User login
Pheochromocytoma: An Incidental Finding in an Asymptomatic Older Adult With Renal Oncocytoma
A high index of suspicion for pheochromocytoma is necessary during the workup of secondary hypertension as untreated pheochromocytoma may lead to significant morbidity and mortality, especially in patients who require any surgical treatment.
Pheochromocytoma is a rare catecholamine-secreting tumor of chromaffin cells of the adrenal medulla or sympathetic ganglia, occurring in about 0.2 to 0.5% of patients with hypertension.1-3 However, in a review of 54 autopsy-proven cases of pheochromocytoma, about 50% of the patients with hypertension were not clinically suspected for pheochromocytoma.4
Pheochromocytoma is usually diagnosed based on symptoms of hyperadrenergic spells, resistant hypertension, especially in the young, with a pressor response to the anesthesia stress test and adrenal incidentaloma.
The classic triad of symptoms associated with pheochromocytoma includes episodic headache (90%), sweating (60-70%), and palpitations (70%).2,5 Sustained or paroxysmal hypertension is the most common symptom reported in about 95% of patients with pheochromocytoma. Other symptoms include pallor, tremors, dyspnea, generalized weakness, orthostatic hypotension, cardiomyopathy, or hyperglycemia.6 However, about 10% of patients with pheochromocytoma are asymptomatic or mildly symptomatic.7 Secondary causes of hypertension are usually suspected in multidrug resistant or sudden early onset of hypertension.8
Approximately 10% of catecholamine-secreting tumors are malignant.9-11 Benign and malignant pheochromocytoma have a similar biochemical and histologic presentation and are differentiated based on local invasion into the surrounding tissues and organs (eg, kidney, liver) or distant metastasis.
A typical workup of a suspected patient with pheochromocytoma includes biochemical tests, including measurements of urinary and fractionated plasma metanephrines and catecholamine. Patients with positive biochemical tests should undergo localization of the tumor with an imaging study either with an adrenal/abdominal magnetic resonance imaging (MRI) or computed tomography (CT) scan. If a patient has paraganglioma or an adrenal mass > 10 cm or negative abdominal imaging with a positive biochemical test, further imaging with an iobenguane I-123 scan is needed (Figure 1).

In this article, we present an unusual case of asymptomatic pheochromocytoma in a patient with right-sided renal oncocytoma who underwent an uneventful nephrectomy and adrenalectomy.
Case Presentation
A 72-year-old male with a medical history of diabetes, hypertension, sensory neuropathy, benign prostatic hypertrophy (BPH) status posttransurethral resection of the prostate, and chronic renal failure presented to establish care with the Arizona Kidney Disease and Hypertension Center. His medications included losartan 50 mg by mouth daily, diltiazem 180 mg extended-release by mouth daily, carvedilol 6.25 mg by mouth twice a day, and tamsulosin 0.4 mg by mouth daily. His presenting vitals were blood pressure (BP), 112/74 left arm sitting, pulse, 63/beats per min, and body mass index, 34. On physical examination, the patient was alert and oriented, and the chest was clear to auscultation without wheeze or rhonchi. On cardiac examination, heart rate and rhythm were regular; S1 and S2 were normal with no added murmurs, rubs or gallops, and no jugular venous distension. The abdomen was soft, nontender, with no palpable mass. His laboratory results showed sodium, 142 mmol/L; potassium, 5.3 mmol/L; chloride, 101 mmol/L; carbon dioxide, 24 mmol/L; albumin, 4.3 g/dL; creatinine, 1.89 mg/dL; blood urea nitrogen, 29 mg/dL; estimated glomerular filtration rate non-African American, 35 mL/min/1.73; 24-h urine creatinine clearance, 105 mL/min; protein, 1306 mg/24 h (Table).
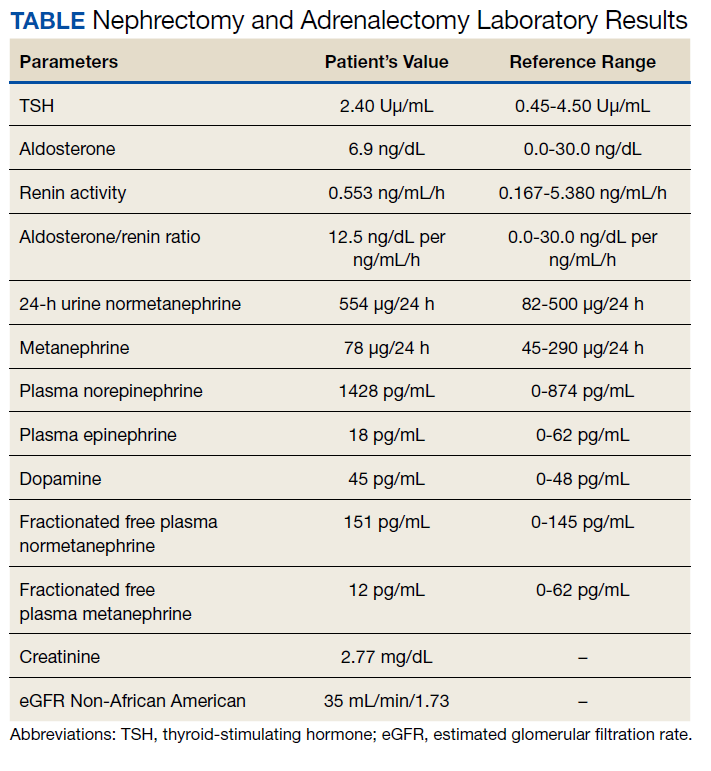
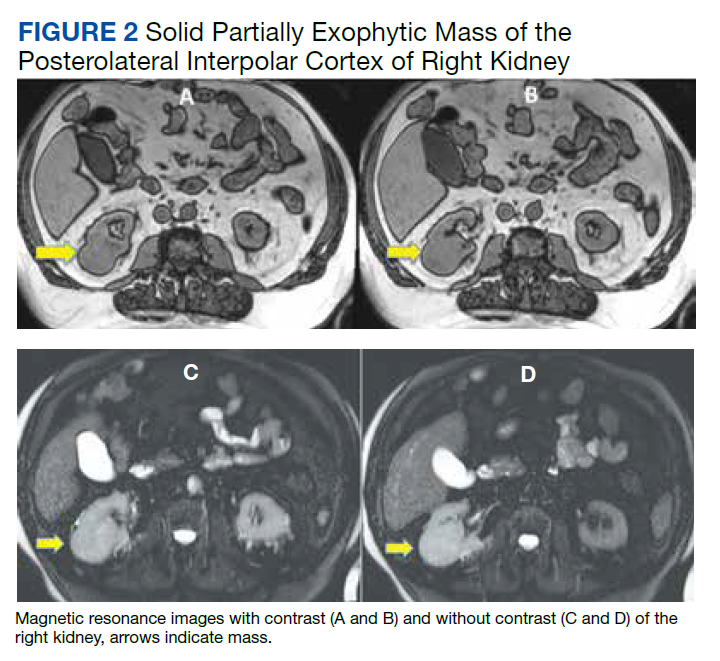
His renal ultrasound showed an exophytic isoechoic mass or complex cyst at the lateral aspect of the lower pole of the right kidney, measuring 45 mm in diameter. An MRI of the abdomen with and without contrast showed a solid partially exophytic mass of the posterolateral interpolar cortex of the right kidney, measuring 5.9 cm in the greatest dimension (Figure 2). No definite involvement of Gerota fascia was noted, a 1-cm metastasis to the right adrenal gland was present, renal veins were patent, and there was no upper retroperitoneal lymphadenopathy.
Treatment and Follow-up
The patient underwent right-hand-assisted lap-aroscopic radical nephrectomy and right adre-nalectomy without any complications. However, the surgical pathology report showed oncocytoma of the kidney (5.7 cm), pheochromocytoma of the adrenal gland (1.4 cm), and papillary adenoma of the kidney (0.7 cm). Right kidney nephrectomy showed non-neoplastic renal parenchyma, diabetic glomerulosclerosis (Renal Pathology Society 2010 diabetic nephropathy class IIb), severe mesangial expansion, moderate interstitial fibrosis, moderate arteriosclerosis, and mild arteriolosclerosis.
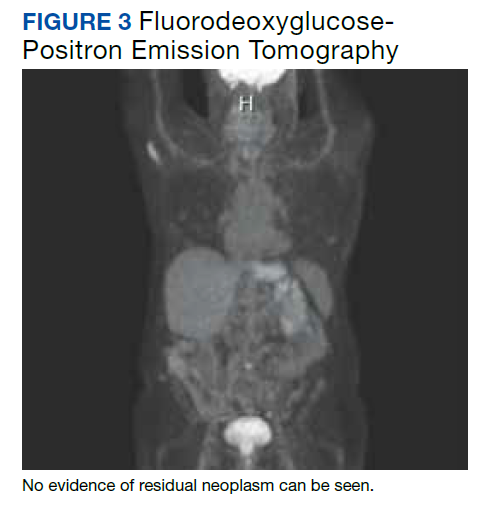
A fluorodeoxyglucose-positron emission tomography (FDG-PET) scan was significant for right nephrectomy and adrenalectomy and showed no significant evidence of residual neoplasm or local or distant metastases. A nuclear medicine (iobenguane I-123) tumor and single positron emission computed tomography (SPECT) scan showed normal activity throughout the body and no evidence of abnormal activity (Figure 3).
Discussion
Pheochromocytoma is a rare cause of secondary hypertension. However, the real numbers are thought to be > 0.2 to 0.5%.1,2,4 Patients with pheochromocytoma should undergo surgical adrenal resection after appropriate medical preparation. Patients with pheochromocytoma who are not diagnosed preoperatively have increased surgical mortality rates due to fatal hypertensive crises, malignant arrhythmia, and multiorgan failure as a result of hypertensive crisis.15 Anesthetic drugs during surgery also can exacerbate the cardiotoxic effects of catecholamines. Short-acting anesthetic agents, such as fentanyl, are used in patients with pheochromocytoma.16
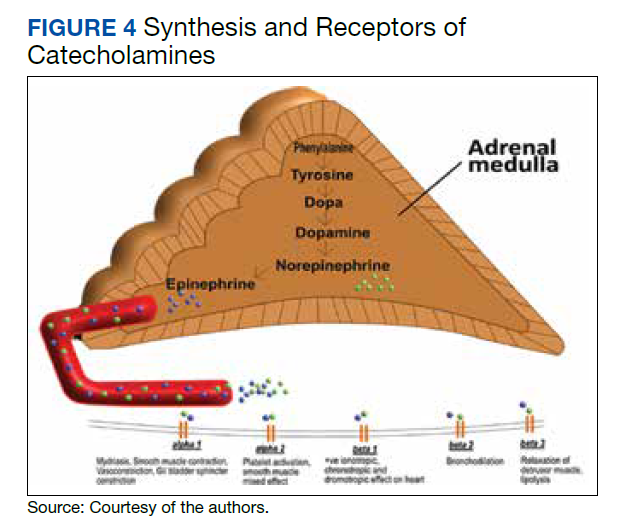
This case of pheochromocytoma illustrated no classic symptoms of episodic headache, sweating, and tachycardia, and the patient was otherwise asymptomatic. BP was well controlled with losartan, diltiazem, and a β-blocker with α-blocking activity (carvedilol). As the patient was not known to have pheochromocytoma, he did not undergo preoperative medical therapy. Figure 4 illustrates the receptors stimulate catecholamines, and the drugs blocking these receptors prevent hypertensive crisis during surgery. However, the surgery was without potential complications (ie, hypertensive crisis, malignant arrhythmia, or multiorgan failure). The patient was diagnosed incidentally on histopathology after right radical nephrectomy and adrenalectomy due to solid partially exophytic right renal mass (5.9 cm) with right adrenal metastasis. About 10% of patients are asymptomatic or mildly symptomatic.7 Sometimes, the symptoms may be ignored because of the episodic nature. Other possible reasons can be small, nonfunctional tumors or the use of antihypertensive medications suppressing the symptoms.7
The adrenal mass that was initially thought to be a metastasis of right kidney mass was later confirmed as pheochromocytoma. One possible explanation for uneventful surgery could be the use of β-blocker with α-blocking activity (carvedilol), α-1 adrenergic blocker (tamsulosin) along with nondihydropyridine calcium channel blocker (diltiazem) as part of the patient’s antihypertensive and BPH medication regimen. Another possible explanation could be silent or episodically secreting pheochromocytoma with a small functional portion.
Subsequent workup after adrenalectomy, including urinary and fractionated plasma metanephrines and catecholamines, were not consistent with catecholamine hypersecretion. A 24-hour urine fractionated metanephrines test has about 98% sensitivity and 98% specificity. Elevated plasma norepinephrine was thought to be due to renal failure because it was < 3-fold the upper limit of normal, which is considered to be a possible indication of pheochromocytoma.17,18 The nuclear medicine (iobenguane I-123) tumor, SPECT, and FDG-PET CT studies were negative for residual pheochromocytoma. Other imaging studies to consider in patients with suspected catecholamine-secreting tumor with positive biochemical test and negative abdominal imaging are a whole-body MRI scan, 68-Ga DOTATATE (gallium 68 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid-octreotate) or FDG-PET scan.19
In a review of 54 autopsy-proven pheochromocytoma cases by Sutton and colleagues in 1981, 74% of the patients were not clinically suspected for pheochromocytoma in their life.4 Similarly, in a retrospective study of hospital autopsies by McNeil and colleagues, one incidental pheochromocytoma was detected in every 2031 autopsies (0.05%).20 In another case series of 41 patients with pheochromocytoma-related adrenalectomy, almost 50% of the pheochromocytomas were detected incidentally on imaging studies.21 Although the number of incidental findings are decreasing due to advances in screening techniques, a significant number of patients remain undiagnosed. Multiple cases of diagnosis of pheochromocytoma on autopsy of patients who died of hemodynamic instability (ie, hypertensive crisis, hypotension crisis precipitated by surgery for adrenal or nonadrenal conditions) are reported.3 To the best of our knowledge, there are no case reports published on the diagnosis of pheochromocytoma after adrenalectomy in an asymptomatic patient without intraoperative complications.
The goal of preoperative medical therapy includes BP control, prevention of tachycardia, and volume expansion. The preoperative medications regimens are combined α- and β-adrenergic blockade, calcium channel blockers, and metyrosine. According to clinical practice guidelines of the Endocrine Society in 2014, the α-adrenergic blockers should be started first at least 7 days before surgery to control BP and to cause vasodilation. Early use of α-blockers is required to prevent cardiotoxicity. The β-adrenergic blockers should be started after the adequate α-adrenergic blockade, typically 2 to 3 days before surgery, as early use can cause vasoconstriction in patients with pheochromocytoma. The α-adrenergic blockers include phenoxybenzamine (nonselective long-acting nonspecific α-adrenergic blocking agent), and selective α-1 adrenergic blockers (doxazosin, prazosin, terazosin). The β-adrenergic blocker (ie, propranolol, metoprolol) should be started cautiously with a low dose and slowly titrated to control heart rate. A high sodium diet and increased fluid intake also are recommended 7 to 14 days before surgery. A sudden drop in catecholamines can cause hypotension during an operation. Continuous fluid infusions are given to prevent hypotension.22 Similarly, anesthetic agents also should be modified to prevent cardiotoxic effects. Rocuronium and vecuronium are less cardiotoxic compared with other sympathomimetic muscle relaxants. Short-acting anesthetic agents, such as fentanyl, are preferred. α-blockers are continued throughout the operation. Biochemical testing with fractionated metanephrines is performed about 1 to 2 weeks postoperatively to look for recurrence of the disease.23
Secondary causes of hypertension are suspected in multidrug resistant or sudden early onset of hypertension before aged 40 years. Pheochromocytoma is a rare cause of secondary hypertension, and older adult patients are rarely diagnosed with pheochromocytoma.24 In this report, pheochromocytoma was detected in a 72-year-old hypertensive patient. Therefore, a pheochromocytoma diagnosis should not be ignored in the older adult patient with adrenal mass and hypertension treated with more than one drug. The authors recommend any patient undergoing surgery with adrenal lesion should be considered for the screening of possible pheochromocytoma and prepared preoperatively, especially any patient with renal cell carcinoma with adrenal metastasis.
Conclusions
Asymptomatic pheochromocytoma is an unusual but serious condition, especially for patients undergoing a surgical procedure. An adrenal mass may be ignored in asymptomatic or mildly symptomatic older adult patients and is mostly considered as adrenal metastasis when present with other malignancies. Fortunately, the nephrectomy and adrenalectomy in our case of asymptomatic pheochromocytoma was uneventful, but pheochromocytoma should be ruled out before a surgical procedure, as an absence of medical pretreatment can lead to serious consequences. Therefore, we suggest a more careful screening of pheochromocytoma in patients with an adrenal mass (primary or metastatic) and hypertension treated with multiple antihypertensive drugs, even in older adult patients.
1. Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193-202. doi:10.1291/hypres.27.193
2. Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma: a review of the literature and report of one institution’s experience. Medicine (Baltimore). 1991;70(1):46-66. doi:10.1097/00005792-199101000-00004
3. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58(12):802-804.
4. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma: review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354-360.
5. Manger WM, Gifford RW Jr. Pheochromocytoma. J Clin Hypertens (Greenwich). 2002;4(1):62-72. doi:10.1111/j.1524-6175.2002.01452.x
6. Kassim TA, Clarke DD, Mai VQ, Clyde PW, Mohamed Shakir KM. Catecholamine-induced cardiomyopathy. Endocr Pract. 2008;14(9):1137-1149. doi:10.4158/EP.14.9.1137
7. Kudva YC, Young WF, Thompson GB, Grant CS, Van Heerden JA. Adrenal incidentaloma: an important component of the clinical presentation spectrum of benign sporadic adrenal pheochromocytoma. The Endocrinologist. 1999;9(2):77-80. doi:10.1097/00019616-199903000-00002
8. Puar TH, Mok Y, Debajyoti R, Khoo J, How CH, Ng AK. Secondary hypertension in adults. Singapore Med J. 2016;57(5):228-232. doi:10.11622/smedj.2016087
9. Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991;40(3):544-556. doi:10.1038/ki.1991.244
10. Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29(5):1133-1139. doi:10.1161/01.hyp.29.5.1133
11. Hamidi O, Young WF Jr, Iñiguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102(9):3296-3305. doi:10.1210/jc.2017-00992
12. Kenny L, Rizzo V, Trevis J, Assimakopoulou E, Timon D. The unexpected diagnosis of phaeochromocytoma in the anaesthetic room. Ann Card Anaesth. 2018;21(3):307-310. doi:10.4103/aca.ACA_206_17
13. Johnston PC, Silversides JA, Wallace H, et al. Phaeochromocytoma crisis: two cases of undiagnosed phaeochromocytoma presenting after elective nonrelated surgical procedures. Case Rep Anesthesiol. 2013;2013:514714. doi:10.1155/2013/514714
14. Shen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J. 2005;28(1):44-50.
15. Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212-215. doi:10.1016/s0002-9610(00)00296-8
16. Myklejord DJ. Undiagnosed pheochromocytoma: the anesthesiologist nightmare. Clin Med Res. 2004;2(1):59-62. doi:10.3121/cmr.2.1.59
17. Stumvoll M, Radjaipour M, Seif F. Diagnostic considerations in pheochromocytoma and chronic hemodialysis: case report and review of the literature. Am J Nephrol. 1995;15(2):147-151. doi:10.1159/000168820
18. Morioka M, Yuihama S, Nakajima T, et al. Incidentally discovered pheochromocytoma in long-term hemodialysis patients. Int J Urol. 2002;9(12):700-703. doi:10.1046/j.1442-2042.2002.00553.x
19. ˇCtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15(4):3151-3160. doi:10.3892/etm.2018.5871
20. McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30(6):648-652. doi:10.1111/j.1445-5994.2000.tb04358.x
21. Baguet JP, Hammer L, Mazzuco TL, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol. 2004;150(5):681-686. doi:10.1530/eje.0.1500681
22. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. doi:10.1210/jc.2014-1498
23. Dortzbach K, Gainsburg DM, Frost EA. Variants of pheochromocytoma and their anesthetic implications--a case report and literature review. Middle East J Anaesthesiol. 2010;20(6):897-905.
24. Januszewicz W, Chodakowska J, Styczy´nski G. Secondary hypertension in the elderly. J Hum Hypertens. 1998;12(9):603-606. doi:10.1038/sj.jhh.1000673
A high index of suspicion for pheochromocytoma is necessary during the workup of secondary hypertension as untreated pheochromocytoma may lead to significant morbidity and mortality, especially in patients who require any surgical treatment.
A high index of suspicion for pheochromocytoma is necessary during the workup of secondary hypertension as untreated pheochromocytoma may lead to significant morbidity and mortality, especially in patients who require any surgical treatment.
Pheochromocytoma is a rare catecholamine-secreting tumor of chromaffin cells of the adrenal medulla or sympathetic ganglia, occurring in about 0.2 to 0.5% of patients with hypertension.1-3 However, in a review of 54 autopsy-proven cases of pheochromocytoma, about 50% of the patients with hypertension were not clinically suspected for pheochromocytoma.4
Pheochromocytoma is usually diagnosed based on symptoms of hyperadrenergic spells, resistant hypertension, especially in the young, with a pressor response to the anesthesia stress test and adrenal incidentaloma.
The classic triad of symptoms associated with pheochromocytoma includes episodic headache (90%), sweating (60-70%), and palpitations (70%).2,5 Sustained or paroxysmal hypertension is the most common symptom reported in about 95% of patients with pheochromocytoma. Other symptoms include pallor, tremors, dyspnea, generalized weakness, orthostatic hypotension, cardiomyopathy, or hyperglycemia.6 However, about 10% of patients with pheochromocytoma are asymptomatic or mildly symptomatic.7 Secondary causes of hypertension are usually suspected in multidrug resistant or sudden early onset of hypertension.8
Approximately 10% of catecholamine-secreting tumors are malignant.9-11 Benign and malignant pheochromocytoma have a similar biochemical and histologic presentation and are differentiated based on local invasion into the surrounding tissues and organs (eg, kidney, liver) or distant metastasis.
A typical workup of a suspected patient with pheochromocytoma includes biochemical tests, including measurements of urinary and fractionated plasma metanephrines and catecholamine. Patients with positive biochemical tests should undergo localization of the tumor with an imaging study either with an adrenal/abdominal magnetic resonance imaging (MRI) or computed tomography (CT) scan. If a patient has paraganglioma or an adrenal mass > 10 cm or negative abdominal imaging with a positive biochemical test, further imaging with an iobenguane I-123 scan is needed (Figure 1).
In this article, we present an unusual case of asymptomatic pheochromocytoma in a patient with right-sided renal oncocytoma who underwent an uneventful nephrectomy and adrenalectomy.
Case Presentation
A 72-year-old male with a medical history of diabetes, hypertension, sensory neuropathy, benign prostatic hypertrophy (BPH) status posttransurethral resection of the prostate, and chronic renal failure presented to establish care with the Arizona Kidney Disease and Hypertension Center. His medications included losartan 50 mg by mouth daily, diltiazem 180 mg extended-release by mouth daily, carvedilol 6.25 mg by mouth twice a day, and tamsulosin 0.4 mg by mouth daily. His presenting vitals were blood pressure (BP), 112/74 left arm sitting, pulse, 63/beats per min, and body mass index, 34. On physical examination, the patient was alert and oriented, and the chest was clear to auscultation without wheeze or rhonchi. On cardiac examination, heart rate and rhythm were regular; S1 and S2 were normal with no added murmurs, rubs or gallops, and no jugular venous distension. The abdomen was soft, nontender, with no palpable mass. His laboratory results showed sodium, 142 mmol/L; potassium, 5.3 mmol/L; chloride, 101 mmol/L; carbon dioxide, 24 mmol/L; albumin, 4.3 g/dL; creatinine, 1.89 mg/dL; blood urea nitrogen, 29 mg/dL; estimated glomerular filtration rate non-African American, 35 mL/min/1.73; 24-h urine creatinine clearance, 105 mL/min; protein, 1306 mg/24 h (Table).
His renal ultrasound showed an exophytic isoechoic mass or complex cyst at the lateral aspect of the lower pole of the right kidney, measuring 45 mm in diameter. An MRI of the abdomen with and without contrast showed a solid partially exophytic mass of the posterolateral interpolar cortex of the right kidney, measuring 5.9 cm in the greatest dimension (Figure 2). No definite involvement of Gerota fascia was noted, a 1-cm metastasis to the right adrenal gland was present, renal veins were patent, and there was no upper retroperitoneal lymphadenopathy.
Treatment and Follow-up
The patient underwent right-hand-assisted lap-aroscopic radical nephrectomy and right adre-nalectomy without any complications. However, the surgical pathology report showed oncocytoma of the kidney (5.7 cm), pheochromocytoma of the adrenal gland (1.4 cm), and papillary adenoma of the kidney (0.7 cm). Right kidney nephrectomy showed non-neoplastic renal parenchyma, diabetic glomerulosclerosis (Renal Pathology Society 2010 diabetic nephropathy class IIb), severe mesangial expansion, moderate interstitial fibrosis, moderate arteriosclerosis, and mild arteriolosclerosis.
A fluorodeoxyglucose-positron emission tomography (FDG-PET) scan was significant for right nephrectomy and adrenalectomy and showed no significant evidence of residual neoplasm or local or distant metastases. A nuclear medicine (iobenguane I-123) tumor and single positron emission computed tomography (SPECT) scan showed normal activity throughout the body and no evidence of abnormal activity (Figure 3).
Discussion
Pheochromocytoma is a rare cause of secondary hypertension. However, the real numbers are thought to be > 0.2 to 0.5%.1,2,4 Patients with pheochromocytoma should undergo surgical adrenal resection after appropriate medical preparation. Patients with pheochromocytoma who are not diagnosed preoperatively have increased surgical mortality rates due to fatal hypertensive crises, malignant arrhythmia, and multiorgan failure as a result of hypertensive crisis.15 Anesthetic drugs during surgery also can exacerbate the cardiotoxic effects of catecholamines. Short-acting anesthetic agents, such as fentanyl, are used in patients with pheochromocytoma.16
This case of pheochromocytoma illustrated no classic symptoms of episodic headache, sweating, and tachycardia, and the patient was otherwise asymptomatic. BP was well controlled with losartan, diltiazem, and a β-blocker with α-blocking activity (carvedilol). As the patient was not known to have pheochromocytoma, he did not undergo preoperative medical therapy. Figure 4 illustrates the receptors stimulate catecholamines, and the drugs blocking these receptors prevent hypertensive crisis during surgery. However, the surgery was without potential complications (ie, hypertensive crisis, malignant arrhythmia, or multiorgan failure). The patient was diagnosed incidentally on histopathology after right radical nephrectomy and adrenalectomy due to solid partially exophytic right renal mass (5.9 cm) with right adrenal metastasis. About 10% of patients are asymptomatic or mildly symptomatic.7 Sometimes, the symptoms may be ignored because of the episodic nature. Other possible reasons can be small, nonfunctional tumors or the use of antihypertensive medications suppressing the symptoms.7
The adrenal mass that was initially thought to be a metastasis of right kidney mass was later confirmed as pheochromocytoma. One possible explanation for uneventful surgery could be the use of β-blocker with α-blocking activity (carvedilol), α-1 adrenergic blocker (tamsulosin) along with nondihydropyridine calcium channel blocker (diltiazem) as part of the patient’s antihypertensive and BPH medication regimen. Another possible explanation could be silent or episodically secreting pheochromocytoma with a small functional portion.
Subsequent workup after adrenalectomy, including urinary and fractionated plasma metanephrines and catecholamines, were not consistent with catecholamine hypersecretion. A 24-hour urine fractionated metanephrines test has about 98% sensitivity and 98% specificity. Elevated plasma norepinephrine was thought to be due to renal failure because it was < 3-fold the upper limit of normal, which is considered to be a possible indication of pheochromocytoma.17,18 The nuclear medicine (iobenguane I-123) tumor, SPECT, and FDG-PET CT studies were negative for residual pheochromocytoma. Other imaging studies to consider in patients with suspected catecholamine-secreting tumor with positive biochemical test and negative abdominal imaging are a whole-body MRI scan, 68-Ga DOTATATE (gallium 68 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid-octreotate) or FDG-PET scan.19
In a review of 54 autopsy-proven pheochromocytoma cases by Sutton and colleagues in 1981, 74% of the patients were not clinically suspected for pheochromocytoma in their life.4 Similarly, in a retrospective study of hospital autopsies by McNeil and colleagues, one incidental pheochromocytoma was detected in every 2031 autopsies (0.05%).20 In another case series of 41 patients with pheochromocytoma-related adrenalectomy, almost 50% of the pheochromocytomas were detected incidentally on imaging studies.21 Although the number of incidental findings are decreasing due to advances in screening techniques, a significant number of patients remain undiagnosed. Multiple cases of diagnosis of pheochromocytoma on autopsy of patients who died of hemodynamic instability (ie, hypertensive crisis, hypotension crisis precipitated by surgery for adrenal or nonadrenal conditions) are reported.3 To the best of our knowledge, there are no case reports published on the diagnosis of pheochromocytoma after adrenalectomy in an asymptomatic patient without intraoperative complications.
The goal of preoperative medical therapy includes BP control, prevention of tachycardia, and volume expansion. The preoperative medications regimens are combined α- and β-adrenergic blockade, calcium channel blockers, and metyrosine. According to clinical practice guidelines of the Endocrine Society in 2014, the α-adrenergic blockers should be started first at least 7 days before surgery to control BP and to cause vasodilation. Early use of α-blockers is required to prevent cardiotoxicity. The β-adrenergic blockers should be started after the adequate α-adrenergic blockade, typically 2 to 3 days before surgery, as early use can cause vasoconstriction in patients with pheochromocytoma. The α-adrenergic blockers include phenoxybenzamine (nonselective long-acting nonspecific α-adrenergic blocking agent), and selective α-1 adrenergic blockers (doxazosin, prazosin, terazosin). The β-adrenergic blocker (ie, propranolol, metoprolol) should be started cautiously with a low dose and slowly titrated to control heart rate. A high sodium diet and increased fluid intake also are recommended 7 to 14 days before surgery. A sudden drop in catecholamines can cause hypotension during an operation. Continuous fluid infusions are given to prevent hypotension.22 Similarly, anesthetic agents also should be modified to prevent cardiotoxic effects. Rocuronium and vecuronium are less cardiotoxic compared with other sympathomimetic muscle relaxants. Short-acting anesthetic agents, such as fentanyl, are preferred. α-blockers are continued throughout the operation. Biochemical testing with fractionated metanephrines is performed about 1 to 2 weeks postoperatively to look for recurrence of the disease.23
Secondary causes of hypertension are suspected in multidrug resistant or sudden early onset of hypertension before aged 40 years. Pheochromocytoma is a rare cause of secondary hypertension, and older adult patients are rarely diagnosed with pheochromocytoma.24 In this report, pheochromocytoma was detected in a 72-year-old hypertensive patient. Therefore, a pheochromocytoma diagnosis should not be ignored in the older adult patient with adrenal mass and hypertension treated with more than one drug. The authors recommend any patient undergoing surgery with adrenal lesion should be considered for the screening of possible pheochromocytoma and prepared preoperatively, especially any patient with renal cell carcinoma with adrenal metastasis.
Conclusions
Asymptomatic pheochromocytoma is an unusual but serious condition, especially for patients undergoing a surgical procedure. An adrenal mass may be ignored in asymptomatic or mildly symptomatic older adult patients and is mostly considered as adrenal metastasis when present with other malignancies. Fortunately, the nephrectomy and adrenalectomy in our case of asymptomatic pheochromocytoma was uneventful, but pheochromocytoma should be ruled out before a surgical procedure, as an absence of medical pretreatment can lead to serious consequences. Therefore, we suggest a more careful screening of pheochromocytoma in patients with an adrenal mass (primary or metastatic) and hypertension treated with multiple antihypertensive drugs, even in older adult patients.
Pheochromocytoma is a rare catecholamine-secreting tumor of chromaffin cells of the adrenal medulla or sympathetic ganglia, occurring in about 0.2 to 0.5% of patients with hypertension.1-3 However, in a review of 54 autopsy-proven cases of pheochromocytoma, about 50% of the patients with hypertension were not clinically suspected for pheochromocytoma.4
Pheochromocytoma is usually diagnosed based on symptoms of hyperadrenergic spells, resistant hypertension, especially in the young, with a pressor response to the anesthesia stress test and adrenal incidentaloma.
The classic triad of symptoms associated with pheochromocytoma includes episodic headache (90%), sweating (60-70%), and palpitations (70%).2,5 Sustained or paroxysmal hypertension is the most common symptom reported in about 95% of patients with pheochromocytoma. Other symptoms include pallor, tremors, dyspnea, generalized weakness, orthostatic hypotension, cardiomyopathy, or hyperglycemia.6 However, about 10% of patients with pheochromocytoma are asymptomatic or mildly symptomatic.7 Secondary causes of hypertension are usually suspected in multidrug resistant or sudden early onset of hypertension.8
Approximately 10% of catecholamine-secreting tumors are malignant.9-11 Benign and malignant pheochromocytoma have a similar biochemical and histologic presentation and are differentiated based on local invasion into the surrounding tissues and organs (eg, kidney, liver) or distant metastasis.
A typical workup of a suspected patient with pheochromocytoma includes biochemical tests, including measurements of urinary and fractionated plasma metanephrines and catecholamine. Patients with positive biochemical tests should undergo localization of the tumor with an imaging study either with an adrenal/abdominal magnetic resonance imaging (MRI) or computed tomography (CT) scan. If a patient has paraganglioma or an adrenal mass > 10 cm or negative abdominal imaging with a positive biochemical test, further imaging with an iobenguane I-123 scan is needed (Figure 1).
In this article, we present an unusual case of asymptomatic pheochromocytoma in a patient with right-sided renal oncocytoma who underwent an uneventful nephrectomy and adrenalectomy.
Case Presentation
A 72-year-old male with a medical history of diabetes, hypertension, sensory neuropathy, benign prostatic hypertrophy (BPH) status posttransurethral resection of the prostate, and chronic renal failure presented to establish care with the Arizona Kidney Disease and Hypertension Center. His medications included losartan 50 mg by mouth daily, diltiazem 180 mg extended-release by mouth daily, carvedilol 6.25 mg by mouth twice a day, and tamsulosin 0.4 mg by mouth daily. His presenting vitals were blood pressure (BP), 112/74 left arm sitting, pulse, 63/beats per min, and body mass index, 34. On physical examination, the patient was alert and oriented, and the chest was clear to auscultation without wheeze or rhonchi. On cardiac examination, heart rate and rhythm were regular; S1 and S2 were normal with no added murmurs, rubs or gallops, and no jugular venous distension. The abdomen was soft, nontender, with no palpable mass. His laboratory results showed sodium, 142 mmol/L; potassium, 5.3 mmol/L; chloride, 101 mmol/L; carbon dioxide, 24 mmol/L; albumin, 4.3 g/dL; creatinine, 1.89 mg/dL; blood urea nitrogen, 29 mg/dL; estimated glomerular filtration rate non-African American, 35 mL/min/1.73; 24-h urine creatinine clearance, 105 mL/min; protein, 1306 mg/24 h (Table).
His renal ultrasound showed an exophytic isoechoic mass or complex cyst at the lateral aspect of the lower pole of the right kidney, measuring 45 mm in diameter. An MRI of the abdomen with and without contrast showed a solid partially exophytic mass of the posterolateral interpolar cortex of the right kidney, measuring 5.9 cm in the greatest dimension (Figure 2). No definite involvement of Gerota fascia was noted, a 1-cm metastasis to the right adrenal gland was present, renal veins were patent, and there was no upper retroperitoneal lymphadenopathy.
Treatment and Follow-up
The patient underwent right-hand-assisted lap-aroscopic radical nephrectomy and right adre-nalectomy without any complications. However, the surgical pathology report showed oncocytoma of the kidney (5.7 cm), pheochromocytoma of the adrenal gland (1.4 cm), and papillary adenoma of the kidney (0.7 cm). Right kidney nephrectomy showed non-neoplastic renal parenchyma, diabetic glomerulosclerosis (Renal Pathology Society 2010 diabetic nephropathy class IIb), severe mesangial expansion, moderate interstitial fibrosis, moderate arteriosclerosis, and mild arteriolosclerosis.
A fluorodeoxyglucose-positron emission tomography (FDG-PET) scan was significant for right nephrectomy and adrenalectomy and showed no significant evidence of residual neoplasm or local or distant metastases. A nuclear medicine (iobenguane I-123) tumor and single positron emission computed tomography (SPECT) scan showed normal activity throughout the body and no evidence of abnormal activity (Figure 3).
Discussion
Pheochromocytoma is a rare cause of secondary hypertension. However, the real numbers are thought to be > 0.2 to 0.5%.1,2,4 Patients with pheochromocytoma should undergo surgical adrenal resection after appropriate medical preparation. Patients with pheochromocytoma who are not diagnosed preoperatively have increased surgical mortality rates due to fatal hypertensive crises, malignant arrhythmia, and multiorgan failure as a result of hypertensive crisis.15 Anesthetic drugs during surgery also can exacerbate the cardiotoxic effects of catecholamines. Short-acting anesthetic agents, such as fentanyl, are used in patients with pheochromocytoma.16
This case of pheochromocytoma illustrated no classic symptoms of episodic headache, sweating, and tachycardia, and the patient was otherwise asymptomatic. BP was well controlled with losartan, diltiazem, and a β-blocker with α-blocking activity (carvedilol). As the patient was not known to have pheochromocytoma, he did not undergo preoperative medical therapy. Figure 4 illustrates the receptors stimulate catecholamines, and the drugs blocking these receptors prevent hypertensive crisis during surgery. However, the surgery was without potential complications (ie, hypertensive crisis, malignant arrhythmia, or multiorgan failure). The patient was diagnosed incidentally on histopathology after right radical nephrectomy and adrenalectomy due to solid partially exophytic right renal mass (5.9 cm) with right adrenal metastasis. About 10% of patients are asymptomatic or mildly symptomatic.7 Sometimes, the symptoms may be ignored because of the episodic nature. Other possible reasons can be small, nonfunctional tumors or the use of antihypertensive medications suppressing the symptoms.7
The adrenal mass that was initially thought to be a metastasis of right kidney mass was later confirmed as pheochromocytoma. One possible explanation for uneventful surgery could be the use of β-blocker with α-blocking activity (carvedilol), α-1 adrenergic blocker (tamsulosin) along with nondihydropyridine calcium channel blocker (diltiazem) as part of the patient’s antihypertensive and BPH medication regimen. Another possible explanation could be silent or episodically secreting pheochromocytoma with a small functional portion.
Subsequent workup after adrenalectomy, including urinary and fractionated plasma metanephrines and catecholamines, were not consistent with catecholamine hypersecretion. A 24-hour urine fractionated metanephrines test has about 98% sensitivity and 98% specificity. Elevated plasma norepinephrine was thought to be due to renal failure because it was < 3-fold the upper limit of normal, which is considered to be a possible indication of pheochromocytoma.17,18 The nuclear medicine (iobenguane I-123) tumor, SPECT, and FDG-PET CT studies were negative for residual pheochromocytoma. Other imaging studies to consider in patients with suspected catecholamine-secreting tumor with positive biochemical test and negative abdominal imaging are a whole-body MRI scan, 68-Ga DOTATATE (gallium 68 1,4,7,10-tetraazacyclododecane-1,4,7,10 tetraacetic acid-octreotate) or FDG-PET scan.19
In a review of 54 autopsy-proven pheochromocytoma cases by Sutton and colleagues in 1981, 74% of the patients were not clinically suspected for pheochromocytoma in their life.4 Similarly, in a retrospective study of hospital autopsies by McNeil and colleagues, one incidental pheochromocytoma was detected in every 2031 autopsies (0.05%).20 In another case series of 41 patients with pheochromocytoma-related adrenalectomy, almost 50% of the pheochromocytomas were detected incidentally on imaging studies.21 Although the number of incidental findings are decreasing due to advances in screening techniques, a significant number of patients remain undiagnosed. Multiple cases of diagnosis of pheochromocytoma on autopsy of patients who died of hemodynamic instability (ie, hypertensive crisis, hypotension crisis precipitated by surgery for adrenal or nonadrenal conditions) are reported.3 To the best of our knowledge, there are no case reports published on the diagnosis of pheochromocytoma after adrenalectomy in an asymptomatic patient without intraoperative complications.
The goal of preoperative medical therapy includes BP control, prevention of tachycardia, and volume expansion. The preoperative medications regimens are combined α- and β-adrenergic blockade, calcium channel blockers, and metyrosine. According to clinical practice guidelines of the Endocrine Society in 2014, the α-adrenergic blockers should be started first at least 7 days before surgery to control BP and to cause vasodilation. Early use of α-blockers is required to prevent cardiotoxicity. The β-adrenergic blockers should be started after the adequate α-adrenergic blockade, typically 2 to 3 days before surgery, as early use can cause vasoconstriction in patients with pheochromocytoma. The α-adrenergic blockers include phenoxybenzamine (nonselective long-acting nonspecific α-adrenergic blocking agent), and selective α-1 adrenergic blockers (doxazosin, prazosin, terazosin). The β-adrenergic blocker (ie, propranolol, metoprolol) should be started cautiously with a low dose and slowly titrated to control heart rate. A high sodium diet and increased fluid intake also are recommended 7 to 14 days before surgery. A sudden drop in catecholamines can cause hypotension during an operation. Continuous fluid infusions are given to prevent hypotension.22 Similarly, anesthetic agents also should be modified to prevent cardiotoxic effects. Rocuronium and vecuronium are less cardiotoxic compared with other sympathomimetic muscle relaxants. Short-acting anesthetic agents, such as fentanyl, are preferred. α-blockers are continued throughout the operation. Biochemical testing with fractionated metanephrines is performed about 1 to 2 weeks postoperatively to look for recurrence of the disease.23
Secondary causes of hypertension are suspected in multidrug resistant or sudden early onset of hypertension before aged 40 years. Pheochromocytoma is a rare cause of secondary hypertension, and older adult patients are rarely diagnosed with pheochromocytoma.24 In this report, pheochromocytoma was detected in a 72-year-old hypertensive patient. Therefore, a pheochromocytoma diagnosis should not be ignored in the older adult patient with adrenal mass and hypertension treated with more than one drug. The authors recommend any patient undergoing surgery with adrenal lesion should be considered for the screening of possible pheochromocytoma and prepared preoperatively, especially any patient with renal cell carcinoma with adrenal metastasis.
Conclusions
Asymptomatic pheochromocytoma is an unusual but serious condition, especially for patients undergoing a surgical procedure. An adrenal mass may be ignored in asymptomatic or mildly symptomatic older adult patients and is mostly considered as adrenal metastasis when present with other malignancies. Fortunately, the nephrectomy and adrenalectomy in our case of asymptomatic pheochromocytoma was uneventful, but pheochromocytoma should be ruled out before a surgical procedure, as an absence of medical pretreatment can lead to serious consequences. Therefore, we suggest a more careful screening of pheochromocytoma in patients with an adrenal mass (primary or metastatic) and hypertension treated with multiple antihypertensive drugs, even in older adult patients.
1. Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193-202. doi:10.1291/hypres.27.193
2. Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma: a review of the literature and report of one institution’s experience. Medicine (Baltimore). 1991;70(1):46-66. doi:10.1097/00005792-199101000-00004
3. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58(12):802-804.
4. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma: review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354-360.
5. Manger WM, Gifford RW Jr. Pheochromocytoma. J Clin Hypertens (Greenwich). 2002;4(1):62-72. doi:10.1111/j.1524-6175.2002.01452.x
6. Kassim TA, Clarke DD, Mai VQ, Clyde PW, Mohamed Shakir KM. Catecholamine-induced cardiomyopathy. Endocr Pract. 2008;14(9):1137-1149. doi:10.4158/EP.14.9.1137
7. Kudva YC, Young WF, Thompson GB, Grant CS, Van Heerden JA. Adrenal incidentaloma: an important component of the clinical presentation spectrum of benign sporadic adrenal pheochromocytoma. The Endocrinologist. 1999;9(2):77-80. doi:10.1097/00019616-199903000-00002
8. Puar TH, Mok Y, Debajyoti R, Khoo J, How CH, Ng AK. Secondary hypertension in adults. Singapore Med J. 2016;57(5):228-232. doi:10.11622/smedj.2016087
9. Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991;40(3):544-556. doi:10.1038/ki.1991.244
10. Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29(5):1133-1139. doi:10.1161/01.hyp.29.5.1133
11. Hamidi O, Young WF Jr, Iñiguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102(9):3296-3305. doi:10.1210/jc.2017-00992
12. Kenny L, Rizzo V, Trevis J, Assimakopoulou E, Timon D. The unexpected diagnosis of phaeochromocytoma in the anaesthetic room. Ann Card Anaesth. 2018;21(3):307-310. doi:10.4103/aca.ACA_206_17
13. Johnston PC, Silversides JA, Wallace H, et al. Phaeochromocytoma crisis: two cases of undiagnosed phaeochromocytoma presenting after elective nonrelated surgical procedures. Case Rep Anesthesiol. 2013;2013:514714. doi:10.1155/2013/514714
14. Shen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J. 2005;28(1):44-50.
15. Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212-215. doi:10.1016/s0002-9610(00)00296-8
16. Myklejord DJ. Undiagnosed pheochromocytoma: the anesthesiologist nightmare. Clin Med Res. 2004;2(1):59-62. doi:10.3121/cmr.2.1.59
17. Stumvoll M, Radjaipour M, Seif F. Diagnostic considerations in pheochromocytoma and chronic hemodialysis: case report and review of the literature. Am J Nephrol. 1995;15(2):147-151. doi:10.1159/000168820
18. Morioka M, Yuihama S, Nakajima T, et al. Incidentally discovered pheochromocytoma in long-term hemodialysis patients. Int J Urol. 2002;9(12):700-703. doi:10.1046/j.1442-2042.2002.00553.x
19. ˇCtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15(4):3151-3160. doi:10.3892/etm.2018.5871
20. McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30(6):648-652. doi:10.1111/j.1445-5994.2000.tb04358.x
21. Baguet JP, Hammer L, Mazzuco TL, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol. 2004;150(5):681-686. doi:10.1530/eje.0.1500681
22. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. doi:10.1210/jc.2014-1498
23. Dortzbach K, Gainsburg DM, Frost EA. Variants of pheochromocytoma and their anesthetic implications--a case report and literature review. Middle East J Anaesthesiol. 2010;20(6):897-905.
24. Januszewicz W, Chodakowska J, Styczy´nski G. Secondary hypertension in the elderly. J Hum Hypertens. 1998;12(9):603-606. doi:10.1038/sj.jhh.1000673
1. Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27(3):193-202. doi:10.1291/hypres.27.193
2. Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma: a review of the literature and report of one institution’s experience. Medicine (Baltimore). 1991;70(1):46-66. doi:10.1097/00005792-199101000-00004
3. Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983;58(12):802-804.
4. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma: review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354-360.
5. Manger WM, Gifford RW Jr. Pheochromocytoma. J Clin Hypertens (Greenwich). 2002;4(1):62-72. doi:10.1111/j.1524-6175.2002.01452.x
6. Kassim TA, Clarke DD, Mai VQ, Clyde PW, Mohamed Shakir KM. Catecholamine-induced cardiomyopathy. Endocr Pract. 2008;14(9):1137-1149. doi:10.4158/EP.14.9.1137
7. Kudva YC, Young WF, Thompson GB, Grant CS, Van Heerden JA. Adrenal incidentaloma: an important component of the clinical presentation spectrum of benign sporadic adrenal pheochromocytoma. The Endocrinologist. 1999;9(2):77-80. doi:10.1097/00019616-199903000-00002
8. Puar TH, Mok Y, Debajyoti R, Khoo J, How CH, Ng AK. Secondary hypertension in adults. Singapore Med J. 2016;57(5):228-232. doi:10.11622/smedj.2016087
9. Bravo EL. Pheochromocytoma: new concepts and future trends. Kidney Int. 1991;40(3):544-556. doi:10.1038/ki.1991.244
10. Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29(5):1133-1139. doi:10.1161/01.hyp.29.5.1133
11. Hamidi O, Young WF Jr, Iñiguez-Ariza NM, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102(9):3296-3305. doi:10.1210/jc.2017-00992
12. Kenny L, Rizzo V, Trevis J, Assimakopoulou E, Timon D. The unexpected diagnosis of phaeochromocytoma in the anaesthetic room. Ann Card Anaesth. 2018;21(3):307-310. doi:10.4103/aca.ACA_206_17
13. Johnston PC, Silversides JA, Wallace H, et al. Phaeochromocytoma crisis: two cases of undiagnosed phaeochromocytoma presenting after elective nonrelated surgical procedures. Case Rep Anesthesiol. 2013;2013:514714. doi:10.1155/2013/514714
14. Shen SJ, Cheng HM, Chiu AW, Chou CW, Chen JY. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J. 2005;28(1):44-50.
15. Lo CY, Lam KY, Wat MS, Lam KS. Adrenal pheochromocytoma remains a frequently overlooked diagnosis. Am J Surg. 2000;179(3):212-215. doi:10.1016/s0002-9610(00)00296-8
16. Myklejord DJ. Undiagnosed pheochromocytoma: the anesthesiologist nightmare. Clin Med Res. 2004;2(1):59-62. doi:10.3121/cmr.2.1.59
17. Stumvoll M, Radjaipour M, Seif F. Diagnostic considerations in pheochromocytoma and chronic hemodialysis: case report and review of the literature. Am J Nephrol. 1995;15(2):147-151. doi:10.1159/000168820
18. Morioka M, Yuihama S, Nakajima T, et al. Incidentally discovered pheochromocytoma in long-term hemodialysis patients. Int J Urol. 2002;9(12):700-703. doi:10.1046/j.1442-2042.2002.00553.x
19. ˇCtvrtlík F, Koranda P, Schovánek J, Škarda J, Hartmann I, Tüdös Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15(4):3151-3160. doi:10.3892/etm.2018.5871
20. McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30(6):648-652. doi:10.1111/j.1445-5994.2000.tb04358.x
21. Baguet JP, Hammer L, Mazzuco TL, et al. Circumstances of discovery of phaeochromocytoma: a retrospective study of 41 consecutive patients. Eur J Endocrinol. 2004;150(5):681-686. doi:10.1530/eje.0.1500681
22. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915-1942. doi:10.1210/jc.2014-1498
23. Dortzbach K, Gainsburg DM, Frost EA. Variants of pheochromocytoma and their anesthetic implications--a case report and literature review. Middle East J Anaesthesiol. 2010;20(6):897-905.
24. Januszewicz W, Chodakowska J, Styczy´nski G. Secondary hypertension in the elderly. J Hum Hypertens. 1998;12(9):603-606. doi:10.1038/sj.jhh.1000673