User login
An A-Peeling Diagnosis
This icon represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 39-year-old previously healthy man presented to the emergency department (ED) with abrupt-onset fever, headache, back pain, myalgias, chills, and photophobia. His past medical history included seasonal allergies and an episode of aseptic meningitis 8 years prior. He denied cough, dysuria, weakness, numbness, or visual changes. He denied using tobacco or injection drugs and rarely drank alcohol. His only medication was acetaminophen for fever.
The patient’s sudden fever indicates the rapid onset of an inflammatory state. While the headache and photophobia might be a result of an underlying systemic infection or an irritant like blood in the cerebral spinal fluid (CSF), one must consider meningitis. Potential sources for sudden meningitis include infectious, autoimmune (rheumatoid arthritis, systemic lupus erythematosus [SLE]), or drug-induced aseptic meningitis, and structural etiologies (ruptured cyst). Recrudescence of prior disease may also present acutely (Mollaret meningitis). Malignant etiologies, being more indolent, seem less likely. Back pain may indicate an epidural inflammatory process like epidural abscess; however, the patient denies risk factors such as injection drug use or recent procedures.
The patient’s temperature was 101.2 °F; blood pressure, 120/72 mm Hg; and heart rate, 112 bpm. He appeared comfortable, without meningismus or spinal tenderness. Pupils were reactive; eyes were without icterus, injection, or suffusion. Cardiac exam was normal. Lungs were clear to auscultation. He had no abdominal tenderness, hepatosplenomegaly, or lymphadenopathy. Cranial nerves II through XII, balance, coordination, strength, and sensation were intact. No rash was noted. Complete blood count (CBC), basic and hepatic chemistry panels, urinalysis, and serum lactate tests were within normal limits. Erythrocyte sedimentation rate (ESR) was elevated to 15 mm/h (normal range, 3-10 mm/h), C-reactive protein (CRP) to 2.4 mg/dL (normal range, <0.5 mg/dL), and procalcitonin to 0.07 ng/mL (normal range, <0.05 ng/mL). The patient was treated with intravenous (IV) fluids, ketorolac, dexamethasone, and acetaminophen, with resolution of symptoms. Given his rapid improvement, absence of meningismus, and lack of immunocompromise, lumbar puncture was deferred. A diagnosis of nonspecific viral syndrome was made. He was discharged home.
Certainly, a systemic infection (eg, influenza, adenovirus, arbovirus-related infection, HIV) could be a cause of this patient’s presentation. Notably, less than two-thirds of patients with meningitis present with the classic triad of fever, neck stiffness, and altered mental status. In this patient with fever, headache, and photophobia, aseptic meningitis should still be considered. While the negative procalcitonin and rapid clinical improvement without antibiotics make acute bacterial meningitis unlikely, nonbacterial causes of meningeal irritation can be severe and life-threatening. An assessment for jolt accentuation of the headache might have been helpful. Information about time of year, geographic exposures (vector-borne infections), and sick contacts (viral illness) can inform the clinical decision to pursue lumbar puncture. Additional history regarding his previous aseptic meningitis would be helpful, as it could suggest a recurrent inflammatory process. Causes of recurrent aseptic meningitis include infectious (herpes simplex virus [HSV], Epstein-Barr virus [EBV], syphilis), drug-related (nonsteroidal anti-inflammatory drugs [NSAIDs]), structural (epidermoid cyst with rupture), and autoimmune (lupus, Sjögren syndrome, Behçet disease) etiologies.
The mildly elevated inflammatory markers are nonspecific and reflect the patient’s known inflammatory state. The dexamethasone given for symptomatic management may have had some therapeutic effect in the setting of an autoimmune process, with additional contribution from ketorolac and acetaminophen.
He returned to the ED 3 days later with a pruritic, disseminated rash involving his palms and soles, accompanied by hand swelling and tingling. Although his headache and photophobia resolved, he reported a productive cough, nasal congestion, and sore throat. He also reported orange-pink urine without dysuria or urinary frequency. Additional questioning revealed a recent motorcycle trip to the Great Lakes region. During this trip, he did not camp, interact with animals or ticks, or swim in streams or lakes. He did not eat any raw, undercooked, or locally hunted meats. He denied new medications, soaps or detergents, or sexual contacts. He had started taking acetaminophen and ibuprofen around the clock since prior discharge.
The orange-pink urine and acute-onset palmoplantar rash with recent fever help narrow the differential. Orange-pink urine might suggest bilirubinuria from liver injury, hemolysis with hemoglobinuria, or myoglobinuria. Most concerning would be hematuria associated with glomerular injury and a systemic vasculopathy.
The rash on the palms and soles should be further characterized as blanching or nonblanching. Blanching, indicating vasodilation of intact blood vessels, is seen with many drug eruptions and viral exanthems. Nonblanching, suggesting broken capillaries (petechiae or purpura), would suggest vasculitis or vasculopathy from emboli, infection, or inflammation. A palmoplantar rash in febrile illness should first prompt evaluation for life-threatening conditions, followed by consideration of both infectious and noninfectious etiologies. Acutely fatal infections include Rocky Mountain spotted fever (RMSF), meningococcemia, toxic shock syndrome, infective endocarditis, and rat-bite fever. The rash, fever, headache, and outdoor exposure raise the possibility of a rickettsial infection, including RMSF, which can be contracted rarely around the Great Lakes. Other life-threatening infections seem unlikely, as the patient would have significantly deteriorated without proper medical care by now. Palmoplantar rash with fever can also be seen in other bacterial infections (eg, secondary syphilis, arbovirus infections, typhus) and in viral infections (eg, cytomegalovirus [CMV], EBV, human herpesvirus-6 [HHV-6], HIV, coxsackievirus, and papular-purpuric gloves and socks syndrome caused by parvovirus B19). Noninfectious considerations include drug hypersensitivity rashes, neoplasm (eg, cutaneous T-cell lymphoma), or inflammatory conditions (eg, SLE, vasculitis). Drug reaction with eosinophilia and systemic symptoms (may also present with severe illness.
The acetaminophen and ibuprofen may be masking ongoing fevers. The cough, nasal congestion, and sore throat might be part of a viral prodrome or, in tandem with fever, associated with a vasculitis such as granulomatosis with polyangiitis.

Vital signs were normal, and the patient appeared nontoxic. Physical examination demonstrated mildly cracked lips, oropharyngeal erythema with small petechiae on the soft palate, a morbilliform rash throughout his extremities and trunk (Figure 1), and confluent, brightly erythematous patches on his palms and soles with associated edema (Figure 2 and Appendix Figure). No lymphadenopathy, hepatosplenomegaly, or joint swelling was noted. CBC and basic chemistry panel remained normal; however, hepatic chemistries were notable for alanine aminotransferase (ALT) of 128 U/L, aspartate aminotransferase (AST) of 49 U/L, total bilirubin of 3.7 mg/dL, direct bilirubin of 2.4 mg/dL, total protein of 7.1 g/dL, albumin of 4.1 g/dL, and alkaline phosphatase of 197 U/L. Urinalysis detected bilirubin without blood, protein, bacteria, cells, or casts. The patient was admitted to the hospital.

The patient now has acute-onset upper respiratory symptoms with oral mucosal erythema, edema and erythema of the hands and feet with morbilliform rash of the extremities, and liver injury causing bilirubinuria. The patient’s initial symptoms may have had some response to therapy, but the current presentation suggests ongoing evolution of disease. Reactive infectious mucocutaneous eruptions include chlamydia, influenza, parainfluenza, and enteroviruses. Measles is possible given its recent resurgence; however, absence of coryza or Koplik spots and the peripheral distribution of the rash without initial truncal involvement make this less likely. Mycoplasma pneumonia–induced rash and mucositis might present with respiratory symptoms and this rash distribution, but typically involves two or more mucosal sites.
Iatrogenic causes are important to consider given the recent exposure to NSAIDs, specifically Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN). In this patient, however, SJS/TEN is unlikely as it typically presents 1 to 3 weeks after exposure, with a truncal-predominant rash rarely involving the palms and soles.
Despite the absence of conjunctivitis and cervical lymphadenopathy, one additional consideration is Kawasaki disease (KD). Though more common in children, it may rarely present in adulthood. The time course of manifesting symptoms with potential steroid responsiveness raises suspicion for this diagnosis.
During a 4-day hospitalization, he developed mild bilateral conjunctivitis, peeling lips, and scleral icterus. CBC remained within normal limits. A peripheral smear demonstrated toxic neutrophilic granulation with normal erythrocytes and platelets. HIV and hepatitis A, B, and C serologies were negative. Blood cultures were negative. CRP and ESR increased to 4.3 mg/dL and 56 mm/h, respectively. Hepatic chemistries increased to ALT 155 U/L, AST 101 U/L, total bilirubin 5.1 mg/dL, direct bilirubin 3.3 mg/dL, and alkaline phosphatase 211 U/L. Right upper-quadrant ultrasound demonstrated gallbladder distention (11.3 cm × 5.0 cm; normal, 10.0 cm × 4.0 cm) without stones, wall thickening, or pericholecystic fluid; sonographic Murphy sign was negative. The liver was unremarkable with normal flow in the portal vein.
The patient’s persistent reactive neutrophilic granulation and rising CRP and ESR indicate ongoing inflammation. The largely direct hyperbilirubinemia with hepatitis, minimal findings on ultrasound imaging, and lack of Murphy sign suggest either direct infection of the liver or cholestasis. Viral serologies for EBV, HSV, and CMV should be sent, although these viruses are less commonly associated with oral rash and conjunctivitis. The marked degree of cholestasis makes adenovirus and mycoplasma less likely. Leptospirosis should be considered given the degree of liver injury with potential conjunctival suffusion. However, oral involvement would be atypical; renal injury is absent; and the patient denied pertinent exposures, vomiting, diarrhea, or persistent myalgias.
It is important to know whether the patient continued to receive
Low-grade fevers resolved without intervention. Tests were sent for tick-borne (ehrlichiosis, babesiosis, RMSF, anaplasmosis), viral (EBV, West Nile virus, parvovirus, CMV, coxsackievirus, adenovirus), other bacterial and protozoal (syphilis, Coxiella, leptospirosis, Lyme, Giardia), and autoimmune (antinuclear antibody, perinuclear antineutrophil cytoplasmic antibody, double-stranded DNA) diseases. Topical steroids and antihistamines were prescribed for a suspected viral exanthem. Empiric doxycycline was prescribed to treat possible tick-borne disease, and the patient was discharged home. At home, progressive darkening of the urine was noted. Outpatient testing demonstrated rising ALT to 377 U/L, AST to 183 U/L, total bilirubin to 5.9 mg/dL, direct bilirubin to 3.5 mg/dL, and alkaline phosphatase to 301 U/L. The patient was readmitted for further evaluation.
Despite concerns of the treating physicians, features of this case make tick-borne infections less likely. Lyme disease does not typically cause significant laboratory abnormalities and is classically associated with erythema migrans rather than a mucocutaneous rash. Relapsing fever, ehrlichioses, and rickettsial infections are associated with leukopenia and thrombocytopenia in addition to hepatocellular, rather than cholestatic, liver injury. The lack of response to doxycycline is helpful diagnostically: most tick-borne infections, in addition to leptospirosis, respond well to treatment. While babesiosis, tularemia, and Powassan or Heartland viruses transmitted by ticks are not treated with doxycycline, babesiosis often involves a hemolytic anemia (not seen in this case), and this patient’s laboratory abnormalities and rash are not characteristic of tularemia or viral tick-borne infections.
Either a new or reactivated viral infection with liver inflammation or an autoimmune etiology, specifically KD, remain the most likely etiology of the patient’s symptoms.
He remained asymptomatic during a 6-day hospitalization. His oral lesions resolved. The morbilliform rash coalesced into confluent macules with fine desquamation on the extremities and trunk. There was prominent periungual and palmar/plantar desquamation (Figure 3 and Figure 4). CBC demonstrated hemoglobin of 12.6 g/dL and platelets of 399,000/μL. CRP was undetectable at <0.5 mg/dL; however, ESR increased to 110 mm/h. Transaminases increased to ALT 551 U/L and AST 219 U/L. Serum alkaline phosphatase and bilirubin decreased without intervention. Albumin and total protein remained unchanged. All infectious and autoimmune testing sent from the prior admission returned negative.

An acute-onset viral-like prodrome with fevers potentially responsive to steroids, followed by conjunctivitis, oral erythema and cracked lips, morbilliform rash with hand and foot erythema and edema, cholestatic hepatitis, and subsequent periungual desquamation is highly suggestive of KD. It would be interesting to revisit the patient’s prior episode of aseptic meningitis to see whether any other symptoms were suggestive of KD. While intravenous immunoglobulin (IVIg) and aspirin are standard therapies for the acute febrile phase of KD, the patient is now nearly 2 weeks into his clinical course, rendering their utility uncertain. Nonetheless, screening for coronary aneurysms should be pursued, which may help confirm the diagnosis.

Upon reviewing the evolution of the findings, a diagnosis of adult-onset KD was made. IVIg 2g/kg and aspirin 325 mg were administered. Echocardiogram did not show any evidence of coronary artery aneurysm, myocarditis, pericarditis, wall motion abnormalities, or pericardial effusion. Computed tomography (CT) coronary angiogram confirmed normal coronary arteries without aneurysm. The patient was discharged home without fever on daily aspirin, and all hepatic chemistries and inflammatory markers normalized. Follow-up cardiac magnetic resonance imaging at 3 months and CT angiogram at 6 months remained normal. The patient remains well now 2 years after the original diagnosis and treatment.
DISCUSSION
KD, also known as mucocutaneous lymph node syndrome, is a vasculitis that typically affects children younger than 5 years.1 Having a sibling with KD confers a 10- to 15-fold higher risk, suggesting a genetic component to the disease.2 The highest incidence of KD is in persons of East Asian descent, but KD can affect patients of all races and ethnicities. In the United States, the majority of patients with KD are non-Hispanic White, followed by Black, Hispanic, and Asian.3 The etiology is still unknown, but it is posited that an unidentified, ubiquitous infectious agent may trigger KD in genetically susceptible individuals.4
KD can cause aneurysms and thromboses in medium-sized blood vessels throughout the body.5,6 The classic presentation involves 5 days of high fever plus four or more of the symptoms in the mnemonic CRASH: conjunctival injection, rash (polymorphous), adenopathy (cervical), strawberry tongue (or red, cracked lips and oropharyngeal edema), hand (erythema and induration of hands or feet, followed by periungual desquamation).7 Multiple organ systems may be affected, manifesting as abdominal pain, arthritis, pneumonitis, aseptic meningitis, and acalculous distention of the gallbladder (hydrops).7 The most feared consequence is coronary artery involvement, which leads to aneurysm, thrombosis, and sudden death.
Though no definitive diagnostic test exists, certain laboratory findings support the diagnosis, such as sterile pyuria, thrombocytosis, elevated CRP and ESR, transaminitis, and hypoalbuminemia.7 Diagnosis requires exclusion of illnesses with similar presentations, such as bacterial, viral, and tick-borne infections; drug hypersensitivity reactions; toxic shock syndrome; scarlet fever; juvenile rheumatoid arthritis; and other rheumatologic conditions. Some cases of KD present with fewer than four of the principal (CRASH) symptoms—these are termed “incomplete” KD. The combination of supportive laboratory findings and echocardiogram can facilitate diagnosis of incomplete KD, which carries a similar risk of coronary artery aneurysm.7
Though primarily a disease of childhood, KD can present in adults.8 Adults, compared with children, are less likely to have thrombocytosis and more likely to have cervical adenopathy, arthralgias, and hepatic test abnormalities.8 Although coronary artery aneurysms occur less frequently in adults compared with children, timely diagnosis and treatment is key to preventing this life-threatening complication.8
In children, treatment is IVIg 2 g/kg and aspirin 80 to 100 mg/kg daily until afebrile for several days.9 Some require a second dose of IVIg.9 Children are then maintained on 3 to 5 mg/kg of aspirin daily for 6 to 8 weeks.9 IVIg, given within 10 days of the onset of fever, is highly effective at preventing coronary artery aneurysms.10,11 When coronary aneurysms do occur, treatment is with aspirin or clopidogrel. Very large aneurysms require systemic anticoagulation. After the acute illness, children are monitored with serial cardiac imaging at 2 weeks and 6 to 8 weeks after diagnosis.7 In adults, the optimal imaging timing is unknown. Echocardiography often cannot visualize the coronary arteries, necessitating coronary CT angiography or cardiac MRI.
Despite the presence of classic features, this patient’s diagnosis was delayed because of the rarity of KD in adults and the need to exclude more common diseases. Furthermore, the administration of dexamethasone likely shortened his febrile period and ameliorated some symptoms,12 affecting the natural history of his illness. The diagnosis relied on three components: ruling out common diagnoses, noting two unusual findings (gallbladder hydrops, desquamating periungual rash), and broadening the differential to include adult presentations of childhood disease. Review of the literature suggests very few causes for gallbladder hydrops: impacted stones, cystic fibrosis, cystic duct narrowing due to tumor or lymph nodes, KD, and bacterial and parasitic disease (eg, salmonella, ascariasis). Gallbladder hydrops and periungual desquamation are seen together only in KD.13 Given the complexity of diagnosis in adults, the time to diagnosis is often delayed compared with that for children. While IVIg treatment is preferred within 10 days of the onset of fever, this patient received IVIg on day 14, given the relatively benign nature of IVIg and the considerable morbidity associated with coronary artery aneurysms. Dosing for aspirin is unclear in adults.8 This patient was started on 325 mg aspirin daily. He recovered fully and remains free of coronary changes at two years after initial diagnosis. This case is an excellent reminder that, after exclusion of common diagnoses, reflection on the most unusual aspects of the case and consideration of childhood diseases is particularly important in our younger patients.
TEACHING POINTS
- Extended fever should broaden the differential to include rheumatologic diagnoses.
- KD is rare in adults but can present with classic findings from childhood.
- Early treatment with IVIg and aspirin can be lifesaving in patients with KD, including adults.
1. Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Article in Japanese. Arerugi. 1967;16(3):178-222.
2. Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. 2005;9(4):185-194. https://doi.org/10.1016/j.ijid.2005.03.002
3. Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis J. 2010;29(6):483-488. https://doi.org/10.1097/INF.0b013e3181cf8705
4. Rowley A, Baker S, Arollo D, et al. A hepacivirus-like protein is targeted by the antibody response to Kawasaki disease (KD) [abstract]. Open Forum Infect Dis. 2019;6(suppl 2):S48.
5. Friedman KG, Gauvreau K, Hamaoka-Okamoto A, et al. Coronary artery aneurysms in Kawasaki disease: risk factors for progressive disease and adverse cardiac events in the US population. J Am Heart Assoc. 2016;5(9):e003289. https://doi.org/10.1161/JAHA.116.003289
6. Zhao QM, Chu C, Wu L, et al. Systemic artery aneurysms and Kawasaki disease. Pediatrics. 2019;144(6):e20192254. https://doi.org/10.1542/peds.2019-2254
7. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114(6):1708-1733. https://doi.org/10.1542/peds.2004-2182
8. Sève P, Stankovic K, Smail A, Durand DV, Marchand G, Broussolle C. Adult Kawasaki disease: report of two cases and literature review. Semin Arthritis Rheum. 2005;34(6):785-792. https://doi.org/10.1016/j.semarthrit.2005.01.012
9. Shulman ST. Intravenous immunoglobulin for the treatment of Kawasaki disease. Pediatr Ann. 2017;46(1):e25-e28. https://doi.org/10.3928/19382359-20161212-01
10. Newburger JW, Takahashi M, Burns JC, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. 1986;315(6):341-347. https://doi.org/10.1056/NEJM198608073150601
11. Rowley AH, Duffy CE, Shulman ST. Prevention of giant coronary artery aneurysms in Kawasaki disease by intravenous gamma globulin therapy. J Pediatr. 1988;113(2):290-294. https://doi/org/10.1016/s0022-3476(88)80267-1
12. Lim YJ, Jung JW. Clinical outcomes of initial dexamethasone treatment combined with a single high dose of intravenous immunoglobulin for primary treatment of Kawasaki disease. Yonsei Med J. 2014;55(5):1260-1266. https://doi.org/10.3349/ymj.2014.55.5.1260
13. Sun Q, Zhang J, Yang Y. Gallbladder hydrops associated with Kawasaki disease: a case report and literature review. Clin Pediatr (Phila). 2018;57(3):341-343. https://doi.org/10.1177/0009922817696468
This icon represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 39-year-old previously healthy man presented to the emergency department (ED) with abrupt-onset fever, headache, back pain, myalgias, chills, and photophobia. His past medical history included seasonal allergies and an episode of aseptic meningitis 8 years prior. He denied cough, dysuria, weakness, numbness, or visual changes. He denied using tobacco or injection drugs and rarely drank alcohol. His only medication was acetaminophen for fever.
The patient’s sudden fever indicates the rapid onset of an inflammatory state. While the headache and photophobia might be a result of an underlying systemic infection or an irritant like blood in the cerebral spinal fluid (CSF), one must consider meningitis. Potential sources for sudden meningitis include infectious, autoimmune (rheumatoid arthritis, systemic lupus erythematosus [SLE]), or drug-induced aseptic meningitis, and structural etiologies (ruptured cyst). Recrudescence of prior disease may also present acutely (Mollaret meningitis). Malignant etiologies, being more indolent, seem less likely. Back pain may indicate an epidural inflammatory process like epidural abscess; however, the patient denies risk factors such as injection drug use or recent procedures.
The patient’s temperature was 101.2 °F; blood pressure, 120/72 mm Hg; and heart rate, 112 bpm. He appeared comfortable, without meningismus or spinal tenderness. Pupils were reactive; eyes were without icterus, injection, or suffusion. Cardiac exam was normal. Lungs were clear to auscultation. He had no abdominal tenderness, hepatosplenomegaly, or lymphadenopathy. Cranial nerves II through XII, balance, coordination, strength, and sensation were intact. No rash was noted. Complete blood count (CBC), basic and hepatic chemistry panels, urinalysis, and serum lactate tests were within normal limits. Erythrocyte sedimentation rate (ESR) was elevated to 15 mm/h (normal range, 3-10 mm/h), C-reactive protein (CRP) to 2.4 mg/dL (normal range, <0.5 mg/dL), and procalcitonin to 0.07 ng/mL (normal range, <0.05 ng/mL). The patient was treated with intravenous (IV) fluids, ketorolac, dexamethasone, and acetaminophen, with resolution of symptoms. Given his rapid improvement, absence of meningismus, and lack of immunocompromise, lumbar puncture was deferred. A diagnosis of nonspecific viral syndrome was made. He was discharged home.
Certainly, a systemic infection (eg, influenza, adenovirus, arbovirus-related infection, HIV) could be a cause of this patient’s presentation. Notably, less than two-thirds of patients with meningitis present with the classic triad of fever, neck stiffness, and altered mental status. In this patient with fever, headache, and photophobia, aseptic meningitis should still be considered. While the negative procalcitonin and rapid clinical improvement without antibiotics make acute bacterial meningitis unlikely, nonbacterial causes of meningeal irritation can be severe and life-threatening. An assessment for jolt accentuation of the headache might have been helpful. Information about time of year, geographic exposures (vector-borne infections), and sick contacts (viral illness) can inform the clinical decision to pursue lumbar puncture. Additional history regarding his previous aseptic meningitis would be helpful, as it could suggest a recurrent inflammatory process. Causes of recurrent aseptic meningitis include infectious (herpes simplex virus [HSV], Epstein-Barr virus [EBV], syphilis), drug-related (nonsteroidal anti-inflammatory drugs [NSAIDs]), structural (epidermoid cyst with rupture), and autoimmune (lupus, Sjögren syndrome, Behçet disease) etiologies.
The mildly elevated inflammatory markers are nonspecific and reflect the patient’s known inflammatory state. The dexamethasone given for symptomatic management may have had some therapeutic effect in the setting of an autoimmune process, with additional contribution from ketorolac and acetaminophen.
He returned to the ED 3 days later with a pruritic, disseminated rash involving his palms and soles, accompanied by hand swelling and tingling. Although his headache and photophobia resolved, he reported a productive cough, nasal congestion, and sore throat. He also reported orange-pink urine without dysuria or urinary frequency. Additional questioning revealed a recent motorcycle trip to the Great Lakes region. During this trip, he did not camp, interact with animals or ticks, or swim in streams or lakes. He did not eat any raw, undercooked, or locally hunted meats. He denied new medications, soaps or detergents, or sexual contacts. He had started taking acetaminophen and ibuprofen around the clock since prior discharge.
The orange-pink urine and acute-onset palmoplantar rash with recent fever help narrow the differential. Orange-pink urine might suggest bilirubinuria from liver injury, hemolysis with hemoglobinuria, or myoglobinuria. Most concerning would be hematuria associated with glomerular injury and a systemic vasculopathy.
The rash on the palms and soles should be further characterized as blanching or nonblanching. Blanching, indicating vasodilation of intact blood vessels, is seen with many drug eruptions and viral exanthems. Nonblanching, suggesting broken capillaries (petechiae or purpura), would suggest vasculitis or vasculopathy from emboli, infection, or inflammation. A palmoplantar rash in febrile illness should first prompt evaluation for life-threatening conditions, followed by consideration of both infectious and noninfectious etiologies. Acutely fatal infections include Rocky Mountain spotted fever (RMSF), meningococcemia, toxic shock syndrome, infective endocarditis, and rat-bite fever. The rash, fever, headache, and outdoor exposure raise the possibility of a rickettsial infection, including RMSF, which can be contracted rarely around the Great Lakes. Other life-threatening infections seem unlikely, as the patient would have significantly deteriorated without proper medical care by now. Palmoplantar rash with fever can also be seen in other bacterial infections (eg, secondary syphilis, arbovirus infections, typhus) and in viral infections (eg, cytomegalovirus [CMV], EBV, human herpesvirus-6 [HHV-6], HIV, coxsackievirus, and papular-purpuric gloves and socks syndrome caused by parvovirus B19). Noninfectious considerations include drug hypersensitivity rashes, neoplasm (eg, cutaneous T-cell lymphoma), or inflammatory conditions (eg, SLE, vasculitis). Drug reaction with eosinophilia and systemic symptoms (may also present with severe illness.
The acetaminophen and ibuprofen may be masking ongoing fevers. The cough, nasal congestion, and sore throat might be part of a viral prodrome or, in tandem with fever, associated with a vasculitis such as granulomatosis with polyangiitis.
Vital signs were normal, and the patient appeared nontoxic. Physical examination demonstrated mildly cracked lips, oropharyngeal erythema with small petechiae on the soft palate, a morbilliform rash throughout his extremities and trunk (Figure 1), and confluent, brightly erythematous patches on his palms and soles with associated edema (Figure 2 and Appendix Figure). No lymphadenopathy, hepatosplenomegaly, or joint swelling was noted. CBC and basic chemistry panel remained normal; however, hepatic chemistries were notable for alanine aminotransferase (ALT) of 128 U/L, aspartate aminotransferase (AST) of 49 U/L, total bilirubin of 3.7 mg/dL, direct bilirubin of 2.4 mg/dL, total protein of 7.1 g/dL, albumin of 4.1 g/dL, and alkaline phosphatase of 197 U/L. Urinalysis detected bilirubin without blood, protein, bacteria, cells, or casts. The patient was admitted to the hospital.
The patient now has acute-onset upper respiratory symptoms with oral mucosal erythema, edema and erythema of the hands and feet with morbilliform rash of the extremities, and liver injury causing bilirubinuria. The patient’s initial symptoms may have had some response to therapy, but the current presentation suggests ongoing evolution of disease. Reactive infectious mucocutaneous eruptions include chlamydia, influenza, parainfluenza, and enteroviruses. Measles is possible given its recent resurgence; however, absence of coryza or Koplik spots and the peripheral distribution of the rash without initial truncal involvement make this less likely. Mycoplasma pneumonia–induced rash and mucositis might present with respiratory symptoms and this rash distribution, but typically involves two or more mucosal sites.
Iatrogenic causes are important to consider given the recent exposure to NSAIDs, specifically Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN). In this patient, however, SJS/TEN is unlikely as it typically presents 1 to 3 weeks after exposure, with a truncal-predominant rash rarely involving the palms and soles.
Despite the absence of conjunctivitis and cervical lymphadenopathy, one additional consideration is Kawasaki disease (KD). Though more common in children, it may rarely present in adulthood. The time course of manifesting symptoms with potential steroid responsiveness raises suspicion for this diagnosis.
During a 4-day hospitalization, he developed mild bilateral conjunctivitis, peeling lips, and scleral icterus. CBC remained within normal limits. A peripheral smear demonstrated toxic neutrophilic granulation with normal erythrocytes and platelets. HIV and hepatitis A, B, and C serologies were negative. Blood cultures were negative. CRP and ESR increased to 4.3 mg/dL and 56 mm/h, respectively. Hepatic chemistries increased to ALT 155 U/L, AST 101 U/L, total bilirubin 5.1 mg/dL, direct bilirubin 3.3 mg/dL, and alkaline phosphatase 211 U/L. Right upper-quadrant ultrasound demonstrated gallbladder distention (11.3 cm × 5.0 cm; normal, 10.0 cm × 4.0 cm) without stones, wall thickening, or pericholecystic fluid; sonographic Murphy sign was negative. The liver was unremarkable with normal flow in the portal vein.
The patient’s persistent reactive neutrophilic granulation and rising CRP and ESR indicate ongoing inflammation. The largely direct hyperbilirubinemia with hepatitis, minimal findings on ultrasound imaging, and lack of Murphy sign suggest either direct infection of the liver or cholestasis. Viral serologies for EBV, HSV, and CMV should be sent, although these viruses are less commonly associated with oral rash and conjunctivitis. The marked degree of cholestasis makes adenovirus and mycoplasma less likely. Leptospirosis should be considered given the degree of liver injury with potential conjunctival suffusion. However, oral involvement would be atypical; renal injury is absent; and the patient denied pertinent exposures, vomiting, diarrhea, or persistent myalgias.
It is important to know whether the patient continued to receive
Low-grade fevers resolved without intervention. Tests were sent for tick-borne (ehrlichiosis, babesiosis, RMSF, anaplasmosis), viral (EBV, West Nile virus, parvovirus, CMV, coxsackievirus, adenovirus), other bacterial and protozoal (syphilis, Coxiella, leptospirosis, Lyme, Giardia), and autoimmune (antinuclear antibody, perinuclear antineutrophil cytoplasmic antibody, double-stranded DNA) diseases. Topical steroids and antihistamines were prescribed for a suspected viral exanthem. Empiric doxycycline was prescribed to treat possible tick-borne disease, and the patient was discharged home. At home, progressive darkening of the urine was noted. Outpatient testing demonstrated rising ALT to 377 U/L, AST to 183 U/L, total bilirubin to 5.9 mg/dL, direct bilirubin to 3.5 mg/dL, and alkaline phosphatase to 301 U/L. The patient was readmitted for further evaluation.
Despite concerns of the treating physicians, features of this case make tick-borne infections less likely. Lyme disease does not typically cause significant laboratory abnormalities and is classically associated with erythema migrans rather than a mucocutaneous rash. Relapsing fever, ehrlichioses, and rickettsial infections are associated with leukopenia and thrombocytopenia in addition to hepatocellular, rather than cholestatic, liver injury. The lack of response to doxycycline is helpful diagnostically: most tick-borne infections, in addition to leptospirosis, respond well to treatment. While babesiosis, tularemia, and Powassan or Heartland viruses transmitted by ticks are not treated with doxycycline, babesiosis often involves a hemolytic anemia (not seen in this case), and this patient’s laboratory abnormalities and rash are not characteristic of tularemia or viral tick-borne infections.
Either a new or reactivated viral infection with liver inflammation or an autoimmune etiology, specifically KD, remain the most likely etiology of the patient’s symptoms.
He remained asymptomatic during a 6-day hospitalization. His oral lesions resolved. The morbilliform rash coalesced into confluent macules with fine desquamation on the extremities and trunk. There was prominent periungual and palmar/plantar desquamation (Figure 3 and Figure 4). CBC demonstrated hemoglobin of 12.6 g/dL and platelets of 399,000/μL. CRP was undetectable at <0.5 mg/dL; however, ESR increased to 110 mm/h. Transaminases increased to ALT 551 U/L and AST 219 U/L. Serum alkaline phosphatase and bilirubin decreased without intervention. Albumin and total protein remained unchanged. All infectious and autoimmune testing sent from the prior admission returned negative.
An acute-onset viral-like prodrome with fevers potentially responsive to steroids, followed by conjunctivitis, oral erythema and cracked lips, morbilliform rash with hand and foot erythema and edema, cholestatic hepatitis, and subsequent periungual desquamation is highly suggestive of KD. It would be interesting to revisit the patient’s prior episode of aseptic meningitis to see whether any other symptoms were suggestive of KD. While intravenous immunoglobulin (IVIg) and aspirin are standard therapies for the acute febrile phase of KD, the patient is now nearly 2 weeks into his clinical course, rendering their utility uncertain. Nonetheless, screening for coronary aneurysms should be pursued, which may help confirm the diagnosis.
Upon reviewing the evolution of the findings, a diagnosis of adult-onset KD was made. IVIg 2g/kg and aspirin 325 mg were administered. Echocardiogram did not show any evidence of coronary artery aneurysm, myocarditis, pericarditis, wall motion abnormalities, or pericardial effusion. Computed tomography (CT) coronary angiogram confirmed normal coronary arteries without aneurysm. The patient was discharged home without fever on daily aspirin, and all hepatic chemistries and inflammatory markers normalized. Follow-up cardiac magnetic resonance imaging at 3 months and CT angiogram at 6 months remained normal. The patient remains well now 2 years after the original diagnosis and treatment.
DISCUSSION
KD, also known as mucocutaneous lymph node syndrome, is a vasculitis that typically affects children younger than 5 years.1 Having a sibling with KD confers a 10- to 15-fold higher risk, suggesting a genetic component to the disease.2 The highest incidence of KD is in persons of East Asian descent, but KD can affect patients of all races and ethnicities. In the United States, the majority of patients with KD are non-Hispanic White, followed by Black, Hispanic, and Asian.3 The etiology is still unknown, but it is posited that an unidentified, ubiquitous infectious agent may trigger KD in genetically susceptible individuals.4
KD can cause aneurysms and thromboses in medium-sized blood vessels throughout the body.5,6 The classic presentation involves 5 days of high fever plus four or more of the symptoms in the mnemonic CRASH: conjunctival injection, rash (polymorphous), adenopathy (cervical), strawberry tongue (or red, cracked lips and oropharyngeal edema), hand (erythema and induration of hands or feet, followed by periungual desquamation).7 Multiple organ systems may be affected, manifesting as abdominal pain, arthritis, pneumonitis, aseptic meningitis, and acalculous distention of the gallbladder (hydrops).7 The most feared consequence is coronary artery involvement, which leads to aneurysm, thrombosis, and sudden death.
Though no definitive diagnostic test exists, certain laboratory findings support the diagnosis, such as sterile pyuria, thrombocytosis, elevated CRP and ESR, transaminitis, and hypoalbuminemia.7 Diagnosis requires exclusion of illnesses with similar presentations, such as bacterial, viral, and tick-borne infections; drug hypersensitivity reactions; toxic shock syndrome; scarlet fever; juvenile rheumatoid arthritis; and other rheumatologic conditions. Some cases of KD present with fewer than four of the principal (CRASH) symptoms—these are termed “incomplete” KD. The combination of supportive laboratory findings and echocardiogram can facilitate diagnosis of incomplete KD, which carries a similar risk of coronary artery aneurysm.7
Though primarily a disease of childhood, KD can present in adults.8 Adults, compared with children, are less likely to have thrombocytosis and more likely to have cervical adenopathy, arthralgias, and hepatic test abnormalities.8 Although coronary artery aneurysms occur less frequently in adults compared with children, timely diagnosis and treatment is key to preventing this life-threatening complication.8
In children, treatment is IVIg 2 g/kg and aspirin 80 to 100 mg/kg daily until afebrile for several days.9 Some require a second dose of IVIg.9 Children are then maintained on 3 to 5 mg/kg of aspirin daily for 6 to 8 weeks.9 IVIg, given within 10 days of the onset of fever, is highly effective at preventing coronary artery aneurysms.10,11 When coronary aneurysms do occur, treatment is with aspirin or clopidogrel. Very large aneurysms require systemic anticoagulation. After the acute illness, children are monitored with serial cardiac imaging at 2 weeks and 6 to 8 weeks after diagnosis.7 In adults, the optimal imaging timing is unknown. Echocardiography often cannot visualize the coronary arteries, necessitating coronary CT angiography or cardiac MRI.
Despite the presence of classic features, this patient’s diagnosis was delayed because of the rarity of KD in adults and the need to exclude more common diseases. Furthermore, the administration of dexamethasone likely shortened his febrile period and ameliorated some symptoms,12 affecting the natural history of his illness. The diagnosis relied on three components: ruling out common diagnoses, noting two unusual findings (gallbladder hydrops, desquamating periungual rash), and broadening the differential to include adult presentations of childhood disease. Review of the literature suggests very few causes for gallbladder hydrops: impacted stones, cystic fibrosis, cystic duct narrowing due to tumor or lymph nodes, KD, and bacterial and parasitic disease (eg, salmonella, ascariasis). Gallbladder hydrops and periungual desquamation are seen together only in KD.13 Given the complexity of diagnosis in adults, the time to diagnosis is often delayed compared with that for children. While IVIg treatment is preferred within 10 days of the onset of fever, this patient received IVIg on day 14, given the relatively benign nature of IVIg and the considerable morbidity associated with coronary artery aneurysms. Dosing for aspirin is unclear in adults.8 This patient was started on 325 mg aspirin daily. He recovered fully and remains free of coronary changes at two years after initial diagnosis. This case is an excellent reminder that, after exclusion of common diagnoses, reflection on the most unusual aspects of the case and consideration of childhood diseases is particularly important in our younger patients.
TEACHING POINTS
- Extended fever should broaden the differential to include rheumatologic diagnoses.
- KD is rare in adults but can present with classic findings from childhood.
- Early treatment with IVIg and aspirin can be lifesaving in patients with KD, including adults.
This icon represents the patient’s case. Each paragraph that follows represents the discussant’s thoughts.
A 39-year-old previously healthy man presented to the emergency department (ED) with abrupt-onset fever, headache, back pain, myalgias, chills, and photophobia. His past medical history included seasonal allergies and an episode of aseptic meningitis 8 years prior. He denied cough, dysuria, weakness, numbness, or visual changes. He denied using tobacco or injection drugs and rarely drank alcohol. His only medication was acetaminophen for fever.
The patient’s sudden fever indicates the rapid onset of an inflammatory state. While the headache and photophobia might be a result of an underlying systemic infection or an irritant like blood in the cerebral spinal fluid (CSF), one must consider meningitis. Potential sources for sudden meningitis include infectious, autoimmune (rheumatoid arthritis, systemic lupus erythematosus [SLE]), or drug-induced aseptic meningitis, and structural etiologies (ruptured cyst). Recrudescence of prior disease may also present acutely (Mollaret meningitis). Malignant etiologies, being more indolent, seem less likely. Back pain may indicate an epidural inflammatory process like epidural abscess; however, the patient denies risk factors such as injection drug use or recent procedures.
The patient’s temperature was 101.2 °F; blood pressure, 120/72 mm Hg; and heart rate, 112 bpm. He appeared comfortable, without meningismus or spinal tenderness. Pupils were reactive; eyes were without icterus, injection, or suffusion. Cardiac exam was normal. Lungs were clear to auscultation. He had no abdominal tenderness, hepatosplenomegaly, or lymphadenopathy. Cranial nerves II through XII, balance, coordination, strength, and sensation were intact. No rash was noted. Complete blood count (CBC), basic and hepatic chemistry panels, urinalysis, and serum lactate tests were within normal limits. Erythrocyte sedimentation rate (ESR) was elevated to 15 mm/h (normal range, 3-10 mm/h), C-reactive protein (CRP) to 2.4 mg/dL (normal range, <0.5 mg/dL), and procalcitonin to 0.07 ng/mL (normal range, <0.05 ng/mL). The patient was treated with intravenous (IV) fluids, ketorolac, dexamethasone, and acetaminophen, with resolution of symptoms. Given his rapid improvement, absence of meningismus, and lack of immunocompromise, lumbar puncture was deferred. A diagnosis of nonspecific viral syndrome was made. He was discharged home.
Certainly, a systemic infection (eg, influenza, adenovirus, arbovirus-related infection, HIV) could be a cause of this patient’s presentation. Notably, less than two-thirds of patients with meningitis present with the classic triad of fever, neck stiffness, and altered mental status. In this patient with fever, headache, and photophobia, aseptic meningitis should still be considered. While the negative procalcitonin and rapid clinical improvement without antibiotics make acute bacterial meningitis unlikely, nonbacterial causes of meningeal irritation can be severe and life-threatening. An assessment for jolt accentuation of the headache might have been helpful. Information about time of year, geographic exposures (vector-borne infections), and sick contacts (viral illness) can inform the clinical decision to pursue lumbar puncture. Additional history regarding his previous aseptic meningitis would be helpful, as it could suggest a recurrent inflammatory process. Causes of recurrent aseptic meningitis include infectious (herpes simplex virus [HSV], Epstein-Barr virus [EBV], syphilis), drug-related (nonsteroidal anti-inflammatory drugs [NSAIDs]), structural (epidermoid cyst with rupture), and autoimmune (lupus, Sjögren syndrome, Behçet disease) etiologies.
The mildly elevated inflammatory markers are nonspecific and reflect the patient’s known inflammatory state. The dexamethasone given for symptomatic management may have had some therapeutic effect in the setting of an autoimmune process, with additional contribution from ketorolac and acetaminophen.
He returned to the ED 3 days later with a pruritic, disseminated rash involving his palms and soles, accompanied by hand swelling and tingling. Although his headache and photophobia resolved, he reported a productive cough, nasal congestion, and sore throat. He also reported orange-pink urine without dysuria or urinary frequency. Additional questioning revealed a recent motorcycle trip to the Great Lakes region. During this trip, he did not camp, interact with animals or ticks, or swim in streams or lakes. He did not eat any raw, undercooked, or locally hunted meats. He denied new medications, soaps or detergents, or sexual contacts. He had started taking acetaminophen and ibuprofen around the clock since prior discharge.
The orange-pink urine and acute-onset palmoplantar rash with recent fever help narrow the differential. Orange-pink urine might suggest bilirubinuria from liver injury, hemolysis with hemoglobinuria, or myoglobinuria. Most concerning would be hematuria associated with glomerular injury and a systemic vasculopathy.
The rash on the palms and soles should be further characterized as blanching or nonblanching. Blanching, indicating vasodilation of intact blood vessels, is seen with many drug eruptions and viral exanthems. Nonblanching, suggesting broken capillaries (petechiae or purpura), would suggest vasculitis or vasculopathy from emboli, infection, or inflammation. A palmoplantar rash in febrile illness should first prompt evaluation for life-threatening conditions, followed by consideration of both infectious and noninfectious etiologies. Acutely fatal infections include Rocky Mountain spotted fever (RMSF), meningococcemia, toxic shock syndrome, infective endocarditis, and rat-bite fever. The rash, fever, headache, and outdoor exposure raise the possibility of a rickettsial infection, including RMSF, which can be contracted rarely around the Great Lakes. Other life-threatening infections seem unlikely, as the patient would have significantly deteriorated without proper medical care by now. Palmoplantar rash with fever can also be seen in other bacterial infections (eg, secondary syphilis, arbovirus infections, typhus) and in viral infections (eg, cytomegalovirus [CMV], EBV, human herpesvirus-6 [HHV-6], HIV, coxsackievirus, and papular-purpuric gloves and socks syndrome caused by parvovirus B19). Noninfectious considerations include drug hypersensitivity rashes, neoplasm (eg, cutaneous T-cell lymphoma), or inflammatory conditions (eg, SLE, vasculitis). Drug reaction with eosinophilia and systemic symptoms (may also present with severe illness.
The acetaminophen and ibuprofen may be masking ongoing fevers. The cough, nasal congestion, and sore throat might be part of a viral prodrome or, in tandem with fever, associated with a vasculitis such as granulomatosis with polyangiitis.
Vital signs were normal, and the patient appeared nontoxic. Physical examination demonstrated mildly cracked lips, oropharyngeal erythema with small petechiae on the soft palate, a morbilliform rash throughout his extremities and trunk (Figure 1), and confluent, brightly erythematous patches on his palms and soles with associated edema (Figure 2 and Appendix Figure). No lymphadenopathy, hepatosplenomegaly, or joint swelling was noted. CBC and basic chemistry panel remained normal; however, hepatic chemistries were notable for alanine aminotransferase (ALT) of 128 U/L, aspartate aminotransferase (AST) of 49 U/L, total bilirubin of 3.7 mg/dL, direct bilirubin of 2.4 mg/dL, total protein of 7.1 g/dL, albumin of 4.1 g/dL, and alkaline phosphatase of 197 U/L. Urinalysis detected bilirubin without blood, protein, bacteria, cells, or casts. The patient was admitted to the hospital.
The patient now has acute-onset upper respiratory symptoms with oral mucosal erythema, edema and erythema of the hands and feet with morbilliform rash of the extremities, and liver injury causing bilirubinuria. The patient’s initial symptoms may have had some response to therapy, but the current presentation suggests ongoing evolution of disease. Reactive infectious mucocutaneous eruptions include chlamydia, influenza, parainfluenza, and enteroviruses. Measles is possible given its recent resurgence; however, absence of coryza or Koplik spots and the peripheral distribution of the rash without initial truncal involvement make this less likely. Mycoplasma pneumonia–induced rash and mucositis might present with respiratory symptoms and this rash distribution, but typically involves two or more mucosal sites.
Iatrogenic causes are important to consider given the recent exposure to NSAIDs, specifically Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN). In this patient, however, SJS/TEN is unlikely as it typically presents 1 to 3 weeks after exposure, with a truncal-predominant rash rarely involving the palms and soles.
Despite the absence of conjunctivitis and cervical lymphadenopathy, one additional consideration is Kawasaki disease (KD). Though more common in children, it may rarely present in adulthood. The time course of manifesting symptoms with potential steroid responsiveness raises suspicion for this diagnosis.
During a 4-day hospitalization, he developed mild bilateral conjunctivitis, peeling lips, and scleral icterus. CBC remained within normal limits. A peripheral smear demonstrated toxic neutrophilic granulation with normal erythrocytes and platelets. HIV and hepatitis A, B, and C serologies were negative. Blood cultures were negative. CRP and ESR increased to 4.3 mg/dL and 56 mm/h, respectively. Hepatic chemistries increased to ALT 155 U/L, AST 101 U/L, total bilirubin 5.1 mg/dL, direct bilirubin 3.3 mg/dL, and alkaline phosphatase 211 U/L. Right upper-quadrant ultrasound demonstrated gallbladder distention (11.3 cm × 5.0 cm; normal, 10.0 cm × 4.0 cm) without stones, wall thickening, or pericholecystic fluid; sonographic Murphy sign was negative. The liver was unremarkable with normal flow in the portal vein.
The patient’s persistent reactive neutrophilic granulation and rising CRP and ESR indicate ongoing inflammation. The largely direct hyperbilirubinemia with hepatitis, minimal findings on ultrasound imaging, and lack of Murphy sign suggest either direct infection of the liver or cholestasis. Viral serologies for EBV, HSV, and CMV should be sent, although these viruses are less commonly associated with oral rash and conjunctivitis. The marked degree of cholestasis makes adenovirus and mycoplasma less likely. Leptospirosis should be considered given the degree of liver injury with potential conjunctival suffusion. However, oral involvement would be atypical; renal injury is absent; and the patient denied pertinent exposures, vomiting, diarrhea, or persistent myalgias.
It is important to know whether the patient continued to receive
Low-grade fevers resolved without intervention. Tests were sent for tick-borne (ehrlichiosis, babesiosis, RMSF, anaplasmosis), viral (EBV, West Nile virus, parvovirus, CMV, coxsackievirus, adenovirus), other bacterial and protozoal (syphilis, Coxiella, leptospirosis, Lyme, Giardia), and autoimmune (antinuclear antibody, perinuclear antineutrophil cytoplasmic antibody, double-stranded DNA) diseases. Topical steroids and antihistamines were prescribed for a suspected viral exanthem. Empiric doxycycline was prescribed to treat possible tick-borne disease, and the patient was discharged home. At home, progressive darkening of the urine was noted. Outpatient testing demonstrated rising ALT to 377 U/L, AST to 183 U/L, total bilirubin to 5.9 mg/dL, direct bilirubin to 3.5 mg/dL, and alkaline phosphatase to 301 U/L. The patient was readmitted for further evaluation.
Despite concerns of the treating physicians, features of this case make tick-borne infections less likely. Lyme disease does not typically cause significant laboratory abnormalities and is classically associated with erythema migrans rather than a mucocutaneous rash. Relapsing fever, ehrlichioses, and rickettsial infections are associated with leukopenia and thrombocytopenia in addition to hepatocellular, rather than cholestatic, liver injury. The lack of response to doxycycline is helpful diagnostically: most tick-borne infections, in addition to leptospirosis, respond well to treatment. While babesiosis, tularemia, and Powassan or Heartland viruses transmitted by ticks are not treated with doxycycline, babesiosis often involves a hemolytic anemia (not seen in this case), and this patient’s laboratory abnormalities and rash are not characteristic of tularemia or viral tick-borne infections.
Either a new or reactivated viral infection with liver inflammation or an autoimmune etiology, specifically KD, remain the most likely etiology of the patient’s symptoms.
He remained asymptomatic during a 6-day hospitalization. His oral lesions resolved. The morbilliform rash coalesced into confluent macules with fine desquamation on the extremities and trunk. There was prominent periungual and palmar/plantar desquamation (Figure 3 and Figure 4). CBC demonstrated hemoglobin of 12.6 g/dL and platelets of 399,000/μL. CRP was undetectable at <0.5 mg/dL; however, ESR increased to 110 mm/h. Transaminases increased to ALT 551 U/L and AST 219 U/L. Serum alkaline phosphatase and bilirubin decreased without intervention. Albumin and total protein remained unchanged. All infectious and autoimmune testing sent from the prior admission returned negative.
An acute-onset viral-like prodrome with fevers potentially responsive to steroids, followed by conjunctivitis, oral erythema and cracked lips, morbilliform rash with hand and foot erythema and edema, cholestatic hepatitis, and subsequent periungual desquamation is highly suggestive of KD. It would be interesting to revisit the patient’s prior episode of aseptic meningitis to see whether any other symptoms were suggestive of KD. While intravenous immunoglobulin (IVIg) and aspirin are standard therapies for the acute febrile phase of KD, the patient is now nearly 2 weeks into his clinical course, rendering their utility uncertain. Nonetheless, screening for coronary aneurysms should be pursued, which may help confirm the diagnosis.
Upon reviewing the evolution of the findings, a diagnosis of adult-onset KD was made. IVIg 2g/kg and aspirin 325 mg were administered. Echocardiogram did not show any evidence of coronary artery aneurysm, myocarditis, pericarditis, wall motion abnormalities, or pericardial effusion. Computed tomography (CT) coronary angiogram confirmed normal coronary arteries without aneurysm. The patient was discharged home without fever on daily aspirin, and all hepatic chemistries and inflammatory markers normalized. Follow-up cardiac magnetic resonance imaging at 3 months and CT angiogram at 6 months remained normal. The patient remains well now 2 years after the original diagnosis and treatment.
DISCUSSION
KD, also known as mucocutaneous lymph node syndrome, is a vasculitis that typically affects children younger than 5 years.1 Having a sibling with KD confers a 10- to 15-fold higher risk, suggesting a genetic component to the disease.2 The highest incidence of KD is in persons of East Asian descent, but KD can affect patients of all races and ethnicities. In the United States, the majority of patients with KD are non-Hispanic White, followed by Black, Hispanic, and Asian.3 The etiology is still unknown, but it is posited that an unidentified, ubiquitous infectious agent may trigger KD in genetically susceptible individuals.4
KD can cause aneurysms and thromboses in medium-sized blood vessels throughout the body.5,6 The classic presentation involves 5 days of high fever plus four or more of the symptoms in the mnemonic CRASH: conjunctival injection, rash (polymorphous), adenopathy (cervical), strawberry tongue (or red, cracked lips and oropharyngeal edema), hand (erythema and induration of hands or feet, followed by periungual desquamation).7 Multiple organ systems may be affected, manifesting as abdominal pain, arthritis, pneumonitis, aseptic meningitis, and acalculous distention of the gallbladder (hydrops).7 The most feared consequence is coronary artery involvement, which leads to aneurysm, thrombosis, and sudden death.
Though no definitive diagnostic test exists, certain laboratory findings support the diagnosis, such as sterile pyuria, thrombocytosis, elevated CRP and ESR, transaminitis, and hypoalbuminemia.7 Diagnosis requires exclusion of illnesses with similar presentations, such as bacterial, viral, and tick-borne infections; drug hypersensitivity reactions; toxic shock syndrome; scarlet fever; juvenile rheumatoid arthritis; and other rheumatologic conditions. Some cases of KD present with fewer than four of the principal (CRASH) symptoms—these are termed “incomplete” KD. The combination of supportive laboratory findings and echocardiogram can facilitate diagnosis of incomplete KD, which carries a similar risk of coronary artery aneurysm.7
Though primarily a disease of childhood, KD can present in adults.8 Adults, compared with children, are less likely to have thrombocytosis and more likely to have cervical adenopathy, arthralgias, and hepatic test abnormalities.8 Although coronary artery aneurysms occur less frequently in adults compared with children, timely diagnosis and treatment is key to preventing this life-threatening complication.8
In children, treatment is IVIg 2 g/kg and aspirin 80 to 100 mg/kg daily until afebrile for several days.9 Some require a second dose of IVIg.9 Children are then maintained on 3 to 5 mg/kg of aspirin daily for 6 to 8 weeks.9 IVIg, given within 10 days of the onset of fever, is highly effective at preventing coronary artery aneurysms.10,11 When coronary aneurysms do occur, treatment is with aspirin or clopidogrel. Very large aneurysms require systemic anticoagulation. After the acute illness, children are monitored with serial cardiac imaging at 2 weeks and 6 to 8 weeks after diagnosis.7 In adults, the optimal imaging timing is unknown. Echocardiography often cannot visualize the coronary arteries, necessitating coronary CT angiography or cardiac MRI.
Despite the presence of classic features, this patient’s diagnosis was delayed because of the rarity of KD in adults and the need to exclude more common diseases. Furthermore, the administration of dexamethasone likely shortened his febrile period and ameliorated some symptoms,12 affecting the natural history of his illness. The diagnosis relied on three components: ruling out common diagnoses, noting two unusual findings (gallbladder hydrops, desquamating periungual rash), and broadening the differential to include adult presentations of childhood disease. Review of the literature suggests very few causes for gallbladder hydrops: impacted stones, cystic fibrosis, cystic duct narrowing due to tumor or lymph nodes, KD, and bacterial and parasitic disease (eg, salmonella, ascariasis). Gallbladder hydrops and periungual desquamation are seen together only in KD.13 Given the complexity of diagnosis in adults, the time to diagnosis is often delayed compared with that for children. While IVIg treatment is preferred within 10 days of the onset of fever, this patient received IVIg on day 14, given the relatively benign nature of IVIg and the considerable morbidity associated with coronary artery aneurysms. Dosing for aspirin is unclear in adults.8 This patient was started on 325 mg aspirin daily. He recovered fully and remains free of coronary changes at two years after initial diagnosis. This case is an excellent reminder that, after exclusion of common diagnoses, reflection on the most unusual aspects of the case and consideration of childhood diseases is particularly important in our younger patients.
TEACHING POINTS
- Extended fever should broaden the differential to include rheumatologic diagnoses.
- KD is rare in adults but can present with classic findings from childhood.
- Early treatment with IVIg and aspirin can be lifesaving in patients with KD, including adults.
1. Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Article in Japanese. Arerugi. 1967;16(3):178-222.
2. Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. 2005;9(4):185-194. https://doi.org/10.1016/j.ijid.2005.03.002
3. Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis J. 2010;29(6):483-488. https://doi.org/10.1097/INF.0b013e3181cf8705
4. Rowley A, Baker S, Arollo D, et al. A hepacivirus-like protein is targeted by the antibody response to Kawasaki disease (KD) [abstract]. Open Forum Infect Dis. 2019;6(suppl 2):S48.
5. Friedman KG, Gauvreau K, Hamaoka-Okamoto A, et al. Coronary artery aneurysms in Kawasaki disease: risk factors for progressive disease and adverse cardiac events in the US population. J Am Heart Assoc. 2016;5(9):e003289. https://doi.org/10.1161/JAHA.116.003289
6. Zhao QM, Chu C, Wu L, et al. Systemic artery aneurysms and Kawasaki disease. Pediatrics. 2019;144(6):e20192254. https://doi.org/10.1542/peds.2019-2254
7. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114(6):1708-1733. https://doi.org/10.1542/peds.2004-2182
8. Sève P, Stankovic K, Smail A, Durand DV, Marchand G, Broussolle C. Adult Kawasaki disease: report of two cases and literature review. Semin Arthritis Rheum. 2005;34(6):785-792. https://doi.org/10.1016/j.semarthrit.2005.01.012
9. Shulman ST. Intravenous immunoglobulin for the treatment of Kawasaki disease. Pediatr Ann. 2017;46(1):e25-e28. https://doi.org/10.3928/19382359-20161212-01
10. Newburger JW, Takahashi M, Burns JC, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. 1986;315(6):341-347. https://doi.org/10.1056/NEJM198608073150601
11. Rowley AH, Duffy CE, Shulman ST. Prevention of giant coronary artery aneurysms in Kawasaki disease by intravenous gamma globulin therapy. J Pediatr. 1988;113(2):290-294. https://doi/org/10.1016/s0022-3476(88)80267-1
12. Lim YJ, Jung JW. Clinical outcomes of initial dexamethasone treatment combined with a single high dose of intravenous immunoglobulin for primary treatment of Kawasaki disease. Yonsei Med J. 2014;55(5):1260-1266. https://doi.org/10.3349/ymj.2014.55.5.1260
13. Sun Q, Zhang J, Yang Y. Gallbladder hydrops associated with Kawasaki disease: a case report and literature review. Clin Pediatr (Phila). 2018;57(3):341-343. https://doi.org/10.1177/0009922817696468
1. Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Article in Japanese. Arerugi. 1967;16(3):178-222.
2. Burgner D, Harnden A. Kawasaki disease: what is the epidemiology telling us about the etiology? Int J Infect Dis. 2005;9(4):185-194. https://doi.org/10.1016/j.ijid.2005.03.002
3. Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997-2007. Pediatr Infect Dis J. 2010;29(6):483-488. https://doi.org/10.1097/INF.0b013e3181cf8705
4. Rowley A, Baker S, Arollo D, et al. A hepacivirus-like protein is targeted by the antibody response to Kawasaki disease (KD) [abstract]. Open Forum Infect Dis. 2019;6(suppl 2):S48.
5. Friedman KG, Gauvreau K, Hamaoka-Okamoto A, et al. Coronary artery aneurysms in Kawasaki disease: risk factors for progressive disease and adverse cardiac events in the US population. J Am Heart Assoc. 2016;5(9):e003289. https://doi.org/10.1161/JAHA.116.003289
6. Zhao QM, Chu C, Wu L, et al. Systemic artery aneurysms and Kawasaki disease. Pediatrics. 2019;144(6):e20192254. https://doi.org/10.1542/peds.2019-2254
7. Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114(6):1708-1733. https://doi.org/10.1542/peds.2004-2182
8. Sève P, Stankovic K, Smail A, Durand DV, Marchand G, Broussolle C. Adult Kawasaki disease: report of two cases and literature review. Semin Arthritis Rheum. 2005;34(6):785-792. https://doi.org/10.1016/j.semarthrit.2005.01.012
9. Shulman ST. Intravenous immunoglobulin for the treatment of Kawasaki disease. Pediatr Ann. 2017;46(1):e25-e28. https://doi.org/10.3928/19382359-20161212-01
10. Newburger JW, Takahashi M, Burns JC, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. 1986;315(6):341-347. https://doi.org/10.1056/NEJM198608073150601
11. Rowley AH, Duffy CE, Shulman ST. Prevention of giant coronary artery aneurysms in Kawasaki disease by intravenous gamma globulin therapy. J Pediatr. 1988;113(2):290-294. https://doi/org/10.1016/s0022-3476(88)80267-1
12. Lim YJ, Jung JW. Clinical outcomes of initial dexamethasone treatment combined with a single high dose of intravenous immunoglobulin for primary treatment of Kawasaki disease. Yonsei Med J. 2014;55(5):1260-1266. https://doi.org/10.3349/ymj.2014.55.5.1260
13. Sun Q, Zhang J, Yang Y. Gallbladder hydrops associated with Kawasaki disease: a case report and literature review. Clin Pediatr (Phila). 2018;57(3):341-343. https://doi.org/10.1177/0009922817696468
© 2021 Society of Hospital Medicine
Metastatic pulmonary calcification and end-stage renal disease
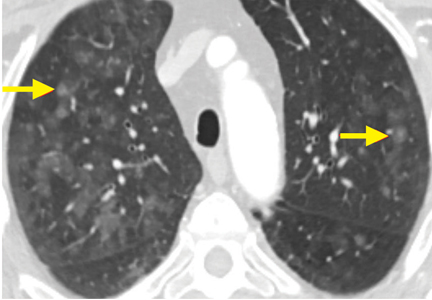
A 64-year-old man with end-stage renal disease was evaluated in the pulmonary clinic for persistent abnormalities on axial computed tomography (CT) of the chest. He was a lifelong nonsmoker and had no history of exposure to occupational dust or fumes. His oxygen saturation was 100% on room air, and he denied any respiratory symptoms.

, as did plain radiography of the elbow.")
WHEN TO CONSIDER METASTATIC PULMONARY CALCIFICATION
The differential diagnosis for chronic upper-lobe-predominant ground-glass nodules is broad and includes atypical infections, recurrent alveolar hemorrhage, hypersensitivity pneumonitis, vasculitis, sarcoidosis, chronic eosinophilic pneumonia, occupational lung disease, and pulmonary alveolar microlithiasis. However, several aspects of our patient’s case suggested an often overlooked diagnosis, metastatic pulmonary calcification.
Metastatic pulmonary calcification is caused by deposition of calcium salts in lung tissue and is most commonly seen in patients on dialysis,1,2 and our patient had been dependent on dialysis for many years. The chronically elevated calcium-phosphorus product and secondary hyperparathyroidism often seen with end-stage renal disease may explain this association.
Our patient’s lack of symptoms is also an important diagnostic clue. Unlike many other causes of chronic upper-lobe-predominant ground-glass nodules, metastatic pulmonary calcification does not usually cause symptoms and is often identified only at autopsy.3 Results of pulmonary function testing are often normal.4
Metastatic pulmonary calcification can appear as diffusely calcified nodules or high-attenuation areas of consolidation on CT. However, as in our patient’s case, CT may demonstrate fluffy, centrilobular ground-glass nodules due to the microscopic size of the deposited calcium crystals.1 Identifying calcified vessels on imaging supports the diagnosis.4
Treatment of metastatic pulmonary calcification in a patient with end-stage renal disease is focused on correcting underlying metabolic abnormalities with phosphate binders, vitamin D supplementation, and dialysis.
- Chan ED, Morales DV, Welsh CH, McDermott MT, Schwarz MI. Calcium deposition with or without bone formation in the lung. Am J Respir Crit Care Med 2002; 165:1654–1669.
- Beyzaei A, Francis J, Knight H, Simon DB, Finkelstein FO. Metabolic lung disease: diffuse metastatic pulmonary calcifications with progression to calciphylaxis in end-stage renal disease. Adv Perit Dial 2007; 23:112–117.
- Conger JD, Hammond WS, Alfrey AC, Contiguglia SR, Stanford RE, Huffer WE. Pulmonary calcification in chronic dialysis patients. Clinical and pathologic studies. Ann Intern Med 1975; 83:330–336.
- Belem LC, Zanetti G, Souza AS Jr, et al. Metastatic pulmonary calcification: state-of-the-art review focused on imaging findings. Respir Med 2014; 108:668–676.
A 64-year-old man with end-stage renal disease was evaluated in the pulmonary clinic for persistent abnormalities on axial computed tomography (CT) of the chest. He was a lifelong nonsmoker and had no history of exposure to occupational dust or fumes. His oxygen saturation was 100% on room air, and he denied any respiratory symptoms.
WHEN TO CONSIDER METASTATIC PULMONARY CALCIFICATION
The differential diagnosis for chronic upper-lobe-predominant ground-glass nodules is broad and includes atypical infections, recurrent alveolar hemorrhage, hypersensitivity pneumonitis, vasculitis, sarcoidosis, chronic eosinophilic pneumonia, occupational lung disease, and pulmonary alveolar microlithiasis. However, several aspects of our patient’s case suggested an often overlooked diagnosis, metastatic pulmonary calcification.
Metastatic pulmonary calcification is caused by deposition of calcium salts in lung tissue and is most commonly seen in patients on dialysis,1,2 and our patient had been dependent on dialysis for many years. The chronically elevated calcium-phosphorus product and secondary hyperparathyroidism often seen with end-stage renal disease may explain this association.
Our patient’s lack of symptoms is also an important diagnostic clue. Unlike many other causes of chronic upper-lobe-predominant ground-glass nodules, metastatic pulmonary calcification does not usually cause symptoms and is often identified only at autopsy.3 Results of pulmonary function testing are often normal.4
Metastatic pulmonary calcification can appear as diffusely calcified nodules or high-attenuation areas of consolidation on CT. However, as in our patient’s case, CT may demonstrate fluffy, centrilobular ground-glass nodules due to the microscopic size of the deposited calcium crystals.1 Identifying calcified vessels on imaging supports the diagnosis.4
Treatment of metastatic pulmonary calcification in a patient with end-stage renal disease is focused on correcting underlying metabolic abnormalities with phosphate binders, vitamin D supplementation, and dialysis.
A 64-year-old man with end-stage renal disease was evaluated in the pulmonary clinic for persistent abnormalities on axial computed tomography (CT) of the chest. He was a lifelong nonsmoker and had no history of exposure to occupational dust or fumes. His oxygen saturation was 100% on room air, and he denied any respiratory symptoms.
WHEN TO CONSIDER METASTATIC PULMONARY CALCIFICATION
The differential diagnosis for chronic upper-lobe-predominant ground-glass nodules is broad and includes atypical infections, recurrent alveolar hemorrhage, hypersensitivity pneumonitis, vasculitis, sarcoidosis, chronic eosinophilic pneumonia, occupational lung disease, and pulmonary alveolar microlithiasis. However, several aspects of our patient’s case suggested an often overlooked diagnosis, metastatic pulmonary calcification.
Metastatic pulmonary calcification is caused by deposition of calcium salts in lung tissue and is most commonly seen in patients on dialysis,1,2 and our patient had been dependent on dialysis for many years. The chronically elevated calcium-phosphorus product and secondary hyperparathyroidism often seen with end-stage renal disease may explain this association.
Our patient’s lack of symptoms is also an important diagnostic clue. Unlike many other causes of chronic upper-lobe-predominant ground-glass nodules, metastatic pulmonary calcification does not usually cause symptoms and is often identified only at autopsy.3 Results of pulmonary function testing are often normal.4
Metastatic pulmonary calcification can appear as diffusely calcified nodules or high-attenuation areas of consolidation on CT. However, as in our patient’s case, CT may demonstrate fluffy, centrilobular ground-glass nodules due to the microscopic size of the deposited calcium crystals.1 Identifying calcified vessels on imaging supports the diagnosis.4
Treatment of metastatic pulmonary calcification in a patient with end-stage renal disease is focused on correcting underlying metabolic abnormalities with phosphate binders, vitamin D supplementation, and dialysis.
- Chan ED, Morales DV, Welsh CH, McDermott MT, Schwarz MI. Calcium deposition with or without bone formation in the lung. Am J Respir Crit Care Med 2002; 165:1654–1669.
- Beyzaei A, Francis J, Knight H, Simon DB, Finkelstein FO. Metabolic lung disease: diffuse metastatic pulmonary calcifications with progression to calciphylaxis in end-stage renal disease. Adv Perit Dial 2007; 23:112–117.
- Conger JD, Hammond WS, Alfrey AC, Contiguglia SR, Stanford RE, Huffer WE. Pulmonary calcification in chronic dialysis patients. Clinical and pathologic studies. Ann Intern Med 1975; 83:330–336.
- Belem LC, Zanetti G, Souza AS Jr, et al. Metastatic pulmonary calcification: state-of-the-art review focused on imaging findings. Respir Med 2014; 108:668–676.
- Chan ED, Morales DV, Welsh CH, McDermott MT, Schwarz MI. Calcium deposition with or without bone formation in the lung. Am J Respir Crit Care Med 2002; 165:1654–1669.
- Beyzaei A, Francis J, Knight H, Simon DB, Finkelstein FO. Metabolic lung disease: diffuse metastatic pulmonary calcifications with progression to calciphylaxis in end-stage renal disease. Adv Perit Dial 2007; 23:112–117.
- Conger JD, Hammond WS, Alfrey AC, Contiguglia SR, Stanford RE, Huffer WE. Pulmonary calcification in chronic dialysis patients. Clinical and pathologic studies. Ann Intern Med 1975; 83:330–336.
- Belem LC, Zanetti G, Souza AS Jr, et al. Metastatic pulmonary calcification: state-of-the-art review focused on imaging findings. Respir Med 2014; 108:668–676.