User login
Massage therapy seems to benefit cancer patients

Photo courtesy of Barbara
E. Carver/New York College
of Health Professions
Massage therapy can reduce pain, fatigue, and anxiety in cancer patients, according to a review and meta-analysis published in Pain Medicine.
Massage therapy is commonly used among people seeking pain management, and research has generally supported its use.
However, there has been no published, rigorous review of the available research and evidence for massage therapy’s efficacy for pain populations, especially for cancer populations.
So Courtney Boyd, of the Samueli Institute in Alexandria, Virginia, and her colleagues set out to conduct just such a review.
The team assessed the quality of massage therapy research and evidence for its efficacy in treating pain, function-related quality of life, and health-related quality of life in cancer patients.
The researchers reviewed data from 12 high-quality studies and 4 low-quality studies.
The team said they could not assess health-related quality of life, emotional stress, or activity outcomes because too few of the studies examined these outcomes.
However, the data suggested that massage therapy can effectively reduce pain intensity or severity when compared to no treatment (standardized mean difference [SMD]=-0.20) or active comparators (SMD=-0.55).
Massage therapy also proved effective for reducing fatigue (SMD=-1.06) and anxiety (SMD=-1.24) when compared to active comparators.
Based on these results, the researchers concluded that massage therapy may have beneficial effects in cancer patients, so they should consider it as an option.
However, before definitive clinical conclusions and recommendations can be made at a policy level, specific factors surrounding the massage protocol, as well as selection of appropriate controls and standard outcomes, need to be well-understood. ![]()
Photo courtesy of Barbara
E. Carver/New York College
of Health Professions
Massage therapy can reduce pain, fatigue, and anxiety in cancer patients, according to a review and meta-analysis published in Pain Medicine.
Massage therapy is commonly used among people seeking pain management, and research has generally supported its use.
However, there has been no published, rigorous review of the available research and evidence for massage therapy’s efficacy for pain populations, especially for cancer populations.
So Courtney Boyd, of the Samueli Institute in Alexandria, Virginia, and her colleagues set out to conduct just such a review.
The team assessed the quality of massage therapy research and evidence for its efficacy in treating pain, function-related quality of life, and health-related quality of life in cancer patients.
The researchers reviewed data from 12 high-quality studies and 4 low-quality studies.
The team said they could not assess health-related quality of life, emotional stress, or activity outcomes because too few of the studies examined these outcomes.
However, the data suggested that massage therapy can effectively reduce pain intensity or severity when compared to no treatment (standardized mean difference [SMD]=-0.20) or active comparators (SMD=-0.55).
Massage therapy also proved effective for reducing fatigue (SMD=-1.06) and anxiety (SMD=-1.24) when compared to active comparators.
Based on these results, the researchers concluded that massage therapy may have beneficial effects in cancer patients, so they should consider it as an option.
However, before definitive clinical conclusions and recommendations can be made at a policy level, specific factors surrounding the massage protocol, as well as selection of appropriate controls and standard outcomes, need to be well-understood. ![]()
Photo courtesy of Barbara
E. Carver/New York College
of Health Professions
Massage therapy can reduce pain, fatigue, and anxiety in cancer patients, according to a review and meta-analysis published in Pain Medicine.
Massage therapy is commonly used among people seeking pain management, and research has generally supported its use.
However, there has been no published, rigorous review of the available research and evidence for massage therapy’s efficacy for pain populations, especially for cancer populations.
So Courtney Boyd, of the Samueli Institute in Alexandria, Virginia, and her colleagues set out to conduct just such a review.
The team assessed the quality of massage therapy research and evidence for its efficacy in treating pain, function-related quality of life, and health-related quality of life in cancer patients.
The researchers reviewed data from 12 high-quality studies and 4 low-quality studies.
The team said they could not assess health-related quality of life, emotional stress, or activity outcomes because too few of the studies examined these outcomes.
However, the data suggested that massage therapy can effectively reduce pain intensity or severity when compared to no treatment (standardized mean difference [SMD]=-0.20) or active comparators (SMD=-0.55).
Massage therapy also proved effective for reducing fatigue (SMD=-1.06) and anxiety (SMD=-1.24) when compared to active comparators.
Based on these results, the researchers concluded that massage therapy may have beneficial effects in cancer patients, so they should consider it as an option.
However, before definitive clinical conclusions and recommendations can be made at a policy level, specific factors surrounding the massage protocol, as well as selection of appropriate controls and standard outcomes, need to be well-understood. ![]()
Drug granted breakthrough designation for BPDCN
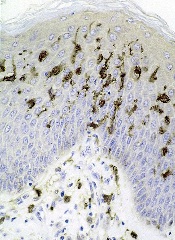
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for SL-401 in the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 is a targeted therapy directed to the interleukin-3 receptor (IL-3R), which is present in BPDCN and other hematologic malignancies.
SL-401 is composed of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
The drug is under development by Stemline Therapeutics, Inc.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Phase 2 trial of SL-401
The breakthrough designation for SL-401 was supported by data from a phase 2 trial of patients with BPDCN or acute myeloid leukemia. Results observed in the BPDCN patients were presented at the 2016 EHA Congress (abstract S812).
The study consists of a lead-in dose-escalation stage (stage 1) and subsequent expansion stage (stage 2). In stage 1, patients received SL-401 as a daily intravenous infusion for up to 5 days (7, 9, 12, or 16 μg/kg/day) every 21 days. In stage 2, patients received SL-401 at the optimal stage 1 dose—12 μg/kg.
At the data cut-off, 18 BPDCN patients had received SL-401 at 7 μg/kg (n=3, stage 1) or 12 μg/kg (n=15, 6 in stage 1, 9 in stage 2).
Fifteen of these patients were evaluable for response. The overall response rate was 87% (13/15). All 10 previously untreated BPDCN patients achieved a response, including 7 complete responses (CRs).
All 8 previously untreated BPDCN patients treated at the optimal dose (12 μg/kg) achieved a response, including 6 CRs. Four of these patients were still on SL-401 and in CR at the data cutoff, and 2 had gone on to stem cell transplant.
The most common treatment-related adverse events in the BPDCN patients were transient transaminase elevation (57%), hypoalbuminemia (40%), and transient thrombocytopenia (15%).
In stage 1, two patients had capillary leak syndrome (CLS)—one grade 5 (7 μg/kg) and one grade 4 (12 μg/kg). After this, safety precautions were implemented to minimize the risk of severe CLS. There have been no cases of severe CLS since then. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for SL-401 in the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 is a targeted therapy directed to the interleukin-3 receptor (IL-3R), which is present in BPDCN and other hematologic malignancies.
SL-401 is composed of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
The drug is under development by Stemline Therapeutics, Inc.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Phase 2 trial of SL-401
The breakthrough designation for SL-401 was supported by data from a phase 2 trial of patients with BPDCN or acute myeloid leukemia. Results observed in the BPDCN patients were presented at the 2016 EHA Congress (abstract S812).
The study consists of a lead-in dose-escalation stage (stage 1) and subsequent expansion stage (stage 2). In stage 1, patients received SL-401 as a daily intravenous infusion for up to 5 days (7, 9, 12, or 16 μg/kg/day) every 21 days. In stage 2, patients received SL-401 at the optimal stage 1 dose—12 μg/kg.
At the data cut-off, 18 BPDCN patients had received SL-401 at 7 μg/kg (n=3, stage 1) or 12 μg/kg (n=15, 6 in stage 1, 9 in stage 2).
Fifteen of these patients were evaluable for response. The overall response rate was 87% (13/15). All 10 previously untreated BPDCN patients achieved a response, including 7 complete responses (CRs).
All 8 previously untreated BPDCN patients treated at the optimal dose (12 μg/kg) achieved a response, including 6 CRs. Four of these patients were still on SL-401 and in CR at the data cutoff, and 2 had gone on to stem cell transplant.
The most common treatment-related adverse events in the BPDCN patients were transient transaminase elevation (57%), hypoalbuminemia (40%), and transient thrombocytopenia (15%).
In stage 1, two patients had capillary leak syndrome (CLS)—one grade 5 (7 μg/kg) and one grade 4 (12 μg/kg). After this, safety precautions were implemented to minimize the risk of severe CLS. There have been no cases of severe CLS since then. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for SL-401 in the treatment of blastic plasmacytoid dendritic cell neoplasm (BPDCN).
SL-401 is a targeted therapy directed to the interleukin-3 receptor (IL-3R), which is present in BPDCN and other hematologic malignancies.
SL-401 is composed of human IL-3 coupled to a truncated diphtheria toxin payload that inhibits protein synthesis.
The drug is under development by Stemline Therapeutics, Inc.
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Phase 2 trial of SL-401
The breakthrough designation for SL-401 was supported by data from a phase 2 trial of patients with BPDCN or acute myeloid leukemia. Results observed in the BPDCN patients were presented at the 2016 EHA Congress (abstract S812).
The study consists of a lead-in dose-escalation stage (stage 1) and subsequent expansion stage (stage 2). In stage 1, patients received SL-401 as a daily intravenous infusion for up to 5 days (7, 9, 12, or 16 μg/kg/day) every 21 days. In stage 2, patients received SL-401 at the optimal stage 1 dose—12 μg/kg.
At the data cut-off, 18 BPDCN patients had received SL-401 at 7 μg/kg (n=3, stage 1) or 12 μg/kg (n=15, 6 in stage 1, 9 in stage 2).
Fifteen of these patients were evaluable for response. The overall response rate was 87% (13/15). All 10 previously untreated BPDCN patients achieved a response, including 7 complete responses (CRs).
All 8 previously untreated BPDCN patients treated at the optimal dose (12 μg/kg) achieved a response, including 6 CRs. Four of these patients were still on SL-401 and in CR at the data cutoff, and 2 had gone on to stem cell transplant.
The most common treatment-related adverse events in the BPDCN patients were transient transaminase elevation (57%), hypoalbuminemia (40%), and transient thrombocytopenia (15%).
In stage 1, two patients had capillary leak syndrome (CLS)—one grade 5 (7 μg/kg) and one grade 4 (12 μg/kg). After this, safety precautions were implemented to minimize the risk of severe CLS. There have been no cases of severe CLS since then. ![]()
Reasons for high cost of prescription drugs in US

Photo courtesy of the CDC
High prescription drug prices in the US have a few causes, according to researchers.
They said causes include the approach the US has taken to granting government-protected monopolies to drug manufacturers, strategies that delay access to generic drugs, and the restriction of price negotiation at a level not observed in other industrialized nations.
The researchers outlined these issues in JAMA.
Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues conducted this research.
The team reviewed the medical and health policy literature from January 2005 to July 2016, looking for articles addressing the sources of drug prices in the US, the justifications and consequences of high prices, and possible solutions.
The researchers found that per-capita prescription drug spending is higher in the US than in all other countries. In 2013, per-capita spending on prescription drugs was $858 in the US, compared with an average of $400 for 19 other industrialized nations.
Dr Kesselheim and his colleagues said prescription drug spending in the US is largely driven by brand-name drug prices that have been increasing in recent years. And drug prices are higher in the US because the US healthcare system allows manufacturers to set their own price for a given product.
In countries with national health insurance systems, on the other hand, a delegated body negotiates drug prices or rejects coverage of products if the price demanded by the manufacturer is excessive in light of the benefit provided. Manufacturers may then decide to offer the drug at a lower price.
The researchers said the most important factor that allows US manufacturers to set high drug prices is market exclusivity. And although cheaper generic drugs can be made available after an exclusivity period has passed, there are strategies for keeping these drugs off the market.
Furthermore, the negotiating power of the payer is constrained by several factors, including the requirement that most government drug payment plans cover nearly all products.
Considering these findings together, Dr Kesselheim and his colleagues said the most realistic short-term strategies to address high drug prices in the US include:
- Enforcing more stringent requirements for market exclusivity rights
- Ensuring timely generic drug availability
- Providing greater opportunities for price negotiation by governmental payers
- Generating more evidence about comparative cost-effectiveness of therapeutic alternatives
- Educating patients, prescribers, payers, and policy makers about these choices.
The researchers said there is little evidence that such policies would hamper innovation. In fact, they might even drive the development of more valuable new therapies rather than rewarding the persistence of older ones. ![]()
Photo courtesy of the CDC
High prescription drug prices in the US have a few causes, according to researchers.
They said causes include the approach the US has taken to granting government-protected monopolies to drug manufacturers, strategies that delay access to generic drugs, and the restriction of price negotiation at a level not observed in other industrialized nations.
The researchers outlined these issues in JAMA.
Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues conducted this research.
The team reviewed the medical and health policy literature from January 2005 to July 2016, looking for articles addressing the sources of drug prices in the US, the justifications and consequences of high prices, and possible solutions.
The researchers found that per-capita prescription drug spending is higher in the US than in all other countries. In 2013, per-capita spending on prescription drugs was $858 in the US, compared with an average of $400 for 19 other industrialized nations.
Dr Kesselheim and his colleagues said prescription drug spending in the US is largely driven by brand-name drug prices that have been increasing in recent years. And drug prices are higher in the US because the US healthcare system allows manufacturers to set their own price for a given product.
In countries with national health insurance systems, on the other hand, a delegated body negotiates drug prices or rejects coverage of products if the price demanded by the manufacturer is excessive in light of the benefit provided. Manufacturers may then decide to offer the drug at a lower price.
The researchers said the most important factor that allows US manufacturers to set high drug prices is market exclusivity. And although cheaper generic drugs can be made available after an exclusivity period has passed, there are strategies for keeping these drugs off the market.
Furthermore, the negotiating power of the payer is constrained by several factors, including the requirement that most government drug payment plans cover nearly all products.
Considering these findings together, Dr Kesselheim and his colleagues said the most realistic short-term strategies to address high drug prices in the US include:
- Enforcing more stringent requirements for market exclusivity rights
- Ensuring timely generic drug availability
- Providing greater opportunities for price negotiation by governmental payers
- Generating more evidence about comparative cost-effectiveness of therapeutic alternatives
- Educating patients, prescribers, payers, and policy makers about these choices.
The researchers said there is little evidence that such policies would hamper innovation. In fact, they might even drive the development of more valuable new therapies rather than rewarding the persistence of older ones. ![]()
Photo courtesy of the CDC
High prescription drug prices in the US have a few causes, according to researchers.
They said causes include the approach the US has taken to granting government-protected monopolies to drug manufacturers, strategies that delay access to generic drugs, and the restriction of price negotiation at a level not observed in other industrialized nations.
The researchers outlined these issues in JAMA.
Aaron S. Kesselheim, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues conducted this research.
The team reviewed the medical and health policy literature from January 2005 to July 2016, looking for articles addressing the sources of drug prices in the US, the justifications and consequences of high prices, and possible solutions.
The researchers found that per-capita prescription drug spending is higher in the US than in all other countries. In 2013, per-capita spending on prescription drugs was $858 in the US, compared with an average of $400 for 19 other industrialized nations.
Dr Kesselheim and his colleagues said prescription drug spending in the US is largely driven by brand-name drug prices that have been increasing in recent years. And drug prices are higher in the US because the US healthcare system allows manufacturers to set their own price for a given product.
In countries with national health insurance systems, on the other hand, a delegated body negotiates drug prices or rejects coverage of products if the price demanded by the manufacturer is excessive in light of the benefit provided. Manufacturers may then decide to offer the drug at a lower price.
The researchers said the most important factor that allows US manufacturers to set high drug prices is market exclusivity. And although cheaper generic drugs can be made available after an exclusivity period has passed, there are strategies for keeping these drugs off the market.
Furthermore, the negotiating power of the payer is constrained by several factors, including the requirement that most government drug payment plans cover nearly all products.
Considering these findings together, Dr Kesselheim and his colleagues said the most realistic short-term strategies to address high drug prices in the US include:
- Enforcing more stringent requirements for market exclusivity rights
- Ensuring timely generic drug availability
- Providing greater opportunities for price negotiation by governmental payers
- Generating more evidence about comparative cost-effectiveness of therapeutic alternatives
- Educating patients, prescribers, payers, and policy makers about these choices.
The researchers said there is little evidence that such policies would hamper innovation. In fact, they might even drive the development of more valuable new therapies rather than rewarding the persistence of older ones. ![]()
Predicting outcomes in relapsed BCP-ALL

Image by Vashi Donsk
Screening for genetic abnormalities can provide a more accurate prediction of outcomes in children with relapsed B-cell precursor acute lymphoblastic leukemia (BCP-ALL), according to a study published in Blood.
Researchers found that mutations or deletions in TP53, NR3C1, BTG1, and NRAS were associated with inferior outcomes in relapsed BCP-ALL.
And screening for these abnormalities could improve upon the predictive accuracy of clinical risk factors.
“Current methods used to guide treatment for relapsed leukemia are not accurate enough, with some children believed to have a good chance of survival actually responding very poorly to chemotherapy,” said study author Anthony Moorman, PhD, of Newcastle University in Newcastle upon Tyne, UK.
“Screening patients at relapse for key genetic abnormalities that influence outcome will ensure that treatment can be personalized, thereby improving their chances of survival.”
For this study, Dr Moorman and his colleagues analyzed cytogenetic data from 427 children with relapsed BCP-ALL and screened 238 patients with a marrow relapse for certain copy number alterations and mutations.
According to univariate analysis, alterations in TP53, NR3C1 deletions, and BTG1 deletions were significantly associated with patient outcomes.
Patients with TP53 alterations had a higher risk of progression (hazard ratio [HR]=2.36, P<0.001) and death (HR=2.56, P<0.001), as did patients with deletions in NR3C1 and BTG1.
Because both NR3C1 and BTG1 are implicated in resistance to glucocorticoids and the deletions are mutually exclusive, the researchers considered the effect of the deletions together. So for patients with NR3C1 and BTG1 deletions, the HR for progression was 2.15 (P=0.002), and the HR for death was 1.91 (P=0.015).
Patients with NRAS mutations had an increased risk of progression and death as well, but this did not reach statistical significance.
The researchers also found that patients who were standard risk according to clinical characteristics but, at the time of relapse, had one or more high-risk genetic abnormalities had poorer outcomes.
Standard-risk patients with a TP53 alteration had an increased risk of death (HR=2.56, P<0.001), as did standard-risk patients with NR3C1 and BTG1 deletions (HR=1.91, P=0.015).
Standard-risk patients with NRAS mutations and high hyperdiploidy had an increased risk of progression (HR=3.17, P=0.026) and death (HR=3.41, P=0.032).
The researchers concluded that the outcomes of clinical standard-risk patients with high-risk cytogenetics were equivalent to outcomes of clinical high-risk patients.
The team therefore believes that screening BCP-ALL patients for the aforementioned genetic abnormalities at relapse will improve patient stratification and outcomes. ![]()
Image by Vashi Donsk
Screening for genetic abnormalities can provide a more accurate prediction of outcomes in children with relapsed B-cell precursor acute lymphoblastic leukemia (BCP-ALL), according to a study published in Blood.
Researchers found that mutations or deletions in TP53, NR3C1, BTG1, and NRAS were associated with inferior outcomes in relapsed BCP-ALL.
And screening for these abnormalities could improve upon the predictive accuracy of clinical risk factors.
“Current methods used to guide treatment for relapsed leukemia are not accurate enough, with some children believed to have a good chance of survival actually responding very poorly to chemotherapy,” said study author Anthony Moorman, PhD, of Newcastle University in Newcastle upon Tyne, UK.
“Screening patients at relapse for key genetic abnormalities that influence outcome will ensure that treatment can be personalized, thereby improving their chances of survival.”
For this study, Dr Moorman and his colleagues analyzed cytogenetic data from 427 children with relapsed BCP-ALL and screened 238 patients with a marrow relapse for certain copy number alterations and mutations.
According to univariate analysis, alterations in TP53, NR3C1 deletions, and BTG1 deletions were significantly associated with patient outcomes.
Patients with TP53 alterations had a higher risk of progression (hazard ratio [HR]=2.36, P<0.001) and death (HR=2.56, P<0.001), as did patients with deletions in NR3C1 and BTG1.
Because both NR3C1 and BTG1 are implicated in resistance to glucocorticoids and the deletions are mutually exclusive, the researchers considered the effect of the deletions together. So for patients with NR3C1 and BTG1 deletions, the HR for progression was 2.15 (P=0.002), and the HR for death was 1.91 (P=0.015).
Patients with NRAS mutations had an increased risk of progression and death as well, but this did not reach statistical significance.
The researchers also found that patients who were standard risk according to clinical characteristics but, at the time of relapse, had one or more high-risk genetic abnormalities had poorer outcomes.
Standard-risk patients with a TP53 alteration had an increased risk of death (HR=2.56, P<0.001), as did standard-risk patients with NR3C1 and BTG1 deletions (HR=1.91, P=0.015).
Standard-risk patients with NRAS mutations and high hyperdiploidy had an increased risk of progression (HR=3.17, P=0.026) and death (HR=3.41, P=0.032).
The researchers concluded that the outcomes of clinical standard-risk patients with high-risk cytogenetics were equivalent to outcomes of clinical high-risk patients.
The team therefore believes that screening BCP-ALL patients for the aforementioned genetic abnormalities at relapse will improve patient stratification and outcomes. ![]()
Image by Vashi Donsk
Screening for genetic abnormalities can provide a more accurate prediction of outcomes in children with relapsed B-cell precursor acute lymphoblastic leukemia (BCP-ALL), according to a study published in Blood.
Researchers found that mutations or deletions in TP53, NR3C1, BTG1, and NRAS were associated with inferior outcomes in relapsed BCP-ALL.
And screening for these abnormalities could improve upon the predictive accuracy of clinical risk factors.
“Current methods used to guide treatment for relapsed leukemia are not accurate enough, with some children believed to have a good chance of survival actually responding very poorly to chemotherapy,” said study author Anthony Moorman, PhD, of Newcastle University in Newcastle upon Tyne, UK.
“Screening patients at relapse for key genetic abnormalities that influence outcome will ensure that treatment can be personalized, thereby improving their chances of survival.”
For this study, Dr Moorman and his colleagues analyzed cytogenetic data from 427 children with relapsed BCP-ALL and screened 238 patients with a marrow relapse for certain copy number alterations and mutations.
According to univariate analysis, alterations in TP53, NR3C1 deletions, and BTG1 deletions were significantly associated with patient outcomes.
Patients with TP53 alterations had a higher risk of progression (hazard ratio [HR]=2.36, P<0.001) and death (HR=2.56, P<0.001), as did patients with deletions in NR3C1 and BTG1.
Because both NR3C1 and BTG1 are implicated in resistance to glucocorticoids and the deletions are mutually exclusive, the researchers considered the effect of the deletions together. So for patients with NR3C1 and BTG1 deletions, the HR for progression was 2.15 (P=0.002), and the HR for death was 1.91 (P=0.015).
Patients with NRAS mutations had an increased risk of progression and death as well, but this did not reach statistical significance.
The researchers also found that patients who were standard risk according to clinical characteristics but, at the time of relapse, had one or more high-risk genetic abnormalities had poorer outcomes.
Standard-risk patients with a TP53 alteration had an increased risk of death (HR=2.56, P<0.001), as did standard-risk patients with NR3C1 and BTG1 deletions (HR=1.91, P=0.015).
Standard-risk patients with NRAS mutations and high hyperdiploidy had an increased risk of progression (HR=3.17, P=0.026) and death (HR=3.41, P=0.032).
The researchers concluded that the outcomes of clinical standard-risk patients with high-risk cytogenetics were equivalent to outcomes of clinical high-risk patients.
The team therefore believes that screening BCP-ALL patients for the aforementioned genetic abnormalities at relapse will improve patient stratification and outcomes. ![]()
Apparent Zika transmission via platelet transfusion
![]()
Photo from Flickr
Researchers have reported 2 cases in which the Zika virus seems to have been transmitted via platelet transfusion.
Brazilian health officials previously announced 2 cases of Zika virus—in a liver transplant recipient and a gunshot victim—that likely resulted from blood transfusions performed in 2015.
Now, researchers have reported possible transmission via transfusion in 2 more Brazilians—a patient with primary myelofibrosis (PMF) and one with acute myeloid leukemia (AML).
The researchers described these cases in a letter to NEJM.
An individual who donated platelets via apheresis on January 16, 2016, later tested positive for the Zika virus.
On January 19, platelets from that donor were transfused into a 54-year-old woman with PMF and a 14-year-old girl with AML who had also received a haploidentical bone marrow transplant on January 6.
The platelet donor called the blood bank on January 21 to report worrying symptoms—a cutaneous rash, retro-orbital pain, and pain in both knees—that had developed on January 18.
Subsequent testing revealed that the donor was negative for chikungunya and dengue virus. However, both plasma and urine samples tested positive for Zika virus.
Pre-transfusion samples collected from both of the recipients were negative for chikungunya, dengue, and Zika. However, samples collected after the transfusions—6 days after for the woman with PMF and 23 to 51 days after for the girl with AML—tested positive for Zika.
Researchers performed molecular sequencing and phylogenetic analysis of Zika virus RNA isolated from the donor and the recipients. Isolates from all 3 parties had nucleotide changes in the envelope gene (codons 11 and 186) that were not observed among other available isolates from Brazil.
The researchers said these results suggest the platelet transfusions were the source of Zika virus infection in the PMF patient and the AML patient.
Although both patients could have been exposed to Zika-carrying mosquitoes, both the RNA results and the timing of Zika infection suggest the transfusions were the cause. ![]()
![]()
Photo from Flickr
Researchers have reported 2 cases in which the Zika virus seems to have been transmitted via platelet transfusion.
Brazilian health officials previously announced 2 cases of Zika virus—in a liver transplant recipient and a gunshot victim—that likely resulted from blood transfusions performed in 2015.
Now, researchers have reported possible transmission via transfusion in 2 more Brazilians—a patient with primary myelofibrosis (PMF) and one with acute myeloid leukemia (AML).
The researchers described these cases in a letter to NEJM.
An individual who donated platelets via apheresis on January 16, 2016, later tested positive for the Zika virus.
On January 19, platelets from that donor were transfused into a 54-year-old woman with PMF and a 14-year-old girl with AML who had also received a haploidentical bone marrow transplant on January 6.
The platelet donor called the blood bank on January 21 to report worrying symptoms—a cutaneous rash, retro-orbital pain, and pain in both knees—that had developed on January 18.
Subsequent testing revealed that the donor was negative for chikungunya and dengue virus. However, both plasma and urine samples tested positive for Zika virus.
Pre-transfusion samples collected from both of the recipients were negative for chikungunya, dengue, and Zika. However, samples collected after the transfusions—6 days after for the woman with PMF and 23 to 51 days after for the girl with AML—tested positive for Zika.
Researchers performed molecular sequencing and phylogenetic analysis of Zika virus RNA isolated from the donor and the recipients. Isolates from all 3 parties had nucleotide changes in the envelope gene (codons 11 and 186) that were not observed among other available isolates from Brazil.
The researchers said these results suggest the platelet transfusions were the source of Zika virus infection in the PMF patient and the AML patient.
Although both patients could have been exposed to Zika-carrying mosquitoes, both the RNA results and the timing of Zika infection suggest the transfusions were the cause. ![]()
![]()
Photo from Flickr
Researchers have reported 2 cases in which the Zika virus seems to have been transmitted via platelet transfusion.
Brazilian health officials previously announced 2 cases of Zika virus—in a liver transplant recipient and a gunshot victim—that likely resulted from blood transfusions performed in 2015.
Now, researchers have reported possible transmission via transfusion in 2 more Brazilians—a patient with primary myelofibrosis (PMF) and one with acute myeloid leukemia (AML).
The researchers described these cases in a letter to NEJM.
An individual who donated platelets via apheresis on January 16, 2016, later tested positive for the Zika virus.
On January 19, platelets from that donor were transfused into a 54-year-old woman with PMF and a 14-year-old girl with AML who had also received a haploidentical bone marrow transplant on January 6.
The platelet donor called the blood bank on January 21 to report worrying symptoms—a cutaneous rash, retro-orbital pain, and pain in both knees—that had developed on January 18.
Subsequent testing revealed that the donor was negative for chikungunya and dengue virus. However, both plasma and urine samples tested positive for Zika virus.
Pre-transfusion samples collected from both of the recipients were negative for chikungunya, dengue, and Zika. However, samples collected after the transfusions—6 days after for the woman with PMF and 23 to 51 days after for the girl with AML—tested positive for Zika.
Researchers performed molecular sequencing and phylogenetic analysis of Zika virus RNA isolated from the donor and the recipients. Isolates from all 3 parties had nucleotide changes in the envelope gene (codons 11 and 186) that were not observed among other available isolates from Brazil.
The researchers said these results suggest the platelet transfusions were the source of Zika virus infection in the PMF patient and the AML patient.
Although both patients could have been exposed to Zika-carrying mosquitoes, both the RNA results and the timing of Zika infection suggest the transfusions were the cause. ![]()
Study supports expanded prenatal genetic testing

Photo by Nina Matthews
New research suggests expanded prenatal genetic testing may increase the detection of carrier status for potentially serious genetic conditions, including hemoglobinopathies.
Researchers analyzed nearly 350,000 adults of diverse racial and ethnic backgrounds and found evidence to suggest that expanded screening for up to 94 conditions can increase the detection of carrier status when compared with current genetic testing recommendations from professional societies.
Imran S. Haque, PhD, of Counsyl in San Francisco, California, and his colleagues reported these findings in JAMA. The study was funded by Counsyl, a laboratory providing expanded carrier screening.
Genetic testing of prospective parents to detect carriers of specific inherited recessive diseases is part of routine obstetrical practice. The current recommendations are to test for a limited number of individual diseases, in part based on self-reported racial/ethnic background.
Dr Haque and his colleagues wanted to determine if recent advances in genetic testing could facilitate screening for an expanded number of conditions independent of racial/ethnic background.
The researchers analyzed results from expanded carrier screening in 346,790 reproductive-aged individuals, primarily from the US, without known indication for specific genetic testing.
The individuals were tested for carrier status for up to 94 conditions. Tests were offered by clinicians providing reproductive care.
Risk was defined as the probability that a hypothetical fetus created from a random pairing of individuals (within or across 15 self-reported racial/ethnic categories) would be homozygous or compound heterozygous for 2 mutations presumed to cause severe or profound disease.
Severe conditions were defined as those that, if left untreated, cause intellectual disability or a substantially shortened lifespan. Profound conditions were those causing both intellectual disability and a shortened lifespan.
The researchers found that, in most racial/ethnic categories, expanded carrier screening modeled more hypothetical fetuses at risk for severe or profound conditions than did screening based on current professional guidelines.
Overall, relative to expanded carrier screening, guideline-based screening ranged from identifying 6% of hypothetical fetuses affected for East Asian couples to 87% for African or African American couples.
Though this study suggests expanded screening could be beneficial, the researchers said their findings should be confirmed with prospective studies comparing current carrier screening with expanded screening in at-risk populations. ![]()
Photo by Nina Matthews
New research suggests expanded prenatal genetic testing may increase the detection of carrier status for potentially serious genetic conditions, including hemoglobinopathies.
Researchers analyzed nearly 350,000 adults of diverse racial and ethnic backgrounds and found evidence to suggest that expanded screening for up to 94 conditions can increase the detection of carrier status when compared with current genetic testing recommendations from professional societies.
Imran S. Haque, PhD, of Counsyl in San Francisco, California, and his colleagues reported these findings in JAMA. The study was funded by Counsyl, a laboratory providing expanded carrier screening.
Genetic testing of prospective parents to detect carriers of specific inherited recessive diseases is part of routine obstetrical practice. The current recommendations are to test for a limited number of individual diseases, in part based on self-reported racial/ethnic background.
Dr Haque and his colleagues wanted to determine if recent advances in genetic testing could facilitate screening for an expanded number of conditions independent of racial/ethnic background.
The researchers analyzed results from expanded carrier screening in 346,790 reproductive-aged individuals, primarily from the US, without known indication for specific genetic testing.
The individuals were tested for carrier status for up to 94 conditions. Tests were offered by clinicians providing reproductive care.
Risk was defined as the probability that a hypothetical fetus created from a random pairing of individuals (within or across 15 self-reported racial/ethnic categories) would be homozygous or compound heterozygous for 2 mutations presumed to cause severe or profound disease.
Severe conditions were defined as those that, if left untreated, cause intellectual disability or a substantially shortened lifespan. Profound conditions were those causing both intellectual disability and a shortened lifespan.
The researchers found that, in most racial/ethnic categories, expanded carrier screening modeled more hypothetical fetuses at risk for severe or profound conditions than did screening based on current professional guidelines.
Overall, relative to expanded carrier screening, guideline-based screening ranged from identifying 6% of hypothetical fetuses affected for East Asian couples to 87% for African or African American couples.
Though this study suggests expanded screening could be beneficial, the researchers said their findings should be confirmed with prospective studies comparing current carrier screening with expanded screening in at-risk populations. ![]()
Photo by Nina Matthews
New research suggests expanded prenatal genetic testing may increase the detection of carrier status for potentially serious genetic conditions, including hemoglobinopathies.
Researchers analyzed nearly 350,000 adults of diverse racial and ethnic backgrounds and found evidence to suggest that expanded screening for up to 94 conditions can increase the detection of carrier status when compared with current genetic testing recommendations from professional societies.
Imran S. Haque, PhD, of Counsyl in San Francisco, California, and his colleagues reported these findings in JAMA. The study was funded by Counsyl, a laboratory providing expanded carrier screening.
Genetic testing of prospective parents to detect carriers of specific inherited recessive diseases is part of routine obstetrical practice. The current recommendations are to test for a limited number of individual diseases, in part based on self-reported racial/ethnic background.
Dr Haque and his colleagues wanted to determine if recent advances in genetic testing could facilitate screening for an expanded number of conditions independent of racial/ethnic background.
The researchers analyzed results from expanded carrier screening in 346,790 reproductive-aged individuals, primarily from the US, without known indication for specific genetic testing.
The individuals were tested for carrier status for up to 94 conditions. Tests were offered by clinicians providing reproductive care.
Risk was defined as the probability that a hypothetical fetus created from a random pairing of individuals (within or across 15 self-reported racial/ethnic categories) would be homozygous or compound heterozygous for 2 mutations presumed to cause severe or profound disease.
Severe conditions were defined as those that, if left untreated, cause intellectual disability or a substantially shortened lifespan. Profound conditions were those causing both intellectual disability and a shortened lifespan.
The researchers found that, in most racial/ethnic categories, expanded carrier screening modeled more hypothetical fetuses at risk for severe or profound conditions than did screening based on current professional guidelines.
Overall, relative to expanded carrier screening, guideline-based screening ranged from identifying 6% of hypothetical fetuses affected for East Asian couples to 87% for African or African American couples.
Though this study suggests expanded screening could be beneficial, the researchers said their findings should be confirmed with prospective studies comparing current carrier screening with expanded screening in at-risk populations. ![]()
Immunogene therapy granted conditional authorization

Image by NIAID
The European Commission (EC) has granted conditional marketing authorization for an immunogene therapy known as Zalmoxis.
This means Zalmoxis can be marketed in the European Economic Area as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults with high-risk hematologic malignancies who are receiving a haploidentical hematopoietic stem cell transplant (haplo-HSCT).
Zalmoxis consists of allogeneic T cells genetically modified to express both a truncated form of the human low-affinity nerve growth factor receptor (ΔLNGFR), as a cell-surface selectable marker, and the suicide gene herpes simplex I virus thymidine kinase (HSV-TK Mut2).
The modified T cells are given to haplo-HSCT recipients to help fight off infection, enhance the success of the transplant, and support long-lasting anticancer effects.
Because the T cells can also cause GVHD, they are equipped with the suicide gene, which makes them susceptible to treatment with ganciclovir or valganciclovir. So if a patient develops GVHD, he or she can receive ganciclovir/valganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A. The company said the treatment should become available in the first European market during the first half of 2017.
About conditional marketing authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
Under the provisions of the conditional marketing authorization for Zalmoxis, MolMed will be required to complete a post-marketing study aimed at confirming the clinical benefit of the treatment.
The European Medicines Agency’s Committee for Medicinal Products for Human Use has accepted the ongoing phase 3 TK008 trial as a post-marketing confirmatory study.
Trials of Zalmoxis
The EC’s decision to grant Zalmoxis conditional marketing authorization was based on cumulative efficacy and safety data collected from patients enrolled in a phase 1/2 trial (TK007) and an ongoing phase 3 trial (TK008).
The Zalmoxis group comprised 30 patients from the TK007 trial and 15 patients from the experimental arm of the TK008 trial. The TK007 trial included haplo-HSCT recipients with various high-risk hematologic malignancies, and the TK008 trial is enrolling haplo-HSCT recipients with high-risk acute leukemia.
The data thus far have indicated that Zalmoxis can provide rapid immune reconstitution, an anti-leukemia effect, and complete control of GVHD, in the absence of any post-transplant immunosuppression.
Overall, these effects led to a clinically meaningful increase in survival rates in Zalmoxis-treated patients, when compared to historical controls from the European Society for Blood and Marrow Transplantation (EBMT) database.
The only adverse event related to Zalmoxis treatment was GVHD, which was fully resolved by the activation of the suicide gene system with ganciclovir treatment, without any GVHD-related death.
Detailed results of this analysis are set to be presented during the MolMed-sponsored symposium “A new era of haplo-transplantation” at the EBMT International Transplant Course, which is scheduled to take place September 9-11 in Barcelona, Spain. ![]()
Image by NIAID
The European Commission (EC) has granted conditional marketing authorization for an immunogene therapy known as Zalmoxis.
This means Zalmoxis can be marketed in the European Economic Area as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults with high-risk hematologic malignancies who are receiving a haploidentical hematopoietic stem cell transplant (haplo-HSCT).
Zalmoxis consists of allogeneic T cells genetically modified to express both a truncated form of the human low-affinity nerve growth factor receptor (ΔLNGFR), as a cell-surface selectable marker, and the suicide gene herpes simplex I virus thymidine kinase (HSV-TK Mut2).
The modified T cells are given to haplo-HSCT recipients to help fight off infection, enhance the success of the transplant, and support long-lasting anticancer effects.
Because the T cells can also cause GVHD, they are equipped with the suicide gene, which makes them susceptible to treatment with ganciclovir or valganciclovir. So if a patient develops GVHD, he or she can receive ganciclovir/valganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A. The company said the treatment should become available in the first European market during the first half of 2017.
About conditional marketing authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
Under the provisions of the conditional marketing authorization for Zalmoxis, MolMed will be required to complete a post-marketing study aimed at confirming the clinical benefit of the treatment.
The European Medicines Agency’s Committee for Medicinal Products for Human Use has accepted the ongoing phase 3 TK008 trial as a post-marketing confirmatory study.
Trials of Zalmoxis
The EC’s decision to grant Zalmoxis conditional marketing authorization was based on cumulative efficacy and safety data collected from patients enrolled in a phase 1/2 trial (TK007) and an ongoing phase 3 trial (TK008).
The Zalmoxis group comprised 30 patients from the TK007 trial and 15 patients from the experimental arm of the TK008 trial. The TK007 trial included haplo-HSCT recipients with various high-risk hematologic malignancies, and the TK008 trial is enrolling haplo-HSCT recipients with high-risk acute leukemia.
The data thus far have indicated that Zalmoxis can provide rapid immune reconstitution, an anti-leukemia effect, and complete control of GVHD, in the absence of any post-transplant immunosuppression.
Overall, these effects led to a clinically meaningful increase in survival rates in Zalmoxis-treated patients, when compared to historical controls from the European Society for Blood and Marrow Transplantation (EBMT) database.
The only adverse event related to Zalmoxis treatment was GVHD, which was fully resolved by the activation of the suicide gene system with ganciclovir treatment, without any GVHD-related death.
Detailed results of this analysis are set to be presented during the MolMed-sponsored symposium “A new era of haplo-transplantation” at the EBMT International Transplant Course, which is scheduled to take place September 9-11 in Barcelona, Spain. ![]()
Image by NIAID
The European Commission (EC) has granted conditional marketing authorization for an immunogene therapy known as Zalmoxis.
This means Zalmoxis can be marketed in the European Economic Area as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults with high-risk hematologic malignancies who are receiving a haploidentical hematopoietic stem cell transplant (haplo-HSCT).
Zalmoxis consists of allogeneic T cells genetically modified to express both a truncated form of the human low-affinity nerve growth factor receptor (ΔLNGFR), as a cell-surface selectable marker, and the suicide gene herpes simplex I virus thymidine kinase (HSV-TK Mut2).
The modified T cells are given to haplo-HSCT recipients to help fight off infection, enhance the success of the transplant, and support long-lasting anticancer effects.
Because the T cells can also cause GVHD, they are equipped with the suicide gene, which makes them susceptible to treatment with ganciclovir or valganciclovir. So if a patient develops GVHD, he or she can receive ganciclovir/valganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A. The company said the treatment should become available in the first European market during the first half of 2017.
About conditional marketing authorization
Conditional marketing authorization represents an expedited path for approval. The EC grants this type of authorization before pivotal registration studies are completed.
Conditional marketing authorization is granted to products whose benefits are thought to outweigh their risks, products that address unmet needs, and products that are expected to provide a significant public health benefit.
Under the provisions of the conditional marketing authorization for Zalmoxis, MolMed will be required to complete a post-marketing study aimed at confirming the clinical benefit of the treatment.
The European Medicines Agency’s Committee for Medicinal Products for Human Use has accepted the ongoing phase 3 TK008 trial as a post-marketing confirmatory study.
Trials of Zalmoxis
The EC’s decision to grant Zalmoxis conditional marketing authorization was based on cumulative efficacy and safety data collected from patients enrolled in a phase 1/2 trial (TK007) and an ongoing phase 3 trial (TK008).
The Zalmoxis group comprised 30 patients from the TK007 trial and 15 patients from the experimental arm of the TK008 trial. The TK007 trial included haplo-HSCT recipients with various high-risk hematologic malignancies, and the TK008 trial is enrolling haplo-HSCT recipients with high-risk acute leukemia.
The data thus far have indicated that Zalmoxis can provide rapid immune reconstitution, an anti-leukemia effect, and complete control of GVHD, in the absence of any post-transplant immunosuppression.
Overall, these effects led to a clinically meaningful increase in survival rates in Zalmoxis-treated patients, when compared to historical controls from the European Society for Blood and Marrow Transplantation (EBMT) database.
The only adverse event related to Zalmoxis treatment was GVHD, which was fully resolved by the activation of the suicide gene system with ganciclovir treatment, without any GVHD-related death.
Detailed results of this analysis are set to be presented during the MolMed-sponsored symposium “A new era of haplo-transplantation” at the EBMT International Transplant Course, which is scheduled to take place September 9-11 in Barcelona, Spain.
Sociodemographic factors impact OS in MM

Photo courtesy of NCI
New research has revealed sociodemographic factors that appear to influence survival in younger patients with multiple myeloma (MM).
The study, which included more than 10,000 MM patients under the age of 65, suggested that marital status, insurance status, and county-level income all affect a patient’s chance of survival.
However, race/ethnicity and county-level educational achievement were not associated with overall survival (OS).
Luciano Costa, MD, PhD, of the University of Alabama at Birmingham, and his colleagues reported these findings in Cancer.
The researchers analyzed data on patients who were diagnosed with MM before the age of 65, between 2007 and 2012. There were 10,161 cases of MM, and the median follow-up was 27 months (range, 0-71 months).
The sociodemographic variables assessed were marital status, insurance status, median household income in the county of residence, and county-level educational achievement.
The researchers also looked at race/ethnicity, which was defined as a self-reported construct including Hispanic, non-Hispanic black, non-Hispanic white, and “other.”
The researchers found that patients had an increased risk of death if they were not married, did not have private insurance (were uninsured or on Medicaid), or lived in a low-income area (belonging to the lowest 2 income quartiles).
The 4-year estimated OS rate was 71.1% for patients who did not have any of these adverse sociodemographic factors, but it was 63.2% for patients with 1 factor, 53.4% for patients with 2 factors, and 46.5% for patients with all 3 factors (P<0.001).
The researchers did find that Hispanic and non-Hispanic black individuals had more adverse sociodemographic factors and worse OS than non-Hispanic whites and people belonging to the “other” category.
However, when the population was stratified by the cumulative number of sociodemographic factors and the researchers adjusted for confounders, there was no consistent association between race/ethnicity and OS.
The researchers said this suggests the apparent impact of race/ethnicity on OS may be due to the disparate distribution of sociodemographic factors observed among the different races/ethnicities rather than the race/ethnicity construct itself.
“This [study] strongly suggests that there is a huge disparity in outcomes that could potentially be overcome by improving access and affordability of treatments,” Dr Costa said.
“With the recent emphasis on comparative effectiveness in oncology, it also becomes crucial that all variables affecting outcomes—including sociodemographic factors—are accounted for when comparisons between different therapeutic approaches and healthcare systems are made.”
Photo courtesy of NCI
New research has revealed sociodemographic factors that appear to influence survival in younger patients with multiple myeloma (MM).
The study, which included more than 10,000 MM patients under the age of 65, suggested that marital status, insurance status, and county-level income all affect a patient’s chance of survival.
However, race/ethnicity and county-level educational achievement were not associated with overall survival (OS).
Luciano Costa, MD, PhD, of the University of Alabama at Birmingham, and his colleagues reported these findings in Cancer.
The researchers analyzed data on patients who were diagnosed with MM before the age of 65, between 2007 and 2012. There were 10,161 cases of MM, and the median follow-up was 27 months (range, 0-71 months).
The sociodemographic variables assessed were marital status, insurance status, median household income in the county of residence, and county-level educational achievement.
The researchers also looked at race/ethnicity, which was defined as a self-reported construct including Hispanic, non-Hispanic black, non-Hispanic white, and “other.”
The researchers found that patients had an increased risk of death if they were not married, did not have private insurance (were uninsured or on Medicaid), or lived in a low-income area (belonging to the lowest 2 income quartiles).
The 4-year estimated OS rate was 71.1% for patients who did not have any of these adverse sociodemographic factors, but it was 63.2% for patients with 1 factor, 53.4% for patients with 2 factors, and 46.5% for patients with all 3 factors (P<0.001).
The researchers did find that Hispanic and non-Hispanic black individuals had more adverse sociodemographic factors and worse OS than non-Hispanic whites and people belonging to the “other” category.
However, when the population was stratified by the cumulative number of sociodemographic factors and the researchers adjusted for confounders, there was no consistent association between race/ethnicity and OS.
The researchers said this suggests the apparent impact of race/ethnicity on OS may be due to the disparate distribution of sociodemographic factors observed among the different races/ethnicities rather than the race/ethnicity construct itself.
“This [study] strongly suggests that there is a huge disparity in outcomes that could potentially be overcome by improving access and affordability of treatments,” Dr Costa said.
“With the recent emphasis on comparative effectiveness in oncology, it also becomes crucial that all variables affecting outcomes—including sociodemographic factors—are accounted for when comparisons between different therapeutic approaches and healthcare systems are made.”
Photo courtesy of NCI
New research has revealed sociodemographic factors that appear to influence survival in younger patients with multiple myeloma (MM).
The study, which included more than 10,000 MM patients under the age of 65, suggested that marital status, insurance status, and county-level income all affect a patient’s chance of survival.
However, race/ethnicity and county-level educational achievement were not associated with overall survival (OS).
Luciano Costa, MD, PhD, of the University of Alabama at Birmingham, and his colleagues reported these findings in Cancer.
The researchers analyzed data on patients who were diagnosed with MM before the age of 65, between 2007 and 2012. There were 10,161 cases of MM, and the median follow-up was 27 months (range, 0-71 months).
The sociodemographic variables assessed were marital status, insurance status, median household income in the county of residence, and county-level educational achievement.
The researchers also looked at race/ethnicity, which was defined as a self-reported construct including Hispanic, non-Hispanic black, non-Hispanic white, and “other.”
The researchers found that patients had an increased risk of death if they were not married, did not have private insurance (were uninsured or on Medicaid), or lived in a low-income area (belonging to the lowest 2 income quartiles).
The 4-year estimated OS rate was 71.1% for patients who did not have any of these adverse sociodemographic factors, but it was 63.2% for patients with 1 factor, 53.4% for patients with 2 factors, and 46.5% for patients with all 3 factors (P<0.001).
The researchers did find that Hispanic and non-Hispanic black individuals had more adverse sociodemographic factors and worse OS than non-Hispanic whites and people belonging to the “other” category.
However, when the population was stratified by the cumulative number of sociodemographic factors and the researchers adjusted for confounders, there was no consistent association between race/ethnicity and OS.
The researchers said this suggests the apparent impact of race/ethnicity on OS may be due to the disparate distribution of sociodemographic factors observed among the different races/ethnicities rather than the race/ethnicity construct itself.
“This [study] strongly suggests that there is a huge disparity in outcomes that could potentially be overcome by improving access and affordability of treatments,” Dr Costa said.
“With the recent emphasis on comparative effectiveness in oncology, it also becomes crucial that all variables affecting outcomes—including sociodemographic factors—are accounted for when comparisons between different therapeutic approaches and healthcare systems are made.”
Drug granted orphan designation for MAS
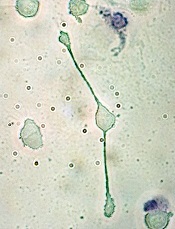
Image from Flickr
The US Food and Drug Administration (FDA) has granted orphan drug designation for dusquetide as a treatment for macrophage activation syndrome (MAS).
Dusquetide is an innate defense regulator, a new class of short, synthetic peptides that accelerate bacterial clearance and resolution of tissue damage while modulating inflammation following exposure to bacterial pathogens, radiation, chemotherapy, and other agents.
According to researchers, dusquetide has demonstrated preclinical efficacy and safety in several animal models.
In a mouse model of MAS, dusquetide was shown to reduce pancytopenia, inhibit IL-12 responses, and improve body weight maintenance.
SGX942, the drug product containing dusquetide, has demonstrated safety in a phase 1 study of 84 healthy volunteers.
In addition, SGX942 has demonstrated preliminary efficacy and safety in an exploratory phase 2 study of 111 patients with oral mucositis due to chemoradiation therapy for head and neck cancer.
SGX942 is being developed by Solgenix, Inc.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved.
About MAS
MAS is a life-threatening complication of rheumatic disease that, for unknown reasons, frequently occurs in individuals with systemic juvenile idiopathic arthritis. MAS also occurs in patients with systemic lupus erythematosus, Kawasaki disease, adult-onset Still’s disease, and various vasculitic syndromes.
MAS is characterized by pancytopenia, liver insufficiency, coagulopathy, and neurologic symptoms.
MAS is thought to be caused by the activation and uncontrolled proliferation of T lymphocytes and well-differentiated macrophages, leading to widespread hemophagocytosis and cytokine overproduction.
Image from Flickr
The US Food and Drug Administration (FDA) has granted orphan drug designation for dusquetide as a treatment for macrophage activation syndrome (MAS).
Dusquetide is an innate defense regulator, a new class of short, synthetic peptides that accelerate bacterial clearance and resolution of tissue damage while modulating inflammation following exposure to bacterial pathogens, radiation, chemotherapy, and other agents.
According to researchers, dusquetide has demonstrated preclinical efficacy and safety in several animal models.
In a mouse model of MAS, dusquetide was shown to reduce pancytopenia, inhibit IL-12 responses, and improve body weight maintenance.
SGX942, the drug product containing dusquetide, has demonstrated safety in a phase 1 study of 84 healthy volunteers.
In addition, SGX942 has demonstrated preliminary efficacy and safety in an exploratory phase 2 study of 111 patients with oral mucositis due to chemoradiation therapy for head and neck cancer.
SGX942 is being developed by Solgenix, Inc.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved.
About MAS
MAS is a life-threatening complication of rheumatic disease that, for unknown reasons, frequently occurs in individuals with systemic juvenile idiopathic arthritis. MAS also occurs in patients with systemic lupus erythematosus, Kawasaki disease, adult-onset Still’s disease, and various vasculitic syndromes.
MAS is characterized by pancytopenia, liver insufficiency, coagulopathy, and neurologic symptoms.
MAS is thought to be caused by the activation and uncontrolled proliferation of T lymphocytes and well-differentiated macrophages, leading to widespread hemophagocytosis and cytokine overproduction.
Image from Flickr
The US Food and Drug Administration (FDA) has granted orphan drug designation for dusquetide as a treatment for macrophage activation syndrome (MAS).
Dusquetide is an innate defense regulator, a new class of short, synthetic peptides that accelerate bacterial clearance and resolution of tissue damage while modulating inflammation following exposure to bacterial pathogens, radiation, chemotherapy, and other agents.
According to researchers, dusquetide has demonstrated preclinical efficacy and safety in several animal models.
In a mouse model of MAS, dusquetide was shown to reduce pancytopenia, inhibit IL-12 responses, and improve body weight maintenance.
SGX942, the drug product containing dusquetide, has demonstrated safety in a phase 1 study of 84 healthy volunteers.
In addition, SGX942 has demonstrated preliminary efficacy and safety in an exploratory phase 2 study of 111 patients with oral mucositis due to chemoradiation therapy for head and neck cancer.
SGX942 is being developed by Solgenix, Inc.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved.
About MAS
MAS is a life-threatening complication of rheumatic disease that, for unknown reasons, frequently occurs in individuals with systemic juvenile idiopathic arthritis. MAS also occurs in patients with systemic lupus erythematosus, Kawasaki disease, adult-onset Still’s disease, and various vasculitic syndromes.
MAS is characterized by pancytopenia, liver insufficiency, coagulopathy, and neurologic symptoms.
MAS is thought to be caused by the activation and uncontrolled proliferation of T lymphocytes and well-differentiated macrophages, leading to widespread hemophagocytosis and cytokine overproduction.
VTE linked to permanent work-related disability

Image by Andre E.X. Brown
Unprovoked venous thromboembolism (VTE) may increase the risk of permanent work-related disability, according to research published in the Journal of Thrombosis and Haemostasis.
Researchers evaluated data from more than 60,000 people and found that individuals with unprovoked VTE had a 52% higher risk of work-related disability than those without VTE.
However, there was no association between provoked VTE and work-related disability.
In addition, only deep vein thrombosis (DVT)—not pulmonary embolism (PE)—was significantly associated with an increased risk of work-related disability.
For this study, the researchers analyzed data from the Tromsø Study and the Nord-Trøndelag Health Study, which enrolled 66,005 subjects from 1994 to 1997. The subjects’ mean age at inclusion was 41.3 (range, 20-65), and about half (51.2%, n=33,901) were women.
The subjects were followed through 2008. During the follow-up period, 384 individuals had a first VTE, and 9862 received a disability pension due to work-related disability.
Compared to individuals without VTE, those who had a VTE were slightly older and had a higher body mass index. VTE patients were more likely to have a history of cancer and cardiovascular disease, and they were more likely to have paying jobs.
In addition, subjects with VTE were more likely to have work-related disability. The crude incidence rate of disability was 37.5 per 1000 person-years among VTE patients and 13.5 per 1000 person-years among subjects without VTE.
VTE patients who received disability pension were slightly older than those who did not, and a higher proportion of the VTEs were DVTs rather than PEs. Half of the VTEs were unprovoked.
Multivariable analysis suggested that subjects with unprovoked VTE had a 52% higher risk of work-related disability than individuals without VTE (hazard ratio [HR]=1.52). However, there was no association between provoked VTE and work-related disability (HR=0.83).
When the researchers analyzed DVT and PE separately, they found that subjects with DVT had an 80% higher risk of work-related disability than those without DVT (HR=1.80). Subjects with PE had a moderately increased risk of work-related disability that was not statistically significant (HR=1.28).
When the researchers adjusted for baseline characteristics other than self-rated health, the association between work-related disability and DVT remained strong (HR=1.66), and there was no association between work-related disability and PE (HR=1.06).
The researchers said this study is the first of its kind to document a relationship between VTE and subsequent work-related disability. And the findings suggest the economic burden of VTE goes beyond costs to the healthcare system. The loss of economic output from people who are unable to work due to VTE-related complications is also a substantial economic cost of VTE.
Image by Andre E.X. Brown
Unprovoked venous thromboembolism (VTE) may increase the risk of permanent work-related disability, according to research published in the Journal of Thrombosis and Haemostasis.
Researchers evaluated data from more than 60,000 people and found that individuals with unprovoked VTE had a 52% higher risk of work-related disability than those without VTE.
However, there was no association between provoked VTE and work-related disability.
In addition, only deep vein thrombosis (DVT)—not pulmonary embolism (PE)—was significantly associated with an increased risk of work-related disability.
For this study, the researchers analyzed data from the Tromsø Study and the Nord-Trøndelag Health Study, which enrolled 66,005 subjects from 1994 to 1997. The subjects’ mean age at inclusion was 41.3 (range, 20-65), and about half (51.2%, n=33,901) were women.
The subjects were followed through 2008. During the follow-up period, 384 individuals had a first VTE, and 9862 received a disability pension due to work-related disability.
Compared to individuals without VTE, those who had a VTE were slightly older and had a higher body mass index. VTE patients were more likely to have a history of cancer and cardiovascular disease, and they were more likely to have paying jobs.
In addition, subjects with VTE were more likely to have work-related disability. The crude incidence rate of disability was 37.5 per 1000 person-years among VTE patients and 13.5 per 1000 person-years among subjects without VTE.
VTE patients who received disability pension were slightly older than those who did not, and a higher proportion of the VTEs were DVTs rather than PEs. Half of the VTEs were unprovoked.
Multivariable analysis suggested that subjects with unprovoked VTE had a 52% higher risk of work-related disability than individuals without VTE (hazard ratio [HR]=1.52). However, there was no association between provoked VTE and work-related disability (HR=0.83).
When the researchers analyzed DVT and PE separately, they found that subjects with DVT had an 80% higher risk of work-related disability than those without DVT (HR=1.80). Subjects with PE had a moderately increased risk of work-related disability that was not statistically significant (HR=1.28).
When the researchers adjusted for baseline characteristics other than self-rated health, the association between work-related disability and DVT remained strong (HR=1.66), and there was no association between work-related disability and PE (HR=1.06).
The researchers said this study is the first of its kind to document a relationship between VTE and subsequent work-related disability. And the findings suggest the economic burden of VTE goes beyond costs to the healthcare system. The loss of economic output from people who are unable to work due to VTE-related complications is also a substantial economic cost of VTE.
Image by Andre E.X. Brown
Unprovoked venous thromboembolism (VTE) may increase the risk of permanent work-related disability, according to research published in the Journal of Thrombosis and Haemostasis.
Researchers evaluated data from more than 60,000 people and found that individuals with unprovoked VTE had a 52% higher risk of work-related disability than those without VTE.
However, there was no association between provoked VTE and work-related disability.
In addition, only deep vein thrombosis (DVT)—not pulmonary embolism (PE)—was significantly associated with an increased risk of work-related disability.
For this study, the researchers analyzed data from the Tromsø Study and the Nord-Trøndelag Health Study, which enrolled 66,005 subjects from 1994 to 1997. The subjects’ mean age at inclusion was 41.3 (range, 20-65), and about half (51.2%, n=33,901) were women.
The subjects were followed through 2008. During the follow-up period, 384 individuals had a first VTE, and 9862 received a disability pension due to work-related disability.
Compared to individuals without VTE, those who had a VTE were slightly older and had a higher body mass index. VTE patients were more likely to have a history of cancer and cardiovascular disease, and they were more likely to have paying jobs.
In addition, subjects with VTE were more likely to have work-related disability. The crude incidence rate of disability was 37.5 per 1000 person-years among VTE patients and 13.5 per 1000 person-years among subjects without VTE.
VTE patients who received disability pension were slightly older than those who did not, and a higher proportion of the VTEs were DVTs rather than PEs. Half of the VTEs were unprovoked.
Multivariable analysis suggested that subjects with unprovoked VTE had a 52% higher risk of work-related disability than individuals without VTE (hazard ratio [HR]=1.52). However, there was no association between provoked VTE and work-related disability (HR=0.83).
When the researchers analyzed DVT and PE separately, they found that subjects with DVT had an 80% higher risk of work-related disability than those without DVT (HR=1.80). Subjects with PE had a moderately increased risk of work-related disability that was not statistically significant (HR=1.28).
When the researchers adjusted for baseline characteristics other than self-rated health, the association between work-related disability and DVT remained strong (HR=1.66), and there was no association between work-related disability and PE (HR=1.06).
The researchers said this study is the first of its kind to document a relationship between VTE and subsequent work-related disability. And the findings suggest the economic burden of VTE goes beyond costs to the healthcare system. The loss of economic output from people who are unable to work due to VTE-related complications is also a substantial economic cost of VTE.