User login
Signal strength may limit potency of CAR T-cell therapy
Contrary to what might be expected, chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model, investigators reported.
Intracellular signaling strength was a key determinant of T cell fate in the study, which was published in the journal Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
Based on those findings, tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center, Seattle, Wash.
In a press conference, Mr. Salter described results of the study, which used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T cell signaling pathways, Mr. Salter said.
That overlap was somewhat surprising, according to Mr. Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Mr. Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor, but the speed and strength of signaling, he added. In particular, the CARs that evoked stronger signals also had increased T cell dysfunction, decreasing their potency in the mouse lymphoma model.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model of lymphoma; by contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, he said.
Those findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Mr. Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense. “This is a modification that we think should be considered in future CAR design,” Mr. Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at the efficacy, noted Stanley Riddell, MD, scientific director of the Immunotherapy Integrated Research Center at Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said during the press conference. “But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics, and together with Mr. Salter, he has filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
SOURCE: Salter AI et al., Sci Signal. 2018 Aug 21;11. pii:eaat6753.
Contrary to what might be expected, chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model, investigators reported.
Intracellular signaling strength was a key determinant of T cell fate in the study, which was published in the journal Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
Based on those findings, tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center, Seattle, Wash.
In a press conference, Mr. Salter described results of the study, which used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T cell signaling pathways, Mr. Salter said.
That overlap was somewhat surprising, according to Mr. Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Mr. Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor, but the speed and strength of signaling, he added. In particular, the CARs that evoked stronger signals also had increased T cell dysfunction, decreasing their potency in the mouse lymphoma model.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model of lymphoma; by contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, he said.
Those findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Mr. Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense. “This is a modification that we think should be considered in future CAR design,” Mr. Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at the efficacy, noted Stanley Riddell, MD, scientific director of the Immunotherapy Integrated Research Center at Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said during the press conference. “But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics, and together with Mr. Salter, he has filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
SOURCE: Salter AI et al., Sci Signal. 2018 Aug 21;11. pii:eaat6753.
Contrary to what might be expected, chimeric antigen receptor (CAR) T cells with stronger signaling capabilities were less effective against lymphoma cells in a mouse model, investigators reported.
Intracellular signaling strength was a key determinant of T cell fate in the study, which was published in the journal Science Signaling.
By contrast, CAR signaling pathways could not be predicted solely by the costimulatory domains used to construct the receptor, investigators said.
Based on those findings, tailoring CAR design based on signal strength might improve the efficacy and reduce the toxicity of CAR T-cell therapy, according to Alexander Salter, an MD/PhD student at Fred Hutchinson Cancer Research Center, Seattle, Wash.
In a press conference, Mr. Salter described results of the study, which used mass spectrometry to evaluate CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells.
While CARs with CD28 domains elicited more robust intracellular signaling than those with 4-1BB domains, there was considerable overlap in activation of T cell signaling pathways, Mr. Salter said.
That overlap was somewhat surprising, according to Mr. Salter, since researchers have generally assumed that CARs with CD28 and 4-1BB costimulatory domains will primarily signal through those respective pathways.
“No matter what costimulatory domain was encoded by the receptor, both CARs… activated both CD28 and 41BB signaling pathways,” Mr. Salter said.
The major determinant of efficacy in the study turned out to be not the domain used to construct the receptor, but the speed and strength of signaling, he added. In particular, the CARs that evoked stronger signals also had increased T cell dysfunction, decreasing their potency in the mouse lymphoma model.
The T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in the mouse model of lymphoma; by contrast, the “slower burning” 4-1BB CAR signal led to T cells that better retained their function in vivo and were associated with longer median survival in the model, he said.
Those findings suggest tailoring CAR design based on signal strength may improve clinical efficacy and reduce toxicity.
As part of the study, Mr. Salter and his co-investigators were able to modify the CAR CD28 domain to make the signaling of the CD28 CARs less intense. “This is a modification that we think should be considered in future CAR design,” Mr. Salter said.
While the alterations in the CD28 signaling domain were able to reduce levels of cytokines produced by T cells, the study was primarily designed to look at the efficacy, noted Stanley Riddell, MD, scientific director of the Immunotherapy Integrated Research Center at Fred Hutchinson Cancer Research Center.
“Our models were not set up to address the question of toxicity, so we can’t directly say this would translate to what we would see in patients,” Dr. Riddell said during the press conference. “But I think we gleaned a lot of insights as to why cytokines are produced at greater or lesser levels with various CAR designs, and insights as to how to redesign these receptors to lower the levels of cytokines they make without compromising their ability to kill.”
Dr. Riddell is a founder, shareholder, and scientific advisor of Juno Therapeutics, and together with Mr. Salter, he has filed a patent application on the use of mutant CD28 CARs for cellular therapy. Co-author Raphael Gottardo, PhD, also with Fred Hutchinson Cancer Research Center, is a consultant for Juno Therapeutics. No other competing interests were reported.
SOURCE: Salter AI et al., Sci Signal. 2018 Aug 21;11. pii:eaat6753.
FROM SCIENCE SIGNALING
Key clinical point:
Major finding: T cells with a CD28 CAR had very strong initial antitumor function that quickly waned in a mouse model of lymphoma, while the 4-1BB CAR signal led to T cells that better retained their function in vivo and had a longer median survival in the model.
Study details: Analysis of CARs encoding CD28 or 4-1BB costimulatory domains in primary human T cells using mass spectrometry, plus analysis of efficacy in a mouse model of lymphoma.
Disclosures: Study authors reported disclosures related to Juno therapeutics and a patent application related to use of mutant CD28 CARs for cellular therapy.
Source: Salter AI et al., Sci Signal. 2018 Aug 21;11. pii:eaat6753.

Role of SES in childhood cancer survival disparities

Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
Socioeconomic status (SES) may explain some racial/ethnic disparities in childhood cancer survival, according to new research.
The study showed that whites had a significant survival advantage over blacks and Hispanics for several childhood cancers.
SES significantly mediated the association between race/ethnicity and survival for acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML), neuroblastoma, and non-Hodgkin lymphoma (NHL).
Rebecca Kehm, PhD, of Columbia University in New York, New York, and her colleagues reported these findings in Cancer alongside a related editorial.
The researchers examined population-based cancer survival data from the Surveillance, Epidemiology, and End Results database.
The team collected information on 31,866 patients, ages 0 to 19, who were diagnosed with cancer between 2000 and 2011.
Survival differences by race/ethnicity
The researchers found that whites had a significant survival advantage over blacks for the cancers listed in the following table.
| Survival—black vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.43 | 1.15-1.77 | <0.01 |
| AML | 1.68 | 1.36-2.07 | <0.001 |
| Neuroblastoma | 1.38 | 1.08-1.75 | 0.01 |
| NHL | 1.53 | 1.14-2.07 | 0.01 |
| Hodgkin lymphoma | 1.66 | 1.06-2.60 | 0.03 |
| Astrocytoma | 1.95 | 1.57-2.43 | <0.001 |
| Non-astrocytoma CNS tumor | 1.53 | 1.25-1.88 | <0.001 |
| Non-rhabdomyosarcoma STS | 1.40 | 1.06-1.84 | 0.02 |
| Rhabdomyosarcoma | 1.44 | 1.10-1.88 | 0.01 |
In addition, whites had a significant survival advantage over Hispanics for the following cancers.
| Survival—Hispanic vs white | |||
| Cancer | Mortality hazard ratio | 95% confidence interval | P value |
| ALL | 1.63 | 1.43-1.86 | <0.001 |
| Neuroblastoma | 1.31 | 1.04-1.65 | 0.02 |
| NHL | 1.65 | 1.29-2.12 | <0.001 |
| Astrocytoma | 1.34 | 1.10-1.64 | <0.01 |
| Wilms tumor | 1.60 | 1.04-2.45 | 0.03 |
| Germ cell tumor | 1.63 | 1.19-2.24 | <0.01 |
Impact of SES
SES significantly mediated the association between race/ethnicity and survival for ALL, AML, neuroblastoma, and NHL but not for Hodgkin lymphoma or other cancers.
For black versus white patients, SES reduced the original association between race/ethnicity and survival by:
- 44% for ALL
- 28% for AML
- 49% for neuroblastoma
- 34% for NHL.
For Hispanics versus whites, SES reduced the original association between race/ethnicity and survival by:
- 31% for ALL
- 73% for AML
- 48% for neuroblastoma
- 28% for NHL.
“These findings provide insight for future intervention efforts aimed at closing the survival gap,” Dr Kehm said.
“For cancers in which socioeconomic status is a key factor in explaining racial and ethnic survival disparities, behavioral and supportive interventions that address social and economic barriers to effective care are warranted. However, for cancers in which survival is less influenced by socioeconomic status, more research is needed on underlying differences in tumor biology and drug processing.”
This research was supported by a grant from the National Institutes of Health, and the study’s authors made no disclosures.
CPI-613 receives orphan designation for PTCL
The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals, Inc., is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma.
Results from this trial were presented at the 2016 ASH Annual Meeting.*
CPI-613 was given at escalating doses starting at 2000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to 6 cycles. There was no intra-patient dose-escalation.
The ASH presentation included safety data on 8 patients. The most common grade 3 or higher toxicities—lymphopenia and neutropenia—occurred in 4 patients.
A patient dosed at 2750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose-escalation at doses of 2750 mg/m2 or higher and to expand the 2500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were 3 complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
*The data presented differ from the abstract.
The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals, Inc., is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma.
Results from this trial were presented at the 2016 ASH Annual Meeting.*
CPI-613 was given at escalating doses starting at 2000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to 6 cycles. There was no intra-patient dose-escalation.
The ASH presentation included safety data on 8 patients. The most common grade 3 or higher toxicities—lymphopenia and neutropenia—occurred in 4 patients.
A patient dosed at 2750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose-escalation at doses of 2750 mg/m2 or higher and to expand the 2500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were 3 complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
*The data presented differ from the abstract.
The US Food and Drug Administration (FDA) has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals, Inc., is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma.
Results from this trial were presented at the 2016 ASH Annual Meeting.*
CPI-613 was given at escalating doses starting at 2000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to 6 cycles. There was no intra-patient dose-escalation.
The ASH presentation included safety data on 8 patients. The most common grade 3 or higher toxicities—lymphopenia and neutropenia—occurred in 4 patients.
A patient dosed at 2750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose-escalation at doses of 2750 mg/m2 or higher and to expand the 2500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were 3 complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
*The data presented differ from the abstract.
Meta-analysis supports rituximab maintenance in MCL
albeit with the trade-off of higher risk of neutropenia, according to results of a meta-analysis reported in HemaSphere.
Investigators led by Liat Vidal, MD, of Tel-Aviv University, analyzed data from six randomized controlled trials of maintenance therapy including 858 patients with MCL who had a complete or partial response to induction therapy. The maintenance therapy was rituximab in five trials and bortezomib (Velcade) in one trial. The median duration of follow-up was 26-59 months across trials.
Main results showed that, compared with patients who were simply observed or given maintenance interferon-alfa, those given maintenance rituximab had a significantly reduced risk of progression or death (pooled hazard ratio, 0.58; 95% confidence interval, 0.45-0.73) and a nonsignificantly reduced risk of death (pHR, 0.79; 95% CI, 0.58-1.06).
Rituximab maintenance therapy was associated with a doubling of the risk of grade 3 or 4 neutropenia (risk ratio, 2.02; 95% CI, 1.50-2.73). However, there was no significant difference between groups with respect to risks of infection, or grade 3 or 4 anemia or thrombocythemia.
None of the included trials reported on quality of life outcomes.
The lone trial of bortezomib maintenance did not find any significant event-free survival or overall survival benefit.
“Based on our results, rituximab maintenance is recommended after immunochemotherapy with R-CHOP or cytarabine-containing induction in the front-line setting for transplant-eligible and -ineligible patients, and after R-CHOP in the relapse setting. It is unclear if maintenance is of benefit after different induction chemotherapy such as bendamustine or fludarabine,” Dr. Vidal and coauthors conclude. “By contrast, current data does not support improved outcomes with bortezomib maintenance for MCL patients.”
Dr. Vidal disclosed that she is an employee of Syneos Health. The study received no funding.
SOURCE: Vidal L et al. HemaSphere. 2018 Aug;2(4):e136.
albeit with the trade-off of higher risk of neutropenia, according to results of a meta-analysis reported in HemaSphere.
Investigators led by Liat Vidal, MD, of Tel-Aviv University, analyzed data from six randomized controlled trials of maintenance therapy including 858 patients with MCL who had a complete or partial response to induction therapy. The maintenance therapy was rituximab in five trials and bortezomib (Velcade) in one trial. The median duration of follow-up was 26-59 months across trials.
Main results showed that, compared with patients who were simply observed or given maintenance interferon-alfa, those given maintenance rituximab had a significantly reduced risk of progression or death (pooled hazard ratio, 0.58; 95% confidence interval, 0.45-0.73) and a nonsignificantly reduced risk of death (pHR, 0.79; 95% CI, 0.58-1.06).
Rituximab maintenance therapy was associated with a doubling of the risk of grade 3 or 4 neutropenia (risk ratio, 2.02; 95% CI, 1.50-2.73). However, there was no significant difference between groups with respect to risks of infection, or grade 3 or 4 anemia or thrombocythemia.
None of the included trials reported on quality of life outcomes.
The lone trial of bortezomib maintenance did not find any significant event-free survival or overall survival benefit.
“Based on our results, rituximab maintenance is recommended after immunochemotherapy with R-CHOP or cytarabine-containing induction in the front-line setting for transplant-eligible and -ineligible patients, and after R-CHOP in the relapse setting. It is unclear if maintenance is of benefit after different induction chemotherapy such as bendamustine or fludarabine,” Dr. Vidal and coauthors conclude. “By contrast, current data does not support improved outcomes with bortezomib maintenance for MCL patients.”
Dr. Vidal disclosed that she is an employee of Syneos Health. The study received no funding.
SOURCE: Vidal L et al. HemaSphere. 2018 Aug;2(4):e136.
albeit with the trade-off of higher risk of neutropenia, according to results of a meta-analysis reported in HemaSphere.
Investigators led by Liat Vidal, MD, of Tel-Aviv University, analyzed data from six randomized controlled trials of maintenance therapy including 858 patients with MCL who had a complete or partial response to induction therapy. The maintenance therapy was rituximab in five trials and bortezomib (Velcade) in one trial. The median duration of follow-up was 26-59 months across trials.
Main results showed that, compared with patients who were simply observed or given maintenance interferon-alfa, those given maintenance rituximab had a significantly reduced risk of progression or death (pooled hazard ratio, 0.58; 95% confidence interval, 0.45-0.73) and a nonsignificantly reduced risk of death (pHR, 0.79; 95% CI, 0.58-1.06).
Rituximab maintenance therapy was associated with a doubling of the risk of grade 3 or 4 neutropenia (risk ratio, 2.02; 95% CI, 1.50-2.73). However, there was no significant difference between groups with respect to risks of infection, or grade 3 or 4 anemia or thrombocythemia.
None of the included trials reported on quality of life outcomes.
The lone trial of bortezomib maintenance did not find any significant event-free survival or overall survival benefit.
“Based on our results, rituximab maintenance is recommended after immunochemotherapy with R-CHOP or cytarabine-containing induction in the front-line setting for transplant-eligible and -ineligible patients, and after R-CHOP in the relapse setting. It is unclear if maintenance is of benefit after different induction chemotherapy such as bendamustine or fludarabine,” Dr. Vidal and coauthors conclude. “By contrast, current data does not support improved outcomes with bortezomib maintenance for MCL patients.”
Dr. Vidal disclosed that she is an employee of Syneos Health. The study received no funding.
SOURCE: Vidal L et al. HemaSphere. 2018 Aug;2(4):e136.
FROM HEMASPHERE
Key clinical point: Rituximab maintenance therapy improves outcomes in patients with MCL.
Major finding: Compared with observation or maintenance interferon-alfa, maintenance rituximab was associated with reduced risk of progression-free survival events (HR, 0.58) and increased risk of grade 3 or 4 neutropenia (RR, 2.02).
Study details: A meta-analysis of six randomized controlled trials including 858 patients with MCL who had a response to induction therapy.
Disclosures: Dr. Vidal disclosed that she is an employee of Syneos Health. The study received no funding.
Source: Vidal L et al. HemaSphere. 2018 Aug;2(4):e136.
Real-world bleeding risk with ibrutinib

The Bruton tyrosine kinase inhibitor ibrutinib has been linked to a 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Joseph Mock, MD, of the University of Virginia Health System in Charlottesville, and his colleagues.
Their report was published in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting, but the authors suggested this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria,” the researchers wrote. “These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice.”
The researchers conducted a review of patients treated within the University of Virginia Health System between January 2012 and May 2016.
The team identified 70 patients, with an average age of 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%), mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström’s macroglobulinemia (1%).
Bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis.
However, major bleeding, defined as grade 3 or higher, occurred in 19% of patients (n=13). Seven of these patients were taking combined antiplatelet and anticoagulant therapy, 4 were taking antiplatelet agents alone, 1 was taking an anticoagulant agent alone, and 1 was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding were antiplatelet or anticoagulant medication, the combination of the 2 medications, interacting medications, anemia (hemoglobin less than 12 g/dL), and an elevated international normalized ratio (INR, > 1.5).
In a multivariate analysis, only the following factors were associated with an increased risk of major bleeding:
- Concomitant antiplatelet and anticoagulant use—hazard ratio=20.0 (95% CI, 2.1-200.0; P=0.0005) vs no antiplatelet/anticoagulant therapy
- Elevated INR—hazard ratio=4.6 (95% CI, 1.1-19.6; P=0.0409).
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, the team said their findings confirm “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.”
They noted that this study was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive.”
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to a 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Joseph Mock, MD, of the University of Virginia Health System in Charlottesville, and his colleagues.
Their report was published in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting, but the authors suggested this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria,” the researchers wrote. “These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice.”
The researchers conducted a review of patients treated within the University of Virginia Health System between January 2012 and May 2016.
The team identified 70 patients, with an average age of 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%), mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström’s macroglobulinemia (1%).
Bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis.
However, major bleeding, defined as grade 3 or higher, occurred in 19% of patients (n=13). Seven of these patients were taking combined antiplatelet and anticoagulant therapy, 4 were taking antiplatelet agents alone, 1 was taking an anticoagulant agent alone, and 1 was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding were antiplatelet or anticoagulant medication, the combination of the 2 medications, interacting medications, anemia (hemoglobin less than 12 g/dL), and an elevated international normalized ratio (INR, > 1.5).
In a multivariate analysis, only the following factors were associated with an increased risk of major bleeding:
- Concomitant antiplatelet and anticoagulant use—hazard ratio=20.0 (95% CI, 2.1-200.0; P=0.0005) vs no antiplatelet/anticoagulant therapy
- Elevated INR—hazard ratio=4.6 (95% CI, 1.1-19.6; P=0.0409).
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, the team said their findings confirm “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.”
They noted that this study was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive.”
The Bruton tyrosine kinase inhibitor ibrutinib has been linked to a 20-fold increased risk of major bleeding in blood cancer patients taking concomitant antiplatelet and anticoagulation therapy in a clinical setting.
Caution should be used when weighing the risks and benefits of ibrutinib for patients already taking antiplatelet or anticoagulation therapy, or both, wrote Joseph Mock, MD, of the University of Virginia Health System in Charlottesville, and his colleagues.
Their report was published in Clinical Lymphoma, Myeloma & Leukemia.
Ibrutinib had been associated with an increased risk of bleeding, albeit low, in the clinical trial setting, but the authors suggested this rate could be higher in everyday clinical practice.
“Much of the information [from clinical trials] on the bleeding risk with ibrutinib, included pooled analyses, was from patients exclusively treated in clinical trials with specific exclusion criteria,” the researchers wrote. “These criteria have generally excluded patients with significant comorbidities. However, these patients are seen in clinical practice.”
The researchers conducted a review of patients treated within the University of Virginia Health System between January 2012 and May 2016.
The team identified 70 patients, with an average age of 72, who were taking ibrutinib for chronic lymphocytic leukemia (64%), mantle cell lymphoma (27%), diffuse large B-cell lymphoma (4%), lymphoblastic lymphoma (3%), and Waldenström’s macroglobulinemia (1%).
Bleeding of any grade occurred in 56% of patients, mostly grade 1-2 bruising and epistaxis.
However, major bleeding, defined as grade 3 or higher, occurred in 19% of patients (n=13). Seven of these patients were taking combined antiplatelet and anticoagulant therapy, 4 were taking antiplatelet agents alone, 1 was taking an anticoagulant agent alone, and 1 was taking only ibrutinib.
Univariate analysis showed that the factors associated with an increased risk of major bleeding were antiplatelet or anticoagulant medication, the combination of the 2 medications, interacting medications, anemia (hemoglobin less than 12 g/dL), and an elevated international normalized ratio (INR, > 1.5).
In a multivariate analysis, only the following factors were associated with an increased risk of major bleeding:
- Concomitant antiplatelet and anticoagulant use—hazard ratio=20.0 (95% CI, 2.1-200.0; P=0.0005) vs no antiplatelet/anticoagulant therapy
- Elevated INR—hazard ratio=4.6 (95% CI, 1.1-19.6; P=0.0409).
The researchers said the risk of major bleeding in patients taking both antiplatelet and anticoagulant therapy was “unacceptably high” and “medications other than ibrutinib should be considered” in this patient population.
Overall, the team said their findings confirm “the increasingly recognized risk of major bleeding complications with ibrutinib compared with what was originally reported in the clinical trial setting.”
They noted that this study was limited by the relatively small population size. Their finding that platelet count was not associated with bleeding risk was also “counterintuitive.”
Phase 1 CAR T trial for NHL launches in Cleveland
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
Key clinical point: A phase 1 trial of CAR T therapy is enrolling adult patients with NHL who have not responded to standard therapies.
Major finding: The trial site has the ability to manufacture the cells on site, saving patients time.
Study details: A phase 1 trial to evaluate safety.
Disclosures: The study will be funded by University Hospitals Seidman Cancer Center.
First CAR T-cell therapy approvals bolster booming immunotherapy market
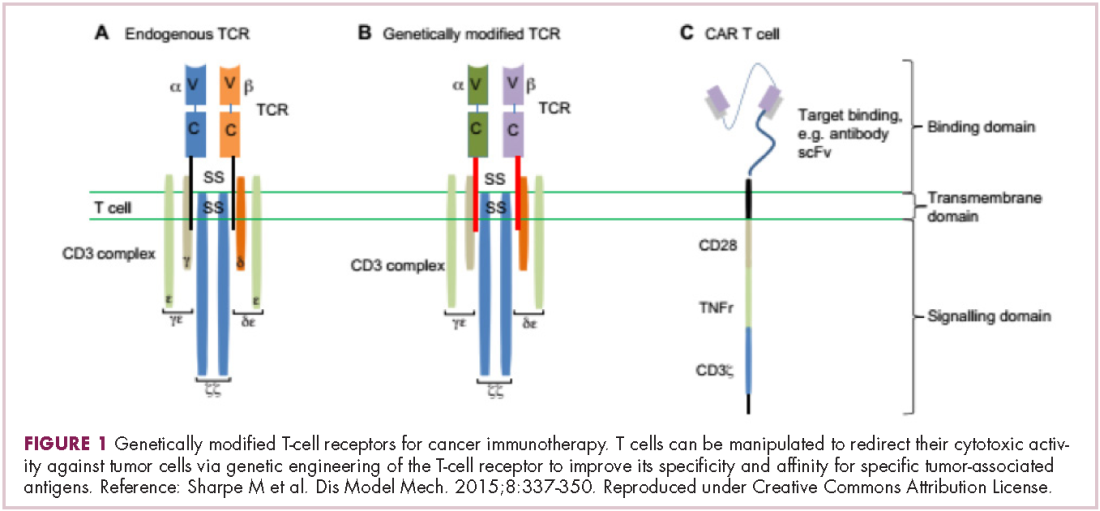
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
An unusual case of primary cardiac prosthetic valve-associated lymphoma
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
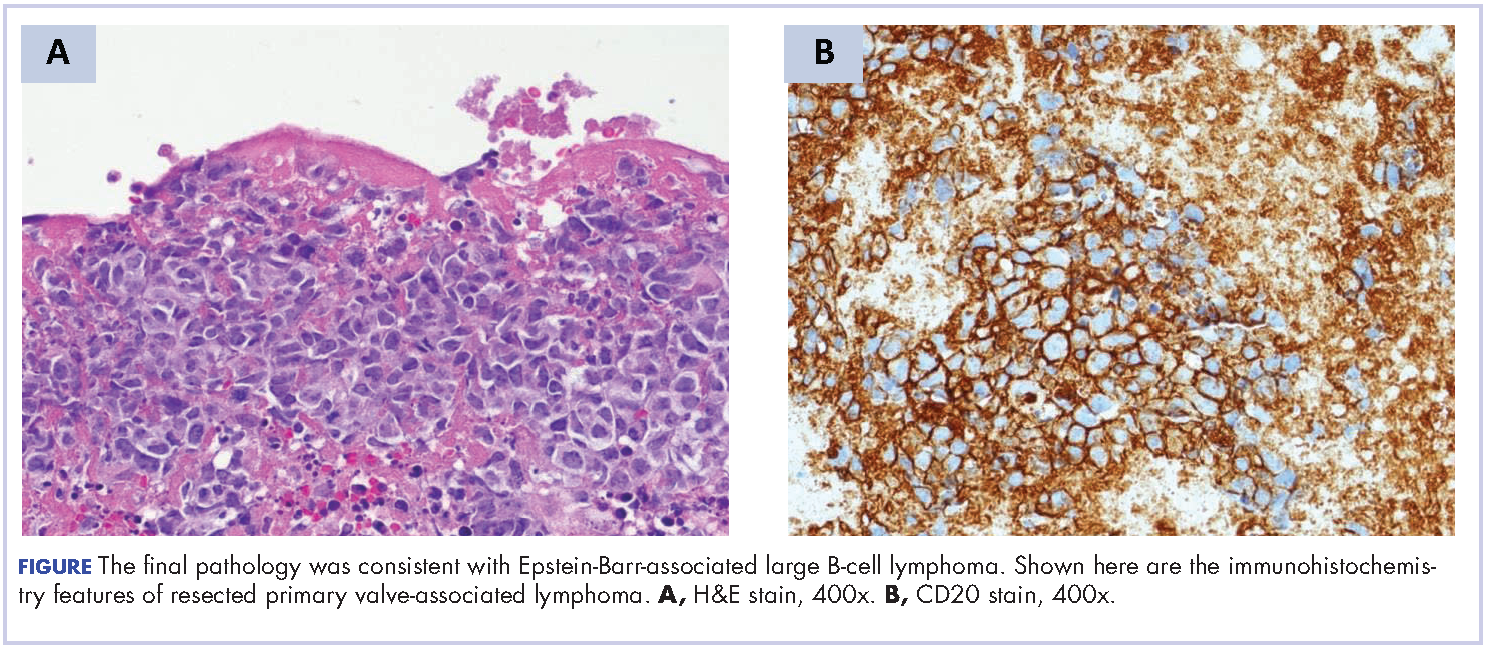
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
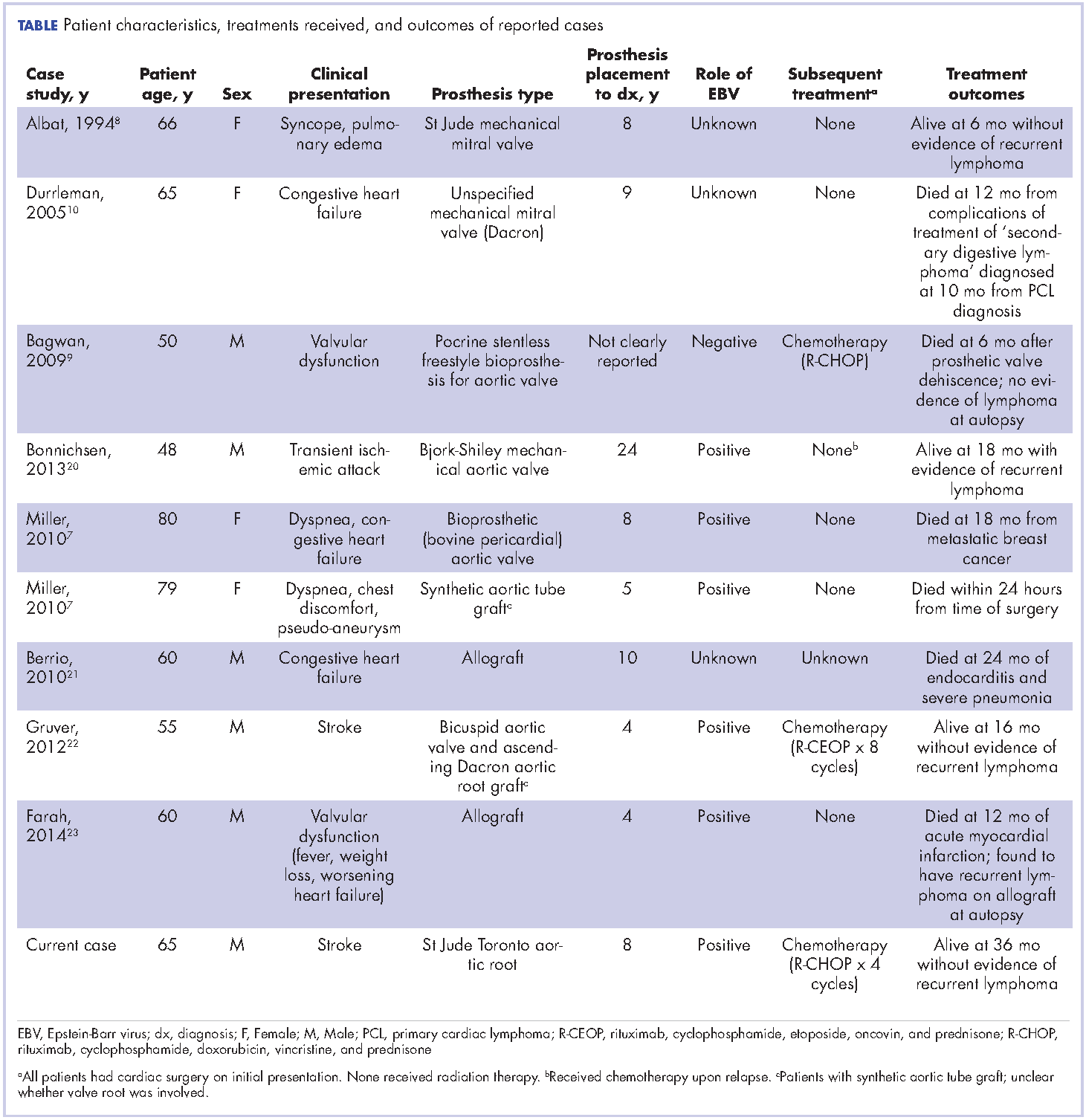
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
Durable response to pralatrexate for aggressive PTCL subtypes
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.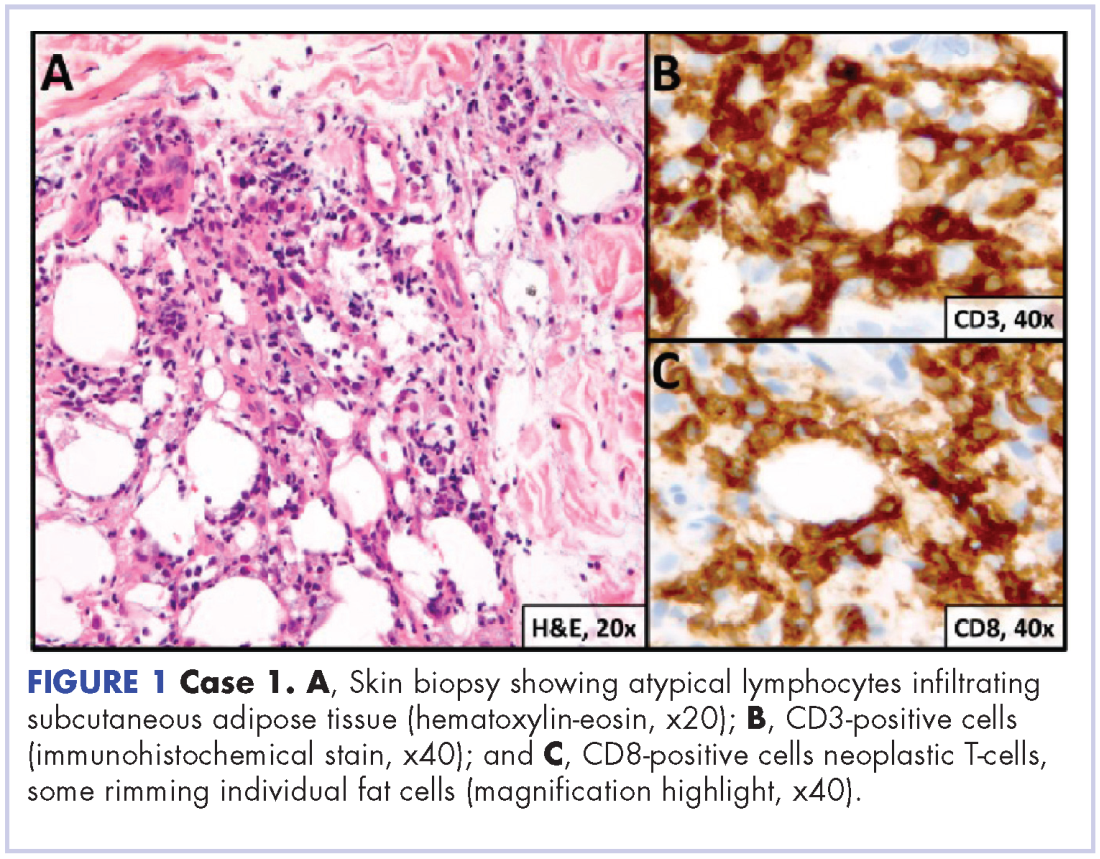
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.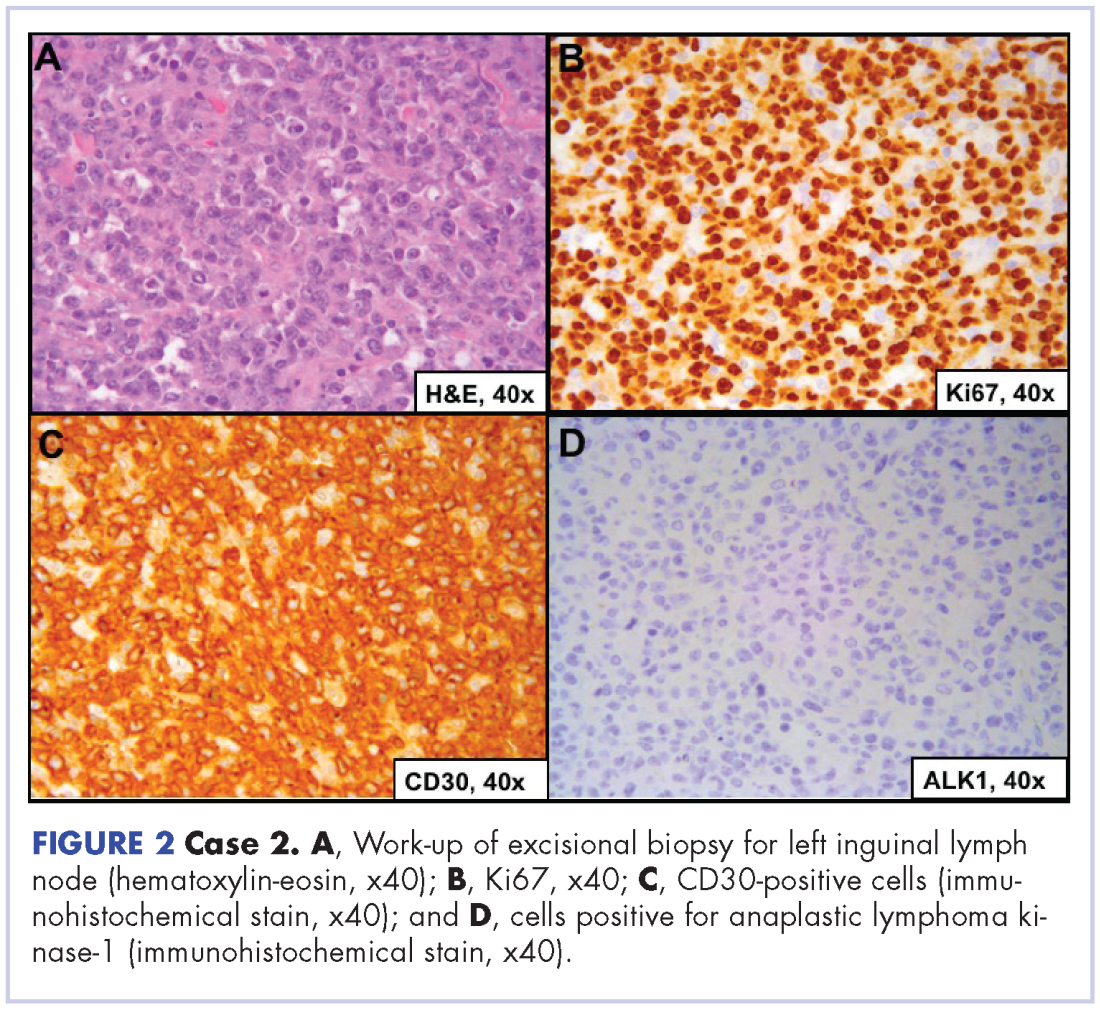
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).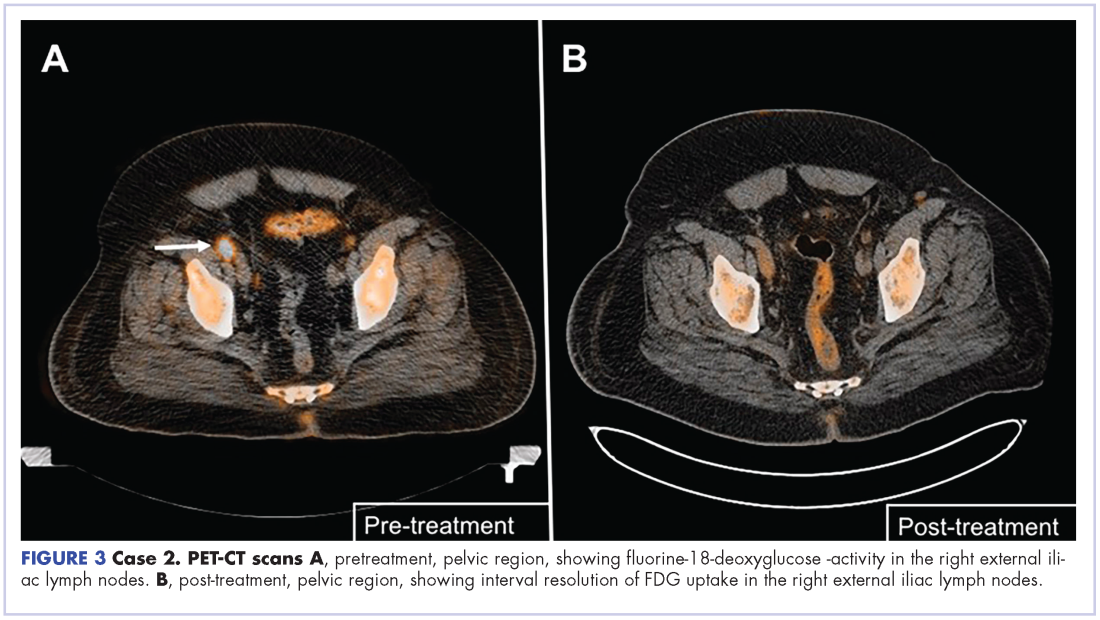
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
Immunotherapies shape the treatment landscape for hematologic malignancies
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.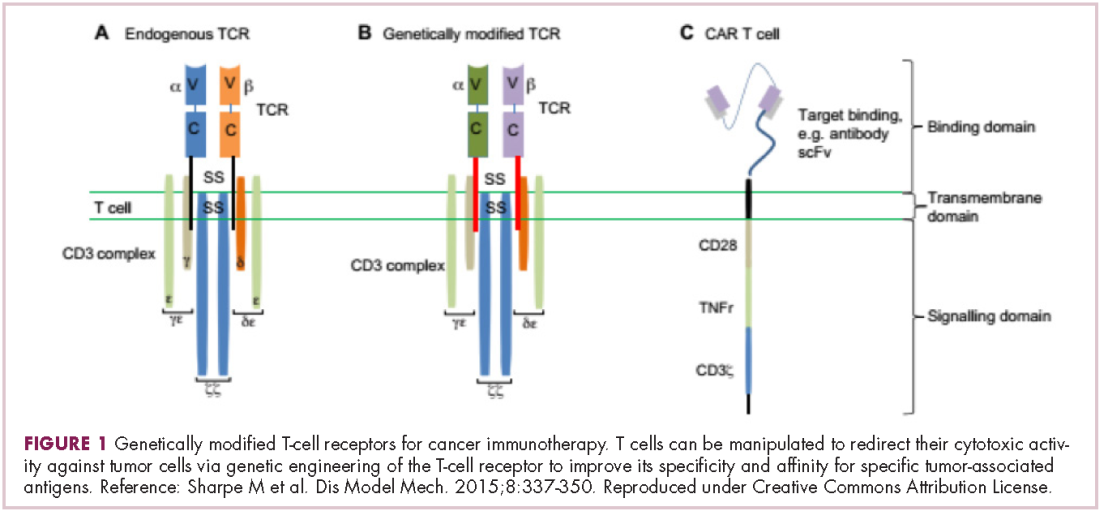
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5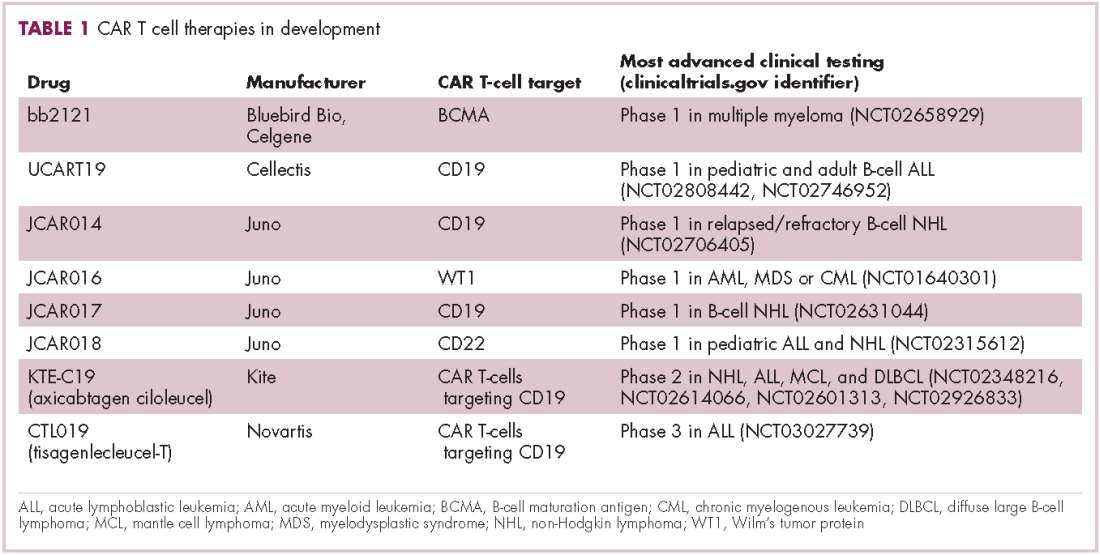
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12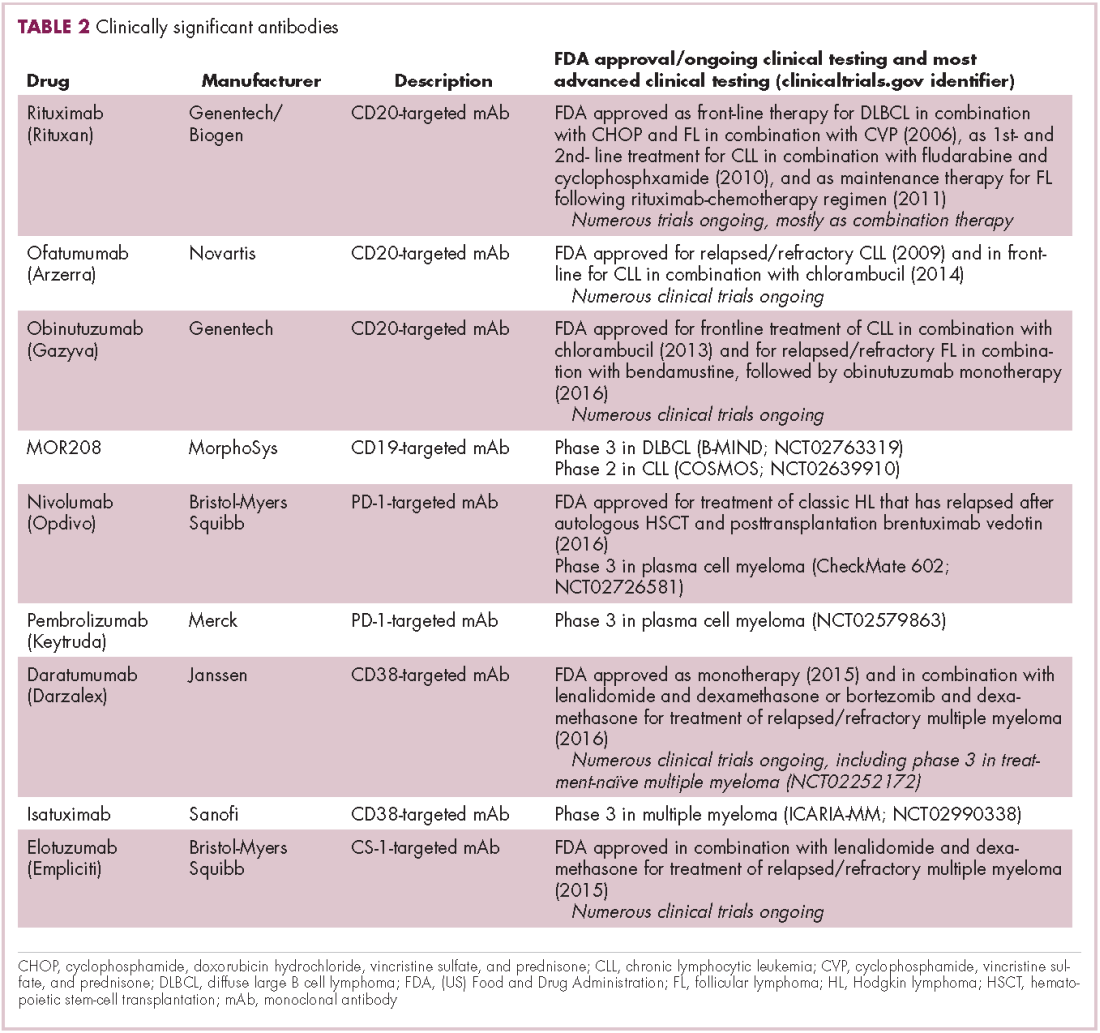
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20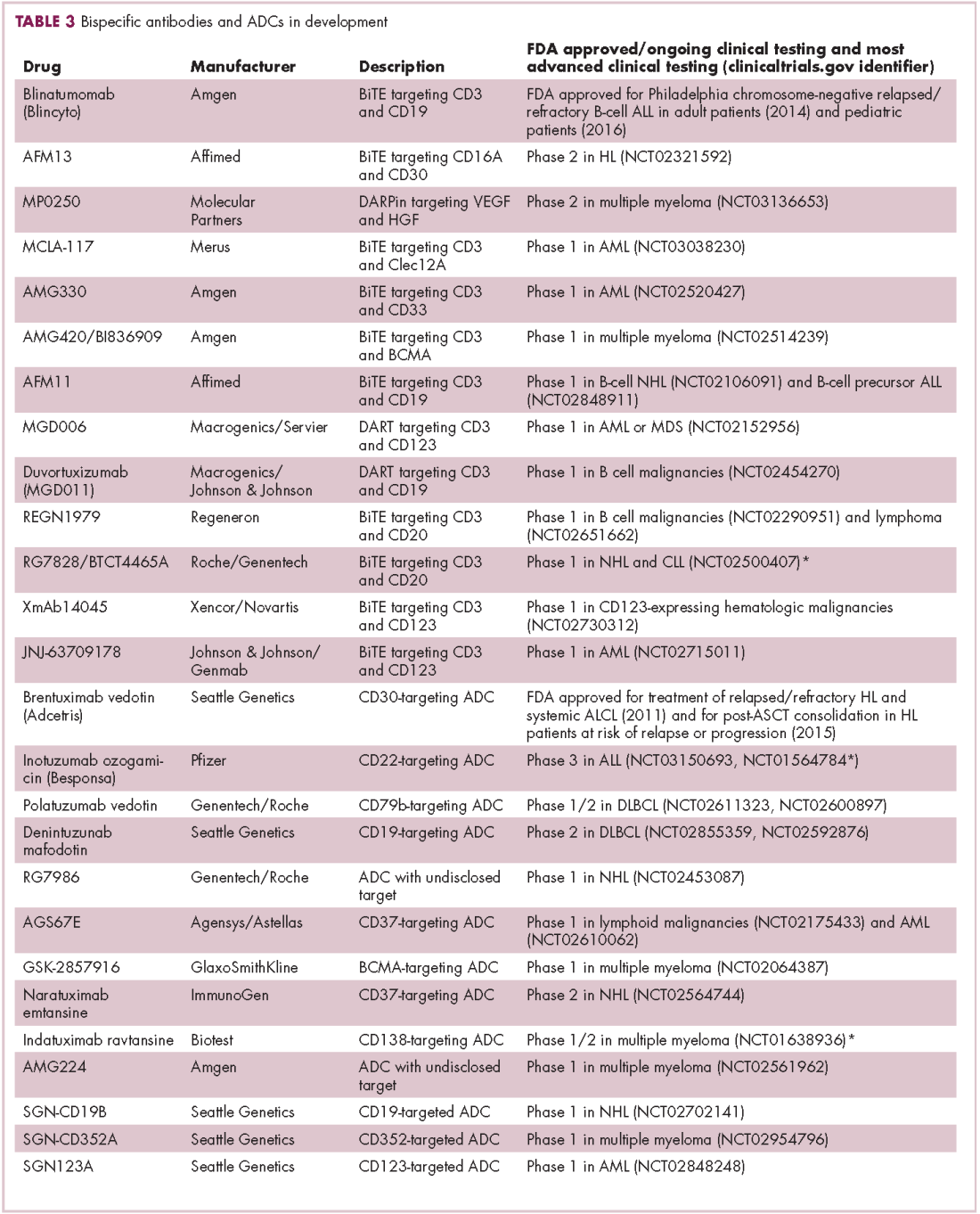
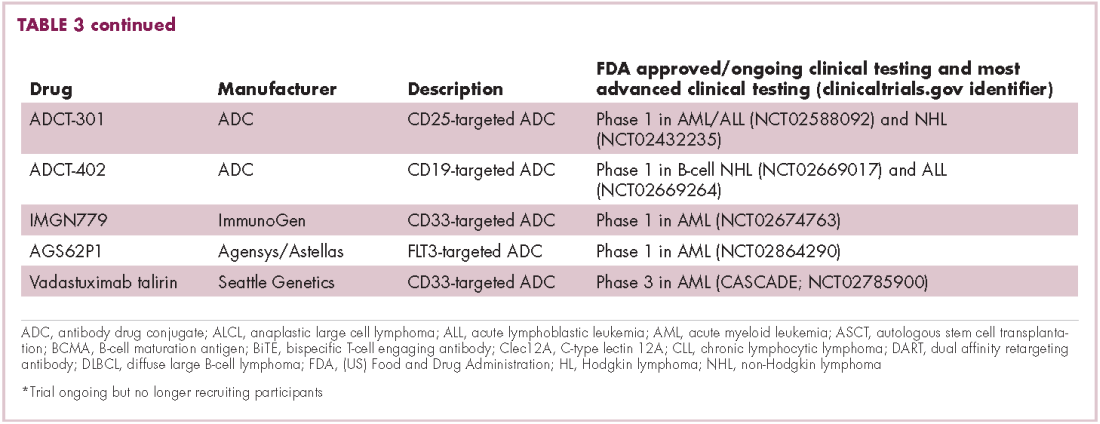
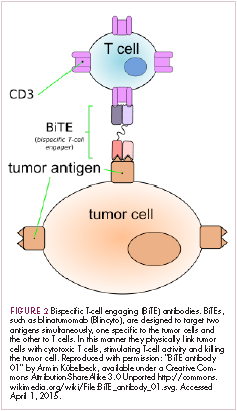
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.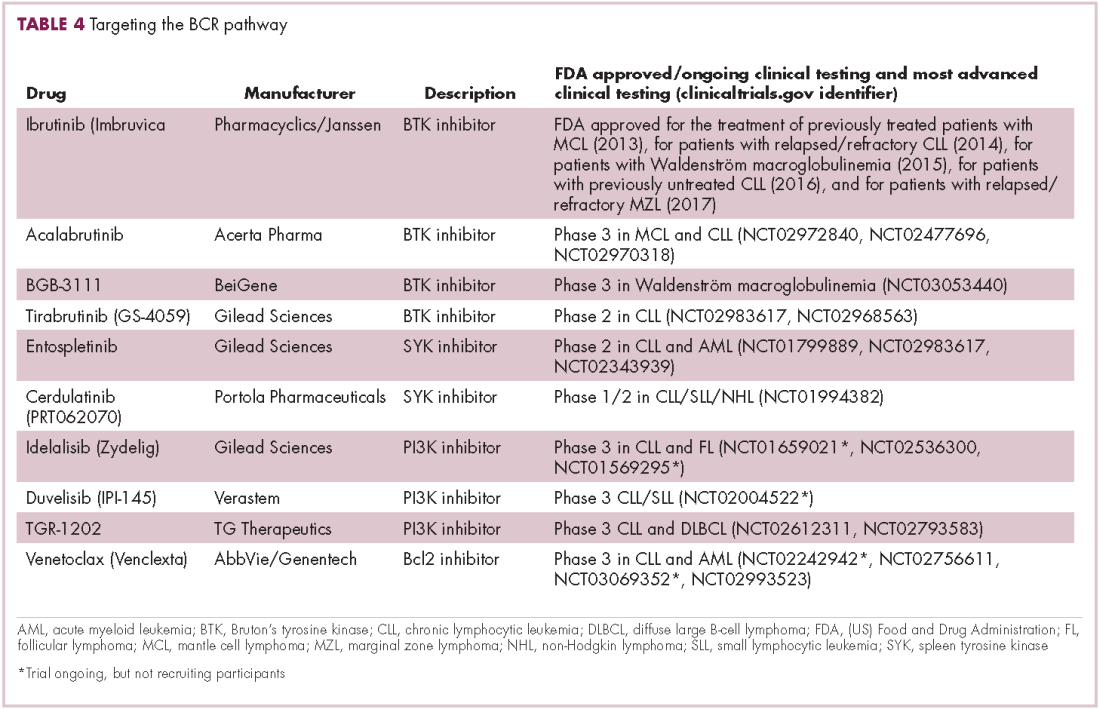
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.