User login
ESRD treatments linked to different cancers
Photo by Anna Frodesiak
Patients with end-stage renal disease (ESRD) may have different cancer risks according to the treatment they are receiving, a new study suggests.
Researchers found that patients had a higher risk of developing infection-related and immune-related cancers—including Hodgkin and non-Hodgkin lymphoma (NHL)—after receiving a kidney transplant.
But patients had a higher risk of ESRD-related cancers when they were on dialysis.
Elizabeth Yanik, PhD, of the National Cancer Institute in Bethesda, Maryland, and her colleagues reported these results in the Journal of the American Society of Nephrology.
The researchers theorized that assessing patterns in ESRD patients across periods of dialysis and kidney transplant might inform cancer etiology.
So the team studied registry data on 202,195 kidney transplant candidates and recipients, comparing the incidence of cancers during kidney function intervals (time with a transplant) to the incidence during nonfunction intervals (waitlist or time after transplant failure [dialysis]). The analysis was adjusted for demographic characteristics.
Results showed the incidence of infection-related and immune-related cancers was higher during kidney function intervals than nonfunction intervals.
Cancers with a significantly higher incidence included Kaposi’s sarcoma (hazard ratio [HR]=9.1, P<0.001), NHL (HR=3.2, P<0.001), Hodgkin lymphoma (HR=3.0, P<0.001), lip cancer (HR=3.4, P<0.001), nonepithelial skin cancers (HR=3.8, P<0.001), melanoma (HR=1.9, P<0.001), prostate cancer (HR=1.2, P=0.003), anal cancer (HR=1.8, P=0.01), other genital cancers (HR=1.5, P=0.03), lung cancer (HR=1.3 P<0.001), and pancreatic cancer (HR=1.5, P=0.004).
Dr Yanik and her colleagues noted that, of these cancers, NHL, anal cancer, lung cancer, melanoma, nonepithelial skin cancers, and pancreatic cancer consistently increased in incidence with each transition to a kidney function interval and decreased with each transition to a nonfunction interval.
And the 2 types of transitions were significant for NHL, lung cancer, melanoma, nonepithelial skin cancers, and pancreatic cancer.
The researchers also identified cancers with a significantly lower incidence during kidney function intervals. This included kidney cancer (HR=0.77, P<0.001), thyroid cancer (HR=0.67, P<0.001), breast cancer (HR=0.81, P=0.002), and liver cancer (HR=0.59, P=0.001).
The team noted that, among cancers with a lower incidence during kidney function intervals, kidney cancer, thyroid cancer, and myeloma consistently decreased in incidence with each transition to a kidney function interval and increased in incidence with each transition to a nonfunction interval. But both transitions were only significant for kidney and thyroid cancers.
“Our study indicates that the needs of individuals with end-stage renal disease, in terms of cancer prevention and cancer screening, will likely differ over time,” Dr Yanik said.
“Vigilance for kidney cancer and thyroid cancer may be of particular importance while these individuals are on dialysis. Extra consideration for screening for melanoma or lung cancer may be called for while taking immunosuppressant medications following a kidney transplant.” ![]()
Photo by Anna Frodesiak
Patients with end-stage renal disease (ESRD) may have different cancer risks according to the treatment they are receiving, a new study suggests.
Researchers found that patients had a higher risk of developing infection-related and immune-related cancers—including Hodgkin and non-Hodgkin lymphoma (NHL)—after receiving a kidney transplant.
But patients had a higher risk of ESRD-related cancers when they were on dialysis.
Elizabeth Yanik, PhD, of the National Cancer Institute in Bethesda, Maryland, and her colleagues reported these results in the Journal of the American Society of Nephrology.
The researchers theorized that assessing patterns in ESRD patients across periods of dialysis and kidney transplant might inform cancer etiology.
So the team studied registry data on 202,195 kidney transplant candidates and recipients, comparing the incidence of cancers during kidney function intervals (time with a transplant) to the incidence during nonfunction intervals (waitlist or time after transplant failure [dialysis]). The analysis was adjusted for demographic characteristics.
Results showed the incidence of infection-related and immune-related cancers was higher during kidney function intervals than nonfunction intervals.
Cancers with a significantly higher incidence included Kaposi’s sarcoma (hazard ratio [HR]=9.1, P<0.001), NHL (HR=3.2, P<0.001), Hodgkin lymphoma (HR=3.0, P<0.001), lip cancer (HR=3.4, P<0.001), nonepithelial skin cancers (HR=3.8, P<0.001), melanoma (HR=1.9, P<0.001), prostate cancer (HR=1.2, P=0.003), anal cancer (HR=1.8, P=0.01), other genital cancers (HR=1.5, P=0.03), lung cancer (HR=1.3 P<0.001), and pancreatic cancer (HR=1.5, P=0.004).
Dr Yanik and her colleagues noted that, of these cancers, NHL, anal cancer, lung cancer, melanoma, nonepithelial skin cancers, and pancreatic cancer consistently increased in incidence with each transition to a kidney function interval and decreased with each transition to a nonfunction interval.
And the 2 types of transitions were significant for NHL, lung cancer, melanoma, nonepithelial skin cancers, and pancreatic cancer.
The researchers also identified cancers with a significantly lower incidence during kidney function intervals. This included kidney cancer (HR=0.77, P<0.001), thyroid cancer (HR=0.67, P<0.001), breast cancer (HR=0.81, P=0.002), and liver cancer (HR=0.59, P=0.001).
The team noted that, among cancers with a lower incidence during kidney function intervals, kidney cancer, thyroid cancer, and myeloma consistently decreased in incidence with each transition to a kidney function interval and increased in incidence with each transition to a nonfunction interval. But both transitions were only significant for kidney and thyroid cancers.
“Our study indicates that the needs of individuals with end-stage renal disease, in terms of cancer prevention and cancer screening, will likely differ over time,” Dr Yanik said.
“Vigilance for kidney cancer and thyroid cancer may be of particular importance while these individuals are on dialysis. Extra consideration for screening for melanoma or lung cancer may be called for while taking immunosuppressant medications following a kidney transplant.” ![]()
Photo by Anna Frodesiak
Patients with end-stage renal disease (ESRD) may have different cancer risks according to the treatment they are receiving, a new study suggests.
Researchers found that patients had a higher risk of developing infection-related and immune-related cancers—including Hodgkin and non-Hodgkin lymphoma (NHL)—after receiving a kidney transplant.
But patients had a higher risk of ESRD-related cancers when they were on dialysis.
Elizabeth Yanik, PhD, of the National Cancer Institute in Bethesda, Maryland, and her colleagues reported these results in the Journal of the American Society of Nephrology.
The researchers theorized that assessing patterns in ESRD patients across periods of dialysis and kidney transplant might inform cancer etiology.
So the team studied registry data on 202,195 kidney transplant candidates and recipients, comparing the incidence of cancers during kidney function intervals (time with a transplant) to the incidence during nonfunction intervals (waitlist or time after transplant failure [dialysis]). The analysis was adjusted for demographic characteristics.
Results showed the incidence of infection-related and immune-related cancers was higher during kidney function intervals than nonfunction intervals.
Cancers with a significantly higher incidence included Kaposi’s sarcoma (hazard ratio [HR]=9.1, P<0.001), NHL (HR=3.2, P<0.001), Hodgkin lymphoma (HR=3.0, P<0.001), lip cancer (HR=3.4, P<0.001), nonepithelial skin cancers (HR=3.8, P<0.001), melanoma (HR=1.9, P<0.001), prostate cancer (HR=1.2, P=0.003), anal cancer (HR=1.8, P=0.01), other genital cancers (HR=1.5, P=0.03), lung cancer (HR=1.3 P<0.001), and pancreatic cancer (HR=1.5, P=0.004).
Dr Yanik and her colleagues noted that, of these cancers, NHL, anal cancer, lung cancer, melanoma, nonepithelial skin cancers, and pancreatic cancer consistently increased in incidence with each transition to a kidney function interval and decreased with each transition to a nonfunction interval.
And the 2 types of transitions were significant for NHL, lung cancer, melanoma, nonepithelial skin cancers, and pancreatic cancer.
The researchers also identified cancers with a significantly lower incidence during kidney function intervals. This included kidney cancer (HR=0.77, P<0.001), thyroid cancer (HR=0.67, P<0.001), breast cancer (HR=0.81, P=0.002), and liver cancer (HR=0.59, P=0.001).
The team noted that, among cancers with a lower incidence during kidney function intervals, kidney cancer, thyroid cancer, and myeloma consistently decreased in incidence with each transition to a kidney function interval and increased in incidence with each transition to a nonfunction interval. But both transitions were only significant for kidney and thyroid cancers.
“Our study indicates that the needs of individuals with end-stage renal disease, in terms of cancer prevention and cancer screening, will likely differ over time,” Dr Yanik said.
“Vigilance for kidney cancer and thyroid cancer may be of particular importance while these individuals are on dialysis. Extra consideration for screening for melanoma or lung cancer may be called for while taking immunosuppressant medications following a kidney transplant.” ![]()
Pharma companies fail to disclose trial information

Photo by Esther Dyson
New research indicates that pharmaceutical companies are still failing to disclose trial information publicly, despite efforts made over the last several years to increase transparency.
Investigators examined publicly available information for 15 drugs approved by the US Food and Drug Administration (FDA) in 2012.
They found that two-thirds of clinical trials per drug were disclosed publicly, which is below legal standards.
In an attempt to fix this problem, the investigators developed the “Good Pharma Scorecard,” which ranks companies according to their adherence to transparency practices.
Jennifer Miller, PhD, of NYU Langone Medical Center in New York, New York, and her colleagues described the scorecard and reported their research results in BMJ Open.
“Selectively disclosing trial information can distort the medical evidence and challenge the abilities of physicians, prescription guideline writers, payers, and formulary decision-makers to recommend and provide the right drugs for the right patients,” Dr Miller said.
She also noted that selectively disclosing information violates the rights of human research subjects laid out in the US Common Rule, a rule of ethics that requires that human-subject experiments have the potential to contribute to generalizable knowledge.
Study details
Dr Miller and her colleagues examined publicly available information for all drugs approved by the FDA in 2012 that were sponsored by the 20 pharmaceutical companies with the highest market value.
The information was gathered from a variety of publicly available documents, including Drugs@FDA, ClinicalTrials.gov, and journals indexed in Medline.
From this information, the team identified 15 drugs from 10 companies with more than 318 associated clinical trials involving 99,599 research subjects.
Almost half of all the drugs reviewed had at least 1 undisclosed phase 2 or 3 trial. A median of 57% of trials per drug were properly registered, 20% of final results were reported on ClinicalTrials.gov, and 56% of trials were published in academic journals.
A median of 65% of trial results were publicly available—either on ClinicalTrials.gov or in the medical literature. But the investigators found “considerable variation” between companies.
Per drug, a median of 17% of trials were subject to public disclosure by the 2007 US Food and Drug Administration Amendments Act (FDAAA). Of these trials, a median of 67% were FDAAA-compliant.
A majority of the research subjects—68%—participated in trials subject to the FDAAA, and 51% of these subjects were enrolled in trials that were noncompliant with the act.
Good Pharma Scorecard
Dr Miller and her colleagues believe they have devised a solution to help fix the transparency problem—the Good Pharma Scorecard.
The scorecard ranks drug sponsors according to 5 elements of transparency:
- Trial registration
- Results reporting
- Publication of trial data in a medical journal
- Compliance with legal disclosure requirements
- Adherence with the ethics standards enshrined in the Common Rule.
“The scorecard and rankings have the potential to benefit consumers by helping assure the integrity and completeness of clinical trial information,” Dr Miller said. “Full transparency of clinical trials would also strengthen the protection of human research subjects by avoiding their unknowing recruitment into already failed experiments.”
The pilot rankings on the Good Pharma Scorecard score the largest pharmaceutical companies for drugs approved by the FDA in 2012.
But Dr Miller and her team plan to publish this scorecard annually, ranking each new group of FDA-approved drugs going forward. A ranking of drugs approved in 2015 and their sponsors is scheduled to be released in 2016. ![]()
Photo by Esther Dyson
New research indicates that pharmaceutical companies are still failing to disclose trial information publicly, despite efforts made over the last several years to increase transparency.
Investigators examined publicly available information for 15 drugs approved by the US Food and Drug Administration (FDA) in 2012.
They found that two-thirds of clinical trials per drug were disclosed publicly, which is below legal standards.
In an attempt to fix this problem, the investigators developed the “Good Pharma Scorecard,” which ranks companies according to their adherence to transparency practices.
Jennifer Miller, PhD, of NYU Langone Medical Center in New York, New York, and her colleagues described the scorecard and reported their research results in BMJ Open.
“Selectively disclosing trial information can distort the medical evidence and challenge the abilities of physicians, prescription guideline writers, payers, and formulary decision-makers to recommend and provide the right drugs for the right patients,” Dr Miller said.
She also noted that selectively disclosing information violates the rights of human research subjects laid out in the US Common Rule, a rule of ethics that requires that human-subject experiments have the potential to contribute to generalizable knowledge.
Study details
Dr Miller and her colleagues examined publicly available information for all drugs approved by the FDA in 2012 that were sponsored by the 20 pharmaceutical companies with the highest market value.
The information was gathered from a variety of publicly available documents, including Drugs@FDA, ClinicalTrials.gov, and journals indexed in Medline.
From this information, the team identified 15 drugs from 10 companies with more than 318 associated clinical trials involving 99,599 research subjects.
Almost half of all the drugs reviewed had at least 1 undisclosed phase 2 or 3 trial. A median of 57% of trials per drug were properly registered, 20% of final results were reported on ClinicalTrials.gov, and 56% of trials were published in academic journals.
A median of 65% of trial results were publicly available—either on ClinicalTrials.gov or in the medical literature. But the investigators found “considerable variation” between companies.
Per drug, a median of 17% of trials were subject to public disclosure by the 2007 US Food and Drug Administration Amendments Act (FDAAA). Of these trials, a median of 67% were FDAAA-compliant.
A majority of the research subjects—68%—participated in trials subject to the FDAAA, and 51% of these subjects were enrolled in trials that were noncompliant with the act.
Good Pharma Scorecard
Dr Miller and her colleagues believe they have devised a solution to help fix the transparency problem—the Good Pharma Scorecard.
The scorecard ranks drug sponsors according to 5 elements of transparency:
- Trial registration
- Results reporting
- Publication of trial data in a medical journal
- Compliance with legal disclosure requirements
- Adherence with the ethics standards enshrined in the Common Rule.
“The scorecard and rankings have the potential to benefit consumers by helping assure the integrity and completeness of clinical trial information,” Dr Miller said. “Full transparency of clinical trials would also strengthen the protection of human research subjects by avoiding their unknowing recruitment into already failed experiments.”
The pilot rankings on the Good Pharma Scorecard score the largest pharmaceutical companies for drugs approved by the FDA in 2012.
But Dr Miller and her team plan to publish this scorecard annually, ranking each new group of FDA-approved drugs going forward. A ranking of drugs approved in 2015 and their sponsors is scheduled to be released in 2016. ![]()
Photo by Esther Dyson
New research indicates that pharmaceutical companies are still failing to disclose trial information publicly, despite efforts made over the last several years to increase transparency.
Investigators examined publicly available information for 15 drugs approved by the US Food and Drug Administration (FDA) in 2012.
They found that two-thirds of clinical trials per drug were disclosed publicly, which is below legal standards.
In an attempt to fix this problem, the investigators developed the “Good Pharma Scorecard,” which ranks companies according to their adherence to transparency practices.
Jennifer Miller, PhD, of NYU Langone Medical Center in New York, New York, and her colleagues described the scorecard and reported their research results in BMJ Open.
“Selectively disclosing trial information can distort the medical evidence and challenge the abilities of physicians, prescription guideline writers, payers, and formulary decision-makers to recommend and provide the right drugs for the right patients,” Dr Miller said.
She also noted that selectively disclosing information violates the rights of human research subjects laid out in the US Common Rule, a rule of ethics that requires that human-subject experiments have the potential to contribute to generalizable knowledge.
Study details
Dr Miller and her colleagues examined publicly available information for all drugs approved by the FDA in 2012 that were sponsored by the 20 pharmaceutical companies with the highest market value.
The information was gathered from a variety of publicly available documents, including Drugs@FDA, ClinicalTrials.gov, and journals indexed in Medline.
From this information, the team identified 15 drugs from 10 companies with more than 318 associated clinical trials involving 99,599 research subjects.
Almost half of all the drugs reviewed had at least 1 undisclosed phase 2 or 3 trial. A median of 57% of trials per drug were properly registered, 20% of final results were reported on ClinicalTrials.gov, and 56% of trials were published in academic journals.
A median of 65% of trial results were publicly available—either on ClinicalTrials.gov or in the medical literature. But the investigators found “considerable variation” between companies.
Per drug, a median of 17% of trials were subject to public disclosure by the 2007 US Food and Drug Administration Amendments Act (FDAAA). Of these trials, a median of 67% were FDAAA-compliant.
A majority of the research subjects—68%—participated in trials subject to the FDAAA, and 51% of these subjects were enrolled in trials that were noncompliant with the act.
Good Pharma Scorecard
Dr Miller and her colleagues believe they have devised a solution to help fix the transparency problem—the Good Pharma Scorecard.
The scorecard ranks drug sponsors according to 5 elements of transparency:
- Trial registration
- Results reporting
- Publication of trial data in a medical journal
- Compliance with legal disclosure requirements
- Adherence with the ethics standards enshrined in the Common Rule.
“The scorecard and rankings have the potential to benefit consumers by helping assure the integrity and completeness of clinical trial information,” Dr Miller said. “Full transparency of clinical trials would also strengthen the protection of human research subjects by avoiding their unknowing recruitment into already failed experiments.”
The pilot rankings on the Good Pharma Scorecard score the largest pharmaceutical companies for drugs approved by the FDA in 2012.
But Dr Miller and her team plan to publish this scorecard annually, ranking each new group of FDA-approved drugs going forward. A ranking of drugs approved in 2015 and their sponsors is scheduled to be released in 2016. ![]()
Team evaluates long-term tolerability of ticagrelor

Photo courtesy of AstraZeneca
ORLANDO, FL—A subanalysis of the PEGASUS-TIMI 54 trial has provided a clearer picture of the risks and benefits associated with long-term use of the antiplatelet drug ticagrelor, according to investigators.
The goals of the analysis were to evaluate the long-term efficacy of ticagrelor in patients with a history of myocardial infarction and to determine the rates of, and reasons for, discontinuing ticagrelor in these patients.
The PEGASUS-TIMI 54 trial included 21,162 patients (age 50 and older) who had experienced a heart attack in the previous 1 to 3 years. The patients were randomized to receive aspirin plus twice-daily doses of ticagrelor at 90 mg, ticagrelor at 60 mg, or placebo.
Earlier results from this study showed that both ticagrelor doses reduced the rate of the primary endpoint, which was a composite of cardiovascular death, heart attack, and stroke.
However, the rates of TIMI major bleeding and dyspnea were higher in patients who received either dose of ticagrelor than in those who received placebo. And premature discontinuation was higher in the ticagrelor arms.
To investigate this further, Marc Bonaca, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues conducted their subanalysis. The results were presented in a late-breaking clinical trials session at the American Heart Association (AHA) Scientific Sessions 2015.
The trial lasted a median of 33 months. During this time, the rate of discontinuation (of ticagrelor or placebo) was 32% in the 90 mg ticagrelor arm, 29% in the 60 mg ticagrelor arm, and 21% in the placebo arm (P<0.001).
The rates of discontinuation due to patient decision or an administrative reason were similar between the arms.
However, patients were more likely to stop taking the study drug due to an adverse event (AE) if they were receiving ticagrelor rather than placebo. Discontinuation due to an AE was 19% in the 90 mg ticagrelor arm, 16.4% in the 60 mg ticagrelor arm, and 8.9% in the placebo arm (P<0.01).
The most frequent AEs leading to discontinuation were bleeding—6.5% in the 90 mg arm, 5.1% in the 60 mg arm, and 1.2% in the placebo arm (P<0.001)—and dyspnea—6.2%, 4.3%, and 0.7%, respectively (P<0.001).
Rates of AEs leading to discontinuation were highest in the first year—16% in the 90 mg arm, 13% in the 60 mg arm, and 6% in the placebo arm.
In those patients who stayed on therapy, discontinuation rates over the subsequent 2 years were 6.5% in the 90 mg arm, 6.0% in the 60 mg arm, 4.6% in the placebo arm.
In patients who stayed on therapy, ticagrelor reduced the rate of the composite efficacy endpoint of cardiovascular death, myocardial infarction, or stroke at 3 years (hazard ratio=0.79), which is consistent with the results of the overall population of the study.
“This analysis pointed to important patterns with regards to common AEs associated with ticagrelor in the context of clinical benefit,” Dr Bonaca said.
“Physicians must consider the overall risks, including higher rates of bleeding and dyspnea, particularly within the first year. For patients at increased risk for recurrent cardiovascular events in the long-term, ticagrelor can provide an important benefit.”
PEGASUS-TIMI 54 was sponsored by AstraZeneca, the company developing ticagrelor. Investigators involved in the subanalysis reported relationships with a range of other companies as well. ![]()
Photo courtesy of AstraZeneca
ORLANDO, FL—A subanalysis of the PEGASUS-TIMI 54 trial has provided a clearer picture of the risks and benefits associated with long-term use of the antiplatelet drug ticagrelor, according to investigators.
The goals of the analysis were to evaluate the long-term efficacy of ticagrelor in patients with a history of myocardial infarction and to determine the rates of, and reasons for, discontinuing ticagrelor in these patients.
The PEGASUS-TIMI 54 trial included 21,162 patients (age 50 and older) who had experienced a heart attack in the previous 1 to 3 years. The patients were randomized to receive aspirin plus twice-daily doses of ticagrelor at 90 mg, ticagrelor at 60 mg, or placebo.
Earlier results from this study showed that both ticagrelor doses reduced the rate of the primary endpoint, which was a composite of cardiovascular death, heart attack, and stroke.
However, the rates of TIMI major bleeding and dyspnea were higher in patients who received either dose of ticagrelor than in those who received placebo. And premature discontinuation was higher in the ticagrelor arms.
To investigate this further, Marc Bonaca, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues conducted their subanalysis. The results were presented in a late-breaking clinical trials session at the American Heart Association (AHA) Scientific Sessions 2015.
The trial lasted a median of 33 months. During this time, the rate of discontinuation (of ticagrelor or placebo) was 32% in the 90 mg ticagrelor arm, 29% in the 60 mg ticagrelor arm, and 21% in the placebo arm (P<0.001).
The rates of discontinuation due to patient decision or an administrative reason were similar between the arms.
However, patients were more likely to stop taking the study drug due to an adverse event (AE) if they were receiving ticagrelor rather than placebo. Discontinuation due to an AE was 19% in the 90 mg ticagrelor arm, 16.4% in the 60 mg ticagrelor arm, and 8.9% in the placebo arm (P<0.01).
The most frequent AEs leading to discontinuation were bleeding—6.5% in the 90 mg arm, 5.1% in the 60 mg arm, and 1.2% in the placebo arm (P<0.001)—and dyspnea—6.2%, 4.3%, and 0.7%, respectively (P<0.001).
Rates of AEs leading to discontinuation were highest in the first year—16% in the 90 mg arm, 13% in the 60 mg arm, and 6% in the placebo arm.
In those patients who stayed on therapy, discontinuation rates over the subsequent 2 years were 6.5% in the 90 mg arm, 6.0% in the 60 mg arm, 4.6% in the placebo arm.
In patients who stayed on therapy, ticagrelor reduced the rate of the composite efficacy endpoint of cardiovascular death, myocardial infarction, or stroke at 3 years (hazard ratio=0.79), which is consistent with the results of the overall population of the study.
“This analysis pointed to important patterns with regards to common AEs associated with ticagrelor in the context of clinical benefit,” Dr Bonaca said.
“Physicians must consider the overall risks, including higher rates of bleeding and dyspnea, particularly within the first year. For patients at increased risk for recurrent cardiovascular events in the long-term, ticagrelor can provide an important benefit.”
PEGASUS-TIMI 54 was sponsored by AstraZeneca, the company developing ticagrelor. Investigators involved in the subanalysis reported relationships with a range of other companies as well. ![]()
Photo courtesy of AstraZeneca
ORLANDO, FL—A subanalysis of the PEGASUS-TIMI 54 trial has provided a clearer picture of the risks and benefits associated with long-term use of the antiplatelet drug ticagrelor, according to investigators.
The goals of the analysis were to evaluate the long-term efficacy of ticagrelor in patients with a history of myocardial infarction and to determine the rates of, and reasons for, discontinuing ticagrelor in these patients.
The PEGASUS-TIMI 54 trial included 21,162 patients (age 50 and older) who had experienced a heart attack in the previous 1 to 3 years. The patients were randomized to receive aspirin plus twice-daily doses of ticagrelor at 90 mg, ticagrelor at 60 mg, or placebo.
Earlier results from this study showed that both ticagrelor doses reduced the rate of the primary endpoint, which was a composite of cardiovascular death, heart attack, and stroke.
However, the rates of TIMI major bleeding and dyspnea were higher in patients who received either dose of ticagrelor than in those who received placebo. And premature discontinuation was higher in the ticagrelor arms.
To investigate this further, Marc Bonaca, MD, of Brigham and Women’s Hospital in Boston, Massachusetts, and his colleagues conducted their subanalysis. The results were presented in a late-breaking clinical trials session at the American Heart Association (AHA) Scientific Sessions 2015.
The trial lasted a median of 33 months. During this time, the rate of discontinuation (of ticagrelor or placebo) was 32% in the 90 mg ticagrelor arm, 29% in the 60 mg ticagrelor arm, and 21% in the placebo arm (P<0.001).
The rates of discontinuation due to patient decision or an administrative reason were similar between the arms.
However, patients were more likely to stop taking the study drug due to an adverse event (AE) if they were receiving ticagrelor rather than placebo. Discontinuation due to an AE was 19% in the 90 mg ticagrelor arm, 16.4% in the 60 mg ticagrelor arm, and 8.9% in the placebo arm (P<0.01).
The most frequent AEs leading to discontinuation were bleeding—6.5% in the 90 mg arm, 5.1% in the 60 mg arm, and 1.2% in the placebo arm (P<0.001)—and dyspnea—6.2%, 4.3%, and 0.7%, respectively (P<0.001).
Rates of AEs leading to discontinuation were highest in the first year—16% in the 90 mg arm, 13% in the 60 mg arm, and 6% in the placebo arm.
In those patients who stayed on therapy, discontinuation rates over the subsequent 2 years were 6.5% in the 90 mg arm, 6.0% in the 60 mg arm, 4.6% in the placebo arm.
In patients who stayed on therapy, ticagrelor reduced the rate of the composite efficacy endpoint of cardiovascular death, myocardial infarction, or stroke at 3 years (hazard ratio=0.79), which is consistent with the results of the overall population of the study.
“This analysis pointed to important patterns with regards to common AEs associated with ticagrelor in the context of clinical benefit,” Dr Bonaca said.
“Physicians must consider the overall risks, including higher rates of bleeding and dyspnea, particularly within the first year. For patients at increased risk for recurrent cardiovascular events in the long-term, ticagrelor can provide an important benefit.”
PEGASUS-TIMI 54 was sponsored by AstraZeneca, the company developing ticagrelor. Investigators involved in the subanalysis reported relationships with a range of other companies as well. ![]()
Findings may help advance treatment of malaria
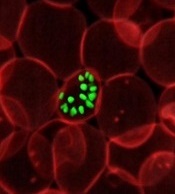
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
Researchers have found that a screening model can classify antimalarial drugs and reveal drug targets for Plasmodium falciparum, according to a paper published in Scientific Reports.
The team performed chemogenomic profiling of P falciparum for the first time.
They used a collection of malaria parasite mutants that each had altered metabolism linked to a defect in a single P falciparum gene.
They then screened 53 drugs and compounds against 71 of these P falciparum piggyBac single-insertion mutant parasites.
Computational analysis of the response patterns linked the different antimalarial drug candidates and metabolic inhibitors to the specific gene defect.
This revealed new insights into the drugs’ mechanism of action and uncovered 6 new genes that were involved in P falciparum’s response to artemisinin but were associated with increased susceptibility to the drugs tested.
“That represents 6 new targets potentially as effective as artemisinin for killing the malaria parasite,” said study author Dennis Kyle, PhD, of the University of South Florida in Tampa.
“There is definitely a sense of urgency for discovering new antimalarial drugs that may replace artemisinin, or work better with artemisinin, to prevent or delay drug resistance.”
P falciparum, which is becoming increasingly resistant to artemisinin, causes three-quarters of all malaria cases in Africa and 95% of malaria deaths worldwide. ![]()
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
Researchers have found that a screening model can classify antimalarial drugs and reveal drug targets for Plasmodium falciparum, according to a paper published in Scientific Reports.
The team performed chemogenomic profiling of P falciparum for the first time.
They used a collection of malaria parasite mutants that each had altered metabolism linked to a defect in a single P falciparum gene.
They then screened 53 drugs and compounds against 71 of these P falciparum piggyBac single-insertion mutant parasites.
Computational analysis of the response patterns linked the different antimalarial drug candidates and metabolic inhibitors to the specific gene defect.
This revealed new insights into the drugs’ mechanism of action and uncovered 6 new genes that were involved in P falciparum’s response to artemisinin but were associated with increased susceptibility to the drugs tested.
“That represents 6 new targets potentially as effective as artemisinin for killing the malaria parasite,” said study author Dennis Kyle, PhD, of the University of South Florida in Tampa.
“There is definitely a sense of urgency for discovering new antimalarial drugs that may replace artemisinin, or work better with artemisinin, to prevent or delay drug resistance.”
P falciparum, which is becoming increasingly resistant to artemisinin, causes three-quarters of all malaria cases in Africa and 95% of malaria deaths worldwide. ![]()
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
Researchers have found that a screening model can classify antimalarial drugs and reveal drug targets for Plasmodium falciparum, according to a paper published in Scientific Reports.
The team performed chemogenomic profiling of P falciparum for the first time.
They used a collection of malaria parasite mutants that each had altered metabolism linked to a defect in a single P falciparum gene.
They then screened 53 drugs and compounds against 71 of these P falciparum piggyBac single-insertion mutant parasites.
Computational analysis of the response patterns linked the different antimalarial drug candidates and metabolic inhibitors to the specific gene defect.
This revealed new insights into the drugs’ mechanism of action and uncovered 6 new genes that were involved in P falciparum’s response to artemisinin but were associated with increased susceptibility to the drugs tested.
“That represents 6 new targets potentially as effective as artemisinin for killing the malaria parasite,” said study author Dennis Kyle, PhD, of the University of South Florida in Tampa.
“There is definitely a sense of urgency for discovering new antimalarial drugs that may replace artemisinin, or work better with artemisinin, to prevent or delay drug resistance.”
P falciparum, which is becoming increasingly resistant to artemisinin, causes three-quarters of all malaria cases in Africa and 95% of malaria deaths worldwide. ![]()
Inhibitor exhibits ‘modest’ activity in lymphoma
A small-molecule inhibitor has shown modest anticancer activity in a phase 1 trial of patients with relapsed/refractory lymphoma or multiple myeloma (MM), according to an investigator involved in the study.
A majority of patients who recieved the drug, pevonedistat, achieved stable disease, and a few patients with lymphoma experienced partial responses.
Investigators said pevonedistat could be given safely, although 100% of patients experienced adverse events (AEs), and a majority experienced grade 3 or higher AEs.
“The most important findings from our study are that pevonedistat hits its target in cancer cells in patients, can be given safely, and has modest activity in heavily pretreated patients with relapsed/refractory lymphoma, suggesting that we are on the right path,” said Jatin J. Shah, MD, of The University of Texas MD Anderson Cancer Center in Houston.
Dr Shah and his colleagues reported these findings in Clinical Cancer Research. The trial was sponsored by Millennium Pharmaceuticals, Inc., the company developing pevonedistat.
Pevonedistat is a first-in-class, investigational, small-molecule inhibitor of the NEDD8-activating enzyme.
“This enzyme is part of the ubiquitin-proteasome system, which is the target of a number of FDA-approved anticancer therapeutics, including bortezomib . . . ,” Dr Shah explained. “Pevonedistat also alters the ability of cancer cells to repair damaged DNA.”
Patient and treatment characteristics
Dr Shah and his colleagues enrolled 44 patients on this trial. Seventeen patients had relapsed/refractory MM, and 27 had relapsed/refractory lymphoma.
The types of lymphoma were diffuse large B-cell lymphoma (n=10), follicular lymphoma (n=5), Hodgkin lymphoma (n=5), small lymphocytic lymphoma/chronic lymphocytic leukemia (n=2), mantle cell lymphoma (n=1), lymphoplasmacytic lymphoma (n=1), peripheral T-cell lymphoma (n=1), splenic marginal zone B-cell lymphoma (n=1), and “other” (n=1).
Twenty-seven patients received escalating doses of pevonedistat on schedule A, which was days 1, 2, 8, and 9 of a 21-day cycle, and 17 patients received escalating doses of the drug on schedule B, which was days 1, 4, 8, and 11 of a 21-day cycle.
Safety
The maximum tolerated doses were 110 mg/m2 and 196 mg/m2 on schedule A and B, respectively. The dose-limiting toxicities were febrile neutropenia, transaminase elevations, and muscle cramps on schedule A, and thrombocytopenia on schedule B.
On schedule A, 100% of patients experienced AEs, and 59% had AEs of grade 3 or higher. The grade 3 or higher AEs were anemia (19%), thrombocytopenia (4%), neutropenia (7%), fatigue (7%), ALT increase (4%), AST increase (7%), diarrhea (4%), pain (4%), dyspnea (4%), hypercalcemia (7%), hypophosphatemia (11%), hyperkalemia (4%), muscle spasms (4%), abdominal discomfort (4%), and pneumonia (4%).
On schedule B, 100% of patients experienced AEs, and 71% had grade 3 or higher AEs. The grade 3 or higher AEs were anemia (6%), thrombocytopenia (6%), neutropenia (12%), fatigue (6%), ALT increase (6%), pyrexia (6%), pain (6%), dyspnea (6%), hypophosphatemia (6%), muscle spasms (6%), upper respiratory tract infection (6%), dehydration (6%), hyperbilirubinemia (6%), and pneumonia (12%).
Efficacy
Three patients had a partial response to treatment, including 1 patient with relapsed nodular sclerosis Hodgkin lymphoma, 1 with relapsed diffuse large B-cell lymphoma, and 1 with relapsed peripheral T-cell lymphoma.
Another 30 patients, 17 with lymphoma and 13 with MM, had stable disease.
“Although pevonedistat had modest activity as a single-agent treatment, we expect greater activity when it is given in combination with standard therapy,” Dr Shah said.
“The pharmacodynamics data showed that pevonedistat hit its target in cancer cells in patients at low doses. This is important because it may mean that we do not need to escalate the dose in future trials to increase anticancer activity. This has the potential to increase the risk-benefit ratio of pevonedistat.”
Dr Shah said a limitation of this study is that it included a small number of patients, all of whom were very heavily pretreated, which may limit assessment of how active pevonedistat could be. ![]()
A small-molecule inhibitor has shown modest anticancer activity in a phase 1 trial of patients with relapsed/refractory lymphoma or multiple myeloma (MM), according to an investigator involved in the study.
A majority of patients who recieved the drug, pevonedistat, achieved stable disease, and a few patients with lymphoma experienced partial responses.
Investigators said pevonedistat could be given safely, although 100% of patients experienced adverse events (AEs), and a majority experienced grade 3 or higher AEs.
“The most important findings from our study are that pevonedistat hits its target in cancer cells in patients, can be given safely, and has modest activity in heavily pretreated patients with relapsed/refractory lymphoma, suggesting that we are on the right path,” said Jatin J. Shah, MD, of The University of Texas MD Anderson Cancer Center in Houston.
Dr Shah and his colleagues reported these findings in Clinical Cancer Research. The trial was sponsored by Millennium Pharmaceuticals, Inc., the company developing pevonedistat.
Pevonedistat is a first-in-class, investigational, small-molecule inhibitor of the NEDD8-activating enzyme.
“This enzyme is part of the ubiquitin-proteasome system, which is the target of a number of FDA-approved anticancer therapeutics, including bortezomib . . . ,” Dr Shah explained. “Pevonedistat also alters the ability of cancer cells to repair damaged DNA.”
Patient and treatment characteristics
Dr Shah and his colleagues enrolled 44 patients on this trial. Seventeen patients had relapsed/refractory MM, and 27 had relapsed/refractory lymphoma.
The types of lymphoma were diffuse large B-cell lymphoma (n=10), follicular lymphoma (n=5), Hodgkin lymphoma (n=5), small lymphocytic lymphoma/chronic lymphocytic leukemia (n=2), mantle cell lymphoma (n=1), lymphoplasmacytic lymphoma (n=1), peripheral T-cell lymphoma (n=1), splenic marginal zone B-cell lymphoma (n=1), and “other” (n=1).
Twenty-seven patients received escalating doses of pevonedistat on schedule A, which was days 1, 2, 8, and 9 of a 21-day cycle, and 17 patients received escalating doses of the drug on schedule B, which was days 1, 4, 8, and 11 of a 21-day cycle.
Safety
The maximum tolerated doses were 110 mg/m2 and 196 mg/m2 on schedule A and B, respectively. The dose-limiting toxicities were febrile neutropenia, transaminase elevations, and muscle cramps on schedule A, and thrombocytopenia on schedule B.
On schedule A, 100% of patients experienced AEs, and 59% had AEs of grade 3 or higher. The grade 3 or higher AEs were anemia (19%), thrombocytopenia (4%), neutropenia (7%), fatigue (7%), ALT increase (4%), AST increase (7%), diarrhea (4%), pain (4%), dyspnea (4%), hypercalcemia (7%), hypophosphatemia (11%), hyperkalemia (4%), muscle spasms (4%), abdominal discomfort (4%), and pneumonia (4%).
On schedule B, 100% of patients experienced AEs, and 71% had grade 3 or higher AEs. The grade 3 or higher AEs were anemia (6%), thrombocytopenia (6%), neutropenia (12%), fatigue (6%), ALT increase (6%), pyrexia (6%), pain (6%), dyspnea (6%), hypophosphatemia (6%), muscle spasms (6%), upper respiratory tract infection (6%), dehydration (6%), hyperbilirubinemia (6%), and pneumonia (12%).
Efficacy
Three patients had a partial response to treatment, including 1 patient with relapsed nodular sclerosis Hodgkin lymphoma, 1 with relapsed diffuse large B-cell lymphoma, and 1 with relapsed peripheral T-cell lymphoma.
Another 30 patients, 17 with lymphoma and 13 with MM, had stable disease.
“Although pevonedistat had modest activity as a single-agent treatment, we expect greater activity when it is given in combination with standard therapy,” Dr Shah said.
“The pharmacodynamics data showed that pevonedistat hit its target in cancer cells in patients at low doses. This is important because it may mean that we do not need to escalate the dose in future trials to increase anticancer activity. This has the potential to increase the risk-benefit ratio of pevonedistat.”
Dr Shah said a limitation of this study is that it included a small number of patients, all of whom were very heavily pretreated, which may limit assessment of how active pevonedistat could be. ![]()
A small-molecule inhibitor has shown modest anticancer activity in a phase 1 trial of patients with relapsed/refractory lymphoma or multiple myeloma (MM), according to an investigator involved in the study.
A majority of patients who recieved the drug, pevonedistat, achieved stable disease, and a few patients with lymphoma experienced partial responses.
Investigators said pevonedistat could be given safely, although 100% of patients experienced adverse events (AEs), and a majority experienced grade 3 or higher AEs.
“The most important findings from our study are that pevonedistat hits its target in cancer cells in patients, can be given safely, and has modest activity in heavily pretreated patients with relapsed/refractory lymphoma, suggesting that we are on the right path,” said Jatin J. Shah, MD, of The University of Texas MD Anderson Cancer Center in Houston.
Dr Shah and his colleagues reported these findings in Clinical Cancer Research. The trial was sponsored by Millennium Pharmaceuticals, Inc., the company developing pevonedistat.
Pevonedistat is a first-in-class, investigational, small-molecule inhibitor of the NEDD8-activating enzyme.
“This enzyme is part of the ubiquitin-proteasome system, which is the target of a number of FDA-approved anticancer therapeutics, including bortezomib . . . ,” Dr Shah explained. “Pevonedistat also alters the ability of cancer cells to repair damaged DNA.”
Patient and treatment characteristics
Dr Shah and his colleagues enrolled 44 patients on this trial. Seventeen patients had relapsed/refractory MM, and 27 had relapsed/refractory lymphoma.
The types of lymphoma were diffuse large B-cell lymphoma (n=10), follicular lymphoma (n=5), Hodgkin lymphoma (n=5), small lymphocytic lymphoma/chronic lymphocytic leukemia (n=2), mantle cell lymphoma (n=1), lymphoplasmacytic lymphoma (n=1), peripheral T-cell lymphoma (n=1), splenic marginal zone B-cell lymphoma (n=1), and “other” (n=1).
Twenty-seven patients received escalating doses of pevonedistat on schedule A, which was days 1, 2, 8, and 9 of a 21-day cycle, and 17 patients received escalating doses of the drug on schedule B, which was days 1, 4, 8, and 11 of a 21-day cycle.
Safety
The maximum tolerated doses were 110 mg/m2 and 196 mg/m2 on schedule A and B, respectively. The dose-limiting toxicities were febrile neutropenia, transaminase elevations, and muscle cramps on schedule A, and thrombocytopenia on schedule B.
On schedule A, 100% of patients experienced AEs, and 59% had AEs of grade 3 or higher. The grade 3 or higher AEs were anemia (19%), thrombocytopenia (4%), neutropenia (7%), fatigue (7%), ALT increase (4%), AST increase (7%), diarrhea (4%), pain (4%), dyspnea (4%), hypercalcemia (7%), hypophosphatemia (11%), hyperkalemia (4%), muscle spasms (4%), abdominal discomfort (4%), and pneumonia (4%).
On schedule B, 100% of patients experienced AEs, and 71% had grade 3 or higher AEs. The grade 3 or higher AEs were anemia (6%), thrombocytopenia (6%), neutropenia (12%), fatigue (6%), ALT increase (6%), pyrexia (6%), pain (6%), dyspnea (6%), hypophosphatemia (6%), muscle spasms (6%), upper respiratory tract infection (6%), dehydration (6%), hyperbilirubinemia (6%), and pneumonia (12%).
Efficacy
Three patients had a partial response to treatment, including 1 patient with relapsed nodular sclerosis Hodgkin lymphoma, 1 with relapsed diffuse large B-cell lymphoma, and 1 with relapsed peripheral T-cell lymphoma.
Another 30 patients, 17 with lymphoma and 13 with MM, had stable disease.
“Although pevonedistat had modest activity as a single-agent treatment, we expect greater activity when it is given in combination with standard therapy,” Dr Shah said.
“The pharmacodynamics data showed that pevonedistat hit its target in cancer cells in patients at low doses. This is important because it may mean that we do not need to escalate the dose in future trials to increase anticancer activity. This has the potential to increase the risk-benefit ratio of pevonedistat.”
Dr Shah said a limitation of this study is that it included a small number of patients, all of whom were very heavily pretreated, which may limit assessment of how active pevonedistat could be. ![]()
Drug ‘life-changing’ for CLL patients in phase 1 trial

Photo courtesy of NIH
A novel Bruton’s tyrosine kinase inhibitor has proven life-changing for patients with chronic lymphocytic leukemia (CLL) who received the drug as part of a phase 1 trial, according to the study’s lead author.
The inhibitor, ONO/GS-4059, produced a response in 96% of evaluable CLL patients.
Most CLL patients are still on the study after 3 years, although a handful withdrew due to adverse events (AEs) or disease progression.
“These patients were confronted with a cruel reality: they had failed multiple chemotherapy lines, and there were no other treatment options available for them,” said lead study author Harriet Walter, MBChB, of the University of Leicester in the UK.
“This drug has changed their lives. From desperate and tired, they are now leading a normal and really active life. This is hugely rewarding and encouraging.”
Dr Walter and her colleagues reported these results in Blood. The trial was funded by ONO Pharmaceuticals, the company developing ONO/GS-4059.
This study opened in January 2012, and 90 patients were enrolled at centers in the UK and France. There were 28 patients with CLL and 62 with non-Hodgkin lymphoma (NHL), including 16 with mantle cell lymphoma (MCL) and 35 with diffuse large B-cell lymphoma (DLBCL).
The study also included patients with follicular lymphoma, marginal zone lymphoma, small lymphocytic lymphoma, and Waldenstrom’s macroglobulinemia, but patient numbers were small for these groups, so the results were not discussed in detail.
There were 9 dose-escalation cohorts in this study. ONO/GS-4059 was given once-daily at doses ranging from 20 mg to 600 mg. Or the drug was given twice daily at doses of 240 mg or 300 mg.
Results
The maximum tolerated dose was not reached in the CLL cohort, but it was 480 mg once-daily in the NHL cohort. Four NHL patients had a dose-limiting toxicity.
In the CLL cohort, 2 patients went off study due to progression and 5 due to AEs.
In the NHL cohort, 49 patients discontinued treatment, 32 due to progression and 5 due to dose-limiting toxicities or AEs. The other 12 NHL patients discontinued due to patient or investigator decision, proceeding to transplant (n=1), or death due to progressive disease.
The median duration of follow-up was 560 days for CLL patients, 309 days for MCL patients, and 60 days for DLBCL patients.
The overall estimated mean progression-free survival was 874 days for CLL patients, 341 days for MCL patients, and 54 days for DLBCL patients.
CLL patients
Of all 28 CLL patients, 16 had relapsed CLL, 11 had refractory disease, and 1 had unknown status. The median number of prior therapies was 3.5 (range, 2-7).
Twenty-five patients were evaluable. Of the 3 who were not evaluable, 1 had not reached cycle 3 disease assessment at the time of data analysis, 1 progressed during cycle 1, and 1 was withdrawn due to an AE (idiopathic thrombocytopenia).
Of the 25 evaluable patients, 24 (96%) responded to ONO/GS-4059. The researchers said they observed rapid resolution of bulky lymphadenopathy within the first 3 months of treatment, but improvement in lymphadenopathy continued for up to 18 months in most patients.
The median treatment duration for these patients is 80 weeks, and 21 patients are still on treatment. Two of the evaluable patients progressed during therapy, one at cycle 3 and one at cycle 12.
MCL patients
Of the 16 MCL patients enrolled, 7 were refractory to their last course of immuno-chemotherapy. The median number of prior therapies was 3 (range, 2-7).
Eleven of 12 (92%) evaluable patients with MCL responded to ONO/GS-4059. Six patients had a partial response, and 5 had a complete response (CR) or unconfirmed CR.
Three patients progressed after an initial response. Four patients were not evaluable because they progressed.
The median treatment duration for MCL patients is 40 weeks, and 8 patients remain on study.
DLBCL patients
All 35 DLBCL patients had relapsed or refractory disease. The median number of prior treatments was 3
(range, 2-10), and 30 patients were refractory to their last line of chemotherapy.
Eleven of 31 (35%) patients with non-germinal center B-cell (non-GCB) DLBCL responded to ONO/GS-4059. Two non-GCB DLBCL patients had a confirmed CR, 1 had an unconfirmed CR, and the rest had partial responses.
The median duration of response was 54 days. And, among responders, the median treatment duration was 12 weeks.
The majority of non-GCB DLBCL patients progressed. There were no responses among the 2 patients with GCB DLBCL, and there were no responses among patients with primary mediastinal B-cell lymphoma or plasmablastic DLBCL.
Toxicity
AEs in this study were mostly grade 1/2—75% in the CLL cohort and 50% in the NHL cohort. However, treatment-related grade 3/4 AEs occurred in 14.3% of CLL patients and 16.1% of NHL patients.
Grade 3/4 events were mainly hematologic in nature and included neutropenia (10%), anemia (13.3%), and thrombocytopenia (13.3%).
There was a grade 3 episode of drug-related hemorrhage in a CLL patient, which resulted in a psoas hematoma (with concomitant CLL progression) in the presence of a normal platelet count. This patient was among those taken off the study.
“The next step is now to see how best we can improve on these outstanding results,” said study author Martin Dyer, DPhil, of the University of Leicester.
“A further study using this drug in combination with additional targeted agents is shortly to open in Leicester with the aim of achieving cure. In parallel with the clinical development of the drug, our team of scientists at the Haematological Research Institute are studying how this drug is working and how to overcome potential resistance.” ![]()
Photo courtesy of NIH
A novel Bruton’s tyrosine kinase inhibitor has proven life-changing for patients with chronic lymphocytic leukemia (CLL) who received the drug as part of a phase 1 trial, according to the study’s lead author.
The inhibitor, ONO/GS-4059, produced a response in 96% of evaluable CLL patients.
Most CLL patients are still on the study after 3 years, although a handful withdrew due to adverse events (AEs) or disease progression.
“These patients were confronted with a cruel reality: they had failed multiple chemotherapy lines, and there were no other treatment options available for them,” said lead study author Harriet Walter, MBChB, of the University of Leicester in the UK.
“This drug has changed their lives. From desperate and tired, they are now leading a normal and really active life. This is hugely rewarding and encouraging.”
Dr Walter and her colleagues reported these results in Blood. The trial was funded by ONO Pharmaceuticals, the company developing ONO/GS-4059.
This study opened in January 2012, and 90 patients were enrolled at centers in the UK and France. There were 28 patients with CLL and 62 with non-Hodgkin lymphoma (NHL), including 16 with mantle cell lymphoma (MCL) and 35 with diffuse large B-cell lymphoma (DLBCL).
The study also included patients with follicular lymphoma, marginal zone lymphoma, small lymphocytic lymphoma, and Waldenstrom’s macroglobulinemia, but patient numbers were small for these groups, so the results were not discussed in detail.
There were 9 dose-escalation cohorts in this study. ONO/GS-4059 was given once-daily at doses ranging from 20 mg to 600 mg. Or the drug was given twice daily at doses of 240 mg or 300 mg.
Results
The maximum tolerated dose was not reached in the CLL cohort, but it was 480 mg once-daily in the NHL cohort. Four NHL patients had a dose-limiting toxicity.
In the CLL cohort, 2 patients went off study due to progression and 5 due to AEs.
In the NHL cohort, 49 patients discontinued treatment, 32 due to progression and 5 due to dose-limiting toxicities or AEs. The other 12 NHL patients discontinued due to patient or investigator decision, proceeding to transplant (n=1), or death due to progressive disease.
The median duration of follow-up was 560 days for CLL patients, 309 days for MCL patients, and 60 days for DLBCL patients.
The overall estimated mean progression-free survival was 874 days for CLL patients, 341 days for MCL patients, and 54 days for DLBCL patients.
CLL patients
Of all 28 CLL patients, 16 had relapsed CLL, 11 had refractory disease, and 1 had unknown status. The median number of prior therapies was 3.5 (range, 2-7).
Twenty-five patients were evaluable. Of the 3 who were not evaluable, 1 had not reached cycle 3 disease assessment at the time of data analysis, 1 progressed during cycle 1, and 1 was withdrawn due to an AE (idiopathic thrombocytopenia).
Of the 25 evaluable patients, 24 (96%) responded to ONO/GS-4059. The researchers said they observed rapid resolution of bulky lymphadenopathy within the first 3 months of treatment, but improvement in lymphadenopathy continued for up to 18 months in most patients.
The median treatment duration for these patients is 80 weeks, and 21 patients are still on treatment. Two of the evaluable patients progressed during therapy, one at cycle 3 and one at cycle 12.
MCL patients
Of the 16 MCL patients enrolled, 7 were refractory to their last course of immuno-chemotherapy. The median number of prior therapies was 3 (range, 2-7).
Eleven of 12 (92%) evaluable patients with MCL responded to ONO/GS-4059. Six patients had a partial response, and 5 had a complete response (CR) or unconfirmed CR.
Three patients progressed after an initial response. Four patients were not evaluable because they progressed.
The median treatment duration for MCL patients is 40 weeks, and 8 patients remain on study.
DLBCL patients
All 35 DLBCL patients had relapsed or refractory disease. The median number of prior treatments was 3
(range, 2-10), and 30 patients were refractory to their last line of chemotherapy.
Eleven of 31 (35%) patients with non-germinal center B-cell (non-GCB) DLBCL responded to ONO/GS-4059. Two non-GCB DLBCL patients had a confirmed CR, 1 had an unconfirmed CR, and the rest had partial responses.
The median duration of response was 54 days. And, among responders, the median treatment duration was 12 weeks.
The majority of non-GCB DLBCL patients progressed. There were no responses among the 2 patients with GCB DLBCL, and there were no responses among patients with primary mediastinal B-cell lymphoma or plasmablastic DLBCL.
Toxicity
AEs in this study were mostly grade 1/2—75% in the CLL cohort and 50% in the NHL cohort. However, treatment-related grade 3/4 AEs occurred in 14.3% of CLL patients and 16.1% of NHL patients.
Grade 3/4 events were mainly hematologic in nature and included neutropenia (10%), anemia (13.3%), and thrombocytopenia (13.3%).
There was a grade 3 episode of drug-related hemorrhage in a CLL patient, which resulted in a psoas hematoma (with concomitant CLL progression) in the presence of a normal platelet count. This patient was among those taken off the study.
“The next step is now to see how best we can improve on these outstanding results,” said study author Martin Dyer, DPhil, of the University of Leicester.
“A further study using this drug in combination with additional targeted agents is shortly to open in Leicester with the aim of achieving cure. In parallel with the clinical development of the drug, our team of scientists at the Haematological Research Institute are studying how this drug is working and how to overcome potential resistance.” ![]()
Photo courtesy of NIH
A novel Bruton’s tyrosine kinase inhibitor has proven life-changing for patients with chronic lymphocytic leukemia (CLL) who received the drug as part of a phase 1 trial, according to the study’s lead author.
The inhibitor, ONO/GS-4059, produced a response in 96% of evaluable CLL patients.
Most CLL patients are still on the study after 3 years, although a handful withdrew due to adverse events (AEs) or disease progression.
“These patients were confronted with a cruel reality: they had failed multiple chemotherapy lines, and there were no other treatment options available for them,” said lead study author Harriet Walter, MBChB, of the University of Leicester in the UK.
“This drug has changed their lives. From desperate and tired, they are now leading a normal and really active life. This is hugely rewarding and encouraging.”
Dr Walter and her colleagues reported these results in Blood. The trial was funded by ONO Pharmaceuticals, the company developing ONO/GS-4059.
This study opened in January 2012, and 90 patients were enrolled at centers in the UK and France. There were 28 patients with CLL and 62 with non-Hodgkin lymphoma (NHL), including 16 with mantle cell lymphoma (MCL) and 35 with diffuse large B-cell lymphoma (DLBCL).
The study also included patients with follicular lymphoma, marginal zone lymphoma, small lymphocytic lymphoma, and Waldenstrom’s macroglobulinemia, but patient numbers were small for these groups, so the results were not discussed in detail.
There were 9 dose-escalation cohorts in this study. ONO/GS-4059 was given once-daily at doses ranging from 20 mg to 600 mg. Or the drug was given twice daily at doses of 240 mg or 300 mg.
Results
The maximum tolerated dose was not reached in the CLL cohort, but it was 480 mg once-daily in the NHL cohort. Four NHL patients had a dose-limiting toxicity.
In the CLL cohort, 2 patients went off study due to progression and 5 due to AEs.
In the NHL cohort, 49 patients discontinued treatment, 32 due to progression and 5 due to dose-limiting toxicities or AEs. The other 12 NHL patients discontinued due to patient or investigator decision, proceeding to transplant (n=1), or death due to progressive disease.
The median duration of follow-up was 560 days for CLL patients, 309 days for MCL patients, and 60 days for DLBCL patients.
The overall estimated mean progression-free survival was 874 days for CLL patients, 341 days for MCL patients, and 54 days for DLBCL patients.
CLL patients
Of all 28 CLL patients, 16 had relapsed CLL, 11 had refractory disease, and 1 had unknown status. The median number of prior therapies was 3.5 (range, 2-7).
Twenty-five patients were evaluable. Of the 3 who were not evaluable, 1 had not reached cycle 3 disease assessment at the time of data analysis, 1 progressed during cycle 1, and 1 was withdrawn due to an AE (idiopathic thrombocytopenia).
Of the 25 evaluable patients, 24 (96%) responded to ONO/GS-4059. The researchers said they observed rapid resolution of bulky lymphadenopathy within the first 3 months of treatment, but improvement in lymphadenopathy continued for up to 18 months in most patients.
The median treatment duration for these patients is 80 weeks, and 21 patients are still on treatment. Two of the evaluable patients progressed during therapy, one at cycle 3 and one at cycle 12.
MCL patients
Of the 16 MCL patients enrolled, 7 were refractory to their last course of immuno-chemotherapy. The median number of prior therapies was 3 (range, 2-7).
Eleven of 12 (92%) evaluable patients with MCL responded to ONO/GS-4059. Six patients had a partial response, and 5 had a complete response (CR) or unconfirmed CR.
Three patients progressed after an initial response. Four patients were not evaluable because they progressed.
The median treatment duration for MCL patients is 40 weeks, and 8 patients remain on study.
DLBCL patients
All 35 DLBCL patients had relapsed or refractory disease. The median number of prior treatments was 3
(range, 2-10), and 30 patients were refractory to their last line of chemotherapy.
Eleven of 31 (35%) patients with non-germinal center B-cell (non-GCB) DLBCL responded to ONO/GS-4059. Two non-GCB DLBCL patients had a confirmed CR, 1 had an unconfirmed CR, and the rest had partial responses.
The median duration of response was 54 days. And, among responders, the median treatment duration was 12 weeks.
The majority of non-GCB DLBCL patients progressed. There were no responses among the 2 patients with GCB DLBCL, and there were no responses among patients with primary mediastinal B-cell lymphoma or plasmablastic DLBCL.
Toxicity
AEs in this study were mostly grade 1/2—75% in the CLL cohort and 50% in the NHL cohort. However, treatment-related grade 3/4 AEs occurred in 14.3% of CLL patients and 16.1% of NHL patients.
Grade 3/4 events were mainly hematologic in nature and included neutropenia (10%), anemia (13.3%), and thrombocytopenia (13.3%).
There was a grade 3 episode of drug-related hemorrhage in a CLL patient, which resulted in a psoas hematoma (with concomitant CLL progression) in the presence of a normal platelet count. This patient was among those taken off the study.
“The next step is now to see how best we can improve on these outstanding results,” said study author Martin Dyer, DPhil, of the University of Leicester.
“A further study using this drug in combination with additional targeted agents is shortly to open in Leicester with the aim of achieving cure. In parallel with the clinical development of the drug, our team of scientists at the Haematological Research Institute are studying how this drug is working and how to overcome potential resistance.” ![]()
Antidote reverses effects of apixaban, rivaroxaban in healthy subjects

An investigational antidote to factor Xa inhibitors has proven succesful in reversing the effects of apixaban and rivaroxaban in a pair of phase 3 studies of healthy
volunteers.
In the ANNEXA-R and ANNEXA-A studies, researchers evaluated the safety and efficacy of the antidote, andexanet alfa, in volunteers receiving rivaroxaban and apixaban, respectively.
In both studies, andexanet alfa met all efficacy endpoints.
There were no serious or severe adverse events and no thrombotic events in either study.
“The findings of [these studies] are an advance towards resolving major bleeding complications effectively within minutes,” said Deborah Siegal, MD, of McMaster University in Hamilton, Ontario, Canada.
Dr Siegal and her colleagues reported the findings in NEJM. The studies were funded by Portola Pharmaceuticals, the company developing andexanet alfa.
The randomized, double-blind, placebo-controlled ANNEXA-R and ANNEXA-A studies were conducted to evaluate the safety and efficacy of andexanet alfa in reversing the anticoagulant effect of rivaroxaban and apixaban, respectively, in healthy volunteers ages 50 to 68.
The primary endpoint was reduction in anti-factor Xa levels. Secondary endpoints included reduction in plasma levels of unbound rivaroxaban or apixaban and restoration of the endogenous thrombin potential, a measure of thrombin generation.
ANNEXA-R efficacy
In part 1 of this study, 41 healthy volunteers received rivaroxaban at 20 mg once daily for 4 days. They were then randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg intravenous (IV) bolus (n=27) or placebo (n=14).
Within 2 to 5 minutes of bolus completion, andexanet alfa significantly reduced the anti-factor Xa activity of rivaroxaban compared with placebo—92% and 18%, respectively (P<0.001).
And andexanet alfa significantly reduced the level of unbound rivaroxaban in the plasma compared with placebo—23.4 ng/mL and 4.2 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 96% of subjects who received andexanet alfa and 7% of placebo-treated subjects (P<0.001).
In part 2 of the study, 39 healthy volunteers received rivaroxaban at 20 mg once daily for 4 days. They were then randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus followed by a continuous infusion of 8 mg/min for 120 minutes (n=26) or placebo (n=13).
Andexanet alfa significantly reduced anti-factor Xa activity compared with placebo—97% and 45%, respectively (P<0.001). And reversal persisted in andexanet alfa-treated subjects for 1 to 2 hours after the infusion was complete.
The reduction in unbound rivaroxaban was sustained with the bolus plus infusion, which significantly reduced the mean plasma concentration of unbound rivaroxaban compared with placebo—30.3 ng/mL and 12.1 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 0% of placebo-treated subjects (P<0.001).
ANNEXA-A efficacy
In part 1 of this study, 33 subjects received apixaban at 5 mg twice daily for 3.5 days. They were then randomized in a 3:1 ratio to receive either andexanet alfa administered as a 400 mg IV bolus (n=24) or placebo (n=9).
Within 2 to 5 minutes of bolus completion, andexanet alfa reduced the anti-factor Xa activity of apixaban compared with placebo—94% and 21%, respectively (P<0.001).
Andexanet alfa significantly reduced the level of unbound apixaban in the plasma compared with placebo—9.3 ng/mL and 1.9 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 11% of placebo-treated subjects (P<0.001).
In part 2, 31 healthy volunteers received apixaban at 5 mg twice daily for 4 days. They were then randomized in a 3:1 ratio to receive either andexanet alfa administered as a 400 mg IV bolus followed by a continuous infusion of 4 mg/min for 120 minutes (n=24) or placebo (n=8).
Andexanet alfa significantly reduced anti-factor Xa activity compared with placebo—92% and 33%, respectively (P<0.001). And reversal persisted in andexanet alfa-treated subjects for 1 to 2 hours after the infusion was complete.
The reduction in unbound apixaban was sustained with the bolus plus infusion, which significantly reduced the mean plasma concentration of unbound apixaban compared with placebo—6.5 ng/mL and 3.0 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 25% of placebo-treated subjects (P<0.001).
ANNEXA safety results
There were no serious or severe adverse events and no thrombotic events in either study. All adverse events related to andexanet alfa were considered mild.
Among subjects who received andexanet alfa in the ANNEXA-A study, there were 4 cases of gastrointestinal disorders—2 cases of constipation and 2 cases of dysgeusia. There were 3 cases in which subjects felt hot after administration of the drug, 4 cases of flushing, and 1 case of urticaria.
Among subjects who received andexanet alfa in the ANNEXA-R study, there were 2 cases of flushing and 1 case of urticaria.
Among subjects who received placebo in either study, there was 1 case of flushing.
One subject with a history of hives developed erythematous hives after receiving andexanet alfa. The infusion was stopped after 35 minutes, the subject received a single dose of diphenhydramine, and the hives resolved.
None of the subjects developed antibodies to factor X or factor Xa, and there were no neutralizing antibodies against andexanet alfa.
However, 1 subject who received placebo (2%) and 17 subjects who received andexanet alfa (17%) had non-neutralizing antibodies against andexanet alfa. Two of the subjects had non-neutralizing antibodies before andexanet alfa administration.
The antibodies tended to appear within 15 to 30 days of andexanet administration, and the titers were generally low (at or below 1:640). The exception was 1 subject who had a titer of 1:2560.
About andexanet alfa
Andexanet alfa is a modified human factor Xa molecule that acts as a decoy to target and sequester both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
Portola Pharmaceuticals is currently evaluating andexanet alfa in ANNEXA-4, a phase 4, single-arm, confirmatory study in patients receiving apixaban, rivaroxaban, edoxaban, or enoxaparin who present with an acute major bleed.
Data from a small number of patients from ANNEXA-4, as well as data from ANNEXA-A and ANNEXA-R, will serve as the clinical basis for the biologics license application to the US Food and Drug Administration.
A rolling submission of the application has been initiated under accelerated approval, and the submission package is expected to be complete by the end of this year. The Food and Drug Administration has already granted andexanet alfa orphan drug designation and breakthrough therapy designation. ![]()
An investigational antidote to factor Xa inhibitors has proven succesful in reversing the effects of apixaban and rivaroxaban in a pair of phase 3 studies of healthy
volunteers.
In the ANNEXA-R and ANNEXA-A studies, researchers evaluated the safety and efficacy of the antidote, andexanet alfa, in volunteers receiving rivaroxaban and apixaban, respectively.
In both studies, andexanet alfa met all efficacy endpoints.
There were no serious or severe adverse events and no thrombotic events in either study.
“The findings of [these studies] are an advance towards resolving major bleeding complications effectively within minutes,” said Deborah Siegal, MD, of McMaster University in Hamilton, Ontario, Canada.
Dr Siegal and her colleagues reported the findings in NEJM. The studies were funded by Portola Pharmaceuticals, the company developing andexanet alfa.
The randomized, double-blind, placebo-controlled ANNEXA-R and ANNEXA-A studies were conducted to evaluate the safety and efficacy of andexanet alfa in reversing the anticoagulant effect of rivaroxaban and apixaban, respectively, in healthy volunteers ages 50 to 68.
The primary endpoint was reduction in anti-factor Xa levels. Secondary endpoints included reduction in plasma levels of unbound rivaroxaban or apixaban and restoration of the endogenous thrombin potential, a measure of thrombin generation.
ANNEXA-R efficacy
In part 1 of this study, 41 healthy volunteers received rivaroxaban at 20 mg once daily for 4 days. They were then randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg intravenous (IV) bolus (n=27) or placebo (n=14).
Within 2 to 5 minutes of bolus completion, andexanet alfa significantly reduced the anti-factor Xa activity of rivaroxaban compared with placebo—92% and 18%, respectively (P<0.001).
And andexanet alfa significantly reduced the level of unbound rivaroxaban in the plasma compared with placebo—23.4 ng/mL and 4.2 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 96% of subjects who received andexanet alfa and 7% of placebo-treated subjects (P<0.001).
In part 2 of the study, 39 healthy volunteers received rivaroxaban at 20 mg once daily for 4 days. They were then randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus followed by a continuous infusion of 8 mg/min for 120 minutes (n=26) or placebo (n=13).
Andexanet alfa significantly reduced anti-factor Xa activity compared with placebo—97% and 45%, respectively (P<0.001). And reversal persisted in andexanet alfa-treated subjects for 1 to 2 hours after the infusion was complete.
The reduction in unbound rivaroxaban was sustained with the bolus plus infusion, which significantly reduced the mean plasma concentration of unbound rivaroxaban compared with placebo—30.3 ng/mL and 12.1 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 0% of placebo-treated subjects (P<0.001).
ANNEXA-A efficacy
In part 1 of this study, 33 subjects received apixaban at 5 mg twice daily for 3.5 days. They were then randomized in a 3:1 ratio to receive either andexanet alfa administered as a 400 mg IV bolus (n=24) or placebo (n=9).
Within 2 to 5 minutes of bolus completion, andexanet alfa reduced the anti-factor Xa activity of apixaban compared with placebo—94% and 21%, respectively (P<0.001).
Andexanet alfa significantly reduced the level of unbound apixaban in the plasma compared with placebo—9.3 ng/mL and 1.9 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 11% of placebo-treated subjects (P<0.001).
In part 2, 31 healthy volunteers received apixaban at 5 mg twice daily for 4 days. They were then randomized in a 3:1 ratio to receive either andexanet alfa administered as a 400 mg IV bolus followed by a continuous infusion of 4 mg/min for 120 minutes (n=24) or placebo (n=8).
Andexanet alfa significantly reduced anti-factor Xa activity compared with placebo—92% and 33%, respectively (P<0.001). And reversal persisted in andexanet alfa-treated subjects for 1 to 2 hours after the infusion was complete.
The reduction in unbound apixaban was sustained with the bolus plus infusion, which significantly reduced the mean plasma concentration of unbound apixaban compared with placebo—6.5 ng/mL and 3.0 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 25% of placebo-treated subjects (P<0.001).
ANNEXA safety results
There were no serious or severe adverse events and no thrombotic events in either study. All adverse events related to andexanet alfa were considered mild.
Among subjects who received andexanet alfa in the ANNEXA-A study, there were 4 cases of gastrointestinal disorders—2 cases of constipation and 2 cases of dysgeusia. There were 3 cases in which subjects felt hot after administration of the drug, 4 cases of flushing, and 1 case of urticaria.
Among subjects who received andexanet alfa in the ANNEXA-R study, there were 2 cases of flushing and 1 case of urticaria.
Among subjects who received placebo in either study, there was 1 case of flushing.
One subject with a history of hives developed erythematous hives after receiving andexanet alfa. The infusion was stopped after 35 minutes, the subject received a single dose of diphenhydramine, and the hives resolved.
None of the subjects developed antibodies to factor X or factor Xa, and there were no neutralizing antibodies against andexanet alfa.
However, 1 subject who received placebo (2%) and 17 subjects who received andexanet alfa (17%) had non-neutralizing antibodies against andexanet alfa. Two of the subjects had non-neutralizing antibodies before andexanet alfa administration.
The antibodies tended to appear within 15 to 30 days of andexanet administration, and the titers were generally low (at or below 1:640). The exception was 1 subject who had a titer of 1:2560.
About andexanet alfa
Andexanet alfa is a modified human factor Xa molecule that acts as a decoy to target and sequester both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
Portola Pharmaceuticals is currently evaluating andexanet alfa in ANNEXA-4, a phase 4, single-arm, confirmatory study in patients receiving apixaban, rivaroxaban, edoxaban, or enoxaparin who present with an acute major bleed.
Data from a small number of patients from ANNEXA-4, as well as data from ANNEXA-A and ANNEXA-R, will serve as the clinical basis for the biologics license application to the US Food and Drug Administration.
A rolling submission of the application has been initiated under accelerated approval, and the submission package is expected to be complete by the end of this year. The Food and Drug Administration has already granted andexanet alfa orphan drug designation and breakthrough therapy designation. ![]()
An investigational antidote to factor Xa inhibitors has proven succesful in reversing the effects of apixaban and rivaroxaban in a pair of phase 3 studies of healthy
volunteers.
In the ANNEXA-R and ANNEXA-A studies, researchers evaluated the safety and efficacy of the antidote, andexanet alfa, in volunteers receiving rivaroxaban and apixaban, respectively.
In both studies, andexanet alfa met all efficacy endpoints.
There were no serious or severe adverse events and no thrombotic events in either study.
“The findings of [these studies] are an advance towards resolving major bleeding complications effectively within minutes,” said Deborah Siegal, MD, of McMaster University in Hamilton, Ontario, Canada.
Dr Siegal and her colleagues reported the findings in NEJM. The studies were funded by Portola Pharmaceuticals, the company developing andexanet alfa.
The randomized, double-blind, placebo-controlled ANNEXA-R and ANNEXA-A studies were conducted to evaluate the safety and efficacy of andexanet alfa in reversing the anticoagulant effect of rivaroxaban and apixaban, respectively, in healthy volunteers ages 50 to 68.
The primary endpoint was reduction in anti-factor Xa levels. Secondary endpoints included reduction in plasma levels of unbound rivaroxaban or apixaban and restoration of the endogenous thrombin potential, a measure of thrombin generation.
ANNEXA-R efficacy
In part 1 of this study, 41 healthy volunteers received rivaroxaban at 20 mg once daily for 4 days. They were then randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg intravenous (IV) bolus (n=27) or placebo (n=14).
Within 2 to 5 minutes of bolus completion, andexanet alfa significantly reduced the anti-factor Xa activity of rivaroxaban compared with placebo—92% and 18%, respectively (P<0.001).
And andexanet alfa significantly reduced the level of unbound rivaroxaban in the plasma compared with placebo—23.4 ng/mL and 4.2 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 96% of subjects who received andexanet alfa and 7% of placebo-treated subjects (P<0.001).
In part 2 of the study, 39 healthy volunteers received rivaroxaban at 20 mg once daily for 4 days. They were then randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus followed by a continuous infusion of 8 mg/min for 120 minutes (n=26) or placebo (n=13).
Andexanet alfa significantly reduced anti-factor Xa activity compared with placebo—97% and 45%, respectively (P<0.001). And reversal persisted in andexanet alfa-treated subjects for 1 to 2 hours after the infusion was complete.
The reduction in unbound rivaroxaban was sustained with the bolus plus infusion, which significantly reduced the mean plasma concentration of unbound rivaroxaban compared with placebo—30.3 ng/mL and 12.1 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 0% of placebo-treated subjects (P<0.001).
ANNEXA-A efficacy
In part 1 of this study, 33 subjects received apixaban at 5 mg twice daily for 3.5 days. They were then randomized in a 3:1 ratio to receive either andexanet alfa administered as a 400 mg IV bolus (n=24) or placebo (n=9).
Within 2 to 5 minutes of bolus completion, andexanet alfa reduced the anti-factor Xa activity of apixaban compared with placebo—94% and 21%, respectively (P<0.001).
Andexanet alfa significantly reduced the level of unbound apixaban in the plasma compared with placebo—9.3 ng/mL and 1.9 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 11% of placebo-treated subjects (P<0.001).
In part 2, 31 healthy volunteers received apixaban at 5 mg twice daily for 4 days. They were then randomized in a 3:1 ratio to receive either andexanet alfa administered as a 400 mg IV bolus followed by a continuous infusion of 4 mg/min for 120 minutes (n=24) or placebo (n=8).
Andexanet alfa significantly reduced anti-factor Xa activity compared with placebo—92% and 33%, respectively (P<0.001). And reversal persisted in andexanet alfa-treated subjects for 1 to 2 hours after the infusion was complete.
The reduction in unbound apixaban was sustained with the bolus plus infusion, which significantly reduced the mean plasma concentration of unbound apixaban compared with placebo—6.5 ng/mL and 3.0 ng/mL, respectively (P<0.001).
Thrombin generation was fully restored in 100% of subjects who received andexanet alfa and 25% of placebo-treated subjects (P<0.001).
ANNEXA safety results
There were no serious or severe adverse events and no thrombotic events in either study. All adverse events related to andexanet alfa were considered mild.
Among subjects who received andexanet alfa in the ANNEXA-A study, there were 4 cases of gastrointestinal disorders—2 cases of constipation and 2 cases of dysgeusia. There were 3 cases in which subjects felt hot after administration of the drug, 4 cases of flushing, and 1 case of urticaria.
Among subjects who received andexanet alfa in the ANNEXA-R study, there were 2 cases of flushing and 1 case of urticaria.
Among subjects who received placebo in either study, there was 1 case of flushing.
One subject with a history of hives developed erythematous hives after receiving andexanet alfa. The infusion was stopped after 35 minutes, the subject received a single dose of diphenhydramine, and the hives resolved.
None of the subjects developed antibodies to factor X or factor Xa, and there were no neutralizing antibodies against andexanet alfa.
However, 1 subject who received placebo (2%) and 17 subjects who received andexanet alfa (17%) had non-neutralizing antibodies against andexanet alfa. Two of the subjects had non-neutralizing antibodies before andexanet alfa administration.
The antibodies tended to appear within 15 to 30 days of andexanet administration, and the titers were generally low (at or below 1:640). The exception was 1 subject who had a titer of 1:2560.
About andexanet alfa
Andexanet alfa is a modified human factor Xa molecule that acts as a decoy to target and sequester both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
Portola Pharmaceuticals is currently evaluating andexanet alfa in ANNEXA-4, a phase 4, single-arm, confirmatory study in patients receiving apixaban, rivaroxaban, edoxaban, or enoxaparin who present with an acute major bleed.
Data from a small number of patients from ANNEXA-4, as well as data from ANNEXA-A and ANNEXA-R, will serve as the clinical basis for the biologics license application to the US Food and Drug Administration.
A rolling submission of the application has been initiated under accelerated approval, and the submission package is expected to be complete by the end of this year. The Food and Drug Administration has already granted andexanet alfa orphan drug designation and breakthrough therapy designation.
Analysis suggests dabigatran is safer than warfarin

ORLANDO, FL—Interim results from a long-term, “real world” study suggest dabigatran etexilate mesylate (Pradaxa) may be safer than warfarin in routine
clinical practice.
The data, which come from a pooled analysis of 2 large US commercial health insurance databases, showed that patients with non-valvular atrial fibrillation (NVAF) who received dabigatran had fewer strokes and major bleeding events than NVAF patients who received warfarin.
The results were presented at the American Heart Association (AHA) Scientific Sessions 2015 (abstract M 2129*).
The research was supported by Boehringer Ingelheim, the company developing dabigatran, and employees from the company were involved in the study.
“Beyond clinical trials, there is a wealth of available health insurance data that provides an excellent opportunity to grow our knowledge of oral anticoagulant use and outcomes for patients,” said study investigator John Seeger, PharmD, DrPH, of Brigham and Women’s Hospital in Boston, Massachusetts.
“These real-world data further define the safety and effectiveness of dabigatran for patients and its use in routine care, and are consistent with the results of the pivotal RE-LY clinical trial.”
Dr Seeger and his colleagues analyzed data collected over 32 months and encompassing 44,672 NVAF patients—22,336 propensity-score-matched patients receiving dabigatran or warfarin for the first time.
The data were collected from 2 insurance databases—Truven MarketScan (18,276 patients per group) and Optum Clinformatics (4060 patients per group).
Patients were followed until they switched or discontinued anticoagulant treatment, experienced an outcome event, or discontinued enrollment. The average follow-up period was 5 months for dabigatran-treated patients and 4 months for warfarin-treated patients.
The primary outcomes of the study were stroke and major bleeding rates.
There were 65 strokes among dabigatran-treated patients (0.73 incidence rate per 100 patient-years) and 78 strokes among warfarin-treated patients (1.08 incidence rate per 100 patient-years).
The investigators said this represents a 28% reduction in stroke risk for dabigatran compared to warfarin. (The hazard ratio was 0.72.)
There were 395 major bleeding events among dabigatran-treated patients (4.47 incidence rate per 100 patient-years) and 459 events for warfarin-treated patients (6.42 incidence rate per 100 patient-years).
This represents a 26% reduction in the risk of major bleeding events with dabigatran compared to warfarin. (The hazard ratio was 0.74.)
The investigators said effectiveness assessments beyond the first 6 months of therapy were limited by the short average follow-up time. But future analyses from this long-term study program will yield more stable estimates.
This study is part of an ongoing research program to evaluate prescribing patterns and real-world safety and effectiveness of novel oral anticoagulants, including dabigatran, for reducing stroke risk.
*Data in the abstract differ from data presented at the meeting.
ORLANDO, FL—Interim results from a long-term, “real world” study suggest dabigatran etexilate mesylate (Pradaxa) may be safer than warfarin in routine
clinical practice.
The data, which come from a pooled analysis of 2 large US commercial health insurance databases, showed that patients with non-valvular atrial fibrillation (NVAF) who received dabigatran had fewer strokes and major bleeding events than NVAF patients who received warfarin.
The results were presented at the American Heart Association (AHA) Scientific Sessions 2015 (abstract M 2129*).
The research was supported by Boehringer Ingelheim, the company developing dabigatran, and employees from the company were involved in the study.
“Beyond clinical trials, there is a wealth of available health insurance data that provides an excellent opportunity to grow our knowledge of oral anticoagulant use and outcomes for patients,” said study investigator John Seeger, PharmD, DrPH, of Brigham and Women’s Hospital in Boston, Massachusetts.
“These real-world data further define the safety and effectiveness of dabigatran for patients and its use in routine care, and are consistent with the results of the pivotal RE-LY clinical trial.”
Dr Seeger and his colleagues analyzed data collected over 32 months and encompassing 44,672 NVAF patients—22,336 propensity-score-matched patients receiving dabigatran or warfarin for the first time.
The data were collected from 2 insurance databases—Truven MarketScan (18,276 patients per group) and Optum Clinformatics (4060 patients per group).
Patients were followed until they switched or discontinued anticoagulant treatment, experienced an outcome event, or discontinued enrollment. The average follow-up period was 5 months for dabigatran-treated patients and 4 months for warfarin-treated patients.
The primary outcomes of the study were stroke and major bleeding rates.
There were 65 strokes among dabigatran-treated patients (0.73 incidence rate per 100 patient-years) and 78 strokes among warfarin-treated patients (1.08 incidence rate per 100 patient-years).
The investigators said this represents a 28% reduction in stroke risk for dabigatran compared to warfarin. (The hazard ratio was 0.72.)
There were 395 major bleeding events among dabigatran-treated patients (4.47 incidence rate per 100 patient-years) and 459 events for warfarin-treated patients (6.42 incidence rate per 100 patient-years).
This represents a 26% reduction in the risk of major bleeding events with dabigatran compared to warfarin. (The hazard ratio was 0.74.)
The investigators said effectiveness assessments beyond the first 6 months of therapy were limited by the short average follow-up time. But future analyses from this long-term study program will yield more stable estimates.
This study is part of an ongoing research program to evaluate prescribing patterns and real-world safety and effectiveness of novel oral anticoagulants, including dabigatran, for reducing stroke risk.
*Data in the abstract differ from data presented at the meeting.
ORLANDO, FL—Interim results from a long-term, “real world” study suggest dabigatran etexilate mesylate (Pradaxa) may be safer than warfarin in routine
clinical practice.
The data, which come from a pooled analysis of 2 large US commercial health insurance databases, showed that patients with non-valvular atrial fibrillation (NVAF) who received dabigatran had fewer strokes and major bleeding events than NVAF patients who received warfarin.
The results were presented at the American Heart Association (AHA) Scientific Sessions 2015 (abstract M 2129*).
The research was supported by Boehringer Ingelheim, the company developing dabigatran, and employees from the company were involved in the study.
“Beyond clinical trials, there is a wealth of available health insurance data that provides an excellent opportunity to grow our knowledge of oral anticoagulant use and outcomes for patients,” said study investigator John Seeger, PharmD, DrPH, of Brigham and Women’s Hospital in Boston, Massachusetts.
“These real-world data further define the safety and effectiveness of dabigatran for patients and its use in routine care, and are consistent with the results of the pivotal RE-LY clinical trial.”
Dr Seeger and his colleagues analyzed data collected over 32 months and encompassing 44,672 NVAF patients—22,336 propensity-score-matched patients receiving dabigatran or warfarin for the first time.
The data were collected from 2 insurance databases—Truven MarketScan (18,276 patients per group) and Optum Clinformatics (4060 patients per group).
Patients were followed until they switched or discontinued anticoagulant treatment, experienced an outcome event, or discontinued enrollment. The average follow-up period was 5 months for dabigatran-treated patients and 4 months for warfarin-treated patients.
The primary outcomes of the study were stroke and major bleeding rates.
There were 65 strokes among dabigatran-treated patients (0.73 incidence rate per 100 patient-years) and 78 strokes among warfarin-treated patients (1.08 incidence rate per 100 patient-years).
The investigators said this represents a 28% reduction in stroke risk for dabigatran compared to warfarin. (The hazard ratio was 0.72.)
There were 395 major bleeding events among dabigatran-treated patients (4.47 incidence rate per 100 patient-years) and 459 events for warfarin-treated patients (6.42 incidence rate per 100 patient-years).
This represents a 26% reduction in the risk of major bleeding events with dabigatran compared to warfarin. (The hazard ratio was 0.74.)
The investigators said effectiveness assessments beyond the first 6 months of therapy were limited by the short average follow-up time. But future analyses from this long-term study program will yield more stable estimates.
This study is part of an ongoing research program to evaluate prescribing patterns and real-world safety and effectiveness of novel oral anticoagulants, including dabigatran, for reducing stroke risk.
*Data in the abstract differ from data presented at the meeting.
CCSs have increased risk of autoimmune diseases

Photo by Bill Branson
Childhood cancer survivors (CCSs) have an increased risk of developing autoimmune diseases, according to research published in the Annals of the Rheumatic Diseases.
CCSs had a significantly increased risk for 11 of 33 autoimmune diseases studied, and the highest risk was observed for autoimmune hemolytic anemia.
Survivors of leukemia and Hodgkin lymphoma were among those CCSs at the greatest risk of developing
an autoimmune disease.
Anna Sällfors Holmqvist, MD, of Lund University in Sweden, and her colleagues conducted this research.
They used national cancer registry data from Denmark, Iceland, and Sweden, spanning the period from the 1940s to 2008, to identify subjects who had cancer as a child.
The researchers identified 20,361 adults who had cancer before the age of 20 and survived for at least a year. These subjects were matched (for age, gender, and country of birth) to 125,794 individuals who had not had cancer as children.
The health of all participants was tracked for an average of 15 to 19 years. The researchers used hospital records to determine the difference between the expected and excess number of autoimmune diseases, expressed as a standardized hospitalization rate ratio (SHRR).
In all, 724 (3.6%) CCSs had at least 1 episode of hospital treatment for any autoimmune condition, but only 516 would have been expected.
So CCSs had an SHRR for autoimmune diseases of 1.4. This corresponds to an absolute excess risk of 67 per 100,000 person-years.
SHRRs were significantly higher for 11 autoimmune diseases, including autoimmune hemolytic anemia (16.3), Addison’s disease (13.9), polyarteritis nodosa (5.8), chronic rheumatic heart disease (4.5), localized scleroderma (3.6), idiopathic thrombocytopenia (3.4), Hashimoto’s thyroiditis (3.1), pernicious anemia (2.7), sarcoidosis (2.2), Sjögren’s syndrome (2.0), and insulin-dependent diabetes mellitus (1.6).
SHRRs for any autoimmune disease were significantly increased for survivors of leukemia (1.6), Hodgkin lymphoma (1.6), renal tumors (1.6), and central nervous system neoplasms (1.4).
The excess risk for all autoimmune diseases combined peaked in the first 5 years after a cancer diagnosis. However, the risk persisted for up to 30 years later for most conditions and up to 50 years later for some conditions.
The researchers said the peak observed in the first 5 years may be a consequence of closer medical monitoring during this time period.
They added that a possible explanation for these findings is that persistent immune abnormalities after chemotherapy predispose CCSs to develop autoantibodies, which are central to the pathogenesis of many autoimmune diseases.
The team said the cancer itself, immunosuppressive treatment, and the increased number and types of infections during cancer treatment could alter the immune system as a whole and result in immunologically different antigens, leading to the production of autoantibodies.
Photo by Bill Branson
Childhood cancer survivors (CCSs) have an increased risk of developing autoimmune diseases, according to research published in the Annals of the Rheumatic Diseases.
CCSs had a significantly increased risk for 11 of 33 autoimmune diseases studied, and the highest risk was observed for autoimmune hemolytic anemia.
Survivors of leukemia and Hodgkin lymphoma were among those CCSs at the greatest risk of developing
an autoimmune disease.
Anna Sällfors Holmqvist, MD, of Lund University in Sweden, and her colleagues conducted this research.
They used national cancer registry data from Denmark, Iceland, and Sweden, spanning the period from the 1940s to 2008, to identify subjects who had cancer as a child.
The researchers identified 20,361 adults who had cancer before the age of 20 and survived for at least a year. These subjects were matched (for age, gender, and country of birth) to 125,794 individuals who had not had cancer as children.
The health of all participants was tracked for an average of 15 to 19 years. The researchers used hospital records to determine the difference between the expected and excess number of autoimmune diseases, expressed as a standardized hospitalization rate ratio (SHRR).
In all, 724 (3.6%) CCSs had at least 1 episode of hospital treatment for any autoimmune condition, but only 516 would have been expected.
So CCSs had an SHRR for autoimmune diseases of 1.4. This corresponds to an absolute excess risk of 67 per 100,000 person-years.
SHRRs were significantly higher for 11 autoimmune diseases, including autoimmune hemolytic anemia (16.3), Addison’s disease (13.9), polyarteritis nodosa (5.8), chronic rheumatic heart disease (4.5), localized scleroderma (3.6), idiopathic thrombocytopenia (3.4), Hashimoto’s thyroiditis (3.1), pernicious anemia (2.7), sarcoidosis (2.2), Sjögren’s syndrome (2.0), and insulin-dependent diabetes mellitus (1.6).
SHRRs for any autoimmune disease were significantly increased for survivors of leukemia (1.6), Hodgkin lymphoma (1.6), renal tumors (1.6), and central nervous system neoplasms (1.4).
The excess risk for all autoimmune diseases combined peaked in the first 5 years after a cancer diagnosis. However, the risk persisted for up to 30 years later for most conditions and up to 50 years later for some conditions.
The researchers said the peak observed in the first 5 years may be a consequence of closer medical monitoring during this time period.
They added that a possible explanation for these findings is that persistent immune abnormalities after chemotherapy predispose CCSs to develop autoantibodies, which are central to the pathogenesis of many autoimmune diseases.
The team said the cancer itself, immunosuppressive treatment, and the increased number and types of infections during cancer treatment could alter the immune system as a whole and result in immunologically different antigens, leading to the production of autoantibodies.
Photo by Bill Branson
Childhood cancer survivors (CCSs) have an increased risk of developing autoimmune diseases, according to research published in the Annals of the Rheumatic Diseases.
CCSs had a significantly increased risk for 11 of 33 autoimmune diseases studied, and the highest risk was observed for autoimmune hemolytic anemia.
Survivors of leukemia and Hodgkin lymphoma were among those CCSs at the greatest risk of developing
an autoimmune disease.
Anna Sällfors Holmqvist, MD, of Lund University in Sweden, and her colleagues conducted this research.
They used national cancer registry data from Denmark, Iceland, and Sweden, spanning the period from the 1940s to 2008, to identify subjects who had cancer as a child.
The researchers identified 20,361 adults who had cancer before the age of 20 and survived for at least a year. These subjects were matched (for age, gender, and country of birth) to 125,794 individuals who had not had cancer as children.
The health of all participants was tracked for an average of 15 to 19 years. The researchers used hospital records to determine the difference between the expected and excess number of autoimmune diseases, expressed as a standardized hospitalization rate ratio (SHRR).
In all, 724 (3.6%) CCSs had at least 1 episode of hospital treatment for any autoimmune condition, but only 516 would have been expected.
So CCSs had an SHRR for autoimmune diseases of 1.4. This corresponds to an absolute excess risk of 67 per 100,000 person-years.
SHRRs were significantly higher for 11 autoimmune diseases, including autoimmune hemolytic anemia (16.3), Addison’s disease (13.9), polyarteritis nodosa (5.8), chronic rheumatic heart disease (4.5), localized scleroderma (3.6), idiopathic thrombocytopenia (3.4), Hashimoto’s thyroiditis (3.1), pernicious anemia (2.7), sarcoidosis (2.2), Sjögren’s syndrome (2.0), and insulin-dependent diabetes mellitus (1.6).
SHRRs for any autoimmune disease were significantly increased for survivors of leukemia (1.6), Hodgkin lymphoma (1.6), renal tumors (1.6), and central nervous system neoplasms (1.4).
The excess risk for all autoimmune diseases combined peaked in the first 5 years after a cancer diagnosis. However, the risk persisted for up to 30 years later for most conditions and up to 50 years later for some conditions.
The researchers said the peak observed in the first 5 years may be a consequence of closer medical monitoring during this time period.
They added that a possible explanation for these findings is that persistent immune abnormalities after chemotherapy predispose CCSs to develop autoantibodies, which are central to the pathogenesis of many autoimmune diseases.
The team said the cancer itself, immunosuppressive treatment, and the increased number and types of infections during cancer treatment could alter the immune system as a whole and result in immunologically different antigens, leading to the production of autoantibodies.
Relapses ‘critical’ for sustained malaria transmission
Plasmodium vivax
Image by Mae Melvin
New research indicates that most childhood malaria infections in Papua New Guinea (PNG) are the result of relapsed, not new, infections.
The data suggest that, among children ages 5 to 10 living in a malaria-hyperendemic region of PNG, relapses cause about 4 of every 5 Plasmodium vivax infections and 3 of every 5 Plasmodium ovale infections.
Investigators therefore concluded that relapses are important for sustaining malaria transmission in PNG.
The team reported their findings in PLOS Medicine.
“Our research has shown that one of the biggest problems in realizing malaria eradication is relapsing P vivax infections, which are critical for sustained transmission in the region,” said study author Leanne Robinson, PhD, of the Walter and Eliza Hall Institute of Medical Research in Parkville, Victoria, Australia.
“P vivax parasites are able to hide in the liver for long periods of time before ‘reawakening’ to cause disease and continue the transmission cycle. Mass drug administration that includes a drug that kills parasites in the liver is likely to be a highly effective strategy for eliminating malaria in PNG.”
To investigate this possibility, Dr Robinson and her colleagues analyzed 524 children, ages 5 to 10, living in a region of PNG where Plasmodium falciparum and P vivax are hyperendemic.
Roughly half the children (n=261) received an antimalarial treatment regimen that targets both blood-stage and liver-stage parasites (3 days of chloroquine, 3 days of artemether-lumefantrine, and 20 days of primaquine).
The other half (n=263) received antimalarial treatment targeting only blood-stage parasites (3 days of chloroquine, 3 days of artemether-lumefantrine, and 20 days of placebo).
The subjects were followed for 8 months. Compared to children in the placebo arm, those in the primaquine arm had a reduced risk of having at least 1 P vivax or P ovale infection during the follow-up period. The hazard ratio was 0.18 for P vivax (P<0.001) and 0.31 for P ovale (P=0.011).
“Children treated with drugs that targeted the liver and blood stages of infection had 80% fewer malaria infections than those treated with drugs that only targeted the blood stage of infection,” Dr Robinson said.
Children in the primaquine arm also had a reduced risk of having at least 1 clinical P vivax episode. The hazard ratio was 0.25 (P=0.002).
In addition, primaquine reduced the molecular force of P vivax blood-stage infection in the first 3 months of follow-up. The incidence rate ratio was 0.21 (P<0.001).
And children who received primaquine were less likely to carry P vivax gametocytes than children who received placebo, with an incidence rate ratio of 0.27 (P<0.001).
To build upon these findings, the investigators fed the trial data into a mathematical transmission model.
The model predicted that a mass drug administration program using blood-stage treatment alone would have only a transient effect on P vivax transmission levels. But a mass drug administration program that includes blood- and liver-stage treatment would be an effective strategy for P vivax elimination.
“We need a better way of identifying children who are chronically infected with malaria so that they can be treated,” said study author Ivo Mueller, PhD, of the Walter and Eliza Hall Institute.
“It is the only way to stop the malaria transmission cycle in PNG and is likely to be the case for eliminating malaria in other parts of the Asia-Pacific and Americas.”
Dr Mueller and an international team of collaborators are currently developing a test that identifies people with dormant malaria parasites in their liver.
Plasmodium vivax
Image by Mae Melvin
New research indicates that most childhood malaria infections in Papua New Guinea (PNG) are the result of relapsed, not new, infections.
The data suggest that, among children ages 5 to 10 living in a malaria-hyperendemic region of PNG, relapses cause about 4 of every 5 Plasmodium vivax infections and 3 of every 5 Plasmodium ovale infections.
Investigators therefore concluded that relapses are important for sustaining malaria transmission in PNG.
The team reported their findings in PLOS Medicine.
“Our research has shown that one of the biggest problems in realizing malaria eradication is relapsing P vivax infections, which are critical for sustained transmission in the region,” said study author Leanne Robinson, PhD, of the Walter and Eliza Hall Institute of Medical Research in Parkville, Victoria, Australia.
“P vivax parasites are able to hide in the liver for long periods of time before ‘reawakening’ to cause disease and continue the transmission cycle. Mass drug administration that includes a drug that kills parasites in the liver is likely to be a highly effective strategy for eliminating malaria in PNG.”
To investigate this possibility, Dr Robinson and her colleagues analyzed 524 children, ages 5 to 10, living in a region of PNG where Plasmodium falciparum and P vivax are hyperendemic.
Roughly half the children (n=261) received an antimalarial treatment regimen that targets both blood-stage and liver-stage parasites (3 days of chloroquine, 3 days of artemether-lumefantrine, and 20 days of primaquine).
The other half (n=263) received antimalarial treatment targeting only blood-stage parasites (3 days of chloroquine, 3 days of artemether-lumefantrine, and 20 days of placebo).
The subjects were followed for 8 months. Compared to children in the placebo arm, those in the primaquine arm had a reduced risk of having at least 1 P vivax or P ovale infection during the follow-up period. The hazard ratio was 0.18 for P vivax (P<0.001) and 0.31 for P ovale (P=0.011).
“Children treated with drugs that targeted the liver and blood stages of infection had 80% fewer malaria infections than those treated with drugs that only targeted the blood stage of infection,” Dr Robinson said.
Children in the primaquine arm also had a reduced risk of having at least 1 clinical P vivax episode. The hazard ratio was 0.25 (P=0.002).
In addition, primaquine reduced the molecular force of P vivax blood-stage infection in the first 3 months of follow-up. The incidence rate ratio was 0.21 (P<0.001).
And children who received primaquine were less likely to carry P vivax gametocytes than children who received placebo, with an incidence rate ratio of 0.27 (P<0.001).
To build upon these findings, the investigators fed the trial data into a mathematical transmission model.
The model predicted that a mass drug administration program using blood-stage treatment alone would have only a transient effect on P vivax transmission levels. But a mass drug administration program that includes blood- and liver-stage treatment would be an effective strategy for P vivax elimination.
“We need a better way of identifying children who are chronically infected with malaria so that they can be treated,” said study author Ivo Mueller, PhD, of the Walter and Eliza Hall Institute.
“It is the only way to stop the malaria transmission cycle in PNG and is likely to be the case for eliminating malaria in other parts of the Asia-Pacific and Americas.”
Dr Mueller and an international team of collaborators are currently developing a test that identifies people with dormant malaria parasites in their liver.
Plasmodium vivax
Image by Mae Melvin
New research indicates that most childhood malaria infections in Papua New Guinea (PNG) are the result of relapsed, not new, infections.
The data suggest that, among children ages 5 to 10 living in a malaria-hyperendemic region of PNG, relapses cause about 4 of every 5 Plasmodium vivax infections and 3 of every 5 Plasmodium ovale infections.
Investigators therefore concluded that relapses are important for sustaining malaria transmission in PNG.
The team reported their findings in PLOS Medicine.
“Our research has shown that one of the biggest problems in realizing malaria eradication is relapsing P vivax infections, which are critical for sustained transmission in the region,” said study author Leanne Robinson, PhD, of the Walter and Eliza Hall Institute of Medical Research in Parkville, Victoria, Australia.
“P vivax parasites are able to hide in the liver for long periods of time before ‘reawakening’ to cause disease and continue the transmission cycle. Mass drug administration that includes a drug that kills parasites in the liver is likely to be a highly effective strategy for eliminating malaria in PNG.”
To investigate this possibility, Dr Robinson and her colleagues analyzed 524 children, ages 5 to 10, living in a region of PNG where Plasmodium falciparum and P vivax are hyperendemic.
Roughly half the children (n=261) received an antimalarial treatment regimen that targets both blood-stage and liver-stage parasites (3 days of chloroquine, 3 days of artemether-lumefantrine, and 20 days of primaquine).
The other half (n=263) received antimalarial treatment targeting only blood-stage parasites (3 days of chloroquine, 3 days of artemether-lumefantrine, and 20 days of placebo).
The subjects were followed for 8 months. Compared to children in the placebo arm, those in the primaquine arm had a reduced risk of having at least 1 P vivax or P ovale infection during the follow-up period. The hazard ratio was 0.18 for P vivax (P<0.001) and 0.31 for P ovale (P=0.011).
“Children treated with drugs that targeted the liver and blood stages of infection had 80% fewer malaria infections than those treated with drugs that only targeted the blood stage of infection,” Dr Robinson said.
Children in the primaquine arm also had a reduced risk of having at least 1 clinical P vivax episode. The hazard ratio was 0.25 (P=0.002).
In addition, primaquine reduced the molecular force of P vivax blood-stage infection in the first 3 months of follow-up. The incidence rate ratio was 0.21 (P<0.001).
And children who received primaquine were less likely to carry P vivax gametocytes than children who received placebo, with an incidence rate ratio of 0.27 (P<0.001).
To build upon these findings, the investigators fed the trial data into a mathematical transmission model.
The model predicted that a mass drug administration program using blood-stage treatment alone would have only a transient effect on P vivax transmission levels. But a mass drug administration program that includes blood- and liver-stage treatment would be an effective strategy for P vivax elimination.
“We need a better way of identifying children who are chronically infected with malaria so that they can be treated,” said study author Ivo Mueller, PhD, of the Walter and Eliza Hall Institute.
“It is the only way to stop the malaria transmission cycle in PNG and is likely to be the case for eliminating malaria in other parts of the Asia-Pacific and Americas.”
Dr Mueller and an international team of collaborators are currently developing a test that identifies people with dormant malaria parasites in their liver.