User login
Rhabdomyolysis Occurring After Use of Cocaine Contaminated With Fentanyl Causing Bilateral Brachial Plexopathy
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.

Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
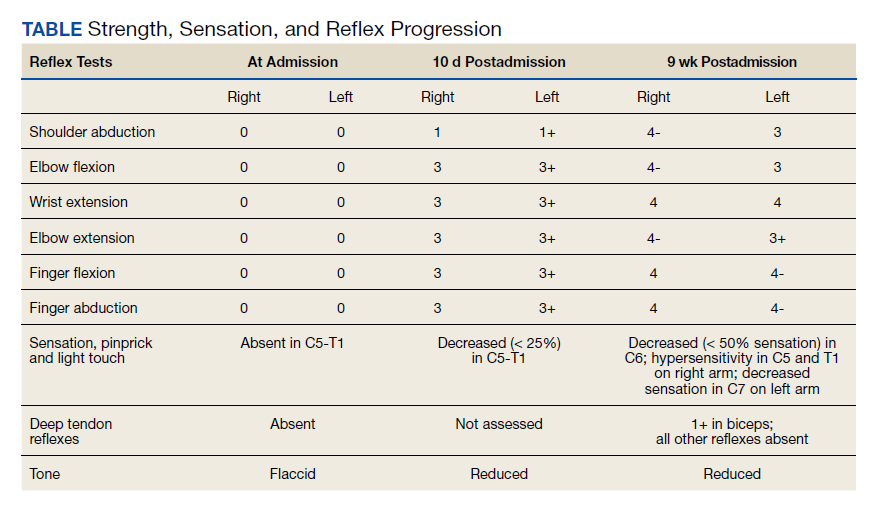
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.
Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.
Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
Clinical Presentation of Subacute Combined Degeneration in a Patient With Chronic B12 Deficiency
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
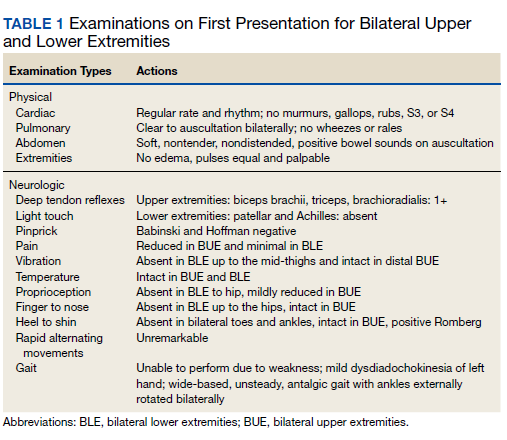
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
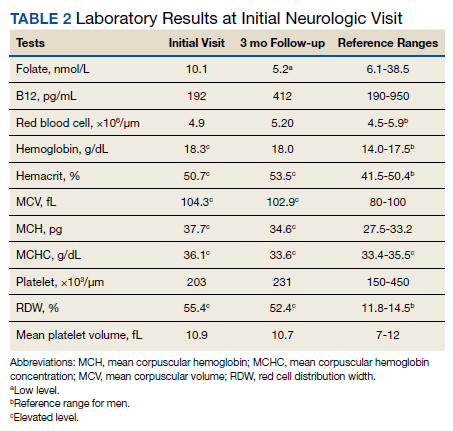
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
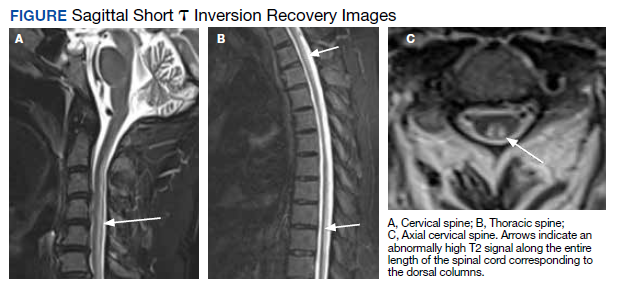
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
Hemiballismus in Patients With Poorly Controlled Type 2 Diabetes Mellitus
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
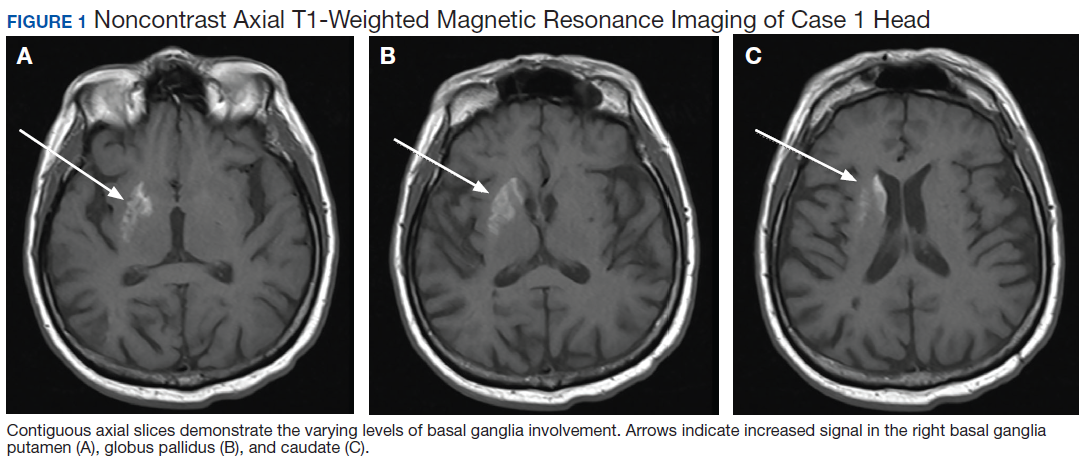
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
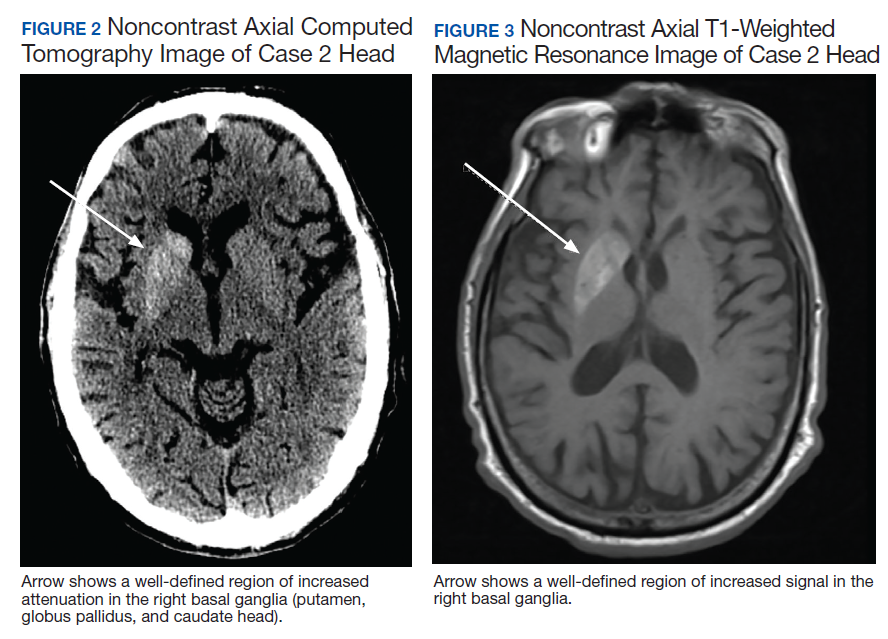
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
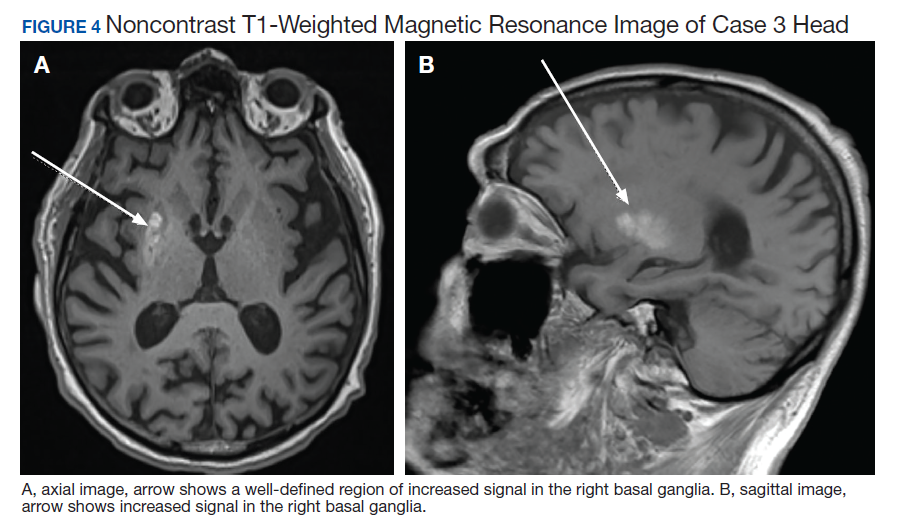
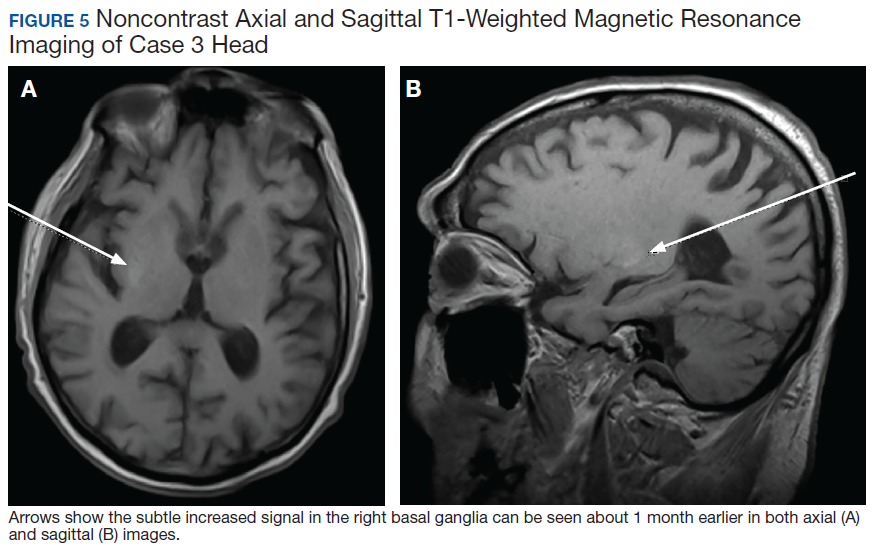
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
Hemolytic Uremic Syndrome With Severe Neurologic Complications in an Adult (FULL)
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
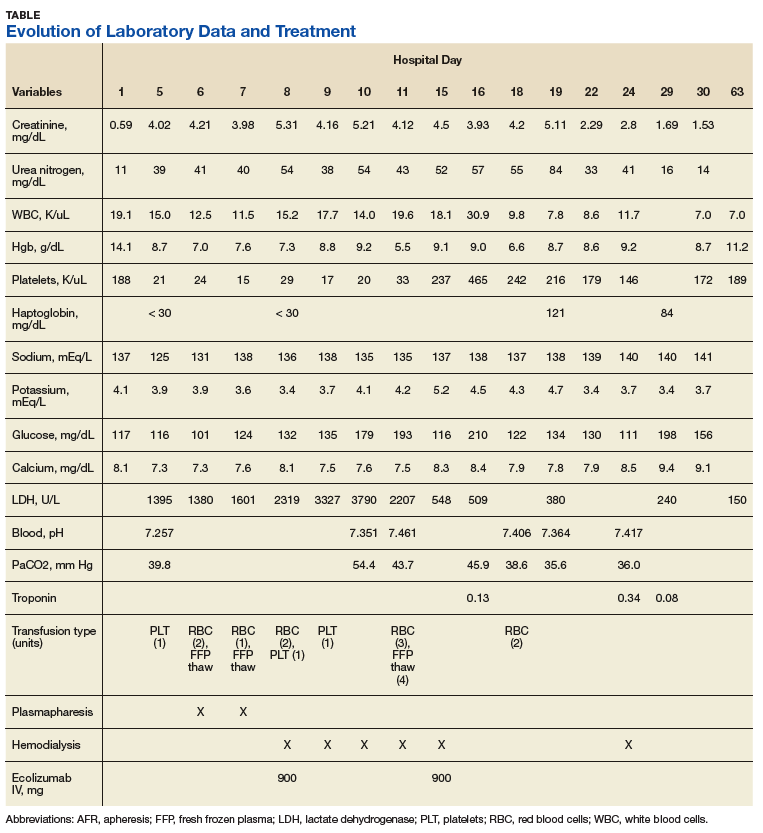
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
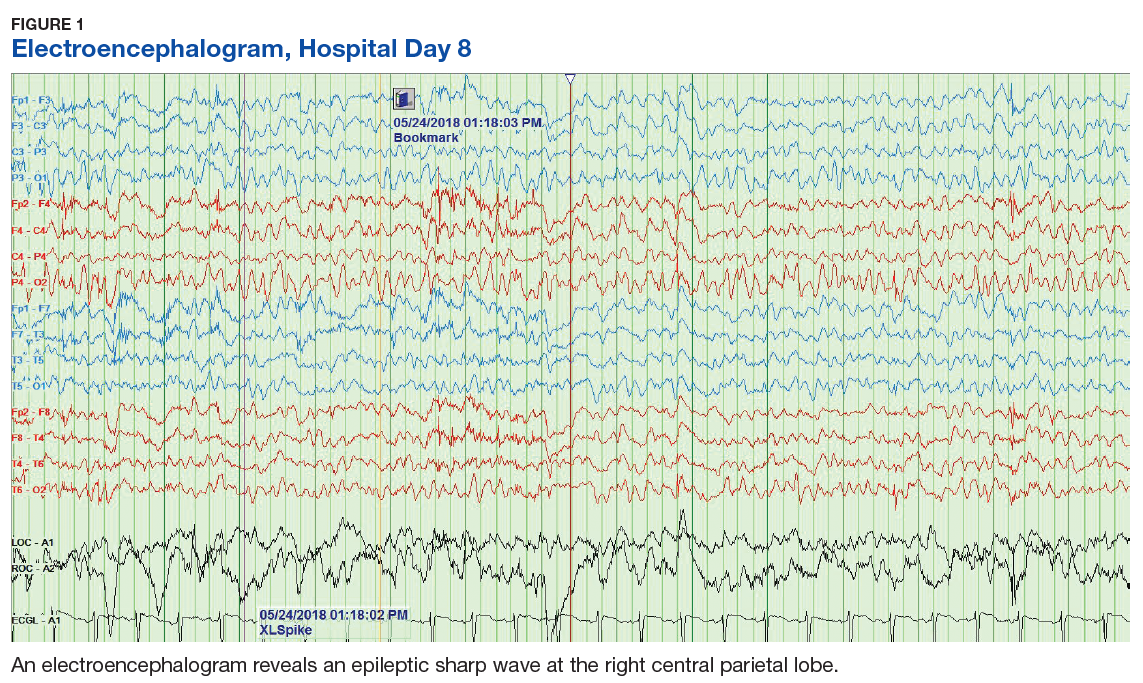
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
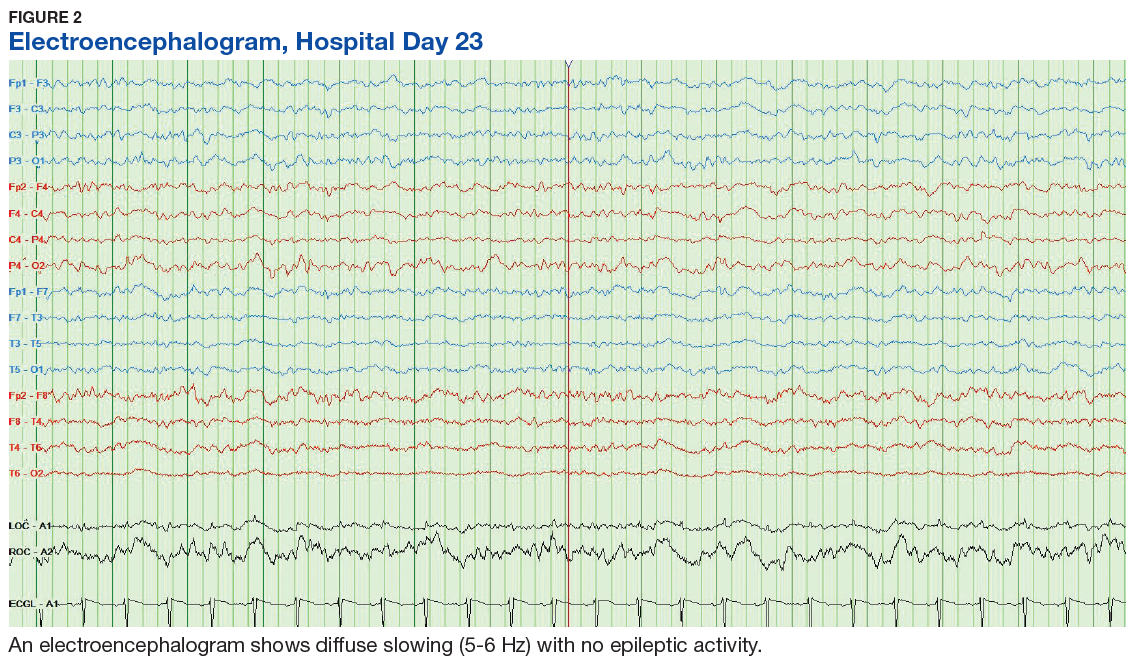
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
The case of a female presenting with Shiga toxin-producing Escherichia coli and hemolytic uremic syndrome highlights a severe neurologic complication that canbe associated with these conditions.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
Hemolytic uremic syndrome (HUS) is a rare illness that can be acquired through the consumption of food products contaminated with strains of Shiga toxin-producing Escherichia coli (E coli; STEC).1 Between 6% and 15% of individuals infected with STEC develop HUS, with children affected more frequently than adults.2,3 This strain of E coli releases Shiga toxin into the systemic circulation, which causes a thrombotic microangiopathy resulting in the characteristic HUS triad of symptoms: acute renal insufficiency, thrombocytopenia, and hemolytic anemia.4-6
Although neurologic features are common in HUS, they have not been extensively studied, particularly in adults. We report a case of STEC 0157:H7 subtype HUS in an adult with severe neurologic complications. This case highlights the neurological sequelae in an adult with typical STEC-HUS. The use of treatment modalities, such as plasmapheresis and eculizumab, and their use in adult typical STEC-HUS also is explored.
Case
A 53-year-old white woman with no pertinent past medical history presented to the Bay Pines Veterans Affairs Healthcare System Emergency Department with a 2-day history of abdominal pain, vomiting, nausea, diarrhea, and bright bloody stools. She returned from a cruise to the Bahamas 3 days prior, where she ate local foods, including salads. She reported no fever, shortness of breath, chest pain, headache, and cognitive difficulties. She presented with a normal mental status and neurologic exam. Apart from leukocytosis and elevated glucose level, her laboratory results at initial presentation were normal, (Table). A stool sample showed occult blood with white blood cell counts (WBCs) but was negative for Clostridium difficile. She was started on ciprofloxacin 400 mg and metronidazole 500 mg on the day of admission.
Hematuria was found on hospital day 2. On hospital day 4, the patient exhibited word finding difficulties. Blood studies revealed anemia, thrombocytopenia, leukocytosis, and increasing blood urea nitrogen (BUN) and creatinine. A computed tomography scan of the head was normal. Laboratory analysis showed schistocytes in the peripheral blood smear.
The patient’s cognitive functioning deteriorated on hospital day 5. She was not oriented to time or place. Her laboratory results showed complement level C3 at 70 mg/dL (ref: 83-193 mg/dL) complement C4 at 12 mg/dL (ref: 15-57mg/dL). The patient exhibited oliguria and hyponatremia, as well as both metabolic and respiratory acidosis; dialysis was then initiated. Results from the stool sample that was collected on hospital day 1 were received and tested positive for Shiga toxin.
At this point, the patient’s presentation of hemolytic anemia and thrombocytopenia in the setting of acute bloody diarrheal illness with known Shiga toxin, schistocytes on blood smear, and lack of pertinent medical history for other causes of this presentation made STEC-HUS the leading differential diagnosis. Plasmapheresis was ordered and performed on hospital day 6 and 7. Shiga toxin was no longer detected in the stool and antibiotics were stopped on hospital day 7.
The patient’s progressive deterioration in mental status continued on hospital day 8. She was not oriented to time or place, unable to perform simple calculations, and could not spell the word “hand” backwards. Physicians observed repetitive jerking motions of the upper extremities that were worse on the left side. An electroencephalogram (EEG) revealed right hemispheric sharp waves that were thought to be epileptiform (Figure 1). The patient began taking levetiracetam 1500 mg IV with 750 mg bid maintenance for seizure control. Plasmapheresis was discontinued due to her continued neurologic deterioration on this therapy. Consequently, eculizumab 900 mg IV was given along with the Neisseria meningitidis (N meningitidis) vaccine and a 19-day course of azithromycin 250 mg po as prophylaxis for encapsulated bacteria.
The patient continued to seize on hospital days 10 through 13. Oculocephalic maneuvers showed a tendency to keep her eyes deviated to the right. Her pupils continued to react to light. A repeat EEG showed diffuse slowing (5-6 Hz) with no epileptic activity seen (Figure 2). A second dose of eculizumab 900 mg IV was administered on hospital day 15. The patient experienced cardiac arrest on hospital day 16 and was successfully resuscitated. On hospital day 25 (10 days after receiving her second dose of eculizumab), the patient was able to speak and follow simple commands but exhibited difficulty concentrating and poor impulse control.
The patient was alert and oriented to person, place, time, and situation on hospital day 28 (6 days after the third and final dose of eculizumab). A neurologic exam was significant only for a slight intention tremor. She was continued on levetiracetam with a plan to be maintained on the medication for the next 6 months for seizure control. She was discharged on hospital day 30.
Twenty-eight days postdischarge (57 days postadmission), the patient showed marked recovery. She had returned to her previous employment as a business administrator on a part-time basis and exhibited no deficiencies in executive functioning or handling activities of daily living. Although she had been very active prior to this illness, she now experienced decreased physical and mental endurance; however, this gradually improved with physical therapy. She planned on returning to work on a full-time basis when she had regained her stamina. She also noticed difficulties in retaining short term memory since her discharge but believed that these symptoms were remitting. On examination her mental status and neurologic exam was significant for inability to continue serial 7s, left sided 4/5 muscle strength in quadriceps and thumb to 5th metacarpal adduction, bilateral 1+ reflexes in muscle groups tested (triceps, biceps, brachioradialis, patellar, and Achilles), loss of dull pinprick sensation bilaterally at web of hands, deficit in tandem gait while looking away, and slight intention tremor on finger to nose testing bilaterally (with left hand tremor more pronounced than right). Her complete blood count was normal. Her recovery continues to be monitored in an outpatient setting.
Discussion
HUS is characterized by 3 core clinical features: microangiopathic hemolytic anemia, thrombocytopenia, and acute kidney injury.4 Schistocytes are seen on peripheral blood smear and occur due to the passage of red blood cells over the microvascular thrombi induced by the disease. HUS can be classified as typical, atypical, or occurring with a coexisting disease. Typical HUS is associated with STEC 0157:H7 subtype, a bacterium known to be acquired through contaminated food and via human-to-human transmission.6-8 In the case of typical STEC 0157:H7, the bacterium releases a verotoxin that damages the vascular endothelium, thereby leading to activation of the coagulation cascade and eventually the formation of thrombi.4 It has been hypothesized that the Shiga toxin also activates the alternative complement pathway directly, which could contribute to thrombosis.9 This would explain the findings of low complement levels in our patient. Atypical HUS is primarily attributable to mutations in the alternative complement pathway. Causes for the third type of HUS can include Streptococcus pneumoniae, HIV, drug toxicity, and alterations in the metabolism of cobalamin C.
Epidemiologically, 15.3% of children aged < 5 years develop typical HUS after exposure to STEC compared with 1.2% of adults aged 18 to 59 years. The median age of patients who developed HUS from STEC exposure was 4 years compared with 16 years for those who did not develop HUS.2
Neurologic manifestations increase mortality for HUS patients.10 These have been described in the pediatric population as alteration in consciousness (85%), seizures (71%), pyramidal syndrome (52%), and extrapyramidal syndrome with hypertonia (42%).11 Brain imaging in children has demonstrated hemorrhagic lesions involving the pons, basal ganglia, and occipital cortex.11 Blood flow to areas such as the cerebellum, brainstem, and orbitofrontal area can be compromised.10 Adult patients with HUS can present without lesions on cranial magnetic resonance imaging (MRI), but instead with transient symmetric vasogenic edema of the central brain stem.12 Unfortunately in this case, MRI was not performed because it was thought to provide limited aid in diagnosis and to avoid unnecessary testing for the acutely ill patient.
The underlying pathophysiology of neurologic manifestations in patients may be due to a metabolic disturbance, toxin-mediated damage of the vascular endothelium, or toxin-induced cytokine release resulting in death of neural cells and subsequent neuroinflammation. However, the most likely mechanism is parenchymal ischemic changes related to microangiopathy.11,13 Pediatric patients often experience seizures and altered mental status, and their EEGs display delta waves.13 This patient’s diffuse slowing on her second EEG and altered mental status suggests that the neuropathologic mechanisms for typical HUS in adults may be similar to those in children.
HUS Treatment
The treatment and management of adults with typical STEC-HUS is evolving. The patient was first suspected to have an infectious colitis and empiric antibiotics were initiated. Some studies suggest that antibiotic administration may worsen the course of HUS in children as it may lead to release and subsequent absorption of Shiga toxin in the intestine.9,14 However, there is little evidence to suggest harm or efficacy of administration in adults. It is unclear what role antibiotic administration played in the recovery time of HUS given the co-administration of other treatments such as eculizumab and plasmapheresis, but it does appear to have helped with the initial E coli infection.
Plasmapheresis was subsequently administered, due to its documented benefit in the treatment of HUS.15 However, it should be noted that even though plasmapheresis is currently used in patients with CNS involvement, it remains unproven with conflicting information on its efficacy.3,16 The mechanism of action is unclear, but it has been hypothesized that plasmapheresis prevents microangiopathy caused by microthrombi.3,16 For this reason, eculizumab is becoming the mainstay for treatment of STEC-HUS with neurologic complications given the lack of well researched alternative treatments. In this case study, the use of plasmapheresis did not result in clinical improvement, and was abandoned after 2 days of treatment.
Eculizumab is a humanized, recombinant monoclonal IgG antibody that is a terminal complement inhibitor of the alternative complement system at the final step to cleave C5.17 The Shiga toxin may directly activate the complement system via the alternative pathway, which can result in uncontrolled platelet and white blood cell activation and depletion, endothelial cell damage, and hemolysis. The galvanized complement system leads to a series of cascading events that contribute to organ damage and death.9 Eculizumab is FDA approved for use in atypical HUS.18 It also can be used off-label to treat typical-HUS in adults with neurologic complications.
Eculizumab interferes with the immune response against encapsulated bacteria because it inhibits the alternative complement pathway. Thus, vaccination against N meningitides is recommended 2 weeks prior to the administration of eculizumab. However, in situations where the risks of delaying eculizumab for 2 weeks are greater than the risk of developing an N meningitides infection, eculizumab may be given without delay.18 Given the rapid deterioration of our patient’s condition, the vaccine and eculizumab were given together with prophylactic azithromycin. Although penicillin is the standard for prophylaxis in this situation, the patient’s penicillin allergy led to the use of azithromycin 250 mg po once a day. Literature also suggests azithromycin reduces the carriage duration of E coli-induced colitis.19 As such, it is possible that some improvement in the patient’s condition could be attributed to the elimination of the pathogen and toxin.
Conclusion
Three doses of eculizumab were administered at weekly intervals, with the first dose on hospital day 8 and the final dose on hospital day 22. Prior to the first dose, the patient displayed significant decline in mental status with EEG findings of right hemisphere epileptogenic discharges. After her third dose, she was found to have a drastically improved mental status exam and a normal EEG. One week later, she was discharged home. At the time of her 1-month follow-up, she was independent in all activities of daily living and had returned to part-time work. Apart from subtle cognitive changes, the remainder of her neurologic exam was normal.
There is evidence that supports the efficacy of eculizumab in children with HUS with neurologic symptoms on dialysis.20 However, its use in adults is not well established.21 This patient required dialysis and had neurologic symptoms similar to pediatric patients described in the literature, and responded similarly to the eculizumab. The rationale for the use of eculizumab in STEC-HUS also is evidenced by in vitro demonstrations of complement activation in STEC-HUS.22-25 This case report adds to the literature supporting the use of eculizumab in adult patients with typical HUS with neurological complications. Further research is necessary to develop guidelines in the treatment of adult STEC-HUS with regards to neurologic complications.
Acknowledgments
The authors would like to thank Pete DiStaso, REEGT for his work on obtaining the electroencephalograms and Anthony Rinaldi, PsyD; Julie Cessnapalas, PsyD; and Syed Faizan Sagheer for proof-reading the article.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
1. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
2. Gould LH, Demma L, Jones TF, et al. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis. 2009;49(10):1480-1485.
3. Boyce TG, Swerdlow DL, Griffin PM. Escherichia coli O157:H7 and the hemolytic-uremic syndrome. N Engl J Med. 1995;333(6):364-368.
4. Rondeau E, Peraldi MN. Escherichia coli and the hemolytic-uremic syndrome. N Engl J Med. 1996;335(9):660-662.
5. Te Loo DM, van Hinsbergh VW, van den Heuvel LP, Monnens LA. Detection of verocytotoxin bound to circulating polymorphonuclear leukocytes of patients with hemolytic uremic syndrome. J Am Soc Nephrol. 2001;12(4):800-806.
6. Tran SL, Jenkins C, Livrelli V, Schüller S. Shiga toxin 2 translocation across intestinal epithelium is linked to virulence of Shiga toxin-producing Escherichia coli in humans. Microbiology. 2018;164(4):509-516.
7. Jokiranta TS. HUS and atypical HUS. Blood. 2017;129(21):2847-2856.
8. Ferens WA, Hovde CJ. Escherichia coli O157:H7: animal reservoir and sources of human infection. Foodborne Pathog Dis. 2011;8(4):465-487.
9. Percheron L, Gramada R, Tellier S, et al. Eculizumab treatment in severe pediatric STEC-HUS: a multicenter retrospective study. Pediatr Nephrol. 2018;33(8):1385-1394.
10. Hosaka T, Nakamagoe K, Tamaoka A. Hemolytic uremic syndrome-associated encephalopathy successfully treated with corticosteroids. Intern Med. 2017;56(21):2937-2941.
11. Nathanson S, Kwon T, Elmaleh M, et al. Acute neurological involvement in diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2010;5(7):1218-1228.
12. Wengenroth M, Hoeltje J, Repenthin J, et al. Central nervous system involvement in adults with epidemic hemolytic uremic syndrome. AJNR Am J Neuroradiol. 2013;34(5):1016-1021, S1.
13. Eriksson KJ, Boyd SG, Tasker RC. Acute neurology and neurophysiology of haemolytic-uraemic syndrome. Arch Dis Child. 2001;84(5):434-435.
14. Wong CS, Jelacic S, Habeeb RL, Watkins SL, Tarr PI. The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N Engl J Med. 2000;342(26):1930-1936.
15. Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28(3):453-468, vii.
16. Loos S, Ahlenstiel T, Kranz B, et al. An outbreak of Shiga toxin-producing Escherichia coli O104:H4 hemolytic uremic syndrome in Germany: presentation and short-term outcome in children. Clin Infect Dis. 2012;55(6):753-759.
17. Hossain MA, Cheema A, Kalathil S, et al. Atypical hemolytic uremic syndrome: Laboratory characteristics, complement-amplifying conditions, renal biopsy, and genetic mutations. Saudi J Kidney Dis Transpl. 2018;29(2):276-283.
18. Soliris (eculizumab) [package insert]. Cheshire, CT: Alexion Pharmaceuticals, Inc; 2011.
19. Keenswijk W, Raes A, Vande Walle J. Is eculizumab efficacious in Shigatoxin-associated hemolytic uremic syndrome? A narrative review of current evidence. Eur J Pediatr. 2018;177(3):311-318.
20. Lapeyraque AL, Malina M, Fremeaux-Bacchi V, et al. Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med. 2011;364(26):2561-2563.
21. Pape L, Hartmann H, Bange FC, Suerbaum S, Bueltmann E, Ahlenstiel-Grunow T. Eculizumab in typical hemolytic uremic syndrome (HUS) with neurological involvement. Medicine (Baltimore). 2015;94(24):e1000.
22. Kim Y, Miller K, Michael AF. Breakdown products of C3 and factor B in hemolytic-uremic syndrome. J Lab Clin Med. 1977;89(4):845-850.
23. Monnens L, Molenaar J, Lambert PH, Proesmans W, van Munster P. The complement system in hemolytic-uremic syndrome in childhood. Clin Nephrol. 1980;13(4):168-171.
24. Thurman JM, Marians R, Emlen W, et al. Alternative pathway of complement in children with diarrhea-associated hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(12):1920-1924.
25. Ståhl AL, Sartz L, Karpman D. Complement activation on platelet-leukocyte complexes and microparticles in enterohemorrhagic Escherichia coli-induced hemolytic uremic syndrome. Blood. 2011;117(20):5503-5513.
Acute Encephalopathy Following Hyperbaric Oxygen Therapy in a Patient on Metronidazole
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2). 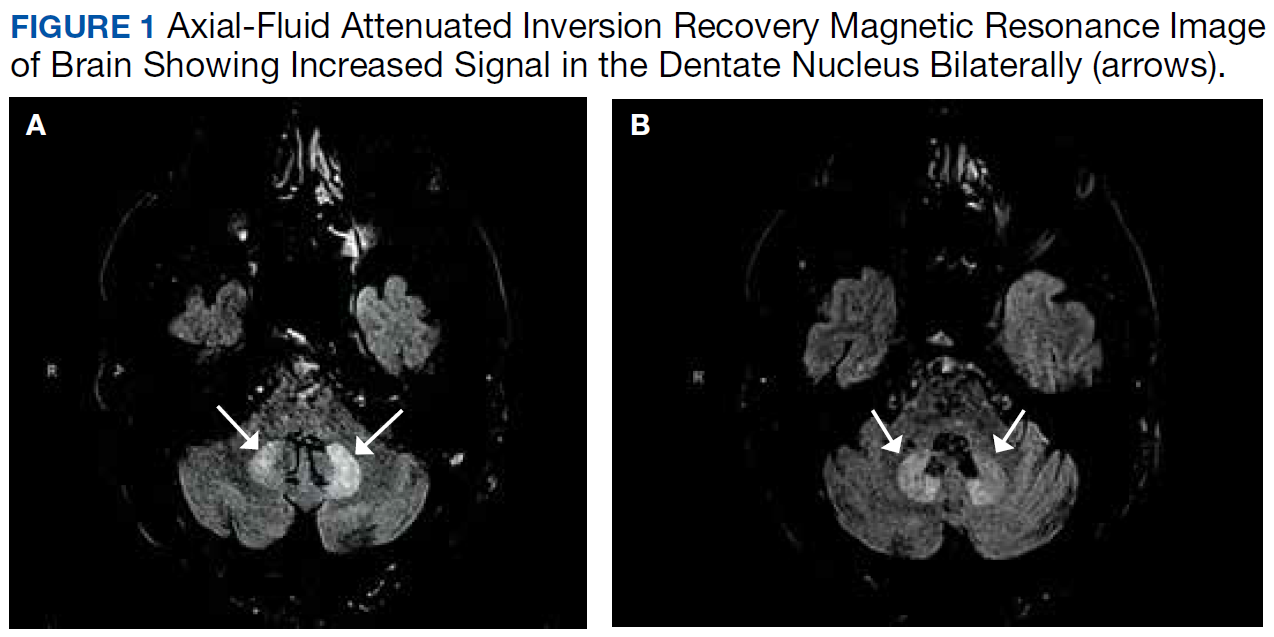
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14 
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2).
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2).
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.