User login
Caution urged over real-world bleeding risk with ibrutinib
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to an almost 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Paul R. Kunk, MD, of University of Virginia, Charlottesville, and his colleagues. Their report is in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting but the authors suggested that this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria. These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice,” the researchers wrote.
They conducted a review of patients attending their center and associated regional clinics between January 2012 and May 2016. They identified 70 patients, average age 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%) and mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström macroglobulinemia (1%).
The analysis showed that bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis. However, major bleeding, defined as grade 3, occurred in 13 patients (19%), a figure that the authors noted was greater than the rate of around 7% reported by clinical trials.
Of these patients, seven were taking combined antiplatelet and anticoagulant therapy, four were taking antiplatelets alone, one was taking an anticoagulant agent alone, and one was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding included antiplatelet or anticoagulant medication, the combination of the two medications or interacting medications, anemia (hemoglobin less than 12 g/dL) and an elevated international normalized ratio (greater than 1.5).
However, in a multivariate analysis, only combined antiplatelet and anticoagulant use (hazard ratio, 20.0; 95% confidence interval, 2.1-200.0; P less than .01) and an elevated INR (HR, 4.6; 95% CI, 1.1-19.6; P less than .01) remained statistically significant.
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, they said their findings confirmed “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.
“As ibrutinib increases in use, it is paramount to increase awareness of the known adverse events. This is especially important given the association of ibrutinib use with atrial fibrillation,” they wrote.
They noted that their trial was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive,” they noted.
SOURCE: Kunk PR et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 15. doi: 10.1016/j.clml.2018.07.287.
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to an almost 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Paul R. Kunk, MD, of University of Virginia, Charlottesville, and his colleagues. Their report is in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting but the authors suggested that this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria. These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice,” the researchers wrote.
They conducted a review of patients attending their center and associated regional clinics between January 2012 and May 2016. They identified 70 patients, average age 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%) and mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström macroglobulinemia (1%).
The analysis showed that bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis. However, major bleeding, defined as grade 3, occurred in 13 patients (19%), a figure that the authors noted was greater than the rate of around 7% reported by clinical trials.
Of these patients, seven were taking combined antiplatelet and anticoagulant therapy, four were taking antiplatelets alone, one was taking an anticoagulant agent alone, and one was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding included antiplatelet or anticoagulant medication, the combination of the two medications or interacting medications, anemia (hemoglobin less than 12 g/dL) and an elevated international normalized ratio (greater than 1.5).
However, in a multivariate analysis, only combined antiplatelet and anticoagulant use (hazard ratio, 20.0; 95% confidence interval, 2.1-200.0; P less than .01) and an elevated INR (HR, 4.6; 95% CI, 1.1-19.6; P less than .01) remained statistically significant.
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, they said their findings confirmed “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.
“As ibrutinib increases in use, it is paramount to increase awareness of the known adverse events. This is especially important given the association of ibrutinib use with atrial fibrillation,” they wrote.
They noted that their trial was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive,” they noted.
SOURCE: Kunk PR et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 15. doi: 10.1016/j.clml.2018.07.287.
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to an almost 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Paul R. Kunk, MD, of University of Virginia, Charlottesville, and his colleagues. Their report is in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting but the authors suggested that this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria. These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice,” the researchers wrote.
They conducted a review of patients attending their center and associated regional clinics between January 2012 and May 2016. They identified 70 patients, average age 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%) and mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström macroglobulinemia (1%).
The analysis showed that bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis. However, major bleeding, defined as grade 3, occurred in 13 patients (19%), a figure that the authors noted was greater than the rate of around 7% reported by clinical trials.
Of these patients, seven were taking combined antiplatelet and anticoagulant therapy, four were taking antiplatelets alone, one was taking an anticoagulant agent alone, and one was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding included antiplatelet or anticoagulant medication, the combination of the two medications or interacting medications, anemia (hemoglobin less than 12 g/dL) and an elevated international normalized ratio (greater than 1.5).
However, in a multivariate analysis, only combined antiplatelet and anticoagulant use (hazard ratio, 20.0; 95% confidence interval, 2.1-200.0; P less than .01) and an elevated INR (HR, 4.6; 95% CI, 1.1-19.6; P less than .01) remained statistically significant.
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, they said their findings confirmed “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.
“As ibrutinib increases in use, it is paramount to increase awareness of the known adverse events. This is especially important given the association of ibrutinib use with atrial fibrillation,” they wrote.
They noted that their trial was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive,” they noted.
SOURCE: Kunk PR et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 15. doi: 10.1016/j.clml.2018.07.287.
FROM CLINICAL LYMPHOMA, MYELOMA & LEUKEMIA
Key clinical point: Clinicians should exercise caution when prescribing antiplatelet and anticoagulant medications in people taking the Bruton tyrosine kinase inhibitor ibrutinib.
Major finding: The use of both antiplatelet and anticoagulant therapy significantly increased the risk of a major bleed event (HR, 19.2; 95% CI, 2.3-166.7; P less than .01) in patients also taking ibrutinib.
Study details: A retrospective analysis of prescription data from 70 patients seen at a single U.S. cancer center and its regional clinics between January 2012 and May 2016.
Disclosures: Two of the authors reported receiving clinical trial support from Acerta and Abbvie.
Source: Kunk PR et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 15. doi: 10.1016/j.clml.2018.07.287.
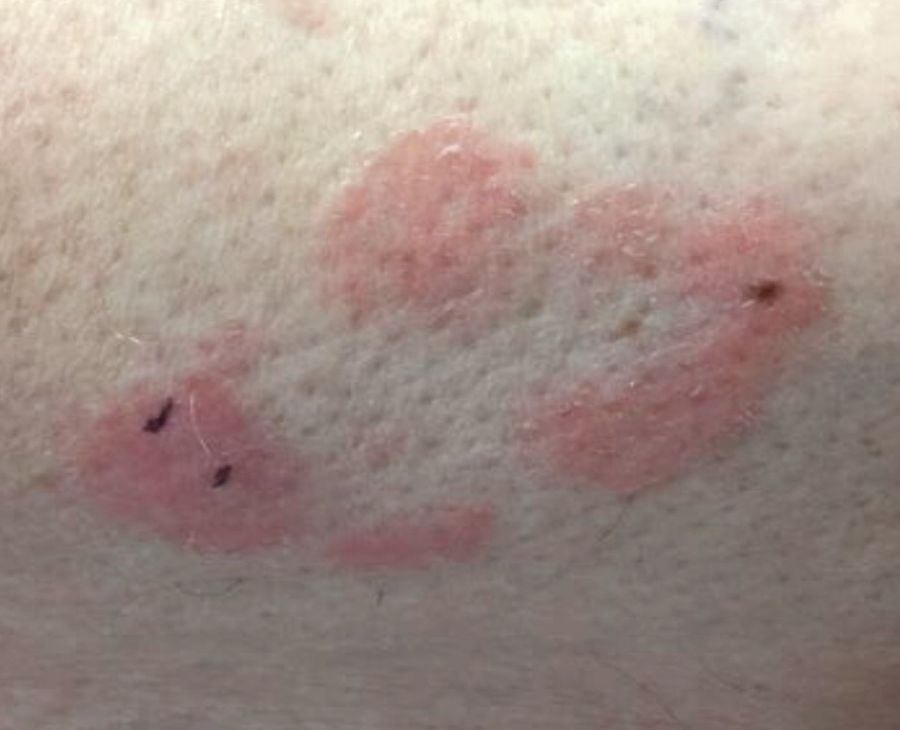
Make The Diagnosis - September 2018
Some have postulated an infectious agent as the cause. Atopic dermatitis may confer an increased risk because of the chronic stimulation of T cells. Males are more commonly affected than females by a 2:1 ratio. A worse prognosis is associated with advanced age. Children and adolescents may be affected as well.
With mycosis fungoides, there are three main types of skin lesions: patch, plaque, and tumor. Patients will progress from patch to plaque to tumor stage in classic MF. Often, lesions begin as scaly, erythematous patches that resemble eczema. Because of the nonspecific nature of early lesions, the median duration from the onset of skin lesions to the diagnosis of MF is 4-6 years. Patch stage lesions may be pruritic or asymptomatic. Commonly, they present in non–sun-exposed areas, such as the buttocks. Annular, infiltrated, red-brown or violaceous plaques can develop, which represent malignant T-cell infiltration. Many patients never progress past the plaque stage. Tumor stage MF is more aggressive, with nodules that may undergo necrosis and ulceration.
The leukemic form of MF is Sézary syndrome. Patients present with pruritic erythroderma and lymphadenopathy. Nail dystrophy, scaling of palms and soles, and alopecia may be present. A peripheral blood smear reveals Sézary cells, which are large, hyperconvoluted lymphocytes. The count of Sézary cells is usually greater than 1000 cells/mm3.
Histology of early lesions may not be diagnostic for CTCL. Often, biopsies will be read as eczematous or psoriasiform for years before the diagnosis of MF is made. Classically, epidermotropism (single-cell exocytosis of lymphocytes into the epidermis) is present. Advanced stages may show a dense infiltrate of lymphocytes in the dermis. Groups of lymphocytes in the epidermis form Pautrier’s microabscesses. Mycosis cells may exhibit cerebriform nuclei. Neoplastic cells in MF are CD3+, CD4+, CD45RO+, CD8–. Tissue can be sent for T-cell gene rearrangement polymerase chain reaction. The presence of monoclonal T-cell gene receptor rearrangements can aid in the diagnosis of MF.
Treatment includes topical steroids, mechlorethamine (nitrogen mustard) or bexarotene gel, PUVA therapy, and narrow-band UVB light for limited and/or patch disease. Localized radiotherapy can be used for more resistant lesions. Topical therapies are preferred in the early stages in MF. Systemic treatments for patients who do not respond to local therapy, or in more advanced disease include methotrexate, interferon-alpha, oral bexarotene, denileukin diftitox, and combination chemotherapy. Photopheresis is reserved for erythrodermic disease.

This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Some have postulated an infectious agent as the cause. Atopic dermatitis may confer an increased risk because of the chronic stimulation of T cells. Males are more commonly affected than females by a 2:1 ratio. A worse prognosis is associated with advanced age. Children and adolescents may be affected as well.
With mycosis fungoides, there are three main types of skin lesions: patch, plaque, and tumor. Patients will progress from patch to plaque to tumor stage in classic MF. Often, lesions begin as scaly, erythematous patches that resemble eczema. Because of the nonspecific nature of early lesions, the median duration from the onset of skin lesions to the diagnosis of MF is 4-6 years. Patch stage lesions may be pruritic or asymptomatic. Commonly, they present in non–sun-exposed areas, such as the buttocks. Annular, infiltrated, red-brown or violaceous plaques can develop, which represent malignant T-cell infiltration. Many patients never progress past the plaque stage. Tumor stage MF is more aggressive, with nodules that may undergo necrosis and ulceration.
The leukemic form of MF is Sézary syndrome. Patients present with pruritic erythroderma and lymphadenopathy. Nail dystrophy, scaling of palms and soles, and alopecia may be present. A peripheral blood smear reveals Sézary cells, which are large, hyperconvoluted lymphocytes. The count of Sézary cells is usually greater than 1000 cells/mm3.
Histology of early lesions may not be diagnostic for CTCL. Often, biopsies will be read as eczematous or psoriasiform for years before the diagnosis of MF is made. Classically, epidermotropism (single-cell exocytosis of lymphocytes into the epidermis) is present. Advanced stages may show a dense infiltrate of lymphocytes in the dermis. Groups of lymphocytes in the epidermis form Pautrier’s microabscesses. Mycosis cells may exhibit cerebriform nuclei. Neoplastic cells in MF are CD3+, CD4+, CD45RO+, CD8–. Tissue can be sent for T-cell gene rearrangement polymerase chain reaction. The presence of monoclonal T-cell gene receptor rearrangements can aid in the diagnosis of MF.
Treatment includes topical steroids, mechlorethamine (nitrogen mustard) or bexarotene gel, PUVA therapy, and narrow-band UVB light for limited and/or patch disease. Localized radiotherapy can be used for more resistant lesions. Topical therapies are preferred in the early stages in MF. Systemic treatments for patients who do not respond to local therapy, or in more advanced disease include methotrexate, interferon-alpha, oral bexarotene, denileukin diftitox, and combination chemotherapy. Photopheresis is reserved for erythrodermic disease.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Some have postulated an infectious agent as the cause. Atopic dermatitis may confer an increased risk because of the chronic stimulation of T cells. Males are more commonly affected than females by a 2:1 ratio. A worse prognosis is associated with advanced age. Children and adolescents may be affected as well.
With mycosis fungoides, there are three main types of skin lesions: patch, plaque, and tumor. Patients will progress from patch to plaque to tumor stage in classic MF. Often, lesions begin as scaly, erythematous patches that resemble eczema. Because of the nonspecific nature of early lesions, the median duration from the onset of skin lesions to the diagnosis of MF is 4-6 years. Patch stage lesions may be pruritic or asymptomatic. Commonly, they present in non–sun-exposed areas, such as the buttocks. Annular, infiltrated, red-brown or violaceous plaques can develop, which represent malignant T-cell infiltration. Many patients never progress past the plaque stage. Tumor stage MF is more aggressive, with nodules that may undergo necrosis and ulceration.
The leukemic form of MF is Sézary syndrome. Patients present with pruritic erythroderma and lymphadenopathy. Nail dystrophy, scaling of palms and soles, and alopecia may be present. A peripheral blood smear reveals Sézary cells, which are large, hyperconvoluted lymphocytes. The count of Sézary cells is usually greater than 1000 cells/mm3.
Histology of early lesions may not be diagnostic for CTCL. Often, biopsies will be read as eczematous or psoriasiform for years before the diagnosis of MF is made. Classically, epidermotropism (single-cell exocytosis of lymphocytes into the epidermis) is present. Advanced stages may show a dense infiltrate of lymphocytes in the dermis. Groups of lymphocytes in the epidermis form Pautrier’s microabscesses. Mycosis cells may exhibit cerebriform nuclei. Neoplastic cells in MF are CD3+, CD4+, CD45RO+, CD8–. Tissue can be sent for T-cell gene rearrangement polymerase chain reaction. The presence of monoclonal T-cell gene receptor rearrangements can aid in the diagnosis of MF.
Treatment includes topical steroids, mechlorethamine (nitrogen mustard) or bexarotene gel, PUVA therapy, and narrow-band UVB light for limited and/or patch disease. Localized radiotherapy can be used for more resistant lesions. Topical therapies are preferred in the early stages in MF. Systemic treatments for patients who do not respond to local therapy, or in more advanced disease include methotrexate, interferon-alpha, oral bexarotene, denileukin diftitox, and combination chemotherapy. Photopheresis is reserved for erythrodermic disease.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
Mogamulizumab prolongs PFS in CTCL

Mogamulizumab is an effective treatment option for relapsed/refractory cutaneous T-cell lymphoma (CTCL), according to researchers.
In the phase 3 MAVORIC trial, mogamulizumab prolonged progression-free survival (PFS) and produced better overall response rates (ORRs) than vorinostat in patients with relapsed/refractory CTCL.
The most common adverse events (AEs) in patients treated with mogamulizumab were infusion-related reactions, diarrhea, fatigue, and drug eruptions.
Youn Kim MD, of the Stanford Cancer Institute in Palo Alto, California, and her colleagues reported these results in The Lancet Oncology.
The results supported the recent approval of mogamulizumab by the US Food and Drug Administration.
The study was sponsored by Kyowa Hakko Kirin Co., Ltd., the company developing/marketing mogamulizumab.
Treatment
For MAVORIC, researchers compared mogamulizumab and vorinostat in adults with mycosis fungoides (MF) or Sézary syndrome (SS) who had received at least 1 prior systemic therapy.
The trial included 372 patients who were randomized to receive mogamulizumab at 1.0 mg/kg (weekly for the first cycle and then every 2 weeks) or vorinostat at 400 mg daily for 28-day cycles.
Patients were treated until disease progression or unacceptable toxicity. Patients on vorinostat who progressed or experienced intolerable toxicity after 2 cycles, despite dose reduction and appropriate management of AEs, could cross over to treatment with mogamulizumab.
There were 184 patients in the mogamulizumab arm and 186 in the vorinostat arm who received treatment.
The median duration of follow-up was 17.0 months.
Most patients (n=157) ultimately discontinued mogamulizumab. Reasons included:
- Disease progression (n=76 by CTCL criteria and 22 by clinical criteria)
- AEs (n=28)
- Withdrawn consent (n=13)
- Investigator decision (n=9)
- Patient decision (n=6)
- Death (n=2)
- Noncompliance (n=1).
Most patients (n=136) in the vorinostat arm crossed over to the mogamulizumab arm, 109 due to disease progression and 27 due to treatment intolerance.
Of the 40 patients who did not cross over to mogamulizumab, reasons for stopping vorinostat included:
- Progressive disease (n=10 by CTCL criteria and 8 by clinical criteria)
- Patient decision (n=9)
- Withdrawn consent (n=5)
- AEs (n=5)
- Death (n=2)
- Lost to follow-up (n=1).
At the data cutoff, there were 27 patients assigned to mogamulizumab and 10 assigned to vorinostat who remained on treatment. There were 31 patients still on treatment who had crossed over from vorinostat to mogamulizumab.
Patient characteristics
Baseline characteristics were similar between the treatment arms.
| Mogamulizumab (n=186) | Vorinostat (n=186) | |
| Median age | 64 (range, 54-73) | 65 (range, 56-72) |
| Male | 109 (59%) | 107 (58%) |
| Female | 77 (41%) | 79 (42%) |
| MF | 105 (56%) | 99 (53%) |
| SS | 81 (44%) | 87 (47%) |
| Time from diagnosis, months | 41.0 (range, 17.4-78.8) | 35.4 (range, 16.2-68.2) |
| Median number of previous systemic regimens | 3 (range, 2-5) | 3 (range, 2-5) |
PFS and ORR
Mogamulizumab provided a significant improvement in PFS, the study’s primary endpoint.
According to investigators, the median PFS was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio=0.53, P<0.0001).
According to independent review, the median PFS was 6.7 months and 3.8 months, respectively (hazard ratio=0.64, P<0.0007).
There was a significant improvement in ORR with mogamulizumab.
According to independent review, the global ORR was 23% (43/186) in the mogamulizumab arm and 4% (7/186) in the vorinostat arm (risk ratio=19.4, P<0.0001).
According to investigators, the global ORR was 28% (52/186) and 5% (9/186), respectively (risk ratio=23.1, P<0.0001).
For patients with MF, the investigator-assessed ORR was 21% (22/105) with mogamulizumab and 7% (7/99) with vorinostat.
For SS patients, the investigator-assessed ORR was 37% (30/81) and 2% (2/87), respectively.
Responses by disease compartment were superior with mogamulizumab as well.
The investigator-assessed blood ORR was 68% (83/122) with mogamulizumab and 19% (23/123) with vorinostat. The skin ORR was 42% (78/186) and 16% (29/186), respectively.
The lymph node ORR was 17% (21/124) and 4% (5/122), respectively. The viscera ORR was 0% in both arms.
Crossover
Among patients who crossed over from vorinostat to mogamulizumab, the ORR was 31% (41/133). In these patients, the median PFS was 8.9 months.
In the 319 patients who were assigned to mogamulizumab or crossed over to that arm, the median PFS was 8.4 months.
Safety
The most common treatment-emergent, grade 1-2 AEs, occurring in at least 20% of patients in either arm (mogamulizumab and vorinostat, respectively), were:
- Thrombocytopenia (14% vs 34%)
- Diarrhea (23% vs 57%)
- Nausea (15% vs 41%)
- Fatigue (22% vs 32%)
- Increased blood creatinine (3% vs 28%)
- Decreased appetite (7% vs 24%)
- Dysgeusia (3% vs 28%)
- Drug eruptions (20% vs 1%)
- Infusion-related reactions (32% vs 1%).
Grade 3 AEs in the mogamulizumab arm included drug eruptions (n=8), hypertension (n=8), pneumonia (n=6), fatigue (n=3), cellulitis (n=3), infusion-related reactions (n=3), sepsis (n=2), decreased appetite (n=2), AST increase (n=2), weight decrease (n=1), pyrexia (n=1), constipation (n=1), nausea (n=1), and diarrhea (n=1).
Grade 4 AEs with mogamulizumab were cellulitis (n=1) and pneumonia (n=1). Grade 5 AEs included pneumonia (n=1) and sepsis (n=1).
Mogamulizumab is an effective treatment option for relapsed/refractory cutaneous T-cell lymphoma (CTCL), according to researchers.
In the phase 3 MAVORIC trial, mogamulizumab prolonged progression-free survival (PFS) and produced better overall response rates (ORRs) than vorinostat in patients with relapsed/refractory CTCL.
The most common adverse events (AEs) in patients treated with mogamulizumab were infusion-related reactions, diarrhea, fatigue, and drug eruptions.
Youn Kim MD, of the Stanford Cancer Institute in Palo Alto, California, and her colleagues reported these results in The Lancet Oncology.
The results supported the recent approval of mogamulizumab by the US Food and Drug Administration.
The study was sponsored by Kyowa Hakko Kirin Co., Ltd., the company developing/marketing mogamulizumab.
Treatment
For MAVORIC, researchers compared mogamulizumab and vorinostat in adults with mycosis fungoides (MF) or Sézary syndrome (SS) who had received at least 1 prior systemic therapy.
The trial included 372 patients who were randomized to receive mogamulizumab at 1.0 mg/kg (weekly for the first cycle and then every 2 weeks) or vorinostat at 400 mg daily for 28-day cycles.
Patients were treated until disease progression or unacceptable toxicity. Patients on vorinostat who progressed or experienced intolerable toxicity after 2 cycles, despite dose reduction and appropriate management of AEs, could cross over to treatment with mogamulizumab.
There were 184 patients in the mogamulizumab arm and 186 in the vorinostat arm who received treatment.
The median duration of follow-up was 17.0 months.
Most patients (n=157) ultimately discontinued mogamulizumab. Reasons included:
- Disease progression (n=76 by CTCL criteria and 22 by clinical criteria)
- AEs (n=28)
- Withdrawn consent (n=13)
- Investigator decision (n=9)
- Patient decision (n=6)
- Death (n=2)
- Noncompliance (n=1).
Most patients (n=136) in the vorinostat arm crossed over to the mogamulizumab arm, 109 due to disease progression and 27 due to treatment intolerance.
Of the 40 patients who did not cross over to mogamulizumab, reasons for stopping vorinostat included:
- Progressive disease (n=10 by CTCL criteria and 8 by clinical criteria)
- Patient decision (n=9)
- Withdrawn consent (n=5)
- AEs (n=5)
- Death (n=2)
- Lost to follow-up (n=1).
At the data cutoff, there were 27 patients assigned to mogamulizumab and 10 assigned to vorinostat who remained on treatment. There were 31 patients still on treatment who had crossed over from vorinostat to mogamulizumab.
Patient characteristics
Baseline characteristics were similar between the treatment arms.
| Mogamulizumab (n=186) | Vorinostat (n=186) | |
| Median age | 64 (range, 54-73) | 65 (range, 56-72) |
| Male | 109 (59%) | 107 (58%) |
| Female | 77 (41%) | 79 (42%) |
| MF | 105 (56%) | 99 (53%) |
| SS | 81 (44%) | 87 (47%) |
| Time from diagnosis, months | 41.0 (range, 17.4-78.8) | 35.4 (range, 16.2-68.2) |
| Median number of previous systemic regimens | 3 (range, 2-5) | 3 (range, 2-5) |
PFS and ORR
Mogamulizumab provided a significant improvement in PFS, the study’s primary endpoint.
According to investigators, the median PFS was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio=0.53, P<0.0001).
According to independent review, the median PFS was 6.7 months and 3.8 months, respectively (hazard ratio=0.64, P<0.0007).
There was a significant improvement in ORR with mogamulizumab.
According to independent review, the global ORR was 23% (43/186) in the mogamulizumab arm and 4% (7/186) in the vorinostat arm (risk ratio=19.4, P<0.0001).
According to investigators, the global ORR was 28% (52/186) and 5% (9/186), respectively (risk ratio=23.1, P<0.0001).
For patients with MF, the investigator-assessed ORR was 21% (22/105) with mogamulizumab and 7% (7/99) with vorinostat.
For SS patients, the investigator-assessed ORR was 37% (30/81) and 2% (2/87), respectively.
Responses by disease compartment were superior with mogamulizumab as well.
The investigator-assessed blood ORR was 68% (83/122) with mogamulizumab and 19% (23/123) with vorinostat. The skin ORR was 42% (78/186) and 16% (29/186), respectively.
The lymph node ORR was 17% (21/124) and 4% (5/122), respectively. The viscera ORR was 0% in both arms.
Crossover
Among patients who crossed over from vorinostat to mogamulizumab, the ORR was 31% (41/133). In these patients, the median PFS was 8.9 months.
In the 319 patients who were assigned to mogamulizumab or crossed over to that arm, the median PFS was 8.4 months.
Safety
The most common treatment-emergent, grade 1-2 AEs, occurring in at least 20% of patients in either arm (mogamulizumab and vorinostat, respectively), were:
- Thrombocytopenia (14% vs 34%)
- Diarrhea (23% vs 57%)
- Nausea (15% vs 41%)
- Fatigue (22% vs 32%)
- Increased blood creatinine (3% vs 28%)
- Decreased appetite (7% vs 24%)
- Dysgeusia (3% vs 28%)
- Drug eruptions (20% vs 1%)
- Infusion-related reactions (32% vs 1%).
Grade 3 AEs in the mogamulizumab arm included drug eruptions (n=8), hypertension (n=8), pneumonia (n=6), fatigue (n=3), cellulitis (n=3), infusion-related reactions (n=3), sepsis (n=2), decreased appetite (n=2), AST increase (n=2), weight decrease (n=1), pyrexia (n=1), constipation (n=1), nausea (n=1), and diarrhea (n=1).
Grade 4 AEs with mogamulizumab were cellulitis (n=1) and pneumonia (n=1). Grade 5 AEs included pneumonia (n=1) and sepsis (n=1).
Mogamulizumab is an effective treatment option for relapsed/refractory cutaneous T-cell lymphoma (CTCL), according to researchers.
In the phase 3 MAVORIC trial, mogamulizumab prolonged progression-free survival (PFS) and produced better overall response rates (ORRs) than vorinostat in patients with relapsed/refractory CTCL.
The most common adverse events (AEs) in patients treated with mogamulizumab were infusion-related reactions, diarrhea, fatigue, and drug eruptions.
Youn Kim MD, of the Stanford Cancer Institute in Palo Alto, California, and her colleagues reported these results in The Lancet Oncology.
The results supported the recent approval of mogamulizumab by the US Food and Drug Administration.
The study was sponsored by Kyowa Hakko Kirin Co., Ltd., the company developing/marketing mogamulizumab.
Treatment
For MAVORIC, researchers compared mogamulizumab and vorinostat in adults with mycosis fungoides (MF) or Sézary syndrome (SS) who had received at least 1 prior systemic therapy.
The trial included 372 patients who were randomized to receive mogamulizumab at 1.0 mg/kg (weekly for the first cycle and then every 2 weeks) or vorinostat at 400 mg daily for 28-day cycles.
Patients were treated until disease progression or unacceptable toxicity. Patients on vorinostat who progressed or experienced intolerable toxicity after 2 cycles, despite dose reduction and appropriate management of AEs, could cross over to treatment with mogamulizumab.
There were 184 patients in the mogamulizumab arm and 186 in the vorinostat arm who received treatment.
The median duration of follow-up was 17.0 months.
Most patients (n=157) ultimately discontinued mogamulizumab. Reasons included:
- Disease progression (n=76 by CTCL criteria and 22 by clinical criteria)
- AEs (n=28)
- Withdrawn consent (n=13)
- Investigator decision (n=9)
- Patient decision (n=6)
- Death (n=2)
- Noncompliance (n=1).
Most patients (n=136) in the vorinostat arm crossed over to the mogamulizumab arm, 109 due to disease progression and 27 due to treatment intolerance.
Of the 40 patients who did not cross over to mogamulizumab, reasons for stopping vorinostat included:
- Progressive disease (n=10 by CTCL criteria and 8 by clinical criteria)
- Patient decision (n=9)
- Withdrawn consent (n=5)
- AEs (n=5)
- Death (n=2)
- Lost to follow-up (n=1).
At the data cutoff, there were 27 patients assigned to mogamulizumab and 10 assigned to vorinostat who remained on treatment. There were 31 patients still on treatment who had crossed over from vorinostat to mogamulizumab.
Patient characteristics
Baseline characteristics were similar between the treatment arms.
| Mogamulizumab (n=186) | Vorinostat (n=186) | |
| Median age | 64 (range, 54-73) | 65 (range, 56-72) |
| Male | 109 (59%) | 107 (58%) |
| Female | 77 (41%) | 79 (42%) |
| MF | 105 (56%) | 99 (53%) |
| SS | 81 (44%) | 87 (47%) |
| Time from diagnosis, months | 41.0 (range, 17.4-78.8) | 35.4 (range, 16.2-68.2) |
| Median number of previous systemic regimens | 3 (range, 2-5) | 3 (range, 2-5) |
PFS and ORR
Mogamulizumab provided a significant improvement in PFS, the study’s primary endpoint.
According to investigators, the median PFS was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio=0.53, P<0.0001).
According to independent review, the median PFS was 6.7 months and 3.8 months, respectively (hazard ratio=0.64, P<0.0007).
There was a significant improvement in ORR with mogamulizumab.
According to independent review, the global ORR was 23% (43/186) in the mogamulizumab arm and 4% (7/186) in the vorinostat arm (risk ratio=19.4, P<0.0001).
According to investigators, the global ORR was 28% (52/186) and 5% (9/186), respectively (risk ratio=23.1, P<0.0001).
For patients with MF, the investigator-assessed ORR was 21% (22/105) with mogamulizumab and 7% (7/99) with vorinostat.
For SS patients, the investigator-assessed ORR was 37% (30/81) and 2% (2/87), respectively.
Responses by disease compartment were superior with mogamulizumab as well.
The investigator-assessed blood ORR was 68% (83/122) with mogamulizumab and 19% (23/123) with vorinostat. The skin ORR was 42% (78/186) and 16% (29/186), respectively.
The lymph node ORR was 17% (21/124) and 4% (5/122), respectively. The viscera ORR was 0% in both arms.
Crossover
Among patients who crossed over from vorinostat to mogamulizumab, the ORR was 31% (41/133). In these patients, the median PFS was 8.9 months.
In the 319 patients who were assigned to mogamulizumab or crossed over to that arm, the median PFS was 8.4 months.
Safety
The most common treatment-emergent, grade 1-2 AEs, occurring in at least 20% of patients in either arm (mogamulizumab and vorinostat, respectively), were:
- Thrombocytopenia (14% vs 34%)
- Diarrhea (23% vs 57%)
- Nausea (15% vs 41%)
- Fatigue (22% vs 32%)
- Increased blood creatinine (3% vs 28%)
- Decreased appetite (7% vs 24%)
- Dysgeusia (3% vs 28%)
- Drug eruptions (20% vs 1%)
- Infusion-related reactions (32% vs 1%).
Grade 3 AEs in the mogamulizumab arm included drug eruptions (n=8), hypertension (n=8), pneumonia (n=6), fatigue (n=3), cellulitis (n=3), infusion-related reactions (n=3), sepsis (n=2), decreased appetite (n=2), AST increase (n=2), weight decrease (n=1), pyrexia (n=1), constipation (n=1), nausea (n=1), and diarrhea (n=1).
Grade 4 AEs with mogamulizumab were cellulitis (n=1) and pneumonia (n=1). Grade 5 AEs included pneumonia (n=1) and sepsis (n=1).
Groups release guidelines for CAR T treatment in children
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.

Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.

There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.
Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.
There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
emphasize the need for a flexible approach to detect early signs of serious complications for younger patients treated with this emerging class of medicines.
Researchers at the University of Texas MD Anderson Cancer Center, Houston, and the Pediatric Acute Lung Injury and Sepsis Investigators Network (PALISI) developed the guidelines, which were published in Nature Reviews Clinical Oncology. The recommendations build on the guidelines for more general use of these medicines from MD Anderson’s CARTOX Program, which Nature Reviews Clinical Oncology published in 2017.
Among the chief concerns with this new class of medicines are cytokine-release syndrome (CRS) and CAR T cell-related encephalopathy syndrome (CRES), according to Kris Michael Mahadeo, MD MPH, of the MD Anderson Cancer Center and his coauthors of the new paper.
Some of the tools used for older patients in screening for complications with CAR T drugs don’t work as well with younger ones, Dr. Mahadeo said in an interview. For instance, at MD Anderson, a handwriting sample is used to monitor patients for CAR T cell-related encephalopathy syndrome, which has symptoms of confusion and delirium. Patients provide a baseline handwriting sample of a single sentence that’s scanned into the medical record, and then they are asked to write this again during their time in the hospital, he said. But this tool may not work for children too young to write well.
The new guidelines suggest using the Cornell Assessment of Pediatric Delirium (CAPD) or to evaluate a child’s mental state, asking questions about eye contact, and level of awareness and mood, Dr. Mahadeo said. An alternative for patients aged 12 years and older with greater cognitive ability is the CARTOX-10 grading system.
“The nurses who spent most of the day with these patients will observe them over their shift and kind of get an idea of what was normal and answer a series of questions” through the CAPD tool, which is already used in ICUs, Dr. Mahadeo said. “It takes into consideration both the nurses’ perception and the parents, or whoever is at the bedside with the child. So that if they have a concern, it gives them a point that actually escalates things upward.”
The newly published recommendations also remind physicians and others caring for young patients to pay attention to these reports.
“Parent and/or caregiver concerns should be addressed because early signs or symptoms of CRS can be subtle and best recognized by those who know the child best,” Dr. Mahadeo and his colleagues wrote in a summary of key recommendations in the paper.
The recommendations also noted a need for close monitoring for complications such as hypotension, hypocalcemia, and catheter-related pain in young patients who require a leukapheresis catheter for cell collection. Infant and younger children “might not verbalize these symptoms,” according to the researchers.
Other recommendations include:
- Obtaining the child’s assent when appropriate, with psychological services often aiding in this goal. Dr. Mahadeo and his colleagues recommend considering “age-appropriate advance directives.”
- Maintaining high vigilance for sinus tachycardia as an early sign of CRS, using age-specific normal range or baseline values.
- Giving pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
- Considering participation with a prospective collaboration with intensive-care registries that could allow accurate data entry of cell-therapy variables into the Center for International Blood and Marrow Transplant Research registry by cell-therapy programs.
The Food and Drug Administration approved the first two CAR T-cell therapies in the United States in 2017: Novartis’ tisagenlecleucel (Kymriah) for children and young adults with B-cell precursor acute lymphoblastic leukemia and later for adults with large B-cell lymphoma; and axicabtagene ciloleucel (Yescarta), sold by Gilead, for adults with large B-cell lymphoma. The therapies involve reengineering a patient’s T cells such that they recognize the threat of cancer, and then introducing them back into the body. The European Medicines Agency’s Committee for Medicinal Products for Human Use in June recommended granting marketing authorization to these drugs.
In the new pediatric guidelines, Dr. Mahadeo and his colleagues noted the use of CAR T-cell therapies for treatment of solid tumors and other malignancies in children already “is being explored.” “Moreover, consideration of earlier or upfront use of CAR T-cell therapy might spare patients the acute and long-term toxicities associated with traditional chemotherapy and/or radiation regimens,” they wrote.
There’s been great interest in learning how to most safely use the CAR T cell therapies, said Helen Heslop, MD, of Baylor College of Medicine.
She pointed to a 2014 publication in the journal Blood from Daniel W. Lee and his colleagues as an earlier example of this research. By now, cancer centers will have worked out their own procedures for pediatric use of CAR T therapies, hewing to standards set by the Foundation for the Accreditation of Cellular Therapy (FACT), Dr. Heslop said.
Dr. Heslop also stressed the role of the FDA in requiring risk evaluation and management strategy programs for these drugs. All of this, including the new guidelines from Dr. Mahadeo and his colleagues, is part of a growing body of research into safe use of CAR T therapies, Dr. Heslop said.
“It’s an active area of research,” she said. “Most centers will look at all of it and then develop what works best in their own individual center for providing the best care for the patients.”
The newly published guidelines could prove an “important contribution” to managing the risk of CAR T therapies, Phyllis I. Warkentin, MD, chief medical officer for FACT, said in an interview, while stressing that they were not more or less important than other similar efforts. Physicians learning how to use the CAR T therapies may welcome new input, as most of what’s been published has been about adults, she said.
“You don’t have the luxury of a lot of time to be learning on the job, so to speak,” with CAR T therapies, she said. “Many of the toxicities are fairly severe and fairly sudden.”
Dr. Heslop has been on advisory board for Gilead and Novartis. Dr. Warkentin and Dr. Mahadeo each reported having no financial disclosures. Other authors of the guidelines paper reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
SOURCE: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
FROM NATURE REVIEWS CLINICAL ONCOLOGY
Key clinical point: Multidisciplinary approach aids in managing CAR T-cell therapy’s severe potential toxicities in children.
Major finding: The guideline calls for pediatric dosing of tocilizumab, with patients weighing less than 30 kg receiving 12 mg/kg, and those weighing 30 kg or greater receiving 8 mg/kg.
Study details: Consensus guidelines on the care of children receiving CAR T-cell therapy from the Pediatric Acute Lung Injury and Sepsis Investigators and the MD Anderson Cancer Center CARTOX program.
Disclosures: Dr. Mahadeo reported having no financial disclosures. Other coauthors reported a patent with applications in the field of gene-modified T cell therapy for cancer, as well as financial ties to Cellectis, NexImmune, Torque Pharma, Kite Pharma (a Gilead company), Poseida Therapeutics, Celgene, Novartis, and Unum Therapeutics.
Source: Mahadeo KM et al. Nat Rev Clin Oncol. 2018 Aug 6. doi: 10.1038/s41571-018-0075-2.
Team recommends melanoma screening in CLL

Patients with chronic lymphocytic leukemia (CLL) should be routinely monitored for melanoma, according to researchers.
A study of 470 CLL patients showed they have a significantly higher risk of invasive melanoma than the general population.
Most of the melanomas reported in this study were detected via routine surveillance, and most were discovered before they reached an advanced stage.
Clive Zent, MD, of Wilmot Cancer Institute at the University of Rochester Medical Center in Rochester, New York, and his colleagues described this study in Leukemia Research.
The researchers analyzed data on 470 CLL patients followed for 2849 person-years. Eighteen of these patients developed 22 melanomas. This included 14 cases of invasive melanoma in 13 patients.
The rate of invasive melanoma was significantly higher in this CLL cohort than the rate observed in the age- and sex-matched general population. The standardized incidence ratio was 6.32.
“We do not for sure know why CLL patients are more susceptible to melanoma, but the most likely cause is a suppressed immune system,” Dr Zent noted.
“Normally, in people with healthy immune systems, malignant skin cells might be detected and destroyed before they become a problem. But in CLL patients, failure of this control system increases the rate at which cancer cells can grow into tumors and also the likelihood that they will become invasive or spread to distant sites.”
Detection and management
Fifteen of the 22 melanomas (68.2%) in the CLL cohort were detected via surveillance in a dermatology clinic, and 2 (9.1%) were detected at the CLL/lymphoma clinic.
Three cases of melanoma (14.3%) were detected within the first year of a patient’s CLL diagnosis.
Seven melanomas (33.3%) were detected at pathologic stage 0, 8 (38.1%) at stage I, 2 (9.5%) at stage II, 3 (14.3%) at stage III, and 1 (4.8%) at stage IV. Detailed data were not available for the remaining case.
Melanomas were managed with wide local excision (n=19), sentinel node biopsies (n=6), Mohs surgery (n=1), drugs (n=2), palliative care (n=1), and comfort care (n=1).
The 4 patients who received drugs, palliative care, or comfort care had advanced melanoma.
The patient who received palliative care was still alive at 2.4 years of follow-up. The patient who received comfort care died of metastatic melanoma 1.4 years after diagnosis.
The third patient with advanced melanoma received 2 cycles of dacarbazine and palliative radiation to lung and brain metastases. This patient died 3.6 years after melanoma diagnosis.
The fourth patient received ipilimumab for the melanoma while also receiving ibrutinib to treat her CLL. When the ipilimumab failed, the patient proceeded to pembrolizumab and achieved a near-complete response within 3 months. Then, an intensely hypermetabolic abdominal node was detected and successfully treated with radiation.
The patient continued on pembrolizumab, and her melanoma was in sustained remission at last follow-up, after 23 cycles of pembrolizumab. Her CLL was still responding to ibrutinib at that point as well.
Based on these data, Dr Zent and his colleagues recommend routine melanoma screening for CLL patients. The team believes such surveillance might decrease morbidity and mortality in these patients, although more research is needed to confirm this theory.
Patients with chronic lymphocytic leukemia (CLL) should be routinely monitored for melanoma, according to researchers.
A study of 470 CLL patients showed they have a significantly higher risk of invasive melanoma than the general population.
Most of the melanomas reported in this study were detected via routine surveillance, and most were discovered before they reached an advanced stage.
Clive Zent, MD, of Wilmot Cancer Institute at the University of Rochester Medical Center in Rochester, New York, and his colleagues described this study in Leukemia Research.
The researchers analyzed data on 470 CLL patients followed for 2849 person-years. Eighteen of these patients developed 22 melanomas. This included 14 cases of invasive melanoma in 13 patients.
The rate of invasive melanoma was significantly higher in this CLL cohort than the rate observed in the age- and sex-matched general population. The standardized incidence ratio was 6.32.
“We do not for sure know why CLL patients are more susceptible to melanoma, but the most likely cause is a suppressed immune system,” Dr Zent noted.
“Normally, in people with healthy immune systems, malignant skin cells might be detected and destroyed before they become a problem. But in CLL patients, failure of this control system increases the rate at which cancer cells can grow into tumors and also the likelihood that they will become invasive or spread to distant sites.”
Detection and management
Fifteen of the 22 melanomas (68.2%) in the CLL cohort were detected via surveillance in a dermatology clinic, and 2 (9.1%) were detected at the CLL/lymphoma clinic.
Three cases of melanoma (14.3%) were detected within the first year of a patient’s CLL diagnosis.
Seven melanomas (33.3%) were detected at pathologic stage 0, 8 (38.1%) at stage I, 2 (9.5%) at stage II, 3 (14.3%) at stage III, and 1 (4.8%) at stage IV. Detailed data were not available for the remaining case.
Melanomas were managed with wide local excision (n=19), sentinel node biopsies (n=6), Mohs surgery (n=1), drugs (n=2), palliative care (n=1), and comfort care (n=1).
The 4 patients who received drugs, palliative care, or comfort care had advanced melanoma.
The patient who received palliative care was still alive at 2.4 years of follow-up. The patient who received comfort care died of metastatic melanoma 1.4 years after diagnosis.
The third patient with advanced melanoma received 2 cycles of dacarbazine and palliative radiation to lung and brain metastases. This patient died 3.6 years after melanoma diagnosis.
The fourth patient received ipilimumab for the melanoma while also receiving ibrutinib to treat her CLL. When the ipilimumab failed, the patient proceeded to pembrolizumab and achieved a near-complete response within 3 months. Then, an intensely hypermetabolic abdominal node was detected and successfully treated with radiation.
The patient continued on pembrolizumab, and her melanoma was in sustained remission at last follow-up, after 23 cycles of pembrolizumab. Her CLL was still responding to ibrutinib at that point as well.
Based on these data, Dr Zent and his colleagues recommend routine melanoma screening for CLL patients. The team believes such surveillance might decrease morbidity and mortality in these patients, although more research is needed to confirm this theory.
Patients with chronic lymphocytic leukemia (CLL) should be routinely monitored for melanoma, according to researchers.
A study of 470 CLL patients showed they have a significantly higher risk of invasive melanoma than the general population.
Most of the melanomas reported in this study were detected via routine surveillance, and most were discovered before they reached an advanced stage.
Clive Zent, MD, of Wilmot Cancer Institute at the University of Rochester Medical Center in Rochester, New York, and his colleagues described this study in Leukemia Research.
The researchers analyzed data on 470 CLL patients followed for 2849 person-years. Eighteen of these patients developed 22 melanomas. This included 14 cases of invasive melanoma in 13 patients.
The rate of invasive melanoma was significantly higher in this CLL cohort than the rate observed in the age- and sex-matched general population. The standardized incidence ratio was 6.32.
“We do not for sure know why CLL patients are more susceptible to melanoma, but the most likely cause is a suppressed immune system,” Dr Zent noted.
“Normally, in people with healthy immune systems, malignant skin cells might be detected and destroyed before they become a problem. But in CLL patients, failure of this control system increases the rate at which cancer cells can grow into tumors and also the likelihood that they will become invasive or spread to distant sites.”
Detection and management
Fifteen of the 22 melanomas (68.2%) in the CLL cohort were detected via surveillance in a dermatology clinic, and 2 (9.1%) were detected at the CLL/lymphoma clinic.
Three cases of melanoma (14.3%) were detected within the first year of a patient’s CLL diagnosis.
Seven melanomas (33.3%) were detected at pathologic stage 0, 8 (38.1%) at stage I, 2 (9.5%) at stage II, 3 (14.3%) at stage III, and 1 (4.8%) at stage IV. Detailed data were not available for the remaining case.
Melanomas were managed with wide local excision (n=19), sentinel node biopsies (n=6), Mohs surgery (n=1), drugs (n=2), palliative care (n=1), and comfort care (n=1).
The 4 patients who received drugs, palliative care, or comfort care had advanced melanoma.
The patient who received palliative care was still alive at 2.4 years of follow-up. The patient who received comfort care died of metastatic melanoma 1.4 years after diagnosis.
The third patient with advanced melanoma received 2 cycles of dacarbazine and palliative radiation to lung and brain metastases. This patient died 3.6 years after melanoma diagnosis.
The fourth patient received ipilimumab for the melanoma while also receiving ibrutinib to treat her CLL. When the ipilimumab failed, the patient proceeded to pembrolizumab and achieved a near-complete response within 3 months. Then, an intensely hypermetabolic abdominal node was detected and successfully treated with radiation.
The patient continued on pembrolizumab, and her melanoma was in sustained remission at last follow-up, after 23 cycles of pembrolizumab. Her CLL was still responding to ibrutinib at that point as well.
Based on these data, Dr Zent and his colleagues recommend routine melanoma screening for CLL patients. The team believes such surveillance might decrease morbidity and mortality in these patients, although more research is needed to confirm this theory.
Frequent BCCs linked to blood cancers

New research suggests people who develop frequent cases of basal cell carcinoma (BCC) have an increased risk of leukemias, lymphomas, and other cancers.
“We discovered that people who develop 6 or more basal cell carcinomas during a 10-year period are about 3 times more likely than the general population to develop other, unrelated cancers,” said Kavita Sarin, MD, PhD, of Stanford University School of Medicine in California.
“We’re hopeful that this finding could be a way to identify people at an increased risk for a life-threatening malignancy before those cancers develop.”
Dr Sarin and her colleagues reported their findings in JCI Insight.
Stanford cohort
The researchers first studied 61 patients treated at Stanford Health Care for unusually frequent BCCs—an average of 11 per patient over a 10-year period. The team investigated whether these patients may have mutations in 29 genes that code for DNA damage repair proteins.
“We found that about 20% of the people with frequent basal cell carcinomas have a mutation in one of the genes responsible for repairing DNA damage, versus about 3% of the general population,” Dr Sarin said. “That’s shockingly high.”
Specifically, there were 12 BCC patients (19.7%) who had 13 pathogenic mutations in 12 genes—APC, BARD1, BRCA1, BRCA2, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, and PALB2. And 3.0% of non-Finnish European subjects in the Exome Aggregation Consortium had pathogenic mutations in these 12 genes.
Furthermore, 21 of the 61 BCC patients (64.4%) had a history of additional cancers. This included 5 hematologic malignancies (leukemia/lymphoma), 5 invasive melanomas, and 2 breast, 2 colon, and 5 prostate cancers.
When the researchers compared the cancer prevalence in these patients to the Surveillance, Epidemiology, and End Results-estimated prevalence of cancer in the 60- to 69-year-old population of European descent, the BCC cohort had an increased risk of any cancer—a relative risk (RR) of 3.5 (P<0.001).
The RR was 3.5 for leukemia and lymphoma (P=0.004), 11.9 for invasive melanoma (P<0.001), 4.5 for colon cancer (P=0.030), 5.6 for breast cancer (P=0.009), and 4.7 for prostate cancer (P<0.001).
Insurance cohort
To confirm the findings in the Stanford cohort, the researchers applied a similar analysis to a large medical insurance claims database, Truven MarketScan.
The database contained 111,562 patients with 1 case of BCC, 13,264 patients with 6 or more BCCs, and 2920 patients with 12 or more BCCs. Truven patients with no history of BCC served as controls.
The researchers adjusted for age and sex and found that patients with 1 BCC, 6 or more BCCs, and 12 or more BCCs had an increased risk of any cancer compared to controls.
The odds ratio (OR) for any cancer was 1.61 for patients with 1 BCC, 3.12 for those with 6 or more BCCs, and 4.15 for patients with 12 or more BCCs.
The OR for Hodgkin lymphoma was 2.27 for patients with 1 BCC, 8.94 for patients with 6 or more BCCs, and 15.41 for patients with 12 or more BCCs.
The OR for non-Hodgkin lymphoma was 1.40 for patients with 1 BCC, 2.59 for patients with 6 or more BCCs, and 3.10 for patients with 12 or more BCCs.
The OR for leukemia was 1.76 for patients with 1 BCC, 3.23 for patients with 6 or more BCCs, and 5.78 for patients with 12 or more BCCs.
The researchers pointed out that, the more BCCs an individual had, the more likely that person was to have had other cancers as well.
“I was surprised to see such a strong correlation, but it’s also very gratifying,” Dr Sarin said. “Now, we can ask patients with repeated basal cell carcinomas whether they have family members with other types of cancers and perhaps suggest that they consider genetic testing and increased screening.”
The researchers are continuing to enroll Stanford patients in their study to learn whether particular mutations in genes responsible for repairing DNA damage are linked to the development of specific malignancies. The team would also like to conduct a similar study in patients with frequent melanomas.
The current study was supported by the Dermatology Foundation, the Stanford Society of Physician Scholars, the American Skin Association, and Pellepharm Inc.
New research suggests people who develop frequent cases of basal cell carcinoma (BCC) have an increased risk of leukemias, lymphomas, and other cancers.
“We discovered that people who develop 6 or more basal cell carcinomas during a 10-year period are about 3 times more likely than the general population to develop other, unrelated cancers,” said Kavita Sarin, MD, PhD, of Stanford University School of Medicine in California.
“We’re hopeful that this finding could be a way to identify people at an increased risk for a life-threatening malignancy before those cancers develop.”
Dr Sarin and her colleagues reported their findings in JCI Insight.
Stanford cohort
The researchers first studied 61 patients treated at Stanford Health Care for unusually frequent BCCs—an average of 11 per patient over a 10-year period. The team investigated whether these patients may have mutations in 29 genes that code for DNA damage repair proteins.
“We found that about 20% of the people with frequent basal cell carcinomas have a mutation in one of the genes responsible for repairing DNA damage, versus about 3% of the general population,” Dr Sarin said. “That’s shockingly high.”
Specifically, there were 12 BCC patients (19.7%) who had 13 pathogenic mutations in 12 genes—APC, BARD1, BRCA1, BRCA2, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, and PALB2. And 3.0% of non-Finnish European subjects in the Exome Aggregation Consortium had pathogenic mutations in these 12 genes.
Furthermore, 21 of the 61 BCC patients (64.4%) had a history of additional cancers. This included 5 hematologic malignancies (leukemia/lymphoma), 5 invasive melanomas, and 2 breast, 2 colon, and 5 prostate cancers.
When the researchers compared the cancer prevalence in these patients to the Surveillance, Epidemiology, and End Results-estimated prevalence of cancer in the 60- to 69-year-old population of European descent, the BCC cohort had an increased risk of any cancer—a relative risk (RR) of 3.5 (P<0.001).
The RR was 3.5 for leukemia and lymphoma (P=0.004), 11.9 for invasive melanoma (P<0.001), 4.5 for colon cancer (P=0.030), 5.6 for breast cancer (P=0.009), and 4.7 for prostate cancer (P<0.001).
Insurance cohort
To confirm the findings in the Stanford cohort, the researchers applied a similar analysis to a large medical insurance claims database, Truven MarketScan.
The database contained 111,562 patients with 1 case of BCC, 13,264 patients with 6 or more BCCs, and 2920 patients with 12 or more BCCs. Truven patients with no history of BCC served as controls.
The researchers adjusted for age and sex and found that patients with 1 BCC, 6 or more BCCs, and 12 or more BCCs had an increased risk of any cancer compared to controls.
The odds ratio (OR) for any cancer was 1.61 for patients with 1 BCC, 3.12 for those with 6 or more BCCs, and 4.15 for patients with 12 or more BCCs.
The OR for Hodgkin lymphoma was 2.27 for patients with 1 BCC, 8.94 for patients with 6 or more BCCs, and 15.41 for patients with 12 or more BCCs.
The OR for non-Hodgkin lymphoma was 1.40 for patients with 1 BCC, 2.59 for patients with 6 or more BCCs, and 3.10 for patients with 12 or more BCCs.
The OR for leukemia was 1.76 for patients with 1 BCC, 3.23 for patients with 6 or more BCCs, and 5.78 for patients with 12 or more BCCs.
The researchers pointed out that, the more BCCs an individual had, the more likely that person was to have had other cancers as well.
“I was surprised to see such a strong correlation, but it’s also very gratifying,” Dr Sarin said. “Now, we can ask patients with repeated basal cell carcinomas whether they have family members with other types of cancers and perhaps suggest that they consider genetic testing and increased screening.”
The researchers are continuing to enroll Stanford patients in their study to learn whether particular mutations in genes responsible for repairing DNA damage are linked to the development of specific malignancies. The team would also like to conduct a similar study in patients with frequent melanomas.
The current study was supported by the Dermatology Foundation, the Stanford Society of Physician Scholars, the American Skin Association, and Pellepharm Inc.
New research suggests people who develop frequent cases of basal cell carcinoma (BCC) have an increased risk of leukemias, lymphomas, and other cancers.
“We discovered that people who develop 6 or more basal cell carcinomas during a 10-year period are about 3 times more likely than the general population to develop other, unrelated cancers,” said Kavita Sarin, MD, PhD, of Stanford University School of Medicine in California.
“We’re hopeful that this finding could be a way to identify people at an increased risk for a life-threatening malignancy before those cancers develop.”
Dr Sarin and her colleagues reported their findings in JCI Insight.
Stanford cohort
The researchers first studied 61 patients treated at Stanford Health Care for unusually frequent BCCs—an average of 11 per patient over a 10-year period. The team investigated whether these patients may have mutations in 29 genes that code for DNA damage repair proteins.
“We found that about 20% of the people with frequent basal cell carcinomas have a mutation in one of the genes responsible for repairing DNA damage, versus about 3% of the general population,” Dr Sarin said. “That’s shockingly high.”
Specifically, there were 12 BCC patients (19.7%) who had 13 pathogenic mutations in 12 genes—APC, BARD1, BRCA1, BRCA2, CDH1, CHEK2, MLH1, MSH2, MSH6, MUTYH, NBN, and PALB2. And 3.0% of non-Finnish European subjects in the Exome Aggregation Consortium had pathogenic mutations in these 12 genes.
Furthermore, 21 of the 61 BCC patients (64.4%) had a history of additional cancers. This included 5 hematologic malignancies (leukemia/lymphoma), 5 invasive melanomas, and 2 breast, 2 colon, and 5 prostate cancers.
When the researchers compared the cancer prevalence in these patients to the Surveillance, Epidemiology, and End Results-estimated prevalence of cancer in the 60- to 69-year-old population of European descent, the BCC cohort had an increased risk of any cancer—a relative risk (RR) of 3.5 (P<0.001).
The RR was 3.5 for leukemia and lymphoma (P=0.004), 11.9 for invasive melanoma (P<0.001), 4.5 for colon cancer (P=0.030), 5.6 for breast cancer (P=0.009), and 4.7 for prostate cancer (P<0.001).
Insurance cohort
To confirm the findings in the Stanford cohort, the researchers applied a similar analysis to a large medical insurance claims database, Truven MarketScan.
The database contained 111,562 patients with 1 case of BCC, 13,264 patients with 6 or more BCCs, and 2920 patients with 12 or more BCCs. Truven patients with no history of BCC served as controls.
The researchers adjusted for age and sex and found that patients with 1 BCC, 6 or more BCCs, and 12 or more BCCs had an increased risk of any cancer compared to controls.
The odds ratio (OR) for any cancer was 1.61 for patients with 1 BCC, 3.12 for those with 6 or more BCCs, and 4.15 for patients with 12 or more BCCs.
The OR for Hodgkin lymphoma was 2.27 for patients with 1 BCC, 8.94 for patients with 6 or more BCCs, and 15.41 for patients with 12 or more BCCs.
The OR for non-Hodgkin lymphoma was 1.40 for patients with 1 BCC, 2.59 for patients with 6 or more BCCs, and 3.10 for patients with 12 or more BCCs.
The OR for leukemia was 1.76 for patients with 1 BCC, 3.23 for patients with 6 or more BCCs, and 5.78 for patients with 12 or more BCCs.
The researchers pointed out that, the more BCCs an individual had, the more likely that person was to have had other cancers as well.
“I was surprised to see such a strong correlation, but it’s also very gratifying,” Dr Sarin said. “Now, we can ask patients with repeated basal cell carcinomas whether they have family members with other types of cancers and perhaps suggest that they consider genetic testing and increased screening.”
The researchers are continuing to enroll Stanford patients in their study to learn whether particular mutations in genes responsible for repairing DNA damage are linked to the development of specific malignancies. The team would also like to conduct a similar study in patients with frequent melanomas.
The current study was supported by the Dermatology Foundation, the Stanford Society of Physician Scholars, the American Skin Association, and Pellepharm Inc.
Increased B-cell lymphoma risk with JAK1/2 inhibitors
Patients with myeloproliferative neoplasms treated with Janus-kinase (JAK) 1/2 inhibitors may be at significantly increased risk of aggressive B cell non-Hodgkin lymphomas, according to a study published in Blood.
A retrospective cohort study of 626 Viennese patients with myeloproliferative neoplasms – 69 of whom were treated with JAK1/2 inhibitors – found that 4 of the 69 patients (5.8%) developed aggressive B-cell lymphoma, compared with just 2 patients (0.36%) in the rest of the group. This represented a significant, 16-fold higher risk of aggressive B cell lymphoma associated with JAK1/2 inhibitor therapy (P = .0017).
The lymphoma was diagnosed within 13-35 months of starting JAK1/2 inhibitors. In three patients, the disease was in the bone marrow and peripheral blood, one patient had it in mammary tissue, and another had it in mucosal tissue. All four lymphomas showed positive MYC and p53 staining.
All four patients had been treated with ruxolitinib, one was also treated with fedratinib, and three of the four had been pretreated with alkylating agents.
Meanwhile, a second retrospective cohort study in Paris of 929 patients with myeloproliferative neoplasms, reported in the same paper, found that 3.51% of those treated with ruxolitinib developed lymphoma, compared with 0.23% of conventionally-treated patients.
Using archived bone marrow samples from 54 of the 69 patients treated with JAK1/2 inhibitors, researchers discovered that 15.9% of them – including three of the B-cell lymphoma patients (the fourth was not tested) – had a preexisting B cell clone. This was present as early as 47-70 months before the lymphoma diagnosis.
“In patients, the clonal B-cell population was present as long as 6 years before overt lymphoma and preceded JAK1/2 inhibition which offers the opportunity to determine patients at risk,” wrote Edit Porpaczy, MD, of the Comprehensive Cancer Center at the Medical University of Vienna, and her coauthors. “Targeted inhibition of JAK-STAT signaling appears to be required to trigger the appearance of the B-cell clone as other treatments eliminating the myeloid cell load in men do not exert a comparable effect.”
In the Viennese cohort, three of the lymphomas were aggressive CD19+ B-cell type, and the fourth was a nonspecified high-grade B-cell lymphoma.
Researchers also looked at the effects of JAK1/2 inhibition in STAT1-/- mice, and found that two-thirds developed a spontaneous myeloid hyperplasia with the concomitant presence of aberrant B-cells.
“Upon STAT1-deficiency myeloid hyperplasia is paralleled by the occurrence of a malignant B-cell clone, which evolves into disease upon bone-marrow transplantation and gives rise to a leukemic lymphoma phenotype,” the authors wrote.
The study was supported by the Austrian Science Fund, the Anniversary Fund of the Austrian National Bank and the WWTF Precision Medicine Program. Several authors reported support, funding or advisory board positions with the pharmaceutical industry.
SOURCE: Porpaczy E et al. Blood. 2018 Jun 14. doi: 10.1182/blood-2017-10-810739.
Patients with myeloproliferative neoplasms treated with Janus-kinase (JAK) 1/2 inhibitors may be at significantly increased risk of aggressive B cell non-Hodgkin lymphomas, according to a study published in Blood.
A retrospective cohort study of 626 Viennese patients with myeloproliferative neoplasms – 69 of whom were treated with JAK1/2 inhibitors – found that 4 of the 69 patients (5.8%) developed aggressive B-cell lymphoma, compared with just 2 patients (0.36%) in the rest of the group. This represented a significant, 16-fold higher risk of aggressive B cell lymphoma associated with JAK1/2 inhibitor therapy (P = .0017).
The lymphoma was diagnosed within 13-35 months of starting JAK1/2 inhibitors. In three patients, the disease was in the bone marrow and peripheral blood, one patient had it in mammary tissue, and another had it in mucosal tissue. All four lymphomas showed positive MYC and p53 staining.
All four patients had been treated with ruxolitinib, one was also treated with fedratinib, and three of the four had been pretreated with alkylating agents.
Meanwhile, a second retrospective cohort study in Paris of 929 patients with myeloproliferative neoplasms, reported in the same paper, found that 3.51% of those treated with ruxolitinib developed lymphoma, compared with 0.23% of conventionally-treated patients.
Using archived bone marrow samples from 54 of the 69 patients treated with JAK1/2 inhibitors, researchers discovered that 15.9% of them – including three of the B-cell lymphoma patients (the fourth was not tested) – had a preexisting B cell clone. This was present as early as 47-70 months before the lymphoma diagnosis.
“In patients, the clonal B-cell population was present as long as 6 years before overt lymphoma and preceded JAK1/2 inhibition which offers the opportunity to determine patients at risk,” wrote Edit Porpaczy, MD, of the Comprehensive Cancer Center at the Medical University of Vienna, and her coauthors. “Targeted inhibition of JAK-STAT signaling appears to be required to trigger the appearance of the B-cell clone as other treatments eliminating the myeloid cell load in men do not exert a comparable effect.”
In the Viennese cohort, three of the lymphomas were aggressive CD19+ B-cell type, and the fourth was a nonspecified high-grade B-cell lymphoma.
Researchers also looked at the effects of JAK1/2 inhibition in STAT1-/- mice, and found that two-thirds developed a spontaneous myeloid hyperplasia with the concomitant presence of aberrant B-cells.
“Upon STAT1-deficiency myeloid hyperplasia is paralleled by the occurrence of a malignant B-cell clone, which evolves into disease upon bone-marrow transplantation and gives rise to a leukemic lymphoma phenotype,” the authors wrote.
The study was supported by the Austrian Science Fund, the Anniversary Fund of the Austrian National Bank and the WWTF Precision Medicine Program. Several authors reported support, funding or advisory board positions with the pharmaceutical industry.
SOURCE: Porpaczy E et al. Blood. 2018 Jun 14. doi: 10.1182/blood-2017-10-810739.
Patients with myeloproliferative neoplasms treated with Janus-kinase (JAK) 1/2 inhibitors may be at significantly increased risk of aggressive B cell non-Hodgkin lymphomas, according to a study published in Blood.
A retrospective cohort study of 626 Viennese patients with myeloproliferative neoplasms – 69 of whom were treated with JAK1/2 inhibitors – found that 4 of the 69 patients (5.8%) developed aggressive B-cell lymphoma, compared with just 2 patients (0.36%) in the rest of the group. This represented a significant, 16-fold higher risk of aggressive B cell lymphoma associated with JAK1/2 inhibitor therapy (P = .0017).
The lymphoma was diagnosed within 13-35 months of starting JAK1/2 inhibitors. In three patients, the disease was in the bone marrow and peripheral blood, one patient had it in mammary tissue, and another had it in mucosal tissue. All four lymphomas showed positive MYC and p53 staining.
All four patients had been treated with ruxolitinib, one was also treated with fedratinib, and three of the four had been pretreated with alkylating agents.
Meanwhile, a second retrospective cohort study in Paris of 929 patients with myeloproliferative neoplasms, reported in the same paper, found that 3.51% of those treated with ruxolitinib developed lymphoma, compared with 0.23% of conventionally-treated patients.
Using archived bone marrow samples from 54 of the 69 patients treated with JAK1/2 inhibitors, researchers discovered that 15.9% of them – including three of the B-cell lymphoma patients (the fourth was not tested) – had a preexisting B cell clone. This was present as early as 47-70 months before the lymphoma diagnosis.
“In patients, the clonal B-cell population was present as long as 6 years before overt lymphoma and preceded JAK1/2 inhibition which offers the opportunity to determine patients at risk,” wrote Edit Porpaczy, MD, of the Comprehensive Cancer Center at the Medical University of Vienna, and her coauthors. “Targeted inhibition of JAK-STAT signaling appears to be required to trigger the appearance of the B-cell clone as other treatments eliminating the myeloid cell load in men do not exert a comparable effect.”
In the Viennese cohort, three of the lymphomas were aggressive CD19+ B-cell type, and the fourth was a nonspecified high-grade B-cell lymphoma.
Researchers also looked at the effects of JAK1/2 inhibition in STAT1-/- mice, and found that two-thirds developed a spontaneous myeloid hyperplasia with the concomitant presence of aberrant B-cells.
“Upon STAT1-deficiency myeloid hyperplasia is paralleled by the occurrence of a malignant B-cell clone, which evolves into disease upon bone-marrow transplantation and gives rise to a leukemic lymphoma phenotype,” the authors wrote.
The study was supported by the Austrian Science Fund, the Anniversary Fund of the Austrian National Bank and the WWTF Precision Medicine Program. Several authors reported support, funding or advisory board positions with the pharmaceutical industry.
SOURCE: Porpaczy E et al. Blood. 2018 Jun 14. doi: 10.1182/blood-2017-10-810739.
FROM BLOOD
Key clinical point:
Major finding: Patients with myeloproliferative neoplasms treated with JAK1/2 inhibitors have a 16-fold higher incidence of lymphoma.
Study details: A retrospective cohort study of 626 patients with myeloproliferative neoplasms.
Disclosures: The study was supported by the Austrian Science Fund, the Anniversary Fund of the Austrian National Bank, and the WWTF Precision Medicine Program. Several authors reported support, funding, or advisory board positions with the pharmaceutical industry.
Source: Porpaczy E et al. Blood. 2018 Jun 14. doi: 10.1182/blood-2017-10-810739.
Company narrows focus of development for tazemetostat

Epizyme, Inc., has announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with diffuse large B-cell lymphoma (DLBCL).
However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Tazemetostat is an EZH2 inhibitor being developed to treat multiple hematologic and solid tumor malignancies.
Epizyme has been conducting a phase 1/2 trial of tazemetostat in patients with relapsed and/or refractory DLBCL as well as other B-cell lymphomas and solid tumors (NCT01897571).
The trial includes DLBCL patients with and without EZH2 activating mutations. Some patients were assigned to receive tazemetostat monotherapy, and some were assigned to tazemetostat in combination with prednisolone.
Epizyme has conducted an interim assessment of data from this trial and concluded that the clinical activity observed “is not sufficient to warrant further development of tazemetostat in DLBCL as a monotherapy or in combination with prednisolone.”
Epizyme said it plans to present data from this trial at a medical meeting in the second half of 2018.
The company is still conducting other studies of tazemetostat in patients with DLBCL.
In one study (NCT02889523), Epizyme and the Lymphoma Academic Research Organisation are evaluating tazemetostat in combination with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisolone) in patients with newly diagnosed DLBCL.
In another study (NCT03028103), Epizyme is evaluating tazemetostat in combination with fluconazole or omeprazole and repaglinide in patients with relapsed/refractory DLBCL, other B-cell lymphomas, or solid tumor malignancies.
Epizyme, Inc., has announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with diffuse large B-cell lymphoma (DLBCL).
However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Tazemetostat is an EZH2 inhibitor being developed to treat multiple hematologic and solid tumor malignancies.
Epizyme has been conducting a phase 1/2 trial of tazemetostat in patients with relapsed and/or refractory DLBCL as well as other B-cell lymphomas and solid tumors (NCT01897571).
The trial includes DLBCL patients with and without EZH2 activating mutations. Some patients were assigned to receive tazemetostat monotherapy, and some were assigned to tazemetostat in combination with prednisolone.
Epizyme has conducted an interim assessment of data from this trial and concluded that the clinical activity observed “is not sufficient to warrant further development of tazemetostat in DLBCL as a monotherapy or in combination with prednisolone.”
Epizyme said it plans to present data from this trial at a medical meeting in the second half of 2018.
The company is still conducting other studies of tazemetostat in patients with DLBCL.
In one study (NCT02889523), Epizyme and the Lymphoma Academic Research Organisation are evaluating tazemetostat in combination with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisolone) in patients with newly diagnosed DLBCL.
In another study (NCT03028103), Epizyme is evaluating tazemetostat in combination with fluconazole or omeprazole and repaglinide in patients with relapsed/refractory DLBCL, other B-cell lymphomas, or solid tumor malignancies.
Epizyme, Inc., has announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with diffuse large B-cell lymphoma (DLBCL).
However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Tazemetostat is an EZH2 inhibitor being developed to treat multiple hematologic and solid tumor malignancies.
Epizyme has been conducting a phase 1/2 trial of tazemetostat in patients with relapsed and/or refractory DLBCL as well as other B-cell lymphomas and solid tumors (NCT01897571).
The trial includes DLBCL patients with and without EZH2 activating mutations. Some patients were assigned to receive tazemetostat monotherapy, and some were assigned to tazemetostat in combination with prednisolone.
Epizyme has conducted an interim assessment of data from this trial and concluded that the clinical activity observed “is not sufficient to warrant further development of tazemetostat in DLBCL as a monotherapy or in combination with prednisolone.”
Epizyme said it plans to present data from this trial at a medical meeting in the second half of 2018.
The company is still conducting other studies of tazemetostat in patients with DLBCL.
In one study (NCT02889523), Epizyme and the Lymphoma Academic Research Organisation are evaluating tazemetostat in combination with R-CHOP (rituximab, cyclophosphamide, vincristine, doxorubicin, and prednisolone) in patients with newly diagnosed DLBCL.
In another study (NCT03028103), Epizyme is evaluating tazemetostat in combination with fluconazole or omeprazole and repaglinide in patients with relapsed/refractory DLBCL, other B-cell lymphomas, or solid tumor malignancies.
PET/CT accurately predicts MCL stage
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
REPORTING FROM CLINICAL LYMPHOMA, MYELOMA & LEUKEMIA
Key clinical point:
Major finding: If greater than 38% of the voxels demonstrated an standard uptake value of less than 0.95, there was a sensitivity of 100% and a specificity of 80%.
Study details: A retrospective cohort study of 23 patients with mantle cell leukemia and 5 controls.
Disclosures: There was no external funding for the study and the researchers reported having no financial disclosures.
Source: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Treatment improves PFS in early stage FL

A multidrug regimen can improve upon involved-field radiotherapy (IFRT) in patients with early stage follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
FL patients who received IFRT plus cyclophosphamide, vincristine, and prednisolone (CVP)—with or without rituximab—had a significant improvement in progression-free survival (PFS) compared to patients who received standard treatment with IFRT alone.
However, there was no significant difference in overall survival (OS) between the treatment arms.
“This is the first successful randomized study ever to be conducted in early stage follicular lymphoma comparing standard therapy to standard therapy plus effective chemotherapy or immunochemotherapy,” said Michael MacManus, MBBCh, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“It shows that the initial treatment received by patients can significantly affect their long-term chance of staying free from disease. Moving forward, we are interested in determining whether there is a benefit in overall long-term survival for patients treated with the combination with further follow-up, and if there is any way to predict if a person will benefit from combined treatment based on analyses of blood or biopsy specimens.”
Dr MacManus and his colleagues studied 150 patients with stage I to II, low-grade FL who were enrolled in this trial between 2000 and 2012.
At randomization, the patients’ median age was 57, 52% were male, 75% had stage I disease, and 48% had PET staging.
Half of patients (n=75) were randomized to receive IFRT (30-36 Gy) alone, and half were randomized to IFRT (30-36 Gy) plus 6 cycles of CVP. From 2006 on, patients in the CVP arm received rituximab (R) as well (n=31).
Baseline characteristics were well-balanced between the treatment arms.
Efficacy
The median follow-up was 9.6 years (range, 3.1 to 15.8 years).
PFS was significantly better among patients randomized to receive CVP±R (hazard ratio [HR]=0.57; P=0.033). The estimated 10-year PFS rate was 41% in the IFRT arm and 59% in the CVP±R arm.
Patients randomized to receive CVP plus R (n=31) had significantly better PFS than patients randomized to receive IFRT alone (n=31) over the same time period (HR=0.26; P=0.045).
There were 10 deaths in the IRFT arm and 5 in the CVP±R arm, but there was no significant difference in OS between the arms (HR=0.62; P=0.40). The 10-year OS rate was 86% in the IFRT arm and 95% in the CVP±R arm.
There was no significant between-arm difference in transformation to aggressive lymphoma (P=0.1). Transformation occurred in 10 patients in the IFRT arm and 4 in the CVP±R arm.
Safety
There were 148 patients from both arms who ultimately received IFRT, and 69 patients who received CVP±R.
Grade 2 toxicities occurring in more than 10% of IFRT recipients included upper gastrointestinal (n=27; 18%), skin (n=21; 14%), and mucous membrane (n=19; 12%) toxicity. One IFRT recipient had grade 3 mucositis, and 1 had grade 4 esophageal/pharyngeal mucosal toxicity.
Grade 3 toxicities occurring in at least 2 patients in the CVP±R arm included neutropenia (n=10; 14%), infection (n=8; 12%), diarrhea (n=3; 4%), elevated gamma-glutamyl transferase (n=3; 4%), fatigue (n=3; 4%), and febrile neutropenia (n=3; 4%).
Three patients (4%) in the CVP±R arm had acute grade 3 neuropathy related to vincristine. Ten patients (14%) had grade 4 neutropenia.
The most common late toxicities for the entire patient cohort were salivary gland (n=8; 5%) and skin (n=4; 3%) toxicities.
Grade 3 lung and menopausal toxicities occurred in 1 patient each. Two patients had late grade 3 vincristine neuropathy. One patient who had grade 3 neuropathy during chemotherapy progressed to grade 4.
A multidrug regimen can improve upon involved-field radiotherapy (IFRT) in patients with early stage follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
FL patients who received IFRT plus cyclophosphamide, vincristine, and prednisolone (CVP)—with or without rituximab—had a significant improvement in progression-free survival (PFS) compared to patients who received standard treatment with IFRT alone.
However, there was no significant difference in overall survival (OS) between the treatment arms.
“This is the first successful randomized study ever to be conducted in early stage follicular lymphoma comparing standard therapy to standard therapy plus effective chemotherapy or immunochemotherapy,” said Michael MacManus, MBBCh, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“It shows that the initial treatment received by patients can significantly affect their long-term chance of staying free from disease. Moving forward, we are interested in determining whether there is a benefit in overall long-term survival for patients treated with the combination with further follow-up, and if there is any way to predict if a person will benefit from combined treatment based on analyses of blood or biopsy specimens.”
Dr MacManus and his colleagues studied 150 patients with stage I to II, low-grade FL who were enrolled in this trial between 2000 and 2012.
At randomization, the patients’ median age was 57, 52% were male, 75% had stage I disease, and 48% had PET staging.
Half of patients (n=75) were randomized to receive IFRT (30-36 Gy) alone, and half were randomized to IFRT (30-36 Gy) plus 6 cycles of CVP. From 2006 on, patients in the CVP arm received rituximab (R) as well (n=31).
Baseline characteristics were well-balanced between the treatment arms.
Efficacy
The median follow-up was 9.6 years (range, 3.1 to 15.8 years).
PFS was significantly better among patients randomized to receive CVP±R (hazard ratio [HR]=0.57; P=0.033). The estimated 10-year PFS rate was 41% in the IFRT arm and 59% in the CVP±R arm.
Patients randomized to receive CVP plus R (n=31) had significantly better PFS than patients randomized to receive IFRT alone (n=31) over the same time period (HR=0.26; P=0.045).
There were 10 deaths in the IRFT arm and 5 in the CVP±R arm, but there was no significant difference in OS between the arms (HR=0.62; P=0.40). The 10-year OS rate was 86% in the IFRT arm and 95% in the CVP±R arm.
There was no significant between-arm difference in transformation to aggressive lymphoma (P=0.1). Transformation occurred in 10 patients in the IFRT arm and 4 in the CVP±R arm.
Safety
There were 148 patients from both arms who ultimately received IFRT, and 69 patients who received CVP±R.
Grade 2 toxicities occurring in more than 10% of IFRT recipients included upper gastrointestinal (n=27; 18%), skin (n=21; 14%), and mucous membrane (n=19; 12%) toxicity. One IFRT recipient had grade 3 mucositis, and 1 had grade 4 esophageal/pharyngeal mucosal toxicity.
Grade 3 toxicities occurring in at least 2 patients in the CVP±R arm included neutropenia (n=10; 14%), infection (n=8; 12%), diarrhea (n=3; 4%), elevated gamma-glutamyl transferase (n=3; 4%), fatigue (n=3; 4%), and febrile neutropenia (n=3; 4%).
Three patients (4%) in the CVP±R arm had acute grade 3 neuropathy related to vincristine. Ten patients (14%) had grade 4 neutropenia.
The most common late toxicities for the entire patient cohort were salivary gland (n=8; 5%) and skin (n=4; 3%) toxicities.
Grade 3 lung and menopausal toxicities occurred in 1 patient each. Two patients had late grade 3 vincristine neuropathy. One patient who had grade 3 neuropathy during chemotherapy progressed to grade 4.
A multidrug regimen can improve upon involved-field radiotherapy (IFRT) in patients with early stage follicular lymphoma (FL), according to research published in the Journal of Clinical Oncology.
FL patients who received IFRT plus cyclophosphamide, vincristine, and prednisolone (CVP)—with or without rituximab—had a significant improvement in progression-free survival (PFS) compared to patients who received standard treatment with IFRT alone.
However, there was no significant difference in overall survival (OS) between the treatment arms.
“This is the first successful randomized study ever to be conducted in early stage follicular lymphoma comparing standard therapy to standard therapy plus effective chemotherapy or immunochemotherapy,” said Michael MacManus, MBBCh, of Peter MacCallum Cancer Centre in Melbourne, Victoria, Australia.
“It shows that the initial treatment received by patients can significantly affect their long-term chance of staying free from disease. Moving forward, we are interested in determining whether there is a benefit in overall long-term survival for patients treated with the combination with further follow-up, and if there is any way to predict if a person will benefit from combined treatment based on analyses of blood or biopsy specimens.”
Dr MacManus and his colleagues studied 150 patients with stage I to II, low-grade FL who were enrolled in this trial between 2000 and 2012.
At randomization, the patients’ median age was 57, 52% were male, 75% had stage I disease, and 48% had PET staging.
Half of patients (n=75) were randomized to receive IFRT (30-36 Gy) alone, and half were randomized to IFRT (30-36 Gy) plus 6 cycles of CVP. From 2006 on, patients in the CVP arm received rituximab (R) as well (n=31).
Baseline characteristics were well-balanced between the treatment arms.
Efficacy
The median follow-up was 9.6 years (range, 3.1 to 15.8 years).
PFS was significantly better among patients randomized to receive CVP±R (hazard ratio [HR]=0.57; P=0.033). The estimated 10-year PFS rate was 41% in the IFRT arm and 59% in the CVP±R arm.
Patients randomized to receive CVP plus R (n=31) had significantly better PFS than patients randomized to receive IFRT alone (n=31) over the same time period (HR=0.26; P=0.045).
There were 10 deaths in the IRFT arm and 5 in the CVP±R arm, but there was no significant difference in OS between the arms (HR=0.62; P=0.40). The 10-year OS rate was 86% in the IFRT arm and 95% in the CVP±R arm.
There was no significant between-arm difference in transformation to aggressive lymphoma (P=0.1). Transformation occurred in 10 patients in the IFRT arm and 4 in the CVP±R arm.
Safety
There were 148 patients from both arms who ultimately received IFRT, and 69 patients who received CVP±R.
Grade 2 toxicities occurring in more than 10% of IFRT recipients included upper gastrointestinal (n=27; 18%), skin (n=21; 14%), and mucous membrane (n=19; 12%) toxicity. One IFRT recipient had grade 3 mucositis, and 1 had grade 4 esophageal/pharyngeal mucosal toxicity.
Grade 3 toxicities occurring in at least 2 patients in the CVP±R arm included neutropenia (n=10; 14%), infection (n=8; 12%), diarrhea (n=3; 4%), elevated gamma-glutamyl transferase (n=3; 4%), fatigue (n=3; 4%), and febrile neutropenia (n=3; 4%).
Three patients (4%) in the CVP±R arm had acute grade 3 neuropathy related to vincristine. Ten patients (14%) had grade 4 neutropenia.
The most common late toxicities for the entire patient cohort were salivary gland (n=8; 5%) and skin (n=4; 3%) toxicities.
Grade 3 lung and menopausal toxicities occurred in 1 patient each. Two patients had late grade 3 vincristine neuropathy. One patient who had grade 3 neuropathy during chemotherapy progressed to grade 4.