. Genet Med 2008; 10:167–172.
- Watson M, Kash KM, Homewood J, Ebbs S, Murday V, Eeles R. Does genetic counseling have any impact on management of breast cancer risk? Genet Test 2005; 9:167–174.
- National Conference of State Legislatures. Genetic counselor licensing. www.ncsl.org/issues-research/health/genetic-counselor-licensing-laws.aspx. Accessed June 4, 2012.
- Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010; 5:40.
- Morrison PJ, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet 2011; 80:281–286.
- Bernhardt C, Schwan AM, Kraus P, Epplen JT, Kunstmann E. Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). Eur J Hum Genet 2009; 17:295–300.
- Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Suicide risk in Huntington’s disease. J Med Genet 1993; 30:293–295.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- International Huntington Association and the World Federation of Neurology Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. J Med Genet 1994; 31:555–559.
- Fiedorowicz JG, Mills JA, Ruggle A, Langbehn D, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. Suicidal behavior in prodromal Huntington disease. Neurodegener Dis 2011; 8:483–490.
- National Institutes of Health. Definition of genetic discrimination. www.genome.gov/Glossary/index.cfm?id=80. Accessed June 4, 2012.
- Powell KP, Cogswell WA, Christianson CA, et al. Primary care physicians’ awareness, experience, and opinions of direct-to-consumer genetic testing. J Genet Couns 2011; (Epub ahead of print.)
- Dietz HC. Marfan syndrome. In:Pagon RA, Bird TD, Dolan CR, et aleditors. GeneReviews. Seattle, WA: University of Washington; 1993.
- Williams JA, Loeys BL, Nwakanma LU, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg 2007; 83:S757–5763.
- Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg 2005; 42:98–106.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343:692–695.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Johns Hopkins University. OMIM entry statistics. http://omim.org/statistics/entries. Accessed June 4, 2012.
- Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet 1988; 42:677–693.
- Berry RJ, Buehler JW, Strauss LT, Hogue CJ, Smith JC. Birth weight-specific infant mortality due to congenital anomalies, 1960 and 1980. Public Health Rep 1987; 102:171–181.
- Hoyert DL, Freedman MA, Strobino DM, Guyer B. Annual summary of vital statistics: 2000. Pediatrics 2001; 108:1241–1255.
- Scheuner MT, McNeel TS, Freedman AN. Population prevalence of familial cancer and common hereditary cancer syndromes. The 2005 California Health Interview Survey. Genet Med 2010; 12:726–735.
Suppose a new patient walks into your office for a routine physical examination. As part of your discussion, you ask about her family history. She relates that her grandmother and uncle had colon cancer.
You know that colon cancer can be hereditary, but you are unsure whether this patient’s family history is significant. You know genetic testing can be ordered, but you only have 15 minutes with the patient and you are unsure which test is appropriate and how it can be ordered. What should you do next?
With advances in genetics and genomics have come expectations that health care providers understand and apply these discoveries to patient care. Identification of a genetic diagnosis can lead to personalized treatment and intensive screening, which can reduce the patient’s risk of contracting the disease in question or dying of it.1,2 But genetic testing may also take patients on an emotional journey as they adjust to learning new information about themselves and the health care implications such a diagnosis may have for themselves and their family members.
Genetic counseling is an important component of risk assessment and testing. With increasing demands and shorter appointment times, physicians are finding it harder to provide comprehensive risk assessment and genetic counseling.3–5 Just as “physician extenders” have helped streamline various aspects of health care, genetic counselors can serve as partners to physicians, from helping determine which testing to consider to helping guide follow-up care after test results are received.
Genetic counselors can help not only patients who have a personal or family history of a hereditary condition, but also their physicians and family members. This article will explain the process of genetic counseling and testing, highlight complexities through case examples, and provide a brief review outlining which patients should be referred for genetic counseling.
WHAT IS GENETIC COUNSELING?
The National Society of Genetic Counselors defines genetic counseling as “the process of helping people understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease.”6 The process includes:
- Interpretation of family and medical histories to assess the chance of disease occurrence or recurrence
- Education about inheritance, testing, management, prevention, resources, and research
- Counseling to promote informed choices and adaptation to the risk or condition.6
WHAT HAPPENS DURING A COUNSELING SESSION?
The goals and outcomes of a successful genetic counseling session (Table 1) reflect the need for genetic counselors to not only give patients enough information to understand what is being discussed, but also to monitor their emotional responses and respond to their needs for support.7 The components of a typical genetic counseling session include:
- Contracting (reviewing why the patient is here)
- Reviewing the patient’s personal medical history
- Documenting relevant diagnoses in the family history
- Educating about the condition in question and relevant basic information about genetics
- If testing is indicated, educating about what the test will and will not tell the patient
- If test results are being discussed, discussing the implications of the results for the patient’s management and the utility of testing for relatives
- Identifying additional sources of support and education for patients, such as disease-specific support groups
- Making sure the patient understands the information provided
- Monitoring the patient’s emotional and psychological reactions and responding appropriately.
Before the visit, which may last from 30 minutes to several hours, the genetic counselor reviews the patient’s available medical information, performs a literature search covering relevant topics, and prepares supporting educational resources such as visual aids. After the visit, the genetic counselor contacts the patient to discuss the results of any tests ordered, makes sure the follow-up plan is clear, and arranges return visits if these are indicated. Studies have shown that these nonbillable patient-related activities take at least as much time as the actual patient visit.8,9
EVIDENCE THAT GENETIC COUNSELING IS BENEFICIAL
Although genetic counseling may be time-consuming, its benefits to patients have been proven in a number of studies.
Improved patient knowledge. Three controlled trials found a significant increase in knowledge about cancer genetics in patients who received genetic counseling as part of their clinical services.10–12 Additionally, a large prospective multicenter study found a continued significant increase in cancer genetics knowledge in women who had received genetic counseling for inherited breast cancer risk 1 year earlier.13
More accurate perception of risk. A meta-analysis of three studies found a significant increase in the accuracy of breast cancer risk perceptions among women who had received genetic counseling.14
Improved psychosocial outcomes. Anxiety was reduced in 82% of parents who received genetic counseling after screening of their newborn was positive for hemoglobinopathy trait.15 And 1 year after genetic counseling, parents of patients with psychotic disorders reported reduced anxiety as a result of an increased understanding of accurate recurrence risks.16
Improved risk-reducing behaviors. Increased genetic counseling support led to improved communication and increased contact with genetics services for at-risk family members.17 Genetic counseling also led to higher rates of mammography, clinical breast examination, and breast self-examination.18
WHO ARE GENETIC COUNSELORS?
Genetic counselors are allied health professionals with a master’s degree and with specific expertise in identifying and educating patients at risk for inherited conditions. They are certified through the American Board of Genetic Counseling. Genetic counseling is a licensed profession in many states,19 and licensure legislation is pending in several others.
HOW GENETIC COUNSELORS FACILITATE DIFFICULT COMPONENTS OF GENETIC TESTING
Genetic counselors can serve as complementary practitioners who possess the time and expertise to discuss some of the more complex components of the genetic testing process, further discussed here.
Making sure that testing is appropriate and that the right test is ordered
Let us revisit our introductory scenario—a patient presents to your office and relates a family history of colon cancer. What would you do if she then says, “I know there’s a gene for colon cancer; I want that test today so I can know if I’m at risk.” You get the sense that the patient is anxious and determined to get this testing done today. Which of the following would you do?
- Say “OK,” enter “colon cancer gene” in your hospital’s laboratory ordering system, and pray that the results are normal.
- Remember that a representative from a genetic testing company came by your office and left sample collection kits. Say “OK,” draw the patient’s blood in the tubes provided, check off testing for “comprehensive colorectal genetics panel,” and pray the results are normal.
- Tell the patient: “Most colon cancers are not necessarily caused by an inherited syndrome. However, a detailed analysis of your family history seems warranted. There are many genes that can play a role in inherited colon cancer risk, and I want to make sure the right test is done for the right person in your family. I’m going to refer you to a genetic counselor who can take a detailed family history and discuss the risks and benefits of genetic testing with you.” You make the referral and within 1 or 2 weeks, your patient is seen for genetic counseling.
If you chose ‘colon cancer gene’ testing
The phlebotomy and laboratory personnel at your facility are likely unsure what kind of sample to draw and where it should be sent. As of this writing, at least 14 genes have been associated with a risk of colorectal cancer, and testing for these genes is available through dozens of laboratories across the country.
In this scenario, your hospital does not have sufficient information to follow through on your orders, and someone pages you to discuss it. However, you are in the midst of a busy clinic and are not able to return the page promptly, so the laboratory informs the patient that it cannot draw her blood for testing today. The patient leaves feeling angry and upset.
If you chose commercial genetic testing
You may have just ordered testing for four of the genes known to cause Lynch syndrome, an inherited condition predisposing to colon, uterine, and a few other cancer types. While testing like this may be labeled as “comprehensive,” it may not include all disorders associated with colon cancer. Such shotgun approaches to patient care without consideration of family history can often lead to ordering genetic testing that may be not only medically unnecessary, but also not reimbursable by insurance companies.
Continuing with the case above, the patient’s insurance company determines that testing is not medically necessary, and she is billed for the entire cost of more than $4,400. Her results are normal, and she feels reassured that she is not at increased risk of colon cancer.
A year later, the patient phones you to say that her uncle had genetic testing with positive results. She sends you the letter she received along with the genetic counselor’s clinic note—the uncle’s mutation is in a completely different gene from the ones you tested. While she was previously told she was at low risk, the appropriate site-specific genetic test (average cost range $185–$450) to target the specific mutation is positive, and she is at increased risk of colon cancer, but is now able to pursue increased screening to reduce her risks of developing and dying from this disease.
If the patient had not been made aware of her uncle’s results, she may not have received this screening. If she were diagnosed with later-stage colon cancer after developing symptoms, she may feel you are liable for this diagnosis based on her perception that she was not at risk according to the previously negative genetic testing results ordered by you. After learning about her family history and that the right test was not ordered for her, the patient pursues legal action.
If you chose genetic counseling
If you chose to refer the patient for genetic counseling, congratulations! Your patient is seen for risk assessment and genetic counseling.
As part of the genetic counseling session, a comprehensive family history identifies the patient’s uncle who was diagnosed with colon cancer. He was previously seen for genetics assessment and was found to have a mutation in the APC gene, predisposing him to familial adenomatous polyposis. Site-specific testing, which the genetic counselor is able to get covered by the patient’s insurance through a letter of medical necessity, reveals that your patient shares her uncle’s mutation. As indicated by national guidelines, you refer the patient to a gastroenterologist for medical management, which will significantly reduce her chances of developing and dying of colorectal cancer.
It is preferable to see the family member at highest risk for an inherited condition—usually, but not always the affected relative—for genetic consultation first. During the consultation the genetic counselor would decide which syndrome, if any, is the best fit for the family.
If the affected relative tests positive, targeted and less costly testing for the specific mutation identified (ie, site-specific testing) can then be offered to family members to provide a yes-or-no answer as to their risk status.
If the relative most likely to be gene-positive tests negative, no genetic testing would be recommended for family members, as the genetic cause of the cancer in the family is unknown. In this situation, family members may be advised to pursue the same screening measures as those with a positive gene test due to their strong family history.
INFORMED CONSENT FOR GENETIC TESTING
Genetic testing consists of much more than a simple blood draw. Obtaining informed consent for genetic testing is a crucial step in the testing process, as the results can be complex and often affect multiple family members. When predictive genetic testing is being discussed, special conversations need to take place to make sure that decisions are well informed. Genetic counselors can facilitate these discussions and guide patients and families through the decision-making process.
Example: Huntington disease
The need for genetic counseling before predictive testing is best illustrated by Huntington disease, a progressive neurodegenerative disorder with typical onset in the third or fourth decade of life. Over the disease course, patients experience decreases in motor control (leading to the aptly named “Huntington chorea”), cognitive decline, and changes in psychiatric state. Ultimately, most patients die 15 to 20 years after the onset of symptoms. Treatment is palliative and symptom-based.
Huntington disease is inherited in an autosomal dominant manner, meaning that each child of an affected person has a 50% risk of inheriting the gene change responsible for this condition and of eventually developing the disease. It is caused by an expansion within the HD gene; this expansion may grow with successive generations, leading to earlier onset of symptoms.20
The availability of predictive testing—which enables people who are at risk but who are without symptoms to find out their genetic status—ultimately leads each at-risk person to ask herself or himself, Do I want to know? Studies have found that only 15% to 67% of offspring of parents with Huntington disease (offspring are at 50% risk of the disease) elected to be tested, and in one longitudinal study, this rate of “uptake” decreased over time.21,22 However, any estimates of uptake may be falsely elevated, given the likelihood that those not wishing to consider testing may not feel the need for a clinical visit focused on this subject.
After predictive testing became available, an increased risk of suicide in persons at risk of Huntington disease was documented.23,24 In view of this risk and the careful decision-making support that people at risk need, predictive testing guidelines were developed by a committee of medical experts and members of Huntington disease family organizations.25 As part of the guidelines, a multivisit pretesting process was established that includes extensive education and counseling. Delay of testing is recommended when contraindications are identified, such as evidence of coercion or a serious psychiatric condition. Most genetic testing companies offering predictive testing require a signature from the ordering clinician verifying that pretest counseling has been completed; some also include a provision that the ordering clinician will relay results to the patient in person.
More than 15 years after these guidelines were adopted, a study of suicide risk in at-risk persons continued to find rates higher than in the general population, but lower than in earlier studies.26 Whether this careful pretest counseling protocol is directly related to a possible decrease in suicide risk has yet to be established, but its successful use in patients undergoing predictive Huntington disease testing has led to its adoption in other neurodegenerative diseases such as Alzheimer disease and Parkinson disease.
EXPLAINING POSITIVE GENETIC TESTING RESULTS
If genetic testing identifies a mutation, genetic counselors can help patients understand the implications of the results for themselves and for their relatives. Some patients become quite inquisitive, and the genetic counseling session morphs into a graduate-level discussion of genes, DNA, disease pathways, genetic-environmental interactions, availability of gene therapy, and clinical trials. The genetic counselor also makes the patient aware of other resources, such as disease-specific support groups, which may be developed by patients and families to provide support and practical knowledge.
In some cases, attention turns to at-risk relatives, and the genetic counselor may role-play with the patient to rehearse ways to share information with them. Genetic counselors may give patients a letter to distribute to family members with a copy of the patient’s test results, briefly explaining the condition identified and how relatives may find a genetic counselor in their area for their own risk assessment.
WHAT ABOUT GENETIC DISCRIMINATION?
Genetic discrimination is addressed in many genetic counseling sessions.
As defined by the National Human Genome Research Institute, genetic discrimination is “prejudice directed against people who have or may have a genetic disease.”27
In May 2008, the Genetic Information Nondiscrimination Act (GINA) was signed into law, providing some legal protections against genetic discrimination for patients undergoing predictive genetic testing. The law applies to most employers and health insurers but does not protect against discrimination by life or disability insurers. When discussing genetic testing, genetic counselors ensure that patients are aware of their rights and protections.
GINA would not be relevant for a patient who has a medical condition that may affect his or her insurability. For example, someone with thyroid cancer who is found to have an underlying gene mutation may still be denied any type of insurance coverage on the basis of his or her personal cancer diagnosis. However, should that person’s son who has not been diagnosed with cancer opt to undergo predictive testing, GINA would provide protection against employment and health insurance discrimination, as described above.
DIRECT-TO-CONSUMER GENETIC TESTING
As DNA technology has become increasingly complex, so has the task of understanding new tests and their clinical relevance to patients.
In the last several years, more companies have begun to offer direct-to-consumer genetic testing, which may be ordered without the involvement of a health care professional. While some companies hire or work closely with genetic counselors to conduct pretest and posttest genetic counseling, others do not, and preliminary research has found that only a minority of primary care physicians feel prepared to answer patients’ questions about direct-to-consumer genetic testing.28
Genetic counselors stay abreast of emerging technologies and are prepared to answer questions from patients who are considering or have already undergone such testing and from physicians who may wonder if a patient’s direct-to-consumer genetic testing results should affect his or her management.
Direct-to-consumer genetic testing will be discussed in depth in a future article in this series.
EXPLAINING ‘NORMAL’ (NEGATIVE) GENETIC TEST RESULTS
When testing results are normal, patients are educated about the meaning of “normal” results, the residual risk, and screening that might be appropriate in each person’s situation.
Sometimes a normal result does not mean the patient is not at risk for disease—for most diseases, genetic testing is not perfect and cannot identify a mutation in every at-risk family. Patients who have a family history of certain conditions may still face a higher risk despite normal test results. In these situations it is imperative that the family continue to adhere to follow-up recommendations even with normal test results.
Example: Marfan syndrome
Marfan syndrome is an autosomal dominant connective tissue disorder that, if unrecognized, is associated with significant morbidity and mortality. People with Marfan syndrome are at increased risk of aortic aneurysms, which can rupture spontaneously, leading to sudden death.
Although at least 70% of patients with Marfan syndrome have a mutation in FBN1, other patients meeting the clinical diagnostic criteria do not. Despite a normal genetic test result, they should adhere to the same screening guidelines as a person who tests positive.29
This concept—that screening should still be done despite a normal “Marfan test”—may be difficult for patients to grasp without a discussion of the imperfect sensitivity of genetic testing and of their real ongoing risks. Even more difficult to understand is the idea that the patient’s family members should also be screened as though they have the disease, given that the family’s mutation is unknown and predictive testing cannot be conducted.
Further complicating matters, other disorders such as Loeys-Dietz and vascular Ehlers-Danlos syndrome can mimic Marfan syndrome by causing aortic aneurysms, but management recommendations for them are very different.30,31
The appropriate genetic diagnosis for patients with aortic aneurysms can be facilitated by referring them to genetic counselors, who can identify appropriate testing. In this way, physicians can personalize medical management and give screening recommendations specific to the genetic disorder present.
EXPLAINING UNCERTAIN RESULTS
There are three possible results for most genetic tests—positive (a pathogenic or disease-causing mutation was found), negative (normal), and the frustrating “variant of uncertain significance” (VUS).
A VUS result means that an abnormality was detected in the gene, but that there are insufficient data about whether the abnormality alters the function of the gene in question and, thus, leads to disease. Since some gene variants are known to be common in the general population and not linked to disease and others are known to definitely alter a gene’s function and cause disease, a VUS that is clearly unknown poses a challenge not only to patient management, but also to family members seeking personal risk assessments.
Knowledge of how or if specific variants relate to disease is emerging. In time, some variants become reclassified as either disease-causing mutations or benign polymorphisms. However, careful consideration needs to be given to how to explain the abnormal result to the patient and to at-risk family members, as well as to how to explain the clinical implications of the VUS.
Example: Hereditary breast and ovarian cancer syndrome
People with hereditary breast and ovarian cancer syndrome face a lifetime risk of breast cancer of up to 87% and a risk of ovarian cancer of up to 44%. Most families with this syndrome have an inherited change in either the BRCA1 or BRCA2 gene.32,33 Given these risks, prophylactic mastectomy and oophorectomy are among the management options for mutation-positive patients. In the absence of clear genetic counseling, a patient with a VUS might see the “abnormal” test result and believe herself to be mutation-positive and thus at very high cancer risk.
An important role for the genetic counselor is to clarify the pathogenicity of a particular VUS. When a VUS is found, genetic counselors search for information about the variant by reviewing the medical literature, discussing it with the testing laboratory, arranging for family studies when appropriate, and contacting researchers whose work focuses on the gene in question.
Failure to properly research a particular VUS can lead to unnecessary and risky surgical procedures, as well as to falsely labelling relatives as being at risk. Until a VUS is reclassified as a disease-causing mutation, testing for it should not be offered to family members (unless through a research study), nor should medical management be based solely on the results of a particular VUS. In time, a VUS may be reclassified as either a benign polymorphism or a disease-causing mutation, and the genetic counselor will recontact the patient and physician with updated information and recommendations.
WHOM SHOULD I REFER?
Genetic counseling is available for patients and families in diverse settings within health systems. The six primary areas of practice are general, cardiovascular, cancer, preconception, prenatal, and pediatrics.
Patients with a personal or family history of a hereditary condition can benefit from genetic counseling regardless of whether they would be considered appropriate for genetic testing.34
At current count, there are 4,424 genetic disorders for which the underlying cause has been identified.35 Individually, each disorder is rare, but when they are considered as a whole, they affect a significant minority of the general population. It is estimated that before age 25 years, 53 (5.3%) of every 1,000 people will be diagnosed with a disease that has an important genetic component.36 From 20% to 30% of infant deaths are related to a genetic disorder,37,38 and 22% of unaffected adults have a family history of cancer significant enough to warrant a genetics referral.39 See Table 2 for a list of common indications for referral.
HOW CAN I FIND GENETIC COUNSELING SERVICES?
The National Society of Genetic Counselors (www.nsgc.org) and American Board of Genetic Counseling (www.abgc.net) both provide searchable databases of registered genetic counselors.
KNOWLEDGE CONTINUES TO EXPAND
Genetic knowledge continues to expand, and testing is becoming available for a growing number of medical conditions. Appropriate identification of individuals with and at risk for genetic disorders through the use of genetic testing and screening is a cornerstone of personalized medicine, with the ultimate goal of improving patient outcomes. However, in this era of value-based medicine and fewer health care dollars, genetic testing must be used in a way that maximizes its clinical impact with a careful fiscal approach.
Genetic counselors are specially trained health care professionals with expertise in genetic and genomic medicine who work in collaboration with physicians to guide patients through the complexities of heritable conditions and emerging technologies. They are trained to personalize, interpret, and communicate complex science into data that will assure best outcomes for patients and their families. Developing a partnership with the genetic counselors in your area can provide multiple benefits to your patients as well as to your own practice.
Suppose a new patient walks into your office for a routine physical examination. As part of your discussion, you ask about her family history. She relates that her grandmother and uncle had colon cancer.
You know that colon cancer can be hereditary, but you are unsure whether this patient’s family history is significant. You know genetic testing can be ordered, but you only have 15 minutes with the patient and you are unsure which test is appropriate and how it can be ordered. What should you do next?
With advances in genetics and genomics have come expectations that health care providers understand and apply these discoveries to patient care. Identification of a genetic diagnosis can lead to personalized treatment and intensive screening, which can reduce the patient’s risk of contracting the disease in question or dying of it.1,2 But genetic testing may also take patients on an emotional journey as they adjust to learning new information about themselves and the health care implications such a diagnosis may have for themselves and their family members.
Genetic counseling is an important component of risk assessment and testing. With increasing demands and shorter appointment times, physicians are finding it harder to provide comprehensive risk assessment and genetic counseling.3–5 Just as “physician extenders” have helped streamline various aspects of health care, genetic counselors can serve as partners to physicians, from helping determine which testing to consider to helping guide follow-up care after test results are received.
Genetic counselors can help not only patients who have a personal or family history of a hereditary condition, but also their physicians and family members. This article will explain the process of genetic counseling and testing, highlight complexities through case examples, and provide a brief review outlining which patients should be referred for genetic counseling.
WHAT IS GENETIC COUNSELING?
The National Society of Genetic Counselors defines genetic counseling as “the process of helping people understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease.”6 The process includes:
- Interpretation of family and medical histories to assess the chance of disease occurrence or recurrence
- Education about inheritance, testing, management, prevention, resources, and research
- Counseling to promote informed choices and adaptation to the risk or condition.6
WHAT HAPPENS DURING A COUNSELING SESSION?
The goals and outcomes of a successful genetic counseling session (Table 1) reflect the need for genetic counselors to not only give patients enough information to understand what is being discussed, but also to monitor their emotional responses and respond to their needs for support.7 The components of a typical genetic counseling session include:
- Contracting (reviewing why the patient is here)
- Reviewing the patient’s personal medical history
- Documenting relevant diagnoses in the family history
- Educating about the condition in question and relevant basic information about genetics
- If testing is indicated, educating about what the test will and will not tell the patient
- If test results are being discussed, discussing the implications of the results for the patient’s management and the utility of testing for relatives
- Identifying additional sources of support and education for patients, such as disease-specific support groups
- Making sure the patient understands the information provided
- Monitoring the patient’s emotional and psychological reactions and responding appropriately.
Before the visit, which may last from 30 minutes to several hours, the genetic counselor reviews the patient’s available medical information, performs a literature search covering relevant topics, and prepares supporting educational resources such as visual aids. After the visit, the genetic counselor contacts the patient to discuss the results of any tests ordered, makes sure the follow-up plan is clear, and arranges return visits if these are indicated. Studies have shown that these nonbillable patient-related activities take at least as much time as the actual patient visit.8,9
EVIDENCE THAT GENETIC COUNSELING IS BENEFICIAL
Although genetic counseling may be time-consuming, its benefits to patients have been proven in a number of studies.
Improved patient knowledge. Three controlled trials found a significant increase in knowledge about cancer genetics in patients who received genetic counseling as part of their clinical services.10–12 Additionally, a large prospective multicenter study found a continued significant increase in cancer genetics knowledge in women who had received genetic counseling for inherited breast cancer risk 1 year earlier.13
More accurate perception of risk. A meta-analysis of three studies found a significant increase in the accuracy of breast cancer risk perceptions among women who had received genetic counseling.14
Improved psychosocial outcomes. Anxiety was reduced in 82% of parents who received genetic counseling after screening of their newborn was positive for hemoglobinopathy trait.15 And 1 year after genetic counseling, parents of patients with psychotic disorders reported reduced anxiety as a result of an increased understanding of accurate recurrence risks.16
Improved risk-reducing behaviors. Increased genetic counseling support led to improved communication and increased contact with genetics services for at-risk family members.17 Genetic counseling also led to higher rates of mammography, clinical breast examination, and breast self-examination.18
WHO ARE GENETIC COUNSELORS?
Genetic counselors are allied health professionals with a master’s degree and with specific expertise in identifying and educating patients at risk for inherited conditions. They are certified through the American Board of Genetic Counseling. Genetic counseling is a licensed profession in many states,19 and licensure legislation is pending in several others.
HOW GENETIC COUNSELORS FACILITATE DIFFICULT COMPONENTS OF GENETIC TESTING
Genetic counselors can serve as complementary practitioners who possess the time and expertise to discuss some of the more complex components of the genetic testing process, further discussed here.
Making sure that testing is appropriate and that the right test is ordered
Let us revisit our introductory scenario—a patient presents to your office and relates a family history of colon cancer. What would you do if she then says, “I know there’s a gene for colon cancer; I want that test today so I can know if I’m at risk.” You get the sense that the patient is anxious and determined to get this testing done today. Which of the following would you do?
- Say “OK,” enter “colon cancer gene” in your hospital’s laboratory ordering system, and pray that the results are normal.
- Remember that a representative from a genetic testing company came by your office and left sample collection kits. Say “OK,” draw the patient’s blood in the tubes provided, check off testing for “comprehensive colorectal genetics panel,” and pray the results are normal.
- Tell the patient: “Most colon cancers are not necessarily caused by an inherited syndrome. However, a detailed analysis of your family history seems warranted. There are many genes that can play a role in inherited colon cancer risk, and I want to make sure the right test is done for the right person in your family. I’m going to refer you to a genetic counselor who can take a detailed family history and discuss the risks and benefits of genetic testing with you.” You make the referral and within 1 or 2 weeks, your patient is seen for genetic counseling.
If you chose ‘colon cancer gene’ testing
The phlebotomy and laboratory personnel at your facility are likely unsure what kind of sample to draw and where it should be sent. As of this writing, at least 14 genes have been associated with a risk of colorectal cancer, and testing for these genes is available through dozens of laboratories across the country.
In this scenario, your hospital does not have sufficient information to follow through on your orders, and someone pages you to discuss it. However, you are in the midst of a busy clinic and are not able to return the page promptly, so the laboratory informs the patient that it cannot draw her blood for testing today. The patient leaves feeling angry and upset.
If you chose commercial genetic testing
You may have just ordered testing for four of the genes known to cause Lynch syndrome, an inherited condition predisposing to colon, uterine, and a few other cancer types. While testing like this may be labeled as “comprehensive,” it may not include all disorders associated with colon cancer. Such shotgun approaches to patient care without consideration of family history can often lead to ordering genetic testing that may be not only medically unnecessary, but also not reimbursable by insurance companies.
Continuing with the case above, the patient’s insurance company determines that testing is not medically necessary, and she is billed for the entire cost of more than $4,400. Her results are normal, and she feels reassured that she is not at increased risk of colon cancer.
A year later, the patient phones you to say that her uncle had genetic testing with positive results. She sends you the letter she received along with the genetic counselor’s clinic note—the uncle’s mutation is in a completely different gene from the ones you tested. While she was previously told she was at low risk, the appropriate site-specific genetic test (average cost range $185–$450) to target the specific mutation is positive, and she is at increased risk of colon cancer, but is now able to pursue increased screening to reduce her risks of developing and dying from this disease.
If the patient had not been made aware of her uncle’s results, she may not have received this screening. If she were diagnosed with later-stage colon cancer after developing symptoms, she may feel you are liable for this diagnosis based on her perception that she was not at risk according to the previously negative genetic testing results ordered by you. After learning about her family history and that the right test was not ordered for her, the patient pursues legal action.
If you chose genetic counseling
If you chose to refer the patient for genetic counseling, congratulations! Your patient is seen for risk assessment and genetic counseling.
As part of the genetic counseling session, a comprehensive family history identifies the patient’s uncle who was diagnosed with colon cancer. He was previously seen for genetics assessment and was found to have a mutation in the APC gene, predisposing him to familial adenomatous polyposis. Site-specific testing, which the genetic counselor is able to get covered by the patient’s insurance through a letter of medical necessity, reveals that your patient shares her uncle’s mutation. As indicated by national guidelines, you refer the patient to a gastroenterologist for medical management, which will significantly reduce her chances of developing and dying of colorectal cancer.
It is preferable to see the family member at highest risk for an inherited condition—usually, but not always the affected relative—for genetic consultation first. During the consultation the genetic counselor would decide which syndrome, if any, is the best fit for the family.
If the affected relative tests positive, targeted and less costly testing for the specific mutation identified (ie, site-specific testing) can then be offered to family members to provide a yes-or-no answer as to their risk status.
If the relative most likely to be gene-positive tests negative, no genetic testing would be recommended for family members, as the genetic cause of the cancer in the family is unknown. In this situation, family members may be advised to pursue the same screening measures as those with a positive gene test due to their strong family history.
INFORMED CONSENT FOR GENETIC TESTING
Genetic testing consists of much more than a simple blood draw. Obtaining informed consent for genetic testing is a crucial step in the testing process, as the results can be complex and often affect multiple family members. When predictive genetic testing is being discussed, special conversations need to take place to make sure that decisions are well informed. Genetic counselors can facilitate these discussions and guide patients and families through the decision-making process.
Example: Huntington disease
The need for genetic counseling before predictive testing is best illustrated by Huntington disease, a progressive neurodegenerative disorder with typical onset in the third or fourth decade of life. Over the disease course, patients experience decreases in motor control (leading to the aptly named “Huntington chorea”), cognitive decline, and changes in psychiatric state. Ultimately, most patients die 15 to 20 years after the onset of symptoms. Treatment is palliative and symptom-based.
Huntington disease is inherited in an autosomal dominant manner, meaning that each child of an affected person has a 50% risk of inheriting the gene change responsible for this condition and of eventually developing the disease. It is caused by an expansion within the HD gene; this expansion may grow with successive generations, leading to earlier onset of symptoms.20
The availability of predictive testing—which enables people who are at risk but who are without symptoms to find out their genetic status—ultimately leads each at-risk person to ask herself or himself, Do I want to know? Studies have found that only 15% to 67% of offspring of parents with Huntington disease (offspring are at 50% risk of the disease) elected to be tested, and in one longitudinal study, this rate of “uptake” decreased over time.21,22 However, any estimates of uptake may be falsely elevated, given the likelihood that those not wishing to consider testing may not feel the need for a clinical visit focused on this subject.
After predictive testing became available, an increased risk of suicide in persons at risk of Huntington disease was documented.23,24 In view of this risk and the careful decision-making support that people at risk need, predictive testing guidelines were developed by a committee of medical experts and members of Huntington disease family organizations.25 As part of the guidelines, a multivisit pretesting process was established that includes extensive education and counseling. Delay of testing is recommended when contraindications are identified, such as evidence of coercion or a serious psychiatric condition. Most genetic testing companies offering predictive testing require a signature from the ordering clinician verifying that pretest counseling has been completed; some also include a provision that the ordering clinician will relay results to the patient in person.
More than 15 years after these guidelines were adopted, a study of suicide risk in at-risk persons continued to find rates higher than in the general population, but lower than in earlier studies.26 Whether this careful pretest counseling protocol is directly related to a possible decrease in suicide risk has yet to be established, but its successful use in patients undergoing predictive Huntington disease testing has led to its adoption in other neurodegenerative diseases such as Alzheimer disease and Parkinson disease.
EXPLAINING POSITIVE GENETIC TESTING RESULTS
If genetic testing identifies a mutation, genetic counselors can help patients understand the implications of the results for themselves and for their relatives. Some patients become quite inquisitive, and the genetic counseling session morphs into a graduate-level discussion of genes, DNA, disease pathways, genetic-environmental interactions, availability of gene therapy, and clinical trials. The genetic counselor also makes the patient aware of other resources, such as disease-specific support groups, which may be developed by patients and families to provide support and practical knowledge.
In some cases, attention turns to at-risk relatives, and the genetic counselor may role-play with the patient to rehearse ways to share information with them. Genetic counselors may give patients a letter to distribute to family members with a copy of the patient’s test results, briefly explaining the condition identified and how relatives may find a genetic counselor in their area for their own risk assessment.
WHAT ABOUT GENETIC DISCRIMINATION?
Genetic discrimination is addressed in many genetic counseling sessions.
As defined by the National Human Genome Research Institute, genetic discrimination is “prejudice directed against people who have or may have a genetic disease.”27
In May 2008, the Genetic Information Nondiscrimination Act (GINA) was signed into law, providing some legal protections against genetic discrimination for patients undergoing predictive genetic testing. The law applies to most employers and health insurers but does not protect against discrimination by life or disability insurers. When discussing genetic testing, genetic counselors ensure that patients are aware of their rights and protections.
GINA would not be relevant for a patient who has a medical condition that may affect his or her insurability. For example, someone with thyroid cancer who is found to have an underlying gene mutation may still be denied any type of insurance coverage on the basis of his or her personal cancer diagnosis. However, should that person’s son who has not been diagnosed with cancer opt to undergo predictive testing, GINA would provide protection against employment and health insurance discrimination, as described above.
DIRECT-TO-CONSUMER GENETIC TESTING
As DNA technology has become increasingly complex, so has the task of understanding new tests and their clinical relevance to patients.
In the last several years, more companies have begun to offer direct-to-consumer genetic testing, which may be ordered without the involvement of a health care professional. While some companies hire or work closely with genetic counselors to conduct pretest and posttest genetic counseling, others do not, and preliminary research has found that only a minority of primary care physicians feel prepared to answer patients’ questions about direct-to-consumer genetic testing.28
Genetic counselors stay abreast of emerging technologies and are prepared to answer questions from patients who are considering or have already undergone such testing and from physicians who may wonder if a patient’s direct-to-consumer genetic testing results should affect his or her management.
Direct-to-consumer genetic testing will be discussed in depth in a future article in this series.
EXPLAINING ‘NORMAL’ (NEGATIVE) GENETIC TEST RESULTS
When testing results are normal, patients are educated about the meaning of “normal” results, the residual risk, and screening that might be appropriate in each person’s situation.
Sometimes a normal result does not mean the patient is not at risk for disease—for most diseases, genetic testing is not perfect and cannot identify a mutation in every at-risk family. Patients who have a family history of certain conditions may still face a higher risk despite normal test results. In these situations it is imperative that the family continue to adhere to follow-up recommendations even with normal test results.
Example: Marfan syndrome
Marfan syndrome is an autosomal dominant connective tissue disorder that, if unrecognized, is associated with significant morbidity and mortality. People with Marfan syndrome are at increased risk of aortic aneurysms, which can rupture spontaneously, leading to sudden death.
Although at least 70% of patients with Marfan syndrome have a mutation in FBN1, other patients meeting the clinical diagnostic criteria do not. Despite a normal genetic test result, they should adhere to the same screening guidelines as a person who tests positive.29
This concept—that screening should still be done despite a normal “Marfan test”—may be difficult for patients to grasp without a discussion of the imperfect sensitivity of genetic testing and of their real ongoing risks. Even more difficult to understand is the idea that the patient’s family members should also be screened as though they have the disease, given that the family’s mutation is unknown and predictive testing cannot be conducted.
Further complicating matters, other disorders such as Loeys-Dietz and vascular Ehlers-Danlos syndrome can mimic Marfan syndrome by causing aortic aneurysms, but management recommendations for them are very different.30,31
The appropriate genetic diagnosis for patients with aortic aneurysms can be facilitated by referring them to genetic counselors, who can identify appropriate testing. In this way, physicians can personalize medical management and give screening recommendations specific to the genetic disorder present.
EXPLAINING UNCERTAIN RESULTS
There are three possible results for most genetic tests—positive (a pathogenic or disease-causing mutation was found), negative (normal), and the frustrating “variant of uncertain significance” (VUS).
A VUS result means that an abnormality was detected in the gene, but that there are insufficient data about whether the abnormality alters the function of the gene in question and, thus, leads to disease. Since some gene variants are known to be common in the general population and not linked to disease and others are known to definitely alter a gene’s function and cause disease, a VUS that is clearly unknown poses a challenge not only to patient management, but also to family members seeking personal risk assessments.
Knowledge of how or if specific variants relate to disease is emerging. In time, some variants become reclassified as either disease-causing mutations or benign polymorphisms. However, careful consideration needs to be given to how to explain the abnormal result to the patient and to at-risk family members, as well as to how to explain the clinical implications of the VUS.
Example: Hereditary breast and ovarian cancer syndrome
People with hereditary breast and ovarian cancer syndrome face a lifetime risk of breast cancer of up to 87% and a risk of ovarian cancer of up to 44%. Most families with this syndrome have an inherited change in either the BRCA1 or BRCA2 gene.32,33 Given these risks, prophylactic mastectomy and oophorectomy are among the management options for mutation-positive patients. In the absence of clear genetic counseling, a patient with a VUS might see the “abnormal” test result and believe herself to be mutation-positive and thus at very high cancer risk.
An important role for the genetic counselor is to clarify the pathogenicity of a particular VUS. When a VUS is found, genetic counselors search for information about the variant by reviewing the medical literature, discussing it with the testing laboratory, arranging for family studies when appropriate, and contacting researchers whose work focuses on the gene in question.
Failure to properly research a particular VUS can lead to unnecessary and risky surgical procedures, as well as to falsely labelling relatives as being at risk. Until a VUS is reclassified as a disease-causing mutation, testing for it should not be offered to family members (unless through a research study), nor should medical management be based solely on the results of a particular VUS. In time, a VUS may be reclassified as either a benign polymorphism or a disease-causing mutation, and the genetic counselor will recontact the patient and physician with updated information and recommendations.
WHOM SHOULD I REFER?
Genetic counseling is available for patients and families in diverse settings within health systems. The six primary areas of practice are general, cardiovascular, cancer, preconception, prenatal, and pediatrics.
Patients with a personal or family history of a hereditary condition can benefit from genetic counseling regardless of whether they would be considered appropriate for genetic testing.34
At current count, there are 4,424 genetic disorders for which the underlying cause has been identified.35 Individually, each disorder is rare, but when they are considered as a whole, they affect a significant minority of the general population. It is estimated that before age 25 years, 53 (5.3%) of every 1,000 people will be diagnosed with a disease that has an important genetic component.36 From 20% to 30% of infant deaths are related to a genetic disorder,37,38 and 22% of unaffected adults have a family history of cancer significant enough to warrant a genetics referral.39 See Table 2 for a list of common indications for referral.
HOW CAN I FIND GENETIC COUNSELING SERVICES?
The National Society of Genetic Counselors (www.nsgc.org) and American Board of Genetic Counseling (www.abgc.net) both provide searchable databases of registered genetic counselors.
KNOWLEDGE CONTINUES TO EXPAND
Genetic knowledge continues to expand, and testing is becoming available for a growing number of medical conditions. Appropriate identification of individuals with and at risk for genetic disorders through the use of genetic testing and screening is a cornerstone of personalized medicine, with the ultimate goal of improving patient outcomes. However, in this era of value-based medicine and fewer health care dollars, genetic testing must be used in a way that maximizes its clinical impact with a careful fiscal approach.
Genetic counselors are specially trained health care professionals with expertise in genetic and genomic medicine who work in collaboration with physicians to guide patients through the complexities of heritable conditions and emerging technologies. They are trained to personalize, interpret, and communicate complex science into data that will assure best outcomes for patients and their families. Developing a partnership with the genetic counselors in your area can provide multiple benefits to your patients as well as to your own practice.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Hunt SC, Gwinn M, Adams TD. Family history assessment: strategies for prevention of cardiovascular disease. Am J Prev Med 2003; 24:136–142.
- Wood ME, Stockdale A, Flynn BS. Interviews with primary care physicians regarding taking and interpreting the cancer family history. Fam Pract 2008; 25:334–340.
- Bellcross CA, Kolor K, Goddard KA, Coates RJ, Reyes M, Khoury MJ. Awareness and utilization of BRCA1/2 testing among U.S. primary care physicians. Am J Prev Med 2011; 40:61–66.
- Hindorff LA, Burke W, Laberge AM, et al. Motivating factors for physician ordering of factor V Leiden genetic tests. Arch Intern Med 2009; 169:68–74.
- National Society of Genetic Counselors. Definition of genetic counseling. www.nsgc.org/About/FAQsDefinitions/tabid/97/Default.aspx. Accessed June 4, 2012.
- Bernhardt BA, Biesecker BB, Mastromarino CL. Goals, benefits, and outcomes of genetic counseling: client and genetic counselor assessment. Am J Med Genet 2000; 94:189–197.
- Bernhardt BA, Pyeritz RE. The economics of clinical genetics services. III. Cognitive genetics services are not self-supporting. Am J Hum Genet 1989; 44:288–293.
- McPherson E, Zaleski C, Benishek K, et al. Clinical genetics provider real-time workflow study. Genet Med 2008; 10:699–706.
- Brain K, Gray J, Norman P, et al. Randomized trial of a specialist genetic assessment service for familial breast cancer. J Natl Cancer Inst 2000; 92:1345–1351.
- Lerman C, Biesecker B, Benkendorf JL, et al. Controlled trial of pretest education approaches to enhance informed decision-making for BRCA1 gene testing. J Natl Cancer Inst 1997; 89:148–157.
- Randall J, Butow P, Kirk J, Tucker K. Psychological impact of genetic counselling and testing in women previously diagnosed with breast cancer. Intern Med J 2001; 31:397–405.
- Meiser B, Butow PN, Barratt AL, et al; Psychological Impact Collaborative Group. Long-term outcomes of genetic counseling in women at increased risk of developing hereditary breast cancer. Patient Educ Couns 2001; 44:215–225.
- Meiser B, Halliday JL. What is the impact of genetic counselling in women at increased risk of developing hereditary breast cancer? A meta-analytic review. Soc Sci Med 2002; 54:1463–1470.
- Kladny B, Williams A, Gupta A, Gettig EA, Krishnamurti L. Genetic counseling following the detection of hemoglobinopathy trait on the newborn screen is well received, improves knowledge, and relieves anxiety. Genet Med 2011; 13:658–661.
- Austin JC, Honer WG. Psychiatric genetic counselling for parents of individuals affected with psychotic disorders: a pilot study. Early Interv Psychiatry 2008; 2:80–89.
- Forrest LE, Burke J, Bacic S, Amor DJ. Increased genetic counseling support improves communication of genetic information in families. Genet Med 2008; 10:167–172.
- Watson M, Kash KM, Homewood J, Ebbs S, Murday V, Eeles R. Does genetic counseling have any impact on management of breast cancer risk? Genet Test 2005; 9:167–174.
- National Conference of State Legislatures. Genetic counselor licensing. www.ncsl.org/issues-research/health/genetic-counselor-licensing-laws.aspx. Accessed June 4, 2012.
- Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010; 5:40.
- Morrison PJ, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet 2011; 80:281–286.
- Bernhardt C, Schwan AM, Kraus P, Epplen JT, Kunstmann E. Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). Eur J Hum Genet 2009; 17:295–300.
- Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Suicide risk in Huntington’s disease. J Med Genet 1993; 30:293–295.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- International Huntington Association and the World Federation of Neurology Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. J Med Genet 1994; 31:555–559.
- Fiedorowicz JG, Mills JA, Ruggle A, Langbehn D, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. Suicidal behavior in prodromal Huntington disease. Neurodegener Dis 2011; 8:483–490.
- National Institutes of Health. Definition of genetic discrimination. www.genome.gov/Glossary/index.cfm?id=80. Accessed June 4, 2012.
- Powell KP, Cogswell WA, Christianson CA, et al. Primary care physicians’ awareness, experience, and opinions of direct-to-consumer genetic testing. J Genet Couns 2011; (Epub ahead of print.)
- Dietz HC. Marfan syndrome. In:Pagon RA, Bird TD, Dolan CR, et aleditors. GeneReviews. Seattle, WA: University of Washington; 1993.
- Williams JA, Loeys BL, Nwakanma LU, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg 2007; 83:S757–5763.
- Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg 2005; 42:98–106.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343:692–695.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Johns Hopkins University. OMIM entry statistics. http://omim.org/statistics/entries. Accessed June 4, 2012.
- Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet 1988; 42:677–693.
- Berry RJ, Buehler JW, Strauss LT, Hogue CJ, Smith JC. Birth weight-specific infant mortality due to congenital anomalies, 1960 and 1980. Public Health Rep 1987; 102:171–181.
- Hoyert DL, Freedman MA, Strobino DM, Guyer B. Annual summary of vital statistics: 2000. Pediatrics 2001; 108:1241–1255.
- Scheuner MT, McNeel TS, Freedman AN. Population prevalence of familial cancer and common hereditary cancer syndromes. The 2005 California Health Interview Survey. Genet Med 2010; 12:726–735.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Hunt SC, Gwinn M, Adams TD. Family history assessment: strategies for prevention of cardiovascular disease. Am J Prev Med 2003; 24:136–142.
- Wood ME, Stockdale A, Flynn BS. Interviews with primary care physicians regarding taking and interpreting the cancer family history. Fam Pract 2008; 25:334–340.
- Bellcross CA, Kolor K, Goddard KA, Coates RJ, Reyes M, Khoury MJ. Awareness and utilization of BRCA1/2 testing among U.S. primary care physicians. Am J Prev Med 2011; 40:61–66.
- Hindorff LA, Burke W, Laberge AM, et al. Motivating factors for physician ordering of factor V Leiden genetic tests. Arch Intern Med 2009; 169:68–74.
- National Society of Genetic Counselors. Definition of genetic counseling. www.nsgc.org/About/FAQsDefinitions/tabid/97/Default.aspx. Accessed June 4, 2012.
- Bernhardt BA, Biesecker BB, Mastromarino CL. Goals, benefits, and outcomes of genetic counseling: client and genetic counselor assessment. Am J Med Genet 2000; 94:189–197.
- Bernhardt BA, Pyeritz RE. The economics of clinical genetics services. III. Cognitive genetics services are not self-supporting. Am J Hum Genet 1989; 44:288–293.
- McPherson E, Zaleski C, Benishek K, et al. Clinical genetics provider real-time workflow study. Genet Med 2008; 10:699–706.
- Brain K, Gray J, Norman P, et al. Randomized trial of a specialist genetic assessment service for familial breast cancer. J Natl Cancer Inst 2000; 92:1345–1351.
- Lerman C, Biesecker B, Benkendorf JL, et al. Controlled trial of pretest education approaches to enhance informed decision-making for BRCA1 gene testing. J Natl Cancer Inst 1997; 89:148–157.
- Randall J, Butow P, Kirk J, Tucker K. Psychological impact of genetic counselling and testing in women previously diagnosed with breast cancer. Intern Med J 2001; 31:397–405.
- Meiser B, Butow PN, Barratt AL, et al; Psychological Impact Collaborative Group. Long-term outcomes of genetic counseling in women at increased risk of developing hereditary breast cancer. Patient Educ Couns 2001; 44:215–225.
- Meiser B, Halliday JL. What is the impact of genetic counselling in women at increased risk of developing hereditary breast cancer? A meta-analytic review. Soc Sci Med 2002; 54:1463–1470.
- Kladny B, Williams A, Gupta A, Gettig EA, Krishnamurti L. Genetic counseling following the detection of hemoglobinopathy trait on the newborn screen is well received, improves knowledge, and relieves anxiety. Genet Med 2011; 13:658–661.
- Austin JC, Honer WG. Psychiatric genetic counselling for parents of individuals affected with psychotic disorders: a pilot study. Early Interv Psychiatry 2008; 2:80–89.
- Forrest LE, Burke J, Bacic S, Amor DJ. Increased genetic counseling support improves communication of genetic information in families. Genet Med 2008; 10:167–172.
- Watson M, Kash KM, Homewood J, Ebbs S, Murday V, Eeles R. Does genetic counseling have any impact on management of breast cancer risk? Genet Test 2005; 9:167–174.
- National Conference of State Legislatures. Genetic counselor licensing. www.ncsl.org/issues-research/health/genetic-counselor-licensing-laws.aspx. Accessed June 4, 2012.
- Roos RA. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010; 5:40.
- Morrison PJ, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet 2011; 80:281–286.
- Bernhardt C, Schwan AM, Kraus P, Epplen JT, Kunstmann E. Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). Eur J Hum Genet 2009; 17:295–300.
- Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Suicide risk in Huntington’s disease. J Med Genet 1993; 30:293–295.
- Schoenfeld M, Myers RH, Cupples LA, Berkman B, Sax DS, Clark E. Increased rate of suicide among patients with Huntington’s disease. J Neurol Neurosurg Psychiatry 1984; 47:1283–1287.
- International Huntington Association and the World Federation of Neurology Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. J Med Genet 1994; 31:555–559.
- Fiedorowicz JG, Mills JA, Ruggle A, Langbehn D, Paulsen JS; PREDICT-HD Investigators of the Huntington Study Group. Suicidal behavior in prodromal Huntington disease. Neurodegener Dis 2011; 8:483–490.
- National Institutes of Health. Definition of genetic discrimination. www.genome.gov/Glossary/index.cfm?id=80. Accessed June 4, 2012.
- Powell KP, Cogswell WA, Christianson CA, et al. Primary care physicians’ awareness, experience, and opinions of direct-to-consumer genetic testing. J Genet Couns 2011; (Epub ahead of print.)
- Dietz HC. Marfan syndrome. In:Pagon RA, Bird TD, Dolan CR, et aleditors. GeneReviews. Seattle, WA: University of Washington; 1993.
- Williams JA, Loeys BL, Nwakanma LU, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg 2007; 83:S757–5763.
- Oderich GS, Panneton JM, Bower TC, et al. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg 2005; 42:98–106.
- Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62:676–689.
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet 1994; 343:692–695.
- Trepanier A, Ahrens M, McKinnon W, et al; National Society of Genetic Counselors. Genetic cancer risk assessment and counseling: recommendations of the National Society of Genetic Counselors. J Genet Couns 2004; 13:83–114.
- Johns Hopkins University. OMIM entry statistics. http://omim.org/statistics/entries. Accessed June 4, 2012.
- Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet 1988; 42:677–693.
- Berry RJ, Buehler JW, Strauss LT, Hogue CJ, Smith JC. Birth weight-specific infant mortality due to congenital anomalies, 1960 and 1980. Public Health Rep 1987; 102:171–181.
- Hoyert DL, Freedman MA, Strobino DM, Guyer B. Annual summary of vital statistics: 2000. Pediatrics 2001; 108:1241–1255.
- Scheuner MT, McNeel TS, Freedman AN. Population prevalence of familial cancer and common hereditary cancer syndromes. The 2005 California Health Interview Survey. Genet Med 2010; 12:726–735.
KEY POINTS
- The sequencing of the human genome has provided valuable information about the genetic causes of many conditions, but it has also uncovered tremendous complexities.
- Genetic counselors are master’s-trained allied health care professionals with specific expertise in identifying and educating patients at risk for inherited conditions.
- Genetic testing should not be ordered without informed consent and without appropriate counseling before and after the test.
- Huntington disease, which is inherited in an autosomal dominant manner, illustrates the need for genetic counseling before predictive testing.
- The National Society of Genetic Counselors (www.nsgc.org) and the American Board of Genetic Counseling (www.abgc.net) provide searchable databases of genetic counselors.
Family history: Still relevant in the genomics era
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
At the dawn of the genomics era, is the family history still relevant? The answer is a resounding yes.1,2
The family history is clinically useful because it is a proxy for genetic, environmental, and behavioral risks to health. It can be used to inform risk stratification, allowing for judicious use of screening and opening the door to early and even prophylactic treatment.3–8 As people live longer, we will need to detect common chronic conditions early in their course so that we can continue to improve health outcomes. Family history can help physicians personalize preventive care for conditions such as diabetes, osteoporosis, and cancers of the breast, colon, and prostate.2,9–15
However, there is ample evidence that the family history is underused. Most practitioners ask about it infrequently and inconsistently.16,17 Why is this, and how can we encourage the use of this powerful tool to enhance our daily clinical practice and improve care?
We will discuss here some of the challenges that make it difficult for physicians to collect and use the family history in clinical practice, and review strategies for collecting and using the family history in a more consistent manner. We anticipate that this discussion will be helpful to clinicians, as the family history is an essential input to personalized, preventive care plans.
CHALLENGE 1: ARE PATIENTS’ REPORTS RELIABLE?
A question that often arises when discussing the utility of the family history is the reliability of patients’ reports. Can we trust that patients can accurately report their family history? For many conditions, the answer is yes.18,19
Ziogas and Anton-Culverl20 asked 1,111 cancer patients whether their relatives had ever had cancer and verified their answers. In more than 95% of cases, if the patient said that a first-degree or second-degree relative did not have cancer of any type, that relative truly did not have cancer. Overall, over-reporting of cancer was rare, occurring in 2.4% of cases.
If the patient said that a relative did have cancer, that statement was usually true as well. The reliability of a report of cancer in first-degree relatives was greater than 75% for most types of cancer (female breast, ovarian, esophageal, colorectal, pancreas, lung, melanoma, brain, thyroid, lymphoma, leukemia). For several of these types of cancer (female breast, colorectal, and brain), the reliability was 90% or higher. For second-degree relatives, the reliability of a reported positive history was moderate (50% to 80%) for the same types of cancer, and for third-degree relatives, the reliability dropped further for all types of cancer except female breast, brain, pancreas, and leukemia, for which the reliability of a positive report remained at 70%.
Wideroff et al21 had similar findings in a study of more than 1,000 patients and more than 20,000 of their relatives.
Yoon et al,18 at the US Centers for Disease Control and Prevention, developed a Web-based risk-assessment tool called Family Healthware, currently undergoing validation trials. They found that patients’ reports were highly reliable for coronary heart disease, stroke, diabetes, and breast, ovarian, and colorectal cancers. They also calculated the degree of risk associated with a positive family history and the prevalence of a family history of each of these diseases.
For the primary care physician, these studies support the reliability of patients’ reports and provide guidance for targeting specific conditions when obtaining a family history.
CHALLENGE 2: NO TIME OR REIMBURSEMENT
Perhaps the most obvious barriers to collecting a family history are lack of time and reimbursement.
Acheson et al,17 in an observational study of 138 primary care physicians and 4,454 patient visits, found that family history was discussed during 51% of new patient visits and 22% of established patient visits. The rate at which the family history was taken varied from 0% (some physicians never asked) to 81% of all patient visits. On average, physicians spent less than 2.5 minutes collecting the family history.
Not surprisingly, the family history was discussed more often at well-care visits than at illness visits, as the former type of visit tends to be longer and, by definition, to be spent partly on preventive care. A difficulty with this strategy is that, given the shortage of primary care physicians, limited access, and patient preference, most preventive-care visits are combined with problem-focused visits, further decreasing the time available to collect and discuss a family history. While some argue that the family history should routinely be obtained and discussed during preventive-care visits regardless of reimbursement and time, the reality is that it may simply drop on the list of priorities for each visit.
Rich et al3 estimated that taking a family history would increase reimbursement for only one new patient evaluation and management code (99202) and one return-visit code (99213) in Current Procedural Terminology. This action would increase reimbursement enough to support about 10 minutes of physician effort for collecting, documenting, and analyzing the family history. While this is certainly better than the average of less than 2.5 minutes observed by Acheson et al,17 doctors would probably do it more if they were paid more for it.
Electronic solutions
Given that a lack of time is a barrier, what are some ways to minimize the time it takes to collect a family history?
With more physicians using electronic health records and with increasing use of Internet-based tools in the population at large, information-technology systems have been developed to help obtain the family history. One of the most widely used is the US surgeon general’s My Family Health Portrait, available free at https://familyhistory.hhs.gov. It is one of the broadest electronic family-history collection tools and has been validated for use in risk assessment for diabetes and cancer of the colon, breast, and ovaries.22
However, electronic solutions have their own challenges. Not all patients have access to the Internet, many need help using these programs, and these tools may not work well with existing electronic medical records systems.23 Ideally, these programs would also provide built-in decision support for the provider, thereby maximizing data use for final patient risk assessment.23 Furthermore, electronic solutions are not a one-time-only risk assessment— periodic re-review of family history and reassessment of familial risk are required.24
Does taking a family history improve outcomes? Lessons from breast cancer
One of the reasons physicians don’t get reimbursed for collecting a family history is that it has been difficult to measure any improvement in outcomes associated with risk prediction through family history.
The best examples of improvement in outcomes associated with family history-based risk prediction come from studies of breast cancer. From 5% to 10% of cases of breast cancer are part of hereditary cancer syndromes, many of which have a known genetic cause. The most prevalent of these genetic syndromes is the hereditary breast and ovarian cancer (HBOC) syndrome, caused by mutations in the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) genes. Clinical testing for BRCA mutations has been available since 1998.25 Women with a BRCA mutation have up to a 65% lifetime risk of developing breast cancer and up to a 40% lifetime risk of developing ovarian cancer.26 Men with a BRCA mutation are at 10 to 100 times the risk of the general population (1% to 10% vs 0.1%) for developing breast cancer, and are also at higher risk of prostate and other cancers.27
People who have a relative who developed breast cancer at a young age are more likely to harbor one of these mutations. For example, based on genetic testing in more than 185,000 people, the prevalence of BRCA mutations among people without cancer, not of Ashkenazi Jewish ancestry (a risk factor for breast cancer), and with no family history of early breast cancer or of ovarian cancer in any relative is 1.5%.28 In contrast, people with no personal history of cancer who have a family history of breast cancer before age 50 have a 5.6% prevalence of BRCA mutation, and if they are of Ashkenazi Jewish ancestry, this number is 16.4%.28
Medical and surgical interventions are available to reduce the risk of cancer in people with hereditary cancer syndromes such as HBOC. Options include screening more often, using advanced screening tests,29 giving preventive drugs such as tamoxifen (Nolvadex), and prophylactic surgery.30–32 What is the evidence that early screening and intervention in these people improve outcomes?
Domcheck et al33 prospectively followed more than 2,400 women who had BRCA mutations to assess the effect of prophylactic mastectomy or salpingo-oophorectomy on cancer outcomes. Mastectomy was indeed associated with a lower risk of breast cancer: 0 cases of breast cancer were diagnosed in 3 years of prospective follow-up in the 247 women who elected to undergo mastectomy, compared with 98 cases diagnosed in the 1,372 women who did not elect it over a similar period.
Women who elected to undergo salpingo-oophorectomy had a similarly lower rate of ovarian cancer compared with those who did not elect surgery (1% vs 6%). Additionally, fewer women who elected prophylactic salpingo-oophorectomy died of any cause (3% vs 10%), died of breast cancer (2% vs 6%), or died of ovarian cancer (0.4% vs 3%) compared with women who did not elect surgery.
Taking a family history reduces costs
What is the evidence that appropriate use of the family history decreases health care costs? Let us continue with the example of HBOC syndrome due to BRCA mutations.
Given that germline mutations account for 5% to 10% of cases of breast cancer in the United States and that the women who develop cancer associated with such mutations do so at a relatively young age, these mutations account for a disproportionate share of life-years lost due to cancer.34 Through taking a family history, these women at high risk can be identified and referred for genetic testing. Genetic testing, though costly, is more cost-effective than diagnosing and treating cancer.
Anderson et al,34 in 2006, estimated that cost-effective policies on testing and preventive treatment for persons at high risk of breast cancer could save up to $800 million of the more than $8 billion spent each year on breast cancer diagnosis, prevention, and treatment.
Kwon et al,35 in a simulation model (not a study in real patients), compared four different criteria for BRCA testing in women with ovarian cancer to see which strategy would be most cost-effective in preventing breast and ovarian cancers in their first-degree relatives. The best strategy, according to this analysis, is to test women with ovarian cancer for BRCA mutations if they also have a personal history of breast cancer, have a family history of breast or ovarian cancer, or are of Ashkenazi Jewish ancestry. The estimated cost per life-year gained with this strategy was $32,018, much lower than the widely accepted threshold for cost-effectiveness of $50,000 per life-year gained.
Although many professional organizations, including the US Preventive Services Task Force, have endorsed family-history-based eligibility criteria for genetic counseling and BRCA testing, awareness of the value of genetic testing in people who have been prescreened by family history has been relatively slow in seeping out to insurance carriers, especially Medicaid.12,36 As evidence continues to accumulate showing that this approach can improve outcomes for at-risk family members, reimbursement and time allotted for obtaining and using the family history should be adjusted.
CHALLENGE 3: A KNOWLEDGE GAP IN CLINICIANS
Another challenge often cited as a cause of the underuse of the family history as a predictor of disease risk is that clinicians may not know enough about the topic. Several studies indicated that even when physicians had obtained some components of the family history, they did not document risk appropriately or recognize the significance of the pattern of inheritance observed.37–39
In a study comparing primary care physicians and gastroenterologists in their use of the family history to predict the risk of hereditary colon cancer, gastroenterologists were more likely to elicit a family history of colorectal cancer and implement appropriate screening strategies, but overall compliance with screening guidelines was suboptimal in both groups.40
A 2011 report by an advisory committee to the secretary of the US Department of Health and Human Services concluded that lack of genetics education in medical school limits the integration of genetics into clinical care.41
How can we close this knowledge gap?
Recognizing a need, the National Coalition for Health Professional Education in Genetics was established in 1996 by the American Medical Association, the American Nurses Association, and the National Human Genome Research Institute (www.nchpeg.org). Its mission is to promote the education of health professionals and access to information about advances in human genetics to improve the health care of the nation. It offers educational materials, including a newly updated “Core Principles in Family History” program, which can be used to educate medical providers and their patients about various concepts related to genetics and family history.
In addition, physicians can use many risk assessment tools based on family history in patient care. Two of the best known are:
- The Gail breast cancer risk assessment model, hosted by the National Cancer Institute (www.cancer.gov/bcrisktool/)
- The FRAX osteoporosis risk assessment model, developed by the World Health Organization (www.shef.ac.uk/FRAX).
As we continue to educate the medical community about the value of the family history in predicting disease, it will be important to increase efforts in medical schools and residency programs and to find new, more interactive ways of teaching these concepts.
A possible strategy is to highlight the use of pedigree drawing to recognize patterns of inheritance.2 In a study of physician attitudes toward using patient-generated pedigrees in practice, such as those produced by the US surgeon general’s My Family Health Portrait, 73% of physicians stated that the patient-generated pedigree would improve their ability to assess the risk of disease, and the majority also agreed that it would not extend the time of the assessment.16
Is this information clinically useful?
Furthermore, knowing they are at risk might empower people and encourage them to engage with the medical system. For example, counseling people at risk of diabetes as reflected in the family history has been shown to increase their understanding of the risk and of preventive behaviors. Further study is needed to determine if such messages can engender lasting changes in behavior across many diseases.42–46
TOWARD PERSONALIZED CARE
Especially now that caregivers are striving to provide value-based health care with emphasis on preventive care, the family history remains an important tool for detecting risk of disease. The evidence clearly indicates that medical providers have room for improvement in taking a family history and in using it.
We hope that asking patients about family history and recognizing patterns of disease will help us create personalized preventive-care plans, providing greater opportunity to educate and motivate our patients to work with us towards better health. Future solutions need to focus on time-effective ways to collect and analyze family history and on innovative methods of teaching medical providers at all levels to apply the family history to clinical practice.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
- Guttmacher AE, Collins FS, Carmona RH. The family history—more important than ever. N Engl J Med 2004; 351:2333–2336.
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 478: Family history as a risk assessment tool. Obstet Gynecol 2011; 117:747–750.
- Rich EC, Burke W, Heaton CJ, et al. Reconsidering the family history in primary care. J Gen Intern Med 2004; 19:273–280.
- Green RF. Summary of workgroup meeting on use of family history information in pediatric primary care and public health. Pediatrics 2007; 120(suppl 2):S87–S100.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009; 113:957–966.
- Scheuner MT, Setodji CM, Pankow JS, Blumenthal RS, Keeler E. General Cardiovascular Risk Profile identifies advanced coronary artery calcium and is improved by family history: the multiethnic study of atherosclerosis. Circ Cardiovasc Genet 2010; 3:97–105.
- Yang Q, Liu T, Valdez R, Moonesinghe R, Khoury MJ. Improvements in ability to detect undiagnosed diabetes by using information on family history among adults in the United States. Am J Epidemiol 2010; 171:1079–1089.
- Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. A comprehensive survey. Drug Des Devel Ther 2011; 5:325–380.
- Rex DK, Johnson DA, Anderson JC, Schoenfeld PS, Burke CA, Inadomi JM; American College of Gastroenterology. American College of Gastroenterology guidelines for colorectal cancer screening 2009 (corrected). Am J Gastroenterol 2009; 104:739–750.
- American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Kanis JA, Johansson H, Oden A, McCloskey EV. Assessment of fracture risk. Eur J Radiol 2009; 71:392–397.
- US Preventive Services Task Force. Genetic risk assessment and BRCA mutation testing for breast and ovarian cancer susceptibility: recommendation statement. Ann Intern Med 2005; 143:355–361.
- Williams SB, Salami S, Regan MM, et al. Selective detection of histologically aggressive prostate cancer: An Early Detection Research Network Prediction model to reduce unnecessary prostate biopsies with validation in the Prostate Cancer Prevention Trial. Cancer 2011; Oct 17(Epub ahead of print.)
- Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res (Phila) 2011; 4:9–22.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011; 29:2247–2252.
- Fuller M, Myers M, Webb T, Tabangin M, Prows C. Primary care providers’ responses to patient-generated family history. J Genet Couns 2010; 19:84–96.
- Acheson LS, Wiesner GL, Zyzanski SJ, Goodwin MA, Stange KC. Family history-taking in community family practice: implications for genetic screening. Genet Med 2000; 2:180–185.
- Yoon PW, Scheuner MT, Jorgensen C, Khoury MJ. Developing Family Healthware, a family history screening tool to prevent common chronic diseases. Prev Chronic Dis 2009; 6:A33.
- Valdez R, Yoon PW, Qureshi N, Green RF, Khoury MJ. Family history in public health practice: a genomic tool for disease prevention and health promotion. Annu Rev Public Health 2010; 31:69–87.
- Ziogas A, Anton-Culver H. Validation of My Family Health Portrait for six common heritable conditions. Am J Prev Med 2003; 24:190–198.
- Wideroff L, Garceau AO, Greene MH, et al. Coherence and completeness of population-based family cancer reports. Cancer Epidemiol Biomarkers Prev 2010; 19:799–810.
- Facio FM, Feero WG, Linn A, Oden N, Manickam K, Biesecker LG. Validation of My Family Health Portrait for six common heritable conditions. Genet Med 2010; 12:370–375.
- Owens KM, Marvin ML, Gelehrter TD, Ruffin MT, Uhlmann WR. Clinical use of the Surgeon General’s “My Family Health Portrait” (MFHP) tool: opinions of future health care providers. J Genet Couns 2011; 20:510–525.
- Tyler CV, Snyder CW. Cancer risk assessment: examining the family physician’s role. J Am Board Fam Med 2006; 19:468–477.
- Rubenstein WS. The genetics of breast cancer. In:Vogel VG, editor. Management of Patients at High Risk for Breast Cancer. Malden, MA: Blackwell Science; 2001:19–55.
- Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 2003; 72:1117–1130.
- Korde LA, Zujewski JA, Kamin L, et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 2010; 28:2114–2122.
- Myriad Genetic Laboratories, Inc. Mutation prevalence tables. http://www.myriad.com/lib/brac/brca-prevalence-tables.pdf. Accessed April 2, 2012.
- Schousboe JT, Kerlikowske K, Loh A, Cummings SR. Personalizing mammography by breast density and other risk factors for breast cancer: analysis of health benefits and cost-effectiveness. Ann Intern Med 2011; 155:10–20.
- National Cancer Institute. http://www.cancer.gov. Accessed January 20, 2012.
- Saslow D, Boetes C, Burke W, et al; American Cancer Society Breast Cancer Advisory Group. American Cancer Society guidelines for breast screening with MRI as an adjunct to mammography. CA Cancer J Clin 2007; 57:75–89.
- Agency for Healthcare Research and Quality; John M. Medications to reduce the risk of primary breast cancer in women: clinician’s guide. http://www.effectivehealthcare.ahrq.gov/index.cfm/searchfor-guides-reviews-and-reports/?productid=390&pageaction=displayproduct. Accessed April 2, 2012.
- Domchek SM, Friebel TM, Singer CF, et al. Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 2010; 304:967–975.
- Anderson K, Jacobson JS, Heitjan DF, et al. Cost-effectiveness of preventive strategies for women with a BRCA1 or a BRCA2 mutation. Ann Intern Med 2006; 144:397–406.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2009; 28:675–682.
- Wang G, Beattie MS, Ponce NA, Phillips KA. Eligibility criteria in private and public coverage policies for BRCA genetic testing and genetic counseling. Genet Med 2011; 13:1045–1050.
- Hinton RB. The family history: reemergence of an established tool. Crit Care Nurs Clin North Am 2008; 20:149–158.
- Murff HJ, Greevy RA, Syngal S. The comprehensiveness of family cancer history assessments in primary care. Community Genet 2007; 10:174–180.
- Wallace E, Hinds A, Campbell H, Mackay J, Cetnarskyj R, Porteous ME. A cross-sectional survey to estimate the prevalence of family history of colorectal, breast and ovarian cancer in a Scottish general practice population. Br J Cancer 2004; 91:1575–1579.
- Schroy PC, Barrison AF, Ling BS, Wilson S, Geller AC. Family history and colorectal cancer screening: a survey of physician knowledge and practice patterns. Am J Gastroenterol 2002; 97:1031–1036.
- Department of Health and Human Services. Genetics education and training: report of the Secretary’s Advisory Committee on Genetics, Health, and Society; 2011. http://oba.od.nih.gov/oba/SACGHS/reports/SACGHS_education_report_2011.pdf. Accessed April 2, 2012.
- Qureshi N, Kai J. Informing patients of familial diabetes mellitus risk: How do they respond? A cross-sectional survey. BMC Health Serv Res 2008; 8:37.
- Zlot AI, Bland MP, Silvey K, Epstein B, Mielke B, Leman RF. Influence of family history of diabetes on health care provider practice and patient behavior among nondiabetic Oregonians. Prev Chronic Dis 2009; 6:A27.
- Pijl M, Timmermans DR, Claassen L, et al. Impact of communicating familial risk of diabetes on illness perceptions and self-reported behavioral outcomes: a randomized controlled trial. Diabetes Care 2009; 32:597–599.
- Ruffin MT, Nease DE, Sen A, et al; Family History Impact Trial (FHITr) Group. Effect of preventive messages tailored to family history on health behaviors: the Family Healthware Impact Trial. Ann Fam Med 2011; 9:3–11.
- Claassen L, Henneman L, Janssens AC, et al. Using family history information to promote healthy lifestyles and prevent diseases; a discussion of the evidence. BMC Public Health 2010; 10:248.
KEY POINTS
- The family history is an underused tool for predicting the risk of disease and for personalizing preventive care.
- Barriers to the appropriate collection and use of the family history include concerns over the reliability of patient reporting, a lack of time and reimbursement, and provider knowledge gaps.
- Use of family history to inform genetic testing for hereditary cancer syndromes has been shown to improve outcomes and may reduce overall health care costs.
- Future solutions need to focus on creating time-effective ways to collect and analyze the family history, and on developing innovative methods of educating medical providers at all levels of training as to how to apply the family history in clinical practice.