User login
In reply: Short QT syndrome
In Reply: We thank Dr. Ratanapo and colleagues for their interest in our article. As we mentioned in our paper, and as they emphasized, the QT interval response to heart rate variation seems to be minimal. They wonder if using a beta-blockers in addition to Holter monitoring can provide a better estimate of the “true corrected QT interval” since it will allow the measurement of corrected QT with slower heart rates. While we agree that Holter monitoring may provide an opportunity to observe the lack of prolongation of the QT interval when the heart rate slows down naturally (eg, during sleep), we have reservations about the other points.
First, we prefer not to use the term “true corrected QT interval” because, as we mentioned in our article, the correction formulas do not perform well in short QT syndrome. A better thing would be to use the QT interval itself, no matter what the heart rate is.
Second, whether beta-blockers would alter the heart rate without altering the QT interval is something that deserves to be evaluated in patients with an established diagnosis of short QT syndrome. Since catecholamines can cause shortening of the QT interval,1 could beta-blockers have a different effect on the QT interval in patients with and without short QT syndrome? To our knowledge, there are no data that specifically address this question.
The last point we would like to emphasize is the complexity of making the diagnosis of short QT syndrome. Electrocardiographic criteria, especially when equivocal, should probably not be the sole diagnostic basis for short QT syndrome. A personal or family history of arrhythmias, with or without genetic testing, has additive value as demonstrated by the excellent paper by Gollob et al.2
- Bjerregaard P, Gusak I. Short QT syndrome: mechanism, diagnosis, and treatment. Nat Clin Pract Cardiovasc Med 2005; 2:84–87.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
In Reply: We thank Dr. Ratanapo and colleagues for their interest in our article. As we mentioned in our paper, and as they emphasized, the QT interval response to heart rate variation seems to be minimal. They wonder if using a beta-blockers in addition to Holter monitoring can provide a better estimate of the “true corrected QT interval” since it will allow the measurement of corrected QT with slower heart rates. While we agree that Holter monitoring may provide an opportunity to observe the lack of prolongation of the QT interval when the heart rate slows down naturally (eg, during sleep), we have reservations about the other points.
First, we prefer not to use the term “true corrected QT interval” because, as we mentioned in our article, the correction formulas do not perform well in short QT syndrome. A better thing would be to use the QT interval itself, no matter what the heart rate is.
Second, whether beta-blockers would alter the heart rate without altering the QT interval is something that deserves to be evaluated in patients with an established diagnosis of short QT syndrome. Since catecholamines can cause shortening of the QT interval,1 could beta-blockers have a different effect on the QT interval in patients with and without short QT syndrome? To our knowledge, there are no data that specifically address this question.
The last point we would like to emphasize is the complexity of making the diagnosis of short QT syndrome. Electrocardiographic criteria, especially when equivocal, should probably not be the sole diagnostic basis for short QT syndrome. A personal or family history of arrhythmias, with or without genetic testing, has additive value as demonstrated by the excellent paper by Gollob et al.2
In Reply: We thank Dr. Ratanapo and colleagues for their interest in our article. As we mentioned in our paper, and as they emphasized, the QT interval response to heart rate variation seems to be minimal. They wonder if using a beta-blockers in addition to Holter monitoring can provide a better estimate of the “true corrected QT interval” since it will allow the measurement of corrected QT with slower heart rates. While we agree that Holter monitoring may provide an opportunity to observe the lack of prolongation of the QT interval when the heart rate slows down naturally (eg, during sleep), we have reservations about the other points.
First, we prefer not to use the term “true corrected QT interval” because, as we mentioned in our article, the correction formulas do not perform well in short QT syndrome. A better thing would be to use the QT interval itself, no matter what the heart rate is.
Second, whether beta-blockers would alter the heart rate without altering the QT interval is something that deserves to be evaluated in patients with an established diagnosis of short QT syndrome. Since catecholamines can cause shortening of the QT interval,1 could beta-blockers have a different effect on the QT interval in patients with and without short QT syndrome? To our knowledge, there are no data that specifically address this question.
The last point we would like to emphasize is the complexity of making the diagnosis of short QT syndrome. Electrocardiographic criteria, especially when equivocal, should probably not be the sole diagnostic basis for short QT syndrome. A personal or family history of arrhythmias, with or without genetic testing, has additive value as demonstrated by the excellent paper by Gollob et al.2
- Bjerregaard P, Gusak I. Short QT syndrome: mechanism, diagnosis, and treatment. Nat Clin Pract Cardiovasc Med 2005; 2:84–87.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Bjerregaard P, Gusak I. Short QT syndrome: mechanism, diagnosis, and treatment. Nat Clin Pract Cardiovasc Med 2005; 2:84–87.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
A short story of the short QT syndrome
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
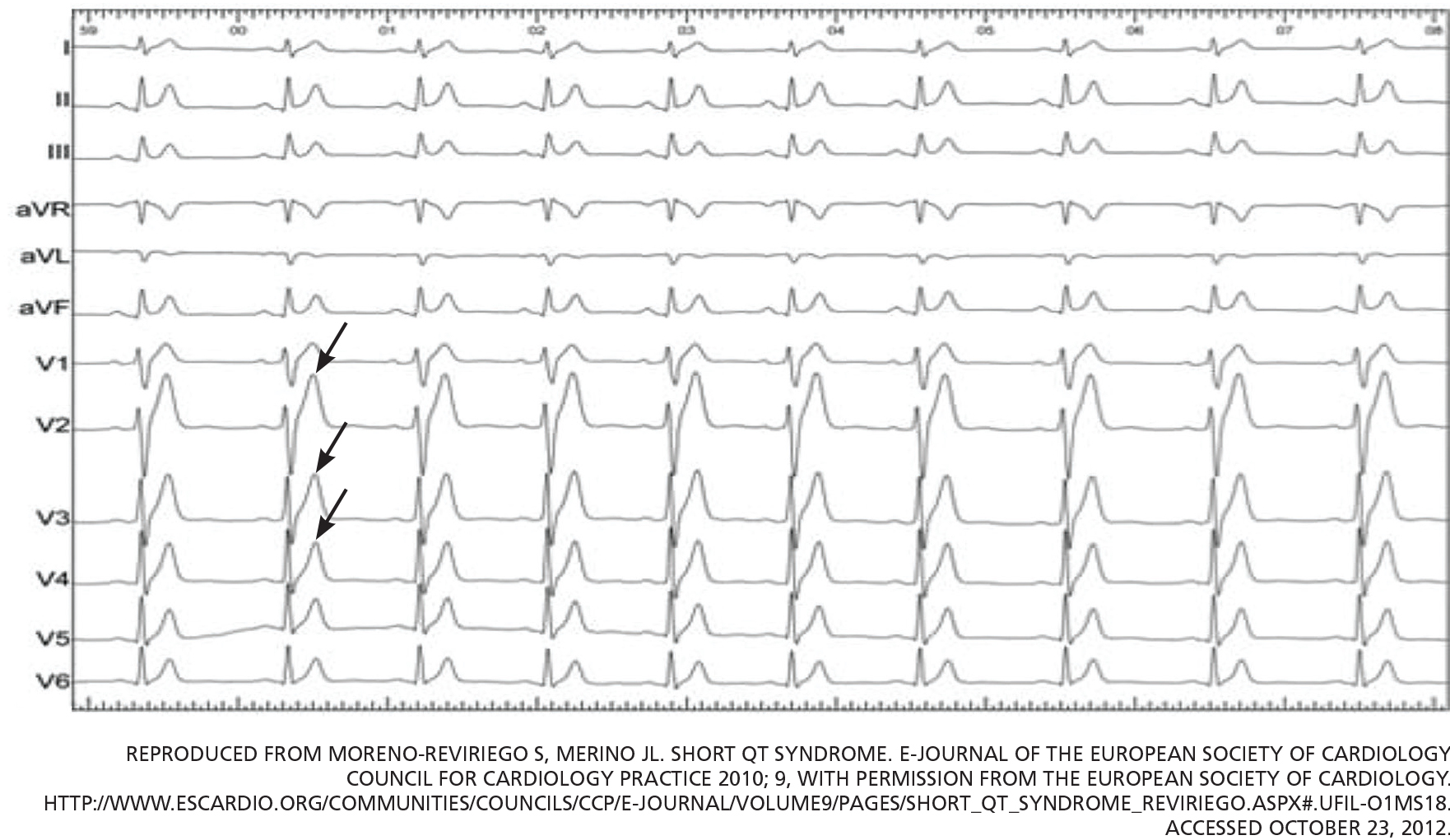
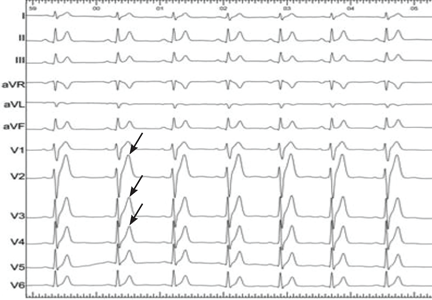
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
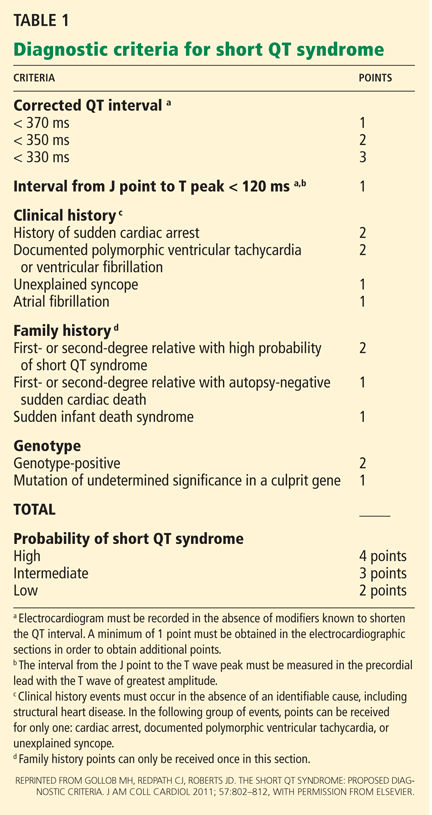
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
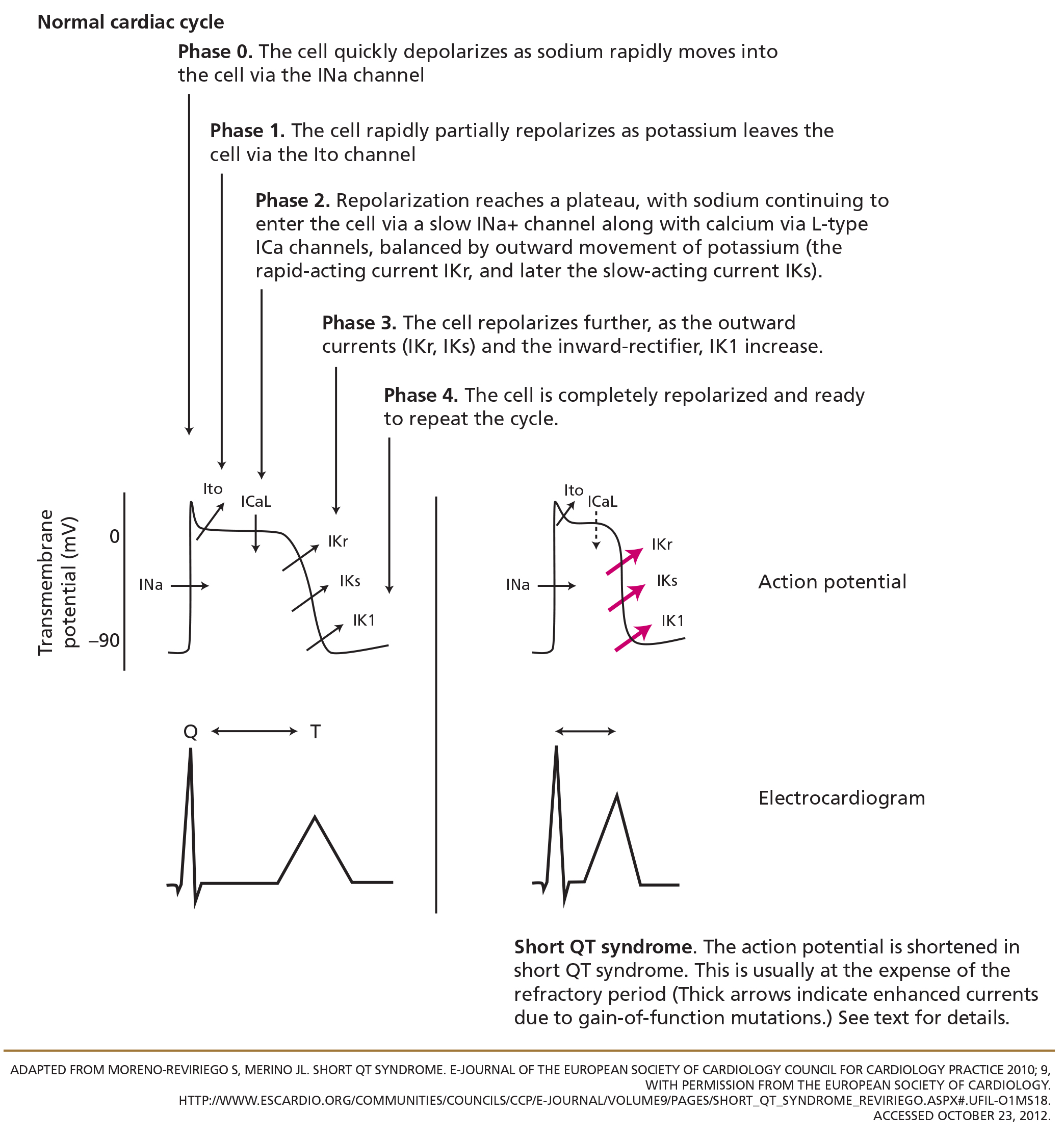
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
Sudden cardiac death in a young person is a devastating event that has puzzled physicians for decades. In recent years, many of the underlying cardiac pathologies have been identified. These include structural abnormalities such as hypertrophic cardiomyopathy and nonstructural disorders associated with unstable rhythms that lead to sudden cardiac death.
The best known of these “channelopathies” are the long QT syndromes, which result from abnormal potassium and sodium channels in myocytes. Recently, interest has been growing in a disorder that may carry a similarly grim prognosis but that has an opposite finding on electrocardiography (ECG).
Short QT syndrome is a recently described heterogeneous genetic channelopathy that causes both atrial and ventricular arrhythmias and that has been documented to cause sudden cardiac death.
In 1996, a 37-year-old woman from Spain died suddenly; ECG several days earlier had shown a short QT interval of 266 ms.1 Two years later, an unrelated 17-year-old American woman undergoing laparoscopic cholecystectomy suddenly developed atrial fibrillation with a rapid ventricular response.1 Her QT interval was 225 ms. Her brother had a QT interval of 240 ms, and her mother’s was 230 ms. The patient’s maternal grandfather had a history of atrial fibrillation, and his QT interval was 245 ms. These cases led to the description of this new clinical syndrome (see below).2
CLINICAL FEATURES
Short QT syndrome has been associated with both atrial and ventricular arrhythmias. Atrial fibrillation, polymorphic ventricular tachycardia, and ventricular fibrillation have all been well described. Patients who have symptoms usually present with palpitations, presyncope, syncope, or sudden or aborted cardiac death.3,4
ELECTROCARDIOGRAPHIC FEATURES
The primary finding on ECG is a short QT interval. However, others have been noted (Figure 1):
Short or absent ST segment
This finding is not merely a consequence of the short QT interval. In 10 patients with short QT syndrome, the distance from the J point to the peak T wave ranged from 80 to 120 ms. In 12 healthy people whose QT interval was less than 320 ms, this distance ranged from 150 ms to 240 ms.5
Tall and peaked T wave
A tall and peaked T wave is a common feature in short QT syndrome. However, it was also evident in people with short QT intervals who had no other features of the syndrome.5
QT response to heart rate
Normally, the QT interval is inversely related to the heart rate, but this is not true in short QT syndrome: the QT interval remains relatively fixed with changes in heart rate.6,7 This feature is less helpful in the office setting but may be found with Holter monitoring by measuring the QT interval at different heart rates.
BUT WHAT IS CONSIDERED A SHORT QT INTERVAL?
In clinical practice, the QT interval is corrected for the heart rate by the Bazett formula:
Corrected QT (QTc) = [QT interval/square root of the RR interval]
Review of ECGs from large populations in Finland (n = 10,822), Japan (n = 12,149), the United States (n = 79,743), and Switzerland (n = 41,676) revealed that a QTc value of 350 ms in males and 365 ms in females was 2.0 standard deviations (SD) below the mean.8–11 However, a QTc less than the 2.0 SD cutoff did not necessarily equal arrhythmogenic potential. This was illustrated in a 29-year follow-up study of Finnish patients with QTc values as short as 320 ms, in whom no arrhythmias were documented.8 Conversely, some patients with purported short QT syndrome had QTc intervals as long as 381 ms.12
Similar problems with uncertainty of values have plagued the diagnosis of long QT syndrome.13 The lack of reference ranges and the overlap between healthy and affected people called for the development of a scoring system that involves criteria based on ECG and on the clinical evaluation.14,15
ESTABLISHING THE DIAGNOSIS OF SHORT QT SYNDROME
Clearly, the diagnosis of short QT syndrome can be challenging to establish. The first step is to rule out other causes of a short QT interval.
Differential diagnosis of short QT interval
In addition to genetic channelopathies, other causes of short QT interval must be ruled out before entertaining the diagnosis of short QT syndrome.
- Hypercalcemia is the most important of these: there is usually an accompanying prolonged PR interval and a wide QRS complex16
- Hyperkalemia17
- Acidosis17
- Increased vagal tone17
- After ventricular fibrillation (thought to be related to increased intracellular calcium)18
- Digitalis use19
- Androgen use.20
Interestingly, a shorter-than-expected QT interval was noted in patients with chronic fatigue syndrome.21
Which interval to use: QT or QTc?
Unfortunately, most population-based studies that searched for a short QT interval on ECG have used QTc as the main search parameter.8–11 As already mentioned, in patients with short QT syndrome, the QT interval is, uniquely, not shortened if the heart beats faster. In contrast, the QTc often overestimates the QT interval in patients with short QT syndrome, especially when the heart rate is in the 80s to 90s.16
In a review of cases of short QT syndrome worldwide, Bjerregaard et al22 found that the QT interval ranged from 210 ms to 340 ms with a mean ± 2 SD of 282 ± 62 ms. On the other hand, the QTc ranged from 248 ms to 345 ms with a mean ± 2 SD of 305 ± 42 ms.
Therefore, correction formulas (such as the Bazett formula) do not perform well in ruling in the diagnosis of short QT syndrome—and they do even worse in ruling it out.16,22
To establish a diagnosis of short QT syndrome in someone with prior evidence of atrial or ventricular fibrillation, a QT interval less than 340 ms or a QTc less than 345 ms is usually sufficient.22 In borderline cases in which the QT interval is slightly longer, some experts recommend other tests, although strong evidence validating their predictive value does not exist. These tests include genotyping, analysis of T wave morphology, and electrophysiologic studies.16
Recently, Gollob et al23 proposed a scoring system for short QT syndrome (Table 1). After reviewing the literature and comparing the diagnostic markers, the investigators determined diagnostic criteria that, when applied to the previously reported cases, were able to identify 58 (95.08%) of 61 patients with short QT syndrome (ie, a sensitivity of 95%).
For patients with intermediate probability, the authors recommended continued medical and ECG surveillance as well as ECGs for first-degree relatives, to further clarify the diagnosis.
Again, a principal caveat about this system is that it relies on the QTc interval rather than the QT interval to diagnose short QT syndrome.
THE SCOPE OF THE DISEASE
In a recent review of the literature, Gollob et al23 found a total of 61 cases of short QT syndrome reported in English. The cohort was predominantly male (75.4%), and most of the symptomatic patients presented during late adolescence and early adulthood. However, there have been reports of infants (4 and 8 months old), and of a man who presented for the first time at the age of 70. Of note, the authors only considered short QT syndrome types 1, 2, and 3 (see below) in their search for cases.
Whether the syndrome is truly this rare or, rather, whether many physicians are not aware of it is still to be determined. In addition, it is possible that incorrectly measuring the QT interval contributes to the lack of identification of this entity. Both of these factors were implicated in the rarity of reported long QT syndrome early after its discovery.14,15
MUTATIONS IN CARDIAC ION CHANNELS
Five distinct genetic defects have been associated with short QT syndrome. As in long QT syndrome, these give rise to subtypes of short QT syndrome, which are numbered 1 to 5 (see below).
The cardiac action potential
To understand how the mutations shorten the QT interval, we will briefly review of the cardiac myocyte action potential.24 In nonpacemaker cells of the heart, the activation of the cell membrane initiates a series of changes in ion channels that allow the movement of ions along an electrical gradient. This movement occurs in five phases and is repeated with every cardiac cycle (Figure 2).
In phase 0, the cardiac cell rapidly depolarizes.
Repolarization occurs in phases 1, 2, and 3 and is largely a function of potassium ions leaving the cell. During phase 2, calcium and sodium ions enter the cell and balance the outward potassium flow, creating the “flat” portion of the repolarization curve. Phase 3 is the main phase of repolarization in which the membrane potential rapidly falls back to its resting state (–90 mV). During phases 1 and 2, the cell membrane is completely refractory to stimulation, whereas phase 3 is divided into three parts:
- The effective refractory period: the cell is able to generate a potential that is too weak to be propagated
- The relative refractory period: the cell can respond to a stimulus that is stronger than normal
- The supernormal phase: the last small portion of phase 3, in which a less-than-normal stimulus can yield a response in the cell.
In phase 4, the cell is completely repolarized, and the cycle can start again.
Five types of short QT syndrome
Short QT syndrome 1. In 2004, Brugada et al25 identified the first mutation that causes abnormal shortening of the action potential duration. In contrast to the mutations that underlie long QT syndrome, this mutation actually causes a gain of function in the gene coding the rapidly acting delayed potassium current (IKr) channel proteins KCNH2 or HERG. Potassium leaving at a more rapid rate causes the cell to repolarize more quickly and shortens the QT interval. The clinical syndrome associated with KCNH2 gene gain-of-function mutation is called short QT syndrome 1.
Short QT syndromes 2 and 3. Other IK (potassium channel) proteins have been implicated as well. Gain-of-function mutations in the KCNQ1 and KCNJ2 genes are believed to account for short QT syndromes 2 and 3, respectively. KCNQ1 codes for the IKs protein, and KCNJ2 codes for the IK1 protein.26,27
Short QT syndromes 4 and 5 were identified by Antzelevitch et al,28 who described several patients who had a combination of channel abnormalities and ECG findings. Their ECGs showed “Brugada-syndrome-like” ST elevation in the right precordial leads, but with a short QT interval. These new syndromes were found to be associated with genetic abnormalities distinct from those of Brugada syndrome and other short QT syndromes. These abnormalities involved loss-of-function mutations in the CACNA1C gene (which codes for the alpha-1 subunit of the L-type cardiac calcium channel) and in the CACNB2 gene (which codes for the beta-2b subunit of the same channel). The two defects correspond to the clinical syndromes short QT syndrome 4 and short QT syndrome 5, respectively.28
MECHANISM OF ARRHYTHMOGENESIS IN SHORT QT SYNDROME
The myocardium is made of different layers: the epicardium, the endocardium, and the middle layer of myocytes composed mainly of M cells. Cells in the different layers differ in the concentration of their channels and can be affected differently in various syndromes. When cells in one or two of the layers repolarize at a rate different from cells in another layer, they create different degrees of refractoriness, which establishes the potential for reentry circuits to form.
It is believed that in short QT syndrome the endocardial cells and M cells repolarize faster than the epicardial cells, predisposing to reentry and arrhythmias. This accentuation of “transmural dispersion of repolarization” accounts for arrhythmogenesis in short QT syndrome as well as in long QT syndrome and the Brugada syndromes. The difference between these syndromes appears to be the layer or area of the myocardium that is affected more by the channelopathy (the M cells in long QT syndrome and the epicardium of the right ventricle in the Brugada syndrome).29
WHEN TO THINK OF SHORT QT SYNDROME
In any survivor of sudden cardiac death, the QT interval should be thoroughly scrutinized, and family members should undergo ECG. Patients in whom a short QT interval is incidentally discovered and for which other reasons are ruled out (see differential diagnosis) should be encouraged to have family members undergo ECG. Other potential patients are young people who develop atrial fibrillation and patients who have idiopathic ventricular fibrillation.4
TREATMENT AND PROGNOSIS
Evidence-based recommendations for the management of short QT syndrome do not yet exist, mainly because the number of patients identified to date is small.
Implantable cardioverter-defibrillators
Although placing an implantable cardioverter-defibrillator (ICD) seems to be warranted in patients who experience ventricular fibrillation, ventricular tachycardia, or aborted cardiac death, or in patients who have a family history of the same symptoms, the best management option is less clear for patients who have no symptoms and no family history.30 In addition, some patients may not want an ICD or may even not qualify for this therapy.
A unique problem with ICDs in short QT syndrome stems from one of the syndrome’s main features on ECG: the tall and peaked T wave that closely follows the R wave can sometimes be interpreted as a short R-R interval, provoking an inappropriate shock from the ICD.31
For the above reasons, we strongly encourage consulting a center with expertise in QT-interval-related disorders before placing an ICD in a patient suspected of having short QT syndrome.
Antiarrhythmic drugs
Prolongation of the QT interval (and the effective refractory period) with drugs has been an interesting area of research. Gaita et al32 studied the effect of four antiarrhythmics—flecainide (Tambocor), sotalol (Betapace), ibutilide (Corvert), and quinidine—in six patients with short QT syndrome. Only quinidine was associated with significant QT prolongation, from 263 ± 12 ms to 362 ms ± 25 ms. This resulted in a longer ventricular effective refractory period (> 200 ms), and ventricular fibrillation was no longer inducible during provocative testing.
In a recent study of long-term outcomes of 53 patients with short QT syndrome, Giustetto et al33 noticed that none of the patients taking quinidine, including those with a history of cardiac arrest, had any further arrhythmsic events. On the other hand, the incidence of arrhythmic events during the follow-up was 4.9% per year in patients not taking this drug. Quinidine had a stronger effect on the QT interval in patients with the HERG mutation than in those without.
RESEARCH MAY LEAD TO A BETTER UNDERSTANDING OF OTHER DISEASES
The short QT syndrome is one of the most recently recognized cardiac channelopathies associated with malignant arrhythmias. As with long QT syndrome, research in short QT syndrome may lead to a better understanding of the pathogenesis of more common but still poorly understood arrhythmias such as lone atrial fibrillation and idiopathic ventricular fibrillation.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
- The Short QT Syndrome http://www.shortqtsyndrome.org/short_qt_history.htm. Accessed October 30, 2012.
- Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology 2000; 94:99–102.
- Giustetto C, Di Monte F, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J 2006; 27:2440–2447.
- Viskin S, Zeltser D, Ish-Shalom M, et al. Is idiopathic ventricular fibrillation a short QT syndrome? Comparison of QT intervals of patients with idiopathic ventricular fibrillation and healthy controls. Heart Rhythm 2004; 1:587–591.
- Anttonen O, Junttila MJ, Maury P, et al. Differences in twelve-lead electrocardiogram between symptomatic and asymptomatic subjects with short QT interval. Heart Rhythm 2009; 6:267–271.
- Redpath CJ, Green MS, Birnie DH, Gollob MH. Rapid genetic testing facilitating the diagnosis of short QT syndrome. Can J Cardiol 2009; 25:e133–e135.
- Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol 2005; 16:54–58.
- Anttonen O, Junttila MJ, Rissanen H, Reunanen A, Viitasalo M, Huikuri HV. Prevalence and prognostic significance of short QT interval in a middle-aged Finnish population. Circulation 2007; 116:714–720.
- Funada A, Hayashi K, Ino H, et al. Assessment of QT intervals and prevalence of short QT syndrome in Japan. Clin Cardiol 2008; 31:270–274.
- Mason JW, Ramseth DJ, Chanter DO, Moon TE, Goodman DB, Mendzelevski B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J Electrocardiol 2007; 40:228–234.
- Kobza R, Roos M, Niggli B, et al. Prevalence of long and short QT in a young population of 41,767 predominantly male Swiss conscripts. Heart Rhythm 2009; 6:652–657.
- Itoh H, Sakaguchi T, Ashihara T, et al. A novel KCNH2 mutation as a modifier for short QT interval. Int J Cardiol 2009; 137:83–85.
- Vincent GM, Timothy KW, Leppert M, Keating M. The spectrum of symptoms and QT intervals in carriers of the gene for the long-QT syndrome. N Engl J Med 1992; 327:846–852.
- Schwartz PJ. Idiopathic long QT syndrome: progress and questions. Am Heart J 1985; 109:399–411.
- Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88:782–784.
- Bjerregaard P, Nallapaneni H, Gussak I. Short QT interval in clinical practice. J Electrocardiol 2010; 43:390–395.
- Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome. Update on a recent entity. Arch Cardiovasc Dis 2008; 101:779–786.
- Kontny F, Dale J. Self-terminating idiopathic ventricular fibrillation presenting as syncope: a 40-year follow-up report. J Intern Med 1990; 227:211–213.
- Cheng TO. Digitalis administration: an underappreciated but common cause of short QT interval. Circulation 2004; 109:e152.
- Hancox JC, Choisy SC, James AF. Short QT interval linked to androgen misuse: wider significance and possible basis. Ann Noninvasive Electrocardiol 2009; 14:311–312.
- Naschitz J, Fields M, Isseroff H, Sharif D, Sabo E, Rosner I. Shortened QT interval: a distinctive feature of the dysautonomia of chronic fatigue syndrome. J Electrocardiol 2006; 39:389–394.
- Bjerregaard P, Collier JL, Gussak I. Upper limits of QT/QTc intervals in the short QT syndrome. Review of the world-wide short QT syndrome population and 3 new USA families. Heart Rhythm 2008; 5:AB43.
- Gollob MH, Redpath CJ, Roberts JD. The short QT syndrome: proposed diagnostic criteria. J Am Coll Cardiol 2011; 57:802–812.
- Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J 1994; 21:30–41.
- Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109:30–35.
- Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 2004; 109:2394–2397.
- Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res 2005; 96:800–807.
- Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115:442–449.
- Antzelevitch C. Heterogeneity and cardiac arrhythmias: an overview. Heart Rhythm 2007; 4:964–972.
- Lunati M, Bongiorni MG, Boriani G, et al. Linee guida AIAC 2006 all’impianto di pacemaker, dispositivi per la resincronizzazione cardiaca (CRT) e defibrillatori automatici impiantabili (ICD). GIAC 2005; 8:1–58.
- Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol 2003; 14:1273–1277.
- Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol 2004; 43:1494–1499.
- Giustetto C, Schimpf R, Mazzanti A, et al. Long-term follow-up of patients with short QT syndrome. J Am Coll Cardiol 2011; 58:587–595.
KEY POINTS
- Short QT syndrome is a genetic disease described initially in young patients who had atrial fibrillation or who died suddenly with no apparent structural heart disease.
- The diagnosis is established by the finding of a short QT interval. However, other factors including personal and family history are also important in establishing the diagnosis.
- The current recommendations for managing patients with short QT syndrome are not evidence-based. We encourage consultation with centers that have special interest in QT-interval-related disorders.
- Placement of an implantable cardioverter-defibrillator is considered the standard of care, especially in survivors of sudden cardiac death, ventricular fibrillation, or ventricular tachycardia. Unfortunately, a higher incidence of inappropriate shocks adds to the challenges of managing this potentially deadly disease.