User login
EMA says plasma/urine-derived meds are safe from Zika

Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Photo by Cristina Granados
Patients who take plasma-derived or urine-derived medicines do not have to worry about these products being contaminated with Zika virus, according to the European Medicines Agency (EMA).
The agency said assessments have confirmed that manufacturing processes for these medicines—which include coagulation factors, immunoglobulins, and urokinase products—successfully inactivate or remove the Zika virus.
These medicines are produced from body fluids that might be sourced in parts of the world where the Zika virus is prevalent. So regulators in the European Union (EU) sought reassurance that there is no risk of the virus contaminating the final product and thus affecting the patients taking these medicines.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) investigated the potential risk with plasma-derived medicinal products.
And the Co-ordination Group for Mutual Recognition and Decentralised Procedures—Human (CMDh) has coordinated the assessment by EU member states on the potential risk with urine-derived medicinal products.
The CHMP concluded at its meeting last week that the manufacturing processes used for plasma-derived products—including, for example, the solvent/detergent method to inactivate viruses, pasteurization, and virus filtration—inactivate or remove the Zika virus from the finished product.
The CHMP therefore concluded that no additional safety measures, such as the testing or exclusion of certain plasma donors, were necessary.
The CMDh, following an assessment of data, concluded that the manufacturing processes for urine-derived products contain complementary steps with inactivation/removal capacity for enveloped viruses, which are considered sufficient for eliminating Zika virus.
Additional safety measures, such as screening urine donors/donations or deferring donors returning from Zika-affected areas, are not considered necessary.
The findings from these assessments are available in a report from the CHMP’s Biologics Working Party.
The Biologics Working Party recommendation on plasma-derived products is in line with the guidance published in July 2016 by the European Centre for Disease Prevention and Control. ![]()
Gene therapy accepted into PRIME program
![]()
Photo by Chad McNeeley
An investigational gene therapy known as LentiGlobin BB305 has been accepted into the European Medicines Agency’s (EMA’s) Priority Medicines (PRIME) program as a treatment for patients with transfusion-dependent beta-thalassemia (TDT).
LentiGlobin BB305 is created by inserting a functional human beta-globin gene into a patient’s hematopoietic stem cells ex vivo. The cells are then returned to the patient via transplant.
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs. The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate potential to benefit patients with unmet medical need through early clinical or nonclinical data.
Bluebird bio, the company developing LentiGlobin BB305, is also participating in the EMA’s Adaptive Pathways pilot program.
Like PRIME, the Adaptive Pathways program aims to expedite patient access to therapies with the potential to treat serious conditions with unmet need. It uses the existing European Union (EU) regulatory framework for medicines, including conditional approval.
“PRIME designation will allow bluebird bio to further improve our communication with European regulators as we continue to refine our evidence generation plan in the context of adaptive biomedical innovation,” said David Davidson, MD, chief medical officer of bluebird bio.
“Overall, we believe this will enable us to accelerate development of LentiGlobin drug product for patients with transfusion-dependent beta-thalassemia, a life-shortening disease with significant unmet medical need.”
“Earlier this year, we completed enrollment in the Northstar (HGB-204) global clinical study of LentiGlobin drug product in patients with TDT, which, along with the supporting HGB-205 study, will form the basis of our eventual application for conditional approval in the EU under the Adaptive Pathways pilot program.” ![]()
![]()
Photo by Chad McNeeley
An investigational gene therapy known as LentiGlobin BB305 has been accepted into the European Medicines Agency’s (EMA’s) Priority Medicines (PRIME) program as a treatment for patients with transfusion-dependent beta-thalassemia (TDT).
LentiGlobin BB305 is created by inserting a functional human beta-globin gene into a patient’s hematopoietic stem cells ex vivo. The cells are then returned to the patient via transplant.
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs. The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate potential to benefit patients with unmet medical need through early clinical or nonclinical data.
Bluebird bio, the company developing LentiGlobin BB305, is also participating in the EMA’s Adaptive Pathways pilot program.
Like PRIME, the Adaptive Pathways program aims to expedite patient access to therapies with the potential to treat serious conditions with unmet need. It uses the existing European Union (EU) regulatory framework for medicines, including conditional approval.
“PRIME designation will allow bluebird bio to further improve our communication with European regulators as we continue to refine our evidence generation plan in the context of adaptive biomedical innovation,” said David Davidson, MD, chief medical officer of bluebird bio.
“Overall, we believe this will enable us to accelerate development of LentiGlobin drug product for patients with transfusion-dependent beta-thalassemia, a life-shortening disease with significant unmet medical need.”
“Earlier this year, we completed enrollment in the Northstar (HGB-204) global clinical study of LentiGlobin drug product in patients with TDT, which, along with the supporting HGB-205 study, will form the basis of our eventual application for conditional approval in the EU under the Adaptive Pathways pilot program.” ![]()
![]()
Photo by Chad McNeeley
An investigational gene therapy known as LentiGlobin BB305 has been accepted into the European Medicines Agency’s (EMA’s) Priority Medicines (PRIME) program as a treatment for patients with transfusion-dependent beta-thalassemia (TDT).
LentiGlobin BB305 is created by inserting a functional human beta-globin gene into a patient’s hematopoietic stem cells ex vivo. The cells are then returned to the patient via transplant.
The goal of the EMA’s PRIME program is to accelerate the development of therapies that target unmet medical needs. The program provides enhanced EMA support and increased interaction to developers, in order to optimize development plans and speed regulatory evaluations to potentially bring these therapies to patients more quickly.
To be accepted for PRIME, a therapy must demonstrate potential to benefit patients with unmet medical need through early clinical or nonclinical data.
Bluebird bio, the company developing LentiGlobin BB305, is also participating in the EMA’s Adaptive Pathways pilot program.
Like PRIME, the Adaptive Pathways program aims to expedite patient access to therapies with the potential to treat serious conditions with unmet need. It uses the existing European Union (EU) regulatory framework for medicines, including conditional approval.
“PRIME designation will allow bluebird bio to further improve our communication with European regulators as we continue to refine our evidence generation plan in the context of adaptive biomedical innovation,” said David Davidson, MD, chief medical officer of bluebird bio.
“Overall, we believe this will enable us to accelerate development of LentiGlobin drug product for patients with transfusion-dependent beta-thalassemia, a life-shortening disease with significant unmet medical need.”
“Earlier this year, we completed enrollment in the Northstar (HGB-204) global clinical study of LentiGlobin drug product in patients with TDT, which, along with the supporting HGB-205 study, will form the basis of our eventual application for conditional approval in the EU under the Adaptive Pathways pilot program.” ![]()
Compound could treat resistant malaria
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above). ![]()
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above). ![]()
of the P falciparum parasite
Image by Mae Melvin/CDC
A novel compound could be effective against malaria infections that are resistant to currently available antimalarial drugs, according to researchers.
In a phase 2 study, the compound, KAF156, proved active against Plasmodium vivax and Plasmodium falciparum malaria.
KAF156 was able to clear both blood and liver stages of malaria parasites, including artemisinin-resistant parasites.
Most of the patients in this study had at least 1 adverse event (AE). However, most were grade 1, and there were no grade 4 or serious AEs.
Results from this study were published in NEJM. The trial was funded, in part, by Novartis, the company developing KAF156.
KAF156 is the first compound from a novel class of drugs called imidazolopiperazines. The drugs’ mechanism of action is still being characterized, but it may be related to a previously uncharacterized gene—Plasmodium falciparum cyclic amine resistance locus (Pfcarl).
From March to August 2013, researchers conducted a phase 2 trial of KAF156 at 5 centers in Thailand and Vietnam.
Twenty-one of the patients in this study had acute, uncomplicated malaria—11 with P vivax and 10 with P falciparum malaria. They received multiple doses of KAF156—400 mg once daily for 3 days.
Twenty-two of the patients studied had uncomplicated, P falciparum malaria and received a single dose of KAF156 at 800 mg.
All patients were assessed for fever and parasite clearance as well as AEs. Patients in the single-dose cohort were also followed for 28 days to assess the cure rate.
Two patients were excluded from the efficacy analysis. One was a P vivax patient receiving the 400 mg dose who turned out to have a mixed infection. And the other was a P falciparum patient who vomited repeatedly after receiving 800 mg of KAF156 (after 3 attempts at dosing).
Efficacy
In the multiple-dose cohorts, the median time to fever clearance after receiving KAF156 was 14 hours (range, 4 to 30) in patients with P vivax malaria and 6 hours (range, 4 to 24) in patients with P falciparum malaria. In the single-dose cohort, the median time to fever clearance was 4 hours (range, 4 to 66).
In the multiple-dose cohorts, the median time to parasite clearance was 24 hours (range, 16 to 36) in patients with P vivax and 45 hours (range, 36 to 66) in patients with P falciparum.
In the single-dose cohort, the median time to parasite clearance was 49 hours (range, 16 to 68). One of these patients had parasite clearance at 66 hours but an asymptomatic recurrence at 84 hours. In all of the other patients, parasitemia cleared.
During follow-up of the single-dose cohort, 1 patient had reinfection, and 7 had recrudescent infections.
Safety
Most patients had at least 1 AE, although none were serious. Seventy-two percent of patients had grade 1 AEs, 35% had grade 2 AEs, and 14% had grade 3 AEs.
Sixty percent of AEs were considered related to treatment. In the multiple-dose cohorts, 14 of the 31 AEs (45%) reported in 7 patients (1 with P vivax and 6 with P falciparum malaria) were considered drug-related. All 22 P falciparum patients in the single-dose cohort had at least 1 AE that was considered drug-related.
The most common AEs were sinus bradycardia, thrombocytopenia, hypokalemia, anemia, and hyperbilirubinemia.
Two patients experienced vomiting of grade 2 or higher. One of these patients discontinued treatment because of repeated vomiting after the 800 mg dose (mentioned above). ![]()
Statins linked to lower risk of death in MM

Photo courtesy of CDC
Results of a retrospective study suggest statins may decrease the risk of death in patients with multiple myeloma (MM).
Researchers analyzed data from nearly 5000 patients with MM and found that patients who took statins had a significant reduction in all-cause mortality and MM-specific mortality when compared to patients who did not take these drugs.
Kristen Marie Sanfilippo, MD, of Washington University School of Medicine in St Louis, Missouri, and her colleagues reported these findings in the Journal of Clinical Oncology.
The researchers analyzed data from the Veterans Administration Central Cancer Registry and identified 4957 patients who were diagnosed with MM between 1999 and 2013.
Of these patients, 2294 were classified as statin users. The researchers defined statin use as the presence of any prescription for a statin within 3 months before MM diagnosis or any time thereafter.
The data showed that statin users had a longer median survival than non-users—39.5 months and 27 months, respectively.
When the researchers adjusted for potential confounders, they found that statin users had a 21% reduction in the risk of all-cause mortality (adjusted hazard ratio [aHR]=0.79, P<0.001) and a 24% reduction in the risk of MM-specific mortality (aHR=0.76, P<0.001).
In addition, statin users had a 31% reduction in the risk of developing a skeletal-related event (aHR=0.69, P<0.001).
In a 12-month landmark analysis, statin use was associated with a significant reduction in the risk of all-cause mortality (aHR=0.86, P=0.001) and MM-specific mortality (aHR=0.83, P=0.01).
The reduction in all-cause mortality was 12% (P=0.004) for patients taking statins for at least 3 months, 16% (P<0.001) for patients taking statins for at least 6 months, and 18% (P=0.003) for patients taking statins for at least 9 months.
Patients with less than 365 daily defined doses (DDDs) of statins had a 20% reduction in the risk of all-cause mortality (aHR=0.80, P<0.001). And patients with ≥ 365 DDDs had a 22% reduction in the risk of all-cause mortality (aHR=0.78, P<0.001).
The reductions in MM-specific mortality according to DDDs were 22% (aHR=0.78, P=0.001) and 28% (aHR=0.72, P<0.001), respectively.
The researchers further adjusted for baseline differences between statin users and non-users with propensity-score matching. And statin use was still associated with a reduction in all-cause mortality (aHR=0.78, P<0.001) and MM-specific mortality (aHR=0.79, P=0.007).
The researchers said these results suggest a potential role for statin therapy in patients with MM, although the findings should be corroborated in prospective studies. ![]()
Photo courtesy of CDC
Results of a retrospective study suggest statins may decrease the risk of death in patients with multiple myeloma (MM).
Researchers analyzed data from nearly 5000 patients with MM and found that patients who took statins had a significant reduction in all-cause mortality and MM-specific mortality when compared to patients who did not take these drugs.
Kristen Marie Sanfilippo, MD, of Washington University School of Medicine in St Louis, Missouri, and her colleagues reported these findings in the Journal of Clinical Oncology.
The researchers analyzed data from the Veterans Administration Central Cancer Registry and identified 4957 patients who were diagnosed with MM between 1999 and 2013.
Of these patients, 2294 were classified as statin users. The researchers defined statin use as the presence of any prescription for a statin within 3 months before MM diagnosis or any time thereafter.
The data showed that statin users had a longer median survival than non-users—39.5 months and 27 months, respectively.
When the researchers adjusted for potential confounders, they found that statin users had a 21% reduction in the risk of all-cause mortality (adjusted hazard ratio [aHR]=0.79, P<0.001) and a 24% reduction in the risk of MM-specific mortality (aHR=0.76, P<0.001).
In addition, statin users had a 31% reduction in the risk of developing a skeletal-related event (aHR=0.69, P<0.001).
In a 12-month landmark analysis, statin use was associated with a significant reduction in the risk of all-cause mortality (aHR=0.86, P=0.001) and MM-specific mortality (aHR=0.83, P=0.01).
The reduction in all-cause mortality was 12% (P=0.004) for patients taking statins for at least 3 months, 16% (P<0.001) for patients taking statins for at least 6 months, and 18% (P=0.003) for patients taking statins for at least 9 months.
Patients with less than 365 daily defined doses (DDDs) of statins had a 20% reduction in the risk of all-cause mortality (aHR=0.80, P<0.001). And patients with ≥ 365 DDDs had a 22% reduction in the risk of all-cause mortality (aHR=0.78, P<0.001).
The reductions in MM-specific mortality according to DDDs were 22% (aHR=0.78, P=0.001) and 28% (aHR=0.72, P<0.001), respectively.
The researchers further adjusted for baseline differences between statin users and non-users with propensity-score matching. And statin use was still associated with a reduction in all-cause mortality (aHR=0.78, P<0.001) and MM-specific mortality (aHR=0.79, P=0.007).
The researchers said these results suggest a potential role for statin therapy in patients with MM, although the findings should be corroborated in prospective studies. ![]()
Photo courtesy of CDC
Results of a retrospective study suggest statins may decrease the risk of death in patients with multiple myeloma (MM).
Researchers analyzed data from nearly 5000 patients with MM and found that patients who took statins had a significant reduction in all-cause mortality and MM-specific mortality when compared to patients who did not take these drugs.
Kristen Marie Sanfilippo, MD, of Washington University School of Medicine in St Louis, Missouri, and her colleagues reported these findings in the Journal of Clinical Oncology.
The researchers analyzed data from the Veterans Administration Central Cancer Registry and identified 4957 patients who were diagnosed with MM between 1999 and 2013.
Of these patients, 2294 were classified as statin users. The researchers defined statin use as the presence of any prescription for a statin within 3 months before MM diagnosis or any time thereafter.
The data showed that statin users had a longer median survival than non-users—39.5 months and 27 months, respectively.
When the researchers adjusted for potential confounders, they found that statin users had a 21% reduction in the risk of all-cause mortality (adjusted hazard ratio [aHR]=0.79, P<0.001) and a 24% reduction in the risk of MM-specific mortality (aHR=0.76, P<0.001).
In addition, statin users had a 31% reduction in the risk of developing a skeletal-related event (aHR=0.69, P<0.001).
In a 12-month landmark analysis, statin use was associated with a significant reduction in the risk of all-cause mortality (aHR=0.86, P=0.001) and MM-specific mortality (aHR=0.83, P=0.01).
The reduction in all-cause mortality was 12% (P=0.004) for patients taking statins for at least 3 months, 16% (P<0.001) for patients taking statins for at least 6 months, and 18% (P=0.003) for patients taking statins for at least 9 months.
Patients with less than 365 daily defined doses (DDDs) of statins had a 20% reduction in the risk of all-cause mortality (aHR=0.80, P<0.001). And patients with ≥ 365 DDDs had a 22% reduction in the risk of all-cause mortality (aHR=0.78, P<0.001).
The reductions in MM-specific mortality according to DDDs were 22% (aHR=0.78, P=0.001) and 28% (aHR=0.72, P<0.001), respectively.
The researchers further adjusted for baseline differences between statin users and non-users with propensity-score matching. And statin use was still associated with a reduction in all-cause mortality (aHR=0.78, P<0.001) and MM-specific mortality (aHR=0.79, P=0.007).
The researchers said these results suggest a potential role for statin therapy in patients with MM, although the findings should be corroborated in prospective studies. ![]()
Cancer report details progress, predicts problems

Photo by Rhoda Baer
A new report highlights recent advances made in the fight against cancer but suggests the burden of cancer in the US is still on the rise.
The AACR Cancer Progress Report 2016 states that the number of cancer survivors in the US rose by 1 million from 2014 to 2016, reaching an estimated 15.5 million.
Meanwhile, the US Food and Drug Administration (FDA) approved new treatments for a range of cancers.
Between August 1, 2015, and July 31, 2016, the FDA approved 13 new anticancer therapies and new uses for 11 previously approved anticancer therapies.
Six of these drugs were approved to treat hematologic malignancies:
- Venetoclax for chronic lymphocytic leukemia
- Daratumumab for multiple myeloma
- Elotuzumab for multiple myeloma
- Ixazomib for multiple myeloma
- Obinutuzumab for follicular lymphoma
- Nivolumab for classical Hodgkin lymphoma.
The report notes that the use of immunotherapy, in particular, is on the rise. For example, on August 1, 2015, checkpoint inhibitors were approved for just 2 cancers—melanoma and lung cancer.
As of September 1, 2016, checkpoint inhibitors have been approved for 6 cancers—Hodgkin lymphoma, bladder cancer, head and neck cancer, kidney cancer, lung cancer, and melanoma.
“The promise of immunotherapy for cancer therapy has never been greater, and the opportunity to make significant progress in this critical area is real,” said Nancy E. Davidson, MD, president of the AACR and director of the University of Pittsburgh Cancer Institute in Pennsylvania.
“However, continued progress is going to require a sustained federal commitment to the research agenda. And in fact, if the necessary funding is provided, we will accelerate the pace of progress and, in turn, markedly reduce morbidity and mortality from cancer.”
Growing burden of cancer
The report emphasizes that although advances are being made against cancers, these diseases continue to exert an immense personal and economic toll, and the burden of cancer is expected to grow in the coming decades.
According to the report:
- More than 595,000 people in the US are projected to die from cancer in 2016
- Cancer is the number one cause of disease-related death among US children
- The number of new cancer cases in the US is predicted to rise from 1.7 million in 2015 to 2.4 million in 2035
- It is estimated that the direct medical costs of cancer care in the US in 2010 were nearly $125 billion, and these costs will rise to $156 billion in 2020.
The report states that the increasing economic and personal burden of cancer underscores the need for more research to develop new approaches to cancer prevention and treatment.
The report also highlights the recommendations of the National Cancer Moonshot Initiative Blue Ribbon Panel for accelerating the pace of progress in cancer research.
“Research has made tremendous advances against cancer,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
“However, we need to accelerate the pace of progress because it is unacceptable that 1 American will die of cancer every minute of every day this year.” ![]()
Photo by Rhoda Baer
A new report highlights recent advances made in the fight against cancer but suggests the burden of cancer in the US is still on the rise.
The AACR Cancer Progress Report 2016 states that the number of cancer survivors in the US rose by 1 million from 2014 to 2016, reaching an estimated 15.5 million.
Meanwhile, the US Food and Drug Administration (FDA) approved new treatments for a range of cancers.
Between August 1, 2015, and July 31, 2016, the FDA approved 13 new anticancer therapies and new uses for 11 previously approved anticancer therapies.
Six of these drugs were approved to treat hematologic malignancies:
- Venetoclax for chronic lymphocytic leukemia
- Daratumumab for multiple myeloma
- Elotuzumab for multiple myeloma
- Ixazomib for multiple myeloma
- Obinutuzumab for follicular lymphoma
- Nivolumab for classical Hodgkin lymphoma.
The report notes that the use of immunotherapy, in particular, is on the rise. For example, on August 1, 2015, checkpoint inhibitors were approved for just 2 cancers—melanoma and lung cancer.
As of September 1, 2016, checkpoint inhibitors have been approved for 6 cancers—Hodgkin lymphoma, bladder cancer, head and neck cancer, kidney cancer, lung cancer, and melanoma.
“The promise of immunotherapy for cancer therapy has never been greater, and the opportunity to make significant progress in this critical area is real,” said Nancy E. Davidson, MD, president of the AACR and director of the University of Pittsburgh Cancer Institute in Pennsylvania.
“However, continued progress is going to require a sustained federal commitment to the research agenda. And in fact, if the necessary funding is provided, we will accelerate the pace of progress and, in turn, markedly reduce morbidity and mortality from cancer.”
Growing burden of cancer
The report emphasizes that although advances are being made against cancers, these diseases continue to exert an immense personal and economic toll, and the burden of cancer is expected to grow in the coming decades.
According to the report:
- More than 595,000 people in the US are projected to die from cancer in 2016
- Cancer is the number one cause of disease-related death among US children
- The number of new cancer cases in the US is predicted to rise from 1.7 million in 2015 to 2.4 million in 2035
- It is estimated that the direct medical costs of cancer care in the US in 2010 were nearly $125 billion, and these costs will rise to $156 billion in 2020.
The report states that the increasing economic and personal burden of cancer underscores the need for more research to develop new approaches to cancer prevention and treatment.
The report also highlights the recommendations of the National Cancer Moonshot Initiative Blue Ribbon Panel for accelerating the pace of progress in cancer research.
“Research has made tremendous advances against cancer,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
“However, we need to accelerate the pace of progress because it is unacceptable that 1 American will die of cancer every minute of every day this year.” ![]()
Photo by Rhoda Baer
A new report highlights recent advances made in the fight against cancer but suggests the burden of cancer in the US is still on the rise.
The AACR Cancer Progress Report 2016 states that the number of cancer survivors in the US rose by 1 million from 2014 to 2016, reaching an estimated 15.5 million.
Meanwhile, the US Food and Drug Administration (FDA) approved new treatments for a range of cancers.
Between August 1, 2015, and July 31, 2016, the FDA approved 13 new anticancer therapies and new uses for 11 previously approved anticancer therapies.
Six of these drugs were approved to treat hematologic malignancies:
- Venetoclax for chronic lymphocytic leukemia
- Daratumumab for multiple myeloma
- Elotuzumab for multiple myeloma
- Ixazomib for multiple myeloma
- Obinutuzumab for follicular lymphoma
- Nivolumab for classical Hodgkin lymphoma.
The report notes that the use of immunotherapy, in particular, is on the rise. For example, on August 1, 2015, checkpoint inhibitors were approved for just 2 cancers—melanoma and lung cancer.
As of September 1, 2016, checkpoint inhibitors have been approved for 6 cancers—Hodgkin lymphoma, bladder cancer, head and neck cancer, kidney cancer, lung cancer, and melanoma.
“The promise of immunotherapy for cancer therapy has never been greater, and the opportunity to make significant progress in this critical area is real,” said Nancy E. Davidson, MD, president of the AACR and director of the University of Pittsburgh Cancer Institute in Pennsylvania.
“However, continued progress is going to require a sustained federal commitment to the research agenda. And in fact, if the necessary funding is provided, we will accelerate the pace of progress and, in turn, markedly reduce morbidity and mortality from cancer.”
Growing burden of cancer
The report emphasizes that although advances are being made against cancers, these diseases continue to exert an immense personal and economic toll, and the burden of cancer is expected to grow in the coming decades.
According to the report:
- More than 595,000 people in the US are projected to die from cancer in 2016
- Cancer is the number one cause of disease-related death among US children
- The number of new cancer cases in the US is predicted to rise from 1.7 million in 2015 to 2.4 million in 2035
- It is estimated that the direct medical costs of cancer care in the US in 2010 were nearly $125 billion, and these costs will rise to $156 billion in 2020.
The report states that the increasing economic and personal burden of cancer underscores the need for more research to develop new approaches to cancer prevention and treatment.
The report also highlights the recommendations of the National Cancer Moonshot Initiative Blue Ribbon Panel for accelerating the pace of progress in cancer research.
“Research has made tremendous advances against cancer,” said Margaret Foti, PhD, MD, chief executive officer of the AACR.
“However, we need to accelerate the pace of progress because it is unacceptable that 1 American will die of cancer every minute of every day this year.” ![]()
Dying cancer patients may be under-treated for pain

Photo courtesy of CDC
New research suggests that many patients who die of cancer do not receive strong opioid medications in their last year of life, despite the fact that these drugs are the recommended treatment for cancer-related pain.
Researchers used UK Cancer Registry Data to study more than 6000 cancer patients who died over a 7-year period.
Less than half of these patients received prescriptions for strong opioid medications in their last year of life.
Among those patients who did receive such prescriptions, many received them late.
Lucy Ziegler, PhD, of the University of Leeds in the UK, and her colleagues conducted this study and reported the results in PAIN.
The study included 6080 cancer patients who died between 2005 and 2012.
About 76% (n=4610) of these patients had received one or more prescriptions for analgesics, including 48% (n=2919) who received a strong opioid and 28% (n=1691) who received a non-opioid or weak opioid. The remaining 24% (n=1470) did not receive any prescription analgesic.
The chance of receiving strong opioids was not affected by patients’ age or sex.
However, patients who died in a hospital were 60% less likely to have a prescription for strong opioids during the last year of life, when compared with those who died in hospice.
And patients who received chemotherapy in the last year of life were 30% more likely to receive a strong opioid than patients who did not receive chemotherapy.
Timing of therapy
Among the patients who did receive strong opioids, the median time between receiving the medication and death was 9 weeks. By 6 weeks before death, just 30% of patients had been prescribed a strong opioid.
The researchers noted that these figures don’t match up with previous studies reporting that severe pain can occur “much earlier in the cancer trajectory.”
Older patients were more likely to receive their strong opioid prescription late (defined as later than 9 weeks before death). After other factors were taken into account, patients age 60 or older were about 2 to 4 times more likely to be in the late-prescribing group, compared with those age 50 or younger.
Compared to patients who died in hospice, patients who died in a hospital were 40% more likely to receive a late prescription for a strong opioid. Patients who died in their own home were 2.6 times more likely to receive a late prescription, and patients who died in a care home were 2.8 times more likely to receive a late prescription.
Patients who had surgery were 40% more likely to receive a late prescription than patients who did not undergo surgery.
But patients who received chemotherapy and/or radiotherapy were 30% more likely to have received an early prescription for a strong opioid than patients who did not receive chemo/radiotherapy.
Dr Ziegler and her colleagues noted that this study had its limitations; in particular, the lack of data on pain severity.
Still, the researchers believe their results support the hypothesis of potential under-treatment of cancer pain and suggest that many more patients with advanced cancer and pain may benefit from a strong opioid analgesic. ![]()
Photo courtesy of CDC
New research suggests that many patients who die of cancer do not receive strong opioid medications in their last year of life, despite the fact that these drugs are the recommended treatment for cancer-related pain.
Researchers used UK Cancer Registry Data to study more than 6000 cancer patients who died over a 7-year period.
Less than half of these patients received prescriptions for strong opioid medications in their last year of life.
Among those patients who did receive such prescriptions, many received them late.
Lucy Ziegler, PhD, of the University of Leeds in the UK, and her colleagues conducted this study and reported the results in PAIN.
The study included 6080 cancer patients who died between 2005 and 2012.
About 76% (n=4610) of these patients had received one or more prescriptions for analgesics, including 48% (n=2919) who received a strong opioid and 28% (n=1691) who received a non-opioid or weak opioid. The remaining 24% (n=1470) did not receive any prescription analgesic.
The chance of receiving strong opioids was not affected by patients’ age or sex.
However, patients who died in a hospital were 60% less likely to have a prescription for strong opioids during the last year of life, when compared with those who died in hospice.
And patients who received chemotherapy in the last year of life were 30% more likely to receive a strong opioid than patients who did not receive chemotherapy.
Timing of therapy
Among the patients who did receive strong opioids, the median time between receiving the medication and death was 9 weeks. By 6 weeks before death, just 30% of patients had been prescribed a strong opioid.
The researchers noted that these figures don’t match up with previous studies reporting that severe pain can occur “much earlier in the cancer trajectory.”
Older patients were more likely to receive their strong opioid prescription late (defined as later than 9 weeks before death). After other factors were taken into account, patients age 60 or older were about 2 to 4 times more likely to be in the late-prescribing group, compared with those age 50 or younger.
Compared to patients who died in hospice, patients who died in a hospital were 40% more likely to receive a late prescription for a strong opioid. Patients who died in their own home were 2.6 times more likely to receive a late prescription, and patients who died in a care home were 2.8 times more likely to receive a late prescription.
Patients who had surgery were 40% more likely to receive a late prescription than patients who did not undergo surgery.
But patients who received chemotherapy and/or radiotherapy were 30% more likely to have received an early prescription for a strong opioid than patients who did not receive chemo/radiotherapy.
Dr Ziegler and her colleagues noted that this study had its limitations; in particular, the lack of data on pain severity.
Still, the researchers believe their results support the hypothesis of potential under-treatment of cancer pain and suggest that many more patients with advanced cancer and pain may benefit from a strong opioid analgesic. ![]()
Photo courtesy of CDC
New research suggests that many patients who die of cancer do not receive strong opioid medications in their last year of life, despite the fact that these drugs are the recommended treatment for cancer-related pain.
Researchers used UK Cancer Registry Data to study more than 6000 cancer patients who died over a 7-year period.
Less than half of these patients received prescriptions for strong opioid medications in their last year of life.
Among those patients who did receive such prescriptions, many received them late.
Lucy Ziegler, PhD, of the University of Leeds in the UK, and her colleagues conducted this study and reported the results in PAIN.
The study included 6080 cancer patients who died between 2005 and 2012.
About 76% (n=4610) of these patients had received one or more prescriptions for analgesics, including 48% (n=2919) who received a strong opioid and 28% (n=1691) who received a non-opioid or weak opioid. The remaining 24% (n=1470) did not receive any prescription analgesic.
The chance of receiving strong opioids was not affected by patients’ age or sex.
However, patients who died in a hospital were 60% less likely to have a prescription for strong opioids during the last year of life, when compared with those who died in hospice.
And patients who received chemotherapy in the last year of life were 30% more likely to receive a strong opioid than patients who did not receive chemotherapy.
Timing of therapy
Among the patients who did receive strong opioids, the median time between receiving the medication and death was 9 weeks. By 6 weeks before death, just 30% of patients had been prescribed a strong opioid.
The researchers noted that these figures don’t match up with previous studies reporting that severe pain can occur “much earlier in the cancer trajectory.”
Older patients were more likely to receive their strong opioid prescription late (defined as later than 9 weeks before death). After other factors were taken into account, patients age 60 or older were about 2 to 4 times more likely to be in the late-prescribing group, compared with those age 50 or younger.
Compared to patients who died in hospice, patients who died in a hospital were 40% more likely to receive a late prescription for a strong opioid. Patients who died in their own home were 2.6 times more likely to receive a late prescription, and patients who died in a care home were 2.8 times more likely to receive a late prescription.
Patients who had surgery were 40% more likely to receive a late prescription than patients who did not undergo surgery.
But patients who received chemotherapy and/or radiotherapy were 30% more likely to have received an early prescription for a strong opioid than patients who did not receive chemo/radiotherapy.
Dr Ziegler and her colleagues noted that this study had its limitations; in particular, the lack of data on pain severity.
Still, the researchers believe their results support the hypothesis of potential under-treatment of cancer pain and suggest that many more patients with advanced cancer and pain may benefit from a strong opioid analgesic. ![]()
Adding rituximab to chemo may improve EFS in ALL

Image by Vashi Donsk
Adding rituximab to a chemotherapy regimen can improve event-free survival (EFS) in adults with newly diagnosed, CD20-positive acute lymphoblastic leukemia (ALL), according to the GRAALL-2005/R study.
The 2-year EFS was significantly higher for patients who received rituximab than for those who received chemotherapy alone.
However, there was no significant difference between the groups in 2-year overall survival.
Sébastien Maury, MD, PhD, of Hȏpital Hénri Mondor in Creteil, France, and his colleagues reported these results in NEJM. Results from this study were previously presented at the 2015 ASH Annual Meeting.
The study included 209 patients with newly diagnosed, Ph-negative, B-cell precursor ALL with 20% or more CD20-positive leukemic blasts.
The patients’ median age was 40.2 (range, 24.5–52.6), 13% had an ECOG performance status greater than 1, 6% had CNS involvement, and 21% had a white blood cell count of 30 x 109/L or higher.
Half of the patients (n=104) were randomized to receive the GRAALL-2005 regimen, and the other half (n=105) were randomized to receive the same regimen plus rituximab. Details on the regimens are available in the supplementary material published with the NEJM paper.
Baseline patient characteristics were well-balanced between the treatment groups.
Results
At a median follow-up of 30 months, 101 patients (48%) had at least 1 event, including 44 (42%) in the rituximab group and 57 (55%) in the control group.
There were 17 induction failures (8 in the rituximab group and 9 in the control group), 57 relapses (22 and 35, respectively), and 27 deaths during remission (14 and 13, respectively). Two patients in the rituximab group were lost to follow-up during the first 12 months.
The 2-year EFS was significantly higher in the rituximab group than the control group—65% and 52%, respectively (hazard ratio [HR]=0.66, P=0.04).
However, the EFS benefit did not translate into a significant improvement in overall survival. The 2-year overall survival was 71% in the rituximab group and 64% in the control group (HR=0.70, P=0.10).
Similarly, the cumulative incidence of death during first remission was not significantly different between the treatment groups—12% for both (HR=0.98, P=0.96).
The researchers said the difference in EFS was mostly due to a lower incidence of relapse in the rituximab group. The 2-year incidence of relapse was 18% in the rituximab group and 32% in the control group (HR=0.52, P=0.02).
In a multivariate analysis, receiving the control treatment, older age, higher white blood cell count at baseline, and CNS involvement were all significantly associated with poor EFS.
There were 245 severe adverse events (AEs) reported in 124 patients—67 patients with 1 event, 26 with 2 events, 13 with 3 events, and 18 with 4 or more events.
The overall incidence of severe AEs did not differ significantly between the treatment groups—96% in the rituximab group and 92% in the control group.
Severe AEs included infection, laboratory abnormalities, allergic reactions, neurologic events, pulmonary events, coagulopathy, cardiac events, gastrointestinal events, and “other” events.
The only severe AE for which there was a significant difference between the treatment groups was allergic reactions. There were 2 severe allergic events in the rituximab group and 14 in the control group (P=0.002). Of these 16 events, all but 1 were due to asparaginase.
The researchers therefore theorized that rituximab might inhibit B-cell protection of antibodies against asparaginase, although they could not confirm this hypothesis. ![]()
Image by Vashi Donsk
Adding rituximab to a chemotherapy regimen can improve event-free survival (EFS) in adults with newly diagnosed, CD20-positive acute lymphoblastic leukemia (ALL), according to the GRAALL-2005/R study.
The 2-year EFS was significantly higher for patients who received rituximab than for those who received chemotherapy alone.
However, there was no significant difference between the groups in 2-year overall survival.
Sébastien Maury, MD, PhD, of Hȏpital Hénri Mondor in Creteil, France, and his colleagues reported these results in NEJM. Results from this study were previously presented at the 2015 ASH Annual Meeting.
The study included 209 patients with newly diagnosed, Ph-negative, B-cell precursor ALL with 20% or more CD20-positive leukemic blasts.
The patients’ median age was 40.2 (range, 24.5–52.6), 13% had an ECOG performance status greater than 1, 6% had CNS involvement, and 21% had a white blood cell count of 30 x 109/L or higher.
Half of the patients (n=104) were randomized to receive the GRAALL-2005 regimen, and the other half (n=105) were randomized to receive the same regimen plus rituximab. Details on the regimens are available in the supplementary material published with the NEJM paper.
Baseline patient characteristics were well-balanced between the treatment groups.
Results
At a median follow-up of 30 months, 101 patients (48%) had at least 1 event, including 44 (42%) in the rituximab group and 57 (55%) in the control group.
There were 17 induction failures (8 in the rituximab group and 9 in the control group), 57 relapses (22 and 35, respectively), and 27 deaths during remission (14 and 13, respectively). Two patients in the rituximab group were lost to follow-up during the first 12 months.
The 2-year EFS was significantly higher in the rituximab group than the control group—65% and 52%, respectively (hazard ratio [HR]=0.66, P=0.04).
However, the EFS benefit did not translate into a significant improvement in overall survival. The 2-year overall survival was 71% in the rituximab group and 64% in the control group (HR=0.70, P=0.10).
Similarly, the cumulative incidence of death during first remission was not significantly different between the treatment groups—12% for both (HR=0.98, P=0.96).
The researchers said the difference in EFS was mostly due to a lower incidence of relapse in the rituximab group. The 2-year incidence of relapse was 18% in the rituximab group and 32% in the control group (HR=0.52, P=0.02).
In a multivariate analysis, receiving the control treatment, older age, higher white blood cell count at baseline, and CNS involvement were all significantly associated with poor EFS.
There were 245 severe adverse events (AEs) reported in 124 patients—67 patients with 1 event, 26 with 2 events, 13 with 3 events, and 18 with 4 or more events.
The overall incidence of severe AEs did not differ significantly between the treatment groups—96% in the rituximab group and 92% in the control group.
Severe AEs included infection, laboratory abnormalities, allergic reactions, neurologic events, pulmonary events, coagulopathy, cardiac events, gastrointestinal events, and “other” events.
The only severe AE for which there was a significant difference between the treatment groups was allergic reactions. There were 2 severe allergic events in the rituximab group and 14 in the control group (P=0.002). Of these 16 events, all but 1 were due to asparaginase.
The researchers therefore theorized that rituximab might inhibit B-cell protection of antibodies against asparaginase, although they could not confirm this hypothesis. ![]()
Image by Vashi Donsk
Adding rituximab to a chemotherapy regimen can improve event-free survival (EFS) in adults with newly diagnosed, CD20-positive acute lymphoblastic leukemia (ALL), according to the GRAALL-2005/R study.
The 2-year EFS was significantly higher for patients who received rituximab than for those who received chemotherapy alone.
However, there was no significant difference between the groups in 2-year overall survival.
Sébastien Maury, MD, PhD, of Hȏpital Hénri Mondor in Creteil, France, and his colleagues reported these results in NEJM. Results from this study were previously presented at the 2015 ASH Annual Meeting.
The study included 209 patients with newly diagnosed, Ph-negative, B-cell precursor ALL with 20% or more CD20-positive leukemic blasts.
The patients’ median age was 40.2 (range, 24.5–52.6), 13% had an ECOG performance status greater than 1, 6% had CNS involvement, and 21% had a white blood cell count of 30 x 109/L or higher.
Half of the patients (n=104) were randomized to receive the GRAALL-2005 regimen, and the other half (n=105) were randomized to receive the same regimen plus rituximab. Details on the regimens are available in the supplementary material published with the NEJM paper.
Baseline patient characteristics were well-balanced between the treatment groups.
Results
At a median follow-up of 30 months, 101 patients (48%) had at least 1 event, including 44 (42%) in the rituximab group and 57 (55%) in the control group.
There were 17 induction failures (8 in the rituximab group and 9 in the control group), 57 relapses (22 and 35, respectively), and 27 deaths during remission (14 and 13, respectively). Two patients in the rituximab group were lost to follow-up during the first 12 months.
The 2-year EFS was significantly higher in the rituximab group than the control group—65% and 52%, respectively (hazard ratio [HR]=0.66, P=0.04).
However, the EFS benefit did not translate into a significant improvement in overall survival. The 2-year overall survival was 71% in the rituximab group and 64% in the control group (HR=0.70, P=0.10).
Similarly, the cumulative incidence of death during first remission was not significantly different between the treatment groups—12% for both (HR=0.98, P=0.96).
The researchers said the difference in EFS was mostly due to a lower incidence of relapse in the rituximab group. The 2-year incidence of relapse was 18% in the rituximab group and 32% in the control group (HR=0.52, P=0.02).
In a multivariate analysis, receiving the control treatment, older age, higher white blood cell count at baseline, and CNS involvement were all significantly associated with poor EFS.
There were 245 severe adverse events (AEs) reported in 124 patients—67 patients with 1 event, 26 with 2 events, 13 with 3 events, and 18 with 4 or more events.
The overall incidence of severe AEs did not differ significantly between the treatment groups—96% in the rituximab group and 92% in the control group.
Severe AEs included infection, laboratory abnormalities, allergic reactions, neurologic events, pulmonary events, coagulopathy, cardiac events, gastrointestinal events, and “other” events.
The only severe AE for which there was a significant difference between the treatment groups was allergic reactions. There were 2 severe allergic events in the rituximab group and 14 in the control group (P=0.002). Of these 16 events, all but 1 were due to asparaginase.
The researchers therefore theorized that rituximab might inhibit B-cell protection of antibodies against asparaginase, although they could not confirm this hypothesis.
PAFC provides early embolus detection in mice
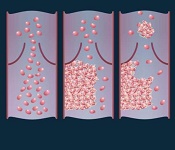
vein (left), thrombus formation
(middle), and a thrombus (right)
detaching from a vessel wall,
leading to thromboembolism.
Image by Moscow Institute
of Physics and Technology
Photoacoustic flow cytometry (PAFC) can provide real-time, non-invasive detection of emboli, according to research published in PLOS ONE.
Experiments in mice showed that PAFC can reveal emboli triggered by melanoma and medical procedures by detecting transient changes in blood absorption.
Investigators believe that by providing early embolus detection, PAFC could enable well-timed anticoagulant therapy and potentially prevent lethal complications.
For this study, the team used PAFC to monitor embolus formation in 4 groups of mice.
Two of the groups had melanoma. In one, the implanted tumor underwent compression. In the other, the investigators performed a surgical excision of the tumor.
The remaining 2 groups consisted of healthy mice. In one of these groups, mice had their limbs clamped to produce vessel stasis. In the other, the mice underwent surgery.
The investigators found that PAFC could detect a single embolus, and the method allowed them to distinguish between erythrocyte-rich and leukocyte/platelet-rich emboli.
They also observed a correlation between the presence of white emboli and melanoma.
The level of circulating emboli was significantly higher in the melanoma-bearing mice than in the healthy ones (P=0.0013).
However, the number of circulating emboli temporarily increased in the healthy mice during vessel stasis (P=0.033) and after surgical excisions (P=0.031).
The melanoma-bearing mice also experienced increases in the number of circulating emboli during tumor compression (P=0.013) and after tumor excision (P=0.012).
“We showed that it is possible to detect emboli in the bloodstream using photoacoustic flow cytometry,” said study author Alexander Melerzanov, PhD, of the Moscow Institute of Physics and Technology in Russia.
“PAFC may also be used to destroy blood clots, and we hope to work on this in our next experiments.”
vein (left), thrombus formation
(middle), and a thrombus (right)
detaching from a vessel wall,
leading to thromboembolism.
Image by Moscow Institute
of Physics and Technology
Photoacoustic flow cytometry (PAFC) can provide real-time, non-invasive detection of emboli, according to research published in PLOS ONE.
Experiments in mice showed that PAFC can reveal emboli triggered by melanoma and medical procedures by detecting transient changes in blood absorption.
Investigators believe that by providing early embolus detection, PAFC could enable well-timed anticoagulant therapy and potentially prevent lethal complications.
For this study, the team used PAFC to monitor embolus formation in 4 groups of mice.
Two of the groups had melanoma. In one, the implanted tumor underwent compression. In the other, the investigators performed a surgical excision of the tumor.
The remaining 2 groups consisted of healthy mice. In one of these groups, mice had their limbs clamped to produce vessel stasis. In the other, the mice underwent surgery.
The investigators found that PAFC could detect a single embolus, and the method allowed them to distinguish between erythrocyte-rich and leukocyte/platelet-rich emboli.
They also observed a correlation between the presence of white emboli and melanoma.
The level of circulating emboli was significantly higher in the melanoma-bearing mice than in the healthy ones (P=0.0013).
However, the number of circulating emboli temporarily increased in the healthy mice during vessel stasis (P=0.033) and after surgical excisions (P=0.031).
The melanoma-bearing mice also experienced increases in the number of circulating emboli during tumor compression (P=0.013) and after tumor excision (P=0.012).
“We showed that it is possible to detect emboli in the bloodstream using photoacoustic flow cytometry,” said study author Alexander Melerzanov, PhD, of the Moscow Institute of Physics and Technology in Russia.
“PAFC may also be used to destroy blood clots, and we hope to work on this in our next experiments.”
vein (left), thrombus formation
(middle), and a thrombus (right)
detaching from a vessel wall,
leading to thromboembolism.
Image by Moscow Institute
of Physics and Technology
Photoacoustic flow cytometry (PAFC) can provide real-time, non-invasive detection of emboli, according to research published in PLOS ONE.
Experiments in mice showed that PAFC can reveal emboli triggered by melanoma and medical procedures by detecting transient changes in blood absorption.
Investigators believe that by providing early embolus detection, PAFC could enable well-timed anticoagulant therapy and potentially prevent lethal complications.
For this study, the team used PAFC to monitor embolus formation in 4 groups of mice.
Two of the groups had melanoma. In one, the implanted tumor underwent compression. In the other, the investigators performed a surgical excision of the tumor.
The remaining 2 groups consisted of healthy mice. In one of these groups, mice had their limbs clamped to produce vessel stasis. In the other, the mice underwent surgery.
The investigators found that PAFC could detect a single embolus, and the method allowed them to distinguish between erythrocyte-rich and leukocyte/platelet-rich emboli.
They also observed a correlation between the presence of white emboli and melanoma.
The level of circulating emboli was significantly higher in the melanoma-bearing mice than in the healthy ones (P=0.0013).
However, the number of circulating emboli temporarily increased in the healthy mice during vessel stasis (P=0.033) and after surgical excisions (P=0.031).
The melanoma-bearing mice also experienced increases in the number of circulating emboli during tumor compression (P=0.013) and after tumor excision (P=0.012).
“We showed that it is possible to detect emboli in the bloodstream using photoacoustic flow cytometry,” said study author Alexander Melerzanov, PhD, of the Moscow Institute of Physics and Technology in Russia.
“PAFC may also be used to destroy blood clots, and we hope to work on this in our next experiments.”
Team creates method for predicting drug toxicity

for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.”
for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.”
for a clinical trial
Photo by Esther Dyson
Researchers have devised a method for predicting whether experimental drugs will fail clinical trials due to excessive toxicity.
They say the method, known as PrOCTOR, upends conventional wisdom about the criteria on which to evaluate new drugs’ safety.
Rather than exclusively examining molecular structure to determine viability, PrOCTOR combines a host of structural features and features related to how the drug binds to molecules in the body.
“We looked more broadly at drug molecule features that drug developers thought were unimportant in predicting drug safety in the past. Then, we let the data speak for itself,” said study author Olivier Elemento, PhD, of Weill Cornell Medicine in New York, New York.
Dr Elemento and his colleagues described PrOCTOR in Cell Chemical Biology.
PrOCTOR was inspired by an approach that baseball statisticians adopted to better predict which players would be successful offensively. Instead of relying on collective wisdom from baseball insiders, statisticians decided to use an objective numbers analysis to measure in-game productivity, a strategy known as “Moneyball.”
Similarly, Dr Elemento and his colleagues developed a computational method that analyzes data from 48 different features of a drug—from molecular weight to details about its target—to determine whether it would be safe for clinical use.
Using machine learning, the researchers trained PrOCTOR on drugs approved by the US Food and Drug Administration (FDA) as well as drugs that failed clinical trials due to toxicity problems.
Based on this information, the researchers created “PrOCTOR scores” that could help distinguish drugs approved by the FDA from those that failed for toxicity.
They tested PrOCTOR on hundreds of additional drugs approved in Europe and Japan and using side effect data on approved drugs collected by the FDA.
The researchers said PrOCTOR was able to accurately recognize toxic side effects that were a consequence of a drug’s chemical features and its target. Records revealing that many of these drugs had failed clinical trials supported PrOCTOR’s accuracy.
“We were able to find several features that led to a very predictive model,” Dr Elemento said. “Hopefully, this approach could be used to determine whether it’s worth pursuing a drug prior to starting human trials.”
Targeting AML’s dependence on fat

New research has revealed a potential treatment strategy for acute myeloid leukemia (AML) and other malignancies that show a preference for metabolizing fat over sugar.
The study suggests the protein prolyl hydroxylase 3 (PHD3) is a key regulator of fatty acid oxidation, and PHD3 expression is low in certain malignancies—particularly AML—but overexpressing PHD3 can have anticancer effects.
Researchers believe this finding might aid the development of therapies that work by starving tumors of their fuel.
“This really represents a new frontier in looking at the metabolism of cancer,” said study author Marcia Haigis, PhD, of Harvard Medical School in Boston, Massachusetts.
“Understanding the molecular handle of this pathway is the first step toward translating the basic work into therapy.”
Dr Haigis and her colleagues described the work in Molecular Cell.
The researchers knew that when cells run low on nutrients, they switch from sugar to fat as their fuel source to sustain function.
When cells have low energy, the protein AMPK targets the enzyme ACC to activate fat oxidation, which helps cells burn fats to make energy. However, when cells have enough resources, they seek to maintain energy balance.
Dr Haigis and her colleagues were searching for precisely how cells turn off fat oxidation and homed in on PHD3. Recent studies had suggested that PHD3 plays a part in cell metabolism, but,
until now, its precise role has remained unclear.
In a series of experiments, Dr Haigis’s team showed that PHD3 suppressed fat-burning by chemically modifying and activating ACC2—a version of the same enzyme responsible for keeping cellular fat-burning in check.
To determine PHD3’s role in cancer, the researchers combed through databases of all human cancers. The team theorized that sugar-craving malignancies would have high levels of PDH3, and cancers that relied on fat for their energy would have low levels.
The researchers found the lowest levels of PHD3 in AML, so they decided to examine the effects of restoring PHD3 in AML cells and a mouse model of the disease.
The experiments showed that restoring PHD3 expression reduces AML potency, and ACC2 is required for the inhibitory effects of PHD3. Overexpressing PHD3 limited fatty acid oxidation via regulation of ACC2, which decreased leukemia cell proliferation and enhanced survival in the mouse model of AML.
Dr Haigis noted that more basic research is needed before this work can move ahead to the clinic, as it’s still unclear why certain cancers depend on fat.
“What do fats provide to tumors that other fuels don’t?” Dr Haigis asked. “That’s one open question, and this is only the first chapter in the story.”
New research has revealed a potential treatment strategy for acute myeloid leukemia (AML) and other malignancies that show a preference for metabolizing fat over sugar.
The study suggests the protein prolyl hydroxylase 3 (PHD3) is a key regulator of fatty acid oxidation, and PHD3 expression is low in certain malignancies—particularly AML—but overexpressing PHD3 can have anticancer effects.
Researchers believe this finding might aid the development of therapies that work by starving tumors of their fuel.
“This really represents a new frontier in looking at the metabolism of cancer,” said study author Marcia Haigis, PhD, of Harvard Medical School in Boston, Massachusetts.
“Understanding the molecular handle of this pathway is the first step toward translating the basic work into therapy.”
Dr Haigis and her colleagues described the work in Molecular Cell.
The researchers knew that when cells run low on nutrients, they switch from sugar to fat as their fuel source to sustain function.
When cells have low energy, the protein AMPK targets the enzyme ACC to activate fat oxidation, which helps cells burn fats to make energy. However, when cells have enough resources, they seek to maintain energy balance.
Dr Haigis and her colleagues were searching for precisely how cells turn off fat oxidation and homed in on PHD3. Recent studies had suggested that PHD3 plays a part in cell metabolism, but,
until now, its precise role has remained unclear.
In a series of experiments, Dr Haigis’s team showed that PHD3 suppressed fat-burning by chemically modifying and activating ACC2—a version of the same enzyme responsible for keeping cellular fat-burning in check.
To determine PHD3’s role in cancer, the researchers combed through databases of all human cancers. The team theorized that sugar-craving malignancies would have high levels of PDH3, and cancers that relied on fat for their energy would have low levels.
The researchers found the lowest levels of PHD3 in AML, so they decided to examine the effects of restoring PHD3 in AML cells and a mouse model of the disease.
The experiments showed that restoring PHD3 expression reduces AML potency, and ACC2 is required for the inhibitory effects of PHD3. Overexpressing PHD3 limited fatty acid oxidation via regulation of ACC2, which decreased leukemia cell proliferation and enhanced survival in the mouse model of AML.
Dr Haigis noted that more basic research is needed before this work can move ahead to the clinic, as it’s still unclear why certain cancers depend on fat.
“What do fats provide to tumors that other fuels don’t?” Dr Haigis asked. “That’s one open question, and this is only the first chapter in the story.”
New research has revealed a potential treatment strategy for acute myeloid leukemia (AML) and other malignancies that show a preference for metabolizing fat over sugar.
The study suggests the protein prolyl hydroxylase 3 (PHD3) is a key regulator of fatty acid oxidation, and PHD3 expression is low in certain malignancies—particularly AML—but overexpressing PHD3 can have anticancer effects.
Researchers believe this finding might aid the development of therapies that work by starving tumors of their fuel.
“This really represents a new frontier in looking at the metabolism of cancer,” said study author Marcia Haigis, PhD, of Harvard Medical School in Boston, Massachusetts.
“Understanding the molecular handle of this pathway is the first step toward translating the basic work into therapy.”
Dr Haigis and her colleagues described the work in Molecular Cell.
The researchers knew that when cells run low on nutrients, they switch from sugar to fat as their fuel source to sustain function.
When cells have low energy, the protein AMPK targets the enzyme ACC to activate fat oxidation, which helps cells burn fats to make energy. However, when cells have enough resources, they seek to maintain energy balance.
Dr Haigis and her colleagues were searching for precisely how cells turn off fat oxidation and homed in on PHD3. Recent studies had suggested that PHD3 plays a part in cell metabolism, but,
until now, its precise role has remained unclear.
In a series of experiments, Dr Haigis’s team showed that PHD3 suppressed fat-burning by chemically modifying and activating ACC2—a version of the same enzyme responsible for keeping cellular fat-burning in check.
To determine PHD3’s role in cancer, the researchers combed through databases of all human cancers. The team theorized that sugar-craving malignancies would have high levels of PDH3, and cancers that relied on fat for their energy would have low levels.
The researchers found the lowest levels of PHD3 in AML, so they decided to examine the effects of restoring PHD3 in AML cells and a mouse model of the disease.
The experiments showed that restoring PHD3 expression reduces AML potency, and ACC2 is required for the inhibitory effects of PHD3. Overexpressing PHD3 limited fatty acid oxidation via regulation of ACC2, which decreased leukemia cell proliferation and enhanced survival in the mouse model of AML.
Dr Haigis noted that more basic research is needed before this work can move ahead to the clinic, as it’s still unclear why certain cancers depend on fat.
“What do fats provide to tumors that other fuels don’t?” Dr Haigis asked. “That’s one open question, and this is only the first chapter in the story.”