User login
FDA warns of anticoagulant/antidepressant mix-up

Photo courtesy of AstraZeneca
The US Food and Drug Administration (FDA) has announced that confusion between the anticoagulant Brilinta (ticagrelor) and the antidepressant Brintellix (vortioxetine) is resulting in the wrong medication being prescribed or dispensed.
The FDA found the main reason for the confusion is the similarity of the drugs’ brand names.
None of the reports the FDA received indicate that a patient ingested the wrong medication. However, the FDA continues to receive reports of prescribing and dispensing errors.
As of June 2015, the agency has received 50 reports of name confusion between Brintellix and Brilinta. The FDA confirmed that the wrong medication was dispensed to a patient in 12 of the cases and may have been dispensed in 3 additional cases.
About the medications
Brilinta is a P2Y12 platelet inhibitor approved for use in patients with acute coronary syndrome to reduce the rate of thrombotic cardiovascular events. The drug comes in the form of a round, yellow tablet with a “90” above a “T” stamped on one side.
Brintellix is a selective serotonin reuptake inhibitor used to treat major depressive disorder in adults. The medication comes in the form of a tear-shaped tablet stamped with “TL” on one side of the tablet and a number that indicates the tablet strength on the other side. It varies in color depending upon the strength prescribed.
About the error reports
As of June 2015, the FDA has received 50 medication error reports describing brand name confusion with Brintellix (vortioxetine) and Brilinta (ticagrelor). In most cases, Brintellix was mistaken as Brilinta.
Some of the contributing factors to the name confusion included the following:
- Both brand names begin with the same 3 letters
- Both brand names are presented when selecting medications in a computerized physician order entry (CPOE) system
- The pharmacist was not familiar with the new medication Brintellix and so dispensed Brilinta
- The brand names look and sound similar.
Of these 50 reports, the wrong medication was actually dispensed in 12 cases and was possibly dispensed in 3 additional cases but could not be confirmed based on the case narrative information. None of the reports indicated a patient had ingested the wrong medication.
However, in one case, Brintellix was misinterpreted as Brilinta, and the pharmacist did not dispense any medication because the patient had a contraindication to antiplatelet therapy. As a result, the patient went untreated for the psychiatric indication for an unreported period.
In the 12 cases where a wrong medication was actually dispensed, the reports showed that, in 6 cases, the error occurred when prescribing the medication.
Five of these prescribing errors occurred during CPOE. Some CPOE systems auto-populate or present a drop-down menu after the first 3 letters are typed, at which point a prescriber can select the wrong medication.
In the other 6 cases, the error occurred during dispensing of the medication.
FDA recommendations
The FDA is recommending that healthcare professionals reduce the risk of name confusion when prescribing these medications by including the generic names, indications for use, correct dosage, and directions for use.
Healthcare professionals should also ensure that patients understand what their medication is used to treat and encourage patients and their caregivers to read the Medication Guides provided with their Brintellix and Brilinta prescriptions.
Healthcare professionals and patients can report name confusion and medication errors involving Brintellix and Brilinta to the FDA MedWatch Program. ![]()
Photo courtesy of AstraZeneca
The US Food and Drug Administration (FDA) has announced that confusion between the anticoagulant Brilinta (ticagrelor) and the antidepressant Brintellix (vortioxetine) is resulting in the wrong medication being prescribed or dispensed.
The FDA found the main reason for the confusion is the similarity of the drugs’ brand names.
None of the reports the FDA received indicate that a patient ingested the wrong medication. However, the FDA continues to receive reports of prescribing and dispensing errors.
As of June 2015, the agency has received 50 reports of name confusion between Brintellix and Brilinta. The FDA confirmed that the wrong medication was dispensed to a patient in 12 of the cases and may have been dispensed in 3 additional cases.
About the medications
Brilinta is a P2Y12 platelet inhibitor approved for use in patients with acute coronary syndrome to reduce the rate of thrombotic cardiovascular events. The drug comes in the form of a round, yellow tablet with a “90” above a “T” stamped on one side.
Brintellix is a selective serotonin reuptake inhibitor used to treat major depressive disorder in adults. The medication comes in the form of a tear-shaped tablet stamped with “TL” on one side of the tablet and a number that indicates the tablet strength on the other side. It varies in color depending upon the strength prescribed.
About the error reports
As of June 2015, the FDA has received 50 medication error reports describing brand name confusion with Brintellix (vortioxetine) and Brilinta (ticagrelor). In most cases, Brintellix was mistaken as Brilinta.
Some of the contributing factors to the name confusion included the following:
- Both brand names begin with the same 3 letters
- Both brand names are presented when selecting medications in a computerized physician order entry (CPOE) system
- The pharmacist was not familiar with the new medication Brintellix and so dispensed Brilinta
- The brand names look and sound similar.
Of these 50 reports, the wrong medication was actually dispensed in 12 cases and was possibly dispensed in 3 additional cases but could not be confirmed based on the case narrative information. None of the reports indicated a patient had ingested the wrong medication.
However, in one case, Brintellix was misinterpreted as Brilinta, and the pharmacist did not dispense any medication because the patient had a contraindication to antiplatelet therapy. As a result, the patient went untreated for the psychiatric indication for an unreported period.
In the 12 cases where a wrong medication was actually dispensed, the reports showed that, in 6 cases, the error occurred when prescribing the medication.
Five of these prescribing errors occurred during CPOE. Some CPOE systems auto-populate or present a drop-down menu after the first 3 letters are typed, at which point a prescriber can select the wrong medication.
In the other 6 cases, the error occurred during dispensing of the medication.
FDA recommendations
The FDA is recommending that healthcare professionals reduce the risk of name confusion when prescribing these medications by including the generic names, indications for use, correct dosage, and directions for use.
Healthcare professionals should also ensure that patients understand what their medication is used to treat and encourage patients and their caregivers to read the Medication Guides provided with their Brintellix and Brilinta prescriptions.
Healthcare professionals and patients can report name confusion and medication errors involving Brintellix and Brilinta to the FDA MedWatch Program. ![]()
Photo courtesy of AstraZeneca
The US Food and Drug Administration (FDA) has announced that confusion between the anticoagulant Brilinta (ticagrelor) and the antidepressant Brintellix (vortioxetine) is resulting in the wrong medication being prescribed or dispensed.
The FDA found the main reason for the confusion is the similarity of the drugs’ brand names.
None of the reports the FDA received indicate that a patient ingested the wrong medication. However, the FDA continues to receive reports of prescribing and dispensing errors.
As of June 2015, the agency has received 50 reports of name confusion between Brintellix and Brilinta. The FDA confirmed that the wrong medication was dispensed to a patient in 12 of the cases and may have been dispensed in 3 additional cases.
About the medications
Brilinta is a P2Y12 platelet inhibitor approved for use in patients with acute coronary syndrome to reduce the rate of thrombotic cardiovascular events. The drug comes in the form of a round, yellow tablet with a “90” above a “T” stamped on one side.
Brintellix is a selective serotonin reuptake inhibitor used to treat major depressive disorder in adults. The medication comes in the form of a tear-shaped tablet stamped with “TL” on one side of the tablet and a number that indicates the tablet strength on the other side. It varies in color depending upon the strength prescribed.
About the error reports
As of June 2015, the FDA has received 50 medication error reports describing brand name confusion with Brintellix (vortioxetine) and Brilinta (ticagrelor). In most cases, Brintellix was mistaken as Brilinta.
Some of the contributing factors to the name confusion included the following:
- Both brand names begin with the same 3 letters
- Both brand names are presented when selecting medications in a computerized physician order entry (CPOE) system
- The pharmacist was not familiar with the new medication Brintellix and so dispensed Brilinta
- The brand names look and sound similar.
Of these 50 reports, the wrong medication was actually dispensed in 12 cases and was possibly dispensed in 3 additional cases but could not be confirmed based on the case narrative information. None of the reports indicated a patient had ingested the wrong medication.
However, in one case, Brintellix was misinterpreted as Brilinta, and the pharmacist did not dispense any medication because the patient had a contraindication to antiplatelet therapy. As a result, the patient went untreated for the psychiatric indication for an unreported period.
In the 12 cases where a wrong medication was actually dispensed, the reports showed that, in 6 cases, the error occurred when prescribing the medication.
Five of these prescribing errors occurred during CPOE. Some CPOE systems auto-populate or present a drop-down menu after the first 3 letters are typed, at which point a prescriber can select the wrong medication.
In the other 6 cases, the error occurred during dispensing of the medication.
FDA recommendations
The FDA is recommending that healthcare professionals reduce the risk of name confusion when prescribing these medications by including the generic names, indications for use, correct dosage, and directions for use.
Healthcare professionals should also ensure that patients understand what their medication is used to treat and encourage patients and their caregivers to read the Medication Guides provided with their Brintellix and Brilinta prescriptions.
Healthcare professionals and patients can report name confusion and medication errors involving Brintellix and Brilinta to the FDA MedWatch Program. ![]()
Companies ‘underinvest’ in long-term cancer research

for a clinical trial
Photo by Esther Dyson
Pharmaceutical companies “underinvest” in long-term research to develop new anticancer drugs, according to a study published in American Economic Review.
Investigators used historical data to show that companies are more likely to develop drugs for late-stage cancers than early stage cancers or cancer prevention, and this is likely because late-stage cancer drugs can be brought to market faster.
The team found that late-stage drugs extend patient survival for shorter periods so that clinical trials for these drugs get wrapped up more quickly. This, in turn, gives drug manufacturers more time to control patented drugs in the marketplace.
“There is a pattern where we get more investment in drugs that take a short time to complete and less investment in drugs that take a longer time to complete,” said study author Heidi Williams, PhD, of the Massachusetts Institute of Technology in Cambridge.
To conduct this study, Dr Williams and her colleagues analyzed 4 decades of clinical trial data from a variety of sources, including the National Cancer Institute and the US Food and Drug Administration. The study encompassed more than 200 subcategories of cancers detected at different stages of development.
In analyzing the data, the investigators divided research and development (R&D) into 2 stages: invention (developing the basic idea for a product to the point where it is patentable) and commercialization (bringing an invented product to market).
They defined the “commercialization lag” of an R&D project as the amount of time between invention and commercialization.
The data showed that patient groups with longer commercialization lags (as proxied by longer survival times) tended to have lower levels of R&D investment than groups with shorter commercialization lags (and survival times).
The investigators also found that when surrogate endpoints (endpoints other than survival) were allowed, there were more trials and money poured into research. This supports the idea that companies are more likely to invest in drugs that will have shorter trials and take less time to develop.
Dr Williams and her colleagues used the surrogate endpoint variation to estimate improvements in cancer survival rates that would have been observed if commercialization lags were reduced. The team estimated that, among US cancer patients diagnosed in 2003, longer commercialization lags resulted in around 890,000 lost life-years.
The investigators noted that commercialization lags reduce both public and private R&D investments, but they found the commercialization lag-R&D correlation is significantly more negative for privately financed trials than publicly financed trials.
The team said that, due to either excessive discounting or the fixed patent term, private incentives decline more rapidly than public incentives, which is what gives rise to the distortion.
Based on their findings, Dr Williams and her colleagues devised 3 new policy approaches that could potentially spark the development of more drugs for early stage cancers or cancer prevention.
The first is expanded use of surrogate endpoints or more research to determine if wider use of surrogate endpoints is valid.
A second possible policy change is more public funding of R&D for anticancer drugs, since such funding is free of short-term, private-sector shareholder pressure to produce returns.
A third potential new policy would be changing the terms of drug patents, which typically run from the time of patent filing to when the drug hits the market. ![]()
for a clinical trial
Photo by Esther Dyson
Pharmaceutical companies “underinvest” in long-term research to develop new anticancer drugs, according to a study published in American Economic Review.
Investigators used historical data to show that companies are more likely to develop drugs for late-stage cancers than early stage cancers or cancer prevention, and this is likely because late-stage cancer drugs can be brought to market faster.
The team found that late-stage drugs extend patient survival for shorter periods so that clinical trials for these drugs get wrapped up more quickly. This, in turn, gives drug manufacturers more time to control patented drugs in the marketplace.
“There is a pattern where we get more investment in drugs that take a short time to complete and less investment in drugs that take a longer time to complete,” said study author Heidi Williams, PhD, of the Massachusetts Institute of Technology in Cambridge.
To conduct this study, Dr Williams and her colleagues analyzed 4 decades of clinical trial data from a variety of sources, including the National Cancer Institute and the US Food and Drug Administration. The study encompassed more than 200 subcategories of cancers detected at different stages of development.
In analyzing the data, the investigators divided research and development (R&D) into 2 stages: invention (developing the basic idea for a product to the point where it is patentable) and commercialization (bringing an invented product to market).
They defined the “commercialization lag” of an R&D project as the amount of time between invention and commercialization.
The data showed that patient groups with longer commercialization lags (as proxied by longer survival times) tended to have lower levels of R&D investment than groups with shorter commercialization lags (and survival times).
The investigators also found that when surrogate endpoints (endpoints other than survival) were allowed, there were more trials and money poured into research. This supports the idea that companies are more likely to invest in drugs that will have shorter trials and take less time to develop.
Dr Williams and her colleagues used the surrogate endpoint variation to estimate improvements in cancer survival rates that would have been observed if commercialization lags were reduced. The team estimated that, among US cancer patients diagnosed in 2003, longer commercialization lags resulted in around 890,000 lost life-years.
The investigators noted that commercialization lags reduce both public and private R&D investments, but they found the commercialization lag-R&D correlation is significantly more negative for privately financed trials than publicly financed trials.
The team said that, due to either excessive discounting or the fixed patent term, private incentives decline more rapidly than public incentives, which is what gives rise to the distortion.
Based on their findings, Dr Williams and her colleagues devised 3 new policy approaches that could potentially spark the development of more drugs for early stage cancers or cancer prevention.
The first is expanded use of surrogate endpoints or more research to determine if wider use of surrogate endpoints is valid.
A second possible policy change is more public funding of R&D for anticancer drugs, since such funding is free of short-term, private-sector shareholder pressure to produce returns.
A third potential new policy would be changing the terms of drug patents, which typically run from the time of patent filing to when the drug hits the market. ![]()
for a clinical trial
Photo by Esther Dyson
Pharmaceutical companies “underinvest” in long-term research to develop new anticancer drugs, according to a study published in American Economic Review.
Investigators used historical data to show that companies are more likely to develop drugs for late-stage cancers than early stage cancers or cancer prevention, and this is likely because late-stage cancer drugs can be brought to market faster.
The team found that late-stage drugs extend patient survival for shorter periods so that clinical trials for these drugs get wrapped up more quickly. This, in turn, gives drug manufacturers more time to control patented drugs in the marketplace.
“There is a pattern where we get more investment in drugs that take a short time to complete and less investment in drugs that take a longer time to complete,” said study author Heidi Williams, PhD, of the Massachusetts Institute of Technology in Cambridge.
To conduct this study, Dr Williams and her colleagues analyzed 4 decades of clinical trial data from a variety of sources, including the National Cancer Institute and the US Food and Drug Administration. The study encompassed more than 200 subcategories of cancers detected at different stages of development.
In analyzing the data, the investigators divided research and development (R&D) into 2 stages: invention (developing the basic idea for a product to the point where it is patentable) and commercialization (bringing an invented product to market).
They defined the “commercialization lag” of an R&D project as the amount of time between invention and commercialization.
The data showed that patient groups with longer commercialization lags (as proxied by longer survival times) tended to have lower levels of R&D investment than groups with shorter commercialization lags (and survival times).
The investigators also found that when surrogate endpoints (endpoints other than survival) were allowed, there were more trials and money poured into research. This supports the idea that companies are more likely to invest in drugs that will have shorter trials and take less time to develop.
Dr Williams and her colleagues used the surrogate endpoint variation to estimate improvements in cancer survival rates that would have been observed if commercialization lags were reduced. The team estimated that, among US cancer patients diagnosed in 2003, longer commercialization lags resulted in around 890,000 lost life-years.
The investigators noted that commercialization lags reduce both public and private R&D investments, but they found the commercialization lag-R&D correlation is significantly more negative for privately financed trials than publicly financed trials.
The team said that, due to either excessive discounting or the fixed patent term, private incentives decline more rapidly than public incentives, which is what gives rise to the distortion.
Based on their findings, Dr Williams and her colleagues devised 3 new policy approaches that could potentially spark the development of more drugs for early stage cancers or cancer prevention.
The first is expanded use of surrogate endpoints or more research to determine if wider use of surrogate endpoints is valid.
A second possible policy change is more public funding of R&D for anticancer drugs, since such funding is free of short-term, private-sector shareholder pressure to produce returns.
A third potential new policy would be changing the terms of drug patents, which typically run from the time of patent filing to when the drug hits the market. ![]()
Health Canada grants drug conditional approval for MCL

Photo courtesy of
Janssen Biotech, Inc.
Health Canada has granted conditional approval for the BTK inhibitor ibrutinib (Imbruvica) to treat patients with relapsed or refractory mantle cell lymphoma (MCL).
This approval was based on data from a phase 2 trial in which ibrutinib conferred an overall response rate of 68% in patients with relapsed/refractory MCL.
For ibrutinib to gain full approval, Health Canada must receive additional data confirming the drug provides a clinical benefit.
Ibrutinib was first approved in Canada in November 2014 for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL with 17p deletion. For this use, ibrutinib was issued marketing authorization without conditions.
Now, Health Canada has issued ibrutinib conditional marketing authorization for the treatment of relapsed/refractory MCL. This decision was based on data from the phase 2 PCYC-1104 trial, which was presented at ASH 2012 and published in NEJM in August 2013.
The study included 111 MCL patients who had received at least one prior therapy. The primary endpoint of the study was overall response rate according to the revised International Working Group criteria for non-Hodgkin lymphoma.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With a median follow-up of 15.3 months, the median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
Eight patients (7%) had an adverse event that led to treatment discontinuation.
Sixteen patients (14%) died during the trial, 12 due to disease progression and 4 due to an adverse event. Two patients died of pneumonia, 1 from sepsis, and 1 from a cardiac arrest that was not considered drug-related.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
Photo courtesy of
Janssen Biotech, Inc.
Health Canada has granted conditional approval for the BTK inhibitor ibrutinib (Imbruvica) to treat patients with relapsed or refractory mantle cell lymphoma (MCL).
This approval was based on data from a phase 2 trial in which ibrutinib conferred an overall response rate of 68% in patients with relapsed/refractory MCL.
For ibrutinib to gain full approval, Health Canada must receive additional data confirming the drug provides a clinical benefit.
Ibrutinib was first approved in Canada in November 2014 for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL with 17p deletion. For this use, ibrutinib was issued marketing authorization without conditions.
Now, Health Canada has issued ibrutinib conditional marketing authorization for the treatment of relapsed/refractory MCL. This decision was based on data from the phase 2 PCYC-1104 trial, which was presented at ASH 2012 and published in NEJM in August 2013.
The study included 111 MCL patients who had received at least one prior therapy. The primary endpoint of the study was overall response rate according to the revised International Working Group criteria for non-Hodgkin lymphoma.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With a median follow-up of 15.3 months, the median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
Eight patients (7%) had an adverse event that led to treatment discontinuation.
Sixteen patients (14%) died during the trial, 12 due to disease progression and 4 due to an adverse event. Two patients died of pneumonia, 1 from sepsis, and 1 from a cardiac arrest that was not considered drug-related.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
Photo courtesy of
Janssen Biotech, Inc.
Health Canada has granted conditional approval for the BTK inhibitor ibrutinib (Imbruvica) to treat patients with relapsed or refractory mantle cell lymphoma (MCL).
This approval was based on data from a phase 2 trial in which ibrutinib conferred an overall response rate of 68% in patients with relapsed/refractory MCL.
For ibrutinib to gain full approval, Health Canada must receive additional data confirming the drug provides a clinical benefit.
Ibrutinib was first approved in Canada in November 2014 for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL with 17p deletion. For this use, ibrutinib was issued marketing authorization without conditions.
Now, Health Canada has issued ibrutinib conditional marketing authorization for the treatment of relapsed/refractory MCL. This decision was based on data from the phase 2 PCYC-1104 trial, which was presented at ASH 2012 and published in NEJM in August 2013.
The study included 111 MCL patients who had received at least one prior therapy. The primary endpoint of the study was overall response rate according to the revised International Working Group criteria for non-Hodgkin lymphoma.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With a median follow-up of 15.3 months, the median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
Eight patients (7%) had an adverse event that led to treatment discontinuation.
Sixteen patients (14%) died during the trial, 12 due to disease progression and 4 due to an adverse event. Two patients died of pneumonia, 1 from sepsis, and 1 from a cardiac arrest that was not considered drug-related.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
VTE-related penalties may be unfair

Photo courtesy of the CDC
Imposing financial penalties on hospitals based on the incidence of hospital-acquired venous thromboembolism (VTE) may be unfair, according to researchers.
They argue that pay-for-performance systems should take VTE prevention efforts into account, instead of simply tallying the number of hospital-acquired VTEs.
They say the current system fails to account for VTEs that occur despite appropriate use of preventive therapies.
The researchers expressed this viewpoint and disclosed research supporting it in a letter to JAMA Surgery.
“We have a big problem with current pay-for-performance systems based on ‘numbers-only’ total counts of clots, because even when hospitals do everything they can to prevent venous thromboembolism events, they are still being dinged for patients who develop these clots,” said Elliott R. Haut, MD, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“Our study of patients just at The Johns Hopkins Hospital identifies a need to dramatically re-evaluate the venous thromboembolism outcome and process measures. Nearly half of the venous thromboembolism events identified by the state program in the records we reviewed were not truly preventable because patients received best practice prevention and still developed blood clots.”
Dr Haut and his colleagues reviewed case records for 128 patients treated between July 2010 and June 2011 at The Johns Hopkins Hospital and who developed hospital-acquired VTE. All 128 were flagged by the Maryland Hospital Acquired Conditions pay-for-performance program.
The researchers looked for evidence that all of the VTEs could have been prevented. They found that 36 patients (28%) had nonpreventable, catheter-related upper extremity deep vein thrombosis (DVT), leaving 92 patients (72%) with clots that were potentially preventable.
Of those 92 patients, 45 had a DVT, 43 had a pulmonary embolism (PE), and 4 had a DVT and PE.
Seventy-nine of the 92 patients (86%) were prescribed optimal thromboprophylaxis, yet only 43 (47%) received “defect-free care,” the researchers found.
Of the 49 patients (53%) who received suboptimal care, 13 (27%) were not prescribed risk-appropriate anticoagulants, and 36 (73%) missed at least one dose of appropriately prescribed medication.
Dr Haut noted that a team of physicians, nurses, quality care researchers, and pharmacists at Johns Hopkins has been studying VTE prevention for the past decade.
Team members have implemented programs to monitor patients in need of VTE prophylaxis through the hospital’s electronic health record system, and they conducted special training for nurses and patients to stress the importance of taking every dose of prescribed medication.
“We know we’re not going to get the VTE rate to 0, but my goal is to have every single one of these events—when they happen—occur when the patient receives best-practice, defect-free care,” Dr Haut said.
The current VTE care goal set by agencies like the Joint Commission and the Centers for Medicare and Medicaid Services is that one dose of VTE prophylaxis is given to patients within the first day of hospitalization. But Dr Haut said this is not enough.
“To reduce preventable harm, policymakers need to re-evaluate how they penalize hospitals and improve the measures they use to assess VTE prevention performance,” he said. “In addition, clinicians need to ensure that patients receive all prescribed preventive therapies.” ![]()
Photo courtesy of the CDC
Imposing financial penalties on hospitals based on the incidence of hospital-acquired venous thromboembolism (VTE) may be unfair, according to researchers.
They argue that pay-for-performance systems should take VTE prevention efforts into account, instead of simply tallying the number of hospital-acquired VTEs.
They say the current system fails to account for VTEs that occur despite appropriate use of preventive therapies.
The researchers expressed this viewpoint and disclosed research supporting it in a letter to JAMA Surgery.
“We have a big problem with current pay-for-performance systems based on ‘numbers-only’ total counts of clots, because even when hospitals do everything they can to prevent venous thromboembolism events, they are still being dinged for patients who develop these clots,” said Elliott R. Haut, MD, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“Our study of patients just at The Johns Hopkins Hospital identifies a need to dramatically re-evaluate the venous thromboembolism outcome and process measures. Nearly half of the venous thromboembolism events identified by the state program in the records we reviewed were not truly preventable because patients received best practice prevention and still developed blood clots.”
Dr Haut and his colleagues reviewed case records for 128 patients treated between July 2010 and June 2011 at The Johns Hopkins Hospital and who developed hospital-acquired VTE. All 128 were flagged by the Maryland Hospital Acquired Conditions pay-for-performance program.
The researchers looked for evidence that all of the VTEs could have been prevented. They found that 36 patients (28%) had nonpreventable, catheter-related upper extremity deep vein thrombosis (DVT), leaving 92 patients (72%) with clots that were potentially preventable.
Of those 92 patients, 45 had a DVT, 43 had a pulmonary embolism (PE), and 4 had a DVT and PE.
Seventy-nine of the 92 patients (86%) were prescribed optimal thromboprophylaxis, yet only 43 (47%) received “defect-free care,” the researchers found.
Of the 49 patients (53%) who received suboptimal care, 13 (27%) were not prescribed risk-appropriate anticoagulants, and 36 (73%) missed at least one dose of appropriately prescribed medication.
Dr Haut noted that a team of physicians, nurses, quality care researchers, and pharmacists at Johns Hopkins has been studying VTE prevention for the past decade.
Team members have implemented programs to monitor patients in need of VTE prophylaxis through the hospital’s electronic health record system, and they conducted special training for nurses and patients to stress the importance of taking every dose of prescribed medication.
“We know we’re not going to get the VTE rate to 0, but my goal is to have every single one of these events—when they happen—occur when the patient receives best-practice, defect-free care,” Dr Haut said.
The current VTE care goal set by agencies like the Joint Commission and the Centers for Medicare and Medicaid Services is that one dose of VTE prophylaxis is given to patients within the first day of hospitalization. But Dr Haut said this is not enough.
“To reduce preventable harm, policymakers need to re-evaluate how they penalize hospitals and improve the measures they use to assess VTE prevention performance,” he said. “In addition, clinicians need to ensure that patients receive all prescribed preventive therapies.” ![]()
Photo courtesy of the CDC
Imposing financial penalties on hospitals based on the incidence of hospital-acquired venous thromboembolism (VTE) may be unfair, according to researchers.
They argue that pay-for-performance systems should take VTE prevention efforts into account, instead of simply tallying the number of hospital-acquired VTEs.
They say the current system fails to account for VTEs that occur despite appropriate use of preventive therapies.
The researchers expressed this viewpoint and disclosed research supporting it in a letter to JAMA Surgery.
“We have a big problem with current pay-for-performance systems based on ‘numbers-only’ total counts of clots, because even when hospitals do everything they can to prevent venous thromboembolism events, they are still being dinged for patients who develop these clots,” said Elliott R. Haut, MD, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
“Our study of patients just at The Johns Hopkins Hospital identifies a need to dramatically re-evaluate the venous thromboembolism outcome and process measures. Nearly half of the venous thromboembolism events identified by the state program in the records we reviewed were not truly preventable because patients received best practice prevention and still developed blood clots.”
Dr Haut and his colleagues reviewed case records for 128 patients treated between July 2010 and June 2011 at The Johns Hopkins Hospital and who developed hospital-acquired VTE. All 128 were flagged by the Maryland Hospital Acquired Conditions pay-for-performance program.
The researchers looked for evidence that all of the VTEs could have been prevented. They found that 36 patients (28%) had nonpreventable, catheter-related upper extremity deep vein thrombosis (DVT), leaving 92 patients (72%) with clots that were potentially preventable.
Of those 92 patients, 45 had a DVT, 43 had a pulmonary embolism (PE), and 4 had a DVT and PE.
Seventy-nine of the 92 patients (86%) were prescribed optimal thromboprophylaxis, yet only 43 (47%) received “defect-free care,” the researchers found.
Of the 49 patients (53%) who received suboptimal care, 13 (27%) were not prescribed risk-appropriate anticoagulants, and 36 (73%) missed at least one dose of appropriately prescribed medication.
Dr Haut noted that a team of physicians, nurses, quality care researchers, and pharmacists at Johns Hopkins has been studying VTE prevention for the past decade.
Team members have implemented programs to monitor patients in need of VTE prophylaxis through the hospital’s electronic health record system, and they conducted special training for nurses and patients to stress the importance of taking every dose of prescribed medication.
“We know we’re not going to get the VTE rate to 0, but my goal is to have every single one of these events—when they happen—occur when the patient receives best-practice, defect-free care,” Dr Haut said.
The current VTE care goal set by agencies like the Joint Commission and the Centers for Medicare and Medicaid Services is that one dose of VTE prophylaxis is given to patients within the first day of hospitalization. But Dr Haut said this is not enough.
“To reduce preventable harm, policymakers need to re-evaluate how they penalize hospitals and improve the measures they use to assess VTE prevention performance,” he said. “In addition, clinicians need to ensure that patients receive all prescribed preventive therapies.” ![]()
Tool identifies optimal TKI for cancers

Photo courtesy of the
University of Colorado
Researchers say they have developed a tool that allows us to determine which tyrosine kinase inhibitor (TKI) will be most effective against a certain type of cancer.
The tool, known as the Kinase Addiction Ranker (KAR), predicts the genetic abnormalities that are driving the cancer in any population of cells and chooses the best TKI or combination of TKIs to target these abnormalities.
The researchers described the tool in Bioinformatics.
“A lot of [TKIs] inhibit a lot more than what they’re supposed to inhibit,” said study author Aik Choon Tan, PhD, of the University of Colorado Anschutz Medical Campus in Aurora.
“Maybe drug A was designed to inhibit kinase B, but it also inhibits kinase C and D as well. Our approach centers on exploiting the promiscuity of these drugs, the ‘drug spillover.’”
For each TKI, there is a signature describing the kinases each drug fully or partially inhibits. Dr Tan and his colleagues combined these kinase inhibition signatures with the results of high-throughput screening. They used the Genomics of Drug Sensitivity in Cancer database to determine which TKIs have already proven active against which cancer cell lines.
The result is KAR, which does 2 things. For any cancer cell line, the program ranks the kinases that are most important to the growth of the disease. Then, the program recommends the combination of existing TKIs that is likely to do the most good against the implicated kinases.
Dr Tan and his colleagues tested KAR using samples from 151 leukemia patients and found that, among the kinases analyzed, FLT3 had the highest variance in sensitivity to TKIs.
But EPHA5, EPHA3, and BTK were the kinases most commonly associated with drug sensitivity. They had significant associations in 72%, 58%, and 54% of the patient samples, respectively.
The researchers said the frequency of BTK dependence they observed is interesting given the fact that the BTK inhibitor ibrutinib produced favorable results in a phase 1b/2 trial of patients with chronic lymphocytic leukemia (CLL). The progression-free survival rate at 26 months was 75% in that trial.
Dr Tan and his colleagues said this was consistent with their findings, which showed that 70% of CLL patient data had a significant association between BTK inhibition and drug sensitivity.
The researchers also found that KAR could predict TKI sensitivity in 21 lung cancer cell lines. In addition, the tool was able to recommend a combination of TKIs that hindered proliferation in the lung cancer cell line H1581. KAR suggested ponatinib and the experimental anticancer agent AZD8055, and experiments showed that these drugs synergistically reduced proliferation in H1581.
KAR is available for download on the Tan lab’s website. ![]()
Photo courtesy of the
University of Colorado
Researchers say they have developed a tool that allows us to determine which tyrosine kinase inhibitor (TKI) will be most effective against a certain type of cancer.
The tool, known as the Kinase Addiction Ranker (KAR), predicts the genetic abnormalities that are driving the cancer in any population of cells and chooses the best TKI or combination of TKIs to target these abnormalities.
The researchers described the tool in Bioinformatics.
“A lot of [TKIs] inhibit a lot more than what they’re supposed to inhibit,” said study author Aik Choon Tan, PhD, of the University of Colorado Anschutz Medical Campus in Aurora.
“Maybe drug A was designed to inhibit kinase B, but it also inhibits kinase C and D as well. Our approach centers on exploiting the promiscuity of these drugs, the ‘drug spillover.’”
For each TKI, there is a signature describing the kinases each drug fully or partially inhibits. Dr Tan and his colleagues combined these kinase inhibition signatures with the results of high-throughput screening. They used the Genomics of Drug Sensitivity in Cancer database to determine which TKIs have already proven active against which cancer cell lines.
The result is KAR, which does 2 things. For any cancer cell line, the program ranks the kinases that are most important to the growth of the disease. Then, the program recommends the combination of existing TKIs that is likely to do the most good against the implicated kinases.
Dr Tan and his colleagues tested KAR using samples from 151 leukemia patients and found that, among the kinases analyzed, FLT3 had the highest variance in sensitivity to TKIs.
But EPHA5, EPHA3, and BTK were the kinases most commonly associated with drug sensitivity. They had significant associations in 72%, 58%, and 54% of the patient samples, respectively.
The researchers said the frequency of BTK dependence they observed is interesting given the fact that the BTK inhibitor ibrutinib produced favorable results in a phase 1b/2 trial of patients with chronic lymphocytic leukemia (CLL). The progression-free survival rate at 26 months was 75% in that trial.
Dr Tan and his colleagues said this was consistent with their findings, which showed that 70% of CLL patient data had a significant association between BTK inhibition and drug sensitivity.
The researchers also found that KAR could predict TKI sensitivity in 21 lung cancer cell lines. In addition, the tool was able to recommend a combination of TKIs that hindered proliferation in the lung cancer cell line H1581. KAR suggested ponatinib and the experimental anticancer agent AZD8055, and experiments showed that these drugs synergistically reduced proliferation in H1581.
KAR is available for download on the Tan lab’s website. ![]()
Photo courtesy of the
University of Colorado
Researchers say they have developed a tool that allows us to determine which tyrosine kinase inhibitor (TKI) will be most effective against a certain type of cancer.
The tool, known as the Kinase Addiction Ranker (KAR), predicts the genetic abnormalities that are driving the cancer in any population of cells and chooses the best TKI or combination of TKIs to target these abnormalities.
The researchers described the tool in Bioinformatics.
“A lot of [TKIs] inhibit a lot more than what they’re supposed to inhibit,” said study author Aik Choon Tan, PhD, of the University of Colorado Anschutz Medical Campus in Aurora.
“Maybe drug A was designed to inhibit kinase B, but it also inhibits kinase C and D as well. Our approach centers on exploiting the promiscuity of these drugs, the ‘drug spillover.’”
For each TKI, there is a signature describing the kinases each drug fully or partially inhibits. Dr Tan and his colleagues combined these kinase inhibition signatures with the results of high-throughput screening. They used the Genomics of Drug Sensitivity in Cancer database to determine which TKIs have already proven active against which cancer cell lines.
The result is KAR, which does 2 things. For any cancer cell line, the program ranks the kinases that are most important to the growth of the disease. Then, the program recommends the combination of existing TKIs that is likely to do the most good against the implicated kinases.
Dr Tan and his colleagues tested KAR using samples from 151 leukemia patients and found that, among the kinases analyzed, FLT3 had the highest variance in sensitivity to TKIs.
But EPHA5, EPHA3, and BTK were the kinases most commonly associated with drug sensitivity. They had significant associations in 72%, 58%, and 54% of the patient samples, respectively.
The researchers said the frequency of BTK dependence they observed is interesting given the fact that the BTK inhibitor ibrutinib produced favorable results in a phase 1b/2 trial of patients with chronic lymphocytic leukemia (CLL). The progression-free survival rate at 26 months was 75% in that trial.
Dr Tan and his colleagues said this was consistent with their findings, which showed that 70% of CLL patient data had a significant association between BTK inhibition and drug sensitivity.
The researchers also found that KAR could predict TKI sensitivity in 21 lung cancer cell lines. In addition, the tool was able to recommend a combination of TKIs that hindered proliferation in the lung cancer cell line H1581. KAR suggested ponatinib and the experimental anticancer agent AZD8055, and experiments showed that these drugs synergistically reduced proliferation in H1581.
KAR is available for download on the Tan lab’s website. ![]()
Analysis reveals ‘distinctive biology’ of CTCL
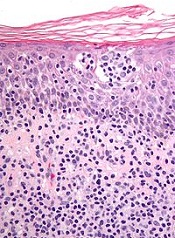
New research suggests cutaneous T-cell lymphoma (CTCL) is driven by a plethora of genetic mutations.
Investigators conducted a genomic analysis of normal and cancer cells from patients with CTCL and identified mutations in 17 genes that are implicated in CTCL pathogenesis.
They also found that somatic copy number variants (SCNVs) driving CTCL outnumbered somatic single-nucleotide variants (SSNVs) by more than 10 to 1.
The team reported these findings in Nature Genetics.
They performed exome and whole-genome DNA sequencing and RNA sequencing on purified CTCL cells and matched normal cells. And they identified genes implicated in CTCL pathogenesis by looking for:
- Genes with recurrent SSNVs altering the same amino acid more often than expected by chance
- Genes with SSNVs previously identified as recurrent mutations in other cancers
- Genes having a significantly increased burden of protein-altering SSNVs
- SCNVs that occurred more often than expected by chance.
This revealed mutations in 17 genes that are implicated in CTCL pathogenesis—TP53, ZEB1, ARID1A, DNMT3A, CDKN2A, FAS, NFKB2, CD28, RHOA, PLCG1, STAT5B, BRAF, ATM, CTCF, TNFAIP3, PRKCQ, and IRF4.
The investigators noted that these are genes involved in T-cell activation, apoptosis, NF-κB signaling, chromatin remodeling, and DNA damage response.
The team also discovered “a striking bias” for SCNVs as drivers of CTCL. They identified 12 statistically significant chromosome-arm SCNVs and 36 significant focal SCNVs.
Collectively, these SCNVs occurred 473 times in the CTCL samples analyzed—a mean of 7.5 focal deletions, 1.6 broad deletions, 1.0 focal amplification, and 1.8 broad amplifications per CTCL.
On the other hand, there were 38 SSNVs in CTCL driver genes—1.0 per tumor.
So, according to these data, SCNVs comprise 92% of all driver mutations in CTCL—a mean of 11.8 pathogenic SCNVs vs 1.0 SSNV per CTCL.
“This cancer has a very distinctive biology,” said Jaehyuk Choi, MD, PhD, of the Yale School of Medicine in New Haven, Connecticut.
And decoding this biology has revealed potential treatment approaches, according to Dr Choi and his colleagues.
For example, the presence of mutations activating the NF-κB pathway suggests NF-κB inhibitors such as bortezomib may have therapeutic potential in CTCL, and the presence of CD28 mutations suggests inhibitors such as abatacept may be effective against the disease. ![]()
New research suggests cutaneous T-cell lymphoma (CTCL) is driven by a plethora of genetic mutations.
Investigators conducted a genomic analysis of normal and cancer cells from patients with CTCL and identified mutations in 17 genes that are implicated in CTCL pathogenesis.
They also found that somatic copy number variants (SCNVs) driving CTCL outnumbered somatic single-nucleotide variants (SSNVs) by more than 10 to 1.
The team reported these findings in Nature Genetics.
They performed exome and whole-genome DNA sequencing and RNA sequencing on purified CTCL cells and matched normal cells. And they identified genes implicated in CTCL pathogenesis by looking for:
- Genes with recurrent SSNVs altering the same amino acid more often than expected by chance
- Genes with SSNVs previously identified as recurrent mutations in other cancers
- Genes having a significantly increased burden of protein-altering SSNVs
- SCNVs that occurred more often than expected by chance.
This revealed mutations in 17 genes that are implicated in CTCL pathogenesis—TP53, ZEB1, ARID1A, DNMT3A, CDKN2A, FAS, NFKB2, CD28, RHOA, PLCG1, STAT5B, BRAF, ATM, CTCF, TNFAIP3, PRKCQ, and IRF4.
The investigators noted that these are genes involved in T-cell activation, apoptosis, NF-κB signaling, chromatin remodeling, and DNA damage response.
The team also discovered “a striking bias” for SCNVs as drivers of CTCL. They identified 12 statistically significant chromosome-arm SCNVs and 36 significant focal SCNVs.
Collectively, these SCNVs occurred 473 times in the CTCL samples analyzed—a mean of 7.5 focal deletions, 1.6 broad deletions, 1.0 focal amplification, and 1.8 broad amplifications per CTCL.
On the other hand, there were 38 SSNVs in CTCL driver genes—1.0 per tumor.
So, according to these data, SCNVs comprise 92% of all driver mutations in CTCL—a mean of 11.8 pathogenic SCNVs vs 1.0 SSNV per CTCL.
“This cancer has a very distinctive biology,” said Jaehyuk Choi, MD, PhD, of the Yale School of Medicine in New Haven, Connecticut.
And decoding this biology has revealed potential treatment approaches, according to Dr Choi and his colleagues.
For example, the presence of mutations activating the NF-κB pathway suggests NF-κB inhibitors such as bortezomib may have therapeutic potential in CTCL, and the presence of CD28 mutations suggests inhibitors such as abatacept may be effective against the disease. ![]()
New research suggests cutaneous T-cell lymphoma (CTCL) is driven by a plethora of genetic mutations.
Investigators conducted a genomic analysis of normal and cancer cells from patients with CTCL and identified mutations in 17 genes that are implicated in CTCL pathogenesis.
They also found that somatic copy number variants (SCNVs) driving CTCL outnumbered somatic single-nucleotide variants (SSNVs) by more than 10 to 1.
The team reported these findings in Nature Genetics.
They performed exome and whole-genome DNA sequencing and RNA sequencing on purified CTCL cells and matched normal cells. And they identified genes implicated in CTCL pathogenesis by looking for:
- Genes with recurrent SSNVs altering the same amino acid more often than expected by chance
- Genes with SSNVs previously identified as recurrent mutations in other cancers
- Genes having a significantly increased burden of protein-altering SSNVs
- SCNVs that occurred more often than expected by chance.
This revealed mutations in 17 genes that are implicated in CTCL pathogenesis—TP53, ZEB1, ARID1A, DNMT3A, CDKN2A, FAS, NFKB2, CD28, RHOA, PLCG1, STAT5B, BRAF, ATM, CTCF, TNFAIP3, PRKCQ, and IRF4.
The investigators noted that these are genes involved in T-cell activation, apoptosis, NF-κB signaling, chromatin remodeling, and DNA damage response.
The team also discovered “a striking bias” for SCNVs as drivers of CTCL. They identified 12 statistically significant chromosome-arm SCNVs and 36 significant focal SCNVs.
Collectively, these SCNVs occurred 473 times in the CTCL samples analyzed—a mean of 7.5 focal deletions, 1.6 broad deletions, 1.0 focal amplification, and 1.8 broad amplifications per CTCL.
On the other hand, there were 38 SSNVs in CTCL driver genes—1.0 per tumor.
So, according to these data, SCNVs comprise 92% of all driver mutations in CTCL—a mean of 11.8 pathogenic SCNVs vs 1.0 SSNV per CTCL.
“This cancer has a very distinctive biology,” said Jaehyuk Choi, MD, PhD, of the Yale School of Medicine in New Haven, Connecticut.
And decoding this biology has revealed potential treatment approaches, according to Dr Choi and his colleagues.
For example, the presence of mutations activating the NF-κB pathway suggests NF-κB inhibitors such as bortezomib may have therapeutic potential in CTCL, and the presence of CD28 mutations suggests inhibitors such as abatacept may be effective against the disease. ![]()
Drones can transport blood samples

Robert Chalmers launch drone
Photo courtesy of
Johns Hopkins Medicine
Small drones can safely transport blood samples, according to a study published in PLOS ONE.
Investigators found that 40 minutes of travel on hobby-sized drones did not affect the results of common and routine blood tests.
The team said that’s promising news for people living in rural and economically impoverished areas that lack passable roads because drones can give healthcare workers quick access to lab tests needed for diagnoses and treatments.
“Biological samples can be very sensitive and fragile,” said study author Timothy Amukele, MD, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
That sensitivity makes even the pneumatic tube systems used by many hospitals, for example, unsuitable for transporting blood for certain purposes.
Of particular concern related to the use of drones, Dr Amukele noted, is the sudden acceleration that marks the launch of the vehicle and the jostling when the drone lands on its belly.
“Such movements could have destroyed blood cells or prompted blood to coagulate, and I thought all kinds of blood tests might be affected, but our study shows they weren’t . . . ,” he said.
For the study, which Dr Amukele believes is the first rigorous examination of the impact of drone transport on biological samples, his team collected 6 blood samples from each of 56 healthy adult volunteers at The Johns Hopkins Hospital.
The samples were then driven to a flight site an hour’s drive from the hospital on days when the temperature was in the 70s. There, half of the samples were packaged for flight, with a view to protecting them for the in-flight environment and preventing leakage.
Those samples were then loaded into a hand-launched, fixed-wing drone and flown around for periods of 6 to 38 minutes. Owing to Federal Aviation Administration rules, the flights were conducted in an unpopulated area, stayed below 100 meters (328 feet) and were in the line of sight of the certified pilot.
The other samples were driven back from the drone flight field to The Johns Hopkins Hospital’s Core Laboratory, where they underwent the 33 most common laboratory tests that account for around 80% of all such tests done. A few of the tests performed were for sodium, glucose, and red blood cell count.
Comparing lab results of the flown versus nonflown blood samples, the investigators found that the flight did not have a significant impact.
Dr Amukele and his team noted that one blood test—the bicarbonate test—did yield different results for some of the flown versus nonflown samples. Dr Amukele said the team isn’t sure why, but the reason could be because the blood sat around for up to 8 hours before being tested.
There were no consistent differences between flown versus nonflown blood, he said, and it’s unknown whether the out-of-range results were due to the time lag or because of the drone transport.
“The ideal way to test that would be to fly the blood around immediately after drawing it, but neither the FAA nor Johns Hopkins would like drones flying around the hospital,” he said.
Given the successful results of this proof-of-concept study, Dr Amukele said the likely next step is a pilot study in a location in Africa where healthcare clinics are sometimes 60 or more miles away from labs.
“A drone could go 100 kilometers in 40 minutes,” he noted. “They’re less expensive than motorcycles and are not subject to traffic delays, and the technology already exists for the drone to be programmed to home to certain GPS coordinates, like a carrier pigeon.”
Drones have already been tested as carriers of medicines to clinics in remote areas, but whether and how drones will be used in flights over populated areas will depend on laws and regulations. ![]()
Robert Chalmers launch drone
Photo courtesy of
Johns Hopkins Medicine
Small drones can safely transport blood samples, according to a study published in PLOS ONE.
Investigators found that 40 minutes of travel on hobby-sized drones did not affect the results of common and routine blood tests.
The team said that’s promising news for people living in rural and economically impoverished areas that lack passable roads because drones can give healthcare workers quick access to lab tests needed for diagnoses and treatments.
“Biological samples can be very sensitive and fragile,” said study author Timothy Amukele, MD, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
That sensitivity makes even the pneumatic tube systems used by many hospitals, for example, unsuitable for transporting blood for certain purposes.
Of particular concern related to the use of drones, Dr Amukele noted, is the sudden acceleration that marks the launch of the vehicle and the jostling when the drone lands on its belly.
“Such movements could have destroyed blood cells or prompted blood to coagulate, and I thought all kinds of blood tests might be affected, but our study shows they weren’t . . . ,” he said.
For the study, which Dr Amukele believes is the first rigorous examination of the impact of drone transport on biological samples, his team collected 6 blood samples from each of 56 healthy adult volunteers at The Johns Hopkins Hospital.
The samples were then driven to a flight site an hour’s drive from the hospital on days when the temperature was in the 70s. There, half of the samples were packaged for flight, with a view to protecting them for the in-flight environment and preventing leakage.
Those samples were then loaded into a hand-launched, fixed-wing drone and flown around for periods of 6 to 38 minutes. Owing to Federal Aviation Administration rules, the flights were conducted in an unpopulated area, stayed below 100 meters (328 feet) and were in the line of sight of the certified pilot.
The other samples were driven back from the drone flight field to The Johns Hopkins Hospital’s Core Laboratory, where they underwent the 33 most common laboratory tests that account for around 80% of all such tests done. A few of the tests performed were for sodium, glucose, and red blood cell count.
Comparing lab results of the flown versus nonflown blood samples, the investigators found that the flight did not have a significant impact.
Dr Amukele and his team noted that one blood test—the bicarbonate test—did yield different results for some of the flown versus nonflown samples. Dr Amukele said the team isn’t sure why, but the reason could be because the blood sat around for up to 8 hours before being tested.
There were no consistent differences between flown versus nonflown blood, he said, and it’s unknown whether the out-of-range results were due to the time lag or because of the drone transport.
“The ideal way to test that would be to fly the blood around immediately after drawing it, but neither the FAA nor Johns Hopkins would like drones flying around the hospital,” he said.
Given the successful results of this proof-of-concept study, Dr Amukele said the likely next step is a pilot study in a location in Africa where healthcare clinics are sometimes 60 or more miles away from labs.
“A drone could go 100 kilometers in 40 minutes,” he noted. “They’re less expensive than motorcycles and are not subject to traffic delays, and the technology already exists for the drone to be programmed to home to certain GPS coordinates, like a carrier pigeon.”
Drones have already been tested as carriers of medicines to clinics in remote areas, but whether and how drones will be used in flights over populated areas will depend on laws and regulations. ![]()
Robert Chalmers launch drone
Photo courtesy of
Johns Hopkins Medicine
Small drones can safely transport blood samples, according to a study published in PLOS ONE.
Investigators found that 40 minutes of travel on hobby-sized drones did not affect the results of common and routine blood tests.
The team said that’s promising news for people living in rural and economically impoverished areas that lack passable roads because drones can give healthcare workers quick access to lab tests needed for diagnoses and treatments.
“Biological samples can be very sensitive and fragile,” said study author Timothy Amukele, MD, PhD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
That sensitivity makes even the pneumatic tube systems used by many hospitals, for example, unsuitable for transporting blood for certain purposes.
Of particular concern related to the use of drones, Dr Amukele noted, is the sudden acceleration that marks the launch of the vehicle and the jostling when the drone lands on its belly.
“Such movements could have destroyed blood cells or prompted blood to coagulate, and I thought all kinds of blood tests might be affected, but our study shows they weren’t . . . ,” he said.
For the study, which Dr Amukele believes is the first rigorous examination of the impact of drone transport on biological samples, his team collected 6 blood samples from each of 56 healthy adult volunteers at The Johns Hopkins Hospital.
The samples were then driven to a flight site an hour’s drive from the hospital on days when the temperature was in the 70s. There, half of the samples were packaged for flight, with a view to protecting them for the in-flight environment and preventing leakage.
Those samples were then loaded into a hand-launched, fixed-wing drone and flown around for periods of 6 to 38 minutes. Owing to Federal Aviation Administration rules, the flights were conducted in an unpopulated area, stayed below 100 meters (328 feet) and were in the line of sight of the certified pilot.
The other samples were driven back from the drone flight field to The Johns Hopkins Hospital’s Core Laboratory, where they underwent the 33 most common laboratory tests that account for around 80% of all such tests done. A few of the tests performed were for sodium, glucose, and red blood cell count.
Comparing lab results of the flown versus nonflown blood samples, the investigators found that the flight did not have a significant impact.
Dr Amukele and his team noted that one blood test—the bicarbonate test—did yield different results for some of the flown versus nonflown samples. Dr Amukele said the team isn’t sure why, but the reason could be because the blood sat around for up to 8 hours before being tested.
There were no consistent differences between flown versus nonflown blood, he said, and it’s unknown whether the out-of-range results were due to the time lag or because of the drone transport.
“The ideal way to test that would be to fly the blood around immediately after drawing it, but neither the FAA nor Johns Hopkins would like drones flying around the hospital,” he said.
Given the successful results of this proof-of-concept study, Dr Amukele said the likely next step is a pilot study in a location in Africa where healthcare clinics are sometimes 60 or more miles away from labs.
“A drone could go 100 kilometers in 40 minutes,” he noted. “They’re less expensive than motorcycles and are not subject to traffic delays, and the technology already exists for the drone to be programmed to home to certain GPS coordinates, like a carrier pigeon.”
Drones have already been tested as carriers of medicines to clinics in remote areas, but whether and how drones will be used in flights over populated areas will depend on laws and regulations.
Study suggests hemophilia therapies are too costly

Hemophilia therapies account for the largest portion of pharmacy expenditures among publicly insured children with serious chronic illnesses in California, according to a study published in JAMA.
Hemophilia therapies accounted for 41% of expenditures for these children, even though hemophilia patients made up only 0.4% of the group studied.
Researchers said this finding suggests a need to improve pricing for hemophilia therapies and other high-cost medications. However, they noted that pricing varies from state to state.
Sonja M. Swenson, of Stanford University in California, and her colleagues conducted this research, analyzing paid claims for children (ages 0-21 years) using the California Children’s Services (CCS) paid claims data set (2010-2012).
CCS provides insurance coverage, care coordination, and a regionalized system of pediatric specialty care facilities for approximately 180,000 publicly insured children with serious chronic illnesses.
The data set includes age, sex, race/ethnicity, county of residence, enrollment dates, primary and secondary eligible diagnoses, claim diagnoses, and procedures for every enrollee. This study included children enrolled through fee-for-service care for at least 6 continuous months.
The researchers examined the records of 34,330 children. Outpatient pharmacy expenditures totaled $475,718,130 (20% of total healthcare expenditures).
Per-child pharmacy expenditures ranged from $0.16 to $56,849,034. The average and median per-child expenditures were $13,857 and $791, respectively.
Expenditures for all products analyzed were as follows:
| Product Class | Expenditures | % of Total
Expenditures |
No. of Children | Expenditures/
Child |
|---|---|---|---|---|
| Blood formation, coagulation, and thrombosis agents | $199,498,843 | 41.9% | 3499 | $57,016 |
| Central nervous system agents | $43,633,418 | 9.2% | 23,351 | $1869 |
| Electrolytic, caloric, and water balance | $39,617,776 | 8.3% | 10,959 | $3615 |
| Anti-infective agents | $35,827,958 | 7.5% | 26,165 | $1369 |
| Respiratory agents | $29,614,645 | 6.2% | 16,706 | $1173 |
| Hormones and synthetic substitutes | $24,722,256 | 5.2% | 8542 | $2894 |
| Enzymes | $13,294,509 | 2.8% | 27 | $492,389 |
| Gastrointestinal drugs | $12,500,330 | 2.6% | 11,817 | $1058 |
| Heavy metal antagonists | $6,983,828 | 1.5% | 108 | $64,665 |
| Cardiovascular drugs | $6,173,792 | 1.3% | 4031 | $1532 |
Hemophilia expenditures
As seen in the above table, the product class of blood formation, coagulation, and thrombosis agents accounted for the greatest share of outpatient pharmacy expenditures (42%).
Antihemophilic factors represented 98% of this class’s expenditures, or 41% of total pharmacy expenditures. Children with an antihemophilic factor paid claim were 0.4% of the entire cohort (n=145). And the average per-child expenditure for antihemophilic factor was $1,343,262.
Among children with antihemophilic factor claims who were enrolled for all 3 years studied, the average and median per-child annualized expenditures were $634,054 and $152,280, respectively.
The researchers said these results suggest a need for better pricing for hemophilia therapies, but it’s important to note that expenditures vary from state to state.
For instance, CCS’s mean per-child antihemophilic factor annual expenditure ($634,054) significantly surpassed that of North Carolina’s Medicaid program ($233,968 in fiscal year 2012) and Medicaid programs in 10 other states ($148,215 in 2008).
Hemophilia therapies account for the largest portion of pharmacy expenditures among publicly insured children with serious chronic illnesses in California, according to a study published in JAMA.
Hemophilia therapies accounted for 41% of expenditures for these children, even though hemophilia patients made up only 0.4% of the group studied.
Researchers said this finding suggests a need to improve pricing for hemophilia therapies and other high-cost medications. However, they noted that pricing varies from state to state.
Sonja M. Swenson, of Stanford University in California, and her colleagues conducted this research, analyzing paid claims for children (ages 0-21 years) using the California Children’s Services (CCS) paid claims data set (2010-2012).
CCS provides insurance coverage, care coordination, and a regionalized system of pediatric specialty care facilities for approximately 180,000 publicly insured children with serious chronic illnesses.
The data set includes age, sex, race/ethnicity, county of residence, enrollment dates, primary and secondary eligible diagnoses, claim diagnoses, and procedures for every enrollee. This study included children enrolled through fee-for-service care for at least 6 continuous months.
The researchers examined the records of 34,330 children. Outpatient pharmacy expenditures totaled $475,718,130 (20% of total healthcare expenditures).
Per-child pharmacy expenditures ranged from $0.16 to $56,849,034. The average and median per-child expenditures were $13,857 and $791, respectively.
Expenditures for all products analyzed were as follows:
| Product Class | Expenditures | % of Total
Expenditures |
No. of Children | Expenditures/
Child |
|---|---|---|---|---|
| Blood formation, coagulation, and thrombosis agents | $199,498,843 | 41.9% | 3499 | $57,016 |
| Central nervous system agents | $43,633,418 | 9.2% | 23,351 | $1869 |
| Electrolytic, caloric, and water balance | $39,617,776 | 8.3% | 10,959 | $3615 |
| Anti-infective agents | $35,827,958 | 7.5% | 26,165 | $1369 |
| Respiratory agents | $29,614,645 | 6.2% | 16,706 | $1173 |
| Hormones and synthetic substitutes | $24,722,256 | 5.2% | 8542 | $2894 |
| Enzymes | $13,294,509 | 2.8% | 27 | $492,389 |
| Gastrointestinal drugs | $12,500,330 | 2.6% | 11,817 | $1058 |
| Heavy metal antagonists | $6,983,828 | 1.5% | 108 | $64,665 |
| Cardiovascular drugs | $6,173,792 | 1.3% | 4031 | $1532 |
Hemophilia expenditures
As seen in the above table, the product class of blood formation, coagulation, and thrombosis agents accounted for the greatest share of outpatient pharmacy expenditures (42%).
Antihemophilic factors represented 98% of this class’s expenditures, or 41% of total pharmacy expenditures. Children with an antihemophilic factor paid claim were 0.4% of the entire cohort (n=145). And the average per-child expenditure for antihemophilic factor was $1,343,262.
Among children with antihemophilic factor claims who were enrolled for all 3 years studied, the average and median per-child annualized expenditures were $634,054 and $152,280, respectively.
The researchers said these results suggest a need for better pricing for hemophilia therapies, but it’s important to note that expenditures vary from state to state.
For instance, CCS’s mean per-child antihemophilic factor annual expenditure ($634,054) significantly surpassed that of North Carolina’s Medicaid program ($233,968 in fiscal year 2012) and Medicaid programs in 10 other states ($148,215 in 2008).
Hemophilia therapies account for the largest portion of pharmacy expenditures among publicly insured children with serious chronic illnesses in California, according to a study published in JAMA.
Hemophilia therapies accounted for 41% of expenditures for these children, even though hemophilia patients made up only 0.4% of the group studied.
Researchers said this finding suggests a need to improve pricing for hemophilia therapies and other high-cost medications. However, they noted that pricing varies from state to state.
Sonja M. Swenson, of Stanford University in California, and her colleagues conducted this research, analyzing paid claims for children (ages 0-21 years) using the California Children’s Services (CCS) paid claims data set (2010-2012).
CCS provides insurance coverage, care coordination, and a regionalized system of pediatric specialty care facilities for approximately 180,000 publicly insured children with serious chronic illnesses.
The data set includes age, sex, race/ethnicity, county of residence, enrollment dates, primary and secondary eligible diagnoses, claim diagnoses, and procedures for every enrollee. This study included children enrolled through fee-for-service care for at least 6 continuous months.
The researchers examined the records of 34,330 children. Outpatient pharmacy expenditures totaled $475,718,130 (20% of total healthcare expenditures).
Per-child pharmacy expenditures ranged from $0.16 to $56,849,034. The average and median per-child expenditures were $13,857 and $791, respectively.
Expenditures for all products analyzed were as follows:
| Product Class | Expenditures | % of Total
Expenditures |
No. of Children | Expenditures/
Child |
|---|---|---|---|---|
| Blood formation, coagulation, and thrombosis agents | $199,498,843 | 41.9% | 3499 | $57,016 |
| Central nervous system agents | $43,633,418 | 9.2% | 23,351 | $1869 |
| Electrolytic, caloric, and water balance | $39,617,776 | 8.3% | 10,959 | $3615 |
| Anti-infective agents | $35,827,958 | 7.5% | 26,165 | $1369 |
| Respiratory agents | $29,614,645 | 6.2% | 16,706 | $1173 |
| Hormones and synthetic substitutes | $24,722,256 | 5.2% | 8542 | $2894 |
| Enzymes | $13,294,509 | 2.8% | 27 | $492,389 |
| Gastrointestinal drugs | $12,500,330 | 2.6% | 11,817 | $1058 |
| Heavy metal antagonists | $6,983,828 | 1.5% | 108 | $64,665 |
| Cardiovascular drugs | $6,173,792 | 1.3% | 4031 | $1532 |
Hemophilia expenditures
As seen in the above table, the product class of blood formation, coagulation, and thrombosis agents accounted for the greatest share of outpatient pharmacy expenditures (42%).
Antihemophilic factors represented 98% of this class’s expenditures, or 41% of total pharmacy expenditures. Children with an antihemophilic factor paid claim were 0.4% of the entire cohort (n=145). And the average per-child expenditure for antihemophilic factor was $1,343,262.
Among children with antihemophilic factor claims who were enrolled for all 3 years studied, the average and median per-child annualized expenditures were $634,054 and $152,280, respectively.
The researchers said these results suggest a need for better pricing for hemophilia therapies, but it’s important to note that expenditures vary from state to state.
For instance, CCS’s mean per-child antihemophilic factor annual expenditure ($634,054) significantly surpassed that of North Carolina’s Medicaid program ($233,968 in fiscal year 2012) and Medicaid programs in 10 other states ($148,215 in 2008).
Inhibitor could treat range of hematologic disorders

A small molecule that targets the sonic Hedgehog signaling pathway has advanced to phase 2 trials in a range of hematologic disorders.
In a phase 1 study, the inhibitor, PF-04449913, exhibited activity in adults with leukemias, myelodysplastic syndromes (MDS), and myelofibrosis (MF).
Sixty percent of the patients studied experienced treatment-related adverse events (AEs), but there were no treatment-related deaths. Most deaths were disease-related.
Researchers detailed the results of this trial in The Lancet Haematology. The study was funded by Pfizer, the company developing PF-04449913, as well as the California Institute for Regenerative Medicine and European Leukemia Net.
Preclinical research showed that PF-04449913 forces dormant cancer stem cells in the bone marrow to begin differentiating and exit into the blood stream where they can be destroyed by chemotherapy agents targeting dividing cells.
“This drug gets that unwanted house guests to leave and never come back,” said Catriona Jamieson, MD, PhD, of University of California, San Diego School of Medicine.
“It’s a significant step forward in treating people with refractory or resistant myeloid leukemia, myelodysplastic syndrome, and myelofibrosis. It’s a bonus that the drug can be administered as easily as an aspirin, in a single, daily, oral tablet.”
For the first-in-human study, Dr Jamieson and her colleagues evaluated PF-04449913 in 47 adult patients. Twenty-eight of them had acute myeloid leukemia (AML), 6 had MDS, 5 had chronic myeloid leukemia (CML), 1 had chronic myelomonocytic leukemia (CMML), and 7 had MF.
Eighty-five percent of patients (n=40) had an ECOG performance status of 0-1. Eighty-one percent (n=38) had received previous systemic treatment, and 47% (n=22) had received 3 or more previous treatment regimens.
Patients received escalating daily doses of PF-04449913 in 28-day cycles. Treatment cycles were repeated until a patient experienced unacceptable AEs without evidence of clinical improvement. Patients who showed clinical activity without experiencing serious AEs received additional treatment cycles.
Dosing and AEs
Patients received PF-04449913 once daily at 5 mg (n=3), 10 mg (n=3), 20 mg (n=4), 40 mg (n=4), 80 mg (n=8), 120 mg (n=3), 180 mg (n=3), 270 mg (n=5), 400 mg (n=9), or 600 mg (n=5).
The researchers found the maximum-tolerated dose to be 400 mg once daily. The mean half-life was 23.9 hours in this dose group, and pharmacokinetics seemed to be dose-proportional.
Two patients experienced dose-limiting toxicities, 1 in the 80 mg group (grade 3 hypoxia and grade 3 pleural effusion), and 1 in the 600 mg group (grade 3 peripheral edema).
In all, 60% of patients (n=28) experienced treatment-related AEs. The most common were dysgeusia (28%), decreased appetite (19%), and alopecia (15%). There were 3 grade 4 AEs—1 case of neutropenia and 2 cases of thrombocytopenia.
There were 15 deaths, none of which were treatment-related. Eleven deaths were disease-related, and the remaining 4 were related to infection.
Clinical activity
The researchers said there was “some suggestion of clinical activity” in 23 patients (49%).
Of the 5 patients with CML (2 chronic phase and 3 blast phase), 1 patient with blast phase CML had a partial cytogenetic response to PF-04449913.
Of the 6 patients with MDS and 1 with CMML, 4 had stable disease after treatment. Two of these patients had hematologic improvement.
Two of the 7 patients with MF had clinical improvement.
Of the 28 patients with AML, 16 showed evidence of possible biological activity. One patient had a complete response and 4 had a partial response with incomplete hematologic recovery. Four AML patients had minor responses, and 7 had stable disease.
Given these results, PF-04449913 is now being investigated in 5 phase 2 trials of hematologic disorders, 4 of which are recruiting participants.
“Our hope is that this drug will enable more effective treatment to begin earlier and that, with earlier intervention, we can alter the course of disease and remove the need for, or improve the chances of success with, bone marrow transplantation,” Dr Jamieson said. “It’s all about reducing the burden of disease by intervening early.”
A small molecule that targets the sonic Hedgehog signaling pathway has advanced to phase 2 trials in a range of hematologic disorders.
In a phase 1 study, the inhibitor, PF-04449913, exhibited activity in adults with leukemias, myelodysplastic syndromes (MDS), and myelofibrosis (MF).
Sixty percent of the patients studied experienced treatment-related adverse events (AEs), but there were no treatment-related deaths. Most deaths were disease-related.
Researchers detailed the results of this trial in The Lancet Haematology. The study was funded by Pfizer, the company developing PF-04449913, as well as the California Institute for Regenerative Medicine and European Leukemia Net.
Preclinical research showed that PF-04449913 forces dormant cancer stem cells in the bone marrow to begin differentiating and exit into the blood stream where they can be destroyed by chemotherapy agents targeting dividing cells.
“This drug gets that unwanted house guests to leave and never come back,” said Catriona Jamieson, MD, PhD, of University of California, San Diego School of Medicine.
“It’s a significant step forward in treating people with refractory or resistant myeloid leukemia, myelodysplastic syndrome, and myelofibrosis. It’s a bonus that the drug can be administered as easily as an aspirin, in a single, daily, oral tablet.”
For the first-in-human study, Dr Jamieson and her colleagues evaluated PF-04449913 in 47 adult patients. Twenty-eight of them had acute myeloid leukemia (AML), 6 had MDS, 5 had chronic myeloid leukemia (CML), 1 had chronic myelomonocytic leukemia (CMML), and 7 had MF.
Eighty-five percent of patients (n=40) had an ECOG performance status of 0-1. Eighty-one percent (n=38) had received previous systemic treatment, and 47% (n=22) had received 3 or more previous treatment regimens.
Patients received escalating daily doses of PF-04449913 in 28-day cycles. Treatment cycles were repeated until a patient experienced unacceptable AEs without evidence of clinical improvement. Patients who showed clinical activity without experiencing serious AEs received additional treatment cycles.
Dosing and AEs
Patients received PF-04449913 once daily at 5 mg (n=3), 10 mg (n=3), 20 mg (n=4), 40 mg (n=4), 80 mg (n=8), 120 mg (n=3), 180 mg (n=3), 270 mg (n=5), 400 mg (n=9), or 600 mg (n=5).
The researchers found the maximum-tolerated dose to be 400 mg once daily. The mean half-life was 23.9 hours in this dose group, and pharmacokinetics seemed to be dose-proportional.
Two patients experienced dose-limiting toxicities, 1 in the 80 mg group (grade 3 hypoxia and grade 3 pleural effusion), and 1 in the 600 mg group (grade 3 peripheral edema).
In all, 60% of patients (n=28) experienced treatment-related AEs. The most common were dysgeusia (28%), decreased appetite (19%), and alopecia (15%). There were 3 grade 4 AEs—1 case of neutropenia and 2 cases of thrombocytopenia.
There were 15 deaths, none of which were treatment-related. Eleven deaths were disease-related, and the remaining 4 were related to infection.
Clinical activity
The researchers said there was “some suggestion of clinical activity” in 23 patients (49%).
Of the 5 patients with CML (2 chronic phase and 3 blast phase), 1 patient with blast phase CML had a partial cytogenetic response to PF-04449913.
Of the 6 patients with MDS and 1 with CMML, 4 had stable disease after treatment. Two of these patients had hematologic improvement.
Two of the 7 patients with MF had clinical improvement.
Of the 28 patients with AML, 16 showed evidence of possible biological activity. One patient had a complete response and 4 had a partial response with incomplete hematologic recovery. Four AML patients had minor responses, and 7 had stable disease.
Given these results, PF-04449913 is now being investigated in 5 phase 2 trials of hematologic disorders, 4 of which are recruiting participants.
“Our hope is that this drug will enable more effective treatment to begin earlier and that, with earlier intervention, we can alter the course of disease and remove the need for, or improve the chances of success with, bone marrow transplantation,” Dr Jamieson said. “It’s all about reducing the burden of disease by intervening early.”
A small molecule that targets the sonic Hedgehog signaling pathway has advanced to phase 2 trials in a range of hematologic disorders.
In a phase 1 study, the inhibitor, PF-04449913, exhibited activity in adults with leukemias, myelodysplastic syndromes (MDS), and myelofibrosis (MF).
Sixty percent of the patients studied experienced treatment-related adverse events (AEs), but there were no treatment-related deaths. Most deaths were disease-related.
Researchers detailed the results of this trial in The Lancet Haematology. The study was funded by Pfizer, the company developing PF-04449913, as well as the California Institute for Regenerative Medicine and European Leukemia Net.
Preclinical research showed that PF-04449913 forces dormant cancer stem cells in the bone marrow to begin differentiating and exit into the blood stream where they can be destroyed by chemotherapy agents targeting dividing cells.
“This drug gets that unwanted house guests to leave and never come back,” said Catriona Jamieson, MD, PhD, of University of California, San Diego School of Medicine.
“It’s a significant step forward in treating people with refractory or resistant myeloid leukemia, myelodysplastic syndrome, and myelofibrosis. It’s a bonus that the drug can be administered as easily as an aspirin, in a single, daily, oral tablet.”
For the first-in-human study, Dr Jamieson and her colleagues evaluated PF-04449913 in 47 adult patients. Twenty-eight of them had acute myeloid leukemia (AML), 6 had MDS, 5 had chronic myeloid leukemia (CML), 1 had chronic myelomonocytic leukemia (CMML), and 7 had MF.
Eighty-five percent of patients (n=40) had an ECOG performance status of 0-1. Eighty-one percent (n=38) had received previous systemic treatment, and 47% (n=22) had received 3 or more previous treatment regimens.
Patients received escalating daily doses of PF-04449913 in 28-day cycles. Treatment cycles were repeated until a patient experienced unacceptable AEs without evidence of clinical improvement. Patients who showed clinical activity without experiencing serious AEs received additional treatment cycles.
Dosing and AEs
Patients received PF-04449913 once daily at 5 mg (n=3), 10 mg (n=3), 20 mg (n=4), 40 mg (n=4), 80 mg (n=8), 120 mg (n=3), 180 mg (n=3), 270 mg (n=5), 400 mg (n=9), or 600 mg (n=5).
The researchers found the maximum-tolerated dose to be 400 mg once daily. The mean half-life was 23.9 hours in this dose group, and pharmacokinetics seemed to be dose-proportional.
Two patients experienced dose-limiting toxicities, 1 in the 80 mg group (grade 3 hypoxia and grade 3 pleural effusion), and 1 in the 600 mg group (grade 3 peripheral edema).
In all, 60% of patients (n=28) experienced treatment-related AEs. The most common were dysgeusia (28%), decreased appetite (19%), and alopecia (15%). There were 3 grade 4 AEs—1 case of neutropenia and 2 cases of thrombocytopenia.
There were 15 deaths, none of which were treatment-related. Eleven deaths were disease-related, and the remaining 4 were related to infection.
Clinical activity
The researchers said there was “some suggestion of clinical activity” in 23 patients (49%).
Of the 5 patients with CML (2 chronic phase and 3 blast phase), 1 patient with blast phase CML had a partial cytogenetic response to PF-04449913.
Of the 6 patients with MDS and 1 with CMML, 4 had stable disease after treatment. Two of these patients had hematologic improvement.
Two of the 7 patients with MF had clinical improvement.
Of the 28 patients with AML, 16 showed evidence of possible biological activity. One patient had a complete response and 4 had a partial response with incomplete hematologic recovery. Four AML patients had minor responses, and 7 had stable disease.
Given these results, PF-04449913 is now being investigated in 5 phase 2 trials of hematologic disorders, 4 of which are recruiting participants.
“Our hope is that this drug will enable more effective treatment to begin earlier and that, with earlier intervention, we can alter the course of disease and remove the need for, or improve the chances of success with, bone marrow transplantation,” Dr Jamieson said. “It’s all about reducing the burden of disease by intervening early.”
CHMP recommends drug for acquired hemophilia A
Photo courtesy of
Baxter International Inc.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for Obizur, a recombinant porcine
factor VIII (FVIII) product, to treat bleeding episodes in adults with acquired hemophilia A.
If the European Commission approves Obizur, it will be the first recombinant porcine FVIII treatment available in the European Union (EU) for this patient population. Obizur already has orphan designation in the EU.
The European Commission is expected to make a decision on Obizur later this year. The decision will be applicable to all 28 EU member states plus Iceland, Norway, and Liechtenstein.
About Obizur
Acquired hemophilia A is caused by the formation of antibodies directed against the body’s own FVIII. The underlying cause of this may be pregnancy, cancer, or the use of certain medications, but the cause is often unknown.
Obizur replaces inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies. Physicians can monitor patients’ response to Obizur by measuring FVIII activity levels.
The CHMP’s positive opinion of Obizur is based on a phase 2/3 trial in which patients with acquired hemophilia A received the drug as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The product is already approved in the US and is under regulatory review in Canada, Switzerland, Australia, and Colombia.
Photo courtesy of
Baxter International Inc.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for Obizur, a recombinant porcine
factor VIII (FVIII) product, to treat bleeding episodes in adults with acquired hemophilia A.
If the European Commission approves Obizur, it will be the first recombinant porcine FVIII treatment available in the European Union (EU) for this patient population. Obizur already has orphan designation in the EU.
The European Commission is expected to make a decision on Obizur later this year. The decision will be applicable to all 28 EU member states plus Iceland, Norway, and Liechtenstein.
About Obizur
Acquired hemophilia A is caused by the formation of antibodies directed against the body’s own FVIII. The underlying cause of this may be pregnancy, cancer, or the use of certain medications, but the cause is often unknown.
Obizur replaces inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies. Physicians can monitor patients’ response to Obizur by measuring FVIII activity levels.
The CHMP’s positive opinion of Obizur is based on a phase 2/3 trial in which patients with acquired hemophilia A received the drug as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The product is already approved in the US and is under regulatory review in Canada, Switzerland, Australia, and Colombia.
Photo courtesy of
Baxter International Inc.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for Obizur, a recombinant porcine
factor VIII (FVIII) product, to treat bleeding episodes in adults with acquired hemophilia A.
If the European Commission approves Obizur, it will be the first recombinant porcine FVIII treatment available in the European Union (EU) for this patient population. Obizur already has orphan designation in the EU.
The European Commission is expected to make a decision on Obizur later this year. The decision will be applicable to all 28 EU member states plus Iceland, Norway, and Liechtenstein.
About Obizur
Acquired hemophilia A is caused by the formation of antibodies directed against the body’s own FVIII. The underlying cause of this may be pregnancy, cancer, or the use of certain medications, but the cause is often unknown.
Obizur replaces inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies. Physicians can monitor patients’ response to Obizur by measuring FVIII activity levels.
The CHMP’s positive opinion of Obizur is based on a phase 2/3 trial in which patients with acquired hemophilia A received the drug as treatment for serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or higher.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse event most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur.
Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
Obizur is under development by Baxalta Incorporated. The product is already approved in the US and is under regulatory review in Canada, Switzerland, Australia, and Colombia.