User login
Berry-derived compound can fight AML

A compound derived from the berries of the Bloodhorn tree has demonstrated activity against acute myeloid leukemia (AML), according to preclinical research published in Investigational New Drugs.
The compound, 7-formyl-10-methylisoellipticine, induced apoptosis in AML cells in a dose- and time-dependent manner.
It also significantly slowed tumor growth and reduced tumor mass in a mouse model of AML.
7-formyl-10-methylisoellipticine is derived from an ellipticine, which has been isolated from the berries of the Ochrosia Elliptica tree. The tree, also known as the Bloodhorn tree due to the shape and color of the berries, grows on the northeast coast of Australia and in the rainforests of Brazil.
“[We have] taken the natural product and restyled it with unique features to improve the potency and solubility,” explained Florence McCarthy, PhD, of University College Cork in Ireland.
“What is truly exceptional is that these features are not common in drugs, and so we aim to exploit this fully. There is also significant potential to apply this approach to other drugs in a similar fashion.”
For this study, Dr McCarthy and his colleagues first tested 7-formyl-10-methylisoellipticine in the AML cell line MV4-11. They tested a range of concentrations in an attempt to identify the minimum concentration that would cause significant cytotoxicity. It turned out to be 5 μM.
Over a period of 24 hours, 5 μM of 7-formyl-10-methylisoellipticine killed up to 40% of MV4-11 cells. And over 96 hours, 5 μM of 7-formyl-10-methylisoellipticine killed more than 90% of cells.
Further investigation revealed that 5 μM of 7-formyl-10-methylisoellipticine increases the sub-G1 phase of the MV4-11 cell cycle. And the compound functions, at least in part, by generating mitochondrial-derived reactive oxygen species.
The researchers then found that 7-formyl-10-methylisoellipticine is not toxic to BALB/c mice. The team injected the mice with 7-formyl-10-methylisoellipticine at a range of doses—5 mg/kg, 10 mg/kg, 25 mg/kg, and 50 mg/kg.
Regardless of the dose, the compound did not cause a change in body weight, significantly increase levels of alanine aminotransferase or aspartate aminotransferase relative to negative control, or significantly change cell morphology or tissue structure in specified major organs.
Finally, the researchers found that 7-formyl-10-methylisoellipticine has antitumor activity in an AML xenograft mouse model. Based on the toxicity experiments, the team used a dose of 25 mg/kg in these mice.
At this dose, 7-formyl-10-methylisoellipticine significantly slowed tumor growth and reduced tumor mass. Tumor growth was 4 times slower in mice treated with 7-formyl-10-methylisoellipticine than in control mice. And tumor mass was up to 7 times greater in controls than it was in treated mice.
Based on these results, the researchers said they plan to continue investigating the mechanism of action of ellipticines, which “have a clear potential clinical application.” ![]()
A compound derived from the berries of the Bloodhorn tree has demonstrated activity against acute myeloid leukemia (AML), according to preclinical research published in Investigational New Drugs.
The compound, 7-formyl-10-methylisoellipticine, induced apoptosis in AML cells in a dose- and time-dependent manner.
It also significantly slowed tumor growth and reduced tumor mass in a mouse model of AML.
7-formyl-10-methylisoellipticine is derived from an ellipticine, which has been isolated from the berries of the Ochrosia Elliptica tree. The tree, also known as the Bloodhorn tree due to the shape and color of the berries, grows on the northeast coast of Australia and in the rainforests of Brazil.
“[We have] taken the natural product and restyled it with unique features to improve the potency and solubility,” explained Florence McCarthy, PhD, of University College Cork in Ireland.
“What is truly exceptional is that these features are not common in drugs, and so we aim to exploit this fully. There is also significant potential to apply this approach to other drugs in a similar fashion.”
For this study, Dr McCarthy and his colleagues first tested 7-formyl-10-methylisoellipticine in the AML cell line MV4-11. They tested a range of concentrations in an attempt to identify the minimum concentration that would cause significant cytotoxicity. It turned out to be 5 μM.
Over a period of 24 hours, 5 μM of 7-formyl-10-methylisoellipticine killed up to 40% of MV4-11 cells. And over 96 hours, 5 μM of 7-formyl-10-methylisoellipticine killed more than 90% of cells.
Further investigation revealed that 5 μM of 7-formyl-10-methylisoellipticine increases the sub-G1 phase of the MV4-11 cell cycle. And the compound functions, at least in part, by generating mitochondrial-derived reactive oxygen species.
The researchers then found that 7-formyl-10-methylisoellipticine is not toxic to BALB/c mice. The team injected the mice with 7-formyl-10-methylisoellipticine at a range of doses—5 mg/kg, 10 mg/kg, 25 mg/kg, and 50 mg/kg.
Regardless of the dose, the compound did not cause a change in body weight, significantly increase levels of alanine aminotransferase or aspartate aminotransferase relative to negative control, or significantly change cell morphology or tissue structure in specified major organs.
Finally, the researchers found that 7-formyl-10-methylisoellipticine has antitumor activity in an AML xenograft mouse model. Based on the toxicity experiments, the team used a dose of 25 mg/kg in these mice.
At this dose, 7-formyl-10-methylisoellipticine significantly slowed tumor growth and reduced tumor mass. Tumor growth was 4 times slower in mice treated with 7-formyl-10-methylisoellipticine than in control mice. And tumor mass was up to 7 times greater in controls than it was in treated mice.
Based on these results, the researchers said they plan to continue investigating the mechanism of action of ellipticines, which “have a clear potential clinical application.” ![]()
A compound derived from the berries of the Bloodhorn tree has demonstrated activity against acute myeloid leukemia (AML), according to preclinical research published in Investigational New Drugs.
The compound, 7-formyl-10-methylisoellipticine, induced apoptosis in AML cells in a dose- and time-dependent manner.
It also significantly slowed tumor growth and reduced tumor mass in a mouse model of AML.
7-formyl-10-methylisoellipticine is derived from an ellipticine, which has been isolated from the berries of the Ochrosia Elliptica tree. The tree, also known as the Bloodhorn tree due to the shape and color of the berries, grows on the northeast coast of Australia and in the rainforests of Brazil.
“[We have] taken the natural product and restyled it with unique features to improve the potency and solubility,” explained Florence McCarthy, PhD, of University College Cork in Ireland.
“What is truly exceptional is that these features are not common in drugs, and so we aim to exploit this fully. There is also significant potential to apply this approach to other drugs in a similar fashion.”
For this study, Dr McCarthy and his colleagues first tested 7-formyl-10-methylisoellipticine in the AML cell line MV4-11. They tested a range of concentrations in an attempt to identify the minimum concentration that would cause significant cytotoxicity. It turned out to be 5 μM.
Over a period of 24 hours, 5 μM of 7-formyl-10-methylisoellipticine killed up to 40% of MV4-11 cells. And over 96 hours, 5 μM of 7-formyl-10-methylisoellipticine killed more than 90% of cells.
Further investigation revealed that 5 μM of 7-formyl-10-methylisoellipticine increases the sub-G1 phase of the MV4-11 cell cycle. And the compound functions, at least in part, by generating mitochondrial-derived reactive oxygen species.
The researchers then found that 7-formyl-10-methylisoellipticine is not toxic to BALB/c mice. The team injected the mice with 7-formyl-10-methylisoellipticine at a range of doses—5 mg/kg, 10 mg/kg, 25 mg/kg, and 50 mg/kg.
Regardless of the dose, the compound did not cause a change in body weight, significantly increase levels of alanine aminotransferase or aspartate aminotransferase relative to negative control, or significantly change cell morphology or tissue structure in specified major organs.
Finally, the researchers found that 7-formyl-10-methylisoellipticine has antitumor activity in an AML xenograft mouse model. Based on the toxicity experiments, the team used a dose of 25 mg/kg in these mice.
At this dose, 7-formyl-10-methylisoellipticine significantly slowed tumor growth and reduced tumor mass. Tumor growth was 4 times slower in mice treated with 7-formyl-10-methylisoellipticine than in control mice. And tumor mass was up to 7 times greater in controls than it was in treated mice.
Based on these results, the researchers said they plan to continue investigating the mechanism of action of ellipticines, which “have a clear potential clinical application.” ![]()
Risk of anaphylaxis with IV iron products
Researchers have compared the risk of anaphylaxis with different intravenous (IV) iron products and found evidence to suggest that iron dextran poses the greatest risk.
Compared with nondextran formulations, iron dextran was associated with a higher cumulative risk of anaphylaxis and an increased risk of anaphylaxis at first administration.
Iron sucrose was associated with the lowest risk of anaphylaxis, both cumulative and at first administration.
Cunlin Wang, MD, PhD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues conducted this research and reported the results in JAMA.
The researchers studied 688,183 recipients of IV iron enrolled in the fee-for-service Medicare program from January 2003 to December 2013.
The team examined administrations of IV iron dextran, gluconate, sucrose, or ferumoxytol. They identified 247,500 iron dextran and 440,683 nondextran users during the study period.
Overall, there were 274 cases of anaphylaxis at first exposure to IV iron and an additional 170 cases during subsequent iron administrations.
At first administration, iron dextran was associated with a higher anaphylaxis risk than nondextran formulations. The risk of anaphylaxis was 68 per 100,000 persons for iron dextran and 24 per 100,000 persons for all nondextran products combined. The odds ratio—adjusted for age, indication, history of coronary heart disease, and hypertension—was 2.6 (P<0.001).
Among the nondextran products, the risk of anaphylaxis at first administration was higher with both iron gluconate and ferumoxytol than with iron sucrose. When compared with iron sucrose, the adjusted odds ratio of anaphylaxis was 3.6 for iron dextran, 2.0 for iron gluconate, and 2.2 for ferumoxytol.
Because each IV iron product has a specific recommended dose and schedule of administration, the researchers also calculated the cumulative risk of anaphylaxis based on both the number of administrations and the clinically relevant repletion level of iron (1000 mg) achieved within 12 weeks.
The cumulative risk of anaphylaxis over multiple administrations was highest for iron dextran, followed by ferumoxytol, iron gluconate, and iron sucrose.
The estimated cumulative anaphylaxis risk following total iron repletion of 1000 mg administered within a 12-week period was highest with iron dextran (82 per 100,000 persons) and lowest with iron sucrose (21 per 100,000 persons).
The researchers noted that the mechanism of anaphylactic reaction after IV iron remains unknown. ![]()
Researchers have compared the risk of anaphylaxis with different intravenous (IV) iron products and found evidence to suggest that iron dextran poses the greatest risk.
Compared with nondextran formulations, iron dextran was associated with a higher cumulative risk of anaphylaxis and an increased risk of anaphylaxis at first administration.
Iron sucrose was associated with the lowest risk of anaphylaxis, both cumulative and at first administration.
Cunlin Wang, MD, PhD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues conducted this research and reported the results in JAMA.
The researchers studied 688,183 recipients of IV iron enrolled in the fee-for-service Medicare program from January 2003 to December 2013.
The team examined administrations of IV iron dextran, gluconate, sucrose, or ferumoxytol. They identified 247,500 iron dextran and 440,683 nondextran users during the study period.
Overall, there were 274 cases of anaphylaxis at first exposure to IV iron and an additional 170 cases during subsequent iron administrations.
At first administration, iron dextran was associated with a higher anaphylaxis risk than nondextran formulations. The risk of anaphylaxis was 68 per 100,000 persons for iron dextran and 24 per 100,000 persons for all nondextran products combined. The odds ratio—adjusted for age, indication, history of coronary heart disease, and hypertension—was 2.6 (P<0.001).
Among the nondextran products, the risk of anaphylaxis at first administration was higher with both iron gluconate and ferumoxytol than with iron sucrose. When compared with iron sucrose, the adjusted odds ratio of anaphylaxis was 3.6 for iron dextran, 2.0 for iron gluconate, and 2.2 for ferumoxytol.
Because each IV iron product has a specific recommended dose and schedule of administration, the researchers also calculated the cumulative risk of anaphylaxis based on both the number of administrations and the clinically relevant repletion level of iron (1000 mg) achieved within 12 weeks.
The cumulative risk of anaphylaxis over multiple administrations was highest for iron dextran, followed by ferumoxytol, iron gluconate, and iron sucrose.
The estimated cumulative anaphylaxis risk following total iron repletion of 1000 mg administered within a 12-week period was highest with iron dextran (82 per 100,000 persons) and lowest with iron sucrose (21 per 100,000 persons).
The researchers noted that the mechanism of anaphylactic reaction after IV iron remains unknown. ![]()
Researchers have compared the risk of anaphylaxis with different intravenous (IV) iron products and found evidence to suggest that iron dextran poses the greatest risk.
Compared with nondextran formulations, iron dextran was associated with a higher cumulative risk of anaphylaxis and an increased risk of anaphylaxis at first administration.
Iron sucrose was associated with the lowest risk of anaphylaxis, both cumulative and at first administration.
Cunlin Wang, MD, PhD, of the US Food and Drug Administration in Silver Spring, Maryland, and his colleagues conducted this research and reported the results in JAMA.
The researchers studied 688,183 recipients of IV iron enrolled in the fee-for-service Medicare program from January 2003 to December 2013.
The team examined administrations of IV iron dextran, gluconate, sucrose, or ferumoxytol. They identified 247,500 iron dextran and 440,683 nondextran users during the study period.
Overall, there were 274 cases of anaphylaxis at first exposure to IV iron and an additional 170 cases during subsequent iron administrations.
At first administration, iron dextran was associated with a higher anaphylaxis risk than nondextran formulations. The risk of anaphylaxis was 68 per 100,000 persons for iron dextran and 24 per 100,000 persons for all nondextran products combined. The odds ratio—adjusted for age, indication, history of coronary heart disease, and hypertension—was 2.6 (P<0.001).
Among the nondextran products, the risk of anaphylaxis at first administration was higher with both iron gluconate and ferumoxytol than with iron sucrose. When compared with iron sucrose, the adjusted odds ratio of anaphylaxis was 3.6 for iron dextran, 2.0 for iron gluconate, and 2.2 for ferumoxytol.
Because each IV iron product has a specific recommended dose and schedule of administration, the researchers also calculated the cumulative risk of anaphylaxis based on both the number of administrations and the clinically relevant repletion level of iron (1000 mg) achieved within 12 weeks.
The cumulative risk of anaphylaxis over multiple administrations was highest for iron dextran, followed by ferumoxytol, iron gluconate, and iron sucrose.
The estimated cumulative anaphylaxis risk following total iron repletion of 1000 mg administered within a 12-week period was highest with iron dextran (82 per 100,000 persons) and lowest with iron sucrose (21 per 100,000 persons).
The researchers noted that the mechanism of anaphylactic reaction after IV iron remains unknown. ![]()
New insight into infection-driven thrombosis
invading cultured human cells
Image courtesy of Rocky
Mountain Laboratories/NIAID
Preclinical research has shown how Salmonella infections that spread to the blood and organs can lead to life-threatening thrombosis.
Experiments in mice demonstrated that systemic infections prompt inflammation, which leads to thrombosis.
However, the sustained threat from thrombosis is independent of the continued presence of infection and instead parallels the regulation of inflammation within the host.
These findings, published in The Journal of Clinical Investigation, shed light on a poorly understood area of clinical medicine.
While some of the mechanisms that underpin the process of infection-driven thrombosis are known, particularly for Gram-positive organisms such as Staphylococci or Streptococci, they are not universally applicable.
Moreover, during sepsis, the causative pathogen is often never isolated or identified. The new study helps to explain why this is the case.
“For all of the advances we’ve made in this field, it is not always clear why people die from infection,” said study author Adam Cunningham, PhD, of the University of Birmingham in the UK.
“We think complications of thrombosis may be one reason. In Salmonella infections, severity is typically associated with the presence of bacteria in the blood, called bacteremia, even though the actual numbers of bacteria in the blood are very low. This suggested to us that the host response was crucial in determining the outcome.”
Dr Cunningham and his colleagues found that thrombi developed within the liver of murine models infected with Salmonella Typhimurium, and these thrombi persisted for many weeks.
The infection caused inflammation in the liver tissue, which then triggered thrombosis within vessels. This occurred via the ligation of C-type lectin-like receptor-2 (CLEC-2) on platelets by podoplanin, a molecule that is ordinarily absent in blood vessels but is expressed by macrophages responding to the infection.
The regulation and amplification of thrombosis was triggered by TLR4, a protein essential for the activation of the inflammatory cascade and the control of infection during its early stages.
Thrombosis remained at peak levels even when bacteria were absent from the blood and largely cleared from the infected organs.
“A little, controlled thrombosis is probably a good thing, as it helps to clear bacteria from the blood,” said study author Steve Watson, PhD, of the University of Birmingham.
“Therefore, any intervention would need to control, rather than deny, the host response. The problem only comes when it develops into a clot. Most of the current approaches to counter the development of these life-threatening thrombi do not account for the non-classical mechanism that we have shown to be at work.”
The researchers are now working to further understand how to manipulate and control this response, and how it can contribute to the complications of other infections and diseases. ![]()
invading cultured human cells
Image courtesy of Rocky
Mountain Laboratories/NIAID
Preclinical research has shown how Salmonella infections that spread to the blood and organs can lead to life-threatening thrombosis.
Experiments in mice demonstrated that systemic infections prompt inflammation, which leads to thrombosis.
However, the sustained threat from thrombosis is independent of the continued presence of infection and instead parallels the regulation of inflammation within the host.
These findings, published in The Journal of Clinical Investigation, shed light on a poorly understood area of clinical medicine.
While some of the mechanisms that underpin the process of infection-driven thrombosis are known, particularly for Gram-positive organisms such as Staphylococci or Streptococci, they are not universally applicable.
Moreover, during sepsis, the causative pathogen is often never isolated or identified. The new study helps to explain why this is the case.
“For all of the advances we’ve made in this field, it is not always clear why people die from infection,” said study author Adam Cunningham, PhD, of the University of Birmingham in the UK.
“We think complications of thrombosis may be one reason. In Salmonella infections, severity is typically associated with the presence of bacteria in the blood, called bacteremia, even though the actual numbers of bacteria in the blood are very low. This suggested to us that the host response was crucial in determining the outcome.”
Dr Cunningham and his colleagues found that thrombi developed within the liver of murine models infected with Salmonella Typhimurium, and these thrombi persisted for many weeks.
The infection caused inflammation in the liver tissue, which then triggered thrombosis within vessels. This occurred via the ligation of C-type lectin-like receptor-2 (CLEC-2) on platelets by podoplanin, a molecule that is ordinarily absent in blood vessels but is expressed by macrophages responding to the infection.
The regulation and amplification of thrombosis was triggered by TLR4, a protein essential for the activation of the inflammatory cascade and the control of infection during its early stages.
Thrombosis remained at peak levels even when bacteria were absent from the blood and largely cleared from the infected organs.
“A little, controlled thrombosis is probably a good thing, as it helps to clear bacteria from the blood,” said study author Steve Watson, PhD, of the University of Birmingham.
“Therefore, any intervention would need to control, rather than deny, the host response. The problem only comes when it develops into a clot. Most of the current approaches to counter the development of these life-threatening thrombi do not account for the non-classical mechanism that we have shown to be at work.”
The researchers are now working to further understand how to manipulate and control this response, and how it can contribute to the complications of other infections and diseases. ![]()
invading cultured human cells
Image courtesy of Rocky
Mountain Laboratories/NIAID
Preclinical research has shown how Salmonella infections that spread to the blood and organs can lead to life-threatening thrombosis.
Experiments in mice demonstrated that systemic infections prompt inflammation, which leads to thrombosis.
However, the sustained threat from thrombosis is independent of the continued presence of infection and instead parallels the regulation of inflammation within the host.
These findings, published in The Journal of Clinical Investigation, shed light on a poorly understood area of clinical medicine.
While some of the mechanisms that underpin the process of infection-driven thrombosis are known, particularly for Gram-positive organisms such as Staphylococci or Streptococci, they are not universally applicable.
Moreover, during sepsis, the causative pathogen is often never isolated or identified. The new study helps to explain why this is the case.
“For all of the advances we’ve made in this field, it is not always clear why people die from infection,” said study author Adam Cunningham, PhD, of the University of Birmingham in the UK.
“We think complications of thrombosis may be one reason. In Salmonella infections, severity is typically associated with the presence of bacteria in the blood, called bacteremia, even though the actual numbers of bacteria in the blood are very low. This suggested to us that the host response was crucial in determining the outcome.”
Dr Cunningham and his colleagues found that thrombi developed within the liver of murine models infected with Salmonella Typhimurium, and these thrombi persisted for many weeks.
The infection caused inflammation in the liver tissue, which then triggered thrombosis within vessels. This occurred via the ligation of C-type lectin-like receptor-2 (CLEC-2) on platelets by podoplanin, a molecule that is ordinarily absent in blood vessels but is expressed by macrophages responding to the infection.
The regulation and amplification of thrombosis was triggered by TLR4, a protein essential for the activation of the inflammatory cascade and the control of infection during its early stages.
Thrombosis remained at peak levels even when bacteria were absent from the blood and largely cleared from the infected organs.
“A little, controlled thrombosis is probably a good thing, as it helps to clear bacteria from the blood,” said study author Steve Watson, PhD, of the University of Birmingham.
“Therefore, any intervention would need to control, rather than deny, the host response. The problem only comes when it develops into a clot. Most of the current approaches to counter the development of these life-threatening thrombi do not account for the non-classical mechanism that we have shown to be at work.”
The researchers are now working to further understand how to manipulate and control this response, and how it can contribute to the complications of other infections and diseases. ![]()
FDA approves first monoclonal antibody for MM

Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted accelerated approval for daratumumab (Darzalex).
The drug is now approved to treat patients with multiple myeloma (MM) who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is the first monoclonal antibody approved to treat MM.
The drug works by binding to CD38 on the surface of MM cells. It triggers the patient’s own immune system to attack MM cells, resulting in cell death through multiple mechanisms of action.
Daratumumab is being developed by Janssen Biotech. The drug was previously granted breakthrough designation, orphan designation, and priority review from the FDA.
Daratumumab was approved under the FDA’s accelerated approval program, which allows the agency to approve a drug to treat a serious or life-threatening disease based on clinical data showing the drug has an effect on a surrogate endpoint reasonably likely to predict clinical benefit to patients.
As a condition of this accelerated approval, Janssen is required to conduct a multicenter, randomized trial establishing the superiority of daratumumab over standard therapy to verify and describe the clinical benefit of daratumumab. Janssen has several ongoing, multicenter, randomized trials with a primary endpoint of progression-free survival.
The recommended dose and schedule for daratumumab is 16 mg/kg once every week for 8 weeks, then once every 2 weeks for 16 weeks, then once every 4 weeks until disease progression.
The FDA said blood banks should be informed that patients are receiving daratumumab because the drug may interfere with tests such as antibody screening.
In addition, women who are pregnant should not use daratumumab, and women planning to become pregnant should use effective contraceptives during and for at least 3 months after stopping daratumumab.
Trial data
The FDA’s approval of daratumumab was based on results of 2 studies—the phase 2 MMY2002 (SIRIUS) study and the phase 1/2 GEN501 study.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested that daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%.
Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 of the patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted accelerated approval for daratumumab (Darzalex).
The drug is now approved to treat patients with multiple myeloma (MM) who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is the first monoclonal antibody approved to treat MM.
The drug works by binding to CD38 on the surface of MM cells. It triggers the patient’s own immune system to attack MM cells, resulting in cell death through multiple mechanisms of action.
Daratumumab is being developed by Janssen Biotech. The drug was previously granted breakthrough designation, orphan designation, and priority review from the FDA.
Daratumumab was approved under the FDA’s accelerated approval program, which allows the agency to approve a drug to treat a serious or life-threatening disease based on clinical data showing the drug has an effect on a surrogate endpoint reasonably likely to predict clinical benefit to patients.
As a condition of this accelerated approval, Janssen is required to conduct a multicenter, randomized trial establishing the superiority of daratumumab over standard therapy to verify and describe the clinical benefit of daratumumab. Janssen has several ongoing, multicenter, randomized trials with a primary endpoint of progression-free survival.
The recommended dose and schedule for daratumumab is 16 mg/kg once every week for 8 weeks, then once every 2 weeks for 16 weeks, then once every 4 weeks until disease progression.
The FDA said blood banks should be informed that patients are receiving daratumumab because the drug may interfere with tests such as antibody screening.
In addition, women who are pregnant should not use daratumumab, and women planning to become pregnant should use effective contraceptives during and for at least 3 months after stopping daratumumab.
Trial data
The FDA’s approval of daratumumab was based on results of 2 studies—the phase 2 MMY2002 (SIRIUS) study and the phase 1/2 GEN501 study.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested that daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%.
Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 of the patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted accelerated approval for daratumumab (Darzalex).
The drug is now approved to treat patients with multiple myeloma (MM) who have received at least 3 prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double-refractory to a proteasome inhibitor and an immunomodulatory agent.
Daratumumab is the first monoclonal antibody approved to treat MM.
The drug works by binding to CD38 on the surface of MM cells. It triggers the patient’s own immune system to attack MM cells, resulting in cell death through multiple mechanisms of action.
Daratumumab is being developed by Janssen Biotech. The drug was previously granted breakthrough designation, orphan designation, and priority review from the FDA.
Daratumumab was approved under the FDA’s accelerated approval program, which allows the agency to approve a drug to treat a serious or life-threatening disease based on clinical data showing the drug has an effect on a surrogate endpoint reasonably likely to predict clinical benefit to patients.
As a condition of this accelerated approval, Janssen is required to conduct a multicenter, randomized trial establishing the superiority of daratumumab over standard therapy to verify and describe the clinical benefit of daratumumab. Janssen has several ongoing, multicenter, randomized trials with a primary endpoint of progression-free survival.
The recommended dose and schedule for daratumumab is 16 mg/kg once every week for 8 weeks, then once every 2 weeks for 16 weeks, then once every 4 weeks until disease progression.
The FDA said blood banks should be informed that patients are receiving daratumumab because the drug may interfere with tests such as antibody screening.
In addition, women who are pregnant should not use daratumumab, and women planning to become pregnant should use effective contraceptives during and for at least 3 months after stopping daratumumab.
Trial data
The FDA’s approval of daratumumab was based on results of 2 studies—the phase 2 MMY2002 (SIRIUS) study and the phase 1/2 GEN501 study.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested that daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%.
Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 of the patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events. ![]()
Strategy could slow spread of resistant malaria
Plasmodium falciparum
Image by Mae Melvin
A varied treatment approach could slow the spread of artemisinin-resistant malaria, according to research published in The Lancet Global Health.
Computer simulations suggested that giving a population multiple artemisinin-based combination therapies simultaneously, along with a non-artemisinin therapy, is the best way to combat malaria and reduce the spread of resistant disease.
Investigators found this approach worked best even when the non-artemisinin drug was only effective in treating malaria 85% of the time.
The team ran their computer simulations to determine if there was an optimal strategy that could stop the spread of drug-resistant Plasmodium falciparum parasites across populations while still effectively treating malaria in individual patients.
The simulations showed that simultaneously dosing a population with several artemisinin-combination therapies—for example, by prescribing artemisinin in combination with different partner drugs on different days of the week—was more effective than either cycling between different artemisinin combination therapies or sticking to one specific combination until that combination started failing.
The simulations also showed that if this simultaneous dosing included a combination without artemisinin, malaria parasites that were resistant to artemisinin were slower to emerge and slower to spread.
Including this potentially less effective treatment option didn’t necessarily mean that many more people would not recover from malaria.
In the worst-case scenario of the non-artemisinin treatment being only 75% as effective as artemisinin-based combination therapy, fewer than 7% of patients would still have post-treatment malaria parasites in their blood as a result of not receiving an artemisinin-based therapy.
“For this subgroup of patients, second-line treatment with an artemisinin combination therapy would be recommended,” said study author Maciej Boni, PhD, of the Hospital for Tropical Diseases in Ho Chi Minh City, Vietnam.
“The ethical implications of such a treatment policy will need to be weighed against the benefit of delaying and slowing down the spread of artemisinin resistance. But the nightmare we all want to avoid is the establishment of artemisinin resistance in Africa, where hundreds of millions of individuals rely on artemisinin-based therapies as their first-line antimalarial treatment.”
“By deploying different antimalarial therapies simultaneously—including non-artemisinin-based therapies—national malaria control programs in Africa should be able to slow down the spread of artemisinin-resistant parasites when they are imported into the continent.” ![]()
Plasmodium falciparum
Image by Mae Melvin
A varied treatment approach could slow the spread of artemisinin-resistant malaria, according to research published in The Lancet Global Health.
Computer simulations suggested that giving a population multiple artemisinin-based combination therapies simultaneously, along with a non-artemisinin therapy, is the best way to combat malaria and reduce the spread of resistant disease.
Investigators found this approach worked best even when the non-artemisinin drug was only effective in treating malaria 85% of the time.
The team ran their computer simulations to determine if there was an optimal strategy that could stop the spread of drug-resistant Plasmodium falciparum parasites across populations while still effectively treating malaria in individual patients.
The simulations showed that simultaneously dosing a population with several artemisinin-combination therapies—for example, by prescribing artemisinin in combination with different partner drugs on different days of the week—was more effective than either cycling between different artemisinin combination therapies or sticking to one specific combination until that combination started failing.
The simulations also showed that if this simultaneous dosing included a combination without artemisinin, malaria parasites that were resistant to artemisinin were slower to emerge and slower to spread.
Including this potentially less effective treatment option didn’t necessarily mean that many more people would not recover from malaria.
In the worst-case scenario of the non-artemisinin treatment being only 75% as effective as artemisinin-based combination therapy, fewer than 7% of patients would still have post-treatment malaria parasites in their blood as a result of not receiving an artemisinin-based therapy.
“For this subgroup of patients, second-line treatment with an artemisinin combination therapy would be recommended,” said study author Maciej Boni, PhD, of the Hospital for Tropical Diseases in Ho Chi Minh City, Vietnam.
“The ethical implications of such a treatment policy will need to be weighed against the benefit of delaying and slowing down the spread of artemisinin resistance. But the nightmare we all want to avoid is the establishment of artemisinin resistance in Africa, where hundreds of millions of individuals rely on artemisinin-based therapies as their first-line antimalarial treatment.”
“By deploying different antimalarial therapies simultaneously—including non-artemisinin-based therapies—national malaria control programs in Africa should be able to slow down the spread of artemisinin-resistant parasites when they are imported into the continent.” ![]()
Plasmodium falciparum
Image by Mae Melvin
A varied treatment approach could slow the spread of artemisinin-resistant malaria, according to research published in The Lancet Global Health.
Computer simulations suggested that giving a population multiple artemisinin-based combination therapies simultaneously, along with a non-artemisinin therapy, is the best way to combat malaria and reduce the spread of resistant disease.
Investigators found this approach worked best even when the non-artemisinin drug was only effective in treating malaria 85% of the time.
The team ran their computer simulations to determine if there was an optimal strategy that could stop the spread of drug-resistant Plasmodium falciparum parasites across populations while still effectively treating malaria in individual patients.
The simulations showed that simultaneously dosing a population with several artemisinin-combination therapies—for example, by prescribing artemisinin in combination with different partner drugs on different days of the week—was more effective than either cycling between different artemisinin combination therapies or sticking to one specific combination until that combination started failing.
The simulations also showed that if this simultaneous dosing included a combination without artemisinin, malaria parasites that were resistant to artemisinin were slower to emerge and slower to spread.
Including this potentially less effective treatment option didn’t necessarily mean that many more people would not recover from malaria.
In the worst-case scenario of the non-artemisinin treatment being only 75% as effective as artemisinin-based combination therapy, fewer than 7% of patients would still have post-treatment malaria parasites in their blood as a result of not receiving an artemisinin-based therapy.
“For this subgroup of patients, second-line treatment with an artemisinin combination therapy would be recommended,” said study author Maciej Boni, PhD, of the Hospital for Tropical Diseases in Ho Chi Minh City, Vietnam.
“The ethical implications of such a treatment policy will need to be weighed against the benefit of delaying and slowing down the spread of artemisinin resistance. But the nightmare we all want to avoid is the establishment of artemisinin resistance in Africa, where hundreds of millions of individuals rely on artemisinin-based therapies as their first-line antimalarial treatment.”
“By deploying different antimalarial therapies simultaneously—including non-artemisinin-based therapies—national malaria control programs in Africa should be able to slow down the spread of artemisinin-resistant parasites when they are imported into the continent.” ![]()
Role of inflammation and aging in leukemia

Image by Michael Zangani
Previous research has suggested the accumulation of cancer-causing mutations is to blame for the increased risk of cancer in the aging population.
But a study published in The Journal of Clinical Investigation tells another story.
Investigators found that, without age-associated inflammation, older mice developed leukemia no faster than young mice.
The study focused primarily on the “ecosystem” of B-cell progenitor pools.
The investigators wanted to determine what allows a population of healthy B-cell progenitors to be replaced over time with a population of cancerous B-cell progenitors.
“We chose to focus on the role of inflammation in the bone marrow—one of the hallmarks of age-associated tissue changes—where these B-cell progenitor pools live,” said study author Curtis Henry, PhD, of the University of Colorado Anschutz Medical Campus in Aurora, Colorado.
He and his colleagues found that inflammation hurts the growth and maintenance of B-progenitor cells, but that’s not all. Cancerous mutations tend to alter cells in ways that help them survive conditions of inflammation in the bone marrow.
“Suddenly, the healthy cells that were the fittest are no longer the most fit,” Dr Henry explained. “Because the tissue changed, cancer cells have a selective advantage.”
The investigators were able to observe this inflammation-driven natural selection in mouse models. The group worked with mice engineered to prevent inflammation and set out to determine how healthy and leukemia-initiated cells would fare in these conditions.
“Basically, without the effects of inflammation, B-cell progenitor pools stayed fit,” Dr Henry said.
And stopping inflammation reduced the ability of cells expressing the oncogene NRAS from taking over the bone marrow niche.
This study suggests that an increase in cancer risk with age may not be inevitable. Instead of simply being a matter of the passage of time, cancer development in aged populations may be partially dependent on inflammation-associated tissue changes.
“Despite the fact that cancer is largely a disease of old age, almost all cancer modeling in mice employs only young mice,” noted study author James DeGregori, PhD, of the University of Colorado Anschutz Medical Campus.
“This is based on the view that finding the genetic mutation that causes cancer should be enough to understand the disease.”
In these studies, the investigators tested both young and old mice. The older mice were more likely to develop leukemia, but only in the presence of age-associated inflammation. If age-associated inflammation was blocked, the older mice were no more likely than young mice to develop leukemia.
The work implies that stopping the effects of inflammation on tissue could stop cancers from forming. However, inflammation can be necessary in some circumstances. So the investigators said more work is needed to understand how to “tune” inflammation in the elderly to maximize its beneficial effects while minimizing negative effects.
“While it’s premature to suggest that people should take medicines to fight inflammation as they age, we believe our results warrant further study into this potential strategy to combat the age-associated increase in cancer risk,” Dr Henry concluded. ![]()
Image by Michael Zangani
Previous research has suggested the accumulation of cancer-causing mutations is to blame for the increased risk of cancer in the aging population.
But a study published in The Journal of Clinical Investigation tells another story.
Investigators found that, without age-associated inflammation, older mice developed leukemia no faster than young mice.
The study focused primarily on the “ecosystem” of B-cell progenitor pools.
The investigators wanted to determine what allows a population of healthy B-cell progenitors to be replaced over time with a population of cancerous B-cell progenitors.
“We chose to focus on the role of inflammation in the bone marrow—one of the hallmarks of age-associated tissue changes—where these B-cell progenitor pools live,” said study author Curtis Henry, PhD, of the University of Colorado Anschutz Medical Campus in Aurora, Colorado.
He and his colleagues found that inflammation hurts the growth and maintenance of B-progenitor cells, but that’s not all. Cancerous mutations tend to alter cells in ways that help them survive conditions of inflammation in the bone marrow.
“Suddenly, the healthy cells that were the fittest are no longer the most fit,” Dr Henry explained. “Because the tissue changed, cancer cells have a selective advantage.”
The investigators were able to observe this inflammation-driven natural selection in mouse models. The group worked with mice engineered to prevent inflammation and set out to determine how healthy and leukemia-initiated cells would fare in these conditions.
“Basically, without the effects of inflammation, B-cell progenitor pools stayed fit,” Dr Henry said.
And stopping inflammation reduced the ability of cells expressing the oncogene NRAS from taking over the bone marrow niche.
This study suggests that an increase in cancer risk with age may not be inevitable. Instead of simply being a matter of the passage of time, cancer development in aged populations may be partially dependent on inflammation-associated tissue changes.
“Despite the fact that cancer is largely a disease of old age, almost all cancer modeling in mice employs only young mice,” noted study author James DeGregori, PhD, of the University of Colorado Anschutz Medical Campus.
“This is based on the view that finding the genetic mutation that causes cancer should be enough to understand the disease.”
In these studies, the investigators tested both young and old mice. The older mice were more likely to develop leukemia, but only in the presence of age-associated inflammation. If age-associated inflammation was blocked, the older mice were no more likely than young mice to develop leukemia.
The work implies that stopping the effects of inflammation on tissue could stop cancers from forming. However, inflammation can be necessary in some circumstances. So the investigators said more work is needed to understand how to “tune” inflammation in the elderly to maximize its beneficial effects while minimizing negative effects.
“While it’s premature to suggest that people should take medicines to fight inflammation as they age, we believe our results warrant further study into this potential strategy to combat the age-associated increase in cancer risk,” Dr Henry concluded. ![]()
Image by Michael Zangani
Previous research has suggested the accumulation of cancer-causing mutations is to blame for the increased risk of cancer in the aging population.
But a study published in The Journal of Clinical Investigation tells another story.
Investigators found that, without age-associated inflammation, older mice developed leukemia no faster than young mice.
The study focused primarily on the “ecosystem” of B-cell progenitor pools.
The investigators wanted to determine what allows a population of healthy B-cell progenitors to be replaced over time with a population of cancerous B-cell progenitors.
“We chose to focus on the role of inflammation in the bone marrow—one of the hallmarks of age-associated tissue changes—where these B-cell progenitor pools live,” said study author Curtis Henry, PhD, of the University of Colorado Anschutz Medical Campus in Aurora, Colorado.
He and his colleagues found that inflammation hurts the growth and maintenance of B-progenitor cells, but that’s not all. Cancerous mutations tend to alter cells in ways that help them survive conditions of inflammation in the bone marrow.
“Suddenly, the healthy cells that were the fittest are no longer the most fit,” Dr Henry explained. “Because the tissue changed, cancer cells have a selective advantage.”
The investigators were able to observe this inflammation-driven natural selection in mouse models. The group worked with mice engineered to prevent inflammation and set out to determine how healthy and leukemia-initiated cells would fare in these conditions.
“Basically, without the effects of inflammation, B-cell progenitor pools stayed fit,” Dr Henry said.
And stopping inflammation reduced the ability of cells expressing the oncogene NRAS from taking over the bone marrow niche.
This study suggests that an increase in cancer risk with age may not be inevitable. Instead of simply being a matter of the passage of time, cancer development in aged populations may be partially dependent on inflammation-associated tissue changes.
“Despite the fact that cancer is largely a disease of old age, almost all cancer modeling in mice employs only young mice,” noted study author James DeGregori, PhD, of the University of Colorado Anschutz Medical Campus.
“This is based on the view that finding the genetic mutation that causes cancer should be enough to understand the disease.”
In these studies, the investigators tested both young and old mice. The older mice were more likely to develop leukemia, but only in the presence of age-associated inflammation. If age-associated inflammation was blocked, the older mice were no more likely than young mice to develop leukemia.
The work implies that stopping the effects of inflammation on tissue could stop cancers from forming. However, inflammation can be necessary in some circumstances. So the investigators said more work is needed to understand how to “tune” inflammation in the elderly to maximize its beneficial effects while minimizing negative effects.
“While it’s premature to suggest that people should take medicines to fight inflammation as they age, we believe our results warrant further study into this potential strategy to combat the age-associated increase in cancer risk,” Dr Henry concluded. ![]()
FDA approves pegylated product for hemophilia A

The US Food and Drug Administration (FDA) has approved Adynovate, a recombinant pegylated factor VIII (FVIII) product, for use in patients age 12 and older with hemophilia A.
The product can be used as routine prophylaxis and for on-demand treatment and control of bleeding episodes.
Adynovate will be available in the US in the coming weeks, according to Baxalta US Inc., the company developing the product.
Adynovate (formerly BAX 855) is built on the full-length Advate, a recombinant antihemophilic factor product that was approved by the FDA in 2003.
Adynovate consists of the full-length FVIII molecule linked to other molecules, known as polyethylene glycol (pegylated). This link extends the circulating half-life of the product and therefore extends the time between treatments.
So patients on Adynovate can receive twice-weekly doses rather than the 3 to 4 weekly doses typically required with other full-length FVIII products.
“The approval of Adynovate provides an important therapeutic option for use in the care of patients with hemophilia A and reduces the frequency of FVIII infusions needed to avoid bleeding,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA approved Adynovate based on results of a phase 2/3 trial. The study included 137 previously treated hemophilia A patients who were 12 to 65 years of age.
Patients were assigned to either twice-weekly prophylaxis (40-50 IU/kg, n=120) or on-demand treatment (10-60 IU/kg, n=17) with Adynovate.
Patients in the twice-weekly prophylaxis arm had far fewer annual bleeds than patients treated on-demand. The median annual bleed rates were 1.9 and 41.5, respectively.
Nearly all (96%) bleeding episodes (n=591) were controlled with 1 or 2 infusions of Adynovate.
None of the patients developed inhibitors to the treatment. However, there were 171 adverse events in the 73 patients who received Adynovate for about 6 months.
There were 7 events (occurring in 6 patients) that were considered possibly related to Adynovate. These included diarrhea, nausea, headache, and flushing.
Studies of Adynovate are ongoing in previously treated patients with severe hemophilia A undergoing surgery and in previously treated patients with severe hemophilia A who are under the age of 12. Baxalta is also planning to initiate a study in previously untreated patients with severe hemophilia A.
The company has filed for regulatory approval of Adynovate in Japan and expects to file for marketing authorization in Europe once the pediatric study is complete. ![]()
The US Food and Drug Administration (FDA) has approved Adynovate, a recombinant pegylated factor VIII (FVIII) product, for use in patients age 12 and older with hemophilia A.
The product can be used as routine prophylaxis and for on-demand treatment and control of bleeding episodes.
Adynovate will be available in the US in the coming weeks, according to Baxalta US Inc., the company developing the product.
Adynovate (formerly BAX 855) is built on the full-length Advate, a recombinant antihemophilic factor product that was approved by the FDA in 2003.
Adynovate consists of the full-length FVIII molecule linked to other molecules, known as polyethylene glycol (pegylated). This link extends the circulating half-life of the product and therefore extends the time between treatments.
So patients on Adynovate can receive twice-weekly doses rather than the 3 to 4 weekly doses typically required with other full-length FVIII products.
“The approval of Adynovate provides an important therapeutic option for use in the care of patients with hemophilia A and reduces the frequency of FVIII infusions needed to avoid bleeding,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA approved Adynovate based on results of a phase 2/3 trial. The study included 137 previously treated hemophilia A patients who were 12 to 65 years of age.
Patients were assigned to either twice-weekly prophylaxis (40-50 IU/kg, n=120) or on-demand treatment (10-60 IU/kg, n=17) with Adynovate.
Patients in the twice-weekly prophylaxis arm had far fewer annual bleeds than patients treated on-demand. The median annual bleed rates were 1.9 and 41.5, respectively.
Nearly all (96%) bleeding episodes (n=591) were controlled with 1 or 2 infusions of Adynovate.
None of the patients developed inhibitors to the treatment. However, there were 171 adverse events in the 73 patients who received Adynovate for about 6 months.
There were 7 events (occurring in 6 patients) that were considered possibly related to Adynovate. These included diarrhea, nausea, headache, and flushing.
Studies of Adynovate are ongoing in previously treated patients with severe hemophilia A undergoing surgery and in previously treated patients with severe hemophilia A who are under the age of 12. Baxalta is also planning to initiate a study in previously untreated patients with severe hemophilia A.
The company has filed for regulatory approval of Adynovate in Japan and expects to file for marketing authorization in Europe once the pediatric study is complete. ![]()
The US Food and Drug Administration (FDA) has approved Adynovate, a recombinant pegylated factor VIII (FVIII) product, for use in patients age 12 and older with hemophilia A.
The product can be used as routine prophylaxis and for on-demand treatment and control of bleeding episodes.
Adynovate will be available in the US in the coming weeks, according to Baxalta US Inc., the company developing the product.
Adynovate (formerly BAX 855) is built on the full-length Advate, a recombinant antihemophilic factor product that was approved by the FDA in 2003.
Adynovate consists of the full-length FVIII molecule linked to other molecules, known as polyethylene glycol (pegylated). This link extends the circulating half-life of the product and therefore extends the time between treatments.
So patients on Adynovate can receive twice-weekly doses rather than the 3 to 4 weekly doses typically required with other full-length FVIII products.
“The approval of Adynovate provides an important therapeutic option for use in the care of patients with hemophilia A and reduces the frequency of FVIII infusions needed to avoid bleeding,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA approved Adynovate based on results of a phase 2/3 trial. The study included 137 previously treated hemophilia A patients who were 12 to 65 years of age.
Patients were assigned to either twice-weekly prophylaxis (40-50 IU/kg, n=120) or on-demand treatment (10-60 IU/kg, n=17) with Adynovate.
Patients in the twice-weekly prophylaxis arm had far fewer annual bleeds than patients treated on-demand. The median annual bleed rates were 1.9 and 41.5, respectively.
Nearly all (96%) bleeding episodes (n=591) were controlled with 1 or 2 infusions of Adynovate.
None of the patients developed inhibitors to the treatment. However, there were 171 adverse events in the 73 patients who received Adynovate for about 6 months.
There were 7 events (occurring in 6 patients) that were considered possibly related to Adynovate. These included diarrhea, nausea, headache, and flushing.
Studies of Adynovate are ongoing in previously treated patients with severe hemophilia A undergoing surgery and in previously treated patients with severe hemophilia A who are under the age of 12. Baxalta is also planning to initiate a study in previously untreated patients with severe hemophilia A.
The company has filed for regulatory approval of Adynovate in Japan and expects to file for marketing authorization in Europe once the pediatric study is complete.
Patients may benefit from managing own VKA treatment

Photo courtesy of NIGMS
Allowing patients to self-manage anticoagulant therapy after heart valve surgery may lower the risk of death, according to a study published in the Annals of Thoracic Surgery.
Some of the patients studied were required to complete an educational program before they could manage their own treatment with a vitamin K antagonist (VKA).
For the remaining patients, VKA treatment was managed by a healthcare provider.
At 5 years, patients who managed their own treatment had a lower risk of death than patients on standard management.
“Oral anticoagulation therapy is usually monitored by laboratory analysis of the patient’s blood, which healthcare providers use to determine the appropriate dosage of medication,” said study author Thomas Decker Christensen, MD, PhD, of Aarhus University Hospital in Denmark.
“We believe that allowing patients to have more control over their own treatment can improve the standard of care in this patient group.”
Dr Christensen and his colleagues evaluated patients treated at 2 hospitals in Denmark between 1996 and 2012. There were 615 patients with mechanical heart valves who self-managed VKA treatment and 3075 control patients (a 1:5 ratio) who received standard VKA management following valve surgery.
Patients were required to attend an educational program consisting of a minimum of 3 lessons to be eligible for self-management. Over a period of 27 weeks following the educational training, patients gradually became self-managed.
“Once patients were approved for self-management, they were allowed to continue as such but could contact the treatment center with questions or concerns,” Dr Christensen noted.
“Patients were provided with a portable coagulometer, which they used to analyze medication levels and monitor dosage. Blood levels were measured and tracked weekly.”
Patients compared their own measurements against the International Normalized Ratio and adjusted their dosage levels according to what they had been taught in the educational training.
Bleeding and VTE
Over 5 years of follow-up, the risk of venous thromboembolism (VTE) and major bleeding was similar between self-managed patients and patients on standard management.
The rate of thromboembolic events requiring hospitalization was 1.6 events per 100 patient-years in the self-management group and 2.0 per 100 patient-years in the standard group. At 1 year of follow-up, the rates were 2.0 and 2.1, respectively.
When the researchers adjusted for potential confounders, there was a statistically nonsignificant higher risk of VTE among self-managed patients after 1 year (hazard ratio [HR]=1.29) and a statistically nonsignificant lower risk after 5 years (HR=0.91).
The rate of major bleeding was about 1.0 per 100 patient-years in the self-management group at both 1 year and 5 years. The corresponding rates in the standard management group were 1.9 at 1 year and 1.4 at 5 years.
When the researchers adjusted for potential confounders, there was a statistically nonsignificant lower risk of major bleeding in the self-management group at 1 year (HR=0.54) and at 5 years (HR=0.83).
Mortality
After 5 years, the rate of all-cause mortality was 1.1 per 100 patient-years in the self-management group and 2.5 in the standard management group. The adjusted HR was 0.49 in favor of the self-management group.
A landmark analysis restricting the survival analysis to patients alive 1 year after study inclusion resulted in an adjusted HR of 0.57.
“There are several reasons that patients who self-manage treatment have better outcomes than those who follow standard management,” Dr Christensen explained.
“Self-management patients receive more detailed information about oral anticoagulation therapy. They also learn more about the influence that diet, infectious diseases, alcohol, and other drug interactions can have on their treatment than do patients receiving standard management.”
He added that he hopes this study will have an impact on the management and care of heart valve patients.
“We believe that the majority of patients who have a mechanical heart valve inserted during surgery should be able to manage their oral anticoagulant therapy and recommend this as the standard treatment approach for these patients,” he concluded.
Photo courtesy of NIGMS
Allowing patients to self-manage anticoagulant therapy after heart valve surgery may lower the risk of death, according to a study published in the Annals of Thoracic Surgery.
Some of the patients studied were required to complete an educational program before they could manage their own treatment with a vitamin K antagonist (VKA).
For the remaining patients, VKA treatment was managed by a healthcare provider.
At 5 years, patients who managed their own treatment had a lower risk of death than patients on standard management.
“Oral anticoagulation therapy is usually monitored by laboratory analysis of the patient’s blood, which healthcare providers use to determine the appropriate dosage of medication,” said study author Thomas Decker Christensen, MD, PhD, of Aarhus University Hospital in Denmark.
“We believe that allowing patients to have more control over their own treatment can improve the standard of care in this patient group.”
Dr Christensen and his colleagues evaluated patients treated at 2 hospitals in Denmark between 1996 and 2012. There were 615 patients with mechanical heart valves who self-managed VKA treatment and 3075 control patients (a 1:5 ratio) who received standard VKA management following valve surgery.
Patients were required to attend an educational program consisting of a minimum of 3 lessons to be eligible for self-management. Over a period of 27 weeks following the educational training, patients gradually became self-managed.
“Once patients were approved for self-management, they were allowed to continue as such but could contact the treatment center with questions or concerns,” Dr Christensen noted.
“Patients were provided with a portable coagulometer, which they used to analyze medication levels and monitor dosage. Blood levels were measured and tracked weekly.”
Patients compared their own measurements against the International Normalized Ratio and adjusted their dosage levels according to what they had been taught in the educational training.
Bleeding and VTE
Over 5 years of follow-up, the risk of venous thromboembolism (VTE) and major bleeding was similar between self-managed patients and patients on standard management.
The rate of thromboembolic events requiring hospitalization was 1.6 events per 100 patient-years in the self-management group and 2.0 per 100 patient-years in the standard group. At 1 year of follow-up, the rates were 2.0 and 2.1, respectively.
When the researchers adjusted for potential confounders, there was a statistically nonsignificant higher risk of VTE among self-managed patients after 1 year (hazard ratio [HR]=1.29) and a statistically nonsignificant lower risk after 5 years (HR=0.91).
The rate of major bleeding was about 1.0 per 100 patient-years in the self-management group at both 1 year and 5 years. The corresponding rates in the standard management group were 1.9 at 1 year and 1.4 at 5 years.
When the researchers adjusted for potential confounders, there was a statistically nonsignificant lower risk of major bleeding in the self-management group at 1 year (HR=0.54) and at 5 years (HR=0.83).
Mortality
After 5 years, the rate of all-cause mortality was 1.1 per 100 patient-years in the self-management group and 2.5 in the standard management group. The adjusted HR was 0.49 in favor of the self-management group.
A landmark analysis restricting the survival analysis to patients alive 1 year after study inclusion resulted in an adjusted HR of 0.57.
“There are several reasons that patients who self-manage treatment have better outcomes than those who follow standard management,” Dr Christensen explained.
“Self-management patients receive more detailed information about oral anticoagulation therapy. They also learn more about the influence that diet, infectious diseases, alcohol, and other drug interactions can have on their treatment than do patients receiving standard management.”
He added that he hopes this study will have an impact on the management and care of heart valve patients.
“We believe that the majority of patients who have a mechanical heart valve inserted during surgery should be able to manage their oral anticoagulant therapy and recommend this as the standard treatment approach for these patients,” he concluded.
Photo courtesy of NIGMS
Allowing patients to self-manage anticoagulant therapy after heart valve surgery may lower the risk of death, according to a study published in the Annals of Thoracic Surgery.
Some of the patients studied were required to complete an educational program before they could manage their own treatment with a vitamin K antagonist (VKA).
For the remaining patients, VKA treatment was managed by a healthcare provider.
At 5 years, patients who managed their own treatment had a lower risk of death than patients on standard management.
“Oral anticoagulation therapy is usually monitored by laboratory analysis of the patient’s blood, which healthcare providers use to determine the appropriate dosage of medication,” said study author Thomas Decker Christensen, MD, PhD, of Aarhus University Hospital in Denmark.
“We believe that allowing patients to have more control over their own treatment can improve the standard of care in this patient group.”
Dr Christensen and his colleagues evaluated patients treated at 2 hospitals in Denmark between 1996 and 2012. There were 615 patients with mechanical heart valves who self-managed VKA treatment and 3075 control patients (a 1:5 ratio) who received standard VKA management following valve surgery.
Patients were required to attend an educational program consisting of a minimum of 3 lessons to be eligible for self-management. Over a period of 27 weeks following the educational training, patients gradually became self-managed.
“Once patients were approved for self-management, they were allowed to continue as such but could contact the treatment center with questions or concerns,” Dr Christensen noted.
“Patients were provided with a portable coagulometer, which they used to analyze medication levels and monitor dosage. Blood levels were measured and tracked weekly.”
Patients compared their own measurements against the International Normalized Ratio and adjusted their dosage levels according to what they had been taught in the educational training.
Bleeding and VTE
Over 5 years of follow-up, the risk of venous thromboembolism (VTE) and major bleeding was similar between self-managed patients and patients on standard management.
The rate of thromboembolic events requiring hospitalization was 1.6 events per 100 patient-years in the self-management group and 2.0 per 100 patient-years in the standard group. At 1 year of follow-up, the rates were 2.0 and 2.1, respectively.
When the researchers adjusted for potential confounders, there was a statistically nonsignificant higher risk of VTE among self-managed patients after 1 year (hazard ratio [HR]=1.29) and a statistically nonsignificant lower risk after 5 years (HR=0.91).
The rate of major bleeding was about 1.0 per 100 patient-years in the self-management group at both 1 year and 5 years. The corresponding rates in the standard management group were 1.9 at 1 year and 1.4 at 5 years.
When the researchers adjusted for potential confounders, there was a statistically nonsignificant lower risk of major bleeding in the self-management group at 1 year (HR=0.54) and at 5 years (HR=0.83).
Mortality
After 5 years, the rate of all-cause mortality was 1.1 per 100 patient-years in the self-management group and 2.5 in the standard management group. The adjusted HR was 0.49 in favor of the self-management group.
A landmark analysis restricting the survival analysis to patients alive 1 year after study inclusion resulted in an adjusted HR of 0.57.
“There are several reasons that patients who self-manage treatment have better outcomes than those who follow standard management,” Dr Christensen explained.
“Self-management patients receive more detailed information about oral anticoagulation therapy. They also learn more about the influence that diet, infectious diseases, alcohol, and other drug interactions can have on their treatment than do patients receiving standard management.”
He added that he hopes this study will have an impact on the management and care of heart valve patients.
“We believe that the majority of patients who have a mechanical heart valve inserted during surgery should be able to manage their oral anticoagulant therapy and recommend this as the standard treatment approach for these patients,” he concluded.
EC approves drug for acquired hemophilia A
Photo courtesy of
Baxter International Inc.
The European Commission (EC) has approved a recombinant porcine factor VIII (FVIII) product, Obizur, to treat bleeding episodes in adults with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Europe.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
The EC’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Twenty-eight patients were evaluated for efficacy, as researchers determined that one of the patients did not actually have acquired hemophilia A.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is approved for use in the US and Canada as well as the European Union. It is under regulatory review in Switzerland, Australia, and Colombia.
Photo courtesy of
Baxter International Inc.
The European Commission (EC) has approved a recombinant porcine factor VIII (FVIII) product, Obizur, to treat bleeding episodes in adults with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Europe.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
The EC’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Twenty-eight patients were evaluated for efficacy, as researchers determined that one of the patients did not actually have acquired hemophilia A.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is approved for use in the US and Canada as well as the European Union. It is under regulatory review in Switzerland, Australia, and Colombia.
Photo courtesy of
Baxter International Inc.
The European Commission (EC) has approved a recombinant porcine factor VIII (FVIII) product, Obizur, to treat bleeding episodes in adults with acquired hemophilia A caused by autoantibodies to FVIII.
Obizur is the first recombinant porcine treatment to be made available for acquired hemophilia A in Europe.
It is specifically designed so physicians can monitor treatment response by measuring FVIII activity levels in addition to making clinical assessments.
The EC’s approval is based on a phase 2/3 trial in which patients with acquired hemophilia A received Obizur as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled in this trial and evaluated for safety. Twenty-eight patients were evaluated for efficacy, as researchers determined that one of the patients did not actually have acquired hemophilia A.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The drug is approved for use in the US and Canada as well as the European Union. It is under regulatory review in Switzerland, Australia, and Colombia.
Role of cyclins in malaria parasite development
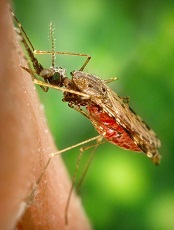
Photo by James Gathany
Researchers say they have characterized the cyclin protein family in the rodent malaria parasite Plasmodium berghei.
The team found there are only 3 cyclins in this parasite, but one of these, the single P-type cyclin CYC3, plays a “vital role” in parasite development in the mosquito.
The researchers believe this work, published in PLoS Pathogens, could pave the way to a better understanding of malaria parasites and lead to potential new treatments.
“This first functional study of cyclin in the malaria parasite and its consequences in parasite development within pathogen-carrying mosquitoes will definitely further our understanding of parasite cell division, which I hope will lead to the elimination of this disease in the future,” said study author Magali Roques, PhD, of the University of Nottingham in the UK.
Photo by James Gathany
Researchers say they have characterized the cyclin protein family in the rodent malaria parasite Plasmodium berghei.
The team found there are only 3 cyclins in this parasite, but one of these, the single P-type cyclin CYC3, plays a “vital role” in parasite development in the mosquito.
The researchers believe this work, published in PLoS Pathogens, could pave the way to a better understanding of malaria parasites and lead to potential new treatments.
“This first functional study of cyclin in the malaria parasite and its consequences in parasite development within pathogen-carrying mosquitoes will definitely further our understanding of parasite cell division, which I hope will lead to the elimination of this disease in the future,” said study author Magali Roques, PhD, of the University of Nottingham in the UK.
Photo by James Gathany
Researchers say they have characterized the cyclin protein family in the rodent malaria parasite Plasmodium berghei.
The team found there are only 3 cyclins in this parasite, but one of these, the single P-type cyclin CYC3, plays a “vital role” in parasite development in the mosquito.
The researchers believe this work, published in PLoS Pathogens, could pave the way to a better understanding of malaria parasites and lead to potential new treatments.
“This first functional study of cyclin in the malaria parasite and its consequences in parasite development within pathogen-carrying mosquitoes will definitely further our understanding of parasite cell division, which I hope will lead to the elimination of this disease in the future,” said study author Magali Roques, PhD, of the University of Nottingham in the UK.