User login
New Paradigms in Lymphoma Treatment
Emerging concepts, a growing knowledge base, and new targets are changing the way mantle cell lymphoma is treated and managed, according to Mark Roschewski, MD, of the Center for Cancer Research National Cancer Institute at the National Institutes of Health.
"With more understanding of the biology about what makes this lymphoma different than others, we see changes," Roschewski said. "We have seen a lot of changes in this particular disease even in the last year. I think this bodes well for the future of how we are going to manage patients."
Emerging concepts, a growing knowledge base, and new targets are changing the way mantle cell lymphoma is treated and managed, according to Mark Roschewski, MD, of the Center for Cancer Research National Cancer Institute at the National Institutes of Health.
"With more understanding of the biology about what makes this lymphoma different than others, we see changes," Roschewski said. "We have seen a lot of changes in this particular disease even in the last year. I think this bodes well for the future of how we are going to manage patients."
Emerging concepts, a growing knowledge base, and new targets are changing the way mantle cell lymphoma is treated and managed, according to Mark Roschewski, MD, of the Center for Cancer Research National Cancer Institute at the National Institutes of Health.
"With more understanding of the biology about what makes this lymphoma different than others, we see changes," Roschewski said. "We have seen a lot of changes in this particular disease even in the last year. I think this bodes well for the future of how we are going to manage patients."

Bone Metastasis: Concise Overview
Bone metastasis is a relatively common complication of cancer, often developing as they advance, especially in prostate cancer and breast cancer. Bone metastasis can profoundly affect patients’ daily activities and quality of life (QOL) due to severe pain and associated major complications. Prompt palliative therapy is required for symptomatic pain relief and prevention of the devastating complications of bone metastasis.
Epidemiology
Bone is the most common and preferred site for metastatic involvement of cancer. Advanced cancers frequently develop metastases to the bone during the later phases of cancer progression. At least 100,000 patients develop bone metastases every year, although the exact number of bone metastases is not known.1 Multiple myeloma (MM), breast cancer, and prostate cancer are responsible for up to 70% of bone metastases cases.2 Gastrointestinal cancers contribute least to bone metastases: < 15% of all cases.2
Related: Effective Treatment Options for Metastatic Pancreatic Cancer
The prognosis of bone metastases is generally poor, although it partly depends on the primary site of the original cancer and on the presence of any additional metastases to visceral organs. For example, it is known that survival times are longer for patients with primary prostate or breast cancer than for patients with lung cancer primary tumors.3,4
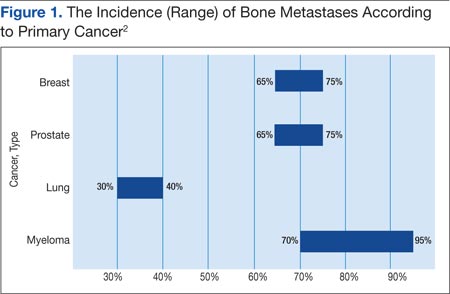
Prostate and breast cancers are the most common primary cancers of bone metastases. At postmortem studies, patients who died of prostate cancer or breast cancer revealed evidence of bone metastases in up to 75% of cases (Figure 1). Regardless of their survival expectancy, however, most patients with bone metastasis need immediate medical attention and active palliative therapy to prevent devastating complications related to bone metastasis, such as pathologic bone fractures and severe bone pain.
Clinical Features
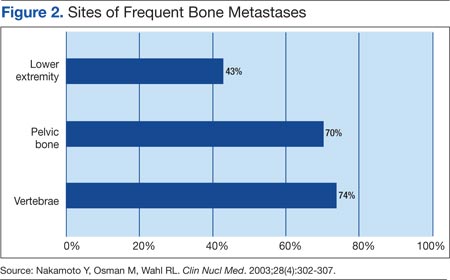
Multiple Myeloma
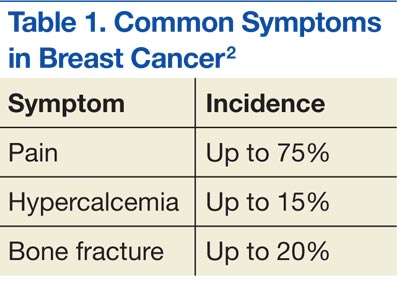
Multiple myeloma is the second most common hematologic malignancy and is caused by an abnormal accumulation of clonal plasma cells in the bone marrow. Characteristic clinical manifestations include bony destruction and related features of bone pain, anemia (80% of cases), hypocalcemia, and renal dysfunction. Pathologic fractures, renal failure, or hyperviscosity syndrome often develops. More than 20,000 new patients are diagnosed with MM and about 11,000 patients in the U.S. die of MM every year. Multiple myeloma and is twice as likely to develop in men as it is in women. A large number of MM cases are under the care of VAMCs (about 10%-12% of all MM cases).7,8
Abnormal laboratory tests show an elevated total protein level in the blood and/or urine (Bence Jones proteinuria). Serum electrophoresis detects M-protein in about 80% to 90% of patients. Patients may also present with renal failure. The differential diagnosis includes other malignancies, such as metastatic carcinoma, lymphoma, leukemia, and monoclonal gammopathy.
Pathophysiology
Normal bone tissue is made up of 2 different types of cells: osteoblasts and osteoclasts. New bone is constantly being produced while old bone is broken down. When tumor cells invade bone, the cancer cells produce 1 of 2 distinct substances; as a result, either osteoclasts or osteoblasts are stimulated, depending on tumor type metastasized to the bone. The activated osteoclasts then dissolve the bone, weakening the bone (osteolytic phenomenon), and the osteoblasts stimulate bone formation, hardening the bone (osteoblastic or sclerotic process).
Diagnosis and Evaluation
The most important first step in evaluating bone metastasis in a patient is to take a thorough, careful medical history and perform a physical examination. The examination not only helps locate suspected sites of bone metastases, but also helps determine necessary diagnostic studies.
The radiographic appearance of bone metastasis can be classified into 4 groups: osteolytic, osteoblastic, osteoporotic, and mixed. Imaging characteristics of osteolytic lesions include the destruction/thinning of bone, whereas osteoblastic (osteosclerotic) lesions appear with excess deposition of new bones. In contrast to malignant osteolytic lesions, osteoporotic lesions look like faded bone without cortical destruction or increased density.
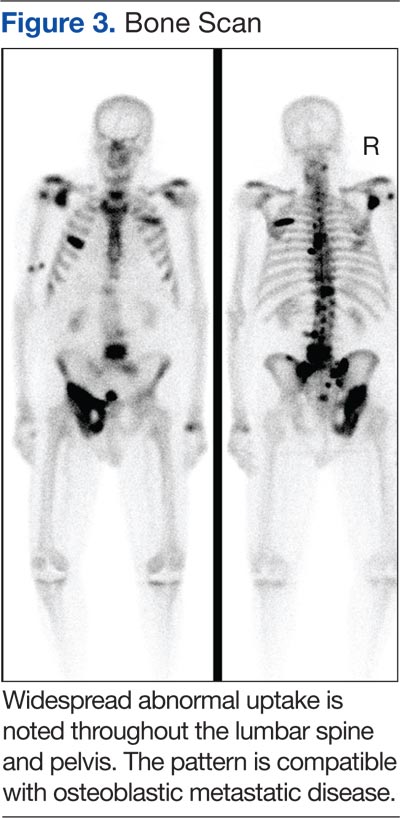
The main choice of imaging study for screening suspected bone metastases is usually the bone scan (Figure 3). Plain radiographs are not useful in the early detection of bone metastases, because bone lesions do not show up on plain films until 30% to 50% of the bone mineral is lost.5,9 Although most metastatic bone lesions represent a mixture of osteoblastic and -lytic processes, metastatic lesions of lung cancer and breast cancer are predominantly osteolytic in contrast to mainly osteoblastic lesions of prostate cancer metastases.10
The osteoblastic process of bone metastases is best demonstrated on a bone scan; however, a positive bone scan does not necessarily indicate bone metastases, because it is not highly specific of metastatic disease. Several benign bone lesions (such as osteoarthritis, traumatic injury, and Paget disease) also show positive readings. Magnetic resonance imaging (MRI) is not useful in screening for bone metastases, but it is better in assessing bone metastases compared with a bone scan, because it is more sensitive, especially for spinal lesions. The reported sensitivity of MRI is 91% to 100%, whereas bone scan sensitivity is only 62% to 85%.11,12
Even though the bone scan has been assumed to be the best imaging study for bone metastases, positron emission tomography (PET) scans can be more useful in detecting osteolytic bone metastases, as they can light up areas of increased metabolic activity. Positron emission tomography scans, however, are less sensitive for osteoblastic metastases. An additional advantage of PET scans is that they can be used for whole-body scanning/surveillance to rule out visceral involvement.
Published studies indicate that bone scans better detect sclerotic bone metastases and PET scans are superior in revealing osteolytic metastases.13-15 Furthermore, in contrast to bone scans, PET scans can identify additional lesions in addition to bone lesion. According to recent reports, PET provides higher sensitivity and specificity in demonstrating lytic and sclerotic metastases compared with that of the bone scan.16
Breast Cancer
The role of PET for breast cancer is controversial. A study by Lonneux and colleagues found that PET is highly sensitive in confirming distant metastasis from breast cancer, whereas researchers reported a similar sensitivity but higher specificity.17 Ohta and colleagues reported that PET and bone scan had identical sensitivity (77.7%), but PET was more specific than the bone scan (97.6% vs 80.9%, respectively).14 The study conclusion by Cook and colleagues was that PET is superior to bone scan in the detection of metastatic osteolytic bone lesions from breast cancer, whereas osteoblastic metastatic bone lesions from breast cancer are less likely to be demonstrated on a PET scan.18
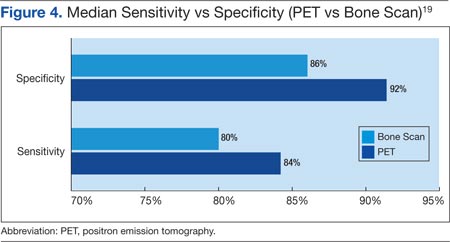
Houssami and Costelloe conducted a systematic review of 16 reported studies that comparatively tested the accuracy of imaging modalities for bone metastases in breast cancer.19 Sensitivity was generally similar between PET and bone scans in most studies reviewed. Four studies reported similar sensitivity but higher specificity for PET; the median specificity for PET and bone scan was 92% vs 85.5%, respectively (Figure 4).
Prostate Cancer
Prostate cancer is now established as the “classic” cancer for false-negative results on PET. Positron emission tomography does not perform well in the identification of osteoblastic skeletal metastases from prostate cancer. Yeh and colleagues reported only 18% positivity with PET.20 Interestingly, however, progressive metastatic prostate cancer showed a higher yield of 77% sensitivity with PET, perhaps because active osseous disease can be better picked up by PET scans.21
Related: Prostate Cancer Survivorship Care
Lung Cancer
For non-small cell lung cancer, both bone scan and PET showed a similar sensitivity for bone metastases detection, but the PET scan was more specific than the bone scan. Lung cancer often metastasizes to bone: up to 36% of patients at postmortem study. Lung cancer with bone metastases has a poor prognosis with median survival time typically measured in months. Most patients with bone metastases develop complications, such as severe pain, bone fracture, hypercalcemia, and spinal cord compression. Bone-targeted therapies play a greater role in the management of lung cancer patients, aiming for delaying disease progression and preserving QOL.22,23
Therapeutic Strategy and Management
Major morbidities associated with bone metastases include severe pain, hypercalcemia, bone fractures, spinal compression fractures, and cord or nerve root compression. This section reviews appropriate management techniques reported in the literature, particularly external beam radiation therapy.
Radiation Therapy
Pain is the most serious complication of bone metastases. Radiation therapy has been established as standard therapy and an effective pain palliation modality. Up to 80% of patients achieve partial pain relief, and > 33% of patients experience complete pain relief after radiation (Figure 5).24,25 Although a 3,000 cGy given over a 2-week period has been commonly used, a standard dose-fraction radiation treatment regimen has not been established.

The RTOG study was a randomized clinical study comparing various radiation schedules; 1,500 cGyin 1 week; vs 2,000 cGy in 1 week; vs 2,500 cGy in 1 week; vs 3,000 cGy in 2 weeks; or 4,050 cGy in 3 weeks. The conclusion was that local radiotherapy was an effective therapy for symptomatic and palliative therapy of bone metastases. Furthermore, low-dose radiotherapy was as good as various higher dose protracted courses of radiation treatments in terms of overall response rates (ORRs).24
Nearly 96% of patients eventually reported minimal pain relief to their palliative course of radiotherapy and experienced at least some pain relief within 4 weeks of radiation therapy. Complete pain relief was attained in 54% of patients regardless of the radiation dose-fraction schedules used. The median duration of complete pain response was about 12 weeks; > 70% of patients did not experience relapse of pain.26
Hartsell and colleagues investigated the efficacy of 800 cGy in a single fraction compared with 3,000 cGy in 10 fractions as part of a phase 3 randomized study of symptomatic therapy for pain palliation.27 The results showed 66% ORRs with similar complete and partial response rates (RRs) for both radiation groups. The complete RRs were 15% in the 800 cGy single-fraction arm vs 18% in the 3,000 cGy therapy arm, whereas partial RRs were 50% and 48% in the single vs the 3,000 cGy arms, respectively. However, there was a higher rate of retreatment for patients treated with the 800 cGy single-fraction radiotherapy. The 800 cGy single-fraction radiotherapy program seems rather popular in Canada and in European countries but is currently not widely used in the U.S.
Surgical Therapy
The surgical indications for managing bone metastases can vary, depending on disease location, surgeon’s preference, and patient’s overall disease status and related morbidities. Pain relief of fractured long bones (humerus, femur, or tibia) is crucial. The main goals of surgical intervention in these cases include the restoration of stability and functional mobility, pain control, and improving QOL. Weight-bearing bones (humerus/tibia) are especially at risk of bone fracture, and compromise of these is an indication of surgery. Postoperative external-beam radiation is recommended in most cases to eradicate residual microscopic disease or tumor progression.28
Radiopharmaceutical Therapy
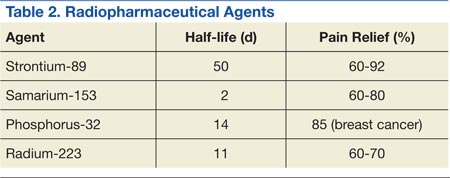
Bone-seeking radiopharmaceuticals are effective and have been widely used for pain palliation. The usual indications for radiopharmaceutical therapy include diffuse osteoblastic skeletal metastases demonstrated on bone scan, painful bone metastases not responding well to analgesics, and hormone-refractory metastatic prostate cancer. At present, strontium-89 (Sr-89), samarium-153 (Sm-153), phosphorus-32 (P-32), and radium 223 dichloride are radionuclides currently accepted as attractive therapeutic modalities for pain management (Table 2).
The clinical response is not immediate, and the average time to response is 1 to 2 weeks, but sometimes much longer. The main adverse reaction of systemic radiopharmaceutical therapy is myelotoxicity, such as thrombocytopenia and/or leukopenia. Occasionally, a so-called flare phenomenon of a transient pain increase may develop as well.29,30
Systemic Pharmacotherapy
Bisphosphonates are drugs commonly used to treat bone metastases. The benefits of bisphosphonate therapy are bone pain relief, the reduction of bone destruction, and the prevention of hypercalcemia and bone fractures. Bisphosphonates are typically more effective in osteolytic metastases and easily bind to bone, inhibiting bone resorption and increasing mineralization.31,32 Also, recent clinical studies suggest that bisphosphonates may inhibit tumor progression of bone metastases.
Related: Cancer Drugs Increase Rate of Preventable Hospital Admissions
Zoledronic acid is currently one of the most potent bisphosphonates and is effective in most types of metastatic bone lesions.33 Denosumab, another drug, diminishes osteoclast activity, leading to decreased bone resorption and increased bone mass.34,35 Denosumab is useful in preventing complications as a result of bone metastases from solid tumors and has been recently approved by the FDA for treatment of postmenopausal osteoporosis and the prevention of skeletal-related events (SREs) in cancer patients with bone metastases.
Adverse Effects
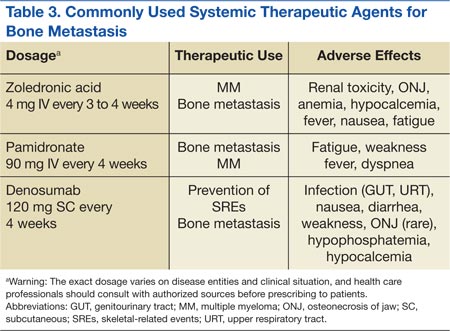
Zoledronate and bisphosphonates in general are not recommended for patients with kidney disease, including hypocalcaemia and severe renal impairment. A rare but well-known complication of bisphosphonate administration is osteonecrosis of the jaw, which is somewhat more common in MM, especially after dental extractions. General nonspecific adverse effects include fatigue, anemia, muscle aches, fever, and/or edema in the feet or legs. Flulike symptoms and generalized bone discomfort can also be seen shortly after the first infusion (Table 3).
Breast Cancer
Bisphosphonates have been shown to effectively prevent SREs in breast cancer patients with bone metastases.36 For example, zoledronic acid is the most effective bisphosphonate and has been demonstrated to significantly delay the time to development of a first SRE, reducing the overall SRE rate by 43%.37
Lung Cancer
According to Rosen and colleagues, lung cancer patients with bone metastases who received zoledronic acid (4 mg every 3 weeks) experienced a 9% reduction in SREs, a relative delay in median time to a first SRE, and a significantly reduced incidence of SREs.37
Prostate Cancer
Zoledronic acid is the only bisphosphonate that proved effective in the treatment of prostate cancer patients with bone metastases. Zoledronic acid significantly reduced the risk of SREs (36%) and bone pain as well as delayed the median time to first SRE (nearly 6 months).38,39
Multiple Myeloma
Bisphosphonates are recommended for bone metastases to prevent new bone lesions. Studies have shown pamidronate (90 mg every 4 weeks) resulted in a 41% reduction in SREs at 9 months and a 25% reduction at 21 months.40,41 Oral clodronate, another agent, also significantly reduced SREs and pain in patients with MM.42
Conclusion
Metastatic cancer with bone metastases occurs as cancer advances and spreads to the bone from the primary site of the original solid cancer. Nearly 70% of patients with prostate and breast cancers and about 30% to 40% of patients with lung cancer develop bone metastases. In addition, up to 95% of MMs involve bone. The most frequent and important symptom of bone metastasis is pain. In addition, bone metastasis causes bone fractures, hypercalcemia, and spinal cord and nerve compression. Imaging studies, such as bone scans and PET studies, are useful tools in diagnosing bone metastases.
Therapeutic management of bone metastases is expanding and rapidly evolving. For better therapy outcomes, treatment should be both individualized and coordinated among the care team, including a medical oncologist, radiation oncologist, surgeon, and radiologist. Available therapeutic modalities include radiation therapy, radiopharmaceutical therapy, surgery, and systemic pharmacotherapy (zoledronate, pamidronate, and denosumab).
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225-249.
2. Cooleman RE. Metastatic bone disease: Clinical features, pathophysiology, and treatment strategies. Cancer Treat Rev. 2001;27(3):165-1763.
3. Hirabayashi H, Ebara S, Kinoshita T, et al. Clinical outcome and survival after palliative surgery for spinal metastases. Cancer. 2003;97(2):476-84.
4. van der Linden YM, Dijkstra SPDS, Vonk EJA, Marijnen CA, Leer JW; Dutch Bone Metastasis Study Group. Prediction of survival in patients with metastases in the spinal column. Cancer. 2005;103(2):320-328.
5. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12(20, pt 2):6243S-6249S.
6. Body JJ. Metastatic bone disease: Clinical and therapeutic aspects. Bone. 1992;13(suppl 1):S57-S62.
7. Siegel RS, Ma J, Zou Z, Jermal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
8. National Cancer Institute. SEER stat fact sheets: Myeloma. National Cancer Institute Website. http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed January 12, 2015.
9. Lentle BC, McGowan DG, Dierich H. Technetium-99M polyphosphate bone scanning in carcinoma of the prostate. Br J Urol. 1974;46(5):543-548.
10. Söderlund V. Radiological diagnosis of skeletal metastases. Eur Radiol. 1996;6(5):587-595.
11. Flickinger FW, Sanal SM. Bone marrow MRI: Techniques and accuracy for detecting breast cancer metastases. Magn Reson Imaging. 1994;12(6):829-35.
12. Hamaoka T, Madewell JE, Podoloff DA, Hortobagyi GN, Ueno NT. Bone imaging in metastatic breast cancer. J Clin Oncol. 2004;22(14):2942-2953.
13. Daldrup-Link HE, Franzius C, Link TM et al. Whole-body MR imaging for detection of bone metastases in children and young adults: Comparison with skeletal scintigraphy and FDG PET. AJR Am J Roentgenol. 2001;177(1):229-236.
14. Ohta M, Tokuda Y, Suzuki Y, et al. Whole body PET for the evaluation of bony metastases in patients with breast cancer: Comparison with 99Tcm-MDP bone scintigraphy. Nucl Med Commun. 2001;22(8):875-879.
15. Koolen BB, Vegt E, Rutgers EJ, et al. FDG-avid sclerotic bone metastases in breast cancer patients: A PET/CT case series. Ann Nucl Med. 2012;26(1):86-91.
16. Even-Sapir E, Metser U, Flusser G, et al. Assessment of malignant skeletal disease: Initial experience with 18F-fluoride PET/CT and comparison between 18F-fluoride PET and 18F-fluoride PET/CT. J Nucl Med. 2004;45(2):272-278.
17. Lonneux M, Borbath II, Berlière M, Kirkove C, Pauwels S. The place of whole-body PET FDG for the diagnosis of distant recurrence of breast cancer. Clin Positron Imaging. 2000;3(2):45-49.
18. Cook GJ, Houston S, Rubens R, Maisey MN, Fogelman I. Detection of bone metastases in breast cancer by 18FDG PET: Differing metabolic activity in osteoblastic and osteolytic lesions. J Clin Oncol. 1998;16(10):3375-3379.
19. Houssami N, Costelloe CM. Imaging bone metastases in breast cancer: Evidence on comparative test accuracy. Ann Oncol. 2012;23(4):834-843.
20. Yeh SD, Imbriaco M, Larson SM, et al. Detection of bony metastases of androgen-independent prostate cancer by PET-FDG. Nucl Med Biol. 1996;23(6):693-697.
21. Morris MJ, Akhurst T, Osman I, et al. Fluorinated deoxyglucose positron emission tomography imaging in progressive metastatic prostate cancer. Urology. 2002;59(6):913-918.
22. Rosen LS, Gordon D, Tchekmedyian S, et al. Zoledronic acid versus placebo in the treatment of skeletal metastases in patients with lung cancer and other solid tumors: A phase III, double-blind, randomized trial—the Zoledronic Acid Lung Cancer and Other Solid Tumors Study Group. J Clin Oncol. 2003;21(16):3150-3157.
23. Hillner BE, Ingle JN, Chlebowski RT, et al; American Society of Clinical Psychology. American Society of Clinical Oncology 2003 update on the role of bisphosphonates and bone health issues in women with breast cancer. J Clin Oncol. 2003;21(21):4042-4057.
24. Chow E, Harris K, Fan G, Tsao M, Size WM. Palliative radiotherapy trials for bone metastases: A systematic review. J Clin Oncol. 2007;25(11):1423-1436.
25. Wu JS, Wong R, Johnston M, Bezjak A, Whelan T; Cancer Care Ontario Practice Guidelines Initiative Supportive Care Group. Meta-analysis of dose-fractionation radiotherapy trials for the palliation of painful bone metastases. Int J Radiat Oncol Biol Phys. 2003;55(3):594-605.
26. Tong D, Gillick L, Hendrickson FR. The palliation of symptomatic osseous metastases. Final results of the study by the Radiation Therapy Oncology Group. Cancer. 1982;50(5):893-899.
27. Hartsell WF, Scott CB, Bruner DW, et al. Randomized trial of short- versus long-course radiotherapy for palliation of painful bone metastases. J Natl Cancer Inst. 2005;97(11):798-804.
28. Frassica DA. General principles of external beam radiation therapy for skeletal metastases. Clin Orthop Relat Res. 2003;415(suppl):S158-S164.
29. Silberstein EB. Systemic radiopharmaceutical therapy of painful osteoblastic metastases. Semin Radiat Oncol. 2000;10(3):240-249.
30. Neville-Webbe HL, Gnant M, Coleman RE. Potential anticancer properties of bisphosphonates. Semin Oncol. 2010;37(suppl 1):S53-S65.
31. Loftus LS, Edwards-Bennett S, Sokol GH. Systemic therapy for bone metastases. Cancer Control. 2012;19(2):145-153.
32. Rosen L, Harland SJ, Oosterlinck W. Broad clinical activity of zoledronic acid in osteolytic to osteoblastic bone lesions in patients with a broad range of solid tumors. Am J Clin Oncol. 2002;25(6)(suppl 1):S19-S24.
33. Fornier MN. Denosumab: Second chapter in controlling bone metastases or a new book? J Clin Oncol. 2010;28(35):5127-5131.
34. Mortimer JE, Pal SK. Safety considerations for use of bone-targeted agents in patients with cancer. Semin Oncol. 2010;37(suppl 1):S66-S72.
35. Pavlakis N, Schmidt R, Stockler M. Bisphosphonates for breast cancer. Cochrane Database Syst Rev. 2005;3:CD003474.
36. Kohno N, Aogi K, Minami H, et al. Zoledronic acid significantly reduces skeletal complications compared with placebo in Japanese women with bone metastases from breast cancer: A randomized, placebo-controlled trial. J Clin Oncol. 2005;23(15):3314-3321.
37. Rosen LS, Gordon D, Tchekmedyian NS, et al. Long-term efficacy and safety of zoledronic acid in the treatment of skeletal metastases in patients with nonsmall cell lung carcinoma and other solid tumors: A randomized, phase III, double-blind, placebo-controlled trial. Cancer. 2004;100(12):2613-2621.
38. Saad F, Gleason DM, Murray R, et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst. 2004;96(11):879-882.
39. Saad F, Eastham J. Zoledronic acid improves clinical outcomes when administered before onset of bone pain in patients with prostate cancer. Urology. 2010;76(5):1175-1181.
40. Berenson JR, Lichtenstein A, Porter L, et al. Efficacy of pamidronate in reducing skeletal events in patients with advanced multiple myeloma. Myeloma Aredia Study Group. N Engl J Med. 1996;334(8):488-493.
41. Berenson JR, Lichtenstein A, Porter L, et al. Long-term pamidronate treatment of advanced multiple myeloma patients reduces skeletal events. Myeloma Aredia Study Group. J Clin Oncol. 1998;16(2):593-602.
42. Lahtinen R, Laakso M, Palva I, Virkkunen P, Elomaa I. Randomised, placebo-controlled multicentre trial of clodronate in multiple myeloma. Finnish Leukaemia Group. Lancet. 1992;340(8827):1049-1052.
Bone metastasis is a relatively common complication of cancer, often developing as they advance, especially in prostate cancer and breast cancer. Bone metastasis can profoundly affect patients’ daily activities and quality of life (QOL) due to severe pain and associated major complications. Prompt palliative therapy is required for symptomatic pain relief and prevention of the devastating complications of bone metastasis.
Epidemiology
Bone is the most common and preferred site for metastatic involvement of cancer. Advanced cancers frequently develop metastases to the bone during the later phases of cancer progression. At least 100,000 patients develop bone metastases every year, although the exact number of bone metastases is not known.1 Multiple myeloma (MM), breast cancer, and prostate cancer are responsible for up to 70% of bone metastases cases.2 Gastrointestinal cancers contribute least to bone metastases: < 15% of all cases.2
Related: Effective Treatment Options for Metastatic Pancreatic Cancer
The prognosis of bone metastases is generally poor, although it partly depends on the primary site of the original cancer and on the presence of any additional metastases to visceral organs. For example, it is known that survival times are longer for patients with primary prostate or breast cancer than for patients with lung cancer primary tumors.3,4
Prostate and breast cancers are the most common primary cancers of bone metastases. At postmortem studies, patients who died of prostate cancer or breast cancer revealed evidence of bone metastases in up to 75% of cases (Figure 1). Regardless of their survival expectancy, however, most patients with bone metastasis need immediate medical attention and active palliative therapy to prevent devastating complications related to bone metastasis, such as pathologic bone fractures and severe bone pain.
Clinical Features
Multiple Myeloma
Multiple myeloma is the second most common hematologic malignancy and is caused by an abnormal accumulation of clonal plasma cells in the bone marrow. Characteristic clinical manifestations include bony destruction and related features of bone pain, anemia (80% of cases), hypocalcemia, and renal dysfunction. Pathologic fractures, renal failure, or hyperviscosity syndrome often develops. More than 20,000 new patients are diagnosed with MM and about 11,000 patients in the U.S. die of MM every year. Multiple myeloma and is twice as likely to develop in men as it is in women. A large number of MM cases are under the care of VAMCs (about 10%-12% of all MM cases).7,8
Abnormal laboratory tests show an elevated total protein level in the blood and/or urine (Bence Jones proteinuria). Serum electrophoresis detects M-protein in about 80% to 90% of patients. Patients may also present with renal failure. The differential diagnosis includes other malignancies, such as metastatic carcinoma, lymphoma, leukemia, and monoclonal gammopathy.
Pathophysiology
Normal bone tissue is made up of 2 different types of cells: osteoblasts and osteoclasts. New bone is constantly being produced while old bone is broken down. When tumor cells invade bone, the cancer cells produce 1 of 2 distinct substances; as a result, either osteoclasts or osteoblasts are stimulated, depending on tumor type metastasized to the bone. The activated osteoclasts then dissolve the bone, weakening the bone (osteolytic phenomenon), and the osteoblasts stimulate bone formation, hardening the bone (osteoblastic or sclerotic process).
Diagnosis and Evaluation
The most important first step in evaluating bone metastasis in a patient is to take a thorough, careful medical history and perform a physical examination. The examination not only helps locate suspected sites of bone metastases, but also helps determine necessary diagnostic studies.
The radiographic appearance of bone metastasis can be classified into 4 groups: osteolytic, osteoblastic, osteoporotic, and mixed. Imaging characteristics of osteolytic lesions include the destruction/thinning of bone, whereas osteoblastic (osteosclerotic) lesions appear with excess deposition of new bones. In contrast to malignant osteolytic lesions, osteoporotic lesions look like faded bone without cortical destruction or increased density.
The main choice of imaging study for screening suspected bone metastases is usually the bone scan (Figure 3). Plain radiographs are not useful in the early detection of bone metastases, because bone lesions do not show up on plain films until 30% to 50% of the bone mineral is lost.5,9 Although most metastatic bone lesions represent a mixture of osteoblastic and -lytic processes, metastatic lesions of lung cancer and breast cancer are predominantly osteolytic in contrast to mainly osteoblastic lesions of prostate cancer metastases.10
The osteoblastic process of bone metastases is best demonstrated on a bone scan; however, a positive bone scan does not necessarily indicate bone metastases, because it is not highly specific of metastatic disease. Several benign bone lesions (such as osteoarthritis, traumatic injury, and Paget disease) also show positive readings. Magnetic resonance imaging (MRI) is not useful in screening for bone metastases, but it is better in assessing bone metastases compared with a bone scan, because it is more sensitive, especially for spinal lesions. The reported sensitivity of MRI is 91% to 100%, whereas bone scan sensitivity is only 62% to 85%.11,12
Even though the bone scan has been assumed to be the best imaging study for bone metastases, positron emission tomography (PET) scans can be more useful in detecting osteolytic bone metastases, as they can light up areas of increased metabolic activity. Positron emission tomography scans, however, are less sensitive for osteoblastic metastases. An additional advantage of PET scans is that they can be used for whole-body scanning/surveillance to rule out visceral involvement.
Published studies indicate that bone scans better detect sclerotic bone metastases and PET scans are superior in revealing osteolytic metastases.13-15 Furthermore, in contrast to bone scans, PET scans can identify additional lesions in addition to bone lesion. According to recent reports, PET provides higher sensitivity and specificity in demonstrating lytic and sclerotic metastases compared with that of the bone scan.16
Breast Cancer
The role of PET for breast cancer is controversial. A study by Lonneux and colleagues found that PET is highly sensitive in confirming distant metastasis from breast cancer, whereas researchers reported a similar sensitivity but higher specificity.17 Ohta and colleagues reported that PET and bone scan had identical sensitivity (77.7%), but PET was more specific than the bone scan (97.6% vs 80.9%, respectively).14 The study conclusion by Cook and colleagues was that PET is superior to bone scan in the detection of metastatic osteolytic bone lesions from breast cancer, whereas osteoblastic metastatic bone lesions from breast cancer are less likely to be demonstrated on a PET scan.18
Houssami and Costelloe conducted a systematic review of 16 reported studies that comparatively tested the accuracy of imaging modalities for bone metastases in breast cancer.19 Sensitivity was generally similar between PET and bone scans in most studies reviewed. Four studies reported similar sensitivity but higher specificity for PET; the median specificity for PET and bone scan was 92% vs 85.5%, respectively (Figure 4).
Prostate Cancer
Prostate cancer is now established as the “classic” cancer for false-negative results on PET. Positron emission tomography does not perform well in the identification of osteoblastic skeletal metastases from prostate cancer. Yeh and colleagues reported only 18% positivity with PET.20 Interestingly, however, progressive metastatic prostate cancer showed a higher yield of 77% sensitivity with PET, perhaps because active osseous disease can be better picked up by PET scans.21
Related: Prostate Cancer Survivorship Care
Lung Cancer
For non-small cell lung cancer, both bone scan and PET showed a similar sensitivity for bone metastases detection, but the PET scan was more specific than the bone scan. Lung cancer often metastasizes to bone: up to 36% of patients at postmortem study. Lung cancer with bone metastases has a poor prognosis with median survival time typically measured in months. Most patients with bone metastases develop complications, such as severe pain, bone fracture, hypercalcemia, and spinal cord compression. Bone-targeted therapies play a greater role in the management of lung cancer patients, aiming for delaying disease progression and preserving QOL.22,23
Therapeutic Strategy and Management
Major morbidities associated with bone metastases include severe pain, hypercalcemia, bone fractures, spinal compression fractures, and cord or nerve root compression. This section reviews appropriate management techniques reported in the literature, particularly external beam radiation therapy.
Radiation Therapy
Pain is the most serious complication of bone metastases. Radiation therapy has been established as standard therapy and an effective pain palliation modality. Up to 80% of patients achieve partial pain relief, and > 33% of patients experience complete pain relief after radiation (Figure 5).24,25 Although a 3,000 cGy given over a 2-week period has been commonly used, a standard dose-fraction radiation treatment regimen has not been established.
The RTOG study was a randomized clinical study comparing various radiation schedules; 1,500 cGyin 1 week; vs 2,000 cGy in 1 week; vs 2,500 cGy in 1 week; vs 3,000 cGy in 2 weeks; or 4,050 cGy in 3 weeks. The conclusion was that local radiotherapy was an effective therapy for symptomatic and palliative therapy of bone metastases. Furthermore, low-dose radiotherapy was as good as various higher dose protracted courses of radiation treatments in terms of overall response rates (ORRs).24
Nearly 96% of patients eventually reported minimal pain relief to their palliative course of radiotherapy and experienced at least some pain relief within 4 weeks of radiation therapy. Complete pain relief was attained in 54% of patients regardless of the radiation dose-fraction schedules used. The median duration of complete pain response was about 12 weeks; > 70% of patients did not experience relapse of pain.26
Hartsell and colleagues investigated the efficacy of 800 cGy in a single fraction compared with 3,000 cGy in 10 fractions as part of a phase 3 randomized study of symptomatic therapy for pain palliation.27 The results showed 66% ORRs with similar complete and partial response rates (RRs) for both radiation groups. The complete RRs were 15% in the 800 cGy single-fraction arm vs 18% in the 3,000 cGy therapy arm, whereas partial RRs were 50% and 48% in the single vs the 3,000 cGy arms, respectively. However, there was a higher rate of retreatment for patients treated with the 800 cGy single-fraction radiotherapy. The 800 cGy single-fraction radiotherapy program seems rather popular in Canada and in European countries but is currently not widely used in the U.S.
Surgical Therapy
The surgical indications for managing bone metastases can vary, depending on disease location, surgeon’s preference, and patient’s overall disease status and related morbidities. Pain relief of fractured long bones (humerus, femur, or tibia) is crucial. The main goals of surgical intervention in these cases include the restoration of stability and functional mobility, pain control, and improving QOL. Weight-bearing bones (humerus/tibia) are especially at risk of bone fracture, and compromise of these is an indication of surgery. Postoperative external-beam radiation is recommended in most cases to eradicate residual microscopic disease or tumor progression.28
Radiopharmaceutical Therapy
Bone-seeking radiopharmaceuticals are effective and have been widely used for pain palliation. The usual indications for radiopharmaceutical therapy include diffuse osteoblastic skeletal metastases demonstrated on bone scan, painful bone metastases not responding well to analgesics, and hormone-refractory metastatic prostate cancer. At present, strontium-89 (Sr-89), samarium-153 (Sm-153), phosphorus-32 (P-32), and radium 223 dichloride are radionuclides currently accepted as attractive therapeutic modalities for pain management (Table 2).
The clinical response is not immediate, and the average time to response is 1 to 2 weeks, but sometimes much longer. The main adverse reaction of systemic radiopharmaceutical therapy is myelotoxicity, such as thrombocytopenia and/or leukopenia. Occasionally, a so-called flare phenomenon of a transient pain increase may develop as well.29,30
Systemic Pharmacotherapy
Bisphosphonates are drugs commonly used to treat bone metastases. The benefits of bisphosphonate therapy are bone pain relief, the reduction of bone destruction, and the prevention of hypercalcemia and bone fractures. Bisphosphonates are typically more effective in osteolytic metastases and easily bind to bone, inhibiting bone resorption and increasing mineralization.31,32 Also, recent clinical studies suggest that bisphosphonates may inhibit tumor progression of bone metastases.
Related: Cancer Drugs Increase Rate of Preventable Hospital Admissions
Zoledronic acid is currently one of the most potent bisphosphonates and is effective in most types of metastatic bone lesions.33 Denosumab, another drug, diminishes osteoclast activity, leading to decreased bone resorption and increased bone mass.34,35 Denosumab is useful in preventing complications as a result of bone metastases from solid tumors and has been recently approved by the FDA for treatment of postmenopausal osteoporosis and the prevention of skeletal-related events (SREs) in cancer patients with bone metastases.
Adverse Effects
Zoledronate and bisphosphonates in general are not recommended for patients with kidney disease, including hypocalcaemia and severe renal impairment. A rare but well-known complication of bisphosphonate administration is osteonecrosis of the jaw, which is somewhat more common in MM, especially after dental extractions. General nonspecific adverse effects include fatigue, anemia, muscle aches, fever, and/or edema in the feet or legs. Flulike symptoms and generalized bone discomfort can also be seen shortly after the first infusion (Table 3).
Breast Cancer
Bisphosphonates have been shown to effectively prevent SREs in breast cancer patients with bone metastases.36 For example, zoledronic acid is the most effective bisphosphonate and has been demonstrated to significantly delay the time to development of a first SRE, reducing the overall SRE rate by 43%.37
Lung Cancer
According to Rosen and colleagues, lung cancer patients with bone metastases who received zoledronic acid (4 mg every 3 weeks) experienced a 9% reduction in SREs, a relative delay in median time to a first SRE, and a significantly reduced incidence of SREs.37
Prostate Cancer
Zoledronic acid is the only bisphosphonate that proved effective in the treatment of prostate cancer patients with bone metastases. Zoledronic acid significantly reduced the risk of SREs (36%) and bone pain as well as delayed the median time to first SRE (nearly 6 months).38,39
Multiple Myeloma
Bisphosphonates are recommended for bone metastases to prevent new bone lesions. Studies have shown pamidronate (90 mg every 4 weeks) resulted in a 41% reduction in SREs at 9 months and a 25% reduction at 21 months.40,41 Oral clodronate, another agent, also significantly reduced SREs and pain in patients with MM.42
Conclusion
Metastatic cancer with bone metastases occurs as cancer advances and spreads to the bone from the primary site of the original solid cancer. Nearly 70% of patients with prostate and breast cancers and about 30% to 40% of patients with lung cancer develop bone metastases. In addition, up to 95% of MMs involve bone. The most frequent and important symptom of bone metastasis is pain. In addition, bone metastasis causes bone fractures, hypercalcemia, and spinal cord and nerve compression. Imaging studies, such as bone scans and PET studies, are useful tools in diagnosing bone metastases.
Therapeutic management of bone metastases is expanding and rapidly evolving. For better therapy outcomes, treatment should be both individualized and coordinated among the care team, including a medical oncologist, radiation oncologist, surgeon, and radiologist. Available therapeutic modalities include radiation therapy, radiopharmaceutical therapy, surgery, and systemic pharmacotherapy (zoledronate, pamidronate, and denosumab).
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Bone metastasis is a relatively common complication of cancer, often developing as they advance, especially in prostate cancer and breast cancer. Bone metastasis can profoundly affect patients’ daily activities and quality of life (QOL) due to severe pain and associated major complications. Prompt palliative therapy is required for symptomatic pain relief and prevention of the devastating complications of bone metastasis.
Epidemiology
Bone is the most common and preferred site for metastatic involvement of cancer. Advanced cancers frequently develop metastases to the bone during the later phases of cancer progression. At least 100,000 patients develop bone metastases every year, although the exact number of bone metastases is not known.1 Multiple myeloma (MM), breast cancer, and prostate cancer are responsible for up to 70% of bone metastases cases.2 Gastrointestinal cancers contribute least to bone metastases: < 15% of all cases.2
Related: Effective Treatment Options for Metastatic Pancreatic Cancer
The prognosis of bone metastases is generally poor, although it partly depends on the primary site of the original cancer and on the presence of any additional metastases to visceral organs. For example, it is known that survival times are longer for patients with primary prostate or breast cancer than for patients with lung cancer primary tumors.3,4
Prostate and breast cancers are the most common primary cancers of bone metastases. At postmortem studies, patients who died of prostate cancer or breast cancer revealed evidence of bone metastases in up to 75% of cases (Figure 1). Regardless of their survival expectancy, however, most patients with bone metastasis need immediate medical attention and active palliative therapy to prevent devastating complications related to bone metastasis, such as pathologic bone fractures and severe bone pain.
Clinical Features
Multiple Myeloma
Multiple myeloma is the second most common hematologic malignancy and is caused by an abnormal accumulation of clonal plasma cells in the bone marrow. Characteristic clinical manifestations include bony destruction and related features of bone pain, anemia (80% of cases), hypocalcemia, and renal dysfunction. Pathologic fractures, renal failure, or hyperviscosity syndrome often develops. More than 20,000 new patients are diagnosed with MM and about 11,000 patients in the U.S. die of MM every year. Multiple myeloma and is twice as likely to develop in men as it is in women. A large number of MM cases are under the care of VAMCs (about 10%-12% of all MM cases).7,8
Abnormal laboratory tests show an elevated total protein level in the blood and/or urine (Bence Jones proteinuria). Serum electrophoresis detects M-protein in about 80% to 90% of patients. Patients may also present with renal failure. The differential diagnosis includes other malignancies, such as metastatic carcinoma, lymphoma, leukemia, and monoclonal gammopathy.
Pathophysiology
Normal bone tissue is made up of 2 different types of cells: osteoblasts and osteoclasts. New bone is constantly being produced while old bone is broken down. When tumor cells invade bone, the cancer cells produce 1 of 2 distinct substances; as a result, either osteoclasts or osteoblasts are stimulated, depending on tumor type metastasized to the bone. The activated osteoclasts then dissolve the bone, weakening the bone (osteolytic phenomenon), and the osteoblasts stimulate bone formation, hardening the bone (osteoblastic or sclerotic process).
Diagnosis and Evaluation
The most important first step in evaluating bone metastasis in a patient is to take a thorough, careful medical history and perform a physical examination. The examination not only helps locate suspected sites of bone metastases, but also helps determine necessary diagnostic studies.
The radiographic appearance of bone metastasis can be classified into 4 groups: osteolytic, osteoblastic, osteoporotic, and mixed. Imaging characteristics of osteolytic lesions include the destruction/thinning of bone, whereas osteoblastic (osteosclerotic) lesions appear with excess deposition of new bones. In contrast to malignant osteolytic lesions, osteoporotic lesions look like faded bone without cortical destruction or increased density.
The main choice of imaging study for screening suspected bone metastases is usually the bone scan (Figure 3). Plain radiographs are not useful in the early detection of bone metastases, because bone lesions do not show up on plain films until 30% to 50% of the bone mineral is lost.5,9 Although most metastatic bone lesions represent a mixture of osteoblastic and -lytic processes, metastatic lesions of lung cancer and breast cancer are predominantly osteolytic in contrast to mainly osteoblastic lesions of prostate cancer metastases.10
The osteoblastic process of bone metastases is best demonstrated on a bone scan; however, a positive bone scan does not necessarily indicate bone metastases, because it is not highly specific of metastatic disease. Several benign bone lesions (such as osteoarthritis, traumatic injury, and Paget disease) also show positive readings. Magnetic resonance imaging (MRI) is not useful in screening for bone metastases, but it is better in assessing bone metastases compared with a bone scan, because it is more sensitive, especially for spinal lesions. The reported sensitivity of MRI is 91% to 100%, whereas bone scan sensitivity is only 62% to 85%.11,12
Even though the bone scan has been assumed to be the best imaging study for bone metastases, positron emission tomography (PET) scans can be more useful in detecting osteolytic bone metastases, as they can light up areas of increased metabolic activity. Positron emission tomography scans, however, are less sensitive for osteoblastic metastases. An additional advantage of PET scans is that they can be used for whole-body scanning/surveillance to rule out visceral involvement.
Published studies indicate that bone scans better detect sclerotic bone metastases and PET scans are superior in revealing osteolytic metastases.13-15 Furthermore, in contrast to bone scans, PET scans can identify additional lesions in addition to bone lesion. According to recent reports, PET provides higher sensitivity and specificity in demonstrating lytic and sclerotic metastases compared with that of the bone scan.16
Breast Cancer
The role of PET for breast cancer is controversial. A study by Lonneux and colleagues found that PET is highly sensitive in confirming distant metastasis from breast cancer, whereas researchers reported a similar sensitivity but higher specificity.17 Ohta and colleagues reported that PET and bone scan had identical sensitivity (77.7%), but PET was more specific than the bone scan (97.6% vs 80.9%, respectively).14 The study conclusion by Cook and colleagues was that PET is superior to bone scan in the detection of metastatic osteolytic bone lesions from breast cancer, whereas osteoblastic metastatic bone lesions from breast cancer are less likely to be demonstrated on a PET scan.18
Houssami and Costelloe conducted a systematic review of 16 reported studies that comparatively tested the accuracy of imaging modalities for bone metastases in breast cancer.19 Sensitivity was generally similar between PET and bone scans in most studies reviewed. Four studies reported similar sensitivity but higher specificity for PET; the median specificity for PET and bone scan was 92% vs 85.5%, respectively (Figure 4).
Prostate Cancer
Prostate cancer is now established as the “classic” cancer for false-negative results on PET. Positron emission tomography does not perform well in the identification of osteoblastic skeletal metastases from prostate cancer. Yeh and colleagues reported only 18% positivity with PET.20 Interestingly, however, progressive metastatic prostate cancer showed a higher yield of 77% sensitivity with PET, perhaps because active osseous disease can be better picked up by PET scans.21
Related: Prostate Cancer Survivorship Care
Lung Cancer
For non-small cell lung cancer, both bone scan and PET showed a similar sensitivity for bone metastases detection, but the PET scan was more specific than the bone scan. Lung cancer often metastasizes to bone: up to 36% of patients at postmortem study. Lung cancer with bone metastases has a poor prognosis with median survival time typically measured in months. Most patients with bone metastases develop complications, such as severe pain, bone fracture, hypercalcemia, and spinal cord compression. Bone-targeted therapies play a greater role in the management of lung cancer patients, aiming for delaying disease progression and preserving QOL.22,23
Therapeutic Strategy and Management
Major morbidities associated with bone metastases include severe pain, hypercalcemia, bone fractures, spinal compression fractures, and cord or nerve root compression. This section reviews appropriate management techniques reported in the literature, particularly external beam radiation therapy.
Radiation Therapy
Pain is the most serious complication of bone metastases. Radiation therapy has been established as standard therapy and an effective pain palliation modality. Up to 80% of patients achieve partial pain relief, and > 33% of patients experience complete pain relief after radiation (Figure 5).24,25 Although a 3,000 cGy given over a 2-week period has been commonly used, a standard dose-fraction radiation treatment regimen has not been established.
The RTOG study was a randomized clinical study comparing various radiation schedules; 1,500 cGyin 1 week; vs 2,000 cGy in 1 week; vs 2,500 cGy in 1 week; vs 3,000 cGy in 2 weeks; or 4,050 cGy in 3 weeks. The conclusion was that local radiotherapy was an effective therapy for symptomatic and palliative therapy of bone metastases. Furthermore, low-dose radiotherapy was as good as various higher dose protracted courses of radiation treatments in terms of overall response rates (ORRs).24
Nearly 96% of patients eventually reported minimal pain relief to their palliative course of radiotherapy and experienced at least some pain relief within 4 weeks of radiation therapy. Complete pain relief was attained in 54% of patients regardless of the radiation dose-fraction schedules used. The median duration of complete pain response was about 12 weeks; > 70% of patients did not experience relapse of pain.26
Hartsell and colleagues investigated the efficacy of 800 cGy in a single fraction compared with 3,000 cGy in 10 fractions as part of a phase 3 randomized study of symptomatic therapy for pain palliation.27 The results showed 66% ORRs with similar complete and partial response rates (RRs) for both radiation groups. The complete RRs were 15% in the 800 cGy single-fraction arm vs 18% in the 3,000 cGy therapy arm, whereas partial RRs were 50% and 48% in the single vs the 3,000 cGy arms, respectively. However, there was a higher rate of retreatment for patients treated with the 800 cGy single-fraction radiotherapy. The 800 cGy single-fraction radiotherapy program seems rather popular in Canada and in European countries but is currently not widely used in the U.S.
Surgical Therapy
The surgical indications for managing bone metastases can vary, depending on disease location, surgeon’s preference, and patient’s overall disease status and related morbidities. Pain relief of fractured long bones (humerus, femur, or tibia) is crucial. The main goals of surgical intervention in these cases include the restoration of stability and functional mobility, pain control, and improving QOL. Weight-bearing bones (humerus/tibia) are especially at risk of bone fracture, and compromise of these is an indication of surgery. Postoperative external-beam radiation is recommended in most cases to eradicate residual microscopic disease or tumor progression.28
Radiopharmaceutical Therapy
Bone-seeking radiopharmaceuticals are effective and have been widely used for pain palliation. The usual indications for radiopharmaceutical therapy include diffuse osteoblastic skeletal metastases demonstrated on bone scan, painful bone metastases not responding well to analgesics, and hormone-refractory metastatic prostate cancer. At present, strontium-89 (Sr-89), samarium-153 (Sm-153), phosphorus-32 (P-32), and radium 223 dichloride are radionuclides currently accepted as attractive therapeutic modalities for pain management (Table 2).
The clinical response is not immediate, and the average time to response is 1 to 2 weeks, but sometimes much longer. The main adverse reaction of systemic radiopharmaceutical therapy is myelotoxicity, such as thrombocytopenia and/or leukopenia. Occasionally, a so-called flare phenomenon of a transient pain increase may develop as well.29,30
Systemic Pharmacotherapy
Bisphosphonates are drugs commonly used to treat bone metastases. The benefits of bisphosphonate therapy are bone pain relief, the reduction of bone destruction, and the prevention of hypercalcemia and bone fractures. Bisphosphonates are typically more effective in osteolytic metastases and easily bind to bone, inhibiting bone resorption and increasing mineralization.31,32 Also, recent clinical studies suggest that bisphosphonates may inhibit tumor progression of bone metastases.
Related: Cancer Drugs Increase Rate of Preventable Hospital Admissions
Zoledronic acid is currently one of the most potent bisphosphonates and is effective in most types of metastatic bone lesions.33 Denosumab, another drug, diminishes osteoclast activity, leading to decreased bone resorption and increased bone mass.34,35 Denosumab is useful in preventing complications as a result of bone metastases from solid tumors and has been recently approved by the FDA for treatment of postmenopausal osteoporosis and the prevention of skeletal-related events (SREs) in cancer patients with bone metastases.
Adverse Effects
Zoledronate and bisphosphonates in general are not recommended for patients with kidney disease, including hypocalcaemia and severe renal impairment. A rare but well-known complication of bisphosphonate administration is osteonecrosis of the jaw, which is somewhat more common in MM, especially after dental extractions. General nonspecific adverse effects include fatigue, anemia, muscle aches, fever, and/or edema in the feet or legs. Flulike symptoms and generalized bone discomfort can also be seen shortly after the first infusion (Table 3).
Breast Cancer
Bisphosphonates have been shown to effectively prevent SREs in breast cancer patients with bone metastases.36 For example, zoledronic acid is the most effective bisphosphonate and has been demonstrated to significantly delay the time to development of a first SRE, reducing the overall SRE rate by 43%.37
Lung Cancer
According to Rosen and colleagues, lung cancer patients with bone metastases who received zoledronic acid (4 mg every 3 weeks) experienced a 9% reduction in SREs, a relative delay in median time to a first SRE, and a significantly reduced incidence of SREs.37
Prostate Cancer
Zoledronic acid is the only bisphosphonate that proved effective in the treatment of prostate cancer patients with bone metastases. Zoledronic acid significantly reduced the risk of SREs (36%) and bone pain as well as delayed the median time to first SRE (nearly 6 months).38,39
Multiple Myeloma
Bisphosphonates are recommended for bone metastases to prevent new bone lesions. Studies have shown pamidronate (90 mg every 4 weeks) resulted in a 41% reduction in SREs at 9 months and a 25% reduction at 21 months.40,41 Oral clodronate, another agent, also significantly reduced SREs and pain in patients with MM.42
Conclusion
Metastatic cancer with bone metastases occurs as cancer advances and spreads to the bone from the primary site of the original solid cancer. Nearly 70% of patients with prostate and breast cancers and about 30% to 40% of patients with lung cancer develop bone metastases. In addition, up to 95% of MMs involve bone. The most frequent and important symptom of bone metastasis is pain. In addition, bone metastasis causes bone fractures, hypercalcemia, and spinal cord and nerve compression. Imaging studies, such as bone scans and PET studies, are useful tools in diagnosing bone metastases.
Therapeutic management of bone metastases is expanding and rapidly evolving. For better therapy outcomes, treatment should be both individualized and coordinated among the care team, including a medical oncologist, radiation oncologist, surgeon, and radiologist. Available therapeutic modalities include radiation therapy, radiopharmaceutical therapy, surgery, and systemic pharmacotherapy (zoledronate, pamidronate, and denosumab).
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225-249.
2. Cooleman RE. Metastatic bone disease: Clinical features, pathophysiology, and treatment strategies. Cancer Treat Rev. 2001;27(3):165-1763.
3. Hirabayashi H, Ebara S, Kinoshita T, et al. Clinical outcome and survival after palliative surgery for spinal metastases. Cancer. 2003;97(2):476-84.
4. van der Linden YM, Dijkstra SPDS, Vonk EJA, Marijnen CA, Leer JW; Dutch Bone Metastasis Study Group. Prediction of survival in patients with metastases in the spinal column. Cancer. 2005;103(2):320-328.
5. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12(20, pt 2):6243S-6249S.
6. Body JJ. Metastatic bone disease: Clinical and therapeutic aspects. Bone. 1992;13(suppl 1):S57-S62.
7. Siegel RS, Ma J, Zou Z, Jermal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
8. National Cancer Institute. SEER stat fact sheets: Myeloma. National Cancer Institute Website. http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed January 12, 2015.
9. Lentle BC, McGowan DG, Dierich H. Technetium-99M polyphosphate bone scanning in carcinoma of the prostate. Br J Urol. 1974;46(5):543-548.
10. Söderlund V. Radiological diagnosis of skeletal metastases. Eur Radiol. 1996;6(5):587-595.
11. Flickinger FW, Sanal SM. Bone marrow MRI: Techniques and accuracy for detecting breast cancer metastases. Magn Reson Imaging. 1994;12(6):829-35.
12. Hamaoka T, Madewell JE, Podoloff DA, Hortobagyi GN, Ueno NT. Bone imaging in metastatic breast cancer. J Clin Oncol. 2004;22(14):2942-2953.
13. Daldrup-Link HE, Franzius C, Link TM et al. Whole-body MR imaging for detection of bone metastases in children and young adults: Comparison with skeletal scintigraphy and FDG PET. AJR Am J Roentgenol. 2001;177(1):229-236.
14. Ohta M, Tokuda Y, Suzuki Y, et al. Whole body PET for the evaluation of bony metastases in patients with breast cancer: Comparison with 99Tcm-MDP bone scintigraphy. Nucl Med Commun. 2001;22(8):875-879.
15. Koolen BB, Vegt E, Rutgers EJ, et al. FDG-avid sclerotic bone metastases in breast cancer patients: A PET/CT case series. Ann Nucl Med. 2012;26(1):86-91.
16. Even-Sapir E, Metser U, Flusser G, et al. Assessment of malignant skeletal disease: Initial experience with 18F-fluoride PET/CT and comparison between 18F-fluoride PET and 18F-fluoride PET/CT. J Nucl Med. 2004;45(2):272-278.
17. Lonneux M, Borbath II, Berlière M, Kirkove C, Pauwels S. The place of whole-body PET FDG for the diagnosis of distant recurrence of breast cancer. Clin Positron Imaging. 2000;3(2):45-49.
18. Cook GJ, Houston S, Rubens R, Maisey MN, Fogelman I. Detection of bone metastases in breast cancer by 18FDG PET: Differing metabolic activity in osteoblastic and osteolytic lesions. J Clin Oncol. 1998;16(10):3375-3379.
19. Houssami N, Costelloe CM. Imaging bone metastases in breast cancer: Evidence on comparative test accuracy. Ann Oncol. 2012;23(4):834-843.
20. Yeh SD, Imbriaco M, Larson SM, et al. Detection of bony metastases of androgen-independent prostate cancer by PET-FDG. Nucl Med Biol. 1996;23(6):693-697.
21. Morris MJ, Akhurst T, Osman I, et al. Fluorinated deoxyglucose positron emission tomography imaging in progressive metastatic prostate cancer. Urology. 2002;59(6):913-918.
22. Rosen LS, Gordon D, Tchekmedyian S, et al. Zoledronic acid versus placebo in the treatment of skeletal metastases in patients with lung cancer and other solid tumors: A phase III, double-blind, randomized trial—the Zoledronic Acid Lung Cancer and Other Solid Tumors Study Group. J Clin Oncol. 2003;21(16):3150-3157.
23. Hillner BE, Ingle JN, Chlebowski RT, et al; American Society of Clinical Psychology. American Society of Clinical Oncology 2003 update on the role of bisphosphonates and bone health issues in women with breast cancer. J Clin Oncol. 2003;21(21):4042-4057.
24. Chow E, Harris K, Fan G, Tsao M, Size WM. Palliative radiotherapy trials for bone metastases: A systematic review. J Clin Oncol. 2007;25(11):1423-1436.
25. Wu JS, Wong R, Johnston M, Bezjak A, Whelan T; Cancer Care Ontario Practice Guidelines Initiative Supportive Care Group. Meta-analysis of dose-fractionation radiotherapy trials for the palliation of painful bone metastases. Int J Radiat Oncol Biol Phys. 2003;55(3):594-605.
26. Tong D, Gillick L, Hendrickson FR. The palliation of symptomatic osseous metastases. Final results of the study by the Radiation Therapy Oncology Group. Cancer. 1982;50(5):893-899.
27. Hartsell WF, Scott CB, Bruner DW, et al. Randomized trial of short- versus long-course radiotherapy for palliation of painful bone metastases. J Natl Cancer Inst. 2005;97(11):798-804.
28. Frassica DA. General principles of external beam radiation therapy for skeletal metastases. Clin Orthop Relat Res. 2003;415(suppl):S158-S164.
29. Silberstein EB. Systemic radiopharmaceutical therapy of painful osteoblastic metastases. Semin Radiat Oncol. 2000;10(3):240-249.
30. Neville-Webbe HL, Gnant M, Coleman RE. Potential anticancer properties of bisphosphonates. Semin Oncol. 2010;37(suppl 1):S53-S65.
31. Loftus LS, Edwards-Bennett S, Sokol GH. Systemic therapy for bone metastases. Cancer Control. 2012;19(2):145-153.
32. Rosen L, Harland SJ, Oosterlinck W. Broad clinical activity of zoledronic acid in osteolytic to osteoblastic bone lesions in patients with a broad range of solid tumors. Am J Clin Oncol. 2002;25(6)(suppl 1):S19-S24.
33. Fornier MN. Denosumab: Second chapter in controlling bone metastases or a new book? J Clin Oncol. 2010;28(35):5127-5131.
34. Mortimer JE, Pal SK. Safety considerations for use of bone-targeted agents in patients with cancer. Semin Oncol. 2010;37(suppl 1):S66-S72.
35. Pavlakis N, Schmidt R, Stockler M. Bisphosphonates for breast cancer. Cochrane Database Syst Rev. 2005;3:CD003474.
36. Kohno N, Aogi K, Minami H, et al. Zoledronic acid significantly reduces skeletal complications compared with placebo in Japanese women with bone metastases from breast cancer: A randomized, placebo-controlled trial. J Clin Oncol. 2005;23(15):3314-3321.
37. Rosen LS, Gordon D, Tchekmedyian NS, et al. Long-term efficacy and safety of zoledronic acid in the treatment of skeletal metastases in patients with nonsmall cell lung carcinoma and other solid tumors: A randomized, phase III, double-blind, placebo-controlled trial. Cancer. 2004;100(12):2613-2621.
38. Saad F, Gleason DM, Murray R, et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst. 2004;96(11):879-882.
39. Saad F, Eastham J. Zoledronic acid improves clinical outcomes when administered before onset of bone pain in patients with prostate cancer. Urology. 2010;76(5):1175-1181.
40. Berenson JR, Lichtenstein A, Porter L, et al. Efficacy of pamidronate in reducing skeletal events in patients with advanced multiple myeloma. Myeloma Aredia Study Group. N Engl J Med. 1996;334(8):488-493.
41. Berenson JR, Lichtenstein A, Porter L, et al. Long-term pamidronate treatment of advanced multiple myeloma patients reduces skeletal events. Myeloma Aredia Study Group. J Clin Oncol. 1998;16(2):593-602.
42. Lahtinen R, Laakso M, Palva I, Virkkunen P, Elomaa I. Randomised, placebo-controlled multicentre trial of clodronate in multiple myeloma. Finnish Leukaemia Group. Lancet. 1992;340(8827):1049-1052.
1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225-249.
2. Cooleman RE. Metastatic bone disease: Clinical features, pathophysiology, and treatment strategies. Cancer Treat Rev. 2001;27(3):165-1763.
3. Hirabayashi H, Ebara S, Kinoshita T, et al. Clinical outcome and survival after palliative surgery for spinal metastases. Cancer. 2003;97(2):476-84.
4. van der Linden YM, Dijkstra SPDS, Vonk EJA, Marijnen CA, Leer JW; Dutch Bone Metastasis Study Group. Prediction of survival in patients with metastases in the spinal column. Cancer. 2005;103(2):320-328.
5. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12(20, pt 2):6243S-6249S.
6. Body JJ. Metastatic bone disease: Clinical and therapeutic aspects. Bone. 1992;13(suppl 1):S57-S62.
7. Siegel RS, Ma J, Zou Z, Jermal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9-29.
8. National Cancer Institute. SEER stat fact sheets: Myeloma. National Cancer Institute Website. http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed January 12, 2015.
9. Lentle BC, McGowan DG, Dierich H. Technetium-99M polyphosphate bone scanning in carcinoma of the prostate. Br J Urol. 1974;46(5):543-548.
10. Söderlund V. Radiological diagnosis of skeletal metastases. Eur Radiol. 1996;6(5):587-595.
11. Flickinger FW, Sanal SM. Bone marrow MRI: Techniques and accuracy for detecting breast cancer metastases. Magn Reson Imaging. 1994;12(6):829-35.
12. Hamaoka T, Madewell JE, Podoloff DA, Hortobagyi GN, Ueno NT. Bone imaging in metastatic breast cancer. J Clin Oncol. 2004;22(14):2942-2953.
13. Daldrup-Link HE, Franzius C, Link TM et al. Whole-body MR imaging for detection of bone metastases in children and young adults: Comparison with skeletal scintigraphy and FDG PET. AJR Am J Roentgenol. 2001;177(1):229-236.
14. Ohta M, Tokuda Y, Suzuki Y, et al. Whole body PET for the evaluation of bony metastases in patients with breast cancer: Comparison with 99Tcm-MDP bone scintigraphy. Nucl Med Commun. 2001;22(8):875-879.
15. Koolen BB, Vegt E, Rutgers EJ, et al. FDG-avid sclerotic bone metastases in breast cancer patients: A PET/CT case series. Ann Nucl Med. 2012;26(1):86-91.
16. Even-Sapir E, Metser U, Flusser G, et al. Assessment of malignant skeletal disease: Initial experience with 18F-fluoride PET/CT and comparison between 18F-fluoride PET and 18F-fluoride PET/CT. J Nucl Med. 2004;45(2):272-278.
17. Lonneux M, Borbath II, Berlière M, Kirkove C, Pauwels S. The place of whole-body PET FDG for the diagnosis of distant recurrence of breast cancer. Clin Positron Imaging. 2000;3(2):45-49.
18. Cook GJ, Houston S, Rubens R, Maisey MN, Fogelman I. Detection of bone metastases in breast cancer by 18FDG PET: Differing metabolic activity in osteoblastic and osteolytic lesions. J Clin Oncol. 1998;16(10):3375-3379.
19. Houssami N, Costelloe CM. Imaging bone metastases in breast cancer: Evidence on comparative test accuracy. Ann Oncol. 2012;23(4):834-843.
20. Yeh SD, Imbriaco M, Larson SM, et al. Detection of bony metastases of androgen-independent prostate cancer by PET-FDG. Nucl Med Biol. 1996;23(6):693-697.
21. Morris MJ, Akhurst T, Osman I, et al. Fluorinated deoxyglucose positron emission tomography imaging in progressive metastatic prostate cancer. Urology. 2002;59(6):913-918.
22. Rosen LS, Gordon D, Tchekmedyian S, et al. Zoledronic acid versus placebo in the treatment of skeletal metastases in patients with lung cancer and other solid tumors: A phase III, double-blind, randomized trial—the Zoledronic Acid Lung Cancer and Other Solid Tumors Study Group. J Clin Oncol. 2003;21(16):3150-3157.
23. Hillner BE, Ingle JN, Chlebowski RT, et al; American Society of Clinical Psychology. American Society of Clinical Oncology 2003 update on the role of bisphosphonates and bone health issues in women with breast cancer. J Clin Oncol. 2003;21(21):4042-4057.
24. Chow E, Harris K, Fan G, Tsao M, Size WM. Palliative radiotherapy trials for bone metastases: A systematic review. J Clin Oncol. 2007;25(11):1423-1436.
25. Wu JS, Wong R, Johnston M, Bezjak A, Whelan T; Cancer Care Ontario Practice Guidelines Initiative Supportive Care Group. Meta-analysis of dose-fractionation radiotherapy trials for the palliation of painful bone metastases. Int J Radiat Oncol Biol Phys. 2003;55(3):594-605.
26. Tong D, Gillick L, Hendrickson FR. The palliation of symptomatic osseous metastases. Final results of the study by the Radiation Therapy Oncology Group. Cancer. 1982;50(5):893-899.
27. Hartsell WF, Scott CB, Bruner DW, et al. Randomized trial of short- versus long-course radiotherapy for palliation of painful bone metastases. J Natl Cancer Inst. 2005;97(11):798-804.
28. Frassica DA. General principles of external beam radiation therapy for skeletal metastases. Clin Orthop Relat Res. 2003;415(suppl):S158-S164.
29. Silberstein EB. Systemic radiopharmaceutical therapy of painful osteoblastic metastases. Semin Radiat Oncol. 2000;10(3):240-249.
30. Neville-Webbe HL, Gnant M, Coleman RE. Potential anticancer properties of bisphosphonates. Semin Oncol. 2010;37(suppl 1):S53-S65.
31. Loftus LS, Edwards-Bennett S, Sokol GH. Systemic therapy for bone metastases. Cancer Control. 2012;19(2):145-153.
32. Rosen L, Harland SJ, Oosterlinck W. Broad clinical activity of zoledronic acid in osteolytic to osteoblastic bone lesions in patients with a broad range of solid tumors. Am J Clin Oncol. 2002;25(6)(suppl 1):S19-S24.
33. Fornier MN. Denosumab: Second chapter in controlling bone metastases or a new book? J Clin Oncol. 2010;28(35):5127-5131.
34. Mortimer JE, Pal SK. Safety considerations for use of bone-targeted agents in patients with cancer. Semin Oncol. 2010;37(suppl 1):S66-S72.
35. Pavlakis N, Schmidt R, Stockler M. Bisphosphonates for breast cancer. Cochrane Database Syst Rev. 2005;3:CD003474.
36. Kohno N, Aogi K, Minami H, et al. Zoledronic acid significantly reduces skeletal complications compared with placebo in Japanese women with bone metastases from breast cancer: A randomized, placebo-controlled trial. J Clin Oncol. 2005;23(15):3314-3321.
37. Rosen LS, Gordon D, Tchekmedyian NS, et al. Long-term efficacy and safety of zoledronic acid in the treatment of skeletal metastases in patients with nonsmall cell lung carcinoma and other solid tumors: A randomized, phase III, double-blind, placebo-controlled trial. Cancer. 2004;100(12):2613-2621.
38. Saad F, Gleason DM, Murray R, et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J Natl Cancer Inst. 2004;96(11):879-882.
39. Saad F, Eastham J. Zoledronic acid improves clinical outcomes when administered before onset of bone pain in patients with prostate cancer. Urology. 2010;76(5):1175-1181.
40. Berenson JR, Lichtenstein A, Porter L, et al. Efficacy of pamidronate in reducing skeletal events in patients with advanced multiple myeloma. Myeloma Aredia Study Group. N Engl J Med. 1996;334(8):488-493.
41. Berenson JR, Lichtenstein A, Porter L, et al. Long-term pamidronate treatment of advanced multiple myeloma patients reduces skeletal events. Myeloma Aredia Study Group. J Clin Oncol. 1998;16(2):593-602.
42. Lahtinen R, Laakso M, Palva I, Virkkunen P, Elomaa I. Randomised, placebo-controlled multicentre trial of clodronate in multiple myeloma. Finnish Leukaemia Group. Lancet. 1992;340(8827):1049-1052.
Targeted Therapy for Chronic Lymphocytic Leukemia
This presentation by Adrian Wiestner, MD, PhD, from the 2014 AVAHO Meeting in Portland, Oregon, provides an overview of new insights into the pathogenesis and treatment of CLL, how to interpret molecular targets during treatment, and the advantages and disadvantages of these treatment options for patients.
"The standard of care today is really chemo-immunotherapy," Wiestner said. "Ideally, we would like to have a more disease-directed therapy that is tolerable and active."
This presentation by Adrian Wiestner, MD, PhD, from the 2014 AVAHO Meeting in Portland, Oregon, provides an overview of new insights into the pathogenesis and treatment of CLL, how to interpret molecular targets during treatment, and the advantages and disadvantages of these treatment options for patients.
"The standard of care today is really chemo-immunotherapy," Wiestner said. "Ideally, we would like to have a more disease-directed therapy that is tolerable and active."
This presentation by Adrian Wiestner, MD, PhD, from the 2014 AVAHO Meeting in Portland, Oregon, provides an overview of new insights into the pathogenesis and treatment of CLL, how to interpret molecular targets during treatment, and the advantages and disadvantages of these treatment options for patients.
"The standard of care today is really chemo-immunotherapy," Wiestner said. "Ideally, we would like to have a more disease-directed therapy that is tolerable and active."

Treating Hodgkin Lymphoma
Christopher Flowers, MD, discusses current management strategies for newly diagnosed and relapsed patients with Hodgkin Lymphoma (HL). He also discusses emerging opportunities for the use of novel approaches to treat HL and surveillance of patients with this type of cancer.
"Stem cell transplant still remains the standard approach for patients with relapsed Hodgkin Lymphoma," Flowers said during his presentation during the 2014 AVAHO Meeting's Lymphoma Mini-Symposium. "Turning to the novel agents... there are a number of potential approaches that can be used."
Christopher Flowers, MD, discusses current management strategies for newly diagnosed and relapsed patients with Hodgkin Lymphoma (HL). He also discusses emerging opportunities for the use of novel approaches to treat HL and surveillance of patients with this type of cancer.
"Stem cell transplant still remains the standard approach for patients with relapsed Hodgkin Lymphoma," Flowers said during his presentation during the 2014 AVAHO Meeting's Lymphoma Mini-Symposium. "Turning to the novel agents... there are a number of potential approaches that can be used."
Christopher Flowers, MD, discusses current management strategies for newly diagnosed and relapsed patients with Hodgkin Lymphoma (HL). He also discusses emerging opportunities for the use of novel approaches to treat HL and surveillance of patients with this type of cancer.
"Stem cell transplant still remains the standard approach for patients with relapsed Hodgkin Lymphoma," Flowers said during his presentation during the 2014 AVAHO Meeting's Lymphoma Mini-Symposium. "Turning to the novel agents... there are a number of potential approaches that can be used."

AVAHO 2014 Meeting: Lymphoma Mini-Symposium Preview
Federal Practitioner recently talked with Dr. Adrian Weistner and Dr. Mark Roschewski of the National Institutes of Health. Both doctors will be presenting during the September 12, 2014 Lymphoma Mini-Symposium and panel discussion that kicks off this weekend’s 2014 AVAHO Meeting in Portland, Oregon.
Federal Practitioner recently talked with Dr. Adrian Weistner and Dr. Mark Roschewski of the National Institutes of Health. Both doctors will be presenting during the September 12, 2014 Lymphoma Mini-Symposium and panel discussion that kicks off this weekend’s 2014 AVAHO Meeting in Portland, Oregon.
Federal Practitioner recently talked with Dr. Adrian Weistner and Dr. Mark Roschewski of the National Institutes of Health. Both doctors will be presenting during the September 12, 2014 Lymphoma Mini-Symposium and panel discussion that kicks off this weekend’s 2014 AVAHO Meeting in Portland, Oregon.

Preventing Skeletal-Related Events in Veterans on Bisphosphonates for Bone Metastases
Purpose: Multiple myeloma and solid tumor metastases can cause bone disease leading to skeletal-related events (SREs) such as bone pain, fractures, and spinal cord compression. Intravenous bisphosphonate therapy—which is indicated in such cases—can lead to osteonecrosis of the jaw and hypocalcemia further putting patients at risk for SREs. These risks can be avoided by dental evaluation before bisphosphonate therapy and calcium and vitamin D supplementation throughout treatment. Our study of veterans treated with bisphosphonates for bone metastases or multiple myeloma aimed to (1) assess screening dental evaluation prior to treatment; and (2) measure effectiveness of calcium and vitamin D supplementation.
Methods: We performed a retrospective chart review at the James J. Peters VAMC of 117 veterans with multiple myeloma or bone metastases who received intravenous bisphosphonate therapy between January 2008 and November 2013. Those receiving bisphosphonates for other morbidities such as osteoporosis or hypercalcemia were excluded. Those getting dental clearance before intravenous bisphosphonate therapy and supplementation of vitamin D and calcium were assessed. Charts were further reviewed to gather outcomes data on incidence of osteonecrosis of the jaw and SREs such as bone pain, pathologic and traumatic fractures, orthopedic surgery, spine or nerve root compression. These data were analyzed using descriptive statistics to calculate frequencies, mean/median, and proportions. Odds ratios were calculated to assess differences in SRE outcomes for those who received supplementation as compared to those who did not get supplementation with calcium and vitamin D.
Results: Of the 117 patients included in the study, 97% were males aged from 58 to 92 years. Of these, 55 (47%) had prostate cancer, 21 (17%) had multiple myeloma, and 16 (14%) had lung cancer. All patients receiving bisphosphonates for bone metastases had undergone a dental evaluation prior to starting therapy; none were reported to have osteonecrosis of the jaw. However, only 78% had vitamin D levels checked before therapy; 69% of these were vitamin D deficient and received vitamin D supplementation. Overall, rates of calcium and vitamin D supplementation were very low (34% and 41%, respectively). Fifty-four percent of the patients reported an SRE; 49% with bone pain, 13% with pathological fractures, 7% with traumatic fractures, and 8% with nerve root compression. Vitamin D supplementation significantly reduced the odds of an SRE for our patients (OR 0.37, 95% CI = 0.19- 0.74, P < .05).
Conclusions: Onset of SREs can be reduced or delayed with bisphosphonates; however, patients need prior screening for osteonecrosis of the jaw and optimized calcium and vitamin D levels. Our study showed that although screening for osteonecrosis of the jaw was at optimum levels, supplementation with calcium and vitamin D was lacking in patients on bisphosphonates. In our study, vitamin D supplementation reduced the risk of an SRE by 63%. Hence, adequate prevention with vitamin D supplementation can improve bone health among veterans with multiple myeloma or bone metastases. Data-based policies and practices need to be incorporated to provide care to ensure adequate bone health.
Purpose: Multiple myeloma and solid tumor metastases can cause bone disease leading to skeletal-related events (SREs) such as bone pain, fractures, and spinal cord compression. Intravenous bisphosphonate therapy—which is indicated in such cases—can lead to osteonecrosis of the jaw and hypocalcemia further putting patients at risk for SREs. These risks can be avoided by dental evaluation before bisphosphonate therapy and calcium and vitamin D supplementation throughout treatment. Our study of veterans treated with bisphosphonates for bone metastases or multiple myeloma aimed to (1) assess screening dental evaluation prior to treatment; and (2) measure effectiveness of calcium and vitamin D supplementation.
Methods: We performed a retrospective chart review at the James J. Peters VAMC of 117 veterans with multiple myeloma or bone metastases who received intravenous bisphosphonate therapy between January 2008 and November 2013. Those receiving bisphosphonates for other morbidities such as osteoporosis or hypercalcemia were excluded. Those getting dental clearance before intravenous bisphosphonate therapy and supplementation of vitamin D and calcium were assessed. Charts were further reviewed to gather outcomes data on incidence of osteonecrosis of the jaw and SREs such as bone pain, pathologic and traumatic fractures, orthopedic surgery, spine or nerve root compression. These data were analyzed using descriptive statistics to calculate frequencies, mean/median, and proportions. Odds ratios were calculated to assess differences in SRE outcomes for those who received supplementation as compared to those who did not get supplementation with calcium and vitamin D.
Results: Of the 117 patients included in the study, 97% were males aged from 58 to 92 years. Of these, 55 (47%) had prostate cancer, 21 (17%) had multiple myeloma, and 16 (14%) had lung cancer. All patients receiving bisphosphonates for bone metastases had undergone a dental evaluation prior to starting therapy; none were reported to have osteonecrosis of the jaw. However, only 78% had vitamin D levels checked before therapy; 69% of these were vitamin D deficient and received vitamin D supplementation. Overall, rates of calcium and vitamin D supplementation were very low (34% and 41%, respectively). Fifty-four percent of the patients reported an SRE; 49% with bone pain, 13% with pathological fractures, 7% with traumatic fractures, and 8% with nerve root compression. Vitamin D supplementation significantly reduced the odds of an SRE for our patients (OR 0.37, 95% CI = 0.19- 0.74, P < .05).
Conclusions: Onset of SREs can be reduced or delayed with bisphosphonates; however, patients need prior screening for osteonecrosis of the jaw and optimized calcium and vitamin D levels. Our study showed that although screening for osteonecrosis of the jaw was at optimum levels, supplementation with calcium and vitamin D was lacking in patients on bisphosphonates. In our study, vitamin D supplementation reduced the risk of an SRE by 63%. Hence, adequate prevention with vitamin D supplementation can improve bone health among veterans with multiple myeloma or bone metastases. Data-based policies and practices need to be incorporated to provide care to ensure adequate bone health.
Purpose: Multiple myeloma and solid tumor metastases can cause bone disease leading to skeletal-related events (SREs) such as bone pain, fractures, and spinal cord compression. Intravenous bisphosphonate therapy—which is indicated in such cases—can lead to osteonecrosis of the jaw and hypocalcemia further putting patients at risk for SREs. These risks can be avoided by dental evaluation before bisphosphonate therapy and calcium and vitamin D supplementation throughout treatment. Our study of veterans treated with bisphosphonates for bone metastases or multiple myeloma aimed to (1) assess screening dental evaluation prior to treatment; and (2) measure effectiveness of calcium and vitamin D supplementation.
Methods: We performed a retrospective chart review at the James J. Peters VAMC of 117 veterans with multiple myeloma or bone metastases who received intravenous bisphosphonate therapy between January 2008 and November 2013. Those receiving bisphosphonates for other morbidities such as osteoporosis or hypercalcemia were excluded. Those getting dental clearance before intravenous bisphosphonate therapy and supplementation of vitamin D and calcium were assessed. Charts were further reviewed to gather outcomes data on incidence of osteonecrosis of the jaw and SREs such as bone pain, pathologic and traumatic fractures, orthopedic surgery, spine or nerve root compression. These data were analyzed using descriptive statistics to calculate frequencies, mean/median, and proportions. Odds ratios were calculated to assess differences in SRE outcomes for those who received supplementation as compared to those who did not get supplementation with calcium and vitamin D.
Results: Of the 117 patients included in the study, 97% were males aged from 58 to 92 years. Of these, 55 (47%) had prostate cancer, 21 (17%) had multiple myeloma, and 16 (14%) had lung cancer. All patients receiving bisphosphonates for bone metastases had undergone a dental evaluation prior to starting therapy; none were reported to have osteonecrosis of the jaw. However, only 78% had vitamin D levels checked before therapy; 69% of these were vitamin D deficient and received vitamin D supplementation. Overall, rates of calcium and vitamin D supplementation were very low (34% and 41%, respectively). Fifty-four percent of the patients reported an SRE; 49% with bone pain, 13% with pathological fractures, 7% with traumatic fractures, and 8% with nerve root compression. Vitamin D supplementation significantly reduced the odds of an SRE for our patients (OR 0.37, 95% CI = 0.19- 0.74, P < .05).
Conclusions: Onset of SREs can be reduced or delayed with bisphosphonates; however, patients need prior screening for osteonecrosis of the jaw and optimized calcium and vitamin D levels. Our study showed that although screening for osteonecrosis of the jaw was at optimum levels, supplementation with calcium and vitamin D was lacking in patients on bisphosphonates. In our study, vitamin D supplementation reduced the risk of an SRE by 63%. Hence, adequate prevention with vitamin D supplementation can improve bone health among veterans with multiple myeloma or bone metastases. Data-based policies and practices need to be incorporated to provide care to ensure adequate bone health.
A Familial Cluster of Myelodysplasia and Myelofibrosis
Purpose: Familial clusters of either myelodysplasia (MDS) or myelofibrosis (MF) are well documented although uncommon. The inheritance of a somatic driver mutation presumably accounts for these kindreds, and DNA sequencing has revealed multiple candidate mutations (JAK2, ASXL1, TET2, EZH2, SRSF2) that are shared across the spectrum of these disorders. Given this overlap of nonrandom mutations in MDS and MF, it is surprising that clusters of both MDS and MF within the same family seem to be very rare. Recently, however, we observed a woman with MDS who reported a deceased sibling with MDS and a deceased paternal uncle with MF.
Methods: A careful family history and a review of the medical records, archived pathology, and clinical course were performed and compared with that of the index case.
Results: The index case is a woman aged 49 years presenting with severe anemia in July 2013. A bone marrow (BM) biopsy was mildly hypocellular with reduced erythroid maturation, dyspoietic hypolobated megakaryocytes and no increase in blasts. Cytogenetics revealed an isolated del(5)(q13q31) (in 20/20 cells) with del(7q) in 3/20 cells. Lenalidomide therapy resulted in initial transfusion independence. The deceased brother presented April 1994 with weakness at age 39. A CBC showed pancytopenia with a few blasts and nucleated rbc. A BM biopsy revealed predominantly extreme erythroid megaloblastosis with marked nuclear atypia, hypolobated megakaryocytes without fibrosis, mildly dyspoietic myeloid maturation, and 5% nonerythroid CD34+ blasts. Complex cytogenetic changes included monosomy 7 and der(5), likely a functional 5q deletion or duplication. Despite transfusion support, the patient died of infection and CNS hemorrhage after several months. The paternal uncle presented in June 1996 at age 60 with anemia. The CBC showed leukoerythro-blastosis with prominent dacrocytes, mild thrombo-cytopenia but no dyspoiesis. A BM biopsy revealed marked fibrosis, prominent osteosclerosis, and large hyperlobated hyperchromatic megakaryo-cytes. Overall the history, blood, and biopsy findings were consistent with primary MF but not MDS with fibrosis. Death occurred after 3 years of transfusion support.
Conclusions: Despite the extreme rarity of reported MDS and MF cases within a single family, the kindred reported here suggests the existence of an inherited gene defect that increases the risk of developing either MDS or MF. Presumably, the onset of clinically evident disease and its eventual phenotype is determined by the accumulation of additional different secondary genetic changes. The lack of disease in 6 other siblings and the deceased father aged 75 years, however, argues that any hypothetical driver mutation has incomplete penetrance, ie, a reduced likelihood of either disorder developing within a lifetime. Moreover, since this report cannot rule out either chance alone or a common environmental etiology despite substantial age and household differences, DNA sequencing studies will be necessary to identify a putative inherited gene mutation driving the development of both MDS or MF in this unusual kindred.
Purpose: Familial clusters of either myelodysplasia (MDS) or myelofibrosis (MF) are well documented although uncommon. The inheritance of a somatic driver mutation presumably accounts for these kindreds, and DNA sequencing has revealed multiple candidate mutations (JAK2, ASXL1, TET2, EZH2, SRSF2) that are shared across the spectrum of these disorders. Given this overlap of nonrandom mutations in MDS and MF, it is surprising that clusters of both MDS and MF within the same family seem to be very rare. Recently, however, we observed a woman with MDS who reported a deceased sibling with MDS and a deceased paternal uncle with MF.
Methods: A careful family history and a review of the medical records, archived pathology, and clinical course were performed and compared with that of the index case.
Results: The index case is a woman aged 49 years presenting with severe anemia in July 2013. A bone marrow (BM) biopsy was mildly hypocellular with reduced erythroid maturation, dyspoietic hypolobated megakaryocytes and no increase in blasts. Cytogenetics revealed an isolated del(5)(q13q31) (in 20/20 cells) with del(7q) in 3/20 cells. Lenalidomide therapy resulted in initial transfusion independence. The deceased brother presented April 1994 with weakness at age 39. A CBC showed pancytopenia with a few blasts and nucleated rbc. A BM biopsy revealed predominantly extreme erythroid megaloblastosis with marked nuclear atypia, hypolobated megakaryocytes without fibrosis, mildly dyspoietic myeloid maturation, and 5% nonerythroid CD34+ blasts. Complex cytogenetic changes included monosomy 7 and der(5), likely a functional 5q deletion or duplication. Despite transfusion support, the patient died of infection and CNS hemorrhage after several months. The paternal uncle presented in June 1996 at age 60 with anemia. The CBC showed leukoerythro-blastosis with prominent dacrocytes, mild thrombo-cytopenia but no dyspoiesis. A BM biopsy revealed marked fibrosis, prominent osteosclerosis, and large hyperlobated hyperchromatic megakaryo-cytes. Overall the history, blood, and biopsy findings were consistent with primary MF but not MDS with fibrosis. Death occurred after 3 years of transfusion support.
Conclusions: Despite the extreme rarity of reported MDS and MF cases within a single family, the kindred reported here suggests the existence of an inherited gene defect that increases the risk of developing either MDS or MF. Presumably, the onset of clinically evident disease and its eventual phenotype is determined by the accumulation of additional different secondary genetic changes. The lack of disease in 6 other siblings and the deceased father aged 75 years, however, argues that any hypothetical driver mutation has incomplete penetrance, ie, a reduced likelihood of either disorder developing within a lifetime. Moreover, since this report cannot rule out either chance alone or a common environmental etiology despite substantial age and household differences, DNA sequencing studies will be necessary to identify a putative inherited gene mutation driving the development of both MDS or MF in this unusual kindred.
Purpose: Familial clusters of either myelodysplasia (MDS) or myelofibrosis (MF) are well documented although uncommon. The inheritance of a somatic driver mutation presumably accounts for these kindreds, and DNA sequencing has revealed multiple candidate mutations (JAK2, ASXL1, TET2, EZH2, SRSF2) that are shared across the spectrum of these disorders. Given this overlap of nonrandom mutations in MDS and MF, it is surprising that clusters of both MDS and MF within the same family seem to be very rare. Recently, however, we observed a woman with MDS who reported a deceased sibling with MDS and a deceased paternal uncle with MF.
Methods: A careful family history and a review of the medical records, archived pathology, and clinical course were performed and compared with that of the index case.
Results: The index case is a woman aged 49 years presenting with severe anemia in July 2013. A bone marrow (BM) biopsy was mildly hypocellular with reduced erythroid maturation, dyspoietic hypolobated megakaryocytes and no increase in blasts. Cytogenetics revealed an isolated del(5)(q13q31) (in 20/20 cells) with del(7q) in 3/20 cells. Lenalidomide therapy resulted in initial transfusion independence. The deceased brother presented April 1994 with weakness at age 39. A CBC showed pancytopenia with a few blasts and nucleated rbc. A BM biopsy revealed predominantly extreme erythroid megaloblastosis with marked nuclear atypia, hypolobated megakaryocytes without fibrosis, mildly dyspoietic myeloid maturation, and 5% nonerythroid CD34+ blasts. Complex cytogenetic changes included monosomy 7 and der(5), likely a functional 5q deletion or duplication. Despite transfusion support, the patient died of infection and CNS hemorrhage after several months. The paternal uncle presented in June 1996 at age 60 with anemia. The CBC showed leukoerythro-blastosis with prominent dacrocytes, mild thrombo-cytopenia but no dyspoiesis. A BM biopsy revealed marked fibrosis, prominent osteosclerosis, and large hyperlobated hyperchromatic megakaryo-cytes. Overall the history, blood, and biopsy findings were consistent with primary MF but not MDS with fibrosis. Death occurred after 3 years of transfusion support.
Conclusions: Despite the extreme rarity of reported MDS and MF cases within a single family, the kindred reported here suggests the existence of an inherited gene defect that increases the risk of developing either MDS or MF. Presumably, the onset of clinically evident disease and its eventual phenotype is determined by the accumulation of additional different secondary genetic changes. The lack of disease in 6 other siblings and the deceased father aged 75 years, however, argues that any hypothetical driver mutation has incomplete penetrance, ie, a reduced likelihood of either disorder developing within a lifetime. Moreover, since this report cannot rule out either chance alone or a common environmental etiology despite substantial age and household differences, DNA sequencing studies will be necessary to identify a putative inherited gene mutation driving the development of both MDS or MF in this unusual kindred.
Treatment Failure Patterns in Patients With Chronic Lymphocytic Leukemia: Results of a Large U.S. Observational Study
Purpose: Treatment for patients with chronic lymphocytic leukemia (CLL) is often not curative. Therefore, patients with CLL eventually experience disease progression with limited therapeutic options, even if the duration of remission can be long. Median overall survival for patients with CLL is usually 8 to 12 years, but there is a large variation in survival among individual patients, ranging from several months to a normal life expectancy. This retrospective observational study describes course of treatment and occurrence of treatment failure in patients with CLL.
Methods: Adult patients with ≥ 1 diagnosis of CLL and ≥ 1 claim for a medication used to treat CLL were identified in the IMS PharMetrics Plus database (January 2008 to September 2013). Patients were excluded if they had evidence of a nonhematologic malignancy, used a non-CLL antineoplastic agent, or received a stem cell transplant during the 12-month baseline period. Initial therapy was defined as the single agent or the combination of medications used to treat CLL that was given to patients in the first 30 days following the first claim for a medication (index date) used to treat CLL. Treatment failure was identified based on earliest occurrence of one of the following events: initiation of a new treatment for CLL that was not part of the initial therapy, resumption of any CLL treatment following a minimum of 3-month break in treatment, radiotherapy, stem cell transplant, hospital mortality, or hospice care.
Results: A total of 6,015 patients with CLL were identified (mean patient aged 63 years; proportion female: 36%). Patients were observed for 22.7 months on average following treatment initiation. Median time between first CLL diagnosis and first claim for a medication used to treat CLL was 102 days. Mean Charlson comorbidity index was 3.2. More frequent comorbidities were hypertension (44%), anemias (37%), coagulation deficiency (21%), diabetes without chronic complications (18%), and chronic pulmonary disease (18%). Of the 6,015 patients with CLL, 2,734 (45%) experienced treatment failure. Main observed measures for treatment failure were initiation of a new CLL treatment that was not part of the initial therapy (41%; mean time to treatment failure [TTF]: 286 days), resumption of any CLL treatment (39%; TTF: 346 days), radiotherapy (14%; TTF: 199 days), and stem cell transplant (5%; TTF: 116 days). Very few patients experienced treatment failure observed through hospice care (0.4%; TTF: 387 days).
Conclusions: A notable number (45%) of patients with CLL experienced treatment failure, which was mainly observed through initiation of alternative therapies or the need for a holiday after initial therapy. These data help in our understanding of CLL treatment failure patterns, which is a first step toward reducing the burden of disease in patients with CLL. Further research evaluating the reasons and economic implications of treatment failure is warranted.
Purpose: Treatment for patients with chronic lymphocytic leukemia (CLL) is often not curative. Therefore, patients with CLL eventually experience disease progression with limited therapeutic options, even if the duration of remission can be long. Median overall survival for patients with CLL is usually 8 to 12 years, but there is a large variation in survival among individual patients, ranging from several months to a normal life expectancy. This retrospective observational study describes course of treatment and occurrence of treatment failure in patients with CLL.
Methods: Adult patients with ≥ 1 diagnosis of CLL and ≥ 1 claim for a medication used to treat CLL were identified in the IMS PharMetrics Plus database (January 2008 to September 2013). Patients were excluded if they had evidence of a nonhematologic malignancy, used a non-CLL antineoplastic agent, or received a stem cell transplant during the 12-month baseline period. Initial therapy was defined as the single agent or the combination of medications used to treat CLL that was given to patients in the first 30 days following the first claim for a medication (index date) used to treat CLL. Treatment failure was identified based on earliest occurrence of one of the following events: initiation of a new treatment for CLL that was not part of the initial therapy, resumption of any CLL treatment following a minimum of 3-month break in treatment, radiotherapy, stem cell transplant, hospital mortality, or hospice care.
Results: A total of 6,015 patients with CLL were identified (mean patient aged 63 years; proportion female: 36%). Patients were observed for 22.7 months on average following treatment initiation. Median time between first CLL diagnosis and first claim for a medication used to treat CLL was 102 days. Mean Charlson comorbidity index was 3.2. More frequent comorbidities were hypertension (44%), anemias (37%), coagulation deficiency (21%), diabetes without chronic complications (18%), and chronic pulmonary disease (18%). Of the 6,015 patients with CLL, 2,734 (45%) experienced treatment failure. Main observed measures for treatment failure were initiation of a new CLL treatment that was not part of the initial therapy (41%; mean time to treatment failure [TTF]: 286 days), resumption of any CLL treatment (39%; TTF: 346 days), radiotherapy (14%; TTF: 199 days), and stem cell transplant (5%; TTF: 116 days). Very few patients experienced treatment failure observed through hospice care (0.4%; TTF: 387 days).
Conclusions: A notable number (45%) of patients with CLL experienced treatment failure, which was mainly observed through initiation of alternative therapies or the need for a holiday after initial therapy. These data help in our understanding of CLL treatment failure patterns, which is a first step toward reducing the burden of disease in patients with CLL. Further research evaluating the reasons and economic implications of treatment failure is warranted.
Purpose: Treatment for patients with chronic lymphocytic leukemia (CLL) is often not curative. Therefore, patients with CLL eventually experience disease progression with limited therapeutic options, even if the duration of remission can be long. Median overall survival for patients with CLL is usually 8 to 12 years, but there is a large variation in survival among individual patients, ranging from several months to a normal life expectancy. This retrospective observational study describes course of treatment and occurrence of treatment failure in patients with CLL.
Methods: Adult patients with ≥ 1 diagnosis of CLL and ≥ 1 claim for a medication used to treat CLL were identified in the IMS PharMetrics Plus database (January 2008 to September 2013). Patients were excluded if they had evidence of a nonhematologic malignancy, used a non-CLL antineoplastic agent, or received a stem cell transplant during the 12-month baseline period. Initial therapy was defined as the single agent or the combination of medications used to treat CLL that was given to patients in the first 30 days following the first claim for a medication (index date) used to treat CLL. Treatment failure was identified based on earliest occurrence of one of the following events: initiation of a new treatment for CLL that was not part of the initial therapy, resumption of any CLL treatment following a minimum of 3-month break in treatment, radiotherapy, stem cell transplant, hospital mortality, or hospice care.
Results: A total of 6,015 patients with CLL were identified (mean patient aged 63 years; proportion female: 36%). Patients were observed for 22.7 months on average following treatment initiation. Median time between first CLL diagnosis and first claim for a medication used to treat CLL was 102 days. Mean Charlson comorbidity index was 3.2. More frequent comorbidities were hypertension (44%), anemias (37%), coagulation deficiency (21%), diabetes without chronic complications (18%), and chronic pulmonary disease (18%). Of the 6,015 patients with CLL, 2,734 (45%) experienced treatment failure. Main observed measures for treatment failure were initiation of a new CLL treatment that was not part of the initial therapy (41%; mean time to treatment failure [TTF]: 286 days), resumption of any CLL treatment (39%; TTF: 346 days), radiotherapy (14%; TTF: 199 days), and stem cell transplant (5%; TTF: 116 days). Very few patients experienced treatment failure observed through hospice care (0.4%; TTF: 387 days).
Conclusions: A notable number (45%) of patients with CLL experienced treatment failure, which was mainly observed through initiation of alternative therapies or the need for a holiday after initial therapy. These data help in our understanding of CLL treatment failure patterns, which is a first step toward reducing the burden of disease in patients with CLL. Further research evaluating the reasons and economic implications of treatment failure is warranted.
Association Between Rituximab Use and Progressive Multifocal Leukoencephalopathy Among Non-HIV Non-Hodgkin Lymphoma VA Patients
Purpose: Progressive multifocal leukoencephalopathy (PML) is a rare, fatal disease that results from activation of a highly prevalent, dormant John Cunningham (JC) virus—a variant of human polyomavirus—during immunosuppressed states. Rituximab, a CD20 monoclonal antibody immunomodulator, has been approved for non-Hodgkin lymphoma (NHL).
Methods: Using electronic medical records from the VA, ICD-9 codes were used to identify patients diagnosed with NHL, PML, and HIV within the VA from 2003 to 2011. Pharmacy records were used to identify patients receiving rituximab, cyclophosphamide, hydroxydaunorubicin, and vncristine. This project was conducted inside the Veterans Affairs Informatics and Computing Infrastructure after obtaining approvals from the VA IRB and other oversight groups.
Results: We identified 57,041 non-HIV NHL patients. A total of 14 out of 57,041 (0.025%) patients had developed PML; 7 out of 8,895 (7.8 per 10,000) were NHL patients who received rituximab subsequently developed PML; 7 out of 48,146 (1.5 per 10,000) were NHL patients who did not receive rituximab subsequently developed PML, which results in a statistically significant unadjusted relative risk of 5.4 (95% CI: 1.9 - 15.4) and an attributable risk of 6.3 per 10,000. Univariate analyses of other outcomes were not statistically significant between patients who received and those that did not receive rituximab.
Conclusions: These results show that among lymphoma patients, the use of rituximab is associated with a statistically significant relative risk for documented PML of 5.4. Measurement of JC virus prior to initiation of rituximab therapy as well as during rituximab therapy should be considered.
Purpose: Progressive multifocal leukoencephalopathy (PML) is a rare, fatal disease that results from activation of a highly prevalent, dormant John Cunningham (JC) virus—a variant of human polyomavirus—during immunosuppressed states. Rituximab, a CD20 monoclonal antibody immunomodulator, has been approved for non-Hodgkin lymphoma (NHL).
Methods: Using electronic medical records from the VA, ICD-9 codes were used to identify patients diagnosed with NHL, PML, and HIV within the VA from 2003 to 2011. Pharmacy records were used to identify patients receiving rituximab, cyclophosphamide, hydroxydaunorubicin, and vncristine. This project was conducted inside the Veterans Affairs Informatics and Computing Infrastructure after obtaining approvals from the VA IRB and other oversight groups.
Results: We identified 57,041 non-HIV NHL patients. A total of 14 out of 57,041 (0.025%) patients had developed PML; 7 out of 8,895 (7.8 per 10,000) were NHL patients who received rituximab subsequently developed PML; 7 out of 48,146 (1.5 per 10,000) were NHL patients who did not receive rituximab subsequently developed PML, which results in a statistically significant unadjusted relative risk of 5.4 (95% CI: 1.9 - 15.4) and an attributable risk of 6.3 per 10,000. Univariate analyses of other outcomes were not statistically significant between patients who received and those that did not receive rituximab.
Conclusions: These results show that among lymphoma patients, the use of rituximab is associated with a statistically significant relative risk for documented PML of 5.4. Measurement of JC virus prior to initiation of rituximab therapy as well as during rituximab therapy should be considered.
Purpose: Progressive multifocal leukoencephalopathy (PML) is a rare, fatal disease that results from activation of a highly prevalent, dormant John Cunningham (JC) virus—a variant of human polyomavirus—during immunosuppressed states. Rituximab, a CD20 monoclonal antibody immunomodulator, has been approved for non-Hodgkin lymphoma (NHL).
Methods: Using electronic medical records from the VA, ICD-9 codes were used to identify patients diagnosed with NHL, PML, and HIV within the VA from 2003 to 2011. Pharmacy records were used to identify patients receiving rituximab, cyclophosphamide, hydroxydaunorubicin, and vncristine. This project was conducted inside the Veterans Affairs Informatics and Computing Infrastructure after obtaining approvals from the VA IRB and other oversight groups.
Results: We identified 57,041 non-HIV NHL patients. A total of 14 out of 57,041 (0.025%) patients had developed PML; 7 out of 8,895 (7.8 per 10,000) were NHL patients who received rituximab subsequently developed PML; 7 out of 48,146 (1.5 per 10,000) were NHL patients who did not receive rituximab subsequently developed PML, which results in a statistically significant unadjusted relative risk of 5.4 (95% CI: 1.9 - 15.4) and an attributable risk of 6.3 per 10,000. Univariate analyses of other outcomes were not statistically significant between patients who received and those that did not receive rituximab.
Conclusions: These results show that among lymphoma patients, the use of rituximab is associated with a statistically significant relative risk for documented PML of 5.4. Measurement of JC virus prior to initiation of rituximab therapy as well as during rituximab therapy should be considered.
Case Report of Dasatinib-Induced Follicular Lymphoid Hyperplasia
Purpose: Dasatinib is a tyrosine kinase inhibitor indicated for the treatment of chronic myeloid leukemia (CML). In October 2013, reversible lymph node follicular hyperplasia associated with dasatinib was first reported. The purpose of this case report is to describe a reactive lymphoid process with follicular and interfollicular hyperplasia associated with dasatinib treatment.
Methods: This is a case report of a white male, aged 37 years, receiving oral dasatinib 100 mg once daily for chronic phase CML for 8 months following failure of imatinib and intolerance to nilotinib. A Pubmed literature review revealed only 1 other report of 9 cases developing cervical lymphadenopathy after a median of 20 months on dasatinib (range 9 to 35 months).
Results: Following 8 months of dasatinib therapy, the patient presented with facial swelling on his right jaw and right cheek bone. A CT scan revealed multiple mildly enlarged lymph nodes within the parotid gland and right level II nodal stations without any definite mucosal lesions seen. On physical exam, he was found to have a 1.5-cm right parotid nontender mass and a 3-cm right level II nontender nodular mass with no overlying erythema or edema. He failed a trial of antibiotics with amoxicillin 875 mg/clavulanate 125 mg for 10 days. Fine needle aspiration of both the right parotid mass and the right nodular mass revealed reactive lymphoid cells. An excisional biopsy of the right parotid mass with flow cytometry demonstrated fragmented benign lymphoid tissue with reactive follicles and expansion of the interfollicular region with no evidence of leukemic involvement. He then underwent right parotidectomy due to progressive swelling and discomfort. Pathology review was consistent with reactive lymphoid process with florid follicular and interfollicular hyperplasia. There was again no evidence of lymphoid or lymphomatous involvement. Dasatinib therapy was subsequently discontinued and resolution of follicular hyperplasia occurred within 1 month.
Conclusions: Follicular hyperplasia is a rare adverse effect associated with the use of dasatinib. Our patient presented much earlier (within 8 months of dasatinib therapy) than previously reported cases. Patients on dasatinib experiencing cervical or other lymph node enlargement should be evaluated for follicular hyperplasia. It is recommended that dasatinib be discontinued in the presence of follicular lymphoid hyperplasia; however, use of another tyrosine kinase inhibitor should be considered. Our patient was subsequently switched to bosutinib.
Purpose: Dasatinib is a tyrosine kinase inhibitor indicated for the treatment of chronic myeloid leukemia (CML). In October 2013, reversible lymph node follicular hyperplasia associated with dasatinib was first reported. The purpose of this case report is to describe a reactive lymphoid process with follicular and interfollicular hyperplasia associated with dasatinib treatment.
Methods: This is a case report of a white male, aged 37 years, receiving oral dasatinib 100 mg once daily for chronic phase CML for 8 months following failure of imatinib and intolerance to nilotinib. A Pubmed literature review revealed only 1 other report of 9 cases developing cervical lymphadenopathy after a median of 20 months on dasatinib (range 9 to 35 months).
Results: Following 8 months of dasatinib therapy, the patient presented with facial swelling on his right jaw and right cheek bone. A CT scan revealed multiple mildly enlarged lymph nodes within the parotid gland and right level II nodal stations without any definite mucosal lesions seen. On physical exam, he was found to have a 1.5-cm right parotid nontender mass and a 3-cm right level II nontender nodular mass with no overlying erythema or edema. He failed a trial of antibiotics with amoxicillin 875 mg/clavulanate 125 mg for 10 days. Fine needle aspiration of both the right parotid mass and the right nodular mass revealed reactive lymphoid cells. An excisional biopsy of the right parotid mass with flow cytometry demonstrated fragmented benign lymphoid tissue with reactive follicles and expansion of the interfollicular region with no evidence of leukemic involvement. He then underwent right parotidectomy due to progressive swelling and discomfort. Pathology review was consistent with reactive lymphoid process with florid follicular and interfollicular hyperplasia. There was again no evidence of lymphoid or lymphomatous involvement. Dasatinib therapy was subsequently discontinued and resolution of follicular hyperplasia occurred within 1 month.
Conclusions: Follicular hyperplasia is a rare adverse effect associated with the use of dasatinib. Our patient presented much earlier (within 8 months of dasatinib therapy) than previously reported cases. Patients on dasatinib experiencing cervical or other lymph node enlargement should be evaluated for follicular hyperplasia. It is recommended that dasatinib be discontinued in the presence of follicular lymphoid hyperplasia; however, use of another tyrosine kinase inhibitor should be considered. Our patient was subsequently switched to bosutinib.
Purpose: Dasatinib is a tyrosine kinase inhibitor indicated for the treatment of chronic myeloid leukemia (CML). In October 2013, reversible lymph node follicular hyperplasia associated with dasatinib was first reported. The purpose of this case report is to describe a reactive lymphoid process with follicular and interfollicular hyperplasia associated with dasatinib treatment.
Methods: This is a case report of a white male, aged 37 years, receiving oral dasatinib 100 mg once daily for chronic phase CML for 8 months following failure of imatinib and intolerance to nilotinib. A Pubmed literature review revealed only 1 other report of 9 cases developing cervical lymphadenopathy after a median of 20 months on dasatinib (range 9 to 35 months).
Results: Following 8 months of dasatinib therapy, the patient presented with facial swelling on his right jaw and right cheek bone. A CT scan revealed multiple mildly enlarged lymph nodes within the parotid gland and right level II nodal stations without any definite mucosal lesions seen. On physical exam, he was found to have a 1.5-cm right parotid nontender mass and a 3-cm right level II nontender nodular mass with no overlying erythema or edema. He failed a trial of antibiotics with amoxicillin 875 mg/clavulanate 125 mg for 10 days. Fine needle aspiration of both the right parotid mass and the right nodular mass revealed reactive lymphoid cells. An excisional biopsy of the right parotid mass with flow cytometry demonstrated fragmented benign lymphoid tissue with reactive follicles and expansion of the interfollicular region with no evidence of leukemic involvement. He then underwent right parotidectomy due to progressive swelling and discomfort. Pathology review was consistent with reactive lymphoid process with florid follicular and interfollicular hyperplasia. There was again no evidence of lymphoid or lymphomatous involvement. Dasatinib therapy was subsequently discontinued and resolution of follicular hyperplasia occurred within 1 month.
Conclusions: Follicular hyperplasia is a rare adverse effect associated with the use of dasatinib. Our patient presented much earlier (within 8 months of dasatinib therapy) than previously reported cases. Patients on dasatinib experiencing cervical or other lymph node enlargement should be evaluated for follicular hyperplasia. It is recommended that dasatinib be discontinued in the presence of follicular lymphoid hyperplasia; however, use of another tyrosine kinase inhibitor should be considered. Our patient was subsequently switched to bosutinib.