User login
Cellulitis unresponsive to antibiotics
A 36-year-old white woman sought care at our outpatient clinic for 2 days of right forearm and hand redness, swelling, and severe pain at a previous intravenous (IV) catheter site. The patient had received intramuscular antibiotics for culture-positive streptococcal pharyngitis 2 weeks earlier, and then IV fluids and steroids due to poor oral intake from odynophagia.
She indicated that since the time of the pharyngitis diagnosis, she’d had a persistent fever. At our clinic, she was given a diagnosis of cellulitis and started on a course of oral cephalexin.
Three days later, she returned. She had a temperature of 100°F and her right forearm and wrist were exquisitely sensitive to touch over a warm and indurated violaceous rash with pseudovesicles over the most edematous portions (FIGURE 1). This rash extended onto her right hand and proximal phalanges. Her fingers hurt when she moved them; sensation and pulses were intact.
Her left wrist was now mildly swollen, with associated warmth and erythema, but no vesicles. Her left ankle was also warm, erythematous, and moderately swollen with significant tenderness to palpation. She had a few scattered nodular, erythematous lesions on her upper right arm and chest.
We (RW and VK) admitted the patient and started her on IV vancomycin and clindamycin to treat presumed refractory cellulitis. On Day 2 she hadn’t improved, so we added gatifloxacin for gram-negative coverage. On Day 3, her lesions worsened, so we transferred her to a higher-level facility.
FIGURE 1
Worsening lesions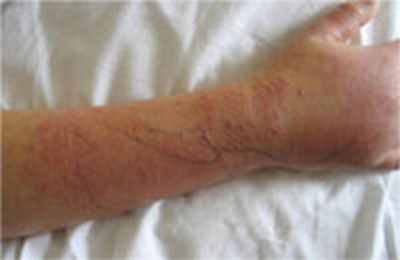
The patient’s right forearm and wrist were exquisitely sensitive to touch over the rash, with pseudovesicles. Ink markings denote original boundaries of the lesion.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Sweet’s syndrome
A skin biopsy revealed subepidermal bullae with marked dermal and mixed lymphocytic and neutrophilic infiltrate consistent with Sweet’s syndrome.
Sweet’s syndrome, also known as acute febrile neutrophilic dermatoses, is a condition that was originally described by Dr. Robert Sweet in 1964.1 The condition can be classified based on etiology as "classical" or idiopathic, malignancy-associated, or drug-induced.2 Classical Sweet’s syndrome typically affects women 30 to 50 years of age, but has been reported in men and women of all ages.3-5 More than 500 cases have been reported in the literature, but incidence and prevalence data are limited.6 Conditions associated with classical Sweet’s syndrome include upper respiratory infection, gastrointestinal infection, inflammatory bowel disease, and pregnancy.5
Cellulitis and lupus erythematosus are part of the differential
The differential diagnosis for erythematous, indurated, tender skin and soft tissue lesions includes cellulitis, erysipelas, thrombophlebitis, urticaria, shingles, drug-induced eruptions, and herpes simplex virus infections.4-6
Less common conditions include erythema multiforme, erythema nodusum, tuberculosis, leukemia cutis, lupus erythematosus, vasculitis, pyoderma gangrenosa, Behçet’s disease, erythema elevatum diutinum, familial Mediterranean fever, and Sweet’s syndrome.4-6
Diagnosis hinges on these criteria
Two major criteria are required to make a diagnosis of Sweet’s syndrome. They are:2
- the abrupt onset of tender, erythematous plaques or nodules (FIGURE 2)
- a predominately neutrophilic infiltration in the dermis without leukocytoclastic vasculitis.
At least 2 of 4 minor criteria must also be present:2
- a precedent respiratory or gastrointestinal (GI) infection, vaccination, inflammatory disease, malignancy, or pregnancy
- malaise and fever
- elevated erythrocyte sedimentation rate, C-reactive protein, and leukocytosis with a left shift. (Our patient’s erythrocyte sedimentation rate was 93 mm/h; her C-reactive protein was 11.47 mg/dL.)
- excellent response to corticosteroids or potassium iodide.
Although skin manifestations are a hallmark sign (which makes Sweet’s syndrome an important differential diagnosis for an unusual case of cellulitis, or one that is unresponsive to antibiotics), many possible coincident manifestations can occur. These include arthralgia, myalgia, headache, and malaise.5 Multi-system involvement has also been reported, affecting bone, the central nervous system, eyes, kidneys, heart, lungs, and GI tract.5
FIGURE 2
Another presentation of Sweet’s syndrome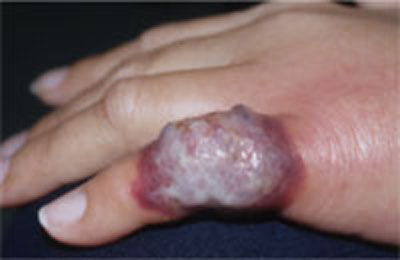
Some Sweet’s syndrome lesions appear "juicy." This lesion occurred at the site of minor trauma (pathergy) in a patient who was febrile and systemically ill.
Treatment: Corticosteroids, not antibiotics
A high clinical suspicion for Sweet’s syndrome is critical to management decisions because the primary treatment is corticosteroids, rather than antibiotics.5,6 Oral prednisone at 0.5 to 1.5 mg/kg per day with taper over 2 to 6 weeks is a standard regimen.2 IV methylprednisolone of up to 1000 mg per day for 3 to 5 days, followed by a tapered oral dose of corticosteroids over several weeks is another option.5 Other first-line treatment options include colchicine (0.5 mg 3 times a day) and oral potassium iodide (300 mg enteric-coated tablets 3 times a day).5,6 Indomethacin, cyclosporine, surgery, and dapsone are other options/adjuncts.
Recurrences of Sweet’s syndrome may occur, regardless of treatment. They are more likely to occur in patients with an underlying malignancy and may herald recurrence of malignancy in a previously treated patient.5 Treating an underlying malignancy (or discontinuing the causative medication in drug-induced Sweet’s syndrome) may resolve symptoms. IV administration of methylprednisolone, as discussed earlier, has been successful for refractory cases.5
Our patient responded well to treatment
Our patient was started on prednisone 60 mg daily and her antibiotics were discontinued. Within 24 hours, her skin lesions regressed. She was discharged on a tapering course of the prednisone.
After several weeks, her lesions completely cleared without scarring or recurrence.
CORRESPONDENCE
Ryan A. Withrow, DO, 4-2807 Reilly Road, Family Medicine Residency Clinic, Fort Bragg, NC 28314; [email protected]
1. Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
2. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh, Scotland: Mosby/Elsevier; 2010.
3. Kemmett D, Hunter JA. Sweet’s syndrome: a clinicopathologic review of twenty-nine cases. J Am Acad Dermatol. 1990;23:503-507.
4. Boatman BW, Taylor RC, Klein LE, et al. Sweet’s syndrome in children. South Med J. 1994;87:193-196.
5. Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
6. Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
A 36-year-old white woman sought care at our outpatient clinic for 2 days of right forearm and hand redness, swelling, and severe pain at a previous intravenous (IV) catheter site. The patient had received intramuscular antibiotics for culture-positive streptococcal pharyngitis 2 weeks earlier, and then IV fluids and steroids due to poor oral intake from odynophagia.
She indicated that since the time of the pharyngitis diagnosis, she’d had a persistent fever. At our clinic, she was given a diagnosis of cellulitis and started on a course of oral cephalexin.
Three days later, she returned. She had a temperature of 100°F and her right forearm and wrist were exquisitely sensitive to touch over a warm and indurated violaceous rash with pseudovesicles over the most edematous portions (FIGURE 1). This rash extended onto her right hand and proximal phalanges. Her fingers hurt when she moved them; sensation and pulses were intact.
Her left wrist was now mildly swollen, with associated warmth and erythema, but no vesicles. Her left ankle was also warm, erythematous, and moderately swollen with significant tenderness to palpation. She had a few scattered nodular, erythematous lesions on her upper right arm and chest.
We (RW and VK) admitted the patient and started her on IV vancomycin and clindamycin to treat presumed refractory cellulitis. On Day 2 she hadn’t improved, so we added gatifloxacin for gram-negative coverage. On Day 3, her lesions worsened, so we transferred her to a higher-level facility.
FIGURE 1
Worsening lesions
The patient’s right forearm and wrist were exquisitely sensitive to touch over the rash, with pseudovesicles. Ink markings denote original boundaries of the lesion.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Sweet’s syndrome
A skin biopsy revealed subepidermal bullae with marked dermal and mixed lymphocytic and neutrophilic infiltrate consistent with Sweet’s syndrome.
Sweet’s syndrome, also known as acute febrile neutrophilic dermatoses, is a condition that was originally described by Dr. Robert Sweet in 1964.1 The condition can be classified based on etiology as "classical" or idiopathic, malignancy-associated, or drug-induced.2 Classical Sweet’s syndrome typically affects women 30 to 50 years of age, but has been reported in men and women of all ages.3-5 More than 500 cases have been reported in the literature, but incidence and prevalence data are limited.6 Conditions associated with classical Sweet’s syndrome include upper respiratory infection, gastrointestinal infection, inflammatory bowel disease, and pregnancy.5
Cellulitis and lupus erythematosus are part of the differential
The differential diagnosis for erythematous, indurated, tender skin and soft tissue lesions includes cellulitis, erysipelas, thrombophlebitis, urticaria, shingles, drug-induced eruptions, and herpes simplex virus infections.4-6
Less common conditions include erythema multiforme, erythema nodusum, tuberculosis, leukemia cutis, lupus erythematosus, vasculitis, pyoderma gangrenosa, Behçet’s disease, erythema elevatum diutinum, familial Mediterranean fever, and Sweet’s syndrome.4-6
Diagnosis hinges on these criteria
Two major criteria are required to make a diagnosis of Sweet’s syndrome. They are:2
- the abrupt onset of tender, erythematous plaques or nodules (FIGURE 2)
- a predominately neutrophilic infiltration in the dermis without leukocytoclastic vasculitis.
At least 2 of 4 minor criteria must also be present:2
- a precedent respiratory or gastrointestinal (GI) infection, vaccination, inflammatory disease, malignancy, or pregnancy
- malaise and fever
- elevated erythrocyte sedimentation rate, C-reactive protein, and leukocytosis with a left shift. (Our patient’s erythrocyte sedimentation rate was 93 mm/h; her C-reactive protein was 11.47 mg/dL.)
- excellent response to corticosteroids or potassium iodide.
Although skin manifestations are a hallmark sign (which makes Sweet’s syndrome an important differential diagnosis for an unusual case of cellulitis, or one that is unresponsive to antibiotics), many possible coincident manifestations can occur. These include arthralgia, myalgia, headache, and malaise.5 Multi-system involvement has also been reported, affecting bone, the central nervous system, eyes, kidneys, heart, lungs, and GI tract.5
FIGURE 2
Another presentation of Sweet’s syndrome
Some Sweet’s syndrome lesions appear "juicy." This lesion occurred at the site of minor trauma (pathergy) in a patient who was febrile and systemically ill.
Treatment: Corticosteroids, not antibiotics
A high clinical suspicion for Sweet’s syndrome is critical to management decisions because the primary treatment is corticosteroids, rather than antibiotics.5,6 Oral prednisone at 0.5 to 1.5 mg/kg per day with taper over 2 to 6 weeks is a standard regimen.2 IV methylprednisolone of up to 1000 mg per day for 3 to 5 days, followed by a tapered oral dose of corticosteroids over several weeks is another option.5 Other first-line treatment options include colchicine (0.5 mg 3 times a day) and oral potassium iodide (300 mg enteric-coated tablets 3 times a day).5,6 Indomethacin, cyclosporine, surgery, and dapsone are other options/adjuncts.
Recurrences of Sweet’s syndrome may occur, regardless of treatment. They are more likely to occur in patients with an underlying malignancy and may herald recurrence of malignancy in a previously treated patient.5 Treating an underlying malignancy (or discontinuing the causative medication in drug-induced Sweet’s syndrome) may resolve symptoms. IV administration of methylprednisolone, as discussed earlier, has been successful for refractory cases.5
Our patient responded well to treatment
Our patient was started on prednisone 60 mg daily and her antibiotics were discontinued. Within 24 hours, her skin lesions regressed. She was discharged on a tapering course of the prednisone.
After several weeks, her lesions completely cleared without scarring or recurrence.
CORRESPONDENCE
Ryan A. Withrow, DO, 4-2807 Reilly Road, Family Medicine Residency Clinic, Fort Bragg, NC 28314; [email protected]
A 36-year-old white woman sought care at our outpatient clinic for 2 days of right forearm and hand redness, swelling, and severe pain at a previous intravenous (IV) catheter site. The patient had received intramuscular antibiotics for culture-positive streptococcal pharyngitis 2 weeks earlier, and then IV fluids and steroids due to poor oral intake from odynophagia.
She indicated that since the time of the pharyngitis diagnosis, she’d had a persistent fever. At our clinic, she was given a diagnosis of cellulitis and started on a course of oral cephalexin.
Three days later, she returned. She had a temperature of 100°F and her right forearm and wrist were exquisitely sensitive to touch over a warm and indurated violaceous rash with pseudovesicles over the most edematous portions (FIGURE 1). This rash extended onto her right hand and proximal phalanges. Her fingers hurt when she moved them; sensation and pulses were intact.
Her left wrist was now mildly swollen, with associated warmth and erythema, but no vesicles. Her left ankle was also warm, erythematous, and moderately swollen with significant tenderness to palpation. She had a few scattered nodular, erythematous lesions on her upper right arm and chest.
We (RW and VK) admitted the patient and started her on IV vancomycin and clindamycin to treat presumed refractory cellulitis. On Day 2 she hadn’t improved, so we added gatifloxacin for gram-negative coverage. On Day 3, her lesions worsened, so we transferred her to a higher-level facility.
FIGURE 1
Worsening lesions
The patient’s right forearm and wrist were exquisitely sensitive to touch over the rash, with pseudovesicles. Ink markings denote original boundaries of the lesion.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Sweet’s syndrome
A skin biopsy revealed subepidermal bullae with marked dermal and mixed lymphocytic and neutrophilic infiltrate consistent with Sweet’s syndrome.
Sweet’s syndrome, also known as acute febrile neutrophilic dermatoses, is a condition that was originally described by Dr. Robert Sweet in 1964.1 The condition can be classified based on etiology as "classical" or idiopathic, malignancy-associated, or drug-induced.2 Classical Sweet’s syndrome typically affects women 30 to 50 years of age, but has been reported in men and women of all ages.3-5 More than 500 cases have been reported in the literature, but incidence and prevalence data are limited.6 Conditions associated with classical Sweet’s syndrome include upper respiratory infection, gastrointestinal infection, inflammatory bowel disease, and pregnancy.5
Cellulitis and lupus erythematosus are part of the differential
The differential diagnosis for erythematous, indurated, tender skin and soft tissue lesions includes cellulitis, erysipelas, thrombophlebitis, urticaria, shingles, drug-induced eruptions, and herpes simplex virus infections.4-6
Less common conditions include erythema multiforme, erythema nodusum, tuberculosis, leukemia cutis, lupus erythematosus, vasculitis, pyoderma gangrenosa, Behçet’s disease, erythema elevatum diutinum, familial Mediterranean fever, and Sweet’s syndrome.4-6
Diagnosis hinges on these criteria
Two major criteria are required to make a diagnosis of Sweet’s syndrome. They are:2
- the abrupt onset of tender, erythematous plaques or nodules (FIGURE 2)
- a predominately neutrophilic infiltration in the dermis without leukocytoclastic vasculitis.
At least 2 of 4 minor criteria must also be present:2
- a precedent respiratory or gastrointestinal (GI) infection, vaccination, inflammatory disease, malignancy, or pregnancy
- malaise and fever
- elevated erythrocyte sedimentation rate, C-reactive protein, and leukocytosis with a left shift. (Our patient’s erythrocyte sedimentation rate was 93 mm/h; her C-reactive protein was 11.47 mg/dL.)
- excellent response to corticosteroids or potassium iodide.
Although skin manifestations are a hallmark sign (which makes Sweet’s syndrome an important differential diagnosis for an unusual case of cellulitis, or one that is unresponsive to antibiotics), many possible coincident manifestations can occur. These include arthralgia, myalgia, headache, and malaise.5 Multi-system involvement has also been reported, affecting bone, the central nervous system, eyes, kidneys, heart, lungs, and GI tract.5
FIGURE 2
Another presentation of Sweet’s syndrome
Some Sweet’s syndrome lesions appear "juicy." This lesion occurred at the site of minor trauma (pathergy) in a patient who was febrile and systemically ill.
Treatment: Corticosteroids, not antibiotics
A high clinical suspicion for Sweet’s syndrome is critical to management decisions because the primary treatment is corticosteroids, rather than antibiotics.5,6 Oral prednisone at 0.5 to 1.5 mg/kg per day with taper over 2 to 6 weeks is a standard regimen.2 IV methylprednisolone of up to 1000 mg per day for 3 to 5 days, followed by a tapered oral dose of corticosteroids over several weeks is another option.5 Other first-line treatment options include colchicine (0.5 mg 3 times a day) and oral potassium iodide (300 mg enteric-coated tablets 3 times a day).5,6 Indomethacin, cyclosporine, surgery, and dapsone are other options/adjuncts.
Recurrences of Sweet’s syndrome may occur, regardless of treatment. They are more likely to occur in patients with an underlying malignancy and may herald recurrence of malignancy in a previously treated patient.5 Treating an underlying malignancy (or discontinuing the causative medication in drug-induced Sweet’s syndrome) may resolve symptoms. IV administration of methylprednisolone, as discussed earlier, has been successful for refractory cases.5
Our patient responded well to treatment
Our patient was started on prednisone 60 mg daily and her antibiotics were discontinued. Within 24 hours, her skin lesions regressed. She was discharged on a tapering course of the prednisone.
After several weeks, her lesions completely cleared without scarring or recurrence.
CORRESPONDENCE
Ryan A. Withrow, DO, 4-2807 Reilly Road, Family Medicine Residency Clinic, Fort Bragg, NC 28314; [email protected]
1. Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
2. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh, Scotland: Mosby/Elsevier; 2010.
3. Kemmett D, Hunter JA. Sweet’s syndrome: a clinicopathologic review of twenty-nine cases. J Am Acad Dermatol. 1990;23:503-507.
4. Boatman BW, Taylor RC, Klein LE, et al. Sweet’s syndrome in children. South Med J. 1994;87:193-196.
5. Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
6. Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.
1. Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
2. Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Edinburgh, Scotland: Mosby/Elsevier; 2010.
3. Kemmett D, Hunter JA. Sweet’s syndrome: a clinicopathologic review of twenty-nine cases. J Am Acad Dermatol. 1990;23:503-507.
4. Boatman BW, Taylor RC, Klein LE, et al. Sweet’s syndrome in children. South Med J. 1994;87:193-196.
5. Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
6. Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556.