User login
Jennifer Smith is the editor of Oncology Practice, part of MDedge Hematology/Oncology. She was previously the editor of Hematology Times, an editor at Principal Investigators Association, and a reporter at The Oneida Daily Dispatch. She has a BS in journalism.
Stand Up To Cancer adds research muscle
Two researchers have joined Stand Up To Cancer’s scientific advisory committee, which directs the organization’s research initiatives, reviews grant proposals, and oversees active grants.
One new committee member is John D. Carpten, PhD, of the University of Southern California in Los Angeles. Dr. Carpten’s research encompasses several cancer types and focuses on the use of genomic technologies and bioinformatics analysis.

The other new committee member is Roderic I. Pettigrew, MD, PhD, of Texas A&M University in College Station. Dr. Pettigrew is known for pioneering four-dimensional imaging of the cardiovascular system using MRI.

In other news, Fox Chase Cancer Center in Philadelphia has appointed five new endowed chairs. The chair holders “will have new resources to strengthen [the center’s] efforts in various areas of cancer research and to implement more effective cancer treatments,” according to Fox Chase.
Hossein Borghaei, DO, now holds the Gloria and Edmund M. Dunn Chair in Thoracic Oncology. Dr. Borghaei conducts research focused on the development of new cancer treatments, particularly immunotherapies and monoclonal antibodies.

Margie L. Clapper, PhD, holds the Samuel M.V. Hamilton Chair in Cancer Prevention. Dr. Clapper is known for creating one of the first programs in the United States to focus on reducing the risk of cancer in high-risk patients and supporting the early detection and prevention of cancers.

Erica Golemis, PhD, holds the William Wikoff Smith Chair in Cancer Research. Dr. Golemis studies changes in cell signaling associated with tumor development and progression, as well as cancer treatment resistance.

Mariusz A. Wasik, MD, holds the Donald E. and Shirley C. Morel, Stanley and Stella Bayster Chair in Molecular Diagnostics. Dr. Wasik’s research is focused on aberrant cell signaling and oncogenic mutations in lymphomas, as well as cell transformation driven by ALK.

Johnathan R. Whetstine, PhD, holds the Jack Schultz Chair in Basic Science. Dr. Whetstine studies epigenetic mechanisms that regulate cell cycle progression, impact cancer therapy, and drive copy gain, amplifications, and drug resistance.

Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at [email protected], and you could be featured in Movers in Medicine.
Two researchers have joined Stand Up To Cancer’s scientific advisory committee, which directs the organization’s research initiatives, reviews grant proposals, and oversees active grants.
One new committee member is John D. Carpten, PhD, of the University of Southern California in Los Angeles. Dr. Carpten’s research encompasses several cancer types and focuses on the use of genomic technologies and bioinformatics analysis.
The other new committee member is Roderic I. Pettigrew, MD, PhD, of Texas A&M University in College Station. Dr. Pettigrew is known for pioneering four-dimensional imaging of the cardiovascular system using MRI.
In other news, Fox Chase Cancer Center in Philadelphia has appointed five new endowed chairs. The chair holders “will have new resources to strengthen [the center’s] efforts in various areas of cancer research and to implement more effective cancer treatments,” according to Fox Chase.
Hossein Borghaei, DO, now holds the Gloria and Edmund M. Dunn Chair in Thoracic Oncology. Dr. Borghaei conducts research focused on the development of new cancer treatments, particularly immunotherapies and monoclonal antibodies.
Margie L. Clapper, PhD, holds the Samuel M.V. Hamilton Chair in Cancer Prevention. Dr. Clapper is known for creating one of the first programs in the United States to focus on reducing the risk of cancer in high-risk patients and supporting the early detection and prevention of cancers.
Erica Golemis, PhD, holds the William Wikoff Smith Chair in Cancer Research. Dr. Golemis studies changes in cell signaling associated with tumor development and progression, as well as cancer treatment resistance.
Mariusz A. Wasik, MD, holds the Donald E. and Shirley C. Morel, Stanley and Stella Bayster Chair in Molecular Diagnostics. Dr. Wasik’s research is focused on aberrant cell signaling and oncogenic mutations in lymphomas, as well as cell transformation driven by ALK.
Johnathan R. Whetstine, PhD, holds the Jack Schultz Chair in Basic Science. Dr. Whetstine studies epigenetic mechanisms that regulate cell cycle progression, impact cancer therapy, and drive copy gain, amplifications, and drug resistance.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at [email protected], and you could be featured in Movers in Medicine.
Two researchers have joined Stand Up To Cancer’s scientific advisory committee, which directs the organization’s research initiatives, reviews grant proposals, and oversees active grants.
One new committee member is John D. Carpten, PhD, of the University of Southern California in Los Angeles. Dr. Carpten’s research encompasses several cancer types and focuses on the use of genomic technologies and bioinformatics analysis.
The other new committee member is Roderic I. Pettigrew, MD, PhD, of Texas A&M University in College Station. Dr. Pettigrew is known for pioneering four-dimensional imaging of the cardiovascular system using MRI.
In other news, Fox Chase Cancer Center in Philadelphia has appointed five new endowed chairs. The chair holders “will have new resources to strengthen [the center’s] efforts in various areas of cancer research and to implement more effective cancer treatments,” according to Fox Chase.
Hossein Borghaei, DO, now holds the Gloria and Edmund M. Dunn Chair in Thoracic Oncology. Dr. Borghaei conducts research focused on the development of new cancer treatments, particularly immunotherapies and monoclonal antibodies.
Margie L. Clapper, PhD, holds the Samuel M.V. Hamilton Chair in Cancer Prevention. Dr. Clapper is known for creating one of the first programs in the United States to focus on reducing the risk of cancer in high-risk patients and supporting the early detection and prevention of cancers.
Erica Golemis, PhD, holds the William Wikoff Smith Chair in Cancer Research. Dr. Golemis studies changes in cell signaling associated with tumor development and progression, as well as cancer treatment resistance.
Mariusz A. Wasik, MD, holds the Donald E. and Shirley C. Morel, Stanley and Stella Bayster Chair in Molecular Diagnostics. Dr. Wasik’s research is focused on aberrant cell signaling and oncogenic mutations in lymphomas, as well as cell transformation driven by ALK.
Johnathan R. Whetstine, PhD, holds the Jack Schultz Chair in Basic Science. Dr. Whetstine studies epigenetic mechanisms that regulate cell cycle progression, impact cancer therapy, and drive copy gain, amplifications, and drug resistance.
Movers in Medicine highlights career moves and personal achievements by hematologists and oncologists. Did you switch jobs, take on a new role, climb a mountain? Tell us all about it at [email protected], and you could be featured in Movers in Medicine.
R2 appears active in high-risk FL and MZL
CHICAGO – Lenalidomide plus rituximab (R2) demonstrated activity against relapsed or refractory follicular lymphoma (FL) and marginal zone lymphoma (MZL) in the phase 3b MAGNIFY trial.

R2 produced responses in FL and MZL patients, including those who had previously experienced early relapse and patients who were refractory to rituximab or both lenalidomide and rituximab at baseline.
David Jacob Andorsky, MD, of Rocky Mountain Cancer Centers in Boulder, Colo., and colleagues presented these results in a poster at the annual meeting of the American Society of Clinical Oncology.
The ongoing MAGNIFY trial has enrolled 370 patients with relapsed/refractory FL (grade 1-3a) or MZL.
For induction, patients receive lenalidomide (20 mg per day on days 1-21 for 12 cycles) and rituximab (375 mg/m2 per week in cycle 1 and then on day 1 of cycles 3, 5, 7, 9, and 11). Patients who achieve stable disease or better on R2 induction are randomized to maintenance with R2 or rituximab alone.
Dr. Andorsky and colleagues presented results of R2 induction in 310 evaluable patients – 247 with FL and 63 with MZL.
The patients had a median age of 66 years (range, 35-91 years) at baseline, and they had received a median of two prior therapies (range, one to eight). Some patients had experienced early relapse (37%, n = 115), were refractory to rituximab (36%, n = 113), or were refractory to both rituximab and lenalidomide (20%, n = 63) at baseline.
Results
At a median follow-up of 16.7 months, the overall response rate was 73%, and the complete response rate was 45%. The overall response rate was 74% in FL patients, 65% in MZL patients, 63% in rituximab-refractory patients, 51% in double-refractory patients, and 68% in patients with an early relapse.
The median duration of response was 36.8 months in all patients, 35.8 months in MZL patients, and not reached in FL patients. The median duration of response was 35.8 months in patients who were rituximab refractory and was not reached in patients who were not refractory to rituximab.
The median progression-free survival was 36 months overall, 30 months in FL patients, 38 months in MZL patients, 23 months in patients with early relapse, and 15.5 months in double-refractory patients.
“While these [subgroup analyses of efficacy] were exploratory endpoints, I think this suggests that [R2] is a promising regimen for patients that are in the high-risk subgroup,” said Carla Casulo, MD, of the University of Rochester (N.Y.), who reviewed this study in a poster discussion session.
The most common adverse events in this trial were fatigue (48%), neutropenia (40%), diarrhea (35%), nausea (30%), and constipation (29%). The most common grade 3/4 adverse event was neutropenia (34%).
The MAGNIFY trial is sponsored by Celgene. Dr. Andorsky reported financial relationships with Celgene, CTI BioPharma, and Gilead Sciences. Dr. Casulo reported financial relationships with Gilead Sciences, Celgene, and Roche.
SOURCE: Andorsky DJ et al. ASCO 2019, Abstract 7513.
CHICAGO – Lenalidomide plus rituximab (R2) demonstrated activity against relapsed or refractory follicular lymphoma (FL) and marginal zone lymphoma (MZL) in the phase 3b MAGNIFY trial.
R2 produced responses in FL and MZL patients, including those who had previously experienced early relapse and patients who were refractory to rituximab or both lenalidomide and rituximab at baseline.
David Jacob Andorsky, MD, of Rocky Mountain Cancer Centers in Boulder, Colo., and colleagues presented these results in a poster at the annual meeting of the American Society of Clinical Oncology.
The ongoing MAGNIFY trial has enrolled 370 patients with relapsed/refractory FL (grade 1-3a) or MZL.
For induction, patients receive lenalidomide (20 mg per day on days 1-21 for 12 cycles) and rituximab (375 mg/m2 per week in cycle 1 and then on day 1 of cycles 3, 5, 7, 9, and 11). Patients who achieve stable disease or better on R2 induction are randomized to maintenance with R2 or rituximab alone.
Dr. Andorsky and colleagues presented results of R2 induction in 310 evaluable patients – 247 with FL and 63 with MZL.
The patients had a median age of 66 years (range, 35-91 years) at baseline, and they had received a median of two prior therapies (range, one to eight). Some patients had experienced early relapse (37%, n = 115), were refractory to rituximab (36%, n = 113), or were refractory to both rituximab and lenalidomide (20%, n = 63) at baseline.
Results
At a median follow-up of 16.7 months, the overall response rate was 73%, and the complete response rate was 45%. The overall response rate was 74% in FL patients, 65% in MZL patients, 63% in rituximab-refractory patients, 51% in double-refractory patients, and 68% in patients with an early relapse.
The median duration of response was 36.8 months in all patients, 35.8 months in MZL patients, and not reached in FL patients. The median duration of response was 35.8 months in patients who were rituximab refractory and was not reached in patients who were not refractory to rituximab.
The median progression-free survival was 36 months overall, 30 months in FL patients, 38 months in MZL patients, 23 months in patients with early relapse, and 15.5 months in double-refractory patients.
“While these [subgroup analyses of efficacy] were exploratory endpoints, I think this suggests that [R2] is a promising regimen for patients that are in the high-risk subgroup,” said Carla Casulo, MD, of the University of Rochester (N.Y.), who reviewed this study in a poster discussion session.
The most common adverse events in this trial were fatigue (48%), neutropenia (40%), diarrhea (35%), nausea (30%), and constipation (29%). The most common grade 3/4 adverse event was neutropenia (34%).
The MAGNIFY trial is sponsored by Celgene. Dr. Andorsky reported financial relationships with Celgene, CTI BioPharma, and Gilead Sciences. Dr. Casulo reported financial relationships with Gilead Sciences, Celgene, and Roche.
SOURCE: Andorsky DJ et al. ASCO 2019, Abstract 7513.
CHICAGO – Lenalidomide plus rituximab (R2) demonstrated activity against relapsed or refractory follicular lymphoma (FL) and marginal zone lymphoma (MZL) in the phase 3b MAGNIFY trial.
R2 produced responses in FL and MZL patients, including those who had previously experienced early relapse and patients who were refractory to rituximab or both lenalidomide and rituximab at baseline.
David Jacob Andorsky, MD, of Rocky Mountain Cancer Centers in Boulder, Colo., and colleagues presented these results in a poster at the annual meeting of the American Society of Clinical Oncology.
The ongoing MAGNIFY trial has enrolled 370 patients with relapsed/refractory FL (grade 1-3a) or MZL.
For induction, patients receive lenalidomide (20 mg per day on days 1-21 for 12 cycles) and rituximab (375 mg/m2 per week in cycle 1 and then on day 1 of cycles 3, 5, 7, 9, and 11). Patients who achieve stable disease or better on R2 induction are randomized to maintenance with R2 or rituximab alone.
Dr. Andorsky and colleagues presented results of R2 induction in 310 evaluable patients – 247 with FL and 63 with MZL.
The patients had a median age of 66 years (range, 35-91 years) at baseline, and they had received a median of two prior therapies (range, one to eight). Some patients had experienced early relapse (37%, n = 115), were refractory to rituximab (36%, n = 113), or were refractory to both rituximab and lenalidomide (20%, n = 63) at baseline.
Results
At a median follow-up of 16.7 months, the overall response rate was 73%, and the complete response rate was 45%. The overall response rate was 74% in FL patients, 65% in MZL patients, 63% in rituximab-refractory patients, 51% in double-refractory patients, and 68% in patients with an early relapse.
The median duration of response was 36.8 months in all patients, 35.8 months in MZL patients, and not reached in FL patients. The median duration of response was 35.8 months in patients who were rituximab refractory and was not reached in patients who were not refractory to rituximab.
The median progression-free survival was 36 months overall, 30 months in FL patients, 38 months in MZL patients, 23 months in patients with early relapse, and 15.5 months in double-refractory patients.
“While these [subgroup analyses of efficacy] were exploratory endpoints, I think this suggests that [R2] is a promising regimen for patients that are in the high-risk subgroup,” said Carla Casulo, MD, of the University of Rochester (N.Y.), who reviewed this study in a poster discussion session.
The most common adverse events in this trial were fatigue (48%), neutropenia (40%), diarrhea (35%), nausea (30%), and constipation (29%). The most common grade 3/4 adverse event was neutropenia (34%).
The MAGNIFY trial is sponsored by Celgene. Dr. Andorsky reported financial relationships with Celgene, CTI BioPharma, and Gilead Sciences. Dr. Casulo reported financial relationships with Gilead Sciences, Celgene, and Roche.
SOURCE: Andorsky DJ et al. ASCO 2019, Abstract 7513.
REPORTING FROM ASCO 2019
Triplet offers longest PFS yet seen in relapsed/refractory myeloma
CHICAGO – Adding isatuximab, an anti-CD38 monoclonal antibody, to pomalidomide and dexamethasone can prolong progression-free survival (PFS) in patients with relapsed/refractory multiple myeloma, a phase 3 trial suggests.

The median PFS was 11.53 months among patients who received isatuximab (isa) plus pomalidomide and dexamethasone (Pd), compared with 6.47 months in patients who received Pd alone (P = .001).
“[W]e now have the first randomized, phase 3 study demonstrating a significant prolonged PFS benefit of CD38 targeting combined with pomalidomide and dexamethasone in relapsed/refractory myeloma,” said Paul Richardson, MD, of the Dana-Farber Cancer Institute in Boston.
“The PFS was the longest observed in this population to date ..., and ... this PFS benefit was consistent among subgroups.”
Dr. Richardson presented these results from the ICARIA-MM trial at the annual meeting of the American Society of Clinical Oncology.
Researchers enrolled 307 patients with relapsed/refractory multiple myeloma. The patients had a median age of 67 years (range, 36-86 years) and a median time from diagnosis of 4.23 years (range, 0.5-20.5 years).
The patients had received a median of three prior therapies (range, 2-11 for the isa-Pd arm and 2-10 for the Pd arm). All patients had previously received a proteasome inhibitor and an immunomodulatory agent.
The patients received isatuximab at 10 mg/kg on days 1, 8, 15, and 22 in cycle 1, then on days 1 and 15 for subsequent cycles. Pomalidomide and dexamethasone were given at standard doses in both treatment arms.
At a median follow-up of 11.6 months, 42.2% of patients were still receiving isa-Pd, and 22.9% of patients were still on Pd alone. The most common reason for stopping either treatment was disease progression (42.9% in the isa-Pd arm and 57.5% in the Pd arm).
Efficacy
The overall response rate was 60.4% in the isa-Pd arm and 35.3% in the Pd arm (P less than .0001). The rates of complete response/stringent complete response were 4.5% and 2.0%, respectively. The rates of minimal residual disease negativity were 5.2% and 0%, respectively.
The median time to next treatment was 9.1 months in the Pd arm and was not reached in the isa-Pd arm (hazard ratio, 0.538).
The median PFS was 11.53 months in the isa-Pd arm and 6.47 months in the Pd arm (HR, 0.596; P = .001). There was a PFS benefit with isa-Pd across subgroups, including in patients with renal dysfunction, those with high-risk cytogenetics, and patients who were refractory to lenalidomide.
The median overall survival was not reached in either treatment arm. The overall survival rate was 72% in the isa-Pd arm and 63% in the Pd arm (HR, 0.687).
Safety
There were more grade 3 or higher treatment-emergent adverse events (TEAEs) with isa-Pd. However, as Dr. Richardson noted, adding isa to Pd did not increase the rates of TEAEs leading to treatment discontinuation or death.
The incidence of grade 3 or higher TEAEs was 86.8% in the isa-Pd arm and 70.5% in the Pd arm. The rate of serious TEAEs was 61.8% and 53.7%, respectively.
TEAEs leading to treatment discontinuation occurred in 7.2% of patients in the isa-Pd arm and 12.8% in the Pd arm. TEAEs leading to death occurred in 7.9% and 9.4%, respectively.
Grade 3 TEAEs (in the isa-Pd and Pd arms, respectively) included upper respiratory tract infection (3.3% and 0.7%), diarrhea (2.0% and 0.7%), bronchitis (3.3% and 0.7%), pneumonia (15.1% and 13.4%), fatigue (3.9% and 0%), back pain (2.0% and 1.3%), asthenia (3.3% and 2.7%), and dyspnea (3.9% and 1.3%).
The only grade 4 TEAE was pneumonia (1.3% in both arms).
Based on the safety and efficacy results, Dr. Richardson concluded that isa-Pd “is an important new treatment option for our patients with relapsed/refractory myeloma.”
This study was funded by Sanofi. Dr. Richardson reported financial relationships with Celgene, Janssen, and Takeda. His coinvestigators reported relationships with a range of companies.
SOURCE: Richardson P et al. ASCO 2019, Abstract 8004.
CHICAGO – Adding isatuximab, an anti-CD38 monoclonal antibody, to pomalidomide and dexamethasone can prolong progression-free survival (PFS) in patients with relapsed/refractory multiple myeloma, a phase 3 trial suggests.
The median PFS was 11.53 months among patients who received isatuximab (isa) plus pomalidomide and dexamethasone (Pd), compared with 6.47 months in patients who received Pd alone (P = .001).
“[W]e now have the first randomized, phase 3 study demonstrating a significant prolonged PFS benefit of CD38 targeting combined with pomalidomide and dexamethasone in relapsed/refractory myeloma,” said Paul Richardson, MD, of the Dana-Farber Cancer Institute in Boston.
“The PFS was the longest observed in this population to date ..., and ... this PFS benefit was consistent among subgroups.”
Dr. Richardson presented these results from the ICARIA-MM trial at the annual meeting of the American Society of Clinical Oncology.
Researchers enrolled 307 patients with relapsed/refractory multiple myeloma. The patients had a median age of 67 years (range, 36-86 years) and a median time from diagnosis of 4.23 years (range, 0.5-20.5 years).
The patients had received a median of three prior therapies (range, 2-11 for the isa-Pd arm and 2-10 for the Pd arm). All patients had previously received a proteasome inhibitor and an immunomodulatory agent.
The patients received isatuximab at 10 mg/kg on days 1, 8, 15, and 22 in cycle 1, then on days 1 and 15 for subsequent cycles. Pomalidomide and dexamethasone were given at standard doses in both treatment arms.
At a median follow-up of 11.6 months, 42.2% of patients were still receiving isa-Pd, and 22.9% of patients were still on Pd alone. The most common reason for stopping either treatment was disease progression (42.9% in the isa-Pd arm and 57.5% in the Pd arm).
Efficacy
The overall response rate was 60.4% in the isa-Pd arm and 35.3% in the Pd arm (P less than .0001). The rates of complete response/stringent complete response were 4.5% and 2.0%, respectively. The rates of minimal residual disease negativity were 5.2% and 0%, respectively.
The median time to next treatment was 9.1 months in the Pd arm and was not reached in the isa-Pd arm (hazard ratio, 0.538).
The median PFS was 11.53 months in the isa-Pd arm and 6.47 months in the Pd arm (HR, 0.596; P = .001). There was a PFS benefit with isa-Pd across subgroups, including in patients with renal dysfunction, those with high-risk cytogenetics, and patients who were refractory to lenalidomide.
The median overall survival was not reached in either treatment arm. The overall survival rate was 72% in the isa-Pd arm and 63% in the Pd arm (HR, 0.687).
Safety
There were more grade 3 or higher treatment-emergent adverse events (TEAEs) with isa-Pd. However, as Dr. Richardson noted, adding isa to Pd did not increase the rates of TEAEs leading to treatment discontinuation or death.
The incidence of grade 3 or higher TEAEs was 86.8% in the isa-Pd arm and 70.5% in the Pd arm. The rate of serious TEAEs was 61.8% and 53.7%, respectively.
TEAEs leading to treatment discontinuation occurred in 7.2% of patients in the isa-Pd arm and 12.8% in the Pd arm. TEAEs leading to death occurred in 7.9% and 9.4%, respectively.
Grade 3 TEAEs (in the isa-Pd and Pd arms, respectively) included upper respiratory tract infection (3.3% and 0.7%), diarrhea (2.0% and 0.7%), bronchitis (3.3% and 0.7%), pneumonia (15.1% and 13.4%), fatigue (3.9% and 0%), back pain (2.0% and 1.3%), asthenia (3.3% and 2.7%), and dyspnea (3.9% and 1.3%).
The only grade 4 TEAE was pneumonia (1.3% in both arms).
Based on the safety and efficacy results, Dr. Richardson concluded that isa-Pd “is an important new treatment option for our patients with relapsed/refractory myeloma.”
This study was funded by Sanofi. Dr. Richardson reported financial relationships with Celgene, Janssen, and Takeda. His coinvestigators reported relationships with a range of companies.
SOURCE: Richardson P et al. ASCO 2019, Abstract 8004.
CHICAGO – Adding isatuximab, an anti-CD38 monoclonal antibody, to pomalidomide and dexamethasone can prolong progression-free survival (PFS) in patients with relapsed/refractory multiple myeloma, a phase 3 trial suggests.
The median PFS was 11.53 months among patients who received isatuximab (isa) plus pomalidomide and dexamethasone (Pd), compared with 6.47 months in patients who received Pd alone (P = .001).
“[W]e now have the first randomized, phase 3 study demonstrating a significant prolonged PFS benefit of CD38 targeting combined with pomalidomide and dexamethasone in relapsed/refractory myeloma,” said Paul Richardson, MD, of the Dana-Farber Cancer Institute in Boston.
“The PFS was the longest observed in this population to date ..., and ... this PFS benefit was consistent among subgroups.”
Dr. Richardson presented these results from the ICARIA-MM trial at the annual meeting of the American Society of Clinical Oncology.
Researchers enrolled 307 patients with relapsed/refractory multiple myeloma. The patients had a median age of 67 years (range, 36-86 years) and a median time from diagnosis of 4.23 years (range, 0.5-20.5 years).
The patients had received a median of three prior therapies (range, 2-11 for the isa-Pd arm and 2-10 for the Pd arm). All patients had previously received a proteasome inhibitor and an immunomodulatory agent.
The patients received isatuximab at 10 mg/kg on days 1, 8, 15, and 22 in cycle 1, then on days 1 and 15 for subsequent cycles. Pomalidomide and dexamethasone were given at standard doses in both treatment arms.
At a median follow-up of 11.6 months, 42.2% of patients were still receiving isa-Pd, and 22.9% of patients were still on Pd alone. The most common reason for stopping either treatment was disease progression (42.9% in the isa-Pd arm and 57.5% in the Pd arm).
Efficacy
The overall response rate was 60.4% in the isa-Pd arm and 35.3% in the Pd arm (P less than .0001). The rates of complete response/stringent complete response were 4.5% and 2.0%, respectively. The rates of minimal residual disease negativity were 5.2% and 0%, respectively.
The median time to next treatment was 9.1 months in the Pd arm and was not reached in the isa-Pd arm (hazard ratio, 0.538).
The median PFS was 11.53 months in the isa-Pd arm and 6.47 months in the Pd arm (HR, 0.596; P = .001). There was a PFS benefit with isa-Pd across subgroups, including in patients with renal dysfunction, those with high-risk cytogenetics, and patients who were refractory to lenalidomide.
The median overall survival was not reached in either treatment arm. The overall survival rate was 72% in the isa-Pd arm and 63% in the Pd arm (HR, 0.687).
Safety
There were more grade 3 or higher treatment-emergent adverse events (TEAEs) with isa-Pd. However, as Dr. Richardson noted, adding isa to Pd did not increase the rates of TEAEs leading to treatment discontinuation or death.
The incidence of grade 3 or higher TEAEs was 86.8% in the isa-Pd arm and 70.5% in the Pd arm. The rate of serious TEAEs was 61.8% and 53.7%, respectively.
TEAEs leading to treatment discontinuation occurred in 7.2% of patients in the isa-Pd arm and 12.8% in the Pd arm. TEAEs leading to death occurred in 7.9% and 9.4%, respectively.
Grade 3 TEAEs (in the isa-Pd and Pd arms, respectively) included upper respiratory tract infection (3.3% and 0.7%), diarrhea (2.0% and 0.7%), bronchitis (3.3% and 0.7%), pneumonia (15.1% and 13.4%), fatigue (3.9% and 0%), back pain (2.0% and 1.3%), asthenia (3.3% and 2.7%), and dyspnea (3.9% and 1.3%).
The only grade 4 TEAE was pneumonia (1.3% in both arms).
Based on the safety and efficacy results, Dr. Richardson concluded that isa-Pd “is an important new treatment option for our patients with relapsed/refractory myeloma.”
This study was funded by Sanofi. Dr. Richardson reported financial relationships with Celgene, Janssen, and Takeda. His coinvestigators reported relationships with a range of companies.
SOURCE: Richardson P et al. ASCO 2019, Abstract 8004.
REPORTING FROM ASCO 2019
Four-drug combo bests triplet in newly diagnosed myeloma
CHICAGO – Daratumumab plus bortezomib, thalidomide, and dexamethasone (D-VTd) provided a “robust clinical benefit” over bortezomib, thalidomide, and dexamethasone alone (VTd) in a phase 3 trial of patients with newly diagnosed multiple myeloma, according to the trial’s principal investigator.
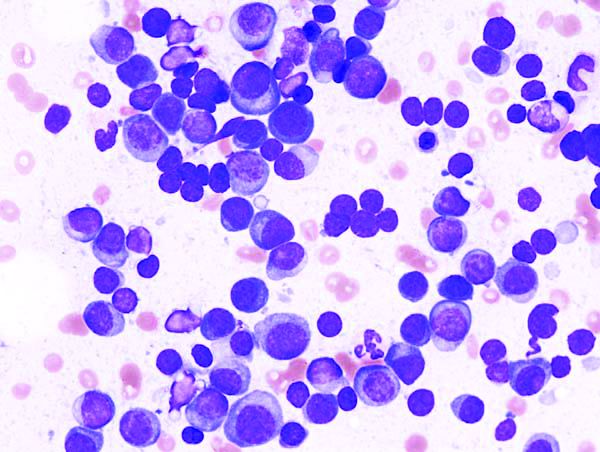
D-VTd produced significantly higher rates of stringent complete response (sCR) and progression-free survival (PFS) than did VTd, said Philippe Moreau, MD, of the University Hospital of Nantes (France).
Dr. Moreau presented these results, from the CASSIOPEIA trial, at the annual meeting of the American Society of Clinical Oncology. The findings were simultaneously published in the Lancet (2019 Jun 3. doi: 10.1016/S0140-6736[19]31240-1).
CASSIOPEIA enrolled 1,085 patients with newly diagnosed multiple myeloma who were eligible for high-dose chemotherapy and hematopoietic stem cell transplant (HSCT). This is a two-part trial, and Dr. Moreau presented final results from part 1.
In part 1, patients were randomized to induction with D-VTd or VTd, followed by autologous HSCT and consolidation with D-VTd or VTd. In part 2, which is ongoing, patients who achieve a partial response or better are randomized to observation or maintenance with daratumumab for up to 2 years.
Baseline characteristics were well balanced between the D-VTd and VTd arms. The median age was 59 years (range, 22-65) and 58 years (range, 26-65), respectively. Most patients had an Eastern Cooperative Oncology Group performance status of 0 or 1, and most had an International Staging System stage of I or II.
The median follow-up was 18.8 months. Most patients completed induction – 85% in the D-VTd arm and 81% in the VTd arm – and went on to HSCT – 90% and 89%, respectively. The most common reasons for treatment discontinuation in both arms were adverse events and progression.
Response and survival
The primary endpoint for part 1 was the rate of sCR at 100 days after HSCT. The sCR rate was significantly higher in the D-VTd arm than it was in the VTd arm – 29% and 20%, respectively (odds ratio, 1.60; P less than .0010).
The overall response rate was significantly higher in the D-VTd arm than in the VTd arm – 93% and 90%, respectively (P less than .0001) – as was the rate of minimal residual disease (MRD) negativity – 64% and 44%, respectively (P less than .0001).
Dr. Moreau noted that sCR and MRD negativity rates were superior with D-VTd across all subgroups except among patients with high-risk cytogenetics and International Staging System stage III disease.
“Dara-VTd resulted in a robust clinical benefit with a higher rate of response, including stringent CR, including MRD negativity,” Dr. Moreau said. “And this translated into a better progression-free survival, with a 53% reduction in the risk of progression or death.”
The 18-month PFS was 93% in the D-VTd arm and 85% in the VTd arm (hazard ratio, 0.47; P less than .0001). D-VTd reduced the risk of progression or death across all subgroups.
The median overall survival (OS) was not reached in either treatment arm. The 18-month OS rate was 98% in the D-VTd arm and 95% in the VTd arm. The 24-month OS rate was 97% and 93%, respectively.
“These results are the best ever reported in the setting of stem cell transplantation,” Dr. Moreau said.
Safety
The most common grade 3/4 treatment-emergent adverse events (in the D-VTd and VTd arms, respectively) were neutropenia (28% and 15%), lymphopenia (17% and 10%), stomatitis (13% and 16%), and thrombocytopenia (11% and 7%).
The rate of infusion-related reactions was 35% in the D-VTd arm and 0% in the VTd arm. The rate of infections was 66% and 57%, respectively. The most common serious infection was pneumonia, which occurred in 4% and 2% of patients, respectively. The rate of second primary malignancies was 2% in both arms.
Based on the safety and efficacy results, Dr. Moreau concluded that D-VTd “should be considered a valid treatment option” for newly diagnosed multiple myeloma patients who are eligible for HSCT.
Dr. Moreau reported relationships with Amgen, Celgene, Janssen-Cilag, Novartis, and Takeda. The study is sponsored by the French Intergroupe Francophone du Myelome in collaboration with the Dutch-Belgian Cooperative Trial Group for Hematology Oncology and Janssen Research & Development.
SOURCE: Moreau P et al. ASCO 2019, Abstract 8003.
CHICAGO – Daratumumab plus bortezomib, thalidomide, and dexamethasone (D-VTd) provided a “robust clinical benefit” over bortezomib, thalidomide, and dexamethasone alone (VTd) in a phase 3 trial of patients with newly diagnosed multiple myeloma, according to the trial’s principal investigator.
D-VTd produced significantly higher rates of stringent complete response (sCR) and progression-free survival (PFS) than did VTd, said Philippe Moreau, MD, of the University Hospital of Nantes (France).
Dr. Moreau presented these results, from the CASSIOPEIA trial, at the annual meeting of the American Society of Clinical Oncology. The findings were simultaneously published in the Lancet (2019 Jun 3. doi: 10.1016/S0140-6736[19]31240-1).
CASSIOPEIA enrolled 1,085 patients with newly diagnosed multiple myeloma who were eligible for high-dose chemotherapy and hematopoietic stem cell transplant (HSCT). This is a two-part trial, and Dr. Moreau presented final results from part 1.
In part 1, patients were randomized to induction with D-VTd or VTd, followed by autologous HSCT and consolidation with D-VTd or VTd. In part 2, which is ongoing, patients who achieve a partial response or better are randomized to observation or maintenance with daratumumab for up to 2 years.
Baseline characteristics were well balanced between the D-VTd and VTd arms. The median age was 59 years (range, 22-65) and 58 years (range, 26-65), respectively. Most patients had an Eastern Cooperative Oncology Group performance status of 0 or 1, and most had an International Staging System stage of I or II.
The median follow-up was 18.8 months. Most patients completed induction – 85% in the D-VTd arm and 81% in the VTd arm – and went on to HSCT – 90% and 89%, respectively. The most common reasons for treatment discontinuation in both arms were adverse events and progression.
Response and survival
The primary endpoint for part 1 was the rate of sCR at 100 days after HSCT. The sCR rate was significantly higher in the D-VTd arm than it was in the VTd arm – 29% and 20%, respectively (odds ratio, 1.60; P less than .0010).
The overall response rate was significantly higher in the D-VTd arm than in the VTd arm – 93% and 90%, respectively (P less than .0001) – as was the rate of minimal residual disease (MRD) negativity – 64% and 44%, respectively (P less than .0001).
Dr. Moreau noted that sCR and MRD negativity rates were superior with D-VTd across all subgroups except among patients with high-risk cytogenetics and International Staging System stage III disease.
“Dara-VTd resulted in a robust clinical benefit with a higher rate of response, including stringent CR, including MRD negativity,” Dr. Moreau said. “And this translated into a better progression-free survival, with a 53% reduction in the risk of progression or death.”
The 18-month PFS was 93% in the D-VTd arm and 85% in the VTd arm (hazard ratio, 0.47; P less than .0001). D-VTd reduced the risk of progression or death across all subgroups.
The median overall survival (OS) was not reached in either treatment arm. The 18-month OS rate was 98% in the D-VTd arm and 95% in the VTd arm. The 24-month OS rate was 97% and 93%, respectively.
“These results are the best ever reported in the setting of stem cell transplantation,” Dr. Moreau said.
Safety
The most common grade 3/4 treatment-emergent adverse events (in the D-VTd and VTd arms, respectively) were neutropenia (28% and 15%), lymphopenia (17% and 10%), stomatitis (13% and 16%), and thrombocytopenia (11% and 7%).
The rate of infusion-related reactions was 35% in the D-VTd arm and 0% in the VTd arm. The rate of infections was 66% and 57%, respectively. The most common serious infection was pneumonia, which occurred in 4% and 2% of patients, respectively. The rate of second primary malignancies was 2% in both arms.
Based on the safety and efficacy results, Dr. Moreau concluded that D-VTd “should be considered a valid treatment option” for newly diagnosed multiple myeloma patients who are eligible for HSCT.
Dr. Moreau reported relationships with Amgen, Celgene, Janssen-Cilag, Novartis, and Takeda. The study is sponsored by the French Intergroupe Francophone du Myelome in collaboration with the Dutch-Belgian Cooperative Trial Group for Hematology Oncology and Janssen Research & Development.
SOURCE: Moreau P et al. ASCO 2019, Abstract 8003.
CHICAGO – Daratumumab plus bortezomib, thalidomide, and dexamethasone (D-VTd) provided a “robust clinical benefit” over bortezomib, thalidomide, and dexamethasone alone (VTd) in a phase 3 trial of patients with newly diagnosed multiple myeloma, according to the trial’s principal investigator.
D-VTd produced significantly higher rates of stringent complete response (sCR) and progression-free survival (PFS) than did VTd, said Philippe Moreau, MD, of the University Hospital of Nantes (France).
Dr. Moreau presented these results, from the CASSIOPEIA trial, at the annual meeting of the American Society of Clinical Oncology. The findings were simultaneously published in the Lancet (2019 Jun 3. doi: 10.1016/S0140-6736[19]31240-1).
CASSIOPEIA enrolled 1,085 patients with newly diagnosed multiple myeloma who were eligible for high-dose chemotherapy and hematopoietic stem cell transplant (HSCT). This is a two-part trial, and Dr. Moreau presented final results from part 1.
In part 1, patients were randomized to induction with D-VTd or VTd, followed by autologous HSCT and consolidation with D-VTd or VTd. In part 2, which is ongoing, patients who achieve a partial response or better are randomized to observation or maintenance with daratumumab for up to 2 years.
Baseline characteristics were well balanced between the D-VTd and VTd arms. The median age was 59 years (range, 22-65) and 58 years (range, 26-65), respectively. Most patients had an Eastern Cooperative Oncology Group performance status of 0 or 1, and most had an International Staging System stage of I or II.
The median follow-up was 18.8 months. Most patients completed induction – 85% in the D-VTd arm and 81% in the VTd arm – and went on to HSCT – 90% and 89%, respectively. The most common reasons for treatment discontinuation in both arms were adverse events and progression.
Response and survival
The primary endpoint for part 1 was the rate of sCR at 100 days after HSCT. The sCR rate was significantly higher in the D-VTd arm than it was in the VTd arm – 29% and 20%, respectively (odds ratio, 1.60; P less than .0010).
The overall response rate was significantly higher in the D-VTd arm than in the VTd arm – 93% and 90%, respectively (P less than .0001) – as was the rate of minimal residual disease (MRD) negativity – 64% and 44%, respectively (P less than .0001).
Dr. Moreau noted that sCR and MRD negativity rates were superior with D-VTd across all subgroups except among patients with high-risk cytogenetics and International Staging System stage III disease.
“Dara-VTd resulted in a robust clinical benefit with a higher rate of response, including stringent CR, including MRD negativity,” Dr. Moreau said. “And this translated into a better progression-free survival, with a 53% reduction in the risk of progression or death.”
The 18-month PFS was 93% in the D-VTd arm and 85% in the VTd arm (hazard ratio, 0.47; P less than .0001). D-VTd reduced the risk of progression or death across all subgroups.
The median overall survival (OS) was not reached in either treatment arm. The 18-month OS rate was 98% in the D-VTd arm and 95% in the VTd arm. The 24-month OS rate was 97% and 93%, respectively.
“These results are the best ever reported in the setting of stem cell transplantation,” Dr. Moreau said.
Safety
The most common grade 3/4 treatment-emergent adverse events (in the D-VTd and VTd arms, respectively) were neutropenia (28% and 15%), lymphopenia (17% and 10%), stomatitis (13% and 16%), and thrombocytopenia (11% and 7%).
The rate of infusion-related reactions was 35% in the D-VTd arm and 0% in the VTd arm. The rate of infections was 66% and 57%, respectively. The most common serious infection was pneumonia, which occurred in 4% and 2% of patients, respectively. The rate of second primary malignancies was 2% in both arms.
Based on the safety and efficacy results, Dr. Moreau concluded that D-VTd “should be considered a valid treatment option” for newly diagnosed multiple myeloma patients who are eligible for HSCT.
Dr. Moreau reported relationships with Amgen, Celgene, Janssen-Cilag, Novartis, and Takeda. The study is sponsored by the French Intergroupe Francophone du Myelome in collaboration with the Dutch-Belgian Cooperative Trial Group for Hematology Oncology and Janssen Research & Development.
SOURCE: Moreau P et al. ASCO 2019, Abstract 8003.
REPORTING FROM ASCO 2019
Adding ipilimumab to nivolumab provides no benefit in SCC trial
CHICAGO – Phase 3 results suggest ipilimumab plus nivolumab is no more effective than nivolumab alone in previously treated patients with metastatic squamous cell lung cancer and no matching biomarker.

However, there is evidence to suggest that patients with a high tumor mutational burden (TMB) and low programmed death–ligand 1 (PD-L1) tumor proportion score (TPS) may derive a benefit from the combination.
Lyudmila Bazhenova, MD, of the University of California, San Diego, and her colleagues presented results from this trial (NCT02785952) in a poster at the annual meeting of the American Society for Clinical Oncology. Kathryn C. Arbour, MD, of Memorial Sloan Kettering Cancer Center in New York reviewed the data in a poster discussion session.
Patients and treatment
The researchers reported on 275 previously treated patients with stage IV or recurrent squamous cell lung cancer who were naive to checkpoint inhibitors. Patients were randomized to receive nivolumab (nivo) at 3 mg/m2 once every 2 weeks (n = 137) or the same dose of nivolumab plus ipilimumab (ipi + nivo) at 1 mg/m2 once every 6 weeks (n = 138).
The patients were stratified by gender and number of prior therapies (one vs. two or more), but they were not stratified by TMB or PD-L1 expression.
The PD-L1 TPS was unknown in 36% of patients, less than 5% in 57%, and 5% or greater in 43% of patients. TMB was unknown in 8% of patients, less than 10 mutations per megabase in 52%, and 10 mutations per megabase or greater in 48%.
Baseline characteristics were similar between the treatment arms. The median age was 67.5 years (range, 42-83 years) in the ipi + nivo arm and 68.1 years (range, 49-90 years) in the nivo arm. Most patients had received only one prior therapy – 85% and 83%, respectively – and most had a performance status of 1 – 71% and 72%, respectively.
Efficacy
There were no significant differences in outcomes between the treatment arms, and the study was closed early for futility.
The overall response rate was 18% in the ipi + nivo arm and 17% in the nivo arm, with one complete response occurring in each arm. The median duration of response was 9.1 months in the ipi + nivo arm and 8.6 months in the nivo arm.
The median progression-free survival was 3.8 months in the ipi + nivo arm and 2.9 months in the nivo arm (hazard ratio, 0.84; P = .19). The 24-month progression-free survival was 8.2% and 5.9%, respectively.
The median overall survival was 10.0 months in the ipi + nivo arm and 11.0 months in the nivo arm (HR, 0.97; P = .82). The 24-month overall survival was 27.6% and 20.1%, respectively.
There were no significant differences in outcomes by TMB or PD-L1 with the cutoffs used in this study, according to Dr. Bazhenova and colleagues, but different cutoffs are being explored.

The median progression-free survival was 4.4 months in TMB-high/PD-L1-low patients in the ipi + nivo arm, compared with 1.7 months in the TMB-high/PD-L1-low patients in the nivo arm. The median overall survival was 15.9 months and 10.3 months, respectively.
“It is slightly challenging to interpret the results without knowing the PD-L1 data of all patients in the cohort, and biomarker selection remains crucial for this combination,” Dr. Arbour said.
Safety
There were no differences in individual toxicities between the treatment arms, but cumulative toxicities were higher in the combination arm, according to the researchers.
The incidence of treatment-related adverse events (AEs) was 88% in the ipi + nivo arm and 90% in the nivo arm. The incidence of grade 3-5 treatment-related AEs was 39% and 31%, respectively.
The incidence of immune-mediated AEs was 65% in the ipi + nivo arm and 57% in the nivo arm. The incidence of immune-mediated grade 3-5 AEs was 20% and 11%, respectively.
There were six AEs leading to death in the ipi + nivo arm – two due to dyspnea, one due to colitis, and one due to respiratory failure. The attribution of one death is under review. For the remaining death, the exact cause is unknown.
There were two AEs leading to death in the nivo arm, both due to pneumonitis.
This study was supported by grants from the National Institutes of Health and by AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, and Pfizer through the Foundation for the National Institutes of Health in partnership with Friends of Cancer Research.
Dr. Bazhenova reported relationships with Epic Sciences, AbbVie, AstraZeneca, Boston Biomedical, Genentech/Roche, Lilly, Loxo, Pfizer, Takeda, and BeyondSpring Pharmaceuticals. Her colleagues reported relationships with these and other companies. Dr. Arbour reported a relationship with AstraZeneca.
SOURCE: Bazhenova L et al. ASCO 2019, Abstract 9014.
CHICAGO – Phase 3 results suggest ipilimumab plus nivolumab is no more effective than nivolumab alone in previously treated patients with metastatic squamous cell lung cancer and no matching biomarker.
However, there is evidence to suggest that patients with a high tumor mutational burden (TMB) and low programmed death–ligand 1 (PD-L1) tumor proportion score (TPS) may derive a benefit from the combination.
Lyudmila Bazhenova, MD, of the University of California, San Diego, and her colleagues presented results from this trial (NCT02785952) in a poster at the annual meeting of the American Society for Clinical Oncology. Kathryn C. Arbour, MD, of Memorial Sloan Kettering Cancer Center in New York reviewed the data in a poster discussion session.
Patients and treatment
The researchers reported on 275 previously treated patients with stage IV or recurrent squamous cell lung cancer who were naive to checkpoint inhibitors. Patients were randomized to receive nivolumab (nivo) at 3 mg/m2 once every 2 weeks (n = 137) or the same dose of nivolumab plus ipilimumab (ipi + nivo) at 1 mg/m2 once every 6 weeks (n = 138).
The patients were stratified by gender and number of prior therapies (one vs. two or more), but they were not stratified by TMB or PD-L1 expression.
The PD-L1 TPS was unknown in 36% of patients, less than 5% in 57%, and 5% or greater in 43% of patients. TMB was unknown in 8% of patients, less than 10 mutations per megabase in 52%, and 10 mutations per megabase or greater in 48%.
Baseline characteristics were similar between the treatment arms. The median age was 67.5 years (range, 42-83 years) in the ipi + nivo arm and 68.1 years (range, 49-90 years) in the nivo arm. Most patients had received only one prior therapy – 85% and 83%, respectively – and most had a performance status of 1 – 71% and 72%, respectively.
Efficacy
There were no significant differences in outcomes between the treatment arms, and the study was closed early for futility.
The overall response rate was 18% in the ipi + nivo arm and 17% in the nivo arm, with one complete response occurring in each arm. The median duration of response was 9.1 months in the ipi + nivo arm and 8.6 months in the nivo arm.
The median progression-free survival was 3.8 months in the ipi + nivo arm and 2.9 months in the nivo arm (hazard ratio, 0.84; P = .19). The 24-month progression-free survival was 8.2% and 5.9%, respectively.
The median overall survival was 10.0 months in the ipi + nivo arm and 11.0 months in the nivo arm (HR, 0.97; P = .82). The 24-month overall survival was 27.6% and 20.1%, respectively.
There were no significant differences in outcomes by TMB or PD-L1 with the cutoffs used in this study, according to Dr. Bazhenova and colleagues, but different cutoffs are being explored.
The median progression-free survival was 4.4 months in TMB-high/PD-L1-low patients in the ipi + nivo arm, compared with 1.7 months in the TMB-high/PD-L1-low patients in the nivo arm. The median overall survival was 15.9 months and 10.3 months, respectively.
“It is slightly challenging to interpret the results without knowing the PD-L1 data of all patients in the cohort, and biomarker selection remains crucial for this combination,” Dr. Arbour said.
Safety
There were no differences in individual toxicities between the treatment arms, but cumulative toxicities were higher in the combination arm, according to the researchers.
The incidence of treatment-related adverse events (AEs) was 88% in the ipi + nivo arm and 90% in the nivo arm. The incidence of grade 3-5 treatment-related AEs was 39% and 31%, respectively.
The incidence of immune-mediated AEs was 65% in the ipi + nivo arm and 57% in the nivo arm. The incidence of immune-mediated grade 3-5 AEs was 20% and 11%, respectively.
There were six AEs leading to death in the ipi + nivo arm – two due to dyspnea, one due to colitis, and one due to respiratory failure. The attribution of one death is under review. For the remaining death, the exact cause is unknown.
There were two AEs leading to death in the nivo arm, both due to pneumonitis.
This study was supported by grants from the National Institutes of Health and by AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, and Pfizer through the Foundation for the National Institutes of Health in partnership with Friends of Cancer Research.
Dr. Bazhenova reported relationships with Epic Sciences, AbbVie, AstraZeneca, Boston Biomedical, Genentech/Roche, Lilly, Loxo, Pfizer, Takeda, and BeyondSpring Pharmaceuticals. Her colleagues reported relationships with these and other companies. Dr. Arbour reported a relationship with AstraZeneca.
SOURCE: Bazhenova L et al. ASCO 2019, Abstract 9014.
CHICAGO – Phase 3 results suggest ipilimumab plus nivolumab is no more effective than nivolumab alone in previously treated patients with metastatic squamous cell lung cancer and no matching biomarker.
However, there is evidence to suggest that patients with a high tumor mutational burden (TMB) and low programmed death–ligand 1 (PD-L1) tumor proportion score (TPS) may derive a benefit from the combination.
Lyudmila Bazhenova, MD, of the University of California, San Diego, and her colleagues presented results from this trial (NCT02785952) in a poster at the annual meeting of the American Society for Clinical Oncology. Kathryn C. Arbour, MD, of Memorial Sloan Kettering Cancer Center in New York reviewed the data in a poster discussion session.
Patients and treatment
The researchers reported on 275 previously treated patients with stage IV or recurrent squamous cell lung cancer who were naive to checkpoint inhibitors. Patients were randomized to receive nivolumab (nivo) at 3 mg/m2 once every 2 weeks (n = 137) or the same dose of nivolumab plus ipilimumab (ipi + nivo) at 1 mg/m2 once every 6 weeks (n = 138).
The patients were stratified by gender and number of prior therapies (one vs. two or more), but they were not stratified by TMB or PD-L1 expression.
The PD-L1 TPS was unknown in 36% of patients, less than 5% in 57%, and 5% or greater in 43% of patients. TMB was unknown in 8% of patients, less than 10 mutations per megabase in 52%, and 10 mutations per megabase or greater in 48%.
Baseline characteristics were similar between the treatment arms. The median age was 67.5 years (range, 42-83 years) in the ipi + nivo arm and 68.1 years (range, 49-90 years) in the nivo arm. Most patients had received only one prior therapy – 85% and 83%, respectively – and most had a performance status of 1 – 71% and 72%, respectively.
Efficacy
There were no significant differences in outcomes between the treatment arms, and the study was closed early for futility.
The overall response rate was 18% in the ipi + nivo arm and 17% in the nivo arm, with one complete response occurring in each arm. The median duration of response was 9.1 months in the ipi + nivo arm and 8.6 months in the nivo arm.
The median progression-free survival was 3.8 months in the ipi + nivo arm and 2.9 months in the nivo arm (hazard ratio, 0.84; P = .19). The 24-month progression-free survival was 8.2% and 5.9%, respectively.
The median overall survival was 10.0 months in the ipi + nivo arm and 11.0 months in the nivo arm (HR, 0.97; P = .82). The 24-month overall survival was 27.6% and 20.1%, respectively.
There were no significant differences in outcomes by TMB or PD-L1 with the cutoffs used in this study, according to Dr. Bazhenova and colleagues, but different cutoffs are being explored.
The median progression-free survival was 4.4 months in TMB-high/PD-L1-low patients in the ipi + nivo arm, compared with 1.7 months in the TMB-high/PD-L1-low patients in the nivo arm. The median overall survival was 15.9 months and 10.3 months, respectively.
“It is slightly challenging to interpret the results without knowing the PD-L1 data of all patients in the cohort, and biomarker selection remains crucial for this combination,” Dr. Arbour said.
Safety
There were no differences in individual toxicities between the treatment arms, but cumulative toxicities were higher in the combination arm, according to the researchers.
The incidence of treatment-related adverse events (AEs) was 88% in the ipi + nivo arm and 90% in the nivo arm. The incidence of grade 3-5 treatment-related AEs was 39% and 31%, respectively.
The incidence of immune-mediated AEs was 65% in the ipi + nivo arm and 57% in the nivo arm. The incidence of immune-mediated grade 3-5 AEs was 20% and 11%, respectively.
There were six AEs leading to death in the ipi + nivo arm – two due to dyspnea, one due to colitis, and one due to respiratory failure. The attribution of one death is under review. For the remaining death, the exact cause is unknown.
There were two AEs leading to death in the nivo arm, both due to pneumonitis.
This study was supported by grants from the National Institutes of Health and by AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, and Pfizer through the Foundation for the National Institutes of Health in partnership with Friends of Cancer Research.
Dr. Bazhenova reported relationships with Epic Sciences, AbbVie, AstraZeneca, Boston Biomedical, Genentech/Roche, Lilly, Loxo, Pfizer, Takeda, and BeyondSpring Pharmaceuticals. Her colleagues reported relationships with these and other companies. Dr. Arbour reported a relationship with AstraZeneca.
SOURCE: Bazhenova L et al. ASCO 2019, Abstract 9014.
REPORTING FROM ASCO 2019
Key clinical point: Ipilimumab plus nivolumab appears no more effective than nivolumab alone in previously treated patients with metastatic squamous cell lung cancer and no matching biomarker.
Major finding: The median progression-free survival was 3.8 months in the ipilimumab plus nivolumab arm and 2.9 months in the nivolumab arm (P = .19). The median overall survival was 10.0 months and 11.0 months, respectively (P = .82).
Study details: A phase 3 trial of 275 previously treated patients with stage IV or recurrent squamous cell lung cancer.
Disclosures: This study was supported by grants from the National Institutes of Health and by AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Genentech, and Pfizer through the Foundation for the National Institutes of Health in partnership with Friends of Cancer Research. The researchers reported relationships with a range of companies. Source: Bazhenova L et al. ASCO 2019, Abstract 9014.
Pregnancy deemed safe in BRCA-mutated breast cancer survivors
CHICAGO – Pregnancy after breast cancer is safe in BRCA-mutated patients, according to a retrospective study.
Pregnancy did not affect disease-free or overall survival in a cohort of BRCA-mutated breast cancer patients. Additionally, fetal and pregnancy complications in this cohort were similar to complications observed in the general population.
“We believe that our findings provide reassurance for counseling young BRCA-mutated breast cancer patients inquiring about the feasibility and safety of future conception,” said Matteo Lambertini, MD, PhD, of Policlinico San Martino Hospital in Genova, Italy.
Dr. Lambertini presented the findings at the annual meeting of the American Society of Clinical Oncology.
He and his colleagues conducted an international, multicenter, retrospective cohort study of 1,252 patients. The patients had been diagnosed with stage I-III breast cancer between January 2000 and December 2012 at age 40 years or younger. All patients had BRCA mutations – 811 with BRCA1 alone, 430 with BRCA2 alone, and 11 with both.
Pregnant versus nonpregnant patients
At a median of 4.5 years after diagnosis, 195 patients (16%) had experienced a pregnancy.
Compared with the nonpregnant women, pregnant patients were younger (P less than .001), more likely to have a BRCA1 mutation (P = .01), have smaller tumors (P = .04), have node-negative disease (P = .003), and have hormone receptor–negative tumors (P = .002). Roughly 95% of patients in both cohorts had received chemotherapy, and the most common regimens were anthracycline or taxane based.
Compared with patients in the nonpregnancy cohort, those in the pregnancy cohort were less likely to receive tamoxifen alone as endocrine therapy (P = .002), were more likely to have a shorter duration of endocrine therapy (P less than .001), and were less likely to undergo salpingo-oophorectomy (P less than .001).
Pregnancy outcomes
“In terms of pregnancy, fetal, and obstetrical outcomes, no alarming signals were observed,” Dr. Lambertini said.
Most pregnant patients had a spontaneous pregnancy (82.1%), completed the pregnancy (76.9%), delivered at term (90.8%), and had no complications (86.6%). However, 10.3% of patients had a spontaneous abortion, 9.2% of pregnancies were pre term, and 1.8% of babies had congenital abnormalities.
“All these rates were highly comparable to rates that are expected in the general healthy population,” Dr. Lambertini said.
Survival analyses
The researchers performed two survival analyses. The first was a case-control approach in which they matched each pregnant patient with three controls (patients without pregnancy) according to the following:
- Disease-free interval from breast cancer diagnosis (equal to or longer than that of pregnant patients).
- Year at diagnosis (plus or minus 2.5 years).
- Nodal status (negative vs. positive).
- Hormone receptor status (positive vs. negative).
- Type of BRCA mutation (BRCA1 vs. BRCA2).
The second survival analysis was an extended Cox model with pregnancy as a time-varying covariate.
Survival outcomes
At a median follow-up of 8.3 years, pregnant patients had better disease-free survival than nonpregnant patients in the case-control analysis, with a hazard ratio of 0.71 (P = .045). With the extended Cox model, the adjusted HR was 0.87 (P = .41). The analysis was adjusted for age, tumor size, nodal status, type of endocrine therapy, hormone receptor status, breast surgery, and BRCA mutation.
There was a significant interaction between type of BRCA mutation and pregnancy, with better disease-free survival observed in the BRCA1-mutated cohort. The HR was 0.53 in the BRCA1 cohort and 1.60 in the BRCA2 cohort (P less than .01). However, as Dr. Lambertini pointed out, only 44 pregnant patients had a BRCA1 mutation.
There was no significant interaction between hormone receptor status and pregnancy (P = .28).
Furthermore, there was no significant difference in overall survival between the pregnant and nonpregnant cohorts. In the case-control analysis, the HR was 0.86 (P = .65). In the extended Cox model, the adjusted HR was 0.88 (P = .66).
Dr. Lambertini disclosed a relationship with Teva.
SOURCE: Lambertini M et al. ASCO 2019, Abstract 11506.
CHICAGO – Pregnancy after breast cancer is safe in BRCA-mutated patients, according to a retrospective study.
Pregnancy did not affect disease-free or overall survival in a cohort of BRCA-mutated breast cancer patients. Additionally, fetal and pregnancy complications in this cohort were similar to complications observed in the general population.
“We believe that our findings provide reassurance for counseling young BRCA-mutated breast cancer patients inquiring about the feasibility and safety of future conception,” said Matteo Lambertini, MD, PhD, of Policlinico San Martino Hospital in Genova, Italy.
Dr. Lambertini presented the findings at the annual meeting of the American Society of Clinical Oncology.
He and his colleagues conducted an international, multicenter, retrospective cohort study of 1,252 patients. The patients had been diagnosed with stage I-III breast cancer between January 2000 and December 2012 at age 40 years or younger. All patients had BRCA mutations – 811 with BRCA1 alone, 430 with BRCA2 alone, and 11 with both.
Pregnant versus nonpregnant patients
At a median of 4.5 years after diagnosis, 195 patients (16%) had experienced a pregnancy.
Compared with the nonpregnant women, pregnant patients were younger (P less than .001), more likely to have a BRCA1 mutation (P = .01), have smaller tumors (P = .04), have node-negative disease (P = .003), and have hormone receptor–negative tumors (P = .002). Roughly 95% of patients in both cohorts had received chemotherapy, and the most common regimens were anthracycline or taxane based.
Compared with patients in the nonpregnancy cohort, those in the pregnancy cohort were less likely to receive tamoxifen alone as endocrine therapy (P = .002), were more likely to have a shorter duration of endocrine therapy (P less than .001), and were less likely to undergo salpingo-oophorectomy (P less than .001).
Pregnancy outcomes
“In terms of pregnancy, fetal, and obstetrical outcomes, no alarming signals were observed,” Dr. Lambertini said.
Most pregnant patients had a spontaneous pregnancy (82.1%), completed the pregnancy (76.9%), delivered at term (90.8%), and had no complications (86.6%). However, 10.3% of patients had a spontaneous abortion, 9.2% of pregnancies were pre term, and 1.8% of babies had congenital abnormalities.
“All these rates were highly comparable to rates that are expected in the general healthy population,” Dr. Lambertini said.
Survival analyses
The researchers performed two survival analyses. The first was a case-control approach in which they matched each pregnant patient with three controls (patients without pregnancy) according to the following:
- Disease-free interval from breast cancer diagnosis (equal to or longer than that of pregnant patients).
- Year at diagnosis (plus or minus 2.5 years).
- Nodal status (negative vs. positive).
- Hormone receptor status (positive vs. negative).
- Type of BRCA mutation (BRCA1 vs. BRCA2).
The second survival analysis was an extended Cox model with pregnancy as a time-varying covariate.
Survival outcomes
At a median follow-up of 8.3 years, pregnant patients had better disease-free survival than nonpregnant patients in the case-control analysis, with a hazard ratio of 0.71 (P = .045). With the extended Cox model, the adjusted HR was 0.87 (P = .41). The analysis was adjusted for age, tumor size, nodal status, type of endocrine therapy, hormone receptor status, breast surgery, and BRCA mutation.
There was a significant interaction between type of BRCA mutation and pregnancy, with better disease-free survival observed in the BRCA1-mutated cohort. The HR was 0.53 in the BRCA1 cohort and 1.60 in the BRCA2 cohort (P less than .01). However, as Dr. Lambertini pointed out, only 44 pregnant patients had a BRCA1 mutation.
There was no significant interaction between hormone receptor status and pregnancy (P = .28).
Furthermore, there was no significant difference in overall survival between the pregnant and nonpregnant cohorts. In the case-control analysis, the HR was 0.86 (P = .65). In the extended Cox model, the adjusted HR was 0.88 (P = .66).
Dr. Lambertini disclosed a relationship with Teva.
SOURCE: Lambertini M et al. ASCO 2019, Abstract 11506.
CHICAGO – Pregnancy after breast cancer is safe in BRCA-mutated patients, according to a retrospective study.
Pregnancy did not affect disease-free or overall survival in a cohort of BRCA-mutated breast cancer patients. Additionally, fetal and pregnancy complications in this cohort were similar to complications observed in the general population.
“We believe that our findings provide reassurance for counseling young BRCA-mutated breast cancer patients inquiring about the feasibility and safety of future conception,” said Matteo Lambertini, MD, PhD, of Policlinico San Martino Hospital in Genova, Italy.
Dr. Lambertini presented the findings at the annual meeting of the American Society of Clinical Oncology.
He and his colleagues conducted an international, multicenter, retrospective cohort study of 1,252 patients. The patients had been diagnosed with stage I-III breast cancer between January 2000 and December 2012 at age 40 years or younger. All patients had BRCA mutations – 811 with BRCA1 alone, 430 with BRCA2 alone, and 11 with both.
Pregnant versus nonpregnant patients
At a median of 4.5 years after diagnosis, 195 patients (16%) had experienced a pregnancy.
Compared with the nonpregnant women, pregnant patients were younger (P less than .001), more likely to have a BRCA1 mutation (P = .01), have smaller tumors (P = .04), have node-negative disease (P = .003), and have hormone receptor–negative tumors (P = .002). Roughly 95% of patients in both cohorts had received chemotherapy, and the most common regimens were anthracycline or taxane based.
Compared with patients in the nonpregnancy cohort, those in the pregnancy cohort were less likely to receive tamoxifen alone as endocrine therapy (P = .002), were more likely to have a shorter duration of endocrine therapy (P less than .001), and were less likely to undergo salpingo-oophorectomy (P less than .001).
Pregnancy outcomes
“In terms of pregnancy, fetal, and obstetrical outcomes, no alarming signals were observed,” Dr. Lambertini said.
Most pregnant patients had a spontaneous pregnancy (82.1%), completed the pregnancy (76.9%), delivered at term (90.8%), and had no complications (86.6%). However, 10.3% of patients had a spontaneous abortion, 9.2% of pregnancies were pre term, and 1.8% of babies had congenital abnormalities.
“All these rates were highly comparable to rates that are expected in the general healthy population,” Dr. Lambertini said.
Survival analyses
The researchers performed two survival analyses. The first was a case-control approach in which they matched each pregnant patient with three controls (patients without pregnancy) according to the following:
- Disease-free interval from breast cancer diagnosis (equal to or longer than that of pregnant patients).
- Year at diagnosis (plus or minus 2.5 years).
- Nodal status (negative vs. positive).
- Hormone receptor status (positive vs. negative).
- Type of BRCA mutation (BRCA1 vs. BRCA2).
The second survival analysis was an extended Cox model with pregnancy as a time-varying covariate.
Survival outcomes
At a median follow-up of 8.3 years, pregnant patients had better disease-free survival than nonpregnant patients in the case-control analysis, with a hazard ratio of 0.71 (P = .045). With the extended Cox model, the adjusted HR was 0.87 (P = .41). The analysis was adjusted for age, tumor size, nodal status, type of endocrine therapy, hormone receptor status, breast surgery, and BRCA mutation.
There was a significant interaction between type of BRCA mutation and pregnancy, with better disease-free survival observed in the BRCA1-mutated cohort. The HR was 0.53 in the BRCA1 cohort and 1.60 in the BRCA2 cohort (P less than .01). However, as Dr. Lambertini pointed out, only 44 pregnant patients had a BRCA1 mutation.
There was no significant interaction between hormone receptor status and pregnancy (P = .28).
Furthermore, there was no significant difference in overall survival between the pregnant and nonpregnant cohorts. In the case-control analysis, the HR was 0.86 (P = .65). In the extended Cox model, the adjusted HR was 0.88 (P = .66).
Dr. Lambertini disclosed a relationship with Teva.
SOURCE: Lambertini M et al. ASCO 2019, Abstract 11506.
REPORTING FROM ASCO 2019
Inhibitor produces high response rate in relapsed/refractory FL
CHICAGO – The phosphoinositide 3-kinase–delta inhibitor ME-401, given with or without rituximab, produced an overall response rate of 80% in a phase 1b trial of patients with relapsed or refractory follicular lymphoma.

Response rates were similar between patients who received ME-401 alone and those who received it in combination with rituximab.
Response rates were also similar between patients on an intermittent dosing schedule and those on a continuous dosing schedule. However, intermittent dosing decreased the rate of delayed grade 3 adverse events (AEs).
“The idea that continuous inhibition of target is absolutely essential for activity of this class of drugs has not been proven,” said Andrew Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York.
“The promising results of this somewhat novel intermittent schedule that we used with ME-401 suggests to me that we can maintain efficacy and reduce toxicity.”
Dr. Zelenetz and colleagues presented these results in a poster at the annual meeting of the American Society of Clinical Oncology.
Patients and dosing
Data were presented for 54 patients with relapsed/refractory follicular lymphoma enrolled on this study. The patients had a median age of 63.5 years, and 80% were male. They had received a median of 2 prior therapies (range, 1-10).
Initially, patients received ME-401 at 60 mg, 120 mg, or 180 mg once daily continuously on a 28-day cycle. However, the dose-escalation portion of the study was closed because response rates were comparable among the three doses, the safety profile was similar, and there were no dose-limiting toxicities.
Two additional groups of patients received ME-401 at 60 mg daily for two cycles, followed by an intermittent schedule (IS) of 60 mg on days 1-7, repeated every 28 days.
The researchers had observed delayed grade 3 AEs on the continuous schedule (CS), and they hypothesized that the IS might prevent these events. Patients could revert to the CS if they had stable disease or progressed on the IS.
“One of the advantages of this particular agent is the very long half-life,” Dr. Zelenetz said. “So, essentially, we have 2 weeks on drug and 2 weeks off [with the IS]. It takes about a week to clear the drug because it has about a 30-hour half-life.”
In all, 40 patients received ME-401 monotherapy, and 14 received ME-401 plus rituximab at 375 mg/m2 weekly for 4 weeks and then on day 1 of cycles 3-6. There were 31 patients who received ME-401 on the CS and 23 who received ME-401 on the IS.
Results
A total of 50 patients were evaluable for efficacy. The overall response rate in these patients was 80% (40/50), and 20% (10/50) achieved a complete response.
The overall response rate was 79% (30/38) in patients who received ME-401 alone, 83% (10/12) in those who received ME-401 plus rituximab, 83% (25/30) in patients on the CS, and 75% (15/20) in those on the IS.
The median duration of response and median progression-free survival have not been reached. The median follow-up for response duration is 8.8 months in the IS group and 8.3 months in the CS group. The median follow-up for progression-free survival is 5.5 months and 6.5 months, respectively.
A total of 18 patients on the IS (78%) were still on therapy at the data cutoff, as were 14 patients (45%) on the CS.
Seven patients (23%) on the CS and two patients (9%) on the IS discontinued treatment due to progression. Four patients in the IS group and two in the CS group who were switched to IS dosing reverted to CS dosing after experiencing progression.
Four CS patients (13%) discontinued treatment because of AEs, but none of the IS patients did.
AEs occurring in at least 15% of patients were diarrhea/colitis (40.7%), fatigue (35.2%), cough (33.3%), rash (24.1%), ALT increase (24.1%), nausea (24.1%), AST increase (22.2%), and decreased appetite (16.7%).
There were no grade 4-5 AEs. Grade 3 drug-related AEs of special interest (in the CS and IS groups, respectively) were diarrhea/colitis (16.1% and 8.7%), rash (12.9% and 0%), ALT increase (6.5% and 4.3%), AST increase (6.5% and 0%), pneumonia (6.5% and 0%), and mucositis (1.9% and 0%).
“[W]hile the grade 3 immune-related events seem to be very consistent in terms of class effects, they did seem to improve with transition to intermittent schedule,” said Carla Casulo, MD, of the University of Rochester (N.Y.), who reviewed this study in a poster discussion session.
“And I think that this novel design helps to create an opportunity to limit treatment and mitigate toxicity without necessarily compromising efficacy.”
The phase 1b trial is sponsored by MEI Pharma. Dr. Zelenetz reported relationships with MEI Pharma and several other companies. Dr. Casulo reported relationships with Gilead Sciences, Celgene, and Roche.
SOURCE: Zelenetz A et al. ASCO 2019, Abstract 7512.
CHICAGO – The phosphoinositide 3-kinase–delta inhibitor ME-401, given with or without rituximab, produced an overall response rate of 80% in a phase 1b trial of patients with relapsed or refractory follicular lymphoma.
Response rates were similar between patients who received ME-401 alone and those who received it in combination with rituximab.
Response rates were also similar between patients on an intermittent dosing schedule and those on a continuous dosing schedule. However, intermittent dosing decreased the rate of delayed grade 3 adverse events (AEs).
“The idea that continuous inhibition of target is absolutely essential for activity of this class of drugs has not been proven,” said Andrew Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York.
“The promising results of this somewhat novel intermittent schedule that we used with ME-401 suggests to me that we can maintain efficacy and reduce toxicity.”
Dr. Zelenetz and colleagues presented these results in a poster at the annual meeting of the American Society of Clinical Oncology.
Patients and dosing
Data were presented for 54 patients with relapsed/refractory follicular lymphoma enrolled on this study. The patients had a median age of 63.5 years, and 80% were male. They had received a median of 2 prior therapies (range, 1-10).
Initially, patients received ME-401 at 60 mg, 120 mg, or 180 mg once daily continuously on a 28-day cycle. However, the dose-escalation portion of the study was closed because response rates were comparable among the three doses, the safety profile was similar, and there were no dose-limiting toxicities.
Two additional groups of patients received ME-401 at 60 mg daily for two cycles, followed by an intermittent schedule (IS) of 60 mg on days 1-7, repeated every 28 days.
The researchers had observed delayed grade 3 AEs on the continuous schedule (CS), and they hypothesized that the IS might prevent these events. Patients could revert to the CS if they had stable disease or progressed on the IS.
“One of the advantages of this particular agent is the very long half-life,” Dr. Zelenetz said. “So, essentially, we have 2 weeks on drug and 2 weeks off [with the IS]. It takes about a week to clear the drug because it has about a 30-hour half-life.”
In all, 40 patients received ME-401 monotherapy, and 14 received ME-401 plus rituximab at 375 mg/m2 weekly for 4 weeks and then on day 1 of cycles 3-6. There were 31 patients who received ME-401 on the CS and 23 who received ME-401 on the IS.
Results
A total of 50 patients were evaluable for efficacy. The overall response rate in these patients was 80% (40/50), and 20% (10/50) achieved a complete response.
The overall response rate was 79% (30/38) in patients who received ME-401 alone, 83% (10/12) in those who received ME-401 plus rituximab, 83% (25/30) in patients on the CS, and 75% (15/20) in those on the IS.
The median duration of response and median progression-free survival have not been reached. The median follow-up for response duration is 8.8 months in the IS group and 8.3 months in the CS group. The median follow-up for progression-free survival is 5.5 months and 6.5 months, respectively.
A total of 18 patients on the IS (78%) were still on therapy at the data cutoff, as were 14 patients (45%) on the CS.
Seven patients (23%) on the CS and two patients (9%) on the IS discontinued treatment due to progression. Four patients in the IS group and two in the CS group who were switched to IS dosing reverted to CS dosing after experiencing progression.
Four CS patients (13%) discontinued treatment because of AEs, but none of the IS patients did.
AEs occurring in at least 15% of patients were diarrhea/colitis (40.7%), fatigue (35.2%), cough (33.3%), rash (24.1%), ALT increase (24.1%), nausea (24.1%), AST increase (22.2%), and decreased appetite (16.7%).
There were no grade 4-5 AEs. Grade 3 drug-related AEs of special interest (in the CS and IS groups, respectively) were diarrhea/colitis (16.1% and 8.7%), rash (12.9% and 0%), ALT increase (6.5% and 4.3%), AST increase (6.5% and 0%), pneumonia (6.5% and 0%), and mucositis (1.9% and 0%).
“[W]hile the grade 3 immune-related events seem to be very consistent in terms of class effects, they did seem to improve with transition to intermittent schedule,” said Carla Casulo, MD, of the University of Rochester (N.Y.), who reviewed this study in a poster discussion session.
“And I think that this novel design helps to create an opportunity to limit treatment and mitigate toxicity without necessarily compromising efficacy.”
The phase 1b trial is sponsored by MEI Pharma. Dr. Zelenetz reported relationships with MEI Pharma and several other companies. Dr. Casulo reported relationships with Gilead Sciences, Celgene, and Roche.
SOURCE: Zelenetz A et al. ASCO 2019, Abstract 7512.
CHICAGO – The phosphoinositide 3-kinase–delta inhibitor ME-401, given with or without rituximab, produced an overall response rate of 80% in a phase 1b trial of patients with relapsed or refractory follicular lymphoma.
Response rates were similar between patients who received ME-401 alone and those who received it in combination with rituximab.
Response rates were also similar between patients on an intermittent dosing schedule and those on a continuous dosing schedule. However, intermittent dosing decreased the rate of delayed grade 3 adverse events (AEs).
“The idea that continuous inhibition of target is absolutely essential for activity of this class of drugs has not been proven,” said Andrew Zelenetz, MD, PhD, of Memorial Sloan Kettering Cancer Center in New York.
“The promising results of this somewhat novel intermittent schedule that we used with ME-401 suggests to me that we can maintain efficacy and reduce toxicity.”
Dr. Zelenetz and colleagues presented these results in a poster at the annual meeting of the American Society of Clinical Oncology.
Patients and dosing
Data were presented for 54 patients with relapsed/refractory follicular lymphoma enrolled on this study. The patients had a median age of 63.5 years, and 80% were male. They had received a median of 2 prior therapies (range, 1-10).
Initially, patients received ME-401 at 60 mg, 120 mg, or 180 mg once daily continuously on a 28-day cycle. However, the dose-escalation portion of the study was closed because response rates were comparable among the three doses, the safety profile was similar, and there were no dose-limiting toxicities.
Two additional groups of patients received ME-401 at 60 mg daily for two cycles, followed by an intermittent schedule (IS) of 60 mg on days 1-7, repeated every 28 days.
The researchers had observed delayed grade 3 AEs on the continuous schedule (CS), and they hypothesized that the IS might prevent these events. Patients could revert to the CS if they had stable disease or progressed on the IS.
“One of the advantages of this particular agent is the very long half-life,” Dr. Zelenetz said. “So, essentially, we have 2 weeks on drug and 2 weeks off [with the IS]. It takes about a week to clear the drug because it has about a 30-hour half-life.”
In all, 40 patients received ME-401 monotherapy, and 14 received ME-401 plus rituximab at 375 mg/m2 weekly for 4 weeks and then on day 1 of cycles 3-6. There were 31 patients who received ME-401 on the CS and 23 who received ME-401 on the IS.
Results
A total of 50 patients were evaluable for efficacy. The overall response rate in these patients was 80% (40/50), and 20% (10/50) achieved a complete response.
The overall response rate was 79% (30/38) in patients who received ME-401 alone, 83% (10/12) in those who received ME-401 plus rituximab, 83% (25/30) in patients on the CS, and 75% (15/20) in those on the IS.
The median duration of response and median progression-free survival have not been reached. The median follow-up for response duration is 8.8 months in the IS group and 8.3 months in the CS group. The median follow-up for progression-free survival is 5.5 months and 6.5 months, respectively.
A total of 18 patients on the IS (78%) were still on therapy at the data cutoff, as were 14 patients (45%) on the CS.
Seven patients (23%) on the CS and two patients (9%) on the IS discontinued treatment due to progression. Four patients in the IS group and two in the CS group who were switched to IS dosing reverted to CS dosing after experiencing progression.
Four CS patients (13%) discontinued treatment because of AEs, but none of the IS patients did.
AEs occurring in at least 15% of patients were diarrhea/colitis (40.7%), fatigue (35.2%), cough (33.3%), rash (24.1%), ALT increase (24.1%), nausea (24.1%), AST increase (22.2%), and decreased appetite (16.7%).
There were no grade 4-5 AEs. Grade 3 drug-related AEs of special interest (in the CS and IS groups, respectively) were diarrhea/colitis (16.1% and 8.7%), rash (12.9% and 0%), ALT increase (6.5% and 4.3%), AST increase (6.5% and 0%), pneumonia (6.5% and 0%), and mucositis (1.9% and 0%).
“[W]hile the grade 3 immune-related events seem to be very consistent in terms of class effects, they did seem to improve with transition to intermittent schedule,” said Carla Casulo, MD, of the University of Rochester (N.Y.), who reviewed this study in a poster discussion session.
“And I think that this novel design helps to create an opportunity to limit treatment and mitigate toxicity without necessarily compromising efficacy.”
The phase 1b trial is sponsored by MEI Pharma. Dr. Zelenetz reported relationships with MEI Pharma and several other companies. Dr. Casulo reported relationships with Gilead Sciences, Celgene, and Roche.
SOURCE: Zelenetz A et al. ASCO 2019, Abstract 7512.
REPORTING FROM ASCO 2019
Olanzapine improves upon standard antiemetic therapy
CHICAGO – Olanzapine plus aprepitant, palonosetron, and dexamethasone (APD) can be considered a new standard antiemetic therapy for patients receiving cisplatin-based chemotherapy, according to a speaker at the annual meeting of the American Society for Clinical Oncology.
A 5 mg dose of olanzapine plus APD produced significantly higher complete response (CR) rates than APD plus placebo in a phase 3 trial, said Hironobu Hashimoto, BPharm, of the National Cancer Center Hospital in Tokyo.
In addition, olanzapine plus APD had a significantly longer time to treatment failure and produced higher rates of complete control and total control. However, rates of somnolence, dizziness, and dry mouth were significantly higher with olanzapine plus APD.
In the phase 3 J-FORCE study, Mr. Hashimoto and his colleagues evaluated olanzapine at 5 mg plus standard antiemetic therapy (APD) for the prevention of chemotherapy-induced nausea and vomiting.
The trial enrolled 710 patients, ages 22-75 years, who had malignant solid tumors and were receiving cisplatin-based chemotherapy for the first time. The patients were randomized to olanzapine plus APD or APD plus placebo.
There were 354 patients evaluable for efficacy in the olanzapine arm and 351 in the placebo arm. The primary endpoint was CR, which was defined as no vomiting and no rescue medications, in the delayed phase (24 hours to 120 hours).
The CR rate in the delayed phase was 79% in the olanzapine arm and 66% in the placebo arm (P less than .001). The CR rate in the acute phase (0 to 24 hours) was 95% and 89%, respectively (P equal to .002), and the overall CR rate was 78% and 64%, respectively (P less than .001).
The time to treatment failure was significantly longer in the olanzapine arm (P less than .001).
The rate of complete control in the overall period (0 to 120 hours) was 76% in the olanzapine arm and 61% in the placebo arm (P less than .001). Complete control was defined as no emesis, no rescue medication, and no nausea or low-grade nausea.
The rate of total control in the overall period was 59% in the olanzapine arm and 48% in the placebo arm (P = .005). Total control was defined as no emesis, no rescue medication, and no nausea.
Treatment-related adverse events (in the olanzapine and placebo arms, respectively) included constipation (15% and 11%), hiccups (10% and 6%), somnolence (43% and 33%), insomnia (5% and 7%), dizziness (8% and 3%), and dry mouth (21% and 9%).
Adverse events that occurred significantly more often with olanzapine were somnolence (P = .011), dizziness (P = .004), and dry mouth (P less than .001).
This study was sponsored by the Japan Agency for Medical Research and Development (AMED). Mr. Hashimoto said he had nothing to disclose. Other investigators involved in this study disclosed relationships with numerous pharmaceutical companies.
SOURCE: Hashimoto H et al. ASCO 2019. Abstract 11503.
CHICAGO – Olanzapine plus aprepitant, palonosetron, and dexamethasone (APD) can be considered a new standard antiemetic therapy for patients receiving cisplatin-based chemotherapy, according to a speaker at the annual meeting of the American Society for Clinical Oncology.
A 5 mg dose of olanzapine plus APD produced significantly higher complete response (CR) rates than APD plus placebo in a phase 3 trial, said Hironobu Hashimoto, BPharm, of the National Cancer Center Hospital in Tokyo.
In addition, olanzapine plus APD had a significantly longer time to treatment failure and produced higher rates of complete control and total control. However, rates of somnolence, dizziness, and dry mouth were significantly higher with olanzapine plus APD.
In the phase 3 J-FORCE study, Mr. Hashimoto and his colleagues evaluated olanzapine at 5 mg plus standard antiemetic therapy (APD) for the prevention of chemotherapy-induced nausea and vomiting.
The trial enrolled 710 patients, ages 22-75 years, who had malignant solid tumors and were receiving cisplatin-based chemotherapy for the first time. The patients were randomized to olanzapine plus APD or APD plus placebo.
There were 354 patients evaluable for efficacy in the olanzapine arm and 351 in the placebo arm. The primary endpoint was CR, which was defined as no vomiting and no rescue medications, in the delayed phase (24 hours to 120 hours).
The CR rate in the delayed phase was 79% in the olanzapine arm and 66% in the placebo arm (P less than .001). The CR rate in the acute phase (0 to 24 hours) was 95% and 89%, respectively (P equal to .002), and the overall CR rate was 78% and 64%, respectively (P less than .001).
The time to treatment failure was significantly longer in the olanzapine arm (P less than .001).
The rate of complete control in the overall period (0 to 120 hours) was 76% in the olanzapine arm and 61% in the placebo arm (P less than .001). Complete control was defined as no emesis, no rescue medication, and no nausea or low-grade nausea.
The rate of total control in the overall period was 59% in the olanzapine arm and 48% in the placebo arm (P = .005). Total control was defined as no emesis, no rescue medication, and no nausea.
Treatment-related adverse events (in the olanzapine and placebo arms, respectively) included constipation (15% and 11%), hiccups (10% and 6%), somnolence (43% and 33%), insomnia (5% and 7%), dizziness (8% and 3%), and dry mouth (21% and 9%).
Adverse events that occurred significantly more often with olanzapine were somnolence (P = .011), dizziness (P = .004), and dry mouth (P less than .001).
This study was sponsored by the Japan Agency for Medical Research and Development (AMED). Mr. Hashimoto said he had nothing to disclose. Other investigators involved in this study disclosed relationships with numerous pharmaceutical companies.
SOURCE: Hashimoto H et al. ASCO 2019. Abstract 11503.
CHICAGO – Olanzapine plus aprepitant, palonosetron, and dexamethasone (APD) can be considered a new standard antiemetic therapy for patients receiving cisplatin-based chemotherapy, according to a speaker at the annual meeting of the American Society for Clinical Oncology.
A 5 mg dose of olanzapine plus APD produced significantly higher complete response (CR) rates than APD plus placebo in a phase 3 trial, said Hironobu Hashimoto, BPharm, of the National Cancer Center Hospital in Tokyo.
In addition, olanzapine plus APD had a significantly longer time to treatment failure and produced higher rates of complete control and total control. However, rates of somnolence, dizziness, and dry mouth were significantly higher with olanzapine plus APD.
In the phase 3 J-FORCE study, Mr. Hashimoto and his colleagues evaluated olanzapine at 5 mg plus standard antiemetic therapy (APD) for the prevention of chemotherapy-induced nausea and vomiting.
The trial enrolled 710 patients, ages 22-75 years, who had malignant solid tumors and were receiving cisplatin-based chemotherapy for the first time. The patients were randomized to olanzapine plus APD or APD plus placebo.
There were 354 patients evaluable for efficacy in the olanzapine arm and 351 in the placebo arm. The primary endpoint was CR, which was defined as no vomiting and no rescue medications, in the delayed phase (24 hours to 120 hours).
The CR rate in the delayed phase was 79% in the olanzapine arm and 66% in the placebo arm (P less than .001). The CR rate in the acute phase (0 to 24 hours) was 95% and 89%, respectively (P equal to .002), and the overall CR rate was 78% and 64%, respectively (P less than .001).
The time to treatment failure was significantly longer in the olanzapine arm (P less than .001).
The rate of complete control in the overall period (0 to 120 hours) was 76% in the olanzapine arm and 61% in the placebo arm (P less than .001). Complete control was defined as no emesis, no rescue medication, and no nausea or low-grade nausea.
The rate of total control in the overall period was 59% in the olanzapine arm and 48% in the placebo arm (P = .005). Total control was defined as no emesis, no rescue medication, and no nausea.
Treatment-related adverse events (in the olanzapine and placebo arms, respectively) included constipation (15% and 11%), hiccups (10% and 6%), somnolence (43% and 33%), insomnia (5% and 7%), dizziness (8% and 3%), and dry mouth (21% and 9%).
Adverse events that occurred significantly more often with olanzapine were somnolence (P = .011), dizziness (P = .004), and dry mouth (P less than .001).
This study was sponsored by the Japan Agency for Medical Research and Development (AMED). Mr. Hashimoto said he had nothing to disclose. Other investigators involved in this study disclosed relationships with numerous pharmaceutical companies.
SOURCE: Hashimoto H et al. ASCO 2019. Abstract 11503.
REPORTING FROM ASCO 2019
Pembrolizumab improves 5-year OS in advanced NSCLC
CHICAGO – New data suggest pembrolizumab can increase 5-year overall survival (OS) for patients with advanced non–small cell lung cancer (NSCLC).

In the phase 1b KEYNOTE-001 trial, the 5-year OS rate was 23.2% in treatment-naive patients and 15.5% in previously treated patients. This is in comparison to the 5.5% average 5-year OS rate observed in NSCLC patients who receive standard chemotherapy (Noone AM et al. SEER Cancer Statistics Review, 1975-2015).
“In total, the data confirm that pembrolizumab has the potential to improve long-term outcomes for both treatment-naive and previously treated patients with advanced non–small cell lung cancer,” said Edward B. Garon, MD, of the University of California, Los Angeles.
Dr. Garon and colleagues presented these results in a poster at the annual meeting of the American Society for Clinical Oncology, and the data were simultaneously published in the Journal of Clinical Oncology.
KEYNOTE-001 (NCT01295827) enrolled 550 patients with advanced NSCLC who had received no prior therapy (n = 101) or at least one prior line of therapy (n = 449). Initially, patients received pembrolizumab at varying doses depending on body weight, but the protocol was changed to a single dose of pembrolizumab at 200 mg every 3 weeks.
At a median follow-up of 60.6 months, 100 patients were still alive. Sixty patients had received at least 2 years of pembrolizumab, 14 of whom were treatment-naive at baseline, and 46 of whom were previously treated at baseline.
Five-year OS rates were best among patients who had high PD-L1 expression, which was defined as 50% or greater.
Among treatment-naive patients, the 5-year OS rate was 29.6% in PD-L1–high patients and 15.7% in PD-L1–low patients (expression of 1% to 49%). The median OS was 35.4 months and 19.5 months, respectively.
Among previously treated patients, the 5-year OS rate was 25.0% in PD-L1–high patients, 12.6% in patients with PD-L1 expression of 1%-49%, and 3.5% in patients with PD-L1 expression less than 1%. The median OS was 15.4 months, 8.5 months, and 8.6 months, respectively.
Among patients who received at least 2 years of pembrolizumab, the 5-year OS rate was 78.6% in the treatment-naive group and 75.8% in the previously treated group. The objective response rate was 86% and 91%, respectively. The rate of ongoing response at the data cutoff was 58% and 71%, respectively.
“The safety data did not show any unanticipated late toxicity, which I consider encouraging,” Dr. Garon noted.
He said rates of immune-mediated adverse events were similar at 3 years and 5 years of follow-up. At 5 years, 17% of patients (n = 92) had experienced an immune-related adverse event, the most common of which were hypothyroidism (9%), pneumonitis (5%), and hyperthyroidism (2%).
Dr. Garon disclosed relationships with AstraZeneca, Bristol-Myers Squibb, Dracen, Dynavax, Genentech, Iovance Biotherapeutics, Lilly, Merck, Mirati Therapeutics, Neon Therapeutics, and Novartis. KEYNOTE-001 was sponsored by Merck Sharp & Dohme Corp.
SOURCES: Garon E. et al. ASCO 2019, Abstract LBA9015; J Clin Oncol. 2019 June 2. doi: 10.1200/JCO.19.00934
CHICAGO – New data suggest pembrolizumab can increase 5-year overall survival (OS) for patients with advanced non–small cell lung cancer (NSCLC).
In the phase 1b KEYNOTE-001 trial, the 5-year OS rate was 23.2% in treatment-naive patients and 15.5% in previously treated patients. This is in comparison to the 5.5% average 5-year OS rate observed in NSCLC patients who receive standard chemotherapy (Noone AM et al. SEER Cancer Statistics Review, 1975-2015).
“In total, the data confirm that pembrolizumab has the potential to improve long-term outcomes for both treatment-naive and previously treated patients with advanced non–small cell lung cancer,” said Edward B. Garon, MD, of the University of California, Los Angeles.
Dr. Garon and colleagues presented these results in a poster at the annual meeting of the American Society for Clinical Oncology, and the data were simultaneously published in the Journal of Clinical Oncology.
KEYNOTE-001 (NCT01295827) enrolled 550 patients with advanced NSCLC who had received no prior therapy (n = 101) or at least one prior line of therapy (n = 449). Initially, patients received pembrolizumab at varying doses depending on body weight, but the protocol was changed to a single dose of pembrolizumab at 200 mg every 3 weeks.
At a median follow-up of 60.6 months, 100 patients were still alive. Sixty patients had received at least 2 years of pembrolizumab, 14 of whom were treatment-naive at baseline, and 46 of whom were previously treated at baseline.
Five-year OS rates were best among patients who had high PD-L1 expression, which was defined as 50% or greater.
Among treatment-naive patients, the 5-year OS rate was 29.6% in PD-L1–high patients and 15.7% in PD-L1–low patients (expression of 1% to 49%). The median OS was 35.4 months and 19.5 months, respectively.
Among previously treated patients, the 5-year OS rate was 25.0% in PD-L1–high patients, 12.6% in patients with PD-L1 expression of 1%-49%, and 3.5% in patients with PD-L1 expression less than 1%. The median OS was 15.4 months, 8.5 months, and 8.6 months, respectively.
Among patients who received at least 2 years of pembrolizumab, the 5-year OS rate was 78.6% in the treatment-naive group and 75.8% in the previously treated group. The objective response rate was 86% and 91%, respectively. The rate of ongoing response at the data cutoff was 58% and 71%, respectively.
“The safety data did not show any unanticipated late toxicity, which I consider encouraging,” Dr. Garon noted.
He said rates of immune-mediated adverse events were similar at 3 years and 5 years of follow-up. At 5 years, 17% of patients (n = 92) had experienced an immune-related adverse event, the most common of which were hypothyroidism (9%), pneumonitis (5%), and hyperthyroidism (2%).
Dr. Garon disclosed relationships with AstraZeneca, Bristol-Myers Squibb, Dracen, Dynavax, Genentech, Iovance Biotherapeutics, Lilly, Merck, Mirati Therapeutics, Neon Therapeutics, and Novartis. KEYNOTE-001 was sponsored by Merck Sharp & Dohme Corp.
SOURCES: Garon E. et al. ASCO 2019, Abstract LBA9015; J Clin Oncol. 2019 June 2. doi: 10.1200/JCO.19.00934
CHICAGO – New data suggest pembrolizumab can increase 5-year overall survival (OS) for patients with advanced non–small cell lung cancer (NSCLC).
In the phase 1b KEYNOTE-001 trial, the 5-year OS rate was 23.2% in treatment-naive patients and 15.5% in previously treated patients. This is in comparison to the 5.5% average 5-year OS rate observed in NSCLC patients who receive standard chemotherapy (Noone AM et al. SEER Cancer Statistics Review, 1975-2015).
“In total, the data confirm that pembrolizumab has the potential to improve long-term outcomes for both treatment-naive and previously treated patients with advanced non–small cell lung cancer,” said Edward B. Garon, MD, of the University of California, Los Angeles.
Dr. Garon and colleagues presented these results in a poster at the annual meeting of the American Society for Clinical Oncology, and the data were simultaneously published in the Journal of Clinical Oncology.
KEYNOTE-001 (NCT01295827) enrolled 550 patients with advanced NSCLC who had received no prior therapy (n = 101) or at least one prior line of therapy (n = 449). Initially, patients received pembrolizumab at varying doses depending on body weight, but the protocol was changed to a single dose of pembrolizumab at 200 mg every 3 weeks.
At a median follow-up of 60.6 months, 100 patients were still alive. Sixty patients had received at least 2 years of pembrolizumab, 14 of whom were treatment-naive at baseline, and 46 of whom were previously treated at baseline.
Five-year OS rates were best among patients who had high PD-L1 expression, which was defined as 50% or greater.
Among treatment-naive patients, the 5-year OS rate was 29.6% in PD-L1–high patients and 15.7% in PD-L1–low patients (expression of 1% to 49%). The median OS was 35.4 months and 19.5 months, respectively.
Among previously treated patients, the 5-year OS rate was 25.0% in PD-L1–high patients, 12.6% in patients with PD-L1 expression of 1%-49%, and 3.5% in patients with PD-L1 expression less than 1%. The median OS was 15.4 months, 8.5 months, and 8.6 months, respectively.
Among patients who received at least 2 years of pembrolizumab, the 5-year OS rate was 78.6% in the treatment-naive group and 75.8% in the previously treated group. The objective response rate was 86% and 91%, respectively. The rate of ongoing response at the data cutoff was 58% and 71%, respectively.
“The safety data did not show any unanticipated late toxicity, which I consider encouraging,” Dr. Garon noted.
He said rates of immune-mediated adverse events were similar at 3 years and 5 years of follow-up. At 5 years, 17% of patients (n = 92) had experienced an immune-related adverse event, the most common of which were hypothyroidism (9%), pneumonitis (5%), and hyperthyroidism (2%).
Dr. Garon disclosed relationships with AstraZeneca, Bristol-Myers Squibb, Dracen, Dynavax, Genentech, Iovance Biotherapeutics, Lilly, Merck, Mirati Therapeutics, Neon Therapeutics, and Novartis. KEYNOTE-001 was sponsored by Merck Sharp & Dohme Corp.
SOURCES: Garon E. et al. ASCO 2019, Abstract LBA9015; J Clin Oncol. 2019 June 2. doi: 10.1200/JCO.19.00934
REPORTING FROM ASCO 2019
SC-PEG comparable to pegaspargase in young ALL/LL patients
CHICAGO – Calaspargase pegol (SC-PEG) produces similar outcomes as standard pegaspargase in pediatric and young adult patients with newly diagnosed acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LL), according to a phase 2 trial.

Patients who received SC-PEG every 3 weeks had similar serum asparaginase activity (SAA), toxicities, and survival rates as patients who received standard pegaspargase every 2 weeks.
Lynda M. Vrooman, MD, of Dana-Farber Cancer Institute in Boston, presented these results at the annual meeting of the American Society of Clinical Oncology.
The trial (NCT01574274) enrolled 239 patients, 230 with ALL and 9 with LL. Most patients had B-cell (n = 207) disease. The patients’ median age was 5.2 years (range, 1.0-20.9 years).
“There were no differences in presenting features by randomization,” Dr. Vrooman noted.
The patients were randomized to receive pegaspargase (n = 120) or SC-PEG (n = 119), a pegylated asparaginase formulation with longer half-life. SC-PEG was given at 2,500 IU/m2 every 3 weeks, and pegaspargase was given at 2,500 IU/m2 every 2 weeks.
Either asparaginase product was given as part of a 4-week induction regimen (vincristine, prednisone, doxorubicin, and methotrexate), a 3-week intensification regimen (intrathecal chemotherapy with or without radiotherapy) for central nervous system disease, and a 27-week second consolidation regimen (mercaptopurine, methotrexate, and, in high-risk patients, doxorubicin).
SAA
The researchers observed significantly longer SAA with SC-PEG during induction but not after.
During induction, at 25 days after the first asparaginase dose, 88% of patients on SC-PEG and 17% of those on pegaspargase had SAA of at least 0.10 IU/mL (P less than .001). Post-induction, at week 25, 100% of patients in each group had a nadir SAA of at least 0.10 IU/mL.
“The high nadir serum asparaginase activity levels observed for both preparations suggest dosing strategies could be further optimized,” Dr. Vrooman noted.
Safety
There were no significant differences in adverse events between the SC-PEG and pegaspargase arms during or after induction.
Adverse events during induction (in the SC-PEG and pegaspargase arms, respectively) included grade 2 or higher asparaginase allergy (0% and 1%), grade 2 or higher pancreatitis (3% in both), grade 2 or higher thrombosis (3% and 9%), grade 4 hyperbilirubinemia (3% and 1%), grade 3 or higher bacterial infection (12% and 9%), and grade 3 or higher fungal infection (4% and 5%).
Adverse events after induction (in the SC-PEG and pegaspargase arms, respectively) included grade 2 or higher asparaginase allergy (17% and 14%), grade 2 or higher pancreatitis (15% in both), grade 2 or higher thrombosis (18% and 13%), grade 4 hyperbilirubinemia (4% and 3%), grade 3 or higher bacterial infection (12% and 15%), grade 3 or higher fungal infection (2% and 1%), grade 2 or higher bone fracture (3% and 8%), and grade 2 or higher osteonecrosis (3% and 4%).
Response and survival
The complete response rate was 95% (109/115) in the SC-PEG arm and 99% (114/115) in the pegaspargase arm. Rates of induction failure were 3% (n = 4) and 1% (n = 1), respectively, and rates of relapse were 3% (n = 5) and 8% (n = 10), respectively.
There were two induction deaths and two remission deaths in the SC-PEG arm but no induction or remission deaths in the pegaspargase arm.
The median follow-up was 4 years. The 4-year event-free survival rate was 87.7% with SC-PEG and 90.2% with pegaspargase (P = .78). The 4-year overall survival rate was 94.8% and 95.6%, respectively (P = .74).
In closing, Dr. Vrooman said these data suggest SC-PEG provides similar results as standard pegaspargase. She noted that these data informed the U.S. approval of SC-PEG for pediatric and young adult ALL.
This trial was sponsored by the Dana-Farber Cancer Institute in collaboration with Shire and the National Cancer Institute. Dr. Vrooman said she had no relationships to disclose.
SOURCE: Vrooman LM et al. ASCO 2019. Abstract 10006.
CHICAGO – Calaspargase pegol (SC-PEG) produces similar outcomes as standard pegaspargase in pediatric and young adult patients with newly diagnosed acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LL), according to a phase 2 trial.
Patients who received SC-PEG every 3 weeks had similar serum asparaginase activity (SAA), toxicities, and survival rates as patients who received standard pegaspargase every 2 weeks.
Lynda M. Vrooman, MD, of Dana-Farber Cancer Institute in Boston, presented these results at the annual meeting of the American Society of Clinical Oncology.
The trial (NCT01574274) enrolled 239 patients, 230 with ALL and 9 with LL. Most patients had B-cell (n = 207) disease. The patients’ median age was 5.2 years (range, 1.0-20.9 years).
“There were no differences in presenting features by randomization,” Dr. Vrooman noted.
The patients were randomized to receive pegaspargase (n = 120) or SC-PEG (n = 119), a pegylated asparaginase formulation with longer half-life. SC-PEG was given at 2,500 IU/m2 every 3 weeks, and pegaspargase was given at 2,500 IU/m2 every 2 weeks.
Either asparaginase product was given as part of a 4-week induction regimen (vincristine, prednisone, doxorubicin, and methotrexate), a 3-week intensification regimen (intrathecal chemotherapy with or without radiotherapy) for central nervous system disease, and a 27-week second consolidation regimen (mercaptopurine, methotrexate, and, in high-risk patients, doxorubicin).
SAA
The researchers observed significantly longer SAA with SC-PEG during induction but not after.
During induction, at 25 days after the first asparaginase dose, 88% of patients on SC-PEG and 17% of those on pegaspargase had SAA of at least 0.10 IU/mL (P less than .001). Post-induction, at week 25, 100% of patients in each group had a nadir SAA of at least 0.10 IU/mL.
“The high nadir serum asparaginase activity levels observed for both preparations suggest dosing strategies could be further optimized,” Dr. Vrooman noted.
Safety
There were no significant differences in adverse events between the SC-PEG and pegaspargase arms during or after induction.
Adverse events during induction (in the SC-PEG and pegaspargase arms, respectively) included grade 2 or higher asparaginase allergy (0% and 1%), grade 2 or higher pancreatitis (3% in both), grade 2 or higher thrombosis (3% and 9%), grade 4 hyperbilirubinemia (3% and 1%), grade 3 or higher bacterial infection (12% and 9%), and grade 3 or higher fungal infection (4% and 5%).
Adverse events after induction (in the SC-PEG and pegaspargase arms, respectively) included grade 2 or higher asparaginase allergy (17% and 14%), grade 2 or higher pancreatitis (15% in both), grade 2 or higher thrombosis (18% and 13%), grade 4 hyperbilirubinemia (4% and 3%), grade 3 or higher bacterial infection (12% and 15%), grade 3 or higher fungal infection (2% and 1%), grade 2 or higher bone fracture (3% and 8%), and grade 2 or higher osteonecrosis (3% and 4%).
Response and survival
The complete response rate was 95% (109/115) in the SC-PEG arm and 99% (114/115) in the pegaspargase arm. Rates of induction failure were 3% (n = 4) and 1% (n = 1), respectively, and rates of relapse were 3% (n = 5) and 8% (n = 10), respectively.
There were two induction deaths and two remission deaths in the SC-PEG arm but no induction or remission deaths in the pegaspargase arm.
The median follow-up was 4 years. The 4-year event-free survival rate was 87.7% with SC-PEG and 90.2% with pegaspargase (P = .78). The 4-year overall survival rate was 94.8% and 95.6%, respectively (P = .74).
In closing, Dr. Vrooman said these data suggest SC-PEG provides similar results as standard pegaspargase. She noted that these data informed the U.S. approval of SC-PEG for pediatric and young adult ALL.
This trial was sponsored by the Dana-Farber Cancer Institute in collaboration with Shire and the National Cancer Institute. Dr. Vrooman said she had no relationships to disclose.
SOURCE: Vrooman LM et al. ASCO 2019. Abstract 10006.
CHICAGO – Calaspargase pegol (SC-PEG) produces similar outcomes as standard pegaspargase in pediatric and young adult patients with newly diagnosed acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LL), according to a phase 2 trial.
Patients who received SC-PEG every 3 weeks had similar serum asparaginase activity (SAA), toxicities, and survival rates as patients who received standard pegaspargase every 2 weeks.
Lynda M. Vrooman, MD, of Dana-Farber Cancer Institute in Boston, presented these results at the annual meeting of the American Society of Clinical Oncology.
The trial (NCT01574274) enrolled 239 patients, 230 with ALL and 9 with LL. Most patients had B-cell (n = 207) disease. The patients’ median age was 5.2 years (range, 1.0-20.9 years).
“There were no differences in presenting features by randomization,” Dr. Vrooman noted.
The patients were randomized to receive pegaspargase (n = 120) or SC-PEG (n = 119), a pegylated asparaginase formulation with longer half-life. SC-PEG was given at 2,500 IU/m2 every 3 weeks, and pegaspargase was given at 2,500 IU/m2 every 2 weeks.
Either asparaginase product was given as part of a 4-week induction regimen (vincristine, prednisone, doxorubicin, and methotrexate), a 3-week intensification regimen (intrathecal chemotherapy with or without radiotherapy) for central nervous system disease, and a 27-week second consolidation regimen (mercaptopurine, methotrexate, and, in high-risk patients, doxorubicin).
SAA
The researchers observed significantly longer SAA with SC-PEG during induction but not after.
During induction, at 25 days after the first asparaginase dose, 88% of patients on SC-PEG and 17% of those on pegaspargase had SAA of at least 0.10 IU/mL (P less than .001). Post-induction, at week 25, 100% of patients in each group had a nadir SAA of at least 0.10 IU/mL.
“The high nadir serum asparaginase activity levels observed for both preparations suggest dosing strategies could be further optimized,” Dr. Vrooman noted.
Safety
There were no significant differences in adverse events between the SC-PEG and pegaspargase arms during or after induction.
Adverse events during induction (in the SC-PEG and pegaspargase arms, respectively) included grade 2 or higher asparaginase allergy (0% and 1%), grade 2 or higher pancreatitis (3% in both), grade 2 or higher thrombosis (3% and 9%), grade 4 hyperbilirubinemia (3% and 1%), grade 3 or higher bacterial infection (12% and 9%), and grade 3 or higher fungal infection (4% and 5%).
Adverse events after induction (in the SC-PEG and pegaspargase arms, respectively) included grade 2 or higher asparaginase allergy (17% and 14%), grade 2 or higher pancreatitis (15% in both), grade 2 or higher thrombosis (18% and 13%), grade 4 hyperbilirubinemia (4% and 3%), grade 3 or higher bacterial infection (12% and 15%), grade 3 or higher fungal infection (2% and 1%), grade 2 or higher bone fracture (3% and 8%), and grade 2 or higher osteonecrosis (3% and 4%).
Response and survival
The complete response rate was 95% (109/115) in the SC-PEG arm and 99% (114/115) in the pegaspargase arm. Rates of induction failure were 3% (n = 4) and 1% (n = 1), respectively, and rates of relapse were 3% (n = 5) and 8% (n = 10), respectively.
There were two induction deaths and two remission deaths in the SC-PEG arm but no induction or remission deaths in the pegaspargase arm.
The median follow-up was 4 years. The 4-year event-free survival rate was 87.7% with SC-PEG and 90.2% with pegaspargase (P = .78). The 4-year overall survival rate was 94.8% and 95.6%, respectively (P = .74).
In closing, Dr. Vrooman said these data suggest SC-PEG provides similar results as standard pegaspargase. She noted that these data informed the U.S. approval of SC-PEG for pediatric and young adult ALL.
This trial was sponsored by the Dana-Farber Cancer Institute in collaboration with Shire and the National Cancer Institute. Dr. Vrooman said she had no relationships to disclose.
SOURCE: Vrooman LM et al. ASCO 2019. Abstract 10006.
REPORTING FROM ASCO 2019