User login
Meeting the potential of immunotherapy: new targets provide rational combinations
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.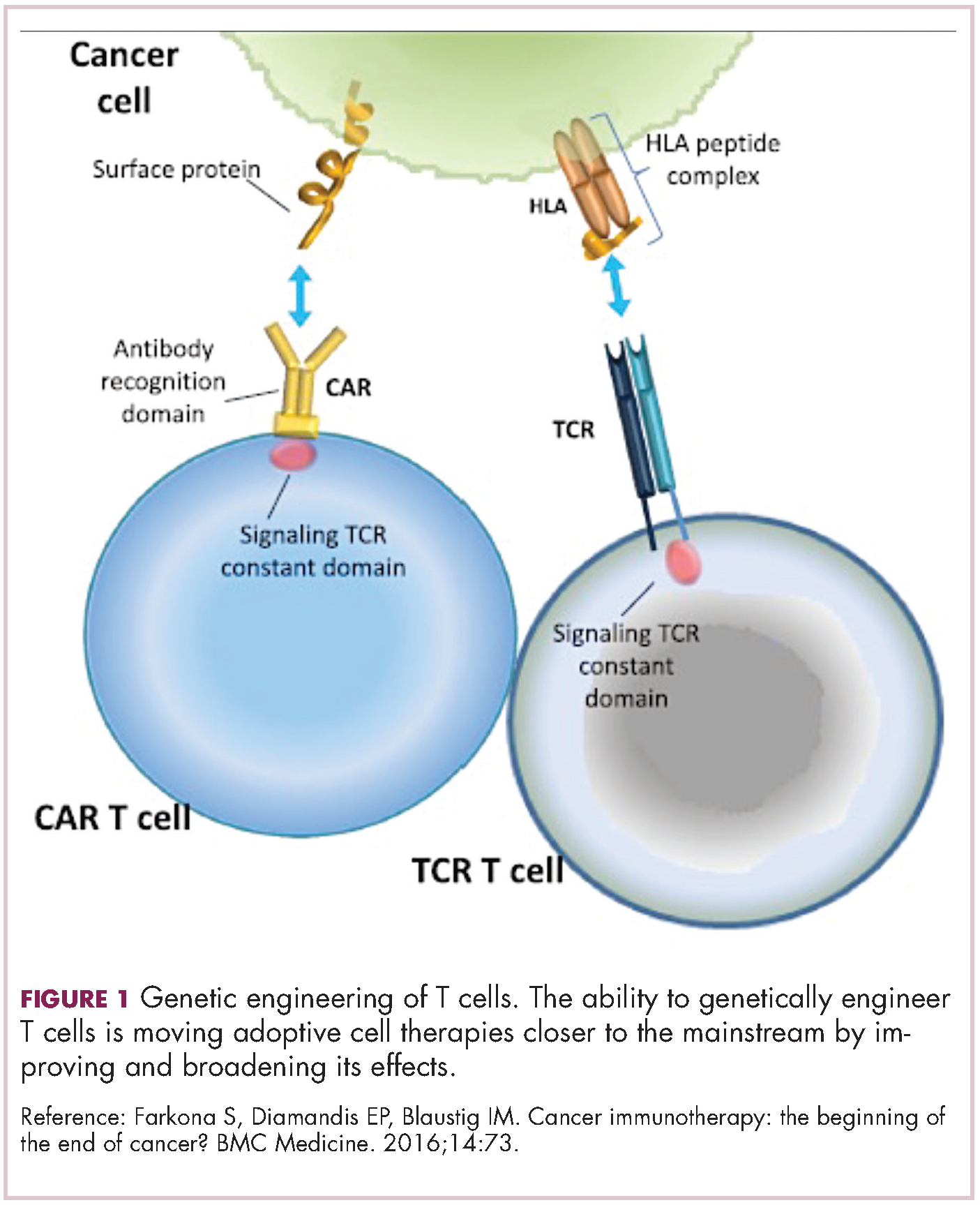
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
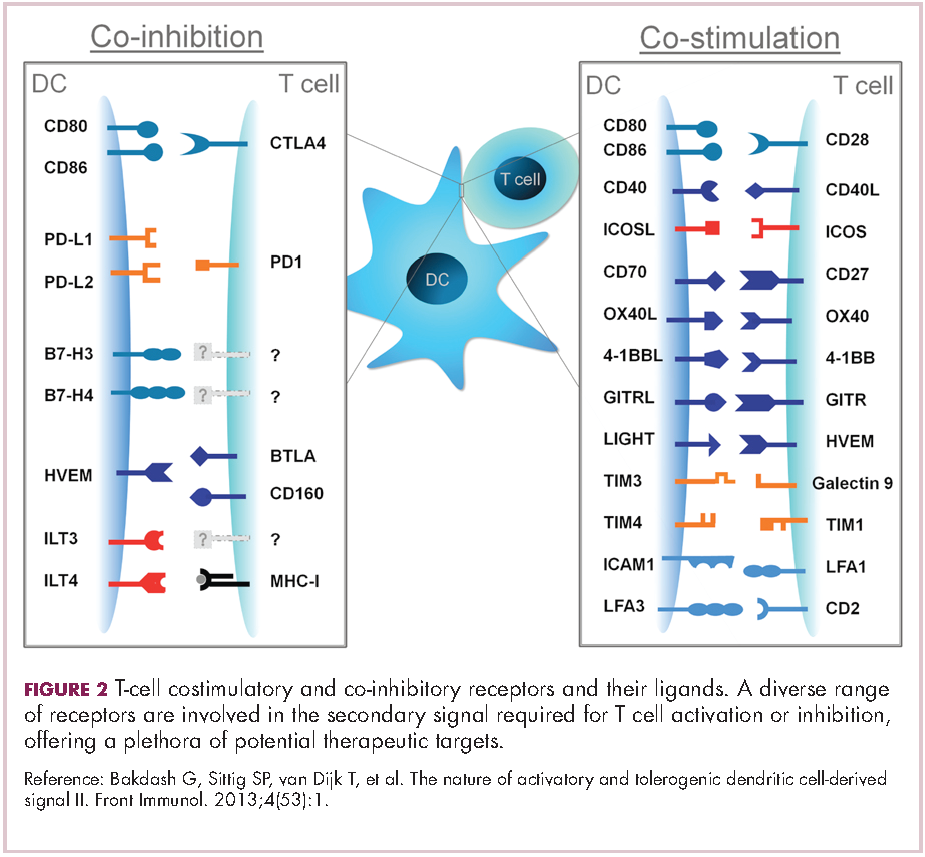
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17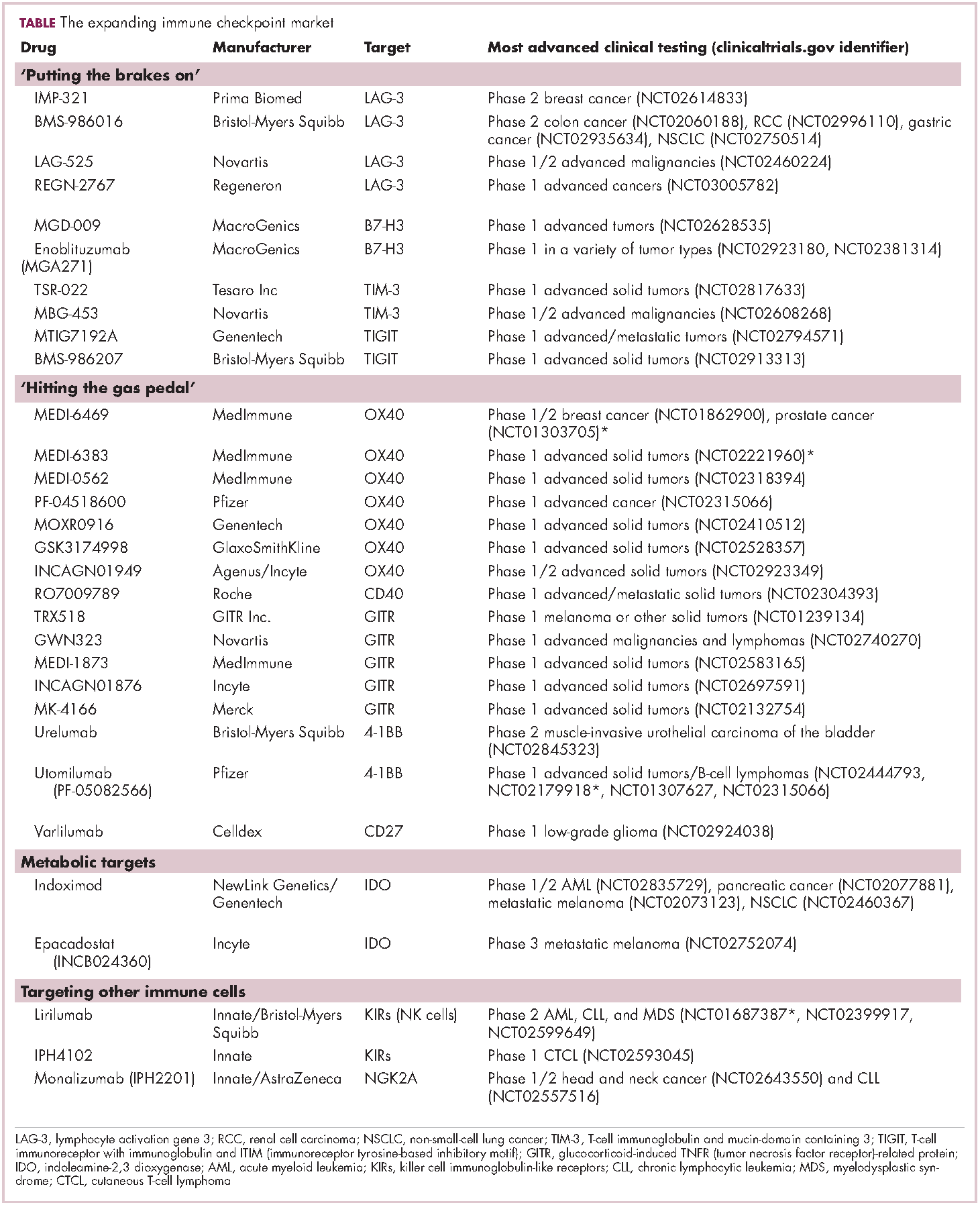
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).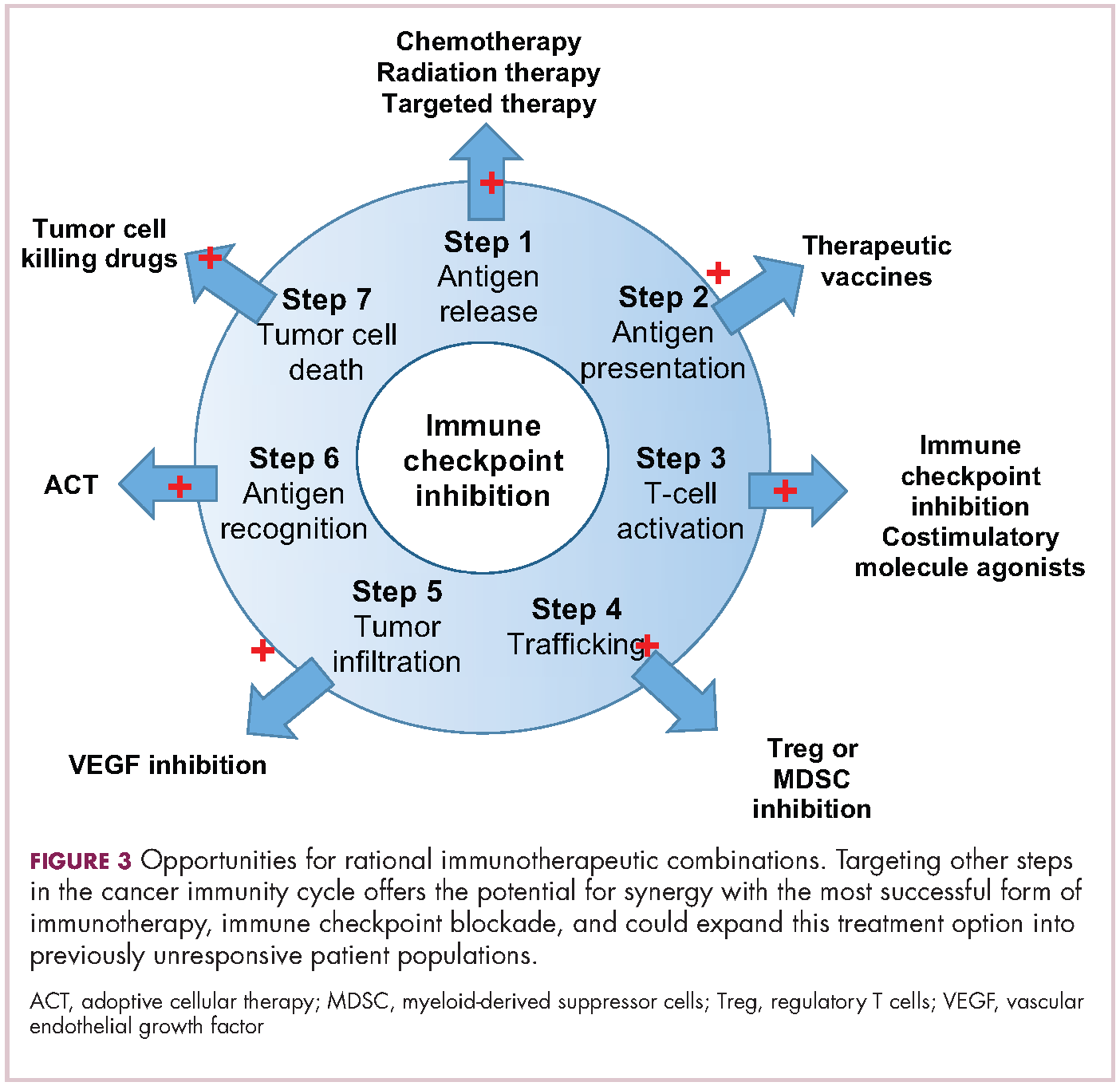
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
Immunotherapies shape the treatment landscape for hematologic malignancies
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.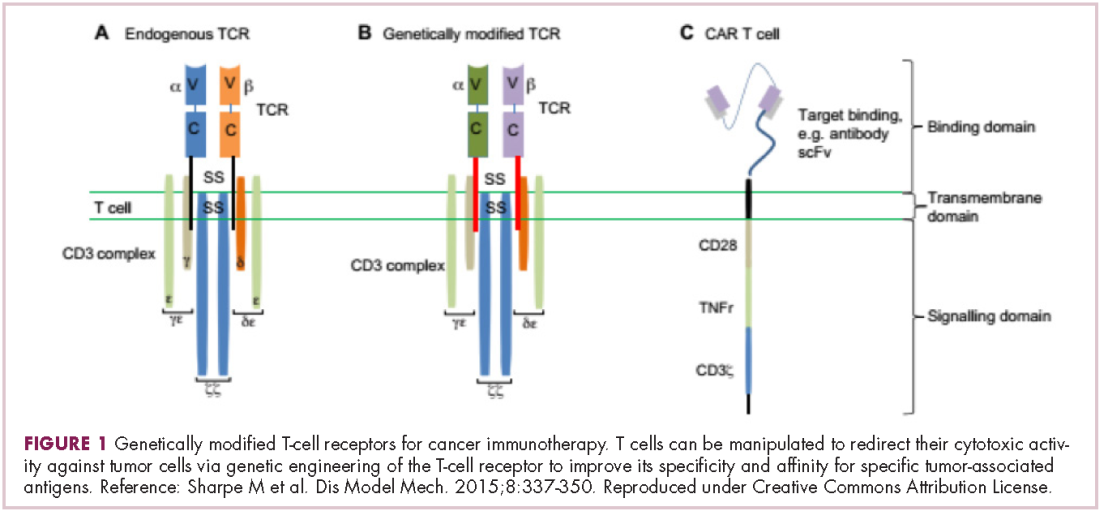
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5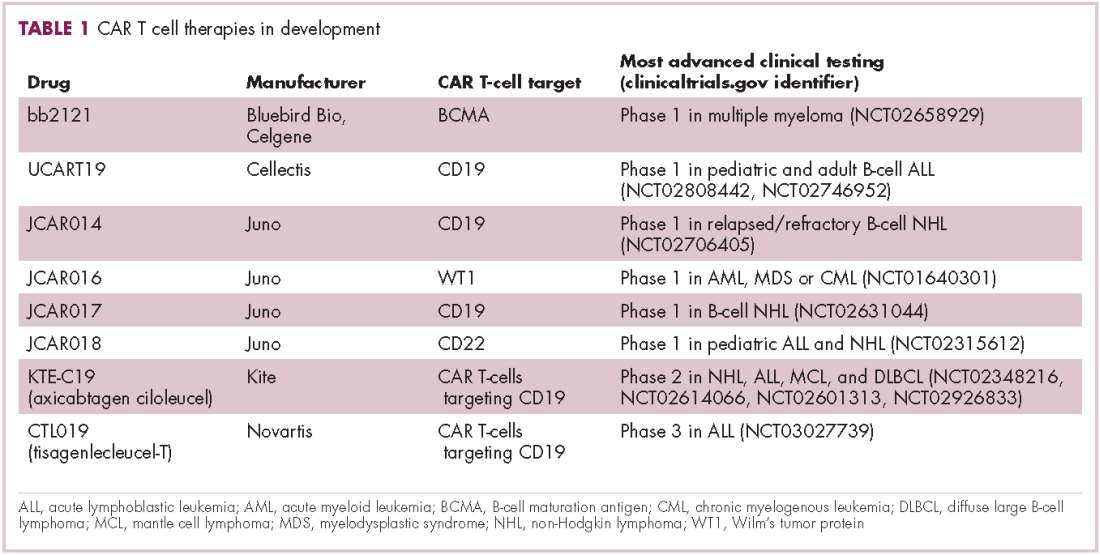
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12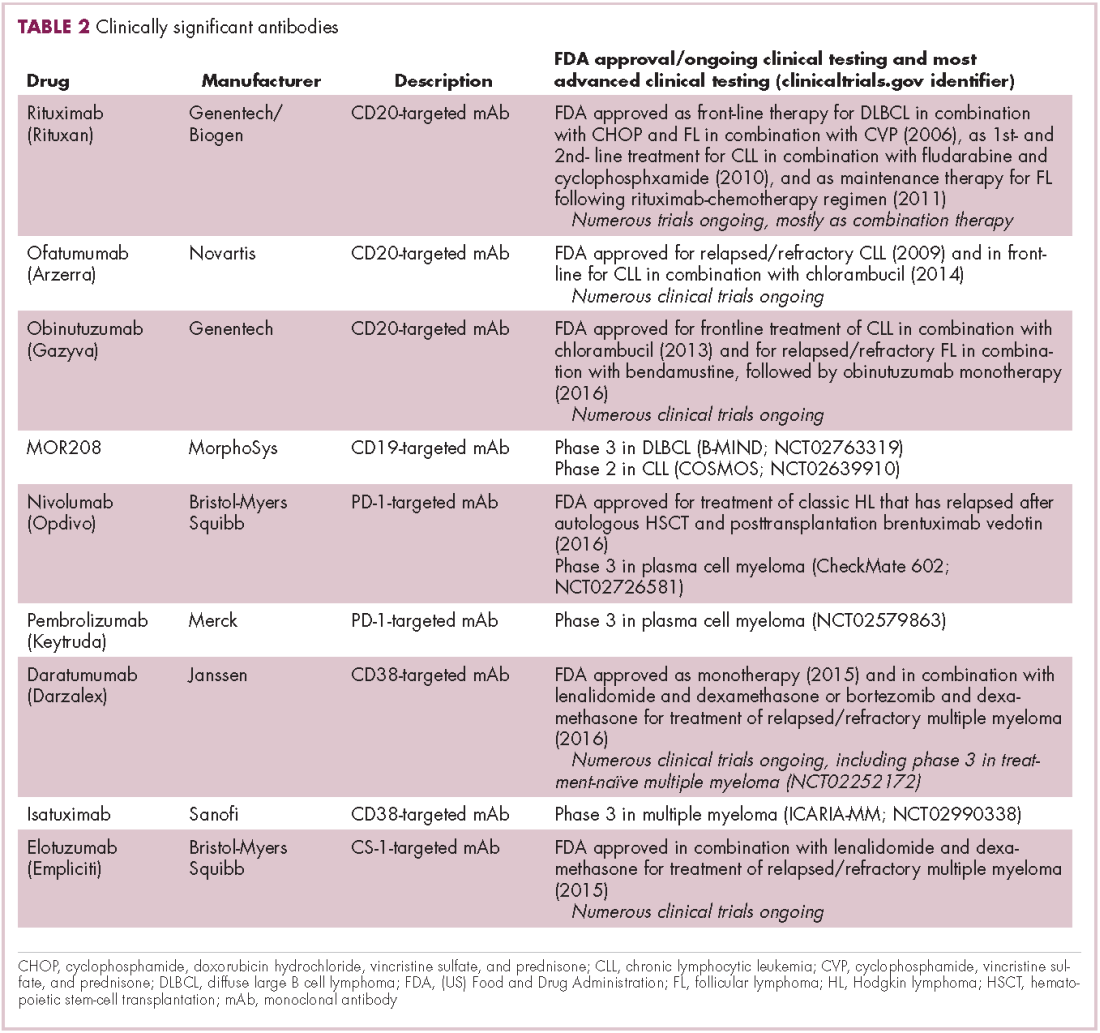
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20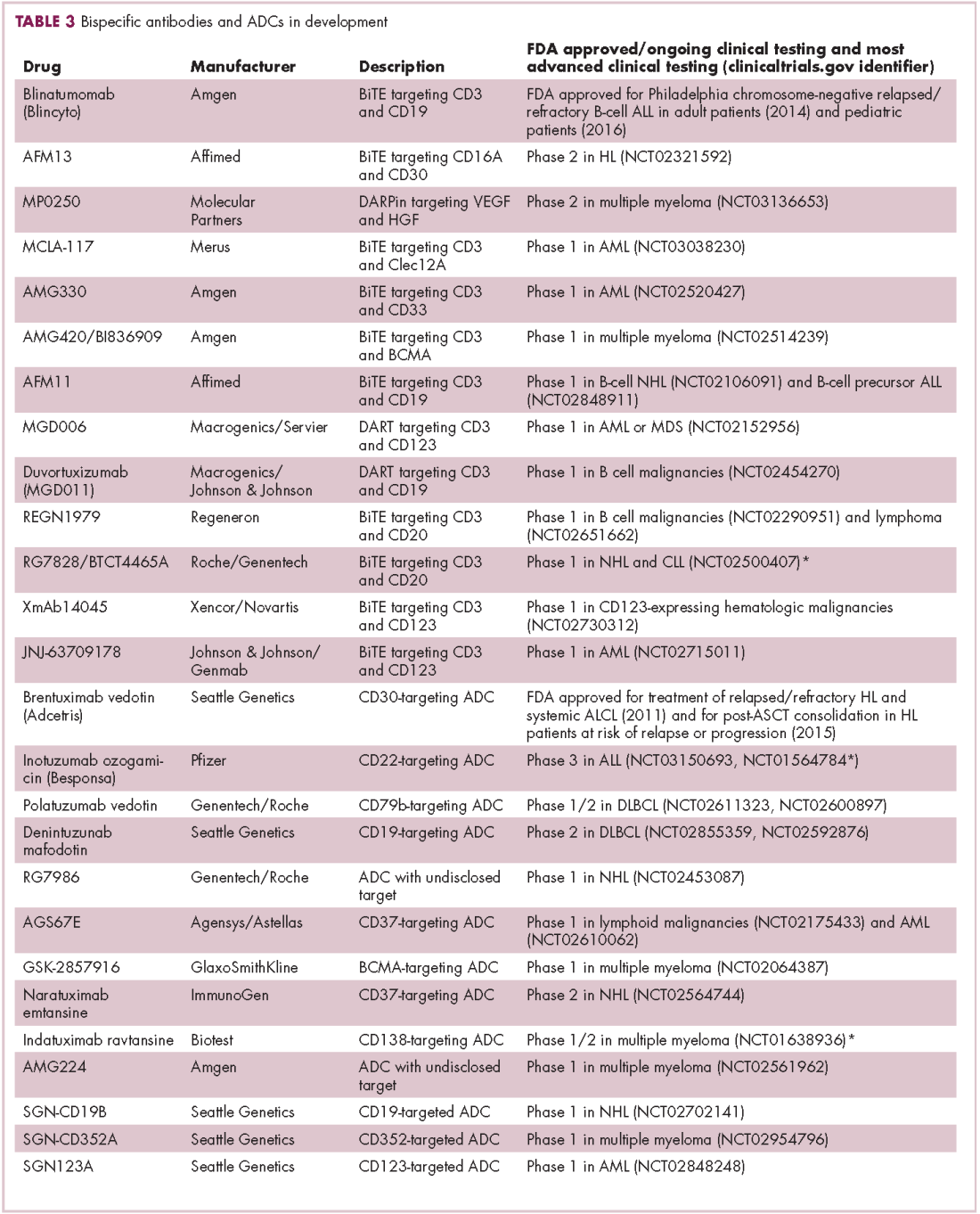
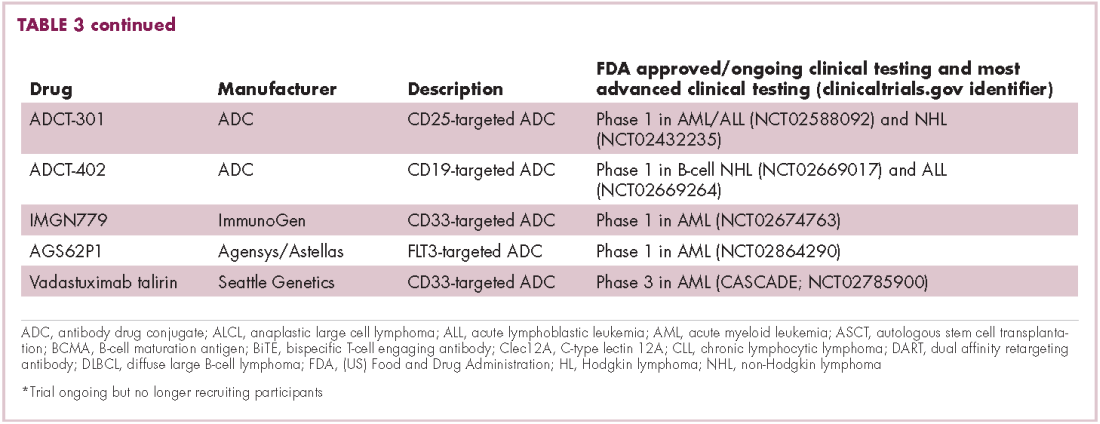
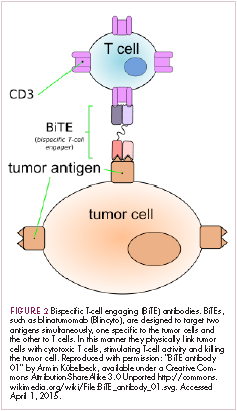
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.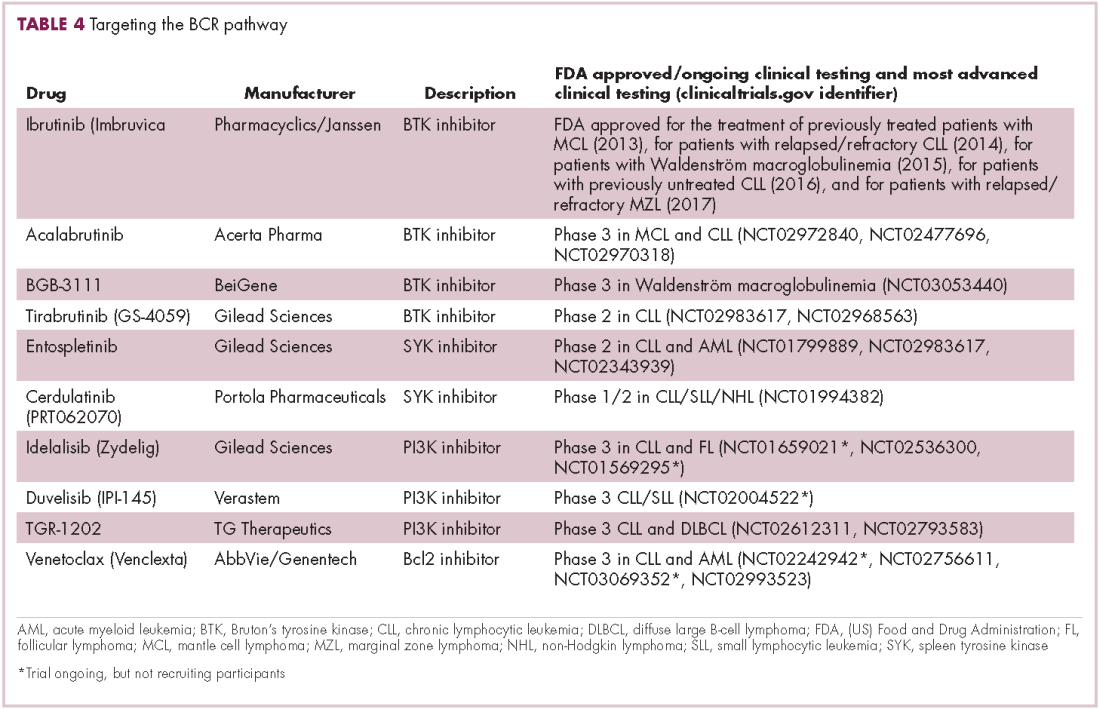
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
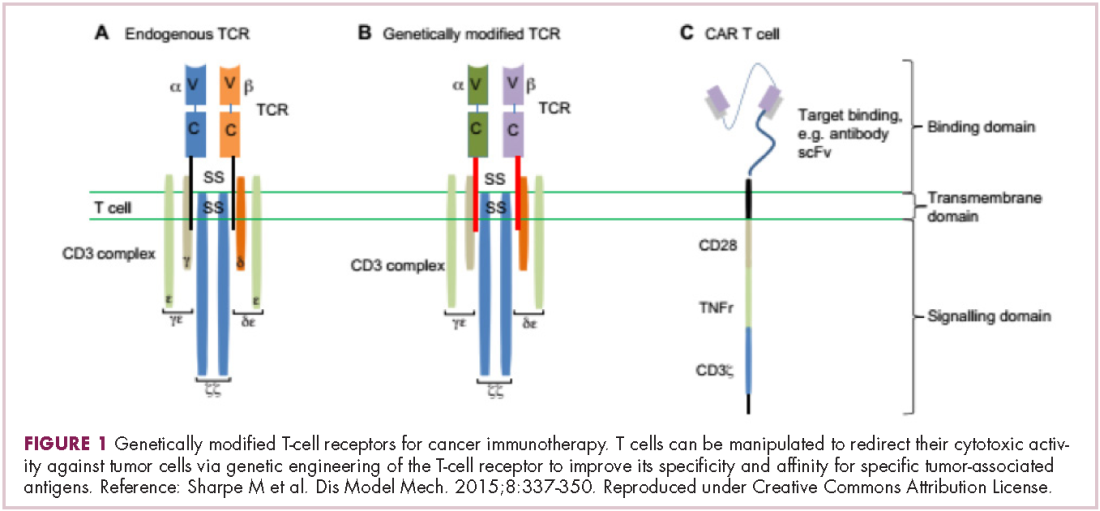
Bevacizumab-awwb becomes first biosimilar approved for cancer treatment
Targeted therapies have revolutionized the treatment of numerous different cancer types and ushered in an era of personalized medicine, yet they can be prohibitively costly. As patent protection expires on many of the first FDA-approved monoclonal antibodies developed for oncologic indications, the doors are opened for other companies to develop their own version of these drugs, known as biosimilars. The price of biosimilars is expected to be considerably lower than the original drugs upon which they are based.
Bevacizumab-awwb, marketed as Mvasi by Amgen and Allergen, became the first such drug to receive approval by the US Food and Drug Administration for the treatment of cancer in fall last year.1 It is a biosimilar of Genentech’s anti-angiogenesis drug, bevacizumab (Avastin), a monoclonal antibody that targets vascular endothelial growth factor-A (VEGF-A).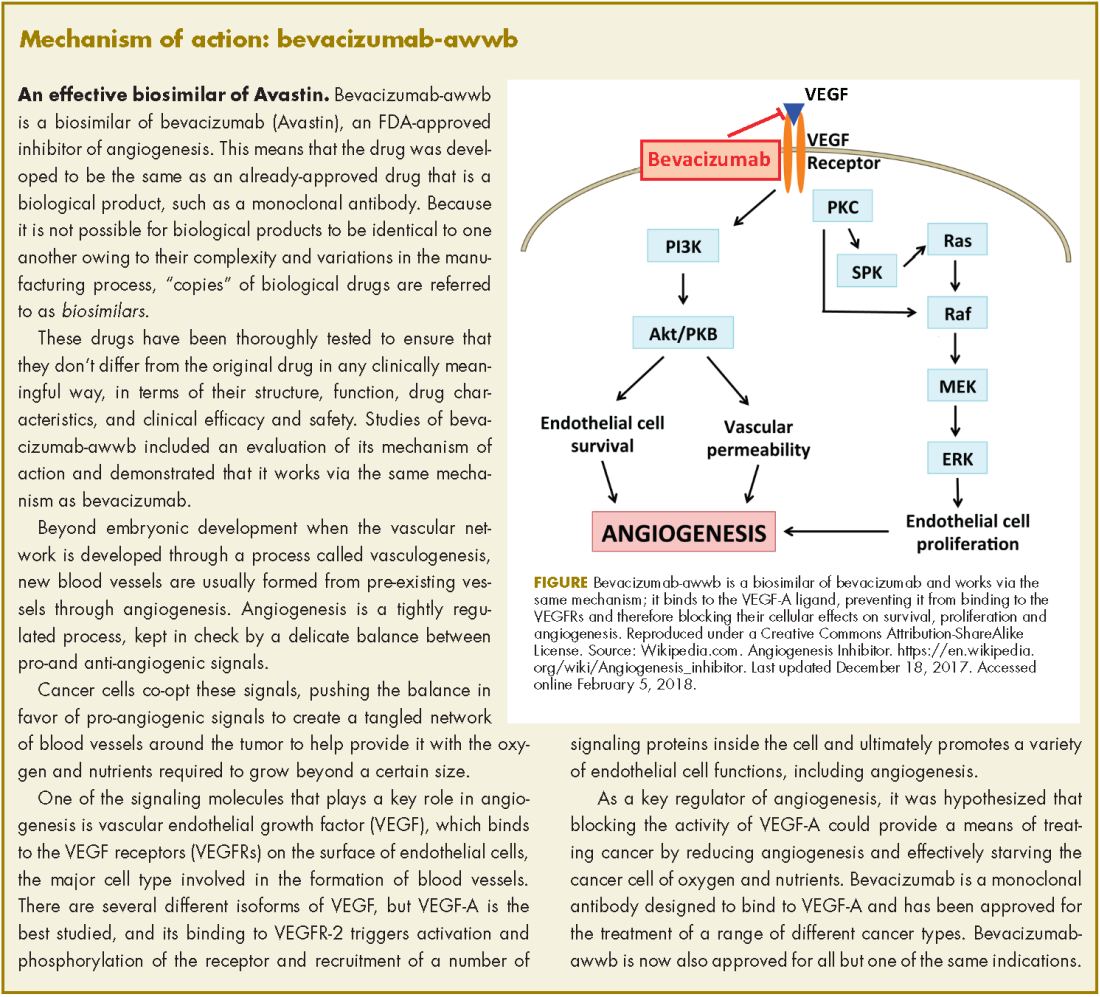
The approval of biosimilars is based on rigorous demonstration of a high level of similarity between the biosimilar and the already-approved reference drug, in terms of structure, function, pharmacokinetics, pharmacodynamics, and clinical efficacy and safety.
Bevacizumab-awwb was approved for the first- or second-line treatment of metastatic colorectal cancer (mCRC) in combination with 5-fluorouracil-based chemotherapy; the second-line treatment of mCRC in combination with fluoropyrimidine-oxaliplatin chemotherapy in patients who progressed on first-line bevacizumab; the first-line treatment of unresectable, locally advanced, recurrent or metastatic nonsquamous non-small cell lung cancer (NSCLC) in combination with carboplatin and paclitaxel; the second-line treatment of glioblastoma (GBM) as monotherapy; and in patients with persistent, recurrent, or metastatic cervical cancer in combination with paclitaxel and cisplatin or paclitaxel and topotecan. It was not approved for the treatment of ovarian cancer, for which bevacizumab is indicated.
The majority of the data used to support approval came from 2 studies – a 3-arm, single-dose pharmacokinetics study, and a comparative clinical study in patients with advanced/metastatic NSCLC. In the pharmacokinetics study, 202 healthy men received an infusion of 3 mg/kg of bevacizumab-awwb, US-approved bevacizumab, or EU-approved bevacizumab. Bevacizumab-awwb was shown to have pharmacokinetic similarity to both approved forms of bevacizumab, and safety and tolerability were comparable, with none of the participants developing binding or neutralizing antidrug antibodies.2
In the clinical study, 648 patients received an infusion of bevacizumab-awwb or EU-approved bevacizumab at a dose of 15 mg/kg every 3 weeks in combination with 6 AUC carboplatin and 200 mg/m2 paclitaxel for 6 cycles. The overall response rate was 39% for bevacizumab-awwb, compared with 41.7% for EU-bevacizumab, and there were 2 complete responses in each group. The median duration of response for bevacizumab-awwb compared with EU-bevacizumab was 5.8 months versus 5.6 months, respectively, and median progression-free survival was 6.6 months versus 7.9 months.3
In terms of safety, the rates of grade 3/4 adverse events (AEs) were 42.9% in the biosimilar arm, compared with 44.3% for the reference drug. Overall, there were no clinically meaningful differences in AEs, serious AEs, deaths, or treatment discontinuations.
The recommended dose for bevacizumab-awwb in patients with mCRC is a 5 mg/kg intravenous dose administered every 2 weeks with bolus-IFL, a 10 mg/kg IV dose administered every 2 weeks with FOLFOX4, or a 5 mg/kg IV dose administered every 2 weeks or 7.5 mg/kg IV dose administered every 3 weeks with fluoropyrimidine-irinotecan or fluoropyrimidine-oxaliplatin-based chemotherapy.
For patients with NSCLC, bevacizumab-awwb should be administered at a 15 mg/kg IV dose every 3 weeks with the carboplatin–paclitaxel combination; for GBM patients, a 10 mg/kg IV dose should be administered every 3 weeks; and for patients with cervical cancer, an IV dose of 15 mg/kg every 3 weeks in combination with paclitaxel–cisplatin or paclitaxel–topotecan is recommended.
The prescribing information outlines warnings and precautions to advise clinicians administering the new biosimilar of the risks of gastrointestinal (GI) perforations, surgery and wound healing complications, and severe and potentially fatal pulmonary, GI, central nervous system, and vaginal bleeding.4
Treatment should be discontinued if GI perforation occurs. Patients should not take bevacizumab-awwb in the 28 days before elective surgery and after surgery until the wound is healed, and treatment should be discontinued if the surgical wound breaks open. Bevacizumab-awwb should not be administered to patients with severe hemorrhage or those with hemoptysis.
Blood pressure should be monitored every 2-3 weeks during treatment and hypertension treated with antihypertensive therapy. Treatment should be temporarily suspended in patients with severe hypertension that is not controlled with antihypertensive therapy and discontinued in patients who experience hypertensive crisis or hypertensive encephalopathy.
Proteinuria should be monitored by dipstick urine analysis during treatment, and patients with a 2+ or greater reading (concentration, 100 mg/dL) should undergo further assessment with 24-hour urine collection. Treatment should be suspended if proteinuria levels are ≥2 g/24h and can be resumed when they fall below that level, but should be discontinued in patients with nephrotic syndrome. Treatment should also be discontinued in patients who develop posterior reversible encephalopathy syndrome, and patients should be advised of the potential for fetal harm
1. FDA approves first biosimilar for the treatment of cancer. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm576112.htm. September 14, 2017. Accessed January 31, 2018.
2. Markus R, Chow V, Pan X, and Hanes V. A phase I, randomized, single-dose study evaluating the pharmacokinetic equivalence of biosimilar ABP 215 and bevacizumab in healthy adult men. Cancer Chemother. Pharmacol. 2017;80:755-763.
3. Thatcher N, Thomas M, Ostoros G, et al. Randomized, double-blind, phase 3 study comparing biosimilar candidate ABP-215 with bevacizumab in patients with non-squamous NSCLC. J Thorac Oncol. 2017;12(1):S902-S903.
4. Mvasi (bevacizumab-awwb) solution, for intravenous infusion. Prescribing information. Amgen Inc, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761028s000lbl.pdf. September 2017. Accessed January 31, 2018.
Targeted therapies have revolutionized the treatment of numerous different cancer types and ushered in an era of personalized medicine, yet they can be prohibitively costly. As patent protection expires on many of the first FDA-approved monoclonal antibodies developed for oncologic indications, the doors are opened for other companies to develop their own version of these drugs, known as biosimilars. The price of biosimilars is expected to be considerably lower than the original drugs upon which they are based.
Bevacizumab-awwb, marketed as Mvasi by Amgen and Allergen, became the first such drug to receive approval by the US Food and Drug Administration for the treatment of cancer in fall last year.1 It is a biosimilar of Genentech’s anti-angiogenesis drug, bevacizumab (Avastin), a monoclonal antibody that targets vascular endothelial growth factor-A (VEGF-A).
The approval of biosimilars is based on rigorous demonstration of a high level of similarity between the biosimilar and the already-approved reference drug, in terms of structure, function, pharmacokinetics, pharmacodynamics, and clinical efficacy and safety.
Bevacizumab-awwb was approved for the first- or second-line treatment of metastatic colorectal cancer (mCRC) in combination with 5-fluorouracil-based chemotherapy; the second-line treatment of mCRC in combination with fluoropyrimidine-oxaliplatin chemotherapy in patients who progressed on first-line bevacizumab; the first-line treatment of unresectable, locally advanced, recurrent or metastatic nonsquamous non-small cell lung cancer (NSCLC) in combination with carboplatin and paclitaxel; the second-line treatment of glioblastoma (GBM) as monotherapy; and in patients with persistent, recurrent, or metastatic cervical cancer in combination with paclitaxel and cisplatin or paclitaxel and topotecan. It was not approved for the treatment of ovarian cancer, for which bevacizumab is indicated.
The majority of the data used to support approval came from 2 studies – a 3-arm, single-dose pharmacokinetics study, and a comparative clinical study in patients with advanced/metastatic NSCLC. In the pharmacokinetics study, 202 healthy men received an infusion of 3 mg/kg of bevacizumab-awwb, US-approved bevacizumab, or EU-approved bevacizumab. Bevacizumab-awwb was shown to have pharmacokinetic similarity to both approved forms of bevacizumab, and safety and tolerability were comparable, with none of the participants developing binding or neutralizing antidrug antibodies.2
In the clinical study, 648 patients received an infusion of bevacizumab-awwb or EU-approved bevacizumab at a dose of 15 mg/kg every 3 weeks in combination with 6 AUC carboplatin and 200 mg/m2 paclitaxel for 6 cycles. The overall response rate was 39% for bevacizumab-awwb, compared with 41.7% for EU-bevacizumab, and there were 2 complete responses in each group. The median duration of response for bevacizumab-awwb compared with EU-bevacizumab was 5.8 months versus 5.6 months, respectively, and median progression-free survival was 6.6 months versus 7.9 months.3
In terms of safety, the rates of grade 3/4 adverse events (AEs) were 42.9% in the biosimilar arm, compared with 44.3% for the reference drug. Overall, there were no clinically meaningful differences in AEs, serious AEs, deaths, or treatment discontinuations.
The recommended dose for bevacizumab-awwb in patients with mCRC is a 5 mg/kg intravenous dose administered every 2 weeks with bolus-IFL, a 10 mg/kg IV dose administered every 2 weeks with FOLFOX4, or a 5 mg/kg IV dose administered every 2 weeks or 7.5 mg/kg IV dose administered every 3 weeks with fluoropyrimidine-irinotecan or fluoropyrimidine-oxaliplatin-based chemotherapy.
For patients with NSCLC, bevacizumab-awwb should be administered at a 15 mg/kg IV dose every 3 weeks with the carboplatin–paclitaxel combination; for GBM patients, a 10 mg/kg IV dose should be administered every 3 weeks; and for patients with cervical cancer, an IV dose of 15 mg/kg every 3 weeks in combination with paclitaxel–cisplatin or paclitaxel–topotecan is recommended.
The prescribing information outlines warnings and precautions to advise clinicians administering the new biosimilar of the risks of gastrointestinal (GI) perforations, surgery and wound healing complications, and severe and potentially fatal pulmonary, GI, central nervous system, and vaginal bleeding.4
Treatment should be discontinued if GI perforation occurs. Patients should not take bevacizumab-awwb in the 28 days before elective surgery and after surgery until the wound is healed, and treatment should be discontinued if the surgical wound breaks open. Bevacizumab-awwb should not be administered to patients with severe hemorrhage or those with hemoptysis.
Blood pressure should be monitored every 2-3 weeks during treatment and hypertension treated with antihypertensive therapy. Treatment should be temporarily suspended in patients with severe hypertension that is not controlled with antihypertensive therapy and discontinued in patients who experience hypertensive crisis or hypertensive encephalopathy.
Proteinuria should be monitored by dipstick urine analysis during treatment, and patients with a 2+ or greater reading (concentration, 100 mg/dL) should undergo further assessment with 24-hour urine collection. Treatment should be suspended if proteinuria levels are ≥2 g/24h and can be resumed when they fall below that level, but should be discontinued in patients with nephrotic syndrome. Treatment should also be discontinued in patients who develop posterior reversible encephalopathy syndrome, and patients should be advised of the potential for fetal harm
Targeted therapies have revolutionized the treatment of numerous different cancer types and ushered in an era of personalized medicine, yet they can be prohibitively costly. As patent protection expires on many of the first FDA-approved monoclonal antibodies developed for oncologic indications, the doors are opened for other companies to develop their own version of these drugs, known as biosimilars. The price of biosimilars is expected to be considerably lower than the original drugs upon which they are based.
Bevacizumab-awwb, marketed as Mvasi by Amgen and Allergen, became the first such drug to receive approval by the US Food and Drug Administration for the treatment of cancer in fall last year.1 It is a biosimilar of Genentech’s anti-angiogenesis drug, bevacizumab (Avastin), a monoclonal antibody that targets vascular endothelial growth factor-A (VEGF-A).
The approval of biosimilars is based on rigorous demonstration of a high level of similarity between the biosimilar and the already-approved reference drug, in terms of structure, function, pharmacokinetics, pharmacodynamics, and clinical efficacy and safety.
Bevacizumab-awwb was approved for the first- or second-line treatment of metastatic colorectal cancer (mCRC) in combination with 5-fluorouracil-based chemotherapy; the second-line treatment of mCRC in combination with fluoropyrimidine-oxaliplatin chemotherapy in patients who progressed on first-line bevacizumab; the first-line treatment of unresectable, locally advanced, recurrent or metastatic nonsquamous non-small cell lung cancer (NSCLC) in combination with carboplatin and paclitaxel; the second-line treatment of glioblastoma (GBM) as monotherapy; and in patients with persistent, recurrent, or metastatic cervical cancer in combination with paclitaxel and cisplatin or paclitaxel and topotecan. It was not approved for the treatment of ovarian cancer, for which bevacizumab is indicated.
The majority of the data used to support approval came from 2 studies – a 3-arm, single-dose pharmacokinetics study, and a comparative clinical study in patients with advanced/metastatic NSCLC. In the pharmacokinetics study, 202 healthy men received an infusion of 3 mg/kg of bevacizumab-awwb, US-approved bevacizumab, or EU-approved bevacizumab. Bevacizumab-awwb was shown to have pharmacokinetic similarity to both approved forms of bevacizumab, and safety and tolerability were comparable, with none of the participants developing binding or neutralizing antidrug antibodies.2
In the clinical study, 648 patients received an infusion of bevacizumab-awwb or EU-approved bevacizumab at a dose of 15 mg/kg every 3 weeks in combination with 6 AUC carboplatin and 200 mg/m2 paclitaxel for 6 cycles. The overall response rate was 39% for bevacizumab-awwb, compared with 41.7% for EU-bevacizumab, and there were 2 complete responses in each group. The median duration of response for bevacizumab-awwb compared with EU-bevacizumab was 5.8 months versus 5.6 months, respectively, and median progression-free survival was 6.6 months versus 7.9 months.3
In terms of safety, the rates of grade 3/4 adverse events (AEs) were 42.9% in the biosimilar arm, compared with 44.3% for the reference drug. Overall, there were no clinically meaningful differences in AEs, serious AEs, deaths, or treatment discontinuations.
The recommended dose for bevacizumab-awwb in patients with mCRC is a 5 mg/kg intravenous dose administered every 2 weeks with bolus-IFL, a 10 mg/kg IV dose administered every 2 weeks with FOLFOX4, or a 5 mg/kg IV dose administered every 2 weeks or 7.5 mg/kg IV dose administered every 3 weeks with fluoropyrimidine-irinotecan or fluoropyrimidine-oxaliplatin-based chemotherapy.
For patients with NSCLC, bevacizumab-awwb should be administered at a 15 mg/kg IV dose every 3 weeks with the carboplatin–paclitaxel combination; for GBM patients, a 10 mg/kg IV dose should be administered every 3 weeks; and for patients with cervical cancer, an IV dose of 15 mg/kg every 3 weeks in combination with paclitaxel–cisplatin or paclitaxel–topotecan is recommended.
The prescribing information outlines warnings and precautions to advise clinicians administering the new biosimilar of the risks of gastrointestinal (GI) perforations, surgery and wound healing complications, and severe and potentially fatal pulmonary, GI, central nervous system, and vaginal bleeding.4
Treatment should be discontinued if GI perforation occurs. Patients should not take bevacizumab-awwb in the 28 days before elective surgery and after surgery until the wound is healed, and treatment should be discontinued if the surgical wound breaks open. Bevacizumab-awwb should not be administered to patients with severe hemorrhage or those with hemoptysis.
Blood pressure should be monitored every 2-3 weeks during treatment and hypertension treated with antihypertensive therapy. Treatment should be temporarily suspended in patients with severe hypertension that is not controlled with antihypertensive therapy and discontinued in patients who experience hypertensive crisis or hypertensive encephalopathy.
Proteinuria should be monitored by dipstick urine analysis during treatment, and patients with a 2+ or greater reading (concentration, 100 mg/dL) should undergo further assessment with 24-hour urine collection. Treatment should be suspended if proteinuria levels are ≥2 g/24h and can be resumed when they fall below that level, but should be discontinued in patients with nephrotic syndrome. Treatment should also be discontinued in patients who develop posterior reversible encephalopathy syndrome, and patients should be advised of the potential for fetal harm
1. FDA approves first biosimilar for the treatment of cancer. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm576112.htm. September 14, 2017. Accessed January 31, 2018.
2. Markus R, Chow V, Pan X, and Hanes V. A phase I, randomized, single-dose study evaluating the pharmacokinetic equivalence of biosimilar ABP 215 and bevacizumab in healthy adult men. Cancer Chemother. Pharmacol. 2017;80:755-763.
3. Thatcher N, Thomas M, Ostoros G, et al. Randomized, double-blind, phase 3 study comparing biosimilar candidate ABP-215 with bevacizumab in patients with non-squamous NSCLC. J Thorac Oncol. 2017;12(1):S902-S903.
4. Mvasi (bevacizumab-awwb) solution, for intravenous infusion. Prescribing information. Amgen Inc, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761028s000lbl.pdf. September 2017. Accessed January 31, 2018.
1. FDA approves first biosimilar for the treatment of cancer. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm576112.htm. September 14, 2017. Accessed January 31, 2018.
2. Markus R, Chow V, Pan X, and Hanes V. A phase I, randomized, single-dose study evaluating the pharmacokinetic equivalence of biosimilar ABP 215 and bevacizumab in healthy adult men. Cancer Chemother. Pharmacol. 2017;80:755-763.
3. Thatcher N, Thomas M, Ostoros G, et al. Randomized, double-blind, phase 3 study comparing biosimilar candidate ABP-215 with bevacizumab in patients with non-squamous NSCLC. J Thorac Oncol. 2017;12(1):S902-S903.
4. Mvasi (bevacizumab-awwb) solution, for intravenous infusion. Prescribing information. Amgen Inc, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761028s000lbl.pdf. September 2017. Accessed January 31, 2018.

Liquid gold: blood-based biopsies make headway
Pathologic and, increasingly, molecular analysis of tumor tissue biopsies is the gold standard in initial diagnosis of cancer, but liquid biopsies, which analyze tumor-derived material circulating in the bloodstream are gaining traction. Here, we discuss the current state of development of this complementary and potentially alternative approach to tumor analysis.
Liquid biopsy gaining traction
Biopsies enable oncologists to gather information about a potential or established tumor, including confirmation of the presence of cancerous tissue and determination of its histological characteristics, such as tumor grade and stage, as well as its molecular features, such as the presence of certain gene mutations. Ultimately, this information can be put to use in determining the most appropriate course of treatment.
The current gold standard is a tissue biopsy that typically involves an invasive procedure to permit the collection of a piece of tumor tissue. Yet, tissue biopsies are not always feasible because of the location of the tumor or the poor performance status of many patients with advanced disease. They also provide only a snapshot of the disease at the time at which they were taken and don’t necessarily reflect the genetic heterogeneity or evolution of a tumor over time.
The detection of components that are derived from the tumor circulating in the blood of cancer patients had fueled the idea of blood-based diagnostics in oncology – so-called liquid biopsies. These have rapidly gained traction in the past several decades as a less expensive (the cost of performing genomic analyses on blood samples is at least an order of magnitude less than on tissue samples), less invasive (requiring only a simple blood draw) alternative source of information about tumors.1
As researchers have refined the ability to exploit liquid biopsies, commercial interest has been piqued. More than 35 companies within the United States alone are developing liquid biopsies, and it’s easy to see why with a market projected to be in the many billions of dollars.2
Seeking out tumor clues in the blood
Liquid biopsies consist of a 10-15 mL blood sample drawn into a tube that contains an anticoagulant and it can contain several different types of tumor-associated material. Thus far, two components – circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) – have formed the cornerstone of liquid biopsies. At present, it is not clear whether these components are released randomly, as a by-product of tumor cell death or if they are released as part of a specific biologic process, such as for the colonization of metastatic sites. It reality, it may be a little of both, and active dissemination may be particularly relevant for CTCs, among which are postulated to be a population of cancer stem cells that can initiate distant metastases.3,4
The discovery of CTCs dates back to the 1860s, when cells that were morphologically identical to the tumor were identified in the blood of a patient with metastatic cancer. Their potential significance was not fully realized until a few decades ago, when they were found to exist from early on in the course of disease.3,4
CTCs, which can be either single cells or clusters of cells known as microemboli, have a short half-life in the bloodstream – less than 2 ½ hours – and are also extremely rare (1 mL of blood contains 1-10 CTCs) against a background of many millions of normal cells. Thus the detection and isolation of CTCs presents a significant challenge. More than 40 different platforms are being developed for the isolation and enrichment of CTCs. For the most part, these use a method called positive selection to pick out CTCs.1,3,4
Positive selection exploits the biological or physical properties that are specific to CTCs and absent in normal cells, for example, the presence of a specific tumor-associated antigen on their surface or differences in size, density or electric charge. The limitations of this method are that, not only do you need to know something about CTCs to begin to understand what makes them truly unique and ensure only isolation of CTCs, but their phenotype is also thought to be continually changing.1,3,4
In recent years, the focus has shifted toward technologies that use negative depletion, meaning that they target the other types of cells in the blood sample and filter those away until only the CTCs are left behind. The most advanced are devices that use microfluidic technology to sort the cells, such as the CTC-iChip system being developed by researchers at Massachusetts General Hospital in Boston.5
ctDNA consists of small fragments of nucleic acids that are not contained within a cell or associated with cell fragments and is thought to be present in 50%-90% of patients, depending on the type of cancer they have. ctDNA has a similarly short half-life in the circulation to CTCs and, like CTCs, ctDNA is present at very low levels in the bloodstream. Although levels of ctDNA have been shown to increase with increasing tumor burden, it is still often obscured by the presence of other cell-free DNA derived from non-tumor cells.
ctDNA can be distinguished from other cell-free DNA by the presence of somatic mutations and a number of highly sensitive methods have been developed to detect them, including the amplification-refractory mutation system (ARMS); digital polymerase chain reaction; and the beads, emulsification, amplification, and magnetics (BEAMing) system. Next-generation sequencing technologies, including tagged-amplicon deep sequencing (TAm-Seq), the Safe-Sequencing System (Safe-SeqS), and cancer personalized profiling by deep sequencing (CAPP-seq), can also be used and the race for ever more sensitive analytical tools is ongoing.1,3,4,6
Applying liquid biopsies now and in the future
There are a plethora of potential applications for liquid biopsies3,7 (Figure 1), and probably the most exciting among them is the potential for screening for and early detection of cancer. The fact that ctDNA and CTCs have both been shown to be present from the earliest stages of disease has sparked interest in the possibility of developing simple blood tests to identify tumors before they become detectable by other methods and at a point at which they may be curable.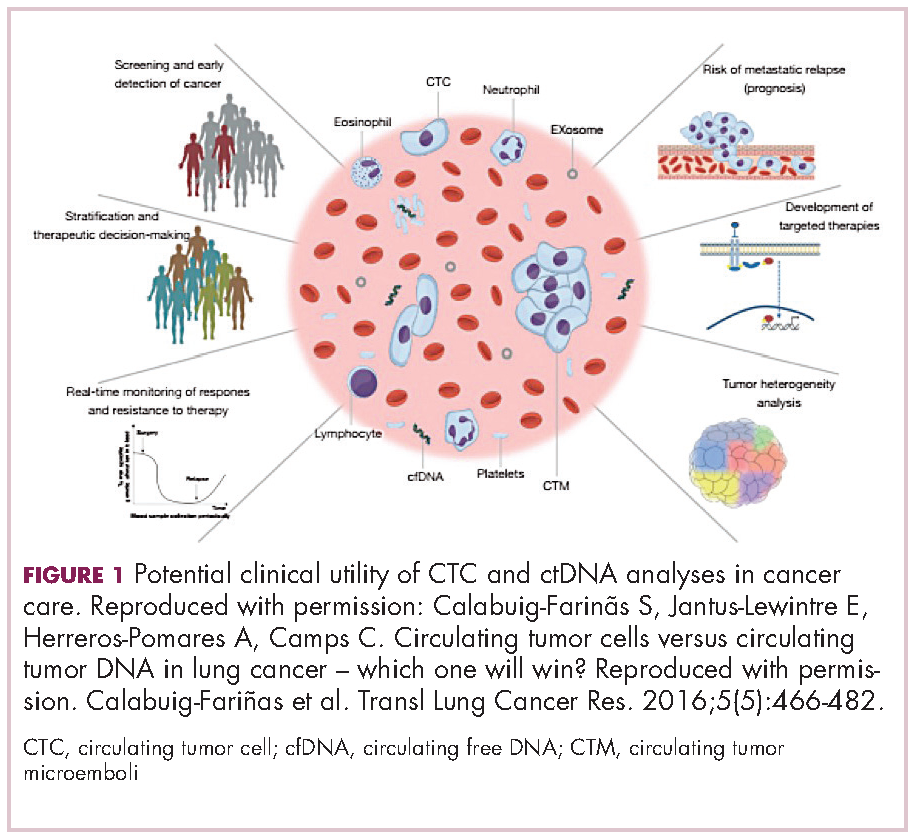
Given that both are present at such low levels within the circulation and are particularly sparse at earlier stages of disease, current technologies may lack the specificity and sensitivity for this application at present. However, numerous clinical trials are ongoing.
For CTCs, simple enumeration has been the most extensively investigated application to date. Numerous studies have shown that the number of CTCs in the bloodstream has prognostic significance in various different tumor types. Three such studies led to the first regulatory approval for a CTC detection system (Table 1 and Table 2).8-10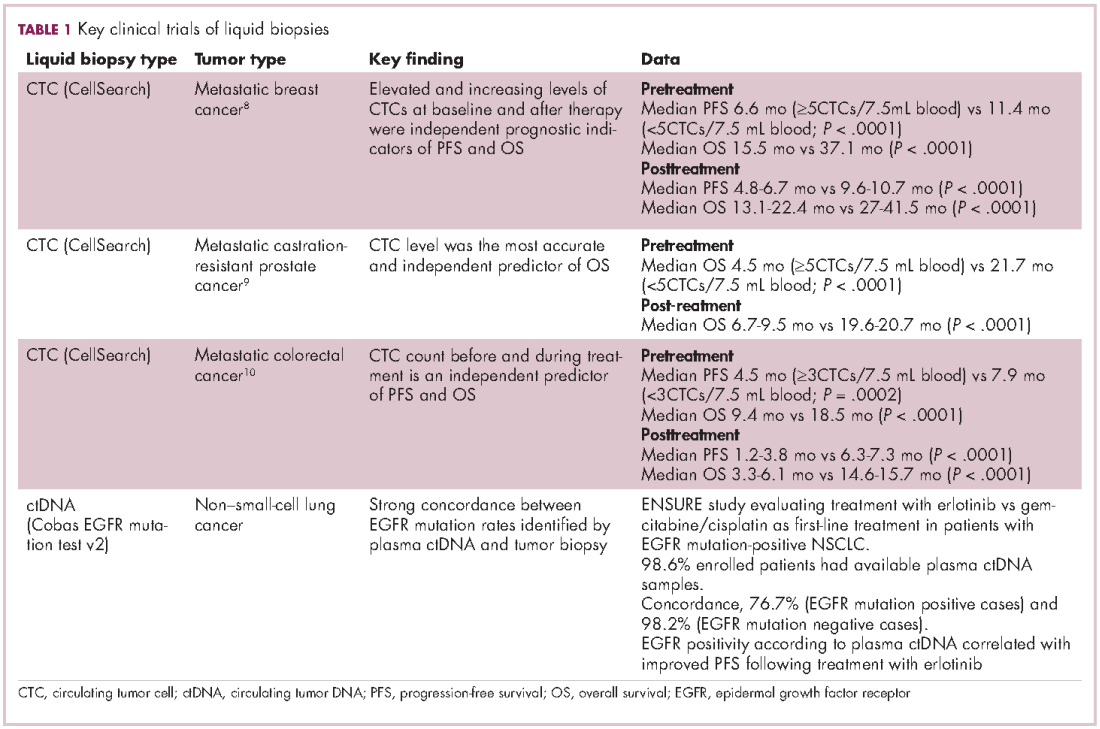
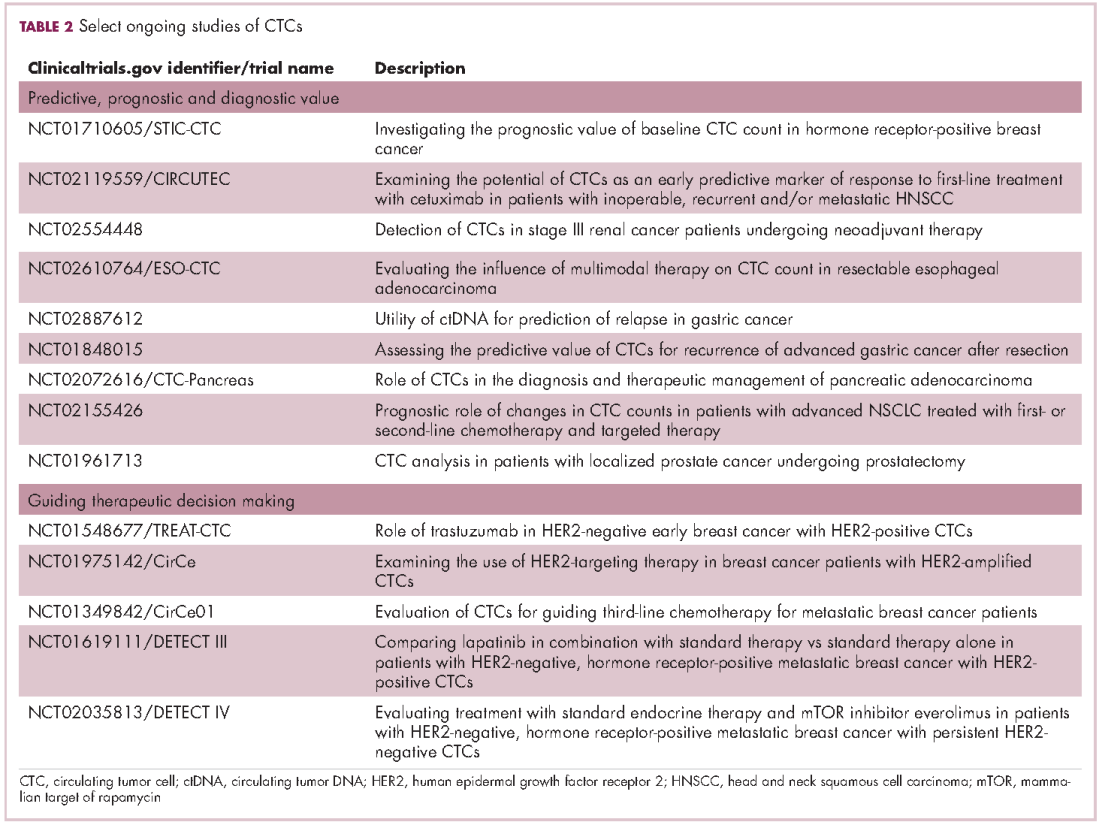
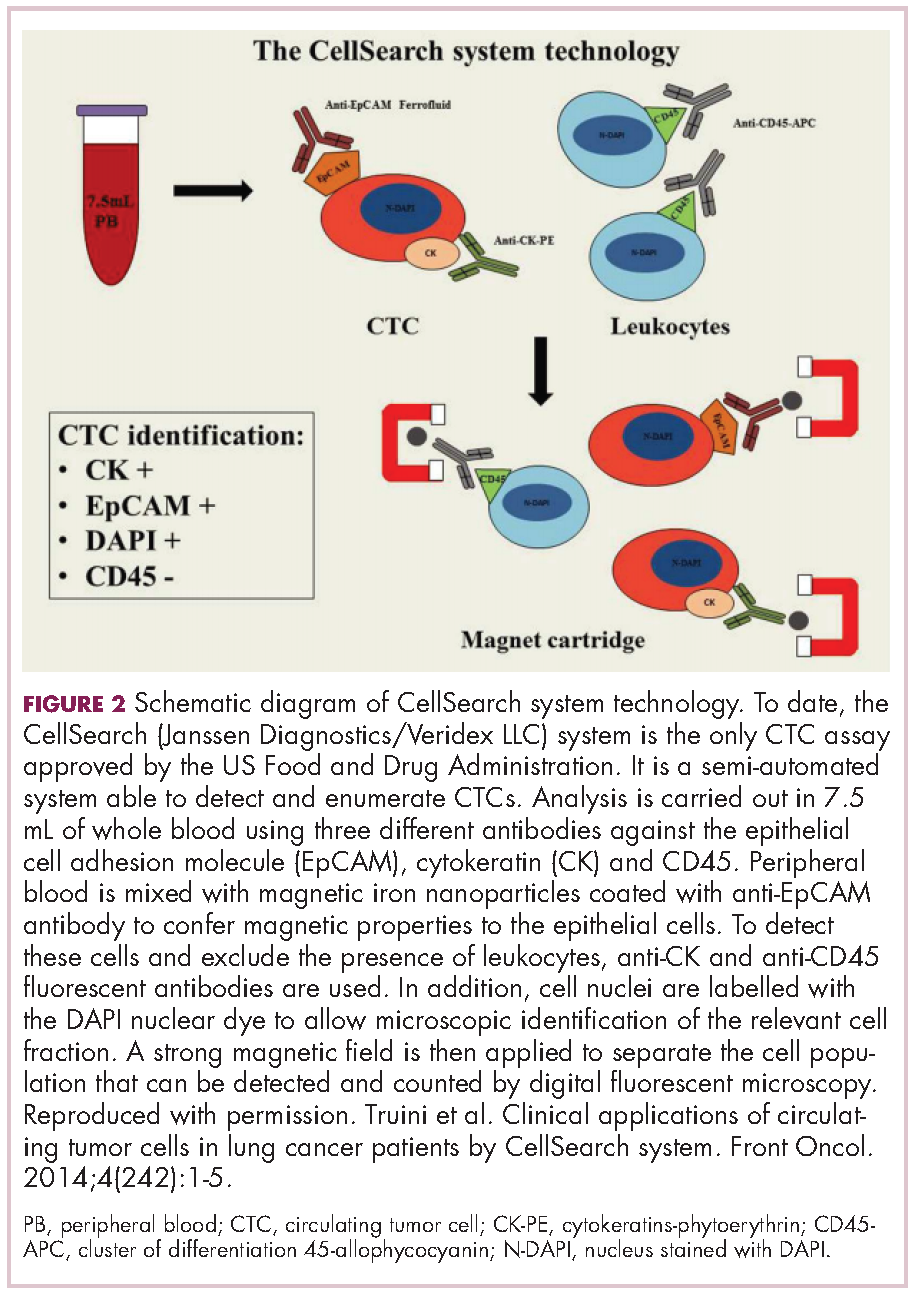
One area in which liquid biopsies could really come into their own is in providing more real-time analysis of tumors. This is something that has proven particularly challenging with tissue biopsies because repeating these invasive procedures is problematic. But the ease of repeat blood draws means that serial liquid biopsies could be performed and might offer the possibility of monitoring disease progression and evolution over the course of disease and particularly in response to treatment.
Indeed, studies have shown that in addition to baseline CTC counts, changes in CTC number during treatment are also prognostic. There was improved survival among patients whose CTC counts decreased below a threshold value during treatment and vice versa. This is also an approved use for CellSearch though at present it is not widely clinically implemented.12
Clinical utility remains elusive
The ultimate goal would be for liquid biopsies to have an impact on treatment decisions, allowing oncologists to change management strategy based on predicted sensitivity or resistance to therapy, so-called clinical utility. Thus far, clinical utility has proved elusive, though liquid biopsies using ctDNA to evaluate tumor genotype have come closest.
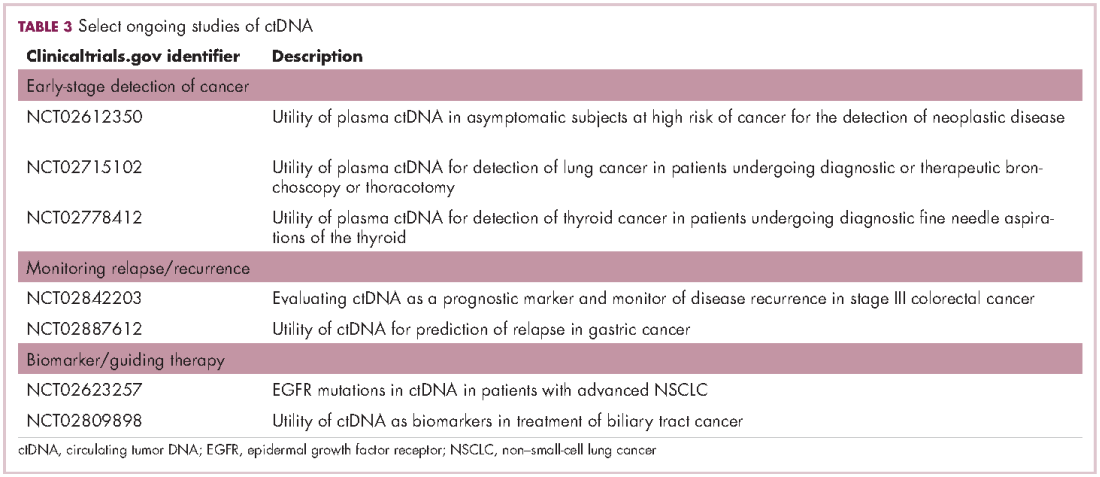
The Cobas EGFR Mutation Test v2 recently became the first ctDNA-based liquid biopsy to receive regulatory approval. It was approved as a companion diagnostic to identify patients with advanced non–small-cell lung cancer (NSCLC) who have specific mutations in the epidermal growth factor receptor (EGFR) gene and are therefore eligible for treatment with the EGFR inhibitor erlotinib.13
Approval was based on comparison of EGFR mutation identification rates using plasma ctDNA samples and tumor tissue samples from patients enrolled in the phase 3 ENSURE trial, which compared the efficacy of erlotinib with chemotherapy as first-line therapy in patients with advanced NSCLC. Of the 217 patients enrolled in the trial, 98.6% of patients had both tumor biopsy and plasma ctDNA samples available for testing. Concordance between the two types of biopsy in identifying patients with EGFR mutations was high and patients with EGFR positivity according to liquid biopsy results demonstrated improved progression-free survival when treated with erlotinib.14
The results of a large-scale genomic analysis of various different types of tumors using ctDNA were also recently presented at the 2016 American Society of Clinical Oncology meeting. Blood samples from more than 15,000 patients with 50 different tumor types, including advanced lung cancer (37%), breast cancer (14%), and CRC (10%), were collected and compared with either available tumor biopsy samples from the same cases (n = 398) or, in the majority of cases, with The Cancer Genome Atlas database, which uses tumor biopsies to perform genome-wide sequencing studies. Both types of biopsy revealed very similar mutation patterns when the Guardant360 next-generation sequencing test, which targets 70 genes, was applied. In particular, when EGFR, BRAF, KRAS, ALK, RET, and ROS1 mutations were identified by tumor tissue biopsy, the same mutations were reported in 94%-100% of plasma samples.15
Studies of the clinical utility of ctDNA and CTCs are among ongoing clinical trials of liquid biopsies (Tables 2 and 3). The potential for using CTCs to guide treatment decisions has become particularly relevant in breast cancer in light of results showing that patients with primary tumors that are negative for human epidermal growth factor receptor 2 (HER2) amplification, an important biomarker in breast cancer, may have CTCs that are HER2-positive, in up to 30% of cases. These patients may therefore still benefit from HER2-targeted therapy.16
The DETECT studies are the first phase 3 trials in which treatment decisions are being based on the phenotypic characteristics of CTCs. DETECT III (NCT01619111) is comparing lapatinib in combination with standard therapy with standard therapy alone in patients with HER2-negative metastatic breast cancer who have HER2-positive CTCs, whereas DETECT IV (NCT02035813) is enrolling patients with HER2-negative, hormone receptor-positive metastatic breast cancer and persistent HER2-negative CTCs to receive standard endocrine therapy and the mammalian target of rapamycin inhibitor everolimus.
Other targets and sources for liquid biopsy
Another approach to liquid biopsies that is also beginning to take off is to collect tumor-derived exosomes from the bloodstream. Exosomes are tiny, fluid-filled, membrane-bound sacks that bud off from the surface of a cell to expel waste or to transport cargo from one cell to another. DNA, RNA, and protein can be extracted from tumor-derived exosomes and could also serve as molecular biomarkers relating to the cancer cells from which they came.6,7
Exosome Diagnostics is bringing the first exosome-based diagnostic tests to the market and recently teamed up with Amgen for the development of these liquid biopsies.17 In January 2016, they launched ExoDx Lung (ALK), for detection of EML4-ALK gene fusions in patients with NSCLC, using a proprietary platform for the isolation of RNA from exosomes. Data that was presented at several different conferences in 2015 demonstrated a sensitivity of 88% and specificity of 100% for this diagnostic when compared with tissue ALK status in NSCLC patients receiving a second-generation ALK inhibitor following progression on prior ALK inhibitor therapy.18
In September, they subsequently announced the launch of a test that analyses genetic information from exosomes collected from a urine sample taken from prostate cancer patients. Using a 3-gene signature, in combination with a proprietary algorithm, this diagnostic generates a score assessing a prostate cancer patient’s risk for higher grade, more aggressive disease. It is designed to complement the prostate-specific antigen score and has demonstrated accuracy in ruling out the presence of high-grade cancer before an initial biopsy in more than 1,
1. Lennon NK, Adalsteinsson VA, Gabriel SB. Technological considerations for genome-guided diagnosis and management of cancer. Genome Med. 2016;8:112.
2. MIT Technology Review website. Liquid biopsy: fast DNA-sequencing machines are leading to simple blood tests for cancer. https://www.technologyreview.com/s/534991/liquid-biopsy/. Published 2015. Accessed December 19, 2016.
3. Alix-Panabières C and Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6(5):479-491.
4. Calabuig-Farinãs S, Jantus-Lewintre E, Herreros-Pomares A, Camps C. Circulating tumor cells versus circulating tumor DNA in lung cancer – which one will win? Transl Lung Cancer Res. 2016;5(5):466-482.
5. Karabacak, NM, Spuhler PS, Fachin F, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat Protoc. 2014;9:694-710.
6. Buder A, Tomuta C, and Filipits M. The potential of liquid biopsies. Curr Opin Oncol. 2016;28:130-134.
7. Hofman P, Popper HH. Pathologists and liquid biopsies: to be or not to be? Virchows Arch. 2016;469:601-609.
8. Bidard FC, Peeters DJ, Fehm T, et al. Clinical validity of circulating tumor cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014;15(4):406-414.
9. de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302-6309.
10. Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213-3221.
11. CellSearch Web site. What is the CELLSEARCH® System? https://www.cellsearchctc.com/product-systems-overview/cellsearch-system-overview. Last updated December 5th, 2016. Accessed online December 19th, 2016.
12. CellSearch Web site [advertisement]. https://www.cellsearchctc.com/clinical-applications/clinical-applications-overview. Last updated December 5, 2016. Accessed December 19, 2016.
13. US Food and Drug Administration. cobas EGFR Mutation Test v2 – P150047. http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm519922.htm. Last updated September 9, 2016. Accessed December 19, 2016.
14. Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015;26(9):1883-1889.
15. Zill OA, Mortimer S, Banks KC, et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. J Clin Oncol. 2016;34(suppl;abstr LBA11501).
16. Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537:102-106.
17. Exosome Diagnostics. Exosome diagnostics enters agreement with Amgen. http://www.exosomedx.com/news-events/press-releases/exosome-diagnostics-enters-agreement-amgen. Published October 3, 2016. Accessed December 19, 2016.
18. Brinkman K, Emenegger J, Tannous B, et al. Exosomal RNA-based liquid biopsy detection of EML4-ALK in plasma from NSCLC patients [2015 World Conference on Lung Cancer, Denver, CO; abstract 2591]. http://library.iaslc.org/search-speaker?search_speaker=30493. Accessed January 6, 2017.
19. Exosome Diagnostics website. Prostate cancer. http://www.exosomedx.com/prostate-cancer-0. Last updated 2017. Accessed online December 19, 2016.
Pathologic and, increasingly, molecular analysis of tumor tissue biopsies is the gold standard in initial diagnosis of cancer, but liquid biopsies, which analyze tumor-derived material circulating in the bloodstream are gaining traction. Here, we discuss the current state of development of this complementary and potentially alternative approach to tumor analysis.
Liquid biopsy gaining traction
Biopsies enable oncologists to gather information about a potential or established tumor, including confirmation of the presence of cancerous tissue and determination of its histological characteristics, such as tumor grade and stage, as well as its molecular features, such as the presence of certain gene mutations. Ultimately, this information can be put to use in determining the most appropriate course of treatment.
The current gold standard is a tissue biopsy that typically involves an invasive procedure to permit the collection of a piece of tumor tissue. Yet, tissue biopsies are not always feasible because of the location of the tumor or the poor performance status of many patients with advanced disease. They also provide only a snapshot of the disease at the time at which they were taken and don’t necessarily reflect the genetic heterogeneity or evolution of a tumor over time.
The detection of components that are derived from the tumor circulating in the blood of cancer patients had fueled the idea of blood-based diagnostics in oncology – so-called liquid biopsies. These have rapidly gained traction in the past several decades as a less expensive (the cost of performing genomic analyses on blood samples is at least an order of magnitude less than on tissue samples), less invasive (requiring only a simple blood draw) alternative source of information about tumors.1
As researchers have refined the ability to exploit liquid biopsies, commercial interest has been piqued. More than 35 companies within the United States alone are developing liquid biopsies, and it’s easy to see why with a market projected to be in the many billions of dollars.2
Seeking out tumor clues in the blood
Liquid biopsies consist of a 10-15 mL blood sample drawn into a tube that contains an anticoagulant and it can contain several different types of tumor-associated material. Thus far, two components – circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) – have formed the cornerstone of liquid biopsies. At present, it is not clear whether these components are released randomly, as a by-product of tumor cell death or if they are released as part of a specific biologic process, such as for the colonization of metastatic sites. It reality, it may be a little of both, and active dissemination may be particularly relevant for CTCs, among which are postulated to be a population of cancer stem cells that can initiate distant metastases.3,4
The discovery of CTCs dates back to the 1860s, when cells that were morphologically identical to the tumor were identified in the blood of a patient with metastatic cancer. Their potential significance was not fully realized until a few decades ago, when they were found to exist from early on in the course of disease.3,4
CTCs, which can be either single cells or clusters of cells known as microemboli, have a short half-life in the bloodstream – less than 2 ½ hours – and are also extremely rare (1 mL of blood contains 1-10 CTCs) against a background of many millions of normal cells. Thus the detection and isolation of CTCs presents a significant challenge. More than 40 different platforms are being developed for the isolation and enrichment of CTCs. For the most part, these use a method called positive selection to pick out CTCs.1,3,4
Positive selection exploits the biological or physical properties that are specific to CTCs and absent in normal cells, for example, the presence of a specific tumor-associated antigen on their surface or differences in size, density or electric charge. The limitations of this method are that, not only do you need to know something about CTCs to begin to understand what makes them truly unique and ensure only isolation of CTCs, but their phenotype is also thought to be continually changing.1,3,4
In recent years, the focus has shifted toward technologies that use negative depletion, meaning that they target the other types of cells in the blood sample and filter those away until only the CTCs are left behind. The most advanced are devices that use microfluidic technology to sort the cells, such as the CTC-iChip system being developed by researchers at Massachusetts General Hospital in Boston.5
ctDNA consists of small fragments of nucleic acids that are not contained within a cell or associated with cell fragments and is thought to be present in 50%-90% of patients, depending on the type of cancer they have. ctDNA has a similarly short half-life in the circulation to CTCs and, like CTCs, ctDNA is present at very low levels in the bloodstream. Although levels of ctDNA have been shown to increase with increasing tumor burden, it is still often obscured by the presence of other cell-free DNA derived from non-tumor cells.
ctDNA can be distinguished from other cell-free DNA by the presence of somatic mutations and a number of highly sensitive methods have been developed to detect them, including the amplification-refractory mutation system (ARMS); digital polymerase chain reaction; and the beads, emulsification, amplification, and magnetics (BEAMing) system. Next-generation sequencing technologies, including tagged-amplicon deep sequencing (TAm-Seq), the Safe-Sequencing System (Safe-SeqS), and cancer personalized profiling by deep sequencing (CAPP-seq), can also be used and the race for ever more sensitive analytical tools is ongoing.1,3,4,6
Applying liquid biopsies now and in the future
There are a plethora of potential applications for liquid biopsies3,7 (Figure 1), and probably the most exciting among them is the potential for screening for and early detection of cancer. The fact that ctDNA and CTCs have both been shown to be present from the earliest stages of disease has sparked interest in the possibility of developing simple blood tests to identify tumors before they become detectable by other methods and at a point at which they may be curable.
Given that both are present at such low levels within the circulation and are particularly sparse at earlier stages of disease, current technologies may lack the specificity and sensitivity for this application at present. However, numerous clinical trials are ongoing.
For CTCs, simple enumeration has been the most extensively investigated application to date. Numerous studies have shown that the number of CTCs in the bloodstream has prognostic significance in various different tumor types. Three such studies led to the first regulatory approval for a CTC detection system (Table 1 and Table 2).8-10
One area in which liquid biopsies could really come into their own is in providing more real-time analysis of tumors. This is something that has proven particularly challenging with tissue biopsies because repeating these invasive procedures is problematic. But the ease of repeat blood draws means that serial liquid biopsies could be performed and might offer the possibility of monitoring disease progression and evolution over the course of disease and particularly in response to treatment.
Indeed, studies have shown that in addition to baseline CTC counts, changes in CTC number during treatment are also prognostic. There was improved survival among patients whose CTC counts decreased below a threshold value during treatment and vice versa. This is also an approved use for CellSearch though at present it is not widely clinically implemented.12
Clinical utility remains elusive
The ultimate goal would be for liquid biopsies to have an impact on treatment decisions, allowing oncologists to change management strategy based on predicted sensitivity or resistance to therapy, so-called clinical utility. Thus far, clinical utility has proved elusive, though liquid biopsies using ctDNA to evaluate tumor genotype have come closest.
The Cobas EGFR Mutation Test v2 recently became the first ctDNA-based liquid biopsy to receive regulatory approval. It was approved as a companion diagnostic to identify patients with advanced non–small-cell lung cancer (NSCLC) who have specific mutations in the epidermal growth factor receptor (EGFR) gene and are therefore eligible for treatment with the EGFR inhibitor erlotinib.13
Approval was based on comparison of EGFR mutation identification rates using plasma ctDNA samples and tumor tissue samples from patients enrolled in the phase 3 ENSURE trial, which compared the efficacy of erlotinib with chemotherapy as first-line therapy in patients with advanced NSCLC. Of the 217 patients enrolled in the trial, 98.6% of patients had both tumor biopsy and plasma ctDNA samples available for testing. Concordance between the two types of biopsy in identifying patients with EGFR mutations was high and patients with EGFR positivity according to liquid biopsy results demonstrated improved progression-free survival when treated with erlotinib.14
The results of a large-scale genomic analysis of various different types of tumors using ctDNA were also recently presented at the 2016 American Society of Clinical Oncology meeting. Blood samples from more than 15,000 patients with 50 different tumor types, including advanced lung cancer (37%), breast cancer (14%), and CRC (10%), were collected and compared with either available tumor biopsy samples from the same cases (n = 398) or, in the majority of cases, with The Cancer Genome Atlas database, which uses tumor biopsies to perform genome-wide sequencing studies. Both types of biopsy revealed very similar mutation patterns when the Guardant360 next-generation sequencing test, which targets 70 genes, was applied. In particular, when EGFR, BRAF, KRAS, ALK, RET, and ROS1 mutations were identified by tumor tissue biopsy, the same mutations were reported in 94%-100% of plasma samples.15
Studies of the clinical utility of ctDNA and CTCs are among ongoing clinical trials of liquid biopsies (Tables 2 and 3). The potential for using CTCs to guide treatment decisions has become particularly relevant in breast cancer in light of results showing that patients with primary tumors that are negative for human epidermal growth factor receptor 2 (HER2) amplification, an important biomarker in breast cancer, may have CTCs that are HER2-positive, in up to 30% of cases. These patients may therefore still benefit from HER2-targeted therapy.16
The DETECT studies are the first phase 3 trials in which treatment decisions are being based on the phenotypic characteristics of CTCs. DETECT III (NCT01619111) is comparing lapatinib in combination with standard therapy with standard therapy alone in patients with HER2-negative metastatic breast cancer who have HER2-positive CTCs, whereas DETECT IV (NCT02035813) is enrolling patients with HER2-negative, hormone receptor-positive metastatic breast cancer and persistent HER2-negative CTCs to receive standard endocrine therapy and the mammalian target of rapamycin inhibitor everolimus.
Other targets and sources for liquid biopsy
Another approach to liquid biopsies that is also beginning to take off is to collect tumor-derived exosomes from the bloodstream. Exosomes are tiny, fluid-filled, membrane-bound sacks that bud off from the surface of a cell to expel waste or to transport cargo from one cell to another. DNA, RNA, and protein can be extracted from tumor-derived exosomes and could also serve as molecular biomarkers relating to the cancer cells from which they came.6,7
Exosome Diagnostics is bringing the first exosome-based diagnostic tests to the market and recently teamed up with Amgen for the development of these liquid biopsies.17 In January 2016, they launched ExoDx Lung (ALK), for detection of EML4-ALK gene fusions in patients with NSCLC, using a proprietary platform for the isolation of RNA from exosomes. Data that was presented at several different conferences in 2015 demonstrated a sensitivity of 88% and specificity of 100% for this diagnostic when compared with tissue ALK status in NSCLC patients receiving a second-generation ALK inhibitor following progression on prior ALK inhibitor therapy.18
In September, they subsequently announced the launch of a test that analyses genetic information from exosomes collected from a urine sample taken from prostate cancer patients. Using a 3-gene signature, in combination with a proprietary algorithm, this diagnostic generates a score assessing a prostate cancer patient’s risk for higher grade, more aggressive disease. It is designed to complement the prostate-specific antigen score and has demonstrated accuracy in ruling out the presence of high-grade cancer before an initial biopsy in more than 1,
Pathologic and, increasingly, molecular analysis of tumor tissue biopsies is the gold standard in initial diagnosis of cancer, but liquid biopsies, which analyze tumor-derived material circulating in the bloodstream are gaining traction. Here, we discuss the current state of development of this complementary and potentially alternative approach to tumor analysis.
Liquid biopsy gaining traction
Biopsies enable oncologists to gather information about a potential or established tumor, including confirmation of the presence of cancerous tissue and determination of its histological characteristics, such as tumor grade and stage, as well as its molecular features, such as the presence of certain gene mutations. Ultimately, this information can be put to use in determining the most appropriate course of treatment.
The current gold standard is a tissue biopsy that typically involves an invasive procedure to permit the collection of a piece of tumor tissue. Yet, tissue biopsies are not always feasible because of the location of the tumor or the poor performance status of many patients with advanced disease. They also provide only a snapshot of the disease at the time at which they were taken and don’t necessarily reflect the genetic heterogeneity or evolution of a tumor over time.
The detection of components that are derived from the tumor circulating in the blood of cancer patients had fueled the idea of blood-based diagnostics in oncology – so-called liquid biopsies. These have rapidly gained traction in the past several decades as a less expensive (the cost of performing genomic analyses on blood samples is at least an order of magnitude less than on tissue samples), less invasive (requiring only a simple blood draw) alternative source of information about tumors.1
As researchers have refined the ability to exploit liquid biopsies, commercial interest has been piqued. More than 35 companies within the United States alone are developing liquid biopsies, and it’s easy to see why with a market projected to be in the many billions of dollars.2
Seeking out tumor clues in the blood
Liquid biopsies consist of a 10-15 mL blood sample drawn into a tube that contains an anticoagulant and it can contain several different types of tumor-associated material. Thus far, two components – circulating tumor cells (CTCs) and circulating tumor DNA (ctDNA) – have formed the cornerstone of liquid biopsies. At present, it is not clear whether these components are released randomly, as a by-product of tumor cell death or if they are released as part of a specific biologic process, such as for the colonization of metastatic sites. It reality, it may be a little of both, and active dissemination may be particularly relevant for CTCs, among which are postulated to be a population of cancer stem cells that can initiate distant metastases.3,4
The discovery of CTCs dates back to the 1860s, when cells that were morphologically identical to the tumor were identified in the blood of a patient with metastatic cancer. Their potential significance was not fully realized until a few decades ago, when they were found to exist from early on in the course of disease.3,4
CTCs, which can be either single cells or clusters of cells known as microemboli, have a short half-life in the bloodstream – less than 2 ½ hours – and are also extremely rare (1 mL of blood contains 1-10 CTCs) against a background of many millions of normal cells. Thus the detection and isolation of CTCs presents a significant challenge. More than 40 different platforms are being developed for the isolation and enrichment of CTCs. For the most part, these use a method called positive selection to pick out CTCs.1,3,4
Positive selection exploits the biological or physical properties that are specific to CTCs and absent in normal cells, for example, the presence of a specific tumor-associated antigen on their surface or differences in size, density or electric charge. The limitations of this method are that, not only do you need to know something about CTCs to begin to understand what makes them truly unique and ensure only isolation of CTCs, but their phenotype is also thought to be continually changing.1,3,4
In recent years, the focus has shifted toward technologies that use negative depletion, meaning that they target the other types of cells in the blood sample and filter those away until only the CTCs are left behind. The most advanced are devices that use microfluidic technology to sort the cells, such as the CTC-iChip system being developed by researchers at Massachusetts General Hospital in Boston.5
ctDNA consists of small fragments of nucleic acids that are not contained within a cell or associated with cell fragments and is thought to be present in 50%-90% of patients, depending on the type of cancer they have. ctDNA has a similarly short half-life in the circulation to CTCs and, like CTCs, ctDNA is present at very low levels in the bloodstream. Although levels of ctDNA have been shown to increase with increasing tumor burden, it is still often obscured by the presence of other cell-free DNA derived from non-tumor cells.
ctDNA can be distinguished from other cell-free DNA by the presence of somatic mutations and a number of highly sensitive methods have been developed to detect them, including the amplification-refractory mutation system (ARMS); digital polymerase chain reaction; and the beads, emulsification, amplification, and magnetics (BEAMing) system. Next-generation sequencing technologies, including tagged-amplicon deep sequencing (TAm-Seq), the Safe-Sequencing System (Safe-SeqS), and cancer personalized profiling by deep sequencing (CAPP-seq), can also be used and the race for ever more sensitive analytical tools is ongoing.1,3,4,6
Applying liquid biopsies now and in the future
There are a plethora of potential applications for liquid biopsies3,7 (Figure 1), and probably the most exciting among them is the potential for screening for and early detection of cancer. The fact that ctDNA and CTCs have both been shown to be present from the earliest stages of disease has sparked interest in the possibility of developing simple blood tests to identify tumors before they become detectable by other methods and at a point at which they may be curable.
Given that both are present at such low levels within the circulation and are particularly sparse at earlier stages of disease, current technologies may lack the specificity and sensitivity for this application at present. However, numerous clinical trials are ongoing.
For CTCs, simple enumeration has been the most extensively investigated application to date. Numerous studies have shown that the number of CTCs in the bloodstream has prognostic significance in various different tumor types. Three such studies led to the first regulatory approval for a CTC detection system (Table 1 and Table 2).8-10
One area in which liquid biopsies could really come into their own is in providing more real-time analysis of tumors. This is something that has proven particularly challenging with tissue biopsies because repeating these invasive procedures is problematic. But the ease of repeat blood draws means that serial liquid biopsies could be performed and might offer the possibility of monitoring disease progression and evolution over the course of disease and particularly in response to treatment.
Indeed, studies have shown that in addition to baseline CTC counts, changes in CTC number during treatment are also prognostic. There was improved survival among patients whose CTC counts decreased below a threshold value during treatment and vice versa. This is also an approved use for CellSearch though at present it is not widely clinically implemented.12
Clinical utility remains elusive
The ultimate goal would be for liquid biopsies to have an impact on treatment decisions, allowing oncologists to change management strategy based on predicted sensitivity or resistance to therapy, so-called clinical utility. Thus far, clinical utility has proved elusive, though liquid biopsies using ctDNA to evaluate tumor genotype have come closest.
The Cobas EGFR Mutation Test v2 recently became the first ctDNA-based liquid biopsy to receive regulatory approval. It was approved as a companion diagnostic to identify patients with advanced non–small-cell lung cancer (NSCLC) who have specific mutations in the epidermal growth factor receptor (EGFR) gene and are therefore eligible for treatment with the EGFR inhibitor erlotinib.13
Approval was based on comparison of EGFR mutation identification rates using plasma ctDNA samples and tumor tissue samples from patients enrolled in the phase 3 ENSURE trial, which compared the efficacy of erlotinib with chemotherapy as first-line therapy in patients with advanced NSCLC. Of the 217 patients enrolled in the trial, 98.6% of patients had both tumor biopsy and plasma ctDNA samples available for testing. Concordance between the two types of biopsy in identifying patients with EGFR mutations was high and patients with EGFR positivity according to liquid biopsy results demonstrated improved progression-free survival when treated with erlotinib.14
The results of a large-scale genomic analysis of various different types of tumors using ctDNA were also recently presented at the 2016 American Society of Clinical Oncology meeting. Blood samples from more than 15,000 patients with 50 different tumor types, including advanced lung cancer (37%), breast cancer (14%), and CRC (10%), were collected and compared with either available tumor biopsy samples from the same cases (n = 398) or, in the majority of cases, with The Cancer Genome Atlas database, which uses tumor biopsies to perform genome-wide sequencing studies. Both types of biopsy revealed very similar mutation patterns when the Guardant360 next-generation sequencing test, which targets 70 genes, was applied. In particular, when EGFR, BRAF, KRAS, ALK, RET, and ROS1 mutations were identified by tumor tissue biopsy, the same mutations were reported in 94%-100% of plasma samples.15
Studies of the clinical utility of ctDNA and CTCs are among ongoing clinical trials of liquid biopsies (Tables 2 and 3). The potential for using CTCs to guide treatment decisions has become particularly relevant in breast cancer in light of results showing that patients with primary tumors that are negative for human epidermal growth factor receptor 2 (HER2) amplification, an important biomarker in breast cancer, may have CTCs that are HER2-positive, in up to 30% of cases. These patients may therefore still benefit from HER2-targeted therapy.16
The DETECT studies are the first phase 3 trials in which treatment decisions are being based on the phenotypic characteristics of CTCs. DETECT III (NCT01619111) is comparing lapatinib in combination with standard therapy with standard therapy alone in patients with HER2-negative metastatic breast cancer who have HER2-positive CTCs, whereas DETECT IV (NCT02035813) is enrolling patients with HER2-negative, hormone receptor-positive metastatic breast cancer and persistent HER2-negative CTCs to receive standard endocrine therapy and the mammalian target of rapamycin inhibitor everolimus.
Other targets and sources for liquid biopsy
Another approach to liquid biopsies that is also beginning to take off is to collect tumor-derived exosomes from the bloodstream. Exosomes are tiny, fluid-filled, membrane-bound sacks that bud off from the surface of a cell to expel waste or to transport cargo from one cell to another. DNA, RNA, and protein can be extracted from tumor-derived exosomes and could also serve as molecular biomarkers relating to the cancer cells from which they came.6,7
Exosome Diagnostics is bringing the first exosome-based diagnostic tests to the market and recently teamed up with Amgen for the development of these liquid biopsies.17 In January 2016, they launched ExoDx Lung (ALK), for detection of EML4-ALK gene fusions in patients with NSCLC, using a proprietary platform for the isolation of RNA from exosomes. Data that was presented at several different conferences in 2015 demonstrated a sensitivity of 88% and specificity of 100% for this diagnostic when compared with tissue ALK status in NSCLC patients receiving a second-generation ALK inhibitor following progression on prior ALK inhibitor therapy.18
In September, they subsequently announced the launch of a test that analyses genetic information from exosomes collected from a urine sample taken from prostate cancer patients. Using a 3-gene signature, in combination with a proprietary algorithm, this diagnostic generates a score assessing a prostate cancer patient’s risk for higher grade, more aggressive disease. It is designed to complement the prostate-specific antigen score and has demonstrated accuracy in ruling out the presence of high-grade cancer before an initial biopsy in more than 1,
1. Lennon NK, Adalsteinsson VA, Gabriel SB. Technological considerations for genome-guided diagnosis and management of cancer. Genome Med. 2016;8:112.
2. MIT Technology Review website. Liquid biopsy: fast DNA-sequencing machines are leading to simple blood tests for cancer. https://www.technologyreview.com/s/534991/liquid-biopsy/. Published 2015. Accessed December 19, 2016.
3. Alix-Panabières C and Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6(5):479-491.
4. Calabuig-Farinãs S, Jantus-Lewintre E, Herreros-Pomares A, Camps C. Circulating tumor cells versus circulating tumor DNA in lung cancer – which one will win? Transl Lung Cancer Res. 2016;5(5):466-482.
5. Karabacak, NM, Spuhler PS, Fachin F, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat Protoc. 2014;9:694-710.
6. Buder A, Tomuta C, and Filipits M. The potential of liquid biopsies. Curr Opin Oncol. 2016;28:130-134.
7. Hofman P, Popper HH. Pathologists and liquid biopsies: to be or not to be? Virchows Arch. 2016;469:601-609.
8. Bidard FC, Peeters DJ, Fehm T, et al. Clinical validity of circulating tumor cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014;15(4):406-414.
9. de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302-6309.
10. Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213-3221.
11. CellSearch Web site. What is the CELLSEARCH® System? https://www.cellsearchctc.com/product-systems-overview/cellsearch-system-overview. Last updated December 5th, 2016. Accessed online December 19th, 2016.
12. CellSearch Web site [advertisement]. https://www.cellsearchctc.com/clinical-applications/clinical-applications-overview. Last updated December 5, 2016. Accessed December 19, 2016.
13. US Food and Drug Administration. cobas EGFR Mutation Test v2 – P150047. http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm519922.htm. Last updated September 9, 2016. Accessed December 19, 2016.
14. Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015;26(9):1883-1889.
15. Zill OA, Mortimer S, Banks KC, et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. J Clin Oncol. 2016;34(suppl;abstr LBA11501).
16. Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537:102-106.
17. Exosome Diagnostics. Exosome diagnostics enters agreement with Amgen. http://www.exosomedx.com/news-events/press-releases/exosome-diagnostics-enters-agreement-amgen. Published October 3, 2016. Accessed December 19, 2016.
18. Brinkman K, Emenegger J, Tannous B, et al. Exosomal RNA-based liquid biopsy detection of EML4-ALK in plasma from NSCLC patients [2015 World Conference on Lung Cancer, Denver, CO; abstract 2591]. http://library.iaslc.org/search-speaker?search_speaker=30493. Accessed January 6, 2017.
19. Exosome Diagnostics website. Prostate cancer. http://www.exosomedx.com/prostate-cancer-0. Last updated 2017. Accessed online December 19, 2016.
1. Lennon NK, Adalsteinsson VA, Gabriel SB. Technological considerations for genome-guided diagnosis and management of cancer. Genome Med. 2016;8:112.
2. MIT Technology Review website. Liquid biopsy: fast DNA-sequencing machines are leading to simple blood tests for cancer. https://www.technologyreview.com/s/534991/liquid-biopsy/. Published 2015. Accessed December 19, 2016.
3. Alix-Panabières C and Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6(5):479-491.
4. Calabuig-Farinãs S, Jantus-Lewintre E, Herreros-Pomares A, Camps C. Circulating tumor cells versus circulating tumor DNA in lung cancer – which one will win? Transl Lung Cancer Res. 2016;5(5):466-482.
5. Karabacak, NM, Spuhler PS, Fachin F, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat Protoc. 2014;9:694-710.
6. Buder A, Tomuta C, and Filipits M. The potential of liquid biopsies. Curr Opin Oncol. 2016;28:130-134.
7. Hofman P, Popper HH. Pathologists and liquid biopsies: to be or not to be? Virchows Arch. 2016;469:601-609.
8. Bidard FC, Peeters DJ, Fehm T, et al. Clinical validity of circulating tumor cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014;15(4):406-414.
9. de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14(19):6302-6309.
10. Cohen SJ, Punt CJ, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213-3221.
11. CellSearch Web site. What is the CELLSEARCH® System? https://www.cellsearchctc.com/product-systems-overview/cellsearch-system-overview. Last updated December 5th, 2016. Accessed online December 19th, 2016.
12. CellSearch Web site [advertisement]. https://www.cellsearchctc.com/clinical-applications/clinical-applications-overview. Last updated December 5, 2016. Accessed December 19, 2016.
13. US Food and Drug Administration. cobas EGFR Mutation Test v2 – P150047. http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm519922.htm. Last updated September 9, 2016. Accessed December 19, 2016.
14. Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small cell lung cancer: analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol. 2015;26(9):1883-1889.
15. Zill OA, Mortimer S, Banks KC, et al. Somatic genomic landscape of over 15,000 patients with advanced-stage cancer from clinical next-generation sequencing analysis of circulating tumor DNA. J Clin Oncol. 2016;34(suppl;abstr LBA11501).
16. Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537:102-106.
17. Exosome Diagnostics. Exosome diagnostics enters agreement with Amgen. http://www.exosomedx.com/news-events/press-releases/exosome-diagnostics-enters-agreement-amgen. Published October 3, 2016. Accessed December 19, 2016.
18. Brinkman K, Emenegger J, Tannous B, et al. Exosomal RNA-based liquid biopsy detection of EML4-ALK in plasma from NSCLC patients [2015 World Conference on Lung Cancer, Denver, CO; abstract 2591]. http://library.iaslc.org/search-speaker?search_speaker=30493. Accessed January 6, 2017.
19. Exosome Diagnostics website. Prostate cancer. http://www.exosomedx.com/prostate-cancer-0. Last updated 2017. Accessed online December 19, 2016.
Expanding treatment options and ongoing challenges for urologic cancers
Urologic cancers are those that form in organs of the urinary and male reproductive systems, the most significant among them being cancers of the bladder, kidney, prostate, and testicles. Collectively, they are diagnosed in close to 400,000 Americans each year and are responsible for almost 60,000 deaths annually.1 Here, we describe the most recent developments in treating these malignancies.
Update/related article
Atezolizumab approval marks first new treatment option for bladder cancer in more than 3 decades
Click on the PDF icon at the top of this introduction to read the full article.
Urologic cancers are those that form in organs of the urinary and male reproductive systems, the most significant among them being cancers of the bladder, kidney, prostate, and testicles. Collectively, they are diagnosed in close to 400,000 Americans each year and are responsible for almost 60,000 deaths annually.1 Here, we describe the most recent developments in treating these malignancies.
Update/related article
Atezolizumab approval marks first new treatment option for bladder cancer in more than 3 decades
Click on the PDF icon at the top of this introduction to read the full article.
Urologic cancers are those that form in organs of the urinary and male reproductive systems, the most significant among them being cancers of the bladder, kidney, prostate, and testicles. Collectively, they are diagnosed in close to 400,000 Americans each year and are responsible for almost 60,000 deaths annually.1 Here, we describe the most recent developments in treating these malignancies.
Update/related article
Atezolizumab approval marks first new treatment option for bladder cancer in more than 3 decades
Click on the PDF icon at the top of this introduction to read the full article.
Evolving therapeutic strategies maintain clinical momentum in melanoma
The past 5 years have witnessed a watershed moment in the management of metastatic melanoma. The successes of molecularly targeted and immune-based therapies have transformed it from an aggressively lethal malignancy into one that is readily treatable. Here, we discuss continued efforts to find new therapies and broaden the clinical impact of existing options to maintain the unprecedented momentum of improving patient outcomes.
Click on the PDF icon at the top of this introduction to read the full article.
The past 5 years have witnessed a watershed moment in the management of metastatic melanoma. The successes of molecularly targeted and immune-based therapies have transformed it from an aggressively lethal malignancy into one that is readily treatable. Here, we discuss continued efforts to find new therapies and broaden the clinical impact of existing options to maintain the unprecedented momentum of improving patient outcomes.
Click on the PDF icon at the top of this introduction to read the full article.
The past 5 years have witnessed a watershed moment in the management of metastatic melanoma. The successes of molecularly targeted and immune-based therapies have transformed it from an aggressively lethal malignancy into one that is readily treatable. Here, we discuss continued efforts to find new therapies and broaden the clinical impact of existing options to maintain the unprecedented momentum of improving patient outcomes.
Click on the PDF icon at the top of this introduction to read the full article.
Old theories spark new ideas about cancer cell metabolism
It has long been understood that cancer cells have a different metabolism from that of normal cells, but this was widely thought to be a by-product of the genetic alterations that are the suspected drivers of cancer development.
Though early proponents of tumor metabolism argued that cancer was fundamentally a metabolic disease, this idea was sidetracked as we entered the genome-sequencing era and focused on precision medicine. In the past decade, however, we’ve come full circle as many researchers have again begun to advocate for a revival of the idea that tumor metabolism might represent a root cause of cancer.
“Altered metabolism can be both a cause and an effect that is essential for cancer progression,” Dr. Chi Van Dang, director of the Abramson Cancer Center at the University of Pennsylvania, Philadelphia, said in an interview.
Cancer’s sweet tooth
The unique metabolism of cancer cells was first highlighted almost a century ago by the German physiologist Dr. Otto Warburg. He noticed that cancer cells change the way they derive energy from glucose. Typically, cells use glycolysis to convert glucose into pyruvate in the cytoplasm. The pyruvate is then fed into the mitochondria to produce energy through oxidative phosphorylation, which requires oxygen. When oxygen levels are low, cells are able to make energy through glycolysis, but this is much less efficient, yielding far less energy for each glucose molecule consumed.

Cancer cells, however, seem to prefer to use glycolysis, even when oxygen is plentiful, and in order to compensate for the fact that glycolysis produces significantly less energy, the cancer cells take up much more glucose than their normal counterparts.
Dr. Warburg’s rationale was that cancer cells use glycolysis because their mitochondria are damaged, and he even proposed that this defect was “the root cause of cancer,” that the genetic dysregulation of cancer cells could be traced to impaired respiration and energy metabolism. In fact, it turned out that mitochondria, for the most part, are intact in cancer cells, and, because the “Warburg effect,” as it has come to be known, didn’t fully explain how cancer develops, it was written off as an effect of cancer, rather than the cause.
Beyond the Warburg effect
As we have added to our understanding of how cancer develops, it has become increasingly clear that many of the drivers of tumorigenesis are intricately linked to cellular metabolism, which has renewed interest in Warburg’s hypothesis and in tumor metabolism.
Because the Warburg effect didn’t appear to be an adaptation to defective respiration, the question remained as to why cancer cells would use a less efficient method of energy production. The answer, at least in part, is that they aren’t just using it to make energy.
Cancer cells are growing and dividing very rapidly, and they need a quick and efficient means of producing all of the metabolic building blocks they need to do this. Using glycolysis is a trade-off between making less energy and making it more quickly. Furthermore, the conversion of glucose into pyruvate via glycolysis occurs through several intermediates that can be diverted into other biosynthetic pathways that produce some of those much needed building blocks required to produce essential cellular components.
Although the Warburg effect is still the best characterized metabolic distinction in cancer cells, it can now be added to a growing number of dysregulated metabolic processes.
“Cancer’s sweet tooth is not the entire picture,” said Dr. Dang, “because cells are made of more than just carbon, hydrogen, and oxygen.” Research has shown that there is a fundamental change in the metabolism of all four major classes of macromolecules – carbohydrates, proteins, lipids, and nucleic acids. Cancer-associated alterations have been shown to encompass all aspects of cellular metabolism, increasing the ability of tumors to acquire nutrients, but also dictating how those nutrients are used.
As the understanding of tumor metabolism is growing, so too is an appreciation of its heterogeneity. Not all tumor types and, indeed, not all cells within the same tumor, will display the same metabolic profile. The metabolic needs of a tumor can depend upon nutrient availability, the influence of its surrounding environment, and the distance to the nearest blood vessels.
The selfish cell
Many researchers have also begun to reexamine the idea that tumor metabolism is more than just a hallmark of cancer. On the one hand, Dr. Dang explained, “there are mutations in genes that affect mitochondrial function that leads to cancer and in metabolic enzymes that in turn trigger cancer development through altering the epigenome.” On the other hand, “cancer genes – oncogenes and tumor suppressor genes that drive cancer development – can alter cancer cell metabolism to support cancer cell growth.” Hence, one can argue for a role of tumor metabolism as both a cause of tumor growth and one of the many effects of the genetic mutability of cancer.
An example of a widely studied gene that has turned out to be vital to tumor metabolism is the MYC oncogene that encodes a transcription factor with a well-known role in cell cycle progression. It turns out, said Dr. Dang, that “MYC opens up the fuel line to drive cell growth; it drives genes that import nutrients into cells and metabolize nutrients to make the building blocks for the growing cell.”
Many of the growth factor signaling pathways that are commonly dysregulated in cancer don’t just regulate cell growth and proliferation, but are also intimately involved in dictating the response to nutrients. A prominent example is the PI3K/Akt pathway; among the most highly mutated pathways in human cancer, it acts as a master regulator of glucose uptake.
Some researchers propose that alterations in growth factor signaling pathways drive cancer formation because they allow the cell to flaunt the rules of the dinner table. Essentially, instead of acting like a cell that forms part of a multicellular organism, taking cues from the surrounding cells about when and how to respond to nutrients, cancer cells are able to ignore these signals and take up whatever nutrients they can find. This may also explain the observation that cancer development is closely linked to obesity and diabetes.
Targeting metabolism
These discoveries have led researchers to consider the possibility of targeting metabolism to prevent cancer cell growth. Some commonly used chemotherapy drugs have a metabolic mechanism of action; the antifolates methotrexate and pemetrexed, for example, target one-carbon metabolism.
Although no metabolism-based small-molecule drugs are approved yet, many are in preclinical development, and some are beginning to move forward into clinical trials. Dr. Dang highlighted the development of drugs targeting metabolic enzymes: “Drugs being developed against the mutant enzymes IDH1/2 have demonstrated clinical responses in acute myelocytic leukemia, and other drugs, such as CB-839 – an inhibitor of glutaminase – are undergoing clinical studies.”
Several existing drugs have also been repurposed in the fight against cancer, including some not previously thought of in this context, such as the antidiabetic drug metformin, which has shown significant promise. Dr. Dang concluded: “The future outlook is the generation of a new class of drugs that could, in combination with other drugs, result in better clinical treatments for cancers.”
It has long been understood that cancer cells have a different metabolism from that of normal cells, but this was widely thought to be a by-product of the genetic alterations that are the suspected drivers of cancer development.
Though early proponents of tumor metabolism argued that cancer was fundamentally a metabolic disease, this idea was sidetracked as we entered the genome-sequencing era and focused on precision medicine. In the past decade, however, we’ve come full circle as many researchers have again begun to advocate for a revival of the idea that tumor metabolism might represent a root cause of cancer.
“Altered metabolism can be both a cause and an effect that is essential for cancer progression,” Dr. Chi Van Dang, director of the Abramson Cancer Center at the University of Pennsylvania, Philadelphia, said in an interview.
Cancer’s sweet tooth
The unique metabolism of cancer cells was first highlighted almost a century ago by the German physiologist Dr. Otto Warburg. He noticed that cancer cells change the way they derive energy from glucose. Typically, cells use glycolysis to convert glucose into pyruvate in the cytoplasm. The pyruvate is then fed into the mitochondria to produce energy through oxidative phosphorylation, which requires oxygen. When oxygen levels are low, cells are able to make energy through glycolysis, but this is much less efficient, yielding far less energy for each glucose molecule consumed.
Cancer cells, however, seem to prefer to use glycolysis, even when oxygen is plentiful, and in order to compensate for the fact that glycolysis produces significantly less energy, the cancer cells take up much more glucose than their normal counterparts.
Dr. Warburg’s rationale was that cancer cells use glycolysis because their mitochondria are damaged, and he even proposed that this defect was “the root cause of cancer,” that the genetic dysregulation of cancer cells could be traced to impaired respiration and energy metabolism. In fact, it turned out that mitochondria, for the most part, are intact in cancer cells, and, because the “Warburg effect,” as it has come to be known, didn’t fully explain how cancer develops, it was written off as an effect of cancer, rather than the cause.
Beyond the Warburg effect
As we have added to our understanding of how cancer develops, it has become increasingly clear that many of the drivers of tumorigenesis are intricately linked to cellular metabolism, which has renewed interest in Warburg’s hypothesis and in tumor metabolism.
Because the Warburg effect didn’t appear to be an adaptation to defective respiration, the question remained as to why cancer cells would use a less efficient method of energy production. The answer, at least in part, is that they aren’t just using it to make energy.
Cancer cells are growing and dividing very rapidly, and they need a quick and efficient means of producing all of the metabolic building blocks they need to do this. Using glycolysis is a trade-off between making less energy and making it more quickly. Furthermore, the conversion of glucose into pyruvate via glycolysis occurs through several intermediates that can be diverted into other biosynthetic pathways that produce some of those much needed building blocks required to produce essential cellular components.
Although the Warburg effect is still the best characterized metabolic distinction in cancer cells, it can now be added to a growing number of dysregulated metabolic processes.
“Cancer’s sweet tooth is not the entire picture,” said Dr. Dang, “because cells are made of more than just carbon, hydrogen, and oxygen.” Research has shown that there is a fundamental change in the metabolism of all four major classes of macromolecules – carbohydrates, proteins, lipids, and nucleic acids. Cancer-associated alterations have been shown to encompass all aspects of cellular metabolism, increasing the ability of tumors to acquire nutrients, but also dictating how those nutrients are used.
As the understanding of tumor metabolism is growing, so too is an appreciation of its heterogeneity. Not all tumor types and, indeed, not all cells within the same tumor, will display the same metabolic profile. The metabolic needs of a tumor can depend upon nutrient availability, the influence of its surrounding environment, and the distance to the nearest blood vessels.
The selfish cell
Many researchers have also begun to reexamine the idea that tumor metabolism is more than just a hallmark of cancer. On the one hand, Dr. Dang explained, “there are mutations in genes that affect mitochondrial function that leads to cancer and in metabolic enzymes that in turn trigger cancer development through altering the epigenome.” On the other hand, “cancer genes – oncogenes and tumor suppressor genes that drive cancer development – can alter cancer cell metabolism to support cancer cell growth.” Hence, one can argue for a role of tumor metabolism as both a cause of tumor growth and one of the many effects of the genetic mutability of cancer.
An example of a widely studied gene that has turned out to be vital to tumor metabolism is the MYC oncogene that encodes a transcription factor with a well-known role in cell cycle progression. It turns out, said Dr. Dang, that “MYC opens up the fuel line to drive cell growth; it drives genes that import nutrients into cells and metabolize nutrients to make the building blocks for the growing cell.”
Many of the growth factor signaling pathways that are commonly dysregulated in cancer don’t just regulate cell growth and proliferation, but are also intimately involved in dictating the response to nutrients. A prominent example is the PI3K/Akt pathway; among the most highly mutated pathways in human cancer, it acts as a master regulator of glucose uptake.
Some researchers propose that alterations in growth factor signaling pathways drive cancer formation because they allow the cell to flaunt the rules of the dinner table. Essentially, instead of acting like a cell that forms part of a multicellular organism, taking cues from the surrounding cells about when and how to respond to nutrients, cancer cells are able to ignore these signals and take up whatever nutrients they can find. This may also explain the observation that cancer development is closely linked to obesity and diabetes.
Targeting metabolism
These discoveries have led researchers to consider the possibility of targeting metabolism to prevent cancer cell growth. Some commonly used chemotherapy drugs have a metabolic mechanism of action; the antifolates methotrexate and pemetrexed, for example, target one-carbon metabolism.
Although no metabolism-based small-molecule drugs are approved yet, many are in preclinical development, and some are beginning to move forward into clinical trials. Dr. Dang highlighted the development of drugs targeting metabolic enzymes: “Drugs being developed against the mutant enzymes IDH1/2 have demonstrated clinical responses in acute myelocytic leukemia, and other drugs, such as CB-839 – an inhibitor of glutaminase – are undergoing clinical studies.”
Several existing drugs have also been repurposed in the fight against cancer, including some not previously thought of in this context, such as the antidiabetic drug metformin, which has shown significant promise. Dr. Dang concluded: “The future outlook is the generation of a new class of drugs that could, in combination with other drugs, result in better clinical treatments for cancers.”
It has long been understood that cancer cells have a different metabolism from that of normal cells, but this was widely thought to be a by-product of the genetic alterations that are the suspected drivers of cancer development.
Though early proponents of tumor metabolism argued that cancer was fundamentally a metabolic disease, this idea was sidetracked as we entered the genome-sequencing era and focused on precision medicine. In the past decade, however, we’ve come full circle as many researchers have again begun to advocate for a revival of the idea that tumor metabolism might represent a root cause of cancer.
“Altered metabolism can be both a cause and an effect that is essential for cancer progression,” Dr. Chi Van Dang, director of the Abramson Cancer Center at the University of Pennsylvania, Philadelphia, said in an interview.
Cancer’s sweet tooth
The unique metabolism of cancer cells was first highlighted almost a century ago by the German physiologist Dr. Otto Warburg. He noticed that cancer cells change the way they derive energy from glucose. Typically, cells use glycolysis to convert glucose into pyruvate in the cytoplasm. The pyruvate is then fed into the mitochondria to produce energy through oxidative phosphorylation, which requires oxygen. When oxygen levels are low, cells are able to make energy through glycolysis, but this is much less efficient, yielding far less energy for each glucose molecule consumed.
Cancer cells, however, seem to prefer to use glycolysis, even when oxygen is plentiful, and in order to compensate for the fact that glycolysis produces significantly less energy, the cancer cells take up much more glucose than their normal counterparts.
Dr. Warburg’s rationale was that cancer cells use glycolysis because their mitochondria are damaged, and he even proposed that this defect was “the root cause of cancer,” that the genetic dysregulation of cancer cells could be traced to impaired respiration and energy metabolism. In fact, it turned out that mitochondria, for the most part, are intact in cancer cells, and, because the “Warburg effect,” as it has come to be known, didn’t fully explain how cancer develops, it was written off as an effect of cancer, rather than the cause.
Beyond the Warburg effect
As we have added to our understanding of how cancer develops, it has become increasingly clear that many of the drivers of tumorigenesis are intricately linked to cellular metabolism, which has renewed interest in Warburg’s hypothesis and in tumor metabolism.
Because the Warburg effect didn’t appear to be an adaptation to defective respiration, the question remained as to why cancer cells would use a less efficient method of energy production. The answer, at least in part, is that they aren’t just using it to make energy.
Cancer cells are growing and dividing very rapidly, and they need a quick and efficient means of producing all of the metabolic building blocks they need to do this. Using glycolysis is a trade-off between making less energy and making it more quickly. Furthermore, the conversion of glucose into pyruvate via glycolysis occurs through several intermediates that can be diverted into other biosynthetic pathways that produce some of those much needed building blocks required to produce essential cellular components.
Although the Warburg effect is still the best characterized metabolic distinction in cancer cells, it can now be added to a growing number of dysregulated metabolic processes.
“Cancer’s sweet tooth is not the entire picture,” said Dr. Dang, “because cells are made of more than just carbon, hydrogen, and oxygen.” Research has shown that there is a fundamental change in the metabolism of all four major classes of macromolecules – carbohydrates, proteins, lipids, and nucleic acids. Cancer-associated alterations have been shown to encompass all aspects of cellular metabolism, increasing the ability of tumors to acquire nutrients, but also dictating how those nutrients are used.
As the understanding of tumor metabolism is growing, so too is an appreciation of its heterogeneity. Not all tumor types and, indeed, not all cells within the same tumor, will display the same metabolic profile. The metabolic needs of a tumor can depend upon nutrient availability, the influence of its surrounding environment, and the distance to the nearest blood vessels.
The selfish cell
Many researchers have also begun to reexamine the idea that tumor metabolism is more than just a hallmark of cancer. On the one hand, Dr. Dang explained, “there are mutations in genes that affect mitochondrial function that leads to cancer and in metabolic enzymes that in turn trigger cancer development through altering the epigenome.” On the other hand, “cancer genes – oncogenes and tumor suppressor genes that drive cancer development – can alter cancer cell metabolism to support cancer cell growth.” Hence, one can argue for a role of tumor metabolism as both a cause of tumor growth and one of the many effects of the genetic mutability of cancer.
An example of a widely studied gene that has turned out to be vital to tumor metabolism is the MYC oncogene that encodes a transcription factor with a well-known role in cell cycle progression. It turns out, said Dr. Dang, that “MYC opens up the fuel line to drive cell growth; it drives genes that import nutrients into cells and metabolize nutrients to make the building blocks for the growing cell.”
Many of the growth factor signaling pathways that are commonly dysregulated in cancer don’t just regulate cell growth and proliferation, but are also intimately involved in dictating the response to nutrients. A prominent example is the PI3K/Akt pathway; among the most highly mutated pathways in human cancer, it acts as a master regulator of glucose uptake.
Some researchers propose that alterations in growth factor signaling pathways drive cancer formation because they allow the cell to flaunt the rules of the dinner table. Essentially, instead of acting like a cell that forms part of a multicellular organism, taking cues from the surrounding cells about when and how to respond to nutrients, cancer cells are able to ignore these signals and take up whatever nutrients they can find. This may also explain the observation that cancer development is closely linked to obesity and diabetes.
Targeting metabolism
These discoveries have led researchers to consider the possibility of targeting metabolism to prevent cancer cell growth. Some commonly used chemotherapy drugs have a metabolic mechanism of action; the antifolates methotrexate and pemetrexed, for example, target one-carbon metabolism.
Although no metabolism-based small-molecule drugs are approved yet, many are in preclinical development, and some are beginning to move forward into clinical trials. Dr. Dang highlighted the development of drugs targeting metabolic enzymes: “Drugs being developed against the mutant enzymes IDH1/2 have demonstrated clinical responses in acute myelocytic leukemia, and other drugs, such as CB-839 – an inhibitor of glutaminase – are undergoing clinical studies.”
Several existing drugs have also been repurposed in the fight against cancer, including some not previously thought of in this context, such as the antidiabetic drug metformin, which has shown significant promise. Dr. Dang concluded: “The future outlook is the generation of a new class of drugs that could, in combination with other drugs, result in better clinical treatments for cancers.”

On the road to harnessing CRISPR gene editing to treat cancer
CRISPR technology, a simple, yet incredibly powerful tool for genetic engineering, is not only allowing cancer researchers to screen for drug targets more efficiently, but is also opening the door for direct cancer treatment through gene interference or activation.
“The pace at which this technology is developing is astounding and almost every cancer research lab is now using some version of it in their studies,” Dr. Scott A. Armstrong, director of Memorial Sloan Kettering Leukemia Center, New York, said in an interview.

Ancient defense mechanism
While the term CRISPR is now synonymous with the editing of human genes, it actually refers to a sort of primitive immune system used by bacteria for billions of years. CRISPR is an acronym for Clustered Regularly Interspaced Short Palindromic Repeats, but the complex terminology belies an elegantly simple mechanism by which bacterial cells destroy invading pathogens.
The bacterial genome contains regions with short repetitive stretches of DNA that are separated by spacers. Researchers made the startling discovery that the spacers are often composed of bits of foreign DNA and it transpired that bacteria use it as a molecular memory of prior infection.
When the same pathogen is encountered again, the stretches of repeats and spacers are transcribed to form CRISPR RNAs (crRNA). Together with a transactivating RNA (tracrRNA), it forms a kind of GPS system for a series of CRISPR-associated (Cas) proteins that function like molecular scissors, destroying the target DNA sequence in the invader’s genome.
There are three CRISPR systems – type I, II, and III – that are associated with different sets of Cas proteins and each employ unique methods for achieving the same ultimate function. The type II system that pairs with Cas9 has received the most attention.
Cut and paste gene editing
The discovery that CRISPR could be exploited as a tool for genetic manipulation in mammalian cells sparked a revolution in the genome editing field. “The CRISPR-Cas9 system has been adapted to specifically edit the genomes of mammalian cells, allowing one to make targeted changes to almost any gene,” Dr. Armstrong said.
The use of CRISPR-Cas9 as a genome editing tool is simplified by joining the crRNA and tracrRNA together so they are transcribed in a single guide RNA (gRNA). The GPS coordinates of the gRNA can be preprogrammed to target a gene of interest, specifically directing the co-transcribed Cas9 protein to cut at that location, and introducing a double-strand break (DSB) in the DNA. Cells employ a number of different mechanisms to repair DSBs and these can then be exploited for genome editing purposes, allowing researchers to introduce changes to the DNA as it is repaired.
The CRISPR-Cas9 system excels in its simplicity – allowing alterations to be made to the genome much more easily, quickly, and cheaply than ever before, plagued by far fewer off-target effects. It also allows researchers to examine the function of multiple genes at once, where before they were mostly limited to a single gene.

“Cancer genomics has identified a large number of genes that are mutated in human cancer,” Tyler Jacks, Ph.D., director of the Koch Institute for Integrative Cancer Research at MIT, Boston, said in an interview. “CRISPR allows us to study these genes in cancer cells and in whole animals much more efficiently than the methods that were in use just a few years ago.”
But the potential of the CRISPR-Cas9 system doesn’t stop there. “At the moment, a particularly exciting application is the use of this approach to inactive genes in very specific fashion to assess the function of a given protein in a cancer cell, which should speed the identification of proteins that are important for cancer cells and thus potentially aid drug discovery efforts,” Dr. Armstrong said.
CRISPR at AACR
The latest developments in the use of the CRISPR-Cas9 system were highlighted at the annual meeting of the American Association of Cancer Research. Dr. David Sabatini, professor of biology at MIT, Boston, described his own lab’s method for using CRISPR-Cas9 to seek out the essential genes involved in different types of cancers. In a study recently published in Science, he and his colleagues employed this method in chronic myelogenous leukemia and Burkitt’s lymphoma cell lines. The gRNA library targeted just over 18,000 genes and roughly 10% of these proved to be essential. Mostly, these genes were linked to key cellular processes (Science 2015;350[6264]:1096-1101).
Dr. Christopher Vakoc of Cold Spring Harbor (N.Y.) Laboratory, presented a slightly different kind of CRISPR screen for drug targets. Most commonly, CRISPR introduces edits at the start of the gene, which may or may not change the DNA enough to produce a nonfunctional protein. Dr. Vakoc’s lab has developed a system that instead edits functional protein domains, which present ideal drug targets. Mutated domains can be identified that are essential for cancer cell survival and small molecule inhibitors designed that bind to them to kill cancer cells.
The technique has already been used to identify such a domain on the BRD4 protein and inhibitors that bind to this domain had significant antitumor activity in leukemia, Dr. Vakoc reported. A screen targeting 192 chromatin regulatory domains expressed in mouse acute myeloid leukemia cells was subsequently performed and identified 25 domains that impacted survival, 6 that are already being therapeutically targeted, and 19 novel potential targets.
Another development in CRISPR-Cas9 technology creates an inactive version of the Cas9 enzyme, one that has lost the ability to cut DNA. Though it seems counterintuitive, this has opened up a wealth of new possible uses. Jonathan S. Weissman, Ph.D., professor of cellular and molecular pharmacology, University of California, San Francisco, part of the group to develop this ‘dead’ Cas9 (dCas9), published a description of the use of two new tools dubbed CRISPR interference and CRISPR activation (Cell 2013;152[5]:1173-83).
Essentially, by fusing dCas9 with different proteins, such as epigenetic modifiers or transcriptional activators or repressors, it can be used as a delivery system to fine-tune gene expression, instead of editing the gene sequence.
Treating cancer?
Ultimately, CRISPR-Cas9 could be used to treat cancers by cutting out defective genes and replacing them with a wild-type version, or by repairing mutations, though for the time being this is theoretical. Studies have suggested it is possible with other types of diseases, however.
“It is not clear exactly how the CRISPR system would be used to directly treat cancer, but the discoveries that come from its use will likely lead to new ways to treat cancer,” said Dr Armstrong.
Dr Jacks highlighted the technical challenges that will need to be overcome first. “In principle, CRISPR-based genome editing could be used to correct cancer-causing mutations in tumors in vivo or to inactivate activated cancer genes,” he said. “At this point, however, we lack the technology necessary to deliver the CRISPR system to all cancer cells in the body. Improvements in this so-called ‘delivery problem’ may allow CRISPR to become a powerful anticancer therapy strategy.”
CRISPR technology, a simple, yet incredibly powerful tool for genetic engineering, is not only allowing cancer researchers to screen for drug targets more efficiently, but is also opening the door for direct cancer treatment through gene interference or activation.
“The pace at which this technology is developing is astounding and almost every cancer research lab is now using some version of it in their studies,” Dr. Scott A. Armstrong, director of Memorial Sloan Kettering Leukemia Center, New York, said in an interview.
Ancient defense mechanism
While the term CRISPR is now synonymous with the editing of human genes, it actually refers to a sort of primitive immune system used by bacteria for billions of years. CRISPR is an acronym for Clustered Regularly Interspaced Short Palindromic Repeats, but the complex terminology belies an elegantly simple mechanism by which bacterial cells destroy invading pathogens.
The bacterial genome contains regions with short repetitive stretches of DNA that are separated by spacers. Researchers made the startling discovery that the spacers are often composed of bits of foreign DNA and it transpired that bacteria use it as a molecular memory of prior infection.
When the same pathogen is encountered again, the stretches of repeats and spacers are transcribed to form CRISPR RNAs (crRNA). Together with a transactivating RNA (tracrRNA), it forms a kind of GPS system for a series of CRISPR-associated (Cas) proteins that function like molecular scissors, destroying the target DNA sequence in the invader’s genome.
There are three CRISPR systems – type I, II, and III – that are associated with different sets of Cas proteins and each employ unique methods for achieving the same ultimate function. The type II system that pairs with Cas9 has received the most attention.
Cut and paste gene editing
The discovery that CRISPR could be exploited as a tool for genetic manipulation in mammalian cells sparked a revolution in the genome editing field. “The CRISPR-Cas9 system has been adapted to specifically edit the genomes of mammalian cells, allowing one to make targeted changes to almost any gene,” Dr. Armstrong said.
The use of CRISPR-Cas9 as a genome editing tool is simplified by joining the crRNA and tracrRNA together so they are transcribed in a single guide RNA (gRNA). The GPS coordinates of the gRNA can be preprogrammed to target a gene of interest, specifically directing the co-transcribed Cas9 protein to cut at that location, and introducing a double-strand break (DSB) in the DNA. Cells employ a number of different mechanisms to repair DSBs and these can then be exploited for genome editing purposes, allowing researchers to introduce changes to the DNA as it is repaired.
The CRISPR-Cas9 system excels in its simplicity – allowing alterations to be made to the genome much more easily, quickly, and cheaply than ever before, plagued by far fewer off-target effects. It also allows researchers to examine the function of multiple genes at once, where before they were mostly limited to a single gene.
“Cancer genomics has identified a large number of genes that are mutated in human cancer,” Tyler Jacks, Ph.D., director of the Koch Institute for Integrative Cancer Research at MIT, Boston, said in an interview. “CRISPR allows us to study these genes in cancer cells and in whole animals much more efficiently than the methods that were in use just a few years ago.”
But the potential of the CRISPR-Cas9 system doesn’t stop there. “At the moment, a particularly exciting application is the use of this approach to inactive genes in very specific fashion to assess the function of a given protein in a cancer cell, which should speed the identification of proteins that are important for cancer cells and thus potentially aid drug discovery efforts,” Dr. Armstrong said.
CRISPR at AACR
The latest developments in the use of the CRISPR-Cas9 system were highlighted at the annual meeting of the American Association of Cancer Research. Dr. David Sabatini, professor of biology at MIT, Boston, described his own lab’s method for using CRISPR-Cas9 to seek out the essential genes involved in different types of cancers. In a study recently published in Science, he and his colleagues employed this method in chronic myelogenous leukemia and Burkitt’s lymphoma cell lines. The gRNA library targeted just over 18,000 genes and roughly 10% of these proved to be essential. Mostly, these genes were linked to key cellular processes (Science 2015;350[6264]:1096-1101).
Dr. Christopher Vakoc of Cold Spring Harbor (N.Y.) Laboratory, presented a slightly different kind of CRISPR screen for drug targets. Most commonly, CRISPR introduces edits at the start of the gene, which may or may not change the DNA enough to produce a nonfunctional protein. Dr. Vakoc’s lab has developed a system that instead edits functional protein domains, which present ideal drug targets. Mutated domains can be identified that are essential for cancer cell survival and small molecule inhibitors designed that bind to them to kill cancer cells.
The technique has already been used to identify such a domain on the BRD4 protein and inhibitors that bind to this domain had significant antitumor activity in leukemia, Dr. Vakoc reported. A screen targeting 192 chromatin regulatory domains expressed in mouse acute myeloid leukemia cells was subsequently performed and identified 25 domains that impacted survival, 6 that are already being therapeutically targeted, and 19 novel potential targets.
Another development in CRISPR-Cas9 technology creates an inactive version of the Cas9 enzyme, one that has lost the ability to cut DNA. Though it seems counterintuitive, this has opened up a wealth of new possible uses. Jonathan S. Weissman, Ph.D., professor of cellular and molecular pharmacology, University of California, San Francisco, part of the group to develop this ‘dead’ Cas9 (dCas9), published a description of the use of two new tools dubbed CRISPR interference and CRISPR activation (Cell 2013;152[5]:1173-83).
Essentially, by fusing dCas9 with different proteins, such as epigenetic modifiers or transcriptional activators or repressors, it can be used as a delivery system to fine-tune gene expression, instead of editing the gene sequence.
Treating cancer?
Ultimately, CRISPR-Cas9 could be used to treat cancers by cutting out defective genes and replacing them with a wild-type version, or by repairing mutations, though for the time being this is theoretical. Studies have suggested it is possible with other types of diseases, however.
“It is not clear exactly how the CRISPR system would be used to directly treat cancer, but the discoveries that come from its use will likely lead to new ways to treat cancer,” said Dr Armstrong.
Dr Jacks highlighted the technical challenges that will need to be overcome first. “In principle, CRISPR-based genome editing could be used to correct cancer-causing mutations in tumors in vivo or to inactivate activated cancer genes,” he said. “At this point, however, we lack the technology necessary to deliver the CRISPR system to all cancer cells in the body. Improvements in this so-called ‘delivery problem’ may allow CRISPR to become a powerful anticancer therapy strategy.”
CRISPR technology, a simple, yet incredibly powerful tool for genetic engineering, is not only allowing cancer researchers to screen for drug targets more efficiently, but is also opening the door for direct cancer treatment through gene interference or activation.
“The pace at which this technology is developing is astounding and almost every cancer research lab is now using some version of it in their studies,” Dr. Scott A. Armstrong, director of Memorial Sloan Kettering Leukemia Center, New York, said in an interview.
Ancient defense mechanism
While the term CRISPR is now synonymous with the editing of human genes, it actually refers to a sort of primitive immune system used by bacteria for billions of years. CRISPR is an acronym for Clustered Regularly Interspaced Short Palindromic Repeats, but the complex terminology belies an elegantly simple mechanism by which bacterial cells destroy invading pathogens.
The bacterial genome contains regions with short repetitive stretches of DNA that are separated by spacers. Researchers made the startling discovery that the spacers are often composed of bits of foreign DNA and it transpired that bacteria use it as a molecular memory of prior infection.
When the same pathogen is encountered again, the stretches of repeats and spacers are transcribed to form CRISPR RNAs (crRNA). Together with a transactivating RNA (tracrRNA), it forms a kind of GPS system for a series of CRISPR-associated (Cas) proteins that function like molecular scissors, destroying the target DNA sequence in the invader’s genome.
There are three CRISPR systems – type I, II, and III – that are associated with different sets of Cas proteins and each employ unique methods for achieving the same ultimate function. The type II system that pairs with Cas9 has received the most attention.
Cut and paste gene editing
The discovery that CRISPR could be exploited as a tool for genetic manipulation in mammalian cells sparked a revolution in the genome editing field. “The CRISPR-Cas9 system has been adapted to specifically edit the genomes of mammalian cells, allowing one to make targeted changes to almost any gene,” Dr. Armstrong said.
The use of CRISPR-Cas9 as a genome editing tool is simplified by joining the crRNA and tracrRNA together so they are transcribed in a single guide RNA (gRNA). The GPS coordinates of the gRNA can be preprogrammed to target a gene of interest, specifically directing the co-transcribed Cas9 protein to cut at that location, and introducing a double-strand break (DSB) in the DNA. Cells employ a number of different mechanisms to repair DSBs and these can then be exploited for genome editing purposes, allowing researchers to introduce changes to the DNA as it is repaired.
The CRISPR-Cas9 system excels in its simplicity – allowing alterations to be made to the genome much more easily, quickly, and cheaply than ever before, plagued by far fewer off-target effects. It also allows researchers to examine the function of multiple genes at once, where before they were mostly limited to a single gene.
“Cancer genomics has identified a large number of genes that are mutated in human cancer,” Tyler Jacks, Ph.D., director of the Koch Institute for Integrative Cancer Research at MIT, Boston, said in an interview. “CRISPR allows us to study these genes in cancer cells and in whole animals much more efficiently than the methods that were in use just a few years ago.”
But the potential of the CRISPR-Cas9 system doesn’t stop there. “At the moment, a particularly exciting application is the use of this approach to inactive genes in very specific fashion to assess the function of a given protein in a cancer cell, which should speed the identification of proteins that are important for cancer cells and thus potentially aid drug discovery efforts,” Dr. Armstrong said.
CRISPR at AACR
The latest developments in the use of the CRISPR-Cas9 system were highlighted at the annual meeting of the American Association of Cancer Research. Dr. David Sabatini, professor of biology at MIT, Boston, described his own lab’s method for using CRISPR-Cas9 to seek out the essential genes involved in different types of cancers. In a study recently published in Science, he and his colleagues employed this method in chronic myelogenous leukemia and Burkitt’s lymphoma cell lines. The gRNA library targeted just over 18,000 genes and roughly 10% of these proved to be essential. Mostly, these genes were linked to key cellular processes (Science 2015;350[6264]:1096-1101).
Dr. Christopher Vakoc of Cold Spring Harbor (N.Y.) Laboratory, presented a slightly different kind of CRISPR screen for drug targets. Most commonly, CRISPR introduces edits at the start of the gene, which may or may not change the DNA enough to produce a nonfunctional protein. Dr. Vakoc’s lab has developed a system that instead edits functional protein domains, which present ideal drug targets. Mutated domains can be identified that are essential for cancer cell survival and small molecule inhibitors designed that bind to them to kill cancer cells.
The technique has already been used to identify such a domain on the BRD4 protein and inhibitors that bind to this domain had significant antitumor activity in leukemia, Dr. Vakoc reported. A screen targeting 192 chromatin regulatory domains expressed in mouse acute myeloid leukemia cells was subsequently performed and identified 25 domains that impacted survival, 6 that are already being therapeutically targeted, and 19 novel potential targets.
Another development in CRISPR-Cas9 technology creates an inactive version of the Cas9 enzyme, one that has lost the ability to cut DNA. Though it seems counterintuitive, this has opened up a wealth of new possible uses. Jonathan S. Weissman, Ph.D., professor of cellular and molecular pharmacology, University of California, San Francisco, part of the group to develop this ‘dead’ Cas9 (dCas9), published a description of the use of two new tools dubbed CRISPR interference and CRISPR activation (Cell 2013;152[5]:1173-83).
Essentially, by fusing dCas9 with different proteins, such as epigenetic modifiers or transcriptional activators or repressors, it can be used as a delivery system to fine-tune gene expression, instead of editing the gene sequence.
Treating cancer?
Ultimately, CRISPR-Cas9 could be used to treat cancers by cutting out defective genes and replacing them with a wild-type version, or by repairing mutations, though for the time being this is theoretical. Studies have suggested it is possible with other types of diseases, however.
“It is not clear exactly how the CRISPR system would be used to directly treat cancer, but the discoveries that come from its use will likely lead to new ways to treat cancer,” said Dr Armstrong.
Dr Jacks highlighted the technical challenges that will need to be overcome first. “In principle, CRISPR-based genome editing could be used to correct cancer-causing mutations in tumors in vivo or to inactivate activated cancer genes,” he said. “At this point, however, we lack the technology necessary to deliver the CRISPR system to all cancer cells in the body. Improvements in this so-called ‘delivery problem’ may allow CRISPR to become a powerful anticancer therapy strategy.”

Multiple myeloma: newly approved drugs forge paradigm shift toward chronic disease
The pace of drug development for multiple myeloma was dizzying in 2015, with 5 regulatory approvals for the treatment of relapsed/refractory disease, 3 in a single month. As we stand on the brink of another paradigm shift in the management of this disease, we discuss the new classes of drugs and how they are shaping standard of care with the potential to make multiple myeloma a chronic disease.
Click on the PDF icon at the top of this introduction to read the full article.
The pace of drug development for multiple myeloma was dizzying in 2015, with 5 regulatory approvals for the treatment of relapsed/refractory disease, 3 in a single month. As we stand on the brink of another paradigm shift in the management of this disease, we discuss the new classes of drugs and how they are shaping standard of care with the potential to make multiple myeloma a chronic disease.
Click on the PDF icon at the top of this introduction to read the full article.
The pace of drug development for multiple myeloma was dizzying in 2015, with 5 regulatory approvals for the treatment of relapsed/refractory disease, 3 in a single month. As we stand on the brink of another paradigm shift in the management of this disease, we discuss the new classes of drugs and how they are shaping standard of care with the potential to make multiple myeloma a chronic disease.
Click on the PDF icon at the top of this introduction to read the full article.
New GI therapies bring hope after much frustration
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.
Click on the PDF icon at the top of this introduction to read the full article.