User login
Expanded approval for daratumumab in multiple myeloma
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.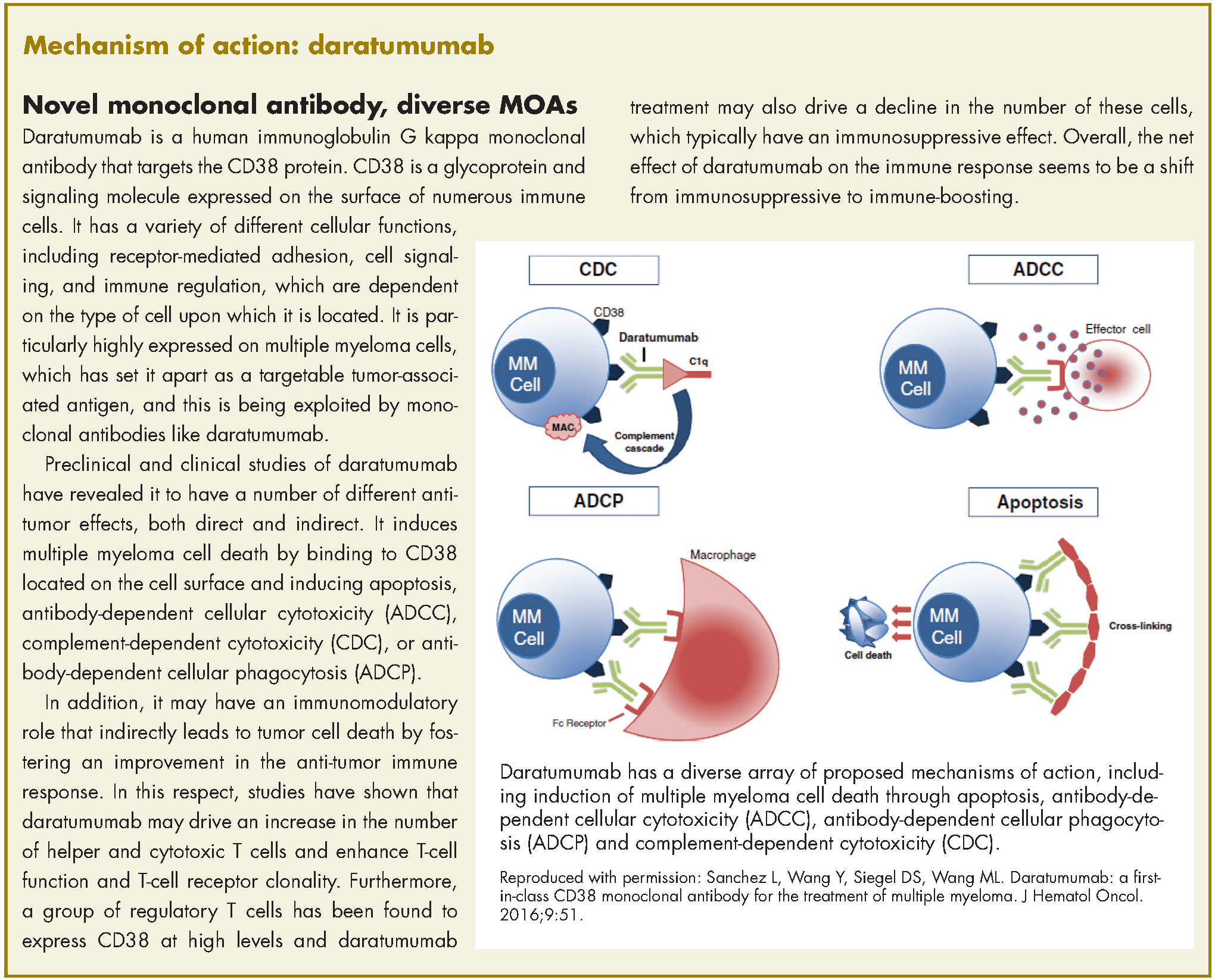
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
Ribociclib: another CDK inhibitor hits the mark in breast cancer
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
This spring, the US Food and Drug Administration approved a second cyclin-dependent kinase (CDK) inhibitor for the treatment of postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced/metastatic breast cancer in combination with aromatase inhibitors (AIs).1 The drug, ribociclib, joins palbociclib as the second drug in this class, which targets key regulators of the mammalian cell cycle and can help to overcome resistance to endocrine therapy–like AIs, a standard front-line treatment option in this group of patients. Palbociclib (Ibrance) was approved last year in combination with the AI letrozole, which was recently expanded to include its use in combination with all AIs, the same indication for which ribociclib received approval.
The ribociclib approval was based on the results of a phase 3, randomized, double-blind, placebo-controlled, international clinical trial called MONALEESA-2.2 The trial, conducted in 29 countries, compared the effects of ribociclib plus letrozole with letrozole plus placebo in 668 postmenopausal women with locally confirmed, HR-positive, HER2-negative, recurrent or metastatic breast cancer.
Patients had not received previous systemic therapy for advanced disease, had measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (range, 1-5; 0, fully active and 5, dead), and had adequate bone marrow and organ function.
Patients were excluded if they had received previous CDK4/6 therapy, any previous systemic chemotherapy, endocrine therapy for advanced disease, previous neoadjuvant or adjuvant therapy with any nonsteroidal AI (unless they had been disease free for more than 12 months), and had inflammatory breast cancer, central nervous system metastases, history of cardiac disease or dysfunction, or impaired gastrointestinal function that alters drug absorption.
Patients were treated with ribociclib at a dose of 600 mg daily on a 3-weeks-on, 1-week-off schedule in 28-day cycles or placebo, which were combined with letrozole at a dose of 2.5 mg a day on a continuous schedule. Randomization was stratified according to the presence or absence of liver or lung metastases and treatment was continued until disease progression, unacceptable toxicity, death or discontinuation of treatment. Dose reductions of ribociclib were allowed, to manage adverse events (AEs), but treatment crossover was not permitted.
Tumor assessments were performed at screening, every 8 weeks during the first 18 months, every 12 weeks thereafter until disease progression, and at the end of treatment, and were assessed by an independent review committee. The baseline characteristics of the patient population were well balanced; patients had a median age of 62 years, all were HR positive except 1 patient who was HER2 positive.
The trial was ended prematurely after an initial interim analysis demonstrated a significant benefit in favor of ribociclib in the primary endpoint, progression-free survival (PFS). Over a median duration of follow-up of 15.3 months, the median PFS was not yet reached in the ribociclib arm, compared with 14.7 months in the placebo arm (hazard ratio, 0.556; P < .0001). In a subsequent analysis with 11 months of additional follow-up, the median PFS was 25.3 months in the combination arm, compared with 16 months in the placebo arm, which translated into a 44% reduction in the risk of disease progression or death. The PFS benefit with ribociclib was observed across all preplanned subgroup analyses. The objective response rates were 52.7% in the ribociclib arm, compared with 37.1% in the placebo arm, but overall survival data were immature.
The frequency and severity of AEs were increased in the combination arm; most common were neutropenia, nausea, fatigue, diarrhea, leukopenia, alopecia, vomiting, constipation, headache, and back pain. The most common grade 3 or 4 AEs experienced with ribociclib were neutropenia, leukopenia, abnormal liver function tests, lymphopenia, and vomiting.
Ribociclib is accompanied by warnings and precautions about QT interval prolongation, hepatobiliary toxicity, and neutropenia. Clinicians are advised to monitor electrocardiograms and electrolytes before the start of ribociclib therapy and to begin treatment only in patients with QTcF values <450 ms and in whom electrolyte abnormalities have been corrected. ECG should be repeated at around day 14 of the first cycle, the beginning of the second cycle, and as deemed clinically necessary.
Liver function tests should be performed before starting treatment, every 2 weeks for the first 2 cycles, at the beginning of each of the subsequent 4 cycles, and as clinically indicated. For aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) levels greater than 3-5 times the upper limit of normal (ULN, grade 2), ribociclib should be interrupted until recovery to baseline or lower. For levels >5-20 times the ULN (grade 3) or recurring grade 2 increases, treatment should be interrupted until recovery to baseline or lower and then resumed at the next lowest dose level. Treatment with ribociclib should be discontinued in the event of recurring grade 3 elevations or for AST/ALT elevations >3 times ULN in combination with total bilirubin >2 times ULN.
Complete blood counts should be performed before starting treatment and monitored every 2 weeks for the first 2 cycles, at the beginning of each of the 4 subsequent cycles, and as clinically needed. If absolute neutrophil counts are 500-1,000 mm3 (grade 3), treatment should be discontinued until recovery to grade 2 or lower. If grade 3 neutropenia recurs or for grade 3 febrile neutropenia or grade 4 neutropenia, treatment should resume at a lower dose level upon recovery to grade 2 or lower.
Pregnant women and those of reproductive age should be warned of the risk of fetal harm and the need for effective contraception during treatment and for at least 3 weeks after the last dose. Ribociclib is marketed as Kisqali by Novartis.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
1. Ribociclib (Kisqali). US Food and Drug Administration website. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm546438.htm. Last updated March 14, 2017. Accessed April 3, 2017.
2. Kisqali (ribociclib) tables, for oral use. Prescribing information. Novartis Pharmaceuticals Corp. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/kisqali.pdf. March 2017. Accessed April 3, 2017.
3. Horobagyi GN, Stemmer SN, Burris HA, et al. Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med. 2016;375:1738-1748.
Approval makes olaratumab the first first-line treatment option for soft tissue sarcoma in more than 40 years
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
When the US Food and Drug Administration approved olaratumab as a first-line treatment for patients with soft tissue sarcoma (STS) in the fall of 2016, it marked the first approval since the chemotherapy drug doxorubicin became standard of care more than 40 years ago.1 Though rare, STS, which comprises a host of different histologic subtypes, has proven difficult to treat. Like pazopanib, which was approved in 2012 for the treatment of STS in the second-line setting, olaratumab targets the platelet-derived growth factor receptor alpha (PDGFRα), a tyrosine kinase receptor involved in cell signaling pathways that promotes key hallmark abilities in both cancer cells and the cells of the tumor microenvironment. Olaratumab, however, is a much more specific inhibitor of PDGFRα compared with pazopanib.
Accelerated approval was granted for the treatment of patients with STS that is not amenable to curative treatment with radiotherapy or surgery and with a subtype that cannot be treated effectively with an anthracycline-containing regimen. The approval was based on the phase 2 JGDG study, a randomized, active-controlled clinical trial in which 133 patients were randomized 1:1 to receive olaratumab plus doxorubicin, or doxorubicin alone.2
Eligible patients included those aged 18 years and over, with histologically confirmed diagnosis of locally advanced or metastatic STS not previously treated with an anthracycline, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2 (range, 1-5; 0, fully active and 5, dead), and with available tumor tissue for determination of PDGFRα expression by immunohistochemistry. Patients were enrolled at 16 clinical sites in 16 cities and 15 states in the United States from October 2010 to January 2013.
Patients were excluded if they had histologically or cytologically confirmed Kaposi sarcoma; untreated central nervous system metastases; received prior treatment with doxorubicin or other anthracyclines and anthracenediones, or any drug targeting PDGF or the PDGFRs; received concurrent treatment with other anticancer therapy within 4 weeks before study entry; unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months before study entry; HIV infection; or if they were pregnant or lactating.
Olaratumab was administered at 15 mg/kg as an intravenous infusion on days 1 and 8 of each 21-day cycle, and doxorubicin at 75 mg/m2 as an intravenous infusion on day 1 of each cycle, for a maximum of 8 cycles. Patients were permitted to receive dexarozoxane on cycles 5-8 and crossover was permitted. Tumor response was assessed by Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1) every 6 weeks, and survival assessed every 2 months, until study completion. PDGFR expression was assessed by immunohistochemistry at a central academic laboratory before randomization.
The primary endpoint of the study was progression-free survival (PFS) and the combination of olaratumab–doxorubicin significantly extended PFS in this patient population: median PFS was 6.6 months in the combination arm, compared with 4.1 months in the doxorubicin-alone arm (hazard ratio [HR], 0.672; P = .0615). The objective response rate (ORR) and median overall survival (OS), which were secondary endpoints in the trial, were also significantly improved with combination therapy compared with doxorubicin alone (ORR, 18.2% vs 11.9%, respectively; median OS, 26.5 months vs 14.7 months). The benefits of combination therapy were observed across prespecified subgroups, including histological tumor type, number of previous treatments, and PDGFRα expression level.
The most common adverse events (AEs) in the patients taking olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. Grade 3/4 AEs were also higher for the combination than for doxorubicin alone. The most common AE leading to discontinuation of olaratumab was infusion-related reactions, which occurred in 13% of patients.
According to the prescribing information, the recommended dose for olaratumab is 15 mg/kg as an intravenous infusion over 60 minutes on days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity, in combination with doxorubicin for the first 8 cycles. Patients should be premedicated with dexamethasone and diphenhydramine, to help protect against infusion-related reactions.
Olaratumab, marketed as Lartruvo by Lilly Oncology, has warnings and precautions relating to infusion-related reactions and embryofetal toxicity. Patients should be monitored for signs and symptoms of the former during and after infusion and olaratumab should be administered in a setting with available resuscitation equipment. Olaratumab should be permanently discontinued in the event of grade 3/4 infusion-related reactions. Olaratumab can cause fetal harm and female patients should be advised of the potential risk to a fetus and the need for effective contraception during treatment and for 3 months after the last dose.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
1. FDA grants accelerated approval to new treatment for advanced soft tissue sarcoma. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm525878.htm. Last updated October 19, 2016. Accessed March 6, 2017.
2. Tap WD, Jones RL, Van Tine BA, et al. Olaratumumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
3. Lartruvo (olaratumumab) injection, for intravenous use. Prescribing information. Eli Lilly and Co. http://pi.lilly.com/us/lartruvo-uspi.pdf. Last update October 2016. Accessed March 6, 2017.
Rucaparib – second PARP inhibitor hits the market for ovarian cancer
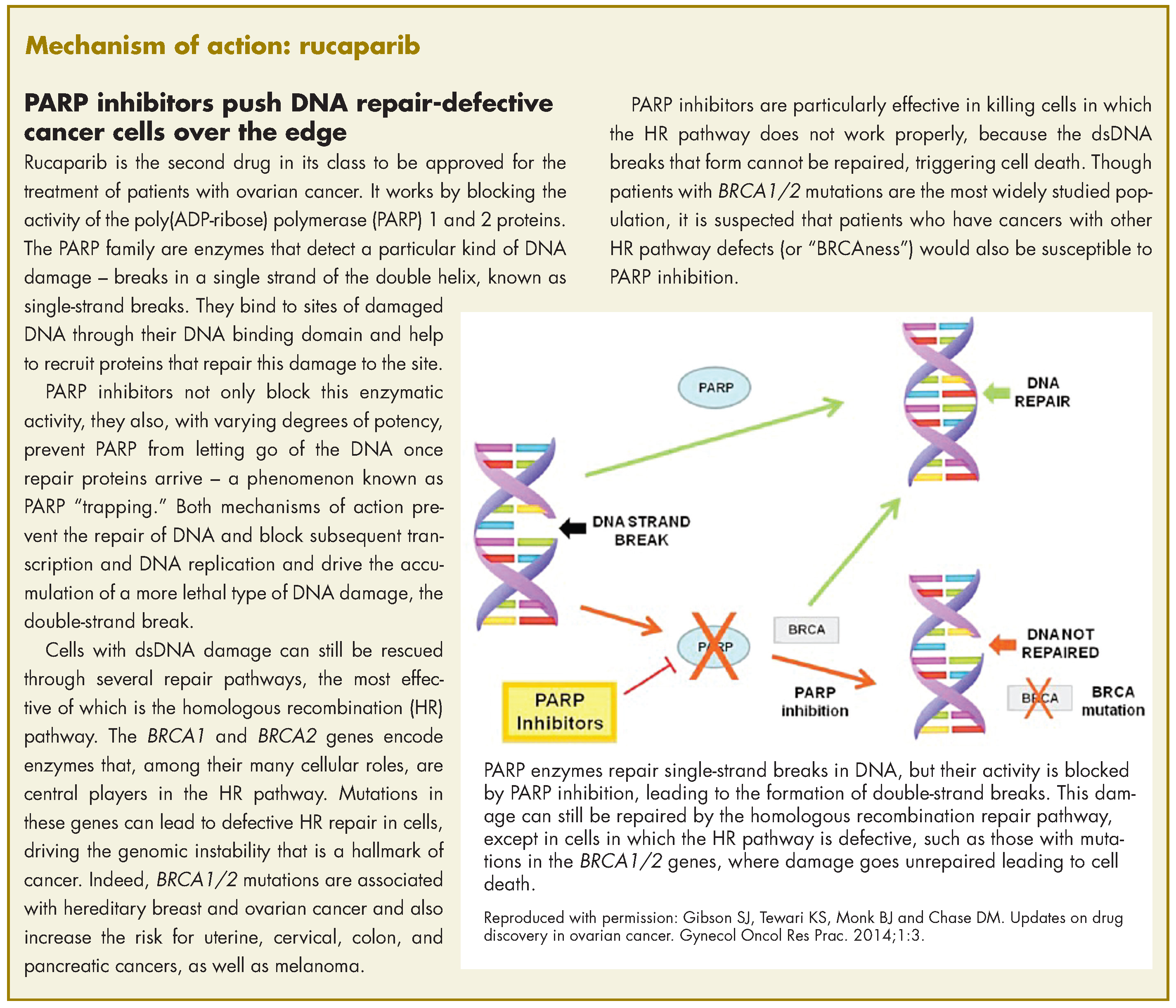
Rucaparib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with BRCA1/2 mutant advanced ovarian cancer in January this year, making it the second drug in its class for this indication. It is a poly(ADP-ribose) polymerase inhibitor that works by blocking the repair of damaged DNA in cancer cells and triggering cell death.
The approval was based on findings from 2 single-arm clinical trials in which rucaparib led to complete or partial tumor shrinkage in more than half of the patients enrolled. A pooled analysis included 106 patients from the phase 2 trials, Study 10 (NCT01482715; N = 42) and ARIEL2 (NCT01891344; N = 64), in which patients with BRCA1/2 mutation-positive ovarian cancer who had progressed on 2 or more previous chemotherapy regimens, received 600 mg rucaparib twice daily.
Study 10 included only patients with platinum-sensitive disease and eligible patients were aged 18 years or older, with a known deleterious BRCA mutation, evidence of measurable disease as defined by Response Evaluation Criteria in Solid Tumors (version 1.1), sufficient archival tumor tissue, histologically confirmed high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer and relapsed disease confirmed by radiologic assessment. Meanwhile, ARIEL2 had similar eligibility criteria, except that patients with platinum-sensitive, resistant, and refractory disease were included.
Both studies excluded patients with active second malignancies, and for those with a history of prior cancer that had been curatively treated, no evidence of current disease was required and chemotherapy should have been completed more than 6 months or bone marrow transplant more than 2 years before the first dose of rucaparib. Patients who had previously been treated with a PARP inhibitor, with symptomatic and/or untreated central nervous system metastases, or who had been hospitalized for bowel obstruction within the previous 3 months, were also ineligible.
Across the 2 trials, the median age of trial participants was 59 years, 78% were white, and all had an Eastern Cooperative Oncology Group performance status of 0 (fully active, able to carry on all pre-disease performance without restriction) or 1 (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature). Both trials used a surrogate endpoint for approval, measuring the percentage of patients who experienced complete or partial tumor shrinkage, the overall response rate (ORR), while taking rucaparib.
In Study 10, the ORR was 60%, including a complete response (CR) rate of 10% and a partial response (PR) rate of 50%, over a median duration of response (DoR) of 7.9 months, while in ARIEL2, the ORR was 50%, including a CR of 8% and a PR of 42%, over a median DoR of 11.6 months. The pooled analysis demonstrated an ORR of 54%, CR of 9% and PR of 45%, over a median DoR of 9.2 months. In separate data reported in the prescribing information, the ORR as assessed by independent radiology review was 42%, with a median DoR of 6.7 months, while ORR according to investigator assessment was 66%. In all analyses, the response rate was similar for patients having BRCA1 versus BRCA2 gene mutations.
Safety analyses were performed in 377 patients across the 2 studies who received 600 mg rucaparib twice daily. The most common adverse events (AEs) of any grade included nausea, fatigue, vomiting, anemia, abdominal pain, dysgeusia, constipation, decreased appetite, diarrhea, thrombocytopenia, and dyspnea. The most common serious AEs (grade 3 or 4) were anemia (25%), fatigue/asthenia (11%), and increased alanine aminotransferase or aspartate aminotransferase levels (11%). Overall, 8% of patients discontinued treatment because of AEs.
The recommended dose according to the prescribing information is 600 mg, in the form of two 300-mg tablets taken orally twice daily with or without food. Phy
If hematologic toxicities occur while taking rucaparib, treatment should be interrupted and blood counts monitored until recovery and failure to recover to grade 1 or higher after 4 weeks should prompt referral to a hematologist for further investigation, while confirmed diagnosis of myelodysplastic syndromes or acute myeloid leukemia should lead to discontinuation of rucaparib. Pregnant women and those of reproductive potential should be advised of the potential risk to a fetus or the need for effective contraception during treatment and for 6 months after the last dose of rucaparib.
Rucaparib is indicated only for the treatment of patients with confirmed BRCA1/2 mutations, so the drug was approved in conjunction with a companion diagnostic. FoundationFocus CDxBRCA is the first next-generation sequencing-based test to receive FDA approval and detects the presence of deleterious BRCA gene mutations in tumor tissue samples. Rucaparib is marketed as Rubraca by Clovis Oncology Inc, and the companion diagnostic by Foundation Medicine Inc.
1. Rubraca (rucaparib) capsules, for oral use. Prescribing information. Clovis Oncology Inc. http://clovisoncology.com/files/rubraca-prescribing-info.pdf. Released December 2016. Accessed January 8th, 2017.
2. FDA grants accelerated approval to new treatment for advanced ovarian cancer. FDA News Release. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm533873.htm. Last updated December 19, 2016. Accessed January 8, 2017.
3. [No author listed.] Rucaparib approved for ovarian cancer. Cancer Discov. Epub ahead of print. January 5, 2017. doi: 10.1158/2159-8290. CD-NB2016-164.
Rucaparib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with BRCA1/2 mutant advanced ovarian cancer in January this year, making it the second drug in its class for this indication. It is a poly(ADP-ribose) polymerase inhibitor that works by blocking the repair of damaged DNA in cancer cells and triggering cell death.
The approval was based on findings from 2 single-arm clinical trials in which rucaparib led to complete or partial tumor shrinkage in more than half of the patients enrolled. A pooled analysis included 106 patients from the phase 2 trials, Study 10 (NCT01482715; N = 42) and ARIEL2 (NCT01891344; N = 64), in which patients with BRCA1/2 mutation-positive ovarian cancer who had progressed on 2 or more previous chemotherapy regimens, received 600 mg rucaparib twice daily.
Study 10 included only patients with platinum-sensitive disease and eligible patients were aged 18 years or older, with a known deleterious BRCA mutation, evidence of measurable disease as defined by Response Evaluation Criteria in Solid Tumors (version 1.1), sufficient archival tumor tissue, histologically confirmed high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer and relapsed disease confirmed by radiologic assessment. Meanwhile, ARIEL2 had similar eligibility criteria, except that patients with platinum-sensitive, resistant, and refractory disease were included.
Both studies excluded patients with active second malignancies, and for those with a history of prior cancer that had been curatively treated, no evidence of current disease was required and chemotherapy should have been completed more than 6 months or bone marrow transplant more than 2 years before the first dose of rucaparib. Patients who had previously been treated with a PARP inhibitor, with symptomatic and/or untreated central nervous system metastases, or who had been hospitalized for bowel obstruction within the previous 3 months, were also ineligible.
Across the 2 trials, the median age of trial participants was 59 years, 78% were white, and all had an Eastern Cooperative Oncology Group performance status of 0 (fully active, able to carry on all pre-disease performance without restriction) or 1 (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature). Both trials used a surrogate endpoint for approval, measuring the percentage of patients who experienced complete or partial tumor shrinkage, the overall response rate (ORR), while taking rucaparib.
In Study 10, the ORR was 60%, including a complete response (CR) rate of 10% and a partial response (PR) rate of 50%, over a median duration of response (DoR) of 7.9 months, while in ARIEL2, the ORR was 50%, including a CR of 8% and a PR of 42%, over a median DoR of 11.6 months. The pooled analysis demonstrated an ORR of 54%, CR of 9% and PR of 45%, over a median DoR of 9.2 months. In separate data reported in the prescribing information, the ORR as assessed by independent radiology review was 42%, with a median DoR of 6.7 months, while ORR according to investigator assessment was 66%. In all analyses, the response rate was similar for patients having BRCA1 versus BRCA2 gene mutations.
Safety analyses were performed in 377 patients across the 2 studies who received 600 mg rucaparib twice daily. The most common adverse events (AEs) of any grade included nausea, fatigue, vomiting, anemia, abdominal pain, dysgeusia, constipation, decreased appetite, diarrhea, thrombocytopenia, and dyspnea. The most common serious AEs (grade 3 or 4) were anemia (25%), fatigue/asthenia (11%), and increased alanine aminotransferase or aspartate aminotransferase levels (11%). Overall, 8% of patients discontinued treatment because of AEs.
The recommended dose according to the prescribing information is 600 mg, in the form of two 300-mg tablets taken orally twice daily with or without food. Phy
If hematologic toxicities occur while taking rucaparib, treatment should be interrupted and blood counts monitored until recovery and failure to recover to grade 1 or higher after 4 weeks should prompt referral to a hematologist for further investigation, while confirmed diagnosis of myelodysplastic syndromes or acute myeloid leukemia should lead to discontinuation of rucaparib. Pregnant women and those of reproductive potential should be advised of the potential risk to a fetus or the need for effective contraception during treatment and for 6 months after the last dose of rucaparib.
Rucaparib is indicated only for the treatment of patients with confirmed BRCA1/2 mutations, so the drug was approved in conjunction with a companion diagnostic. FoundationFocus CDxBRCA is the first next-generation sequencing-based test to receive FDA approval and detects the presence of deleterious BRCA gene mutations in tumor tissue samples. Rucaparib is marketed as Rubraca by Clovis Oncology Inc, and the companion diagnostic by Foundation Medicine Inc.
Rucaparib was granted accelerated approval by the US Food and Drug Administration for the treatment of patients with BRCA1/2 mutant advanced ovarian cancer in January this year, making it the second drug in its class for this indication. It is a poly(ADP-ribose) polymerase inhibitor that works by blocking the repair of damaged DNA in cancer cells and triggering cell death.
The approval was based on findings from 2 single-arm clinical trials in which rucaparib led to complete or partial tumor shrinkage in more than half of the patients enrolled. A pooled analysis included 106 patients from the phase 2 trials, Study 10 (NCT01482715; N = 42) and ARIEL2 (NCT01891344; N = 64), in which patients with BRCA1/2 mutation-positive ovarian cancer who had progressed on 2 or more previous chemotherapy regimens, received 600 mg rucaparib twice daily.
Study 10 included only patients with platinum-sensitive disease and eligible patients were aged 18 years or older, with a known deleterious BRCA mutation, evidence of measurable disease as defined by Response Evaluation Criteria in Solid Tumors (version 1.1), sufficient archival tumor tissue, histologically confirmed high-grade epithelial ovarian, fallopian tube or primary peritoneal cancer and relapsed disease confirmed by radiologic assessment. Meanwhile, ARIEL2 had similar eligibility criteria, except that patients with platinum-sensitive, resistant, and refractory disease were included.
Both studies excluded patients with active second malignancies, and for those with a history of prior cancer that had been curatively treated, no evidence of current disease was required and chemotherapy should have been completed more than 6 months or bone marrow transplant more than 2 years before the first dose of rucaparib. Patients who had previously been treated with a PARP inhibitor, with symptomatic and/or untreated central nervous system metastases, or who had been hospitalized for bowel obstruction within the previous 3 months, were also ineligible.
Across the 2 trials, the median age of trial participants was 59 years, 78% were white, and all had an Eastern Cooperative Oncology Group performance status of 0 (fully active, able to carry on all pre-disease performance without restriction) or 1 (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature). Both trials used a surrogate endpoint for approval, measuring the percentage of patients who experienced complete or partial tumor shrinkage, the overall response rate (ORR), while taking rucaparib.
In Study 10, the ORR was 60%, including a complete response (CR) rate of 10% and a partial response (PR) rate of 50%, over a median duration of response (DoR) of 7.9 months, while in ARIEL2, the ORR was 50%, including a CR of 8% and a PR of 42%, over a median DoR of 11.6 months. The pooled analysis demonstrated an ORR of 54%, CR of 9% and PR of 45%, over a median DoR of 9.2 months. In separate data reported in the prescribing information, the ORR as assessed by independent radiology review was 42%, with a median DoR of 6.7 months, while ORR according to investigator assessment was 66%. In all analyses, the response rate was similar for patients having BRCA1 versus BRCA2 gene mutations.
Safety analyses were performed in 377 patients across the 2 studies who received 600 mg rucaparib twice daily. The most common adverse events (AEs) of any grade included nausea, fatigue, vomiting, anemia, abdominal pain, dysgeusia, constipation, decreased appetite, diarrhea, thrombocytopenia, and dyspnea. The most common serious AEs (grade 3 or 4) were anemia (25%), fatigue/asthenia (11%), and increased alanine aminotransferase or aspartate aminotransferase levels (11%). Overall, 8% of patients discontinued treatment because of AEs.
The recommended dose according to the prescribing information is 600 mg, in the form of two 300-mg tablets taken orally twice daily with or without food. Phy
If hematologic toxicities occur while taking rucaparib, treatment should be interrupted and blood counts monitored until recovery and failure to recover to grade 1 or higher after 4 weeks should prompt referral to a hematologist for further investigation, while confirmed diagnosis of myelodysplastic syndromes or acute myeloid leukemia should lead to discontinuation of rucaparib. Pregnant women and those of reproductive potential should be advised of the potential risk to a fetus or the need for effective contraception during treatment and for 6 months after the last dose of rucaparib.
Rucaparib is indicated only for the treatment of patients with confirmed BRCA1/2 mutations, so the drug was approved in conjunction with a companion diagnostic. FoundationFocus CDxBRCA is the first next-generation sequencing-based test to receive FDA approval and detects the presence of deleterious BRCA gene mutations in tumor tissue samples. Rucaparib is marketed as Rubraca by Clovis Oncology Inc, and the companion diagnostic by Foundation Medicine Inc.
1. Rubraca (rucaparib) capsules, for oral use. Prescribing information. Clovis Oncology Inc. http://clovisoncology.com/files/rubraca-prescribing-info.pdf. Released December 2016. Accessed January 8th, 2017.
2. FDA grants accelerated approval to new treatment for advanced ovarian cancer. FDA News Release. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm533873.htm. Last updated December 19, 2016. Accessed January 8, 2017.
3. [No author listed.] Rucaparib approved for ovarian cancer. Cancer Discov. Epub ahead of print. January 5, 2017. doi: 10.1158/2159-8290. CD-NB2016-164.
1. Rubraca (rucaparib) capsules, for oral use. Prescribing information. Clovis Oncology Inc. http://clovisoncology.com/files/rubraca-prescribing-info.pdf. Released December 2016. Accessed January 8th, 2017.
2. FDA grants accelerated approval to new treatment for advanced ovarian cancer. FDA News Release. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm533873.htm. Last updated December 19, 2016. Accessed January 8, 2017.
3. [No author listed.] Rucaparib approved for ovarian cancer. Cancer Discov. Epub ahead of print. January 5, 2017. doi: 10.1158/2159-8290. CD-NB2016-164.
Lenvatinib expands its reach into renal cell carcinoma
The US Food and Drug Administration (FDA) expanded the approval of the multitargeted tyrosine kinase inhibitor lenvatinib to a second indication in 2016. In addition to thyroid cancer, the drug is now approved in combination with the mammalian target of rapamycin (mTOR) inhibitor everolimus for the treatment of advanced renal cell carcinoma (RCC) after one prior anti-angiogenic therapy.
The current approval was based on the demonstration of synergistic efficacy and a manageable toxicity profile for the combination in a randomized, open-label, phase 2 clinical trial performed at 37 centers in 5 countries. Patients were eligible for the study if they were aged 18 years or older and had histologically verified clear cell RCC, measurable disease as assessed by RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1, radiographic evidence of progression or metastasis
From March 16, 2012 to June 19, 2013, 153 patients were randomly assigned in a 1:1:1 ratio to 3 treatment arms; lenvatinib 18 mg plus everolimus 5 mg, lenvatinib 24 mg monotherapy, or everolimus 10 mg monotherapy, all administered once daily. Randomization was stratified according to hemoglobin (men ≤130 g/L and >130 g/L; women ≤115 g/L and >115 g/L) and corrected serum calcium (≥2.5 mmol/L and <2.5 mmol/L).
Radiographic tumor response assessments were performed every 8 weeks from randomization until disease progression or the start of another anticancer treatment. To enable pharmacokinetic analyses, 6 blood samples were obtained on day 1 of the first 3 treatment cycles for all patients. In addition, 9 samples were obtained over a 24-hour period for 9-12 patients in each treatment group to provide intensive samples.
The primary endpoint of the study was progression-free survival (PFS), which was significantly improved with a combination of lenvatinib and everolimus, compared with single-agent everolimus. Median PFS was 14.6 months, compared with 5.5 months, respectively (hazard ratio [HR], 0.40; P = .0005), translating into a 63% reduction in the risk of disease progression or death. In the lenvatinib monotherapy group, median PFS was 7.4 months.
Over a median follow-up of 24.2 months there was also a significant difference in overall survival (OS) between the combination arm and single-agent everolimus (24.2 months vs 15.4 months, respectively). Objective responses were seen in 43% of patients in the combination arm and 6% and 27% of patients in the everolimus and lenvatinib monotherapy arms, respectively. The median duration of response was 13 months, 8.5 months, and 7.5 months in the 3 treatment arms, respectively.
All patients had at least 1 treatment-related adverse event (AE), almost all considered to be related to the study drug. Among patients treated with lenvatinib and everolimus, 24% discontinued therapy because of AEs, whereas the rate of discontinuation was 12% and 25% among patients treated with everolimus or lenvatinib monotherapy, respectively. There was 1 instance of a trans-arterial embolization leading to death that was judged to be probably treatment related in the combination arm, compared with 2 in the everolimus arm, neither judged treatment-related, and 3 in the lenvatinib arm, 1 of which was considered to be possibly treatment related.
The rates of grade 3/4 AEs were 71% for combination therapy, compared with 50% and 79%, respectively, among patients treated with everolimus or lenvatinib alone. Most commonly, in the combination arm, these included renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%), and dyspnea (5%).
The prescribing information carries warnings and precautions about hypertension, cardiac dysfunction, arterial thromboembolic events, hepatotoxicity, proteinuria, diarrhea, renal failure and impairment, gastrointestinal perforation, and fistula formation, QT interval prolongation, hypocalcemia, reversible posterior leukoencephalopathy syndrome, hemorrhagic events, and impairment of thyroid stimulating hormone suppression or thyroid dysfunction, all of which have been reported in clinical trials of lenvatinib and everolimus. Patients should also be warned about the risk of fetal harm.
Blood pressure should be closely monitored prior to treatment, after 1 week and then every 2 weeks for the first 2 months, then at least monthly thereafter during treatment. Patients should be monitored for signs of cardiac decompensation and proteinuria. Liver function should be monitored before initiating therapy, every 2 weeks for the first 2 months and then at least monthly while treatment continues. Electrolyte abnormalities should be monitored and corrected, blood calcium levels should be monitored at least monthly, and thyroid function should be evaluated before and at least monthly during treatment.
The prescribing information details dose reductions and modifications for AEs. Treatment should be withheld for grade 3 hypertension, grade 3 cardiac dysfunction, grade 3 or greater hepatotoxicity, proteinuria >2 g/24 hours, grade 3 diarrhea, grade 3/4 renal failure or impairment, corrected QT interval prolongation >500 ms, hypocalcemia as necessary, reversible posterior leukoencephalopathy syndrome confirmed by magnetic resonance imaging, and grade 3 hemorrhagic events.
Treatment discontinuation should occur in the event of life-threatening hypertension, grade 4 cardiac dysfunction, arterial thromboembolic events, hepatic failure, grade 3 diarrhea that persists despite medical management, severe or persistent renal impairment, gastrointestinal perforation or life-threatening fistula formation, severe and persistent neurologic symptoms, and grade 4 hemorrhage. The recommended dose for lenvatinib, which is marketed as Lenvima by Eisai Inc, is 18 mg (1 x 10 mg capsule and 2 x 4 mg capsules) in combination with 5 mg everolimus orally taken daily, with or without food, until disease progression or unacceptable toxicity.
1. Lenvima (lenvatinib) capsules, for oral use. Prescribing information. Woodcliff Lake, NJ: Eisai Inc: 2016. http://www.lenvima.com/pdfs/prescribing-information.pdf. Accessed November 17, 2016.
2. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473-1482.
3. US Food and Drug Administration. Lenvatinib in combination with everolimus. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm501070.htm. Last updated May 16, 2016. Accessed November 17, 2016.
The US Food and Drug Administration (FDA) expanded the approval of the multitargeted tyrosine kinase inhibitor lenvatinib to a second indication in 2016. In addition to thyroid cancer, the drug is now approved in combination with the mammalian target of rapamycin (mTOR) inhibitor everolimus for the treatment of advanced renal cell carcinoma (RCC) after one prior anti-angiogenic therapy.
The current approval was based on the demonstration of synergistic efficacy and a manageable toxicity profile for the combination in a randomized, open-label, phase 2 clinical trial performed at 37 centers in 5 countries. Patients were eligible for the study if they were aged 18 years or older and had histologically verified clear cell RCC, measurable disease as assessed by RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1, radiographic evidence of progression or metastasis
From March 16, 2012 to June 19, 2013, 153 patients were randomly assigned in a 1:1:1 ratio to 3 treatment arms; lenvatinib 18 mg plus everolimus 5 mg, lenvatinib 24 mg monotherapy, or everolimus 10 mg monotherapy, all administered once daily. Randomization was stratified according to hemoglobin (men ≤130 g/L and >130 g/L; women ≤115 g/L and >115 g/L) and corrected serum calcium (≥2.5 mmol/L and <2.5 mmol/L).
Radiographic tumor response assessments were performed every 8 weeks from randomization until disease progression or the start of another anticancer treatment. To enable pharmacokinetic analyses, 6 blood samples were obtained on day 1 of the first 3 treatment cycles for all patients. In addition, 9 samples were obtained over a 24-hour period for 9-12 patients in each treatment group to provide intensive samples.
The primary endpoint of the study was progression-free survival (PFS), which was significantly improved with a combination of lenvatinib and everolimus, compared with single-agent everolimus. Median PFS was 14.6 months, compared with 5.5 months, respectively (hazard ratio [HR], 0.40; P = .0005), translating into a 63% reduction in the risk of disease progression or death. In the lenvatinib monotherapy group, median PFS was 7.4 months.
Over a median follow-up of 24.2 months there was also a significant difference in overall survival (OS) between the combination arm and single-agent everolimus (24.2 months vs 15.4 months, respectively). Objective responses were seen in 43% of patients in the combination arm and 6% and 27% of patients in the everolimus and lenvatinib monotherapy arms, respectively. The median duration of response was 13 months, 8.5 months, and 7.5 months in the 3 treatment arms, respectively.
All patients had at least 1 treatment-related adverse event (AE), almost all considered to be related to the study drug. Among patients treated with lenvatinib and everolimus, 24% discontinued therapy because of AEs, whereas the rate of discontinuation was 12% and 25% among patients treated with everolimus or lenvatinib monotherapy, respectively. There was 1 instance of a trans-arterial embolization leading to death that was judged to be probably treatment related in the combination arm, compared with 2 in the everolimus arm, neither judged treatment-related, and 3 in the lenvatinib arm, 1 of which was considered to be possibly treatment related.
The rates of grade 3/4 AEs were 71% for combination therapy, compared with 50% and 79%, respectively, among patients treated with everolimus or lenvatinib alone. Most commonly, in the combination arm, these included renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%), and dyspnea (5%).
The prescribing information carries warnings and precautions about hypertension, cardiac dysfunction, arterial thromboembolic events, hepatotoxicity, proteinuria, diarrhea, renal failure and impairment, gastrointestinal perforation, and fistula formation, QT interval prolongation, hypocalcemia, reversible posterior leukoencephalopathy syndrome, hemorrhagic events, and impairment of thyroid stimulating hormone suppression or thyroid dysfunction, all of which have been reported in clinical trials of lenvatinib and everolimus. Patients should also be warned about the risk of fetal harm.
Blood pressure should be closely monitored prior to treatment, after 1 week and then every 2 weeks for the first 2 months, then at least monthly thereafter during treatment. Patients should be monitored for signs of cardiac decompensation and proteinuria. Liver function should be monitored before initiating therapy, every 2 weeks for the first 2 months and then at least monthly while treatment continues. Electrolyte abnormalities should be monitored and corrected, blood calcium levels should be monitored at least monthly, and thyroid function should be evaluated before and at least monthly during treatment.
The prescribing information details dose reductions and modifications for AEs. Treatment should be withheld for grade 3 hypertension, grade 3 cardiac dysfunction, grade 3 or greater hepatotoxicity, proteinuria >2 g/24 hours, grade 3 diarrhea, grade 3/4 renal failure or impairment, corrected QT interval prolongation >500 ms, hypocalcemia as necessary, reversible posterior leukoencephalopathy syndrome confirmed by magnetic resonance imaging, and grade 3 hemorrhagic events.
Treatment discontinuation should occur in the event of life-threatening hypertension, grade 4 cardiac dysfunction, arterial thromboembolic events, hepatic failure, grade 3 diarrhea that persists despite medical management, severe or persistent renal impairment, gastrointestinal perforation or life-threatening fistula formation, severe and persistent neurologic symptoms, and grade 4 hemorrhage. The recommended dose for lenvatinib, which is marketed as Lenvima by Eisai Inc, is 18 mg (1 x 10 mg capsule and 2 x 4 mg capsules) in combination with 5 mg everolimus orally taken daily, with or without food, until disease progression or unacceptable toxicity.
The US Food and Drug Administration (FDA) expanded the approval of the multitargeted tyrosine kinase inhibitor lenvatinib to a second indication in 2016. In addition to thyroid cancer, the drug is now approved in combination with the mammalian target of rapamycin (mTOR) inhibitor everolimus for the treatment of advanced renal cell carcinoma (RCC) after one prior anti-angiogenic therapy.
The current approval was based on the demonstration of synergistic efficacy and a manageable toxicity profile for the combination in a randomized, open-label, phase 2 clinical trial performed at 37 centers in 5 countries. Patients were eligible for the study if they were aged 18 years or older and had histologically verified clear cell RCC, measurable disease as assessed by RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1, radiographic evidence of progression or metastasis
From March 16, 2012 to June 19, 2013, 153 patients were randomly assigned in a 1:1:1 ratio to 3 treatment arms; lenvatinib 18 mg plus everolimus 5 mg, lenvatinib 24 mg monotherapy, or everolimus 10 mg monotherapy, all administered once daily. Randomization was stratified according to hemoglobin (men ≤130 g/L and >130 g/L; women ≤115 g/L and >115 g/L) and corrected serum calcium (≥2.5 mmol/L and <2.5 mmol/L).
Radiographic tumor response assessments were performed every 8 weeks from randomization until disease progression or the start of another anticancer treatment. To enable pharmacokinetic analyses, 6 blood samples were obtained on day 1 of the first 3 treatment cycles for all patients. In addition, 9 samples were obtained over a 24-hour period for 9-12 patients in each treatment group to provide intensive samples.
The primary endpoint of the study was progression-free survival (PFS), which was significantly improved with a combination of lenvatinib and everolimus, compared with single-agent everolimus. Median PFS was 14.6 months, compared with 5.5 months, respectively (hazard ratio [HR], 0.40; P = .0005), translating into a 63% reduction in the risk of disease progression or death. In the lenvatinib monotherapy group, median PFS was 7.4 months.
Over a median follow-up of 24.2 months there was also a significant difference in overall survival (OS) between the combination arm and single-agent everolimus (24.2 months vs 15.4 months, respectively). Objective responses were seen in 43% of patients in the combination arm and 6% and 27% of patients in the everolimus and lenvatinib monotherapy arms, respectively. The median duration of response was 13 months, 8.5 months, and 7.5 months in the 3 treatment arms, respectively.
All patients had at least 1 treatment-related adverse event (AE), almost all considered to be related to the study drug. Among patients treated with lenvatinib and everolimus, 24% discontinued therapy because of AEs, whereas the rate of discontinuation was 12% and 25% among patients treated with everolimus or lenvatinib monotherapy, respectively. There was 1 instance of a trans-arterial embolization leading to death that was judged to be probably treatment related in the combination arm, compared with 2 in the everolimus arm, neither judged treatment-related, and 3 in the lenvatinib arm, 1 of which was considered to be possibly treatment related.
The rates of grade 3/4 AEs were 71% for combination therapy, compared with 50% and 79%, respectively, among patients treated with everolimus or lenvatinib alone. Most commonly, in the combination arm, these included renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%), and dyspnea (5%).
The prescribing information carries warnings and precautions about hypertension, cardiac dysfunction, arterial thromboembolic events, hepatotoxicity, proteinuria, diarrhea, renal failure and impairment, gastrointestinal perforation, and fistula formation, QT interval prolongation, hypocalcemia, reversible posterior leukoencephalopathy syndrome, hemorrhagic events, and impairment of thyroid stimulating hormone suppression or thyroid dysfunction, all of which have been reported in clinical trials of lenvatinib and everolimus. Patients should also be warned about the risk of fetal harm.
Blood pressure should be closely monitored prior to treatment, after 1 week and then every 2 weeks for the first 2 months, then at least monthly thereafter during treatment. Patients should be monitored for signs of cardiac decompensation and proteinuria. Liver function should be monitored before initiating therapy, every 2 weeks for the first 2 months and then at least monthly while treatment continues. Electrolyte abnormalities should be monitored and corrected, blood calcium levels should be monitored at least monthly, and thyroid function should be evaluated before and at least monthly during treatment.
The prescribing information details dose reductions and modifications for AEs. Treatment should be withheld for grade 3 hypertension, grade 3 cardiac dysfunction, grade 3 or greater hepatotoxicity, proteinuria >2 g/24 hours, grade 3 diarrhea, grade 3/4 renal failure or impairment, corrected QT interval prolongation >500 ms, hypocalcemia as necessary, reversible posterior leukoencephalopathy syndrome confirmed by magnetic resonance imaging, and grade 3 hemorrhagic events.
Treatment discontinuation should occur in the event of life-threatening hypertension, grade 4 cardiac dysfunction, arterial thromboembolic events, hepatic failure, grade 3 diarrhea that persists despite medical management, severe or persistent renal impairment, gastrointestinal perforation or life-threatening fistula formation, severe and persistent neurologic symptoms, and grade 4 hemorrhage. The recommended dose for lenvatinib, which is marketed as Lenvima by Eisai Inc, is 18 mg (1 x 10 mg capsule and 2 x 4 mg capsules) in combination with 5 mg everolimus orally taken daily, with or without food, until disease progression or unacceptable toxicity.
1. Lenvima (lenvatinib) capsules, for oral use. Prescribing information. Woodcliff Lake, NJ: Eisai Inc: 2016. http://www.lenvima.com/pdfs/prescribing-information.pdf. Accessed November 17, 2016.
2. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473-1482.
3. US Food and Drug Administration. Lenvatinib in combination with everolimus. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm501070.htm. Last updated May 16, 2016. Accessed November 17, 2016.
1. Lenvima (lenvatinib) capsules, for oral use. Prescribing information. Woodcliff Lake, NJ: Eisai Inc: 2016. http://www.lenvima.com/pdfs/prescribing-information.pdf. Accessed November 17, 2016.
2. Motzer RJ, Hutson TE, Glen H, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473-1482.
3. US Food and Drug Administration. Lenvatinib in combination with everolimus. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm501070.htm. Last updated May 16, 2016. Accessed November 17, 2016.
Pembrolizumab is the first immune checkpoint inhibitor to receive approval for head and neck cancer
The first immune checkpoint inhibitor was approved for the treatment of head and neck cancer approved in August 2016. Pembrolizumab, which targets the programmed cell death 1 (PD-1) protein, is designed to reinstate the anti-tumor immune response to kill cancer cells and was approved for the treatment of recurrent or metastatic disease that progressed during or after platinum-containing chemotherapy.
Click on the PDF icon at the top of this introduction to read the full article.
The first immune checkpoint inhibitor was approved for the treatment of head and neck cancer approved in August 2016. Pembrolizumab, which targets the programmed cell death 1 (PD-1) protein, is designed to reinstate the anti-tumor immune response to kill cancer cells and was approved for the treatment of recurrent or metastatic disease that progressed during or after platinum-containing chemotherapy.
Click on the PDF icon at the top of this introduction to read the full article.
The first immune checkpoint inhibitor was approved for the treatment of head and neck cancer approved in August 2016. Pembrolizumab, which targets the programmed cell death 1 (PD-1) protein, is designed to reinstate the anti-tumor immune response to kill cancer cells and was approved for the treatment of recurrent or metastatic disease that progressed during or after platinum-containing chemotherapy.
Click on the PDF icon at the top of this introduction to read the full article.
Fourth approved indication for ofatumumab in chronic lymphocytic leukemia
The recent decision by the US Food and Drug Administration to approve ofatumumab in combination with fludarabine and cyclophosphamide in relapsed disease marks a fourth approved indication for this drug in patients with chronic lymphocytic leukemia (CLL). Ofatumumab is a fully human monoclonal antibody that targets the CD20 protein on the surface of B cells, first approved for the treatment of CLL back in 2009.
Click on the PDF icon at the top of this introduction to read the full article.
The recent decision by the US Food and Drug Administration to approve ofatumumab in combination with fludarabine and cyclophosphamide in relapsed disease marks a fourth approved indication for this drug in patients with chronic lymphocytic leukemia (CLL). Ofatumumab is a fully human monoclonal antibody that targets the CD20 protein on the surface of B cells, first approved for the treatment of CLL back in 2009.
Click on the PDF icon at the top of this introduction to read the full article.
The recent decision by the US Food and Drug Administration to approve ofatumumab in combination with fludarabine and cyclophosphamide in relapsed disease marks a fourth approved indication for this drug in patients with chronic lymphocytic leukemia (CLL). Ofatumumab is a fully human monoclonal antibody that targets the CD20 protein on the surface of B cells, first approved for the treatment of CLL back in 2009.
Click on the PDF icon at the top of this introduction to read the full article.
Renal cell carcinoma approval adds another notch to cabozantinib’s belt
In April this year, the US Food and Drug Administration awarded regulatory approval to cabozantinib for the treatment of advanced renal cell carcinoma patients previously treated with anti-angiogenic therapy.1 The small-molecule inhibitor, which targets multiple kinases, including the vascular endothelial growth factor receptors (VEGFRs) and the hepatocyte growth factor receptor (MET), had previously been approved for the treatment of medullary thyroid carcinoma in 2012.
Click on the PDF icon below for the full article.
In April this year, the US Food and Drug Administration awarded regulatory approval to cabozantinib for the treatment of advanced renal cell carcinoma patients previously treated with anti-angiogenic therapy.1 The small-molecule inhibitor, which targets multiple kinases, including the vascular endothelial growth factor receptors (VEGFRs) and the hepatocyte growth factor receptor (MET), had previously been approved for the treatment of medullary thyroid carcinoma in 2012.
Click on the PDF icon below for the full article.
In April this year, the US Food and Drug Administration awarded regulatory approval to cabozantinib for the treatment of advanced renal cell carcinoma patients previously treated with anti-angiogenic therapy.1 The small-molecule inhibitor, which targets multiple kinases, including the vascular endothelial growth factor receptors (VEGFRs) and the hepatocyte growth factor receptor (MET), had previously been approved for the treatment of medullary thyroid carcinoma in 2012.
Click on the PDF icon below for the full article.
Alectinib provides a new option for ALK-positive NSCLC patients after progression on crizotinib
In December 2015, alectinib became the third ALK inhibitor approved by the United States Food and Drug Administration for the treatment of non-small-cell lung cancer (NSCLC) that displays rearrangements of the anaplastic lymphoma kinase (ALK) gene. Alectinib is a second-generation small molecule inhibitor of the ALK protein that joins ceritinib in providing a useful treatment option for patients who have progressed on crizotinib, as a result of its ability to target crizotinib-resistant mutant forms of the ALK protein. Alectinib also displays enhanced penetrance of the blood-brain barrier, which improves efficacy against central nervous system (CNS) metastases.
Click on the PDF icon at the top of this introduction to read the full article.
In December 2015, alectinib became the third ALK inhibitor approved by the United States Food and Drug Administration for the treatment of non-small-cell lung cancer (NSCLC) that displays rearrangements of the anaplastic lymphoma kinase (ALK) gene. Alectinib is a second-generation small molecule inhibitor of the ALK protein that joins ceritinib in providing a useful treatment option for patients who have progressed on crizotinib, as a result of its ability to target crizotinib-resistant mutant forms of the ALK protein. Alectinib also displays enhanced penetrance of the blood-brain barrier, which improves efficacy against central nervous system (CNS) metastases.
Click on the PDF icon at the top of this introduction to read the full article.
In December 2015, alectinib became the third ALK inhibitor approved by the United States Food and Drug Administration for the treatment of non-small-cell lung cancer (NSCLC) that displays rearrangements of the anaplastic lymphoma kinase (ALK) gene. Alectinib is a second-generation small molecule inhibitor of the ALK protein that joins ceritinib in providing a useful treatment option for patients who have progressed on crizotinib, as a result of its ability to target crizotinib-resistant mutant forms of the ALK protein. Alectinib also displays enhanced penetrance of the blood-brain barrier, which improves efficacy against central nervous system (CNS) metastases.
Click on the PDF icon at the top of this introduction to read the full article.
Panobinostat: a novel mechanism of action shows promise in multiple myeloma
Following an initial “no” vote from the Oncologic Drugs Advisory Committee (ODAC) in late 2014, the US Food and Drug Administration eventually awarded accelerated approval in February 2015 to the histone deacetylase (HDAC) inhibitor panobinostat for use in select patients with relapsed multiple myeloma. Panobinostat has a novel mechanism of action that demonstrates synergy with the proteasome inhibitor bortezomib and the immunomodulatory agent dexamethasone, which translated into improved progression-free survival (PFS) for patients with multiple myeloma who had received at least 2 prior therapies, according to data from a prespecified subgroup analysis from the Panorama-1 trial.
Click on the PDF icon at the top of this introduction to read the full article.
Following an initial “no” vote from the Oncologic Drugs Advisory Committee (ODAC) in late 2014, the US Food and Drug Administration eventually awarded accelerated approval in February 2015 to the histone deacetylase (HDAC) inhibitor panobinostat for use in select patients with relapsed multiple myeloma. Panobinostat has a novel mechanism of action that demonstrates synergy with the proteasome inhibitor bortezomib and the immunomodulatory agent dexamethasone, which translated into improved progression-free survival (PFS) for patients with multiple myeloma who had received at least 2 prior therapies, according to data from a prespecified subgroup analysis from the Panorama-1 trial.
Click on the PDF icon at the top of this introduction to read the full article.
Following an initial “no” vote from the Oncologic Drugs Advisory Committee (ODAC) in late 2014, the US Food and Drug Administration eventually awarded accelerated approval in February 2015 to the histone deacetylase (HDAC) inhibitor panobinostat for use in select patients with relapsed multiple myeloma. Panobinostat has a novel mechanism of action that demonstrates synergy with the proteasome inhibitor bortezomib and the immunomodulatory agent dexamethasone, which translated into improved progression-free survival (PFS) for patients with multiple myeloma who had received at least 2 prior therapies, according to data from a prespecified subgroup analysis from the Panorama-1 trial.
Click on the PDF icon at the top of this introduction to read the full article.