User login
US agencies update regulations on research subjects

tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and 15 other federal agencies have issued a final rule to update regulations intended to safeguard individuals who participate in research.
Most provisions in the new rule will go into effect in 2018.
The HHS says the new rule strengthens protections for people who volunteer to participate in research, while ensuring that the oversight system does not add inappropriate administrative burdens.
The current regulations, which have been in place since 1991, are often referred to as the “Common Rule.”
In September 2015, HHS and the other Common Rule agencies published a proposed new rule regarding research subjects—a notice of proposed rulemaking (NPRM)—which drew more than 2100 comments.
In response to concerns raised during the review process, the final rule contains a number of significant changes from the NPRM.
Research covered
The final rule does not cover clinical trials that are not federally funded.
The Common Rule has historically applied only to research conducted or supported by a Common Rule department or agency. And, although the NPRM proposed changing this policy, the final rule remains in line with the Common Rule.
Consent
The final rule requires consent forms to provide potential research subjects with a better understanding of a project’s scope so they can make a more fully informed decision about whether to participate.
Consent forms should include a concise explanation—at the beginning of the document—of the information that would be most important to individuals contemplating participation in a particular study, including the purpose of the research, the risks and benefits, and appropriate alternative treatments that might be beneficial to the prospective subject.
The rule also requires that consent forms for certain federally funded clinical trials be posted on a public website.
Institutional review boards
The final rule requires, in many cases, use of a single institutional review board (IRB) for multi-institutional research studies.
However, the final rule has been modified from the NPRM to add substantial increased flexibility in now allowing broad groups of studies (instead of just specific studies) to be removed from this requirement.
Privacy
The final rule does not include the standardized privacy safeguards for identifiable private information and identifiable biospecimens that were proposed in the NPRM.
In most respects, the final rule retains the current approach to privacy standards.
For studies on stored identifiable data or identifiable biospecimens, researchers will have the option of relying on broad consent obtained for future research as an alternative to seeking IRB approval to waive the consent requirement.
As under the current rule, researchers will not have to obtain consent for studies on non-identified stored data or biospecimens.
Exemptions
The final rule establishes new exempt categories of research based on the level of risk they pose to participants.
For example, to reduce unnecessary regulatory burden and allow IRBs to focus their attention on higher-risk studies, there is a new exemption for secondary research involving identifiable private information if the research is regulated by and participants are protected under the HIPAA rules.
Review
The final rule removes the requirement to conduct continuing review of ongoing research studies in certain instances where such review does little to protect subjects.
For more details, see the final rule. ![]()
tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and 15 other federal agencies have issued a final rule to update regulations intended to safeguard individuals who participate in research.
Most provisions in the new rule will go into effect in 2018.
The HHS says the new rule strengthens protections for people who volunteer to participate in research, while ensuring that the oversight system does not add inappropriate administrative burdens.
The current regulations, which have been in place since 1991, are often referred to as the “Common Rule.”
In September 2015, HHS and the other Common Rule agencies published a proposed new rule regarding research subjects—a notice of proposed rulemaking (NPRM)—which drew more than 2100 comments.
In response to concerns raised during the review process, the final rule contains a number of significant changes from the NPRM.
Research covered
The final rule does not cover clinical trials that are not federally funded.
The Common Rule has historically applied only to research conducted or supported by a Common Rule department or agency. And, although the NPRM proposed changing this policy, the final rule remains in line with the Common Rule.
Consent
The final rule requires consent forms to provide potential research subjects with a better understanding of a project’s scope so they can make a more fully informed decision about whether to participate.
Consent forms should include a concise explanation—at the beginning of the document—of the information that would be most important to individuals contemplating participation in a particular study, including the purpose of the research, the risks and benefits, and appropriate alternative treatments that might be beneficial to the prospective subject.
The rule also requires that consent forms for certain federally funded clinical trials be posted on a public website.
Institutional review boards
The final rule requires, in many cases, use of a single institutional review board (IRB) for multi-institutional research studies.
However, the final rule has been modified from the NPRM to add substantial increased flexibility in now allowing broad groups of studies (instead of just specific studies) to be removed from this requirement.
Privacy
The final rule does not include the standardized privacy safeguards for identifiable private information and identifiable biospecimens that were proposed in the NPRM.
In most respects, the final rule retains the current approach to privacy standards.
For studies on stored identifiable data or identifiable biospecimens, researchers will have the option of relying on broad consent obtained for future research as an alternative to seeking IRB approval to waive the consent requirement.
As under the current rule, researchers will not have to obtain consent for studies on non-identified stored data or biospecimens.
Exemptions
The final rule establishes new exempt categories of research based on the level of risk they pose to participants.
For example, to reduce unnecessary regulatory burden and allow IRBs to focus their attention on higher-risk studies, there is a new exemption for secondary research involving identifiable private information if the research is regulated by and participants are protected under the HIPAA rules.
Review
The final rule removes the requirement to conduct continuing review of ongoing research studies in certain instances where such review does little to protect subjects.
For more details, see the final rule. ![]()
tumor in a test tube
Photo by Rhoda Baer
The US Department of Health and Human Services (HHS) and 15 other federal agencies have issued a final rule to update regulations intended to safeguard individuals who participate in research.
Most provisions in the new rule will go into effect in 2018.
The HHS says the new rule strengthens protections for people who volunteer to participate in research, while ensuring that the oversight system does not add inappropriate administrative burdens.
The current regulations, which have been in place since 1991, are often referred to as the “Common Rule.”
In September 2015, HHS and the other Common Rule agencies published a proposed new rule regarding research subjects—a notice of proposed rulemaking (NPRM)—which drew more than 2100 comments.
In response to concerns raised during the review process, the final rule contains a number of significant changes from the NPRM.
Research covered
The final rule does not cover clinical trials that are not federally funded.
The Common Rule has historically applied only to research conducted or supported by a Common Rule department or agency. And, although the NPRM proposed changing this policy, the final rule remains in line with the Common Rule.
Consent
The final rule requires consent forms to provide potential research subjects with a better understanding of a project’s scope so they can make a more fully informed decision about whether to participate.
Consent forms should include a concise explanation—at the beginning of the document—of the information that would be most important to individuals contemplating participation in a particular study, including the purpose of the research, the risks and benefits, and appropriate alternative treatments that might be beneficial to the prospective subject.
The rule also requires that consent forms for certain federally funded clinical trials be posted on a public website.
Institutional review boards
The final rule requires, in many cases, use of a single institutional review board (IRB) for multi-institutional research studies.
However, the final rule has been modified from the NPRM to add substantial increased flexibility in now allowing broad groups of studies (instead of just specific studies) to be removed from this requirement.
Privacy
The final rule does not include the standardized privacy safeguards for identifiable private information and identifiable biospecimens that were proposed in the NPRM.
In most respects, the final rule retains the current approach to privacy standards.
For studies on stored identifiable data or identifiable biospecimens, researchers will have the option of relying on broad consent obtained for future research as an alternative to seeking IRB approval to waive the consent requirement.
As under the current rule, researchers will not have to obtain consent for studies on non-identified stored data or biospecimens.
Exemptions
The final rule establishes new exempt categories of research based on the level of risk they pose to participants.
For example, to reduce unnecessary regulatory burden and allow IRBs to focus their attention on higher-risk studies, there is a new exemption for secondary research involving identifiable private information if the research is regulated by and participants are protected under the HIPAA rules.
Review
The final rule removes the requirement to conduct continuing review of ongoing research studies in certain instances where such review does little to protect subjects.
For more details, see the final rule. ![]()
Pharma is gaming the system for orphan drugs, investigation suggests

Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs. ![]()
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs. ![]()
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs. ![]()
FDA releases draft guidances on biosimilars, medical communications

Photo by Bill Branson
The US Food and Drug Administration (FDA) has released 3 new draft guidance documents for industry.
One document provides an overview of scientific considerations when attempting to demonstrate that a biosimilar product is interchangeable with a reference product.
The other 2 guidance documents are intended to clarify the FDA’s position regarding communications about medical products.
The first draft guidance, “Considerations in Demonstrating Interchangeability With a Reference Product,” is intended to assist applicants in demonstrating that a proposed therapeutic protein product (eg, monoclonal antibodies) is interchangeable with a reference product under section 351(k) of the Public Health Service Act.
An interchangeable biological product is biosimilar to the reference product and can be expected to produce the same clinical result as the reference product in any given patient.
For a biological product that is administered more than once, the risk in terms of safety or diminished efficacy of alternating or switching between the biological product and the reference product must not be greater than the risk of using the reference product without such alternating/switching.
The approval pathway for biosimilar and interchangeable products was established by the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the Affordable Care Act in March 2010.
The FDA’s draft guidance on biosimilars contains information on:
- Factors impacting the type and amount of data and information needed to support a demonstration of interchangeability
- The data and information needed to support a demonstration of interchangeability
- Considerations for the design and analysis of a switching study or studies to support a demonstration of interchangeability
- Recommendations regarding the use of US-licensed reference products in a switching study or studies
- Considerations for developing presentations (eg, container closure systems) for proposed interchangeable products.
In addition to soliciting comments on this draft guidance, the FDA is inviting comments on questions posed in the notice of availability about interchangeability in general, the regulation of interchangeable products over their life cycle, and questions about considerations regarding post-approval manufacturing changes.
The FDA also released a Center for Drug Evaluation and Research “From Our Perspective,” by Leah Christl, describing aspects of this draft guidance.
For more information on how to submit comments on the draft guidance and questions posed in the notice of availability, see the Federal Register notice.
Medical communications
The FDA also released 2 draft guidance documents that, the agency says, will each help provide clarity for medical product companies, as well as other interested parties, on the FDA’s current thinking and recommendations for a few different types of communications about medical products.
The first draft guidance, “Drug and Device Manufacturer Communications with Payors, Formulary Committees, and Similar Entities,” explains the FDA’s current thinking and recommendations on firms’ communication of healthcare economic information about approved drugs under section 502(a) of the Federal Food, Drug, and Cosmetic Act, which was recently amended by the 21st Century Cures Act.
The guidance also answers common questions and provides the FDA’s recommendations regarding firms’ communications to payors about investigational drugs and devices that are not yet approved or cleared for any use.
The second draft guidance, “Medical Product Communications That Are Consistent With the FDA-Required Labeling,” explains the FDA’s current thinking about firms’ medical product communications that include data and information that are not contained in their products’ FDA-required labeling, but that concern the approved or cleared uses of their products.
The FDA has opened a public comment period for each draft guidance.
The agency is also asking for stakeholder input on another, distinct topic—communications about unapproved uses of approved or cleared medical products.
The FDA held a Part 15 hearing in November 2016 to hear from a broad range of stakeholders regarding this topic. The agency has now reopened the comment period for the docket opened in connection with that public hearing for an additional 90 days (until April 10, 2017) to allow interested parties an opportunity to review the 2 draft guidances before submitting comments to any of the relevant dockets.
The FDA also added a document to the docket for the public hearing titled, “Memorandum: Public Health Interests and First Amendment Considerations Related to Manufacturer Communications Regarding Unapproved Uses of Approved or Cleared Medical Products.”
This document provides additional background on the issues the FDA is considering as part of its review of the agency’s rules and policies relating to firm communications regarding unapproved uses of approved or cleared medical products, including a discussion of First Amendment considerations.
The FDA is requesting input on the memorandum as it relates to the questions set forth in the initial notice of public hearing. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has released 3 new draft guidance documents for industry.
One document provides an overview of scientific considerations when attempting to demonstrate that a biosimilar product is interchangeable with a reference product.
The other 2 guidance documents are intended to clarify the FDA’s position regarding communications about medical products.
The first draft guidance, “Considerations in Demonstrating Interchangeability With a Reference Product,” is intended to assist applicants in demonstrating that a proposed therapeutic protein product (eg, monoclonal antibodies) is interchangeable with a reference product under section 351(k) of the Public Health Service Act.
An interchangeable biological product is biosimilar to the reference product and can be expected to produce the same clinical result as the reference product in any given patient.
For a biological product that is administered more than once, the risk in terms of safety or diminished efficacy of alternating or switching between the biological product and the reference product must not be greater than the risk of using the reference product without such alternating/switching.
The approval pathway for biosimilar and interchangeable products was established by the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the Affordable Care Act in March 2010.
The FDA’s draft guidance on biosimilars contains information on:
- Factors impacting the type and amount of data and information needed to support a demonstration of interchangeability
- The data and information needed to support a demonstration of interchangeability
- Considerations for the design and analysis of a switching study or studies to support a demonstration of interchangeability
- Recommendations regarding the use of US-licensed reference products in a switching study or studies
- Considerations for developing presentations (eg, container closure systems) for proposed interchangeable products.
In addition to soliciting comments on this draft guidance, the FDA is inviting comments on questions posed in the notice of availability about interchangeability in general, the regulation of interchangeable products over their life cycle, and questions about considerations regarding post-approval manufacturing changes.
The FDA also released a Center for Drug Evaluation and Research “From Our Perspective,” by Leah Christl, describing aspects of this draft guidance.
For more information on how to submit comments on the draft guidance and questions posed in the notice of availability, see the Federal Register notice.
Medical communications
The FDA also released 2 draft guidance documents that, the agency says, will each help provide clarity for medical product companies, as well as other interested parties, on the FDA’s current thinking and recommendations for a few different types of communications about medical products.
The first draft guidance, “Drug and Device Manufacturer Communications with Payors, Formulary Committees, and Similar Entities,” explains the FDA’s current thinking and recommendations on firms’ communication of healthcare economic information about approved drugs under section 502(a) of the Federal Food, Drug, and Cosmetic Act, which was recently amended by the 21st Century Cures Act.
The guidance also answers common questions and provides the FDA’s recommendations regarding firms’ communications to payors about investigational drugs and devices that are not yet approved or cleared for any use.
The second draft guidance, “Medical Product Communications That Are Consistent With the FDA-Required Labeling,” explains the FDA’s current thinking about firms’ medical product communications that include data and information that are not contained in their products’ FDA-required labeling, but that concern the approved or cleared uses of their products.
The FDA has opened a public comment period for each draft guidance.
The agency is also asking for stakeholder input on another, distinct topic—communications about unapproved uses of approved or cleared medical products.
The FDA held a Part 15 hearing in November 2016 to hear from a broad range of stakeholders regarding this topic. The agency has now reopened the comment period for the docket opened in connection with that public hearing for an additional 90 days (until April 10, 2017) to allow interested parties an opportunity to review the 2 draft guidances before submitting comments to any of the relevant dockets.
The FDA also added a document to the docket for the public hearing titled, “Memorandum: Public Health Interests and First Amendment Considerations Related to Manufacturer Communications Regarding Unapproved Uses of Approved or Cleared Medical Products.”
This document provides additional background on the issues the FDA is considering as part of its review of the agency’s rules and policies relating to firm communications regarding unapproved uses of approved or cleared medical products, including a discussion of First Amendment considerations.
The FDA is requesting input on the memorandum as it relates to the questions set forth in the initial notice of public hearing. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has released 3 new draft guidance documents for industry.
One document provides an overview of scientific considerations when attempting to demonstrate that a biosimilar product is interchangeable with a reference product.
The other 2 guidance documents are intended to clarify the FDA’s position regarding communications about medical products.
The first draft guidance, “Considerations in Demonstrating Interchangeability With a Reference Product,” is intended to assist applicants in demonstrating that a proposed therapeutic protein product (eg, monoclonal antibodies) is interchangeable with a reference product under section 351(k) of the Public Health Service Act.
An interchangeable biological product is biosimilar to the reference product and can be expected to produce the same clinical result as the reference product in any given patient.
For a biological product that is administered more than once, the risk in terms of safety or diminished efficacy of alternating or switching between the biological product and the reference product must not be greater than the risk of using the reference product without such alternating/switching.
The approval pathway for biosimilar and interchangeable products was established by the Biologics Price Competition and Innovation Act of 2009, which was enacted as part of the Affordable Care Act in March 2010.
The FDA’s draft guidance on biosimilars contains information on:
- Factors impacting the type and amount of data and information needed to support a demonstration of interchangeability
- The data and information needed to support a demonstration of interchangeability
- Considerations for the design and analysis of a switching study or studies to support a demonstration of interchangeability
- Recommendations regarding the use of US-licensed reference products in a switching study or studies
- Considerations for developing presentations (eg, container closure systems) for proposed interchangeable products.
In addition to soliciting comments on this draft guidance, the FDA is inviting comments on questions posed in the notice of availability about interchangeability in general, the regulation of interchangeable products over their life cycle, and questions about considerations regarding post-approval manufacturing changes.
The FDA also released a Center for Drug Evaluation and Research “From Our Perspective,” by Leah Christl, describing aspects of this draft guidance.
For more information on how to submit comments on the draft guidance and questions posed in the notice of availability, see the Federal Register notice.
Medical communications
The FDA also released 2 draft guidance documents that, the agency says, will each help provide clarity for medical product companies, as well as other interested parties, on the FDA’s current thinking and recommendations for a few different types of communications about medical products.
The first draft guidance, “Drug and Device Manufacturer Communications with Payors, Formulary Committees, and Similar Entities,” explains the FDA’s current thinking and recommendations on firms’ communication of healthcare economic information about approved drugs under section 502(a) of the Federal Food, Drug, and Cosmetic Act, which was recently amended by the 21st Century Cures Act.
The guidance also answers common questions and provides the FDA’s recommendations regarding firms’ communications to payors about investigational drugs and devices that are not yet approved or cleared for any use.
The second draft guidance, “Medical Product Communications That Are Consistent With the FDA-Required Labeling,” explains the FDA’s current thinking about firms’ medical product communications that include data and information that are not contained in their products’ FDA-required labeling, but that concern the approved or cleared uses of their products.
The FDA has opened a public comment period for each draft guidance.
The agency is also asking for stakeholder input on another, distinct topic—communications about unapproved uses of approved or cleared medical products.
The FDA held a Part 15 hearing in November 2016 to hear from a broad range of stakeholders regarding this topic. The agency has now reopened the comment period for the docket opened in connection with that public hearing for an additional 90 days (until April 10, 2017) to allow interested parties an opportunity to review the 2 draft guidances before submitting comments to any of the relevant dockets.
The FDA also added a document to the docket for the public hearing titled, “Memorandum: Public Health Interests and First Amendment Considerations Related to Manufacturer Communications Regarding Unapproved Uses of Approved or Cleared Medical Products.”
This document provides additional background on the issues the FDA is considering as part of its review of the agency’s rules and policies relating to firm communications regarding unapproved uses of approved or cleared medical products, including a discussion of First Amendment considerations.
The FDA is requesting input on the memorandum as it relates to the questions set forth in the initial notice of public hearing. ![]()
Considering cattle could help fight malaria

Photo by Ilya Mauter
The goal of eliminating malaria in countries like India could be more achievable if mosquito-control efforts take into account the relationship between mosquitoes and cattle, according to an international team of researchers.
The group analyzed 2 mosquito
species found in Odisha, the state with the highest number of malaria cases in
India, and found that both species fed on cattle as well as humans.
“In many parts of the world, the mosquitoes responsible for transmitting malaria are specialist feeders on humans and often rest within human houses,” said Matthew Thomas, PhD, of Pennsylvania State University in University Park, Pennsylvania.
“We found that, in an area of India that has a high burden of malaria, most of the mosquitoes that are known to transmit malaria rest in cattle sheds and feed on both cows and humans.”
Dr Thomas and his colleagues reported these findings in Scientific Reports.
According to the researchers, cattle sheds in India are often next to, and sometimes even share a wall with, human houses. However, current malaria control efforts are restricted to domestic dwellings only.
“Given this cattle-shed ‘refuge’ for mosquitoes, focusing only on humans with regard to malaria control is a bit like treating the tip of an iceberg,” said study author Jessica Waite, PhD, also of Pennsylvania State University.
She, Dr Thomas, and their colleagues determined the importance of cows in the malaria-control problem by capturing adult mosquitoes in different habitats within 6 villages in Odisha state—which has the highest number of malaria cases in the country—and noting where the mosquitoes had been resting.
The team then used molecular techniques to determine which species of mosquitoes had been captured and which hosts they had been feeding on.
The researchers collected 1774 Anopheles culicifacies mosquitoes and 169 Anopheles fluviatilis mosquitoes across all study sites.
Both species were denser in cattle sheds than in human dwellings, and both were feeding on humans as well as cattle.
Next, the researchers used their field-collected data to help build a computer model that simulated the life of an adult mosquito. The team used the model to explore how best to control the mosquitoes to have maximum impact on malaria transmission in these villages.
“Our model analysis suggests that conventional control tools—such as insecticide-treated bed nets and indoor insecticide sprays—are less effective when mosquitoes exhibit ‘zoophilic’ behaviors (having an attraction to nonhuman animals),” Dr Thomas said.
“However, extending controls to better target the zoophilic mosquitoes—for example, by broadening coverage of non-repellant insecticide sprays to include cattle sheds—could help reduce transmission dramatically.”
Dr Waite added that the model suggests very little cattle-based vector control effort would be required to drive malaria transmission in the region to elimination.
“We show that directing even modest amounts of effort to specifically increase mosquito mortality associated with zoophilic behavior can shift the balance towards elimination,” she said.
“Understanding the dynamic between humans, cattle, and mosquitoes could have major implications for malaria control policy and practice, not only in India, but in other areas where transmission is sustained by zoophilic vectors,” Dr Thomas said.
“Specifically, optimizing use of existing tools will be essential to achieving the ambitious 2030 elimination target set by the World Health Organization, which aims to decrease malaria cases globally by 90% compared to 2015 levels and eliminate malaria in at least 35 additional countries by 2030.” ![]()
Photo by Ilya Mauter
The goal of eliminating malaria in countries like India could be more achievable if mosquito-control efforts take into account the relationship between mosquitoes and cattle, according to an international team of researchers.
The group analyzed 2 mosquito
species found in Odisha, the state with the highest number of malaria cases in
India, and found that both species fed on cattle as well as humans.
“In many parts of the world, the mosquitoes responsible for transmitting malaria are specialist feeders on humans and often rest within human houses,” said Matthew Thomas, PhD, of Pennsylvania State University in University Park, Pennsylvania.
“We found that, in an area of India that has a high burden of malaria, most of the mosquitoes that are known to transmit malaria rest in cattle sheds and feed on both cows and humans.”
Dr Thomas and his colleagues reported these findings in Scientific Reports.
According to the researchers, cattle sheds in India are often next to, and sometimes even share a wall with, human houses. However, current malaria control efforts are restricted to domestic dwellings only.
“Given this cattle-shed ‘refuge’ for mosquitoes, focusing only on humans with regard to malaria control is a bit like treating the tip of an iceberg,” said study author Jessica Waite, PhD, also of Pennsylvania State University.
She, Dr Thomas, and their colleagues determined the importance of cows in the malaria-control problem by capturing adult mosquitoes in different habitats within 6 villages in Odisha state—which has the highest number of malaria cases in the country—and noting where the mosquitoes had been resting.
The team then used molecular techniques to determine which species of mosquitoes had been captured and which hosts they had been feeding on.
The researchers collected 1774 Anopheles culicifacies mosquitoes and 169 Anopheles fluviatilis mosquitoes across all study sites.
Both species were denser in cattle sheds than in human dwellings, and both were feeding on humans as well as cattle.
Next, the researchers used their field-collected data to help build a computer model that simulated the life of an adult mosquito. The team used the model to explore how best to control the mosquitoes to have maximum impact on malaria transmission in these villages.
“Our model analysis suggests that conventional control tools—such as insecticide-treated bed nets and indoor insecticide sprays—are less effective when mosquitoes exhibit ‘zoophilic’ behaviors (having an attraction to nonhuman animals),” Dr Thomas said.
“However, extending controls to better target the zoophilic mosquitoes—for example, by broadening coverage of non-repellant insecticide sprays to include cattle sheds—could help reduce transmission dramatically.”
Dr Waite added that the model suggests very little cattle-based vector control effort would be required to drive malaria transmission in the region to elimination.
“We show that directing even modest amounts of effort to specifically increase mosquito mortality associated with zoophilic behavior can shift the balance towards elimination,” she said.
“Understanding the dynamic between humans, cattle, and mosquitoes could have major implications for malaria control policy and practice, not only in India, but in other areas where transmission is sustained by zoophilic vectors,” Dr Thomas said.
“Specifically, optimizing use of existing tools will be essential to achieving the ambitious 2030 elimination target set by the World Health Organization, which aims to decrease malaria cases globally by 90% compared to 2015 levels and eliminate malaria in at least 35 additional countries by 2030.” ![]()
Photo by Ilya Mauter
The goal of eliminating malaria in countries like India could be more achievable if mosquito-control efforts take into account the relationship between mosquitoes and cattle, according to an international team of researchers.
The group analyzed 2 mosquito
species found in Odisha, the state with the highest number of malaria cases in
India, and found that both species fed on cattle as well as humans.
“In many parts of the world, the mosquitoes responsible for transmitting malaria are specialist feeders on humans and often rest within human houses,” said Matthew Thomas, PhD, of Pennsylvania State University in University Park, Pennsylvania.
“We found that, in an area of India that has a high burden of malaria, most of the mosquitoes that are known to transmit malaria rest in cattle sheds and feed on both cows and humans.”
Dr Thomas and his colleagues reported these findings in Scientific Reports.
According to the researchers, cattle sheds in India are often next to, and sometimes even share a wall with, human houses. However, current malaria control efforts are restricted to domestic dwellings only.
“Given this cattle-shed ‘refuge’ for mosquitoes, focusing only on humans with regard to malaria control is a bit like treating the tip of an iceberg,” said study author Jessica Waite, PhD, also of Pennsylvania State University.
She, Dr Thomas, and their colleagues determined the importance of cows in the malaria-control problem by capturing adult mosquitoes in different habitats within 6 villages in Odisha state—which has the highest number of malaria cases in the country—and noting where the mosquitoes had been resting.
The team then used molecular techniques to determine which species of mosquitoes had been captured and which hosts they had been feeding on.
The researchers collected 1774 Anopheles culicifacies mosquitoes and 169 Anopheles fluviatilis mosquitoes across all study sites.
Both species were denser in cattle sheds than in human dwellings, and both were feeding on humans as well as cattle.
Next, the researchers used their field-collected data to help build a computer model that simulated the life of an adult mosquito. The team used the model to explore how best to control the mosquitoes to have maximum impact on malaria transmission in these villages.
“Our model analysis suggests that conventional control tools—such as insecticide-treated bed nets and indoor insecticide sprays—are less effective when mosquitoes exhibit ‘zoophilic’ behaviors (having an attraction to nonhuman animals),” Dr Thomas said.
“However, extending controls to better target the zoophilic mosquitoes—for example, by broadening coverage of non-repellant insecticide sprays to include cattle sheds—could help reduce transmission dramatically.”
Dr Waite added that the model suggests very little cattle-based vector control effort would be required to drive malaria transmission in the region to elimination.
“We show that directing even modest amounts of effort to specifically increase mosquito mortality associated with zoophilic behavior can shift the balance towards elimination,” she said.
“Understanding the dynamic between humans, cattle, and mosquitoes could have major implications for malaria control policy and practice, not only in India, but in other areas where transmission is sustained by zoophilic vectors,” Dr Thomas said.
“Specifically, optimizing use of existing tools will be essential to achieving the ambitious 2030 elimination target set by the World Health Organization, which aims to decrease malaria cases globally by 90% compared to 2015 levels and eliminate malaria in at least 35 additional countries by 2030.” ![]()
FDA issues CRL for IV formulation of antiemetic agent

chemotherapy
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) regarding the new drug application (NDA) for an intravenous (IV) formulation of rolapitant.
An oral formulation of rolapitant, marketed as VARUBI®, is FDA-approved for use in combination with other antiemetic agents to prevent delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy in adults.
The NDA for rolapitant IV is for the same indication.
The FDA requested additional information regarding the in vitro method utilized to demonstrate comparability of drug product produced at the 2 proposed commercial manufacturers for rolapitant IV that were included in the NDA.
TESARO Inc., the company developing rolapitant IV, said it is working to provide the requested information.
The CRL did not identify concerns related to the safety or efficacy of rolapitant IV or request additional clinical studies. No concerns were raised regarding the active pharmaceutical ingredient, which is also used for VARUBI®.
TESARO identified potential deficiencies at the original contract manufacturer for rolapitant IV, secured a second drug product supplier, and included data from this manufacturer in the NDA.
During the NDA review, the FDA requested and TESARO provided in vitro data to demonstrate comparability of drug product made at the 2 manufacturing sites.
“TESARO is committed to bringing this new intravenous formulation of rolapitant to physicians and patients to enable additional flexibility and choice of antiemetic regimens, and we plan to address FDA’s questions expeditiously and complete this application, which we expect to enable approval in the first half of 2017,” said Mary Lynne Hedley, PhD, president and chief operating officer of TESARO. ![]()
chemotherapy
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) regarding the new drug application (NDA) for an intravenous (IV) formulation of rolapitant.
An oral formulation of rolapitant, marketed as VARUBI®, is FDA-approved for use in combination with other antiemetic agents to prevent delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy in adults.
The NDA for rolapitant IV is for the same indication.
The FDA requested additional information regarding the in vitro method utilized to demonstrate comparability of drug product produced at the 2 proposed commercial manufacturers for rolapitant IV that were included in the NDA.
TESARO Inc., the company developing rolapitant IV, said it is working to provide the requested information.
The CRL did not identify concerns related to the safety or efficacy of rolapitant IV or request additional clinical studies. No concerns were raised regarding the active pharmaceutical ingredient, which is also used for VARUBI®.
TESARO identified potential deficiencies at the original contract manufacturer for rolapitant IV, secured a second drug product supplier, and included data from this manufacturer in the NDA.
During the NDA review, the FDA requested and TESARO provided in vitro data to demonstrate comparability of drug product made at the 2 manufacturing sites.
“TESARO is committed to bringing this new intravenous formulation of rolapitant to physicians and patients to enable additional flexibility and choice of antiemetic regimens, and we plan to address FDA’s questions expeditiously and complete this application, which we expect to enable approval in the first half of 2017,” said Mary Lynne Hedley, PhD, president and chief operating officer of TESARO. ![]()
chemotherapy
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has issued a complete response letter (CRL) regarding the new drug application (NDA) for an intravenous (IV) formulation of rolapitant.
An oral formulation of rolapitant, marketed as VARUBI®, is FDA-approved for use in combination with other antiemetic agents to prevent delayed nausea and vomiting associated with initial and repeat courses of emetogenic cancer chemotherapy in adults.
The NDA for rolapitant IV is for the same indication.
The FDA requested additional information regarding the in vitro method utilized to demonstrate comparability of drug product produced at the 2 proposed commercial manufacturers for rolapitant IV that were included in the NDA.
TESARO Inc., the company developing rolapitant IV, said it is working to provide the requested information.
The CRL did not identify concerns related to the safety or efficacy of rolapitant IV or request additional clinical studies. No concerns were raised regarding the active pharmaceutical ingredient, which is also used for VARUBI®.
TESARO identified potential deficiencies at the original contract manufacturer for rolapitant IV, secured a second drug product supplier, and included data from this manufacturer in the NDA.
During the NDA review, the FDA requested and TESARO provided in vitro data to demonstrate comparability of drug product made at the 2 manufacturing sites.
“TESARO is committed to bringing this new intravenous formulation of rolapitant to physicians and patients to enable additional flexibility and choice of antiemetic regimens, and we plan to address FDA’s questions expeditiously and complete this application, which we expect to enable approval in the first half of 2017,” said Mary Lynne Hedley, PhD, president and chief operating officer of TESARO. ![]()
Malaria infection depends on number of parasites in mosquitoes
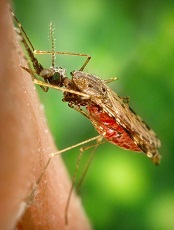
Photo by James Gathany
Mosquitoes carrying a greater number of malaria-causing parasites may be more likely to cause infection, according to a study published in PLOS Pathogens.
More than 100 years have passed since scientists first discovered that infectious mosquitoes inject malaria parasites when they bite people.
However, it hasn’t been clear whether injecting more parasites with each bite increases a person’s chances of infection or if all infectious bites are equally dangerous.
In the new study, researchers set out to determine whether the number of parasites found in the salivary glands of malaria-carrying mosquitos impacts disease transmission.
Via experiments in mice, the team determined that the more parasites present in a mosquito’s salivary glands, the more likely it was to be infectious and the faster an infection would develop.
“It is surprising that the relationship between parasite density and infectiousness has not been properly investigated before, but the studies are quite complex to carry out,” noted Andrew Blagborough, PhD, of Imperial College London in the UK.
For this study, he and his colleagues set up repeated cycles of infection so that groups of infected mosquitoes containing variable numbers of parasites repeatedly bit sedated mice, transmitting malaria to them under a range of transmission settings.
This allowed the researchers to track how many individual parasites different mosquitoes harbored, how many mice were infected as a result of exposure to them, and how long it took the mice to develop malaria.
The team also analyzed data from human volunteers who were exposed to bites from infectious mosquitoes.
Dissection of these mosquitoes revealed that infection was significantly more likely—and occurred sooner—after bites from mosquitoes with more than 1000 individual parasites in their salivary glands.
By conducting further studies with mice and human volunteers, the researchers were able to provide an explanation for why the malaria vaccine RTS,S is effective only some of the time and why any protection rapidly drops off after 3 years.
The vaccine was less effective when mice or humans were bitten by mosquitoes carrying a greater number of parasites. The researchers think this is because the vaccine can only kill a certain proportion of the parasites and is overwhelmed when the parasite population is too large.
“Vaccine development has come a long way, and this new insight should help future vaccine studies to be tested more rigorously,” said study author Thomas Churcher, PhD, of Imperial College London.
“However, in the end, it is unlikely that one magic bullet will eradicate malaria, and we should continue to seek and apply combinations of strategies for reducing the burden of this disease.” ![]()
Photo by James Gathany
Mosquitoes carrying a greater number of malaria-causing parasites may be more likely to cause infection, according to a study published in PLOS Pathogens.
More than 100 years have passed since scientists first discovered that infectious mosquitoes inject malaria parasites when they bite people.
However, it hasn’t been clear whether injecting more parasites with each bite increases a person’s chances of infection or if all infectious bites are equally dangerous.
In the new study, researchers set out to determine whether the number of parasites found in the salivary glands of malaria-carrying mosquitos impacts disease transmission.
Via experiments in mice, the team determined that the more parasites present in a mosquito’s salivary glands, the more likely it was to be infectious and the faster an infection would develop.
“It is surprising that the relationship between parasite density and infectiousness has not been properly investigated before, but the studies are quite complex to carry out,” noted Andrew Blagborough, PhD, of Imperial College London in the UK.
For this study, he and his colleagues set up repeated cycles of infection so that groups of infected mosquitoes containing variable numbers of parasites repeatedly bit sedated mice, transmitting malaria to them under a range of transmission settings.
This allowed the researchers to track how many individual parasites different mosquitoes harbored, how many mice were infected as a result of exposure to them, and how long it took the mice to develop malaria.
The team also analyzed data from human volunteers who were exposed to bites from infectious mosquitoes.
Dissection of these mosquitoes revealed that infection was significantly more likely—and occurred sooner—after bites from mosquitoes with more than 1000 individual parasites in their salivary glands.
By conducting further studies with mice and human volunteers, the researchers were able to provide an explanation for why the malaria vaccine RTS,S is effective only some of the time and why any protection rapidly drops off after 3 years.
The vaccine was less effective when mice or humans were bitten by mosquitoes carrying a greater number of parasites. The researchers think this is because the vaccine can only kill a certain proportion of the parasites and is overwhelmed when the parasite population is too large.
“Vaccine development has come a long way, and this new insight should help future vaccine studies to be tested more rigorously,” said study author Thomas Churcher, PhD, of Imperial College London.
“However, in the end, it is unlikely that one magic bullet will eradicate malaria, and we should continue to seek and apply combinations of strategies for reducing the burden of this disease.” ![]()
Photo by James Gathany
Mosquitoes carrying a greater number of malaria-causing parasites may be more likely to cause infection, according to a study published in PLOS Pathogens.
More than 100 years have passed since scientists first discovered that infectious mosquitoes inject malaria parasites when they bite people.
However, it hasn’t been clear whether injecting more parasites with each bite increases a person’s chances of infection or if all infectious bites are equally dangerous.
In the new study, researchers set out to determine whether the number of parasites found in the salivary glands of malaria-carrying mosquitos impacts disease transmission.
Via experiments in mice, the team determined that the more parasites present in a mosquito’s salivary glands, the more likely it was to be infectious and the faster an infection would develop.
“It is surprising that the relationship between parasite density and infectiousness has not been properly investigated before, but the studies are quite complex to carry out,” noted Andrew Blagborough, PhD, of Imperial College London in the UK.
For this study, he and his colleagues set up repeated cycles of infection so that groups of infected mosquitoes containing variable numbers of parasites repeatedly bit sedated mice, transmitting malaria to them under a range of transmission settings.
This allowed the researchers to track how many individual parasites different mosquitoes harbored, how many mice were infected as a result of exposure to them, and how long it took the mice to develop malaria.
The team also analyzed data from human volunteers who were exposed to bites from infectious mosquitoes.
Dissection of these mosquitoes revealed that infection was significantly more likely—and occurred sooner—after bites from mosquitoes with more than 1000 individual parasites in their salivary glands.
By conducting further studies with mice and human volunteers, the researchers were able to provide an explanation for why the malaria vaccine RTS,S is effective only some of the time and why any protection rapidly drops off after 3 years.
The vaccine was less effective when mice or humans were bitten by mosquitoes carrying a greater number of parasites. The researchers think this is because the vaccine can only kill a certain proportion of the parasites and is overwhelmed when the parasite population is too large.
“Vaccine development has come a long way, and this new insight should help future vaccine studies to be tested more rigorously,” said study author Thomas Churcher, PhD, of Imperial College London.
“However, in the end, it is unlikely that one magic bullet will eradicate malaria, and we should continue to seek and apply combinations of strategies for reducing the burden of this disease.” ![]()
FDA warns against use of ‘anticancer’ product

Photo by James Heilman
The US Food and Drug Administration (FDA) is warning consumers not to purchase or use PNC-27, a product being promoted and sold through PNC27.com as a treatment for all cancers.
The FDA has not evaluated or approved PNC-27 as safe and effective to treat any disease, including any form of cancer.
In addition, an FDA laboratory discovered the bacteria Variovorax paradoxus in a PNC-27 solution sample for inhalation.
The FDA has not received reports of illnesses or serious adverse events related to PNC-27.
However, the agency said consumers who use a contaminated product are at risk for serious, potentially life-threatening infections.
Individuals at higher risk include vulnerable populations, such as young children, elderly people, pregnant women, and individuals with weakened immune systems.
PNC-27 may be available in various dosage forms, such as a nebulized solution, intravenous solution, vaginal suppository, or rectal suppository.
The FDA recommends that patients who have used any PNC-27 product and have concerns contact their healthcare provider as soon as possible.
The agency is also encouraging healthcare professionals and consumers to report any adverse events possibly related to the use of a PNC-27 product to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Photo by James Heilman
The US Food and Drug Administration (FDA) is warning consumers not to purchase or use PNC-27, a product being promoted and sold through PNC27.com as a treatment for all cancers.
The FDA has not evaluated or approved PNC-27 as safe and effective to treat any disease, including any form of cancer.
In addition, an FDA laboratory discovered the bacteria Variovorax paradoxus in a PNC-27 solution sample for inhalation.
The FDA has not received reports of illnesses or serious adverse events related to PNC-27.
However, the agency said consumers who use a contaminated product are at risk for serious, potentially life-threatening infections.
Individuals at higher risk include vulnerable populations, such as young children, elderly people, pregnant women, and individuals with weakened immune systems.
PNC-27 may be available in various dosage forms, such as a nebulized solution, intravenous solution, vaginal suppository, or rectal suppository.
The FDA recommends that patients who have used any PNC-27 product and have concerns contact their healthcare provider as soon as possible.
The agency is also encouraging healthcare professionals and consumers to report any adverse events possibly related to the use of a PNC-27 product to the FDA’s MedWatch Adverse Event Reporting Program. ![]()
Photo by James Heilman
The US Food and Drug Administration (FDA) is warning consumers not to purchase or use PNC-27, a product being promoted and sold through PNC27.com as a treatment for all cancers.
The FDA has not evaluated or approved PNC-27 as safe and effective to treat any disease, including any form of cancer.
In addition, an FDA laboratory discovered the bacteria Variovorax paradoxus in a PNC-27 solution sample for inhalation.
The FDA has not received reports of illnesses or serious adverse events related to PNC-27.
However, the agency said consumers who use a contaminated product are at risk for serious, potentially life-threatening infections.
Individuals at higher risk include vulnerable populations, such as young children, elderly people, pregnant women, and individuals with weakened immune systems.
PNC-27 may be available in various dosage forms, such as a nebulized solution, intravenous solution, vaginal suppository, or rectal suppository.
The FDA recommends that patients who have used any PNC-27 product and have concerns contact their healthcare provider as soon as possible.
The agency is also encouraging healthcare professionals and consumers to report any adverse events possibly related to the use of a PNC-27 product to the FDA’s MedWatch Adverse Event Reporting Program.
NCI launches program to expedite drug research
Photo by Bill Branson
The National Cancer Institute (NCI) has launched a new drug formulary designed to provide investigators at NCI-designated cancer centers with quicker access to approved and investigational agents for use in preclinical studies and clinical trials.
The formulary will enable NCI to act as an intermediary between investigators and participating pharmaceutical companies, facilitating the arrangements for access to and use of pharmaceutical agents.
Following company approval, investigators will be able to obtain agents from the formulary and test them in preclinical or clinical studies, including studies combining formulary agents from different companies.
The NCI says having agents available through the formulary will expedite the start of clinical trials by alleviating the lengthy negotiation process—sometimes up to 18 months—that has been required for investigators to access such agents on their own.
“The NCI Formulary will help researchers begin testing promising drug combinations more quickly, potentially helping patients much sooner,” said NCI Acting Director Douglas Lowy, MD.
“Rather than spending time negotiating agreements, investigators will be able to focus on the important research that can ultimately lead to improved cancer care.”
The NCI Formulary includes 15 targeted agents:
- Alectinib (ALK inhibitor, tyrosine kinase inhibitor)
- Atezolizumab (PD-L1 blocking monoclonal antibody)
- Bevacizumab (anti-angiogenesis inhibitor, monoclonal antibody)
- Cobimetinib (MEK1/2 inhibitor)
- Ensartinib (ALK inhibitor)
- Ipilimumab (anti-CTLA-4 monoclonal antibody)
- Larotrectinib (tyrosine kinase inhibitor)
- LY3039478 (Notch inhibitor)
- Nivolumab (PD-1 blocking monoclonal antibody)
- Obinutuzumab (anti-CD20 monoclonal antibody)
- Pertuzumab (anti-HER2 monoclonal antibody)
- Prexasertib (checkpoint kinase 1 inhibitor)
- Trastuzumab (anti-HER2 monoclonal antibody)
- Vemurafenib (BRAF mutant v600 inhibitor)
- Vismodegib (Hedgehog inhibitor)
The agents are products of 6 different pharmaceutical companies: Bristol-Myers Squibb, Eli Lilly and Company, Genentech, Kyowa Hakko Kirin, Loxo Oncology, and Xcovery Holding Company LLC.
“The agreements with these companies demonstrate our shared commitment to expedite cancer clinical trials and improve outcomes for patients,” said James Doroshow, MD, NCI deputy director for clinical and translational research.
“We are very pleased that several additional pharmaceutical companies have already pledged a willingness to participate and are in various stages of negotiation with NCI. By the end of 2017, we expect to have doubled the number of partnerships and drugs available in the NCI Formulary.”
Photo by Bill Branson
The National Cancer Institute (NCI) has launched a new drug formulary designed to provide investigators at NCI-designated cancer centers with quicker access to approved and investigational agents for use in preclinical studies and clinical trials.
The formulary will enable NCI to act as an intermediary between investigators and participating pharmaceutical companies, facilitating the arrangements for access to and use of pharmaceutical agents.
Following company approval, investigators will be able to obtain agents from the formulary and test them in preclinical or clinical studies, including studies combining formulary agents from different companies.
The NCI says having agents available through the formulary will expedite the start of clinical trials by alleviating the lengthy negotiation process—sometimes up to 18 months—that has been required for investigators to access such agents on their own.
“The NCI Formulary will help researchers begin testing promising drug combinations more quickly, potentially helping patients much sooner,” said NCI Acting Director Douglas Lowy, MD.
“Rather than spending time negotiating agreements, investigators will be able to focus on the important research that can ultimately lead to improved cancer care.”
The NCI Formulary includes 15 targeted agents:
- Alectinib (ALK inhibitor, tyrosine kinase inhibitor)
- Atezolizumab (PD-L1 blocking monoclonal antibody)
- Bevacizumab (anti-angiogenesis inhibitor, monoclonal antibody)
- Cobimetinib (MEK1/2 inhibitor)
- Ensartinib (ALK inhibitor)
- Ipilimumab (anti-CTLA-4 monoclonal antibody)
- Larotrectinib (tyrosine kinase inhibitor)
- LY3039478 (Notch inhibitor)
- Nivolumab (PD-1 blocking monoclonal antibody)
- Obinutuzumab (anti-CD20 monoclonal antibody)
- Pertuzumab (anti-HER2 monoclonal antibody)
- Prexasertib (checkpoint kinase 1 inhibitor)
- Trastuzumab (anti-HER2 monoclonal antibody)
- Vemurafenib (BRAF mutant v600 inhibitor)
- Vismodegib (Hedgehog inhibitor)
The agents are products of 6 different pharmaceutical companies: Bristol-Myers Squibb, Eli Lilly and Company, Genentech, Kyowa Hakko Kirin, Loxo Oncology, and Xcovery Holding Company LLC.
“The agreements with these companies demonstrate our shared commitment to expedite cancer clinical trials and improve outcomes for patients,” said James Doroshow, MD, NCI deputy director for clinical and translational research.
“We are very pleased that several additional pharmaceutical companies have already pledged a willingness to participate and are in various stages of negotiation with NCI. By the end of 2017, we expect to have doubled the number of partnerships and drugs available in the NCI Formulary.”
Photo by Bill Branson
The National Cancer Institute (NCI) has launched a new drug formulary designed to provide investigators at NCI-designated cancer centers with quicker access to approved and investigational agents for use in preclinical studies and clinical trials.
The formulary will enable NCI to act as an intermediary between investigators and participating pharmaceutical companies, facilitating the arrangements for access to and use of pharmaceutical agents.
Following company approval, investigators will be able to obtain agents from the formulary and test them in preclinical or clinical studies, including studies combining formulary agents from different companies.
The NCI says having agents available through the formulary will expedite the start of clinical trials by alleviating the lengthy negotiation process—sometimes up to 18 months—that has been required for investigators to access such agents on their own.
“The NCI Formulary will help researchers begin testing promising drug combinations more quickly, potentially helping patients much sooner,” said NCI Acting Director Douglas Lowy, MD.
“Rather than spending time negotiating agreements, investigators will be able to focus on the important research that can ultimately lead to improved cancer care.”
The NCI Formulary includes 15 targeted agents:
- Alectinib (ALK inhibitor, tyrosine kinase inhibitor)
- Atezolizumab (PD-L1 blocking monoclonal antibody)
- Bevacizumab (anti-angiogenesis inhibitor, monoclonal antibody)
- Cobimetinib (MEK1/2 inhibitor)
- Ensartinib (ALK inhibitor)
- Ipilimumab (anti-CTLA-4 monoclonal antibody)
- Larotrectinib (tyrosine kinase inhibitor)
- LY3039478 (Notch inhibitor)
- Nivolumab (PD-1 blocking monoclonal antibody)
- Obinutuzumab (anti-CD20 monoclonal antibody)
- Pertuzumab (anti-HER2 monoclonal antibody)
- Prexasertib (checkpoint kinase 1 inhibitor)
- Trastuzumab (anti-HER2 monoclonal antibody)
- Vemurafenib (BRAF mutant v600 inhibitor)
- Vismodegib (Hedgehog inhibitor)
The agents are products of 6 different pharmaceutical companies: Bristol-Myers Squibb, Eli Lilly and Company, Genentech, Kyowa Hakko Kirin, Loxo Oncology, and Xcovery Holding Company LLC.
“The agreements with these companies demonstrate our shared commitment to expedite cancer clinical trials and improve outcomes for patients,” said James Doroshow, MD, NCI deputy director for clinical and translational research.
“We are very pleased that several additional pharmaceutical companies have already pledged a willingness to participate and are in various stages of negotiation with NCI. By the end of 2017, we expect to have doubled the number of partnerships and drugs available in the NCI Formulary.”
Team uses light to launch drugs from RBCs

Researchers say they’ve developed a technique that uses light to activate a drug stored in circulating red blood cells (RBCs) so the drug is released exactly when and where it’s needed.
The group believes the work could have profound implications for the field of drug delivery.
They say the technique could drastically reduce the amount of drug needed to treat diseases and therefore decrease the risk of side effects.
“Using light to treat a disease site has a lot of benefits beyond the ‘isn’t-that-cool’ factor,” said study author David Lawrence, PhD, of the University of North Carolina at Chapel Hill.
“Those benefits could include avoiding surgery and the risk of infection, making anesthesia unnecessary, and allowing people to treat themselves by shining a light on a problem area, such as an arthritic knee.”
Dr Lawrence and his colleagues described their technique in Angewandte Chemie.
The researchers attached various drug molecules (methotrexate, colchicine, and paclitaxel) to vitamin B12 and loaded the compounds into RBCs, which can circulate for up to 4 months, potentially providing a lasting reservoir of treatment that could be tapped as needed.
The team then demonstrated their ability to overcome a long-time technical hurdle: using long-wavelength light to penetrate deep enough into the body to break molecular bonds; in this case, the drug linked to vitamin B12.
Long-wavelength light can penetrate much more deeply into the body, but it doesn’t carry as much energy as short-wavelength light and cannot typically break molecular bonds.
To activate the drugs with long-wavelength light, the researchers had to determine how to do it in a way that required less energy.
“That’s the trick, and that’s where we’ve been successful,” Dr Lawrence said.
The team solved the energy problem by introducing a weak energy bond between vitamin B12 and the drug and then attaching a fluorescent molecule to the bond.
The fluorescent molecule acts as an antenna, capturing long-wavelength light and using it to cut the bond between the drug and the vitamin carrier.
Dr Lawrence noted that this technique could prove useful in treating cancers for which patients may need to receive a wide array of anticancer agents.
“The problem is, when you start using 4 or 5 very toxic drugs, you’re going to have intolerable side effects,” he said. “However, by focusing powerful drugs at a specific site, it may be possible to significantly reduce or eliminate the side effects that commonly accompany cancer chemotherapy.”
Dr Lawrence has created a company in partnership with the University of North Carolina, Iris BioMed, to further develop the technology to be used in humans.
Researchers say they’ve developed a technique that uses light to activate a drug stored in circulating red blood cells (RBCs) so the drug is released exactly when and where it’s needed.
The group believes the work could have profound implications for the field of drug delivery.
They say the technique could drastically reduce the amount of drug needed to treat diseases and therefore decrease the risk of side effects.
“Using light to treat a disease site has a lot of benefits beyond the ‘isn’t-that-cool’ factor,” said study author David Lawrence, PhD, of the University of North Carolina at Chapel Hill.
“Those benefits could include avoiding surgery and the risk of infection, making anesthesia unnecessary, and allowing people to treat themselves by shining a light on a problem area, such as an arthritic knee.”
Dr Lawrence and his colleagues described their technique in Angewandte Chemie.
The researchers attached various drug molecules (methotrexate, colchicine, and paclitaxel) to vitamin B12 and loaded the compounds into RBCs, which can circulate for up to 4 months, potentially providing a lasting reservoir of treatment that could be tapped as needed.
The team then demonstrated their ability to overcome a long-time technical hurdle: using long-wavelength light to penetrate deep enough into the body to break molecular bonds; in this case, the drug linked to vitamin B12.
Long-wavelength light can penetrate much more deeply into the body, but it doesn’t carry as much energy as short-wavelength light and cannot typically break molecular bonds.
To activate the drugs with long-wavelength light, the researchers had to determine how to do it in a way that required less energy.
“That’s the trick, and that’s where we’ve been successful,” Dr Lawrence said.
The team solved the energy problem by introducing a weak energy bond between vitamin B12 and the drug and then attaching a fluorescent molecule to the bond.
The fluorescent molecule acts as an antenna, capturing long-wavelength light and using it to cut the bond between the drug and the vitamin carrier.
Dr Lawrence noted that this technique could prove useful in treating cancers for which patients may need to receive a wide array of anticancer agents.
“The problem is, when you start using 4 or 5 very toxic drugs, you’re going to have intolerable side effects,” he said. “However, by focusing powerful drugs at a specific site, it may be possible to significantly reduce or eliminate the side effects that commonly accompany cancer chemotherapy.”
Dr Lawrence has created a company in partnership with the University of North Carolina, Iris BioMed, to further develop the technology to be used in humans.
Researchers say they’ve developed a technique that uses light to activate a drug stored in circulating red blood cells (RBCs) so the drug is released exactly when and where it’s needed.
The group believes the work could have profound implications for the field of drug delivery.
They say the technique could drastically reduce the amount of drug needed to treat diseases and therefore decrease the risk of side effects.
“Using light to treat a disease site has a lot of benefits beyond the ‘isn’t-that-cool’ factor,” said study author David Lawrence, PhD, of the University of North Carolina at Chapel Hill.
“Those benefits could include avoiding surgery and the risk of infection, making anesthesia unnecessary, and allowing people to treat themselves by shining a light on a problem area, such as an arthritic knee.”
Dr Lawrence and his colleagues described their technique in Angewandte Chemie.
The researchers attached various drug molecules (methotrexate, colchicine, and paclitaxel) to vitamin B12 and loaded the compounds into RBCs, which can circulate for up to 4 months, potentially providing a lasting reservoir of treatment that could be tapped as needed.
The team then demonstrated their ability to overcome a long-time technical hurdle: using long-wavelength light to penetrate deep enough into the body to break molecular bonds; in this case, the drug linked to vitamin B12.
Long-wavelength light can penetrate much more deeply into the body, but it doesn’t carry as much energy as short-wavelength light and cannot typically break molecular bonds.
To activate the drugs with long-wavelength light, the researchers had to determine how to do it in a way that required less energy.
“That’s the trick, and that’s where we’ve been successful,” Dr Lawrence said.
The team solved the energy problem by introducing a weak energy bond between vitamin B12 and the drug and then attaching a fluorescent molecule to the bond.
The fluorescent molecule acts as an antenna, capturing long-wavelength light and using it to cut the bond between the drug and the vitamin carrier.
Dr Lawrence noted that this technique could prove useful in treating cancers for which patients may need to receive a wide array of anticancer agents.
“The problem is, when you start using 4 or 5 very toxic drugs, you’re going to have intolerable side effects,” he said. “However, by focusing powerful drugs at a specific site, it may be possible to significantly reduce or eliminate the side effects that commonly accompany cancer chemotherapy.”
Dr Lawrence has created a company in partnership with the University of North Carolina, Iris BioMed, to further develop the technology to be used in humans.
Iron deficiency anemia protects kids from malaria better than sickle cell trait

Photo by Aurimas Rimsa
New research suggests that, for young children, iron deficiency anemia offers a
greater protective effect against malaria than sickle cell trait.
The
study indicates that iron deficiency anemia can protect children age 2 and
younger from the blood stage of Plasmodium falciparum malaria, and
treating the anemia with iron supplementation removes this protective
effect.
The researchers reported these findings in EBioMedicine.
“This study is elegant in its simplicity yet remains one of the most
substantial and systematic attempts to unveil the cellular-level
relationship between anemia, iron supplementation, and malaria risk,”
said study author Carla Cerami, MD, PhD, of the Medical Research Council
Unit The Gambia in Banjul, Gambia.
For this study, she and her colleagues analyzed the red blood cells of 135 anemic children who were participating in an iron supplementation trial.
The children ranged in age from 6 months to 24 months and lived in a malaria-endemic region of The Gambia where sickle cell trait was also common.
The children received iron (12 mg/day) through micronutrient powder for 84 days, and the researchers analyzed the children’s red blood cells at baseline, day 49, and day 84.
The team conducted in vitro growth and invasion assays with P falciparum laboratory and field strains. The study’s primary endpoint was in vitro parasite growth in the children’s RBCs.
The researchers found that “anemia substantially reduced the invasion and growth of both laboratory and field strains of P falciparum.” The team noted a roughly 10% growth reduction per standard deviation shift in hemoglobin.
On a population-wide basis, anemia reduced the blood stage of malaria by 15.9%, while the sickle cell trait reduced it by 3.5%.
“Our finding that anemia offers greater natural protection against blood-stage malaria infection than sickle cell trait has led us to formulate the interesting hypothesis that the widespread prevalence of anemia in people of African descent is a genetic signature of malaria,” said study author Morgan Goheen, PhD, of University of North Carolina in Chapel Hill.
The researchers also found that deficits in invasion and growth for blood-stage P falciparum were reversed when anemic children had received 7 weeks of iron supplementation. Parasite growth was 2.4-fold higher after supplementation than it was at baseline (P<0.001).
Prior work by the same research group suggested the increased invasion and growth rates following iron supplementation are caused by the parasites’ strong preference for young red blood cells.
The researchers said these new field results consolidate the evidence that iron supplementation increases the risk of P falciparum malaria and provide support for the use of malaria prophylaxis in
conjunction with iron supplementation, especially during the early
phases of erythroid recovery.
Photo by Aurimas Rimsa
New research suggests that, for young children, iron deficiency anemia offers a
greater protective effect against malaria than sickle cell trait.
The
study indicates that iron deficiency anemia can protect children age 2 and
younger from the blood stage of Plasmodium falciparum malaria, and
treating the anemia with iron supplementation removes this protective
effect.
The researchers reported these findings in EBioMedicine.
“This study is elegant in its simplicity yet remains one of the most
substantial and systematic attempts to unveil the cellular-level
relationship between anemia, iron supplementation, and malaria risk,”
said study author Carla Cerami, MD, PhD, of the Medical Research Council
Unit The Gambia in Banjul, Gambia.
For this study, she and her colleagues analyzed the red blood cells of 135 anemic children who were participating in an iron supplementation trial.
The children ranged in age from 6 months to 24 months and lived in a malaria-endemic region of The Gambia where sickle cell trait was also common.
The children received iron (12 mg/day) through micronutrient powder for 84 days, and the researchers analyzed the children’s red blood cells at baseline, day 49, and day 84.
The team conducted in vitro growth and invasion assays with P falciparum laboratory and field strains. The study’s primary endpoint was in vitro parasite growth in the children’s RBCs.
The researchers found that “anemia substantially reduced the invasion and growth of both laboratory and field strains of P falciparum.” The team noted a roughly 10% growth reduction per standard deviation shift in hemoglobin.
On a population-wide basis, anemia reduced the blood stage of malaria by 15.9%, while the sickle cell trait reduced it by 3.5%.
“Our finding that anemia offers greater natural protection against blood-stage malaria infection than sickle cell trait has led us to formulate the interesting hypothesis that the widespread prevalence of anemia in people of African descent is a genetic signature of malaria,” said study author Morgan Goheen, PhD, of University of North Carolina in Chapel Hill.
The researchers also found that deficits in invasion and growth for blood-stage P falciparum were reversed when anemic children had received 7 weeks of iron supplementation. Parasite growth was 2.4-fold higher after supplementation than it was at baseline (P<0.001).
Prior work by the same research group suggested the increased invasion and growth rates following iron supplementation are caused by the parasites’ strong preference for young red blood cells.
The researchers said these new field results consolidate the evidence that iron supplementation increases the risk of P falciparum malaria and provide support for the use of malaria prophylaxis in
conjunction with iron supplementation, especially during the early
phases of erythroid recovery.
Photo by Aurimas Rimsa
New research suggests that, for young children, iron deficiency anemia offers a
greater protective effect against malaria than sickle cell trait.
The
study indicates that iron deficiency anemia can protect children age 2 and
younger from the blood stage of Plasmodium falciparum malaria, and
treating the anemia with iron supplementation removes this protective
effect.
The researchers reported these findings in EBioMedicine.
“This study is elegant in its simplicity yet remains one of the most
substantial and systematic attempts to unveil the cellular-level
relationship between anemia, iron supplementation, and malaria risk,”
said study author Carla Cerami, MD, PhD, of the Medical Research Council
Unit The Gambia in Banjul, Gambia.
For this study, she and her colleagues analyzed the red blood cells of 135 anemic children who were participating in an iron supplementation trial.
The children ranged in age from 6 months to 24 months and lived in a malaria-endemic region of The Gambia where sickle cell trait was also common.
The children received iron (12 mg/day) through micronutrient powder for 84 days, and the researchers analyzed the children’s red blood cells at baseline, day 49, and day 84.
The team conducted in vitro growth and invasion assays with P falciparum laboratory and field strains. The study’s primary endpoint was in vitro parasite growth in the children’s RBCs.
The researchers found that “anemia substantially reduced the invasion and growth of both laboratory and field strains of P falciparum.” The team noted a roughly 10% growth reduction per standard deviation shift in hemoglobin.
On a population-wide basis, anemia reduced the blood stage of malaria by 15.9%, while the sickle cell trait reduced it by 3.5%.
“Our finding that anemia offers greater natural protection against blood-stage malaria infection than sickle cell trait has led us to formulate the interesting hypothesis that the widespread prevalence of anemia in people of African descent is a genetic signature of malaria,” said study author Morgan Goheen, PhD, of University of North Carolina in Chapel Hill.
The researchers also found that deficits in invasion and growth for blood-stage P falciparum were reversed when anemic children had received 7 weeks of iron supplementation. Parasite growth was 2.4-fold higher after supplementation than it was at baseline (P<0.001).
Prior work by the same research group suggested the increased invasion and growth rates following iron supplementation are caused by the parasites’ strong preference for young red blood cells.
The researchers said these new field results consolidate the evidence that iron supplementation increases the risk of P falciparum malaria and provide support for the use of malaria prophylaxis in
conjunction with iron supplementation, especially during the early
phases of erythroid recovery.