User login
Diet and Cancer: Here's What I Tell Patients
Diet and Cancer: Here's What I Tell Patients
One of the most common questions my patients ask is, “What diet can help me beat this cancer?” It is a profoundly important question that is worthy of our efforts to answer. In this brief essay, I will take a deep dive into this question in depth and explore the broader clinical and scientific themes it brings into play.
Low-Hanging Fruit: Nutrition Science
A cancer diagnosis can be a deeply disempowering experience. Although I have not lived with cancer myself, I have seen this play out repeatedly over the past 5 years in my role as an oncologist treating patients with hematologic malignancies.
Our diet is an important part of our personal identity, culturally and spiritually. If lifestyle changes, such as a modified diet or more exercise, can contribute to cancer treatment, it may help us regain a sense of control over our lives, one that cancer so often cruelly strips away. I hypothesize that, among other factors, this is why diet is so important to our patients.
Another factor is exposure to a compelling diet-cancer narrative. Nearly every day, a media headline appears claiming that eating a particular food, or drinking coffee, can either increase or decrease your risk for a certain disease.
These claims, however, are often based on studies of large observational datasets where individuals fill out surveys about their dietary habits and are subsequently assessed for disease outcomes. In these studies, people aren’t asked to eat a particular diet; instead, their dietary habits are analyzed by researchers who have endless permutations to explore. This, in a nutshell, is the field of nutritional epidemiology.
In my opinion, nutritional epidemiology represents the collision of the well-intentioned effort to answer clinically meaningful questions with the ease — and near-infinite permutations — of dietary questions that can be asked from an increasingly larger number of different datasets.
Now, factor in the never-ending appetite (pun intended) of journalism and the public’s desire for dietary studies, and you create the perfect storm of incentives that drives a flood of low-quality nutritional science. These studies are highly malleable to analytical choices and can essentially produce results consistent with your prior beliefs, regardless of the philosophical inclination you have (pro keto-diet, pro-vegan, etc.). I love quoting this study to my trainees that, depending on what variables are included and how the analysis is conducted, the same dataset could be used to show that red meat either increases, decreases, or has no effect on all-cause mortality. Unfortunately, much of the evidence base for diet in cancer comes from similarly confounded, low-quality studies.
Diet and Cancer
So, what do randomized trials show for diet and cancer?
The highest-quality evidence is generated from randomized controlled trials. One of their key advantages is the ability to control both measured and unmeasured confounders.
Unfortunately, the evidence supporting diet as an anticancer modality in randomized trials in patients with cancer is bleak. We did a systematic review of all randomized trials of dietary intervention ever done in patients with cancer. Most of the trials measured outcomes such as feasibility (often small pilot studies that measure variables such as weight changes or lab values). The trials that measure clinical endpoints, such as survival, were largely negative and demonstrated no meaningful effect of diet on outcomes. Take trials exploring whether a Mediterranean diet helps prevent breast cancer recurrence, or whether a diet rich in fruits and vegetables improves prostate cancer outcomes. Although these diets may offer benefits, these studies found that specific diets did not change the natural history of cancer.
Myeloma and Diet
In my specialty, multiple myeloma, I am thankful that some trials are beginning to shed light on whether diet influences cancer outcomes.
One study, which was recently published in Cancer Discovery, explored whether a high-fiber, plant-based diet could potentially slow or delay progression from myeloma precursor conditions toward full-blown multiple myeloma. The trial enrolled 23 participants, with the primary endpoints of dietary adherence and changes to BMI. Measures of progression to multiple myeloma were exploratory at best. Yet, the media coverage, as well as the majority of the discussion and results sections of this study manuscript, claimed that the diet changes can prevent progression to myeloma.
However, the study design and conclusions were flawed. The paper focused on two patients who had some improvement in disease trajectory, while descriptions of patients who had an increase in their bone marrow plasma cell percentage were relegated to the supplemental section.
As a primary investigator of a trial in smoldering myeloma where we use advanced imaging as an alternative to pharmacologic treatment, I frequently see myeloma markers fluctuate and often decrease. I attribute these changes to random variation, or possibly regression to the mean, rather than the effect of any intervention.
Future randomized studies by this group used primary endpoints of stool butyrate level and implement dietary interventions for a limited period— 2 weeks in one study and 12 weeks in another — to again assess the impact of a high-fiber, plant-based diet on progression to myeloma. Although there are no data yet, the limited timeframes in these studies severely limits generalizability for outcomes that would truly matter, such as cancer control and longevity. There is also no evidence that changes in stool butyrate levels influence patient outcomes.
High-quality science — whether it is evaluating diet or other interventions—requires high-quality data, effort, funding, and time. It is not impossible.
We can draw inspiration from the CHALLENGE trial. This large, randomized trial, which took over a decade to complete, assessed the benefit of a structured exercise program in the adjuvant setting for colon cancer. The endpoint of this study was disease-free survival, and the intervention was deployed over a much longer period: 3 years, as opposed to a 2-week intervention. This trial took years from inception to completion, but it yielded a conclusive result and will probably lead to more dedicated efforts to facilitate exercise programs for patients with cancer.
Our patients deserve the same effort as the CHALLENGE trial to answer their important dietary questions. Until such trials are completed, we must acknowledge, with humility, that despite the common sense and feel-good factor that many diets offer us, their impact on cancer remains uncertain.
Conversely, we must recognize that even if diet does not cure or alter the course of a certain cancer, it can still impact quality of life, treatment tolerance, and other supportive care outcomes, making it an important factor in patient care.
This is what I tell my patients that it is unlikely any one diet will change the trajectory of your cancer. Focus on eating healthy, and remember that most things in moderation are fine. Your diet remains an important risk factor and determinant for health outcomes beyond cancer. Eat what makes you happy. You are going through a tough time, and this is not the moment to impose stringent restrictions on yourself.
A version of this article first appeared on Medscape.com.
One of the most common questions my patients ask is, “What diet can help me beat this cancer?” It is a profoundly important question that is worthy of our efforts to answer. In this brief essay, I will take a deep dive into this question in depth and explore the broader clinical and scientific themes it brings into play.
Low-Hanging Fruit: Nutrition Science
A cancer diagnosis can be a deeply disempowering experience. Although I have not lived with cancer myself, I have seen this play out repeatedly over the past 5 years in my role as an oncologist treating patients with hematologic malignancies.
Our diet is an important part of our personal identity, culturally and spiritually. If lifestyle changes, such as a modified diet or more exercise, can contribute to cancer treatment, it may help us regain a sense of control over our lives, one that cancer so often cruelly strips away. I hypothesize that, among other factors, this is why diet is so important to our patients.
Another factor is exposure to a compelling diet-cancer narrative. Nearly every day, a media headline appears claiming that eating a particular food, or drinking coffee, can either increase or decrease your risk for a certain disease.
These claims, however, are often based on studies of large observational datasets where individuals fill out surveys about their dietary habits and are subsequently assessed for disease outcomes. In these studies, people aren’t asked to eat a particular diet; instead, their dietary habits are analyzed by researchers who have endless permutations to explore. This, in a nutshell, is the field of nutritional epidemiology.
In my opinion, nutritional epidemiology represents the collision of the well-intentioned effort to answer clinically meaningful questions with the ease — and near-infinite permutations — of dietary questions that can be asked from an increasingly larger number of different datasets.
Now, factor in the never-ending appetite (pun intended) of journalism and the public’s desire for dietary studies, and you create the perfect storm of incentives that drives a flood of low-quality nutritional science. These studies are highly malleable to analytical choices and can essentially produce results consistent with your prior beliefs, regardless of the philosophical inclination you have (pro keto-diet, pro-vegan, etc.). I love quoting this study to my trainees that, depending on what variables are included and how the analysis is conducted, the same dataset could be used to show that red meat either increases, decreases, or has no effect on all-cause mortality. Unfortunately, much of the evidence base for diet in cancer comes from similarly confounded, low-quality studies.
Diet and Cancer
So, what do randomized trials show for diet and cancer?
The highest-quality evidence is generated from randomized controlled trials. One of their key advantages is the ability to control both measured and unmeasured confounders.
Unfortunately, the evidence supporting diet as an anticancer modality in randomized trials in patients with cancer is bleak. We did a systematic review of all randomized trials of dietary intervention ever done in patients with cancer. Most of the trials measured outcomes such as feasibility (often small pilot studies that measure variables such as weight changes or lab values). The trials that measure clinical endpoints, such as survival, were largely negative and demonstrated no meaningful effect of diet on outcomes. Take trials exploring whether a Mediterranean diet helps prevent breast cancer recurrence, or whether a diet rich in fruits and vegetables improves prostate cancer outcomes. Although these diets may offer benefits, these studies found that specific diets did not change the natural history of cancer.
Myeloma and Diet
In my specialty, multiple myeloma, I am thankful that some trials are beginning to shed light on whether diet influences cancer outcomes.
One study, which was recently published in Cancer Discovery, explored whether a high-fiber, plant-based diet could potentially slow or delay progression from myeloma precursor conditions toward full-blown multiple myeloma. The trial enrolled 23 participants, with the primary endpoints of dietary adherence and changes to BMI. Measures of progression to multiple myeloma were exploratory at best. Yet, the media coverage, as well as the majority of the discussion and results sections of this study manuscript, claimed that the diet changes can prevent progression to myeloma.
However, the study design and conclusions were flawed. The paper focused on two patients who had some improvement in disease trajectory, while descriptions of patients who had an increase in their bone marrow plasma cell percentage were relegated to the supplemental section.
As a primary investigator of a trial in smoldering myeloma where we use advanced imaging as an alternative to pharmacologic treatment, I frequently see myeloma markers fluctuate and often decrease. I attribute these changes to random variation, or possibly regression to the mean, rather than the effect of any intervention.
Future randomized studies by this group used primary endpoints of stool butyrate level and implement dietary interventions for a limited period— 2 weeks in one study and 12 weeks in another — to again assess the impact of a high-fiber, plant-based diet on progression to myeloma. Although there are no data yet, the limited timeframes in these studies severely limits generalizability for outcomes that would truly matter, such as cancer control and longevity. There is also no evidence that changes in stool butyrate levels influence patient outcomes.
High-quality science — whether it is evaluating diet or other interventions—requires high-quality data, effort, funding, and time. It is not impossible.
We can draw inspiration from the CHALLENGE trial. This large, randomized trial, which took over a decade to complete, assessed the benefit of a structured exercise program in the adjuvant setting for colon cancer. The endpoint of this study was disease-free survival, and the intervention was deployed over a much longer period: 3 years, as opposed to a 2-week intervention. This trial took years from inception to completion, but it yielded a conclusive result and will probably lead to more dedicated efforts to facilitate exercise programs for patients with cancer.
Our patients deserve the same effort as the CHALLENGE trial to answer their important dietary questions. Until such trials are completed, we must acknowledge, with humility, that despite the common sense and feel-good factor that many diets offer us, their impact on cancer remains uncertain.
Conversely, we must recognize that even if diet does not cure or alter the course of a certain cancer, it can still impact quality of life, treatment tolerance, and other supportive care outcomes, making it an important factor in patient care.
This is what I tell my patients that it is unlikely any one diet will change the trajectory of your cancer. Focus on eating healthy, and remember that most things in moderation are fine. Your diet remains an important risk factor and determinant for health outcomes beyond cancer. Eat what makes you happy. You are going through a tough time, and this is not the moment to impose stringent restrictions on yourself.
A version of this article first appeared on Medscape.com.
One of the most common questions my patients ask is, “What diet can help me beat this cancer?” It is a profoundly important question that is worthy of our efforts to answer. In this brief essay, I will take a deep dive into this question in depth and explore the broader clinical and scientific themes it brings into play.
Low-Hanging Fruit: Nutrition Science
A cancer diagnosis can be a deeply disempowering experience. Although I have not lived with cancer myself, I have seen this play out repeatedly over the past 5 years in my role as an oncologist treating patients with hematologic malignancies.
Our diet is an important part of our personal identity, culturally and spiritually. If lifestyle changes, such as a modified diet or more exercise, can contribute to cancer treatment, it may help us regain a sense of control over our lives, one that cancer so often cruelly strips away. I hypothesize that, among other factors, this is why diet is so important to our patients.
Another factor is exposure to a compelling diet-cancer narrative. Nearly every day, a media headline appears claiming that eating a particular food, or drinking coffee, can either increase or decrease your risk for a certain disease.
These claims, however, are often based on studies of large observational datasets where individuals fill out surveys about their dietary habits and are subsequently assessed for disease outcomes. In these studies, people aren’t asked to eat a particular diet; instead, their dietary habits are analyzed by researchers who have endless permutations to explore. This, in a nutshell, is the field of nutritional epidemiology.
In my opinion, nutritional epidemiology represents the collision of the well-intentioned effort to answer clinically meaningful questions with the ease — and near-infinite permutations — of dietary questions that can be asked from an increasingly larger number of different datasets.
Now, factor in the never-ending appetite (pun intended) of journalism and the public’s desire for dietary studies, and you create the perfect storm of incentives that drives a flood of low-quality nutritional science. These studies are highly malleable to analytical choices and can essentially produce results consistent with your prior beliefs, regardless of the philosophical inclination you have (pro keto-diet, pro-vegan, etc.). I love quoting this study to my trainees that, depending on what variables are included and how the analysis is conducted, the same dataset could be used to show that red meat either increases, decreases, or has no effect on all-cause mortality. Unfortunately, much of the evidence base for diet in cancer comes from similarly confounded, low-quality studies.
Diet and Cancer
So, what do randomized trials show for diet and cancer?
The highest-quality evidence is generated from randomized controlled trials. One of their key advantages is the ability to control both measured and unmeasured confounders.
Unfortunately, the evidence supporting diet as an anticancer modality in randomized trials in patients with cancer is bleak. We did a systematic review of all randomized trials of dietary intervention ever done in patients with cancer. Most of the trials measured outcomes such as feasibility (often small pilot studies that measure variables such as weight changes or lab values). The trials that measure clinical endpoints, such as survival, were largely negative and demonstrated no meaningful effect of diet on outcomes. Take trials exploring whether a Mediterranean diet helps prevent breast cancer recurrence, or whether a diet rich in fruits and vegetables improves prostate cancer outcomes. Although these diets may offer benefits, these studies found that specific diets did not change the natural history of cancer.
Myeloma and Diet
In my specialty, multiple myeloma, I am thankful that some trials are beginning to shed light on whether diet influences cancer outcomes.
One study, which was recently published in Cancer Discovery, explored whether a high-fiber, plant-based diet could potentially slow or delay progression from myeloma precursor conditions toward full-blown multiple myeloma. The trial enrolled 23 participants, with the primary endpoints of dietary adherence and changes to BMI. Measures of progression to multiple myeloma were exploratory at best. Yet, the media coverage, as well as the majority of the discussion and results sections of this study manuscript, claimed that the diet changes can prevent progression to myeloma.
However, the study design and conclusions were flawed. The paper focused on two patients who had some improvement in disease trajectory, while descriptions of patients who had an increase in their bone marrow plasma cell percentage were relegated to the supplemental section.
As a primary investigator of a trial in smoldering myeloma where we use advanced imaging as an alternative to pharmacologic treatment, I frequently see myeloma markers fluctuate and often decrease. I attribute these changes to random variation, or possibly regression to the mean, rather than the effect of any intervention.
Future randomized studies by this group used primary endpoints of stool butyrate level and implement dietary interventions for a limited period— 2 weeks in one study and 12 weeks in another — to again assess the impact of a high-fiber, plant-based diet on progression to myeloma. Although there are no data yet, the limited timeframes in these studies severely limits generalizability for outcomes that would truly matter, such as cancer control and longevity. There is also no evidence that changes in stool butyrate levels influence patient outcomes.
High-quality science — whether it is evaluating diet or other interventions—requires high-quality data, effort, funding, and time. It is not impossible.
We can draw inspiration from the CHALLENGE trial. This large, randomized trial, which took over a decade to complete, assessed the benefit of a structured exercise program in the adjuvant setting for colon cancer. The endpoint of this study was disease-free survival, and the intervention was deployed over a much longer period: 3 years, as opposed to a 2-week intervention. This trial took years from inception to completion, but it yielded a conclusive result and will probably lead to more dedicated efforts to facilitate exercise programs for patients with cancer.
Our patients deserve the same effort as the CHALLENGE trial to answer their important dietary questions. Until such trials are completed, we must acknowledge, with humility, that despite the common sense and feel-good factor that many diets offer us, their impact on cancer remains uncertain.
Conversely, we must recognize that even if diet does not cure or alter the course of a certain cancer, it can still impact quality of life, treatment tolerance, and other supportive care outcomes, making it an important factor in patient care.
This is what I tell my patients that it is unlikely any one diet will change the trajectory of your cancer. Focus on eating healthy, and remember that most things in moderation are fine. Your diet remains an important risk factor and determinant for health outcomes beyond cancer. Eat what makes you happy. You are going through a tough time, and this is not the moment to impose stringent restrictions on yourself.
A version of this article first appeared on Medscape.com.
Diet and Cancer: Here's What I Tell Patients
Diet and Cancer: Here's What I Tell Patients
ASH 2024 Myeloma Studies: My Top 10 Picks
First, let me place my selected studies in context by acknowledging my biases. As a clinician, I’m prone to choosing clinical rather than basic science or translational work — even if translational work might well end up exerting a pivotal impact on practice in the future. And now — in no particular order — here are my picks:
IFM 2017-03 Phase 3 Study
Frail patients are underrepresented in most myeloma studies, yet in this randomized trial for newly diagnosed myeloma, exclusively frail patients were enrolled. The trial compared daratumumab/lenalidomide to lenalidomide/dexamethasone, and the most recent follow/up shows a progression-free survival (PFS) (48.5 vs 21 months) and overall survival (OS) (not reached vs 36 months) benefit to daratumumab/lenalidomide. What I see in practice is that anti-CD38 monoclonal antibodies are the best-tolerated drugs in this space and should be the backbone of any regimen for frail patients. Steroids should be omitted as early as possible. Future trials may optimize what to give in addition to the anti-CD38 therapy, and how to adapt/escalate therapy to frailty and clinical status, as lenalidomide remains difficult for such patients to tolerate.
AQUILA Study
This is a randomized, phase 3 comparison of daratumumab to observation for patients with smoldering myeloma. The endpoint was PFS. For context, similar studies done in asymptomatic CLL have shown improved PFS, but not OS, and the authors of such studies have concluded that improvement in PFS alone should not justify a change in approach.

This study shows that daratumumab can improve laboratory markers and reduce progression (60-month PFS rates of 63.1% for daratumumab vs 40.8% with observation). However, several important caveats remain. The protocol only mandated spine/pelvis MRI imaging, not whole-body MRI imaging, and such imaging was only performed once a year, which may not be frequent enough to catch lesions at an earlier stage. These details have important implications, as previous research shows that up to half of lesions can be missed by doing only a spine MRI, as opposed to a whole-body MRI.
All of this means that had more comprehensive imaging been done, many more patients may have been diagnosed with myeloma. Such patients may have been undertreated, and single agent daratumumab, with a response rate of just 63%, may not have been enough. Conversely, some patients may also have been overtreated using this approach, as the protocol allowed patients who had been diagnosed with smoldering myeloma up to 5 years earlier to be enrolled. Many of these people could have had indolent disease for years prior to enrollment and may not have ever progressed.
Further information is needed to help us understand this study better. What was the nature of the progression events: asymptomatic lab changes or morbid end organ damage? Was daratumumab given when patients in the control arm progressed to myeloma? My concern is that if patients in the control arm do not universally receive modern daratumumab-containing therapies when they develop myeloma, then an overall survival advantage may be shown simply because patients in the intervention arm are getting a good drug earlier in the disease, while those in the control arm are not getting a good drug at all. Nevertheless, despite these limitations, it is likely this trial will lead to regulatory approval of daratumumab in this space, and lots of discussions from patients and clinicians alike.
Extended Follow-Up of Anito-Cel in its First In-Human study
Two chimeric antigen receptor therapy (CAR-T) products are currently approved for myeloma. Cilta-cel is clearly effective but is associated with problematic late-onset neurological toxicities. Ide-cel appears much less effective. There is clearly a need for a product that is both effective and safe.
Anito-cel has two relevant abstracts this meeting that show much promise. Extended follow-up of anito-cel from its first in human study shows a promising 27-month PFS of 52%, and with no cases of delayed neurotoxicity. I also eagerly await further information from the registrational single-arm study of anti-cel being presented at ASH 2024, which should (hopefully) lead to its accelerated approval.
Screening for Myeloma for all People With Vertebral Fractures Likely Unnecessary
This elegant study of over 9,000 people with vertebral fractures shows that absolute risks for myeloma were 0.43% and 0.63% in women and men with grade 2-3 fractures, respectively, indicating that there is likely little benefit in evaluating asymptomatic individuals with incidentally discovered vertebral fractures for myeloma, unless other features are present. Spread the word.
Further Information on Cevostamab, Another Bispecific Option
We do need effective treatments for targets beyond just BCMA and GPRC5D. Cevostamab, a bispecific targeting FCRH5, represents another option, with updated data on 167 patients. With an overall response rate of 43% (duration of response, 10 months), and a response rate of about 30% in those with prior bispecific exposure, this data helps us contextualize expected benefits as we look forward to the eventual approval of this drug. The efficacy is relatively modest in those who have already progressed on bispecifics, but cevostamab would still be a welcome addition to our arsenal.
Is GPRC5D a Better Target for Car T Rather Than Bispecifics?
Our currently available GPRC5D bispecific (talquetamab) leads to high rates of skin, oral, and nail toxicity. This drug can also bring significant weight loss. These side effects make me consider that continuous targeting of GPRC5D through a bispecific may not be ideal, and that GPRC5D may be better as a one-time CAR T target. At ASH 2024, we will have 15-month follow-up data from BMS-986393, a GPRC5D CAR T. Response rates for this heavily pretreated population (76% of whom had triple refractory disease) were at 87%, with a median PFS of 14.5 months. Only 6% of patients experienced treatment-related weight loss, and nail (19%), skin (30%), and oral (31%) toxicities were relatively low. I look forward to updated data, as well as data on the resolution of the toxicities seen.
Daratumumab, a Game-Changer for AL Amyloidosis
A truly effective drug given early can change the natural history of disease, even if patients in the control arm only receive the drug later. A case in point is daratumumab. The 5-year survival rate was 76.1% for the daratumumab/cyclophosphamide/bortezomib/dexamethasone arm and 64.7% for cyclophosphamide/bortezomib/dexamethasone arm. This happened despite the fact that 67% of the control arm patients (among those who received therapy) went on to receive daratumumab later in the disease course.
Understanding how SMM Progresses to MM
We often hear that we should treat SMM and not just watch carefully because fractures may suddenly happen, or a patient may end up on dialysis. What this retrospective study tells us that amongst 427 patients with SMM, 42 had progression to myeloma, and amongst those 42, only 1 developed renal dysfunction (unclear if this resolved), and only 1 had lytic lesions that were symptomatic. The remainder were all asymptomatic lab and imaging changes. This is a retrospective study, and one should assume that follow-up was thus highly variable. It does not appear that diffusion weighted whole-body MRI imaging (our most sensitive imaging test) was employed universally or very frequently. Nevertheless, these powerful findings reassure us that, with close observation, morbidity is unlikely. Our group has designed a prospective study incorporating frequent diffusion weighted whole body MRI imaging to formally test this hypothesis (SPOTLIGHT, NCT06212323).
The Underperformance of Daratumumab/Lenalidomide/Dexamethasone in the Real World
At every major meeting I am reminded of the disconnect between real-world efficacy and clinical trial efficacy. Case in point: In this Austrian experience, daratumumab/lenalidomide/dexamethasone led to a PFS of 22.7 months vs 61.9 months in the MAIA trial of daratumumab/lenalidomide/dexamethasone. Such a sobering difference! And if you think this is an isolated experience, even in a US real-world cohort, consider that in a recently published comparative study dara/len/dex underperformed, although the time to next treatment or death was longer (37.8 months).
Delayed Neurotoxicity may not be Just a Consequence of high tumor burden
We currently think that some of the scariest side effects of cilta-cel, such as delayed neurotoxicity, may be a consequence of a high number of cancer cells and may be prevented by better disease control at the time of infusion. This study, a sobering analysis of 52 patients with delayed neurotoxicity occurring after CAR T, included 8 patients (15%) who were not heavily pretreated, and all had less than 5% plasma cells at the time of infusion. None of these patients had extramedullary disease. This result worries me, especially because cilta-cel is being studied and is poised for future approval in earlier line settings. It suggests that this toxicity may not always be a product of disease burden, contrary to our current belief.
I will be paying close attention to these 10 myeloma studies at ASH 2024, where I look forward to meeting you and learning more.Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah, Salt Lake City.
First, let me place my selected studies in context by acknowledging my biases. As a clinician, I’m prone to choosing clinical rather than basic science or translational work — even if translational work might well end up exerting a pivotal impact on practice in the future. And now — in no particular order — here are my picks:
IFM 2017-03 Phase 3 Study
Frail patients are underrepresented in most myeloma studies, yet in this randomized trial for newly diagnosed myeloma, exclusively frail patients were enrolled. The trial compared daratumumab/lenalidomide to lenalidomide/dexamethasone, and the most recent follow/up shows a progression-free survival (PFS) (48.5 vs 21 months) and overall survival (OS) (not reached vs 36 months) benefit to daratumumab/lenalidomide. What I see in practice is that anti-CD38 monoclonal antibodies are the best-tolerated drugs in this space and should be the backbone of any regimen for frail patients. Steroids should be omitted as early as possible. Future trials may optimize what to give in addition to the anti-CD38 therapy, and how to adapt/escalate therapy to frailty and clinical status, as lenalidomide remains difficult for such patients to tolerate.
AQUILA Study
This is a randomized, phase 3 comparison of daratumumab to observation for patients with smoldering myeloma. The endpoint was PFS. For context, similar studies done in asymptomatic CLL have shown improved PFS, but not OS, and the authors of such studies have concluded that improvement in PFS alone should not justify a change in approach.
This study shows that daratumumab can improve laboratory markers and reduce progression (60-month PFS rates of 63.1% for daratumumab vs 40.8% with observation). However, several important caveats remain. The protocol only mandated spine/pelvis MRI imaging, not whole-body MRI imaging, and such imaging was only performed once a year, which may not be frequent enough to catch lesions at an earlier stage. These details have important implications, as previous research shows that up to half of lesions can be missed by doing only a spine MRI, as opposed to a whole-body MRI.
All of this means that had more comprehensive imaging been done, many more patients may have been diagnosed with myeloma. Such patients may have been undertreated, and single agent daratumumab, with a response rate of just 63%, may not have been enough. Conversely, some patients may also have been overtreated using this approach, as the protocol allowed patients who had been diagnosed with smoldering myeloma up to 5 years earlier to be enrolled. Many of these people could have had indolent disease for years prior to enrollment and may not have ever progressed.
Further information is needed to help us understand this study better. What was the nature of the progression events: asymptomatic lab changes or morbid end organ damage? Was daratumumab given when patients in the control arm progressed to myeloma? My concern is that if patients in the control arm do not universally receive modern daratumumab-containing therapies when they develop myeloma, then an overall survival advantage may be shown simply because patients in the intervention arm are getting a good drug earlier in the disease, while those in the control arm are not getting a good drug at all. Nevertheless, despite these limitations, it is likely this trial will lead to regulatory approval of daratumumab in this space, and lots of discussions from patients and clinicians alike.
Extended Follow-Up of Anito-Cel in its First In-Human study
Two chimeric antigen receptor therapy (CAR-T) products are currently approved for myeloma. Cilta-cel is clearly effective but is associated with problematic late-onset neurological toxicities. Ide-cel appears much less effective. There is clearly a need for a product that is both effective and safe.
Anito-cel has two relevant abstracts this meeting that show much promise. Extended follow-up of anito-cel from its first in human study shows a promising 27-month PFS of 52%, and with no cases of delayed neurotoxicity. I also eagerly await further information from the registrational single-arm study of anti-cel being presented at ASH 2024, which should (hopefully) lead to its accelerated approval.
Screening for Myeloma for all People With Vertebral Fractures Likely Unnecessary
This elegant study of over 9,000 people with vertebral fractures shows that absolute risks for myeloma were 0.43% and 0.63% in women and men with grade 2-3 fractures, respectively, indicating that there is likely little benefit in evaluating asymptomatic individuals with incidentally discovered vertebral fractures for myeloma, unless other features are present. Spread the word.
Further Information on Cevostamab, Another Bispecific Option
We do need effective treatments for targets beyond just BCMA and GPRC5D. Cevostamab, a bispecific targeting FCRH5, represents another option, with updated data on 167 patients. With an overall response rate of 43% (duration of response, 10 months), and a response rate of about 30% in those with prior bispecific exposure, this data helps us contextualize expected benefits as we look forward to the eventual approval of this drug. The efficacy is relatively modest in those who have already progressed on bispecifics, but cevostamab would still be a welcome addition to our arsenal.
Is GPRC5D a Better Target for Car T Rather Than Bispecifics?
Our currently available GPRC5D bispecific (talquetamab) leads to high rates of skin, oral, and nail toxicity. This drug can also bring significant weight loss. These side effects make me consider that continuous targeting of GPRC5D through a bispecific may not be ideal, and that GPRC5D may be better as a one-time CAR T target. At ASH 2024, we will have 15-month follow-up data from BMS-986393, a GPRC5D CAR T. Response rates for this heavily pretreated population (76% of whom had triple refractory disease) were at 87%, with a median PFS of 14.5 months. Only 6% of patients experienced treatment-related weight loss, and nail (19%), skin (30%), and oral (31%) toxicities were relatively low. I look forward to updated data, as well as data on the resolution of the toxicities seen.
Daratumumab, a Game-Changer for AL Amyloidosis
A truly effective drug given early can change the natural history of disease, even if patients in the control arm only receive the drug later. A case in point is daratumumab. The 5-year survival rate was 76.1% for the daratumumab/cyclophosphamide/bortezomib/dexamethasone arm and 64.7% for cyclophosphamide/bortezomib/dexamethasone arm. This happened despite the fact that 67% of the control arm patients (among those who received therapy) went on to receive daratumumab later in the disease course.
Understanding how SMM Progresses to MM
We often hear that we should treat SMM and not just watch carefully because fractures may suddenly happen, or a patient may end up on dialysis. What this retrospective study tells us that amongst 427 patients with SMM, 42 had progression to myeloma, and amongst those 42, only 1 developed renal dysfunction (unclear if this resolved), and only 1 had lytic lesions that were symptomatic. The remainder were all asymptomatic lab and imaging changes. This is a retrospective study, and one should assume that follow-up was thus highly variable. It does not appear that diffusion weighted whole-body MRI imaging (our most sensitive imaging test) was employed universally or very frequently. Nevertheless, these powerful findings reassure us that, with close observation, morbidity is unlikely. Our group has designed a prospective study incorporating frequent diffusion weighted whole body MRI imaging to formally test this hypothesis (SPOTLIGHT, NCT06212323).
The Underperformance of Daratumumab/Lenalidomide/Dexamethasone in the Real World
At every major meeting I am reminded of the disconnect between real-world efficacy and clinical trial efficacy. Case in point: In this Austrian experience, daratumumab/lenalidomide/dexamethasone led to a PFS of 22.7 months vs 61.9 months in the MAIA trial of daratumumab/lenalidomide/dexamethasone. Such a sobering difference! And if you think this is an isolated experience, even in a US real-world cohort, consider that in a recently published comparative study dara/len/dex underperformed, although the time to next treatment or death was longer (37.8 months).
Delayed Neurotoxicity may not be Just a Consequence of high tumor burden
We currently think that some of the scariest side effects of cilta-cel, such as delayed neurotoxicity, may be a consequence of a high number of cancer cells and may be prevented by better disease control at the time of infusion. This study, a sobering analysis of 52 patients with delayed neurotoxicity occurring after CAR T, included 8 patients (15%) who were not heavily pretreated, and all had less than 5% plasma cells at the time of infusion. None of these patients had extramedullary disease. This result worries me, especially because cilta-cel is being studied and is poised for future approval in earlier line settings. It suggests that this toxicity may not always be a product of disease burden, contrary to our current belief.
I will be paying close attention to these 10 myeloma studies at ASH 2024, where I look forward to meeting you and learning more.Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah, Salt Lake City.
First, let me place my selected studies in context by acknowledging my biases. As a clinician, I’m prone to choosing clinical rather than basic science or translational work — even if translational work might well end up exerting a pivotal impact on practice in the future. And now — in no particular order — here are my picks:
IFM 2017-03 Phase 3 Study
Frail patients are underrepresented in most myeloma studies, yet in this randomized trial for newly diagnosed myeloma, exclusively frail patients were enrolled. The trial compared daratumumab/lenalidomide to lenalidomide/dexamethasone, and the most recent follow/up shows a progression-free survival (PFS) (48.5 vs 21 months) and overall survival (OS) (not reached vs 36 months) benefit to daratumumab/lenalidomide. What I see in practice is that anti-CD38 monoclonal antibodies are the best-tolerated drugs in this space and should be the backbone of any regimen for frail patients. Steroids should be omitted as early as possible. Future trials may optimize what to give in addition to the anti-CD38 therapy, and how to adapt/escalate therapy to frailty and clinical status, as lenalidomide remains difficult for such patients to tolerate.
AQUILA Study
This is a randomized, phase 3 comparison of daratumumab to observation for patients with smoldering myeloma. The endpoint was PFS. For context, similar studies done in asymptomatic CLL have shown improved PFS, but not OS, and the authors of such studies have concluded that improvement in PFS alone should not justify a change in approach.
This study shows that daratumumab can improve laboratory markers and reduce progression (60-month PFS rates of 63.1% for daratumumab vs 40.8% with observation). However, several important caveats remain. The protocol only mandated spine/pelvis MRI imaging, not whole-body MRI imaging, and such imaging was only performed once a year, which may not be frequent enough to catch lesions at an earlier stage. These details have important implications, as previous research shows that up to half of lesions can be missed by doing only a spine MRI, as opposed to a whole-body MRI.
All of this means that had more comprehensive imaging been done, many more patients may have been diagnosed with myeloma. Such patients may have been undertreated, and single agent daratumumab, with a response rate of just 63%, may not have been enough. Conversely, some patients may also have been overtreated using this approach, as the protocol allowed patients who had been diagnosed with smoldering myeloma up to 5 years earlier to be enrolled. Many of these people could have had indolent disease for years prior to enrollment and may not have ever progressed.
Further information is needed to help us understand this study better. What was the nature of the progression events: asymptomatic lab changes or morbid end organ damage? Was daratumumab given when patients in the control arm progressed to myeloma? My concern is that if patients in the control arm do not universally receive modern daratumumab-containing therapies when they develop myeloma, then an overall survival advantage may be shown simply because patients in the intervention arm are getting a good drug earlier in the disease, while those in the control arm are not getting a good drug at all. Nevertheless, despite these limitations, it is likely this trial will lead to regulatory approval of daratumumab in this space, and lots of discussions from patients and clinicians alike.
Extended Follow-Up of Anito-Cel in its First In-Human study
Two chimeric antigen receptor therapy (CAR-T) products are currently approved for myeloma. Cilta-cel is clearly effective but is associated with problematic late-onset neurological toxicities. Ide-cel appears much less effective. There is clearly a need for a product that is both effective and safe.
Anito-cel has two relevant abstracts this meeting that show much promise. Extended follow-up of anito-cel from its first in human study shows a promising 27-month PFS of 52%, and with no cases of delayed neurotoxicity. I also eagerly await further information from the registrational single-arm study of anti-cel being presented at ASH 2024, which should (hopefully) lead to its accelerated approval.
Screening for Myeloma for all People With Vertebral Fractures Likely Unnecessary
This elegant study of over 9,000 people with vertebral fractures shows that absolute risks for myeloma were 0.43% and 0.63% in women and men with grade 2-3 fractures, respectively, indicating that there is likely little benefit in evaluating asymptomatic individuals with incidentally discovered vertebral fractures for myeloma, unless other features are present. Spread the word.
Further Information on Cevostamab, Another Bispecific Option
We do need effective treatments for targets beyond just BCMA and GPRC5D. Cevostamab, a bispecific targeting FCRH5, represents another option, with updated data on 167 patients. With an overall response rate of 43% (duration of response, 10 months), and a response rate of about 30% in those with prior bispecific exposure, this data helps us contextualize expected benefits as we look forward to the eventual approval of this drug. The efficacy is relatively modest in those who have already progressed on bispecifics, but cevostamab would still be a welcome addition to our arsenal.
Is GPRC5D a Better Target for Car T Rather Than Bispecifics?
Our currently available GPRC5D bispecific (talquetamab) leads to high rates of skin, oral, and nail toxicity. This drug can also bring significant weight loss. These side effects make me consider that continuous targeting of GPRC5D through a bispecific may not be ideal, and that GPRC5D may be better as a one-time CAR T target. At ASH 2024, we will have 15-month follow-up data from BMS-986393, a GPRC5D CAR T. Response rates for this heavily pretreated population (76% of whom had triple refractory disease) were at 87%, with a median PFS of 14.5 months. Only 6% of patients experienced treatment-related weight loss, and nail (19%), skin (30%), and oral (31%) toxicities were relatively low. I look forward to updated data, as well as data on the resolution of the toxicities seen.
Daratumumab, a Game-Changer for AL Amyloidosis
A truly effective drug given early can change the natural history of disease, even if patients in the control arm only receive the drug later. A case in point is daratumumab. The 5-year survival rate was 76.1% for the daratumumab/cyclophosphamide/bortezomib/dexamethasone arm and 64.7% for cyclophosphamide/bortezomib/dexamethasone arm. This happened despite the fact that 67% of the control arm patients (among those who received therapy) went on to receive daratumumab later in the disease course.
Understanding how SMM Progresses to MM
We often hear that we should treat SMM and not just watch carefully because fractures may suddenly happen, or a patient may end up on dialysis. What this retrospective study tells us that amongst 427 patients with SMM, 42 had progression to myeloma, and amongst those 42, only 1 developed renal dysfunction (unclear if this resolved), and only 1 had lytic lesions that were symptomatic. The remainder were all asymptomatic lab and imaging changes. This is a retrospective study, and one should assume that follow-up was thus highly variable. It does not appear that diffusion weighted whole-body MRI imaging (our most sensitive imaging test) was employed universally or very frequently. Nevertheless, these powerful findings reassure us that, with close observation, morbidity is unlikely. Our group has designed a prospective study incorporating frequent diffusion weighted whole body MRI imaging to formally test this hypothesis (SPOTLIGHT, NCT06212323).
The Underperformance of Daratumumab/Lenalidomide/Dexamethasone in the Real World
At every major meeting I am reminded of the disconnect between real-world efficacy and clinical trial efficacy. Case in point: In this Austrian experience, daratumumab/lenalidomide/dexamethasone led to a PFS of 22.7 months vs 61.9 months in the MAIA trial of daratumumab/lenalidomide/dexamethasone. Such a sobering difference! And if you think this is an isolated experience, even in a US real-world cohort, consider that in a recently published comparative study dara/len/dex underperformed, although the time to next treatment or death was longer (37.8 months).
Delayed Neurotoxicity may not be Just a Consequence of high tumor burden
We currently think that some of the scariest side effects of cilta-cel, such as delayed neurotoxicity, may be a consequence of a high number of cancer cells and may be prevented by better disease control at the time of infusion. This study, a sobering analysis of 52 patients with delayed neurotoxicity occurring after CAR T, included 8 patients (15%) who were not heavily pretreated, and all had less than 5% plasma cells at the time of infusion. None of these patients had extramedullary disease. This result worries me, especially because cilta-cel is being studied and is poised for future approval in earlier line settings. It suggests that this toxicity may not always be a product of disease burden, contrary to our current belief.
I will be paying close attention to these 10 myeloma studies at ASH 2024, where I look forward to meeting you and learning more.Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah, Salt Lake City.
ASCO 2024: Treating Myeloma Just Got More Complicated
For brevity’s sake, I’ll focus on trials about newly diagnosed MM and myeloma at first relapse. Here’s my take on how to interpret those studies in light of broader evidence, what I view as their key limitations, and how what came out of ASCO 2024 changes my approach.
The Return of Belantamab
Belantamab, a BCMA targeting antibody-drug conjugate, previously had shown a response rate of 34% in a single-arm, heavily pretreated population, albeit with modest progression free survival (PFS), only to fail its confirmatory randomized study against pomalidomide/dexamethasone. Given the ocular toxicity associated with belantamab, many — including myself — had written off this drug (save in exceptional/unique circumstances), especially with the rise of novel immunotherapies targeting BCMA, such as chimeric antigen receptor (CAR T-cell) therapy and bispecific antibodies.
However, this year at ASCO, two key randomized trials were presented with concurrent publications, a trial of belantamab/bortezomib/dexamethasone versus daratumumab/bortezomib/dexamethasone (DVd) (DREAMM-7), and a trial of belantamab/pomalidomide/dexamethasone versus bortezomib/pomalidomide/dexamethasone (DREAMM-8). Both trials evaluated patients with myeloma who had relapsed disease and had received at least one prior line of therapy.
In both trials, the belantamab triplet beat the other triplets for the endpoint of PFS (median PFS 36.6 vs 13 months for DREAMM-7, and 12 months PFS 71% vs 51% for DREAMM-8). We must commend the bold three-versus-three design and a convincing result.
What are the caveats? Some censoring of information happened in DREAMM-7, which helped make the intervention arm look better than reality and the control arm look even worse than reality. To illustrate this point: the control arm of DVd (PFS 13 months) underperformed, compared to the CASTOR trial, where DVd led to a PFS of 16.7 months. The drug remains toxic, with high rates of keratopathy and vision problems in its current dosing schema. (Perhaps the future lies in less frequent dosing.) This toxicity is almost always reversible, but it is a huge problem to deal with, and our current quality-of-life instruments fail miserably at capturing this.
Furthermore, DVd is now emerging as perhaps the weakest daratumumab triplet that exists. Almost all patients in this trial had disease sensitivity to lenalidomide, and daratumumab/lenalidomide/dexamethasone (PFS of 45 months in the POLLUX trial) is unequivocally easier to use and handle (in my opinion) than this belantamab triplet--which is quite literally “an eyesore.” Would belantamab-based triplets beat dara/len/dex for patients with lenalidomide sensitive disease? Or, for that matter, would belantamab combos beat anti-CD38+carfilzomib+dex combinations, or cilta-cel (which is also now approved for first relapse)?
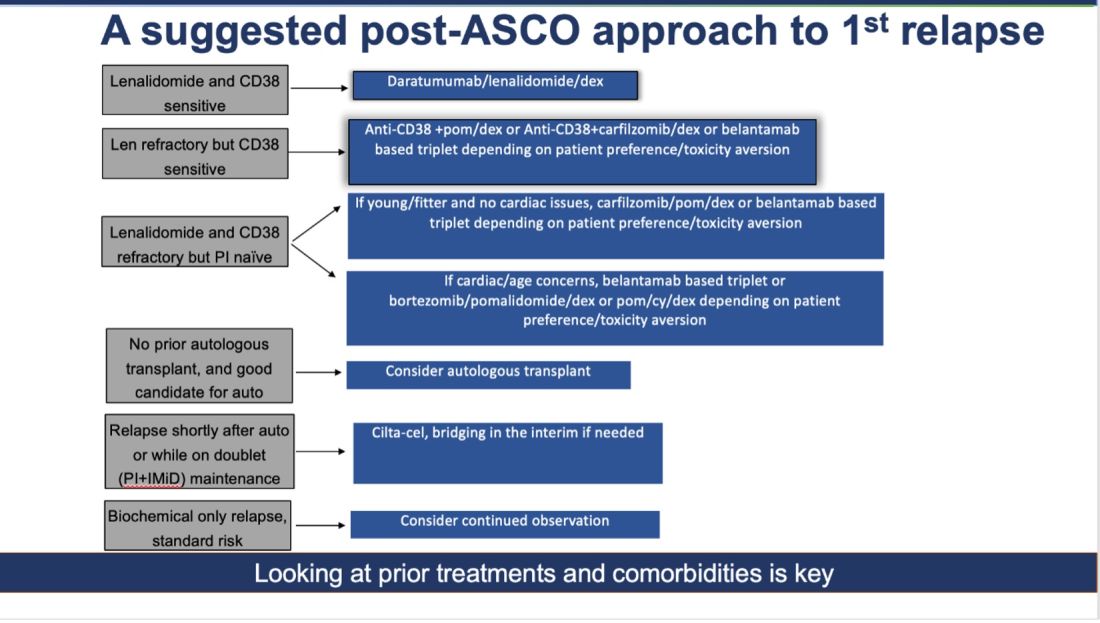
How do I foresee the future of belantamab? Despite these unequivocally positive results, I am not enthused about using it for most patients at first relapse. When trials for bispecifics at first relapse read out, my enthusiasm will likely wane even more. Still, it is useful to have belantamab in the armamentarium. For some patients perceived to be at very high risk of infection, belantamab-based triplets may indeed prove to be a better option than bispecifics. However, I suspect that with better dosing strategies for bispecifics, perhaps even that trend may be mitigated. Since we do not yet have bispecifics available in this line, my suggested algorithm for first relapse is as follows:
Newly Diagnosed MM: The Era of Quads Solidifies
At ASCO 2024, two key trials with concurrent publications assessed the role of quadruplets (without the use of transplant): the IMROZ trial of a quadruplet of isatuximab/bortezomib/lenalidomide/dexamethasone versus bortezomib/lenalidomide/dexamethasone (VRd), and the BENEFIT trial (isatuximab/lenalidomide/bortezomib/dexamethasone versus isatuximab/lenalidomide/dexamethasone).
The IMROZ trial tested the addition of an anti-CD38 antibody to a triplet backbone, and the results are compelling. The PFS was not reached for the quad vs 54 months for VRd. Unlike in the belantamab trial (where the control arm underperformed), here the control arm really overperformed. In this case, we have never seen such a compelling PFS of 54 months for VRd before. (Based on other trials, VRd PFS has been more in the ballpark of 35-43 months.) This speaks to the fitness and biology of the patients enrolled in this trial, and perhaps to how we will not see such stellar results with this quad recreated in real life.
The addition of isatuximab did not seem to impair quality of life, and although there were more treatment-related deaths with isatuximab, those higher numbers seem to have been driven by longer treatment durations. For this study, the upper age limit was 80 years, and most patients enrolled had an excellent functional status--making it clear that frail patients were greatly underrepresented.
What can we conclude from this study? For fit, older patients (who would have been transplant-eligible in the United States), this study provides excellent proof of concept that very good outcomes can be obtained without the use of transplantation. In treating frail patients, we do not know if quads are safe (or even necessary, compared to gentler sequencing), so these data are not applicable.
High-risk cytogenetics were underrepresented, and although the subgroup analysis for such patients did not show a benefit, it is hard to draw conclusions either way. For me, this trial is further evidence that for many older patients with MM, even if you “can” do a transplant, you probably “shouldn’t, they will experience increasingly better outcomes.
The standard for newly diagnosed MM in older patients for whom transplant is not intended is currently dara/len/dex. Is isa/bort/len/dex better? I do not know. It may give a better PFS, but the addition of bortezomib will lead to more neuropathy: 60% of patients developed neuropathy here, with 7% developing Grade III/IV peripheral neuropathy.
To resolve this issue, highly individualized discussions with patients will be needed. The BENEFIT trial evaluated this question more directly, with a randomized comparison of Isa-VRd versus Isa-Rd (the role of bortezomib being the main variable assessed here) with a primary endpoint of MRD negativity at 10-5 at 18 months. Although MRD negativity allows for a quick read-out, having MRD as an endpoint is a foregone conclusion. Adding another drug will almost certainly lead to deeper responses. But is it worth it?
In the BENEFIT trial, the MRD negativity at 10-5 was 26% versus 53% with the quad. However, peripheral neuropathy rates were much higher with the quad (28% vs 52%). Without longer-term data such as PFS and OS, I do not know whether it is worth the extra risks of neuropathy for older patients. Their priority may not be eradication of cancer cells at all costs. Instead, it may be better quality of life and functioning while preserving survival.
To sum up: Post-ASCO 2024, the approach to newly diagnosed MM just got a lot more complicated. For fit, older patients willing to endure extra toxicities of neuropathy (and acknowledging that we do not know whether survival will be any better with this approach), a quad is a very reasonable option to offer while forgoing transplant, in resource-rich areas of the world, such as the United States. Omitting a transplant now seems very reasonable for most older adults. However, a nuanced and individualized approach remains paramount. And given the speed of new developments, even this suggested approach will be outdated soon!
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
For brevity’s sake, I’ll focus on trials about newly diagnosed MM and myeloma at first relapse. Here’s my take on how to interpret those studies in light of broader evidence, what I view as their key limitations, and how what came out of ASCO 2024 changes my approach.
The Return of Belantamab
Belantamab, a BCMA targeting antibody-drug conjugate, previously had shown a response rate of 34% in a single-arm, heavily pretreated population, albeit with modest progression free survival (PFS), only to fail its confirmatory randomized study against pomalidomide/dexamethasone. Given the ocular toxicity associated with belantamab, many — including myself — had written off this drug (save in exceptional/unique circumstances), especially with the rise of novel immunotherapies targeting BCMA, such as chimeric antigen receptor (CAR T-cell) therapy and bispecific antibodies.
However, this year at ASCO, two key randomized trials were presented with concurrent publications, a trial of belantamab/bortezomib/dexamethasone versus daratumumab/bortezomib/dexamethasone (DVd) (DREAMM-7), and a trial of belantamab/pomalidomide/dexamethasone versus bortezomib/pomalidomide/dexamethasone (DREAMM-8). Both trials evaluated patients with myeloma who had relapsed disease and had received at least one prior line of therapy.
In both trials, the belantamab triplet beat the other triplets for the endpoint of PFS (median PFS 36.6 vs 13 months for DREAMM-7, and 12 months PFS 71% vs 51% for DREAMM-8). We must commend the bold three-versus-three design and a convincing result.
What are the caveats? Some censoring of information happened in DREAMM-7, which helped make the intervention arm look better than reality and the control arm look even worse than reality. To illustrate this point: the control arm of DVd (PFS 13 months) underperformed, compared to the CASTOR trial, where DVd led to a PFS of 16.7 months. The drug remains toxic, with high rates of keratopathy and vision problems in its current dosing schema. (Perhaps the future lies in less frequent dosing.) This toxicity is almost always reversible, but it is a huge problem to deal with, and our current quality-of-life instruments fail miserably at capturing this.
Furthermore, DVd is now emerging as perhaps the weakest daratumumab triplet that exists. Almost all patients in this trial had disease sensitivity to lenalidomide, and daratumumab/lenalidomide/dexamethasone (PFS of 45 months in the POLLUX trial) is unequivocally easier to use and handle (in my opinion) than this belantamab triplet--which is quite literally “an eyesore.” Would belantamab-based triplets beat dara/len/dex for patients with lenalidomide sensitive disease? Or, for that matter, would belantamab combos beat anti-CD38+carfilzomib+dex combinations, or cilta-cel (which is also now approved for first relapse)?
How do I foresee the future of belantamab? Despite these unequivocally positive results, I am not enthused about using it for most patients at first relapse. When trials for bispecifics at first relapse read out, my enthusiasm will likely wane even more. Still, it is useful to have belantamab in the armamentarium. For some patients perceived to be at very high risk of infection, belantamab-based triplets may indeed prove to be a better option than bispecifics. However, I suspect that with better dosing strategies for bispecifics, perhaps even that trend may be mitigated. Since we do not yet have bispecifics available in this line, my suggested algorithm for first relapse is as follows:
Newly Diagnosed MM: The Era of Quads Solidifies
At ASCO 2024, two key trials with concurrent publications assessed the role of quadruplets (without the use of transplant): the IMROZ trial of a quadruplet of isatuximab/bortezomib/lenalidomide/dexamethasone versus bortezomib/lenalidomide/dexamethasone (VRd), and the BENEFIT trial (isatuximab/lenalidomide/bortezomib/dexamethasone versus isatuximab/lenalidomide/dexamethasone).
The IMROZ trial tested the addition of an anti-CD38 antibody to a triplet backbone, and the results are compelling. The PFS was not reached for the quad vs 54 months for VRd. Unlike in the belantamab trial (where the control arm underperformed), here the control arm really overperformed. In this case, we have never seen such a compelling PFS of 54 months for VRd before. (Based on other trials, VRd PFS has been more in the ballpark of 35-43 months.) This speaks to the fitness and biology of the patients enrolled in this trial, and perhaps to how we will not see such stellar results with this quad recreated in real life.
The addition of isatuximab did not seem to impair quality of life, and although there were more treatment-related deaths with isatuximab, those higher numbers seem to have been driven by longer treatment durations. For this study, the upper age limit was 80 years, and most patients enrolled had an excellent functional status--making it clear that frail patients were greatly underrepresented.
What can we conclude from this study? For fit, older patients (who would have been transplant-eligible in the United States), this study provides excellent proof of concept that very good outcomes can be obtained without the use of transplantation. In treating frail patients, we do not know if quads are safe (or even necessary, compared to gentler sequencing), so these data are not applicable.
High-risk cytogenetics were underrepresented, and although the subgroup analysis for such patients did not show a benefit, it is hard to draw conclusions either way. For me, this trial is further evidence that for many older patients with MM, even if you “can” do a transplant, you probably “shouldn’t, they will experience increasingly better outcomes.
The standard for newly diagnosed MM in older patients for whom transplant is not intended is currently dara/len/dex. Is isa/bort/len/dex better? I do not know. It may give a better PFS, but the addition of bortezomib will lead to more neuropathy: 60% of patients developed neuropathy here, with 7% developing Grade III/IV peripheral neuropathy.
To resolve this issue, highly individualized discussions with patients will be needed. The BENEFIT trial evaluated this question more directly, with a randomized comparison of Isa-VRd versus Isa-Rd (the role of bortezomib being the main variable assessed here) with a primary endpoint of MRD negativity at 10-5 at 18 months. Although MRD negativity allows for a quick read-out, having MRD as an endpoint is a foregone conclusion. Adding another drug will almost certainly lead to deeper responses. But is it worth it?
In the BENEFIT trial, the MRD negativity at 10-5 was 26% versus 53% with the quad. However, peripheral neuropathy rates were much higher with the quad (28% vs 52%). Without longer-term data such as PFS and OS, I do not know whether it is worth the extra risks of neuropathy for older patients. Their priority may not be eradication of cancer cells at all costs. Instead, it may be better quality of life and functioning while preserving survival.
To sum up: Post-ASCO 2024, the approach to newly diagnosed MM just got a lot more complicated. For fit, older patients willing to endure extra toxicities of neuropathy (and acknowledging that we do not know whether survival will be any better with this approach), a quad is a very reasonable option to offer while forgoing transplant, in resource-rich areas of the world, such as the United States. Omitting a transplant now seems very reasonable for most older adults. However, a nuanced and individualized approach remains paramount. And given the speed of new developments, even this suggested approach will be outdated soon!
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
For brevity’s sake, I’ll focus on trials about newly diagnosed MM and myeloma at first relapse. Here’s my take on how to interpret those studies in light of broader evidence, what I view as their key limitations, and how what came out of ASCO 2024 changes my approach.
The Return of Belantamab
Belantamab, a BCMA targeting antibody-drug conjugate, previously had shown a response rate of 34% in a single-arm, heavily pretreated population, albeit with modest progression free survival (PFS), only to fail its confirmatory randomized study against pomalidomide/dexamethasone. Given the ocular toxicity associated with belantamab, many — including myself — had written off this drug (save in exceptional/unique circumstances), especially with the rise of novel immunotherapies targeting BCMA, such as chimeric antigen receptor (CAR T-cell) therapy and bispecific antibodies.
However, this year at ASCO, two key randomized trials were presented with concurrent publications, a trial of belantamab/bortezomib/dexamethasone versus daratumumab/bortezomib/dexamethasone (DVd) (DREAMM-7), and a trial of belantamab/pomalidomide/dexamethasone versus bortezomib/pomalidomide/dexamethasone (DREAMM-8). Both trials evaluated patients with myeloma who had relapsed disease and had received at least one prior line of therapy.
In both trials, the belantamab triplet beat the other triplets for the endpoint of PFS (median PFS 36.6 vs 13 months for DREAMM-7, and 12 months PFS 71% vs 51% for DREAMM-8). We must commend the bold three-versus-three design and a convincing result.
What are the caveats? Some censoring of information happened in DREAMM-7, which helped make the intervention arm look better than reality and the control arm look even worse than reality. To illustrate this point: the control arm of DVd (PFS 13 months) underperformed, compared to the CASTOR trial, where DVd led to a PFS of 16.7 months. The drug remains toxic, with high rates of keratopathy and vision problems in its current dosing schema. (Perhaps the future lies in less frequent dosing.) This toxicity is almost always reversible, but it is a huge problem to deal with, and our current quality-of-life instruments fail miserably at capturing this.
Furthermore, DVd is now emerging as perhaps the weakest daratumumab triplet that exists. Almost all patients in this trial had disease sensitivity to lenalidomide, and daratumumab/lenalidomide/dexamethasone (PFS of 45 months in the POLLUX trial) is unequivocally easier to use and handle (in my opinion) than this belantamab triplet--which is quite literally “an eyesore.” Would belantamab-based triplets beat dara/len/dex for patients with lenalidomide sensitive disease? Or, for that matter, would belantamab combos beat anti-CD38+carfilzomib+dex combinations, or cilta-cel (which is also now approved for first relapse)?
How do I foresee the future of belantamab? Despite these unequivocally positive results, I am not enthused about using it for most patients at first relapse. When trials for bispecifics at first relapse read out, my enthusiasm will likely wane even more. Still, it is useful to have belantamab in the armamentarium. For some patients perceived to be at very high risk of infection, belantamab-based triplets may indeed prove to be a better option than bispecifics. However, I suspect that with better dosing strategies for bispecifics, perhaps even that trend may be mitigated. Since we do not yet have bispecifics available in this line, my suggested algorithm for first relapse is as follows:
Newly Diagnosed MM: The Era of Quads Solidifies
At ASCO 2024, two key trials with concurrent publications assessed the role of quadruplets (without the use of transplant): the IMROZ trial of a quadruplet of isatuximab/bortezomib/lenalidomide/dexamethasone versus bortezomib/lenalidomide/dexamethasone (VRd), and the BENEFIT trial (isatuximab/lenalidomide/bortezomib/dexamethasone versus isatuximab/lenalidomide/dexamethasone).
The IMROZ trial tested the addition of an anti-CD38 antibody to a triplet backbone, and the results are compelling. The PFS was not reached for the quad vs 54 months for VRd. Unlike in the belantamab trial (where the control arm underperformed), here the control arm really overperformed. In this case, we have never seen such a compelling PFS of 54 months for VRd before. (Based on other trials, VRd PFS has been more in the ballpark of 35-43 months.) This speaks to the fitness and biology of the patients enrolled in this trial, and perhaps to how we will not see such stellar results with this quad recreated in real life.
The addition of isatuximab did not seem to impair quality of life, and although there were more treatment-related deaths with isatuximab, those higher numbers seem to have been driven by longer treatment durations. For this study, the upper age limit was 80 years, and most patients enrolled had an excellent functional status--making it clear that frail patients were greatly underrepresented.
What can we conclude from this study? For fit, older patients (who would have been transplant-eligible in the United States), this study provides excellent proof of concept that very good outcomes can be obtained without the use of transplantation. In treating frail patients, we do not know if quads are safe (or even necessary, compared to gentler sequencing), so these data are not applicable.
High-risk cytogenetics were underrepresented, and although the subgroup analysis for such patients did not show a benefit, it is hard to draw conclusions either way. For me, this trial is further evidence that for many older patients with MM, even if you “can” do a transplant, you probably “shouldn’t, they will experience increasingly better outcomes.
The standard for newly diagnosed MM in older patients for whom transplant is not intended is currently dara/len/dex. Is isa/bort/len/dex better? I do not know. It may give a better PFS, but the addition of bortezomib will lead to more neuropathy: 60% of patients developed neuropathy here, with 7% developing Grade III/IV peripheral neuropathy.
To resolve this issue, highly individualized discussions with patients will be needed. The BENEFIT trial evaluated this question more directly, with a randomized comparison of Isa-VRd versus Isa-Rd (the role of bortezomib being the main variable assessed here) with a primary endpoint of MRD negativity at 10-5 at 18 months. Although MRD negativity allows for a quick read-out, having MRD as an endpoint is a foregone conclusion. Adding another drug will almost certainly lead to deeper responses. But is it worth it?
In the BENEFIT trial, the MRD negativity at 10-5 was 26% versus 53% with the quad. However, peripheral neuropathy rates were much higher with the quad (28% vs 52%). Without longer-term data such as PFS and OS, I do not know whether it is worth the extra risks of neuropathy for older patients. Their priority may not be eradication of cancer cells at all costs. Instead, it may be better quality of life and functioning while preserving survival.
To sum up: Post-ASCO 2024, the approach to newly diagnosed MM just got a lot more complicated. For fit, older patients willing to endure extra toxicities of neuropathy (and acknowledging that we do not know whether survival will be any better with this approach), a quad is a very reasonable option to offer while forgoing transplant, in resource-rich areas of the world, such as the United States. Omitting a transplant now seems very reasonable for most older adults. However, a nuanced and individualized approach remains paramount. And given the speed of new developments, even this suggested approach will be outdated soon!
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
Do No Harm: What Smoldering Myeloma Teaches Us
My approach to treating SMM takes into account what its history can teach us about 1) how advancements in imaging and diagnostic reclassifications can revise the entire natural history of a disease, and 2) how evidence generated by even the best of studies may have an expiration date.
Much of what we know about SMM today dates to a pivotal study by Robert A. Kyle, MD, and colleagues, published in 2007. That inspirational team of investigators followed people diagnosed with SMM from 1970 to 1995 and established the first natural history of the condition. Their monumental effort and the data and conclusions it generated (eg,10% risk annually of SMM becoming MM for the first 5 years) are still cited today in references, papers, and slide sets.
Despite the seminal importance of this work, from today’s perspective the 2007 study might just as well have been describing a different disease. Back then people were diagnosed with SMM if their blood work detected a monoclonal protein and a follow-up bone marrow biopsy found at least 10% plasma cells (or a monoclonal protein exceeding 3g/dL). If there were no signs of end-organ damage (ie, no anemia or kidney problems) and an x-ray showed no fractures or lesions in the bones, the diagnosis was determined to be SMM.
What’s different in 2024? First and foremost: advanced, highly sensitive imaging techniques. MRIs can pick up small lytic lesions (and even the precursor to lytic lesions) that would not appear on an x-ray. In fact, relying solely on x-rays risks missing half of the lytic lesions.
Therefore, using the same criteria, many people who in the past were diagnosed with SMM would today be diagnosed with MM. Furthermore, in 2014 a diagnostic change reclassified people’s diagnosis from the highest risk category of SMM to the category of active MM.
Due to these scientific advances and classification changes, I believe that the natural history of SMM is unknown. Risk stratification models for SMM derived from data sets of people who had not undergone rigorous advanced imaging likely are skewed by data from people who had MM. In addition, current risk stratification models have very poor concordance with each other. I routinely see people whose 2-year risk according to different models varies by more than 30%-40%.
All this information tells us that SMM today is more indolent than the SMM of the past. Paradoxically, however, our therapies keep getting more and more aggressive, exposing this vulnerable group of people to intense treatment regimens that they may not require. Therapies tested on people diagnosed with SMM include an aggressive three-drug regimen, autologous stem cell transplant, and 2 years of additional therapy, as well as, more recently CAR T-cell therapy which so far has at least a 4%-5% treatment-related mortality risk in people with myeloma and a strong signal for secondary cancer risk. Other trials are testing bispecific therapies such as talquetamab, a drug which in my experience causes horrendous skin toxicity, profound weight loss, and one’s nails to fall off.
Doctors routinely keep showing slides from Kyle’s pivotal work to describe the natural history of SMM and to justify the need for treatment, and trials continue to use outdated progression prediction models. In my opinion, as people with MM keep living longer and treatments for MM keep getting better, the threshold for intervening with asymptomatic, healthy people with SMM should be getting higher, not lower.
I strongly believe that the current landscape of SMM treatment exemplifies good intentions leading to bad outcomes. A routine blood test in a completely healthy person that finds elevated total protein in the blood could culminate in well-intentioned but aggressive therapies that can lead to many serious side effects. (I repeat: Secondary cancers and deaths from infections have all occurred in SMM trials.)
With no control arm, we simply don’t know how well these people might have fared without any therapy. For all we know, treatment may have shortened their lives due to complications up to and including death — all because of a blood test often conducted for reasons that have no evidentiary basis.
For example, plasma cell diseases are not linked to low bone density or auto-immune diseases, yet these labs are sent routinely as part of a workup for those conditions, leading to increasing anxiety and costs.
So, what is my approach? When treating people with SMM, I hold nuanced discussions of this data to help prioritize and reach informed decisions. After our honest conversation about the limitations of SMM models, older data, and the limitations of prospective data studying pharmacological treatment, almost no one signs up for treatment.
I want these people to stay safe, and I’m proud to be a part of a trial (SPOTLIGHT, NCT06212323) that aims to show prospectively that these people can be watched off treatment with monitoring via advanced imaging modalities.
In conclusion: SMM teaches us how, even in the absence of pharmacological interventions, the natural history of a disease can change over time, simply via better imaging techniques and changes in diagnostic classifications. Unfortunately, SMM also illustrates how good intentions can lead to harm.
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
My approach to treating SMM takes into account what its history can teach us about 1) how advancements in imaging and diagnostic reclassifications can revise the entire natural history of a disease, and 2) how evidence generated by even the best of studies may have an expiration date.
Much of what we know about SMM today dates to a pivotal study by Robert A. Kyle, MD, and colleagues, published in 2007. That inspirational team of investigators followed people diagnosed with SMM from 1970 to 1995 and established the first natural history of the condition. Their monumental effort and the data and conclusions it generated (eg,10% risk annually of SMM becoming MM for the first 5 years) are still cited today in references, papers, and slide sets.
Despite the seminal importance of this work, from today’s perspective the 2007 study might just as well have been describing a different disease. Back then people were diagnosed with SMM if their blood work detected a monoclonal protein and a follow-up bone marrow biopsy found at least 10% plasma cells (or a monoclonal protein exceeding 3g/dL). If there were no signs of end-organ damage (ie, no anemia or kidney problems) and an x-ray showed no fractures or lesions in the bones, the diagnosis was determined to be SMM.
What’s different in 2024? First and foremost: advanced, highly sensitive imaging techniques. MRIs can pick up small lytic lesions (and even the precursor to lytic lesions) that would not appear on an x-ray. In fact, relying solely on x-rays risks missing half of the lytic lesions.
Therefore, using the same criteria, many people who in the past were diagnosed with SMM would today be diagnosed with MM. Furthermore, in 2014 a diagnostic change reclassified people’s diagnosis from the highest risk category of SMM to the category of active MM.
Due to these scientific advances and classification changes, I believe that the natural history of SMM is unknown. Risk stratification models for SMM derived from data sets of people who had not undergone rigorous advanced imaging likely are skewed by data from people who had MM. In addition, current risk stratification models have very poor concordance with each other. I routinely see people whose 2-year risk according to different models varies by more than 30%-40%.
All this information tells us that SMM today is more indolent than the SMM of the past. Paradoxically, however, our therapies keep getting more and more aggressive, exposing this vulnerable group of people to intense treatment regimens that they may not require. Therapies tested on people diagnosed with SMM include an aggressive three-drug regimen, autologous stem cell transplant, and 2 years of additional therapy, as well as, more recently CAR T-cell therapy which so far has at least a 4%-5% treatment-related mortality risk in people with myeloma and a strong signal for secondary cancer risk. Other trials are testing bispecific therapies such as talquetamab, a drug which in my experience causes horrendous skin toxicity, profound weight loss, and one’s nails to fall off.
Doctors routinely keep showing slides from Kyle’s pivotal work to describe the natural history of SMM and to justify the need for treatment, and trials continue to use outdated progression prediction models. In my opinion, as people with MM keep living longer and treatments for MM keep getting better, the threshold for intervening with asymptomatic, healthy people with SMM should be getting higher, not lower.
I strongly believe that the current landscape of SMM treatment exemplifies good intentions leading to bad outcomes. A routine blood test in a completely healthy person that finds elevated total protein in the blood could culminate in well-intentioned but aggressive therapies that can lead to many serious side effects. (I repeat: Secondary cancers and deaths from infections have all occurred in SMM trials.)
With no control arm, we simply don’t know how well these people might have fared without any therapy. For all we know, treatment may have shortened their lives due to complications up to and including death — all because of a blood test often conducted for reasons that have no evidentiary basis.
For example, plasma cell diseases are not linked to low bone density or auto-immune diseases, yet these labs are sent routinely as part of a workup for those conditions, leading to increasing anxiety and costs.
So, what is my approach? When treating people with SMM, I hold nuanced discussions of this data to help prioritize and reach informed decisions. After our honest conversation about the limitations of SMM models, older data, and the limitations of prospective data studying pharmacological treatment, almost no one signs up for treatment.
I want these people to stay safe, and I’m proud to be a part of a trial (SPOTLIGHT, NCT06212323) that aims to show prospectively that these people can be watched off treatment with monitoring via advanced imaging modalities.
In conclusion: SMM teaches us how, even in the absence of pharmacological interventions, the natural history of a disease can change over time, simply via better imaging techniques and changes in diagnostic classifications. Unfortunately, SMM also illustrates how good intentions can lead to harm.
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
My approach to treating SMM takes into account what its history can teach us about 1) how advancements in imaging and diagnostic reclassifications can revise the entire natural history of a disease, and 2) how evidence generated by even the best of studies may have an expiration date.
Much of what we know about SMM today dates to a pivotal study by Robert A. Kyle, MD, and colleagues, published in 2007. That inspirational team of investigators followed people diagnosed with SMM from 1970 to 1995 and established the first natural history of the condition. Their monumental effort and the data and conclusions it generated (eg,10% risk annually of SMM becoming MM for the first 5 years) are still cited today in references, papers, and slide sets.
Despite the seminal importance of this work, from today’s perspective the 2007 study might just as well have been describing a different disease. Back then people were diagnosed with SMM if their blood work detected a monoclonal protein and a follow-up bone marrow biopsy found at least 10% plasma cells (or a monoclonal protein exceeding 3g/dL). If there were no signs of end-organ damage (ie, no anemia or kidney problems) and an x-ray showed no fractures or lesions in the bones, the diagnosis was determined to be SMM.
What’s different in 2024? First and foremost: advanced, highly sensitive imaging techniques. MRIs can pick up small lytic lesions (and even the precursor to lytic lesions) that would not appear on an x-ray. In fact, relying solely on x-rays risks missing half of the lytic lesions.
Therefore, using the same criteria, many people who in the past were diagnosed with SMM would today be diagnosed with MM. Furthermore, in 2014 a diagnostic change reclassified people’s diagnosis from the highest risk category of SMM to the category of active MM.
Due to these scientific advances and classification changes, I believe that the natural history of SMM is unknown. Risk stratification models for SMM derived from data sets of people who had not undergone rigorous advanced imaging likely are skewed by data from people who had MM. In addition, current risk stratification models have very poor concordance with each other. I routinely see people whose 2-year risk according to different models varies by more than 30%-40%.
All this information tells us that SMM today is more indolent than the SMM of the past. Paradoxically, however, our therapies keep getting more and more aggressive, exposing this vulnerable group of people to intense treatment regimens that they may not require. Therapies tested on people diagnosed with SMM include an aggressive three-drug regimen, autologous stem cell transplant, and 2 years of additional therapy, as well as, more recently CAR T-cell therapy which so far has at least a 4%-5% treatment-related mortality risk in people with myeloma and a strong signal for secondary cancer risk. Other trials are testing bispecific therapies such as talquetamab, a drug which in my experience causes horrendous skin toxicity, profound weight loss, and one’s nails to fall off.
Doctors routinely keep showing slides from Kyle’s pivotal work to describe the natural history of SMM and to justify the need for treatment, and trials continue to use outdated progression prediction models. In my opinion, as people with MM keep living longer and treatments for MM keep getting better, the threshold for intervening with asymptomatic, healthy people with SMM should be getting higher, not lower.
I strongly believe that the current landscape of SMM treatment exemplifies good intentions leading to bad outcomes. A routine blood test in a completely healthy person that finds elevated total protein in the blood could culminate in well-intentioned but aggressive therapies that can lead to many serious side effects. (I repeat: Secondary cancers and deaths from infections have all occurred in SMM trials.)
With no control arm, we simply don’t know how well these people might have fared without any therapy. For all we know, treatment may have shortened their lives due to complications up to and including death — all because of a blood test often conducted for reasons that have no evidentiary basis.
For example, plasma cell diseases are not linked to low bone density or auto-immune diseases, yet these labs are sent routinely as part of a workup for those conditions, leading to increasing anxiety and costs.
So, what is my approach? When treating people with SMM, I hold nuanced discussions of this data to help prioritize and reach informed decisions. After our honest conversation about the limitations of SMM models, older data, and the limitations of prospective data studying pharmacological treatment, almost no one signs up for treatment.
I want these people to stay safe, and I’m proud to be a part of a trial (SPOTLIGHT, NCT06212323) that aims to show prospectively that these people can be watched off treatment with monitoring via advanced imaging modalities.
In conclusion: SMM teaches us how, even in the absence of pharmacological interventions, the natural history of a disease can change over time, simply via better imaging techniques and changes in diagnostic classifications. Unfortunately, SMM also illustrates how good intentions can lead to harm.
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
‘Less is More’ in Myeloma
Among those that intrigue me most are the pioneering “less is more” trials that challenged conventional practices and remain relevant today. One such trial was inspired by a patient’s dissatisfaction with high doses of dexamethasone and its side effects.
Unlike the prevailing norm of frequent high doses, this trial compared a steroid dose administered weekly (as opposed to doses given several days a week). Lo and behold, the lower steroid dosage was associated with significantly better survival rates. At 1-year follow-up, 96% of patients in the lower-dose group were alive, compared with 87% in the higher-dose group.
Another noteworthy “less-is more” trial that I love, spearheaded by an Italian team, also focused on steroid dosage. This trial investigated discontinuing dexamethasone after nine cycles, along with reducing the dose of lenalidomide, versus maintaining long-term treatment without reductions. The findings revealed comparable progression-free survival with reduced toxicity, highlighting the potential benefits of this less-is-more approach.
While these trials are inspirational, a closer examination of myeloma trial history, especially those that led to regulatory approvals, reveals a preponderance of “add-on” trials. You add a potentially effective drug to an existing backbone, and you get an improvement in an outcome such as response rate (shrinking cancer) or duration of remission or progression free survival (amount of time alive and in remission).
Such trials have led to an abundance of effective options. But these same trials have almost always been a comparison of three drugs versus two drugs, and almost never three drugs versus three. And the drugs are often given continuously, especially the “newer” added drug, without a break. As a result, we are left completely unsure of how to sequence our drugs, and whether a finite course of the new drug would be equivalent to administering that new drug forever.
This problem is not unique to myeloma. Yet it is very apparent in myeloma, because we have been lucky to have so many good drugs (or at least “potentially” good drugs) that make it to phase 3 trials.
Unfortunately, the landscape of clinical trials is heavily influenced by the pharmaceutical industry, with limited funding available from alternative sources. As a result, there is a scarcity of trials exploring “less is more” approaches, despite their potential to optimize treatment outcomes and quality of life.
Even government-funded trials run by cooperative groups require industry buy-in or are run by people who have very close contacts and conflicts of interest with industry. We need so many more of these less-is-more trials, but we have limited means to fund them.
These are the kinds of discussions I have with my patients daily. We grapple with questions about the necessity of lifelong (or any) maintenance therapy or the feasibility of treatment breaks for patients with stable disease. While we strive to provide the best care possible, the lack of definitive data often leaves us making tough decisions in the clinic.
I am grateful to those who are working tirelessly to facilitate trials that prioritize quality of life and “less is more” approaches. Your efforts are invaluable. Looking forward, I aspire to contribute to this important work.
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
Among those that intrigue me most are the pioneering “less is more” trials that challenged conventional practices and remain relevant today. One such trial was inspired by a patient’s dissatisfaction with high doses of dexamethasone and its side effects.
Unlike the prevailing norm of frequent high doses, this trial compared a steroid dose administered weekly (as opposed to doses given several days a week). Lo and behold, the lower steroid dosage was associated with significantly better survival rates. At 1-year follow-up, 96% of patients in the lower-dose group were alive, compared with 87% in the higher-dose group.
Another noteworthy “less-is more” trial that I love, spearheaded by an Italian team, also focused on steroid dosage. This trial investigated discontinuing dexamethasone after nine cycles, along with reducing the dose of lenalidomide, versus maintaining long-term treatment without reductions. The findings revealed comparable progression-free survival with reduced toxicity, highlighting the potential benefits of this less-is-more approach.
While these trials are inspirational, a closer examination of myeloma trial history, especially those that led to regulatory approvals, reveals a preponderance of “add-on” trials. You add a potentially effective drug to an existing backbone, and you get an improvement in an outcome such as response rate (shrinking cancer) or duration of remission or progression free survival (amount of time alive and in remission).
Such trials have led to an abundance of effective options. But these same trials have almost always been a comparison of three drugs versus two drugs, and almost never three drugs versus three. And the drugs are often given continuously, especially the “newer” added drug, without a break. As a result, we are left completely unsure of how to sequence our drugs, and whether a finite course of the new drug would be equivalent to administering that new drug forever.
This problem is not unique to myeloma. Yet it is very apparent in myeloma, because we have been lucky to have so many good drugs (or at least “potentially” good drugs) that make it to phase 3 trials.
Unfortunately, the landscape of clinical trials is heavily influenced by the pharmaceutical industry, with limited funding available from alternative sources. As a result, there is a scarcity of trials exploring “less is more” approaches, despite their potential to optimize treatment outcomes and quality of life.
Even government-funded trials run by cooperative groups require industry buy-in or are run by people who have very close contacts and conflicts of interest with industry. We need so many more of these less-is-more trials, but we have limited means to fund them.
These are the kinds of discussions I have with my patients daily. We grapple with questions about the necessity of lifelong (or any) maintenance therapy or the feasibility of treatment breaks for patients with stable disease. While we strive to provide the best care possible, the lack of definitive data often leaves us making tough decisions in the clinic.
I am grateful to those who are working tirelessly to facilitate trials that prioritize quality of life and “less is more” approaches. Your efforts are invaluable. Looking forward, I aspire to contribute to this important work.
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.
Among those that intrigue me most are the pioneering “less is more” trials that challenged conventional practices and remain relevant today. One such trial was inspired by a patient’s dissatisfaction with high doses of dexamethasone and its side effects.
Unlike the prevailing norm of frequent high doses, this trial compared a steroid dose administered weekly (as opposed to doses given several days a week). Lo and behold, the lower steroid dosage was associated with significantly better survival rates. At 1-year follow-up, 96% of patients in the lower-dose group were alive, compared with 87% in the higher-dose group.
Another noteworthy “less-is more” trial that I love, spearheaded by an Italian team, also focused on steroid dosage. This trial investigated discontinuing dexamethasone after nine cycles, along with reducing the dose of lenalidomide, versus maintaining long-term treatment without reductions. The findings revealed comparable progression-free survival with reduced toxicity, highlighting the potential benefits of this less-is-more approach.
While these trials are inspirational, a closer examination of myeloma trial history, especially those that led to regulatory approvals, reveals a preponderance of “add-on” trials. You add a potentially effective drug to an existing backbone, and you get an improvement in an outcome such as response rate (shrinking cancer) or duration of remission or progression free survival (amount of time alive and in remission).
Such trials have led to an abundance of effective options. But these same trials have almost always been a comparison of three drugs versus two drugs, and almost never three drugs versus three. And the drugs are often given continuously, especially the “newer” added drug, without a break. As a result, we are left completely unsure of how to sequence our drugs, and whether a finite course of the new drug would be equivalent to administering that new drug forever.
This problem is not unique to myeloma. Yet it is very apparent in myeloma, because we have been lucky to have so many good drugs (or at least “potentially” good drugs) that make it to phase 3 trials.
Unfortunately, the landscape of clinical trials is heavily influenced by the pharmaceutical industry, with limited funding available from alternative sources. As a result, there is a scarcity of trials exploring “less is more” approaches, despite their potential to optimize treatment outcomes and quality of life.
Even government-funded trials run by cooperative groups require industry buy-in or are run by people who have very close contacts and conflicts of interest with industry. We need so many more of these less-is-more trials, but we have limited means to fund them.
These are the kinds of discussions I have with my patients daily. We grapple with questions about the necessity of lifelong (or any) maintenance therapy or the feasibility of treatment breaks for patients with stable disease. While we strive to provide the best care possible, the lack of definitive data often leaves us making tough decisions in the clinic.
I am grateful to those who are working tirelessly to facilitate trials that prioritize quality of life and “less is more” approaches. Your efforts are invaluable. Looking forward, I aspire to contribute to this important work.
Dr. Mohyuddin is assistant professor in the multiple myeloma program at the Huntsman Cancer Institute at the University of Utah in Salt Lake City.