User login
FDA approves Doptelet for liver disease patients undergoing procedures
Doptelet (avatrombopag) is the first drug to be approved by the Food and Drug Administration for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure, the FDA announced in a statement.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
The safety and efficacy of two different doses of Doptelet administered orally over 5 days, as compared with placebo, was studied in the ADAPT trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. At both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to 7 days following the procedure as compared with those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and edema in the hands or feet. People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet, the FDA said in a press release announcing the approval.
The FDA granted the Doptelet approval to AkaRx.
Doptelet (avatrombopag) is the first drug to be approved by the Food and Drug Administration for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure, the FDA announced in a statement.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
The safety and efficacy of two different doses of Doptelet administered orally over 5 days, as compared with placebo, was studied in the ADAPT trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. At both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to 7 days following the procedure as compared with those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and edema in the hands or feet. People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet, the FDA said in a press release announcing the approval.
The FDA granted the Doptelet approval to AkaRx.
Doptelet (avatrombopag) is the first drug to be approved by the Food and Drug Administration for thrombocytopenia in adults with chronic liver disease who are scheduled to undergo a medical or dental procedure, the FDA announced in a statement.
“Patients with chronic liver disease who have low platelet counts and require a procedure are at increased risk of bleeding,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Doptelet was demonstrated to safely increase the platelet count. This drug may decrease or eliminate the need for platelet transfusions, which are associated with risk of infection and other adverse reactions.”
The safety and efficacy of two different doses of Doptelet administered orally over 5 days, as compared with placebo, was studied in the ADAPT trials (ADAPT-1 and ADAPT-2) involving 435 patients with chronic liver disease and severe thrombocytopenia who were scheduled to undergo a procedure that would typically require platelet transfusion. At both dose levels of Doptelet, a higher proportion of patients had increased platelet counts and did not require platelet transfusion or any rescue therapy on the day of the procedure and up to 7 days following the procedure as compared with those treated with placebo.
The most common side effects reported by clinical trial participants who received Doptelet were fever, stomach (abdominal) pain, nausea, headache, fatigue and edema in the hands or feet. People with chronic liver disease and people with certain blood clotting conditions may have an increased risk of developing blood clots when taking Doptelet, the FDA said in a press release announcing the approval.
The FDA granted the Doptelet approval to AkaRx.
Emicizumab reduces bleeding in hemophilia A

GLASGOW—Final results from the HAVEN 3 study suggest emicizumab prophylaxis can reduce bleeding in hemophilia A patients without factor VIII inhibitors.
Compared to patients who did not receive prophylaxis, those who received emicizumab prophylaxis had a 96% to 97% reduction in treated bleeds and a 94% to 95% reduction in all bleeds.
An intra-patient comparison showed a 68% reduction in treated bleeds with once-weekly emicizumab, compared to prior factor VIII prophylaxis.
“[Emicizumab] is the first medicine to show superior efficacy to prior factor VIII prophylaxis, the current standard of care therapy, as demonstrated by a statistically significant reduction in treated bleeds in the HAVEN 3 study intra-patient comparison,” said Johnny Mahlangu, MB BCh, of the University of the Witwatersrand in Johannesburg, South Africa.
Dr Mahlangu presented these results at the World Federation of Hemophilia (WFH) 2018 World Congress during the late-breaking abstract session on Monday. HAVEN 3 was sponsored by Hoffmann-La Roche.
Patients and treatment
In this phase 3 trial, researchers evaluated emicizumab in patients with hemophilia A without factor VIII inhibitors. The study included 152 patients who were 12 years of age or older and were previously treated with factor VIII therapy on-demand or as prophylaxis.
Patients previously treated with on-demand factor VIII were randomized in a 2:2:1 fashion to receive:
- Emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study (arm A, n=36)
- Emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 3 mg/kg/2wks for at least 24 weeks (arm B, n=35)
- No prophylaxis, only episodic/on-demand factor VIII treatment (arm C, n=18).
Patients previously treated with factor VIII prophylaxis received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study (arm D, n=63).
Episodic treatment of breakthrough bleeds with factor VIII therapy was allowed per protocol.
Emicizumab vs no prophylaxis
The model-based (negative binomial regression model) annualized bleeding rate (ABR) for treated bleeds was 1.5 in arm A, 1.3 in arm B, and 38.2 in arm C. The median ABR for treated bleeds was 0 in arms A and B and 40.4 in arm C.
Compared to patients in arm C, those in arm A had a 96% (P<0.0001) reduction in treated bleeds, and those in arm B had a 97% (P<0.0001) reduction in treated bleeds.
None of the patients in arm C had 0 treated bleeds, compared to 55.6% of patients in arm A and 60% of patients in arm B.
The model-based ABR for all bleeds was 2.5 in arm A, 2.6 in arm B, and 47.6 in arm C.
Patients in arm A had a 95% reduction in all bleeds (P<0.0001), and patients in arm B had a 94% reduction in all bleeds (P<0.0001), compared to patients in arm C.
Fifty percent of patients in arm A had 0 total bleeds, as did 40% of patients in arm B and 0% of patients in arm C.
Intra-patient comparison
The researchers compared previous prophylaxis to once-weekly emicizumab prophylaxis in 48 patients from arm D.
The model-based ABR for treated bleeds was 4.8 with prior prophylaxis and 1.5 with emicizumab. The median ABR for treated bleeds was 1.8 and 0.0, respectively.
Patients had a 68% reduction in treated bleeds with emicizumab (P<0.0001).
With prior prophylaxis, 39.6% of patients had 0 treated bleeds. With emicizumab, 54.2% of patients had 0 treated bleeds.
Safety
There were no serious adverse events (AEs) related to emicizumab, no anti-drug antibodies detected, and none of the patients on emicizumab developed de novo factor VIII inhibitors.
Injection-site reactions occurred in 25.3% of all patients (38/150), 25% of patients in arm A (9/36), 20% in arm B (7/35), 12.5% in arm C (2/16), and 31.7% in arm D (20/63).
An additional patient in arm D (who was included in the total) reported an “injection-site erythema,” not an “injection-site reaction.”
Upper respiratory tract infections occurred in 10.7% of all patients (n=16), 11.1% (n=4) of those in arm A, 11.4% (n=4) of those in arm B, 0% of those in arm C, and 12.7% (n=8) of those in arm D.
Other AEs occurring in at least 5% of patients were arthralgia (19%), nasopharyngitis (12%), headache (11%), and influenza (6%).
One patient in arm B discontinued emicizumab due to multiple mild AEs—insomnia, hair loss, nightmare, lethargy, depressed mood, headache, and pruritus.
Two patients were lost to follow-up—1 in arm A and 1 in arm C.
GLASGOW—Final results from the HAVEN 3 study suggest emicizumab prophylaxis can reduce bleeding in hemophilia A patients without factor VIII inhibitors.
Compared to patients who did not receive prophylaxis, those who received emicizumab prophylaxis had a 96% to 97% reduction in treated bleeds and a 94% to 95% reduction in all bleeds.
An intra-patient comparison showed a 68% reduction in treated bleeds with once-weekly emicizumab, compared to prior factor VIII prophylaxis.
“[Emicizumab] is the first medicine to show superior efficacy to prior factor VIII prophylaxis, the current standard of care therapy, as demonstrated by a statistically significant reduction in treated bleeds in the HAVEN 3 study intra-patient comparison,” said Johnny Mahlangu, MB BCh, of the University of the Witwatersrand in Johannesburg, South Africa.
Dr Mahlangu presented these results at the World Federation of Hemophilia (WFH) 2018 World Congress during the late-breaking abstract session on Monday. HAVEN 3 was sponsored by Hoffmann-La Roche.
Patients and treatment
In this phase 3 trial, researchers evaluated emicizumab in patients with hemophilia A without factor VIII inhibitors. The study included 152 patients who were 12 years of age or older and were previously treated with factor VIII therapy on-demand or as prophylaxis.
Patients previously treated with on-demand factor VIII were randomized in a 2:2:1 fashion to receive:
- Emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study (arm A, n=36)
- Emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 3 mg/kg/2wks for at least 24 weeks (arm B, n=35)
- No prophylaxis, only episodic/on-demand factor VIII treatment (arm C, n=18).
Patients previously treated with factor VIII prophylaxis received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study (arm D, n=63).
Episodic treatment of breakthrough bleeds with factor VIII therapy was allowed per protocol.
Emicizumab vs no prophylaxis
The model-based (negative binomial regression model) annualized bleeding rate (ABR) for treated bleeds was 1.5 in arm A, 1.3 in arm B, and 38.2 in arm C. The median ABR for treated bleeds was 0 in arms A and B and 40.4 in arm C.
Compared to patients in arm C, those in arm A had a 96% (P<0.0001) reduction in treated bleeds, and those in arm B had a 97% (P<0.0001) reduction in treated bleeds.
None of the patients in arm C had 0 treated bleeds, compared to 55.6% of patients in arm A and 60% of patients in arm B.
The model-based ABR for all bleeds was 2.5 in arm A, 2.6 in arm B, and 47.6 in arm C.
Patients in arm A had a 95% reduction in all bleeds (P<0.0001), and patients in arm B had a 94% reduction in all bleeds (P<0.0001), compared to patients in arm C.
Fifty percent of patients in arm A had 0 total bleeds, as did 40% of patients in arm B and 0% of patients in arm C.
Intra-patient comparison
The researchers compared previous prophylaxis to once-weekly emicizumab prophylaxis in 48 patients from arm D.
The model-based ABR for treated bleeds was 4.8 with prior prophylaxis and 1.5 with emicizumab. The median ABR for treated bleeds was 1.8 and 0.0, respectively.
Patients had a 68% reduction in treated bleeds with emicizumab (P<0.0001).
With prior prophylaxis, 39.6% of patients had 0 treated bleeds. With emicizumab, 54.2% of patients had 0 treated bleeds.
Safety
There were no serious adverse events (AEs) related to emicizumab, no anti-drug antibodies detected, and none of the patients on emicizumab developed de novo factor VIII inhibitors.
Injection-site reactions occurred in 25.3% of all patients (38/150), 25% of patients in arm A (9/36), 20% in arm B (7/35), 12.5% in arm C (2/16), and 31.7% in arm D (20/63).
An additional patient in arm D (who was included in the total) reported an “injection-site erythema,” not an “injection-site reaction.”
Upper respiratory tract infections occurred in 10.7% of all patients (n=16), 11.1% (n=4) of those in arm A, 11.4% (n=4) of those in arm B, 0% of those in arm C, and 12.7% (n=8) of those in arm D.
Other AEs occurring in at least 5% of patients were arthralgia (19%), nasopharyngitis (12%), headache (11%), and influenza (6%).
One patient in arm B discontinued emicizumab due to multiple mild AEs—insomnia, hair loss, nightmare, lethargy, depressed mood, headache, and pruritus.
Two patients were lost to follow-up—1 in arm A and 1 in arm C.
GLASGOW—Final results from the HAVEN 3 study suggest emicizumab prophylaxis can reduce bleeding in hemophilia A patients without factor VIII inhibitors.
Compared to patients who did not receive prophylaxis, those who received emicizumab prophylaxis had a 96% to 97% reduction in treated bleeds and a 94% to 95% reduction in all bleeds.
An intra-patient comparison showed a 68% reduction in treated bleeds with once-weekly emicizumab, compared to prior factor VIII prophylaxis.
“[Emicizumab] is the first medicine to show superior efficacy to prior factor VIII prophylaxis, the current standard of care therapy, as demonstrated by a statistically significant reduction in treated bleeds in the HAVEN 3 study intra-patient comparison,” said Johnny Mahlangu, MB BCh, of the University of the Witwatersrand in Johannesburg, South Africa.
Dr Mahlangu presented these results at the World Federation of Hemophilia (WFH) 2018 World Congress during the late-breaking abstract session on Monday. HAVEN 3 was sponsored by Hoffmann-La Roche.
Patients and treatment
In this phase 3 trial, researchers evaluated emicizumab in patients with hemophilia A without factor VIII inhibitors. The study included 152 patients who were 12 years of age or older and were previously treated with factor VIII therapy on-demand or as prophylaxis.
Patients previously treated with on-demand factor VIII were randomized in a 2:2:1 fashion to receive:
- Emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study (arm A, n=36)
- Emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 3 mg/kg/2wks for at least 24 weeks (arm B, n=35)
- No prophylaxis, only episodic/on-demand factor VIII treatment (arm C, n=18).
Patients previously treated with factor VIII prophylaxis received emicizumab prophylaxis at 3 mg/kg/wk for 4 weeks, followed by 1.5 mg/kg/wk until the end of study (arm D, n=63).
Episodic treatment of breakthrough bleeds with factor VIII therapy was allowed per protocol.
Emicizumab vs no prophylaxis
The model-based (negative binomial regression model) annualized bleeding rate (ABR) for treated bleeds was 1.5 in arm A, 1.3 in arm B, and 38.2 in arm C. The median ABR for treated bleeds was 0 in arms A and B and 40.4 in arm C.
Compared to patients in arm C, those in arm A had a 96% (P<0.0001) reduction in treated bleeds, and those in arm B had a 97% (P<0.0001) reduction in treated bleeds.
None of the patients in arm C had 0 treated bleeds, compared to 55.6% of patients in arm A and 60% of patients in arm B.
The model-based ABR for all bleeds was 2.5 in arm A, 2.6 in arm B, and 47.6 in arm C.
Patients in arm A had a 95% reduction in all bleeds (P<0.0001), and patients in arm B had a 94% reduction in all bleeds (P<0.0001), compared to patients in arm C.
Fifty percent of patients in arm A had 0 total bleeds, as did 40% of patients in arm B and 0% of patients in arm C.
Intra-patient comparison
The researchers compared previous prophylaxis to once-weekly emicizumab prophylaxis in 48 patients from arm D.
The model-based ABR for treated bleeds was 4.8 with prior prophylaxis and 1.5 with emicizumab. The median ABR for treated bleeds was 1.8 and 0.0, respectively.
Patients had a 68% reduction in treated bleeds with emicizumab (P<0.0001).
With prior prophylaxis, 39.6% of patients had 0 treated bleeds. With emicizumab, 54.2% of patients had 0 treated bleeds.
Safety
There were no serious adverse events (AEs) related to emicizumab, no anti-drug antibodies detected, and none of the patients on emicizumab developed de novo factor VIII inhibitors.
Injection-site reactions occurred in 25.3% of all patients (38/150), 25% of patients in arm A (9/36), 20% in arm B (7/35), 12.5% in arm C (2/16), and 31.7% in arm D (20/63).
An additional patient in arm D (who was included in the total) reported an “injection-site erythema,” not an “injection-site reaction.”
Upper respiratory tract infections occurred in 10.7% of all patients (n=16), 11.1% (n=4) of those in arm A, 11.4% (n=4) of those in arm B, 0% of those in arm C, and 12.7% (n=8) of those in arm D.
Other AEs occurring in at least 5% of patients were arthralgia (19%), nasopharyngitis (12%), headache (11%), and influenza (6%).
One patient in arm B discontinued emicizumab due to multiple mild AEs—insomnia, hair loss, nightmare, lethargy, depressed mood, headache, and pruritus.
Two patients were lost to follow-up—1 in arm A and 1 in arm C.
Structured PPH management cuts severe hemorrhage
AUSTIN, TEX. – Taking a page from critical care, an obstetrical team that implemented a checklist-based management protocol for postpartum hemorrhage saw a significant drop in severe obstetric hemorrhage, with numeric reductions in other maternal outcomes.
The protocol, piloted in a single hospital, is now being rolled out in all 28 hospitals of a large, multistate health care system.
“Our medical critical care colleagues long ago abandoned the notion that physician judgment should guide the provision of basic and advanced cardiac life support in favor of highly specific and uniform protocols,” wrote first author Rachael Smith, DO, and her coauthors in the poster accompanying the presentation at the annual clinical and scientific meeting of the American Society of Obstetricians and Gynecologists.
“While existing guidelines outlining a general approach to postpartum hemorrhage are useful, recent data suggest that greater specificity is necessary to significantly impact morbidity and mortality,” they wrote.
When comparing outcomes for 9 matched months before and after implementation of the protocol, Dr. Smith and her collaborators found that rates of severe postpartum hemorrhage (PPH), defined as estimated blood loss (EBL) of at least 2,500 cc, were halved, dropping from 18% to 9% (P = .035).

“Patients with life-threatening illnesses seem to do better when their providers are following very structured, regimented protocols, and [advanced cardiac life support protocols] is probably the best example of that,” said Ms. Hermann.
They then produced a training video to educate nursing and house staff and attending physicians about the new checklist-based protocol. In this way, each team member would understand the rationale behind the checklist, know the steps in the care pathway, and understand his or her specific role.
The protocol, which begins when uterine atony is suspected, first calls the physician to the patient room, along with a second nurse to be the recorder and timekeeper. Among other duties, this individual tracks blood loss during a maternal bleeding event, weighing linens and sponges, and alerting the team when EBL exceeds 500, 1,000, and 1,500 cc, or when pulse or blood pressure fall outside of designated parameters.
“Having a second nurse in the room who is keeping the team on track, saying ‘Hey, we’re at this much blood loss; these are the next steps,’ and who is recording everything” can avert the sense of chaos that sometimes occurs in critical scenarios, said Ms. Hermann.
When stage 1 PPH (EBL of at least 500 cc) has occurred, a team lead is called. At this point, a PPH cart containing necessary equipment and medication, including uterotonics, is brought to the room.
Having the uterotonic kit in the room, said Ms. Hermann, is a key component of the protocol. “Having a kit you can wheel into the room, and having everything you need to manage PPH” saves critical time, she said. “The nurses aren’t running back and forth to the Pyxis to get the next uterotonic that you need.”
If EBL of at least 1,500 cc is reached, a third nurse is called and the obstetric rapid-response team is activated, meaning that a code cart and additional supportive equipment are also brought to the patient.
The checklist paperwork lays out all interventions, including uterotonic dosing, timing, and contraindications. It also includes differential diagnoses for PPH, and provides directions for visual estimation of blood loss.
Finally, a structured debrief takes place after each PPH, said Ms. Hermann.
The study included women who experienced PPH during matched 9-month periods before and after the PPH protocol implementation. PPH was defined as EBL of at least 500 cc for vaginal delivery, and 1,000 cc for cesarean delivery. Women were excluded if they delivered before 22 weeks’ gestation, or if there was a diagnosis of or suspicion for placenta accreta, increta, or percreta.
A total of 147 women were in the preintervention group; of these, 98 (66%) had vaginal deliveries. In the postintervention group, 110 out of150 women (73%) had vaginal deliveries.
In addition to the significant reduction in severe PPH that followed implementation of the protocol, numeric reductions were also seen in other surrogate measures of maternal morbidity, including stage 1 hemorrhage, the need for transfusion, surgical interventions, intensive care admissions, and length of stay.
“Across all of these surrogates, we saw an improvement in our postprotocol patients,” said Ms. Hermann. “We think that the reason the rest of them weren’t statistically significant was due to lack of power” in the single-center study, she said. “The clinical trend speaks for itself.”
Once the protocol is rolled out in all 28 hospitals, she anticipates seeing statistics that confirm what the investigators are already seeing clinically.
SOURCE: Smith R et al. ACOG 2018, Abstract 26R.
AUSTIN, TEX. – Taking a page from critical care, an obstetrical team that implemented a checklist-based management protocol for postpartum hemorrhage saw a significant drop in severe obstetric hemorrhage, with numeric reductions in other maternal outcomes.
The protocol, piloted in a single hospital, is now being rolled out in all 28 hospitals of a large, multistate health care system.
“Our medical critical care colleagues long ago abandoned the notion that physician judgment should guide the provision of basic and advanced cardiac life support in favor of highly specific and uniform protocols,” wrote first author Rachael Smith, DO, and her coauthors in the poster accompanying the presentation at the annual clinical and scientific meeting of the American Society of Obstetricians and Gynecologists.
“While existing guidelines outlining a general approach to postpartum hemorrhage are useful, recent data suggest that greater specificity is necessary to significantly impact morbidity and mortality,” they wrote.
When comparing outcomes for 9 matched months before and after implementation of the protocol, Dr. Smith and her collaborators found that rates of severe postpartum hemorrhage (PPH), defined as estimated blood loss (EBL) of at least 2,500 cc, were halved, dropping from 18% to 9% (P = .035).
“Patients with life-threatening illnesses seem to do better when their providers are following very structured, regimented protocols, and [advanced cardiac life support protocols] is probably the best example of that,” said Ms. Hermann.
They then produced a training video to educate nursing and house staff and attending physicians about the new checklist-based protocol. In this way, each team member would understand the rationale behind the checklist, know the steps in the care pathway, and understand his or her specific role.
The protocol, which begins when uterine atony is suspected, first calls the physician to the patient room, along with a second nurse to be the recorder and timekeeper. Among other duties, this individual tracks blood loss during a maternal bleeding event, weighing linens and sponges, and alerting the team when EBL exceeds 500, 1,000, and 1,500 cc, or when pulse or blood pressure fall outside of designated parameters.
“Having a second nurse in the room who is keeping the team on track, saying ‘Hey, we’re at this much blood loss; these are the next steps,’ and who is recording everything” can avert the sense of chaos that sometimes occurs in critical scenarios, said Ms. Hermann.
When stage 1 PPH (EBL of at least 500 cc) has occurred, a team lead is called. At this point, a PPH cart containing necessary equipment and medication, including uterotonics, is brought to the room.
Having the uterotonic kit in the room, said Ms. Hermann, is a key component of the protocol. “Having a kit you can wheel into the room, and having everything you need to manage PPH” saves critical time, she said. “The nurses aren’t running back and forth to the Pyxis to get the next uterotonic that you need.”
If EBL of at least 1,500 cc is reached, a third nurse is called and the obstetric rapid-response team is activated, meaning that a code cart and additional supportive equipment are also brought to the patient.
The checklist paperwork lays out all interventions, including uterotonic dosing, timing, and contraindications. It also includes differential diagnoses for PPH, and provides directions for visual estimation of blood loss.
Finally, a structured debrief takes place after each PPH, said Ms. Hermann.
The study included women who experienced PPH during matched 9-month periods before and after the PPH protocol implementation. PPH was defined as EBL of at least 500 cc for vaginal delivery, and 1,000 cc for cesarean delivery. Women were excluded if they delivered before 22 weeks’ gestation, or if there was a diagnosis of or suspicion for placenta accreta, increta, or percreta.
A total of 147 women were in the preintervention group; of these, 98 (66%) had vaginal deliveries. In the postintervention group, 110 out of150 women (73%) had vaginal deliveries.
In addition to the significant reduction in severe PPH that followed implementation of the protocol, numeric reductions were also seen in other surrogate measures of maternal morbidity, including stage 1 hemorrhage, the need for transfusion, surgical interventions, intensive care admissions, and length of stay.
“Across all of these surrogates, we saw an improvement in our postprotocol patients,” said Ms. Hermann. “We think that the reason the rest of them weren’t statistically significant was due to lack of power” in the single-center study, she said. “The clinical trend speaks for itself.”
Once the protocol is rolled out in all 28 hospitals, she anticipates seeing statistics that confirm what the investigators are already seeing clinically.
SOURCE: Smith R et al. ACOG 2018, Abstract 26R.
AUSTIN, TEX. – Taking a page from critical care, an obstetrical team that implemented a checklist-based management protocol for postpartum hemorrhage saw a significant drop in severe obstetric hemorrhage, with numeric reductions in other maternal outcomes.
The protocol, piloted in a single hospital, is now being rolled out in all 28 hospitals of a large, multistate health care system.
“Our medical critical care colleagues long ago abandoned the notion that physician judgment should guide the provision of basic and advanced cardiac life support in favor of highly specific and uniform protocols,” wrote first author Rachael Smith, DO, and her coauthors in the poster accompanying the presentation at the annual clinical and scientific meeting of the American Society of Obstetricians and Gynecologists.
“While existing guidelines outlining a general approach to postpartum hemorrhage are useful, recent data suggest that greater specificity is necessary to significantly impact morbidity and mortality,” they wrote.
When comparing outcomes for 9 matched months before and after implementation of the protocol, Dr. Smith and her collaborators found that rates of severe postpartum hemorrhage (PPH), defined as estimated blood loss (EBL) of at least 2,500 cc, were halved, dropping from 18% to 9% (P = .035).
“Patients with life-threatening illnesses seem to do better when their providers are following very structured, regimented protocols, and [advanced cardiac life support protocols] is probably the best example of that,” said Ms. Hermann.
They then produced a training video to educate nursing and house staff and attending physicians about the new checklist-based protocol. In this way, each team member would understand the rationale behind the checklist, know the steps in the care pathway, and understand his or her specific role.
The protocol, which begins when uterine atony is suspected, first calls the physician to the patient room, along with a second nurse to be the recorder and timekeeper. Among other duties, this individual tracks blood loss during a maternal bleeding event, weighing linens and sponges, and alerting the team when EBL exceeds 500, 1,000, and 1,500 cc, or when pulse or blood pressure fall outside of designated parameters.
“Having a second nurse in the room who is keeping the team on track, saying ‘Hey, we’re at this much blood loss; these are the next steps,’ and who is recording everything” can avert the sense of chaos that sometimes occurs in critical scenarios, said Ms. Hermann.
When stage 1 PPH (EBL of at least 500 cc) has occurred, a team lead is called. At this point, a PPH cart containing necessary equipment and medication, including uterotonics, is brought to the room.
Having the uterotonic kit in the room, said Ms. Hermann, is a key component of the protocol. “Having a kit you can wheel into the room, and having everything you need to manage PPH” saves critical time, she said. “The nurses aren’t running back and forth to the Pyxis to get the next uterotonic that you need.”
If EBL of at least 1,500 cc is reached, a third nurse is called and the obstetric rapid-response team is activated, meaning that a code cart and additional supportive equipment are also brought to the patient.
The checklist paperwork lays out all interventions, including uterotonic dosing, timing, and contraindications. It also includes differential diagnoses for PPH, and provides directions for visual estimation of blood loss.
Finally, a structured debrief takes place after each PPH, said Ms. Hermann.
The study included women who experienced PPH during matched 9-month periods before and after the PPH protocol implementation. PPH was defined as EBL of at least 500 cc for vaginal delivery, and 1,000 cc for cesarean delivery. Women were excluded if they delivered before 22 weeks’ gestation, or if there was a diagnosis of or suspicion for placenta accreta, increta, or percreta.
A total of 147 women were in the preintervention group; of these, 98 (66%) had vaginal deliveries. In the postintervention group, 110 out of150 women (73%) had vaginal deliveries.
In addition to the significant reduction in severe PPH that followed implementation of the protocol, numeric reductions were also seen in other surrogate measures of maternal morbidity, including stage 1 hemorrhage, the need for transfusion, surgical interventions, intensive care admissions, and length of stay.
“Across all of these surrogates, we saw an improvement in our postprotocol patients,” said Ms. Hermann. “We think that the reason the rest of them weren’t statistically significant was due to lack of power” in the single-center study, she said. “The clinical trend speaks for itself.”
Once the protocol is rolled out in all 28 hospitals, she anticipates seeing statistics that confirm what the investigators are already seeing clinically.
SOURCE: Smith R et al. ACOG 2018, Abstract 26R.
REPORTING FROM ACOG 2018
Key clinical point: Indicators of maternal morbidity decreased after a postpartum hemorrhage checklist was implemented.
Major finding: Severe postpartum hemorrhage rates fell from 18% to 9% (P = .035).
Study details: A prospective pre/post implementation study of 297 women experiencing postpartum hemorrhage.
Disclosures: The study authors reported no relevant financial conflicts of interest.
Source: Smith R et al. ACOG 2018, Abstract 26R.
Hydroxyurea in infancy yields better SCD outcomes
PITTSBURGH – Children with sickle cell disease (SCD) who were started on hydroxyurea in infancy had significantly better outcomes than do children started on the drug as toddlers, researchers report.
Among 65 children with SCD, those who started on hydroxyurea before age 1 year had significantly fewer hospitalizations, pain crises, and transfusions in the first 2 years of life, compared with patients started on the drug during 1-2 years of age or after age 2 years, found Sarah B. Schuchard, PharmD, from the SSM Health Cardinal Glennon Children’s Hospital in St. Louis and her colleagues.

At the Children’s Hospital and Clinics of Minnesota in Minneapolis, where Dr. Schuchard recently completed her training and conducted the research, the goal is to initiate all infants with sickle cell anemia and sickle-beta0-thalassemia on hydroxyurea within this first year of life.
To evaluate outcomes associated with this practice, the investigators conducted a retrospective review of all children with SCD who began hydroxyurea therapy at their center during 2008-2016.
They divided the population into three cohorts. Patients in cohort 1 were started on hydroxyurea before age 1 year (35 patients; mean age, 7.2 months), those in cohort 2 started between 1 and 2 years of age (13 patients; mean age, 19.5 months) and those in cohort 3 were started after 2 years of age (three patients; mean age, 35.5 months).
All patients had been diagnosed with either sickle cell anemia or sickle-beta0-thalassemia, and all had at least two laboratory assessments at 6 months, 12 months, 18 months, or 24 months of age.
For the coprimary endpoint of laboratory data, the investigators found that patients in cohort 1, the early starters, had significantly higher hemoglobin (P = .0003), lower absolute reticulocyte counts (P = .0304), and mean corpuscular volume (P = .0199) than did the patients in cohort 3.
Infants in cohort 1 had significantly lower white blood cell counts than did the patients in either cohorts 2 or 3 (P = .0007 and P less than .0001, respectively) and lower absolute neutrophil counts (P = .0364 and .0025, respectively), although no patients required hydroxyurea therapy to be held because of low ANC.
Clinical events, the other coprimary endpoint, were also significantly better among patients in cohort 1, who had significantly fewer hospitalizations (P = .0025), a trend toward fewer painful events (P = .0618), and significantly fewer transfusions (P = .0426) than did patients in the other two cohorts.
“Early hydroxyurea also appears to contribute to fewer pain crises requiring admission,” the investigators noted.
They noted that in their study, the hematologic response was greater than that seen in the BABY HUG study, which studied the protective effects of hydroxyurea in children aged 9-18 months. The mean age of hydroxyurea initiation was 13.6 months in that study, compared with 7.2 months in the study by Dr. Schuchard and her colleagues.
“It would be interesting to see if the splenic and renal function (the unmet primary endpoints of BABY HUG) are preserved in patients starting hydroxyurea at this younger age,” they wrote.
The study was internally funded. Dr. Schuchard reported having no conflicts of interest.
SOURCE: Schuchard S et al. ASPHO 2018, Poster 342.
PITTSBURGH – Children with sickle cell disease (SCD) who were started on hydroxyurea in infancy had significantly better outcomes than do children started on the drug as toddlers, researchers report.
Among 65 children with SCD, those who started on hydroxyurea before age 1 year had significantly fewer hospitalizations, pain crises, and transfusions in the first 2 years of life, compared with patients started on the drug during 1-2 years of age or after age 2 years, found Sarah B. Schuchard, PharmD, from the SSM Health Cardinal Glennon Children’s Hospital in St. Louis and her colleagues.
At the Children’s Hospital and Clinics of Minnesota in Minneapolis, where Dr. Schuchard recently completed her training and conducted the research, the goal is to initiate all infants with sickle cell anemia and sickle-beta0-thalassemia on hydroxyurea within this first year of life.
To evaluate outcomes associated with this practice, the investigators conducted a retrospective review of all children with SCD who began hydroxyurea therapy at their center during 2008-2016.
They divided the population into three cohorts. Patients in cohort 1 were started on hydroxyurea before age 1 year (35 patients; mean age, 7.2 months), those in cohort 2 started between 1 and 2 years of age (13 patients; mean age, 19.5 months) and those in cohort 3 were started after 2 years of age (three patients; mean age, 35.5 months).
All patients had been diagnosed with either sickle cell anemia or sickle-beta0-thalassemia, and all had at least two laboratory assessments at 6 months, 12 months, 18 months, or 24 months of age.
For the coprimary endpoint of laboratory data, the investigators found that patients in cohort 1, the early starters, had significantly higher hemoglobin (P = .0003), lower absolute reticulocyte counts (P = .0304), and mean corpuscular volume (P = .0199) than did the patients in cohort 3.
Infants in cohort 1 had significantly lower white blood cell counts than did the patients in either cohorts 2 or 3 (P = .0007 and P less than .0001, respectively) and lower absolute neutrophil counts (P = .0364 and .0025, respectively), although no patients required hydroxyurea therapy to be held because of low ANC.
Clinical events, the other coprimary endpoint, were also significantly better among patients in cohort 1, who had significantly fewer hospitalizations (P = .0025), a trend toward fewer painful events (P = .0618), and significantly fewer transfusions (P = .0426) than did patients in the other two cohorts.
“Early hydroxyurea also appears to contribute to fewer pain crises requiring admission,” the investigators noted.
They noted that in their study, the hematologic response was greater than that seen in the BABY HUG study, which studied the protective effects of hydroxyurea in children aged 9-18 months. The mean age of hydroxyurea initiation was 13.6 months in that study, compared with 7.2 months in the study by Dr. Schuchard and her colleagues.
“It would be interesting to see if the splenic and renal function (the unmet primary endpoints of BABY HUG) are preserved in patients starting hydroxyurea at this younger age,” they wrote.
The study was internally funded. Dr. Schuchard reported having no conflicts of interest.
SOURCE: Schuchard S et al. ASPHO 2018, Poster 342.
PITTSBURGH – Children with sickle cell disease (SCD) who were started on hydroxyurea in infancy had significantly better outcomes than do children started on the drug as toddlers, researchers report.
Among 65 children with SCD, those who started on hydroxyurea before age 1 year had significantly fewer hospitalizations, pain crises, and transfusions in the first 2 years of life, compared with patients started on the drug during 1-2 years of age or after age 2 years, found Sarah B. Schuchard, PharmD, from the SSM Health Cardinal Glennon Children’s Hospital in St. Louis and her colleagues.
At the Children’s Hospital and Clinics of Minnesota in Minneapolis, where Dr. Schuchard recently completed her training and conducted the research, the goal is to initiate all infants with sickle cell anemia and sickle-beta0-thalassemia on hydroxyurea within this first year of life.
To evaluate outcomes associated with this practice, the investigators conducted a retrospective review of all children with SCD who began hydroxyurea therapy at their center during 2008-2016.
They divided the population into three cohorts. Patients in cohort 1 were started on hydroxyurea before age 1 year (35 patients; mean age, 7.2 months), those in cohort 2 started between 1 and 2 years of age (13 patients; mean age, 19.5 months) and those in cohort 3 were started after 2 years of age (three patients; mean age, 35.5 months).
All patients had been diagnosed with either sickle cell anemia or sickle-beta0-thalassemia, and all had at least two laboratory assessments at 6 months, 12 months, 18 months, or 24 months of age.
For the coprimary endpoint of laboratory data, the investigators found that patients in cohort 1, the early starters, had significantly higher hemoglobin (P = .0003), lower absolute reticulocyte counts (P = .0304), and mean corpuscular volume (P = .0199) than did the patients in cohort 3.
Infants in cohort 1 had significantly lower white blood cell counts than did the patients in either cohorts 2 or 3 (P = .0007 and P less than .0001, respectively) and lower absolute neutrophil counts (P = .0364 and .0025, respectively), although no patients required hydroxyurea therapy to be held because of low ANC.
Clinical events, the other coprimary endpoint, were also significantly better among patients in cohort 1, who had significantly fewer hospitalizations (P = .0025), a trend toward fewer painful events (P = .0618), and significantly fewer transfusions (P = .0426) than did patients in the other two cohorts.
“Early hydroxyurea also appears to contribute to fewer pain crises requiring admission,” the investigators noted.
They noted that in their study, the hematologic response was greater than that seen in the BABY HUG study, which studied the protective effects of hydroxyurea in children aged 9-18 months. The mean age of hydroxyurea initiation was 13.6 months in that study, compared with 7.2 months in the study by Dr. Schuchard and her colleagues.
“It would be interesting to see if the splenic and renal function (the unmet primary endpoints of BABY HUG) are preserved in patients starting hydroxyurea at this younger age,” they wrote.
The study was internally funded. Dr. Schuchard reported having no conflicts of interest.
SOURCE: Schuchard S et al. ASPHO 2018, Poster 342.
REPORTING FROM ASPHO 2018
Key clinical point: Early initiation of hydroxyurea is associated with better SCD outcomes.
Major finding: Patients started on hydroxyurea at a mean of 7.2 months had significantly fewer admissions, pain crises, and transfusions than did patients started after age 1 year.
Study details: Retrospective review of data on 65 children with SCD in a single center.
Disclosures: The study was internally funded. Dr. Schuchard reported having no conflicts of interest.
Source: Schuchard S et al. ASPHO 2018, Poster 342.
Adding vasopressin in distributive shock may cut AF risk
In patients with distributive shock, the risk of atrial fibrillation may be lower when vasopressin is administered along with catecholamine vasopressors, results of a recent systematic review and meta-analysis suggest.
The relative risk of atrial fibrillation was reduced for the combination of vasopressin and catecholamines versus the current standard of care, which is catecholamines alone, according to study results published in JAMA.
Beyond atrial fibrillation, however, findings of the meta-analysis were consistent with regard to other endpoints, including mortality, according to William F. McIntyre, MD, of McMaster University, Hamilton, Ont., and his coinvestigators.
Mortality was lower with the combination approach when all studies were analyzed together. Yet, when the analysis was limited to the studies with the lowest risk of bias, the difference in mortality versus catecholamines alone was not statistically significant, investigators said.
Nevertheless, the meta-analysis does suggest that vasopressin may offer a clinical advantage regarding prevention of atrial fibrillation in patients with distributive shock, a frequently fatal condition most often seen in patients with sepsis.
Vasopressin is an endogenous peptide hormone that decreases stimulation of certain myocardial receptors associated with cardiac arrhythmia, the authors noted.
“This, among other mechanisms, may translate into a reduction in adverse events, including atrial fibrillation, injury to other organs, and death,” they said in their report.
Dr. McIntyre and his colleagues included 23 trials that had enrolled a total of 3,088 patients with distributive shock, a condition in which widespread vasodilation lowers vascular resistances and mean arterial pressure. Sepsis is its most common cause. The current study is one of the first to directly compare the combination of vasopressin and catecholamine to catecholamines alone, which is the current standard of care, the investigators wrote.
They found that the administration of vasopressin was associated with a significant 23% reduction in risk of atrial fibrillation.
“The absolute effect is that 68 fewer people per 1,000 patients will experience atrial fibrillation when vasopressin is added to catecholaminergic vasopressors,” Dr. McIntyre and his coauthors said of the results.
The atrial fibrillation finding was judged to be high-quality evidence, they said, noting that two separate sensitivity analyses confirmed the benefit.
Mortality data were less consistent, they said.
Pooled data showed administration of vasopressin along with catecholamines was associated an 11% relative reduction in mortality. In absolute terms, 45 lives would be saved for every 1,000 patients receiving vasopressin, they noted.
However, the mortality findings were different when the analysis was limited to the two studies with low risk of bias. That analysis yielded a relative risk of 0.96 and was not statistically significant.
Studies show patients with distributive shock have a relative vasopressin deficiency, providing a theoretical basis for vasopressin administration as part of care, investigators said.
The current Surviving Sepsis guidelines suggest either adding vasopressin to norepinephrine to help raise mean arterial pressure to target or adding vasopressin to decrease the dosage of norepinephrine. Those are considered weak recommendations based on moderate quality of evidence, Dr. McIntyre and colleagues noted in their report.
Authors of the study reported disclosures related to Tenax Therapeutics, Orion Pharma, Ferring Pharmaceuticals, GlaxoSmithKline, and Bristol-Myers Squibb, among other entities.
SOURCE: McIntyre WF et al. JAMA. 2018;319(18):1889-900.
In patients with distributive shock, the risk of atrial fibrillation may be lower when vasopressin is administered along with catecholamine vasopressors, results of a recent systematic review and meta-analysis suggest.
The relative risk of atrial fibrillation was reduced for the combination of vasopressin and catecholamines versus the current standard of care, which is catecholamines alone, according to study results published in JAMA.
Beyond atrial fibrillation, however, findings of the meta-analysis were consistent with regard to other endpoints, including mortality, according to William F. McIntyre, MD, of McMaster University, Hamilton, Ont., and his coinvestigators.
Mortality was lower with the combination approach when all studies were analyzed together. Yet, when the analysis was limited to the studies with the lowest risk of bias, the difference in mortality versus catecholamines alone was not statistically significant, investigators said.
Nevertheless, the meta-analysis does suggest that vasopressin may offer a clinical advantage regarding prevention of atrial fibrillation in patients with distributive shock, a frequently fatal condition most often seen in patients with sepsis.
Vasopressin is an endogenous peptide hormone that decreases stimulation of certain myocardial receptors associated with cardiac arrhythmia, the authors noted.
“This, among other mechanisms, may translate into a reduction in adverse events, including atrial fibrillation, injury to other organs, and death,” they said in their report.
Dr. McIntyre and his colleagues included 23 trials that had enrolled a total of 3,088 patients with distributive shock, a condition in which widespread vasodilation lowers vascular resistances and mean arterial pressure. Sepsis is its most common cause. The current study is one of the first to directly compare the combination of vasopressin and catecholamine to catecholamines alone, which is the current standard of care, the investigators wrote.
They found that the administration of vasopressin was associated with a significant 23% reduction in risk of atrial fibrillation.
“The absolute effect is that 68 fewer people per 1,000 patients will experience atrial fibrillation when vasopressin is added to catecholaminergic vasopressors,” Dr. McIntyre and his coauthors said of the results.
The atrial fibrillation finding was judged to be high-quality evidence, they said, noting that two separate sensitivity analyses confirmed the benefit.
Mortality data were less consistent, they said.
Pooled data showed administration of vasopressin along with catecholamines was associated an 11% relative reduction in mortality. In absolute terms, 45 lives would be saved for every 1,000 patients receiving vasopressin, they noted.
However, the mortality findings were different when the analysis was limited to the two studies with low risk of bias. That analysis yielded a relative risk of 0.96 and was not statistically significant.
Studies show patients with distributive shock have a relative vasopressin deficiency, providing a theoretical basis for vasopressin administration as part of care, investigators said.
The current Surviving Sepsis guidelines suggest either adding vasopressin to norepinephrine to help raise mean arterial pressure to target or adding vasopressin to decrease the dosage of norepinephrine. Those are considered weak recommendations based on moderate quality of evidence, Dr. McIntyre and colleagues noted in their report.
Authors of the study reported disclosures related to Tenax Therapeutics, Orion Pharma, Ferring Pharmaceuticals, GlaxoSmithKline, and Bristol-Myers Squibb, among other entities.
SOURCE: McIntyre WF et al. JAMA. 2018;319(18):1889-900.
In patients with distributive shock, the risk of atrial fibrillation may be lower when vasopressin is administered along with catecholamine vasopressors, results of a recent systematic review and meta-analysis suggest.
The relative risk of atrial fibrillation was reduced for the combination of vasopressin and catecholamines versus the current standard of care, which is catecholamines alone, according to study results published in JAMA.
Beyond atrial fibrillation, however, findings of the meta-analysis were consistent with regard to other endpoints, including mortality, according to William F. McIntyre, MD, of McMaster University, Hamilton, Ont., and his coinvestigators.
Mortality was lower with the combination approach when all studies were analyzed together. Yet, when the analysis was limited to the studies with the lowest risk of bias, the difference in mortality versus catecholamines alone was not statistically significant, investigators said.
Nevertheless, the meta-analysis does suggest that vasopressin may offer a clinical advantage regarding prevention of atrial fibrillation in patients with distributive shock, a frequently fatal condition most often seen in patients with sepsis.
Vasopressin is an endogenous peptide hormone that decreases stimulation of certain myocardial receptors associated with cardiac arrhythmia, the authors noted.
“This, among other mechanisms, may translate into a reduction in adverse events, including atrial fibrillation, injury to other organs, and death,” they said in their report.
Dr. McIntyre and his colleagues included 23 trials that had enrolled a total of 3,088 patients with distributive shock, a condition in which widespread vasodilation lowers vascular resistances and mean arterial pressure. Sepsis is its most common cause. The current study is one of the first to directly compare the combination of vasopressin and catecholamine to catecholamines alone, which is the current standard of care, the investigators wrote.
They found that the administration of vasopressin was associated with a significant 23% reduction in risk of atrial fibrillation.
“The absolute effect is that 68 fewer people per 1,000 patients will experience atrial fibrillation when vasopressin is added to catecholaminergic vasopressors,” Dr. McIntyre and his coauthors said of the results.
The atrial fibrillation finding was judged to be high-quality evidence, they said, noting that two separate sensitivity analyses confirmed the benefit.
Mortality data were less consistent, they said.
Pooled data showed administration of vasopressin along with catecholamines was associated an 11% relative reduction in mortality. In absolute terms, 45 lives would be saved for every 1,000 patients receiving vasopressin, they noted.
However, the mortality findings were different when the analysis was limited to the two studies with low risk of bias. That analysis yielded a relative risk of 0.96 and was not statistically significant.
Studies show patients with distributive shock have a relative vasopressin deficiency, providing a theoretical basis for vasopressin administration as part of care, investigators said.
The current Surviving Sepsis guidelines suggest either adding vasopressin to norepinephrine to help raise mean arterial pressure to target or adding vasopressin to decrease the dosage of norepinephrine. Those are considered weak recommendations based on moderate quality of evidence, Dr. McIntyre and colleagues noted in their report.
Authors of the study reported disclosures related to Tenax Therapeutics, Orion Pharma, Ferring Pharmaceuticals, GlaxoSmithKline, and Bristol-Myers Squibb, among other entities.
SOURCE: McIntyre WF et al. JAMA. 2018;319(18):1889-900.
FROM JAMA
Key clinical point: For patients with distributive shock, the addition of vasopressin to catecholamine vasopressors may reduce atrial fibrillation risk, compared with catecholamines alone.
Major finding: Vasopressin was associated with a 23% lower risk of atrial fibrillation.
Study details: A systematic review and meta-analysis including 23 randomized clinical trials enrolling a total of 3,088 patients.
Disclosures: Authors reported disclosures related to Tenax Therapeutics, Orion Pharma, Ferring Pharmaceuticals, GlaxoSmithKline, and Bristol-Myers Squibb, among other entities.
Source: McIntyre WF et al. JAMA. 2018;319(18):1889-900.
NHLBI seeks to accelerate hemostasis/thrombosis research
SAN DIEGO – The National Heart, Lung, and Blood Institute is looking to jump-start research into hemostasis and thrombosis. Donna DiMichele, MD, offered two reasons for the research push.
“The first is to stimulate research in areas that are undersubscribed through investigator-initiated research,” Dr. DiMichele, deputy director of the NHLBI’s Division of Blood Diseases and Resources, said at the biennial summit of the Thrombosis & Hemostasis Societies of North America. “We also do it sometimes to steer research in directions that are not developing organically through investigator-initiated efforts. In hemostasis and thrombosis, 50% of the investigator-led research is basic in nature. Nonetheless, there were several basic research initiatives that NHLBI released in the last few years that stimulated the field in a new direction.”

The centers will be required to look beyond current active science disciplines in this field, to include emerging sciences and technologies not currently being exploited in this research area. The initiative is also intended to cross-train the next generation of physicians/scientists with interdisciplinary skill sets. “The research teams with successful applications are going to begin their work by the summer of 2018, and hopefully we’ll soon gain some new insights into Factor VIII immunogenicity,” Dr. DiMichele said.
One novel scientific area that successful applicants will be able to take advantage of is the Trans-Omics for Precision Medicine Program (TOPMed). Launched in 2014, this program facilitates whole-genome sequencing across cohorts in heart, lung, blood, and sleep science. TOPMed now includes more than 120,000 whole genomes, with more than 5,000 of those in hemophilia.
In basic science, two NHLBI-funded initiatives aim to improve the understanding of thrombosis.
One, Sex Hormone Induced Thromboembolism in Premenopausal Women (R61/R33), is meant to elucidate the mechanisms by which female sex hormones and sex hormone-based therapies can increase the risk of venous and arterial thromboembolism in premenopausal women. The other initiative, Consortium Linking Oncology with Thrombosis (U01), is aimed at encouraging studies in individual cancer types that expand investigation into the intersection between cancer and thrombotic pathways. It also aims to help researchers identify and develop biomarkers of thrombotic risk or cancer progression, and new strategies for preventing or treating the deleterious interplay between cancer, cancer therapy, and hemostasis/thrombosis.
In the translational research arena, the NHLBI released an initiative on Perinatal Stroke (R01) in 2017. It solicits applications that propose basic and/or translational research studies related to the developing neurovascular unit, perinatal injury/repair response, and/or stroke-related etiologies and risk factors. The purpose is to stimulate research that will identify therapeutic targets in perinatal stroke.
Other NHLBI-sponsored efforts include the Trans-Agency Research Consortium for Trauma-Induced Coagulopathy (TACTIC), as well as the Translational Research Centers in Thrombotic and Hemostatic Disorders program, which set out to enhance the translation of basic research discoveries that could lead to improved prevention, diagnosis, and treatment for thrombotic and hemostatic disorders. This program ended but it led to the development of several projects that are moving into the commercialization phase.
Finally, to further stimulate research in FVIII immunogenicity, on May 15-16, 2018, the NHBLI will host a free workshop entitled “Factor VIII Inhibitors: Generating a Blueprint for Future Research.” For information, visit https://factorviiinhibitors.eventbrite.com.
Dr. DiMichele reported having no financial disclosures.
SAN DIEGO – The National Heart, Lung, and Blood Institute is looking to jump-start research into hemostasis and thrombosis. Donna DiMichele, MD, offered two reasons for the research push.
“The first is to stimulate research in areas that are undersubscribed through investigator-initiated research,” Dr. DiMichele, deputy director of the NHLBI’s Division of Blood Diseases and Resources, said at the biennial summit of the Thrombosis & Hemostasis Societies of North America. “We also do it sometimes to steer research in directions that are not developing organically through investigator-initiated efforts. In hemostasis and thrombosis, 50% of the investigator-led research is basic in nature. Nonetheless, there were several basic research initiatives that NHLBI released in the last few years that stimulated the field in a new direction.”
The centers will be required to look beyond current active science disciplines in this field, to include emerging sciences and technologies not currently being exploited in this research area. The initiative is also intended to cross-train the next generation of physicians/scientists with interdisciplinary skill sets. “The research teams with successful applications are going to begin their work by the summer of 2018, and hopefully we’ll soon gain some new insights into Factor VIII immunogenicity,” Dr. DiMichele said.
One novel scientific area that successful applicants will be able to take advantage of is the Trans-Omics for Precision Medicine Program (TOPMed). Launched in 2014, this program facilitates whole-genome sequencing across cohorts in heart, lung, blood, and sleep science. TOPMed now includes more than 120,000 whole genomes, with more than 5,000 of those in hemophilia.
In basic science, two NHLBI-funded initiatives aim to improve the understanding of thrombosis.
One, Sex Hormone Induced Thromboembolism in Premenopausal Women (R61/R33), is meant to elucidate the mechanisms by which female sex hormones and sex hormone-based therapies can increase the risk of venous and arterial thromboembolism in premenopausal women. The other initiative, Consortium Linking Oncology with Thrombosis (U01), is aimed at encouraging studies in individual cancer types that expand investigation into the intersection between cancer and thrombotic pathways. It also aims to help researchers identify and develop biomarkers of thrombotic risk or cancer progression, and new strategies for preventing or treating the deleterious interplay between cancer, cancer therapy, and hemostasis/thrombosis.
In the translational research arena, the NHLBI released an initiative on Perinatal Stroke (R01) in 2017. It solicits applications that propose basic and/or translational research studies related to the developing neurovascular unit, perinatal injury/repair response, and/or stroke-related etiologies and risk factors. The purpose is to stimulate research that will identify therapeutic targets in perinatal stroke.
Other NHLBI-sponsored efforts include the Trans-Agency Research Consortium for Trauma-Induced Coagulopathy (TACTIC), as well as the Translational Research Centers in Thrombotic and Hemostatic Disorders program, which set out to enhance the translation of basic research discoveries that could lead to improved prevention, diagnosis, and treatment for thrombotic and hemostatic disorders. This program ended but it led to the development of several projects that are moving into the commercialization phase.
Finally, to further stimulate research in FVIII immunogenicity, on May 15-16, 2018, the NHBLI will host a free workshop entitled “Factor VIII Inhibitors: Generating a Blueprint for Future Research.” For information, visit https://factorviiinhibitors.eventbrite.com.
Dr. DiMichele reported having no financial disclosures.
SAN DIEGO – The National Heart, Lung, and Blood Institute is looking to jump-start research into hemostasis and thrombosis. Donna DiMichele, MD, offered two reasons for the research push.
“The first is to stimulate research in areas that are undersubscribed through investigator-initiated research,” Dr. DiMichele, deputy director of the NHLBI’s Division of Blood Diseases and Resources, said at the biennial summit of the Thrombosis & Hemostasis Societies of North America. “We also do it sometimes to steer research in directions that are not developing organically through investigator-initiated efforts. In hemostasis and thrombosis, 50% of the investigator-led research is basic in nature. Nonetheless, there were several basic research initiatives that NHLBI released in the last few years that stimulated the field in a new direction.”
The centers will be required to look beyond current active science disciplines in this field, to include emerging sciences and technologies not currently being exploited in this research area. The initiative is also intended to cross-train the next generation of physicians/scientists with interdisciplinary skill sets. “The research teams with successful applications are going to begin their work by the summer of 2018, and hopefully we’ll soon gain some new insights into Factor VIII immunogenicity,” Dr. DiMichele said.
One novel scientific area that successful applicants will be able to take advantage of is the Trans-Omics for Precision Medicine Program (TOPMed). Launched in 2014, this program facilitates whole-genome sequencing across cohorts in heart, lung, blood, and sleep science. TOPMed now includes more than 120,000 whole genomes, with more than 5,000 of those in hemophilia.
In basic science, two NHLBI-funded initiatives aim to improve the understanding of thrombosis.
One, Sex Hormone Induced Thromboembolism in Premenopausal Women (R61/R33), is meant to elucidate the mechanisms by which female sex hormones and sex hormone-based therapies can increase the risk of venous and arterial thromboembolism in premenopausal women. The other initiative, Consortium Linking Oncology with Thrombosis (U01), is aimed at encouraging studies in individual cancer types that expand investigation into the intersection between cancer and thrombotic pathways. It also aims to help researchers identify and develop biomarkers of thrombotic risk or cancer progression, and new strategies for preventing or treating the deleterious interplay between cancer, cancer therapy, and hemostasis/thrombosis.
In the translational research arena, the NHLBI released an initiative on Perinatal Stroke (R01) in 2017. It solicits applications that propose basic and/or translational research studies related to the developing neurovascular unit, perinatal injury/repair response, and/or stroke-related etiologies and risk factors. The purpose is to stimulate research that will identify therapeutic targets in perinatal stroke.
Other NHLBI-sponsored efforts include the Trans-Agency Research Consortium for Trauma-Induced Coagulopathy (TACTIC), as well as the Translational Research Centers in Thrombotic and Hemostatic Disorders program, which set out to enhance the translation of basic research discoveries that could lead to improved prevention, diagnosis, and treatment for thrombotic and hemostatic disorders. This program ended but it led to the development of several projects that are moving into the commercialization phase.
Finally, to further stimulate research in FVIII immunogenicity, on May 15-16, 2018, the NHBLI will host a free workshop entitled “Factor VIII Inhibitors: Generating a Blueprint for Future Research.” For information, visit https://factorviiinhibitors.eventbrite.com.
Dr. DiMichele reported having no financial disclosures.
EXPERT ANALYSIS FROM THSNA 2018
Single injection could treat hemophilia B long-term
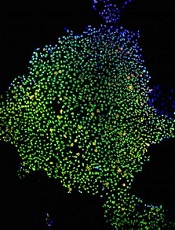
Cell therapy could produce lasting effects in hemophilia B, according to a group of researchers.
They genetically modified induced pluripotent cells (iPSCs) derived from patients with hemophilia B and converted those iPSCs into hepatocyte-like cells (HLCs).
When transplanted into mouse models of hemophilia B, the HLCs remained functional for months, increasing levels of factor IX (FIX) and clotting efficiency.
However, the HLCs were substantially less effective than cryopreserved human hepatocytes (hHeps), which were able to correct clotting defects in mice long-term.
“The appeal of a cell-based approach is that you minimize the number of treatments that a patient needs,” explained Suvasini Ramaswamy, PhD, of The Boston Consulting Group in Massachusetts.
“Rather than constant injections, you can do this in one shot.”
Dr Ramaswamy and her colleagues described their results with this approach in Cell Reports. Study authors include employees of Shire Therapeutics, Vertex Pharmaceuticals, and Thermo Fisher Scientific.
The researchers first developed a quadruple knockout mouse model of hemophilia B that allows for the engraftment and expansion of hHeps. The team crossed transgenic FIX-/--deficient mice with Rag2-/- IL2rg -/- Fah-/- mice to create this model.
The researchers transplanted cryopreserved hHeps into the mice and observed a sustained increase in circulating levels of human albumin, human FIX, and clusters of FIX- or Fah-positive cells in the liver.
The team said the hHeps were able to restore clotting function to wild-type levels in the mice—at least 10-fold higher than levels needed for a significant improvement in hemophilia B.
The hHeps remained functional for up to a year, and the researchers believe the cells could persist even longer. The team noted that there was no difference in the efficacy of hHeps from different donors or vendors.
The researchers then tested the cell therapy they developed using iPSCs. They collected blood samples from 2 patients with severe hemophilia B, reprogrammed peripheral blood-derived mononuclear cells into iPSCs, and used CRISPR/Cas9 to repair the mutations in each patient’s FIX gene.
The team coaxed those repaired cells into HLCs and tested the HLCs in the mouse model of hemophilia B. The HLCs were transplanted through the spleen so the cells were distributed uniformly in the liver.
The researchers said the HLCs were present and functional in the liver for up to a year, but they were less effective than hHeps. Mice that received HLCs experienced “modest” increases in clotting efficiency, from less than 10% to about 25% of wild-type activity.
The researchers believe this work demonstrates the value of combining stem cell reprogramming and gene-modifying approaches to treat genetic diseases. However, more work is needed to optimize this approach for hemophilia B.
Cell therapy could produce lasting effects in hemophilia B, according to a group of researchers.
They genetically modified induced pluripotent cells (iPSCs) derived from patients with hemophilia B and converted those iPSCs into hepatocyte-like cells (HLCs).
When transplanted into mouse models of hemophilia B, the HLCs remained functional for months, increasing levels of factor IX (FIX) and clotting efficiency.
However, the HLCs were substantially less effective than cryopreserved human hepatocytes (hHeps), which were able to correct clotting defects in mice long-term.
“The appeal of a cell-based approach is that you minimize the number of treatments that a patient needs,” explained Suvasini Ramaswamy, PhD, of The Boston Consulting Group in Massachusetts.
“Rather than constant injections, you can do this in one shot.”
Dr Ramaswamy and her colleagues described their results with this approach in Cell Reports. Study authors include employees of Shire Therapeutics, Vertex Pharmaceuticals, and Thermo Fisher Scientific.
The researchers first developed a quadruple knockout mouse model of hemophilia B that allows for the engraftment and expansion of hHeps. The team crossed transgenic FIX-/--deficient mice with Rag2-/- IL2rg -/- Fah-/- mice to create this model.
The researchers transplanted cryopreserved hHeps into the mice and observed a sustained increase in circulating levels of human albumin, human FIX, and clusters of FIX- or Fah-positive cells in the liver.
The team said the hHeps were able to restore clotting function to wild-type levels in the mice—at least 10-fold higher than levels needed for a significant improvement in hemophilia B.
The hHeps remained functional for up to a year, and the researchers believe the cells could persist even longer. The team noted that there was no difference in the efficacy of hHeps from different donors or vendors.
The researchers then tested the cell therapy they developed using iPSCs. They collected blood samples from 2 patients with severe hemophilia B, reprogrammed peripheral blood-derived mononuclear cells into iPSCs, and used CRISPR/Cas9 to repair the mutations in each patient’s FIX gene.
The team coaxed those repaired cells into HLCs and tested the HLCs in the mouse model of hemophilia B. The HLCs were transplanted through the spleen so the cells were distributed uniformly in the liver.
The researchers said the HLCs were present and functional in the liver for up to a year, but they were less effective than hHeps. Mice that received HLCs experienced “modest” increases in clotting efficiency, from less than 10% to about 25% of wild-type activity.
The researchers believe this work demonstrates the value of combining stem cell reprogramming and gene-modifying approaches to treat genetic diseases. However, more work is needed to optimize this approach for hemophilia B.
Cell therapy could produce lasting effects in hemophilia B, according to a group of researchers.
They genetically modified induced pluripotent cells (iPSCs) derived from patients with hemophilia B and converted those iPSCs into hepatocyte-like cells (HLCs).
When transplanted into mouse models of hemophilia B, the HLCs remained functional for months, increasing levels of factor IX (FIX) and clotting efficiency.
However, the HLCs were substantially less effective than cryopreserved human hepatocytes (hHeps), which were able to correct clotting defects in mice long-term.
“The appeal of a cell-based approach is that you minimize the number of treatments that a patient needs,” explained Suvasini Ramaswamy, PhD, of The Boston Consulting Group in Massachusetts.
“Rather than constant injections, you can do this in one shot.”
Dr Ramaswamy and her colleagues described their results with this approach in Cell Reports. Study authors include employees of Shire Therapeutics, Vertex Pharmaceuticals, and Thermo Fisher Scientific.
The researchers first developed a quadruple knockout mouse model of hemophilia B that allows for the engraftment and expansion of hHeps. The team crossed transgenic FIX-/--deficient mice with Rag2-/- IL2rg -/- Fah-/- mice to create this model.
The researchers transplanted cryopreserved hHeps into the mice and observed a sustained increase in circulating levels of human albumin, human FIX, and clusters of FIX- or Fah-positive cells in the liver.
The team said the hHeps were able to restore clotting function to wild-type levels in the mice—at least 10-fold higher than levels needed for a significant improvement in hemophilia B.
The hHeps remained functional for up to a year, and the researchers believe the cells could persist even longer. The team noted that there was no difference in the efficacy of hHeps from different donors or vendors.
The researchers then tested the cell therapy they developed using iPSCs. They collected blood samples from 2 patients with severe hemophilia B, reprogrammed peripheral blood-derived mononuclear cells into iPSCs, and used CRISPR/Cas9 to repair the mutations in each patient’s FIX gene.
The team coaxed those repaired cells into HLCs and tested the HLCs in the mouse model of hemophilia B. The HLCs were transplanted through the spleen so the cells were distributed uniformly in the liver.
The researchers said the HLCs were present and functional in the liver for up to a year, but they were less effective than hHeps. Mice that received HLCs experienced “modest” increases in clotting efficiency, from less than 10% to about 25% of wild-type activity.
The researchers believe this work demonstrates the value of combining stem cell reprogramming and gene-modifying approaches to treat genetic diseases. However, more work is needed to optimize this approach for hemophilia B.
PI3K inhibitors could treat HHT

Preclinical research suggests PI3K inhibitors could treat hereditary hemorrhagic telangiectasia (HHT).
Experiments in mice and patient samples revealed that loss of ALK1 function induces vascular hyperplasia and increases activity of the PI3K pathway.
Pharmacological inhibition of PI3K was able to eliminate vascular hyperplasia in mouse models.
Francesc Viñals, PhD, of Institut Catala d’Oncologia in Barcelona, Spain, and his colleagues described this research in Arteriosclerosis, Thrombosis and Vascular Biology.
“Our group has been working with endothelial cells for a long time, focusing on how they are affected by changes in the TGF-beta signaling pathway from a basic research perspective,” Dr Viñals noted.
The group was especially interested in ALK1, a receptor for the TGF-beta factor BMP9 that is expressed by endothelial cells. Mutations in ALK1 have been associated with HHT.
The researchers developed a model for the formation of blood vessels in the retina of mice that lacked a copy of the gene for the ALK1 receptor. These models exhibited an excess of endothelial cells and errors in the formation of blood vessels.
These results corresponded with the researchers’ theory. The team thought that, if members of the TGF-beta family usually act as a “brake” for cellular proliferation, mutations in one of the family’s components should lead to uncontrolled proliferation. The researchers replicated and confirmed their results in in vitro cultures.
Further investigation revealed the role of the PI3K pathway. The researchers found that stimulation of PI3K acts as an “accelerator” for endothelial cells to proliferate.
BMP9 (through ALK1) acts as a brake, inhibiting VEGF-mediated PI3K signaling by increasing PTEN activity. When ALK1 is mutated, there is no brake, and endothelial cells proliferate more thanks to the PI3K pathway.
The researchers discovered this mechanism via experiments in human umbilical vein endothelial cells. However, experiments in HHT2 patient samples supported these findings.
The team found that mutations in ALK1 increased endothelial cell proliferation in vessels from patients with HHT2. The researchers also found overexpression of genes linked to PI3K/AKT signaling in telangiectasial tissue from patients with HHT2, as compared to normal tissue from either HHT2 patients or healthy individuals.
Going back to murine experiments, the researchers found that loss of both PI3K and ALK1 function stabilizes the proliferation of endothelial cells.
The team tested LY294002, a pan-PI3K inhibitor, in mice and found that PI3K inhibition can reverse the vascular hyperplasia induced by loss of ALK1 function.
“Now, the next step is clear,” Dr Viñals said. “We have to assess the effects of PI3K inhibitors in patients. PI3K inhibitors are currently used in the treatment of patients with cancer or patients who require immunosuppression after organ transplantation, and it has been observed that, in patients with these pathologies and HHT, treatment results in a reduction of the issues caused by HHT, which is a very encouraging fact.”
Preclinical research suggests PI3K inhibitors could treat hereditary hemorrhagic telangiectasia (HHT).
Experiments in mice and patient samples revealed that loss of ALK1 function induces vascular hyperplasia and increases activity of the PI3K pathway.
Pharmacological inhibition of PI3K was able to eliminate vascular hyperplasia in mouse models.
Francesc Viñals, PhD, of Institut Catala d’Oncologia in Barcelona, Spain, and his colleagues described this research in Arteriosclerosis, Thrombosis and Vascular Biology.
“Our group has been working with endothelial cells for a long time, focusing on how they are affected by changes in the TGF-beta signaling pathway from a basic research perspective,” Dr Viñals noted.
The group was especially interested in ALK1, a receptor for the TGF-beta factor BMP9 that is expressed by endothelial cells. Mutations in ALK1 have been associated with HHT.
The researchers developed a model for the formation of blood vessels in the retina of mice that lacked a copy of the gene for the ALK1 receptor. These models exhibited an excess of endothelial cells and errors in the formation of blood vessels.
These results corresponded with the researchers’ theory. The team thought that, if members of the TGF-beta family usually act as a “brake” for cellular proliferation, mutations in one of the family’s components should lead to uncontrolled proliferation. The researchers replicated and confirmed their results in in vitro cultures.
Further investigation revealed the role of the PI3K pathway. The researchers found that stimulation of PI3K acts as an “accelerator” for endothelial cells to proliferate.
BMP9 (through ALK1) acts as a brake, inhibiting VEGF-mediated PI3K signaling by increasing PTEN activity. When ALK1 is mutated, there is no brake, and endothelial cells proliferate more thanks to the PI3K pathway.
The researchers discovered this mechanism via experiments in human umbilical vein endothelial cells. However, experiments in HHT2 patient samples supported these findings.
The team found that mutations in ALK1 increased endothelial cell proliferation in vessels from patients with HHT2. The researchers also found overexpression of genes linked to PI3K/AKT signaling in telangiectasial tissue from patients with HHT2, as compared to normal tissue from either HHT2 patients or healthy individuals.
Going back to murine experiments, the researchers found that loss of both PI3K and ALK1 function stabilizes the proliferation of endothelial cells.
The team tested LY294002, a pan-PI3K inhibitor, in mice and found that PI3K inhibition can reverse the vascular hyperplasia induced by loss of ALK1 function.
“Now, the next step is clear,” Dr Viñals said. “We have to assess the effects of PI3K inhibitors in patients. PI3K inhibitors are currently used in the treatment of patients with cancer or patients who require immunosuppression after organ transplantation, and it has been observed that, in patients with these pathologies and HHT, treatment results in a reduction of the issues caused by HHT, which is a very encouraging fact.”
Preclinical research suggests PI3K inhibitors could treat hereditary hemorrhagic telangiectasia (HHT).
Experiments in mice and patient samples revealed that loss of ALK1 function induces vascular hyperplasia and increases activity of the PI3K pathway.
Pharmacological inhibition of PI3K was able to eliminate vascular hyperplasia in mouse models.
Francesc Viñals, PhD, of Institut Catala d’Oncologia in Barcelona, Spain, and his colleagues described this research in Arteriosclerosis, Thrombosis and Vascular Biology.
“Our group has been working with endothelial cells for a long time, focusing on how they are affected by changes in the TGF-beta signaling pathway from a basic research perspective,” Dr Viñals noted.
The group was especially interested in ALK1, a receptor for the TGF-beta factor BMP9 that is expressed by endothelial cells. Mutations in ALK1 have been associated with HHT.
The researchers developed a model for the formation of blood vessels in the retina of mice that lacked a copy of the gene for the ALK1 receptor. These models exhibited an excess of endothelial cells and errors in the formation of blood vessels.
These results corresponded with the researchers’ theory. The team thought that, if members of the TGF-beta family usually act as a “brake” for cellular proliferation, mutations in one of the family’s components should lead to uncontrolled proliferation. The researchers replicated and confirmed their results in in vitro cultures.
Further investigation revealed the role of the PI3K pathway. The researchers found that stimulation of PI3K acts as an “accelerator” for endothelial cells to proliferate.
BMP9 (through ALK1) acts as a brake, inhibiting VEGF-mediated PI3K signaling by increasing PTEN activity. When ALK1 is mutated, there is no brake, and endothelial cells proliferate more thanks to the PI3K pathway.
The researchers discovered this mechanism via experiments in human umbilical vein endothelial cells. However, experiments in HHT2 patient samples supported these findings.
The team found that mutations in ALK1 increased endothelial cell proliferation in vessels from patients with HHT2. The researchers also found overexpression of genes linked to PI3K/AKT signaling in telangiectasial tissue from patients with HHT2, as compared to normal tissue from either HHT2 patients or healthy individuals.
Going back to murine experiments, the researchers found that loss of both PI3K and ALK1 function stabilizes the proliferation of endothelial cells.
The team tested LY294002, a pan-PI3K inhibitor, in mice and found that PI3K inhibition can reverse the vascular hyperplasia induced by loss of ALK1 function.
“Now, the next step is clear,” Dr Viñals said. “We have to assess the effects of PI3K inhibitors in patients. PI3K inhibitors are currently used in the treatment of patients with cancer or patients who require immunosuppression after organ transplantation, and it has been observed that, in patients with these pathologies and HHT, treatment results in a reduction of the issues caused by HHT, which is a very encouraging fact.”
Consider caregiver oral health for children with bleeding disorders
SAN DIEGO – Caregiver oral health status is an identifiable risk factor that could be used to screen for poor oral health among children and young adults with bleeding disorders, results from a single-center suggest.
“We ask parents one simple question: ‘Have you had a cavity in the last year?’ If they say yes, we would be more concerned that their children would be more likely to have poor oral health,” Elizabeth Hastie said in an interview during a poster session at the biennial summit of the Thrombosis & Hemostasis Societies of North America.
Proper oral health may prevent joint disease and other conditions that predispose patients to bleeds, according to Ms. Hastie, a fourth-year medical student at Emory University, Atlanta. However, of the 147 hemophilia treatment centers in the United States, just 30% have a dentist on staff, while 90% of centers have expressed interest in increasing patient education in oral health.
In an effort to evaluate the dental habits, needs, and oral health issues of children and young adults up to age 18 with bleeding disorders, Ms. Hastie and her associates conducted a cross-sectional study of 226 patients who were evaluated by a staff dental hygienist at Children’s Healthcare of Atlanta Comprehensive Bleeding Disorders Clinic from May 2016 to October 2017.
The evaluation consisted of a 14-question survey derived from the American Academy of Pediatric Dentistry Caries–Risk Assessment Tool completed by the primary caregiver present during the visit and oral screening. The researchers extracted demographic and clinical characteristics from the patient’s chart and included age, race, county of residence, and bleeding disorder type and severity.

Nearly half of the patients (44%) reported they did not brush their teeth twice a day. Children younger than age 5 years were more likely to not brush their teeth twice a day, compared with children aged 5-14 years and young adults aged 15-18 years (57% vs. 44% and 31% respectively, P = .08).
More than one-quarter of patients (27%) reported not having a current dentist and 15% reported specific challenges with access to dental care including burdens related to distance, insurance coverage, and finding a provider willing to treat in the setting of their medical condition. Those who were Medicaid eligible or of low socioeconomic status were significantly more likely to report dental care access issues, compared with other patients (20% vs. 9%; P = .01).
Oral screening performed by the dental hygienist demonstrated significant oral pathology: 89% of patients had plaque accumulation, 57% had white spots or decalcifications, 37% had gingivitis, and 8% had suspicious lesions suggestive of dental caries.
The researchers also found that having a caregiver with active oral disease in the past 12 months increased the odds of suspicious lesions (odds ratio, 4.34), increased the odds of gingivitis (OR, 3.80), and decreased the odds of the patients’ brushing their teeth at least twice per day (OR, 0.17).
“Hopefully, if we can target those high-risk patients in clinic, we could reduce costs, the number of bleeds, the number of products and factor used, and potentially even morbidity in the future,” Ms. Hastie said.
She acknowledged certain limitations of the study, including its single-center design and the fact that a dental hygienist performed the majority of evaluations. She reported having no financial disclosures.
SAN DIEGO – Caregiver oral health status is an identifiable risk factor that could be used to screen for poor oral health among children and young adults with bleeding disorders, results from a single-center suggest.
“We ask parents one simple question: ‘Have you had a cavity in the last year?’ If they say yes, we would be more concerned that their children would be more likely to have poor oral health,” Elizabeth Hastie said in an interview during a poster session at the biennial summit of the Thrombosis & Hemostasis Societies of North America.
Proper oral health may prevent joint disease and other conditions that predispose patients to bleeds, according to Ms. Hastie, a fourth-year medical student at Emory University, Atlanta. However, of the 147 hemophilia treatment centers in the United States, just 30% have a dentist on staff, while 90% of centers have expressed interest in increasing patient education in oral health.
In an effort to evaluate the dental habits, needs, and oral health issues of children and young adults up to age 18 with bleeding disorders, Ms. Hastie and her associates conducted a cross-sectional study of 226 patients who were evaluated by a staff dental hygienist at Children’s Healthcare of Atlanta Comprehensive Bleeding Disorders Clinic from May 2016 to October 2017.
The evaluation consisted of a 14-question survey derived from the American Academy of Pediatric Dentistry Caries–Risk Assessment Tool completed by the primary caregiver present during the visit and oral screening. The researchers extracted demographic and clinical characteristics from the patient’s chart and included age, race, county of residence, and bleeding disorder type and severity.
Nearly half of the patients (44%) reported they did not brush their teeth twice a day. Children younger than age 5 years were more likely to not brush their teeth twice a day, compared with children aged 5-14 years and young adults aged 15-18 years (57% vs. 44% and 31% respectively, P = .08).
More than one-quarter of patients (27%) reported not having a current dentist and 15% reported specific challenges with access to dental care including burdens related to distance, insurance coverage, and finding a provider willing to treat in the setting of their medical condition. Those who were Medicaid eligible or of low socioeconomic status were significantly more likely to report dental care access issues, compared with other patients (20% vs. 9%; P = .01).
Oral screening performed by the dental hygienist demonstrated significant oral pathology: 89% of patients had plaque accumulation, 57% had white spots or decalcifications, 37% had gingivitis, and 8% had suspicious lesions suggestive of dental caries.
The researchers also found that having a caregiver with active oral disease in the past 12 months increased the odds of suspicious lesions (odds ratio, 4.34), increased the odds of gingivitis (OR, 3.80), and decreased the odds of the patients’ brushing their teeth at least twice per day (OR, 0.17).
“Hopefully, if we can target those high-risk patients in clinic, we could reduce costs, the number of bleeds, the number of products and factor used, and potentially even morbidity in the future,” Ms. Hastie said.
She acknowledged certain limitations of the study, including its single-center design and the fact that a dental hygienist performed the majority of evaluations. She reported having no financial disclosures.
SAN DIEGO – Caregiver oral health status is an identifiable risk factor that could be used to screen for poor oral health among children and young adults with bleeding disorders, results from a single-center suggest.
“We ask parents one simple question: ‘Have you had a cavity in the last year?’ If they say yes, we would be more concerned that their children would be more likely to have poor oral health,” Elizabeth Hastie said in an interview during a poster session at the biennial summit of the Thrombosis & Hemostasis Societies of North America.
Proper oral health may prevent joint disease and other conditions that predispose patients to bleeds, according to Ms. Hastie, a fourth-year medical student at Emory University, Atlanta. However, of the 147 hemophilia treatment centers in the United States, just 30% have a dentist on staff, while 90% of centers have expressed interest in increasing patient education in oral health.
In an effort to evaluate the dental habits, needs, and oral health issues of children and young adults up to age 18 with bleeding disorders, Ms. Hastie and her associates conducted a cross-sectional study of 226 patients who were evaluated by a staff dental hygienist at Children’s Healthcare of Atlanta Comprehensive Bleeding Disorders Clinic from May 2016 to October 2017.
The evaluation consisted of a 14-question survey derived from the American Academy of Pediatric Dentistry Caries–Risk Assessment Tool completed by the primary caregiver present during the visit and oral screening. The researchers extracted demographic and clinical characteristics from the patient’s chart and included age, race, county of residence, and bleeding disorder type and severity.
Nearly half of the patients (44%) reported they did not brush their teeth twice a day. Children younger than age 5 years were more likely to not brush their teeth twice a day, compared with children aged 5-14 years and young adults aged 15-18 years (57% vs. 44% and 31% respectively, P = .08).
More than one-quarter of patients (27%) reported not having a current dentist and 15% reported specific challenges with access to dental care including burdens related to distance, insurance coverage, and finding a provider willing to treat in the setting of their medical condition. Those who were Medicaid eligible or of low socioeconomic status were significantly more likely to report dental care access issues, compared with other patients (20% vs. 9%; P = .01).
Oral screening performed by the dental hygienist demonstrated significant oral pathology: 89% of patients had plaque accumulation, 57% had white spots or decalcifications, 37% had gingivitis, and 8% had suspicious lesions suggestive of dental caries.
The researchers also found that having a caregiver with active oral disease in the past 12 months increased the odds of suspicious lesions (odds ratio, 4.34), increased the odds of gingivitis (OR, 3.80), and decreased the odds of the patients’ brushing their teeth at least twice per day (OR, 0.17).
“Hopefully, if we can target those high-risk patients in clinic, we could reduce costs, the number of bleeds, the number of products and factor used, and potentially even morbidity in the future,” Ms. Hastie said.
She acknowledged certain limitations of the study, including its single-center design and the fact that a dental hygienist performed the majority of evaluations. She reported having no financial disclosures.
REPORTING FROM THSNA 2018
Key clinical point:
Major finding: Having a caregiver with active oral disease in the past 12 months increased the odds of the child having a suspicious lesion (OR 4.34) and gingivitis (OR 3.80).
Study details: A cross-sectional study of 226 pediatric patients who were evaluated by a dental hygienist.
Disclosures: Ms. Hastie reported having no financial disclosures.
Source: Hastie E et al. THSNA 2018, Poster 150.
Fostamatinib produces responses in ITP
Fostamatinib has produced “clinically meaningful” responses in adults with persistent or chronic immune thrombocytopenia (ITP), according to researchers.
In a pair of phase 3 trials, 18% of patients who received fostamatinib had a stable response, which was defined as having a platelet count of at least 50,000/µL for at least 4 of 6 clinic visits.
In comparison, 2% of patients who received placebo achieved a stable response.
The most common adverse events (AEs) in these trials were diarrhea, hypertension, and nausea. Most AEs were deemed mild or moderate.
These results were published in the American Journal of Hematology. The trials—known as FIT1 and FIT2—were sponsored by Rigel Pharmaceuticals, Inc., the company marketing fostamatinib.
Fostamatinib is an oral Syk inhibitor that was recently approved by the US Food and Drug Administration.
Patients and treatment
Researchers evaluated fostamatinib in the parallel FIT1 and FIT2 trials, which included 150 patients with persistent or chronic ITP who had an insufficient response to previous treatment.
In each study, patients were randomized 2:1 to receive fostamatinib or placebo for 24 weeks. In FIT1, 76 patients were randomized—51 to fostamatinib and 25 to placebo. In FIT2, 74 patients were randomized—50 to fostamatinib and 24 to placebo.
All patients initially received fostamatinib at 100 mg twice daily. Most (88%) were escalated to 150 mg twice daily at week 4 or later. Patients could also receive stable concurrent ITP therapy—glucocorticoids (< 20 mg prednisone equivalent per day), azathioprine, or danazol—and rescue therapy if needed.
The median age was 54 (range, 20-88) in the fostamatinib recipients and 53 (range, 20-78) in the placebo recipients. Sixty-one percent and 60%, respectively, were female. And 93% and 92%, respectively, were white.
The median duration of ITP was 8.7 years for fostamatinib recipients and 7.8 years for placebo recipients. Both fostamatinib and placebo recipients had a median of 3 prior unique treatments for ITP (range, 1-13 and 1-10, respectively).
Prior ITP treatments (in the fostamatinib and placebo arms, respectively) included corticosteroids (93% vs 96%), immunoglobulins (51% vs 55%), thrombopoietin receptor agonists (47% vs 51%), immunosuppressants (44% vs 45%), splenectomy (34% vs 39%), and rituximab (34% vs 29%), among other treatments.
Overall, the patients’ median platelet count at baseline was 16,000/µL. Mean baseline platelet counts were 16,052/µL (range, 1000-51,000) in fostamatinib recipients and 19,818/µL (range, 1000-156,000) in placebo recipients.
Response
The efficacy of fostamatinib was based on stable platelet response, defined as a platelet count of at least 50,000/µL on at least 4 of 6 biweekly clinic visits between weeks 14 and 24, without the use of rescue therapy.
Overall, 18% (18/101) of patients on fostamatinib and 2% (1/49) of those on placebo achieved this endpoint (P=0.0003).
In FIT1, 18% of patients in the fostamatinib arm and 0% of those in the placebo arm achieved a stable platelet response (P=0.026). In FIT2, rates of stable response were 18% and 4%, respectively (P=0.152).
A secondary endpoint was overall response, which was defined retrospectively as at least 1 platelet count ≥ 50,000/μL within the first 12 weeks on treatment.
Forty-three percent (43/101) of fostamatinib recipients achieved an overall response, as did 14% (7/49) of patients on placebo (P=0.0006).
In FIT1, the rate of overall response was 37% in the fostamatinib arm and 8% in the placebo arm (P=0.007). In FIT2, overall response rates were 48% and 21%, respectively (P=0.025).
The researchers said they observed responses to fostamatinib across all patient subgroups, regardless of age, sex, prior therapy, baseline platelet count, or duration of ITP at study entry.
Role of concomitant therapy
The researchers noted that 2 of the 18 patients with a stable platelet response were on concomitant treatment. Both were on steroids, one of them for 62 days before the first dose of fostamatinib and the other for 14 years.
Four of the 43 patients with an overall platelet response were on concomitant therapy. Three were on steroids—for 62 days, 67 days, and 14 years prior to first dose of fostamatinib—and 1 was on azathioprine—for 197 days prior to the first dose of fostamatinib.
The researchers said the effects of these therapies probably would have been evident prior to study entry, so they aren’t likely to have influenced the results.
Safety
In both trials, the rate of AEs was 83% in the fostamatinib recipients (32% mild, 35% moderate, and 16% severe AEs) and 75% in the placebo recipients (42% mild, 19% moderate, and 15% severe AEs.).
AEs occurring in at least 5% of patients (in the fostamatinib and placebo arms, respectively) included diarrhea (31% vs 15%), hypertension (28% vs 13%), nausea (19% vs 8%), dizziness (11% vs 8%), ALT increase (11% vs 0%), AST increase (9% vs 0%), respiratory infection (11% vs 6%), rash (9% vs 2%), abdominal pain (6% vs 2%), fatigue (6% vs 2%), chest pain (6% vs 2%), and neutropenia (6% vs 0%).
Moderate or severe bleeding-related AEs occurred in 9% of patients who had an overall response to fostamatinib and 16% of patients on placebo.
Serious AEs considered related to study drug occurred in 4 patients on fostamatinib and 1 patient on placebo. The placebo recipient experienced a bleeding event, and the fostamatinib-related events were consistent with the AE profile of the drug (hypertension, diarrhea, etc.), according to Rigel Pharmaceuticals.
There were 2 deaths. One placebo recipient died of probable sepsis 19 days after withdrawing from the study due to epistaxis. One fostamatinib recipient developed plasma cell myeloma, stopped treatment on day 19, and died 71 days later.
Fostamatinib has produced “clinically meaningful” responses in adults with persistent or chronic immune thrombocytopenia (ITP), according to researchers.
In a pair of phase 3 trials, 18% of patients who received fostamatinib had a stable response, which was defined as having a platelet count of at least 50,000/µL for at least 4 of 6 clinic visits.
In comparison, 2% of patients who received placebo achieved a stable response.
The most common adverse events (AEs) in these trials were diarrhea, hypertension, and nausea. Most AEs were deemed mild or moderate.
These results were published in the American Journal of Hematology. The trials—known as FIT1 and FIT2—were sponsored by Rigel Pharmaceuticals, Inc., the company marketing fostamatinib.
Fostamatinib is an oral Syk inhibitor that was recently approved by the US Food and Drug Administration.
Patients and treatment
Researchers evaluated fostamatinib in the parallel FIT1 and FIT2 trials, which included 150 patients with persistent or chronic ITP who had an insufficient response to previous treatment.
In each study, patients were randomized 2:1 to receive fostamatinib or placebo for 24 weeks. In FIT1, 76 patients were randomized—51 to fostamatinib and 25 to placebo. In FIT2, 74 patients were randomized—50 to fostamatinib and 24 to placebo.
All patients initially received fostamatinib at 100 mg twice daily. Most (88%) were escalated to 150 mg twice daily at week 4 or later. Patients could also receive stable concurrent ITP therapy—glucocorticoids (< 20 mg prednisone equivalent per day), azathioprine, or danazol—and rescue therapy if needed.
The median age was 54 (range, 20-88) in the fostamatinib recipients and 53 (range, 20-78) in the placebo recipients. Sixty-one percent and 60%, respectively, were female. And 93% and 92%, respectively, were white.
The median duration of ITP was 8.7 years for fostamatinib recipients and 7.8 years for placebo recipients. Both fostamatinib and placebo recipients had a median of 3 prior unique treatments for ITP (range, 1-13 and 1-10, respectively).
Prior ITP treatments (in the fostamatinib and placebo arms, respectively) included corticosteroids (93% vs 96%), immunoglobulins (51% vs 55%), thrombopoietin receptor agonists (47% vs 51%), immunosuppressants (44% vs 45%), splenectomy (34% vs 39%), and rituximab (34% vs 29%), among other treatments.
Overall, the patients’ median platelet count at baseline was 16,000/µL. Mean baseline platelet counts were 16,052/µL (range, 1000-51,000) in fostamatinib recipients and 19,818/µL (range, 1000-156,000) in placebo recipients.
Response
The efficacy of fostamatinib was based on stable platelet response, defined as a platelet count of at least 50,000/µL on at least 4 of 6 biweekly clinic visits between weeks 14 and 24, without the use of rescue therapy.
Overall, 18% (18/101) of patients on fostamatinib and 2% (1/49) of those on placebo achieved this endpoint (P=0.0003).
In FIT1, 18% of patients in the fostamatinib arm and 0% of those in the placebo arm achieved a stable platelet response (P=0.026). In FIT2, rates of stable response were 18% and 4%, respectively (P=0.152).
A secondary endpoint was overall response, which was defined retrospectively as at least 1 platelet count ≥ 50,000/μL within the first 12 weeks on treatment.
Forty-three percent (43/101) of fostamatinib recipients achieved an overall response, as did 14% (7/49) of patients on placebo (P=0.0006).
In FIT1, the rate of overall response was 37% in the fostamatinib arm and 8% in the placebo arm (P=0.007). In FIT2, overall response rates were 48% and 21%, respectively (P=0.025).
The researchers said they observed responses to fostamatinib across all patient subgroups, regardless of age, sex, prior therapy, baseline platelet count, or duration of ITP at study entry.
Role of concomitant therapy
The researchers noted that 2 of the 18 patients with a stable platelet response were on concomitant treatment. Both were on steroids, one of them for 62 days before the first dose of fostamatinib and the other for 14 years.
Four of the 43 patients with an overall platelet response were on concomitant therapy. Three were on steroids—for 62 days, 67 days, and 14 years prior to first dose of fostamatinib—and 1 was on azathioprine—for 197 days prior to the first dose of fostamatinib.
The researchers said the effects of these therapies probably would have been evident prior to study entry, so they aren’t likely to have influenced the results.
Safety
In both trials, the rate of AEs was 83% in the fostamatinib recipients (32% mild, 35% moderate, and 16% severe AEs) and 75% in the placebo recipients (42% mild, 19% moderate, and 15% severe AEs.).
AEs occurring in at least 5% of patients (in the fostamatinib and placebo arms, respectively) included diarrhea (31% vs 15%), hypertension (28% vs 13%), nausea (19% vs 8%), dizziness (11% vs 8%), ALT increase (11% vs 0%), AST increase (9% vs 0%), respiratory infection (11% vs 6%), rash (9% vs 2%), abdominal pain (6% vs 2%), fatigue (6% vs 2%), chest pain (6% vs 2%), and neutropenia (6% vs 0%).
Moderate or severe bleeding-related AEs occurred in 9% of patients who had an overall response to fostamatinib and 16% of patients on placebo.
Serious AEs considered related to study drug occurred in 4 patients on fostamatinib and 1 patient on placebo. The placebo recipient experienced a bleeding event, and the fostamatinib-related events were consistent with the AE profile of the drug (hypertension, diarrhea, etc.), according to Rigel Pharmaceuticals.
There were 2 deaths. One placebo recipient died of probable sepsis 19 days after withdrawing from the study due to epistaxis. One fostamatinib recipient developed plasma cell myeloma, stopped treatment on day 19, and died 71 days later.
Fostamatinib has produced “clinically meaningful” responses in adults with persistent or chronic immune thrombocytopenia (ITP), according to researchers.
In a pair of phase 3 trials, 18% of patients who received fostamatinib had a stable response, which was defined as having a platelet count of at least 50,000/µL for at least 4 of 6 clinic visits.
In comparison, 2% of patients who received placebo achieved a stable response.
The most common adverse events (AEs) in these trials were diarrhea, hypertension, and nausea. Most AEs were deemed mild or moderate.
These results were published in the American Journal of Hematology. The trials—known as FIT1 and FIT2—were sponsored by Rigel Pharmaceuticals, Inc., the company marketing fostamatinib.
Fostamatinib is an oral Syk inhibitor that was recently approved by the US Food and Drug Administration.
Patients and treatment
Researchers evaluated fostamatinib in the parallel FIT1 and FIT2 trials, which included 150 patients with persistent or chronic ITP who had an insufficient response to previous treatment.
In each study, patients were randomized 2:1 to receive fostamatinib or placebo for 24 weeks. In FIT1, 76 patients were randomized—51 to fostamatinib and 25 to placebo. In FIT2, 74 patients were randomized—50 to fostamatinib and 24 to placebo.
All patients initially received fostamatinib at 100 mg twice daily. Most (88%) were escalated to 150 mg twice daily at week 4 or later. Patients could also receive stable concurrent ITP therapy—glucocorticoids (< 20 mg prednisone equivalent per day), azathioprine, or danazol—and rescue therapy if needed.
The median age was 54 (range, 20-88) in the fostamatinib recipients and 53 (range, 20-78) in the placebo recipients. Sixty-one percent and 60%, respectively, were female. And 93% and 92%, respectively, were white.
The median duration of ITP was 8.7 years for fostamatinib recipients and 7.8 years for placebo recipients. Both fostamatinib and placebo recipients had a median of 3 prior unique treatments for ITP (range, 1-13 and 1-10, respectively).
Prior ITP treatments (in the fostamatinib and placebo arms, respectively) included corticosteroids (93% vs 96%), immunoglobulins (51% vs 55%), thrombopoietin receptor agonists (47% vs 51%), immunosuppressants (44% vs 45%), splenectomy (34% vs 39%), and rituximab (34% vs 29%), among other treatments.
Overall, the patients’ median platelet count at baseline was 16,000/µL. Mean baseline platelet counts were 16,052/µL (range, 1000-51,000) in fostamatinib recipients and 19,818/µL (range, 1000-156,000) in placebo recipients.
Response
The efficacy of fostamatinib was based on stable platelet response, defined as a platelet count of at least 50,000/µL on at least 4 of 6 biweekly clinic visits between weeks 14 and 24, without the use of rescue therapy.
Overall, 18% (18/101) of patients on fostamatinib and 2% (1/49) of those on placebo achieved this endpoint (P=0.0003).
In FIT1, 18% of patients in the fostamatinib arm and 0% of those in the placebo arm achieved a stable platelet response (P=0.026). In FIT2, rates of stable response were 18% and 4%, respectively (P=0.152).
A secondary endpoint was overall response, which was defined retrospectively as at least 1 platelet count ≥ 50,000/μL within the first 12 weeks on treatment.
Forty-three percent (43/101) of fostamatinib recipients achieved an overall response, as did 14% (7/49) of patients on placebo (P=0.0006).
In FIT1, the rate of overall response was 37% in the fostamatinib arm and 8% in the placebo arm (P=0.007). In FIT2, overall response rates were 48% and 21%, respectively (P=0.025).
The researchers said they observed responses to fostamatinib across all patient subgroups, regardless of age, sex, prior therapy, baseline platelet count, or duration of ITP at study entry.
Role of concomitant therapy
The researchers noted that 2 of the 18 patients with a stable platelet response were on concomitant treatment. Both were on steroids, one of them for 62 days before the first dose of fostamatinib and the other for 14 years.
Four of the 43 patients with an overall platelet response were on concomitant therapy. Three were on steroids—for 62 days, 67 days, and 14 years prior to first dose of fostamatinib—and 1 was on azathioprine—for 197 days prior to the first dose of fostamatinib.
The researchers said the effects of these therapies probably would have been evident prior to study entry, so they aren’t likely to have influenced the results.
Safety
In both trials, the rate of AEs was 83% in the fostamatinib recipients (32% mild, 35% moderate, and 16% severe AEs) and 75% in the placebo recipients (42% mild, 19% moderate, and 15% severe AEs.).
AEs occurring in at least 5% of patients (in the fostamatinib and placebo arms, respectively) included diarrhea (31% vs 15%), hypertension (28% vs 13%), nausea (19% vs 8%), dizziness (11% vs 8%), ALT increase (11% vs 0%), AST increase (9% vs 0%), respiratory infection (11% vs 6%), rash (9% vs 2%), abdominal pain (6% vs 2%), fatigue (6% vs 2%), chest pain (6% vs 2%), and neutropenia (6% vs 0%).
Moderate or severe bleeding-related AEs occurred in 9% of patients who had an overall response to fostamatinib and 16% of patients on placebo.
Serious AEs considered related to study drug occurred in 4 patients on fostamatinib and 1 patient on placebo. The placebo recipient experienced a bleeding event, and the fostamatinib-related events were consistent with the AE profile of the drug (hypertension, diarrhea, etc.), according to Rigel Pharmaceuticals.
There were 2 deaths. One placebo recipient died of probable sepsis 19 days after withdrawing from the study due to epistaxis. One fostamatinib recipient developed plasma cell myeloma, stopped treatment on day 19, and died 71 days later.