User login
Warty papule and scaling around finger
A 35-year-old Caucasian man was referred to our clinic for treatment of a nonhealing “wart-like” growth on his left index finger. He said that the lesion had been there for at least 2 years and complained of extensive periungual erythema and scaling of the same finger. The patient was immunocompetent and denied trauma, chronic nail infection, arsenic exposure, or radiation to this particular finger. On several occasions, liquid nitrogen cryotherapy had been used on the growth, without improvement.
Physical examination revealed a well-demarcated erythematous patch with scaling involving the medial and proximal periungual areas of the left index finger (FIGURE). There was also a distinct, rough-surfaced 5×4 mm papule with scales, crusts, and small fissures in the middle of the patch. In addition, there was onychodystrophy with keratotic debris on the medial aspect of the same finger. Our attempt to scrape this papule failed and was painful for the patient.
The other nails were normal. Review of the patient’s systems, family history, and personal history were otherwise unremarkable. We biopsied the lesion.
FIGURE
Erythematous patch and rough-surfaced papule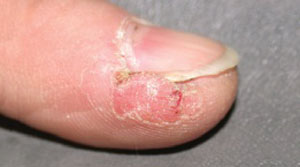
What is your diagnosis?
How would you manage this condition?
Dx: Bowen’s disease of the nail
Bowen’s disease (BD), a form of intraepidermal (in situ) squamous cell carcinoma (SCC), may affect the skin but also the nail unit. It presents as periungual or subungual verrucous plaques, erosions, and ulcerations, with nail discoloration, dystrophy, or onycholysis.
Our patient was younger than the norm. BD of the nail—which typically involves the fingers—occurs in both men and women, but is most common in men in their 50s. It often presents with verrucous, scaly, crusting, erythematous, or fissuring lesions that may involve any portion of the nail apparatus with associated onycholysis, nail dystrophy, or longitudinal erythronychia or melanonychia (red or black longitudinal nail streak).1-6 BD of the nail may involve more than one digit on multiple extremities, simultaneously or sequentially.5 Initial BD of the nail may spread to periungual or subungual areas or vice versa. Bleeding, ulceration, or a nodule may signal the development of invasive SCC.3,6
Radiation and HPV linked to BD of the nail
While the etiology of this condition is unclear, trauma, chronic paronychia, ionizing radiation, infectious agents, ultraviolet exposure, arsenic or pesticide exposure, and immunosuppression are a few proposed predisposing factors.1,3,4,6
Human papilloma virus (HPV) subtypes 16, 34, and 35 have been identified in BD and SCC lesions. These subtypes with oncogenic potential may play a role in the development of BD and invasive SCC of the nail and other cutaneous regions.
In cases where there has been HPV-16 infection in a digit and the genital region, researchers have suggested autoinoculation as a transmission mode from the anogenital area to the digit, or vice versa.6-8
The pathogenesis behind polydactylous BD of the nail has been linked to factors such as trauma, radiation, and immunosuppression (eg, post-transplant, oncological, and HIV patients).5,8
Differential includes onychomycosis, eczema
The differential for BD of the nail includes onychomycosis, paronychia, verruca vulgaris, eczema, pyogenic granuloma, glomus tumor, and verrucous tuberculosis. The differential also includes: subungual exostosis, onychomatricoma, amelanotic malignant melanoma, keratoacanthoma, fibrokeratoma, and gouty tophus.
Irregular borders and the presence of a scaly patch with papules should raise your suspicion of BD. Biopsy is necessary to confirm your suspicions. In fact, all chronic and recalcitrant lesions of the nail apparatus should be biopsied to rule out BD.9
Biopsy with care
The matrix is a germinating portion of the nail and requires special care, because damage to it may permanently affect nail formation and function. Proper anesthesia and hemostasis are also key, given that the nail apparatus is very vascular and well innervated.9
The histopathology of BD lesions is characterized by hyperkeratosis; parakeratosis; loss of orderly maturation, polarity, and a granular layer; and keratinocytic atypia involving the entire acanthotic epithelial layer. The atypia and dyskeratosis are confined to the epidermis. However, some microscopic specimens of BD may simultaneously demonstrate features of invasive SCC in other areas of the lesion. Microinvasion is common in long-term BD with reports of invasive carcinoma in approximately 15% of cases.4-6,9
Consider CO2 laser therapy, Mohs surgery
Various therapeutic modalities have been used for BD of the nail, including electrodesiccation and curettage, 5% fluorouracil cream (Efudex), cryosurgery, and radiotherapy. Most of these treatments have not been successful and are associated with high recurrence rates.10-12
Simple excision of nail bed and matrix is successful in small and localized BD lesions. CO2 laser therapy for periungual BD has been reported to have up to an 80% cure rate, with less scarring and contractures when compared with surgical excision.10 Recently, imiquimod (Aldara 5% cream) has been used with success for BD of the nail—especially recurrent disease.11
Mohs surgery is considered the best treatment approach, with cure rates of up to 96%.12 Mohs surgery allows for adequate depth of tumor resection, great preservation of normal digit function, and excellent cosmetic results, with healing by secondary intention. Although the 5-year recurrence rate after Mohs surgery is small (about 3%), it is still a good idea to follow the patient closely to assess for a potential relapse.
If invasive SCC is suspected, the patient’s regional lymph nodes should be evaluated for possible metastasis. Radiologic study of the digit(s) to assess for bone invasion should also be considered. Amputation of the affected digit, although a drastic measure, is an option if there is evidence of bone involvement.13
Our patient’s course. After discussing the different treatment options with our patient, we referred him to plastic surgery for wide excision. He was subsequently lost to follow up.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected]
1. Fleckman P, Allan C. Surgical anatomy of the nail unit. Dermatol Surg. 2001;27:257-260.
2. Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol. 2000;143:132-135.
3. Kaiser JF, Proctor-Shipman L. Squamous cell carcinoma in situ (Bowen’s disease) mimicking subungual verruca vulgaris. J Fam Pract. 1994;39:384-387.
4. Ongenae K, Kerckhove MV, Naeyaert J. Bowen’s disease of the nail. Dermatology. 2002;204:348-350.
5. Koch A, Schonlebe J, Haroske G, et al. Polydactylous Bowen’s disease. J Eur Acad Dermatol Venereol. 2003;17:213-215.
6. Ratner D. Recurrent squamous cell carcinoma in situ of the finger. SKINmed. 2003;2:251-252.
7. McHugh RW, Hazen P, Eliezri YD, et al. Metastatic periungual squamous cell carcinoma: detection of human papillomavirus type 35 RNA in the digital tumor and axillary lymph node metastases. J Am Acad Dermatol. 1996;34:1080-1082.
8. Moy RL, Eliezri YD, Nuovo GJ, et al. Human papilloma type 16 DNA in periungual squamous cell carcinomas. JAMA. 1989;261:2669-2673.
9. Rich P. Nail biopsy: indications and methods. Dermatol Surg. 2001;27:229-234.
10. Gordon KB, Garden JM, Robinson JK. Bowen’s disease of the distal digit. Outcome of treatment with carbon dioxide laser vaporization. Dermatol Surg. 1996;22:723-728.
11. Laffitte E, Saurat JH. Recurrent Bowen’s disease of the nail: treatment by topical imiquimod (Aldara). Ann Dermatol Venereol. 2003;130:211-213.
12. Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
13. Peterson SR, Layton EG, Joseph AK. Squamous cell carcinoma of the nail unit with evidence of bony involvement: a multidisciplinary approach to resection and reconstruction. Dermatol Surg. 2004;30:218-221.
A 35-year-old Caucasian man was referred to our clinic for treatment of a nonhealing “wart-like” growth on his left index finger. He said that the lesion had been there for at least 2 years and complained of extensive periungual erythema and scaling of the same finger. The patient was immunocompetent and denied trauma, chronic nail infection, arsenic exposure, or radiation to this particular finger. On several occasions, liquid nitrogen cryotherapy had been used on the growth, without improvement.
Physical examination revealed a well-demarcated erythematous patch with scaling involving the medial and proximal periungual areas of the left index finger (FIGURE). There was also a distinct, rough-surfaced 5×4 mm papule with scales, crusts, and small fissures in the middle of the patch. In addition, there was onychodystrophy with keratotic debris on the medial aspect of the same finger. Our attempt to scrape this papule failed and was painful for the patient.
The other nails were normal. Review of the patient’s systems, family history, and personal history were otherwise unremarkable. We biopsied the lesion.
FIGURE
Erythematous patch and rough-surfaced papule
What is your diagnosis?
How would you manage this condition?
Dx: Bowen’s disease of the nail
Bowen’s disease (BD), a form of intraepidermal (in situ) squamous cell carcinoma (SCC), may affect the skin but also the nail unit. It presents as periungual or subungual verrucous plaques, erosions, and ulcerations, with nail discoloration, dystrophy, or onycholysis.
Our patient was younger than the norm. BD of the nail—which typically involves the fingers—occurs in both men and women, but is most common in men in their 50s. It often presents with verrucous, scaly, crusting, erythematous, or fissuring lesions that may involve any portion of the nail apparatus with associated onycholysis, nail dystrophy, or longitudinal erythronychia or melanonychia (red or black longitudinal nail streak).1-6 BD of the nail may involve more than one digit on multiple extremities, simultaneously or sequentially.5 Initial BD of the nail may spread to periungual or subungual areas or vice versa. Bleeding, ulceration, or a nodule may signal the development of invasive SCC.3,6
Radiation and HPV linked to BD of the nail
While the etiology of this condition is unclear, trauma, chronic paronychia, ionizing radiation, infectious agents, ultraviolet exposure, arsenic or pesticide exposure, and immunosuppression are a few proposed predisposing factors.1,3,4,6
Human papilloma virus (HPV) subtypes 16, 34, and 35 have been identified in BD and SCC lesions. These subtypes with oncogenic potential may play a role in the development of BD and invasive SCC of the nail and other cutaneous regions.
In cases where there has been HPV-16 infection in a digit and the genital region, researchers have suggested autoinoculation as a transmission mode from the anogenital area to the digit, or vice versa.6-8
The pathogenesis behind polydactylous BD of the nail has been linked to factors such as trauma, radiation, and immunosuppression (eg, post-transplant, oncological, and HIV patients).5,8
Differential includes onychomycosis, eczema
The differential for BD of the nail includes onychomycosis, paronychia, verruca vulgaris, eczema, pyogenic granuloma, glomus tumor, and verrucous tuberculosis. The differential also includes: subungual exostosis, onychomatricoma, amelanotic malignant melanoma, keratoacanthoma, fibrokeratoma, and gouty tophus.
Irregular borders and the presence of a scaly patch with papules should raise your suspicion of BD. Biopsy is necessary to confirm your suspicions. In fact, all chronic and recalcitrant lesions of the nail apparatus should be biopsied to rule out BD.9
Biopsy with care
The matrix is a germinating portion of the nail and requires special care, because damage to it may permanently affect nail formation and function. Proper anesthesia and hemostasis are also key, given that the nail apparatus is very vascular and well innervated.9
The histopathology of BD lesions is characterized by hyperkeratosis; parakeratosis; loss of orderly maturation, polarity, and a granular layer; and keratinocytic atypia involving the entire acanthotic epithelial layer. The atypia and dyskeratosis are confined to the epidermis. However, some microscopic specimens of BD may simultaneously demonstrate features of invasive SCC in other areas of the lesion. Microinvasion is common in long-term BD with reports of invasive carcinoma in approximately 15% of cases.4-6,9
Consider CO2 laser therapy, Mohs surgery
Various therapeutic modalities have been used for BD of the nail, including electrodesiccation and curettage, 5% fluorouracil cream (Efudex), cryosurgery, and radiotherapy. Most of these treatments have not been successful and are associated with high recurrence rates.10-12
Simple excision of nail bed and matrix is successful in small and localized BD lesions. CO2 laser therapy for periungual BD has been reported to have up to an 80% cure rate, with less scarring and contractures when compared with surgical excision.10 Recently, imiquimod (Aldara 5% cream) has been used with success for BD of the nail—especially recurrent disease.11
Mohs surgery is considered the best treatment approach, with cure rates of up to 96%.12 Mohs surgery allows for adequate depth of tumor resection, great preservation of normal digit function, and excellent cosmetic results, with healing by secondary intention. Although the 5-year recurrence rate after Mohs surgery is small (about 3%), it is still a good idea to follow the patient closely to assess for a potential relapse.
If invasive SCC is suspected, the patient’s regional lymph nodes should be evaluated for possible metastasis. Radiologic study of the digit(s) to assess for bone invasion should also be considered. Amputation of the affected digit, although a drastic measure, is an option if there is evidence of bone involvement.13
Our patient’s course. After discussing the different treatment options with our patient, we referred him to plastic surgery for wide excision. He was subsequently lost to follow up.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected]
A 35-year-old Caucasian man was referred to our clinic for treatment of a nonhealing “wart-like” growth on his left index finger. He said that the lesion had been there for at least 2 years and complained of extensive periungual erythema and scaling of the same finger. The patient was immunocompetent and denied trauma, chronic nail infection, arsenic exposure, or radiation to this particular finger. On several occasions, liquid nitrogen cryotherapy had been used on the growth, without improvement.
Physical examination revealed a well-demarcated erythematous patch with scaling involving the medial and proximal periungual areas of the left index finger (FIGURE). There was also a distinct, rough-surfaced 5×4 mm papule with scales, crusts, and small fissures in the middle of the patch. In addition, there was onychodystrophy with keratotic debris on the medial aspect of the same finger. Our attempt to scrape this papule failed and was painful for the patient.
The other nails were normal. Review of the patient’s systems, family history, and personal history were otherwise unremarkable. We biopsied the lesion.
FIGURE
Erythematous patch and rough-surfaced papule
What is your diagnosis?
How would you manage this condition?
Dx: Bowen’s disease of the nail
Bowen’s disease (BD), a form of intraepidermal (in situ) squamous cell carcinoma (SCC), may affect the skin but also the nail unit. It presents as periungual or subungual verrucous plaques, erosions, and ulcerations, with nail discoloration, dystrophy, or onycholysis.
Our patient was younger than the norm. BD of the nail—which typically involves the fingers—occurs in both men and women, but is most common in men in their 50s. It often presents with verrucous, scaly, crusting, erythematous, or fissuring lesions that may involve any portion of the nail apparatus with associated onycholysis, nail dystrophy, or longitudinal erythronychia or melanonychia (red or black longitudinal nail streak).1-6 BD of the nail may involve more than one digit on multiple extremities, simultaneously or sequentially.5 Initial BD of the nail may spread to periungual or subungual areas or vice versa. Bleeding, ulceration, or a nodule may signal the development of invasive SCC.3,6
Radiation and HPV linked to BD of the nail
While the etiology of this condition is unclear, trauma, chronic paronychia, ionizing radiation, infectious agents, ultraviolet exposure, arsenic or pesticide exposure, and immunosuppression are a few proposed predisposing factors.1,3,4,6
Human papilloma virus (HPV) subtypes 16, 34, and 35 have been identified in BD and SCC lesions. These subtypes with oncogenic potential may play a role in the development of BD and invasive SCC of the nail and other cutaneous regions.
In cases where there has been HPV-16 infection in a digit and the genital region, researchers have suggested autoinoculation as a transmission mode from the anogenital area to the digit, or vice versa.6-8
The pathogenesis behind polydactylous BD of the nail has been linked to factors such as trauma, radiation, and immunosuppression (eg, post-transplant, oncological, and HIV patients).5,8
Differential includes onychomycosis, eczema
The differential for BD of the nail includes onychomycosis, paronychia, verruca vulgaris, eczema, pyogenic granuloma, glomus tumor, and verrucous tuberculosis. The differential also includes: subungual exostosis, onychomatricoma, amelanotic malignant melanoma, keratoacanthoma, fibrokeratoma, and gouty tophus.
Irregular borders and the presence of a scaly patch with papules should raise your suspicion of BD. Biopsy is necessary to confirm your suspicions. In fact, all chronic and recalcitrant lesions of the nail apparatus should be biopsied to rule out BD.9
Biopsy with care
The matrix is a germinating portion of the nail and requires special care, because damage to it may permanently affect nail formation and function. Proper anesthesia and hemostasis are also key, given that the nail apparatus is very vascular and well innervated.9
The histopathology of BD lesions is characterized by hyperkeratosis; parakeratosis; loss of orderly maturation, polarity, and a granular layer; and keratinocytic atypia involving the entire acanthotic epithelial layer. The atypia and dyskeratosis are confined to the epidermis. However, some microscopic specimens of BD may simultaneously demonstrate features of invasive SCC in other areas of the lesion. Microinvasion is common in long-term BD with reports of invasive carcinoma in approximately 15% of cases.4-6,9
Consider CO2 laser therapy, Mohs surgery
Various therapeutic modalities have been used for BD of the nail, including electrodesiccation and curettage, 5% fluorouracil cream (Efudex), cryosurgery, and radiotherapy. Most of these treatments have not been successful and are associated with high recurrence rates.10-12
Simple excision of nail bed and matrix is successful in small and localized BD lesions. CO2 laser therapy for periungual BD has been reported to have up to an 80% cure rate, with less scarring and contractures when compared with surgical excision.10 Recently, imiquimod (Aldara 5% cream) has been used with success for BD of the nail—especially recurrent disease.11
Mohs surgery is considered the best treatment approach, with cure rates of up to 96%.12 Mohs surgery allows for adequate depth of tumor resection, great preservation of normal digit function, and excellent cosmetic results, with healing by secondary intention. Although the 5-year recurrence rate after Mohs surgery is small (about 3%), it is still a good idea to follow the patient closely to assess for a potential relapse.
If invasive SCC is suspected, the patient’s regional lymph nodes should be evaluated for possible metastasis. Radiologic study of the digit(s) to assess for bone invasion should also be considered. Amputation of the affected digit, although a drastic measure, is an option if there is evidence of bone involvement.13
Our patient’s course. After discussing the different treatment options with our patient, we referred him to plastic surgery for wide excision. He was subsequently lost to follow up.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected]
1. Fleckman P, Allan C. Surgical anatomy of the nail unit. Dermatol Surg. 2001;27:257-260.
2. Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol. 2000;143:132-135.
3. Kaiser JF, Proctor-Shipman L. Squamous cell carcinoma in situ (Bowen’s disease) mimicking subungual verruca vulgaris. J Fam Pract. 1994;39:384-387.
4. Ongenae K, Kerckhove MV, Naeyaert J. Bowen’s disease of the nail. Dermatology. 2002;204:348-350.
5. Koch A, Schonlebe J, Haroske G, et al. Polydactylous Bowen’s disease. J Eur Acad Dermatol Venereol. 2003;17:213-215.
6. Ratner D. Recurrent squamous cell carcinoma in situ of the finger. SKINmed. 2003;2:251-252.
7. McHugh RW, Hazen P, Eliezri YD, et al. Metastatic periungual squamous cell carcinoma: detection of human papillomavirus type 35 RNA in the digital tumor and axillary lymph node metastases. J Am Acad Dermatol. 1996;34:1080-1082.
8. Moy RL, Eliezri YD, Nuovo GJ, et al. Human papilloma type 16 DNA in periungual squamous cell carcinomas. JAMA. 1989;261:2669-2673.
9. Rich P. Nail biopsy: indications and methods. Dermatol Surg. 2001;27:229-234.
10. Gordon KB, Garden JM, Robinson JK. Bowen’s disease of the distal digit. Outcome of treatment with carbon dioxide laser vaporization. Dermatol Surg. 1996;22:723-728.
11. Laffitte E, Saurat JH. Recurrent Bowen’s disease of the nail: treatment by topical imiquimod (Aldara). Ann Dermatol Venereol. 2003;130:211-213.
12. Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
13. Peterson SR, Layton EG, Joseph AK. Squamous cell carcinoma of the nail unit with evidence of bony involvement: a multidisciplinary approach to resection and reconstruction. Dermatol Surg. 2004;30:218-221.
1. Fleckman P, Allan C. Surgical anatomy of the nail unit. Dermatol Surg. 2001;27:257-260.
2. Baran R, Perrin C. Longitudinal erythronychia with distal subungual keratosis: onychopapilloma of the nail bed and Bowen’s disease. Br J Dermatol. 2000;143:132-135.
3. Kaiser JF, Proctor-Shipman L. Squamous cell carcinoma in situ (Bowen’s disease) mimicking subungual verruca vulgaris. J Fam Pract. 1994;39:384-387.
4. Ongenae K, Kerckhove MV, Naeyaert J. Bowen’s disease of the nail. Dermatology. 2002;204:348-350.
5. Koch A, Schonlebe J, Haroske G, et al. Polydactylous Bowen’s disease. J Eur Acad Dermatol Venereol. 2003;17:213-215.
6. Ratner D. Recurrent squamous cell carcinoma in situ of the finger. SKINmed. 2003;2:251-252.
7. McHugh RW, Hazen P, Eliezri YD, et al. Metastatic periungual squamous cell carcinoma: detection of human papillomavirus type 35 RNA in the digital tumor and axillary lymph node metastases. J Am Acad Dermatol. 1996;34:1080-1082.
8. Moy RL, Eliezri YD, Nuovo GJ, et al. Human papilloma type 16 DNA in periungual squamous cell carcinomas. JAMA. 1989;261:2669-2673.
9. Rich P. Nail biopsy: indications and methods. Dermatol Surg. 2001;27:229-234.
10. Gordon KB, Garden JM, Robinson JK. Bowen’s disease of the distal digit. Outcome of treatment with carbon dioxide laser vaporization. Dermatol Surg. 1996;22:723-728.
11. Laffitte E, Saurat JH. Recurrent Bowen’s disease of the nail: treatment by topical imiquimod (Aldara). Ann Dermatol Venereol. 2003;130:211-213.
12. Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
13. Peterson SR, Layton EG, Joseph AK. Squamous cell carcinoma of the nail unit with evidence of bony involvement: a multidisciplinary approach to resection and reconstruction. Dermatol Surg. 2004;30:218-221.
Tumor with central crusting
A 63-year-old man came into our dermatologic surgery clinic with a growth on his left cheek just anterior to his sideburn (FIGURE). Our patient indicated that the lesion, which appeared 6 weeks earlier, started as a small, hard papule with a central depression and rapidly grew to reach its current size and shape.
The patient’s face was sun damaged, and he had a prominent 2.1×1.8 cm well-circumscribed, skin-colored tumor on his cheek. The tumor had a central depression covered by a crust that appeared to conceal a deep keratinous plug. The tumor also had a volcano-like shape and was firm in texture, but tender to palpation and pressure. No lymphadenopathy was present.
The patient had a history of extensive sun exposure, and he’d had previous nonmelanoma skin cancers treated with various medical and surgical techniques. The rest of his history and exam were within normal limits. We performed a biopsy to confirm our clinical diagnosis.
FIGURE
Rapidly growing skin-colored tumor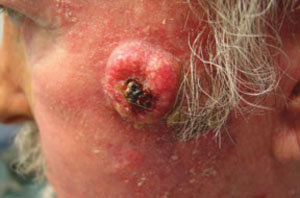
What is your diagnosis?
How would you manage this condition?
Diagnosis: Solitary keratoacanthoma
Our patient had a solitary keratoacanthoma, a unique epidermal tumor that’s characterized by rapid, abundant growth and spontaneous resolution. This tumor goes by many names—molluscum sebaceum, molluscum pseudocarcinomatosum, cutaneous sebaceous neoplasm, and self-healing squamous epithelioma—but keratoacanthoma is the preferred term.1,2
There are several types of keratoacanthoma, but solitary keratoacanthoma remains the most common. It is typically found in light-skinned people in hair-bearing, sun-exposed areas. Peak incidence occurs between the ages of 50 and 69, although the tumors have been reported in patients of all ages. Both sexes are about equally affected, although there is a slight predilection for males. Keratoacanthomas mainly develop on the face (lower lip, cheek, nose, and eyelid), neck, and hands.
The tumors are often considered benign, but they become aggressive in 20% of cases—showing signs of perineural, perivascular, and intravascular invasion and metastases to regional lymph nodes. As a result, some clinicians consider keratoacanthoma to be a pseudomalignancy with self-regressing potential, while others view it as a pseudo-benign tumor progressing into an invasive squamous cell carcinoma (SCC).1,2
The exact etiology of keratoacanthoma is unknown. However, several precipitating factors have been implicated. Primary among them is exposure to ultraviolet light. Other etiological factors include:3-6
- chemical carcinogens (tar, pitch, mineral oil, and cigarette smoking)
- trauma (body peel, carbon dioxide laser resurfacing, megavoltage radiation therapy, and cryosurgery)
- immunosuppression
- surgical scar
- human papilloma virus (HPV).
A tumor with 3 stages
Keratoacanthoma undergoes a proliferative, mature, and involution stage.
In the proliferative stage, there is a rapid increase in tumor size; the tumor can get as big as 10 to 25 mm in diameter in 6 to 8 weeks.1,7
In the mature stage, the tumor stops growing and maintains a typical volcano-like form with a central keratin-filled crater.
In the involution stage, up to 50% of keratoacanthomas undergo spontaneous resolution with expulsion of the keratin plug and resorption of the tumoral mass. The process lasts 4 to 6 weeks, on average, but may take up to 1 year. What’s left behind is a residual atrophic and hypopigmented scar.8
Some lesions persist for a year or more, although the entire process from start to spontaneous resolution usually takes about 4 to 9 months.2,7,8
Is it an SCC or keratoacanthoma?
Histology is the gold standard in diagnosing a keratoacanthoma. A deep biopsy specimen that preferably includes part or full subcutaneous fat with excision of the entire lesion should allow for good histologic interpretation and diagnosis. Keratoacanthoma presents as a downgrowth of well-differentiated squamous epithelium. However, even with a well-performed biopsy, the diagnosis of keratoacanthoma remains challenging due to the lack of sufficient sensitive or specific histological features that can distinguish between keratoacanthomas and SCCs.
As a rule, a normal surface epithelium surrounding the keratin plug with sharp demarcation between tumor and stroma favors keratoacanthoma, whereas ulceration, numerous mitoses, and marked pleomorphism/anaplasia favor SCC. Because of the lack of a universal diagnostic criterion, several experts recommend that all keratoacanthomas be considered potential SCCs and thus treated as such.1,2
When in doubt, cut it out
Although the natural course of a keratoacanthoma is spontaneous regression, the lack of reliable criteria to differentiate it from an SCC with confidence renders therapeutic intervention the safest approach. Solitary keratoacanthomas respond well to surgical excision and may require aggressive procedures if they become too large or invade other structures. Since Mohs’ micrographic surgery is tissue sparing, consider it the treatment of choice if the keratoacanthoma is located in a sensitive area, such as the face.
Cryotherapy with liquid nitrogen, electrodessication and curettage, radiation therapy, and CO2 laser surgery have all been used in small solitary keratoacanthomas with good success.9,10 Other treatment options include intralesional and/or topical treatment using several compounds, such as 5-fluorouracil, corticosteroids, bleomycin, imiquimod, interferon alpha 2b, and methotrexate.7,9-13
Keratoacanthoma patients are often UV light sensitive, so they must avoid excessive sun exposure and use sunscreen with high SPF at all times to prevent recurrence and minimize scarring.
We opted for Mohs’ surgery for our patient
Given the cosmetically sensitive location of our patient’s keratoacanthoma, the size of it, and the patient’s history of skin cancers, we decided to use Mohs’ micrographic surgery for the management of this tumor, with good clinical outcome. There were no new lesions or recurrence on follow-up visit 6 months later.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
1. Beham A, Regauer S, Soyer HP, Beham-Schmid C. Keratoacanthoma: a clinically distinct variant of well differentiated squamous cell carcinoma. Ad Anat Pathol. 1998;5:269-280.
2. Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30(2 Pt 2):326-333; discussion 333.
3. Miot HA, Miot LD, da Costa AL, Matsuo CY, Stolf HO, Marques ME. Association between solitary keratoacanthoma and cigarette smoking: a case-control study. Dermatol Online J. 2006;12(2):2.-
4. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
5. Goldberg LH, Silapunt S, Beyrau KK, Peterson SR, Friedman PM, Alam M. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
6. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198:122-125.
7. Oh CK, Son HS, Lee JB, Jang HS, Kwon KS. Intralesional interferon alfa-2b treatment of keratoacanthomas. J Am Acad Dermatol. 2004;51(5 suppl):S177-S180.
8. Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501.
9. Caccialanza M, Sopelana N. Radiation therapy of keratoacanthomas: results in 55 patients. Int J Radiat Oncol Biol Phys. 1989;16:475-477.
10. Gray RJ, Meland NB. Topical 5-fluorouracil as primary therapy for keratoacanthoma. Ann Plast Surg. 2000;44:82-85.
11. Sanders S, Busam KJ, Halpern AC, Nehal KS. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958.
12. Di Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
13. Cohen PR, Schulze KE, Teller CF, Nelson BR. Intralesional methotrexate for keratoacanthoma of the nose. Skinmed. 2005;4:393-395.
A 63-year-old man came into our dermatologic surgery clinic with a growth on his left cheek just anterior to his sideburn (FIGURE). Our patient indicated that the lesion, which appeared 6 weeks earlier, started as a small, hard papule with a central depression and rapidly grew to reach its current size and shape.
The patient’s face was sun damaged, and he had a prominent 2.1×1.8 cm well-circumscribed, skin-colored tumor on his cheek. The tumor had a central depression covered by a crust that appeared to conceal a deep keratinous plug. The tumor also had a volcano-like shape and was firm in texture, but tender to palpation and pressure. No lymphadenopathy was present.
The patient had a history of extensive sun exposure, and he’d had previous nonmelanoma skin cancers treated with various medical and surgical techniques. The rest of his history and exam were within normal limits. We performed a biopsy to confirm our clinical diagnosis.
FIGURE
Rapidly growing skin-colored tumor
What is your diagnosis?
How would you manage this condition?
Diagnosis: Solitary keratoacanthoma
Our patient had a solitary keratoacanthoma, a unique epidermal tumor that’s characterized by rapid, abundant growth and spontaneous resolution. This tumor goes by many names—molluscum sebaceum, molluscum pseudocarcinomatosum, cutaneous sebaceous neoplasm, and self-healing squamous epithelioma—but keratoacanthoma is the preferred term.1,2
There are several types of keratoacanthoma, but solitary keratoacanthoma remains the most common. It is typically found in light-skinned people in hair-bearing, sun-exposed areas. Peak incidence occurs between the ages of 50 and 69, although the tumors have been reported in patients of all ages. Both sexes are about equally affected, although there is a slight predilection for males. Keratoacanthomas mainly develop on the face (lower lip, cheek, nose, and eyelid), neck, and hands.
The tumors are often considered benign, but they become aggressive in 20% of cases—showing signs of perineural, perivascular, and intravascular invasion and metastases to regional lymph nodes. As a result, some clinicians consider keratoacanthoma to be a pseudomalignancy with self-regressing potential, while others view it as a pseudo-benign tumor progressing into an invasive squamous cell carcinoma (SCC).1,2
The exact etiology of keratoacanthoma is unknown. However, several precipitating factors have been implicated. Primary among them is exposure to ultraviolet light. Other etiological factors include:3-6
- chemical carcinogens (tar, pitch, mineral oil, and cigarette smoking)
- trauma (body peel, carbon dioxide laser resurfacing, megavoltage radiation therapy, and cryosurgery)
- immunosuppression
- surgical scar
- human papilloma virus (HPV).
A tumor with 3 stages
Keratoacanthoma undergoes a proliferative, mature, and involution stage.
In the proliferative stage, there is a rapid increase in tumor size; the tumor can get as big as 10 to 25 mm in diameter in 6 to 8 weeks.1,7
In the mature stage, the tumor stops growing and maintains a typical volcano-like form with a central keratin-filled crater.
In the involution stage, up to 50% of keratoacanthomas undergo spontaneous resolution with expulsion of the keratin plug and resorption of the tumoral mass. The process lasts 4 to 6 weeks, on average, but may take up to 1 year. What’s left behind is a residual atrophic and hypopigmented scar.8
Some lesions persist for a year or more, although the entire process from start to spontaneous resolution usually takes about 4 to 9 months.2,7,8
Is it an SCC or keratoacanthoma?
Histology is the gold standard in diagnosing a keratoacanthoma. A deep biopsy specimen that preferably includes part or full subcutaneous fat with excision of the entire lesion should allow for good histologic interpretation and diagnosis. Keratoacanthoma presents as a downgrowth of well-differentiated squamous epithelium. However, even with a well-performed biopsy, the diagnosis of keratoacanthoma remains challenging due to the lack of sufficient sensitive or specific histological features that can distinguish between keratoacanthomas and SCCs.
As a rule, a normal surface epithelium surrounding the keratin plug with sharp demarcation between tumor and stroma favors keratoacanthoma, whereas ulceration, numerous mitoses, and marked pleomorphism/anaplasia favor SCC. Because of the lack of a universal diagnostic criterion, several experts recommend that all keratoacanthomas be considered potential SCCs and thus treated as such.1,2
When in doubt, cut it out
Although the natural course of a keratoacanthoma is spontaneous regression, the lack of reliable criteria to differentiate it from an SCC with confidence renders therapeutic intervention the safest approach. Solitary keratoacanthomas respond well to surgical excision and may require aggressive procedures if they become too large or invade other structures. Since Mohs’ micrographic surgery is tissue sparing, consider it the treatment of choice if the keratoacanthoma is located in a sensitive area, such as the face.
Cryotherapy with liquid nitrogen, electrodessication and curettage, radiation therapy, and CO2 laser surgery have all been used in small solitary keratoacanthomas with good success.9,10 Other treatment options include intralesional and/or topical treatment using several compounds, such as 5-fluorouracil, corticosteroids, bleomycin, imiquimod, interferon alpha 2b, and methotrexate.7,9-13
Keratoacanthoma patients are often UV light sensitive, so they must avoid excessive sun exposure and use sunscreen with high SPF at all times to prevent recurrence and minimize scarring.
We opted for Mohs’ surgery for our patient
Given the cosmetically sensitive location of our patient’s keratoacanthoma, the size of it, and the patient’s history of skin cancers, we decided to use Mohs’ micrographic surgery for the management of this tumor, with good clinical outcome. There were no new lesions or recurrence on follow-up visit 6 months later.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
A 63-year-old man came into our dermatologic surgery clinic with a growth on his left cheek just anterior to his sideburn (FIGURE). Our patient indicated that the lesion, which appeared 6 weeks earlier, started as a small, hard papule with a central depression and rapidly grew to reach its current size and shape.
The patient’s face was sun damaged, and he had a prominent 2.1×1.8 cm well-circumscribed, skin-colored tumor on his cheek. The tumor had a central depression covered by a crust that appeared to conceal a deep keratinous plug. The tumor also had a volcano-like shape and was firm in texture, but tender to palpation and pressure. No lymphadenopathy was present.
The patient had a history of extensive sun exposure, and he’d had previous nonmelanoma skin cancers treated with various medical and surgical techniques. The rest of his history and exam were within normal limits. We performed a biopsy to confirm our clinical diagnosis.
FIGURE
Rapidly growing skin-colored tumor
What is your diagnosis?
How would you manage this condition?
Diagnosis: Solitary keratoacanthoma
Our patient had a solitary keratoacanthoma, a unique epidermal tumor that’s characterized by rapid, abundant growth and spontaneous resolution. This tumor goes by many names—molluscum sebaceum, molluscum pseudocarcinomatosum, cutaneous sebaceous neoplasm, and self-healing squamous epithelioma—but keratoacanthoma is the preferred term.1,2
There are several types of keratoacanthoma, but solitary keratoacanthoma remains the most common. It is typically found in light-skinned people in hair-bearing, sun-exposed areas. Peak incidence occurs between the ages of 50 and 69, although the tumors have been reported in patients of all ages. Both sexes are about equally affected, although there is a slight predilection for males. Keratoacanthomas mainly develop on the face (lower lip, cheek, nose, and eyelid), neck, and hands.
The tumors are often considered benign, but they become aggressive in 20% of cases—showing signs of perineural, perivascular, and intravascular invasion and metastases to regional lymph nodes. As a result, some clinicians consider keratoacanthoma to be a pseudomalignancy with self-regressing potential, while others view it as a pseudo-benign tumor progressing into an invasive squamous cell carcinoma (SCC).1,2
The exact etiology of keratoacanthoma is unknown. However, several precipitating factors have been implicated. Primary among them is exposure to ultraviolet light. Other etiological factors include:3-6
- chemical carcinogens (tar, pitch, mineral oil, and cigarette smoking)
- trauma (body peel, carbon dioxide laser resurfacing, megavoltage radiation therapy, and cryosurgery)
- immunosuppression
- surgical scar
- human papilloma virus (HPV).
A tumor with 3 stages
Keratoacanthoma undergoes a proliferative, mature, and involution stage.
In the proliferative stage, there is a rapid increase in tumor size; the tumor can get as big as 10 to 25 mm in diameter in 6 to 8 weeks.1,7
In the mature stage, the tumor stops growing and maintains a typical volcano-like form with a central keratin-filled crater.
In the involution stage, up to 50% of keratoacanthomas undergo spontaneous resolution with expulsion of the keratin plug and resorption of the tumoral mass. The process lasts 4 to 6 weeks, on average, but may take up to 1 year. What’s left behind is a residual atrophic and hypopigmented scar.8
Some lesions persist for a year or more, although the entire process from start to spontaneous resolution usually takes about 4 to 9 months.2,7,8
Is it an SCC or keratoacanthoma?
Histology is the gold standard in diagnosing a keratoacanthoma. A deep biopsy specimen that preferably includes part or full subcutaneous fat with excision of the entire lesion should allow for good histologic interpretation and diagnosis. Keratoacanthoma presents as a downgrowth of well-differentiated squamous epithelium. However, even with a well-performed biopsy, the diagnosis of keratoacanthoma remains challenging due to the lack of sufficient sensitive or specific histological features that can distinguish between keratoacanthomas and SCCs.
As a rule, a normal surface epithelium surrounding the keratin plug with sharp demarcation between tumor and stroma favors keratoacanthoma, whereas ulceration, numerous mitoses, and marked pleomorphism/anaplasia favor SCC. Because of the lack of a universal diagnostic criterion, several experts recommend that all keratoacanthomas be considered potential SCCs and thus treated as such.1,2
When in doubt, cut it out
Although the natural course of a keratoacanthoma is spontaneous regression, the lack of reliable criteria to differentiate it from an SCC with confidence renders therapeutic intervention the safest approach. Solitary keratoacanthomas respond well to surgical excision and may require aggressive procedures if they become too large or invade other structures. Since Mohs’ micrographic surgery is tissue sparing, consider it the treatment of choice if the keratoacanthoma is located in a sensitive area, such as the face.
Cryotherapy with liquid nitrogen, electrodessication and curettage, radiation therapy, and CO2 laser surgery have all been used in small solitary keratoacanthomas with good success.9,10 Other treatment options include intralesional and/or topical treatment using several compounds, such as 5-fluorouracil, corticosteroids, bleomycin, imiquimod, interferon alpha 2b, and methotrexate.7,9-13
Keratoacanthoma patients are often UV light sensitive, so they must avoid excessive sun exposure and use sunscreen with high SPF at all times to prevent recurrence and minimize scarring.
We opted for Mohs’ surgery for our patient
Given the cosmetically sensitive location of our patient’s keratoacanthoma, the size of it, and the patient’s history of skin cancers, we decided to use Mohs’ micrographic surgery for the management of this tumor, with good clinical outcome. There were no new lesions or recurrence on follow-up visit 6 months later.
Correspondence
Amor Khachemoune, MD, CWS, Assistant Professor, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
1. Beham A, Regauer S, Soyer HP, Beham-Schmid C. Keratoacanthoma: a clinically distinct variant of well differentiated squamous cell carcinoma. Ad Anat Pathol. 1998;5:269-280.
2. Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30(2 Pt 2):326-333; discussion 333.
3. Miot HA, Miot LD, da Costa AL, Matsuo CY, Stolf HO, Marques ME. Association between solitary keratoacanthoma and cigarette smoking: a case-control study. Dermatol Online J. 2006;12(2):2.-
4. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
5. Goldberg LH, Silapunt S, Beyrau KK, Peterson SR, Friedman PM, Alam M. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
6. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198:122-125.
7. Oh CK, Son HS, Lee JB, Jang HS, Kwon KS. Intralesional interferon alfa-2b treatment of keratoacanthomas. J Am Acad Dermatol. 2004;51(5 suppl):S177-S180.
8. Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501.
9. Caccialanza M, Sopelana N. Radiation therapy of keratoacanthomas: results in 55 patients. Int J Radiat Oncol Biol Phys. 1989;16:475-477.
10. Gray RJ, Meland NB. Topical 5-fluorouracil as primary therapy for keratoacanthoma. Ann Plast Surg. 2000;44:82-85.
11. Sanders S, Busam KJ, Halpern AC, Nehal KS. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958.
12. Di Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
13. Cohen PR, Schulze KE, Teller CF, Nelson BR. Intralesional methotrexate for keratoacanthoma of the nose. Skinmed. 2005;4:393-395.
1. Beham A, Regauer S, Soyer HP, Beham-Schmid C. Keratoacanthoma: a clinically distinct variant of well differentiated squamous cell carcinoma. Ad Anat Pathol. 1998;5:269-280.
2. Schwartz RA. Keratoacanthoma: a clinico-pathologic enigma. Dermatol Surg. 2004;30(2 Pt 2):326-333; discussion 333.
3. Miot HA, Miot LD, da Costa AL, Matsuo CY, Stolf HO, Marques ME. Association between solitary keratoacanthoma and cigarette smoking: a case-control study. Dermatol Online J. 2006;12(2):2.-
4. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
5. Goldberg LH, Silapunt S, Beyrau KK, Peterson SR, Friedman PM, Alam M. Keratoacanthoma as a postoperative complication of skin cancer excision. J Am Acad Dermatol. 2004;50:753-758.
6. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198:122-125.
7. Oh CK, Son HS, Lee JB, Jang HS, Kwon KS. Intralesional interferon alfa-2b treatment of keratoacanthomas. J Am Acad Dermatol. 2004;51(5 suppl):S177-S180.
8. Griffiths RW. Keratoacanthoma observed. Br J Plast Surg. 2004;57:485-501.
9. Caccialanza M, Sopelana N. Radiation therapy of keratoacanthomas: results in 55 patients. Int J Radiat Oncol Biol Phys. 1989;16:475-477.
10. Gray RJ, Meland NB. Topical 5-fluorouracil as primary therapy for keratoacanthoma. Ann Plast Surg. 2000;44:82-85.
11. Sanders S, Busam KJ, Halpern AC, Nehal KS. Intralesional corticosteroid treatment of multiple eruptive keratoacanthomas: case report and review of a controversial therapy. Dermatol Surg. 2002;28:954-958.
12. Di Lernia V, Ricci C, Albertini G. Spontaneous regression of keratoacanthoma can be promoted by topical treatment with imiquimod cream. J Eur Acad Dermatol Venereol. 2004;18:626-629.
13. Cohen PR, Schulze KE, Teller CF, Nelson BR. Intralesional methotrexate for keratoacanthoma of the nose. Skinmed. 2005;4:393-395.
Dusky plaque on the knee
A progressively growing lesion on the left knee prompted a 35-year-old woman to visit our clinic. She reported that about 3 months earlier, she had developed a small ulceration on the knee following a fall. With local wound care, the ulceration healed with a scar. The scar, however, continued to grow and she developed distinct papules outside the original scar.
FIGURE
Scar develops into a dusky plaque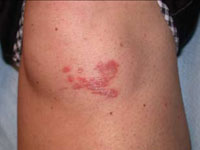
The scar subsequently became raised with violaceous discoloration. The patient reported having no history of excessive scarring or keloid forming after skin surgery or trauma. There was neither a personal nor family history of inflammatory or infectious granulomatous diseases.
On physical examination, there was an erythematous to dusky plaque with well-defined irregular borders. There were also discrete papules on the anterior and medial aspect of the knee. The plaque measured approximately 2.8 cm by 3.8 cm. There were no tender nodules on the shins, nor was lymphadenopathy present. A routine chest x-ray was normal.
To support our clinical diagnosis, we took a 4-mm punch biopsy from the center of the plaque. The histologic examination revealed changes in the dermis termed noncaseating “naked” granulomas.
What is your diagnosis?
How would you manage this condition?
Diagnosis: Scar sarcoidosis
Sarcoidosis is a systemic granulomatous disease that may affect any organ system, and therefore may present with various clinical manifestations.1 Sarcoidosis can be an incidental finding on chest x-ray or be discovered in patients that present with respiratory or constitutional symptoms.2
Cutaneous sarcoidosis occurs in up to one third of patients with systemic sarcoidosis.2 The classic skin lesions are “erythema nodosum”—an acute, nodular, erythematous eruption that usually is limited to the shins, and “lupus pernio”—red-to-purple or violaceous indurated nodules affecting the nose, cheeks, ears, and lips. There are other uncommon skin presentations of sarcoidosis ranging from scattered papules and annular lesions to erythrodermic skin manifestations.3
In scar sarcoidosis, there is spontaneous development of livid or reddish-brown plaques on scars that were previously atrophic for the most part. Scar sarcoidosis may be caused by:4-6
- venipuncture
- tuberculin skin tests
- herpes zoster
- tattoos
- cosmetic fillers such as hyaluronic acid injection.
Infection and other factors may be at work
Although the precise cause of sarcoidosis remains unknown, various infectious, noninfectious, environmental, and genetic factors may be at work. Researchers have theorized that immune dysregulation may be involved. Contact with a persistent antigen that is poorly cleared by the immune system may lead to T lymphocytes and mononuclear phagocytes accumulating in the granulomas of sarcoidosis.7 Researchers have proposed that inoculation of foreign matter from minor trauma may be one type of pathogenic mechanism in cutaneous sarcoid.8
Granuloma annulare is part of the differential
A wide range of diseases comprise the differential diagnosis of sarcoidosis. These diseases include:9
- Granuloma annulare. It is also a granulomatous skin disease, but it appears as single or multiple rings.
- Rheumatoid nodules. These usually appear in the context of a diagnosis of rheumatoid arthritis.
- Granulomatous mycosis fungoides. This type of cutaneous lymphoma has many clinical forms, including granuloma formation.
- Syringoma. On inspection, you’ll see small, firm adnexal benign tumors that usually appear around the upper cheeks and lower eyelids
- Xanthelasma. These are benign, yellow macules, papules, or plaques that tend to appear on the eyelids. Patients with xanthelasma often have a lipid disorder.
- Lichen planus. This is a very pruritic skin involvement with pink to violaceous papules and plaques. It may present in different locations, but the most common areas are the wrists and ankles.
- Granulomatous rosacea. This is a variant of rosacea characterized by uniform papules on the face.
Clinical findings, biopsy clinch the diagnosis
The diagnosis of sarcoidosis is made by a combination of clinical and histologic findings.1
- Clinical findings. Cutaneous involvement is either “specific” or “nonspecific.”
- With specific cutaneous involvement, which our patient had, you’ll see typical noncaseating granulomas, with no evidence of infection or a foreign body. It may be disfiguring, but it’s almost always nontender and it is rarely ulcerative.
- With nonspecific cutaneous involvement, you’ll see erythema nodosum lesions, especially on the legs. The serum angiotensin-converting enzyme (ACE) level is elevated in many of these patients.
Histologic findings. Skin biopsy demonstrating noncaseating granulomas provides definitive evidence of skin involvement. Typical sarcoid lesions are characterized by circumscribed granulomas of epithelioid cells with little or no necrosis. (The term “naked” granuloma refers to the absence, or small number, of surrounding lymphocytes.)
Other granulomatous diseases, such as berylliosis and tuberculosis, must be excluded since they often present the same way as scar sarcoidosis.7
Steroids control symptoms, slow disease progression
Topical, intralesional, and systemic corticosteroids are used to treat scar sarcoidosis, as are systemic medications such as chloroquine10 and allopurinol.11 Corticosteroids (local and systemic) are effective in controlling all sarcoid symptoms; they also slow disease progression.1
For localized skin involvement, intralesional corticosteroids are typically more effective than topical steroids. Systemic corticosteroids are reserved for widespread, progressive lesions or those that impair function.1,12,13 A starting dose of 1 mg/kg of prednisone is appropriate.
In general, the prognosis of cutaneous sarcoidosis is good.2 The course is variable, ranging from self-limited acute episodes to a chronic debilitating disease that may result in death.2 Spontaneous remissions occur in nearly two thirds of patients, but 10% to 30% have a more chronic or progressive course.1,2,13
Our patient responds to treatment
Our patient declined intralesional corticosteroid injections, so we started her on potent topical corticosteroid tapes (Cordran). She had significant improvement 6 weeks later.
Correspondence
Amor Khachemoune, MD, CWS, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203; [email protected]
1. Howard A, White CR, Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo, JL, Rapini RP, eds. Dermatology. London: Mosby; 2003:1455-1460.
2. Giuffrida TJ, Kerdel FA. Sarcoidosis. Dermatol Clin 2002;20:435-447.
3. Okamoto H. Cutaneous sarcoidosis [in Japanese]. Nippon Rinsho 2002;60:1801-1806.
4. Barrazza V. Post-herpes zoster scar sarcoidosis. Acta Derm Venereol 1999;79:495.-
5. Dal Sacco D, Cozzani E, Parodi A, Rebora A. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol 2005;44:411-412.
6. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005;141:869-872.
7. Gal AA, Koss MN. The pathology of sarcoidosis. Curr Opin Pulm Med 2002;8:445-451.
8. Marcoval J, Mañà J, Moreno A, Gallego I, Fortuño Y, Peyrí J. Foreign bodies in granulomatous cutaneous lesions of patients with systemic sarcoidosis. Arch Dermatol 2001;137:427-430.
9. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician 2002;65:1581-1584.
10. Wallace DJ. The use of chloroquine and hydroxychloroquine for non-infectious conditions other than rheumatoid arthritis or lupus: a critical review. Lupus 1996;5 suppl 1:s59-64.
11. Bregnhoej A, Jemec GB. Low-dose allopurinol in the treatment of cutaneous sarcoidosis: response in four of seven patients. J Dermatolog Treat 2005;16:125-127.
12. Wu JJ, Schiff KR. Sarcoidosis. Am Fam Physician 2004;70:312-322.
13. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease? J Am Acad Dermatol 2006;54:55-60.
A progressively growing lesion on the left knee prompted a 35-year-old woman to visit our clinic. She reported that about 3 months earlier, she had developed a small ulceration on the knee following a fall. With local wound care, the ulceration healed with a scar. The scar, however, continued to grow and she developed distinct papules outside the original scar.
FIGURE
Scar develops into a dusky plaque
The scar subsequently became raised with violaceous discoloration. The patient reported having no history of excessive scarring or keloid forming after skin surgery or trauma. There was neither a personal nor family history of inflammatory or infectious granulomatous diseases.
On physical examination, there was an erythematous to dusky plaque with well-defined irregular borders. There were also discrete papules on the anterior and medial aspect of the knee. The plaque measured approximately 2.8 cm by 3.8 cm. There were no tender nodules on the shins, nor was lymphadenopathy present. A routine chest x-ray was normal.
To support our clinical diagnosis, we took a 4-mm punch biopsy from the center of the plaque. The histologic examination revealed changes in the dermis termed noncaseating “naked” granulomas.
What is your diagnosis?
How would you manage this condition?
Diagnosis: Scar sarcoidosis
Sarcoidosis is a systemic granulomatous disease that may affect any organ system, and therefore may present with various clinical manifestations.1 Sarcoidosis can be an incidental finding on chest x-ray or be discovered in patients that present with respiratory or constitutional symptoms.2
Cutaneous sarcoidosis occurs in up to one third of patients with systemic sarcoidosis.2 The classic skin lesions are “erythema nodosum”—an acute, nodular, erythematous eruption that usually is limited to the shins, and “lupus pernio”—red-to-purple or violaceous indurated nodules affecting the nose, cheeks, ears, and lips. There are other uncommon skin presentations of sarcoidosis ranging from scattered papules and annular lesions to erythrodermic skin manifestations.3
In scar sarcoidosis, there is spontaneous development of livid or reddish-brown plaques on scars that were previously atrophic for the most part. Scar sarcoidosis may be caused by:4-6
- venipuncture
- tuberculin skin tests
- herpes zoster
- tattoos
- cosmetic fillers such as hyaluronic acid injection.
Infection and other factors may be at work
Although the precise cause of sarcoidosis remains unknown, various infectious, noninfectious, environmental, and genetic factors may be at work. Researchers have theorized that immune dysregulation may be involved. Contact with a persistent antigen that is poorly cleared by the immune system may lead to T lymphocytes and mononuclear phagocytes accumulating in the granulomas of sarcoidosis.7 Researchers have proposed that inoculation of foreign matter from minor trauma may be one type of pathogenic mechanism in cutaneous sarcoid.8
Granuloma annulare is part of the differential
A wide range of diseases comprise the differential diagnosis of sarcoidosis. These diseases include:9
- Granuloma annulare. It is also a granulomatous skin disease, but it appears as single or multiple rings.
- Rheumatoid nodules. These usually appear in the context of a diagnosis of rheumatoid arthritis.
- Granulomatous mycosis fungoides. This type of cutaneous lymphoma has many clinical forms, including granuloma formation.
- Syringoma. On inspection, you’ll see small, firm adnexal benign tumors that usually appear around the upper cheeks and lower eyelids
- Xanthelasma. These are benign, yellow macules, papules, or plaques that tend to appear on the eyelids. Patients with xanthelasma often have a lipid disorder.
- Lichen planus. This is a very pruritic skin involvement with pink to violaceous papules and plaques. It may present in different locations, but the most common areas are the wrists and ankles.
- Granulomatous rosacea. This is a variant of rosacea characterized by uniform papules on the face.
Clinical findings, biopsy clinch the diagnosis
The diagnosis of sarcoidosis is made by a combination of clinical and histologic findings.1
- Clinical findings. Cutaneous involvement is either “specific” or “nonspecific.”
- With specific cutaneous involvement, which our patient had, you’ll see typical noncaseating granulomas, with no evidence of infection or a foreign body. It may be disfiguring, but it’s almost always nontender and it is rarely ulcerative.
- With nonspecific cutaneous involvement, you’ll see erythema nodosum lesions, especially on the legs. The serum angiotensin-converting enzyme (ACE) level is elevated in many of these patients.
Histologic findings. Skin biopsy demonstrating noncaseating granulomas provides definitive evidence of skin involvement. Typical sarcoid lesions are characterized by circumscribed granulomas of epithelioid cells with little or no necrosis. (The term “naked” granuloma refers to the absence, or small number, of surrounding lymphocytes.)
Other granulomatous diseases, such as berylliosis and tuberculosis, must be excluded since they often present the same way as scar sarcoidosis.7
Steroids control symptoms, slow disease progression
Topical, intralesional, and systemic corticosteroids are used to treat scar sarcoidosis, as are systemic medications such as chloroquine10 and allopurinol.11 Corticosteroids (local and systemic) are effective in controlling all sarcoid symptoms; they also slow disease progression.1
For localized skin involvement, intralesional corticosteroids are typically more effective than topical steroids. Systemic corticosteroids are reserved for widespread, progressive lesions or those that impair function.1,12,13 A starting dose of 1 mg/kg of prednisone is appropriate.
In general, the prognosis of cutaneous sarcoidosis is good.2 The course is variable, ranging from self-limited acute episodes to a chronic debilitating disease that may result in death.2 Spontaneous remissions occur in nearly two thirds of patients, but 10% to 30% have a more chronic or progressive course.1,2,13
Our patient responds to treatment
Our patient declined intralesional corticosteroid injections, so we started her on potent topical corticosteroid tapes (Cordran). She had significant improvement 6 weeks later.
Correspondence
Amor Khachemoune, MD, CWS, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203; [email protected]
A progressively growing lesion on the left knee prompted a 35-year-old woman to visit our clinic. She reported that about 3 months earlier, she had developed a small ulceration on the knee following a fall. With local wound care, the ulceration healed with a scar. The scar, however, continued to grow and she developed distinct papules outside the original scar.
FIGURE
Scar develops into a dusky plaque
The scar subsequently became raised with violaceous discoloration. The patient reported having no history of excessive scarring or keloid forming after skin surgery or trauma. There was neither a personal nor family history of inflammatory or infectious granulomatous diseases.
On physical examination, there was an erythematous to dusky plaque with well-defined irregular borders. There were also discrete papules on the anterior and medial aspect of the knee. The plaque measured approximately 2.8 cm by 3.8 cm. There were no tender nodules on the shins, nor was lymphadenopathy present. A routine chest x-ray was normal.
To support our clinical diagnosis, we took a 4-mm punch biopsy from the center of the plaque. The histologic examination revealed changes in the dermis termed noncaseating “naked” granulomas.
What is your diagnosis?
How would you manage this condition?
Diagnosis: Scar sarcoidosis
Sarcoidosis is a systemic granulomatous disease that may affect any organ system, and therefore may present with various clinical manifestations.1 Sarcoidosis can be an incidental finding on chest x-ray or be discovered in patients that present with respiratory or constitutional symptoms.2
Cutaneous sarcoidosis occurs in up to one third of patients with systemic sarcoidosis.2 The classic skin lesions are “erythema nodosum”—an acute, nodular, erythematous eruption that usually is limited to the shins, and “lupus pernio”—red-to-purple or violaceous indurated nodules affecting the nose, cheeks, ears, and lips. There are other uncommon skin presentations of sarcoidosis ranging from scattered papules and annular lesions to erythrodermic skin manifestations.3
In scar sarcoidosis, there is spontaneous development of livid or reddish-brown plaques on scars that were previously atrophic for the most part. Scar sarcoidosis may be caused by:4-6
- venipuncture
- tuberculin skin tests
- herpes zoster
- tattoos
- cosmetic fillers such as hyaluronic acid injection.
Infection and other factors may be at work
Although the precise cause of sarcoidosis remains unknown, various infectious, noninfectious, environmental, and genetic factors may be at work. Researchers have theorized that immune dysregulation may be involved. Contact with a persistent antigen that is poorly cleared by the immune system may lead to T lymphocytes and mononuclear phagocytes accumulating in the granulomas of sarcoidosis.7 Researchers have proposed that inoculation of foreign matter from minor trauma may be one type of pathogenic mechanism in cutaneous sarcoid.8
Granuloma annulare is part of the differential
A wide range of diseases comprise the differential diagnosis of sarcoidosis. These diseases include:9
- Granuloma annulare. It is also a granulomatous skin disease, but it appears as single or multiple rings.
- Rheumatoid nodules. These usually appear in the context of a diagnosis of rheumatoid arthritis.
- Granulomatous mycosis fungoides. This type of cutaneous lymphoma has many clinical forms, including granuloma formation.
- Syringoma. On inspection, you’ll see small, firm adnexal benign tumors that usually appear around the upper cheeks and lower eyelids
- Xanthelasma. These are benign, yellow macules, papules, or plaques that tend to appear on the eyelids. Patients with xanthelasma often have a lipid disorder.
- Lichen planus. This is a very pruritic skin involvement with pink to violaceous papules and plaques. It may present in different locations, but the most common areas are the wrists and ankles.
- Granulomatous rosacea. This is a variant of rosacea characterized by uniform papules on the face.
Clinical findings, biopsy clinch the diagnosis
The diagnosis of sarcoidosis is made by a combination of clinical and histologic findings.1
- Clinical findings. Cutaneous involvement is either “specific” or “nonspecific.”
- With specific cutaneous involvement, which our patient had, you’ll see typical noncaseating granulomas, with no evidence of infection or a foreign body. It may be disfiguring, but it’s almost always nontender and it is rarely ulcerative.
- With nonspecific cutaneous involvement, you’ll see erythema nodosum lesions, especially on the legs. The serum angiotensin-converting enzyme (ACE) level is elevated in many of these patients.
Histologic findings. Skin biopsy demonstrating noncaseating granulomas provides definitive evidence of skin involvement. Typical sarcoid lesions are characterized by circumscribed granulomas of epithelioid cells with little or no necrosis. (The term “naked” granuloma refers to the absence, or small number, of surrounding lymphocytes.)
Other granulomatous diseases, such as berylliosis and tuberculosis, must be excluded since they often present the same way as scar sarcoidosis.7
Steroids control symptoms, slow disease progression
Topical, intralesional, and systemic corticosteroids are used to treat scar sarcoidosis, as are systemic medications such as chloroquine10 and allopurinol.11 Corticosteroids (local and systemic) are effective in controlling all sarcoid symptoms; they also slow disease progression.1
For localized skin involvement, intralesional corticosteroids are typically more effective than topical steroids. Systemic corticosteroids are reserved for widespread, progressive lesions or those that impair function.1,12,13 A starting dose of 1 mg/kg of prednisone is appropriate.
In general, the prognosis of cutaneous sarcoidosis is good.2 The course is variable, ranging from self-limited acute episodes to a chronic debilitating disease that may result in death.2 Spontaneous remissions occur in nearly two thirds of patients, but 10% to 30% have a more chronic or progressive course.1,2,13
Our patient responds to treatment
Our patient declined intralesional corticosteroid injections, so we started her on potent topical corticosteroid tapes (Cordran). She had significant improvement 6 weeks later.
Correspondence
Amor Khachemoune, MD, CWS, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203; [email protected]
1. Howard A, White CR, Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo, JL, Rapini RP, eds. Dermatology. London: Mosby; 2003:1455-1460.
2. Giuffrida TJ, Kerdel FA. Sarcoidosis. Dermatol Clin 2002;20:435-447.
3. Okamoto H. Cutaneous sarcoidosis [in Japanese]. Nippon Rinsho 2002;60:1801-1806.
4. Barrazza V. Post-herpes zoster scar sarcoidosis. Acta Derm Venereol 1999;79:495.-
5. Dal Sacco D, Cozzani E, Parodi A, Rebora A. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol 2005;44:411-412.
6. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005;141:869-872.
7. Gal AA, Koss MN. The pathology of sarcoidosis. Curr Opin Pulm Med 2002;8:445-451.
8. Marcoval J, Mañà J, Moreno A, Gallego I, Fortuño Y, Peyrí J. Foreign bodies in granulomatous cutaneous lesions of patients with systemic sarcoidosis. Arch Dermatol 2001;137:427-430.
9. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician 2002;65:1581-1584.
10. Wallace DJ. The use of chloroquine and hydroxychloroquine for non-infectious conditions other than rheumatoid arthritis or lupus: a critical review. Lupus 1996;5 suppl 1:s59-64.
11. Bregnhoej A, Jemec GB. Low-dose allopurinol in the treatment of cutaneous sarcoidosis: response in four of seven patients. J Dermatolog Treat 2005;16:125-127.
12. Wu JJ, Schiff KR. Sarcoidosis. Am Fam Physician 2004;70:312-322.
13. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease? J Am Acad Dermatol 2006;54:55-60.
1. Howard A, White CR, Jr. Non-infectious granulomas. In: Bolognia JL, Jorizzo, JL, Rapini RP, eds. Dermatology. London: Mosby; 2003:1455-1460.
2. Giuffrida TJ, Kerdel FA. Sarcoidosis. Dermatol Clin 2002;20:435-447.
3. Okamoto H. Cutaneous sarcoidosis [in Japanese]. Nippon Rinsho 2002;60:1801-1806.
4. Barrazza V. Post-herpes zoster scar sarcoidosis. Acta Derm Venereol 1999;79:495.-
5. Dal Sacco D, Cozzani E, Parodi A, Rebora A. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol 2005;44:411-412.
6. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005;141:869-872.
7. Gal AA, Koss MN. The pathology of sarcoidosis. Curr Opin Pulm Med 2002;8:445-451.
8. Marcoval J, Mañà J, Moreno A, Gallego I, Fortuño Y, Peyrí J. Foreign bodies in granulomatous cutaneous lesions of patients with systemic sarcoidosis. Arch Dermatol 2001;137:427-430.
9. Katta R. Cutaneous sarcoidosis: a dermatologic masquerader. Am Fam Physician 2002;65:1581-1584.
10. Wallace DJ. The use of chloroquine and hydroxychloroquine for non-infectious conditions other than rheumatoid arthritis or lupus: a critical review. Lupus 1996;5 suppl 1:s59-64.
11. Bregnhoej A, Jemec GB. Low-dose allopurinol in the treatment of cutaneous sarcoidosis: response in four of seven patients. J Dermatolog Treat 2005;16:125-127.
12. Wu JJ, Schiff KR. Sarcoidosis. Am Fam Physician 2004;70:312-322.
13. Ahmed I, Harshad SR. Subcutaneous sarcoidosis: is it a specific subset of cutaneous sarcoidosis frequently associated with systemic disease? J Am Acad Dermatol 2006;54:55-60.
A young girl with blisters on her forehead
A 5-year-old girl came into the office complaining of severe burning and tingling sensation of her right forehead. She’d had fever, chills, myalgia, and a relentless headache for 3 days. The morning of her appointment, a few “bumps” and water-filled blisters began to appear on the right side of the forehead; the lesions then started to multiply and grow (FIGURE). The patient’s mother expressed concern over the rapid development of the lesions, which were accompanied by marked edema of the forehead and right eyelid. The mother indicated that no one else in the family was affected at the time of presentation.
The child appeared ill and pale at the time of presentation, but there was no history of immediate antecedent illness or any drug intake prior to the current eruption. Her growth and development were in the lower normal range. The child had a history of recurrent bacterial infections; she was not vaccinated for varicella. There was, however, a personal (and family) history of varicella when the child was about 3 years old.
FIGURE
Vesicles and bullae on forehead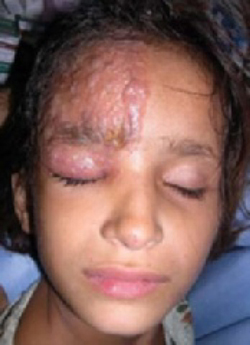
On physical examination, there were multiple vesicles and bullae that varied in size from 2 mm to 1 cm in diameter on the right side of the forehead; there were areas of dried yellowish serous exudates limited to the right eyebrow. The forehead and periorbital areas were edematous with underlying erythematous skin. The entire eruption appeared to be restricted to the right upper part of the face, extending from the eyelid and medial canthus up to the frontal scalp. On close examination, several vesicles and crusts were present on the right side of the nasal tip. Serology for HIV was negative.
What is your diagnosis?
How would you treat?
Diagnosis: Herpes zoster ophthalmicus
This case of herpes zoster ophthalmicus (HZO) was unusual—not because of the way the patient presented, but because of her age. This condition is rarely seen in children, though it is not uncommon in adults—specifically, patients who are 60 years of age and older.1-4
Herpes zoster ophthalmicus, which refers to the involvement of the ophthalmic branch of the trigeminal nerve or the fifth cranial nerve, usually manifests with a typical vesicular or bullous eruption of the forehead, often in unilateral fashion. The lesions of herpes zoster (shingles) are similar to those of varicella (chickenpox) but in herpes zoster, painful unilateral vesicular eruptions are usually limited to one dermatome. (If hematogenous dissemination occurs, more than 20 vesicles will form in skin areas away from the affected dermatome.1)
Patients typically present with fever, headache, and abnormal sensations that precede the development of cutaneous lesions by a few days. The eruptions may be pustular and hemorrhagic initially, and within 10 days evolve into crusts.5,6
Differential diagnosis: Pain sets HZO apart
In its classical presentation, HZO does not pose a diagnostic challenge for practitioners well-versed in skin disease management. The clinical differential diagnosis of herpes zoster will include other causes of blisters or vesicular eruptions of autoimmune etiologies, viral infections, or hypersensitivity reactions.
The most common blistering diseases that may be mistaken for zoster include herpes simplex, contact dermatitis, erythema multiforme, and cellulitis. The prodromal stage and the characteristics of pain usually set herpes zoster apart from the other diagnoses.
Beware of complications
Eye complications. HZO may result in paralytic ptosis, conjunctivitis, keratitis, cataracts, glaucoma, retinitis, and optic neuritis and atrophy.2,5,7
Herpetic lesions on the tip of the nose are believed to herald ocular involvement and may precede typical dermatomal eruption of HZO.2 Such cutaneous involvement along the distribution of the nasociliary nerve is called Hutchinson’s sign. It should prompt a complete ophthalmologic evaluation, as it did with our patient.
Neurologic complications. The most common complication of herpes zoster infection in general is postherpetic neuralgia (PHN), which results in severe pain that persists for months—or years—after the skin lesions have completely healed. Patients over 70 have a 70% chance of developing PHN.4,7
Neurological complications such as encephalitis, myelitis, and Guillain-Barré syndrome have also been reported.
Other complications. Other complications include secondary bacterial infections, pneumonitis, and polyradiculitis.
Management: Antivirals and pain meds
Patients with HZO have a 50% chance of having eye complications (iritis and keratitis) without antiviral treatment.2,5,6 Therefore, treatment is recommended for all HZO patients.
Antivirals ASAP
Start antiviral drugs within 72 hours of clinical presentation, and when new lesions are still appearing on the skin, to achieve optimal effect. Acyclovir, famciclovir, and valacyclovir are the antivirals of choice.6 The usual dosages for adults are: acyclovir (800 mg orally 5 times per day for 7–10 days), valacyclovir (1000 mg orally 3 times daily for 7 days) and famciclovir (500 mg orally 3 times daily for 7 days). The suggested dose of acyclovir for children is 10 to 20 mg/kg/dose qid for 5 days; not to exceed 800 mg per day.2,5,6
Preventing postherpetic neuralgia
The role of systemic corticosteroids in the prevention of PHN, decreasing duration and severity of the acute symptoms in the initial days of herpes zoster infection, remains controversial.8
Immunosuppressed individuals may be treated with acyclovir, interferon-alpha, and vidarabine. In this population the live, attenuated vaccine is safer and is preferred to the varicella zoster immunoglobulins (VZIG).1,6,8,9
PHN can be reduced by treating the patient within the first 24 hours of symptom onset. Pain usually resolves within 3 months in 50% of patients and within 1 year in 75% of patients.1,6,7
Pain therapies for PHN
Therapeutic approaches to the management of PHN include topical anesthetic creams (lidocaine plus prilocaine), capsaicin, and oral medications such as tricyclic antidepressants (amitriptyline and desipramine), carbamazepine, and gabapentin, as well as nerve blocks.6,7
Gloves and handwashing are key for caregivers
Individuals who have not had a confirmed varicella infection should avoid contact with those who have shingles unless a varicella zoster virus antibody is satisfactory and shows immunity.6,7
Gloves should be worn when touching the lesions or infectious drainage, and hands should be washed after glove removal.5-9
Patients with disseminated disease require hospitalization with airborne precautions, which include a negative air pressure room and ensuring that caregivers wear N95 respirators.
What put our patient at risk?
In the case of our young patient, low immunity may have played a role in her contracting HZO. She had a history of recurrent bacterial infections and suffered from generally poor health.
She was referred to an ophthalmologist, who did not find any dendritic corneal ulcer or other complications on weekly follow up. The patient was successfully managed with oral acyclovir 10 mg/kg/dose for 5 days, and she responded well to treatment. We also prescribed acyclovir eye ointment, and local wet compresses with good outcome.
Acknowledgments
The authors thank Dr Shahbaz Janjua for his assistance in the preparation of this manuscript.
Correspondence
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
1. Trent JT, Kirsner RS. Herpesvirus infections and herpetic wounds. Adv Skin Wound Care 2003;16:236-243.
2. Zaal MJ, Volker-Dieben HJ, D’Amaro J. Prognostic value of Hutchinson’s sign in acute herpes zoster ophthalmicus. Graefes Arch Clin Exp Ophthalmol 2003;241:187-191.
3. Binder NR, Holland GN, Hosea S, Silverberg ML. Herpes zoster ophthalmicus in an otherwisehealthy child. JAAPOS 2005;9:597-598.
4. Gebo KA, Kalyani R, Moore RD, Polydefkis MJ. The incidence of, risk factors for, and sequelae of herpes zoster among HIV patients in the highly active antiretroviral therapy era. JAIDS 2005;40:169-174.
5. Liesegang TJ. Herpes zoster virus infection. Curr Opin Ophthalmol 2004;15:531-536.
6. McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol 1999;41:1-14.
7. Cohen JI, Brunell PA, Straus SE, Krause PR. Recent advances in varicella-zoster virus infection. Ann Intern Med 1999;130:922-932.
8. Santee JA. Corticosteroids for herpes zoster: what do they accomplish? Am J Clin Dermatol 2002;3:517-524.
9. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005;352:2271-2284.
A 5-year-old girl came into the office complaining of severe burning and tingling sensation of her right forehead. She’d had fever, chills, myalgia, and a relentless headache for 3 days. The morning of her appointment, a few “bumps” and water-filled blisters began to appear on the right side of the forehead; the lesions then started to multiply and grow (FIGURE). The patient’s mother expressed concern over the rapid development of the lesions, which were accompanied by marked edema of the forehead and right eyelid. The mother indicated that no one else in the family was affected at the time of presentation.
The child appeared ill and pale at the time of presentation, but there was no history of immediate antecedent illness or any drug intake prior to the current eruption. Her growth and development were in the lower normal range. The child had a history of recurrent bacterial infections; she was not vaccinated for varicella. There was, however, a personal (and family) history of varicella when the child was about 3 years old.
FIGURE
Vesicles and bullae on forehead
On physical examination, there were multiple vesicles and bullae that varied in size from 2 mm to 1 cm in diameter on the right side of the forehead; there were areas of dried yellowish serous exudates limited to the right eyebrow. The forehead and periorbital areas were edematous with underlying erythematous skin. The entire eruption appeared to be restricted to the right upper part of the face, extending from the eyelid and medial canthus up to the frontal scalp. On close examination, several vesicles and crusts were present on the right side of the nasal tip. Serology for HIV was negative.
What is your diagnosis?
How would you treat?
Diagnosis: Herpes zoster ophthalmicus
This case of herpes zoster ophthalmicus (HZO) was unusual—not because of the way the patient presented, but because of her age. This condition is rarely seen in children, though it is not uncommon in adults—specifically, patients who are 60 years of age and older.1-4
Herpes zoster ophthalmicus, which refers to the involvement of the ophthalmic branch of the trigeminal nerve or the fifth cranial nerve, usually manifests with a typical vesicular or bullous eruption of the forehead, often in unilateral fashion. The lesions of herpes zoster (shingles) are similar to those of varicella (chickenpox) but in herpes zoster, painful unilateral vesicular eruptions are usually limited to one dermatome. (If hematogenous dissemination occurs, more than 20 vesicles will form in skin areas away from the affected dermatome.1)
Patients typically present with fever, headache, and abnormal sensations that precede the development of cutaneous lesions by a few days. The eruptions may be pustular and hemorrhagic initially, and within 10 days evolve into crusts.5,6
Differential diagnosis: Pain sets HZO apart
In its classical presentation, HZO does not pose a diagnostic challenge for practitioners well-versed in skin disease management. The clinical differential diagnosis of herpes zoster will include other causes of blisters or vesicular eruptions of autoimmune etiologies, viral infections, or hypersensitivity reactions.
The most common blistering diseases that may be mistaken for zoster include herpes simplex, contact dermatitis, erythema multiforme, and cellulitis. The prodromal stage and the characteristics of pain usually set herpes zoster apart from the other diagnoses.
Beware of complications
Eye complications. HZO may result in paralytic ptosis, conjunctivitis, keratitis, cataracts, glaucoma, retinitis, and optic neuritis and atrophy.2,5,7
Herpetic lesions on the tip of the nose are believed to herald ocular involvement and may precede typical dermatomal eruption of HZO.2 Such cutaneous involvement along the distribution of the nasociliary nerve is called Hutchinson’s sign. It should prompt a complete ophthalmologic evaluation, as it did with our patient.
Neurologic complications. The most common complication of herpes zoster infection in general is postherpetic neuralgia (PHN), which results in severe pain that persists for months—or years—after the skin lesions have completely healed. Patients over 70 have a 70% chance of developing PHN.4,7
Neurological complications such as encephalitis, myelitis, and Guillain-Barré syndrome have also been reported.
Other complications. Other complications include secondary bacterial infections, pneumonitis, and polyradiculitis.
Management: Antivirals and pain meds
Patients with HZO have a 50% chance of having eye complications (iritis and keratitis) without antiviral treatment.2,5,6 Therefore, treatment is recommended for all HZO patients.
Antivirals ASAP
Start antiviral drugs within 72 hours of clinical presentation, and when new lesions are still appearing on the skin, to achieve optimal effect. Acyclovir, famciclovir, and valacyclovir are the antivirals of choice.6 The usual dosages for adults are: acyclovir (800 mg orally 5 times per day for 7–10 days), valacyclovir (1000 mg orally 3 times daily for 7 days) and famciclovir (500 mg orally 3 times daily for 7 days). The suggested dose of acyclovir for children is 10 to 20 mg/kg/dose qid for 5 days; not to exceed 800 mg per day.2,5,6
Preventing postherpetic neuralgia
The role of systemic corticosteroids in the prevention of PHN, decreasing duration and severity of the acute symptoms in the initial days of herpes zoster infection, remains controversial.8
Immunosuppressed individuals may be treated with acyclovir, interferon-alpha, and vidarabine. In this population the live, attenuated vaccine is safer and is preferred to the varicella zoster immunoglobulins (VZIG).1,6,8,9
PHN can be reduced by treating the patient within the first 24 hours of symptom onset. Pain usually resolves within 3 months in 50% of patients and within 1 year in 75% of patients.1,6,7
Pain therapies for PHN
Therapeutic approaches to the management of PHN include topical anesthetic creams (lidocaine plus prilocaine), capsaicin, and oral medications such as tricyclic antidepressants (amitriptyline and desipramine), carbamazepine, and gabapentin, as well as nerve blocks.6,7
Gloves and handwashing are key for caregivers
Individuals who have not had a confirmed varicella infection should avoid contact with those who have shingles unless a varicella zoster virus antibody is satisfactory and shows immunity.6,7
Gloves should be worn when touching the lesions or infectious drainage, and hands should be washed after glove removal.5-9
Patients with disseminated disease require hospitalization with airborne precautions, which include a negative air pressure room and ensuring that caregivers wear N95 respirators.
What put our patient at risk?
In the case of our young patient, low immunity may have played a role in her contracting HZO. She had a history of recurrent bacterial infections and suffered from generally poor health.
She was referred to an ophthalmologist, who did not find any dendritic corneal ulcer or other complications on weekly follow up. The patient was successfully managed with oral acyclovir 10 mg/kg/dose for 5 days, and she responded well to treatment. We also prescribed acyclovir eye ointment, and local wet compresses with good outcome.
Acknowledgments
The authors thank Dr Shahbaz Janjua for his assistance in the preparation of this manuscript.
Correspondence
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
A 5-year-old girl came into the office complaining of severe burning and tingling sensation of her right forehead. She’d had fever, chills, myalgia, and a relentless headache for 3 days. The morning of her appointment, a few “bumps” and water-filled blisters began to appear on the right side of the forehead; the lesions then started to multiply and grow (FIGURE). The patient’s mother expressed concern over the rapid development of the lesions, which were accompanied by marked edema of the forehead and right eyelid. The mother indicated that no one else in the family was affected at the time of presentation.
The child appeared ill and pale at the time of presentation, but there was no history of immediate antecedent illness or any drug intake prior to the current eruption. Her growth and development were in the lower normal range. The child had a history of recurrent bacterial infections; she was not vaccinated for varicella. There was, however, a personal (and family) history of varicella when the child was about 3 years old.
FIGURE
Vesicles and bullae on forehead
On physical examination, there were multiple vesicles and bullae that varied in size from 2 mm to 1 cm in diameter on the right side of the forehead; there were areas of dried yellowish serous exudates limited to the right eyebrow. The forehead and periorbital areas were edematous with underlying erythematous skin. The entire eruption appeared to be restricted to the right upper part of the face, extending from the eyelid and medial canthus up to the frontal scalp. On close examination, several vesicles and crusts were present on the right side of the nasal tip. Serology for HIV was negative.
What is your diagnosis?
How would you treat?
Diagnosis: Herpes zoster ophthalmicus
This case of herpes zoster ophthalmicus (HZO) was unusual—not because of the way the patient presented, but because of her age. This condition is rarely seen in children, though it is not uncommon in adults—specifically, patients who are 60 years of age and older.1-4
Herpes zoster ophthalmicus, which refers to the involvement of the ophthalmic branch of the trigeminal nerve or the fifth cranial nerve, usually manifests with a typical vesicular or bullous eruption of the forehead, often in unilateral fashion. The lesions of herpes zoster (shingles) are similar to those of varicella (chickenpox) but in herpes zoster, painful unilateral vesicular eruptions are usually limited to one dermatome. (If hematogenous dissemination occurs, more than 20 vesicles will form in skin areas away from the affected dermatome.1)
Patients typically present with fever, headache, and abnormal sensations that precede the development of cutaneous lesions by a few days. The eruptions may be pustular and hemorrhagic initially, and within 10 days evolve into crusts.5,6
Differential diagnosis: Pain sets HZO apart
In its classical presentation, HZO does not pose a diagnostic challenge for practitioners well-versed in skin disease management. The clinical differential diagnosis of herpes zoster will include other causes of blisters or vesicular eruptions of autoimmune etiologies, viral infections, or hypersensitivity reactions.
The most common blistering diseases that may be mistaken for zoster include herpes simplex, contact dermatitis, erythema multiforme, and cellulitis. The prodromal stage and the characteristics of pain usually set herpes zoster apart from the other diagnoses.
Beware of complications
Eye complications. HZO may result in paralytic ptosis, conjunctivitis, keratitis, cataracts, glaucoma, retinitis, and optic neuritis and atrophy.2,5,7
Herpetic lesions on the tip of the nose are believed to herald ocular involvement and may precede typical dermatomal eruption of HZO.2 Such cutaneous involvement along the distribution of the nasociliary nerve is called Hutchinson’s sign. It should prompt a complete ophthalmologic evaluation, as it did with our patient.
Neurologic complications. The most common complication of herpes zoster infection in general is postherpetic neuralgia (PHN), which results in severe pain that persists for months—or years—after the skin lesions have completely healed. Patients over 70 have a 70% chance of developing PHN.4,7
Neurological complications such as encephalitis, myelitis, and Guillain-Barré syndrome have also been reported.
Other complications. Other complications include secondary bacterial infections, pneumonitis, and polyradiculitis.
Management: Antivirals and pain meds
Patients with HZO have a 50% chance of having eye complications (iritis and keratitis) without antiviral treatment.2,5,6 Therefore, treatment is recommended for all HZO patients.
Antivirals ASAP
Start antiviral drugs within 72 hours of clinical presentation, and when new lesions are still appearing on the skin, to achieve optimal effect. Acyclovir, famciclovir, and valacyclovir are the antivirals of choice.6 The usual dosages for adults are: acyclovir (800 mg orally 5 times per day for 7–10 days), valacyclovir (1000 mg orally 3 times daily for 7 days) and famciclovir (500 mg orally 3 times daily for 7 days). The suggested dose of acyclovir for children is 10 to 20 mg/kg/dose qid for 5 days; not to exceed 800 mg per day.2,5,6
Preventing postherpetic neuralgia
The role of systemic corticosteroids in the prevention of PHN, decreasing duration and severity of the acute symptoms in the initial days of herpes zoster infection, remains controversial.8
Immunosuppressed individuals may be treated with acyclovir, interferon-alpha, and vidarabine. In this population the live, attenuated vaccine is safer and is preferred to the varicella zoster immunoglobulins (VZIG).1,6,8,9
PHN can be reduced by treating the patient within the first 24 hours of symptom onset. Pain usually resolves within 3 months in 50% of patients and within 1 year in 75% of patients.1,6,7
Pain therapies for PHN
Therapeutic approaches to the management of PHN include topical anesthetic creams (lidocaine plus prilocaine), capsaicin, and oral medications such as tricyclic antidepressants (amitriptyline and desipramine), carbamazepine, and gabapentin, as well as nerve blocks.6,7
Gloves and handwashing are key for caregivers
Individuals who have not had a confirmed varicella infection should avoid contact with those who have shingles unless a varicella zoster virus antibody is satisfactory and shows immunity.6,7
Gloves should be worn when touching the lesions or infectious drainage, and hands should be washed after glove removal.5-9
Patients with disseminated disease require hospitalization with airborne precautions, which include a negative air pressure room and ensuring that caregivers wear N95 respirators.
What put our patient at risk?
In the case of our young patient, low immunity may have played a role in her contracting HZO. She had a history of recurrent bacterial infections and suffered from generally poor health.
She was referred to an ophthalmologist, who did not find any dendritic corneal ulcer or other complications on weekly follow up. The patient was successfully managed with oral acyclovir 10 mg/kg/dose for 5 days, and she responded well to treatment. We also prescribed acyclovir eye ointment, and local wet compresses with good outcome.
Acknowledgments
The authors thank Dr Shahbaz Janjua for his assistance in the preparation of this manuscript.
Correspondence
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016; [email protected].
1. Trent JT, Kirsner RS. Herpesvirus infections and herpetic wounds. Adv Skin Wound Care 2003;16:236-243.
2. Zaal MJ, Volker-Dieben HJ, D’Amaro J. Prognostic value of Hutchinson’s sign in acute herpes zoster ophthalmicus. Graefes Arch Clin Exp Ophthalmol 2003;241:187-191.
3. Binder NR, Holland GN, Hosea S, Silverberg ML. Herpes zoster ophthalmicus in an otherwisehealthy child. JAAPOS 2005;9:597-598.
4. Gebo KA, Kalyani R, Moore RD, Polydefkis MJ. The incidence of, risk factors for, and sequelae of herpes zoster among HIV patients in the highly active antiretroviral therapy era. JAIDS 2005;40:169-174.
5. Liesegang TJ. Herpes zoster virus infection. Curr Opin Ophthalmol 2004;15:531-536.
6. McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol 1999;41:1-14.
7. Cohen JI, Brunell PA, Straus SE, Krause PR. Recent advances in varicella-zoster virus infection. Ann Intern Med 1999;130:922-932.
8. Santee JA. Corticosteroids for herpes zoster: what do they accomplish? Am J Clin Dermatol 2002;3:517-524.
9. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005;352:2271-2284.
1. Trent JT, Kirsner RS. Herpesvirus infections and herpetic wounds. Adv Skin Wound Care 2003;16:236-243.
2. Zaal MJ, Volker-Dieben HJ, D’Amaro J. Prognostic value of Hutchinson’s sign in acute herpes zoster ophthalmicus. Graefes Arch Clin Exp Ophthalmol 2003;241:187-191.
3. Binder NR, Holland GN, Hosea S, Silverberg ML. Herpes zoster ophthalmicus in an otherwisehealthy child. JAAPOS 2005;9:597-598.
4. Gebo KA, Kalyani R, Moore RD, Polydefkis MJ. The incidence of, risk factors for, and sequelae of herpes zoster among HIV patients in the highly active antiretroviral therapy era. JAIDS 2005;40:169-174.
5. Liesegang TJ. Herpes zoster virus infection. Curr Opin Ophthalmol 2004;15:531-536.
6. McCrary ML, Severson J, Tyring SK. Varicella zoster virus. J Am Acad Dermatol 1999;41:1-14.
7. Cohen JI, Brunell PA, Straus SE, Krause PR. Recent advances in varicella-zoster virus infection. Ann Intern Med 1999;130:922-932.
8. Santee JA. Corticosteroids for herpes zoster: what do they accomplish? Am J Clin Dermatol 2002;3:517-524.
9. Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005;352:2271-2284.
Bilateral lesions on the legs
A 50-year-old woman came to the office for medical advice regarding bilateral erythematous lesions on the inner aspects of both of her legs. She had been aware of the lesions for more than 5 months. She had no other medical complaints, and was not taking any prescribed, over-the-counter, or herbal medications, or using any moisturizers or other medicated topical preparations. She mentioned that she was using a hot water bottle on her legs to keep her warm.
On physical examination, we noted bilateral erythema in a mesh-like distribution on her frontal, inner lower thighs as well as the upper and medial aspects of her calves. There were prominent telangiectatic vessels and “spider veins” surrounding the erythema, but her skin was otherwise normal. The review of her systems showed no problems. Personal and family histories were not contributory.
FIGURE 1
Lesions on the legs
Skin biopsy revealed subepidermal separation accompanied by thinning of the epidermis with loss of rete ridges and the presence of dermal edema. There was an increase in the number of elastic fibers with aberrant disruption. Perivascular infiltration of inflammatory cells was also noted.
FIGURE 2
Close-up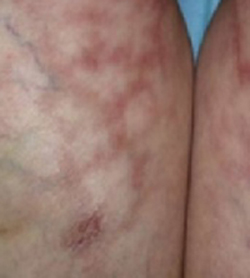
What is your diagnosis?
How would you manage this condition?
Diagnosis: Erythema ab igne
Erythema ab igne is characterized by localized erythema, reticulosis, and hypo-/hyperpigmentation, with telangiectsia and skin atrophy appearing in severe cases. This condition is caused by continuous exposure to sources of heat for prolonged periods of time.1 Patients often do not connect their heat exposure to the rash, so you must recognize the local pattern of erythema ab igne and use your detective skills to identify the source.
History: Old disease, modern causes
Erythema ab igne has been noted for many years, and the sources of heat have changed over time. It used to be seen in women who stayed for long periods in front of open fires or furnaces to cook.2,3 Most of the lesions appeared on the medial side of the thigh and the lower leg.
In modern times it is seen on different parts of the body depending on the heat source that initiated the pathology, the angle of the heat radiation, the morphology of the skin, and the layers of clothing.1 Some of the modern causes of erythema ab igne are repeated application of hot water bottles or heat pads to treat chronic pain, exposure to car heaters and furniture with internal heaters, use of a laptop computer for long periods,4 and cook stoves, for restaurant workers who stand for long periods near the heat.3 Ultrasound physiotherapy was also reported as a cause.5 Recently, a case caused by frequent and prolonged hot bathing was reported.6
Clinical features: Mottled pattern clinically, subtle changes
The pattern of lesions form as a result of multiple exposures to the source of heat. Skin lesions may not appear immediately after the exposure—it might take a month to show up.7 Changes start as a reddish-brown pigmentation distributed as a mottled rash, followed by skin atrophy. Telangiectasias with diffuse hyperpigmentation and subepidermal bullae may also develop.8 Some patients complain of mild pruritus or burning sensation, but most patients are asymptomatic.9 Erythema ab igne should be differentiated from other diseases with skin changes that mimic its presentation (TABLE).
If clinically warranted, perform a biopsy to exclude the possibility of malignant formation. Histopathology results show epidermal atrophy, subepidermal separation, and haziness of the dermoepidermal junction. Dilatation of capillaries and connective tissue disintegration, elastosis, hemosiderin deposition, melanocytosis, and abundance of inflammatory cells are all seen in the dermis.10 Some of these lesions might progress to actinic keratosis, which could be a precursor for squamous cell carcinoma of the skin.11 In some rare cases, Merkel cell carcinoma has developed in areas of erythema ab igne.12
Treatment: Eliminate heat source
The first goal of treatment is to identify the source of heat radiation to avoid further exposure. For mild lesions, no intervention is needed after the heat source is removed; the probability of full resolution is good. In this case, the patient was advised to stop using the hot water bottle on her skin. Over 4 months her lesions started to clear with no further intervention.
Topical meds help with cosmesis
Topical retinoids, vitamin A derivatives, hydroquinone, and 5-fluorouracil can be prescribed to treat abnormal skin pigmentation.13 Laser therapy has been used to even out the skin color.
TABLE
Differential diagnosis for erythema ab igne
| DISEASE | CLINICAL FEATURES |
|---|---|
| Acanthosis nigricans | Velvety, light-brown-to-black markings usually on the neck, under the arms, or in the groin |
| Most often associated with being overweight | |
| More common in people with darker skin pigmentation | |
| May begin at any age | |
| May be inherited as a primary condition or associated with various underlying syndromes | |
| Livedo reticularis | Reticular cyanotic cutaneous discoloration surrounding pale central areas due to dilation of capillary blood vessels and stagnation of blood |
| Occurs mostly on the legs, arms, and trunk | |
| More pronounced in cold weather | |
| Idiopathic condition that may be associated with systemic diseases | |
| Poikiloderma atrophicans vasculare | Circumscribed violaceous erythema |
| Occurs mostly in posterior shoulders, back, buttocks, V-shaped area of anterior neck, and chest | |
| May be asymptomatic or mildly pruritic | |
| May remain stable in size or gradually increase | |
| Numerous atypical lymphocytes are observed around dermal blood vessels and someepidermotropism is observed | |
| A variant of mycosis fungoides |
CORRESPONDENCE
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016. E-mail: [email protected]
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol 1988;18:1003-1019.
2. Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol 1996;34:516-517.
3. Helm TN, Spigel GT, Helm KF. Erythema ab igne caused by a car heater. Cutis 1997;59:81-82.
4. Bilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol 2004;50:973-974.
5. Weber MB, Ponzio HA, Costa FB, Camini L. An Bras Dermatol 2005;80:187-188.
6. Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol 2002;82:478-479.
7. Galvin SA, Buchness MR. Rectangular reticulate patches on the pretibial areas. Erythema ab igne. Arch Dermatol 1990;126:386-387,389.
8. Flanagan N, Watson R, Sweeney E, Barnes L. Bullous erythema ab igne. Br J Dermatol 1996;134:1159-1160.
9. Shahrad P, Marks R. The wages of warmth: changes in erythema ab igne. Br J Dermatol 1977;97:179-186.
10. Milligan A, Graham-Brown RA. Erythema ab igne affecting the palms. Clin Exp Dermatol 1989;14:168-169.
11. Arrington JH, 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. Arch Dermatol 1979;115:1226-1228.
12. Hewitt JB, Sherif A, Kerr KM, Stankler L. Merkel cell and squamous cell carcinomas arising in erythema ab igne. Br J Dermatol 1993;128:591-592.
13. Sahl WJ, Jr, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol 1992;27:109-110.
A 50-year-old woman came to the office for medical advice regarding bilateral erythematous lesions on the inner aspects of both of her legs. She had been aware of the lesions for more than 5 months. She had no other medical complaints, and was not taking any prescribed, over-the-counter, or herbal medications, or using any moisturizers or other medicated topical preparations. She mentioned that she was using a hot water bottle on her legs to keep her warm.
On physical examination, we noted bilateral erythema in a mesh-like distribution on her frontal, inner lower thighs as well as the upper and medial aspects of her calves. There were prominent telangiectatic vessels and “spider veins” surrounding the erythema, but her skin was otherwise normal. The review of her systems showed no problems. Personal and family histories were not contributory.
FIGURE 1
Lesions on the legs
Skin biopsy revealed subepidermal separation accompanied by thinning of the epidermis with loss of rete ridges and the presence of dermal edema. There was an increase in the number of elastic fibers with aberrant disruption. Perivascular infiltration of inflammatory cells was also noted.
FIGURE 2
Close-up
What is your diagnosis?
How would you manage this condition?
Diagnosis: Erythema ab igne
Erythema ab igne is characterized by localized erythema, reticulosis, and hypo-/hyperpigmentation, with telangiectsia and skin atrophy appearing in severe cases. This condition is caused by continuous exposure to sources of heat for prolonged periods of time.1 Patients often do not connect their heat exposure to the rash, so you must recognize the local pattern of erythema ab igne and use your detective skills to identify the source.
History: Old disease, modern causes
Erythema ab igne has been noted for many years, and the sources of heat have changed over time. It used to be seen in women who stayed for long periods in front of open fires or furnaces to cook.2,3 Most of the lesions appeared on the medial side of the thigh and the lower leg.
In modern times it is seen on different parts of the body depending on the heat source that initiated the pathology, the angle of the heat radiation, the morphology of the skin, and the layers of clothing.1 Some of the modern causes of erythema ab igne are repeated application of hot water bottles or heat pads to treat chronic pain, exposure to car heaters and furniture with internal heaters, use of a laptop computer for long periods,4 and cook stoves, for restaurant workers who stand for long periods near the heat.3 Ultrasound physiotherapy was also reported as a cause.5 Recently, a case caused by frequent and prolonged hot bathing was reported.6
Clinical features: Mottled pattern clinically, subtle changes
The pattern of lesions form as a result of multiple exposures to the source of heat. Skin lesions may not appear immediately after the exposure—it might take a month to show up.7 Changes start as a reddish-brown pigmentation distributed as a mottled rash, followed by skin atrophy. Telangiectasias with diffuse hyperpigmentation and subepidermal bullae may also develop.8 Some patients complain of mild pruritus or burning sensation, but most patients are asymptomatic.9 Erythema ab igne should be differentiated from other diseases with skin changes that mimic its presentation (TABLE).
If clinically warranted, perform a biopsy to exclude the possibility of malignant formation. Histopathology results show epidermal atrophy, subepidermal separation, and haziness of the dermoepidermal junction. Dilatation of capillaries and connective tissue disintegration, elastosis, hemosiderin deposition, melanocytosis, and abundance of inflammatory cells are all seen in the dermis.10 Some of these lesions might progress to actinic keratosis, which could be a precursor for squamous cell carcinoma of the skin.11 In some rare cases, Merkel cell carcinoma has developed in areas of erythema ab igne.12
Treatment: Eliminate heat source
The first goal of treatment is to identify the source of heat radiation to avoid further exposure. For mild lesions, no intervention is needed after the heat source is removed; the probability of full resolution is good. In this case, the patient was advised to stop using the hot water bottle on her skin. Over 4 months her lesions started to clear with no further intervention.
Topical meds help with cosmesis
Topical retinoids, vitamin A derivatives, hydroquinone, and 5-fluorouracil can be prescribed to treat abnormal skin pigmentation.13 Laser therapy has been used to even out the skin color.
TABLE
Differential diagnosis for erythema ab igne
| DISEASE | CLINICAL FEATURES |
|---|---|
| Acanthosis nigricans | Velvety, light-brown-to-black markings usually on the neck, under the arms, or in the groin |
| Most often associated with being overweight | |
| More common in people with darker skin pigmentation | |
| May begin at any age | |
| May be inherited as a primary condition or associated with various underlying syndromes | |
| Livedo reticularis | Reticular cyanotic cutaneous discoloration surrounding pale central areas due to dilation of capillary blood vessels and stagnation of blood |
| Occurs mostly on the legs, arms, and trunk | |
| More pronounced in cold weather | |
| Idiopathic condition that may be associated with systemic diseases | |
| Poikiloderma atrophicans vasculare | Circumscribed violaceous erythema |
| Occurs mostly in posterior shoulders, back, buttocks, V-shaped area of anterior neck, and chest | |
| May be asymptomatic or mildly pruritic | |
| May remain stable in size or gradually increase | |
| Numerous atypical lymphocytes are observed around dermal blood vessels and someepidermotropism is observed | |
| A variant of mycosis fungoides |
CORRESPONDENCE
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016. E-mail: [email protected]
A 50-year-old woman came to the office for medical advice regarding bilateral erythematous lesions on the inner aspects of both of her legs. She had been aware of the lesions for more than 5 months. She had no other medical complaints, and was not taking any prescribed, over-the-counter, or herbal medications, or using any moisturizers or other medicated topical preparations. She mentioned that she was using a hot water bottle on her legs to keep her warm.
On physical examination, we noted bilateral erythema in a mesh-like distribution on her frontal, inner lower thighs as well as the upper and medial aspects of her calves. There were prominent telangiectatic vessels and “spider veins” surrounding the erythema, but her skin was otherwise normal. The review of her systems showed no problems. Personal and family histories were not contributory.
FIGURE 1
Lesions on the legs
Skin biopsy revealed subepidermal separation accompanied by thinning of the epidermis with loss of rete ridges and the presence of dermal edema. There was an increase in the number of elastic fibers with aberrant disruption. Perivascular infiltration of inflammatory cells was also noted.
FIGURE 2
Close-up
What is your diagnosis?
How would you manage this condition?
Diagnosis: Erythema ab igne
Erythema ab igne is characterized by localized erythema, reticulosis, and hypo-/hyperpigmentation, with telangiectsia and skin atrophy appearing in severe cases. This condition is caused by continuous exposure to sources of heat for prolonged periods of time.1 Patients often do not connect their heat exposure to the rash, so you must recognize the local pattern of erythema ab igne and use your detective skills to identify the source.
History: Old disease, modern causes
Erythema ab igne has been noted for many years, and the sources of heat have changed over time. It used to be seen in women who stayed for long periods in front of open fires or furnaces to cook.2,3 Most of the lesions appeared on the medial side of the thigh and the lower leg.
In modern times it is seen on different parts of the body depending on the heat source that initiated the pathology, the angle of the heat radiation, the morphology of the skin, and the layers of clothing.1 Some of the modern causes of erythema ab igne are repeated application of hot water bottles or heat pads to treat chronic pain, exposure to car heaters and furniture with internal heaters, use of a laptop computer for long periods,4 and cook stoves, for restaurant workers who stand for long periods near the heat.3 Ultrasound physiotherapy was also reported as a cause.5 Recently, a case caused by frequent and prolonged hot bathing was reported.6
Clinical features: Mottled pattern clinically, subtle changes
The pattern of lesions form as a result of multiple exposures to the source of heat. Skin lesions may not appear immediately after the exposure—it might take a month to show up.7 Changes start as a reddish-brown pigmentation distributed as a mottled rash, followed by skin atrophy. Telangiectasias with diffuse hyperpigmentation and subepidermal bullae may also develop.8 Some patients complain of mild pruritus or burning sensation, but most patients are asymptomatic.9 Erythema ab igne should be differentiated from other diseases with skin changes that mimic its presentation (TABLE).
If clinically warranted, perform a biopsy to exclude the possibility of malignant formation. Histopathology results show epidermal atrophy, subepidermal separation, and haziness of the dermoepidermal junction. Dilatation of capillaries and connective tissue disintegration, elastosis, hemosiderin deposition, melanocytosis, and abundance of inflammatory cells are all seen in the dermis.10 Some of these lesions might progress to actinic keratosis, which could be a precursor for squamous cell carcinoma of the skin.11 In some rare cases, Merkel cell carcinoma has developed in areas of erythema ab igne.12
Treatment: Eliminate heat source
The first goal of treatment is to identify the source of heat radiation to avoid further exposure. For mild lesions, no intervention is needed after the heat source is removed; the probability of full resolution is good. In this case, the patient was advised to stop using the hot water bottle on her skin. Over 4 months her lesions started to clear with no further intervention.
Topical meds help with cosmesis
Topical retinoids, vitamin A derivatives, hydroquinone, and 5-fluorouracil can be prescribed to treat abnormal skin pigmentation.13 Laser therapy has been used to even out the skin color.
TABLE
Differential diagnosis for erythema ab igne
| DISEASE | CLINICAL FEATURES |
|---|---|
| Acanthosis nigricans | Velvety, light-brown-to-black markings usually on the neck, under the arms, or in the groin |
| Most often associated with being overweight | |
| More common in people with darker skin pigmentation | |
| May begin at any age | |
| May be inherited as a primary condition or associated with various underlying syndromes | |
| Livedo reticularis | Reticular cyanotic cutaneous discoloration surrounding pale central areas due to dilation of capillary blood vessels and stagnation of blood |
| Occurs mostly on the legs, arms, and trunk | |
| More pronounced in cold weather | |
| Idiopathic condition that may be associated with systemic diseases | |
| Poikiloderma atrophicans vasculare | Circumscribed violaceous erythema |
| Occurs mostly in posterior shoulders, back, buttocks, V-shaped area of anterior neck, and chest | |
| May be asymptomatic or mildly pruritic | |
| May remain stable in size or gradually increase | |
| Numerous atypical lymphocytes are observed around dermal blood vessels and someepidermotropism is observed | |
| A variant of mycosis fungoides |
CORRESPONDENCE
Amor Khachemoune, MD, CWS, Ronald O. Perelman Department of Dermatology, New York University School of Medicine, 530 First Avenue, Suite 7R, New York, NY 10016. E-mail: [email protected]
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol 1988;18:1003-1019.
2. Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol 1996;34:516-517.
3. Helm TN, Spigel GT, Helm KF. Erythema ab igne caused by a car heater. Cutis 1997;59:81-82.
4. Bilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol 2004;50:973-974.
5. Weber MB, Ponzio HA, Costa FB, Camini L. An Bras Dermatol 2005;80:187-188.
6. Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol 2002;82:478-479.
7. Galvin SA, Buchness MR. Rectangular reticulate patches on the pretibial areas. Erythema ab igne. Arch Dermatol 1990;126:386-387,389.
8. Flanagan N, Watson R, Sweeney E, Barnes L. Bullous erythema ab igne. Br J Dermatol 1996;134:1159-1160.
9. Shahrad P, Marks R. The wages of warmth: changes in erythema ab igne. Br J Dermatol 1977;97:179-186.
10. Milligan A, Graham-Brown RA. Erythema ab igne affecting the palms. Clin Exp Dermatol 1989;14:168-169.
11. Arrington JH, 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. Arch Dermatol 1979;115:1226-1228.
12. Hewitt JB, Sherif A, Kerr KM, Stankler L. Merkel cell and squamous cell carcinomas arising in erythema ab igne. Br J Dermatol 1993;128:591-592.
13. Sahl WJ, Jr, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol 1992;27:109-110.
1. Page EH, Shear NH. Temperature-dependent skin disorders. J Am Acad Dermatol 1988;18:1003-1019.
2. Meffert JJ, Davis BM. Furniture-induced erythema ab igne. J Am Acad Dermatol 1996;34:516-517.
3. Helm TN, Spigel GT, Helm KF. Erythema ab igne caused by a car heater. Cutis 1997;59:81-82.
4. Bilic M, Adams BB. Erythema ab igne induced by a laptop computer. J Am Acad Dermatol 2004;50:973-974.
5. Weber MB, Ponzio HA, Costa FB, Camini L. An Bras Dermatol 2005;80:187-188.
6. Lin SJ, Hsu CJ, Chiu HC. Erythema ab igne caused by frequent hot bathing. Acta Derm Venereol 2002;82:478-479.
7. Galvin SA, Buchness MR. Rectangular reticulate patches on the pretibial areas. Erythema ab igne. Arch Dermatol 1990;126:386-387,389.
8. Flanagan N, Watson R, Sweeney E, Barnes L. Bullous erythema ab igne. Br J Dermatol 1996;134:1159-1160.
9. Shahrad P, Marks R. The wages of warmth: changes in erythema ab igne. Br J Dermatol 1977;97:179-186.
10. Milligan A, Graham-Brown RA. Erythema ab igne affecting the palms. Clin Exp Dermatol 1989;14:168-169.
11. Arrington JH, 3rd, Lockman DS. Thermal keratoses and squamous cell carcinoma in situ associated with erythema ab igne. Arch Dermatol 1979;115:1226-1228.
12. Hewitt JB, Sherif A, Kerr KM, Stankler L. Merkel cell and squamous cell carcinomas arising in erythema ab igne. Br J Dermatol 1993;128:591-592.
13. Sahl WJ, Jr, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol 1992;27:109-110.
Dark brown scaly plaques on face and ears
A 27-year-old woman, otherwise healthy, presented for evaluation of a mildly pruritic eruption of plaques on her face and ears. The eruption started as a few small, scaly red papules on her cheeks and nose about 3 months earlier. The papules slowly expanded to form well-demarcated, rounded, dark brown plaques covered with superficial scale. Similar lesions appeared on the concha of both ears. The older lesions started to regress after 2 months. Complete regression was followed by residual scarring and hyperpigmentation.
Physical examination revealed multiple well-defined, erythematous, hyperpigmented, round plaques covered with adherent scale affecting her nose, cheeks, and the concha of both ears (FIGURE 1). The mucosae and the rest of her skin and adnexa were unaffected. She had no personal or family history of similar skin findings or autoimmune disorders. She also had no history of any drug intake prior to the eruption. Her routine blood tests and urinalysis results were unremarkable, and the serological analysis for antinuclear antibodies had negative results.
A punch biopsy was taken from one of the lesions for histopathology. The epidermal histopathological findings were remarkable for hyperkeratosis, follicular plugging, pigment incontinence, and vacuolization of the basal cell layer. There was a predominantly lymphocytic infiltrate of the subepidermal and perivascular dermal areas.
FIGURE 1
Facial plaques
What is your diagnosis?
How would you treat this patient?
Diagnosis: Discoid lupus erythematosus
Discoid lupus erythematosus (DLE) is an autoimmune inflammatory disorder of the skin that often leads to scarring and alopecia. It may be localized to sun-exposed areas such as the face, ears, and scalp but occasionally is much more extensive, involving the trunk and extremities. Most patients are otherwise healthy, and DLE may be the only clinical finding.
Although its prevalence is not known, DLE is not uncommon. It affects females twice as often as males, and patients fall between the ages of 25 to 45 years, with no racial predilection. While about 15% to 20% of patients with systemic lupus erythematosus (SLE) manifest DLE lesions, only about 5% to 10% of patients with DLE go on to develop SLE.1
Like SLE, DLE is believed to be an autoimmune disorder. Unlike SLE, however, DLE patients do not have similar serologic abnormalities. Skin trauma and ultraviolet light exposure have been reported to induce or exacerbate the lesions of DLE. Sex hormones may also play a role: exacerbation may occur during pregnancy, during menstrual or premenstrual periods, or while taking oral contraceptives. Drugs such as procainamide, hydralazine, isoniazid, diphenylhydantoin, methyldopa, penicillamine, guanides, and lithium may also precipitate DLE lesions.2
DLE usually begins as dull red macules with adherent scales on sun-exposed areas. If the overlying scales are removed, an undersurface of horny plugs is revealed. These plugs fill the follicles and resemble carpet tacks or a cat’s tongue (langue au chat). The macules slowly expand to form large plaques.
Usually only a mild pruritus and tenderness is seen with DLE lesions. As the lesions progress, the scale may thicken and pigmentary changes become evident. The lesions heal with atrophy, scarring, telangiectasias, and changes in pigmentation. Morphological appearance of older lesions may vary from erythematous plaques to hyperkeratotic, dark gray plaques with centrally depressed scars.3 Scalp involvement in DLE results in more sclerotic and depressed scars with subsequent scarring alopecia.4
Differential diagnosis is wide. The differential diagnosis of DLE is extensive and includes actinic keratosis, dermatomyositis, granuloma annulare, granuloma faciale, keratoacanthoma, lichen planus, subacute cutaneous lupus erythematosus, psoriasis, rosacea, sarcoidosis, squamous cell carcinoma, syphilis, and nongenital warts.
Histopathology
Histopathological findings include hyperkeratosis, parakeratosis, follicular plugging, telangiectasias, and atrophy of the epidermis. Liquefaction or hydropic degeneration of the basal layer leads to pigmentary incontinence. A perivascular and perifollicular mononuclear inflammatory cell infiltrate is present in the superficial and deep dermis.5,6
Direct immunofluorescence demonstrates immunoglobulins and complement deposits at the dermoepidermal junction.5 This test was not recommended for this patient, as the diagnosis of DLE had already been confirmed on clinical and histopathologic grounds.
Workup: Exam, biopsy
In addition to a routine history and physical examination, the workup for DLE should include a complete blood count, antinuclear antibody levels, anti-Ro, anti-La, hepatic and renal function tests, and urinalysis. Consider a diagnosis of SLE by using the American College of Rheumatology criteria. A biopsy for histopathology of a fresh lesion or a biopsy for immunofluorescence of an old lesion can confirm the diagnosis.
Therapeutic options are varied
Effective early therapies for DLE are available, but patients who do not respond appropriately may end up with deep scars, alopecia, and pigmentary changes that are considerably disfiguring, especially for dark-skinned people. Therefore, the goal of treatment is not only to improve the appearance of the skin by minimizing the scarring and preventing further lesions, but also to prevent future complications.
Avoiding sunlight. The primary therapeutic approach is to educate patients regarding exposure to sunlight. Sun-protective measures include the use of high-SPF sunscreen lotions and protective clothing, such as baseball caps without vent holes and wide-brimmed hats.7
Medication options. Current treatment options for DLE include antimalarial agents such as chloroquine, topical and intralesional glucocorticoids, and thalidomide. The use of potent topical steroids may prevent significant scarring and deformity, especially of the face. Common side effects include steroid withdrawal syndrome, perioral dermatitis, steroid acne, and rosacea. All of these side effects can be treated and result in less long-term deformity than untreated DLE (FIGURE 2).
Systemic agents. Widespread disease may require you use systemic agents, such as antimalarials. Chloroquine has long been considered the gold standard in the treatment of DLE.8 Due to the frequency of ocular side effects with chloroquine, hydroxychloroquine is by far the most widely used agent. Hydroxychloroquine at a dose of 6.5 mg/kg/d for 3 months may lead to resolution of lesions for many patients.7 In resistant cases, higher dosages (eg, 400 mg/d) or combinations (eg, hydroxychloroquine 200 mg/d plus quinacrine 50 to 100 mg/d) may be required for months or even years. An ophthalmological evaluation is advisable before starting antimalarial treatment, and you should repeat it at 4- to 6-month intervals during treatment.9 Systemic corticosteroids may be needed to obtain timely initial control, especially for widespread and disfiguring lesions.
When this therapy is inadequate, other treatments for DLE include azathioprine, retinoids, and dapsone. Recent reports confirm the efficacy of thalidomide for cutaneous lupus erythematosus in dosages ranging from 50 to 200 mg/d.10 Twice-daily application of tacrolimus 0.01% has also been shown to be effective in a few clinical trials.11
Surgery. Surgical intervention is useful to remove scarred lesions, and laser therapy has also been effective, especially for lesions with prominent telangiectasias.12
Patient education. Patient education plays an important role in the treatment of DLE. Advising patients on how to avoid the sun and instructing them in the proper use of sunscreens is extremely important. Smoking cessation has also been shown to be beneficial.13
Patients with DLE generally have a favorable prognosis with regards to morbidity and mortality. Because DLE is usually self-limited, the course is most often benign; therefore, early recognition and adequate therapy may prevent clinical complications. Many patients with DLE go on to develop destructive or deforming scarring or pigmentary disturbances,14 especially in the spring and summer months when the sun is the strongest.
FIGURE 2
After treatment
This patient’s outcome
Our patient was treated with topical fluocinolone acetonide 0.025% ointment and oral hydroxychloroquine 200 mg/d for 1 month. The patient responded well to the treatment and the skin lesion regressed perceptibly (FIGURE 2). The treatment was nevertheless continued for another 5 months, and resulted in complete regression of the lesions, with minimal residual scarring.
CORRESPONDENCE
Amor Khachemoune, MD, CWS, Department of Dermatology, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203. E-mail: [email protected]
1. Callen JP. Systemic lupus erythematosus in patients with chronic cutaneous (discoid) lupus erythematosus. Clinical and laboratory findings in seventeen patients. J Am Acad Dermatol 1985;12(2 Pt 1):278-288.
2. Hess E. Drug-related lupus. N Engl J Med 1988;318:1460-1462.
3. Callen JP, Fowler JF, Kulick KB. Serologic and clinical features of patients with discoid lupus erythematosus: relationship of antibodies to single-stranded deoxyribonucleic acid and of other antinuclear antibody subsets to clinical manifestations. J Am Acad Dermatol 1985;13(5 Pt 1):748-755.
4. Wilson CL, Burge SM, Dean D, Dawber RP. Scarring alopecia in discoid lupus erythematosus. Br J Dermatol 1992;126:307-314.
5. Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin 2002;20:373-385, v.
6. Biesla I, et al. Histopathologic findings in cutaneous lupus erythematosus. Arch Dermatol 1994;130:54-58.
7. Callen JP. Treatment of cutaneous lesions in patients with lupus erythematosus. Dermatol Clin 1994;12:201-206.
8. Goldman L, Cole DP, Preston RH. Chloroquine diphosphate in treatment of discoid lupus erythematosus. J Am Med Assoc 1953;152:1428-1429.
9. Rynes RI. Ophthalmologic safety of long-term hydroxychloroquine sulfate treatment. Am J Med 1983;75:35-39.
10. Kyriakis KP, Kontochristopoulos GJ, Panteleos DN. Experience with low-dose thalidomide therapy in chronic discoid lupus erythematosus. Int J Dermatol 2000;39:218-222.
11. Heffernan MP, Nelson MM, Smith DI, Chung JH. 0.1% tacrolimus ointment in the treatment of discoid lupus erythematosus. Arch Dermatol 2005;141:1170-1171.
12. Nunez M, Boixeda P, Miralles ES, et al. Pulsed dye laser treatment of telangiectatic chronic erythema of cutaneous lupus erythematosus. Arch Dermatol 1996;132:354-355.
13. Miot HA, Bartoli Miot LD, Haddad GR. Association between discoid lupus erythematosus and cigarette smoking. Dermatology 2005;211:118-122.
14. de Berker D, Dissaneyeka M, Burge S. The sequelae of chronic cutaneous lupus erythematosus. Lupus 1992;1:181-186.
A 27-year-old woman, otherwise healthy, presented for evaluation of a mildly pruritic eruption of plaques on her face and ears. The eruption started as a few small, scaly red papules on her cheeks and nose about 3 months earlier. The papules slowly expanded to form well-demarcated, rounded, dark brown plaques covered with superficial scale. Similar lesions appeared on the concha of both ears. The older lesions started to regress after 2 months. Complete regression was followed by residual scarring and hyperpigmentation.
Physical examination revealed multiple well-defined, erythematous, hyperpigmented, round plaques covered with adherent scale affecting her nose, cheeks, and the concha of both ears (FIGURE 1). The mucosae and the rest of her skin and adnexa were unaffected. She had no personal or family history of similar skin findings or autoimmune disorders. She also had no history of any drug intake prior to the eruption. Her routine blood tests and urinalysis results were unremarkable, and the serological analysis for antinuclear antibodies had negative results.
A punch biopsy was taken from one of the lesions for histopathology. The epidermal histopathological findings were remarkable for hyperkeratosis, follicular plugging, pigment incontinence, and vacuolization of the basal cell layer. There was a predominantly lymphocytic infiltrate of the subepidermal and perivascular dermal areas.
FIGURE 1
Facial plaques
What is your diagnosis?
How would you treat this patient?
Diagnosis: Discoid lupus erythematosus
Discoid lupus erythematosus (DLE) is an autoimmune inflammatory disorder of the skin that often leads to scarring and alopecia. It may be localized to sun-exposed areas such as the face, ears, and scalp but occasionally is much more extensive, involving the trunk and extremities. Most patients are otherwise healthy, and DLE may be the only clinical finding.
Although its prevalence is not known, DLE is not uncommon. It affects females twice as often as males, and patients fall between the ages of 25 to 45 years, with no racial predilection. While about 15% to 20% of patients with systemic lupus erythematosus (SLE) manifest DLE lesions, only about 5% to 10% of patients with DLE go on to develop SLE.1
Like SLE, DLE is believed to be an autoimmune disorder. Unlike SLE, however, DLE patients do not have similar serologic abnormalities. Skin trauma and ultraviolet light exposure have been reported to induce or exacerbate the lesions of DLE. Sex hormones may also play a role: exacerbation may occur during pregnancy, during menstrual or premenstrual periods, or while taking oral contraceptives. Drugs such as procainamide, hydralazine, isoniazid, diphenylhydantoin, methyldopa, penicillamine, guanides, and lithium may also precipitate DLE lesions.2
DLE usually begins as dull red macules with adherent scales on sun-exposed areas. If the overlying scales are removed, an undersurface of horny plugs is revealed. These plugs fill the follicles and resemble carpet tacks or a cat’s tongue (langue au chat). The macules slowly expand to form large plaques.
Usually only a mild pruritus and tenderness is seen with DLE lesions. As the lesions progress, the scale may thicken and pigmentary changes become evident. The lesions heal with atrophy, scarring, telangiectasias, and changes in pigmentation. Morphological appearance of older lesions may vary from erythematous plaques to hyperkeratotic, dark gray plaques with centrally depressed scars.3 Scalp involvement in DLE results in more sclerotic and depressed scars with subsequent scarring alopecia.4
Differential diagnosis is wide. The differential diagnosis of DLE is extensive and includes actinic keratosis, dermatomyositis, granuloma annulare, granuloma faciale, keratoacanthoma, lichen planus, subacute cutaneous lupus erythematosus, psoriasis, rosacea, sarcoidosis, squamous cell carcinoma, syphilis, and nongenital warts.
Histopathology
Histopathological findings include hyperkeratosis, parakeratosis, follicular plugging, telangiectasias, and atrophy of the epidermis. Liquefaction or hydropic degeneration of the basal layer leads to pigmentary incontinence. A perivascular and perifollicular mononuclear inflammatory cell infiltrate is present in the superficial and deep dermis.5,6
Direct immunofluorescence demonstrates immunoglobulins and complement deposits at the dermoepidermal junction.5 This test was not recommended for this patient, as the diagnosis of DLE had already been confirmed on clinical and histopathologic grounds.
Workup: Exam, biopsy
In addition to a routine history and physical examination, the workup for DLE should include a complete blood count, antinuclear antibody levels, anti-Ro, anti-La, hepatic and renal function tests, and urinalysis. Consider a diagnosis of SLE by using the American College of Rheumatology criteria. A biopsy for histopathology of a fresh lesion or a biopsy for immunofluorescence of an old lesion can confirm the diagnosis.
Therapeutic options are varied
Effective early therapies for DLE are available, but patients who do not respond appropriately may end up with deep scars, alopecia, and pigmentary changes that are considerably disfiguring, especially for dark-skinned people. Therefore, the goal of treatment is not only to improve the appearance of the skin by minimizing the scarring and preventing further lesions, but also to prevent future complications.
Avoiding sunlight. The primary therapeutic approach is to educate patients regarding exposure to sunlight. Sun-protective measures include the use of high-SPF sunscreen lotions and protective clothing, such as baseball caps without vent holes and wide-brimmed hats.7
Medication options. Current treatment options for DLE include antimalarial agents such as chloroquine, topical and intralesional glucocorticoids, and thalidomide. The use of potent topical steroids may prevent significant scarring and deformity, especially of the face. Common side effects include steroid withdrawal syndrome, perioral dermatitis, steroid acne, and rosacea. All of these side effects can be treated and result in less long-term deformity than untreated DLE (FIGURE 2).
Systemic agents. Widespread disease may require you use systemic agents, such as antimalarials. Chloroquine has long been considered the gold standard in the treatment of DLE.8 Due to the frequency of ocular side effects with chloroquine, hydroxychloroquine is by far the most widely used agent. Hydroxychloroquine at a dose of 6.5 mg/kg/d for 3 months may lead to resolution of lesions for many patients.7 In resistant cases, higher dosages (eg, 400 mg/d) or combinations (eg, hydroxychloroquine 200 mg/d plus quinacrine 50 to 100 mg/d) may be required for months or even years. An ophthalmological evaluation is advisable before starting antimalarial treatment, and you should repeat it at 4- to 6-month intervals during treatment.9 Systemic corticosteroids may be needed to obtain timely initial control, especially for widespread and disfiguring lesions.
When this therapy is inadequate, other treatments for DLE include azathioprine, retinoids, and dapsone. Recent reports confirm the efficacy of thalidomide for cutaneous lupus erythematosus in dosages ranging from 50 to 200 mg/d.10 Twice-daily application of tacrolimus 0.01% has also been shown to be effective in a few clinical trials.11
Surgery. Surgical intervention is useful to remove scarred lesions, and laser therapy has also been effective, especially for lesions with prominent telangiectasias.12
Patient education. Patient education plays an important role in the treatment of DLE. Advising patients on how to avoid the sun and instructing them in the proper use of sunscreens is extremely important. Smoking cessation has also been shown to be beneficial.13
Patients with DLE generally have a favorable prognosis with regards to morbidity and mortality. Because DLE is usually self-limited, the course is most often benign; therefore, early recognition and adequate therapy may prevent clinical complications. Many patients with DLE go on to develop destructive or deforming scarring or pigmentary disturbances,14 especially in the spring and summer months when the sun is the strongest.
FIGURE 2
After treatment
This patient’s outcome
Our patient was treated with topical fluocinolone acetonide 0.025% ointment and oral hydroxychloroquine 200 mg/d for 1 month. The patient responded well to the treatment and the skin lesion regressed perceptibly (FIGURE 2). The treatment was nevertheless continued for another 5 months, and resulted in complete regression of the lesions, with minimal residual scarring.
CORRESPONDENCE
Amor Khachemoune, MD, CWS, Department of Dermatology, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203. E-mail: [email protected]
A 27-year-old woman, otherwise healthy, presented for evaluation of a mildly pruritic eruption of plaques on her face and ears. The eruption started as a few small, scaly red papules on her cheeks and nose about 3 months earlier. The papules slowly expanded to form well-demarcated, rounded, dark brown plaques covered with superficial scale. Similar lesions appeared on the concha of both ears. The older lesions started to regress after 2 months. Complete regression was followed by residual scarring and hyperpigmentation.
Physical examination revealed multiple well-defined, erythematous, hyperpigmented, round plaques covered with adherent scale affecting her nose, cheeks, and the concha of both ears (FIGURE 1). The mucosae and the rest of her skin and adnexa were unaffected. She had no personal or family history of similar skin findings or autoimmune disorders. She also had no history of any drug intake prior to the eruption. Her routine blood tests and urinalysis results were unremarkable, and the serological analysis for antinuclear antibodies had negative results.
A punch biopsy was taken from one of the lesions for histopathology. The epidermal histopathological findings were remarkable for hyperkeratosis, follicular plugging, pigment incontinence, and vacuolization of the basal cell layer. There was a predominantly lymphocytic infiltrate of the subepidermal and perivascular dermal areas.
FIGURE 1
Facial plaques
What is your diagnosis?
How would you treat this patient?
Diagnosis: Discoid lupus erythematosus
Discoid lupus erythematosus (DLE) is an autoimmune inflammatory disorder of the skin that often leads to scarring and alopecia. It may be localized to sun-exposed areas such as the face, ears, and scalp but occasionally is much more extensive, involving the trunk and extremities. Most patients are otherwise healthy, and DLE may be the only clinical finding.
Although its prevalence is not known, DLE is not uncommon. It affects females twice as often as males, and patients fall between the ages of 25 to 45 years, with no racial predilection. While about 15% to 20% of patients with systemic lupus erythematosus (SLE) manifest DLE lesions, only about 5% to 10% of patients with DLE go on to develop SLE.1
Like SLE, DLE is believed to be an autoimmune disorder. Unlike SLE, however, DLE patients do not have similar serologic abnormalities. Skin trauma and ultraviolet light exposure have been reported to induce or exacerbate the lesions of DLE. Sex hormones may also play a role: exacerbation may occur during pregnancy, during menstrual or premenstrual periods, or while taking oral contraceptives. Drugs such as procainamide, hydralazine, isoniazid, diphenylhydantoin, methyldopa, penicillamine, guanides, and lithium may also precipitate DLE lesions.2
DLE usually begins as dull red macules with adherent scales on sun-exposed areas. If the overlying scales are removed, an undersurface of horny plugs is revealed. These plugs fill the follicles and resemble carpet tacks or a cat’s tongue (langue au chat). The macules slowly expand to form large plaques.
Usually only a mild pruritus and tenderness is seen with DLE lesions. As the lesions progress, the scale may thicken and pigmentary changes become evident. The lesions heal with atrophy, scarring, telangiectasias, and changes in pigmentation. Morphological appearance of older lesions may vary from erythematous plaques to hyperkeratotic, dark gray plaques with centrally depressed scars.3 Scalp involvement in DLE results in more sclerotic and depressed scars with subsequent scarring alopecia.4
Differential diagnosis is wide. The differential diagnosis of DLE is extensive and includes actinic keratosis, dermatomyositis, granuloma annulare, granuloma faciale, keratoacanthoma, lichen planus, subacute cutaneous lupus erythematosus, psoriasis, rosacea, sarcoidosis, squamous cell carcinoma, syphilis, and nongenital warts.
Histopathology
Histopathological findings include hyperkeratosis, parakeratosis, follicular plugging, telangiectasias, and atrophy of the epidermis. Liquefaction or hydropic degeneration of the basal layer leads to pigmentary incontinence. A perivascular and perifollicular mononuclear inflammatory cell infiltrate is present in the superficial and deep dermis.5,6
Direct immunofluorescence demonstrates immunoglobulins and complement deposits at the dermoepidermal junction.5 This test was not recommended for this patient, as the diagnosis of DLE had already been confirmed on clinical and histopathologic grounds.
Workup: Exam, biopsy
In addition to a routine history and physical examination, the workup for DLE should include a complete blood count, antinuclear antibody levels, anti-Ro, anti-La, hepatic and renal function tests, and urinalysis. Consider a diagnosis of SLE by using the American College of Rheumatology criteria. A biopsy for histopathology of a fresh lesion or a biopsy for immunofluorescence of an old lesion can confirm the diagnosis.
Therapeutic options are varied
Effective early therapies for DLE are available, but patients who do not respond appropriately may end up with deep scars, alopecia, and pigmentary changes that are considerably disfiguring, especially for dark-skinned people. Therefore, the goal of treatment is not only to improve the appearance of the skin by minimizing the scarring and preventing further lesions, but also to prevent future complications.
Avoiding sunlight. The primary therapeutic approach is to educate patients regarding exposure to sunlight. Sun-protective measures include the use of high-SPF sunscreen lotions and protective clothing, such as baseball caps without vent holes and wide-brimmed hats.7
Medication options. Current treatment options for DLE include antimalarial agents such as chloroquine, topical and intralesional glucocorticoids, and thalidomide. The use of potent topical steroids may prevent significant scarring and deformity, especially of the face. Common side effects include steroid withdrawal syndrome, perioral dermatitis, steroid acne, and rosacea. All of these side effects can be treated and result in less long-term deformity than untreated DLE (FIGURE 2).
Systemic agents. Widespread disease may require you use systemic agents, such as antimalarials. Chloroquine has long been considered the gold standard in the treatment of DLE.8 Due to the frequency of ocular side effects with chloroquine, hydroxychloroquine is by far the most widely used agent. Hydroxychloroquine at a dose of 6.5 mg/kg/d for 3 months may lead to resolution of lesions for many patients.7 In resistant cases, higher dosages (eg, 400 mg/d) or combinations (eg, hydroxychloroquine 200 mg/d plus quinacrine 50 to 100 mg/d) may be required for months or even years. An ophthalmological evaluation is advisable before starting antimalarial treatment, and you should repeat it at 4- to 6-month intervals during treatment.9 Systemic corticosteroids may be needed to obtain timely initial control, especially for widespread and disfiguring lesions.
When this therapy is inadequate, other treatments for DLE include azathioprine, retinoids, and dapsone. Recent reports confirm the efficacy of thalidomide for cutaneous lupus erythematosus in dosages ranging from 50 to 200 mg/d.10 Twice-daily application of tacrolimus 0.01% has also been shown to be effective in a few clinical trials.11
Surgery. Surgical intervention is useful to remove scarred lesions, and laser therapy has also been effective, especially for lesions with prominent telangiectasias.12
Patient education. Patient education plays an important role in the treatment of DLE. Advising patients on how to avoid the sun and instructing them in the proper use of sunscreens is extremely important. Smoking cessation has also been shown to be beneficial.13
Patients with DLE generally have a favorable prognosis with regards to morbidity and mortality. Because DLE is usually self-limited, the course is most often benign; therefore, early recognition and adequate therapy may prevent clinical complications. Many patients with DLE go on to develop destructive or deforming scarring or pigmentary disturbances,14 especially in the spring and summer months when the sun is the strongest.
FIGURE 2
After treatment
This patient’s outcome
Our patient was treated with topical fluocinolone acetonide 0.025% ointment and oral hydroxychloroquine 200 mg/d for 1 month. The patient responded well to the treatment and the skin lesion regressed perceptibly (FIGURE 2). The treatment was nevertheless continued for another 5 months, and resulted in complete regression of the lesions, with minimal residual scarring.
CORRESPONDENCE
Amor Khachemoune, MD, CWS, Department of Dermatology, 450 Clarkson Avenue Box 46, Brooklyn, NY 11203. E-mail: [email protected]
1. Callen JP. Systemic lupus erythematosus in patients with chronic cutaneous (discoid) lupus erythematosus. Clinical and laboratory findings in seventeen patients. J Am Acad Dermatol 1985;12(2 Pt 1):278-288.
2. Hess E. Drug-related lupus. N Engl J Med 1988;318:1460-1462.
3. Callen JP, Fowler JF, Kulick KB. Serologic and clinical features of patients with discoid lupus erythematosus: relationship of antibodies to single-stranded deoxyribonucleic acid and of other antinuclear antibody subsets to clinical manifestations. J Am Acad Dermatol 1985;13(5 Pt 1):748-755.
4. Wilson CL, Burge SM, Dean D, Dawber RP. Scarring alopecia in discoid lupus erythematosus. Br J Dermatol 1992;126:307-314.
5. Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin 2002;20:373-385, v.
6. Biesla I, et al. Histopathologic findings in cutaneous lupus erythematosus. Arch Dermatol 1994;130:54-58.
7. Callen JP. Treatment of cutaneous lesions in patients with lupus erythematosus. Dermatol Clin 1994;12:201-206.
8. Goldman L, Cole DP, Preston RH. Chloroquine diphosphate in treatment of discoid lupus erythematosus. J Am Med Assoc 1953;152:1428-1429.
9. Rynes RI. Ophthalmologic safety of long-term hydroxychloroquine sulfate treatment. Am J Med 1983;75:35-39.
10. Kyriakis KP, Kontochristopoulos GJ, Panteleos DN. Experience with low-dose thalidomide therapy in chronic discoid lupus erythematosus. Int J Dermatol 2000;39:218-222.
11. Heffernan MP, Nelson MM, Smith DI, Chung JH. 0.1% tacrolimus ointment in the treatment of discoid lupus erythematosus. Arch Dermatol 2005;141:1170-1171.
12. Nunez M, Boixeda P, Miralles ES, et al. Pulsed dye laser treatment of telangiectatic chronic erythema of cutaneous lupus erythematosus. Arch Dermatol 1996;132:354-355.
13. Miot HA, Bartoli Miot LD, Haddad GR. Association between discoid lupus erythematosus and cigarette smoking. Dermatology 2005;211:118-122.
14. de Berker D, Dissaneyeka M, Burge S. The sequelae of chronic cutaneous lupus erythematosus. Lupus 1992;1:181-186.
1. Callen JP. Systemic lupus erythematosus in patients with chronic cutaneous (discoid) lupus erythematosus. Clinical and laboratory findings in seventeen patients. J Am Acad Dermatol 1985;12(2 Pt 1):278-288.
2. Hess E. Drug-related lupus. N Engl J Med 1988;318:1460-1462.
3. Callen JP, Fowler JF, Kulick KB. Serologic and clinical features of patients with discoid lupus erythematosus: relationship of antibodies to single-stranded deoxyribonucleic acid and of other antinuclear antibody subsets to clinical manifestations. J Am Acad Dermatol 1985;13(5 Pt 1):748-755.
4. Wilson CL, Burge SM, Dean D, Dawber RP. Scarring alopecia in discoid lupus erythematosus. Br J Dermatol 1992;126:307-314.
5. Patel P, Werth V. Cutaneous lupus erythematosus: a review. Dermatol Clin 2002;20:373-385, v.
6. Biesla I, et al. Histopathologic findings in cutaneous lupus erythematosus. Arch Dermatol 1994;130:54-58.
7. Callen JP. Treatment of cutaneous lesions in patients with lupus erythematosus. Dermatol Clin 1994;12:201-206.
8. Goldman L, Cole DP, Preston RH. Chloroquine diphosphate in treatment of discoid lupus erythematosus. J Am Med Assoc 1953;152:1428-1429.
9. Rynes RI. Ophthalmologic safety of long-term hydroxychloroquine sulfate treatment. Am J Med 1983;75:35-39.
10. Kyriakis KP, Kontochristopoulos GJ, Panteleos DN. Experience with low-dose thalidomide therapy in chronic discoid lupus erythematosus. Int J Dermatol 2000;39:218-222.
11. Heffernan MP, Nelson MM, Smith DI, Chung JH. 0.1% tacrolimus ointment in the treatment of discoid lupus erythematosus. Arch Dermatol 2005;141:1170-1171.
12. Nunez M, Boixeda P, Miralles ES, et al. Pulsed dye laser treatment of telangiectatic chronic erythema of cutaneous lupus erythematosus. Arch Dermatol 1996;132:354-355.
13. Miot HA, Bartoli Miot LD, Haddad GR. Association between discoid lupus erythematosus and cigarette smoking. Dermatology 2005;211:118-122.
14. de Berker D, Dissaneyeka M, Burge S. The sequelae of chronic cutaneous lupus erythematosus. Lupus 1992;1:181-186.
A man with a pigmented growth on his chest
A 50-year-old man came to the office with a 7-year history of a slowly growing lesion on his chest. The patient, an outdoor worker with excessive sun exposure for 20 years, had skin phototype IV (burns minimally, always tans well to moderately brown). He reported that this lesion has never healed, has bled on occasion, and that regular wound care over several months was ineffective. He had suffered no trauma, nor had he applied any caustic products to this area. The patient was otherwise healthy and was not taking any medications. His family history was not contributory.
Physical examination revealed a large plaque about 4.5 by 3 cm with irregular borders (FIGURE 1). It seemed to have been expanding centrifugally for a long time, with areas of skipping, individual nodules, and papules. Close exam showed central ulcerations and crusting on scattered papules and nodules (FIGURE 2). The borders were rolled up and had a somewhat pearly appearance with erythema and telangiectasia. The area immediately surrounding the plaque was also remarkable for faint erythema. The central part of the lesion was atrophic with pinkish discoloration; on finger pinching of the central, the skin seemed to be sclerotic. The rest of the skin, mucosal, adnexal examination and review of systems was unremarkable. No lymphadenopathy was present. A skin biopsy was done to confirm the clinical impression.
FIGURE 1
Pigmented plaque on chest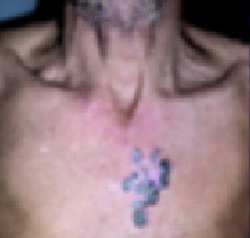
FIGURE 2
Close-up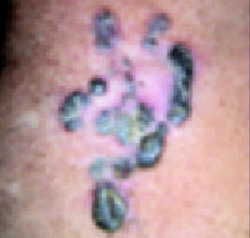
What is your diagnosis?
Diagnosis: Basal cell carcinoma, pigmented subtype
Basal cell carcinoma (BCC) is the most common cancer among Caucasians1 and accounts for approximately 75% of all skin cancers. Resultant mortality is very low, but it may cause destruction to local and surrounding structures. Metastasis to lymph nodes and other organs and subsequent death have been reported with BCC.2,3 Other names for BCC are basalioma, basal cell epithelioma, rodent ulcer, and Jacobs’ ulcer.
Clinical presentations: Location, subtypes, risk of recurrence
The face (particularly the nose) is the most common site for BCC. A small percentage of BCCs occur on the trunk; it is rare on areas not exposed to the sun, such as the penis, vulva, perianal areas and axilla. Many subtypes of BCCs exist, including nodular, superficial, cystic, micronodular, morpheaform, and pigmented. In addition to features seen in lesions of nodular BCC, the pigmented subtype contains increased brown or black pigment, and it is seen more commonly in persons with dark skin.
Histology and new investigations
BCC arises from the basal layer of the epidermis. There are many histologic subtypes. The basaloid cells form tumor aggregates or nests of varying sizes. Cells tend to align more densely in a palisade pattern at the periphery of these nests. In the morpheaform subtype, the cells are embedded in fibrous stroma.
In a recent article, Goldberg et al4 used histopathology and special staining to show that BCC lesions had melanin pigment (positive for Fontana-Masson stain and negative for Perl’s stain) within nests of tumor cells. The authors concluded that speckled pigmentation of a basal cell carcinoma is a distinguishing feature, which may be useful in differentiating this tumor from other discrete skin tumors.
More recently, endothelins (ETs) have been implicated as participating in the pigmentation process of BCC. Enhanced ET-1 expression in pigmented BCC plays an important role in the hyperpigmentation of this tumor.5
Risk factors for BCC: Environment, genetics
Solar radiation is the chief environmental cause of BCC. Therapeutic radiation, such as PUVA for psoriasis or radiation for the treatment of acne or tinea capitis may result in BCC many years after exposure. Genetic characteristics such as fair skin, light-colored eyes, and red hair are also important risk factors. Another reported risk factor is chronic arsenic exposure, which may result from ingestion of contaminated water or seafood.
Basal cell nevus syndrome is an autosomal dominant genetic disorder with multiple characteristic clinical features. These features include the development of multiple BCCs at a relatively young age, macrocephaly, frontal bossing, hypertelorism, bifid ribs, palmar and plantar pitting, and bone cysts in the mandible. These patients have a mutation in the tumor suppressor gene patched (PTCH) on chromosome 9.6 Xeroderma pigmentosum and other rare genetic diseases also increase the likelihood of BCC tumors. Risk of BCC recurrence is related to tumor location and size, histologic type, and treatment modality and history of UV exposure.
Differential diagnosis for pigmented BCC
- Malignant melanoma
- Melanocytic nevus
- Spitz nevus
- Pigmented seborrheic keratosis
- Pigmented dermatofibroma
- Squamous cell carcinoma
- Pigmented Bowen’s disease (squamous cell carcinoma in situ)
- Keratoacanthoma
- Trichoepithelioma
- Sebaceous hyperplasia
- Fibrous papule of the nose
Treatment: Local destruction, photodynamic therapy, immune modulators
Treatment options available for BCC depend on characteristics of the tumor type, location, individual patient, and economic resources.
Cryosurgery, electrodessication and curettage, CO2 laser destruction, surgical excision, Mohs micrographic surgery, radiation therapy, topical therapy such as 5-fluorouracil (5-FU) and imiquimod (Aldara), intralesional interferon, and photodynamic therapy are used to treat BCC.
Older treatments for low-risk BCCs in cosmetically less significant areas
Cryosurgery induces cytotoxicity by production of extracellular and intracellular ice crystals. It is rapid and cost-effective, and demonstrates high cure rates.
Electrosurgery can be used for superficial and nodular BCC on the trunk and extremities.
Surgical excision is an effective method for BCCs located on low-risk areas without aggressive histologic features.
Radiation therapy is an option for the treatment of both primary and recurrent BCCs.
Topical chemotherapeutic agent 5-fluorouracil (Efudex, Carac, Fluoroplex) is also effective against BCC.
Interferon (interferon alpha 2b) has also been used as intralesional injections to treat superficial and nodular BCCs.
Treatment for high-risk tumors
Mohs micrographic surgery is currently the standard of treatment for high-risk BCCs and BCCs located in cosmetically sensitive locations such as the nasolabial fold and periorbital areas. It has a higher cure rate than other modalities.
Relatively new methods
Imiquimod is used for primary superficial BCC (not on head or neck) in adults with normal immune systems. It is used for tumors 2 cm or smaller in diameter on certain areas of the body. Imiquimod treatment is indicated only when surgical methods are not appropriate.7
Photodynamic therapy includes the use of various intravenous and topical photosensitizers—ie, intravenous verteporfin, topical 5-aminolevulinic acid (ALA), topical methyl aminolevulinate (mALA)—combined with different sources of visible light. Photodynamic therapy is being used for some BCCs but is not recommended for pigmented BCC.8
Given the ill-defined nature of this lesion, and its size, we recommended surgical excision using either regular excision with staining or marking of margins, or Mohs micrographic surgery. Unfortunately, the patient was lost to follow-up.
Acknowledgments
The author wants to thank Dr. Shahbaz A. Janjua with his assistance in the preparation of this manuscript.
CORRESPONDING AUTHOR
Amor Khachemoune, MD, CWS, SUNY Downstate Medical Center, Department of Dermatology Box 46, 450 Clarkson Ave, Brooklyn, NY 11203. E-mail: [email protected]
1. Marks R. An overview of skin cancers: incidence and causation. Cancer 1995;75:607-612.
2. Robinson JK, Dahiya M. Basal cell carcinoma with pulmonary and lymph node metastasis causing death. Arch Dermatol 2003;139:643-648.
3. Tilli CM, Van Steensel MA, Krekels GA, Neumann HA, Ramaekers FC. Molecular aetiology and pathogenesis of basal cell carcinoma. Br J Dermatol 2005;152:1108-1124.
4. Goldberg LH, Friedman RH, Silapunt S. Pigmented speckling as a sign of basal cell carcinoma. Dermatol Surg 2004;30:1553-1555.
5. Lan CC, Wu CS, Cheng CM, Yu CL, Chen GS, Yu HS. Pigmentation in basal cell carcinoma involves enhanced endothelin-1 expression. Exp Dermatol 2005;14:528-534.
6. Blyumin ML, Khachemoune A. Self-assessment examination. Man with multiple papulonodules on the back. Identification no. 804-203. J Am Acad Dermatol 2004;50:498-494.
7. Oldfield V, Keating GM, Perry CM. Imiquimod: in superficial basal cell carcinoma. Am J Clin Dermatol 2005;6:195-200.
8. Kaviani A, Ataie-Fashtami L, Fateh M, Sheikhbahaee N, Ghodsi M, Zand N, Djavid GE. Photodynamic therapy of head and neck basal cell carcinoma according to different clinicopathologic features. Lasers Surg Med 2005;36:377-382.
A 50-year-old man came to the office with a 7-year history of a slowly growing lesion on his chest. The patient, an outdoor worker with excessive sun exposure for 20 years, had skin phototype IV (burns minimally, always tans well to moderately brown). He reported that this lesion has never healed, has bled on occasion, and that regular wound care over several months was ineffective. He had suffered no trauma, nor had he applied any caustic products to this area. The patient was otherwise healthy and was not taking any medications. His family history was not contributory.
Physical examination revealed a large plaque about 4.5 by 3 cm with irregular borders (FIGURE 1). It seemed to have been expanding centrifugally for a long time, with areas of skipping, individual nodules, and papules. Close exam showed central ulcerations and crusting on scattered papules and nodules (FIGURE 2). The borders were rolled up and had a somewhat pearly appearance with erythema and telangiectasia. The area immediately surrounding the plaque was also remarkable for faint erythema. The central part of the lesion was atrophic with pinkish discoloration; on finger pinching of the central, the skin seemed to be sclerotic. The rest of the skin, mucosal, adnexal examination and review of systems was unremarkable. No lymphadenopathy was present. A skin biopsy was done to confirm the clinical impression.
FIGURE 1
Pigmented plaque on chest
FIGURE 2
Close-up
What is your diagnosis?
Diagnosis: Basal cell carcinoma, pigmented subtype
Basal cell carcinoma (BCC) is the most common cancer among Caucasians1 and accounts for approximately 75% of all skin cancers. Resultant mortality is very low, but it may cause destruction to local and surrounding structures. Metastasis to lymph nodes and other organs and subsequent death have been reported with BCC.2,3 Other names for BCC are basalioma, basal cell epithelioma, rodent ulcer, and Jacobs’ ulcer.
Clinical presentations: Location, subtypes, risk of recurrence
The face (particularly the nose) is the most common site for BCC. A small percentage of BCCs occur on the trunk; it is rare on areas not exposed to the sun, such as the penis, vulva, perianal areas and axilla. Many subtypes of BCCs exist, including nodular, superficial, cystic, micronodular, morpheaform, and pigmented. In addition to features seen in lesions of nodular BCC, the pigmented subtype contains increased brown or black pigment, and it is seen more commonly in persons with dark skin.
Histology and new investigations
BCC arises from the basal layer of the epidermis. There are many histologic subtypes. The basaloid cells form tumor aggregates or nests of varying sizes. Cells tend to align more densely in a palisade pattern at the periphery of these nests. In the morpheaform subtype, the cells are embedded in fibrous stroma.
In a recent article, Goldberg et al4 used histopathology and special staining to show that BCC lesions had melanin pigment (positive for Fontana-Masson stain and negative for Perl’s stain) within nests of tumor cells. The authors concluded that speckled pigmentation of a basal cell carcinoma is a distinguishing feature, which may be useful in differentiating this tumor from other discrete skin tumors.
More recently, endothelins (ETs) have been implicated as participating in the pigmentation process of BCC. Enhanced ET-1 expression in pigmented BCC plays an important role in the hyperpigmentation of this tumor.5
Risk factors for BCC: Environment, genetics
Solar radiation is the chief environmental cause of BCC. Therapeutic radiation, such as PUVA for psoriasis or radiation for the treatment of acne or tinea capitis may result in BCC many years after exposure. Genetic characteristics such as fair skin, light-colored eyes, and red hair are also important risk factors. Another reported risk factor is chronic arsenic exposure, which may result from ingestion of contaminated water or seafood.
Basal cell nevus syndrome is an autosomal dominant genetic disorder with multiple characteristic clinical features. These features include the development of multiple BCCs at a relatively young age, macrocephaly, frontal bossing, hypertelorism, bifid ribs, palmar and plantar pitting, and bone cysts in the mandible. These patients have a mutation in the tumor suppressor gene patched (PTCH) on chromosome 9.6 Xeroderma pigmentosum and other rare genetic diseases also increase the likelihood of BCC tumors. Risk of BCC recurrence is related to tumor location and size, histologic type, and treatment modality and history of UV exposure.
Differential diagnosis for pigmented BCC
- Malignant melanoma
- Melanocytic nevus
- Spitz nevus
- Pigmented seborrheic keratosis
- Pigmented dermatofibroma
- Squamous cell carcinoma
- Pigmented Bowen’s disease (squamous cell carcinoma in situ)
- Keratoacanthoma
- Trichoepithelioma
- Sebaceous hyperplasia
- Fibrous papule of the nose
Treatment: Local destruction, photodynamic therapy, immune modulators
Treatment options available for BCC depend on characteristics of the tumor type, location, individual patient, and economic resources.
Cryosurgery, electrodessication and curettage, CO2 laser destruction, surgical excision, Mohs micrographic surgery, radiation therapy, topical therapy such as 5-fluorouracil (5-FU) and imiquimod (Aldara), intralesional interferon, and photodynamic therapy are used to treat BCC.
Older treatments for low-risk BCCs in cosmetically less significant areas
Cryosurgery induces cytotoxicity by production of extracellular and intracellular ice crystals. It is rapid and cost-effective, and demonstrates high cure rates.
Electrosurgery can be used for superficial and nodular BCC on the trunk and extremities.
Surgical excision is an effective method for BCCs located on low-risk areas without aggressive histologic features.
Radiation therapy is an option for the treatment of both primary and recurrent BCCs.
Topical chemotherapeutic agent 5-fluorouracil (Efudex, Carac, Fluoroplex) is also effective against BCC.
Interferon (interferon alpha 2b) has also been used as intralesional injections to treat superficial and nodular BCCs.
Treatment for high-risk tumors
Mohs micrographic surgery is currently the standard of treatment for high-risk BCCs and BCCs located in cosmetically sensitive locations such as the nasolabial fold and periorbital areas. It has a higher cure rate than other modalities.
Relatively new methods
Imiquimod is used for primary superficial BCC (not on head or neck) in adults with normal immune systems. It is used for tumors 2 cm or smaller in diameter on certain areas of the body. Imiquimod treatment is indicated only when surgical methods are not appropriate.7
Photodynamic therapy includes the use of various intravenous and topical photosensitizers—ie, intravenous verteporfin, topical 5-aminolevulinic acid (ALA), topical methyl aminolevulinate (mALA)—combined with different sources of visible light. Photodynamic therapy is being used for some BCCs but is not recommended for pigmented BCC.8
Given the ill-defined nature of this lesion, and its size, we recommended surgical excision using either regular excision with staining or marking of margins, or Mohs micrographic surgery. Unfortunately, the patient was lost to follow-up.
Acknowledgments
The author wants to thank Dr. Shahbaz A. Janjua with his assistance in the preparation of this manuscript.
CORRESPONDING AUTHOR
Amor Khachemoune, MD, CWS, SUNY Downstate Medical Center, Department of Dermatology Box 46, 450 Clarkson Ave, Brooklyn, NY 11203. E-mail: [email protected]
A 50-year-old man came to the office with a 7-year history of a slowly growing lesion on his chest. The patient, an outdoor worker with excessive sun exposure for 20 years, had skin phototype IV (burns minimally, always tans well to moderately brown). He reported that this lesion has never healed, has bled on occasion, and that regular wound care over several months was ineffective. He had suffered no trauma, nor had he applied any caustic products to this area. The patient was otherwise healthy and was not taking any medications. His family history was not contributory.
Physical examination revealed a large plaque about 4.5 by 3 cm with irregular borders (FIGURE 1). It seemed to have been expanding centrifugally for a long time, with areas of skipping, individual nodules, and papules. Close exam showed central ulcerations and crusting on scattered papules and nodules (FIGURE 2). The borders were rolled up and had a somewhat pearly appearance with erythema and telangiectasia. The area immediately surrounding the plaque was also remarkable for faint erythema. The central part of the lesion was atrophic with pinkish discoloration; on finger pinching of the central, the skin seemed to be sclerotic. The rest of the skin, mucosal, adnexal examination and review of systems was unremarkable. No lymphadenopathy was present. A skin biopsy was done to confirm the clinical impression.
FIGURE 1
Pigmented plaque on chest
FIGURE 2
Close-up
What is your diagnosis?
Diagnosis: Basal cell carcinoma, pigmented subtype
Basal cell carcinoma (BCC) is the most common cancer among Caucasians1 and accounts for approximately 75% of all skin cancers. Resultant mortality is very low, but it may cause destruction to local and surrounding structures. Metastasis to lymph nodes and other organs and subsequent death have been reported with BCC.2,3 Other names for BCC are basalioma, basal cell epithelioma, rodent ulcer, and Jacobs’ ulcer.
Clinical presentations: Location, subtypes, risk of recurrence
The face (particularly the nose) is the most common site for BCC. A small percentage of BCCs occur on the trunk; it is rare on areas not exposed to the sun, such as the penis, vulva, perianal areas and axilla. Many subtypes of BCCs exist, including nodular, superficial, cystic, micronodular, morpheaform, and pigmented. In addition to features seen in lesions of nodular BCC, the pigmented subtype contains increased brown or black pigment, and it is seen more commonly in persons with dark skin.
Histology and new investigations
BCC arises from the basal layer of the epidermis. There are many histologic subtypes. The basaloid cells form tumor aggregates or nests of varying sizes. Cells tend to align more densely in a palisade pattern at the periphery of these nests. In the morpheaform subtype, the cells are embedded in fibrous stroma.
In a recent article, Goldberg et al4 used histopathology and special staining to show that BCC lesions had melanin pigment (positive for Fontana-Masson stain and negative for Perl’s stain) within nests of tumor cells. The authors concluded that speckled pigmentation of a basal cell carcinoma is a distinguishing feature, which may be useful in differentiating this tumor from other discrete skin tumors.
More recently, endothelins (ETs) have been implicated as participating in the pigmentation process of BCC. Enhanced ET-1 expression in pigmented BCC plays an important role in the hyperpigmentation of this tumor.5
Risk factors for BCC: Environment, genetics
Solar radiation is the chief environmental cause of BCC. Therapeutic radiation, such as PUVA for psoriasis or radiation for the treatment of acne or tinea capitis may result in BCC many years after exposure. Genetic characteristics such as fair skin, light-colored eyes, and red hair are also important risk factors. Another reported risk factor is chronic arsenic exposure, which may result from ingestion of contaminated water or seafood.
Basal cell nevus syndrome is an autosomal dominant genetic disorder with multiple characteristic clinical features. These features include the development of multiple BCCs at a relatively young age, macrocephaly, frontal bossing, hypertelorism, bifid ribs, palmar and plantar pitting, and bone cysts in the mandible. These patients have a mutation in the tumor suppressor gene patched (PTCH) on chromosome 9.6 Xeroderma pigmentosum and other rare genetic diseases also increase the likelihood of BCC tumors. Risk of BCC recurrence is related to tumor location and size, histologic type, and treatment modality and history of UV exposure.
Differential diagnosis for pigmented BCC
- Malignant melanoma
- Melanocytic nevus
- Spitz nevus
- Pigmented seborrheic keratosis
- Pigmented dermatofibroma
- Squamous cell carcinoma
- Pigmented Bowen’s disease (squamous cell carcinoma in situ)
- Keratoacanthoma
- Trichoepithelioma
- Sebaceous hyperplasia
- Fibrous papule of the nose
Treatment: Local destruction, photodynamic therapy, immune modulators
Treatment options available for BCC depend on characteristics of the tumor type, location, individual patient, and economic resources.
Cryosurgery, electrodessication and curettage, CO2 laser destruction, surgical excision, Mohs micrographic surgery, radiation therapy, topical therapy such as 5-fluorouracil (5-FU) and imiquimod (Aldara), intralesional interferon, and photodynamic therapy are used to treat BCC.
Older treatments for low-risk BCCs in cosmetically less significant areas
Cryosurgery induces cytotoxicity by production of extracellular and intracellular ice crystals. It is rapid and cost-effective, and demonstrates high cure rates.
Electrosurgery can be used for superficial and nodular BCC on the trunk and extremities.
Surgical excision is an effective method for BCCs located on low-risk areas without aggressive histologic features.
Radiation therapy is an option for the treatment of both primary and recurrent BCCs.
Topical chemotherapeutic agent 5-fluorouracil (Efudex, Carac, Fluoroplex) is also effective against BCC.
Interferon (interferon alpha 2b) has also been used as intralesional injections to treat superficial and nodular BCCs.
Treatment for high-risk tumors
Mohs micrographic surgery is currently the standard of treatment for high-risk BCCs and BCCs located in cosmetically sensitive locations such as the nasolabial fold and periorbital areas. It has a higher cure rate than other modalities.
Relatively new methods
Imiquimod is used for primary superficial BCC (not on head or neck) in adults with normal immune systems. It is used for tumors 2 cm or smaller in diameter on certain areas of the body. Imiquimod treatment is indicated only when surgical methods are not appropriate.7
Photodynamic therapy includes the use of various intravenous and topical photosensitizers—ie, intravenous verteporfin, topical 5-aminolevulinic acid (ALA), topical methyl aminolevulinate (mALA)—combined with different sources of visible light. Photodynamic therapy is being used for some BCCs but is not recommended for pigmented BCC.8
Given the ill-defined nature of this lesion, and its size, we recommended surgical excision using either regular excision with staining or marking of margins, or Mohs micrographic surgery. Unfortunately, the patient was lost to follow-up.
Acknowledgments
The author wants to thank Dr. Shahbaz A. Janjua with his assistance in the preparation of this manuscript.
CORRESPONDING AUTHOR
Amor Khachemoune, MD, CWS, SUNY Downstate Medical Center, Department of Dermatology Box 46, 450 Clarkson Ave, Brooklyn, NY 11203. E-mail: [email protected]
1. Marks R. An overview of skin cancers: incidence and causation. Cancer 1995;75:607-612.
2. Robinson JK, Dahiya M. Basal cell carcinoma with pulmonary and lymph node metastasis causing death. Arch Dermatol 2003;139:643-648.
3. Tilli CM, Van Steensel MA, Krekels GA, Neumann HA, Ramaekers FC. Molecular aetiology and pathogenesis of basal cell carcinoma. Br J Dermatol 2005;152:1108-1124.
4. Goldberg LH, Friedman RH, Silapunt S. Pigmented speckling as a sign of basal cell carcinoma. Dermatol Surg 2004;30:1553-1555.
5. Lan CC, Wu CS, Cheng CM, Yu CL, Chen GS, Yu HS. Pigmentation in basal cell carcinoma involves enhanced endothelin-1 expression. Exp Dermatol 2005;14:528-534.
6. Blyumin ML, Khachemoune A. Self-assessment examination. Man with multiple papulonodules on the back. Identification no. 804-203. J Am Acad Dermatol 2004;50:498-494.
7. Oldfield V, Keating GM, Perry CM. Imiquimod: in superficial basal cell carcinoma. Am J Clin Dermatol 2005;6:195-200.
8. Kaviani A, Ataie-Fashtami L, Fateh M, Sheikhbahaee N, Ghodsi M, Zand N, Djavid GE. Photodynamic therapy of head and neck basal cell carcinoma according to different clinicopathologic features. Lasers Surg Med 2005;36:377-382.
1. Marks R. An overview of skin cancers: incidence and causation. Cancer 1995;75:607-612.
2. Robinson JK, Dahiya M. Basal cell carcinoma with pulmonary and lymph node metastasis causing death. Arch Dermatol 2003;139:643-648.
3. Tilli CM, Van Steensel MA, Krekels GA, Neumann HA, Ramaekers FC. Molecular aetiology and pathogenesis of basal cell carcinoma. Br J Dermatol 2005;152:1108-1124.
4. Goldberg LH, Friedman RH, Silapunt S. Pigmented speckling as a sign of basal cell carcinoma. Dermatol Surg 2004;30:1553-1555.
5. Lan CC, Wu CS, Cheng CM, Yu CL, Chen GS, Yu HS. Pigmentation in basal cell carcinoma involves enhanced endothelin-1 expression. Exp Dermatol 2005;14:528-534.
6. Blyumin ML, Khachemoune A. Self-assessment examination. Man with multiple papulonodules on the back. Identification no. 804-203. J Am Acad Dermatol 2004;50:498-494.
7. Oldfield V, Keating GM, Perry CM. Imiquimod: in superficial basal cell carcinoma. Am J Clin Dermatol 2005;6:195-200.
8. Kaviani A, Ataie-Fashtami L, Fateh M, Sheikhbahaee N, Ghodsi M, Zand N, Djavid GE. Photodynamic therapy of head and neck basal cell carcinoma according to different clinicopathologic features. Lasers Surg Med 2005;36:377-382.
A young girl with scaly skin plaques
An otherwise healthy 12-year-old girl came to the office with a 1-year history of a symmetric, generalized scaly eruption. These skin plaques did not itch; she had no recent history of sore throat. She also had no personal or family history of atopy or any similar eruption. She mentioned that the appearance of her skin and the “flaking” was making her very self-conscious and she wanted to have some intervention to make the skin clear.
The patient had multiple red, scaly, thick plaques on her back and the extensor surfaces of her elbows, knees, legs, and forearms (FIGURES 1 AND 2). There were no scalp, mucosal, or nail involvements. Rheumatologic examination and review of systems were unremarkable.
FIGURE 1
Plaques on the arm…
FIGURE 2
…and on the back
What is your diagnosis?
How would you manage this case?
Diagnosis: Chronic plaque psoriasis
Psoriasis is a noninfectious inflammatory skin disorder characterized by well-defined erythematous plaques that bear large, adherent silvery scales. It can appear at any age, but 75% of patients have an onset before the age of 40 years.1
Psoriasis was once considered a hyperproliferative disorder, but is now recognized as an autoimmune phenomenon involving activation of T-cells. As a result, new immunosuppressive agents have been added to the list of traditional therapies, opening a new chapter of immunomodulatory therapy.
Clinical presentations of psoriasis
In its classic presentation, psoriasis does not pose a diagnosis challenge to most clinicians—it presents as a sharply demarcated erythematous plaques with silvery white scales. Although many classification systems exist, a concise classification is included in TABLE 1. Skin conditions to be considered for differential diagnosis are summarized in TABLE 2. The US Food and Drug Administration defines “severe” psoriasis as involving more than 20% of body surface area.2
TABLE 1
Classification of psoriasis
| Chronic plaque psoriasis | Symmetrical plaques up to 20 cm in diameter, with a predilection for the elbows, knees, presacrum, scalp, hands, and feet |
| 5%–30% of patients develop a seronegative arthropathy | |
| Guttate psoriasis | Numerous small papules and plaques over the upper trunk and proximal extremities |
| Most common form of psoriasis in children, and may be triggered by any streptococcal infection including streptococcal perianal dermatitis | |
| Spontaneous remission is the rule | |
| Generalized pustular and erythrodermic psoriasis | Uncommon variants associated with high morbidity that may be fatal |
TABLE 2
Differential diagnosis
| Fungal infections |
| Squamous cell carcinoma in situ |
| Cutaneous T-cell lymphoma |
| Discoid eczema |
| Seborrhoeic eczema |
| Pityriasis rosea |
| Secondary syphilis |
| Hypertrophic lichen planus |
| Nummular dermatitis |
Pathogenesis of psoriasis
Psoriasis is a polygenic disorder. Susceptibility is determined by a large number of genes, each with a low penetrance. Important genetic associations are with HLA-Cw6, HLA-B27, and the genes PSOR-S1 and PSOR-S2 on chromosome 6 and 17, respectively.
Several triggering factors are identified, of which streptococcal antigens and certain drugs seem important, but the specific antigens are still unknown. The end result is activation of T-cells, overexpression of cytokines, and an inflammatory response.
Review of the classic treatments for psoriasis
Topical therapy
Topical therapy is indicated when psoriasis is limited to less than 20% of the body surface. Potent class I and II topical corticosteroids are the most widely used treatment for mild disease. After plaque clearance they can be given intermittently for maintenance.
The vitamin D3 derivative calcipotriene (Dovonex) is another first-line agent. Tazarotene (Tazorac), a topical retinoid prodrug, is a second-line agent used as monotherapy or in combination. Many combined regimens use topical corticosteroids, calcipotriene, and tazarotene. Coal tar—different concentrations in liquid form—is useful in treating extensive areas of the body and scalp psoriasis.
Phototherapy
Failure of topical therapy or extensive disease are indications for phototherapy or systemic medications. The trend is to use phototherapy in the form of narrowband UVB, which has proven more effective than broadband UVB and to have fewer adverse effects than psoralen UVA therapy (PUVA).3 Other light sources for home use are being developed.
Systemic agents
Methotrexate, given as a single weekly dose or in divided doses, has been used for more than 30 years; it inhibits the enzyme dihydrofolate reductase. An alternative immunosuppressive agent is cyclosporine. These 2 agents have high efficacy, but due to potential adverse effects they require careful patient selection and close follow-up.
Acitretin (Soriatane), an oral retinoid, is the treatment of choice for generalized pustular and erythrodermic psoriasis. It is also used in chronic plaque psoriasis, often in combination with phototherapy, which has a synergistic effect.New treatments
Our understanding of the immunopathogenesis of psoriasis has led to the development of therapies designed specifically to interfere with T-cell activation and effector functions. Three new immunomodulatory biologics are FDA-approved for the treatment of moderate to severe psoriasis. Typical cost of this therapy is more than $1000/month.
Anti-TNF-α strategies
Etanercept (Enbrel) is an antibody against the cytokine tumor necrosis factor alpha (TNF-α). It is self-administered at 25 mg to 50 mg subcutaneously twice weekly. Studies with 50-mg injections have shown a 75% clinical improvement in 49% of patients at 12 weeks and 59% of patients at 24 weeks.4
The most common side effect is reaction at the injection site. It was reported to produce dramatic remission of psoriatic arthritis and prevent radiographic progression of the disease.4-5 Due to raised concern about the risk of opportunistic infections, a purified protein derivative (PPD) test is advised before initiation of therapy to detect potential latent tuberculosis. Other risks include sepsis, pancytopenia, and worsening of multiple sclerosis.
Antibodies against T-lymphocyte surface molecules
Alefacept (Amevive) is a fusion protein that blocks T-cell activation and triggers apoptosis of activated T-cells. It is given as 15mg weekly intramuscular injections. Thirty-three percent of patients reported a 75% clinical improvement in their psoriasis within 12 weeks.6 Alefacept also decreases synovial tissue T-cell count, and may have a future role in psoriatic arthritis.
It has few side effects, but patients need weekly monitoring of their CD4+ count. Ongoing studies on combining it with ultraviolet light or extending the dose to 16 weeks are showing promise.6
Antibodies against adhesion molecules
Efalizumab (Raptiva) is a monoclonal antibody that blocks T-cell activation and migration. It is self-administered by the patient as a 1 mg/kg weekly subcutaneous injection. Forty-four percent of patients achieve a 75% clinical improvement in their psoriasis by 24 weeks.
The most common side effects are headache, fever, nausea, vomiting, and myalgia. A “rebound” phenomenon after discontinuation is observed in 14%, and it may worsen psoriasis in those unresponsive to treatment.7
Other anti-TNF medications such as infliximab (Remicade), adalimumab (Humira), and onercept are still in clinical trials.
Combination therapy: Achieving goals while reducing adverse effects
Some patients require therapy with several agents to maintain adequate clearing of their psoriasis. The ideal combination therapy should lead to enhanced clinical response with reduction of adverse effects.
It is important to choose agents with synergistic effects without additive toxicities. Examples are combination of a systemic agent with topical calcipotriene, topical steroids, or with phototherapy. PUVA should be used with care for patients taking immunosuppressive agents due to risk of squamous cell carcinoma.8 A combination of 2 immunosuppressive agents is generally avoided due to risk of opportunistic infections, but has proven beneficial in a few therapy-resistant patients.9 Further clinical experience is needed for the inclusion of the new biologics in combination therapy.
New directions in treating psoriasis
A summary of future directions and current investigations in the management of psoriasis is given in TABLE 3.
Our patient’s treatment consisted of topical emollients, mid-potency topical corticosteroids, and tar shampoos/tar baths. She was responding well to the treatment. Introducing calcipotriene and reducing topical steroids is our next step. Regular follow-up visits are scheduled every 4 to 6 weeks.
TABLE 3
Future investigations
| Photodynamic therapy | The use of a photosensitizing drug in combination with a light source is showing promise, and clinical studies are under way for the treatment of psoriasis |
| Excimer lasers | Deliver high-dose narrowband UVB to a localized area sparing uninvolved skin. Clear psoriasis faster than conventional phototherapy, and may become predominant in the future |
| CNTO-1275 | Anti-interleukin-12 antibody that switches the immune response from a T-helper cell 1 cytokine reaction most commonly seen in psoriasis to a T-helper cell 2 cytokine response |
| T-cell receptor vaccines | Have been developed and are undergoing clinical trials in patients with psoriasis |
| Pimecrolimus | Used orally. It is a cytokine-release inhibitor with lesser immunosuppressive effects and side effects than tacrolimus and cyclosporin |
| Angiogenesis | Cutaneous blood vessels in psoriatic plaques are dilated, tortuous, and leaky |
| Vascular endothelial growth factor (VEGF) is overexpressed, and VEGF or its receptors are potential therapeutic targets for psoriasis | |
| Gene therapy | Chromosomes involved in psoriasis are being mapped |
| Gene therapy promises to be one of the most important areas of treatment of psoriasis for the new millennium |
Conclusion
Patients with mild localized psoriasis can easily and effectively be managed by family physicians using topical treatments or combinations modalities as outlined above. Patients with extensive disease or resistant to treatment should be referred to a dermatologist in conjunction with a rheumatologist if psoriatic arthritis is suspected.
With improved understanding of the immunopathogenesis and genetics of psoriasis, advent of the biologic agents, and future strategies under investigation, our approach to treating psoriasis may be very different in the years to come.
CORRESPONDING AUTHOR
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine (BAR 314), Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street, Boston, MA 02114. E-mail: [email protected]
1. Khachemoune A, Phillips TJ. Current treatment options in psoriasis. Hosp Pract (Off Ed) 2000;35:93-96,101-104,107.-
2. Winterfield LS, Menter A, Gordon A, Gottlieb A. Psoriasis treatment: current and emerging directed therapies. Ann Rheum Dis 2005;64:ii87-ii90.
3. Zanolli M. Phototherapy arsenal in the treatment of psoriasis. Dermatol Clin 2004;22:397-406.
4. Leonardi CL, Powers JL, Matheson RT, et al. Etanercept Psoriasis Study Group. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2005;349:2014-22.
5. Lebwohl M. Innovations in the treatment of psoriasis. J Am Acad Dermatol 2004;51:40-41.
6. Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE. Alefacept Clinical Study Group. An international, randomized, double blind placebo-controlled phase 3 trial of intramuscular Alefacept in patients with chronic plaque psoriasis. Arch Dermatol 2003;139:719-727.
7. Gordon KB, Papp KA, Hamilton TK, et al. Efalizumab Study Group. Efalizumab for patients with moderate to severe plaque psoriasis: a randomized controlled trial. JAMA 2003;290:3073-3080.
8. Marcil I, Stern RS. Squamous-cell cancer of the skin in patients given PUVA and cyclosporin: nested cohort crossover study. Lancet 2001;358:1942-1945.
9. Clark CM, Kirby B, Morris AO, et al. Combination treatment with methotrexate and cyclosporin for severe and recalcitrant psoriasis. Br J Dermatol 1999;141:279-282.
An otherwise healthy 12-year-old girl came to the office with a 1-year history of a symmetric, generalized scaly eruption. These skin plaques did not itch; she had no recent history of sore throat. She also had no personal or family history of atopy or any similar eruption. She mentioned that the appearance of her skin and the “flaking” was making her very self-conscious and she wanted to have some intervention to make the skin clear.
The patient had multiple red, scaly, thick plaques on her back and the extensor surfaces of her elbows, knees, legs, and forearms (FIGURES 1 AND 2). There were no scalp, mucosal, or nail involvements. Rheumatologic examination and review of systems were unremarkable.
FIGURE 1
Plaques on the arm…
FIGURE 2
…and on the back
What is your diagnosis?
How would you manage this case?
Diagnosis: Chronic plaque psoriasis
Psoriasis is a noninfectious inflammatory skin disorder characterized by well-defined erythematous plaques that bear large, adherent silvery scales. It can appear at any age, but 75% of patients have an onset before the age of 40 years.1
Psoriasis was once considered a hyperproliferative disorder, but is now recognized as an autoimmune phenomenon involving activation of T-cells. As a result, new immunosuppressive agents have been added to the list of traditional therapies, opening a new chapter of immunomodulatory therapy.
Clinical presentations of psoriasis
In its classic presentation, psoriasis does not pose a diagnosis challenge to most clinicians—it presents as a sharply demarcated erythematous plaques with silvery white scales. Although many classification systems exist, a concise classification is included in TABLE 1. Skin conditions to be considered for differential diagnosis are summarized in TABLE 2. The US Food and Drug Administration defines “severe” psoriasis as involving more than 20% of body surface area.2
TABLE 1
Classification of psoriasis
| Chronic plaque psoriasis | Symmetrical plaques up to 20 cm in diameter, with a predilection for the elbows, knees, presacrum, scalp, hands, and feet |
| 5%–30% of patients develop a seronegative arthropathy | |
| Guttate psoriasis | Numerous small papules and plaques over the upper trunk and proximal extremities |
| Most common form of psoriasis in children, and may be triggered by any streptococcal infection including streptococcal perianal dermatitis | |
| Spontaneous remission is the rule | |
| Generalized pustular and erythrodermic psoriasis | Uncommon variants associated with high morbidity that may be fatal |
TABLE 2
Differential diagnosis
| Fungal infections |
| Squamous cell carcinoma in situ |
| Cutaneous T-cell lymphoma |
| Discoid eczema |
| Seborrhoeic eczema |
| Pityriasis rosea |
| Secondary syphilis |
| Hypertrophic lichen planus |
| Nummular dermatitis |
Pathogenesis of psoriasis
Psoriasis is a polygenic disorder. Susceptibility is determined by a large number of genes, each with a low penetrance. Important genetic associations are with HLA-Cw6, HLA-B27, and the genes PSOR-S1 and PSOR-S2 on chromosome 6 and 17, respectively.
Several triggering factors are identified, of which streptococcal antigens and certain drugs seem important, but the specific antigens are still unknown. The end result is activation of T-cells, overexpression of cytokines, and an inflammatory response.
Review of the classic treatments for psoriasis
Topical therapy
Topical therapy is indicated when psoriasis is limited to less than 20% of the body surface. Potent class I and II topical corticosteroids are the most widely used treatment for mild disease. After plaque clearance they can be given intermittently for maintenance.
The vitamin D3 derivative calcipotriene (Dovonex) is another first-line agent. Tazarotene (Tazorac), a topical retinoid prodrug, is a second-line agent used as monotherapy or in combination. Many combined regimens use topical corticosteroids, calcipotriene, and tazarotene. Coal tar—different concentrations in liquid form—is useful in treating extensive areas of the body and scalp psoriasis.
Phototherapy
Failure of topical therapy or extensive disease are indications for phototherapy or systemic medications. The trend is to use phototherapy in the form of narrowband UVB, which has proven more effective than broadband UVB and to have fewer adverse effects than psoralen UVA therapy (PUVA).3 Other light sources for home use are being developed.
Systemic agents
Methotrexate, given as a single weekly dose or in divided doses, has been used for more than 30 years; it inhibits the enzyme dihydrofolate reductase. An alternative immunosuppressive agent is cyclosporine. These 2 agents have high efficacy, but due to potential adverse effects they require careful patient selection and close follow-up.
Acitretin (Soriatane), an oral retinoid, is the treatment of choice for generalized pustular and erythrodermic psoriasis. It is also used in chronic plaque psoriasis, often in combination with phototherapy, which has a synergistic effect.New treatments
Our understanding of the immunopathogenesis of psoriasis has led to the development of therapies designed specifically to interfere with T-cell activation and effector functions. Three new immunomodulatory biologics are FDA-approved for the treatment of moderate to severe psoriasis. Typical cost of this therapy is more than $1000/month.
Anti-TNF-α strategies
Etanercept (Enbrel) is an antibody against the cytokine tumor necrosis factor alpha (TNF-α). It is self-administered at 25 mg to 50 mg subcutaneously twice weekly. Studies with 50-mg injections have shown a 75% clinical improvement in 49% of patients at 12 weeks and 59% of patients at 24 weeks.4
The most common side effect is reaction at the injection site. It was reported to produce dramatic remission of psoriatic arthritis and prevent radiographic progression of the disease.4-5 Due to raised concern about the risk of opportunistic infections, a purified protein derivative (PPD) test is advised before initiation of therapy to detect potential latent tuberculosis. Other risks include sepsis, pancytopenia, and worsening of multiple sclerosis.
Antibodies against T-lymphocyte surface molecules
Alefacept (Amevive) is a fusion protein that blocks T-cell activation and triggers apoptosis of activated T-cells. It is given as 15mg weekly intramuscular injections. Thirty-three percent of patients reported a 75% clinical improvement in their psoriasis within 12 weeks.6 Alefacept also decreases synovial tissue T-cell count, and may have a future role in psoriatic arthritis.
It has few side effects, but patients need weekly monitoring of their CD4+ count. Ongoing studies on combining it with ultraviolet light or extending the dose to 16 weeks are showing promise.6
Antibodies against adhesion molecules
Efalizumab (Raptiva) is a monoclonal antibody that blocks T-cell activation and migration. It is self-administered by the patient as a 1 mg/kg weekly subcutaneous injection. Forty-four percent of patients achieve a 75% clinical improvement in their psoriasis by 24 weeks.
The most common side effects are headache, fever, nausea, vomiting, and myalgia. A “rebound” phenomenon after discontinuation is observed in 14%, and it may worsen psoriasis in those unresponsive to treatment.7
Other anti-TNF medications such as infliximab (Remicade), adalimumab (Humira), and onercept are still in clinical trials.
Combination therapy: Achieving goals while reducing adverse effects
Some patients require therapy with several agents to maintain adequate clearing of their psoriasis. The ideal combination therapy should lead to enhanced clinical response with reduction of adverse effects.
It is important to choose agents with synergistic effects without additive toxicities. Examples are combination of a systemic agent with topical calcipotriene, topical steroids, or with phototherapy. PUVA should be used with care for patients taking immunosuppressive agents due to risk of squamous cell carcinoma.8 A combination of 2 immunosuppressive agents is generally avoided due to risk of opportunistic infections, but has proven beneficial in a few therapy-resistant patients.9 Further clinical experience is needed for the inclusion of the new biologics in combination therapy.
New directions in treating psoriasis
A summary of future directions and current investigations in the management of psoriasis is given in TABLE 3.
Our patient’s treatment consisted of topical emollients, mid-potency topical corticosteroids, and tar shampoos/tar baths. She was responding well to the treatment. Introducing calcipotriene and reducing topical steroids is our next step. Regular follow-up visits are scheduled every 4 to 6 weeks.
TABLE 3
Future investigations
| Photodynamic therapy | The use of a photosensitizing drug in combination with a light source is showing promise, and clinical studies are under way for the treatment of psoriasis |
| Excimer lasers | Deliver high-dose narrowband UVB to a localized area sparing uninvolved skin. Clear psoriasis faster than conventional phototherapy, and may become predominant in the future |
| CNTO-1275 | Anti-interleukin-12 antibody that switches the immune response from a T-helper cell 1 cytokine reaction most commonly seen in psoriasis to a T-helper cell 2 cytokine response |
| T-cell receptor vaccines | Have been developed and are undergoing clinical trials in patients with psoriasis |
| Pimecrolimus | Used orally. It is a cytokine-release inhibitor with lesser immunosuppressive effects and side effects than tacrolimus and cyclosporin |
| Angiogenesis | Cutaneous blood vessels in psoriatic plaques are dilated, tortuous, and leaky |
| Vascular endothelial growth factor (VEGF) is overexpressed, and VEGF or its receptors are potential therapeutic targets for psoriasis | |
| Gene therapy | Chromosomes involved in psoriasis are being mapped |
| Gene therapy promises to be one of the most important areas of treatment of psoriasis for the new millennium |
Conclusion
Patients with mild localized psoriasis can easily and effectively be managed by family physicians using topical treatments or combinations modalities as outlined above. Patients with extensive disease or resistant to treatment should be referred to a dermatologist in conjunction with a rheumatologist if psoriatic arthritis is suspected.
With improved understanding of the immunopathogenesis and genetics of psoriasis, advent of the biologic agents, and future strategies under investigation, our approach to treating psoriasis may be very different in the years to come.
CORRESPONDING AUTHOR
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine (BAR 314), Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street, Boston, MA 02114. E-mail: [email protected]
An otherwise healthy 12-year-old girl came to the office with a 1-year history of a symmetric, generalized scaly eruption. These skin plaques did not itch; she had no recent history of sore throat. She also had no personal or family history of atopy or any similar eruption. She mentioned that the appearance of her skin and the “flaking” was making her very self-conscious and she wanted to have some intervention to make the skin clear.
The patient had multiple red, scaly, thick plaques on her back and the extensor surfaces of her elbows, knees, legs, and forearms (FIGURES 1 AND 2). There were no scalp, mucosal, or nail involvements. Rheumatologic examination and review of systems were unremarkable.
FIGURE 1
Plaques on the arm…
FIGURE 2
…and on the back
What is your diagnosis?
How would you manage this case?
Diagnosis: Chronic plaque psoriasis
Psoriasis is a noninfectious inflammatory skin disorder characterized by well-defined erythematous plaques that bear large, adherent silvery scales. It can appear at any age, but 75% of patients have an onset before the age of 40 years.1
Psoriasis was once considered a hyperproliferative disorder, but is now recognized as an autoimmune phenomenon involving activation of T-cells. As a result, new immunosuppressive agents have been added to the list of traditional therapies, opening a new chapter of immunomodulatory therapy.
Clinical presentations of psoriasis
In its classic presentation, psoriasis does not pose a diagnosis challenge to most clinicians—it presents as a sharply demarcated erythematous plaques with silvery white scales. Although many classification systems exist, a concise classification is included in TABLE 1. Skin conditions to be considered for differential diagnosis are summarized in TABLE 2. The US Food and Drug Administration defines “severe” psoriasis as involving more than 20% of body surface area.2
TABLE 1
Classification of psoriasis
| Chronic plaque psoriasis | Symmetrical plaques up to 20 cm in diameter, with a predilection for the elbows, knees, presacrum, scalp, hands, and feet |
| 5%–30% of patients develop a seronegative arthropathy | |
| Guttate psoriasis | Numerous small papules and plaques over the upper trunk and proximal extremities |
| Most common form of psoriasis in children, and may be triggered by any streptococcal infection including streptococcal perianal dermatitis | |
| Spontaneous remission is the rule | |
| Generalized pustular and erythrodermic psoriasis | Uncommon variants associated with high morbidity that may be fatal |
TABLE 2
Differential diagnosis
| Fungal infections |
| Squamous cell carcinoma in situ |
| Cutaneous T-cell lymphoma |
| Discoid eczema |
| Seborrhoeic eczema |
| Pityriasis rosea |
| Secondary syphilis |
| Hypertrophic lichen planus |
| Nummular dermatitis |
Pathogenesis of psoriasis
Psoriasis is a polygenic disorder. Susceptibility is determined by a large number of genes, each with a low penetrance. Important genetic associations are with HLA-Cw6, HLA-B27, and the genes PSOR-S1 and PSOR-S2 on chromosome 6 and 17, respectively.
Several triggering factors are identified, of which streptococcal antigens and certain drugs seem important, but the specific antigens are still unknown. The end result is activation of T-cells, overexpression of cytokines, and an inflammatory response.
Review of the classic treatments for psoriasis
Topical therapy
Topical therapy is indicated when psoriasis is limited to less than 20% of the body surface. Potent class I and II topical corticosteroids are the most widely used treatment for mild disease. After plaque clearance they can be given intermittently for maintenance.
The vitamin D3 derivative calcipotriene (Dovonex) is another first-line agent. Tazarotene (Tazorac), a topical retinoid prodrug, is a second-line agent used as monotherapy or in combination. Many combined regimens use topical corticosteroids, calcipotriene, and tazarotene. Coal tar—different concentrations in liquid form—is useful in treating extensive areas of the body and scalp psoriasis.
Phototherapy
Failure of topical therapy or extensive disease are indications for phototherapy or systemic medications. The trend is to use phototherapy in the form of narrowband UVB, which has proven more effective than broadband UVB and to have fewer adverse effects than psoralen UVA therapy (PUVA).3 Other light sources for home use are being developed.
Systemic agents
Methotrexate, given as a single weekly dose or in divided doses, has been used for more than 30 years; it inhibits the enzyme dihydrofolate reductase. An alternative immunosuppressive agent is cyclosporine. These 2 agents have high efficacy, but due to potential adverse effects they require careful patient selection and close follow-up.
Acitretin (Soriatane), an oral retinoid, is the treatment of choice for generalized pustular and erythrodermic psoriasis. It is also used in chronic plaque psoriasis, often in combination with phototherapy, which has a synergistic effect.New treatments
Our understanding of the immunopathogenesis of psoriasis has led to the development of therapies designed specifically to interfere with T-cell activation and effector functions. Three new immunomodulatory biologics are FDA-approved for the treatment of moderate to severe psoriasis. Typical cost of this therapy is more than $1000/month.
Anti-TNF-α strategies
Etanercept (Enbrel) is an antibody against the cytokine tumor necrosis factor alpha (TNF-α). It is self-administered at 25 mg to 50 mg subcutaneously twice weekly. Studies with 50-mg injections have shown a 75% clinical improvement in 49% of patients at 12 weeks and 59% of patients at 24 weeks.4
The most common side effect is reaction at the injection site. It was reported to produce dramatic remission of psoriatic arthritis and prevent radiographic progression of the disease.4-5 Due to raised concern about the risk of opportunistic infections, a purified protein derivative (PPD) test is advised before initiation of therapy to detect potential latent tuberculosis. Other risks include sepsis, pancytopenia, and worsening of multiple sclerosis.
Antibodies against T-lymphocyte surface molecules
Alefacept (Amevive) is a fusion protein that blocks T-cell activation and triggers apoptosis of activated T-cells. It is given as 15mg weekly intramuscular injections. Thirty-three percent of patients reported a 75% clinical improvement in their psoriasis within 12 weeks.6 Alefacept also decreases synovial tissue T-cell count, and may have a future role in psoriatic arthritis.
It has few side effects, but patients need weekly monitoring of their CD4+ count. Ongoing studies on combining it with ultraviolet light or extending the dose to 16 weeks are showing promise.6
Antibodies against adhesion molecules
Efalizumab (Raptiva) is a monoclonal antibody that blocks T-cell activation and migration. It is self-administered by the patient as a 1 mg/kg weekly subcutaneous injection. Forty-four percent of patients achieve a 75% clinical improvement in their psoriasis by 24 weeks.
The most common side effects are headache, fever, nausea, vomiting, and myalgia. A “rebound” phenomenon after discontinuation is observed in 14%, and it may worsen psoriasis in those unresponsive to treatment.7
Other anti-TNF medications such as infliximab (Remicade), adalimumab (Humira), and onercept are still in clinical trials.
Combination therapy: Achieving goals while reducing adverse effects
Some patients require therapy with several agents to maintain adequate clearing of their psoriasis. The ideal combination therapy should lead to enhanced clinical response with reduction of adverse effects.
It is important to choose agents with synergistic effects without additive toxicities. Examples are combination of a systemic agent with topical calcipotriene, topical steroids, or with phototherapy. PUVA should be used with care for patients taking immunosuppressive agents due to risk of squamous cell carcinoma.8 A combination of 2 immunosuppressive agents is generally avoided due to risk of opportunistic infections, but has proven beneficial in a few therapy-resistant patients.9 Further clinical experience is needed for the inclusion of the new biologics in combination therapy.
New directions in treating psoriasis
A summary of future directions and current investigations in the management of psoriasis is given in TABLE 3.
Our patient’s treatment consisted of topical emollients, mid-potency topical corticosteroids, and tar shampoos/tar baths. She was responding well to the treatment. Introducing calcipotriene and reducing topical steroids is our next step. Regular follow-up visits are scheduled every 4 to 6 weeks.
TABLE 3
Future investigations
| Photodynamic therapy | The use of a photosensitizing drug in combination with a light source is showing promise, and clinical studies are under way for the treatment of psoriasis |
| Excimer lasers | Deliver high-dose narrowband UVB to a localized area sparing uninvolved skin. Clear psoriasis faster than conventional phototherapy, and may become predominant in the future |
| CNTO-1275 | Anti-interleukin-12 antibody that switches the immune response from a T-helper cell 1 cytokine reaction most commonly seen in psoriasis to a T-helper cell 2 cytokine response |
| T-cell receptor vaccines | Have been developed and are undergoing clinical trials in patients with psoriasis |
| Pimecrolimus | Used orally. It is a cytokine-release inhibitor with lesser immunosuppressive effects and side effects than tacrolimus and cyclosporin |
| Angiogenesis | Cutaneous blood vessels in psoriatic plaques are dilated, tortuous, and leaky |
| Vascular endothelial growth factor (VEGF) is overexpressed, and VEGF or its receptors are potential therapeutic targets for psoriasis | |
| Gene therapy | Chromosomes involved in psoriasis are being mapped |
| Gene therapy promises to be one of the most important areas of treatment of psoriasis for the new millennium |
Conclusion
Patients with mild localized psoriasis can easily and effectively be managed by family physicians using topical treatments or combinations modalities as outlined above. Patients with extensive disease or resistant to treatment should be referred to a dermatologist in conjunction with a rheumatologist if psoriatic arthritis is suspected.
With improved understanding of the immunopathogenesis and genetics of psoriasis, advent of the biologic agents, and future strategies under investigation, our approach to treating psoriasis may be very different in the years to come.
CORRESPONDING AUTHOR
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine (BAR 314), Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street, Boston, MA 02114. E-mail: [email protected]
1. Khachemoune A, Phillips TJ. Current treatment options in psoriasis. Hosp Pract (Off Ed) 2000;35:93-96,101-104,107.-
2. Winterfield LS, Menter A, Gordon A, Gottlieb A. Psoriasis treatment: current and emerging directed therapies. Ann Rheum Dis 2005;64:ii87-ii90.
3. Zanolli M. Phototherapy arsenal in the treatment of psoriasis. Dermatol Clin 2004;22:397-406.
4. Leonardi CL, Powers JL, Matheson RT, et al. Etanercept Psoriasis Study Group. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2005;349:2014-22.
5. Lebwohl M. Innovations in the treatment of psoriasis. J Am Acad Dermatol 2004;51:40-41.
6. Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE. Alefacept Clinical Study Group. An international, randomized, double blind placebo-controlled phase 3 trial of intramuscular Alefacept in patients with chronic plaque psoriasis. Arch Dermatol 2003;139:719-727.
7. Gordon KB, Papp KA, Hamilton TK, et al. Efalizumab Study Group. Efalizumab for patients with moderate to severe plaque psoriasis: a randomized controlled trial. JAMA 2003;290:3073-3080.
8. Marcil I, Stern RS. Squamous-cell cancer of the skin in patients given PUVA and cyclosporin: nested cohort crossover study. Lancet 2001;358:1942-1945.
9. Clark CM, Kirby B, Morris AO, et al. Combination treatment with methotrexate and cyclosporin for severe and recalcitrant psoriasis. Br J Dermatol 1999;141:279-282.
1. Khachemoune A, Phillips TJ. Current treatment options in psoriasis. Hosp Pract (Off Ed) 2000;35:93-96,101-104,107.-
2. Winterfield LS, Menter A, Gordon A, Gottlieb A. Psoriasis treatment: current and emerging directed therapies. Ann Rheum Dis 2005;64:ii87-ii90.
3. Zanolli M. Phototherapy arsenal in the treatment of psoriasis. Dermatol Clin 2004;22:397-406.
4. Leonardi CL, Powers JL, Matheson RT, et al. Etanercept Psoriasis Study Group. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2005;349:2014-22.
5. Lebwohl M. Innovations in the treatment of psoriasis. J Am Acad Dermatol 2004;51:40-41.
6. Lebwohl M, Christophers E, Langley R, Ortonne JP, Roberts J, Griffiths CE. Alefacept Clinical Study Group. An international, randomized, double blind placebo-controlled phase 3 trial of intramuscular Alefacept in patients with chronic plaque psoriasis. Arch Dermatol 2003;139:719-727.
7. Gordon KB, Papp KA, Hamilton TK, et al. Efalizumab Study Group. Efalizumab for patients with moderate to severe plaque psoriasis: a randomized controlled trial. JAMA 2003;290:3073-3080.
8. Marcil I, Stern RS. Squamous-cell cancer of the skin in patients given PUVA and cyclosporin: nested cohort crossover study. Lancet 2001;358:1942-1945.
9. Clark CM, Kirby B, Morris AO, et al. Combination treatment with methotrexate and cyclosporin for severe and recalcitrant psoriasis. Br J Dermatol 1999;141:279-282.
Unilateral rash on a baby girl
An 11-month-old baby girl came to the clinic with a pruritic rash. The rash initially appeared in her popliteal fossa 2 weeks before the visit. The eruption extended to the right leg, arm, and flank the week before the visit, subsequently spreading to the contralateral flank. Three weeks before to the eruption’s appearance, the patient had an upper respiratory infection with a dry nonproductive cough, which resolved spontaneously without antibiotics.
The physical examination revealed a healthy-appearing infant girl with excoriated erythematous papules coalescing into plaques on her right flexural arm that continued to the axilla and down the right flank to the flexural aspect of her leg (FIGURE 1). Her left side was essentially free of any rash (FIGURE 2). No cervical or axillary lymphadenopathy was noted, and the remainder of her exam was normal.
FIGURE 1
The right side has a rash…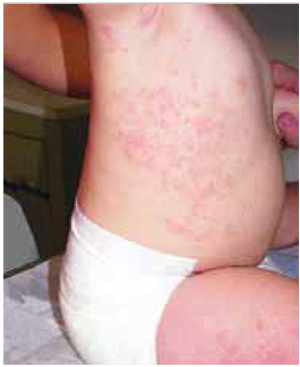
FIGURE 2
…and the left side is clear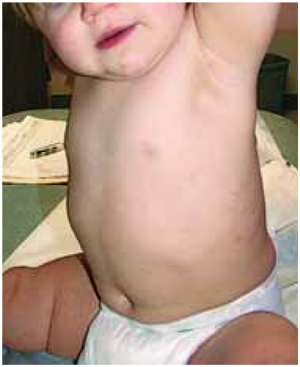
What is your diagnosis?
How Would You Manage This Condition?
Diagnosis: Asymmetric periflexural exanthem of childhood
Asymmetric periflexural exanthem of childhood (APEC) is a diagnosis defined by its unique clinical presentation. Since its original description in 1962 by Brunner 1 as a new papular erythema of childhood, a number of names have been used to describe the same clinical process: unilateral laterothoracic exanthema,2 asymmetric periflexural exanthem of childhood,3 and lichen miliaris.4
Clinical picture of APEC
The initial clinical finding is a unilateral erythematous macular and papular eruption, often beginning in or around the axilla. Over the following 1 to 3 weeks, centrifugal spread involves the upper and lower extremities. Approximately 70% of APEC cases have involvement of the contralateral trunk. Despite the progression to the contralateral side, the eruption remains asymmetric throughout its course.
Additional findings include lymphadenopathy and pruritus in 70% and 65% of cases, respectively. 3-5 In contrast to other exanthems, APEC rarely involves the face.5 A study by Coustou reported that 60% of cases had a preceding prodrome including rhinitis, pharyngitis, otitis, and fever.4,5
Cause is unknown
Although the precise cause of APEC is not known, it has features consistent with a viral exanthem. A viral source is supported by a springtime and pediatric predominance with spontaneous resolution. In addition, 1 adult case of APEC has been attributed to an acute Parvo B19 infection.6
However, consistent serologic evidence supporting a viral cause is lacking,2, 7 and no human transmissions have been documented except for reports of 2 familial cases.8 Some have proposed that this could be a childhood form of pityriasis rosea possibly caused by human herpes virus 7.4
Differential diagnosis
The differential diagnosis for APEC includes viral exanthems, eczema, scabies, pityriasis rosea, contact dermatitis, and miliaria (heat rash). APEC mainly affects children aged 2 to 3 years but can occur at a younger age. There are no laboratory tests that help establish the diagnosis of APEC. The diagnosis is based on the clinical picture of an asymmetric macular and papular exanthem in a young child with a viral-like prodrome.
Treatment and outcome
There is no specific treatment for APEC other than to treat the symptoms. No treatment has been shown to shorten the course of this disease. A low-potency topical steroid along with an antihistamine provides adequate symptomatic treatment.
This child had no significant symptoms and therefore no medications were prescribed. The parents were told they may get 1% hydrocortisone cream over-the-counter if their daughter developed troublesome itching. Reassurance was provided about the limited nature of this exanthem. The parents were advised to bring the child for follow-up if the rash did not completely resolve in 2 months. By the time for the child’s 1-year check-up, the rash was gone.
Corresponding Author
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine (BAR 314), Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street, Boston, MA 02114. E-mail: [email protected].
1. Brunner MJ, Rubin L, Dunlap F. A new papular erythema of childhood. Arch Dermatol 1962;85:539-540.
2. Bodemer C, de Prost Y. Unilateral laterothoracic exanthem in children: a new disease? J Am Acad Dermato 1992;27:693-696.
3. Taïeb A, Megraud F, Le Roy JM, Magne F, Reguilhem MO, Maleville J. Érythème localisé avec adénopathie régionale de l’enfant: Une maladie d’inoculation? Ann Dermatol Venereol 1986;113:1023-1024.
4. Laur WE. Unilateral laterothoracic exanthem in children. J Am Acad Dermatol 1993;29:799-800.
5. Coustou D, Leaute-Labreze C, Bioulac-Sage P, et al. Asymmetric periflexural exanthem of childhood: a clinical, pathologic, and epidemiologic prospective study. Arch Dermatol 1999;135:799-803.
6. Pauluzzi P, Festini G, Gelmetti C. Asymmetric periflexural exanthem of childhood in an adult patient with parvovirus B19. J Eur Acad Dermatol Venereol 2001;15:372-374.
7. Jhin MH, Eidelman M, Cohen SR, Husain S. Unilateral eruption in a child. Arch Dermatol 2002;138:1371-1376.
8. McCuaig CC, Russo P, Powell J, et al. Unilateral laterothoracic exanthem. A clinicopathologic study of forty-eight patients. J Am Acad Dermatol 1996;34:979-984.
An 11-month-old baby girl came to the clinic with a pruritic rash. The rash initially appeared in her popliteal fossa 2 weeks before the visit. The eruption extended to the right leg, arm, and flank the week before the visit, subsequently spreading to the contralateral flank. Three weeks before to the eruption’s appearance, the patient had an upper respiratory infection with a dry nonproductive cough, which resolved spontaneously without antibiotics.
The physical examination revealed a healthy-appearing infant girl with excoriated erythematous papules coalescing into plaques on her right flexural arm that continued to the axilla and down the right flank to the flexural aspect of her leg (FIGURE 1). Her left side was essentially free of any rash (FIGURE 2). No cervical or axillary lymphadenopathy was noted, and the remainder of her exam was normal.
FIGURE 1
The right side has a rash…
FIGURE 2
…and the left side is clear
What is your diagnosis?
How Would You Manage This Condition?
Diagnosis: Asymmetric periflexural exanthem of childhood
Asymmetric periflexural exanthem of childhood (APEC) is a diagnosis defined by its unique clinical presentation. Since its original description in 1962 by Brunner 1 as a new papular erythema of childhood, a number of names have been used to describe the same clinical process: unilateral laterothoracic exanthema,2 asymmetric periflexural exanthem of childhood,3 and lichen miliaris.4
Clinical picture of APEC
The initial clinical finding is a unilateral erythematous macular and papular eruption, often beginning in or around the axilla. Over the following 1 to 3 weeks, centrifugal spread involves the upper and lower extremities. Approximately 70% of APEC cases have involvement of the contralateral trunk. Despite the progression to the contralateral side, the eruption remains asymmetric throughout its course.
Additional findings include lymphadenopathy and pruritus in 70% and 65% of cases, respectively. 3-5 In contrast to other exanthems, APEC rarely involves the face.5 A study by Coustou reported that 60% of cases had a preceding prodrome including rhinitis, pharyngitis, otitis, and fever.4,5
Cause is unknown
Although the precise cause of APEC is not known, it has features consistent with a viral exanthem. A viral source is supported by a springtime and pediatric predominance with spontaneous resolution. In addition, 1 adult case of APEC has been attributed to an acute Parvo B19 infection.6
However, consistent serologic evidence supporting a viral cause is lacking,2, 7 and no human transmissions have been documented except for reports of 2 familial cases.8 Some have proposed that this could be a childhood form of pityriasis rosea possibly caused by human herpes virus 7.4
Differential diagnosis
The differential diagnosis for APEC includes viral exanthems, eczema, scabies, pityriasis rosea, contact dermatitis, and miliaria (heat rash). APEC mainly affects children aged 2 to 3 years but can occur at a younger age. There are no laboratory tests that help establish the diagnosis of APEC. The diagnosis is based on the clinical picture of an asymmetric macular and papular exanthem in a young child with a viral-like prodrome.
Treatment and outcome
There is no specific treatment for APEC other than to treat the symptoms. No treatment has been shown to shorten the course of this disease. A low-potency topical steroid along with an antihistamine provides adequate symptomatic treatment.
This child had no significant symptoms and therefore no medications were prescribed. The parents were told they may get 1% hydrocortisone cream over-the-counter if their daughter developed troublesome itching. Reassurance was provided about the limited nature of this exanthem. The parents were advised to bring the child for follow-up if the rash did not completely resolve in 2 months. By the time for the child’s 1-year check-up, the rash was gone.
Corresponding Author
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine (BAR 314), Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street, Boston, MA 02114. E-mail: [email protected].
An 11-month-old baby girl came to the clinic with a pruritic rash. The rash initially appeared in her popliteal fossa 2 weeks before the visit. The eruption extended to the right leg, arm, and flank the week before the visit, subsequently spreading to the contralateral flank. Three weeks before to the eruption’s appearance, the patient had an upper respiratory infection with a dry nonproductive cough, which resolved spontaneously without antibiotics.
The physical examination revealed a healthy-appearing infant girl with excoriated erythematous papules coalescing into plaques on her right flexural arm that continued to the axilla and down the right flank to the flexural aspect of her leg (FIGURE 1). Her left side was essentially free of any rash (FIGURE 2). No cervical or axillary lymphadenopathy was noted, and the remainder of her exam was normal.
FIGURE 1
The right side has a rash…
FIGURE 2
…and the left side is clear
What is your diagnosis?
How Would You Manage This Condition?
Diagnosis: Asymmetric periflexural exanthem of childhood
Asymmetric periflexural exanthem of childhood (APEC) is a diagnosis defined by its unique clinical presentation. Since its original description in 1962 by Brunner 1 as a new papular erythema of childhood, a number of names have been used to describe the same clinical process: unilateral laterothoracic exanthema,2 asymmetric periflexural exanthem of childhood,3 and lichen miliaris.4
Clinical picture of APEC
The initial clinical finding is a unilateral erythematous macular and papular eruption, often beginning in or around the axilla. Over the following 1 to 3 weeks, centrifugal spread involves the upper and lower extremities. Approximately 70% of APEC cases have involvement of the contralateral trunk. Despite the progression to the contralateral side, the eruption remains asymmetric throughout its course.
Additional findings include lymphadenopathy and pruritus in 70% and 65% of cases, respectively. 3-5 In contrast to other exanthems, APEC rarely involves the face.5 A study by Coustou reported that 60% of cases had a preceding prodrome including rhinitis, pharyngitis, otitis, and fever.4,5
Cause is unknown
Although the precise cause of APEC is not known, it has features consistent with a viral exanthem. A viral source is supported by a springtime and pediatric predominance with spontaneous resolution. In addition, 1 adult case of APEC has been attributed to an acute Parvo B19 infection.6
However, consistent serologic evidence supporting a viral cause is lacking,2, 7 and no human transmissions have been documented except for reports of 2 familial cases.8 Some have proposed that this could be a childhood form of pityriasis rosea possibly caused by human herpes virus 7.4
Differential diagnosis
The differential diagnosis for APEC includes viral exanthems, eczema, scabies, pityriasis rosea, contact dermatitis, and miliaria (heat rash). APEC mainly affects children aged 2 to 3 years but can occur at a younger age. There are no laboratory tests that help establish the diagnosis of APEC. The diagnosis is based on the clinical picture of an asymmetric macular and papular exanthem in a young child with a viral-like prodrome.
Treatment and outcome
There is no specific treatment for APEC other than to treat the symptoms. No treatment has been shown to shorten the course of this disease. A low-potency topical steroid along with an antihistamine provides adequate symptomatic treatment.
This child had no significant symptoms and therefore no medications were prescribed. The parents were told they may get 1% hydrocortisone cream over-the-counter if their daughter developed troublesome itching. Reassurance was provided about the limited nature of this exanthem. The parents were advised to bring the child for follow-up if the rash did not completely resolve in 2 months. By the time for the child’s 1-year check-up, the rash was gone.
Corresponding Author
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine (BAR 314), Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street, Boston, MA 02114. E-mail: [email protected].
1. Brunner MJ, Rubin L, Dunlap F. A new papular erythema of childhood. Arch Dermatol 1962;85:539-540.
2. Bodemer C, de Prost Y. Unilateral laterothoracic exanthem in children: a new disease? J Am Acad Dermato 1992;27:693-696.
3. Taïeb A, Megraud F, Le Roy JM, Magne F, Reguilhem MO, Maleville J. Érythème localisé avec adénopathie régionale de l’enfant: Une maladie d’inoculation? Ann Dermatol Venereol 1986;113:1023-1024.
4. Laur WE. Unilateral laterothoracic exanthem in children. J Am Acad Dermatol 1993;29:799-800.
5. Coustou D, Leaute-Labreze C, Bioulac-Sage P, et al. Asymmetric periflexural exanthem of childhood: a clinical, pathologic, and epidemiologic prospective study. Arch Dermatol 1999;135:799-803.
6. Pauluzzi P, Festini G, Gelmetti C. Asymmetric periflexural exanthem of childhood in an adult patient with parvovirus B19. J Eur Acad Dermatol Venereol 2001;15:372-374.
7. Jhin MH, Eidelman M, Cohen SR, Husain S. Unilateral eruption in a child. Arch Dermatol 2002;138:1371-1376.
8. McCuaig CC, Russo P, Powell J, et al. Unilateral laterothoracic exanthem. A clinicopathologic study of forty-eight patients. J Am Acad Dermatol 1996;34:979-984.
1. Brunner MJ, Rubin L, Dunlap F. A new papular erythema of childhood. Arch Dermatol 1962;85:539-540.
2. Bodemer C, de Prost Y. Unilateral laterothoracic exanthem in children: a new disease? J Am Acad Dermato 1992;27:693-696.
3. Taïeb A, Megraud F, Le Roy JM, Magne F, Reguilhem MO, Maleville J. Érythème localisé avec adénopathie régionale de l’enfant: Une maladie d’inoculation? Ann Dermatol Venereol 1986;113:1023-1024.
4. Laur WE. Unilateral laterothoracic exanthem in children. J Am Acad Dermatol 1993;29:799-800.
5. Coustou D, Leaute-Labreze C, Bioulac-Sage P, et al. Asymmetric periflexural exanthem of childhood: a clinical, pathologic, and epidemiologic prospective study. Arch Dermatol 1999;135:799-803.
6. Pauluzzi P, Festini G, Gelmetti C. Asymmetric periflexural exanthem of childhood in an adult patient with parvovirus B19. J Eur Acad Dermatol Venereol 2001;15:372-374.
7. Jhin MH, Eidelman M, Cohen SR, Husain S. Unilateral eruption in a child. Arch Dermatol 2002;138:1371-1376.
8. McCuaig CC, Russo P, Powell J, et al. Unilateral laterothoracic exanthem. A clinicopathologic study of forty-eight patients. J Am Acad Dermatol 1996;34:979-984.
A sore and sensitive tongue
A 40-year-old woman came into the office with a 1-year history of painful red lesions with white striations on her tongue (Figure 1). The lesions caused burning after she ate spicy food and an increased sensitivity to mouthwash. She reported a slow, progressive onset and worsening of her condition, with intensifying symptoms during periods of emotional stress.
On physical examination, we observed an erythematous and cream-colored reticulated patch with a few small focal areas of erosion covering the dorsal tongue. The remainder of the oral mucosa appeared normal. She also had thinning and ridging of the toenails (Figure 2).
What is the most likely diagnosis?
How would you manage this case?
FIGURE 1
Tongue with painful lesions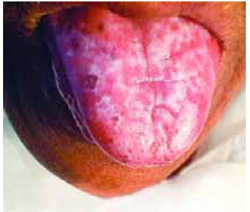
FIGURE 2
Thinning toenails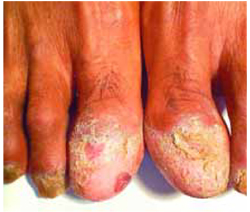
Diagnosis: oral lichen planus
Compared with the more self-limited nature of its cutaneous counterpart, oral lichen planus (OLP) causes more chronic inflammatory lesions, resulting in increased morbidity and a greater therapeutic challenge for physicians. Of the 3 basic clinical morphologies of the disease (reticular, erythematous, and erosive), the last form tends to be more symptomatic and prompts patients to seek treatment.1,2 The more asymptomatic reticular form is the most easily recognizable variant and may cause increased symptoms when involving the tongue, typically manifesting on the dorsal surface.1
In contrast to reticular lesions in other oral sites, tongue lesions do not usually exhibit the characteristic interlacing pattern of white, raised striae; instead they manifest as well-demarcated areas with a patch-like pattern of erythematous, atrophic, and keratotic regions. 3 Erosive OLP more commonly involves the lateral tongue and displays erythematous or ulcerated areas with peripheral keratosis, forming fine centrifugal striae.3
Oral lichen planus affects approximately 1% to 4% of the population 4 and is seen most commonly in older women.1,5,6 It has a predilection for bilateral involvement of the buccal mucosa but may also affect (in descending order of frequency) the tongue, gingiva, lips, floor of the mouth, and palate.1,2 Isolated involvement of only 1 oral site is infrequent, with the exception of gingival lesions.1 Extraoral cutaneous lichen planus has been reported in 16% to 44% of patients with oral disease; it most frequently involves the genital mucosa, as well as the scalp, nails, and esophageal and ocular mucosae.4 A multidisciplinary approach including generalists, dermatologists, otolaryngologists, ophthalmologists, and gynecologists may be necessary for patient evaluation and management.
Pathogenesis of oral lichen planus
Although the precise cause of OLP is unknown, its pathogenesis has been linked to an autoimmune mechanism involving autocytotoxic CD8+ T cells, which trigger apoptosis (programmed cell death) of basal keratinocytes.4,7 An imbalance between T cell helper and suppressor activity has also been observed.1,7
Complications: Carcinoma, chronic liver disease
Malignant transformation to squamous cell carcinoma (SCC) is seen in 0.4% to 5% of patients with OLP, particularly those with erosive and erythematous disease.1,2 Increased risk factors for SCC have not been identified in these patients, but a greater prevalence of Candida albicansmay be associated with carcinogenesis, as may herpes simplex and human papilloma viruses, immunosuppressive therapy, and an inflammatory cytokine-rich microenvironment.1
In Japan and parts of Southern Europe, OLP has been associated with hepatitis C infection and chronic liver disease, but these findings have not been reproduced in patients in the US.
Making the diagnosis: exam, biopsy, immunofluorescence
A detailed history and physical examination are usually sufficient to diagnose OLP, although laboratory studies and biopsy may be required to exclude malignancy or distinguish OLP from conditions such as pemphigus vulgaris and mucous membrane pemphigoid. A biopsy is rarely needed, and should only be performed by physicians that have training in a tongue biopsy.
If necessary, biopsy samples should be obtained from the most representative sample area of the tongue. After administration of local anesthesia (lidocaine 1:100,000 with epinephrine) near the periphery of the site, biopsy is performed using a #15 blade scalpel or biopsy punch. An assistant may be needed to grasp the tongue by wrapping a gauze sponge over the tip and securing it firmly. When tongue biopsy sites are closed, deep sutures should be placed if necessary, and mucosal sutures should be placed relatively close together, as inadequately closed wounds on the tongue tend to reopen, resulting in bleeding and prolonged healing time.
Specimens should be placed in a 10% formalin solution for transportation to a pathologist. Patients should return to the clinic after 7 to 14 days for examination of the biopsy site and suture removal.
Microscopically, OLP demonstrates vacuolar degeneration, basal cell lysis, and liquefactive degeneration1 along with focal hyperkeratosis, a characteristic amorphous eosinophilic band at the basement membrane level, and a dense, bandlike subepithelial lymphocytic infiltrate.5,6 Direct immunofluorescence, displaying granular fibrino-gen and variable immunoglobulin deposited linearly near the basement membrane, and possibly cytoid bodies, is useful in cases of suspected OLP with nondiagnostic histological findings, as well as in those with gingival involvement.1
Differential diagnosis
The clinical differential diagnosis of OLP includes the autoimmune bullous diseases pemphigus vulgaris, mucous membrane pemphigoid, dermatitis herpetiformis, and linear immuno-globulin A disease, which can all be excluded with direct and indirect immunofluorescence studies. When clinically appropriate, biopsy specimens submitted for direct immunofluorescence must be preserved in Michel solution. Reactive keratoses, chronic hyperplastic candidosis, epithelial dysplasia, discoid lupus erythematosus, anemic states, and several gastrointestinal diseases including oral Crohn disease and chronic hepatic disease, must also be ruled out.
Oral lichenoid reactions may develop as a hypersensitivity to dental materials and drugs, and must also be considered.1 Patch testing may help identify a contact allergy to dental prosthesis components, including gold, mercury, and palladium salts.
Management: steroids, immunosuppressants, surgery
Because fewer than 20% of patients experience total remission,5 treatment of OLP is chronic and palliative. Most symptomatic patients are taught to avoid exacerbating factors and given topical steroid therapy (dexamethasone rinse, fluocinolone gel, triamcinolone cream). Observation, intralesional and systemic corticosteroids, immunosuppressants, antifungals, retinoids, antimalarials, dapsone, oral psoralen-ultraviolet-light (PUVA) treatment, and surgical techniques (CO2 and neodymium: yttrium-aluminum-garnet [Nd:YAG] laser therapy, cryotherapy, and excision) are also employed, alone and in combination.5,8
Tacrolimus (Protopic), an immunomodulator, administered topically in 0.03% to 0.3% concentrations using Aquaphor or paraffin ointment base, has been shown effective and well tolerated in controlling symptoms,8,9 and is a less costly alternative to topical cyclosporine.9 It has been demonstrated effective prospective-ly,10 and is safe in long-term therapy11 of erosive OLP, but it has been reported in one case to cause hyperpigmentation of oral mucosa.12
A recent study13 demonstrated the utility of low-dose 308-nm excimer laser radiation for symptomatic OLP.
Patient management
Our patient was instructed to avoid foods and substances that caused irritation of her tongue and oral mucosa. In addition, she was prescribed topical fluocinolone gel 0.025% 3 times daily, and was given information about alternative treatment options, including tacrolimus and surgical therapy. She was instructed to perform gentle yet thorough daily oral hygiene and to follow-up in 6 months for re-examination.
Corresponding author
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street (BAR 314), Boston, MA 02114. E-mail: [email protected].
December 2004’s Photo Rounds, “Rupturing bullae not respondign to antiobiotics,” left out the names of two of the article’s authors. The correct authors of the piece are John Sauret, MD, FAAFP, Sandra Yale, DO, and Ahunna Ahiarah, MD. We regret the error.
1. Eisen D. The clinical features, malignant potential, and systemic associations of oral lichen planus: A study of 723 patients. J Am Acad Dermatol 2002;46:207-214.
2. Gorsky M, Raviv M, Moskona D, Laufer M, Bodner L. Clinical characteristics and treatment of patients with oral lichen planus in Israel. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1996;82:644-649.
3. Allen CM, Blozis GG. Oral mucosal lesions. In Cummings CW, Schuller DE (eds): Otolaryngology: Head and Neck Surgery, vol 2, 2nd ed. St Louis, Mo: Mosby-Yearbook; 1993;1374-1375.
4. Eisen D. The evaluation of cutaneous, genital, scalp, nail, esophageal, and ocular involvement in patients with oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999;88:431-436.
5. Brown RS, Bottomley WK, Puente E, Lavigne GL. A retrospective evaluation of 193 patients with oral lichen planus. J Oral Pathol Med 1993;22:69-72.
6. Thorn JJ, Holmstrup P, Rindum J, Pindborg JJ. Course of various clinical forms of oral lichen planus. A retrospective follow-up study of 611 patients. J Oral Pathol 1998;17:213-218.
7. Porter SR, Kirby A, Olsen I, Barrett W. Immunologic aspects of dermal and oral lichen planus. A review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997;83:358-366.
8. Rozycki TW, Rogers RS, Pittelkow MR, et al. Topical tacrolimus in the treatment of symptomatic oral lichen planus: A series of 13 patients. J Am Acad Dermatol 2002;46:27-34.
9. Kaliakatsou F, Hodgson TA, Lewsey JD, Hegarty AM, Murphy AG, Porter SR. Management of recalcitrant ulcer-ative oral lichen planus with topical tacrolimus. J Am Acad Dermatol 2002;46:35-41.
10. Olivier V, Lacour JP, Mousnier A, Garraffo R, Monteil RA, Ortonne JP. Treatment of chronic erosive oral lichen planus with low concentrations of topical tacrolimus. An open prospective study. Arch Dermatol 2002;138:1335-1338.
11. Hodgson TA, Sahni N, Kaliakatsou F, Buchanan JA, Porter SR. Long-term efficacy and safety of topical tacrolimus in the management of ulcerative/erosive oral lichen planus. Eur J Dermatol 2003;13:466-470.
12. Shen JT, Pedvis-Leftick A. Mucosal staining after using topical tacrolimus to treat erosive oral lichen planus. J Am Acad Dermatol 2004;50-326.
13. Trehan M, Taylor CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol 2004;140:415-420.
A 40-year-old woman came into the office with a 1-year history of painful red lesions with white striations on her tongue (Figure 1). The lesions caused burning after she ate spicy food and an increased sensitivity to mouthwash. She reported a slow, progressive onset and worsening of her condition, with intensifying symptoms during periods of emotional stress.
On physical examination, we observed an erythematous and cream-colored reticulated patch with a few small focal areas of erosion covering the dorsal tongue. The remainder of the oral mucosa appeared normal. She also had thinning and ridging of the toenails (Figure 2).
What is the most likely diagnosis?
How would you manage this case?
FIGURE 1
Tongue with painful lesions
FIGURE 2
Thinning toenails
Diagnosis: oral lichen planus
Compared with the more self-limited nature of its cutaneous counterpart, oral lichen planus (OLP) causes more chronic inflammatory lesions, resulting in increased morbidity and a greater therapeutic challenge for physicians. Of the 3 basic clinical morphologies of the disease (reticular, erythematous, and erosive), the last form tends to be more symptomatic and prompts patients to seek treatment.1,2 The more asymptomatic reticular form is the most easily recognizable variant and may cause increased symptoms when involving the tongue, typically manifesting on the dorsal surface.1
In contrast to reticular lesions in other oral sites, tongue lesions do not usually exhibit the characteristic interlacing pattern of white, raised striae; instead they manifest as well-demarcated areas with a patch-like pattern of erythematous, atrophic, and keratotic regions. 3 Erosive OLP more commonly involves the lateral tongue and displays erythematous or ulcerated areas with peripheral keratosis, forming fine centrifugal striae.3
Oral lichen planus affects approximately 1% to 4% of the population 4 and is seen most commonly in older women.1,5,6 It has a predilection for bilateral involvement of the buccal mucosa but may also affect (in descending order of frequency) the tongue, gingiva, lips, floor of the mouth, and palate.1,2 Isolated involvement of only 1 oral site is infrequent, with the exception of gingival lesions.1 Extraoral cutaneous lichen planus has been reported in 16% to 44% of patients with oral disease; it most frequently involves the genital mucosa, as well as the scalp, nails, and esophageal and ocular mucosae.4 A multidisciplinary approach including generalists, dermatologists, otolaryngologists, ophthalmologists, and gynecologists may be necessary for patient evaluation and management.
Pathogenesis of oral lichen planus
Although the precise cause of OLP is unknown, its pathogenesis has been linked to an autoimmune mechanism involving autocytotoxic CD8+ T cells, which trigger apoptosis (programmed cell death) of basal keratinocytes.4,7 An imbalance between T cell helper and suppressor activity has also been observed.1,7
Complications: Carcinoma, chronic liver disease
Malignant transformation to squamous cell carcinoma (SCC) is seen in 0.4% to 5% of patients with OLP, particularly those with erosive and erythematous disease.1,2 Increased risk factors for SCC have not been identified in these patients, but a greater prevalence of Candida albicansmay be associated with carcinogenesis, as may herpes simplex and human papilloma viruses, immunosuppressive therapy, and an inflammatory cytokine-rich microenvironment.1
In Japan and parts of Southern Europe, OLP has been associated with hepatitis C infection and chronic liver disease, but these findings have not been reproduced in patients in the US.
Making the diagnosis: exam, biopsy, immunofluorescence
A detailed history and physical examination are usually sufficient to diagnose OLP, although laboratory studies and biopsy may be required to exclude malignancy or distinguish OLP from conditions such as pemphigus vulgaris and mucous membrane pemphigoid. A biopsy is rarely needed, and should only be performed by physicians that have training in a tongue biopsy.
If necessary, biopsy samples should be obtained from the most representative sample area of the tongue. After administration of local anesthesia (lidocaine 1:100,000 with epinephrine) near the periphery of the site, biopsy is performed using a #15 blade scalpel or biopsy punch. An assistant may be needed to grasp the tongue by wrapping a gauze sponge over the tip and securing it firmly. When tongue biopsy sites are closed, deep sutures should be placed if necessary, and mucosal sutures should be placed relatively close together, as inadequately closed wounds on the tongue tend to reopen, resulting in bleeding and prolonged healing time.
Specimens should be placed in a 10% formalin solution for transportation to a pathologist. Patients should return to the clinic after 7 to 14 days for examination of the biopsy site and suture removal.
Microscopically, OLP demonstrates vacuolar degeneration, basal cell lysis, and liquefactive degeneration1 along with focal hyperkeratosis, a characteristic amorphous eosinophilic band at the basement membrane level, and a dense, bandlike subepithelial lymphocytic infiltrate.5,6 Direct immunofluorescence, displaying granular fibrino-gen and variable immunoglobulin deposited linearly near the basement membrane, and possibly cytoid bodies, is useful in cases of suspected OLP with nondiagnostic histological findings, as well as in those with gingival involvement.1
Differential diagnosis
The clinical differential diagnosis of OLP includes the autoimmune bullous diseases pemphigus vulgaris, mucous membrane pemphigoid, dermatitis herpetiformis, and linear immuno-globulin A disease, which can all be excluded with direct and indirect immunofluorescence studies. When clinically appropriate, biopsy specimens submitted for direct immunofluorescence must be preserved in Michel solution. Reactive keratoses, chronic hyperplastic candidosis, epithelial dysplasia, discoid lupus erythematosus, anemic states, and several gastrointestinal diseases including oral Crohn disease and chronic hepatic disease, must also be ruled out.
Oral lichenoid reactions may develop as a hypersensitivity to dental materials and drugs, and must also be considered.1 Patch testing may help identify a contact allergy to dental prosthesis components, including gold, mercury, and palladium salts.
Management: steroids, immunosuppressants, surgery
Because fewer than 20% of patients experience total remission,5 treatment of OLP is chronic and palliative. Most symptomatic patients are taught to avoid exacerbating factors and given topical steroid therapy (dexamethasone rinse, fluocinolone gel, triamcinolone cream). Observation, intralesional and systemic corticosteroids, immunosuppressants, antifungals, retinoids, antimalarials, dapsone, oral psoralen-ultraviolet-light (PUVA) treatment, and surgical techniques (CO2 and neodymium: yttrium-aluminum-garnet [Nd:YAG] laser therapy, cryotherapy, and excision) are also employed, alone and in combination.5,8
Tacrolimus (Protopic), an immunomodulator, administered topically in 0.03% to 0.3% concentrations using Aquaphor or paraffin ointment base, has been shown effective and well tolerated in controlling symptoms,8,9 and is a less costly alternative to topical cyclosporine.9 It has been demonstrated effective prospective-ly,10 and is safe in long-term therapy11 of erosive OLP, but it has been reported in one case to cause hyperpigmentation of oral mucosa.12
A recent study13 demonstrated the utility of low-dose 308-nm excimer laser radiation for symptomatic OLP.
Patient management
Our patient was instructed to avoid foods and substances that caused irritation of her tongue and oral mucosa. In addition, she was prescribed topical fluocinolone gel 0.025% 3 times daily, and was given information about alternative treatment options, including tacrolimus and surgical therapy. She was instructed to perform gentle yet thorough daily oral hygiene and to follow-up in 6 months for re-examination.
Corresponding author
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street (BAR 314), Boston, MA 02114. E-mail: [email protected].
December 2004’s Photo Rounds, “Rupturing bullae not respondign to antiobiotics,” left out the names of two of the article’s authors. The correct authors of the piece are John Sauret, MD, FAAFP, Sandra Yale, DO, and Ahunna Ahiarah, MD. We regret the error.
A 40-year-old woman came into the office with a 1-year history of painful red lesions with white striations on her tongue (Figure 1). The lesions caused burning after she ate spicy food and an increased sensitivity to mouthwash. She reported a slow, progressive onset and worsening of her condition, with intensifying symptoms during periods of emotional stress.
On physical examination, we observed an erythematous and cream-colored reticulated patch with a few small focal areas of erosion covering the dorsal tongue. The remainder of the oral mucosa appeared normal. She also had thinning and ridging of the toenails (Figure 2).
What is the most likely diagnosis?
How would you manage this case?
FIGURE 1
Tongue with painful lesions
FIGURE 2
Thinning toenails
Diagnosis: oral lichen planus
Compared with the more self-limited nature of its cutaneous counterpart, oral lichen planus (OLP) causes more chronic inflammatory lesions, resulting in increased morbidity and a greater therapeutic challenge for physicians. Of the 3 basic clinical morphologies of the disease (reticular, erythematous, and erosive), the last form tends to be more symptomatic and prompts patients to seek treatment.1,2 The more asymptomatic reticular form is the most easily recognizable variant and may cause increased symptoms when involving the tongue, typically manifesting on the dorsal surface.1
In contrast to reticular lesions in other oral sites, tongue lesions do not usually exhibit the characteristic interlacing pattern of white, raised striae; instead they manifest as well-demarcated areas with a patch-like pattern of erythematous, atrophic, and keratotic regions. 3 Erosive OLP more commonly involves the lateral tongue and displays erythematous or ulcerated areas with peripheral keratosis, forming fine centrifugal striae.3
Oral lichen planus affects approximately 1% to 4% of the population 4 and is seen most commonly in older women.1,5,6 It has a predilection for bilateral involvement of the buccal mucosa but may also affect (in descending order of frequency) the tongue, gingiva, lips, floor of the mouth, and palate.1,2 Isolated involvement of only 1 oral site is infrequent, with the exception of gingival lesions.1 Extraoral cutaneous lichen planus has been reported in 16% to 44% of patients with oral disease; it most frequently involves the genital mucosa, as well as the scalp, nails, and esophageal and ocular mucosae.4 A multidisciplinary approach including generalists, dermatologists, otolaryngologists, ophthalmologists, and gynecologists may be necessary for patient evaluation and management.
Pathogenesis of oral lichen planus
Although the precise cause of OLP is unknown, its pathogenesis has been linked to an autoimmune mechanism involving autocytotoxic CD8+ T cells, which trigger apoptosis (programmed cell death) of basal keratinocytes.4,7 An imbalance between T cell helper and suppressor activity has also been observed.1,7
Complications: Carcinoma, chronic liver disease
Malignant transformation to squamous cell carcinoma (SCC) is seen in 0.4% to 5% of patients with OLP, particularly those with erosive and erythematous disease.1,2 Increased risk factors for SCC have not been identified in these patients, but a greater prevalence of Candida albicansmay be associated with carcinogenesis, as may herpes simplex and human papilloma viruses, immunosuppressive therapy, and an inflammatory cytokine-rich microenvironment.1
In Japan and parts of Southern Europe, OLP has been associated with hepatitis C infection and chronic liver disease, but these findings have not been reproduced in patients in the US.
Making the diagnosis: exam, biopsy, immunofluorescence
A detailed history and physical examination are usually sufficient to diagnose OLP, although laboratory studies and biopsy may be required to exclude malignancy or distinguish OLP from conditions such as pemphigus vulgaris and mucous membrane pemphigoid. A biopsy is rarely needed, and should only be performed by physicians that have training in a tongue biopsy.
If necessary, biopsy samples should be obtained from the most representative sample area of the tongue. After administration of local anesthesia (lidocaine 1:100,000 with epinephrine) near the periphery of the site, biopsy is performed using a #15 blade scalpel or biopsy punch. An assistant may be needed to grasp the tongue by wrapping a gauze sponge over the tip and securing it firmly. When tongue biopsy sites are closed, deep sutures should be placed if necessary, and mucosal sutures should be placed relatively close together, as inadequately closed wounds on the tongue tend to reopen, resulting in bleeding and prolonged healing time.
Specimens should be placed in a 10% formalin solution for transportation to a pathologist. Patients should return to the clinic after 7 to 14 days for examination of the biopsy site and suture removal.
Microscopically, OLP demonstrates vacuolar degeneration, basal cell lysis, and liquefactive degeneration1 along with focal hyperkeratosis, a characteristic amorphous eosinophilic band at the basement membrane level, and a dense, bandlike subepithelial lymphocytic infiltrate.5,6 Direct immunofluorescence, displaying granular fibrino-gen and variable immunoglobulin deposited linearly near the basement membrane, and possibly cytoid bodies, is useful in cases of suspected OLP with nondiagnostic histological findings, as well as in those with gingival involvement.1
Differential diagnosis
The clinical differential diagnosis of OLP includes the autoimmune bullous diseases pemphigus vulgaris, mucous membrane pemphigoid, dermatitis herpetiformis, and linear immuno-globulin A disease, which can all be excluded with direct and indirect immunofluorescence studies. When clinically appropriate, biopsy specimens submitted for direct immunofluorescence must be preserved in Michel solution. Reactive keratoses, chronic hyperplastic candidosis, epithelial dysplasia, discoid lupus erythematosus, anemic states, and several gastrointestinal diseases including oral Crohn disease and chronic hepatic disease, must also be ruled out.
Oral lichenoid reactions may develop as a hypersensitivity to dental materials and drugs, and must also be considered.1 Patch testing may help identify a contact allergy to dental prosthesis components, including gold, mercury, and palladium salts.
Management: steroids, immunosuppressants, surgery
Because fewer than 20% of patients experience total remission,5 treatment of OLP is chronic and palliative. Most symptomatic patients are taught to avoid exacerbating factors and given topical steroid therapy (dexamethasone rinse, fluocinolone gel, triamcinolone cream). Observation, intralesional and systemic corticosteroids, immunosuppressants, antifungals, retinoids, antimalarials, dapsone, oral psoralen-ultraviolet-light (PUVA) treatment, and surgical techniques (CO2 and neodymium: yttrium-aluminum-garnet [Nd:YAG] laser therapy, cryotherapy, and excision) are also employed, alone and in combination.5,8
Tacrolimus (Protopic), an immunomodulator, administered topically in 0.03% to 0.3% concentrations using Aquaphor or paraffin ointment base, has been shown effective and well tolerated in controlling symptoms,8,9 and is a less costly alternative to topical cyclosporine.9 It has been demonstrated effective prospective-ly,10 and is safe in long-term therapy11 of erosive OLP, but it has been reported in one case to cause hyperpigmentation of oral mucosa.12
A recent study13 demonstrated the utility of low-dose 308-nm excimer laser radiation for symptomatic OLP.
Patient management
Our patient was instructed to avoid foods and substances that caused irritation of her tongue and oral mucosa. In addition, she was prescribed topical fluocinolone gel 0.025% 3 times daily, and was given information about alternative treatment options, including tacrolimus and surgical therapy. She was instructed to perform gentle yet thorough daily oral hygiene and to follow-up in 6 months for re-examination.
Corresponding author
Amor Khachemoune, MD, CWS, Wellman Center for Photomedicine Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, 40 Blossom Street (BAR 314), Boston, MA 02114. E-mail: [email protected].
December 2004’s Photo Rounds, “Rupturing bullae not respondign to antiobiotics,” left out the names of two of the article’s authors. The correct authors of the piece are John Sauret, MD, FAAFP, Sandra Yale, DO, and Ahunna Ahiarah, MD. We regret the error.
1. Eisen D. The clinical features, malignant potential, and systemic associations of oral lichen planus: A study of 723 patients. J Am Acad Dermatol 2002;46:207-214.
2. Gorsky M, Raviv M, Moskona D, Laufer M, Bodner L. Clinical characteristics and treatment of patients with oral lichen planus in Israel. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1996;82:644-649.
3. Allen CM, Blozis GG. Oral mucosal lesions. In Cummings CW, Schuller DE (eds): Otolaryngology: Head and Neck Surgery, vol 2, 2nd ed. St Louis, Mo: Mosby-Yearbook; 1993;1374-1375.
4. Eisen D. The evaluation of cutaneous, genital, scalp, nail, esophageal, and ocular involvement in patients with oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999;88:431-436.
5. Brown RS, Bottomley WK, Puente E, Lavigne GL. A retrospective evaluation of 193 patients with oral lichen planus. J Oral Pathol Med 1993;22:69-72.
6. Thorn JJ, Holmstrup P, Rindum J, Pindborg JJ. Course of various clinical forms of oral lichen planus. A retrospective follow-up study of 611 patients. J Oral Pathol 1998;17:213-218.
7. Porter SR, Kirby A, Olsen I, Barrett W. Immunologic aspects of dermal and oral lichen planus. A review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997;83:358-366.
8. Rozycki TW, Rogers RS, Pittelkow MR, et al. Topical tacrolimus in the treatment of symptomatic oral lichen planus: A series of 13 patients. J Am Acad Dermatol 2002;46:27-34.
9. Kaliakatsou F, Hodgson TA, Lewsey JD, Hegarty AM, Murphy AG, Porter SR. Management of recalcitrant ulcer-ative oral lichen planus with topical tacrolimus. J Am Acad Dermatol 2002;46:35-41.
10. Olivier V, Lacour JP, Mousnier A, Garraffo R, Monteil RA, Ortonne JP. Treatment of chronic erosive oral lichen planus with low concentrations of topical tacrolimus. An open prospective study. Arch Dermatol 2002;138:1335-1338.
11. Hodgson TA, Sahni N, Kaliakatsou F, Buchanan JA, Porter SR. Long-term efficacy and safety of topical tacrolimus in the management of ulcerative/erosive oral lichen planus. Eur J Dermatol 2003;13:466-470.
12. Shen JT, Pedvis-Leftick A. Mucosal staining after using topical tacrolimus to treat erosive oral lichen planus. J Am Acad Dermatol 2004;50-326.
13. Trehan M, Taylor CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol 2004;140:415-420.
1. Eisen D. The clinical features, malignant potential, and systemic associations of oral lichen planus: A study of 723 patients. J Am Acad Dermatol 2002;46:207-214.
2. Gorsky M, Raviv M, Moskona D, Laufer M, Bodner L. Clinical characteristics and treatment of patients with oral lichen planus in Israel. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1996;82:644-649.
3. Allen CM, Blozis GG. Oral mucosal lesions. In Cummings CW, Schuller DE (eds): Otolaryngology: Head and Neck Surgery, vol 2, 2nd ed. St Louis, Mo: Mosby-Yearbook; 1993;1374-1375.
4. Eisen D. The evaluation of cutaneous, genital, scalp, nail, esophageal, and ocular involvement in patients with oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999;88:431-436.
5. Brown RS, Bottomley WK, Puente E, Lavigne GL. A retrospective evaluation of 193 patients with oral lichen planus. J Oral Pathol Med 1993;22:69-72.
6. Thorn JJ, Holmstrup P, Rindum J, Pindborg JJ. Course of various clinical forms of oral lichen planus. A retrospective follow-up study of 611 patients. J Oral Pathol 1998;17:213-218.
7. Porter SR, Kirby A, Olsen I, Barrett W. Immunologic aspects of dermal and oral lichen planus. A review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997;83:358-366.
8. Rozycki TW, Rogers RS, Pittelkow MR, et al. Topical tacrolimus in the treatment of symptomatic oral lichen planus: A series of 13 patients. J Am Acad Dermatol 2002;46:27-34.
9. Kaliakatsou F, Hodgson TA, Lewsey JD, Hegarty AM, Murphy AG, Porter SR. Management of recalcitrant ulcer-ative oral lichen planus with topical tacrolimus. J Am Acad Dermatol 2002;46:35-41.
10. Olivier V, Lacour JP, Mousnier A, Garraffo R, Monteil RA, Ortonne JP. Treatment of chronic erosive oral lichen planus with low concentrations of topical tacrolimus. An open prospective study. Arch Dermatol 2002;138:1335-1338.
11. Hodgson TA, Sahni N, Kaliakatsou F, Buchanan JA, Porter SR. Long-term efficacy and safety of topical tacrolimus in the management of ulcerative/erosive oral lichen planus. Eur J Dermatol 2003;13:466-470.
12. Shen JT, Pedvis-Leftick A. Mucosal staining after using topical tacrolimus to treat erosive oral lichen planus. J Am Acad Dermatol 2004;50-326.
13. Trehan M, Taylor CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol 2004;140:415-420.