User login
Drug gets priority review as CLL treatment

Image by Mary Ann Thompson
Despite previous safety concerns, the US Food and Drug Administration (FDA) has granted priority review for the BCL-2 inhibitor venetoclax.
The FDA is reviewing the drug as a potential treatment for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least 1 prior therapy.
A priority review designation is granted to drugs thought to have the potential to provide significant improvements in the treatment, prevention, or diagnosis of a disease.
The designation means the FDA’s goal is to take action on a drug application within 6 months, compared to 10 months under standard review.
Venetoclax has proven active against CLL and other hematologic malignancies, but it is known to induce tumor lysis syndrome (TLS). In fact, TLS-related deaths temporarily halted enrollment in trials of venetoclax. But researchers discovered ways to reduce the risk of TLS, and the trials continued.
Venetoclax received breakthrough therapy designation from the FDA last year for the treatment of patients with relapsed or refractory CLL and 17p deletion. This designation is designed to expedite the development and review of medicines intended to treat serious or life-threatening diseases.
The new drug application for venetoclax is based, in part, on data from the phase 2 M13-982 study, which were just presented at the 2015 ASH Annual Meeting.
Phase 2 trial
M13-982 is an open-label, single-arm, multicenter study in which researchers are evaluating the efficacy and safety of venetoclax in patients with relapsed, refractory, or previously untreated CLL with 17p deletion.
The study included 107 patients with relapsed or refractory disease, and all but 1 had 17p deletion. An additional 50 patients with relapsed, refractory, or previously untreated disease have been enrolled in the safety expansion cohort.
The primary endpoint of the study is overall response rate as determined by an independent review committee, and secondary endpoints include complete response, partial response, duration of response, progression-free survival, and overall survival. The level of minimal residual disease (MRD) in peripheral blood and/or bone marrow was assessed in a subset of patients.
The study met its primary endpoint, with an overall response rate of 79.4% among the 107 patients with relapsed or refractory disease. In addition, 7.5% of patients achieved a complete response, with or without complete recovery of blood counts in the bone marrow.
Forty-five patients had an assessment for MRD in the blood. Of these, 18 patients achieved MRD-negativity. Ten of these 18 patients also had bone marrow assessments, and 6 were MRD-negative.
At 1 year, 84.7% of all responses and 94.4% of MRD-negative responses were maintained. The 1-year progression-free survival and overall survival rates were 72% and 86.7%, respectively.
The most common serious adverse events were pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%). The most common grade 3-4 adverse events were neutropenia (40%), infection (20%), anemia (18%), and thrombocytopenia (15%).
Laboratory TLS was reported in 5 patients. None had clinical consequences.
Venetoclax is under development by AbbVie and Genentech/Roche. ![]()
Image by Mary Ann Thompson
Despite previous safety concerns, the US Food and Drug Administration (FDA) has granted priority review for the BCL-2 inhibitor venetoclax.
The FDA is reviewing the drug as a potential treatment for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least 1 prior therapy.
A priority review designation is granted to drugs thought to have the potential to provide significant improvements in the treatment, prevention, or diagnosis of a disease.
The designation means the FDA’s goal is to take action on a drug application within 6 months, compared to 10 months under standard review.
Venetoclax has proven active against CLL and other hematologic malignancies, but it is known to induce tumor lysis syndrome (TLS). In fact, TLS-related deaths temporarily halted enrollment in trials of venetoclax. But researchers discovered ways to reduce the risk of TLS, and the trials continued.
Venetoclax received breakthrough therapy designation from the FDA last year for the treatment of patients with relapsed or refractory CLL and 17p deletion. This designation is designed to expedite the development and review of medicines intended to treat serious or life-threatening diseases.
The new drug application for venetoclax is based, in part, on data from the phase 2 M13-982 study, which were just presented at the 2015 ASH Annual Meeting.
Phase 2 trial
M13-982 is an open-label, single-arm, multicenter study in which researchers are evaluating the efficacy and safety of venetoclax in patients with relapsed, refractory, or previously untreated CLL with 17p deletion.
The study included 107 patients with relapsed or refractory disease, and all but 1 had 17p deletion. An additional 50 patients with relapsed, refractory, or previously untreated disease have been enrolled in the safety expansion cohort.
The primary endpoint of the study is overall response rate as determined by an independent review committee, and secondary endpoints include complete response, partial response, duration of response, progression-free survival, and overall survival. The level of minimal residual disease (MRD) in peripheral blood and/or bone marrow was assessed in a subset of patients.
The study met its primary endpoint, with an overall response rate of 79.4% among the 107 patients with relapsed or refractory disease. In addition, 7.5% of patients achieved a complete response, with or without complete recovery of blood counts in the bone marrow.
Forty-five patients had an assessment for MRD in the blood. Of these, 18 patients achieved MRD-negativity. Ten of these 18 patients also had bone marrow assessments, and 6 were MRD-negative.
At 1 year, 84.7% of all responses and 94.4% of MRD-negative responses were maintained. The 1-year progression-free survival and overall survival rates were 72% and 86.7%, respectively.
The most common serious adverse events were pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%). The most common grade 3-4 adverse events were neutropenia (40%), infection (20%), anemia (18%), and thrombocytopenia (15%).
Laboratory TLS was reported in 5 patients. None had clinical consequences.
Venetoclax is under development by AbbVie and Genentech/Roche. ![]()
Image by Mary Ann Thompson
Despite previous safety concerns, the US Food and Drug Administration (FDA) has granted priority review for the BCL-2 inhibitor venetoclax.
The FDA is reviewing the drug as a potential treatment for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least 1 prior therapy.
A priority review designation is granted to drugs thought to have the potential to provide significant improvements in the treatment, prevention, or diagnosis of a disease.
The designation means the FDA’s goal is to take action on a drug application within 6 months, compared to 10 months under standard review.
Venetoclax has proven active against CLL and other hematologic malignancies, but it is known to induce tumor lysis syndrome (TLS). In fact, TLS-related deaths temporarily halted enrollment in trials of venetoclax. But researchers discovered ways to reduce the risk of TLS, and the trials continued.
Venetoclax received breakthrough therapy designation from the FDA last year for the treatment of patients with relapsed or refractory CLL and 17p deletion. This designation is designed to expedite the development and review of medicines intended to treat serious or life-threatening diseases.
The new drug application for venetoclax is based, in part, on data from the phase 2 M13-982 study, which were just presented at the 2015 ASH Annual Meeting.
Phase 2 trial
M13-982 is an open-label, single-arm, multicenter study in which researchers are evaluating the efficacy and safety of venetoclax in patients with relapsed, refractory, or previously untreated CLL with 17p deletion.
The study included 107 patients with relapsed or refractory disease, and all but 1 had 17p deletion. An additional 50 patients with relapsed, refractory, or previously untreated disease have been enrolled in the safety expansion cohort.
The primary endpoint of the study is overall response rate as determined by an independent review committee, and secondary endpoints include complete response, partial response, duration of response, progression-free survival, and overall survival. The level of minimal residual disease (MRD) in peripheral blood and/or bone marrow was assessed in a subset of patients.
The study met its primary endpoint, with an overall response rate of 79.4% among the 107 patients with relapsed or refractory disease. In addition, 7.5% of patients achieved a complete response, with or without complete recovery of blood counts in the bone marrow.
Forty-five patients had an assessment for MRD in the blood. Of these, 18 patients achieved MRD-negativity. Ten of these 18 patients also had bone marrow assessments, and 6 were MRD-negative.
At 1 year, 84.7% of all responses and 94.4% of MRD-negative responses were maintained. The 1-year progression-free survival and overall survival rates were 72% and 86.7%, respectively.
The most common serious adverse events were pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%). The most common grade 3-4 adverse events were neutropenia (40%), infection (20%), anemia (18%), and thrombocytopenia (15%).
Laboratory TLS was reported in 5 patients. None had clinical consequences.
Venetoclax is under development by AbbVie and Genentech/Roche. ![]()
Increasing eligibility for engineered T-cell therapy

Image courtesy of NIAID
A new study suggests that having a certain type of cancer or receiving certain chemotherapeutic agents
can affect T-cell function and make patients ineligible for engineered T-cell therapy.
However, researchers found that proper timing of T-cell collection can increase the number of patients eiligible for the therapy.
And the team developed a culture technique that can boost T cells’ fitness for expansion, which can increase eligibility as well.
Nathan Singh, MD, of the University of Pennsylvania in Philadelphia, and his colleagues described this work in Science Translational Medicine.
The researchers set out to determine why some patients’ T cells fail to multiply in culture. The team studied T cells from children with acute lymphoblastic leukemia (ALL) or non-Hodgkin lymphoma (NHL) who were undergoing chemotherapy. (NHL subtypes included Burkitt lymphoma, diffuse large B-cell lymphoma, primary mediastinal large B-cell lymphoma, primary lymphoma of bone, and follicular lymphoma.)
The researchers found that T cells from patients with ALL expanded better in culture than those from patients with NHL.
The team said a threshold of greater than 5-fold expansion during test expansion was associated with a high likelihood of successful clinical expansion.
Nearly 80% of patients with ALL met this threshold at diagnosis, but the rate declined over the course of therapy, falling to about 40% during maintenance therapy.
About 25% of NHL patients met the threshold at diagnosis, but few samples demonstrated any expansion after therapy began (12.5% of samples at all remaining time points tested).
The researchers said the difference in the proportion of ALL and NHL samples that met the expansion threshold was significant at all time points tested.
Analysis revealed that ALL patients had higher numbers of naïve T cells and stem central memory T cells, T cell subtypes known to be highly potent and proliferative with an enhanced capacity for self-renewal.
The researchers also found that certain chemotherapy drugs—namely, cyclophosphamide and cytarabine—selectively depleted early lineage T cells.
Fortunately, the team discovered that poor expansion can be rescued by exposing T cells to signaling molecules that stimulate T-cell activity. Culture with IL-7 and IL-15 boosted the expansion capacity of T cells from patients with NHL and those with ALL.
The researchers therefore concluded that using this culture technique or collecting T cells prior to chemotherapy can increase the number of patients eligible for engineered T-cell therapy. ![]()
Image courtesy of NIAID
A new study suggests that having a certain type of cancer or receiving certain chemotherapeutic agents
can affect T-cell function and make patients ineligible for engineered T-cell therapy.
However, researchers found that proper timing of T-cell collection can increase the number of patients eiligible for the therapy.
And the team developed a culture technique that can boost T cells’ fitness for expansion, which can increase eligibility as well.
Nathan Singh, MD, of the University of Pennsylvania in Philadelphia, and his colleagues described this work in Science Translational Medicine.
The researchers set out to determine why some patients’ T cells fail to multiply in culture. The team studied T cells from children with acute lymphoblastic leukemia (ALL) or non-Hodgkin lymphoma (NHL) who were undergoing chemotherapy. (NHL subtypes included Burkitt lymphoma, diffuse large B-cell lymphoma, primary mediastinal large B-cell lymphoma, primary lymphoma of bone, and follicular lymphoma.)
The researchers found that T cells from patients with ALL expanded better in culture than those from patients with NHL.
The team said a threshold of greater than 5-fold expansion during test expansion was associated with a high likelihood of successful clinical expansion.
Nearly 80% of patients with ALL met this threshold at diagnosis, but the rate declined over the course of therapy, falling to about 40% during maintenance therapy.
About 25% of NHL patients met the threshold at diagnosis, but few samples demonstrated any expansion after therapy began (12.5% of samples at all remaining time points tested).
The researchers said the difference in the proportion of ALL and NHL samples that met the expansion threshold was significant at all time points tested.
Analysis revealed that ALL patients had higher numbers of naïve T cells and stem central memory T cells, T cell subtypes known to be highly potent and proliferative with an enhanced capacity for self-renewal.
The researchers also found that certain chemotherapy drugs—namely, cyclophosphamide and cytarabine—selectively depleted early lineage T cells.
Fortunately, the team discovered that poor expansion can be rescued by exposing T cells to signaling molecules that stimulate T-cell activity. Culture with IL-7 and IL-15 boosted the expansion capacity of T cells from patients with NHL and those with ALL.
The researchers therefore concluded that using this culture technique or collecting T cells prior to chemotherapy can increase the number of patients eligible for engineered T-cell therapy. ![]()
Image courtesy of NIAID
A new study suggests that having a certain type of cancer or receiving certain chemotherapeutic agents
can affect T-cell function and make patients ineligible for engineered T-cell therapy.
However, researchers found that proper timing of T-cell collection can increase the number of patients eiligible for the therapy.
And the team developed a culture technique that can boost T cells’ fitness for expansion, which can increase eligibility as well.
Nathan Singh, MD, of the University of Pennsylvania in Philadelphia, and his colleagues described this work in Science Translational Medicine.
The researchers set out to determine why some patients’ T cells fail to multiply in culture. The team studied T cells from children with acute lymphoblastic leukemia (ALL) or non-Hodgkin lymphoma (NHL) who were undergoing chemotherapy. (NHL subtypes included Burkitt lymphoma, diffuse large B-cell lymphoma, primary mediastinal large B-cell lymphoma, primary lymphoma of bone, and follicular lymphoma.)
The researchers found that T cells from patients with ALL expanded better in culture than those from patients with NHL.
The team said a threshold of greater than 5-fold expansion during test expansion was associated with a high likelihood of successful clinical expansion.
Nearly 80% of patients with ALL met this threshold at diagnosis, but the rate declined over the course of therapy, falling to about 40% during maintenance therapy.
About 25% of NHL patients met the threshold at diagnosis, but few samples demonstrated any expansion after therapy began (12.5% of samples at all remaining time points tested).
The researchers said the difference in the proportion of ALL and NHL samples that met the expansion threshold was significant at all time points tested.
Analysis revealed that ALL patients had higher numbers of naïve T cells and stem central memory T cells, T cell subtypes known to be highly potent and proliferative with an enhanced capacity for self-renewal.
The researchers also found that certain chemotherapy drugs—namely, cyclophosphamide and cytarabine—selectively depleted early lineage T cells.
Fortunately, the team discovered that poor expansion can be rescued by exposing T cells to signaling molecules that stimulate T-cell activity. Culture with IL-7 and IL-15 boosted the expansion capacity of T cells from patients with NHL and those with ALL.
The researchers therefore concluded that using this culture technique or collecting T cells prior to chemotherapy can increase the number of patients eligible for engineered T-cell therapy. ![]()
How microbes drive progression of CTCL
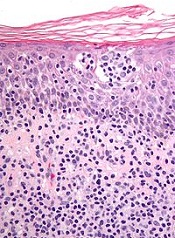
New research indicates that toxins in Staphylococcus bacteria help malignant cells gain control over healthy cells in patients with cutaneous T-cell lymphoma (CTCL).
Investigators found that staphylococcal enterotoxin-A (SEA) induces STAT3 activation and IL-17 expression in malignant T cells via engagement of non-malignant CD4 T cells.
As STAT3 activation has been implicated in CTCL pathogenesis, the discovery suggests bacterial toxins play a key role in activating an oncogenic pathway in CTCL.
“We have gained important insight into the processes that activate cancer cells and make them grow,” said Niels Oedum, MD, of the University of Copenhagen in Denmark.
“[CTCL] patients’ frequent bacterial infections might not be a mere side effect of the disease. On the contrary, toxins in the bacteria actually ‘benefit’ cancer cells. Our next step is examining whether combatting infections can slow down the growth of cancer cells and thus stop the disease.”
Dr Oedum and his colleagues described their research in Blood.
The investigators knew that, in CTCL, CD4 T cells become malignant and turn parasitic on the rest of the immune system. In addition to using healthy cells to do their work for them, the malignant cells slowly destroy the skin’s immune defense mechanism.
The team’s new discoveries indicate that bacterial toxins in some patients enable malignant cells to send off signals that obstruct and change the immune defense mechanism, which would otherwise fight the malignant cells. What was believed to be an overly active immune defense mechanism could, in other words, turn out to be a malignant infection brought on by bacteria, which only worsens the disease.
Dr Oedum and his colleagues found that SEA-positive bacteria isolatated from the skin of CTCL patients stimulated activation of STAT3 and upregulation of IL-17 in malignant and non-malignant T cells.
Malignant T cells expressing an SEA non-responsive T-cell receptor V beta chain did not respond to SEA when cultured alone but exhibited STAT3 activation and IL-17 expression in co-cultures with SEA-responsive, non-malignant T cells.
The investigators found evidence to suggest the response is induced via IL-2Rg cytokines and a JAK3-dependent pathway in malignant T cells. The JAK3 inhibitor tofacitinib inhibited SEA-induced IL-17 production in co-cultures of malignant and non-malignant T cells.
Dr Oedum and his colleagues plan to continue their work investigating how bacteria might affect the balance between the immune defense mechanism and the disease in patients with CTCL.
In the long-term, the investigators’ aim is to understand how bacteria and their toxins can worsen CTCL, knowledge that may be used to develop new targeted treatments.
As only some of the bacteria produce toxins, the team said it will also be important to develop methods to determine which patients may benefit from treatment with antibiotics. ![]()
New research indicates that toxins in Staphylococcus bacteria help malignant cells gain control over healthy cells in patients with cutaneous T-cell lymphoma (CTCL).
Investigators found that staphylococcal enterotoxin-A (SEA) induces STAT3 activation and IL-17 expression in malignant T cells via engagement of non-malignant CD4 T cells.
As STAT3 activation has been implicated in CTCL pathogenesis, the discovery suggests bacterial toxins play a key role in activating an oncogenic pathway in CTCL.
“We have gained important insight into the processes that activate cancer cells and make them grow,” said Niels Oedum, MD, of the University of Copenhagen in Denmark.
“[CTCL] patients’ frequent bacterial infections might not be a mere side effect of the disease. On the contrary, toxins in the bacteria actually ‘benefit’ cancer cells. Our next step is examining whether combatting infections can slow down the growth of cancer cells and thus stop the disease.”
Dr Oedum and his colleagues described their research in Blood.
The investigators knew that, in CTCL, CD4 T cells become malignant and turn parasitic on the rest of the immune system. In addition to using healthy cells to do their work for them, the malignant cells slowly destroy the skin’s immune defense mechanism.
The team’s new discoveries indicate that bacterial toxins in some patients enable malignant cells to send off signals that obstruct and change the immune defense mechanism, which would otherwise fight the malignant cells. What was believed to be an overly active immune defense mechanism could, in other words, turn out to be a malignant infection brought on by bacteria, which only worsens the disease.
Dr Oedum and his colleagues found that SEA-positive bacteria isolatated from the skin of CTCL patients stimulated activation of STAT3 and upregulation of IL-17 in malignant and non-malignant T cells.
Malignant T cells expressing an SEA non-responsive T-cell receptor V beta chain did not respond to SEA when cultured alone but exhibited STAT3 activation and IL-17 expression in co-cultures with SEA-responsive, non-malignant T cells.
The investigators found evidence to suggest the response is induced via IL-2Rg cytokines and a JAK3-dependent pathway in malignant T cells. The JAK3 inhibitor tofacitinib inhibited SEA-induced IL-17 production in co-cultures of malignant and non-malignant T cells.
Dr Oedum and his colleagues plan to continue their work investigating how bacteria might affect the balance between the immune defense mechanism and the disease in patients with CTCL.
In the long-term, the investigators’ aim is to understand how bacteria and their toxins can worsen CTCL, knowledge that may be used to develop new targeted treatments.
As only some of the bacteria produce toxins, the team said it will also be important to develop methods to determine which patients may benefit from treatment with antibiotics. ![]()
New research indicates that toxins in Staphylococcus bacteria help malignant cells gain control over healthy cells in patients with cutaneous T-cell lymphoma (CTCL).
Investigators found that staphylococcal enterotoxin-A (SEA) induces STAT3 activation and IL-17 expression in malignant T cells via engagement of non-malignant CD4 T cells.
As STAT3 activation has been implicated in CTCL pathogenesis, the discovery suggests bacterial toxins play a key role in activating an oncogenic pathway in CTCL.
“We have gained important insight into the processes that activate cancer cells and make them grow,” said Niels Oedum, MD, of the University of Copenhagen in Denmark.
“[CTCL] patients’ frequent bacterial infections might not be a mere side effect of the disease. On the contrary, toxins in the bacteria actually ‘benefit’ cancer cells. Our next step is examining whether combatting infections can slow down the growth of cancer cells and thus stop the disease.”
Dr Oedum and his colleagues described their research in Blood.
The investigators knew that, in CTCL, CD4 T cells become malignant and turn parasitic on the rest of the immune system. In addition to using healthy cells to do their work for them, the malignant cells slowly destroy the skin’s immune defense mechanism.
The team’s new discoveries indicate that bacterial toxins in some patients enable malignant cells to send off signals that obstruct and change the immune defense mechanism, which would otherwise fight the malignant cells. What was believed to be an overly active immune defense mechanism could, in other words, turn out to be a malignant infection brought on by bacteria, which only worsens the disease.
Dr Oedum and his colleagues found that SEA-positive bacteria isolatated from the skin of CTCL patients stimulated activation of STAT3 and upregulation of IL-17 in malignant and non-malignant T cells.
Malignant T cells expressing an SEA non-responsive T-cell receptor V beta chain did not respond to SEA when cultured alone but exhibited STAT3 activation and IL-17 expression in co-cultures with SEA-responsive, non-malignant T cells.
The investigators found evidence to suggest the response is induced via IL-2Rg cytokines and a JAK3-dependent pathway in malignant T cells. The JAK3 inhibitor tofacitinib inhibited SEA-induced IL-17 production in co-cultures of malignant and non-malignant T cells.
Dr Oedum and his colleagues plan to continue their work investigating how bacteria might affect the balance between the immune defense mechanism and the disease in patients with CTCL.
In the long-term, the investigators’ aim is to understand how bacteria and their toxins can worsen CTCL, knowledge that may be used to develop new targeted treatments.
As only some of the bacteria produce toxins, the team said it will also be important to develop methods to determine which patients may benefit from treatment with antibiotics. ![]()
Novel delivery system could treat MCL
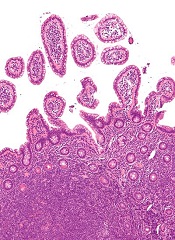
Researchers say they have developed a system for delivering therapy at the site of mantle cell lymphoma (MCL).
The system harnesses nanoparticles coated with “GPS” antibodies that navigate toward cancerous cells, where they offload cyclin D1-blockers in the form of small interfering RNAs (siRNAs).
Experiments showed the system can halt the proliferation of cyclin D1 in both animal models and samples from MCL patients.
Dan Peer, PhD, of Tel Aviv University in Israel, and his colleagues reported these results in PNAS.
“MCL has a genetic hallmark,” Dr Peer noted. “In 85% of cases, the characteristic that defines this aggressive and prototypic B-cell lymphoma is the heightened activity of the gene CCND1, which leads to the extreme overexpression—a 3000- to 15,000-fold increase—of cyclin D1, a protein that controls the proliferation of cells. Downregulation of cyclin D1 using siRNAs is a potential therapeutic approach to this malignancy.”
For this research, Dr Peer and his colleagues designed lipid-based nanoparticles (LNPs) coated with anti-CD38 monoclonal antibodies that were taken up by human MCL cells in the bone marrow of affected mice.
When loaded with siRNAs against cyclin D1, the targeting LNPs induced gene silencing in MCL cells and prolonged the survival of tumor-bearing mice, with no observed adverse effects.
“In MCL, cyclin D1 is the exclusive cause of the overproduction of B lymphocytes, the cells responsible for generating antibodies,” Dr Peer said. “This makes the protein a perfect target for RNA therapy by siRNAs.”
“Normal, healthy cells don’t express the gene, so therapies that destroy the gene will only attack cancer cells. The RNA interference we have developed targets the faulty cyclin D1 within the cancerous cells. And when the cells are inhibited from proliferating, they sense they are being targeted and begin to die off.”
Dr Peer and his colleagues believe this work presents new opportunities for treating MCL and other similar B-cell malignancies.
“This research makes a definite contribution to the revolution of personalized medicine, whereby you tailor the drug based on the genetic profile of patient,” Dr Peer said. “In this case, MCL is a disease with a specific genetic hallmark, so you can sequence the patient to identify the mutation(s) and design RNA blockers to be placed inside a nanovehicle.”
“While the targeting antibodies—the ‘GPS’—can be used to target many different B-cell malignancies, the drug itself is designed to silence this specific disease. However, the delivery system can be used to accommodate any disease with a genetic profile. This could be the future. We are seeing it happen before our very eyes.” ![]()
Researchers say they have developed a system for delivering therapy at the site of mantle cell lymphoma (MCL).
The system harnesses nanoparticles coated with “GPS” antibodies that navigate toward cancerous cells, where they offload cyclin D1-blockers in the form of small interfering RNAs (siRNAs).
Experiments showed the system can halt the proliferation of cyclin D1 in both animal models and samples from MCL patients.
Dan Peer, PhD, of Tel Aviv University in Israel, and his colleagues reported these results in PNAS.
“MCL has a genetic hallmark,” Dr Peer noted. “In 85% of cases, the characteristic that defines this aggressive and prototypic B-cell lymphoma is the heightened activity of the gene CCND1, which leads to the extreme overexpression—a 3000- to 15,000-fold increase—of cyclin D1, a protein that controls the proliferation of cells. Downregulation of cyclin D1 using siRNAs is a potential therapeutic approach to this malignancy.”
For this research, Dr Peer and his colleagues designed lipid-based nanoparticles (LNPs) coated with anti-CD38 monoclonal antibodies that were taken up by human MCL cells in the bone marrow of affected mice.
When loaded with siRNAs against cyclin D1, the targeting LNPs induced gene silencing in MCL cells and prolonged the survival of tumor-bearing mice, with no observed adverse effects.
“In MCL, cyclin D1 is the exclusive cause of the overproduction of B lymphocytes, the cells responsible for generating antibodies,” Dr Peer said. “This makes the protein a perfect target for RNA therapy by siRNAs.”
“Normal, healthy cells don’t express the gene, so therapies that destroy the gene will only attack cancer cells. The RNA interference we have developed targets the faulty cyclin D1 within the cancerous cells. And when the cells are inhibited from proliferating, they sense they are being targeted and begin to die off.”
Dr Peer and his colleagues believe this work presents new opportunities for treating MCL and other similar B-cell malignancies.
“This research makes a definite contribution to the revolution of personalized medicine, whereby you tailor the drug based on the genetic profile of patient,” Dr Peer said. “In this case, MCL is a disease with a specific genetic hallmark, so you can sequence the patient to identify the mutation(s) and design RNA blockers to be placed inside a nanovehicle.”
“While the targeting antibodies—the ‘GPS’—can be used to target many different B-cell malignancies, the drug itself is designed to silence this specific disease. However, the delivery system can be used to accommodate any disease with a genetic profile. This could be the future. We are seeing it happen before our very eyes.” ![]()
Researchers say they have developed a system for delivering therapy at the site of mantle cell lymphoma (MCL).
The system harnesses nanoparticles coated with “GPS” antibodies that navigate toward cancerous cells, where they offload cyclin D1-blockers in the form of small interfering RNAs (siRNAs).
Experiments showed the system can halt the proliferation of cyclin D1 in both animal models and samples from MCL patients.
Dan Peer, PhD, of Tel Aviv University in Israel, and his colleagues reported these results in PNAS.
“MCL has a genetic hallmark,” Dr Peer noted. “In 85% of cases, the characteristic that defines this aggressive and prototypic B-cell lymphoma is the heightened activity of the gene CCND1, which leads to the extreme overexpression—a 3000- to 15,000-fold increase—of cyclin D1, a protein that controls the proliferation of cells. Downregulation of cyclin D1 using siRNAs is a potential therapeutic approach to this malignancy.”
For this research, Dr Peer and his colleagues designed lipid-based nanoparticles (LNPs) coated with anti-CD38 monoclonal antibodies that were taken up by human MCL cells in the bone marrow of affected mice.
When loaded with siRNAs against cyclin D1, the targeting LNPs induced gene silencing in MCL cells and prolonged the survival of tumor-bearing mice, with no observed adverse effects.
“In MCL, cyclin D1 is the exclusive cause of the overproduction of B lymphocytes, the cells responsible for generating antibodies,” Dr Peer said. “This makes the protein a perfect target for RNA therapy by siRNAs.”
“Normal, healthy cells don’t express the gene, so therapies that destroy the gene will only attack cancer cells. The RNA interference we have developed targets the faulty cyclin D1 within the cancerous cells. And when the cells are inhibited from proliferating, they sense they are being targeted and begin to die off.”
Dr Peer and his colleagues believe this work presents new opportunities for treating MCL and other similar B-cell malignancies.
“This research makes a definite contribution to the revolution of personalized medicine, whereby you tailor the drug based on the genetic profile of patient,” Dr Peer said. “In this case, MCL is a disease with a specific genetic hallmark, so you can sequence the patient to identify the mutation(s) and design RNA blockers to be placed inside a nanovehicle.”
“While the targeting antibodies—the ‘GPS’—can be used to target many different B-cell malignancies, the drug itself is designed to silence this specific disease. However, the delivery system can be used to accommodate any disease with a genetic profile. This could be the future. We are seeing it happen before our very eyes.” ![]()
Mutations could be therapeutic target for FL

Mutations in the RRAGC gene appear to be an “excellent candidate for therapeutic targeting” in follicular lymphoma (FL), according to investigators.
They analyzed mutations found in tumors with multiple relapses of FL without transformation to diffuse large B-cell lymphoma.
And they found that one commonly mutated gene encodes the protein RagC, which is essential for activating the amino-acid sensing mTORC1 pathway.
Although mutations in genes in the mTORC1 pathway have been associated with various cancers, this is the first time a genetic mutation in any of the 4 Rag proteins has been identified in malignancy.
“One of the mutations that we have identified allows follicular lymphoma tumors to turn on growth signals regardless of whether nutrients are available, thereby evading normal restrictions on its growth,” said study author Jessica Okosun, MB BChir, PhD, of Barts Cancer Institute at Queen Mary University of London in the UK.
“Remarkably, the mutations we have discovered have not been seen in other cancer types. However, drugs that directly target this nutrient-sensing mechanism are currently used to treat other types of cancer and may benefit patients with follicular lymphoma.”
Dr Okosun and her colleagues reported these findings in a letter to Nature Genetics.
In experiments with cell lines, the investigators found that expression of the mutated RagC proteins activate mTORC1 signaling in the absence of amino acids and increase binding to an important part of the mTORC1 complex, consistent with the established role of RagC in the mTORC1 pathway.
Because this research was performed exclusively in cell lines, the investigators have not yet deciphered the mutations’ mechanistic effect in patients. However, study author Rachel Wolfson, of the Whitehead Institute for Biomedical Research and Massachusetts Institute of Technology in Cambridge, Massachusetts, said there are clues to the mutations’ significance.
“mTORC1 is linked to cell growth, so it is not surprising that activation of the pathway could lead to some growth advantage for cancer cells,” Wolfson said. “But it leads to an interesting question: When is it a proliferative advantage versus a disadvantage to no longer be able to accurately sense amino acid levels? That is something we would need to investigate further, likely in vivo.”
The investigators would also like to know how the drug rapamycin affects FL with RagC mutations. Rapamycin binds to mTORC1 and inhibits its activity. If the drug interferes with mTORC1 dysregulation caused by RagC mutations, perhaps the drug could be used in FL treatment.
“If so, maybe these RagC mutations could be used as biomarkers to predict sensitivity to rapamycin treatment in follicular lymphoma patients,” Wolfson said. “That would be very exciting, and it’s something that should be investigated further.” ![]()
Mutations in the RRAGC gene appear to be an “excellent candidate for therapeutic targeting” in follicular lymphoma (FL), according to investigators.
They analyzed mutations found in tumors with multiple relapses of FL without transformation to diffuse large B-cell lymphoma.
And they found that one commonly mutated gene encodes the protein RagC, which is essential for activating the amino-acid sensing mTORC1 pathway.
Although mutations in genes in the mTORC1 pathway have been associated with various cancers, this is the first time a genetic mutation in any of the 4 Rag proteins has been identified in malignancy.
“One of the mutations that we have identified allows follicular lymphoma tumors to turn on growth signals regardless of whether nutrients are available, thereby evading normal restrictions on its growth,” said study author Jessica Okosun, MB BChir, PhD, of Barts Cancer Institute at Queen Mary University of London in the UK.
“Remarkably, the mutations we have discovered have not been seen in other cancer types. However, drugs that directly target this nutrient-sensing mechanism are currently used to treat other types of cancer and may benefit patients with follicular lymphoma.”
Dr Okosun and her colleagues reported these findings in a letter to Nature Genetics.
In experiments with cell lines, the investigators found that expression of the mutated RagC proteins activate mTORC1 signaling in the absence of amino acids and increase binding to an important part of the mTORC1 complex, consistent with the established role of RagC in the mTORC1 pathway.
Because this research was performed exclusively in cell lines, the investigators have not yet deciphered the mutations’ mechanistic effect in patients. However, study author Rachel Wolfson, of the Whitehead Institute for Biomedical Research and Massachusetts Institute of Technology in Cambridge, Massachusetts, said there are clues to the mutations’ significance.
“mTORC1 is linked to cell growth, so it is not surprising that activation of the pathway could lead to some growth advantage for cancer cells,” Wolfson said. “But it leads to an interesting question: When is it a proliferative advantage versus a disadvantage to no longer be able to accurately sense amino acid levels? That is something we would need to investigate further, likely in vivo.”
The investigators would also like to know how the drug rapamycin affects FL with RagC mutations. Rapamycin binds to mTORC1 and inhibits its activity. If the drug interferes with mTORC1 dysregulation caused by RagC mutations, perhaps the drug could be used in FL treatment.
“If so, maybe these RagC mutations could be used as biomarkers to predict sensitivity to rapamycin treatment in follicular lymphoma patients,” Wolfson said. “That would be very exciting, and it’s something that should be investigated further.” ![]()
Mutations in the RRAGC gene appear to be an “excellent candidate for therapeutic targeting” in follicular lymphoma (FL), according to investigators.
They analyzed mutations found in tumors with multiple relapses of FL without transformation to diffuse large B-cell lymphoma.
And they found that one commonly mutated gene encodes the protein RagC, which is essential for activating the amino-acid sensing mTORC1 pathway.
Although mutations in genes in the mTORC1 pathway have been associated with various cancers, this is the first time a genetic mutation in any of the 4 Rag proteins has been identified in malignancy.
“One of the mutations that we have identified allows follicular lymphoma tumors to turn on growth signals regardless of whether nutrients are available, thereby evading normal restrictions on its growth,” said study author Jessica Okosun, MB BChir, PhD, of Barts Cancer Institute at Queen Mary University of London in the UK.
“Remarkably, the mutations we have discovered have not been seen in other cancer types. However, drugs that directly target this nutrient-sensing mechanism are currently used to treat other types of cancer and may benefit patients with follicular lymphoma.”
Dr Okosun and her colleagues reported these findings in a letter to Nature Genetics.
In experiments with cell lines, the investigators found that expression of the mutated RagC proteins activate mTORC1 signaling in the absence of amino acids and increase binding to an important part of the mTORC1 complex, consistent with the established role of RagC in the mTORC1 pathway.
Because this research was performed exclusively in cell lines, the investigators have not yet deciphered the mutations’ mechanistic effect in patients. However, study author Rachel Wolfson, of the Whitehead Institute for Biomedical Research and Massachusetts Institute of Technology in Cambridge, Massachusetts, said there are clues to the mutations’ significance.
“mTORC1 is linked to cell growth, so it is not surprising that activation of the pathway could lead to some growth advantage for cancer cells,” Wolfson said. “But it leads to an interesting question: When is it a proliferative advantage versus a disadvantage to no longer be able to accurately sense amino acid levels? That is something we would need to investigate further, likely in vivo.”
The investigators would also like to know how the drug rapamycin affects FL with RagC mutations. Rapamycin binds to mTORC1 and inhibits its activity. If the drug interferes with mTORC1 dysregulation caused by RagC mutations, perhaps the drug could be used in FL treatment.
“If so, maybe these RagC mutations could be used as biomarkers to predict sensitivity to rapamycin treatment in follicular lymphoma patients,” Wolfson said. “That would be very exciting, and it’s something that should be investigated further.” ![]()
EZH2 inhibitor can produce durable responses

ORLANDO, FL—Updated results of a phase 1 study suggest the EZH2 inhibitor tazemetostat (EPZ-6438) can produce durable responses in patients with advanced non-Hodgkin lymphoma (NHL).
The drug has demonstrated activity against diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), and marginal zone lymphoma (MZL).
The overall response rate among NHL patients in this study was 56%, and 1 patient has maintained a response for more than 21 months.
In addition, the drug’s safety profile is “still acceptable,” according to Vincent Ribrag, MD, of Institut Gustave Roussy in Villejuif, France.
Dr Ribrag presented the results of this study at the 2015 ASH Annual Meeting (abstract 473*). The research, which was previously presented at the 13th International Conference on Malignant Lymphoma, was sponsored by Epizyme, Inc., the company developing tazemetostat.
The trial has enrolled 58 patients, 21 with relapsed or refractory B-cell NHL and 37 with advanced solid tumors. The NHL cohort includes 5 patients with germinal center B-cell (GCB) DLBCL, 6 cases of non-GCB DLBCL, 3 DLBCL cases of an undetermined subtype, 6 patients with FL, and 1 case of MZL.
At baseline, the NHL patients had a median age of 63 (range, 24-84) and were heavily pretreated. Eighty-five percent of patients had received 3 or more prior therapies, and 33% had received 5 or more. Thirty-eight percent of patients had undergone an autologous transplant, and 57% had received radiotherapy.
The patients received tazemetostat twice daily at a range of doses. For the dose-escalation portion of the trial, they received 100 mg, 200 mg, 400 mg, 800 mg, or 1600 mg. For the dose-expansion phase, they received 800 mg or 1600 mg.
The researchers are now conducting a drug-drug interaction substudy in which patients receive 800 mg of tazemetostat twice daily and a food-effect substudy in which patients receive the drug at 400 mg twice daily.
Dr Ribrag said the recommended phase 2 dose of tazemetostat is 800 mg twice daily.
Safety
At the data cutoff point (November 7, 2015), 55 patients—20 with NHL and 35 with solid tumors—were evaluable for safety.
Treatment-related adverse events in these patients included asthenia (n=13), nausea (n=8), thrombocytopenia (n=7), dysgeusia (n=5), vomiting (n=5), dry skin (n=4), decreased appetite (n=4), diarrhea (n=4), muscle spasms (n=3), neutropenia (n=3), anemia (n=3), night sweats (n=3), hypertension (n=2), constipation (n=2), peripheral edema (n=2), hypophosphatemia (n=1), anxiety (n=1), depression (n=1), abdominal pain (n=1), and hepatocellular injury (n=1).
There were 4 grade 3 or higher adverse events that were considered treatment-related, including nausea, hypertension, neutropenia, and hepatocellular injury.
Efficacy
Sixteen of the NHL patients were evaluable for efficacy. Nine patients responded to treatment, 2 with complete responses (CRs) and 7 with partial responses (PRs).
Five of the 10 DLBCL patients responded, 4 with PRs and 1 with a CR. Three of the 5 FL patients responded, 2 with PRs and 1 with a CR. The patient with MZL achieved a PR.
Four responders remain on study—2 with DLBCL and 2 with FL.
One DLBCL patient with an EZH2 mutation (Y646H) had relapsed after or was refractory to 6 previous treatment regimens. This patient achieved a PR after 16 weeks of tazemetostat. The patient is still in PR at week 44 and remains on study.
Based on these results, Epizyme is currently enrolling patients in a phase 2 study of tazemetostat monotherapy. The trial is open to patients with DLBCL or FL in France, Australia, and the UK. ![]()
*Data in the abstract differ from the presentation.
ORLANDO, FL—Updated results of a phase 1 study suggest the EZH2 inhibitor tazemetostat (EPZ-6438) can produce durable responses in patients with advanced non-Hodgkin lymphoma (NHL).
The drug has demonstrated activity against diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), and marginal zone lymphoma (MZL).
The overall response rate among NHL patients in this study was 56%, and 1 patient has maintained a response for more than 21 months.
In addition, the drug’s safety profile is “still acceptable,” according to Vincent Ribrag, MD, of Institut Gustave Roussy in Villejuif, France.
Dr Ribrag presented the results of this study at the 2015 ASH Annual Meeting (abstract 473*). The research, which was previously presented at the 13th International Conference on Malignant Lymphoma, was sponsored by Epizyme, Inc., the company developing tazemetostat.
The trial has enrolled 58 patients, 21 with relapsed or refractory B-cell NHL and 37 with advanced solid tumors. The NHL cohort includes 5 patients with germinal center B-cell (GCB) DLBCL, 6 cases of non-GCB DLBCL, 3 DLBCL cases of an undetermined subtype, 6 patients with FL, and 1 case of MZL.
At baseline, the NHL patients had a median age of 63 (range, 24-84) and were heavily pretreated. Eighty-five percent of patients had received 3 or more prior therapies, and 33% had received 5 or more. Thirty-eight percent of patients had undergone an autologous transplant, and 57% had received radiotherapy.
The patients received tazemetostat twice daily at a range of doses. For the dose-escalation portion of the trial, they received 100 mg, 200 mg, 400 mg, 800 mg, or 1600 mg. For the dose-expansion phase, they received 800 mg or 1600 mg.
The researchers are now conducting a drug-drug interaction substudy in which patients receive 800 mg of tazemetostat twice daily and a food-effect substudy in which patients receive the drug at 400 mg twice daily.
Dr Ribrag said the recommended phase 2 dose of tazemetostat is 800 mg twice daily.
Safety
At the data cutoff point (November 7, 2015), 55 patients—20 with NHL and 35 with solid tumors—were evaluable for safety.
Treatment-related adverse events in these patients included asthenia (n=13), nausea (n=8), thrombocytopenia (n=7), dysgeusia (n=5), vomiting (n=5), dry skin (n=4), decreased appetite (n=4), diarrhea (n=4), muscle spasms (n=3), neutropenia (n=3), anemia (n=3), night sweats (n=3), hypertension (n=2), constipation (n=2), peripheral edema (n=2), hypophosphatemia (n=1), anxiety (n=1), depression (n=1), abdominal pain (n=1), and hepatocellular injury (n=1).
There were 4 grade 3 or higher adverse events that were considered treatment-related, including nausea, hypertension, neutropenia, and hepatocellular injury.
Efficacy
Sixteen of the NHL patients were evaluable for efficacy. Nine patients responded to treatment, 2 with complete responses (CRs) and 7 with partial responses (PRs).
Five of the 10 DLBCL patients responded, 4 with PRs and 1 with a CR. Three of the 5 FL patients responded, 2 with PRs and 1 with a CR. The patient with MZL achieved a PR.
Four responders remain on study—2 with DLBCL and 2 with FL.
One DLBCL patient with an EZH2 mutation (Y646H) had relapsed after or was refractory to 6 previous treatment regimens. This patient achieved a PR after 16 weeks of tazemetostat. The patient is still in PR at week 44 and remains on study.
Based on these results, Epizyme is currently enrolling patients in a phase 2 study of tazemetostat monotherapy. The trial is open to patients with DLBCL or FL in France, Australia, and the UK. ![]()
*Data in the abstract differ from the presentation.
ORLANDO, FL—Updated results of a phase 1 study suggest the EZH2 inhibitor tazemetostat (EPZ-6438) can produce durable responses in patients with advanced non-Hodgkin lymphoma (NHL).
The drug has demonstrated activity against diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), and marginal zone lymphoma (MZL).
The overall response rate among NHL patients in this study was 56%, and 1 patient has maintained a response for more than 21 months.
In addition, the drug’s safety profile is “still acceptable,” according to Vincent Ribrag, MD, of Institut Gustave Roussy in Villejuif, France.
Dr Ribrag presented the results of this study at the 2015 ASH Annual Meeting (abstract 473*). The research, which was previously presented at the 13th International Conference on Malignant Lymphoma, was sponsored by Epizyme, Inc., the company developing tazemetostat.
The trial has enrolled 58 patients, 21 with relapsed or refractory B-cell NHL and 37 with advanced solid tumors. The NHL cohort includes 5 patients with germinal center B-cell (GCB) DLBCL, 6 cases of non-GCB DLBCL, 3 DLBCL cases of an undetermined subtype, 6 patients with FL, and 1 case of MZL.
At baseline, the NHL patients had a median age of 63 (range, 24-84) and were heavily pretreated. Eighty-five percent of patients had received 3 or more prior therapies, and 33% had received 5 or more. Thirty-eight percent of patients had undergone an autologous transplant, and 57% had received radiotherapy.
The patients received tazemetostat twice daily at a range of doses. For the dose-escalation portion of the trial, they received 100 mg, 200 mg, 400 mg, 800 mg, or 1600 mg. For the dose-expansion phase, they received 800 mg or 1600 mg.
The researchers are now conducting a drug-drug interaction substudy in which patients receive 800 mg of tazemetostat twice daily and a food-effect substudy in which patients receive the drug at 400 mg twice daily.
Dr Ribrag said the recommended phase 2 dose of tazemetostat is 800 mg twice daily.
Safety
At the data cutoff point (November 7, 2015), 55 patients—20 with NHL and 35 with solid tumors—were evaluable for safety.
Treatment-related adverse events in these patients included asthenia (n=13), nausea (n=8), thrombocytopenia (n=7), dysgeusia (n=5), vomiting (n=5), dry skin (n=4), decreased appetite (n=4), diarrhea (n=4), muscle spasms (n=3), neutropenia (n=3), anemia (n=3), night sweats (n=3), hypertension (n=2), constipation (n=2), peripheral edema (n=2), hypophosphatemia (n=1), anxiety (n=1), depression (n=1), abdominal pain (n=1), and hepatocellular injury (n=1).
There were 4 grade 3 or higher adverse events that were considered treatment-related, including nausea, hypertension, neutropenia, and hepatocellular injury.
Efficacy
Sixteen of the NHL patients were evaluable for efficacy. Nine patients responded to treatment, 2 with complete responses (CRs) and 7 with partial responses (PRs).
Five of the 10 DLBCL patients responded, 4 with PRs and 1 with a CR. Three of the 5 FL patients responded, 2 with PRs and 1 with a CR. The patient with MZL achieved a PR.
Four responders remain on study—2 with DLBCL and 2 with FL.
One DLBCL patient with an EZH2 mutation (Y646H) had relapsed after or was refractory to 6 previous treatment regimens. This patient achieved a PR after 16 weeks of tazemetostat. The patient is still in PR at week 44 and remains on study.
Based on these results, Epizyme is currently enrolling patients in a phase 2 study of tazemetostat monotherapy. The trial is open to patients with DLBCL or FL in France, Australia, and the UK. ![]()
*Data in the abstract differ from the presentation.
Antibody shows promise for treating CLL

Preclinical research suggests a humanized monoclonal antibody called cirmtuzumab (UC-961) might be an effective treatment for chronic lymphocytic leukemia (CLL).
Experiments revealed that the Wnt5a protein acts on the tumor-surface proteins ROR1 and ROR2 to accelerate the proliferation and spread of CLL cells.
But cirmtuzumab, which is specific for ROR1, can block the effects of Wnt5a and inhibit the growth and spread of CLL cells both in vitro and in vivo.
Investigators reported these results in The Journal of Clinical Investigation.
They noted that ROR1 and ROR2 are considered orphan receptors, which are expressed primarily during embryonic development. The expression of these proteins, particularly ROR1, becomes suppressed during fetal development and is negligible on normal adult tissues. However, CLL and many solid tissue cancers re-express these orphan receptors.
“Our findings show that ROR1 and ROR2 team up to stimulate tumor cell growth and metastasis in response to Wnt5a, which appears overexpressed in patients with CLL and can act as a survival/growth factor for leukemia cells,” said study author Thomas J. Kipps, MD, PhD, of Moores Cancer Center at the University of California, San Diego.
“By blocking the capacity of Wnt5a to stimulate tumor cells, cirmtuzumab can inhibit the growth and spread of cancer cells. We now have better insight into how cirmtuzumab works against leukemia cells. This should help find better ways to treat patients who have other cancers with cirmtuzumab, which currently is being evaluated in a phase 1 clinical trial for patients with CLL.”
The JCI paper follows a series of related findings by Dr Kipps and his colleagues in recent years.
In 2008, they reported that patients vaccinated with their own CLL cells could make antibodies against ROR1, some of which had the ability to reduce CLL cell survival. They found ROR1 on CLL cells but not on all normal adult tissues examined.
In 2012, the investigators reported finding ROR1 on many different types of cancer, particularly cancers that appear less differentiated and more likely to spread to other parts in the body. Because this protein was not found on normal adult tissues, these findings made ROR1 a new target for anticancer drug research.
In June 2013, the team linked ROR1 to a process used in early development, suggesting cancer cells hijack an embryological process called epithelial-mesenchymal transition to spread or metastasize more quickly.
In January 2014, the investigators reported expression of ROR1 resulted in a faster-developing, more aggressive form of CLL in mice.
In September 2014, the team launched a phase 1 trial of cirmtuzumab in patients with CLL. The trial is ongoing.
In November 2014, the investigators described cellular experiments indicating that cirmtuzumab might be effective against cancer stem cells, which appear responsible for the relapse and spread of cancer after conventional therapy.
The latest research more precisely defines the roles of ROR1 and ROR2 in CLL development.
Both are evolutionarily conserved proteins that are found in many species and are most active in the early stages of embryogenesis when cells are migrating to form organs and parts of the body. The lack of either during this process results in severe developmental abnormalities.
Low levels of ROR2 remain in some adult tissues, but ROR1 is found only in cancer cells. The investigators found that, in response to signaling by Wnt5a, ROR1 and ROR2 come together to signal the growth and migration of CLL cells.
But treating mice with cirmtuzumab disrupted the process, inhibiting the engraftment of CLL cells and slowing or stopping the disease from spreading. ![]()
Preclinical research suggests a humanized monoclonal antibody called cirmtuzumab (UC-961) might be an effective treatment for chronic lymphocytic leukemia (CLL).
Experiments revealed that the Wnt5a protein acts on the tumor-surface proteins ROR1 and ROR2 to accelerate the proliferation and spread of CLL cells.
But cirmtuzumab, which is specific for ROR1, can block the effects of Wnt5a and inhibit the growth and spread of CLL cells both in vitro and in vivo.
Investigators reported these results in The Journal of Clinical Investigation.
They noted that ROR1 and ROR2 are considered orphan receptors, which are expressed primarily during embryonic development. The expression of these proteins, particularly ROR1, becomes suppressed during fetal development and is negligible on normal adult tissues. However, CLL and many solid tissue cancers re-express these orphan receptors.
“Our findings show that ROR1 and ROR2 team up to stimulate tumor cell growth and metastasis in response to Wnt5a, which appears overexpressed in patients with CLL and can act as a survival/growth factor for leukemia cells,” said study author Thomas J. Kipps, MD, PhD, of Moores Cancer Center at the University of California, San Diego.
“By blocking the capacity of Wnt5a to stimulate tumor cells, cirmtuzumab can inhibit the growth and spread of cancer cells. We now have better insight into how cirmtuzumab works against leukemia cells. This should help find better ways to treat patients who have other cancers with cirmtuzumab, which currently is being evaluated in a phase 1 clinical trial for patients with CLL.”
The JCI paper follows a series of related findings by Dr Kipps and his colleagues in recent years.
In 2008, they reported that patients vaccinated with their own CLL cells could make antibodies against ROR1, some of which had the ability to reduce CLL cell survival. They found ROR1 on CLL cells but not on all normal adult tissues examined.
In 2012, the investigators reported finding ROR1 on many different types of cancer, particularly cancers that appear less differentiated and more likely to spread to other parts in the body. Because this protein was not found on normal adult tissues, these findings made ROR1 a new target for anticancer drug research.
In June 2013, the team linked ROR1 to a process used in early development, suggesting cancer cells hijack an embryological process called epithelial-mesenchymal transition to spread or metastasize more quickly.
In January 2014, the investigators reported expression of ROR1 resulted in a faster-developing, more aggressive form of CLL in mice.
In September 2014, the team launched a phase 1 trial of cirmtuzumab in patients with CLL. The trial is ongoing.
In November 2014, the investigators described cellular experiments indicating that cirmtuzumab might be effective against cancer stem cells, which appear responsible for the relapse and spread of cancer after conventional therapy.
The latest research more precisely defines the roles of ROR1 and ROR2 in CLL development.
Both are evolutionarily conserved proteins that are found in many species and are most active in the early stages of embryogenesis when cells are migrating to form organs and parts of the body. The lack of either during this process results in severe developmental abnormalities.
Low levels of ROR2 remain in some adult tissues, but ROR1 is found only in cancer cells. The investigators found that, in response to signaling by Wnt5a, ROR1 and ROR2 come together to signal the growth and migration of CLL cells.
But treating mice with cirmtuzumab disrupted the process, inhibiting the engraftment of CLL cells and slowing or stopping the disease from spreading. ![]()
Preclinical research suggests a humanized monoclonal antibody called cirmtuzumab (UC-961) might be an effective treatment for chronic lymphocytic leukemia (CLL).
Experiments revealed that the Wnt5a protein acts on the tumor-surface proteins ROR1 and ROR2 to accelerate the proliferation and spread of CLL cells.
But cirmtuzumab, which is specific for ROR1, can block the effects of Wnt5a and inhibit the growth and spread of CLL cells both in vitro and in vivo.
Investigators reported these results in The Journal of Clinical Investigation.
They noted that ROR1 and ROR2 are considered orphan receptors, which are expressed primarily during embryonic development. The expression of these proteins, particularly ROR1, becomes suppressed during fetal development and is negligible on normal adult tissues. However, CLL and many solid tissue cancers re-express these orphan receptors.
“Our findings show that ROR1 and ROR2 team up to stimulate tumor cell growth and metastasis in response to Wnt5a, which appears overexpressed in patients with CLL and can act as a survival/growth factor for leukemia cells,” said study author Thomas J. Kipps, MD, PhD, of Moores Cancer Center at the University of California, San Diego.
“By blocking the capacity of Wnt5a to stimulate tumor cells, cirmtuzumab can inhibit the growth and spread of cancer cells. We now have better insight into how cirmtuzumab works against leukemia cells. This should help find better ways to treat patients who have other cancers with cirmtuzumab, which currently is being evaluated in a phase 1 clinical trial for patients with CLL.”
The JCI paper follows a series of related findings by Dr Kipps and his colleagues in recent years.
In 2008, they reported that patients vaccinated with their own CLL cells could make antibodies against ROR1, some of which had the ability to reduce CLL cell survival. They found ROR1 on CLL cells but not on all normal adult tissues examined.
In 2012, the investigators reported finding ROR1 on many different types of cancer, particularly cancers that appear less differentiated and more likely to spread to other parts in the body. Because this protein was not found on normal adult tissues, these findings made ROR1 a new target for anticancer drug research.
In June 2013, the team linked ROR1 to a process used in early development, suggesting cancer cells hijack an embryological process called epithelial-mesenchymal transition to spread or metastasize more quickly.
In January 2014, the investigators reported expression of ROR1 resulted in a faster-developing, more aggressive form of CLL in mice.
In September 2014, the team launched a phase 1 trial of cirmtuzumab in patients with CLL. The trial is ongoing.
In November 2014, the investigators described cellular experiments indicating that cirmtuzumab might be effective against cancer stem cells, which appear responsible for the relapse and spread of cancer after conventional therapy.
The latest research more precisely defines the roles of ROR1 and ROR2 in CLL development.
Both are evolutionarily conserved proteins that are found in many species and are most active in the early stages of embryogenesis when cells are migrating to form organs and parts of the body. The lack of either during this process results in severe developmental abnormalities.
Low levels of ROR2 remain in some adult tissues, but ROR1 is found only in cancer cells. The investigators found that, in response to signaling by Wnt5a, ROR1 and ROR2 come together to signal the growth and migration of CLL cells.
But treating mice with cirmtuzumab disrupted the process, inhibiting the engraftment of CLL cells and slowing or stopping the disease from spreading.
Venetoclax produces deep responses in ultra-high-risk CLL

Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review.
Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review.
Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review.
Surprising finding in upfront use of idelalisib monotherapy

Photo courtesy of ASH
ORLANDO, FL—Investigators have observed early fulminant hepatotoxicity in a subset of primarily younger chronic lymphocytic leukemia (CLL) patients treated with idelalisib monotherapy in the frontline setting.
In a phase 2 study of idelalisib plus ofatumumab, 52% of the 24 patients enrolled experienced grade 3 or higher hepatotoxicity shortly after idelalisib was started.
The investigators say this may occur because a proportion of regulatory T cells in the peripheral blood decreases while patients are on idelalisib. The team believes this early hepatotoxicity is immune-mediated.
Benjamin Lampson, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, described these surprising findings at the 2015 ASH Annual Meeting as abstract 497.*
Study design and patient demographics
Patients received 150 mg of idelalisib twice daily as monotherapy on days 1 through 56. They then received combination therapy with idelalisib plus ofatumumab for 8 weekly infusions, followed by 4 monthly infusions through day 225, and then idelalisib monotherapy indefinitely.
The primary endpoint of overall response rate was assessed 2 months after the completion of combination therapy.
“This dosing strategy is slightly different than what has been previously used in trials combining these particular drugs,” Dr Lampson said. “Specifically, previously reported trials started these agents simultaneously without a lead-in period of monotherapy.”
The investigators monitored the patients weekly for toxicities during the 2-month monotherapy lead-in period.
Three-quarters of the patients are male. Their median age is 67.4 years (range, 57.6–84.9), 54% have unmutated IgHV, 17% have deletion 17p or TP53 mutation, 4% have deletion 11q, and 54% have deletion 13q.
The patients received no prior therapies.
Results
The trial is currently ongoing.
The 24 patients enrolled as of early November have been on therapy a median of 7.7 months, for a median follow-up time of 14.7 months.
“What we began to notice after enrolling just a few subjects on the trial was that severe hepatotoxicity was occurring shortly after initiating idelalisib,” Dr Lampson said.
In the first 2 months of therapy, 52% of patients developed transaminitis, and 13% developed colitis or diarrhea, all grade 3 or higher. Thirteen percent developed pneumonitis of any grade.
Younger age is a risk factor for early hepatotoxicity, Dr Lampson said, with a significance of P=0.02. All subjects age 65 or younger (n=7) required systemic steroids to treat their toxicities.
Hepatotoxicity developed in a median of 28 days, he said, “and the hepatotoxicity is typically occurring before the first dose of ofatumumab is administered at week 8, suggesting that idelalisib alone is the cause of the hepatotoxicity.”
Dr Lampson noted that toxicities resolved rapidly with steroids.
“I do want to point out that all subjects evaluable for a response have had a response,” he added. “Additionally, in all subjects where treatment has been discontinued, the discontinuation was due to adverse events rather than disease progression.”
Twelve patients with grade 2 or higher transaminitis were re-challenged with idelalisib after holding the drug for toxicity.
Five patients were re-challenged while off steroids, and 4 developed recurrent transaminitis within 4 days. Seven patients were re-challenged while on steroids, and 2 developed recurrent transaminitis within 4 days.
“In general, our experience has been, if idelalisib is resumed while the subject remains on steroids, the drug is more likely to be tolerated and the subject can eventually be taken off steroids,” Dr Lampson said.
Comparison with earlier studies
The investigators compared the frequency of toxicity in their trial to earlier studies of idelalisib (Brown, Blood 2014; Coutre, EHA 2015, abstr P588; O’Brien, Blood 2015).
They found that grade 3 or higher transaminitis (52%) and any grade pneumonitis (13%) were higher in their trial than in the 3 other trials.
Colitis/diarrhea was about the same in 2 of the 3 other trials. But in the paper by O’Brien et al, 42% of patients experienced grade 3 or greater colitis/diarrhea.
The lower rate of colitis in the present trial may be due to the shorter follow-up, Dr Lampson said, as colitis is a late adverse event.
The O’Brien trial was also an upfront study, so patients had no prior therapies. The investigators observed that toxicities appeared to be more common in less heavily pretreated patients.
“As the median number of prior therapies decreases,” Dr Lampson said, “the frequency of adverse events increases.”
He noted that, in the O’Brien trial, idelalisib was started simultaneously with the other drugs, perhaps accounting for its somewhat lower rate (21%) of grade 3 or higher transaminitis.
Additionally, the patient population in the O’Brien trial was older than the population in the current trial, which could account for the higher rate of transaminitis, as younger age is a risk factor.
Decrease in regulatory T cells
Investigators noted a decrease in regulatory T cells while patients were on therapy. Eleven of 15 patients (73%) with matched samples had a significant (P<0.05) decrease in the percentage of T cells over time.
This, they say, could provide a possible explanation for the development of early hepatotoxicity.
The trial is investigator-initiated and funded by Gilead Sciences.
*Data in the presentation differs from the abstract.
Photo courtesy of ASH
ORLANDO, FL—Investigators have observed early fulminant hepatotoxicity in a subset of primarily younger chronic lymphocytic leukemia (CLL) patients treated with idelalisib monotherapy in the frontline setting.
In a phase 2 study of idelalisib plus ofatumumab, 52% of the 24 patients enrolled experienced grade 3 or higher hepatotoxicity shortly after idelalisib was started.
The investigators say this may occur because a proportion of regulatory T cells in the peripheral blood decreases while patients are on idelalisib. The team believes this early hepatotoxicity is immune-mediated.
Benjamin Lampson, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, described these surprising findings at the 2015 ASH Annual Meeting as abstract 497.*
Study design and patient demographics
Patients received 150 mg of idelalisib twice daily as monotherapy on days 1 through 56. They then received combination therapy with idelalisib plus ofatumumab for 8 weekly infusions, followed by 4 monthly infusions through day 225, and then idelalisib monotherapy indefinitely.
The primary endpoint of overall response rate was assessed 2 months after the completion of combination therapy.
“This dosing strategy is slightly different than what has been previously used in trials combining these particular drugs,” Dr Lampson said. “Specifically, previously reported trials started these agents simultaneously without a lead-in period of monotherapy.”
The investigators monitored the patients weekly for toxicities during the 2-month monotherapy lead-in period.
Three-quarters of the patients are male. Their median age is 67.4 years (range, 57.6–84.9), 54% have unmutated IgHV, 17% have deletion 17p or TP53 mutation, 4% have deletion 11q, and 54% have deletion 13q.
The patients received no prior therapies.
Results
The trial is currently ongoing.
The 24 patients enrolled as of early November have been on therapy a median of 7.7 months, for a median follow-up time of 14.7 months.
“What we began to notice after enrolling just a few subjects on the trial was that severe hepatotoxicity was occurring shortly after initiating idelalisib,” Dr Lampson said.
In the first 2 months of therapy, 52% of patients developed transaminitis, and 13% developed colitis or diarrhea, all grade 3 or higher. Thirteen percent developed pneumonitis of any grade.
Younger age is a risk factor for early hepatotoxicity, Dr Lampson said, with a significance of P=0.02. All subjects age 65 or younger (n=7) required systemic steroids to treat their toxicities.
Hepatotoxicity developed in a median of 28 days, he said, “and the hepatotoxicity is typically occurring before the first dose of ofatumumab is administered at week 8, suggesting that idelalisib alone is the cause of the hepatotoxicity.”
Dr Lampson noted that toxicities resolved rapidly with steroids.
“I do want to point out that all subjects evaluable for a response have had a response,” he added. “Additionally, in all subjects where treatment has been discontinued, the discontinuation was due to adverse events rather than disease progression.”
Twelve patients with grade 2 or higher transaminitis were re-challenged with idelalisib after holding the drug for toxicity.
Five patients were re-challenged while off steroids, and 4 developed recurrent transaminitis within 4 days. Seven patients were re-challenged while on steroids, and 2 developed recurrent transaminitis within 4 days.
“In general, our experience has been, if idelalisib is resumed while the subject remains on steroids, the drug is more likely to be tolerated and the subject can eventually be taken off steroids,” Dr Lampson said.
Comparison with earlier studies
The investigators compared the frequency of toxicity in their trial to earlier studies of idelalisib (Brown, Blood 2014; Coutre, EHA 2015, abstr P588; O’Brien, Blood 2015).
They found that grade 3 or higher transaminitis (52%) and any grade pneumonitis (13%) were higher in their trial than in the 3 other trials.
Colitis/diarrhea was about the same in 2 of the 3 other trials. But in the paper by O’Brien et al, 42% of patients experienced grade 3 or greater colitis/diarrhea.
The lower rate of colitis in the present trial may be due to the shorter follow-up, Dr Lampson said, as colitis is a late adverse event.
The O’Brien trial was also an upfront study, so patients had no prior therapies. The investigators observed that toxicities appeared to be more common in less heavily pretreated patients.
“As the median number of prior therapies decreases,” Dr Lampson said, “the frequency of adverse events increases.”
He noted that, in the O’Brien trial, idelalisib was started simultaneously with the other drugs, perhaps accounting for its somewhat lower rate (21%) of grade 3 or higher transaminitis.
Additionally, the patient population in the O’Brien trial was older than the population in the current trial, which could account for the higher rate of transaminitis, as younger age is a risk factor.
Decrease in regulatory T cells
Investigators noted a decrease in regulatory T cells while patients were on therapy. Eleven of 15 patients (73%) with matched samples had a significant (P<0.05) decrease in the percentage of T cells over time.
This, they say, could provide a possible explanation for the development of early hepatotoxicity.
The trial is investigator-initiated and funded by Gilead Sciences.
*Data in the presentation differs from the abstract.
Photo courtesy of ASH
ORLANDO, FL—Investigators have observed early fulminant hepatotoxicity in a subset of primarily younger chronic lymphocytic leukemia (CLL) patients treated with idelalisib monotherapy in the frontline setting.
In a phase 2 study of idelalisib plus ofatumumab, 52% of the 24 patients enrolled experienced grade 3 or higher hepatotoxicity shortly after idelalisib was started.
The investigators say this may occur because a proportion of regulatory T cells in the peripheral blood decreases while patients are on idelalisib. The team believes this early hepatotoxicity is immune-mediated.
Benjamin Lampson, MD, PhD, of the Dana-Farber Cancer Institute in Boston, Massachusetts, described these surprising findings at the 2015 ASH Annual Meeting as abstract 497.*
Study design and patient demographics
Patients received 150 mg of idelalisib twice daily as monotherapy on days 1 through 56. They then received combination therapy with idelalisib plus ofatumumab for 8 weekly infusions, followed by 4 monthly infusions through day 225, and then idelalisib monotherapy indefinitely.
The primary endpoint of overall response rate was assessed 2 months after the completion of combination therapy.
“This dosing strategy is slightly different than what has been previously used in trials combining these particular drugs,” Dr Lampson said. “Specifically, previously reported trials started these agents simultaneously without a lead-in period of monotherapy.”
The investigators monitored the patients weekly for toxicities during the 2-month monotherapy lead-in period.
Three-quarters of the patients are male. Their median age is 67.4 years (range, 57.6–84.9), 54% have unmutated IgHV, 17% have deletion 17p or TP53 mutation, 4% have deletion 11q, and 54% have deletion 13q.
The patients received no prior therapies.
Results
The trial is currently ongoing.
The 24 patients enrolled as of early November have been on therapy a median of 7.7 months, for a median follow-up time of 14.7 months.
“What we began to notice after enrolling just a few subjects on the trial was that severe hepatotoxicity was occurring shortly after initiating idelalisib,” Dr Lampson said.
In the first 2 months of therapy, 52% of patients developed transaminitis, and 13% developed colitis or diarrhea, all grade 3 or higher. Thirteen percent developed pneumonitis of any grade.
Younger age is a risk factor for early hepatotoxicity, Dr Lampson said, with a significance of P=0.02. All subjects age 65 or younger (n=7) required systemic steroids to treat their toxicities.
Hepatotoxicity developed in a median of 28 days, he said, “and the hepatotoxicity is typically occurring before the first dose of ofatumumab is administered at week 8, suggesting that idelalisib alone is the cause of the hepatotoxicity.”
Dr Lampson noted that toxicities resolved rapidly with steroids.
“I do want to point out that all subjects evaluable for a response have had a response,” he added. “Additionally, in all subjects where treatment has been discontinued, the discontinuation was due to adverse events rather than disease progression.”
Twelve patients with grade 2 or higher transaminitis were re-challenged with idelalisib after holding the drug for toxicity.
Five patients were re-challenged while off steroids, and 4 developed recurrent transaminitis within 4 days. Seven patients were re-challenged while on steroids, and 2 developed recurrent transaminitis within 4 days.
“In general, our experience has been, if idelalisib is resumed while the subject remains on steroids, the drug is more likely to be tolerated and the subject can eventually be taken off steroids,” Dr Lampson said.
Comparison with earlier studies
The investigators compared the frequency of toxicity in their trial to earlier studies of idelalisib (Brown, Blood 2014; Coutre, EHA 2015, abstr P588; O’Brien, Blood 2015).
They found that grade 3 or higher transaminitis (52%) and any grade pneumonitis (13%) were higher in their trial than in the 3 other trials.
Colitis/diarrhea was about the same in 2 of the 3 other trials. But in the paper by O’Brien et al, 42% of patients experienced grade 3 or greater colitis/diarrhea.
The lower rate of colitis in the present trial may be due to the shorter follow-up, Dr Lampson said, as colitis is a late adverse event.
The O’Brien trial was also an upfront study, so patients had no prior therapies. The investigators observed that toxicities appeared to be more common in less heavily pretreated patients.
“As the median number of prior therapies decreases,” Dr Lampson said, “the frequency of adverse events increases.”
He noted that, in the O’Brien trial, idelalisib was started simultaneously with the other drugs, perhaps accounting for its somewhat lower rate (21%) of grade 3 or higher transaminitis.
Additionally, the patient population in the O’Brien trial was older than the population in the current trial, which could account for the higher rate of transaminitis, as younger age is a risk factor.
Decrease in regulatory T cells
Investigators noted a decrease in regulatory T cells while patients were on therapy. Eleven of 15 patients (73%) with matched samples had a significant (P<0.05) decrease in the percentage of T cells over time.
This, they say, could provide a possible explanation for the development of early hepatotoxicity.
The trial is investigator-initiated and funded by Gilead Sciences.
*Data in the presentation differs from the abstract.
Combination offers ‘important new option’ for CLL, team says

Photo courtesy of ASH
ORLANDO, FL—Idelalisib, the first-in-class PI3Kδ inhibitor, combined with bendamustine and rituximab (BR) for relapsed/refractory chronic
lymphocytic leukemia (CLL) offers “an important new option over the standard of care,” according to Andrew Zelenetz, MD, a member of the
international research team that conducted the phase 3 study of this combination.
Patients who received idelalisib plus BR experienced a much longer progression-free survival (PFS) than those who received BR alone, 23.1
months versus 11.1 months, respectively.
“And the benefit was seen across risk groups,” Dr Zelenetz said.
He pointed out that the trial was stopped early in October because of the “overwhelming benefit” of idelalisib compared to the conventional therapy arm.
Dr Zelenetz, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the findings at the 2015 ASH Annual Meeting as LBA-5.
Idelalisib had already been approved by the US Food and Drug Administration for the treatment of relapsed/refractory CLL.
“Many people refer to this [idelalisib] as a B-cell receptor drug,” Dr Zelenetz said, “but it is more than that. It is involved in signaling of very key pathways in cell survival and migration.”
The investigators hoped that by combining idelalisib with BR, they would be able to improve PFS and maintain tolerable toxicity. So they conducted Study 115 to find out.
Study 115 design and population
Study 115 was a double-blind, placebo-controlled phase 3 study.
The idelalisib arm consisted of 207 patients randomized to receive bendamustine at 70 mg/m2 on days 1 and 2 every 4 weeks for 6 cycles, rituximab at 375 mg/m2 during cycle 1 and 500 mg/m2 cycles 2 through 6, and idelalisib at 150 mg twice daily until progression.
The BR arm consisted of 209 patients randomized to the same BR regimen plus placebo twice daily until progression.
Investigators stratified patients according to 17p deletion and/or TP52 mutation, IGHV mutation status, and refractory versus relapsed disease.
The primary endpoint was PFS and the secondary endpoints were overall response rate (ORR), nodal response, overall survival (OS), and complete response (CR) rate.
Patients had to have disease progression within less than 36 months from their last therapy, measurable disease, and no history of CLL transformation. They could not have progressed in less than 6 months from their last bendamustine treatment and they could not have had any prior inhibitors of BTK, PI3Kδ, or SYK.
Patient disposition and demographics
One hundred fifteen patients (56%) in the idelalisib arm are still on study, and 52% are on treatment. In the BR arm, 63 patients (30%) are still on study, and 29% are on treatment.
Patient characteristics were well balanced between the arms. Most patients (76%) were male, 58% were younger than 65 years and 42% were 65 or older. About half were Rai stage III/IV and the median number of prior regimens was 2 (range, 1–13).
The most common prior regimens in both arms were fludarabine/cyclophosphamide/rituximab, fludarabine/cyclophosphamide, and chlorambucil. Fifteen percent of patients in the idelalisib arm and 8% in the BR arm had prior BR.
A third of patients in each arm had either 17p deletion or TP53 mutation, and two-thirds had neither. Most patients did not have IGHV mutation—84% in the idelalisib group and 83% in the BR group.
Thirty-one percent of the idelalisib-treated patients and 29% of the placebo patients had refractory disease, and 69% and 71%, respectively, had relapsed disease.
Efficacy
Median PFS, as assessed by independent review committee, “was highly statistically significant,” Dr Zelenetz said, at 23.1 months for idelalisib and 11.1 for BR (P<0.0001).
In addition, all subgroups analyzed favored idelalisib—refractory or relapsed disease, mutation status, cytogenetics, gender, age, and race.
Patients with neither deletion 17p nor TP53 had a hazard ratio of 0.22 favoring the idelalisib group, and patients with either one or the other of those mutations had a hazard ratio of 0.50 favoring idelalisib.
ORR was 68% and 45% for idelalisib and placebo, respectively, with 5% in the idelalisib arm and none in the placebo arm achieving a CR.
Dr Zelenetz pointed out that the CR rate was low largely due to missing confirmatory biopsies.
Ninety-six percent of patients in the idelalisib arm experienced 50% or more reduction in lymph nodes, compared with 61% in the placebo arm.
Patients in the idelalisib arm also experienced a significant improvement in OS of P=0.008 when stratified and P=0.023 when unstratified. Median OS has not been reached in either arm.
There was no difference in survival benefit in patients with refractory disease.
Safety
All patients in the idelalisib arm and 97% in the BR arm experienced an adverse event (AE), with 93% and 76% grade 3 or higher in the idelalisib and BR arms, respectively.
Serious AEs occurred in 66% of idelalisib-treated patients and 44% of placebo patients.
Fifty-four patients (26%) in the idelalisib arm discontinued the study drug due to AEs, and 22 (11%) required a study drug dose reduction. This was compared with 28 patients (13%) discontinuing and 13 patients requiring dose reductions in the placebo arm.
The most frequent AE occurring in more than 10% of patients was neutropenia. Grade 3 or higher neutropenia occurred in 60% of idelalisib patients and 46% of placebo patients.
Most AEs were higher in the idelalisib arm compared with the BR arm, including grade 3 or higher events, such as febrile neutropenia (20%, 6%), anemia (15%, 12%), thrombocytopenia (13%, 12%), pneumonia (11%, 6%), ALT increase (11%, <1%), pyrexia (7%, 3%), diarrhea (7%, 2%), and rash (3%, 0), among others.
Serious AEs occurring in more than 2% of patients were also higher in the idelalisib arm than the BR arm, and included febrile neutropenia (18%, 5%), pneumonia (14%, 6%), pyrexia (12%, 6%), neutropenia (4%, 1%), sepsis (4%, 1%), anemia (2%, 2%), lower respiratory tract infection (2%, 2%), diarrhea (4%, <1%), and neutropenic sepsis (1%, 3%).
The remainder of the serious AEs—urinary tract infection, bronchitis, septic shock, and squamous cell carcinoma—occurred in 2% or fewer patients in either arm.
Dr Zelenetz pointed out that the safety profile is consistent with previously reported studies.
Gilead Sciences developed idelalisib and funded Study 115.
*Data in the abstract differ slightly from data presented at the meeting.
Photo courtesy of ASH
ORLANDO, FL—Idelalisib, the first-in-class PI3Kδ inhibitor, combined with bendamustine and rituximab (BR) for relapsed/refractory chronic
lymphocytic leukemia (CLL) offers “an important new option over the standard of care,” according to Andrew Zelenetz, MD, a member of the
international research team that conducted the phase 3 study of this combination.
Patients who received idelalisib plus BR experienced a much longer progression-free survival (PFS) than those who received BR alone, 23.1
months versus 11.1 months, respectively.
“And the benefit was seen across risk groups,” Dr Zelenetz said.
He pointed out that the trial was stopped early in October because of the “overwhelming benefit” of idelalisib compared to the conventional therapy arm.
Dr Zelenetz, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the findings at the 2015 ASH Annual Meeting as LBA-5.
Idelalisib had already been approved by the US Food and Drug Administration for the treatment of relapsed/refractory CLL.
“Many people refer to this [idelalisib] as a B-cell receptor drug,” Dr Zelenetz said, “but it is more than that. It is involved in signaling of very key pathways in cell survival and migration.”
The investigators hoped that by combining idelalisib with BR, they would be able to improve PFS and maintain tolerable toxicity. So they conducted Study 115 to find out.
Study 115 design and population
Study 115 was a double-blind, placebo-controlled phase 3 study.
The idelalisib arm consisted of 207 patients randomized to receive bendamustine at 70 mg/m2 on days 1 and 2 every 4 weeks for 6 cycles, rituximab at 375 mg/m2 during cycle 1 and 500 mg/m2 cycles 2 through 6, and idelalisib at 150 mg twice daily until progression.
The BR arm consisted of 209 patients randomized to the same BR regimen plus placebo twice daily until progression.
Investigators stratified patients according to 17p deletion and/or TP52 mutation, IGHV mutation status, and refractory versus relapsed disease.
The primary endpoint was PFS and the secondary endpoints were overall response rate (ORR), nodal response, overall survival (OS), and complete response (CR) rate.
Patients had to have disease progression within less than 36 months from their last therapy, measurable disease, and no history of CLL transformation. They could not have progressed in less than 6 months from their last bendamustine treatment and they could not have had any prior inhibitors of BTK, PI3Kδ, or SYK.
Patient disposition and demographics
One hundred fifteen patients (56%) in the idelalisib arm are still on study, and 52% are on treatment. In the BR arm, 63 patients (30%) are still on study, and 29% are on treatment.
Patient characteristics were well balanced between the arms. Most patients (76%) were male, 58% were younger than 65 years and 42% were 65 or older. About half were Rai stage III/IV and the median number of prior regimens was 2 (range, 1–13).
The most common prior regimens in both arms were fludarabine/cyclophosphamide/rituximab, fludarabine/cyclophosphamide, and chlorambucil. Fifteen percent of patients in the idelalisib arm and 8% in the BR arm had prior BR.
A third of patients in each arm had either 17p deletion or TP53 mutation, and two-thirds had neither. Most patients did not have IGHV mutation—84% in the idelalisib group and 83% in the BR group.
Thirty-one percent of the idelalisib-treated patients and 29% of the placebo patients had refractory disease, and 69% and 71%, respectively, had relapsed disease.
Efficacy
Median PFS, as assessed by independent review committee, “was highly statistically significant,” Dr Zelenetz said, at 23.1 months for idelalisib and 11.1 for BR (P<0.0001).
In addition, all subgroups analyzed favored idelalisib—refractory or relapsed disease, mutation status, cytogenetics, gender, age, and race.
Patients with neither deletion 17p nor TP53 had a hazard ratio of 0.22 favoring the idelalisib group, and patients with either one or the other of those mutations had a hazard ratio of 0.50 favoring idelalisib.
ORR was 68% and 45% for idelalisib and placebo, respectively, with 5% in the idelalisib arm and none in the placebo arm achieving a CR.
Dr Zelenetz pointed out that the CR rate was low largely due to missing confirmatory biopsies.
Ninety-six percent of patients in the idelalisib arm experienced 50% or more reduction in lymph nodes, compared with 61% in the placebo arm.
Patients in the idelalisib arm also experienced a significant improvement in OS of P=0.008 when stratified and P=0.023 when unstratified. Median OS has not been reached in either arm.
There was no difference in survival benefit in patients with refractory disease.
Safety
All patients in the idelalisib arm and 97% in the BR arm experienced an adverse event (AE), with 93% and 76% grade 3 or higher in the idelalisib and BR arms, respectively.
Serious AEs occurred in 66% of idelalisib-treated patients and 44% of placebo patients.
Fifty-four patients (26%) in the idelalisib arm discontinued the study drug due to AEs, and 22 (11%) required a study drug dose reduction. This was compared with 28 patients (13%) discontinuing and 13 patients requiring dose reductions in the placebo arm.
The most frequent AE occurring in more than 10% of patients was neutropenia. Grade 3 or higher neutropenia occurred in 60% of idelalisib patients and 46% of placebo patients.
Most AEs were higher in the idelalisib arm compared with the BR arm, including grade 3 or higher events, such as febrile neutropenia (20%, 6%), anemia (15%, 12%), thrombocytopenia (13%, 12%), pneumonia (11%, 6%), ALT increase (11%, <1%), pyrexia (7%, 3%), diarrhea (7%, 2%), and rash (3%, 0), among others.
Serious AEs occurring in more than 2% of patients were also higher in the idelalisib arm than the BR arm, and included febrile neutropenia (18%, 5%), pneumonia (14%, 6%), pyrexia (12%, 6%), neutropenia (4%, 1%), sepsis (4%, 1%), anemia (2%, 2%), lower respiratory tract infection (2%, 2%), diarrhea (4%, <1%), and neutropenic sepsis (1%, 3%).
The remainder of the serious AEs—urinary tract infection, bronchitis, septic shock, and squamous cell carcinoma—occurred in 2% or fewer patients in either arm.
Dr Zelenetz pointed out that the safety profile is consistent with previously reported studies.
Gilead Sciences developed idelalisib and funded Study 115.
*Data in the abstract differ slightly from data presented at the meeting.
Photo courtesy of ASH
ORLANDO, FL—Idelalisib, the first-in-class PI3Kδ inhibitor, combined with bendamustine and rituximab (BR) for relapsed/refractory chronic
lymphocytic leukemia (CLL) offers “an important new option over the standard of care,” according to Andrew Zelenetz, MD, a member of the
international research team that conducted the phase 3 study of this combination.
Patients who received idelalisib plus BR experienced a much longer progression-free survival (PFS) than those who received BR alone, 23.1
months versus 11.1 months, respectively.
“And the benefit was seen across risk groups,” Dr Zelenetz said.
He pointed out that the trial was stopped early in October because of the “overwhelming benefit” of idelalisib compared to the conventional therapy arm.
Dr Zelenetz, of Memorial Sloan Kettering Cancer Center in New York, New York, presented the findings at the 2015 ASH Annual Meeting as LBA-5.
Idelalisib had already been approved by the US Food and Drug Administration for the treatment of relapsed/refractory CLL.
“Many people refer to this [idelalisib] as a B-cell receptor drug,” Dr Zelenetz said, “but it is more than that. It is involved in signaling of very key pathways in cell survival and migration.”
The investigators hoped that by combining idelalisib with BR, they would be able to improve PFS and maintain tolerable toxicity. So they conducted Study 115 to find out.
Study 115 design and population
Study 115 was a double-blind, placebo-controlled phase 3 study.
The idelalisib arm consisted of 207 patients randomized to receive bendamustine at 70 mg/m2 on days 1 and 2 every 4 weeks for 6 cycles, rituximab at 375 mg/m2 during cycle 1 and 500 mg/m2 cycles 2 through 6, and idelalisib at 150 mg twice daily until progression.
The BR arm consisted of 209 patients randomized to the same BR regimen plus placebo twice daily until progression.
Investigators stratified patients according to 17p deletion and/or TP52 mutation, IGHV mutation status, and refractory versus relapsed disease.
The primary endpoint was PFS and the secondary endpoints were overall response rate (ORR), nodal response, overall survival (OS), and complete response (CR) rate.
Patients had to have disease progression within less than 36 months from their last therapy, measurable disease, and no history of CLL transformation. They could not have progressed in less than 6 months from their last bendamustine treatment and they could not have had any prior inhibitors of BTK, PI3Kδ, or SYK.
Patient disposition and demographics
One hundred fifteen patients (56%) in the idelalisib arm are still on study, and 52% are on treatment. In the BR arm, 63 patients (30%) are still on study, and 29% are on treatment.
Patient characteristics were well balanced between the arms. Most patients (76%) were male, 58% were younger than 65 years and 42% were 65 or older. About half were Rai stage III/IV and the median number of prior regimens was 2 (range, 1–13).
The most common prior regimens in both arms were fludarabine/cyclophosphamide/rituximab, fludarabine/cyclophosphamide, and chlorambucil. Fifteen percent of patients in the idelalisib arm and 8% in the BR arm had prior BR.
A third of patients in each arm had either 17p deletion or TP53 mutation, and two-thirds had neither. Most patients did not have IGHV mutation—84% in the idelalisib group and 83% in the BR group.
Thirty-one percent of the idelalisib-treated patients and 29% of the placebo patients had refractory disease, and 69% and 71%, respectively, had relapsed disease.
Efficacy
Median PFS, as assessed by independent review committee, “was highly statistically significant,” Dr Zelenetz said, at 23.1 months for idelalisib and 11.1 for BR (P<0.0001).
In addition, all subgroups analyzed favored idelalisib—refractory or relapsed disease, mutation status, cytogenetics, gender, age, and race.
Patients with neither deletion 17p nor TP53 had a hazard ratio of 0.22 favoring the idelalisib group, and patients with either one or the other of those mutations had a hazard ratio of 0.50 favoring idelalisib.
ORR was 68% and 45% for idelalisib and placebo, respectively, with 5% in the idelalisib arm and none in the placebo arm achieving a CR.
Dr Zelenetz pointed out that the CR rate was low largely due to missing confirmatory biopsies.
Ninety-six percent of patients in the idelalisib arm experienced 50% or more reduction in lymph nodes, compared with 61% in the placebo arm.
Patients in the idelalisib arm also experienced a significant improvement in OS of P=0.008 when stratified and P=0.023 when unstratified. Median OS has not been reached in either arm.
There was no difference in survival benefit in patients with refractory disease.
Safety
All patients in the idelalisib arm and 97% in the BR arm experienced an adverse event (AE), with 93% and 76% grade 3 or higher in the idelalisib and BR arms, respectively.
Serious AEs occurred in 66% of idelalisib-treated patients and 44% of placebo patients.
Fifty-four patients (26%) in the idelalisib arm discontinued the study drug due to AEs, and 22 (11%) required a study drug dose reduction. This was compared with 28 patients (13%) discontinuing and 13 patients requiring dose reductions in the placebo arm.
The most frequent AE occurring in more than 10% of patients was neutropenia. Grade 3 or higher neutropenia occurred in 60% of idelalisib patients and 46% of placebo patients.
Most AEs were higher in the idelalisib arm compared with the BR arm, including grade 3 or higher events, such as febrile neutropenia (20%, 6%), anemia (15%, 12%), thrombocytopenia (13%, 12%), pneumonia (11%, 6%), ALT increase (11%, <1%), pyrexia (7%, 3%), diarrhea (7%, 2%), and rash (3%, 0), among others.
Serious AEs occurring in more than 2% of patients were also higher in the idelalisib arm than the BR arm, and included febrile neutropenia (18%, 5%), pneumonia (14%, 6%), pyrexia (12%, 6%), neutropenia (4%, 1%), sepsis (4%, 1%), anemia (2%, 2%), lower respiratory tract infection (2%, 2%), diarrhea (4%, <1%), and neutropenic sepsis (1%, 3%).
The remainder of the serious AEs—urinary tract infection, bronchitis, septic shock, and squamous cell carcinoma—occurred in 2% or fewer patients in either arm.
Dr Zelenetz pointed out that the safety profile is consistent with previously reported studies.
Gilead Sciences developed idelalisib and funded Study 115.
*Data in the abstract differ slightly from data presented at the meeting.