User login
Chemo linked to long-term learning problems in ALL survivors

Pediatric acute lymphoblastic leukemia (ALL) patients treated with chemotherapy alone are at risk for attention and learning problems that persist after treatment ends, according to a study published in the Journal of Clinical Oncology.
Researchers said this study is the largest assessment to date of neurocognitive outcomes in pediatric ALL survivors treated with intensive chemotherapy alone rather than in combination with cranial radiation.
“This is an important contribution to the literature because the smaller size and design of previous studies made examining the impact of treatment difficult,” said study author Lisa Jacola, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The findings underscore the need for neurocognitive and academic screening to be included as part of routine survivorship care for all pediatric ALL survivors.”
The study included patients enrolled in the St. Jude Total Therapy Study XV. They underwent neurocognitive assessments at the beginning of induction (n=142), end of maintenance (n=243), and 2 years after completing treatment (n=211).
The subjects completed standardized tests of overall intelligence, attention, learning, and academic performance. In addition, parents and other caregivers rated the subjects’ attention, learning, and behavior.
Two years after treatment completion, subjects performed as expected for their age (compared to national data) on measures of overall intelligence, learning, and memory. However, study subjects had significant attention deficits and a significantly greater frequency of learning problems (all P≤0.005).
The risk of such problems was greatest for survivors who were less than 5 years old when diagnosed with ALL and for those who received more intensive chemotherapy.
Specifically, the younger subjects had a greater risk of difficulties with attention, learning, working memory, and processing speed (all P≤0.05). And subjects who received higher-intensity, CNS-directed chemotherapy had a greater risk of difficulties in attention, processing speed, and academics (all P≤0.01).
Researchers also found that subjects with attention problems at the end of therapy had lower academic scores 2 years later (all P≤0.05).
“These findings provide additional evidence that neurocognitive functioning has improved in survivors of childhood ALL since cranial irradiation was replaced with intensified chemotherapy,” Dr Jacola said.
“But we also show these young people are at an elevated risk for attention problems that have real-world consequences, particularly for learning and school performance. Attention is a building block for learning, and, in this study, attention difficulties predicted academic problems later. If we know attention problems seen at the end of therapy continue and contribute to academic problems, then our goal is to intervene earlier to reduce or prevent such difficulties.” ![]()
Pediatric acute lymphoblastic leukemia (ALL) patients treated with chemotherapy alone are at risk for attention and learning problems that persist after treatment ends, according to a study published in the Journal of Clinical Oncology.
Researchers said this study is the largest assessment to date of neurocognitive outcomes in pediatric ALL survivors treated with intensive chemotherapy alone rather than in combination with cranial radiation.
“This is an important contribution to the literature because the smaller size and design of previous studies made examining the impact of treatment difficult,” said study author Lisa Jacola, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The findings underscore the need for neurocognitive and academic screening to be included as part of routine survivorship care for all pediatric ALL survivors.”
The study included patients enrolled in the St. Jude Total Therapy Study XV. They underwent neurocognitive assessments at the beginning of induction (n=142), end of maintenance (n=243), and 2 years after completing treatment (n=211).
The subjects completed standardized tests of overall intelligence, attention, learning, and academic performance. In addition, parents and other caregivers rated the subjects’ attention, learning, and behavior.
Two years after treatment completion, subjects performed as expected for their age (compared to national data) on measures of overall intelligence, learning, and memory. However, study subjects had significant attention deficits and a significantly greater frequency of learning problems (all P≤0.005).
The risk of such problems was greatest for survivors who were less than 5 years old when diagnosed with ALL and for those who received more intensive chemotherapy.
Specifically, the younger subjects had a greater risk of difficulties with attention, learning, working memory, and processing speed (all P≤0.05). And subjects who received higher-intensity, CNS-directed chemotherapy had a greater risk of difficulties in attention, processing speed, and academics (all P≤0.01).
Researchers also found that subjects with attention problems at the end of therapy had lower academic scores 2 years later (all P≤0.05).
“These findings provide additional evidence that neurocognitive functioning has improved in survivors of childhood ALL since cranial irradiation was replaced with intensified chemotherapy,” Dr Jacola said.
“But we also show these young people are at an elevated risk for attention problems that have real-world consequences, particularly for learning and school performance. Attention is a building block for learning, and, in this study, attention difficulties predicted academic problems later. If we know attention problems seen at the end of therapy continue and contribute to academic problems, then our goal is to intervene earlier to reduce or prevent such difficulties.” ![]()
Pediatric acute lymphoblastic leukemia (ALL) patients treated with chemotherapy alone are at risk for attention and learning problems that persist after treatment ends, according to a study published in the Journal of Clinical Oncology.
Researchers said this study is the largest assessment to date of neurocognitive outcomes in pediatric ALL survivors treated with intensive chemotherapy alone rather than in combination with cranial radiation.
“This is an important contribution to the literature because the smaller size and design of previous studies made examining the impact of treatment difficult,” said study author Lisa Jacola, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The findings underscore the need for neurocognitive and academic screening to be included as part of routine survivorship care for all pediatric ALL survivors.”
The study included patients enrolled in the St. Jude Total Therapy Study XV. They underwent neurocognitive assessments at the beginning of induction (n=142), end of maintenance (n=243), and 2 years after completing treatment (n=211).
The subjects completed standardized tests of overall intelligence, attention, learning, and academic performance. In addition, parents and other caregivers rated the subjects’ attention, learning, and behavior.
Two years after treatment completion, subjects performed as expected for their age (compared to national data) on measures of overall intelligence, learning, and memory. However, study subjects had significant attention deficits and a significantly greater frequency of learning problems (all P≤0.005).
The risk of such problems was greatest for survivors who were less than 5 years old when diagnosed with ALL and for those who received more intensive chemotherapy.
Specifically, the younger subjects had a greater risk of difficulties with attention, learning, working memory, and processing speed (all P≤0.05). And subjects who received higher-intensity, CNS-directed chemotherapy had a greater risk of difficulties in attention, processing speed, and academics (all P≤0.01).
Researchers also found that subjects with attention problems at the end of therapy had lower academic scores 2 years later (all P≤0.05).
“These findings provide additional evidence that neurocognitive functioning has improved in survivors of childhood ALL since cranial irradiation was replaced with intensified chemotherapy,” Dr Jacola said.
“But we also show these young people are at an elevated risk for attention problems that have real-world consequences, particularly for learning and school performance. Attention is a building block for learning, and, in this study, attention difficulties predicted academic problems later. If we know attention problems seen at the end of therapy continue and contribute to academic problems, then our goal is to intervene earlier to reduce or prevent such difficulties.” ![]()
Low doses of iron may cause cell damage
Image courtesy of NIH
Low doses of iron can modify the vascular endothelium and induce a DNA-damage response, researchers have reported in PLOS ONE.
The team observed these phenomena in vitro and said the results must be confirmed via additional research.
However, the findings suggest a need to assess the amount of iron given in standard treatments and the effects this may have on the body, according to Claire Shovlin, PhD, of Imperial College London in the UK.
Dr Shovlin and her colleagues studied human endothelial cells, adding either placebo or an iron solution of 10 micromolar, which is a similar concentration to that seen in the blood after taking an iron tablet.
Within 10 minutes, cells treated with the iron solution had activated DNA repair systems, and these were still activated 6 hours later.
“We already knew that iron could be damaging to cells in very high doses,” Dr Shovlin said. “However, in this study, we found that when we applied the kinds of levels of iron you would find in the bloodstream after taking an iron tablet, this also seemed to be able to trigger cell damage—at least in the laboratory. In other words, cells seem more sensitive to iron than we previously thought.”
“This is very early stage research, and we need more work to confirm these findings and investigate what effects this may have on the body. We are still not sure how these laboratory findings translate to blood vessels in the body.”
“However, this study helps to open the conversation about how much iron people take. At the moment, each standard iron tablet contains almost 10 times the amount of iron men are recommended to eat each day, and these dosages haven’t changed for more than 50 years. This research suggests we may need to think more carefully about how much iron we give to people, and try and tailor the dose to the patient.”
Dr Shovlin and her colleagues initially started researching this area after finding that a small proportion of people using iron tablets for hereditary hemorrhagic telangiectasia, which causes abnormalities in the blood vessels, reported their nose bleeds worsened after iron treatment. ![]()
Image courtesy of NIH
Low doses of iron can modify the vascular endothelium and induce a DNA-damage response, researchers have reported in PLOS ONE.
The team observed these phenomena in vitro and said the results must be confirmed via additional research.
However, the findings suggest a need to assess the amount of iron given in standard treatments and the effects this may have on the body, according to Claire Shovlin, PhD, of Imperial College London in the UK.
Dr Shovlin and her colleagues studied human endothelial cells, adding either placebo or an iron solution of 10 micromolar, which is a similar concentration to that seen in the blood after taking an iron tablet.
Within 10 minutes, cells treated with the iron solution had activated DNA repair systems, and these were still activated 6 hours later.
“We already knew that iron could be damaging to cells in very high doses,” Dr Shovlin said. “However, in this study, we found that when we applied the kinds of levels of iron you would find in the bloodstream after taking an iron tablet, this also seemed to be able to trigger cell damage—at least in the laboratory. In other words, cells seem more sensitive to iron than we previously thought.”
“This is very early stage research, and we need more work to confirm these findings and investigate what effects this may have on the body. We are still not sure how these laboratory findings translate to blood vessels in the body.”
“However, this study helps to open the conversation about how much iron people take. At the moment, each standard iron tablet contains almost 10 times the amount of iron men are recommended to eat each day, and these dosages haven’t changed for more than 50 years. This research suggests we may need to think more carefully about how much iron we give to people, and try and tailor the dose to the patient.”
Dr Shovlin and her colleagues initially started researching this area after finding that a small proportion of people using iron tablets for hereditary hemorrhagic telangiectasia, which causes abnormalities in the blood vessels, reported their nose bleeds worsened after iron treatment. ![]()
Image courtesy of NIH
Low doses of iron can modify the vascular endothelium and induce a DNA-damage response, researchers have reported in PLOS ONE.
The team observed these phenomena in vitro and said the results must be confirmed via additional research.
However, the findings suggest a need to assess the amount of iron given in standard treatments and the effects this may have on the body, according to Claire Shovlin, PhD, of Imperial College London in the UK.
Dr Shovlin and her colleagues studied human endothelial cells, adding either placebo or an iron solution of 10 micromolar, which is a similar concentration to that seen in the blood after taking an iron tablet.
Within 10 minutes, cells treated with the iron solution had activated DNA repair systems, and these were still activated 6 hours later.
“We already knew that iron could be damaging to cells in very high doses,” Dr Shovlin said. “However, in this study, we found that when we applied the kinds of levels of iron you would find in the bloodstream after taking an iron tablet, this also seemed to be able to trigger cell damage—at least in the laboratory. In other words, cells seem more sensitive to iron than we previously thought.”
“This is very early stage research, and we need more work to confirm these findings and investigate what effects this may have on the body. We are still not sure how these laboratory findings translate to blood vessels in the body.”
“However, this study helps to open the conversation about how much iron people take. At the moment, each standard iron tablet contains almost 10 times the amount of iron men are recommended to eat each day, and these dosages haven’t changed for more than 50 years. This research suggests we may need to think more carefully about how much iron we give to people, and try and tailor the dose to the patient.”
Dr Shovlin and her colleagues initially started researching this area after finding that a small proportion of people using iron tablets for hereditary hemorrhagic telangiectasia, which causes abnormalities in the blood vessels, reported their nose bleeds worsened after iron treatment. ![]()
NICE publishes guideline for multiple myeloma

Photo courtesy of NIH
The National Institute for Health and Care Excellence (NICE) has published a guideline containing recommendations for diagnosing, treating, and monitoring patients with multiple myeloma (MM).
The aim of the guideline is to help the National Health Service provide optimal care for MM patients over the age of 16 in England.
The guideline complements existing NICE guidance on the treatment of MM.
“Although there is no cure for myeloma, several novel drug treatments have been licensed in the past 10 years that have led to substantial improvements in the quality and length of time it is possible to live with the disease,” said Mark Baker, clinical practice director for NICE.
“However, there is still variation across the country in terms of providing a coherent and consistent approach to the management of myeloma. Myeloma is also a difficult condition to diagnose because many of the symptoms are non-specific. Our guideline sets out best-practice care to ensure people live as normal a life as possible for as long as possible.”
Some of the recommendations in the guideline include:
Communication and support: Offer prompt psychological assessment and support to MM patients at diagnosis and, as appropriate, at the beginning and end of each treatment, whenever the disease progresses, and when patients require end-of-life care.
Laboratory investigations to provide prognostic information: Use the same sample for all diagnostic and prognostic tests on bone marrow so patients only have to have one bone marrow aspirate and trephine biopsy.
Imaging for people with suspected MM: Offer imaging to all people with a plasma cell disorder suspected to be myeloma. Doctors should consider whole-body MRI as the first imaging procedure.
Service organizations: Each hospital treating MM patients should provide regional access through its network to facilities for intensive inpatient chemotherapy or transplantation, renal support, spinal disease management, specialized pain management, therapeutic apheresis, radiotherapy, restorative dentistry and oral surgery, and clinical trials, in particular early phase trials.
Managing relapsed MM: Offer a second autologous stem cell transplant to people with relapsed MM who are suitable and who have completed re-induction therapy without disease progression and had a response duration of more than 24 months after their first transplant. A second autologous stem cell transplant should be considered in people who have had a response duration of between 12 and 24 months after their first transplant. ![]()
Photo courtesy of NIH
The National Institute for Health and Care Excellence (NICE) has published a guideline containing recommendations for diagnosing, treating, and monitoring patients with multiple myeloma (MM).
The aim of the guideline is to help the National Health Service provide optimal care for MM patients over the age of 16 in England.
The guideline complements existing NICE guidance on the treatment of MM.
“Although there is no cure for myeloma, several novel drug treatments have been licensed in the past 10 years that have led to substantial improvements in the quality and length of time it is possible to live with the disease,” said Mark Baker, clinical practice director for NICE.
“However, there is still variation across the country in terms of providing a coherent and consistent approach to the management of myeloma. Myeloma is also a difficult condition to diagnose because many of the symptoms are non-specific. Our guideline sets out best-practice care to ensure people live as normal a life as possible for as long as possible.”
Some of the recommendations in the guideline include:
Communication and support: Offer prompt psychological assessment and support to MM patients at diagnosis and, as appropriate, at the beginning and end of each treatment, whenever the disease progresses, and when patients require end-of-life care.
Laboratory investigations to provide prognostic information: Use the same sample for all diagnostic and prognostic tests on bone marrow so patients only have to have one bone marrow aspirate and trephine biopsy.
Imaging for people with suspected MM: Offer imaging to all people with a plasma cell disorder suspected to be myeloma. Doctors should consider whole-body MRI as the first imaging procedure.
Service organizations: Each hospital treating MM patients should provide regional access through its network to facilities for intensive inpatient chemotherapy or transplantation, renal support, spinal disease management, specialized pain management, therapeutic apheresis, radiotherapy, restorative dentistry and oral surgery, and clinical trials, in particular early phase trials.
Managing relapsed MM: Offer a second autologous stem cell transplant to people with relapsed MM who are suitable and who have completed re-induction therapy without disease progression and had a response duration of more than 24 months after their first transplant. A second autologous stem cell transplant should be considered in people who have had a response duration of between 12 and 24 months after their first transplant. ![]()
Photo courtesy of NIH
The National Institute for Health and Care Excellence (NICE) has published a guideline containing recommendations for diagnosing, treating, and monitoring patients with multiple myeloma (MM).
The aim of the guideline is to help the National Health Service provide optimal care for MM patients over the age of 16 in England.
The guideline complements existing NICE guidance on the treatment of MM.
“Although there is no cure for myeloma, several novel drug treatments have been licensed in the past 10 years that have led to substantial improvements in the quality and length of time it is possible to live with the disease,” said Mark Baker, clinical practice director for NICE.
“However, there is still variation across the country in terms of providing a coherent and consistent approach to the management of myeloma. Myeloma is also a difficult condition to diagnose because many of the symptoms are non-specific. Our guideline sets out best-practice care to ensure people live as normal a life as possible for as long as possible.”
Some of the recommendations in the guideline include:
Communication and support: Offer prompt psychological assessment and support to MM patients at diagnosis and, as appropriate, at the beginning and end of each treatment, whenever the disease progresses, and when patients require end-of-life care.
Laboratory investigations to provide prognostic information: Use the same sample for all diagnostic and prognostic tests on bone marrow so patients only have to have one bone marrow aspirate and trephine biopsy.
Imaging for people with suspected MM: Offer imaging to all people with a plasma cell disorder suspected to be myeloma. Doctors should consider whole-body MRI as the first imaging procedure.
Service organizations: Each hospital treating MM patients should provide regional access through its network to facilities for intensive inpatient chemotherapy or transplantation, renal support, spinal disease management, specialized pain management, therapeutic apheresis, radiotherapy, restorative dentistry and oral surgery, and clinical trials, in particular early phase trials.
Managing relapsed MM: Offer a second autologous stem cell transplant to people with relapsed MM who are suitable and who have completed re-induction therapy without disease progression and had a response duration of more than 24 months after their first transplant. A second autologous stem cell transplant should be considered in people who have had a response duration of between 12 and 24 months after their first transplant. ![]()
Method may reduce toxicity of anticancer agents

Researchers believe they have found a way to make certain anticancer agents safer without compromising their efficacy.
The team noted that Aurora B kinase inhibitors and other agents targeting the cell cycle have proven effective but highly toxic in clinical trials.
In an attempt to solve this problem, the researchers turned to nanotechnology. They encapsulated the Aurora B kinase inhibitor AZD2811 in polymeric nanoparticles called Accurins.
Susan Ashton, of AstraZeneca in Macclesfield, Cheshire, UK, and her colleagues developed the Accurins and described the work in Science Translational Medicine. The work was funded by AstraZeneca.
The Accurins consist of block copolymers of poly-D,L-lactide and poly(ethylene glycol). The researchers used an ion pairing approach to efficiently encapsulate AZD2811 and control release of the drug.
They found the Accurins could release AZD2811 continuously for more than 1 week in vitro. The nanoparticles also reduced tumor phosphorylated histone H3 levels in vivo for up to 96 hours after a single administration.
The researchers tested the AZD2811 Accurins in mice with diffuse large B-cell lymphoma and rats with colorectal tumors. The nanoparticles accumulated specifically in tumors, where they slowly released AZD2811 to cancer cells.
Compared to AZD1152 (a water-soluble prodrug of AZD2811), the AZD2811 Accurins blocked tumor growth more effectively at one-half the drug dose and caused fewer side effects in the rodents.
Based on these results, the researchers said Accurins could provide efficacy and tolerability using a more convenient dosing regimen, which may extend the utility of Aurora B kinase inhibition to a broader range of hematologic and solid tumor malignancies.
A phase 1 study (NCT02579226) testing AZD2811 Accurins in advanced solid tumors is currently recruiting patients.
A related Focus article published in Science Translational Medicine offers more insights on how Accurin nanoparticles may help enhance the safety and antitumor activity of Aurora kinase inhibitors and other molecularly targeted drugs. ![]()
Researchers believe they have found a way to make certain anticancer agents safer without compromising their efficacy.
The team noted that Aurora B kinase inhibitors and other agents targeting the cell cycle have proven effective but highly toxic in clinical trials.
In an attempt to solve this problem, the researchers turned to nanotechnology. They encapsulated the Aurora B kinase inhibitor AZD2811 in polymeric nanoparticles called Accurins.
Susan Ashton, of AstraZeneca in Macclesfield, Cheshire, UK, and her colleagues developed the Accurins and described the work in Science Translational Medicine. The work was funded by AstraZeneca.
The Accurins consist of block copolymers of poly-D,L-lactide and poly(ethylene glycol). The researchers used an ion pairing approach to efficiently encapsulate AZD2811 and control release of the drug.
They found the Accurins could release AZD2811 continuously for more than 1 week in vitro. The nanoparticles also reduced tumor phosphorylated histone H3 levels in vivo for up to 96 hours after a single administration.
The researchers tested the AZD2811 Accurins in mice with diffuse large B-cell lymphoma and rats with colorectal tumors. The nanoparticles accumulated specifically in tumors, where they slowly released AZD2811 to cancer cells.
Compared to AZD1152 (a water-soluble prodrug of AZD2811), the AZD2811 Accurins blocked tumor growth more effectively at one-half the drug dose and caused fewer side effects in the rodents.
Based on these results, the researchers said Accurins could provide efficacy and tolerability using a more convenient dosing regimen, which may extend the utility of Aurora B kinase inhibition to a broader range of hematologic and solid tumor malignancies.
A phase 1 study (NCT02579226) testing AZD2811 Accurins in advanced solid tumors is currently recruiting patients.
A related Focus article published in Science Translational Medicine offers more insights on how Accurin nanoparticles may help enhance the safety and antitumor activity of Aurora kinase inhibitors and other molecularly targeted drugs. ![]()
Researchers believe they have found a way to make certain anticancer agents safer without compromising their efficacy.
The team noted that Aurora B kinase inhibitors and other agents targeting the cell cycle have proven effective but highly toxic in clinical trials.
In an attempt to solve this problem, the researchers turned to nanotechnology. They encapsulated the Aurora B kinase inhibitor AZD2811 in polymeric nanoparticles called Accurins.
Susan Ashton, of AstraZeneca in Macclesfield, Cheshire, UK, and her colleagues developed the Accurins and described the work in Science Translational Medicine. The work was funded by AstraZeneca.
The Accurins consist of block copolymers of poly-D,L-lactide and poly(ethylene glycol). The researchers used an ion pairing approach to efficiently encapsulate AZD2811 and control release of the drug.
They found the Accurins could release AZD2811 continuously for more than 1 week in vitro. The nanoparticles also reduced tumor phosphorylated histone H3 levels in vivo for up to 96 hours after a single administration.
The researchers tested the AZD2811 Accurins in mice with diffuse large B-cell lymphoma and rats with colorectal tumors. The nanoparticles accumulated specifically in tumors, where they slowly released AZD2811 to cancer cells.
Compared to AZD1152 (a water-soluble prodrug of AZD2811), the AZD2811 Accurins blocked tumor growth more effectively at one-half the drug dose and caused fewer side effects in the rodents.
Based on these results, the researchers said Accurins could provide efficacy and tolerability using a more convenient dosing regimen, which may extend the utility of Aurora B kinase inhibition to a broader range of hematologic and solid tumor malignancies.
A phase 1 study (NCT02579226) testing AZD2811 Accurins in advanced solid tumors is currently recruiting patients.
A related Focus article published in Science Translational Medicine offers more insights on how Accurin nanoparticles may help enhance the safety and antitumor activity of Aurora kinase inhibitors and other molecularly targeted drugs. ![]()
Dendritic cells appear to promote T-ALL
Dendritic cells may play a key role in T-cell acute lymphoblastic leukemia (T-ALL), according to research published in PNAS.
Investigators identified tumor-associated dendritic cells that appeared to promote T-ALL growth and survival at primary and metastatic tumor sites in mice.
Analyses of samples from patients with T-ALL suggested dendritic cells are positioned to support T-ALL growth in humans as well.
“It’s only more recently that people have really appreciated that tumors are complex organs in and of themselves, with all of the heterogenous cell types that can talk to each other and promote each other’s survival and proliferation,” said study author Lauren Ehrlich, PhD, of the University of Texas at Austin.
Dr Ehrlich and her colleagues first found that primary T-ALL cells required tumor stroma for survival ex vivo. When T-ALL cells were cultured alone or in wild-type thymic stroma, the cells died off. Only T-ALL cells cultured with tumor-associated stroma survived.
Subsequent experiments suggested it was tumor-associated dendritic cells that spurred T-ALL growth, both for newly developing T-ALL cells and tumors that had spread to distant organs in mouse models. Tissue samples from pediatric T-ALL patients had similar growth environments with abundant dendritic cells.
To determine the mechanism by which dendritic cells support T-ALL, the investigators performed gene expression profiling. They found upregulation of PDGFRB and IGF1R on T-ALL cells, with concomitant expression of their ligands by tumor-associated dendritic cells.
The team said PDGFRB and IGF1R were activated in T-ALL cells ex vivo. And when they cocultured T-ALL cells with tumor-associated dendritic cells, they observed sustained IGF1R activation. However, they did not see this activation when they cocultured T-ALL cells with normal thymic dendritic cells.
Finally, the investigators found that IGF1R signaling was necessary for dendritic cell-mediated T-ALL survival.
The team said this is the first evidence that endogenous tumor-associated dendritic cells supply signals driving T-ALL growth.
“We hope this study will be a catalyst to spur other research groups to further elucidate the roles of dendritic cells in supporting T-ALL,” said study author Todd Triplett, PhD, also of the University of Texas at Austin.
“[T]hat could ultimately lead to the discovery of novel therapeutic targets that are more effective and less toxic than current treatment regimens.” ![]()
Dendritic cells may play a key role in T-cell acute lymphoblastic leukemia (T-ALL), according to research published in PNAS.
Investigators identified tumor-associated dendritic cells that appeared to promote T-ALL growth and survival at primary and metastatic tumor sites in mice.
Analyses of samples from patients with T-ALL suggested dendritic cells are positioned to support T-ALL growth in humans as well.
“It’s only more recently that people have really appreciated that tumors are complex organs in and of themselves, with all of the heterogenous cell types that can talk to each other and promote each other’s survival and proliferation,” said study author Lauren Ehrlich, PhD, of the University of Texas at Austin.
Dr Ehrlich and her colleagues first found that primary T-ALL cells required tumor stroma for survival ex vivo. When T-ALL cells were cultured alone or in wild-type thymic stroma, the cells died off. Only T-ALL cells cultured with tumor-associated stroma survived.
Subsequent experiments suggested it was tumor-associated dendritic cells that spurred T-ALL growth, both for newly developing T-ALL cells and tumors that had spread to distant organs in mouse models. Tissue samples from pediatric T-ALL patients had similar growth environments with abundant dendritic cells.
To determine the mechanism by which dendritic cells support T-ALL, the investigators performed gene expression profiling. They found upregulation of PDGFRB and IGF1R on T-ALL cells, with concomitant expression of their ligands by tumor-associated dendritic cells.
The team said PDGFRB and IGF1R were activated in T-ALL cells ex vivo. And when they cocultured T-ALL cells with tumor-associated dendritic cells, they observed sustained IGF1R activation. However, they did not see this activation when they cocultured T-ALL cells with normal thymic dendritic cells.
Finally, the investigators found that IGF1R signaling was necessary for dendritic cell-mediated T-ALL survival.
The team said this is the first evidence that endogenous tumor-associated dendritic cells supply signals driving T-ALL growth.
“We hope this study will be a catalyst to spur other research groups to further elucidate the roles of dendritic cells in supporting T-ALL,” said study author Todd Triplett, PhD, also of the University of Texas at Austin.
“[T]hat could ultimately lead to the discovery of novel therapeutic targets that are more effective and less toxic than current treatment regimens.” ![]()
Dendritic cells may play a key role in T-cell acute lymphoblastic leukemia (T-ALL), according to research published in PNAS.
Investigators identified tumor-associated dendritic cells that appeared to promote T-ALL growth and survival at primary and metastatic tumor sites in mice.
Analyses of samples from patients with T-ALL suggested dendritic cells are positioned to support T-ALL growth in humans as well.
“It’s only more recently that people have really appreciated that tumors are complex organs in and of themselves, with all of the heterogenous cell types that can talk to each other and promote each other’s survival and proliferation,” said study author Lauren Ehrlich, PhD, of the University of Texas at Austin.
Dr Ehrlich and her colleagues first found that primary T-ALL cells required tumor stroma for survival ex vivo. When T-ALL cells were cultured alone or in wild-type thymic stroma, the cells died off. Only T-ALL cells cultured with tumor-associated stroma survived.
Subsequent experiments suggested it was tumor-associated dendritic cells that spurred T-ALL growth, both for newly developing T-ALL cells and tumors that had spread to distant organs in mouse models. Tissue samples from pediatric T-ALL patients had similar growth environments with abundant dendritic cells.
To determine the mechanism by which dendritic cells support T-ALL, the investigators performed gene expression profiling. They found upregulation of PDGFRB and IGF1R on T-ALL cells, with concomitant expression of their ligands by tumor-associated dendritic cells.
The team said PDGFRB and IGF1R were activated in T-ALL cells ex vivo. And when they cocultured T-ALL cells with tumor-associated dendritic cells, they observed sustained IGF1R activation. However, they did not see this activation when they cocultured T-ALL cells with normal thymic dendritic cells.
Finally, the investigators found that IGF1R signaling was necessary for dendritic cell-mediated T-ALL survival.
The team said this is the first evidence that endogenous tumor-associated dendritic cells supply signals driving T-ALL growth.
“We hope this study will be a catalyst to spur other research groups to further elucidate the roles of dendritic cells in supporting T-ALL,” said study author Todd Triplett, PhD, also of the University of Texas at Austin.
“[T]hat could ultimately lead to the discovery of novel therapeutic targets that are more effective and less toxic than current treatment regimens.” ![]()
Development of myelofibrosis drug on hold
The US Food and Drug Administration (FDA) has placed a full clinical hold on trials conducted under the investigational new drug application for pacritinib, a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The hold means all patients currently on pacritinib must stop taking the drug immediately, and no patients can be enrolled on a pacritinib trial or start pacritinib as initial or crossover treatment.
In addition, CTI BioPharma has withdrawn the new drug application for pacritinib while the company reviews data from the phase 3 PERSIST-2 trial.
The FDA’s decision to place a full clinical hold on pacritinib trials was due to interim results from PERSIST-2. The aim of this trial was to compare pacritinib to best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The overall survival results from PERSIST-2 indicate that pacritinib had a detrimental effect on survival, which is consistent with results from the PERSIST-1 trial. The deaths in pacritinib-treated patients on PERSIST-2 include intracranial hemorrhage, cardiac failure, and cardiac arrest.
Based on these results, the FDA has made recommendations for CTI BioPharma that supersede the agency’s previous recommendations.
On February 4, 2016, the FDA placed a partial clinical hold on pacritinib trials and made related recommendations for CTI BioPharma, advising that the company modify trial protocols and take other actions in compliance with the partial clinical hold.
Now that pacritinib trials are on full clinical hold, the FDA is recommending that CTI BioPharma conduct dose exploration studies for pacritinib in patients with MF and submit final study reports and datasets for PERSIST-1 and PERSIST-2.
The FDA is also recommending that CTI BioPharma provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, make certain modifications to protocols, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma said all clinical investigators worldwide have been notified of the hold. ![]()
The US Food and Drug Administration (FDA) has placed a full clinical hold on trials conducted under the investigational new drug application for pacritinib, a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The hold means all patients currently on pacritinib must stop taking the drug immediately, and no patients can be enrolled on a pacritinib trial or start pacritinib as initial or crossover treatment.
In addition, CTI BioPharma has withdrawn the new drug application for pacritinib while the company reviews data from the phase 3 PERSIST-2 trial.
The FDA’s decision to place a full clinical hold on pacritinib trials was due to interim results from PERSIST-2. The aim of this trial was to compare pacritinib to best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The overall survival results from PERSIST-2 indicate that pacritinib had a detrimental effect on survival, which is consistent with results from the PERSIST-1 trial. The deaths in pacritinib-treated patients on PERSIST-2 include intracranial hemorrhage, cardiac failure, and cardiac arrest.
Based on these results, the FDA has made recommendations for CTI BioPharma that supersede the agency’s previous recommendations.
On February 4, 2016, the FDA placed a partial clinical hold on pacritinib trials and made related recommendations for CTI BioPharma, advising that the company modify trial protocols and take other actions in compliance with the partial clinical hold.
Now that pacritinib trials are on full clinical hold, the FDA is recommending that CTI BioPharma conduct dose exploration studies for pacritinib in patients with MF and submit final study reports and datasets for PERSIST-1 and PERSIST-2.
The FDA is also recommending that CTI BioPharma provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, make certain modifications to protocols, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma said all clinical investigators worldwide have been notified of the hold. ![]()
The US Food and Drug Administration (FDA) has placed a full clinical hold on trials conducted under the investigational new drug application for pacritinib, a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The hold means all patients currently on pacritinib must stop taking the drug immediately, and no patients can be enrolled on a pacritinib trial or start pacritinib as initial or crossover treatment.
In addition, CTI BioPharma has withdrawn the new drug application for pacritinib while the company reviews data from the phase 3 PERSIST-2 trial.
The FDA’s decision to place a full clinical hold on pacritinib trials was due to interim results from PERSIST-2. The aim of this trial was to compare pacritinib to best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The overall survival results from PERSIST-2 indicate that pacritinib had a detrimental effect on survival, which is consistent with results from the PERSIST-1 trial. The deaths in pacritinib-treated patients on PERSIST-2 include intracranial hemorrhage, cardiac failure, and cardiac arrest.
Based on these results, the FDA has made recommendations for CTI BioPharma that supersede the agency’s previous recommendations.
On February 4, 2016, the FDA placed a partial clinical hold on pacritinib trials and made related recommendations for CTI BioPharma, advising that the company modify trial protocols and take other actions in compliance with the partial clinical hold.
Now that pacritinib trials are on full clinical hold, the FDA is recommending that CTI BioPharma conduct dose exploration studies for pacritinib in patients with MF and submit final study reports and datasets for PERSIST-1 and PERSIST-2.
The FDA is also recommending that CTI BioPharma provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, make certain modifications to protocols, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma said all clinical investigators worldwide have been notified of the hold. ![]()
Method could improve diagnosis of platelet disorders
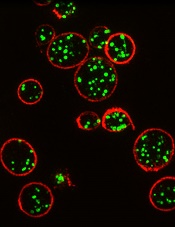
Courtesy of Dan Cutler
A proof-of-concept study suggests structured illumination microscopy (SIM) enables accurate diagnosis of Hermansky-Pudlak syndrome (HPS), an autosomal recessive disorder characterized by platelet dysfunction and prolonged bleeding.
Researchers therefore believe SIM could provide a more accessible and cost-effective method for diagnosing platelet disorders.
They also said SIM provides detailed data that may enable personalized treatments.
The researchers described their results with SIM in the Journal of Thrombosis and Haemostasis.
The team noted that electron microscopy can be used to diagnose HPS and other disorders characterized by platelet dysfunction. But the method is costly, requires fresh samples, gives limited information, and is not widely available.
“We’ve found that SIM has a lot of advantages over whole mount electron microscopy as a diagnosis method,” said study author David Westmoreland, of University College London in the UK.
“Samples don’t need to be analyzed live and can be reanalyzed, and automation means analysis is unbiased and less time-consuming. Given about 75% of patients with a bleeding disorder such as Hermansky-Pudlak syndrome are initially misdiagnosed, and 28% need to see between 4 to 6 specialists before receiving the correct diagnosis, there is a demand for a new method of analysis.”
For this proof-of-concept study, Westmoreland and his colleagues used SIM to image platelet granules in blood samples using a marker protein, CD63.
The imaging technology was custom-built by the team to automatically count the number of granules per platelet, thereby identifying patients with HPS, a rare disorder thought to affect 1 in 500,000 people.
The researchers distinguished the 3 patients with HPS from 7 normal controls with 99% confidence. Automated counting of granules showed that individuals with the disorder had a third as many granules as controls.
“Our limited analysis of this new method is extremely promising, and we hope to take it forward to test for multiple parameters with different markers,” said Dan Cutler, PhD, also of University College London.
“In this way, we could use a single super-resolution image to screen for many different platelet-based blood disorders.” ![]()
Courtesy of Dan Cutler
A proof-of-concept study suggests structured illumination microscopy (SIM) enables accurate diagnosis of Hermansky-Pudlak syndrome (HPS), an autosomal recessive disorder characterized by platelet dysfunction and prolonged bleeding.
Researchers therefore believe SIM could provide a more accessible and cost-effective method for diagnosing platelet disorders.
They also said SIM provides detailed data that may enable personalized treatments.
The researchers described their results with SIM in the Journal of Thrombosis and Haemostasis.
The team noted that electron microscopy can be used to diagnose HPS and other disorders characterized by platelet dysfunction. But the method is costly, requires fresh samples, gives limited information, and is not widely available.
“We’ve found that SIM has a lot of advantages over whole mount electron microscopy as a diagnosis method,” said study author David Westmoreland, of University College London in the UK.
“Samples don’t need to be analyzed live and can be reanalyzed, and automation means analysis is unbiased and less time-consuming. Given about 75% of patients with a bleeding disorder such as Hermansky-Pudlak syndrome are initially misdiagnosed, and 28% need to see between 4 to 6 specialists before receiving the correct diagnosis, there is a demand for a new method of analysis.”
For this proof-of-concept study, Westmoreland and his colleagues used SIM to image platelet granules in blood samples using a marker protein, CD63.
The imaging technology was custom-built by the team to automatically count the number of granules per platelet, thereby identifying patients with HPS, a rare disorder thought to affect 1 in 500,000 people.
The researchers distinguished the 3 patients with HPS from 7 normal controls with 99% confidence. Automated counting of granules showed that individuals with the disorder had a third as many granules as controls.
“Our limited analysis of this new method is extremely promising, and we hope to take it forward to test for multiple parameters with different markers,” said Dan Cutler, PhD, also of University College London.
“In this way, we could use a single super-resolution image to screen for many different platelet-based blood disorders.” ![]()
Courtesy of Dan Cutler
A proof-of-concept study suggests structured illumination microscopy (SIM) enables accurate diagnosis of Hermansky-Pudlak syndrome (HPS), an autosomal recessive disorder characterized by platelet dysfunction and prolonged bleeding.
Researchers therefore believe SIM could provide a more accessible and cost-effective method for diagnosing platelet disorders.
They also said SIM provides detailed data that may enable personalized treatments.
The researchers described their results with SIM in the Journal of Thrombosis and Haemostasis.
The team noted that electron microscopy can be used to diagnose HPS and other disorders characterized by platelet dysfunction. But the method is costly, requires fresh samples, gives limited information, and is not widely available.
“We’ve found that SIM has a lot of advantages over whole mount electron microscopy as a diagnosis method,” said study author David Westmoreland, of University College London in the UK.
“Samples don’t need to be analyzed live and can be reanalyzed, and automation means analysis is unbiased and less time-consuming. Given about 75% of patients with a bleeding disorder such as Hermansky-Pudlak syndrome are initially misdiagnosed, and 28% need to see between 4 to 6 specialists before receiving the correct diagnosis, there is a demand for a new method of analysis.”
For this proof-of-concept study, Westmoreland and his colleagues used SIM to image platelet granules in blood samples using a marker protein, CD63.
The imaging technology was custom-built by the team to automatically count the number of granules per platelet, thereby identifying patients with HPS, a rare disorder thought to affect 1 in 500,000 people.
The researchers distinguished the 3 patients with HPS from 7 normal controls with 99% confidence. Automated counting of granules showed that individuals with the disorder had a third as many granules as controls.
“Our limited analysis of this new method is extremely promising, and we hope to take it forward to test for multiple parameters with different markers,” said Dan Cutler, PhD, also of University College London.
“In this way, we could use a single super-resolution image to screen for many different platelet-based blood disorders.”
EBV-CTLs get orphan designation for EBV-PTLD
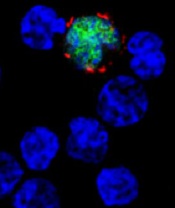
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The US Food and Drug Administration (FDA) has granted orphan designation for cytotoxic T lymphocytes activated against Epstein-Barr virus (EBV-CTLs) to treat EBV post-transplant lymphoproliferative disorder (EBV-PTLD) occurring after solid organ or hematopoietic stem cell transplant.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases, which may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and a 7-year period of marketing exclusivity if the product is approved.
The EBV-CTL product is under development by Atara Biotherapeutics, Inc. It is produced by collecting T cells from third-party donors and exposing the cells to EBV antigens.
The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient, providing an “off-the-shelf,” allogeneic, cellular therapeutic option for patients.
In the context of EBV-PTLD, Atara’s EBV-CTLs find the cancer cells expressing EBV and kill them. EBV-CTLs are currently being studied in phase 2 trials.
Results of a phase 1/2 study were presented at the APHON 37th Annual Conference and Exhibit and at the 2015 ASCO Annual Meeting.
The FDA previously granted EBV-CTLs breakthrough designation to treat EBV-PTLD. This designation is intended to expedite the development and review of new drugs for serious or life-threatening conditions.
To qualify for breakthrough designation, a drug must show credible evidence of a substantial improvement on a clinically significant endpoint over available therapies, or over placebo if there is no available therapy, or in a study that compares the new treatment plus the standard of care to the standard alone.
The designation confers several benefits, including intensive FDA guidance and eligibility for submission of a rolling biologic license application.
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The US Food and Drug Administration (FDA) has granted orphan designation for cytotoxic T lymphocytes activated against Epstein-Barr virus (EBV-CTLs) to treat EBV post-transplant lymphoproliferative disorder (EBV-PTLD) occurring after solid organ or hematopoietic stem cell transplant.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases, which may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and a 7-year period of marketing exclusivity if the product is approved.
The EBV-CTL product is under development by Atara Biotherapeutics, Inc. It is produced by collecting T cells from third-party donors and exposing the cells to EBV antigens.
The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient, providing an “off-the-shelf,” allogeneic, cellular therapeutic option for patients.
In the context of EBV-PTLD, Atara’s EBV-CTLs find the cancer cells expressing EBV and kill them. EBV-CTLs are currently being studied in phase 2 trials.
Results of a phase 1/2 study were presented at the APHON 37th Annual Conference and Exhibit and at the 2015 ASCO Annual Meeting.
The FDA previously granted EBV-CTLs breakthrough designation to treat EBV-PTLD. This designation is intended to expedite the development and review of new drugs for serious or life-threatening conditions.
To qualify for breakthrough designation, a drug must show credible evidence of a substantial improvement on a clinically significant endpoint over available therapies, or over placebo if there is no available therapy, or in a study that compares the new treatment plus the standard of care to the standard alone.
The designation confers several benefits, including intensive FDA guidance and eligibility for submission of a rolling biologic license application.
among uninfected cells (blue)
Image courtesy of
Benjamin Chaigne-Delalande
The US Food and Drug Administration (FDA) has granted orphan designation for cytotoxic T lymphocytes activated against Epstein-Barr virus (EBV-CTLs) to treat EBV post-transplant lymphoproliferative disorder (EBV-PTLD) occurring after solid organ or hematopoietic stem cell transplant.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases, which may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and a 7-year period of marketing exclusivity if the product is approved.
The EBV-CTL product is under development by Atara Biotherapeutics, Inc. It is produced by collecting T cells from third-party donors and exposing the cells to EBV antigens.
The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient, providing an “off-the-shelf,” allogeneic, cellular therapeutic option for patients.
In the context of EBV-PTLD, Atara’s EBV-CTLs find the cancer cells expressing EBV and kill them. EBV-CTLs are currently being studied in phase 2 trials.
Results of a phase 1/2 study were presented at the APHON 37th Annual Conference and Exhibit and at the 2015 ASCO Annual Meeting.
The FDA previously granted EBV-CTLs breakthrough designation to treat EBV-PTLD. This designation is intended to expedite the development and review of new drugs for serious or life-threatening conditions.
To qualify for breakthrough designation, a drug must show credible evidence of a substantial improvement on a clinically significant endpoint over available therapies, or over placebo if there is no available therapy, or in a study that compares the new treatment plus the standard of care to the standard alone.
The designation confers several benefits, including intensive FDA guidance and eligibility for submission of a rolling biologic license application.
Gut microbiota linked to severity of malaria
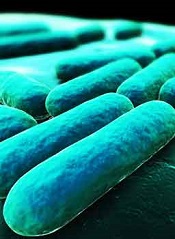
Microorganisms in the gut play a role in the severity of malaria, according to a study published in PNAS.
Investigators examined the gut microbiomes of mice and found evidence to suggest that malaria severity is not only a function of the parasite or the host. It is also influenced by the microbes in the infected organism.
And 2 types of bacteria—Bifidobacterium and Lactobacillus—were associated with reduced malaria severity.
“The research provides a potential new avenue to investigate factors that control the severity of malaria,” said study author Steven Wilhelm, PhD, of the University of Tennessee at Knoxville.
“With 1 million people dying [of malaria] each year, many of whom are young children, any approach that may save even a few lives is worth following up on.”
With this study, Dr Wilhelm and his colleagues found that genetically similar mice acquired from different vendors had differences in pathology after malaria infection. There were significant differences in both parasite burden and mortality after infection with multiple Plasmodium species.
The investigators measured gut microbiomes in the mice by sequencing bacteria in the digestive tract and noted significant differences within the different mouse populations.
So the team transferred cecal content from the first set of mice into germ-free mice and found that differences in malaria severity were transferred.
The mice that received transplants from donors that were more resistant to malaria had low parasite burdens. And mice that received transplants from donors that were more susceptible to malaria had high parasite burdens.
The investigators also observed an increased abundance of Bifidobacterium and Lactobacillus bacteria in the mice that exhibited reduced malaria pathology.
So the team took mice that were more susceptible to malaria, treated them with antibiotics, and fed them yogurt containing Bifidobacterium and Lactobacillus. As expected, the severity of malaria in these mice decreased.
“These results demonstrate the possibility of modifying the gut microbiome to prevent severe malaria,” said study author Nathan Schmidt, PhD, of the University of Louisville in Kentucky.
Dr Wilhelm noted that, although the research interventions lessened the severity of malaria in mice, it did not prevent or cure it. And the investigators are a long way from perfecting similar treatments in humans but are working on understanding the mechanism.
“A way to help people who are infected—and especially a simple and cheap way, as much of the infection occurs in the developing world—would be a great service to society,” Dr Wilhelm said.
Microorganisms in the gut play a role in the severity of malaria, according to a study published in PNAS.
Investigators examined the gut microbiomes of mice and found evidence to suggest that malaria severity is not only a function of the parasite or the host. It is also influenced by the microbes in the infected organism.
And 2 types of bacteria—Bifidobacterium and Lactobacillus—were associated with reduced malaria severity.
“The research provides a potential new avenue to investigate factors that control the severity of malaria,” said study author Steven Wilhelm, PhD, of the University of Tennessee at Knoxville.
“With 1 million people dying [of malaria] each year, many of whom are young children, any approach that may save even a few lives is worth following up on.”
With this study, Dr Wilhelm and his colleagues found that genetically similar mice acquired from different vendors had differences in pathology after malaria infection. There were significant differences in both parasite burden and mortality after infection with multiple Plasmodium species.
The investigators measured gut microbiomes in the mice by sequencing bacteria in the digestive tract and noted significant differences within the different mouse populations.
So the team transferred cecal content from the first set of mice into germ-free mice and found that differences in malaria severity were transferred.
The mice that received transplants from donors that were more resistant to malaria had low parasite burdens. And mice that received transplants from donors that were more susceptible to malaria had high parasite burdens.
The investigators also observed an increased abundance of Bifidobacterium and Lactobacillus bacteria in the mice that exhibited reduced malaria pathology.
So the team took mice that were more susceptible to malaria, treated them with antibiotics, and fed them yogurt containing Bifidobacterium and Lactobacillus. As expected, the severity of malaria in these mice decreased.
“These results demonstrate the possibility of modifying the gut microbiome to prevent severe malaria,” said study author Nathan Schmidt, PhD, of the University of Louisville in Kentucky.
Dr Wilhelm noted that, although the research interventions lessened the severity of malaria in mice, it did not prevent or cure it. And the investigators are a long way from perfecting similar treatments in humans but are working on understanding the mechanism.
“A way to help people who are infected—and especially a simple and cheap way, as much of the infection occurs in the developing world—would be a great service to society,” Dr Wilhelm said.
Microorganisms in the gut play a role in the severity of malaria, according to a study published in PNAS.
Investigators examined the gut microbiomes of mice and found evidence to suggest that malaria severity is not only a function of the parasite or the host. It is also influenced by the microbes in the infected organism.
And 2 types of bacteria—Bifidobacterium and Lactobacillus—were associated with reduced malaria severity.
“The research provides a potential new avenue to investigate factors that control the severity of malaria,” said study author Steven Wilhelm, PhD, of the University of Tennessee at Knoxville.
“With 1 million people dying [of malaria] each year, many of whom are young children, any approach that may save even a few lives is worth following up on.”
With this study, Dr Wilhelm and his colleagues found that genetically similar mice acquired from different vendors had differences in pathology after malaria infection. There were significant differences in both parasite burden and mortality after infection with multiple Plasmodium species.
The investigators measured gut microbiomes in the mice by sequencing bacteria in the digestive tract and noted significant differences within the different mouse populations.
So the team transferred cecal content from the first set of mice into germ-free mice and found that differences in malaria severity were transferred.
The mice that received transplants from donors that were more resistant to malaria had low parasite burdens. And mice that received transplants from donors that were more susceptible to malaria had high parasite burdens.
The investigators also observed an increased abundance of Bifidobacterium and Lactobacillus bacteria in the mice that exhibited reduced malaria pathology.
So the team took mice that were more susceptible to malaria, treated them with antibiotics, and fed them yogurt containing Bifidobacterium and Lactobacillus. As expected, the severity of malaria in these mice decreased.
“These results demonstrate the possibility of modifying the gut microbiome to prevent severe malaria,” said study author Nathan Schmidt, PhD, of the University of Louisville in Kentucky.
Dr Wilhelm noted that, although the research interventions lessened the severity of malaria in mice, it did not prevent or cure it. And the investigators are a long way from perfecting similar treatments in humans but are working on understanding the mechanism.
“A way to help people who are infected—and especially a simple and cheap way, as much of the infection occurs in the developing world—would be a great service to society,” Dr Wilhelm said.
New insight into Ph-like ALL could lead to new treatment

Photo courtesy of St. Jude
Children’s Research Hospital
Research published in Cancer Cell appears to explain how the abnormal breakage and rearrangement of chromosomes in white blood cells triggers Philadelphia chromosome-like (Ph-like) acute lymphoblastic leukemia (ALL).
Genomic analysis revealed 4 chromosomal rearrangements that all resulted in a truncated version of the erythropoietin receptor (EPOR) gene and drove white blood cells to proliferate out of control.
“To our knowledge, this is a previously unknown mechanism for leukemia,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our search of cancer genomic data has shown that there are many other examples of chromosomal rearrangements that alter genes’ structure, but this type—where a truncating rearrangement leads to activation—is new.”
Although Dr Mullighan and his colleagues had previously identified an abnormal chromosome rearrangement in Ph-like ALL, little was known about the biological effects of that rearrangement. So they set out to pinpoint those effects by studying human leukemic cells and mouse cells engineered to mimic Ph-like ALL.
The investigators discovered the 4 rearrangements of EPOR, all of which resulted in truncation of the cytoplasmic tail of EPOR at residues similar to those mutated in primary familial congenital polycythemia. The proximal tyrosine essential for receptor activation was preserved, but distal regulatory residues were lost.
The team said these rearrangements resulted in deregulated EPOR expression, hypersensitivity to erythropoietin stimulation, and heightened JAK-STAT activation.
The investigators noted that the rearrangements were present in all of the leukemic cells from patients, which suggests these changes were fundamental to Ph-like ALL development. The team also showed that introducing truncated EPOR in mouse B-cell progenitors gave rise to ALL in mice.
Further investigation revealed that EPOR rearrangements arise early in the development of Ph-like ALL and persist as the disease progresses.
“That finding was important because it suggests that treatments for this leukemia targeting this receptor won’t just impact a subset of the leukemia cells, allowing others to keep proliferating,” said study author Ilaria Iacobucci, PhD, of St. Jude Children’s Research Hospital.
The investigators then found that human leukemic cells with EPOR rearrangements were sensitive to JAK-STAT inhibition via treatment with ruxolitinib.
The team also cited the case of an adult patient treated at MD Anderson Cancer Research Center in Houston, Texas, whose genetic analysis revealed EPOR-rearranged ALL. That patient had not responded significantly to other chemotherapy drugs. But, when given ruxolitinib, the patient showed a major drop in leukemia cells.
In experiments with leukemic cells, the investigators found that ruxolitinib worked synergistically with 3 chemotherapeutic agents—dexamethasone, vincristine, and daunorubicin.
“We think these findings provide a useful road map for planning more accurate testing of combination chemotherapies,” Dr Mullighan said.
“These findings expand the number of ALL patients who should be amenable to precision medicine therapies that add targeted inhibitors to chemotherapy for ALL patents with specific genetic changes in the leukemia cells,” added study author Stephen Hunger, MD, of Children’s Hospital of Philadelphia in Pennsylvania.
Dr Hunger said the Children’s Oncology Group has developed a clinical trial testing this strategy with ruxolitinib, which will begin treating patients in mid-2016. Based on the results of the Cancer Cell research, the trial will include children with ALL and EPOR rearrangements.
Photo courtesy of St. Jude
Children’s Research Hospital
Research published in Cancer Cell appears to explain how the abnormal breakage and rearrangement of chromosomes in white blood cells triggers Philadelphia chromosome-like (Ph-like) acute lymphoblastic leukemia (ALL).
Genomic analysis revealed 4 chromosomal rearrangements that all resulted in a truncated version of the erythropoietin receptor (EPOR) gene and drove white blood cells to proliferate out of control.
“To our knowledge, this is a previously unknown mechanism for leukemia,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our search of cancer genomic data has shown that there are many other examples of chromosomal rearrangements that alter genes’ structure, but this type—where a truncating rearrangement leads to activation—is new.”
Although Dr Mullighan and his colleagues had previously identified an abnormal chromosome rearrangement in Ph-like ALL, little was known about the biological effects of that rearrangement. So they set out to pinpoint those effects by studying human leukemic cells and mouse cells engineered to mimic Ph-like ALL.
The investigators discovered the 4 rearrangements of EPOR, all of which resulted in truncation of the cytoplasmic tail of EPOR at residues similar to those mutated in primary familial congenital polycythemia. The proximal tyrosine essential for receptor activation was preserved, but distal regulatory residues were lost.
The team said these rearrangements resulted in deregulated EPOR expression, hypersensitivity to erythropoietin stimulation, and heightened JAK-STAT activation.
The investigators noted that the rearrangements were present in all of the leukemic cells from patients, which suggests these changes were fundamental to Ph-like ALL development. The team also showed that introducing truncated EPOR in mouse B-cell progenitors gave rise to ALL in mice.
Further investigation revealed that EPOR rearrangements arise early in the development of Ph-like ALL and persist as the disease progresses.
“That finding was important because it suggests that treatments for this leukemia targeting this receptor won’t just impact a subset of the leukemia cells, allowing others to keep proliferating,” said study author Ilaria Iacobucci, PhD, of St. Jude Children’s Research Hospital.
The investigators then found that human leukemic cells with EPOR rearrangements were sensitive to JAK-STAT inhibition via treatment with ruxolitinib.
The team also cited the case of an adult patient treated at MD Anderson Cancer Research Center in Houston, Texas, whose genetic analysis revealed EPOR-rearranged ALL. That patient had not responded significantly to other chemotherapy drugs. But, when given ruxolitinib, the patient showed a major drop in leukemia cells.
In experiments with leukemic cells, the investigators found that ruxolitinib worked synergistically with 3 chemotherapeutic agents—dexamethasone, vincristine, and daunorubicin.
“We think these findings provide a useful road map for planning more accurate testing of combination chemotherapies,” Dr Mullighan said.
“These findings expand the number of ALL patients who should be amenable to precision medicine therapies that add targeted inhibitors to chemotherapy for ALL patents with specific genetic changes in the leukemia cells,” added study author Stephen Hunger, MD, of Children’s Hospital of Philadelphia in Pennsylvania.
Dr Hunger said the Children’s Oncology Group has developed a clinical trial testing this strategy with ruxolitinib, which will begin treating patients in mid-2016. Based on the results of the Cancer Cell research, the trial will include children with ALL and EPOR rearrangements.
Photo courtesy of St. Jude
Children’s Research Hospital
Research published in Cancer Cell appears to explain how the abnormal breakage and rearrangement of chromosomes in white blood cells triggers Philadelphia chromosome-like (Ph-like) acute lymphoblastic leukemia (ALL).
Genomic analysis revealed 4 chromosomal rearrangements that all resulted in a truncated version of the erythropoietin receptor (EPOR) gene and drove white blood cells to proliferate out of control.
“To our knowledge, this is a previously unknown mechanism for leukemia,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“Our search of cancer genomic data has shown that there are many other examples of chromosomal rearrangements that alter genes’ structure, but this type—where a truncating rearrangement leads to activation—is new.”
Although Dr Mullighan and his colleagues had previously identified an abnormal chromosome rearrangement in Ph-like ALL, little was known about the biological effects of that rearrangement. So they set out to pinpoint those effects by studying human leukemic cells and mouse cells engineered to mimic Ph-like ALL.
The investigators discovered the 4 rearrangements of EPOR, all of which resulted in truncation of the cytoplasmic tail of EPOR at residues similar to those mutated in primary familial congenital polycythemia. The proximal tyrosine essential for receptor activation was preserved, but distal regulatory residues were lost.
The team said these rearrangements resulted in deregulated EPOR expression, hypersensitivity to erythropoietin stimulation, and heightened JAK-STAT activation.
The investigators noted that the rearrangements were present in all of the leukemic cells from patients, which suggests these changes were fundamental to Ph-like ALL development. The team also showed that introducing truncated EPOR in mouse B-cell progenitors gave rise to ALL in mice.
Further investigation revealed that EPOR rearrangements arise early in the development of Ph-like ALL and persist as the disease progresses.
“That finding was important because it suggests that treatments for this leukemia targeting this receptor won’t just impact a subset of the leukemia cells, allowing others to keep proliferating,” said study author Ilaria Iacobucci, PhD, of St. Jude Children’s Research Hospital.
The investigators then found that human leukemic cells with EPOR rearrangements were sensitive to JAK-STAT inhibition via treatment with ruxolitinib.
The team also cited the case of an adult patient treated at MD Anderson Cancer Research Center in Houston, Texas, whose genetic analysis revealed EPOR-rearranged ALL. That patient had not responded significantly to other chemotherapy drugs. But, when given ruxolitinib, the patient showed a major drop in leukemia cells.
In experiments with leukemic cells, the investigators found that ruxolitinib worked synergistically with 3 chemotherapeutic agents—dexamethasone, vincristine, and daunorubicin.
“We think these findings provide a useful road map for planning more accurate testing of combination chemotherapies,” Dr Mullighan said.
“These findings expand the number of ALL patients who should be amenable to precision medicine therapies that add targeted inhibitors to chemotherapy for ALL patents with specific genetic changes in the leukemia cells,” added study author Stephen Hunger, MD, of Children’s Hospital of Philadelphia in Pennsylvania.
Dr Hunger said the Children’s Oncology Group has developed a clinical trial testing this strategy with ruxolitinib, which will begin treating patients in mid-2016. Based on the results of the Cancer Cell research, the trial will include children with ALL and EPOR rearrangements.