User login
Diffuse abdominal pain, vomiting
• Use the APACHE-II scoring system early on to help predict the severity of pancreatitis.
• Consider early enteral nutrition in patients with severe disease; taking this step has been linked to lower infection rates and shorter lengths of stay.
• Consider patient factors and the risk of severe infection when deciding whether or not to use prophylactic antibiotics in cases of severe necrotizing pancreatitis.
CASE A 57-year-old Caucasian woman sought care at our emergency department (ED) for diffuse abdominal pain and nausea. She said that the pain began after eating lunch earlier that day, and localized periumbilically, with radiation to the back. She had several episodes of nonbilious, nonbloody vomiting, but denied fever, chills, or diarrhea.
Her past medical history was notable only for an episode of gallstone pancreatitis 11 years earlier, after which she underwent a cholecystectomy. Her only medications were ibandronate sodium (Boniva) taken for osteoporosis (diagnosed 2 years earlier), a multivitamin, calcium, magnesium, and vitamin E supplements. Her family history was notable for a brother who had pancreatic cancer in his 50s. The patient reported infrequent alcohol use.
The abdominal exam was notable for diffuse tenderness to palpation, most prominent in the epigastric region. The patient exhibited voluntary guarding, without rebound, and positive bowel sounds throughout.
The patient’s laboratory studies on admission included leukocytosis of 21,300 cells/mcL and hemoglobin and hematocrit of 17.3 g/dL and 52.1%, respectively. She had an amylase of 1733 U/L and lipase of 4288 U/L. Lactate and lactic dehydrogenase were 1.83 mg/dL and 265 U/L, respectively. Liver function tests and a basic metabolic panel were within normal limits. A noncontrast computed tomography (CT) scan of the abdomen and pelvis was notable for an enlarged pancreas with peripancreatic edema and free fluid in the abdomen.
The patient underwent aggressive fluid resuscitation throughout the first 6 hours of her hospital stay. Urine output was noted to be incongruent with fluid intake, at just over 60 cc/h. Over the next 4 hours, she became progressively tachycardic, tachypneic, and somnolent, with increasing abdominal tenderness. Her serum potassium level rose to 4.9 mEq/L, while serum bicarbonate declined to 13 mEq/L and serum calcium, to 6.2 mg/dL. Arterial blood gas revealed metabolic acidosis with a pH of 7.22.
Our patient was subsequently transferred to the medical intensive care unit, where she required endotracheal intubation.
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Acute necrotizing pancreatitis
A repeat CT scan of the abdomen and pelvis with IV contrast taken on the second day of admission revealed extensive pancreatitis with complete disintegration of the pancreatic tissue and absence of pancreatic enhancement (FIGURE), as well as a large amount of abdominal ascites.
Pancreatitis is a common inpatient diagnosis, with approximately 200,000 hospitalizations yearly.1 Most cases are mild and self-limiting, requiring minimal intervention including parenteral fluid resuscitation, pain control, and restriction of oral intake. Most cases can be attributed to gallstones or excessive alcohol use, but approximately 25% of cases are idiopathic.1 Other causes include hypertriglyceridemia, infection, hypercalcemia, and medications such as azathioprine, 6-mercaptopurine, trimethoprim sulfa-methoxazole, and furosemide. Severe necrotizing pancreatitis represents about 20% of all cases, but carries a mortality rate of between 10% and 30%.1
Diagnosis is based on clinical features in conjunction with biochemical markers. Amylase is nonspecific, but levels 3 times the upper limit of normal are usually diagnostic of acute pancreatitis. Lipase is 85% to 100% sensitive for pancreatitis, and is more specific than amylase. Alanine aminotransferase >150 IU/L is 96% specific for gallstone pancreatitis.2 Of note: there is no evidence to support daily monitoring of these enzyme levels as predictors of clinical improvement or disease severity.
FIGURE
CT scan of abdomen taken on second day of admission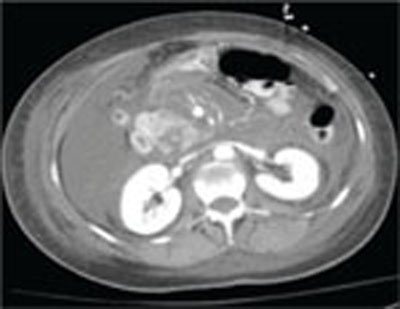
Predicting severity at time of presentation can be difficult
As was true with our patient, predicting the severity of acute pancreatitis at the time of presentation can be difficult. Scoring systems that are commonly used to evaluate disease severity include Ranson’s score, APACHE-II (Acute Physiology and Chronic Health Evaluation-II), and CT severity index, among others (TABLE). Of these, the APACHE-II score has been found to be most predictive of progression to severe disease, with accuracy of up to 75%.3
Recent studies have shown that a body mass index >30 kg/m2 is an independent risk factor for progression to severe pancreatitis.4 Other clinical predictors include poor urine output, rising hematocrit, agitation or confusion, and lack of improvement in symptoms within 48 hours.1
Though our patient came in with symptoms that were initially mild, she quickly manifested several clinical predictors for severe pancreatitis, including poor urine output and increasing confusion, as well as an APACHE-II score of 12 at 6 hours after presentation (values ≥8 indicate high risk for progression to severe disease).
TABLE
Predictors for progression to severe pancreatitis1
| Ranson score ≥3 |
| APACHE-II score ≥8 |
| CT severity index (CT grade + necrosis score) >6 |
| Body mass index >30 kg/m2 |
| Hematocrit >44% (clearly increases risk for pancreatic necrosis) |
Clinical findings:
|
| Lack of improvement in symptoms within the first 48 hours |
| APACHE, Acute Physiology and Chronic Health Evaluation; CT, computed tomography. |
Role of antibiotics? A source of debate
Infection represents the leading cause of morbidity and mortality in patients with pancreatic necrosis. Approximately 40% of patients with necrosis develop infection, with a 20% mortality rate.5 Signs of infection usually develop relatively late in the clinical course and rates increase drastically each week a patient remains hospitalized (71% of patients have signs of infection at 3 weeks).5
Interestingly, the role for antibiotics in such patients has been a source of debate in practice, as well as in the medical literature. Two recent large meta-analyses came to different conclusions regarding the use of antibiotics. A 2006 study by Heinrich et al concluded that patients with pancreatic necrosis demonstrated by contrast-enhanced CT scans should receive antibiotic prophylaxis with imipenem or meropenem for 14 days, and that prophylactic antibiotics do not increase rates of subsequent fungal infection.6 Conversely, as noted in a 2008 study published in the American Journal of Gastroenterology, “prophylactic antibiotics cannot reduce infected pancreatic necrosis and mortality in patients with acute necrotizing pancreatitis.”7
Two leading professional groups have similarly contradictory recommendations on the topic, with the American Gastroenterological Association (AGA) supporting antibiotic use for patients with >30% pancreatic necrosis noted on CT and the American College of Gastroenterology (ACG) recommending against the use of prophylactic antibiotics.8
As with any clinical dilemma, it seems prudent to make the decision for or against prophylactic antibiotics based on available clinical information and the particular patient’s risk factors. Clearly, in the most high-risk patients, it would be difficult to justify withholding antibiotic therapy.
Complete bowel rest—or not?
In the past, it was thought necessary to allow for complete bowel rest and suppression of pancreatic exocrine secretion during acute pancreatitis by providing total parenteral nutrition.6,9 More recently, though, the use of early nasojejunal enteral feeding (which was initiated for our patient) has been advocated by several large meta-analyses,6 as well as by the AGA and ACG.2
The use of enteral feeding has been associated with improved outcomes, including lower infection rates (due to maintenance of the intestinal barrier and prevention of bacterial translocation), decreased length of stay, reduced rates of organ failure, and fewer deaths among patients who require surgical intervention.6
A lengthy road to recovery for our patient
After 7 days of mechanical ventilation, our patient was extubated. However, she developed significant bilateral pleural effusions as a result of fluid third spacing, and required thoracentesis.
She completed a 14-day course of imipenem, followed by an additional 10-day course due to hypotension and a suspected infected pseudocyst. Subsequent imaging studies confirmed our suspicions: She had developed a large pseudocyst (>13 cm), which remained under observation by both a gastroenterologist and general surgeon. Six weeks after admission, our patient was discharged to home with family.
But what was the cause? Although we were unable to clearly delineate an inciting cause for her pancreatitis during the admission, she was to undergo further investigation as an outpatient. There were also plans to drain the pseudocyst 6 weeks after discharge.
A learning opportunity. This patient’s case provided an excellent opportunity for our team to review the important clinical predictors for progression to severe pancreatitis, and the rapid nature of clinical decline in such patients. In hindsight, the predictors of severity in our patient were few, but included the rapid onset and clinical progression of her symptoms, as well as her elevated hematocrit on presentation and poor urine output over the first 6 hours of admission.
1. Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med. 2006;354:2142-2150.
2. Vege SS, Whitcomb DC, Ginsburg CH. Clinical manifestations and diagnosis of acute pancreatitis. In: Basow DS. ed. UpTo-Date [online database]. Version 18.2. Waltham, Mass: UpTo-Date; 2010.
3. Vege SS, Whitcomb DC, Ginsburg CH. Predicting severity of acute pancreatitis. In: Basow DS, ed. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
4. Skipworth JRA, Pereira SP. Acute pancreatitis. Curr Opin Crit Care. 2008;14:172-178.
5. Windsor JA, Schweder P. Complications of acute pancreatitis (including pseudocysts). In: Zinner MJ, Ashley SW, eds. Main-got’s Abdominal Operations. 11th ed. New York: McGraw-Hill; 2007:chap 37. Available at: http://www.accesssurgery.com/content.aspx?filename="6003JFP_HospitalRounds" aid=130125. Accessed November 30, 2010.
6. Heinrich S, Shafer M, Rousson V, et al. Evidenced-based treatment of acute pancreatitis: a look at established paradigms. Ann Surg. 2006;243:154-168.
7. Bai Y, Gao J, Zou DW, et al. Prophylactic antibiotics cannot reduce infected pancreatic necrosis and mortality in acute necrotizing pancreatitis: evidence from a meta-analysis of randomized controlled trials. Am J Gastroenterol. 2008;103:104-110.
8. Vege SS, Whitcomb DC, Ginsburg CH. Treatment of acute pancreatitis. In: Basow DS, ed. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
9. Haney JC, Pappas TN. Necrotizing pancreatitis: diagnosis and management. Surg Clin North Am. 2007;87:1431-1446.
• Use the APACHE-II scoring system early on to help predict the severity of pancreatitis.
• Consider early enteral nutrition in patients with severe disease; taking this step has been linked to lower infection rates and shorter lengths of stay.
• Consider patient factors and the risk of severe infection when deciding whether or not to use prophylactic antibiotics in cases of severe necrotizing pancreatitis.
CASE A 57-year-old Caucasian woman sought care at our emergency department (ED) for diffuse abdominal pain and nausea. She said that the pain began after eating lunch earlier that day, and localized periumbilically, with radiation to the back. She had several episodes of nonbilious, nonbloody vomiting, but denied fever, chills, or diarrhea.
Her past medical history was notable only for an episode of gallstone pancreatitis 11 years earlier, after which she underwent a cholecystectomy. Her only medications were ibandronate sodium (Boniva) taken for osteoporosis (diagnosed 2 years earlier), a multivitamin, calcium, magnesium, and vitamin E supplements. Her family history was notable for a brother who had pancreatic cancer in his 50s. The patient reported infrequent alcohol use.
The abdominal exam was notable for diffuse tenderness to palpation, most prominent in the epigastric region. The patient exhibited voluntary guarding, without rebound, and positive bowel sounds throughout.
The patient’s laboratory studies on admission included leukocytosis of 21,300 cells/mcL and hemoglobin and hematocrit of 17.3 g/dL and 52.1%, respectively. She had an amylase of 1733 U/L and lipase of 4288 U/L. Lactate and lactic dehydrogenase were 1.83 mg/dL and 265 U/L, respectively. Liver function tests and a basic metabolic panel were within normal limits. A noncontrast computed tomography (CT) scan of the abdomen and pelvis was notable for an enlarged pancreas with peripancreatic edema and free fluid in the abdomen.
The patient underwent aggressive fluid resuscitation throughout the first 6 hours of her hospital stay. Urine output was noted to be incongruent with fluid intake, at just over 60 cc/h. Over the next 4 hours, she became progressively tachycardic, tachypneic, and somnolent, with increasing abdominal tenderness. Her serum potassium level rose to 4.9 mEq/L, while serum bicarbonate declined to 13 mEq/L and serum calcium, to 6.2 mg/dL. Arterial blood gas revealed metabolic acidosis with a pH of 7.22.
Our patient was subsequently transferred to the medical intensive care unit, where she required endotracheal intubation.
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Acute necrotizing pancreatitis
A repeat CT scan of the abdomen and pelvis with IV contrast taken on the second day of admission revealed extensive pancreatitis with complete disintegration of the pancreatic tissue and absence of pancreatic enhancement (FIGURE), as well as a large amount of abdominal ascites.
Pancreatitis is a common inpatient diagnosis, with approximately 200,000 hospitalizations yearly.1 Most cases are mild and self-limiting, requiring minimal intervention including parenteral fluid resuscitation, pain control, and restriction of oral intake. Most cases can be attributed to gallstones or excessive alcohol use, but approximately 25% of cases are idiopathic.1 Other causes include hypertriglyceridemia, infection, hypercalcemia, and medications such as azathioprine, 6-mercaptopurine, trimethoprim sulfa-methoxazole, and furosemide. Severe necrotizing pancreatitis represents about 20% of all cases, but carries a mortality rate of between 10% and 30%.1
Diagnosis is based on clinical features in conjunction with biochemical markers. Amylase is nonspecific, but levels 3 times the upper limit of normal are usually diagnostic of acute pancreatitis. Lipase is 85% to 100% sensitive for pancreatitis, and is more specific than amylase. Alanine aminotransferase >150 IU/L is 96% specific for gallstone pancreatitis.2 Of note: there is no evidence to support daily monitoring of these enzyme levels as predictors of clinical improvement or disease severity.
FIGURE
CT scan of abdomen taken on second day of admission
Predicting severity at time of presentation can be difficult
As was true with our patient, predicting the severity of acute pancreatitis at the time of presentation can be difficult. Scoring systems that are commonly used to evaluate disease severity include Ranson’s score, APACHE-II (Acute Physiology and Chronic Health Evaluation-II), and CT severity index, among others (TABLE). Of these, the APACHE-II score has been found to be most predictive of progression to severe disease, with accuracy of up to 75%.3
Recent studies have shown that a body mass index >30 kg/m2 is an independent risk factor for progression to severe pancreatitis.4 Other clinical predictors include poor urine output, rising hematocrit, agitation or confusion, and lack of improvement in symptoms within 48 hours.1
Though our patient came in with symptoms that were initially mild, she quickly manifested several clinical predictors for severe pancreatitis, including poor urine output and increasing confusion, as well as an APACHE-II score of 12 at 6 hours after presentation (values ≥8 indicate high risk for progression to severe disease).
TABLE
Predictors for progression to severe pancreatitis1
| Ranson score ≥3 |
| APACHE-II score ≥8 |
| CT severity index (CT grade + necrosis score) >6 |
| Body mass index >30 kg/m2 |
| Hematocrit >44% (clearly increases risk for pancreatic necrosis) |
Clinical findings:
|
| Lack of improvement in symptoms within the first 48 hours |
| APACHE, Acute Physiology and Chronic Health Evaluation; CT, computed tomography. |
Role of antibiotics? A source of debate
Infection represents the leading cause of morbidity and mortality in patients with pancreatic necrosis. Approximately 40% of patients with necrosis develop infection, with a 20% mortality rate.5 Signs of infection usually develop relatively late in the clinical course and rates increase drastically each week a patient remains hospitalized (71% of patients have signs of infection at 3 weeks).5
Interestingly, the role for antibiotics in such patients has been a source of debate in practice, as well as in the medical literature. Two recent large meta-analyses came to different conclusions regarding the use of antibiotics. A 2006 study by Heinrich et al concluded that patients with pancreatic necrosis demonstrated by contrast-enhanced CT scans should receive antibiotic prophylaxis with imipenem or meropenem for 14 days, and that prophylactic antibiotics do not increase rates of subsequent fungal infection.6 Conversely, as noted in a 2008 study published in the American Journal of Gastroenterology, “prophylactic antibiotics cannot reduce infected pancreatic necrosis and mortality in patients with acute necrotizing pancreatitis.”7
Two leading professional groups have similarly contradictory recommendations on the topic, with the American Gastroenterological Association (AGA) supporting antibiotic use for patients with >30% pancreatic necrosis noted on CT and the American College of Gastroenterology (ACG) recommending against the use of prophylactic antibiotics.8
As with any clinical dilemma, it seems prudent to make the decision for or against prophylactic antibiotics based on available clinical information and the particular patient’s risk factors. Clearly, in the most high-risk patients, it would be difficult to justify withholding antibiotic therapy.
Complete bowel rest—or not?
In the past, it was thought necessary to allow for complete bowel rest and suppression of pancreatic exocrine secretion during acute pancreatitis by providing total parenteral nutrition.6,9 More recently, though, the use of early nasojejunal enteral feeding (which was initiated for our patient) has been advocated by several large meta-analyses,6 as well as by the AGA and ACG.2
The use of enteral feeding has been associated with improved outcomes, including lower infection rates (due to maintenance of the intestinal barrier and prevention of bacterial translocation), decreased length of stay, reduced rates of organ failure, and fewer deaths among patients who require surgical intervention.6
A lengthy road to recovery for our patient
After 7 days of mechanical ventilation, our patient was extubated. However, she developed significant bilateral pleural effusions as a result of fluid third spacing, and required thoracentesis.
She completed a 14-day course of imipenem, followed by an additional 10-day course due to hypotension and a suspected infected pseudocyst. Subsequent imaging studies confirmed our suspicions: She had developed a large pseudocyst (>13 cm), which remained under observation by both a gastroenterologist and general surgeon. Six weeks after admission, our patient was discharged to home with family.
But what was the cause? Although we were unable to clearly delineate an inciting cause for her pancreatitis during the admission, she was to undergo further investigation as an outpatient. There were also plans to drain the pseudocyst 6 weeks after discharge.
A learning opportunity. This patient’s case provided an excellent opportunity for our team to review the important clinical predictors for progression to severe pancreatitis, and the rapid nature of clinical decline in such patients. In hindsight, the predictors of severity in our patient were few, but included the rapid onset and clinical progression of her symptoms, as well as her elevated hematocrit on presentation and poor urine output over the first 6 hours of admission.
• Use the APACHE-II scoring system early on to help predict the severity of pancreatitis.
• Consider early enteral nutrition in patients with severe disease; taking this step has been linked to lower infection rates and shorter lengths of stay.
• Consider patient factors and the risk of severe infection when deciding whether or not to use prophylactic antibiotics in cases of severe necrotizing pancreatitis.
CASE A 57-year-old Caucasian woman sought care at our emergency department (ED) for diffuse abdominal pain and nausea. She said that the pain began after eating lunch earlier that day, and localized periumbilically, with radiation to the back. She had several episodes of nonbilious, nonbloody vomiting, but denied fever, chills, or diarrhea.
Her past medical history was notable only for an episode of gallstone pancreatitis 11 years earlier, after which she underwent a cholecystectomy. Her only medications were ibandronate sodium (Boniva) taken for osteoporosis (diagnosed 2 years earlier), a multivitamin, calcium, magnesium, and vitamin E supplements. Her family history was notable for a brother who had pancreatic cancer in his 50s. The patient reported infrequent alcohol use.
The abdominal exam was notable for diffuse tenderness to palpation, most prominent in the epigastric region. The patient exhibited voluntary guarding, without rebound, and positive bowel sounds throughout.
The patient’s laboratory studies on admission included leukocytosis of 21,300 cells/mcL and hemoglobin and hematocrit of 17.3 g/dL and 52.1%, respectively. She had an amylase of 1733 U/L and lipase of 4288 U/L. Lactate and lactic dehydrogenase were 1.83 mg/dL and 265 U/L, respectively. Liver function tests and a basic metabolic panel were within normal limits. A noncontrast computed tomography (CT) scan of the abdomen and pelvis was notable for an enlarged pancreas with peripancreatic edema and free fluid in the abdomen.
The patient underwent aggressive fluid resuscitation throughout the first 6 hours of her hospital stay. Urine output was noted to be incongruent with fluid intake, at just over 60 cc/h. Over the next 4 hours, she became progressively tachycardic, tachypneic, and somnolent, with increasing abdominal tenderness. Her serum potassium level rose to 4.9 mEq/L, while serum bicarbonate declined to 13 mEq/L and serum calcium, to 6.2 mg/dL. Arterial blood gas revealed metabolic acidosis with a pH of 7.22.
Our patient was subsequently transferred to the medical intensive care unit, where she required endotracheal intubation.
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Acute necrotizing pancreatitis
A repeat CT scan of the abdomen and pelvis with IV contrast taken on the second day of admission revealed extensive pancreatitis with complete disintegration of the pancreatic tissue and absence of pancreatic enhancement (FIGURE), as well as a large amount of abdominal ascites.
Pancreatitis is a common inpatient diagnosis, with approximately 200,000 hospitalizations yearly.1 Most cases are mild and self-limiting, requiring minimal intervention including parenteral fluid resuscitation, pain control, and restriction of oral intake. Most cases can be attributed to gallstones or excessive alcohol use, but approximately 25% of cases are idiopathic.1 Other causes include hypertriglyceridemia, infection, hypercalcemia, and medications such as azathioprine, 6-mercaptopurine, trimethoprim sulfa-methoxazole, and furosemide. Severe necrotizing pancreatitis represents about 20% of all cases, but carries a mortality rate of between 10% and 30%.1
Diagnosis is based on clinical features in conjunction with biochemical markers. Amylase is nonspecific, but levels 3 times the upper limit of normal are usually diagnostic of acute pancreatitis. Lipase is 85% to 100% sensitive for pancreatitis, and is more specific than amylase. Alanine aminotransferase >150 IU/L is 96% specific for gallstone pancreatitis.2 Of note: there is no evidence to support daily monitoring of these enzyme levels as predictors of clinical improvement or disease severity.
FIGURE
CT scan of abdomen taken on second day of admission
Predicting severity at time of presentation can be difficult
As was true with our patient, predicting the severity of acute pancreatitis at the time of presentation can be difficult. Scoring systems that are commonly used to evaluate disease severity include Ranson’s score, APACHE-II (Acute Physiology and Chronic Health Evaluation-II), and CT severity index, among others (TABLE). Of these, the APACHE-II score has been found to be most predictive of progression to severe disease, with accuracy of up to 75%.3
Recent studies have shown that a body mass index >30 kg/m2 is an independent risk factor for progression to severe pancreatitis.4 Other clinical predictors include poor urine output, rising hematocrit, agitation or confusion, and lack of improvement in symptoms within 48 hours.1
Though our patient came in with symptoms that were initially mild, she quickly manifested several clinical predictors for severe pancreatitis, including poor urine output and increasing confusion, as well as an APACHE-II score of 12 at 6 hours after presentation (values ≥8 indicate high risk for progression to severe disease).
TABLE
Predictors for progression to severe pancreatitis1
| Ranson score ≥3 |
| APACHE-II score ≥8 |
| CT severity index (CT grade + necrosis score) >6 |
| Body mass index >30 kg/m2 |
| Hematocrit >44% (clearly increases risk for pancreatic necrosis) |
Clinical findings:
|
| Lack of improvement in symptoms within the first 48 hours |
| APACHE, Acute Physiology and Chronic Health Evaluation; CT, computed tomography. |
Role of antibiotics? A source of debate
Infection represents the leading cause of morbidity and mortality in patients with pancreatic necrosis. Approximately 40% of patients with necrosis develop infection, with a 20% mortality rate.5 Signs of infection usually develop relatively late in the clinical course and rates increase drastically each week a patient remains hospitalized (71% of patients have signs of infection at 3 weeks).5
Interestingly, the role for antibiotics in such patients has been a source of debate in practice, as well as in the medical literature. Two recent large meta-analyses came to different conclusions regarding the use of antibiotics. A 2006 study by Heinrich et al concluded that patients with pancreatic necrosis demonstrated by contrast-enhanced CT scans should receive antibiotic prophylaxis with imipenem or meropenem for 14 days, and that prophylactic antibiotics do not increase rates of subsequent fungal infection.6 Conversely, as noted in a 2008 study published in the American Journal of Gastroenterology, “prophylactic antibiotics cannot reduce infected pancreatic necrosis and mortality in patients with acute necrotizing pancreatitis.”7
Two leading professional groups have similarly contradictory recommendations on the topic, with the American Gastroenterological Association (AGA) supporting antibiotic use for patients with >30% pancreatic necrosis noted on CT and the American College of Gastroenterology (ACG) recommending against the use of prophylactic antibiotics.8
As with any clinical dilemma, it seems prudent to make the decision for or against prophylactic antibiotics based on available clinical information and the particular patient’s risk factors. Clearly, in the most high-risk patients, it would be difficult to justify withholding antibiotic therapy.
Complete bowel rest—or not?
In the past, it was thought necessary to allow for complete bowel rest and suppression of pancreatic exocrine secretion during acute pancreatitis by providing total parenteral nutrition.6,9 More recently, though, the use of early nasojejunal enteral feeding (which was initiated for our patient) has been advocated by several large meta-analyses,6 as well as by the AGA and ACG.2
The use of enteral feeding has been associated with improved outcomes, including lower infection rates (due to maintenance of the intestinal barrier and prevention of bacterial translocation), decreased length of stay, reduced rates of organ failure, and fewer deaths among patients who require surgical intervention.6
A lengthy road to recovery for our patient
After 7 days of mechanical ventilation, our patient was extubated. However, she developed significant bilateral pleural effusions as a result of fluid third spacing, and required thoracentesis.
She completed a 14-day course of imipenem, followed by an additional 10-day course due to hypotension and a suspected infected pseudocyst. Subsequent imaging studies confirmed our suspicions: She had developed a large pseudocyst (>13 cm), which remained under observation by both a gastroenterologist and general surgeon. Six weeks after admission, our patient was discharged to home with family.
But what was the cause? Although we were unable to clearly delineate an inciting cause for her pancreatitis during the admission, she was to undergo further investigation as an outpatient. There were also plans to drain the pseudocyst 6 weeks after discharge.
A learning opportunity. This patient’s case provided an excellent opportunity for our team to review the important clinical predictors for progression to severe pancreatitis, and the rapid nature of clinical decline in such patients. In hindsight, the predictors of severity in our patient were few, but included the rapid onset and clinical progression of her symptoms, as well as her elevated hematocrit on presentation and poor urine output over the first 6 hours of admission.
1. Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med. 2006;354:2142-2150.
2. Vege SS, Whitcomb DC, Ginsburg CH. Clinical manifestations and diagnosis of acute pancreatitis. In: Basow DS. ed. UpTo-Date [online database]. Version 18.2. Waltham, Mass: UpTo-Date; 2010.
3. Vege SS, Whitcomb DC, Ginsburg CH. Predicting severity of acute pancreatitis. In: Basow DS, ed. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
4. Skipworth JRA, Pereira SP. Acute pancreatitis. Curr Opin Crit Care. 2008;14:172-178.
5. Windsor JA, Schweder P. Complications of acute pancreatitis (including pseudocysts). In: Zinner MJ, Ashley SW, eds. Main-got’s Abdominal Operations. 11th ed. New York: McGraw-Hill; 2007:chap 37. Available at: http://www.accesssurgery.com/content.aspx?filename="6003JFP_HospitalRounds" aid=130125. Accessed November 30, 2010.
6. Heinrich S, Shafer M, Rousson V, et al. Evidenced-based treatment of acute pancreatitis: a look at established paradigms. Ann Surg. 2006;243:154-168.
7. Bai Y, Gao J, Zou DW, et al. Prophylactic antibiotics cannot reduce infected pancreatic necrosis and mortality in acute necrotizing pancreatitis: evidence from a meta-analysis of randomized controlled trials. Am J Gastroenterol. 2008;103:104-110.
8. Vege SS, Whitcomb DC, Ginsburg CH. Treatment of acute pancreatitis. In: Basow DS, ed. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
9. Haney JC, Pappas TN. Necrotizing pancreatitis: diagnosis and management. Surg Clin North Am. 2007;87:1431-1446.
1. Whitcomb DC. Clinical practice. Acute pancreatitis. N Engl J Med. 2006;354:2142-2150.
2. Vege SS, Whitcomb DC, Ginsburg CH. Clinical manifestations and diagnosis of acute pancreatitis. In: Basow DS. ed. UpTo-Date [online database]. Version 18.2. Waltham, Mass: UpTo-Date; 2010.
3. Vege SS, Whitcomb DC, Ginsburg CH. Predicting severity of acute pancreatitis. In: Basow DS, ed. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
4. Skipworth JRA, Pereira SP. Acute pancreatitis. Curr Opin Crit Care. 2008;14:172-178.
5. Windsor JA, Schweder P. Complications of acute pancreatitis (including pseudocysts). In: Zinner MJ, Ashley SW, eds. Main-got’s Abdominal Operations. 11th ed. New York: McGraw-Hill; 2007:chap 37. Available at: http://www.accesssurgery.com/content.aspx?filename="6003JFP_HospitalRounds" aid=130125. Accessed November 30, 2010.
6. Heinrich S, Shafer M, Rousson V, et al. Evidenced-based treatment of acute pancreatitis: a look at established paradigms. Ann Surg. 2006;243:154-168.
7. Bai Y, Gao J, Zou DW, et al. Prophylactic antibiotics cannot reduce infected pancreatic necrosis and mortality in acute necrotizing pancreatitis: evidence from a meta-analysis of randomized controlled trials. Am J Gastroenterol. 2008;103:104-110.
8. Vege SS, Whitcomb DC, Ginsburg CH. Treatment of acute pancreatitis. In: Basow DS, ed. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
9. Haney JC, Pappas TN. Necrotizing pancreatitis: diagnosis and management. Surg Clin North Am. 2007;87:1431-1446.
Fever, rash, and peeling skin
• Ask about inciting factors for SJS/TEN— anti-gout agents, anti-epileptics, NSAIDs, sulfonamides, penicillins, and cephalosporins—when a patient seeks care for a painful rash involving mucous membranes.
• Rule out look-alike conditions, including erythema multiforme, sunburn, toxic shock syndrome, staphylococcal scalded skin syndrome, and paraneoplastic pemphigus.
• Withdraw the causative agent as the first step in the treatment of SJS/TEN.
• Refer patients with SJS/TEN to a burn unit for supportive treatment, including wound care, fluid replacement, and electrolyte and ocular monitoring.
CASE A 21-year-old woman sought care at our emergency department (ED) for an extensive rash that began approximately 1 week earlier and hurt “like a sunburn.” She said that 2 weeks earlier, she’d been seen at another hospital for an incision and drainage of a thigh abscess. She was started on sulfamethoxazole/trimethoprim (Bactrim) at that time.
She returned to that ED several times, complaining of headache and fever, then went to a different ED because her lips were swollen and she was developing a rash (FIGURES 1A AND 1B). Initially the rash was red, slightly rough, and covered most of her body (except for her palms and soles). Providers at the second ED discontinued the Bactrim, started her on a course of steroids by mouth, and sent her home.
On this particular morning, she woke up feeling that her mouth was burning and her throat was “closing up.” She was admitted to our intensive care unit (ICU) for monitoring of respiratory compromise related to angioedema. She did not require intubation, but her skin began to desquamate.
FIGURE 1
Swollen lips and rash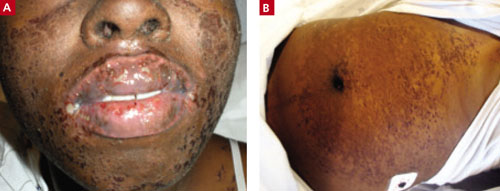
This 21-year-old patient had swollen lips and a rash that hurt “like a sunburn.” She said that when she woke up that morning, her mouth was burning and her throat felt like it was “closing up.”
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Stevens-Johnson syndrome
A painful rash involving mucous membranes, along with fever, should alert the clinician to the possibility of Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN). The 2 conditions are distinguished by how much of the body surface area (BSA) is involved: If the rash covers <10% of the BSA (as it did with our patient), the condition is called SJS; if >90%, it’s referred to as TEN.1 Skin involvement that falls between these 2 parameters is referred to as SJS/TEN overlap syndrome.
Mucous membranes are involved in more than 90% of SJS cases and in virtually all cases of TEN.1 Illnesses on this spectrum typically start with prodromal fever and malaise, followed by the onset of a maculopapular rash. The rash then becomes vesicular and eventually progresses to desquamation and epidermal necrolysis.2
Common inciting factors. SJS/TEN is most commonly caused by an infection or a reaction to medication. In adults, medication reaction is the more common etiology. In children, medications are still the leading cause but a larger proportion of pediatric cases are associated with infection. Multiple drug classifications can lead to the disorder, including anti-gout agents, anti-epileptics, nonsteroidal anti-inflammatory drugs (NSAIDs), and certain antibiotics—sulfonamides, penicillins, and cephalosporins (TABLE).3
TABLE
Agents implicated in SJS/TEN
| Allopurinol |
| Amoxicillin |
| Ampicillin |
| Lamotrigine |
| Phenylbutazone |
| Piroxicam |
| Sulfadiazine |
| Trimethoprim/sulfamethoxazole |
| SJS/TEN, Stevens-Johnson syndrome/toxic epidermal necrolysis. Adapted from: Sharma VK et al. Indian J Dermatol Venerol Leprol. 2008.3 |
Slow metabolizers may be at risk
The pathophysiology that underlies these conditions is not entirely understood. Recent research indicates a possible role of granulysin, a product of cytotoxic T and natural killer cells.4 People with slow medication metabolism due to lower rates of N-acetylation may be at increased risk because of the greater accumulation of potentially toxic metabolites.5
In addition, the risk for SJS/TEN is 3 times higher in patients who are positive for human immunodeficiency virus (HIV). If an HIV patient is exposed to Bactrim—the off ending agent in this case—the relative risk increases to 40-fold.6 (Our patient was not infected with HIV.)
Other conditions associated with higher risk are malignancy, systemic lupus erythematosus (SLE), rapid titration or high doses of medication, and radiation exposure. Patients with HLA B 1502 haplotype are at increased risk when exposed to aromatic anticonvulsant agents, such as phenobarbital and phenytoin.7
The differential was large, but the diagnosis was clear
Conditions with a presentation similar to SJS/ TEN include erythema multiforme, erythematous drug reaction, pustular drug eruptions, sunburn, toxic shock syndrome, staphylococcal scalded skin syndrome, and paraneoplastic pemphigus.
While the differential diagnosis for acute rash is large, the diagnosis in this case was clear because of the combination of prodromal symptoms, pain that was out of proportion to the appearance of the skin, and skin sloughing. In addition, a classic inciting drug (Bactrim) was easily identified and the timing of the drug exposure—1 to 3 weeks before the onset of the rash—was consistent with SJS/ TEN. Sparing of the palms and soles helped us differentiate SJS/TEN from toxic shock syndrome.
Why so long to diagnosis?
We suspected that the diagnosis was missed during our patient’s earlier ED visits because she initially had nonspecific symptoms and did not have mucosal involvement early on. Further complicating matters: She went to multiple EDs.
Labwork is of limited value
No laboratory values or pathologic tests are pathognomonic for SJS/TEN. Anemia and lymphopenia are possible findings, and neutropenia is an indicator for a poor prognosis.8 Elevated liver enzymes are common, reaching levels approximately 2 to 3 times the upper limit of normal.8 All of these laboratory findings become increasingly more likely as BSA involved increases.8 Our patient’s laboratory values were unremarkable.
Tx: Discontinue drug, replace fluids
This syndrome is potentially deadly and must be treated as an emergency as soon as it is recognized. The mortality rate is 1% to 3% for SJS and 25% to 35% for TEN.9
Discontinuing the causative agent is the first step in treatment. In our patient’s case, Bactrim had been discontinued before she came to our ED. The next step, ideally, is to transfer the patient to a burn unit where she can receive the same type of supportive care burn victims require. Diligent wound care, fluid replacement, electrolyte monitoring, and raised ambient temperature are vital elements in proper care. The patient should also be evaluated throughout the treatment period for any ocular involvement, such as conjunctivitis, keratitis, or severe dryness.10
As is the case with burn victims, the major risks are secondary infection and sepsis. Skin, blood, and access-line cultures should be gathered throughout the hospitalization to evaluate for infection. Some authorities believe glucocorticoids are helpful in children with SJS/TEN, but the adverse effects are sufficiently significant that the risks may outweigh the benefits. In adults, the evidence favors the use of glucocorticoids in SJS, but not in TEN, where the increased risk of sepsis following immunosuppression may outweigh the benefits.11
Intravenous immunoglobulin (IVIG) may benefit both children and adults, and the benefit appears to outweigh risks associated with this therapy.12 Plasmaphoresis to remove toxic metabolites from the circulation is an additional treatment option, but there is no strong evidence to support this approach.13
A positive outcome for our patient
Our patient was transferred to a facility with an inpatient burn unit and a consulting dermatology service. She was maintained on steroids, but neither plasmapheresis nor IVIG was necessary.
She was discharged to her home with prescriptions for topical emollients and an oral steroid solution for her mouth irritation. Systemic steroids were not continued, as the relatively small area of desquamation made this unnecessary.
Staying safe hinges on education. Our patient was advised that going forward, she needed to avoid Bactrim and any other sulfa drugs. She was told that when medications are prescribed for her in the future, she must consult with the pharmacist to make sure sulfonamides are not included.11
CORRESPONDENCE Bryan Cairns, MD, 2123 Auburn Avenue, Cincinnati, OH 45219; [email protected]
1. Bastuji-Garin S, Rzany B, Stern RS, et al. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol. 1993;129:92-96.
2. Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med. 1994;331:1272-1285.
3. Sharma VK, Sethuraman G, Minz A. Stevens Johnson syndrome, toxic epidermal necrolysis, and SJS-TEN overlap: a retrospective study of causative drugs and clinical outcome. Indian J Dermatol Venereol Leprol. 2008;74:238-240.
4. Chung WH, Hung SI, Yang JY, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med. 2008;14:1343-1350.
5. Dietrich A, Kawakubo Y, Rzany B, et al. Low N-acetylating capacity in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. Exp Dermatol. 1995;4:313-316.
6. Rotunda A, Hirsch RJ, Scheinfeld N, et al. Severe cutaneous reactions associated with the use of human immunodeficiency virus medications. Acta Derm Venerol. 2003;83:1-9.
7. US Food and Drug administration. Information for healthcare professionals: dangerous or even fatal skin reactions—carbamazepine (marketed as Carbatrol, Equetro, Tegretol, and generics). Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketdrugsafetyinformationforPatientsandProviders/ucm124718.htm. Page last updated: January 25, 2010. Accessed December 20, 2010.
8. Roujeau JC, Chosidow O, Saiag P, et al. Toxic epidermal necrolysis (Lyell syndrome). J Am Acad Dermatol. 1990;23:1039-1058.
9. Letko E, Papaliodis GN, Daoud YJ, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of the literature. Ann Allergy Asthma Immunol. 2005;94:419-436.
10. Williams PM, Conklin RJ. Erythema multiforme: a review and contrast from Stevens-Johnson syndrome/toxic epidermal necrolysis. Dent Clin North Am. 2005;49:67-76, viii.
11. Nirken M, High W. Stevens-Johnson syndrome and toxic epidermal necrolysis: clinical manifestations, pathogenesis, and diagnosis. In: Ofori A, Levy M, Adkinson NF, eds. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
12. French LE, Trent JT, Kerdel FA. Use of intravenous immunoglobulin in toxic epidermal necrolysis and Stevens-Johnson syndrome: our current understanding. Int Immunopharmacol. 2006;6:543-549.
13. Furubacke A, Berlin G, Anderson C, et al. Lack of significant treatment effect of plasma exchange in the treatment of drug-induced toxic epidermal necrolysis? Intensive Care Med. 1999;25:1307-1310.
• Ask about inciting factors for SJS/TEN— anti-gout agents, anti-epileptics, NSAIDs, sulfonamides, penicillins, and cephalosporins—when a patient seeks care for a painful rash involving mucous membranes.
• Rule out look-alike conditions, including erythema multiforme, sunburn, toxic shock syndrome, staphylococcal scalded skin syndrome, and paraneoplastic pemphigus.
• Withdraw the causative agent as the first step in the treatment of SJS/TEN.
• Refer patients with SJS/TEN to a burn unit for supportive treatment, including wound care, fluid replacement, and electrolyte and ocular monitoring.
CASE A 21-year-old woman sought care at our emergency department (ED) for an extensive rash that began approximately 1 week earlier and hurt “like a sunburn.” She said that 2 weeks earlier, she’d been seen at another hospital for an incision and drainage of a thigh abscess. She was started on sulfamethoxazole/trimethoprim (Bactrim) at that time.
She returned to that ED several times, complaining of headache and fever, then went to a different ED because her lips were swollen and she was developing a rash (FIGURES 1A AND 1B). Initially the rash was red, slightly rough, and covered most of her body (except for her palms and soles). Providers at the second ED discontinued the Bactrim, started her on a course of steroids by mouth, and sent her home.
On this particular morning, she woke up feeling that her mouth was burning and her throat was “closing up.” She was admitted to our intensive care unit (ICU) for monitoring of respiratory compromise related to angioedema. She did not require intubation, but her skin began to desquamate.
FIGURE 1
Swollen lips and rash
This 21-year-old patient had swollen lips and a rash that hurt “like a sunburn.” She said that when she woke up that morning, her mouth was burning and her throat felt like it was “closing up.”
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Stevens-Johnson syndrome
A painful rash involving mucous membranes, along with fever, should alert the clinician to the possibility of Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN). The 2 conditions are distinguished by how much of the body surface area (BSA) is involved: If the rash covers <10% of the BSA (as it did with our patient), the condition is called SJS; if >90%, it’s referred to as TEN.1 Skin involvement that falls between these 2 parameters is referred to as SJS/TEN overlap syndrome.
Mucous membranes are involved in more than 90% of SJS cases and in virtually all cases of TEN.1 Illnesses on this spectrum typically start with prodromal fever and malaise, followed by the onset of a maculopapular rash. The rash then becomes vesicular and eventually progresses to desquamation and epidermal necrolysis.2
Common inciting factors. SJS/TEN is most commonly caused by an infection or a reaction to medication. In adults, medication reaction is the more common etiology. In children, medications are still the leading cause but a larger proportion of pediatric cases are associated with infection. Multiple drug classifications can lead to the disorder, including anti-gout agents, anti-epileptics, nonsteroidal anti-inflammatory drugs (NSAIDs), and certain antibiotics—sulfonamides, penicillins, and cephalosporins (TABLE).3
TABLE
Agents implicated in SJS/TEN
| Allopurinol |
| Amoxicillin |
| Ampicillin |
| Lamotrigine |
| Phenylbutazone |
| Piroxicam |
| Sulfadiazine |
| Trimethoprim/sulfamethoxazole |
| SJS/TEN, Stevens-Johnson syndrome/toxic epidermal necrolysis. Adapted from: Sharma VK et al. Indian J Dermatol Venerol Leprol. 2008.3 |
Slow metabolizers may be at risk
The pathophysiology that underlies these conditions is not entirely understood. Recent research indicates a possible role of granulysin, a product of cytotoxic T and natural killer cells.4 People with slow medication metabolism due to lower rates of N-acetylation may be at increased risk because of the greater accumulation of potentially toxic metabolites.5
In addition, the risk for SJS/TEN is 3 times higher in patients who are positive for human immunodeficiency virus (HIV). If an HIV patient is exposed to Bactrim—the off ending agent in this case—the relative risk increases to 40-fold.6 (Our patient was not infected with HIV.)
Other conditions associated with higher risk are malignancy, systemic lupus erythematosus (SLE), rapid titration or high doses of medication, and radiation exposure. Patients with HLA B 1502 haplotype are at increased risk when exposed to aromatic anticonvulsant agents, such as phenobarbital and phenytoin.7
The differential was large, but the diagnosis was clear
Conditions with a presentation similar to SJS/ TEN include erythema multiforme, erythematous drug reaction, pustular drug eruptions, sunburn, toxic shock syndrome, staphylococcal scalded skin syndrome, and paraneoplastic pemphigus.
While the differential diagnosis for acute rash is large, the diagnosis in this case was clear because of the combination of prodromal symptoms, pain that was out of proportion to the appearance of the skin, and skin sloughing. In addition, a classic inciting drug (Bactrim) was easily identified and the timing of the drug exposure—1 to 3 weeks before the onset of the rash—was consistent with SJS/ TEN. Sparing of the palms and soles helped us differentiate SJS/TEN from toxic shock syndrome.
Why so long to diagnosis?
We suspected that the diagnosis was missed during our patient’s earlier ED visits because she initially had nonspecific symptoms and did not have mucosal involvement early on. Further complicating matters: She went to multiple EDs.
Labwork is of limited value
No laboratory values or pathologic tests are pathognomonic for SJS/TEN. Anemia and lymphopenia are possible findings, and neutropenia is an indicator for a poor prognosis.8 Elevated liver enzymes are common, reaching levels approximately 2 to 3 times the upper limit of normal.8 All of these laboratory findings become increasingly more likely as BSA involved increases.8 Our patient’s laboratory values were unremarkable.
Tx: Discontinue drug, replace fluids
This syndrome is potentially deadly and must be treated as an emergency as soon as it is recognized. The mortality rate is 1% to 3% for SJS and 25% to 35% for TEN.9
Discontinuing the causative agent is the first step in treatment. In our patient’s case, Bactrim had been discontinued before she came to our ED. The next step, ideally, is to transfer the patient to a burn unit where she can receive the same type of supportive care burn victims require. Diligent wound care, fluid replacement, electrolyte monitoring, and raised ambient temperature are vital elements in proper care. The patient should also be evaluated throughout the treatment period for any ocular involvement, such as conjunctivitis, keratitis, or severe dryness.10
As is the case with burn victims, the major risks are secondary infection and sepsis. Skin, blood, and access-line cultures should be gathered throughout the hospitalization to evaluate for infection. Some authorities believe glucocorticoids are helpful in children with SJS/TEN, but the adverse effects are sufficiently significant that the risks may outweigh the benefits. In adults, the evidence favors the use of glucocorticoids in SJS, but not in TEN, where the increased risk of sepsis following immunosuppression may outweigh the benefits.11
Intravenous immunoglobulin (IVIG) may benefit both children and adults, and the benefit appears to outweigh risks associated with this therapy.12 Plasmaphoresis to remove toxic metabolites from the circulation is an additional treatment option, but there is no strong evidence to support this approach.13
A positive outcome for our patient
Our patient was transferred to a facility with an inpatient burn unit and a consulting dermatology service. She was maintained on steroids, but neither plasmapheresis nor IVIG was necessary.
She was discharged to her home with prescriptions for topical emollients and an oral steroid solution for her mouth irritation. Systemic steroids were not continued, as the relatively small area of desquamation made this unnecessary.
Staying safe hinges on education. Our patient was advised that going forward, she needed to avoid Bactrim and any other sulfa drugs. She was told that when medications are prescribed for her in the future, she must consult with the pharmacist to make sure sulfonamides are not included.11
CORRESPONDENCE Bryan Cairns, MD, 2123 Auburn Avenue, Cincinnati, OH 45219; [email protected]
• Ask about inciting factors for SJS/TEN— anti-gout agents, anti-epileptics, NSAIDs, sulfonamides, penicillins, and cephalosporins—when a patient seeks care for a painful rash involving mucous membranes.
• Rule out look-alike conditions, including erythema multiforme, sunburn, toxic shock syndrome, staphylococcal scalded skin syndrome, and paraneoplastic pemphigus.
• Withdraw the causative agent as the first step in the treatment of SJS/TEN.
• Refer patients with SJS/TEN to a burn unit for supportive treatment, including wound care, fluid replacement, and electrolyte and ocular monitoring.
CASE A 21-year-old woman sought care at our emergency department (ED) for an extensive rash that began approximately 1 week earlier and hurt “like a sunburn.” She said that 2 weeks earlier, she’d been seen at another hospital for an incision and drainage of a thigh abscess. She was started on sulfamethoxazole/trimethoprim (Bactrim) at that time.
She returned to that ED several times, complaining of headache and fever, then went to a different ED because her lips were swollen and she was developing a rash (FIGURES 1A AND 1B). Initially the rash was red, slightly rough, and covered most of her body (except for her palms and soles). Providers at the second ED discontinued the Bactrim, started her on a course of steroids by mouth, and sent her home.
On this particular morning, she woke up feeling that her mouth was burning and her throat was “closing up.” She was admitted to our intensive care unit (ICU) for monitoring of respiratory compromise related to angioedema. She did not require intubation, but her skin began to desquamate.
FIGURE 1
Swollen lips and rash
This 21-year-old patient had swollen lips and a rash that hurt “like a sunburn.” She said that when she woke up that morning, her mouth was burning and her throat felt like it was “closing up.”
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Stevens-Johnson syndrome
A painful rash involving mucous membranes, along with fever, should alert the clinician to the possibility of Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN). The 2 conditions are distinguished by how much of the body surface area (BSA) is involved: If the rash covers <10% of the BSA (as it did with our patient), the condition is called SJS; if >90%, it’s referred to as TEN.1 Skin involvement that falls between these 2 parameters is referred to as SJS/TEN overlap syndrome.
Mucous membranes are involved in more than 90% of SJS cases and in virtually all cases of TEN.1 Illnesses on this spectrum typically start with prodromal fever and malaise, followed by the onset of a maculopapular rash. The rash then becomes vesicular and eventually progresses to desquamation and epidermal necrolysis.2
Common inciting factors. SJS/TEN is most commonly caused by an infection or a reaction to medication. In adults, medication reaction is the more common etiology. In children, medications are still the leading cause but a larger proportion of pediatric cases are associated with infection. Multiple drug classifications can lead to the disorder, including anti-gout agents, anti-epileptics, nonsteroidal anti-inflammatory drugs (NSAIDs), and certain antibiotics—sulfonamides, penicillins, and cephalosporins (TABLE).3
TABLE
Agents implicated in SJS/TEN
| Allopurinol |
| Amoxicillin |
| Ampicillin |
| Lamotrigine |
| Phenylbutazone |
| Piroxicam |
| Sulfadiazine |
| Trimethoprim/sulfamethoxazole |
| SJS/TEN, Stevens-Johnson syndrome/toxic epidermal necrolysis. Adapted from: Sharma VK et al. Indian J Dermatol Venerol Leprol. 2008.3 |
Slow metabolizers may be at risk
The pathophysiology that underlies these conditions is not entirely understood. Recent research indicates a possible role of granulysin, a product of cytotoxic T and natural killer cells.4 People with slow medication metabolism due to lower rates of N-acetylation may be at increased risk because of the greater accumulation of potentially toxic metabolites.5
In addition, the risk for SJS/TEN is 3 times higher in patients who are positive for human immunodeficiency virus (HIV). If an HIV patient is exposed to Bactrim—the off ending agent in this case—the relative risk increases to 40-fold.6 (Our patient was not infected with HIV.)
Other conditions associated with higher risk are malignancy, systemic lupus erythematosus (SLE), rapid titration or high doses of medication, and radiation exposure. Patients with HLA B 1502 haplotype are at increased risk when exposed to aromatic anticonvulsant agents, such as phenobarbital and phenytoin.7
The differential was large, but the diagnosis was clear
Conditions with a presentation similar to SJS/ TEN include erythema multiforme, erythematous drug reaction, pustular drug eruptions, sunburn, toxic shock syndrome, staphylococcal scalded skin syndrome, and paraneoplastic pemphigus.
While the differential diagnosis for acute rash is large, the diagnosis in this case was clear because of the combination of prodromal symptoms, pain that was out of proportion to the appearance of the skin, and skin sloughing. In addition, a classic inciting drug (Bactrim) was easily identified and the timing of the drug exposure—1 to 3 weeks before the onset of the rash—was consistent with SJS/ TEN. Sparing of the palms and soles helped us differentiate SJS/TEN from toxic shock syndrome.
Why so long to diagnosis?
We suspected that the diagnosis was missed during our patient’s earlier ED visits because she initially had nonspecific symptoms and did not have mucosal involvement early on. Further complicating matters: She went to multiple EDs.
Labwork is of limited value
No laboratory values or pathologic tests are pathognomonic for SJS/TEN. Anemia and lymphopenia are possible findings, and neutropenia is an indicator for a poor prognosis.8 Elevated liver enzymes are common, reaching levels approximately 2 to 3 times the upper limit of normal.8 All of these laboratory findings become increasingly more likely as BSA involved increases.8 Our patient’s laboratory values were unremarkable.
Tx: Discontinue drug, replace fluids
This syndrome is potentially deadly and must be treated as an emergency as soon as it is recognized. The mortality rate is 1% to 3% for SJS and 25% to 35% for TEN.9
Discontinuing the causative agent is the first step in treatment. In our patient’s case, Bactrim had been discontinued before she came to our ED. The next step, ideally, is to transfer the patient to a burn unit where she can receive the same type of supportive care burn victims require. Diligent wound care, fluid replacement, electrolyte monitoring, and raised ambient temperature are vital elements in proper care. The patient should also be evaluated throughout the treatment period for any ocular involvement, such as conjunctivitis, keratitis, or severe dryness.10
As is the case with burn victims, the major risks are secondary infection and sepsis. Skin, blood, and access-line cultures should be gathered throughout the hospitalization to evaluate for infection. Some authorities believe glucocorticoids are helpful in children with SJS/TEN, but the adverse effects are sufficiently significant that the risks may outweigh the benefits. In adults, the evidence favors the use of glucocorticoids in SJS, but not in TEN, where the increased risk of sepsis following immunosuppression may outweigh the benefits.11
Intravenous immunoglobulin (IVIG) may benefit both children and adults, and the benefit appears to outweigh risks associated with this therapy.12 Plasmaphoresis to remove toxic metabolites from the circulation is an additional treatment option, but there is no strong evidence to support this approach.13
A positive outcome for our patient
Our patient was transferred to a facility with an inpatient burn unit and a consulting dermatology service. She was maintained on steroids, but neither plasmapheresis nor IVIG was necessary.
She was discharged to her home with prescriptions for topical emollients and an oral steroid solution for her mouth irritation. Systemic steroids were not continued, as the relatively small area of desquamation made this unnecessary.
Staying safe hinges on education. Our patient was advised that going forward, she needed to avoid Bactrim and any other sulfa drugs. She was told that when medications are prescribed for her in the future, she must consult with the pharmacist to make sure sulfonamides are not included.11
CORRESPONDENCE Bryan Cairns, MD, 2123 Auburn Avenue, Cincinnati, OH 45219; [email protected]
1. Bastuji-Garin S, Rzany B, Stern RS, et al. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol. 1993;129:92-96.
2. Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med. 1994;331:1272-1285.
3. Sharma VK, Sethuraman G, Minz A. Stevens Johnson syndrome, toxic epidermal necrolysis, and SJS-TEN overlap: a retrospective study of causative drugs and clinical outcome. Indian J Dermatol Venereol Leprol. 2008;74:238-240.
4. Chung WH, Hung SI, Yang JY, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med. 2008;14:1343-1350.
5. Dietrich A, Kawakubo Y, Rzany B, et al. Low N-acetylating capacity in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. Exp Dermatol. 1995;4:313-316.
6. Rotunda A, Hirsch RJ, Scheinfeld N, et al. Severe cutaneous reactions associated with the use of human immunodeficiency virus medications. Acta Derm Venerol. 2003;83:1-9.
7. US Food and Drug administration. Information for healthcare professionals: dangerous or even fatal skin reactions—carbamazepine (marketed as Carbatrol, Equetro, Tegretol, and generics). Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketdrugsafetyinformationforPatientsandProviders/ucm124718.htm. Page last updated: January 25, 2010. Accessed December 20, 2010.
8. Roujeau JC, Chosidow O, Saiag P, et al. Toxic epidermal necrolysis (Lyell syndrome). J Am Acad Dermatol. 1990;23:1039-1058.
9. Letko E, Papaliodis GN, Daoud YJ, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of the literature. Ann Allergy Asthma Immunol. 2005;94:419-436.
10. Williams PM, Conklin RJ. Erythema multiforme: a review and contrast from Stevens-Johnson syndrome/toxic epidermal necrolysis. Dent Clin North Am. 2005;49:67-76, viii.
11. Nirken M, High W. Stevens-Johnson syndrome and toxic epidermal necrolysis: clinical manifestations, pathogenesis, and diagnosis. In: Ofori A, Levy M, Adkinson NF, eds. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
12. French LE, Trent JT, Kerdel FA. Use of intravenous immunoglobulin in toxic epidermal necrolysis and Stevens-Johnson syndrome: our current understanding. Int Immunopharmacol. 2006;6:543-549.
13. Furubacke A, Berlin G, Anderson C, et al. Lack of significant treatment effect of plasma exchange in the treatment of drug-induced toxic epidermal necrolysis? Intensive Care Med. 1999;25:1307-1310.
1. Bastuji-Garin S, Rzany B, Stern RS, et al. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol. 1993;129:92-96.
2. Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med. 1994;331:1272-1285.
3. Sharma VK, Sethuraman G, Minz A. Stevens Johnson syndrome, toxic epidermal necrolysis, and SJS-TEN overlap: a retrospective study of causative drugs and clinical outcome. Indian J Dermatol Venereol Leprol. 2008;74:238-240.
4. Chung WH, Hung SI, Yang JY, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med. 2008;14:1343-1350.
5. Dietrich A, Kawakubo Y, Rzany B, et al. Low N-acetylating capacity in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. Exp Dermatol. 1995;4:313-316.
6. Rotunda A, Hirsch RJ, Scheinfeld N, et al. Severe cutaneous reactions associated with the use of human immunodeficiency virus medications. Acta Derm Venerol. 2003;83:1-9.
7. US Food and Drug administration. Information for healthcare professionals: dangerous or even fatal skin reactions—carbamazepine (marketed as Carbatrol, Equetro, Tegretol, and generics). Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketdrugsafetyinformationforPatientsandProviders/ucm124718.htm. Page last updated: January 25, 2010. Accessed December 20, 2010.
8. Roujeau JC, Chosidow O, Saiag P, et al. Toxic epidermal necrolysis (Lyell syndrome). J Am Acad Dermatol. 1990;23:1039-1058.
9. Letko E, Papaliodis GN, Daoud YJ, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: a review of the literature. Ann Allergy Asthma Immunol. 2005;94:419-436.
10. Williams PM, Conklin RJ. Erythema multiforme: a review and contrast from Stevens-Johnson syndrome/toxic epidermal necrolysis. Dent Clin North Am. 2005;49:67-76, viii.
11. Nirken M, High W. Stevens-Johnson syndrome and toxic epidermal necrolysis: clinical manifestations, pathogenesis, and diagnosis. In: Ofori A, Levy M, Adkinson NF, eds. UpToDate [online database]. Version 18.2. Waltham, Mass: UpToDate; 2010.
12. French LE, Trent JT, Kerdel FA. Use of intravenous immunoglobulin in toxic epidermal necrolysis and Stevens-Johnson syndrome: our current understanding. Int Immunopharmacol. 2006;6:543-549.
13. Furubacke A, Berlin G, Anderson C, et al. Lack of significant treatment effect of plasma exchange in the treatment of drug-induced toxic epidermal necrolysis? Intensive Care Med. 1999;25:1307-1310.
Sudden onset of amnesia in a healthy woman
CASE A 63-year-old woman came to our emergency department with her fiancé following an abrupt onset of confusion that began 1 hour earlier. The patient had been working outside in the yard when she approached her fiancé, repeatedly asking where she was and what she was doing. She remained conscious of her identity, however, and exhibited no other neurologic symptoms, such as muscle weakness, gait imbalance, sensory loss, vision changes, slurred speech, or facial droop. The fiancé did not witness any loss of consciousness, head trauma, or seizure-like activity.
Before the event, the patient was feeling well, without any fever, headache, emesis, or vertigo. She denied using tobacco, alcohol, or illicit drugs. Her medical history was unremarkable, including an absence of diabetes, hypertension, and hyperlipidemia. The only significant finding in her family history was a stroke her mother experienced at an advanced age. During our interview, the patient remained confused about where she was and what was happening. She was aware of her confusion and distressed by it.
On examination, the patient was alert and oriented to self and year. She appeared appropriately anxious about her situation. She was afebrile and slightly hypertensive. Her other vital signs were normal. She could not recall events immediately preceding her arrival at the emergency department, but could recall events of the day before and earlier. There was no evidence of trauma. Head, neck, cardiovascular, lung, and abdominal exams were within normal limits.
Her neurologic exam revealed intact cranial nerves, symmetric face, 5/5 muscle strength in all extremities, intact sensation, and normal gait. Grossly, visual fields were intact. There was no Babinski sign, clonus, or pronator drift. She had 3/3 immediate recall of named objects, but 0/3 recall at 5 minutes. Results for complete blood count, basic metabolic panel, and urinalysis were within normal limits, including a blood glucose level of 77 mg/dL and a low-density lipoprotein level of 161 mg/dL. The result for cardiac enzymes was negative. Noncontrast computed tomography of the head revealed a remote pontine lacunar infarct.
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Transient global amnesia
We admitted the patient for further evaluation with a presumptive diagnosis of transient global amnesia (TGA).
With a chief complaint of amnesia, the differential diagnosis is broad (TABLE 1).1-3 In this case, a stroke was unlikely given the absence of neurologic deficits, specifically the lack of visual field defects. The elapsed time of her symptoms was too long for a transient ischemic attack or seizure. There was no supporting evidence for encephalitis, intracranial bleed, or hypoglycemia. While delirium could be considered, its characteristic features of inattention and a waxing and waning course were not present, nor was there any obvious underlying cause, such as infection or polypharmacy. The patient had no loss of self-identity that would suggest a psychogenic cause. The time course and the patient’s symptoms were congruent with the clinical criteria for TGA, and we confidently based our diagnosis on this.
TABLE 1
Rule out these disorders with acute anterograde amnesia1-3
| Transient ischemic attack |
| Delirium |
| Intoxication or alcohol/drug withdrawal |
| Concussion |
| Intracranial bleed |
| Complex partial seizures |
| Postictal state |
| Hypoglycemia |
| Encephalitis |
| Transient global amnesia |
| Psychogenic amnesia |
| Wernicke’s encephalopathy |
Type of memory loss as a clue to cause
Amnesia occurs when memory and learning in an alert person are impaired to a degree out of proportion to the person’s overall neurologic status. It may affect the formation of new memories (anterograde amnesia) or the recall of past memories (retrograde amnesia).
How memory works. Memory can be broken down into categories (TABLE 2).1 Explicit memory requires a conscious effort to recall. An example is episodic memory, in which memories are framed within a context, such as recalling what was served for dinner the night before. Its function is critical to creating new memories. Other forms of explicit memory are semantic memory—memorized facts that are independent of a context—and working memory, in which focused attention is used to manipulate information. Implicit memory operates subconsciously. The prime example is procedural memory, involving the ability to learn new skills and perform them without total concentration.
Memory function affected in TGA. In TGA, episodic memory—critical in the laying down of new memories—is most affected. Episodic memory relies heavily on the hippocampus to function correctly. When it dysfunctions, a person cannot consolidate and retain new information, thus resulting in anterograde amnesia.1
Retrograde amnesia generally requires dysfunction of the frontal lobe in addition to the temporal lobe.3 However, it may be present concurrently with anterograde amnesia when a lesion is isolated to the hippocampus; it is usually limited to more recent memories. That recent memories tend to be the more vulnerable is known as Ribot’s law. If retrograde amnesia is present, it usually resolves before anterograde amnesia.4
In TGA, procedural memory is unaffected. Thus, activities of daily living and instrumental activities of daily living remain intact—eg, the patient retains the necessary skills to drive a car.
TABLE 2
Categories of memory function1
Explicit memory: requiring conscious effort to recall information.
|
Implicit memory: recall is done subconsciously.
|
Most often the prognosis is good
TGA is an unusual manifestation of anterograde amnesia that is self-limited and tends not to recur.5 An episode typically lasts 1 to 8 hours.6 Although the disorder was first described in 1956, a set of clinical criteria (TABLE 3) was not defined until 1990.7 The highlights of these criteria are that self-identity is preserved and no evidence exists for neurologic deficit or seizure activity.6 The incidence of TGA is 3 to 10 in 100,000.5 TGA usually affects patients in their early 60s,2 and men and women are affected equally.
Interestingly, more than half of patients with TGA report a precipitating event, usually involving physical activity or a Valsalva maneuver.6 Classically, the patient repeatedly asks the same questions. The most common associated symptoms are headache, dizziness, and nausea.2,6
Generally, the patient’s prognosis is good, without long-term sequelae. Importantly, reassure patients and their families that there will be no memories of the event itself, as their memory-making ability was impaired.2
TABLE 3
Clinical criteria for transient global amnesia, as defined by Hodges and Warlow7
| Amnesia must be witnessed by another |
| Acute onset of anterograde amnesia |
| Patient is alert—no change in consciousness |
| No loss of personal identity |
| No focal neurologic deficits |
| No recent history of head trauma or seizure |
| Amnesia resolves in 24 hours |
If episodes do recur
A small subset of people may have recurrent episodes. Recurrence rates over a 5-year span have been reported as 3% to 26%; however, this range includes cases and studies recorded before the diagnostic criteria were developed in 1990.6 Although the clinical criteria for TGA can be helpful in diagnosing the disorder, there is no standardized workup because TGA has no clear etiology or known underlying mechanism. Many causal theories exist, however, and have evidence to support them.
Possible underlying conditions. One proposed explanation is ischemia of the hippocampus. This raises questions of whether vascular risk factors place people at higher risk.8 Recent studies have not confirmed this theory, and patients with diabetes, hypertension, or hyperlipidemia appear not to be at higher risk of TGA. Still, it is interesting that TGA is a disease affecting older adults and that evidence of small-vessel ischemia is often discovered incidentally.6,8
On the other hand, some experts take into account the high association of TGA with migraines documented in multiple studies, and therefore propose a spreading depression as the cause.5 Another hypothesis is a valvular insufficiency of the jugular veins that allows reflux, resulting in venous ischemia of the hippocampal area, especially during a Valsalva maneuver.9 Indeed, jugular valve insufficiency has been noted in up to two-thirds of TGA patients. However, if valvular insufficiency is truly the mechanism of disease, why do recurrence rates remain so low?10
MRI may be helpful. Given the many theories of TGA origin, several imaging mechanisms have been tried with mixed results: single photon emission computed tomography, magnetic resonance imaging (MRI) with diffusion-weighted imaging, and positron emission tomography.
The lack of reliable results makes it difficult to establish diagnostic criteria. Some generalized guidelines are as follows:
If there are any neurologic findings or concern about a transient ischemic attack or cerebrovascular accident, obtain an MRI. This should include diffusion-weighted imaging, which may reveal a transient lesion in the hippocampus.6 If the patient has recurrent episodes, or has episodes that last less than 1 hour, suspect the possibility of seizure and consider arranging for an electroencephalogram.4,6 Likewise, recurrence may also be due to a patent foramen ovale (PFO) causing paradoxical emboli and transient ischemia of the hippocampus. In 1 study, the rate of PFO in the TGA arm was 55%; it was 50% in those with recurrent episodes.11
- Order an MRI if your patient with a suspected case of TGA has any neurologic findings or if you are concerned about transient ischemic attack or cerebrovascular accident.
- If the patient has had recurrent episodes, or has episodes that last less than 1 hour, suspect the possibility of seizure and consider an electroencephalogram.
- Reassure TGA patients that there will be no memories of the event itself, as their memory-making ability was impaired, and that there are no long-term sequelae.
Our patient’s outcome
In the 24 hours after admission, the patient’s anterograde amnesia gradually resolved. She was able to remember the medical staff caring for her and retain orientation to her situation. However, she was unable to regain memories of the events immediately surrounding the onset of amnesia. During her hospitalization, the patient underwent a thorough work-up, including carotid artery Doppler ultrasound and echocardiogram with agitated saline (bubble study), both of which yielded normal results. Her MR angiography showed patent cerebral vessels. As mentioned, an MRI of the head showed a remote lacunar infarct of her left upper pons and nonspecific subcortical white matter disease was noted, consistent with chronic small vessel disease. The patient was discharged with reassurance, and she has done well.
CORRESPONDENCE
Chris Bernheisel, MD, director, Family Medicine Inpatient Service, The University of Cincinnati, 2123 Auburn Ave, Suite 340, Cincinnati, OH 45219; [email protected]
1. Budson AE, Price BH. Memory dysfunction. N Engl J Med. 2005;352:692-699.
2. Owen D, Paranandi B, Sivakumar R, et al. Classical diseases revisited: transient global amnesia. Postgrad Med J. 2007;83:236-239.
3. Kopelman MD. Disorders of memory. Brain. 2002;125:2152-2190.
4. Guillery-Girard B, Desgrandes B, Urban C, et al. The dynamic time course of memory recovery in transient global amnesia. J Neurol Neurosurg Psychiatry. 2004;75:1532-1540.
5. Pantoni L, Lamassa M, Inzitari D. Transient global amnesia: a review emphasizing pathogenic aspects. Acta Neurol Scand. 2000;102:275-283.
6. Quinette P, Guillery-Girard B, Dayan J, et al. What does transient global amnesia really mean? Review of the literature and thorough study of 142 cases. Brain. 2006;129:1640-1658.
7. Hodges JR, Warlow CP. Syndromes of transient amnesia: towards a classification. A study of 153 cases. J Neurol Neurosurg Psychiatry. 1990;53:834-843.
8. Sander K, Sander D. New insights into transient global amnesia: recent imaging and clinical findings. Lancet Neurol. 2005;4:437-444.
9. Menendez Gonzalez M, Rivera MM. Transient global amnesia: Increasing evidence of a venous etiology. Arch Neurol. 2006;63:1334-1335.
10. Bettermann K. Transient global amnesia: the continuing quest for a source. Arch Neurol. 2006;63:1336-1338.
11. Klotzsch C, Sliwka U, Berlit P, et al. An increased frequency of patent foramen ovale in patients with transient global amnesia. Arch Neurol. 1996;53:504-508.
CASE A 63-year-old woman came to our emergency department with her fiancé following an abrupt onset of confusion that began 1 hour earlier. The patient had been working outside in the yard when she approached her fiancé, repeatedly asking where she was and what she was doing. She remained conscious of her identity, however, and exhibited no other neurologic symptoms, such as muscle weakness, gait imbalance, sensory loss, vision changes, slurred speech, or facial droop. The fiancé did not witness any loss of consciousness, head trauma, or seizure-like activity.
Before the event, the patient was feeling well, without any fever, headache, emesis, or vertigo. She denied using tobacco, alcohol, or illicit drugs. Her medical history was unremarkable, including an absence of diabetes, hypertension, and hyperlipidemia. The only significant finding in her family history was a stroke her mother experienced at an advanced age. During our interview, the patient remained confused about where she was and what was happening. She was aware of her confusion and distressed by it.
On examination, the patient was alert and oriented to self and year. She appeared appropriately anxious about her situation. She was afebrile and slightly hypertensive. Her other vital signs were normal. She could not recall events immediately preceding her arrival at the emergency department, but could recall events of the day before and earlier. There was no evidence of trauma. Head, neck, cardiovascular, lung, and abdominal exams were within normal limits.
Her neurologic exam revealed intact cranial nerves, symmetric face, 5/5 muscle strength in all extremities, intact sensation, and normal gait. Grossly, visual fields were intact. There was no Babinski sign, clonus, or pronator drift. She had 3/3 immediate recall of named objects, but 0/3 recall at 5 minutes. Results for complete blood count, basic metabolic panel, and urinalysis were within normal limits, including a blood glucose level of 77 mg/dL and a low-density lipoprotein level of 161 mg/dL. The result for cardiac enzymes was negative. Noncontrast computed tomography of the head revealed a remote pontine lacunar infarct.
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Transient global amnesia
We admitted the patient for further evaluation with a presumptive diagnosis of transient global amnesia (TGA).
With a chief complaint of amnesia, the differential diagnosis is broad (TABLE 1).1-3 In this case, a stroke was unlikely given the absence of neurologic deficits, specifically the lack of visual field defects. The elapsed time of her symptoms was too long for a transient ischemic attack or seizure. There was no supporting evidence for encephalitis, intracranial bleed, or hypoglycemia. While delirium could be considered, its characteristic features of inattention and a waxing and waning course were not present, nor was there any obvious underlying cause, such as infection or polypharmacy. The patient had no loss of self-identity that would suggest a psychogenic cause. The time course and the patient’s symptoms were congruent with the clinical criteria for TGA, and we confidently based our diagnosis on this.
TABLE 1
Rule out these disorders with acute anterograde amnesia1-3
| Transient ischemic attack |
| Delirium |
| Intoxication or alcohol/drug withdrawal |
| Concussion |
| Intracranial bleed |
| Complex partial seizures |
| Postictal state |
| Hypoglycemia |
| Encephalitis |
| Transient global amnesia |
| Psychogenic amnesia |
| Wernicke’s encephalopathy |
Type of memory loss as a clue to cause
Amnesia occurs when memory and learning in an alert person are impaired to a degree out of proportion to the person’s overall neurologic status. It may affect the formation of new memories (anterograde amnesia) or the recall of past memories (retrograde amnesia).
How memory works. Memory can be broken down into categories (TABLE 2).1 Explicit memory requires a conscious effort to recall. An example is episodic memory, in which memories are framed within a context, such as recalling what was served for dinner the night before. Its function is critical to creating new memories. Other forms of explicit memory are semantic memory—memorized facts that are independent of a context—and working memory, in which focused attention is used to manipulate information. Implicit memory operates subconsciously. The prime example is procedural memory, involving the ability to learn new skills and perform them without total concentration.
Memory function affected in TGA. In TGA, episodic memory—critical in the laying down of new memories—is most affected. Episodic memory relies heavily on the hippocampus to function correctly. When it dysfunctions, a person cannot consolidate and retain new information, thus resulting in anterograde amnesia.1
Retrograde amnesia generally requires dysfunction of the frontal lobe in addition to the temporal lobe.3 However, it may be present concurrently with anterograde amnesia when a lesion is isolated to the hippocampus; it is usually limited to more recent memories. That recent memories tend to be the more vulnerable is known as Ribot’s law. If retrograde amnesia is present, it usually resolves before anterograde amnesia.4
In TGA, procedural memory is unaffected. Thus, activities of daily living and instrumental activities of daily living remain intact—eg, the patient retains the necessary skills to drive a car.
TABLE 2
Categories of memory function1
Explicit memory: requiring conscious effort to recall information.
|
Implicit memory: recall is done subconsciously.
|
Most often the prognosis is good
TGA is an unusual manifestation of anterograde amnesia that is self-limited and tends not to recur.5 An episode typically lasts 1 to 8 hours.6 Although the disorder was first described in 1956, a set of clinical criteria (TABLE 3) was not defined until 1990.7 The highlights of these criteria are that self-identity is preserved and no evidence exists for neurologic deficit or seizure activity.6 The incidence of TGA is 3 to 10 in 100,000.5 TGA usually affects patients in their early 60s,2 and men and women are affected equally.
Interestingly, more than half of patients with TGA report a precipitating event, usually involving physical activity or a Valsalva maneuver.6 Classically, the patient repeatedly asks the same questions. The most common associated symptoms are headache, dizziness, and nausea.2,6
Generally, the patient’s prognosis is good, without long-term sequelae. Importantly, reassure patients and their families that there will be no memories of the event itself, as their memory-making ability was impaired.2
TABLE 3
Clinical criteria for transient global amnesia, as defined by Hodges and Warlow7
| Amnesia must be witnessed by another |
| Acute onset of anterograde amnesia |
| Patient is alert—no change in consciousness |
| No loss of personal identity |
| No focal neurologic deficits |
| No recent history of head trauma or seizure |
| Amnesia resolves in 24 hours |
If episodes do recur
A small subset of people may have recurrent episodes. Recurrence rates over a 5-year span have been reported as 3% to 26%; however, this range includes cases and studies recorded before the diagnostic criteria were developed in 1990.6 Although the clinical criteria for TGA can be helpful in diagnosing the disorder, there is no standardized workup because TGA has no clear etiology or known underlying mechanism. Many causal theories exist, however, and have evidence to support them.
Possible underlying conditions. One proposed explanation is ischemia of the hippocampus. This raises questions of whether vascular risk factors place people at higher risk.8 Recent studies have not confirmed this theory, and patients with diabetes, hypertension, or hyperlipidemia appear not to be at higher risk of TGA. Still, it is interesting that TGA is a disease affecting older adults and that evidence of small-vessel ischemia is often discovered incidentally.6,8
On the other hand, some experts take into account the high association of TGA with migraines documented in multiple studies, and therefore propose a spreading depression as the cause.5 Another hypothesis is a valvular insufficiency of the jugular veins that allows reflux, resulting in venous ischemia of the hippocampal area, especially during a Valsalva maneuver.9 Indeed, jugular valve insufficiency has been noted in up to two-thirds of TGA patients. However, if valvular insufficiency is truly the mechanism of disease, why do recurrence rates remain so low?10
MRI may be helpful. Given the many theories of TGA origin, several imaging mechanisms have been tried with mixed results: single photon emission computed tomography, magnetic resonance imaging (MRI) with diffusion-weighted imaging, and positron emission tomography.
The lack of reliable results makes it difficult to establish diagnostic criteria. Some generalized guidelines are as follows:
If there are any neurologic findings or concern about a transient ischemic attack or cerebrovascular accident, obtain an MRI. This should include diffusion-weighted imaging, which may reveal a transient lesion in the hippocampus.6 If the patient has recurrent episodes, or has episodes that last less than 1 hour, suspect the possibility of seizure and consider arranging for an electroencephalogram.4,6 Likewise, recurrence may also be due to a patent foramen ovale (PFO) causing paradoxical emboli and transient ischemia of the hippocampus. In 1 study, the rate of PFO in the TGA arm was 55%; it was 50% in those with recurrent episodes.11
- Order an MRI if your patient with a suspected case of TGA has any neurologic findings or if you are concerned about transient ischemic attack or cerebrovascular accident.
- If the patient has had recurrent episodes, or has episodes that last less than 1 hour, suspect the possibility of seizure and consider an electroencephalogram.
- Reassure TGA patients that there will be no memories of the event itself, as their memory-making ability was impaired, and that there are no long-term sequelae.
Our patient’s outcome
In the 24 hours after admission, the patient’s anterograde amnesia gradually resolved. She was able to remember the medical staff caring for her and retain orientation to her situation. However, she was unable to regain memories of the events immediately surrounding the onset of amnesia. During her hospitalization, the patient underwent a thorough work-up, including carotid artery Doppler ultrasound and echocardiogram with agitated saline (bubble study), both of which yielded normal results. Her MR angiography showed patent cerebral vessels. As mentioned, an MRI of the head showed a remote lacunar infarct of her left upper pons and nonspecific subcortical white matter disease was noted, consistent with chronic small vessel disease. The patient was discharged with reassurance, and she has done well.
CORRESPONDENCE
Chris Bernheisel, MD, director, Family Medicine Inpatient Service, The University of Cincinnati, 2123 Auburn Ave, Suite 340, Cincinnati, OH 45219; [email protected]
CASE A 63-year-old woman came to our emergency department with her fiancé following an abrupt onset of confusion that began 1 hour earlier. The patient had been working outside in the yard when she approached her fiancé, repeatedly asking where she was and what she was doing. She remained conscious of her identity, however, and exhibited no other neurologic symptoms, such as muscle weakness, gait imbalance, sensory loss, vision changes, slurred speech, or facial droop. The fiancé did not witness any loss of consciousness, head trauma, or seizure-like activity.
Before the event, the patient was feeling well, without any fever, headache, emesis, or vertigo. She denied using tobacco, alcohol, or illicit drugs. Her medical history was unremarkable, including an absence of diabetes, hypertension, and hyperlipidemia. The only significant finding in her family history was a stroke her mother experienced at an advanced age. During our interview, the patient remained confused about where she was and what was happening. She was aware of her confusion and distressed by it.
On examination, the patient was alert and oriented to self and year. She appeared appropriately anxious about her situation. She was afebrile and slightly hypertensive. Her other vital signs were normal. She could not recall events immediately preceding her arrival at the emergency department, but could recall events of the day before and earlier. There was no evidence of trauma. Head, neck, cardiovascular, lung, and abdominal exams were within normal limits.
Her neurologic exam revealed intact cranial nerves, symmetric face, 5/5 muscle strength in all extremities, intact sensation, and normal gait. Grossly, visual fields were intact. There was no Babinski sign, clonus, or pronator drift. She had 3/3 immediate recall of named objects, but 0/3 recall at 5 minutes. Results for complete blood count, basic metabolic panel, and urinalysis were within normal limits, including a blood glucose level of 77 mg/dL and a low-density lipoprotein level of 161 mg/dL. The result for cardiac enzymes was negative. Noncontrast computed tomography of the head revealed a remote pontine lacunar infarct.
WHAT IS THE MOST LIKELY EXPLANATION FOR HER CONDITION?
Transient global amnesia
We admitted the patient for further evaluation with a presumptive diagnosis of transient global amnesia (TGA).
With a chief complaint of amnesia, the differential diagnosis is broad (TABLE 1).1-3 In this case, a stroke was unlikely given the absence of neurologic deficits, specifically the lack of visual field defects. The elapsed time of her symptoms was too long for a transient ischemic attack or seizure. There was no supporting evidence for encephalitis, intracranial bleed, or hypoglycemia. While delirium could be considered, its characteristic features of inattention and a waxing and waning course were not present, nor was there any obvious underlying cause, such as infection or polypharmacy. The patient had no loss of self-identity that would suggest a psychogenic cause. The time course and the patient’s symptoms were congruent with the clinical criteria for TGA, and we confidently based our diagnosis on this.
TABLE 1
Rule out these disorders with acute anterograde amnesia1-3
| Transient ischemic attack |
| Delirium |
| Intoxication or alcohol/drug withdrawal |
| Concussion |
| Intracranial bleed |
| Complex partial seizures |
| Postictal state |
| Hypoglycemia |
| Encephalitis |
| Transient global amnesia |
| Psychogenic amnesia |
| Wernicke’s encephalopathy |
Type of memory loss as a clue to cause
Amnesia occurs when memory and learning in an alert person are impaired to a degree out of proportion to the person’s overall neurologic status. It may affect the formation of new memories (anterograde amnesia) or the recall of past memories (retrograde amnesia).
How memory works. Memory can be broken down into categories (TABLE 2).1 Explicit memory requires a conscious effort to recall. An example is episodic memory, in which memories are framed within a context, such as recalling what was served for dinner the night before. Its function is critical to creating new memories. Other forms of explicit memory are semantic memory—memorized facts that are independent of a context—and working memory, in which focused attention is used to manipulate information. Implicit memory operates subconsciously. The prime example is procedural memory, involving the ability to learn new skills and perform them without total concentration.
Memory function affected in TGA. In TGA, episodic memory—critical in the laying down of new memories—is most affected. Episodic memory relies heavily on the hippocampus to function correctly. When it dysfunctions, a person cannot consolidate and retain new information, thus resulting in anterograde amnesia.1
Retrograde amnesia generally requires dysfunction of the frontal lobe in addition to the temporal lobe.3 However, it may be present concurrently with anterograde amnesia when a lesion is isolated to the hippocampus; it is usually limited to more recent memories. That recent memories tend to be the more vulnerable is known as Ribot’s law. If retrograde amnesia is present, it usually resolves before anterograde amnesia.4
In TGA, procedural memory is unaffected. Thus, activities of daily living and instrumental activities of daily living remain intact—eg, the patient retains the necessary skills to drive a car.
TABLE 2
Categories of memory function1
Explicit memory: requiring conscious effort to recall information.
|
Implicit memory: recall is done subconsciously.
|
Most often the prognosis is good
TGA is an unusual manifestation of anterograde amnesia that is self-limited and tends not to recur.5 An episode typically lasts 1 to 8 hours.6 Although the disorder was first described in 1956, a set of clinical criteria (TABLE 3) was not defined until 1990.7 The highlights of these criteria are that self-identity is preserved and no evidence exists for neurologic deficit or seizure activity.6 The incidence of TGA is 3 to 10 in 100,000.5 TGA usually affects patients in their early 60s,2 and men and women are affected equally.
Interestingly, more than half of patients with TGA report a precipitating event, usually involving physical activity or a Valsalva maneuver.6 Classically, the patient repeatedly asks the same questions. The most common associated symptoms are headache, dizziness, and nausea.2,6
Generally, the patient’s prognosis is good, without long-term sequelae. Importantly, reassure patients and their families that there will be no memories of the event itself, as their memory-making ability was impaired.2
TABLE 3
Clinical criteria for transient global amnesia, as defined by Hodges and Warlow7
| Amnesia must be witnessed by another |
| Acute onset of anterograde amnesia |
| Patient is alert—no change in consciousness |
| No loss of personal identity |
| No focal neurologic deficits |
| No recent history of head trauma or seizure |
| Amnesia resolves in 24 hours |
If episodes do recur
A small subset of people may have recurrent episodes. Recurrence rates over a 5-year span have been reported as 3% to 26%; however, this range includes cases and studies recorded before the diagnostic criteria were developed in 1990.6 Although the clinical criteria for TGA can be helpful in diagnosing the disorder, there is no standardized workup because TGA has no clear etiology or known underlying mechanism. Many causal theories exist, however, and have evidence to support them.
Possible underlying conditions. One proposed explanation is ischemia of the hippocampus. This raises questions of whether vascular risk factors place people at higher risk.8 Recent studies have not confirmed this theory, and patients with diabetes, hypertension, or hyperlipidemia appear not to be at higher risk of TGA. Still, it is interesting that TGA is a disease affecting older adults and that evidence of small-vessel ischemia is often discovered incidentally.6,8
On the other hand, some experts take into account the high association of TGA with migraines documented in multiple studies, and therefore propose a spreading depression as the cause.5 Another hypothesis is a valvular insufficiency of the jugular veins that allows reflux, resulting in venous ischemia of the hippocampal area, especially during a Valsalva maneuver.9 Indeed, jugular valve insufficiency has been noted in up to two-thirds of TGA patients. However, if valvular insufficiency is truly the mechanism of disease, why do recurrence rates remain so low?10
MRI may be helpful. Given the many theories of TGA origin, several imaging mechanisms have been tried with mixed results: single photon emission computed tomography, magnetic resonance imaging (MRI) with diffusion-weighted imaging, and positron emission tomography.
The lack of reliable results makes it difficult to establish diagnostic criteria. Some generalized guidelines are as follows:
If there are any neurologic findings or concern about a transient ischemic attack or cerebrovascular accident, obtain an MRI. This should include diffusion-weighted imaging, which may reveal a transient lesion in the hippocampus.6 If the patient has recurrent episodes, or has episodes that last less than 1 hour, suspect the possibility of seizure and consider arranging for an electroencephalogram.4,6 Likewise, recurrence may also be due to a patent foramen ovale (PFO) causing paradoxical emboli and transient ischemia of the hippocampus. In 1 study, the rate of PFO in the TGA arm was 55%; it was 50% in those with recurrent episodes.11
- Order an MRI if your patient with a suspected case of TGA has any neurologic findings or if you are concerned about transient ischemic attack or cerebrovascular accident.
- If the patient has had recurrent episodes, or has episodes that last less than 1 hour, suspect the possibility of seizure and consider an electroencephalogram.
- Reassure TGA patients that there will be no memories of the event itself, as their memory-making ability was impaired, and that there are no long-term sequelae.
Our patient’s outcome
In the 24 hours after admission, the patient’s anterograde amnesia gradually resolved. She was able to remember the medical staff caring for her and retain orientation to her situation. However, she was unable to regain memories of the events immediately surrounding the onset of amnesia. During her hospitalization, the patient underwent a thorough work-up, including carotid artery Doppler ultrasound and echocardiogram with agitated saline (bubble study), both of which yielded normal results. Her MR angiography showed patent cerebral vessels. As mentioned, an MRI of the head showed a remote lacunar infarct of her left upper pons and nonspecific subcortical white matter disease was noted, consistent with chronic small vessel disease. The patient was discharged with reassurance, and she has done well.
CORRESPONDENCE
Chris Bernheisel, MD, director, Family Medicine Inpatient Service, The University of Cincinnati, 2123 Auburn Ave, Suite 340, Cincinnati, OH 45219; [email protected]
1. Budson AE, Price BH. Memory dysfunction. N Engl J Med. 2005;352:692-699.
2. Owen D, Paranandi B, Sivakumar R, et al. Classical diseases revisited: transient global amnesia. Postgrad Med J. 2007;83:236-239.
3. Kopelman MD. Disorders of memory. Brain. 2002;125:2152-2190.
4. Guillery-Girard B, Desgrandes B, Urban C, et al. The dynamic time course of memory recovery in transient global amnesia. J Neurol Neurosurg Psychiatry. 2004;75:1532-1540.
5. Pantoni L, Lamassa M, Inzitari D. Transient global amnesia: a review emphasizing pathogenic aspects. Acta Neurol Scand. 2000;102:275-283.
6. Quinette P, Guillery-Girard B, Dayan J, et al. What does transient global amnesia really mean? Review of the literature and thorough study of 142 cases. Brain. 2006;129:1640-1658.
7. Hodges JR, Warlow CP. Syndromes of transient amnesia: towards a classification. A study of 153 cases. J Neurol Neurosurg Psychiatry. 1990;53:834-843.
8. Sander K, Sander D. New insights into transient global amnesia: recent imaging and clinical findings. Lancet Neurol. 2005;4:437-444.
9. Menendez Gonzalez M, Rivera MM. Transient global amnesia: Increasing evidence of a venous etiology. Arch Neurol. 2006;63:1334-1335.
10. Bettermann K. Transient global amnesia: the continuing quest for a source. Arch Neurol. 2006;63:1336-1338.
11. Klotzsch C, Sliwka U, Berlit P, et al. An increased frequency of patent foramen ovale in patients with transient global amnesia. Arch Neurol. 1996;53:504-508.
1. Budson AE, Price BH. Memory dysfunction. N Engl J Med. 2005;352:692-699.
2. Owen D, Paranandi B, Sivakumar R, et al. Classical diseases revisited: transient global amnesia. Postgrad Med J. 2007;83:236-239.
3. Kopelman MD. Disorders of memory. Brain. 2002;125:2152-2190.
4. Guillery-Girard B, Desgrandes B, Urban C, et al. The dynamic time course of memory recovery in transient global amnesia. J Neurol Neurosurg Psychiatry. 2004;75:1532-1540.
5. Pantoni L, Lamassa M, Inzitari D. Transient global amnesia: a review emphasizing pathogenic aspects. Acta Neurol Scand. 2000;102:275-283.
6. Quinette P, Guillery-Girard B, Dayan J, et al. What does transient global amnesia really mean? Review of the literature and thorough study of 142 cases. Brain. 2006;129:1640-1658.
7. Hodges JR, Warlow CP. Syndromes of transient amnesia: towards a classification. A study of 153 cases. J Neurol Neurosurg Psychiatry. 1990;53:834-843.
8. Sander K, Sander D. New insights into transient global amnesia: recent imaging and clinical findings. Lancet Neurol. 2005;4:437-444.
9. Menendez Gonzalez M, Rivera MM. Transient global amnesia: Increasing evidence of a venous etiology. Arch Neurol. 2006;63:1334-1335.
10. Bettermann K. Transient global amnesia: the continuing quest for a source. Arch Neurol. 2006;63:1336-1338.
11. Klotzsch C, Sliwka U, Berlit P, et al. An increased frequency of patent foramen ovale in patients with transient global amnesia. Arch Neurol. 1996;53:504-508.