User login
Review reveals lack of data on mild hemophilia A
A literature review has failed to provide new insights regarding the burden of mild hemophilia A.

In the 17 studies reviewed, mean annual bleeding rates (ABRs) were largely unreported. Data on joint pain and damage, quality of life (QOL), societal impacts, and costs of care were limited and inconsistent across the studies.
The review “revealed a lack of evidence” in adults with mild hemophilia A, Flora Peyvandi, MD, PhD, of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan and colleagues wrote in Haemophilia.
The researchers reviewed data from 10 studies conducted in Europe, 6 in North America, and 1 in Japan. Six studies were prospective cohort or registry studies, six were retrospective, and five studies were surveys or outcomes research.
The studies included 3,213 patients with mild hemophilia A aged 13 years or older. There were few details on treatment protocols, but patients received factor VIII concentrates, recombinant factor VIII, and desmopressin.
Most studies did not report mean ABRs. For the three that did, the mean ABRs were 0.44, 0.56, and 4.5. Six studies reported the percentage of patients with bleeding events, and those numbers ranged from 5.5% (1/18) to 90.7% (68/75).
Data on joint pain and damage were not standardized across studies, so the researchers were unable to draw any conclusions. One study showed no significant difference in Health Assessment Questionnaire pain score between patients with mild hemophilia A and control subjects. In another study, 5% of patients with mild hemophilia A reported having severe joint pain in the previous year, and 15% of patients reported moderate joint pain.
The researchers also found it difficult to draw conclusions about QOL. Three studies reported QOL data, and all used a different instrument.
In a study using the SF-36, general health and emotional role functioning were both significantly lower for patients with mild hemophilia A than for age-matched healthy control subjects (P less than .05). In a study using the SF-12, the physical component summary was significantly higher for patients with mild hemophilia A than for those with severe disease (P = .014).
In a study using the Haemo-QOL-A, there were no significant differences between patients with mild and severe hemophilia A. However, Dr. Peyvandi and colleagues noted that this study required long-term use of factor VIII concentrate, so the mild hemophilia A patients in this group were “probably not representative” of the overall mild hemophilia A population.
Societal impacts were difficult to assess because of a lack of standardization across studies. One study showed no significant difference in employment between patients with mild hemophilia A and healthy controls. In a U.S.-based study, patients with mild hemophilia A missed an average of 6.2 workdays per year, and 4.7 days were caused by their hemophilia. A study in Italy showed that patients with mild hemophilia A missed an average of 3.4 workdays per year.
Just two studies included data on health care costs for patients with mild hemophilia A. The mean cost of care was €793 per year in a study from Portugal published in 2015. In a U.S. study published in 1995, the annual cost of care was $22,182.
“Considering the limitations of the current body of evidence, higher-quality studies in this area are needed,” Dr. Peyvandi and colleagues wrote. “Such studies would report both bleeding and other clinical outcomes based on common definitions and for a representative population of mild [hemophilia A] adults. Areas for further research include more robust comparison to healthy controls or population norms, especially for QOL and other patient-reported outcomes.”
Seven of the eight researchers reported relationships, including employment, with BioMarin. Dr. Peyvandi reported relationships with Sanofi, Grifols, Novo Nordisk, Roche, Takeda, Sobi, Bioverativ, Spark Therapeutics, Sysmex, and CSL Behring.
SOURCE: Peyvandi F et al. Haemophilia. 2019 Jul 11. doi: 10.1111/hae.13777.
A literature review has failed to provide new insights regarding the burden of mild hemophilia A.
In the 17 studies reviewed, mean annual bleeding rates (ABRs) were largely unreported. Data on joint pain and damage, quality of life (QOL), societal impacts, and costs of care were limited and inconsistent across the studies.
The review “revealed a lack of evidence” in adults with mild hemophilia A, Flora Peyvandi, MD, PhD, of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan and colleagues wrote in Haemophilia.
The researchers reviewed data from 10 studies conducted in Europe, 6 in North America, and 1 in Japan. Six studies were prospective cohort or registry studies, six were retrospective, and five studies were surveys or outcomes research.
The studies included 3,213 patients with mild hemophilia A aged 13 years or older. There were few details on treatment protocols, but patients received factor VIII concentrates, recombinant factor VIII, and desmopressin.
Most studies did not report mean ABRs. For the three that did, the mean ABRs were 0.44, 0.56, and 4.5. Six studies reported the percentage of patients with bleeding events, and those numbers ranged from 5.5% (1/18) to 90.7% (68/75).
Data on joint pain and damage were not standardized across studies, so the researchers were unable to draw any conclusions. One study showed no significant difference in Health Assessment Questionnaire pain score between patients with mild hemophilia A and control subjects. In another study, 5% of patients with mild hemophilia A reported having severe joint pain in the previous year, and 15% of patients reported moderate joint pain.
The researchers also found it difficult to draw conclusions about QOL. Three studies reported QOL data, and all used a different instrument.
In a study using the SF-36, general health and emotional role functioning were both significantly lower for patients with mild hemophilia A than for age-matched healthy control subjects (P less than .05). In a study using the SF-12, the physical component summary was significantly higher for patients with mild hemophilia A than for those with severe disease (P = .014).
In a study using the Haemo-QOL-A, there were no significant differences between patients with mild and severe hemophilia A. However, Dr. Peyvandi and colleagues noted that this study required long-term use of factor VIII concentrate, so the mild hemophilia A patients in this group were “probably not representative” of the overall mild hemophilia A population.
Societal impacts were difficult to assess because of a lack of standardization across studies. One study showed no significant difference in employment between patients with mild hemophilia A and healthy controls. In a U.S.-based study, patients with mild hemophilia A missed an average of 6.2 workdays per year, and 4.7 days were caused by their hemophilia. A study in Italy showed that patients with mild hemophilia A missed an average of 3.4 workdays per year.
Just two studies included data on health care costs for patients with mild hemophilia A. The mean cost of care was €793 per year in a study from Portugal published in 2015. In a U.S. study published in 1995, the annual cost of care was $22,182.
“Considering the limitations of the current body of evidence, higher-quality studies in this area are needed,” Dr. Peyvandi and colleagues wrote. “Such studies would report both bleeding and other clinical outcomes based on common definitions and for a representative population of mild [hemophilia A] adults. Areas for further research include more robust comparison to healthy controls or population norms, especially for QOL and other patient-reported outcomes.”
Seven of the eight researchers reported relationships, including employment, with BioMarin. Dr. Peyvandi reported relationships with Sanofi, Grifols, Novo Nordisk, Roche, Takeda, Sobi, Bioverativ, Spark Therapeutics, Sysmex, and CSL Behring.
SOURCE: Peyvandi F et al. Haemophilia. 2019 Jul 11. doi: 10.1111/hae.13777.
A literature review has failed to provide new insights regarding the burden of mild hemophilia A.
In the 17 studies reviewed, mean annual bleeding rates (ABRs) were largely unreported. Data on joint pain and damage, quality of life (QOL), societal impacts, and costs of care were limited and inconsistent across the studies.
The review “revealed a lack of evidence” in adults with mild hemophilia A, Flora Peyvandi, MD, PhD, of Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico in Milan and colleagues wrote in Haemophilia.
The researchers reviewed data from 10 studies conducted in Europe, 6 in North America, and 1 in Japan. Six studies were prospective cohort or registry studies, six were retrospective, and five studies were surveys or outcomes research.
The studies included 3,213 patients with mild hemophilia A aged 13 years or older. There were few details on treatment protocols, but patients received factor VIII concentrates, recombinant factor VIII, and desmopressin.
Most studies did not report mean ABRs. For the three that did, the mean ABRs were 0.44, 0.56, and 4.5. Six studies reported the percentage of patients with bleeding events, and those numbers ranged from 5.5% (1/18) to 90.7% (68/75).
Data on joint pain and damage were not standardized across studies, so the researchers were unable to draw any conclusions. One study showed no significant difference in Health Assessment Questionnaire pain score between patients with mild hemophilia A and control subjects. In another study, 5% of patients with mild hemophilia A reported having severe joint pain in the previous year, and 15% of patients reported moderate joint pain.
The researchers also found it difficult to draw conclusions about QOL. Three studies reported QOL data, and all used a different instrument.
In a study using the SF-36, general health and emotional role functioning were both significantly lower for patients with mild hemophilia A than for age-matched healthy control subjects (P less than .05). In a study using the SF-12, the physical component summary was significantly higher for patients with mild hemophilia A than for those with severe disease (P = .014).
In a study using the Haemo-QOL-A, there were no significant differences between patients with mild and severe hemophilia A. However, Dr. Peyvandi and colleagues noted that this study required long-term use of factor VIII concentrate, so the mild hemophilia A patients in this group were “probably not representative” of the overall mild hemophilia A population.
Societal impacts were difficult to assess because of a lack of standardization across studies. One study showed no significant difference in employment between patients with mild hemophilia A and healthy controls. In a U.S.-based study, patients with mild hemophilia A missed an average of 6.2 workdays per year, and 4.7 days were caused by their hemophilia. A study in Italy showed that patients with mild hemophilia A missed an average of 3.4 workdays per year.
Just two studies included data on health care costs for patients with mild hemophilia A. The mean cost of care was €793 per year in a study from Portugal published in 2015. In a U.S. study published in 1995, the annual cost of care was $22,182.
“Considering the limitations of the current body of evidence, higher-quality studies in this area are needed,” Dr. Peyvandi and colleagues wrote. “Such studies would report both bleeding and other clinical outcomes based on common definitions and for a representative population of mild [hemophilia A] adults. Areas for further research include more robust comparison to healthy controls or population norms, especially for QOL and other patient-reported outcomes.”
Seven of the eight researchers reported relationships, including employment, with BioMarin. Dr. Peyvandi reported relationships with Sanofi, Grifols, Novo Nordisk, Roche, Takeda, Sobi, Bioverativ, Spark Therapeutics, Sysmex, and CSL Behring.
SOURCE: Peyvandi F et al. Haemophilia. 2019 Jul 11. doi: 10.1111/hae.13777.
FROM HAEMOPHILIA
Concizumab looks feasible in hemophilia A and B treatment
MELBOURNE – A once-daily subcutaneous treatment that inhibits the tissue factor 4 pathway inhibitor has shown significant reductions in bleeding rates in patients with hemophilia A and B, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Jan Astermark, MD, PhD, of the Centre for Thrombosis and Haemostasis at Lund University in Sweden, presented data from two phase 2, dose-escalation trials of the monoclonal antibody concizumab.
The explorer 5 trial involved 36 adults with severe hemophilia A without inhibitors who were started on 0.15 mg/kg of concizumab for 24 weeks. If they experienced three or more bleeds during that time, they were escalated to 0.20 mg/kg, and then to 0.25mg/kg if they experienced an additional three bleeds. The initial 24-week treatment period was then extended by more than 52 weeks.
In the explorer 4 trial, 16 adults with hemophilia A and 10 adults with hemophilia B – all with inhibitors – were initially randomized 2:1 to either 24 weeks of 0.15mg/kg of concizumab, including a loading dose, or placebo, with similar dose escalation in response to breakthrough bleeds. After 24 weeks, the study continued with a 52-week extension, during which all patients were treated with concizumab.
Both studies saw reductions in bleeding rates associated with concizumab treatment.
In patients with hemophilia A and B with inhibitors, the mean annualized bleeding rate for all bleeds declined from 20.4 to 4.5 bleeds, spontaneous bleeds declined from 18.5 to 2.5, and joint bleeds declined from 15 to 3.2. All three of the reductions were statistically significant.
Almost all patients achieved a concizumab concentration of 100 ng/mL, which was the expected level based on data from the phase 1 trial. Some patients showed anticoncizumab antibodies, but these were transient and did not appear to have any effect on clinical outcomes, according to Dr. Astermark.
Most patients also reached a normal level of thrombin generation, although Dr. Astermark noted that there were some patients with hemophilia B with inhibitors who produced a lower amount of thrombin than normal.
Despite the increase in thrombin generation, there were no thromboembolic events, and no significant safety concerns emerged during the study, he reported.
“Importantly and interestingly, all patients completing the main phase went into the extension phase of this trial, indicating that it was something they think was a contribution to their treatment,” Dr. Astermark said.
Earlier in 2019, concizumab was granted breakthrough designation by the Food and Drug Administration for the treatment of patients with hemophilia B and inhibitors, allowing it to receive an accelerated review by the agency.
“What the FDA based their decision on was the B patients with inhibitors, because this is truly a group where we do not have so many options,” Dr. Astermark said in an interview. He also noted that the subcutaneous treatment, delivered via a pen-like device, was much more convenient for patients who, until now, had required repeated intravenous infusions.
Two phase 3 trials are now scheduled, in which patients will receive a higher loading dose than what was used in the phase 2 trials.
Novo Nordisk sponsored both studies. Dr. Astermark reported consultancies and research funding unrelated to the study.
SOURCE: Astermark J et al. 2019 ISTH Congress, Abstract LB 01.1.
MELBOURNE – A once-daily subcutaneous treatment that inhibits the tissue factor 4 pathway inhibitor has shown significant reductions in bleeding rates in patients with hemophilia A and B, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Jan Astermark, MD, PhD, of the Centre for Thrombosis and Haemostasis at Lund University in Sweden, presented data from two phase 2, dose-escalation trials of the monoclonal antibody concizumab.
The explorer 5 trial involved 36 adults with severe hemophilia A without inhibitors who were started on 0.15 mg/kg of concizumab for 24 weeks. If they experienced three or more bleeds during that time, they were escalated to 0.20 mg/kg, and then to 0.25mg/kg if they experienced an additional three bleeds. The initial 24-week treatment period was then extended by more than 52 weeks.
In the explorer 4 trial, 16 adults with hemophilia A and 10 adults with hemophilia B – all with inhibitors – were initially randomized 2:1 to either 24 weeks of 0.15mg/kg of concizumab, including a loading dose, or placebo, with similar dose escalation in response to breakthrough bleeds. After 24 weeks, the study continued with a 52-week extension, during which all patients were treated with concizumab.
Both studies saw reductions in bleeding rates associated with concizumab treatment.
In patients with hemophilia A and B with inhibitors, the mean annualized bleeding rate for all bleeds declined from 20.4 to 4.5 bleeds, spontaneous bleeds declined from 18.5 to 2.5, and joint bleeds declined from 15 to 3.2. All three of the reductions were statistically significant.
Almost all patients achieved a concizumab concentration of 100 ng/mL, which was the expected level based on data from the phase 1 trial. Some patients showed anticoncizumab antibodies, but these were transient and did not appear to have any effect on clinical outcomes, according to Dr. Astermark.
Most patients also reached a normal level of thrombin generation, although Dr. Astermark noted that there were some patients with hemophilia B with inhibitors who produced a lower amount of thrombin than normal.
Despite the increase in thrombin generation, there were no thromboembolic events, and no significant safety concerns emerged during the study, he reported.
“Importantly and interestingly, all patients completing the main phase went into the extension phase of this trial, indicating that it was something they think was a contribution to their treatment,” Dr. Astermark said.
Earlier in 2019, concizumab was granted breakthrough designation by the Food and Drug Administration for the treatment of patients with hemophilia B and inhibitors, allowing it to receive an accelerated review by the agency.
“What the FDA based their decision on was the B patients with inhibitors, because this is truly a group where we do not have so many options,” Dr. Astermark said in an interview. He also noted that the subcutaneous treatment, delivered via a pen-like device, was much more convenient for patients who, until now, had required repeated intravenous infusions.
Two phase 3 trials are now scheduled, in which patients will receive a higher loading dose than what was used in the phase 2 trials.
Novo Nordisk sponsored both studies. Dr. Astermark reported consultancies and research funding unrelated to the study.
SOURCE: Astermark J et al. 2019 ISTH Congress, Abstract LB 01.1.
MELBOURNE – A once-daily subcutaneous treatment that inhibits the tissue factor 4 pathway inhibitor has shown significant reductions in bleeding rates in patients with hemophilia A and B, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Jan Astermark, MD, PhD, of the Centre for Thrombosis and Haemostasis at Lund University in Sweden, presented data from two phase 2, dose-escalation trials of the monoclonal antibody concizumab.
The explorer 5 trial involved 36 adults with severe hemophilia A without inhibitors who were started on 0.15 mg/kg of concizumab for 24 weeks. If they experienced three or more bleeds during that time, they were escalated to 0.20 mg/kg, and then to 0.25mg/kg if they experienced an additional three bleeds. The initial 24-week treatment period was then extended by more than 52 weeks.
In the explorer 4 trial, 16 adults with hemophilia A and 10 adults with hemophilia B – all with inhibitors – were initially randomized 2:1 to either 24 weeks of 0.15mg/kg of concizumab, including a loading dose, or placebo, with similar dose escalation in response to breakthrough bleeds. After 24 weeks, the study continued with a 52-week extension, during which all patients were treated with concizumab.
Both studies saw reductions in bleeding rates associated with concizumab treatment.
In patients with hemophilia A and B with inhibitors, the mean annualized bleeding rate for all bleeds declined from 20.4 to 4.5 bleeds, spontaneous bleeds declined from 18.5 to 2.5, and joint bleeds declined from 15 to 3.2. All three of the reductions were statistically significant.
Almost all patients achieved a concizumab concentration of 100 ng/mL, which was the expected level based on data from the phase 1 trial. Some patients showed anticoncizumab antibodies, but these were transient and did not appear to have any effect on clinical outcomes, according to Dr. Astermark.
Most patients also reached a normal level of thrombin generation, although Dr. Astermark noted that there were some patients with hemophilia B with inhibitors who produced a lower amount of thrombin than normal.
Despite the increase in thrombin generation, there were no thromboembolic events, and no significant safety concerns emerged during the study, he reported.
“Importantly and interestingly, all patients completing the main phase went into the extension phase of this trial, indicating that it was something they think was a contribution to their treatment,” Dr. Astermark said.
Earlier in 2019, concizumab was granted breakthrough designation by the Food and Drug Administration for the treatment of patients with hemophilia B and inhibitors, allowing it to receive an accelerated review by the agency.
“What the FDA based their decision on was the B patients with inhibitors, because this is truly a group where we do not have so many options,” Dr. Astermark said in an interview. He also noted that the subcutaneous treatment, delivered via a pen-like device, was much more convenient for patients who, until now, had required repeated intravenous infusions.
Two phase 3 trials are now scheduled, in which patients will receive a higher loading dose than what was used in the phase 2 trials.
Novo Nordisk sponsored both studies. Dr. Astermark reported consultancies and research funding unrelated to the study.
SOURCE: Astermark J et al. 2019 ISTH Congress, Abstract LB 01.1.
REPORTING FROM 2019 ISTH CONGRESS
RNA interference drug fitusiran looks effective in both hemophilia A and B
MELBOURNE – An investigational RNA interference therapeutic that suppresses the production of antithrombin has shown significant reductions in bleeding rates with no major safety events, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Fitusiran is a once-monthly, fixed-dose subcutaneous therapy that uses RNA interference to silence the gene for the endogenous anticoagulant antithrombin.

“The therapeutic hypothesis is based on the fact that hemophilia A and B are essentially thrombin-deficiency disorders, so if we lack factor VIII or factor IX, we can’t generate enough thrombin and we can’t produce a significant and substantial blood clot,” John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust. “If we, however, administer fitusiran, which will suppress antithrombin production, we can rebalance coagulation, generate more thrombin and form a much more substantial clot.”
Dr. Pasi presented results of an interim analysis of safety and efficacy data from an open-label, phase 2 extension study in 34 individuals with hemophilia A or B, with or without inhibitors, who were treated either with 50-mg or 80-mg doses of fitusiran for a median of at least 2 years.
Researchers saw significant declines in annualized bleeding rates in patients with hemophilia A and B, with and without inhibitors. Among those without inhibitors, the median annualized bleeding rate declined from 2.00 in patients already on hemophilia prophylaxis and 12.00 in those using on-demand treatment to 1.08 overall. In patients with inhibitors, the median annualized bleeding rate dropped from 42.00 to 1.04.
The treatment was also associated with substantial reductions in antithrombin production and increases in thrombin generation.
One patient in the phase 1 study experienced a fatal cerebral venous sinus thrombosis, which subsequently led to introduction of a bleed management protocol.
“Following that last case, we revised and reviewed the bleed management guidelines in view of the fact that there might potentially be an interaction between the amount of replacement therapy and thrombin generation,” Dr. Pasi said. Since introduction of that protocol, there have been no related thrombotic events.
The majority of adverse events reported were mild and deemed not related to the study drug, Dr. Pasi said. These included headache, injection site erythema, and arthralgia. A total of 14 subjects – all of whom were positive for hepatitis C at baseline – experienced rises in ALT levels but these were asymptomatic and resolved spontaneously.
One patient with chronic active hepatitis C infection also showed significant ALT/AST elevation which led to discontinuation of treatment.
In an interview, Dr. Pasi said one of the biggest advantages of fitusiran was that it could be used in patients with hemophilia A and B. “You’ve got patients with hemophilia B who’ve got no options at the moment. That would be an obvious specific group that would gain from this.”
Another advantage was fitusiran’s stability and dosing, he said, pointing out that the treatment was fixed dosing and stable at room temperature. Fitusiran is now undergoing phase 3 trials.
The study was funded by Sanofi Genzyme and Alnylam Pharmaceuticals, and six authors were employees of Sanofi Genzyme. Dr. Pasi reported financial relationships with pharmaceutical companies, including Alnylam.
SOURCE: Pasi J et al. 2019 ISTH Congress, Abstract OC 11.3.
MELBOURNE – An investigational RNA interference therapeutic that suppresses the production of antithrombin has shown significant reductions in bleeding rates with no major safety events, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Fitusiran is a once-monthly, fixed-dose subcutaneous therapy that uses RNA interference to silence the gene for the endogenous anticoagulant antithrombin.
“The therapeutic hypothesis is based on the fact that hemophilia A and B are essentially thrombin-deficiency disorders, so if we lack factor VIII or factor IX, we can’t generate enough thrombin and we can’t produce a significant and substantial blood clot,” John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust. “If we, however, administer fitusiran, which will suppress antithrombin production, we can rebalance coagulation, generate more thrombin and form a much more substantial clot.”
Dr. Pasi presented results of an interim analysis of safety and efficacy data from an open-label, phase 2 extension study in 34 individuals with hemophilia A or B, with or without inhibitors, who were treated either with 50-mg or 80-mg doses of fitusiran for a median of at least 2 years.
Researchers saw significant declines in annualized bleeding rates in patients with hemophilia A and B, with and without inhibitors. Among those without inhibitors, the median annualized bleeding rate declined from 2.00 in patients already on hemophilia prophylaxis and 12.00 in those using on-demand treatment to 1.08 overall. In patients with inhibitors, the median annualized bleeding rate dropped from 42.00 to 1.04.
The treatment was also associated with substantial reductions in antithrombin production and increases in thrombin generation.
One patient in the phase 1 study experienced a fatal cerebral venous sinus thrombosis, which subsequently led to introduction of a bleed management protocol.
“Following that last case, we revised and reviewed the bleed management guidelines in view of the fact that there might potentially be an interaction between the amount of replacement therapy and thrombin generation,” Dr. Pasi said. Since introduction of that protocol, there have been no related thrombotic events.
The majority of adverse events reported were mild and deemed not related to the study drug, Dr. Pasi said. These included headache, injection site erythema, and arthralgia. A total of 14 subjects – all of whom were positive for hepatitis C at baseline – experienced rises in ALT levels but these were asymptomatic and resolved spontaneously.
One patient with chronic active hepatitis C infection also showed significant ALT/AST elevation which led to discontinuation of treatment.
In an interview, Dr. Pasi said one of the biggest advantages of fitusiran was that it could be used in patients with hemophilia A and B. “You’ve got patients with hemophilia B who’ve got no options at the moment. That would be an obvious specific group that would gain from this.”
Another advantage was fitusiran’s stability and dosing, he said, pointing out that the treatment was fixed dosing and stable at room temperature. Fitusiran is now undergoing phase 3 trials.
The study was funded by Sanofi Genzyme and Alnylam Pharmaceuticals, and six authors were employees of Sanofi Genzyme. Dr. Pasi reported financial relationships with pharmaceutical companies, including Alnylam.
SOURCE: Pasi J et al. 2019 ISTH Congress, Abstract OC 11.3.
MELBOURNE – An investigational RNA interference therapeutic that suppresses the production of antithrombin has shown significant reductions in bleeding rates with no major safety events, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Fitusiran is a once-monthly, fixed-dose subcutaneous therapy that uses RNA interference to silence the gene for the endogenous anticoagulant antithrombin.
“The therapeutic hypothesis is based on the fact that hemophilia A and B are essentially thrombin-deficiency disorders, so if we lack factor VIII or factor IX, we can’t generate enough thrombin and we can’t produce a significant and substantial blood clot,” John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust. “If we, however, administer fitusiran, which will suppress antithrombin production, we can rebalance coagulation, generate more thrombin and form a much more substantial clot.”
Dr. Pasi presented results of an interim analysis of safety and efficacy data from an open-label, phase 2 extension study in 34 individuals with hemophilia A or B, with or without inhibitors, who were treated either with 50-mg or 80-mg doses of fitusiran for a median of at least 2 years.
Researchers saw significant declines in annualized bleeding rates in patients with hemophilia A and B, with and without inhibitors. Among those without inhibitors, the median annualized bleeding rate declined from 2.00 in patients already on hemophilia prophylaxis and 12.00 in those using on-demand treatment to 1.08 overall. In patients with inhibitors, the median annualized bleeding rate dropped from 42.00 to 1.04.
The treatment was also associated with substantial reductions in antithrombin production and increases in thrombin generation.
One patient in the phase 1 study experienced a fatal cerebral venous sinus thrombosis, which subsequently led to introduction of a bleed management protocol.
“Following that last case, we revised and reviewed the bleed management guidelines in view of the fact that there might potentially be an interaction between the amount of replacement therapy and thrombin generation,” Dr. Pasi said. Since introduction of that protocol, there have been no related thrombotic events.
The majority of adverse events reported were mild and deemed not related to the study drug, Dr. Pasi said. These included headache, injection site erythema, and arthralgia. A total of 14 subjects – all of whom were positive for hepatitis C at baseline – experienced rises in ALT levels but these were asymptomatic and resolved spontaneously.
One patient with chronic active hepatitis C infection also showed significant ALT/AST elevation which led to discontinuation of treatment.
In an interview, Dr. Pasi said one of the biggest advantages of fitusiran was that it could be used in patients with hemophilia A and B. “You’ve got patients with hemophilia B who’ve got no options at the moment. That would be an obvious specific group that would gain from this.”
Another advantage was fitusiran’s stability and dosing, he said, pointing out that the treatment was fixed dosing and stable at room temperature. Fitusiran is now undergoing phase 3 trials.
The study was funded by Sanofi Genzyme and Alnylam Pharmaceuticals, and six authors were employees of Sanofi Genzyme. Dr. Pasi reported financial relationships with pharmaceutical companies, including Alnylam.
SOURCE: Pasi J et al. 2019 ISTH Congress, Abstract OC 11.3.
REPORTING FROM 2019 ISTH CONGRESS
Early phase trial shows durable responses to gene therapy for hemophilia A
MELBOURNE – A gene therapy treatment for hemophilia A has shown sustained reductions in bleeding rates 3 years after treatment, with no major safety issues, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Valoctocogene roxaparvovec is an investigational gene therapy that involves using an adenovirus-associated virus to deliver the gene for clotting factor VIII.

John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust, presented the 3-year efficacy and safety results from the phase 1/2 trial of the therapy, involving 15 men with hemophilia A without inhibitors who received a single intravenous dose – either 4 x 1013 vector genomes (vg) per kg or 6 x 1013 vg/kg – of the therapy.
Participants’ mean annualized bleeding rate at baseline ranged from 6.5 among men who had been receiving prophylactic therapy to 25 among those who had been historically been treated on demand.
The treatment was associated with a substantial, significant reduction in mean annualized bleed rates; a 96% reduction in the 6 x 1013 vg/kg group by year 3, and 92% reduction in the 4 x 1013 vg/kg group by year 2.
By year 3, 86% of patients in the higher dose group had not experienced a bleed in the prior 12 months, all patients were off prophylaxis, and all had experienced resolution of target joints.
Mean factor VIII usage also decreased significantly, with a 96% reduction by year 3 in the higher dose cohort, and a 97% reduction by year 2 in the lower dose cohort.
The study also showed significant improvements in quality of life across all domains, Dr. Pasi reported.
There were no significant safety concerns raised during the study. Several patients experienced mild to moderate, transient rises in alanine aminotransferase levels at around 8-16 weeks after treatment, but there was no significant impact on liver function or on corticosteroid use. Two patients reported mild infusion reactions, which resolved within 48 hours with altering treatment.
The researchers also examined durability of factor VIII activity levels following the gene therapy, which was monitored using chromogenic assays. This revealed that after the initial increase following therapy, the factor VIII levels plateaued between years 2 and 3.
“We’ve got what we feel is really good clinical evidence of a persistent effect and we think this is dramatic,” Dr. Pasi said. A phase 3 trial is now underway.
A commenter from the audience, who remarked that the data were incredible and would make a huge difference for patients, asked about whether this represented a possible cure for the disease.
It’s premature to talk about a cure, Dr. Pasi said.
“It’s like watching paint dry; it’s going to take years before we know where we are,” he said in an interview.
However, this could represent massive and transformational change in the management of hemophilia A, he added.
On the question of whether this approach might also work in patients with inhibitors, Dr. Pasi said there were animal data suggesting that gene therapy could work in individuals with inhibitors, but the focus for the moment was on patients without inhibitors.
“But for patients that previously had a history of inhibitors and are now tolerant, that’s quite a significant group of patients that we were going to have to think about how we deal with that in due course,” he said.
The study was sponsored by manufacturer BioMarin Pharmaceutical. Dr. Pasi reported financial relationships with the study sponsor and other companies.
SOURCE: Pasi KJ et al. 2019 ISTH Congress, Abstract LB 01.2.
MELBOURNE – A gene therapy treatment for hemophilia A has shown sustained reductions in bleeding rates 3 years after treatment, with no major safety issues, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Valoctocogene roxaparvovec is an investigational gene therapy that involves using an adenovirus-associated virus to deliver the gene for clotting factor VIII.
John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust, presented the 3-year efficacy and safety results from the phase 1/2 trial of the therapy, involving 15 men with hemophilia A without inhibitors who received a single intravenous dose – either 4 x 1013 vector genomes (vg) per kg or 6 x 1013 vg/kg – of the therapy.
Participants’ mean annualized bleeding rate at baseline ranged from 6.5 among men who had been receiving prophylactic therapy to 25 among those who had been historically been treated on demand.
The treatment was associated with a substantial, significant reduction in mean annualized bleed rates; a 96% reduction in the 6 x 1013 vg/kg group by year 3, and 92% reduction in the 4 x 1013 vg/kg group by year 2.
By year 3, 86% of patients in the higher dose group had not experienced a bleed in the prior 12 months, all patients were off prophylaxis, and all had experienced resolution of target joints.
Mean factor VIII usage also decreased significantly, with a 96% reduction by year 3 in the higher dose cohort, and a 97% reduction by year 2 in the lower dose cohort.
The study also showed significant improvements in quality of life across all domains, Dr. Pasi reported.
There were no significant safety concerns raised during the study. Several patients experienced mild to moderate, transient rises in alanine aminotransferase levels at around 8-16 weeks after treatment, but there was no significant impact on liver function or on corticosteroid use. Two patients reported mild infusion reactions, which resolved within 48 hours with altering treatment.
The researchers also examined durability of factor VIII activity levels following the gene therapy, which was monitored using chromogenic assays. This revealed that after the initial increase following therapy, the factor VIII levels plateaued between years 2 and 3.
“We’ve got what we feel is really good clinical evidence of a persistent effect and we think this is dramatic,” Dr. Pasi said. A phase 3 trial is now underway.
A commenter from the audience, who remarked that the data were incredible and would make a huge difference for patients, asked about whether this represented a possible cure for the disease.
It’s premature to talk about a cure, Dr. Pasi said.
“It’s like watching paint dry; it’s going to take years before we know where we are,” he said in an interview.
However, this could represent massive and transformational change in the management of hemophilia A, he added.
On the question of whether this approach might also work in patients with inhibitors, Dr. Pasi said there were animal data suggesting that gene therapy could work in individuals with inhibitors, but the focus for the moment was on patients without inhibitors.
“But for patients that previously had a history of inhibitors and are now tolerant, that’s quite a significant group of patients that we were going to have to think about how we deal with that in due course,” he said.
The study was sponsored by manufacturer BioMarin Pharmaceutical. Dr. Pasi reported financial relationships with the study sponsor and other companies.
SOURCE: Pasi KJ et al. 2019 ISTH Congress, Abstract LB 01.2.
MELBOURNE – A gene therapy treatment for hemophilia A has shown sustained reductions in bleeding rates 3 years after treatment, with no major safety issues, according to findings presented at the International Society on Thrombosis and Haemostasis congress.
Valoctocogene roxaparvovec is an investigational gene therapy that involves using an adenovirus-associated virus to deliver the gene for clotting factor VIII.
John Pasi, MBChB, PhD, of the Royal London Haemophilia Centre, Barts Health NHS Trust, presented the 3-year efficacy and safety results from the phase 1/2 trial of the therapy, involving 15 men with hemophilia A without inhibitors who received a single intravenous dose – either 4 x 1013 vector genomes (vg) per kg or 6 x 1013 vg/kg – of the therapy.
Participants’ mean annualized bleeding rate at baseline ranged from 6.5 among men who had been receiving prophylactic therapy to 25 among those who had been historically been treated on demand.
The treatment was associated with a substantial, significant reduction in mean annualized bleed rates; a 96% reduction in the 6 x 1013 vg/kg group by year 3, and 92% reduction in the 4 x 1013 vg/kg group by year 2.
By year 3, 86% of patients in the higher dose group had not experienced a bleed in the prior 12 months, all patients were off prophylaxis, and all had experienced resolution of target joints.
Mean factor VIII usage also decreased significantly, with a 96% reduction by year 3 in the higher dose cohort, and a 97% reduction by year 2 in the lower dose cohort.
The study also showed significant improvements in quality of life across all domains, Dr. Pasi reported.
There were no significant safety concerns raised during the study. Several patients experienced mild to moderate, transient rises in alanine aminotransferase levels at around 8-16 weeks after treatment, but there was no significant impact on liver function or on corticosteroid use. Two patients reported mild infusion reactions, which resolved within 48 hours with altering treatment.
The researchers also examined durability of factor VIII activity levels following the gene therapy, which was monitored using chromogenic assays. This revealed that after the initial increase following therapy, the factor VIII levels plateaued between years 2 and 3.
“We’ve got what we feel is really good clinical evidence of a persistent effect and we think this is dramatic,” Dr. Pasi said. A phase 3 trial is now underway.
A commenter from the audience, who remarked that the data were incredible and would make a huge difference for patients, asked about whether this represented a possible cure for the disease.
It’s premature to talk about a cure, Dr. Pasi said.
“It’s like watching paint dry; it’s going to take years before we know where we are,” he said in an interview.
However, this could represent massive and transformational change in the management of hemophilia A, he added.
On the question of whether this approach might also work in patients with inhibitors, Dr. Pasi said there were animal data suggesting that gene therapy could work in individuals with inhibitors, but the focus for the moment was on patients without inhibitors.
“But for patients that previously had a history of inhibitors and are now tolerant, that’s quite a significant group of patients that we were going to have to think about how we deal with that in due course,” he said.
The study was sponsored by manufacturer BioMarin Pharmaceutical. Dr. Pasi reported financial relationships with the study sponsor and other companies.
SOURCE: Pasi KJ et al. 2019 ISTH Congress, Abstract LB 01.2.
REPORTING FROM 2019 ISTH CONGRESS
Emicizumab follow-up shows further bleeding declines
MELBOURNE – according to data presented at the International Society on Thrombosis and Haemostasis congress.
Michael Callaghan, MD, of the Children’s Hospital of Michigan, Detroit, reported on a pooled analysis of data from 399 patients with hemophilia A who were treated with emicizumab (Hemlibra) for a median duration of 83.1 weeks, representing 650 patient-years of exposure. The studies included pediatric and adult patients, both with and without factor VIII inhibitors.
Patients enrolled in the studies had a median of eight bleeds in the 24 weeks before enrollment, but in the first 24 weeks of treatment with emicizumab, the mean annualized bleed rate dropped to 1.9. During weeks 25-48, this dropped further to 0.8, remained at that level in weeks 49-72, then declined further to 0.3 during weeks 73-96.
During the first 24 weeks of treatment, 70.8% of patients experienced zero bleeds, and 22.5% experienced 1-3 bleeds. By week 96, the number of patients experiencing zero bleeds had increased to 88.6% and nearly 100% of patients had had fewer than three bleeds during that 24-week period.
The study also reported on target joint bleeds and showed the mean annualized bleed rate in target joints decreased from 1.4 in the first 24 weeks of treatment to 0.3 in weeks 73-96, by which time 90.4% of patients reported no target joint bleeds at all. Overall, 99.2% of target joints resolved, which was defined as two or fewer spontaneous bleeding events into a target joint in a year.
“The bleed rate seemed to converge on a low number, suggesting that maybe patients that came with preexisting synovitis or inflamed joints improved over time to resemble the patients who had better joint health at the beginning of the study,” Dr. Callaghan said.
The long-term follow-up did not reveal any major safety concerns. The most common drug-related adverse event was injection site reactions, which just over one-quarter of patients reported. The main serious adverse events were bleeding related.
“With any biologic agent, we were concerned about antidrug antibodies,” Dr. Callaghan told the conference. “At this follow-up point, less than 1% of patients treated with emicizumab in this group have had neutralizing antidrug antibodies.” Most of these antibodies were detected with routine screening, but there was one patient with antidrug antibodies who developed breakthrough bleeding during the study.
In an interview, Dr. Callaghan said emicizumab was “game-changing” therapy, and that the data showed it was efficacious even long term. However, he said there were still some questions to be answered about which patients were most likely to benefit.
“How early do we start this? Do we put previously untreated patients on this, and if we do, how do we expose them to factor VIII?” he said. Other challenging questions are whether to do immune tolerance induction for patients with factor VIII inhibitors and how the drug would work for other patient groups, such as those with comorbidities or who were very active.
The study was sponsored by F. Hoffman-La Roche and Chugai Pharmaceutical. Dr. Callaghan declared consultancies, grants, clinical trial involvement, speakers bureau engagements, and shares with the pharmaceutical sector.
SOURCE: Callaghan M et al. 2019 ISTH Congress, Abstract OC 60.2.
MELBOURNE – according to data presented at the International Society on Thrombosis and Haemostasis congress.
Michael Callaghan, MD, of the Children’s Hospital of Michigan, Detroit, reported on a pooled analysis of data from 399 patients with hemophilia A who were treated with emicizumab (Hemlibra) for a median duration of 83.1 weeks, representing 650 patient-years of exposure. The studies included pediatric and adult patients, both with and without factor VIII inhibitors.
Patients enrolled in the studies had a median of eight bleeds in the 24 weeks before enrollment, but in the first 24 weeks of treatment with emicizumab, the mean annualized bleed rate dropped to 1.9. During weeks 25-48, this dropped further to 0.8, remained at that level in weeks 49-72, then declined further to 0.3 during weeks 73-96.
During the first 24 weeks of treatment, 70.8% of patients experienced zero bleeds, and 22.5% experienced 1-3 bleeds. By week 96, the number of patients experiencing zero bleeds had increased to 88.6% and nearly 100% of patients had had fewer than three bleeds during that 24-week period.
The study also reported on target joint bleeds and showed the mean annualized bleed rate in target joints decreased from 1.4 in the first 24 weeks of treatment to 0.3 in weeks 73-96, by which time 90.4% of patients reported no target joint bleeds at all. Overall, 99.2% of target joints resolved, which was defined as two or fewer spontaneous bleeding events into a target joint in a year.
“The bleed rate seemed to converge on a low number, suggesting that maybe patients that came with preexisting synovitis or inflamed joints improved over time to resemble the patients who had better joint health at the beginning of the study,” Dr. Callaghan said.
The long-term follow-up did not reveal any major safety concerns. The most common drug-related adverse event was injection site reactions, which just over one-quarter of patients reported. The main serious adverse events were bleeding related.
“With any biologic agent, we were concerned about antidrug antibodies,” Dr. Callaghan told the conference. “At this follow-up point, less than 1% of patients treated with emicizumab in this group have had neutralizing antidrug antibodies.” Most of these antibodies were detected with routine screening, but there was one patient with antidrug antibodies who developed breakthrough bleeding during the study.
In an interview, Dr. Callaghan said emicizumab was “game-changing” therapy, and that the data showed it was efficacious even long term. However, he said there were still some questions to be answered about which patients were most likely to benefit.
“How early do we start this? Do we put previously untreated patients on this, and if we do, how do we expose them to factor VIII?” he said. Other challenging questions are whether to do immune tolerance induction for patients with factor VIII inhibitors and how the drug would work for other patient groups, such as those with comorbidities or who were very active.
The study was sponsored by F. Hoffman-La Roche and Chugai Pharmaceutical. Dr. Callaghan declared consultancies, grants, clinical trial involvement, speakers bureau engagements, and shares with the pharmaceutical sector.
SOURCE: Callaghan M et al. 2019 ISTH Congress, Abstract OC 60.2.
MELBOURNE – according to data presented at the International Society on Thrombosis and Haemostasis congress.
Michael Callaghan, MD, of the Children’s Hospital of Michigan, Detroit, reported on a pooled analysis of data from 399 patients with hemophilia A who were treated with emicizumab (Hemlibra) for a median duration of 83.1 weeks, representing 650 patient-years of exposure. The studies included pediatric and adult patients, both with and without factor VIII inhibitors.
Patients enrolled in the studies had a median of eight bleeds in the 24 weeks before enrollment, but in the first 24 weeks of treatment with emicizumab, the mean annualized bleed rate dropped to 1.9. During weeks 25-48, this dropped further to 0.8, remained at that level in weeks 49-72, then declined further to 0.3 during weeks 73-96.
During the first 24 weeks of treatment, 70.8% of patients experienced zero bleeds, and 22.5% experienced 1-3 bleeds. By week 96, the number of patients experiencing zero bleeds had increased to 88.6% and nearly 100% of patients had had fewer than three bleeds during that 24-week period.
The study also reported on target joint bleeds and showed the mean annualized bleed rate in target joints decreased from 1.4 in the first 24 weeks of treatment to 0.3 in weeks 73-96, by which time 90.4% of patients reported no target joint bleeds at all. Overall, 99.2% of target joints resolved, which was defined as two or fewer spontaneous bleeding events into a target joint in a year.
“The bleed rate seemed to converge on a low number, suggesting that maybe patients that came with preexisting synovitis or inflamed joints improved over time to resemble the patients who had better joint health at the beginning of the study,” Dr. Callaghan said.
The long-term follow-up did not reveal any major safety concerns. The most common drug-related adverse event was injection site reactions, which just over one-quarter of patients reported. The main serious adverse events were bleeding related.
“With any biologic agent, we were concerned about antidrug antibodies,” Dr. Callaghan told the conference. “At this follow-up point, less than 1% of patients treated with emicizumab in this group have had neutralizing antidrug antibodies.” Most of these antibodies were detected with routine screening, but there was one patient with antidrug antibodies who developed breakthrough bleeding during the study.
In an interview, Dr. Callaghan said emicizumab was “game-changing” therapy, and that the data showed it was efficacious even long term. However, he said there were still some questions to be answered about which patients were most likely to benefit.
“How early do we start this? Do we put previously untreated patients on this, and if we do, how do we expose them to factor VIII?” he said. Other challenging questions are whether to do immune tolerance induction for patients with factor VIII inhibitors and how the drug would work for other patient groups, such as those with comorbidities or who were very active.
The study was sponsored by F. Hoffman-La Roche and Chugai Pharmaceutical. Dr. Callaghan declared consultancies, grants, clinical trial involvement, speakers bureau engagements, and shares with the pharmaceutical sector.
SOURCE: Callaghan M et al. 2019 ISTH Congress, Abstract OC 60.2.
REPORTING FROM 2019 ISTH CONGRESS
Minor surgeries appear safe for hemophilia patients on emicizumab
MELBOURNE – A majority of minor surgeries can be performed in hemophilia A patients receiving emicizumab therapy without requiring prophylactic treatment with coagulation factors, according to data presented at the International Society on Thrombosis and Haemostasis congress.

Elena Santagostino, MD, PhD, from the Hemophilia and Thrombosis Center at Ospedale Maggiore Policlinico in Milan presented data from 399 patients involved in the four HAVEN trials of the humanized bispecific monoclonal antibody emicizumab (Hemlibra), which is Food and Drug Administration–approved for the prevention of bleeding episodes in individuals with hemophilia A, with or without inhibitors.
The analysis focused on the 126 patients (31.6%) who underwent at least one surgical procedure during the studies. Of the 233 surgeries, there were 215 minor procedures performed in 115 patients, and 18 major surgeries in 18 patients. All patients were receiving ongoing treatment with emicizumab, and there was no change to that treatment regimen during surgery.
“It is clear that surgery is a challenge for hemophilia,” Dr. Santagostino said. “It is a challenge for bleeding, it is a challenge for thrombosis, it is a challenge for any new drug, and this is why there is a lot of interest around this topic.”
Overall, 65.6% of minor surgeries were performed without any prophylactic coagulation factor treatment, and 90.8% of minor surgeries were conducted without postoperative bleeds requiring treatment. There were no cases of thrombosis reported.
The surgeries that did not require prophylactic coagulation factor included 42 dental procedures, 25 central venous access devices, 17 endoscopic procedures, and 12 joint procedures.
While the HAVEN studies did not allow for elective major surgery, there were still 18 unplanned major surgical situations that arose during the course of the studies. These included three hip, one knee, and one ankle arthroplasties; three synovectomies; and some dental, central venous line, and endoscopic biopsy procedures.
Of these, 15 involved prophylactic coagulant factor administration, but three procedures – including one synovectomy – were performed without prophylaxis and none resulted in a bleed.
There was one complicated bleed that occurred in a patient undergoing multiple procedures including a synovectomy, joint debridement and chondroplasty, who received prolonged treatment with recombinant Factor VIIa.
Dr. Santagostino said the findings showed surgery could be safely performed in patients who were being treated with emicizumab, both with and without inhibitors.
“A large number of minor procedures can be done without adding coagulation factors,” she said in an interview. “This is true for less invasive surgeries, such as catheter-related central venous line procedures. Even several endoscopic procedures, like a single biopsy, can be done reasonably safely.”
However she said there was still a lack of experience in dealing with hemophilia A patients who were undergoing cancer surgery, or who had significant comorbidities that might put them at higher risk of thrombosis.
“These are special patients populations that are still not investigated in the trial setting,” she said.
Commenting on the data, session cochair Liane Khoo, MD, from the Haemophilia Treatment Centre at Royal Prince Alfred Hospital in Sydney, said the results showed surgery could be performed in hemophilia A patients with and without inhibitors.
“The more we have the medication and the more experience we have, then we become more confident in using it,” she said.
The study was funded by F. Hoffman-La Roche and Chugai Pharmaceutical. Dr. Santagostino reported consultancies and speakers bureau engagements with the pharmaceutical sector.
SOURCE: Santagostino E et al. 2019 ISTH Congress, Abstract OC 60.1.
MELBOURNE – A majority of minor surgeries can be performed in hemophilia A patients receiving emicizumab therapy without requiring prophylactic treatment with coagulation factors, according to data presented at the International Society on Thrombosis and Haemostasis congress.
Elena Santagostino, MD, PhD, from the Hemophilia and Thrombosis Center at Ospedale Maggiore Policlinico in Milan presented data from 399 patients involved in the four HAVEN trials of the humanized bispecific monoclonal antibody emicizumab (Hemlibra), which is Food and Drug Administration–approved for the prevention of bleeding episodes in individuals with hemophilia A, with or without inhibitors.
The analysis focused on the 126 patients (31.6%) who underwent at least one surgical procedure during the studies. Of the 233 surgeries, there were 215 minor procedures performed in 115 patients, and 18 major surgeries in 18 patients. All patients were receiving ongoing treatment with emicizumab, and there was no change to that treatment regimen during surgery.
“It is clear that surgery is a challenge for hemophilia,” Dr. Santagostino said. “It is a challenge for bleeding, it is a challenge for thrombosis, it is a challenge for any new drug, and this is why there is a lot of interest around this topic.”
Overall, 65.6% of minor surgeries were performed without any prophylactic coagulation factor treatment, and 90.8% of minor surgeries were conducted without postoperative bleeds requiring treatment. There were no cases of thrombosis reported.
The surgeries that did not require prophylactic coagulation factor included 42 dental procedures, 25 central venous access devices, 17 endoscopic procedures, and 12 joint procedures.
While the HAVEN studies did not allow for elective major surgery, there were still 18 unplanned major surgical situations that arose during the course of the studies. These included three hip, one knee, and one ankle arthroplasties; three synovectomies; and some dental, central venous line, and endoscopic biopsy procedures.
Of these, 15 involved prophylactic coagulant factor administration, but three procedures – including one synovectomy – were performed without prophylaxis and none resulted in a bleed.
There was one complicated bleed that occurred in a patient undergoing multiple procedures including a synovectomy, joint debridement and chondroplasty, who received prolonged treatment with recombinant Factor VIIa.
Dr. Santagostino said the findings showed surgery could be safely performed in patients who were being treated with emicizumab, both with and without inhibitors.
“A large number of minor procedures can be done without adding coagulation factors,” she said in an interview. “This is true for less invasive surgeries, such as catheter-related central venous line procedures. Even several endoscopic procedures, like a single biopsy, can be done reasonably safely.”
However she said there was still a lack of experience in dealing with hemophilia A patients who were undergoing cancer surgery, or who had significant comorbidities that might put them at higher risk of thrombosis.
“These are special patients populations that are still not investigated in the trial setting,” she said.
Commenting on the data, session cochair Liane Khoo, MD, from the Haemophilia Treatment Centre at Royal Prince Alfred Hospital in Sydney, said the results showed surgery could be performed in hemophilia A patients with and without inhibitors.
“The more we have the medication and the more experience we have, then we become more confident in using it,” she said.
The study was funded by F. Hoffman-La Roche and Chugai Pharmaceutical. Dr. Santagostino reported consultancies and speakers bureau engagements with the pharmaceutical sector.
SOURCE: Santagostino E et al. 2019 ISTH Congress, Abstract OC 60.1.
MELBOURNE – A majority of minor surgeries can be performed in hemophilia A patients receiving emicizumab therapy without requiring prophylactic treatment with coagulation factors, according to data presented at the International Society on Thrombosis and Haemostasis congress.
Elena Santagostino, MD, PhD, from the Hemophilia and Thrombosis Center at Ospedale Maggiore Policlinico in Milan presented data from 399 patients involved in the four HAVEN trials of the humanized bispecific monoclonal antibody emicizumab (Hemlibra), which is Food and Drug Administration–approved for the prevention of bleeding episodes in individuals with hemophilia A, with or without inhibitors.
The analysis focused on the 126 patients (31.6%) who underwent at least one surgical procedure during the studies. Of the 233 surgeries, there were 215 minor procedures performed in 115 patients, and 18 major surgeries in 18 patients. All patients were receiving ongoing treatment with emicizumab, and there was no change to that treatment regimen during surgery.
“It is clear that surgery is a challenge for hemophilia,” Dr. Santagostino said. “It is a challenge for bleeding, it is a challenge for thrombosis, it is a challenge for any new drug, and this is why there is a lot of interest around this topic.”
Overall, 65.6% of minor surgeries were performed without any prophylactic coagulation factor treatment, and 90.8% of minor surgeries were conducted without postoperative bleeds requiring treatment. There were no cases of thrombosis reported.
The surgeries that did not require prophylactic coagulation factor included 42 dental procedures, 25 central venous access devices, 17 endoscopic procedures, and 12 joint procedures.
While the HAVEN studies did not allow for elective major surgery, there were still 18 unplanned major surgical situations that arose during the course of the studies. These included three hip, one knee, and one ankle arthroplasties; three synovectomies; and some dental, central venous line, and endoscopic biopsy procedures.
Of these, 15 involved prophylactic coagulant factor administration, but three procedures – including one synovectomy – were performed without prophylaxis and none resulted in a bleed.
There was one complicated bleed that occurred in a patient undergoing multiple procedures including a synovectomy, joint debridement and chondroplasty, who received prolonged treatment with recombinant Factor VIIa.
Dr. Santagostino said the findings showed surgery could be safely performed in patients who were being treated with emicizumab, both with and without inhibitors.
“A large number of minor procedures can be done without adding coagulation factors,” she said in an interview. “This is true for less invasive surgeries, such as catheter-related central venous line procedures. Even several endoscopic procedures, like a single biopsy, can be done reasonably safely.”
However she said there was still a lack of experience in dealing with hemophilia A patients who were undergoing cancer surgery, or who had significant comorbidities that might put them at higher risk of thrombosis.
“These are special patients populations that are still not investigated in the trial setting,” she said.
Commenting on the data, session cochair Liane Khoo, MD, from the Haemophilia Treatment Centre at Royal Prince Alfred Hospital in Sydney, said the results showed surgery could be performed in hemophilia A patients with and without inhibitors.
“The more we have the medication and the more experience we have, then we become more confident in using it,” she said.
The study was funded by F. Hoffman-La Roche and Chugai Pharmaceutical. Dr. Santagostino reported consultancies and speakers bureau engagements with the pharmaceutical sector.
SOURCE: Santagostino E et al. 2019 ISTH Congress, Abstract OC 60.1.
REPORTING FROM 2019 ISTH CONGRESS
Labor outcomes unchanged by alternate anesthesia in bleeding disorder patients
Among pregnant women with inherited bleeding disorders, having a contraindication to regional anesthesia appears not to impact labor outcomes, according to a retrospective analysis.
“The purpose of this study was to determine the anesthetic use in labour in a cohort of women with inherited bleeding disorders,” wrote Sean C. Boyd, of Coombe Women & Infants University Hospital in Dublin, Ireland, and colleagues. The findings were reported in the European Journal of Obstetrics & Gynecology and Reproductive Biology.
The study comprised 97 pregnant women with an inherited bleeding disorder and outcomes related to 130 delivered newborns.
The researchers reviewed medical records of patients with a variety of inherited bleeding disorders: type 1 von Willebrand disease (VWD), deficiencies of factors VII, VIII, IX, X, and XI, combined deficiencies, and others.
Various clinical data, including both obstetric and anesthetic outcomes, were collected from January 2011 to December 2016.
When researchers compared pregnancies where regional anesthesia was contraindicated to those in which it was considered safe, women with a contraindication were more likely to receive general anesthesia for cesarean section (20% vs. 1%), more likely to use a remifentanil infusion (31% vs. 0), and more likely to require prophylactic hemostatic support for delivery (61% vs. 1%).
Vaginal (71% vs. 65%; P = .4) and caesarean section (29% vs. 32%; P = .28) delivery rates were similar between the two groups.
Rates of postpartum hemorrhage were greater in pregnancies where regional anesthesia was contraindicated (24% vs. 12%), but not significantly different (P = .07). No cases of vertebral canal hematoma or neonatal hemorrhage were reported among participants.
“Women are anxious about analgesia and anesthesia in labour and understandably, more so, when they are aware that they are unable to have an epidural or spinal,” the researchers wrote. “This study shows what alternative analgesia is used in labour and, reassuringly, that labour outcome is the same.”
Two key limitations of the study were the small sample size and the wide range of included bleeding disorders.
No funding sources were reported. The authors did not report conflicts of interest.
SOURCE: Boyd SC et al. Eur J Obstet Gynecol Reprod Biol. 2019 Jun 3. doi: 10.1016/j.ejogrb.2019.05.043.
Among pregnant women with inherited bleeding disorders, having a contraindication to regional anesthesia appears not to impact labor outcomes, according to a retrospective analysis.
“The purpose of this study was to determine the anesthetic use in labour in a cohort of women with inherited bleeding disorders,” wrote Sean C. Boyd, of Coombe Women & Infants University Hospital in Dublin, Ireland, and colleagues. The findings were reported in the European Journal of Obstetrics & Gynecology and Reproductive Biology.
The study comprised 97 pregnant women with an inherited bleeding disorder and outcomes related to 130 delivered newborns.
The researchers reviewed medical records of patients with a variety of inherited bleeding disorders: type 1 von Willebrand disease (VWD), deficiencies of factors VII, VIII, IX, X, and XI, combined deficiencies, and others.
Various clinical data, including both obstetric and anesthetic outcomes, were collected from January 2011 to December 2016.
When researchers compared pregnancies where regional anesthesia was contraindicated to those in which it was considered safe, women with a contraindication were more likely to receive general anesthesia for cesarean section (20% vs. 1%), more likely to use a remifentanil infusion (31% vs. 0), and more likely to require prophylactic hemostatic support for delivery (61% vs. 1%).
Vaginal (71% vs. 65%; P = .4) and caesarean section (29% vs. 32%; P = .28) delivery rates were similar between the two groups.
Rates of postpartum hemorrhage were greater in pregnancies where regional anesthesia was contraindicated (24% vs. 12%), but not significantly different (P = .07). No cases of vertebral canal hematoma or neonatal hemorrhage were reported among participants.
“Women are anxious about analgesia and anesthesia in labour and understandably, more so, when they are aware that they are unable to have an epidural or spinal,” the researchers wrote. “This study shows what alternative analgesia is used in labour and, reassuringly, that labour outcome is the same.”
Two key limitations of the study were the small sample size and the wide range of included bleeding disorders.
No funding sources were reported. The authors did not report conflicts of interest.
SOURCE: Boyd SC et al. Eur J Obstet Gynecol Reprod Biol. 2019 Jun 3. doi: 10.1016/j.ejogrb.2019.05.043.
Among pregnant women with inherited bleeding disorders, having a contraindication to regional anesthesia appears not to impact labor outcomes, according to a retrospective analysis.
“The purpose of this study was to determine the anesthetic use in labour in a cohort of women with inherited bleeding disorders,” wrote Sean C. Boyd, of Coombe Women & Infants University Hospital in Dublin, Ireland, and colleagues. The findings were reported in the European Journal of Obstetrics & Gynecology and Reproductive Biology.
The study comprised 97 pregnant women with an inherited bleeding disorder and outcomes related to 130 delivered newborns.
The researchers reviewed medical records of patients with a variety of inherited bleeding disorders: type 1 von Willebrand disease (VWD), deficiencies of factors VII, VIII, IX, X, and XI, combined deficiencies, and others.
Various clinical data, including both obstetric and anesthetic outcomes, were collected from January 2011 to December 2016.
When researchers compared pregnancies where regional anesthesia was contraindicated to those in which it was considered safe, women with a contraindication were more likely to receive general anesthesia for cesarean section (20% vs. 1%), more likely to use a remifentanil infusion (31% vs. 0), and more likely to require prophylactic hemostatic support for delivery (61% vs. 1%).
Vaginal (71% vs. 65%; P = .4) and caesarean section (29% vs. 32%; P = .28) delivery rates were similar between the two groups.
Rates of postpartum hemorrhage were greater in pregnancies where regional anesthesia was contraindicated (24% vs. 12%), but not significantly different (P = .07). No cases of vertebral canal hematoma or neonatal hemorrhage were reported among participants.
“Women are anxious about analgesia and anesthesia in labour and understandably, more so, when they are aware that they are unable to have an epidural or spinal,” the researchers wrote. “This study shows what alternative analgesia is used in labour and, reassuringly, that labour outcome is the same.”
Two key limitations of the study were the small sample size and the wide range of included bleeding disorders.
No funding sources were reported. The authors did not report conflicts of interest.
SOURCE: Boyd SC et al. Eur J Obstet Gynecol Reprod Biol. 2019 Jun 3. doi: 10.1016/j.ejogrb.2019.05.043.
FROM THE EUROPEAN JOURNAL OF OBSTETRICS & GYNECOLOGY AND REPRODUCTIVE BIOLOGY
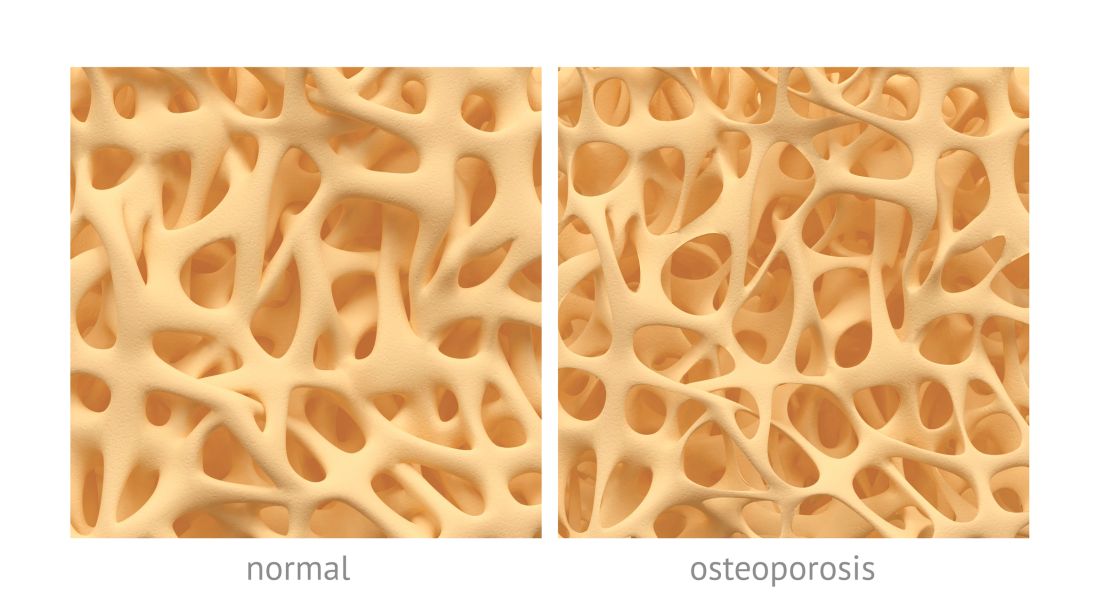
Osteoporotic fracture risk appears higher in adults with hemophilia
Adults with newly diagnosed hemophilia may be at a higher risk of developing osteoporotic fractures, according to results from a retrospective study.
Sheng-Hui Tuan, MD, of Cishan Hospital in Kaohsiung, Taiwan, and colleagues conducted a population-based nationwide cohort study that included 75 patients with hemophilia and 300 control subjects without hemophilia matched for age and sex. Data was obtained from a national insurance database in Taiwan from January 2000 to December 2013. The findings were published in Haemophilia.
The primary outcome measured was newly diagnosed osteoporotic fractures, defined as wrist, vertebral, and hip fractures among individuals from both groups. Patients with osteoporotic fractures before hemophilia diagnosis were excluded.
In the analysis, the team calculated hazard ratios and incidence rates of new-onset osteoporotic fractures in both cohorts.
After analysis, the researchers found that the risk of developing new-onset osteoporotic fractures was greater in the hemophilia group versus the comparison group (HR, 5.41; 95% confidence interval, 2.42-12.1; P less than .001).
After adjusting for covariates, such as socioeconomic status, age, sex, and other comorbidities, patients with hemophilia had a 337% higher risk of developing osteoporotic fractures post diagnosis versus matched controls (95% CI, 1.88-10.17; P = .001).
“The risk of osteoporotic fractures following haemophilia increased with time and was significantly higher at 5 years after the diagnosis,” the researchers wrote.
The underlying mechanisms driving these associations remain unknown, according to the authors. Possible risk factors include reduced physical activity, HIV and hepatitis C virus infections, and arthropathy.
The researchers acknowledged that a key limitation of the study was the absence of some relevant clinical information within the database. As a result, information bias could have lowered the accuracy of the analysis.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Tuan S-H et al. Haemophilia. 2019 Jul 7. doi: 10.1111/hae.13814.
Adults with newly diagnosed hemophilia may be at a higher risk of developing osteoporotic fractures, according to results from a retrospective study.
Sheng-Hui Tuan, MD, of Cishan Hospital in Kaohsiung, Taiwan, and colleagues conducted a population-based nationwide cohort study that included 75 patients with hemophilia and 300 control subjects without hemophilia matched for age and sex. Data was obtained from a national insurance database in Taiwan from January 2000 to December 2013. The findings were published in Haemophilia.
The primary outcome measured was newly diagnosed osteoporotic fractures, defined as wrist, vertebral, and hip fractures among individuals from both groups. Patients with osteoporotic fractures before hemophilia diagnosis were excluded.
In the analysis, the team calculated hazard ratios and incidence rates of new-onset osteoporotic fractures in both cohorts.
After analysis, the researchers found that the risk of developing new-onset osteoporotic fractures was greater in the hemophilia group versus the comparison group (HR, 5.41; 95% confidence interval, 2.42-12.1; P less than .001).
After adjusting for covariates, such as socioeconomic status, age, sex, and other comorbidities, patients with hemophilia had a 337% higher risk of developing osteoporotic fractures post diagnosis versus matched controls (95% CI, 1.88-10.17; P = .001).
“The risk of osteoporotic fractures following haemophilia increased with time and was significantly higher at 5 years after the diagnosis,” the researchers wrote.
The underlying mechanisms driving these associations remain unknown, according to the authors. Possible risk factors include reduced physical activity, HIV and hepatitis C virus infections, and arthropathy.
The researchers acknowledged that a key limitation of the study was the absence of some relevant clinical information within the database. As a result, information bias could have lowered the accuracy of the analysis.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Tuan S-H et al. Haemophilia. 2019 Jul 7. doi: 10.1111/hae.13814.
Adults with newly diagnosed hemophilia may be at a higher risk of developing osteoporotic fractures, according to results from a retrospective study.
Sheng-Hui Tuan, MD, of Cishan Hospital in Kaohsiung, Taiwan, and colleagues conducted a population-based nationwide cohort study that included 75 patients with hemophilia and 300 control subjects without hemophilia matched for age and sex. Data was obtained from a national insurance database in Taiwan from January 2000 to December 2013. The findings were published in Haemophilia.
The primary outcome measured was newly diagnosed osteoporotic fractures, defined as wrist, vertebral, and hip fractures among individuals from both groups. Patients with osteoporotic fractures before hemophilia diagnosis were excluded.
In the analysis, the team calculated hazard ratios and incidence rates of new-onset osteoporotic fractures in both cohorts.
After analysis, the researchers found that the risk of developing new-onset osteoporotic fractures was greater in the hemophilia group versus the comparison group (HR, 5.41; 95% confidence interval, 2.42-12.1; P less than .001).
After adjusting for covariates, such as socioeconomic status, age, sex, and other comorbidities, patients with hemophilia had a 337% higher risk of developing osteoporotic fractures post diagnosis versus matched controls (95% CI, 1.88-10.17; P = .001).
“The risk of osteoporotic fractures following haemophilia increased with time and was significantly higher at 5 years after the diagnosis,” the researchers wrote.
The underlying mechanisms driving these associations remain unknown, according to the authors. Possible risk factors include reduced physical activity, HIV and hepatitis C virus infections, and arthropathy.
The researchers acknowledged that a key limitation of the study was the absence of some relevant clinical information within the database. As a result, information bias could have lowered the accuracy of the analysis.
No funding sources were reported. The authors reported having no conflicts of interest.
SOURCE: Tuan S-H et al. Haemophilia. 2019 Jul 7. doi: 10.1111/hae.13814.
FROM HAEMOPHILIA
Novel point-of-care assay appears accurate in monitoring hemophilia A
A novel point-of-care assay demonstrated sensitivity to monitor coagulation factor replacement therapy in patients with severe hemophilia A, with and without inhibitors, according to recent study findings.
The ClotChip assay is a novel factor-replacement therapy monitoring tool that uses a dielectric microsensor to assess whole-blood coagulation in patients with severe hemophilia A.
Debnath Maji of Case Western Reserve University, Cleveland, and colleagues evaluated the novel assay in attempt to address the unmet need of reliable monitoring of coagulation factor–replacement therapy. While global haemostasis assays are available to measure the missing function, these tools require expert interpretation, are technically challenging to operate, and may be unavailable at the bedside, they wrote in Haemophilia.
They evaluated the analytical capabilities of the assay in 6 patients with inhibitors and 12 patients without inhibitors. A total of 50 healthy controls were included in the study.
The team collected whole blood samples pre- and postcoagulation factor–replacement therapy. Measurements were also conducted on the thrombin generation assay (TGA), thromboelastography (TEG), and rotational thromboelastometry (ROTEM) global assays for comparison.
After analysis, the researchers found that ClotChip T-peak parameters showed a significant reduction for samples obtained post–factor replacement therapy versus pretherapy among patients with and without inhibitors (P = .028 and P = .0001, respectively).
Additionally, ClotChip T-peak values exhibited strong correlations with factor VIII clotting activity, clotting time parameters of ROTEM, and peak thrombin and endogenous thrombin potential of TGA.
“Taken together, these data suggest that the ClotChip T-peak parameter is sensitive to detection and correction of coagulopathy in children with haemophilia with and without inhibitors, as indicated by T-peak reaching reference‐range values after coagulation factor replacement therapy,” the researchers wrote.
One key limitation of the study was the small sample size, which may limit the generalizability of the findings.
The novel assay may be an appropriate tool for “real-world” decision making around the use of coagulation factor replacement therapy in patients with severe hemophilia A, they added.
The study was funded by the National Institutes of Health, the American Heart Association, and a Veterans Affairs Research Center of Excellence at Case Western Reserve University. The authors reported financial affiliations with Bayer, Bioverativ, Shire, and XaTek.
SOURCE: Maji D et al. Haemophilia. 2019 Jul 7. doi: 10.1111/hae.13799.
A novel point-of-care assay demonstrated sensitivity to monitor coagulation factor replacement therapy in patients with severe hemophilia A, with and without inhibitors, according to recent study findings.
The ClotChip assay is a novel factor-replacement therapy monitoring tool that uses a dielectric microsensor to assess whole-blood coagulation in patients with severe hemophilia A.
Debnath Maji of Case Western Reserve University, Cleveland, and colleagues evaluated the novel assay in attempt to address the unmet need of reliable monitoring of coagulation factor–replacement therapy. While global haemostasis assays are available to measure the missing function, these tools require expert interpretation, are technically challenging to operate, and may be unavailable at the bedside, they wrote in Haemophilia.
They evaluated the analytical capabilities of the assay in 6 patients with inhibitors and 12 patients without inhibitors. A total of 50 healthy controls were included in the study.
The team collected whole blood samples pre- and postcoagulation factor–replacement therapy. Measurements were also conducted on the thrombin generation assay (TGA), thromboelastography (TEG), and rotational thromboelastometry (ROTEM) global assays for comparison.
After analysis, the researchers found that ClotChip T-peak parameters showed a significant reduction for samples obtained post–factor replacement therapy versus pretherapy among patients with and without inhibitors (P = .028 and P = .0001, respectively).
Additionally, ClotChip T-peak values exhibited strong correlations with factor VIII clotting activity, clotting time parameters of ROTEM, and peak thrombin and endogenous thrombin potential of TGA.
“Taken together, these data suggest that the ClotChip T-peak parameter is sensitive to detection and correction of coagulopathy in children with haemophilia with and without inhibitors, as indicated by T-peak reaching reference‐range values after coagulation factor replacement therapy,” the researchers wrote.
One key limitation of the study was the small sample size, which may limit the generalizability of the findings.
The novel assay may be an appropriate tool for “real-world” decision making around the use of coagulation factor replacement therapy in patients with severe hemophilia A, they added.
The study was funded by the National Institutes of Health, the American Heart Association, and a Veterans Affairs Research Center of Excellence at Case Western Reserve University. The authors reported financial affiliations with Bayer, Bioverativ, Shire, and XaTek.
SOURCE: Maji D et al. Haemophilia. 2019 Jul 7. doi: 10.1111/hae.13799.
A novel point-of-care assay demonstrated sensitivity to monitor coagulation factor replacement therapy in patients with severe hemophilia A, with and without inhibitors, according to recent study findings.
The ClotChip assay is a novel factor-replacement therapy monitoring tool that uses a dielectric microsensor to assess whole-blood coagulation in patients with severe hemophilia A.
Debnath Maji of Case Western Reserve University, Cleveland, and colleagues evaluated the novel assay in attempt to address the unmet need of reliable monitoring of coagulation factor–replacement therapy. While global haemostasis assays are available to measure the missing function, these tools require expert interpretation, are technically challenging to operate, and may be unavailable at the bedside, they wrote in Haemophilia.
They evaluated the analytical capabilities of the assay in 6 patients with inhibitors and 12 patients without inhibitors. A total of 50 healthy controls were included in the study.
The team collected whole blood samples pre- and postcoagulation factor–replacement therapy. Measurements were also conducted on the thrombin generation assay (TGA), thromboelastography (TEG), and rotational thromboelastometry (ROTEM) global assays for comparison.
After analysis, the researchers found that ClotChip T-peak parameters showed a significant reduction for samples obtained post–factor replacement therapy versus pretherapy among patients with and without inhibitors (P = .028 and P = .0001, respectively).
Additionally, ClotChip T-peak values exhibited strong correlations with factor VIII clotting activity, clotting time parameters of ROTEM, and peak thrombin and endogenous thrombin potential of TGA.
“Taken together, these data suggest that the ClotChip T-peak parameter is sensitive to detection and correction of coagulopathy in children with haemophilia with and without inhibitors, as indicated by T-peak reaching reference‐range values after coagulation factor replacement therapy,” the researchers wrote.
One key limitation of the study was the small sample size, which may limit the generalizability of the findings.
The novel assay may be an appropriate tool for “real-world” decision making around the use of coagulation factor replacement therapy in patients with severe hemophilia A, they added.
The study was funded by the National Institutes of Health, the American Heart Association, and a Veterans Affairs Research Center of Excellence at Case Western Reserve University. The authors reported financial affiliations with Bayer, Bioverativ, Shire, and XaTek.
SOURCE: Maji D et al. Haemophilia. 2019 Jul 7. doi: 10.1111/hae.13799.
FROM HAEMOPHILIA
Subcutaneous marstacimab appears safe in hemophilia A and B
MELBOURNE – Subcutaneous administration of the monoclonal antibody marstacimab (PF-06741086) provides significant reductions in bleeding rates for patients with hemophilia A and B, with reasonably safety and tolerability, according to research presented at the International Society on Thrombosis and Haemostasis congress.

Johnny Mahlangu, MBBCh, of the University of Witwatersrand in Johannesburg, South Africa, presented data from a multicenter, international phase 1B/2 open-label study involving 26 patients with severe hemophilia, who had experienced at least six acute bleeding episodes in the 6 months prior to enrollment. Twenty-three patients had hemophilia A, the remaining three patients had hemophilia B, and all were receiving on-demand treatment.
Patients were divided into one of four cohorts. Cohort 1 received a weekly 300-mg dose subcutaneously for 30 days, cohort 2 received a 300-mg loading dose followed by 150 mg weekly for 30 days, cohort 3 received 450 mg weekly for 30 days, and cohort 4 also received a 300-mg weekly dose for 30 days but consisted of patients with inhibitors.
With the primary outcome being safety, the researchers reported no treatment-related serious adverse events. There were four grade 3/4 adverse events, including two subjects who reported injection site reactions, and some generalized pruritus and erythematous rash.
Two patients discontinued treatment after reaching prespecified dose-limiting toxicity related to decreasing fibrinogen levels, compared with baseline. However, Dr. Mahlangu pointed out that, in one of these patients, the fibrinogen levels were still within normal levels but protocol required removing the patient from the study.
The study did see a significant 85%-97% reduction in annualized bleed rates across the four cohorts, including among patients with inhibitors.
“Most patients who were exposed to marstacimab actually did not bleed at all when they were receiving marstacimab, compared to when they weren’t receiving marstacimab,” Dr. Mahlangu said.
While three patients developed antidrug antibodies, this did not appear to impact the pharmacokinetics, pharmacodynamics, or safety, he said. No patients developed neutralizing antibodies, and the pharmacodynamics showed no difference between patients with hemophilia A and B.
“I would like to believe that the results of this study are fairly promising in terms of the safety, the efficacy, the pharmoacokinetics, and the pharmacodynamics,” Dr. Mahlangu said.
There is an unmet need for therapies that can be used in patients with either hemophilia A or B, and with or without inhibitors, Dr. Mahlangu said in an interview. Another unmet need that marstacimab could potentially address is for subcutaneous treatment options, he added.
“We are particularly pleased by the fact that injection-site reactions are very low, and they seem not to carry on every time the patients have injected,” Dr. Mahlangu said.
Commenting on the presentation, session cochair Julia Phillips, MD, from PathLab in Waikato, New Zealand, said subcutaneous treatments offered a huge advantage to patients with hemophilia and their families.
“Often more than one member of the family is affected. So [for] a family with children, if they have several sons with hemophilia, then doing IV injections before school on a regular basis can be quite a big burden on the family,” she said in an interview.
The study was sponsored by Pfizer. Dr. Mahlangu reported research support from and scientific advisory board and speakers bureau roles with several pharmaceutical companies, including Pfizer.
SOURCE: Mahlangu J et al. 2019. ISTH CONGRESS, Abstract OC 11.2.
MELBOURNE – Subcutaneous administration of the monoclonal antibody marstacimab (PF-06741086) provides significant reductions in bleeding rates for patients with hemophilia A and B, with reasonably safety and tolerability, according to research presented at the International Society on Thrombosis and Haemostasis congress.
Johnny Mahlangu, MBBCh, of the University of Witwatersrand in Johannesburg, South Africa, presented data from a multicenter, international phase 1B/2 open-label study involving 26 patients with severe hemophilia, who had experienced at least six acute bleeding episodes in the 6 months prior to enrollment. Twenty-three patients had hemophilia A, the remaining three patients had hemophilia B, and all were receiving on-demand treatment.
Patients were divided into one of four cohorts. Cohort 1 received a weekly 300-mg dose subcutaneously for 30 days, cohort 2 received a 300-mg loading dose followed by 150 mg weekly for 30 days, cohort 3 received 450 mg weekly for 30 days, and cohort 4 also received a 300-mg weekly dose for 30 days but consisted of patients with inhibitors.
With the primary outcome being safety, the researchers reported no treatment-related serious adverse events. There were four grade 3/4 adverse events, including two subjects who reported injection site reactions, and some generalized pruritus and erythematous rash.
Two patients discontinued treatment after reaching prespecified dose-limiting toxicity related to decreasing fibrinogen levels, compared with baseline. However, Dr. Mahlangu pointed out that, in one of these patients, the fibrinogen levels were still within normal levels but protocol required removing the patient from the study.
The study did see a significant 85%-97% reduction in annualized bleed rates across the four cohorts, including among patients with inhibitors.
“Most patients who were exposed to marstacimab actually did not bleed at all when they were receiving marstacimab, compared to when they weren’t receiving marstacimab,” Dr. Mahlangu said.
While three patients developed antidrug antibodies, this did not appear to impact the pharmacokinetics, pharmacodynamics, or safety, he said. No patients developed neutralizing antibodies, and the pharmacodynamics showed no difference between patients with hemophilia A and B.
“I would like to believe that the results of this study are fairly promising in terms of the safety, the efficacy, the pharmoacokinetics, and the pharmacodynamics,” Dr. Mahlangu said.
There is an unmet need for therapies that can be used in patients with either hemophilia A or B, and with or without inhibitors, Dr. Mahlangu said in an interview. Another unmet need that marstacimab could potentially address is for subcutaneous treatment options, he added.
“We are particularly pleased by the fact that injection-site reactions are very low, and they seem not to carry on every time the patients have injected,” Dr. Mahlangu said.
Commenting on the presentation, session cochair Julia Phillips, MD, from PathLab in Waikato, New Zealand, said subcutaneous treatments offered a huge advantage to patients with hemophilia and their families.
“Often more than one member of the family is affected. So [for] a family with children, if they have several sons with hemophilia, then doing IV injections before school on a regular basis can be quite a big burden on the family,” she said in an interview.
The study was sponsored by Pfizer. Dr. Mahlangu reported research support from and scientific advisory board and speakers bureau roles with several pharmaceutical companies, including Pfizer.
SOURCE: Mahlangu J et al. 2019. ISTH CONGRESS, Abstract OC 11.2.
MELBOURNE – Subcutaneous administration of the monoclonal antibody marstacimab (PF-06741086) provides significant reductions in bleeding rates for patients with hemophilia A and B, with reasonably safety and tolerability, according to research presented at the International Society on Thrombosis and Haemostasis congress.
Johnny Mahlangu, MBBCh, of the University of Witwatersrand in Johannesburg, South Africa, presented data from a multicenter, international phase 1B/2 open-label study involving 26 patients with severe hemophilia, who had experienced at least six acute bleeding episodes in the 6 months prior to enrollment. Twenty-three patients had hemophilia A, the remaining three patients had hemophilia B, and all were receiving on-demand treatment.
Patients were divided into one of four cohorts. Cohort 1 received a weekly 300-mg dose subcutaneously for 30 days, cohort 2 received a 300-mg loading dose followed by 150 mg weekly for 30 days, cohort 3 received 450 mg weekly for 30 days, and cohort 4 also received a 300-mg weekly dose for 30 days but consisted of patients with inhibitors.
With the primary outcome being safety, the researchers reported no treatment-related serious adverse events. There were four grade 3/4 adverse events, including two subjects who reported injection site reactions, and some generalized pruritus and erythematous rash.
Two patients discontinued treatment after reaching prespecified dose-limiting toxicity related to decreasing fibrinogen levels, compared with baseline. However, Dr. Mahlangu pointed out that, in one of these patients, the fibrinogen levels were still within normal levels but protocol required removing the patient from the study.
The study did see a significant 85%-97% reduction in annualized bleed rates across the four cohorts, including among patients with inhibitors.
“Most patients who were exposed to marstacimab actually did not bleed at all when they were receiving marstacimab, compared to when they weren’t receiving marstacimab,” Dr. Mahlangu said.
While three patients developed antidrug antibodies, this did not appear to impact the pharmacokinetics, pharmacodynamics, or safety, he said. No patients developed neutralizing antibodies, and the pharmacodynamics showed no difference between patients with hemophilia A and B.
“I would like to believe that the results of this study are fairly promising in terms of the safety, the efficacy, the pharmoacokinetics, and the pharmacodynamics,” Dr. Mahlangu said.
There is an unmet need for therapies that can be used in patients with either hemophilia A or B, and with or without inhibitors, Dr. Mahlangu said in an interview. Another unmet need that marstacimab could potentially address is for subcutaneous treatment options, he added.
“We are particularly pleased by the fact that injection-site reactions are very low, and they seem not to carry on every time the patients have injected,” Dr. Mahlangu said.
Commenting on the presentation, session cochair Julia Phillips, MD, from PathLab in Waikato, New Zealand, said subcutaneous treatments offered a huge advantage to patients with hemophilia and their families.
“Often more than one member of the family is affected. So [for] a family with children, if they have several sons with hemophilia, then doing IV injections before school on a regular basis can be quite a big burden on the family,” she said in an interview.
The study was sponsored by Pfizer. Dr. Mahlangu reported research support from and scientific advisory board and speakers bureau roles with several pharmaceutical companies, including Pfizer.
SOURCE: Mahlangu J et al. 2019. ISTH CONGRESS, Abstract OC 11.2.
REPORTING FROM 2019 ISTH CONGRESS