User login
Drug granted fast track designation for PNH

The US Food and Drug Administration (FDA) has granted fast track designation to Coversin™ for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) in patients who have polymorphisms conferring eculizumab resistance.
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is a second-generation complement inhibitor that acts on complement component C5, preventing release of C5a and formation of C5b-9, and also independently inhibits LTB4 activity.
Coversin is being developed by Akari Therapeutics.
Akari is evaluating Coversin in a pair of phase 2 trials.
In the first trial, researchers are evaluating Coversin in patients with PNH who have never received a complement-blocking therapy. Interim results from this ongoing trial are scheduled to be presented at Akari’s Research and Development Day on April 24 in New York, New York.
In the second phase 2 trial, researchers are evaluating Coversin in patients with PNH who have C5 polymorphisms that confer resistance to eculizumab.
One patient has been enrolled in this trial and has received Coversin for over a year. The treatment has resulted in significant lactate dehydrogenase reduction and complete complement blockade.
About fast track designation
The FDA created the fast track program to facilitate the development and expedite the review of drugs that show promise for treating serious or life-threatening diseases and address unmet medical needs.
Companies developing drugs that receive fast track designation benefit from more frequent communications and meetings with the FDA to review their drug’s development plan, including the design of proposed clinical trials, use of biomarkers, and the extent of data needed for approval.
Drugs with fast track designation may qualify for priority review as well, if relevant criteria are met. Priority review shortens the FDA review process from 10 months to 6 months.
Fast track designation also allows for a rolling review process, whereby completed sections of the investigational new drug application can be submitted for FDA review as they become available, instead of waiting for all sections to be completed. ![]()
The US Food and Drug Administration (FDA) has granted fast track designation to Coversin™ for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) in patients who have polymorphisms conferring eculizumab resistance.
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is a second-generation complement inhibitor that acts on complement component C5, preventing release of C5a and formation of C5b-9, and also independently inhibits LTB4 activity.
Coversin is being developed by Akari Therapeutics.
Akari is evaluating Coversin in a pair of phase 2 trials.
In the first trial, researchers are evaluating Coversin in patients with PNH who have never received a complement-blocking therapy. Interim results from this ongoing trial are scheduled to be presented at Akari’s Research and Development Day on April 24 in New York, New York.
In the second phase 2 trial, researchers are evaluating Coversin in patients with PNH who have C5 polymorphisms that confer resistance to eculizumab.
One patient has been enrolled in this trial and has received Coversin for over a year. The treatment has resulted in significant lactate dehydrogenase reduction and complete complement blockade.
About fast track designation
The FDA created the fast track program to facilitate the development and expedite the review of drugs that show promise for treating serious or life-threatening diseases and address unmet medical needs.
Companies developing drugs that receive fast track designation benefit from more frequent communications and meetings with the FDA to review their drug’s development plan, including the design of proposed clinical trials, use of biomarkers, and the extent of data needed for approval.
Drugs with fast track designation may qualify for priority review as well, if relevant criteria are met. Priority review shortens the FDA review process from 10 months to 6 months.
Fast track designation also allows for a rolling review process, whereby completed sections of the investigational new drug application can be submitted for FDA review as they become available, instead of waiting for all sections to be completed. ![]()
The US Food and Drug Administration (FDA) has granted fast track designation to Coversin™ for the treatment of paroxysmal nocturnal hemoglobinuria (PNH) in patients who have polymorphisms conferring eculizumab resistance.
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is a second-generation complement inhibitor that acts on complement component C5, preventing release of C5a and formation of C5b-9, and also independently inhibits LTB4 activity.
Coversin is being developed by Akari Therapeutics.
Akari is evaluating Coversin in a pair of phase 2 trials.
In the first trial, researchers are evaluating Coversin in patients with PNH who have never received a complement-blocking therapy. Interim results from this ongoing trial are scheduled to be presented at Akari’s Research and Development Day on April 24 in New York, New York.
In the second phase 2 trial, researchers are evaluating Coversin in patients with PNH who have C5 polymorphisms that confer resistance to eculizumab.
One patient has been enrolled in this trial and has received Coversin for over a year. The treatment has resulted in significant lactate dehydrogenase reduction and complete complement blockade.
About fast track designation
The FDA created the fast track program to facilitate the development and expedite the review of drugs that show promise for treating serious or life-threatening diseases and address unmet medical needs.
Companies developing drugs that receive fast track designation benefit from more frequent communications and meetings with the FDA to review their drug’s development plan, including the design of proposed clinical trials, use of biomarkers, and the extent of data needed for approval.
Drugs with fast track designation may qualify for priority review as well, if relevant criteria are met. Priority review shortens the FDA review process from 10 months to 6 months.
Fast track designation also allows for a rolling review process, whereby completed sections of the investigational new drug application can be submitted for FDA review as they become available, instead of waiting for all sections to be completed. ![]()
Replacement factors, bypassing agents safely manage fitusiran bleed events
Fitusiran appears to promote hemostasis and reduce the frequency of bleeding in patients with hemophilia. In a phase I trial of the investigational agent, breakthrough bleeds were treated effectively and safely with replacement factor or bypassing agent.
Bleed events were rare among patients achieving target antithrombin lowering of greater than 75% on fitusiran. Those that did occur were treated with factor concentrates, including recombinant Factor VIII or recombinant Factor IX, or with bypassing agents, including recombinant Factor VIIa or activated prothrombin complex–concentrates, Savita Rangarajan, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The study included 41 patients with hemophilia A or B – 25 patients with inhibitors and 16 without inhibitors – who received either 50 mg or 80 mg of fitusiran. Early multiple ascending dose–cohorts received weekly subcutaneous dosing, and later cohorts received monthly dosing. All patients tolerated treatment well, with no serious adverse events related to the study drug. No thromboembolic events occurred, and the majority of adverse events were mild or moderate in severity, she noted.
Among patients with inhibitors, eight bleeds occurred in five patients with hemophilia A who were treated with Factor VIII, and three bleeds occurred in two patients with hemophilia B who were treated with Factor IX. Among those without inhibitors, six bleeds occurred in three patients treated with activated prothrombin complex–concentrates, and four occurred in three patients treated with recombinant Factor VIIa, said Dr. Rangarajan of Hampshire Hospitals NHS Foundation Trust, Basingstoke, England.
The ranges of factor replacement used per injection were 7-32 IU/kg of Factor VIII and 7-43 IU/kg of Factor IX.
The ranges of bypassing agents used per injection were 14-75 U/kg of activated prothrombin complex–concentrates (mean, 2.2 administrations per bleed) and 93-133 μg/kg of recombinant Factor VIIa (mean, 1.5 administrations per bleed), she said.
Doses of the factor concentrates and bypassing agents used were at or below those recommended by the World Federation of Hemophilia.
This phase I study of fitusiran, which targets and lowers antithrombin to improve thrombin generation and promote hemostasis in patients with hemophilia, is being conducted in four parts: Part A with healthy volunteers, parts B and C with patients with moderate to severe hemophilia A or B, and part D with patients with hemophilia A or B with inhibitors.
Findings from the current exploratory analysis of the data are encouraging as they demonstrate good treatment effect in the absence of identified safety concerns, Dr. Rangarajan said, noting that fitusiran should advance to pivotal studies in 2017 and that data on bleed management from a phase I and phase II open label extension will guide protocol on bleed management in phase III.
Dr. Rangarajan has received grant or research support from Alnylam Pharmaceuticals, BioMarin Pharmaceutical, Novo Nordisk, Pfizer, and Shire.
Fitusiran appears to promote hemostasis and reduce the frequency of bleeding in patients with hemophilia. In a phase I trial of the investigational agent, breakthrough bleeds were treated effectively and safely with replacement factor or bypassing agent.
Bleed events were rare among patients achieving target antithrombin lowering of greater than 75% on fitusiran. Those that did occur were treated with factor concentrates, including recombinant Factor VIII or recombinant Factor IX, or with bypassing agents, including recombinant Factor VIIa or activated prothrombin complex–concentrates, Savita Rangarajan, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The study included 41 patients with hemophilia A or B – 25 patients with inhibitors and 16 without inhibitors – who received either 50 mg or 80 mg of fitusiran. Early multiple ascending dose–cohorts received weekly subcutaneous dosing, and later cohorts received monthly dosing. All patients tolerated treatment well, with no serious adverse events related to the study drug. No thromboembolic events occurred, and the majority of adverse events were mild or moderate in severity, she noted.
Among patients with inhibitors, eight bleeds occurred in five patients with hemophilia A who were treated with Factor VIII, and three bleeds occurred in two patients with hemophilia B who were treated with Factor IX. Among those without inhibitors, six bleeds occurred in three patients treated with activated prothrombin complex–concentrates, and four occurred in three patients treated with recombinant Factor VIIa, said Dr. Rangarajan of Hampshire Hospitals NHS Foundation Trust, Basingstoke, England.
The ranges of factor replacement used per injection were 7-32 IU/kg of Factor VIII and 7-43 IU/kg of Factor IX.
The ranges of bypassing agents used per injection were 14-75 U/kg of activated prothrombin complex–concentrates (mean, 2.2 administrations per bleed) and 93-133 μg/kg of recombinant Factor VIIa (mean, 1.5 administrations per bleed), she said.
Doses of the factor concentrates and bypassing agents used were at or below those recommended by the World Federation of Hemophilia.
This phase I study of fitusiran, which targets and lowers antithrombin to improve thrombin generation and promote hemostasis in patients with hemophilia, is being conducted in four parts: Part A with healthy volunteers, parts B and C with patients with moderate to severe hemophilia A or B, and part D with patients with hemophilia A or B with inhibitors.
Findings from the current exploratory analysis of the data are encouraging as they demonstrate good treatment effect in the absence of identified safety concerns, Dr. Rangarajan said, noting that fitusiran should advance to pivotal studies in 2017 and that data on bleed management from a phase I and phase II open label extension will guide protocol on bleed management in phase III.
Dr. Rangarajan has received grant or research support from Alnylam Pharmaceuticals, BioMarin Pharmaceutical, Novo Nordisk, Pfizer, and Shire.
Fitusiran appears to promote hemostasis and reduce the frequency of bleeding in patients with hemophilia. In a phase I trial of the investigational agent, breakthrough bleeds were treated effectively and safely with replacement factor or bypassing agent.
Bleed events were rare among patients achieving target antithrombin lowering of greater than 75% on fitusiran. Those that did occur were treated with factor concentrates, including recombinant Factor VIII or recombinant Factor IX, or with bypassing agents, including recombinant Factor VIIa or activated prothrombin complex–concentrates, Savita Rangarajan, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The study included 41 patients with hemophilia A or B – 25 patients with inhibitors and 16 without inhibitors – who received either 50 mg or 80 mg of fitusiran. Early multiple ascending dose–cohorts received weekly subcutaneous dosing, and later cohorts received monthly dosing. All patients tolerated treatment well, with no serious adverse events related to the study drug. No thromboembolic events occurred, and the majority of adverse events were mild or moderate in severity, she noted.
Among patients with inhibitors, eight bleeds occurred in five patients with hemophilia A who were treated with Factor VIII, and three bleeds occurred in two patients with hemophilia B who were treated with Factor IX. Among those without inhibitors, six bleeds occurred in three patients treated with activated prothrombin complex–concentrates, and four occurred in three patients treated with recombinant Factor VIIa, said Dr. Rangarajan of Hampshire Hospitals NHS Foundation Trust, Basingstoke, England.
The ranges of factor replacement used per injection were 7-32 IU/kg of Factor VIII and 7-43 IU/kg of Factor IX.
The ranges of bypassing agents used per injection were 14-75 U/kg of activated prothrombin complex–concentrates (mean, 2.2 administrations per bleed) and 93-133 μg/kg of recombinant Factor VIIa (mean, 1.5 administrations per bleed), she said.
Doses of the factor concentrates and bypassing agents used were at or below those recommended by the World Federation of Hemophilia.
This phase I study of fitusiran, which targets and lowers antithrombin to improve thrombin generation and promote hemostasis in patients with hemophilia, is being conducted in four parts: Part A with healthy volunteers, parts B and C with patients with moderate to severe hemophilia A or B, and part D with patients with hemophilia A or B with inhibitors.
Findings from the current exploratory analysis of the data are encouraging as they demonstrate good treatment effect in the absence of identified safety concerns, Dr. Rangarajan said, noting that fitusiran should advance to pivotal studies in 2017 and that data on bleed management from a phase I and phase II open label extension will guide protocol on bleed management in phase III.
Dr. Rangarajan has received grant or research support from Alnylam Pharmaceuticals, BioMarin Pharmaceutical, Novo Nordisk, Pfizer, and Shire.
Key clinical point:
Major finding: 21 bleeds occurred in 13 patients, and all were treated effectively and safely.
Data source: An exploratory analysis of data from a four-part phase I trial.
Disclosures: Dr. Rangarajan has received grant or research support from Alnylam Pharmaceuticals, BioMarin Pharmaceutical, Novo Nordisk, Pfizer, and Shire.
Hemophilia A and B: An Overview
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).

MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
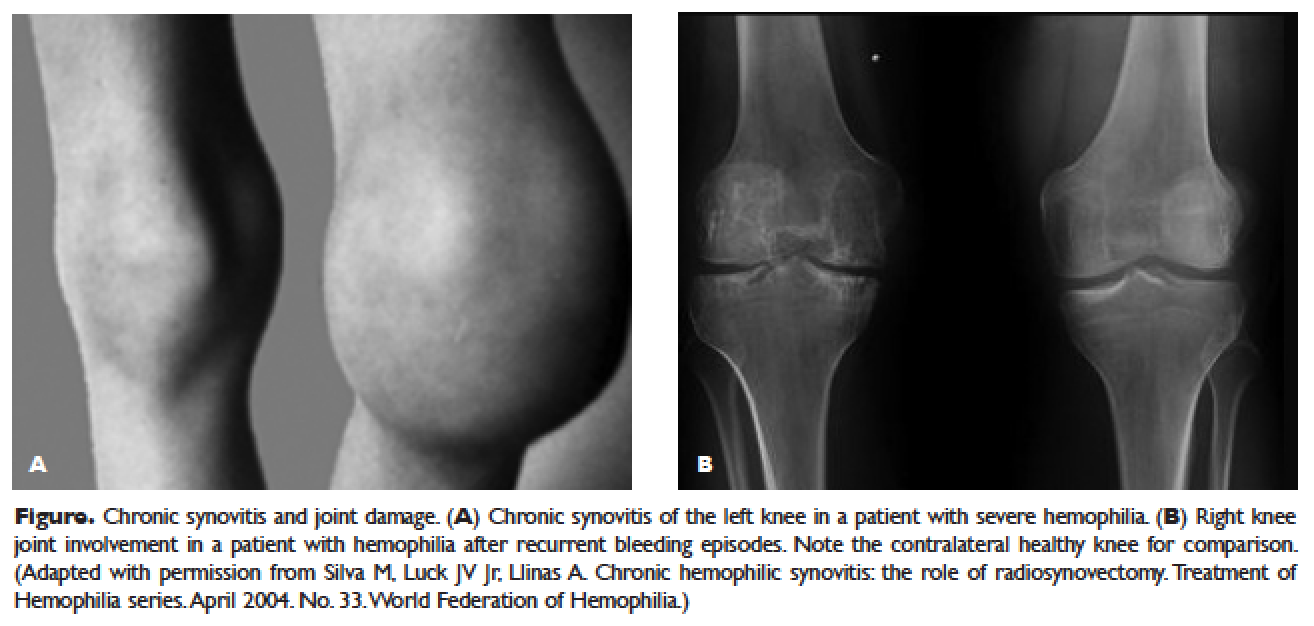
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.

Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82

ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).
MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.
LIFE-THREATENING HEMORRHAGE
Central Nervous System Bleeding
Most central nervous system (CNS) events, which involve bleeding inside the skull or spinal canal, are caused by trauma. CNS hemorrhage is the most common form of severe hemophilic trauma. However, since patients with hemophilia can experience bleeding even weeks after a minor head injury, a history of head trauma may be hard to determine, particularly in children. Spontaneous CNS bleeding in individuals with hemophilia is rare except when there has been a recent antecedent CNS hemorrhage (ie, a recurrent bleed at a previously injured site) or when there is an associated anatomic lesion that predisposes to acute hemorrhage (eg, aneurysm or arteriovenous malformation). Data from the Universal Data Collection Project of the U.S. Centers for Disease Control and Prevention indicates that predisposing risk factors for intracranial hemorrhage include HIV infection, presence of inhibitory antibodies, and age younger than 5 years or older than 51 years.16 Neonatal intracranial hemorrhage is most commonly due to birth trauma. Difficult vaginal deliveries (often requiring the application of forceps or vacuum extraction) are predisposing factors for intracranial hemorrhage in hemophilic newborns.
The site of intracranial CNS bleeding can be subdural, epidural, or intraparenchymal. Bleeding at any of these sites can cause rapidly deteriorating CNS brain function, associated brain swelling, and, in the most extreme circumstances, herniation of the brainstem and rapid death. If the bleeding is stopped with rapid clotting factor replacement, adverse clinical effects can be avoided. However, with intraparenchymal hemorrhage, even small hemorrhages can induce permanent structural and/or neurologic sequelae (in particular, if the anatomic site of the bleed is essential for routine brain function).17
Throat and Neck Hemorrhage
An acute neck injury or a retropharyngeal hemorrhage induced by dental or oral surgical instrumentation can lead to a dissecting facial plane hematoma. This in turn can sometimes lead to compression and acute airway compromise. Bleeding from these injuries that is compressing or compromising the airway may require a rapid clinical response.18 The time from the injury until the trachea is compressed may be long, sometimes many hours. However, once the compression is sufficient to cause difficulty breathing, there may be a short amount of time to stop the bleeding and prevent complete respiratory obstruction.
MUCOCUTANEOUS BLEEDING
One of the common manifestations of hemophilia is oral bleeding. Tooth extraction poses a specific problem, and bleeding following extraction can be the first symptom that leads to the diagnosis of hemophilia. Bleeding after circumcision may also suggest the diagnosis. In 1 study cohort looking at sites of initial bleeding episodes in babies with hemophilia diagnosed before the age of 2 years, bleeding from circumcision and other iatrogenic causes tended to be most common in the neonatal period. Circumcision bleeding events occurred more often in infants with no family history (43%) as compared to those born to known maternal carriers (9.2%) or to mothers with some other family history of hemophilia (14.3%).19
Gastrointestinal (GI) bleeding occurs occasionally in hemophilia, and a wide spectrum of esophageal and GI bleeding may occur. A review of 41 episodes of GI bleeding in hemophilia patients who presented to 1 institution over 10 years implicated duodenal ulcer (22%), unknown site (22%), and gastritis (14%) as the most common sources.20 Mallory-Weiss syndrome has also been cited as a cause for upper GI bleeding in hemophilia patients.21
PRINCIPLES OF TREATMENT
Understanding the pathophysiology of hemophilia as well as the type and severity of hemophilia and the inhibitor status in an individual patient are paramount in the management of a patient with hemophilia. In the past, management mainly focused on the treatment of acute bleeding episodes (Table 2). With data showing the benefit of bleed prevention, the management of hemophilia now focuses on prophylaxis of bleeding episodes, which prevents chronic arthropathy and improves quality of life.
ACUTE BLEEDING EPISODES
Dosing of Factor VIII Products
Dosing for factor VIII concentrate is as follows: 1 IU of factor VIII concentrate per kg will increase the circulating factor VIII level by 2% (ie, patient weight in kg × 50 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor VIII needs an infusion of 1500 IU of factor VIII (30 kg × 50 IU/kg).
Dosing of Factor IX Products
Dosing for factor IX concentrate is as follows: 1 IU of factor IX concentrate per kg will increase the circulating factor IX level by 1% (ie, patient weight in kg × 100 IU/kg = 100% correction). For example, a 30-kg patient requiring 100% correction of factor IX needs an infusion of 3000 IU of factor IX (30 kg × 100 IU/kg). Higher doses (120 to 130 IU/kg) of the recombinant factor IX product BeneFIX (Pfizer) may be needed to reach the 100% circulating factor IX level.
ADJUVANT THERAPY
Desmopressin
Desmopressin is a synthetic vasopressin analogue that increases plasma factor VIII and von Willebrand factor (VWF) levels; it is used to prevent and treat bleeding episodes associated with dental and surgical procedures in patients with mild and moderate hemophilia A and von Willebrand disease.22 Desmopressin causes the release of VWF and factor VIII from storage in the Weibel–Palade bodies of the endothelial cells that line the blood vessels. Individual response to desmopressin varies, with factor VIII level increasing between 2 and 15 times baseline level in patients with mild or moderate hemophilia A.23 It is therefore recommended that patients undergo a therapeutic trial of desmopressin with laboratory measurement of response to factor VIII before it is used for treatment of bleeding episodes or as prophylactic therapy before dental and other surgical procedures. A similar response is generally seen in an individual patient with subsequent doses, and thus the factor VIII level attained after a trial dose can be used to predict the response to future therapy.24
The recommended intravenous dosage of desmopressin is 0.3 µg/kg, administered in 25 to 50 mL of normal saline, over a period of 20 to 30 minutes.25 A concentrated form of desmopressin is available for intranasal administration to treat bleeding disorders. The appropriate dose of concentrated intranasal desmopressin is 150 µg (1 puff) for persons weighing less than 50 kg, and 300 µg (1 puff in each nostril) for persons weighing more than 50 kg.26
Antifibrinolytic Therapy
Antifibrinolytics (both epsilon-aminocaproic acid [EACA] and tranexamic acid) reversibly block the lysine binding sites of plasminogen, preventing its activation to plasmin and thus inhibiting the lysis of polymerized fibrin. EACA is also believed to stabilize the active form of thrombin activatable fibrinolysis inhibitor (TAFIa). It is believed that inactivation of TAFIa is due to conformational rearrangements in the TAFIa molecule; EACA has been shown to slow down spontaneous inactivation of TAFIa, thus curtailing fibrinolysis.27 Although hemostasis is generally achieved with either factor VIII replacement or desmopressin, the risk of recurrent bleeding from oral mucosal surfaces is dramatically reduced with the use of antifibrinolytic agents. These agents are typically contraindicated in patients with hematuria because they can cause a clot to form in the urinary bladder or ureters, leading to obstruction.
EACA is available in intravenous, oral tablet, and elixir formulations; the oral dose is 100 to 200 mg/kg initially (maximum dose, 10 g), followed by 50 to 100 mg/kg per dose every 6 hours (maximum dose, 5 g). Tranexamic acid is available in 650-mg capsules; the dose is 25 mg/kg every 6 to 8 hours.28,29 To treat spontaneous oral hemorrhage or to prevent bleeding from dental procedures in patients with hemophilia, either drug is usually begun in conjunction with desmopressin or factor replacement therapy immediately prior to the procedure and continued for up to 7 days or until mucosal healing is complete. Nonsteroidal anti-inflammatory drugs and aspirin affect platelet function and hence are contraindicated in affected individuals.30
PROPHYLAXIS
Patients with mild to moderate hemophilia typically bleed only after trauma, although the trauma needed to induce bleeding may be more minor than that which would cause bleeding in a normal individual. They usually do not suffer from significant morbidities, whereas patients with severe hemophilia often have spontaneous severe muscle and joint bleeds and can develop early crippling hemophilic arthropathy. Hence, routine prophylaxis has now become the standard of care in the United States and other developed countries in the management of patients with severe hemophilia. Prophylactic replacement therapy with cryoprecipitate in boys with severe hemophilia was first used nearly 50 years ago in Sweden31 and the Netherlands,32 and was shown to reduce the number and the severity of bleeds.32 Moreover, it was observed that early prophylaxis was more effective in preventing arthropathy compared to starting later in life, and that radiologic joint damage could not be reversed by prophylaxis. Subsequently, primary prophylaxis, defined as the start of regular, continuous treatment before the age of 2 years or after the occurrence of first joint bleed,33 was recommended and eventually became the standard treatment; it is currently recommended by the World Health Organization/World Federation of Hemophilia (WFH).34
The timing to begin prophylaxis is somewhat controversial, but many authors suggest starting prophylaxis before the first hemarthrosis occurs. Several studies have reported a wide variation in the age at first joint bleed, ranging from 0.2 to 5.8 years, with medians of 1.6 to 1.7 years.35,36 It has been suggested that arthropathy is best prevented if prophylaxis is started before the second or third joint bleed, but the benefits of starting before the occurrence of first bleed have not been established.37,38 The Swedish experience provides strong support for early prophylaxis.39 In an analysis of 121 patients with severe hemophilia, age at initiation of prophylaxis was an independent predictor of the development of arthropathy, but dose and interval of prophylaxis at the start of prophylactic treatment were not.39
In the Italian ESPRIT study, it was shown that children randomly assigned to prophylaxis had significantly fewer total bleeding episodes and joint bleeding episodes compared with those assigned to episodic therapy. Eleven of 21 patients (52%) in the prophylaxis group had on average less than 1 hemarthrosis per year, whereas only 4 of 19 patients in the episodic therapy group (21%) had the same low frequency of bleeding (P < 0.05).40 In a study of long-term prophylaxis versus on-demand treatment comparing age-matched Danish and Russian patients, the median annual number of joint bleeds in patients on prophylaxis was 1, while patients managed with on-demand treatment experienced a median of 37 joint bleeds. Patients taking prophylaxis also had a statistically significantly better quality of life estimate (P < 0.001) and better functional independence.41 In another trial, prophylaxis was initiated between the ages of 6 and 30 months based on a history of joint hemorrhage rather than age. Radiologic evidence of preserved joint architecture was found in 93% of participants in the prophylaxis group at 6 years of age. In this group, 18 of 32 (56%) children had 1 or 2 bleeds into one or more index joints before prophylaxis, and 17 (53%) had 1 to 5 hemorrhages into 1 or more index joints during prophylaxis. Prophylaxis was efficacious in decreasing bleeding and joint damage after up to 5 hemarthroses.42
Optimal Prophylactic Regimen
Although the benefits of prophylactic replacement therapy are firmly established, the optimal dose and frequency remain unclear. The half-life of clotting factor concentrates is short: about 8 hours for factor VIII in children, and about 12 hours for factor IX. As a result, prophylactic therapy is most effective when given frequently. The most common factor VIII concentrate dosing regimen for prophylaxis in hemophilia A is 25 to 40 IU/kg 3 times per week; for hemophilia B, a dose of 80 to 100 IU/kg is given twice weekly. This is aimed at a pre-infusion level > 1% to mimic the clinical phenotype of moderate hemophilia.
Recently, the US Food and Drug Administration (FDA) approved the first long-lasting antihemophilic factor (recombinant) Fc fusion protein for use in adults and children with hemophilia A. This medication contains the Fc region of human immunoglobulin G1 (IgG1), which binds to the neonatal Fc receptor (FcRn). FcRn is part of a naturally occurring pathway that delays lysosomal degradation of immunoglobulins by cycling them back into circulation and prolonging their plasma half-life. Dosing for routine prophylaxis is 50 IU/kg every 4 days; it may be adjusted based on patient response, with dosing in the range of 25 to 65 IU/kg at 3- to 5-day intervals. More frequent or higher doses up to 80 IU/kg may be required in children younger than 6 years.43
DEVELOPMENT OF INHIBITORS
FACTOR VIII INHIBITORS
Despite the success in the clinical management of hemophilia A, treated patients remain at risk for developing neutralizing antibodies that inhibit factor VIII activity. An inhibitor is a polyclonal high-affinity IgG that is directed against the factor VIII protein and renders exogenous factor ineffective. IgG4 antibodies are predominant and do not fix complement.
Risk Factors
The pathophysiology underlying the development of factor VIII inhibitors is a T-helper (Th)–cell dependent event that involves antigen-presenting cells and B lymphocytes; why only a fraction of patients experience this adverse effect of factor therapy is not known. Patients with mild/moderate hemophilia have a lower risk for inhibitor development than those with severe hemophilia A. The estimated prevalence of inhibitors ranges from 3% to 13% in mild to moderate disease,44–46 and up to 36% in severe hemophilia A.47,48 Usually the presence of an inhibitor in patients with mild/moderate hemophilia is suggested by a change in bleeding pattern: patients who previously used to bleed only after trauma or surgery suddenly start to experience severe spontaneous bleeding. This change in bleeding pattern is explained by cross-reactivity of the inhibitor with the mutated factor VIII of the patient, resulting in a residual factor level of < 0.01 IU/dL.49–51 Occasionally, there is no change in the residual factor VIII level but an inhibitor is detected in the Bethesda assay and/or there is lack of efficacy of factor VIII trans-fusions.51–53
Genetic factors. Data indicate that the risk of developing neutralizing antibodies is to a large extent determined by patient-related genetic factors.54,55 The immune response to factor VIII is similar in up to 80% of family members, significantly higher than expected compared with data from unrelated subjects. In a meta-analysis of patients with severe hemophilia A, the inhibitor incidence was twice as high in African American patients as compared with white patients.56 One study showed that patients of Hispanic ancestry with severe hemophilia A have a higher prevalence of neutralizing inhibitors than non-Hispanic white patients.57
Type of causative mutation. In severe hemophilia A, the risk of inhibitor formation is associated with the type of mutation. More disruptive mutations in the factor VIII gene, such as the intron 22 inversion, large gene deletions, and stop codons are associated with an approximately 35% risk of inhibitor formation, compared with only about 5% in those with missense mutation and small deletions.58 Persons with mutations involving large gene deletions, nonsense mutations, and intrachromosomal aberrations are usually at higher risk for the development of inhibitors than persons with missense mutations, small deletions/insertions, and splice site mutations.59,60 A relatively high risk is also encountered in patients with splicing errors and frame-shift mutations.61
Major histocompatibility complex. The HLA class I alleles A3, B7, and C7, as well as the class II alleles DQA0102, DQB0602, and DR15 have all been associated with a slightly higher risk for inhibitor development in unrelated patients, whereas the HLA C2, DQA0103, DQB0603, and DR13 alleles might be protective.62,63
Immune-regulatory molecules. In the Malmö International Brother Study, polymorphic sites in the genes coding for interleukin 10 (IL-10), tumor necrosis factor-α, and cytotoxic T lymphocyte–associated protein 4 were all associated with the risk of developing inhibitors.64–66 In this study, a 134 bp–long variant of a CAA microsatellite in the promoter region (IL-10.G) was identified in 26.8% of patients with hemophilia A. Thirty-two of these patients (72.7%) developed inhibitors as compared with 37.5% of those without the allele.65
Intensive exposure to factor VIII. Inhibitors in mild/moderate hemophilia seem to occur more commonly later in life, and an episode of intensive treatment with factor VIII concentrate has been reported to precede detection in most reported cases. In the series reported by Hay et al,67 16 out of 26 inhibitors were detected after such intensive replacement therapy, and no particular concentrate was implicated.
INHIBITORS TO FACTOR IX
Factor IX inhibitors are relatively uncommon, occurring in only 1% to 3% of persons with hemophilia B. This is in striking contrast to hemophilia A, where approximately 30% of patients develop inhibitors. The majority of patients with hemophilia B who develop inhibitors have severe hemophilia B.
Risk Factors
Certain mutations in the factor IX gene are associated with an increased incidence of inhibitor development. Large deletions and frame-shift mutations leading to the loss of coding information are much more likely to be associated with inhibitor development. Large deletions account for only 1% to 3% of all hemophilia B patients, but account for 50% of inhibitor patients.68 Patients with hemophilia B who develop inhibitors are at risk for developing anaphylactic reactions to factor IX–containing products. Anaphylaxis occurred more frequently in families with null mutations (large deletions, frame-shift mutations, or nonsense mutations) than in those with missense mutations.69 With hemophilia A, approximately 40% to 50% of black individuals develop inhibitors, but no such association has been found in hemophilia B. Individuals who develop an inhibitor to factor IX do so relatively early in life (within the first 4 to 5 years), after a median of 9 to 11 exposure days to any factor IX–containing products. Because of the severity of a potential anaphylactic reaction occurring early in life after very few exposures to factor IX, all infants and small children with severe hemophilia B should be closely followed over their first 10 infusions with any factor IX–containing products in a facility equipped to treat anaphylactic shock.70–72 A comparison of inhibitors in hemophilia A and B is shown in Table 3.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR VIII INHIBITORS
The available therapeutic agents for treatment of acute hemorrhage in children with hemophilia A with an inhibitor include high-dose recombinant or plasma-derived factor VIII concentrate, activated prothrombin complex concentrates (aPCCs), and recombinant activated factor VII (rFVIIa). In addition, antifibrinolytics may be used as an adjunct therapy.
Patient response to each treatment varies widely, with some patients responding well to one treatment and less well to another. Neither the patient's history nor standard lab tests can assist in making the best choice for the patient. A personalized approach to factor selection is used, and the dosing of that particular agent is often determined primarily by clinical assessment. Inhibitors are quantitated using the Bethesda inhibitor assay and clinically are classified as low- and high-responding inhibitors (Table 4). Inhibitor screening should be done prior to invasive procedures and periodically during the first 50 days of treatment since the risk for inhibitor development is highest during this period.
Low-Responding Inhibitors
A low-responding inhibitor is one in which inhibitor titers are < 5 Bethesda units (BU)/mL; patients with low-responding inhibitors can generally be treated with factor VIII concentrates at higher doses.73 Because the effect of factor VIII inhibitor is usually delayed, the Bethesda titer in plasma is determined after a 2-hour incubation period. As a result of this time delay, continuous administration of factor VIII is usually found to be effective.74 For a serious limb- or life-threatening bleeding episode, a bolus infusion of 100 IU of factor VIII per kg of body weight is administered, and the level is maintained by treatment at a rate of 20 IU/kg/hr. An assay for factor VIII should be performed 1 hour after the bolus infusion and at least daily thereafter. As the antibody titer drops, the daily level of factor VIII may rise and thus downward adjustment of the continuous infusion rate may be required. For routine joint and muscle hemorrhage, patients can usually be managed with infusions at twice the usual dosage. Routine inhibitor assays should be performed after exposure to factor VIII to determine whether an anamnestic response has occurred.
High-Responding Inhibitors
Most clinicians caring for patients with limb- or life-threatening bleeding episodes prefer to use products for which therapeutic levels can be monitored. As described earlier, continuous admin-istration of factor VIII is often effective because of the time delay in inhibition by the antibody. An initial dose of 100 to 200 IU/kg can be administered, and factor VIII levels can be determined 1 hour after initiation of continuous infusion at a rate of 20 to 40 IU/kg/hr. If a factor VIII level cannot be obtained (ie, patients with inhibitor titers > 5 to 10 BU/mL), alternative approaches include the bypassing agents aPCC and rFVIIa.
First used in the 1970s, aPCCs represented a significant improvement in the management in patients with hemophilia with inhibitors. They contain multiple activated serine protease molecules; activated factor X and prothrombin are the main active components in FEIBA (factor eight inhibitor bypassing activity), the most commonly used aPCC in the United States. FEIBA is a pooled plasma product that contains activated factors II, VII, IX and X, and has a duration of action of about 6 to 12 hours. For treatment of acute bleeds, the recommended dose of FEIBA is 50 to 100 IU/kg infused every 8 to 12 hours (maximum daily dose of 200 IU/kg). There is a risk of thrombosis/disseminated intravascular coagulation (DIC) with very large doses given frequently (> 200 IU/kg/day).
rFVIIa directly activates factor X and increases thrombin production on the surface of activated platelets in the absence of factor VIII or factor IX. Standard dosing of rFVIIa is 90 to 120 µg/kg, and many hemophilia treatment centers use higher doses (270 µg/kg/dose), especially in children and young adults. The half-life is about 1.5 to 3 hours, and therefore frequent administration (every 2–6 hours) is required. In one study that assessed the safety and efficacy of fixed-dose rFVIIa in the home setting, hemostasis was achieved in 566 (92%) of evaluable bleeding episodes, and following administration of the additional maintenance dose, hemostasis was maintained in 95% of successfully treated cases.75 As with aPCCs, there is no standardized quantitative laboratory test for measuring the effectiveness of rFVIIa therapy.
All currently used bypassing agents are associated with a risk of thrombotic complications including thromboembolism, DIC, and myocardial infarction. These complications are very rare in patients with hemophilia, however. In general, bypassing agents work for most bleeds and for most patients, but are not as predictable as factor replacement therapy and cannot be monitored by laboratory assays.
TREATMENT OF ACUTE BLEEDS IN PATIENTS WITH FACTOR IX INHIBITORS
rFVIIa and FEIBA are the mainstays of treatment of bleeding episodes in individuals with hemophilia B complicated by an inhibitor to factor IX. Treatment of hemorrhagic episodes in these patients depends on the type of bleeding episode experienced, the inhibitor classification (high- versus low-responding [Table 4]), and the history and severity of infusion reactions. Patients with low-responding inhibitors who have not experienced infusion reactions may be treated with doses of factor IX concentrate calculated to overcome the inhibitor titer and achieve a hemostatic level. In patients with high-responding inhibitors, the use of factor IX concentrates is impractical because of the inhibitor titer or the anamnestic response. Regardless of inhibitor titer, in patients with a history of an anaphylactic event, factor IX usage is contraindicated.
The most commonly used therapy for hemostatic control in patients with high-responding inhibitors with factor IX deficiency and a history of infusion reaction is rFVIIa; the standard dosing regimen is 90 to 120 µg/kg/dose administered every 2 to 3 hours, with a maximum dose of 270 µg/kg/dose. aPCCs, which contain factor IX, can be utilized if the patient has not experienced prior infusion reactions. Repeated exposures to products containing factor IX may stimulate the inhibitor titer and prevent its natural decline over time. This can pose a problem in cases of life- or limb-threatening hemorrhage unresponsive to rFVIIa as these patients will not have factor IX available as an effective mode of therapy. The dosing of FEIBA ranges from 50 to 100 IU/kg every 12 hours, with daily dosing not to exceed 200 IU/kg.
IMMUNE TOLERANCE INDUCTION
Because of the associated inhibitor-related morbidity resulting from limited treatment options, antibody eradication is the ultimate goal in inhibitor management. The only proven strategy for achieving antigen-specific tolerance to factor VIII or factor IX is immune tolerance induction (ITI) therapy. Successful ITI in hemophilia A is currently defined as both an undetectable inhibitor titer (< 0.6 BU), and normalized factor VIII pharmacokinetics, which in turn is defined as plasma factor VIII recovery > 66% of expected and a half-life > 6 hours, determined following a 72-hour factor VIII exposure-free period (Consensus Proceedings from the Second International Conference on Immune Tolerance Therapy, Bonn, Germany, 1997 [unpublished]). Once successful immune tolerance is achieved, long-term prophylaxis is commonly instituted. Using conclusions drawn from international consensus criteria and analysis of the International Immune Tolerance Registry, the I-ITI study has defined ITI failure by the presence of either of 2 criteria:
1. Failure to attain the definition of success within 33 months of uninterrupted ITI;
2. Failure to demonstrate a progressive 20% reduction in inhibitor titer over each 6-month period of uninterrupted ITI, beginning 3 months after initiation to allow for expected anamnesis.76–78
This definition implies a minimum ITI trial period of 9 months before failure is declared.
The European Hemophilia Standardization Board (EHSB), the International Consensus Panel (ICP), and the United Kingdom Hemophilia Center Doctors’ Organization (UKHCDO) have agreed that it is preferable to initiate ITI at a titer of < 10 BU/mL, unless, per the ICP, the titer does not decline over a period of 1 to 2 years and/or inhibitor development is associated with severe or life-threatening bleeding. The ICP noted that for “poor-risk” ITI patients (defined by a historical titer of > 200 BU/mL and/or a pre-ITI inhibitor titer of > 10 BU/mL and/or an interval of > 5 years since inhibitor diagnosis), published efficacy data are limited to dosing regimens > 200 IU/kg/day. The groups all independently concluded that ITI has been successfully performed using recombinant and plasma-derived factor VIII replacement therapy (usually the product on which they developed the inhibitor), and that there are no data to support the superiority of any single product type.79–81 However, both EHSB and ICP have suggested that VWF-containing concentrates be considered for patients who fail ITI using high-purity factor VIII.79,80
The recommendations from US guidelines for ITI in patients with hemophilia A and inhibitors are listed in Table 5.82
ARTHROPATHY
Before the advent of factor products for the treatment of hemophilia, hemarthrosis was one of the leading causes of morbidity. Today, the routine use of prophylactic treatment has resulted in a significant improvement in the lifestyle, quality of life, and life expectancy of these patients. However, despite best efforts, some patients will have severe joint destruction as a result of repeated articular bleeding episodes during their early years. This leads to pain and significant functional disability, thus impairing the quality of life. The basic pathology behind hemophilic arthropathy is chronic synovitis.
It is common to observe a pattern of repeated bleeding (chronic hemarthrosis), especially in patients with severe hemophilia, that can lead to chronic synovitis, inflammatory arthritis, and progressive arthropathy. Therefore, the key to preventing hemophilic arthropathy is aggressive management of the initial hemarthrosis. This is generally accomplished with the use of clotting factor replacement, restorative physiotherapy, and close clinical follow-up. If chronic synovitis develops, synovectomy may be considered in order to slow the progression of the hemophilic arthropathy and to prevent the development of major articular surface erosions that can lead to end-stage arthropathy.83 Primary prophylaxis is discussed earlier and is the mainstay of prevention of chronic hemophilic arthropathy.
SYNOVECTOMY
The emergence of chronic hemophilic hemarthrosis is incited by a hypertrophic and highly vascular synovium. Removal of the synovium prevents further joint damage,84 and can be accomplished through surgical and nonsurgical procedures.
Surgical excision of the hypertrophic synovium can be performed through open or arthroscopic procedures. The open approach has largely been replaced by arthroscopic synovectomies. Regardless of the approach, these patients need prolonged hospitalization, extensive factor replacement, and exhaustive physiotherapy. Moreover, patients with inhibitors are usually not considered candidates for surgical synovectomy.
Chemical and radioactive agents injected intra-articularly can decrease the volume and activity of the synovial tissue. Due to the minimally invasive nature of these procedures, nonsurgical synovectomies are of special importance for hemophilic patients with inhibitors to clotting factors.
Chemical Synovectomy
Chemical synovectomies, using thiotepa, osmic acid, D-penicillamine and other agents, have been used in the distant past. Rifampicin, which is used an antibiotic, is now the most commonly used chemical for the purpose of synovectomy, and the one that has shown better results in terms of decreasing hemarthrosis.85 Each one of the injections should be accompanied by prophylactic administration of clotting factor concentrate. Excellent results (no synovitis and restoration of previous function) have been reported in up to 83% of patients at an average of 2.4 years after the intra-articular injection of rifampicin. As the pathology of the joint becomes more severe, however, the number of injections required to achieve improvement increases. Younger patients and smaller joints benefit more from this procedure.
Radiation Synovectomy
Radiosynovectomy (RS) and radiosynoviorthesis are common terms used to describe the synovial ablation accomplished by intra-articular injection of radioisotopes. Isotopes of gold, yttrium, rhenium, and dysprosium have been used to perform radiation synovectomies in patients with hemophilia. Yttrium-90, a pure beta emitter with adequate particle size and depth penetration, has been used successfully for the treatment of hemophilic synovitis.
The local (growth plate and articular cartilage) and remote effects of radiation are a concern. There have been no reported cases of growth plate disturbance after radiosynovectomy, even after the use of beta emitters such as gold-198.86 Articular cartilage is highly resistant to radiation, and although damage is theoretically possible, none has been reported. Progressive degeneration of treated joints does occur, but the rate is slower than that expected without radiosynovectomy. The principal concern is the potential for late, radiation-induced neoplasia. However, the safety of intra-articular radioisotopes is supported by a long-term follow-up study of more than 5000 RS procedures performed for rheumatoid arthritis, which found no reported radiation-induced malignancies.87
One review analyzed the safety of RS in pediatric patients with hemophilia to provide a risk-benefit assessment. During knee RS, patients receive a radiation dose of approximately 0.74 mSv, and during elbow and ankle RS, a dose of approximately 0.32 mSv. The radiation dose from natural sources is approximately 2 mSv per year and the recommended limit for patients (apart from natural sources) is 1 mSv per year. The lifetime cancer risk increases about 0.5% per 100 mSv per year. Considering the risks and benefits of RS, the authors recommend that clinicians consider this procedure in children with inhibitors or in patients without inhibitors when bleeding is recurrent and persistent despite aggressive factor replacement.88 External-beam radiation has been extensively studied and carries a small risk of osteosarcoma induction.
ACQUIRED INHIBITORS TO FACTOR VIII
Acquired hemophilia (AH) has an estimated prevalence of 1.48 cases per million per year, and a reported mortality between 9% and 22%.89,90 AH is uncommon in children younger than 16 years (prevalence estimated at 0.045/million/year), and may be underdiagnosed in persons older than age 85 (prevalence estimated at 14.7/million/year).89 In the largest published population series, 50% to 60% of diagnosed individuals were previously healthy with no identified underlying disease state.90–91 Underlying conditions consistently associated with AH include pregnancy, evolving or pre-existing autoimmune or malignant disorders, and rarely medications. Primary among the autoimmune disorders are collagen vascular disorders, including systemic lupus erythematosus, rheumatoid arthritis, myasthenia gravis, multiple sclerosis, and autoimmune hemolytic anemia. Most antibodies are mixtures of polyclonal IgG1 and IgG4 immunoglobulins, with the IgG4 molecules mainly responsible for inhibiting clotting activity. The clinical picture of AH is characterized by acute onset of severe bleeding in individuals who previously had no history of bleeding diathesis. Patients generally present with mucocutaneous bleeding (eg, epistaxis and gastrointestinal bleeding), as well as soft tissue bleeding (eg, extensive ecchymoses and hematomas).
The 2 major goals of treatment of AH are the immediate control of acute and chronic bleeding and the long-term suppression/eradication of the autoantibody inhibitor. For patients with an inhibitor titer < 5 BU/mL, administration of desmopressin and concentrates of human recombinant factor VIII may raise the factor VIII activity levels in plasma. If the inhibitor titer is > 5 BU/mL, or if bleeding persists despite infusions of factor VIII concentrates, then factor VIII bypassing agents, such as aPCCs or rFVIIa, are indicated. Local measures for treatment of mucosal hemorrhage, such as antifibrinolytic agents or topical fibrin glues, are helpful.
The primary aim in long-term management of AH is to eradicate the factor VIII autoantibodies so that further bleeding can be averted. Although in some clinical situations (postpartum women and drug-related AH) factor VIII antibodies may remit spontaneously, most published guidelines and algorithms recommend early initiation of eradication therapy. This is usually achieved through immunosuppressive medications or immunomodulation. Successful immunosuppression regimens in AH have most frequently used corticosteroids as the cornerstone, either as a single agent or in combination with cyclophosphamide. In a prospective randomized trial involving 31 participants treated with prednisone 1 mg/kg/day for 3 weeks, 32% achieved complete remission. In participants with antibody persistence after 3 weeks, switching to oral cyclophosphamide 2 mg/kg/day as second-line therapy appeared more effective than continuing prednisone (complete remission rate 50% versus 42%).92
Other immunosuppressive medications have been employed for eradication of refractory autoantibody inhibitors, including azathioprine, cyclosporine, tacrolimus, mycophenolate motefil, and sirolimus. Controlled studies have not been performed to confirm their comparative safety and efficacy in sufficiently large populations. Anti-CD20 antibody has been used to treat inhibitors in patients with both congenital and acquired hemophilia.93,94 Other less frequently used treatment options include administration of intravenous immunoglobulins (IVIG) in large doses. IVIG by itself rarely is able to induce a complete remission, but may be useful adjunctive therapy along with immunosuppressants, as part of an ITI regimen, or with extracorporeal plasmapheresis.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
- Brinkhous KM. Clotting defect in hemophilia: deficiency in a plasma factor required for platelet utilization. Proc Soc Exp Biol Med 1947;66:117–20.
- Quick AJ. Studies on the enigma of the haemostatic dysfunction of hemophilia. Am J Med Sci 1947;214:272–80.
- Ingram JIC. The history of hemophilia. J Clin Path 1976;29:469–79.
- Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature 1984;312:326–30.
- Vehar GA, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature 1984;312:337–42.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Yoshitake S, Schach BG, Foster DC, et al. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry 1985;24:3736–50.
Vander VP, Giles AR. A detailed morphological evaluation of the evolution of the haemostatic plug in normal, factor VII and factor VIII deficient dogs. Br J Haematol 1988;70:345–55.
Gitschier J, Wood WI, Goralka TM, et al. Characterizartion of the human factor VIII gene. Nature 1984;312:326–30.
Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature 1984;312:342–7.
Naylor J, Brinke A, Hassock S, Green PM, Gianelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993;2:1773–8.
Lakich D, Kazazian HH Jr., Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe hemophilia A. Nat Genet 1993;5:236–41.
Stenson PD, Ball E, Howells K, et al. Gene mutation database. J Med Genet 2008;45:124–6.
Mauser Bunschoten EP, van Houwelingen JC, Sjamsoedin Visser EJM, et al. Bleeding symptoms in carriers of hemophilia A and B. Thromb Haemost 1988;59: 349–52.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, et al. Bleeding in carriers of hemophilia. Blood 2006;108:52–6.
Nuss R, Soucie JM, Evatt B, and the Hemophilia Surveillance System Project Investigators. Changes in the occurrence of and risk factors for hemophilia-associated intracranial hemorrhage. Am J Hematol 2001;68:37–42.
Yoffe G, Buchanan G. Intracranial hemorrhage in newborn and young infants with hemophilia. J Pediatr 1988;113:333–6.
Roderick PJ, Robinson AC. Life-threatening oropharyngeal bleeding in a haemophiliac with factor VIII inhibitors. Clin Lab Haemat 1988;10:217–9.
Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from the Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Hemophilia 2009; 15:1281–90.
Mittal R, Spero J, Lewis JH, Taylor F, et al. Patterns of gastrointestinal hemorrhage in hemophilia. Gastroenterol 1985;88:515–22.
Lander E, Pechlaner C, Mayr A, et al. Mallory-Weiss syndrome in a patient with hemophilia A and chronic liver disease. Ital J Gastroenterol 1995;27:73–4.
de la Fuente B, Kasper CK, Rickles FR, et al. Response of patients with mild and moderate hemophilia A and von Willebrand disease to treatment with desmopressin. Ann Intern Med 1985; 103:6–14.
Prowse CV, Sas G, Gader AM, et al. Specificity in the factor VIII response to vasopressin infusion in man. Br J Haematol 1979;41:437–47.
Rodeghiero F, Castaman G, Di Bona E, et al. Consistency of responses to repeated DDAVP infusions in patients with von Willebrand disease and hemophilia A. Blood 1989;74:1997–2000.
Warrier AI, Lusher JM. DDAVP: a useful alternative to blood components in moderate hemophilia A and von Willebrand disease. J Pediatr 1983;102:228–33.
Nilsson IM, Mikaelsson M, Vilhardt H. The effect of intranasal DDAVP on coagulation and fibrinolytic activity in normal persons. Scand J Haematol 1982;29:70–4.
Kogan A. Thrombin activatable fibrinolysis inhibitor (TAFI): a new marker of cardiovascular disease. Clinical Laboratory International. June 2004.
Vinckier F, Vermylen J. Dental extractions in hemophilia: reflections on 10 years’ experience. Oral Surg Oral Med Oral Pathol 1985;59:6–9.
Evans BE. The use of epsilon-aminocaproic acid for the management of hemophilia in dental and oral surgery patients. J Am Dent Assoc 1977;94:21.
Kaneshiro MM, Mielke CH Jr, Kasper CK, et al. Bleeding time after aspirin in disorders of intrinsic clotting. N Engl J Med 1969;281:1039–42.
Nilsson IM, Blomback M, Ahlberg A. Our experience in Sweden with prophylaxis on haemophilia. Bibl Haematol 1970;34:111–24.
Van Creveld S. Prophylaxis of joint hemorrhages in hemophilia. Acta Haematol 1969; 41:206–14.
Ljung R, Aronis-Vournas S, Kurnik-Auberger K, et al. Treatment of children with haemophilia in Europe: A survey of 20 centers in 16 countries. Haemophilia 2001;7:446–52.
Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: Summary statement. Haemophilia 2003; 9(Suppl. 1):1–4.
Pollman H, Richter H, Ringkamp H, Jurgens H. When are children diagnosed as having severe hemophilia and when do they start to bleed? A 10-year single-center PUP study. Eur J Pediatr 1999;158:166–70.
Van Dijk K, Fischer K, Van Der Bom JG, et al. Variability in clinical phenotype of severe haemophilia: the role of the first joint bleed. Haemophilia 2005;11:438–43.
Kreuz W, Escuriola-Ettingshausen C, Funk M, et al. When should prophylactic treatment in patients with Hemophilia A and B start? The German experience. Hemophilia 1998;4:413–7.
Fischer K, Van der Bom JG, Mauser-Bunschoten EP, et al. Effects of postponing prophylactic treatment on long-term outcome in patients with severe Hemophilia. Blood 2002;99:2337–41.
Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe hemophilia should be started at an early age but can be individualized. Br J Haematol 1999;105:1109–13.
Gringeri A, Lundin B, von mackensen S, et al; ESPRIT study group. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011;9:700–10.
Ingerslev J, Lethagen S, Hvitfeldt Poulsen L, et al. Long-standing prophylactic therapy vs. episodic treatment in young people with severe haemophilia: a comparison of age-matched Danish and Russian patients. Haemophilia 2014;20: 58–64.
Maco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357:535–44.
Eloctate [package insert]. Cambridge [MA]: Biogen, Inc.
Lusher JM, Arkin S, Abildgaard CF, et al. Recombinant factor VIII for the treatment of previously untreated patients with henophilia A. N Engl J Med 1993;328:453–9.
Sultan Y, and the French Hemophilia Study Group. Prevalence of inhibitors in a population of 3435 hemophilia patients in France. Thromb Haemost 1992;67:600–2.
Rizza CR, Spooner RGD. Treatment of hemophilia and related disorders in Britain and Northern Ireland during 1976-80: report on behalf of the directors of hemophilia centers in the United Kingdom. Br Med J 1983;286:929–32.
Darby SC, Keeling DM, Spooner RJ, et al. The incidence of factor VIII and factor IX inhibitors in the hemophilia population of the UK and their effect on subsequent mortality, 1977-99. J Thromb Haemost 2004;2:1047–54.
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet 1992;339:594–8.
Fijnvandraat K, Turenhout EAM, van den Brink EN, et al. The missense mutation Arg593à Cys is related to antibody formation in a patient with mild Hemophilia A. Blood 1997;89:4371–7.
Vlot AJ, Wittebol S, Strengers PFW, et al. Factor VIII inhibitor in a patient with mild Hemophilia A and an Asn618-Ser mutation responsive to immune tolerance induction and cyclophosphamide. Br J Hematolol 2002;117:136–40.
Santagostino E, Gringeri A, Tagliavacca L, et al. Inhibitors to factor VIII in a family with mild hemophilia: Molecular characterization and response to factor VIII and desmopressin. Throm Haemost 1995;74:61–21.
Peerlinck K, Jacquemin M, Arnout J, et al. Antifactor VIII antibody inhibiting allogenic but not autologous factor VIII in patients with mild hemophilia A. Blood 1999;93:2267–73.
Kesteven PJ, Holland LJ, Lawrie AS, et al. Inhibitor to factor VIII in mild hemophilia. Thromb Haemost 1984;52:50–2.
Gill JC. The role of genetics in inhibitor formation. Thromb Hemostat 1999;82:500–4.
Astermark J, Berntorp E, White GC, Kroner BL; MIBS Study group. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patient. Hemophilia 2001;7:267–72.
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in Hemophilia A patients- A review of recent studies of recombinant and plasma-derived factor VIII concentrates. Hemophilia 1999;5:145–54.
Carpenter SL, Michael Soucie J, Sterner S, Presley R; Hemophilia Treatment Center Network (HTCN) Investigators. Increased prevalence of inhibitors in Hispanic patients with severe haemophilia A enrolled in the Universal Data Collection databse. Haemophilia 2012;18:e260–5.
Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Haemost 2003;29:23–30.
Schwaab R, Brackman HH, Meyer C, et al. Hemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:1402–6.
Oldenburg J, El-Maari O, Schwaab R. Inhibitor development in correlation to Factor VIII genotypes. Hemophilia 2002;8(Suppl. 2):23–9.
Boekhorst J, Lari GR, D’oiron R, et al. Factor VIII genotype and inhibitor development in patients with hemophilia A: Highest risk in patients with splice site mutation. Haemophilia 2008;14:729–35.
Oldenburg J, Picard JK, Schwaab R, et al. HLA genotype of patients with severe hemophilia A due to intron 22 inversion with and without inhibitors of factor VIII. Thromb Haemost 1997;77:238–42.
Hay CR, Ollier W, Pepper L, et al. HLA class II profile: A weak determinant of factor VIII inhibitor development in severe hemophilia A. UKHCDO Inhibitor Working Party. Thromb Haemost 1997;77:234–7.
Astermark J, Olderburg J, Carlson J, et al. Polymorphisms in the TNFA gene and the risk of inhibitor development in severe Hemophilia A. Blood 2006;108:3739–45.
Astemark J, Olderburg J, Pavlova A, et al. Polymorphisms in the IL10 but not in the IL1Beta and IL4 genes are associated with inhibitor development in patients with Hemophilia A. Blood 2006;107:3167–72.
Astermark J, Wang X, Olderburg, et al. MIBS Study group. Polymorphisms in the CTLA-4 gene and inhibitor development in patients with Hemophilia A. J Throm Haemost 2007;5:263–5.
Hay CR, Ludlam CA, Colvin BT, et al. Factor VIII inhibitors in mild and moderate-severity hemophilia A. Thromb Haemost 1998;79:762–6.
High HA. Factor IX molecular structure, epitopes and mutations associated with inhibitor formation. In: Aledort LM, Hoyer LW, Lusher JM, et al, eds. Inhibitors to coagulation factors. New York: Plenum Press; 1995:79-86.
Thorland ED, Drost JB, Lusher JM, et al. Anaphylactic response to factor IX replacement therapy in hemophilia B patients: Complete gene deletions confer the highest risk. Hemophilia 1999;5:101–5.
Warrier I. ITI in hemophilia B: Possibilities and problems. International Monitor on Hemophilia 2003:20–3.
Warrier I, Ewenstein B, Koerper M, et al. FIX Inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol 1997;19:23–7.
Warrier I. Management of hemophilia B patients with inhibitors and anaphylaxis. In: Varon D, Martinowitz U, Heim M, eds. Haemophilia and related disorders. Vol. 4. Oxford: Blackwell Science; 1998:574–6.
Kasper CK, Aledort L, Aronson D, et al: Proceedings: a more uniform measurement of factor VIII inhibitors. Thromb Diath Haemorrh 1975;34:612.
White GC, Taylor RE, Blatt PM, et al. Treatment of a high titer anti–factor-VIII antibody by continuous factor VIII administration: report of a case. Blood 1983;62:141–5.
Key NS, Aledort LM, Beardsley D, et al. Home treatment of mild to moderate bleeding episodes using recombinant factor VIIa (Novoseven) in haemophiliacs with inhibitors. Thromb Haemost 1998;80:912–8.
Mariani G, Scheibel E, Nogao T, et al. Immune tolerance as treatment of alloantibodies to factor VIII in hemophilia. The international registry of Immunetolerance Protocols. Semin Hematol 1994;31(Suppl. 4):62–4.
DiMichele D, Kroner B. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Hemostasis. The maintenance of tolerance after successful immune tolerance induction in hemophilia A and B. The North American Registry. Factor VIII/IX Subcommittee of the International Society for Thrombosis and Haemostasis. Haematologica 2000;85(Suppl. 10):40–4.
DiMichele DM, Hay CRM. The international immune tolerance study: A multicenter prospective randomized trial in progress. J Thromb Haemost 2006;4:2271–3000.
Astermark J, Morado M, Rocino A, et al. Current European practice in immune tolerance induction therapy in patients with hemophilia and inhibitors. Hemophilia 2006;12:363–71.
DiMichele DM, Hoots WK, Pipe SW, et al. International workshop on immune tolerance induction: Consensus recommendations. Hemophilia 2007;13:(Suppl. 1):1–22.
Hay CRM, Brown S, Collins PW, et al. The diagnosis and management of factor VIII and IX inhibitors: A guideline from the United Kingdom Center Doctors Organization. Br J Haematol 2006;133:591–605.
Valentino LA, Kempton CL, Kruse-Jarres R, et al. US Guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia 2015;21:559–67.
Silva M, Luck JV Jr. Chronic hemophilic synovitis: the role of radiosynovectomy. World Federation of Hemophilia. Treatment of Hemophilia. April 2004 (no 33). http://www1.wfh.org/publications/files/pdf-1176.pdf.
Storti E, Traldi A, Tosatti E, Davoli PG. Synovectomy, a new approach to hemophilic arthropathy. Acta Haemotol 1969;41:193–205.
Caviglia HA, Fernandez-Palazzi F, Maffei E, et al. Chemical synoviorthesis for hemophilic synovitis. Clin Orthop 1997;343:30–6.
Ahlberg A, Pettersson H. Synoviorthesis with radioactive gold in hemophiliacs. Clinical and radiological follow-up. Acta Orthop Scand 1979;50:513–7.
Lee P. The efficacy and safety of radiosynovectomy. J Rheumatol 1982;9:165–8.
Rodriguez-Merchan EC, Valentino LA. Safety of radiation exposure after radiosynovectomy in paediatric patients with haemophilia. Haemophilia 2015;21:411–8.
Green D, Lechner K. A survey of 215 non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost 1981;45:200–3.
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: A 2 year national surveillance study by the United Kingdom Haemophilia Center Doctors’ Organisation. Blood 2007;109:1870–7.
Kessler CM, Ludlam CA. The treatment of acquired factor VIII inhibitors: Worldwide experience with porcine factor VIII concentrate. International Acquired Hemophilia Study Group. Semin Hematol 1993;30(Suppl. 1):22–7.
Green D, Rademaker AW, Briet E. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thromb Haemost 1993;70:753–7.
Stasi R, Brunetti M, Stipa E, Amadori S. Selective B-cell depletion with rituximab for the treatment of patients with acquired hemophilia. Blood 2004;103:4424–8.
Franchini M. Rituximab in the treatment of adult acquired hemophilia A: a systematic review. Crit Rev Oncol Hematol 2007;63:47–52.
Specific polymorphisms excluded in hemophilic arthropathy
Carriers of a hemochromatosis (HFE) gene mutation or a long (GT)n-repeat length within the HMOX1 promoter regions did not appear to have an increase in hemophilic arthropathy.
In 201 blood samples from patients with severe hemophilia A or B and 37 from patients with moderate disease, neither the presence of an HFE mutation nor a long (GT)n-repeat length was associated with an increase in joint damage. The assessment was based on Pettersson score after adjustment for disease severity, presence of inhibitors, annual joint bleeding rate (AJBR), age at Pettersson score and at clinic entrance, and birth cohort (standardized beta = 0.033 and -0.022, respectively), Lize F. van Vulpen, MD, of University Medical Center Utrecht, the Netherlands, reported at the annual meeting of the European Association of Haemophilia and Allied Disorders.
Study subjects had a median age of 43 years, median AJBRs of 2.5 and 0.5 for severe and moderate disease, respectively, and median Pettersson scores of 22 and 4 in the groups, respectively. An HFE mutation was detected in 91 patients, and their levels of ferritin, iron, and transferrin saturation were significantly increased, but other baseline characteristic were similar in those with and without HFE mutation, and regardless of (GT)n-repeat length.
The marked heterogeneity in joint damage seen among hemophilia patients was hypothesized in this study to be associated with differences in iron handling, but the findings failed to support that hypothesis, Dr. van Vulpen concluded.
Dr. van Vulpen reported having no disclosures.
Carriers of a hemochromatosis (HFE) gene mutation or a long (GT)n-repeat length within the HMOX1 promoter regions did not appear to have an increase in hemophilic arthropathy.
In 201 blood samples from patients with severe hemophilia A or B and 37 from patients with moderate disease, neither the presence of an HFE mutation nor a long (GT)n-repeat length was associated with an increase in joint damage. The assessment was based on Pettersson score after adjustment for disease severity, presence of inhibitors, annual joint bleeding rate (AJBR), age at Pettersson score and at clinic entrance, and birth cohort (standardized beta = 0.033 and -0.022, respectively), Lize F. van Vulpen, MD, of University Medical Center Utrecht, the Netherlands, reported at the annual meeting of the European Association of Haemophilia and Allied Disorders.
Study subjects had a median age of 43 years, median AJBRs of 2.5 and 0.5 for severe and moderate disease, respectively, and median Pettersson scores of 22 and 4 in the groups, respectively. An HFE mutation was detected in 91 patients, and their levels of ferritin, iron, and transferrin saturation were significantly increased, but other baseline characteristic were similar in those with and without HFE mutation, and regardless of (GT)n-repeat length.
The marked heterogeneity in joint damage seen among hemophilia patients was hypothesized in this study to be associated with differences in iron handling, but the findings failed to support that hypothesis, Dr. van Vulpen concluded.
Dr. van Vulpen reported having no disclosures.
Carriers of a hemochromatosis (HFE) gene mutation or a long (GT)n-repeat length within the HMOX1 promoter regions did not appear to have an increase in hemophilic arthropathy.
In 201 blood samples from patients with severe hemophilia A or B and 37 from patients with moderate disease, neither the presence of an HFE mutation nor a long (GT)n-repeat length was associated with an increase in joint damage. The assessment was based on Pettersson score after adjustment for disease severity, presence of inhibitors, annual joint bleeding rate (AJBR), age at Pettersson score and at clinic entrance, and birth cohort (standardized beta = 0.033 and -0.022, respectively), Lize F. van Vulpen, MD, of University Medical Center Utrecht, the Netherlands, reported at the annual meeting of the European Association of Haemophilia and Allied Disorders.
Study subjects had a median age of 43 years, median AJBRs of 2.5 and 0.5 for severe and moderate disease, respectively, and median Pettersson scores of 22 and 4 in the groups, respectively. An HFE mutation was detected in 91 patients, and their levels of ferritin, iron, and transferrin saturation were significantly increased, but other baseline characteristic were similar in those with and without HFE mutation, and regardless of (GT)n-repeat length.
The marked heterogeneity in joint damage seen among hemophilia patients was hypothesized in this study to be associated with differences in iron handling, but the findings failed to support that hypothesis, Dr. van Vulpen concluded.
Dr. van Vulpen reported having no disclosures.
FROM EAHAD 2017
Key clinical point:
Major finding: Neither the presence of an HFE mutation, nor a long (GT)n-repeat length was associated with an increase in joint damage as assessed by Pettersson score (standardized beta = 0.033 and –0.022, respectively).
Data source: An evaluation of blood samples from a cohort of 238 patients.
Disclosures: Dr. van Vulpen reported having no disclosures.
Low inhibitor incidence seen with new generation rhFVIII
Human-cl rhFVIII, a new generation recombinant factor VIII of human origin, appears to be associated with a low inhibitor incidence in patients with severe hemophilia A, according to interim findings from the ongoing NuProtect study.
In 66 evaluable previously untreated patients with at least 20 days of exposure to human-cl rhFVIII, the cumulative incidence of high-titer and low-titer inhibitor development was 12.8% and 8.4%, respectively, Ellis J. Neufeld, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The median age at first treatment was 13 months (range of 3-135 months). Of 59 patients with available F8 gene analysis, 44 had high-risk mutations and 47 had null mutations; 1 had no mutation identified. After a median of 11.5 treatment exposure days, 8 of the 66 patients developed high-titer inhibitors and 5 developed low-titer inhibitors; 4 of those were transient, said Dr. Neufeld of Harvard Medical School, Boston.
Only two of the patients developed an inhibitor, including one high- and one low-titer inhibitor, after 20 exposure days. Among the 13 inhibitor patients, 12 had an F8 mutation identified; all were null, and 11 were high risk.
In a prior study of 201 patients with previously treated severe hemophilia A, no inhibitors were reported with human-cl rhFVIII – which is produced in human cells without chemical modification or protein fusion. The NuProtect study is looking at immunogenicity, efficacy, and safety in previously untreated patients. Final data are expected in 2018, Dr. Neufeld noted.
Dr. Neufeld disclosed financial ties to Octapharma, the sponsor of NuProtect, and numerous other pharmaceutical companies.
Human-cl rhFVIII, a new generation recombinant factor VIII of human origin, appears to be associated with a low inhibitor incidence in patients with severe hemophilia A, according to interim findings from the ongoing NuProtect study.
In 66 evaluable previously untreated patients with at least 20 days of exposure to human-cl rhFVIII, the cumulative incidence of high-titer and low-titer inhibitor development was 12.8% and 8.4%, respectively, Ellis J. Neufeld, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The median age at first treatment was 13 months (range of 3-135 months). Of 59 patients with available F8 gene analysis, 44 had high-risk mutations and 47 had null mutations; 1 had no mutation identified. After a median of 11.5 treatment exposure days, 8 of the 66 patients developed high-titer inhibitors and 5 developed low-titer inhibitors; 4 of those were transient, said Dr. Neufeld of Harvard Medical School, Boston.
Only two of the patients developed an inhibitor, including one high- and one low-titer inhibitor, after 20 exposure days. Among the 13 inhibitor patients, 12 had an F8 mutation identified; all were null, and 11 were high risk.
In a prior study of 201 patients with previously treated severe hemophilia A, no inhibitors were reported with human-cl rhFVIII – which is produced in human cells without chemical modification or protein fusion. The NuProtect study is looking at immunogenicity, efficacy, and safety in previously untreated patients. Final data are expected in 2018, Dr. Neufeld noted.
Dr. Neufeld disclosed financial ties to Octapharma, the sponsor of NuProtect, and numerous other pharmaceutical companies.
Human-cl rhFVIII, a new generation recombinant factor VIII of human origin, appears to be associated with a low inhibitor incidence in patients with severe hemophilia A, according to interim findings from the ongoing NuProtect study.
In 66 evaluable previously untreated patients with at least 20 days of exposure to human-cl rhFVIII, the cumulative incidence of high-titer and low-titer inhibitor development was 12.8% and 8.4%, respectively, Ellis J. Neufeld, MD, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
The median age at first treatment was 13 months (range of 3-135 months). Of 59 patients with available F8 gene analysis, 44 had high-risk mutations and 47 had null mutations; 1 had no mutation identified. After a median of 11.5 treatment exposure days, 8 of the 66 patients developed high-titer inhibitors and 5 developed low-titer inhibitors; 4 of those were transient, said Dr. Neufeld of Harvard Medical School, Boston.
Only two of the patients developed an inhibitor, including one high- and one low-titer inhibitor, after 20 exposure days. Among the 13 inhibitor patients, 12 had an F8 mutation identified; all were null, and 11 were high risk.
In a prior study of 201 patients with previously treated severe hemophilia A, no inhibitors were reported with human-cl rhFVIII – which is produced in human cells without chemical modification or protein fusion. The NuProtect study is looking at immunogenicity, efficacy, and safety in previously untreated patients. Final data are expected in 2018, Dr. Neufeld noted.
Dr. Neufeld disclosed financial ties to Octapharma, the sponsor of NuProtect, and numerous other pharmaceutical companies.
Key clinical point:
Major finding: The cumulative incidence of high-titer and low-titer inhibitor development was 12.8% and 8.4%, respectively.
Data source: The ongoing NuProtect study of 66 patients.
Disclosures: Dr. Neufeld disclosed financial ties to Octapharma, the sponsor of NuProtect, and numerous other pharmaceutical companies.
Study highlights importance of genotyping in von Willebrand disease
Patients with genetically confirmed von Willebrand disease (VWD) type 2M have a relatively mild clinical phenotype, according to findings from a retrospective cross-sectional study.
In fact, three of 31 patients included in the study had a near normal laboratory phenotype. Additionally, patients with the p.Val1360Ala mutation had significantly higher values of all von Willebrand factor (VWF)-related laboratory parameters, compared with those with the p.Arg1374Cys or p.Phe1293Leu mutation, Dominique Maas reported at the European Association for Haemophilia and Allied Disorders annual meeting.
The findings underscore the importance of genotyping for making a correct diagnosis of VWD type 2M, said Ms. Maas of Radboud University Medical Center, Nijmegen, The Netherlands.
The study subjects, who had a least one VWD type 2M mutation, had a median age of 34 years and a median bleeding score of 6. Most suffered from mucocutaneous bleeding, but with low frequency compared with other VWD subtypes. The incidence of muscle hematomas, postpartum hemorrhage, and postsurgery bleeding were relatively high, however. Age and bleeding score were strongly positively correlated.
Subjects had a median VWF antigen level of 24 IU dL-1, median VWF ristocetin cofactor activity of 6 IU dL-1, and median VSF:RCo/VWF:Ag ratio of 0.29. The VSF collagen binding activity and VWF:Ag ratio was normal, she said.
Genotyping is a powerful diagnostic tool for making an appropriate diagnosis and classification of VWD. The current findings are important because understanding the correlation between genotype and phenotype improves the understanding of VWD pathogenesis and has important implications for treatment, follow-up, and genetic counseling, but has not been throughly investigated in fully genotyped patients with VWD type 2M, she said.
Ms. Maas reported having no disclosures.
Patients with genetically confirmed von Willebrand disease (VWD) type 2M have a relatively mild clinical phenotype, according to findings from a retrospective cross-sectional study.
In fact, three of 31 patients included in the study had a near normal laboratory phenotype. Additionally, patients with the p.Val1360Ala mutation had significantly higher values of all von Willebrand factor (VWF)-related laboratory parameters, compared with those with the p.Arg1374Cys or p.Phe1293Leu mutation, Dominique Maas reported at the European Association for Haemophilia and Allied Disorders annual meeting.
The findings underscore the importance of genotyping for making a correct diagnosis of VWD type 2M, said Ms. Maas of Radboud University Medical Center, Nijmegen, The Netherlands.
The study subjects, who had a least one VWD type 2M mutation, had a median age of 34 years and a median bleeding score of 6. Most suffered from mucocutaneous bleeding, but with low frequency compared with other VWD subtypes. The incidence of muscle hematomas, postpartum hemorrhage, and postsurgery bleeding were relatively high, however. Age and bleeding score were strongly positively correlated.
Subjects had a median VWF antigen level of 24 IU dL-1, median VWF ristocetin cofactor activity of 6 IU dL-1, and median VSF:RCo/VWF:Ag ratio of 0.29. The VSF collagen binding activity and VWF:Ag ratio was normal, she said.
Genotyping is a powerful diagnostic tool for making an appropriate diagnosis and classification of VWD. The current findings are important because understanding the correlation between genotype and phenotype improves the understanding of VWD pathogenesis and has important implications for treatment, follow-up, and genetic counseling, but has not been throughly investigated in fully genotyped patients with VWD type 2M, she said.
Ms. Maas reported having no disclosures.
Patients with genetically confirmed von Willebrand disease (VWD) type 2M have a relatively mild clinical phenotype, according to findings from a retrospective cross-sectional study.
In fact, three of 31 patients included in the study had a near normal laboratory phenotype. Additionally, patients with the p.Val1360Ala mutation had significantly higher values of all von Willebrand factor (VWF)-related laboratory parameters, compared with those with the p.Arg1374Cys or p.Phe1293Leu mutation, Dominique Maas reported at the European Association for Haemophilia and Allied Disorders annual meeting.
The findings underscore the importance of genotyping for making a correct diagnosis of VWD type 2M, said Ms. Maas of Radboud University Medical Center, Nijmegen, The Netherlands.
The study subjects, who had a least one VWD type 2M mutation, had a median age of 34 years and a median bleeding score of 6. Most suffered from mucocutaneous bleeding, but with low frequency compared with other VWD subtypes. The incidence of muscle hematomas, postpartum hemorrhage, and postsurgery bleeding were relatively high, however. Age and bleeding score were strongly positively correlated.
Subjects had a median VWF antigen level of 24 IU dL-1, median VWF ristocetin cofactor activity of 6 IU dL-1, and median VSF:RCo/VWF:Ag ratio of 0.29. The VSF collagen binding activity and VWF:Ag ratio was normal, she said.
Genotyping is a powerful diagnostic tool for making an appropriate diagnosis and classification of VWD. The current findings are important because understanding the correlation between genotype and phenotype improves the understanding of VWD pathogenesis and has important implications for treatment, follow-up, and genetic counseling, but has not been throughly investigated in fully genotyped patients with VWD type 2M, she said.
Ms. Maas reported having no disclosures.
FROM EAHAD 2017
Key clinical point:
Major finding: Three of 31 patients included in the study had a near normal laboratory phenotype.
Data source: A retrospective cross-sectional study of 31 patients.
Disclosures: Ms. Maas reported having no disclosures.
Solulin variants activate TAFI in vitro
A new generation of solulin variants is showing promise for the treatment of severe hemophilia A.
The in vitro production and characterization of these solulin variants – F376A-, M388A-, and F376A/M388A-solulin – showed that while they lost their abilities to activate protein C (an inhibitor of thrombin generation), they were still capable of activating thrombin activatable fibrinolysis inhibitor (TAFI), Yohann Repesse, PhD reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Additionally, the double mutant F376A/M388A-solulin, which was tested ex vivo using blood samples from hemophilic A patients, was found on thromboelastography to increase clot firmness and stability. As opposed to wild type solulin, the effects were maintained even at high concentrations of F376A/M388A-solulin, said Dr. Repesse of the National Institute of Health and Medical Research, Caen, France.
An important aggravating factor when it comes to treating hemophilia involves premature fibrinolysis, which means that antifibrinolytics are of interest, he explained. Thrombomodulin is a key player in the coagulation cascade, because it activates protein C, and also in the fibrinolytic cascade, as it activates TAFI. Solulin, a soluble form of thrombomodulin, has both capabilities.
The findings with respect to the new generation of solulin variants tested in this study open new opportunities for the development of specific medications for hemophilia patients, he concluded.
Dr. Repesse reported having no disclosures.
A new generation of solulin variants is showing promise for the treatment of severe hemophilia A.
The in vitro production and characterization of these solulin variants – F376A-, M388A-, and F376A/M388A-solulin – showed that while they lost their abilities to activate protein C (an inhibitor of thrombin generation), they were still capable of activating thrombin activatable fibrinolysis inhibitor (TAFI), Yohann Repesse, PhD reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Additionally, the double mutant F376A/M388A-solulin, which was tested ex vivo using blood samples from hemophilic A patients, was found on thromboelastography to increase clot firmness and stability. As opposed to wild type solulin, the effects were maintained even at high concentrations of F376A/M388A-solulin, said Dr. Repesse of the National Institute of Health and Medical Research, Caen, France.
An important aggravating factor when it comes to treating hemophilia involves premature fibrinolysis, which means that antifibrinolytics are of interest, he explained. Thrombomodulin is a key player in the coagulation cascade, because it activates protein C, and also in the fibrinolytic cascade, as it activates TAFI. Solulin, a soluble form of thrombomodulin, has both capabilities.
The findings with respect to the new generation of solulin variants tested in this study open new opportunities for the development of specific medications for hemophilia patients, he concluded.
Dr. Repesse reported having no disclosures.
A new generation of solulin variants is showing promise for the treatment of severe hemophilia A.
The in vitro production and characterization of these solulin variants – F376A-, M388A-, and F376A/M388A-solulin – showed that while they lost their abilities to activate protein C (an inhibitor of thrombin generation), they were still capable of activating thrombin activatable fibrinolysis inhibitor (TAFI), Yohann Repesse, PhD reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Additionally, the double mutant F376A/M388A-solulin, which was tested ex vivo using blood samples from hemophilic A patients, was found on thromboelastography to increase clot firmness and stability. As opposed to wild type solulin, the effects were maintained even at high concentrations of F376A/M388A-solulin, said Dr. Repesse of the National Institute of Health and Medical Research, Caen, France.
An important aggravating factor when it comes to treating hemophilia involves premature fibrinolysis, which means that antifibrinolytics are of interest, he explained. Thrombomodulin is a key player in the coagulation cascade, because it activates protein C, and also in the fibrinolytic cascade, as it activates TAFI. Solulin, a soluble form of thrombomodulin, has both capabilities.
The findings with respect to the new generation of solulin variants tested in this study open new opportunities for the development of specific medications for hemophilia patients, he concluded.
Dr. Repesse reported having no disclosures.
FROM EAHAD 2017
Key clinical point:
Major finding: F376A-, M388A-, and F376A/M388A-solulin lost their abilities to activate protein C but were still capable of activating TAFI.
Data source: An in vitro and ex vivo evaluation of solulin variants.
Disclosures: Dr. Repesse reported having no disclosures.
Boys with severe hemophilia have good physical function
Children with severe hemophilia in a U.K. study had generally good self-reported health-related quality of life and physical functioning, but impairment on both measures was greater in those who reported pain.
Of 123 boys with a mean age of 12 years who were included in the multicenter SO-FIT study, 110 had hemophilia A, 25 had a past inhibitor, and 9 had a current inhibitor. Prophylactic treatment was given in 120 of the boys, Kate Khair, PhD of Great Ormond Street Hospital for Children, London, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
In the preceding 6 months, participants experienced a median of 0 joint bleeds (range of 0-8), and a median Hemophilia Joint Health Score (HJHS) of 1 (range, 0-37). Physical function scores were good; the Haemophilia & Exercise Project questionnaire (HEP-Test-Q) mean score was 80.82, with the highest impairments seen in the domain of “endurance” (mean, 72.21), and the Paediatric Haemophilia Activities List (PedHAL) mean score was 85.13, with the highest impairments seen in the domains of “leisure activities and sports” and “lying/sitting/kneeling/standing” (mean, 82.04 and 82.97, respectively).
Health-related quality of life as assessed by the Haemo-QoL Short Form was also generally good (mean, 23.07), with the highest impairments seen in the domains of “friends” and “family” (mean, 27.08 and 26.59, respectively).
In the 27% of participants with pain, a high correlation was seen between the HEP-Test-Q and Haemo-QoL, and moderate correlation was seen between the PedHAL and Haemo-QoL, which implies that good physical functioning is related to good health-related quality of life, Dr. Khair explained.
In fact, patients who said they often or always had pain reported significantly worse subjective physical functioning in all HEP-Test-Q and PedHAL domains, as well as higher impairments in their HRQoL vs. those who said they sometimes, rarely, or never experienced pain, she said.
Interestingly, data completion by study participants was improved with use of an iPad vs. “paper and pencil” for questionnaire completion, Dr. Khair said in an interview.
“They preferred this method – it was more in keeping with how they communicate and learn at school,” she said, noting that the iPad database was set up to give real time scores, which could then be used to tailor assessments.
This is an approach that could be undertaken at home pre-clinic visits, she said.
“Gaining the views of children and young adults in patient-reported outcomes is complex, but reveals their views of their care and their lives – an area we should address more,” she added.
Dr. Khair received grant or research support from Pfizer ASPIRE.
Children with severe hemophilia in a U.K. study had generally good self-reported health-related quality of life and physical functioning, but impairment on both measures was greater in those who reported pain.
Of 123 boys with a mean age of 12 years who were included in the multicenter SO-FIT study, 110 had hemophilia A, 25 had a past inhibitor, and 9 had a current inhibitor. Prophylactic treatment was given in 120 of the boys, Kate Khair, PhD of Great Ormond Street Hospital for Children, London, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
In the preceding 6 months, participants experienced a median of 0 joint bleeds (range of 0-8), and a median Hemophilia Joint Health Score (HJHS) of 1 (range, 0-37). Physical function scores were good; the Haemophilia & Exercise Project questionnaire (HEP-Test-Q) mean score was 80.82, with the highest impairments seen in the domain of “endurance” (mean, 72.21), and the Paediatric Haemophilia Activities List (PedHAL) mean score was 85.13, with the highest impairments seen in the domains of “leisure activities and sports” and “lying/sitting/kneeling/standing” (mean, 82.04 and 82.97, respectively).
Health-related quality of life as assessed by the Haemo-QoL Short Form was also generally good (mean, 23.07), with the highest impairments seen in the domains of “friends” and “family” (mean, 27.08 and 26.59, respectively).
In the 27% of participants with pain, a high correlation was seen between the HEP-Test-Q and Haemo-QoL, and moderate correlation was seen between the PedHAL and Haemo-QoL, which implies that good physical functioning is related to good health-related quality of life, Dr. Khair explained.
In fact, patients who said they often or always had pain reported significantly worse subjective physical functioning in all HEP-Test-Q and PedHAL domains, as well as higher impairments in their HRQoL vs. those who said they sometimes, rarely, or never experienced pain, she said.
Interestingly, data completion by study participants was improved with use of an iPad vs. “paper and pencil” for questionnaire completion, Dr. Khair said in an interview.
“They preferred this method – it was more in keeping with how they communicate and learn at school,” she said, noting that the iPad database was set up to give real time scores, which could then be used to tailor assessments.
This is an approach that could be undertaken at home pre-clinic visits, she said.
“Gaining the views of children and young adults in patient-reported outcomes is complex, but reveals their views of their care and their lives – an area we should address more,” she added.
Dr. Khair received grant or research support from Pfizer ASPIRE.
Children with severe hemophilia in a U.K. study had generally good self-reported health-related quality of life and physical functioning, but impairment on both measures was greater in those who reported pain.
Of 123 boys with a mean age of 12 years who were included in the multicenter SO-FIT study, 110 had hemophilia A, 25 had a past inhibitor, and 9 had a current inhibitor. Prophylactic treatment was given in 120 of the boys, Kate Khair, PhD of Great Ormond Street Hospital for Children, London, reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
In the preceding 6 months, participants experienced a median of 0 joint bleeds (range of 0-8), and a median Hemophilia Joint Health Score (HJHS) of 1 (range, 0-37). Physical function scores were good; the Haemophilia & Exercise Project questionnaire (HEP-Test-Q) mean score was 80.82, with the highest impairments seen in the domain of “endurance” (mean, 72.21), and the Paediatric Haemophilia Activities List (PedHAL) mean score was 85.13, with the highest impairments seen in the domains of “leisure activities and sports” and “lying/sitting/kneeling/standing” (mean, 82.04 and 82.97, respectively).
Health-related quality of life as assessed by the Haemo-QoL Short Form was also generally good (mean, 23.07), with the highest impairments seen in the domains of “friends” and “family” (mean, 27.08 and 26.59, respectively).
In the 27% of participants with pain, a high correlation was seen between the HEP-Test-Q and Haemo-QoL, and moderate correlation was seen between the PedHAL and Haemo-QoL, which implies that good physical functioning is related to good health-related quality of life, Dr. Khair explained.
In fact, patients who said they often or always had pain reported significantly worse subjective physical functioning in all HEP-Test-Q and PedHAL domains, as well as higher impairments in their HRQoL vs. those who said they sometimes, rarely, or never experienced pain, she said.
Interestingly, data completion by study participants was improved with use of an iPad vs. “paper and pencil” for questionnaire completion, Dr. Khair said in an interview.
“They preferred this method – it was more in keeping with how they communicate and learn at school,” she said, noting that the iPad database was set up to give real time scores, which could then be used to tailor assessments.
This is an approach that could be undertaken at home pre-clinic visits, she said.
“Gaining the views of children and young adults in patient-reported outcomes is complex, but reveals their views of their care and their lives – an area we should address more,” she added.
Dr. Khair received grant or research support from Pfizer ASPIRE.
FROM EAHAD 2017
Key clinical point:
Major finding: Physical function score (HEP-Test-Q) was a mean of 80.82, with the highest impairments seen in the domain of “endurance” (mean, 72.21).
Data source: The SO-FIT study of 123 boys with severe hemophilia.
Disclosures: Dr. Khair received grant or research support from Pfizer ASPIRE.
ITI protects against bleeding in hemophilia A with factor VIII inhibitors
Immune tolerance induction, or ITI, in hemophilia A patients with inhibitor development against factor VIII (FVIII) appears to provide some protection against bleeding, as bleeding during ITI in patients from the PedNet registry was dependent on inhibitor titer and ITI regimen, according to Kathelijn Fischer, MD.
Of 218 registry patients who were born during 1990-2009 and who had clinically relevant inhibitors and available data on joint bleeding, 157 had bleeding for at least 3 months. They were followed for a median of 26 months starting at age 16 months, and most had high titer inhibitors (52% had titers of 5-199 BU, and 32% had titers greater than 200 BU).
The bleeding rates in 12 patients without ITI who were on prophylaxis were similar to those in 89 such patients without prophylaxis (0.3 and 0.4 bleeds/month, respectively); no significant difference was seen between the groups based on inhibitor titer, Dr. Fischer of University Medical Center Utrecht, The Netherlands reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Similarly, the bleeding rate during ITI among those on prophylaxis did not differ significantly from the rate in those without prophylaxis (0.4 vs. 0.3/month, respectively), but the rate did increase significantly according to inhibitor titer: The rate increased from 0.05/month in the group with less than 5 BU, to 0.2/month in those with 5-199 BU, and to 0.5/month in the group with greater than 200 BU.
Further, while ITI dose did not affect bleeding, dosing frequency had a strong effect. The median rate with nondaily dosing in 36 patients vs. daily dosing in 63 patients was 0.4/month vs. 0.2/month, respectively, Dr. Fischer said.
She reported having no disclosures.
Immune tolerance induction, or ITI, in hemophilia A patients with inhibitor development against factor VIII (FVIII) appears to provide some protection against bleeding, as bleeding during ITI in patients from the PedNet registry was dependent on inhibitor titer and ITI regimen, according to Kathelijn Fischer, MD.
Of 218 registry patients who were born during 1990-2009 and who had clinically relevant inhibitors and available data on joint bleeding, 157 had bleeding for at least 3 months. They were followed for a median of 26 months starting at age 16 months, and most had high titer inhibitors (52% had titers of 5-199 BU, and 32% had titers greater than 200 BU).
The bleeding rates in 12 patients without ITI who were on prophylaxis were similar to those in 89 such patients without prophylaxis (0.3 and 0.4 bleeds/month, respectively); no significant difference was seen between the groups based on inhibitor titer, Dr. Fischer of University Medical Center Utrecht, The Netherlands reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Similarly, the bleeding rate during ITI among those on prophylaxis did not differ significantly from the rate in those without prophylaxis (0.4 vs. 0.3/month, respectively), but the rate did increase significantly according to inhibitor titer: The rate increased from 0.05/month in the group with less than 5 BU, to 0.2/month in those with 5-199 BU, and to 0.5/month in the group with greater than 200 BU.
Further, while ITI dose did not affect bleeding, dosing frequency had a strong effect. The median rate with nondaily dosing in 36 patients vs. daily dosing in 63 patients was 0.4/month vs. 0.2/month, respectively, Dr. Fischer said.
She reported having no disclosures.
Immune tolerance induction, or ITI, in hemophilia A patients with inhibitor development against factor VIII (FVIII) appears to provide some protection against bleeding, as bleeding during ITI in patients from the PedNet registry was dependent on inhibitor titer and ITI regimen, according to Kathelijn Fischer, MD.
Of 218 registry patients who were born during 1990-2009 and who had clinically relevant inhibitors and available data on joint bleeding, 157 had bleeding for at least 3 months. They were followed for a median of 26 months starting at age 16 months, and most had high titer inhibitors (52% had titers of 5-199 BU, and 32% had titers greater than 200 BU).
The bleeding rates in 12 patients without ITI who were on prophylaxis were similar to those in 89 such patients without prophylaxis (0.3 and 0.4 bleeds/month, respectively); no significant difference was seen between the groups based on inhibitor titer, Dr. Fischer of University Medical Center Utrecht, The Netherlands reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Similarly, the bleeding rate during ITI among those on prophylaxis did not differ significantly from the rate in those without prophylaxis (0.4 vs. 0.3/month, respectively), but the rate did increase significantly according to inhibitor titer: The rate increased from 0.05/month in the group with less than 5 BU, to 0.2/month in those with 5-199 BU, and to 0.5/month in the group with greater than 200 BU.
Further, while ITI dose did not affect bleeding, dosing frequency had a strong effect. The median rate with nondaily dosing in 36 patients vs. daily dosing in 63 patients was 0.4/month vs. 0.2/month, respectively, Dr. Fischer said.
She reported having no disclosures.
FROM EAHAD 2017
Key clinical point:
Major finding: Median bleeding rate with nondaily vs. daily ITI dosing was 0.4/month vs. 0.2/month, respectively.
Data source: An analysis of 218 patients from the PedNet registry.
Disclosures: Dr. Fischer reported having no disclosures.
Study: No link between vaccines, inhibitor development
The administration of pediatric vaccinations in close proximity to factor VIII (FVIII) exposure was not associated with inhibitor development in previously untreated patients with severe hemophilia A in the PedNet Registry.
Similarly, no association was seen between recurrent vaccinations and inhibitor development, Dr. van den Berg reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Inhibitor development in this patient population is a multifactorial event, but these findings show no association between vaccinations administered early in life and increased inhibitor risk, she concluded.
Dr. van den Berg received grant/research support from Bayer, Baxalta, Pfizer, CSI, and Grifols.
The administration of pediatric vaccinations in close proximity to factor VIII (FVIII) exposure was not associated with inhibitor development in previously untreated patients with severe hemophilia A in the PedNet Registry.
Similarly, no association was seen between recurrent vaccinations and inhibitor development, Dr. van den Berg reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Inhibitor development in this patient population is a multifactorial event, but these findings show no association between vaccinations administered early in life and increased inhibitor risk, she concluded.
Dr. van den Berg received grant/research support from Bayer, Baxalta, Pfizer, CSI, and Grifols.
The administration of pediatric vaccinations in close proximity to factor VIII (FVIII) exposure was not associated with inhibitor development in previously untreated patients with severe hemophilia A in the PedNet Registry.
Similarly, no association was seen between recurrent vaccinations and inhibitor development, Dr. van den Berg reported at the annual meeting of the European Association for Haemophilia and Allied Disorders.
Inhibitor development in this patient population is a multifactorial event, but these findings show no association between vaccinations administered early in life and increased inhibitor risk, she concluded.
Dr. van den Berg received grant/research support from Bayer, Baxalta, Pfizer, CSI, and Grifols.
FROM EAHAD 2017
Key clinical point:
Major finding: The adjusted hazard ratio for any inhibitor development related to vaccinations in close proximity to FVIII was 0.65.
Data source: A review of data from 375 children in the PedNet Registry.
Disclosures: Dr. van den Berg received grant/research support from Bayer, Baxalta, Pfizer, CSI, and Grifols.