User login
Evolution drives cancer development, scientists say

Photo courtesy of University
of Colorado Cancer Center
Oncogenesis does not depend only on the accumulation of mutations but on evolutionary pressures acting on cell populations, according to a paper published in PNAS.
The authors say the ecosystem of a healthy tissue landscape lets healthy cells outcompete cells with cancerous mutations.
It is when the tissue ecosystem changes due to aging, smoking, or other stressors that cells with cancerous mutations can suddenly find themselves the most fit.
And this allows the cell population to expand over generations of natural selection.
This model of oncogenesis has profound implications for cancer therapy and drug design, according to the authors.
“We’ve been trying to make drugs that target mutations in cancer cells,” said author James DeGregori, PhD, of University of Colorado School of Medicine in Aurora.
“But if it’s the ecosystem of the body, and not only cancer-causing mutations, that allows the growth of cancer, we should also be prioritizing interventions and lifestyle choices that promote the fitness of healthy cells in order to suppress the emergence of cancer.”
The proposed model helps to answer a long-standing question known as Peto’s Paradox. If cancer is due to random activating mutation, larger animals with more cells should be at greater risk of developing cancer earlier in their lives. Why then do mammals of vastly different sizes and lifespans all seem to develop cancer mostly late in life?
“Blue whales have more than a million times more cells and live about 50 times longer than a mouse, but the whale has no more risk than a mouse of developing cancer over its lifespan,” Dr DeGregori noted.
The answer he and colleague Andrii Rozhok, PhD, propose is that, in addition to activating mutations, cancer may require age-associated changes to the tissue landscape in order for evolution to favor the survival and growth of cancer cells over the competition of healthy cells.
“Healthy cells are optimized for the ecosystem of the healthy body,” Dr DeGregori said. “But when the tissue ecosystem changes, such as with aging or smoking, cancer-causing mutations are often very good at exploiting the conditions of a damaged tissue landscape.”
This model is supported by studies showing that mutations that can cause cancer do not necessarily increase a cell’s fitness.
“In fact, healthy cells are so optimized to the healthy tissue landscape that almost any mutation makes them less fit,” Dr DeGregori said.
For example, some cancer cells mutate in a way that allows them to survive in the oxygen-poor tissue environments found in the center of developing tumors. But this adaptation only confers a fitness benefit in oxygen-poor tissues.
In a healthy, oxygen-rich tissue, this mutation would not confer this advantage. In healthy tissue, cells with this mutation lose the evolutionary race to the healthy cells. Cancer cells are outcompeted and die, or, at least, their population is held in check and remains insignificantly small.
But what happens when the tissue landscape changes?
“When the body changes due to aging, smoking, inherited genetic differences, or other factors, it changes the tissue ecosystem, allowing a new kind of cell to replace the healthy ones,” Dr DeGregori said.
Certainly, cancer development requires mutations and other genetic alterations. But how do these mutations cause cancer?
It may not be that these mutations create accidental “super cells” that immediately run amok. Instead, it may be that oncogenic mutations are often or always present in the body but are kept at bay by selection pressures set against them.
That is, until the tissue ecosystem and its pressures change in ways that make cells with cancerous mutations more likely to survive than healthy cells. Over time, this allows the population of cancer cells to overcome that of healthy cells.
People can avoid some of these tissue changes by lifestyle choices, Dr DeGregori noted, but aging cannot be stopped. Still, there may be features of the tissue landscape that, with new therapies and new understanding, could be reinforced in ways to resist cancer better for longer. ![]()
Photo courtesy of University
of Colorado Cancer Center
Oncogenesis does not depend only on the accumulation of mutations but on evolutionary pressures acting on cell populations, according to a paper published in PNAS.
The authors say the ecosystem of a healthy tissue landscape lets healthy cells outcompete cells with cancerous mutations.
It is when the tissue ecosystem changes due to aging, smoking, or other stressors that cells with cancerous mutations can suddenly find themselves the most fit.
And this allows the cell population to expand over generations of natural selection.
This model of oncogenesis has profound implications for cancer therapy and drug design, according to the authors.
“We’ve been trying to make drugs that target mutations in cancer cells,” said author James DeGregori, PhD, of University of Colorado School of Medicine in Aurora.
“But if it’s the ecosystem of the body, and not only cancer-causing mutations, that allows the growth of cancer, we should also be prioritizing interventions and lifestyle choices that promote the fitness of healthy cells in order to suppress the emergence of cancer.”
The proposed model helps to answer a long-standing question known as Peto’s Paradox. If cancer is due to random activating mutation, larger animals with more cells should be at greater risk of developing cancer earlier in their lives. Why then do mammals of vastly different sizes and lifespans all seem to develop cancer mostly late in life?
“Blue whales have more than a million times more cells and live about 50 times longer than a mouse, but the whale has no more risk than a mouse of developing cancer over its lifespan,” Dr DeGregori noted.
The answer he and colleague Andrii Rozhok, PhD, propose is that, in addition to activating mutations, cancer may require age-associated changes to the tissue landscape in order for evolution to favor the survival and growth of cancer cells over the competition of healthy cells.
“Healthy cells are optimized for the ecosystem of the healthy body,” Dr DeGregori said. “But when the tissue ecosystem changes, such as with aging or smoking, cancer-causing mutations are often very good at exploiting the conditions of a damaged tissue landscape.”
This model is supported by studies showing that mutations that can cause cancer do not necessarily increase a cell’s fitness.
“In fact, healthy cells are so optimized to the healthy tissue landscape that almost any mutation makes them less fit,” Dr DeGregori said.
For example, some cancer cells mutate in a way that allows them to survive in the oxygen-poor tissue environments found in the center of developing tumors. But this adaptation only confers a fitness benefit in oxygen-poor tissues.
In a healthy, oxygen-rich tissue, this mutation would not confer this advantage. In healthy tissue, cells with this mutation lose the evolutionary race to the healthy cells. Cancer cells are outcompeted and die, or, at least, their population is held in check and remains insignificantly small.
But what happens when the tissue landscape changes?
“When the body changes due to aging, smoking, inherited genetic differences, or other factors, it changes the tissue ecosystem, allowing a new kind of cell to replace the healthy ones,” Dr DeGregori said.
Certainly, cancer development requires mutations and other genetic alterations. But how do these mutations cause cancer?
It may not be that these mutations create accidental “super cells” that immediately run amok. Instead, it may be that oncogenic mutations are often or always present in the body but are kept at bay by selection pressures set against them.
That is, until the tissue ecosystem and its pressures change in ways that make cells with cancerous mutations more likely to survive than healthy cells. Over time, this allows the population of cancer cells to overcome that of healthy cells.
People can avoid some of these tissue changes by lifestyle choices, Dr DeGregori noted, but aging cannot be stopped. Still, there may be features of the tissue landscape that, with new therapies and new understanding, could be reinforced in ways to resist cancer better for longer. ![]()
Photo courtesy of University
of Colorado Cancer Center
Oncogenesis does not depend only on the accumulation of mutations but on evolutionary pressures acting on cell populations, according to a paper published in PNAS.
The authors say the ecosystem of a healthy tissue landscape lets healthy cells outcompete cells with cancerous mutations.
It is when the tissue ecosystem changes due to aging, smoking, or other stressors that cells with cancerous mutations can suddenly find themselves the most fit.
And this allows the cell population to expand over generations of natural selection.
This model of oncogenesis has profound implications for cancer therapy and drug design, according to the authors.
“We’ve been trying to make drugs that target mutations in cancer cells,” said author James DeGregori, PhD, of University of Colorado School of Medicine in Aurora.
“But if it’s the ecosystem of the body, and not only cancer-causing mutations, that allows the growth of cancer, we should also be prioritizing interventions and lifestyle choices that promote the fitness of healthy cells in order to suppress the emergence of cancer.”
The proposed model helps to answer a long-standing question known as Peto’s Paradox. If cancer is due to random activating mutation, larger animals with more cells should be at greater risk of developing cancer earlier in their lives. Why then do mammals of vastly different sizes and lifespans all seem to develop cancer mostly late in life?
“Blue whales have more than a million times more cells and live about 50 times longer than a mouse, but the whale has no more risk than a mouse of developing cancer over its lifespan,” Dr DeGregori noted.
The answer he and colleague Andrii Rozhok, PhD, propose is that, in addition to activating mutations, cancer may require age-associated changes to the tissue landscape in order for evolution to favor the survival and growth of cancer cells over the competition of healthy cells.
“Healthy cells are optimized for the ecosystem of the healthy body,” Dr DeGregori said. “But when the tissue ecosystem changes, such as with aging or smoking, cancer-causing mutations are often very good at exploiting the conditions of a damaged tissue landscape.”
This model is supported by studies showing that mutations that can cause cancer do not necessarily increase a cell’s fitness.
“In fact, healthy cells are so optimized to the healthy tissue landscape that almost any mutation makes them less fit,” Dr DeGregori said.
For example, some cancer cells mutate in a way that allows them to survive in the oxygen-poor tissue environments found in the center of developing tumors. But this adaptation only confers a fitness benefit in oxygen-poor tissues.
In a healthy, oxygen-rich tissue, this mutation would not confer this advantage. In healthy tissue, cells with this mutation lose the evolutionary race to the healthy cells. Cancer cells are outcompeted and die, or, at least, their population is held in check and remains insignificantly small.
But what happens when the tissue landscape changes?
“When the body changes due to aging, smoking, inherited genetic differences, or other factors, it changes the tissue ecosystem, allowing a new kind of cell to replace the healthy ones,” Dr DeGregori said.
Certainly, cancer development requires mutations and other genetic alterations. But how do these mutations cause cancer?
It may not be that these mutations create accidental “super cells” that immediately run amok. Instead, it may be that oncogenic mutations are often or always present in the body but are kept at bay by selection pressures set against them.
That is, until the tissue ecosystem and its pressures change in ways that make cells with cancerous mutations more likely to survive than healthy cells. Over time, this allows the population of cancer cells to overcome that of healthy cells.
People can avoid some of these tissue changes by lifestyle choices, Dr DeGregori noted, but aging cannot be stopped. Still, there may be features of the tissue landscape that, with new therapies and new understanding, could be reinforced in ways to resist cancer better for longer. ![]()
Team discovers ‘new avenue’ for TTP treatment
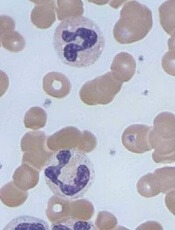
Image by Erhabor Osaro
Researchers say they have uncovered a new avenue for therapeutic intervention in thrombotic thrombocytopenic purpura (TTP).
The team discovered how autoimmune antibodies in a TTP patient recognize and bind to ADAMTS13.
They believe this knowledge could help them alter ADAMTS13 to produce a therapeutic enzyme that can elude recognition by autoimmune antibodies yet still retain its ability to cleave von Willebrand factor.
Such an enzyme could be given to TTP patients in the hospital to speed recovery and cut the cost of treatment.
Long Zheng, MD, PhD, of the University of Alabama at Birmingham, and his colleagues described this work in PNAS.
The researchers found that 5 small loops in ADAMTS13’s amino acid sequence are necessary for autoantibodies to bind to ADAMTS13.
Cutting or substituting several amino acids out of any of the 5 loops prevented binding. Small deletions in a loop also left the enzyme unable to cleave von Willebrand factor.
“This was really surprising,” Dr Zheng said. “It’s like a table with 5 legs. If you take 1 away, it should still stand, but, somehow, it collapsed. This suggests that you need the coordinated activity of all 5.”
Thus, it appears that the autoimmune antibodies in TTP patients inhibit the enzyme by physically blocking the recognition site of ADAMTS13 for von Willebrand factor.
Analyses of autoantibodies from 23 more TTP patients revealed that most use the same binding site. This suggests modifying the ADAMTS13 enzyme by protein engineering may be able to help a wide range of TTP patients.
Details of the research
Dr Zheng and his colleagues first isolated messenger RNAs that code single chains of variable regions of monoclonal antibodies from B cells collected from patients with acquired TTP.
The team used phage display to select the messenger RNAs that code specific antibodies that bind and inhibit ADAMTS13. These monoclonal antibodies were then expressed in E coli cells, purified, and biochemically characterized.
Three inhibitory monoclonal antibodies were selected for further study by hydrogen-deuterium exchange coupled with mass spectrometry. This technology uses amine hydrogen exchange with deuterium on each amino acid residue except proline.
After the reaction was stopped, the protein was cut into small pieces (or peptide fragments) and run through high-performance liquid chromatography for separation and mass spectrometry for identification.
Antibody binding sites were detected by their ability to block the hydrogen and deuterium exchange, as compared with ADAMTS13 that was unbound.
One of the 3 high-affinity probes selected by phage display was used for the competition experiments against polyclonal autoimmune antibodies from 23 TTP patients.
The results show that this particular binding epitope is common among patients with acquired autoimmune TTP. ![]()
Image by Erhabor Osaro
Researchers say they have uncovered a new avenue for therapeutic intervention in thrombotic thrombocytopenic purpura (TTP).
The team discovered how autoimmune antibodies in a TTP patient recognize and bind to ADAMTS13.
They believe this knowledge could help them alter ADAMTS13 to produce a therapeutic enzyme that can elude recognition by autoimmune antibodies yet still retain its ability to cleave von Willebrand factor.
Such an enzyme could be given to TTP patients in the hospital to speed recovery and cut the cost of treatment.
Long Zheng, MD, PhD, of the University of Alabama at Birmingham, and his colleagues described this work in PNAS.
The researchers found that 5 small loops in ADAMTS13’s amino acid sequence are necessary for autoantibodies to bind to ADAMTS13.
Cutting or substituting several amino acids out of any of the 5 loops prevented binding. Small deletions in a loop also left the enzyme unable to cleave von Willebrand factor.
“This was really surprising,” Dr Zheng said. “It’s like a table with 5 legs. If you take 1 away, it should still stand, but, somehow, it collapsed. This suggests that you need the coordinated activity of all 5.”
Thus, it appears that the autoimmune antibodies in TTP patients inhibit the enzyme by physically blocking the recognition site of ADAMTS13 for von Willebrand factor.
Analyses of autoantibodies from 23 more TTP patients revealed that most use the same binding site. This suggests modifying the ADAMTS13 enzyme by protein engineering may be able to help a wide range of TTP patients.
Details of the research
Dr Zheng and his colleagues first isolated messenger RNAs that code single chains of variable regions of monoclonal antibodies from B cells collected from patients with acquired TTP.
The team used phage display to select the messenger RNAs that code specific antibodies that bind and inhibit ADAMTS13. These monoclonal antibodies were then expressed in E coli cells, purified, and biochemically characterized.
Three inhibitory monoclonal antibodies were selected for further study by hydrogen-deuterium exchange coupled with mass spectrometry. This technology uses amine hydrogen exchange with deuterium on each amino acid residue except proline.
After the reaction was stopped, the protein was cut into small pieces (or peptide fragments) and run through high-performance liquid chromatography for separation and mass spectrometry for identification.
Antibody binding sites were detected by their ability to block the hydrogen and deuterium exchange, as compared with ADAMTS13 that was unbound.
One of the 3 high-affinity probes selected by phage display was used for the competition experiments against polyclonal autoimmune antibodies from 23 TTP patients.
The results show that this particular binding epitope is common among patients with acquired autoimmune TTP. ![]()
Image by Erhabor Osaro
Researchers say they have uncovered a new avenue for therapeutic intervention in thrombotic thrombocytopenic purpura (TTP).
The team discovered how autoimmune antibodies in a TTP patient recognize and bind to ADAMTS13.
They believe this knowledge could help them alter ADAMTS13 to produce a therapeutic enzyme that can elude recognition by autoimmune antibodies yet still retain its ability to cleave von Willebrand factor.
Such an enzyme could be given to TTP patients in the hospital to speed recovery and cut the cost of treatment.
Long Zheng, MD, PhD, of the University of Alabama at Birmingham, and his colleagues described this work in PNAS.
The researchers found that 5 small loops in ADAMTS13’s amino acid sequence are necessary for autoantibodies to bind to ADAMTS13.
Cutting or substituting several amino acids out of any of the 5 loops prevented binding. Small deletions in a loop also left the enzyme unable to cleave von Willebrand factor.
“This was really surprising,” Dr Zheng said. “It’s like a table with 5 legs. If you take 1 away, it should still stand, but, somehow, it collapsed. This suggests that you need the coordinated activity of all 5.”
Thus, it appears that the autoimmune antibodies in TTP patients inhibit the enzyme by physically blocking the recognition site of ADAMTS13 for von Willebrand factor.
Analyses of autoantibodies from 23 more TTP patients revealed that most use the same binding site. This suggests modifying the ADAMTS13 enzyme by protein engineering may be able to help a wide range of TTP patients.
Details of the research
Dr Zheng and his colleagues first isolated messenger RNAs that code single chains of variable regions of monoclonal antibodies from B cells collected from patients with acquired TTP.
The team used phage display to select the messenger RNAs that code specific antibodies that bind and inhibit ADAMTS13. These monoclonal antibodies were then expressed in E coli cells, purified, and biochemically characterized.
Three inhibitory monoclonal antibodies were selected for further study by hydrogen-deuterium exchange coupled with mass spectrometry. This technology uses amine hydrogen exchange with deuterium on each amino acid residue except proline.
After the reaction was stopped, the protein was cut into small pieces (or peptide fragments) and run through high-performance liquid chromatography for separation and mass spectrometry for identification.
Antibody binding sites were detected by their ability to block the hydrogen and deuterium exchange, as compared with ADAMTS13 that was unbound.
One of the 3 high-affinity probes selected by phage display was used for the competition experiments against polyclonal autoimmune antibodies from 23 TTP patients.
The results show that this particular binding epitope is common among patients with acquired autoimmune TTP. ![]()
Pediatric cancer specialist passes away

Photo courtesy of University
of Chicago Medicine
Charles M. Rubin, MD, an associate professor of pediatrics at the University of Chicago Medicine, has passed away at the age of 62.
Dr Rubin died while at work on July 17.
He had just arrived at the pediatric clinic at the University of Chicago Medicine Comprehensive Cancer Center at Silver Cross Hospital in New Lenox when his heart stopped.
Resuscitation efforts were unsuccessful.
An authority on pediatric cancers, Dr Rubin had a particular interest in brain tumors and cancer occurring in children with genetic syndromes. He combined experience in basic laboratory research on the genetics of cancer with broad clinical expertise.
“I can’t put into words how much I respected him,” said colleague Tara Henderson, MD, associate professor of pediatrics and director of the Childhood Cancer Survivors Center at the University of Chicago’s Comer Children’s Hospital.
“He was amazingly knowledgeable, compassionate, and thoughtful—traits at the core of our program. I take his influence with me as I care for my patients. He could also be funny, with a dry, quiet sense of humor. We never knew when it was coming. We miss him dearly.”
Dr Rubin was born February 10, 1953, in Long Branch, New Jersey. He earned his bachelor’s degree from the University of Pennsylvania in 1975 and his medical degree from Tufts University School of Medicine in 1979.
Dr Rubin completed his pediatric residency at the Children’s Hospital of Philadelphia in 1982, followed by a fellowship in pediatric hematology/oncology at the University of Minnesota in 1985.
He went to the University of Chicago in 1985 as a cytogenetics and molecular biology fellow in the laboratory of Janet Rowley, MD, an internationally recognized pioneer in understanding the genetics of cancer.
Dr Rubin joined the faculty as an assistant professor of pediatrics and medicine and a member of the university’s Cancer Research Center in 1987. In 1991, he and adult oncologist Funmi Olopade, MD, co-founded the university’s Cancer Risk Clinic.
“Chuck Rubin was one of the finest individuals I have ever known,” said Michelle Le Beau, PhD, a former colleague in the Rowley laboratory and now director of the University of Chicago Medicine Comprehensive Cancer Center.
“He was a consummate academician and physician who blended compassion and sensitivity with brilliant clinical acumen. His dedication to his family, his patients, and the University of Chicago was selfless and unparalleled. It was a privilege to work with him and an honor to learn from his example.”
Although Dr Rubin continued to work closely with his basic-science colleagues, contributing to more than 50 original reports in academic journals, his interests increasingly focused on patient care.
At the same time, he took on several administrative roles. He served as course director for pediatric grand rounds and the medical center’s pediatric tumor board.
He directed the pediatric hematology/oncology fellowship for 7 years and the pediatric neuro-oncology program for 10 years. He also volunteered for medical staff positions in various educational and rehabilitative summer camps for children with cancer.
Dr Rubin was a leader in the University of Chicago Medicine’s efforts to take a research-driven approach to pediatric cancer care into the community, serving as director of pediatric hematology/oncology outreach since 2008.
Dr Rubin is survived by his wife, Gretchen; their 4 daughters, Elizabeth, Jane, Lucy, and Claire; brothers Michael, Peter, and Richard; and many nieces and nephews. ![]()
Photo courtesy of University
of Chicago Medicine
Charles M. Rubin, MD, an associate professor of pediatrics at the University of Chicago Medicine, has passed away at the age of 62.
Dr Rubin died while at work on July 17.
He had just arrived at the pediatric clinic at the University of Chicago Medicine Comprehensive Cancer Center at Silver Cross Hospital in New Lenox when his heart stopped.
Resuscitation efforts were unsuccessful.
An authority on pediatric cancers, Dr Rubin had a particular interest in brain tumors and cancer occurring in children with genetic syndromes. He combined experience in basic laboratory research on the genetics of cancer with broad clinical expertise.
“I can’t put into words how much I respected him,” said colleague Tara Henderson, MD, associate professor of pediatrics and director of the Childhood Cancer Survivors Center at the University of Chicago’s Comer Children’s Hospital.
“He was amazingly knowledgeable, compassionate, and thoughtful—traits at the core of our program. I take his influence with me as I care for my patients. He could also be funny, with a dry, quiet sense of humor. We never knew when it was coming. We miss him dearly.”
Dr Rubin was born February 10, 1953, in Long Branch, New Jersey. He earned his bachelor’s degree from the University of Pennsylvania in 1975 and his medical degree from Tufts University School of Medicine in 1979.
Dr Rubin completed his pediatric residency at the Children’s Hospital of Philadelphia in 1982, followed by a fellowship in pediatric hematology/oncology at the University of Minnesota in 1985.
He went to the University of Chicago in 1985 as a cytogenetics and molecular biology fellow in the laboratory of Janet Rowley, MD, an internationally recognized pioneer in understanding the genetics of cancer.
Dr Rubin joined the faculty as an assistant professor of pediatrics and medicine and a member of the university’s Cancer Research Center in 1987. In 1991, he and adult oncologist Funmi Olopade, MD, co-founded the university’s Cancer Risk Clinic.
“Chuck Rubin was one of the finest individuals I have ever known,” said Michelle Le Beau, PhD, a former colleague in the Rowley laboratory and now director of the University of Chicago Medicine Comprehensive Cancer Center.
“He was a consummate academician and physician who blended compassion and sensitivity with brilliant clinical acumen. His dedication to his family, his patients, and the University of Chicago was selfless and unparalleled. It was a privilege to work with him and an honor to learn from his example.”
Although Dr Rubin continued to work closely with his basic-science colleagues, contributing to more than 50 original reports in academic journals, his interests increasingly focused on patient care.
At the same time, he took on several administrative roles. He served as course director for pediatric grand rounds and the medical center’s pediatric tumor board.
He directed the pediatric hematology/oncology fellowship for 7 years and the pediatric neuro-oncology program for 10 years. He also volunteered for medical staff positions in various educational and rehabilitative summer camps for children with cancer.
Dr Rubin was a leader in the University of Chicago Medicine’s efforts to take a research-driven approach to pediatric cancer care into the community, serving as director of pediatric hematology/oncology outreach since 2008.
Dr Rubin is survived by his wife, Gretchen; their 4 daughters, Elizabeth, Jane, Lucy, and Claire; brothers Michael, Peter, and Richard; and many nieces and nephews. ![]()
Photo courtesy of University
of Chicago Medicine
Charles M. Rubin, MD, an associate professor of pediatrics at the University of Chicago Medicine, has passed away at the age of 62.
Dr Rubin died while at work on July 17.
He had just arrived at the pediatric clinic at the University of Chicago Medicine Comprehensive Cancer Center at Silver Cross Hospital in New Lenox when his heart stopped.
Resuscitation efforts were unsuccessful.
An authority on pediatric cancers, Dr Rubin had a particular interest in brain tumors and cancer occurring in children with genetic syndromes. He combined experience in basic laboratory research on the genetics of cancer with broad clinical expertise.
“I can’t put into words how much I respected him,” said colleague Tara Henderson, MD, associate professor of pediatrics and director of the Childhood Cancer Survivors Center at the University of Chicago’s Comer Children’s Hospital.
“He was amazingly knowledgeable, compassionate, and thoughtful—traits at the core of our program. I take his influence with me as I care for my patients. He could also be funny, with a dry, quiet sense of humor. We never knew when it was coming. We miss him dearly.”
Dr Rubin was born February 10, 1953, in Long Branch, New Jersey. He earned his bachelor’s degree from the University of Pennsylvania in 1975 and his medical degree from Tufts University School of Medicine in 1979.
Dr Rubin completed his pediatric residency at the Children’s Hospital of Philadelphia in 1982, followed by a fellowship in pediatric hematology/oncology at the University of Minnesota in 1985.
He went to the University of Chicago in 1985 as a cytogenetics and molecular biology fellow in the laboratory of Janet Rowley, MD, an internationally recognized pioneer in understanding the genetics of cancer.
Dr Rubin joined the faculty as an assistant professor of pediatrics and medicine and a member of the university’s Cancer Research Center in 1987. In 1991, he and adult oncologist Funmi Olopade, MD, co-founded the university’s Cancer Risk Clinic.
“Chuck Rubin was one of the finest individuals I have ever known,” said Michelle Le Beau, PhD, a former colleague in the Rowley laboratory and now director of the University of Chicago Medicine Comprehensive Cancer Center.
“He was a consummate academician and physician who blended compassion and sensitivity with brilliant clinical acumen. His dedication to his family, his patients, and the University of Chicago was selfless and unparalleled. It was a privilege to work with him and an honor to learn from his example.”
Although Dr Rubin continued to work closely with his basic-science colleagues, contributing to more than 50 original reports in academic journals, his interests increasingly focused on patient care.
At the same time, he took on several administrative roles. He served as course director for pediatric grand rounds and the medical center’s pediatric tumor board.
He directed the pediatric hematology/oncology fellowship for 7 years and the pediatric neuro-oncology program for 10 years. He also volunteered for medical staff positions in various educational and rehabilitative summer camps for children with cancer.
Dr Rubin was a leader in the University of Chicago Medicine’s efforts to take a research-driven approach to pediatric cancer care into the community, serving as director of pediatric hematology/oncology outreach since 2008.
Dr Rubin is survived by his wife, Gretchen; their 4 daughters, Elizabeth, Jane, Lucy, and Claire; brothers Michael, Peter, and Richard; and many nieces and nephews. ![]()
SNP linked to poor survival in MM

Photo courtesy of NIGMS
Investigators have identified a single nucleotide polymorphism (SNP) that seems to confer shorter survival in patients with multiple myeloma (MM).
The group found a significant association between survival and a SNP near the gene FOPNL on chromosome 16p13.
On average, MM patients with this SNP (rs72773978) died 1 to 3 years sooner than patients without it.
The investigators pinpointed the SNP via genome-wide association studies and verified its impact on survival in patient populations from North America and Europe.
The research, which is published in Nature Communications, included 1635 MM patients.
“This is the largest study of inherited genetics and myeloma survival to date,” said Nicola Camp, PhD, of the Huntsman Cancer Institute in Salt Lake City, Utah.
“We were able to identify the FOPNL variant because it has quite a large effect on survival. With even larger collaborative studies, we hope to add to this. The ability to stratify patients based on their genetic make-up opens the door to personalizing their treatment and care.”
For this study, Dr Camp and her colleagues first conducted a meta-analysis of 306 MM patients treated at University of California, San Francisco and 239 patients treated at the Mayo Clinic.
The investigators found a significant association between rs72773978 and survival. Patients with the minor allele had an increased risk of mortality compared to patients who were homozygous for the major allele (hazard ratio=2.65).
The team then conducted a replication meta-analysis of 1090 MM cases, including 772 European patients from the IMMEnSE consortium and 318 from the Utah cohort. Again, there was a significant association between rs72773978 and survival (hazard ratio=1.34).
Although the investigators don’t yet understand why the SNP is associated with poor prognosis, there are clues that it could be involved in disease progression through centrosome amplification.
Analyses of the different MM patient datasets showed that individuals with the worst outcomes have abnormal amounts of FOPNL and carry another sign of poor prognosis—a high centrosome index. The implication is that disruptions in FOPNL could affect fundamental mechanisms controlling the distribution of genetic material to newly made cells.
“The results point us to a previously unrecognized gene as a determinant of myeloma prognosis,” said Elad Ziv, MD, of the University of California, San Francisco.
“If we understand what about this gene is causing poor prognosis, that may lead to a better fundamental grasp of the pathways that are important in multiple myeloma progression. Such knowledge could ultimately lead to better therapies.” ![]()
Photo courtesy of NIGMS
Investigators have identified a single nucleotide polymorphism (SNP) that seems to confer shorter survival in patients with multiple myeloma (MM).
The group found a significant association between survival and a SNP near the gene FOPNL on chromosome 16p13.
On average, MM patients with this SNP (rs72773978) died 1 to 3 years sooner than patients without it.
The investigators pinpointed the SNP via genome-wide association studies and verified its impact on survival in patient populations from North America and Europe.
The research, which is published in Nature Communications, included 1635 MM patients.
“This is the largest study of inherited genetics and myeloma survival to date,” said Nicola Camp, PhD, of the Huntsman Cancer Institute in Salt Lake City, Utah.
“We were able to identify the FOPNL variant because it has quite a large effect on survival. With even larger collaborative studies, we hope to add to this. The ability to stratify patients based on their genetic make-up opens the door to personalizing their treatment and care.”
For this study, Dr Camp and her colleagues first conducted a meta-analysis of 306 MM patients treated at University of California, San Francisco and 239 patients treated at the Mayo Clinic.
The investigators found a significant association between rs72773978 and survival. Patients with the minor allele had an increased risk of mortality compared to patients who were homozygous for the major allele (hazard ratio=2.65).
The team then conducted a replication meta-analysis of 1090 MM cases, including 772 European patients from the IMMEnSE consortium and 318 from the Utah cohort. Again, there was a significant association between rs72773978 and survival (hazard ratio=1.34).
Although the investigators don’t yet understand why the SNP is associated with poor prognosis, there are clues that it could be involved in disease progression through centrosome amplification.
Analyses of the different MM patient datasets showed that individuals with the worst outcomes have abnormal amounts of FOPNL and carry another sign of poor prognosis—a high centrosome index. The implication is that disruptions in FOPNL could affect fundamental mechanisms controlling the distribution of genetic material to newly made cells.
“The results point us to a previously unrecognized gene as a determinant of myeloma prognosis,” said Elad Ziv, MD, of the University of California, San Francisco.
“If we understand what about this gene is causing poor prognosis, that may lead to a better fundamental grasp of the pathways that are important in multiple myeloma progression. Such knowledge could ultimately lead to better therapies.” ![]()
Photo courtesy of NIGMS
Investigators have identified a single nucleotide polymorphism (SNP) that seems to confer shorter survival in patients with multiple myeloma (MM).
The group found a significant association between survival and a SNP near the gene FOPNL on chromosome 16p13.
On average, MM patients with this SNP (rs72773978) died 1 to 3 years sooner than patients without it.
The investigators pinpointed the SNP via genome-wide association studies and verified its impact on survival in patient populations from North America and Europe.
The research, which is published in Nature Communications, included 1635 MM patients.
“This is the largest study of inherited genetics and myeloma survival to date,” said Nicola Camp, PhD, of the Huntsman Cancer Institute in Salt Lake City, Utah.
“We were able to identify the FOPNL variant because it has quite a large effect on survival. With even larger collaborative studies, we hope to add to this. The ability to stratify patients based on their genetic make-up opens the door to personalizing their treatment and care.”
For this study, Dr Camp and her colleagues first conducted a meta-analysis of 306 MM patients treated at University of California, San Francisco and 239 patients treated at the Mayo Clinic.
The investigators found a significant association between rs72773978 and survival. Patients with the minor allele had an increased risk of mortality compared to patients who were homozygous for the major allele (hazard ratio=2.65).
The team then conducted a replication meta-analysis of 1090 MM cases, including 772 European patients from the IMMEnSE consortium and 318 from the Utah cohort. Again, there was a significant association between rs72773978 and survival (hazard ratio=1.34).
Although the investigators don’t yet understand why the SNP is associated with poor prognosis, there are clues that it could be involved in disease progression through centrosome amplification.
Analyses of the different MM patient datasets showed that individuals with the worst outcomes have abnormal amounts of FOPNL and carry another sign of poor prognosis—a high centrosome index. The implication is that disruptions in FOPNL could affect fundamental mechanisms controlling the distribution of genetic material to newly made cells.
“The results point us to a previously unrecognized gene as a determinant of myeloma prognosis,” said Elad Ziv, MD, of the University of California, San Francisco.
“If we understand what about this gene is causing poor prognosis, that may lead to a better fundamental grasp of the pathways that are important in multiple myeloma progression. Such knowledge could ultimately lead to better therapies.” ![]()
Team synthesizes compounds that induce rapid apoptosis in leukemia
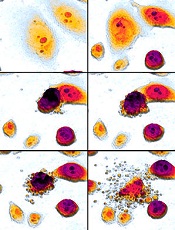
apoptosis in cancer cells
For the first time, researchers have synthesized compounds that induce rapid apoptosis in leukemic cells.
The team synthesized several members of the family of dimeric nuphar alkaloids, which are compounds previously isolated from the yellow pond lily.
These structurally complex molecules have proven capable of inducing apoptosis in human leukemia cell lines faster than any other small molecule tested to date.
The researchers were also able to synthesize some related structures that they predict might exist in nature but have not yet been found.
Their work is published in Angewandte Chemie International Edition.
“We anticipate that these compounds will serve as useful tools for dissecting an important, but as yet undefined, step in the regulation of apoptosis,” said study author Jimmy Wu, PhD, of Dartmouth College in Hanover, New Hampshire.
The research also provides a means to a steady supply of the active compounds for further study.
Preliminary biological tests conducted by Alan Eastman, PhD, also of Dartmouth College, suggest the compounds, both the naturally occurring ones and those predicted to exist in nature, are capable of inducing extremely rapid apoptosis in leukemic cells.
“Studies to clarify the biological mechanism by which they operate are ongoing,” Dr Wu said.
He noted that there have been 2 reports that attempt to explain the molecules’ mechanism of action. But these are incomplete, and more research is required to fully reveal how these compounds work.
“A better understanding of the biological basis of how the dimeric nuphar alkaloids can so rapidly induce cell death may lead to novel points of intervention for the design of prospective therapeutics,” Dr Wu concluded. ![]()
apoptosis in cancer cells
For the first time, researchers have synthesized compounds that induce rapid apoptosis in leukemic cells.
The team synthesized several members of the family of dimeric nuphar alkaloids, which are compounds previously isolated from the yellow pond lily.
These structurally complex molecules have proven capable of inducing apoptosis in human leukemia cell lines faster than any other small molecule tested to date.
The researchers were also able to synthesize some related structures that they predict might exist in nature but have not yet been found.
Their work is published in Angewandte Chemie International Edition.
“We anticipate that these compounds will serve as useful tools for dissecting an important, but as yet undefined, step in the regulation of apoptosis,” said study author Jimmy Wu, PhD, of Dartmouth College in Hanover, New Hampshire.
The research also provides a means to a steady supply of the active compounds for further study.
Preliminary biological tests conducted by Alan Eastman, PhD, also of Dartmouth College, suggest the compounds, both the naturally occurring ones and those predicted to exist in nature, are capable of inducing extremely rapid apoptosis in leukemic cells.
“Studies to clarify the biological mechanism by which they operate are ongoing,” Dr Wu said.
He noted that there have been 2 reports that attempt to explain the molecules’ mechanism of action. But these are incomplete, and more research is required to fully reveal how these compounds work.
“A better understanding of the biological basis of how the dimeric nuphar alkaloids can so rapidly induce cell death may lead to novel points of intervention for the design of prospective therapeutics,” Dr Wu concluded. ![]()
apoptosis in cancer cells
For the first time, researchers have synthesized compounds that induce rapid apoptosis in leukemic cells.
The team synthesized several members of the family of dimeric nuphar alkaloids, which are compounds previously isolated from the yellow pond lily.
These structurally complex molecules have proven capable of inducing apoptosis in human leukemia cell lines faster than any other small molecule tested to date.
The researchers were also able to synthesize some related structures that they predict might exist in nature but have not yet been found.
Their work is published in Angewandte Chemie International Edition.
“We anticipate that these compounds will serve as useful tools for dissecting an important, but as yet undefined, step in the regulation of apoptosis,” said study author Jimmy Wu, PhD, of Dartmouth College in Hanover, New Hampshire.
The research also provides a means to a steady supply of the active compounds for further study.
Preliminary biological tests conducted by Alan Eastman, PhD, also of Dartmouth College, suggest the compounds, both the naturally occurring ones and those predicted to exist in nature, are capable of inducing extremely rapid apoptosis in leukemic cells.
“Studies to clarify the biological mechanism by which they operate are ongoing,” Dr Wu said.
He noted that there have been 2 reports that attempt to explain the molecules’ mechanism of action. But these are incomplete, and more research is required to fully reveal how these compounds work.
“A better understanding of the biological basis of how the dimeric nuphar alkaloids can so rapidly induce cell death may lead to novel points of intervention for the design of prospective therapeutics,” Dr Wu concluded. ![]()
Lipids aid engraftment of HSPCs
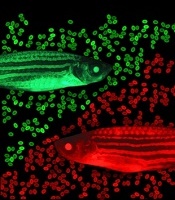
zebrafish used in the study
Image by Jonathan Henninger
and Vera Binder
Using zebrafish drug-screening models, researchers have identified a family of lipids that aid the engraftment of hematopoietic stem and progenitor cells (HSPCs).
The lipids, known as epoxyeicosatrienoic acids (EETs), boosted HSPC engraftment in zebrafish and mice.
The researchers therefore believe EETs could help make human HSPC transplants, particularly umbilical cord blood transplants, more efficient and effective.
“Ninety percent of cord blood units can’t be used because they’re too small,” said Leonard Zon, MD, of Boston Children’s Hospital in Massachusetts.
“If you add these chemicals, you might be able to use more units. Being able to get engraftment allows you to pick a smaller cord blood sample that might be a better match.”
Dr Zon and his colleagues described their work with EETs in Nature.
EETs appear to work by stimulating cell migration. They were among the top hits in a screen of 500 known compounds the researchers conducted.
In the past, such screens have led Dr Zon’s team to compounds that boost HSPC numbers, such as prostaglandin. But the new drug screen was designed to assess HSPCs’ transplantability and engraftment.
The screen was done in a lab-created strain of zebrafish called Casper. Because Casper is translucent, Dr Zon and his colleagues could visually compare the engraftment of transplanted HSPCs chemically tagged to glow green or red.
The researchers first used tagging to color the fishes’ marrow either red or green, then removed HSPCs for transplantation. The green cells were incubated with various chemicals, while the red cells were left untreated.
The team then injected a mixture of green and red HSPCs into other groups of zebrafish (10 fish per test chemical). And they visually tracked the cells’ activity, measuring the green-to-red ratio.
“The expectation was that if a chemical didn’t increase engraftment, all the fish would be equal parts red and green,” Dr Zon said. “But if it was effective, green marrow would predominate.”
That was the case for green marrow incubated with EETs, a finding that held up over thousands of transplants.
“In a mouse system, this experiment would cost $3 million,” Dr Zon noted. “In fish, it cost about $150,000.”
In a smaller-scale set of mouse experiments, the team confirmed EETs’ efficacy in promoting homing and engraftment of HSPCs.
EETs are chemical cousins of prostaglandin. Both are made from arachidonic acid, and both are made during inflammation. But EETs work in a different way, by activating the PI3K pathway. EETs also enhanced PI3K activity in human blood vessel cells in vitro.
After more studies in human cells to determine exactly how EETs work, Dr Zon hopes to begin clinical trials of EETs within the next 2 years, likely in the setting of cord blood transplant. The lab is also investigating its other top hits from the zebrafish screen.
“Every new pathway that we find has the chance of making stem cell engraftment and migration even better,” Dr Zon said. “I think we’ll end up being able to manipulate this process.” ![]()
zebrafish used in the study
Image by Jonathan Henninger
and Vera Binder
Using zebrafish drug-screening models, researchers have identified a family of lipids that aid the engraftment of hematopoietic stem and progenitor cells (HSPCs).
The lipids, known as epoxyeicosatrienoic acids (EETs), boosted HSPC engraftment in zebrafish and mice.
The researchers therefore believe EETs could help make human HSPC transplants, particularly umbilical cord blood transplants, more efficient and effective.
“Ninety percent of cord blood units can’t be used because they’re too small,” said Leonard Zon, MD, of Boston Children’s Hospital in Massachusetts.
“If you add these chemicals, you might be able to use more units. Being able to get engraftment allows you to pick a smaller cord blood sample that might be a better match.”
Dr Zon and his colleagues described their work with EETs in Nature.
EETs appear to work by stimulating cell migration. They were among the top hits in a screen of 500 known compounds the researchers conducted.
In the past, such screens have led Dr Zon’s team to compounds that boost HSPC numbers, such as prostaglandin. But the new drug screen was designed to assess HSPCs’ transplantability and engraftment.
The screen was done in a lab-created strain of zebrafish called Casper. Because Casper is translucent, Dr Zon and his colleagues could visually compare the engraftment of transplanted HSPCs chemically tagged to glow green or red.
The researchers first used tagging to color the fishes’ marrow either red or green, then removed HSPCs for transplantation. The green cells were incubated with various chemicals, while the red cells were left untreated.
The team then injected a mixture of green and red HSPCs into other groups of zebrafish (10 fish per test chemical). And they visually tracked the cells’ activity, measuring the green-to-red ratio.
“The expectation was that if a chemical didn’t increase engraftment, all the fish would be equal parts red and green,” Dr Zon said. “But if it was effective, green marrow would predominate.”
That was the case for green marrow incubated with EETs, a finding that held up over thousands of transplants.
“In a mouse system, this experiment would cost $3 million,” Dr Zon noted. “In fish, it cost about $150,000.”
In a smaller-scale set of mouse experiments, the team confirmed EETs’ efficacy in promoting homing and engraftment of HSPCs.
EETs are chemical cousins of prostaglandin. Both are made from arachidonic acid, and both are made during inflammation. But EETs work in a different way, by activating the PI3K pathway. EETs also enhanced PI3K activity in human blood vessel cells in vitro.
After more studies in human cells to determine exactly how EETs work, Dr Zon hopes to begin clinical trials of EETs within the next 2 years, likely in the setting of cord blood transplant. The lab is also investigating its other top hits from the zebrafish screen.
“Every new pathway that we find has the chance of making stem cell engraftment and migration even better,” Dr Zon said. “I think we’ll end up being able to manipulate this process.” ![]()
zebrafish used in the study
Image by Jonathan Henninger
and Vera Binder
Using zebrafish drug-screening models, researchers have identified a family of lipids that aid the engraftment of hematopoietic stem and progenitor cells (HSPCs).
The lipids, known as epoxyeicosatrienoic acids (EETs), boosted HSPC engraftment in zebrafish and mice.
The researchers therefore believe EETs could help make human HSPC transplants, particularly umbilical cord blood transplants, more efficient and effective.
“Ninety percent of cord blood units can’t be used because they’re too small,” said Leonard Zon, MD, of Boston Children’s Hospital in Massachusetts.
“If you add these chemicals, you might be able to use more units. Being able to get engraftment allows you to pick a smaller cord blood sample that might be a better match.”
Dr Zon and his colleagues described their work with EETs in Nature.
EETs appear to work by stimulating cell migration. They were among the top hits in a screen of 500 known compounds the researchers conducted.
In the past, such screens have led Dr Zon’s team to compounds that boost HSPC numbers, such as prostaglandin. But the new drug screen was designed to assess HSPCs’ transplantability and engraftment.
The screen was done in a lab-created strain of zebrafish called Casper. Because Casper is translucent, Dr Zon and his colleagues could visually compare the engraftment of transplanted HSPCs chemically tagged to glow green or red.
The researchers first used tagging to color the fishes’ marrow either red or green, then removed HSPCs for transplantation. The green cells were incubated with various chemicals, while the red cells were left untreated.
The team then injected a mixture of green and red HSPCs into other groups of zebrafish (10 fish per test chemical). And they visually tracked the cells’ activity, measuring the green-to-red ratio.
“The expectation was that if a chemical didn’t increase engraftment, all the fish would be equal parts red and green,” Dr Zon said. “But if it was effective, green marrow would predominate.”
That was the case for green marrow incubated with EETs, a finding that held up over thousands of transplants.
“In a mouse system, this experiment would cost $3 million,” Dr Zon noted. “In fish, it cost about $150,000.”
In a smaller-scale set of mouse experiments, the team confirmed EETs’ efficacy in promoting homing and engraftment of HSPCs.
EETs are chemical cousins of prostaglandin. Both are made from arachidonic acid, and both are made during inflammation. But EETs work in a different way, by activating the PI3K pathway. EETs also enhanced PI3K activity in human blood vessel cells in vitro.
After more studies in human cells to determine exactly how EETs work, Dr Zon hopes to begin clinical trials of EETs within the next 2 years, likely in the setting of cord blood transplant. The lab is also investigating its other top hits from the zebrafish screen.
“Every new pathway that we find has the chance of making stem cell engraftment and migration even better,” Dr Zon said. “I think we’ll end up being able to manipulate this process.” ![]()
Tests improve treatment of malaria

artemisinin-based combination
therapy. Photo courtesy of
The Global Fund
Introducing rapid diagnostic tests in drug shops can improve the treatment of malaria, according to research published in PLOS ONE.
Most of the 15,000 patients in this study, all of whom visited drug shops with a fever, chose to buy a rapid diagnostic test when offered one.
Test results showed that less than 60% of the patients had malaria, and vendors usually complied with the results, which reduced overprescription of malaria drugs by 73%.
Investigators conducted this study because the private sector is a common source of treatment in many malaria-endemic areas, especially where there is poor access to public health facilities.
Patients buy antimalarial drugs in shops to medicate themselves, but malaria is not always the cause of their fever, so inappropriate treatment is common.
“Our findings show that it is feasible to collaborate with the private health sector and introduce malaria rapid diagnostic tests in drug shops,” said study author Anthony Mbonye, PhD, of the Ugandan Ministry of Health in Kampala, Uganda.
“The next step is to refine the strategy and understand the cost implications of scaling it up in Uganda. Our long-term aim is to provide evidence to help the World Health Organization develop guidance to improve malaria treatment in the private sector.”
For this study, Dr Mbonye and his colleagues introduced rapid diagnostic tests in 10 clusters of drug shops in the Mukono district of central Uganda.
The team compared results at these shops to results at 10 other clusters of shops in the control arm, where treatment was given based on patients’ signs and symptoms.
The vendors’ decision of whether to treat a patient with artemisinin-based combination therapy was validated by confirming the presence of malaria parasites in the patient’s blood through microscopy carried out by the research team.
The rapid diagnostic tests reduced overdiagnosis and overprescription of malaria treatment by 73%, increasing appropriate treatment with artemisinin-based combination therapy by 36%.
“This study shows that rapid diagnostic tests can improve the use of artemisinin-based combination therapies—the most effective treatment for malaria—in drug shops, but it’s not without its challenges,” said Sian Clarke, PhD, of the London School of Hygiene & Tropical Medicine in the UK.
“These tests alone will not improve the treatment of other diseases. We now need to continue working with the Ministry of Health to investigate how to improve our approach and expand it to other common illnesses.”
An investigation conducted alongside this trial showed that, despite their popularity, rapid diagnostic tests for malaria were not a simple fix in the private sector.
Patients welcomed the rapid diagnostic tests as well as government involvement in improving drug shops. And vendors felt more akin to qualified health workers in the public sector for being allowed to test blood.
But investigators warn that this could give a false impression of vendors’ other skills and services, and regulation by authorities is needed.
This report was published in Critical Public Health. ![]()
artemisinin-based combination
therapy. Photo courtesy of
The Global Fund
Introducing rapid diagnostic tests in drug shops can improve the treatment of malaria, according to research published in PLOS ONE.
Most of the 15,000 patients in this study, all of whom visited drug shops with a fever, chose to buy a rapid diagnostic test when offered one.
Test results showed that less than 60% of the patients had malaria, and vendors usually complied with the results, which reduced overprescription of malaria drugs by 73%.
Investigators conducted this study because the private sector is a common source of treatment in many malaria-endemic areas, especially where there is poor access to public health facilities.
Patients buy antimalarial drugs in shops to medicate themselves, but malaria is not always the cause of their fever, so inappropriate treatment is common.
“Our findings show that it is feasible to collaborate with the private health sector and introduce malaria rapid diagnostic tests in drug shops,” said study author Anthony Mbonye, PhD, of the Ugandan Ministry of Health in Kampala, Uganda.
“The next step is to refine the strategy and understand the cost implications of scaling it up in Uganda. Our long-term aim is to provide evidence to help the World Health Organization develop guidance to improve malaria treatment in the private sector.”
For this study, Dr Mbonye and his colleagues introduced rapid diagnostic tests in 10 clusters of drug shops in the Mukono district of central Uganda.
The team compared results at these shops to results at 10 other clusters of shops in the control arm, where treatment was given based on patients’ signs and symptoms.
The vendors’ decision of whether to treat a patient with artemisinin-based combination therapy was validated by confirming the presence of malaria parasites in the patient’s blood through microscopy carried out by the research team.
The rapid diagnostic tests reduced overdiagnosis and overprescription of malaria treatment by 73%, increasing appropriate treatment with artemisinin-based combination therapy by 36%.
“This study shows that rapid diagnostic tests can improve the use of artemisinin-based combination therapies—the most effective treatment for malaria—in drug shops, but it’s not without its challenges,” said Sian Clarke, PhD, of the London School of Hygiene & Tropical Medicine in the UK.
“These tests alone will not improve the treatment of other diseases. We now need to continue working with the Ministry of Health to investigate how to improve our approach and expand it to other common illnesses.”
An investigation conducted alongside this trial showed that, despite their popularity, rapid diagnostic tests for malaria were not a simple fix in the private sector.
Patients welcomed the rapid diagnostic tests as well as government involvement in improving drug shops. And vendors felt more akin to qualified health workers in the public sector for being allowed to test blood.
But investigators warn that this could give a false impression of vendors’ other skills and services, and regulation by authorities is needed.
This report was published in Critical Public Health. ![]()
artemisinin-based combination
therapy. Photo courtesy of
The Global Fund
Introducing rapid diagnostic tests in drug shops can improve the treatment of malaria, according to research published in PLOS ONE.
Most of the 15,000 patients in this study, all of whom visited drug shops with a fever, chose to buy a rapid diagnostic test when offered one.
Test results showed that less than 60% of the patients had malaria, and vendors usually complied with the results, which reduced overprescription of malaria drugs by 73%.
Investigators conducted this study because the private sector is a common source of treatment in many malaria-endemic areas, especially where there is poor access to public health facilities.
Patients buy antimalarial drugs in shops to medicate themselves, but malaria is not always the cause of their fever, so inappropriate treatment is common.
“Our findings show that it is feasible to collaborate with the private health sector and introduce malaria rapid diagnostic tests in drug shops,” said study author Anthony Mbonye, PhD, of the Ugandan Ministry of Health in Kampala, Uganda.
“The next step is to refine the strategy and understand the cost implications of scaling it up in Uganda. Our long-term aim is to provide evidence to help the World Health Organization develop guidance to improve malaria treatment in the private sector.”
For this study, Dr Mbonye and his colleagues introduced rapid diagnostic tests in 10 clusters of drug shops in the Mukono district of central Uganda.
The team compared results at these shops to results at 10 other clusters of shops in the control arm, where treatment was given based on patients’ signs and symptoms.
The vendors’ decision of whether to treat a patient with artemisinin-based combination therapy was validated by confirming the presence of malaria parasites in the patient’s blood through microscopy carried out by the research team.
The rapid diagnostic tests reduced overdiagnosis and overprescription of malaria treatment by 73%, increasing appropriate treatment with artemisinin-based combination therapy by 36%.
“This study shows that rapid diagnostic tests can improve the use of artemisinin-based combination therapies—the most effective treatment for malaria—in drug shops, but it’s not without its challenges,” said Sian Clarke, PhD, of the London School of Hygiene & Tropical Medicine in the UK.
“These tests alone will not improve the treatment of other diseases. We now need to continue working with the Ministry of Health to investigate how to improve our approach and expand it to other common illnesses.”
An investigation conducted alongside this trial showed that, despite their popularity, rapid diagnostic tests for malaria were not a simple fix in the private sector.
Patients welcomed the rapid diagnostic tests as well as government involvement in improving drug shops. And vendors felt more akin to qualified health workers in the public sector for being allowed to test blood.
But investigators warn that this could give a false impression of vendors’ other skills and services, and regulation by authorities is needed.
This report was published in Critical Public Health.
Database may help predict cancer patients’ survival

Photo by Darren Baker
A newly developed database may help physicians predict survival outcomes in patients with hematologic and solid tumor malignancies, according to a paper published in Nature Medicine.
The database, known as PRECOG, integrates gene expression patterns of 39 types of cancer from nearly 18,000 patients with data about how long those patients lived.
By combining these data, researchers were able to see broad patterns that correlate with survival. They also believe this information could help them pinpoint potential therapeutic targets for a range of cancers.
“We were able to identify key pathways that can dramatically stratify survival across diverse cancer types,” said Ash Alizadeh, MD, PhD, of Stanford University in California.
“The patterns were very striking, especially because few such examples are currently available for the use of genes or immune cells for cancer prognosis.”
In addition to identifying potentially useful gene expression patterns, the researchers used an analytical tool called CIBERSORT to determine the composition of leukocytes that flock to a tumor.
“We were able to infer which immune cells are present or absent in individual solid tumors, to estimate their prevalence, and to correlate that information with patient survival,” said Aaron Newman, PhD, of Stanford University.
“We found you can even broadly distinguish cancer types just based on what kind of immune cells have infiltrated the tumor.”
Compiling the data
Researchers have tried for years to identify specific patterns of gene expression in cancerous tumors that differ from those in normal tissue. But the extreme variability among individual patients and tumors has made the process difficult, even when focused on particular cancer types.
“There are many more genes in a cell than there are patients with any one type of cancer, and this makes discovering the important genes for cancer outcomes a tough problem,” said Andrew Gentles, PhD, of Stanford University.
“Because it’s easy to find spurious associations that don’t hold up in follow-up studies, we combined information from a vast array of cancer types to better see meaningful correlations.”
The researchers first collected publicly available data on gene expression patterns of many types of cancers.
They then matched the gene expression profiles with clinical information about the patients, including their age, disease status, and how long they survived after diagnosis. Finally, the team combined the studies in a database.
“We wanted to be able to connect gene expression data with patient outcome for thousands of people at once,” Dr Alizadeh said. “Then, we could ask what we could learn more broadly.”
Surprising findings
The researchers were surprised to find that prognostic genes were often shared among distinct cancer types, suggesting that similar biological programs impact survival across cancers.
They were able to identify the top 10 genes that seemed to confer adverse outcomes—FOXM1, BIRC5, TOP2A, TPX2, NME1, CCNB1, CEP55, TYMS, CENPF, and CDKN3—and the top 10 genes associated with more positive outcomes—KLRB1, ITM2B, CBX7, CD2, CREBL2, SATB1, NR3C1, TMEM66, KLRK1, and FUCA1.
Many of these genes are involved in aspects of cell division or are associated with distinct leukocytes that flood a tumor.
The researchers were also able to identify combinations of leukocytes that appear to be correlated with outcomes.
In particular, elevated numbers of plasma cells and certain types of T cells correlated with better patient survival rates across many different solid tumors. But a high proportion of granulocytes was associated with adverse outcomes.
The researchers hope that PRECOG and CIBERSORT will increase our understanding of cancer biology and aid the development of new therapies for cancer patients. The team is applying these tools to better predict which patients will respond to new and emerging anticancer therapies.
Dr Alizadeh said this is especially important given recent advances in the development of drugs that engage immune responses but work well only for a subset of cancer patients.
Photo by Darren Baker
A newly developed database may help physicians predict survival outcomes in patients with hematologic and solid tumor malignancies, according to a paper published in Nature Medicine.
The database, known as PRECOG, integrates gene expression patterns of 39 types of cancer from nearly 18,000 patients with data about how long those patients lived.
By combining these data, researchers were able to see broad patterns that correlate with survival. They also believe this information could help them pinpoint potential therapeutic targets for a range of cancers.
“We were able to identify key pathways that can dramatically stratify survival across diverse cancer types,” said Ash Alizadeh, MD, PhD, of Stanford University in California.
“The patterns were very striking, especially because few such examples are currently available for the use of genes or immune cells for cancer prognosis.”
In addition to identifying potentially useful gene expression patterns, the researchers used an analytical tool called CIBERSORT to determine the composition of leukocytes that flock to a tumor.
“We were able to infer which immune cells are present or absent in individual solid tumors, to estimate their prevalence, and to correlate that information with patient survival,” said Aaron Newman, PhD, of Stanford University.
“We found you can even broadly distinguish cancer types just based on what kind of immune cells have infiltrated the tumor.”
Compiling the data
Researchers have tried for years to identify specific patterns of gene expression in cancerous tumors that differ from those in normal tissue. But the extreme variability among individual patients and tumors has made the process difficult, even when focused on particular cancer types.
“There are many more genes in a cell than there are patients with any one type of cancer, and this makes discovering the important genes for cancer outcomes a tough problem,” said Andrew Gentles, PhD, of Stanford University.
“Because it’s easy to find spurious associations that don’t hold up in follow-up studies, we combined information from a vast array of cancer types to better see meaningful correlations.”
The researchers first collected publicly available data on gene expression patterns of many types of cancers.
They then matched the gene expression profiles with clinical information about the patients, including their age, disease status, and how long they survived after diagnosis. Finally, the team combined the studies in a database.
“We wanted to be able to connect gene expression data with patient outcome for thousands of people at once,” Dr Alizadeh said. “Then, we could ask what we could learn more broadly.”
Surprising findings
The researchers were surprised to find that prognostic genes were often shared among distinct cancer types, suggesting that similar biological programs impact survival across cancers.
They were able to identify the top 10 genes that seemed to confer adverse outcomes—FOXM1, BIRC5, TOP2A, TPX2, NME1, CCNB1, CEP55, TYMS, CENPF, and CDKN3—and the top 10 genes associated with more positive outcomes—KLRB1, ITM2B, CBX7, CD2, CREBL2, SATB1, NR3C1, TMEM66, KLRK1, and FUCA1.
Many of these genes are involved in aspects of cell division or are associated with distinct leukocytes that flood a tumor.
The researchers were also able to identify combinations of leukocytes that appear to be correlated with outcomes.
In particular, elevated numbers of plasma cells and certain types of T cells correlated with better patient survival rates across many different solid tumors. But a high proportion of granulocytes was associated with adverse outcomes.
The researchers hope that PRECOG and CIBERSORT will increase our understanding of cancer biology and aid the development of new therapies for cancer patients. The team is applying these tools to better predict which patients will respond to new and emerging anticancer therapies.
Dr Alizadeh said this is especially important given recent advances in the development of drugs that engage immune responses but work well only for a subset of cancer patients.
Photo by Darren Baker
A newly developed database may help physicians predict survival outcomes in patients with hematologic and solid tumor malignancies, according to a paper published in Nature Medicine.
The database, known as PRECOG, integrates gene expression patterns of 39 types of cancer from nearly 18,000 patients with data about how long those patients lived.
By combining these data, researchers were able to see broad patterns that correlate with survival. They also believe this information could help them pinpoint potential therapeutic targets for a range of cancers.
“We were able to identify key pathways that can dramatically stratify survival across diverse cancer types,” said Ash Alizadeh, MD, PhD, of Stanford University in California.
“The patterns were very striking, especially because few such examples are currently available for the use of genes or immune cells for cancer prognosis.”
In addition to identifying potentially useful gene expression patterns, the researchers used an analytical tool called CIBERSORT to determine the composition of leukocytes that flock to a tumor.
“We were able to infer which immune cells are present or absent in individual solid tumors, to estimate their prevalence, and to correlate that information with patient survival,” said Aaron Newman, PhD, of Stanford University.
“We found you can even broadly distinguish cancer types just based on what kind of immune cells have infiltrated the tumor.”
Compiling the data
Researchers have tried for years to identify specific patterns of gene expression in cancerous tumors that differ from those in normal tissue. But the extreme variability among individual patients and tumors has made the process difficult, even when focused on particular cancer types.
“There are many more genes in a cell than there are patients with any one type of cancer, and this makes discovering the important genes for cancer outcomes a tough problem,” said Andrew Gentles, PhD, of Stanford University.
“Because it’s easy to find spurious associations that don’t hold up in follow-up studies, we combined information from a vast array of cancer types to better see meaningful correlations.”
The researchers first collected publicly available data on gene expression patterns of many types of cancers.
They then matched the gene expression profiles with clinical information about the patients, including their age, disease status, and how long they survived after diagnosis. Finally, the team combined the studies in a database.
“We wanted to be able to connect gene expression data with patient outcome for thousands of people at once,” Dr Alizadeh said. “Then, we could ask what we could learn more broadly.”
Surprising findings
The researchers were surprised to find that prognostic genes were often shared among distinct cancer types, suggesting that similar biological programs impact survival across cancers.
They were able to identify the top 10 genes that seemed to confer adverse outcomes—FOXM1, BIRC5, TOP2A, TPX2, NME1, CCNB1, CEP55, TYMS, CENPF, and CDKN3—and the top 10 genes associated with more positive outcomes—KLRB1, ITM2B, CBX7, CD2, CREBL2, SATB1, NR3C1, TMEM66, KLRK1, and FUCA1.
Many of these genes are involved in aspects of cell division or are associated with distinct leukocytes that flood a tumor.
The researchers were also able to identify combinations of leukocytes that appear to be correlated with outcomes.
In particular, elevated numbers of plasma cells and certain types of T cells correlated with better patient survival rates across many different solid tumors. But a high proportion of granulocytes was associated with adverse outcomes.
The researchers hope that PRECOG and CIBERSORT will increase our understanding of cancer biology and aid the development of new therapies for cancer patients. The team is applying these tools to better predict which patients will respond to new and emerging anticancer therapies.
Dr Alizadeh said this is especially important given recent advances in the development of drugs that engage immune responses but work well only for a subset of cancer patients.
Antibiotic can affect INR levels

Treatment with the antibiotic dicloxacillin may cause a significant decrease in international normalized ratio (INR) levels among patients taking vitamin K antagonists (VKAs), according to research published in JAMA.
Researchers studied 7400 patients on VKA therapy and found that 61% of patients taking warfarin and dicloxacillin experienced a decrease in INR after dicloxacillin exposure.
Forty-one percent of patients taking phenprocoumon had a decrease in INR after exposure to the antibiotic.
Anton Pottegard, PhD, of the University of Southern Denmark, Odense, and his colleagues conducted this research using Thrombobase, a clinical database of all VKA-treated patients followed up by 3 outpatient clinics and 50 general practitioners in Funen, Denmark.
The researchers included all patients who filled a prescription for dicloxacillin while receiving warfarin or phenprocoumon between March 1998 and November 2012.
INR results were grouped by the week relative to dicloxacillin exposure. The last INR measurement before dicloxacillin exposure was compared with the first measurement within weeks 2 to 4 after dicloxacillin exposure.
Of the 519 patients taking warfarin and initiating treatment with dicloxacillin, 236 met inclusion criteria. The average INR level was 2.6 prior to dicloxacillin exposure and 2 at two to four weeks after dicloxacillin exposure.
In total, 61% of patients (n=144) experienced sub-therapeutic INR levels (<2.0) within 2 to 4 weeks of dicloxacillin exposure.
Among patients taking phenprocoumon (n=64), average INR levels were 2.6 before exposure to dicloxacillin and 2.3 at two to four weeks after exposure. The proportion of patients with sub-therapeutic INR levels after dicloxacillin exposure was 41% (n=26).
The researchers said these results suggest treatment with dicloxacillin can cause a significant decrease in INR levels among patients taking VKAs.
Treatment with the antibiotic dicloxacillin may cause a significant decrease in international normalized ratio (INR) levels among patients taking vitamin K antagonists (VKAs), according to research published in JAMA.
Researchers studied 7400 patients on VKA therapy and found that 61% of patients taking warfarin and dicloxacillin experienced a decrease in INR after dicloxacillin exposure.
Forty-one percent of patients taking phenprocoumon had a decrease in INR after exposure to the antibiotic.
Anton Pottegard, PhD, of the University of Southern Denmark, Odense, and his colleagues conducted this research using Thrombobase, a clinical database of all VKA-treated patients followed up by 3 outpatient clinics and 50 general practitioners in Funen, Denmark.
The researchers included all patients who filled a prescription for dicloxacillin while receiving warfarin or phenprocoumon between March 1998 and November 2012.
INR results were grouped by the week relative to dicloxacillin exposure. The last INR measurement before dicloxacillin exposure was compared with the first measurement within weeks 2 to 4 after dicloxacillin exposure.
Of the 519 patients taking warfarin and initiating treatment with dicloxacillin, 236 met inclusion criteria. The average INR level was 2.6 prior to dicloxacillin exposure and 2 at two to four weeks after dicloxacillin exposure.
In total, 61% of patients (n=144) experienced sub-therapeutic INR levels (<2.0) within 2 to 4 weeks of dicloxacillin exposure.
Among patients taking phenprocoumon (n=64), average INR levels were 2.6 before exposure to dicloxacillin and 2.3 at two to four weeks after exposure. The proportion of patients with sub-therapeutic INR levels after dicloxacillin exposure was 41% (n=26).
The researchers said these results suggest treatment with dicloxacillin can cause a significant decrease in INR levels among patients taking VKAs.
Treatment with the antibiotic dicloxacillin may cause a significant decrease in international normalized ratio (INR) levels among patients taking vitamin K antagonists (VKAs), according to research published in JAMA.
Researchers studied 7400 patients on VKA therapy and found that 61% of patients taking warfarin and dicloxacillin experienced a decrease in INR after dicloxacillin exposure.
Forty-one percent of patients taking phenprocoumon had a decrease in INR after exposure to the antibiotic.
Anton Pottegard, PhD, of the University of Southern Denmark, Odense, and his colleagues conducted this research using Thrombobase, a clinical database of all VKA-treated patients followed up by 3 outpatient clinics and 50 general practitioners in Funen, Denmark.
The researchers included all patients who filled a prescription for dicloxacillin while receiving warfarin or phenprocoumon between March 1998 and November 2012.
INR results were grouped by the week relative to dicloxacillin exposure. The last INR measurement before dicloxacillin exposure was compared with the first measurement within weeks 2 to 4 after dicloxacillin exposure.
Of the 519 patients taking warfarin and initiating treatment with dicloxacillin, 236 met inclusion criteria. The average INR level was 2.6 prior to dicloxacillin exposure and 2 at two to four weeks after dicloxacillin exposure.
In total, 61% of patients (n=144) experienced sub-therapeutic INR levels (<2.0) within 2 to 4 weeks of dicloxacillin exposure.
Among patients taking phenprocoumon (n=64), average INR levels were 2.6 before exposure to dicloxacillin and 2.3 at two to four weeks after exposure. The proportion of patients with sub-therapeutic INR levels after dicloxacillin exposure was 41% (n=26).
The researchers said these results suggest treatment with dicloxacillin can cause a significant decrease in INR levels among patients taking VKAs.
Common chemicals may increase cancer risk

Common environmental chemicals assumed to be safe at low doses may act separately or together to induce cancer development, according to research published in Carcinogenesis.
Investigators studied low-dose effects of 85 common chemicals not considered to be carcinogenic to humans, reviewing the actions of these chemicals against a long list of mechanisms that are important for cancer development.
The team compared the chemicals’ biological activity patterns to 11 known hallmarks of cancer—distinctive patterns of cellular and genetic disruption associated with early cancer development.
The chemicals included the pain reliever acetaminophen; bisphenol A (BPA), which is used in plastic food and beverage containers; rotenone, a broad-spectrum insecticide; paraquat, an agricultural herbicide; and triclosan, an antibacterial agent used in soaps and cosmetics.
The investigators learned that 50 of the 85 chemicals they analyzed can disrupt cell function in ways that correlate with known early patterns of cancer, even at the low, presumably benign levels at which most people are exposed.
For 13 of the chemicals, the team found evidence of a dose-response threshold—a level of exposure at which a chemical is considered toxic by regulators. For 22 chemicals, there was no toxicity information at all.
“Our findings also suggest these molecules may be acting in synergy to increase cancer activity,” said William Bisson, PhD, of Oregon State University in Corvallis.
“For example, EDTA, a metal-ion-binding compound used in manufacturing and medicine, interferes with the body’s repair of damaged genes. EDTA doesn’t cause genetic mutations itself, but if you’re exposed to it along with some substance that is mutagenic, it enhances the effect because it disrupts DNA repair, a key layer of cancer defense.”
Dr Bisson said the main purpose of this study was to highlight gaps in knowledge of environmentally influenced cancers and to set forth a research agenda for the next few years. He added that more research is still necessary to assess early exposure and to understand early stages of cancer development.
Traditional risk assessment, Dr Bisson said, has historically focused on a quest for single chemicals and single modes of action—approaches that may underestimate cancer risk. With this study, investigators took a different tack, examining the interplay over time of independent molecular processes triggered by low-dose exposures to chemicals.
“Cancer is a disease of diseases,” Dr Bisson said. “It follows multi-step development patterns, and, in most cases, it has a long latency period. It has to be tackled from an angle that considers the complexity of these patterns. A better understanding of what’s driving things to the point where they get uncontrollable will be key for the development of effective strategies for prevention and early detection.”
Common environmental chemicals assumed to be safe at low doses may act separately or together to induce cancer development, according to research published in Carcinogenesis.
Investigators studied low-dose effects of 85 common chemicals not considered to be carcinogenic to humans, reviewing the actions of these chemicals against a long list of mechanisms that are important for cancer development.
The team compared the chemicals’ biological activity patterns to 11 known hallmarks of cancer—distinctive patterns of cellular and genetic disruption associated with early cancer development.
The chemicals included the pain reliever acetaminophen; bisphenol A (BPA), which is used in plastic food and beverage containers; rotenone, a broad-spectrum insecticide; paraquat, an agricultural herbicide; and triclosan, an antibacterial agent used in soaps and cosmetics.
The investigators learned that 50 of the 85 chemicals they analyzed can disrupt cell function in ways that correlate with known early patterns of cancer, even at the low, presumably benign levels at which most people are exposed.
For 13 of the chemicals, the team found evidence of a dose-response threshold—a level of exposure at which a chemical is considered toxic by regulators. For 22 chemicals, there was no toxicity information at all.
“Our findings also suggest these molecules may be acting in synergy to increase cancer activity,” said William Bisson, PhD, of Oregon State University in Corvallis.
“For example, EDTA, a metal-ion-binding compound used in manufacturing and medicine, interferes with the body’s repair of damaged genes. EDTA doesn’t cause genetic mutations itself, but if you’re exposed to it along with some substance that is mutagenic, it enhances the effect because it disrupts DNA repair, a key layer of cancer defense.”
Dr Bisson said the main purpose of this study was to highlight gaps in knowledge of environmentally influenced cancers and to set forth a research agenda for the next few years. He added that more research is still necessary to assess early exposure and to understand early stages of cancer development.
Traditional risk assessment, Dr Bisson said, has historically focused on a quest for single chemicals and single modes of action—approaches that may underestimate cancer risk. With this study, investigators took a different tack, examining the interplay over time of independent molecular processes triggered by low-dose exposures to chemicals.
“Cancer is a disease of diseases,” Dr Bisson said. “It follows multi-step development patterns, and, in most cases, it has a long latency period. It has to be tackled from an angle that considers the complexity of these patterns. A better understanding of what’s driving things to the point where they get uncontrollable will be key for the development of effective strategies for prevention and early detection.”
Common environmental chemicals assumed to be safe at low doses may act separately or together to induce cancer development, according to research published in Carcinogenesis.
Investigators studied low-dose effects of 85 common chemicals not considered to be carcinogenic to humans, reviewing the actions of these chemicals against a long list of mechanisms that are important for cancer development.
The team compared the chemicals’ biological activity patterns to 11 known hallmarks of cancer—distinctive patterns of cellular and genetic disruption associated with early cancer development.
The chemicals included the pain reliever acetaminophen; bisphenol A (BPA), which is used in plastic food and beverage containers; rotenone, a broad-spectrum insecticide; paraquat, an agricultural herbicide; and triclosan, an antibacterial agent used in soaps and cosmetics.
The investigators learned that 50 of the 85 chemicals they analyzed can disrupt cell function in ways that correlate with known early patterns of cancer, even at the low, presumably benign levels at which most people are exposed.
For 13 of the chemicals, the team found evidence of a dose-response threshold—a level of exposure at which a chemical is considered toxic by regulators. For 22 chemicals, there was no toxicity information at all.
“Our findings also suggest these molecules may be acting in synergy to increase cancer activity,” said William Bisson, PhD, of Oregon State University in Corvallis.
“For example, EDTA, a metal-ion-binding compound used in manufacturing and medicine, interferes with the body’s repair of damaged genes. EDTA doesn’t cause genetic mutations itself, but if you’re exposed to it along with some substance that is mutagenic, it enhances the effect because it disrupts DNA repair, a key layer of cancer defense.”
Dr Bisson said the main purpose of this study was to highlight gaps in knowledge of environmentally influenced cancers and to set forth a research agenda for the next few years. He added that more research is still necessary to assess early exposure and to understand early stages of cancer development.
Traditional risk assessment, Dr Bisson said, has historically focused on a quest for single chemicals and single modes of action—approaches that may underestimate cancer risk. With this study, investigators took a different tack, examining the interplay over time of independent molecular processes triggered by low-dose exposures to chemicals.
“Cancer is a disease of diseases,” Dr Bisson said. “It follows multi-step development patterns, and, in most cases, it has a long latency period. It has to be tackled from an angle that considers the complexity of these patterns. A better understanding of what’s driving things to the point where they get uncontrollable will be key for the development of effective strategies for prevention and early detection.”