User login
Blocking the ‘munchies’ receptor: A novel approach to obesity
Marijuana has long been known to stimulate appetite, particularly for sweet foods.1 The naughty boys in my fraternity called it “the munchies;” the professionals call it hyperphagia. Cannabinoid receptor (CB1) stimulation by marijuana’s main active component—9-THC—is believed to induce this behavior. Clinicians have successfully used this effect to treat AIDS-related wasting syndrome and other anorexic conditions.2
CB1 is widely expressed throughout the brain and seems to inhibit release of various neurotransmitters.3 How this effect leads to increased appetite is unclear, but it may result from a decrease in the appetite-suppressing effects of hormones such as leptin. In other words, tweaking the CB1 receptor may take the “brakes” off appetite.
Some researchers have speculated that if stimulating CB1 triggers appetite, blocking the receptor might inhibit it (Figure 1).
THE ‘MUNCHIES’ IN MICE
Rimonabant (SR141716), an experimental agent, is a potent and selective CB1 antagonist.
Ravinet Trillou et al fed mice a high-fat diet known to induce obesity.4 The mice were randomized to receive rimonabant or placebo while maintained on the highly palatable diet. The authors asked: Would rimonabant help the mice lose weight even when they could eat as much delicious fatty food as they wanted?
Figure 1 Blocking CB1 may prevent weight gain
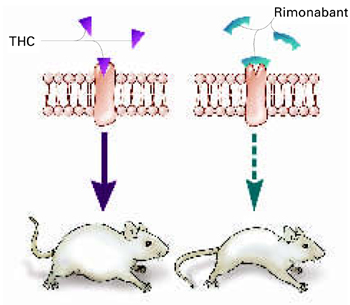
Δ9-THC activates the cannabinoid receptor (CB1), stimulating appetite and leading to weight gain in mice (left). When the same receptor is blocked, appetite is controlled (right).
Source: Illustration for CURRENT PSYCHIATRY by Marcia HartsockRimonabant induced a sustained body weight reduction of approximately 20% in the treatment group compared with the placebo group across 5 weeks (Figure 2). Estimated fat stores among the treatment group were depleted by slightly more than 50%.
The authors noted that the mice in the treatment group had decreased their food intake, but the decrease was not sufficient to explain the weight loss. They speculate that rimonabant could activate metabolic processes and decrease intake.
RIMONABANT’S ROLE IN PSYCHIATRY
Phase III human trials of rimonabant are under way for obesity as well as smoking cessation.5 In uncontrolled studies, rimonabant has been shown to help people avoid weight gain while quitting smoking.5
If rimonabant shows effectiveness in controlled trials and is safe in humans, it could be most valuable. Obesity in industrial countries is epidemic and causes serious secondary morbidity, including diabetes, arthritis, and hypertension. Rimonabant, if approved by the FDA, could reach the market by early 2006.6
It is unknown whether rimonabant’s metabolic effects could offset those of many psychotropics. As psychiatrists, we often must stop an effective antipsychotic or antidepressant because it is causing significant weight gain. A treatment that would prevent medication-induced weight gain could improve patient compliance and, ultimately, outcomes.
MANAGING SCHIZOPHRENIA
Some evidence also suggests that rimonabant may offer additional benefits for patients with schizophrenia beyond weight reduction or smoking cessation.
Figure 2 Rimonabant’s effects on weight in mice on a high-fat diet
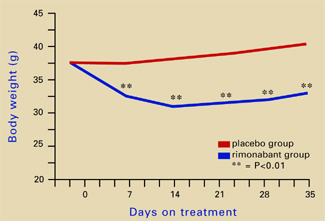
Source: Adapted from reference 4.Leweke et al found increased endogenous cannabinoids in the CSF of patients with schizophrenia, suggesting that a cannabinoid signaling imbalance may contribute to the disorder’s pathogenesis.7 However, 72 patients with schizophrenia or schizoaffective disorder who took rimonabant for 6 weeks showed no improvement compared with a placebo group.8
1. Abel EL. Cannabis: effects on hunger and thirst. Behav Biol 1975;15:255-81.
2. Beal JE, Olson R, Laubenstein L, et al. Dronabinol as a treatment for anorexia associated with weight loss in patients with AIDS. J Pain Symptom Manage 1995;10:89-97.
3. Iversen L. Cannabis and the brain. Brain 2003;126:1252-70.
4. Ravinet Trillou C, Arnone M, Delgorge C, et al. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am J Physiol Regul Integr Comp Physiol 2003;284:R345-53.
5. Fernandez JR, Allison DB. Rimonabant Sanofi-Synthelabo. Curr Opin Investig Drugs 2004;5:430-5.
6. The Website for the Drug Development Industry. Acomplia (rimonabant)—investigational agent for the management of obesity. London: SPGMedia. Available at: http://www. drugdevelopment-technology.com/projects/rimonabant/. Accessed Oct. 14, 2004.
7. Leweke FM, Giuffrida A, Wurster U, et al. Elevated endogenous cannabinoids in schizophrenia. Neuroreport 1999;10:1665-9.
8. Meltzer HY, Arvanitis L, Bauer D, et al. Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry 2004;161:975-84.
Marijuana has long been known to stimulate appetite, particularly for sweet foods.1 The naughty boys in my fraternity called it “the munchies;” the professionals call it hyperphagia. Cannabinoid receptor (CB1) stimulation by marijuana’s main active component—9-THC—is believed to induce this behavior. Clinicians have successfully used this effect to treat AIDS-related wasting syndrome and other anorexic conditions.2
CB1 is widely expressed throughout the brain and seems to inhibit release of various neurotransmitters.3 How this effect leads to increased appetite is unclear, but it may result from a decrease in the appetite-suppressing effects of hormones such as leptin. In other words, tweaking the CB1 receptor may take the “brakes” off appetite.
Some researchers have speculated that if stimulating CB1 triggers appetite, blocking the receptor might inhibit it (Figure 1).
THE ‘MUNCHIES’ IN MICE
Rimonabant (SR141716), an experimental agent, is a potent and selective CB1 antagonist.
Ravinet Trillou et al fed mice a high-fat diet known to induce obesity.4 The mice were randomized to receive rimonabant or placebo while maintained on the highly palatable diet. The authors asked: Would rimonabant help the mice lose weight even when they could eat as much delicious fatty food as they wanted?
Figure 1 Blocking CB1 may prevent weight gain
Δ9-THC activates the cannabinoid receptor (CB1), stimulating appetite and leading to weight gain in mice (left). When the same receptor is blocked, appetite is controlled (right).
Source: Illustration for CURRENT PSYCHIATRY by Marcia HartsockRimonabant induced a sustained body weight reduction of approximately 20% in the treatment group compared with the placebo group across 5 weeks (Figure 2). Estimated fat stores among the treatment group were depleted by slightly more than 50%.
The authors noted that the mice in the treatment group had decreased their food intake, but the decrease was not sufficient to explain the weight loss. They speculate that rimonabant could activate metabolic processes and decrease intake.
RIMONABANT’S ROLE IN PSYCHIATRY
Phase III human trials of rimonabant are under way for obesity as well as smoking cessation.5 In uncontrolled studies, rimonabant has been shown to help people avoid weight gain while quitting smoking.5
If rimonabant shows effectiveness in controlled trials and is safe in humans, it could be most valuable. Obesity in industrial countries is epidemic and causes serious secondary morbidity, including diabetes, arthritis, and hypertension. Rimonabant, if approved by the FDA, could reach the market by early 2006.6
It is unknown whether rimonabant’s metabolic effects could offset those of many psychotropics. As psychiatrists, we often must stop an effective antipsychotic or antidepressant because it is causing significant weight gain. A treatment that would prevent medication-induced weight gain could improve patient compliance and, ultimately, outcomes.
MANAGING SCHIZOPHRENIA
Some evidence also suggests that rimonabant may offer additional benefits for patients with schizophrenia beyond weight reduction or smoking cessation.
Figure 2 Rimonabant’s effects on weight in mice on a high-fat diet
Source: Adapted from reference 4.Leweke et al found increased endogenous cannabinoids in the CSF of patients with schizophrenia, suggesting that a cannabinoid signaling imbalance may contribute to the disorder’s pathogenesis.7 However, 72 patients with schizophrenia or schizoaffective disorder who took rimonabant for 6 weeks showed no improvement compared with a placebo group.8
Marijuana has long been known to stimulate appetite, particularly for sweet foods.1 The naughty boys in my fraternity called it “the munchies;” the professionals call it hyperphagia. Cannabinoid receptor (CB1) stimulation by marijuana’s main active component—9-THC—is believed to induce this behavior. Clinicians have successfully used this effect to treat AIDS-related wasting syndrome and other anorexic conditions.2
CB1 is widely expressed throughout the brain and seems to inhibit release of various neurotransmitters.3 How this effect leads to increased appetite is unclear, but it may result from a decrease in the appetite-suppressing effects of hormones such as leptin. In other words, tweaking the CB1 receptor may take the “brakes” off appetite.
Some researchers have speculated that if stimulating CB1 triggers appetite, blocking the receptor might inhibit it (Figure 1).
THE ‘MUNCHIES’ IN MICE
Rimonabant (SR141716), an experimental agent, is a potent and selective CB1 antagonist.
Ravinet Trillou et al fed mice a high-fat diet known to induce obesity.4 The mice were randomized to receive rimonabant or placebo while maintained on the highly palatable diet. The authors asked: Would rimonabant help the mice lose weight even when they could eat as much delicious fatty food as they wanted?
Figure 1 Blocking CB1 may prevent weight gain
Δ9-THC activates the cannabinoid receptor (CB1), stimulating appetite and leading to weight gain in mice (left). When the same receptor is blocked, appetite is controlled (right).
Source: Illustration for CURRENT PSYCHIATRY by Marcia HartsockRimonabant induced a sustained body weight reduction of approximately 20% in the treatment group compared with the placebo group across 5 weeks (Figure 2). Estimated fat stores among the treatment group were depleted by slightly more than 50%.
The authors noted that the mice in the treatment group had decreased their food intake, but the decrease was not sufficient to explain the weight loss. They speculate that rimonabant could activate metabolic processes and decrease intake.
RIMONABANT’S ROLE IN PSYCHIATRY
Phase III human trials of rimonabant are under way for obesity as well as smoking cessation.5 In uncontrolled studies, rimonabant has been shown to help people avoid weight gain while quitting smoking.5
If rimonabant shows effectiveness in controlled trials and is safe in humans, it could be most valuable. Obesity in industrial countries is epidemic and causes serious secondary morbidity, including diabetes, arthritis, and hypertension. Rimonabant, if approved by the FDA, could reach the market by early 2006.6
It is unknown whether rimonabant’s metabolic effects could offset those of many psychotropics. As psychiatrists, we often must stop an effective antipsychotic or antidepressant because it is causing significant weight gain. A treatment that would prevent medication-induced weight gain could improve patient compliance and, ultimately, outcomes.
MANAGING SCHIZOPHRENIA
Some evidence also suggests that rimonabant may offer additional benefits for patients with schizophrenia beyond weight reduction or smoking cessation.
Figure 2 Rimonabant’s effects on weight in mice on a high-fat diet
Source: Adapted from reference 4.Leweke et al found increased endogenous cannabinoids in the CSF of patients with schizophrenia, suggesting that a cannabinoid signaling imbalance may contribute to the disorder’s pathogenesis.7 However, 72 patients with schizophrenia or schizoaffective disorder who took rimonabant for 6 weeks showed no improvement compared with a placebo group.8
1. Abel EL. Cannabis: effects on hunger and thirst. Behav Biol 1975;15:255-81.
2. Beal JE, Olson R, Laubenstein L, et al. Dronabinol as a treatment for anorexia associated with weight loss in patients with AIDS. J Pain Symptom Manage 1995;10:89-97.
3. Iversen L. Cannabis and the brain. Brain 2003;126:1252-70.
4. Ravinet Trillou C, Arnone M, Delgorge C, et al. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am J Physiol Regul Integr Comp Physiol 2003;284:R345-53.
5. Fernandez JR, Allison DB. Rimonabant Sanofi-Synthelabo. Curr Opin Investig Drugs 2004;5:430-5.
6. The Website for the Drug Development Industry. Acomplia (rimonabant)—investigational agent for the management of obesity. London: SPGMedia. Available at: http://www. drugdevelopment-technology.com/projects/rimonabant/. Accessed Oct. 14, 2004.
7. Leweke FM, Giuffrida A, Wurster U, et al. Elevated endogenous cannabinoids in schizophrenia. Neuroreport 1999;10:1665-9.
8. Meltzer HY, Arvanitis L, Bauer D, et al. Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry 2004;161:975-84.
1. Abel EL. Cannabis: effects on hunger and thirst. Behav Biol 1975;15:255-81.
2. Beal JE, Olson R, Laubenstein L, et al. Dronabinol as a treatment for anorexia associated with weight loss in patients with AIDS. J Pain Symptom Manage 1995;10:89-97.
3. Iversen L. Cannabis and the brain. Brain 2003;126:1252-70.
4. Ravinet Trillou C, Arnone M, Delgorge C, et al. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in diet-induced obese mice. Am J Physiol Regul Integr Comp Physiol 2003;284:R345-53.
5. Fernandez JR, Allison DB. Rimonabant Sanofi-Synthelabo. Curr Opin Investig Drugs 2004;5:430-5.
6. The Website for the Drug Development Industry. Acomplia (rimonabant)—investigational agent for the management of obesity. London: SPGMedia. Available at: http://www. drugdevelopment-technology.com/projects/rimonabant/. Accessed Oct. 14, 2004.
7. Leweke FM, Giuffrida A, Wurster U, et al. Elevated endogenous cannabinoids in schizophrenia. Neuroreport 1999;10:1665-9.
8. Meltzer HY, Arvanitis L, Bauer D, et al. Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry 2004;161:975-84.
Is depression neurochemical or neurodegenerative?
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
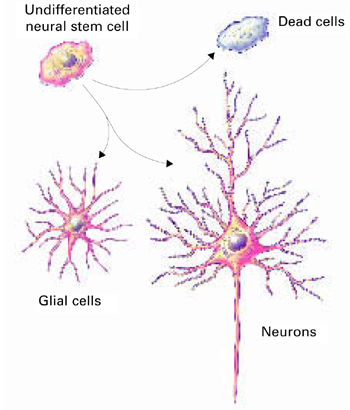
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
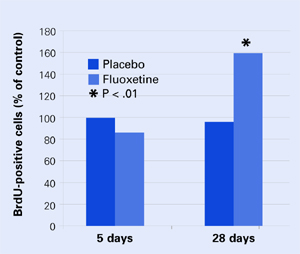
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
A relative lack of important neurotransmitters is popularly believed to cause depression. This “monoamine hypothesis” makes sense: We give patients “chemicals,” and many depression symptoms improve. Just because an antidepressant works, however, does not mean that a “chemical imbalance” is causing the depression.
For one, 40 years of research has not consistently found diminished neurotransmitters or their metabolites in depressed persons. Also, the monoamine hypothesis fails to explain why clinical improvement can be delayed for weeks, even though antidepressants rapidly increase extracellular serotonin and norepinephrine.
Figure 1 How neural cells differentiate in the brain
In neurogenesis, undifferentiated neural stem cells proliferate and migrate in the brain. Approximately one-half develop into useful cells, such as neurons and glial cells, and one-half die.
Source: Ilustration for CURRENTPSYCHIATRY by Maura Flynn
Increasing evidence suggests that depression may be a subtle neurodegenerative disorder. Postmortem and imaging studies have consistently found atrophy or neuron loss in the prefrontal cortices and hippocampi of depressed patients.1 Some studies suggest that antidepressants prevent the atrophy.2
Figure 2 Neural cell development may explain delayed antidepressant effect
In mouse brains, fluoxetine—but not placebo—increased the number of recently developed neuralcells (as shown by the injected marker 5-bromo-2’- deoxyuridine [BrdU]), but only after 28 days.
Source: Adapted from reference 4.
NEW VIEWS ON NEUROGENESIS
Until recently, the brain was believed incapable of generating nerve cells. We were thought to be born with our entire allotment of brain cells and we could only lose them because of age, trauma, or toxins. Within the past 5 years, however, it has become clear that human neuronal stem cells are capable of neurogenesis (Figure 1).3
Neurogenesis is an ongoing process in the brain, and depression may result from a relative decrease in new neuron development. Effective depression treatments may work by stimulating neurogenesis, and Santarelli et al4 offer compelling support for this idea.
A group of mice was treated orally with fluoxetine or placebo. Several from each group were sacrificed after 5 days and others at 28 days. Twenty-four hours before being sacrificed, each mouse was injected with 5-bromo-2’-deoxyuridine (BrdU), which is incorporated into DNA and serves as a marker for recently developed nerve cells.
None of the mice at 5 days showed any change in BrdU-positive cells, and only the mice who received fluoxetine showed an increase in new cells after 28 days Figure 2. These results correlated with a greater willingness after 28 days by those mice on fluoxetine to venture into open, lighted areas—a behavioral change that was not seen at 5 days or in the placebo group.
This study shows that an antidepressant can increase development of new neurons and does so in a time course similar to the onset of efficacy seen in human clinical trials.
CHEMICAL OR CELLULAR?
One can speculate that insufficient neurogenesis is a possible cause of depression and that effective depression treatments may reverse that problem. Supporting this concept are other studies showing that lithium and electroconvulsive therapy—well-known treatments for depression—also increase neurogenesis.5
All this evidence makes depression look more like a cellular than a chemical imbalance.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.
1. Manji HK, Quiroz JA, Sporn J, et al. Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 2003;53:707-42.
2. Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry 2003;160:1516-8.
3. Gage FH. Brain, repair yourself. Sci Am 2003;289:46-53.
4. Santarelli L, Saxe M, Gross C, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-9.
5. Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry 2003;54:499-503.
Understanding the ‘joy’ of aggression
One of the most perplexing and shocking aspects of human behavior is the cruelty we can inflict on one another. Infamous events such as the Spanish Inquisition, slavery, lynching, conquest of the native Americans, and Nazi concentration camps are so sickening that many of us can barely tolerate hearing about them. How can people behave this way?
Recently, we’ve been shocked by photographs from the Abu Ghraib military prison in Iraq. American soldiers with unremarkable backgrounds—not Saddam’s henchmen—were shown humiliating and abusing Iraqi prisoners. Most remarkable was the joy on the Americans’ faces (Figure 1).
‘Good’ people, ‘bad’ circumstances
In experiments with college students, psychologists have shown that “regular” people can become sadistic under the right (or wrong) circumstances.1 One explanation is that it’s not just psychopaths who perpetrate crimes against humanity. Somehow, the psychopath within us all becomes unleashed.
Artificial situations such as the Stanford Prison Experiment (www.prisonexp.org) show that normal people can dissolve into cruelty but don’t explain why. A recent neuroscience experiment suggests a mechanism for this kind of aggression.
Figure 1 Deriving pleasure from abuse?

American soldiers appearing to enjoy themselves as they humiliate Iraqi prisoners in the Abu Ghraib military prison.
Source: Reprinted with permission of The New Yorker, which first published this photo.
Dopamine and aggression
The nucleus accumbens has been called the brain’s “pleasure center,” and dopamine is the neurotransmitter that activates it.2 Activities and substances that stimulate dopamine release include sex, gambling, and smoking as well as cocaine and alcohol. The good feeling a person gets from these activities/substances reinforces the behavior that produced the feeling. In some cases, problems develop when people cannot resist the urge for more.
Ferrari et al3 placed micropipettes in rats’ nucleus accumbens to measure extracellular dopamine before, during, and after an aggressive confrontation. When the rats were confronted with an intruder rat for 10 minutes, they attacked and bit the intruder an average of 5 times, despite being implanted, tethered, and sampled. During and after the fight, dopamine was increased in the rats’ nucleus accumbens (Figure 2). Clearly, fighting gave them a “squirt” of pleasure that lasted almost 2 hours.
If we can extrapolate from this study to humans, we may understand why people become aggressive. At some level, they enjoy it. The bully on the playground, the wife beater, the mean boss—they get pleasure from being aggressive. It’s not just serial killers.
It is important to acknowledge that other variables such as poor supervision and too much power affected the actions of American soldiers working as prison guards in Iraq. However, the neuroscientific studies show us that aggression can be pleasurable, and people often have a hard time resisting what feels good. This knowledge may help us treat war veterans struggling not only with traumatic memories of violence but also with socially and personally unacceptable feelings of pleasure.
Figure 2 A ‘squirt’ of dopamine during violence
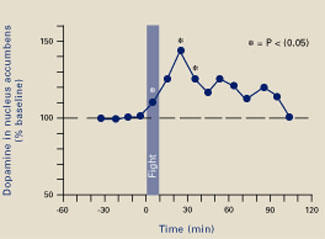
A 10-minute fight increased extracellular dopamine levels in rats’ nucleus accumbens for approximately 2 hours, suggesting that aggressive behavior produced pleasure.
Source: Adapted and reprinted with permission from reference 3. Copyright 2003, Blackwell Publishing.
1. Shermer M. The science of good and evil: Why people cheat, gossip, care, share, and follow the Golden Rule. New York: Times Books, 2004.
2. Ikemoto S, Panksepp J. The role of nucleus accumbens dopamine in motivated behavior: a unifying interpretation with special reference to reward-seeking. Brain Res Brain Res Rev 1999;31(1):6-41.
3. Ferrari PF, van Erp AM, Tornatzky W, Miczek KA. Accumbal dopamine and serotonin in anticipation of the next aggressive episode in rats. Eur J Neurosci 2003;17:371-8.
One of the most perplexing and shocking aspects of human behavior is the cruelty we can inflict on one another. Infamous events such as the Spanish Inquisition, slavery, lynching, conquest of the native Americans, and Nazi concentration camps are so sickening that many of us can barely tolerate hearing about them. How can people behave this way?
Recently, we’ve been shocked by photographs from the Abu Ghraib military prison in Iraq. American soldiers with unremarkable backgrounds—not Saddam’s henchmen—were shown humiliating and abusing Iraqi prisoners. Most remarkable was the joy on the Americans’ faces (Figure 1).
‘Good’ people, ‘bad’ circumstances
In experiments with college students, psychologists have shown that “regular” people can become sadistic under the right (or wrong) circumstances.1 One explanation is that it’s not just psychopaths who perpetrate crimes against humanity. Somehow, the psychopath within us all becomes unleashed.
Artificial situations such as the Stanford Prison Experiment (www.prisonexp.org) show that normal people can dissolve into cruelty but don’t explain why. A recent neuroscience experiment suggests a mechanism for this kind of aggression.
Figure 1 Deriving pleasure from abuse?
American soldiers appearing to enjoy themselves as they humiliate Iraqi prisoners in the Abu Ghraib military prison.
Source: Reprinted with permission of The New Yorker, which first published this photo.
Dopamine and aggression
The nucleus accumbens has been called the brain’s “pleasure center,” and dopamine is the neurotransmitter that activates it.2 Activities and substances that stimulate dopamine release include sex, gambling, and smoking as well as cocaine and alcohol. The good feeling a person gets from these activities/substances reinforces the behavior that produced the feeling. In some cases, problems develop when people cannot resist the urge for more.
Ferrari et al3 placed micropipettes in rats’ nucleus accumbens to measure extracellular dopamine before, during, and after an aggressive confrontation. When the rats were confronted with an intruder rat for 10 minutes, they attacked and bit the intruder an average of 5 times, despite being implanted, tethered, and sampled. During and after the fight, dopamine was increased in the rats’ nucleus accumbens (Figure 2). Clearly, fighting gave them a “squirt” of pleasure that lasted almost 2 hours.
If we can extrapolate from this study to humans, we may understand why people become aggressive. At some level, they enjoy it. The bully on the playground, the wife beater, the mean boss—they get pleasure from being aggressive. It’s not just serial killers.
It is important to acknowledge that other variables such as poor supervision and too much power affected the actions of American soldiers working as prison guards in Iraq. However, the neuroscientific studies show us that aggression can be pleasurable, and people often have a hard time resisting what feels good. This knowledge may help us treat war veterans struggling not only with traumatic memories of violence but also with socially and personally unacceptable feelings of pleasure.
Figure 2 A ‘squirt’ of dopamine during violence
A 10-minute fight increased extracellular dopamine levels in rats’ nucleus accumbens for approximately 2 hours, suggesting that aggressive behavior produced pleasure.
Source: Adapted and reprinted with permission from reference 3. Copyright 2003, Blackwell Publishing.
One of the most perplexing and shocking aspects of human behavior is the cruelty we can inflict on one another. Infamous events such as the Spanish Inquisition, slavery, lynching, conquest of the native Americans, and Nazi concentration camps are so sickening that many of us can barely tolerate hearing about them. How can people behave this way?
Recently, we’ve been shocked by photographs from the Abu Ghraib military prison in Iraq. American soldiers with unremarkable backgrounds—not Saddam’s henchmen—were shown humiliating and abusing Iraqi prisoners. Most remarkable was the joy on the Americans’ faces (Figure 1).
‘Good’ people, ‘bad’ circumstances
In experiments with college students, psychologists have shown that “regular” people can become sadistic under the right (or wrong) circumstances.1 One explanation is that it’s not just psychopaths who perpetrate crimes against humanity. Somehow, the psychopath within us all becomes unleashed.
Artificial situations such as the Stanford Prison Experiment (www.prisonexp.org) show that normal people can dissolve into cruelty but don’t explain why. A recent neuroscience experiment suggests a mechanism for this kind of aggression.
Figure 1 Deriving pleasure from abuse?
American soldiers appearing to enjoy themselves as they humiliate Iraqi prisoners in the Abu Ghraib military prison.
Source: Reprinted with permission of The New Yorker, which first published this photo.
Dopamine and aggression
The nucleus accumbens has been called the brain’s “pleasure center,” and dopamine is the neurotransmitter that activates it.2 Activities and substances that stimulate dopamine release include sex, gambling, and smoking as well as cocaine and alcohol. The good feeling a person gets from these activities/substances reinforces the behavior that produced the feeling. In some cases, problems develop when people cannot resist the urge for more.
Ferrari et al3 placed micropipettes in rats’ nucleus accumbens to measure extracellular dopamine before, during, and after an aggressive confrontation. When the rats were confronted with an intruder rat for 10 minutes, they attacked and bit the intruder an average of 5 times, despite being implanted, tethered, and sampled. During and after the fight, dopamine was increased in the rats’ nucleus accumbens (Figure 2). Clearly, fighting gave them a “squirt” of pleasure that lasted almost 2 hours.
If we can extrapolate from this study to humans, we may understand why people become aggressive. At some level, they enjoy it. The bully on the playground, the wife beater, the mean boss—they get pleasure from being aggressive. It’s not just serial killers.
It is important to acknowledge that other variables such as poor supervision and too much power affected the actions of American soldiers working as prison guards in Iraq. However, the neuroscientific studies show us that aggression can be pleasurable, and people often have a hard time resisting what feels good. This knowledge may help us treat war veterans struggling not only with traumatic memories of violence but also with socially and personally unacceptable feelings of pleasure.
Figure 2 A ‘squirt’ of dopamine during violence
A 10-minute fight increased extracellular dopamine levels in rats’ nucleus accumbens for approximately 2 hours, suggesting that aggressive behavior produced pleasure.
Source: Adapted and reprinted with permission from reference 3. Copyright 2003, Blackwell Publishing.
1. Shermer M. The science of good and evil: Why people cheat, gossip, care, share, and follow the Golden Rule. New York: Times Books, 2004.
2. Ikemoto S, Panksepp J. The role of nucleus accumbens dopamine in motivated behavior: a unifying interpretation with special reference to reward-seeking. Brain Res Brain Res Rev 1999;31(1):6-41.
3. Ferrari PF, van Erp AM, Tornatzky W, Miczek KA. Accumbal dopamine and serotonin in anticipation of the next aggressive episode in rats. Eur J Neurosci 2003;17:371-8.
1. Shermer M. The science of good and evil: Why people cheat, gossip, care, share, and follow the Golden Rule. New York: Times Books, 2004.
2. Ikemoto S, Panksepp J. The role of nucleus accumbens dopamine in motivated behavior: a unifying interpretation with special reference to reward-seeking. Brain Res Brain Res Rev 1999;31(1):6-41.
3. Ferrari PF, van Erp AM, Tornatzky W, Miczek KA. Accumbal dopamine and serotonin in anticipation of the next aggressive episode in rats. Eur J Neurosci 2003;17:371-8.
Posttraumatic stress disorder: Nature and nurture?
Posttraumatic stress disorder (PTSD) can be one of the most frustrating anxiety disorders for both the patient and clinician. Asymptomatic persons become haunted by an experience they can’t forget. Their resulting anxiety can sour what were once healthy relationships or disable someone who previously was productive.
In some cases, despite aggressive psychopharmacology and psychotherapy, the patient remains incapacitated by inappropriate and unremitting fear. The trauma seems to have broken something—changed something inside the brain—that can’t be fixed.
Brain imaging studies of patients with PTSD—combat veterans and women with histories of childhood sexual abuse—have shown smaller hippocampal volumes compared with patients without PTSD.1,2 This finding has led to speculation that stress hormones (glucocorticoids) adversely affect the hippocampus (Figure 1).
This line of reasoning suggests that prolonged stress causes increased production of glucocorticoids that are neurotoxic to the hippocampus, resulting in hippocampal atrophy.3 Studies of rodents and patients with Cushing’s syndrome support this hypothesis. The hippocampus, therefore, may have been irreversibly damaged in patients with severe PTSD.
Figure 1
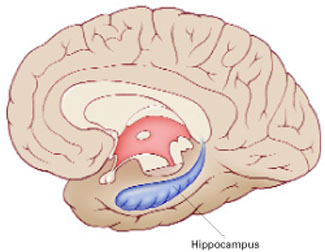
The hippocampus, a specialized type of cortex, is key to memory and emotion. As this medial view shows, it extends along the lateral ventricle floor on each side of the brain.
Illustration for Current Psychiatry by Marcia Hartsock, CMI Hippocampus
Intuitively, this theory makes sense, as the hippocampus is crucial for memory and emotion. However, a recent study of identical twins raises doubts.
Surprising evidence
Gilbertson et al recruited 40 pairs of twins, in which one was a Vietnam combat veteran and the other stayed home.4 Using MRI, the researchers measured hippocampal volume in each twin and assessed the presence and severity of PTSD in the combat-exposed twin.
Consistent with earlier reports, the authors found smaller hippocampal volumes in combat-exposed individuals diagnosed with PTSD. However, they found an almost identical correlation between the noncombat-exposed twin’s hippocampal volume and the combat-exposed twin’s PTSD score (Figure 2). In other words, the twin’s hippocampus size was a better predictor of the veteran’s hippocampus size than was the veteran’s trauma exposure or PTSD symptoms.
This finding puts a new spin on the association between small hippocampal volume and PTSD. The authors stated, “these data indicate that smaller hippocampi in PTSD represents a pre-existing, familial vulnerability factor rather than the neurotoxic product of trauma exposure per se.” Put another way, the small hippocampus is not created by stress and trauma but is a preexisting condition. Further, this study suggests that a larger hippocampus may protect a person from developing PTSD.
This study may help explain why different individuals exposed to the same trauma are frequently left with different symptoms.5,6 PTSD would seem to be an excellent example of the combined effects of nature (small hippocampus) and nurture (traumatic experience).
Figure 2 Hippocampal volume correlates with posttraumatic symptoms
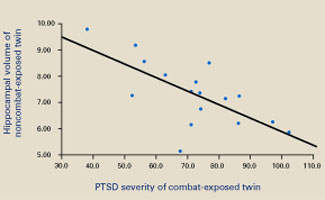
Smaller hippocampal volume in identical twins not exposed to combat was related to more-severe PTSD symptoms in their combat-exposed brothers (P = 0.002). Symptom severity was measured using Clinician-Administered PTSD Scale (CAPS) total scores.
Source: Reprinted with permission from Gilbertson MW, Shenton ME, Ciszewski A, et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nature Neurosci 2002;5:1242-7.
1. Bremner JD, Randall P, Scott TM, et al. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 1995;152:973-81.
2. Bremner JD, Vythilingam M, Vermetten E, et al. MRI and PET study of deficits in hippocampal structure and function in women with childhood sexual abuse and posttraumatic stress disorder. Am J Psychiatry 2003;160:924-32.
3. Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 2000;57:925-35.
4. Gilbertson MW, Shenton ME, Ciszewski A, et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci 2002;5:1242-7.
5. Macklin ML, Metzger LJ, Litz BT, et al. Lower precombat intelligence is a risk factor for posttraumatic stress disorder. J Consult Clin Psychol 1998;66:323-6.
6. Schlenger WE, Caddell JM, Ebert L, et al. Psychological reactions to terrorist attacks: findings from the National Study of Americans’ Reactions to September 11. JAMA 2002;288:581-8.
Posttraumatic stress disorder (PTSD) can be one of the most frustrating anxiety disorders for both the patient and clinician. Asymptomatic persons become haunted by an experience they can’t forget. Their resulting anxiety can sour what were once healthy relationships or disable someone who previously was productive.
In some cases, despite aggressive psychopharmacology and psychotherapy, the patient remains incapacitated by inappropriate and unremitting fear. The trauma seems to have broken something—changed something inside the brain—that can’t be fixed.
Brain imaging studies of patients with PTSD—combat veterans and women with histories of childhood sexual abuse—have shown smaller hippocampal volumes compared with patients without PTSD.1,2 This finding has led to speculation that stress hormones (glucocorticoids) adversely affect the hippocampus (Figure 1).
This line of reasoning suggests that prolonged stress causes increased production of glucocorticoids that are neurotoxic to the hippocampus, resulting in hippocampal atrophy.3 Studies of rodents and patients with Cushing’s syndrome support this hypothesis. The hippocampus, therefore, may have been irreversibly damaged in patients with severe PTSD.
Figure 1
The hippocampus, a specialized type of cortex, is key to memory and emotion. As this medial view shows, it extends along the lateral ventricle floor on each side of the brain.
Illustration for Current Psychiatry by Marcia Hartsock, CMI Hippocampus
Intuitively, this theory makes sense, as the hippocampus is crucial for memory and emotion. However, a recent study of identical twins raises doubts.
Surprising evidence
Gilbertson et al recruited 40 pairs of twins, in which one was a Vietnam combat veteran and the other stayed home.4 Using MRI, the researchers measured hippocampal volume in each twin and assessed the presence and severity of PTSD in the combat-exposed twin.
Consistent with earlier reports, the authors found smaller hippocampal volumes in combat-exposed individuals diagnosed with PTSD. However, they found an almost identical correlation between the noncombat-exposed twin’s hippocampal volume and the combat-exposed twin’s PTSD score (Figure 2). In other words, the twin’s hippocampus size was a better predictor of the veteran’s hippocampus size than was the veteran’s trauma exposure or PTSD symptoms.
This finding puts a new spin on the association between small hippocampal volume and PTSD. The authors stated, “these data indicate that smaller hippocampi in PTSD represents a pre-existing, familial vulnerability factor rather than the neurotoxic product of trauma exposure per se.” Put another way, the small hippocampus is not created by stress and trauma but is a preexisting condition. Further, this study suggests that a larger hippocampus may protect a person from developing PTSD.
This study may help explain why different individuals exposed to the same trauma are frequently left with different symptoms.5,6 PTSD would seem to be an excellent example of the combined effects of nature (small hippocampus) and nurture (traumatic experience).
Figure 2 Hippocampal volume correlates with posttraumatic symptoms
Smaller hippocampal volume in identical twins not exposed to combat was related to more-severe PTSD symptoms in their combat-exposed brothers (P = 0.002). Symptom severity was measured using Clinician-Administered PTSD Scale (CAPS) total scores.
Source: Reprinted with permission from Gilbertson MW, Shenton ME, Ciszewski A, et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nature Neurosci 2002;5:1242-7.
Posttraumatic stress disorder (PTSD) can be one of the most frustrating anxiety disorders for both the patient and clinician. Asymptomatic persons become haunted by an experience they can’t forget. Their resulting anxiety can sour what were once healthy relationships or disable someone who previously was productive.
In some cases, despite aggressive psychopharmacology and psychotherapy, the patient remains incapacitated by inappropriate and unremitting fear. The trauma seems to have broken something—changed something inside the brain—that can’t be fixed.
Brain imaging studies of patients with PTSD—combat veterans and women with histories of childhood sexual abuse—have shown smaller hippocampal volumes compared with patients without PTSD.1,2 This finding has led to speculation that stress hormones (glucocorticoids) adversely affect the hippocampus (Figure 1).
This line of reasoning suggests that prolonged stress causes increased production of glucocorticoids that are neurotoxic to the hippocampus, resulting in hippocampal atrophy.3 Studies of rodents and patients with Cushing’s syndrome support this hypothesis. The hippocampus, therefore, may have been irreversibly damaged in patients with severe PTSD.
Figure 1
The hippocampus, a specialized type of cortex, is key to memory and emotion. As this medial view shows, it extends along the lateral ventricle floor on each side of the brain.
Illustration for Current Psychiatry by Marcia Hartsock, CMI Hippocampus
Intuitively, this theory makes sense, as the hippocampus is crucial for memory and emotion. However, a recent study of identical twins raises doubts.
Surprising evidence
Gilbertson et al recruited 40 pairs of twins, in which one was a Vietnam combat veteran and the other stayed home.4 Using MRI, the researchers measured hippocampal volume in each twin and assessed the presence and severity of PTSD in the combat-exposed twin.
Consistent with earlier reports, the authors found smaller hippocampal volumes in combat-exposed individuals diagnosed with PTSD. However, they found an almost identical correlation between the noncombat-exposed twin’s hippocampal volume and the combat-exposed twin’s PTSD score (Figure 2). In other words, the twin’s hippocampus size was a better predictor of the veteran’s hippocampus size than was the veteran’s trauma exposure or PTSD symptoms.
This finding puts a new spin on the association between small hippocampal volume and PTSD. The authors stated, “these data indicate that smaller hippocampi in PTSD represents a pre-existing, familial vulnerability factor rather than the neurotoxic product of trauma exposure per se.” Put another way, the small hippocampus is not created by stress and trauma but is a preexisting condition. Further, this study suggests that a larger hippocampus may protect a person from developing PTSD.
This study may help explain why different individuals exposed to the same trauma are frequently left with different symptoms.5,6 PTSD would seem to be an excellent example of the combined effects of nature (small hippocampus) and nurture (traumatic experience).
Figure 2 Hippocampal volume correlates with posttraumatic symptoms
Smaller hippocampal volume in identical twins not exposed to combat was related to more-severe PTSD symptoms in their combat-exposed brothers (P = 0.002). Symptom severity was measured using Clinician-Administered PTSD Scale (CAPS) total scores.
Source: Reprinted with permission from Gilbertson MW, Shenton ME, Ciszewski A, et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nature Neurosci 2002;5:1242-7.
1. Bremner JD, Randall P, Scott TM, et al. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 1995;152:973-81.
2. Bremner JD, Vythilingam M, Vermetten E, et al. MRI and PET study of deficits in hippocampal structure and function in women with childhood sexual abuse and posttraumatic stress disorder. Am J Psychiatry 2003;160:924-32.
3. Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 2000;57:925-35.
4. Gilbertson MW, Shenton ME, Ciszewski A, et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci 2002;5:1242-7.
5. Macklin ML, Metzger LJ, Litz BT, et al. Lower precombat intelligence is a risk factor for posttraumatic stress disorder. J Consult Clin Psychol 1998;66:323-6.
6. Schlenger WE, Caddell JM, Ebert L, et al. Psychological reactions to terrorist attacks: findings from the National Study of Americans’ Reactions to September 11. JAMA 2002;288:581-8.
1. Bremner JD, Randall P, Scott TM, et al. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 1995;152:973-81.
2. Bremner JD, Vythilingam M, Vermetten E, et al. MRI and PET study of deficits in hippocampal structure and function in women with childhood sexual abuse and posttraumatic stress disorder. Am J Psychiatry 2003;160:924-32.
3. Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 2000;57:925-35.
4. Gilbertson MW, Shenton ME, Ciszewski A, et al. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci 2002;5:1242-7.
5. Macklin ML, Metzger LJ, Litz BT, et al. Lower precombat intelligence is a risk factor for posttraumatic stress disorder. J Consult Clin Psychol 1998;66:323-6.
6. Schlenger WE, Caddell JM, Ebert L, et al. Psychological reactions to terrorist attacks: findings from the National Study of Americans’ Reactions to September 11. JAMA 2002;288:581-8.
ADHD and substance abuse: 4 therapeutic options for patients with addictions
Should you prescribe a stimulant to treat attention and hyperactivity problems in teenagers and adults with a history of substance abuse? Evidence suggests that using a stimulant to treat attention-deficit/hyperactivity disorder (ADHD) may place such patients at risk for stimulant abuse or for relapse into abuse of other substances. But a stimulant may be the only option for patients whose ADHD symptoms do not respond to alternate medications, such as antidepressants.
Growing numbers of adults are being treated for ADHD. Because substance abuse problems are common in adults with ADHD (Box 1 ),1-3 prescribing an antidepressant instead of a stimulant in some cases may be prudent. Consider the following factors when choosing ADHD therapy for patients with a history of substance abuse.
Prevalence of stimulant use and abuse
In the United States, more than 95% of medications prescribed for children and adults with ADHD are stimulants—usually methylphenidate.4 Stimulant use has increased as more children and adults are diagnosed with ADHD. Methylphenidate prescriptions increased five-fold from 1990 to 1995.5 Visits to psychiatrists and physicians that included stimulant prescriptions grew from 570,000 to 2.86 million from 1985 to 1994, with most of that increase occurring during visits to primary care and other physicians.6
When used as prescribed, methylphenidate is safe and effective for treating most children and adults with ADHD. Methylphenidate’s pharmacologic properties, however, are similar to those of amphetamines and cocaine (Box 2, Figure 1),7,8 which is why methylphenidate is a schedule-II controlled substance.
Published data. Fifteen reports of methylphenidate abuse were published in the medical literature between 1960 and 1999,7 but little is known about the prevalence of stimulant abuse among patients with ADHD. Banov and colleagues recently published what may be the only data available, when they reported that 3 of 37 (8%) patients abused the stimulants they were prescribed for ADHD.9 The three patients who abused stimulants had histories of drug and alcohol abuse at study entry. In all three cases, stimulant abuse did not develop immediately but became apparent within 6 months after the study began.
In a study of 651 students ages 11 to 18 in Wisconsin and Minnesota, more than one-third of those taking stimulants reported being asked to sell or trade their medications. More than one-half of those not taking ADHD medications said they knew someone who sold or gave away his or her medication.10
Stimulant theft, recreational use. Methylphenidate has been identified as the third most abused prescribed substance in the United States.11 It was the 10th most frequently stolen controlled drug from pharmacies between 1990 and 1995, and 700,000 dosage units were reported stolen in 1996 and 1997.12
As many as 50% of adults with ADHD have substance abuse problems (including alcohol, cocaine, and marijuana), and as many as 30% have antisocial personality disorder (with increased potential for drug-seeking behaviors).1 Compared with the general population, persons with ADHD have an earlier onset of substance abuse that is less responsive to treatment and more likely to progress from alcohol to other drugs.2
The elevated risk of substance abuse in ADHD may be related to a subtle lack of response to normal positive and negative reinforcements. Hunt has outlined four neurobehavioral deficits that define ADHD.3 Besides inattention, hyperarousal, and impulsiveness, he proposes that persons with ADHD have a reward system deficit. They may gravitate toward substance abuse because drugs, alcohol, and nicotine provide stronger rewards than life’s more subtle social interactions.
The popular media have reported recreational use of methylphenidate—with street names such as “R-Ball” and “Vitamin R”—among teens and college students.13 Illegal stimulants are perceived to be easily accessible on college campuses, but no data have been reported.
The use of stimulant medication for ADHD patients with substance abuse problems remains controversial. For such patients, this author reserves stimulant medication for those:
- whose ADHD symptoms have not responded adequately to alternate treatments
- who have been reliable with prescription medications
- and whose functional level is seriously impaired by their ADHD.
Antidepressants vs. stimulants
Although few well-designed controlled studies have been published, four antidepressants appear to be reasonably equivalent in effectiveness for adults with ADHD and do not carry potential for stimulant abuse.14
Desipraime, bupropion, venlafaxine, and the experimental drug atomoxetine (Table 1) all increase norepinephrine at the synapse by inhibiting presynaptic reuptake. Though dopamine has traditionally been considered the neurotransmitter of choice for ADHD treatment, norepinephrine may be equally potent.
Impulse control center. Several lines of research have recently established a connection between the prefrontal cortex, norepinephrine, and ADHD.15 This evidence suggests that the prefrontal cortex plays a major role in inhibiting impulses and responses to distractions:
Figure 1
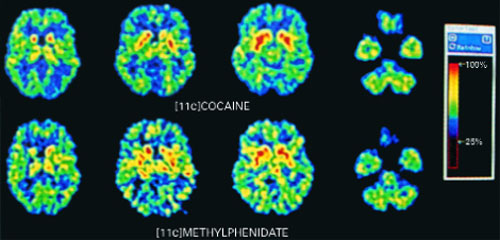
PET scans of the brain using carbon 11 (11C)-labeled cocaine and methylphenidate HCl show similar distributions in the striatium when the drugs are administered intravenously.
Source: Reproduced with permission from Volkow ND, Ding YS, Fowler JS, et al. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch Gen Psychiatry 1995;52:456-63.
Stimulants are classified as schedule-II drugs because they produce powerful reinforcing effects by increasing synaptic dopamine.7 Positron-emission tomography (PET) scans using carbon labeling have shown similar distributions of methylphenidate and cocaine in the brain (Figure 1).8 When administered intravenously, both drugs occupy the same receptors in the striatum and produce a “high” that parallels rapid neuronal uptake. Methylphenidate and cocaine similarly increase stimulation of the postsynaptic neuron by blocking the dopamine reuptake pump (dopamine transporter).
How a substance gets to the brain’s euphoric receptors greatly affects its addictive properties. Delivery systems with rapid onset—smoking, “snorting,” or IV injection—have much greater ability to produce a “high” than do oral or transdermal routes. The greater the “high,” the greater the potential for abuse.
Because methylphenidate is prescribed for oral use, the potential for abuse is minimal. However, we need to be extremely cautious when giving methylphenidate or similar stimulant medications to patients who have shown they are unable to control their abuse of other substances.
Stimulants can also re-ignite a dormant substance abuse problem. Though little has been written about this in the medical literature, Elizabeth Wurtzel, author of the controversial Prozac Nation, chronicles the resumption of her cocaine abuse in More, Now, Again: A Memoir of Addiction. She contends that after her doctor added methylphenidate to augment treatment of partially remitting depression, she began abusing it and eventually was using 40 tablets per day before slipping back into cocaine dependence.
- Patients with prefrontal cortex deficits can have problems with inattention and poor impulse control.
- Patients with ADHD have frontal lobe impairments, as neuropsychological testing and imaging studies have shown.
- Norepinephrine neurons, with cell bodies in the locus coeruleus, have projections that terminate in the prefrontal cortex.
- Agents with norepinephrine activity, but without mood-altering properties (e.g., clonidine), have been shown to improve ADHD symptoms.
Table 1
ADHD IN ADULTS: ANTIDEPRESSANT DOSAGES AND SIDE EFFECTS
| Medication | Class | Effective dosage | Side effects |
|---|---|---|---|
| Desipramine | Tricyclic | 100 to 200 mg/d | Sedation, weight gain, dry mouth, constipation, orthostatic hypotension, prolonged cardiac conduction time; may be lethal in overdose |
| Bupropion SR | Norepinephrine and dopamine reuptake inhibitor | 150 mg bid to 200 mg bid | Headaches, insomnia, agitation, increased risk of seizures |
| Venlafaxine XR | Serotonin and norepinephrine reuptake inhibitor | 75 to 225 mg/d | Nausea, sexual side effects, agitation, increased blood pressure at higher dosages |
| Atomoxetine* | Norepinephrine reuptake inhibitor | To be determined | To be determined |
| * Investigational agent; not FDA-approved | |||
Additional evidence suggests that the prefrontal cortex has projections back to the locus coeruleus, which may explain the relationship between the two areas. It may be that the brain’s higher-functioning areas, such as the prefrontal cortex, provide intelligent screening of impulses from the brain’s older areas, such as the locus coeruleus. Therefore, increased prefrontal cortex activity may modulate some impulses that ADHD patients cannot control otherwise.

No ‘high’ with antidepressants. Patients with ADHD who have experienced the powerful effects of street drugs such as cocaine, methamphetamine, or even alcohol may report that antidepressants do not provide the effect they desire. It is difficult to know if these patients are reporting a lack of benefit or simply the absence of a euphoric “high” they are used to experiencing with substances of abuse. The newer antidepressants do not activate the brain’s euphoric receptors to an appreciable degree.
Patients who take stimulants as prescribed also do not report a “high” but can detect the medication’s presence and absence. Most do not crave this feeling, but substance abusers tend to like it. A patient recently told me he didn’t think stimulants improved his ADHD, but said, “I just liked the way they made me feel.”
Desipramine
The tricyclic antidepressant desipramine is a potent norepinephrine reuptake inhibitor that is effective in treating ADHD in children and adults. In a double-blind, placebo-controlled study of adults with ADHD, subjects receiving desipramine showed robust improvement in symptom scores on the ADHD Rating Scale,16 compared with those receiving the placebo (Figure 2).17
During the 6-week trial, 41 adults with ADHD received desipramine, 200 mg/d, or a placebo. Those receiving desipramine showed significant improvement in 12 of 14 ADHD symptoms and less hyperactivity, impulsivity, and inattentiveness, whereas those receiving the placebo showed no improvement. According to the study criteria, 68% of those who received desipramine and none who received the placebo were considered positive responders.
Though no head-to-head studies have compared desipramine with methylphenidate, the same researchers conducted a similar placebo-controlled study with methylphenidate. The 6-week symptom score on the ADHD Rating Scale was 12.5 for methylphenidate, compared with a score of 12 for desipramine.18
Recommendation. Desipramine may be the most effective of the antidepressant treatments for patients with ADHD. Because of its side effects, however, it is not this author’s first choice and is usually reserved for patients whose symptoms fail to respond to other antidepressants. Desipramine can cause sedation, dry mouth, and constipation, which are related to blockade of adrenergic, histamine, and muscarinic cholinergic receptors. It also can be lethal in overdose.
Some substance abusers lose confidence in a medication that they cannot feel working. The side effects of desipramine, which can be intolerable for some patients, can reassure others with a history of substance abuse that they are being medicated.
Bupropion
Bupropion is a unique antidepressant that inhibits the presynaptic reuptake of dopamine and norepinephrine. Open-label studies demonstrate good responses to bupropion by adults with ADHD. One placebo-controlled, double-blind study found improved ADHD symptoms in 76% of patients receiving bupropion SR, compared with 37% of those receiving a placebo; the difference was statistically significant.19
Figure 2 IMPROVED ADHD SYMPTOMS WITH DESIPRAMINE
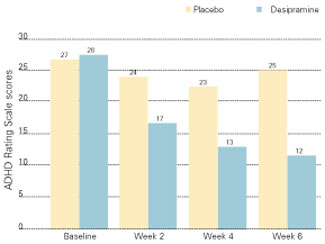
Adults with ADHD who received desipramine, 200 mg/d, in a double-blind trial showed significantly less hyperactivity, impulsivity, and inattentiveness after 6 weeks of therapy than a control group that received a placebo.
Source: Adapted with permission from Wilens TE, Biederman J, Prince J, et al. Six-week, double-blind, placebo-controlled study of desipramine for adult attention-deficit/hyperactivity disorder. Am J Psychiatry 1996;153:1147-53.Another study of adults with ADHD compared bupropion SR to methylphenidate and a placebo.20 Using a primary outcome of the Clinical Global Impression (CGI) scale, response rates were 64% for bupropion SR, 50% for methylphenidate, and 27% for the placebo. The difference in response rates between the two agents was not statistically significant (p = 0.14).
Recommendation. The risk of seizures with bupropion is about 1 in 1,000. Therefore, bupropion should not be given to patients with a seizure disorder or to those with conditions that alter the seizure threshold (e.g., eating disorders, recent head trauma, or benzodiazepine withdrawal).21
This author uses bupropion as first-line treatment for appropriate patients with ADHD and a substance abuse history. Bupropion’s mild benefit with smoking cessation may provide some crossover effect for other substances of abuse. The low incidence of sexual side effects is another benefit. Drawbacks include twice-daily dosing and lack of a robust effect on attention and concentration.
Venlafaxine
Venlafaxine is a potent inhibitor of serotonin reuptake, a moderate inhibitor of norepinephrine, and a mild inhibitor of dopamine. Venlafaxine has displayed response rates similar to those of desipramine and bupropion in open-label studies in adults with ADHD,14 but no placebo-controlled studies exist.
As noted above, these antidepressants are believed to improve ADHD symptoms by making norepinephrine more available at the synapse. Hypothetically, then, one would need to administer venlafaxine at dosages that adequately inhibit the norepinephrine reuptake receptor. Venlafaxine XR, 150 mg/d, provides significant norepinephrine activity, according to several lines of evidence.
Recently, Upadhyaya et al reported the use of venlafaxine in one of the few treatment studies of patients with ADHD and comorbid alcohol/cocaine abuse. In an open-label trial, 10 subjects received venlafaxine, up to 300 mg/d, along with psychotherapy and attendance at Alcoholics Anonymous meetings. The nine who completed 4 weeks of treatment showed significantly improved ADHD symptoms and decreased alcohol craving.22
Recommendation. Venlafaxine is this author’s second choice for patients with ADHD and substance abuse problems. Sexual side effects that some patients experience with venlafaxine can limit its use. Some clinicians are concerned about increases in blood pressure associated with venlafaxine, although significant changes do not seem to occur at dosages below 300 mg/d.23
Atomoxetine
Atomoxetine is an investigational antidepressant in phase-III trials as a treatment for ADHD. Evidence shows atomoxetine to be a potent inhibitor of the presynaptic norepinephrine transporter, with minimal affinity for other neurotransmitter receptors. Initial studies suggest that atomoxetine is effective for adults and children with ADHD:
- In a small, double-blind, placebo-controlled, crossover trial, 11 of 20 adults showed improvement in ADHD symptoms within 3 weeks of starting atomoxetine.24
- In 297 children and adolescents, atomoxetine at dosages averaging approximately 1.2 mg/kg/d was more effective than a placebo in reducing ADHD symptoms and improving social and family functioning. Treatment was well tolerated and without significant side effects.25
- In a randomized open-label trial, 228 children received atomoxetine or methylphenidate. Both treatments significantly reduced inattention and hyperactive/impulsive symptoms.26
Related resources
- Arnsten AF. Genetics of childhood disorders (XVIII). ADHD, Part 2: Norepinephrine has a critical modulatory influence on prefrontal cortical function. J Am Acad Child Adolesc Psychiatry. 2000;39:1201-3.
- Ward MF, Wender PH, Reimherr FW. The Wender Utah Rating Scale: An aid in the retrospective diagnosis of childhood attention-deficit/hyperactivity disorder. Am J Psychiatry. 1993;150:885-90.
- Research Report: Prescription Drugs—abuse and addiction. National Institute on Drug Abuse, Substance Abuse and Mental Health Services Administration.
Drug brand names
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Methylphenidate • Ritalin, Concerta
- Venlafaxine • Effexor
Disclosure
Dr. Higgins reports that he is on the speakers’ bureaus for Wyeth-Ayerst Pharmaceuticals and Eli Lilly and Co.
1. Mannuzza S, Klein RG, Bessler A, et al. Adult outcome of hyperactive boys. Educational achievement, occupational rank, and psychiatric status. Arch Gen Psychiatry 1993;50:565-76.
2. Sullivan MA, Rudnik-Levin F. Attention deficit/hyperactivity disorder and substance abuse. Diagnostic and therapeutic considerations. Ann NY Acad Sci 2001;931:251-70.
3. Hunt RD. Nosology, neurobiology, and clinical patterns of ADHD in adults. Psychiatric Ann 1997;27:572-81.
4. Taylor MA. Evaluation and management of attention-deficit hyperactivity disorder. Am Fam Phys 1997;55:887-904.
5. Diller LH. The run on Ritalin. Attention deficit disorder and stimulant treatment in the 1990s. Hasting Center Report 1996;26:12-18.
6. Pincus HA, Tanielian TL, Marcus SC, et al. Prescribing trends in psychotrophic medications: primary care, psychiatry, and other medical specialties. JAMA 1998;279:526-31.
7. Mortan WA, Stockton GG. Methylphenidate abuse and psychiatric side effects. J Clin Psychiatry (Primary Care) 2000;2:159-64.
8. Volkow ND, Ding YS, Fowler JS, et al. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch Gen Psychiatry 1995;52:456-63.
9. Banov MD, Palmer T, Brody E. Antidepressants are as effective as stimulants in the long-term treatment of ADHD in Adults. Primary Psychiatry 2001;8:54-7.
10. Moline S, Frankenberger W. Use of stimulant medication for treatment of attention-deficit/hyperactivity disorder. A survey of middle and high school students attitudes. Psychology in the Schools 2001;38:569-84.
11. Prescription drugs: abuse and addiction. National Institute on Drug Abuse Research Report Series. NIH publication number 01-4881, July 2001.
12. Mann A. Illicit methylphenidate trade appears widespread. Clinical Psychiatry News June 2000;28(6):5-
13. ABCNews.com. http://more/abcnews.go.com/sections/living/DailyNews/ritalin0505.html
14. Higgins ES. A comparative analysis of antidepressants and stimulants for the treatment of adults with attention-deficit hyperactivity disorder. J Fam Pract 1999;48:15-20.
15. Arnsten AF, Steere JC, Hunt RD. The contribution of alpha2-noradrenergic mechanisms to prefrontal cortical cognitive function. Arch Gen Psychiatry 1996;53:448-55.
16. DuPaul G. The ADHD Rating Scale: normative data, reliability, and validity. Worcester, MA: University of Massachusetts Medical School, 1990.
17. Wilens TE, Biederman J, Prince J, et al. Six-week, double-blind, placebo-controlled study of desipramine for adult attention-deficit/hyperactivity disorder. Am J Psychiatry 1996;153:1147-53.
18. Spencer T, Wilens T, Biederman J, Faraone SV, Ablon JS, Lapey K. A double-blind, crossover comparison of methylphenidate and placebo in adults with childhood-onset attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 1995;52:434-43.
19. Wilens TE, Spencer TJ, Biederman J, et al. A controlled clinical trial of bupropion for attention-deficit/hyperactivity disorder in adults. Am J Psychiatry 2001;158:282-8.
20. Kuperman S, Perry PJ, Gaffney GR, et al. Bupropion SR vs. methylphenidate vs. placebo for attention-deficit/hyperactivity disorder in adults. Ann Clin Psychiatry 2001;13:129-34.
21. Wooltorton E. Bupropion (Zyban, Wellbutrin SR): reports of death, seizures, serum sickness. Can Med J 2002;166:68.-
22. Upadhyaya HP, Brady KT, Sethuraman G, et al. Venlafaxine treatment of patients with comorbid alcohol/cocaine abuse and attention-deficit/hyperactivity disorder: a pilot study. J Clin Psychopharmacol 2001;21:116-8.
23. Thase ME. Effects of venlafaxine on blood pressure: a meta-analysis of original data from 3744 depressed patients. J Clin Psychiatry 1998;59:502-8.
24. Spencer T, Biederman J, Wilens T, Prince J, Hatch M. Effectiveness and tolerability of tomoxetine in adults with attention-deficit/hyperactivity disorder. Am J Psychiatry 1998;155:693-5.
25. Michelson D, Faries D, Wernicke J, et al. Atomoxetine in the treatment of children and adolescents with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled, dose-response study. Pediatrics 2001;108:E83.-
26. Kratochvil C, Heiligenstein JH, Dittmann R, et al. Atomoxetine and methylphenidate treatment in ADHD children: a randomized, open-label trial (presentation). Honolulu: American Academy of Child and Adolescent Psychiatry, October, 2001.
Should you prescribe a stimulant to treat attention and hyperactivity problems in teenagers and adults with a history of substance abuse? Evidence suggests that using a stimulant to treat attention-deficit/hyperactivity disorder (ADHD) may place such patients at risk for stimulant abuse or for relapse into abuse of other substances. But a stimulant may be the only option for patients whose ADHD symptoms do not respond to alternate medications, such as antidepressants.
Growing numbers of adults are being treated for ADHD. Because substance abuse problems are common in adults with ADHD (Box 1 ),1-3 prescribing an antidepressant instead of a stimulant in some cases may be prudent. Consider the following factors when choosing ADHD therapy for patients with a history of substance abuse.
Prevalence of stimulant use and abuse
In the United States, more than 95% of medications prescribed for children and adults with ADHD are stimulants—usually methylphenidate.4 Stimulant use has increased as more children and adults are diagnosed with ADHD. Methylphenidate prescriptions increased five-fold from 1990 to 1995.5 Visits to psychiatrists and physicians that included stimulant prescriptions grew from 570,000 to 2.86 million from 1985 to 1994, with most of that increase occurring during visits to primary care and other physicians.6
When used as prescribed, methylphenidate is safe and effective for treating most children and adults with ADHD. Methylphenidate’s pharmacologic properties, however, are similar to those of amphetamines and cocaine (Box 2, Figure 1),7,8 which is why methylphenidate is a schedule-II controlled substance.
Published data. Fifteen reports of methylphenidate abuse were published in the medical literature between 1960 and 1999,7 but little is known about the prevalence of stimulant abuse among patients with ADHD. Banov and colleagues recently published what may be the only data available, when they reported that 3 of 37 (8%) patients abused the stimulants they were prescribed for ADHD.9 The three patients who abused stimulants had histories of drug and alcohol abuse at study entry. In all three cases, stimulant abuse did not develop immediately but became apparent within 6 months after the study began.
In a study of 651 students ages 11 to 18 in Wisconsin and Minnesota, more than one-third of those taking stimulants reported being asked to sell or trade their medications. More than one-half of those not taking ADHD medications said they knew someone who sold or gave away his or her medication.10
Stimulant theft, recreational use. Methylphenidate has been identified as the third most abused prescribed substance in the United States.11 It was the 10th most frequently stolen controlled drug from pharmacies between 1990 and 1995, and 700,000 dosage units were reported stolen in 1996 and 1997.12
As many as 50% of adults with ADHD have substance abuse problems (including alcohol, cocaine, and marijuana), and as many as 30% have antisocial personality disorder (with increased potential for drug-seeking behaviors).1 Compared with the general population, persons with ADHD have an earlier onset of substance abuse that is less responsive to treatment and more likely to progress from alcohol to other drugs.2
The elevated risk of substance abuse in ADHD may be related to a subtle lack of response to normal positive and negative reinforcements. Hunt has outlined four neurobehavioral deficits that define ADHD.3 Besides inattention, hyperarousal, and impulsiveness, he proposes that persons with ADHD have a reward system deficit. They may gravitate toward substance abuse because drugs, alcohol, and nicotine provide stronger rewards than life’s more subtle social interactions.
The popular media have reported recreational use of methylphenidate—with street names such as “R-Ball” and “Vitamin R”—among teens and college students.13 Illegal stimulants are perceived to be easily accessible on college campuses, but no data have been reported.
The use of stimulant medication for ADHD patients with substance abuse problems remains controversial. For such patients, this author reserves stimulant medication for those:
- whose ADHD symptoms have not responded adequately to alternate treatments
- who have been reliable with prescription medications
- and whose functional level is seriously impaired by their ADHD.
Antidepressants vs. stimulants
Although few well-designed controlled studies have been published, four antidepressants appear to be reasonably equivalent in effectiveness for adults with ADHD and do not carry potential for stimulant abuse.14
Desipraime, bupropion, venlafaxine, and the experimental drug atomoxetine (Table 1) all increase norepinephrine at the synapse by inhibiting presynaptic reuptake. Though dopamine has traditionally been considered the neurotransmitter of choice for ADHD treatment, norepinephrine may be equally potent.
Impulse control center. Several lines of research have recently established a connection between the prefrontal cortex, norepinephrine, and ADHD.15 This evidence suggests that the prefrontal cortex plays a major role in inhibiting impulses and responses to distractions:
Figure 1
PET scans of the brain using carbon 11 (11C)-labeled cocaine and methylphenidate HCl show similar distributions in the striatium when the drugs are administered intravenously.
Source: Reproduced with permission from Volkow ND, Ding YS, Fowler JS, et al. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch Gen Psychiatry 1995;52:456-63.
Stimulants are classified as schedule-II drugs because they produce powerful reinforcing effects by increasing synaptic dopamine.7 Positron-emission tomography (PET) scans using carbon labeling have shown similar distributions of methylphenidate and cocaine in the brain (Figure 1).8 When administered intravenously, both drugs occupy the same receptors in the striatum and produce a “high” that parallels rapid neuronal uptake. Methylphenidate and cocaine similarly increase stimulation of the postsynaptic neuron by blocking the dopamine reuptake pump (dopamine transporter).
How a substance gets to the brain’s euphoric receptors greatly affects its addictive properties. Delivery systems with rapid onset—smoking, “snorting,” or IV injection—have much greater ability to produce a “high” than do oral or transdermal routes. The greater the “high,” the greater the potential for abuse.
Because methylphenidate is prescribed for oral use, the potential for abuse is minimal. However, we need to be extremely cautious when giving methylphenidate or similar stimulant medications to patients who have shown they are unable to control their abuse of other substances.
Stimulants can also re-ignite a dormant substance abuse problem. Though little has been written about this in the medical literature, Elizabeth Wurtzel, author of the controversial Prozac Nation, chronicles the resumption of her cocaine abuse in More, Now, Again: A Memoir of Addiction. She contends that after her doctor added methylphenidate to augment treatment of partially remitting depression, she began abusing it and eventually was using 40 tablets per day before slipping back into cocaine dependence.
- Patients with prefrontal cortex deficits can have problems with inattention and poor impulse control.
- Patients with ADHD have frontal lobe impairments, as neuropsychological testing and imaging studies have shown.
- Norepinephrine neurons, with cell bodies in the locus coeruleus, have projections that terminate in the prefrontal cortex.
- Agents with norepinephrine activity, but without mood-altering properties (e.g., clonidine), have been shown to improve ADHD symptoms.
Table 1
ADHD IN ADULTS: ANTIDEPRESSANT DOSAGES AND SIDE EFFECTS
| Medication | Class | Effective dosage | Side effects |
|---|---|---|---|
| Desipramine | Tricyclic | 100 to 200 mg/d | Sedation, weight gain, dry mouth, constipation, orthostatic hypotension, prolonged cardiac conduction time; may be lethal in overdose |
| Bupropion SR | Norepinephrine and dopamine reuptake inhibitor | 150 mg bid to 200 mg bid | Headaches, insomnia, agitation, increased risk of seizures |
| Venlafaxine XR | Serotonin and norepinephrine reuptake inhibitor | 75 to 225 mg/d | Nausea, sexual side effects, agitation, increased blood pressure at higher dosages |
| Atomoxetine* | Norepinephrine reuptake inhibitor | To be determined | To be determined |
| * Investigational agent; not FDA-approved | |||
Additional evidence suggests that the prefrontal cortex has projections back to the locus coeruleus, which may explain the relationship between the two areas. It may be that the brain’s higher-functioning areas, such as the prefrontal cortex, provide intelligent screening of impulses from the brain’s older areas, such as the locus coeruleus. Therefore, increased prefrontal cortex activity may modulate some impulses that ADHD patients cannot control otherwise.
No ‘high’ with antidepressants. Patients with ADHD who have experienced the powerful effects of street drugs such as cocaine, methamphetamine, or even alcohol may report that antidepressants do not provide the effect they desire. It is difficult to know if these patients are reporting a lack of benefit or simply the absence of a euphoric “high” they are used to experiencing with substances of abuse. The newer antidepressants do not activate the brain’s euphoric receptors to an appreciable degree.
Patients who take stimulants as prescribed also do not report a “high” but can detect the medication’s presence and absence. Most do not crave this feeling, but substance abusers tend to like it. A patient recently told me he didn’t think stimulants improved his ADHD, but said, “I just liked the way they made me feel.”
Desipramine
The tricyclic antidepressant desipramine is a potent norepinephrine reuptake inhibitor that is effective in treating ADHD in children and adults. In a double-blind, placebo-controlled study of adults with ADHD, subjects receiving desipramine showed robust improvement in symptom scores on the ADHD Rating Scale,16 compared with those receiving the placebo (Figure 2).17
During the 6-week trial, 41 adults with ADHD received desipramine, 200 mg/d, or a placebo. Those receiving desipramine showed significant improvement in 12 of 14 ADHD symptoms and less hyperactivity, impulsivity, and inattentiveness, whereas those receiving the placebo showed no improvement. According to the study criteria, 68% of those who received desipramine and none who received the placebo were considered positive responders.
Though no head-to-head studies have compared desipramine with methylphenidate, the same researchers conducted a similar placebo-controlled study with methylphenidate. The 6-week symptom score on the ADHD Rating Scale was 12.5 for methylphenidate, compared with a score of 12 for desipramine.18
Recommendation. Desipramine may be the most effective of the antidepressant treatments for patients with ADHD. Because of its side effects, however, it is not this author’s first choice and is usually reserved for patients whose symptoms fail to respond to other antidepressants. Desipramine can cause sedation, dry mouth, and constipation, which are related to blockade of adrenergic, histamine, and muscarinic cholinergic receptors. It also can be lethal in overdose.
Some substance abusers lose confidence in a medication that they cannot feel working. The side effects of desipramine, which can be intolerable for some patients, can reassure others with a history of substance abuse that they are being medicated.
Bupropion
Bupropion is a unique antidepressant that inhibits the presynaptic reuptake of dopamine and norepinephrine. Open-label studies demonstrate good responses to bupropion by adults with ADHD. One placebo-controlled, double-blind study found improved ADHD symptoms in 76% of patients receiving bupropion SR, compared with 37% of those receiving a placebo; the difference was statistically significant.19
Figure 2 IMPROVED ADHD SYMPTOMS WITH DESIPRAMINE
Adults with ADHD who received desipramine, 200 mg/d, in a double-blind trial showed significantly less hyperactivity, impulsivity, and inattentiveness after 6 weeks of therapy than a control group that received a placebo.
Source: Adapted with permission from Wilens TE, Biederman J, Prince J, et al. Six-week, double-blind, placebo-controlled study of desipramine for adult attention-deficit/hyperactivity disorder. Am J Psychiatry 1996;153:1147-53.Another study of adults with ADHD compared bupropion SR to methylphenidate and a placebo.20 Using a primary outcome of the Clinical Global Impression (CGI) scale, response rates were 64% for bupropion SR, 50% for methylphenidate, and 27% for the placebo. The difference in response rates between the two agents was not statistically significant (p = 0.14).
Recommendation. The risk of seizures with bupropion is about 1 in 1,000. Therefore, bupropion should not be given to patients with a seizure disorder or to those with conditions that alter the seizure threshold (e.g., eating disorders, recent head trauma, or benzodiazepine withdrawal).21
This author uses bupropion as first-line treatment for appropriate patients with ADHD and a substance abuse history. Bupropion’s mild benefit with smoking cessation may provide some crossover effect for other substances of abuse. The low incidence of sexual side effects is another benefit. Drawbacks include twice-daily dosing and lack of a robust effect on attention and concentration.
Venlafaxine
Venlafaxine is a potent inhibitor of serotonin reuptake, a moderate inhibitor of norepinephrine, and a mild inhibitor of dopamine. Venlafaxine has displayed response rates similar to those of desipramine and bupropion in open-label studies in adults with ADHD,14 but no placebo-controlled studies exist.
As noted above, these antidepressants are believed to improve ADHD symptoms by making norepinephrine more available at the synapse. Hypothetically, then, one would need to administer venlafaxine at dosages that adequately inhibit the norepinephrine reuptake receptor. Venlafaxine XR, 150 mg/d, provides significant norepinephrine activity, according to several lines of evidence.
Recently, Upadhyaya et al reported the use of venlafaxine in one of the few treatment studies of patients with ADHD and comorbid alcohol/cocaine abuse. In an open-label trial, 10 subjects received venlafaxine, up to 300 mg/d, along with psychotherapy and attendance at Alcoholics Anonymous meetings. The nine who completed 4 weeks of treatment showed significantly improved ADHD symptoms and decreased alcohol craving.22
Recommendation. Venlafaxine is this author’s second choice for patients with ADHD and substance abuse problems. Sexual side effects that some patients experience with venlafaxine can limit its use. Some clinicians are concerned about increases in blood pressure associated with venlafaxine, although significant changes do not seem to occur at dosages below 300 mg/d.23
Atomoxetine
Atomoxetine is an investigational antidepressant in phase-III trials as a treatment for ADHD. Evidence shows atomoxetine to be a potent inhibitor of the presynaptic norepinephrine transporter, with minimal affinity for other neurotransmitter receptors. Initial studies suggest that atomoxetine is effective for adults and children with ADHD:
- In a small, double-blind, placebo-controlled, crossover trial, 11 of 20 adults showed improvement in ADHD symptoms within 3 weeks of starting atomoxetine.24
- In 297 children and adolescents, atomoxetine at dosages averaging approximately 1.2 mg/kg/d was more effective than a placebo in reducing ADHD symptoms and improving social and family functioning. Treatment was well tolerated and without significant side effects.25
- In a randomized open-label trial, 228 children received atomoxetine or methylphenidate. Both treatments significantly reduced inattention and hyperactive/impulsive symptoms.26
Related resources
- Arnsten AF. Genetics of childhood disorders (XVIII). ADHD, Part 2: Norepinephrine has a critical modulatory influence on prefrontal cortical function. J Am Acad Child Adolesc Psychiatry. 2000;39:1201-3.
- Ward MF, Wender PH, Reimherr FW. The Wender Utah Rating Scale: An aid in the retrospective diagnosis of childhood attention-deficit/hyperactivity disorder. Am J Psychiatry. 1993;150:885-90.
- Research Report: Prescription Drugs—abuse and addiction. National Institute on Drug Abuse, Substance Abuse and Mental Health Services Administration.
Drug brand names
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Methylphenidate • Ritalin, Concerta
- Venlafaxine • Effexor
Disclosure
Dr. Higgins reports that he is on the speakers’ bureaus for Wyeth-Ayerst Pharmaceuticals and Eli Lilly and Co.
Should you prescribe a stimulant to treat attention and hyperactivity problems in teenagers and adults with a history of substance abuse? Evidence suggests that using a stimulant to treat attention-deficit/hyperactivity disorder (ADHD) may place such patients at risk for stimulant abuse or for relapse into abuse of other substances. But a stimulant may be the only option for patients whose ADHD symptoms do not respond to alternate medications, such as antidepressants.
Growing numbers of adults are being treated for ADHD. Because substance abuse problems are common in adults with ADHD (Box 1 ),1-3 prescribing an antidepressant instead of a stimulant in some cases may be prudent. Consider the following factors when choosing ADHD therapy for patients with a history of substance abuse.
Prevalence of stimulant use and abuse
In the United States, more than 95% of medications prescribed for children and adults with ADHD are stimulants—usually methylphenidate.4 Stimulant use has increased as more children and adults are diagnosed with ADHD. Methylphenidate prescriptions increased five-fold from 1990 to 1995.5 Visits to psychiatrists and physicians that included stimulant prescriptions grew from 570,000 to 2.86 million from 1985 to 1994, with most of that increase occurring during visits to primary care and other physicians.6
When used as prescribed, methylphenidate is safe and effective for treating most children and adults with ADHD. Methylphenidate’s pharmacologic properties, however, are similar to those of amphetamines and cocaine (Box 2, Figure 1),7,8 which is why methylphenidate is a schedule-II controlled substance.
Published data. Fifteen reports of methylphenidate abuse were published in the medical literature between 1960 and 1999,7 but little is known about the prevalence of stimulant abuse among patients with ADHD. Banov and colleagues recently published what may be the only data available, when they reported that 3 of 37 (8%) patients abused the stimulants they were prescribed for ADHD.9 The three patients who abused stimulants had histories of drug and alcohol abuse at study entry. In all three cases, stimulant abuse did not develop immediately but became apparent within 6 months after the study began.
In a study of 651 students ages 11 to 18 in Wisconsin and Minnesota, more than one-third of those taking stimulants reported being asked to sell or trade their medications. More than one-half of those not taking ADHD medications said they knew someone who sold or gave away his or her medication.10
Stimulant theft, recreational use. Methylphenidate has been identified as the third most abused prescribed substance in the United States.11 It was the 10th most frequently stolen controlled drug from pharmacies between 1990 and 1995, and 700,000 dosage units were reported stolen in 1996 and 1997.12
As many as 50% of adults with ADHD have substance abuse problems (including alcohol, cocaine, and marijuana), and as many as 30% have antisocial personality disorder (with increased potential for drug-seeking behaviors).1 Compared with the general population, persons with ADHD have an earlier onset of substance abuse that is less responsive to treatment and more likely to progress from alcohol to other drugs.2
The elevated risk of substance abuse in ADHD may be related to a subtle lack of response to normal positive and negative reinforcements. Hunt has outlined four neurobehavioral deficits that define ADHD.3 Besides inattention, hyperarousal, and impulsiveness, he proposes that persons with ADHD have a reward system deficit. They may gravitate toward substance abuse because drugs, alcohol, and nicotine provide stronger rewards than life’s more subtle social interactions.
The popular media have reported recreational use of methylphenidate—with street names such as “R-Ball” and “Vitamin R”—among teens and college students.13 Illegal stimulants are perceived to be easily accessible on college campuses, but no data have been reported.
The use of stimulant medication for ADHD patients with substance abuse problems remains controversial. For such patients, this author reserves stimulant medication for those:
- whose ADHD symptoms have not responded adequately to alternate treatments
- who have been reliable with prescription medications
- and whose functional level is seriously impaired by their ADHD.
Antidepressants vs. stimulants
Although few well-designed controlled studies have been published, four antidepressants appear to be reasonably equivalent in effectiveness for adults with ADHD and do not carry potential for stimulant abuse.14
Desipraime, bupropion, venlafaxine, and the experimental drug atomoxetine (Table 1) all increase norepinephrine at the synapse by inhibiting presynaptic reuptake. Though dopamine has traditionally been considered the neurotransmitter of choice for ADHD treatment, norepinephrine may be equally potent.
Impulse control center. Several lines of research have recently established a connection between the prefrontal cortex, norepinephrine, and ADHD.15 This evidence suggests that the prefrontal cortex plays a major role in inhibiting impulses and responses to distractions:
Figure 1
PET scans of the brain using carbon 11 (11C)-labeled cocaine and methylphenidate HCl show similar distributions in the striatium when the drugs are administered intravenously.
Source: Reproduced with permission from Volkow ND, Ding YS, Fowler JS, et al. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch Gen Psychiatry 1995;52:456-63.
Stimulants are classified as schedule-II drugs because they produce powerful reinforcing effects by increasing synaptic dopamine.7 Positron-emission tomography (PET) scans using carbon labeling have shown similar distributions of methylphenidate and cocaine in the brain (Figure 1).8 When administered intravenously, both drugs occupy the same receptors in the striatum and produce a “high” that parallels rapid neuronal uptake. Methylphenidate and cocaine similarly increase stimulation of the postsynaptic neuron by blocking the dopamine reuptake pump (dopamine transporter).
How a substance gets to the brain’s euphoric receptors greatly affects its addictive properties. Delivery systems with rapid onset—smoking, “snorting,” or IV injection—have much greater ability to produce a “high” than do oral or transdermal routes. The greater the “high,” the greater the potential for abuse.
Because methylphenidate is prescribed for oral use, the potential for abuse is minimal. However, we need to be extremely cautious when giving methylphenidate or similar stimulant medications to patients who have shown they are unable to control their abuse of other substances.
Stimulants can also re-ignite a dormant substance abuse problem. Though little has been written about this in the medical literature, Elizabeth Wurtzel, author of the controversial Prozac Nation, chronicles the resumption of her cocaine abuse in More, Now, Again: A Memoir of Addiction. She contends that after her doctor added methylphenidate to augment treatment of partially remitting depression, she began abusing it and eventually was using 40 tablets per day before slipping back into cocaine dependence.
- Patients with prefrontal cortex deficits can have problems with inattention and poor impulse control.
- Patients with ADHD have frontal lobe impairments, as neuropsychological testing and imaging studies have shown.
- Norepinephrine neurons, with cell bodies in the locus coeruleus, have projections that terminate in the prefrontal cortex.
- Agents with norepinephrine activity, but without mood-altering properties (e.g., clonidine), have been shown to improve ADHD symptoms.
Table 1
ADHD IN ADULTS: ANTIDEPRESSANT DOSAGES AND SIDE EFFECTS
| Medication | Class | Effective dosage | Side effects |
|---|---|---|---|
| Desipramine | Tricyclic | 100 to 200 mg/d | Sedation, weight gain, dry mouth, constipation, orthostatic hypotension, prolonged cardiac conduction time; may be lethal in overdose |
| Bupropion SR | Norepinephrine and dopamine reuptake inhibitor | 150 mg bid to 200 mg bid | Headaches, insomnia, agitation, increased risk of seizures |
| Venlafaxine XR | Serotonin and norepinephrine reuptake inhibitor | 75 to 225 mg/d | Nausea, sexual side effects, agitation, increased blood pressure at higher dosages |
| Atomoxetine* | Norepinephrine reuptake inhibitor | To be determined | To be determined |
| * Investigational agent; not FDA-approved | |||
Additional evidence suggests that the prefrontal cortex has projections back to the locus coeruleus, which may explain the relationship between the two areas. It may be that the brain’s higher-functioning areas, such as the prefrontal cortex, provide intelligent screening of impulses from the brain’s older areas, such as the locus coeruleus. Therefore, increased prefrontal cortex activity may modulate some impulses that ADHD patients cannot control otherwise.
No ‘high’ with antidepressants. Patients with ADHD who have experienced the powerful effects of street drugs such as cocaine, methamphetamine, or even alcohol may report that antidepressants do not provide the effect they desire. It is difficult to know if these patients are reporting a lack of benefit or simply the absence of a euphoric “high” they are used to experiencing with substances of abuse. The newer antidepressants do not activate the brain’s euphoric receptors to an appreciable degree.
Patients who take stimulants as prescribed also do not report a “high” but can detect the medication’s presence and absence. Most do not crave this feeling, but substance abusers tend to like it. A patient recently told me he didn’t think stimulants improved his ADHD, but said, “I just liked the way they made me feel.”
Desipramine
The tricyclic antidepressant desipramine is a potent norepinephrine reuptake inhibitor that is effective in treating ADHD in children and adults. In a double-blind, placebo-controlled study of adults with ADHD, subjects receiving desipramine showed robust improvement in symptom scores on the ADHD Rating Scale,16 compared with those receiving the placebo (Figure 2).17
During the 6-week trial, 41 adults with ADHD received desipramine, 200 mg/d, or a placebo. Those receiving desipramine showed significant improvement in 12 of 14 ADHD symptoms and less hyperactivity, impulsivity, and inattentiveness, whereas those receiving the placebo showed no improvement. According to the study criteria, 68% of those who received desipramine and none who received the placebo were considered positive responders.
Though no head-to-head studies have compared desipramine with methylphenidate, the same researchers conducted a similar placebo-controlled study with methylphenidate. The 6-week symptom score on the ADHD Rating Scale was 12.5 for methylphenidate, compared with a score of 12 for desipramine.18
Recommendation. Desipramine may be the most effective of the antidepressant treatments for patients with ADHD. Because of its side effects, however, it is not this author’s first choice and is usually reserved for patients whose symptoms fail to respond to other antidepressants. Desipramine can cause sedation, dry mouth, and constipation, which are related to blockade of adrenergic, histamine, and muscarinic cholinergic receptors. It also can be lethal in overdose.
Some substance abusers lose confidence in a medication that they cannot feel working. The side effects of desipramine, which can be intolerable for some patients, can reassure others with a history of substance abuse that they are being medicated.
Bupropion
Bupropion is a unique antidepressant that inhibits the presynaptic reuptake of dopamine and norepinephrine. Open-label studies demonstrate good responses to bupropion by adults with ADHD. One placebo-controlled, double-blind study found improved ADHD symptoms in 76% of patients receiving bupropion SR, compared with 37% of those receiving a placebo; the difference was statistically significant.19
Figure 2 IMPROVED ADHD SYMPTOMS WITH DESIPRAMINE
Adults with ADHD who received desipramine, 200 mg/d, in a double-blind trial showed significantly less hyperactivity, impulsivity, and inattentiveness after 6 weeks of therapy than a control group that received a placebo.
Source: Adapted with permission from Wilens TE, Biederman J, Prince J, et al. Six-week, double-blind, placebo-controlled study of desipramine for adult attention-deficit/hyperactivity disorder. Am J Psychiatry 1996;153:1147-53.Another study of adults with ADHD compared bupropion SR to methylphenidate and a placebo.20 Using a primary outcome of the Clinical Global Impression (CGI) scale, response rates were 64% for bupropion SR, 50% for methylphenidate, and 27% for the placebo. The difference in response rates between the two agents was not statistically significant (p = 0.14).
Recommendation. The risk of seizures with bupropion is about 1 in 1,000. Therefore, bupropion should not be given to patients with a seizure disorder or to those with conditions that alter the seizure threshold (e.g., eating disorders, recent head trauma, or benzodiazepine withdrawal).21
This author uses bupropion as first-line treatment for appropriate patients with ADHD and a substance abuse history. Bupropion’s mild benefit with smoking cessation may provide some crossover effect for other substances of abuse. The low incidence of sexual side effects is another benefit. Drawbacks include twice-daily dosing and lack of a robust effect on attention and concentration.
Venlafaxine
Venlafaxine is a potent inhibitor of serotonin reuptake, a moderate inhibitor of norepinephrine, and a mild inhibitor of dopamine. Venlafaxine has displayed response rates similar to those of desipramine and bupropion in open-label studies in adults with ADHD,14 but no placebo-controlled studies exist.
As noted above, these antidepressants are believed to improve ADHD symptoms by making norepinephrine more available at the synapse. Hypothetically, then, one would need to administer venlafaxine at dosages that adequately inhibit the norepinephrine reuptake receptor. Venlafaxine XR, 150 mg/d, provides significant norepinephrine activity, according to several lines of evidence.
Recently, Upadhyaya et al reported the use of venlafaxine in one of the few treatment studies of patients with ADHD and comorbid alcohol/cocaine abuse. In an open-label trial, 10 subjects received venlafaxine, up to 300 mg/d, along with psychotherapy and attendance at Alcoholics Anonymous meetings. The nine who completed 4 weeks of treatment showed significantly improved ADHD symptoms and decreased alcohol craving.22
Recommendation. Venlafaxine is this author’s second choice for patients with ADHD and substance abuse problems. Sexual side effects that some patients experience with venlafaxine can limit its use. Some clinicians are concerned about increases in blood pressure associated with venlafaxine, although significant changes do not seem to occur at dosages below 300 mg/d.23
Atomoxetine
Atomoxetine is an investigational antidepressant in phase-III trials as a treatment for ADHD. Evidence shows atomoxetine to be a potent inhibitor of the presynaptic norepinephrine transporter, with minimal affinity for other neurotransmitter receptors. Initial studies suggest that atomoxetine is effective for adults and children with ADHD:
- In a small, double-blind, placebo-controlled, crossover trial, 11 of 20 adults showed improvement in ADHD symptoms within 3 weeks of starting atomoxetine.24
- In 297 children and adolescents, atomoxetine at dosages averaging approximately 1.2 mg/kg/d was more effective than a placebo in reducing ADHD symptoms and improving social and family functioning. Treatment was well tolerated and without significant side effects.25
- In a randomized open-label trial, 228 children received atomoxetine or methylphenidate. Both treatments significantly reduced inattention and hyperactive/impulsive symptoms.26
Related resources
- Arnsten AF. Genetics of childhood disorders (XVIII). ADHD, Part 2: Norepinephrine has a critical modulatory influence on prefrontal cortical function. J Am Acad Child Adolesc Psychiatry. 2000;39:1201-3.
- Ward MF, Wender PH, Reimherr FW. The Wender Utah Rating Scale: An aid in the retrospective diagnosis of childhood attention-deficit/hyperactivity disorder. Am J Psychiatry. 1993;150:885-90.
- Research Report: Prescription Drugs—abuse and addiction. National Institute on Drug Abuse, Substance Abuse and Mental Health Services Administration.
Drug brand names
- Bupropion • Wellbutrin
- Desipramine • Norpramin
- Methylphenidate • Ritalin, Concerta
- Venlafaxine • Effexor
Disclosure
Dr. Higgins reports that he is on the speakers’ bureaus for Wyeth-Ayerst Pharmaceuticals and Eli Lilly and Co.
1. Mannuzza S, Klein RG, Bessler A, et al. Adult outcome of hyperactive boys. Educational achievement, occupational rank, and psychiatric status. Arch Gen Psychiatry 1993;50:565-76.
2. Sullivan MA, Rudnik-Levin F. Attention deficit/hyperactivity disorder and substance abuse. Diagnostic and therapeutic considerations. Ann NY Acad Sci 2001;931:251-70.
3. Hunt RD. Nosology, neurobiology, and clinical patterns of ADHD in adults. Psychiatric Ann 1997;27:572-81.
4. Taylor MA. Evaluation and management of attention-deficit hyperactivity disorder. Am Fam Phys 1997;55:887-904.
5. Diller LH. The run on Ritalin. Attention deficit disorder and stimulant treatment in the 1990s. Hasting Center Report 1996;26:12-18.
6. Pincus HA, Tanielian TL, Marcus SC, et al. Prescribing trends in psychotrophic medications: primary care, psychiatry, and other medical specialties. JAMA 1998;279:526-31.
7. Mortan WA, Stockton GG. Methylphenidate abuse and psychiatric side effects. J Clin Psychiatry (Primary Care) 2000;2:159-64.
8. Volkow ND, Ding YS, Fowler JS, et al. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch Gen Psychiatry 1995;52:456-63.
9. Banov MD, Palmer T, Brody E. Antidepressants are as effective as stimulants in the long-term treatment of ADHD in Adults. Primary Psychiatry 2001;8:54-7.
10. Moline S, Frankenberger W. Use of stimulant medication for treatment of attention-deficit/hyperactivity disorder. A survey of middle and high school students attitudes. Psychology in the Schools 2001;38:569-84.
11. Prescription drugs: abuse and addiction. National Institute on Drug Abuse Research Report Series. NIH publication number 01-4881, July 2001.
12. Mann A. Illicit methylphenidate trade appears widespread. Clinical Psychiatry News June 2000;28(6):5-
13. ABCNews.com. http://more/abcnews.go.com/sections/living/DailyNews/ritalin0505.html
14. Higgins ES. A comparative analysis of antidepressants and stimulants for the treatment of adults with attention-deficit hyperactivity disorder. J Fam Pract 1999;48:15-20.
15. Arnsten AF, Steere JC, Hunt RD. The contribution of alpha2-noradrenergic mechanisms to prefrontal cortical cognitive function. Arch Gen Psychiatry 1996;53:448-55.
16. DuPaul G. The ADHD Rating Scale: normative data, reliability, and validity. Worcester, MA: University of Massachusetts Medical School, 1990.
17. Wilens TE, Biederman J, Prince J, et al. Six-week, double-blind, placebo-controlled study of desipramine for adult attention-deficit/hyperactivity disorder. Am J Psychiatry 1996;153:1147-53.
18. Spencer T, Wilens T, Biederman J, Faraone SV, Ablon JS, Lapey K. A double-blind, crossover comparison of methylphenidate and placebo in adults with childhood-onset attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 1995;52:434-43.
19. Wilens TE, Spencer TJ, Biederman J, et al. A controlled clinical trial of bupropion for attention-deficit/hyperactivity disorder in adults. Am J Psychiatry 2001;158:282-8.
20. Kuperman S, Perry PJ, Gaffney GR, et al. Bupropion SR vs. methylphenidate vs. placebo for attention-deficit/hyperactivity disorder in adults. Ann Clin Psychiatry 2001;13:129-34.
21. Wooltorton E. Bupropion (Zyban, Wellbutrin SR): reports of death, seizures, serum sickness. Can Med J 2002;166:68.-
22. Upadhyaya HP, Brady KT, Sethuraman G, et al. Venlafaxine treatment of patients with comorbid alcohol/cocaine abuse and attention-deficit/hyperactivity disorder: a pilot study. J Clin Psychopharmacol 2001;21:116-8.
23. Thase ME. Effects of venlafaxine on blood pressure: a meta-analysis of original data from 3744 depressed patients. J Clin Psychiatry 1998;59:502-8.
24. Spencer T, Biederman J, Wilens T, Prince J, Hatch M. Effectiveness and tolerability of tomoxetine in adults with attention-deficit/hyperactivity disorder. Am J Psychiatry 1998;155:693-5.
25. Michelson D, Faries D, Wernicke J, et al. Atomoxetine in the treatment of children and adolescents with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled, dose-response study. Pediatrics 2001;108:E83.-
26. Kratochvil C, Heiligenstein JH, Dittmann R, et al. Atomoxetine and methylphenidate treatment in ADHD children: a randomized, open-label trial (presentation). Honolulu: American Academy of Child and Adolescent Psychiatry, October, 2001.
1. Mannuzza S, Klein RG, Bessler A, et al. Adult outcome of hyperactive boys. Educational achievement, occupational rank, and psychiatric status. Arch Gen Psychiatry 1993;50:565-76.
2. Sullivan MA, Rudnik-Levin F. Attention deficit/hyperactivity disorder and substance abuse. Diagnostic and therapeutic considerations. Ann NY Acad Sci 2001;931:251-70.
3. Hunt RD. Nosology, neurobiology, and clinical patterns of ADHD in adults. Psychiatric Ann 1997;27:572-81.
4. Taylor MA. Evaluation and management of attention-deficit hyperactivity disorder. Am Fam Phys 1997;55:887-904.
5. Diller LH. The run on Ritalin. Attention deficit disorder and stimulant treatment in the 1990s. Hasting Center Report 1996;26:12-18.
6. Pincus HA, Tanielian TL, Marcus SC, et al. Prescribing trends in psychotrophic medications: primary care, psychiatry, and other medical specialties. JAMA 1998;279:526-31.
7. Mortan WA, Stockton GG. Methylphenidate abuse and psychiatric side effects. J Clin Psychiatry (Primary Care) 2000;2:159-64.
8. Volkow ND, Ding YS, Fowler JS, et al. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch Gen Psychiatry 1995;52:456-63.
9. Banov MD, Palmer T, Brody E. Antidepressants are as effective as stimulants in the long-term treatment of ADHD in Adults. Primary Psychiatry 2001;8:54-7.
10. Moline S, Frankenberger W. Use of stimulant medication for treatment of attention-deficit/hyperactivity disorder. A survey of middle and high school students attitudes. Psychology in the Schools 2001;38:569-84.
11. Prescription drugs: abuse and addiction. National Institute on Drug Abuse Research Report Series. NIH publication number 01-4881, July 2001.
12. Mann A. Illicit methylphenidate trade appears widespread. Clinical Psychiatry News June 2000;28(6):5-
13. ABCNews.com. http://more/abcnews.go.com/sections/living/DailyNews/ritalin0505.html
14. Higgins ES. A comparative analysis of antidepressants and stimulants for the treatment of adults with attention-deficit hyperactivity disorder. J Fam Pract 1999;48:15-20.
15. Arnsten AF, Steere JC, Hunt RD. The contribution of alpha2-noradrenergic mechanisms to prefrontal cortical cognitive function. Arch Gen Psychiatry 1996;53:448-55.
16. DuPaul G. The ADHD Rating Scale: normative data, reliability, and validity. Worcester, MA: University of Massachusetts Medical School, 1990.
17. Wilens TE, Biederman J, Prince J, et al. Six-week, double-blind, placebo-controlled study of desipramine for adult attention-deficit/hyperactivity disorder. Am J Psychiatry 1996;153:1147-53.
18. Spencer T, Wilens T, Biederman J, Faraone SV, Ablon JS, Lapey K. A double-blind, crossover comparison of methylphenidate and placebo in adults with childhood-onset attention-deficit/hyperactivity disorder. Arch Gen Psychiatry 1995;52:434-43.
19. Wilens TE, Spencer TJ, Biederman J, et al. A controlled clinical trial of bupropion for attention-deficit/hyperactivity disorder in adults. Am J Psychiatry 2001;158:282-8.
20. Kuperman S, Perry PJ, Gaffney GR, et al. Bupropion SR vs. methylphenidate vs. placebo for attention-deficit/hyperactivity disorder in adults. Ann Clin Psychiatry 2001;13:129-34.
21. Wooltorton E. Bupropion (Zyban, Wellbutrin SR): reports of death, seizures, serum sickness. Can Med J 2002;166:68.-
22. Upadhyaya HP, Brady KT, Sethuraman G, et al. Venlafaxine treatment of patients with comorbid alcohol/cocaine abuse and attention-deficit/hyperactivity disorder: a pilot study. J Clin Psychopharmacol 2001;21:116-8.
23. Thase ME. Effects of venlafaxine on blood pressure: a meta-analysis of original data from 3744 depressed patients. J Clin Psychiatry 1998;59:502-8.
24. Spencer T, Biederman J, Wilens T, Prince J, Hatch M. Effectiveness and tolerability of tomoxetine in adults with attention-deficit/hyperactivity disorder. Am J Psychiatry 1998;155:693-5.
25. Michelson D, Faries D, Wernicke J, et al. Atomoxetine in the treatment of children and adolescents with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled, dose-response study. Pediatrics 2001;108:E83.-
26. Kratochvil C, Heiligenstein JH, Dittmann R, et al. Atomoxetine and methylphenidate treatment in ADHD children: a randomized, open-label trial (presentation). Honolulu: American Academy of Child and Adolescent Psychiatry, October, 2001.