User login
Most patients off transfusions after gene therapy for thalassemia
SALT LAKE CITY – Lentiviral delivery of BB305 gene therapy via autologous hematopoietic stem cell transplant (HSCT) was safe and effective for individuals with transfusion dependent beta thalassemia, according to results of a phase 1/2 study.
None of the study participants died, and the majority of patients are now transfusion independent.
The Northstar study is an international, multicenter open-label, single-arm study of adolescents and adults with transfusion dependent beta thalassemia (TDT). A total of 18 patients at a median 21 years of age – 15 young adults aged 18-35 years and three adolescents aged 12-17 years – have now been treated, Mark Walters, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
Of these, 11 are now transfusion independent, with most patients stopping transfusions within 6 months of receiving gene therapy, said Dr. Walters, director of the blood and marrow transplantation program at the University of California, San Francisco’s Benioff Children’s Hospital, Oakland.
Eight patients had the beta0/beta0 genotype, and had essentially been transfusion dependent from infancy. Six other patients were betaE/beta0, and had become transfusion dependent over time. Four patients had other thalassemia genotypes.
Patients who enrolled in the Northstar study first had peripheral stem cell collection via apheresis after mobilization with granulocyte-colony stimulating factor and plerixafor. Then they received myeloablative conditioning with busulfan. At the same time, selected CD34+ cells were tranduced with the BB305 lentiviral vector and cryopreserved. Patients were infused with the transduced cells and managed through the engraftment process.
As a measure of annualized pre-procedure transfusion requirements, patients had received a median 163.6 mL/kg/year of packed red blood cells, Dr. Walters said. Not unexpectedly, liver iron concentration was a median 5.7 mg/g, though with a wide range among participants (0.4-26.4 mg/g). However, participants did not show signs of cardiac tissue iron on T2* magnetic resonance imaging . Six patients had undergone a splenectomy.
The median vector copy number was 0.7 (range, 0.3-1.5), with a median 31.5 CD34+ cells transduced (range, 17.0-58.0). The final cell dose delivered was a median 8.1 x 106 CD34+ cells/kg (range, 5.2-18.1).
“All 18 patients have had at least 18 months of follow-up,” said Dr. Walters, and data from 10 patients has been analyzed out to 2 years. Three patients have a full 3 years of follow-up, he said.
The self-inactivating lentiviral vector has behaved as expected; no replication-competent lentivirus has been found, with investigators conducting assessments at months 3, 6, and 12, and then annually through year 5.
The study protocol also calls for integration site analysis every 6 months for 5 years, and additional analyses at years 7, 10, and 15. Thus far, all samples have shown a polyclonal vector integration profile without clonal dominance, Dr. Walter said.
The median time to neutrophil engraftment was study day 18.5 (range, 14-30), while platelet engraftment was more variable, and overall slower, with engraftment at a median of study day 39.5 (range, 19-191).
Dr. Walters said that he and his colleagues examined characteristics of the four patients who still had platelet counts at or less than 100,000/microliters at 12 months after HSCT. They found that two of these patients had had splenectomies, but saw no clear relationship between speed of platelet engraftment and platelet count at 12 months. Three of the four patients had drug product cell doses less than the median.
However, two patients had no bleeding events after neutrophil engraftment, and bleeding events were all grade 1 or 2 in the other two patients. The slower-than-expected platelet engraftment rate was likely attributable to the ex vivo manipulation of the stem cells, Dr. Walters noted.
Looking at safety data from the point of neutrophil engraftment to the last follow-up, there have been no graft failures; six patients have had serious adverse events. Two events of veno-occlusive disease were assessed as grade 3 and attributed to the transplant. Two of these three patients had an extended hospital stay. Other grade 3 events including intracardiac thrombus, central catheter thrombosis, and cellulitis, as well as hyperglycemia and infectious diseases.
No grade 4 or 5 infections were reported, and the researchers saw no viral reactivations or opportunistic infections.
The safety profile for autologous HSCT with LentiGlobin was overall as expected for a myeloablative regimen that used single-agent busulfan, Dr. Walters said.
Most patients (11/18) with transfusion dependent beta thalassemia were able to stop transfusions, and the remaining patients had reduced transfusion requirements. Participants’ clinical status has stayed consistent through up to 3 years of follow-up, he said.
Of the patients who were able to stop transfusions, just two had the beta0/beta0 genotype. Among all transfusion independent participants, hemoglobin levels at the last study visit ranged from 8.4-13.7 g/dL. Beta0/beta0 genotype patients still receiving transfusions have seen a 60% median reduction in transfusion volume and a similar reduction in number of transfusions.
In response to an attendee question, Dr. Walters said that an analysis not included in the presentation has shown a fairly direct relationship between vector copy numbers and transfusion independence.
Currently, he said, vector copy numbers are higher, at around 3. With a higher vector copy number, more CD34+ cells will be transduced and infused, so there may be less concern about the dilutional effect of incomplete myeloablation.
“There may be an opportunity in the future to lessen the intensity of the conditioning regimen,” Dr. Walters said.
The study was funded by bluebird bio. Dr. Walters also reported several consulting relationships with pharmaceutical companies and laboratories.
SOURCE: Walters, M et al. 2018 BMT Tandem Meetings, Abstract 62.
SALT LAKE CITY – Lentiviral delivery of BB305 gene therapy via autologous hematopoietic stem cell transplant (HSCT) was safe and effective for individuals with transfusion dependent beta thalassemia, according to results of a phase 1/2 study.
None of the study participants died, and the majority of patients are now transfusion independent.
The Northstar study is an international, multicenter open-label, single-arm study of adolescents and adults with transfusion dependent beta thalassemia (TDT). A total of 18 patients at a median 21 years of age – 15 young adults aged 18-35 years and three adolescents aged 12-17 years – have now been treated, Mark Walters, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
Of these, 11 are now transfusion independent, with most patients stopping transfusions within 6 months of receiving gene therapy, said Dr. Walters, director of the blood and marrow transplantation program at the University of California, San Francisco’s Benioff Children’s Hospital, Oakland.
Eight patients had the beta0/beta0 genotype, and had essentially been transfusion dependent from infancy. Six other patients were betaE/beta0, and had become transfusion dependent over time. Four patients had other thalassemia genotypes.
Patients who enrolled in the Northstar study first had peripheral stem cell collection via apheresis after mobilization with granulocyte-colony stimulating factor and plerixafor. Then they received myeloablative conditioning with busulfan. At the same time, selected CD34+ cells were tranduced with the BB305 lentiviral vector and cryopreserved. Patients were infused with the transduced cells and managed through the engraftment process.
As a measure of annualized pre-procedure transfusion requirements, patients had received a median 163.6 mL/kg/year of packed red blood cells, Dr. Walters said. Not unexpectedly, liver iron concentration was a median 5.7 mg/g, though with a wide range among participants (0.4-26.4 mg/g). However, participants did not show signs of cardiac tissue iron on T2* magnetic resonance imaging . Six patients had undergone a splenectomy.
The median vector copy number was 0.7 (range, 0.3-1.5), with a median 31.5 CD34+ cells transduced (range, 17.0-58.0). The final cell dose delivered was a median 8.1 x 106 CD34+ cells/kg (range, 5.2-18.1).
“All 18 patients have had at least 18 months of follow-up,” said Dr. Walters, and data from 10 patients has been analyzed out to 2 years. Three patients have a full 3 years of follow-up, he said.
The self-inactivating lentiviral vector has behaved as expected; no replication-competent lentivirus has been found, with investigators conducting assessments at months 3, 6, and 12, and then annually through year 5.
The study protocol also calls for integration site analysis every 6 months for 5 years, and additional analyses at years 7, 10, and 15. Thus far, all samples have shown a polyclonal vector integration profile without clonal dominance, Dr. Walter said.
The median time to neutrophil engraftment was study day 18.5 (range, 14-30), while platelet engraftment was more variable, and overall slower, with engraftment at a median of study day 39.5 (range, 19-191).
Dr. Walters said that he and his colleagues examined characteristics of the four patients who still had platelet counts at or less than 100,000/microliters at 12 months after HSCT. They found that two of these patients had had splenectomies, but saw no clear relationship between speed of platelet engraftment and platelet count at 12 months. Three of the four patients had drug product cell doses less than the median.
However, two patients had no bleeding events after neutrophil engraftment, and bleeding events were all grade 1 or 2 in the other two patients. The slower-than-expected platelet engraftment rate was likely attributable to the ex vivo manipulation of the stem cells, Dr. Walters noted.
Looking at safety data from the point of neutrophil engraftment to the last follow-up, there have been no graft failures; six patients have had serious adverse events. Two events of veno-occlusive disease were assessed as grade 3 and attributed to the transplant. Two of these three patients had an extended hospital stay. Other grade 3 events including intracardiac thrombus, central catheter thrombosis, and cellulitis, as well as hyperglycemia and infectious diseases.
No grade 4 or 5 infections were reported, and the researchers saw no viral reactivations or opportunistic infections.
The safety profile for autologous HSCT with LentiGlobin was overall as expected for a myeloablative regimen that used single-agent busulfan, Dr. Walters said.
Most patients (11/18) with transfusion dependent beta thalassemia were able to stop transfusions, and the remaining patients had reduced transfusion requirements. Participants’ clinical status has stayed consistent through up to 3 years of follow-up, he said.
Of the patients who were able to stop transfusions, just two had the beta0/beta0 genotype. Among all transfusion independent participants, hemoglobin levels at the last study visit ranged from 8.4-13.7 g/dL. Beta0/beta0 genotype patients still receiving transfusions have seen a 60% median reduction in transfusion volume and a similar reduction in number of transfusions.
In response to an attendee question, Dr. Walters said that an analysis not included in the presentation has shown a fairly direct relationship between vector copy numbers and transfusion independence.
Currently, he said, vector copy numbers are higher, at around 3. With a higher vector copy number, more CD34+ cells will be transduced and infused, so there may be less concern about the dilutional effect of incomplete myeloablation.
“There may be an opportunity in the future to lessen the intensity of the conditioning regimen,” Dr. Walters said.
The study was funded by bluebird bio. Dr. Walters also reported several consulting relationships with pharmaceutical companies and laboratories.
SOURCE: Walters, M et al. 2018 BMT Tandem Meetings, Abstract 62.
SALT LAKE CITY – Lentiviral delivery of BB305 gene therapy via autologous hematopoietic stem cell transplant (HSCT) was safe and effective for individuals with transfusion dependent beta thalassemia, according to results of a phase 1/2 study.
None of the study participants died, and the majority of patients are now transfusion independent.
The Northstar study is an international, multicenter open-label, single-arm study of adolescents and adults with transfusion dependent beta thalassemia (TDT). A total of 18 patients at a median 21 years of age – 15 young adults aged 18-35 years and three adolescents aged 12-17 years – have now been treated, Mark Walters, MD, reported at the combined annual meetings of the Center for International Blood & Marrow Transplant Research and the American Society for Blood and Marrow Transplantation.
Of these, 11 are now transfusion independent, with most patients stopping transfusions within 6 months of receiving gene therapy, said Dr. Walters, director of the blood and marrow transplantation program at the University of California, San Francisco’s Benioff Children’s Hospital, Oakland.
Eight patients had the beta0/beta0 genotype, and had essentially been transfusion dependent from infancy. Six other patients were betaE/beta0, and had become transfusion dependent over time. Four patients had other thalassemia genotypes.
Patients who enrolled in the Northstar study first had peripheral stem cell collection via apheresis after mobilization with granulocyte-colony stimulating factor and plerixafor. Then they received myeloablative conditioning with busulfan. At the same time, selected CD34+ cells were tranduced with the BB305 lentiviral vector and cryopreserved. Patients were infused with the transduced cells and managed through the engraftment process.
As a measure of annualized pre-procedure transfusion requirements, patients had received a median 163.6 mL/kg/year of packed red blood cells, Dr. Walters said. Not unexpectedly, liver iron concentration was a median 5.7 mg/g, though with a wide range among participants (0.4-26.4 mg/g). However, participants did not show signs of cardiac tissue iron on T2* magnetic resonance imaging . Six patients had undergone a splenectomy.
The median vector copy number was 0.7 (range, 0.3-1.5), with a median 31.5 CD34+ cells transduced (range, 17.0-58.0). The final cell dose delivered was a median 8.1 x 106 CD34+ cells/kg (range, 5.2-18.1).
“All 18 patients have had at least 18 months of follow-up,” said Dr. Walters, and data from 10 patients has been analyzed out to 2 years. Three patients have a full 3 years of follow-up, he said.
The self-inactivating lentiviral vector has behaved as expected; no replication-competent lentivirus has been found, with investigators conducting assessments at months 3, 6, and 12, and then annually through year 5.
The study protocol also calls for integration site analysis every 6 months for 5 years, and additional analyses at years 7, 10, and 15. Thus far, all samples have shown a polyclonal vector integration profile without clonal dominance, Dr. Walter said.
The median time to neutrophil engraftment was study day 18.5 (range, 14-30), while platelet engraftment was more variable, and overall slower, with engraftment at a median of study day 39.5 (range, 19-191).
Dr. Walters said that he and his colleagues examined characteristics of the four patients who still had platelet counts at or less than 100,000/microliters at 12 months after HSCT. They found that two of these patients had had splenectomies, but saw no clear relationship between speed of platelet engraftment and platelet count at 12 months. Three of the four patients had drug product cell doses less than the median.
However, two patients had no bleeding events after neutrophil engraftment, and bleeding events were all grade 1 or 2 in the other two patients. The slower-than-expected platelet engraftment rate was likely attributable to the ex vivo manipulation of the stem cells, Dr. Walters noted.
Looking at safety data from the point of neutrophil engraftment to the last follow-up, there have been no graft failures; six patients have had serious adverse events. Two events of veno-occlusive disease were assessed as grade 3 and attributed to the transplant. Two of these three patients had an extended hospital stay. Other grade 3 events including intracardiac thrombus, central catheter thrombosis, and cellulitis, as well as hyperglycemia and infectious diseases.
No grade 4 or 5 infections were reported, and the researchers saw no viral reactivations or opportunistic infections.
The safety profile for autologous HSCT with LentiGlobin was overall as expected for a myeloablative regimen that used single-agent busulfan, Dr. Walters said.
Most patients (11/18) with transfusion dependent beta thalassemia were able to stop transfusions, and the remaining patients had reduced transfusion requirements. Participants’ clinical status has stayed consistent through up to 3 years of follow-up, he said.
Of the patients who were able to stop transfusions, just two had the beta0/beta0 genotype. Among all transfusion independent participants, hemoglobin levels at the last study visit ranged from 8.4-13.7 g/dL. Beta0/beta0 genotype patients still receiving transfusions have seen a 60% median reduction in transfusion volume and a similar reduction in number of transfusions.
In response to an attendee question, Dr. Walters said that an analysis not included in the presentation has shown a fairly direct relationship between vector copy numbers and transfusion independence.
Currently, he said, vector copy numbers are higher, at around 3. With a higher vector copy number, more CD34+ cells will be transduced and infused, so there may be less concern about the dilutional effect of incomplete myeloablation.
“There may be an opportunity in the future to lessen the intensity of the conditioning regimen,” Dr. Walters said.
The study was funded by bluebird bio. Dr. Walters also reported several consulting relationships with pharmaceutical companies and laboratories.
SOURCE: Walters, M et al. 2018 BMT Tandem Meetings, Abstract 62.
REPORTING FROM THE 2018 BMT TANDEM MEETINGS
Key clinical point: Major finding: Eleven of 18 patients became transfusion independent, and transfusions were reduced for the remainder of patients.
Study details: Open label, international, single-arm phase 1/2 study of 20 patients with transfusion-dependent beta thalassemia.
Disclosures: The study was funded by bluebird bio. Dr. Walters also reported consulting agreements with several pharmaceutical companies and laboratories.
Source: Walters, M et al. 2018 BMT Tandem Meetings, Abstract 62.
Deaths in patients on emicizumab

Two hemophilia organizations have notified the public of 5 deaths in adult patients receiving emicizumab (Hemlibra).
All 5 deaths—occurring in 2016 (n=1), 2017 (n=2), and this year (n=2)—were deemed unrelated to emicizumab by the investigator or treating physician.
The National Hemophilia Foundation and Hemophilia Federation of America reported these deaths after receiving notifications from Genentech.
The company said it has limited information about the circumstances of the deaths.
However, Genentech did say the 2016 death, 1 of the 2017 deaths, and 1 of the 2018 deaths occurred in patients receiving emicizumab via a compassionate use program.
Compassionate use of emicizumab has been available on a case-by-case basis, following a request to Roche from a patient’s treating physician, if the patient has a serious or life-threatening condition, has exhausted all other treatment options, and is unable to participate in a clinical trial.
The other death in 2017 occurred in a patient on the phase 3 HAVEN 1 trial and was reported along with the other results from that trial.
The remaining death in 2018 was in a patient receiving emicizumab via an expanded access protocol.
This protocol, which was reviewed by the US Food and Drug Administration, allowed US patients who were not participating in a clinical trial of emicizumab but who met eligibility criteria similar to key studies to have access to emicizumab prior to approval, which occurred in November 2017.
In response to the deaths, Genentech has pledged to share information on any adverse events that impact the overall benefit/risk profile of emicizumab.
“We are committed to providing timely and transparent updates on the safety profile of Hemlibra to health authorities, healthcare professionals, and the hemophilia community,” the company said.
For more information, patients and healthcare providers can call Genentech’s medical communications line at 1-800-821-8590.
Two hemophilia organizations have notified the public of 5 deaths in adult patients receiving emicizumab (Hemlibra).
All 5 deaths—occurring in 2016 (n=1), 2017 (n=2), and this year (n=2)—were deemed unrelated to emicizumab by the investigator or treating physician.
The National Hemophilia Foundation and Hemophilia Federation of America reported these deaths after receiving notifications from Genentech.
The company said it has limited information about the circumstances of the deaths.
However, Genentech did say the 2016 death, 1 of the 2017 deaths, and 1 of the 2018 deaths occurred in patients receiving emicizumab via a compassionate use program.
Compassionate use of emicizumab has been available on a case-by-case basis, following a request to Roche from a patient’s treating physician, if the patient has a serious or life-threatening condition, has exhausted all other treatment options, and is unable to participate in a clinical trial.
The other death in 2017 occurred in a patient on the phase 3 HAVEN 1 trial and was reported along with the other results from that trial.
The remaining death in 2018 was in a patient receiving emicizumab via an expanded access protocol.
This protocol, which was reviewed by the US Food and Drug Administration, allowed US patients who were not participating in a clinical trial of emicizumab but who met eligibility criteria similar to key studies to have access to emicizumab prior to approval, which occurred in November 2017.
In response to the deaths, Genentech has pledged to share information on any adverse events that impact the overall benefit/risk profile of emicizumab.
“We are committed to providing timely and transparent updates on the safety profile of Hemlibra to health authorities, healthcare professionals, and the hemophilia community,” the company said.
For more information, patients and healthcare providers can call Genentech’s medical communications line at 1-800-821-8590.
Two hemophilia organizations have notified the public of 5 deaths in adult patients receiving emicizumab (Hemlibra).
All 5 deaths—occurring in 2016 (n=1), 2017 (n=2), and this year (n=2)—were deemed unrelated to emicizumab by the investigator or treating physician.
The National Hemophilia Foundation and Hemophilia Federation of America reported these deaths after receiving notifications from Genentech.
The company said it has limited information about the circumstances of the deaths.
However, Genentech did say the 2016 death, 1 of the 2017 deaths, and 1 of the 2018 deaths occurred in patients receiving emicizumab via a compassionate use program.
Compassionate use of emicizumab has been available on a case-by-case basis, following a request to Roche from a patient’s treating physician, if the patient has a serious or life-threatening condition, has exhausted all other treatment options, and is unable to participate in a clinical trial.
The other death in 2017 occurred in a patient on the phase 3 HAVEN 1 trial and was reported along with the other results from that trial.
The remaining death in 2018 was in a patient receiving emicizumab via an expanded access protocol.
This protocol, which was reviewed by the US Food and Drug Administration, allowed US patients who were not participating in a clinical trial of emicizumab but who met eligibility criteria similar to key studies to have access to emicizumab prior to approval, which occurred in November 2017.
In response to the deaths, Genentech has pledged to share information on any adverse events that impact the overall benefit/risk profile of emicizumab.
“We are committed to providing timely and transparent updates on the safety profile of Hemlibra to health authorities, healthcare professionals, and the hemophilia community,” the company said.
For more information, patients and healthcare providers can call Genentech’s medical communications line at 1-800-821-8590.
Von Willebrand Disease: Approach to Diagnosis and Management
Introduction
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types (Table 1).1 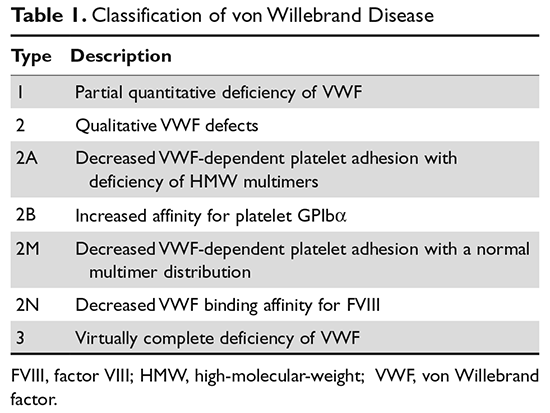
Prevalence
VWD is the most common inherited bleeding disorder. However, because VWF levels are highly variable and disease severity ranges from mild bleeding symptoms to severe or life-threatening bleeds, the reported prevalence of VWD depends on the diagnostic definition used. Two large epidemiologic studies have reported prevalence rates of approximately 1%.2,3 In these studies, healthy school-aged children were screened and diagnosed with VWD based on low VWF activity, measured as ristocetin cofactor, and a personal and family history of bleeding symptoms. At the other extreme, when considering patients whose bleeding symptoms are sufficiently severe to warrant referral to specialized centers, the reported prevalence of VWD ranges from 20 to 113 per million.4 These studies likely over- and underestimate clinically significant VWD. More recent studies suggest that the prevalence of VWD in individuals whose bleeding symptoms are significant enough to present to a primary care physician is approximately 0.1%.5 This figure is likely a more accurate estimate of the true prevalence of symptomatic VWD.
Although VWD is autosomally inherited, females are more likely to present with bleeding symptoms and be diagnosed because of increased exposure to bleeding challenges, such as menorrhagia and childbirth. VWD does not show any geographic or ethnic predilection, but there is an increased prevalence of the recessive forms, such as type 2N and type 3 VWD, in areas with high rates of consanguinity.
VWF Protein Structure and Function
The VWF gene is located on chromosome 12 at p13.3 and spans 178 kb comprising 52 exons.6 The expression of the VWF gene is tightly restricted to endothelial cells, platelets, and megakaryocytes, where VWF is stored in Weibel-Palade bodies and α-granules. VWF is a large multimeric glycoprotein with several important functional domains (Figure). 
Classification, Pathophysiology, and Genetics
The International Society of Thrombosis and Hemostasis (ISTH) classification of VWD was updated in 2006 (Table 1).1 It incorporates important aspects of clinical phenotype, pathophysiological mechanisms, and treatment considerations. The 3 categories are: type 1, which is a partial quantitative deficiency; type 2 with 4 subtypes (2A, 2B, 2M, and 2N), which is a qualitative defect; and type 3, which is a virtual absence of VWF. Although the diagnosis and categorization of VWD can be achieved with widely available laboratory testing, further subcategorization among type 2 VWD subtypes may require referral to a specialized laboratory. The current ISTH classification intentionally does not incorporate genotypic data. In type 2 or type 3 VWD disease, VWF mutations are identified in more than 90% of cases and are completely penetrant, whereas mutations are identified in only approximately 65% of type 1 VWD cases and have been associated with incomplete penetrance and variable expressivity.10 These studies suggest that type 1 VWD is an oligogenic disease with mutations in genes regulating secretion or clearance contributing to a VWD phenotype.
VWD Types
Type 1
Type 1 VWD is caused by a partial quantitative deficiency of VWF and represents approximately 75% of VWD cases. It is the most clinically heterogeneous type, with patients having a mild to moderate bleeding phenotype.11 Bleeding in type 1 VWD results from a decrease in the concentration of VWF. The VWF function is normal without a significant abnormality in the platelet, collagen, or FVIII binding sites or a significant decrease in HMW multimers. Functional assays of VWF, such as VWF ristocetin cofactor (VWF:RCo) or VWF activity (VWF:Act) (see section on Laboratory Testing for further details), are proportionally decreased relative to the VWF antigen level (VWF:Ag), and the ratio of functional activity as compared with the VWF level is normal (ie, VWF:RCo/VWF:Ag ratio is > 0.6). As noted, VWF mutations are identified in only 65% of type 1 VWD cases and have incomplete penetrance and variable expressivity.10 Approximately 70% of mutations identified are missense mutations. Missense mutations may affect VWF levels by affecting any part of the biosynthetic pathway, including trafficking, storage, secretion, and/or clearance of VWF.
Increased VWF clearance is a well-described mechanism for type 1 VWD, known as type 1C. These patients will typically have very low VWF levels, an increased VWF propeptide to antigen ratio (VWFpp/VWF:Ag), and a marked but short-lived response to DDAVP, limiting DDAVP’s clinical applicability.12 On the other hand, the half-life of VWF/FVIII concentrates is normal in these individuals. Type 1C VWD is caused by missense mutations which occur mainly in the D3 domain and reduce the half-life of VWF up to 15-fold. R1205H, known as the “Vicenza” variant, is the most common and severe as well as the best characterized of these mutations.13
Type 2
Accounting for approximately 25% of VWD cases, type 2 VWD is characterized by a qualitative deficiency of VWF activity and is further subcategorized based on the mechanism of VWF dysfunction. Type 2A, 2B, and 2M affect VWF–platelet interactions by way of loss of HMW multimers, a gain of function of the GPIbα binding site, or a loss of function of the same site, respectively. On the other hand, type 2N is caused by defective VWF binding to FVIII. Type 2 VWD is often suspected when investigations demonstrate a function-antigen discordance: the VWF:RCo or VWF:Act is decreased disproportionately to the decrease in VWF:Ag, and the VWF:RCo/VWF:Ag ratio is less than 0.6.
Type 2A VWD is the most common type 2 variant. It is characterized by disproportionately low functional activity compared to antigen level (ie, VWF:RCo/VWF:ag ratio is < 0.6) and a loss of HMW and sometimes intermediate molecular weight (IMW) multimers. Ristocetin-induced platelet agglutination (RIPA) will be decreased with standard doses of ristocetin and absent with low doses. Type 2A VWD is usually inherited as an autosomal dominant trait. This subtype encompasses missense mutations that impair dimerization or multimerization of VWF subunits (CK, D1, and D2 domains); disrupt intersubunit disulphide bonds (D3 and D2 domains); enhance susceptibility to ADAMTS13-mediated proteolysis (A2 and A1 domains); or result in intracellular retention of the HMW multimers (D3, A1, and A2 domains).10 The result is VWF that lacks HMW multimers, thereby possessing fewer GPIbα binding sites, and that is less effective in binding platelets.
Type 2B VWD is the result of gain-of-function mutations within the GPIbα binding site of VWF. Generally, the platelet-binding site of VWF within the A1 domain is only exposed once VWF is immobilized on injured collagen and subjected to shear forces, resulting in a conformational change.7 In type 2B VWD, the gain-of-function mutation results in spontaneous binding of VWF to platelets without the need for a VWF-collagen interaction and unfolding of VWF by shear forces. The VWF–platelet interaction selectively depletes the HMW multimers by the unfolding of the A2 domain and increasing ADAMTS13 proteolysis. The increased binding of mutant VWF to platelets also triggers the formation of platelet aggregates, which are removed from circulation resulting in thrombocytopenia. Increases in endogenous VWF seen with acute stressors or pregnancy can worsen thrombocytopenia and increase the risk of bleeding.14 Certain mutations, such as V1316M, alter megakaryocytopoiesis and are characterized by giant platelets with abnormal ultrastructure and further exacerbate the thrombocytopenia.15 The laboratory profile reveals a VWF:RCo/VWF:Ag ratio of < 0.6 and absence of HMW multimers. In contrast to type 2A, platelets will agglutinate with low-dose ristocetin. Missense mutations are highly penetrant dominant and occur in or close to the A1 domain.16
Type 2M VWD is characterized by loss-of-function mutations within the GPIbα binding site of VWF. Phenotypic characteristics include a reduced ratio of VWF:RCo/VWF:Ag of < 0.6 but a normal multimer pattern.17 Missense mutations are reported in the A1 domain affecting the GPIbα-binding site. In very rare instances, mutations in the A3 domain that impair the VWF/collagen interaction have been described.18 These collagen-binding mutations are not included in the last iteration of the ISTH classification in 2006,1 but fit best in the type 2M category. In these cases, VWF:RCo or VWF:Act, which reflect activity at the GPIbα-binding site, may be normal and the diagnosis requires VWF/collagen binding assays (VWF:CB).
Type 2N VWD results from mutations of the FVIII binding site or conformational changes that impair the VWF–FVIII interaction. Most (~80%) missense mutations are located in domains D’ and D3.19 These mutations are autosomal recessive, and affected individuals are either homozygous or compound heterozygous for type 2N/2N or type 1/2N mutations, or compound heterozygous for a missense mutation and a mutation resulting in a null allele (type 2N/3 mutations). The laboratory phenotype is a disproportionate reduction in the FVIII level relative to the VWF level, which may be low or normal. Most cases of type 2N VWD have a normal multimeric profile, but rare cases will demonstrate loss of HMW multimers. Definitive diagnosis requires evidence of reduced FVIII binding to VWF (VWF:FVIIIB) or the identification of causative mutations in the FVIII binding region of the VWF gene.20
Type 3
Type 3 VWD is defined by a virtual absence of VWF. The inheritance of type 3 VWD has often been reported as autosomal recessive. However, there is emerging evidence that it can also be inherited in a co-dominant pattern: obligate carriers of type 3 VWD mutations have more mucocutaneous bleeding symptoms than normal individuals, and in approximately 50% of cases may carry a diagnosis of type 1 VWD.21 This condition is characterized by prolongation of the activated partial thromboplastin time (aPTT), undetectable levels of VWF:Ag, and VWF:RCo and FVIII levels less than 10 IU/dL (10%). The majority (~80%) of type 3 VWD patients have 2 null alleles as a result of a variety of mutations, with nonsense mutations accounting for about one-third.10 The remainder of the mutational spectrum is made up of missense mutations predominantly located in the D1-D2 (exons 3–11) and D4-CK (exons 37–52) domains that result in intracellular VWF retention, or large deletions, resulting in frameshift mutations affecting 1 or more exons. Because there is little or no circulating VWF, patients with type 3 VWD may develop alloantibodies to VWF, which can complicate treatment.22
Diagnosis
Clinical Manifestations
VWD is a congenital bleeding disorder. The increased risk of bleeding is present from birth, but symptoms may only manifest when there is a hemostatic challenge. Bleeding symptoms become more apparent with increasing age and exposure to hemostatic challenges. As a result, the diagnosis is often delayed into adulthood in mild to moderate forms of VWD. On the other hand, with more severe bleeding phenotypes such as type 3 VWD, the diagnosis is often made in childhood. Individuals with VWD primarily complain of excessive mucocutaneous bleeding, which includes spontaneous bruising, recurrent epistaxis, and bleeding from the gums after brushing, dental cleaning, and extractions. In addition, prolonged or excessive bleeding after surgery or trauma is often reported. Females frequently experience menorrhagia, usually beginning at menarche, and can have prolonged or excessive bleeding after childbirth.23 Musculoskeletal bleeding is unusual, except in type 2N or type 3 VWD when the FVIII:C level may be less than 10 IU/dL.
Mucocutaneous bleeding symptoms such as epistaxis, gum bleeding, ecchymosis, and menorrhagia overlap with those experienced by a normal population, and therefore can be easily overlooked by both patients and physicians.11 The use of bleeding assessment tools (BATs) to standardize the bleeding history and interpretation of the severity of the bleeding phenotype is becoming part of routine clinical practice. Three different BATs, each an adaptation of its predecessor, have been created and validated.24 Each of the scores performs well in an undiagnosed population presenting with bleeding symptoms. The negative predictive value is typically greater than 0.99, meaning that a negative bleeding score nearly excludes a clinically significant bleeding disorder. Thus, the main utility of the current BATs is at the time of new patient assessments: a negative bleeding score will help avoid unnecessary laboratory testing and prevent false-positive diagnoses of VWD (borderline low VWF:Ag without a significant bleeding history). However, the currently available BATs have some limitations. When scoring severe bleeding disorders, BATs become saturated as they take into account the worst episode of bleeding within each category but not the frequency of bleeding. BATs need to be administered by an expert and are time consuming to complete. Finally, they are not useful for monitoring bleeding symptoms or response to therapy because of the cumulative nature of the scores. In an attempt to standardize the BAT and bleeding score, the ISTH/Scientific and Standardization Committee (SSC) Joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group has established a revised BAT, known as the ISTH-BAT, specifically designed to extend the utility of the earlier BATS by incorporating information on both symptom frequency and severity.25,26 The ISTH-BAT has been further modified to a patient- or self-administered BAT (SELF-BAT). The SELF-BAT has been shown to be a reliable and effective tool in the assessment of patients who are being evaluated for VWD.27
Laboratory Testing
Screening tests include a complete blood count (CBC), prothrombin time, aPTT, thrombin time, and fibrinogen concentration to exclude the presence of other hemostatic disorders. The CBC may show thrombocytopenia in type 2B VWD. The aPTT is often normal, but will be prolonged if the FVIII level is below 30 IU/dL, as can be seen in severe type 1, type 2N, or type 3 VWD. The platelet function analyzer (PFA-100) is a system for analyzing primary hemostasis under high shear rates, but its role in the diagnosis of VWD is controversial.11
The evaluation of VWD involves quantitative (VWF:Ag) and qualitative measurements of VWF (VWF:RCo, or one of the novel assays: VWF:Act or VWF:GPIbM) and FVIII activity (FVIII:C). Type 2 VWD is suspected when the VWF activity to VWF:Ag ratio is < 0.6, the FVIII:C is more significantly decreased as compared to VWF:Ag, or with the presence of thrombocytopenia. In these cases, further testing (multimer gel electrophoresis, VWF:CB, RIPA, VWF:FVIIIB, and genotyping) is required to discriminate the type 2 VWD subtype, but such testing may be available only in specialized laboratories. If type 1C VWD is suspected, the VWFpp/VWF:ag ratio may confirm the diagnosis. Table 2 summarizes the results seen with each subtype. These assays are described in detail below.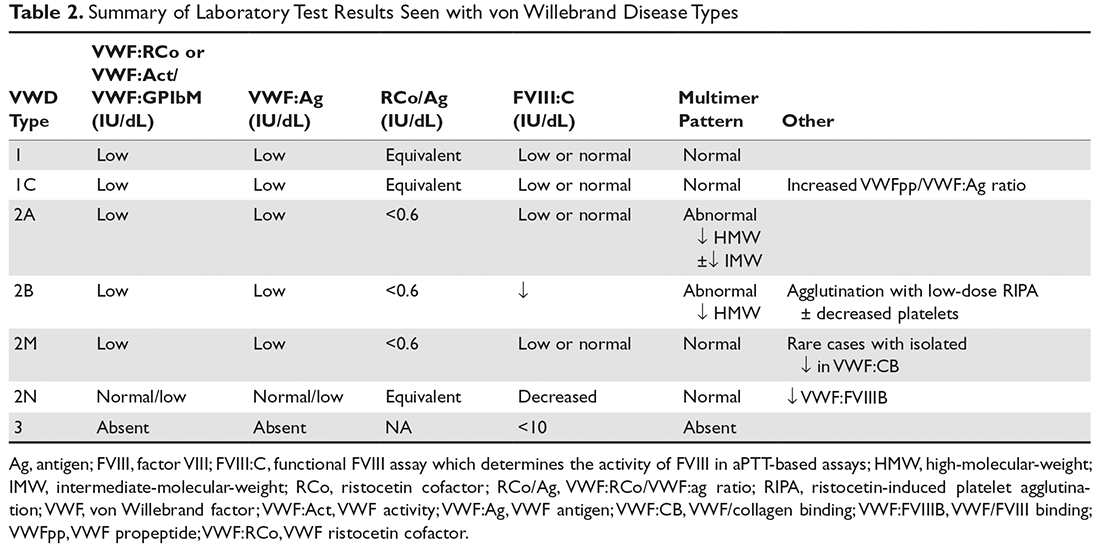
VWD Assays
VWF:Ag represents the quantity of VWF protein (antigen) in the plasma measured using an enzyme-linked immunosorbent assay (ELISA) or latex immunoassay. The normal range is approximately 50 to 200 IU/dL.
VWF:RCo is a functional assay that determines the capacity of VWF to agglutinate platelets via the platelet receptor GPIbα in the presence of ristocetin. The normal range is approximately 50 to 200 IU/dL. Novel methods of measuring VWF’s platelet-binding activity are increasingly being adopted by clinical laboratories and are associated with greater precision and improved lower limits of detection and coefficients of variation.28,29 The first is the VWF:Act, a rapid automated assay that measures VWF activity using an antibody directed to the GPIbα binding site of VWF.28 The second novel assay is VWF:GPIbM, which involves a gain-of-function GPIB construct that binds VWF without ristocetin.30,31 For simplicity, VWF:RCo will be used to refer to VWF platelet-binding activity in the ensuing text. Factor VIII:C is a functional FVIII assay that determines the activity of FVIII in aPTT-based assays. The normal range is approximately 50 to 150 IU/dL.
VWF multimer analysis by SDS-agarose electrophoresis assesses VWF oligomers in plasma.32 Normal plasma contains multimers composed of over 40 VWF dimers, and these multimers are classified as high (HMW), intermediate (IMW), or low molecular weight (LMW). HMW multimers are decreased or missing in types 2A and 2B VWD, and IMW multimers may also be absent in type 2A VWD.
Low-dose RIPA tests the capacity of the patient’s platelets to agglutinate at low concentrations of ristocetin (~0.5 mg/mL). This is in contrast to the VWF:RCo, in which formalin-fixed control platelets are used. With type 2B, the platelet membrane is “overloaded” with high-affinity mutant VWF, resulting in abnormal platelet agglutination at low ristocetin concentrations. In some cases of type 2B VWD, all variables except RIPA may be normal.29
VWF:FVIIIB is an ELISA-based assay that determines the ability of VWF to bind FVIII and is used to make the diagnosis of type 2N VWD.19
VWF:CB is an ELISA-based assay that measures the ability of VWF to bind to collagen, a function of VWF that is dependent on the collagen-binding domain (A3) and on the presence of HMW multimers. VWF:CB helps to distinguish between types 1 and 2 VWD by reflecting the loss of HMW multimer forms (type 2A VWD) or can reflect a specific collagen-binding deficiency (type 2M VWD).33 The normal range is approximately 50 to 200 IU/dL. This assay is not available in most clinical laboratories.
VWFpp/VWF:Ag takes advantage of 2 facts: the VWF propeptide is secreted in a one-to-one ratio to VWF subunits and has a stable half-life in plasma. Thus, an increased ratio identifies patients with mutations that increase VWF clearance, such as type 1C VWD.34 The mean ratio in normal individuals is 1.3, with a normal range of 0.54 to 1.98.
Genotyping should be considered when specialized testing with the VWF:FVIIIB, RIPA, or VWF:CB assays is unavailable and a diagnosis of type 2 VWD is suspected. A guideline on VWD genetic testing has been published by the UK Haemophilia Centre Doctors Organisation.35
Interpretation of Clinical History and Laboratory Investigations
Normal plasma levels of VWF are approximately 100 IU/dL (100%, corresponds to ~10 μg/mL) with a population range of 50 to 200 IU/dL (50%–200%). There are a number of preanalytical variables (patient specific or laboratory specific) that affect the results of VWF laboratory testing. Patient-specific variables that are associated with increased VWF levels include increasing age, African ethnicity, exercise, inflammatory disease states, blood group A or B, increased levels of epinephrine, cocaine use, and neuroendocrine hormone levels. Decreased VWF levels are associated with medications such as valproic acid, hypothyroidism, autoantibodies, and blood group O. Individuals with blood group O have VWF levels that are 25% lower than levels in other blood groups.36 Several analytical variables also can complicate the diagnosis of VWD: methods for established reference ranges, limitations to the sensitivity of assays, and sample handling issues.11 These factors (summarized in Table 3) must be considered when interpreting VWF laboratory results, and at least 2 sets of tests using fresh samples are needed to confirm the diagnosis of VWD. Testing should be avoided in stressed, ill, or pregnant patients.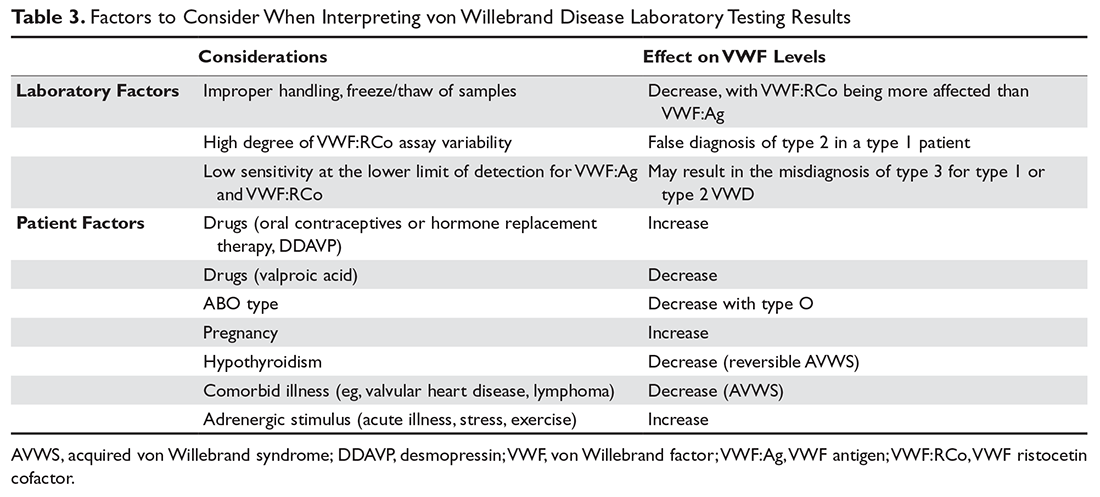
Mild type 1 VWD can be a difficult diagnosis to make because of the overlap of bleeding symptoms among normal individuals and those with mild type 1 VWD, as well as the variability of VWF levels. There is no consensus on the exact VWF levels required to confirm the diagnosis: the NHLBI Expert Panel recommends VWF:Ag and VWF:RCo levels less than 0.30 IU/mL to diagnose type 1 VWD,11 whereas the ISTH-SSC Subcommittee on von Willebrand factor recommends using VWF:RCo and VWF:Ag levels greater than 2 standard deviations below the population mean.37 In the absence of a bleeding history, slightly reduced VWF levels do not predict future significant bleeding events.38 Therefore, regardless of the laboratory cut-off used, the cornerstone of a VWD diagnosis should be a history of excessive mucocutaneous bleeding.
Differential Diagnosis
When considering a diagnosis of VWD, the differential diagnosis must be considered and includes acquired von Willebrand syndrome (AVWS), platelet-type VWD (PT-VWD), and hemophilia A. AVWS is the result of an acquired deficiency or defect of VWF and manifests with a mild to moderate bleeding disorder without a lifelong personal and family history of bleeding. AVWS has diverse pathology. The most common mechanism is proteolytic cleavage of VWF after shear stress–induced unfolding, as seen with aortic stenosis and ventricular assist devices, where as many as 79% of persons with aortic stenosis39 and up to 100% with left ventricular assist devices are affected.40 Other disease mechanisms include autoantibody formation that impairs VWF function or increases its clearance (autoimmune disease or lymphoproliferative disease), adsorption of HMW VWF multimers to malignant cells or platelets (myeloproliferative neoplasms and Wilm’s tumor), or decreased synthesis (hypothyroidism, valproic acid). The median age of diagnosis is 62 years, but the disorder may occur in any age-group (range 2–96 years).41 The approach to management of AVWS should focus on treatment of bleeding and induction of long-term remission. Treatment of bleeding will depend on the underlying mechanism of AVWS and may include a combination of DDAVP or VWF/FVIII concentrates, recombinant factor VIIa, antifibrinolytic agents, intravenous immunoglobulin, or plasmapheresis for AVWS associated with autoantibodies. Treatment of the underlying disorder (eg, aortic valve repair or treatment of a lymphoproliferative disorder) may result in remission of the AVWS.
Mild hemophilia A (caused by mutations in the F8 gene) and type 2N VWD can be difficult to differentiate clinically. Both present with reduced FVIII:C, and type 2N VWD may have normal or borderline low levels of VWF. Although the VWF:FVIIIB assay will distinguish between the 2 disorders, the test is not available in many centers. The pattern of inheritance may be helpful: hemophilia A is an X-linked disorder, whereas type 2N is autosomal recessive. Often, the diagnosis of type 2N VWD is suspected when genotyping of F8 does not identify a mutation in mild hemophilia A, when infused FVIII concentrates have a decreased half-life, or when DDAVP is associated with a brisk but short-lived response. In the absence of VWF:FVIIIB assay availability, genotyping of VWF will confirm the diagnosis, with missense mutations being located in exons 17–20 or 24–27.19
PT-VWD represents the phenocopy of type 2B VWD. The mutation is in the platelet receptor gene GPIBA and causes enhanced VWF-platelet binding. The disorders can be differentiated by RIPA plasma/platelet mixing studies or flow cytometry.42,43 However, these assays are technically challenging. In the absence of mutations in exon 28 of VWF, mutations in exon 2 of GPIBA may be identified in approximately 10% of persons misdiagnosed with type 2B VWD.
Management
Patients with VWD present to medical attention in a number of ways: excessive post-trauma or surgical bleeding, recurrent mucocutaneous bleeding such as epistaxis, menorrhagia, gastrointestinal bleeding, or, in severe cases, recurrent hemarthroses and muscle hematomas. Irrespective of the presentation, the goal is to minimize and control bleeding. Therapeutic options can be divided into 3 main categories: (1) localized measures to stop bleeding; (2) pharmacologic agents with indirect hemostatic benefit; and (3) treatments that directly increase plasma VWF and FVIII levels. A combination of all 3 of these modalities can be used depending on the bleeding location and severity.
Localized Measures
Localized measures to control bleeding in VWD will depend on the site of bleeding. Epistaxis can be particularly problematic for affected children, and patients should be armed with a step-wise action plan that escalates from pressure to packing and includes guidelines regarding how long to wait before seeking medical attention. In selected cases, nasal cautery may be required for prolonged or excessive epistaxis. Topical hemostatic agents such as gelatin foam/matrix, topical thrombin, and fibrin sealants are predominately used to achieve surgical hemostasis and may have a limited role in the treatment of VWD-associated bleeding. In the case of menorrhagia, hormonal treatments (ie, the combined oral contraceptive pill, OCP), levonorgestrel-releasing intrauterine systems, or endometrial ablation all effectively reduce menstrual blood loss through their local effects on the endometrial lining.44 In addition, older generations of OCP are associated with increases in VWF levels. This effect is mediated by the estrogen component and is evident with ethynylestradiol doses of 0.5 μg or higher. Lower estrogen doses, seen in currently used OCP, have little or no effect on VWF levels.11,45
Pharmacologic Therapy
Indirect therapies include the antifibrinolytic agents (eg, tranexamic acid and aminocaproic acid). These agents are used either as the sole therapy at the time of minor surgical and dental procedures, or as an adjunct in combination with DDAVP or VWF/FVIII concentrates. Antifibrinolytics are thought to be particularly useful for controlling mucosal bleeding in areas of high fibrinolytic activity: the oral cavity, gastrointestinal tract, or uterus. Tranexamic acid inhibits the conversion of plasminogen to plasmin, and is the more commonly used antifibrinolytic.11 Tranexamic acid can be administered either intravenously or orally at doses of 10 to 25 mg/kg, respectively. It is usually continued until bleeding is controlled or up to 7 to 10 days postoperatively. The most common adverse events associated with tranexamic acid are headache, back pain, and gastrointestinal side effects.46 Tranexamic acid is contraindicated in disseminated intravascular coagulation and bleeding from the upper urinary tract, where it can lead to urinary tract obstruction by clots.
DDAVP, a synthetic derivative of vasopressin, promotes release of stored VWF from endothelial cells. Most individuals with type 1 VWD and some with type 2A VWD respond to treatment with DDAVP: a therapeutic trial to confirm adequate DDAVP response should be performed prior to its clinical use. Assessment of VWF:Ag, VWF:RCo, and FVIII levels should be performed before and at several time points after the DDAVP administration up to and including 4 hours. Peak VWF levels are achieved 30 and 90 minutes after intravenous and intranasal delivery, respectively. An increase in VWF:Ag/VWF:RCo and FVIII levels to at least 30 IU/dL is adequate for most dental procedures, minor surgery, or the treatment of epistaxis or menorrhagia. DDAVP may be adequate to treat major bleeds or for major surgery when VWF levels increase well above 50 IU/dL. Decisions surrounding the use of DDAVP versus a VWF/FVIII concentrate will depend on the expected DDAVP response, the type of surgery, and the anticipated duration of therapy required to achieve hemostasis. If treatment is required for more than 3 days, concerns regarding tachyphylaxis and side effects may limit its use. Significantly decreased VWF:Ag/VWF:RCo or FVIII at the 4-hour time point of a DDAVP trial may indicate type 1C or type 2N VWD, which are associated with increased clearance of endogenous VWF or FVIII, respectively. Despite the transient response in these patients, DDAVP remains a therapeutic option and its use should be assessed on a case-by-case basis.47
The parenteral dose of DDAVP is 0.3 μg/kg infused in 30 to 50 mL of normal saline over approximately 30 minutes every 12 to 24 hours. The dose of the highly concentrated intranasal preparation is 150 μg for children under 50 kg and 300 μg for larger children and adults (1 spray per naris). It is important to note that the products used to treat VWD (eg, Stimate) deliver 150 μg per spray, a much higher concentration than that used to treat enuresis. Repeated DDAVP dosing is associated with the development of tachyphylaxis: with subsequent dosing, the magnitude of the VWF and FVIII increments can fall to approximately 70% of that obtained with the initial dose.48 DDAVP is safe and generally well tolerated. Side effects include facial flushing, headache, tachycardia, light-headedness, and mild hypotension. The most serious side effects, severe hyponatremia and seizures,49 can be avoided by fluid restriction for 24 hours after DDAVP administration. Serum sodium levels should be monitored with repeated doses. DDAVP is generally avoided in those younger than 2 years of age because of a higher risk of hyponatremia. Patients who are intolerant of DDAVP or have a poor VWF response need to be treated with a VWF/FVIII concentrate.
VWF/FVIII Concentrate
VWF/FVIII concentrates are required for patients who do not have an adequate response to DDAVP, who have side effects from or contraindications to DDAVP, or who require a long duration of treatment, rendering the use of DDAVP impractical. Purified, viral-inactivated, plasma-derived VWF/FVIII concentrates are the products most frequently used (eg, Humate-P, Wilate, Alphanate SD/HT). The quantity of VWF:RCo activity relative to FVIII:C varies by product; Humate-P contains 2.4 VWF:RCo units for each unit of FVIII:C; Wilate contains a 1:1 ratio; and Alphanate contains a 0.5:1 ratio. Both Humate-P and Wilate are reported to contain a full spectrum of VWF multimers, including HMW multimers, and closely resemble normal plasma, but Alphanate SD/HT lacks HMW mutimers.11,50 Thus, the available VWF/FVIII vary in terms of VWF:RCo to FVIII concentrate, HMW multimer composition, reported VWF:RCo, and FVIII half-lives and even approved indications. They should not be considered interchangeable, and further information should be sought from the respective product inserts.
Dosing recommendations are provided either in VWF:RCo (North America) or FVIII:C units (Europe) and are weight-based (Table 4); repeat infusions can be given every 8 to 24 hours depending on the type of surgery/injury and the product used. 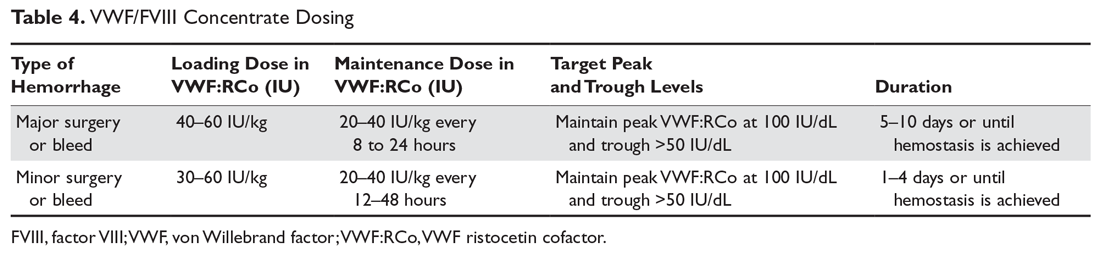
Long-term continuous use of concentrates to prevent bleeds, known as prophylaxis, is the standard of care in severe hemophilia A and B and is now being adopted in severe VWD. Patients with type 3 VWD or severe type 1 or type 2 VWD may experience recurrent bleeds into joints, nasal/oropharynx, or gastrointestinal tract or excessive menstrual bleeding. Retrospective cohort and case series suggest that prophylaxis improves quality of life; reduces the frequency of bleeding, need for transfusions, and hospitalizations; and prevents chronic joint disease.54,55 More recently, a prospective study confirmed that prophylaxis with VWF concentrates at doses ranging from 50 IU VWF RCo/kg 1 to 3 times per week was highly effective at reducing bleeding rates, with annualized bleeding rates decreasing from 25 to 6.1 in 11 participants with either type 2A or type 3 VWD.56
VWF/FVIII concentrates are effective in more than 97% of events.57 Rarely, when infusion of a VWF/FVIII concentrate is ineffective at stopping bleeding, transfusion of platelet concentrates may be beneficial, presumably because they facilitate the delivery of small amounts of platelet VWF to the site of vascular injury. Highly purified FVIII concentrates (monoclonal antibody purified and recombinant) should not be used to treat VWD because they lack VWF.
A recombinant VWF concentrate (Vonvendi) combined initially with recombinant FVIII concentrate in a 1.3:1 ratio of VWF:RCo to FVIII:C has been shown to be safe and efficacious for the on-demand treatment of bleeds.58,59 After the initial FVIII dose, the patients’ endogenous FVIII levels are stabilized within 6 hours and further FVIII administration may not required. A prospective phase 3 trial investigating the efficacy of recombinant VWF in the prophylaxis of severe VWD is ongoing. Vonvendi has been licensed for on-demand treatment in the United States since 2015. For further dosing information, please refer to the product insert.
Conclusion
VWF is a complex protein with several important and distinct functional domains: binding sites to collagen, FVIII, and platelet GPIbα; an ADAMTS13 cleavage site; and domains important for multimer formation. Mutations in any of these sites can result in a dysfunctional protein and as a result, VWD is a heterogeneous disorder with many specific assays available to determine the subtype. Despite this, the treatment of VWD is straightforward with only a small number of therapeutic options: indirect therapies such as antifibrinolytic agents, or direct therapies that increase VWF levels, DDAVP, or VWF/FVIII concentrates. Management focuses on preventing bleeding complications associated with invasive procedures or promptly treating bleeding episodes.
1. Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006;4:2103–14.
2. Rodeghiero F, Castaman G. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987;69:454–9.
3. Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993;123:893–8.
4. Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000;84:160–74.
5. Bowman M, Hopman WM, Rapson D, et al. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost 2010;8:213–6.
6. Mancuso DJ, Tuley EA, Westfield LA, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989;264:19514–27.
7. Kang I, Raghavachari M, Hofmann CM, Marchant RE. Surface-dependent expression in the platelet GPIb binding domain within human von Willebrand factor studied by atomic force microscopy. Thromb Res 2007;119:731–40.
8. Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996;84:289–97.
9. Dong J, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002;100:4033–9.
10. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010;24:123–34.
11. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report (USA). Haemophilia 2008;14:171–232.
12. Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 vwd (MCMDM-1VWD). Blood 2008;111:4979–85.
13. Goodeve A. Vicenza deciphered: modeling the von Willebrand disease enigma: commentary on accelerated clearance alone explains ultralarge multimers in VWD Vicenza. J Thromb Haemost 2010;8:1271–2.
14. Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009;113:526–34.
15. Nurden AT, Federici AB, Nurden P. Altered megakaryocytopoiesis in von Willebrand type 2B disease. J Thromb Haemost 2009;7 Suppl 1:277–81.
16. Ruggeri ZM, Pareti FI, Mannucci PM, et al. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand’s disease. New Engl J Med 1980;302:1047–51.
17. James PD, Notley C, Hegadorn C, et al. Challenges in defining type 2M von Willebrand disease: results from a Canadian cohort study. J Thromb Haemost 2007;5:1914–22.
18. Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost 2010;8:1431–3.
19. Mazurier C, Hilbert L. Type 2N von Willebrand disease. Curr Hematol Rep 2005;4:350–8.
20. Nesbitt IM, Goodeve AC, Guilliatt AM, et al. Characterisation of type 2N von Willebrand disease using phenotypic and molecular techniques. Thromb Haemost 1996;75:959–64.
21. Bowman M, Tuttle A, Notley C, et al. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant inheritance of mutant alleles. J Thromb Haemost 2013;11:512–20.
22. James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013;122:636–40.
23. James AH, Jamison MG. Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost 2007;5:1165–9.
24. Rydz N, James PD. The evolution and value of bleeding assessment tools. J Thromb Haemost 2012;2223–9.
25. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost 2010;8:2063–5.
26. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia 2014;20:831–5.
27. Deforest M, Grabell J, Alberta S et al. Generation and optimization of the self-administered bleeding assessment tool and its validation as a screening test for von Willebrand disease. Haemophilia 2015;21:e384-8.
28. Castaman G, Hillarp A, Goodeve A. Laboratory aspects of von Willebrand disease: test repertoire and options for activity assays and genetic analysis. Haemophilia 2014;20(Suppl. 4):65–70.
29. Favaloro EJ. Von Willebrand disease, type 2B: a diagnosis more elusive than previously thought. Thromb Haemost 2008;99:630–1.
30. Budde U. Diagnosis of von Willebrand disease subtypes: implications for treatment. Haemophilia 2008;14 Suppl 5:27–38.
31. Favaloro EJ. Von Willebrand factor collagen-binding (activity) assay in the diagnosis of von Willebrand disease: a 15-year journey. Sem Thromb Hemost 2002;28:191–202.
32. Patzke J, Budde U, Huber A, et al. Performance evaluation and multicenter study of a von Willebrand factor activity assay based on GPIb binding in the absence of ristocetin. Blood Coagul Fibrinolysis 2014;25:860-70.
33. Graf L, Moffat KA, Carlino SA, et al. Evaluation of an automated method for measuring von Willebrand factor activity in clinical samples without ristocetin. Int J Lab Hematol 2014;36:341–51.
34. Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood 2006;108:3344–51.
35. Keeney S, Bowen D, Cumming A, et al. The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organisation Haemophilia genetics laboratory network. Haemophilia 2008;14:1099–111.
36. Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987;69:1691–5.
37. Sadler JE, Rodeghiero F. Provisional criteria for the diagnosis of VWD type 1. J Thromb Haemost 2005;3:775–7.
38. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM- 1VWD). J Thromb Haemost 2006;4:766–73.
39. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003;349:343–9.
40. Uriel N, Pak S-W, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010;56:1207–13.
41. Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost 2000;84:345–9.
42. Favaloro EJ, Patterson D, Denholm A, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Brit J Haematol 2007;139:621–8.
43. Giannini S, Cecchetti L, Mezzasoma AM, Gresele P. Diagnosis of platelet-type von Willebrand disease by flow cytometry. Haematologica 2010;95:1021–4.
44. Farquhar C, Brown J. Oral contraceptive pill for heavy menstrual bleeding. Cochrane Database Syst Rev 2009 Oct 7;(4):CD000154.
45. Kadir R, Economides DL, Sabin C, et al. Variations in coagulation factors in women: effects of age, ethnicity, menstrual cycle and combined oral contraceptive. Thromb Haemost 1999;82:1456–61.
46. Muse K, Lukes AS, Gersten J, et al. Long-term evaluation of safety and health-related quality of life in women with heavy menstrual bleeding treated with oral tranexamic acid. Womens Health 2011;7:699–707.
47. Castaman G, Tosetto A, Federici AB, Rodeghiero F. Bleeding tendency and efficacy of anti-haemorrhagic treatments in patients with type 1 von Willebrand disease and increased von Willebrand factor clearance. Thromb Haemost 2011;105:647–54.
48. Mannucci PM, Bettega D, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Brit J Haematol 1992;82:87–93.
49. Greaves M, Watson HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost 2007;5 Suppl 1:167–74.
50. Kessler CM, Friedman K, Schwartz BA, Gill JC, Powell JS. The pharmacokinetic diversity of two von Willebrand factor (VWF) / factor VIII (FVIII) concentrates in subjects with congenital von Willebrand disease. results from a prospective, randomised crossover study. Thromb Haemost 2011;106:279–88.
51. Weiss HJ, Sussman II, Hoyer LW. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand’s disease. J Clin Invest 1977;60:390–404.
52. Berntorp E. Haemate P/Humate-P: a systematic review. Thromb Res 2009;124:S11–14.
53. Lillicrap D, Poon MC, Walker I, et al. Efficacy and safety of the factor VIII/von Willebrand factor concentrate, Haemate-P/Humate-P: ristocetin cofactor unit dosing in patients with von Willebrand disease. Thromb Haemost 2002;87:224–30.
54. Halimeh S, Krümpel A, Rott H, et al. Long-term secondary prophylaxis in children, adolescents and young adults with von Willebrand disease. results of a cohort study. Thromb Haemost 2011;105:597–604.
55. Abshire TC, Federici AB, Alvárez MT, et al. Prophylaxis in severe forms of von Willebrand’s disease: results from the von Willebrand disease prophylaxis network (VWD PN). Haemophilia 2013;19:76–81.
56. Abshire T, Cox-Gill J, Kempton CL, et al. Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. J Thromb Haemost 2015;13:1585– 9.
57. Auerswald G, Kreuz W. Haemate P/Humate-P for the treatment of von Willebrand disease: considerations for use and clinical experience. Sem Thromb Hemost 2008;14 (Suppl 5):39–46.
58. Mannucci PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood 2013;122:648–57.
59. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood 2015;126:2038–46.
Introduction
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types (Table 1).1
Prevalence
VWD is the most common inherited bleeding disorder. However, because VWF levels are highly variable and disease severity ranges from mild bleeding symptoms to severe or life-threatening bleeds, the reported prevalence of VWD depends on the diagnostic definition used. Two large epidemiologic studies have reported prevalence rates of approximately 1%.2,3 In these studies, healthy school-aged children were screened and diagnosed with VWD based on low VWF activity, measured as ristocetin cofactor, and a personal and family history of bleeding symptoms. At the other extreme, when considering patients whose bleeding symptoms are sufficiently severe to warrant referral to specialized centers, the reported prevalence of VWD ranges from 20 to 113 per million.4 These studies likely over- and underestimate clinically significant VWD. More recent studies suggest that the prevalence of VWD in individuals whose bleeding symptoms are significant enough to present to a primary care physician is approximately 0.1%.5 This figure is likely a more accurate estimate of the true prevalence of symptomatic VWD.
Although VWD is autosomally inherited, females are more likely to present with bleeding symptoms and be diagnosed because of increased exposure to bleeding challenges, such as menorrhagia and childbirth. VWD does not show any geographic or ethnic predilection, but there is an increased prevalence of the recessive forms, such as type 2N and type 3 VWD, in areas with high rates of consanguinity.
VWF Protein Structure and Function
The VWF gene is located on chromosome 12 at p13.3 and spans 178 kb comprising 52 exons.6 The expression of the VWF gene is tightly restricted to endothelial cells, platelets, and megakaryocytes, where VWF is stored in Weibel-Palade bodies and α-granules. VWF is a large multimeric glycoprotein with several important functional domains (Figure).
Classification, Pathophysiology, and Genetics
The International Society of Thrombosis and Hemostasis (ISTH) classification of VWD was updated in 2006 (Table 1).1 It incorporates important aspects of clinical phenotype, pathophysiological mechanisms, and treatment considerations. The 3 categories are: type 1, which is a partial quantitative deficiency; type 2 with 4 subtypes (2A, 2B, 2M, and 2N), which is a qualitative defect; and type 3, which is a virtual absence of VWF. Although the diagnosis and categorization of VWD can be achieved with widely available laboratory testing, further subcategorization among type 2 VWD subtypes may require referral to a specialized laboratory. The current ISTH classification intentionally does not incorporate genotypic data. In type 2 or type 3 VWD disease, VWF mutations are identified in more than 90% of cases and are completely penetrant, whereas mutations are identified in only approximately 65% of type 1 VWD cases and have been associated with incomplete penetrance and variable expressivity.10 These studies suggest that type 1 VWD is an oligogenic disease with mutations in genes regulating secretion or clearance contributing to a VWD phenotype.
VWD Types
Type 1
Type 1 VWD is caused by a partial quantitative deficiency of VWF and represents approximately 75% of VWD cases. It is the most clinically heterogeneous type, with patients having a mild to moderate bleeding phenotype.11 Bleeding in type 1 VWD results from a decrease in the concentration of VWF. The VWF function is normal without a significant abnormality in the platelet, collagen, or FVIII binding sites or a significant decrease in HMW multimers. Functional assays of VWF, such as VWF ristocetin cofactor (VWF:RCo) or VWF activity (VWF:Act) (see section on Laboratory Testing for further details), are proportionally decreased relative to the VWF antigen level (VWF:Ag), and the ratio of functional activity as compared with the VWF level is normal (ie, VWF:RCo/VWF:Ag ratio is > 0.6). As noted, VWF mutations are identified in only 65% of type 1 VWD cases and have incomplete penetrance and variable expressivity.10 Approximately 70% of mutations identified are missense mutations. Missense mutations may affect VWF levels by affecting any part of the biosynthetic pathway, including trafficking, storage, secretion, and/or clearance of VWF.
Increased VWF clearance is a well-described mechanism for type 1 VWD, known as type 1C. These patients will typically have very low VWF levels, an increased VWF propeptide to antigen ratio (VWFpp/VWF:Ag), and a marked but short-lived response to DDAVP, limiting DDAVP’s clinical applicability.12 On the other hand, the half-life of VWF/FVIII concentrates is normal in these individuals. Type 1C VWD is caused by missense mutations which occur mainly in the D3 domain and reduce the half-life of VWF up to 15-fold. R1205H, known as the “Vicenza” variant, is the most common and severe as well as the best characterized of these mutations.13
Type 2
Accounting for approximately 25% of VWD cases, type 2 VWD is characterized by a qualitative deficiency of VWF activity and is further subcategorized based on the mechanism of VWF dysfunction. Type 2A, 2B, and 2M affect VWF–platelet interactions by way of loss of HMW multimers, a gain of function of the GPIbα binding site, or a loss of function of the same site, respectively. On the other hand, type 2N is caused by defective VWF binding to FVIII. Type 2 VWD is often suspected when investigations demonstrate a function-antigen discordance: the VWF:RCo or VWF:Act is decreased disproportionately to the decrease in VWF:Ag, and the VWF:RCo/VWF:Ag ratio is less than 0.6.
Type 2A VWD is the most common type 2 variant. It is characterized by disproportionately low functional activity compared to antigen level (ie, VWF:RCo/VWF:ag ratio is < 0.6) and a loss of HMW and sometimes intermediate molecular weight (IMW) multimers. Ristocetin-induced platelet agglutination (RIPA) will be decreased with standard doses of ristocetin and absent with low doses. Type 2A VWD is usually inherited as an autosomal dominant trait. This subtype encompasses missense mutations that impair dimerization or multimerization of VWF subunits (CK, D1, and D2 domains); disrupt intersubunit disulphide bonds (D3 and D2 domains); enhance susceptibility to ADAMTS13-mediated proteolysis (A2 and A1 domains); or result in intracellular retention of the HMW multimers (D3, A1, and A2 domains).10 The result is VWF that lacks HMW multimers, thereby possessing fewer GPIbα binding sites, and that is less effective in binding platelets.
Type 2B VWD is the result of gain-of-function mutations within the GPIbα binding site of VWF. Generally, the platelet-binding site of VWF within the A1 domain is only exposed once VWF is immobilized on injured collagen and subjected to shear forces, resulting in a conformational change.7 In type 2B VWD, the gain-of-function mutation results in spontaneous binding of VWF to platelets without the need for a VWF-collagen interaction and unfolding of VWF by shear forces. The VWF–platelet interaction selectively depletes the HMW multimers by the unfolding of the A2 domain and increasing ADAMTS13 proteolysis. The increased binding of mutant VWF to platelets also triggers the formation of platelet aggregates, which are removed from circulation resulting in thrombocytopenia. Increases in endogenous VWF seen with acute stressors or pregnancy can worsen thrombocytopenia and increase the risk of bleeding.14 Certain mutations, such as V1316M, alter megakaryocytopoiesis and are characterized by giant platelets with abnormal ultrastructure and further exacerbate the thrombocytopenia.15 The laboratory profile reveals a VWF:RCo/VWF:Ag ratio of < 0.6 and absence of HMW multimers. In contrast to type 2A, platelets will agglutinate with low-dose ristocetin. Missense mutations are highly penetrant dominant and occur in or close to the A1 domain.16
Type 2M VWD is characterized by loss-of-function mutations within the GPIbα binding site of VWF. Phenotypic characteristics include a reduced ratio of VWF:RCo/VWF:Ag of < 0.6 but a normal multimer pattern.17 Missense mutations are reported in the A1 domain affecting the GPIbα-binding site. In very rare instances, mutations in the A3 domain that impair the VWF/collagen interaction have been described.18 These collagen-binding mutations are not included in the last iteration of the ISTH classification in 2006,1 but fit best in the type 2M category. In these cases, VWF:RCo or VWF:Act, which reflect activity at the GPIbα-binding site, may be normal and the diagnosis requires VWF/collagen binding assays (VWF:CB).
Type 2N VWD results from mutations of the FVIII binding site or conformational changes that impair the VWF–FVIII interaction. Most (~80%) missense mutations are located in domains D’ and D3.19 These mutations are autosomal recessive, and affected individuals are either homozygous or compound heterozygous for type 2N/2N or type 1/2N mutations, or compound heterozygous for a missense mutation and a mutation resulting in a null allele (type 2N/3 mutations). The laboratory phenotype is a disproportionate reduction in the FVIII level relative to the VWF level, which may be low or normal. Most cases of type 2N VWD have a normal multimeric profile, but rare cases will demonstrate loss of HMW multimers. Definitive diagnosis requires evidence of reduced FVIII binding to VWF (VWF:FVIIIB) or the identification of causative mutations in the FVIII binding region of the VWF gene.20
Type 3
Type 3 VWD is defined by a virtual absence of VWF. The inheritance of type 3 VWD has often been reported as autosomal recessive. However, there is emerging evidence that it can also be inherited in a co-dominant pattern: obligate carriers of type 3 VWD mutations have more mucocutaneous bleeding symptoms than normal individuals, and in approximately 50% of cases may carry a diagnosis of type 1 VWD.21 This condition is characterized by prolongation of the activated partial thromboplastin time (aPTT), undetectable levels of VWF:Ag, and VWF:RCo and FVIII levels less than 10 IU/dL (10%). The majority (~80%) of type 3 VWD patients have 2 null alleles as a result of a variety of mutations, with nonsense mutations accounting for about one-third.10 The remainder of the mutational spectrum is made up of missense mutations predominantly located in the D1-D2 (exons 3–11) and D4-CK (exons 37–52) domains that result in intracellular VWF retention, or large deletions, resulting in frameshift mutations affecting 1 or more exons. Because there is little or no circulating VWF, patients with type 3 VWD may develop alloantibodies to VWF, which can complicate treatment.22
Diagnosis
Clinical Manifestations
VWD is a congenital bleeding disorder. The increased risk of bleeding is present from birth, but symptoms may only manifest when there is a hemostatic challenge. Bleeding symptoms become more apparent with increasing age and exposure to hemostatic challenges. As a result, the diagnosis is often delayed into adulthood in mild to moderate forms of VWD. On the other hand, with more severe bleeding phenotypes such as type 3 VWD, the diagnosis is often made in childhood. Individuals with VWD primarily complain of excessive mucocutaneous bleeding, which includes spontaneous bruising, recurrent epistaxis, and bleeding from the gums after brushing, dental cleaning, and extractions. In addition, prolonged or excessive bleeding after surgery or trauma is often reported. Females frequently experience menorrhagia, usually beginning at menarche, and can have prolonged or excessive bleeding after childbirth.23 Musculoskeletal bleeding is unusual, except in type 2N or type 3 VWD when the FVIII:C level may be less than 10 IU/dL.
Mucocutaneous bleeding symptoms such as epistaxis, gum bleeding, ecchymosis, and menorrhagia overlap with those experienced by a normal population, and therefore can be easily overlooked by both patients and physicians.11 The use of bleeding assessment tools (BATs) to standardize the bleeding history and interpretation of the severity of the bleeding phenotype is becoming part of routine clinical practice. Three different BATs, each an adaptation of its predecessor, have been created and validated.24 Each of the scores performs well in an undiagnosed population presenting with bleeding symptoms. The negative predictive value is typically greater than 0.99, meaning that a negative bleeding score nearly excludes a clinically significant bleeding disorder. Thus, the main utility of the current BATs is at the time of new patient assessments: a negative bleeding score will help avoid unnecessary laboratory testing and prevent false-positive diagnoses of VWD (borderline low VWF:Ag without a significant bleeding history). However, the currently available BATs have some limitations. When scoring severe bleeding disorders, BATs become saturated as they take into account the worst episode of bleeding within each category but not the frequency of bleeding. BATs need to be administered by an expert and are time consuming to complete. Finally, they are not useful for monitoring bleeding symptoms or response to therapy because of the cumulative nature of the scores. In an attempt to standardize the BAT and bleeding score, the ISTH/Scientific and Standardization Committee (SSC) Joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group has established a revised BAT, known as the ISTH-BAT, specifically designed to extend the utility of the earlier BATS by incorporating information on both symptom frequency and severity.25,26 The ISTH-BAT has been further modified to a patient- or self-administered BAT (SELF-BAT). The SELF-BAT has been shown to be a reliable and effective tool in the assessment of patients who are being evaluated for VWD.27
Laboratory Testing
Screening tests include a complete blood count (CBC), prothrombin time, aPTT, thrombin time, and fibrinogen concentration to exclude the presence of other hemostatic disorders. The CBC may show thrombocytopenia in type 2B VWD. The aPTT is often normal, but will be prolonged if the FVIII level is below 30 IU/dL, as can be seen in severe type 1, type 2N, or type 3 VWD. The platelet function analyzer (PFA-100) is a system for analyzing primary hemostasis under high shear rates, but its role in the diagnosis of VWD is controversial.11
The evaluation of VWD involves quantitative (VWF:Ag) and qualitative measurements of VWF (VWF:RCo, or one of the novel assays: VWF:Act or VWF:GPIbM) and FVIII activity (FVIII:C). Type 2 VWD is suspected when the VWF activity to VWF:Ag ratio is < 0.6, the FVIII:C is more significantly decreased as compared to VWF:Ag, or with the presence of thrombocytopenia. In these cases, further testing (multimer gel electrophoresis, VWF:CB, RIPA, VWF:FVIIIB, and genotyping) is required to discriminate the type 2 VWD subtype, but such testing may be available only in specialized laboratories. If type 1C VWD is suspected, the VWFpp/VWF:ag ratio may confirm the diagnosis. Table 2 summarizes the results seen with each subtype. These assays are described in detail below.
VWD Assays
VWF:Ag represents the quantity of VWF protein (antigen) in the plasma measured using an enzyme-linked immunosorbent assay (ELISA) or latex immunoassay. The normal range is approximately 50 to 200 IU/dL.
VWF:RCo is a functional assay that determines the capacity of VWF to agglutinate platelets via the platelet receptor GPIbα in the presence of ristocetin. The normal range is approximately 50 to 200 IU/dL. Novel methods of measuring VWF’s platelet-binding activity are increasingly being adopted by clinical laboratories and are associated with greater precision and improved lower limits of detection and coefficients of variation.28,29 The first is the VWF:Act, a rapid automated assay that measures VWF activity using an antibody directed to the GPIbα binding site of VWF.28 The second novel assay is VWF:GPIbM, which involves a gain-of-function GPIB construct that binds VWF without ristocetin.30,31 For simplicity, VWF:RCo will be used to refer to VWF platelet-binding activity in the ensuing text. Factor VIII:C is a functional FVIII assay that determines the activity of FVIII in aPTT-based assays. The normal range is approximately 50 to 150 IU/dL.
VWF multimer analysis by SDS-agarose electrophoresis assesses VWF oligomers in plasma.32 Normal plasma contains multimers composed of over 40 VWF dimers, and these multimers are classified as high (HMW), intermediate (IMW), or low molecular weight (LMW). HMW multimers are decreased or missing in types 2A and 2B VWD, and IMW multimers may also be absent in type 2A VWD.
Low-dose RIPA tests the capacity of the patient’s platelets to agglutinate at low concentrations of ristocetin (~0.5 mg/mL). This is in contrast to the VWF:RCo, in which formalin-fixed control platelets are used. With type 2B, the platelet membrane is “overloaded” with high-affinity mutant VWF, resulting in abnormal platelet agglutination at low ristocetin concentrations. In some cases of type 2B VWD, all variables except RIPA may be normal.29
VWF:FVIIIB is an ELISA-based assay that determines the ability of VWF to bind FVIII and is used to make the diagnosis of type 2N VWD.19
VWF:CB is an ELISA-based assay that measures the ability of VWF to bind to collagen, a function of VWF that is dependent on the collagen-binding domain (A3) and on the presence of HMW multimers. VWF:CB helps to distinguish between types 1 and 2 VWD by reflecting the loss of HMW multimer forms (type 2A VWD) or can reflect a specific collagen-binding deficiency (type 2M VWD).33 The normal range is approximately 50 to 200 IU/dL. This assay is not available in most clinical laboratories.
VWFpp/VWF:Ag takes advantage of 2 facts: the VWF propeptide is secreted in a one-to-one ratio to VWF subunits and has a stable half-life in plasma. Thus, an increased ratio identifies patients with mutations that increase VWF clearance, such as type 1C VWD.34 The mean ratio in normal individuals is 1.3, with a normal range of 0.54 to 1.98.
Genotyping should be considered when specialized testing with the VWF:FVIIIB, RIPA, or VWF:CB assays is unavailable and a diagnosis of type 2 VWD is suspected. A guideline on VWD genetic testing has been published by the UK Haemophilia Centre Doctors Organisation.35
Interpretation of Clinical History and Laboratory Investigations
Normal plasma levels of VWF are approximately 100 IU/dL (100%, corresponds to ~10 μg/mL) with a population range of 50 to 200 IU/dL (50%–200%). There are a number of preanalytical variables (patient specific or laboratory specific) that affect the results of VWF laboratory testing. Patient-specific variables that are associated with increased VWF levels include increasing age, African ethnicity, exercise, inflammatory disease states, blood group A or B, increased levels of epinephrine, cocaine use, and neuroendocrine hormone levels. Decreased VWF levels are associated with medications such as valproic acid, hypothyroidism, autoantibodies, and blood group O. Individuals with blood group O have VWF levels that are 25% lower than levels in other blood groups.36 Several analytical variables also can complicate the diagnosis of VWD: methods for established reference ranges, limitations to the sensitivity of assays, and sample handling issues.11 These factors (summarized in Table 3) must be considered when interpreting VWF laboratory results, and at least 2 sets of tests using fresh samples are needed to confirm the diagnosis of VWD. Testing should be avoided in stressed, ill, or pregnant patients.
Mild type 1 VWD can be a difficult diagnosis to make because of the overlap of bleeding symptoms among normal individuals and those with mild type 1 VWD, as well as the variability of VWF levels. There is no consensus on the exact VWF levels required to confirm the diagnosis: the NHLBI Expert Panel recommends VWF:Ag and VWF:RCo levels less than 0.30 IU/mL to diagnose type 1 VWD,11 whereas the ISTH-SSC Subcommittee on von Willebrand factor recommends using VWF:RCo and VWF:Ag levels greater than 2 standard deviations below the population mean.37 In the absence of a bleeding history, slightly reduced VWF levels do not predict future significant bleeding events.38 Therefore, regardless of the laboratory cut-off used, the cornerstone of a VWD diagnosis should be a history of excessive mucocutaneous bleeding.
Differential Diagnosis
When considering a diagnosis of VWD, the differential diagnosis must be considered and includes acquired von Willebrand syndrome (AVWS), platelet-type VWD (PT-VWD), and hemophilia A. AVWS is the result of an acquired deficiency or defect of VWF and manifests with a mild to moderate bleeding disorder without a lifelong personal and family history of bleeding. AVWS has diverse pathology. The most common mechanism is proteolytic cleavage of VWF after shear stress–induced unfolding, as seen with aortic stenosis and ventricular assist devices, where as many as 79% of persons with aortic stenosis39 and up to 100% with left ventricular assist devices are affected.40 Other disease mechanisms include autoantibody formation that impairs VWF function or increases its clearance (autoimmune disease or lymphoproliferative disease), adsorption of HMW VWF multimers to malignant cells or platelets (myeloproliferative neoplasms and Wilm’s tumor), or decreased synthesis (hypothyroidism, valproic acid). The median age of diagnosis is 62 years, but the disorder may occur in any age-group (range 2–96 years).41 The approach to management of AVWS should focus on treatment of bleeding and induction of long-term remission. Treatment of bleeding will depend on the underlying mechanism of AVWS and may include a combination of DDAVP or VWF/FVIII concentrates, recombinant factor VIIa, antifibrinolytic agents, intravenous immunoglobulin, or plasmapheresis for AVWS associated with autoantibodies. Treatment of the underlying disorder (eg, aortic valve repair or treatment of a lymphoproliferative disorder) may result in remission of the AVWS.
Mild hemophilia A (caused by mutations in the F8 gene) and type 2N VWD can be difficult to differentiate clinically. Both present with reduced FVIII:C, and type 2N VWD may have normal or borderline low levels of VWF. Although the VWF:FVIIIB assay will distinguish between the 2 disorders, the test is not available in many centers. The pattern of inheritance may be helpful: hemophilia A is an X-linked disorder, whereas type 2N is autosomal recessive. Often, the diagnosis of type 2N VWD is suspected when genotyping of F8 does not identify a mutation in mild hemophilia A, when infused FVIII concentrates have a decreased half-life, or when DDAVP is associated with a brisk but short-lived response. In the absence of VWF:FVIIIB assay availability, genotyping of VWF will confirm the diagnosis, with missense mutations being located in exons 17–20 or 24–27.19
PT-VWD represents the phenocopy of type 2B VWD. The mutation is in the platelet receptor gene GPIBA and causes enhanced VWF-platelet binding. The disorders can be differentiated by RIPA plasma/platelet mixing studies or flow cytometry.42,43 However, these assays are technically challenging. In the absence of mutations in exon 28 of VWF, mutations in exon 2 of GPIBA may be identified in approximately 10% of persons misdiagnosed with type 2B VWD.
Management
Patients with VWD present to medical attention in a number of ways: excessive post-trauma or surgical bleeding, recurrent mucocutaneous bleeding such as epistaxis, menorrhagia, gastrointestinal bleeding, or, in severe cases, recurrent hemarthroses and muscle hematomas. Irrespective of the presentation, the goal is to minimize and control bleeding. Therapeutic options can be divided into 3 main categories: (1) localized measures to stop bleeding; (2) pharmacologic agents with indirect hemostatic benefit; and (3) treatments that directly increase plasma VWF and FVIII levels. A combination of all 3 of these modalities can be used depending on the bleeding location and severity.
Localized Measures
Localized measures to control bleeding in VWD will depend on the site of bleeding. Epistaxis can be particularly problematic for affected children, and patients should be armed with a step-wise action plan that escalates from pressure to packing and includes guidelines regarding how long to wait before seeking medical attention. In selected cases, nasal cautery may be required for prolonged or excessive epistaxis. Topical hemostatic agents such as gelatin foam/matrix, topical thrombin, and fibrin sealants are predominately used to achieve surgical hemostasis and may have a limited role in the treatment of VWD-associated bleeding. In the case of menorrhagia, hormonal treatments (ie, the combined oral contraceptive pill, OCP), levonorgestrel-releasing intrauterine systems, or endometrial ablation all effectively reduce menstrual blood loss through their local effects on the endometrial lining.44 In addition, older generations of OCP are associated with increases in VWF levels. This effect is mediated by the estrogen component and is evident with ethynylestradiol doses of 0.5 μg or higher. Lower estrogen doses, seen in currently used OCP, have little or no effect on VWF levels.11,45
Pharmacologic Therapy
Indirect therapies include the antifibrinolytic agents (eg, tranexamic acid and aminocaproic acid). These agents are used either as the sole therapy at the time of minor surgical and dental procedures, or as an adjunct in combination with DDAVP or VWF/FVIII concentrates. Antifibrinolytics are thought to be particularly useful for controlling mucosal bleeding in areas of high fibrinolytic activity: the oral cavity, gastrointestinal tract, or uterus. Tranexamic acid inhibits the conversion of plasminogen to plasmin, and is the more commonly used antifibrinolytic.11 Tranexamic acid can be administered either intravenously or orally at doses of 10 to 25 mg/kg, respectively. It is usually continued until bleeding is controlled or up to 7 to 10 days postoperatively. The most common adverse events associated with tranexamic acid are headache, back pain, and gastrointestinal side effects.46 Tranexamic acid is contraindicated in disseminated intravascular coagulation and bleeding from the upper urinary tract, where it can lead to urinary tract obstruction by clots.
DDAVP, a synthetic derivative of vasopressin, promotes release of stored VWF from endothelial cells. Most individuals with type 1 VWD and some with type 2A VWD respond to treatment with DDAVP: a therapeutic trial to confirm adequate DDAVP response should be performed prior to its clinical use. Assessment of VWF:Ag, VWF:RCo, and FVIII levels should be performed before and at several time points after the DDAVP administration up to and including 4 hours. Peak VWF levels are achieved 30 and 90 minutes after intravenous and intranasal delivery, respectively. An increase in VWF:Ag/VWF:RCo and FVIII levels to at least 30 IU/dL is adequate for most dental procedures, minor surgery, or the treatment of epistaxis or menorrhagia. DDAVP may be adequate to treat major bleeds or for major surgery when VWF levels increase well above 50 IU/dL. Decisions surrounding the use of DDAVP versus a VWF/FVIII concentrate will depend on the expected DDAVP response, the type of surgery, and the anticipated duration of therapy required to achieve hemostasis. If treatment is required for more than 3 days, concerns regarding tachyphylaxis and side effects may limit its use. Significantly decreased VWF:Ag/VWF:RCo or FVIII at the 4-hour time point of a DDAVP trial may indicate type 1C or type 2N VWD, which are associated with increased clearance of endogenous VWF or FVIII, respectively. Despite the transient response in these patients, DDAVP remains a therapeutic option and its use should be assessed on a case-by-case basis.47
The parenteral dose of DDAVP is 0.3 μg/kg infused in 30 to 50 mL of normal saline over approximately 30 minutes every 12 to 24 hours. The dose of the highly concentrated intranasal preparation is 150 μg for children under 50 kg and 300 μg for larger children and adults (1 spray per naris). It is important to note that the products used to treat VWD (eg, Stimate) deliver 150 μg per spray, a much higher concentration than that used to treat enuresis. Repeated DDAVP dosing is associated with the development of tachyphylaxis: with subsequent dosing, the magnitude of the VWF and FVIII increments can fall to approximately 70% of that obtained with the initial dose.48 DDAVP is safe and generally well tolerated. Side effects include facial flushing, headache, tachycardia, light-headedness, and mild hypotension. The most serious side effects, severe hyponatremia and seizures,49 can be avoided by fluid restriction for 24 hours after DDAVP administration. Serum sodium levels should be monitored with repeated doses. DDAVP is generally avoided in those younger than 2 years of age because of a higher risk of hyponatremia. Patients who are intolerant of DDAVP or have a poor VWF response need to be treated with a VWF/FVIII concentrate.
VWF/FVIII Concentrate
VWF/FVIII concentrates are required for patients who do not have an adequate response to DDAVP, who have side effects from or contraindications to DDAVP, or who require a long duration of treatment, rendering the use of DDAVP impractical. Purified, viral-inactivated, plasma-derived VWF/FVIII concentrates are the products most frequently used (eg, Humate-P, Wilate, Alphanate SD/HT). The quantity of VWF:RCo activity relative to FVIII:C varies by product; Humate-P contains 2.4 VWF:RCo units for each unit of FVIII:C; Wilate contains a 1:1 ratio; and Alphanate contains a 0.5:1 ratio. Both Humate-P and Wilate are reported to contain a full spectrum of VWF multimers, including HMW multimers, and closely resemble normal plasma, but Alphanate SD/HT lacks HMW mutimers.11,50 Thus, the available VWF/FVIII vary in terms of VWF:RCo to FVIII concentrate, HMW multimer composition, reported VWF:RCo, and FVIII half-lives and even approved indications. They should not be considered interchangeable, and further information should be sought from the respective product inserts.
Dosing recommendations are provided either in VWF:RCo (North America) or FVIII:C units (Europe) and are weight-based (Table 4); repeat infusions can be given every 8 to 24 hours depending on the type of surgery/injury and the product used.
Long-term continuous use of concentrates to prevent bleeds, known as prophylaxis, is the standard of care in severe hemophilia A and B and is now being adopted in severe VWD. Patients with type 3 VWD or severe type 1 or type 2 VWD may experience recurrent bleeds into joints, nasal/oropharynx, or gastrointestinal tract or excessive menstrual bleeding. Retrospective cohort and case series suggest that prophylaxis improves quality of life; reduces the frequency of bleeding, need for transfusions, and hospitalizations; and prevents chronic joint disease.54,55 More recently, a prospective study confirmed that prophylaxis with VWF concentrates at doses ranging from 50 IU VWF RCo/kg 1 to 3 times per week was highly effective at reducing bleeding rates, with annualized bleeding rates decreasing from 25 to 6.1 in 11 participants with either type 2A or type 3 VWD.56
VWF/FVIII concentrates are effective in more than 97% of events.57 Rarely, when infusion of a VWF/FVIII concentrate is ineffective at stopping bleeding, transfusion of platelet concentrates may be beneficial, presumably because they facilitate the delivery of small amounts of platelet VWF to the site of vascular injury. Highly purified FVIII concentrates (monoclonal antibody purified and recombinant) should not be used to treat VWD because they lack VWF.
A recombinant VWF concentrate (Vonvendi) combined initially with recombinant FVIII concentrate in a 1.3:1 ratio of VWF:RCo to FVIII:C has been shown to be safe and efficacious for the on-demand treatment of bleeds.58,59 After the initial FVIII dose, the patients’ endogenous FVIII levels are stabilized within 6 hours and further FVIII administration may not required. A prospective phase 3 trial investigating the efficacy of recombinant VWF in the prophylaxis of severe VWD is ongoing. Vonvendi has been licensed for on-demand treatment in the United States since 2015. For further dosing information, please refer to the product insert.
Conclusion
VWF is a complex protein with several important and distinct functional domains: binding sites to collagen, FVIII, and platelet GPIbα; an ADAMTS13 cleavage site; and domains important for multimer formation. Mutations in any of these sites can result in a dysfunctional protein and as a result, VWD is a heterogeneous disorder with many specific assays available to determine the subtype. Despite this, the treatment of VWD is straightforward with only a small number of therapeutic options: indirect therapies such as antifibrinolytic agents, or direct therapies that increase VWF levels, DDAVP, or VWF/FVIII concentrates. Management focuses on preventing bleeding complications associated with invasive procedures or promptly treating bleeding episodes.
Introduction
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types (Table 1).1
Prevalence
VWD is the most common inherited bleeding disorder. However, because VWF levels are highly variable and disease severity ranges from mild bleeding symptoms to severe or life-threatening bleeds, the reported prevalence of VWD depends on the diagnostic definition used. Two large epidemiologic studies have reported prevalence rates of approximately 1%.2,3 In these studies, healthy school-aged children were screened and diagnosed with VWD based on low VWF activity, measured as ristocetin cofactor, and a personal and family history of bleeding symptoms. At the other extreme, when considering patients whose bleeding symptoms are sufficiently severe to warrant referral to specialized centers, the reported prevalence of VWD ranges from 20 to 113 per million.4 These studies likely over- and underestimate clinically significant VWD. More recent studies suggest that the prevalence of VWD in individuals whose bleeding symptoms are significant enough to present to a primary care physician is approximately 0.1%.5 This figure is likely a more accurate estimate of the true prevalence of symptomatic VWD.
Although VWD is autosomally inherited, females are more likely to present with bleeding symptoms and be diagnosed because of increased exposure to bleeding challenges, such as menorrhagia and childbirth. VWD does not show any geographic or ethnic predilection, but there is an increased prevalence of the recessive forms, such as type 2N and type 3 VWD, in areas with high rates of consanguinity.
VWF Protein Structure and Function
The VWF gene is located on chromosome 12 at p13.3 and spans 178 kb comprising 52 exons.6 The expression of the VWF gene is tightly restricted to endothelial cells, platelets, and megakaryocytes, where VWF is stored in Weibel-Palade bodies and α-granules. VWF is a large multimeric glycoprotein with several important functional domains (Figure).
Classification, Pathophysiology, and Genetics
The International Society of Thrombosis and Hemostasis (ISTH) classification of VWD was updated in 2006 (Table 1).1 It incorporates important aspects of clinical phenotype, pathophysiological mechanisms, and treatment considerations. The 3 categories are: type 1, which is a partial quantitative deficiency; type 2 with 4 subtypes (2A, 2B, 2M, and 2N), which is a qualitative defect; and type 3, which is a virtual absence of VWF. Although the diagnosis and categorization of VWD can be achieved with widely available laboratory testing, further subcategorization among type 2 VWD subtypes may require referral to a specialized laboratory. The current ISTH classification intentionally does not incorporate genotypic data. In type 2 or type 3 VWD disease, VWF mutations are identified in more than 90% of cases and are completely penetrant, whereas mutations are identified in only approximately 65% of type 1 VWD cases and have been associated with incomplete penetrance and variable expressivity.10 These studies suggest that type 1 VWD is an oligogenic disease with mutations in genes regulating secretion or clearance contributing to a VWD phenotype.
VWD Types
Type 1
Type 1 VWD is caused by a partial quantitative deficiency of VWF and represents approximately 75% of VWD cases. It is the most clinically heterogeneous type, with patients having a mild to moderate bleeding phenotype.11 Bleeding in type 1 VWD results from a decrease in the concentration of VWF. The VWF function is normal without a significant abnormality in the platelet, collagen, or FVIII binding sites or a significant decrease in HMW multimers. Functional assays of VWF, such as VWF ristocetin cofactor (VWF:RCo) or VWF activity (VWF:Act) (see section on Laboratory Testing for further details), are proportionally decreased relative to the VWF antigen level (VWF:Ag), and the ratio of functional activity as compared with the VWF level is normal (ie, VWF:RCo/VWF:Ag ratio is > 0.6). As noted, VWF mutations are identified in only 65% of type 1 VWD cases and have incomplete penetrance and variable expressivity.10 Approximately 70% of mutations identified are missense mutations. Missense mutations may affect VWF levels by affecting any part of the biosynthetic pathway, including trafficking, storage, secretion, and/or clearance of VWF.
Increased VWF clearance is a well-described mechanism for type 1 VWD, known as type 1C. These patients will typically have very low VWF levels, an increased VWF propeptide to antigen ratio (VWFpp/VWF:Ag), and a marked but short-lived response to DDAVP, limiting DDAVP’s clinical applicability.12 On the other hand, the half-life of VWF/FVIII concentrates is normal in these individuals. Type 1C VWD is caused by missense mutations which occur mainly in the D3 domain and reduce the half-life of VWF up to 15-fold. R1205H, known as the “Vicenza” variant, is the most common and severe as well as the best characterized of these mutations.13
Type 2
Accounting for approximately 25% of VWD cases, type 2 VWD is characterized by a qualitative deficiency of VWF activity and is further subcategorized based on the mechanism of VWF dysfunction. Type 2A, 2B, and 2M affect VWF–platelet interactions by way of loss of HMW multimers, a gain of function of the GPIbα binding site, or a loss of function of the same site, respectively. On the other hand, type 2N is caused by defective VWF binding to FVIII. Type 2 VWD is often suspected when investigations demonstrate a function-antigen discordance: the VWF:RCo or VWF:Act is decreased disproportionately to the decrease in VWF:Ag, and the VWF:RCo/VWF:Ag ratio is less than 0.6.
Type 2A VWD is the most common type 2 variant. It is characterized by disproportionately low functional activity compared to antigen level (ie, VWF:RCo/VWF:ag ratio is < 0.6) and a loss of HMW and sometimes intermediate molecular weight (IMW) multimers. Ristocetin-induced platelet agglutination (RIPA) will be decreased with standard doses of ristocetin and absent with low doses. Type 2A VWD is usually inherited as an autosomal dominant trait. This subtype encompasses missense mutations that impair dimerization or multimerization of VWF subunits (CK, D1, and D2 domains); disrupt intersubunit disulphide bonds (D3 and D2 domains); enhance susceptibility to ADAMTS13-mediated proteolysis (A2 and A1 domains); or result in intracellular retention of the HMW multimers (D3, A1, and A2 domains).10 The result is VWF that lacks HMW multimers, thereby possessing fewer GPIbα binding sites, and that is less effective in binding platelets.
Type 2B VWD is the result of gain-of-function mutations within the GPIbα binding site of VWF. Generally, the platelet-binding site of VWF within the A1 domain is only exposed once VWF is immobilized on injured collagen and subjected to shear forces, resulting in a conformational change.7 In type 2B VWD, the gain-of-function mutation results in spontaneous binding of VWF to platelets without the need for a VWF-collagen interaction and unfolding of VWF by shear forces. The VWF–platelet interaction selectively depletes the HMW multimers by the unfolding of the A2 domain and increasing ADAMTS13 proteolysis. The increased binding of mutant VWF to platelets also triggers the formation of platelet aggregates, which are removed from circulation resulting in thrombocytopenia. Increases in endogenous VWF seen with acute stressors or pregnancy can worsen thrombocytopenia and increase the risk of bleeding.14 Certain mutations, such as V1316M, alter megakaryocytopoiesis and are characterized by giant platelets with abnormal ultrastructure and further exacerbate the thrombocytopenia.15 The laboratory profile reveals a VWF:RCo/VWF:Ag ratio of < 0.6 and absence of HMW multimers. In contrast to type 2A, platelets will agglutinate with low-dose ristocetin. Missense mutations are highly penetrant dominant and occur in or close to the A1 domain.16
Type 2M VWD is characterized by loss-of-function mutations within the GPIbα binding site of VWF. Phenotypic characteristics include a reduced ratio of VWF:RCo/VWF:Ag of < 0.6 but a normal multimer pattern.17 Missense mutations are reported in the A1 domain affecting the GPIbα-binding site. In very rare instances, mutations in the A3 domain that impair the VWF/collagen interaction have been described.18 These collagen-binding mutations are not included in the last iteration of the ISTH classification in 2006,1 but fit best in the type 2M category. In these cases, VWF:RCo or VWF:Act, which reflect activity at the GPIbα-binding site, may be normal and the diagnosis requires VWF/collagen binding assays (VWF:CB).
Type 2N VWD results from mutations of the FVIII binding site or conformational changes that impair the VWF–FVIII interaction. Most (~80%) missense mutations are located in domains D’ and D3.19 These mutations are autosomal recessive, and affected individuals are either homozygous or compound heterozygous for type 2N/2N or type 1/2N mutations, or compound heterozygous for a missense mutation and a mutation resulting in a null allele (type 2N/3 mutations). The laboratory phenotype is a disproportionate reduction in the FVIII level relative to the VWF level, which may be low or normal. Most cases of type 2N VWD have a normal multimeric profile, but rare cases will demonstrate loss of HMW multimers. Definitive diagnosis requires evidence of reduced FVIII binding to VWF (VWF:FVIIIB) or the identification of causative mutations in the FVIII binding region of the VWF gene.20
Type 3
Type 3 VWD is defined by a virtual absence of VWF. The inheritance of type 3 VWD has often been reported as autosomal recessive. However, there is emerging evidence that it can also be inherited in a co-dominant pattern: obligate carriers of type 3 VWD mutations have more mucocutaneous bleeding symptoms than normal individuals, and in approximately 50% of cases may carry a diagnosis of type 1 VWD.21 This condition is characterized by prolongation of the activated partial thromboplastin time (aPTT), undetectable levels of VWF:Ag, and VWF:RCo and FVIII levels less than 10 IU/dL (10%). The majority (~80%) of type 3 VWD patients have 2 null alleles as a result of a variety of mutations, with nonsense mutations accounting for about one-third.10 The remainder of the mutational spectrum is made up of missense mutations predominantly located in the D1-D2 (exons 3–11) and D4-CK (exons 37–52) domains that result in intracellular VWF retention, or large deletions, resulting in frameshift mutations affecting 1 or more exons. Because there is little or no circulating VWF, patients with type 3 VWD may develop alloantibodies to VWF, which can complicate treatment.22
Diagnosis
Clinical Manifestations
VWD is a congenital bleeding disorder. The increased risk of bleeding is present from birth, but symptoms may only manifest when there is a hemostatic challenge. Bleeding symptoms become more apparent with increasing age and exposure to hemostatic challenges. As a result, the diagnosis is often delayed into adulthood in mild to moderate forms of VWD. On the other hand, with more severe bleeding phenotypes such as type 3 VWD, the diagnosis is often made in childhood. Individuals with VWD primarily complain of excessive mucocutaneous bleeding, which includes spontaneous bruising, recurrent epistaxis, and bleeding from the gums after brushing, dental cleaning, and extractions. In addition, prolonged or excessive bleeding after surgery or trauma is often reported. Females frequently experience menorrhagia, usually beginning at menarche, and can have prolonged or excessive bleeding after childbirth.23 Musculoskeletal bleeding is unusual, except in type 2N or type 3 VWD when the FVIII:C level may be less than 10 IU/dL.
Mucocutaneous bleeding symptoms such as epistaxis, gum bleeding, ecchymosis, and menorrhagia overlap with those experienced by a normal population, and therefore can be easily overlooked by both patients and physicians.11 The use of bleeding assessment tools (BATs) to standardize the bleeding history and interpretation of the severity of the bleeding phenotype is becoming part of routine clinical practice. Three different BATs, each an adaptation of its predecessor, have been created and validated.24 Each of the scores performs well in an undiagnosed population presenting with bleeding symptoms. The negative predictive value is typically greater than 0.99, meaning that a negative bleeding score nearly excludes a clinically significant bleeding disorder. Thus, the main utility of the current BATs is at the time of new patient assessments: a negative bleeding score will help avoid unnecessary laboratory testing and prevent false-positive diagnoses of VWD (borderline low VWF:Ag without a significant bleeding history). However, the currently available BATs have some limitations. When scoring severe bleeding disorders, BATs become saturated as they take into account the worst episode of bleeding within each category but not the frequency of bleeding. BATs need to be administered by an expert and are time consuming to complete. Finally, they are not useful for monitoring bleeding symptoms or response to therapy because of the cumulative nature of the scores. In an attempt to standardize the BAT and bleeding score, the ISTH/Scientific and Standardization Committee (SSC) Joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group has established a revised BAT, known as the ISTH-BAT, specifically designed to extend the utility of the earlier BATS by incorporating information on both symptom frequency and severity.25,26 The ISTH-BAT has been further modified to a patient- or self-administered BAT (SELF-BAT). The SELF-BAT has been shown to be a reliable and effective tool in the assessment of patients who are being evaluated for VWD.27
Laboratory Testing
Screening tests include a complete blood count (CBC), prothrombin time, aPTT, thrombin time, and fibrinogen concentration to exclude the presence of other hemostatic disorders. The CBC may show thrombocytopenia in type 2B VWD. The aPTT is often normal, but will be prolonged if the FVIII level is below 30 IU/dL, as can be seen in severe type 1, type 2N, or type 3 VWD. The platelet function analyzer (PFA-100) is a system for analyzing primary hemostasis under high shear rates, but its role in the diagnosis of VWD is controversial.11
The evaluation of VWD involves quantitative (VWF:Ag) and qualitative measurements of VWF (VWF:RCo, or one of the novel assays: VWF:Act or VWF:GPIbM) and FVIII activity (FVIII:C). Type 2 VWD is suspected when the VWF activity to VWF:Ag ratio is < 0.6, the FVIII:C is more significantly decreased as compared to VWF:Ag, or with the presence of thrombocytopenia. In these cases, further testing (multimer gel electrophoresis, VWF:CB, RIPA, VWF:FVIIIB, and genotyping) is required to discriminate the type 2 VWD subtype, but such testing may be available only in specialized laboratories. If type 1C VWD is suspected, the VWFpp/VWF:ag ratio may confirm the diagnosis. Table 2 summarizes the results seen with each subtype. These assays are described in detail below.
VWD Assays
VWF:Ag represents the quantity of VWF protein (antigen) in the plasma measured using an enzyme-linked immunosorbent assay (ELISA) or latex immunoassay. The normal range is approximately 50 to 200 IU/dL.
VWF:RCo is a functional assay that determines the capacity of VWF to agglutinate platelets via the platelet receptor GPIbα in the presence of ristocetin. The normal range is approximately 50 to 200 IU/dL. Novel methods of measuring VWF’s platelet-binding activity are increasingly being adopted by clinical laboratories and are associated with greater precision and improved lower limits of detection and coefficients of variation.28,29 The first is the VWF:Act, a rapid automated assay that measures VWF activity using an antibody directed to the GPIbα binding site of VWF.28 The second novel assay is VWF:GPIbM, which involves a gain-of-function GPIB construct that binds VWF without ristocetin.30,31 For simplicity, VWF:RCo will be used to refer to VWF platelet-binding activity in the ensuing text. Factor VIII:C is a functional FVIII assay that determines the activity of FVIII in aPTT-based assays. The normal range is approximately 50 to 150 IU/dL.
VWF multimer analysis by SDS-agarose electrophoresis assesses VWF oligomers in plasma.32 Normal plasma contains multimers composed of over 40 VWF dimers, and these multimers are classified as high (HMW), intermediate (IMW), or low molecular weight (LMW). HMW multimers are decreased or missing in types 2A and 2B VWD, and IMW multimers may also be absent in type 2A VWD.
Low-dose RIPA tests the capacity of the patient’s platelets to agglutinate at low concentrations of ristocetin (~0.5 mg/mL). This is in contrast to the VWF:RCo, in which formalin-fixed control platelets are used. With type 2B, the platelet membrane is “overloaded” with high-affinity mutant VWF, resulting in abnormal platelet agglutination at low ristocetin concentrations. In some cases of type 2B VWD, all variables except RIPA may be normal.29
VWF:FVIIIB is an ELISA-based assay that determines the ability of VWF to bind FVIII and is used to make the diagnosis of type 2N VWD.19
VWF:CB is an ELISA-based assay that measures the ability of VWF to bind to collagen, a function of VWF that is dependent on the collagen-binding domain (A3) and on the presence of HMW multimers. VWF:CB helps to distinguish between types 1 and 2 VWD by reflecting the loss of HMW multimer forms (type 2A VWD) or can reflect a specific collagen-binding deficiency (type 2M VWD).33 The normal range is approximately 50 to 200 IU/dL. This assay is not available in most clinical laboratories.
VWFpp/VWF:Ag takes advantage of 2 facts: the VWF propeptide is secreted in a one-to-one ratio to VWF subunits and has a stable half-life in plasma. Thus, an increased ratio identifies patients with mutations that increase VWF clearance, such as type 1C VWD.34 The mean ratio in normal individuals is 1.3, with a normal range of 0.54 to 1.98.
Genotyping should be considered when specialized testing with the VWF:FVIIIB, RIPA, or VWF:CB assays is unavailable and a diagnosis of type 2 VWD is suspected. A guideline on VWD genetic testing has been published by the UK Haemophilia Centre Doctors Organisation.35
Interpretation of Clinical History and Laboratory Investigations
Normal plasma levels of VWF are approximately 100 IU/dL (100%, corresponds to ~10 μg/mL) with a population range of 50 to 200 IU/dL (50%–200%). There are a number of preanalytical variables (patient specific or laboratory specific) that affect the results of VWF laboratory testing. Patient-specific variables that are associated with increased VWF levels include increasing age, African ethnicity, exercise, inflammatory disease states, blood group A or B, increased levels of epinephrine, cocaine use, and neuroendocrine hormone levels. Decreased VWF levels are associated with medications such as valproic acid, hypothyroidism, autoantibodies, and blood group O. Individuals with blood group O have VWF levels that are 25% lower than levels in other blood groups.36 Several analytical variables also can complicate the diagnosis of VWD: methods for established reference ranges, limitations to the sensitivity of assays, and sample handling issues.11 These factors (summarized in Table 3) must be considered when interpreting VWF laboratory results, and at least 2 sets of tests using fresh samples are needed to confirm the diagnosis of VWD. Testing should be avoided in stressed, ill, or pregnant patients.
Mild type 1 VWD can be a difficult diagnosis to make because of the overlap of bleeding symptoms among normal individuals and those with mild type 1 VWD, as well as the variability of VWF levels. There is no consensus on the exact VWF levels required to confirm the diagnosis: the NHLBI Expert Panel recommends VWF:Ag and VWF:RCo levels less than 0.30 IU/mL to diagnose type 1 VWD,11 whereas the ISTH-SSC Subcommittee on von Willebrand factor recommends using VWF:RCo and VWF:Ag levels greater than 2 standard deviations below the population mean.37 In the absence of a bleeding history, slightly reduced VWF levels do not predict future significant bleeding events.38 Therefore, regardless of the laboratory cut-off used, the cornerstone of a VWD diagnosis should be a history of excessive mucocutaneous bleeding.
Differential Diagnosis
When considering a diagnosis of VWD, the differential diagnosis must be considered and includes acquired von Willebrand syndrome (AVWS), platelet-type VWD (PT-VWD), and hemophilia A. AVWS is the result of an acquired deficiency or defect of VWF and manifests with a mild to moderate bleeding disorder without a lifelong personal and family history of bleeding. AVWS has diverse pathology. The most common mechanism is proteolytic cleavage of VWF after shear stress–induced unfolding, as seen with aortic stenosis and ventricular assist devices, where as many as 79% of persons with aortic stenosis39 and up to 100% with left ventricular assist devices are affected.40 Other disease mechanisms include autoantibody formation that impairs VWF function or increases its clearance (autoimmune disease or lymphoproliferative disease), adsorption of HMW VWF multimers to malignant cells or platelets (myeloproliferative neoplasms and Wilm’s tumor), or decreased synthesis (hypothyroidism, valproic acid). The median age of diagnosis is 62 years, but the disorder may occur in any age-group (range 2–96 years).41 The approach to management of AVWS should focus on treatment of bleeding and induction of long-term remission. Treatment of bleeding will depend on the underlying mechanism of AVWS and may include a combination of DDAVP or VWF/FVIII concentrates, recombinant factor VIIa, antifibrinolytic agents, intravenous immunoglobulin, or plasmapheresis for AVWS associated with autoantibodies. Treatment of the underlying disorder (eg, aortic valve repair or treatment of a lymphoproliferative disorder) may result in remission of the AVWS.
Mild hemophilia A (caused by mutations in the F8 gene) and type 2N VWD can be difficult to differentiate clinically. Both present with reduced FVIII:C, and type 2N VWD may have normal or borderline low levels of VWF. Although the VWF:FVIIIB assay will distinguish between the 2 disorders, the test is not available in many centers. The pattern of inheritance may be helpful: hemophilia A is an X-linked disorder, whereas type 2N is autosomal recessive. Often, the diagnosis of type 2N VWD is suspected when genotyping of F8 does not identify a mutation in mild hemophilia A, when infused FVIII concentrates have a decreased half-life, or when DDAVP is associated with a brisk but short-lived response. In the absence of VWF:FVIIIB assay availability, genotyping of VWF will confirm the diagnosis, with missense mutations being located in exons 17–20 or 24–27.19
PT-VWD represents the phenocopy of type 2B VWD. The mutation is in the platelet receptor gene GPIBA and causes enhanced VWF-platelet binding. The disorders can be differentiated by RIPA plasma/platelet mixing studies or flow cytometry.42,43 However, these assays are technically challenging. In the absence of mutations in exon 28 of VWF, mutations in exon 2 of GPIBA may be identified in approximately 10% of persons misdiagnosed with type 2B VWD.
Management
Patients with VWD present to medical attention in a number of ways: excessive post-trauma or surgical bleeding, recurrent mucocutaneous bleeding such as epistaxis, menorrhagia, gastrointestinal bleeding, or, in severe cases, recurrent hemarthroses and muscle hematomas. Irrespective of the presentation, the goal is to minimize and control bleeding. Therapeutic options can be divided into 3 main categories: (1) localized measures to stop bleeding; (2) pharmacologic agents with indirect hemostatic benefit; and (3) treatments that directly increase plasma VWF and FVIII levels. A combination of all 3 of these modalities can be used depending on the bleeding location and severity.
Localized Measures
Localized measures to control bleeding in VWD will depend on the site of bleeding. Epistaxis can be particularly problematic for affected children, and patients should be armed with a step-wise action plan that escalates from pressure to packing and includes guidelines regarding how long to wait before seeking medical attention. In selected cases, nasal cautery may be required for prolonged or excessive epistaxis. Topical hemostatic agents such as gelatin foam/matrix, topical thrombin, and fibrin sealants are predominately used to achieve surgical hemostasis and may have a limited role in the treatment of VWD-associated bleeding. In the case of menorrhagia, hormonal treatments (ie, the combined oral contraceptive pill, OCP), levonorgestrel-releasing intrauterine systems, or endometrial ablation all effectively reduce menstrual blood loss through their local effects on the endometrial lining.44 In addition, older generations of OCP are associated with increases in VWF levels. This effect is mediated by the estrogen component and is evident with ethynylestradiol doses of 0.5 μg or higher. Lower estrogen doses, seen in currently used OCP, have little or no effect on VWF levels.11,45
Pharmacologic Therapy
Indirect therapies include the antifibrinolytic agents (eg, tranexamic acid and aminocaproic acid). These agents are used either as the sole therapy at the time of minor surgical and dental procedures, or as an adjunct in combination with DDAVP or VWF/FVIII concentrates. Antifibrinolytics are thought to be particularly useful for controlling mucosal bleeding in areas of high fibrinolytic activity: the oral cavity, gastrointestinal tract, or uterus. Tranexamic acid inhibits the conversion of plasminogen to plasmin, and is the more commonly used antifibrinolytic.11 Tranexamic acid can be administered either intravenously or orally at doses of 10 to 25 mg/kg, respectively. It is usually continued until bleeding is controlled or up to 7 to 10 days postoperatively. The most common adverse events associated with tranexamic acid are headache, back pain, and gastrointestinal side effects.46 Tranexamic acid is contraindicated in disseminated intravascular coagulation and bleeding from the upper urinary tract, where it can lead to urinary tract obstruction by clots.
DDAVP, a synthetic derivative of vasopressin, promotes release of stored VWF from endothelial cells. Most individuals with type 1 VWD and some with type 2A VWD respond to treatment with DDAVP: a therapeutic trial to confirm adequate DDAVP response should be performed prior to its clinical use. Assessment of VWF:Ag, VWF:RCo, and FVIII levels should be performed before and at several time points after the DDAVP administration up to and including 4 hours. Peak VWF levels are achieved 30 and 90 minutes after intravenous and intranasal delivery, respectively. An increase in VWF:Ag/VWF:RCo and FVIII levels to at least 30 IU/dL is adequate for most dental procedures, minor surgery, or the treatment of epistaxis or menorrhagia. DDAVP may be adequate to treat major bleeds or for major surgery when VWF levels increase well above 50 IU/dL. Decisions surrounding the use of DDAVP versus a VWF/FVIII concentrate will depend on the expected DDAVP response, the type of surgery, and the anticipated duration of therapy required to achieve hemostasis. If treatment is required for more than 3 days, concerns regarding tachyphylaxis and side effects may limit its use. Significantly decreased VWF:Ag/VWF:RCo or FVIII at the 4-hour time point of a DDAVP trial may indicate type 1C or type 2N VWD, which are associated with increased clearance of endogenous VWF or FVIII, respectively. Despite the transient response in these patients, DDAVP remains a therapeutic option and its use should be assessed on a case-by-case basis.47
The parenteral dose of DDAVP is 0.3 μg/kg infused in 30 to 50 mL of normal saline over approximately 30 minutes every 12 to 24 hours. The dose of the highly concentrated intranasal preparation is 150 μg for children under 50 kg and 300 μg for larger children and adults (1 spray per naris). It is important to note that the products used to treat VWD (eg, Stimate) deliver 150 μg per spray, a much higher concentration than that used to treat enuresis. Repeated DDAVP dosing is associated with the development of tachyphylaxis: with subsequent dosing, the magnitude of the VWF and FVIII increments can fall to approximately 70% of that obtained with the initial dose.48 DDAVP is safe and generally well tolerated. Side effects include facial flushing, headache, tachycardia, light-headedness, and mild hypotension. The most serious side effects, severe hyponatremia and seizures,49 can be avoided by fluid restriction for 24 hours after DDAVP administration. Serum sodium levels should be monitored with repeated doses. DDAVP is generally avoided in those younger than 2 years of age because of a higher risk of hyponatremia. Patients who are intolerant of DDAVP or have a poor VWF response need to be treated with a VWF/FVIII concentrate.
VWF/FVIII Concentrate
VWF/FVIII concentrates are required for patients who do not have an adequate response to DDAVP, who have side effects from or contraindications to DDAVP, or who require a long duration of treatment, rendering the use of DDAVP impractical. Purified, viral-inactivated, plasma-derived VWF/FVIII concentrates are the products most frequently used (eg, Humate-P, Wilate, Alphanate SD/HT). The quantity of VWF:RCo activity relative to FVIII:C varies by product; Humate-P contains 2.4 VWF:RCo units for each unit of FVIII:C; Wilate contains a 1:1 ratio; and Alphanate contains a 0.5:1 ratio. Both Humate-P and Wilate are reported to contain a full spectrum of VWF multimers, including HMW multimers, and closely resemble normal plasma, but Alphanate SD/HT lacks HMW mutimers.11,50 Thus, the available VWF/FVIII vary in terms of VWF:RCo to FVIII concentrate, HMW multimer composition, reported VWF:RCo, and FVIII half-lives and even approved indications. They should not be considered interchangeable, and further information should be sought from the respective product inserts.
Dosing recommendations are provided either in VWF:RCo (North America) or FVIII:C units (Europe) and are weight-based (Table 4); repeat infusions can be given every 8 to 24 hours depending on the type of surgery/injury and the product used.
Long-term continuous use of concentrates to prevent bleeds, known as prophylaxis, is the standard of care in severe hemophilia A and B and is now being adopted in severe VWD. Patients with type 3 VWD or severe type 1 or type 2 VWD may experience recurrent bleeds into joints, nasal/oropharynx, or gastrointestinal tract or excessive menstrual bleeding. Retrospective cohort and case series suggest that prophylaxis improves quality of life; reduces the frequency of bleeding, need for transfusions, and hospitalizations; and prevents chronic joint disease.54,55 More recently, a prospective study confirmed that prophylaxis with VWF concentrates at doses ranging from 50 IU VWF RCo/kg 1 to 3 times per week was highly effective at reducing bleeding rates, with annualized bleeding rates decreasing from 25 to 6.1 in 11 participants with either type 2A or type 3 VWD.56
VWF/FVIII concentrates are effective in more than 97% of events.57 Rarely, when infusion of a VWF/FVIII concentrate is ineffective at stopping bleeding, transfusion of platelet concentrates may be beneficial, presumably because they facilitate the delivery of small amounts of platelet VWF to the site of vascular injury. Highly purified FVIII concentrates (monoclonal antibody purified and recombinant) should not be used to treat VWD because they lack VWF.
A recombinant VWF concentrate (Vonvendi) combined initially with recombinant FVIII concentrate in a 1.3:1 ratio of VWF:RCo to FVIII:C has been shown to be safe and efficacious for the on-demand treatment of bleeds.58,59 After the initial FVIII dose, the patients’ endogenous FVIII levels are stabilized within 6 hours and further FVIII administration may not required. A prospective phase 3 trial investigating the efficacy of recombinant VWF in the prophylaxis of severe VWD is ongoing. Vonvendi has been licensed for on-demand treatment in the United States since 2015. For further dosing information, please refer to the product insert.
Conclusion
VWF is a complex protein with several important and distinct functional domains: binding sites to collagen, FVIII, and platelet GPIbα; an ADAMTS13 cleavage site; and domains important for multimer formation. Mutations in any of these sites can result in a dysfunctional protein and as a result, VWD is a heterogeneous disorder with many specific assays available to determine the subtype. Despite this, the treatment of VWD is straightforward with only a small number of therapeutic options: indirect therapies such as antifibrinolytic agents, or direct therapies that increase VWF levels, DDAVP, or VWF/FVIII concentrates. Management focuses on preventing bleeding complications associated with invasive procedures or promptly treating bleeding episodes.
1. Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006;4:2103–14.
2. Rodeghiero F, Castaman G. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987;69:454–9.
3. Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993;123:893–8.
4. Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000;84:160–74.
5. Bowman M, Hopman WM, Rapson D, et al. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost 2010;8:213–6.
6. Mancuso DJ, Tuley EA, Westfield LA, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989;264:19514–27.
7. Kang I, Raghavachari M, Hofmann CM, Marchant RE. Surface-dependent expression in the platelet GPIb binding domain within human von Willebrand factor studied by atomic force microscopy. Thromb Res 2007;119:731–40.
8. Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996;84:289–97.
9. Dong J, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002;100:4033–9.
10. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010;24:123–34.
11. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report (USA). Haemophilia 2008;14:171–232.
12. Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 vwd (MCMDM-1VWD). Blood 2008;111:4979–85.
13. Goodeve A. Vicenza deciphered: modeling the von Willebrand disease enigma: commentary on accelerated clearance alone explains ultralarge multimers in VWD Vicenza. J Thromb Haemost 2010;8:1271–2.
14. Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009;113:526–34.
15. Nurden AT, Federici AB, Nurden P. Altered megakaryocytopoiesis in von Willebrand type 2B disease. J Thromb Haemost 2009;7 Suppl 1:277–81.
16. Ruggeri ZM, Pareti FI, Mannucci PM, et al. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand’s disease. New Engl J Med 1980;302:1047–51.
17. James PD, Notley C, Hegadorn C, et al. Challenges in defining type 2M von Willebrand disease: results from a Canadian cohort study. J Thromb Haemost 2007;5:1914–22.
18. Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost 2010;8:1431–3.
19. Mazurier C, Hilbert L. Type 2N von Willebrand disease. Curr Hematol Rep 2005;4:350–8.
20. Nesbitt IM, Goodeve AC, Guilliatt AM, et al. Characterisation of type 2N von Willebrand disease using phenotypic and molecular techniques. Thromb Haemost 1996;75:959–64.
21. Bowman M, Tuttle A, Notley C, et al. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant inheritance of mutant alleles. J Thromb Haemost 2013;11:512–20.
22. James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013;122:636–40.
23. James AH, Jamison MG. Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost 2007;5:1165–9.
24. Rydz N, James PD. The evolution and value of bleeding assessment tools. J Thromb Haemost 2012;2223–9.
25. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost 2010;8:2063–5.
26. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia 2014;20:831–5.
27. Deforest M, Grabell J, Alberta S et al. Generation and optimization of the self-administered bleeding assessment tool and its validation as a screening test for von Willebrand disease. Haemophilia 2015;21:e384-8.
28. Castaman G, Hillarp A, Goodeve A. Laboratory aspects of von Willebrand disease: test repertoire and options for activity assays and genetic analysis. Haemophilia 2014;20(Suppl. 4):65–70.
29. Favaloro EJ. Von Willebrand disease, type 2B: a diagnosis more elusive than previously thought. Thromb Haemost 2008;99:630–1.
30. Budde U. Diagnosis of von Willebrand disease subtypes: implications for treatment. Haemophilia 2008;14 Suppl 5:27–38.
31. Favaloro EJ. Von Willebrand factor collagen-binding (activity) assay in the diagnosis of von Willebrand disease: a 15-year journey. Sem Thromb Hemost 2002;28:191–202.
32. Patzke J, Budde U, Huber A, et al. Performance evaluation and multicenter study of a von Willebrand factor activity assay based on GPIb binding in the absence of ristocetin. Blood Coagul Fibrinolysis 2014;25:860-70.
33. Graf L, Moffat KA, Carlino SA, et al. Evaluation of an automated method for measuring von Willebrand factor activity in clinical samples without ristocetin. Int J Lab Hematol 2014;36:341–51.
34. Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood 2006;108:3344–51.
35. Keeney S, Bowen D, Cumming A, et al. The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organisation Haemophilia genetics laboratory network. Haemophilia 2008;14:1099–111.
36. Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987;69:1691–5.
37. Sadler JE, Rodeghiero F. Provisional criteria for the diagnosis of VWD type 1. J Thromb Haemost 2005;3:775–7.
38. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM- 1VWD). J Thromb Haemost 2006;4:766–73.
39. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003;349:343–9.
40. Uriel N, Pak S-W, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010;56:1207–13.
41. Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost 2000;84:345–9.
42. Favaloro EJ, Patterson D, Denholm A, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Brit J Haematol 2007;139:621–8.
43. Giannini S, Cecchetti L, Mezzasoma AM, Gresele P. Diagnosis of platelet-type von Willebrand disease by flow cytometry. Haematologica 2010;95:1021–4.
44. Farquhar C, Brown J. Oral contraceptive pill for heavy menstrual bleeding. Cochrane Database Syst Rev 2009 Oct 7;(4):CD000154.
45. Kadir R, Economides DL, Sabin C, et al. Variations in coagulation factors in women: effects of age, ethnicity, menstrual cycle and combined oral contraceptive. Thromb Haemost 1999;82:1456–61.
46. Muse K, Lukes AS, Gersten J, et al. Long-term evaluation of safety and health-related quality of life in women with heavy menstrual bleeding treated with oral tranexamic acid. Womens Health 2011;7:699–707.
47. Castaman G, Tosetto A, Federici AB, Rodeghiero F. Bleeding tendency and efficacy of anti-haemorrhagic treatments in patients with type 1 von Willebrand disease and increased von Willebrand factor clearance. Thromb Haemost 2011;105:647–54.
48. Mannucci PM, Bettega D, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Brit J Haematol 1992;82:87–93.
49. Greaves M, Watson HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost 2007;5 Suppl 1:167–74.
50. Kessler CM, Friedman K, Schwartz BA, Gill JC, Powell JS. The pharmacokinetic diversity of two von Willebrand factor (VWF) / factor VIII (FVIII) concentrates in subjects with congenital von Willebrand disease. results from a prospective, randomised crossover study. Thromb Haemost 2011;106:279–88.
51. Weiss HJ, Sussman II, Hoyer LW. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand’s disease. J Clin Invest 1977;60:390–404.
52. Berntorp E. Haemate P/Humate-P: a systematic review. Thromb Res 2009;124:S11–14.
53. Lillicrap D, Poon MC, Walker I, et al. Efficacy and safety of the factor VIII/von Willebrand factor concentrate, Haemate-P/Humate-P: ristocetin cofactor unit dosing in patients with von Willebrand disease. Thromb Haemost 2002;87:224–30.
54. Halimeh S, Krümpel A, Rott H, et al. Long-term secondary prophylaxis in children, adolescents and young adults with von Willebrand disease. results of a cohort study. Thromb Haemost 2011;105:597–604.
55. Abshire TC, Federici AB, Alvárez MT, et al. Prophylaxis in severe forms of von Willebrand’s disease: results from the von Willebrand disease prophylaxis network (VWD PN). Haemophilia 2013;19:76–81.
56. Abshire T, Cox-Gill J, Kempton CL, et al. Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. J Thromb Haemost 2015;13:1585– 9.
57. Auerswald G, Kreuz W. Haemate P/Humate-P for the treatment of von Willebrand disease: considerations for use and clinical experience. Sem Thromb Hemost 2008;14 (Suppl 5):39–46.
58. Mannucci PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood 2013;122:648–57.
59. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood 2015;126:2038–46.
1. Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006;4:2103–14.
2. Rodeghiero F, Castaman G. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood 1987;69:454–9.
3. Werner EJ, Broxson EH, Tucker EL, et al. Prevalence of von Willebrand disease in children: a multiethnic study. J Pediatr 1993;123:893–8.
4. Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost 2000;84:160–74.
5. Bowman M, Hopman WM, Rapson D, et al. The prevalence of symptomatic von Willebrand disease in primary care practice. J Thromb Haemost 2010;8:213–6.
6. Mancuso DJ, Tuley EA, Westfield LA, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989;264:19514–27.
7. Kang I, Raghavachari M, Hofmann CM, Marchant RE. Surface-dependent expression in the platelet GPIb binding domain within human von Willebrand factor studied by atomic force microscopy. Thromb Res 2007;119:731–40.
8. Savage B, Saldívar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996;84:289–97.
9. Dong J, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002;100:4033–9.
10. Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010;24:123–34.
11. Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) expert panel report (USA). Haemophilia 2008;14:171–232.
12. Haberichter SL, Castaman G, Budde U, et al. Identification of type 1 von Willebrand disease patients with reduced von Willebrand factor survival by assay of the VWF propeptide in the European study: molecular and clinical markers for the diagnosis and management of type 1 vwd (MCMDM-1VWD). Blood 2008;111:4979–85.
13. Goodeve A. Vicenza deciphered: modeling the von Willebrand disease enigma: commentary on accelerated clearance alone explains ultralarge multimers in VWD Vicenza. J Thromb Haemost 2010;8:1271–2.
14. Federici AB, Mannucci PM, Castaman G, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009;113:526–34.
15. Nurden AT, Federici AB, Nurden P. Altered megakaryocytopoiesis in von Willebrand type 2B disease. J Thromb Haemost 2009;7 Suppl 1:277–81.
16. Ruggeri ZM, Pareti FI, Mannucci PM, et al. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand’s disease. New Engl J Med 1980;302:1047–51.
17. James PD, Notley C, Hegadorn C, et al. Challenges in defining type 2M von Willebrand disease: results from a Canadian cohort study. J Thromb Haemost 2007;5:1914–22.
18. Flood VH, Lederman CA, Wren JS, et al. Absent collagen binding in a VWF A3 domain mutant: utility of the VWF:CB in diagnosis of VWD. J Thromb Haemost 2010;8:1431–3.
19. Mazurier C, Hilbert L. Type 2N von Willebrand disease. Curr Hematol Rep 2005;4:350–8.
20. Nesbitt IM, Goodeve AC, Guilliatt AM, et al. Characterisation of type 2N von Willebrand disease using phenotypic and molecular techniques. Thromb Haemost 1996;75:959–64.
21. Bowman M, Tuttle A, Notley C, et al. The genetics of Canadian type 3 von Willebrand disease: further evidence for co-dominant inheritance of mutant alleles. J Thromb Haemost 2013;11:512–20.
22. James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013;122:636–40.
23. James AH, Jamison MG. Bleeding events and other complications during pregnancy and childbirth in women with von Willebrand disease. J Thromb Haemost 2007;5:1165–9.
24. Rydz N, James PD. The evolution and value of bleeding assessment tools. J Thromb Haemost 2012;2223–9.
25. Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost 2010;8:2063–5.
26. Elbatarny M, Mollah S, Grabell J, et al. Normal range of bleeding scores for the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia 2014;20:831–5.
27. Deforest M, Grabell J, Alberta S et al. Generation and optimization of the self-administered bleeding assessment tool and its validation as a screening test for von Willebrand disease. Haemophilia 2015;21:e384-8.
28. Castaman G, Hillarp A, Goodeve A. Laboratory aspects of von Willebrand disease: test repertoire and options for activity assays and genetic analysis. Haemophilia 2014;20(Suppl. 4):65–70.
29. Favaloro EJ. Von Willebrand disease, type 2B: a diagnosis more elusive than previously thought. Thromb Haemost 2008;99:630–1.
30. Budde U. Diagnosis of von Willebrand disease subtypes: implications for treatment. Haemophilia 2008;14 Suppl 5:27–38.
31. Favaloro EJ. Von Willebrand factor collagen-binding (activity) assay in the diagnosis of von Willebrand disease: a 15-year journey. Sem Thromb Hemost 2002;28:191–202.
32. Patzke J, Budde U, Huber A, et al. Performance evaluation and multicenter study of a von Willebrand factor activity assay based on GPIb binding in the absence of ristocetin. Blood Coagul Fibrinolysis 2014;25:860-70.
33. Graf L, Moffat KA, Carlino SA, et al. Evaluation of an automated method for measuring von Willebrand factor activity in clinical samples without ristocetin. Int J Lab Hematol 2014;36:341–51.
34. Haberichter SL, Balistreri M, Christopherson P, et al. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood 2006;108:3344–51.
35. Keeney S, Bowen D, Cumming A, et al. The molecular analysis of von Willebrand disease: a guideline from the UK Haemophilia Centre Doctors’ Organisation Haemophilia genetics laboratory network. Haemophilia 2008;14:1099–111.
36. Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood 1987;69:1691–5.
37. Sadler JE, Rodeghiero F. Provisional criteria for the diagnosis of VWD type 1. J Thromb Haemost 2005;3:775–7.
38. Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM- 1VWD). J Thromb Haemost 2006;4:766–73.
39. Vincentelli A, Susen S, Le Tourneau T, et al. Acquired von Willebrand syndrome in aortic stenosis. N Engl J Med 2003;349:343–9.
40. Uriel N, Pak S-W, Jorde UP, et al. Acquired von Willebrand syndrome after continuous-flow mechanical device support contributes to a high prevalence of bleeding during long-term support and at the time of transplantation. J Am Coll Cardiol 2010;56:1207–13.
41. Federici AB, Rand JH, Bucciarelli P, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost 2000;84:345–9.
42. Favaloro EJ, Patterson D, Denholm A, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Brit J Haematol 2007;139:621–8.
43. Giannini S, Cecchetti L, Mezzasoma AM, Gresele P. Diagnosis of platelet-type von Willebrand disease by flow cytometry. Haematologica 2010;95:1021–4.
44. Farquhar C, Brown J. Oral contraceptive pill for heavy menstrual bleeding. Cochrane Database Syst Rev 2009 Oct 7;(4):CD000154.
45. Kadir R, Economides DL, Sabin C, et al. Variations in coagulation factors in women: effects of age, ethnicity, menstrual cycle and combined oral contraceptive. Thromb Haemost 1999;82:1456–61.
46. Muse K, Lukes AS, Gersten J, et al. Long-term evaluation of safety and health-related quality of life in women with heavy menstrual bleeding treated with oral tranexamic acid. Womens Health 2011;7:699–707.
47. Castaman G, Tosetto A, Federici AB, Rodeghiero F. Bleeding tendency and efficacy of anti-haemorrhagic treatments in patients with type 1 von Willebrand disease and increased von Willebrand factor clearance. Thromb Haemost 2011;105:647–54.
48. Mannucci PM, Bettega D, Cattaneo M. Patterns of development of tachyphylaxis in patients with haemophilia and von Willebrand disease after repeated doses of desmopressin (DDAVP). Brit J Haematol 1992;82:87–93.
49. Greaves M, Watson HG. Approach to the diagnosis and management of mild bleeding disorders. J Thromb Haemost 2007;5 Suppl 1:167–74.
50. Kessler CM, Friedman K, Schwartz BA, Gill JC, Powell JS. The pharmacokinetic diversity of two von Willebrand factor (VWF) / factor VIII (FVIII) concentrates in subjects with congenital von Willebrand disease. results from a prospective, randomised crossover study. Thromb Haemost 2011;106:279–88.
51. Weiss HJ, Sussman II, Hoyer LW. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand’s disease. J Clin Invest 1977;60:390–404.
52. Berntorp E. Haemate P/Humate-P: a systematic review. Thromb Res 2009;124:S11–14.
53. Lillicrap D, Poon MC, Walker I, et al. Efficacy and safety of the factor VIII/von Willebrand factor concentrate, Haemate-P/Humate-P: ristocetin cofactor unit dosing in patients with von Willebrand disease. Thromb Haemost 2002;87:224–30.
54. Halimeh S, Krümpel A, Rott H, et al. Long-term secondary prophylaxis in children, adolescents and young adults with von Willebrand disease. results of a cohort study. Thromb Haemost 2011;105:597–604.
55. Abshire TC, Federici AB, Alvárez MT, et al. Prophylaxis in severe forms of von Willebrand’s disease: results from the von Willebrand disease prophylaxis network (VWD PN). Haemophilia 2013;19:76–81.
56. Abshire T, Cox-Gill J, Kempton CL, et al. Prophylaxis escalation in severe von Willebrand disease: a prospective study from the von Willebrand Disease Prophylaxis Network. J Thromb Haemost 2015;13:1585– 9.
57. Auerswald G, Kreuz W. Haemate P/Humate-P for the treatment of von Willebrand disease: considerations for use and clinical experience. Sem Thromb Hemost 2008;14 (Suppl 5):39–46.
58. Mannucci PM, Kempton C, Millar C, et al. Pharmacokinetics and safety of a novel recombinant human von Willebrand factor manufactured with a plasma-free method: a prospective clinical trial. Blood 2013;122:648–57.
59. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood 2015;126:2038–46.
Product approved for hemophilia patients in Japan
Japan’s Ministry of Health, Labour and Welfare (MHLW) has approved the bispecific factor IXa- and factor X-directed antibody emicizumab (Hemlibra), according to Chugai Pharmaceutical Co., Ltd.
Emicizumab is now approved for use in Japan as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in patients with hemophilia A and factor VIII inhibitors.
There are a few conditions for this approval, including requirements for a risk management plan, early phase post-marketing vigilance, and post-marketing drug use surveillance.
A risk management plan is intended to assess measures for appropriate management of the risks associated with a drug, either at regular intervals or in response to the progress of post-marketing surveillance.
According to Japan’s Pharmaceuticals and Medical Devices Agency, a risk management plan must consist of 3 elements:
- Safety specification—Important adverse drug reactions and missing information
- Pharmacovigilance activities—Information collection activities performed in the post-marketing period
- Risk-minimization activities—Safety measures taken to minimize risks, which consists of providing information to healthcare professionals and setting the terms of use for a drug.
For the early phase post-marketing vigilance requirement, Chugai must provide safety information to healthcare professionals and collect information on adverse reactions to emicizumab in the early post-marketing phase.
Post-marketing drug use surveillance for emicizumab will include all patients receiving the product and is scheduled to continue until data is collected from approximately 100 people. The goal is to understand background information on patients receiving emicizumab, as well as to collect safety and efficacy data on the product and take necessary measures for the appropriate use of emicizumab.
The data collected via this surveillance effort will be reviewed to determine whether new surveillance or further safety measures are needed. Results of the surveillance will be reported to the regulatory authorities, and the data will be presented at scientific meetings, according to Chugai.
Phase 3 studies
The MHLW’s approval of emicizumab is based on data from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs). Three had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
Japan’s Ministry of Health, Labour and Welfare (MHLW) has approved the bispecific factor IXa- and factor X-directed antibody emicizumab (Hemlibra), according to Chugai Pharmaceutical Co., Ltd.
Emicizumab is now approved for use in Japan as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in patients with hemophilia A and factor VIII inhibitors.
There are a few conditions for this approval, including requirements for a risk management plan, early phase post-marketing vigilance, and post-marketing drug use surveillance.
A risk management plan is intended to assess measures for appropriate management of the risks associated with a drug, either at regular intervals or in response to the progress of post-marketing surveillance.
According to Japan’s Pharmaceuticals and Medical Devices Agency, a risk management plan must consist of 3 elements:
- Safety specification—Important adverse drug reactions and missing information
- Pharmacovigilance activities—Information collection activities performed in the post-marketing period
- Risk-minimization activities—Safety measures taken to minimize risks, which consists of providing information to healthcare professionals and setting the terms of use for a drug.
For the early phase post-marketing vigilance requirement, Chugai must provide safety information to healthcare professionals and collect information on adverse reactions to emicizumab in the early post-marketing phase.
Post-marketing drug use surveillance for emicizumab will include all patients receiving the product and is scheduled to continue until data is collected from approximately 100 people. The goal is to understand background information on patients receiving emicizumab, as well as to collect safety and efficacy data on the product and take necessary measures for the appropriate use of emicizumab.
The data collected via this surveillance effort will be reviewed to determine whether new surveillance or further safety measures are needed. Results of the surveillance will be reported to the regulatory authorities, and the data will be presented at scientific meetings, according to Chugai.
Phase 3 studies
The MHLW’s approval of emicizumab is based on data from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs). Three had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
Japan’s Ministry of Health, Labour and Welfare (MHLW) has approved the bispecific factor IXa- and factor X-directed antibody emicizumab (Hemlibra), according to Chugai Pharmaceutical Co., Ltd.
Emicizumab is now approved for use in Japan as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in patients with hemophilia A and factor VIII inhibitors.
There are a few conditions for this approval, including requirements for a risk management plan, early phase post-marketing vigilance, and post-marketing drug use surveillance.
A risk management plan is intended to assess measures for appropriate management of the risks associated with a drug, either at regular intervals or in response to the progress of post-marketing surveillance.
According to Japan’s Pharmaceuticals and Medical Devices Agency, a risk management plan must consist of 3 elements:
- Safety specification—Important adverse drug reactions and missing information
- Pharmacovigilance activities—Information collection activities performed in the post-marketing period
- Risk-minimization activities—Safety measures taken to minimize risks, which consists of providing information to healthcare professionals and setting the terms of use for a drug.
For the early phase post-marketing vigilance requirement, Chugai must provide safety information to healthcare professionals and collect information on adverse reactions to emicizumab in the early post-marketing phase.
Post-marketing drug use surveillance for emicizumab will include all patients receiving the product and is scheduled to continue until data is collected from approximately 100 people. The goal is to understand background information on patients receiving emicizumab, as well as to collect safety and efficacy data on the product and take necessary measures for the appropriate use of emicizumab.
The data collected via this surveillance effort will be reviewed to determine whether new surveillance or further safety measures are needed. Results of the surveillance will be reported to the regulatory authorities, and the data will be presented at scientific meetings, according to Chugai.
Phase 3 studies
The MHLW’s approval of emicizumab is based on data from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs). Three had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
Bleeding episodes more common in boys with VWD
SAN DIEGO – Among children with types 1 and 2 von Willebrand disease (VWD), a higher proportion of boys than girls reported ever having a bleeding episode and using more treatment products. But the trend did not continue among children with type 3 disease.
Those are some of the key findings from a never-before-published analysis of surveillance data from the Centers for Disease Control and Prevention presented by Karon Abe, PhD, during a poster session at the biennial summit of the Thrombosis & Hemostasis Societies of North America.

The researchers retrieved data from the UDC (Universal Data Collection System), a federally funded surveillance system of people with hemophilia and other bleeding disorders treated at 130 U.S. Hemophilia Treatment Centers (HTCs) during 1998-2011. Although UDC data collection ended in 2011, a current CDC bleeding surveillance project called Community Counts continues and expands on the work of the UDC.
Between 1998 and 2011, data were collected on 2,413 children with VWD aged 2-12 years. Of these, 2,070 had type 1, 224 had type 2, and 119 had type 3 VWD. The researchers used chi-square analysis and Wilcoxon rank sum tests to assess differences in bleeding characteristics by sex and by type of VWD. Next, they used a multivariate regression model to examine the association between demographic and clinical characteristics and a history of ever having had a bleeding episode among type 1 VWD patients.
Nearly two-thirds of children (65%) were non-Hispanic, 17% were Hispanic, 8% were black, and the remainder were from other ethnicities. In addition, 40% of the children had no family history of a bleeding disorder.
The median age of first bleed was lower among children with type 3 VWD, compared with other VWD types, and was lower among boys than girls with type 1 VWD (36 months vs. 48 months, respectively; P less than .001) and type 3 VWD (9 months vs. 12 months; P = .04), Dr. Abe reported.
A higher proportion of boys than girls reported ever having a bleeding episode among children with type 1 VWD (78% vs. 73%; P = .01) and type 2 VWD (90% vs. 75%; P = .01), but not among children with type 3 VWD (97% vs. 96%; P = .77).
A higher prevalence of treatment-product use was reported among children with type 3 VWD, compared with those with the other VWD types (a mean of 95% vs. 79% and 71% among types 2 and 1, respectively). A significantly higher prevalence of the use of treatment product was seen among boys than girls with type 1 VWD (73% vs. 68%, P = .03) and type 2 VWD (87% vs. 72%, P =.01), but not type 3 VWD (94% vs. 96%, P = .87).
The most common sites of the first bleed among all patients regardless of gender or VWD type were epistaxis and oral cavity bleeding.
“To our surprise, the boys were showing more bleeding and were receiving more product than the females,” Dr. Abe said in an interview. “This is a fairly large population.”
Multivariate regression analysis revealed independent associations between the following patient characteristics and ever having a bleed among children with type 1 VWD: male gender (adjusted odds ratio, 1.23); being aged 7-9 years at registration, compared with being aged 2-6 years (aOR, 1.5); being black (aOR, 1.7); being Asian, Native Hawaiian or Pacific Islander (aOR, 2.4), being Hispanic (aOR, 2.8), and being some other race/ethnicity (aOR, 1.8). However, family history of a bleeding disorder was protective (aOR, 0.721).
Dr. Abe said she hopes that the findings will raise awareness and help physicians to educate families about bleeding symptoms and intervene to treat bleeding episodes appropriately. She and her associates are planning to compare the data with Community Counts, “so it’s more up to date,” she said.
Dr. Abe reported having no financial disclosures.
SOURCE: Abe K et al. THSNA 2018, Poster 145.
SAN DIEGO – Among children with types 1 and 2 von Willebrand disease (VWD), a higher proportion of boys than girls reported ever having a bleeding episode and using more treatment products. But the trend did not continue among children with type 3 disease.
Those are some of the key findings from a never-before-published analysis of surveillance data from the Centers for Disease Control and Prevention presented by Karon Abe, PhD, during a poster session at the biennial summit of the Thrombosis & Hemostasis Societies of North America.
The researchers retrieved data from the UDC (Universal Data Collection System), a federally funded surveillance system of people with hemophilia and other bleeding disorders treated at 130 U.S. Hemophilia Treatment Centers (HTCs) during 1998-2011. Although UDC data collection ended in 2011, a current CDC bleeding surveillance project called Community Counts continues and expands on the work of the UDC.
Between 1998 and 2011, data were collected on 2,413 children with VWD aged 2-12 years. Of these, 2,070 had type 1, 224 had type 2, and 119 had type 3 VWD. The researchers used chi-square analysis and Wilcoxon rank sum tests to assess differences in bleeding characteristics by sex and by type of VWD. Next, they used a multivariate regression model to examine the association between demographic and clinical characteristics and a history of ever having had a bleeding episode among type 1 VWD patients.
Nearly two-thirds of children (65%) were non-Hispanic, 17% were Hispanic, 8% were black, and the remainder were from other ethnicities. In addition, 40% of the children had no family history of a bleeding disorder.
The median age of first bleed was lower among children with type 3 VWD, compared with other VWD types, and was lower among boys than girls with type 1 VWD (36 months vs. 48 months, respectively; P less than .001) and type 3 VWD (9 months vs. 12 months; P = .04), Dr. Abe reported.
A higher proportion of boys than girls reported ever having a bleeding episode among children with type 1 VWD (78% vs. 73%; P = .01) and type 2 VWD (90% vs. 75%; P = .01), but not among children with type 3 VWD (97% vs. 96%; P = .77).
A higher prevalence of treatment-product use was reported among children with type 3 VWD, compared with those with the other VWD types (a mean of 95% vs. 79% and 71% among types 2 and 1, respectively). A significantly higher prevalence of the use of treatment product was seen among boys than girls with type 1 VWD (73% vs. 68%, P = .03) and type 2 VWD (87% vs. 72%, P =.01), but not type 3 VWD (94% vs. 96%, P = .87).
The most common sites of the first bleed among all patients regardless of gender or VWD type were epistaxis and oral cavity bleeding.
“To our surprise, the boys were showing more bleeding and were receiving more product than the females,” Dr. Abe said in an interview. “This is a fairly large population.”
Multivariate regression analysis revealed independent associations between the following patient characteristics and ever having a bleed among children with type 1 VWD: male gender (adjusted odds ratio, 1.23); being aged 7-9 years at registration, compared with being aged 2-6 years (aOR, 1.5); being black (aOR, 1.7); being Asian, Native Hawaiian or Pacific Islander (aOR, 2.4), being Hispanic (aOR, 2.8), and being some other race/ethnicity (aOR, 1.8). However, family history of a bleeding disorder was protective (aOR, 0.721).
Dr. Abe said she hopes that the findings will raise awareness and help physicians to educate families about bleeding symptoms and intervene to treat bleeding episodes appropriately. She and her associates are planning to compare the data with Community Counts, “so it’s more up to date,” she said.
Dr. Abe reported having no financial disclosures.
SOURCE: Abe K et al. THSNA 2018, Poster 145.
SAN DIEGO – Among children with types 1 and 2 von Willebrand disease (VWD), a higher proportion of boys than girls reported ever having a bleeding episode and using more treatment products. But the trend did not continue among children with type 3 disease.
Those are some of the key findings from a never-before-published analysis of surveillance data from the Centers for Disease Control and Prevention presented by Karon Abe, PhD, during a poster session at the biennial summit of the Thrombosis & Hemostasis Societies of North America.
The researchers retrieved data from the UDC (Universal Data Collection System), a federally funded surveillance system of people with hemophilia and other bleeding disorders treated at 130 U.S. Hemophilia Treatment Centers (HTCs) during 1998-2011. Although UDC data collection ended in 2011, a current CDC bleeding surveillance project called Community Counts continues and expands on the work of the UDC.
Between 1998 and 2011, data were collected on 2,413 children with VWD aged 2-12 years. Of these, 2,070 had type 1, 224 had type 2, and 119 had type 3 VWD. The researchers used chi-square analysis and Wilcoxon rank sum tests to assess differences in bleeding characteristics by sex and by type of VWD. Next, they used a multivariate regression model to examine the association between demographic and clinical characteristics and a history of ever having had a bleeding episode among type 1 VWD patients.
Nearly two-thirds of children (65%) were non-Hispanic, 17% were Hispanic, 8% were black, and the remainder were from other ethnicities. In addition, 40% of the children had no family history of a bleeding disorder.
The median age of first bleed was lower among children with type 3 VWD, compared with other VWD types, and was lower among boys than girls with type 1 VWD (36 months vs. 48 months, respectively; P less than .001) and type 3 VWD (9 months vs. 12 months; P = .04), Dr. Abe reported.
A higher proportion of boys than girls reported ever having a bleeding episode among children with type 1 VWD (78% vs. 73%; P = .01) and type 2 VWD (90% vs. 75%; P = .01), but not among children with type 3 VWD (97% vs. 96%; P = .77).
A higher prevalence of treatment-product use was reported among children with type 3 VWD, compared with those with the other VWD types (a mean of 95% vs. 79% and 71% among types 2 and 1, respectively). A significantly higher prevalence of the use of treatment product was seen among boys than girls with type 1 VWD (73% vs. 68%, P = .03) and type 2 VWD (87% vs. 72%, P =.01), but not type 3 VWD (94% vs. 96%, P = .87).
The most common sites of the first bleed among all patients regardless of gender or VWD type were epistaxis and oral cavity bleeding.
“To our surprise, the boys were showing more bleeding and were receiving more product than the females,” Dr. Abe said in an interview. “This is a fairly large population.”
Multivariate regression analysis revealed independent associations between the following patient characteristics and ever having a bleed among children with type 1 VWD: male gender (adjusted odds ratio, 1.23); being aged 7-9 years at registration, compared with being aged 2-6 years (aOR, 1.5); being black (aOR, 1.7); being Asian, Native Hawaiian or Pacific Islander (aOR, 2.4), being Hispanic (aOR, 2.8), and being some other race/ethnicity (aOR, 1.8). However, family history of a bleeding disorder was protective (aOR, 0.721).
Dr. Abe said she hopes that the findings will raise awareness and help physicians to educate families about bleeding symptoms and intervene to treat bleeding episodes appropriately. She and her associates are planning to compare the data with Community Counts, “so it’s more up to date,” she said.
Dr. Abe reported having no financial disclosures.
SOURCE: Abe K et al. THSNA 2018, Poster 145.
REPORTING FROM THSNA 2018
Key clinical point:
Major finding: A higher proportion of boys than girls reported ever having a bleeding episode among children with type 1 VWD (78% vs. 73%; P= .01) and type 2 VWD (90% vs. 75%; P= .01), but not among children with type 3 VWD (97% vs. 96%; P= .77).
Study details: An analysis of 2,413 children with VWD aged 2-12 years.
Disclosures: Dr. Abe reported having no financial disclosures.
Source: Abe K et al. THSNA 2018, Poster 145.
Of ‘miracles’ and money: Why hemophilia drugs are so expensive
YUBA CITY, Calif. – When Landon Morris was diagnosed with hemophilia shortly after birth, his mother, Jessica Morris, was devastated. “It was like having your dreams – all the dreams you imagined for your child – just kind of disappear,” she recalled.
Hemophilia, a rare bleeding disorder caused by a gene mutation that prevents blood from clotting properly, is typically passed from mother to son. Ms. Morris’ grandfather had it, and she remembered hearing how painful it was. “It was almost like he was bubble-wrapped,” she said. “He was coddled, because his mom didn’t want him to get hurt.”
But Landon’s life turned out much different than she expected.
“He’s wild. He’s probably sometimes the roughest of them all,” she said, as she watched the 6-year-old race around a park. “He leads a totally normal life. He plays T-ball. He’ll start soccer in the fall. He runs and jumps and wrestles with his brothers.” That’s due almost entirely to his medication – the kind that wasn’t available in his grandfather’s day. For the Morris family, this type of drug – broadly known as clotting factor – is a miracle, helping Landon’s blood clot normally. And its cost is almost entirely covered by his father’s federal employee health plan.
But for the health care system, such drugs are enormously expensive, among the priciest in the nation. Medications to treat hemophilia cost an average of more than $270,000 annually per patient, according to a 2015 Express Scripts report. If complications arise, that annual price tag can soar above $1 million. The U.S. hemophilia drug market, which serves about 20,000 patients, is worth $4.6 billion a year, according to the investment research firm AllianceBernstein.
Examining the stubbornly high cost of these medications opens a window into why some prescription drugs in the United States – especially those for rare diseases – have stratospheric prices. The short answer: Competition doesn’t do its traditional job of tamping down costs.
Vying for patients
The market for hemophilia medicines in the United States is flooded with 28 different drugs, with another 21 drugs in development. Because blood factor drugs are biological products – in this case, a protein – there are no cheaper copies, called biosimilars, available. Not only do prices rise steadily as each new product comes on the market, demand is growing – and pushing costs upward – as more and more clotting factor is used to prevent bleeding episodes, not just to treat them.
Yet competition has not brought prices down in the way someone “operating at the level of undergrad Econ 101 would expect,” said Jerry Avorn, MD, a professor of medicine at Harvard Medical School, Boston, who studies prescription drug costs.
The problem is that companies have no incentive to lower prices. Patients generally don’t push back because insurers pay the bulk of the cost. And insurers tend not to object because the market for the drugs – expensive as they are – is small and the patients are especially vulnerable.
For drug companies, Dr. Avorn said, “it’s a magical formula: Lifesaving drug, child at risk of bleeding to death – it kind of casts anybody who looks at costs into the role of some evil Scrooge-like person.”
“The insurers don’t want to end up on the front page of the newspaper saying Little Timmy bled to death because his drug wasn’t covered,” he said.
Also, because prices are high across the hemophilia market, no drug company wants to be the one to blink first. “They don’t want to get a price war started and end up at a super low price point,” said Edmund Pezalla, a consultant to pharmaceutical companies and former executive at Aetna.
So, these drugmakers compete not on price but clinical benefits – such as how long the drugs’ effects last – and through intensive marketing. The pool of potential customers is so valuable that companies often vie directly for individual patients.
Manufacturers, as well as specialty pharmacies that sell the drugs, hire patients and parents as recruiters and advisers, hold dinners and holiday parties, offer scholarships to patients, and even run summer camps for children with the disease. The Morris family regularly receives such invitations.
Jonathan Ducore, MD, a pediatric hematologist-oncologist at the University of California, Davis, Hemophilia Treatment Center in Sacramento, said some of his patients are persuaded by drug company presentations to switch medications. ”But the real differences between the drugs are limited,” he said.
Dr. Ducore said he tells patients if he thinks they are being misled by drugmakers about what a product will do. “But even though the tactics may seem a little smarmy, if it’s the patient’s choice, you have to go with it,” said Dr. Ducore, who has been Landon’s doctor since the boy was born.
The first clotting factor products, which came onto the market in the mid-1960s, were derived from human blood plasma, with thousands of donations combined to create one batch. This proved disastrous in the 1980s, when donors unwittingly spread HIV into the blood supply. An estimated 4,000 people with hemophilia – about 40 percent of the patient population in the United States – died from AIDS as a result.
In the 1990s, manufacturers introduced a product that did not carry the disease risk of plasma-based drugs – made by cloning human clotting proteins in animal cells. Companies charged a premium for this ever-more-popular “recombinant factor.”
Recombinant factor is difficult and delicate to make, said Steve Garger, a development scientist at Bayer, which produces two popular factor products at its Berkeley, Calif., plant – including Landon Morris’ drug, Kogenate.
Inside a concrete building on the campus, kidney cells from baby hamsters are grown in stainless-steel vessels called bioreactors, and the clotting factor they produce is then purified in steel tanks kept in cold rooms. Working at full capacity, this factory produces less than a pound of clotting factor each year – but when diluted with other ingredients, it’s enough to treat thousands of patients in 80 countries.
The investment in manufacturing and marketing is only part of the reason for the high cost of the drugs, said Kevin O’Leary, vice president of pricing and contracting at Bayer. Bayer does not simply add up the costs, slap on a profit margin and come up with the price, Mr. O’Leary explained.
Instead, he said, the company begins by talking to insurers, doctors, and patients to get a sense of what value its products bring to the market, especially compared with drugs already available. Bayer then sets a price based on both its investment and the product’s perceived worth. In the end, he said, “we’re charging a price that’s competitive with the other factor products on the market.”
Bayer’s annual sales from its hemophilia drugs were 1.166 billion euros in 2016. That’s the equivalent of about $1.45 billion in the United States.
Pushing back on costs
In Europe, hemophilia drugs cost less than half what they cost in the United States. That’s because payers – usually governments – request bids and pick products based on cost and quality.
Without pushback from insurers in the United States, “the price of any drug in the U.S. is whatever the market will bear as seen by the manufacturer,” said Dr. Avorn of Harvard.
Recently, a few insurance companies have quietly started to push back on costs. Bayer’s Mr. O’Leary said several insurers have approached the company and demanded rebates in exchange for offering the drug to their customers. Mr. O’Leary would not discuss the details because he said the contracts are confidential.
State Medicaid programs, which provide health insurance to low-income Americans and cover about half of hemophilia patients, already receive significant rebates from hemophilia drug manufacturers.
Michelle Rice, a senior vice president at the National Hemophilia Foundation, said she has been working with several insurers to help them manage costs safely. “We understand the need to control costs, but they can’t impede access to the product a patient needs,” she said.
It is not yet clear whether such efforts will work, let alone spread.
Sitting at a picnic bench at a park, Jessica Morris pages through Landon’s insurance documents. Over the past year, his care cost over $120,000. She wonders sometimes what would happen if they lost their coverage.
“How much would you be willing to pay to have your child lead a normal life?” she said. “I don’t think that there’s anything we wouldn’t pay or sacrifice for him.”
It’s a problem she prays they’ll never have to face.
Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of the Kaiser Family Foundation that is not affiliated with Kaiser Permanente. KHN’s coverage of prescription drug development, costs, and pricing is supported by the Laura and John Arnold Foundation.
YUBA CITY, Calif. – When Landon Morris was diagnosed with hemophilia shortly after birth, his mother, Jessica Morris, was devastated. “It was like having your dreams – all the dreams you imagined for your child – just kind of disappear,” she recalled.
Hemophilia, a rare bleeding disorder caused by a gene mutation that prevents blood from clotting properly, is typically passed from mother to son. Ms. Morris’ grandfather had it, and she remembered hearing how painful it was. “It was almost like he was bubble-wrapped,” she said. “He was coddled, because his mom didn’t want him to get hurt.”
But Landon’s life turned out much different than she expected.
“He’s wild. He’s probably sometimes the roughest of them all,” she said, as she watched the 6-year-old race around a park. “He leads a totally normal life. He plays T-ball. He’ll start soccer in the fall. He runs and jumps and wrestles with his brothers.” That’s due almost entirely to his medication – the kind that wasn’t available in his grandfather’s day. For the Morris family, this type of drug – broadly known as clotting factor – is a miracle, helping Landon’s blood clot normally. And its cost is almost entirely covered by his father’s federal employee health plan.
But for the health care system, such drugs are enormously expensive, among the priciest in the nation. Medications to treat hemophilia cost an average of more than $270,000 annually per patient, according to a 2015 Express Scripts report. If complications arise, that annual price tag can soar above $1 million. The U.S. hemophilia drug market, which serves about 20,000 patients, is worth $4.6 billion a year, according to the investment research firm AllianceBernstein.
Examining the stubbornly high cost of these medications opens a window into why some prescription drugs in the United States – especially those for rare diseases – have stratospheric prices. The short answer: Competition doesn’t do its traditional job of tamping down costs.
Vying for patients
The market for hemophilia medicines in the United States is flooded with 28 different drugs, with another 21 drugs in development. Because blood factor drugs are biological products – in this case, a protein – there are no cheaper copies, called biosimilars, available. Not only do prices rise steadily as each new product comes on the market, demand is growing – and pushing costs upward – as more and more clotting factor is used to prevent bleeding episodes, not just to treat them.
Yet competition has not brought prices down in the way someone “operating at the level of undergrad Econ 101 would expect,” said Jerry Avorn, MD, a professor of medicine at Harvard Medical School, Boston, who studies prescription drug costs.
The problem is that companies have no incentive to lower prices. Patients generally don’t push back because insurers pay the bulk of the cost. And insurers tend not to object because the market for the drugs – expensive as they are – is small and the patients are especially vulnerable.
For drug companies, Dr. Avorn said, “it’s a magical formula: Lifesaving drug, child at risk of bleeding to death – it kind of casts anybody who looks at costs into the role of some evil Scrooge-like person.”
“The insurers don’t want to end up on the front page of the newspaper saying Little Timmy bled to death because his drug wasn’t covered,” he said.
Also, because prices are high across the hemophilia market, no drug company wants to be the one to blink first. “They don’t want to get a price war started and end up at a super low price point,” said Edmund Pezalla, a consultant to pharmaceutical companies and former executive at Aetna.
So, these drugmakers compete not on price but clinical benefits – such as how long the drugs’ effects last – and through intensive marketing. The pool of potential customers is so valuable that companies often vie directly for individual patients.
Manufacturers, as well as specialty pharmacies that sell the drugs, hire patients and parents as recruiters and advisers, hold dinners and holiday parties, offer scholarships to patients, and even run summer camps for children with the disease. The Morris family regularly receives such invitations.
Jonathan Ducore, MD, a pediatric hematologist-oncologist at the University of California, Davis, Hemophilia Treatment Center in Sacramento, said some of his patients are persuaded by drug company presentations to switch medications. ”But the real differences between the drugs are limited,” he said.
Dr. Ducore said he tells patients if he thinks they are being misled by drugmakers about what a product will do. “But even though the tactics may seem a little smarmy, if it’s the patient’s choice, you have to go with it,” said Dr. Ducore, who has been Landon’s doctor since the boy was born.
The first clotting factor products, which came onto the market in the mid-1960s, were derived from human blood plasma, with thousands of donations combined to create one batch. This proved disastrous in the 1980s, when donors unwittingly spread HIV into the blood supply. An estimated 4,000 people with hemophilia – about 40 percent of the patient population in the United States – died from AIDS as a result.
In the 1990s, manufacturers introduced a product that did not carry the disease risk of plasma-based drugs – made by cloning human clotting proteins in animal cells. Companies charged a premium for this ever-more-popular “recombinant factor.”
Recombinant factor is difficult and delicate to make, said Steve Garger, a development scientist at Bayer, which produces two popular factor products at its Berkeley, Calif., plant – including Landon Morris’ drug, Kogenate.
Inside a concrete building on the campus, kidney cells from baby hamsters are grown in stainless-steel vessels called bioreactors, and the clotting factor they produce is then purified in steel tanks kept in cold rooms. Working at full capacity, this factory produces less than a pound of clotting factor each year – but when diluted with other ingredients, it’s enough to treat thousands of patients in 80 countries.
The investment in manufacturing and marketing is only part of the reason for the high cost of the drugs, said Kevin O’Leary, vice president of pricing and contracting at Bayer. Bayer does not simply add up the costs, slap on a profit margin and come up with the price, Mr. O’Leary explained.
Instead, he said, the company begins by talking to insurers, doctors, and patients to get a sense of what value its products bring to the market, especially compared with drugs already available. Bayer then sets a price based on both its investment and the product’s perceived worth. In the end, he said, “we’re charging a price that’s competitive with the other factor products on the market.”
Bayer’s annual sales from its hemophilia drugs were 1.166 billion euros in 2016. That’s the equivalent of about $1.45 billion in the United States.
Pushing back on costs
In Europe, hemophilia drugs cost less than half what they cost in the United States. That’s because payers – usually governments – request bids and pick products based on cost and quality.
Without pushback from insurers in the United States, “the price of any drug in the U.S. is whatever the market will bear as seen by the manufacturer,” said Dr. Avorn of Harvard.
Recently, a few insurance companies have quietly started to push back on costs. Bayer’s Mr. O’Leary said several insurers have approached the company and demanded rebates in exchange for offering the drug to their customers. Mr. O’Leary would not discuss the details because he said the contracts are confidential.
State Medicaid programs, which provide health insurance to low-income Americans and cover about half of hemophilia patients, already receive significant rebates from hemophilia drug manufacturers.
Michelle Rice, a senior vice president at the National Hemophilia Foundation, said she has been working with several insurers to help them manage costs safely. “We understand the need to control costs, but they can’t impede access to the product a patient needs,” she said.
It is not yet clear whether such efforts will work, let alone spread.
Sitting at a picnic bench at a park, Jessica Morris pages through Landon’s insurance documents. Over the past year, his care cost over $120,000. She wonders sometimes what would happen if they lost their coverage.
“How much would you be willing to pay to have your child lead a normal life?” she said. “I don’t think that there’s anything we wouldn’t pay or sacrifice for him.”
It’s a problem she prays they’ll never have to face.
Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of the Kaiser Family Foundation that is not affiliated with Kaiser Permanente. KHN’s coverage of prescription drug development, costs, and pricing is supported by the Laura and John Arnold Foundation.
YUBA CITY, Calif. – When Landon Morris was diagnosed with hemophilia shortly after birth, his mother, Jessica Morris, was devastated. “It was like having your dreams – all the dreams you imagined for your child – just kind of disappear,” she recalled.
Hemophilia, a rare bleeding disorder caused by a gene mutation that prevents blood from clotting properly, is typically passed from mother to son. Ms. Morris’ grandfather had it, and she remembered hearing how painful it was. “It was almost like he was bubble-wrapped,” she said. “He was coddled, because his mom didn’t want him to get hurt.”
But Landon’s life turned out much different than she expected.
“He’s wild. He’s probably sometimes the roughest of them all,” she said, as she watched the 6-year-old race around a park. “He leads a totally normal life. He plays T-ball. He’ll start soccer in the fall. He runs and jumps and wrestles with his brothers.” That’s due almost entirely to his medication – the kind that wasn’t available in his grandfather’s day. For the Morris family, this type of drug – broadly known as clotting factor – is a miracle, helping Landon’s blood clot normally. And its cost is almost entirely covered by his father’s federal employee health plan.
But for the health care system, such drugs are enormously expensive, among the priciest in the nation. Medications to treat hemophilia cost an average of more than $270,000 annually per patient, according to a 2015 Express Scripts report. If complications arise, that annual price tag can soar above $1 million. The U.S. hemophilia drug market, which serves about 20,000 patients, is worth $4.6 billion a year, according to the investment research firm AllianceBernstein.
Examining the stubbornly high cost of these medications opens a window into why some prescription drugs in the United States – especially those for rare diseases – have stratospheric prices. The short answer: Competition doesn’t do its traditional job of tamping down costs.
Vying for patients
The market for hemophilia medicines in the United States is flooded with 28 different drugs, with another 21 drugs in development. Because blood factor drugs are biological products – in this case, a protein – there are no cheaper copies, called biosimilars, available. Not only do prices rise steadily as each new product comes on the market, demand is growing – and pushing costs upward – as more and more clotting factor is used to prevent bleeding episodes, not just to treat them.
Yet competition has not brought prices down in the way someone “operating at the level of undergrad Econ 101 would expect,” said Jerry Avorn, MD, a professor of medicine at Harvard Medical School, Boston, who studies prescription drug costs.
The problem is that companies have no incentive to lower prices. Patients generally don’t push back because insurers pay the bulk of the cost. And insurers tend not to object because the market for the drugs – expensive as they are – is small and the patients are especially vulnerable.
For drug companies, Dr. Avorn said, “it’s a magical formula: Lifesaving drug, child at risk of bleeding to death – it kind of casts anybody who looks at costs into the role of some evil Scrooge-like person.”
“The insurers don’t want to end up on the front page of the newspaper saying Little Timmy bled to death because his drug wasn’t covered,” he said.
Also, because prices are high across the hemophilia market, no drug company wants to be the one to blink first. “They don’t want to get a price war started and end up at a super low price point,” said Edmund Pezalla, a consultant to pharmaceutical companies and former executive at Aetna.
So, these drugmakers compete not on price but clinical benefits – such as how long the drugs’ effects last – and through intensive marketing. The pool of potential customers is so valuable that companies often vie directly for individual patients.
Manufacturers, as well as specialty pharmacies that sell the drugs, hire patients and parents as recruiters and advisers, hold dinners and holiday parties, offer scholarships to patients, and even run summer camps for children with the disease. The Morris family regularly receives such invitations.
Jonathan Ducore, MD, a pediatric hematologist-oncologist at the University of California, Davis, Hemophilia Treatment Center in Sacramento, said some of his patients are persuaded by drug company presentations to switch medications. ”But the real differences between the drugs are limited,” he said.
Dr. Ducore said he tells patients if he thinks they are being misled by drugmakers about what a product will do. “But even though the tactics may seem a little smarmy, if it’s the patient’s choice, you have to go with it,” said Dr. Ducore, who has been Landon’s doctor since the boy was born.
The first clotting factor products, which came onto the market in the mid-1960s, were derived from human blood plasma, with thousands of donations combined to create one batch. This proved disastrous in the 1980s, when donors unwittingly spread HIV into the blood supply. An estimated 4,000 people with hemophilia – about 40 percent of the patient population in the United States – died from AIDS as a result.
In the 1990s, manufacturers introduced a product that did not carry the disease risk of plasma-based drugs – made by cloning human clotting proteins in animal cells. Companies charged a premium for this ever-more-popular “recombinant factor.”
Recombinant factor is difficult and delicate to make, said Steve Garger, a development scientist at Bayer, which produces two popular factor products at its Berkeley, Calif., plant – including Landon Morris’ drug, Kogenate.
Inside a concrete building on the campus, kidney cells from baby hamsters are grown in stainless-steel vessels called bioreactors, and the clotting factor they produce is then purified in steel tanks kept in cold rooms. Working at full capacity, this factory produces less than a pound of clotting factor each year – but when diluted with other ingredients, it’s enough to treat thousands of patients in 80 countries.
The investment in manufacturing and marketing is only part of the reason for the high cost of the drugs, said Kevin O’Leary, vice president of pricing and contracting at Bayer. Bayer does not simply add up the costs, slap on a profit margin and come up with the price, Mr. O’Leary explained.
Instead, he said, the company begins by talking to insurers, doctors, and patients to get a sense of what value its products bring to the market, especially compared with drugs already available. Bayer then sets a price based on both its investment and the product’s perceived worth. In the end, he said, “we’re charging a price that’s competitive with the other factor products on the market.”
Bayer’s annual sales from its hemophilia drugs were 1.166 billion euros in 2016. That’s the equivalent of about $1.45 billion in the United States.
Pushing back on costs
In Europe, hemophilia drugs cost less than half what they cost in the United States. That’s because payers – usually governments – request bids and pick products based on cost and quality.
Without pushback from insurers in the United States, “the price of any drug in the U.S. is whatever the market will bear as seen by the manufacturer,” said Dr. Avorn of Harvard.
Recently, a few insurance companies have quietly started to push back on costs. Bayer’s Mr. O’Leary said several insurers have approached the company and demanded rebates in exchange for offering the drug to their customers. Mr. O’Leary would not discuss the details because he said the contracts are confidential.
State Medicaid programs, which provide health insurance to low-income Americans and cover about half of hemophilia patients, already receive significant rebates from hemophilia drug manufacturers.
Michelle Rice, a senior vice president at the National Hemophilia Foundation, said she has been working with several insurers to help them manage costs safely. “We understand the need to control costs, but they can’t impede access to the product a patient needs,” she said.
It is not yet clear whether such efforts will work, let alone spread.
Sitting at a picnic bench at a park, Jessica Morris pages through Landon’s insurance documents. Over the past year, his care cost over $120,000. She wonders sometimes what would happen if they lost their coverage.
“How much would you be willing to pay to have your child lead a normal life?” she said. “I don’t think that there’s anything we wouldn’t pay or sacrifice for him.”
It’s a problem she prays they’ll never have to face.
Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of the Kaiser Family Foundation that is not affiliated with Kaiser Permanente. KHN’s coverage of prescription drug development, costs, and pricing is supported by the Laura and John Arnold Foundation.
Hemophilia A treatment gains approval in Europe
The European Commission has approved emicizumab for routine prophylaxis of bleeding episodes in adults and children with hemophilia A with factor VIII inhibitors.
The European Medicines Agency’s Committee for Medicinal Products for Human Use recommended granting marketing authorization to emicizumab (Hemlibra) in January 2018 and it was approved by the Food and Drug Administration in November 2017.
The approvals are based on findings from the HAVEN 1 trial (in 109 adults and adolescents) and the HAVEN 2 trial (in children younger than age 12 years), according to the drug’s maker, Roche. In HAVEN 1 (NCT02622321), emicizumab showed a significant reduction in treatment bleeds (87%), compared with no prophylaxis. And the drug reduced treated bleeds by 79%, compared with previous treatment with bypassing agent prophylaxis. An interim analysis of HAVEN 2 (NCT02795767) of 23 children, showed that 87% of children who received emicizumab prophylaxis had zero treated bleeds, according to Roche.
The European Commission has approved emicizumab for routine prophylaxis of bleeding episodes in adults and children with hemophilia A with factor VIII inhibitors.
The European Medicines Agency’s Committee for Medicinal Products for Human Use recommended granting marketing authorization to emicizumab (Hemlibra) in January 2018 and it was approved by the Food and Drug Administration in November 2017.
The approvals are based on findings from the HAVEN 1 trial (in 109 adults and adolescents) and the HAVEN 2 trial (in children younger than age 12 years), according to the drug’s maker, Roche. In HAVEN 1 (NCT02622321), emicizumab showed a significant reduction in treatment bleeds (87%), compared with no prophylaxis. And the drug reduced treated bleeds by 79%, compared with previous treatment with bypassing agent prophylaxis. An interim analysis of HAVEN 2 (NCT02795767) of 23 children, showed that 87% of children who received emicizumab prophylaxis had zero treated bleeds, according to Roche.
The European Commission has approved emicizumab for routine prophylaxis of bleeding episodes in adults and children with hemophilia A with factor VIII inhibitors.
The European Medicines Agency’s Committee for Medicinal Products for Human Use recommended granting marketing authorization to emicizumab (Hemlibra) in January 2018 and it was approved by the Food and Drug Administration in November 2017.
The approvals are based on findings from the HAVEN 1 trial (in 109 adults and adolescents) and the HAVEN 2 trial (in children younger than age 12 years), according to the drug’s maker, Roche. In HAVEN 1 (NCT02622321), emicizumab showed a significant reduction in treatment bleeds (87%), compared with no prophylaxis. And the drug reduced treated bleeds by 79%, compared with previous treatment with bypassing agent prophylaxis. An interim analysis of HAVEN 2 (NCT02795767) of 23 children, showed that 87% of children who received emicizumab prophylaxis had zero treated bleeds, according to Roche.
States strive to curb costs for a crucial – but exorbitant – hemophilia treatment
The child is well-known in the halls in which state bureaucrats oversee health care for millions of Californians – not by name, but by a number: $21 million.
His medications alone cost state taxpayers that much in a single year, not including other health care. The boy, whose identity has not been released, was California’s most expensive Medicaid patient in recent years. His case was singled out in a tweet last year by the state’s top health care official to highlight the public insurance program’s extraordinary obligations as a backstop for low-income patients.
How on earth can a single child’s treatment cost that much? The answer: He has hemophilia and needs large quantities of a pricey drug – known as clotting factor – that makes blood coagulate.
Hemophilia drugs are among the most costly drugs in the nation, and taxpayers are footing the bill for many patients on Medicaid who could never afford them on their own. Officials in California and other states are doing what they can to manage the costs, but it’s a daunting task that highlights the complexity and secrecy of prescription drug pricing.
Kaiser Health News is examining how America has become a “Medicaid Nation” – where tens of millions of poor and disabled people now rely on the support of the federal and state insurance program. Hemophilia is one those diseases that helps explain its burgeoning cost.
Medications for hemophilia are crucial to patients – overwhelmingly male – with the rare genetic condition that prevents clotting and puts them at great risk of bleeding to death, even from a minor injury. There is no question the drugs prolong and save lives, and state officials are not arguing that they should be withheld.
“It’s a highly vulnerable population,” said Ken Kizer, a veteran federal and state health administrator who formerly oversaw Medi-Cal, California’s version of Medicaid. “If anyone has seen a hemophiliac in crisis, you’re not going to say no.”
But drugmakers profit handsomely, competing vigorously for the limited number of patients.
The U.S. hemophilia market, which serves about 20,000 patients, is worth $4.6 billion a year, according to AllianceBernstein, a research and investment firm.
“There are millions being made out there on these kids – it’s a huge business,” said Doris Quon, MD, medical director of the Orthopaedic Hemophilia Treatment Center at the University of California, Los Angeles.
Contributing to the costs is the fact that there is no cure for hemophilia and no cheaper substitute for blood factor. Factor may be prescribed at high doses for a lifetime, even more so when a patient has an injury or complications.
Nationwide, a third of adults and children living with hemophilia are covered by Medicaid. And the Medicaid program’s three most expensive drugs per prescription are for hemophilia, according to an analysis by the Kaiser Family Foundation. (California Healthline is produced by Kaiser Health News, an editorially independent publication of the foundation.)
In 2015 alone, Medicaid paid about $353 million for prescriptions of Advate, the most commonly prescribed blood-clotting medication for hemophilia – a 273% increase from 2011.
Generally speaking, the price of hemophilia drugs rise as rival drugs hit the market. But, in addition, doctors are prescribing ever more clotting factor for prevention of joint-damaging bleeds and for improved long-term health. The increase in the cost of Advate, for example, was nearly all attributed to increased use.
Tab for 145 kids: $195 million
The California boy whose drugs cost $21 million in a single year was an extreme case, and the circumstances of his care have not been disclosed because of confidentiality protections. Still, medications to treat hemophilia on average annual cost more than $270,000 per patient, according to a 2015 Express Scripts report, and they can easily soar past $1 million annually.
In contrast to more common diseases like hepatitis C, hemophilia treatment is not a state “budget buster” per se: about 4,000 patients live in California. About 1,100 of them are covered by Medi-Cal or two other government-funded programs for chronically ill children in California, according to Jennifer Kent, director of the state Department of Health Care Services and author of last year’s tweet. But the amount of money spent per person dwarfs that spent on people with other serious diseases.
One Stanford University study of 34,000 California kids with severe chronic diseases found that the tiny portion of children who needed blood factor accounted for 41% of the state’s outpatient drug spending on this entire patient population. About $195 million was spent on just 145 kids over a 3-year period, although some of that money came back to the state in rebates from drug companies – a portion of the cost that Medicaid can recoup after purchase.
Caitlin Carroll, director of public affairs for PhRMA, the pharmaceutical industry lobbying group, said high development costs and the complicated and lengthy manufacturing process play a role in how hemophilia drugs are priced. She added that federally mandated rebates significantly reduce the cost of blood factor. They amount to 17% of the average manufacturer price per unit.
Manufacturers also note that some newer and more expensive hemophilia drugs last longer and do not need to be administered as frequently, so they can prove less costly to payers overall.
Even so, some patients require a monumental investment to survive.
‘Extremely fortunate’
Colleen Tuite’s son Kevin, a 7-year-old, has severe hemophilia with a complication known as an inhibitor – an antibody that makes his regular blood-factor infusions less effective. Inhibitors can dramatically increase the cost of care, because massive doses of blood factor or expensive, specialized blood products known as bypassing agents may be needed.
Ms. Tuite and her husband initially were Kevin’s foster parents, then adopted the boy as a toddler. Because he has been a foster child, Kevin qualifies for Medi-Cal until he is 26.
The Monrovia, Calif., family also has private health insurance, which pays for about half of Kevin’s medical bills. These can run upward of $200,000 per month, Ms. Tuite said.
“We definitely would not have been able to adopt him without the help of Medi-Cal,” Ms. Tuite said. “We’ve been extremely fortunate.”
With the support of drug manufacturers and hemophilia advocacy groups, patients and their families have significant political clout. Some experts say they also have a moral claim on public resources: In the early days of the AIDS epidemic, thousands of the nation’s hemophilia patients died after they contracted HIV through transfusions before the virus was eliminated from the blood supply.
State health officials say the costs of hemophilia are hard to anticipate and control, even with rebates.
“We do a really aggressive job of collecting rebates on our pharmacy costs,” said Ms. Kent, California’s top Medicaid official. “But there’s just not any way around blood factor. It is just a very, very expensive product. It’s nonnegotiable for people that require it.”
In 2016, California’s Medicaid program paid at least $205 million for medications used to treat hemophilia, according to a Kaiser Health News analysis of federal Medicaid data. That figure doesn’t account for the federal rebates.
States can negotiate “supplemental” rebates with drugmakers for individual medications – but those must be kept secret under federal and some state laws. Such secrecy is becoming increasingly controversial as states continue to confront spiraling drug prices.
Limited options for states
In 2016, Pfizer sued Texas’ state health agency for giving data on the drug company’s supplemental Medicaid rebates to state lawmakers who requested it. The drugmaker alleged that releasing the confidential information would undermine the company’s competitiveness and give away trade secrets, and warned that the discounts it gave Texas could disappear.
In early October, a judge ruled that lawmakers should be able to obtain some of that data, noting dryly that “in Pfizer’s view, legislators are not necessary to carry out the state’s Medicaid program.”
Instead of seeking additional rebates from manufacturers for blood factor, some states, including Washington and Oregon, have chosen to require patients to get their blood factor from federally designated Hemophilia Treatment Centers only. That allows state Medicaid programs to take advantage of a federal drug-discount program known as “340B.”
However, officials in California said they studied that option and determined it wouldn’t save them any more money than the rebates they negotiate with drugmakers.
Whatever their approach, state health officials say they are struggling against forces they are nearly powerless to change.
“There aren’t a lot of options available to Medicaid programs in terms of controlling costs, because we don’t set the initial costs,” said Deborah Weston, pharmacy program manager for Oregon’s Medicaid program.
Kaiser Health News data correspondent Sydney Lupkin contributed to this report. KHN’s coverage of these topics is supported by Laura and John Arnold Foundation and Heising-Simons Foundation. This story was produced by Kaiser Health News, which publishes California Healthline, a service of the California Health Care Foundation. Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of the Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
The child is well-known in the halls in which state bureaucrats oversee health care for millions of Californians – not by name, but by a number: $21 million.
His medications alone cost state taxpayers that much in a single year, not including other health care. The boy, whose identity has not been released, was California’s most expensive Medicaid patient in recent years. His case was singled out in a tweet last year by the state’s top health care official to highlight the public insurance program’s extraordinary obligations as a backstop for low-income patients.
How on earth can a single child’s treatment cost that much? The answer: He has hemophilia and needs large quantities of a pricey drug – known as clotting factor – that makes blood coagulate.
Hemophilia drugs are among the most costly drugs in the nation, and taxpayers are footing the bill for many patients on Medicaid who could never afford them on their own. Officials in California and other states are doing what they can to manage the costs, but it’s a daunting task that highlights the complexity and secrecy of prescription drug pricing.
Kaiser Health News is examining how America has become a “Medicaid Nation” – where tens of millions of poor and disabled people now rely on the support of the federal and state insurance program. Hemophilia is one those diseases that helps explain its burgeoning cost.
Medications for hemophilia are crucial to patients – overwhelmingly male – with the rare genetic condition that prevents clotting and puts them at great risk of bleeding to death, even from a minor injury. There is no question the drugs prolong and save lives, and state officials are not arguing that they should be withheld.
“It’s a highly vulnerable population,” said Ken Kizer, a veteran federal and state health administrator who formerly oversaw Medi-Cal, California’s version of Medicaid. “If anyone has seen a hemophiliac in crisis, you’re not going to say no.”
But drugmakers profit handsomely, competing vigorously for the limited number of patients.
The U.S. hemophilia market, which serves about 20,000 patients, is worth $4.6 billion a year, according to AllianceBernstein, a research and investment firm.
“There are millions being made out there on these kids – it’s a huge business,” said Doris Quon, MD, medical director of the Orthopaedic Hemophilia Treatment Center at the University of California, Los Angeles.
Contributing to the costs is the fact that there is no cure for hemophilia and no cheaper substitute for blood factor. Factor may be prescribed at high doses for a lifetime, even more so when a patient has an injury or complications.
Nationwide, a third of adults and children living with hemophilia are covered by Medicaid. And the Medicaid program’s three most expensive drugs per prescription are for hemophilia, according to an analysis by the Kaiser Family Foundation. (California Healthline is produced by Kaiser Health News, an editorially independent publication of the foundation.)
In 2015 alone, Medicaid paid about $353 million for prescriptions of Advate, the most commonly prescribed blood-clotting medication for hemophilia – a 273% increase from 2011.
Generally speaking, the price of hemophilia drugs rise as rival drugs hit the market. But, in addition, doctors are prescribing ever more clotting factor for prevention of joint-damaging bleeds and for improved long-term health. The increase in the cost of Advate, for example, was nearly all attributed to increased use.
Tab for 145 kids: $195 million
The California boy whose drugs cost $21 million in a single year was an extreme case, and the circumstances of his care have not been disclosed because of confidentiality protections. Still, medications to treat hemophilia on average annual cost more than $270,000 per patient, according to a 2015 Express Scripts report, and they can easily soar past $1 million annually.
In contrast to more common diseases like hepatitis C, hemophilia treatment is not a state “budget buster” per se: about 4,000 patients live in California. About 1,100 of them are covered by Medi-Cal or two other government-funded programs for chronically ill children in California, according to Jennifer Kent, director of the state Department of Health Care Services and author of last year’s tweet. But the amount of money spent per person dwarfs that spent on people with other serious diseases.
One Stanford University study of 34,000 California kids with severe chronic diseases found that the tiny portion of children who needed blood factor accounted for 41% of the state’s outpatient drug spending on this entire patient population. About $195 million was spent on just 145 kids over a 3-year period, although some of that money came back to the state in rebates from drug companies – a portion of the cost that Medicaid can recoup after purchase.
Caitlin Carroll, director of public affairs for PhRMA, the pharmaceutical industry lobbying group, said high development costs and the complicated and lengthy manufacturing process play a role in how hemophilia drugs are priced. She added that federally mandated rebates significantly reduce the cost of blood factor. They amount to 17% of the average manufacturer price per unit.
Manufacturers also note that some newer and more expensive hemophilia drugs last longer and do not need to be administered as frequently, so they can prove less costly to payers overall.
Even so, some patients require a monumental investment to survive.
‘Extremely fortunate’
Colleen Tuite’s son Kevin, a 7-year-old, has severe hemophilia with a complication known as an inhibitor – an antibody that makes his regular blood-factor infusions less effective. Inhibitors can dramatically increase the cost of care, because massive doses of blood factor or expensive, specialized blood products known as bypassing agents may be needed.
Ms. Tuite and her husband initially were Kevin’s foster parents, then adopted the boy as a toddler. Because he has been a foster child, Kevin qualifies for Medi-Cal until he is 26.
The Monrovia, Calif., family also has private health insurance, which pays for about half of Kevin’s medical bills. These can run upward of $200,000 per month, Ms. Tuite said.
“We definitely would not have been able to adopt him without the help of Medi-Cal,” Ms. Tuite said. “We’ve been extremely fortunate.”
With the support of drug manufacturers and hemophilia advocacy groups, patients and their families have significant political clout. Some experts say they also have a moral claim on public resources: In the early days of the AIDS epidemic, thousands of the nation’s hemophilia patients died after they contracted HIV through transfusions before the virus was eliminated from the blood supply.
State health officials say the costs of hemophilia are hard to anticipate and control, even with rebates.
“We do a really aggressive job of collecting rebates on our pharmacy costs,” said Ms. Kent, California’s top Medicaid official. “But there’s just not any way around blood factor. It is just a very, very expensive product. It’s nonnegotiable for people that require it.”
In 2016, California’s Medicaid program paid at least $205 million for medications used to treat hemophilia, according to a Kaiser Health News analysis of federal Medicaid data. That figure doesn’t account for the federal rebates.
States can negotiate “supplemental” rebates with drugmakers for individual medications – but those must be kept secret under federal and some state laws. Such secrecy is becoming increasingly controversial as states continue to confront spiraling drug prices.
Limited options for states
In 2016, Pfizer sued Texas’ state health agency for giving data on the drug company’s supplemental Medicaid rebates to state lawmakers who requested it. The drugmaker alleged that releasing the confidential information would undermine the company’s competitiveness and give away trade secrets, and warned that the discounts it gave Texas could disappear.
In early October, a judge ruled that lawmakers should be able to obtain some of that data, noting dryly that “in Pfizer’s view, legislators are not necessary to carry out the state’s Medicaid program.”
Instead of seeking additional rebates from manufacturers for blood factor, some states, including Washington and Oregon, have chosen to require patients to get their blood factor from federally designated Hemophilia Treatment Centers only. That allows state Medicaid programs to take advantage of a federal drug-discount program known as “340B.”
However, officials in California said they studied that option and determined it wouldn’t save them any more money than the rebates they negotiate with drugmakers.
Whatever their approach, state health officials say they are struggling against forces they are nearly powerless to change.
“There aren’t a lot of options available to Medicaid programs in terms of controlling costs, because we don’t set the initial costs,” said Deborah Weston, pharmacy program manager for Oregon’s Medicaid program.
Kaiser Health News data correspondent Sydney Lupkin contributed to this report. KHN’s coverage of these topics is supported by Laura and John Arnold Foundation and Heising-Simons Foundation. This story was produced by Kaiser Health News, which publishes California Healthline, a service of the California Health Care Foundation. Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of the Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
The child is well-known in the halls in which state bureaucrats oversee health care for millions of Californians – not by name, but by a number: $21 million.
His medications alone cost state taxpayers that much in a single year, not including other health care. The boy, whose identity has not been released, was California’s most expensive Medicaid patient in recent years. His case was singled out in a tweet last year by the state’s top health care official to highlight the public insurance program’s extraordinary obligations as a backstop for low-income patients.
How on earth can a single child’s treatment cost that much? The answer: He has hemophilia and needs large quantities of a pricey drug – known as clotting factor – that makes blood coagulate.
Hemophilia drugs are among the most costly drugs in the nation, and taxpayers are footing the bill for many patients on Medicaid who could never afford them on their own. Officials in California and other states are doing what they can to manage the costs, but it’s a daunting task that highlights the complexity and secrecy of prescription drug pricing.
Kaiser Health News is examining how America has become a “Medicaid Nation” – where tens of millions of poor and disabled people now rely on the support of the federal and state insurance program. Hemophilia is one those diseases that helps explain its burgeoning cost.
Medications for hemophilia are crucial to patients – overwhelmingly male – with the rare genetic condition that prevents clotting and puts them at great risk of bleeding to death, even from a minor injury. There is no question the drugs prolong and save lives, and state officials are not arguing that they should be withheld.
“It’s a highly vulnerable population,” said Ken Kizer, a veteran federal and state health administrator who formerly oversaw Medi-Cal, California’s version of Medicaid. “If anyone has seen a hemophiliac in crisis, you’re not going to say no.”
But drugmakers profit handsomely, competing vigorously for the limited number of patients.
The U.S. hemophilia market, which serves about 20,000 patients, is worth $4.6 billion a year, according to AllianceBernstein, a research and investment firm.
“There are millions being made out there on these kids – it’s a huge business,” said Doris Quon, MD, medical director of the Orthopaedic Hemophilia Treatment Center at the University of California, Los Angeles.
Contributing to the costs is the fact that there is no cure for hemophilia and no cheaper substitute for blood factor. Factor may be prescribed at high doses for a lifetime, even more so when a patient has an injury or complications.
Nationwide, a third of adults and children living with hemophilia are covered by Medicaid. And the Medicaid program’s three most expensive drugs per prescription are for hemophilia, according to an analysis by the Kaiser Family Foundation. (California Healthline is produced by Kaiser Health News, an editorially independent publication of the foundation.)
In 2015 alone, Medicaid paid about $353 million for prescriptions of Advate, the most commonly prescribed blood-clotting medication for hemophilia – a 273% increase from 2011.
Generally speaking, the price of hemophilia drugs rise as rival drugs hit the market. But, in addition, doctors are prescribing ever more clotting factor for prevention of joint-damaging bleeds and for improved long-term health. The increase in the cost of Advate, for example, was nearly all attributed to increased use.
Tab for 145 kids: $195 million
The California boy whose drugs cost $21 million in a single year was an extreme case, and the circumstances of his care have not been disclosed because of confidentiality protections. Still, medications to treat hemophilia on average annual cost more than $270,000 per patient, according to a 2015 Express Scripts report, and they can easily soar past $1 million annually.
In contrast to more common diseases like hepatitis C, hemophilia treatment is not a state “budget buster” per se: about 4,000 patients live in California. About 1,100 of them are covered by Medi-Cal or two other government-funded programs for chronically ill children in California, according to Jennifer Kent, director of the state Department of Health Care Services and author of last year’s tweet. But the amount of money spent per person dwarfs that spent on people with other serious diseases.
One Stanford University study of 34,000 California kids with severe chronic diseases found that the tiny portion of children who needed blood factor accounted for 41% of the state’s outpatient drug spending on this entire patient population. About $195 million was spent on just 145 kids over a 3-year period, although some of that money came back to the state in rebates from drug companies – a portion of the cost that Medicaid can recoup after purchase.
Caitlin Carroll, director of public affairs for PhRMA, the pharmaceutical industry lobbying group, said high development costs and the complicated and lengthy manufacturing process play a role in how hemophilia drugs are priced. She added that federally mandated rebates significantly reduce the cost of blood factor. They amount to 17% of the average manufacturer price per unit.
Manufacturers also note that some newer and more expensive hemophilia drugs last longer and do not need to be administered as frequently, so they can prove less costly to payers overall.
Even so, some patients require a monumental investment to survive.
‘Extremely fortunate’
Colleen Tuite’s son Kevin, a 7-year-old, has severe hemophilia with a complication known as an inhibitor – an antibody that makes his regular blood-factor infusions less effective. Inhibitors can dramatically increase the cost of care, because massive doses of blood factor or expensive, specialized blood products known as bypassing agents may be needed.
Ms. Tuite and her husband initially were Kevin’s foster parents, then adopted the boy as a toddler. Because he has been a foster child, Kevin qualifies for Medi-Cal until he is 26.
The Monrovia, Calif., family also has private health insurance, which pays for about half of Kevin’s medical bills. These can run upward of $200,000 per month, Ms. Tuite said.
“We definitely would not have been able to adopt him without the help of Medi-Cal,” Ms. Tuite said. “We’ve been extremely fortunate.”
With the support of drug manufacturers and hemophilia advocacy groups, patients and their families have significant political clout. Some experts say they also have a moral claim on public resources: In the early days of the AIDS epidemic, thousands of the nation’s hemophilia patients died after they contracted HIV through transfusions before the virus was eliminated from the blood supply.
State health officials say the costs of hemophilia are hard to anticipate and control, even with rebates.
“We do a really aggressive job of collecting rebates on our pharmacy costs,” said Ms. Kent, California’s top Medicaid official. “But there’s just not any way around blood factor. It is just a very, very expensive product. It’s nonnegotiable for people that require it.”
In 2016, California’s Medicaid program paid at least $205 million for medications used to treat hemophilia, according to a Kaiser Health News analysis of federal Medicaid data. That figure doesn’t account for the federal rebates.
States can negotiate “supplemental” rebates with drugmakers for individual medications – but those must be kept secret under federal and some state laws. Such secrecy is becoming increasingly controversial as states continue to confront spiraling drug prices.
Limited options for states
In 2016, Pfizer sued Texas’ state health agency for giving data on the drug company’s supplemental Medicaid rebates to state lawmakers who requested it. The drugmaker alleged that releasing the confidential information would undermine the company’s competitiveness and give away trade secrets, and warned that the discounts it gave Texas could disappear.
In early October, a judge ruled that lawmakers should be able to obtain some of that data, noting dryly that “in Pfizer’s view, legislators are not necessary to carry out the state’s Medicaid program.”
Instead of seeking additional rebates from manufacturers for blood factor, some states, including Washington and Oregon, have chosen to require patients to get their blood factor from federally designated Hemophilia Treatment Centers only. That allows state Medicaid programs to take advantage of a federal drug-discount program known as “340B.”
However, officials in California said they studied that option and determined it wouldn’t save them any more money than the rebates they negotiate with drugmakers.
Whatever their approach, state health officials say they are struggling against forces they are nearly powerless to change.
“There aren’t a lot of options available to Medicaid programs in terms of controlling costs, because we don’t set the initial costs,” said Deborah Weston, pharmacy program manager for Oregon’s Medicaid program.
Kaiser Health News data correspondent Sydney Lupkin contributed to this report. KHN’s coverage of these topics is supported by Laura and John Arnold Foundation and Heising-Simons Foundation. This story was produced by Kaiser Health News, which publishes California Healthline, a service of the California Health Care Foundation. Kaiser Health News is a nonprofit news service covering health issues. It is an editorially independent program of the Kaiser Family Foundation that is not affiliated with Kaiser Permanente.
PK dosing software available in US

Pharmacokinetic (PK) dosing software intended for use in patients with hemophilia A is now available in the US.
This free, web-based, prescription software—myPKFiT for ADVATE—is intended to aid healthcare professionals in personalizing the dose and schedule of ADVATE, a full-length recombinant factor VIII product.
The software was cleared by the US Food and Drug Administration (FDA) for use in hemophilia A patients age 16 and older who weigh at least 45 kg and are receiving prophylaxis with ADVATE.
The myPKFiT software generates ADVATE dosage amount and frequency recommendations using a patient’s age and body weight information, as well as local laboratory factor VIII one-stage clotting activity measurements of sparse samples collected from the patient.
A minimum of 2 sparse sampling points are required at the recommended 3 to 4 hours (± 30 minutes) and at 24 to 32 hours (±1 hour) post-infusion.
The software output may be used to guide ADVATE use to maintain factor VIII activity levels at or above a user-specified minimum of 1% to 3% above natural baseline, in accordance with the FDA-approved dosing recommendations for ADVATE.
myPKFiT should only be used to evaluate prophylactic dosing regimens for hemophilia A patients treated with ADVATE.
The software should not be used for patients who have developed neutralizing antibodies to factor VIII products and is not indicated for use in patients with von Willebrand disease.
“A version of myPKFiT has already been widely adopted in Europe since 2014, and we’ve seen how important it has been in helping physicians develop personalized dosing regimens tailored to the specific needs of their patients,” said Andreas Busch, global head of research and development at Shire, the company behind myPKFiT for ADVATE.
“As part of our commitment to precision medicine, we are pleased to bring this innovative application to physicians and patients in the United States.”
More information on the myPKFiT software is available on the ADVATE website and the myPKFiT site.
Pharmacokinetic (PK) dosing software intended for use in patients with hemophilia A is now available in the US.
This free, web-based, prescription software—myPKFiT for ADVATE—is intended to aid healthcare professionals in personalizing the dose and schedule of ADVATE, a full-length recombinant factor VIII product.
The software was cleared by the US Food and Drug Administration (FDA) for use in hemophilia A patients age 16 and older who weigh at least 45 kg and are receiving prophylaxis with ADVATE.
The myPKFiT software generates ADVATE dosage amount and frequency recommendations using a patient’s age and body weight information, as well as local laboratory factor VIII one-stage clotting activity measurements of sparse samples collected from the patient.
A minimum of 2 sparse sampling points are required at the recommended 3 to 4 hours (± 30 minutes) and at 24 to 32 hours (±1 hour) post-infusion.
The software output may be used to guide ADVATE use to maintain factor VIII activity levels at or above a user-specified minimum of 1% to 3% above natural baseline, in accordance with the FDA-approved dosing recommendations for ADVATE.
myPKFiT should only be used to evaluate prophylactic dosing regimens for hemophilia A patients treated with ADVATE.
The software should not be used for patients who have developed neutralizing antibodies to factor VIII products and is not indicated for use in patients with von Willebrand disease.
“A version of myPKFiT has already been widely adopted in Europe since 2014, and we’ve seen how important it has been in helping physicians develop personalized dosing regimens tailored to the specific needs of their patients,” said Andreas Busch, global head of research and development at Shire, the company behind myPKFiT for ADVATE.
“As part of our commitment to precision medicine, we are pleased to bring this innovative application to physicians and patients in the United States.”
More information on the myPKFiT software is available on the ADVATE website and the myPKFiT site.
Pharmacokinetic (PK) dosing software intended for use in patients with hemophilia A is now available in the US.
This free, web-based, prescription software—myPKFiT for ADVATE—is intended to aid healthcare professionals in personalizing the dose and schedule of ADVATE, a full-length recombinant factor VIII product.
The software was cleared by the US Food and Drug Administration (FDA) for use in hemophilia A patients age 16 and older who weigh at least 45 kg and are receiving prophylaxis with ADVATE.
The myPKFiT software generates ADVATE dosage amount and frequency recommendations using a patient’s age and body weight information, as well as local laboratory factor VIII one-stage clotting activity measurements of sparse samples collected from the patient.
A minimum of 2 sparse sampling points are required at the recommended 3 to 4 hours (± 30 minutes) and at 24 to 32 hours (±1 hour) post-infusion.
The software output may be used to guide ADVATE use to maintain factor VIII activity levels at or above a user-specified minimum of 1% to 3% above natural baseline, in accordance with the FDA-approved dosing recommendations for ADVATE.
myPKFiT should only be used to evaluate prophylactic dosing regimens for hemophilia A patients treated with ADVATE.
The software should not be used for patients who have developed neutralizing antibodies to factor VIII products and is not indicated for use in patients with von Willebrand disease.
“A version of myPKFiT has already been widely adopted in Europe since 2014, and we’ve seen how important it has been in helping physicians develop personalized dosing regimens tailored to the specific needs of their patients,” said Andreas Busch, global head of research and development at Shire, the company behind myPKFiT for ADVATE.
“As part of our commitment to precision medicine, we are pleased to bring this innovative application to physicians and patients in the United States.”
More information on the myPKFiT software is available on the ADVATE website and the myPKFiT site.
EC approves emicizumab for hemophilia A with inhibitors
The European Commission (EC) has granted marketing authorization for emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
This means emicizumab is approved for use in the European Union for routine prophylaxis of bleeding episodes in patients of all ages who have hemophilia A and factor VIII inhibitors.
The recommended dose of emicizumab is 3 mg/kg once a week for the first 4 weeks, followed by 1.5 mg/kg once a week.
Emicizumab is designed to bring together factor IXa and factor X, proteins required to activate the natural coagulation cascade and restore the blood clotting process for patients with hemophilia A.
Emicizumab was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Chugai, Roche, and Genentech.
The EC’s decision to authorize marketing of emicizumab is based on results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
The European Commission (EC) has granted marketing authorization for emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
This means emicizumab is approved for use in the European Union for routine prophylaxis of bleeding episodes in patients of all ages who have hemophilia A and factor VIII inhibitors.
The recommended dose of emicizumab is 3 mg/kg once a week for the first 4 weeks, followed by 1.5 mg/kg once a week.
Emicizumab is designed to bring together factor IXa and factor X, proteins required to activate the natural coagulation cascade and restore the blood clotting process for patients with hemophilia A.
Emicizumab was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Chugai, Roche, and Genentech.
The EC’s decision to authorize marketing of emicizumab is based on results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
The European Commission (EC) has granted marketing authorization for emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
This means emicizumab is approved for use in the European Union for routine prophylaxis of bleeding episodes in patients of all ages who have hemophilia A and factor VIII inhibitors.
The recommended dose of emicizumab is 3 mg/kg once a week for the first 4 weeks, followed by 1.5 mg/kg once a week.
Emicizumab is designed to bring together factor IXa and factor X, proteins required to activate the natural coagulation cascade and restore the blood clotting process for patients with hemophilia A.
Emicizumab was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Chugai, Roche, and Genentech.
The EC’s decision to authorize marketing of emicizumab is based on results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.