User login
FDA grants drug orphan designation for AML

The US Food and Drug Administration (FDA) has granted orphan drug designation to SY-1425 for the treatment of acute myeloid leukemia (AML).
SY-1425 is an oral, first-in-class, selective retinoic acid receptor alpha (RARα) agonist. It is currently under investigation in a phase 2 trial of patients with AML and myelodysplastic syndromes (MDS).
“We believe that SY-1425 may provide a meaningful benefit for subsets of AML patients whose disease is driven by abnormally high expression of the RARA or IRF8 genes,” said David A. Roth, MD, chief medical officer at Syros Pharmaceuticals, the company developing SY-1425.
“Receiving orphan drug designation is an important regulatory milestone in the development of SY-1425. We’re pleased with the continued progress of the ongoing phase 2 clinical trial, and we look forward to presenting initial clinical data in the fourth quarter of this year.”
Using its gene control platform, Syros discovered subsets of AML and MDS patients with super-enhancers associated with RARA or IRF8. These super-enhancers are believed to drive overexpression of the RARA or IRF8 genes, locking cells in an immature, undifferentiated, and proliferative state and leading to disease.
In preclinical studies, SY-1425 promoted differentiation of AML cells with high RARA or IRF8 expression and inhibited tumor growth and prolonged survival in patient-derived xenograft models of AML with high RARA expression.
In the ongoing phase 2 trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy.
All patients are prospectively selected using biomarkers for high expression of RARA or IRF8. Additional details about the trial can be found at https://clinicaltrials.gov/ct2/show/NCT02807558.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to SY-1425 for the treatment of acute myeloid leukemia (AML).
SY-1425 is an oral, first-in-class, selective retinoic acid receptor alpha (RARα) agonist. It is currently under investigation in a phase 2 trial of patients with AML and myelodysplastic syndromes (MDS).
“We believe that SY-1425 may provide a meaningful benefit for subsets of AML patients whose disease is driven by abnormally high expression of the RARA or IRF8 genes,” said David A. Roth, MD, chief medical officer at Syros Pharmaceuticals, the company developing SY-1425.
“Receiving orphan drug designation is an important regulatory milestone in the development of SY-1425. We’re pleased with the continued progress of the ongoing phase 2 clinical trial, and we look forward to presenting initial clinical data in the fourth quarter of this year.”
Using its gene control platform, Syros discovered subsets of AML and MDS patients with super-enhancers associated with RARA or IRF8. These super-enhancers are believed to drive overexpression of the RARA or IRF8 genes, locking cells in an immature, undifferentiated, and proliferative state and leading to disease.
In preclinical studies, SY-1425 promoted differentiation of AML cells with high RARA or IRF8 expression and inhibited tumor growth and prolonged survival in patient-derived xenograft models of AML with high RARA expression.
In the ongoing phase 2 trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy.
All patients are prospectively selected using biomarkers for high expression of RARA or IRF8. Additional details about the trial can be found at https://clinicaltrials.gov/ct2/show/NCT02807558.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to SY-1425 for the treatment of acute myeloid leukemia (AML).
SY-1425 is an oral, first-in-class, selective retinoic acid receptor alpha (RARα) agonist. It is currently under investigation in a phase 2 trial of patients with AML and myelodysplastic syndromes (MDS).
“We believe that SY-1425 may provide a meaningful benefit for subsets of AML patients whose disease is driven by abnormally high expression of the RARA or IRF8 genes,” said David A. Roth, MD, chief medical officer at Syros Pharmaceuticals, the company developing SY-1425.
“Receiving orphan drug designation is an important regulatory milestone in the development of SY-1425. We’re pleased with the continued progress of the ongoing phase 2 clinical trial, and we look forward to presenting initial clinical data in the fourth quarter of this year.”
Using its gene control platform, Syros discovered subsets of AML and MDS patients with super-enhancers associated with RARA or IRF8. These super-enhancers are believed to drive overexpression of the RARA or IRF8 genes, locking cells in an immature, undifferentiated, and proliferative state and leading to disease.
In preclinical studies, SY-1425 promoted differentiation of AML cells with high RARA or IRF8 expression and inhibited tumor growth and prolonged survival in patient-derived xenograft models of AML with high RARA expression.
In the ongoing phase 2 trial, researchers are assessing the safety and efficacy of SY-1425 as a single agent in 4 AML and MDS patient populations, as well as SY-1425 in combination with azacitidine in newly diagnosed AML patients who are not suitable candidates for standard chemotherapy.
All patients are prospectively selected using biomarkers for high expression of RARA or IRF8. Additional details about the trial can be found at https://clinicaltrials.gov/ct2/show/NCT02807558.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
Inotuzumab ozogamicin tied to sinusoidal obstruction syndrome in ALL
Inotuzumab ozogamicin therapy significantly increased the risk of sinusoidal obstruction syndrome (veno-occlusive disease) among adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL), especially when they also received follow-up hematopoietic stem cell transplantation, according to a safety analysis from the INO-VATE trial.
After a median of 9 weeks of treatment, 13% of 164 patients who received inotuzumab ozogamicin (Besponsa, Wyeth/Pfizer) developed sinusoidal obstruction syndrome, compared with less than 1% of 143 patients who received standard care, reported Hagop M. Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his associates (Lancet Haematol. 2017 Jul 4. doi: 10.1016/ S2352-3026[17]30103-5).
Follow-up treatment with HSCT increased the risk of sinusoidal obstruction syndrome in both the intervention (22%) and standard-care (3%) groups. Among patients who did not undergo HSCT, rates of this adverse event were 3% and 0%, respectively. Five patients died from sinusoidal obstruction syndrome, all of whom received both inotuzumab ozogamicin and HSCT. The findings earned the newly approved regimen a boxed warning for severe hepatotoxicity.
The open-label, phase 3, multicenter INO-VATE study included 326 adults with CD22-positive, Philadelphia chromosome–negative or Philadelphia chromosome–positive relapsed or refractory B-cell precursor ALL. The safety analysis included 305 patients. Rates of treatment-emergent hepatotoxicities, of all grades, were 51% with inotuzumab ozogamicin and 34% with standard care. Most adverse hepatic events were grade 1-2 liver-related laboratory abnormalities, but 8% of inotuzumab ozogamicin recipients developed grade 3 or higher sinusoidal obstruction syndrome, versus less than 1% of the control group.
“After follow-up HSCT, the frequency of sinusoidal obstruction syndrome was 50% or higher in the following subgroups: patients aged 65 years or older, patients with last available pre-HSCT serum bilirubin concentration more than or equal to the upper limit of normal, and patients who received conditioning regimens with two alkylating agents,” Dr. Kantarjian and his fellow investigators wrote. Conditioning regimens that included thiotepa markedly increased the risk of sinusoidal obstruction syndrome. Additional risk factors included HSCT before study enrollment, history of liver disease, and a final pre-HSCT platelet count of less than 100 × 109 platelets per L.
Rates of sinusoidal obstruction syndrome were 42% with four to six cycles of inotuzumab ozogamicin, 23% with three cycles, 19% with two cycles, and 8% with one cycle, said the investigators. In multivariate analysis, conditioning with two alkylating agents (P = .02 compared with one alkylating agent) and pre-HSCT bilirubin of at least the upper limit of normal (P = .01) significantly increased the risk of sinusoidal obstruction syndrome during or after treatment with inotuzumab ozogamicin.
Notably, inotuzumab ozogamicin did not significantly increase the chances of survival compared with standard care among patients who also received follow-up HSCT (hazard ratio, 1.3; 97.5% confidence interval, 0.66 to 2.3; P = 0.77). Among HSCT recipients, the chances of surviving to 24 months were 39% (95% CI, 28%-50%) with inotuzumab ozogamicin and 29% (11%-49%) with standard care. Nonetheless, HSCT “offers possibility of cure in the relapsed or recurrent [ALL] setting,” the researchers wrote. Clinicians should be especially wary of sinusoidal obstruction syndrome if patients are 65 years or older, received HSCT before inotuzumab ozogamicin treatment, or have a baseline history of liver disease, they said. Strategies to minimize risk include shortening the duration of inotuzumab ozogamicin treatment and avoiding conditioning regimens that contain two alkylating agents.
Pfizer funded and collaborated in the trial. Dr. Kantarjian disclosed ties to Pfizer and numerous other pharmaceutical companies.
Inotuzumab ozogamicin therapy significantly increased the risk of sinusoidal obstruction syndrome (veno-occlusive disease) among adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL), especially when they also received follow-up hematopoietic stem cell transplantation, according to a safety analysis from the INO-VATE trial.
After a median of 9 weeks of treatment, 13% of 164 patients who received inotuzumab ozogamicin (Besponsa, Wyeth/Pfizer) developed sinusoidal obstruction syndrome, compared with less than 1% of 143 patients who received standard care, reported Hagop M. Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his associates (Lancet Haematol. 2017 Jul 4. doi: 10.1016/ S2352-3026[17]30103-5).
Follow-up treatment with HSCT increased the risk of sinusoidal obstruction syndrome in both the intervention (22%) and standard-care (3%) groups. Among patients who did not undergo HSCT, rates of this adverse event were 3% and 0%, respectively. Five patients died from sinusoidal obstruction syndrome, all of whom received both inotuzumab ozogamicin and HSCT. The findings earned the newly approved regimen a boxed warning for severe hepatotoxicity.
The open-label, phase 3, multicenter INO-VATE study included 326 adults with CD22-positive, Philadelphia chromosome–negative or Philadelphia chromosome–positive relapsed or refractory B-cell precursor ALL. The safety analysis included 305 patients. Rates of treatment-emergent hepatotoxicities, of all grades, were 51% with inotuzumab ozogamicin and 34% with standard care. Most adverse hepatic events were grade 1-2 liver-related laboratory abnormalities, but 8% of inotuzumab ozogamicin recipients developed grade 3 or higher sinusoidal obstruction syndrome, versus less than 1% of the control group.
“After follow-up HSCT, the frequency of sinusoidal obstruction syndrome was 50% or higher in the following subgroups: patients aged 65 years or older, patients with last available pre-HSCT serum bilirubin concentration more than or equal to the upper limit of normal, and patients who received conditioning regimens with two alkylating agents,” Dr. Kantarjian and his fellow investigators wrote. Conditioning regimens that included thiotepa markedly increased the risk of sinusoidal obstruction syndrome. Additional risk factors included HSCT before study enrollment, history of liver disease, and a final pre-HSCT platelet count of less than 100 × 109 platelets per L.
Rates of sinusoidal obstruction syndrome were 42% with four to six cycles of inotuzumab ozogamicin, 23% with three cycles, 19% with two cycles, and 8% with one cycle, said the investigators. In multivariate analysis, conditioning with two alkylating agents (P = .02 compared with one alkylating agent) and pre-HSCT bilirubin of at least the upper limit of normal (P = .01) significantly increased the risk of sinusoidal obstruction syndrome during or after treatment with inotuzumab ozogamicin.
Notably, inotuzumab ozogamicin did not significantly increase the chances of survival compared with standard care among patients who also received follow-up HSCT (hazard ratio, 1.3; 97.5% confidence interval, 0.66 to 2.3; P = 0.77). Among HSCT recipients, the chances of surviving to 24 months were 39% (95% CI, 28%-50%) with inotuzumab ozogamicin and 29% (11%-49%) with standard care. Nonetheless, HSCT “offers possibility of cure in the relapsed or recurrent [ALL] setting,” the researchers wrote. Clinicians should be especially wary of sinusoidal obstruction syndrome if patients are 65 years or older, received HSCT before inotuzumab ozogamicin treatment, or have a baseline history of liver disease, they said. Strategies to minimize risk include shortening the duration of inotuzumab ozogamicin treatment and avoiding conditioning regimens that contain two alkylating agents.
Pfizer funded and collaborated in the trial. Dr. Kantarjian disclosed ties to Pfizer and numerous other pharmaceutical companies.
Inotuzumab ozogamicin therapy significantly increased the risk of sinusoidal obstruction syndrome (veno-occlusive disease) among adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL), especially when they also received follow-up hematopoietic stem cell transplantation, according to a safety analysis from the INO-VATE trial.
After a median of 9 weeks of treatment, 13% of 164 patients who received inotuzumab ozogamicin (Besponsa, Wyeth/Pfizer) developed sinusoidal obstruction syndrome, compared with less than 1% of 143 patients who received standard care, reported Hagop M. Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his associates (Lancet Haematol. 2017 Jul 4. doi: 10.1016/ S2352-3026[17]30103-5).
Follow-up treatment with HSCT increased the risk of sinusoidal obstruction syndrome in both the intervention (22%) and standard-care (3%) groups. Among patients who did not undergo HSCT, rates of this adverse event were 3% and 0%, respectively. Five patients died from sinusoidal obstruction syndrome, all of whom received both inotuzumab ozogamicin and HSCT. The findings earned the newly approved regimen a boxed warning for severe hepatotoxicity.
The open-label, phase 3, multicenter INO-VATE study included 326 adults with CD22-positive, Philadelphia chromosome–negative or Philadelphia chromosome–positive relapsed or refractory B-cell precursor ALL. The safety analysis included 305 patients. Rates of treatment-emergent hepatotoxicities, of all grades, were 51% with inotuzumab ozogamicin and 34% with standard care. Most adverse hepatic events were grade 1-2 liver-related laboratory abnormalities, but 8% of inotuzumab ozogamicin recipients developed grade 3 or higher sinusoidal obstruction syndrome, versus less than 1% of the control group.
“After follow-up HSCT, the frequency of sinusoidal obstruction syndrome was 50% or higher in the following subgroups: patients aged 65 years or older, patients with last available pre-HSCT serum bilirubin concentration more than or equal to the upper limit of normal, and patients who received conditioning regimens with two alkylating agents,” Dr. Kantarjian and his fellow investigators wrote. Conditioning regimens that included thiotepa markedly increased the risk of sinusoidal obstruction syndrome. Additional risk factors included HSCT before study enrollment, history of liver disease, and a final pre-HSCT platelet count of less than 100 × 109 platelets per L.
Rates of sinusoidal obstruction syndrome were 42% with four to six cycles of inotuzumab ozogamicin, 23% with three cycles, 19% with two cycles, and 8% with one cycle, said the investigators. In multivariate analysis, conditioning with two alkylating agents (P = .02 compared with one alkylating agent) and pre-HSCT bilirubin of at least the upper limit of normal (P = .01) significantly increased the risk of sinusoidal obstruction syndrome during or after treatment with inotuzumab ozogamicin.
Notably, inotuzumab ozogamicin did not significantly increase the chances of survival compared with standard care among patients who also received follow-up HSCT (hazard ratio, 1.3; 97.5% confidence interval, 0.66 to 2.3; P = 0.77). Among HSCT recipients, the chances of surviving to 24 months were 39% (95% CI, 28%-50%) with inotuzumab ozogamicin and 29% (11%-49%) with standard care. Nonetheless, HSCT “offers possibility of cure in the relapsed or recurrent [ALL] setting,” the researchers wrote. Clinicians should be especially wary of sinusoidal obstruction syndrome if patients are 65 years or older, received HSCT before inotuzumab ozogamicin treatment, or have a baseline history of liver disease, they said. Strategies to minimize risk include shortening the duration of inotuzumab ozogamicin treatment and avoiding conditioning regimens that contain two alkylating agents.
Pfizer funded and collaborated in the trial. Dr. Kantarjian disclosed ties to Pfizer and numerous other pharmaceutical companies.
FROM LANCET HAEMATOLOGY
Key clinical point: Treatment with inotuzumab ozogamicin (Besponsa, Wyeth/Pfizer) led to sinusoidal obstructive syndrome (veno-occlusive disease), especially after follow-up hematopoietic stem cell transplantation, compared with standard care for relapsed or refractory acute lymphoblastic leukemia.
Major finding: After a median of 9 weeks of treatment, rates of sinusoidal obstructive syndrome were 13% among inotuzumab ozogamicin recipients overall, 22% among those who also received HSCT, and less than 1% in the standard-care group.
Data source: A prespecified safety analysis of INO-VATE, an open-label, phase 3, multicenter trial of 326 adults with Philadelphia chromosome–negative or Philadelphia chromosome–positive relapsed or refractory B-cell precursor ALL.
Disclosures: Pfizer funded and collaborated in the trial. Dr. Kantarjian disclosed ties to Pfizer and numerous other pharmaceutical companies.
Vitamin C could help treat TET2-mutant leukemias

Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well. ![]()
Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well. ![]()
Preclinical research suggests high-dose vitamin C may be effective against TET2-mutant leukemias.
Investigators found that vitamin C mimics TET2 restoration, thereby suppressing leukemic colony formation, inhibiting leukemia progression in mice, and enhancing leukemia cells’ sensitivity to treatment with a PARP inhibitor.
“We’re excited by the prospect that high-dose vitamin C might become a safe treatment for blood diseases caused by TET2-deficient leukemia stem cells, most likely in combination with other targeted therapies,” said study author Benjamin G. Neel, MD, PhD, of NYU School of Medicine in New York, New York.
Dr Neel and his colleagues reported their findings in Cell.
Previous research had shown that vitamin C could stimulate the activity of TET2 as well as TET1 and TET3.
Because only 1 copy of the TET2 gene in each stem cell is usually affected in TET2-mutant blood diseases, the investigators hypothesized that high doses of vitamin C might reverse the effects of TET2 deficiency by turning up the action of the remaining functional gene.
Indeed, the team found that vitamin C had the same effect as restoring TET2 function genetically. By promoting DNA demethylation, high-dose vitamin C induced stem cells to mature and suppressed the growth of leukemic stem cells (LSCs) implanted in mice.
“Interestingly, we also found that vitamin C treatment had an effect on leukemic stem cells that resembled damage to their DNA,” said study author Luisa Cimmino, PhD, of NYU School of Medicine.
“For this reason, we decided to combine vitamin C with a PARP inhibitor, a drug type known to cause cancer cell death by blocking the repair of DNA damage, and already approved for treating certain patients with ovarian cancer.”
The combination had an enhanced effect on LSCs, further shifting them from self-renewal back toward maturity and cell death.
Dr Cimmino said these results suggest vitamin C might also be effective against leukemias without TET2 mutations. As vitamin C turns up any TET2 activity normally in place, it might drive LSCs without TET2 mutations toward death as well. ![]()
FDA approves inotuzumab ozogamicin for rel/ref ALL

The US Food and Drug Administration (FDA) has approved inotuzumab ozogamicin (Besponsa®) for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The labeling for inotuzumab ozogamicin includes a boxed warning stating that the drug poses a risk of hepatotoxicity, including hepatic veno-occlusive disease (or sinusoidal obstruction syndrome), as well as an increased risk of post-transplant non-relapse mortality.
The full prescribing information for inotuzumab ozogamicin is available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf.
Inotuzumab ozogamicin is an antibody-drug conjugate that consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
The product originates from a collaboration between Pfizer and Celltech (now UCB), but Pfizer has sole responsibility for all manufacturing and clinical development activities.
Inotuzumab ozogamicin was reviewed and approved under the FDA’s breakthrough therapy designation and priority review programs.
The application for inotuzumab ozogamicin was supported by results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell ALL. Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens (high-dose cytarabine, cytarabine plus mitoxantrone, or fludarabine, cytarabine, and granulocyte colony-stimulating factor).
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were thought to be related to chemotherapy. ![]()
The US Food and Drug Administration (FDA) has approved inotuzumab ozogamicin (Besponsa®) for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The labeling for inotuzumab ozogamicin includes a boxed warning stating that the drug poses a risk of hepatotoxicity, including hepatic veno-occlusive disease (or sinusoidal obstruction syndrome), as well as an increased risk of post-transplant non-relapse mortality.
The full prescribing information for inotuzumab ozogamicin is available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf.
Inotuzumab ozogamicin is an antibody-drug conjugate that consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
The product originates from a collaboration between Pfizer and Celltech (now UCB), but Pfizer has sole responsibility for all manufacturing and clinical development activities.
Inotuzumab ozogamicin was reviewed and approved under the FDA’s breakthrough therapy designation and priority review programs.
The application for inotuzumab ozogamicin was supported by results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell ALL. Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens (high-dose cytarabine, cytarabine plus mitoxantrone, or fludarabine, cytarabine, and granulocyte colony-stimulating factor).
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were thought to be related to chemotherapy. ![]()
The US Food and Drug Administration (FDA) has approved inotuzumab ozogamicin (Besponsa®) for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The labeling for inotuzumab ozogamicin includes a boxed warning stating that the drug poses a risk of hepatotoxicity, including hepatic veno-occlusive disease (or sinusoidal obstruction syndrome), as well as an increased risk of post-transplant non-relapse mortality.
The full prescribing information for inotuzumab ozogamicin is available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf.
Inotuzumab ozogamicin is an antibody-drug conjugate that consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
The product originates from a collaboration between Pfizer and Celltech (now UCB), but Pfizer has sole responsibility for all manufacturing and clinical development activities.
Inotuzumab ozogamicin was reviewed and approved under the FDA’s breakthrough therapy designation and priority review programs.
The application for inotuzumab ozogamicin was supported by results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell ALL. Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens (high-dose cytarabine, cytarabine plus mitoxantrone, or fludarabine, cytarabine, and granulocyte colony-stimulating factor).
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were thought to be related to chemotherapy. ![]()
Study reveals elevated cancer risk in Holocaust survivors

A new study indicates that survivors of the Holocaust have experienced a small but consistent increase in the risk of developing cancer.
The findings, published in the journal Cancer, offer an example of how extreme population-level tragedies can have an impact on health.
Holocaust survivors were exposed to a variety of factors that have been linked with cancer.
So researchers set out to investigate whether the starvation, overcrowding, infectious diseases, and psychological stress that survivors endured might have contributed to the development of cancer in some individuals.
The team studied 152,622 Holocaust survivors who were followed for more than 45 years.
The researchers used 2 definitions of exposure to classify the survivors.
One definition was based on an individual’s entitlement for compensation for suffering persecution during the war. The other was based on the country of origin, dividing countries into those that were directly governed by Nazi Germany and those that were not occupied by Nazis.
The cancer incidence was significantly higher in survivors who were granted compensation than in those who were not—21.9% and 16.1%, respectively (P<0.0001).
However, the difference between survivors from occupied and non-occupied countries was not significant—22.7% and 21.4%, respectively.
On the other hand, when the researchers adjusted for confounding factors, survivors who had been exposed by either definition had a significantly increased risk of cancer.
For survivors who were granted compensation, the hazard ratio (HR) was 1.06 (P<0.001). For survivors born in occupied countries, the HR was 1.08 (P<0.001).
There was no increased risk of acute or chronic leukemia among patients who received compensation. And there was no increased risk of acute leukemia for survivors born in occupied countries.
However, there was a significantly increased risk of chronic leukemia among survivors born in occupied countries (HR=1.33, P=0.001)
“The data emphasize the importance of learning about the combined effect of several exposures occurring intensely and contemporaneously on cancer risk, such as those that unfortunately occurred during World War II,” said study author Siegal Sadetzki, MD, of the Chaim Sheba Medical Center in Tel HaShomer, Israel.
An editorial related to this study noted that the association between cancer and the extreme deprivation experienced by Holocaust survivors may also have parallels with other extreme population-level events, including in racial/ethnic minority groups experiencing severe social deprivation over time. ![]()
A new study indicates that survivors of the Holocaust have experienced a small but consistent increase in the risk of developing cancer.
The findings, published in the journal Cancer, offer an example of how extreme population-level tragedies can have an impact on health.
Holocaust survivors were exposed to a variety of factors that have been linked with cancer.
So researchers set out to investigate whether the starvation, overcrowding, infectious diseases, and psychological stress that survivors endured might have contributed to the development of cancer in some individuals.
The team studied 152,622 Holocaust survivors who were followed for more than 45 years.
The researchers used 2 definitions of exposure to classify the survivors.
One definition was based on an individual’s entitlement for compensation for suffering persecution during the war. The other was based on the country of origin, dividing countries into those that were directly governed by Nazi Germany and those that were not occupied by Nazis.
The cancer incidence was significantly higher in survivors who were granted compensation than in those who were not—21.9% and 16.1%, respectively (P<0.0001).
However, the difference between survivors from occupied and non-occupied countries was not significant—22.7% and 21.4%, respectively.
On the other hand, when the researchers adjusted for confounding factors, survivors who had been exposed by either definition had a significantly increased risk of cancer.
For survivors who were granted compensation, the hazard ratio (HR) was 1.06 (P<0.001). For survivors born in occupied countries, the HR was 1.08 (P<0.001).
There was no increased risk of acute or chronic leukemia among patients who received compensation. And there was no increased risk of acute leukemia for survivors born in occupied countries.
However, there was a significantly increased risk of chronic leukemia among survivors born in occupied countries (HR=1.33, P=0.001)
“The data emphasize the importance of learning about the combined effect of several exposures occurring intensely and contemporaneously on cancer risk, such as those that unfortunately occurred during World War II,” said study author Siegal Sadetzki, MD, of the Chaim Sheba Medical Center in Tel HaShomer, Israel.
An editorial related to this study noted that the association between cancer and the extreme deprivation experienced by Holocaust survivors may also have parallels with other extreme population-level events, including in racial/ethnic minority groups experiencing severe social deprivation over time. ![]()
A new study indicates that survivors of the Holocaust have experienced a small but consistent increase in the risk of developing cancer.
The findings, published in the journal Cancer, offer an example of how extreme population-level tragedies can have an impact on health.
Holocaust survivors were exposed to a variety of factors that have been linked with cancer.
So researchers set out to investigate whether the starvation, overcrowding, infectious diseases, and psychological stress that survivors endured might have contributed to the development of cancer in some individuals.
The team studied 152,622 Holocaust survivors who were followed for more than 45 years.
The researchers used 2 definitions of exposure to classify the survivors.
One definition was based on an individual’s entitlement for compensation for suffering persecution during the war. The other was based on the country of origin, dividing countries into those that were directly governed by Nazi Germany and those that were not occupied by Nazis.
The cancer incidence was significantly higher in survivors who were granted compensation than in those who were not—21.9% and 16.1%, respectively (P<0.0001).
However, the difference between survivors from occupied and non-occupied countries was not significant—22.7% and 21.4%, respectively.
On the other hand, when the researchers adjusted for confounding factors, survivors who had been exposed by either definition had a significantly increased risk of cancer.
For survivors who were granted compensation, the hazard ratio (HR) was 1.06 (P<0.001). For survivors born in occupied countries, the HR was 1.08 (P<0.001).
There was no increased risk of acute or chronic leukemia among patients who received compensation. And there was no increased risk of acute leukemia for survivors born in occupied countries.
However, there was a significantly increased risk of chronic leukemia among survivors born in occupied countries (HR=1.33, P=0.001)
“The data emphasize the importance of learning about the combined effect of several exposures occurring intensely and contemporaneously on cancer risk, such as those that unfortunately occurred during World War II,” said study author Siegal Sadetzki, MD, of the Chaim Sheba Medical Center in Tel HaShomer, Israel.
An editorial related to this study noted that the association between cancer and the extreme deprivation experienced by Holocaust survivors may also have parallels with other extreme population-level events, including in racial/ethnic minority groups experiencing severe social deprivation over time. ![]()
Inotuzumab ozogamicin approved for relapsed/refractory ALL
The Food and Drug Administration has approved the antibody drug conjugate inotuzumab ozogamicin for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The treatment, to be marketed by Pfizer as Besponsa, won approval based on the results of the INO-VATE ALL trial, which randomized 326 patients to receive either inotuzumab ozogamicin (164 patients) or a chemotherapy regimen of the investigator’s choice (162 patients). To be considered for inclusion in the trial, patients with Philadelphia chromosome–negative or –positive relapsed or refractory B-cell precursor ALL were required to have at least 5% bone marrow blasts and have received one or two induction chemotherapy regimens.
Adverse events that occurred in more than 20% of patients included thrombocytopenia, neutropenia, anemia, leukopenia, fatigue, hemorrhage, pyrexia, nausea, headache, febrile neutropenia, abdominal pain, and hyperbilirubinemia, as well as increases in gamma-glutamyltransferase and transaminases. Adverse events that led to discontinuation of treatment were infection, thrombocytopenia, hyperbilirubinemia, hemorrhage, and increases in transaminases.
Preliminary results were published in August 2016 (N Engl J Med. 2016;375:740-53).
Inotuzumab ozogamicin was granted orphan drug and breakthrough status, as well as priority review, by the FDA in February 2017.
[email protected]
On Twitter @denisefulton
The Food and Drug Administration has approved the antibody drug conjugate inotuzumab ozogamicin for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The treatment, to be marketed by Pfizer as Besponsa, won approval based on the results of the INO-VATE ALL trial, which randomized 326 patients to receive either inotuzumab ozogamicin (164 patients) or a chemotherapy regimen of the investigator’s choice (162 patients). To be considered for inclusion in the trial, patients with Philadelphia chromosome–negative or –positive relapsed or refractory B-cell precursor ALL were required to have at least 5% bone marrow blasts and have received one or two induction chemotherapy regimens.
Adverse events that occurred in more than 20% of patients included thrombocytopenia, neutropenia, anemia, leukopenia, fatigue, hemorrhage, pyrexia, nausea, headache, febrile neutropenia, abdominal pain, and hyperbilirubinemia, as well as increases in gamma-glutamyltransferase and transaminases. Adverse events that led to discontinuation of treatment were infection, thrombocytopenia, hyperbilirubinemia, hemorrhage, and increases in transaminases.
Preliminary results were published in August 2016 (N Engl J Med. 2016;375:740-53).
Inotuzumab ozogamicin was granted orphan drug and breakthrough status, as well as priority review, by the FDA in February 2017.
[email protected]
On Twitter @denisefulton
The Food and Drug Administration has approved the antibody drug conjugate inotuzumab ozogamicin for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The treatment, to be marketed by Pfizer as Besponsa, won approval based on the results of the INO-VATE ALL trial, which randomized 326 patients to receive either inotuzumab ozogamicin (164 patients) or a chemotherapy regimen of the investigator’s choice (162 patients). To be considered for inclusion in the trial, patients with Philadelphia chromosome–negative or –positive relapsed or refractory B-cell precursor ALL were required to have at least 5% bone marrow blasts and have received one or two induction chemotherapy regimens.
Adverse events that occurred in more than 20% of patients included thrombocytopenia, neutropenia, anemia, leukopenia, fatigue, hemorrhage, pyrexia, nausea, headache, febrile neutropenia, abdominal pain, and hyperbilirubinemia, as well as increases in gamma-glutamyltransferase and transaminases. Adverse events that led to discontinuation of treatment were infection, thrombocytopenia, hyperbilirubinemia, hemorrhage, and increases in transaminases.
Preliminary results were published in August 2016 (N Engl J Med. 2016;375:740-53).
Inotuzumab ozogamicin was granted orphan drug and breakthrough status, as well as priority review, by the FDA in February 2017.
[email protected]
On Twitter @denisefulton
Post-approval trials for accelerated drugs fall short

New research has revealed shortcomings of post-approval studies for drugs granted accelerated approval in the US.
Researchers found that, for drugs granted accelerated approval from 2009 to 2013, both pre-approval and post-approval trials had limitations in their design and the endpoints used.
“One might expect accelerated approval confirmatory trials to be much more rigorous than the pre-approval trials,” said study author Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts.
“But we found that there were few differences in these key design features of the trials conducted before or after approval.”
Dr Kesselheim and his colleagues reported these findings in JAMA.
The researchers examined pre- and post-approval clinical trials of drugs granted accelerated approval by the US Food and Drug Administration (FDA) between 2009 and 2013.
During that time, the FDA granted 22 drugs accelerated approval for 24 indications (15 of them for hematologic disorders).
Fourteen of the indications were approved on the basis of single-intervention-group studies that enrolled a median of 132 patients.
The FDA ordered 38 post-approval studies to confirm the safety and efficacy of the drugs.
Three years after the last drug’s approval, half of those studies (n=19) were not complete. Eight (42%) of the incomplete studies were either terminated or delayed by more than 1 year.
For 14 of the 24 indications (58%), results from the post-approval studies were not available after a median of 5 years of follow-up.
Study comparison
Published reports were available for 18 of the 19 completed post-approval studies. The characteristics of these studies did not differ much from the 30 pre-approval studies.
There were no statistically significant differences with regard to median patient enrollment (P=0.17), the use of randomized (P=0.31) or double-blind trials (P=0.17), the use of placebo as a comparator (P=0.17), or the lack of a comparator (P=0.21).
However, there was a significant difference in the use of an active comparator (P=0.02), with more post-approval studies using an active comparator.
The researchers also found that 17 of the 18 post-approval trials still used surrogate measures of effect as primary endpoints.
There was no significant difference between pre- and post-approval trials when it came to the use of disease response (P=0.17) or most other surrogate measures (P=0.21) as the trials’ primary endpoint.
The same was true for overall survival (P=0.20), although significantly more post-approval studies used progression-free survival (P=0.001) as a primary endpoint.
“It is important to use clinical endpoints in testing investigational drugs whenever possible because there are numerous cases of drugs approved on the basis of a surrogate measure that turn out to later not effect actual clinical outcomes—or even make them worse,” Dr Kesselheim said.
To address these issues and improve the quality of confirmatory studies, Dr Kesselheim suggested the FDA clearly describe the limitations in the pre-approval data that will need to be addressed in post-approval studies.
He also suggested the agency work with manufacturers to ensure that post-approval studies are conducted using design features that will be optimally useful for confirming the efficacy of the drug. ![]()
New research has revealed shortcomings of post-approval studies for drugs granted accelerated approval in the US.
Researchers found that, for drugs granted accelerated approval from 2009 to 2013, both pre-approval and post-approval trials had limitations in their design and the endpoints used.
“One might expect accelerated approval confirmatory trials to be much more rigorous than the pre-approval trials,” said study author Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts.
“But we found that there were few differences in these key design features of the trials conducted before or after approval.”
Dr Kesselheim and his colleagues reported these findings in JAMA.
The researchers examined pre- and post-approval clinical trials of drugs granted accelerated approval by the US Food and Drug Administration (FDA) between 2009 and 2013.
During that time, the FDA granted 22 drugs accelerated approval for 24 indications (15 of them for hematologic disorders).
Fourteen of the indications were approved on the basis of single-intervention-group studies that enrolled a median of 132 patients.
The FDA ordered 38 post-approval studies to confirm the safety and efficacy of the drugs.
Three years after the last drug’s approval, half of those studies (n=19) were not complete. Eight (42%) of the incomplete studies were either terminated or delayed by more than 1 year.
For 14 of the 24 indications (58%), results from the post-approval studies were not available after a median of 5 years of follow-up.
Study comparison
Published reports were available for 18 of the 19 completed post-approval studies. The characteristics of these studies did not differ much from the 30 pre-approval studies.
There were no statistically significant differences with regard to median patient enrollment (P=0.17), the use of randomized (P=0.31) or double-blind trials (P=0.17), the use of placebo as a comparator (P=0.17), or the lack of a comparator (P=0.21).
However, there was a significant difference in the use of an active comparator (P=0.02), with more post-approval studies using an active comparator.
The researchers also found that 17 of the 18 post-approval trials still used surrogate measures of effect as primary endpoints.
There was no significant difference between pre- and post-approval trials when it came to the use of disease response (P=0.17) or most other surrogate measures (P=0.21) as the trials’ primary endpoint.
The same was true for overall survival (P=0.20), although significantly more post-approval studies used progression-free survival (P=0.001) as a primary endpoint.
“It is important to use clinical endpoints in testing investigational drugs whenever possible because there are numerous cases of drugs approved on the basis of a surrogate measure that turn out to later not effect actual clinical outcomes—or even make them worse,” Dr Kesselheim said.
To address these issues and improve the quality of confirmatory studies, Dr Kesselheim suggested the FDA clearly describe the limitations in the pre-approval data that will need to be addressed in post-approval studies.
He also suggested the agency work with manufacturers to ensure that post-approval studies are conducted using design features that will be optimally useful for confirming the efficacy of the drug. ![]()
New research has revealed shortcomings of post-approval studies for drugs granted accelerated approval in the US.
Researchers found that, for drugs granted accelerated approval from 2009 to 2013, both pre-approval and post-approval trials had limitations in their design and the endpoints used.
“One might expect accelerated approval confirmatory trials to be much more rigorous than the pre-approval trials,” said study author Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts.
“But we found that there were few differences in these key design features of the trials conducted before or after approval.”
Dr Kesselheim and his colleagues reported these findings in JAMA.
The researchers examined pre- and post-approval clinical trials of drugs granted accelerated approval by the US Food and Drug Administration (FDA) between 2009 and 2013.
During that time, the FDA granted 22 drugs accelerated approval for 24 indications (15 of them for hematologic disorders).
Fourteen of the indications were approved on the basis of single-intervention-group studies that enrolled a median of 132 patients.
The FDA ordered 38 post-approval studies to confirm the safety and efficacy of the drugs.
Three years after the last drug’s approval, half of those studies (n=19) were not complete. Eight (42%) of the incomplete studies were either terminated or delayed by more than 1 year.
For 14 of the 24 indications (58%), results from the post-approval studies were not available after a median of 5 years of follow-up.
Study comparison
Published reports were available for 18 of the 19 completed post-approval studies. The characteristics of these studies did not differ much from the 30 pre-approval studies.
There were no statistically significant differences with regard to median patient enrollment (P=0.17), the use of randomized (P=0.31) or double-blind trials (P=0.17), the use of placebo as a comparator (P=0.17), or the lack of a comparator (P=0.21).
However, there was a significant difference in the use of an active comparator (P=0.02), with more post-approval studies using an active comparator.
The researchers also found that 17 of the 18 post-approval trials still used surrogate measures of effect as primary endpoints.
There was no significant difference between pre- and post-approval trials when it came to the use of disease response (P=0.17) or most other surrogate measures (P=0.21) as the trials’ primary endpoint.
The same was true for overall survival (P=0.20), although significantly more post-approval studies used progression-free survival (P=0.001) as a primary endpoint.
“It is important to use clinical endpoints in testing investigational drugs whenever possible because there are numerous cases of drugs approved on the basis of a surrogate measure that turn out to later not effect actual clinical outcomes—or even make them worse,” Dr Kesselheim said.
To address these issues and improve the quality of confirmatory studies, Dr Kesselheim suggested the FDA clearly describe the limitations in the pre-approval data that will need to be addressed in post-approval studies.
He also suggested the agency work with manufacturers to ensure that post-approval studies are conducted using design features that will be optimally useful for confirming the efficacy of the drug. ![]()
FDA grants drug orphan designation for AML, CMML
The US Food and Drug Administration (FDA) has granted orphan drug designation to H3B-8800 as a treatment for patients with acute myelogenous leukemia (AML) or chronic myelomonocytic leukemia (CMML).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
About H3B-8800
H3B-8800 is an orally bioavailable small-molecule modulator of wild-type and mutant SF3b complexes. The SF3b complex is a key component of the spliceosome that is found in the cell nucleus and is responsible for the removal of noncoding introns from a transcribed pre-messenger RNA.
Recurrent heterozygous mutations in several core members of the spliceosome (SF3B1, U2AF1, SRSF2, and ZRSR2) have been identified in solid tumor and hematologic malignancies. Mutations in the core spliceosome components lead to aberrant mRNA splicing that may contribute to disease pathogenesis.
Preclinical data have suggested that H3B-8800 modulates RNA splicing and shows preferential antitumor activity in spliceosome-mutant cancer models, including models of AML and CMML. H3B-8800 showed dose-dependent modulation of canonical and aberrant splicing when administered at tolerated doses.
Results from this research were presented at the 2017 AACR Annual Meeting (abstract 1185).
H3B-8800 is currently under investigation in a phase 1 trial of patients with AML, CMML, and myelodysplastic syndromes that may carry mutations in the core spliceosome genes.
H3B-8800 is being developed by H3 Biomedicine Inc. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to H3B-8800 as a treatment for patients with acute myelogenous leukemia (AML) or chronic myelomonocytic leukemia (CMML).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
About H3B-8800
H3B-8800 is an orally bioavailable small-molecule modulator of wild-type and mutant SF3b complexes. The SF3b complex is a key component of the spliceosome that is found in the cell nucleus and is responsible for the removal of noncoding introns from a transcribed pre-messenger RNA.
Recurrent heterozygous mutations in several core members of the spliceosome (SF3B1, U2AF1, SRSF2, and ZRSR2) have been identified in solid tumor and hematologic malignancies. Mutations in the core spliceosome components lead to aberrant mRNA splicing that may contribute to disease pathogenesis.
Preclinical data have suggested that H3B-8800 modulates RNA splicing and shows preferential antitumor activity in spliceosome-mutant cancer models, including models of AML and CMML. H3B-8800 showed dose-dependent modulation of canonical and aberrant splicing when administered at tolerated doses.
Results from this research were presented at the 2017 AACR Annual Meeting (abstract 1185).
H3B-8800 is currently under investigation in a phase 1 trial of patients with AML, CMML, and myelodysplastic syndromes that may carry mutations in the core spliceosome genes.
H3B-8800 is being developed by H3 Biomedicine Inc. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to H3B-8800 as a treatment for patients with acute myelogenous leukemia (AML) or chronic myelomonocytic leukemia (CMML).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
About H3B-8800
H3B-8800 is an orally bioavailable small-molecule modulator of wild-type and mutant SF3b complexes. The SF3b complex is a key component of the spliceosome that is found in the cell nucleus and is responsible for the removal of noncoding introns from a transcribed pre-messenger RNA.
Recurrent heterozygous mutations in several core members of the spliceosome (SF3B1, U2AF1, SRSF2, and ZRSR2) have been identified in solid tumor and hematologic malignancies. Mutations in the core spliceosome components lead to aberrant mRNA splicing that may contribute to disease pathogenesis.
Preclinical data have suggested that H3B-8800 modulates RNA splicing and shows preferential antitumor activity in spliceosome-mutant cancer models, including models of AML and CMML. H3B-8800 showed dose-dependent modulation of canonical and aberrant splicing when administered at tolerated doses.
Results from this research were presented at the 2017 AACR Annual Meeting (abstract 1185).
H3B-8800 is currently under investigation in a phase 1 trial of patients with AML, CMML, and myelodysplastic syndromes that may carry mutations in the core spliceosome genes.
H3B-8800 is being developed by H3 Biomedicine Inc. ![]()
Popular theory of mast cell development is wrong, team says
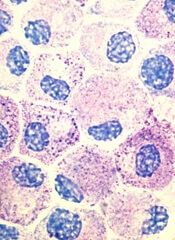
Stem cell factor (SCF) and KIT signaling are not necessary for early mast cell development, according to research published in Blood.
It has been assumed that the differentiation of hematopoietic progenitors to mast cells requires SCF and KIT signaling.
However, researchers found that mast cell progenitors can survive, mature, and proliferate in the absence of SCF and KIT signaling.
The researchers began this work by analyzing mast cell progenitor populations in samples from healthy subjects, patients with chronic myeloid leukemia (CML) or gastrointestinal stromal tumors (GIST) who were treated with imatinib, and patients with systemic mastocytosis carrying the D816V KIT mutation.
Imatinib inhibits KIT signaling, and the D816V KIT mutation causes KIT signaling to be constitutively active.
The researchers found the imatinib-treated CML and GIST patients and the patients with systemic mastocytosis all had mast cell progenitor populations similar to those observed in healthy subjects.
The team therefore concluded that dysfunctional KIT signaling does not affect the frequency of circulating mast cell progenitors in vivo.
On the other hand, the researchers also found that circulating mast cells were sensitive to imatinib in patients with CML. The patients had higher numbers of peripheral blood mast cells at diagnosis than they did after treatment with imatinib.
“When the patients were treated with the drug imatinib, which blocks the effect of stem cell factor, the number of mature mast cells dropped, while the number of progenitor cells did not change,” said study author Gunnar Nilsson, PhD, of Karolinska Institutet in Stockholm, Sweden.
Subsequent experiments showed that mast cell progenitors can survive in vitro without KIT signaling and without SCF. In addition, mast cell progenitors were able to mature and proliferate in vitro without SCF.
In fact, the researchers said they found that interleukin 3 was sufficient to promote the survival of mast cell progenitors in vitro.
“The study increases our understanding of how mast cells are formed and could be important in the development of new therapies, for example, for mastocytosis . . . ,” said study author Joakim Dahlin, PhD, of the University of Cambridge in the UK.
“One hypothesis that we will now test is whether interleukin 3 can be a new target in the treatment of mast cell-driven diseases.” ![]()
Stem cell factor (SCF) and KIT signaling are not necessary for early mast cell development, according to research published in Blood.
It has been assumed that the differentiation of hematopoietic progenitors to mast cells requires SCF and KIT signaling.
However, researchers found that mast cell progenitors can survive, mature, and proliferate in the absence of SCF and KIT signaling.
The researchers began this work by analyzing mast cell progenitor populations in samples from healthy subjects, patients with chronic myeloid leukemia (CML) or gastrointestinal stromal tumors (GIST) who were treated with imatinib, and patients with systemic mastocytosis carrying the D816V KIT mutation.
Imatinib inhibits KIT signaling, and the D816V KIT mutation causes KIT signaling to be constitutively active.
The researchers found the imatinib-treated CML and GIST patients and the patients with systemic mastocytosis all had mast cell progenitor populations similar to those observed in healthy subjects.
The team therefore concluded that dysfunctional KIT signaling does not affect the frequency of circulating mast cell progenitors in vivo.
On the other hand, the researchers also found that circulating mast cells were sensitive to imatinib in patients with CML. The patients had higher numbers of peripheral blood mast cells at diagnosis than they did after treatment with imatinib.
“When the patients were treated with the drug imatinib, which blocks the effect of stem cell factor, the number of mature mast cells dropped, while the number of progenitor cells did not change,” said study author Gunnar Nilsson, PhD, of Karolinska Institutet in Stockholm, Sweden.
Subsequent experiments showed that mast cell progenitors can survive in vitro without KIT signaling and without SCF. In addition, mast cell progenitors were able to mature and proliferate in vitro without SCF.
In fact, the researchers said they found that interleukin 3 was sufficient to promote the survival of mast cell progenitors in vitro.
“The study increases our understanding of how mast cells are formed and could be important in the development of new therapies, for example, for mastocytosis . . . ,” said study author Joakim Dahlin, PhD, of the University of Cambridge in the UK.
“One hypothesis that we will now test is whether interleukin 3 can be a new target in the treatment of mast cell-driven diseases.” ![]()
Stem cell factor (SCF) and KIT signaling are not necessary for early mast cell development, according to research published in Blood.
It has been assumed that the differentiation of hematopoietic progenitors to mast cells requires SCF and KIT signaling.
However, researchers found that mast cell progenitors can survive, mature, and proliferate in the absence of SCF and KIT signaling.
The researchers began this work by analyzing mast cell progenitor populations in samples from healthy subjects, patients with chronic myeloid leukemia (CML) or gastrointestinal stromal tumors (GIST) who were treated with imatinib, and patients with systemic mastocytosis carrying the D816V KIT mutation.
Imatinib inhibits KIT signaling, and the D816V KIT mutation causes KIT signaling to be constitutively active.
The researchers found the imatinib-treated CML and GIST patients and the patients with systemic mastocytosis all had mast cell progenitor populations similar to those observed in healthy subjects.
The team therefore concluded that dysfunctional KIT signaling does not affect the frequency of circulating mast cell progenitors in vivo.
On the other hand, the researchers also found that circulating mast cells were sensitive to imatinib in patients with CML. The patients had higher numbers of peripheral blood mast cells at diagnosis than they did after treatment with imatinib.
“When the patients were treated with the drug imatinib, which blocks the effect of stem cell factor, the number of mature mast cells dropped, while the number of progenitor cells did not change,” said study author Gunnar Nilsson, PhD, of Karolinska Institutet in Stockholm, Sweden.
Subsequent experiments showed that mast cell progenitors can survive in vitro without KIT signaling and without SCF. In addition, mast cell progenitors were able to mature and proliferate in vitro without SCF.
In fact, the researchers said they found that interleukin 3 was sufficient to promote the survival of mast cell progenitors in vitro.
“The study increases our understanding of how mast cells are formed and could be important in the development of new therapies, for example, for mastocytosis . . . ,” said study author Joakim Dahlin, PhD, of the University of Cambridge in the UK.
“One hypothesis that we will now test is whether interleukin 3 can be a new target in the treatment of mast cell-driven diseases.”
Cancer patients perceive their abilities differently than caregivers do

New research suggests older cancer patients and their caregivers often differ in their assessment of the patients’ abilities.
In this study, patients generally rated their physical and mental function higher than caregivers did.
The study also showed the differences in assessment of patients’ physical abilities were associated with greater caregiver burden.
This research was published in The Oncologist.
“Caregivers are such an important part of our healthcare system, particularly for older adults with cancer,” said study author Arti Hurria, MD, of City of Hope National Medical Center in Duarte, California.
“We wanted to further understand the factors that are associated with caregiver burden.”
One factor Dr Hurria and her colleagues thought might be important is differences in assessments of patient health and physical abilities between patients and their caregivers.
“In daily practice, we sometimes see a disconnect between what the patient perceives their general health and abilities to be in comparison to what the caregiver thinks,” Dr Hurria said. “We wanted to see whether this disconnect impacted caregiver burden.”
To do this, Dr Hurria and her colleagues questioned 100 older cancer patients and their caregivers.
Subjects were asked about the patients’ general health and physical function, meaning their ability to perform everyday activities. The researchers then compared the answers given by the patients and their respective caregivers.
The researchers also assessed the level of caregiver burden (defined as a subjective feeling of stress caused by being overwhelmed by the demands of caring) by administering a standard questionnaire on topics such as sleep disturbance, physical effort, and patient behavior.
The 100 cancer patients, ages 65 to 91, were suffering from a variety of cancers. The most common were lymphoma (n=26), breast cancer (n=19), and gastrointestinal cancers (n=15). Twelve patients had leukemia, and 10 had myeloma.
The ages of the caregivers ranged from 28 to 85, and the majority were female (73%). They were mainly either the spouse of the patient (68%) or an adult child (18%).
Results
There was no significant difference in patient and caregiver accounts of the patients’ comorbidities (P=0.68), falls in the last 6 months (P=0.71), or percent weight change in the last 6 months (P=0.21).
However, caregivers consistently rated patients as having poorer physical function and mental health and requiring more social support than the patients themselves did.
There was a significant difference (P<0.05) in caregiver and patient accounts when it came to the following measures:
- Need for help with instrumental activities of daily living
- Karnofsky Performance Status
- Medical Outcomes Study-Physical Function
- Medical Outcomes Study-Social Support Survey
- Mental Health Inventory.
Only the disparity in the assessment of physical function was significantly associated with greater caregiver burden (P<0.001). What is still unclear is the cause of this disparity.
“I think there are 2 possible explanations,” said study author Tina Hsu, MD, of the University of Ottawa in Ontario, Canada.
“One is that older adults with cancer either don’t appreciate how much help they require or, more likely, they are able preserve their sense of independence and dignity through a perception that they feel they can do more than they really can.”
“Alternatively, it is possible that caregivers who are more stressed out perceive their loved one to require more help than they actually do need. Most likely, the truth of how much help the patient actually needs lies somewhere between what patients and caregivers report.”
Based on their findings, Drs Hsu and Hurria and their colleagues advise that clinicians consider assessing caregiver burden in those caregivers who report the patient as being more dependent than the patient does themselves.
“Caregivers play an essential role in supporting older adults with cancer,” Dr Hsu said. “We plan to further explore factors associated with caregiver burden in this population, particularly in those who are frailer and thus require even more hands-on support. We also hope to explore what resources caregivers of older adults with cancer feel they need to better help them with their role.”
New research suggests older cancer patients and their caregivers often differ in their assessment of the patients’ abilities.
In this study, patients generally rated their physical and mental function higher than caregivers did.
The study also showed the differences in assessment of patients’ physical abilities were associated with greater caregiver burden.
This research was published in The Oncologist.
“Caregivers are such an important part of our healthcare system, particularly for older adults with cancer,” said study author Arti Hurria, MD, of City of Hope National Medical Center in Duarte, California.
“We wanted to further understand the factors that are associated with caregiver burden.”
One factor Dr Hurria and her colleagues thought might be important is differences in assessments of patient health and physical abilities between patients and their caregivers.
“In daily practice, we sometimes see a disconnect between what the patient perceives their general health and abilities to be in comparison to what the caregiver thinks,” Dr Hurria said. “We wanted to see whether this disconnect impacted caregiver burden.”
To do this, Dr Hurria and her colleagues questioned 100 older cancer patients and their caregivers.
Subjects were asked about the patients’ general health and physical function, meaning their ability to perform everyday activities. The researchers then compared the answers given by the patients and their respective caregivers.
The researchers also assessed the level of caregiver burden (defined as a subjective feeling of stress caused by being overwhelmed by the demands of caring) by administering a standard questionnaire on topics such as sleep disturbance, physical effort, and patient behavior.
The 100 cancer patients, ages 65 to 91, were suffering from a variety of cancers. The most common were lymphoma (n=26), breast cancer (n=19), and gastrointestinal cancers (n=15). Twelve patients had leukemia, and 10 had myeloma.
The ages of the caregivers ranged from 28 to 85, and the majority were female (73%). They were mainly either the spouse of the patient (68%) or an adult child (18%).
Results
There was no significant difference in patient and caregiver accounts of the patients’ comorbidities (P=0.68), falls in the last 6 months (P=0.71), or percent weight change in the last 6 months (P=0.21).
However, caregivers consistently rated patients as having poorer physical function and mental health and requiring more social support than the patients themselves did.
There was a significant difference (P<0.05) in caregiver and patient accounts when it came to the following measures:
- Need for help with instrumental activities of daily living
- Karnofsky Performance Status
- Medical Outcomes Study-Physical Function
- Medical Outcomes Study-Social Support Survey
- Mental Health Inventory.
Only the disparity in the assessment of physical function was significantly associated with greater caregiver burden (P<0.001). What is still unclear is the cause of this disparity.
“I think there are 2 possible explanations,” said study author Tina Hsu, MD, of the University of Ottawa in Ontario, Canada.
“One is that older adults with cancer either don’t appreciate how much help they require or, more likely, they are able preserve their sense of independence and dignity through a perception that they feel they can do more than they really can.”
“Alternatively, it is possible that caregivers who are more stressed out perceive their loved one to require more help than they actually do need. Most likely, the truth of how much help the patient actually needs lies somewhere between what patients and caregivers report.”
Based on their findings, Drs Hsu and Hurria and their colleagues advise that clinicians consider assessing caregiver burden in those caregivers who report the patient as being more dependent than the patient does themselves.
“Caregivers play an essential role in supporting older adults with cancer,” Dr Hsu said. “We plan to further explore factors associated with caregiver burden in this population, particularly in those who are frailer and thus require even more hands-on support. We also hope to explore what resources caregivers of older adults with cancer feel they need to better help them with their role.”
New research suggests older cancer patients and their caregivers often differ in their assessment of the patients’ abilities.
In this study, patients generally rated their physical and mental function higher than caregivers did.
The study also showed the differences in assessment of patients’ physical abilities were associated with greater caregiver burden.
This research was published in The Oncologist.
“Caregivers are such an important part of our healthcare system, particularly for older adults with cancer,” said study author Arti Hurria, MD, of City of Hope National Medical Center in Duarte, California.
“We wanted to further understand the factors that are associated with caregiver burden.”
One factor Dr Hurria and her colleagues thought might be important is differences in assessments of patient health and physical abilities between patients and their caregivers.
“In daily practice, we sometimes see a disconnect between what the patient perceives their general health and abilities to be in comparison to what the caregiver thinks,” Dr Hurria said. “We wanted to see whether this disconnect impacted caregiver burden.”
To do this, Dr Hurria and her colleagues questioned 100 older cancer patients and their caregivers.
Subjects were asked about the patients’ general health and physical function, meaning their ability to perform everyday activities. The researchers then compared the answers given by the patients and their respective caregivers.
The researchers also assessed the level of caregiver burden (defined as a subjective feeling of stress caused by being overwhelmed by the demands of caring) by administering a standard questionnaire on topics such as sleep disturbance, physical effort, and patient behavior.
The 100 cancer patients, ages 65 to 91, were suffering from a variety of cancers. The most common were lymphoma (n=26), breast cancer (n=19), and gastrointestinal cancers (n=15). Twelve patients had leukemia, and 10 had myeloma.
The ages of the caregivers ranged from 28 to 85, and the majority were female (73%). They were mainly either the spouse of the patient (68%) or an adult child (18%).
Results
There was no significant difference in patient and caregiver accounts of the patients’ comorbidities (P=0.68), falls in the last 6 months (P=0.71), or percent weight change in the last 6 months (P=0.21).
However, caregivers consistently rated patients as having poorer physical function and mental health and requiring more social support than the patients themselves did.
There was a significant difference (P<0.05) in caregiver and patient accounts when it came to the following measures:
- Need for help with instrumental activities of daily living
- Karnofsky Performance Status
- Medical Outcomes Study-Physical Function
- Medical Outcomes Study-Social Support Survey
- Mental Health Inventory.
Only the disparity in the assessment of physical function was significantly associated with greater caregiver burden (P<0.001). What is still unclear is the cause of this disparity.
“I think there are 2 possible explanations,” said study author Tina Hsu, MD, of the University of Ottawa in Ontario, Canada.
“One is that older adults with cancer either don’t appreciate how much help they require or, more likely, they are able preserve their sense of independence and dignity through a perception that they feel they can do more than they really can.”
“Alternatively, it is possible that caregivers who are more stressed out perceive their loved one to require more help than they actually do need. Most likely, the truth of how much help the patient actually needs lies somewhere between what patients and caregivers report.”
Based on their findings, Drs Hsu and Hurria and their colleagues advise that clinicians consider assessing caregiver burden in those caregivers who report the patient as being more dependent than the patient does themselves.
“Caregivers play an essential role in supporting older adults with cancer,” Dr Hsu said. “We plan to further explore factors associated with caregiver burden in this population, particularly in those who are frailer and thus require even more hands-on support. We also hope to explore what resources caregivers of older adults with cancer feel they need to better help them with their role.”