User login
Brentuximab Vedotin in a Patient With Aggressive Systemic Mastocytosis and Pure Red Cell Aplasia
Backround: Mastocytosis is a rare disease with a variable clinical course. Pure red cell aplasia has not been frequently reported in association with aggressive systemic mastocytosis. CD30 is often expressed on mast cells in systemic mastocytosis. The CD30-directed antibody-drug conjugate brentuximab vedotin has shown in vitro activity against CD30-positive neoplastic mast cells. We describe a case of systemic mastocytosis with the development of severe hemolytic anemia, pure red cell aplasia, and evidence of weak bone marrow CD30 positivity. Brentuximab vedotin was administered with some evidence of clinical benefit.
Patient: A 64-year-old man presented with fatigue and a 20 pound weight loss. He was found to have transfusion-dependent anemia, a low reticulocyte count, and splenomegaly. Evaluation for gastrointestinal blood loss was unremarkable. Bone marrow biopsy showed mast cell aggregates, spindle-shaped mast cells, CD-2 and CD25-positivity, increased myelopoiesis and dysmegakaryopoiesis, red cell aplasia, eosinophilia, and increased reticulin fibrosis. The D816V c-kit mutation was present, and serum tryptase level was elevated at 114 ng/mL. He later developed Coombs-positive hemolysis and direct hyperbilirubinemia, concerning for mast cell liver infiltration.
Intervention: The patient received high-dose steroids and red blood cell transfusions. Interferon alpha and cladribine were contraindicated, given severe liver dysfunction. He was started on hydroxyurea. He received a dose of brentuximab vedotin 1.8 mg/kg IV. Thirteen days after starting steroids and 10 days after brentuximab, haptoglobin became detectable, and total bilirubin decreased to 18.4 mg/dL from 40.5 mg/dL. Red blood cell transfusion requirements decreased. He then developed severe neutropenia, pneumonia with sepsis, and respiratory failure, requiring intubation, maximal pressor support, and broad-spectrum antibiotics. Despite these measures, he expired.
Discussion/Conclusion: This is a rare case of pure red cellaplasia in the setting of aggressive systemic mastocytosis. Therapeutic options were limited in the setting of liver dysfunction, and therapy with brentuximab vedotin resulted in initial clinical benefit. Brentuximab vedotin is being investigated in patients with advanced systemic mastocytosis or mast cell leukemia.
Backround: Mastocytosis is a rare disease with a variable clinical course. Pure red cell aplasia has not been frequently reported in association with aggressive systemic mastocytosis. CD30 is often expressed on mast cells in systemic mastocytosis. The CD30-directed antibody-drug conjugate brentuximab vedotin has shown in vitro activity against CD30-positive neoplastic mast cells. We describe a case of systemic mastocytosis with the development of severe hemolytic anemia, pure red cell aplasia, and evidence of weak bone marrow CD30 positivity. Brentuximab vedotin was administered with some evidence of clinical benefit.
Patient: A 64-year-old man presented with fatigue and a 20 pound weight loss. He was found to have transfusion-dependent anemia, a low reticulocyte count, and splenomegaly. Evaluation for gastrointestinal blood loss was unremarkable. Bone marrow biopsy showed mast cell aggregates, spindle-shaped mast cells, CD-2 and CD25-positivity, increased myelopoiesis and dysmegakaryopoiesis, red cell aplasia, eosinophilia, and increased reticulin fibrosis. The D816V c-kit mutation was present, and serum tryptase level was elevated at 114 ng/mL. He later developed Coombs-positive hemolysis and direct hyperbilirubinemia, concerning for mast cell liver infiltration.
Intervention: The patient received high-dose steroids and red blood cell transfusions. Interferon alpha and cladribine were contraindicated, given severe liver dysfunction. He was started on hydroxyurea. He received a dose of brentuximab vedotin 1.8 mg/kg IV. Thirteen days after starting steroids and 10 days after brentuximab, haptoglobin became detectable, and total bilirubin decreased to 18.4 mg/dL from 40.5 mg/dL. Red blood cell transfusion requirements decreased. He then developed severe neutropenia, pneumonia with sepsis, and respiratory failure, requiring intubation, maximal pressor support, and broad-spectrum antibiotics. Despite these measures, he expired.
Discussion/Conclusion: This is a rare case of pure red cellaplasia in the setting of aggressive systemic mastocytosis. Therapeutic options were limited in the setting of liver dysfunction, and therapy with brentuximab vedotin resulted in initial clinical benefit. Brentuximab vedotin is being investigated in patients with advanced systemic mastocytosis or mast cell leukemia.
Backround: Mastocytosis is a rare disease with a variable clinical course. Pure red cell aplasia has not been frequently reported in association with aggressive systemic mastocytosis. CD30 is often expressed on mast cells in systemic mastocytosis. The CD30-directed antibody-drug conjugate brentuximab vedotin has shown in vitro activity against CD30-positive neoplastic mast cells. We describe a case of systemic mastocytosis with the development of severe hemolytic anemia, pure red cell aplasia, and evidence of weak bone marrow CD30 positivity. Brentuximab vedotin was administered with some evidence of clinical benefit.
Patient: A 64-year-old man presented with fatigue and a 20 pound weight loss. He was found to have transfusion-dependent anemia, a low reticulocyte count, and splenomegaly. Evaluation for gastrointestinal blood loss was unremarkable. Bone marrow biopsy showed mast cell aggregates, spindle-shaped mast cells, CD-2 and CD25-positivity, increased myelopoiesis and dysmegakaryopoiesis, red cell aplasia, eosinophilia, and increased reticulin fibrosis. The D816V c-kit mutation was present, and serum tryptase level was elevated at 114 ng/mL. He later developed Coombs-positive hemolysis and direct hyperbilirubinemia, concerning for mast cell liver infiltration.
Intervention: The patient received high-dose steroids and red blood cell transfusions. Interferon alpha and cladribine were contraindicated, given severe liver dysfunction. He was started on hydroxyurea. He received a dose of brentuximab vedotin 1.8 mg/kg IV. Thirteen days after starting steroids and 10 days after brentuximab, haptoglobin became detectable, and total bilirubin decreased to 18.4 mg/dL from 40.5 mg/dL. Red blood cell transfusion requirements decreased. He then developed severe neutropenia, pneumonia with sepsis, and respiratory failure, requiring intubation, maximal pressor support, and broad-spectrum antibiotics. Despite these measures, he expired.
Discussion/Conclusion: This is a rare case of pure red cellaplasia in the setting of aggressive systemic mastocytosis. Therapeutic options were limited in the setting of liver dysfunction, and therapy with brentuximab vedotin resulted in initial clinical benefit. Brentuximab vedotin is being investigated in patients with advanced systemic mastocytosis or mast cell leukemia.
Telehematology: Improving Access for Veterans With Select Hematologic Disorders
Background: Historically the Hematology Clinics at the Robley Rex VA Medical Center (RRVAMC) had a chronically elevated no-show rate (NSR). The major cause of these missed opportunities was patient adherence secondary to transportation, distance traveled, and patients’ lack of concern about their disease. Our hypothesis was that the no-show rates could be significantly reduced by providing virtual hematology services outside the main VA campus.
Methods: A comprehensive review of the hematology patient population was conducted to identify veterans with stable hematologic disorders (SHD), eg, stage 0 chronic lymphocytic leukemia, monoclonal gammopathy of undetermined significance, and chronic anemia. The NSR was determined for 2012. Patients with SHD were identified as potential subjects for the Telehematology Clinic. The clinic was started July 2013 and eventually expanded to a bimonthly clinic that visited 4 of the 6 community-based outpatient clinics. This review compares the RRVAMC Hematology and the Telehematology Clinics’ NSR from March31, 2014, to March 31, 2015.
Results: The Telehematology Clinic has a patient cohort of 57 patients, which will expand to at least 77 patients by May 2016. To date, there have been 157 Telehematology Clinic visits. Most patients have an annual Telehematology Clinic visit and either quarterly or semiannual laboratory studies. The Telehematology Clinic has had a NSR of 3% since 2013. The Hematology Clinic’s NSR decreased from 14% to 12% (P = .09). Comparing the NSR of traditional Hematology Clinics with NSR of Telehematology Clinics is statically significant, P < .0007.
Implications: Telehematology Clinics have proven sustainability with greater adherence and attendance than that of the traditional hematology clinics scheduled within the RRVAMC. The NSR did not decrease significantly and remains greater than our goal of < 10%. Future video supported clinics may increase access and improve NSRs within the veteran population. Our goal in the next year is to increase the frequency of Telehematology Clinics and to expand the clinic population in an effort to reduce our overall NSR.
Background: Historically the Hematology Clinics at the Robley Rex VA Medical Center (RRVAMC) had a chronically elevated no-show rate (NSR). The major cause of these missed opportunities was patient adherence secondary to transportation, distance traveled, and patients’ lack of concern about their disease. Our hypothesis was that the no-show rates could be significantly reduced by providing virtual hematology services outside the main VA campus.
Methods: A comprehensive review of the hematology patient population was conducted to identify veterans with stable hematologic disorders (SHD), eg, stage 0 chronic lymphocytic leukemia, monoclonal gammopathy of undetermined significance, and chronic anemia. The NSR was determined for 2012. Patients with SHD were identified as potential subjects for the Telehematology Clinic. The clinic was started July 2013 and eventually expanded to a bimonthly clinic that visited 4 of the 6 community-based outpatient clinics. This review compares the RRVAMC Hematology and the Telehematology Clinics’ NSR from March31, 2014, to March 31, 2015.
Results: The Telehematology Clinic has a patient cohort of 57 patients, which will expand to at least 77 patients by May 2016. To date, there have been 157 Telehematology Clinic visits. Most patients have an annual Telehematology Clinic visit and either quarterly or semiannual laboratory studies. The Telehematology Clinic has had a NSR of 3% since 2013. The Hematology Clinic’s NSR decreased from 14% to 12% (P = .09). Comparing the NSR of traditional Hematology Clinics with NSR of Telehematology Clinics is statically significant, P < .0007.
Implications: Telehematology Clinics have proven sustainability with greater adherence and attendance than that of the traditional hematology clinics scheduled within the RRVAMC. The NSR did not decrease significantly and remains greater than our goal of < 10%. Future video supported clinics may increase access and improve NSRs within the veteran population. Our goal in the next year is to increase the frequency of Telehematology Clinics and to expand the clinic population in an effort to reduce our overall NSR.
Background: Historically the Hematology Clinics at the Robley Rex VA Medical Center (RRVAMC) had a chronically elevated no-show rate (NSR). The major cause of these missed opportunities was patient adherence secondary to transportation, distance traveled, and patients’ lack of concern about their disease. Our hypothesis was that the no-show rates could be significantly reduced by providing virtual hematology services outside the main VA campus.
Methods: A comprehensive review of the hematology patient population was conducted to identify veterans with stable hematologic disorders (SHD), eg, stage 0 chronic lymphocytic leukemia, monoclonal gammopathy of undetermined significance, and chronic anemia. The NSR was determined for 2012. Patients with SHD were identified as potential subjects for the Telehematology Clinic. The clinic was started July 2013 and eventually expanded to a bimonthly clinic that visited 4 of the 6 community-based outpatient clinics. This review compares the RRVAMC Hematology and the Telehematology Clinics’ NSR from March31, 2014, to March 31, 2015.
Results: The Telehematology Clinic has a patient cohort of 57 patients, which will expand to at least 77 patients by May 2016. To date, there have been 157 Telehematology Clinic visits. Most patients have an annual Telehematology Clinic visit and either quarterly or semiannual laboratory studies. The Telehematology Clinic has had a NSR of 3% since 2013. The Hematology Clinic’s NSR decreased from 14% to 12% (P = .09). Comparing the NSR of traditional Hematology Clinics with NSR of Telehematology Clinics is statically significant, P < .0007.
Implications: Telehematology Clinics have proven sustainability with greater adherence and attendance than that of the traditional hematology clinics scheduled within the RRVAMC. The NSR did not decrease significantly and remains greater than our goal of < 10%. Future video supported clinics may increase access and improve NSRs within the veteran population. Our goal in the next year is to increase the frequency of Telehematology Clinics and to expand the clinic population in an effort to reduce our overall NSR.
Patterns of Initial Treatment in Veteran Patients With Chronic Lymphocytic Leukemia: A National VA Tumor Registry Study
Background: Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults, including elderly veterans, with many new treatment options now available. Data on patterns of treatment in elderly veteran patients with CLL is limited. We sought to assess initial treatment patterns over a 13-year period among veteran patients in the Minneapolis VA Health Care System.
Methods: We identified 6,756 CLL cases diagnosed from 2000 to 2013 and are presenting interim data on 2015. We reviewed clinical data from 2,015 patients with CLL diagnosed from 2000 to 2013 and identified through the National VA Tumor Registry. Baseline demographics and treatment information were collected. The objective of this study was to assess initial treatment patterns, time to initial treatment, and variation of these parameters by age.
Results: At diagnosis, median age was 69 years (range, 37-96 years); 98% were male (1,979); Rai stage was 0 (n = 1,331, 66%), 1 (n = 317, 16%), 2 (n = 156, 8%), 3 (n = 91, 5%), 4 (n = 113, 6%). The majority of patients were white (n = 1,752, 87%); followed by African American (n = 203, 10%); and Hispanic (n = 33, 2%). Of the 2,015 patients, 751 (37%) received therapy over this period of follow-up. Median time from diagnosis to initial treatment was 1.3 years (range, 0-13 years). The most common initial therapies utilized were chlorambucil (39.4%); fludarabine/cyclophosphamide/ritux-imab (FCR) (12.4%); and single-agent fludarabine (10.5%). When examining these parameters by age in decades, we found that there were no differences in Rai stage at diagnosis by age-decade. There was a progressive increase in initial chlorambucil usage by advancing age. Likewise, the majority of FCR usage was in patients aged < 70 years.
Conclusions: In this veteran population, including many elderly patients, the majority of patients requiring therapy initiated it within 2 years of diagnosis. These patients were most commonly treated with chlorambucil. These patterns of care will be changing with the introduction of newer oral agents, such as ibrutinib and idelalisib, but at a significantly higher cost. The National VA Tumor Registry data will allow future opportunity to examine evolving treatment patterns in both an elderly as well as a veteran population. Updated data will be presented at the AVAHO annual meeting.
Background: Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults, including elderly veterans, with many new treatment options now available. Data on patterns of treatment in elderly veteran patients with CLL is limited. We sought to assess initial treatment patterns over a 13-year period among veteran patients in the Minneapolis VA Health Care System.
Methods: We identified 6,756 CLL cases diagnosed from 2000 to 2013 and are presenting interim data on 2015. We reviewed clinical data from 2,015 patients with CLL diagnosed from 2000 to 2013 and identified through the National VA Tumor Registry. Baseline demographics and treatment information were collected. The objective of this study was to assess initial treatment patterns, time to initial treatment, and variation of these parameters by age.
Results: At diagnosis, median age was 69 years (range, 37-96 years); 98% were male (1,979); Rai stage was 0 (n = 1,331, 66%), 1 (n = 317, 16%), 2 (n = 156, 8%), 3 (n = 91, 5%), 4 (n = 113, 6%). The majority of patients were white (n = 1,752, 87%); followed by African American (n = 203, 10%); and Hispanic (n = 33, 2%). Of the 2,015 patients, 751 (37%) received therapy over this period of follow-up. Median time from diagnosis to initial treatment was 1.3 years (range, 0-13 years). The most common initial therapies utilized were chlorambucil (39.4%); fludarabine/cyclophosphamide/ritux-imab (FCR) (12.4%); and single-agent fludarabine (10.5%). When examining these parameters by age in decades, we found that there were no differences in Rai stage at diagnosis by age-decade. There was a progressive increase in initial chlorambucil usage by advancing age. Likewise, the majority of FCR usage was in patients aged < 70 years.
Conclusions: In this veteran population, including many elderly patients, the majority of patients requiring therapy initiated it within 2 years of diagnosis. These patients were most commonly treated with chlorambucil. These patterns of care will be changing with the introduction of newer oral agents, such as ibrutinib and idelalisib, but at a significantly higher cost. The National VA Tumor Registry data will allow future opportunity to examine evolving treatment patterns in both an elderly as well as a veteran population. Updated data will be presented at the AVAHO annual meeting.
Background: Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults, including elderly veterans, with many new treatment options now available. Data on patterns of treatment in elderly veteran patients with CLL is limited. We sought to assess initial treatment patterns over a 13-year period among veteran patients in the Minneapolis VA Health Care System.
Methods: We identified 6,756 CLL cases diagnosed from 2000 to 2013 and are presenting interim data on 2015. We reviewed clinical data from 2,015 patients with CLL diagnosed from 2000 to 2013 and identified through the National VA Tumor Registry. Baseline demographics and treatment information were collected. The objective of this study was to assess initial treatment patterns, time to initial treatment, and variation of these parameters by age.
Results: At diagnosis, median age was 69 years (range, 37-96 years); 98% were male (1,979); Rai stage was 0 (n = 1,331, 66%), 1 (n = 317, 16%), 2 (n = 156, 8%), 3 (n = 91, 5%), 4 (n = 113, 6%). The majority of patients were white (n = 1,752, 87%); followed by African American (n = 203, 10%); and Hispanic (n = 33, 2%). Of the 2,015 patients, 751 (37%) received therapy over this period of follow-up. Median time from diagnosis to initial treatment was 1.3 years (range, 0-13 years). The most common initial therapies utilized were chlorambucil (39.4%); fludarabine/cyclophosphamide/ritux-imab (FCR) (12.4%); and single-agent fludarabine (10.5%). When examining these parameters by age in decades, we found that there were no differences in Rai stage at diagnosis by age-decade. There was a progressive increase in initial chlorambucil usage by advancing age. Likewise, the majority of FCR usage was in patients aged < 70 years.
Conclusions: In this veteran population, including many elderly patients, the majority of patients requiring therapy initiated it within 2 years of diagnosis. These patients were most commonly treated with chlorambucil. These patterns of care will be changing with the introduction of newer oral agents, such as ibrutinib and idelalisib, but at a significantly higher cost. The National VA Tumor Registry data will allow future opportunity to examine evolving treatment patterns in both an elderly as well as a veteran population. Updated data will be presented at the AVAHO annual meeting.
Hypercalcemia in Diffuse Large B-cell Lymphoma
Purpose: Review the clinical presentation and pathophysiology of hypercalcemia in diffuse large B-cell lymphoma.
Methods: Case study of a 59-year-old man with untreateddiffuse large B-cell lymphoma (DLBCL) who presented with anorexia, constipation, and weight loss in the setting of a fungating breast mass.
Results: The patient presented to an outside hospital 1.5 years prior for a left breast mass biopsy that was diagnosed as triple-hit DLBCL. However, he deferred treatment at that time. He subsequently experienced anorexia, nausea, vomiting, consti-pation, and a 20-pound unintentional weight loss. At hospital presentation, a physical examination revealed enlargement of the left breast mass measuring 8 x 14 cm with central ulceration and malodorous purulent discharge and a proximal 7 x 4 cm firm axillary mass. The patient reported no abdominal pain or fever. He was found to have acute kidney injury (creatinine 2.9 mg/dL); hyperuricemia (13.3 mg/dL); and corrected hypercalcemia (17.4 mg/dL). Empiric treatment for tumor lysis syndrome with rasburicase, allopurinol, and aggressive IV hydration was initiated. Further evaluation revealed normal lactate dehydrogenase and K+.The patient was found to have an elevated vitamin D 1,25-OH (244 pg/mL) but low parathyroid hormone (PTH) (6.4 pg/mL); normal PTH-rP; mild hyperphosphatemia (5.5 mg/dL); and negative serum protein electrophoresis/urine protein electrophoresis. Clinical presentation was suggestive of vitamin D-mediated hypercalcemia. The patient received pamidronate and aggressive IV hydration. Electrolyte abnormalities and renal function gradually improved. Computed tomography and positron emission tomography imaging revealed masses localized to the left chest wall with ipsilateral axillary lymphadenopathy. Surgical biopsy of the mass reconfirmed DLBCL ultimately staged as IIE. Following stabilization, the patient declined more aggressive chemotherapy and received R-CHOP with a complete response.
Conclusions: The patient presented with vitamin D-mediated hypercalcemia in the setting of aggressive triple-hit DLBCL. Vitamin D-mediated hypercalcemia occurs in < 1% of lymphomas, and < 0.5% of breast tumors are primary lymphomas. This case study presents a rare constellation of findings at diagnosis. The patient responded to IV hydration and pamidronate and subsequently received R-CHOP for stage IIE DLBCL. While hypercalcemia in the setting of malignancy is associated with poor prognosis, treatment of hypercalcemia does not improve survival.
Purpose: Review the clinical presentation and pathophysiology of hypercalcemia in diffuse large B-cell lymphoma.
Methods: Case study of a 59-year-old man with untreateddiffuse large B-cell lymphoma (DLBCL) who presented with anorexia, constipation, and weight loss in the setting of a fungating breast mass.
Results: The patient presented to an outside hospital 1.5 years prior for a left breast mass biopsy that was diagnosed as triple-hit DLBCL. However, he deferred treatment at that time. He subsequently experienced anorexia, nausea, vomiting, consti-pation, and a 20-pound unintentional weight loss. At hospital presentation, a physical examination revealed enlargement of the left breast mass measuring 8 x 14 cm with central ulceration and malodorous purulent discharge and a proximal 7 x 4 cm firm axillary mass. The patient reported no abdominal pain or fever. He was found to have acute kidney injury (creatinine 2.9 mg/dL); hyperuricemia (13.3 mg/dL); and corrected hypercalcemia (17.4 mg/dL). Empiric treatment for tumor lysis syndrome with rasburicase, allopurinol, and aggressive IV hydration was initiated. Further evaluation revealed normal lactate dehydrogenase and K+.The patient was found to have an elevated vitamin D 1,25-OH (244 pg/mL) but low parathyroid hormone (PTH) (6.4 pg/mL); normal PTH-rP; mild hyperphosphatemia (5.5 mg/dL); and negative serum protein electrophoresis/urine protein electrophoresis. Clinical presentation was suggestive of vitamin D-mediated hypercalcemia. The patient received pamidronate and aggressive IV hydration. Electrolyte abnormalities and renal function gradually improved. Computed tomography and positron emission tomography imaging revealed masses localized to the left chest wall with ipsilateral axillary lymphadenopathy. Surgical biopsy of the mass reconfirmed DLBCL ultimately staged as IIE. Following stabilization, the patient declined more aggressive chemotherapy and received R-CHOP with a complete response.
Conclusions: The patient presented with vitamin D-mediated hypercalcemia in the setting of aggressive triple-hit DLBCL. Vitamin D-mediated hypercalcemia occurs in < 1% of lymphomas, and < 0.5% of breast tumors are primary lymphomas. This case study presents a rare constellation of findings at diagnosis. The patient responded to IV hydration and pamidronate and subsequently received R-CHOP for stage IIE DLBCL. While hypercalcemia in the setting of malignancy is associated with poor prognosis, treatment of hypercalcemia does not improve survival.
Purpose: Review the clinical presentation and pathophysiology of hypercalcemia in diffuse large B-cell lymphoma.
Methods: Case study of a 59-year-old man with untreateddiffuse large B-cell lymphoma (DLBCL) who presented with anorexia, constipation, and weight loss in the setting of a fungating breast mass.
Results: The patient presented to an outside hospital 1.5 years prior for a left breast mass biopsy that was diagnosed as triple-hit DLBCL. However, he deferred treatment at that time. He subsequently experienced anorexia, nausea, vomiting, consti-pation, and a 20-pound unintentional weight loss. At hospital presentation, a physical examination revealed enlargement of the left breast mass measuring 8 x 14 cm with central ulceration and malodorous purulent discharge and a proximal 7 x 4 cm firm axillary mass. The patient reported no abdominal pain or fever. He was found to have acute kidney injury (creatinine 2.9 mg/dL); hyperuricemia (13.3 mg/dL); and corrected hypercalcemia (17.4 mg/dL). Empiric treatment for tumor lysis syndrome with rasburicase, allopurinol, and aggressive IV hydration was initiated. Further evaluation revealed normal lactate dehydrogenase and K+.The patient was found to have an elevated vitamin D 1,25-OH (244 pg/mL) but low parathyroid hormone (PTH) (6.4 pg/mL); normal PTH-rP; mild hyperphosphatemia (5.5 mg/dL); and negative serum protein electrophoresis/urine protein electrophoresis. Clinical presentation was suggestive of vitamin D-mediated hypercalcemia. The patient received pamidronate and aggressive IV hydration. Electrolyte abnormalities and renal function gradually improved. Computed tomography and positron emission tomography imaging revealed masses localized to the left chest wall with ipsilateral axillary lymphadenopathy. Surgical biopsy of the mass reconfirmed DLBCL ultimately staged as IIE. Following stabilization, the patient declined more aggressive chemotherapy and received R-CHOP with a complete response.
Conclusions: The patient presented with vitamin D-mediated hypercalcemia in the setting of aggressive triple-hit DLBCL. Vitamin D-mediated hypercalcemia occurs in < 1% of lymphomas, and < 0.5% of breast tumors are primary lymphomas. This case study presents a rare constellation of findings at diagnosis. The patient responded to IV hydration and pamidronate and subsequently received R-CHOP for stage IIE DLBCL. While hypercalcemia in the setting of malignancy is associated with poor prognosis, treatment of hypercalcemia does not improve survival.
Initial Cytogenetic Features of Veteran Patients With Chronic Lymphocytic Leukemia: A National VA Tumor Registry Study
Background: Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults, including elderly veterans. Some veterans have a history of Agent Orange exposure, which may potentially impact their presentation and disease course. We sought to assess initial patterns of cytogenetic aberrations among patients with CLL within the Minneapolis VA Health Care System (MVAHCS).
Methods: For this interim analysis, we evaluated a subset (30%) of a larger sample (6,756). We reviewed clinical data from 2,015 patients with CLL diagnosed from 2000 to 2013 and identified through the National VA Tumor Registry. Baseline demographics, including bone marrow/cytogenetic findings and treatment information were collected. The objective of this study was to assess initial cytogenetic patterns and variation of these parameters by age and Agent Orange exposure.
Results: Median age at diagnosis was 69 years (range, 37-96 years); 98% were male (1,979); Rai stage was 0 (n = 1,331, 66%), 1 (n = 317, 16%), 2 (n = 156, 8%), 3 (n = 91, 5%), 4 (n = 113, 6%). Cytogenetic data were available on 590 of 2,015 (29%) patients. Cytogenetic findings were normal in 258 (44%) patients. Abnormal cytogenetic findings in the remaining 330 cases included del 13q (28%); trisomy 12 (15%); del 11q (11%); del 17p (6%); and other abnor-malities (13%). Of 330 patients with noted abnormalities, 191 (58%) had 1 abnormality; 60 (18%) had 2; and 79 (24%) had > 2 abnormalities. Out of 2,015 patients, 283 (14%) had a reported exposure to Agent Orange; cytogenetic information was available in 130 (46%). Chromosomal abnormalities were detected in 80 of 130 cases (62%). The most frequent abnormality was del 13q (40%); trisomy 12 (19%); other abnormalities (18%); and del 11q (17%). Of the 80 pa-tients with noted abnormalities, 44 (55%) had 1 abnormality;14 (18%) had 2; and 22 (28%) had > 2 abnormalities.
Conclusions: Cytogenetic abnormalities in CLL play an important role in predicting disease progression and survival. These abnormalities paired with Agent Orange exposure have yet to be explored. Utilization of the National VA Tumor Registry data will allow the opportunity to examine the impact, if any, of Agent Orange exposure on the presentation and disease course of veterans with CLL. Updated cytogenetic findings will be presented at the AVAHO annual meeting.
Background: Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults, including elderly veterans. Some veterans have a history of Agent Orange exposure, which may potentially impact their presentation and disease course. We sought to assess initial patterns of cytogenetic aberrations among patients with CLL within the Minneapolis VA Health Care System (MVAHCS).
Methods: For this interim analysis, we evaluated a subset (30%) of a larger sample (6,756). We reviewed clinical data from 2,015 patients with CLL diagnosed from 2000 to 2013 and identified through the National VA Tumor Registry. Baseline demographics, including bone marrow/cytogenetic findings and treatment information were collected. The objective of this study was to assess initial cytogenetic patterns and variation of these parameters by age and Agent Orange exposure.
Results: Median age at diagnosis was 69 years (range, 37-96 years); 98% were male (1,979); Rai stage was 0 (n = 1,331, 66%), 1 (n = 317, 16%), 2 (n = 156, 8%), 3 (n = 91, 5%), 4 (n = 113, 6%). Cytogenetic data were available on 590 of 2,015 (29%) patients. Cytogenetic findings were normal in 258 (44%) patients. Abnormal cytogenetic findings in the remaining 330 cases included del 13q (28%); trisomy 12 (15%); del 11q (11%); del 17p (6%); and other abnor-malities (13%). Of 330 patients with noted abnormalities, 191 (58%) had 1 abnormality; 60 (18%) had 2; and 79 (24%) had > 2 abnormalities. Out of 2,015 patients, 283 (14%) had a reported exposure to Agent Orange; cytogenetic information was available in 130 (46%). Chromosomal abnormalities were detected in 80 of 130 cases (62%). The most frequent abnormality was del 13q (40%); trisomy 12 (19%); other abnormalities (18%); and del 11q (17%). Of the 80 pa-tients with noted abnormalities, 44 (55%) had 1 abnormality;14 (18%) had 2; and 22 (28%) had > 2 abnormalities.
Conclusions: Cytogenetic abnormalities in CLL play an important role in predicting disease progression and survival. These abnormalities paired with Agent Orange exposure have yet to be explored. Utilization of the National VA Tumor Registry data will allow the opportunity to examine the impact, if any, of Agent Orange exposure on the presentation and disease course of veterans with CLL. Updated cytogenetic findings will be presented at the AVAHO annual meeting.
Background: Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults, including elderly veterans. Some veterans have a history of Agent Orange exposure, which may potentially impact their presentation and disease course. We sought to assess initial patterns of cytogenetic aberrations among patients with CLL within the Minneapolis VA Health Care System (MVAHCS).
Methods: For this interim analysis, we evaluated a subset (30%) of a larger sample (6,756). We reviewed clinical data from 2,015 patients with CLL diagnosed from 2000 to 2013 and identified through the National VA Tumor Registry. Baseline demographics, including bone marrow/cytogenetic findings and treatment information were collected. The objective of this study was to assess initial cytogenetic patterns and variation of these parameters by age and Agent Orange exposure.
Results: Median age at diagnosis was 69 years (range, 37-96 years); 98% were male (1,979); Rai stage was 0 (n = 1,331, 66%), 1 (n = 317, 16%), 2 (n = 156, 8%), 3 (n = 91, 5%), 4 (n = 113, 6%). Cytogenetic data were available on 590 of 2,015 (29%) patients. Cytogenetic findings were normal in 258 (44%) patients. Abnormal cytogenetic findings in the remaining 330 cases included del 13q (28%); trisomy 12 (15%); del 11q (11%); del 17p (6%); and other abnor-malities (13%). Of 330 patients with noted abnormalities, 191 (58%) had 1 abnormality; 60 (18%) had 2; and 79 (24%) had > 2 abnormalities. Out of 2,015 patients, 283 (14%) had a reported exposure to Agent Orange; cytogenetic information was available in 130 (46%). Chromosomal abnormalities were detected in 80 of 130 cases (62%). The most frequent abnormality was del 13q (40%); trisomy 12 (19%); other abnormalities (18%); and del 11q (17%). Of the 80 pa-tients with noted abnormalities, 44 (55%) had 1 abnormality;14 (18%) had 2; and 22 (28%) had > 2 abnormalities.
Conclusions: Cytogenetic abnormalities in CLL play an important role in predicting disease progression and survival. These abnormalities paired with Agent Orange exposure have yet to be explored. Utilization of the National VA Tumor Registry data will allow the opportunity to examine the impact, if any, of Agent Orange exposure on the presentation and disease course of veterans with CLL. Updated cytogenetic findings will be presented at the AVAHO annual meeting.
Link Found Between Agent Orange Exposure and Multiple Myeloma
There was a 2.4-fold increased risk for monoclonal gammopathy of undetermined significance (MGUS), a precursor to multiple myeloma (MM), for Air Force veterans exposed to Agent Orange, according to a study reported in JAMA Oncology. Already, veterans who develop MM and were exposed to Agent Orange during military service are eligible to receive benefits, but the study further highlights the relationship.
Related: Management of Myeloma and Its Precursor Syndromes
The Agent Orange used during aerial spray missions of herbicides in the Vietnam War contained 2, 4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), as well as human carcinogen 2,3,7,8-tetrachlorodibenzo-p-doxin in variable amounts. After obtaining the laboratory data from 958 serum samples from Air Force personnel, the Air Force Health Studies questionnaire, and results from the physical exam from all participants, researchers were able to compare their findings with control group veterans.
Related: Nephrotic Syndrome Is a Marker for Occult Cancer
The researchers created 2 test groups from Air Force veterans. The first were 777 participants of Operation Ranch Hand, who conducted aerial herbicidal missions from 1962 to 1971, and the second group was made up of 1,174 participants, who had similar duties but did not participate in the missions.
The risk of MGUS was more pronounced in veterans aged > 70 years (odds ratio [OR], 3.4; 95% confidence interval [CI], 1.27-4.44; P = .007). Among veterans aged > 70 years, there was not a significant increase in risk (OR, 1.4%; 95% CI, 0.55-3.63; P = .63). The crude prevalence of overall MGUS was 7.1% (34 of 479) in the exposed veterans, compared with 3.1% (15 of 479) in the comparison group.
Source
Landgren O, Shim YK, Michalek J, et al. JAMA Oncol. [Published online ahead of print September 3, 2015.]
doi: 10.1001/jamaoncol.2015.2938.
There was a 2.4-fold increased risk for monoclonal gammopathy of undetermined significance (MGUS), a precursor to multiple myeloma (MM), for Air Force veterans exposed to Agent Orange, according to a study reported in JAMA Oncology. Already, veterans who develop MM and were exposed to Agent Orange during military service are eligible to receive benefits, but the study further highlights the relationship.
Related: Management of Myeloma and Its Precursor Syndromes
The Agent Orange used during aerial spray missions of herbicides in the Vietnam War contained 2, 4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), as well as human carcinogen 2,3,7,8-tetrachlorodibenzo-p-doxin in variable amounts. After obtaining the laboratory data from 958 serum samples from Air Force personnel, the Air Force Health Studies questionnaire, and results from the physical exam from all participants, researchers were able to compare their findings with control group veterans.
Related: Nephrotic Syndrome Is a Marker for Occult Cancer
The researchers created 2 test groups from Air Force veterans. The first were 777 participants of Operation Ranch Hand, who conducted aerial herbicidal missions from 1962 to 1971, and the second group was made up of 1,174 participants, who had similar duties but did not participate in the missions.
The risk of MGUS was more pronounced in veterans aged > 70 years (odds ratio [OR], 3.4; 95% confidence interval [CI], 1.27-4.44; P = .007). Among veterans aged > 70 years, there was not a significant increase in risk (OR, 1.4%; 95% CI, 0.55-3.63; P = .63). The crude prevalence of overall MGUS was 7.1% (34 of 479) in the exposed veterans, compared with 3.1% (15 of 479) in the comparison group.
Source
Landgren O, Shim YK, Michalek J, et al. JAMA Oncol. [Published online ahead of print September 3, 2015.]
doi: 10.1001/jamaoncol.2015.2938.
There was a 2.4-fold increased risk for monoclonal gammopathy of undetermined significance (MGUS), a precursor to multiple myeloma (MM), for Air Force veterans exposed to Agent Orange, according to a study reported in JAMA Oncology. Already, veterans who develop MM and were exposed to Agent Orange during military service are eligible to receive benefits, but the study further highlights the relationship.
Related: Management of Myeloma and Its Precursor Syndromes
The Agent Orange used during aerial spray missions of herbicides in the Vietnam War contained 2, 4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T), as well as human carcinogen 2,3,7,8-tetrachlorodibenzo-p-doxin in variable amounts. After obtaining the laboratory data from 958 serum samples from Air Force personnel, the Air Force Health Studies questionnaire, and results from the physical exam from all participants, researchers were able to compare their findings with control group veterans.
Related: Nephrotic Syndrome Is a Marker for Occult Cancer
The researchers created 2 test groups from Air Force veterans. The first were 777 participants of Operation Ranch Hand, who conducted aerial herbicidal missions from 1962 to 1971, and the second group was made up of 1,174 participants, who had similar duties but did not participate in the missions.
The risk of MGUS was more pronounced in veterans aged > 70 years (odds ratio [OR], 3.4; 95% confidence interval [CI], 1.27-4.44; P = .007). Among veterans aged > 70 years, there was not a significant increase in risk (OR, 1.4%; 95% CI, 0.55-3.63; P = .63). The crude prevalence of overall MGUS was 7.1% (34 of 479) in the exposed veterans, compared with 3.1% (15 of 479) in the comparison group.
Source
Landgren O, Shim YK, Michalek J, et al. JAMA Oncol. [Published online ahead of print September 3, 2015.]
doi: 10.1001/jamaoncol.2015.2938.
Castleman Disease
Castleman disease (CD) is a rare nonclonal lymphoproliferative disorder, also known as angiofollicular lymph-node hyperplasia or giant node hyperplasia. It was first reported in 1954 and in 1956 described by Benjamin Castleman, MD, in a case series of localized mediastinal lymph-node hyperplasia.1 Unicentric Castleman (UCD) disease presents as a localized disease affecting a single lymph node/lymph node chain. Multicentric Castleman disease (MCD) is a more widespread or generalized disease (Table 1). About 4,000 to 6,000 new cases of CD are diagnosed per year of which about 20% to 25% cases are MCD. The estimated incidence rate for CD has recently been calculated as 21 to 25 per million person-years, or about 6,000 new cases annually.2

The clinical presentation of CD often overlaps with autoimmune, infectious, or other malignant diseases. The diagnosis is confirmed by a biopsy of the affected lymph-node tissue. Interleukin-6 (IL-6) and a viral analog of IL-6 play major role in the pathogenesis by stimulating a widespread inflammatory response that results in systemic manifestations. It is often associated with HIV and human herpesvirus-8 (HHV-8) infections. Castleman disease is histologically characterized into the hyaline vascular variant, the plasma-cell variant, and the mixed form. The plasmablastic variety is associated with HIV and HHV-8 infections. The prognosis ranges from good in UCD (91% overall survival [OS] at 5 y) to poor in MCD (65% OS at 5 y).3
Treatment options range from local surgical excision to systemic treatments. Newer therapies include monoclonal antibodies against both IL-6 and CD20 and a few other targets in the inflammatory cascade. This article discusses the updated approach to diagnosis and management of CD.
Unicentric Castleman Disease
Unicentric CD is more common than MCD, presents as a localized lymph node or chain involvement, and is generally diagnosed in the third or fourth decade of life but has been reported in children. The presenting symptoms of UCD vary by site. It presents as nontender lymphadenopathy when confined to peripheral lymph nodes, whereas respiratory symptoms or bowel obstruction may be seen with lymphadenopathy in the chest/mediastinum, neck, or abdomen. The systemic
symptoms, such as fever, night sweats, and weight loss, are uncommon.
Dysplastic follicular dendritic cells characterize UCD. Histologically, it is usually classified as hyaline vascular disease with the follicles comprising small lymphocytes
and dendritic cells forming concentric rings with prominent vascularity.4,5 No association with HIV or HHV-8 has been seen.
Unicentric CD is often amenable to resection, and a complete cure can be achieved.6 Partial resection may be attempted when complete resection is not possible. Radiation therapy is offered for unresectable disease.7 In patients who are not candidates for any intervention, close long-term follow-up is recommended unless patients are symptomatic, in which case systemic treatment should be considered.
Multicentric Castleman Disease
The more widespread MCD is generally diagnosed in the fifth or sixth decade of life. It is more aggressive than UCD and presents a wide spectrum of symptoms and abnormal laboratory findings (Table 2).8
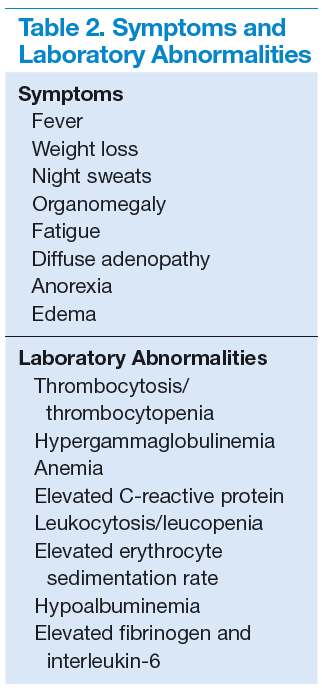
It is histologically classified into (a) plasmablastic or HHV-8 associated: It is often seen in patients with MCD infected with HIV, which can give rise to large B-cell lymphoma, known as HHV-8 plasmablastic lymphoma9; (b) plasmacytic variant has marked paracortical plasmacytosis with retained nodal architecture10; and (c) mixed MCD has abundant plasma cells with features similar to those of the hyaline-vascular variant.
Most patients with HIV-associated MCD are co-infected with HHV-8. The HHV-8 infection is also present in about 50% of HIV-negative cases.11 The incidence of HIVassociated MCD is increasing in the highly active antiretroviral therapy (HAART) era secondary to improved survival of patients infected with HIV.12 To diagnose active HIV MCD, the French Agence Nationale de Recherche sur le SIDA 117 CastlemaB trial group has described criteria based on the clinical symptoms, including fever, a raised serum C-reactive protein > 20 mg/L without any other cause, and 3 of 12 additional clinical findings described as peripheral lymphadenopathy, splenomegaly, ascites, edema, pleural effusion, cough, nasal obstruction, xerostomia, rash, central neurologic symptoms, jaundice, and autoimmune hemolytic anemia.13 The reported 2-year survival of patients who are HIV-negative is 97% compared with HIV-positive cases at 67%.14
Idiopathic MCD is diagnosed when there is no evidence of any underlying infectious, autoimmune, and neoplastic process.15
Patients with MCD are at an increased risk of developing non-Hodgkin and Hodgkin lymphoma, Kaposi sarcoma, primary effusion lymphoma, and follicular dendritic
cell sarcoma. POEMS (peripheral neuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome and paraneoplastic disease, such as paraneoplastic pemphigus myasthenia gravis, may be commonly diagnosed concurrently or sequentially with MCD.16-20
The disease course of MCD ranges from indolent to rapidly progressive, and its 5-year OS is about 65%. When associated with POEMS syndrome, the 5-year survival was estimated to be 90% with the osteosclerotic variant and 27% without osteosclerotic lesions.3 Treatment options for MCD include systemic chemotherapy, including antiviral therapy for HHV-8 positive and HAART for HIV positive and newer monoclonal antibody therapies targeting CD20 or IL-6.
Pathophysiology
Interleukin-6 plays an important role for inflammation in both UCD and MCD (Figure 1). There is dysregulation and overproduction of IL-6, which further stimulates the production of acute-phase reactants, resulting in various systemic manifestations.15,21,22 There is increased expression of IL-1 and IL-6, upregulation of IL-6 secondary to interaction of IL-1 with nuclear factor-kappa B (NF-kappa B), thus stimulating B-cell proliferation. IL-6 binding to IL-6 receptor (IL6-R) results in downstream activation of transcription Janus kinases/signal transducers and activators of the transcription pathway. This promotes the transcription of genes encoding the acute-phase reactant proteins. Hence, interfering with IL-6 transduction by blocking downstream signals are potential therapeutic targets. The mitogen-activated protein kinase cascade, the rapidly accelerated fibrosarcoma kinases, and the overexpression of the endothelial growth factor receptor (EGFR), all contribute to disease pathogenesis by promoting increased B-cell proliferation and vascular EGFR mediated angiogenesis. 23,24
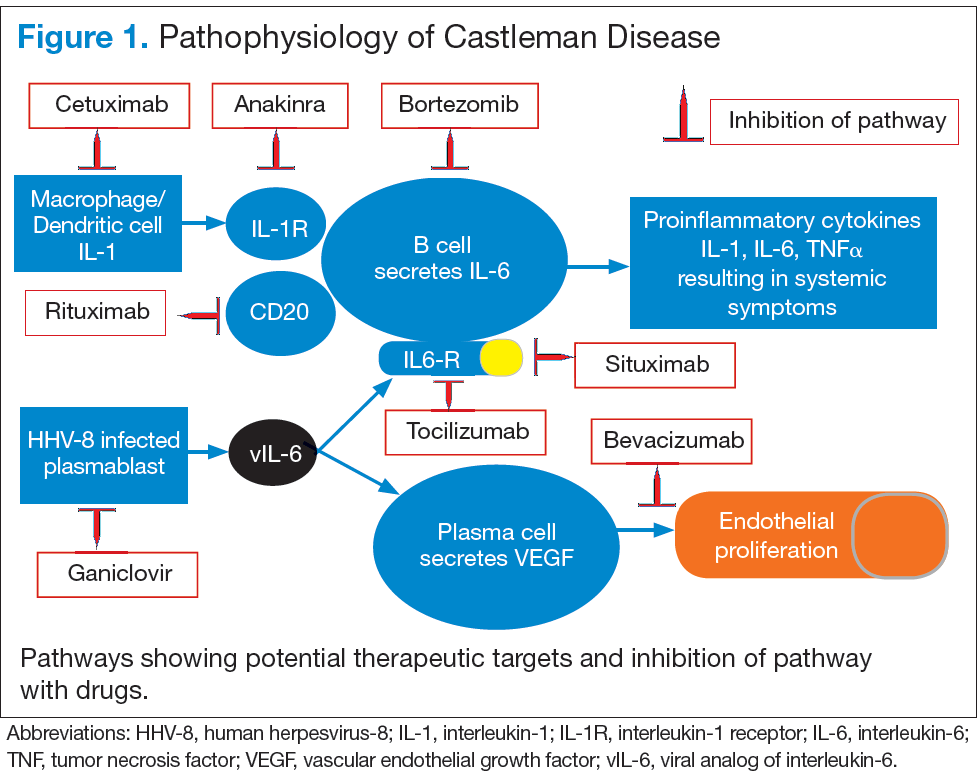
In HHV-8–associated MCD, the virus replicates within lymph node plasmablasts, causing increased production of viral IL-6 analog, human IL-6, and other proinflammatory proteins resulting in B-cell and plasma-cell proliferation, increased vascular endothelial growth factor secretion and angiogenesis.25,26 The HHV-8–infected plasmablasts are marked by variable expression of CD20, and therefore, anti-CD20 is also shown to be an effective treatment. The calmodulin/calcineurin nuclear factor assists in the proliferation of HHV-8, thereby making calcineurin another potential target for the antiviral proliferation.27
Staging
The treatment decisions and prognosis for patients with CD is based on the clinical and histologic staging. The initial workup includes but is not limited to routine laboratory evaluation, imaging, and HIV and HHV-8 testing (Table 3). Routine tests of the levels of cytokines are not recommended. Other relevant tests for known disease associations should be obtained when relevant.

Treatment
Better understanding of the disease process in CD has helped to identify potential therapeutic targets (Figures 2 and 3).
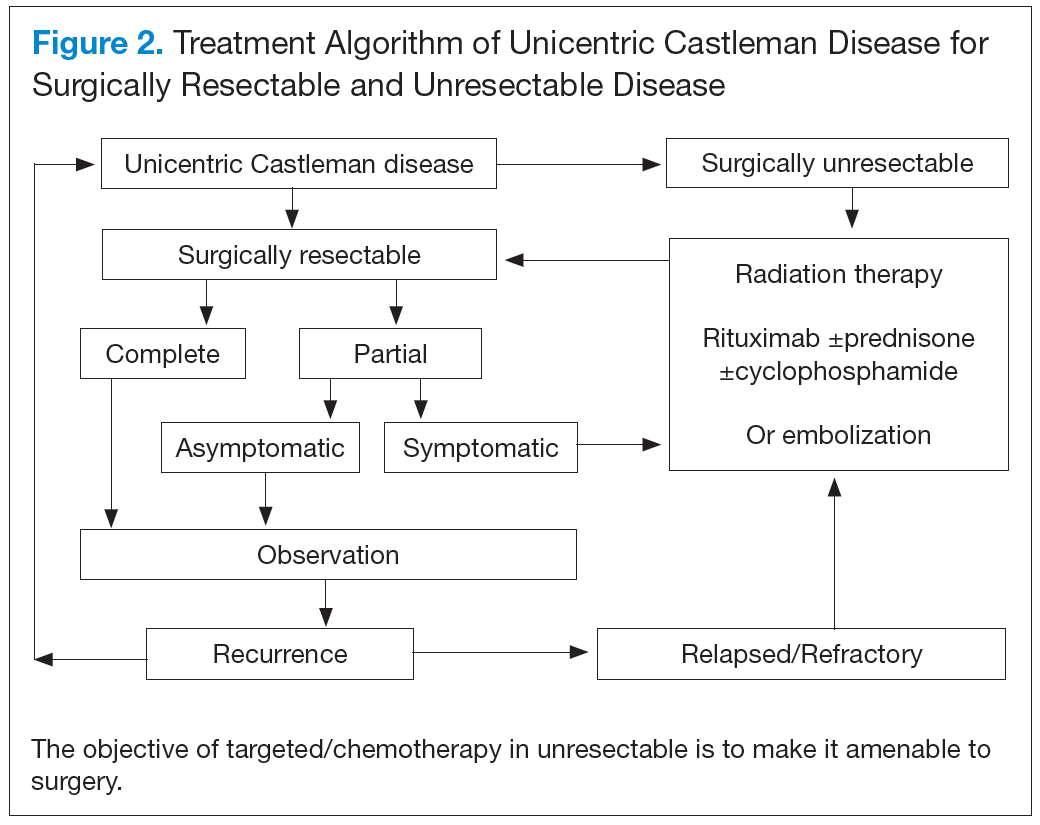
For UCD, surgery is the mainstay of treatment.4,28,29 In surgically unresectable cases, radiation therapy is helpful for local disease control. Alternatively, neoadjuvant
chemotherapy and rituximab are used. Corticosteroids are generally used to treat acute exacerbations and as adjuncts to chemotherapy.
For MCD, the treatment approach depends on the HIV and HHV-8 status of the patient. For patients with HHV-8 infection, both with and without HIV co-infection, antiviral agents, such as ganciclovir, foscarnet, or cidofovir, have shown in vitro activity against HHV-8 but with limited clinical success.30 In patients infected with HIV, the aim of treating with HAART is to control the disease, prevent opportunistic infections, and improve tolerance to chemotherapy.31-33 Rituximab with or without chemotherapy is the standard treatment approach. The additional chemotherapeutic agents are used depending on the presence or absence of organ failure. This approach has improved the OS in HIV-associated MCD.34,35 Treatment with HAART does not decrease the risk of relapse in HIV MCD; therefore, the role of rituximab and antiherpesvirus agents as maintenance therapy has been explored.36 In patients who fail to respond to or relapse rapidly following rituximab monotherapy, the use of either single-agent chemotherapy with or without rituximab or antiherpesvirus therapy with high-dose zidovudine and valganciclovir is recommended.37
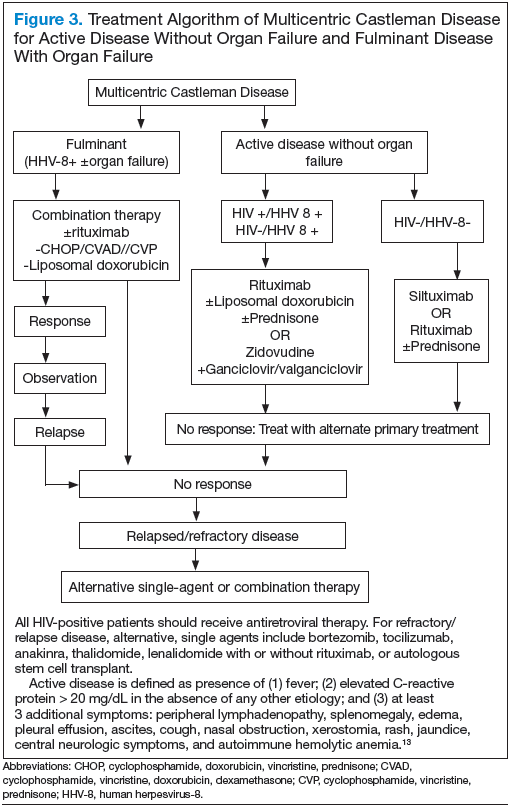
The cytotoxic chemotherapy with single agents, such as etoposide, vinblastine, cyclophosphamide, cladribine, chlorambucil, and liposomal doxorubicin, has been used with limited success.22 The combination chemotherapy with cyclophosphamide/doxorubicin/vincristine/prednisone (CHOP) or cyclophosphamide/vincristine/
prednisone (CVP) without rituximab has been shown to achieve durable remissions. Corticosteroids are usually administered as an initial adjunct to chemotherapy or for acute exacerbations. In patients with MCD, regardless of HIV status, the interferon therapy was found to achieve long-term remission.38,39 The interferon therapy
exerts antiviral effects via downregulation of the IL-6R and inhibition of HHV-8 replication. For patients in remission, maintenance therapy with oral valganciclovir is promising.40
Immunomodulators & Targeted Therapies
For unresectable UCD or MCD with organ failure or relapse, the use of alternativesingle-agent or combination chemotherapies with or without rituximab is recommended. Thalidomide has shown some success, probably secondary to disruption of IL-6 production.41 In cases of progression following second-line therapy, bortezomib, antiherpesvirus therapies, or IL-6–directed therapy with siltuximab or tocilizumab should be considered.
Rituximab is a monoclonal chimeric antibody that targets CD20 on B cells, thus leading to B-cell lymphodepletion via activating complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity. As monotherapy, it has been shown to achieve 2-year progression-free survival in 80% of patients.42 In patients with MCD who are HIV positive, rituximab with and without chemotherapy has shown improved overall and disease-free survival of 70% to 80% at 2 years.43
Siltuximab is a chimeric human-mouse monoclonal antibody to IL-6 that has been approved for treatment of patients with MCD who are both HIV negative and HHV-8 negative.44-46 Tocilizumab targets the IL-6R. The antibody has shown improvement in a study in HIVseronegative adults with MCD.47,48
Bortezomib is a proteasome inhibitor that inhibits the NF-kappa B pathway, which induces the expression of numerous proinflammatory proteins, including IL-6. It is recommended for relapsed or refractory disease.49,50
Anakinra is a recombinant IL-1R antagonist that blocks IL-1 effects and controls disease by decreasing IL-6 production.51
Conculsion
There has been significant progress in disease diagnosis and management as more information is available about the incidence, clinical presentation, and pathophysiology of CD. The understanding of the disease pathogenesis and biology has helped to discover multiple potential therapeutic targets. Successful treatment has been achieved through targeting HHV-8 replication, CD20, and IL-6 and anti– IL-6R antibodies. Although surgical resection continues to be the
standard of therapy for UCD, the management of MCD and relapsed or refractory disease continues to evolve. Exploration of various treatment strategies in different clinical presentations is warranted.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S.
Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information
for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to
patients.
Click here to read the digital edition.
1. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymph-node hyperplasia
resembling thymoma. Cancer. 1956;9(4):822-830.
2. Munshi N, Mehra M, van de Velde H, Desai A, Potluri R, Vermeulen J. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma. 2015;56(5):1252-1260.
3. Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997-1002.
4. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29(3):670-683.
5. Cronin DM, Warnke RA. Castleman disease: an update on classification and the
spectrum of associated lesions. Adv Anat Pathol. 2009;16(4):236-246.
6. Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman’s disease: a systematic review of 404 published cases. Ann Surg. 2012;255(4):677-684.
7. Chronowski GM, Ha CS, Wilder RB, Cabanillas F, Manning J, Cox JD. Treatment of unicentric and multicentric Castleman disease and the role of radiotherapy. Cancer. 2001;92(3):670-676.
8. Herrada J, Cabanillas F, Rice L, Manning J, Pugh W. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med. 1998;128(8):657-662.
9. Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95(4):1406-1412.
10. Ferry JA, Harris NL. Atlas of Lymphoid Hyperplasia and Lymphoma. Philadelphia, PA: W.B. Saunders; 1997.
11. Soulier J, Grollet L, Oksenhendler E, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86(4):1276-1280.
12. Powles T, Stebbing J, Bazeos A, et al. The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman’s disease. Ann Oncol. 2009;20(4):775-779.
13. Gérard L, Bérezné A, Galicier L, et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman’s disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25(22):3350-3356.
14. Casper C, Teltsch DY, Robinson D Jr, et al. Clinical characteristics and healthcare utilization of patients with multicentric Castleman disease. Br J Haematol. 2015;168(1):82-93.
15. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933.
16. Larroche C, Cacoub P, Soulier J, et al. Castleman’s disease and lymphoma: report of eight cases in HIV-negative patients and literature review. Am J Hematol. 2002;69(2):119-126.
17. Dispenzieri A. POEMS syndrome: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89(2):214-223.
18. Andhavarapu S, Jiang L. POEMS syndrome and Castleman disease. Blood. 2013;122(2):159.
19. Bélec L, Mohamed AS, Authier FJ, et al. Human herpesvirus 8 infection in patients with POEMS syndrome-associated multicentric Castleman’s disease. Blood. 1999;93(11):3643-3653.
20. Oksenhendler E, Boulanger E, Galicier L, et al. High incidence of Kaposi sarcomaassociated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood. 2002;99(7):2331-2336.
21. Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood. 1989;74(4):1360-1367.
22. El-Osta HE, Kurzrock R. Castleman’s disease: from basic mechanisms to molecular therapeutics. Oncologist. 2011;16(4):497-511.
23. Akula SM, Ford PW, Whitman AG, et al. B-Raf-dependent expression of vascular endothelial growth factor-A in Kaposi sarcoma-associated herpesvirus-infected human B cells. Blood. 2005;105(11):4516-4522.
24. Sun X, Chang KC, Abruzzo LV, Lai R, Younes A, Jones D. Epidermal growth factor receptor expression in follicular dendritic cells: a shared feature of follicular dendritic cell sarcoma and Castleman’s disease. Hum Pathol. 2003;34(9):835-840.
25. Adam N, Rabe B, Suthaus J, Grötzinger J, Rose-John S, Scheller J. Unraveling viral interleukin-6 binding to gp130 and activation of STAT-signaling pathways independently of the interleukin-6 receptor. J Virol. 2009;83(10):5117-5126.
26. Suda T, Katano H, Delsol G, et al. HHV-8 infection status of AIDS-unrelated and
AIDS-associated multicentric Castleman’s disease. Pathol Int. 2001;51(9):671-679.
27. Zoeteweij JP, Moses AV, Rinderknecht AS, et al. Targeted inhibition of calcineurin signaling blocks calcium-dependent reactivation of Kaposi sarcoma-associated herpesvirus. Blood. 2001;97(8):2374-2380.
28. McCarty MJ, Vukelja SJ, Banks PM, Weiss RB. Angiofollicular lymph node hyperplasia
(Castleman’s disease). Cancer Treat Rev. 1995;21(4):291-310.
29. Bowne WB, Lewis JJ, Filippa DA, et al. The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer. 1999;85(3):706-717.
30. Reddy D, Mitsuyasu R. HIV-associated multicentric Castleman disease. Curr Opin Oncol. 2011;23(5):475-481.
31. Aaron L, Lidove O, Yousry C, Roudiere L, Dupont B, Viard JP. Human herpesvirus 8-positive Castleman disease in human immunodeficiency virus-infected patients: the impact of highly active antiretroviral therapy. Clin Infect Dis. 2002;35(7):880-882.
32. Sprinz E, Jeffman M, Liedke P, Putten A, Schwartsmann G. Successful treatment of AIDS-related Castleman’s disease following the administration of highly active antiretroviral therapy (HAART). Ann Oncol. 2004;15(2):356-358.
33. Lee SM, Edwards SG, Chilton DN, Ramsay A, Miller RF. Highly active antiretroviral therapy alone may be an effective treatment for HIV-associated multi-centric Castleman’s disease. Haematologica. 2010;95(11):1979-1981.
34. Bower M. How I treat HIV-associated multicentric Castleman disease. Blood. 2010;116(22):4415-4421.
35. Bower M, Newsom-Davis T, Naresh K, et al. Clinical features and outcome in HIVassociated
multicentric Castleman’s disease. J Clin Oncol. 2011;29(18):2481-2486.
36. Casper C, Nichols WG, Huang ML, Corey L, Wald A. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood. 2004;103(5):1632-1634.
37. Uldrick TS, Polizzotto MN, Aleman K, et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood. 2011;117(26):6977-6986.
38. Kumari P, Schechter GP, Saini N, Benator DA. Successful treatment of human immunodeficiency virus-related Castleman’s disease with interferon-alpha. Clin Infect Dis. 2000;31(2):602-604.
39. Nord JA, Karter D. Low dose interferon-alpha therapy for HIV-associated multicentric Castleman’s disease. Int J STD AIDS. 2003;14(1):61-62.
40. Oksenhendler E. HIV-associated multicentric Castleman disease. Curr Opin HIV AIDS. 2009;4(1):16-21.
41. Jung CP, Emmerich B, Goebel FD, Bogner JR. Successful treatment of a patient with HIV-associated multicentric Castleman disease (MCD) with thalidomide. Am J Hematol. 2004;75(3):176-177.
42. Ide M, Kawachi Y, Izumi Y, Kasagi K, Ogino T. Long-term remission in HIV negative patients with multicentric Castleman’s disease using rituximab. Eur J Haematol. 2006;76(2):119-123.
43. Marcelin AG, Aaron L, Mateus C, et al. Rituximab therapy for HIV-associated Castleman disease. Blood. 2003;102(8):2786-2788.
44. Van Rhee F, Fayad L, Voorhees P, et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol. 2010;28(23):3701-3708.
45. Wong RS, Casper C, Munshi N, et al. A multicenter, randomized, doubleblind, placebo-controlled study of the efficacy and safety of siltuximab, an antiinterleukin-6 monoclonal antibody, in patients with multicentric Castleman’s disease. Blood. 2013;122(21):505.
46. Van Rhee F, Casper C, Voorhees PM, et al. An open-label, phase 2, multicenter study of the safety of long-term treatment with siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman’s disease. Blood. 2013;122(21):1806.
47. Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627-2632.
48. Müzes G, Sipos F, Csomor J, Sréter L. Successful tocilizumab treatment in a patient with human herpesvirus 8-positive and human immunodeficiency virusnegative multicentric Castleman’s disease of plasma cell type nonresponsive to rituximab-CVP therapy. APMIS. 2013;121(7):668-674.
49. Hess G, Wagner V, Kreft A, Heussel CP, Huber C. Effects of bortezomib on proinflammatory cytokine levels and transfusion dependency in a patient with multicentric Castleman disease. Br J Haematol. 2006;134(5):544-545.
50. Sobas MA, Alonso Vence N, Diaz Arias J, Bendaña Lopez A, Fraga Rodriguez M, Bello Lopez JL. Efficacy of bortezomib in refractory form of multicentric Castleman disease associated to poems syndrome (MCD-POEMS variant). Ann Hematol. 2010;89(2):217-219.
51. El-Osta H, Janku F, Kurzrock R. Successful treatment of Castleman’s disease with interleukin-1 receptor antagonist (Anakinra). Mol Cancer Ther. 2010;9(6):1485-1488.
Castleman disease (CD) is a rare nonclonal lymphoproliferative disorder, also known as angiofollicular lymph-node hyperplasia or giant node hyperplasia. It was first reported in 1954 and in 1956 described by Benjamin Castleman, MD, in a case series of localized mediastinal lymph-node hyperplasia.1 Unicentric Castleman (UCD) disease presents as a localized disease affecting a single lymph node/lymph node chain. Multicentric Castleman disease (MCD) is a more widespread or generalized disease (Table 1). About 4,000 to 6,000 new cases of CD are diagnosed per year of which about 20% to 25% cases are MCD. The estimated incidence rate for CD has recently been calculated as 21 to 25 per million person-years, or about 6,000 new cases annually.2
The clinical presentation of CD often overlaps with autoimmune, infectious, or other malignant diseases. The diagnosis is confirmed by a biopsy of the affected lymph-node tissue. Interleukin-6 (IL-6) and a viral analog of IL-6 play major role in the pathogenesis by stimulating a widespread inflammatory response that results in systemic manifestations. It is often associated with HIV and human herpesvirus-8 (HHV-8) infections. Castleman disease is histologically characterized into the hyaline vascular variant, the plasma-cell variant, and the mixed form. The plasmablastic variety is associated with HIV and HHV-8 infections. The prognosis ranges from good in UCD (91% overall survival [OS] at 5 y) to poor in MCD (65% OS at 5 y).3
Treatment options range from local surgical excision to systemic treatments. Newer therapies include monoclonal antibodies against both IL-6 and CD20 and a few other targets in the inflammatory cascade. This article discusses the updated approach to diagnosis and management of CD.
Unicentric Castleman Disease
Unicentric CD is more common than MCD, presents as a localized lymph node or chain involvement, and is generally diagnosed in the third or fourth decade of life but has been reported in children. The presenting symptoms of UCD vary by site. It presents as nontender lymphadenopathy when confined to peripheral lymph nodes, whereas respiratory symptoms or bowel obstruction may be seen with lymphadenopathy in the chest/mediastinum, neck, or abdomen. The systemic
symptoms, such as fever, night sweats, and weight loss, are uncommon.
Dysplastic follicular dendritic cells characterize UCD. Histologically, it is usually classified as hyaline vascular disease with the follicles comprising small lymphocytes
and dendritic cells forming concentric rings with prominent vascularity.4,5 No association with HIV or HHV-8 has been seen.
Unicentric CD is often amenable to resection, and a complete cure can be achieved.6 Partial resection may be attempted when complete resection is not possible. Radiation therapy is offered for unresectable disease.7 In patients who are not candidates for any intervention, close long-term follow-up is recommended unless patients are symptomatic, in which case systemic treatment should be considered.
Multicentric Castleman Disease
The more widespread MCD is generally diagnosed in the fifth or sixth decade of life. It is more aggressive than UCD and presents a wide spectrum of symptoms and abnormal laboratory findings (Table 2).8
It is histologically classified into (a) plasmablastic or HHV-8 associated: It is often seen in patients with MCD infected with HIV, which can give rise to large B-cell lymphoma, known as HHV-8 plasmablastic lymphoma9; (b) plasmacytic variant has marked paracortical plasmacytosis with retained nodal architecture10; and (c) mixed MCD has abundant plasma cells with features similar to those of the hyaline-vascular variant.
Most patients with HIV-associated MCD are co-infected with HHV-8. The HHV-8 infection is also present in about 50% of HIV-negative cases.11 The incidence of HIVassociated MCD is increasing in the highly active antiretroviral therapy (HAART) era secondary to improved survival of patients infected with HIV.12 To diagnose active HIV MCD, the French Agence Nationale de Recherche sur le SIDA 117 CastlemaB trial group has described criteria based on the clinical symptoms, including fever, a raised serum C-reactive protein > 20 mg/L without any other cause, and 3 of 12 additional clinical findings described as peripheral lymphadenopathy, splenomegaly, ascites, edema, pleural effusion, cough, nasal obstruction, xerostomia, rash, central neurologic symptoms, jaundice, and autoimmune hemolytic anemia.13 The reported 2-year survival of patients who are HIV-negative is 97% compared with HIV-positive cases at 67%.14
Idiopathic MCD is diagnosed when there is no evidence of any underlying infectious, autoimmune, and neoplastic process.15
Patients with MCD are at an increased risk of developing non-Hodgkin and Hodgkin lymphoma, Kaposi sarcoma, primary effusion lymphoma, and follicular dendritic
cell sarcoma. POEMS (peripheral neuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome and paraneoplastic disease, such as paraneoplastic pemphigus myasthenia gravis, may be commonly diagnosed concurrently or sequentially with MCD.16-20
The disease course of MCD ranges from indolent to rapidly progressive, and its 5-year OS is about 65%. When associated with POEMS syndrome, the 5-year survival was estimated to be 90% with the osteosclerotic variant and 27% without osteosclerotic lesions.3 Treatment options for MCD include systemic chemotherapy, including antiviral therapy for HHV-8 positive and HAART for HIV positive and newer monoclonal antibody therapies targeting CD20 or IL-6.
Pathophysiology
Interleukin-6 plays an important role for inflammation in both UCD and MCD (Figure 1). There is dysregulation and overproduction of IL-6, which further stimulates the production of acute-phase reactants, resulting in various systemic manifestations.15,21,22 There is increased expression of IL-1 and IL-6, upregulation of IL-6 secondary to interaction of IL-1 with nuclear factor-kappa B (NF-kappa B), thus stimulating B-cell proliferation. IL-6 binding to IL-6 receptor (IL6-R) results in downstream activation of transcription Janus kinases/signal transducers and activators of the transcription pathway. This promotes the transcription of genes encoding the acute-phase reactant proteins. Hence, interfering with IL-6 transduction by blocking downstream signals are potential therapeutic targets. The mitogen-activated protein kinase cascade, the rapidly accelerated fibrosarcoma kinases, and the overexpression of the endothelial growth factor receptor (EGFR), all contribute to disease pathogenesis by promoting increased B-cell proliferation and vascular EGFR mediated angiogenesis. 23,24
In HHV-8–associated MCD, the virus replicates within lymph node plasmablasts, causing increased production of viral IL-6 analog, human IL-6, and other proinflammatory proteins resulting in B-cell and plasma-cell proliferation, increased vascular endothelial growth factor secretion and angiogenesis.25,26 The HHV-8–infected plasmablasts are marked by variable expression of CD20, and therefore, anti-CD20 is also shown to be an effective treatment. The calmodulin/calcineurin nuclear factor assists in the proliferation of HHV-8, thereby making calcineurin another potential target for the antiviral proliferation.27
Staging
The treatment decisions and prognosis for patients with CD is based on the clinical and histologic staging. The initial workup includes but is not limited to routine laboratory evaluation, imaging, and HIV and HHV-8 testing (Table 3). Routine tests of the levels of cytokines are not recommended. Other relevant tests for known disease associations should be obtained when relevant.
Treatment
Better understanding of the disease process in CD has helped to identify potential therapeutic targets (Figures 2 and 3).
For UCD, surgery is the mainstay of treatment.4,28,29 In surgically unresectable cases, radiation therapy is helpful for local disease control. Alternatively, neoadjuvant
chemotherapy and rituximab are used. Corticosteroids are generally used to treat acute exacerbations and as adjuncts to chemotherapy.
For MCD, the treatment approach depends on the HIV and HHV-8 status of the patient. For patients with HHV-8 infection, both with and without HIV co-infection, antiviral agents, such as ganciclovir, foscarnet, or cidofovir, have shown in vitro activity against HHV-8 but with limited clinical success.30 In patients infected with HIV, the aim of treating with HAART is to control the disease, prevent opportunistic infections, and improve tolerance to chemotherapy.31-33 Rituximab with or without chemotherapy is the standard treatment approach. The additional chemotherapeutic agents are used depending on the presence or absence of organ failure. This approach has improved the OS in HIV-associated MCD.34,35 Treatment with HAART does not decrease the risk of relapse in HIV MCD; therefore, the role of rituximab and antiherpesvirus agents as maintenance therapy has been explored.36 In patients who fail to respond to or relapse rapidly following rituximab monotherapy, the use of either single-agent chemotherapy with or without rituximab or antiherpesvirus therapy with high-dose zidovudine and valganciclovir is recommended.37
The cytotoxic chemotherapy with single agents, such as etoposide, vinblastine, cyclophosphamide, cladribine, chlorambucil, and liposomal doxorubicin, has been used with limited success.22 The combination chemotherapy with cyclophosphamide/doxorubicin/vincristine/prednisone (CHOP) or cyclophosphamide/vincristine/
prednisone (CVP) without rituximab has been shown to achieve durable remissions. Corticosteroids are usually administered as an initial adjunct to chemotherapy or for acute exacerbations. In patients with MCD, regardless of HIV status, the interferon therapy was found to achieve long-term remission.38,39 The interferon therapy
exerts antiviral effects via downregulation of the IL-6R and inhibition of HHV-8 replication. For patients in remission, maintenance therapy with oral valganciclovir is promising.40
Immunomodulators & Targeted Therapies
For unresectable UCD or MCD with organ failure or relapse, the use of alternativesingle-agent or combination chemotherapies with or without rituximab is recommended. Thalidomide has shown some success, probably secondary to disruption of IL-6 production.41 In cases of progression following second-line therapy, bortezomib, antiherpesvirus therapies, or IL-6–directed therapy with siltuximab or tocilizumab should be considered.
Rituximab is a monoclonal chimeric antibody that targets CD20 on B cells, thus leading to B-cell lymphodepletion via activating complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity. As monotherapy, it has been shown to achieve 2-year progression-free survival in 80% of patients.42 In patients with MCD who are HIV positive, rituximab with and without chemotherapy has shown improved overall and disease-free survival of 70% to 80% at 2 years.43
Siltuximab is a chimeric human-mouse monoclonal antibody to IL-6 that has been approved for treatment of patients with MCD who are both HIV negative and HHV-8 negative.44-46 Tocilizumab targets the IL-6R. The antibody has shown improvement in a study in HIVseronegative adults with MCD.47,48
Bortezomib is a proteasome inhibitor that inhibits the NF-kappa B pathway, which induces the expression of numerous proinflammatory proteins, including IL-6. It is recommended for relapsed or refractory disease.49,50
Anakinra is a recombinant IL-1R antagonist that blocks IL-1 effects and controls disease by decreasing IL-6 production.51
Conculsion
There has been significant progress in disease diagnosis and management as more information is available about the incidence, clinical presentation, and pathophysiology of CD. The understanding of the disease pathogenesis and biology has helped to discover multiple potential therapeutic targets. Successful treatment has been achieved through targeting HHV-8 replication, CD20, and IL-6 and anti– IL-6R antibodies. Although surgical resection continues to be the
standard of therapy for UCD, the management of MCD and relapsed or refractory disease continues to evolve. Exploration of various treatment strategies in different clinical presentations is warranted.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S.
Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information
for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to
patients.
Click here to read the digital edition.
Castleman disease (CD) is a rare nonclonal lymphoproliferative disorder, also known as angiofollicular lymph-node hyperplasia or giant node hyperplasia. It was first reported in 1954 and in 1956 described by Benjamin Castleman, MD, in a case series of localized mediastinal lymph-node hyperplasia.1 Unicentric Castleman (UCD) disease presents as a localized disease affecting a single lymph node/lymph node chain. Multicentric Castleman disease (MCD) is a more widespread or generalized disease (Table 1). About 4,000 to 6,000 new cases of CD are diagnosed per year of which about 20% to 25% cases are MCD. The estimated incidence rate for CD has recently been calculated as 21 to 25 per million person-years, or about 6,000 new cases annually.2
The clinical presentation of CD often overlaps with autoimmune, infectious, or other malignant diseases. The diagnosis is confirmed by a biopsy of the affected lymph-node tissue. Interleukin-6 (IL-6) and a viral analog of IL-6 play major role in the pathogenesis by stimulating a widespread inflammatory response that results in systemic manifestations. It is often associated with HIV and human herpesvirus-8 (HHV-8) infections. Castleman disease is histologically characterized into the hyaline vascular variant, the plasma-cell variant, and the mixed form. The plasmablastic variety is associated with HIV and HHV-8 infections. The prognosis ranges from good in UCD (91% overall survival [OS] at 5 y) to poor in MCD (65% OS at 5 y).3
Treatment options range from local surgical excision to systemic treatments. Newer therapies include monoclonal antibodies against both IL-6 and CD20 and a few other targets in the inflammatory cascade. This article discusses the updated approach to diagnosis and management of CD.
Unicentric Castleman Disease
Unicentric CD is more common than MCD, presents as a localized lymph node or chain involvement, and is generally diagnosed in the third or fourth decade of life but has been reported in children. The presenting symptoms of UCD vary by site. It presents as nontender lymphadenopathy when confined to peripheral lymph nodes, whereas respiratory symptoms or bowel obstruction may be seen with lymphadenopathy in the chest/mediastinum, neck, or abdomen. The systemic
symptoms, such as fever, night sweats, and weight loss, are uncommon.
Dysplastic follicular dendritic cells characterize UCD. Histologically, it is usually classified as hyaline vascular disease with the follicles comprising small lymphocytes
and dendritic cells forming concentric rings with prominent vascularity.4,5 No association with HIV or HHV-8 has been seen.
Unicentric CD is often amenable to resection, and a complete cure can be achieved.6 Partial resection may be attempted when complete resection is not possible. Radiation therapy is offered for unresectable disease.7 In patients who are not candidates for any intervention, close long-term follow-up is recommended unless patients are symptomatic, in which case systemic treatment should be considered.
Multicentric Castleman Disease
The more widespread MCD is generally diagnosed in the fifth or sixth decade of life. It is more aggressive than UCD and presents a wide spectrum of symptoms and abnormal laboratory findings (Table 2).8
It is histologically classified into (a) plasmablastic or HHV-8 associated: It is often seen in patients with MCD infected with HIV, which can give rise to large B-cell lymphoma, known as HHV-8 plasmablastic lymphoma9; (b) plasmacytic variant has marked paracortical plasmacytosis with retained nodal architecture10; and (c) mixed MCD has abundant plasma cells with features similar to those of the hyaline-vascular variant.
Most patients with HIV-associated MCD are co-infected with HHV-8. The HHV-8 infection is also present in about 50% of HIV-negative cases.11 The incidence of HIVassociated MCD is increasing in the highly active antiretroviral therapy (HAART) era secondary to improved survival of patients infected with HIV.12 To diagnose active HIV MCD, the French Agence Nationale de Recherche sur le SIDA 117 CastlemaB trial group has described criteria based on the clinical symptoms, including fever, a raised serum C-reactive protein > 20 mg/L without any other cause, and 3 of 12 additional clinical findings described as peripheral lymphadenopathy, splenomegaly, ascites, edema, pleural effusion, cough, nasal obstruction, xerostomia, rash, central neurologic symptoms, jaundice, and autoimmune hemolytic anemia.13 The reported 2-year survival of patients who are HIV-negative is 97% compared with HIV-positive cases at 67%.14
Idiopathic MCD is diagnosed when there is no evidence of any underlying infectious, autoimmune, and neoplastic process.15
Patients with MCD are at an increased risk of developing non-Hodgkin and Hodgkin lymphoma, Kaposi sarcoma, primary effusion lymphoma, and follicular dendritic
cell sarcoma. POEMS (peripheral neuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome and paraneoplastic disease, such as paraneoplastic pemphigus myasthenia gravis, may be commonly diagnosed concurrently or sequentially with MCD.16-20
The disease course of MCD ranges from indolent to rapidly progressive, and its 5-year OS is about 65%. When associated with POEMS syndrome, the 5-year survival was estimated to be 90% with the osteosclerotic variant and 27% without osteosclerotic lesions.3 Treatment options for MCD include systemic chemotherapy, including antiviral therapy for HHV-8 positive and HAART for HIV positive and newer monoclonal antibody therapies targeting CD20 or IL-6.
Pathophysiology
Interleukin-6 plays an important role for inflammation in both UCD and MCD (Figure 1). There is dysregulation and overproduction of IL-6, which further stimulates the production of acute-phase reactants, resulting in various systemic manifestations.15,21,22 There is increased expression of IL-1 and IL-6, upregulation of IL-6 secondary to interaction of IL-1 with nuclear factor-kappa B (NF-kappa B), thus stimulating B-cell proliferation. IL-6 binding to IL-6 receptor (IL6-R) results in downstream activation of transcription Janus kinases/signal transducers and activators of the transcription pathway. This promotes the transcription of genes encoding the acute-phase reactant proteins. Hence, interfering with IL-6 transduction by blocking downstream signals are potential therapeutic targets. The mitogen-activated protein kinase cascade, the rapidly accelerated fibrosarcoma kinases, and the overexpression of the endothelial growth factor receptor (EGFR), all contribute to disease pathogenesis by promoting increased B-cell proliferation and vascular EGFR mediated angiogenesis. 23,24
In HHV-8–associated MCD, the virus replicates within lymph node plasmablasts, causing increased production of viral IL-6 analog, human IL-6, and other proinflammatory proteins resulting in B-cell and plasma-cell proliferation, increased vascular endothelial growth factor secretion and angiogenesis.25,26 The HHV-8–infected plasmablasts are marked by variable expression of CD20, and therefore, anti-CD20 is also shown to be an effective treatment. The calmodulin/calcineurin nuclear factor assists in the proliferation of HHV-8, thereby making calcineurin another potential target for the antiviral proliferation.27
Staging
The treatment decisions and prognosis for patients with CD is based on the clinical and histologic staging. The initial workup includes but is not limited to routine laboratory evaluation, imaging, and HIV and HHV-8 testing (Table 3). Routine tests of the levels of cytokines are not recommended. Other relevant tests for known disease associations should be obtained when relevant.
Treatment
Better understanding of the disease process in CD has helped to identify potential therapeutic targets (Figures 2 and 3).
For UCD, surgery is the mainstay of treatment.4,28,29 In surgically unresectable cases, radiation therapy is helpful for local disease control. Alternatively, neoadjuvant
chemotherapy and rituximab are used. Corticosteroids are generally used to treat acute exacerbations and as adjuncts to chemotherapy.
For MCD, the treatment approach depends on the HIV and HHV-8 status of the patient. For patients with HHV-8 infection, both with and without HIV co-infection, antiviral agents, such as ganciclovir, foscarnet, or cidofovir, have shown in vitro activity against HHV-8 but with limited clinical success.30 In patients infected with HIV, the aim of treating with HAART is to control the disease, prevent opportunistic infections, and improve tolerance to chemotherapy.31-33 Rituximab with or without chemotherapy is the standard treatment approach. The additional chemotherapeutic agents are used depending on the presence or absence of organ failure. This approach has improved the OS in HIV-associated MCD.34,35 Treatment with HAART does not decrease the risk of relapse in HIV MCD; therefore, the role of rituximab and antiherpesvirus agents as maintenance therapy has been explored.36 In patients who fail to respond to or relapse rapidly following rituximab monotherapy, the use of either single-agent chemotherapy with or without rituximab or antiherpesvirus therapy with high-dose zidovudine and valganciclovir is recommended.37
The cytotoxic chemotherapy with single agents, such as etoposide, vinblastine, cyclophosphamide, cladribine, chlorambucil, and liposomal doxorubicin, has been used with limited success.22 The combination chemotherapy with cyclophosphamide/doxorubicin/vincristine/prednisone (CHOP) or cyclophosphamide/vincristine/
prednisone (CVP) without rituximab has been shown to achieve durable remissions. Corticosteroids are usually administered as an initial adjunct to chemotherapy or for acute exacerbations. In patients with MCD, regardless of HIV status, the interferon therapy was found to achieve long-term remission.38,39 The interferon therapy
exerts antiviral effects via downregulation of the IL-6R and inhibition of HHV-8 replication. For patients in remission, maintenance therapy with oral valganciclovir is promising.40
Immunomodulators & Targeted Therapies
For unresectable UCD or MCD with organ failure or relapse, the use of alternativesingle-agent or combination chemotherapies with or without rituximab is recommended. Thalidomide has shown some success, probably secondary to disruption of IL-6 production.41 In cases of progression following second-line therapy, bortezomib, antiherpesvirus therapies, or IL-6–directed therapy with siltuximab or tocilizumab should be considered.
Rituximab is a monoclonal chimeric antibody that targets CD20 on B cells, thus leading to B-cell lymphodepletion via activating complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity. As monotherapy, it has been shown to achieve 2-year progression-free survival in 80% of patients.42 In patients with MCD who are HIV positive, rituximab with and without chemotherapy has shown improved overall and disease-free survival of 70% to 80% at 2 years.43
Siltuximab is a chimeric human-mouse monoclonal antibody to IL-6 that has been approved for treatment of patients with MCD who are both HIV negative and HHV-8 negative.44-46 Tocilizumab targets the IL-6R. The antibody has shown improvement in a study in HIVseronegative adults with MCD.47,48
Bortezomib is a proteasome inhibitor that inhibits the NF-kappa B pathway, which induces the expression of numerous proinflammatory proteins, including IL-6. It is recommended for relapsed or refractory disease.49,50
Anakinra is a recombinant IL-1R antagonist that blocks IL-1 effects and controls disease by decreasing IL-6 production.51
Conculsion
There has been significant progress in disease diagnosis and management as more information is available about the incidence, clinical presentation, and pathophysiology of CD. The understanding of the disease pathogenesis and biology has helped to discover multiple potential therapeutic targets. Successful treatment has been achieved through targeting HHV-8 replication, CD20, and IL-6 and anti– IL-6R antibodies. Although surgical resection continues to be the
standard of therapy for UCD, the management of MCD and relapsed or refractory disease continues to evolve. Exploration of various treatment strategies in different clinical presentations is warranted.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S.
Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information
for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to
patients.
Click here to read the digital edition.
1. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymph-node hyperplasia
resembling thymoma. Cancer. 1956;9(4):822-830.
2. Munshi N, Mehra M, van de Velde H, Desai A, Potluri R, Vermeulen J. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma. 2015;56(5):1252-1260.
3. Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997-1002.
4. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29(3):670-683.
5. Cronin DM, Warnke RA. Castleman disease: an update on classification and the
spectrum of associated lesions. Adv Anat Pathol. 2009;16(4):236-246.
6. Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman’s disease: a systematic review of 404 published cases. Ann Surg. 2012;255(4):677-684.
7. Chronowski GM, Ha CS, Wilder RB, Cabanillas F, Manning J, Cox JD. Treatment of unicentric and multicentric Castleman disease and the role of radiotherapy. Cancer. 2001;92(3):670-676.
8. Herrada J, Cabanillas F, Rice L, Manning J, Pugh W. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med. 1998;128(8):657-662.
9. Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95(4):1406-1412.
10. Ferry JA, Harris NL. Atlas of Lymphoid Hyperplasia and Lymphoma. Philadelphia, PA: W.B. Saunders; 1997.
11. Soulier J, Grollet L, Oksenhendler E, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86(4):1276-1280.
12. Powles T, Stebbing J, Bazeos A, et al. The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman’s disease. Ann Oncol. 2009;20(4):775-779.
13. Gérard L, Bérezné A, Galicier L, et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman’s disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25(22):3350-3356.
14. Casper C, Teltsch DY, Robinson D Jr, et al. Clinical characteristics and healthcare utilization of patients with multicentric Castleman disease. Br J Haematol. 2015;168(1):82-93.
15. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933.
16. Larroche C, Cacoub P, Soulier J, et al. Castleman’s disease and lymphoma: report of eight cases in HIV-negative patients and literature review. Am J Hematol. 2002;69(2):119-126.
17. Dispenzieri A. POEMS syndrome: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89(2):214-223.
18. Andhavarapu S, Jiang L. POEMS syndrome and Castleman disease. Blood. 2013;122(2):159.
19. Bélec L, Mohamed AS, Authier FJ, et al. Human herpesvirus 8 infection in patients with POEMS syndrome-associated multicentric Castleman’s disease. Blood. 1999;93(11):3643-3653.
20. Oksenhendler E, Boulanger E, Galicier L, et al. High incidence of Kaposi sarcomaassociated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood. 2002;99(7):2331-2336.
21. Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood. 1989;74(4):1360-1367.
22. El-Osta HE, Kurzrock R. Castleman’s disease: from basic mechanisms to molecular therapeutics. Oncologist. 2011;16(4):497-511.
23. Akula SM, Ford PW, Whitman AG, et al. B-Raf-dependent expression of vascular endothelial growth factor-A in Kaposi sarcoma-associated herpesvirus-infected human B cells. Blood. 2005;105(11):4516-4522.
24. Sun X, Chang KC, Abruzzo LV, Lai R, Younes A, Jones D. Epidermal growth factor receptor expression in follicular dendritic cells: a shared feature of follicular dendritic cell sarcoma and Castleman’s disease. Hum Pathol. 2003;34(9):835-840.
25. Adam N, Rabe B, Suthaus J, Grötzinger J, Rose-John S, Scheller J. Unraveling viral interleukin-6 binding to gp130 and activation of STAT-signaling pathways independently of the interleukin-6 receptor. J Virol. 2009;83(10):5117-5126.
26. Suda T, Katano H, Delsol G, et al. HHV-8 infection status of AIDS-unrelated and
AIDS-associated multicentric Castleman’s disease. Pathol Int. 2001;51(9):671-679.
27. Zoeteweij JP, Moses AV, Rinderknecht AS, et al. Targeted inhibition of calcineurin signaling blocks calcium-dependent reactivation of Kaposi sarcoma-associated herpesvirus. Blood. 2001;97(8):2374-2380.
28. McCarty MJ, Vukelja SJ, Banks PM, Weiss RB. Angiofollicular lymph node hyperplasia
(Castleman’s disease). Cancer Treat Rev. 1995;21(4):291-310.
29. Bowne WB, Lewis JJ, Filippa DA, et al. The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer. 1999;85(3):706-717.
30. Reddy D, Mitsuyasu R. HIV-associated multicentric Castleman disease. Curr Opin Oncol. 2011;23(5):475-481.
31. Aaron L, Lidove O, Yousry C, Roudiere L, Dupont B, Viard JP. Human herpesvirus 8-positive Castleman disease in human immunodeficiency virus-infected patients: the impact of highly active antiretroviral therapy. Clin Infect Dis. 2002;35(7):880-882.
32. Sprinz E, Jeffman M, Liedke P, Putten A, Schwartsmann G. Successful treatment of AIDS-related Castleman’s disease following the administration of highly active antiretroviral therapy (HAART). Ann Oncol. 2004;15(2):356-358.
33. Lee SM, Edwards SG, Chilton DN, Ramsay A, Miller RF. Highly active antiretroviral therapy alone may be an effective treatment for HIV-associated multi-centric Castleman’s disease. Haematologica. 2010;95(11):1979-1981.
34. Bower M. How I treat HIV-associated multicentric Castleman disease. Blood. 2010;116(22):4415-4421.
35. Bower M, Newsom-Davis T, Naresh K, et al. Clinical features and outcome in HIVassociated
multicentric Castleman’s disease. J Clin Oncol. 2011;29(18):2481-2486.
36. Casper C, Nichols WG, Huang ML, Corey L, Wald A. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood. 2004;103(5):1632-1634.
37. Uldrick TS, Polizzotto MN, Aleman K, et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood. 2011;117(26):6977-6986.
38. Kumari P, Schechter GP, Saini N, Benator DA. Successful treatment of human immunodeficiency virus-related Castleman’s disease with interferon-alpha. Clin Infect Dis. 2000;31(2):602-604.
39. Nord JA, Karter D. Low dose interferon-alpha therapy for HIV-associated multicentric Castleman’s disease. Int J STD AIDS. 2003;14(1):61-62.
40. Oksenhendler E. HIV-associated multicentric Castleman disease. Curr Opin HIV AIDS. 2009;4(1):16-21.
41. Jung CP, Emmerich B, Goebel FD, Bogner JR. Successful treatment of a patient with HIV-associated multicentric Castleman disease (MCD) with thalidomide. Am J Hematol. 2004;75(3):176-177.
42. Ide M, Kawachi Y, Izumi Y, Kasagi K, Ogino T. Long-term remission in HIV negative patients with multicentric Castleman’s disease using rituximab. Eur J Haematol. 2006;76(2):119-123.
43. Marcelin AG, Aaron L, Mateus C, et al. Rituximab therapy for HIV-associated Castleman disease. Blood. 2003;102(8):2786-2788.
44. Van Rhee F, Fayad L, Voorhees P, et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol. 2010;28(23):3701-3708.
45. Wong RS, Casper C, Munshi N, et al. A multicenter, randomized, doubleblind, placebo-controlled study of the efficacy and safety of siltuximab, an antiinterleukin-6 monoclonal antibody, in patients with multicentric Castleman’s disease. Blood. 2013;122(21):505.
46. Van Rhee F, Casper C, Voorhees PM, et al. An open-label, phase 2, multicenter study of the safety of long-term treatment with siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman’s disease. Blood. 2013;122(21):1806.
47. Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627-2632.
48. Müzes G, Sipos F, Csomor J, Sréter L. Successful tocilizumab treatment in a patient with human herpesvirus 8-positive and human immunodeficiency virusnegative multicentric Castleman’s disease of plasma cell type nonresponsive to rituximab-CVP therapy. APMIS. 2013;121(7):668-674.
49. Hess G, Wagner V, Kreft A, Heussel CP, Huber C. Effects of bortezomib on proinflammatory cytokine levels and transfusion dependency in a patient with multicentric Castleman disease. Br J Haematol. 2006;134(5):544-545.
50. Sobas MA, Alonso Vence N, Diaz Arias J, Bendaña Lopez A, Fraga Rodriguez M, Bello Lopez JL. Efficacy of bortezomib in refractory form of multicentric Castleman disease associated to poems syndrome (MCD-POEMS variant). Ann Hematol. 2010;89(2):217-219.
51. El-Osta H, Janku F, Kurzrock R. Successful treatment of Castleman’s disease with interleukin-1 receptor antagonist (Anakinra). Mol Cancer Ther. 2010;9(6):1485-1488.
1. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymph-node hyperplasia
resembling thymoma. Cancer. 1956;9(4):822-830.
2. Munshi N, Mehra M, van de Velde H, Desai A, Potluri R, Vermeulen J. Use of a claims database to characterize and estimate the incidence rate for Castleman disease. Leuk Lymphoma. 2015;56(5):1252-1260.
3. Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997-1002.
4. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29(3):670-683.
5. Cronin DM, Warnke RA. Castleman disease: an update on classification and the
spectrum of associated lesions. Adv Anat Pathol. 2009;16(4):236-246.
6. Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman’s disease: a systematic review of 404 published cases. Ann Surg. 2012;255(4):677-684.
7. Chronowski GM, Ha CS, Wilder RB, Cabanillas F, Manning J, Cox JD. Treatment of unicentric and multicentric Castleman disease and the role of radiotherapy. Cancer. 2001;92(3):670-676.
8. Herrada J, Cabanillas F, Rice L, Manning J, Pugh W. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med. 1998;128(8):657-662.
9. Dupin N, Diss TL, Kellam P, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood. 2000;95(4):1406-1412.
10. Ferry JA, Harris NL. Atlas of Lymphoid Hyperplasia and Lymphoma. Philadelphia, PA: W.B. Saunders; 1997.
11. Soulier J, Grollet L, Oksenhendler E, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86(4):1276-1280.
12. Powles T, Stebbing J, Bazeos A, et al. The role of immune suppression and HHV-8 in the increasing incidence of HIV-associated multicentric Castleman’s disease. Ann Oncol. 2009;20(4):775-779.
13. Gérard L, Bérezné A, Galicier L, et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman’s disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25(22):3350-3356.
14. Casper C, Teltsch DY, Robinson D Jr, et al. Clinical characteristics and healthcare utilization of patients with multicentric Castleman disease. Br J Haematol. 2015;168(1):82-93.
15. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924-2933.
16. Larroche C, Cacoub P, Soulier J, et al. Castleman’s disease and lymphoma: report of eight cases in HIV-negative patients and literature review. Am J Hematol. 2002;69(2):119-126.
17. Dispenzieri A. POEMS syndrome: 2014 update on diagnosis, risk-stratification, and management. Am J Hematol. 2014;89(2):214-223.
18. Andhavarapu S, Jiang L. POEMS syndrome and Castleman disease. Blood. 2013;122(2):159.
19. Bélec L, Mohamed AS, Authier FJ, et al. Human herpesvirus 8 infection in patients with POEMS syndrome-associated multicentric Castleman’s disease. Blood. 1999;93(11):3643-3653.
20. Oksenhendler E, Boulanger E, Galicier L, et al. High incidence of Kaposi sarcomaassociated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood. 2002;99(7):2331-2336.
21. Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood. 1989;74(4):1360-1367.
22. El-Osta HE, Kurzrock R. Castleman’s disease: from basic mechanisms to molecular therapeutics. Oncologist. 2011;16(4):497-511.
23. Akula SM, Ford PW, Whitman AG, et al. B-Raf-dependent expression of vascular endothelial growth factor-A in Kaposi sarcoma-associated herpesvirus-infected human B cells. Blood. 2005;105(11):4516-4522.
24. Sun X, Chang KC, Abruzzo LV, Lai R, Younes A, Jones D. Epidermal growth factor receptor expression in follicular dendritic cells: a shared feature of follicular dendritic cell sarcoma and Castleman’s disease. Hum Pathol. 2003;34(9):835-840.
25. Adam N, Rabe B, Suthaus J, Grötzinger J, Rose-John S, Scheller J. Unraveling viral interleukin-6 binding to gp130 and activation of STAT-signaling pathways independently of the interleukin-6 receptor. J Virol. 2009;83(10):5117-5126.
26. Suda T, Katano H, Delsol G, et al. HHV-8 infection status of AIDS-unrelated and
AIDS-associated multicentric Castleman’s disease. Pathol Int. 2001;51(9):671-679.
27. Zoeteweij JP, Moses AV, Rinderknecht AS, et al. Targeted inhibition of calcineurin signaling blocks calcium-dependent reactivation of Kaposi sarcoma-associated herpesvirus. Blood. 2001;97(8):2374-2380.
28. McCarty MJ, Vukelja SJ, Banks PM, Weiss RB. Angiofollicular lymph node hyperplasia
(Castleman’s disease). Cancer Treat Rev. 1995;21(4):291-310.
29. Bowne WB, Lewis JJ, Filippa DA, et al. The management of unicentric and multicentric Castleman’s disease: a report of 16 cases and a review of the literature. Cancer. 1999;85(3):706-717.
30. Reddy D, Mitsuyasu R. HIV-associated multicentric Castleman disease. Curr Opin Oncol. 2011;23(5):475-481.
31. Aaron L, Lidove O, Yousry C, Roudiere L, Dupont B, Viard JP. Human herpesvirus 8-positive Castleman disease in human immunodeficiency virus-infected patients: the impact of highly active antiretroviral therapy. Clin Infect Dis. 2002;35(7):880-882.
32. Sprinz E, Jeffman M, Liedke P, Putten A, Schwartsmann G. Successful treatment of AIDS-related Castleman’s disease following the administration of highly active antiretroviral therapy (HAART). Ann Oncol. 2004;15(2):356-358.
33. Lee SM, Edwards SG, Chilton DN, Ramsay A, Miller RF. Highly active antiretroviral therapy alone may be an effective treatment for HIV-associated multi-centric Castleman’s disease. Haematologica. 2010;95(11):1979-1981.
34. Bower M. How I treat HIV-associated multicentric Castleman disease. Blood. 2010;116(22):4415-4421.
35. Bower M, Newsom-Davis T, Naresh K, et al. Clinical features and outcome in HIVassociated
multicentric Castleman’s disease. J Clin Oncol. 2011;29(18):2481-2486.
36. Casper C, Nichols WG, Huang ML, Corey L, Wald A. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood. 2004;103(5):1632-1634.
37. Uldrick TS, Polizzotto MN, Aleman K, et al. High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy. Blood. 2011;117(26):6977-6986.
38. Kumari P, Schechter GP, Saini N, Benator DA. Successful treatment of human immunodeficiency virus-related Castleman’s disease with interferon-alpha. Clin Infect Dis. 2000;31(2):602-604.
39. Nord JA, Karter D. Low dose interferon-alpha therapy for HIV-associated multicentric Castleman’s disease. Int J STD AIDS. 2003;14(1):61-62.
40. Oksenhendler E. HIV-associated multicentric Castleman disease. Curr Opin HIV AIDS. 2009;4(1):16-21.
41. Jung CP, Emmerich B, Goebel FD, Bogner JR. Successful treatment of a patient with HIV-associated multicentric Castleman disease (MCD) with thalidomide. Am J Hematol. 2004;75(3):176-177.
42. Ide M, Kawachi Y, Izumi Y, Kasagi K, Ogino T. Long-term remission in HIV negative patients with multicentric Castleman’s disease using rituximab. Eur J Haematol. 2006;76(2):119-123.
43. Marcelin AG, Aaron L, Mateus C, et al. Rituximab therapy for HIV-associated Castleman disease. Blood. 2003;102(8):2786-2788.
44. Van Rhee F, Fayad L, Voorhees P, et al. Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol. 2010;28(23):3701-3708.
45. Wong RS, Casper C, Munshi N, et al. A multicenter, randomized, doubleblind, placebo-controlled study of the efficacy and safety of siltuximab, an antiinterleukin-6 monoclonal antibody, in patients with multicentric Castleman’s disease. Blood. 2013;122(21):505.
46. Van Rhee F, Casper C, Voorhees PM, et al. An open-label, phase 2, multicenter study of the safety of long-term treatment with siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman’s disease. Blood. 2013;122(21):1806.
47. Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627-2632.
48. Müzes G, Sipos F, Csomor J, Sréter L. Successful tocilizumab treatment in a patient with human herpesvirus 8-positive and human immunodeficiency virusnegative multicentric Castleman’s disease of plasma cell type nonresponsive to rituximab-CVP therapy. APMIS. 2013;121(7):668-674.
49. Hess G, Wagner V, Kreft A, Heussel CP, Huber C. Effects of bortezomib on proinflammatory cytokine levels and transfusion dependency in a patient with multicentric Castleman disease. Br J Haematol. 2006;134(5):544-545.
50. Sobas MA, Alonso Vence N, Diaz Arias J, Bendaña Lopez A, Fraga Rodriguez M, Bello Lopez JL. Efficacy of bortezomib in refractory form of multicentric Castleman disease associated to poems syndrome (MCD-POEMS variant). Ann Hematol. 2010;89(2):217-219.
51. El-Osta H, Janku F, Kurzrock R. Successful treatment of Castleman’s disease with interleukin-1 receptor antagonist (Anakinra). Mol Cancer Ther. 2010;9(6):1485-1488.
New Treatments for Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) is a slow-growing malignancy of B lymphocytes (B cells) that tends to affect older people and men more than women. More than 17,000 new cases of CLL are reported every year. Patients with CLL do not need treatment with chemotherapy until they become symptomatic or display evidence of rapid progression of the disease. In multiple studies and a meta-analysis, early initiation of chemotherapy has failed to show benefit in managing CLL; indeed, it may increase mortality.1,2
The combination chemotherapy fludarabine, cyclophosphamide, and rituximab (FCR) is often the initial choice for treatment. Other chemotherapy drugs used are chlorambucil, bendamustine, pentostatin or cladribine, rituximab, ofatumumab, and alemtuzumab. Although chlorambucil is a forgotten drug in the U.S., it is still used first line in elderly, fragile populations in Europe, which make up the bulk of true CLL cases.3
Various combination regimens used in CLL treatment have shown improved response rates in several randomized trials but have failed to show any survival advantage until recently. The treatment of patients with CLL has undergone a dramatic transformation and has changed the management paradigm since the FDA approved new, targeted agents. This article includes a brief discussion of these new agents and the pipeline for new agents.
Newly Approved Treatments
Obinutuzumab is a CD20-directed cytolytic antibody, which on binding to CD20, mediates B-cell lysis. Mediation may be (1) through engagement of immune effector cells; (2) by directly activating intracellular deathsignaling pathways; and/or (3) by activation of the complement. The FDA approved obinutuzumab in November 2013 for previously untreated CLL in combination with chlorambucil based on a pivotal phase 3 trial in 356 previously untreated patients with CLL (mean age, 73 years). Those who received obinutuzumab in combination with chlorambucil had significantly better median progression-free survival (PFS) than did those treated with chlorambucil alone (23 months vs 11.1 months; P < .0001). These results effectively end the use of chlorambucil as monotherapy.4
Ibrutinib is a Bruton’s tyrosine kinase (BTK) inhibitor that forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. The BTK is a signaling molecule of the B-cell antigen receptor and cytokine receptor pathways. Accelerated approval of ibrutinib was based on of a clinical study of participants with CLL who had received 4 previous therapies. At 26 months, estimated PFS was 75%, and the rate of overall survival (OS) was 83%.5 In January 2014, the RESONATE trial was stopped early because of a positive interim analysis showing statistically significant improvement in PFS as well as in OS with oral ibrutinib compared with IV ofatumumab. The RESONATE trial was a phase 3, multicenter study involving 391 patients with relapsed or refractory CLL who had received at least 1 previous therapy.6 At 6 months, 88% of patients treated with ibrutinib were progression free compared with 65% with ofatumumab. At 12 months, the OS rate was 90% for patients treated with ibrutinib compared with 81% for patients in the ofatumumab group. By traditional response criteria, the overall response rate (ORR) with ibrutinib was 43% compared with 4% for ofatumumab. Ibrutinib is now approved for chemotherapy-naïve CLL patients with a 17p13.1 deletion, a genetic abnormality that generally portends a poor prognosis.
On July 23, 2014, the FDA approved an oral PI3-kinase delta (PI3K d) inhibitor called idelalisib in combination with rituximab used as a treatment for patients with high-risk CLL. In supporting data from a phase 3 study, the addition of idelalisib to rituximab improved OS by 72% and PFS by 82% vs placebo and rituximab. At 24 weeks, 90% of patients treated with idelalisib remained progression free compared with 50% of patients treated with the placebo. Approval was based on a placebo-controlled study of 220 patients in which patients treated with idelalisib plus rituximab showed significantly longer PFS (10.7 months) than did those who received placebo plus rituximab (5.5 months).7
Lenalidomide is an immunomodulatory drug (IMiD) currently approved for use in multiple myeloma and myelodysplastic syndrome with deletion of chromosome 5q. Studies have used this medication in treatment of patients with relapsed and refractory CLL. Response rates of 47% to 38% with complete response rates of 9% and elimination of minimal residual disease (MRD) have also been reported.8
CLL Pipeline
Clinical trials continue to explore new agents, with the most promising being the PI3K δ and γ inhibitor duvelisib (IPI-145) and the BCL-2 inhibitor ABT-199. In
a phase 1 study exploring duvelisib in patients with relapsed or refractory CLL, the ORR was 47%.9 Additionally, 98% of patients with refractory disease had nodal responses, which did not differ between patients with or without the 17p deletion or p53 mutation.
ABT-199 demonstrated efficacy in a phase 1b study when administered to patients with relapsed/refractory CLL in combination with rituximab.10 In 18 evaluable patients, 39% achieved complete remission (CR) or CR with incomplete blood count recovery and 39% achieved partial remissions (78% ORR). Altogether, 22% were deemed MRD-negative. In evaluable patients with 17p deletions, 81% achieved a response to ABT-199. In patients with fludarabine-refractory CLL, 78% achieved a response.
Although it is advantageous to have so many newer effective, targeted drugs for relapsed/refractory advanced CLL, in early-stage CLL when a watch and wait approach might be best, it may become a challenge for the patient as well as for the treating physician. All these drugs are expensive and carry risks. Although PFS is promising, physicians have to make a judgment call in balancing cost and toxicity.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to continue reading.
1. Bosch F, Ferrer A, Villamor N, et al. Fludarabine, cyclophosphamide, and mitoxantrone as initial therapy of chronic lymphocytic leukemia: high response rate and disease eradication. Clin Cancer Res. 2008;14(1):155-161.
2. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol. 2006;24(3):437-443.
3. Eichhorst BF, Busch R, Stilgenbauer S, et al; German CLL Study Group (GCLLSG). First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood. 2009;114(16):3382-3391.
4. Robak T, Dmoszynska A, Solal-Céligny P, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. J Clin Oncol. 2010;28(10):1756-1765.
5. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42.
6. Byrd JC, Brown JR, et al; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213-223.
7. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
8. Molica S. Immunomodulatory drugs in chronic lymphocytic leukemia: a new
treatment paradigm. Leuk Lymphoma. 2007;48(5):866-869.
9. O’Brien S, Patel M, et al. Duvelisib (IPI-145), a PI3K-d,g inhibitor, is clinically active in patients with relapsed/refractory chronic lymphocytic leukemia. Paper presented at: the 56th ASH Annual Meeting and Exposition; December 7, 2014; San Francisco, CA. Abstract 3334.
10. Ma S, Seymour JF, Lanasa MC, et al. ABT-199 (GDC-0199) combined with rituximab (R) in patients (pts) with relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL): interim results of a phase 1b study. J Clin Oncol. 2014;32 (suppl; abstr 7013):5s.
Chronic lymphocytic leukemia (CLL) is a slow-growing malignancy of B lymphocytes (B cells) that tends to affect older people and men more than women. More than 17,000 new cases of CLL are reported every year. Patients with CLL do not need treatment with chemotherapy until they become symptomatic or display evidence of rapid progression of the disease. In multiple studies and a meta-analysis, early initiation of chemotherapy has failed to show benefit in managing CLL; indeed, it may increase mortality.1,2
The combination chemotherapy fludarabine, cyclophosphamide, and rituximab (FCR) is often the initial choice for treatment. Other chemotherapy drugs used are chlorambucil, bendamustine, pentostatin or cladribine, rituximab, ofatumumab, and alemtuzumab. Although chlorambucil is a forgotten drug in the U.S., it is still used first line in elderly, fragile populations in Europe, which make up the bulk of true CLL cases.3
Various combination regimens used in CLL treatment have shown improved response rates in several randomized trials but have failed to show any survival advantage until recently. The treatment of patients with CLL has undergone a dramatic transformation and has changed the management paradigm since the FDA approved new, targeted agents. This article includes a brief discussion of these new agents and the pipeline for new agents.
Newly Approved Treatments
Obinutuzumab is a CD20-directed cytolytic antibody, which on binding to CD20, mediates B-cell lysis. Mediation may be (1) through engagement of immune effector cells; (2) by directly activating intracellular deathsignaling pathways; and/or (3) by activation of the complement. The FDA approved obinutuzumab in November 2013 for previously untreated CLL in combination with chlorambucil based on a pivotal phase 3 trial in 356 previously untreated patients with CLL (mean age, 73 years). Those who received obinutuzumab in combination with chlorambucil had significantly better median progression-free survival (PFS) than did those treated with chlorambucil alone (23 months vs 11.1 months; P < .0001). These results effectively end the use of chlorambucil as monotherapy.4
Ibrutinib is a Bruton’s tyrosine kinase (BTK) inhibitor that forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. The BTK is a signaling molecule of the B-cell antigen receptor and cytokine receptor pathways. Accelerated approval of ibrutinib was based on of a clinical study of participants with CLL who had received 4 previous therapies. At 26 months, estimated PFS was 75%, and the rate of overall survival (OS) was 83%.5 In January 2014, the RESONATE trial was stopped early because of a positive interim analysis showing statistically significant improvement in PFS as well as in OS with oral ibrutinib compared with IV ofatumumab. The RESONATE trial was a phase 3, multicenter study involving 391 patients with relapsed or refractory CLL who had received at least 1 previous therapy.6 At 6 months, 88% of patients treated with ibrutinib were progression free compared with 65% with ofatumumab. At 12 months, the OS rate was 90% for patients treated with ibrutinib compared with 81% for patients in the ofatumumab group. By traditional response criteria, the overall response rate (ORR) with ibrutinib was 43% compared with 4% for ofatumumab. Ibrutinib is now approved for chemotherapy-naïve CLL patients with a 17p13.1 deletion, a genetic abnormality that generally portends a poor prognosis.
On July 23, 2014, the FDA approved an oral PI3-kinase delta (PI3K d) inhibitor called idelalisib in combination with rituximab used as a treatment for patients with high-risk CLL. In supporting data from a phase 3 study, the addition of idelalisib to rituximab improved OS by 72% and PFS by 82% vs placebo and rituximab. At 24 weeks, 90% of patients treated with idelalisib remained progression free compared with 50% of patients treated with the placebo. Approval was based on a placebo-controlled study of 220 patients in which patients treated with idelalisib plus rituximab showed significantly longer PFS (10.7 months) than did those who received placebo plus rituximab (5.5 months).7
Lenalidomide is an immunomodulatory drug (IMiD) currently approved for use in multiple myeloma and myelodysplastic syndrome with deletion of chromosome 5q. Studies have used this medication in treatment of patients with relapsed and refractory CLL. Response rates of 47% to 38% with complete response rates of 9% and elimination of minimal residual disease (MRD) have also been reported.8
CLL Pipeline
Clinical trials continue to explore new agents, with the most promising being the PI3K δ and γ inhibitor duvelisib (IPI-145) and the BCL-2 inhibitor ABT-199. In
a phase 1 study exploring duvelisib in patients with relapsed or refractory CLL, the ORR was 47%.9 Additionally, 98% of patients with refractory disease had nodal responses, which did not differ between patients with or without the 17p deletion or p53 mutation.
ABT-199 demonstrated efficacy in a phase 1b study when administered to patients with relapsed/refractory CLL in combination with rituximab.10 In 18 evaluable patients, 39% achieved complete remission (CR) or CR with incomplete blood count recovery and 39% achieved partial remissions (78% ORR). Altogether, 22% were deemed MRD-negative. In evaluable patients with 17p deletions, 81% achieved a response to ABT-199. In patients with fludarabine-refractory CLL, 78% achieved a response.
Although it is advantageous to have so many newer effective, targeted drugs for relapsed/refractory advanced CLL, in early-stage CLL when a watch and wait approach might be best, it may become a challenge for the patient as well as for the treating physician. All these drugs are expensive and carry risks. Although PFS is promising, physicians have to make a judgment call in balancing cost and toxicity.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to continue reading.
Chronic lymphocytic leukemia (CLL) is a slow-growing malignancy of B lymphocytes (B cells) that tends to affect older people and men more than women. More than 17,000 new cases of CLL are reported every year. Patients with CLL do not need treatment with chemotherapy until they become symptomatic or display evidence of rapid progression of the disease. In multiple studies and a meta-analysis, early initiation of chemotherapy has failed to show benefit in managing CLL; indeed, it may increase mortality.1,2
The combination chemotherapy fludarabine, cyclophosphamide, and rituximab (FCR) is often the initial choice for treatment. Other chemotherapy drugs used are chlorambucil, bendamustine, pentostatin or cladribine, rituximab, ofatumumab, and alemtuzumab. Although chlorambucil is a forgotten drug in the U.S., it is still used first line in elderly, fragile populations in Europe, which make up the bulk of true CLL cases.3
Various combination regimens used in CLL treatment have shown improved response rates in several randomized trials but have failed to show any survival advantage until recently. The treatment of patients with CLL has undergone a dramatic transformation and has changed the management paradigm since the FDA approved new, targeted agents. This article includes a brief discussion of these new agents and the pipeline for new agents.
Newly Approved Treatments
Obinutuzumab is a CD20-directed cytolytic antibody, which on binding to CD20, mediates B-cell lysis. Mediation may be (1) through engagement of immune effector cells; (2) by directly activating intracellular deathsignaling pathways; and/or (3) by activation of the complement. The FDA approved obinutuzumab in November 2013 for previously untreated CLL in combination with chlorambucil based on a pivotal phase 3 trial in 356 previously untreated patients with CLL (mean age, 73 years). Those who received obinutuzumab in combination with chlorambucil had significantly better median progression-free survival (PFS) than did those treated with chlorambucil alone (23 months vs 11.1 months; P < .0001). These results effectively end the use of chlorambucil as monotherapy.4
Ibrutinib is a Bruton’s tyrosine kinase (BTK) inhibitor that forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. The BTK is a signaling molecule of the B-cell antigen receptor and cytokine receptor pathways. Accelerated approval of ibrutinib was based on of a clinical study of participants with CLL who had received 4 previous therapies. At 26 months, estimated PFS was 75%, and the rate of overall survival (OS) was 83%.5 In January 2014, the RESONATE trial was stopped early because of a positive interim analysis showing statistically significant improvement in PFS as well as in OS with oral ibrutinib compared with IV ofatumumab. The RESONATE trial was a phase 3, multicenter study involving 391 patients with relapsed or refractory CLL who had received at least 1 previous therapy.6 At 6 months, 88% of patients treated with ibrutinib were progression free compared with 65% with ofatumumab. At 12 months, the OS rate was 90% for patients treated with ibrutinib compared with 81% for patients in the ofatumumab group. By traditional response criteria, the overall response rate (ORR) with ibrutinib was 43% compared with 4% for ofatumumab. Ibrutinib is now approved for chemotherapy-naïve CLL patients with a 17p13.1 deletion, a genetic abnormality that generally portends a poor prognosis.
On July 23, 2014, the FDA approved an oral PI3-kinase delta (PI3K d) inhibitor called idelalisib in combination with rituximab used as a treatment for patients with high-risk CLL. In supporting data from a phase 3 study, the addition of idelalisib to rituximab improved OS by 72% and PFS by 82% vs placebo and rituximab. At 24 weeks, 90% of patients treated with idelalisib remained progression free compared with 50% of patients treated with the placebo. Approval was based on a placebo-controlled study of 220 patients in which patients treated with idelalisib plus rituximab showed significantly longer PFS (10.7 months) than did those who received placebo plus rituximab (5.5 months).7
Lenalidomide is an immunomodulatory drug (IMiD) currently approved for use in multiple myeloma and myelodysplastic syndrome with deletion of chromosome 5q. Studies have used this medication in treatment of patients with relapsed and refractory CLL. Response rates of 47% to 38% with complete response rates of 9% and elimination of minimal residual disease (MRD) have also been reported.8
CLL Pipeline
Clinical trials continue to explore new agents, with the most promising being the PI3K δ and γ inhibitor duvelisib (IPI-145) and the BCL-2 inhibitor ABT-199. In
a phase 1 study exploring duvelisib in patients with relapsed or refractory CLL, the ORR was 47%.9 Additionally, 98% of patients with refractory disease had nodal responses, which did not differ between patients with or without the 17p deletion or p53 mutation.
ABT-199 demonstrated efficacy in a phase 1b study when administered to patients with relapsed/refractory CLL in combination with rituximab.10 In 18 evaluable patients, 39% achieved complete remission (CR) or CR with incomplete blood count recovery and 39% achieved partial remissions (78% ORR). Altogether, 22% were deemed MRD-negative. In evaluable patients with 17p deletions, 81% achieved a response to ABT-199. In patients with fludarabine-refractory CLL, 78% achieved a response.
Although it is advantageous to have so many newer effective, targeted drugs for relapsed/refractory advanced CLL, in early-stage CLL when a watch and wait approach might be best, it may become a challenge for the patient as well as for the treating physician. All these drugs are expensive and carry risks. Although PFS is promising, physicians have to make a judgment call in balancing cost and toxicity.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to continue reading.
1. Bosch F, Ferrer A, Villamor N, et al. Fludarabine, cyclophosphamide, and mitoxantrone as initial therapy of chronic lymphocytic leukemia: high response rate and disease eradication. Clin Cancer Res. 2008;14(1):155-161.
2. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol. 2006;24(3):437-443.
3. Eichhorst BF, Busch R, Stilgenbauer S, et al; German CLL Study Group (GCLLSG). First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood. 2009;114(16):3382-3391.
4. Robak T, Dmoszynska A, Solal-Céligny P, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. J Clin Oncol. 2010;28(10):1756-1765.
5. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42.
6. Byrd JC, Brown JR, et al; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213-223.
7. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
8. Molica S. Immunomodulatory drugs in chronic lymphocytic leukemia: a new
treatment paradigm. Leuk Lymphoma. 2007;48(5):866-869.
9. O’Brien S, Patel M, et al. Duvelisib (IPI-145), a PI3K-d,g inhibitor, is clinically active in patients with relapsed/refractory chronic lymphocytic leukemia. Paper presented at: the 56th ASH Annual Meeting and Exposition; December 7, 2014; San Francisco, CA. Abstract 3334.
10. Ma S, Seymour JF, Lanasa MC, et al. ABT-199 (GDC-0199) combined with rituximab (R) in patients (pts) with relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL): interim results of a phase 1b study. J Clin Oncol. 2014;32 (suppl; abstr 7013):5s.
1. Bosch F, Ferrer A, Villamor N, et al. Fludarabine, cyclophosphamide, and mitoxantrone as initial therapy of chronic lymphocytic leukemia: high response rate and disease eradication. Clin Cancer Res. 2008;14(1):155-161.
2. Byrd JC, Gribben JG, Peterson BL, et al. Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol. 2006;24(3):437-443.
3. Eichhorst BF, Busch R, Stilgenbauer S, et al; German CLL Study Group (GCLLSG). First-line therapy with fludarabine compared with chlorambucil does not result in a major benefit for elderly patients with advanced chronic lymphocytic leukemia. Blood. 2009;114(16):3382-3391.
4. Robak T, Dmoszynska A, Solal-Céligny P, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. J Clin Oncol. 2010;28(10):1756-1765.
5. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42.
6. Byrd JC, Brown JR, et al; RESONATE Investigators. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213-223.
7. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007.
8. Molica S. Immunomodulatory drugs in chronic lymphocytic leukemia: a new
treatment paradigm. Leuk Lymphoma. 2007;48(5):866-869.
9. O’Brien S, Patel M, et al. Duvelisib (IPI-145), a PI3K-d,g inhibitor, is clinically active in patients with relapsed/refractory chronic lymphocytic leukemia. Paper presented at: the 56th ASH Annual Meeting and Exposition; December 7, 2014; San Francisco, CA. Abstract 3334.
10. Ma S, Seymour JF, Lanasa MC, et al. ABT-199 (GDC-0199) combined with rituximab (R) in patients (pts) with relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL): interim results of a phase 1b study. J Clin Oncol. 2014;32 (suppl; abstr 7013):5s.
Mantle Cell Lymphoma: An Evolving Therapeutic Landscape
Mantle cell lymphoma (MCL) is an uncommon B-cell non-Hodgkin lymphoma (NHL) characterized by the translocation, t(11;14), that results in aberrant expression of cyclin D1.1 The clinical presentation varies significantly from asymptomatic to rapidly enlarging lymph nodes, necessitating immediate treatment. Treatment approaches to newly diagnosed MCL correspondingly vary to match the clinical presentation, but they also reflect the bias of individual providers. No treatment is curative, so different treatment philosophies heavily influence management strategies. Mantle cell lymphoma was first described in the 1990s as a unique pathobiologic entity, and it is only now developing its own set of treatment principles distinct from other lymphomas.2 As novel targeted therapies become available, fundamental questions regarding the best treatment approach are certain to evolve.
Traditional Intensive Treatment
The clinical challenge of treating patients with MCL centers on its propensity to relapse quickly after initial therapy. Although most patients will respond to the initial therapy, the duration of their remissions are disappointingly short. The R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) regimens can induce a complete response in the majority of patients, but they invariably relapse within 18 months of finishing therapy. Recognition of this problem led investigators to test highly intensive regimens designed to maximize the depth of remission often followed by autologous stem cell transplant. The highly intensive regimens are successful at extending remission durations to > 5 years in most cases, but at the cost of significant myelotoxicity and a nontrivial risk of death during induction therapy (Table 1).
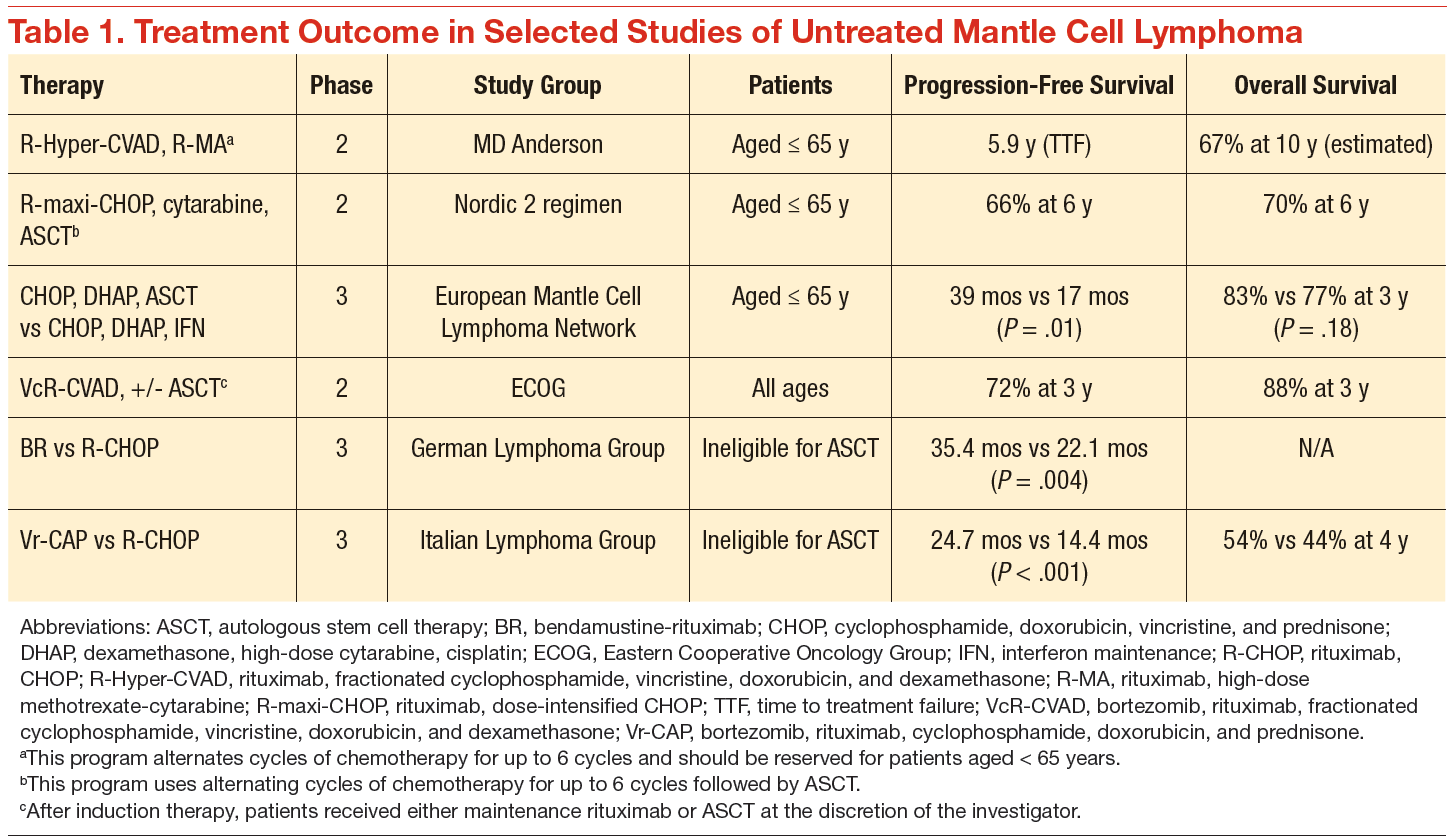
In a subset analysis of a hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) study in MCL, it was observed that patients aged < 65 years had a better survival rate than did older patients.3 Accordingly, these regimens are largely restricted to patients aged < 65 years, which is only a minority of patients with MCL. Importantly, despite initial enthusiasm for the impressive rates of remission, most data suggest that these regimens are not curative.
Nonintensive Treatment
Until recently, patients aged > 65 years or those with significant comorbidities did not have effective treatment options that resulted in durable remissions. Most patients were treated with R-CHOP therapy, and the median survival was only 2 to 3 years.4 Interferon maintenance was frequently used but was largely ineffective at prolongation of remission. Treatment patterns changed quickly after the preliminary results of the randomized StiL (Study Group Indolent Lymphomas) study demonstrated that bendamustine-rituximab (BR) could induce durable remissions with less toxicity than that of R-CHOP therapy (Table 1).5 The superiority of BR over R-CHOP at inducing remissions in MCL was subsequently confirmed in the U.S.-led BRIGHT study; this regimen has become a popular regimen for patients aged > 65 years.6 Despite improved remission rate with less toxicity, however, the BR induction regimen still does not adequately address the problem of short durations of remission.
In a phase 3 study, the European MCL Network demonstrated that the use of maintenance rituximab after induction therapy prolonged both progression-free survival (PFS) as well as overall survival (when given after R-CHOP) compared with interferon maintenance.7 Many providers have extrapolated the benefits seen with rituximab maintenance after R-CHOP to all induction regimens, and rituximab maintenance is increasingly offered to older patients after their induction therapy. An important detail of this study is that rituximab was given indefinitely during maintenance and was not capped at 2 years, as often is done for maintenance treatment in other indolent lymphomas.
A landmark randomized study was recently published that demonstrated an advantage of substituting bortezomib for vincristine in the initial regimen.8 In a phase 3 study of 487 patients, the Vr-CAP (bortezomib in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone) regimen demonstrated an improvement in median PFS over R-CHOP after 40 months of followup (24.7 months vs 14.4 months; hazard radio [HR] 0.63; P < .001) (Table 1). Of particular note, however, is that for patients who achieved a complete remission, the median duration of remission was significantly longer than that of R-CHOP (42.1 months vs 18.0 months; HR 0.63; P < .001). These data suggest that not only is Vr-CAP superior to R-CHOP as a whole, but also that some patients are particularly sensitive to bortezomib, resulting in durable remissions.
Novel Targeted Agents
Since 2006, 3 novel targeted agents have been approved for use in relapsed MCL, and many others are currently in clinical trials (Table 2). Bortezomib is approved for use in both the relapsed and upfront setting and demonstrates clear activity in MCL; however, no biomarker currently exists to help understand which patients will respond. Furthermore, most responses to single agents are partial and short-lived.9
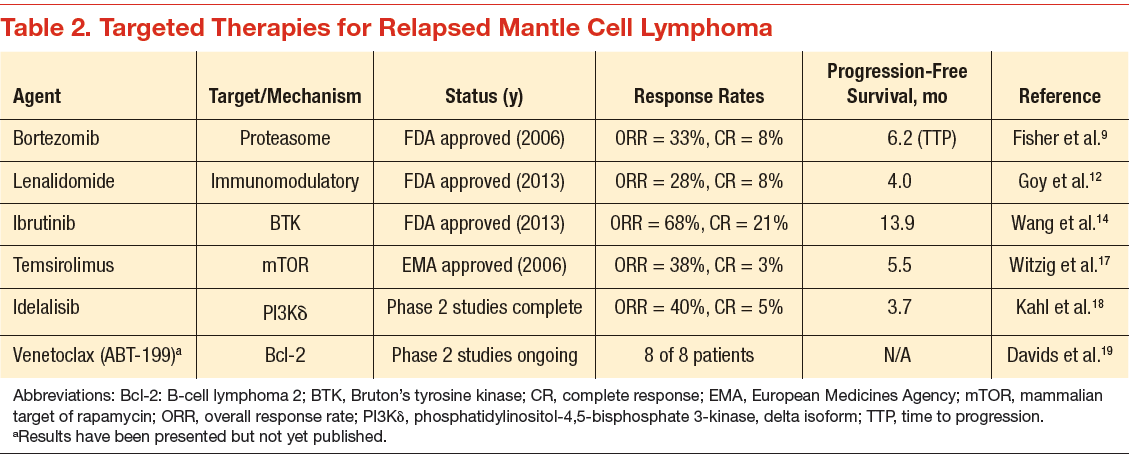
Lenalidomide is an oral immunomodulatory agent that works in NHL through a variety of direct and indirect mechanisms.10 As a single agent, lenalidomide has shown activity in a number of studies and can be combined with rituximab without an apparent increase in the toxicity profile.11-13 The EMERGE trial tested lenalidomide as a single agent at 25 mg (every 21 days out of a 28-day cycle) in patients who had previously been treated with bortezomib. In 128 patients, the overall response rate (RR) was 28% with a complete RR of 7.5%. Although these numbers are modest, long-term data from the NHL-003 trial show a subset of patients with durations of remissions nearing 4 years.11 Thus, a biomarker for lenalidomide that predicts response would be of great
clinical utility.
Perhaps of even greater interest have been the clinical results seen with the use of the oral Bruton’s tyrosine kinase inhibitor, ibrutinib. In 111 patients with relapsed MCL treated with ibrutinib 560 mg, the overall RR was 68% with a complete RR of 21%.14 Most patients tolerated therapy very well, and the duration of remission was estimated at 17.5 months. This was a very impressive result with an agent that is tolerable by most patients with MCL.
Conclusions and Future Directions
Mantle cell lymphoma remains a clinical challenge to many providers due to the heterogeneity in the clinical presentation and the lack of consensus regarding the optimal management strategy. The most commonly recommended approach remains to offer highly intensive chemotherapy programs to patients aged < 65 years, but the introduction of novel active agents into the treatment paradigm is beginning to challenge the assumption that all patients need aggressive therapy.
Future research directions should include predictive biomarkers to enhance treatment decisions. Researchers also should begin to understand the toxicity profile and efficacy of the novel targeted agents when used in combination. Early, informative reports are available regarding ibrutinib when added to both rituximab and BR.15,16 Important research questions will include the long-term effects of extended duration therapy (maintenance) paradigms as well as the introduction of novel molecular monitoring methods, such as circulating tumor DNA and circulating tumor cells, to help guide clinical decisions. In a disease with a reportedly dismal outcome, the near future holds much promise as the MCL treatment landscape evolves at a rapid pace.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to continue reading.
1. Pérez-Galán P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117(1):26-38.
2. Raffeld M, Jaffe ES. bcl-1, t(11;14), and mantle cell-derived lymphomas. Blood. 1991;78(2):259-263.
3. Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol. 2005;23(28):7013-7023.
4. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23(9):1984-1992.
5. Rummel MJ, Niederle N, Maschmeyer G, et al; Study group indolent Lymphomas (StiL). Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203-1210.
6. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustinerituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123(19):2944-2952.
7. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med. 2012;367(6):520-531.
8. Ro bak T, Huang H, Jin J, et al; LYM-3002 Investigators. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372(10):944-953.
9. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867-4874.
10 . Kritharis A, Coyle M, Sharma J, Evens AM. Lenalidomide in non-Hodgkin lymphoma: biologic perspectives and therapeutic opportunities [published online ahead of print March 3, 2015]. Blood. pii: blood-2014-11-567792.
11 . Zinzani PL, Vose JM, Czuczman MS, et al. Long-term follow-up of lenalidomide in relapsed/refractory mantle cell lymphoma: subset analysis of the NHL-003 study. Ann Oncol. 2013;24(11):2892-2897.
12 . Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) Study [published online ahead of print September 3, 2013]. J Clin Oncol. doi: 10.1200/JCO.2013.49.2835.
13 . Chong EA, Ahmadi T, Aqui NA, et al. Combination of lenalidomide and rituximab overcomes rituximab-resistance in patients with indolent B-cell and mantle cell lymphomas [published online ahead of print January 28, 2015]. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-14-2221.
14 . Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507-516.
15 . Wang ML, Hagemeister F, Westin JR, et al. Ibrutinib and rituximab are an efficacious and safe combination in relapsed mantle cell lymphoma: preliminary results from a phase II clinical trial. Blood. 2014;124(21):627.
16 . Maddocks K, Christian B, Jaglowski S, et al. A phase 1/1b study of rituximab, bendamustine, and ibrutinib in patients with untreated and relapsed/refractory non-Hodgkin lymphoma. Blood. 2015;125(2):242-248.
17 . Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23(23):5347-5356.
18 . Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3K inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398-3405.
19. Davids MS, Roberts AW, Anderson MA, et al. The BCL-2-specific BH3-mimetic ABT-199 (GDC-0199) is active and well-tolerated in patients with relapsed nonhodgkin lymphoma: interim results of a phase I study. Blood. 2012;120(21): Abstract 304.
Mantle cell lymphoma (MCL) is an uncommon B-cell non-Hodgkin lymphoma (NHL) characterized by the translocation, t(11;14), that results in aberrant expression of cyclin D1.1 The clinical presentation varies significantly from asymptomatic to rapidly enlarging lymph nodes, necessitating immediate treatment. Treatment approaches to newly diagnosed MCL correspondingly vary to match the clinical presentation, but they also reflect the bias of individual providers. No treatment is curative, so different treatment philosophies heavily influence management strategies. Mantle cell lymphoma was first described in the 1990s as a unique pathobiologic entity, and it is only now developing its own set of treatment principles distinct from other lymphomas.2 As novel targeted therapies become available, fundamental questions regarding the best treatment approach are certain to evolve.
Traditional Intensive Treatment
The clinical challenge of treating patients with MCL centers on its propensity to relapse quickly after initial therapy. Although most patients will respond to the initial therapy, the duration of their remissions are disappointingly short. The R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) regimens can induce a complete response in the majority of patients, but they invariably relapse within 18 months of finishing therapy. Recognition of this problem led investigators to test highly intensive regimens designed to maximize the depth of remission often followed by autologous stem cell transplant. The highly intensive regimens are successful at extending remission durations to > 5 years in most cases, but at the cost of significant myelotoxicity and a nontrivial risk of death during induction therapy (Table 1).
In a subset analysis of a hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) study in MCL, it was observed that patients aged < 65 years had a better survival rate than did older patients.3 Accordingly, these regimens are largely restricted to patients aged < 65 years, which is only a minority of patients with MCL. Importantly, despite initial enthusiasm for the impressive rates of remission, most data suggest that these regimens are not curative.
Nonintensive Treatment
Until recently, patients aged > 65 years or those with significant comorbidities did not have effective treatment options that resulted in durable remissions. Most patients were treated with R-CHOP therapy, and the median survival was only 2 to 3 years.4 Interferon maintenance was frequently used but was largely ineffective at prolongation of remission. Treatment patterns changed quickly after the preliminary results of the randomized StiL (Study Group Indolent Lymphomas) study demonstrated that bendamustine-rituximab (BR) could induce durable remissions with less toxicity than that of R-CHOP therapy (Table 1).5 The superiority of BR over R-CHOP at inducing remissions in MCL was subsequently confirmed in the U.S.-led BRIGHT study; this regimen has become a popular regimen for patients aged > 65 years.6 Despite improved remission rate with less toxicity, however, the BR induction regimen still does not adequately address the problem of short durations of remission.
In a phase 3 study, the European MCL Network demonstrated that the use of maintenance rituximab after induction therapy prolonged both progression-free survival (PFS) as well as overall survival (when given after R-CHOP) compared with interferon maintenance.7 Many providers have extrapolated the benefits seen with rituximab maintenance after R-CHOP to all induction regimens, and rituximab maintenance is increasingly offered to older patients after their induction therapy. An important detail of this study is that rituximab was given indefinitely during maintenance and was not capped at 2 years, as often is done for maintenance treatment in other indolent lymphomas.
A landmark randomized study was recently published that demonstrated an advantage of substituting bortezomib for vincristine in the initial regimen.8 In a phase 3 study of 487 patients, the Vr-CAP (bortezomib in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone) regimen demonstrated an improvement in median PFS over R-CHOP after 40 months of followup (24.7 months vs 14.4 months; hazard radio [HR] 0.63; P < .001) (Table 1). Of particular note, however, is that for patients who achieved a complete remission, the median duration of remission was significantly longer than that of R-CHOP (42.1 months vs 18.0 months; HR 0.63; P < .001). These data suggest that not only is Vr-CAP superior to R-CHOP as a whole, but also that some patients are particularly sensitive to bortezomib, resulting in durable remissions.
Novel Targeted Agents
Since 2006, 3 novel targeted agents have been approved for use in relapsed MCL, and many others are currently in clinical trials (Table 2). Bortezomib is approved for use in both the relapsed and upfront setting and demonstrates clear activity in MCL; however, no biomarker currently exists to help understand which patients will respond. Furthermore, most responses to single agents are partial and short-lived.9
Lenalidomide is an oral immunomodulatory agent that works in NHL through a variety of direct and indirect mechanisms.10 As a single agent, lenalidomide has shown activity in a number of studies and can be combined with rituximab without an apparent increase in the toxicity profile.11-13 The EMERGE trial tested lenalidomide as a single agent at 25 mg (every 21 days out of a 28-day cycle) in patients who had previously been treated with bortezomib. In 128 patients, the overall response rate (RR) was 28% with a complete RR of 7.5%. Although these numbers are modest, long-term data from the NHL-003 trial show a subset of patients with durations of remissions nearing 4 years.11 Thus, a biomarker for lenalidomide that predicts response would be of great
clinical utility.
Perhaps of even greater interest have been the clinical results seen with the use of the oral Bruton’s tyrosine kinase inhibitor, ibrutinib. In 111 patients with relapsed MCL treated with ibrutinib 560 mg, the overall RR was 68% with a complete RR of 21%.14 Most patients tolerated therapy very well, and the duration of remission was estimated at 17.5 months. This was a very impressive result with an agent that is tolerable by most patients with MCL.
Conclusions and Future Directions
Mantle cell lymphoma remains a clinical challenge to many providers due to the heterogeneity in the clinical presentation and the lack of consensus regarding the optimal management strategy. The most commonly recommended approach remains to offer highly intensive chemotherapy programs to patients aged < 65 years, but the introduction of novel active agents into the treatment paradigm is beginning to challenge the assumption that all patients need aggressive therapy.
Future research directions should include predictive biomarkers to enhance treatment decisions. Researchers also should begin to understand the toxicity profile and efficacy of the novel targeted agents when used in combination. Early, informative reports are available regarding ibrutinib when added to both rituximab and BR.15,16 Important research questions will include the long-term effects of extended duration therapy (maintenance) paradigms as well as the introduction of novel molecular monitoring methods, such as circulating tumor DNA and circulating tumor cells, to help guide clinical decisions. In a disease with a reportedly dismal outcome, the near future holds much promise as the MCL treatment landscape evolves at a rapid pace.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to continue reading.
Mantle cell lymphoma (MCL) is an uncommon B-cell non-Hodgkin lymphoma (NHL) characterized by the translocation, t(11;14), that results in aberrant expression of cyclin D1.1 The clinical presentation varies significantly from asymptomatic to rapidly enlarging lymph nodes, necessitating immediate treatment. Treatment approaches to newly diagnosed MCL correspondingly vary to match the clinical presentation, but they also reflect the bias of individual providers. No treatment is curative, so different treatment philosophies heavily influence management strategies. Mantle cell lymphoma was first described in the 1990s as a unique pathobiologic entity, and it is only now developing its own set of treatment principles distinct from other lymphomas.2 As novel targeted therapies become available, fundamental questions regarding the best treatment approach are certain to evolve.
Traditional Intensive Treatment
The clinical challenge of treating patients with MCL centers on its propensity to relapse quickly after initial therapy. Although most patients will respond to the initial therapy, the duration of their remissions are disappointingly short. The R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) regimens can induce a complete response in the majority of patients, but they invariably relapse within 18 months of finishing therapy. Recognition of this problem led investigators to test highly intensive regimens designed to maximize the depth of remission often followed by autologous stem cell transplant. The highly intensive regimens are successful at extending remission durations to > 5 years in most cases, but at the cost of significant myelotoxicity and a nontrivial risk of death during induction therapy (Table 1).
In a subset analysis of a hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) study in MCL, it was observed that patients aged < 65 years had a better survival rate than did older patients.3 Accordingly, these regimens are largely restricted to patients aged < 65 years, which is only a minority of patients with MCL. Importantly, despite initial enthusiasm for the impressive rates of remission, most data suggest that these regimens are not curative.
Nonintensive Treatment
Until recently, patients aged > 65 years or those with significant comorbidities did not have effective treatment options that resulted in durable remissions. Most patients were treated with R-CHOP therapy, and the median survival was only 2 to 3 years.4 Interferon maintenance was frequently used but was largely ineffective at prolongation of remission. Treatment patterns changed quickly after the preliminary results of the randomized StiL (Study Group Indolent Lymphomas) study demonstrated that bendamustine-rituximab (BR) could induce durable remissions with less toxicity than that of R-CHOP therapy (Table 1).5 The superiority of BR over R-CHOP at inducing remissions in MCL was subsequently confirmed in the U.S.-led BRIGHT study; this regimen has become a popular regimen for patients aged > 65 years.6 Despite improved remission rate with less toxicity, however, the BR induction regimen still does not adequately address the problem of short durations of remission.
In a phase 3 study, the European MCL Network demonstrated that the use of maintenance rituximab after induction therapy prolonged both progression-free survival (PFS) as well as overall survival (when given after R-CHOP) compared with interferon maintenance.7 Many providers have extrapolated the benefits seen with rituximab maintenance after R-CHOP to all induction regimens, and rituximab maintenance is increasingly offered to older patients after their induction therapy. An important detail of this study is that rituximab was given indefinitely during maintenance and was not capped at 2 years, as often is done for maintenance treatment in other indolent lymphomas.
A landmark randomized study was recently published that demonstrated an advantage of substituting bortezomib for vincristine in the initial regimen.8 In a phase 3 study of 487 patients, the Vr-CAP (bortezomib in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone) regimen demonstrated an improvement in median PFS over R-CHOP after 40 months of followup (24.7 months vs 14.4 months; hazard radio [HR] 0.63; P < .001) (Table 1). Of particular note, however, is that for patients who achieved a complete remission, the median duration of remission was significantly longer than that of R-CHOP (42.1 months vs 18.0 months; HR 0.63; P < .001). These data suggest that not only is Vr-CAP superior to R-CHOP as a whole, but also that some patients are particularly sensitive to bortezomib, resulting in durable remissions.
Novel Targeted Agents
Since 2006, 3 novel targeted agents have been approved for use in relapsed MCL, and many others are currently in clinical trials (Table 2). Bortezomib is approved for use in both the relapsed and upfront setting and demonstrates clear activity in MCL; however, no biomarker currently exists to help understand which patients will respond. Furthermore, most responses to single agents are partial and short-lived.9
Lenalidomide is an oral immunomodulatory agent that works in NHL through a variety of direct and indirect mechanisms.10 As a single agent, lenalidomide has shown activity in a number of studies and can be combined with rituximab without an apparent increase in the toxicity profile.11-13 The EMERGE trial tested lenalidomide as a single agent at 25 mg (every 21 days out of a 28-day cycle) in patients who had previously been treated with bortezomib. In 128 patients, the overall response rate (RR) was 28% with a complete RR of 7.5%. Although these numbers are modest, long-term data from the NHL-003 trial show a subset of patients with durations of remissions nearing 4 years.11 Thus, a biomarker for lenalidomide that predicts response would be of great
clinical utility.
Perhaps of even greater interest have been the clinical results seen with the use of the oral Bruton’s tyrosine kinase inhibitor, ibrutinib. In 111 patients with relapsed MCL treated with ibrutinib 560 mg, the overall RR was 68% with a complete RR of 21%.14 Most patients tolerated therapy very well, and the duration of remission was estimated at 17.5 months. This was a very impressive result with an agent that is tolerable by most patients with MCL.
Conclusions and Future Directions
Mantle cell lymphoma remains a clinical challenge to many providers due to the heterogeneity in the clinical presentation and the lack of consensus regarding the optimal management strategy. The most commonly recommended approach remains to offer highly intensive chemotherapy programs to patients aged < 65 years, but the introduction of novel active agents into the treatment paradigm is beginning to challenge the assumption that all patients need aggressive therapy.
Future research directions should include predictive biomarkers to enhance treatment decisions. Researchers also should begin to understand the toxicity profile and efficacy of the novel targeted agents when used in combination. Early, informative reports are available regarding ibrutinib when added to both rituximab and BR.15,16 Important research questions will include the long-term effects of extended duration therapy (maintenance) paradigms as well as the introduction of novel molecular monitoring methods, such as circulating tumor DNA and circulating tumor cells, to help guide clinical decisions. In a disease with a reportedly dismal outcome, the near future holds much promise as the MCL treatment landscape evolves at a rapid pace.
Author disclosures
The author reports no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the author and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to continue reading.
1. Pérez-Galán P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117(1):26-38.
2. Raffeld M, Jaffe ES. bcl-1, t(11;14), and mantle cell-derived lymphomas. Blood. 1991;78(2):259-263.
3. Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol. 2005;23(28):7013-7023.
4. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23(9):1984-1992.
5. Rummel MJ, Niederle N, Maschmeyer G, et al; Study group indolent Lymphomas (StiL). Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203-1210.
6. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustinerituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123(19):2944-2952.
7. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med. 2012;367(6):520-531.
8. Ro bak T, Huang H, Jin J, et al; LYM-3002 Investigators. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372(10):944-953.
9. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867-4874.
10 . Kritharis A, Coyle M, Sharma J, Evens AM. Lenalidomide in non-Hodgkin lymphoma: biologic perspectives and therapeutic opportunities [published online ahead of print March 3, 2015]. Blood. pii: blood-2014-11-567792.
11 . Zinzani PL, Vose JM, Czuczman MS, et al. Long-term follow-up of lenalidomide in relapsed/refractory mantle cell lymphoma: subset analysis of the NHL-003 study. Ann Oncol. 2013;24(11):2892-2897.
12 . Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) Study [published online ahead of print September 3, 2013]. J Clin Oncol. doi: 10.1200/JCO.2013.49.2835.
13 . Chong EA, Ahmadi T, Aqui NA, et al. Combination of lenalidomide and rituximab overcomes rituximab-resistance in patients with indolent B-cell and mantle cell lymphomas [published online ahead of print January 28, 2015]. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-14-2221.
14 . Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507-516.
15 . Wang ML, Hagemeister F, Westin JR, et al. Ibrutinib and rituximab are an efficacious and safe combination in relapsed mantle cell lymphoma: preliminary results from a phase II clinical trial. Blood. 2014;124(21):627.
16 . Maddocks K, Christian B, Jaglowski S, et al. A phase 1/1b study of rituximab, bendamustine, and ibrutinib in patients with untreated and relapsed/refractory non-Hodgkin lymphoma. Blood. 2015;125(2):242-248.
17 . Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23(23):5347-5356.
18 . Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3K inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398-3405.
19. Davids MS, Roberts AW, Anderson MA, et al. The BCL-2-specific BH3-mimetic ABT-199 (GDC-0199) is active and well-tolerated in patients with relapsed nonhodgkin lymphoma: interim results of a phase I study. Blood. 2012;120(21): Abstract 304.
1. Pérez-Galán P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117(1):26-38.
2. Raffeld M, Jaffe ES. bcl-1, t(11;14), and mantle cell-derived lymphomas. Blood. 1991;78(2):259-263.
3. Romaguera JE, Fayad L, Rodriguez MA, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol. 2005;23(28):7013-7023.
4. Lenz G, Dreyling M, Hoster E, et al. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG). J Clin Oncol. 2005;23(9):1984-1992.
5. Rummel MJ, Niederle N, Maschmeyer G, et al; Study group indolent Lymphomas (StiL). Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203-1210.
6. Flinn IW, van der Jagt R, Kahl BS, et al. Randomized trial of bendamustinerituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood. 2014;123(19):2944-2952.
7. Kluin-Nelemans HC, Hoster E, Hermine O, et al. Treatment of older patients with mantle-cell lymphoma. N Engl J Med. 2012;367(6):520-531.
8. Ro bak T, Huang H, Jin J, et al; LYM-3002 Investigators. Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med. 2015;372(10):944-953.
9. Fisher RI, Bernstein SH, Kahl BS, et al. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867-4874.
10 . Kritharis A, Coyle M, Sharma J, Evens AM. Lenalidomide in non-Hodgkin lymphoma: biologic perspectives and therapeutic opportunities [published online ahead of print March 3, 2015]. Blood. pii: blood-2014-11-567792.
11 . Zinzani PL, Vose JM, Czuczman MS, et al. Long-term follow-up of lenalidomide in relapsed/refractory mantle cell lymphoma: subset analysis of the NHL-003 study. Ann Oncol. 2013;24(11):2892-2897.
12 . Goy A, Sinha R, Williams ME, et al. Single-agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or were refractory to bortezomib: phase II MCL-001 (EMERGE) Study [published online ahead of print September 3, 2013]. J Clin Oncol. doi: 10.1200/JCO.2013.49.2835.
13 . Chong EA, Ahmadi T, Aqui NA, et al. Combination of lenalidomide and rituximab overcomes rituximab-resistance in patients with indolent B-cell and mantle cell lymphomas [published online ahead of print January 28, 2015]. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-14-2221.
14 . Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507-516.
15 . Wang ML, Hagemeister F, Westin JR, et al. Ibrutinib and rituximab are an efficacious and safe combination in relapsed mantle cell lymphoma: preliminary results from a phase II clinical trial. Blood. 2014;124(21):627.
16 . Maddocks K, Christian B, Jaglowski S, et al. A phase 1/1b study of rituximab, bendamustine, and ibrutinib in patients with untreated and relapsed/refractory non-Hodgkin lymphoma. Blood. 2015;125(2):242-248.
17 . Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23(23):5347-5356.
18 . Kahl BS, Spurgeon SE, Furman RR, et al. A phase 1 study of the PI3K inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood. 2014;123(22):3398-3405.
19. Davids MS, Roberts AW, Anderson MA, et al. The BCL-2-specific BH3-mimetic ABT-199 (GDC-0199) is active and well-tolerated in patients with relapsed nonhodgkin lymphoma: interim results of a phase I study. Blood. 2012;120(21): Abstract 304.
Preventing Skeletal-Related Events
Supplementation with calcium and vitamin D can be effective ways to prevent skeletal or teeth-related issues during treatment for bone metastasis or multiple myeloma, according to Sheetal Malhotra, MD, of the James J. Peters VAMC in the Bronx, New York.
“Data suggest that if you optimize vitamin D levels when you are giving patients bisphosphonates it can further improve response to bisphosphonate therapy,” Malhotra said. “Supplementation should be given to these patients who are on treatment with bisphosphonates unless there is some contraindication.”
Supplementation with calcium and vitamin D can be effective ways to prevent skeletal or teeth-related issues during treatment for bone metastasis or multiple myeloma, according to Sheetal Malhotra, MD, of the James J. Peters VAMC in the Bronx, New York.
“Data suggest that if you optimize vitamin D levels when you are giving patients bisphosphonates it can further improve response to bisphosphonate therapy,” Malhotra said. “Supplementation should be given to these patients who are on treatment with bisphosphonates unless there is some contraindication.”
Supplementation with calcium and vitamin D can be effective ways to prevent skeletal or teeth-related issues during treatment for bone metastasis or multiple myeloma, according to Sheetal Malhotra, MD, of the James J. Peters VAMC in the Bronx, New York.
“Data suggest that if you optimize vitamin D levels when you are giving patients bisphosphonates it can further improve response to bisphosphonate therapy,” Malhotra said. “Supplementation should be given to these patients who are on treatment with bisphosphonates unless there is some contraindication.”
