User login
Consensus document issued on hematology research priorities for Europe
A consensus document that summarizes the status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical needs in Europe has been published in the February 2016 issue of Haematologica.
“For the first time, hematologists in Europe came together to develop a road map to guide hematology research in Europe,” Professor Andreas Engert, chair of the European Hematology Association’s Research Roadmap Task Force, said in a written statement. “Hematology ... must focus and collaborate to be efficient and remain successful in improving patient outcomes.”
Some 300 experts from over 20 countries in Europe helped to draft the road map. A wide variety of stakeholders, such as national hematology societies, patient organizations, hematology trial groups, and other European organizations, were consulted to comment on the final draft.
“The document reflects the views of the hematological research community in Europe, Professor Tony Green, president of the European Hematology Association (EHA), noted in the statement. “This is crucial if we want to convince policy makers to support the realization of this important research.”
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations – quite a few of which involve hematology researchers, Europe faces increased health expenditures while budgets are limited,” Professor Ulrich Jäger, chair of the EHA European Affairs Committee, said in the statement. “So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. ... Now it is up to the policy makers in the EU to deliver, too.”
You may find the full article in Haematologica 2016 Jan. doi: 10.3324/haematol.2015.136739.
In a time of restricted federal budgets, research funding becomes somewhat of a luxury. Yet, research and innovation are the primary movers of change and progress, both of which are needed to drive growth to ease budget restrictions. In order to ensure that precious resources are allocated to the most promising endeavors, federal governments establish bureaucracies charged with the task of allocating funding to the “best” proposals. Unfortunately, the task of defining “best” is imprecise and largely subjective.
To address this systemic deficiency and to improve the efficient allocation of resources, the scientific community often provides guidance to funding agencies to help choose among competing proposals. In 2015, the American Society of Hematology (ASH) announced its “Agenda for Hematology Research.” The agenda included recommendations to prioritize funding to projects in the following domains: genomic profiling and chemical biology, immunologic treatments of hematologic malignancies, genome editing and gene therapy, stem cell biology and regenerative medicine, epigenetic mechanisms, and venous thromboembolic disease.

|
Dr. Matt Kalyacio |
More recently, the European Hematology Association has published its Roadmap for European Hematology Research. Ostensibly similar to the ASH Agenda, the EHA document is a much more detailed policy statement that calls out the most promising research opportunities across the nine major components of hematology: normal hematopoiesis, malignant lymphoid disorders, malignant myeloid disease, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation and other cell based therapies.
I find the two documents complementary in that the ASH Agenda is more accessible to grant reviewers and funding agencies, while the EHA Roadmap seems more directed to the scientific community. Whether a grant writer or a grant reviewer, these documents should help researchers focus their applications on preferred projects and help reviewers prioritize proposals.
While laudable in their goals globally, such consensus documents place much faith in the knowable future and less in the unknowable, disruptive future. Researchers with innovative ideas that do not fall into the prioritizations set forth by the community at large might find themselves struggling for resources. This unintended consequence of consensus building risks the loss of inspired science on the altar of groupthink. The “moonshot” championed by Vice-President Biden will be more likely to succeed when consensus science allows for novel approaches that have yet to be revealed.
Dr. Matt Kalaycio is the editor-in-chief of Hematology News and chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute, Cleveland. Leave your comments on our website or write to Dr. Kalaycio at [email protected].
In a time of restricted federal budgets, research funding becomes somewhat of a luxury. Yet, research and innovation are the primary movers of change and progress, both of which are needed to drive growth to ease budget restrictions. In order to ensure that precious resources are allocated to the most promising endeavors, federal governments establish bureaucracies charged with the task of allocating funding to the “best” proposals. Unfortunately, the task of defining “best” is imprecise and largely subjective.
To address this systemic deficiency and to improve the efficient allocation of resources, the scientific community often provides guidance to funding agencies to help choose among competing proposals. In 2015, the American Society of Hematology (ASH) announced its “Agenda for Hematology Research.” The agenda included recommendations to prioritize funding to projects in the following domains: genomic profiling and chemical biology, immunologic treatments of hematologic malignancies, genome editing and gene therapy, stem cell biology and regenerative medicine, epigenetic mechanisms, and venous thromboembolic disease.
|
|
Dr. Matt Kalyacio |
More recently, the European Hematology Association has published its Roadmap for European Hematology Research. Ostensibly similar to the ASH Agenda, the EHA document is a much more detailed policy statement that calls out the most promising research opportunities across the nine major components of hematology: normal hematopoiesis, malignant lymphoid disorders, malignant myeloid disease, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation and other cell based therapies.
I find the two documents complementary in that the ASH Agenda is more accessible to grant reviewers and funding agencies, while the EHA Roadmap seems more directed to the scientific community. Whether a grant writer or a grant reviewer, these documents should help researchers focus their applications on preferred projects and help reviewers prioritize proposals.
While laudable in their goals globally, such consensus documents place much faith in the knowable future and less in the unknowable, disruptive future. Researchers with innovative ideas that do not fall into the prioritizations set forth by the community at large might find themselves struggling for resources. This unintended consequence of consensus building risks the loss of inspired science on the altar of groupthink. The “moonshot” championed by Vice-President Biden will be more likely to succeed when consensus science allows for novel approaches that have yet to be revealed.
Dr. Matt Kalaycio is the editor-in-chief of Hematology News and chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute, Cleveland. Leave your comments on our website or write to Dr. Kalaycio at [email protected].
In a time of restricted federal budgets, research funding becomes somewhat of a luxury. Yet, research and innovation are the primary movers of change and progress, both of which are needed to drive growth to ease budget restrictions. In order to ensure that precious resources are allocated to the most promising endeavors, federal governments establish bureaucracies charged with the task of allocating funding to the “best” proposals. Unfortunately, the task of defining “best” is imprecise and largely subjective.
To address this systemic deficiency and to improve the efficient allocation of resources, the scientific community often provides guidance to funding agencies to help choose among competing proposals. In 2015, the American Society of Hematology (ASH) announced its “Agenda for Hematology Research.” The agenda included recommendations to prioritize funding to projects in the following domains: genomic profiling and chemical biology, immunologic treatments of hematologic malignancies, genome editing and gene therapy, stem cell biology and regenerative medicine, epigenetic mechanisms, and venous thromboembolic disease.
|
|
Dr. Matt Kalyacio |
More recently, the European Hematology Association has published its Roadmap for European Hematology Research. Ostensibly similar to the ASH Agenda, the EHA document is a much more detailed policy statement that calls out the most promising research opportunities across the nine major components of hematology: normal hematopoiesis, malignant lymphoid disorders, malignant myeloid disease, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation and other cell based therapies.
I find the two documents complementary in that the ASH Agenda is more accessible to grant reviewers and funding agencies, while the EHA Roadmap seems more directed to the scientific community. Whether a grant writer or a grant reviewer, these documents should help researchers focus their applications on preferred projects and help reviewers prioritize proposals.
While laudable in their goals globally, such consensus documents place much faith in the knowable future and less in the unknowable, disruptive future. Researchers with innovative ideas that do not fall into the prioritizations set forth by the community at large might find themselves struggling for resources. This unintended consequence of consensus building risks the loss of inspired science on the altar of groupthink. The “moonshot” championed by Vice-President Biden will be more likely to succeed when consensus science allows for novel approaches that have yet to be revealed.
Dr. Matt Kalaycio is the editor-in-chief of Hematology News and chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute, Cleveland. Leave your comments on our website or write to Dr. Kalaycio at [email protected].
A consensus document that summarizes the status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical needs in Europe has been published in the February 2016 issue of Haematologica.
“For the first time, hematologists in Europe came together to develop a road map to guide hematology research in Europe,” Professor Andreas Engert, chair of the European Hematology Association’s Research Roadmap Task Force, said in a written statement. “Hematology ... must focus and collaborate to be efficient and remain successful in improving patient outcomes.”
Some 300 experts from over 20 countries in Europe helped to draft the road map. A wide variety of stakeholders, such as national hematology societies, patient organizations, hematology trial groups, and other European organizations, were consulted to comment on the final draft.
“The document reflects the views of the hematological research community in Europe, Professor Tony Green, president of the European Hematology Association (EHA), noted in the statement. “This is crucial if we want to convince policy makers to support the realization of this important research.”
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations – quite a few of which involve hematology researchers, Europe faces increased health expenditures while budgets are limited,” Professor Ulrich Jäger, chair of the EHA European Affairs Committee, said in the statement. “So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. ... Now it is up to the policy makers in the EU to deliver, too.”
You may find the full article in Haematologica 2016 Jan. doi: 10.3324/haematol.2015.136739.
A consensus document that summarizes the status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical needs in Europe has been published in the February 2016 issue of Haematologica.
“For the first time, hematologists in Europe came together to develop a road map to guide hematology research in Europe,” Professor Andreas Engert, chair of the European Hematology Association’s Research Roadmap Task Force, said in a written statement. “Hematology ... must focus and collaborate to be efficient and remain successful in improving patient outcomes.”
Some 300 experts from over 20 countries in Europe helped to draft the road map. A wide variety of stakeholders, such as national hematology societies, patient organizations, hematology trial groups, and other European organizations, were consulted to comment on the final draft.
“The document reflects the views of the hematological research community in Europe, Professor Tony Green, president of the European Hematology Association (EHA), noted in the statement. “This is crucial if we want to convince policy makers to support the realization of this important research.”
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations – quite a few of which involve hematology researchers, Europe faces increased health expenditures while budgets are limited,” Professor Ulrich Jäger, chair of the EHA European Affairs Committee, said in the statement. “So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. ... Now it is up to the policy makers in the EU to deliver, too.”
You may find the full article in Haematologica 2016 Jan. doi: 10.3324/haematol.2015.136739.

Blocking two targets boosted fetal hemoglobin expression
Two distinct proteins appear to control the switch from fetal to adult globin, based on studies performed in a humanized mouse model and human cells. The findings suggest therapies that target both proteins might induce a fetal-type globin state, which could prove therapeutically useful in individuals with human hemoglobinopathies such as sickle cell disease and thalassemia.
The leukemia/lymphoma-related factor (LRF) and B-cell lymphoma/leukemia 11A (BCL11A) are independently involved in the switch, and blocking their production may turn on fetal globin expression, Takeshi Masuda, Ph.D., of Brigham and Women’s Hospital and Harvard Medical School, Boston, and his colleagues report (SCIENCE. 2015 Jan 15;351[6270]:285-9)
Using a humanized mouse model, the researchers knocked out the ZBTB7A gene, which is responsible for producing LRF. This action boosted the expression of genes that control fetal but not adult-type hemoglobin. Knocking out the gene in human cells also resulted in an increase in fetal hemoglobin proteins.
The researchers then examined BCL11A, which is involved with fetal hemoglobin but does not suppress it. When genes were knocked out for both ZBTB7A and BCL11A in the mice, fetal hemoglobin represented a 91%-94% greater percentage of total hemoglobin than when either gene alone was knocked out.
The research was supported by awards and/or grants from the National Institute of Diabetes and Digestive and Kidney Disease, the Doris Duke Charitable Foundation, the National Institutes of Health, and the American Society of Hematology. Dr. Masuda, along with two other study authors, is a contributor to a patent application filed on behalf of Brigham and Women’s Hospital related to therapeutic targeting of the pathways.
Click here to read the study at Science.
Two distinct proteins appear to control the switch from fetal to adult globin, based on studies performed in a humanized mouse model and human cells. The findings suggest therapies that target both proteins might induce a fetal-type globin state, which could prove therapeutically useful in individuals with human hemoglobinopathies such as sickle cell disease and thalassemia.
The leukemia/lymphoma-related factor (LRF) and B-cell lymphoma/leukemia 11A (BCL11A) are independently involved in the switch, and blocking their production may turn on fetal globin expression, Takeshi Masuda, Ph.D., of Brigham and Women’s Hospital and Harvard Medical School, Boston, and his colleagues report (SCIENCE. 2015 Jan 15;351[6270]:285-9)
Using a humanized mouse model, the researchers knocked out the ZBTB7A gene, which is responsible for producing LRF. This action boosted the expression of genes that control fetal but not adult-type hemoglobin. Knocking out the gene in human cells also resulted in an increase in fetal hemoglobin proteins.
The researchers then examined BCL11A, which is involved with fetal hemoglobin but does not suppress it. When genes were knocked out for both ZBTB7A and BCL11A in the mice, fetal hemoglobin represented a 91%-94% greater percentage of total hemoglobin than when either gene alone was knocked out.
The research was supported by awards and/or grants from the National Institute of Diabetes and Digestive and Kidney Disease, the Doris Duke Charitable Foundation, the National Institutes of Health, and the American Society of Hematology. Dr. Masuda, along with two other study authors, is a contributor to a patent application filed on behalf of Brigham and Women’s Hospital related to therapeutic targeting of the pathways.
Click here to read the study at Science.
Two distinct proteins appear to control the switch from fetal to adult globin, based on studies performed in a humanized mouse model and human cells. The findings suggest therapies that target both proteins might induce a fetal-type globin state, which could prove therapeutically useful in individuals with human hemoglobinopathies such as sickle cell disease and thalassemia.
The leukemia/lymphoma-related factor (LRF) and B-cell lymphoma/leukemia 11A (BCL11A) are independently involved in the switch, and blocking their production may turn on fetal globin expression, Takeshi Masuda, Ph.D., of Brigham and Women’s Hospital and Harvard Medical School, Boston, and his colleagues report (SCIENCE. 2015 Jan 15;351[6270]:285-9)
Using a humanized mouse model, the researchers knocked out the ZBTB7A gene, which is responsible for producing LRF. This action boosted the expression of genes that control fetal but not adult-type hemoglobin. Knocking out the gene in human cells also resulted in an increase in fetal hemoglobin proteins.
The researchers then examined BCL11A, which is involved with fetal hemoglobin but does not suppress it. When genes were knocked out for both ZBTB7A and BCL11A in the mice, fetal hemoglobin represented a 91%-94% greater percentage of total hemoglobin than when either gene alone was knocked out.
The research was supported by awards and/or grants from the National Institute of Diabetes and Digestive and Kidney Disease, the Doris Duke Charitable Foundation, the National Institutes of Health, and the American Society of Hematology. Dr. Masuda, along with two other study authors, is a contributor to a patent application filed on behalf of Brigham and Women’s Hospital related to therapeutic targeting of the pathways.
Click here to read the study at Science.
FROM SCIENCE
ASH: Novel GBT440 reduces sickle cells, improves hematologic parameters
ORLANDO – An experimental agent that restores plasticity to red blood cells significantly improved hematologic parameters and reduced the deformation of red blood cells from sickle cell disease, suggest preliminary results from a phase I/II randomized clinical trial.
The drug, labeled GBT440, was well tolerated over one month, with no serious drug-related adverse events and no evidence of tissue hypoxia, said Dr. Claire Hemmaway from Queens Hospital in Essex, United Kingdom.
“We have emerging data with 28 days of dosing, and this supports the hypothesis that this drug inhibits polymerization of sickle hemoglobin, improves hemolysis, reduces red blood cell damage even in the micro-circulation, and improves oxygen delivery. Longer-term dosing is clearly required and this will define the optimal hematologic effects and the clinical benefit of this drug,” she said at the American Society of Hematology annual meeting.

Although its mechanism of action is not completely understood, GBT440 is a small-molecule hemoglobin modifier which is known to increase hemoglobin oxygen affinity. This first-in-class agent has been shown in both in vitro and in vivo studies to be a strong and direct anti-sickling agent. The drug has also been shown to inhibit polymerization of hemoglobin S, the central event in sickle cell disease, Dr. Hemmaway said.
“We hypothesized that patients on GBT440 will see a reduction in the red cell damage, see a reduction in hemolysis, and an improvement in the anemia, and with an inhibition of the formation of the sickle cells we’ll also see an improvement in blood flow, and this could potentially modify the course of sickle cell disease in our patients,” she said at a briefing prior to her presentation of the data in an oral abstract session.
To test the safety and efficacy of the drug, the investigators enrolled 64 healthy volunteers and 16 patients with homozygous HbSS sickle-cell disease into a phase I/II randomized, double-placebo controlled, parallel group trial.
The patients all had baseline hemoglobin levels from 6 to 10 g/dL, and had not had a vaso-occlusive crisis or transfusion with 30 days of screening.
The study was divided into parts, with part A testing single ascending doses, and part B testing multiple ascending doses compared with placebo on a 6:2 randomization basis.
As of July 24 2015, 54 healthy volunteers had completed the study, two were discontinued due to mild-to-moderate rash and/or headache, and eight were still on follow-up.
Eight patients with sickle cell disease completed part A, and eight were on follow-up in part B. No patients dropped out of the study, although one had a dose reduction from 700 mg to 400 mg because of abdominal discomfort.
Patients and volunteers generally tolerated the drug well, with generally mild adverse events, no deaths, and just one serious adverse event, an acute painful crisis in a patient on placebo.
The drug was shown to increase hemoglobin in both healthy volunteers and patients, reduce reticulocytosis, and improve biomarkers of hemolysis and inflammation. The hematologic effects correlate with GBT440 blood levels, Dr. Hemmaway said.
“You can see dramatic reductions in the reticulocyte counts, which maximally are reduced by more than 50%, and these are maintained over 28 days. The reduction in reticulocyte counts suggests an improvement in red cell life span,” she said.
One patient who received a 700 mg daily oral dose of GBT440 had evidence of a complete absence of sickled cells 28 days after starting on therapy, she noted.
The investigators have not been able to determine whether the hematologic improvements seen in the study correlate with symptomatic improvements, because they have only 28 days of dosing data and the results are still blinded, Dr. Hemmaway said.
A pediatric hematologist who was not involved in the study said that the early results offer hope for patients.
“I think many of us in the field continue to come back to the painful reality that as of today there is only one FDA-approved medication for sickle cell disease. That fundamentally is something that I think many of us are extremely troubled by, especially at a meeting like this where we see so many other opportunities in other conditions,” commented Dr. Alexis Thompson, a professor of hematology at the Robert H. Lurie Children’s Hospital of Chicago, Illinois.
Although the data are early, they are encouraging enough to justify moving forward with a phase II trial, which is planned to start in early 2016.
“I think the person who is going to be the ultimate beneficiary from many of these advances will be a child who can look forward to not only a longer lifespan but a lifespan that is less hampered by the complications of this disease,” she said.
Dr. Thompson moderated the briefing in which Dr. Hemmaway presented the data.
The study is supported by Global Blood Therapeutics. Dr. Hemmaway had no relevant disclosures. Several co-investigators are employees of the company. Dr, Thompson had no disclosures relevant to the study.
ORLANDO – An experimental agent that restores plasticity to red blood cells significantly improved hematologic parameters and reduced the deformation of red blood cells from sickle cell disease, suggest preliminary results from a phase I/II randomized clinical trial.
The drug, labeled GBT440, was well tolerated over one month, with no serious drug-related adverse events and no evidence of tissue hypoxia, said Dr. Claire Hemmaway from Queens Hospital in Essex, United Kingdom.
“We have emerging data with 28 days of dosing, and this supports the hypothesis that this drug inhibits polymerization of sickle hemoglobin, improves hemolysis, reduces red blood cell damage even in the micro-circulation, and improves oxygen delivery. Longer-term dosing is clearly required and this will define the optimal hematologic effects and the clinical benefit of this drug,” she said at the American Society of Hematology annual meeting.
Although its mechanism of action is not completely understood, GBT440 is a small-molecule hemoglobin modifier which is known to increase hemoglobin oxygen affinity. This first-in-class agent has been shown in both in vitro and in vivo studies to be a strong and direct anti-sickling agent. The drug has also been shown to inhibit polymerization of hemoglobin S, the central event in sickle cell disease, Dr. Hemmaway said.
“We hypothesized that patients on GBT440 will see a reduction in the red cell damage, see a reduction in hemolysis, and an improvement in the anemia, and with an inhibition of the formation of the sickle cells we’ll also see an improvement in blood flow, and this could potentially modify the course of sickle cell disease in our patients,” she said at a briefing prior to her presentation of the data in an oral abstract session.
To test the safety and efficacy of the drug, the investigators enrolled 64 healthy volunteers and 16 patients with homozygous HbSS sickle-cell disease into a phase I/II randomized, double-placebo controlled, parallel group trial.
The patients all had baseline hemoglobin levels from 6 to 10 g/dL, and had not had a vaso-occlusive crisis or transfusion with 30 days of screening.
The study was divided into parts, with part A testing single ascending doses, and part B testing multiple ascending doses compared with placebo on a 6:2 randomization basis.
As of July 24 2015, 54 healthy volunteers had completed the study, two were discontinued due to mild-to-moderate rash and/or headache, and eight were still on follow-up.
Eight patients with sickle cell disease completed part A, and eight were on follow-up in part B. No patients dropped out of the study, although one had a dose reduction from 700 mg to 400 mg because of abdominal discomfort.
Patients and volunteers generally tolerated the drug well, with generally mild adverse events, no deaths, and just one serious adverse event, an acute painful crisis in a patient on placebo.
The drug was shown to increase hemoglobin in both healthy volunteers and patients, reduce reticulocytosis, and improve biomarkers of hemolysis and inflammation. The hematologic effects correlate with GBT440 blood levels, Dr. Hemmaway said.
“You can see dramatic reductions in the reticulocyte counts, which maximally are reduced by more than 50%, and these are maintained over 28 days. The reduction in reticulocyte counts suggests an improvement in red cell life span,” she said.
One patient who received a 700 mg daily oral dose of GBT440 had evidence of a complete absence of sickled cells 28 days after starting on therapy, she noted.
The investigators have not been able to determine whether the hematologic improvements seen in the study correlate with symptomatic improvements, because they have only 28 days of dosing data and the results are still blinded, Dr. Hemmaway said.
A pediatric hematologist who was not involved in the study said that the early results offer hope for patients.
“I think many of us in the field continue to come back to the painful reality that as of today there is only one FDA-approved medication for sickle cell disease. That fundamentally is something that I think many of us are extremely troubled by, especially at a meeting like this where we see so many other opportunities in other conditions,” commented Dr. Alexis Thompson, a professor of hematology at the Robert H. Lurie Children’s Hospital of Chicago, Illinois.
Although the data are early, they are encouraging enough to justify moving forward with a phase II trial, which is planned to start in early 2016.
“I think the person who is going to be the ultimate beneficiary from many of these advances will be a child who can look forward to not only a longer lifespan but a lifespan that is less hampered by the complications of this disease,” she said.
Dr. Thompson moderated the briefing in which Dr. Hemmaway presented the data.
The study is supported by Global Blood Therapeutics. Dr. Hemmaway had no relevant disclosures. Several co-investigators are employees of the company. Dr, Thompson had no disclosures relevant to the study.
ORLANDO – An experimental agent that restores plasticity to red blood cells significantly improved hematologic parameters and reduced the deformation of red blood cells from sickle cell disease, suggest preliminary results from a phase I/II randomized clinical trial.
The drug, labeled GBT440, was well tolerated over one month, with no serious drug-related adverse events and no evidence of tissue hypoxia, said Dr. Claire Hemmaway from Queens Hospital in Essex, United Kingdom.
“We have emerging data with 28 days of dosing, and this supports the hypothesis that this drug inhibits polymerization of sickle hemoglobin, improves hemolysis, reduces red blood cell damage even in the micro-circulation, and improves oxygen delivery. Longer-term dosing is clearly required and this will define the optimal hematologic effects and the clinical benefit of this drug,” she said at the American Society of Hematology annual meeting.
Although its mechanism of action is not completely understood, GBT440 is a small-molecule hemoglobin modifier which is known to increase hemoglobin oxygen affinity. This first-in-class agent has been shown in both in vitro and in vivo studies to be a strong and direct anti-sickling agent. The drug has also been shown to inhibit polymerization of hemoglobin S, the central event in sickle cell disease, Dr. Hemmaway said.
“We hypothesized that patients on GBT440 will see a reduction in the red cell damage, see a reduction in hemolysis, and an improvement in the anemia, and with an inhibition of the formation of the sickle cells we’ll also see an improvement in blood flow, and this could potentially modify the course of sickle cell disease in our patients,” she said at a briefing prior to her presentation of the data in an oral abstract session.
To test the safety and efficacy of the drug, the investigators enrolled 64 healthy volunteers and 16 patients with homozygous HbSS sickle-cell disease into a phase I/II randomized, double-placebo controlled, parallel group trial.
The patients all had baseline hemoglobin levels from 6 to 10 g/dL, and had not had a vaso-occlusive crisis or transfusion with 30 days of screening.
The study was divided into parts, with part A testing single ascending doses, and part B testing multiple ascending doses compared with placebo on a 6:2 randomization basis.
As of July 24 2015, 54 healthy volunteers had completed the study, two were discontinued due to mild-to-moderate rash and/or headache, and eight were still on follow-up.
Eight patients with sickle cell disease completed part A, and eight were on follow-up in part B. No patients dropped out of the study, although one had a dose reduction from 700 mg to 400 mg because of abdominal discomfort.
Patients and volunteers generally tolerated the drug well, with generally mild adverse events, no deaths, and just one serious adverse event, an acute painful crisis in a patient on placebo.
The drug was shown to increase hemoglobin in both healthy volunteers and patients, reduce reticulocytosis, and improve biomarkers of hemolysis and inflammation. The hematologic effects correlate with GBT440 blood levels, Dr. Hemmaway said.
“You can see dramatic reductions in the reticulocyte counts, which maximally are reduced by more than 50%, and these are maintained over 28 days. The reduction in reticulocyte counts suggests an improvement in red cell life span,” she said.
One patient who received a 700 mg daily oral dose of GBT440 had evidence of a complete absence of sickled cells 28 days after starting on therapy, she noted.
The investigators have not been able to determine whether the hematologic improvements seen in the study correlate with symptomatic improvements, because they have only 28 days of dosing data and the results are still blinded, Dr. Hemmaway said.
A pediatric hematologist who was not involved in the study said that the early results offer hope for patients.
“I think many of us in the field continue to come back to the painful reality that as of today there is only one FDA-approved medication for sickle cell disease. That fundamentally is something that I think many of us are extremely troubled by, especially at a meeting like this where we see so many other opportunities in other conditions,” commented Dr. Alexis Thompson, a professor of hematology at the Robert H. Lurie Children’s Hospital of Chicago, Illinois.
Although the data are early, they are encouraging enough to justify moving forward with a phase II trial, which is planned to start in early 2016.
“I think the person who is going to be the ultimate beneficiary from many of these advances will be a child who can look forward to not only a longer lifespan but a lifespan that is less hampered by the complications of this disease,” she said.
Dr. Thompson moderated the briefing in which Dr. Hemmaway presented the data.
The study is supported by Global Blood Therapeutics. Dr. Hemmaway had no relevant disclosures. Several co-investigators are employees of the company. Dr, Thompson had no disclosures relevant to the study.
AT ASH 2015
Key clinical point: A first-in class small molecule drug improved hematologic parameters and reduced sickling of red blood cells.
Major finding: GBT440 inhibits polymerization of sickle hemoglobin, improves hemolysis, reduces red blood cell damage, and improves oxygen delivery.
Data source: Randomized, double blind, placebo-controlled phase I/II trial.
Disclosures: The study is supported by Global Blood Therapeutics. Dr. Hemmaway had no relevant disclosures. Several co-investigators are employees of the company. Dr, Thompson had no disclosures relevant to the study.

ASH: Gene therapy restores immune function to older children with SCID-X1
ORLANDO – – Gene therapy can help restore immune function to older children and young adults with X-linked severe combined immunodeficiency, a team of US investigators reports.
“This is the first demonstration of the use of gene therapy to salvage failed allogeneic hematopoietic stem cell transplants in older SCID-X1 patients,” said Dr. Suk See De Ravin from the Laboratory of Host Defenses at the National Institutes of Health in Bethesda, Maryland.
Although allogeneic hematopoietic stem cell transplantation (HSCT) from a matched sibling donor can be curative, transplants using parental bone marrow or genetically modified autologous transplants without myeloconditiong restore T-cell-mediated immunity but not humoral immunity, Dr. De Ravin said at the American Society of Hematology annual meeting.

Additionally, transplants in older children with SCID-X1 who have persistent immune system defects – despite having received a transplant from a haploidentical donor in infancy – leave the patients with serious medical problems, including the life-long need for immunoglobulin G (IgG) supplementation, recurrent and chronic infections, warts, malnutrition, growth failure, and progressive diseases of the gut and lung.
Previous attempts at using gene therapy to correct mutations in IL2RG, the cause of SCID-X1, used mouse retroviral vectors to insert normal IL2RG into autologous hematopoietic stem cells without chemotherapy conditioning. This treatment restored T-cell immunity, but not B-cell- or natural killer (NK)-cell immunity
Of even greater concern is the fact that among infants with SCID-X1 who received autologous stem cells transduced with murine gamma retrovirus carrying the common gamma chain, 25% developed vector-associated leukemia.
To overcome these problems, investigators in the current study used a lentiviral vector containing an insulator fragment from a chicken beta-globin gene. The insulator fragment allows expression of the gamma chain complementary DNA while protecting against up-regulation of neighboring oncogenes.
Dr. De Ravin reported data on five patients from the ages of 7 to 24 years who had worsening immune dysfunction and complex medical problems, including dependence on immunoglobulin G (IgG) supplementations. All of the patients had previously undergone one or more HSCT from haploidentical donors.
The patients were treated with granulocyte-colony stimulating factor and plerixafor to mobilize peripheral blood cells, and then underwent apheresis to isolate CD34 cells. The cells were transduced in vitro with a lentiviral vector, and reinfused into patients after conditioning with low-dose busulfan (6 mg/kg). The vector was developed by researchers at St Jude Children’s Hospital in Memphis, Tennessee.
At the most recent follow-up, the first two patients treated with the protocol had stable engraftment of gamma-chain expressing cells gene with enhanced expression of B, T, and NK cells, with the cells continuing to show improvements, Early data on the remaining four patients indicates a similar positive trend.
Chimerism studies of the patients’ T-cells showed that the host cells were continuing to increase their contribution, suggesting gradual replacement over time of the T-cell graft, Dr. De Ravin said.
In the second patient treated, an increase in NK cells corresponded with an improvement in chronic warts. In addition, the first two patients began to produce both IgG and antigen-specific responses to vaccination, clearance of chronic norovirus infections, and resolution of their protein-losing enteropathy.
“Gene therapy does not appear to reverse prior organ damage, supporting early intervention to improve immunity in these patients before such damage occurs,” Dr. De Ravin said.
The study was supported by the National Institutes of Health. The viral vector was developed at St. Jude Children’s Research Hospital in Memphis, Tennessee. The authors reported no relevant conflicts of interest.
ORLANDO – – Gene therapy can help restore immune function to older children and young adults with X-linked severe combined immunodeficiency, a team of US investigators reports.
“This is the first demonstration of the use of gene therapy to salvage failed allogeneic hematopoietic stem cell transplants in older SCID-X1 patients,” said Dr. Suk See De Ravin from the Laboratory of Host Defenses at the National Institutes of Health in Bethesda, Maryland.
Although allogeneic hematopoietic stem cell transplantation (HSCT) from a matched sibling donor can be curative, transplants using parental bone marrow or genetically modified autologous transplants without myeloconditiong restore T-cell-mediated immunity but not humoral immunity, Dr. De Ravin said at the American Society of Hematology annual meeting.
Additionally, transplants in older children with SCID-X1 who have persistent immune system defects – despite having received a transplant from a haploidentical donor in infancy – leave the patients with serious medical problems, including the life-long need for immunoglobulin G (IgG) supplementation, recurrent and chronic infections, warts, malnutrition, growth failure, and progressive diseases of the gut and lung.
Previous attempts at using gene therapy to correct mutations in IL2RG, the cause of SCID-X1, used mouse retroviral vectors to insert normal IL2RG into autologous hematopoietic stem cells without chemotherapy conditioning. This treatment restored T-cell immunity, but not B-cell- or natural killer (NK)-cell immunity
Of even greater concern is the fact that among infants with SCID-X1 who received autologous stem cells transduced with murine gamma retrovirus carrying the common gamma chain, 25% developed vector-associated leukemia.
To overcome these problems, investigators in the current study used a lentiviral vector containing an insulator fragment from a chicken beta-globin gene. The insulator fragment allows expression of the gamma chain complementary DNA while protecting against up-regulation of neighboring oncogenes.
Dr. De Ravin reported data on five patients from the ages of 7 to 24 years who had worsening immune dysfunction and complex medical problems, including dependence on immunoglobulin G (IgG) supplementations. All of the patients had previously undergone one or more HSCT from haploidentical donors.
The patients were treated with granulocyte-colony stimulating factor and plerixafor to mobilize peripheral blood cells, and then underwent apheresis to isolate CD34 cells. The cells were transduced in vitro with a lentiviral vector, and reinfused into patients after conditioning with low-dose busulfan (6 mg/kg). The vector was developed by researchers at St Jude Children’s Hospital in Memphis, Tennessee.
At the most recent follow-up, the first two patients treated with the protocol had stable engraftment of gamma-chain expressing cells gene with enhanced expression of B, T, and NK cells, with the cells continuing to show improvements, Early data on the remaining four patients indicates a similar positive trend.
Chimerism studies of the patients’ T-cells showed that the host cells were continuing to increase their contribution, suggesting gradual replacement over time of the T-cell graft, Dr. De Ravin said.
In the second patient treated, an increase in NK cells corresponded with an improvement in chronic warts. In addition, the first two patients began to produce both IgG and antigen-specific responses to vaccination, clearance of chronic norovirus infections, and resolution of their protein-losing enteropathy.
“Gene therapy does not appear to reverse prior organ damage, supporting early intervention to improve immunity in these patients before such damage occurs,” Dr. De Ravin said.
The study was supported by the National Institutes of Health. The viral vector was developed at St. Jude Children’s Research Hospital in Memphis, Tennessee. The authors reported no relevant conflicts of interest.
ORLANDO – – Gene therapy can help restore immune function to older children and young adults with X-linked severe combined immunodeficiency, a team of US investigators reports.
“This is the first demonstration of the use of gene therapy to salvage failed allogeneic hematopoietic stem cell transplants in older SCID-X1 patients,” said Dr. Suk See De Ravin from the Laboratory of Host Defenses at the National Institutes of Health in Bethesda, Maryland.
Although allogeneic hematopoietic stem cell transplantation (HSCT) from a matched sibling donor can be curative, transplants using parental bone marrow or genetically modified autologous transplants without myeloconditiong restore T-cell-mediated immunity but not humoral immunity, Dr. De Ravin said at the American Society of Hematology annual meeting.
Additionally, transplants in older children with SCID-X1 who have persistent immune system defects – despite having received a transplant from a haploidentical donor in infancy – leave the patients with serious medical problems, including the life-long need for immunoglobulin G (IgG) supplementation, recurrent and chronic infections, warts, malnutrition, growth failure, and progressive diseases of the gut and lung.
Previous attempts at using gene therapy to correct mutations in IL2RG, the cause of SCID-X1, used mouse retroviral vectors to insert normal IL2RG into autologous hematopoietic stem cells without chemotherapy conditioning. This treatment restored T-cell immunity, but not B-cell- or natural killer (NK)-cell immunity
Of even greater concern is the fact that among infants with SCID-X1 who received autologous stem cells transduced with murine gamma retrovirus carrying the common gamma chain, 25% developed vector-associated leukemia.
To overcome these problems, investigators in the current study used a lentiviral vector containing an insulator fragment from a chicken beta-globin gene. The insulator fragment allows expression of the gamma chain complementary DNA while protecting against up-regulation of neighboring oncogenes.
Dr. De Ravin reported data on five patients from the ages of 7 to 24 years who had worsening immune dysfunction and complex medical problems, including dependence on immunoglobulin G (IgG) supplementations. All of the patients had previously undergone one or more HSCT from haploidentical donors.
The patients were treated with granulocyte-colony stimulating factor and plerixafor to mobilize peripheral blood cells, and then underwent apheresis to isolate CD34 cells. The cells were transduced in vitro with a lentiviral vector, and reinfused into patients after conditioning with low-dose busulfan (6 mg/kg). The vector was developed by researchers at St Jude Children’s Hospital in Memphis, Tennessee.
At the most recent follow-up, the first two patients treated with the protocol had stable engraftment of gamma-chain expressing cells gene with enhanced expression of B, T, and NK cells, with the cells continuing to show improvements, Early data on the remaining four patients indicates a similar positive trend.
Chimerism studies of the patients’ T-cells showed that the host cells were continuing to increase their contribution, suggesting gradual replacement over time of the T-cell graft, Dr. De Ravin said.
In the second patient treated, an increase in NK cells corresponded with an improvement in chronic warts. In addition, the first two patients began to produce both IgG and antigen-specific responses to vaccination, clearance of chronic norovirus infections, and resolution of their protein-losing enteropathy.
“Gene therapy does not appear to reverse prior organ damage, supporting early intervention to improve immunity in these patients before such damage occurs,” Dr. De Ravin said.
The study was supported by the National Institutes of Health. The viral vector was developed at St. Jude Children’s Research Hospital in Memphis, Tennessee. The authors reported no relevant conflicts of interest.
AT ASH 2015
Key clinical point: Gene therapy can correct B, T, and NK cell immunity in older patients with SCID-X1.
Major finding: The first two patients treated with the protocol had stable engraftment of gamma-chain expressing cells gene with enhanced expression of B, T, and NK cells, with the cells continuing to show improvements.
Data source: Clinical study of 5 patients with X-linked severe combined immunodeficiency syndrome (SCID-X1).
Disclosures: The study was supported by the National Institutes of Health. The viral vector was developed at St. Jude Children’s Research Hospital in Memphis, Tennessee. The authors reported no relevant conflicts of interest.

ASH: Gene therapy reduces transfusion needs in beta-thalassemia major
ORLANDO – Lentiviral gene therapy with LentiGlobin BB305 boosts beta-globin production in patients with beta-thalassemia, but frees only some from lifelong dependence on blood transfusions, updated results of the Northstar study show.
Five patients with non-Beta-0/Beta-0 genotypes were able to stop transfusions shortly after their infusion, and remain transfusion independent for up to 16.4 months.
In four patients with the more severe form of beta-thalassemia, the Beta-0/Beta-0 genotype, red blood cell transfusion volume was reduced by 33% to 100%, with one patient stopping transfusions entirely, Dr. Mark C. Walters of the University of California-San Francisco Benioff Children’s Hospital in Oakland reported at the annual meeting of the American Society of Hematology.
Preliminary findings reported over the last two years have raised hopes that the experimental lentiviral-based therapy could be a functional cure for beta-thalassemia major and severe sickle cell disease.
Patients with beta-thalassemia major, also called Cooley’s anemia, rely on frequent blood transfusions to correct the anemia, with less than a quarter undergoing curative treatment with an allogeneic hematopoietic transplant.
In the ongoing Northstar study, 13 patients with transfusion-dependent beta-thalassemia major have been infused as of Oct. 28, 2015 with autologous CD34-positive cells transduced ex-vivo with LentiGlobin BB305 (Bluebird bio, Cambridge, Mass.), a self-inactivating, second-generation lentiviral vector containing a functioning, engineered beta-globin gene (A-T87Q). Their median age was 21 years and 11 were women.Vector-derived hemoglobin AT87Q was detectable at 6 months in 8 of 9 evaluable patients with at least six months follow-up and in 100% at 9 months, Dr. Walters reported. The median HbAT87Q level was 4.9 g/dL at 6 months, 6.5 g/dL at 9 months, and 4.2 g/dL at 12 months.
The difference in transfusion independence between genotypes is explained by endogenous non-HbAT87Q production, he said during a press briefing. While lentiglobin production was the same in patients with Beta-0/Beta-0 and non-Beta-0/Beta-0 genotypes, the Beta-0/Beta-0 patients made much smaller amounts of native hemoglobin.
Three serious post-infusion events occurred: grade 2 thrombosis, grade 3 skin infection and grade 3 veno-occlusive liver disease.
Importantly, there was no evidence of clonal dominance or replication competent lentivirus with up to 19 months follow-up.
“This is a significant advancement in the treatment of thalassemia for several reasons,” Dr. Walters told reporters. “First, compared to a bone marrow transplant, which is the only curative therapy that’s been approved, this appears to be a safer treatment in that none of these patients had a life-threatening complication. Second, because the treatment uses a thalassemia patient’s own stem cells, this bypasses the need to find a healthy bone marrow donor and thus should be more broadly available to patients affected by this disease.”
Dr. George Daley of Harvard Medical School in Boston and moderator of the press briefing, agreed that the results are an welcome advancement after decades of disappointments in the field of gene therapy including the development of insertional mutigenesis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition to the Northstar study (Ab. 201), results will be presented at the meeting from a second study examining LentiGlobin BB305 gene therapy for severe sickle cell disease and beta-thalassemia major (Ab. 202) and from the recently expanded phase I HGB-206 study in severe sickle cell disease (Ab. 3233).
Sickle cell disease represents a much larger potential market for LentiGlobin BB305, with an estimated 90,000 to 100,000 Americans affected compared with about 15,000 patients in America and Europe living with beta-thalassemia major.
ORLANDO – Lentiviral gene therapy with LentiGlobin BB305 boosts beta-globin production in patients with beta-thalassemia, but frees only some from lifelong dependence on blood transfusions, updated results of the Northstar study show.
Five patients with non-Beta-0/Beta-0 genotypes were able to stop transfusions shortly after their infusion, and remain transfusion independent for up to 16.4 months.
In four patients with the more severe form of beta-thalassemia, the Beta-0/Beta-0 genotype, red blood cell transfusion volume was reduced by 33% to 100%, with one patient stopping transfusions entirely, Dr. Mark C. Walters of the University of California-San Francisco Benioff Children’s Hospital in Oakland reported at the annual meeting of the American Society of Hematology.
Preliminary findings reported over the last two years have raised hopes that the experimental lentiviral-based therapy could be a functional cure for beta-thalassemia major and severe sickle cell disease.
Patients with beta-thalassemia major, also called Cooley’s anemia, rely on frequent blood transfusions to correct the anemia, with less than a quarter undergoing curative treatment with an allogeneic hematopoietic transplant.
In the ongoing Northstar study, 13 patients with transfusion-dependent beta-thalassemia major have been infused as of Oct. 28, 2015 with autologous CD34-positive cells transduced ex-vivo with LentiGlobin BB305 (Bluebird bio, Cambridge, Mass.), a self-inactivating, second-generation lentiviral vector containing a functioning, engineered beta-globin gene (A-T87Q). Their median age was 21 years and 11 were women.Vector-derived hemoglobin AT87Q was detectable at 6 months in 8 of 9 evaluable patients with at least six months follow-up and in 100% at 9 months, Dr. Walters reported. The median HbAT87Q level was 4.9 g/dL at 6 months, 6.5 g/dL at 9 months, and 4.2 g/dL at 12 months.
The difference in transfusion independence between genotypes is explained by endogenous non-HbAT87Q production, he said during a press briefing. While lentiglobin production was the same in patients with Beta-0/Beta-0 and non-Beta-0/Beta-0 genotypes, the Beta-0/Beta-0 patients made much smaller amounts of native hemoglobin.
Three serious post-infusion events occurred: grade 2 thrombosis, grade 3 skin infection and grade 3 veno-occlusive liver disease.
Importantly, there was no evidence of clonal dominance or replication competent lentivirus with up to 19 months follow-up.
“This is a significant advancement in the treatment of thalassemia for several reasons,” Dr. Walters told reporters. “First, compared to a bone marrow transplant, which is the only curative therapy that’s been approved, this appears to be a safer treatment in that none of these patients had a life-threatening complication. Second, because the treatment uses a thalassemia patient’s own stem cells, this bypasses the need to find a healthy bone marrow donor and thus should be more broadly available to patients affected by this disease.”
Dr. George Daley of Harvard Medical School in Boston and moderator of the press briefing, agreed that the results are an welcome advancement after decades of disappointments in the field of gene therapy including the development of insertional mutigenesis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition to the Northstar study (Ab. 201), results will be presented at the meeting from a second study examining LentiGlobin BB305 gene therapy for severe sickle cell disease and beta-thalassemia major (Ab. 202) and from the recently expanded phase I HGB-206 study in severe sickle cell disease (Ab. 3233).
Sickle cell disease represents a much larger potential market for LentiGlobin BB305, with an estimated 90,000 to 100,000 Americans affected compared with about 15,000 patients in America and Europe living with beta-thalassemia major.
ORLANDO – Lentiviral gene therapy with LentiGlobin BB305 boosts beta-globin production in patients with beta-thalassemia, but frees only some from lifelong dependence on blood transfusions, updated results of the Northstar study show.
Five patients with non-Beta-0/Beta-0 genotypes were able to stop transfusions shortly after their infusion, and remain transfusion independent for up to 16.4 months.
In four patients with the more severe form of beta-thalassemia, the Beta-0/Beta-0 genotype, red blood cell transfusion volume was reduced by 33% to 100%, with one patient stopping transfusions entirely, Dr. Mark C. Walters of the University of California-San Francisco Benioff Children’s Hospital in Oakland reported at the annual meeting of the American Society of Hematology.
Preliminary findings reported over the last two years have raised hopes that the experimental lentiviral-based therapy could be a functional cure for beta-thalassemia major and severe sickle cell disease.
Patients with beta-thalassemia major, also called Cooley’s anemia, rely on frequent blood transfusions to correct the anemia, with less than a quarter undergoing curative treatment with an allogeneic hematopoietic transplant.
In the ongoing Northstar study, 13 patients with transfusion-dependent beta-thalassemia major have been infused as of Oct. 28, 2015 with autologous CD34-positive cells transduced ex-vivo with LentiGlobin BB305 (Bluebird bio, Cambridge, Mass.), a self-inactivating, second-generation lentiviral vector containing a functioning, engineered beta-globin gene (A-T87Q). Their median age was 21 years and 11 were women.Vector-derived hemoglobin AT87Q was detectable at 6 months in 8 of 9 evaluable patients with at least six months follow-up and in 100% at 9 months, Dr. Walters reported. The median HbAT87Q level was 4.9 g/dL at 6 months, 6.5 g/dL at 9 months, and 4.2 g/dL at 12 months.
The difference in transfusion independence between genotypes is explained by endogenous non-HbAT87Q production, he said during a press briefing. While lentiglobin production was the same in patients with Beta-0/Beta-0 and non-Beta-0/Beta-0 genotypes, the Beta-0/Beta-0 patients made much smaller amounts of native hemoglobin.
Three serious post-infusion events occurred: grade 2 thrombosis, grade 3 skin infection and grade 3 veno-occlusive liver disease.
Importantly, there was no evidence of clonal dominance or replication competent lentivirus with up to 19 months follow-up.
“This is a significant advancement in the treatment of thalassemia for several reasons,” Dr. Walters told reporters. “First, compared to a bone marrow transplant, which is the only curative therapy that’s been approved, this appears to be a safer treatment in that none of these patients had a life-threatening complication. Second, because the treatment uses a thalassemia patient’s own stem cells, this bypasses the need to find a healthy bone marrow donor and thus should be more broadly available to patients affected by this disease.”
Dr. George Daley of Harvard Medical School in Boston and moderator of the press briefing, agreed that the results are an welcome advancement after decades of disappointments in the field of gene therapy including the development of insertional mutigenesis.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
In addition to the Northstar study (Ab. 201), results will be presented at the meeting from a second study examining LentiGlobin BB305 gene therapy for severe sickle cell disease and beta-thalassemia major (Ab. 202) and from the recently expanded phase I HGB-206 study in severe sickle cell disease (Ab. 3233).
Sickle cell disease represents a much larger potential market for LentiGlobin BB305, with an estimated 90,000 to 100,000 Americans affected compared with about 15,000 patients in America and Europe living with beta-thalassemia major.
AT ASH 2015
Key clinical point: Lentiviral-based gene therapy with LentiGlobin BB305 restarts hemoglobin production and leads to transfusion independence in some patients with beta-thalassemia major.
Major finding: Five patients with the non-Beta-0/Beta-0 genotype were transfusion independent post-infusion.
Data source: Phase I/II study in 13 patients with transfusion-dependent beta-thalassemia major.
Disclosures: Dr. Walters reported financial relationships with ViaCord and AllCells Inc. Several co-authors have financial relationships including employment with Bluebird bio, the study sponsor. Dr. Daley disclosed consultancy with True North Therapeutics and serving as an advisory committee member for Raze Therapeutics, Ocata Therapeutics, MPM Capital, and Solasia.

Imetelstat elicits response in myelofibrosis, thrombocythemia
The telomerase inhibitor imetelstat showed promise against advanced myelofibrosis and essential thrombocythemia in two industry-funded preliminary studies, according to separate reports published online Sept. 3 in the New England Journal of Medicine.
In previous in vitro and animal studies, imetelstat inhibited the proliferation of various types of malignant cells but was not active in normal somatic tissue. Researchers assessed the agent for advanced myelofibrosis in part because, at present, only one available treatment – allogeneic stem-cell transplantation (ASCT) – sometimes induces long-term remission. ASCT carries a relatively high rate of treatment-related death and complications, and is contraindicated in many older patients.
In the first report, researchers conducted a small, single-center cohort study to collect preliminary data on the agent’s efficacy and safety in 33 patients with primary myelofibrosis (18 participants), myelofibrosis that was related to polycythemia (10 participants), or myelofibrosis associated with essential thrombocytopenia (10 participants). Imetelstat was administered in 2-hour intravenous infusions given in 3-week cycles, said Dr. Ayalew Tefferi of the division of hematology, Mayo Clinic, Rochester Minn.
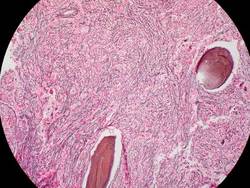
The median duration of treatment was 8.6 months (range, 1.4-21.7 months). Seven patients (21%) had either a complete or partial response; the 4 patients with a complete response had documented complete reversal of bone marrow fibrosis. The time to onset of response was 3.5 months (range, 1.4-7.2 months), and the median duration of response was 18 months (range, 13-20 months).
These remissions “confirm selective anticlonal activity, which has not previously been documented in drug treatment of myelofibrosis,” noted Dr. Tefferi and his associates (N Engl J Med 2015 Sep 3. doi:10.1056/NEJMoa1310523). Three of the seven patients who responded to imetelstat “had been heavily dependent on red-cell transfusions at study entry and became transfusion-independent and sustained a hemoglobin level of more than 10 g/dL for a minimum of 3 months during therapy,” they noted.
In addition, 8 of 10 patients who had marked leukocytosis at baseline had either a complete resolution (3 patients) or a reduction of at least 50% in white-cell counts (5 patients). All 11 participants who had thrombocytosis at baseline had either complete resolution (10 patients) or a reduction in platelet count of at least 50% (1 patient). Of the 27 participants who had leukoerythroblastosis at baseline, 22 showed either complete resolution (13 patients) or a reduction of at least 50% in the percentage of immature myeloid cells and nucleated red cells (9 patients). Also, 17 of the 21 participants who had at least 1% circulating blasts at baseline had either a complete disappearance of circulating blasts (14 patients) or a reduction of at least 50% (3 patients).
The most clinically significant adverse effect of imetelstat, myelosuppression, occurred in 22 patients (67%) and often necessitated dose reductions. Low-grade elevations in liver enzymes also were a concern. One patient died from an intracranial hemorrhage that the treating physician attributed to drug-induced grade 4 thrombocytopenia. Other adverse events that may or may not have been treatment related included fever, epistaxis, bruising, hematoma, lung infection, skin infection, and upper-GI hemorrhage.
These findings not only identify imetelstat as a possible treatment for myelofibrosis, they also suggest that other telomerase-targeting strategies may be beneficial in this disease, Dr. Tefferi and his associates added.
In the second report, researchers performing a phase-II study at seven medical centers in the United States, Germany, and Switzerland found that imetelstat produced rapid and durable hematologic and molecular responses in all 18 patients in their study of essential thrombocythemia refractory to other treatments. This result is particularly encouraging because current standard therapies “induce nonspecific reductions in platelet counts but do not typically eliminate or alter the biologic characteristics of the disease,” said Dr. Gabriela M. Baerlocher of the department of hematology and the Stem Cell Molecular Diagnostics Laboratory, University of Bern, Switzerland.
These study participants had either failed to respond to hydroxyurea, anagrelide, and interferon therapy or were forced to discontinue these agents because of adverse effects. After weekly treatment with imetelstat at one of two doses, 100% of the patients achieved a hematologic response, attaining platelet counts of 250,000-300,000 per cc. Sixteen participants (89%) achieved a complete hematologic response. The median time to complete response was 6.1 weeks, Dr. Baerlocher and her associates said (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMoa1503479).
After a median follow-up of 17 months on a maintenance dose of imetelstat, 10 patients were still receiving treatment. The median duration of response had not been reached as of press time (range, 5-30 months).
The most important adverse events were neutropenia (15 patients) and abnormal results on liver-function tests (14 patients). The treating physicians attributed 18 adverse events of grade 3 or higher to the study drug, including headache, anemia, and one syncopal episode. Other adverse events included fatigue, nausea, diarrhea, infections, and rash.
The results of both of these studies are compelling and certainly warrant further research, given the limited treatment options for myeloproliferative disorders.
Although imetelstat’s mechanism of action remains to be elucidated, both studies hint at the possibility that the agent may actually change the natural history of these debilitating disorders.
More important, assessing imetelstat’s long-term safety profile is a vital next step for researchers.
Dr. Mary Armanios and Carol W. Greider, Ph.D., are at Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, Baltimore. Dr. Armanios reported having no relevant disclosures; Dr. Greider reported patents related to an RNA component of telomerase and telomerase-associated proteins. Dr. Armanios and Dr. Greider made these remarks in an editorial accompanying the two reports on imetelstat (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMe1508740).
The results of both of these studies are compelling and certainly warrant further research, given the limited treatment options for myeloproliferative disorders.
Although imetelstat’s mechanism of action remains to be elucidated, both studies hint at the possibility that the agent may actually change the natural history of these debilitating disorders.
More important, assessing imetelstat’s long-term safety profile is a vital next step for researchers.
Dr. Mary Armanios and Carol W. Greider, Ph.D., are at Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, Baltimore. Dr. Armanios reported having no relevant disclosures; Dr. Greider reported patents related to an RNA component of telomerase and telomerase-associated proteins. Dr. Armanios and Dr. Greider made these remarks in an editorial accompanying the two reports on imetelstat (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMe1508740).
The results of both of these studies are compelling and certainly warrant further research, given the limited treatment options for myeloproliferative disorders.
Although imetelstat’s mechanism of action remains to be elucidated, both studies hint at the possibility that the agent may actually change the natural history of these debilitating disorders.
More important, assessing imetelstat’s long-term safety profile is a vital next step for researchers.
Dr. Mary Armanios and Carol W. Greider, Ph.D., are at Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, Baltimore. Dr. Armanios reported having no relevant disclosures; Dr. Greider reported patents related to an RNA component of telomerase and telomerase-associated proteins. Dr. Armanios and Dr. Greider made these remarks in an editorial accompanying the two reports on imetelstat (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMe1508740).
The telomerase inhibitor imetelstat showed promise against advanced myelofibrosis and essential thrombocythemia in two industry-funded preliminary studies, according to separate reports published online Sept. 3 in the New England Journal of Medicine.
In previous in vitro and animal studies, imetelstat inhibited the proliferation of various types of malignant cells but was not active in normal somatic tissue. Researchers assessed the agent for advanced myelofibrosis in part because, at present, only one available treatment – allogeneic stem-cell transplantation (ASCT) – sometimes induces long-term remission. ASCT carries a relatively high rate of treatment-related death and complications, and is contraindicated in many older patients.
In the first report, researchers conducted a small, single-center cohort study to collect preliminary data on the agent’s efficacy and safety in 33 patients with primary myelofibrosis (18 participants), myelofibrosis that was related to polycythemia (10 participants), or myelofibrosis associated with essential thrombocytopenia (10 participants). Imetelstat was administered in 2-hour intravenous infusions given in 3-week cycles, said Dr. Ayalew Tefferi of the division of hematology, Mayo Clinic, Rochester Minn.
The median duration of treatment was 8.6 months (range, 1.4-21.7 months). Seven patients (21%) had either a complete or partial response; the 4 patients with a complete response had documented complete reversal of bone marrow fibrosis. The time to onset of response was 3.5 months (range, 1.4-7.2 months), and the median duration of response was 18 months (range, 13-20 months).
These remissions “confirm selective anticlonal activity, which has not previously been documented in drug treatment of myelofibrosis,” noted Dr. Tefferi and his associates (N Engl J Med 2015 Sep 3. doi:10.1056/NEJMoa1310523). Three of the seven patients who responded to imetelstat “had been heavily dependent on red-cell transfusions at study entry and became transfusion-independent and sustained a hemoglobin level of more than 10 g/dL for a minimum of 3 months during therapy,” they noted.
In addition, 8 of 10 patients who had marked leukocytosis at baseline had either a complete resolution (3 patients) or a reduction of at least 50% in white-cell counts (5 patients). All 11 participants who had thrombocytosis at baseline had either complete resolution (10 patients) or a reduction in platelet count of at least 50% (1 patient). Of the 27 participants who had leukoerythroblastosis at baseline, 22 showed either complete resolution (13 patients) or a reduction of at least 50% in the percentage of immature myeloid cells and nucleated red cells (9 patients). Also, 17 of the 21 participants who had at least 1% circulating blasts at baseline had either a complete disappearance of circulating blasts (14 patients) or a reduction of at least 50% (3 patients).
The most clinically significant adverse effect of imetelstat, myelosuppression, occurred in 22 patients (67%) and often necessitated dose reductions. Low-grade elevations in liver enzymes also were a concern. One patient died from an intracranial hemorrhage that the treating physician attributed to drug-induced grade 4 thrombocytopenia. Other adverse events that may or may not have been treatment related included fever, epistaxis, bruising, hematoma, lung infection, skin infection, and upper-GI hemorrhage.
These findings not only identify imetelstat as a possible treatment for myelofibrosis, they also suggest that other telomerase-targeting strategies may be beneficial in this disease, Dr. Tefferi and his associates added.
In the second report, researchers performing a phase-II study at seven medical centers in the United States, Germany, and Switzerland found that imetelstat produced rapid and durable hematologic and molecular responses in all 18 patients in their study of essential thrombocythemia refractory to other treatments. This result is particularly encouraging because current standard therapies “induce nonspecific reductions in platelet counts but do not typically eliminate or alter the biologic characteristics of the disease,” said Dr. Gabriela M. Baerlocher of the department of hematology and the Stem Cell Molecular Diagnostics Laboratory, University of Bern, Switzerland.
These study participants had either failed to respond to hydroxyurea, anagrelide, and interferon therapy or were forced to discontinue these agents because of adverse effects. After weekly treatment with imetelstat at one of two doses, 100% of the patients achieved a hematologic response, attaining platelet counts of 250,000-300,000 per cc. Sixteen participants (89%) achieved a complete hematologic response. The median time to complete response was 6.1 weeks, Dr. Baerlocher and her associates said (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMoa1503479).
After a median follow-up of 17 months on a maintenance dose of imetelstat, 10 patients were still receiving treatment. The median duration of response had not been reached as of press time (range, 5-30 months).
The most important adverse events were neutropenia (15 patients) and abnormal results on liver-function tests (14 patients). The treating physicians attributed 18 adverse events of grade 3 or higher to the study drug, including headache, anemia, and one syncopal episode. Other adverse events included fatigue, nausea, diarrhea, infections, and rash.
The telomerase inhibitor imetelstat showed promise against advanced myelofibrosis and essential thrombocythemia in two industry-funded preliminary studies, according to separate reports published online Sept. 3 in the New England Journal of Medicine.
In previous in vitro and animal studies, imetelstat inhibited the proliferation of various types of malignant cells but was not active in normal somatic tissue. Researchers assessed the agent for advanced myelofibrosis in part because, at present, only one available treatment – allogeneic stem-cell transplantation (ASCT) – sometimes induces long-term remission. ASCT carries a relatively high rate of treatment-related death and complications, and is contraindicated in many older patients.
In the first report, researchers conducted a small, single-center cohort study to collect preliminary data on the agent’s efficacy and safety in 33 patients with primary myelofibrosis (18 participants), myelofibrosis that was related to polycythemia (10 participants), or myelofibrosis associated with essential thrombocytopenia (10 participants). Imetelstat was administered in 2-hour intravenous infusions given in 3-week cycles, said Dr. Ayalew Tefferi of the division of hematology, Mayo Clinic, Rochester Minn.
The median duration of treatment was 8.6 months (range, 1.4-21.7 months). Seven patients (21%) had either a complete or partial response; the 4 patients with a complete response had documented complete reversal of bone marrow fibrosis. The time to onset of response was 3.5 months (range, 1.4-7.2 months), and the median duration of response was 18 months (range, 13-20 months).
These remissions “confirm selective anticlonal activity, which has not previously been documented in drug treatment of myelofibrosis,” noted Dr. Tefferi and his associates (N Engl J Med 2015 Sep 3. doi:10.1056/NEJMoa1310523). Three of the seven patients who responded to imetelstat “had been heavily dependent on red-cell transfusions at study entry and became transfusion-independent and sustained a hemoglobin level of more than 10 g/dL for a minimum of 3 months during therapy,” they noted.
In addition, 8 of 10 patients who had marked leukocytosis at baseline had either a complete resolution (3 patients) or a reduction of at least 50% in white-cell counts (5 patients). All 11 participants who had thrombocytosis at baseline had either complete resolution (10 patients) or a reduction in platelet count of at least 50% (1 patient). Of the 27 participants who had leukoerythroblastosis at baseline, 22 showed either complete resolution (13 patients) or a reduction of at least 50% in the percentage of immature myeloid cells and nucleated red cells (9 patients). Also, 17 of the 21 participants who had at least 1% circulating blasts at baseline had either a complete disappearance of circulating blasts (14 patients) or a reduction of at least 50% (3 patients).
The most clinically significant adverse effect of imetelstat, myelosuppression, occurred in 22 patients (67%) and often necessitated dose reductions. Low-grade elevations in liver enzymes also were a concern. One patient died from an intracranial hemorrhage that the treating physician attributed to drug-induced grade 4 thrombocytopenia. Other adverse events that may or may not have been treatment related included fever, epistaxis, bruising, hematoma, lung infection, skin infection, and upper-GI hemorrhage.
These findings not only identify imetelstat as a possible treatment for myelofibrosis, they also suggest that other telomerase-targeting strategies may be beneficial in this disease, Dr. Tefferi and his associates added.
In the second report, researchers performing a phase-II study at seven medical centers in the United States, Germany, and Switzerland found that imetelstat produced rapid and durable hematologic and molecular responses in all 18 patients in their study of essential thrombocythemia refractory to other treatments. This result is particularly encouraging because current standard therapies “induce nonspecific reductions in platelet counts but do not typically eliminate or alter the biologic characteristics of the disease,” said Dr. Gabriela M. Baerlocher of the department of hematology and the Stem Cell Molecular Diagnostics Laboratory, University of Bern, Switzerland.
These study participants had either failed to respond to hydroxyurea, anagrelide, and interferon therapy or were forced to discontinue these agents because of adverse effects. After weekly treatment with imetelstat at one of two doses, 100% of the patients achieved a hematologic response, attaining platelet counts of 250,000-300,000 per cc. Sixteen participants (89%) achieved a complete hematologic response. The median time to complete response was 6.1 weeks, Dr. Baerlocher and her associates said (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMoa1503479).
After a median follow-up of 17 months on a maintenance dose of imetelstat, 10 patients were still receiving treatment. The median duration of response had not been reached as of press time (range, 5-30 months).
The most important adverse events were neutropenia (15 patients) and abnormal results on liver-function tests (14 patients). The treating physicians attributed 18 adverse events of grade 3 or higher to the study drug, including headache, anemia, and one syncopal episode. Other adverse events included fatigue, nausea, diarrhea, infections, and rash.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: The telomerase inhibitor imetelstat showed promise in separate preliminary studies for treatment of myelofibrosis and thrombocythemia.
Major finding: A complete or partial response to imetelstat was seen in 7 of 33 patients with advanced myelofibrosis and 18 of 18 with thrombocythemia.
Data source: An international phase-II open-label study involving 18 patients with essential thrombocythemia and a single-center observational cohort study involving 33 patients with myelofibrosis.
Disclosures: Both studies were funded by Geron.
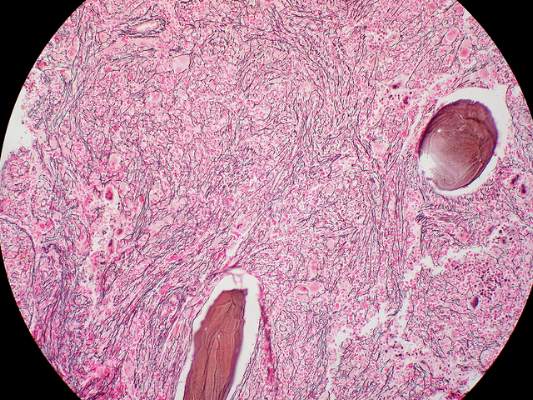
CLL patients achieve remission with CAR-modified T-cells
Treatment with chimeric antigen receptor (CAR)-modified T cells targeting CD19 achieved a response in 8 of 14 patients (57%) with advanced chronic lymphocytic leukemia (CLL), of whom 4 experienced a complete remission without relapse, based on the mature results of a small pilot study.
Of these four patients, two have remained free of their disease for up to 4 years after they received treatment. An analysis of blood samples also showed that these modified T cells can multiply and persist in the body for a period of years, the researchers report in a study published Sept. 2 in Science Translational Medicine
“Both patients remain alive and cancer free and just passed the 5-year anniversary of their treatment this summer,” said Dr. David L. Porter, the Jodi Fisher Horowitz Professor in Leukemia Care Excellence and director of blood and marrow transplantation at the University of Pennsylvania’s Abramson Cancer Center in Philadelphia. “A third patient in remission just passed the 3-year anniversary with no signs of leukemia” (Sci Transl Med. 2015;7:303ra139).
The current study indicates the mature results from this trial, which began in the summer of 2010. In 2011, preliminary findings from the first three patients to enroll in the study were published and showed that two of them had experienced a complete response. Their disease currently remains in remission more than 4 years after beginning treatment. The first patient to receive the therapy has been cancer free for 5 years.
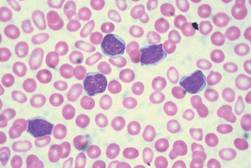
In the current trial, 14 patients with relapsed or refractory CLL received at least one infusion of autologous T cells transduced with a CD19-directed CAR (CTL019) lentiviral vector. All of the patients had active disease at the time they received the experimental treatment, and had received a median of 5 previous therapies (range, 1-11). One participant had undergone two previous autologous stem cell transplants and one had progressed on ibrutinib therapy.
In addition to those who achieved a complete remission, four other patients (29%) had partial responses to the therapy with responses that persisted for a median of 7 months. Two died of disease progression at 10 and 27 months after receiving CTL019, and one died from a pulmonary embolism; the remaining patient remains alive after CLL progressed at 13 months, and is receiving other therapies.
Overall, the CTL019 infusions were well tolerated, with grade less than 2 toxicities that included primarily low-grade fevers and chills. The most frequent related events were associated with complications of neutropenia and delayed cytokine release syndrome, which correlated with in vivo CTL019 expansion. There were two cases of tumor lysis syndrome, and one patient died in remission 21 months after T cell infusion, after developing ecthyma gangrenosum after pseudomonas infection at a skin biopsy site.
Six subjects (43%) had no response and all six progressed within 1-9 months (median, 4 months) of CTL019 therapy. “We are working hard to determine why this therapy may be appropriate for some patients and not others, and trying to optimize either treatment conditions or patient-specific factors so that this might be more effective for more patients,” Dr. Porter wrote.
Minimal residual disease was not detectable in patients who achieved a complete response, suggesting that disease eradication may be possible in some patients with advanced CLL. The activity of CTLO19 seemed to be on par with results achieved with allogeneic stem cell transplantation, suggesting that this therapy could possibly cure CLL. But Dr. Porter pointed out that this study was conducted with a small number of patients and for CLL, a relatively short follow-up.
“However, these patients all had heavily pretreated resistant disease,” he said. “Though we do not know if patients are indeed cured, it is certainly our goal to find a cure for CLL and without the toxicities and limitations of allogeneic stem cell transplantation. Indeed, longer follow-up will be needed but we are quite excited about the results to date.”
Dr. Porter said he and his team have ongoing trials in CLL in progress, where they are working on trying to identify the optimal dose of T cells for this approach. Also, “this research has led to expansion of this approach to other B cell malignancies such as acute lymphocytic leukemia.”
Novartis, the Leukemia and Lymphoma Society (Specialized Center of Research Award), and the National Institutes of Health funded the study. The University of Pennsylvania has licensed technologies involved in this trial to Novartis. Some scientists involved in these trials, including Dr. Porter, are inventors of these technologies. As a result of the licensing relationship with Novartis, the University of Pennsylvania receives significant financial benefit, and these inventors have benefited financially and/or may benefit financially in the future.
Treatment with chimeric antigen receptor (CAR)-modified T cells targeting CD19 achieved a response in 8 of 14 patients (57%) with advanced chronic lymphocytic leukemia (CLL), of whom 4 experienced a complete remission without relapse, based on the mature results of a small pilot study.
Of these four patients, two have remained free of their disease for up to 4 years after they received treatment. An analysis of blood samples also showed that these modified T cells can multiply and persist in the body for a period of years, the researchers report in a study published Sept. 2 in Science Translational Medicine
“Both patients remain alive and cancer free and just passed the 5-year anniversary of their treatment this summer,” said Dr. David L. Porter, the Jodi Fisher Horowitz Professor in Leukemia Care Excellence and director of blood and marrow transplantation at the University of Pennsylvania’s Abramson Cancer Center in Philadelphia. “A third patient in remission just passed the 3-year anniversary with no signs of leukemia” (Sci Transl Med. 2015;7:303ra139).
The current study indicates the mature results from this trial, which began in the summer of 2010. In 2011, preliminary findings from the first three patients to enroll in the study were published and showed that two of them had experienced a complete response. Their disease currently remains in remission more than 4 years after beginning treatment. The first patient to receive the therapy has been cancer free for 5 years.
In the current trial, 14 patients with relapsed or refractory CLL received at least one infusion of autologous T cells transduced with a CD19-directed CAR (CTL019) lentiviral vector. All of the patients had active disease at the time they received the experimental treatment, and had received a median of 5 previous therapies (range, 1-11). One participant had undergone two previous autologous stem cell transplants and one had progressed on ibrutinib therapy.
In addition to those who achieved a complete remission, four other patients (29%) had partial responses to the therapy with responses that persisted for a median of 7 months. Two died of disease progression at 10 and 27 months after receiving CTL019, and one died from a pulmonary embolism; the remaining patient remains alive after CLL progressed at 13 months, and is receiving other therapies.
Overall, the CTL019 infusions were well tolerated, with grade less than 2 toxicities that included primarily low-grade fevers and chills. The most frequent related events were associated with complications of neutropenia and delayed cytokine release syndrome, which correlated with in vivo CTL019 expansion. There were two cases of tumor lysis syndrome, and one patient died in remission 21 months after T cell infusion, after developing ecthyma gangrenosum after pseudomonas infection at a skin biopsy site.
Six subjects (43%) had no response and all six progressed within 1-9 months (median, 4 months) of CTL019 therapy. “We are working hard to determine why this therapy may be appropriate for some patients and not others, and trying to optimize either treatment conditions or patient-specific factors so that this might be more effective for more patients,” Dr. Porter wrote.
Minimal residual disease was not detectable in patients who achieved a complete response, suggesting that disease eradication may be possible in some patients with advanced CLL. The activity of CTLO19 seemed to be on par with results achieved with allogeneic stem cell transplantation, suggesting that this therapy could possibly cure CLL. But Dr. Porter pointed out that this study was conducted with a small number of patients and for CLL, a relatively short follow-up.
“However, these patients all had heavily pretreated resistant disease,” he said. “Though we do not know if patients are indeed cured, it is certainly our goal to find a cure for CLL and without the toxicities and limitations of allogeneic stem cell transplantation. Indeed, longer follow-up will be needed but we are quite excited about the results to date.”
Dr. Porter said he and his team have ongoing trials in CLL in progress, where they are working on trying to identify the optimal dose of T cells for this approach. Also, “this research has led to expansion of this approach to other B cell malignancies such as acute lymphocytic leukemia.”
Novartis, the Leukemia and Lymphoma Society (Specialized Center of Research Award), and the National Institutes of Health funded the study. The University of Pennsylvania has licensed technologies involved in this trial to Novartis. Some scientists involved in these trials, including Dr. Porter, are inventors of these technologies. As a result of the licensing relationship with Novartis, the University of Pennsylvania receives significant financial benefit, and these inventors have benefited financially and/or may benefit financially in the future.
Treatment with chimeric antigen receptor (CAR)-modified T cells targeting CD19 achieved a response in 8 of 14 patients (57%) with advanced chronic lymphocytic leukemia (CLL), of whom 4 experienced a complete remission without relapse, based on the mature results of a small pilot study.
Of these four patients, two have remained free of their disease for up to 4 years after they received treatment. An analysis of blood samples also showed that these modified T cells can multiply and persist in the body for a period of years, the researchers report in a study published Sept. 2 in Science Translational Medicine
“Both patients remain alive and cancer free and just passed the 5-year anniversary of their treatment this summer,” said Dr. David L. Porter, the Jodi Fisher Horowitz Professor in Leukemia Care Excellence and director of blood and marrow transplantation at the University of Pennsylvania’s Abramson Cancer Center in Philadelphia. “A third patient in remission just passed the 3-year anniversary with no signs of leukemia” (Sci Transl Med. 2015;7:303ra139).
The current study indicates the mature results from this trial, which began in the summer of 2010. In 2011, preliminary findings from the first three patients to enroll in the study were published and showed that two of them had experienced a complete response. Their disease currently remains in remission more than 4 years after beginning treatment. The first patient to receive the therapy has been cancer free for 5 years.
In the current trial, 14 patients with relapsed or refractory CLL received at least one infusion of autologous T cells transduced with a CD19-directed CAR (CTL019) lentiviral vector. All of the patients had active disease at the time they received the experimental treatment, and had received a median of 5 previous therapies (range, 1-11). One participant had undergone two previous autologous stem cell transplants and one had progressed on ibrutinib therapy.
In addition to those who achieved a complete remission, four other patients (29%) had partial responses to the therapy with responses that persisted for a median of 7 months. Two died of disease progression at 10 and 27 months after receiving CTL019, and one died from a pulmonary embolism; the remaining patient remains alive after CLL progressed at 13 months, and is receiving other therapies.
Overall, the CTL019 infusions were well tolerated, with grade less than 2 toxicities that included primarily low-grade fevers and chills. The most frequent related events were associated with complications of neutropenia and delayed cytokine release syndrome, which correlated with in vivo CTL019 expansion. There were two cases of tumor lysis syndrome, and one patient died in remission 21 months after T cell infusion, after developing ecthyma gangrenosum after pseudomonas infection at a skin biopsy site.
Six subjects (43%) had no response and all six progressed within 1-9 months (median, 4 months) of CTL019 therapy. “We are working hard to determine why this therapy may be appropriate for some patients and not others, and trying to optimize either treatment conditions or patient-specific factors so that this might be more effective for more patients,” Dr. Porter wrote.
Minimal residual disease was not detectable in patients who achieved a complete response, suggesting that disease eradication may be possible in some patients with advanced CLL. The activity of CTLO19 seemed to be on par with results achieved with allogeneic stem cell transplantation, suggesting that this therapy could possibly cure CLL. But Dr. Porter pointed out that this study was conducted with a small number of patients and for CLL, a relatively short follow-up.
“However, these patients all had heavily pretreated resistant disease,” he said. “Though we do not know if patients are indeed cured, it is certainly our goal to find a cure for CLL and without the toxicities and limitations of allogeneic stem cell transplantation. Indeed, longer follow-up will be needed but we are quite excited about the results to date.”
Dr. Porter said he and his team have ongoing trials in CLL in progress, where they are working on trying to identify the optimal dose of T cells for this approach. Also, “this research has led to expansion of this approach to other B cell malignancies such as acute lymphocytic leukemia.”
Novartis, the Leukemia and Lymphoma Society (Specialized Center of Research Award), and the National Institutes of Health funded the study. The University of Pennsylvania has licensed technologies involved in this trial to Novartis. Some scientists involved in these trials, including Dr. Porter, are inventors of these technologies. As a result of the licensing relationship with Novartis, the University of Pennsylvania receives significant financial benefit, and these inventors have benefited financially and/or may benefit financially in the future.
FROM SCIENCE TRANSLATIONAL MEDICINE
Key clinical point: CAR-modified T cell therapy lacks the toxicities and limitations of allogeneic stem cell transplantation and may be an effective treatment for chronic lymphocytic leukemia.
Major finding: CAR-modified T cell therapy elicited a response in 8 of 14 patients (57%) with relapsed and refractory chronic lymphocytic leukemia, and 4 patients (29%) achieved a complete remission.
Data source: Mature results from a pilot clinical trial.
Disclosures: Novartis, the Leukemia and Lymphoma Society (Specialized Center of Research Award), and the National Institutes of Health funded the study. The University of Pennsylvania has licensed technologies involved in this trial to Novartis. Some scientists involved in these trials, including Dr. Porter, are inventors of these technologies. As a result of the licensing relationship with Novartis, the University of Pennsylvania receives significant financial benefit, and these inventors have benefited financially and/or may benefit financially in the future.
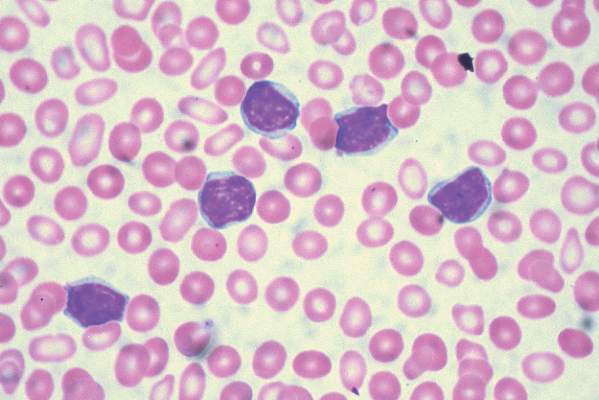
ABT-199 rebounds in advanced CLL after tackling tumor lysis syndrome
MILAN – The investigational Bcl-2 inhibitor ABT-199 continues to impress with substantial activity as single-agent or combination therapy in relapsed or refractory chronic lymphocytic leukemia, following dose-scheduling modifications to address the risk of tumor lysis syndrome.
ABT-199 monotherapy
Updated data from all 105 CLL patients in the phase I trial show the overall response rate remains high at 77%, with 23% of patients achieving complete remission.
Seven of 11 complete responders assessed had no detectable minimal residual disease, Dr. John F. Seymour said at the annual congress of the European Hematology Association.
Overall response rates were sustained in the 75%-80% range for high-risk patients with deletion 17p, fludarabine-refractory, or immunoglobulin heavy-chain variable (IGHV)-unmutated CLL; and complete response rates in these subgroups also did not differ from the overall group at 22% to 29%.
Prior results from the first-in-human trial dazzled the leukemia community, but tumor lysis syndrome (TLS) complications, including two fatal events, temporarily halted ABT-199 clinical trials and gave the advantage to its closest competitor, the recently approved and more tolerable CLL drug ibrutinib (Imbruvica).
Use of a modified, ramp-up dosing scheme and aggressive TLS prophylaxis appear to have ameliorated the risk of TLS, with no further clinically significant or grade 3 or 4 events reported in the 49 patients treated with this schema, said Dr. Seymour, director of hematology and cancer medicine at the Peter MacCallum Cancer Centre, East Melbourne, Australia.
Rather than using a 3-week schedule and 50-mg starting dose, the safety expansion cohort received once-daily oral ABT-199 beginning at 20 mg, with weekly adjustments to 50 mg, 100 mg, 200 mg, and 400 mg over 5 weeks.
The 400-mg dose has been identified as the phase II dose, with 59% of patients free of progression at 18 months and beyond on this dose, he said.
As of April 2014, 37 of the 105 patients discontinued treatment, 22 due to progressive disease and 12 for adverse events; in addition, two proceeded to allogeneic hematopoietic cell transplantation, and one needed Coumadin, which is not permitted on protocol.
The median duration of response has not yet been reached for patients treated at doses of 400 mg or above, he said.
The most common treatment-emergent adverse events of any grade were diarrhea (40% of patients), neutropenia (36%), and nausea (35%).
Neutropenia was the only grade 3/4 event occurring in more than 10% of patients (33%), followed by anemia in 10%.
Combination ABT-199
Of substantial interest to many was a second phase Ib study presented in the same session, evaluating the role of ABT-199 with the anti-CD20 antibody rituximab (Rituxan) in relapsed or refractory CLL.
After a median time on study of just 7.5 months, the overall response rate was 84% among 25 evaluable patients, including 9 complete responses (36%) and 12 partial responses (48%), said Dr. Andrew W. Roberts, with the Royal Melbourne (Australia) Hospital and Walter and Eliza Hall Institute of Medical Research.
Six of eight complete responders tested were negative for minimal residual disease by flow cytometry.
Preliminary pharmacokinetic results suggest no apparent effect of rituximab on ABT-199 exposure, he said.
Three patients discontinued ABT-199 after achieving a complete remission, including one with fludarabine-refractory disease, and all remain in complete remission at 8.6, 8.8, and 11.6 months after cessation.
Dose modifications were also made in this study following a fatal TLS event in December 2012 after a first dose of ABT-199 at 50 mg. Under the modified step-up dosing, ABT-199 was started at 20 mg, escalating up to 600 mg daily over 5 weeks, with rituximab 375 mg/m2 added on day 1 of week 5 and rituximab 500 mg/m2 added on day 1 of months 2-6.
The combination was well tolerated, and no new safety concerns were identified, Dr. Roberts said. The most common grade 3/4 adverse events among 45 patients evaluable for safety were neutropenia in 47%, anemia in 16%, thrombocytopenia in 13%, and febrile neutropenia in 7%. Grade 3/4 neutropenia was more common at 600 mg, with the 400-mg dose selected for the ongoing safety expansion cohort. Two serious TLS events occurred, but both were prior to schedule modifications, he said.
One patient in the combination study and 13 in the monotherapy study received treatment for small lymphocytic lymphoma. Response rates and tolerability were similar between CLL and SLL patients in the monotherapy study, Dr. Roberts said in an interview.
During a press briefing at the meeting, Dr. Seymour said ABT-199 potentially could be combined with ibrutinib and that the combination was very potent in laboratory tests in both CLL and some forms of mantle cell lymphoma. Negotiations with the various companies involved are intricate, but there is agreement and commitment "to begin clinical trials of the combination later this year," he added.
ABT-199 is currently being evaluated in a phase II trial as monotherapy in deletion 17p relapsed CLL, as combination therapy with rituximab versus bendamustine plus rituximab in a phase III trial in relapsed/refractory CLL, and in combination trials with bendamustine/rituximab and obinutuzumab in relapsed/refractory CLL.
AbbVie and Genentech sponsored the trials. Dr. Seymour is a consultant and adviser for AbbVie, Genentech, and Roche. Dr. Roberts reported research funding from AbbVie and Genentech and milestone payments to his institution related to ABT-199.
MILAN – The investigational Bcl-2 inhibitor ABT-199 continues to impress with substantial activity as single-agent or combination therapy in relapsed or refractory chronic lymphocytic leukemia, following dose-scheduling modifications to address the risk of tumor lysis syndrome.
ABT-199 monotherapy
Updated data from all 105 CLL patients in the phase I trial show the overall response rate remains high at 77%, with 23% of patients achieving complete remission.
Seven of 11 complete responders assessed had no detectable minimal residual disease, Dr. John F. Seymour said at the annual congress of the European Hematology Association.
Overall response rates were sustained in the 75%-80% range for high-risk patients with deletion 17p, fludarabine-refractory, or immunoglobulin heavy-chain variable (IGHV)-unmutated CLL; and complete response rates in these subgroups also did not differ from the overall group at 22% to 29%.
Prior results from the first-in-human trial dazzled the leukemia community, but tumor lysis syndrome (TLS) complications, including two fatal events, temporarily halted ABT-199 clinical trials and gave the advantage to its closest competitor, the recently approved and more tolerable CLL drug ibrutinib (Imbruvica).
Use of a modified, ramp-up dosing scheme and aggressive TLS prophylaxis appear to have ameliorated the risk of TLS, with no further clinically significant or grade 3 or 4 events reported in the 49 patients treated with this schema, said Dr. Seymour, director of hematology and cancer medicine at the Peter MacCallum Cancer Centre, East Melbourne, Australia.
Rather than using a 3-week schedule and 50-mg starting dose, the safety expansion cohort received once-daily oral ABT-199 beginning at 20 mg, with weekly adjustments to 50 mg, 100 mg, 200 mg, and 400 mg over 5 weeks.
The 400-mg dose has been identified as the phase II dose, with 59% of patients free of progression at 18 months and beyond on this dose, he said.
As of April 2014, 37 of the 105 patients discontinued treatment, 22 due to progressive disease and 12 for adverse events; in addition, two proceeded to allogeneic hematopoietic cell transplantation, and one needed Coumadin, which is not permitted on protocol.
The median duration of response has not yet been reached for patients treated at doses of 400 mg or above, he said.
The most common treatment-emergent adverse events of any grade were diarrhea (40% of patients), neutropenia (36%), and nausea (35%).
Neutropenia was the only grade 3/4 event occurring in more than 10% of patients (33%), followed by anemia in 10%.
Combination ABT-199
Of substantial interest to many was a second phase Ib study presented in the same session, evaluating the role of ABT-199 with the anti-CD20 antibody rituximab (Rituxan) in relapsed or refractory CLL.
After a median time on study of just 7.5 months, the overall response rate was 84% among 25 evaluable patients, including 9 complete responses (36%) and 12 partial responses (48%), said Dr. Andrew W. Roberts, with the Royal Melbourne (Australia) Hospital and Walter and Eliza Hall Institute of Medical Research.
Six of eight complete responders tested were negative for minimal residual disease by flow cytometry.
Preliminary pharmacokinetic results suggest no apparent effect of rituximab on ABT-199 exposure, he said.
Three patients discontinued ABT-199 after achieving a complete remission, including one with fludarabine-refractory disease, and all remain in complete remission at 8.6, 8.8, and 11.6 months after cessation.
Dose modifications were also made in this study following a fatal TLS event in December 2012 after a first dose of ABT-199 at 50 mg. Under the modified step-up dosing, ABT-199 was started at 20 mg, escalating up to 600 mg daily over 5 weeks, with rituximab 375 mg/m2 added on day 1 of week 5 and rituximab 500 mg/m2 added on day 1 of months 2-6.
The combination was well tolerated, and no new safety concerns were identified, Dr. Roberts said. The most common grade 3/4 adverse events among 45 patients evaluable for safety were neutropenia in 47%, anemia in 16%, thrombocytopenia in 13%, and febrile neutropenia in 7%. Grade 3/4 neutropenia was more common at 600 mg, with the 400-mg dose selected for the ongoing safety expansion cohort. Two serious TLS events occurred, but both were prior to schedule modifications, he said.
One patient in the combination study and 13 in the monotherapy study received treatment for small lymphocytic lymphoma. Response rates and tolerability were similar between CLL and SLL patients in the monotherapy study, Dr. Roberts said in an interview.
During a press briefing at the meeting, Dr. Seymour said ABT-199 potentially could be combined with ibrutinib and that the combination was very potent in laboratory tests in both CLL and some forms of mantle cell lymphoma. Negotiations with the various companies involved are intricate, but there is agreement and commitment "to begin clinical trials of the combination later this year," he added.
ABT-199 is currently being evaluated in a phase II trial as monotherapy in deletion 17p relapsed CLL, as combination therapy with rituximab versus bendamustine plus rituximab in a phase III trial in relapsed/refractory CLL, and in combination trials with bendamustine/rituximab and obinutuzumab in relapsed/refractory CLL.
AbbVie and Genentech sponsored the trials. Dr. Seymour is a consultant and adviser for AbbVie, Genentech, and Roche. Dr. Roberts reported research funding from AbbVie and Genentech and milestone payments to his institution related to ABT-199.
MILAN – The investigational Bcl-2 inhibitor ABT-199 continues to impress with substantial activity as single-agent or combination therapy in relapsed or refractory chronic lymphocytic leukemia, following dose-scheduling modifications to address the risk of tumor lysis syndrome.
ABT-199 monotherapy
Updated data from all 105 CLL patients in the phase I trial show the overall response rate remains high at 77%, with 23% of patients achieving complete remission.
Seven of 11 complete responders assessed had no detectable minimal residual disease, Dr. John F. Seymour said at the annual congress of the European Hematology Association.
Overall response rates were sustained in the 75%-80% range for high-risk patients with deletion 17p, fludarabine-refractory, or immunoglobulin heavy-chain variable (IGHV)-unmutated CLL; and complete response rates in these subgroups also did not differ from the overall group at 22% to 29%.
Prior results from the first-in-human trial dazzled the leukemia community, but tumor lysis syndrome (TLS) complications, including two fatal events, temporarily halted ABT-199 clinical trials and gave the advantage to its closest competitor, the recently approved and more tolerable CLL drug ibrutinib (Imbruvica).
Use of a modified, ramp-up dosing scheme and aggressive TLS prophylaxis appear to have ameliorated the risk of TLS, with no further clinically significant or grade 3 or 4 events reported in the 49 patients treated with this schema, said Dr. Seymour, director of hematology and cancer medicine at the Peter MacCallum Cancer Centre, East Melbourne, Australia.
Rather than using a 3-week schedule and 50-mg starting dose, the safety expansion cohort received once-daily oral ABT-199 beginning at 20 mg, with weekly adjustments to 50 mg, 100 mg, 200 mg, and 400 mg over 5 weeks.
The 400-mg dose has been identified as the phase II dose, with 59% of patients free of progression at 18 months and beyond on this dose, he said.
As of April 2014, 37 of the 105 patients discontinued treatment, 22 due to progressive disease and 12 for adverse events; in addition, two proceeded to allogeneic hematopoietic cell transplantation, and one needed Coumadin, which is not permitted on protocol.
The median duration of response has not yet been reached for patients treated at doses of 400 mg or above, he said.
The most common treatment-emergent adverse events of any grade were diarrhea (40% of patients), neutropenia (36%), and nausea (35%).
Neutropenia was the only grade 3/4 event occurring in more than 10% of patients (33%), followed by anemia in 10%.
Combination ABT-199
Of substantial interest to many was a second phase Ib study presented in the same session, evaluating the role of ABT-199 with the anti-CD20 antibody rituximab (Rituxan) in relapsed or refractory CLL.
After a median time on study of just 7.5 months, the overall response rate was 84% among 25 evaluable patients, including 9 complete responses (36%) and 12 partial responses (48%), said Dr. Andrew W. Roberts, with the Royal Melbourne (Australia) Hospital and Walter and Eliza Hall Institute of Medical Research.
Six of eight complete responders tested were negative for minimal residual disease by flow cytometry.
Preliminary pharmacokinetic results suggest no apparent effect of rituximab on ABT-199 exposure, he said.
Three patients discontinued ABT-199 after achieving a complete remission, including one with fludarabine-refractory disease, and all remain in complete remission at 8.6, 8.8, and 11.6 months after cessation.
Dose modifications were also made in this study following a fatal TLS event in December 2012 after a first dose of ABT-199 at 50 mg. Under the modified step-up dosing, ABT-199 was started at 20 mg, escalating up to 600 mg daily over 5 weeks, with rituximab 375 mg/m2 added on day 1 of week 5 and rituximab 500 mg/m2 added on day 1 of months 2-6.
The combination was well tolerated, and no new safety concerns were identified, Dr. Roberts said. The most common grade 3/4 adverse events among 45 patients evaluable for safety were neutropenia in 47%, anemia in 16%, thrombocytopenia in 13%, and febrile neutropenia in 7%. Grade 3/4 neutropenia was more common at 600 mg, with the 400-mg dose selected for the ongoing safety expansion cohort. Two serious TLS events occurred, but both were prior to schedule modifications, he said.
One patient in the combination study and 13 in the monotherapy study received treatment for small lymphocytic lymphoma. Response rates and tolerability were similar between CLL and SLL patients in the monotherapy study, Dr. Roberts said in an interview.
During a press briefing at the meeting, Dr. Seymour said ABT-199 potentially could be combined with ibrutinib and that the combination was very potent in laboratory tests in both CLL and some forms of mantle cell lymphoma. Negotiations with the various companies involved are intricate, but there is agreement and commitment "to begin clinical trials of the combination later this year," he added.
ABT-199 is currently being evaluated in a phase II trial as monotherapy in deletion 17p relapsed CLL, as combination therapy with rituximab versus bendamustine plus rituximab in a phase III trial in relapsed/refractory CLL, and in combination trials with bendamustine/rituximab and obinutuzumab in relapsed/refractory CLL.
AbbVie and Genentech sponsored the trials. Dr. Seymour is a consultant and adviser for AbbVie, Genentech, and Roche. Dr. Roberts reported research funding from AbbVie and Genentech and milestone payments to his institution related to ABT-199.
AT THE EHA CONGRESS
Major finding: The overall response rate was 77% with ABT-199 monotherapy and 84% with the addition of rituximab in relapsed or refractory CLL.
Key clinical point: ABT-199 alone or as combination therapy with rituximab has substantial activity in relapsed/refractory CLL.
Data source: Two phase I trials in patients with CLL.
Disclosures: AbbVie and Genentech, codevelopers of ABT-199, sponsored the trials. Dr. Seymour is a consultant and adviser for AbbVie, Genentech, and Roche. Dr. Roberts reported research funding from AbbVie and Genentech and milestone payments to his institution related to ABT-199.
Cutaneous erythema predicted low-dose alemtuzumab response in leukemic cutaneous T-cell lymphoma
In patients with leukemic cutaneous T-cell lymphoma, diffuse cutaneous erythema without plaques or tumors predicted complete remission with low-dose alemtuzumab therapy, investigators reported online April 23 in a JAMA Dermatology research letter.
"Initial clinical presentation was more predictive of response than was complex cellular phenotyping of T cells from blood and skin," wrote Dr. Rei Watanabe at Brigham and Women’s Hospital in Boston and associates. "In other words, the eyes of a well-trained dermatologist were more powerful than a comprehensive translational research program in identifying complete responders to LDA [low-dose alemtuzumab] therapy."
The researchers treated 23 patients with leukemic cutaneous T-cell lymphoma with 10 mg subcutaneous LDA three times weekly. Seventeen patients presented with diffuse erythema without superimposed plaques or tumors, of whom 13 experienced complete remission after LDA treatment and 4 had residual or emergent disease that could be controlled with skin-directed therapy (JAMA Dermatol. 2014 [doi:10.1001/jamadermatol.2013.10099]).
However, none of the six patients who presented with discrete patches, plaques, or tumors experienced full remission with LDA, the researchers reported. One patient later remitted with electron beam therapy, while five had their disease recur or progress, including two who developed large cell transformation.
Patients also were more likely to respond to LDA if more than 80% of their malignant T cells had TCM markers (CCR7+/L-selectin+), but clinical presentation better predicted response, the investigators reported.
"On a scientific level, diffuse erythema is likely caused by migrating T cells, and only T cells that migrate into blood are cleared by LDA," noted Dr. Watanabe and her associates. "Fixed skin lesions are more likely to be caused by TRM [nonmigratory skin resident memory T cells], cells that escape LDA clearance by remaining long-term in skin."
Based on the findings, the investigators recommended using LDA with or without adjuvant skin-directed treatments in patients who present with diffuse cutaneous erythema. But "we caution against its use in patients with preexisting plaques and/or tumors," the researchers said.
The Leukemia & Lymphoma Society, the National Institutes of Health, the Damon Runyon Cancer Research Foundation, and a private contribution from Edward P. Lawrence, Esq., funded the study. Coauthor Dr. Clark reported receiving honoraria from Novartis and Stiefel Laboratories. The authors disclosed no other conflicts of interest.
In patients with leukemic cutaneous T-cell lymphoma, diffuse cutaneous erythema without plaques or tumors predicted complete remission with low-dose alemtuzumab therapy, investigators reported online April 23 in a JAMA Dermatology research letter.
"Initial clinical presentation was more predictive of response than was complex cellular phenotyping of T cells from blood and skin," wrote Dr. Rei Watanabe at Brigham and Women’s Hospital in Boston and associates. "In other words, the eyes of a well-trained dermatologist were more powerful than a comprehensive translational research program in identifying complete responders to LDA [low-dose alemtuzumab] therapy."
The researchers treated 23 patients with leukemic cutaneous T-cell lymphoma with 10 mg subcutaneous LDA three times weekly. Seventeen patients presented with diffuse erythema without superimposed plaques or tumors, of whom 13 experienced complete remission after LDA treatment and 4 had residual or emergent disease that could be controlled with skin-directed therapy (JAMA Dermatol. 2014 [doi:10.1001/jamadermatol.2013.10099]).
However, none of the six patients who presented with discrete patches, plaques, or tumors experienced full remission with LDA, the researchers reported. One patient later remitted with electron beam therapy, while five had their disease recur or progress, including two who developed large cell transformation.
Patients also were more likely to respond to LDA if more than 80% of their malignant T cells had TCM markers (CCR7+/L-selectin+), but clinical presentation better predicted response, the investigators reported.
"On a scientific level, diffuse erythema is likely caused by migrating T cells, and only T cells that migrate into blood are cleared by LDA," noted Dr. Watanabe and her associates. "Fixed skin lesions are more likely to be caused by TRM [nonmigratory skin resident memory T cells], cells that escape LDA clearance by remaining long-term in skin."
Based on the findings, the investigators recommended using LDA with or without adjuvant skin-directed treatments in patients who present with diffuse cutaneous erythema. But "we caution against its use in patients with preexisting plaques and/or tumors," the researchers said.
The Leukemia & Lymphoma Society, the National Institutes of Health, the Damon Runyon Cancer Research Foundation, and a private contribution from Edward P. Lawrence, Esq., funded the study. Coauthor Dr. Clark reported receiving honoraria from Novartis and Stiefel Laboratories. The authors disclosed no other conflicts of interest.
In patients with leukemic cutaneous T-cell lymphoma, diffuse cutaneous erythema without plaques or tumors predicted complete remission with low-dose alemtuzumab therapy, investigators reported online April 23 in a JAMA Dermatology research letter.
"Initial clinical presentation was more predictive of response than was complex cellular phenotyping of T cells from blood and skin," wrote Dr. Rei Watanabe at Brigham and Women’s Hospital in Boston and associates. "In other words, the eyes of a well-trained dermatologist were more powerful than a comprehensive translational research program in identifying complete responders to LDA [low-dose alemtuzumab] therapy."
The researchers treated 23 patients with leukemic cutaneous T-cell lymphoma with 10 mg subcutaneous LDA three times weekly. Seventeen patients presented with diffuse erythema without superimposed plaques or tumors, of whom 13 experienced complete remission after LDA treatment and 4 had residual or emergent disease that could be controlled with skin-directed therapy (JAMA Dermatol. 2014 [doi:10.1001/jamadermatol.2013.10099]).
However, none of the six patients who presented with discrete patches, plaques, or tumors experienced full remission with LDA, the researchers reported. One patient later remitted with electron beam therapy, while five had their disease recur or progress, including two who developed large cell transformation.
Patients also were more likely to respond to LDA if more than 80% of their malignant T cells had TCM markers (CCR7+/L-selectin+), but clinical presentation better predicted response, the investigators reported.
"On a scientific level, diffuse erythema is likely caused by migrating T cells, and only T cells that migrate into blood are cleared by LDA," noted Dr. Watanabe and her associates. "Fixed skin lesions are more likely to be caused by TRM [nonmigratory skin resident memory T cells], cells that escape LDA clearance by remaining long-term in skin."
Based on the findings, the investigators recommended using LDA with or without adjuvant skin-directed treatments in patients who present with diffuse cutaneous erythema. But "we caution against its use in patients with preexisting plaques and/or tumors," the researchers said.
The Leukemia & Lymphoma Society, the National Institutes of Health, the Damon Runyon Cancer Research Foundation, and a private contribution from Edward P. Lawrence, Esq., funded the study. Coauthor Dr. Clark reported receiving honoraria from Novartis and Stiefel Laboratories. The authors disclosed no other conflicts of interest.
FROM JAMA DERMATOLOGY
Major finding: Of 17 patients who presented with diffuse cutaneous erythema without superimposed plaques or tumors, 13 fully remitted on low-dose alemtuzumab LDA and 4 had disease that was controllable with skin-directed therapy. None of the six patients who presented with discrete patches, plaques, or tumors experienced full remission with LDA.
Data source: A study of 23 patients with leukemic cutaneous T-cell lymphoma.
Disclosures: The Leukemia & Lymphoma Society, the National Institutes of Health, the Damon Runyon Cancer Research Foundation, and a private contribution from Edward P. Lawrence, Esq., funded the study. Coauthor Dr. Clark reported receiving honoraria from Novartis and Stiefel Laboratories. The authors disclosed no other conflicts of interest.
No survival benefit for routine surveillance scans in classical Hodgkin disease
CHICAGO – Routine surveillance imaging does not improve clinical outcomes in patients with classic Hodgkin disease who are in first complete remission and it sharply increases costs, researchers reported at the annual meeting of the American Society of Clinical Oncology.
The team, led by Dr. Sai Ravi Pingali, retrospectively reviewed the charts of 241 adult patients who achieved a complete remission after first-line therapy.
In 68%, the treating physicians’ planned approach was routine surveillance imaging, which consisted of radiologic imaging with scans every few months, plus clinical exams and laboratory testing. In the other 32%, the planned approach was clinical surveillance, meaning that radiologic imaging was performed only if concerning signs or symptoms occurred.
The two groups had statistically indistinguishable overall survival, and in both groups, all patients experiencing relapse successfully achieved a second complete remission with salvage therapy.
In the routinely imaged group, scanning increased costs by nearly $20,000 per patient and by almost $600,000 per each relapse detected. In addition, patients were exposed to the associated radiation.
"We were unable to detect an overall survival benefit associated with routine surveillance imaging, although I have to acknowledge that our study was limited in power given the small number of deaths and relapses," commented Dr. Pingali, an oncologist with the Medical College of Wisconsin Affiliated Hospitals in Milwaukee.
"Relapses in both ... groups were effectively salvaged with autologous stem cell transplantation, arguing against a critical advantage of detection of asymptomatic relapse. Also, we need to keep in mind that the costs associated with routine surveillance imaging are significant, and it is also associated with potential risks, both in terms of radiation exposure and unnecessary work-up," he added.
"We do not feel that potential risks and costs without overall survival benefit or any other clinical benefit justify the practice of routine surveillance imaging in classical Hodgkin lymphoma patients who have achieved a complete remission after first-line therapy. We recommend that such patients be followed clinically," Dr. Pingali concluded.
Invited discussant Dr. Leo Gordon of Northwestern University in Chicago, agreed that accumulating data argue against routine imaging for surveillance in this context and noted that insurers will likely not continue to cover scans having no proven benefit. The data should prompt a revision of guidelines and reeducation of clinicians and patients, he said.
"For translational researchers and investigators and academics, I think we need to convince journal reviewers that a manuscript is acceptable if scans are not so frequent. And for industry trials, I think we need to discuss with the Food and Drug Administration the endpoint of progression-free survival and that those endpoints may not only be driven by scans but by more mundane parameters," he said—namely, the history and physical examination.
But session comoderator Dr. Gilles A. Salles of Hospices Civils de Lyon, Université Claude Bernard, France, expressed reservations, noting that the study did not provide information on how patients were allocated to groups and the time frame of relapse.
"It may be different whether relapses occur early, in the first year, or they occur later, and that may have some implications for the surveillance," he said. "I understand that you and many others jumped over the idea that we should immediately stop. A few people may think that we need more solid data, despite the provocative and quality data that were presented, to really make this jump in clinical practice. That’s my personal opinion."
Dr. Pingali and his team retrospectively reviewed the charts of adult patients who received a new diagnosis of classical Hodgkin lymphoma between 2000 and 2010 at three institutions, achieved complete remission after first-line therapy and had at least 2 years of follow-up.
The routine surveillance imaging and clinical surveillance groups had similar demographic and disease characteristics, Dr. Pingali reported. The former were significantly more likely to have received ABVD (doxorubicin [Adriamycin], bleomycin, vinblastine, and dacarbazine) and less likely to have received the Stanford V regimen as first-line therapy, and they were significantly less likely to have received radiation therapy.
With a median duration of follow-up of about 4 years, the groups did not differ significantly with respect to overall survival. "When we look at the 5-year time point, when typically the surveillance CT scans are discontinued, the curves are pretty much superimposable," he pointed out.
There were five deaths in the routine surveillance imaging group, one of which was from relapsed disease; the other deaths were from cancer, heart failure, hip fracture, and myelodysplastic syndrome. There were four deaths in the clinical surveillance group: two were from non-Hodgkin lymphoma and two from unknown causes while the patient was in confirmed remission.
All of the six patients in the routinely imaged group and all of the five patients in the clinically followed group experiencing a relapse achieved another complete remission with second-line therapy.
The mean number of scans received was 1.14 in the clinical surveillance group – usually the scan performed after first-line treatment to confirm remission, according to Dr. Pingali – and 4.25 in the routine surveillance imaging group. The ratio of scans to detected relapses was 18 vs. 124.
The extra charges incurred from scans using the routine surveillance imaging approach were $18,896/patient and $593,698/relapse.
"It is important to note that this does not include additional costs from the work-up of the false-positive scans and also the wages lost," he noted.
Dr. Pingali disclosed no relevant conflicts of interest. Dr. Gordon disclosed that he receives honoraria from Genentech and research funding from Millennium and Pharmacyclics. Dr. Salles disclosed serving as an advisor or consultant for Calistoga Pharmaceuticals, Celgene; Genentech, Janssen Pharmaceutica, and Roche. He has served as a speaker or a member of a speakers bureau and has received grants for clinical research from Celgene and Roche.
CHICAGO – Routine surveillance imaging does not improve clinical outcomes in patients with classic Hodgkin disease who are in first complete remission and it sharply increases costs, researchers reported at the annual meeting of the American Society of Clinical Oncology.
The team, led by Dr. Sai Ravi Pingali, retrospectively reviewed the charts of 241 adult patients who achieved a complete remission after first-line therapy.
In 68%, the treating physicians’ planned approach was routine surveillance imaging, which consisted of radiologic imaging with scans every few months, plus clinical exams and laboratory testing. In the other 32%, the planned approach was clinical surveillance, meaning that radiologic imaging was performed only if concerning signs or symptoms occurred.
The two groups had statistically indistinguishable overall survival, and in both groups, all patients experiencing relapse successfully achieved a second complete remission with salvage therapy.
In the routinely imaged group, scanning increased costs by nearly $20,000 per patient and by almost $600,000 per each relapse detected. In addition, patients were exposed to the associated radiation.
"We were unable to detect an overall survival benefit associated with routine surveillance imaging, although I have to acknowledge that our study was limited in power given the small number of deaths and relapses," commented Dr. Pingali, an oncologist with the Medical College of Wisconsin Affiliated Hospitals in Milwaukee.
"Relapses in both ... groups were effectively salvaged with autologous stem cell transplantation, arguing against a critical advantage of detection of asymptomatic relapse. Also, we need to keep in mind that the costs associated with routine surveillance imaging are significant, and it is also associated with potential risks, both in terms of radiation exposure and unnecessary work-up," he added.
"We do not feel that potential risks and costs without overall survival benefit or any other clinical benefit justify the practice of routine surveillance imaging in classical Hodgkin lymphoma patients who have achieved a complete remission after first-line therapy. We recommend that such patients be followed clinically," Dr. Pingali concluded.
Invited discussant Dr. Leo Gordon of Northwestern University in Chicago, agreed that accumulating data argue against routine imaging for surveillance in this context and noted that insurers will likely not continue to cover scans having no proven benefit. The data should prompt a revision of guidelines and reeducation of clinicians and patients, he said.
"For translational researchers and investigators and academics, I think we need to convince journal reviewers that a manuscript is acceptable if scans are not so frequent. And for industry trials, I think we need to discuss with the Food and Drug Administration the endpoint of progression-free survival and that those endpoints may not only be driven by scans but by more mundane parameters," he said—namely, the history and physical examination.
But session comoderator Dr. Gilles A. Salles of Hospices Civils de Lyon, Université Claude Bernard, France, expressed reservations, noting that the study did not provide information on how patients were allocated to groups and the time frame of relapse.
"It may be different whether relapses occur early, in the first year, or they occur later, and that may have some implications for the surveillance," he said. "I understand that you and many others jumped over the idea that we should immediately stop. A few people may think that we need more solid data, despite the provocative and quality data that were presented, to really make this jump in clinical practice. That’s my personal opinion."
Dr. Pingali and his team retrospectively reviewed the charts of adult patients who received a new diagnosis of classical Hodgkin lymphoma between 2000 and 2010 at three institutions, achieved complete remission after first-line therapy and had at least 2 years of follow-up.
The routine surveillance imaging and clinical surveillance groups had similar demographic and disease characteristics, Dr. Pingali reported. The former were significantly more likely to have received ABVD (doxorubicin [Adriamycin], bleomycin, vinblastine, and dacarbazine) and less likely to have received the Stanford V regimen as first-line therapy, and they were significantly less likely to have received radiation therapy.
With a median duration of follow-up of about 4 years, the groups did not differ significantly with respect to overall survival. "When we look at the 5-year time point, when typically the surveillance CT scans are discontinued, the curves are pretty much superimposable," he pointed out.
There were five deaths in the routine surveillance imaging group, one of which was from relapsed disease; the other deaths were from cancer, heart failure, hip fracture, and myelodysplastic syndrome. There were four deaths in the clinical surveillance group: two were from non-Hodgkin lymphoma and two from unknown causes while the patient was in confirmed remission.
All of the six patients in the routinely imaged group and all of the five patients in the clinically followed group experiencing a relapse achieved another complete remission with second-line therapy.
The mean number of scans received was 1.14 in the clinical surveillance group – usually the scan performed after first-line treatment to confirm remission, according to Dr. Pingali – and 4.25 in the routine surveillance imaging group. The ratio of scans to detected relapses was 18 vs. 124.
The extra charges incurred from scans using the routine surveillance imaging approach were $18,896/patient and $593,698/relapse.
"It is important to note that this does not include additional costs from the work-up of the false-positive scans and also the wages lost," he noted.
Dr. Pingali disclosed no relevant conflicts of interest. Dr. Gordon disclosed that he receives honoraria from Genentech and research funding from Millennium and Pharmacyclics. Dr. Salles disclosed serving as an advisor or consultant for Calistoga Pharmaceuticals, Celgene; Genentech, Janssen Pharmaceutica, and Roche. He has served as a speaker or a member of a speakers bureau and has received grants for clinical research from Celgene and Roche.
CHICAGO – Routine surveillance imaging does not improve clinical outcomes in patients with classic Hodgkin disease who are in first complete remission and it sharply increases costs, researchers reported at the annual meeting of the American Society of Clinical Oncology.
The team, led by Dr. Sai Ravi Pingali, retrospectively reviewed the charts of 241 adult patients who achieved a complete remission after first-line therapy.
In 68%, the treating physicians’ planned approach was routine surveillance imaging, which consisted of radiologic imaging with scans every few months, plus clinical exams and laboratory testing. In the other 32%, the planned approach was clinical surveillance, meaning that radiologic imaging was performed only if concerning signs or symptoms occurred.
The two groups had statistically indistinguishable overall survival, and in both groups, all patients experiencing relapse successfully achieved a second complete remission with salvage therapy.
In the routinely imaged group, scanning increased costs by nearly $20,000 per patient and by almost $600,000 per each relapse detected. In addition, patients were exposed to the associated radiation.
"We were unable to detect an overall survival benefit associated with routine surveillance imaging, although I have to acknowledge that our study was limited in power given the small number of deaths and relapses," commented Dr. Pingali, an oncologist with the Medical College of Wisconsin Affiliated Hospitals in Milwaukee.
"Relapses in both ... groups were effectively salvaged with autologous stem cell transplantation, arguing against a critical advantage of detection of asymptomatic relapse. Also, we need to keep in mind that the costs associated with routine surveillance imaging are significant, and it is also associated with potential risks, both in terms of radiation exposure and unnecessary work-up," he added.
"We do not feel that potential risks and costs without overall survival benefit or any other clinical benefit justify the practice of routine surveillance imaging in classical Hodgkin lymphoma patients who have achieved a complete remission after first-line therapy. We recommend that such patients be followed clinically," Dr. Pingali concluded.
Invited discussant Dr. Leo Gordon of Northwestern University in Chicago, agreed that accumulating data argue against routine imaging for surveillance in this context and noted that insurers will likely not continue to cover scans having no proven benefit. The data should prompt a revision of guidelines and reeducation of clinicians and patients, he said.
"For translational researchers and investigators and academics, I think we need to convince journal reviewers that a manuscript is acceptable if scans are not so frequent. And for industry trials, I think we need to discuss with the Food and Drug Administration the endpoint of progression-free survival and that those endpoints may not only be driven by scans but by more mundane parameters," he said—namely, the history and physical examination.
But session comoderator Dr. Gilles A. Salles of Hospices Civils de Lyon, Université Claude Bernard, France, expressed reservations, noting that the study did not provide information on how patients were allocated to groups and the time frame of relapse.
"It may be different whether relapses occur early, in the first year, or they occur later, and that may have some implications for the surveillance," he said. "I understand that you and many others jumped over the idea that we should immediately stop. A few people may think that we need more solid data, despite the provocative and quality data that were presented, to really make this jump in clinical practice. That’s my personal opinion."
Dr. Pingali and his team retrospectively reviewed the charts of adult patients who received a new diagnosis of classical Hodgkin lymphoma between 2000 and 2010 at three institutions, achieved complete remission after first-line therapy and had at least 2 years of follow-up.
The routine surveillance imaging and clinical surveillance groups had similar demographic and disease characteristics, Dr. Pingali reported. The former were significantly more likely to have received ABVD (doxorubicin [Adriamycin], bleomycin, vinblastine, and dacarbazine) and less likely to have received the Stanford V regimen as first-line therapy, and they were significantly less likely to have received radiation therapy.
With a median duration of follow-up of about 4 years, the groups did not differ significantly with respect to overall survival. "When we look at the 5-year time point, when typically the surveillance CT scans are discontinued, the curves are pretty much superimposable," he pointed out.
There were five deaths in the routine surveillance imaging group, one of which was from relapsed disease; the other deaths were from cancer, heart failure, hip fracture, and myelodysplastic syndrome. There were four deaths in the clinical surveillance group: two were from non-Hodgkin lymphoma and two from unknown causes while the patient was in confirmed remission.
All of the six patients in the routinely imaged group and all of the five patients in the clinically followed group experiencing a relapse achieved another complete remission with second-line therapy.
The mean number of scans received was 1.14 in the clinical surveillance group – usually the scan performed after first-line treatment to confirm remission, according to Dr. Pingali – and 4.25 in the routine surveillance imaging group. The ratio of scans to detected relapses was 18 vs. 124.
The extra charges incurred from scans using the routine surveillance imaging approach were $18,896/patient and $593,698/relapse.
"It is important to note that this does not include additional costs from the work-up of the false-positive scans and also the wages lost," he noted.
Dr. Pingali disclosed no relevant conflicts of interest. Dr. Gordon disclosed that he receives honoraria from Genentech and research funding from Millennium and Pharmacyclics. Dr. Salles disclosed serving as an advisor or consultant for Calistoga Pharmaceuticals, Celgene; Genentech, Janssen Pharmaceutica, and Roche. He has served as a speaker or a member of a speakers bureau and has received grants for clinical research from Celgene and Roche.
AT THE ASCO ANNUAL MEETING 2013
Major finding: The ratio of scans to detected relapses was 18 for the clinical surveillance group and 124 for the routine scan group. The extra charges incurred from scans using the routine surveillance imaging approach were $18,896/patient and $593,698/relapse.
Data source: A retrospective analysis of 241 patients with classical Hodgkin lymphoma in first complete remission
Disclosures: Dr. Pingali disclosed no relevant conflicts of interest. Dr. Gordon disclosed that he receives honoraria from Genentech and research funding from Millennium and Pharmacyclics. Dr. Salles disclosed serving as an advisor or consultant for Calistoga Pharmaceuticals, Celgene; Genentech, Janssen Pharmaceutica, and Roche. He has served as a speaker or a member of a speakers bureau and has received grants for clinical research from Celgene and Roche.